US8853390B2 - Processes for preparing 1,2-substituted cyclopropyl derivatives - Google Patents
Processes for preparing 1,2-substituted cyclopropyl derivatives Download PDFInfo
- Publication number
- US8853390B2 US8853390B2 US13/232,751 US201113232751A US8853390B2 US 8853390 B2 US8853390 B2 US 8853390B2 US 201113232751 A US201113232751 A US 201113232751A US 8853390 B2 US8853390 B2 US 8853390B2
- Authority
- US
- United States
- Prior art keywords
- chiral
- amine
- hydrogen
- group
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related, expires
Links
- 0 *C1=C([1*])C([2*])=C([3*])C(C2CC2CN([4*])[5*])=C1* Chemical compound *C1=C([1*])C([2*])=C([3*])C(C2CC2CN([4*])[5*])=C1* 0.000 description 13
- RSRIUYJBNCLNQB-VXGBXAGGSA-N CC(=O)[C@H]1C[C@@H]1C1=CC=C(C)C=C1 Chemical compound CC(=O)[C@H]1C[C@@H]1C1=CC=C(C)C=C1 RSRIUYJBNCLNQB-VXGBXAGGSA-N 0.000 description 4
- GAGYDDATWSBNEG-ZJUUUORDSA-N CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 Chemical compound CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 GAGYDDATWSBNEG-ZJUUUORDSA-N 0.000 description 3
- RMRCDGPZBLGYGJ-JQWIXIFHSA-N CC[C@H]1C[C@@H]1C1=CC=C(C)C=C1 Chemical compound CC[C@H]1C[C@@H]1C1=CC=C(C)C=C1 RMRCDGPZBLGYGJ-JQWIXIFHSA-N 0.000 description 3
- YOAAWXGZILTNEY-UKRRQHHQSA-N BrC1=CC=C([C@H]2C[C@@H]2CN2CCCCC2)C=C1 Chemical compound BrC1=CC=C([C@H]2C[C@@H]2CN2CCCCC2)C=C1 YOAAWXGZILTNEY-UKRRQHHQSA-N 0.000 description 2
- ADXJJDKDQDJSMJ-CBZTYRTBSA-N *.*.*.*.CC(C)(C)OC(=O)CP(C)(C)=O.CC1=CC=C(/C=C/C(=O)OC(C)(C)C)C=C1.CC1=CC=C(C2CC2C(=O)OC(C)(C)C)C=C1.CC1=CC=C(C=O)C=C1.CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 Chemical compound *.*.*.*.CC(C)(C)OC(=O)CP(C)(C)=O.CC1=CC=C(/C=C/C(=O)OC(C)(C)C)C=C1.CC1=CC=C(C2CC2C(=O)OC(C)(C)C)C=C1.CC1=CC=C(C=O)C=C1.CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 ADXJJDKDQDJSMJ-CBZTYRTBSA-N 0.000 description 1
- HQHMZSBHVZJWTN-JFVNGJROSA-N B.C1CCOC1.CC(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1.CCCCNC.CC[C@H]1C[C@@H]1C1=CC=C(Br)C=C1.CC[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1.O=C(O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1.O=C1C=CC=NN1 Chemical compound B.C1CCOC1.CC(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1.CCCCNC.CC[C@H]1C[C@@H]1C1=CC=C(Br)C=C1.CC[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1.O=C(O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1.O=C1C=CC=NN1 HQHMZSBHVZJWTN-JFVNGJROSA-N 0.000 description 1
- XRBKNTXOIJFLEF-TZMCWYRMSA-N BrC1=CC=C([C@H]2C[C@@H]2CN2CCCC2)C=C1 Chemical compound BrC1=CC=C([C@H]2C[C@@H]2CN2CCCC2)C=C1 XRBKNTXOIJFLEF-TZMCWYRMSA-N 0.000 description 1
- IPELTGMLRHJEAJ-TZMCWYRMSA-N BrC1=CC=C([C@H]2C[C@@H]2CN2CCOCC2)C=C1 Chemical compound BrC1=CC=C([C@H]2C[C@@H]2CN2CCOCC2)C=C1 IPELTGMLRHJEAJ-TZMCWYRMSA-N 0.000 description 1
- XCBQXNGVZLLNLT-OXQJLTHESA-N CC(=O)[C@H]1C[C@@H]1C1=CC=C(C)C=C1.CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 Chemical compound CC(=O)[C@H]1C[C@@H]1C1=CC=C(C)C=C1.CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 XCBQXNGVZLLNLT-OXQJLTHESA-N 0.000 description 1
- GVZYQQFHOCMYGC-IZRXFCGDSA-N CC(=O)[C@H]1C[C@@H]1C1=CC=C(C)C=C1.CC[C@H]1C[C@@H]1C1=CC=C(C)C=C1 Chemical compound CC(=O)[C@H]1C[C@@H]1C1=CC=C(C)C=C1.CC[C@H]1C[C@@H]1C1=CC=C(C)C=C1 GVZYQQFHOCMYGC-IZRXFCGDSA-N 0.000 description 1
- IPKDUXLMDVHQFR-NQAVBDFFSA-N CC(C)(C)OC(=O)/C=C/C1=CC=C(Br)C=C1.CC(C)(C)OC(=O)C1CC1C1=CC=C(Br)C=C1.CC(C)(C)OC(=O)CP(C)(C)=O.C[S+](C)(C)=O.O=C(O)C1CC1C1=CC=C(Br)C=C1.O=CC1=CC=C(Br)C=C1.[I-] Chemical compound CC(C)(C)OC(=O)/C=C/C1=CC=C(Br)C=C1.CC(C)(C)OC(=O)C1CC1C1=CC=C(Br)C=C1.CC(C)(C)OC(=O)CP(C)(C)=O.C[S+](C)(C)=O.O=C(O)C1CC1C1=CC=C(Br)C=C1.O=CC1=CC=C(Br)C=C1.[I-] IPKDUXLMDVHQFR-NQAVBDFFSA-N 0.000 description 1
- HXHXLGMTGMUXJY-PWYQZFHGSA-O CC(C)C1=CC=CC=C1.C[C@@H]([NH3+])C1CCCCC1.O=C(O)C1CC1C1=CC=C(Br)C=C1.O=C(O)C1CC1C1=CC=C(Br)C=C1 Chemical compound CC(C)C1=CC=CC=C1.C[C@@H]([NH3+])C1CCCCC1.O=C(O)C1CC1C1=CC=C(Br)C=C1.O=C(O)C1CC1C1=CC=C(Br)C=C1 HXHXLGMTGMUXJY-PWYQZFHGSA-O 0.000 description 1
- GAGYDDATWSBNEG-UHFFFAOYSA-N CC1=CC=C(C2CC2C(=O)O)C=C1 Chemical compound CC1=CC=C(C2CC2C(=O)O)C=C1 GAGYDDATWSBNEG-UHFFFAOYSA-N 0.000 description 1
- WERRACVCHSGTAH-UXQCFNEQSA-N CC1=CC=C(C2CC2C(=O)O)C=C1.CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 Chemical compound CC1=CC=C(C2CC2C(=O)O)C=C1.CC1=CC=C([C@H]2C[C@@H]2C(=O)O)C=C1 WERRACVCHSGTAH-UXQCFNEQSA-N 0.000 description 1
- NYHWHDCODVERDM-OLZOCXBDSA-N CCN(CC)C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound CCN(CC)C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 NYHWHDCODVERDM-OLZOCXBDSA-N 0.000 description 1
- YXSKJHVBNZEUQD-TZMCWYRMSA-N CCN(CC)C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound CCN(CC)C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 YXSKJHVBNZEUQD-TZMCWYRMSA-N 0.000 description 1
- OPGHIXILYXDSIA-NVXWUHKLSA-N CCN(CC)C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 Chemical compound CCN(CC)C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 OPGHIXILYXDSIA-NVXWUHKLSA-N 0.000 description 1
- QLMPMAHXBVXDPB-MNOVXSKESA-N CN(C)C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound CN(C)C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 QLMPMAHXBVXDPB-MNOVXSKESA-N 0.000 description 1
- WYPJAPCMQZBXCK-ZYHUDNBSSA-N CN(C)C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound CN(C)C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 WYPJAPCMQZBXCK-ZYHUDNBSSA-N 0.000 description 1
- CNPJNPISVZTLAF-UKRRQHHQSA-N CN(C)C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 Chemical compound CN(C)C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 CNPJNPISVZTLAF-UKRRQHHQSA-N 0.000 description 1
- KUAATBRUDXUAOF-UZJXMSKXSA-N C[C@@H](N)C1CCCCC1.O=C(O)C1CC1C1=CC=C(Br)C=C1.O=C(O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound C[C@@H](N)C1CCCCC1.O=C(O)C1CC1C1=CC=C(Br)C=C1.O=C(O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 KUAATBRUDXUAOF-UZJXMSKXSA-N 0.000 description 1
- VJCVJUMJJXGRJG-HONMWMINSA-N C[C@@H]1CCCN1C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound C[C@@H]1CCCN1C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 VJCVJUMJJXGRJG-HONMWMINSA-N 0.000 description 1
- NMZRPOCZSZTBNJ-UXIGCNINSA-N C[C@@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound C[C@@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 NMZRPOCZSZTBNJ-UXIGCNINSA-N 0.000 description 1
- YQMKBLZYOGEKLS-QGPMSJSTSA-N C[C@@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 Chemical compound C[C@@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 YQMKBLZYOGEKLS-QGPMSJSTSA-N 0.000 description 1
- VJCVJUMJJXGRJG-GDLCADMTSA-N C[C@H]1CCCN1C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound C[C@H]1CCCN1C(=O)[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 VJCVJUMJJXGRJG-GDLCADMTSA-N 0.000 description 1
- NMZRPOCZSZTBNJ-NJZAAPMLSA-N C[C@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 Chemical compound C[C@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(Br)C=C1 NMZRPOCZSZTBNJ-NJZAAPMLSA-N 0.000 description 1
- YQMKBLZYOGEKLS-YXJHDRRASA-N C[C@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 Chemical compound C[C@H]1CCCN1C[C@H]1C[C@@H]1C1=CC=C(N2N=CC=CC2=O)C=C1 YQMKBLZYOGEKLS-YXJHDRRASA-N 0.000 description 1
- HOVHKJPHKDFKPE-OLZOCXBDSA-N O=C([C@H]1C[C@@H]1C1=CC=C(Br)C=C1)N1CCCC1 Chemical compound O=C([C@H]1C[C@@H]1C1=CC=C(Br)C=C1)N1CCCC1 HOVHKJPHKDFKPE-OLZOCXBDSA-N 0.000 description 1
- ZCVRZVLGWKLDCX-KGLIPLIRSA-N O=C([C@H]1C[C@@H]1C1=CC=C(Br)C=C1)N1CCCCC1 Chemical compound O=C([C@H]1C[C@@H]1C1=CC=C(Br)C=C1)N1CCCCC1 ZCVRZVLGWKLDCX-KGLIPLIRSA-N 0.000 description 1
- PVYODCWWKWZHDP-OLZOCXBDSA-N O=C([C@H]1C[C@@H]1C1=CC=C(Br)C=C1)N1CCOCC1 Chemical compound O=C([C@H]1C[C@@H]1C1=CC=C(Br)C=C1)N1CCOCC1 PVYODCWWKWZHDP-OLZOCXBDSA-N 0.000 description 1
- RRDUNQBJWMNJKE-NVXWUHKLSA-N O=C1C=CC=NN1C1=CC=C([C@H]2C[C@@H]2CN2CCCC2)C=C1 Chemical compound O=C1C=CC=NN1C1=CC=C([C@H]2C[C@@H]2CN2CCCC2)C=C1 RRDUNQBJWMNJKE-NVXWUHKLSA-N 0.000 description 1
- IZIAFQHCCPHXGT-SJLPKXTDSA-N O=C1C=CC=NN1C1=CC=C([C@H]2C[C@@H]2CN2CCCCC2)C=C1 Chemical compound O=C1C=CC=NN1C1=CC=C([C@H]2C[C@@H]2CN2CCCCC2)C=C1 IZIAFQHCCPHXGT-SJLPKXTDSA-N 0.000 description 1
- OOFRFROKBVNNTL-NVXWUHKLSA-N O=C1C=CC=NN1C1=CC=C([C@H]2C[C@@H]2CN2CCOCC2)C=C1 Chemical compound O=C1C=CC=NN1C1=CC=C([C@H]2C[C@@H]2CN2CCOCC2)C=C1 OOFRFROKBVNNTL-NVXWUHKLSA-N 0.000 description 1
- IZIAFQHCCPHXGT-DAFXYXGESA-N O=C1N(c2ccc([C@H]3C(CN4CCCCC4)C3)cc2)N=CC=C1 Chemical compound O=C1N(c2ccc([C@H]3C(CN4CCCCC4)C3)cc2)N=CC=C1 IZIAFQHCCPHXGT-DAFXYXGESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/14—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/16—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of a saturated carbon skeleton containing rings other than six-membered aromatic rings
- C07C211/17—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of a saturated carbon skeleton containing rings other than six-membered aromatic rings containing only non-condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/58—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/41—Preparation of salts of carboxylic acids
- C07C51/412—Preparation of salts of carboxylic acids by conversion of the acids, their salts, esters or anhydrides with the same carboxylic acid part
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/43—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C61/00—Compounds having carboxyl groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C61/16—Unsaturated compounds
- C07C61/40—Unsaturated compounds containing halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C67/347—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by addition to unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/06—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with radicals, containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/06—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by halogen atoms or nitro radicals
- C07D295/073—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by halogen atoms or nitro radicals with the ring nitrogen atoms and the substituents separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/10—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by doubly bound oxygen or sulphur atoms
- C07D295/104—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by doubly bound oxygen or sulphur atoms with the ring nitrogen atoms and the doubly bound oxygen or sulfur atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- the present invention relates to processes for preparing chiral cyclopropyl amine derivatives and salts thereof; intermediates useful for the preparation of such compounds and salts; pharmaceutical compositions comprising the compounds and salts; and method of using such compositions.
- the cyclopropyl amine derivatives are useful for binding to histamine H 3 receptor sites and for providing therapeutic agents for histamine H 3 mediated disease.
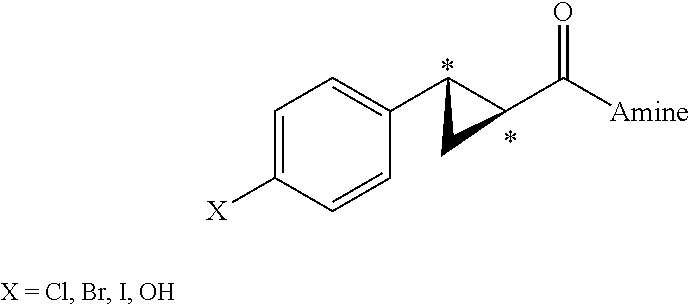
- chiral 1,2-substituted cyclopropyl carboxylic acid and chiral 1,2-substituted cyclopropyl amide derivatives are intermediate compounds useful for the preparation of chiral cyclopropyl amine derivatives of a general formula:
- R 1 , R 2 , R 3 , R 3a , R 3b , R 4 , and R 5 are as defined below.
- Compounds of formula (II) are described in WO 2007150010, published on Dec. 27, 2007, corresponding to U.S. patent application Ser. No. 11/766,987, filed on Jun. 22, 2007, and U.S. patent application Ser. No. 11/956,816, filed on Dec. 14, 2007, each of which are all hereby incorporated by reference.
- the present invention offers a more efficient process to obtain chiral compounds of formula (II) via the chiral resolution of an aryl-cyclopropanecarboxylic acid with chiral amines.
- the chiral resolution step forms a diasteriomeric chiral salt, which is crystallized to obtain an enantiomerically pure salt.
- the enantiomerically pure arylcyclopropyl carboxylic acid is obtained upon breaking up the salt.
- the resulting enantiomerically pure cyclopropyl carboxylic acids can be reacted with various amines to form amides, which can be reduced to form chiral amine derivatives.
- the intermediate chiral amines can be further coupled with a desired aromatic or heteroaromatic reagent to provide compounds of formula (II).
- histamine H 3 receptor antagonists have been identified as histamine H 3 receptor antagonists.
- Various histamine H 3 receptor antagonists are currently in clinical development for treatment of disease.
- Diseases for which histamine H 3 receptor antagonists are under clinical study include, for example, schizophrenia, cognitive deficits of schizophrenia, Alzheimer's disease, narcolepsy, cataplexy, sleep disorder, hyperalgesia, allergic rhinitis, obesity, attention-deficit hyperactivity disorder, and dementia.
- histamine H 3 receptor ligands can demonstrate therapeutic effect are deficits in attention, diseases with deficits of memory or learning, cognitive deficits and dysfunction in psychiatric disorders, mild cognitive impairment, epilepsy, seizures, and asthma, motion sickness, dizziness, Meniere's disease, vestibular disorders, vertigo, diabetes, type II diabetes, Syndrome X, insulin resistance syndrome, metabolic syndrome, pain, including neuropathic pain, neuropathy, pathological sleepiness, jet lag, drug abuse, mood alteration, bipolar disorder, depression, obsessive compulsive disorder, Tourette's syndrome, Parkinson's disease, and medullary thyroid carcinoma, melanoma, and polycystic ovary syndrome.
- the present invention relates to a process for preparing chiral cyclopropyl amine derivatives of formula (II), as described herein.
- the present invention relates to a process for preparing the chiral salts of aryl-cyclopropanecarboxylic acid with a chiral amine.
- the present invention also is directed to the chiral compounds and salts thereof prepared by the process for preparing chiral salts described above.
- the present invention also is directed a chiral aryl-cyclopropanecarboxylic acid salt.
- the present invention also is directed to various intermediates useful for preparing chiral compounds of formula (II).
- compositions including pharmaceutical compositions
- compounds of formula (II) or a salt thereof that are prepared by the above processes.
- the present invention also is directed to methods of using the compositions of the invention.
- aryl as used herein means a monocyclic hydrocarbon aromatic ring system. Representative examples of aryl include, but are not limited to, phenyl.
- aryl groups of this invention are substituted with 0, 1, 2, 3, 4, or 5 substituents independently selected from acyl, acyloxy, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxyimino, alkoxysulfonyl, alkyl, alkylcarbonyl, alkylsulfonyl, alkynyl, amido, carboxy, cyano, cycloalkylcarbonyl, formyl, haloalkoxy, haloalkyl, halogen, hydroxy, hydroxyalkyl, mercapto, nitro, thioalkoxy, NR A R B , and (NR A R B )sulfonyl.
- substituents independently selected from acyl, acyloxy, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxyimino, alkoxysul
- heteroaryl refers to an aromatic ring containing one or more heteroatoms independently selected from nitrogen, oxygen, or sulfur, or a tautomer thereof. Such rings can be monocyclic or bicyclic as further described herein. Heteroaryl rings are connected to the parent molecular moiety.
- heteroaryl or “5- or 6-membered heteroaryl ring”, as used herein, refer to 5- or 6-membered aromatic rings containing 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or a tautomer thereof.
- examples of such rings include, but are not limited to, a ring wherein one carbon is replaced with an O or S atom; one, two, or three N atoms arranged in a suitable manner to provide an aromatic ring; or a ring wherein two carbon atoms in the ring are replaced with one O or S atom and one N atom.
- Such rings can include, but are not limited to, a six-membered aromatic ring wherein one to four of the ring carbon atoms are replaced by nitrogen atoms, five-membered rings containing a sulfur, oxygen, or nitrogen in the ring; five membered rings containing one to four nitrogen atoms; and five membered rings containing an oxygen or sulfur and one to three nitrogen atoms.
- 5- to 6-membered heteroaryl rings include, but are not limited to, furyl, imidazolyl, isoxazolyl, isothiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridinyl, pyrimidinyl, pyrrolyl, tetrazolyl, [1,2,3]thiadiazolyl, [1,2,3]oxadiazolyl, thiazolyl, thienyl, [1,2,3]triazinyl, [1,2,4]triazinyl, [1,3,5]triazinyl, [1,2,3]triazolyl, and [1,2,4]triazolyl.
- bicyclic heteroaryl or “8- to 12-membered bicyclic heteroaryl ring”, as used herein, refers to an 8-, 9-, 10-, 11-, or 12-membered bicyclic aromatic ring containing at least 3 double bonds, and wherein the atoms of the ring include one or more heteroatoms independently selected from oxygen, sulfur, and nitrogen.
- bicyclic heteroaryl rings include indolyl, benzothienyl, benzofuranyl, indazolyl, benzimidazolyl, benzothiazolyl, benzoxazolyl, benzoisothiazolyl, benzoisoxazolyl, quinolinyl, isoquinolinyl, quinazolinyl, quinoxalinyl, phthalazinyl, pteridinyl, purinyl, naphthyridinyl, cinnolinyl, thieno[2,3-d]imidazole, thieno[3,2-b]pyridinyl, and pyrrolopyrimidinyl.
- Heteroaryl groups of the invention may be substituted with hydrogen, or optionally substituted with one or more substituents independently selected from acyl, acyloxy, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxyimino, alkoxysulfonyl, alkyl, alkylcarbonyl, alkylsulfonyl, amido, carboxy, cyano, cycloalkyl, fluoroalkoxy, formyl, haloalkoxy, haloalkyl, halogen, hydroxy, hydroxyalkyl, mercapto, nitro, alkylthio, —NR A R B , and (NR A R B )carbonyl.
- substituents independently selected from acyl, acyloxy, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxyimino, alkoxy
- Monocyclic heteroaryl or 5- or 6-membered heteroaryl rings are substituted with 0, 1, 2, 3, 4, or 5 substituents.
- Bicyclic heteroaryl or 8- to 12-membered bicyclic heteroaryl rings are substituted with 0, 1, 2, 3, 4, 5, 6, 7, 8, or 9 substituents.
- Heteroaryl groups of the present invention may be present as tautomers.
- heterocyclic ring and “heterocycle”, as used herein, refer to a 4- to 12-membered monocyclic or bicyclic ring containing one, two, three, four, or five heteroatoms independently selected from the group consisting of nitrogen, oxygen, and sulfur and also containing either at least one carbon atom attached to four other atoms or one carbon atom substituted with an oxo group and attached to two other atoms.
- Four- and five-membered rings may have zero or one double bond.
- Six-membered rings may have zero, one, or two double bonds.
- Seven- and eight-membered rings may have zero, one, two, or three double bonds.
- the non-aromatic heterocycle groups of the invention can be attached through a carbon atom or a nitrogen atom.
- the non-aromatic heterocycle groups may be present in tautomeric form.
- Representative examples of nitrogen-containing heterocycles include, but are not limited to, azepanyl, azetidinyl, aziridinyl, azocanyl, dihydropyridazinyl, dihydropyridinyl, dihydropyrimidinyl, morpholinyl, piperazinyl, piperidinyl, pyrrolidinyl, pyrrolinyl, dihydrothiazolyl, dihydropyridinyl, and thiomorpholinyl.
- non-nitrogen containing non-aromatic heterocycles include, but are not limited to, dioxanyl, dithianyl, tetrahydrofuryl, dihydropyranyl, tetrahydropyranyl, and [1,3]dioxolanyl.
- the heterocycles of the invention are substituted with hydrogen, or optionally substituted with 0, 1, 2, 3, 4, 5, 6, 7, 8, or 9 substituents independently selected from acyl, acyloxy, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxyimino, alkoxysulfonyl, alkyl, alkylsulfonyl, amido, arylalkyl, arylalkoxycarbonyl, carboxy, cyano, formyl, haloalkoxy, haloalkyl, halogen, hydroxy, hydroxyalkyl, mercapto, nitro, oxo, thioalkoxy, —NR A R B , and (NR A R B )sulfonyl.
- substituents independently selected from acyl, acyloxy, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl,
- heterocycles include, but are not limited to, azetidin-2-one, azepan-2-one, isoindolin-1,3-dione, (Z)-1H-benzo[e][1,4]diazepin-5(4H)-one, pyridazin-3(2H)-one, pyridin-2(1H)-one, pyrimidin-2(1H)-one, pyrimidin-2,4(1H,3H)-dione, pyrrolidin-2-one, benzo[d]thiazol-2(3H)-one, pyridin-4(1H)-one, imidazolidin-2-one, 1H-imidazol-2(3H)-one, piperidin-2-one, tetrahydropyrimidin-2(1H)-one, 1H-benzo[d]imidazol-2(3H)-one, [1,2,4]thiadiazolonyl, [1,2,5]thiadiazol
- room temperature refers to about 25° C.
- room temperature can vary within a few degrees depending on the environment in which any reaction is conducted. For example, temperatures from about 20° C. to about 30° C. are considered to be room temperature.
- R denotes a chiral center that can be designated as a R- or S-stereocenter.
- These stereoisomers are “R” or “S” depending on the configuration of substituents around the chiral carbon atom.
- R and S used herein are configurations as defined in the IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, Pure Appl. Chem., 1976, 45:13-30.
- chiral refers to a compound that is enantiopure or contains only one of a possible two configurations at a designated stereocenter.
- THF for tetrahydrofuran
- CDI 1,1′-carbonyldiimidazole
- NaH for sodium hydride
- NaHCO 3 for sodium bicarbonate
- MTBE for methyl t-butyl ether
- BH 3 for borane
- DMSO for dimethylsulfoxide
- EtOAc for ethyl acetate
- NaOtBu sodium t-butoxide
- OTf for trifluoromethanesulfonate.
- NLT is used to denote “Not Less Than”.
- the present invention relates to a process for preparing a chiral compound of formula:
- R 2 , R 3 , R 3a , R 3b are hydrogen;
- R 1 is a 5- to 6-membered heteroaryl ring, cyanophenyl, a 8- to 12-membered bicyclic heteroaryl ring, or a 4- to 12-membered heterocyclic ring; and
- R 4 and R 5 taken together with the nitrogen atom to which they are attached form an amine moiety represented by structure:
- R 7 , R 8 , R 9 , and R 10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl;
- R 11 , R 12 , R 13 , and R 14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl;
- R x and R y are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino;
- Q is O or S; and
- m is an integer from 1 to 5.
- enantiomerically pure arylcyclopropanecarboxylic acids salt can be broken-up to release the enantiomerically pure arylcyclopropanecarboxylic acids that can be further reacted with amines, reduced, and coupled with a suitable aromatic, heteroaromatic, or heterocycle group to provide compounds of formula (II).
- the chiral amine is selected from the group consisting of (S)-( ⁇ )- ⁇ -methylbenzylamine, (R)-(+)-N-benzyl- ⁇ -methylbenzylamine, (S)-( ⁇ )-N-benzyl- ⁇ -methylbenzylamine, (R)-(+)-N,N-dimethyl-1-phenylethylamine, (S)-( ⁇ )-N,N-dimethyl-1-phenylethylamine, [R—(R*,R*)]-(+)-bis(a-methylbenzyl)amine, [S—(R*,R*)]-( ⁇ )-bis(a-methylbenzyl)amine, (S)-(+)-1-cyclohexylethylamine, (R)-(+)-1-(1-naphthyl)ethylamine, (S)-( ⁇ )-1-(1-naphthyl)ethylamine, (1
