WO1991017161A1 - Isoquinoline amides and esters as 5 ht3 receptor antagonists - Google Patents
Isoquinoline amides and esters as 5 ht3 receptor antagonists Download PDFInfo
- Publication number
- WO1991017161A1 WO1991017161A1 PCT/GB1991/000636 GB9100636W WO9117161A1 WO 1991017161 A1 WO1991017161 A1 WO 1991017161A1 GB 9100636 W GB9100636 W GB 9100636W WO 9117161 A1 WO9117161 A1 WO 9117161A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound according
- formula
- hydrogen
- alkyl
- compound
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/14—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing 9-azabicyclo [3.3.1] nonane ring systems, e.g. granatane, 2-aza-adamantane; Cyclic acetals thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
Definitions
- This invention relates to novel compounds having useful pharmacological properties, to pharmaceutical compositions containing them, to a process and intermediates for their preparation, and to their use as pharmaceuticals.
- GB 2145416A (Sandoz Ltd) describes a group of naphthylene, chromene and quinoline derivatives with saturated
- the present invention provides a compound of formula (I), or a pharmaceutically acceptable salt thereof:
- E is NH or O
- R 1 is hydrogen, halogen, C 1 -4 alkyl, C 1-4 alkoxy, hydroxy or nitro;
- Z is an azacyclic or azabicyclic side chain, such as a group of formula (a), (b) or (c):
- R 3 or R 4 is hydrogen or C 1-4 alkyl, and Y is a group
- R 2 is in the 3-position and is hydrogen, C 1 -6 alkyl or C 1 -6 alkoxy, or R 2 is in the 4-position and is hydrogen, halogen, CF 3 , C 1 -6 alkyl, C 1 -7 acyl, C 1 -7 acylamino, phenyl optionally substituted by one or two C 1 -6 alkyl, C 1 -6 alkoxy or halogen groups, or amino, aminocarbonyl or aminosulphonyl, optionally
- Suitable examples of the group R 1 include hydrogen 7 bromo, chloro, methyl, ethyl, n- and iso-propyl, n-, iso-, sec- and tert-butyl, methoxy, ethoxy, n- and iso-propoxy, and n-, iso-, sec- and tert-butoxy.
- Suitable examples of Z are described in the art relating to 5-HT 3 receptor antagonists, ie. as follows: i) GB 2125398A (Sandoz Limited)
- EP-A-215545 (Beecham Group p. I.e.)
- EP-A-214772 (Beecham Group p. I.e.)
- R is hydrogen or methyl; and X is oxygen or sulphur.
- Side chains Z of particular interest include tropane and oxagranatane, where R is methyl.
- E is preferably NH.
- suitable examples of the group R 2 when in the 4-position include the
- alkanoylamino such as formylamino, acetylamino,
- propionylamino, n- and iso-butyrylamino, aminosulphonyl, and amino and aminosulphonyl optionally substituted by one or two methyl, ethyl, n- or iso-propyl, n-, sec-, iso- or
- tert-butyl or phenyl groups nitro, methoxy, ethoxy, n- and iso-propoxy, methylthio, ethylthio, n- and iso-propylthio, hydroxy, methylsulphonyl and ethylsulphonyl or when R 2 is in the 3-position suitable examples, include the following groups, hydrogen, methyl, ethyl, n- or iso-propyl, methoxy, and ethoxy.
- examples of the group R 2 when in the 1-position include the groups hydrogen, methyl, ethyl, n- or iso- propyl, methoxy and ethoxy, or when R 2 is in the 4-position, suitable examples include the following groups; hydrogen, methoxy and ethoxy.
- R 2 groups in any of the positions specified above, include hydrogen, methyl and methoxy.
- R 3 /R 4 when alkyl are methyl, ethyl, n- and
- p, q and r are 1 or 2.
- the pharmaceutically acceptable salts of the compounds of the formula (I) include acid addition salts with
- the pharmaceutically acceptable salts of the compounds of the formula (I) are usually acid addition salts with acids such as hydrochloric, hydrobromic, phosphoric, sulphuric, citric, tartaric, lactic and acetic acid.
