US20050124583A1 - Prodrugs of phosphonate nucleotide analogues - Google Patents
Prodrugs of phosphonate nucleotide analogues Download PDFInfo
- Publication number
- US20050124583A1 US20050124583A1 US11/031,250 US3125005A US2005124583A1 US 20050124583 A1 US20050124583 A1 US 20050124583A1 US 3125005 A US3125005 A US 3125005A US 2005124583 A1 US2005124583 A1 US 2005124583A1
- Authority
- US
- United States
- Prior art keywords
- pmpa
- prodrugs
- prodrug
- plasma
- tissues
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 229940002612 prodrug Drugs 0.000 title description 98
- 239000000651 prodrug Substances 0.000 title description 98
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 title description 5
- -1 nucleotide analog compound Chemical class 0.000 claims description 34
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 10
- VCMJCVGFSROFHV-WZGZYPNHSA-N tenofovir disoproxil fumarate Chemical compound OC(=O)\C=C\C(O)=O.N1=CN=C2N(C[C@@H](C)OCP(=O)(OCOC(=O)OC(C)C)OCOC(=O)OC(C)C)C=NC2=C1N VCMJCVGFSROFHV-WZGZYPNHSA-N 0.000 abstract description 95
- 238000000034 method Methods 0.000 abstract description 40
- 239000000203 mixture Substances 0.000 abstract description 39
- 150000001875 compounds Chemical class 0.000 abstract description 36
- 125000001424 substituent group Chemical group 0.000 abstract description 9
- 238000002560 therapeutic procedure Methods 0.000 abstract description 5
- 239000000546 pharmaceutical excipient Substances 0.000 abstract description 3
- 238000011321 prophylaxis Methods 0.000 abstract description 2
- 230000001177 retroviral effect Effects 0.000 abstract 1
- 210000001519 tissue Anatomy 0.000 description 81
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 63
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- LDEKQSIMHVQZJK-CAQYMETFSA-N tenofovir alafenamide Chemical compound O([P@@](=O)(CO[C@H](C)CN1C2=NC=NC(N)=C2N=C1)N[C@@H](C)C(=O)OC(C)C)C1=CC=CC=C1 LDEKQSIMHVQZJK-CAQYMETFSA-N 0.000 description 47
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 42
- 210000002381 plasma Anatomy 0.000 description 41
- 229960004946 tenofovir alafenamide Drugs 0.000 description 38
- 210000004027 cell Anatomy 0.000 description 32
- 239000007787 solid Substances 0.000 description 30
- 239000000243 solution Substances 0.000 description 29
- 229940079593 drug Drugs 0.000 description 25
- 239000003814 drug Substances 0.000 description 25
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 25
- 239000002207 metabolite Substances 0.000 description 24
- LDEKQSIMHVQZJK-AZFZMOAFSA-N propan-2-yl (2s)-2-[[[(2r)-1-(6-aminopurin-9-yl)propan-2-yl]oxymethyl-phenoxyphosphoryl]amino]propanoate Chemical compound O([C@H](C)CN1C2=NC=NC(N)=C2N=C1)CP(=O)(N[C@@H](C)C(=O)OC(C)C)OC1=CC=CC=C1 LDEKQSIMHVQZJK-AZFZMOAFSA-N 0.000 description 23
- 230000002829 reductive effect Effects 0.000 description 23
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- 0 [5*][C@H](CN1C=NC2=C1N=C([12*])N=C2[11*])OC[P@@](=O)(N[C@@H]([7*])C(=O)O[6*])O[6*] Chemical compound [5*][C@H](CN1C=NC2=C1N=C([12*])N=C2[11*])OC[P@@](=O)(N[C@@H]([7*])C(=O)O[6*])O[6*] 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 230000000694 effects Effects 0.000 description 20
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 239000012071 phase Substances 0.000 description 18
- 229960004693 tenofovir disoproxil fumarate Drugs 0.000 description 18
- 238000006243 chemical reaction Methods 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 16
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 16
- 230000000840 anti-viral effect Effects 0.000 description 16
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 125000000217 alkyl group Chemical group 0.000 description 15
- 238000001914 filtration Methods 0.000 description 15
- 125000003118 aryl group Chemical group 0.000 description 14
- 229910052736 halogen Inorganic materials 0.000 description 14
- 150000002367 halogens Chemical class 0.000 description 14
- CAAULPUQFIIOTL-UHFFFAOYSA-N methyl dihydrogen phosphate Chemical class COP(O)(O)=O CAAULPUQFIIOTL-UHFFFAOYSA-N 0.000 description 14
- 229910052757 nitrogen Inorganic materials 0.000 description 14
- 238000012216 screening Methods 0.000 description 14
- 238000003556 assay Methods 0.000 description 13
- 239000002585 base Substances 0.000 description 13
- 229910052760 oxygen Inorganic materials 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 12
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 12
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 11
- 241000282472 Canis lupus familiaris Species 0.000 description 11
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 description 11
- 125000000623 heterocyclic group Chemical group 0.000 description 11
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 11
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 11
- 238000012360 testing method Methods 0.000 description 11
- 125000003710 aryl alkyl group Chemical group 0.000 description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 10
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 9
- 125000003342 alkenyl group Chemical group 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 9
- 210000003563 lymphoid tissue Anatomy 0.000 description 9
- 229930024421 Adenine Natural products 0.000 description 8
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- 229960000643 adenine Drugs 0.000 description 8
- 125000000304 alkynyl group Chemical group 0.000 description 8
- 229940024606 amino acid Drugs 0.000 description 8
- 235000001014 amino acid Nutrition 0.000 description 8
- 230000000259 anti-tumor effect Effects 0.000 description 8
- 238000004587 chromatography analysis Methods 0.000 description 8
- 239000000284 extract Substances 0.000 description 8
- 230000003834 intracellular effect Effects 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 238000000926 separation method Methods 0.000 description 8
- 229960004556 tenofovir Drugs 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- 241000725303 Human immunodeficiency virus Species 0.000 description 7
- 229930010555 Inosine Natural products 0.000 description 7
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 7
- 229940059260 amidate Drugs 0.000 description 7
- 239000013078 crystal Substances 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 238000010828 elution Methods 0.000 description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 7
- 229960003786 inosine Drugs 0.000 description 7
- 230000004060 metabolic process Effects 0.000 description 7
- 239000002002 slurry Substances 0.000 description 7
- 238000004679 31P NMR spectroscopy Methods 0.000 description 6
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 6
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- 150000001413 amino acids Chemical class 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 210000003743 erythrocyte Anatomy 0.000 description 6
- 239000012458 free base Substances 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 125000004437 phosphorous atom Chemical group 0.000 description 6
- 229910052698 phosphorus Inorganic materials 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- DKVBOUDTNWVDEP-NJCHZNEYSA-N teicoplanin aglycone Chemical compound N([C@H](C(N[C@@H](C1=CC(O)=CC(O)=C1C=1C(O)=CC=C2C=1)C(O)=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)OC=1C=C3C=C(C=1O)OC1=CC=C(C=C1Cl)C[C@H](C(=O)N1)NC([C@H](N)C=4C=C(O5)C(O)=CC=4)=O)C(=O)[C@@H]2NC(=O)[C@@H]3NC(=O)[C@@H]1C1=CC5=CC(O)=C1 DKVBOUDTNWVDEP-NJCHZNEYSA-N 0.000 description 6
- 229940075420 xanthine Drugs 0.000 description 6
- RYYIULNRIVUMTQ-UHFFFAOYSA-N 6-chloroguanine Chemical compound NC1=NC(Cl)=C2N=CNC2=N1 RYYIULNRIVUMTQ-UHFFFAOYSA-N 0.000 description 5
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000006260 foam Substances 0.000 description 5
- 125000005843 halogen group Chemical group 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 210000004180 plasmocyte Anatomy 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- SGOIRFVFHAKUTI-ZCFIWIBFSA-N tenofovir (anhydrous) Chemical compound N1=CN=C2N(C[C@@H](C)OCP(O)(O)=O)C=NC2=C1N SGOIRFVFHAKUTI-ZCFIWIBFSA-N 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 239000003039 volatile agent Substances 0.000 description 5
- VAQOTZQDXZDBJK-UHFFFAOYSA-N 2-(6-aminopurin-9-yl)ethanol Chemical compound NC1=NC=NC2=C1N=CN2CCO VAQOTZQDXZDBJK-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- 101001126442 Homo sapiens 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase epsilon-1 Proteins 0.000 description 4
- 101000691618 Homo sapiens Inactive phospholipase C-like protein 1 Proteins 0.000 description 4
- 102100026207 Inactive phospholipase C-like protein 1 Human genes 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 229960003767 alanine Drugs 0.000 description 4
- 235000004279 alanine Nutrition 0.000 description 4
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 description 4
- 125000005018 aryl alkenyl group Chemical group 0.000 description 4
- 125000005015 aryl alkynyl group Chemical group 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 description 4
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 210000001165 lymph node Anatomy 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 239000011777 magnesium Substances 0.000 description 4
- 229910052749 magnesium Inorganic materials 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 4
- 235000019799 monosodium phosphate Nutrition 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- 150000005019 2-aminopurines Chemical class 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 3
- RKOTXQYWCBGZLP-UHFFFAOYSA-N N-[(2,4-difluorophenyl)methyl]-2-ethyl-9-hydroxy-3-methoxy-1,8-dioxospiro[3H-pyrido[1,2-a]pyrazine-4,3'-oxolane]-7-carboxamide Chemical compound CCN1C(OC)C2(CCOC2)N2C=C(C(=O)NCC3=C(F)C=C(F)C=C3)C(=O)C(O)=C2C1=O RKOTXQYWCBGZLP-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- IACQCQDWSIQSRP-ZCFIWIBFSA-N [(2r)-1-(6-aminopurin-9-yl)propan-2-yl]oxymethyl-[hydroxy(phosphonooxy)phosphoryl]oxyphosphinic acid Chemical compound N1=CN=C2N(C[C@@H](C)OCP(O)(=O)OP(O)(=O)OP(O)(O)=O)C=NC2=C1N IACQCQDWSIQSRP-ZCFIWIBFSA-N 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- HAXFWIACAGNFHA-UHFFFAOYSA-N aldrithiol Chemical compound C=1C=CC=NC=1SSC1=CC=CC=N1 HAXFWIACAGNFHA-UHFFFAOYSA-N 0.000 description 3
- 125000005036 alkoxyphenyl group Chemical group 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 125000004104 aryloxy group Chemical group 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000012472 biological sample Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 210000001185 bone marrow Anatomy 0.000 description 3
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 125000004663 dialkyl amino group Chemical group 0.000 description 3
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 3
- 239000001177 diphosphate Substances 0.000 description 3
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 3
- 235000011180 diphosphates Nutrition 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical group CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 description 3
- 239000012456 homogeneous solution Substances 0.000 description 3
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- RTKCPZYOLXPARI-UHFFFAOYSA-N magnesium;2-methylpropan-2-olate Chemical compound [Mg+2].CC(C)(C)[O-].CC(C)(C)[O-] RTKCPZYOLXPARI-UHFFFAOYSA-N 0.000 description 3
- ORPJQHHQRCLVIC-UHFFFAOYSA-N magnesium;propan-2-olate Chemical compound CC(C)O[Mg]OC(C)C ORPJQHHQRCLVIC-UHFFFAOYSA-N 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 229940127073 nucleoside analogue Drugs 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000001103 potassium chloride Substances 0.000 description 3
- 235000011164 potassium chloride Nutrition 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 3
- 239000003656 tris buffered saline Substances 0.000 description 3
- MJZYTEBKXLVLMY-RXMQYKEDSA-N (2r)-1-(6-aminopurin-9-yl)propan-2-ol Chemical compound N1=CN=C2N(C[C@H](O)C)C=NC2=C1N MJZYTEBKXLVLMY-RXMQYKEDSA-N 0.000 description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 2
- 125000006732 (C1-C15) alkyl group Chemical group 0.000 description 2
- MJZYTEBKXLVLMY-UHFFFAOYSA-N 1-(6-aminopurin-9-yl)propan-2-ol Chemical compound N1=CN=C2N(CC(O)C)C=NC2=C1N MJZYTEBKXLVLMY-UHFFFAOYSA-N 0.000 description 2
- SGOIRFVFHAKUTI-UHFFFAOYSA-N 1-(6-aminopurin-9-yl)propan-2-yloxymethylphosphonic acid Chemical compound N1=CN=C2N(CC(C)OCP(O)(O)=O)C=NC2=C1N SGOIRFVFHAKUTI-UHFFFAOYSA-N 0.000 description 2
- QSKPIOLLBIHNAC-UHFFFAOYSA-N 2-chloro-acetaldehyde Chemical compound ClCC=O QSKPIOLLBIHNAC-UHFFFAOYSA-N 0.000 description 2
- PNWOYKVCNDZOLS-UHFFFAOYSA-N 6-amino-5-chloro-1h-pyrimidin-2-one Chemical compound NC=1NC(=O)N=CC=1Cl PNWOYKVCNDZOLS-UHFFFAOYSA-N 0.000 description 2
- NZVORGQIEFTOQZ-UHFFFAOYSA-N 9-[2-(phosphonomethoxy)ethyl]guanine Chemical compound N1C(N)=NC(=O)C2=C1N(CCOCP(O)(O)=O)C=N2 NZVORGQIEFTOQZ-UHFFFAOYSA-N 0.000 description 2
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 description 2
- QAGYKUNXZHXKMR-UHFFFAOYSA-N CPD000469186 Natural products CC1=C(O)C=CC=C1C(=O)NC(C(O)CN1C(CC2CCCCC2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-UHFFFAOYSA-N 0.000 description 2
- 208000031886 HIV Infections Diseases 0.000 description 2
- 208000037357 HIV infectious disease Diseases 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical group [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- SUPKOOSCJHTBAH-UHFFFAOYSA-N adefovir Chemical compound NC1=NC=NC2=C1N=CN2CCOCP(O)(O)=O SUPKOOSCJHTBAH-UHFFFAOYSA-N 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 230000004087 circulation Effects 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- UOEFFQWLRUBDME-UHFFFAOYSA-N diethoxyphosphorylmethyl 4-methylbenzenesulfonate Chemical compound CCOP(=O)(OCC)COS(=O)(=O)C1=CC=C(C)C=C1 UOEFFQWLRUBDME-UHFFFAOYSA-N 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- KXSBWGSJFSEIED-YFKPBYRVSA-N ethyl (2s)-2-aminobutanoate Chemical compound CCOC(=O)[C@@H](N)CC KXSBWGSJFSEIED-YFKPBYRVSA-N 0.000 description 2
- 210000003608 fece Anatomy 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 208000006454 hepatitis Diseases 0.000 description 2
- 231100000283 hepatitis Toxicity 0.000 description 2
- 125000004404 heteroalkyl group Chemical group 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 150000004712 monophosphates Chemical class 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- QAGYKUNXZHXKMR-HKWSIXNMSA-N nelfinavir Chemical compound CC1=C(O)C=CC=C1C(=O)N[C@H]([C@H](O)CN1[C@@H](C[C@@H]2CCCC[C@@H]2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-HKWSIXNMSA-N 0.000 description 2
- 229960000884 nelfinavir Drugs 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- 125000003835 nucleoside group Chemical group 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 125000004043 oxo group Chemical group O=* 0.000 description 2
- 238000012856 packing Methods 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 239000008057 potassium phosphate buffer Substances 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- QDQVXVRZVCTVHE-YFKPBYRVSA-N propan-2-yl (2s)-2-aminopropanoate Chemical compound CC(C)OC(=O)[C@H](C)N QDQVXVRZVCTVHE-YFKPBYRVSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000007423 screening assay Methods 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000011282 treatment Methods 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- MFGWMAAZYZSWMY-UHFFFAOYSA-N (2-naphthyl)methanol Chemical compound C1=CC=CC2=CC(CO)=CC=C21 MFGWMAAZYZSWMY-UHFFFAOYSA-N 0.000 description 1
- LVOASXNJWPROPP-UHFFFAOYSA-N (4-methylphenyl)sulfonyloxymethylphosphonic acid Chemical compound CC1=CC=C(S(=O)(=O)OCP(O)(O)=O)C=C1 LVOASXNJWPROPP-UHFFFAOYSA-N 0.000 description 1
- RUOJZAUFBMNUDX-GSVOUGTGSA-N (4r)-4-methyl-1,3-dioxolan-2-one Chemical compound C[C@@H]1COC(=O)O1 RUOJZAUFBMNUDX-GSVOUGTGSA-N 0.000 description 1
- 125000006559 (C1-C3) alkylamino group Chemical group 0.000 description 1
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 1
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 1
- MEJAFWXKUKMUIR-FHPNUNMMSA-N (e)-but-2-enedioic acid;propan-2-yl (2s)-2-[[[(2r)-1-(6-aminopurin-9-yl)propan-2-yl]oxymethyl-phenoxyphosphoryl]amino]propanoate Chemical compound OC(=O)\C=C\C(O)=O.O([P@@](=O)(CO[C@H](C)CN1C2=NC=NC(N)=C2N=C1)N[C@@H](C)C(=O)OC(C)C)C1=CC=CC=C1 MEJAFWXKUKMUIR-FHPNUNMMSA-N 0.000 description 1
- AZUYLZMQTIKGSC-UHFFFAOYSA-N 1-[6-[4-(5-chloro-6-methyl-1H-indazol-4-yl)-5-methyl-3-(1-methylindazol-5-yl)pyrazol-1-yl]-2-azaspiro[3.3]heptan-2-yl]prop-2-en-1-one Chemical compound ClC=1C(=C2C=NNC2=CC=1C)C=1C(=NN(C=1C)C1CC2(CN(C2)C(C=C)=O)C1)C=1C=C2C=NN(C2=CC=1)C AZUYLZMQTIKGSC-UHFFFAOYSA-N 0.000 description 1
- MWBWWFOAEOYUST-UHFFFAOYSA-N 2-aminopurine Chemical compound NC1=NC=C2N=CNC2=N1 MWBWWFOAEOYUST-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000004398 2-methyl-2-butyl group Chemical group CC(C)(CC)* 0.000 description 1
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 1
- 125000004922 2-methyl-3-pentyl group Chemical group CC(C)C(CC)* 0.000 description 1
- CFIBTBBTJWHPQV-UHFFFAOYSA-N 2-methyl-n-(6-oxo-3,7-dihydropurin-2-yl)propanamide Chemical compound N1C(NC(=O)C(C)C)=NC(=O)C2=C1N=CN2 CFIBTBBTJWHPQV-UHFFFAOYSA-N 0.000 description 1
- 125000004917 3-methyl-2-butyl group Chemical group CC(C(C)*)C 0.000 description 1
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 1
- 125000004921 3-methyl-3-pentyl group Chemical group CC(CC)(CC)* 0.000 description 1
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- RZEXJHGIEXQMTI-UHFFFAOYSA-N 5-(methoxymethyl)-1h-pyrimidine-2,4-dione Chemical compound COCC1=CNC(=O)NC1=O RZEXJHGIEXQMTI-UHFFFAOYSA-N 0.000 description 1
- LMNPKIOZMGYQIU-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-pyrimidine-2,4-dione Chemical compound FC(F)(F)C1=CNC(=O)NC1=O LMNPKIOZMGYQIU-UHFFFAOYSA-N 0.000 description 1
- BLXGZIDBSXVMLU-OWOJBTEDSA-N 5-[(e)-2-bromoethenyl]-1h-pyrimidine-2,4-dione Chemical compound Br\C=C\C1=CNC(=O)NC1=O BLXGZIDBSXVMLU-OWOJBTEDSA-N 0.000 description 1
- SVXNJCYYMRMXNM-UHFFFAOYSA-N 5-amino-2h-1,2,4-triazin-3-one Chemical compound NC=1C=NNC(=O)N=1 SVXNJCYYMRMXNM-UHFFFAOYSA-N 0.000 description 1
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical compound BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 1
- ZFTBZKVVGZNMJR-UHFFFAOYSA-N 5-chlorouracil Chemical compound ClC1=CNC(=O)NC1=O ZFTBZKVVGZNMJR-UHFFFAOYSA-N 0.000 description 1
- KSNXJLQDQOIRIP-UHFFFAOYSA-N 5-iodouracil Chemical compound IC1=CNC(=O)NC1=O KSNXJLQDQOIRIP-UHFFFAOYSA-N 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- UJBCLAXPPIDQEE-UHFFFAOYSA-N 5-prop-1-ynyl-1h-pyrimidine-2,4-dione Chemical compound CC#CC1=CNC(=O)NC1=O UJBCLAXPPIDQEE-UHFFFAOYSA-N 0.000 description 1
- QQJXZVKXNSFHRI-UHFFFAOYSA-N 6-Benzamidopurine Chemical compound N=1C=NC=2N=CNC=2C=1NC(=O)C1=CC=CC=C1 QQJXZVKXNSFHRI-UHFFFAOYSA-N 0.000 description 1
- QFVKLKDEXOWFSL-UHFFFAOYSA-N 6-amino-5-bromo-1h-pyrimidin-2-one Chemical compound NC=1NC(=O)N=CC=1Br QFVKLKDEXOWFSL-UHFFFAOYSA-N 0.000 description 1
- UFVWJVAMULFOMC-UHFFFAOYSA-N 6-amino-5-iodo-1h-pyrimidin-2-one Chemical compound NC=1NC(=O)N=CC=1I UFVWJVAMULFOMC-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- ALEIUAGMWPBBMS-UHFFFAOYSA-N BCOCP(=O)(O)O Chemical compound BCOCP(=O)(O)O ALEIUAGMWPBBMS-UHFFFAOYSA-N 0.000 description 1
- POUYUFVYLZGOJZ-FBZFNTLTSA-N C.C(=NC1CCCCC1)=NC1CCCCC1.CC#N.CC(C)OC(=O)[C@H](C)N.CC(C)OC(=O)[C@H](C)NP(=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1.CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1.CCN(CC)CC.CCN(CC)CC.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.ClCCl.II.O=C(O)/C=C/C(=O)O.O=S(Cl)Cl.OC1=CC=CC=C1 Chemical compound C.C(=NC1CCCCC1)=NC1CCCCC1.CC#N.CC(C)OC(=O)[C@H](C)N.CC(C)OC(=O)[C@H](C)NP(=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1.CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1.CCN(CC)CC.CCN(CC)CC.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.ClCCl.II.O=C(O)/C=C/C(=O)O.O=S(Cl)Cl.OC1=CC=CC=C1 POUYUFVYLZGOJZ-FBZFNTLTSA-N 0.000 description 1
- YYCROHVULSQKER-LVGFKTSTSA-N C.CC#N.CCN(CC)CC.CC[C@H](N)C(=O)OC(C)C.CC[C@H](NP(=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1)C(=O)OC(C)C.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.ClCCl.II.O=S(Cl)Cl Chemical compound C.CC#N.CCN(CC)CC.CC[C@H](N)C(=O)OC(C)C.CC[C@H](NP(=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1)C(=O)OC(C)C.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.ClCCl.II.O=S(Cl)Cl YYCROHVULSQKER-LVGFKTSTSA-N 0.000 description 1
- GWQQVFSUUYQOPK-PYCYZAEZSA-N CC#N.CC(C)OC(=O)[C@H](C)N.CCN(C(C)C)C(C)C.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.C[Si](C)(C)OC1=CC=CC=C1.ClCCl.II.O=C(O)/C=C/C(=O)O.O=COO.O=S(Cl)Cl.O=S(Cl)Cl.O=S1(=O)CCCC1.O=S1(=O)CCCC1.[KH] Chemical compound CC#N.CC(C)OC(=O)[C@H](C)N.CCN(C(C)C)C(C)C.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)OC1=CC=CC=C1.C[Si](C)(C)OC1=CC=CC=C1.ClCCl.II.O=C(O)/C=C/C(=O)O.O=COO.O=S(Cl)Cl.O=S(Cl)Cl.O=S1(=O)CCCC1.O=S1(=O)CCCC1.[KH] GWQQVFSUUYQOPK-PYCYZAEZSA-N 0.000 description 1
- MYCQGKYTLHVPKA-UHFFFAOYSA-N CC(C)OC(=O)C(C)NP(=O)(COC([RaH])CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1 Chemical compound CC(C)OC(=O)C(C)NP(=O)(COC([RaH])CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1 MYCQGKYTLHVPKA-UHFFFAOYSA-N 0.000 description 1
- KWEHGGRVJNHWBA-FHPNUNMMSA-N CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1.N/C(C(=O)O)=C(/N)C(=O)O Chemical compound CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1.N/C(C(=O)O)=C(/N)C(=O)O KWEHGGRVJNHWBA-FHPNUNMMSA-N 0.000 description 1
- DSTNEWBVEQPYJY-QLYSAPKRSA-O CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2[NH2+]OC(=O)/C=C/C(=O)O)OC1=CC=CC=C1 Chemical compound CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2[NH2+]OC(=O)/C=C/C(=O)O)OC1=CC=CC=C1 DSTNEWBVEQPYJY-QLYSAPKRSA-O 0.000 description 1
- ADOLFHFKWDQFBI-UHFFFAOYSA-N CCOP(=O)(COCCN1C=NC2=C1N=CN=C2N)OCC.CCOP(=O)(COS(=O)(=O)C1=CC=C(C)C=C1)OCC.C[Si](C)(C)Br.NC1=NC=NC2=C1N=CN2CCO.NC1=NC=NC2=C1N=CN2CCOCP(=O)(O)O Chemical compound CCOP(=O)(COCCN1C=NC2=C1N=CN=C2N)OCC.CCOP(=O)(COS(=O)(=O)C1=CC=C(C)C=C1)OCC.C[Si](C)(C)Br.NC1=NC=NC2=C1N=CN2CCO.NC1=NC=NC2=C1N=CN2CCOCP(=O)(O)O ADOLFHFKWDQFBI-UHFFFAOYSA-N 0.000 description 1
- XOJLOFMHMRWWNA-QOQANMAQSA-N CCOP(=O)(COS(=O)(=O)C1=CC=C(C)C=C1)OCC.C[Si](C)(C)Br.[H][C@@](C)(CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O.[H][C@@](C)(CN1C=NC2=C1N=CN=C2N)OCP(=O)(OCC)OCC.[H][C@](C)(O)CN1C=NC2=C1N=CN=C2N Chemical compound CCOP(=O)(COS(=O)(=O)C1=CC=C(C)C=C1)OCC.C[Si](C)(C)Br.[H][C@@](C)(CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O.[H][C@@](C)(CN1C=NC2=C1N=CN=C2N)OCP(=O)(OCC)OCC.[H][C@](C)(O)CN1C=NC2=C1N=CN=C2N XOJLOFMHMRWWNA-QOQANMAQSA-N 0.000 description 1
- WRXCXOUDSPTXNX-UHFFFAOYSA-N CN1C=NC2=C1N=CN=C2N Chemical compound CN1C=NC2=C1N=CN=C2N WRXCXOUDSPTXNX-UHFFFAOYSA-N 0.000 description 1
- YDBCQGNEXYFIHD-UHFFFAOYSA-N C[n]1nc(C(N)=O)nc1 Chemical compound C[n]1nc(C(N)=O)nc1 YDBCQGNEXYFIHD-UHFFFAOYSA-N 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 229940126656 GS-4224 Drugs 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- 206010062767 Hypophysitis Diseases 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- SHZGCJCMOBCMKK-JFNONXLTSA-N L-rhamnopyranose Chemical compound C[C@@H]1OC(O)[C@H](O)[C@H](O)[C@H]1O SHZGCJCMOBCMKK-JFNONXLTSA-N 0.000 description 1
- PNNNRSAQSRJVSB-UHFFFAOYSA-N L-rhamnose Natural products CC(O)C(O)C(O)C(O)C=O PNNNRSAQSRJVSB-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- UHJKWKRSKOYNIS-UHFFFAOYSA-M NC1=NC=NC2=C1N=CN2.NC1=NC=NC2=C1N=CN2CCO.O=C1OCCO1.O[Na] Chemical compound NC1=NC=NC2=C1N=CN2.NC1=NC=NC2=C1N=CN2CCO.O=C1OCCO1.O[Na] UHJKWKRSKOYNIS-UHFFFAOYSA-M 0.000 description 1
- QQMHNJMWTBMXSW-DIQGLWQBSA-M NC1=NC=NC2=C1N=CN2.O[Na].[H][C@@]1(C)COC(=O)O1.[H][C@](C)(O)CN1C=NC2=C1N=CN=C2N Chemical compound NC1=NC=NC2=C1N=CN2.O[Na].[H][C@@]1(C)COC(=O)O1.[H][C@](C)(O)CN1C=NC2=C1N=CN=C2N QQMHNJMWTBMXSW-DIQGLWQBSA-M 0.000 description 1
- BVMWIXWOIGJRGE-UHFFFAOYSA-N NP(O)=O Chemical class NP(O)=O BVMWIXWOIGJRGE-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- GFPMTTHVYNIDBY-UHFFFAOYSA-N S(=O)(=O)(O)C1=CC=C(C)C=C1.C1(=CC=C(C=C1)S(=O)(=O)OCP(O)(O)=O)C Chemical compound S(=O)(=O)(O)C1=CC=C(C)C=C1.C1(=CC=C(C=C1)S(=O)(=O)OCP(O)(O)=O)C GFPMTTHVYNIDBY-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 210000000579 abdominal fat Anatomy 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 229960001997 adefovir Drugs 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 125000006323 alkenyl amino group Chemical group 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000005257 alkyl acyl group Chemical group 0.000 description 1
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 1
- 125000003275 alpha amino acid group Chemical group 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000001195 anabolic effect Effects 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000000798 anti-retroviral effect Effects 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 125000005127 aryl alkoxy alkyl group Chemical group 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940090047 auto-injector Drugs 0.000 description 1
- 125000005335 azido alkyl group Chemical group 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 230000000981 bystander Effects 0.000 description 1
- SZTWKXQGCGRUHR-UHFFFAOYSA-N c1nc2ncn3cc[nH]c3c2n1 Chemical class c1nc2ncn3cc[nH]c3c2n1 SZTWKXQGCGRUHR-UHFFFAOYSA-N 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000001925 catabolic effect Effects 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 210000004970 cd4 cell Anatomy 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000012824 chemical production Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000004985 dialkyl amino alkyl group Chemical group 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-N diphosphoric acid Chemical class OP(O)(=O)OP(O)(O)=O XPPKVPWEQAFLFU-UHFFFAOYSA-N 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- JOZGNYDSEBIJDH-UHFFFAOYSA-N eniluracil Chemical compound O=C1NC=C(C#C)C(=O)N1 JOZGNYDSEBIJDH-UHFFFAOYSA-N 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- JCXLZWMDXJFOOI-WCCKRBBISA-N ethyl (2s)-2-aminopropanoate;hydrochloride Chemical compound Cl.CCOC(=O)[C@H](C)N JCXLZWMDXJFOOI-WCCKRBBISA-N 0.000 description 1
- NTNZTEQNFHNYBC-UHFFFAOYSA-N ethyl 2-aminoacetate Chemical group CCOC(=O)CN NTNZTEQNFHNYBC-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000013213 extrapolation Methods 0.000 description 1
- 239000012997 ficoll-paque Substances 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 210000003405 ileum Anatomy 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000012606 in vitro cell culture Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 210000001630 jejunum Anatomy 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000005567 liquid scintillation counting Methods 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 230000031852 maintenance of location in cell Effects 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- XBDUZBHKKUFFRH-UHFFFAOYSA-N n-(2-oxo-1h-pyrimidin-6-yl)benzamide Chemical compound OC1=NC=CC(NC(=O)C=2C=CC=CC=2)=N1 XBDUZBHKKUFFRH-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003589 nefrotoxic effect Effects 0.000 description 1
- 231100000381 nephrotoxic Toxicity 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000004971 nitroalkyl group Chemical group 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 210000000578 peripheral nerve Anatomy 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 210000003635 pituitary gland Anatomy 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000006412 propinylene group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 210000005084 renal tissue Anatomy 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 238000011894 semi-preparative HPLC Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000012064 sodium phosphate buffer Substances 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 238000007070 tosylation reaction Methods 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- OJAJJFGMKAZGRZ-UHFFFAOYSA-N trimethyl(phenoxy)silane Chemical compound C[Si](C)(C)OC1=CC=CC=C1 OJAJJFGMKAZGRZ-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000005065 undecenyl group Chemical group C(=CCCCCCCCCC)* 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/70—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving virus or bacteriophage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
- C12Q1/18—Testing for antimicrobial activity of a material
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5011—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing antineoplastic activity
Definitions
- Prodrugs of methoxyphosphonate nucleotide analogues intended for antiviral or antitumor therapy traditionally have been selected for their systemic effect.
- such prodrugs have been selected for enhanced bioavailability, i.e., ability to be absorbed from the gastrointestinal tract and converted rapidly to parent drug to ensure that the parent drug is available to all tissues.
- the objective of this invention is, among other advantages, to produce less toxicity to bystander tissues and greater potency of the parental drug in tissues which are the targets of therapy with the parent methoxyphosphonate nucleotide analogue.
- a screening method for identifying a methoxyphosphonate nucleotide analogue prodrug conferring enhanced activity in a target tissue comprising:
- the target tissue are sites where HIV is actively replicated and/or which serve as an HIV reservoir, and the non-target tissue is an intact animal.
- selecting lymphoid tissue as the target tissue for the practice of this method for HIV led to identification of prodrugs that enhance the delivery of active drug to such tissues.
- a preferred compound of this invention which has been identified by this method has the structure (1),
- a preferred compound of this invention has the structure (2)
- the diastereomers of structure (3) are designated the (S) isomers at the phosphorus chiral center.
- Preferred embodiments of this invention are the diastereomerically enriched compounds having the structure (5a)
- a preferred embodiment of this invention is the compound of structure (6), 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine, also designated herein GS-7340.
- Another preferred embodiment of this invention is the fumarate salt of structure (5) (structure (7)), 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine fumarate (1:1), also designated herein GS-7340-2
- compositions containing pharmaceutically acceptable excipients are used in effective doses in the therapy or prophylaxis of viral (particularly HIV or hepadnaviral) infections.
- a method for the facile manufacture of 9-[2-(phosphonomethoxy)propyl]adenine (hereinafter “PMPA” or 9-[2-(phosphonomethoxy)ethyl]adenine (hereinafter “PMEA”) using magnesium alkoxide comprises combining 9-(2-hydroxypropyl)adenine or 9-(2-hydroxyethyl)adenine, protected p-toluenesulfonyloxymethylphosphonate and magnesium alkoxide, and recovering PMPA or PMEA, respectively.
- FIG. 1 shows HPLC/C-14 traces of PBMC extracts from human blood incubated for 1 h at 37° C. with TDF, GS-7340 and PMPA.
- FIG. 2 shows PMPA and Prodrug concentration in plasma and PBMCs following oral administration of GS 7340-2 to Dogs at 10 mg-eq/kg.
- FIG. 3 depicts Tenofovir exposure in PBMCs and plasma upon administration of 10 mg-eq/kg in dogs.
- the methoxyphosphonate nucleotide analogue parent drugs for use in this screening method are compounds having the structure A-OCH 2 P(O)(OH) 2 wherein A is the residue of a nucleoside analogue. These compounds are known per se and are not part of this invention. More particularly, the parent compounds comprise a heterocyclic base B and an aglycon E, in general having the structure wherein the group B is defined below and group E is defined above. Examples are described in U.S. Pat. Nos.
- the prodrugs for use in the screening method of this invention are covalently modified analogues of the parent methoxyphosphonate nucleotide analogues described in the preceding paragraph.
- the phosphorus atom of the parent drug is the preferred site for prodrug modification, but other sites are found on the heterocyclic base B or the aglycon E.
- Many such prodrugs are already known. Primarily, they are esters or amidates of the phosphorus atom, but also include substitutions on the base and aglycon. None of these modifications per se is part of this invention and none are to be considered limiting on the scope of the invention herein.
- the phosphorus atom of the methoxyphosphonate nucleotide analogues contains two valences for covalent modification such as amidation or esterification (unless one phosphoryl hydroxyl is esterified to an aglycon E hydroxyl substituent, whereupon only one phosphorus valence is free for substitution).
- the esters typically are aryloxy.
- the amidates ordinarily are naturally occurring monoamino acids having free carboxyl group(s) esterified with an alkyl or aryl group, usually phenyl, cycloalkyl, or t-, n- or s-alkyl groups.
- Suitable prodrugs for use in the screening method of this invention are disclosed for example in U.S. Pat. No.
- any prodrug which is potentially believed to be capable of being converted in vivo within target tissue cells to the free methoxyphosphonate nucleotide analogue parent drug, e.g., whether by hydrolysis, oxidation, or other covalent transformation resulting from exposure to biological tissues, is suitable for use in the method of this invention.
- Such prodrugs may not be known at this time but are identified in the future and thus become suitable candidates available for testing in the method of this invention. Since the prodrugs are simply candidates for screening in the methods their structures are not relevant to practicing or enabling the screening method, although of course their structures ultimately are dispositive of whether or not a prodrug will be shown to be selective in the assay.
