WO1984001385A1 - Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament - Google Patents
Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament Download PDFInfo
- Publication number
- WO1984001385A1 WO1984001385A1 PCT/FR1982/000159 FR8200159W WO8401385A1 WO 1984001385 A1 WO1984001385 A1 WO 1984001385A1 FR 8200159 W FR8200159 W FR 8200159W WO 8401385 A1 WO8401385 A1 WO 8401385A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- methyl
- hydrogen
- trityl
- compounds
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/20—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D239/22—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to ring carbon atoms
Definitions
- New acylated derivatives of dihydro orotic acid their preparation and their use as medicaments.
- the invention relates to new derivatives of dihydroorotic acid, one of the nitrogen of which is acylated by a derivative of propionic acid.
- orotic dihydro or thio orotic acid one of the nitrogen atoms of which is acylated by an amidinopropionic chain. It specifically relates to the N-acyl dihydro orotic acids of general formula I.
- R is hydrogen or a lower alkyl radical
- R 1 is oxygen, or sulfur
- R 3 is hydrogen, the benzhydrile group or the trityl group
- R 4 is hydrogen or a lower alkyl radical.
- the invention also relates to the diastereoisomers and to the optical isomers of the compounds of general formula I.
- the molecule of dihydro orotic acid or of dihydrothio orotic acid comprises two asymmetric carbon atoms giving rise to two diastereoisomers which can each be split.
- the propionic chain has a chiral carbon atom when R 4 is different from hydrogen. This new center of symmetry can be split giving rise to two optical isomers.
- the invention also relates to the salts of the compounds of general formula I with a mineral or organic acid, preferably therapeutically compatible.
- the invention also relates to the esters of acids of general formula I corresponding to general formula I ',
- R, R 1 , R 3 and R 4 are defined as above, and R 2 represents a lower alkyl, hydroxy-alkyl or polyhydroxy-lower alkyl, lower aralkyl radical.
- the compounds according to The invention are inhibitors of the enzyme which converts the inactive decapeptide Angiotensin I to the active octapeptide angiotensin II.
- Angiotensin II is the circulating hormone which can be considered to be responsible for essential hypertension when it is in excess.
- the pharmacological tests below have shown that the compounds I of the invention are useful as antihypertensive agents having a prolonged effect or as medicaments for the treatment or prophylaxis of complications of diabetes.
- these compounds can be administered in combination with diuretics such as hydroflumethiazide, furosemide and bumetanide, indapamide or chlortalidone.
- these preparations may contain not only inert excipients, but also other antihypertensive agents such as reserpine, ⁇ - methyldopa, guanethidine, clonidine, hydralazine, etc., or beta-adrenergic receptor blockers such as propanolol, alprenolol pindolol, bufetolol, bupranolol, bunitr ⁇ lol, practolol, oxprenolol, indenolol, timolol, bunolol, etc.
- antihypertensive agents such as reserpine, ⁇ - methyldopa, guanethidine, clonidine, hydralazine, etc.
- beta-adrenergic receptor blockers such as propanolol, alprenolol pindolol, bufeto
- the compounds of formula I are present in the form of pharmaceutical compositions suitable for administration by the oral, parenteral or rectal route.
- the administration forms are tablets, capsules, powders, suppositories, injectable forms, ophthalmic solutions, ophthalmic ointments, etc. These preparations may also contain common excipients.
- the doses are adjusted according to the therapeutic indications, the form of administration and the weight of the subject. In general, the daily doses are between 100 and 5000 mg, preferably between 250 and 1000 mg in a single dose or several divided doses.
- the administration forms preferably contain between 125 and 500 mg of active principle per unit dose.
- the subject of the invention is also a process for preparing the compounds of general formula I, characterized in that a dihydro orotic acid of general formula II is reacted
- R and R 1 are defined as above, and R 3 is a benzhydryl or trityl radical, which is esterified with an alkanol or an arylalkanol to form an ester of general formula IV
- R 2 is an alkyl or aralkyl radical
- R, R 1 and R 3 are defined as above
- the latter is reacted with a halide of an ⁇ -halogenated propionic acid of general formula V
- R 3 is hydrogen or a lower alkyl X is chlorine or bromine
- Hal is a halogen other than fluorine, to form the N-acylated derivative of dihydro orotic acid of general formula VI
- the process of the invention also comprises the additional step which consists in unblocking the second nitrogen atom by acid hydrolysis using acetic acid or hydrochloric acid to form an N-acylated dihydro orotic acid of formula I '.
- R 3 is hydrogen and the substituents R, R 1 , R 2 and R 4 are defined as above.
- the process of the invention also comprises the step of hydrolysis of the esters of formula I 'into acids of formula I
- the compounds of general formula I or I ′ can, if desired, be split into their isomeric diastero or split into their optical isomers by chemical or physical splitting agents.
- the compounds of general formula I or I ′ can also be salified by addition of a mineral or organic acid such as hydrochloric acid, sulfuric acid, acetic acid, benzoic acid, nicotinic acid, methane sulfonic acid, isethionic acid, p. toluene sulfonic or glucose 1 - phosphoric acid.