- the chiral amine is (R)-( ⁇ )-1-cyclohexylethylamine. In another embodiment, the chiral amine is (R)-(+)-a-methylbenzylamine.
- the reaction is carried out in a polar organic solvent.
- the polar organic solvent is an alcohol.
- the alcohol can be selected from the group consisting of methanol, ethanol, isopropyl alcohol, tert-butyl alcohol, and n-butyl alcohol.
- the alcohol is isopropyl alcohol.
- the alcohol is tert-butyl alcohol.
- the reaction is carried at a temperature of from about room temperature to about 75° C.
- the temperature is from about 40° C. to about 60° C. In one particular embodiment, the temperature at which the reaction is carried out is from about 45° C. to about 50° C. Typically, the reaction is accomplished in a period of about 1 to 48 hours, however, the length of the reaction time can vary depending on the particular conditions and quality of the reagents, among other aspects of the reaction. In a preferred embodiment, the reaction is conducted for a time period of about 8 hours.
- the aryl-cyclopropanecarboxylic acid is prepared from arylaldehyde. An illustration of a process for preparing arylcyclopropanecarboxylic acid from aryl aldehyde is shown below in Scheme A.
- aryl aldehyde (A-1) is treated with t-butyldimethylphosphonoacetate to provide tert-butyl arylacrylate (A-2).
- Tert-butyl arylacrylate is treated a sulfoxonium ylide to provide tert-butyl arylcyclopropanecarboxylic acid ester (A-3).
- Hydrolysis of the tert-butyl arylcyclopropanecarboxylic acid ester under basic conditions with lithium hydroxide provides arylcyclopropanecarboxylic acid (A-4).
- aryl aldehyde (A-1) is treated with t-butyldimethylphosphonoacetate with any strong base in a non-polar solvent.
- the strong base is a metal hydride.
- metal hydride base are lithium hydride and sodium hydride.
- the strong base can also be potassium t-butyloxide, sodium t-butyloxide, lithium t-butyloxide.
- sodium hydride (NaH) is the base or potassium t-butyloxide. The reaction is carried out in any non-polar solvent.
- the non-polar solvent is an organic solvent, for example, toluene, hexane, benzene, 1,4-dioxane, chloroform, or diethyl ether.
- the organic solvent is toluene.
- Suitable sulfoxonium ylides for treating tert-butyl arylacrylate (A-2) to provide tert-butyl arylcyclopropanecarboxylic acid ester (A-3) can be dimethylsulfoxonium iodide methylide (Corey-Chaykovsky Regent) and trimethylsulfoxonium iodide reagent.
- the sulfoxonium reagent is trimethylsulfoxonium iodide reagent.
- the sulfoxonium reagent is dimethylsulfoxonium iodide methylide.
- the reaction is carried out in a polar solvent.
- the polar solvent can be any suitable polar aprotic solvent.
- Suitable polar aprotic solvents include, but are not limited to, dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide, or mixtures thereof.
- the solvent is a mixture of dimethyl sulfoxide and dimethyl acetamide.
- the mixture can be in a ratio of from about 1:1 to about 2:1 dimethyl sulfoxide/dimethyl acetamide.
- the reaction can be carried out at any temperature, however, in the reaction mixture is maintained at a temperature of less than about 10° C.
- any base is suitable for use in the hydrolysis of the ester of tert-butyl arylcyclopropanecarboxylic acid (A-3) to provide arylcyclopropanecarboxylic acid (A-4).
- the base is any metal hydroxide base.
- Such base can include, for example, sodium hydroxide, lithium hydroxide, and potassium hydroxide.
- Suitable amine reagents can be represented by the formula:
- R 7 , R 8 , R 9 , and R 10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl;
- R 11 , R 12 , R 13 , and R 14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl;
- R x and R y are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino;
- Q is O or S; and
- m is an integer from 1 to 5.
- Suitable amines for the reaction can include any amine of formula (a), as defined above.
- Such amine can be more particularly selected from pyrrolidine, 2-(S)-methylpyrrolidine, 2-(R)-methylpyrrolidine, 3-methylpyrrolidine, 2-fluoropyrrolidine, 3-fluoropyrrolidine, 2-hydroxypyrrolidine, 3-hydroxypyrrolidine, 2-hydroxymethylpyrrolidine, and 3-hydroxymethylpyrrolidine.
- Other suitable amines can be more particularly selected from pyridine, 2-methylpyridine, 3-methylpyridine, 4-methylpyridine, 2-fluoropyridine, 3-fluoropyridine, 4-fluoropyridine, 2-hydroxypyridine, 3-hydroxypyridine, 4-hydroxymethylpyridine, 2-hydroxymethylpyridine, 3-hydroxymethylpyridine, and 4-hydroxymethylpyridine.
- the amines are those wherein one of the substituents represented by R 7 , R 8 , R 9 , and R 10 is hydrogen or alkyl and the other substituents are hydrogen.
- Particular preferred examples are pyrrolidine, 2-(S)-methylpyrrolidine, and 2-(R)-methylpyrrolidine.
- Suitable amines are those of formula (b), as defined above. Such amine may be more particularly selected from dimethylamine, diethylamine, methylamine, and ethylamine. In one particular embodiment, the amines are those wherein one of the substituents represented by R 7 , R 8 , R 9 , and R 10 is hydrogen or alkyl and the other substituents are hydrogen. Particular preferred examples are dimethylamine and diethylamine.
- Additional reagents having an amine group are those of formula (c), as defined above.
- Such amine may be more particularly selected from morpholine and thiomorpholine.
- the amine reagent is morpholine.
- the reaction is carried out using N,N′-carbonyldiimidazole.
- the arylcyclopropanecarboxylic acid, amine, and N,N′-carbonyldiimidazole are combined in an organic solvent.
- suitable organic solvents are tetrahydrofuran, toluene, 1,2-dimethoxyethane, 1,4-dioxane, N-methyl-pyrrolidinone, dimethylacetamide, and dimethylformamide.
- the solvent is tetrahydrofuran.
- the solvent is toluene. Tetrahydrofuran is the most preferred solvent.
- the reaction can be accomplished at room temperature.
- the reaction is accomplished in a period of about 1 to 48 hours, however, the length of the reaction time can vary depending on the particular conditions and quality of the reagents, among other aspects of the reaction. In a preferred embodiment, the reaction is conducted for a time period of about 8 hours.
- the cyclopropanecarboxylic acid amide is reduced using a reducing agent selected from borane reducing reagents.
- Suitable reducing agents are, for example, borane tetrahydrofuran complex, diborane, borane dimethylsulfide complex, a combination of sodium borohydride and sodium trifluoride.
- the reaction is conducted in a polar, aprotic solvent.
- suitable solvents are tetrahydrofuran, 1,2-dimethoxyethane, 1,2-diethoxyethane, 2-methyltetrahydrofuran, 1,4-dioxane, and methyl-tert-butyl ethers.
- the preferred solvent is tetrahydrofuran.
- the reaction can be conducted at any suitable temperature. Typically, the reaction is conducted at a temperature between 0° C. and 80° C. In a preferred embodiment, the reaction is conducted at a temperature of about 50° C. Typically, the reaction is accomplished in a period of about 1 to 48 hours, however, the length of the reaction time can vary depending on the particular conditions and quality of the reagents, among other aspects of the reaction. In one embodiment, the reaction is conducted for a time period of about 8 hours.
- the compound of formula (I-b) undergoes coupling reactions to provide the compounds of formula (II).
- Coupling conditions commonly referred to as metal-catalyzed reaction including palladium, nickel, iron or copper catalyzed reaction, such as Ullmann reaction conditions, are preferred for the reaction.
- Reagent suitable for providing a moiety within the definition of R 1 can be used.
- Reagents suitable for the reaction can include, for example, 5- to 6-membered heteroaryl, 8- to 12-membered bicyclic heteroaryl, and 4- to 12-membered heterocyclic reagents.
- Examples of particular 5- to 6-membered heteroaryl reagents include, but are not limited to, pyridazin-3(2H)-one, pyridin-2(1H)-one, pyrimidin-2(1H)-one, pyrimidin-2,4(1H,3H)-dione, pyrrolidin-2-one, benzo[d]thiazol-2(3H)-one, pyridin-4(1H)-one, pyrroline, imidazolidin-2-one, 1H-imidazol-2(3H)-one, piperidin-2-one, tetrahydropyrimidin-2(1H)-one, [1,2,4]thiadiazolone, [1,2,5]thiadiazolone, [1,3,4]thiadiazinone, [1,2,4]oxadiazolone, [1,2,5]oxadiazolone, and [1,3,4]oxadiazin-one.
- Examples of particular 4- to 12-membered heterocyclic reagents include, but are not limited to, azepane, azetidine, aziridine, azocane, dihydropyridine, dihydropyrimidine, piperidine, pyrrolidine, dihydrothiazole, dihydropyridine, thiomorpholine, dioxane, dithiane, tetrahydrofuran, dihydropyrane, tetrahydropyran, [1,3]dioxolane, azetidin-2-one, and azepan-2-one.
- Examples of particular 8- to 12-membered bicyclic heteroaryl reagents include, but are not limited to, isoindolin-1,3-dione, (Z)-1H-benzo[e][1,4]diazepin-5(4H)-one, and 1H-benzo[d]imidazol-2(3H)-one.
- the reaction is conducted with a copper catalyst and base in a polar aprotic solvent in the presence of N,N′-dimethylenediamine.
- the copper catalyst can be any copper catalyst.
- the copper catalyst is a copper (I) catalyst. Examples of such catalysts are, for example, copper (I) iodide, copper (I) bromide, and copper (I) chloride. Copper (I) iodide is preferred.
- the base is any suitable organic base.
- Examples of such base can include, for example, potassium carbonate (K 2 CO 3 ), potassium phosphate (K 3 PO 4 ), cesium carbonate (Cs 2 CO 3 ), sodium methoxide (NaOMe), sodium tert-butoxide (NaOt-Bu), sodium acetate (NaOAc), and potassium tert-butoxide (KOt-Bu).
- the base is K 2 CO 3 .
- the base is K 3 PO 4 .
- the basic solvent can be any polar aprotic solvent.
- polar aprotic solvent examples include, for example, dimethyl acetamide, dimethyl formamide, 1-methyl-2-pyrrolidinone, and pyridine.
- the polar aprotic solvent is pyridine.
- the reaction can be conducted at any suitable temperature. Typically, the reaction is conducted at a temperature between 0° C. and 140° C. In a preferred embodiment, the reaction is conducted at a temperature of about 115° C. Typically, the reaction is accomplished in a period of about 1 to 48 hours, however, the length of the reaction time can vary depending on the particular conditions and quality of the reagents, among other aspects of the reaction. In one embodiment, the reaction is conducted for a time period of about 8 hours.
- the present invention in one embodiment, also relates to compounds that are:
- the chiral amines are (S)-( ⁇ )- ⁇ -methylbenzylamine, (R)-(+)-N-benzyl-a-methylbenzylamine, (S)-( ⁇ )-N-benzyl- ⁇ -methylbenzylamine, (R)-(+)-N,N-dimethyl-1-phenylethylamine, (S)-( ⁇ )-N,N-dimethyl-1-phenylethylamine, [R—(R*,R*)]-(+)-bis( ⁇ -methylbenzyl)amine, [S-(R*,R*)]-( ⁇ )-bis(a-methylbenzyl)amine, (S)-(+)-1-cyclohexylethylamine, (R)-(+)-1-(1-naphthyl)ethylamine, (S)-( ⁇ )-1-(1-naphthyl)ethylamine, (1R,2R,3R
- the chiral amines also can be selected from (R)-( ⁇ )-1-cyclohexylethylamine or (R)-(+)- ⁇ -methylbenzylamine.
- the chiral amine is (R)-( ⁇ )-1-cyclohexylethylamine.
- the chiral amine is (R)-(+)- ⁇ -methylbenzylamine.
- the present invention also relates to compounds that are:
- the present invention in another embodiment, relates to a compound of formula:
- R 7 , R 8 , R 9 , and R 10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl;
- R 11 , R 12 , R 13 , and R 14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl;
- R x and R y are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino;
- Q is O or S; and
- m is an integer from 1 to 5.
- the present invention in another embodiment, relates to a compound of formula:
- R 7 , R 8 , R 9 , and R 10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl;
- R 11 , R 12 , R 13 , and R 14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl;
- R x and R y are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino;
- Q is O or S; and
- m is an integer from 1 to 5.
- the present invention also includes isotopically-labeled compounds, which are identical to those recited in Formula (I-a), (I-b), and (II), but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the invention are hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine, and chlorine, such as but not limited to 2 H, 3 H, 13 C, 15 C, 15 N, 18 O, 17 O, 31 P, 32 P, 35 S, 18 F, and 36 Cl, respectively.
- isotopes such as deuterium, i.e., 2 H
- Compounds incorporating positron-emitting isotopes are useful in medical imaging and positron-emitting tomography (PET) studies for determining the distribution of receptors.
- Suitable positron-emitting isotopes that can be incorporated in compounds of formula (I) are 11 C, 13 N, 15 O, and 18 F.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art using appropriate isotopically-labeled reagent in place of non-isotopically-labeled reagent.
- compositions typically also comprise one or more conventional pharmaceutically acceptable carriers, adjuvants, and/or vehicles (together referred to as “excipients”).
- compositions for oral administration and solid dosage forms in particular, are preferred.
- Such solid dosage forms include, for example, capsules, tablets, pills, powders, and granules.
- the compounds or salts are ordinarily combined with one or more excipients.
- the compounds or salts can be mixed with, for example, lactose, sucrose, starch powder, cellulose esters of alkanoic acids, cellulose alkyl esters, talc, stearic acid, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulfuric acids, gelatin, acacia gum, sodium alginate, polyvinylpyrrolidone, and/or polyvinyl alcohol, and then tableted or encapsulated for convenient administration.
- Such capsules or tablets can contain a controlled-release formulation, as can be provided in, for example, a dispersion of the compound or salt in hydroxypropylmethyl cellulose.
- the dosage forms also can comprise buffering agents, such as sodium citrate, or magnesium or calcium carbonate or bicarbonate. Tablets and pills additionally can be prepared with enteric coatings.
- the compounds and compositions of the invention are useful for treating and preventing certain diseases and disorders in humans and animals.
- the compounds described in the invention can affect physiological processes in humans and animals.
- the compounds and compositions described in the invention are useful for treating and preventing diseases and disorders modulated by histamine-3 receptors.
- treatment or prevention of such diseases and disorders can be effected by selectively modulating the histamine-3 receptors in a mammal, by administering a compound or composition of the invention, either alone or in combination with another active agent as part of a therapeutic regimen.
- the compounds of the invention may be useful for the treatment and prevention of diseases or conditions such as attention-deficit hyperactivity disorder (ADHD), deficits in attention, dementia, and diseases with deficits of memory, learning, schizophrenia, cognitive deficits of schizophrenia, cognitive deficits and dysfunction in psychiatric disorders, Alzheimer's disease, mild cognitive impairment, epilepsy, seizures, allergic rhinitis, and asthma, motion sickness, dizziness, Meniere's disease, vestibular disorders, vertigo, obesity, diabetes, type II diabetes, Syndrome X, insulin resistance syndrome, metabolic syndrome, pain, including neuropathic pain, neuropathy, sleep disorders, narcolepsy, pathological sleepiness, jet lag, drug abuse, mood alteration, bipolar disorder, depression, obsessive compulsive disorder, Tourette's syndrome, Parkinson's disease, and medullary thyroid carcinoma, melanoma, and polycystic ovary
- ADHD attention-deficit hyperactivity disorder
- dementia dementia
- diseases with deficits of memory learning
- schizophrenia cognitive deficit
- the preferred total daily dose of a compound or salt is typically from about 0.001 to about 100 mg/kg, more preferably from about 0.001 to about 30 mg/kg, and even more preferably from about 0.01 to about 10 mg/kg (i.e., mg of the compound or salt per kg body weight).
- Dosage unit compositions can contain such amounts or submultiples thereof to make up the daily dose.
- the administration of the compound or salt will be repeated a plurality of times. Multiple doses per day typically may be used to increase the total daily dose, if desired.
- Factors affecting the preferred dosage regimen include the type, age, weight, sex, diet, and condition of the patient; the severity of the pathological condition; the severity of the pathological condition; pharmacological considerations, such as the activity, efficacy, pharmacokinetic, and toxicology profiles of the particular compound or salt used; whether a drug delivery system is utilized; and the specific drug combination.
- the dosage regimen actually employed can vary widely, and therefore, can derive from the preferred dosage regimen set forth above.
- the flask and wetcake were rinsed with the mother liquors, followed by isopropyl alcohol (5 mL).
- the wetcake was dried on the filter under vacuum (1.88 g, 61.5% ee).
- the wetcake was then charged back to the round-bottom flask with isopropyl alcohol (38 mL, 20 mL/g).
- the suspension was heated to 80° C. After 0.5 h, all solids dissolved.
- the solution was then slowly cooled to 50° C. ( ⁇ 2 h), during which time a suspension formed.
- the suspension was stirred at 50° C. for 2 h, and then slowly cooled to room temperature (1-2 h). Stirred the suspension overnight ( ⁇ 15 h) at room temperature.
- the suspension was filtered.
- the chiral salt 5 was first broken up by treating it with citric acid solution, exacted with MTBE and drying to obtain the free acid, 2-(4-bromophenyl)cyclopropanecarboxylic acid 4 (4.82 g, 20 mmol) and mixed with 1,1′-carbonyldiimidazole (4.22 g, 26 mmol) in THF (45 mL, 10 mL/g total volume). A suspension formed after stirring at room temperature for >30 min. The suspension was cooled to ⁇ 15° C. Then a solution of the (S)-2-methylpyrrolidine (2.55 g, 30 mmol) in THF (5 mL) was added over ⁇ 5 min.
- reaction of 5 (3.62 g, 15 mmol) with CDI (3.16 g, 19.5 mmol) and piperidine (2.22 mL, 22.5 mmol) afforded 6b in an assay yield of 4.55 g (14.8 mmol, 99% assay yield, 99% peak area).
- a sample was purified by washing the product solution with 5% NaHCO 3 and concentrating in vacuo to an oil that solidified at room temperature to provide the standard.
- reaction of 5 (3.62 g, 15 mmol) with CDI (3.16 g, 19.5 mmol) and diethylamine (2.33 mL, 22.5 mmol) afforded 6c in an assay yield of 4.18 g (14.1 mmol, 94% assay yield, 98% peak area).
- a sample was purified by washing the product solution with 5% NaHCO 3 and concentrating in vacuo to an oil to provide the standard.
- reaction of 5 (3.62 g, 15 mmol) with CDI (3.16 g, 19.5 mmol) and 2 M dimethylamine in THF (11.25 mL, 22.5 mmol) afforded 6d in an assay yield of 3.67 g (13.7 mmol, 91% assay yield, 99% peak area).
- a sample was purified by washing the product solution with 5% NaHCO 3 and concentrating in vacuo to an oil that solidified at room temperature to provide the standard.
- reaction of 5 (3.62 g, 15 mmol) with CDI (3.16 g, 19.5 mmol) and morpholine (1.96 mL, 22.5 mmol) afforded 6e in an assay yield of 4.48 g (14.45 mmol, 96% assay yield, 100% peak area).
- a sample was purified by washing the product solution with 5% NaHCO 3 and concentrating in vacuo to a white solid to provide the standard.
- reaction of 5 (3.62 g, 15 mmol) with CDI (3.16 g, 19.5 mmol) and pyrrolidine (1.86 mL, 22.5 mmol) afforded 6f in an assay yield of 4.24 g (14.40 mmol, 96% assay yield, 100% peak area).
- a sample was purified by washing the product solution with 5% NaHCO 3 and concentrating in vacuo to a white solid to provide the standard.
- reaction of 5 (3.62 g, 15 mmol) with CDI (3.16 g, 19.5 mmol) and (R)-2-methylpyrrolidine (1.92 g, 22.5 mmol) afforded 6 g in an assay yield of 4.62 g (15.00 mmol, 100% assay yield, 97.5% peak area).
- a sample was purified by washing the product solution with 5% NaHCO 3 and concentrating in vacuo to an oil that solidified at room temperature to provide the standard.
- the product solution is then cooled to room temperature and extracted with t-butyl methyl ether (40 mL, 2 ⁇ ).
- the basic aqueous layer is then extracted with more MTBE (40 mL).
- the organic layers are combined and washed with saturated sodium chloride solution (40 mL).
- the resulting product solution was assayed by HPLC against a known standard. 2.66 g product was assayed (92% assayed yield, >99% peak area).
- a sample was concentrated under high vacuum to an oil to provide a standard.
- reaction of 6b (3.70 g assayed, 12.0 mmol) with 1 M BH 3 in THF (42.0 mL, 42.0 mmol) afforded 7b in an assay yield of 2.77 g (9.4 mmol, 78% assay yield, 99% peak area).
- a sample was concentrated in vacuo to an oil to provide the standard.
- a sample was concentrated in vacuo to an oil to provide the standard.
- a sample was concentrated in vacuo to an oil to provide the standard.
- reaction of 6e (4.20 g assayed, 13.55 mmol) with 1 M BH 3 in THF (47.4 mL, 47.4 mmol) afforded 7e in an assay yield of 2.68 g (9.05 mmol, 67% assay yield, 100% peak area).
- a sample was concentrated in vacuo to an oil to provide the standard.
- a sample was concentrated in vacuo to an oil to provide the standard.
- a sample was concentrated in vacuo to an oil to provide the standard.
- the yield was increased by stepwise addition of isopropyl alcohol alternating with hold times over 10 h at 15° C., reducing the water content to 10% (by volume) in the final solvent composition.
- the solid was filtered and washed with isopropyl alcohol twice (4.5 mL/g free base). Wet cake was dried at 50° C. under vacuum, in humidified environment, with intermittent slight nitrogen bleeding. The isolated solid was used as a standard.
- reaction of 7b (2.67 g assayed, 9.08 mmol) with 3(2H)-pyridazinone (1.14 g, 11.8 mmol) afforded a solution of the free base of 8b in an assayed yield of 2.61 g (93% assay yield, 95% peak area).
- reaction of 7c (2.87 g assayed, 10.2 mmol) with 3(2H)-pyridazinone (1.27 g, 13.2 mmol) afforded a solution of the free base of 8c in an assayed yield of 2.55 g (84% assay yield, 93% peak area).
- reaction of 7d (2.13 g assayed, 8.37 mmol) with 3(2H)-pyridazinone (1.05 g, 10.5 mmol) afforded a solution of the free base of 8d in an assayed yield of 1.57 g (5.8 mmol, 70% assay yield, 93% peak area).
- reaction of 7f (3.05 g assayed, 10.90 mmol) with 3(211)-pyridazinone (1.36 g, 14.17 mmol) afforded a solution of the free base of 8f in an assayed yield of 3.60 g (74% assay yield, 97% peak area).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description