- Examples of pharmaceutically acceptable salts include quaternary derivatives of the compounds of formula (I) such as the compounds quaternised by compounds R a -T wherein R a is C 1 -6 alkyl, phenyl-C 1 -6 alkyl or C 5 -7 cycloalkyl, and T is a radical corresponding to an anion of an acid.
- R a include methyl, ethyl and n- and iso-propyl; and benzyl and phenethyl, preferably methyl.
- Suitable examples of T include halide such as chloride, bromide and iodide.
- Examples of pharmaceutically acceptable salts of compounds of formula (I) also include internal salts such as
- N-oxides pharmaceutically acceptable N-oxides.
- the compounds of the formula (I), their pharmaceutically acceptable salts, (including quaternary derivatives and N-oxides) may also form pharmaceutically acceptable
- a group of compounds within formula (I) is of formula (II):
- a further group of compounds within formula (I) is of formula (III):
- the invention also provides a process for the preparation of a compound of formula (I) which process comprises reacting a compound of formula (V):
- a 1 and A 2 are, for example, as described in the aforementioned patent publications.
- a 1 may be an actived carbonyl function such as an acid chloride or N-hydroxysuccinmide ester and A 2 may be an amino group, when E in formula (I) is NH.
- Intermediates of the formula (V) are generally known or are prepared by analogous methods to those used for structurally related known compounds.
- Intermediates of formula A 2 -Z' may be prepared from the corresponding exocyclic keto derivative of the azabicyclic side chain, prepared by condensation methods, often using a substituted piperidine, as described in the aforementioned patent references.
- the invention also provides a process for the preparation of a compound of formula (I), or a pharmaceutically acceptable salt thereof, which process comprises reacting a compound of formula (VI):
- R 3 ' and R 4 ' respectively is R 3 and R 4
- Examples of leaving groups Q, displaceable by a nucleophile include halogen such as chloro and bromo, C 1-4 alkoxy, such as CH 3 O and C 2 H 5 O-, PhO-, or activated hydrocarbyloxy, such as Cl 5 C 6 O- or COQ forms a mixed anhydride, so that Q is carboxylic acyloxy.
- halogen such as chloro and bromo
- C 1-4 alkoxy such as CH 3 O and C 2 H 5 O-, PhO-
- activated hydrocarbyloxy such as Cl 5 C 6 O- or COQ forms a mixed anhydride, so that Q is carboxylic acyloxy.
- a group Q is a halide or COQ forms a mixed anhydride
- the reaction is preferably carried out at non-extreme temperatures in an inert non-hydroxylic solvent, such as benzene, dichloromethane, toluene, diethyl ether,
- tetrahydrofuran THF
- DMF dimethylformamide
- an acid acceptor such as an organic base, in particular a tertiary amine, such as triethylamine, trimethylamine, pyridine or picoline, some of which can also function as the solvent.
- the acid acceptor can be inorganic, such as calcium carbonate, sodium carbonate or potassium carbonate.
- reaction is preferably carried out in an inert polar solvent, such as toluene or
- a group Q is hydroxy
- the reaction is generally carried out in an inert non-hydroxylic solvent, such as dichloromethane, THF or DMF optionally in the presence of a dehydrating agent such as a carbodiimide, for example dicyclohexylcarbodiimide, optionally in the presence of N- hydroxysuccinimide.
- a dehydrating agent such as a carbodiimide, for example dicyclohexylcarbodiimide, optionally in the presence of N- hydroxysuccinimide.
- the reaction may be carried out at any non-extreme temperature, such as -10 to 100°C, for example, 0 to 80°C. Generally, higher reaction temperatures are employed with less active compounds whereas lower
- acyloxy leaving groups include C 1-4 alkanoyloxy and C 1 -4
- alkoxycarbonyloxy in which case the reaction is preferably carried out in an inert solvent, such as dichloromethane, at a non-extreme temperature for example ambient temperatures in the presence of an acid acceptor, such as triethylamine.
- C 1 -4 alkoxycarbonyloxy leaving groups may be generated in situ by treatment of the corresponding compound wherein Q is hydroxy with a C 1-4 alkyl chloroformate.
- a group Q is activated hydrocarbyloxy then the reaction is preferably carried out in an inert polar solvent, such as dimethylformamide. It is also preferred that the activated hydrocarbyloxy group is a pentachlorophenyl ester and that the reaction is carried out at ambient temperature.