- a mineral or organic acid such as hydrochloric acid, sulfuric acid, acetic acid, benzoic acid, nicotinic acid, methane sulfonic acid, isethionic acid, p. toluene sulfonic or glucose 1 - phosphoric acid.
- Stage B N'_trityl dihydro methyl orotate. 11 g15 of methyl dihydro orotate are dissolved in 40 ml of methylene chloride, then 2 ml of triethylamine and finally gradually a solution of 18g5 of trityl chloride in 60 ml of methylene chloride. The mixture is stirred at temperature for 6 hours. The triphenyl carbinol which has precipitated is separated by filtration and then the filtrate is washed with water until the washings are neutral. The methylene solution is then dried over magnesium sulfate and evaporated to dryness.
- N '- trityl dihydro orotate of methyl is taken up in 50 ml of hot ethyl acetate. By cooling, the trityl derivative separates. The suspension is left to stand for 12 hours in a cooler and then the precipitate is separated by filtration. It is washed with cold ethyl acetate and then dried to constant weight.
- the N '-trityl dihydro orotate of methyl is recrystallized from acetonitrile by hot and cold. It is in the form of colorless crystals melting at 237 - 240o.
- Stage C N '-trityl N - (2 - chloropropionyl) dihydro orotate methyl.
- 16g30 of 2-chloropropionic acid are dissolved in 45 ml of hexamethyl phosphorotriamide and then 20g 5 of freshly redistilled thionyl chloride is added in small portions.
- the reaction mixture is kept at a temperature in the region of 5 ° for 2 hours.
- the hydrochloric acid formed is removed by vacuum distillation.
- the residual solution is added gradually to a solution of 62 g of N'-trityl dihydro orotate methyl in 125 ml of hexaphosphorotriamide previously cooled to 5o. Stirring is continued for 4 hours, then the excess reagent is decomposed by the addition of an aqueous solution of sodium carbonate at 20%. It is left in contact for one hour, then the aqueous phase is exhausted with isopropyl ether three times. The ethereal solutions are combined, washed with water until neutral, then dried and evaporated to dryness.
- the residue of crude methyl N '- trityl N - (2-chloropropionyl) dihydro orotate is purified by dissolving in a minimum of hot ethyl acetate. By cooling, the N'-trityl N - (2-chloropropionyl) dihydro orotate of crystallized methyl is separated, which is filtered, drained and dried under vacuum. This compound melts at 251 - 252o.
- Stage D N'-trityl N - (2-cyanopropionyl) dihydro methyl orotate.
- N'-trityl N - (2-chloropropionyl) dihydro orotate methyl are dissolved in 120 ml of dimethyl sulfoxide. After complete dissolution, a solution of 4 g of potassium cyanide in 20 ml of water is carefully added. The mixture is brought to 40-45 ° and maintained at this temperature for 4 hours. The reaction mixture is allowed to return to room temperature and then diluted with an equal volume of water.
- N '- trityl N - (2-cyanopropionyl) dihydro orotate of methyl precipitates progressively. It is left to stand for 12 hours and then the precipitate is separated, which is washed with water then with sodium bicarbonate and then again with water. It is then dried under vacuum.
- Stage F N - (2-formamidino propionyl) methyl dihydro orotate.
- Stage G N- (2,6 - dioxo 4-carboxy hexahydro pyrimidinyl -3) 2-carbonyl 2-methyl acetamidine.
- hydrochloride or methane sulfonate is obtained.
- salts with organic bases are obtained, for example the dicyclohexylamine salt, the benzothine, N-methyl-D-glucamine, and hydrabamine salts, salts with amino acids such as arginine, lysine, glycine, valine, ornithine or citrulline.
- Example I By operating as in Example I, starting from 5 - methyl dihydro uracil 6-carboxylic acid, ethyl N-trityl 5 - methyl dihydro uracil 6-carboxylate is obtained which is condensed with chloride. ⁇ -chloropropionyl to form: N- trityl N '- ⁇ -chloropropionyl 5 -methyl dihydro uracil 6 - ethyl carboxylate
- compositions compound of Example I 230 g lactose 150 g crystalline cellulose 50 g carboxymethylcellulose calcium 7 g magnesium stearate 3 g
- the acute toxicity of the compounds of general formula I corresponds to an LD 50 of 2g50 / kg
- the LD 50 was determined graphically as follows: 4-week old Rockland mice weighing 19 to 21 g are placed in a breeding room at constant temperature and humidity (23oC ⁇ 1.55oC - 5%) and they are provided with food pellets and water for a week at will. We choose for the experiment mice whose growth is normal. (Method of administration).
- test compound is suspended in a 0.5% gum tragacanth solution and administered orally at a dose of 0.5 ml per 20 g body weight.
- b Study of antihypertensive action.
- compounds that inhibit the angiotensin I transforming enzyme may have curative activity vis-à-vis renal hypertension and essential hypertension.