- pro-moieties bound to the parent drug may be the same or different.
- each prodrug to be used in the screening assay will differ structurally from the other prodrugs to be tested. Distinct, i.e. structurally different, prodrugs generally are selected on the basis of either their stereochemistry or their covalent structure, or these features are varied in combination.
- Each prodrug tested desirably is structurally and stereochemically substantially pure, else the output of the screening assay will be less useful. It is of course within the scope of this invention to test only a single prodrug in an individual embodiment of the method of this invention, although typically then one would compare the results with prior studies with other prodrugs.
- Chiral sites are at the phosphorus atom and are also found in its substituents.
- amino acid used in preparing amidates may be D or L forms, and the phosphonate esters or the amino acid esters can contain chiral centers as well.
- Chiral sites also are found on the nucleoside analogue portion of the molecules, but these typically are already dictated by the stereochemistry of the parent drug and will not be varied as part of the screen.
- the R isomer of PMPA is preferred as it is more active than the corresponding S isomer.
- these diasteromers or enantiomers will be chirally enriched if not pure at each site so that the results of the screen will be more meaningful.
- distinctiveness of stereoisomers is conferred by enriching or purifying the stereoisomer (typically this will be a diastereomer rather than an enantiomer in the case of most methoxyphosphonate nucleotide analogues) free of other stereoisomers at the chiral center in question, so that each test compound is substantially homogeneous.
- substantially homogeneous or chirally enriched we mean that the desired stereoisomer constitutes greater than about 60% by weight of the compound, ordinarily greater than about 80% and preferably greater than about 95%.
- the remaining steps of the screening method of this invention are used to identify a prodrug possessing the required selectivity for the target tissue.
- the prodrugs are labeled with a detectable group, e.g. radiolabeled, in order to facilitate detection later in tissues or cells.
- a label is not required since other suitable assays for the prodrug or its metabolites (including the parent drug) can also be employed. These assays could include mass spectrometry, HPLC, bioassays or immunoassays for instance.
- the assay may detect the prodrug and any one or more of its metabolites, but preferably the assay is conducted to detect only the generation of the parent drug. This is based on the assumption (which may not be warranted in all cases) that the degree and rate of conversion of prodrug to antivirally active parent diphosphate is the same across all tissues tested. Otherwise, one can test for the diphosphate.
- the target tissue preferably will be lymphoid tissue when screening for prodrugs useful in the treatment of HIV infection.
- Lymphoid tissue will be known to the artisan and includes CD4 cells, lymphocytes, lymph nodes, macrophages and macrophage-like cells including monocytes such as peripheral blood monocytic cells (PBMCs) and glial cells.
- Lymphoid tissue also includes non-lymphoid tissues that are enriched in lymphoid tissues or cells, e.g. lung, skin and spleen.
- Other targets for other antiviral drugs of course will be the primary sites of replication or latency for the particular virus concerned, e.g., liver for hepatitis and peripheral nerves for HSV.
- target tissues for tumors will in fact be the tumors themselves. These tissues are all well-known to the artisan and would not require undue experimentation to select. When screening for antiviral compounds, target tissue can be infected by the virus.
- Non-target tissues or cells also are screened as part of the method herein. Any number or identity of such tissues or cells can be employed in this regard. In general, tissues for which the parent drug is expected to be toxic will be used as non-target tissues. The selection of a non-target tissue is entirely dependent upon the nature of the prodrug and the activity of the parent. For example, non-hepatic tissues would be selected for prodrugs against hepatitis, and untransformed cells of the same tissue as the tumor will suffice for the antitumor-selective prodrug screen.
- target tissues to be evaluated in the method of this invention generally do not include the small intestines or, if the intestines are included, then the tissues also include additional tissues other than the small intestines.
- the target and non-target tissues used in the screening method of this invention typically will be in an intact living animal.
- Prodrugs containing esters are more desirably tested in dogs, monkeys or other animals than rodents; mice and rat plasma contains high circulating levels of esterases that may produce a misleading result if the desired therapeutic subject is a human or higher mammal.
- tissue shall not be construed to require organized cellular structures, or the structures of tissues as they may be found in nature, although such would be preferred. Rather, the term “tissue” shall be construed to be synonymous with cells of a particular source, origin or differentiation stage.
- the target and non-target tissue may in fact be the same tissue, but the tissues will be in different biological status.
- the method herein could be used to select for prodrugs that confer activity in virally-infected tissue (target tissue) but which remain substantially inactive in virally-uninfected cells (corresponding non-target tissue).
- target tissue i.e., prodrugs metabolized to antivirally active forms incidental to viral infection but which remain substantially unmetabolized in uninfected cells.
- prodrugs could be screened in transformed cells and the untransformed counterpart tissue. This would be particularly useful in comparative testing to select prodrugs for the treatment of hematological malignancies, e.g. leukemias.
- tissue selective prodrugs are thought to be selectively taken up by target cells and/or selectively metabolized within the cell, as compared to other tissues or cells.
- the unique advantage of the methoxyphosphonate prodrugs herein is that their metabolism to the dianion at physiological pH ensures that they will be unable to diffuse back out of the cell. They therefore remain effective for lengthy periods of time and are maintained at elevated intracellular concentrations, thereby exhibiting increased potency.
- the mechanisms for enhanced activity in the target tissue are believed to include enhanced uptake by the target cells, enhanced intracellular retention, or both mechanisms working together.
- the manner in which selectivity or enhanced delivery occurs in the target tissue is not important. It also is not important that all of the metabolic conversion of the prodrug to the parent compound occurs within the target tissue. Only the final drug activity-conferring conversion need occur in the target tissue; metabolism in other tissues may provide intermediates finally converted to antiviral forms in the target tissue.
- the degree of selectivity or enhanced delivery that is desired will vary with the parent compound and the manner in which it is measured (% dose distribution or parent drug concentration). In general, if the parent drug already possess a generous therapeutic window, a low degree of selectivity may be sufficient for the desired prodrug. On the other hand, toxic compounds may require more extensive screening to identify selective prodrugs. The relative expense of the method of this invention can be reduced by screening only in the target tissue and tissues against which the parent compound is known to be relatively toxic, e.g. for PMEA, which is nephrotoxic at higher doses, the primary focus will be on kidney and lymphoid tissues.
- the step of determining the relative antiviral activity of a prodrug in the selected tissues ordinarily is accomplished by assaying target and non-target tissues for the relative presence or activity of a metabolite of the prodrug, which metabolite is known to have, or is converted to, a metabolite having antiviral or antitumor activity.
- a metabolite of the prodrug which metabolite is known to have, or is converted to, a metabolite having antiviral or antitumor activity.
- the active metabolite is the diphosphate of the phosphonate parent compounds.
- Metabolites of the prodrug can be anabolic metabolites, catabolic metabolites, or the product of anabolism and catabolism together.
- the manner in which the metabolite is produced is not important in the practice of the method of this invention.
- the method of this invention is not limited to assaying a metabolite which per se possesses antiviral or antitumor activity. Instead, one can assay inactive precursors of the active metabolites.
- Precursors of the antivirally active diphosphate metabolite include the monophosphate of the parent drug, monophosphates of other metabolites of the parent drug (e.g., an intermediate modification of a substituent on the heterocyclic base), the parent itself and metabolites generated by the cell in converting the prodrug to the parent prior to phosphorylation.
- the precursor structures may vary considerably as they are the result of cellular metabolism. However, this information is already known or could be readily determined by one skilled in the art.
- step (d) of the method herein calls for determining the activity, activity can be either measured directly or extrapolated. It does not mean that the method herein is limited to only assaying intermediates that are active per se.
- Step (d) only requires assessment of the activity conferred by the prodrug as it interacts with the tissue concerned, and this may be based on extrapolation or other indirect measurement.
- Step (d) of the method of this invention calls for determining the “relative” activity of the prodrug. It will be understood that this does not require that each and every assay or series of assays necessarily must also contain runs with the selected non-target tissue. On the contrary, it is within the scope of this invention to employ historical controls of the non-target tissue or tissues, or algorithms representing results to be expected from such non-target tissues, in order to provide the benchmark non-target activity.
- step (d) The results obtained in step (d) are then used optimally to select or identify a prodrug which produces greater antiviral activity in the target tissue than in the non-target tissue. It is this prodrug that is selected for further development.
- prodrug candidates can be undertaken before the practice of the method of this invention.
- the prodrug will need to be capable of passing largely unmetabolized through the gastrointestinal tract, it will need to be substantially stable in blood, and it should be able to permeate cells at least to some degree. In most cases it also will need to complete a first pass of the hepatic circulation without substantial metabolism.
- Such prestudies are optional, and are well-known to those skilled in the art.
- antiviral activity is applicable to antitumor prodrugs of methoxyphosphonate nucleotide analogues as well.
- these include, for example, prodrugs of PMEG, the guanyl analogue of PMEA.
- cytotoxic phosphonates such as PMEG are worthwhile candidates to pursue as their cytotoxicity in fact confers their antitumor activity.
- a compound identified by this novel screening method then can be entered into a traditional preclinical or clinical program to confirm that the desired objectives have been met.
- a prodrug is considered to be selective if the activity or concentration of parent drug in the target tissue (% dose distribution) is greater than 2 ⁇ , and preferably 5 ⁇ , that of the parent compound in non-target tissue.
- a prodrug candidate can be compared against a benchmark prodrug. In this case, selectivity is relative rather than absolute. Selective prodrugs will be those resulting in greater than about 10 ⁇ concentration or activity in the target tissue as compared with the prototype, although the degree of selectivity is a matter of discretion.
- this method comprises reacting 9-(2-hydroxypropyl)adenine (HPA) or 9-(2-hydroxyethyl)adenine (HEA) with a magnesium alkoxide, thereafter adding the protected aglycon synthon p-toluene-sulfonyloxymethylphosphonate (tosylate) to the reaction mixture, and recovering PMPA or PMEA, respectively.
- HPA is the enriched or isolated R enantiomer. If a chiral HPA mixture is used, R-PMPA can be isolated from the chiral PMPA mixture after the synthesis is completed.
- the tosylate is protected by lower alkyl groups, but other suitable groups will be apparent to the artisan. It may be convenient to employ the tosylate presubstituted with the prodrug phosphonate substituents which are capable of acting as protecting groups in the tosylation reaction, thereby allowing one to bypass the deprotection step and directly recover prodrug or an intermediate therefore.
- the alkyl group of the magnesium alkoxide is not critical and can be any C 1 -C 6 branched or normal alkyl, but is preferably t-butyl (for PMPA) or isopropyl (for PMEA).
- the reaction conditions also are not critical, but preferably comprise heating the reaction mixture at about 70-75° C. with stirring or other moderate agitation.
- the product is deprotected (usually with bromotrimethylsilane where the tosylate protecting group is alkyl), and the product then recovered by crystallization or other conventional method as will be apparent to the artisan.
- heterocyclic base B is selected from the structures
- B also includes both protected and unprotected heterocyclic bases, particularly purine and pyrimidine bases.
- Protecting groups for exocyclic amines and other labile groups are known (Greene et al. “Protective Groups in Organic Synthesis”) and include N-benzoyl, isobutyryl, 4,4′-dimethoxytrityl (DMT) and the like.
- DMT 4,4′-dimethoxytrityl
- the selection of protecting group will be apparent to the ordinary artisan and will depend upon the nature of the labile group and the chemistry which the protecting group is expected to encounter, e.g. acidic, basic, oxidative, reductive or other conditions.
- Exemplary protected species are N 4 -benzoylcytosine, N 6 -benzoyladenine, N 2 -isobutyrylguanine and the like.
- Protected bases have the formulas Xa.1, XIa.1, XIb.1, XIIa.1 or XIIIa.1
- R 39 is present at R 22A or R 23A , both R 39 groups on the same base will generally be the same.
- Exemplary R 36 are phenyl, phenyl substituted with one of the foregoing R 36 aryl substituents, —C 10 H 15 (where C 10 H 15 is 2-adamantoyl), —CH 2 —C 6 H 5 , —C 6 H 5 , —CH(CH 3 ) 2 , —CH 2 CH 3 , methyl, butyl, t-butyl, heptanyl, nonanyl, undecanyl, or undecenyl.
- Specific bases include hypoxanthine, guanine, adenine, cytosine, inosine, thymine, uracil, xanthine, 8-aza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 7-deaza-8-aza derivatives of adenine, guanine, 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 1-deaza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 7-deaza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine
- B is a 9-purinyl residue selected from guanyl, 3-deazaguanyl, 1-deazaguanyl, 8-azaguanyl, 7-deazaguanyl, adenyl, 3-deazaadenyl, 1-dezazadenyl, 8-azaadenyl, 7-deazaadenyl, 2,6-diaminopurinyl, 2-aminopurinyl, 6-chloro-2-aminopurinyl and 6-thio-2-aminopurinyl, or a B′ is a 1-pyrimidinyl residue selected from cytosinyl, 5-halocytosinyl, and 5-(C 1 -C 3 -alkyl)cytosinyl.
- Preferred B groups have the formula
- E groups represent the aglycons employed in the methoxyphosphonate nucleotide analogues.
- the E group is —CH(CH 3 )CH 2 — or —CH 2 CH 2 —.
- the side groups at chiral centers in the aglycon be substantially solely in the (R) configuration (except for hydroxymethyl, which is the enriched (S) enantiomer).
- R 1 is an in vivo hydrolyzable oxyester having the structure —OR 35 or —OR 6 wherein R 35 is defined in column 64, line 49 of U.S. Pat. No. 5,798,340, herein incorporated by reference, and R 6 is defined above.
- R 1 is aryloxy, ordinarily unsubstituted or para-substituted (as defined in R 6 ) phenoxy.
- R 2 is an amino acid residue, optionally provided that any carboxy group linked by less than about 5 atoms to the amidate N is esterified.
- R 2 typically has the structure
- the invention includes metabolites in which the phenoxy and isopropyl esters have been hydrolyzed to —OH.
- the de-esterified enriched phosphonoamidate metabolites of compounds (5a), 5(b) and (6) are included within the scope of this invention.
- Aryl and “0” or “N” substitution are defined in column 16, lines 42-58, of U.S. Pat. No. 5,798,340.
- amino acids are in the natural or l amino acids. Suitable specific examples are set forth in U.S. Pat. No. 5,798,340, for instance Table 4 and col. 8-10 therein.
- the prodrug compounds of this invention are provided in the form of free base or the various salts enumerated in U.S. Pat. No. 5,798,340, and are formulated with pharmaceutically acceptable excipients or solvating diluents for use as pharmaceutical products also as set forth in U.S. Pat. No. 5,798,340.
- These prodrugs have the antiviral and utilities already established for the parent drugs (see U.S. Pat. No. 5,798,340 and other citations relating to the methoxyphosphonate nucleotide analogues). It will be understood that the diastereomer of structure (4) at least is useful as an intermediate in the chemical production of the parent drug by hydrolysis in vitro, regardless of its relatively unselective character as revealed in the studies herein.
- HSA 9-(2-Hydroxyethyl)adenine
- a glass-lined reactor was charged with anhydrous PMPA, (I) (14.6 kg, 50.8 mol), phenyl (9.6 kg, 102 mol), and 1-methyl-2-pyrrolidinone (39 kg). The mixture was heated to 85° C. and triethylamine (6.3 kg, 62.3 mol) added. A solution of 1,3-dicyclohexylcarbodiimide (17.1 kg, 82.9 mol) in 1-methyl-2-pyrrolidinone (1.6 kg) was then added over 6 hours at 100° C. Heating was continued for 16 hours. The reaction was cooled to 45° C., water (29 kg) added, and cooled to 25° C. Solids were removed from the reaction by filtration and rinsed with water (15.3 kg).
- Half the product solution was purified by chromatography over a 38 ⁇ 38 cm bed of 22 kg silica gel 60, 230 to 400 mesh. The column was eluted with 480 kg acetone. The purification was repeated on the second half of the oil using fresh silica gel and acetone. Clean product bearing fractions were concentrated under reduced pressure to an oil. Acetonitrile (19.6 kg) was charged to the oil and the mixture concentrated under reduced pressure. Acetonitrile (66.4 kg) was charged and the solution chilled to 0 to ⁇ 5° C. for 16 hours.
- Monophenyl PMPA (II) A round-bottom flask with reflux condenser and nitrogen inlet was placed in a 70° C. oil bath. The flask was charged with anhydrous PMPA (I) (19.2 g, 67 mmol), N,N-dimethylformamide (0.29 g, 3.3 mmol), and tetramethylene sulfone (40 mL). Thionyl chloride (14.2 g, 119 mmol) was added over 4 hours. Heating was increased to 100° C. over the same time. A homogeneous solution resulted. Phenoxytrimethylsilane (11.7 g, 70 mmol) was added to the solution over 5 minutes. Heating in the 100° C.
- the reaction mixture was warmed to room temperature and washed three times with sodium dihydrogenphosphate solution (10% aq., 10 mL each wash).
- the organic solution was dried over anhydrous sodium sulfate and concentrated under reduced pressure to a oil.
- the oil was combined with fumaric acid (0.77 g, 6.6 mmol) and acetonitrile (40 mL) and heated to reflux to give a homogeneous solution.
- the solution was cooled in an ice bath and solids isolated by filtration.
- the solid GS-7171 fumarate salt was dried under reduced pressure to 3.7 g.
- the salt (3.16 g, 5.3 mmol) was suspended in dichloromethane (30 mL) and stirred with potassium carbonate solution (5 mL, 2.5 M in water) until the solid dissolved. The organic layer was isolated, then washed with water (5 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford 2.4 g III as a tan foam.
- Chiralpak AS is a proprietary packing material manufactured by Diacel and sold in North America by Chiral Technologies, Inc. (U.S. Pat. Nos. 5,202,433, RE 35,919, 5,434,298, 5,434,299 and 5,498,752).
- Chiralpak AS is a chiral stationary phase (CSP) comprised of amylosetris[(S)- ⁇ -methylbenzyl carbamate] coated onto a silica gel support.
- the GS-7171 diastereomeric mixture was dissolved in mobile phase, and approximately 1 g aliquots of GS-7171 were pumped onto the chromatographic system.
- the undesired diastereomer, designated GS-7339 was the first major broad (approx. 15 min. duration) peak to elute from the column.
- the mobile phase was immediately switched to 100% methyl alcohol, which caused the desired diastereomer, designated GS-7340 (IV), to elute as a sharp peak from the column with the methyl alcohol solvent front.
- the methyl alcohol was used to reduce the over-all cycle time.
- both diastereomers were collected as a single large fractions containing one of the purified diastereomers (>99.0% single diastereomer).
- the mobile phase solvents were removed in vacuo to yield the purified diastereomer as a friable foam.
- the GS-7340 fraction comprised about 50% of the total recovered mass.
- GS-7340 IV.
- GS-7171 (III) 2.8 kg, was purified by simulated moving bed chromatography over 10 cm by 5 cm beds of packing (Chiral Technologies Inc., 20 micron Chiralpak AS coated on silica gel) (1.2 kg). The columns were eluted with 30% methanol in acetonitrile. Product bearing fractions were concentrated to a solution of IV in acetonitrile (2.48 kg). The solution solidified to a crystalline mass wet with acetonitrile on standing.
- GS-7171 (III) was chromatographed by reverse phase HPLC to separate the diastereomers using the following summary protocol.
- Mobile Phase A - 0.02% (85%) H 3 PO 4 in water:acetonitrile (95:5)
- a Flow Rate 1.2 mL/min
- Temperature Ambient Detection: UV at 260 nm
- Sample Solution 20 mM sodium phosphat
- GS-7340 IV.
- a solution of GS-7171 (III) in acetonitrile was concentrated to an amber foam (14.9 g) under reduced pressure.
- the foam was dissolved in acetonitrile (20 mL) and seeded with a crystal of IV.
- the mixture was stirred overnight, cooled to 5° C., and solids isolated by filtration.
- GS-7340-02 (V).
- Scheme 1 A glass-lined reactor was charged with GS-7340 (IV), (1.294 kg, 2.71 mol), fumaric acid (284 g, 2.44 mol), and acetonitrile (24.6 kg). The mixture was heated to reflux to dissolve the solids, filtered while hot and cooled to 5° C. for 16 hours. The product was isolated by filtration, rinsed with acetonitrile (9.2 kg), and dried to 1329 g (V) as a white powder: mp 119.7-121.1° C.; [ ⁇ ] D 20 ⁇ 41.7° (c 1.0, acetic acid).
- the oil was purified by chromatography over a 15 ⁇ 13 cm bed of 1.2 kg silica gel 60, 230 to 400 mesh.
- the column was eluted with a gradient of dichloromethane and methanol. Product bearing fractions were concentrated under reduced pressure to afford 211 g VI (Scheme 3) as a tan foam.
- the diastereomeric mixture was purified using the conditions described for GS-7171 in Example 3A except for the following: Mobile Phase (Initial) GS-7120 - Acetonitrile:Isopropyl Alcohol (98:2) (Final) 100% Methyl Alcohol Elution Profile: GS-7341 (diastereomer B) GS-7342 (diastereomer A)
- the diastereomeric mixture was purified using the conditions described for GS-7171 (Example 3A) except for the following: Mobile Phase (Initial) GS-7120 - Acetonitrile:Isopropyl Alcohol (95:5) (Final) 100% Methyl Alcohol Elution Profile GS-7115 (diastereomer B) GS-7114 (diastereomer A)
- Phenyl PMPA Ethyl L-Alanyl Amidate.
- Phenyl PMPA (15.0 g, 41.3 mmol)
- L-alanine ethyl ester hydrochloride 12.6 g, 83 mmol
- triethylamine (11.5 mL, 83 mmol) were slurried together in 500 mL pyridine under dry N 2 .
- This suspension was combined with a solution of triphenylphosphine (37.9 g, 145 mmol), Aldrithiol 2 (2,2′-dipyridyl disulfide) (31.8 g, 145 mmol), and 120 mL pyridine.
- the mixture was heated at an internal temperature of 57° C. for 15 hours.
- the complete reaction was concentrated under vacuum to a yellow paste, 100 g.
- the paste was purified by column chromatography over a 25 ⁇ 11 cm bed of 1.1 kg silica gel 60, 230 to 400 mesh.
- the column was eluted with 8 liters of 2% methanol in dichloromethane followed by a linear gradient over a course of 26 liters eluent up to a final composition of 13% methanol. Clean product bearing fractions were concentrated to yield 12.4 g crude (5), 65% theory. This material was contaminated with about 15% (weight) triethylamine hydrochloride by 1 H NMR.
- the contamination was removed by dissolving the product in 350 mL ethyl acetate, extracting with 20 mL water, drying the organic solution over anhydrous sodium sulfate, and concentrating to yield 11.1 g pure GS-7097 as a white solid, 58% yield.
- the process also is employed to synthesize the diastereomeric mixture of GS-7003a and GS-7003b (the phenylalanyl amidate) and the mixture GS-7119 and GS-7335 (the glycyl amidate). These diastereomers are separated using a batch elution procedure such as shown in Example 3A, 6 and 7.
- GS-7340 shows a 10-fold increase in antiviral activity relative to TDF and a 200-fold increase in plasma stability. This greater plasma stability is expected to result in higher circulating levels of GS-7340 than TDF after oral administration.
- both prodrugs and PMPA were radiolabeled and spiked into intact human whole blood at equimolar concentrations. After 1 hour, plasma, red blood cells (RBCs) and peripheral blood mononuclear cells (PBMCs) were isolated and analyzed by HPLC with radiometric detection. The results are shown in Table 2.
- GS-7340 results in 10 ⁇ and 30 ⁇ the total intracellular concentration of PMPA species in PBMCs as compared to TDF and PMPA, respectively.
- 84% of the radioactivity is due to intact GS-7340, whereas no TDF is detected at 1 hour. Since no intact TDF is detected in plasma, the 10 ⁇ difference at 1 hour between TDF and GS-7340 is the minimum difference expected in vivo.
- the HPLC chromatogram for all three compounds in PBMCs is shown in FIG. 1 . TABLE 2 PMPA Metabolites in Plasma, PBMCs and RBCs After 1 h Incubation of PMPA Prodrugs or PMPA in Human Blood.
- Met. X and Met Y are shown in Table 5. Lower case “p” designates phosphorylation. These results were obtained after 1 hour in human blood. With increasing time, the in vitro differences are expected to increase, since 84% of GS-7340 is still 10 intact in plasma after one hour. Because intact GS-7340 is present in plasma after oral administration, the relative clinical efficacy should be related to the IC 50 values seen in vitro.
- IC 50 values of tenofovir, TDF, GS-7340, several nucleosides and the protease inhibitor nelfinivir are listed. As shown, nelfinavir and GS-7340 are 2-3 orders of magnitude more potent than all other nucleotides or nucleosides.
- “Phe-methylester” is the methylphenylalaninyl monoamidate, phenyl monoester of tenofovir; “gly-methylester” is the methylglycyl monoamidate, phenyl monoester of tenofovir.
- isomer A is believed to have the same absolute stereochemistry as GS-7340 (S), and isomer B is believed to have the same absolute stereochemistry that of GS-7339.
- the in vitro metabolism and stability of separated diastereomers were determined in PLCE, MT-2 extract and human plasma.
- a biological sample listed below, 80 ⁇ L was transferred into a screw-capped centrifuge tube and incubated at 37° C. for 5 min.
- a solution containing 0.2 mg/mL of the test compound in a suitable buffer, 20 ⁇ L was added to the biological sample and mixed.
- the reaction mixture, 20 mL was immediately sampled and mixed with 60 ⁇ L of methanol containing 0.015 mg/mL of 2-hydroxymethylnaphthalene as an internal standard for HPLC analysis. The sample was taken as the time-zero sample.
- reaction mixture 20 ⁇ L
- 60 ⁇ L of methanol containing the internal standard 60 ⁇ L
- the mixture thus obtained was centrifuged at 15,000 G for 5 min and the supernatant was analyzed with HPLC under the conditions described below.
- the biological samples evaluated are as follows.
- PLCE proteoid liver carboxyesterase from Sigma, 160 u/mg protein, 21 mg protein/mL
- PBS phosphated-buffered saline
- MT-2 cell extract was prepared from MT-2 cells according to the published procedure [A. Pompon, I. Lefebvre, J.-L. Imbach, S. Kahn, and D. Farquhar, “Antiviral Chemistry & Chemotherapy”, 5:91-98 (1994)] except for using HEPES buffer described below as the medium.
- the buffer systems used in the studies are as follows.
- PBS phosphate-buffered saline, Sigma
- PBS phosphate-buffered saline, Sigma
- HEPES buffer contains 0.010 M HEPES, 0.05 M potassium chloride, 0.005 M magnesium chloride, and 0.005 M dl-dithiothreitol. pH 7.4 at 37° C.
- TBS tris-buffered saline, Sigma
- TBS contains 0.05 M Tris, 0.0027 M potassium chloride, and 0.138 M sodium chloride. pH 7.5 at 37° C.
- X: Met. Y: A
- GS 7340 The pharmacokinetics of GS 7340 were studied in dogs after oral administration of a 10 mg-eq/kg dose.
- PBMC Peripheral Blood Mononuclear Cell preparation.
- Whole blood (8 ml) drawn at specified time points was mixed in equal proportion with phosphate buffered saline (PBS), layered onto 15 ml of Ficoll-Paque solution (Pharmacia Biotech) and centrifuged at 400 ⁇ g for 40 min.
- PBMC layer was removed and washed once with PBS.
- PMBC pellet was reconstituted in 0.5 ml of PBS, cells were resuspended, counted using hemocytometer and maintained at 70° C. until analyzed. The number of cells multiplied by the mean single-cell volume was used in calculation of intracellular concentrations.
- PMPA and Prodrugs in plasma and PBMCs The concentration of PMPA in dog plasma samples was determined by derivatizing PMPA with chloroacetaldehyde to yield a highly fluorescent N 1 , N 6 -ethenoadenine derivative (L. Naesens, J. Balzarini, and E. De Clercq, Clin. Chem. 38, 480 (1992). Briefly, plasma (100 ⁇ l) was mixed with 200 ⁇ l acetonitrile to precipitate protein. Samples were then evaporated to dryness under reduced pressure at room temperature.
- Dried samples were reconstituted in 200 ⁇ l derivatization cocktail (0.34% chloroacetaldehyde in 100 mM sodium acetate, pH 4.5), vortexed, and centrifuged. Supernatant was then transferred to a clean screw-cap tube and incubated at 95° C. for 40 min. Derivatized samples were then evaporated to dryness and reconstituted in 100 ⁇ l of water for HPLC analysis.
- derivatization cocktail 0.34% chloroacetaldehyde in 100 mM sodium acetate, pH 4.5
- the HPLC system was comprised of a P4000 solvent delivery system with AS3000 autoinjector and F2000 fluorescence detector (Thermo Separation, San Jose, Calif.).
- the column was an Inertsil ODS-2 column (4.6 ⁇ 150 mm).
- the mobile phases used were: A, 5% acetonitrile in 25 mM potassium phosphate buffer with 5 mM tetrabutyl ammonium bromide (TBABr), pH 6.0; B, 60% acetonitrile in 25 mM potassium phosphate buffer with 5 mM TBABr, pH 6.0.
- the flow rate was 2 ml/min and the column temperature was maintained at 35° C. by a column oven.
- the gradient profile was 90% A/10% B for 10 min for PMPA and 65% A/35% B for 10 min for the prodrug. Detection was by fluorescence with excitation at 236 nm and emission at 420 nm, and the injection volume was 10 ⁇ l. Data was acquired and stored by a laboratory data acquisition system (PeakPro, Beckman, Allendale, N.J.).
- FIG. 2 shows the time course of GS 7340-2 metabolism summary of plasma and PBMC exposures following oral administration of pure diastereoisomers of the PMPA prodrugs.
- the bar graph in FIG. 2 shows the AUC (0-24 h) for tenofovir in dog PBMCs and plasma after administration of PMPA s.c., TDF and amidate ester prodrugs. All of the amidate prodrugs exhibited increases in PBMC exposure. For example, GS 7340 results in a ⁇ 21-fold increase in PBMC exposure as compared to PMPA s.c. and TDF; and a 6.25-fold and 1.29-fold decrease in plasma exposure, respectively.
- GS-7340 isopropyl alaninyl monoamidate, phenyl monoester of tenofovir
- GS-7340 isopropyl alaninyl monoamidate, phenyl monoester of tenofovir
- aqueous solution 50 mM citric acid, pH 2.2
- Plasma and peripheral blood mononuclear cells were obtained over the 24-hr period.
- Urine and feces were cage collected over 24 hr. At 24 h after the dose, the animals were sacrificed and tissues removed for analysis. Total radioactivity in tissues was determined by oxidation and liquid scintillation counting.
- the biodistribution of PMPA after 24 hours after a single oral dose of radiolabelled GS 7340 is shown in Table 4 along with the data from a previous study with TDF (GS-4331).
- TDF the prodrug concentration in the plasma is below the level of assay detection, and the main species observed in plasma is the parent drug.
- Levels of PMPA in the lymphatic tissues, bone marrow, and skeletal muscle are increased 10-fold after administration of GS-7340.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Genetics & Genomics (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Virology (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Communicable Diseases (AREA)
- Toxicology (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Biophysics (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Engineering & Computer Science (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Food Science & Technology (AREA)
- Cell Biology (AREA)
Abstract
A novel method has led to the identification of novel mixed ester-amidates of PMPA for retroviral or hepadnaviral therapy, including compounds of structure (5a)