- R 3 ' and R 4 ' when other than R 3 and R 4 respectively, may be a hydrogenolysable protecting group which is benzyl optionally substituted by one or two groups selected from halo, C 1-4 alkoxy and C 1 -4 alkyl. Such benzyl groups may, for example, be removed, by conventional transition metal catalysed hydrogenolysis to give compounds of the formula (VII) or (VIII) respectively:
- This invention also provides a further process for the preparation of a compound of the formula (I) wherein Z is a) or c) or a pharmaceutically acceptable salt thereof, which comprises N-alkylating a compound of formula (VII) or (VIII) respectively, and optionally forming a pharmaceutically acceptable salt of the resulting compound of the formula (I).
- N-alkylation' comprises the substitution of the N-atom depicted in formula (VII) or (VIII) respectively, by a group R 3 or R 4
- Suitable values for Q 3 include groups displaced by
- nucleophiles such as Cl, Br, I, OSO 2 CH 3 or OSO 2 C 6 H 4 pCH 3 .
- Favoured values for Q 3 include Cl, Br and I.
- the reaction may be carried out under conventional
- alkylation conditions for example in an inert solvent such as dimethylformamide in the presence of an acid acceptor such as potassium carbonate.
- an acid acceptor such as potassium carbonate.
- the reaction is carried out at non-extreme temperature such as at ambient or slightly above.
- 'N-alkylation' may be effected under
- the compounds of formula (VI) are known or are preparable analogously to, or routinely from, known isoquinoline compounds.
- the -COE- linkage has an endo orientation with respect to the ring of the bicyclic moiety to which it is attached.
- a mixture of endo and exo isomers of the compound of the formula (I) may be synthesised non-stereospecifically and the desired isomer separated conventionally therefrom e.g. by chromatography; or alternatively the endo isomer may if desired by synthesised from the corresponding endo form of the compound of the formula (II).
- isoquinuclidine isogranatane, oxa/thia-isogranatane and isotropane side chains.
- compositions of this invention may be formed conventionally.
- the acid addition salts may be formed for example by reaction of the base compound of formula (I) with a pharmaceutically acceptable organic or inorganic acid.
- the compounds of the present invention are 5-HT 3 receptor antagonists and it is thus believed may generally be used in the treatment or prophylaxis of pain, emesis, CNS disorders and gastrointestinal disorders. Pain includes migraine, cluster headache, trigeminal neuralgia and visceral pain; emesis, includes in particular that of preventing vomiting and nausea associated with cancer therapy, and motion sickness. Examples of such cancer therapy include that using cytotoxic agents, such as cisplatin, doxorubicin and cyclophosphamide, particularly cisplatin; and also radiation treatment.
- CNS disorders include anxiety, psychosis, senile dementia and drug dependence.
- Gastrointestinal disorders include irritable bowel syndrome and diarrohea.
- 5-HT 3 receptor antagonists may also be of potential use in the treatment of obesity and/or arrhythmia.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- compositions are prepared by admixture and are suitably adapted for oral or parenteral administration, and as such may be in the form of tablets, capsules, oral liquid
- compositions preparations, powders, granules, lozenges, reconstitutable powders, injectable and infusable solutions or suspensions or suppositories.
- Orally administrable compositions are preferred, since they are more convenient for general use.
- Tablets and capsules for oral administration are usually presented in a unit dose, and contain conventional
- excipients such as binding agents, fillers, diluents, tabletting agents, lubricants, disintegrants, colourants, flavourings, and wetting agents.
- the tablets may be coated according to well known methods in the art, for example with an enteric coating.
- Suitable fillers for use include cellulose, mannitol, lactose and other similar agents.
- Suitable disintegrants include starch, polyvinylpolypyrrolidone and starch
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups, or elixirs, or may be
- Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose, carboxymethylcellulose, aluminium stearate gel or hydrogenated edible fats,
- suspending agents for example sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose, carboxymethylcellulose, aluminium stearate gel or hydrogenated edible fats,
- emulsifying agents for example lecithin, sorbitan
- non-aqueous vehicles which may include edible oils, for example, almond oil, fractionated coconut oil, oily esters such as esters of glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and if desired conventional flavouring or colouring agents.