- the compounds of the invention were therefore evaluated as antihypertensive agents according to the following method: Male Wistar rats weighing 200 to 300 g are used. Under ether anesthesia, a polyethylene cannula is placed in the carotid artery and in the jugular vein. The cannula of the jugular vein is connected to a continuous perfusion device.
- angiotensin I is infused intravenously at a dose of 300 g / kg using the continuous infusion set and the hypertensive response is recorded with a polygraph.
- the compounds of the invention are administered in suspension in a 0.5% aqueous solution of tragacanth, orally at a dose of 0.3 ml / 100 g of body weight and the response of hypertension to the blood is measured. intravenous infusion of angiotensin I over time.
- the inhibitory activity of the compounds with respect to the angiotensin I converting enzyme is expressed by the percentage inhibition of the hypertension response to angiotensin I.
- the table shows the variations in the percentages d inhibition over time obtained with the compounds of the invention. (Results).
- the table shows the results of the pharmacological tests when the compounds (I) of the invention and their salts are used as agents inhibiting the angiotensin I converting enzyme.
Abstract
Nouveaux dérivés de l'acide dihydro orotique dont un des atomes d'azote est acylé par un dérivé de l'acide propionique, et leur préparation. L'invention a spécifiquement pour objet les acides N-acyl dihydro orotiques de formule générale (I) dans laquelle R et R4 sont de l'hydrogène ou un radical alcoyle inférieur, R1 est de l'oxygène ou du soufre et R3 est de l'hydrogène, un benzhydryle ou un trityle. Les composés de l'invention trouvent un emploi en thérapeutique comme principes actifs de compositions pharmaceutiques anti-hypertensives, seuls ou associés à un autre principe actif d'action synergistique ou complémentaire.
Description
Nouveaux dérivés acylés de l'acide dihydro orotique, leur préparation et leur emploi comme médicament.
L'invention se rapporte à de nouveaux dérivés de l ' acide dihydro orotique dont un des azotes est acylé par un dérivé de l'acide propionique.
Elle a plus particulièrement pour objet, des dérivés de l'acid dihydro orotique ou thio orotique dont un des atomes d'azote est acylé par une chaîne amidinopropionique. Elle a spécifiquement pour objet les acides N-acyl dihydro orotiques de formule générale I.

L'invention se rapporte également aux sels des composés de formule générale I avec un acide minéral ou organique, de préférence thérapeutiquement compatible.
L'invention se rapporte également aux esters des acides de formule générale I répondant à la formule générale I',

dans laquelle R, R1, R3 et R4 sont définis comme précédemment, et R2 représente un radical alcoyle inférieur, hydroxy-alcoyle ou polyhydroxy-alcoyle inférieur, aralcoyle inférieur.
Parmi les composés de formule générale I, on citera plus particulièrement la N-(2-thio 4 - carboxy 6-oxohexahydropyrimidinyl- 3) carbonylacetamidine, la N-(2,6 dioxo 4-carboxy hexahydropyridimidinyl-3) carbonylacetamidine, la N-(2,6- dioxo 4 - carboxy 5 - methyl hexahydropyrimidinyl - 3) carbonylacetamidine, la N-(2,6-dioxo 4-carboxy 5-methyl hexahydropyrimidinyl -3) 2-carbonyl- 2-methyl acetamidine les composés selon l'invention sont des inhibiteurs de l'enzyme qui convertit le decapeptide inactif Angiotensine I en octapeptide actif l'angiotensine II.
L' angiotensine II est l'hormone circulante qui peut être considérée comme responsable de l'hypertension essentielle lorsqu'elle est en excès. Les tests pharmacologiques ci-après ont montré que les composés I de l'invention sont utiles comme agents antihypertenseurs ayant un effet prolongé ou comme médicaments pour le traitement ou la prophylaxie des complications du diabète. Lorsqu'on utilise ces composés pour abaisser la pression sanguine , on peut les administrer en combinaison avec des diurétiques tels que l'hydrofluméthiazide, le furosémide et le bumétanide, l'indapamide ou la chlortalidone.
Pour le traitement de l'hypertension, ces préparations peuvent contenir non seulement des excipients inertes, mais encore d'autres agents antihypertenseurs tels que la réserpine, l'α - méthyldopa, la guanéthidine, la clonidine, l'hydralazine, etc., ou des bloqueurs des récepteursβadrénergiques tels que le
propanolol, l' alprénolol le pindolol, le bufétolol, le bupranolol, le bunitrόlol, le practolol, l' oxprénolol, l'indénolol, le timolol, le bunolol , etc.
En vue de l'administration en thérapeutique les composés de formule I sont présentes sous forme de compositions pharmaceutiques adaptées pour l'administration par voie buccale, parentérale ou rectale.
Les formes d'administration sont des comprimés, des capsules, des poudres, des suppositoires, des formes injectables, des solutions ophtalmiques, des pommades ophtalmiques, etc. Ces préparations peuvent également contenir des excipients courants. On ajuste les doses selon les indications thérapeutiques, la forme d'administration et le poids du sujet. En général, les doses journalières sont comprises entre 100 et 5000 mg , de préférence entre 250 et 1000 mg en une seule dose ou plusieurs doses fractionnées.