having substituent groups as defined herein. Compositions of these novel compounds in pharmaceutically acceptable excipients and their use in therapy and prophylaxis are provided.
Description
- This application is a continuation of Ser. No. 10/798,692, filed Mar. 11, 2004, now pending, which is a continuation of Ser. No. 10/354,207, filed Jan. 28, 2003, now pending, which is a continuation of Ser. No. 09/909,560, filed Jul. 20, 2001, now abandoned, which claims benefit under 35 U.S.C. § 119(e) to Ser. No. 60/220,021, filed Jul. 21, 2000, all of which are incorporated herein by reference.
- This application relates to prodrugs of methoxyphosphonate nucleotide analogues. In particular it relates to improved methods for making and identifying such prodrugs.
- Many methoxyphosphonate nucleotide analogues are known. In general, such compounds have the structure A-OCH2P(O)(OR)2 where A is the residue of a nucleoside analogue and R independently is hydrogen or various protecting or prodrug functionalities. See U.S. Pat. Nos. 5,663,159, 5,977,061 and 5,798,340, Oliyai et al, “Pharmaceutical Research” 16(11):1687-1693 (1999), Stella et al., “J. Med. Chem.” 23(12):1275-1282 (1980), Aarons, L., Boddy, A. and Petrak, K. (1989) Novel Drug Delivery and Its Therapeutic Application (Prescott, L. F. and Nimmo, W. S., ed.), pp. 121-126; Bundgaard, H. (1985) Design of Prodrugs (Bundgaard, H., ed.) pp. 70-74 and 79-92; Banerjee, P. K. and Amidon, G. L. (1985) Design of Prodrugs (Bundgaard, H., ed.) pp. 118-121; Notari, R. E. (1985) Design of Prodrugs (Bundgaard, H., ed.) pp. 135-156; Stella, V. J. and Himmelstein, K. J. (1985) Design of Prodrugs (Bundgaard, H., ed.) pp. 177-198; Jones, G. (1985) Design of Prodrugs (Bundgaard, H., ed.) pp. 199-241; Connors, T. A. (1985) Design of Prodrugs (Bundgaard, H., ed.) pp. 291-316. All literature and patent citations herein are expressly incorporated by reference.
- Prodrugs of methoxyphosphonate nucleotide analogues intended for antiviral or antitumor therapy, while known, traditionally have been selected for their systemic effect. For example, such prodrugs have been selected for enhanced bioavailability, i.e., ability to be absorbed from the gastrointestinal tract and converted rapidly to parent drug to ensure that the parent drug is available to all tissues. However, applicants now have found that it is possible to select prodrugs that become enriched at therapeutic sites, as illustrated by the studies described herein where the analogues are enriched at localized focal sites of HIV infection. The objective of this invention is, among other advantages, to produce less toxicity to bystander tissues and greater potency of the parental drug in tissues which are the targets of therapy with the parent methoxyphosphonate nucleotide analogue.
- Accordingly, pursuant to these observations, a screening method is provided for identifying a methoxyphosphonate nucleotide analogue prodrug conferring enhanced activity in a target tissue comprising:
-
- (a) providing at least one of said prodrugs;
- (b) selecting at least one therapeutic target tissue and at least one non-target tissue;
- (c) administering the prodrug to the target tissue and to said at least one non-target tissue; and
- (d) determining the relative antiviral activity conferred by the prodrug in the tissues in step (c).
- In preferred embodiments, the target tissue are sites where HIV is actively replicated and/or which serve as an HIV reservoir, and the non-target tissue is an intact animal. Unexpectedly, we found that selecting lymphoid tissue as the target tissue for the practice of this method for HIV led to identification of prodrugs that enhance the delivery of active drug to such tissues.
- A preferred compound of this invention, which has been identified by this method has the structure (1),