wherein R1, R2, R3, R3a, R3b, R4, and R5 are as defined below. Compounds of formula (II) are described in WO 2007150010, published on Dec. 27, 2007, corresponding to U.S. patent application Ser. No. 11/766,987, filed on Jun. 22, 2007, and U.S. patent application Ser. No. 11/956,816, filed on Dec. 14, 2007, each of which are all hereby incorporated by reference. The present invention offers a more efficient process to obtain chiral compounds of formula (II) via the chiral resolution of an aryl-cyclopropanecarboxylic acid with chiral amines. The chiral resolution step forms a diasteriomeric chiral salt, which is crystallized to obtain an enantiomerically pure salt. The enantiomerically pure arylcyclopropyl carboxylic acid is obtained upon breaking up the salt. The resulting enantiomerically pure cyclopropyl carboxylic acids can be reacted with various amines to form amides, which can be reduced to form chiral amine derivatives. The intermediate chiral amines can be further coupled with a desired aromatic or heteroaromatic reagent to provide compounds of formula (II).

wherein R2, R3, R3a, R3b, are hydrogen; R1 is a 5- to 6-membered heteroaryl ring, cyanophenyl, a 8- to 12-membered bicyclic heteroaryl ring, or a 4- to 12-membered heterocyclic ring; and R4 and R5 taken together with the nitrogen atom to which they are attached form an amine moiety represented by structure:

wherein R7, R8, R9, and R10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl; R11, R12, R13, and R14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl; Rx and Ry are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino; Q is O or S; and m is an integer from 1 to 5.
A. Chiral Resolution of Cyclopropanecarboxylic Acid with Chiral Amines

The cyclopropanecarboxylic acids:

is treated with a chiral amine to form a diasteromeric chiral salt, which can be further crystallized to form an enantiomerically pure salt. The enantiomerically pure arylcyclopropanecarboxylic acids salt can be broken-up to release the enantiomerically pure arylcyclopropanecarboxylic acids that can be further reacted with amines, reduced, and coupled with a suitable aromatic, heteroaromatic, or heterocycle group to provide compounds of formula (II). In one embodiment, the chiral amine is selected from the group consisting of (S)-(−)-α-methylbenzylamine, (R)-(+)-N-benzyl-α-methylbenzylamine, (S)-(−)-N-benzyl-α-methylbenzylamine, (R)-(+)-N,N-dimethyl-1-phenylethylamine, (S)-(−)-N,N-dimethyl-1-phenylethylamine, [R—(R*,R*)]-(+)-bis(a-methylbenzyl)amine, [S—(R*,R*)]-(−)-bis(a-methylbenzyl)amine, (S)-(+)-1-cyclohexylethylamine, (R)-(+)-1-(1-naphthyl)ethylamine, (S)-(−)-1-(1-naphthyl)ethylamine, (1R,2R,3R,5S)-(−)-isopinocamphenylamine, (1S,2S,3S,5R)-(+)-isopinocamphenylamine, (1R2R)-(−)-pseudoephedrine, (1S,2S)-(+)-pseudoephedrine, (1R,2S)-(−)-ephedrine, (1S,2R)-(+)-ephedrine, (1R,2S)-(−)-N-methylephedrine, (1S,2R)-(+)-N-methylephedrine, (1R,2S)-(−)-norephedrine, (1S,2R)-(+)-norephedrine, (1R,2S)-(+)-cis-1-amino-2-indanol, (1S,2R)-(−)-cis-1-amino-2-indanol, quinine, and cinchonine. In one embodiment, the chiral amine is (R)-(−)-1-cyclohexylethylamine. In another embodiment, the chiral amine is (R)-(+)-a-methylbenzylamine. In a preferred embodiment, the reaction is carried out in a polar organic solvent. In one embodiment the polar organic solvent is an alcohol. The alcohol can be selected from the group consisting of methanol, ethanol, isopropyl alcohol, tert-butyl alcohol, and n-butyl alcohol. In one embodiment, the alcohol is isopropyl alcohol. In another embodiment, the alcohol is tert-butyl alcohol. In one embodiment, the reaction is carried at a temperature of from about room temperature to about 75° C. In a particular embodiment, the temperature is from about 40° C. to about 60° C. In one particular embodiment, the temperature at which the reaction is carried out is from about 45° C. to about 50° C. Typically, the reaction is accomplished in a period of about 1 to 48 hours, however, the length of the reaction time can vary depending on the particular conditions and quality of the reagents, among other aspects of the reaction. In a preferred embodiment, the reaction is conducted for a time period of about 8 hours.
In another embodiment, the aryl-cyclopropanecarboxylic acid is prepared from arylaldehyde. An illustration of a process for preparing arylcyclopropanecarboxylic acid from aryl aldehyde is shown below in Scheme A.


obtained from the chiral resolution is coupled with an amine. An illustration of the process is shown below in Scheme 2.