En pratique, les formes d'administration renferment de préférence entre 125 et 500 mg de principe actif par prise unitaire. L'invention a encore pour objet un procédé de préparation des composés de formule générale I caractérisé en ce qu'on fait réagir un acide dihydro orotique de formule générale II



dans laquelle R2 est un radical alcoyle ou aralcoyle, et R, R1 et R3 sont définis comme précédemment on fait réagir celui-ci avec un halogenure d'un acide propionique α-halogèné de formule générale V

Hal est un halogène autre que le fluor, pour former le dérivé N-acylé de l'acide dihydro orotique de formule générale VI


dans laquelle les substituants R, R1, R2, R3 et R4 sont définis comme précédemment, que l'on convertit en amidine de formule générale I par action de l'ammoniaque en milieu alcanolique.
Le procédé de l'invention comprend aussi l'étape supplémentaire qui consiste à débloquer le second atome d'azote par hydrolyse acide au moyen d'acide acétique ou d'acide chlorhydrique pour former un acide dihydro orotique N-acylé de formule I'.

Le procédé de l'invention comprend encore l'étape d'hydrolyse des esters de formule I' en acides de formule I
dan
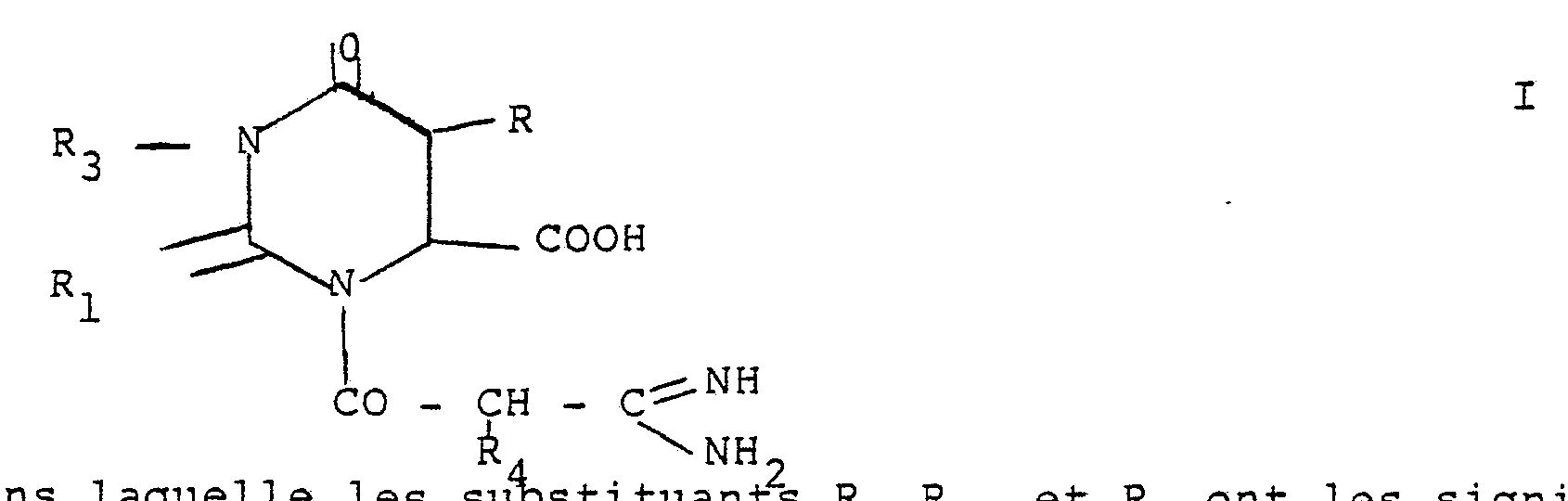 s laquelle les substituants R, R1, et R4 ont les significations antérieures. Les composés de formule générale I ou I' peuvent, si désiré, être dédoublés en leurs diastereo isomères
ou dédoublés en leurs isomères optiques par des agents de dédoublement chimiques ou physiques.
s laquelle les substituants R, R1, et R4 ont les significations antérieures. Les composés de formule générale I ou I' peuvent, si désiré, être dédoublés en leurs diastereo isomères
ou dédoublés en leurs isomères optiques par des agents de dédoublement chimiques ou physiques.
Les composés de formule générale I ou I' peuvent aussi être salifiés par addition d'un acide minéral ou organique comme l'acide chlorhydrique, l'acide sulfurique, l'acide acétique, l'acide benzoîque, l'acide nicotinique, l'acide méthane sulfonique, l'acide isethionique, l'acide p. toluène sulfonique ou l'acide glucose 1 - phosphorique.
Les exemples suivants illustrent l'invention. Ils ne la limitent en aucune façon.
EXEMPLE I
N - (2.6 - dioxo 4 - carboxy hexahydropyrimidinyl - 3) carbonyl acetamidine. Stade A : dihydroorotate de methyle.