-
- where Ra is H or methyl,
- and chirally enriched compositions thereof, salts, their free base and solvates thereof.
- A preferred compound of this invention has the structure (2)

-
- and its enriched diasteromers, salts, free base and solvates.
- In addition, we unexpectedly found that the chirality of substituents on the phosphorous atom and/or the amidate substituent are influential in the enrichment observed in the practice of this invention. Thus, in another embodiment of this invention, we provide diastereomerically enriched compounds of this invention having the structure (3)

-
- which are substantially free of the diastereomer (4)

- wherein
- R1 is an oxyester which is hydrolyzable in vivo, or hydroxyl;
- B is a heterocyclic base;
- R2 is hydroxyl, or the residue of an amino acid bonded to the P atom through an amino group of the amino acid and having each carboxy substituent of the amino acid optionally esterified, but not both of R1 and R2 are hydroxyl;
- E is —(CH2)2—, —CH(CH3)CH2—, —CH(CH2F)CH2—, —CH(CH2OH)CH2, —CH(CH═CH2)CH2—, —CH(C≡CH)CH2—, —CH(CH2N3)CH2—,

—CH(R6)OCH(R6′)—, —CH(R9)CH2O—or —CH(R8)O—, wherein the right hand bond is linked to the heterocyclic base; - the broken line represents an optional double bond;
- R4 and R5 are independently hydrogen, hydroxy, halo, amino or a substituent having 1-5 carbon atoms selected from acyloxy, alkyoxy, alkylthio, alkylamino and dialkylamino;
- R6 and R6′ are independently H, C1-C6 alkyl, C1-C6 hydroxyalkyl, or C2-C7 alkanoyl;
- R7 is independently H, C1-C6 alkyl, or are taken together to form —O— or —CH2—;
- R8 is H, C1-C6 alkyl, C1-C6 hydroxyalkyl or C1-C6 haloalkyl; and
- R9 is H, hydroxymethyl or acyloxymethyl;
- and their salts, free base, and solvates.
- which are substantially free of the diastereomer (4)
- The diastereomers of structure (3) are designated the (S) isomers at the phosphorus chiral center.
- Preferred embodiments of this invention are the diastereomerically enriched compounds having the structure (5a)

-
- which is substantially free of diastereomer (5b)

- wherein
- R5 is methyl or hydrogen;
- R6 independently is H, alkyl, alkenyl, alkynyl, aryl or arylalkyl, or R6 independently is alkyl, alkenyl, alkynyl, aryl or arylalkyl which is substituted with from 1 to 3 substituents selected from alkylamino, alkylaminoalkyl, dialkylaminoalkyl, dialkylamino, hydroxyl, oxo, halo, amino, alkylthio, alkoxy, alkoxyalkyl, aryloxy, aryloxyalkyl, arylalkoxy, arylalkoxyalkyl, haloalkyl, nitro, nitroalkyl, azido, azidoalkyl, alkylacyl, alkylacylalkyl, carboxyl, or alkylacylamino;
- R7 is the side chain of any naturally-occurring or pharmaceutically acceptable amino acid and which, if the side chain comprises carboxyl, the carboxyl group is optionally esterified with an alkyl or aryl group;
- R11 is amino, alkylamino, oxo, or dialkylamino; and
- R12 is amino or H;
- and its salts, tautomers, free base and solvates.
- which is substantially free of diastereomer (5b)
- A preferred embodiment of this invention is the compound of structure (6), 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine, also designated herein GS-7340.

- Another preferred embodiment of this invention is the fumarate salt of structure (5) (structure (7)), 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine fumarate (1:1), also designated herein GS-7340-2

- The compounds of structures (1)-(7) optionally are formulated into compositions containing pharmaceutically acceptable excipients. Such compositions are used in effective doses in the therapy or prophylaxis of viral (particularly HIV or hepadnaviral) infections.
- In a further embodiment, a method is provided for the facile manufacture of 9-[2-(phosphonomethoxy)propyl]adenine (hereinafter “PMPA” or 9-[2-(phosphonomethoxy)ethyl]adenine (hereinafter “PMEA”) using magnesium alkoxide, which comprises combining 9-(2-hydroxypropyl)adenine or 9-(2-hydroxyethyl)adenine, protected p-toluenesulfonyloxymethylphosphonate and magnesium alkoxide, and recovering PMPA or PMEA, respectively.
-
FIG. 1 shows HPLC/C-14 traces of PBMC extracts from human blood incubated for 1 h at 37° C. with TDF, GS-7340 and PMPA. -
FIG. 2 shows PMPA and Prodrug concentration in plasma and PBMCs following oral administration of GS 7340-2 to Dogs at 10 mg-eq/kg. -
FIG. 3 depicts Tenofovir exposure in PBMCs and plasma upon administration of 10 mg-eq/kg in dogs. - The methoxyphosphonate nucleotide analogue parent drugs for use in this screening method are compounds having the structure A-OCH2P(O)(OH)2 wherein A is the residue of a nucleoside analogue. These compounds are known