- Oral liquid preparations are usually in the form of aqueous or oily suspensions, solutions, emulsions, syrups, or elixirs or are presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, emulsifying agents, non-aqueous vehicles (which may include edible oils), preservatives, and flavouring or colouring agents.
- the oral compositions may be prepared by conventional methods of blending, filling or tabletting. Repeated blending operations may be used to distribute the active agent throughout those compositions employing large quantities of fillers. Such operations are, of course, conventional in the art.
- fluid unit dose forms are prepared containing a compound of the present invention and a sterile vehicle.
- the compound depending on the vehicle and the concentration, can be either suspended or dissolved.
- Parenteral solutions are normally prepared by dissolving the compound in a vehicle and filter sterilising before filling into a suitable vial or ampoule and sealing.
- adjuvants such as a local anaesthetic, preservatives and buffering agents are also dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum.
- Parenteral suspensions are prepared in substantially the same manner except that the compound is suspended in the vehicle instead of being dissolved and sterilised by
- a surfactant or wetting agent is included in the composition to facilitate uniform
- the invention further provides a method of treatment or prophylaxis of pain, emesis, CNS disorders and/or
- gastrointestinal disorders in mammals such as humans, which comprises the administration of an effective amount of a compound of the formula (I) or a pharmaceutically acceptable salt thereof.
- a unit dose for a 70kg adult will normally contain 0.05 to 1000mg for example 0.1 to 500mg, of the compound of the invention.
- Unit doses may be administered once or more than once a day, for example, 2, 3 or 4 times a day, more usually 1 to 3 times a day, that is in the range of
- the invention also provides a pharmaceutical composition for use in the treatment and/or prophylaxis of pain, emesis, CNS disorders and/or gastrointestinal disorders which
- composition comprises an effect non-toxic amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof and pharmaceutically acceptable carrier.
- the invention also provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use as an active therapeutic substance, in particular for use in the treatment of pain, emesis, CNS disorders and/or
- the invention further provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for the treatment and/or prophylaxis of pain, emesis, CNS disorders and/or
- Compounds are given intravenously and the concentration required to reduce the 5-HT-evoked response to 50% of the control response (ED 50 ) is then determined.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description







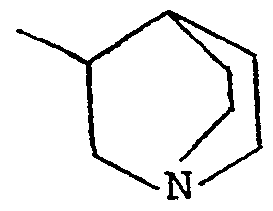












Claims


Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP91508211A JPH05507071A (en) | 1990-04-27 | 1991-04-22 | new compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9009542.3 | 1990-04-27 | ||
| GB909009542A GB9009542D0 (en) | 1990-04-27 | 1990-04-27 | Novel compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1991017161A1 true WO1991017161A1 (en) | 1991-11-14 |
Family
ID=10675130
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB1991/000636 WO1991017161A1 (en) | 1990-04-27 | 1991-04-22 | Isoquinoline amides and esters as 5 ht3 receptor antagonists |
Country Status (11)
| Country | Link |
|---|---|
| EP (1) | EP0526545A1 (en) |
| JP (1) | JPH05507071A (en) |
| AU (1) | AU7753991A (en) |
| CA (1) | CA2081350A1 (en) |
| GB (1) | GB9009542D0 (en) |
| IE (1) | IE911399A1 (en) |
| NZ (1) | NZ237957A (en) |
| PT (1) | PT97490A (en) |
| TW (1) | TW199157B (en) |
| WO (1) | WO1991017161A1 (en) |
| ZA (1) | ZA913123B (en) |
Cited By (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0560604A1 (en) * | 1992-03-12 | 1993-09-15 | Mitsubishi Chemical Corporation | Cinnoline-3-carboxylic acid derivatives as 5-HT3 antagonists |