On dissout 15g8 d'acide dihydro orotique dans 250 ml de methanol. On ajoute 1g50 d'acide p. toluène sulfonique et on porte le mélange au reflux pendant 4 heures. On laisse ensuite refroidir, puis on neutralise le milieu par addition d'une solution saturée de bicarbonate de sodium. On décante la phase aqueuse. La phase organique est séchée sur sulfate de sodium, filtrée puis évaporée à sec. Le dihydro orotate de methyle brut est repris par 25 ml de chlorure de méthylène ; la solution est décolorée au charbon activé, filtrée et évaporée à sec. Le dihydro orotate de methyle est finalement purifié par distillation sous pression réduite. On obtient ainsi 13g67 de dihydro orotate de methyle que l'on utilise tel quel pour la poursuite de la synthèse. Stade B : N'_trityl dihydro orotate de methyle. On dissout 11g15 de dihydro orotate de methyle dans 40 ml de chlorure de méthylène, puis 2ml 5 de triethylamine et enfin progressivement une solution de 18g5 de chlorure de trityle dans 60 ml de chlorure de méthylène. On agite à température pendant 6 heures. On sépare par filtration le triphenyl carbinol qui a précipité puis lave le filtrat à l'eau jusqu'à neutralité des eaux de lavage. La solution methylènique est
alors séchée sur sulfate de magnésium et évaporée à sec. Le résidu de N' - trityl dihydro orotate de methyle est repris par 50 ml d'acétate d'éthyle à chaud. Par refroidissement, le dérivé trityle se sépare. On laisse reposer la suspension pendant 12 heures en glacière puis sépare le précipité par filtration. On le lave à l'acétate d'éthyle froid puis le sèche à poids constant. Le N' -trityl dihydro orotate de methyle est recristallisé de l'acetonitrile par chaud et froid. I1 se présente sous forme de cristaux incolores fondant à 237 - 240º.
Stade C : N' -trityl N -(2 - chloropropionyl) dihydro orotate de methyle.
On dissout 16g30 d'acide 2-ch.loropropionique dans 45 ml d'he xamethyl phosphorotriamide puis on ajoute par petites fractions 20g 5 de chlorure de thionyle fraichement redistillé. On maintient le mélange réactionnel à une température voisine de 5° pendant 2 heures. On chasse l'acide chlorhydrique formé par distillation sous vide. La solution résiduelle est ajoutée progressivement, à une solution de 62 g de N'-trityl dihydro orotate de methyle dans 125 ml d ' hexaphosphorotriamide préalablement refroidi à 5º. On maintient sous agitation pendant 4 heures, puis décompose l'excès de réactif par addition d'une solution aqueuse de carbonate de sodium à 20%. On laisse en contact pendant une heure, puis épuise la phase aqueuse à l'éther isopropylique à trois reprises. Les solutions éthérées sont réunies, lavées à l'eau jusqu'à neutralité, puis séchées et évaporées à sec.
Le résidu de N' - trityl N -( 2-chloropropionyl) dihydro orotate de methyle brut est purifié par dissolution dans le minimum d'acétate d'éthyle chaud. Par refroidissement on sépare le N'-trityl N - ( 2-chloropropionyl) dihydro orotate de methyle cristallisé que l'on filtre, essore et sèche sous vide. Ce composé fond à 251 - 252º .
Stade D : N'-trityl N -( 2-cyanopropionyl) dihydro orotate de methyle.
25g de N'- trityl N - ( 2-chloropropionyl) dihydro orotate de
methyle sont dissouts dans 120 ml de dimethyl suifoxyde. Après complète dissolution, on ajoute avec précaution une solution de 4g de cyanure de pgtassium dans 20 ml d'eau. On porte le mélange à 40 - 45° et on le maintient à cette température pendant 4 heures. On laisse le mélange réactionnel revenir à température ordinaire puis le dilue avec un volume égal d'eau. Le N' - trityl N -( 2-cyanopropionyl) dihydro orotate de methyle précipite progressivement. On laisse reposer pendant 12 heures puis sépare le précipité qu'on lave à l'eau puis au bicarbonate de sodium puis encore à l'eau. On le sèche ensuite sous vide. On obtient ainsi 17g65 de N' - trityl N-(2-cyano propionyl) dihydro orotate de methyle qu'on utilise tel quel pour la suite de la synthèse. Stade E : N' - trityl N -(2 formamidino propionyl) dihydro orotate de methyle.
On dissout 13g85 de N' - trityl N- ( 2 cyano propionyl) dihydro orotate de methyle dans 100 ml d'ethanol et on fait passer un courant d'acide chlorhydrique sec pendant 3 heures tout en chauffant à l'ébullition. Au bout de ce temps on laisse refroidir le mélange au voisinage de zéro par immersion dans un bain d'eau glacée puis on ajoute goutte à goutte 30 ml d'une solution éthanolique à 10% d'ammoniaque. On laisse en contact pendant 2 Heures puis sépare le précipité par filtration. On le lave à l'eau, l'essore et le sèche, on obtient ainsi 8g70 de formamidine.