per se and are not part of this invention. More particularly, the parent compounds comprise a heterocyclic base B and an aglycon E, in general having the structure wherein the group B is defined below and group E is defined above. Examples are described in U.S. Pat. Nos. 4,659,825, 4,808,716, 4,724,233, 5,142,051, 5,130,427, 5,650,510, 5,663,159, 5,302,585, 5,476,938, 5,696,263, 5,744,600, 5,688,778, 5,386,030, 5,733,896, 5,352,786, and 5,798,340, and EP 821,690 and 654,037. - The prodrugs for use in the screening method of this invention are covalently modified analogues of the parent methoxyphosphonate nucleotide analogues described in the preceding paragraph. In general, the phosphorus atom of the parent drug is the preferred site for prodrug modification, but other sites are found on the heterocyclic base B or the aglycon E. Many such prodrugs are already known. Primarily, they are esters or amidates of the phosphorus atom, but also include substitutions on the base and aglycon. None of these modifications per se is part of this invention and none are to be considered limiting on the scope of the invention herein.
- The phosphorus atom of the methoxyphosphonate nucleotide analogues contains two valences for covalent modification such as amidation or esterification (unless one phosphoryl hydroxyl is esterified to an aglycon E hydroxyl substituent, whereupon only one phosphorus valence is free for substitution). The esters typically are aryloxy. The amidates ordinarily are naturally occurring monoamino acids having free carboxyl group(s) esterified with an alkyl or aryl group, usually phenyl, cycloalkyl, or t-, n- or s-alkyl groups. Suitable prodrugs for use in the screening method of this invention are disclosed for example in U.S. Pat. No. 5,798,340. However, any prodrug which is potentially believed to be capable of being converted in vivo within target tissue cells to the free methoxyphosphonate nucleotide analogue parent drug, e.g., whether by hydrolysis, oxidation, or other covalent transformation resulting from exposure to biological tissues, is suitable for use in the method of this invention. Such prodrugs may not be known at this time but are identified in the future and thus become suitable candidates available for testing in the method of this invention. Since the prodrugs are simply candidates for screening in the methods their structures are not relevant to practicing or enabling the screening method, although of course their structures ultimately are dispositive of whether or not a prodrug will be shown to be selective in the assay.
- The pro-moieties bound to the parent drug may be the same or different. However, each prodrug to be used in the screening assay will differ structurally from the other prodrugs to be tested. Distinct, i.e. structurally different, prodrugs generally are selected on the basis of either their stereochemistry or their covalent structure, or these features are varied in combination. Each prodrug tested, however, desirably is structurally and stereochemically substantially pure, else the output of the screening assay will be less useful. It is of course within the scope of this invention to test only a single prodrug in an individual embodiment of the method of this invention, although typically then one would compare the results with prior studies with other prodrugs.
- We have found that the stereochemistry of the prodrugs is capable of influencing the enrichment in target tissues. Chiral sites are at the phosphorus atom and are also found in its substituents. For example, amino acid used in preparing amidates may be D or L forms, and the phosphonate esters or the amino acid esters can contain chiral centers as well. Chiral sites also are found on the nucleoside analogue portion of the molecules, but these typically are already dictated by the stereochemistry of the parent drug and will not be varied as part of the screen. For example the R isomer of PMPA is preferred as it is more active than the corresponding S isomer. Typically these diasteromers or enantiomers will be chirally enriched if not pure at each site so that the results of the screen will be more meaningful. As noted, distinctiveness of stereoisomers is conferred by enriching or purifying the stereoisomer (typically this will be a diastereomer rather than an enantiomer in the case of most methoxyphosphonate nucleotide analogues) free of other stereoisomers at the chiral center in question, so that each test compound is substantially homogeneous. By substantially homogeneous or chirally enriched, we mean that the desired stereoisomer constitutes greater than about 60% by weight of the compound, ordinarily greater than about 80% and preferably greater than about 95%.
- 5.1 Novel Screening Method
- Once at least one candidate prodrug has been selected, the remaining steps of the screening method of this invention are used to identify a prodrug possessing the required selectivity for the target tissue. Most conveniently the prodrugs are labeled with a detectable group, e.g. radiolabeled, in order to facilitate detection later in tissues or cells. However, a label is not required since other suitable assays for the prodrug or its metabolites (including the parent drug) can also be employed. These assays could include mass spectrometry, HPLC, bioassays or immunoassays for instance. The assay may detect the prodrug and any one or more of its metabolites, but preferably the assay is conducted to detect only the generation of the parent drug. This is based on the assumption (which may not be warranted in all cases) that the degree and rate of conversion of prodrug to antivirally active parent diphosphate is the same across all tissues tested. Otherwise, one can test for the diphosphate.
- The target tissue preferably will be lymphoid tissue when screening for prodrugs useful in the treatment of HIV infection. Lymphoid tissue will be known to the artisan and includes CD4 cells, lymphocytes, lymph nodes, macrophages and macrophage-like cells including monocytes such as peripheral blood monocytic cells (PBMCs) and glial cells. Lymphoid tissue also includes non-lymphoid tissues that are enriched in lymphoid tissues or cells, e.g. lung, skin and spleen. Other targets for other antiviral drugs of course will be the primary sites of replication or latency for the particular virus concerned, e.g., liver for hepatitis and peripheral nerves for HSV. Similarly, target tissues for tumors will in fact be the tumors themselves. These tissues are all well-known to the artisan and would not require undue experimentation to select. When screening for antiviral compounds, target tissue can be infected by the virus.
- Non-target tissues or cells also are screened as part of the method herein. Any number or identity of such tissues or cells can be employed in this regard. In general, tissues for which the parent drug is expected to be toxic will be used as non-target tissues. The selection of a non-target tissue is entirely dependent upon the nature of the prodrug and the activity of the parent. For example, non-hepatic tissues would be selected for prodrugs against hepatitis, and untransformed cells of the same tissue as the tumor will suffice for the antitumor-selective prodrug screen.
- It should be noted that the method of this invention is distinct from studies typically undertaken to determine oral bioavailability of prodrugs. In oral bioavailability studies, the objective is to identify a prodrug which passes into the systemic circulation substantially converted to parent drug. In the present invention, the objective is to find prodrugs that are not metabolized in the gastrointestinal tract or circulation. Thus, target tissues to be evaluated in the method of this invention generally do not include the small intestines or, if the intestines are included, then the tissues also include additional tissues other than the small intestines.
- The target and non-target tissues used in the screening method of this invention typically will be in an intact living animal. Prodrugs containing esters are more desirably tested in dogs, monkeys or other animals than rodents; mice and rat plasma contains high circulating levels of esterases that may produce a misleading result if the desired therapeutic subject is a human or higher mammal.
- It is not necessary to practice this method with intact animals. It also is within the scope of this invention to employ perfused organs, in vitro culture of organs (e.g. skin grafts) or cell lines maintained in various forms of cell culture, e.g. roller bottles or zero gravity suspension systems. For example, MT-2 cells can be used as a target tissue for selecting HIV prodrugs. Thus, the term “tissue” shall not be construed to require organized cellular structures, or the structures of tissues as they may be found in nature, although such would be preferred. Rather, the term “tissue” shall be construed to be synonymous with cells of a particular source, origin or differentiation stage.
- The target and non-target tissue may in fact be the same tissue, but the tissues will be in different biological status. For example, the method herein could be used to select for prodrugs that confer activity in virally-infected tissue (target tissue) but which remain substantially inactive in virally-uninfected cells (corresponding non-target tissue). The same strategy would be employed to select prophylactic prodrugs, i.e., prodrugs metabolized to antivirally active forms incidental to viral infection but which remain substantially unmetabolized in uninfected cells. Similarly, prodrugs could be screened in transformed cells and the untransformed counterpart tissue. This would be particularly useful in comparative testing to select prodrugs for the treatment of hematological malignancies, e.g. leukemias.
- Without being limited by any particular theory of operation, tissue selective prodrugs are thought to be selectively taken up by target cells and/or selectively metabolized within the cell, as compared to other tissues or cells. The unique advantage of the methoxyphosphonate prodrugs herein is that their metabolism to the dianion at physiological pH ensures that they will be unable to diffuse back out of the cell. They therefore remain effective for lengthy periods of time and are maintained at elevated intracellular concentrations, thereby exhibiting increased potency. The mechanisms for enhanced activity in the target tissue are believed to include enhanced uptake by the target cells, enhanced intracellular retention, or both mechanisms working together. However, the manner in which selectivity or enhanced delivery occurs in the target tissue is not important. It also is not important that all of the metabolic conversion of the prodrug to the parent compound occurs within the target tissue. Only the final drug activity-conferring conversion need occur in the target tissue; metabolism in other tissues may provide intermediates finally converted to antiviral forms in the target tissue.
- The degree of selectivity or enhanced delivery that is desired will vary with the parent compound and the manner in which it is measured (% dose distribution or parent drug concentration). In general, if the parent drug already possess a generous therapeutic window, a low degree of selectivity may be sufficient for the desired prodrug. On the other hand, toxic compounds may require more extensive screening to identify selective prodrugs. The relative expense of the method of this invention can be reduced by screening only in the target tissue and tissues against which the parent compound is known to be relatively toxic, e.g. for PMEA, which is nephrotoxic at higher doses, the primary focus will be on kidney and lymphoid tissues.
- The step of determining the relative antiviral activity of a prodrug in the selected tissues ordinarily is accomplished by assaying target and non-target tissues for the relative presence or activity of a metabolite of the prodrug, which metabolite is known to have, or is converted to, a metabolite having antiviral or antitumor activity. Thus, typically one would determine the relative amount of the parent drug in the tissues over substantially the same time course in order to identify prodrugs that are preferentially metabolized in the target tissue to an antivirally or antitumor active metabolite or precursor thereof which in the target tissue ultimately produces the active metabolite. In the case of antiviral compounds, the active metabolite is the diphosphate of the phosphonate parent compounds. It is this metabolite that is incorporated into the viral nucleic acid, thereby truncating the elongating nucleic acid strand and halting viral replication. Metabolites of the prodrug can be anabolic metabolites, catabolic metabolites, or the product of anabolism and catabolism together. The manner in which the metabolite is produced is not important in the practice of the method of this invention.
- The method of this invention is not limited to assaying a metabolite which per se possesses antiviral or antitumor activity. Instead, one can assay inactive precursors of the active metabolites. Precursors of the antivirally active diphosphate metabolite include the monophosphate of the parent drug, monophosphates of other metabolites of the parent drug (e.g., an intermediate modification of a substituent on the heterocyclic base), the parent itself and metabolites generated by the cell in converting the prodrug to the parent prior to phosphorylation. The precursor structures may vary considerably as they are the result of cellular metabolism. However, this information is already known or could be readily determined by one skilled in the art.
- If the prodrug being assayed does not exhibit antitumor or antiviral activity per se then adjustments to the raw assay results may be required. For example, if the intracellular processing of the inactive metabolite to an active metabolite occurs at different rates among the tissues being tested, the raw assay results with the inactive metabolite would need to be adjusted to take account of the differences among the cell types because the relevant parameter is the generation of activity in the target tissue, not accumulation of inactive metabolites. However, determining the proper adjustments would be within the ordinary skill. Thus, when step (d) of the method herein calls for determining the activity, activity can be either measured directly or extrapolated. It does not mean that the method herein is limited to only assaying intermediates that are active per se. For instance, the absence or decline of the prodrug in the test tissues also could be assayed. Step (d) only requires assessment of the activity conferred by the prodrug as it interacts with the tissue concerned, and this may be based on extrapolation or other indirect measurement.
- Step (d) of the method of this invention calls for determining the “relative” activity of the prodrug. It will be understood that this does not require that each and every assay or series of assays necessarily must also contain runs with the selected non-target tissue. On the contrary, it is within the scope of this invention to employ historical controls of the non-target tissue or tissues, or algorithms representing results to be expected from such non-target tissues, in order to provide the benchmark non-target activity.
- The results obtained in step (d) are then used optimally to select or identify a prodrug which produces greater antiviral activity in the target tissue than in the non-target tissue. It is this prodrug that is selected for further development.
- It will be appreciated that some preassessment of prodrug candidates can be undertaken before the practice of the method of this invention. For example, the prodrug will need to be capable of passing largely unmetabolized through the gastrointestinal tract, it will need to be substantially stable in blood, and it should be able to permeate cells at least to some degree. In most cases it also will need to complete a first pass of the hepatic circulation without substantial metabolism. Such prestudies are optional, and are well-known to those skilled in the art.
- The same reasoning as is described above for antiviral activity is applicable to antitumor prodrugs of methoxyphosphonate nucleotide analogues as well. These include, for example, prodrugs of PMEG, the guanyl analogue of PMEA. In this case, cytotoxic phosphonates such as PMEG are worthwhile candidates to pursue as their cytotoxicity in fact confers their antitumor activity.
- A compound identified by this novel screening method then can be entered into a traditional preclinical or clinical program to confirm that the desired objectives have been met. Typically, a prodrug is considered to be selective if the activity or concentration of parent drug in the target tissue (% dose distribution) is greater than 2×, and preferably 5×, that of the parent compound in non-target tissue. Alternatively, a prodrug candidate can be compared against a benchmark prodrug. In this case, selectivity is relative rather than absolute. Selective prodrugs will be those resulting in greater than about 10× concentration or activity in the target tissue as compared with the prototype, although the degree of selectivity is a matter of discretion.
- 5.2 Novel Method for Preparation of Starting Materials or Intermediates
- Also included herein is an improved method for manufacture of preferred starting materials (parent drugs) of this invention, PMEA and (R)-PMPA. Typically, this method comprises reacting 9-(2-hydroxypropyl)adenine (HPA) or 9-(2-hydroxyethyl)adenine (HEA) with a magnesium alkoxide, thereafter adding the protected aglycon synthon p-toluene-sulfonyloxymethylphosphonate (tosylate) to the reaction mixture, and recovering PMPA or PMEA, respectively.
- Preferably, HPA is the enriched or isolated R enantiomer. If a chiral HPA mixture is used, R-PMPA can be isolated from the chiral PMPA mixture after the synthesis is completed.
- Typically the tosylate is protected by lower alkyl groups, but other suitable groups will be apparent to the artisan. It may be convenient to employ the tosylate presubstituted with the prodrug phosphonate substituents which are capable of acting as protecting groups in the tosylation reaction, thereby allowing one to bypass the deprotection step and directly recover prodrug or an intermediate therefore.
- The alkyl group of the magnesium alkoxide is not critical and can be any C1-C6 branched or normal alkyl, but is preferably t-butyl (for PMPA) or isopropyl (for PMEA). The reaction conditions also are not critical, but preferably comprise heating the reaction mixture at about 70-75° C. with stirring or other moderate agitation.
- If there is no interest in retaining the phosphonate substituents, the product is deprotected (usually with bromotrimethylsilane where the tosylate protecting group is alkyl), and the product then recovered by crystallization or other conventional method as will be apparent to the artisan.
- 5.3 Heterocyclic Base
- In the compounds of this invention depicted in structures (3) and (4), the heterocyclic base B is selected from the structures
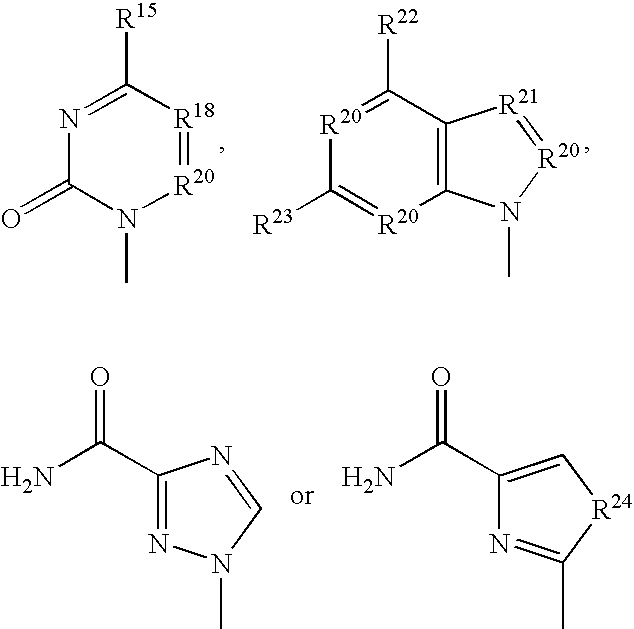
-
- wherein
- R15 is H, OH, F, Cl, Br, I, OR16, SH, SR16, NH2, or NHR17;
- R16 is C1-C6 alkyl or C2-C6 alkenyl including —CH3, —CH2CH3, —CH2C≡CH, —CH2CH═CH2 and —C3H7;
- R17 is C1-C6 alkyl or C2-C6 alkenyl including —CH3, —CH2CH3, —CH2C≡CH, —CH2CH═CH2, and —C3H7;
- R18 is N, CF, CC1, CBr, CI, CR19, CSR19, or COR19;
- R19 is H, C1-C9 alkyl, C2-C9 alkenyl, C2-C9 alkynyl, C1-C9 alkyl-C1-C9 alkoxy, or C7-C9 aryl-alkyl unsubstituted or substituted by OH, F, Cl, Br or I, R19 therefore including —CH3, —CH2CH3, —CHCH2, —CHCHBr, —CH2CH2Cl,
- —CH2CH2F, —CH2CCH, —CH2CHCH2, —C3H7, —CH2OH, —CH2OCH3, —CH2OC2H5, —CH2OCCH, —CH2OCH2CHCH2, —CH2C3H7, —CH2CH2OH, —CH2CH2OCH3,
- —CH2CH2OC2H5, —CH2CH2OCCH, —CH2CH2OCH2CHCH2, and
- —CH2CH2OC3H7;
- R20 is N or CH;
- R21 is N, CH, CCN, CCF3, CC≡CH or CC(O)NH2;
- R22 is H, OH, NH2, SH, SCH3, SCH2CH3, SCH2C≡CH, SCH2CH═CH2, SC3H7, NH(CH3), N(CH3)2, NH(CH2CH3), N(CH2CH3)2, NH(CH2C≡CH), NH(CH2CHCH2), NH(C3H7), halogen (F, Cl, Br or I) or X wherein X is —(CH2)m(O)n(CH2)mN(R10)2 wherein each m is independently 0-2, n is 0-1, and
- R10 independently is H,
- C1-C15 alkyl, C2-C15 alkenyl, C6-C15 arylalkenyl, C6-C15 arylalkynyl, C2-C15 alkynyl, C1-C6-alkylamino-C1-C6 alkyl, C5-C15 aralkyl, C6-C15 heteroaralkyl, C5-C6 aryl, C2-C6 heterocycloalkyl,
- C2-C15 alkyl, C3-C15 alkenyl, C6-C15 arylalkenyl, C3-C15 alkynyl, C7-C15 arylalkynyl, C1-C6-alkylamino-C1-C6 alkyl, C5-C15 aralkyl, C6-C15 heteroalkyl or C3-C6 heterocycloalkyl wherein methylene in the alkyl moiety not adjacent to N6 has been replaced by —O—,
- optionally both R10 are joined together with N to form a saturated or unsaturated C2-C5 heterocycle containing one or two N heteroatoms and optionally an additional O or S heteroatom,
- or one of the foregoing R10 groups which is substituted with 1 to 3 halo, CN or N3; but optionally at least one R10 group is not H;
- R23 is H, OH, F, Cl, Br, I, SCH3, SCH2CH3, SCH2C≡CH, SCH2CHCH2, SC3H7, OR16, NH2, NHR17 or R22; and
- R24 is O, S or Se.
- B also includes both protected and unprotected heterocyclic bases, particularly purine and pyrimidine bases. Protecting groups for exocyclic amines and other labile groups are known (Greene et al. “Protective Groups in Organic Synthesis”) and include N-benzoyl, isobutyryl, 4,4′-dimethoxytrityl (DMT) and the like. The selection of protecting group will be apparent to the ordinary artisan and will depend upon the nature of the labile group and the chemistry which the protecting group is expected to encounter, e.g. acidic, basic, oxidative, reductive or other conditions. Exemplary protected species are N4-benzoylcytosine, N6-benzoyladenine, N2-isobutyrylguanine and the like.
- Protected bases have the formulas Xa.1, XIa.1, XIb.1, XIIa.1 or XIIIa.1
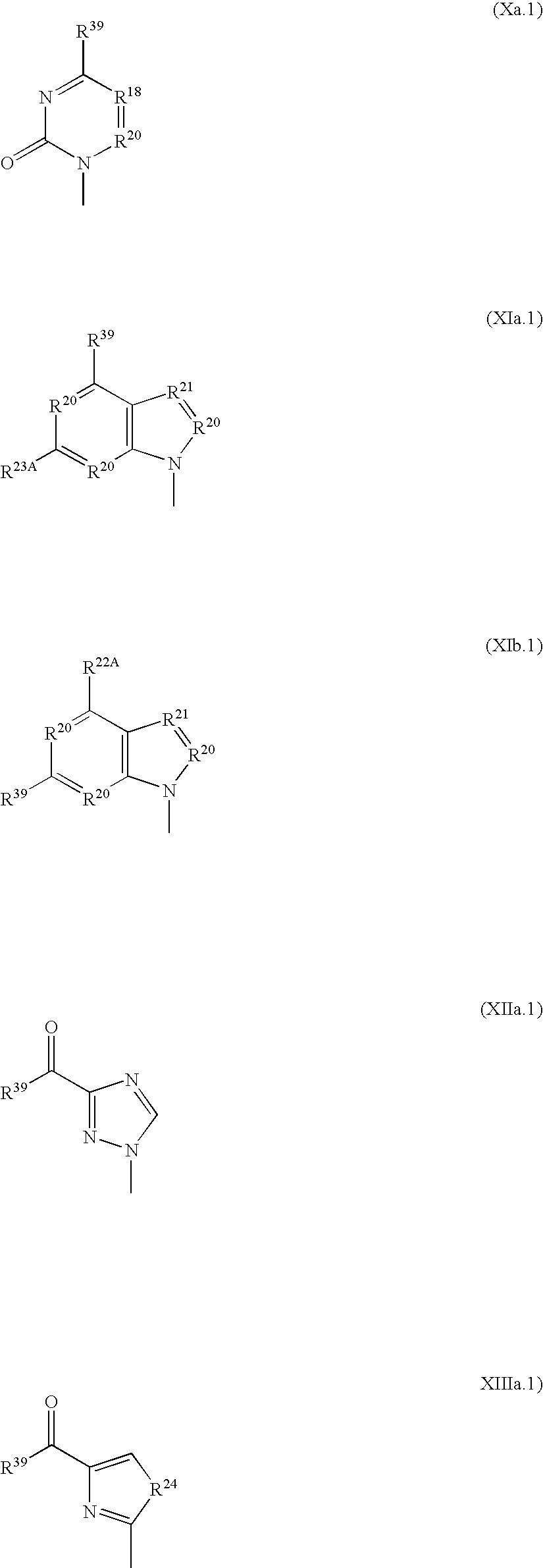
-
- wherein R18, R20, R21, R24 have the meanings previously defined; R22A is R39 or R22 provided that R22 is not NH2; R23A is R39 or R23 provided that R23 is not NH2; R39 is NHR40, NHC(O)R36 or CR41N(R38)2 wherein R36 is C1-C19 alkyl, C1-C19 alkenyl, C3-C10 aryl, adamantoyl, alkylanyl, or C3-C10 aryl substituted with 1 or 2 atoms or groups selected from halogen, methyl, ethyl, methoxy, ethoxy, hydroxy and cyano; R38 is C1-C10 alkyl, or both R38 together are 1-morpholino, 1-piperidine or 1-pyrrolidine; R40 is C1-C10 alkyl, including methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl, pentyl, hexyl, octyl and decanyl; and R41 is hydrogen or CH3.
- For bases of structures XIa.1 and XIb.1, if R39 is present at R22A or R23A, both R39 groups on the same base will generally be the same. Exemplary R36 are phenyl, phenyl substituted with one of the foregoing R36 aryl substituents, —C10H15 (where C10H15 is 2-adamantoyl), —CH2—C6H5, —C6H5, —CH(CH3)2, —CH2CH3, methyl, butyl, t-butyl, heptanyl, nonanyl, undecanyl, or undecenyl.
- Specific bases include hypoxanthine, guanine, adenine, cytosine, inosine, thymine, uracil, xanthine, 8-aza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 7-deaza-8-aza derivatives of adenine, guanine, 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 1-deaza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 7-deaza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 3-deaza derivatives of 2-aminopurine, 2,6-diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 6-azacytosine; 5-fluorocytosine; 5-chlorocytosine; 5-iodocytosine; 5-bromocytosine; 5-methylcytosine; 5-bromovinyluracil; 5-fluorouracil; 5-chlorouracil; 5-iodouracil; 5-bromouracil; 5-trifluoromethyluracil; 5-methoxymethyluracil; 5-ethynyluracil and 5-propynyluracil.
- Preferably, B is a 9-purinyl residue selected from guanyl, 3-deazaguanyl, 1-deazaguanyl, 8-azaguanyl, 7-deazaguanyl, adenyl, 3-deazaadenyl, 1-dezazadenyl, 8-azaadenyl, 7-deazaadenyl, 2,6-diaminopurinyl, 2-aminopurinyl, 6-chloro-2-aminopurinyl and 6-thio-2-aminopurinyl, or a B′ is a 1-pyrimidinyl residue selected from cytosinyl, 5-halocytosinyl, and 5-(C1-C3-alkyl)cytosinyl.
- Preferred B groups have the formula

-
- wherein
- R22 independently is halo, oxygen, NH2, X or H, but optionally at least one R22 is X;
- X is —(CH2)m(O)n(CH2)mN(R10)2 wherein m is 0-2, n is 0-1, and
- R10 independently is H,
- C1-C15 alkyl, C2-C15 alkenyl, C6-C15 arylalkenyl, C6-C15 arylalkynyl, C2-C15 alkynyl, C1-C6-alkylamino-C1-C6 alkyl, C5-C15 aralkyl, C6-C15 heteroaralkyl, C5-C6 aryl, C2-C6 heterocycloalkyl,
- C2-C15 alkyl, C3-C15 alkenyl, C6-C15 arylalkenyl, C3-C15 alkynyl, C7-C15 arylalkynyl, C1-C6-alkylamino-C1-C6 alkyl, C5-C15 aralkyl, C6-C15 heteroalkyl or C3-C6 heterocycloalkyl wherein methylene in the alkyl moiety not adjacent to N6 has been replaced by —O—,
- optionally both R10 are joined together with N to form a saturated or unsaturated C2-C5 heterocycle containing one or two N heteroatoms and optionally an additional O or S heteroatom,
- or one of the foregoing R10 groups is substituted with 1 to 3 halo, CN or N3; but optionally at least one R10 group is not H; and
- Z is N or CH, provided that the heterocyclic nucleus varies from purine by no more than one Z.
- E groups represent the aglycons employed in the methoxyphosphonate nucleotide analogues. Preferably, the E group is —CH(CH3)CH2— or —CH2CH2—. Also, it is preferred that the side groups at chiral centers in the aglycon be substantially solely in the (R) configuration (except for hydroxymethyl, which is the enriched (S) enantiomer).
- R1 is an in vivo hydrolyzable oxyester having the structure —OR35 or —OR6 wherein R35 is defined in column 64, line 49 of U.S. Pat. No. 5,798,340, herein incorporated by reference, and R6 is defined above. Preferably R1 is aryloxy, ordinarily unsubstituted or para-substituted (as defined in R6) phenoxy.
- R2 is an amino acid residue, optionally provided that any carboxy group linked by less than about 5 atoms to the amidate N is esterified. R2 typically has the structure

-
- wherein
- n is 1 or 2;
- R11 is R6 or H; preferably R6═C3-C9 alkyl; C3-C9 alkyl substituted independently with OH, halogen, O or N; C3-C6 aryl; C3-C6 aryl which is independently substituted with OH, halogen, O or N; or C3-C6 arylalkyl which is independently substituted with OH, halogen, O or N;
- R12 independently is H or C1-C9 alkyl which is unsubstituted or substituted by substituents independently selected from the group consisting of OH, O, N, COOR11 and halogen; C3-C6 aryl which is unsubstituted or substituted by substituents independently selected from the group consisting of OH, O, N, COOR11 and halogen; or C3-C9 aryl-alkyl which is unsubstituted or substituted by substituents independently selected from the group consisting of OH, O, N, COOR11 and halogen;
- R13 independently is C(O)—OR11; amino; amide; guanidinyl; imidazolyl; indolyl; sulfoxide; phosphoryl; C1-C3 alkylamino; C1-C3 alkyldiamino; C1-C6 alkenylamino; hydroxy; thiol; C1-C3 alkoxy; C1-C3 alkthiol; (CH2), COOR11; C1-C6 alkyl which is unsubstituted or substituted with OH, halogen, SH, NH2, phenyl, hydroxyphenyl or C7-C10 alkoxyphenyl; C2-C6 alkenyl which is unsubstituted or substituted with OH, halogen, SH, NH2, phenyl, hydroxyphenyl or C7-C10 alkoxyphenyl; and C6-C12 aryl which is unsubstituted or substituted with OH, halogen, SH, NH2, phenyl, hydroxyphenyl or C7-C10 alkoxyphenyl; and
- R14 is H or C1-C9 alkyl or C1-C9 alkyl independently substituted with OH, halogen, COOR11, O or N; C3-C6 aryl; C3-C6 aryl which is independently substituted with OH, halogen, COOR11, O or N; or C3-C6 arylalkyl which is independently substituted with OH, halogen, COOR11, O or N.
- Preferably, R11 is C1-C6 alkyl, most preferably isopropyl, R13 is the side chain of a naturally occurring amino acid, n=1, R12 is H and R14 is H. In the compound of structure (2), the invention includes metabolites in which the phenoxy and isopropyl esters have been hydrolyzed to —OH. Similarly, the de-esterified enriched phosphonoamidate metabolites of compounds (5a), 5(b) and (6) are included within the scope of this invention.
- Aryl and “0” or “N” substitution are defined in column 16, lines 42-58, of U.S. Pat. No. 5,798,340.
- Typically, the amino acids are in the natural or l amino acids. Suitable specific examples are set forth in U.S. Pat. No. 5,798,340, for instance Table 4 and col. 8-10 therein.
- Alkyl as used herein, unless stated to the contrary, is a normal, secondary, tertiary or cyclic hydrocarbon. Unless stated to the contrary alkyl is C1-C12. Examples are —CH3, —CH2CH3, —CH2CH2CH3, —CH(CH3)2, —CH2CH2CH2CH3), —CH2CH(CH3)2, —CH(CH3)CH2CH3, —C(CH3)3, —CH2CH2CH2CH2CH3, —CH(CH3)CH2CH2CH3, —CH(CH2CH3)2, —C(CH3)2CH2CH3), —CH(CH3)CH(CH3)2, —CH2CH2CH(CH3)2), —CH2CH(CH3)CH2CH3, —CH2CH2CH2CH2CH2CH3, —CH(CH3)CH2CH2CH2CH3, —CH(CH2CH3)(CH2CH2CH3), —C(CH3)2CH2CH2CH3, —CH(CH3)CH(CH3)CH2CH3, —CH(CH3)CH2CH(CH3)2, —C(CH3)(CH2CH3)2, —CH(CH2CH3)CH(CH3)2, —C(CH3)2CH(CH3)2, and —CH(CH3)C(CH3)3. Alkenyl and alkynyl are defined in the same fashion, but contain at least one double or triple bond, respectively.
- Where enol or keto groups are disclosed, the corresponding tautomers are to be construed as taught as well.
- The prodrug compounds of this invention are provided in the form of free base or the various salts enumerated in U.S. Pat. No. 5,798,340, and are formulated with pharmaceutically acceptable excipients or solvating diluents for use as pharmaceutical products also as set forth in U.S. Pat. No. 5,798,340. These prodrugs have the antiviral and utilities already established for the parent drugs (see U.S. Pat. No. 5,798,340 and other citations relating to the methoxyphosphonate nucleotide analogues). It will be understood that the diastereomer of structure (4) at least is useful as an intermediate in the chemical production of the parent drug by hydrolysis in vitro, regardless of its relatively unselective character as revealed in the studies herein.
- The invention will be more fully understood by reference to the following examples:
-

- Adenine to PMEA using Magnesium Isopropoxide. To a suspension of adenine (16.8 g, 0.124 mol) in DMF (41.9 ml) was added ethylene carbonate (12.1 g, 0.137 mol) and sodium hydroxide (0.100 g, 0.0025 mol). The mixture was heated at 130° C. overnight. The reaction was cooled to below 50° C. and toluene (62.1 ml) was added. The slurry was further cooled to 5° C. for 2 hours, filtered, and rinsed with toluene (2×). The wet solid was dried in vacuo at 65° C. to yield 20.0 g (90%) of 9-(2-hydroxyethyl)adenine as an off-white solid. Mp: 238-240° C.