| EP1018512A1 (en) * | 1998-10-13 | 2000-07-12 | Rotta Research Laboratorium S.P.A. | Novel basic derivatives of benz(e)isoindol-1-ones and pyrrolo(3,4-c)quinolin-1-ones with 5-HT3-antagonistic activity, their preparation and their therapeutic use |
| WO2003043991A1 (en) * | 2001-11-19 | 2003-05-30 | Bayer Healthcare Ag | Heteroaryl carboxylic acid amides |
| WO2003072578A1 (en) * | 2002-02-20 | 2003-09-04 | Pharmacia & Upjohn Company | Azabicyclic compounds with alfa7 nicotinic acetylcholine receptor activity |
| US6828330B2 (en) | 2001-06-12 | 2004-12-07 | Pharmacia & Upjohn Company | Quinuclidine-substituted hetero-bicyclic aromatic compounds for the treatment of disease |
| US6849620B2 (en) | 2001-10-26 | 2005-02-01 | Pfizer Inc | N-(azabicyclo moieties)-substituted hetero-bicyclic aromatic compounds for the treatment of disease |
| US6858613B2 (en) | 2002-02-19 | 2005-02-22 | Pfizer Inc. | Fused bicyclic-N-bridged-heteroaromatic carboxamides for the treatment of disease |
| US6894042B2 (en) | 2002-02-19 | 2005-05-17 | Pharmacia & Upjohn Company | Azabicyclic compounds for the treatment of disease |
| US6911543B2 (en) | 2001-10-02 | 2005-06-28 | Pfizer Inc. | Azabicyclic-substituted fused-heteroaryl compounds for the treatment of disease |
| US7067515B2 (en) | 2001-06-12 | 2006-06-27 | Pfizer Inc. | Quinuclidines-substituted-multi-cyclic-heteroaryls for the treatment of disease |
| US7683075B2 (en) | 2003-05-12 | 2010-03-23 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US8642604B2 (en) | 2006-04-04 | 2014-02-04 | The Regents Of The University Of California | Substituted pyrazolo[3,2-d]pyrimidines as anti-cancer agents |
| US8697709B2 (en) | 2008-10-16 | 2014-04-15 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8703777B2 (en) | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8703778B2 (en) | 2008-09-26 | 2014-04-22 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US8785454B2 (en) | 2009-05-07 | 2014-07-22 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8809349B2 (en) | 2011-01-10 | 2014-08-19 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9056877B2 (en) | 2011-07-19 | 2015-06-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US9321772B2 (en) | 2011-09-02 | 2016-04-26 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| US9629843B2 (en) | 2008-07-08 | 2017-04-25 | The Regents Of The University Of California | MTOR modulators and uses thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10131668B2 (en) | 2012-09-26 | 2018-11-20 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pYRAZINES for modulation of IRE1 |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11110096B2 (en) | 2014-04-16 | 2021-09-07 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11147818B2 (en) | 2016-06-24 | 2021-10-19 | Infinity Pharmaceuticals, Inc. | Combination therapies |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0041817A1 (en) * | 1980-06-10 | 1981-12-16 | Beecham Group Plc | Benzamide derivatives, process for their preparation and pharmaceutical compositions containing them |
| WO1984000166A1 (en) * | 1982-06-29 | 1984-01-19 | Sandoz Ag | Benzoic acid piperidyl ester derivatives and method of production and utilization thereof |
| WO1984001151A1 (en) * | 1982-09-24 | 1984-03-29 | Beecham Group Plc | Amino-azabicycloalkyl derivatives as dopamine antagonists |
| EP0189002A2 (en) * | 1984-12-20 | 1986-07-30 | Sandoz Ag | Treatment of gastrointestinal disorders using 5-HT3 antagonists |
| EP0200444A2 (en) * | 1985-04-27 | 1986-11-05 | Beecham Group Plc | Azabicyclononyl-indazole-carboxamide having 5-HT antagonist activity |
-
1990
- 1990-04-27 GB GB909009542A patent/GB9009542D0/en active Pending
-
1991
- 1991-04-22 EP EP91908698A patent/EP0526545A1/en not_active Withdrawn
- 1991-04-22 JP JP91508211A patent/JPH05507071A/en active Pending
- 1991-04-22 WO PCT/GB1991/000636 patent/WO1991017161A1/en not_active Application Discontinuation
- 1991-04-22 CA CA002081350A patent/CA2081350A1/en not_active Abandoned
- 1991-04-22 AU AU77539/91A patent/AU7753991A/en not_active Abandoned
- 1991-04-25 IE IE139991A patent/IE911399A1/en unknown
- 1991-04-25 ZA ZA913123A patent/ZA913123B/en unknown
- 1991-04-26 NZ NZ237957A patent/NZ237957A/en unknown
- 1991-04-26 PT PT97490A patent/PT97490A/en not_active Application Discontinuation
- 1991-04-27 TW TW080103317A patent/TW199157B/zh active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0041817A1 (en) * | 1980-06-10 | 1981-12-16 | Beecham Group Plc | Benzamide derivatives, process for their preparation and pharmaceutical compositions containing them |
| WO1984000166A1 (en) * | 1982-06-29 | 1984-01-19 | Sandoz Ag | Benzoic acid piperidyl ester derivatives and method of production and utilization thereof |
| WO1984001151A1 (en) * | 1982-09-24 | 1984-03-29 | Beecham Group Plc | Amino-azabicycloalkyl derivatives as dopamine antagonists |
| EP0189002A2 (en) * | 1984-12-20 | 1986-07-30 | Sandoz Ag | Treatment of gastrointestinal disorders using 5-HT3 antagonists |
| EP0200444A2 (en) * | 1985-04-27 | 1986-11-05 | Beecham Group Plc | Azabicyclononyl-indazole-carboxamide having 5-HT antagonist activity |
Cited By (97)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0560604A1 (en) * | 1992-03-12 | 1993-09-15 | Mitsubishi Chemical Corporation | Cinnoline-3-carboxylic acid derivatives as 5-HT3 antagonists |
| US5391549A (en) * | 1992-03-12 | 1995-02-21 | Mitsubishi Kasei Corporation | Cinnoline-3-carboxylic acid derivatives |
| US5556851A (en) * | 1992-03-12 | 1996-09-17 | Mitsubishi Chemical Corporation | Cinnoline-3-carboxylic acid derivatives |
| EP1018512A1 (en) * | 1998-10-13 | 2000-07-12 | Rotta Research Laboratorium S.P.A. | Novel basic derivatives of benz(e)isoindol-1-ones and pyrrolo(3,4-c)quinolin-1-ones with 5-HT3-antagonistic activity, their preparation and their therapeutic use |
| US6323216B1 (en) | 1998-10-13 | 2001-11-27 | Rotta Research Laboratorium S.P.A | Basic derivatives of benz[E]isoindol-1-ones and pyrrolo[3,4-c]quinolin-1-ones with 5-HT3-antagonistic activity, their preparation and their therapeutic use |
| US6413978B1 (en) | 1998-10-13 | 2002-07-02 | Rotta Research Laboratorium S.P.A. | Basic derivatives of benz[e] isoindol-1-ones and pyrrolo[3,4-c] quinolin-1-ones with 5-HT3-antagonistic activity, their preparation and their therapeutic use |
| US6828330B2 (en) | 2001-06-12 | 2004-12-07 | Pharmacia & Upjohn Company | Quinuclidine-substituted hetero-bicyclic aromatic compounds for the treatment of disease |
| US7067515B2 (en) | 2001-06-12 | 2006-06-27 | Pfizer Inc. | Quinuclidines-substituted-multi-cyclic-heteroaryls for the treatment of disease |
| US6911543B2 (en) | 2001-10-02 | 2005-06-28 | Pfizer Inc. | Azabicyclic-substituted fused-heteroaryl compounds for the treatment of disease |
| US6849620B2 (en) | 2001-10-26 | 2005-02-01 | Pfizer Inc | N-(azabicyclo moieties)-substituted hetero-bicyclic aromatic compounds for the treatment of disease |
| WO2003043991A1 (en) * | 2001-11-19 | 2003-05-30 | Bayer Healthcare Ag | Heteroaryl carboxylic acid amides |
| US8227598B2 (en) | 2001-11-19 | 2012-07-24 | Bayer Schering Pharma Aktiengesellschaft | Heteroaryl carboxamides |
| US9139579B2 (en) | 2001-11-19 | 2015-09-22 | Bayer Intellectual Property Gmbh | Heteroaryl carboxamides |
| US9926311B2 (en) | 2001-11-19 | 2018-03-27 | Bayer Intellectual Property Gmbh | Heteroaryl carboxamides |
| US10487079B2 (en) | 2001-11-19 | 2019-11-26 | Bayer Intellectual Property Gmbh | Heteroaryl carboxamides |
| US7256288B2 (en) | 2001-11-19 | 2007-08-14 | Bayer Healthcare Ag | Heteroaryl carboxylic acid amides |
| US6894042B2 (en) | 2002-02-19 | 2005-05-17 | Pharmacia & Upjohn Company | Azabicyclic compounds for the treatment of disease |
| US6858613B2 (en) | 2002-02-19 | 2005-02-22 | Pfizer Inc. | Fused bicyclic-N-bridged-heteroaromatic carboxamides for the treatment of disease |
| WO2003072578A1 (en) * | 2002-02-20 | 2003-09-04 | Pharmacia & Upjohn Company | Azabicyclic compounds with alfa7 nicotinic acetylcholine receptor activity |
| US7001900B2 (en) | 2002-02-20 | 2006-02-21 | Pfizer Inc. | Azabicyclic compounds for the treatment of disease |
| US7683075B2 (en) | 2003-05-12 | 2010-03-23 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| US9493467B2 (en) | 2006-04-04 | 2016-11-15 | The Regents Of The University Of California | PI3 kinase antagonists |
| US8642604B2 (en) | 2006-04-04 | 2014-02-04 | The Regents Of The University Of California | Substituted pyrazolo[3,2-d]pyrimidines as anti-cancer agents |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US8703777B2 (en) | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9655892B2 (en) | 2008-01-04 | 2017-05-23 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9216982B2 (en) | 2008-01-04 | 2015-12-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8785456B2 (en) | 2008-01-04 | 2014-07-22 | Intellikine Llc | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US11433065B2 (en) | 2008-01-04 | 2022-09-06 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9822131B2 (en) | 2008-01-04 | 2017-11-21 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US9637492B2 (en) | 2008-03-14 | 2017-05-02 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9629843B2 (en) | 2008-07-08 | 2017-04-25 | The Regents Of The University Of California | MTOR modulators and uses thereof |
| US9828378B2 (en) | 2008-07-08 | 2017-11-28 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9790228B2 (en) | 2008-09-26 | 2017-10-17 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US8703778B2 (en) | 2008-09-26 | 2014-04-22 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US9296742B2 (en) | 2008-09-26 | 2016-03-29 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US8697709B2 (en) | 2008-10-16 | 2014-04-15 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| US9315505B2 (en) | 2009-05-07 | 2016-04-19 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8785454B2 (en) | 2009-05-07 | 2014-07-22 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8569323B2 (en) | 2009-07-15 | 2013-10-29 | Intellikine, Llc | Substituted isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US9206182B2 (en) | 2009-07-15 | 2015-12-08 | Intellikine Llc | Substituted isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US9522146B2 (en) | 2009-07-15 | 2016-12-20 | Intellikine Llc | Substituted Isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US9181221B2 (en) | 2010-05-21 | 2015-11-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9738644B2 (en) | 2010-05-21 | 2017-08-22 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9388183B2 (en) | 2010-11-10 | 2016-07-12 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11312718B2 (en) | 2011-01-10 | 2022-04-26 | Infinity Pharmaceuticals, Inc. | Formulations of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one |
| US10550122B2 (en) | 2011-01-10 | 2020-02-04 | Infinity Pharmaceuticals, Inc. | Solid forms of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one and methods of use thereof |
| USRE46621E1 (en) | 2011-01-10 | 2017-12-05 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9290497B2 (en) | 2011-01-10 | 2016-03-22 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9840505B2 (en) | 2011-01-10 | 2017-12-12 | Infinity Pharmaceuticals, Inc. | Solid forms of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1 (2H)-one and methods of use thereof |
| US8809349B2 (en) | 2011-01-10 | 2014-08-19 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US9056877B2 (en) | 2011-07-19 | 2015-06-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9605003B2 (en) | 2011-07-19 | 2017-03-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9718815B2 (en) | 2011-07-19 | 2017-08-01 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9115141B2 (en) | 2011-08-29 | 2015-08-25 | Infinity Pharmaceuticals, Inc. | Substituted isoquinolinones and methods of treatment thereof |
| US9546180B2 (en) | 2011-08-29 | 2017-01-17 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9321772B2 (en) | 2011-09-02 | 2016-04-26 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9895373B2 (en) | 2011-09-02 | 2018-02-20 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9255108B2 (en) | 2012-04-10 | 2016-02-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US9527847B2 (en) | 2012-06-25 | 2016-12-27 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US11613544B2 (en) | 2012-09-26 | 2023-03-28 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pyrazines for modulation of IRE1 |
| US10822340B2 (en) | 2012-09-26 | 2020-11-03 | The Regents Of The University Of California | Substituted imidazolopyrazine compounds and methods of using same |
| US10131668B2 (en) | 2012-09-26 | 2018-11-20 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pYRAZINES for modulation of IRE1 |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9828377B2 (en) | 2013-10-04 | 2017-11-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10329299B2 (en) | 2013-10-04 | 2019-06-25 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10675286B2 (en) | 2014-03-19 | 2020-06-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11541059B2 (en) | 2014-03-19 | 2023-01-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11110096B2 (en) | 2014-04-16 | 2021-09-07 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11944631B2 (en) | 2014-04-16 | 2024-04-02 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US10941162B2 (en) | 2014-10-03 | 2021-03-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US10253047B2 (en) | 2014-10-03 | 2019-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11247995B2 (en) | 2015-09-14 | 2022-02-15 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US11939333B2 (en) | 2015-09-14 | 2024-03-26 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11147818B2 (en) | 2016-06-24 | 2021-10-19 | Infinity Pharmaceuticals, Inc. | Combination therapies |
Also Published As
| Publication number | Publication date |
|---|---|
| NZ237957A (en) | 1993-09-27 |
| ZA913123B (en) | 1992-05-27 |
| TW199157B (en) | 1993-02-01 |
| JPH05507071A (en) | 1993-10-14 |
| AU7753991A (en) | 1991-11-27 |
| GB9009542D0 (en) | 1990-06-20 |
| CA2081350A1 (en) | 1991-10-28 |
| EP0526545A1 (en) | 1993-02-10 |
| PT97490A (en) | 1992-01-31 |
| IE911399A1 (en) | 1991-11-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1991017161A1 (en) | Isoquinoline amides and esters as 5 ht3 receptor antagonists | |
| EP0255297B1 (en) | Azabicyclic compounds, process for their preparation, and their pharmaceutical use | |
| US4937247A (en) | 1-acyl indazoles | |
| EP0315390B1 (en) | Novel 4-oxobenzotriazines and 4-oxoquinazolines | |
| EP0200444B1 (en) | Azabicyclononyl-indazole-carboxamide having 5-ht antagonist activity | |
| AU626614B2 (en) | 9-aza-3(oxa or thio)bicyclo(3.3.1)nonane derivatives | |
| EP0254584B1 (en) | Azabicyclic compounds, process for their preparation, and their pharmaceutical use | |
| IE60111B1 (en) | Indole derivatives having an azabicyclic side chain, process for their preparation, intermediates, and pharmaceutical compositions | |
| EP0223385B1 (en) | 8-Azabicyclo[3,2,1]octane and 9-azabicyclo[3,3,1]nonane derivatives | |
| EP0289170B1 (en) | Azabicyclic compounds, process for their preparation and pharmaceutical compositions containing them | |
| EP0261964A2 (en) | Azabicyclic compounds and their use as 5-ht3 receptor antagonists | |
| WO1993008185A1 (en) | N-aryl-n1-azabicyclo-ureas as 5-ht3 antagonists | |
| US5049556A (en) | Certain N-azabicyclo-2,3-dihydro-indole-1-carboxamides having anti-emetic properties, further found useful in treating headaches, neuralgia and anxiety | |
| WO1992011259A1 (en) | Azabicyclic amides or esters of halogenated benzoic acids | |
| AU651645B2 (en) | Pharmaceuticals | |
| WO1991001316A1 (en) | New 9-azabicyclo[3.3.1]nonane-derivate | |
| WO1993008186A1 (en) | Pyridine-3-carboxylic acid esters or amides useful as 5-ht3 antagonists | |
| WO1993007147A1 (en) | 3,9-diazabicyclo(3.3.1)nonane derivatives with 5-ht3 receptor antagonist activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IT LU NL SE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1991908698 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2081350 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1991908698 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1991908698 Country of ref document: EP |