Stade F : N -( 2-formamidino propionyl) dihydro orotate de methyle.
On met la totalité des 8g 70 de formamidine obtenue au stade précédent, en suspension dans 60 ml d'eau puis on ajoute 5 ml d'acide acétique cristallisé. On maintient l'agitation pendant 30 mn. On sépare ensuite le précipité de triphényl carbinol. Le filtrat est ensuite neutralisé par addition de bicarbonate de sodium. La formamidine précipite. On la lave à l'eau jusqu'à neutralité des eaux de lavage, l'essore et la sèche. On recueille ainsi 3g71 de formamidine que l'on purifie par recristallisation du methanol. La formamidine ainsi obtenue
fond à 195-196º.
Stade G : N-(2,6 - dioxo 4- carboxy hexahydro pyrimidinyl -3) 2- carbonyl 2- méthyl acétamidine.
On met en suspension 3g de formamidine obtenue au stade précé dent dans 25 ml d'eau et on ajoute sous forte agitation 20 ml de potasse éthanolique N. On maintient sous agitation à tempé rature ordinaire pendant 6 heures puis neutralise le milieu par addition d'acide acétique. L' acétamidine qui précipite est séparée par filtration, lavée à plusieurs reprises à l ' eau puis essorée et séchée sous vide. Pour l'analyse, elle est recristallisée de l'acetonitrile. On obtient ainsi 2g25 d'acétamidine fondant à 210 - 211º (avec décomposition). Par neutralisation, à l'aide d'une base appropriée on obtient respectivement le sel de sodium, de potassium, de lithium, de calcium, de magnésium, de strontium, d'aluminium, ou de fer.
Par neutralisation par l'acide chlorhydrique ou l'acide/mseultrboane nique, on obtient le chlorhydrate ou le méthane suifonate. En neutralisant avec une aminé on obtient des sels avec des bases organiques, par exemple le sel de dicyclohexylamine, les sels de benzothine, de N-méthyl-D-glucamine, et d'hydrabamine, des sels avec des amino-acides comme l'arginine, la lysine, la glycine, la valine, l'ornithine ou la citrulline.
Exemple II En opérant comme à l'exemple I au départ de l'acide orotique, puis condensation du N-trityl th-io orotate de methyle avec l'acide α -bromo acétique puis condensation du dérivé bromoacétylé avec le cyanure de potassium, on obtient finalement la N -(2-thio 4- carboxy 6-oxo hexahydro pyrimidinyl -3) carbonyl acétamidine. F = 237 - 238ºC.
Exemple III
En opérant comme à l'exemple I au départ du N-trityl orotate de methyle et de l'acide α -bromo acétique puis condensation du N-trityl N'- α-bromo acétyl orotate de methyle formé avec le cyanure de potassium, on obtient finalement la N- (2,6 dioxo
4-carboxy hexahydropyrimidinyl -3 ) acétamidine . F : 201 - 202° .
Exemple IV
En opérant comme à l'exemple I au départ de l'acide 5 - méthyl dihydro uracile 6 - carboxylique on obtient respectivement :
- le 5 - méthyl dihydro uracile 6-carboxylate d'éthyle,
- le N-trityl 5-méthyl dihydro uracile 6 - carboxylate d'éthyle,
- le N-trityl N'- α- bromoacétyl 5-méthyl dihydro uracile 6- carboxylate d'éthyle, - le N-trityl N'-α- cyanoacétyl 5 - méthyl dihydro uracyle 6- carboxylate d'éthyle,
- le N-trityl N'- (α - formamidino acétyl) 5 - méthyl dihydro uracile 6- carboxylate d'éthyle,
- le N'- ( α - amidino acétyl) 5 - méthyl dihydro uracile 6- carboxylate d'éthyle
- la N - (2,6- dioxo 4-carboxy 5- méthyl hexahydro pyrimidinyl-3 carbonyl acétamidine.
F = 189 - 190°.
Exemple V
En opérant comme à l'exemple I au départ de l'acide 5 - méthyl dihydro uracile 6- carboxylique, on obtient le N- trityl 5 - méthyl dihydro uracile 6 -carboxylate d'éthyle que l'on condense avec le chlorure d' α-chloropropionyle pour former le : N- trityl N' - α -chloropropionyl 5 -méthyl dihydro uracile 6 - carboxylate d'éthyle
N -trityl N' - α-cyano propionyl 5 -méthyl dihydro uracile 6-carboxylate d'éthyle
N -trityl N' - (α-formamidino propionyl) 5 -méthyl dihydro uracile 6-carboxylate d'éthyle
N'- ( α formamidino propionyl) 5- méthyl dihydro uracile 6- carboxylate d'éthyle
N- (2,6 dioxo 5 -méthyl 4 -carboxy hexa hydropyrimidinyl -3)
2- carbonyl 2 méthyl acétamidine. F = 197 - 198º.