- 9-(2-Hydroxyethyl)adenine (HEA) (20.0 g, 0.112 mol) was suspended in DMF (125 ml) and heated to 80° C. Magnesium isopropoxide (11.2 g, 0.0784 mol), or alternatively magnesium t-butoxide, was added to the mixture followed by diethyl p-toluenesulfonyloxymethylphosphonate (66.0 g, 0.162 mol) over one hour. The mixture was stirred at 80° C. for 7 hours. 30 ml of volatiles were removed via vacuum distillation and the reaction was recharged with 30 ml of fresh DMF. After cooling to room temperature, bromotrimethylsilane (69.6 g, 0.450 mol) was added and the mixture heated to 80° C. for 6 hours. The reaction was concentrated to yield a thick gum. The gum was dissolved into 360 ml water, extracted with 120 ml dichloromethane, adjusted to pH 3.2 with sodium hydroxide, and the resulting slurry stirred at room temperature overnight. The slurry was cooled to 4° C. for one hour. The solids were isolated by filtration, washed with water (2×), and dried in vacuo at 56° C. to yield 20 g (65.4%) of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA) as a white solid. Mp: >200° C. dec. 1H NMR (D2O) δ 3.49 (t, 2H); 3.94 (t, 2H); 4.39 (t, 2H); 8.13 (s, 1H); 8.22 (s, 1H).
-

- Adenine to PMPA using Magnesium t-Butoxide. To a suspension of adenine (40 g, 0.296 mol) in DMF (41.9 ml) was added (R)-propylene carbonate (34.5 g, 0.338 mol) and sodium hydroxide (0.480 g, 0.012 mol). The mixture was heated at 130° C. overnight. The reaction was cooled to 100° C. and toluene (138 ml) was added followed by methanesulfonic acid (4.7 g, 0.049 mol) while maintaining the reaction temperature between 100-110° C. Additional toluene (114 ml) was added to create a homogeneous solution. The solution was cooled to 3° C. over 7 hours and then held at 3° C. for one hour. The resulting solid was isolated by filtration and rinsed with acetone (2×). The wet solid was dried in vacuo at 80° C. to yield 42.6 g (75%) of (R)-9-[2-(hydroxy)propyl]adenine (HPA) as an off-white solid. Mp: 188-190° C.

- (R)-9-[2-(hydroxy)propyl]adenine (HPA) (20.0 g, 0.104 mol) was suspended in DMF (44.5 ml) and heated to 65° C. Magnesium t-butoxide (14.2 g, 0.083 mol), or alternatively magnesium isopropoxide, was added to the mixture over one hour followed by diethyl p-toluenesulfonyloxymethylphosphonate (66.0 g, 0.205 mol) over two hours while the temperature was kept at 78° C. The mixture was stirred at 75° C. for 4 hours. After cooling to below 50° C., bromotrimethylsilane (73.9 g, 0.478 mol) was added and the mixture heated to 77° C. for 3 hours. When complete, the reaction was heated to 80° C. and volatiles were removed via atmospheric distillation. The residue was dissolved into water (120 ml) at 50° C. and then extracted with ethyl acetate (101 ml). The pH of the aqueous phase was adjusted to pH 1.1 with sodium hydroxide, seeded with authentic (R)-PMPA, and the pH of the aqueous layer was readjusted to pH 2.1 with sodium hydroxide. The resulting slurry was stirred at room temperature overnight. The slurry was cooled to 4° C. for three hours. The solid was isolated by filtration, washed with water (60 ml), and dried in vacuo at 50° C. to yield 18.9 g (63.5%) of crude(R)-9-[2-(phosphonomethoxy)propyl]adenine (PMPA) as an off-white solid.
- The crude(R)-9-[2-(phosphonomethoxy)propyl]adenine was heated at reflux in water (255 ml) until all solids dissolved. The solution was cooled to room temperature over 4 hours. The resulting slurry was cooled at 4° C. for three hours. The solid was isolated by filtration, washed with water (56 ml) and acetone (56 ml), and dried in vacuo at 50° C. to yield 15.0 g (50.4%) of (R)-9-[2-(phosphonomethoxy)propyl]adenine (PMPA) as a white solid. Mp: 278-280° C.
-


- A glass-lined reactor was charged with anhydrous PMPA, (I) (14.6 kg, 50.8 mol), phenyl (9.6 kg, 102 mol), and 1-methyl-2-pyrrolidinone (39 kg). The mixture was heated to 85° C. and triethylamine (6.3 kg, 62.3 mol) added. A solution of 1,3-dicyclohexylcarbodiimide (17.1 kg, 82.9 mol) in 1-methyl-2-pyrrolidinone (1.6 kg) was then added over 6 hours at 100° C. Heating was continued for 16 hours. The reaction was cooled to 45° C., water (29 kg) added, and cooled to 25° C. Solids were removed from the reaction by filtration and rinsed with water (15.3 kg). The combined filtrate and rinse was concentrated to a tan slurry under reduced pressure, water (24.6 kg) added, and adjusted to pH=11 with NaOH (25% in water). Fines were removed by filtration through diatomaceous earth (2 kg) followed by a water (4.4 kg) rinse. The combined filtrate and rinse was extracted with ethyl acetate (28 kg). The aqueous solution was adjusted to pH=3.1 with HCl (37% in water) (4 kg). Crude II was isolated by filtration and washed with methanol (12.7 kg). The crude II wet cake was slurried in methanol (58 kg). Solids were isolated by filtration, washed with methanol (8.5 kg), and dried under reduced pressure to yield 9.33 kg II as a white powder: 1H NMR (D2O) δ 1.2 (d, 3H), 3.45 (q, 2H), 3.7 (q, 2H), 4 (m, 2H), 4.2 (q, 2H), 4.35 (dd, 2H), 6.6 (d, 2H), 7 (t, 1H), 7.15 (t, 2H), 8.15 (s, 1H), 8.2 (s, 1H); 31P NMR (D2O) δ 15.0 (decoupled).
- GS-7171 (III). (Scheme 1) A glass-lined reactor was charged with monophenyl PMPA, (II), (9.12 kg, 25.1 mol) and acetonitrile (30.7 kg). Thionyl chloride (6.57 kg, 56.7 mol) was added below 50° C. The mixture was heated at 75° C. until solids dissolved. Reaction temperature was increased to 80° C. and volatiles (11.4 kg) collected by atmospheric distillation under nitrogen. The pot residue was cooled to 25° C., dichloromethane (41 kg) added, and cooled to −29° C. A solution of (L)-alanine isopropyl ester (7.1 kg, 54.4 mol) in dichloromethane (36 kg) was added over 60 minutes at −18° C. followed by triethylamine (7.66 kg, 75.7 mol) over 30 minutes at −18 to −11° C. The reaction mixture was warmed to room temperature and washed five times with sodium dihydrogenphosphate solution (10% in water, 15.7 kg each wash). The organic solution was dried with anhydrous sodium sulfate (18.2 kg), filtered, rinsed with dichloromethane (28 kg), and concentrated to an oil under reduced pressure. Acetone (20 kg) was charged to the oil and the mixture concentrated under reduced pressure. Acetone (18.8 kg) was charged to the resulting oil. Half the product solution was purified by chromatography over a 38×38 cm bed of 22 kg silica gel 60, 230 to 400 mesh. The column was eluted with 480 kg acetone. The purification was repeated on the second half of the oil using fresh silica gel and acetone. Clean product bearing fractions were concentrated under reduced pressure to an oil. Acetonitrile (19.6 kg) was charged to the oil and the mixture concentrated under reduced pressure. Acetonitrile (66.4 kg) was charged and the solution chilled to 0 to −5° C. for 16 hours. Solids were removed by filtration and the filtrate concentrated under reduced pressure to 5.6 kg III as a dark oil: 1H NMR (CDCl3) δ 1.1 (m 12H), 3.7 (m, 1H), 4.0 (m, 5H), 4.2 (m, 1H), 5.0 (m, 1H), 6.2 (s, 2H), 7.05 (m, 5H), 8.0 (s, 1H), 8.25 (d, 1H); 31P NMR (CDCl3) δ 21.0, 22.5 (decoupled).
-

- Monophenyl PMPA (II). A round-bottom flask with reflux condenser and nitrogen inlet was placed in a 70° C. oil bath. The flask was charged with anhydrous PMPA (I) (19.2 g, 67 mmol), N,N-dimethylformamide (0.29 g, 3.3 mmol), and tetramethylene sulfone (40 mL). Thionyl chloride (14.2 g, 119 mmol) was added over 4 hours. Heating was increased to 100° C. over the same time. A homogeneous solution resulted. Phenoxytrimethylsilane (11.7 g, 70 mmol) was added to the solution over 5 minutes. Heating in the 100° C. oil bath continued for two hours more. The reaction was poured into rapidly stirring acetone (400 mL) with cooling at 0° C. Solids were isolated by filtration, dried under reduced pressure, and dissolved in methanol (75 mL). The solution pH was adjusted to 3.0 with potassium hydroxide solution (45% aq.) with cooling in ice/water. The resulting solids were isolated by filtration, rinsed with methanol, and dried under reduced pressure to 20.4 g II (Scheme 2) as a white powder.
- GS-7171 (III). Monophenyl PMPA (II) (3 g, 8.3 mmol), tetramethylene sulfone (5 mL), and N,N-dimethylformamide (1 drop) were combined in a round bottom flask in a 40° C. oil bath. Thionyl chloride (1.96 g, 16.5 mmol) was added. After 20 minutes the clear solution was removed from heat, diluted with dichloromethane (10 ml), and added to a solution of (L)-alanine isopropyl ester (5 g, 33 mmol) and diisopropylethylamine (5.33 g, 41 mmol) in dichloromethane (20 mL) at −10° C. The reaction mixture was warmed to room temperature and washed three times with sodium dihydrogenphosphate solution (10% aq., 10 mL each wash). The organic solution was dried over anhydrous sodium sulfate and concentrated under reduced pressure to a oil. The oil was combined with fumaric acid (0.77 g, 6.6 mmol) and acetonitrile (40 mL) and heated to reflux to give a homogeneous solution. The solution was cooled in an ice bath and solids isolated by filtration. The solid GS-7171 fumarate salt was dried under reduced pressure to 3.7 g. The salt (3.16 g, 5.3 mmol) was suspended in dichloromethane (30 mL) and stirred with potassium carbonate solution (5 mL, 2.5 M in water) until the solid dissolved. The organic layer was isolated, then washed with water (5 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford 2.4 g III as a tan foam.
- The diastereomers of GS-7171 (III) were resolved by batch elution chromatography using a commercially available Chiralpak AS, 20 μm, 21×250 mm semi-preparative HPLC column with a Chiralpak AS, 20 μm, 21×50 mm guard column. Chiralpak® AS is a proprietary packing material manufactured by Diacel and sold in North America by Chiral Technologies, Inc. (U.S. Pat. Nos. 5,202,433, RE 35,919, 5,434,298, 5,434,299 and 5,498,752). Chiralpak AS is a chiral stationary phase (CSP) comprised of amylosetris[(S)-α-methylbenzyl carbamate] coated onto a silica gel support.
- The GS-7171 diastereomeric mixture was dissolved in mobile phase, and approximately 1 g aliquots of GS-7171 were pumped onto the chromatographic system. The undesired diastereomer, designated GS-7339, was the first major broad (approx. 15 min. duration) peak to elute from the column. When the GS-7339 peak had finished eluting, the mobile phase was immediately switched to 100% methyl alcohol, which caused the desired diastereomer, designated GS-7340 (IV), to elute as a sharp peak from the column with the methyl alcohol solvent front. The methyl alcohol was used to reduce the over-all cycle time. After the first couple of injections, both diastereomers were collected as a single large fractions containing one of the purified diastereomers (>99.0% single diastereomer). The mobile phase solvents were removed in vacuo to yield the purified diastereomer as a friable foam.
- About 95% of the starting GS-7171 mass was recovered in the two diastereomer fractions. The GS-7340 fraction comprised about 50% of the total recovered mass.
- The chromatographic conditions were as follows:
Mobile Phase (Initial) GS-7171 - Acetonitrile:Isopropyl Alcohol (90:10) (Final) 100% Methyl Alcohol Flow 10 mL/minute Run Time About 45 minute Detection UV at 275 nm Temperature Ambient Elution Profile GS-7339 (diastereomer B) GS-7340 (diastereomer A; (IV))
Diastereomer Separation of GS-7171 by SMB Chromatography - For a general description of simulated moving bed (SMB) chromatography, see Strube et al., “Organic Process Research and Development” 2:305-319 (1998).
- GS-7340 (IV). GS-7171 (III), 2.8 kg, was purified by simulated moving bed chromatography over 10 cm by 5 cm beds of packing (Chiral Technologies Inc., 20 micron Chiralpak AS coated on silica gel) (1.2 kg). The columns were eluted with 30% methanol in acetonitrile. Product bearing fractions were concentrated to a solution of IV in acetonitrile (2.48 kg). The solution solidified to a crystalline mass wet with acetonitrile on standing. The crystalline mass was dried under reduced pressure to a tan crystalline powder, 1.301 kg IV, 98.7% diastereomeric purity: mp 117-120° C.; 1H NMR (CDCl3) δ 1.15 (m 12H), 3.7 (t, 1H), 4.0 (m, 5H), 4.2 (dd, 1H), 5.0 (m, 1H), 6.05 (s, 2H), 7.1 (m, 5H), 8.0 (s, 1H), 8.2 (s, 1H); 31P NMR (CDCl3) δ 21.0 (decoupled).
- Diastereomer Separation by C18 RP-HPLC
- GS-7171 (III) was chromatographed by reverse phase HPLC to separate the diastereomers using the following summary protocol.
Chromatographic Phenomenex Luna ™ C18(2), 5 μm, 100 Å column: pore size, (Phenomenex, Torrance, CA), or equivalent Guard column: Pellicular C18 (Alltech, Deerfield, IL), or equivalent Mobile Phase: A - 0.02% (85%) H3PO4 in water:acetonitrile (95:5) B - 0.02% (85%) H3PO4 in water:acetonitrile (50:50) Mobile Phase Gradient: % Mobile Phase % Mobile Phase Time A B 0 100 0 5 100 0 7 70 30 32 70 30 40 0 100 50 0 100 Run Time: 50 minutes Equilibration Delay: 10 min at 100% mobile phase A Flow Rate: 1.2 mL/min Temperature: Ambient Detection: UV at 260 nm Sample Solution: 20 mM sodium phosphate buffer, pH 6 Retention Times: GS-7339, about 25 minutes GS-7340, about 27 minutes
Diastereomer Separation by Crystallization - GS-7340 (IV). A solution of GS-7171 (III) in acetonitrile was concentrated to an amber foam (14.9 g) under reduced pressure. The foam was dissolved in acetonitrile (20 mL) and seeded with a crystal of IV. The mixture was stirred overnight, cooled to 5° C., and solids isolated by filtration. The solids were dried to 2.3 g IV as white crystals, 98% diastereomeric purity (31P NMR): 1H NMR (CDCl3) δ 1.15 (m 12H), 3.7 (t, 1H), 3.95 (m, 2H), 4.05 (m, 2H), 4.2 (m, 2H), 5.0 (m, 1H), 6.4 (s, 2H), 7.1 (m, 5H), 8.0 (s, 1H), 8.2 (s, 1H); 31P NMR (CDCl3) δ 19.5 (decoupled). X-ray crystal analysis of a single crystal selected from this product yielded the following data:
Crystal Color, Habit colorless, column Crystal Dimensions 0.25 × 0.12 × 0.08 mm Crystal System orthorhombic Lattice Type Primitive Lattice Parameters a = 8.352(1) Å b = 15.574(2) Å c = 18.253(2) Å V = 2374.2(5) Å3 Space Group P212121 (#19) Z value 4 Dcalc 1.333 g/cm3 F000 1008.00 μ(MoKα) 1.60 cm−1 - GS-7340-02 (V). (Scheme 1) A glass-lined reactor was charged with GS-7340 (IV), (1.294 kg, 2.71 mol), fumaric acid (284 g, 2.44 mol), and acetonitrile (24.6 kg). The mixture was heated to reflux to dissolve the solids, filtered while hot and cooled to 5° C. for 16 hours. The product was isolated by filtration, rinsed with acetonitrile (9.2 kg), and dried to 1329 g (V) as a white powder: mp 119.7-121.1° C.; [α]D 20 −41.7° (c 1.0, acetic acid).
-