Suitable amine reagents can be represented by the formula:

wherein R7, R8, R9, and R10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl; R11, R12, R13, and R14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl; Rx and Ry are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino; Q is O or S; and m is an integer from 1 to 5.
is reduced to provide a compound as shown below in Scheme 3.


is reacted with a suitable aromatic or non-aromatic reagent to provide compounds of formula (II). An illustration of this process is shown below in Scheme 4.

In a preferred embodiment, the compound of formula (I-b) undergoes coupling reactions to provide the compounds of formula (II). Coupling conditions, commonly referred to as metal-catalyzed reaction including palladium, nickel, iron or copper catalyzed reaction, such as Ullmann reaction conditions, are preferred for the reaction.

wherein, the chiral amines are (S)-(−)-α-methylbenzylamine, (R)-(+)-N-benzyl-a-methylbenzylamine, (S)-(−)-N-benzyl-α-methylbenzylamine, (R)-(+)-N,N-dimethyl-1-phenylethylamine, (S)-(−)-N,N-dimethyl-1-phenylethylamine, [R—(R*,R*)]-(+)-bis(α-methylbenzyl)amine, [S-(R*,R*)]-(−)-bis(a-methylbenzyl)amine, (S)-(+)-1-cyclohexylethylamine, (R)-(+)-1-(1-naphthyl)ethylamine, (S)-(−)-1-(1-naphthyl)ethylamine, (1R,2R,3R,5S)-(−)-isopinocamphenylamine, (1S,2S,3S,5R)-(+)-isopinocamphenylamine, (1R2R)-(−)-pseudoephedrine, (1S,2S)-(+)-pseudoephedrine, (1R,2S)-(−)-ephedrine, (1S,2R)-(+)-ephedrine, (1R,2S)-(−)-N-methylephedrine, (1S,2R)-(+)-N-methylephedrine, (1R,2S)-(−)-norephedrine, (1S,2R)-(+)-norephedrine, (1R,2S)-(+)-cis-1-amino-2-indanol, (1S,2R)-(−)-cis-1-amino-2-indanol, quinine, or cinchonine. The chiral amines also can be selected from (R)-(−)-1-cyclohexylethylamine or (R)-(+)-α-methylbenzylamine. In one embodiment, the chiral amine is (R)-(−)-1-cyclohexylethylamine. In another embodiment, the chiral amine is (R)-(+)-α-methylbenzylamine.

i.e. 2-(4-bromophenyl)cyclopropanecarboxylic acid R-(−)-1-cyclohexylethylamine salt and 2-(4-bromophenyl)cyclopropanecarboxylic acid (R)-(+)-α-methylbenzylamine salt.

wherein the amine moiety in the structure above is represented by:

wherein R7, R8, R9, and R10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl; R11, R12, R13, and R14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl; Rx and Ry are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino; Q is O or S; and m is an integer from 1 to 5.

wherein the amine moiety in the structure above is represented by:

wherein R7, R8, R9, and R10 at each occurrence are independently selected from the group consisting of hydrogen, hydroxyalkyl, fluoroalkyl, cycloalkyl, and alkyl; R11, R12, R13, and R14 are each independently selected from the group consisting of hydrogen, hydroxyalkyl, alkyl, and fluoroalkyl; Rx and Ry are each independently selected from the group consisting of hydrogen, hydroxy, alkyl, alkoxy, alkylamino, fluoro, and dialkylamino; Q is O or S; and m is an integer from 1 to 5.
F. Isotopically-Labeled Compounds
























Claims (16)









Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/232,751 US8853390B2 (en) | 2010-09-16 | 2011-09-14 | Processes for preparing 1,2-substituted cyclopropyl derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US39770510P | 2010-09-16 | 2010-09-16 | |
| US13/232,751 US8853390B2 (en) | 2010-09-16 | 2011-09-14 | Processes for preparing 1,2-substituted cyclopropyl derivatives |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20120071651A1 US20120071651A1 (en) | 2012-03-22 |
| US8853390B2 true US8853390B2 (en) | 2014-10-07 |
Family
ID=44678063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/232,751 Expired - Fee Related US8853390B2 (en) | 2010-09-16 | 2011-09-14 | Processes for preparing 1,2-substituted cyclopropyl derivatives |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US8853390B2 (en) |
| WO (1) | WO2012037258A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10285998B1 (en) | 2018-04-04 | 2019-05-14 | The Menopause Method, Inc. | Composition and method to aid in hormone replacement therapy |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5203360B2 (en) * | 2006-06-23 | 2013-06-05 | アボット・ラボラトリーズ | Cyclopropylamine derivatives as histamine H3 receptor modulators |
| US9108948B2 (en) * | 2006-06-23 | 2015-08-18 | Abbvie Inc. | Cyclopropyl amine derivatives |
| US9186353B2 (en) | 2009-04-27 | 2015-11-17 | Abbvie Inc. | Treatment of osteoarthritis pain |
| WO2012037258A1 (en) | 2010-09-16 | 2012-03-22 | Abbott Laboratories | Processes for preparing 1,2-substituted cyclopropyl derivatives |
| UA111746C2 (en) | 2011-07-08 | 2016-06-10 | Х. Луннбек А/С | POSITIVE Allosteric Modulators of the Nicotinic Acetylcholine Receptor |
| WO2015027058A2 (en) * | 2013-08-21 | 2015-02-26 | Prexa Pharmaceuticals, Inc. | Cyclolkyl amine compounds |
| CN113277974B (en) * | 2020-02-20 | 2023-04-07 | 上海科技大学 | 2-phenylcyclopropylmethylamine derivative, and preparation method and use thereof |
Citations (139)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL6412766A (en) | 1963-11-05 | 1965-05-06 | ||
| US4507318A (en) | 1981-06-23 | 1985-03-26 | Pierre Fabre S.A. | 1-Aryl 2-aminomethyl cyclopropane carboxylates (Z) as drugs in the treatment of pain |
| EP0188887A1 (en) | 1985-01-17 | 1986-07-30 | Imperial Chemical Industries Plc | Tertiary amine compounds |
| EP0251466A2 (en) | 1986-06-23 | 1988-01-07 | Imperial Chemical Industries Plc | Chemical process for the preparation of esters of substituted cyclopropyl caboxylic acids |
| GB2210364A (en) | 1987-09-29 | 1989-06-07 | Ici Plc | Cyclopropane ring-containing tertiary amine derivatives |
| US5086054A (en) | 1990-07-31 | 1992-02-04 | Sri International | Novel arylcycloalkanepolyalkylamines |
| EP0668270A2 (en) | 1994-02-17 | 1995-08-23 | Rhone-Poulenc Agrochimie | 2-Imidazoline-5-ones derivatives as fungicides |
| JP2000047358A (en) | 1998-07-28 | 2000-02-18 | Fuji Photo Film Co Ltd | Photosensitive material processor |
| US6048876A (en) | 1995-01-23 | 2000-04-11 | Suntory Limited | Medicament for the alleviation or treatment of symptom derived from ischemic disease and compound useful therefor |
| WO2000042023A1 (en) | 1999-01-18 | 2000-07-20 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000044728A1 (en) | 1999-01-27 | 2000-08-03 | Pfizer Products Inc. | Substituted bicyclic derivatives useful as anticancer agents |
| WO2000063208A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000064884A1 (en) | 1999-04-26 | 2000-11-02 | Novo Nordisk A/S | Piperidyl-imidazole derivatives, their preparations and therapeutic uses |
| US6166023A (en) | 1998-05-11 | 2000-12-26 | Arzneimittelwerk Dresden Gmbh | 1,5- and 3-O-substituted 1H-indazoles having anti-asthmatic, anti-allergic, anti-inflammatory, immunomodulating and neuroprotective action, process for their preparation and their use as medicaments |
| US6235791B1 (en) | 1996-07-29 | 2001-05-22 | Sanofi-Synthelabo | Use of amines to produce drugs for preventing tumor cell proliferation |
| WO2002013821A1 (en) | 2000-08-17 | 2002-02-21 | Gliatech, Inc. | Novel alicyclic imidazoles as h3 agents |
| US20020052383A1 (en) | 2000-07-06 | 2002-05-02 | Rajagopal Bakthavatchalam | Melanin concentrating hormone receptor ligands |
| WO2002044128A2 (en) | 2000-11-28 | 2002-06-06 | Sunesis Pharmaceuticals, Inc. | Salicylate analogs as interleukin-4 antagonists |
| JP2002236340A (en) | 2001-02-09 | 2002-08-23 | Konica Corp | Processing method for silver halide photographic sensitive material with automatic processing machine |
| US20020138210A1 (en) | 2000-10-10 | 2002-09-26 | The Government Of The United States Of America | Microbial identification databases |
| US20020169188A1 (en) | 2001-03-16 | 2002-11-14 | Cowart Marlon D. | Novel amines as histamine-3 receptor ligands and their therapeutic applications |
| US6515013B2 (en) | 2000-07-13 | 2003-02-04 | Abbott Laboratories | 1,3-disubstituted and 1,3,3-trisubstituted pyrrolidines as histamine-3 receptor ligands and their therapeutic applications |
| DE10153345A1 (en) | 2001-10-29 | 2003-05-08 | Gruenenthal Gmbh | Substituted 1H-quinoxalin-2-one compounds and substituted 4-aryl and 4-heteroarylcyclohexane compounds |
| DE10153347A1 (en) | 2001-10-29 | 2003-05-08 | Gruenenthal Gmbh | Substituted 1H-quinolin-2-one compounds |
| EP1321169A1 (en) | 2001-12-18 | 2003-06-25 | Biofrontera Pharmaceuticals AG | Combination of a serotonin receptor antagonist with a histidine decarboxylase inhibitor as a medicament |
| US20030119796A1 (en) | 2001-09-21 | 2003-06-26 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| WO2003066604A2 (en) | 2002-02-05 | 2003-08-14 | Novo Nordisk A/S | Novel aryl- and heteroarylpiperazines |
| US6620839B2 (en) | 2000-07-13 | 2003-09-16 | Abbott Laboratories | 1,3-disubstituted and 1,3,3-trisubstituted pyrrolidines as histamine-3 receptor ligands and their therapeutic applications |
| WO2003099276A1 (en) | 2002-05-10 | 2003-12-04 | Bristol-Myers Squibb Company | 1,1-disubstituted cycloalkyl derivatives as factor xa inhibitors |
| WO2003104235A1 (en) | 2002-06-06 | 2003-12-18 | Novo Nordisk A/S | Substituted hexahydropyrrolo[1,2-a]pyrazines, octahydropyrido[1,2-a]pyrazines and decahydropyrazino[1,2-a]azepines |
| WO2004026305A1 (en) | 2002-09-19 | 2004-04-01 | Eli Lilly And Company | Diaryl ethers as opioid receptor antagonist |
| WO2004035556A1 (en) | 2002-10-16 | 2004-04-29 | Glaxo Group Limited | Substituted piperazines, (1,4) diaszepines, and 2,5-diazabicyclo (2.2.1) heptanes as histamine h1 and/or h3 antagonists or histamine h3 reverse antagonists |
| WO2004037813A1 (en) | 2002-10-25 | 2004-05-06 | Merck Frosst Canada & Co. | Pyrrolidin-2-on derivatives as ep4 receptor agonists |
| WO2004037801A1 (en) | 2002-10-23 | 2004-05-06 | Janssen Pharmaceutica, N.V. | Piperazinyl and diazapanyl benzamides and benzthioamides |
| WO2004041776A2 (en) | 2002-05-06 | 2004-05-21 | Bristol-Myers Squibb Company | SULFONYLAMINOVALEROLAC TAMS AND DERIVATIVES THEREOF AS FACTOR Xa INHIBITORS |
| WO2004046110A1 (en) | 2002-11-15 | 2004-06-03 | Yamanouchi Pharmaceutical Co., Ltd. | Antagonist to melanin-concentrating hormone receptor |
| WO2004056369A1 (en) | 2002-12-20 | 2004-07-08 | Glaxo Group Limited | Benzo ‘ d!azepine derivatives for the treatment of neurological disorders |
| WO2004098625A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| WO2004099199A1 (en) | 2003-05-06 | 2004-11-18 | Ranbaxy Laboratories Limited | Oxazolidinone derivatives as antimicrobials |
| WO2004101546A1 (en) | 2003-04-23 | 2004-11-25 | Glaxo Group Limited | Piperazine derivatives and their use for the treatment of neurological and psychiatric diseases |
| FR2856596A1 (en) | 2003-06-27 | 2004-12-31 | Bioprojet Soc Civ | NOVEL PSYCHIATRIC DRUG ASSOCIATION AND THE USE OF AN INVERSE HISTAMINE H3 RECEPTOR ANTAGONIST OR AGONIST TO PREPARE A MEDICAMENT PREVENTING ADVERSE EFFECTS OF PSYCHOTROPES. |
| US6838466B2 (en) | 2001-12-20 | 2005-01-04 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| WO2005009976A1 (en) | 2003-07-29 | 2005-02-03 | Novo Nordisk A/S | Pyridazinyl- piperazines and their use as histamine h3 receptor ligands |
| WO2005009471A1 (en) | 2003-07-28 | 2005-02-03 | Osaka Industrial Promotion Organization | Composition for lowering blood-sugar level |
| WO2005018045A1 (en) | 2003-08-15 | 2005-02-24 | Koninklijke Philips Electronics N.V. | Antenna arrangement and a module and a radio communications apparatus having such an arrangement |
| WO2005032468A2 (en) | 2003-10-01 | 2005-04-14 | Bristol-Myers Squibb Company | Monocyclic and bicyclic lactams as factor xa inhibitors |
| JP2005170934A (en) | 2003-11-21 | 2005-06-30 | Chisso Corp | Liquid crystalline compound with polymerizing ability, which has alkylene fluoride, and polymer thereof |
| WO2005058837A1 (en) | 2003-12-17 | 2005-06-30 | Glaxo Group Limited | Benzazepine derivatives as histamine h3 antagonists |
| US20050171181A1 (en) | 2004-02-02 | 2005-08-04 | Pfizer Inc | Histamine-3 receptor modulators |
| WO2005072740A2 (en) | 2004-01-30 | 2005-08-11 | Japan Tobacco Inc. | Anorectic compounds |
| US20050182045A1 (en) | 2004-02-13 | 2005-08-18 | Banyu Pharmaceutical Co., Ltd. | Fused ring 4-oxopyrimidine derivative |
| WO2005087746A1 (en) | 2004-03-12 | 2005-09-22 | Glaxo Group Limited | Benzazepine derivatives for the treatment of neurological and psychiatric disorders |
| JP2005281223A (en) | 2004-03-30 | 2005-10-13 | Chisso Corp | Optically active compound having 1,2-cyclohexylene ring, liquid crystal composition, liquid crystal display element and molded product |
| WO2005103032A2 (en) | 2004-04-14 | 2005-11-03 | Boehringer Ingelheim International Gmbh | Novel alkyne compounds having an mch-antagonistic effect, and medicaments containing said compounds |
| US20050245529A1 (en) | 2004-04-14 | 2005-11-03 | Boehringer Ingelheim International Gmbh | Alkyne compounds with MCH antagonistic activity and medicaments comprising these compounds |
| EP1595881A1 (en) | 2004-05-12 | 2005-11-16 | Pfizer Limited | Tetrahydronaphthyridine derivates useful as histamine H3 receptor ligands |
| WO2005108384A1 (en) | 2004-05-07 | 2005-11-17 | Warner-Lambert Company Llc | 3- or 4-monosubstituted phenol and thiophenol derivatives useful as h3 ligands |
| WO2005123723A1 (en) | 2004-06-18 | 2005-12-29 | Glaxo Group Limited | 3-cycloalkylbenzazepines as histamine h3 antagonists |
| WO2006004937A2 (en) | 2004-06-30 | 2006-01-12 | Athersys, Inc. | Non-imidazole tertiary amines as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other cns disorders |
| WO2006018260A1 (en) | 2004-08-16 | 2006-02-23 | Glaxo Group Limited | Tetrahydrobenzazepines as antagonists and/or reverse agonists of the histamine h 3 receptor |
| WO2006029906A1 (en) | 2004-09-17 | 2006-03-23 | Glaxo Group Limited | Methylene dipiperidine derivatives |
| US20060074103A1 (en) | 2004-10-06 | 2006-04-06 | Corte James R | Cyclic beta-amino acid derivatives as factor Xa inhibitors |
| WO2006040192A1 (en) | 2004-10-15 | 2006-04-20 | Glaxo Group Limited | Pyrrolidine derivatives as histamine receptors ligands |
| WO2006061193A1 (en) | 2004-12-07 | 2006-06-15 | Glaxo Group Limited | Indenyl derivatives and use thereof for the treatment of neurological disorders |
| EP1675578A2 (en) | 2003-10-15 | 2006-07-05 | Probiodrug AG | Use of effectors of glutaminyl and glutamate cyclases |
| WO2006072596A1 (en) | 2005-01-07 | 2006-07-13 | Glaxo Group Limited | 6- (2 , 3 , 4 , 5-TETRAHYDRO-lH-BENZO [D] AZEPIN-7-YLOXY) -NICOTAMIDE DERIVATIVES AS RADIOLABELLED LIGANDS |
| WO2006085692A1 (en) | 2005-02-14 | 2006-08-17 | Banyu Pharmaceutical Co., Ltd | Crystal of 4(3h)-quinazolinone derivative |
| US7094790B2 (en) | 2003-05-07 | 2006-08-22 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US7098222B2 (en) | 2004-05-12 | 2006-08-29 | Abbott Laboratories | Bicyclic-substituted amines having cyclic-substituted monocyclic substituents |
| WO2006090142A1 (en) | 2005-02-24 | 2006-08-31 | Glaxo Group Limited | l-{4- [ (l-CYCLOBUTYL-4-PIPERIDINYL) OXY] PHENYL] -4-{ [4- (METHYLSULFONYL) PHENYL]CARBONYL PIPERAZINE AS HISTAMINE H3 ANTAGONIST |
| WO2006097691A1 (en) | 2005-03-14 | 2006-09-21 | Glaxo Group Limited | Fused thiazole derivatives having affinity for the histamine h3 receptor |
| WO2006103537A2 (en) | 2005-04-01 | 2006-10-05 | Bioprojet | Treatment of epilepsy with non-imidazole alkylamines histamine h3-receptor ligands |
| WO2006103546A2 (en) | 2005-04-01 | 2006-10-05 | Bioprojet | Treatment of parkinson's disease, obstructive sleep apnea, dementia with lewy bodies, vascular dementia with non-imidazole alkylamines histamine h3-receptor ligands |
| WO2006124687A1 (en) | 2005-05-12 | 2006-11-23 | University Of Medicine And Dentistry Of New Jersey | Opioid receptor subtype-selective agents |
| WO2006123020A1 (en) | 2005-05-18 | 2006-11-23 | Juvantia Pharma Ltd Oy | Peptidomimetics selective for the somatostatin receptor subtypes 1 and/or 4 |
| WO2006132424A1 (en) | 2005-06-07 | 2006-12-14 | Banyu Pharmaceutical Co., Ltd. | Process for production of 4(3h)-quinazolinone derivative |
| WO2006132914A2 (en) | 2005-06-03 | 2006-12-14 | Abbott Laboratories | Cyclobutyl amine derivatives |
| US7153889B2 (en) | 2002-11-12 | 2006-12-26 | Abbott Laboratories | Bicyclic-substituted amines as histamine-3 receptor ligands |
| WO2007004735A1 (en) | 2005-07-05 | 2007-01-11 | Banyu Pharmaceutical Co., Ltd. | Method for producing 4(3h)-quinazolinone derivative |
| WO2007003604A2 (en) | 2005-07-04 | 2007-01-11 | Novo Nordisk A/S | Hists1mine h3 receptor antagonists |
| WO2007025144A1 (en) | 2005-08-24 | 2007-03-01 | University Of Illinois - Chicago | 5-ht2c receptor agonists as anorectic agents |
| WO2007024004A1 (en) | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
| WO2007025596A1 (en) | 2005-07-06 | 2007-03-08 | Glaxo Group Limited | Pyrazolo [3 , 4-d] azepine derivatives as histamine h3 antagonists |
| US20070066644A1 (en) | 2005-09-20 | 2007-03-22 | Schering Corporation | 1-[[1-[(2-Amino-6-methyl-4-pyridinyl)methyl]-4-fluoro-4-piperidinyl]carbonyl]-4-[2-(2-pyridinyl)-3H-imidazo[4,5-b]pyridin-3-yl]piperidine |
| US20070066588A1 (en) | 2005-09-22 | 2007-03-22 | Cowart Marlon D | Benzothiazole cyclobutyl amine derivatives |
| US20070066821A1 (en) | 2005-09-16 | 2007-03-22 | Allison Brett D | Cyclopropyl amines as modulators of the histamine h3 receptor |
| US7205316B2 (en) | 2004-05-12 | 2007-04-17 | Abbott Laboratories | Tri- and bi-cyclic heteroaryl histamine-3 receptor ligands |
| WO2007048595A1 (en) | 2005-10-27 | 2007-05-03 | Ucb Pharma, S.A. | Compounds comprising a lactam or a lactam derivative moiety, processes for making them, and their uses |
| WO2007052124A1 (en) | 2005-11-04 | 2007-05-10 | Pfizer Limited | Tetrahydronaphthyridine derivative |
| WO2007126957A2 (en) | 2006-03-31 | 2007-11-08 | Novartis Ag | New compounds |
| WO2007137968A1 (en) | 2006-05-29 | 2007-12-06 | High Point Pharmaceuticals, Llc | 3- (1, 3-benz0di0x0l-5-yl) -6- (4-cyclopropylpiperazin-1-yl) -pyridazine, its salts and solvates and its use as histamine h3 receptor antagonist |
| WO2007150010A2 (en) * | 2006-06-23 | 2007-12-27 | Abbott Laboratories | Cyclopropyl amine derivatives as histamin h3 receptor modulators |
| US20080027041A1 (en) | 2006-07-25 | 2008-01-31 | Cephalon, Inc. | Pyridizinone derivatives |
| US7345034B2 (en) | 2004-04-07 | 2008-03-18 | Abbott Laboratories | Azacyclosteroid histamine-3 receptor ligands |
| WO2008064310A2 (en) | 2006-11-22 | 2008-05-29 | University Of Medicine And Dentistry Of New Jersey | Mixed opioid receptor active compounds |
| WO2008064318A2 (en) | 2006-11-22 | 2008-05-29 | University Of Medicine And Dentistry Of New Jersey | Peripheral opioid receptor active compounds |
| WO2008064317A1 (en) | 2006-11-22 | 2008-05-29 | University Of Medicine And Dentistry Of New Jersey | Lipophilic opioid receptor active compounds |
| US7381537B2 (en) | 2003-05-05 | 2008-06-03 | Probiodrug Ag | Use of inhibitors of glutaminyl cyclases for treatment and prevention of disease |
| WO2008067257A2 (en) | 2006-11-29 | 2008-06-05 | Abbott Laboratories | Inhibitors of diacylglycerol o-acyltransferase type 1 enzyme |
| US20080176925A1 (en) | 2007-01-22 | 2008-07-24 | Pfizer Inc | Toluene sulfonic acid salt of a therapeutic compound and pharmaceutical compositions thereof |
| WO2008104590A2 (en) | 2007-03-01 | 2008-09-04 | Glaxo Group Limited | Novel dosage form |
| US20080242653A1 (en) | 2006-06-23 | 2008-10-02 | Huaqing Liu | Cyclopropyl amine derivatives |
| WO2008151156A1 (en) | 2007-05-31 | 2008-12-11 | Sepracor Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US20090036425A1 (en) | 2007-08-02 | 2009-02-05 | Pfizer Inc | Substituted bicyclolactam compounds |
| WO2009024823A2 (en) | 2007-08-22 | 2009-02-26 | Astrazeneca Ab | Oxadiazole derivatives as dgat inhibitors |
| WO2009030716A1 (en) | 2007-09-06 | 2009-03-12 | Glaxo Group Limited | Piperazine derivative having affinity for the histamine h3 receptor |
| US20090068699A1 (en) | 2003-05-05 | 2009-03-12 | Probiodrug Ag | Use of glutaminyl cyclase inhibitors |
| US20090075938A1 (en) | 2006-02-10 | 2009-03-19 | Graham Michael Wynne | Treatment of duchenne muscular dystrophy |
| WO2009039431A2 (en) | 2007-09-21 | 2009-03-26 | Neurogen Corporation | Substituted aryl-fused spirocyclic amines |
| WO2009081195A1 (en) | 2007-12-20 | 2009-07-02 | Astrazeneca Ab | Carbamoyl compounds as dgat1 inhibitors 190 |
| WO2009085945A1 (en) | 2007-12-20 | 2009-07-09 | Abbott Laboratories | Benzothiazole and benzoxazole derivatives and methods of use |
| US20090192168A1 (en) | 2008-01-04 | 2009-07-30 | Alex Muci | Compounds, Compositions and Methods |
| WO2009092764A1 (en) | 2008-01-24 | 2009-07-30 | Ucb Pharma, S.A. | Compounds comprising a cyclobutoxy group |
| WO2009100294A2 (en) | 2008-02-07 | 2009-08-13 | Abbott Laboratories | Amide derivatives as positive allosteric modulators and methods of use thereof |
| WO2009100120A2 (en) | 2008-02-04 | 2009-08-13 | Neurogen Corporation | Pyridinyl-substituted piperazinyl oxoethyl tetrahydropyrazolopyridines |
| WO2009115874A2 (en) | 2008-03-17 | 2009-09-24 | Matrix Laboratories Ltd. | Novel heterocyclic compounds, pharmaceutical compositions containing them and processes for their preparation |
| WO2009124553A2 (en) | 2008-04-09 | 2009-10-15 | Neurokey A/S | Use of hypothermia inducing drugs |
| EP2117540A1 (en) | 2007-03-01 | 2009-11-18 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| WO2009147149A1 (en) | 2008-06-06 | 2009-12-10 | Ucb Pharma, S.A. | Compounds comprising a cyclobutoxy group |
| WO2009151991A1 (en) | 2008-06-11 | 2009-12-17 | Merck & Co., Inc. | Pyrazole derivatives useful as inhibitors of faah |
| US20100016344A1 (en) | 2008-07-11 | 2010-01-21 | Abbott Laboratories | Amino heterocyclic linked pyrimidine derivatives |
| WO2010007382A1 (en) | 2008-07-18 | 2010-01-21 | Takeda Pharmaceutical Company Limited. | Benzazepine derivatives and their use as hstamine h3 antagonists |
| US20100040575A1 (en) | 2003-05-05 | 2010-02-18 | Probiodrug Ag | Use of inhibitors of glutaminyl cyclase and glutamate cyclase for treatment and prevention of neurodegenerative diseases |
| CA2734800A1 (en) | 2008-08-20 | 2010-02-25 | Probiodrug Ag | Antibodies directed against pyroglutamate monocyte chemoattractant protein-1 (mcp-1 n1pe) |
| WO2010071822A1 (en) | 2008-12-19 | 2010-06-24 | Schering Corporation | Piperidine and piperazine derivatives and methods of use thereof |
| WO2010080757A2 (en) | 2009-01-07 | 2010-07-15 | Astrazeneca Ab | Combinations with an alpha-4beta-2 nicotinic agonist |
| US20100204205A1 (en) | 2007-06-13 | 2010-08-12 | Nir Barak | Tolerability of mirtazapine and a second active agent by using them in combination |
| US20100216812A1 (en) | 2009-02-20 | 2010-08-26 | Astrazeneca Ab | Cyclopropyl Amide Derivatives |
| US20100227876A1 (en) | 2009-03-06 | 2010-09-09 | Rechfensen Llp | Methods of Reducing Side Effects of Analgesics |
| US20100249144A1 (en) | 2007-06-28 | 2010-09-30 | Demong Duane Eugene | Substituted piperazines as cb1 antagonists |
| US20100267714A1 (en) | 2008-08-18 | 2010-10-21 | Jorgensen William L | Mif modulators |
| US20100273778A1 (en) | 2009-04-27 | 2010-10-28 | Abbott Laboratories | Treatment of osteoarthritis pain |
| US20100286160A1 (en) | 2007-06-28 | 2010-11-11 | Intervet Inc. | Substituted piperazines as cb1 antagonists |
| US20110009430A1 (en) | 2005-12-22 | 2011-01-13 | Moran Magdalene M | Methods and compositions for treating pain |
| WO2011083314A1 (en) | 2010-01-08 | 2011-07-14 | Takeda Pharmaceutical Company Limited | Benzazepine derivatives for the treatment of central nervous system disorders |
| WO2011083315A1 (en) | 2010-01-08 | 2011-07-14 | Takeda Pharmaceutical Company Limited | Compounds and their use |
| WO2011083316A1 (en) | 2010-01-08 | 2011-07-14 | Takeda Pharmaceutical Company Limited | Benzazepine derivatives for the treatment of central nervous system disorders |
| US20110195932A1 (en) | 2007-08-03 | 2011-08-11 | Graham Michael Wynne | Drug Combinations for the Treatment of Duchenne Muscular Dystrophy |
| US20120071651A1 (en) | 2010-09-16 | 2012-03-22 | Abbott Laboratories | Processes for Preparing 1,2-Substituted Cyclopropyl Derivatives |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1766987A (en) | 1923-10-18 | 1930-06-24 | Universal Oil Prod Co | Process of cracking petroleum oil |
-
2011
- 2011-09-14 WO PCT/US2011/051603 patent/WO2012037258A1/en active Application Filing
- 2011-09-14 US US13/232,751 patent/US8853390B2/en not_active Expired - Fee Related
Patent Citations (172)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1086191A (en) | 1963-11-05 | 1967-10-04 | Whitefin Holding Sa | Phenylcyclopropane derivatives |
| NL6412766A (en) | 1963-11-05 | 1965-05-06 | ||
| US4507318A (en) | 1981-06-23 | 1985-03-26 | Pierre Fabre S.A. | 1-Aryl 2-aminomethyl cyclopropane carboxylates (Z) as drugs in the treatment of pain |
| EP0188887A1 (en) | 1985-01-17 | 1986-07-30 | Imperial Chemical Industries Plc | Tertiary amine compounds |
| EP0251466A2 (en) | 1986-06-23 | 1988-01-07 | Imperial Chemical Industries Plc | Chemical process for the preparation of esters of substituted cyclopropyl caboxylic acids |
| GB2210364A (en) | 1987-09-29 | 1989-06-07 | Ici Plc | Cyclopropane ring-containing tertiary amine derivatives |
| US5086054A (en) | 1990-07-31 | 1992-02-04 | Sri International | Novel arylcycloalkanepolyalkylamines |
| EP0668270A2 (en) | 1994-02-17 | 1995-08-23 | Rhone-Poulenc Agrochimie | 2-Imidazoline-5-ones derivatives as fungicides |
| US6048876A (en) | 1995-01-23 | 2000-04-11 | Suntory Limited | Medicament for the alleviation or treatment of symptom derived from ischemic disease and compound useful therefor |
| US6235791B1 (en) | 1996-07-29 | 2001-05-22 | Sanofi-Synthelabo | Use of amines to produce drugs for preventing tumor cell proliferation |
| US6166023A (en) | 1998-05-11 | 2000-12-26 | Arzneimittelwerk Dresden Gmbh | 1,5- and 3-O-substituted 1H-indazoles having anti-asthmatic, anti-allergic, anti-inflammatory, immunomodulating and neuroprotective action, process for their preparation and their use as medicaments |
| JP2000047358A (en) | 1998-07-28 | 2000-02-18 | Fuji Photo Film Co Ltd | Photosensitive material processor |
| WO2000042023A1 (en) | 1999-01-18 | 2000-07-20 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000044728A1 (en) | 1999-01-27 | 2000-08-03 | Pfizer Products Inc. | Substituted bicyclic derivatives useful as anticancer agents |
| WO2000063208A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000064884A1 (en) | 1999-04-26 | 2000-11-02 | Novo Nordisk A/S | Piperidyl-imidazole derivatives, their preparations and therapeutic uses |
| US20020052383A1 (en) | 2000-07-06 | 2002-05-02 | Rajagopal Bakthavatchalam | Melanin concentrating hormone receptor ligands |
| US6515013B2 (en) | 2000-07-13 | 2003-02-04 | Abbott Laboratories | 1,3-disubstituted and 1,3,3-trisubstituted pyrrolidines as histamine-3 receptor ligands and their therapeutic applications |
| US6620839B2 (en) | 2000-07-13 | 2003-09-16 | Abbott Laboratories | 1,3-disubstituted and 1,3,3-trisubstituted pyrrolidines as histamine-3 receptor ligands and their therapeutic applications |
| WO2002013821A1 (en) | 2000-08-17 | 2002-02-21 | Gliatech, Inc. | Novel alicyclic imidazoles as h3 agents |
| US20020138210A1 (en) | 2000-10-10 | 2002-09-26 | The Government Of The United States Of America | Microbial identification databases |
| WO2002044128A2 (en) | 2000-11-28 | 2002-06-06 | Sunesis Pharmaceuticals, Inc. | Salicylate analogs as interleukin-4 antagonists |
| JP2002236340A (en) | 2001-02-09 | 2002-08-23 | Konica Corp | Processing method for silver halide photographic sensitive material with automatic processing machine |
| US20020169188A1 (en) | 2001-03-16 | 2002-11-14 | Cowart Marlon D. | Novel amines as histamine-3 receptor ligands and their therapeutic applications |
| US6969730B2 (en) | 2001-03-16 | 2005-11-29 | Abbott Laboratories | Amines as histamine-3 receptor ligands and their therapeutic applications |
| US20030119796A1 (en) | 2001-09-21 | 2003-06-26 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| DE10153347A1 (en) | 2001-10-29 | 2003-05-08 | Gruenenthal Gmbh | Substituted 1H-quinolin-2-one compounds |
| US20040224954A1 (en) | 2001-10-29 | 2004-11-11 | Michael Sattlegger | Substituted 1H-quinoxalin-2-one compounds and substituted 4-aryl- and 4-heteroarylcyclohexane compounds |
| DE10153345A1 (en) | 2001-10-29 | 2003-05-08 | Gruenenthal Gmbh | Substituted 1H-quinoxalin-2-one compounds and substituted 4-aryl and 4-heteroarylcyclohexane compounds |
| US20040224980A1 (en) | 2001-10-29 | 2004-11-11 | Michael Sattlegger | Substituted 1H-quinolin-2-one compounds |
| EP1321169A1 (en) | 2001-12-18 | 2003-06-25 | Biofrontera Pharmaceuticals AG | Combination of a serotonin receptor antagonist with a histidine decarboxylase inhibitor as a medicament |
| US6838466B2 (en) | 2001-12-20 | 2005-01-04 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| WO2003066604A2 (en) | 2002-02-05 | 2003-08-14 | Novo Nordisk A/S | Novel aryl- and heteroarylpiperazines |
| WO2004041776A2 (en) | 2002-05-06 | 2004-05-21 | Bristol-Myers Squibb Company | SULFONYLAMINOVALEROLAC TAMS AND DERIVATIVES THEREOF AS FACTOR Xa INHIBITORS |
| WO2003099276A1 (en) | 2002-05-10 | 2003-12-04 | Bristol-Myers Squibb Company | 1,1-disubstituted cycloalkyl derivatives as factor xa inhibitors |
| WO2003104235A1 (en) | 2002-06-06 | 2003-12-18 | Novo Nordisk A/S | Substituted hexahydropyrrolo[1,2-a]pyrazines, octahydropyrido[1,2-a]pyrazines and decahydropyrazino[1,2-a]azepines |
| WO2004026305A1 (en) | 2002-09-19 | 2004-04-01 | Eli Lilly And Company | Diaryl ethers as opioid receptor antagonist |
| WO2004035556A1 (en) | 2002-10-16 | 2004-04-29 | Glaxo Group Limited | Substituted piperazines, (1,4) diaszepines, and 2,5-diazabicyclo (2.2.1) heptanes as histamine h1 and/or h3 antagonists or histamine h3 reverse antagonists |
| WO2004037801A1 (en) | 2002-10-23 | 2004-05-06 | Janssen Pharmaceutica, N.V. | Piperazinyl and diazapanyl benzamides and benzthioamides |
| WO2004037813A1 (en) | 2002-10-25 | 2004-05-06 | Merck Frosst Canada & Co. | Pyrrolidin-2-on derivatives as ep4 receptor agonists |
| US7153889B2 (en) | 2002-11-12 | 2006-12-26 | Abbott Laboratories | Bicyclic-substituted amines as histamine-3 receptor ligands |
| WO2004046110A1 (en) | 2002-11-15 | 2004-06-03 | Yamanouchi Pharmaceutical Co., Ltd. | Antagonist to melanin-concentrating hormone receptor |
| WO2004056369A1 (en) | 2002-12-20 | 2004-07-08 | Glaxo Group Limited | Benzo ‘ d!azepine derivatives for the treatment of neurological disorders |
| US20060040918A1 (en) | 2002-12-20 | 2006-02-23 | Bamford Mark J | Benzo d!azepine derivatives for the treatment of neurological disorders |
| US7799773B2 (en) | 2002-12-20 | 2010-09-21 | Glaxo Group Limited | Benzazepine derivatives for the treatment of neurological disorders |
| US7696193B2 (en) | 2002-12-20 | 2010-04-13 | Glaxo Group Limited | Benzazepine derivatives for the treatment of neurological disorders |
| US20070299056A1 (en) | 2002-12-20 | 2007-12-27 | Bamford Mark J | Benzazepine derivatives for the treatment of neurological disorders |
| WO2004101546A1 (en) | 2003-04-23 | 2004-11-25 | Glaxo Group Limited | Piperazine derivatives and their use for the treatment of neurological and psychiatric diseases |
| US7381537B2 (en) | 2003-05-05 | 2008-06-03 | Probiodrug Ag | Use of inhibitors of glutaminyl cyclases for treatment and prevention of disease |
| US20100040575A1 (en) | 2003-05-05 | 2010-02-18 | Probiodrug Ag | Use of inhibitors of glutaminyl cyclase and glutamate cyclase for treatment and prevention of neurodegenerative diseases |
| WO2004098625A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| EP2206496A1 (en) | 2003-05-05 | 2010-07-14 | Probiodrug AG | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| EP1961416A1 (en) | 2003-05-05 | 2008-08-27 | Probiodrug AG | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| US20080286810A1 (en) | 2003-05-05 | 2008-11-20 | Probiodrug Ag | Use of Inhibtors of Glutaminyl Cyclases for Treatment and Prevention of Disease |
| US7732162B2 (en) | 2003-05-05 | 2010-06-08 | Probiodrug Ag | Inhibitors of glutaminyl cyclase for treating neurodegenerative diseases |
| US20090068699A1 (en) | 2003-05-05 | 2009-03-12 | Probiodrug Ag | Use of glutaminyl cyclase inhibitors |
| WO2004099199A1 (en) | 2003-05-06 | 2004-11-18 | Ranbaxy Laboratories Limited | Oxazolidinone derivatives as antimicrobials |
| US7094790B2 (en) | 2003-05-07 | 2006-08-22 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US7358263B2 (en) | 2003-05-07 | 2008-04-15 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| WO2005000315A1 (en) | 2003-06-27 | 2005-01-06 | Bioprojet | Combination product comprising an antagonist or inverse agonist of histamine receptor h3 and an antipsychotic or antidepressant agent, and use thereof for the preparation of a medicament that prevents the adverse effects of psychotropic drugs |
| FR2856596A1 (en) | 2003-06-27 | 2004-12-31 | Bioprojet Soc Civ | NOVEL PSYCHIATRIC DRUG ASSOCIATION AND THE USE OF AN INVERSE HISTAMINE H3 RECEPTOR ANTAGONIST OR AGONIST TO PREPARE A MEDICAMENT PREVENTING ADVERSE EFFECTS OF PSYCHOTROPES. |
| WO2005009471A1 (en) | 2003-07-28 | 2005-02-03 | Osaka Industrial Promotion Organization | Composition for lowering blood-sugar level |
| WO2005009976A1 (en) | 2003-07-29 | 2005-02-03 | Novo Nordisk A/S | Pyridazinyl- piperazines and their use as histamine h3 receptor ligands |
| WO2005018045A1 (en) | 2003-08-15 | 2005-02-24 | Koninklijke Philips Electronics N.V. | Antenna arrangement and a module and a radio communications apparatus having such an arrangement |
| WO2005032468A2 (en) | 2003-10-01 | 2005-04-14 | Bristol-Myers Squibb Company | Monocyclic and bicyclic lactams as factor xa inhibitors |
| US7462599B2 (en) | 2003-10-15 | 2008-12-09 | Probiodrug Ag | Use of effectors of glutaminyl and glutamate cyclases |
| EP1675578A2 (en) | 2003-10-15 | 2006-07-05 | Probiodrug AG | Use of effectors of glutaminyl and glutamate cyclases |
| EP2289498A1 (en) | 2003-10-15 | 2011-03-02 | Probiodrug AG | Use of inhibitors of glutaminyl clyclase |
| JP2005170934A (en) | 2003-11-21 | 2005-06-30 | Chisso Corp | Liquid crystalline compound with polymerizing ability, which has alkylene fluoride, and polymer thereof |
| WO2005058837A1 (en) | 2003-12-17 | 2005-06-30 | Glaxo Group Limited | Benzazepine derivatives as histamine h3 antagonists |
| WO2005072740A2 (en) | 2004-01-30 | 2005-08-11 | Japan Tobacco Inc. | Anorectic compounds |
| US20050171181A1 (en) | 2004-02-02 | 2005-08-04 | Pfizer Inc | Histamine-3 receptor modulators |
| WO2005080361A1 (en) | 2004-02-02 | 2005-09-01 | Pfizer Products Inc. | Histamine-3 receptor modulators |
| US20050182045A1 (en) | 2004-02-13 | 2005-08-18 | Banyu Pharmaceutical Co., Ltd. | Fused ring 4-oxopyrimidine derivative |
| WO2005087746A1 (en) | 2004-03-12 | 2005-09-22 | Glaxo Group Limited | Benzazepine derivatives for the treatment of neurological and psychiatric disorders |
| JP2005281223A (en) | 2004-03-30 | 2005-10-13 | Chisso Corp | Optically active compound having 1,2-cyclohexylene ring, liquid crystal composition, liquid crystal display element and molded product |
| US7345034B2 (en) | 2004-04-07 | 2008-03-18 | Abbott Laboratories | Azacyclosteroid histamine-3 receptor ligands |
| WO2005103032A2 (en) | 2004-04-14 | 2005-11-03 | Boehringer Ingelheim International Gmbh | Novel alkyne compounds having an mch-antagonistic effect, and medicaments containing said compounds |
| US20050245529A1 (en) | 2004-04-14 | 2005-11-03 | Boehringer Ingelheim International Gmbh | Alkyne compounds with MCH antagonistic activity and medicaments comprising these compounds |
| WO2005108384A1 (en) | 2004-05-07 | 2005-11-17 | Warner-Lambert Company Llc | 3- or 4-monosubstituted phenol and thiophenol derivatives useful as h3 ligands |
| US7098222B2 (en) | 2004-05-12 | 2006-08-29 | Abbott Laboratories | Bicyclic-substituted amines having cyclic-substituted monocyclic substituents |
| US7205316B2 (en) | 2004-05-12 | 2007-04-17 | Abbott Laboratories | Tri- and bi-cyclic heteroaryl histamine-3 receptor ligands |
| EP1595881A1 (en) | 2004-05-12 | 2005-11-16 | Pfizer Limited | Tetrahydronaphthyridine derivates useful as histamine H3 receptor ligands |
| WO2005123723A1 (en) | 2004-06-18 | 2005-12-29 | Glaxo Group Limited | 3-cycloalkylbenzazepines as histamine h3 antagonists |
| WO2006004937A2 (en) | 2004-06-30 | 2006-01-12 | Athersys, Inc. | Non-imidazole tertiary amines as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other cns disorders |
| WO2006018260A1 (en) | 2004-08-16 | 2006-02-23 | Glaxo Group Limited | Tetrahydrobenzazepines as antagonists and/or reverse agonists of the histamine h 3 receptor |
| US20070208005A1 (en) | 2004-08-16 | 2007-09-06 | Parr Christopher A | Tetrahydrobenzazepines as antagonists and/or reverse agonists of the histamine h3 receptor |
| WO2006029906A1 (en) | 2004-09-17 | 2006-03-23 | Glaxo Group Limited | Methylene dipiperidine derivatives |
| US20060074103A1 (en) | 2004-10-06 | 2006-04-06 | Corte James R | Cyclic beta-amino acid derivatives as factor Xa inhibitors |
| WO2006040192A1 (en) | 2004-10-15 | 2006-04-20 | Glaxo Group Limited | Pyrrolidine derivatives as histamine receptors ligands |
| WO2006061193A1 (en) | 2004-12-07 | 2006-06-15 | Glaxo Group Limited | Indenyl derivatives and use thereof for the treatment of neurological disorders |
| WO2006072596A1 (en) | 2005-01-07 | 2006-07-13 | Glaxo Group Limited | 6- (2 , 3 , 4 , 5-TETRAHYDRO-lH-BENZO [D] AZEPIN-7-YLOXY) -NICOTAMIDE DERIVATIVES AS RADIOLABELLED LIGANDS |
| WO2006085692A1 (en) | 2005-02-14 | 2006-08-17 | Banyu Pharmaceutical Co., Ltd | Crystal of 4(3h)-quinazolinone derivative |
| US20080139589A1 (en) | 2005-02-14 | 2008-06-12 | Akio Kanatani | Crystal of 4(3H)-Quinazolinone Derivative |
| WO2006090142A1 (en) | 2005-02-24 | 2006-08-31 | Glaxo Group Limited | l-{4- [ (l-CYCLOBUTYL-4-PIPERIDINYL) OXY] PHENYL] -4-{ [4- (METHYLSULFONYL) PHENYL]CARBONYL PIPERAZINE AS HISTAMINE H3 ANTAGONIST |
| WO2006097691A1 (en) | 2005-03-14 | 2006-09-21 | Glaxo Group Limited | Fused thiazole derivatives having affinity for the histamine h3 receptor |
| WO2006103546A2 (en) | 2005-04-01 | 2006-10-05 | Bioprojet | Treatment of parkinson's disease, obstructive sleep apnea, dementia with lewy bodies, vascular dementia with non-imidazole alkylamines histamine h3-receptor ligands |
| WO2006103537A2 (en) | 2005-04-01 | 2006-10-05 | Bioprojet | Treatment of epilepsy with non-imidazole alkylamines histamine h3-receptor ligands |
| WO2006124687A1 (en) | 2005-05-12 | 2006-11-23 | University Of Medicine And Dentistry Of New Jersey | Opioid receptor subtype-selective agents |
| WO2006123020A1 (en) | 2005-05-18 | 2006-11-23 | Juvantia Pharma Ltd Oy | Peptidomimetics selective for the somatostatin receptor subtypes 1 and/or 4 |
| WO2006132914A2 (en) | 2005-06-03 | 2006-12-14 | Abbott Laboratories | Cyclobutyl amine derivatives |
| US20070078133A1 (en) | 2005-06-03 | 2007-04-05 | Huaqing Liu | Cyclobutyl amine derivatives |
| WO2006132914A3 (en) | 2005-06-03 | 2007-03-29 | Abbott Lab | Cyclobutyl amine derivatives |
| WO2006132424A1 (en) | 2005-06-07 | 2006-12-14 | Banyu Pharmaceutical Co., Ltd. | Process for production of 4(3h)-quinazolinone derivative |
| WO2007003604A2 (en) | 2005-07-04 | 2007-01-11 | Novo Nordisk A/S | Hists1mine h3 receptor antagonists |
| WO2007004735A1 (en) | 2005-07-05 | 2007-01-11 | Banyu Pharmaceutical Co., Ltd. | Method for producing 4(3h)-quinazolinone derivative |
| WO2007025596A1 (en) | 2005-07-06 | 2007-03-08 | Glaxo Group Limited | Pyrazolo [3 , 4-d] azepine derivatives as histamine h3 antagonists |
| US20090137587A1 (en) | 2005-08-24 | 2009-05-28 | Akira Naya | Phenylpyridone Derivative |
| WO2007025144A1 (en) | 2005-08-24 | 2007-03-01 | University Of Illinois - Chicago | 5-ht2c receptor agonists as anorectic agents |
| WO2007024004A1 (en) | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
| US20070066821A1 (en) | 2005-09-16 | 2007-03-22 | Allison Brett D | Cyclopropyl amines as modulators of the histamine h3 receptor |
| US20070066644A1 (en) | 2005-09-20 | 2007-03-22 | Schering Corporation | 1-[[1-[(2-Amino-6-methyl-4-pyridinyl)methyl]-4-fluoro-4-piperidinyl]carbonyl]-4-[2-(2-pyridinyl)-3H-imidazo[4,5-b]pyridin-3-yl]piperidine |
| US20070066588A1 (en) | 2005-09-22 | 2007-03-22 | Cowart Marlon D | Benzothiazole cyclobutyl amine derivatives |
| WO2007038074A1 (en) | 2005-09-22 | 2007-04-05 | Abbott Laboratories | Benzothiazole cyclobutyl amine derivatives and their use as histamine-3 receptors ligands |
| US7576110B2 (en) | 2005-09-22 | 2009-08-18 | Abbott Laboratories | Benzothiazole cyclobutyl amine derivatives |
| WO2007048595A1 (en) | 2005-10-27 | 2007-05-03 | Ucb Pharma, S.A. | Compounds comprising a lactam or a lactam derivative moiety, processes for making them, and their uses |
| WO2007052124A1 (en) | 2005-11-04 | 2007-05-10 | Pfizer Limited | Tetrahydronaphthyridine derivative |
| US20110009430A1 (en) | 2005-12-22 | 2011-01-13 | Moran Magdalene M | Methods and compositions for treating pain |
| US20090075938A1 (en) | 2006-02-10 | 2009-03-19 | Graham Michael Wynne | Treatment of duchenne muscular dystrophy |
| WO2007126957A2 (en) | 2006-03-31 | 2007-11-08 | Novartis Ag | New compounds |
| WO2007137968A1 (en) | 2006-05-29 | 2007-12-06 | High Point Pharmaceuticals, Llc | 3- (1, 3-benz0di0x0l-5-yl) -6- (4-cyclopropylpiperazin-1-yl) -pyridazine, its salts and solvates and its use as histamine h3 receptor antagonist |
| US20080242653A1 (en) | 2006-06-23 | 2008-10-02 | Huaqing Liu | Cyclopropyl amine derivatives |
| WO2007150010A2 (en) * | 2006-06-23 | 2007-12-27 | Abbott Laboratories | Cyclopropyl amine derivatives as histamin h3 receptor modulators |
| US20080021081A1 (en) | 2006-06-23 | 2008-01-24 | Huaqing Liu | Cyclopropyl amine derivatives |
| US20080027041A1 (en) | 2006-07-25 | 2008-01-31 | Cephalon, Inc. | Pyridizinone derivatives |
| WO2008064318A2 (en) | 2006-11-22 | 2008-05-29 | University Of Medicine And Dentistry Of New Jersey | Peripheral opioid receptor active compounds |
| WO2008064317A1 (en) | 2006-11-22 | 2008-05-29 | University Of Medicine And Dentistry Of New Jersey | Lipophilic opioid receptor active compounds |
| WO2008064310A2 (en) | 2006-11-22 | 2008-05-29 | University Of Medicine And Dentistry Of New Jersey | Mixed opioid receptor active compounds |
| WO2008067257A2 (en) | 2006-11-29 | 2008-06-05 | Abbott Laboratories | Inhibitors of diacylglycerol o-acyltransferase type 1 enzyme |
| US20080176925A1 (en) | 2007-01-22 | 2008-07-24 | Pfizer Inc | Toluene sulfonic acid salt of a therapeutic compound and pharmaceutical compositions thereof |
| WO2008104590A2 (en) | 2007-03-01 | 2008-09-04 | Glaxo Group Limited | Novel dosage form |
| EP2117540A1 (en) | 2007-03-01 | 2009-11-18 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| WO2008151156A1 (en) | 2007-05-31 | 2008-12-11 | Sepracor Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US20100204205A1 (en) | 2007-06-13 | 2010-08-12 | Nir Barak | Tolerability of mirtazapine and a second active agent by using them in combination |
| US20100249144A1 (en) | 2007-06-28 | 2010-09-30 | Demong Duane Eugene | Substituted piperazines as cb1 antagonists |
| US20100286160A1 (en) | 2007-06-28 | 2010-11-11 | Intervet Inc. | Substituted piperazines as cb1 antagonists |
| US20090036425A1 (en) | 2007-08-02 | 2009-02-05 | Pfizer Inc | Substituted bicyclolactam compounds |
| US20110195932A1 (en) | 2007-08-03 | 2011-08-11 | Graham Michael Wynne | Drug Combinations for the Treatment of Duchenne Muscular Dystrophy |
| WO2009024823A2 (en) | 2007-08-22 | 2009-02-26 | Astrazeneca Ab | Oxadiazole derivatives as dgat inhibitors |
| EP2195293A2 (en) | 2007-08-22 | 2010-06-16 | AstraZeneca AB | Cycloptopyl amide derivatives |
| US20090076020A1 (en) | 2007-08-22 | 2009-03-19 | Astrazeneca Ab | Cyclopropyl Amide Derivatives 978 |
| EP2253615A1 (en) | 2007-08-22 | 2010-11-24 | AstraZeneca AB | Cyclopropyl amide derivatives |
| WO2009030716A1 (en) | 2007-09-06 | 2009-03-12 | Glaxo Group Limited | Piperazine derivative having affinity for the histamine h3 receptor |
| WO2009039431A2 (en) | 2007-09-21 | 2009-03-26 | Neurogen Corporation | Substituted aryl-fused spirocyclic amines |
| WO2009081195A1 (en) | 2007-12-20 | 2009-07-02 | Astrazeneca Ab | Carbamoyl compounds as dgat1 inhibitors 190 |
| WO2009085945A1 (en) | 2007-12-20 | 2009-07-09 | Abbott Laboratories | Benzothiazole and benzoxazole derivatives and methods of use |
| US20090192168A1 (en) | 2008-01-04 | 2009-07-30 | Alex Muci | Compounds, Compositions and Methods |
| US20100292188A1 (en) | 2008-01-24 | 2010-11-18 | Ucb Pharma S.A. | Compounds Comprising A Cyclobutoxy Group |
| WO2009092764A1 (en) | 2008-01-24 | 2009-07-30 | Ucb Pharma, S.A. | Compounds comprising a cyclobutoxy group |
| EP2238144A1 (en) | 2008-01-24 | 2010-10-13 | UCB Pharma, S.A. | Compounds comprising a cyclobutoxy group |
| WO2009100120A2 (en) | 2008-02-04 | 2009-08-13 | Neurogen Corporation | Pyridinyl-substituted piperazinyl oxoethyl tetrahydropyrazolopyridines |
| WO2009100294A2 (en) | 2008-02-07 | 2009-08-13 | Abbott Laboratories | Amide derivatives as positive allosteric modulators and methods of use thereof |
| WO2009115874A2 (en) | 2008-03-17 | 2009-09-24 | Matrix Laboratories Ltd. | Novel heterocyclic compounds, pharmaceutical compositions containing them and processes for their preparation |
| WO2009124553A2 (en) | 2008-04-09 | 2009-10-15 | Neurokey A/S | Use of hypothermia inducing drugs |
| EP2300426A1 (en) | 2008-06-06 | 2011-03-30 | UCB Pharma, S.A. | Compounds comprising a cyclobutoxy group |
| US20110098300A1 (en) | 2008-06-06 | 2011-04-28 | Ucb Pharma, S.A. | Compounds Comprising A Cyclobutoxy Group |
| WO2009147149A1 (en) | 2008-06-06 | 2009-12-10 | Ucb Pharma, S.A. | Compounds comprising a cyclobutoxy group |
| WO2009151991A1 (en) | 2008-06-11 | 2009-12-17 | Merck & Co., Inc. | Pyrazole derivatives useful as inhibitors of faah |
| US20100016344A1 (en) | 2008-07-11 | 2010-01-21 | Abbott Laboratories | Amino heterocyclic linked pyrimidine derivatives |
| WO2010007382A1 (en) | 2008-07-18 | 2010-01-21 | Takeda Pharmaceutical Company Limited. | Benzazepine derivatives and their use as hstamine h3 antagonists |
| US20100267714A1 (en) | 2008-08-18 | 2010-10-21 | Jorgensen William L | Mif modulators |
| CA2734800A1 (en) | 2008-08-20 | 2010-02-25 | Probiodrug Ag | Antibodies directed against pyroglutamate monocyte chemoattractant protein-1 (mcp-1 n1pe) |
| WO2010071822A1 (en) | 2008-12-19 | 2010-06-24 | Schering Corporation | Piperidine and piperazine derivatives and methods of use thereof |
| WO2010080757A2 (en) | 2009-01-07 | 2010-07-15 | Astrazeneca Ab | Combinations with an alpha-4beta-2 nicotinic agonist |
| US20100216812A1 (en) | 2009-02-20 | 2010-08-26 | Astrazeneca Ab | Cyclopropyl Amide Derivatives |
| US20100227876A1 (en) | 2009-03-06 | 2010-09-09 | Rechfensen Llp | Methods of Reducing Side Effects of Analgesics |
| WO2010129242A2 (en) | 2009-04-27 | 2010-11-11 | Abbott Laboratories | Treatment of osteoarthritis pain |
| US20100273778A1 (en) | 2009-04-27 | 2010-10-28 | Abbott Laboratories | Treatment of osteoarthritis pain |
| WO2011083314A1 (en) | 2010-01-08 | 2011-07-14 | Takeda Pharmaceutical Company Limited | Benzazepine derivatives for the treatment of central nervous system disorders |
| WO2011083315A1 (en) | 2010-01-08 | 2011-07-14 | Takeda Pharmaceutical Company Limited | Compounds and their use |
| WO2011083316A1 (en) | 2010-01-08 | 2011-07-14 | Takeda Pharmaceutical Company Limited | Benzazepine derivatives for the treatment of central nervous system disorders |
| US20120071651A1 (en) | 2010-09-16 | 2012-03-22 | Abbott Laboratories | Processes for Preparing 1,2-Substituted Cyclopropyl Derivatives |
Non-Patent Citations (135)
| Title |
|---|
| Airaksinen M.S., et al., "Histamine Neurons in Human Hypothalamus: Anatomy in Normal and Alzheimer Diseased Brains," Neuroscience, 1991, vol. 44 (2), pp. 465-481. |
| Arrang J.M., et al., "Auto-inhibition Of Brain Histamine Release Mediated by a Novel Class (H3) of Histamine Receptor," Nature, 1983, vol. 302, pp. 832-837. |
| Arrang J.M., et al., "Highly Potent and Selective Ligands for Histamine H3-Receptors," Nature, 1987, vol. 327, pp. 117-123. |
| Arrang J.M., et al., "Histamine H3 Receptor Binding Sites in Rat Brain Membranes:Modulations by Guanine Nucleotides and Divalent Cations," European Journal of Pharmacology, 1990, vol. 188, pp. 219-227. |
| Barbier A.J., et al., "Acute Wake-Promoting Actions of JNJ-5207852, a Novel, Diamine-based H3 Antagonist," British Journal of Pharmacology, 2004, vol. 143, pp. 649-661. |
| Barbier A.J., et al., "Histaminergic Control of Sleep-Wake Cycles: Recent Therapeutic Advances for Sleep and Wake Disorders ," CNS and Neurological Disorders-Drug Targets, 2007, vol. 6, pp. 31-43. |
| Berlin M., et al., "Histamine H3 Receptor as a Drug Discovery Target," Journal of Medicinal Chemistry, 2011, vol. 54 (1), pp. 26-53. |
| Berlin M., et al., "Recent Advances in the Development of Histamine H3 Antagonists," Expert Opinion in the Therapeutic Patents, 2007, vol. 17 (6), pp. 675-687. |
| Bernaerts P., et al., "Histamine H3 Antagonist Thioperamide Dose-Dependently Enhances Memory Consolidation and Reverse Amnesia Induced by Dizocilpine or Scopolamine in a One-Trail Inhibitory Avoidance Task in Mice," Behavioural Brain Research, 2004, vol. 154, pp. 211-219. |
| Bjenning C., et al., "Peripherally Administered Ciproxifan Elevates Hypothalamic Histamine levels and Potently Reduces Food Intake in the Sprague Dawley Rat" in: Histamine Research in the New Mellennium, Watanabe T., et al., eds., Elsevier Science, 2001, pp. 449-450. |
| Blandina P., et al., "Histamine Neuronal System as a Therapeutic Target for the Treatment of Cognitive Disorders," Future Neurology, 2010, vol. 5 (4), pp. 543-555. |
| Browman K.E. et al., "Enhancement of Prepulse Inhibition of Startle in Mice by the H3 Receptor Antagonists Thioperamide and Ciproxifan," Behavioiral Brain Research, 2004m vol. 153 (1), pp. 69-76. |
| Burger A., et al., "2-(4-imidazolul)cyclopropylamine," Journal of Medicinal Chemistry, 1970, vol. 13, pp. 33-35. |
| Celanire S., et al., "Keynote Review: Histamine H3 Receptor Antagonists Reach Out for The Clinic," Drug Discovery Today, 2005, vol. 10 (23/24), pp. 1613-1627. |
| Charette A.B. et al., "(2S,3S)-(+)-(3-Phenylcyclopropyl)Methanol," Org Syntheses Coll, 1999, vol. 76, pp. 86-96. |
| Chen Z., et al., "Effects of Histamine on MK-801-induced Memory Deficits in Radial Maze Performance in Rats," Brain Research, 1999, vol. 839, pp. 186-189. |
| Chen Z., et al., "Pharmacological Effects of Carcinine on Histaminergic Neurons in the Brain," British Journal of Pharmacology, 2004, vol. 143, pp. 573-580. |
| Clapham J., et al., "Thioperamide, the Selective Histamine H3 Receptor Antagonist, Attenuates Stimulant Induced Locomotor Acitivity in the Mouse," European Jounal of Pharmacology, 1994, vol. 259 (2), pp. 107-114. |
| Collins S.D., "Emerging Therapies for Neuropathic Pain," Expert Opinion on Emerging Drugs, 2005, vol. 10 (1), pp. 95-108. |
| Cowart M., et al., "4-(2-[2-(2(R)- Methylpyrrolidin-1-yl) ethyl] Benzofuran-5yl) Benzonitrile and Related 2-Aminoethylbenzofuran H3 Receptor Antagonists Potently Enhance Cognition and Attention," Journal of Medicinal Chemistry, 2005, vol. 48 (1), pp. 38-55. |
| Cowart M., et al., "Pharmacological Characterization of A-960656, a Histamine H3 Receptor Antagonist with Efficacy in Animal Models of Osteoarthritis and Neuropathic Pain," European Journal of Pharmacology, 2012, vol. 684 (1-3), pp. 87-94. |
| Cross, L.C. et al., "IUPAC Commission on Nomenclature of Organic Chemistry: Rules for the Nomenclature of Organic Chemistry, Section E: Stereochemistry," Pure and Applied Chemistry, 1976, vol. 45, pp. 13-30. |
| De Almeida M.A., et al., "Memory Facilitation by Histamine," Archives Internationales De Physiologie Et De Biochimie, 1986, vol. 283 (2), pp. 193-198. |
| Delaunois A., et al., "Modulation of Acetylcholine, Capsaicin and subsztance P Effects by Histamine H3 Receptors in Isolated Perfused Rabbit Lungs," European Journal of Pharmacology, 1995, vol. 277 (2-3), pp. 243-250. |
| Dimitriadou V., et al., "Functional Relationship Between Mast Cells and C-Sensitive Nerve Fibres Evidenced by Histamine H3 -Receptor Modulation in Rat Lung and Spleen," Clinical Science, 1994, vol. 87, pp. 151-163. |
| Dray A., et al., "Pharmacology of Chronic Pain," Trends in Pharmacological Sciences, 1994, vol. 15 (6), pp. 190-197. |
| Dray et al., "Arthritis Pain. Future Targets to Control Osteoarthritis Pain," Arthritis Research and Therapy, 2007, vol. 9 (3), pp. 212. |
| Dumery V., et al., "Development of Amygdaloid Cholinergic Mediation of Passive Avoidance Learning in the Rat," Experimental Brain Research , 1987, vol. 67(1), pp. 61-69. |
| Dvorak C.A., et al., "4-Phenoxypiperidines: Potent, Conformationally Restricted, non-Imidazole Histamine H3 Antagonists," Journal of Medicinal Chemistry, 2005, vol. 48 (6), pp. 2229-2238. |
| Dworkin R.H., "An Overview of Neuropathic Pain: Syndromes, Symptoms, Signs, and Several Mechanisms," Clinical Journal of Pain, 2002, vol. 18 (6), pp. 343-349. |
| Esbenshade T.A., et al., "Histamine H3 Receptor Antagonists: Preclinical Promise for Treating Obesity and Cognitive Disorders," Molecular Interventions, 2006, vol. 6 (2), pp. 77-88. |
| Esbenshade T.A., et al., "Pharmacological and Behavioral Properties of A-349821, a Selective and Potent Human Histamine H3 Receptor Antagonist," Biochemical Pharmacology, 2004, vol. 68 (5), pp. 933-945. |
| Esbenshade T.A., et al., "Pharmacological Properties of ABT-239 [4-(2- {2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yObenzonitrile]: I. Potent and Selective Histamine H3 Receptor Antagonist with Drug-Like Properties," Journal of Pharmacology and Experimental Therapeutics, 2005, vol. 313 (1), pp. 165-175. |
| Esbenshade T.A., et al., "The Histamine H3 Receptor: An Attractive Target for the Treatment of Cognitive Disorders," British Journal of Pharmacology, 2008, vol. 154 (6), pp. 1166-1181. |
| Fernihough J., et al., "Pain Related Behaviour in Two Models of Osteoarthritis in the Rat Knee," Pain, 2004, vol. 112 (1-2), pp. 83-93. |
| Final Office Action mailed Aug. 8, 2011 for U.S. Appl. No. 11/766,987, filed Jun. 22, 2007. |
| Fitzsimons C., et al., "Histamine Receptors Signaling in Epidermal Tumor Cell Lines with H-Ras Gene Alterations," Inflammation Research, 1998, vol. 47 (1), pp. S50-S51. |
| Foley A.G., et al., "H3 Receptor Antagonism Enhances NCAM PSA-Mediated Plasticity and Improves Memory Consolidation in Odor Discrimination and Delayed Match-to-Position Paradigms," Neuropsychopharmacology, 2009, vol. 34 (12), pp. 2585-2600. |
| Fox G.B., et al., "Effects of Histamine H3 Receptor Ligands GT2331 and Ciproxifan in a Repeated Acquisition Response in the Spontaneously Hypertensive Rat Pup," Behavioural Brain Research, 2002, vol. 131 (1-2), pp. 151-161. |
| Fox G.B., et al., "Identification of Novel H3 Receptor (H3R) Antagonists with Cognition Enhancing Properties in Rats," Inflammation Research, 2003, vol. 52 (1), pp. S31-S32. |
| Fox G.B., et al., "Pharmacological Properties of ABT-239 [4-(2-{2-[(2R)-2-Methylpyrrolidinty]ethyl}-benzofuran-5-yhbenzonitrile]- : II Neurophysiological Characterization and Broad Preclinical Efficacy in Cognition and Schizophrenia of a Potent and Selective Histamine H3 R," Journal of Pharmacology and Experimental Therapeutics, 2005, vol. 313 (1), pp. 176-190. |
| Fox G.B., et al., "Two Novel and Selective Nonimidazole H3 Receptor Antagonists A-304121 and A-317920: II. In Vivo Behavioral and Neurophysiological Characterization," Journal of Pharmacology and Experimental Therapeutics, 2003, vol. 305 (3), pp. 897-908. |
| Furniss B.S., et al., Vogel's Textbook of Practical Organic Chemistry, 5th Edition, Longman Scientific & Technical, 1989, Table of Contents. |
| Glase S.A., et al., "Attention Deficit Hyperactivity Disorder: Pathophysiology and Design of New Treatments," Annual Reports in Medicinal Chemistry, 2002, vol. 37, pp. 11-20. |
| Haas L., et al., "Subcortical Modulation of Synaptic Plasticity in the Hippocampus," Behavioural Brain Research, 1995, vol. 66 (1-2), pp. 41-44. |
| Halpern, M.T., "GT-2331," Current Opinion in Central and Peripheral Nervous System Investigational Drugs, vol. 1, pp. 524-527, 1999. |
| Hancock A.A., et al., "Antiobesity Effects of A-331440, a Novel Non-Imidazole Histamine H3 Receptor Antagonist," European Journal of Pharmacology, 2004, vol. 487 (1-3), pp. 183-197. |
| Hancock A.A., et al., "Histamine H3 Antagonists in Models of Obesity," Inflammatory Research, 2004, vol. 53 (Suppl. 1), pp. S47-S48. |
| Harada C., et al., "Inhibitory Effect of lodophenpropit, a Selective Histamine H3 Antagonist, on Amygdaloid Kindled Seizures," Brain Research Bulletin, 2004, vol. 63 (2), pp. 143-146. |
| Harada C., et al., "Intracerebroventricular Administration of Histamine H3 Receptor Antagonists Decreases Seizures in Ray Models of Epilepsia," Methods and Findings in Experimental and Clinical Pharmacology, 2004, vol. 26 (4), pp. 263-270. |
| Hriscu A., et al., "Experimental Evaluation of the Analgesic Efficacy of Some Antihistamines as Proof of the Histaminergic Receptor Involvement in Pain," Famacia, 2001, vol. 49 (2), pp. 23-30. |
| Hsieh G.C., et al., "The Histamine H3 Receptor as a Potential Antinociceptive Target: Effects of Selective H3 Antagonists in Several Preclinical Pain Models and the Involvement of Noradrenergic Systems," Global Pharmaceutical Research & Development, 2009, Abbott Laboratories, Abbott Park, IL 60064. |
| Huang Y.W., et al., "Effect of the Histamine H3-antagonist Clobenpropit on Spatial Memory Deficits Induced by MK-801 as Evaluated by Radial Maze in Sprague-Dawley Rats," Behavioural Brain Research, 2004, vol. 151 (1-2), pp. 287-293. |
| International Search Report for Application No. PCT/DK2003/000071, mailed on Jul. 29, 2003, 10 pages. |
| International Search Report for Application No. PCT/EP2006/063753, mailed on Apr. 27, 2007, 11 pages. |
| International Search Report for Application No. PCT/EP2008/052430, mailed on May 25, 2009, 5 pages. |
| International Search Report for Application No. PCT/US2007/071849, mailed on Jan. 29, 2008, 4 pages. |
| International Search Report for Application No. PCT/US2008/077103, mailed on Apr. 27, 2009, 3 pages. |
| International Search Report for Application No. PCT/US2008/085622, mailed on Jun. 8, 2009, 6 pages. |
| International Search Report for Application No. PCT/US2008/085662, mailed on Jun. 8, 2009, 4 pages. |
| International Search Report for Application No. PCT/US2009/033062, mailed on Sep. 1, 2009, 4 pages. |
| International Search Report for Application No. PCT/US2009/033329, mailed on Sep. 17, 2009, 3 pages. |
| International Search Report for Application No. PCT/US2010/032488, mailed on Nov. 2, 2010, 11 pages. |
| International Search Report for Application No. PCT/US2011/051603, mailed on Dec. 5, 2011, 5 pages. |
| Itoh E., et al., "Thioperamide, A Histamine H3 Receptor Antagonist, Powerfully Suppresses Peptide YY-Induced Food Intake in Rats," Biological Psychiatry, 1999, vol. 45 (4), pp. 475-481. |
| Jantzen, et al., Modern Pharmacueticals, 1996, pp. 596. |
| Joshi S.K., et al., "Animal Models of Pain for Drug Discovery," Expert Opinion on Drug Discovery, 2006, vol. 1 (4), pp. 341-352. |
| Kallemeyn J.M., et al., "Asymmetric Synthesis of Di- and Trisubstituted Cyclopropanes Through an Intramolecular Ring Closure," Synlett, 2011, vol. 4, pp. 535-538. |
| Kallemeyn J.M., et al., ChemInform, 2011, vol. 42 (26), 4 pages. |
| Komater V.A., et al., "H3 Receptor Blockade by Thioperamide Enhances Cognition in Rats without Inducing Locomotor Sensitization," Psychopharmacology, 2003, vol. 167 (4), pp. 363-372. |
| Krueger K.M., et al., "G Protein-Dependent Pharmacology of Histamine H3 Receptor Ligands: Evidence for Heterogeneous Active State Receptor Conformations," Journal of Pharmacology and Experimental Therapeutics, 2005, vol. 314 (1), pp. 271-281. |
| Leurs R., et al., "En Route to New Blockbuster Anti-Histamines: Surveying the Offspring of the Expanding Histamine Receptor Family," Trends in Pharmacological Sciences, 2011, vol. 32 (4), pp. 250-257. |
| Leurs R., et al., "Histamine Homologues Discriminating between Two Functional H3 Receptor Assays. Evidence for H3 Receptor," Journal of Pharmacology and Experimental Therapeutics, 1996, vol. 276 (3), pp. 1009-1015. |
| Leurs R., et al., "The Medicinal Chemistry And Therapeutic Potentials Of Ligands Of The Histamine H3 Receptor," Progress In Drug Research, 1995, vol. 45, pp. 107-165. |
| Leurs R., et al., eds., "The Histamine H3 Receptor: A Target for New Drugs," vol. 30, Elsevier Science B.V., 1998, Table of Contents. |
| Leurs R., et al., The Histamine H3-Receptor: A Target For Developing New Drugs, Elsevier Science, 1998, vol. 39, pp. 127-165. |
| Ligneau X., et al., "Neurochemical and Behavioral Effects of Ciproxifan, a Potent Histamine H3-Receptor Antagonist," Journal of Pharmaceutical and Experimental Therapeutics, 1998, vol. 287 (2), pp. 658-666. |
| Lin J.S., et al., "Involvement of Histaminergic Neurons in Arousal Mechanisms Demonstrated with H3 -Receptor Ligands in the Cat," 1990, vol. 523 (2), pp. 325-330. |
| Lozada A.F., et al., "Plasticity of Histamine H3 Receptor Expression and Binding in the Vestibular Nuclei After Labyrinthectomy in Rat," Biomedical Center Neuroscience, 2004, vol. 5, pp. 32. |
| Malmberg Aiello P., et al., "Role of Histamine in Rodent Antinociception," British Journal of Pharmacology, 1994, vol. 111 (4), pp. 1269-1279. |
| Mazurkiewicz-Kwilecki I.M., et al., "Changes in the Regional Brain Histamine and Histidine Levels in Postmortem Brains of Alzheimer Patients," Canadian Journal of Physiology and Pharmacology, 1989, vol. 67 (1), pp. 75-78. |
| McLeod R.L., et al., "Combined Histamine H1 and H3 Receptor Blockade Produces Nasal Decongestion in an Experimental Model of Nasal Congestion," American Journal of Rhinology, 1999, vol. 13 (5), pp. 391-399. |
| McLeod R.L., et al., "Histamine H3 Antagonists," Progress in Respiratory Research, 2001, vol. 31, pp. 133-136. |
| Medhurst a.D., et al., "GSK189254, a Novel H3 Receptor Antagonist that Binds to Histamine H3 Receptors in Alzheimer's Disease Brain and Improves Cognitive Performance in Preclinical Models," Journal of Pharmacology and Experimental Therapeutics, 2007, vol. 321 (3), pp. 1032-1045. |
| Medhurst A.D., et al., "Structually Novel Histamine H3 Receptor Antagonists GSK207040 Secondary Allodynia in Rats," Biochemical Pharmacology, 2007, vol. 73, pp. 1182-1194. |
| Medhurst S.J., et al., "Novel histamine H3 receptor antagonists GSK189254 and GSK334429 are efficacious in surgically-induced and virally-induced rat models of neuropathic pain," Pain, 2008, vol. 138 (1), pp. 61-69. |
| Meguro K., et al., "Effects of Thioperamide, a Histamine H3 Antagonist, on the Step-Through Passive Avoidance Response and Histidine Decarboxylase Activity in Senescence-Accelerated Mice," Pharmacology, Biochemistry and Behavior, 1995, vol. 50 (3), pp. 321-325. |
| Monti J., et al., "Sleep and Waking During Acute Histamine H3 Agonist BP2.94 or H3 Antagonist Carboperamide (MR 16155) Administration in Rats," Neuropsychopharmacology, 1996, vol. 15 (1), pp. 31-35. |
| Morisset S., et al., "Atypical Neuroleptics Enhance Histamine Turnoer in Brain Via 5-Hydroxytryptamino2A Receptor Blockade," Journal Pharmacology and Experimental Therapeutics, 1999, vol. 288 (2), pp. 590-596. |
| Murakami K., et al., "AQ-0145, A Newly Developed Histamine H3 Antagonist, Decreased Seizure Susceptibility of Electrically Induced Convulsions In Mice," Methods and Findings in Experimental and Clinical Pharmacology, 1995, vol. 17 Suppl C, pp. 70-73. |
| Njar, "High-Yields Synthesis of Novel Imidazoles and Triazoles form Alcohols and Phenols," Synthesis, 2000, pp. 2019-2028. |
| Office Action mailed Jan. 7, 2011 for U.S. Appl. No. 11/766,987, filed Jun. 22, 2007. |
| Office Action mailed Jan. 9, 2009 for U.S. Appl. No. 11/766,987, filed Jun. 22, 2007. |
| Office Action mailed Mar. 22, 2010 for U.S. Appl. No. 11/956,816, filed Dec. 14, 2007. |
| Office Action mailed Oct. 19, 2009 for U.S. Appl. No. 11/766,987, filed Jun. 22, 2007. |
| Office Action mailed Oct. 7, 2010 for U.S. Appl. No. 11/956,816, filed Dec. 14, 2007. |
| Office Action mailed Sep. 8, 2008 for U.S. Appl. No. 11/766,987, filed Jun. 22, 2007. |
| O'Neill A.B., et al., "Pharmacological Evaluation of the in Vivo Model of Vestibular Dysfunction in the Rat," Methods and Findings in Experimental and Clinical Pharmacology, 1999, vol. 21 (4), pp. 285-289. |
| Onodera K., et al., "Improvement by FUB 181, A Novel Histamine H 3-Receptor Antagonist, of Learning and Memory in the Elevated Plus-Maze Test in Mice," Naunyn-Schmiedebergs' Archives of Pharmacology, 1998, vol. 357 (5), pp. 508-513. |
| Onodera K., et al., "Neuropharmacology of the Histaminergic Neuron System in the Brain And its Relationship with Behavioral Disorders," Progress in Neurobiology, 1994, vol. 42 (6), pp. 685-702. |
| Pan J.B., et al., "Histaminergic Ligands Attenuate Barrel Rotation in Rats Following Unilateral Labyrinthectomy," Methods and Findings in Experimental and Clinical Pharmacology, 1998, vol. 20 (9), pp. 771-777. |
| Panula P., et al., "Neuronal Histamine Deficit in Alzheimer's Disease," Neuroscience, 1998, vol. 82 (4), pp. 993-997. |
| Passani M.B., et al., "Central Histaminergic System and Cognition," Neuroscience and Biobehavioral Reviews, 2000, vol. 24 (1), pp. 107-113. |
| Penning T.D., et al., "Structure-Activity Relationship Studies on 1-[2-(4-Phenylphenoxy)ethly]pyrrolidine (SC-22716), a Potent Inhibitor of Leukotriene A4 (LTA4) Hydrolase," Journal of Medicinal Chemistry, 2000, vol. 43 (4), pp. 721-735. |
| Perez-Garcia C., et al., "Effects of Histamine H3 Receptor Ligands in Experimental Models Of Anxiety And Depression," Psychopharmacology, 1999, vol. 142 (2), pp. 215-220. |
| Poste G. et al., "Lipid Vesicles as Carriers for Introducing Biologically Active Materials into Cells," Methods in Cell Biology, 1976, vol. 14, pp. 33-71. |
| Prast H., et al., "Histaminergic Neurons Facilitate Social Memory in Rats," Brain Research, 1996, vol. 734 (1-2), pp. 316-318. |
| Pu Y.M., et al., "A Facile and Scaleable Synthesis of ABT-239, A Benzofuranoid H3 Antagonist," Organic Process Research and Development, 2005, vol. 9, pp. 45-50. |
| Roche E.B., ed., Bioreversible Carries in Drug Design Theory and Application, Pergamon Press, 1987, Table of Contents. |
| Rodrigues A.A., et al., "Interaction of Clozapine with the Histamine H3 Receptor in Rat Brain," British Journal of Pharmacology, 1995, vol. 114 (8), pp. 1523-1524. |
| Rubin B.R., "Management of Osteoarthritic Knee Pain," Journal of the American Osteopathic Association, 2005, vol. 105 (9 Suppl. 4), pp. S23-S28. |
| Sakai N., et al., "Effects of Thioperamide, A Histamine H3 Receptor Antagonist, On Locomotor Activity and Brain Histamine Content in Mast Cell-Deficient Wiwv Mice," Life Sciences, 1991, vol. 48 (25), pp. 2397-2404. |
| Sakata T., et al., "Hypothalamic Neuronal Histamine Modulates Ad Libitum Feeding by Rats," Brain research, 1990, vol. 537 (1-2), pp. 303-306. |
| Sanchez-Lemus E., et al., "Histamine H3 Receptor Activation Inhibits Dopamine D1 Receptor-Induced Camp Accumulation in Rat Striatal Slices," Neuroscience Letters, 2004, vol. 364 (3), pp. 179-184. |
| Sander K., et al., "Histamine H3 Receptor Antagonists Go to Clinics," Biological & Pharmaeutical Bulletin, 2008, vol. 31 (12), pp. 2163-2181. |
| Schwartz J., et al., "Histamine", in: Psychopharmacology: The Fourth Generation Of Progress, Chapter 35, Bloom F.E., et al., eds., Raven Press, 1995, pp. 397-405. |
| Schweitzer J.B., et al., "Drugs Under Investigation for Attention-Deficit Hyperactivity Disorder," Current Opinion in Investigational Drugs, 2002, vol. 3 (8), pp. 1207-1211. |
| Shaywitz B.A., et al., "Dopaminergic But Not Noradrenergic Mediation of Hyperactivity and Performance Deficits in the Developing Rat Pup," Psychopharmacology, 1984, vol. 82 (1-2), pp. 73-77. |
| Smith P.A., et al., "Neuropathic Pain and the Electrophsiology and Pharmacology of Nerve Injury," Drug Development Research, 2001, vol. 54 (3), pp. 140-153. |
| Szelag A., "Role of Histamine H3 -Receptors in the Proliferation Neoplastic Cells in Vitro," Medical Science Monitor, 1998, vol. 4 (5), pp. 747-755. |
| Tedford C.E., "Pharmacological Characterization of Gt-2016, A Non-Thiourea-Containing Histamine H3 Antagonist: in Vitro and in Vivo Studies," The Journals Of Pharmacology And Experimental Therapeutics, 1995, vol. 275 (2), pp. 598-604. |
| Tedford et al., "Cognition and Locomotor Activity in the Developing Rat: Comparisons Of Histamine H3 Receptor Antagonists And ADHD Therapeutics," Society For Neuroscience Abstr, vol. 22, pp. 22, 1996. |
| Tozer M., et al., "Histamine H3 Receptor Antagonists," Expert Opinion Therapeutic Patents, 2000, vol. 10 (7), pp. 1045-1055. |
| Vinik A.L., et al., "Diabetic Neuropathies," The Medical Clinics of North America, 2004, vol. 88 (4), pp. 947-999. |
| Vohora D., et al., "Thioperamide, A Selective Histamine H3 Receptor Antagonist, Protects Against PTZ-Induced Seizures in Mice," Life Sciences, 2000, vol. 66 (22), pp. PL297-PL301. |
| Wada H., et al., "Is the Histaminergic Neuron System a Regulatory Center for Whole-Brain Activity", Trends in Neurosciences, 1991, vol. 14 (9), pp. 415-418. |
| Wang Y., et al., "Design and Synthesis of Ether Analogues as Potent and Selective M2 Muscarinic Receptor Antagonists," Bioorganic and Medicinal Chemistry Letters, 2001, vol. 11 (7), pp. 891-894. |
| Witkin J.M., et al., "Selective Histamine H3 Receptor Antagonists for the Treatment of Cognitive Deficiencies and Other Disorders of the Central Nervous System," Pharmacology and Therapeutics, 2004, vol. 103 (1), pp. 1-20. |
| Yates S.L., et al., "Identification and Pharmacological Characterization of a Series of New 1H-4-Substituted-Imidazoyl Histamine H3 Receptor Ligands," Journal of Pharmacology and Experimental Therapeutics, 1999, vol. 289 (2), pp. 1151-1159. |
| Yates, S.L., et al., "Effects of a novel histamine H3 receptor antagonist, GT2394, on food intake and weight gain in Sprague-Dawley rats," Society for Neuroscience, vol. 102 (10), pp. 219, 2000. |
| Yawata I., et al., "Role Of Histaminergic Neurons in Development of Epileptic Seizures in El Mice," Brain Research. Molecular Brain Research, 2004, vol. 132 (1), pp. 13-17. |
| Yokoyama H., et al., "Clobenpropit (Vuf-9153), a New Histamine H3 Receptor Antagonist, Inhibits Electrically Induced Convulsions in Mice," European Journal of Pharmacology, 1994, vol. 260 (1), pp. 23-28. |
| Yokoyama H., et al., "Effect of Thioperamide, a Histamine H3 Receptor Antagonist, on Electrically Induced Convulsions in Mice," Journal of Pharmacology, 1993, vol. 234 (1), pp. 129-133. |
| Yokoyama H., et al., "Histamine And Seizures Implcations For The Treatment Of Epilepsy," CNS Drugs, 1996, vol. 5 (5), pp. 321-330. |
| Zhang X., et al., "Trans-1-[(2-Phenylcyclopropyl)methyl]-4-arylpiperazines: Mixed Dopamine D(2)/D(4) Receptor Antagonists as Potential Antipsychotic Agents," Journal of Medicinal Chemistry, 2000, vol. 43 (21), pp. 3923-3932. |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10285998B1 (en) | 2018-04-04 | 2019-05-14 | The Menopause Method, Inc. | Composition and method to aid in hormone replacement therapy |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2012037258A1 (en) | 2012-03-22 |
| US20120071651A1 (en) | 2012-03-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8853390B2 (en) | Processes for preparing 1,2-substituted cyclopropyl derivatives | |
| US20230303489A1 (en) | Compounds, compositions and methods of use | |
| KR102085557B1 (en) | Prodrugs of fumarates and their use in treating various diseases | |
| AU2023202086A1 (en) | Antagonists of the muscarinic acetylcholine receptor M4 | |
| EP3153167B1 (en) | Substituted 2-(4-heterocyclylbenzyl)isoindolin-1-one analogs as positive allosteric modulators of the muscarinic acetylcholine receptor m1 | |
| ES2902806T3 (en) | Compositions Containing Substituted [1,2,4]triazolo[1,5-a]pyrimidin-7-yl Compounds as PDE2 Inhibitors | |
| US9309199B2 (en) | Inhibitors of catechol O-methyl transferase and their use in the treatment of psychotic disorders | |
| AU2017245125A1 (en) | Heterocyclic compound | |
| JP5562866B2 (en) | Mineralocorticoid receptor antagonists and methods of use | |
| WO2013103931A1 (en) | Substituted 1-benzylindolin-2-one analogs as positive allosteric modulators of muscarinic acetylcholine m1 receptors | |
| US20080280879A1 (en) | Substituted heterocyclic derivatives and their pharmaceutical use and compositions | |
| WO2008029825A1 (en) | Imidazole derivative | |
| JP2020507582A (en) | Aminotriazolopyridines as kinase inhibitors | |
| JP2012524798A (en) | 2-alkylpiperidine MGLUR5 receptor modulator | |
| CA2878006C (en) | Carbamate/urea derivatives | |
| US10246414B2 (en) | Allosteric modulators of CB1 cannabinoid receptors | |
| WO2010059922A1 (en) | Pyrrolidine carboxamide compounds | |
| WO2011163280A1 (en) | Indole compounds as positive allosteric modulators of the muscarinic receptor | |
| US9029563B2 (en) | Substituted 1-benzylindolin-2-one analogs as positive allosteric modulators of muscarinic acetylcholine M1 receptors | |
| AU2021261002A1 (en) | Condensed substituted hydropyrroles as antagonists of the muscarinic acetylcholine receptor m4 | |
| CA2755335C (en) | Process for the preparation of histamine h3 receptor modulators | |
| US9073864B2 (en) | Aromatic ring compound | |
| WO1993020053A1 (en) | Pyridylserine derivative | |
| TW201206901A (en) | Substituted N-heteroaryl bipyrrolidine carboxamides, preparation and therapeutic use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: ABBOTT LABORATORIES, ILLINOIS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:KU, YI-YIN;GRIEME, TIMOTHY A.;KALLEMEYN, JEFFREY M.;AND OTHERS;REEL/FRAME:027072/0677 Effective date: 20111012 |
|
| AS | Assignment |
Owner name: ABBVIE INC., ILLINOIS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:ABBOTT LABORATORIES;REEL/FRAME:030137/0222 Effective date: 20120801 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 4TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1551) Year of fee payment: 4 |
|
| FEPP | Fee payment procedure |
Free format text: MAINTENANCE FEE REMINDER MAILED (ORIGINAL EVENT CODE: REM.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| LAPS | Lapse for failure to pay maintenance fees |
Free format text: PATENT EXPIRED FOR FAILURE TO PAY MAINTENANCE FEES (ORIGINAL EVENT CODE: EXP.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| STCH | Information on status: patent discontinuation |
Free format text: PATENT EXPIRED DUE TO NONPAYMENT OF MAINTENANCE FEES UNDER 37 CFR 1.362 |
|
| FP | Lapsed due to failure to pay maintenance fee |
Effective date: 20221007 |