Exemple VI
Exemples de compositions : composé de l ' exemple I 230 g lactose 150 g cellulose cristalline 50 g carboxyméthylcellulose calcique 7 g stéarate de magnésium 3 g
Total pour 1000 comprimés 410 g
composé de l'exemple II 150 g lactose 120 g cellulose cristalline 50 g carboxyméthylcellulose calcique 7 g stéarate de magnésium 13 g
Total pour 1000 comprimés 340 g
Exemple VII
Etude pharmacologique des composés selon l'invention a/ Test de toxicité
La toxicité aiguë des composés de fromule générale I correspond à une DL50 de 2g50/kg
La DL50 a été déterminée graphiquement de la manière suivante : On place des souris de souche Rockland de 4 semaines pesant 19 à 21 g dans une pièce d'élevage à température et à humidité constantes (23ºC ± 1,55ºC - 5%) et on leur fournit à volonté un aliment en boulettes et de l'eau pendant une semaine. On choisit pour l'expérience des souris dont la croissance est normale. (Méthode d'administration).
On met le composé à étudier en suspension dans une solution à 0,5 % de gomme adragante et on l'administre par voie orale à la dose de 0,5 ml pour 20 g de poids corporel. b/ Etude de l'action antihypertensive. On a récemment mis en évidence que les composés inhibant l'enzyme de transformation de l'angiotensine I peuvent avoir
une activité curative vis-à-vis de l'hypertension rénale et de l'hypertension essentielle. On a donc évalué les composés de l'invention comme agents antihypertenseurs selon la méthode suivante : On utilise des rats Wistar mâles pesant 200 à 300 g. Sous anesthésie à l'éther, on place une canule en polyéthylène dans l'artère carotide et dans la veine jugulaire. On raccorde la canule de la veine jugulaire à un appareil de perfusion continue. Lorsque les effets de l' anesthésie ont totalement cessé, on perfuse de l'angiotensine I par voie intraveineuse à la dose de 300 g/kg au moyen de l'appareil de perfusion continue et on enregistre la réponse hypertensive avec un polygraphe. On administre les composés de l'invention en suspension dans une solution aqueuse à 0,5 % de gomme adragante, par voie orale à la dose de 0,3 ml/100 g de poids corporel et on mesure la réponse d'hypertension à la perfusion intraveineuse d'angiotensine I au cours du temps. On exprime l'activité inhibitrice des composés vis-à-vis de l'enzyme de conversion de l'angiotensine I par le pourcentage d'inhibition de la réponse d'hypertension à l'angiotensine I. Le tableau montre les variations des pourcentages d'inhibition au cours du temps obtenu avec les composés de l'invention. (Résultats). Le tableau montre les résultats des tests pharmacologiques lorsqu'on utilise les composés (I) de l'invention et leurs sels, comme agents inhibant l'enzyme de conversion de l'angiotensine I.
Effet suppresseur des composés de formule I sur la réponse hypertensive à l' angiotensine I. Composé Nº Dose (mg/kg) Inhibitions ( % ) à
25 (min) 65 ( min )
1 0,2 45 55 2 0,4 30 35 3 1,0 15 55 4 0,1 45 55 5 0,3 50 60
Claims
REVENDICATIONS
1/ Les acide N -acyl dihydro orotiques de formule générale I

2/ Les diastereo isomères et les isomères optiques des composés de formule générale I selon la revendication 1/ .
3/ Les sels des composés selon l'une des revendications 1/ ou
2/ avec un acide minéral ou organique.
4/ Les esters des composés de formule générale I selon l'une des revendications 1/ à 3/ répondant à la formule générale I'

5/ La N-(2- thio 4-carboxy 6- oxo hexahydropyrimidinyl -3) carbonyl acétamidine selon la revendication 1/
6/ La N-(2,6- dioxo 4- carboxy hexahydropyrimidinyl -3) carbonyl acétamidine selon la revendication 1/.
7/ La N- (2,6- dioxo 4 -carboxy 5-méthyl hexahydropyrimidinyl-3) carbonyl acétamidine selon la revendication 1/ .
8/ La N-(2,6- dioxo 4-carboxy 5-méthyl hexahydropyrimidinyl -3) 2- carbonyl 2-méthyl acétamidine selon la revendication 1/ 9/ Les compositions pharmaceutiques renfermant à titre de principe actif au moins un composé selon l'une des revendications 1/ à 8/ éventuellement en association avec un autre principe actif d'action synergistique ou complémentaire, mélangé avec un excipient ou un véhicule inerte non toxique pharmaceutiquement-acceptable.