- A 5 L round bottom flask was charged with monophenyl PMPA, (II), (200 g, 0.55 mol) and acetonitrile (0.629 kg). Thionyl chloride (0.144 kg, 1.21 mol) was added below 27° C. The mixture was heated at 70° C. until solids dissolved. Volatiles (0.45 L) were removed by atmospheric distillation under nitrogen. The pot residue was cooled to 25° C., dichloromethane (1.6 kg) was added and the mixture was cooled to −20° C. A solution of (L)-α aminobutyric acid ethyl ester (0.144 kg, 1.1 mol) in dichloromethane (1.33 kg) was added over 18 minutes at −20 to −10° C. followed by triethylamine (0.17 kg, 1.65 mol) over 15 minutes at −8 to −15° C. The reaction mixture was warmed to room temperature and washed four times with sodium dihydrogenphosphate solution (10% aq., 0.3 L each wash). The organic solution was dried with anhydrous sodium sulfate (0.5 kg) and filtered. The solids were rinsed with dichloromethane (0.6 kg) and the combined filtrate and rinse was concentrated to an oil under reduced pressure. The oil was purified by chromatography over a 15×13 cm bed of 1.2 kg silica gel 60, 230 to 400 mesh. The column was eluted with a gradient of dichloromethane and methanol. Product bearing fractions were concentrated under reduced pressure to afford 211 g VI (Scheme 3) as a tan foam.
- The diastereomeric mixture was purified using the conditions described for GS-7171 in Example 3A except for the following:
Mobile Phase (Initial) GS-7120 - Acetonitrile:Isopropyl Alcohol (98:2) (Final) 100% Methyl Alcohol Elution Profile: GS-7341 (diastereomer B) GS-7342 (diastereomer A) - A 1 L round bottom flask was charged with monophenyl PMPA, (II), (50 g, 0.137 mol) and acetonitrile (0.2 L). Thionyl chloride (0.036 kg, 0.303 mol) was added with a 10° C. exotherm. The mixture was heated to reflux until solids dissolved. Volatiles (0.1 L) were removed by atmospheric distillation under nitrogen. The pot residue was cooled to 25° C., dichloromethane (0.2 kg) was added, and the mixture was cooled to −20° C. A solution of (L)-α aminobutyric acid ethyl ester (0.036 kg, 0.275 mol) in dichloromethane (0.67 kg) was added over 30 minutes at −20 to −8° C. followed by triethylamine (0.042 kg, 0.41 mol) over 10 minutes at up to −6° C. The reaction mixture was warmed to room temperature and washed four times with sodium dihydrogenphosphate solution (10% aq., 0.075 L each wash). The organic solution was dried with anhydrous sodium sulfate (0.1 kg) and filtered. The solids were rinsed with ethyl acetate (0.25 L, and the combined filtrate and rinse was concentrated to an oil under reduced pressure. The oil was diluted with ethyl acetate (0.25 L), seeded, stirred overnight, and chilled to −15° C. The solids were isolated by filtration and dried under reduced pressure to afford 17.7 g of GS-7342 (Table 5) as a tan powder: 1H NMR (CDCl3) δ 0.95 (t, 3H), 1.3 (m, 6H), 1.7, (m, 2H), 3.7 (m, 2H), 4.1 (m, 6H), 4.4 (dd, 1H), 5.8 (s, 2H), 7.1 (m, 5H), 8.0 (s, 1H), 8.4 (s, 1H); 31P NMR (CDCl3) δ 21 (decoupled).
- The diastereomeric mixture was purified using the conditions described for GS-7171 (Example 3A) except for the following:
Mobile Phase (Initial) GS-7120 - Acetonitrile:Isopropyl Alcohol (95:5) (Final) 100% Methyl Alcohol Elution Profile GS-7115 (diastereomer B) GS-7114 (diastereomer A) - GS-7097: Phenyl PMPA, Ethyl L-Alanyl Amidate. Phenyl PMPA (15.0 g, 41.3 mmol), L-alanine ethyl ester hydrochloride (12.6 g, 83 mmol) and triethylamine (11.5 mL, 83 mmol) were slurried together in 500 mL pyridine under dry N2. This suspension was combined with a solution of triphenylphosphine (37.9 g, 145 mmol), Aldrithiol 2 (2,2′-dipyridyl disulfide) (31.8 g, 145 mmol), and 120 mL pyridine. The mixture was heated at an internal temperature of 57° C. for 15 hours. The complete reaction was concentrated under vacuum to a yellow paste, 100 g. The paste was purified by column chromatography over a 25×11 cm bed of 1.1 kg silica gel 60, 230 to 400 mesh. The column was eluted with 8 liters of 2% methanol in dichloromethane followed by a linear gradient over a course of 26 liters eluent up to a final composition of 13% methanol. Clean product bearing fractions were concentrated to yield 12.4 g crude (5), 65% theory. This material was contaminated with about 15% (weight) triethylamine hydrochloride by 1H NMR. The contamination was removed by dissolving the product in 350 mL ethyl acetate, extracting with 20 mL water, drying the organic solution over anhydrous sodium sulfate, and concentrating to yield 11.1 g pure GS-7097 as a white solid, 58% yield. The process also is employed to synthesize the diastereomeric mixture of GS-7003a and GS-7003b (the phenylalanyl amidate) and the mixture GS-7119 and GS-7335 (the glycyl amidate). These diastereomers are separated using a batch elution procedure such as shown in Example 3A, 6 and 7.
- The in vitro anti-HIV-1 activity and cytotoxicity in MT-2 cells and stability in human plasma and MT-2 cell extracts of GS-7340 (freebase) and tenofovir disoproxil fumarate (TDF), are shown in Table 1. GS-7340 shows a 10-fold increase in antiviral activity relative to TDF and a 200-fold increase in plasma stability. This greater plasma stability is expected to result in higher circulating levels of GS-7340 than TDF after oral administration.
TABLE 1 In Vitro Activity and Stability Stability T ½ (min) HIV-1 MT-2 Activity Cytotoxicity Human Cell IC50 μM CC50 μM Plasma Extract (P/MT-2) GS 73400.005 >40 90.0 28.3 3.2 TDF 0.05 70 0.41 70.7 0.006 Tenofovir 5 6000 — — — - In order to estimate the relative intracellular PMPA resulting from the intracellular metabolism of TDF as compared to that from GS-7340, both prodrugs and PMPA were radiolabeled and spiked into intact human whole blood at equimolar concentrations. After 1 hour, plasma, red blood cells (RBCs) and peripheral blood mononuclear cells (PBMCs) were isolated and analyzed by HPLC with radiometric detection. The results are shown in Table 2.
- After 1 hour, GS-7340 results in 10× and 30× the total intracellular concentration of PMPA species in PBMCs as compared to TDF and PMPA, respectively. In plasma after 1 hour, 84% of the radioactivity is due to intact GS-7340, whereas no TDF is detected at 1 hour. Since no intact TDF is detected in plasma, the 10× difference at 1 hour between TDF and GS-7340 is the minimum difference expected in vivo. The HPLC chromatogram for all three compounds in PBMCs is shown in
FIG. 1 .TABLE 2 PMPA Metabolites in Plasma, PBMCs and RBCs After 1 h Incubation of PMPA Prodrugs or PMPA in Human Blood. Total Metabolites (% of Total Peak Area) C-14 Recovered, PMPA PMPAp, PMPApp, Met. X, Met. Y, GS 7340,Compound Matrix μg-eq % % % % % % GS-7340 Plasma/ 43.0 1 — — 2 13 84 FP (60 μg-eq) PBMC 1.25 45 16 21 18 — — RBC/FP 12.6 8 — — 24 11 57 Mono- PMPA PMPAp PMPApp POC GS-4331 GS-4331 Plasma/ 48.1 11 — — 89 — (TDF) FP (60 μg-eq) PBMC 0.133 50 25 18 7 — RBC/FP 10.5 93 7.0 — — — PMPA PMPAp PMPApp PMPA Plasma/ 55.7 100 — — FP (60 μg-eq) PBMC 0.033 86 14 — RBC/FP 3.72 74 10 16 - Met. X and Met Y (metabolites X and Y) are shown in Table 5. Lower case “p” designates phosphorylation. These results were obtained after 1 hour in human blood. With increasing time, the in vitro differences are expected to increase, since 84% of GS-7340 is still 10 intact in plasma after one hour. Because intact GS-7340 is present in plasma after oral administration, the relative clinical efficacy should be related to the IC50 values seen in vitro.
- In Table 3 below, IC50 values of tenofovir, TDF, GS-7340, several nucleosides and the protease inhibitor nelfinivir are listed. As shown, nelfinavir and GS-7340 are 2-3 orders of magnitude more potent than all other nucleotides or nucleosides.
TABLE 3 In Vitro Anti-HIV-1 Activities of Antiretroviral Compounds Compound IC50 (μM) Adefovir (PMEA) 13.4 ± 4.21 Tenofovir (PMPA) 6.3 ± 3.31 AZT 0.17 ± 0.081 3TC 1.8 ± 0.251 d4T 8 ± 2.51 Nelfinavir 0.006 ± 0.0021 TDF 0.05 GS 73400.005
1A. S. Mulato and J. M. Cherrington, Antiviral Research 36, 91 (1997)
- Additional studies of the in vitro cell culture anti-HIV-1 activity and CC50 of separated diastereomers of this invention were conducted and the results tabulated below.
TABLE 4 Effect of Diastereomer Diastereomer Fold A/B CC50 Compound residue IC50 (μM) change activity (μM) PMPA — 5 1× — 6000 Ala-methylester Mixture 1:1 0.025 200× 20× 80 GS-6957a A 0.0075 670× GS-6957b 0.15 33× Phe-methylester Mixture 1:1 0.03 170× 10× 60 GS-7003a A 0.01 500× GS-7003b B 0.1 50×× Gly-ethylester Mixture 1:1 0.5 10× 20× GS-7119 A 0.05 100× >100 GS-7335 B 1.0 5× Ala-isopropyl Mixture 1:1 0.01 500× 12× GS-7340 A 0.005 1,000× 40 GS-7339 B 0.06 83× >100 ABA-ethyl Mixture 1:1 0.008 625× 7.5× >100 GS-7342 A 0.004 1,250× GS-7341 B 0.03 170× Ala-ethyl Mixture 1:1 0.02 250× 10× 60 GS-7114 A 0.005 1,000× GS-7115 B 0.05 100×× - Assay reference: Arimilli, M N, et al., (1997) Synthesis, in vitro biological evaluation and oral bioavailability of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA) prodrugs. Antiviral Chemistry and Chemotherapy 8(6):557-564.
- “Phe-methylester” is the methylphenylalaninyl monoamidate, phenyl monoester of tenofovir; “gly-methylester” is the methylglycyl monoamidate, phenyl monoester of tenofovir.
- In each instance above, isomer A is believed to have the same absolute stereochemistry as GS-7340 (S), and isomer B is believed to have the same absolute stereochemistry that of GS-7339.
- The in vitro metabolism and stability of separated diastereomers were determined in PLCE, MT-2 extract and human plasma. A biological sample listed below, 80 μL, was transferred into a screw-capped centrifuge tube and incubated at 37° C. for 5 min. A solution containing 0.2 mg/mL of the test compound in a suitable buffer, 20 μL, was added to the biological sample and mixed. The reaction mixture, 20 mL, was immediately sampled and mixed with 60 μL of methanol containing 0.015 mg/mL of 2-hydroxymethylnaphthalene as an internal standard for HPLC analysis. The sample was taken as the time-zero sample. Then, at specific time points, the reaction mixture, 20 μL, was sampled and mixed with 60 μL of methanol containing the internal standard. The mixture thus obtained was centrifuged at 15,000 G for 5 min and the supernatant was analyzed with HPLC under the conditions described below.
- The biological samples evaluated are as follows.
- (1) PLCE (porcine liver carboxyesterase from Sigma, 160 u/mg protein, 21 mg protein/mL) diluted 20 fold with PBS (phosphated-buffered saline).
- (2) MT-2 cell extract was prepared from MT-2 cells according to the published procedure [A. Pompon, I. Lefebvre, J.-L. Imbach, S. Kahn, and D. Farquhar, “Antiviral Chemistry & Chemotherapy”, 5:91-98 (1994)] except for using HEPES buffer described below as the medium.
- (3) Human plasma (pooled normal human plasma from George King Biomedical Systems, Inc.)
- The buffer systems used in the studies are as follows.
- In the study for PLCE, the test compound was dissolved in PBS. PBS (phosphate-buffered saline, Sigma) contains 0.01 M phosphate, 0.0027 M potassium chloride, and 0.137 M sodium chloride. pH 7.4 at 37° C.
- In the study for MT-2 cell extracts, the test compound was dissolved in HEPES buffer. HEPES buffer contains 0.010 M HEPES, 0.05 M potassium chloride, 0.005 M magnesium chloride, and 0.005 M dl-dithiothreitol. pH 7.4 at 37° C.
- In the study for human plasma, the test compound was dissolved in TBS. TBS (tris-buffered saline, Sigma) contains 0.05 M Tris, 0.0027 M potassium chloride, and 0.138 M sodium chloride. pH 7.5 at 37° C.
- The HPLC analysis was carried out under the following conditions.
Column: Zorbax Rx-C8, 4.6 × 250 mm, 5μ (MAC-MOD Analytical, Inc. Chadds Ford, PA) Detection: UV at 260 nm Flow Rate: 1.0 mL/min Run Time: 30 min Injection Volume: 20 μL Column Temperature: Ambient temperature Mobile Phase A: 50 mM potassium phosphate (pH 6.0)/CH3CN = 95/5 (v/v) Mobile Phase B: 50 mM Potassium phosphate (pH 6.0)/CH3CN = 50/50 (v/v) Gradient Run: 0 min 100% Mobile Phase A 25 min 100% Mobile Phase B 30 min 100% Mobile Phase B - The results are shown below in Table 5 (also including selected IC50 data from Table 4).
TABLE 5 In Vitro Metabolism of Isomers A and B of PMPA monoamidate at 37° C. Human HIV MT-2 extract Plasma PMPA monoamidate IC50 PLCE hydrolysis hydrolysis rate Stability No. structure (□M) rate and product and product (HP) 1 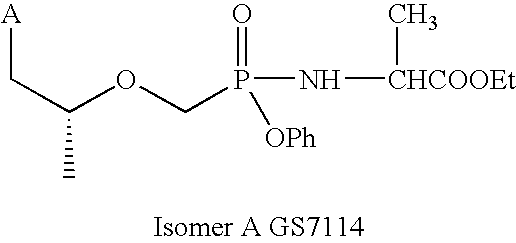
0.005 t½ = 2.9 min Met. X & PMPA t½ = 2.9 min Met. X & PMPA t½ = 148 min Met. Y 2 
0.05 t½ = 8.0 min Met. X & PMPA t½ = 150.6 min Met. X & PMPA t½ = 495 min Met. Y 3 
0.005 t½ = 3.3 min Met. X & PMPA t½ = 28.3 min Met. X & PMPA t½ = 90.0 min Met. Y 4 
0.06 t½ = 10.1 min Met. X & PMPA t½ > 1000 min t½ = 231 min Met. Y 5 
0.004 t½ = 3.9 min Met. X t½ = 49.2 min Met. X & PMPA t½ = 103 min Met. Y 6 
0.03 t½ = 11.3 min Met. X t½ > 1000 min t½ = 257 min Met. Y 7 
0.05 t½ < 0.14 min MonoPOC PMPA t½ = 70.7 min monoPOC PMPA t½ = 0.41 min monoPOC PMPA Met. X: 
Met. Y: 
A = 
- The pharmacokinetics of
GS 7340 were studied in dogs after oral administration of a 10 mg-eq/kg dose. - Formulations. The prodrugs were formulated as solutions in 50 mM citric acid within 0.5 hour prior to dose. All compounds used in the studies were synthesized by Gilead Sciences. The following lots were used:
Amidate Amino Diastereo- GSI acid AA Ester isomer Lot Number GS-7340-2 Alanine i-Propyl Isomer A 1504-187-19 GS-7339 Alanine i-Propyl Isomer B 1509-185-31 GS7114 Alanine Ethyl Isomer A 1509-181-26 GS7115 Alanine Ethyl Isomer B 1509-181-22 GS7119 Glycine Ethyl Isomer A 1428-163-28 GS7342 □-Aminobutyric Ethyl Isomer A 1509-191-12 Acid GS7341 □-Aminobutyric Ethyl Isomer B 1509-191-7 Acid - Dose Administration and Sample Collection. The in-life phase of this study was conducted in accordance with the recommendations of the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health publication 86-23) and was approved by an Institutional Animal Care and Use Committee. Fasted male beagle dogs (10±2 kg) were used for the studies. Each drug was administered as a single dose by oral gavage (1.5-2 ml/kg). The dose was 10 mg-equivalent of PMPA/kg. For PBMCs, blood samples were collected at 0 (pre-dose), 2, 8, and 24 h post-dose. For plasma, blood samples were collected at 0 (pre-dose), 5, 15, and 30 min, and 1, 2, 3, 4, 6, 8, 12 and 24 h post-dose. Blood (1.0 ml) was processed immediately for plasma by centrifugation at 2,000 rpm for 10 min. Plasma samples were frozen and maintained at 70° C. until analyzed.
- Peripheral Blood Mononuclear Cell (PBMC) preparation. Whole blood (8 ml) drawn at specified time points was mixed in equal proportion with phosphate buffered saline (PBS), layered onto 15 ml of Ficoll-Paque solution (Pharmacia Biotech) and centrifuged at 400×g for 40 min. PBMC layer was removed and washed once with PBS. Formed PMBC pellet was reconstituted in 0.5 ml of PBS, cells were resuspended, counted using hemocytometer and maintained at 70° C. until analyzed. The number of cells multiplied by the mean single-cell volume was used in calculation of intracellular concentrations. A reported value of 200 femtoliters/cell was used as the resting PBMC volume (B. L. Robins, R. V. Srinivas, C. Kim, N. Bischofberger, and A. Fridland, Antimicrob. Agents Chemother. 42, 612 (1998).
- Determination of PMPA and Prodrugs in plasma and PBMCs. The concentration of PMPA in dog plasma samples was determined by derivatizing PMPA with chloroacetaldehyde to yield a highly fluorescent N1, N6-ethenoadenine derivative (L. Naesens, J. Balzarini, and E. De Clercq, Clin. Chem. 38, 480 (1992). Briefly, plasma (100 μl) was mixed with 200 μl acetonitrile to precipitate protein. Samples were then evaporated to dryness under reduced pressure at room temperature. Dried samples were reconstituted in 200 μl derivatization cocktail (0.34% chloroacetaldehyde in 100 mM sodium acetate, pH 4.5), vortexed, and centrifuged. Supernatant was then transferred to a clean screw-cap tube and incubated at 95° C. for 40 min. Derivatized samples were then evaporated to dryness and reconstituted in 100 μl of water for HPLC analysis.
- Before intracellular PMPA could be determined by HPLC, the large amounts of adenine related ribonucleotides present in the PBMC extracts had to be removed by selective oxidation. We used a modified procedure of Tanaka et al (K. Tanaka, A. Yoshioka, S. Tanaka, and Y. Wataya, Anal. Biochem., 139, 35 (1984). Briefly, PBMC samples were mixed 1:2 with methanol and evaporated to dryness under reduced pressure. The dried samples were derivatized as described in the plasma assay. The derivatized samples were mixed with 20 μL of 1M rhamnose and 30 μL of 0.1M sodium periodate and incubated at 37° C. for 5 min. Following incubation, 40 μL of 4M methylamine and 20 μL of 0.5M inosine were added. After incubation at 37° C. for 30 min, samples were evaporated to dryness under reduced pressure and reconstituted in water for HPLC analysis.
- No intact prodrug was detected in any PBMC samples. For plasma samples potentially containing intact prodrugs, experiments were performed to verify that no further conversion to PMPA occurred during derivatization. Prodrug standards were added to drug-free plasma and derivatized as described. There were no detectable levels of PMPA present in any of the plasma samples, and the projected % of conversion was less than 1%.
- The HPLC system was comprised of a P4000 solvent delivery system with AS3000 autoinjector and F2000 fluorescence detector (Thermo Separation, San Jose, Calif.). The column was an Inertsil ODS-2 column (4.6×150 mm). The mobile phases used were: A, 5% acetonitrile in 25 mM potassium phosphate buffer with 5 mM tetrabutyl ammonium bromide (TBABr), pH 6.0; B, 60% acetonitrile in 25 mM potassium phosphate buffer with 5 mM TBABr, pH 6.0. The flow rate was 2 ml/min and the column temperature was maintained at 35° C. by a column oven. The gradient profile was 90% A/10% B for 10 min for PMPA and 65% A/35% B for 10 min for the prodrug. Detection was by fluorescence with excitation at 236 nm and emission at 420 nm, and the injection volume was 10 μl. Data was acquired and stored by a laboratory data acquisition system (PeakPro, Beckman, Allendale, N.J.).
- Pharmacokinetic Calculations. PMPA and prodrug exposures were expressed as areas under concentration curves in plasma or PBMC from zero to 24 hours (AUC). The AUC values were calculated using the trapezoidal rule.
- Plasma and PBMC Concentrations. The results of this study is shown in
FIGS. 2 and 3 .FIG. 2 shows the time course of GS 7340-2 metabolism summary of plasma and PBMC exposures following oral administration of pure diastereoisomers of the PMPA prodrugs. - The bar graph in
FIG. 2 shows the AUC (0-24 h) for tenofovir in dog PBMCs and plasma after administration of PMPA s.c., TDF and amidate ester prodrugs. All of the amidate prodrugs exhibited increases in PBMC exposure. For example,GS 7340 results in a ˜21-fold increase in PBMC exposure as compared to PMPA s.c. and TDF; and a 6.25-fold and 1.29-fold decrease in plasma exposure, respectively. - These data establish in vivo that
GS 7340 can be delivered orally, minimizes systemic exposure to PMPA and greatly enhances the intracellular concentration of PMPA in the cells primarily responsible for HIV replication.TABLE 6 PMPA Exposure in PBMC and Plasma from Oral Prodrugs of PMPA in Dogs PBMC/Plasma PMPA AUC in Plasma PMPA AUC in PBMC Prodrug Exposure GS# Moiety Mean StDev N Mean StDev N in Plasma Ratio GS-7114 Mono-Ala-Et-A 5.8 0.9 2 706 331 5 YES 122 GS-7115 Mono-Ala-Et-B 6.6 1.5 2 284 94 5 YES 43 GS-7340-2 Mono-Ala-iPr-A 5.0 1.1 5 805 222 5 YES 161 GS-7339 Mono-Ala-iPr-A 6.4 1.3 2 200 57 5 YES 31 GS-7119 Mono-Gly-Et-A 6.11 1.86 2 530 304 5 YES 87 GS-7342 Mono-ABA-Et-A 4.6 1.2 2 1060 511 5 YES 230 GS7341 Mono-ABA-Et-B 5.8 1.4 2 199 86 5 YES 34 - As part of the preclinical characterization of GS-7340, its biodistribution in dogs was determined. The tissue distribution of GS-7340 (isopropyl alaninyl monoamidate, phenyl monoester of tenofovir) was examined following oral administration to beagle dogs. Two male animals were dosed orally with 14C=GS-7340 (8.85 mg-equiv. of PMPA/kg, 33.2 μCi/kg; the 8-carbon of adenine is labeled) in an aqueous solution (50 mM citric acid, pH 2.2). Plasma and peripheral blood mononuclear cells (PBMCs) were obtained over the 24-hr period. Urine and feces were cage collected over 24 hr. At 24 h after the dose, the animals were sacrificed and tissues removed for analysis. Total radioactivity in tissues was determined by oxidation and liquid scintillation counting.
- The biodistribution of PMPA after 24 hours after a single oral dose of
radiolabelled GS 7340 is shown in Table 4 along with the data from a previous study with TDF (GS-4331). In the case of TDF, the prodrug concentration in the plasma is below the level of assay detection, and the main species observed in plasma is the parent drug. Levels of PMPA in the lymphatic tissues, bone marrow, and skeletal muscle are increased 10-fold after administration of GS-7340. - Accumulation in lymphatic tissues is consistent with the data observed from the PBMC analyses, since these tissues are composed primarily of lymphocytes. Likewise, accumulation in bone marrow is probably due to the high percentage of lymphocytes (70%) in this tissue.
TABLE 7 Excretion and Tissue Distribution of Radiolabelled GS-7340 in Dogs (Mean, N = 2) Following an Oral Dose at 10 mg-eq. PMPA/kg. Tissue Conc. GS-4331 GS-7340 Ratio of GS Conc. Conc. 7340 Tissue/Fluid % Dose (ug-eq/g) % Dose (ug-eq/g) to GS-4331 Liver 12.40 38.30 16.45 52.94 1.4 Kidney 4.58 87.90 3.78 80.21 0.9 Lungs 0.03 0.53 0.34 4.33 8.2 Iliac Lymph Nodes 0.00 0.51 0.01 5.42 10.6 Axillary Lymph Nodes 0.00 0.37 0.01 5.54 14.8 Inguinal Lymph Nodes 0.00 0.28 0.00 4.12 15.0 Mesenteric Lymph 0.00 1.20 0.04 6.88 5.7 Thyroid Gland 0.00 0.30 0.00 4.78 15.8 Pituitary Gland 0.00 0.23 0.00 1.80 7.8 Salivary Gland (L + R) 0.00 0.45 0.03 5.54 12.3 Adrenal Gland 0.00 1.90 0.00 3.47 1.8 Spleen 0.00 0.63 0.17 8.13 12.8 Pancreas 0.00 0.57 0.01 3.51 6.2 Prostate 0.00 0.23 0.00 2.14 9.1 Testes (L + R) 0.02 1.95 0.02 2.01 1.0 Skeletal Muscle 0.00 0.11 0.01 1.12 10.1 Heart 0.03 0.46 0.15 1.97 4.3 Femoral Bone 0.00 0.08 0.00 0.28 3.5 Bone Marrow 0.00 0.20 0.00 2.05 10.2 Skin 0.00 0.13 0.00 0.95 7.2 Abdominal fat 0.00 0.16 0.00 0.90 5.8 Eye (L + R) 0.00 0.06 0.00 0.23 3.7 Brain 0.00 <LOD 0.00 <LOD n.d. Cerebrospinal Fluid 0.00 <LOD 0.00 0.00 n.d. Spinal Cord 0.00 <LOD 0.00 0.04 n.d. Stomach 0.11 1.92 0.26 2.68 1.4 Jejunum 1.34 3.01 0.79 4.16 1.4 Duodenum 0.49 4.96 0.44 8.77 1.8 Ileum 0.01 0.50 0.16 4.61 9.2 Large Intestine 1.63 5.97 2.65 47.20 7.9 Gall bladder 0.00 3.58 0.04 25.02 7.0 Bile 0.00 9.63 0.22 40.48 4.2 Feces 40.96 n.d. 0.19 n.d. n.a. Total GI Tract Contents 5.61 n.d. 21.64 n.d. n.a. Urine 23.72 n.d. 14.73 n.d. n.a. Plasma at 24 h 0.00 0.20 0.00 0.20 1.0 Plasma at 0.25 h n.a. 3.68 n.a. 3.48 0.9 PBMC* 0.00 n.d. 0.00 63.20 n.d. Whole Blood 0.00 0.85 0.16 0.20 0.2 Total Recovery 81.10 68.96
Calculated using typical recovery of 15 × 106 cells total, and mean PBMC volume of 0.2 picoliters/cell
n.s. = no sample,
n.a. = not applicable,
n.d. = not determined.
Claims (4)
1. A fumarate salt of a nucleotide analog compound according to structural formula (Ia):

that comprises at least about 60% by weight a diastereomer according to structure (Ib):