10/ Un procédé d'obtention des composés selon la revendication 1/ caractérisé en ce que l'on fait réagir un acide dihydro orotique de formule générale II




dans laquelle R4 est de l'hydrogène ou un radical alcoyle inférieur
X est du chlore ou du brome et Hal est un halogène autre que le fluor pour former un dérivé N - acylé de l'acide dihydro orotique de formule générale VI


Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19820902820 EP0119995A1 (fr) | 1982-09-28 | 1982-09-28 | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament |
| PCT/FR1982/000159 WO1984001385A1 (fr) | 1982-09-28 | 1982-09-28 | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/FR1982/000159 WO1984001385A1 (fr) | 1982-09-28 | 1982-09-28 | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1984001385A1 true WO1984001385A1 (fr) | 1984-04-12 |
Family
ID=9269774
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR1982/000159 WO1984001385A1 (fr) | 1982-09-28 | 1982-09-28 | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP0119995A1 (fr) |
| WO (1) | WO1984001385A1 (fr) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4855301A (en) * | 1986-10-09 | 1989-08-08 | E. R. Squibb & Sons, Inc. | 1,2,3,4-Tetrahydro-6-substituted-4-aryl(or heterocyclo)-3-((substituted amino)carbonyl)-2-thioxo (or oxo)-5-pyrimidinecarboxylic acids and esters |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2501205A1 (fr) * | 1981-03-09 | 1982-09-10 | Corbiere Jerome | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament |
-
1982
- 1982-09-28 WO PCT/FR1982/000159 patent/WO1984001385A1/fr unknown
- 1982-09-28 EP EP19820902820 patent/EP0119995A1/fr not_active Withdrawn
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2501205A1 (fr) * | 1981-03-09 | 1982-09-10 | Corbiere Jerome | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4855301A (en) * | 1986-10-09 | 1989-08-08 | E. R. Squibb & Sons, Inc. | 1,2,3,4-Tetrahydro-6-substituted-4-aryl(or heterocyclo)-3-((substituted amino)carbonyl)-2-thioxo (or oxo)-5-pyrimidinecarboxylic acids and esters |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0119995A1 (fr) | 1984-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA1209985A (fr) | Procede de preparation de nouveaux imino acides substitues | |
| EP0501892B1 (fr) | Dérivés hétérocycliques diazotés N-substitués par un groupement biphénylméthyle, leur préparation, les compositions pharmaceutiques en contenant | |
| EP0049658A1 (fr) | Iminodiacides substitués, leur préparation et compositions pharmaceutiques les contenant | |
| JPH0587504B2 (fr) | ||
| EP0465323A1 (fr) | Dérivés de pyrimidine antagonistes des récepteurs à l'angiotensine II, leurs procédés de préparation et compositions pharmaceutiques les contenant | |
| EP0296975A1 (fr) | Dérivés du diméthyl-2,2 chromannol-3, procédé d'obtention et compositions pharmaceutiques les contenant | |
| EP0370901A1 (fr) | Nouveaux dérivés du chromanne actifs sur le système nerveux central, leur procédé de préparation et les compositions pharmaceutiques en contenant | |
| EP0202157B1 (fr) | Amino-2 thiazoles N-substitués, leur procédé de préparation et leur application en thérapeutique | |
| EP0051020A1 (fr) | Acides aza bicyclooctane carboxyliques, leur préparation et compositions pharmaceutiques les contenant | |
| EP0532410A1 (fr) | Dérivés hétérocycliques N-substitués, comme inhibiteurs de l'angiotensive II | |
| EP0469984A2 (fr) | Dérivés de la N-sulfonyl indoline, leur préparation, les compositions pharmaceutiques en contenant | |
| FR2491469A1 (fr) | Nouveaux imino diacides substitues, leurs procedes de preparation et leur emploi comme inhibiteur d'enzyme | |
| US4134991A (en) | Derivatives of 2-(3-phenyl-2-aminopropionyloxy)-acetic acid | |
| SU1635899A3 (ru) | Способ получени 3-[(1Н-имидазол-4-ил)метил]-2-оксибензолметанолов | |
| EP0541407B1 (fr) | Dérivés bicycliques azotés, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| JPH0611739B2 (ja) | N―[6―メトキシ―5―(パーフルオロアルキル)―1―ナフトイル―n―メチルグリシンおよびチオナフトイル類似体 | |
| FR2564832A1 (fr) | Nouveaux n-acyl derives d'amino-acides et leurs esters, leur procede de preparation et medicaments les contenant | |
| CA1139763A (fr) | Procede d'obtention de nouveaux aryltrifluoroethanols | |
| JPH0536436B2 (fr) | ||
| FR2470767A1 (fr) | Nouveaux iminoacides substitues, leurs procedes de preparation et leur emploi comme inhibiteur d'enzymes | |
| EP0463944A1 (fr) | Acyl benzoxazolinones, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| WO1984001385A1 (fr) | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament | |
| FR2501205A1 (fr) | Nouveaux derives acyles de l'acide dihydro orotique, leur preparation et leur emploi comme medicament | |
| EP0002635B1 (fr) | Procédé de préparation de dérivés de thiéno (2,3-c) et (3,2-c) pyridines, nouveaux dérivés de la thiéno (2,3-c) pyridine obtenus et leur application thérapeutique | |
| JPH062748B2 (ja) | 新規ヒダントイン誘導体及び該化合物を有効成分として含有する医薬組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Designated state(s): BE CH DE FR GB LU NL |