2. The fumarate salt of claim 1 in which the nucleotide analog compound comprises at least about 80% by weight the diastereomer of structure (Ib).
3. The fumarate salt of claim 1 in which the nucleotide analog compound comprises at least about 95% by weight the diastereomer of structure (Ib).
4. The fumarate salt of claim 1 in which the nucleotide analog compound comprises at least about 99% by wieight the diastereomer of structure (Ib).
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/031,250 US20050124583A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22002100P | 2000-07-21 | 2000-07-21 | |
| US09/909,560 US20020119443A1 (en) | 2000-07-21 | 2001-07-20 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US10/354,207 US20030219727A1 (en) | 2000-07-21 | 2003-01-28 | Prodrugs of phosphonate nucleotide analogues |
| US10/798,692 US7390791B2 (en) | 2000-07-21 | 2004-03-11 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,250 US20050124583A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/798,692 Continuation US7390791B2 (en) | 2000-07-21 | 2004-03-11 | Prodrugs of phosphonate nucleotide analogues |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20050124583A1 true US20050124583A1 (en) | 2005-06-09 |
Family
ID=22821718
Family Applications (10)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/333,107 Abandoned US20040018150A1 (en) | 2000-07-21 | 2001-07-20 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US09/909,560 Abandoned US20020119443A1 (en) | 2000-07-21 | 2001-07-20 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US10/354,207 Abandoned US20030219727A1 (en) | 2000-07-21 | 2003-01-28 | Prodrugs of phosphonate nucleotide analogues |
| US10/785,497 Abandoned US20060024659A1 (en) | 2000-07-21 | 2004-02-24 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US10/798,692 Active 2025-04-17 US7390791B2 (en) | 2000-07-21 | 2004-03-11 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,252 Abandoned US20050124585A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,250 Abandoned US20050124583A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,251 Abandoned US20050124584A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,228 Abandoned US20050159392A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
| US12/110,829 Expired - Fee Related US7803788B2 (en) | 2000-07-21 | 2008-04-28 | Prodrugs of phosphonate nucoleotide analogues |
Family Applications Before (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/333,107 Abandoned US20040018150A1 (en) | 2000-07-21 | 2001-07-20 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US09/909,560 Abandoned US20020119443A1 (en) | 2000-07-21 | 2001-07-20 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US10/354,207 Abandoned US20030219727A1 (en) | 2000-07-21 | 2003-01-28 | Prodrugs of phosphonate nucleotide analogues |
| US10/785,497 Abandoned US20060024659A1 (en) | 2000-07-21 | 2004-02-24 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US10/798,692 Active 2025-04-17 US7390791B2 (en) | 2000-07-21 | 2004-03-11 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,252 Abandoned US20050124585A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/031,251 Abandoned US20050124584A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
| US11/031,228 Abandoned US20050159392A1 (en) | 2000-07-21 | 2005-01-06 | Prodrugs of phosphonate nucleotide analogues |
| US12/110,829 Expired - Fee Related US7803788B2 (en) | 2000-07-21 | 2008-04-28 | Prodrugs of phosphonate nucoleotide analogues |
Country Status (37)
| Country | Link |
|---|---|
| US (10) | US20040018150A1 (en) |
| EP (3) | EP2682397B1 (en) |
| JP (4) | JP4651264B2 (en) |
| KR (2) | KR100749160B1 (en) |
| CN (2) | CN1291994C (en) |
| AP (1) | AP1466A (en) |
| AU (3) | AU2001282941C1 (en) |
| BE (1) | BE2016C018I2 (en) |
| BG (1) | BG66037B1 (en) |
| BR (1) | BRPI0112646B8 (en) |
| CA (3) | CA2416757C (en) |
| CY (2) | CY2016008I1 (en) |
| CZ (2) | CZ304886B6 (en) |
| DK (2) | DK1301519T4 (en) |
| EA (1) | EA004926B1 (en) |
| EE (1) | EE05366B1 (en) |
| ES (2) | ES2627903T3 (en) |
| FR (1) | FR16C0013I2 (en) |
| HK (2) | HK1054238A1 (en) |
| HR (2) | HRP20160074B1 (en) |
| HU (2) | HU230960B1 (en) |
| IL (1) | IL153658A0 (en) |
| IS (1) | IS2985B (en) |
| LT (2) | LT2682397T (en) |
| LU (1) | LU93029I2 (en) |
| MX (1) | MXPA03000587A (en) |
| NL (1) | NL300803I2 (en) |
| NO (6) | NO336718B1 (en) |
| NZ (3) | NZ535408A (en) |
| OA (1) | OA12393A (en) |
| PL (1) | PL213214B1 (en) |
| PT (2) | PT1301519E (en) |
| SI (2) | SI2682397T1 (en) |
| TR (1) | TR200300055T2 (en) |
| UA (1) | UA75889C2 (en) |
| WO (1) | WO2002008241A2 (en) |
| ZA (1) | ZA200210271B (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8411105B1 (en) | 2004-05-14 | 2013-04-02 | Nvidia Corporation | Method and system for computing pixel parameters |
| US8416242B1 (en) | 2004-05-14 | 2013-04-09 | Nvidia Corporation | Method and system for interpolating level-of-detail in graphics processors |
| US8432394B1 (en) | 2004-05-14 | 2013-04-30 | Nvidia Corporation | Method and system for implementing clamped z value interpolation in a raster stage of a graphics pipeline |
| US8441497B1 (en) * | 2007-08-07 | 2013-05-14 | Nvidia Corporation | Interpolation of vertex attributes in a graphics processor |
| US8749576B2 (en) | 2004-05-14 | 2014-06-10 | Nvidia Corporation | Method and system for implementing multiple high precision and low precision interpolators for a graphics pipeline |
| WO2016205141A1 (en) * | 2015-06-17 | 2016-12-22 | Gilead Sciences, Inc. | Co-crystals, salts and solid forms of tenofovir alafenamide |
| US10548846B2 (en) | 2015-11-09 | 2020-02-04 | Gilead Sciences, Inc. | Therapeutic compositions for treatment of human immunodeficiency virus |
Families Citing this family (215)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2001282941C1 (en) * | 2000-07-21 | 2016-12-22 | Gilead Sciences, Inc. | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| CA2466301A1 (en) * | 2001-11-14 | 2003-10-23 | Biocryst Pharmaceuticals, Inc. | Nucleosides preparation thereof and use as inhibitors of rna viral polymerases |
| US7388002B2 (en) * | 2001-11-14 | 2008-06-17 | Biocryst Pharmaceuticals, Inc. | Nucleosides, preparation thereof and use as inhibitors of RNA viral polymerases |
| BR0309557A (en) * | 2002-04-26 | 2005-03-01 | Gilead Sciences Inc | Non-Nucleoside Reverse Transcriptase Inhibitors |
| US20050239054A1 (en) * | 2002-04-26 | 2005-10-27 | Arimilli Murty N | Method and compositions for identifying anti-HIV therapeutic compounds |
| JP4476811B2 (en) | 2002-05-13 | 2010-06-09 | メタバシス・セラピューティクス・インコーポレイテッド | New phosphonic acid prodrugs of PMEA and its analogs |
| ATE398455T1 (en) * | 2003-01-14 | 2008-07-15 | Gilead Sciences Inc | COMPOSITIONS AND METHODS FOR ANTIVIRAL COMBINATION THERAPY |
| WO2005002626A2 (en) * | 2003-04-25 | 2005-01-13 | Gilead Sciences, Inc. | Therapeutic phosphonate compounds |
| US7452901B2 (en) * | 2003-04-25 | 2008-11-18 | Gilead Sciences, Inc. | Anti-cancer phosphonate analogs |
| WO2004096285A2 (en) * | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Anti-infective phosphonate conjugates |
| US7407965B2 (en) * | 2003-04-25 | 2008-08-05 | Gilead Sciences, Inc. | Phosphonate analogs for treating metabolic diseases |
| US7432261B2 (en) * | 2003-04-25 | 2008-10-07 | Gilead Sciences, Inc. | Anti-inflammatory phosphonate compounds |
| EA014685B1 (en) | 2003-04-25 | 2010-12-30 | Джилид Сайэнс, Инк. | Phosphonate-containing antiviral compounds (variants) and pharmaceutical composition based thereon |
| US20090247488A1 (en) * | 2003-04-25 | 2009-10-01 | Carina Cannizzaro | Anti-inflammatory phosphonate compounds |
| US7470724B2 (en) * | 2003-04-25 | 2008-12-30 | Gilead Sciences, Inc. | Phosphonate compounds having immuno-modulatory activity |
| US7427636B2 (en) | 2003-04-25 | 2008-09-23 | Gilead Sciences, Inc. | Inosine monophosphate dehydrogenase inhibitory phosphonate compounds |
| CN101410120A (en) * | 2003-04-25 | 2009-04-15 | 吉里德科学公司 | Anti-inflammatory phosphonate compounds |
| US20050261237A1 (en) * | 2003-04-25 | 2005-11-24 | Boojamra Constantine G | Nucleoside phosphonate analogs |
| AU2004233897A1 (en) | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Kinase inhibitor phosphonate conjugates |
| US7427624B2 (en) | 2003-10-24 | 2008-09-23 | Gilead Sciences, Inc. | Purine nucleoside phosphorylase inhibitory phosphonate compounds |
| US7432273B2 (en) * | 2003-10-24 | 2008-10-07 | Gilead Sciences, Inc. | Phosphonate analogs of antimetabolites |
| EP1678321A1 (en) * | 2003-10-24 | 2006-07-12 | Gilead Sciences, Inc. | Methods and compositions for identifying therapeutic compounds |
| NZ547907A (en) * | 2003-12-22 | 2010-07-30 | Gilead Sciences Inc | 4'-Substituted carbovir-and abacavir-derivatives as well as related compounds with HIV and HCV antiviral activity |
| US20050153990A1 (en) * | 2003-12-22 | 2005-07-14 | Watkins William J. | Phosphonate substituted kinase inhibitors |
| US20070281907A1 (en) * | 2003-12-22 | 2007-12-06 | Watkins William J | Kinase Inhibitor Phosphonate Conjugates |
| DK1716162T3 (en) * | 2003-12-30 | 2011-01-10 | Gilead Sciences Inc | Phosphonates, amidomonophosphonates, amidobis phosphonates for the treatment of viral diseases |
| DE602005017131D1 (en) * | 2004-01-21 | 2009-11-26 | Gilead Sciences Inc | USE OF ADEFOVIR OR TENOFOVIR FOR INHIBITING MMTV-LIKE VIRUSES IN CONNECTION WITH BREAST CANCER AND PRIMARY BILIARY CIRRHOSIS |
| CN1964967B (en) | 2004-06-08 | 2014-04-16 | 症变治疗公司 | Lewis acid mediated synthesis of cyclic esters |
| UA88313C2 (en) * | 2004-07-27 | 2009-10-12 | Гилиад Сайенсиз, Инк. | Phosphonate analogs of hiv inhibitor compounds |
| US8642577B2 (en) | 2005-04-08 | 2014-02-04 | Chimerix, Inc. | Compounds, compositions and methods for the treatment of poxvirus infections |
| WO2006110656A2 (en) * | 2005-04-08 | 2006-10-19 | Chimerix, Inc. | Compounds, compositions and methods for the treatment of viral infections and other medical disorders |
| CN100359315C (en) * | 2005-05-26 | 2008-01-02 | 林维宣 | Animal remedy residual ability verification sample and method for preparing same |
| TWI375560B (en) | 2005-06-13 | 2012-11-01 | Gilead Sciences Inc | Composition comprising dry granulated emtricitabine and tenofovir df and method for making the same |
| TWI471145B (en) | 2005-06-13 | 2015-02-01 | Bristol Myers Squibb & Gilead Sciences Llc | Unitary pharmaceutical dosage form |
| US8076303B2 (en) | 2005-12-13 | 2011-12-13 | Spring Bank Pharmaceuticals, Inc. | Nucleotide and oligonucleotide prodrugs |
| CN100396689C (en) * | 2006-03-07 | 2008-06-25 | 中国医学科学院医药生物技术研究所 | Tenoforv monoester compounds with HIV-1/HBV virus copying inhibiting activity |
| PT2020996E (en) | 2006-05-16 | 2012-02-20 | Gilead Sciences Inc | Method and compositions for treating hematological malignancies |
| ES2532502T3 (en) | 2006-07-12 | 2015-03-27 | Mylan Laboratories Limited | Tenofovir preparation process |
| US7951789B2 (en) | 2006-12-28 | 2011-05-31 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| US8105489B2 (en) * | 2007-06-26 | 2012-01-31 | The University Of Wyoming Research Corporation | Treatment and prevention systems for acid mine drainage and halogenated contaminants |
| EP2254582B1 (en) * | 2008-01-25 | 2016-01-20 | Chimerix, Inc. | Methods of treating viral infections |
| TWI444384B (en) | 2008-02-20 | 2014-07-11 | Gilead Sciences Inc | Nucleotide analogues and their use in the treatment of malignancies |
| JP5757860B2 (en) * | 2008-04-25 | 2015-08-05 | シプラ・リミテッド | Crystalline form of tenofovir disoproxil and process for producing the same |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| EP2476690A1 (en) * | 2008-07-02 | 2012-07-18 | IDENIX Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| EA018308B1 (en) | 2008-07-08 | 2013-07-30 | Джилид Сайэнс, Инк. | Salts of hiv inhibitor compounds |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| SG172363A1 (en) * | 2008-12-23 | 2011-07-28 | Pharmasset Inc | Synthesis of purine nucleosides |
| CL2009002207A1 (en) | 2008-12-23 | 2011-02-18 | Gilead Pharmasset Llc | Compounds derived from 3-hydroxy-5- (9h-purin-9-yl) tetrahydrofuran-2-yl, an inhibitor of the replication of arn-dependent viral arn; pharmaceutical composition; use for the treatment of hepatitis c. |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| TWI583692B (en) | 2009-05-20 | 2017-05-21 | 基利法瑪席特有限責任公司 | Nucleoside phosphoramidates |
| WO2011011519A1 (en) | 2009-07-21 | 2011-01-27 | Chimerix, Inc. | Compounds, compositions and methods for treating ocular conditions |
| MX2012003126A (en) | 2009-09-21 | 2012-06-19 | Gilead Sciences Inc | Processes and intermediates for the preparation of 1'-substituted carba-nucleoside analogs. |
| WO2011100698A2 (en) | 2010-02-12 | 2011-08-18 | Chimerix, Inc. | Methods of treating viral infection |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| AP3515A (en) | 2010-03-31 | 2016-01-11 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| WO2011123586A1 (en) | 2010-04-01 | 2011-10-06 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| EP2563367A4 (en) | 2010-04-26 | 2013-12-04 | Chimerix Inc | Methods of treating retroviral infections and related dosage regimes |
| BR112013001267A2 (en) | 2010-07-19 | 2016-05-17 | Gilead Sciences Inc | Methods for Preparing Diasteromerically Pure Phosphoramidate Prodrugs |
| US20120027752A1 (en) | 2010-07-22 | 2012-02-02 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
| JP6069215B2 (en) | 2010-11-30 | 2017-02-01 | ギリアド ファーマセット エルエルシー | Compound |
| KR101880724B1 (en) | 2010-12-10 | 2018-08-17 | 시그마팜 래보러토리즈, 엘엘씨 | Highly stable compositions of orally active nucleotide analogues or orally active nucleotide analogue prodrugs |
| ZA201103820B (en) | 2010-12-13 | 2012-01-25 | Laurus Labs Private Ltd | Process for the preparation of tenofovir |
| EP2691409B1 (en) | 2011-03-31 | 2018-02-21 | Idenix Pharmaceuticals LLC. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US9550803B2 (en) | 2011-05-06 | 2017-01-24 | University Of Southern California | Method to improve antiviral activity of nucleotide analogue drugs |
| AU2012255029B2 (en) | 2011-05-19 | 2016-04-28 | Gilead Sciences, Inc. | Processes and intermediates for preparing anti-HIV agents |
| AU2014271320B2 (en) * | 2011-08-16 | 2017-02-23 | Gilead Sciences, Inc. | Tenofovir alafenamide hemifumarate |
| US8754065B2 (en) * | 2011-08-16 | 2014-06-17 | Gilead Sciences, Inc. | Tenofovir alafenamide hemifumarate |
| UA116087C2 (en) | 2011-09-16 | 2018-02-12 | Гіліад Фармассет Елелсі | Methods for treating hcv |
| AU2016228317B2 (en) * | 2011-10-07 | 2018-07-19 | Gilead Sciences, Inc. | Methods for preparing anti-viral nucleotide analogs |
| CN107266498B (en) * | 2011-10-07 | 2023-10-03 | 吉利德科学公司 | Method for preparing antiviral nucleotide analogues |
| AU2014215976B2 (en) * | 2011-10-07 | 2016-06-30 | Gilead Sciences, Inc. | Methods for preparing anti-viral nucleotide analogs |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| ES2874774T3 (en) | 2011-12-22 | 2021-11-05 | Geron Corp | Guanine Analogs as Telomerase Substrates and Telomere Length Affects |
| AU2012327170A1 (en) | 2012-02-03 | 2013-08-22 | Gilead Sciences, Inc. | Therapeutic compounds |
| KR20140119177A (en) | 2012-02-03 | 2014-10-08 | 길리애드 사이언시즈, 인코포레이티드 | Combination therapy comprising tenofovir alafenamide hemifumarate and cobicistat for use in the treatment of viral infections |
| CN107312039B (en) | 2012-08-30 | 2019-06-25 | 江苏豪森药业集团有限公司 | A kind of preparation method of tenofovir prodrug |
| GB201215696D0 (en) * | 2012-09-03 | 2012-10-17 | Ithemba Pharmaceuticals Pty Ltd | A process for the preparation of (R)-9-[2-(Phosphonometh-Oxy)propyl]adenine (PMPA) |
| US9227990B2 (en) | 2012-10-29 | 2016-01-05 | Cipla Limited | Antiviral phosphonate analogues and process for preparation thereof |
| CN102899327B (en) * | 2012-11-06 | 2014-06-11 | 清华大学深圳研究生院 | Antiviral small nucleic acid and temperature-sensitive type gel preparation and application thereof |
| EP2920171B1 (en) * | 2012-11-16 | 2018-08-29 | Merck Sharp & Dohme Corp. | Purine inhibitors of human phosphatidylinositol 3-kinase delta |
| CN103848868B (en) * | 2012-12-04 | 2017-04-12 | 蚌埠丰原涂山制药有限公司 | method for preparing tenofovir |
| CN103848869B (en) * | 2012-12-04 | 2016-12-21 | 上海医药工业研究院 | The method preparing tenofovir |
| KR20140119012A (en) | 2013-01-31 | 2014-10-08 | 길리어드 파마셋 엘엘씨 | Combination formulation of two antiviral compounds |
| CN104072539B (en) * | 2013-03-25 | 2017-03-29 | 安徽贝克联合制药有限公司 | Double (4 acetaminophenol epoxide) esters of tenofovir and preparation method thereof and its application |
| EP3000898B1 (en) * | 2013-05-21 | 2020-11-18 | Hitgen Inc. | Drug target capturing method |
| EP3004121A1 (en) | 2013-06-07 | 2016-04-13 | Cipla Limited | An efficient process for separation of diastereomers of 9-[(r)-2-[[(r,s)-[[(s)-1-(isopropoxycarbonyl)ethyl]amino]-phenoxyphosphinyl]methoxy]propyl]adenine |
| ES2900570T3 (en) | 2013-08-27 | 2022-03-17 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| WO2015040640A2 (en) * | 2013-09-20 | 2015-03-26 | Laurus Labs Private Limited | An improved process for the preparation of tenofovir alafenamide or pharmaceutically acceptable salts thereof |
| EP2860185A1 (en) | 2013-10-09 | 2015-04-15 | Zentiva, k.s. | An improved process for the preparation of Tenofovir disoproxil and pharmaceutically acceptable salts thereof |
| WO2015079455A2 (en) * | 2013-11-27 | 2015-06-04 | Laurus Labs Private Limited | A recycling process for preparing tenofovir alafenamide diastereomers |
| ES2842123T3 (en) | 2014-01-14 | 2021-07-12 | Mylan Laboratories Ltd | Purification of tenofovir alafenamide and its intermediates |
| TWI660965B (en) | 2014-01-15 | 2019-06-01 | 美商基利科學股份有限公司 | Solid forms of tenofovir |
| CN104804042B (en) * | 2014-01-24 | 2018-01-19 | 齐鲁制药有限公司 | Phosphonate-nucleotide ester class compound, its pharmaceutical composition, Preparation method and use |
| US9463194B2 (en) | 2014-02-05 | 2016-10-11 | Gilead Sciences, Inc. | Methods of treating patients co-infected with HIV and tuberculosis |
| EP3105238A4 (en) | 2014-02-13 | 2017-11-08 | Ligand Pharmaceuticals, Inc. | Prodrug compounds and their uses |
| CN105814068B (en) * | 2014-02-27 | 2017-08-04 | 四川海思科制药有限公司 | A kind of substituted phosphoramidic acid ester derivative, its preparation method and its application |
| CN105001262B (en) * | 2014-04-18 | 2017-09-01 | 四川海思科制药有限公司 | The phosphonaminate of aryl substitution and its application medically |
| CN105531281B (en) * | 2014-04-21 | 2017-12-15 | 四川海思科制药有限公司 | A kind of preparation method of nucleoside analog and its intermediate |
| CN105085571A (en) * | 2014-05-20 | 2015-11-25 | 四川海思科制药有限公司 | Tenofovir alafenamide compound, preparation method and purpose thereof |
| CN105518012B (en) * | 2014-06-25 | 2018-03-02 | 四川海思科制药有限公司 | A kind of substituted amino acid sulfur ester, its composition and application |
| WO2016003812A1 (en) | 2014-07-02 | 2016-01-07 | Ligand Pharmaceuticals, Inc. | Prodrug compounds and uses therof |
| KR101703258B1 (en) | 2014-12-30 | 2017-02-06 | 한미정밀화학주식회사 | Preparation method for (r)-9-[2-(phosphonomethoxy)propyl]adenine with high purity |
| KR101703257B1 (en) | 2014-09-30 | 2017-02-06 | 한미정밀화학주식회사 | Preparation method for (r)-9-[2-(phosphonomethoxy)propyl]adenine with high purity |
| WO2016052930A1 (en) * | 2014-09-30 | 2016-04-07 | 한미정밀화학주식회사 | Method for preparing high-purity (r)-9-[2-(phosphonomethoxy)propyl]adenine |
| WO2016057866A1 (en) | 2014-10-09 | 2016-04-14 | Board Of Regents Of The University Of Nebraska | Compositions and methods for the delivery of therapeutics |
| TWI687432B (en) | 2014-10-29 | 2020-03-11 | 美商基利科學股份有限公司 | Methods for treating filoviridae virus infections |
| CN105646584B (en) * | 2014-11-12 | 2018-09-28 | 四川海思科制药有限公司 | Tenofovir Chinese mugwort draws phenol amine fumarate crystal form and its preparation method and application |
| CN104558036A (en) * | 2014-12-11 | 2015-04-29 | 杭州和泽医药科技有限公司 | Tenofovir alafenamide hemi-fumarate crystal form and preparation method thereof |
| WO2016108205A1 (en) | 2015-01-03 | 2016-07-07 | Mylan Laboratories Limited | Processes for the preparation of amorphous tenofovir alafenamide hemifumarate and a premix thereof |
| WO2016187160A1 (en) * | 2015-05-16 | 2016-11-24 | Godx, Inc. | Point of need testing device and methods of use thereof |
| CN106188139B (en) * | 2015-05-29 | 2020-02-18 | 江苏天士力帝益药业有限公司 | Tenofovir monobenzyl phosphoric acid amide prodrug, preparation method and application thereof |
| CZ2015384A3 (en) | 2015-06-05 | 2016-12-14 | Zentiva, K.S. | Tenofovir alafenamide solid forms |
| WO2017004244A1 (en) | 2015-06-30 | 2017-01-05 | Gilead Sciences, Inc. | Pharmaceutical formulations comprising tenofovir and emtricitabine |
| CN107849071B (en) | 2015-08-10 | 2021-03-09 | 默沙东公司 | Antiviral beta amino acid ester phosphorodiamidate compounds |
| TWI620754B (en) * | 2015-08-26 | 2018-04-11 | Method for preparing amino phosphate derivative and preparation method thereof | |
| TWI616452B (en) * | 2015-08-26 | 2018-03-01 | Preparation method of nucleoside analog and intermediate thereof | |
| TWI616453B (en) * | 2015-08-27 | 2018-03-01 | Substituted amino acid thioester compounds, materials and uses thereof | |
| WO2017037608A1 (en) * | 2015-08-28 | 2017-03-09 | Laurus Labs Private Limited | Solid forms of tenofovir alafenamide and salts thereof, processes for its preparation and pharmaceutical compositions thereof |
| BR112018005048B8 (en) | 2015-09-16 | 2021-03-23 | Gilead Sciences Inc | use of an antiviral compound or salt thereof for the treatment of a coronaviridae infection |
| CN106800573B (en) * | 2015-11-25 | 2020-03-10 | 四川海思科制药有限公司 | Nucleotide phosphonate monohydrate, preparation method and medical application thereof |
| US10745428B2 (en) | 2015-12-10 | 2020-08-18 | Idenix Pharmaceuticals Llc | Antiviral phosphodiamide prodrugs of tenofovir |
| CN106866737B (en) * | 2015-12-11 | 2020-11-20 | 南京圣和药物研发有限公司 | Phosphonic acid derivatives and their use |
| US10450335B2 (en) | 2015-12-15 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral oxime phosphoramide compounds |
| WO2017118928A1 (en) | 2016-01-06 | 2017-07-13 | Lupin Limited | Process for the separation of diastereomers of tenofovir alafenamide |
| EP3411378B1 (en) | 2016-02-02 | 2020-03-25 | Sandoz AG | Crystalline forms of tenofovir alafenamide monofumarate |
| CN107709288A (en) * | 2016-02-03 | 2018-02-16 | 四川海思科制药有限公司 | A kind of phosphinylidyne amine derivative and preparation method and purposes |
| WO2017148290A1 (en) * | 2016-03-01 | 2017-09-08 | 深圳市塔吉瑞生物医药有限公司 | Substituted adenine compound and pharmaceutical composition thereof |
| CN107179355B (en) * | 2016-03-11 | 2021-08-10 | 广东东阳光药业有限公司 | Method for separating and detecting tenofovir alafenamide and related substances thereof |
| CZ2016156A3 (en) | 2016-03-17 | 2017-09-27 | Zentiva, K.S. | The method of preparation of diastereomerically pure Tenofovir Alafenamide or its salts |
| CN107226826A (en) * | 2016-03-25 | 2017-10-03 | 江苏奥赛康药业股份有限公司 | Tenofovir Chinese mugwort draws phenol amine fumarate compound and its pharmaceutical composition |
| CN109476689B (en) * | 2016-06-05 | 2021-09-03 | 上海诚妙医药科技有限公司 | Novel crystal form of tenofovir alafenamide fumarate, preparation method and application thereof |
| CN106543227B (en) * | 2016-06-20 | 2018-02-02 | 杭州和泽医药科技有限公司 | A kind of phosphonate prodrugs of adenine derivative and its application in medicine |
| WO2017221189A1 (en) * | 2016-06-22 | 2017-12-28 | Laurus Labs Limited | An improved process for the preparation of tenofovir alafenamide or pharmaceutically acceptable salts thereof |
| MX369307B (en) | 2016-08-19 | 2019-11-05 | Gilead Sciences Inc | Therapeutic compounds useful for the prophylactic or therapeutic treatment of an hiv virus infection. |
| CN106317116A (en) * | 2016-08-19 | 2017-01-11 | 张红利 | Phosphamide nucleosides compound, pharmaceutically acceptable salt and application thereof, and pharmaceutical composition |
| US10449208B2 (en) | 2016-08-25 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral prodrugs of tenofovir |
| CN106380484A (en) * | 2016-08-29 | 2017-02-08 | 杭州百诚医药科技股份有限公司 | New crystal form of tenofovir alafenamide and preparation method thereof |
| WO2018042331A1 (en) | 2016-08-31 | 2018-03-08 | Glaxosmithkline Intellectual Property (No.2) Limited | Combinations and uses and treatments thereof |
| WO2018051250A1 (en) | 2016-09-14 | 2018-03-22 | Viiv Healthcare Company | Combination comprising tenofovir alafenamide, bictegravir and 3tc |
| EP3532069A4 (en) | 2016-10-26 | 2020-05-13 | Merck Sharp & Dohme Corp. | Antiviral aryl-amide phosphodiamide compounds |
| CN106565785B (en) * | 2016-11-09 | 2019-11-12 | 周雨恬 | One kind having the active nucleoside phosphoramidate class compound of Anti-HBV activity/HIV and its salt and purposes |
| CN108129514A (en) * | 2016-12-01 | 2018-06-08 | 北京美倍他药物研究有限公司 | The individual isomer and its medical usage of phosphoric acid/phosphonate derivative |
| US10519159B2 (en) | 2016-12-22 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antiviral aliphatic ester prodrugs of tenofovir |
| CA3046029A1 (en) | 2016-12-22 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antiviral benzyl-amine phosphodiamide compounds |
| WO2018115046A1 (en) | 2016-12-23 | 2018-06-28 | Sandoz Ag | Crystalline solid forms of tenofovir alafenamide |
| TWI784370B (en) | 2017-01-31 | 2022-11-21 | 美商基利科學股份有限公司 | Crystalline forms of tenofovir alafenamide |
| WO2018153977A1 (en) | 2017-02-24 | 2018-08-30 | Hexal Ag | Stable composition of tenofovir alafenamide |
| RU2659388C1 (en) | 2017-02-28 | 2018-07-02 | Васильевич Иващенко Александр | Nucleotides including n-[(s)-1-cyclobutoxycarbonyl]phosphoramidate fragment, their analogs and their application |
| US20190374557A1 (en) * | 2017-02-28 | 2019-12-12 | Alexandre Vasilievich Ivachtchenko | Cyclobutyl (S)-2-[[[(R)-2-(6-aminopurin-9-yl)-1-methyl-ethoxy]methyl-phenoxy-phosphoryl]amino]-propanoates, and production process and application thereof |
| CN106866739B (en) * | 2017-03-10 | 2018-11-02 | 华东师范大学 | The preparation method of one kind (R) -1- (6- amino -9H- purine -9- bases) 2- phenyl esters |
| EP4331677A3 (en) | 2017-03-14 | 2024-05-29 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US11191763B2 (en) | 2017-03-20 | 2021-12-07 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | HIV post-exposure prophylaxis |
| CN108794530A (en) * | 2017-04-26 | 2018-11-13 | 上海医药工业研究院 | A kind of the third phenol of tenofovir amidic-salt crystal form and its preparation method and application |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| KR102379965B1 (en) * | 2017-05-19 | 2022-03-29 | 주식회사 종근당 | Efficient preparation method of Tenofovir |
| CN107266499B (en) * | 2017-06-05 | 2019-07-02 | 珠海优润医药科技有限公司 | A kind of antiviral compound and preparation method thereof |
| WO2019003251A1 (en) | 2017-06-30 | 2019-01-03 | Cipla Limited | Pharmaceutical compositions |
| WO2019014247A1 (en) | 2017-07-11 | 2019-01-17 | Gilead Sciences, Inc. | Compositions comprising an rna polymerase inhibitor and cyclodextrin for treating viral infections |
| WO2019021319A1 (en) | 2017-07-27 | 2019-01-31 | Cipla Limited | Pharmaceutical compositions |
| PL3661937T3 (en) | 2017-08-01 | 2021-12-20 | Gilead Sciences, Inc. | Crystalline forms of ethyl ((s)-((((2r,5r)-5-(6-amino-9h-purin-9-yl)-4-fluoro-2,5-dihydrofuran-2-yl)oxy)methyl)(phenoxy)phosphoryl)-l-alaninate (gs-9131) for treating viral infections |
| AR112412A1 (en) | 2017-08-17 | 2019-10-23 | Gilead Sciences Inc | CHOLINE SALT FORMS OF AN HIV CAPSID INHIBITOR |
| CN107655987B (en) * | 2017-09-08 | 2020-11-03 | 厦门蔚扬药业有限公司 | HPLC detection method for tenofovir alafenamide and isomer thereof |
| CN107522743A (en) * | 2017-09-30 | 2017-12-29 | 深圳科兴生物工程有限公司 | A kind of half fumaric acid tenofovir Chinese mugwort draws phenol amine industrial continuous producing method |
| EP3700573A1 (en) | 2017-10-24 | 2020-09-02 | Gilead Sciences, Inc. | Methods of treating patients co-infected with a virus and tuberculosis |
| CN109942632B (en) * | 2017-12-20 | 2021-08-31 | 上海博志研新药物研究有限公司 | Preparation method of tenofovir alafenamide intermediate |
| CN109942633B (en) * | 2017-12-20 | 2021-08-31 | 上海新礼泰药业有限公司 | Preparation method of tenofovir alafenamide intermediate |
| US20200407382A1 (en) | 2017-12-30 | 2020-12-31 | Cipla Limited | Polymorphic forms of (9-[(r)-2-[[(s)-[[(s)-1-(isopropoxycarbonyl)ethyl]amino]phenoxy phosphinyl]methoxy]propyl] adenine and pharmaceutically acceptable salts thereof |
| CN111788196A (en) | 2018-01-09 | 2020-10-16 | 配体药物公司 | Acetal compounds and their therapeutic use |
| CA3088287A1 (en) * | 2018-01-10 | 2019-07-18 | Nucorion Pharmaceuticals, Inc. | Phosphor(n)amidatacetal and phosph(on)atalcetal compounds |
| US11839623B2 (en) * | 2018-01-12 | 2023-12-12 | Board Of Regents Of The University Of Nebraska | Antiviral prodrugs and formulations thereof |
| AU2019221568B2 (en) | 2018-02-15 | 2021-04-22 | Gilead Sciences, Inc. | Pyridine derivatives and their use for treating HIV infection |
| EP3752496B1 (en) | 2018-02-16 | 2023-07-05 | Gilead Sciences, Inc. | Methods and intermediates for preparing a therapeutic compound useful in the treatment of retroviridae viral infection |
| CN108101943B (en) * | 2018-02-28 | 2020-11-24 | 顾世海 | Tenofovir prodrug or pharmaceutically acceptable salt and application thereof in medicine |
| WO2019199756A1 (en) | 2018-04-09 | 2019-10-17 | Board Of Regents Of The University Of Nebraska | Antiviral prodrugs and formulations thereof |
| KR20210033492A (en) | 2018-07-16 | 2021-03-26 | 길리애드 사이언시즈, 인코포레이티드 | Capsid inhibitors for the treatment of HIV |
| WO2020018399A1 (en) | 2018-07-19 | 2020-01-23 | Merck Sharp & Dohme Corp. | Phosphinic amide prodrugs of tenofovir |
| EP3852761A1 (en) | 2018-09-19 | 2021-07-28 | Gilead Sciences, Inc. | Integrase inhibitors for the prevention of hiv |
| US12110311B2 (en) | 2019-07-17 | 2024-10-08 | Nucorion Pharmaceuticals, Inc. | Cyclic deoxyribonucleotide compounds |
| US20220257619A1 (en) | 2019-07-18 | 2022-08-18 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| WO2021015818A1 (en) | 2019-07-19 | 2021-01-28 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Hiv pre-exposure prophylaxis |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| AU2020334152A1 (en) | 2019-08-22 | 2022-03-24 | Emory University | Nucleoside prodrugs and uses related thereto |
| WO2021055808A1 (en) * | 2019-09-20 | 2021-03-25 | Abbott Rapid Diagnostics International Unlimited Company | Antibodies directed against tenofovir and derivatives thereof |
| CN114727999A (en) | 2019-11-26 | 2022-07-08 | 吉利德科学公司 | Capsid inhibitors for the prevention of HIV |
| CA3163424A1 (en) | 2020-01-27 | 2021-08-05 | Gilead Sciences, Inc. | Methods for treating sars cov-2 infections |
| WO2021165995A1 (en) | 2020-02-20 | 2021-08-26 | Cipla Limited | Novel salts and/or co-crystals of tenofovir alafenamide |
| JP7554841B2 (en) | 2020-03-12 | 2024-09-20 | ギリアード サイエンシーズ, インコーポレイテッド | Methods for preparing 1'-cyanonucleosides |
| JP2023518433A (en) | 2020-03-20 | 2023-05-01 | ギリアード サイエンシーズ, インコーポレイテッド | Prodrugs of 4'-C-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using them |
| WO2021202669A2 (en) | 2020-04-01 | 2021-10-07 | Reyoung Corporation | Nucleoside and nucleotide conjugate compounds and uses thereof |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| KR20210125298A (en) | 2020-04-08 | 2021-10-18 | 주식회사 파마코스텍 | New process for the preparation of Tenofovir alafenamide hemi-tartrate |
| WO2021216427A1 (en) | 2020-04-21 | 2021-10-28 | Ligand Pharmaceuticals, Inc. | Nucleotide prodrug compounds |
| KR20230018473A (en) | 2020-05-29 | 2023-02-07 | 길리애드 사이언시즈, 인코포레이티드 | How to treat remdesivir |
| CN115996928A (en) | 2020-06-24 | 2023-04-21 | 吉利德科学公司 | 1' -cyanonucleoside analogs and uses thereof |
| CN115996925A (en) | 2020-06-25 | 2023-04-21 | 吉利德科学公司 | Capsid inhibitors for the treatment of HIV |
| CN113970612B (en) * | 2020-07-22 | 2023-08-01 | 北京四环制药有限公司 | Method for measuring related substances of propiophenone tenofovir by high performance liquid chromatography |
| AU2021331214B2 (en) | 2020-08-27 | 2024-01-04 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| CN112336695B (en) * | 2020-09-28 | 2023-01-03 | 华北制药华坤河北生物技术有限公司 | Propofol fumarate and tenofovir tablet, preparation method thereof and detection method of related substances |
| KR20230107288A (en) | 2020-11-11 | 2023-07-14 | 길리애드 사이언시즈, 인코포레이티드 | Method for identifying HIV patients susceptible to therapy with gp120 CD4 binding site-directed antibody |
| US11667656B2 (en) | 2021-01-27 | 2023-06-06 | Apotex Inc. | Crystalline forms of Tenofovir alafenamide |
| CN113214322B (en) * | 2021-04-30 | 2022-10-25 | 山东立新制药有限公司 | Green and environment-friendly preparation method of tenofovir |
| WO2022251594A1 (en) * | 2021-05-27 | 2022-12-01 | Antios Therapeutics, Inc. | Pharmacokinetics and dose-related improvments in subjects treated with phosphoramidate clevudine prodrugs |
| EP4440702A1 (en) | 2021-12-03 | 2024-10-09 | Gilead Sciences, Inc. | Therapeutic compounds for hiv virus infection |
| KR20240117588A (en) | 2021-12-03 | 2024-08-01 | 길리애드 사이언시즈, 인코포레이티드 | Compounds for the treatment of HIV viral infection |
| WO2023102523A1 (en) | 2021-12-03 | 2023-06-08 | Gilead Sciences, Inc. | Therapeutic compounds for hiv virus infection |
| CN114369120A (en) * | 2022-01-28 | 2022-04-19 | 石家庄龙泽制药股份有限公司 | Preparation method of key intermediate of prophenoltenofovir |
| TW202400185A (en) | 2022-03-02 | 2024-01-01 | 美商基利科學股份有限公司 | Compounds and methods for treatment of viral infections |
| TWI843506B (en) | 2022-04-06 | 2024-05-21 | 美商基利科學股份有限公司 | Bridged tricyclic carbamoylpyridone compounds and uses thereof |
| WO2024006982A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Therapeutic compounds useful for the prophylactic or therapeutic treatment of an hiv virus infection |
| WO2024044477A1 (en) | 2022-08-26 | 2024-02-29 | Gilead Sciences, Inc. | Dosing and scheduling regimen for broadly neutralizing antibodies |
| WO2024076915A1 (en) | 2022-10-04 | 2024-04-11 | Gilead Sciences, Inc. | 4'-thionucleoside analogues and their pharmaceutical use |
| WO2024196814A1 (en) | 2023-03-17 | 2024-09-26 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methods for treatment of age-related macular degeneration |
| CN118652277A (en) * | 2024-08-19 | 2024-09-17 | 成都工业学院 | Preparation method of compound for treating cancer diseases |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CS233665B1 (en) | 1983-01-06 | 1985-03-14 | Antonin Holy | Processing of isomere o-phosphonylmethylderivative of anantiomere racemic vicinal diene |
| CS263951B1 (en) | 1985-04-25 | 1989-05-12 | Antonin Holy | 9-(phosponylmethoxyalkyl)adenines and method of their preparation |
| CS263952B1 (en) | 1985-04-25 | 1989-05-12 | Holy Antonin | Remedy with antiviral effect |
| CS264222B1 (en) | 1986-07-18 | 1989-06-13 | Holy Antonin | N-phosphonylmethoxyalkylderivatives of bases of pytimidine and purine and method of use them |
| US5650510A (en) | 1986-11-18 | 1997-07-22 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Antiviral phosphonomethoxyalkylene purine and pyrimidine derivatives |
| US5057301A (en) | 1988-04-06 | 1991-10-15 | Neorx Corporation | Modified cellular substrates used as linkers for increased cell retention of diagnostic and therapeutic agents |
| US5053215A (en) * | 1988-05-26 | 1991-10-01 | University Of Florida | NMR-assayable ligand-labelled trifluorothymidine containing composition and method for diagnosis of HSV infection |
| US5744600A (en) | 1988-11-14 | 1998-04-28 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Phosphonomethoxy carbocyclic nucleosides and nucleotides |
| US5688778A (en) | 1989-05-15 | 1997-11-18 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Nucleoside analogs |
| JP2648516B2 (en) | 1989-07-27 | 1997-09-03 | ダイセル化学工業株式会社 | Separation of stereoisomers |
| US5624898A (en) * | 1989-12-05 | 1997-04-29 | Ramsey Foundation | Method for administering neurologic agents to the brain |
| JP2925753B2 (en) | 1990-02-23 | 1999-07-28 | ダイセル化学工業株式会社 | Optical isomer separation method |
| US5302585A (en) | 1990-04-20 | 1994-04-12 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Use of chiral 2-(phosphonomethoxy)propyl guanines as antiviral agents |
| ATE124050T1 (en) * | 1990-04-20 | 1995-07-15 | Acad Of Science Czech Republic | CHIRAL 2-(PHOSPHONOMETHOXY)PROPYL-GUANINE AS ANTIVIRAL AGENTS. |
| CZ285420B6 (en) | 1990-04-24 | 1999-08-11 | Ústav Organické Chemie A Biochemie Avčr | N-(3-fluoro-2-phosphonylmethoxypropyl) derivatives of purine and pyrimidine heterocyclic bases, processes of their preparation and use |
| US5627165A (en) * | 1990-06-13 | 1997-05-06 | Drug Innovation & Design, Inc. | Phosphorous prodrugs and therapeutic delivery systems using same |
| US5177064A (en) | 1990-07-13 | 1993-01-05 | University Of Florida | Targeted drug delivery via phosphonate derivatives |
| CS276072B6 (en) | 1990-08-06 | 1992-03-18 | Ustav Organicke Chemie A Bioch | (2R)-2-/DI(2-PROPYL)PHOSPHONYLMETHOXY/-3-p-TOLUENESULFONYLOXY -1- TRIMETHYLACETOXYPROPANE AND PROCESS FOR PREPARING THEREOF |
| WO1992002511A1 (en) | 1990-08-10 | 1992-02-20 | Bristol-Myers Squibb Company | Novel process for the preparation of nucleotides |
| EP0481214B1 (en) * | 1990-09-14 | 1998-06-24 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Prodrugs of phosphonates |
| US5827819A (en) * | 1990-11-01 | 1998-10-27 | Oregon Health Sciences University | Covalent polar lipid conjugates with neurologically active compounds for targeting |
| US5208221A (en) * | 1990-11-29 | 1993-05-04 | Bristol-Myers Squibb Company | Antiviral (phosphonomethoxy) methoxy purine/pyrimidine derivatives |
| CZ284678B6 (en) | 1991-05-20 | 1999-01-13 | Ústav Organické Chemie A Biochemie Avčr | Di(2-propyl)esters of 1-fluoro-2-phosphonomethoxy-3-p-toluenesulfonyloxypropanes, process of their preparation and use |
| US5498752A (en) | 1991-08-22 | 1996-03-12 | Daicel Chemical Industries, Ltd. | Process for recovering optical isomers and solvent, process for using solvent by circulation and process for reusing optical isomers in optical resolution |
| JP3010816B2 (en) | 1991-08-22 | 2000-02-21 | ダイセル化学工業株式会社 | Method for recovering optical isomer and solvent in optical resolution, method for recycling solvent, and method for reusing optical isomer |
| JP3497505B2 (en) | 1991-10-11 | 2004-02-16 | インスティテュート オブ オルガニック ケミストリー アンド バイオケミストリー オブ ザ アカデミー オブ サイエンシズ オブ ザ チェコ パブリック | Antiviral acyclic phosphonomethoxyalkyl-substituted alkenyl and alkynylpurine and pyrimidine derivatives |
| US6057305A (en) | 1992-08-05 | 2000-05-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Antiretroviral enantiomeric nucleotide analogs |
| IL106998A0 (en) * | 1992-09-17 | 1993-12-28 | Univ Florida | Brain-enhanced delivery of neuroactive peptides by sequential metabolism |
| US6413949B1 (en) * | 1995-06-07 | 2002-07-02 | D-Pharm, Ltd. | Prodrugs with enhanced penetration into cells |
| EP0719274A1 (en) * | 1993-09-17 | 1996-07-03 | Gilead Sciences, Inc. | Method for dosing therapeutic compounds |
| US5656745A (en) * | 1993-09-17 | 1997-08-12 | Gilead Sciences, Inc. | Nucleotide analogs |
| AU691527B2 (en) | 1993-09-17 | 1998-05-21 | Gilead Sciences, Inc. | Nucleotide analogs |
| US5798340A (en) * | 1993-09-17 | 1998-08-25 | Gilead Sciences, Inc. | Nucleotide analogs |
| GB9505025D0 (en) | 1995-03-13 | 1995-05-03 | Medical Res Council | Chemical compounds |
| US5977061A (en) | 1995-04-21 | 1999-11-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | N6 - substituted nucleotide analagues and their use |
| CA2222048A1 (en) * | 1995-05-26 | 1996-11-28 | Polska Akademia Nauk | Compositions and methods for the synthesis of organophosphorus derivatives |
| CA2239020A1 (en) | 1995-12-29 | 1997-07-10 | Norbert W. Bischofberger | Nucleotide analogs |
| US5717095A (en) * | 1995-12-29 | 1998-02-10 | Gilead Sciences, Inc. | Nucleotide analogs |
| US5874577A (en) * | 1996-04-03 | 1999-02-23 | Medichem Research, Inc. | Method for the preparing 9-12-(Diethoxyphosphonomethoxy)ethyl!adenine and analogues thereof |
| US5922695A (en) * | 1996-07-26 | 1999-07-13 | Gilead Sciences, Inc. | Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavarilability |
| JP4033494B2 (en) * | 1996-07-26 | 2008-01-16 | ギリヤド サイエンシーズ, インコーポレイテッド | Nucleotide analogs |
| US5739314A (en) | 1997-04-25 | 1998-04-14 | Hybridon, Inc. | Method for synthesizing 2'-O-substituted pyrimidine nucleosides |
| US5935946A (en) | 1997-07-25 | 1999-08-10 | Gilead Sciences, Inc. | Nucleotide analog composition and synthesis method |
| PT1256584E (en) | 1997-07-25 | 2004-12-31 | Gilead Sciences Inc | PROCESS FOR THE PREPARATION OF ADEFOVIR-DIPIVOXYL |
| BR9811045A (en) † | 1997-07-25 | 2000-08-22 | Gilead Sciences Inc | Composition of nucleotide analog and synthesis process |
| WO1999030727A1 (en) * | 1997-12-17 | 1999-06-24 | Enzon, Inc. | Polymeric prodrugs of amino- and hydroxyl-containing bioactive agents |
| IL137164A0 (en) * | 1998-01-23 | 2001-07-24 | Newbiotics Inc | Enzyme catalyzed therapeutic agents |
| US6169078B1 (en) * | 1998-05-12 | 2001-01-02 | University Of Florida | Materials and methods for the intracellular delivery of substances |
| JP2002518521A (en) * | 1998-06-20 | 2002-06-25 | ワシントン・ユニバーシティ | Membrane permeable peptide complexes for medical image analysis, diagnosis and therapy |
| US6169879B1 (en) * | 1998-09-16 | 2001-01-02 | Webtv Networks, Inc. | System and method of interconnecting and using components of home entertainment system |
| GB9821058D0 (en) † | 1998-09-28 | 1998-11-18 | Univ Cardiff | Chemical compound |
| TWI230618B (en) * | 1998-12-15 | 2005-04-11 | Gilead Sciences Inc | Pharmaceutical compositions of 9-[2-[[bis[(pivaloyloxy)methyl]phosphono]methoxy]ethyl]adenine and tablets or capsules containing the same |
| IL144647A0 (en) † | 1999-02-12 | 2002-05-23 | Glaxo Group Ltd | Phosphoramidate, and mono-, di-, and tri-phosphate esters of (ir, cis)-4-(6-amino-9h-purin-9-yl) 2-cyclopentene-1-methanol as antiviral agents |
| AU2001282941C1 (en) * | 2000-07-21 | 2016-12-22 | Gilead Sciences, Inc. | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| EP1345956A2 (en) * | 2000-10-13 | 2003-09-24 | University of Lausanne | Intracellular delivery of biological effectors by novel transporter peptide sequences |
| US20020119433A1 (en) * | 2000-12-15 | 2002-08-29 | Callender Thomas J. | Process and system for creating and administering interview or test |
-
2001
- 2001-07-20 AU AU2001282941A patent/AU2001282941C1/en active Active
- 2001-07-20 ES ES13164300.9T patent/ES2627903T3/en not_active Expired - Lifetime
- 2001-07-20 NZ NZ535408A patent/NZ535408A/en not_active IP Right Cessation
- 2001-07-20 EP EP13164300.9A patent/EP2682397B1/en not_active Expired - Lifetime
- 2001-07-20 BR BR0112646A patent/BRPI0112646B8/en active IP Right Grant
- 2001-07-20 NZ NZ523438A patent/NZ523438A/en not_active IP Right Cessation
- 2001-07-20 CZ CZ2003-413A patent/CZ304886B6/en unknown
- 2001-07-20 DK DK01961695.2T patent/DK1301519T4/en active
- 2001-07-20 CN CNB018131611A patent/CN1291994C/en not_active Expired - Lifetime
- 2001-07-20 KR KR1020067020061A patent/KR100749160B1/en active IP Right Grant
- 2001-07-20 JP JP2002514146A patent/JP4651264B2/en not_active Expired - Lifetime
- 2001-07-20 CA CA2416757A patent/CA2416757C/en not_active Expired - Lifetime
- 2001-07-20 HR HRP20160074AA patent/HRP20160074B1/en not_active IP Right Cessation
- 2001-07-20 PT PT1961695T patent/PT1301519E/en unknown
- 2001-07-20 US US10/333,107 patent/US20040018150A1/en not_active Abandoned
- 2001-07-20 NZ NZ536942A patent/NZ536942A/en not_active IP Right Cessation
- 2001-07-20 US US09/909,560 patent/US20020119443A1/en not_active Abandoned
- 2001-07-20 EE EEP200300029A patent/EE05366B1/en active Protection Beyond IP Right Term
- 2001-07-20 CZ CZ2013-310A patent/CZ304734B6/en not_active IP Right Cessation
- 2001-07-20 WO PCT/US2001/023104 patent/WO2002008241A2/en active Application Filing
- 2001-07-20 AU AU8294101A patent/AU8294101A/en active Pending
- 2001-07-20 CA CA2893174A patent/CA2893174A1/en not_active Abandoned
- 2001-07-20 EP EP17160970.4A patent/EP3235823A1/en not_active Withdrawn
- 2001-07-20 PL PL360490A patent/PL213214B1/en unknown
- 2001-07-20 EP EP01961695.2A patent/EP1301519B2/en not_active Expired - Lifetime
- 2001-07-20 LT LTEP13164300.9T patent/LT2682397T/en unknown
- 2001-07-20 SI SI200131061T patent/SI2682397T1/en unknown
- 2001-07-20 OA OA1200300003A patent/OA12393A/en unknown
- 2001-07-20 PT PT131643009T patent/PT2682397T/en unknown
- 2001-07-20 IL IL15365801A patent/IL153658A0/en active Protection Beyond IP Right Term
- 2001-07-20 CA CA2725819A patent/CA2725819C/en not_active Expired - Lifetime
- 2001-07-20 CN CNB2004100978453A patent/CN100402539C/en not_active Expired - Lifetime
- 2001-07-20 MX MXPA03000587A patent/MXPA03000587A/en active IP Right Grant
- 2001-07-20 ES ES01961695T patent/ES2536972T5/en not_active Expired - Lifetime
- 2001-07-20 SI SI200131040T patent/SI1301519T1/en unknown
- 2001-07-20 UA UA2003021482A patent/UA75889C2/en unknown
- 2001-07-20 KR KR1020037000872A patent/KR100767432B1/en active Protection Beyond IP Right Term
- 2001-07-20 HU HU0301307A patent/HU230960B1/en active Protection Beyond IP Right Term
- 2001-07-20 EA EA200300188A patent/EA004926B1/en active Protection Beyond IP Right Term
- 2001-07-20 AP APAP/P/2003/002724A patent/AP1466A/en active
- 2001-07-20 DK DK13164300.9T patent/DK2682397T3/en active
- 2001-07-20 TR TR2003/00055T patent/TR200300055T2/en unknown
-
2002
- 2002-12-19 ZA ZA2002/10271A patent/ZA200210271B/en unknown
-
2003
- 2003-01-17 IS IS6689A patent/IS2985B/en unknown
- 2003-01-20 NO NO20030270A patent/NO336718B1/en active Protection Beyond IP Right Term
- 2003-01-24 HR HRP20030047AA patent/HRP20030047B1/en not_active IP Right Cessation
- 2003-01-28 US US10/354,207 patent/US20030219727A1/en not_active Abandoned
- 2003-02-19 BG BG107572A patent/BG66037B1/en unknown
- 2003-08-15 HK HK03105871.0A patent/HK1054238A1/en active IP Right Maintenance
-
2004
- 2004-02-24 US US10/785,497 patent/US20060024659A1/en not_active Abandoned
- 2004-03-11 US US10/798,692 patent/US7390791B2/en active Active
-
2005
- 2005-01-06 US US11/031,252 patent/US20050124585A1/en not_active Abandoned
- 2005-01-06 US US11/031,250 patent/US20050124583A1/en not_active Abandoned
- 2005-01-06 US US11/031,251 patent/US20050124584A1/en not_active Abandoned
- 2005-01-06 US US11/031,228 patent/US20050159392A1/en not_active Abandoned
- 2005-10-18 AU AU2005225039A patent/AU2005225039B2/en not_active Expired
-
2008
- 2008-04-28 US US12/110,829 patent/US7803788B2/en not_active Expired - Fee Related
- 2008-10-16 JP JP2008267991A patent/JP5063554B2/en not_active Expired - Lifetime
-
2010
- 2010-04-16 JP JP2010095542A patent/JP5111551B2/en not_active Expired - Lifetime
-
2011
- 2011-03-08 JP JP2011050854A patent/JP2011140506A/en not_active Withdrawn
-
2012
- 2012-04-23 NO NO20120466A patent/NO20120466L/en not_active Application Discontinuation
-
2013
- 2013-12-20 NO NO20131717A patent/NO20131717L/en unknown
-
2015
- 2015-07-10 NO NO20150909A patent/NO20150909L/en not_active Application Discontinuation
-
2016
- 2016-04-05 LT LTPA2016009C patent/LTC1301519I2/en unknown
- 2016-04-07 BE BE2016C018C patent/BE2016C018I2/fr unknown
- 2016-04-11 FR FR16C0013C patent/FR16C0013I2/en active Active
- 2016-04-12 CY CY2016008C patent/CY2016008I1/en unknown
- 2016-04-14 LU LU93029C patent/LU93029I2/en unknown
- 2016-04-19 NO NO2016006C patent/NO2016006I2/en unknown
- 2016-04-22 NL NL300803C patent/NL300803I2/nl unknown
-
2017
- 2017-06-28 CY CY20171100687T patent/CY1119411T1/en unknown
-
2018
- 2018-03-09 HK HK18103347.4A patent/HK1243711A1/en unknown
-
2019
- 2019-05-09 HU HUS1900027C patent/HUS1900027I1/en unknown
-
2023
- 2023-02-03 NO NO2023006C patent/NO2023006I1/en unknown
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8411105B1 (en) | 2004-05-14 | 2013-04-02 | Nvidia Corporation | Method and system for computing pixel parameters |
| US8416242B1 (en) | 2004-05-14 | 2013-04-09 | Nvidia Corporation | Method and system for interpolating level-of-detail in graphics processors |
| US8432394B1 (en) | 2004-05-14 | 2013-04-30 | Nvidia Corporation | Method and system for implementing clamped z value interpolation in a raster stage of a graphics pipeline |
| US8749576B2 (en) | 2004-05-14 | 2014-06-10 | Nvidia Corporation | Method and system for implementing multiple high precision and low precision interpolators for a graphics pipeline |
| US8441497B1 (en) * | 2007-08-07 | 2013-05-14 | Nvidia Corporation | Interpolation of vertex attributes in a graphics processor |
| WO2016205141A1 (en) * | 2015-06-17 | 2016-12-22 | Gilead Sciences, Inc. | Co-crystals, salts and solid forms of tenofovir alafenamide |
| US9777028B2 (en) | 2015-06-17 | 2017-10-03 | Gilead Sciences, Inc. | Co-crystals, salts and solid forms of tenofovir alafenamide |
| US10155781B2 (en) | 2015-06-17 | 2018-12-18 | Gilead Sciences, Inc. | Co-crystals, salts and solid forms of tenofovir alafenamide |
| AU2016277859B2 (en) * | 2015-06-17 | 2019-08-01 | Gilead Sciences, Inc. | Co-crystals, salts and solid forms of tenofovir alafenamide |
| EP4092037A1 (en) * | 2015-06-17 | 2022-11-23 | Gilead Sciences, Inc. | Co-crystals, salts and solid forms of tenofovir alafenamide |
| US10548846B2 (en) | 2015-11-09 | 2020-02-04 | Gilead Sciences, Inc. | Therapeutic compositions for treatment of human immunodeficiency virus |
| US11744802B2 (en) | 2015-11-09 | 2023-09-05 | Gilead Sciences, Inc. | Therapeutic compositions for treatment of human immunodeficiency virus |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7803788B2 (en) | Prodrugs of phosphonate nucoleotide analogues | |
| AU2001282941A1 (en) | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |