US20040224917A1 - Compositions and methods for combination antiviral therapy - Google Patents
Compositions and methods for combination antiviral therapy Download PDFInfo
- Publication number
- US20040224917A1 US20040224917A1 US10/757,141 US75714104A US2004224917A1 US 20040224917 A1 US20040224917 A1 US 20040224917A1 US 75714104 A US75714104 A US 75714104A US 2004224917 A1 US2004224917 A1 US 2004224917A1
- Authority
- US
- United States
- Prior art keywords
- emtricitabine
- tenofovir disoproxil
- physiologically functional
- disoproxil fumarate
- functional derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 [3*]C(CN1C([5*])=NC2=C1N=C([4*])N=C2N([6*])[7*])OCP(C)(C)=O Chemical compound [3*]C(CN1C([5*])=NC2=C1N=C([4*])N=C2N([6*])[7*])OCP(C)(C)=O 0.000 description 5
- RRBSKZVTTAAQPV-UHFFFAOYSA-N BC1CSC(CC)O1 Chemical compound BC1CSC(CC)O1 RRBSKZVTTAAQPV-UHFFFAOYSA-N 0.000 description 3
- JFVZFKDSXNQEJW-CQSZACIVSA-N CC(C)OC(=O)OCOP(=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OCOC(=O)OC(C)C Chemical compound CC(C)OC(=O)OCOP(=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OCOC(=O)OC(C)C JFVZFKDSXNQEJW-CQSZACIVSA-N 0.000 description 1
- LDEKQSIMHVQZJK-CAQYMETFSA-N CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1 Chemical compound CC(C)OC(=O)[C@H](C)N[P@](=O)(CO[C@H](C)CN1C=NC2=C1N=CN=C2N)OC1=CC=CC=C1 LDEKQSIMHVQZJK-CAQYMETFSA-N 0.000 description 1
- SGOIRFVFHAKUTI-ZCFIWIBFSA-N C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O Chemical compound C[C@H](CN1C=NC2=C1N=CN=C2N)OCP(=O)(O)O SGOIRFVFHAKUTI-ZCFIWIBFSA-N 0.000 description 1
- UZJQFWKPDGUPJU-RIHPBJNCSA-N SSSSS.[H][C@@]1(N2C=C(F)C(N)=NC2=O)CS[C@]([H])(CO)O1 Chemical compound SSSSS.[H][C@@]1(N2C=C(F)C(N)=NC2=O)CS[C@]([H])(CO)O1 UZJQFWKPDGUPJU-RIHPBJNCSA-N 0.000 description 1
- TWWVFOBDTTXRPW-UOERWJHTSA-N SSSSS.[H][C@@]1(N2C=CC(N)=NC2=O)CS[C@]([H])(CO)O1 Chemical compound SSSSS.[H][C@@]1(N2C=CC(N)=NC2=O)CS[C@]([H])(CO)O1 TWWVFOBDTTXRPW-UOERWJHTSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the invention relates generally to combinations of compounds with antiviral activity and more specifically with anti-HIV properties. In particular, it relates to chemically stable combinations of structurally diverse anti-viral agents.
- HIV-1 Human immunodeficiency virus type 1
- RT reverse transcriptase
- Prt protease
- Int integrase
- drugs targeting reverse transcriptase and protease are in wide use and have shown effectiveness, particularly when employed in combination, toxicity and development of resistant strains have limited their usefulness (Palella, et al N. Engl. J. Med . (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001).
- HIV-1 protease Prt
- the HIV Prt cleaves the viral Gag and Gag-Pol polyproteins to produce viral structural proteins (p17, p24, p7 and p6) and the three viral enzymes.
- Combination therapy with RT inhibitors has proven to be highly effective in suppressing viral replication to unquantifiable levels for a sustained period of time. Also, combination therapy with RT and Prt inhibitors (PI) have shown synergistic effects in suppressing HIV replication.
- AZT zidovudineTM, 3′-azido, 3′-deoxythymidine
- AZT demonstrates synergistic antiviral activity in vitro in combination with agents that act at HIV-1 replicative steps other than reverse transcription, including recombinant soluble CD4 castanospermine and recombinant interferon- ⁇ .
- agents that act at HIV-1 replicative steps other than reverse transcription including recombinant soluble CD4 castanospermine and recombinant interferon- ⁇ .
- combinations of compounds can give rise to increased cytotoxicity.
- AZT and recombinant interferon- ⁇ have an increased cytotoxic effect on normal human bone marrow progenitor cells.
- the present invention provides combinations of antiviral compounds, in particular compositions and methods for inhibition of HIV.
- the invention includes a composition including tenofovir disoproxil fumarate and emtricitabine which has anti-HIV activity.
- the composition of tenofovir DF and emtricitabine is both chemically stable and either synergistic and/or reduces the side effects of one or both of tenofovir DF and emtricitabine. Increased patient compliance is likely in view of the lower pill burden and simplified dosing schedule.
- the present invention relates to therapeutic combinations of [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate, tenofovir DF, TDF, Viread®) and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine, EmtrivaTM, ( ⁇ )-cis FTC) and their use in the treatment of HIV infections including infections with HIV mutants bearing resistance to nucleoside and/or non-nucleoside inhibitors.
- the present invention is also concerned with pharmaceutical compositions and formulations of said combinations of tenofovir disoproxil fumarate and emtricitabine.
- Another aspect of the invention is a pharmaceutical formulation comprising a physiologically functional derivative of tenofovir disoproxil fumarate or a physiologically functional derivative of emtricitabine.
- Therapeutic combinations and pharmaceutical compositions and formulations of the invention include the combination of PMEA or PMPA (tenofovir) compounds with emtricitabine or (2R, 5S, cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (3TC, lamivudine, EpivirTM), and their use in the treatment of HIV infections.
- PMEA or PMPA tenofovir
- One aspect of the invention is a method for the treatment or prevention of the symptoms or effects of an HIV infection in an infected animal which comprises administering to, i.e. treating, said animal with a therapeutically effective amount of a combination comprising [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir DF, TDF) or a physiologically functional derivative thereof, and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof.
- Another aspect of the invention is a unit dosage form of a therapeutic combination comprising tenofovir disoproxil fumarate and emtricitabine, or physiological functional derivatives thereof.
- the unit dosage form may be formulated for administration by oral or other routes and is unexpectedly chemically stable in view of the properties of the structurally diverse components.
- Another aspect of the invention is directed to chemically stable combination antiviral compositions comprising tenofovir disoproxil fumarate and emtricitabine.
- the chemically stable combinations of tenofovir disoproxil fumarate and emtricitabine further comprise a third antiviral agent.
- the unique chemical stability of tenofovir disoproxil fumarate and emtricitabine is taken advantage of in order to enable the combination with the third antiviral agent.
- Particularly useful third agents include, by way of example and not limitation, those of Table A.
- the third component is an agent approved for antiviral use in humans, more preferably, it is an NNRTI or a protease inhibitor (PI), more preferably yet, it is an NNRTI.
- the invention is directed to a combination of the chemically stable mixture of tenofovir disoproxil fumarate and emtricitabine together with efavirenz.
- Another aspect of the invention is a patient pack comprising at least one, typically two, and optionally, three active ingredients and other antiviral agents selected from tenofovir disoproxil fumarate and emtricitabine, and an information insert containing directions on the use of tenofovir disoproxil fumarate and emtricitabine together in combination.
- Another aspect of the invention is a process for preparing the combinations hereinbefore described, which comprises bringing into association tenofovir DF and emtricitabine of the combination in a medicament to provide an antiviral effect.
- a combination of the present invention in the manufacture of a medicament for the treatment of any of the aforementioned viral infections or conditions.
- chemical stability means that the two primary antiviral agents in combination are substantially stable to chemical degradation. Preferably, they are sufficiently stable in physical combination to permit commercially useful shelf life of the combination product.
- “chemically stable” means that a first component of the mixture does not act to degrade a second component when the two are brought into physical combination to form a pharmaceutical dosage form. More typically, “chemically stable” means that the acidity of a first component does not catalyzes or otherwise accelerate the acid decomposition of a second component.
- “chemically stable” means that tenofovir disoproxil fumarate is not substantially degraded by the acidity of emtricitabine.
- “Substantially” in this context means at least about less than 10%, preferably less than 1%, more preferably less than 0.1%, more preferably yet, less than 0.01% acid degradation of tenofovir disoproxil fumarate over a 24-hour period when the products are in a pharmaceutical dosage form.
- synergy and “synergistic” mean that the effect achieved with the compounds used together is greater than the sum of the effects that results from using the compounds separately, i.e. greater than what would be predicted based on the two active ingredients administered separately.
- a synergistic effect may be attained when the compounds are: (1) co-formulated and administered or delivered simultaneously in a combined formulation; (2) delivered by alternation or in parallel as separate formulations; or (3) by some other regimen.
- a synergistic effect may be attained when the compounds are administered or delivered sequentially, e.g. in separate tablets, pills or capsules, or by different injections in separate syringes.
- an effective dosage of each active ingredient is administered sequentially, i.e. serially, whereas in combination therapy, effective dosages of two or more active ingredients are administered together.
- a synergistic antiviral effect denotes an antiviral effect which is greater than the predicted purely additive effects of the individual compounds of the combination.
- physiologically functional derivative means a pharmaceutically active compound with equivalent or near equivalent physiological functionality to tenofovir DF or emtricitabine when administered in combination with another pharmaceutically active compound in a combination of the invention.
- physiologically functional derivative includes any: physiologically acceptable salt, ether, ester, prodrug, solvate, stereoisomer including enantiomer, diastereomer or stereoisomerically enriched or racemic mixture, and any other compound which upon administration to the recipient, is capable of providing (directly or indirectly) such a compound or an antivirally active metabolite or residue thereof.
- Bioavailability is the degree to which the pharmaceutically active agent becomes available to the target tissue after the agent's introduction into the body. Enhancement of the bioavailability of a pharmaceutically active agent can provide a more efficient and effective treatment for patients because, for a given dose, more of the pharmaceutically active agent will be available at the targeted tissue sites.
- the compounds of the combinations of the invention may be referred to as “active ingredients” or “pharmaceutically active agents.”
- prodrug refers to any compound that when administered to a biological system generates the drug substance, i.e. active ingredient, as a result of spontaneous chemical reaction(s), enzyme catalyzed chemical reaction(s), and/or metabolic chemical reaction(s).
- Prodrug moiety means a labile functional group which separates from the active inhibitory compound during metabolism, systemically, inside a cell, by hydrolysis, enzymatic cleavage, or by some other process (Bundgaard, Hans, “Design and Application of Prodrugs” in Textbook of Drug Design and Development (1991), P. Krogsgaard-Larsen and H. Bundgaard, Eds. Harwood Academic Publishers, pp. 113-191). Prodrug moieties can serve to enhance solubility, absorption and lipophilicity to optimize drug delivery, bioavailability and efficacy. A “prodrug” is thus a covalently modified analog of a therapeutically-active compound.
- Alkyl means a saturated or unsaturated, branched, straight-chain, branched, or cyclic hydrocarbon radical derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane, alkene, or alkyne.
- Typical alkyl groups consist of 1-18 saturated and/or unsaturated carbons, such as normal, secondary, tertiary or cyclic carbon atoms.
- Examples include, but are not limited to: methyl, Me (—CH 3 ), ethyl, Et (—CH 2 CH 3 ), acetylenic (—C ⁇ CH), ethylene, vinyl (—CH ⁇ CH 2 ), 1-propyl, n-Pr, n-propyl (—CH 2 CH 2 CH 3 ), 2-propyl, i-Pr, i-propyl (—CH(CH 3 ) 2 ), allyl (—CH 2 CH ⁇ CH 2 ), propargyl (—CH 2 C ⁇ CH), cyclopropyl (—C 3 H 5 ), 1-butyl, n-Bu, n-butyl (—CH 2 CH 2 CH 2 CH 3 ), 2-methyl-1-propyl, i-Bu, i-butyl (—CH 2 CH(CH 3 ) 2 ), 2-butyl, s-Bu, s-butyl (—CH(CH 3 )CH 2 CH 3 ), 2-methyl-2-propyl, t-Bu
- Aryl means a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system.
- Typical aryl groups include, but are not limited to, radicals derived from benzene, substituted benzene, naphthalene, anthracene, biphenyl, and the like.
- Arylalkyl refers to an acyclic alkyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp 3 carbon atom, is replaced with an aryl radical.
- Typical arylalkyl groups include, but are not limited to, benzyl, 2-phenylethan-1-yl, 2-phenylethen-1-yl, naphthylmethyl, 2-naphthylethan-1-yl, 2-naphthylethen-1-yl, naphthobenzyl, 2-naphthophenylethan-1-yl and the like.
- the arylalkyl group 6 to 20 carbon atoms e.g., the alkyl moiety, including alkanyl, alkenyl or alkynyl groups, of the arylalkyl group is 1 to 6 carbon atoms and the aryl moiety is 5 to 14 carbon atoms.
- Substituted alkyl mean alkyl, aryl, and arylalkyl respectively, in which one or more hydrogen atoms are each independently replaced with a substituent.
- Typical substituents include, but are not limited to, —X, —R, —O ⁇ , —OR, —SR, —S ⁇ , —NR 2 , —NR 3 , ⁇ NR, —CX 3 , —CN, —OCN, —SCN, —N ⁇ C ⁇ O, —NCS, —NO, —NO 2 , ⁇ N 2 , —N 3 , NC( ⁇ O)R, —C( ⁇ O)R, —C( ⁇ O)NRR —S( ⁇ O) 2 O ⁇ , —S( ⁇ O) 2 OH, —S( ⁇ O) 2 R, —OS( ⁇ O) 2 OR, —S( ⁇ O) 2 NR, —S( ⁇ O)R, —OP( ⁇ O)O 2 RR, —P( ⁇ O)O 2 RR —P( ⁇ O)(O ⁇ ) 2 , —P( ⁇ O)(OH) 2
- Heteroaryl and “Heterocycle” refer to a ring system in which one or more ring atoms is a heteroatom, e.g. nitrogen, oxygen, and sulfur. Heterocycles are described in: Katritzky, Alan R., Rees, C. W., and Scriven, E. Comprehensive Heterocyclic Chemistry (1996) Pergamon Press; Paquette, Leo A Principles of Modern Heterocyclic Chemistry W. A. Benjamin, New York, (1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28.
- heterocycles include but are not limited to substituents, i.e. radicals, derived from pyrrole, indole, furan, benzofuran, thiophene, benzothiophene, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 2-imidazole, 4-imidazole, 3-pyrazole, 4-pyrazole, pyridazine, pyrimidine, pyrazine, purine, cinnoline, pthalazine, quinazoline, quinoxaline, 3-(1,2,4-N)-triazolyl, 5-(1,2,4-N)-triazolyl, 5-tetrazolyl, 4-(1-0,3-N)-oxazole, 5-(1-O, 3-N)-oxazole, 4-(1-S, 3-N)-thiazole, 5-(1-S, 3-N)-thiazole, 2-(
- d and 1 or (+) and ( ⁇ ) are employed to designate the sign of rotation of plane-polarized light by the compound, with ( ⁇ ) or 1 meaning that the compound is levorotatory.
- a compound prefixed with (+) or d is dextrorotatory.
- these compounds, called stereoisomers are identical except that they are mirror images of one another.
- a specific stereoisomer is also referred to as an enantiomer, and a mixture of such isomers is often called an enantiomeric mixture.
- a 50:50 mixture of enantiomers is referred to as a racemic mixture or a racemate.
- racemic mixture and “racemate” refer to an equimolar mixture of two enantiomeric species, devoid of optical activity.
- chiral refers to molecules which have the property of non-superimposability of the mirror image partner, while the term “achiral” refers to molecules which are superimposable on their mirror image partner.
- stereoisomers refers to compounds which have identical chemical constitution, but differ with regard to the arrangement of the atoms or groups in space.
- Diastereomer refers to a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities. Mixtures of diastereomers may separate under high resolution analytical procedures such as electrophoresis and chromatography.
- Enantiomers refer to two stereoisomers of a compound which are non-superimposable mirror images of one another.
- the present invention provides novel combinations of two or more active ingredients being employed together.
- a synergistic antiviral effect is achieved.
- a chemically stable combination is obtained.
- the combinations include at least one active ingredient selected from (1) tenofovir disoproxil fumarate and physiologically functional derivatives, and at least one active ingredient selected from (2) emtricitabine and physiologically functional derivatives.
- the term “synergistic antiviral effect” is used herein to denote an antiviral effect which is greater than the predicted purely additive effects of the individual components (a) and (b) of the combination.
- Tenofovir disoproxil fumarate also known as Viread®, Tenofovir DF, Tenofovir disoproxil, TDF, Bis-POC-PMPA (U.S. Pat. Nos. 5,935,946, 5,922,695, 5,977,089, 6,043,230, 6,069,249) is a prodrug of tenofovir, and has the structure:
- Tenofovir disoproxil include: [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester; 9-[(R)-2-[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine; and 2,4,6,8-tetraoxa-5-phosphanonanedioic acid, 5-[[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]-, bis(1-methylethyl)ester, 5-oxide.
- the CAS Registry numbers include: 201341-05-1; 202138-50-9; 206184-49-8. It should be noted that the ethoxymethyl unit of tenofovir has a chiral center.
- the R (rectus, right handed configuration) enantiomer is shown. However, the invention also includes the S isomer.
- the invention includes all enantiomers, diastereomers, racemates, and enriched stereoisomer mixtures of tenofovir (PMPA) and physiologically functional derivatives thereof.
- PMPA or tenofovir (U.S. Pat. Nos. 4,808,716, 5,733,788, 6,057,305) has the structure:
- the chemical names of PMPA, tenofovir include: (R)-9-(2-phosphonylmethoxypropyl)adenine; and phosphonic acid, [[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl].
- the CAS Registry number is 147127-20-6.
- Tenofovir disoproxil fumarate is a nucleotide reverse transcriptase inhibitor approved in the United States in 2001 for the treatment of HIV-1 infection in combination with other antiretroviral agents.
- Tenofovir disoproxil fumarate or Viread® is the fumarate salt of tenofovir disoproxil.
- Viread® may be named as: 9-[(R)-2-[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1); or 2,4,6,8-tetraoxa-5-phosphanonanedioic acid, 5-[[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]-, bis(1-methylethyl)ester, 5-oxide, (2E)-2-butenedioate (1:1).
- the CAS Registry number is 202138-50-9.
- Physiologically functional derivatives of tenofovir disoproxil fumarate include PMEA (adefovir, 9-((R)-2-(phosphonomethoxy)ethyl)adenine) and PMPA compounds.
- PMEA adefovir, 9-((R)-2-(phosphonomethoxy)ethyl)adenine
- PMPA compounds Exemplary combinations include a PMEA or PMPA compound in combination with emtricitabine or 3TC.
- PMEA and PMPA compounds have the structures:
- PMEA R 3 is H
- PMPA R 3 is C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, or CH 2 OR 8 where R 8 is C 1 -C 6 alkyl, C 1 -C 6 hydroxyalkyl or C 1 -C 6 haloalkyl.
- R 6 and R 7 are independently H or C 1 -C 6 alkyl.
- R 4 and R 5 are independently H, NH 2 , NHR or NR 2 where R is C 1 -C 6 alkyl.
- R 1 and R 2 are independently H, C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl, C 6 -C 20 substituted aryl, C 6 -C 20 arylalkyl, C 6 -C 20 substituted arylalkyl, acyloxymethyl esters —CH 2 OC( ⁇ O)R 9 (e.g. POM) or acyloxymethyl carbonates —CH 2 OC( ⁇ O)OR 9 (e.g. POC) where R 9 is C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl or C 6 -C 20 substituted aryl.
- R 1 and R 2 may be pivaloyloxymethoxy, POM, —CH 2 OC( ⁇ O)C(CH 3 ) 3 ; —CH 2 OC( ⁇ O)OC(CH 3 ) 3 ; or POC, —CH 2 OC( ⁇ O)OCH(CH 3 ) 2 .
- tenofovir has the structure where R 3 is CH 3 , and R 1 , R 2 , R 4 , R 5 , R 6 and R 7 are H.
- Dialkyl phosphonates may be prepared according to the methods of: Quast et al (1974) Synthesis 490; Stowell et al (1990) Tetrahedron Lett. 3261; U.S. Pat. No. 5,663,159.
- the PMPA compound may be enantiomerically-enriched or purified (single stereoisomer) where the carbon atom bearing R 3 may be the R or S enantiomer.
- the PMPA compound may be a racemate, i.e. a mixture of R and S stereoisomers.
- Adefovir (9-(2-phosphonomethoxyethyl)adenine where R 1 —R 7 ⁇ H) is an exemplary PMEA compound (U.S. Pat. Nos. 4,808,716, 4,724,233).
- Adefovir dipivoxil also known as bis-POM PMEA, (R 3 —R 7 ⁇ H, R 1 and R 2 ⁇ —CH 2 OC( ⁇ O)C(CH 3 ) 3 , pivoxil, POM, pivaloyloxymethoxy), is effective against HIV and Hepatitis B infections (U.S. Pat. Nos. 5,663,159, 6,451,340).
- Adefovir dipivoxil has demonstrated minor to moderate synergistic inhibition of HIV replication in combination with other compounds with anti-HIV activity including PMPA, d4T, ddC, nelfinavir, ritonavir, and saquinavir (Mulato et al (1997) Antiviral Research 36:91-97).
- the invention includes all enantiomers, diastereomers, racemates, and enriched stereoisomer mixtures of PMEA and PMPA, and physiologically functional derivatives thereof.
- Emtricitabine ( ⁇ )-cis-FTC, EmtrivaTM), a single enantiomer of FTC, is a potent nucleoside reverse transcriptase inhibitor approved for the treatment of HIV (U.S. Pat. Nos. 5,047,407, 5,179,104, 5,204,466, 5,210,085, 5,486,520, 5,538,975, 5,587,480, 5618820, 5,763,606, 5,814,639, 5,914,331, 6,114,343, 6180639, 6215004; WO 02/070518).
- the single enantiomer emtricitabine has the structure:
- the chemical names for emtricitabine include: ( ⁇ )-cis-FTC; ⁇ -L-hydroxymethyl-5-(5-fluorocytosin-1-yl)-1,3-oxathiolane; (2R,5S)-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine; and 4-amino-5-fluoro-1-(2-hydroxymethyl-[1,3]-(2R,5S)-oxathiolan-5-yl)-1H-pyrimidin-2-one.
- the CAS Registry numbers include: 143491-57-0; 143491-54-7.
- FTC contains two chiral centers, at the 2 and 5 positions of the oxathiolane ring, and therefore can exist in the form of two pairs of optical isomers (i.e. enantiomers) and mixtures thereof including racemic mixtures.
- FTC may be either a cis or a trans isomer or mixtures thereof.
- Mixtures of cis and trans isomers are diastereomers with different physical properties.
- Each cis and trans isomer can exist as one of two enantiomers or mixtures thereof including racemic mixtures.
- the invention includes all enantiomers, diastereomers, racemates, and enriched stereoisomer mixtures of emtricitabine and physiologically functional derivatives thereof.
- the invention includes physiological functional derivatives such as the 1:1 racemic mixture of the enantiomers (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) and its mirror image (2S, 5R, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one, or mixtures of the two enantiomers in any relative amount.
- the invention also includes mixtures of cis and trans forms of FTC.
- Physiologically functional derivatives of emtricitabine include 1,3 oxathiolane nucleosides having the structure:
- B is a nucleobase including any nitrogen-containing heterocyclic moiety capable of forming Watson-Crick hydrogen bonds in pairing with a complementary nucleobase or nucleobase analog, e.g. a purine, a 7-deazapurine, or a pyrimidine.
- a complementary nucleobase or nucleobase analog e.g. a purine, a 7-deazapurine, or a pyrimidine.
- B include the naturally occurring nucleobases: adenine, guanine, cytosine, uracil, thymine, and minor constituents and analogs of the naturally occurring nucleobases, e.g.
- Nucleobases B may be attached in the configurations of naturally-occurring nucleic acids to the 1,3 oxathiolane moiety through a covalent bond between the N-9 of purines, e.g. adenin-9-yl and guanin-9-yl, or N-1 of pyrimidines, e.g. thymin-1-yl and cytosin-1-yl (Blackburn, G. and Gait, M. Eds. “DNA and RNA structure” in Nucleic Acids in Chemistry and Biology, 2nd Edition, (1996) Oxford University Press, pp. 15-81).
- purines e.g. adenin-9-yl and guanin-9-yl
- N-1 of pyrimidines e.g. thymin-1-yl and cytosin-1-yl
- R is H, C 1 -C 18 alkyl, C 1 -C 18 substituted alkyl, C 2 -C 18 alkenyl, C 2 -C 18 substituted alkenyl, C 2 -C 18 alkynyl, C 2 -C 18 substituted alkynyl, C 6 -C 20 aryl, C 6 -C 20 substituted aryl, C 2 -C 20 heterocycle, C 2 -C 20 substituted heterocycle, phosphonate, phosphophosphonate, diphosphophosphonate, phosphate, diphosphate, triphosphate, polyethyleneoxy, or a prodrug moiety.
- Physiologically functional derivatives of emtricitabine also include 3TC (lamivudine, Epivir®), a reverse transcriptase inhibitor approved in the United States for the treatment of HIV-1 infection in combination with AZT as Combivir® (GlaxoSmithKline).
- 3TC lamivudine, Epivir®
- AZT AZT
- Combivir® GaxoSmithKline
- Lamivudine U.S. Pat. Nos. 5,587,480, 5,696,254, 5,618,820, 5,756,706, 5,744,596, 568,164, 5,466,806, 5,151,426) has the structure:
- 3TC may be a physiologically functional derivative of emtricitabine in combination with tenofovir DF or a physiologically functional derivative of tenofovir DF.
- tenofovir DF and emtricitabine may exist in keto or enol tautomeric forms and the use of any tautomeric form thereof is within the scope of this invention.
- Tenofovir DF and emtricitabine will normally be utilized in the combinations of the invention substantially free of the corresponding enantiomer, that is to say no more than about 5% w/w of the corresponding enantiomer will be present.
- the invention includes all prodrugs of tenofovir and emtricitabine.
- An exemplary prodrug of tenofovir is tenofovir disoproxil fumarate (TDF, Viread®).
- TDF tenofovir disoproxil fumarate
- a large number of structurally-diverse prodrugs have been described for phosphonic acids (Freeman and Ross in Progress in Medicinal Chemistry 34: 112-147 (1997).
- a commonly used prodrug class is the acyloxyalkyl ester, which was first used as a prodrug strategy for carboxylic acids and then applied to phosphates and phosphonates by Farquhar et al (1983) J. Pharm. Sci. 72: 324; also U.S. Pat. Nos.
- acyloxyalkyl ester was used to deliver phosphonic acids across cell membranes and to enhance oral bioavailability.
- a close variant of the acyloxyalkyl ester strategy, the alkoxycarbonyloxyalkyl ester, may also enhance oral bioavailability as a prodrug moiety in the compounds of the combinations of the invention.
- Aryl esters of phosphorus groups, especially phenyl esters, are reported to enhance oral bioavailability (DeLambert et al (1994) J. Med. Chem. 37: 498).
- Phenyl esters containing a carboxylic ester ortho to the phosphate have also been described (Khamnei and Torrence, (1996) J. Med. Chem. 39:4109-4115). Benzyl esters are reported to generate the parent phosphonic acid. In some cases, substituents at the ortho- or para-position may accelerate the hydrolysis. Benzyl analogs with an acylated phenol or an alkylated phenol may generate the phenolic compound through the action of enzymes, e.g. esterases, oxidases, etc., which in turn undergoes cleavage at the benzylic C—O bond to generate the phosphoric acid and the quinone methide intermediate.
- enzymes e.g. esterases, oxidases, etc.
- prodrugs examples include Mitchell et al (1992) J. Chem. Soc. Perkin Trans. I 2345; Brook et al WO 91/19721. Still other benzylic prodrugs have been described containing a carboxylic ester-containing group attached to the benzylic methylene (Glazier et al WO 91/19721). Thio-containing prodrugs are reported to be useful for the intracellular delivery of phosphonate drugs. These proesters contain an ethylthio group in which the thiol group is either esterified with an acyl group or combined with another thiol group to form a disulfide.
- Prodrug esters in accordance with the invention are independently selected from the following groups: (1) mono-, di-, and tri-phosphate esters of tenofovir or emtricitabine or any other compound which upon administration to a human subject is capable of providing (directly or indirectly) said mono-, di, or triphosphate ester; (2) carboxylic acid esters (3) sulphonate esters, such as alkyl- or aralkylsulphonyl (for example, methanesulphonyl); (4) amino acid esters (for example, alanine, L-valyl or L-isoleucyl); (5) phosphonate; and (6) phosphonamidate esters.
- Ester groups (1)-(6) may be substituted with; straight or branched chain C 1 -C 18 alkyl (for example, methyl, n-propyl, t-butyl, or n-butyl); C 3 -C 12 cycloalkyl; alkoxyalkyl (for example, methoxymethyl); arylalkyl (for example, benzyl); aryloxyalkyl (for example, phenoxymethyl); C 5 -C 20 aryl (for example, phenyl optionally substituted by, for example, halogen, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, or amino; acyloxymethyl esters —CH 2 OC( ⁇ O)R 9 (e.g.
- ester groups may be: —CH 2 OC( ⁇ O)C(CH 3 ) 3 , —CH 2 OC( ⁇ O)OC(CH 3 ) 3 or —CH 2 OC( ⁇ O)OCH(CH 3 ) 2 .
- An exemplary aryl moiety present in such esters comprises a phenyl or substituted phenyl group.
- Many phosphate prodrug moieties are described in U.S. Pat. No. 6,312,662; Jones et al (1995) Antiviral Research 27:1-17; Kucera et al (1990) AIDS Res. Hum. Retro Viruses 6:491-501; Piantadosi et al (1991) J. Med. Chem. 34:1408-14; Hosteller et al (1992) Antimicrob. Agents Chemother. 36:2025-29; Hostetler et al (1990) J. Biol. Chem. 265:611127; and Siddiqui et al (1999) J. Med. Chem. 42:4122-28.
- prodrugs refer to a compound that is metabolized in the host, for example hydrolyzed or oxidized, by either enzymatic action or by general acid or base solvolysis, to form an active ingredient.
- Typical examples of prodrugs of the active ingredients of the combinations of the invention have biologically labile protecting groups on a functional moiety of the active compound.
- Prodrugs include compounds that can be oxidized, reduced, aminated, deaminated, esterified, deesterified, alkylated, dealkylated, acylated, deacylated, phosphorylated, dephosphorylated, or other functional group change or conversion involving forming or breaking chemical bonds on the prodrug.
- the chemical stability of the active ingredients in a pharmaceutical formulation is of concern to minimize the generation of impurities and ensure adequate shelf-life.
- the active ingredients, tenofovir disoproxil fumarate and emtricitabine, in the pharmaceutical formulations of the invention have relatively low pKa values, indicative of the potential to cause acidic hydrolysis of the active ingredients.
- Emtricitabine, with a pKa of 2.65 (EmtrivaTM Product Insert, Gilead Sciences, Inc. 2003, available at gilead.com) is subject to hydrolytic deamination of the 5-fluoro cytosine nucleobase to form the 5-fluoro uridine nucleobase.
- Tenofovir disoproxil fumarate with a pKa of 3.75 (Yuan L. et al “Degradation Kinetics of Oxycarbonyloxymethyl Prodrugs of Phosphonates in Solution”, Pharmaceutical Research (2001) Vol. 18, No. 2, 234-237), is subject also to hydrolytic deamination of the exocyclic amine of the adenine nucleobase, and to hydrolysis of one or both of the POC ester groups (U.S. Pat. No. 5,922,695). It is desirable to formulate a therapeutic combination of tenofovir disoproxil fumarate and emtricitabine, and the physiological functional derivatives thereof, with a minimum of impurities and adequate stability.
- the combinations of the present invention provide combination pharmaceutical dosage forms which are chemically stable to acid degradation of: (1) a first component (such as tenofovir disoproxil fumarate, and physiological functional derivatives; (2) a second component (such as emtricitabine, and physiological functional derivatives; and (3) optionally a third component having antiviral activity.
- the third component includes anti-HIV agents and include: protease inhibitors (PI), nucleoside reverse transcriptase inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI), and integrase inhibitors.
- Exemplary third active ingredients to be administered in combination with first and second components are shown in Table A. First and second components are as defined in the above section entitled: ACTIVE INGREDIENTS OF THE COMBINATIONS.
- any reference to any of the compounds in the compositions of the invention also includes any physiologically acceptable salt thereof.
- physiologically acceptable salts of tenofovir DF, emtricitabine and their physiologically functional derivatives include salts derived from an appropriate base, such as an alkali metal (for example, sodium), an alkaline earth (for example, magnesium), ammonium and NX 4 + (wherein X is C 1 -C 4 alkyl), or an organic acid such as fumaric acid, acetic acid, succinic acid.
- Physiologically acceptable salts of an hydrogen atom or an amino group include salts of organic carboxylic acids such as acetic, benzoic, lactic, fumaric, tartaric, maleic, malonic, malic, isethionic, lactobionic and succinic acids; organic sulfonic acids, such as methanesulfonic, ethanesulfonic, benzenesulfonic and p-toluenesulfonic acids; and inorganic acids, such as hydrochloric, sulfuric, phosphoric and sulfamic acids.
- organic carboxylic acids such as acetic, benzoic, lactic, fumaric, tartaric, maleic, malonic, malic, isethionic, lactobionic and succinic acids
- organic sulfonic acids such as methanesulfonic, ethanesulfonic, benzenesulfonic and p-toluenesulfonic acids
- Physiologically acceptable salts of a compound of an hydroxy group include the anion of said compound in combination with a suitable cation such as Na + and NX 4 + (wherein X is independently selected from H or a C 1 -C 4 alkyl group).
- salts of active ingredients of the combinations of the invention will be physiologically acceptable, i.e. they will be salts derived from a physiologically acceptable acid or base.
- salts of acids or bases which are not physiologically acceptable may also find use, for example, in the preparation or purification of a physiologically acceptable compound. All salts, whether or not derived from a physiologically acceptable acid or base, are within the scope of the present invention.
- a two-part or three-part combination may be administered simultaneously or sequentially.
- the combination may be administered in one, two, or three administrations.
- two-part or three-part combinations are administered in a single pharmaceutical dosage form. More preferably, a two-part combination is administered as a single oral dosage form and a three-part combination is administered as two identical oral dosage forms. Examples include a single tablet of tenofovir disoproxil fumarate and emtricitabine, or two tablets of tenofovir disoproxil fumarate, emtricitabine, and efavirenz.
- the compounds of the combination may be administered: (1) simultaneously by combination of the compounds in a co-formulation or (2) by alternation, i.e. delivering the compounds serially, sequentially, in parallel or simultaneously in separate pharmaceutical formulations.
- alternation therapy the delay in administering the second, and optionally a third active ingredient, should not be such as to lose the benefit of a synergistic therapeutic effect of the combination of the active ingredients.
- the combination should be administered to achieve peak plasma concentrations of each of the active ingredients.
- a one pill once-per-day regimen by administration of a combination co-formulation may be feasible for some HIV-positive patients.
- Effective peak plasma concentrations of the active ingredients of the combination will be in the range of approximately 0.001 to 100 ⁇ M. Optimal peak plasma concentrations may be achieved by a formulation and dosing regimen prescribed for a particular patient. It will also be understood that tenofovir DF and emtricitabine, or the physiologically functional derivatives of either thereof, whether presented simultaneously or sequentially, may be administered individually, in multiples, or in any combination thereof. In general, during alternation therapy (2), an effective dosage of each compound is administered serially, where in co-formulation therapy (1), effective dosages of two or more compounds are administered together.
- the individual components of the combination are administered separately they are generally each presented as a pharmaceutical formulation.
- the references hereinafter to formulations refer unless otherwise stated to formulations containing either the combination or a component compound thereof. It will be understood that the administration of the combination of the invention by means of a single patient pack, or patient packs of each formulation, within a package insert diverting the patient to the correct use of the invention is a desirable additional feature of this invention.
- the invention also includes a double pack comprising in association for separate administration, formulations of tenofovir disoproxil fumarate and emtricitabine, or a physiologically functional derivative of either or both thereof.
- the combination therapies of the invention include: (1) a combination of tenofovir DF and emtricitabine or (2) a combination containing a physiologically functional derivative of either or both thereof.
- the combination may be formulated in a unit dosage formulation comprising a fixed amount of each active pharmaceutical ingredient for a periodic, e.g. daily, dose or subdose of the active ingredients.
- compositions according to the present invention comprise a combination according to the invention together with one or more pharmaceutically acceptable carriers or excipients and optionally other therapeutic agents.
- Pharmaceutical formulations containing the active ingredient may be in any form suitable for the intended method of administration. When used for oral use for example, tablets, troches, lozenges, aqueous or oil suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups or elixirs may be prepared ( Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, Pa.).
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents including antioxidants, sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation.
- Tablets containing the active ingredient in admixture with non-toxic pharmaceutically acceptable excipient which are suitable for manufacture of tablets are acceptable.
- excipients may be, for example, inert diluents, such as calcium or sodium carbonate, lactose, lactose monohydrate, croscarmellose sodium, povidone, calcium or sodium phosphate; granulating and disintegrating agents, such as maize starch, or alginic acid; binding agents, such as cellulose, microcrystalline cellulose, starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- inert diluents such as calcium or sodium carbonate, lactose, lactose monohydrate, croscarmellose sodium
- Formulations for oral use may be also presented as hard gelatin capsules where the active ingredient is mixed with an inert solid diluent, for example pregelatinized starch, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example pregelatinized starch, calcium phosphate or kaolin
- an oil medium such as peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions of the invention contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients include a suspending agent, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia, and dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadecaethyleneoxycetanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene sorbitan monooleate).
- a suspending agent
- the aqueous suspension may also contain one or more preservatives such as ethyl or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents and one or more sweetening agents, such as sucrose, sucralose or saccharin.
- preservatives such as ethyl or n-propyl p-hydroxybenzoate
- coloring agents such as a coloring agent
- flavoring agents such as sucrose, sucralose or saccharin.
- sweetening agents such as sucrose, sucralose or saccharin.
- Oil suspensions may be formulated by suspending the active ingredient in a vegetable oil, such as arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oral suspensions may contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol.
- Sweetening agents, such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation.
- These compositions may be preserved by the addition of an antioxidant such as ascorbic acid, BHT, etc.
- Dispersible powders and granules of the invention suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, a suspending agent, and one or more preservatives.
- a dispersing or wetting agent and suspending agents are exemplified by those disclosed above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
- the pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions or liposome formulations.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these.
- Suitable emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth, naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids and hexitol anhydrides, such as sorbitan monooleate, and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan monooleate.
- the emulsion may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose.
- Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- the pharmaceutical compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous or oleaginous suspension.
- a sterile injectable preparation such as a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1,3-butane-diol or prepared as a lyophilized powder.
- a non-toxic parenterally acceptable diluent or solvent such as a solution in 1,3-butane-diol or prepared as a lyophilized powder.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile fixed oils may conventionally be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- compositions of the invention may be injected parenterally, for example, intravenously, intraperitoneally, intrathecally, intraventricularly, intrastemally, intracranially, intramuscularly or subcutaneously, or they may be administered by infusion techniques. They are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood.
- aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary.
- suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
- compositions of the invention may also be administered intranasally or by inhalation and are conveniently delivered in the form of a dry powder inhaler or an aerosol spray presentation from a pressurized container or a nebuliser with the use of a suitable propellant, e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoroalkane such as 1,1,1,2-tetrafluoroethane (HFC 134a), carbon dioxide or other suitable gas.
- a suitable propellant e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoroalkane such as 1,1,1,2-tetrafluoroethane (HFC 134a), carbon dioxide or other suitable gas.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- the pressurized container or nebuliser may contain a solution or suspension of the composition, e.g. using a mixture of ethanol and the propellant as the solvent, which may additional contain a lubricant, e.g. sorbitan trioleate.
- Capsules and cartridges (made, for example, from gelatin) for use in an inhaler or insufflator may be formulated to contain a powder mix of a compound of the formula (I) and a suitable powder base such as lactose or starch. Aerosol or dry powder formulations are preferably arranged so that each metered dose or “puff” contains from 20 ⁇ g to 20 mg of a composition for delivery to the patient.
- the overall daily dose with an aerosol will be in the range of from 20 ⁇ g to 20 mg which may be administered in a single dose or, more usually, in divided doses throughout the day.
- a time-release formulation intended for oral administration to humans may contain approximately 1 to 1000 mg of active material compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total compositions (weight:weight).
- the pharmaceutical composition can be prepared to provide easily measurable amounts for administration.
- an aqueous solution intended for intravenous infusion may contain from about 3 to 500 ⁇ g of the active ingredient per milliliter of solution in order that infusion of a suitable volume at a rate of about 30 mL/hr can occur.
- formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be administered as a bolus, electuary or paste.
- the combinations of the invention may conveniently be presented as a pharmaceutical formulation in a unitary dosage form.
- a convenient unitary dosage formulation contains the active ingredients in any amount from 1 mg to 1 g each, for example but not limited to, 10 mg to 300 mg.
- the synergistic effects of tenofovir DF in combination with emtricitabine may be realized over a wide ratio, for example 1:50 to 50:1 (tenofovir DF:emtricitabine). In one embodiment, the ratio may range from about 1:10 to 10:1. In another embodiment, the weight/weight ratio of tenofovir to emtricitabine in a co-formulated combination dosage form, such as a pill, tablet, caplet or capsule will be about 1, i.e.
- tenofovir DF an approximately equal amount of tenofovir DF and emtricitabine.
- 300 mg tenofovir DF and 200 mg emtricitabine can be co-formulated in a ratio of 1.5:1 (tenofovir DF: emtricitabine).
- each compound will be employed in the combination in an amount at which it exhibits antiviral activity when used alone.
- Exemplary Formulations A, B, C, D, E, and F have ratios of 12:1 to 1:1 (tenofovir DF: emtricitabine).
- Exemplary Formulations A, B, C, D, E, and F use amounts of tenofovir DF and emtricitabine ranging from 25 mg to 300 mg. Other ratios and amounts of the compounds of said combinations are contemplated within the scope of the invention.
- a unitary dosage form may further comprise tenofovir DF and emtricitabine, or physiologically functional derivatives of either thereof, and a pharmaceutically acceptable carrier.
- the amount of active ingredients in the combinations of the invention required for use in treatment will vary according to a variety of factors, including the nature of the condition being treated and the age and condition of the patient, and will ultimately be at the discretion of the attending physician or health care practitioner.
- the factors to be considered include the route of administration and nature of the formulation, the animal's body weight, age and general condition and the nature and severity of the disease to be treated.
- doses ranging from 25 mg to 200 mg twice-a-day for two weeks. At each dose regimen greater or equal to 200 mg, a 98-percent (1.75 log 10) or greater viral suppression was observed.
- Viread® tenofovir DF
- EmtrivaTM emtricitabine
- any two of the active ingredients in a unitary dosage form for simultaneous or sequential administration with a third active ingredient.
- the three-part combination may be administered simultaneously or sequentially. When administered sequentially, the combination may be administered in two or three administrations.
- Third active ingredients have anti-HIV activity and include protease inhibitors (PI), nucleoside reverse transcriptase inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI), and integrase inhibitors.
- PI protease inhibitors
- NRTI nucleoside reverse transcriptase inhibitors
- NRTI non-nucleoside reverse transcriptase inhibitors
- integrase inhibitors Exemplary third active ingredients to be administered in combination with tenofovir DF, emtricitabine, and their physiological functional derivatives, are shown in Table A.
- Another aspect of the present invention is a three-part combination comprising tenofovir DF, FTC, and 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine, also designated herein as GS-7340, which has the structure:
- GS-7340 is a prodrug of tenofovir and the subject of commonly owned, pending application, U.S. Ser. No. 09/909,560, filed Jul. 20, 2001 and Becker et al WO 02/08241.
- a ternary unitary dosage may contain 1 mg to 1000 mg of tenofovir disoproxil fumarate, 1 mg to 1000 mg of emtricitabine, and 1 mg to 1000 mg of the third active ingredient.
- a unitary dosage form may further comprise tenofovir DF, emtricitabine, the third active ingredient, or physiologically functional derivatives of the three active ingredients thereof, and a pharmaceutically acceptable carrier.
- Combinations of the present invention enable patients greater freedom from multiple dosage medication regimens and ease the needed diligence required in remembering and complying with complex daily dosing times and schedules.
- the desired daily regimen may be presented in a single dose or as two or more sub-doses per day.
- the combination of co-formulated tenofovir DF and emtricitabine may be administered as a single pill, once per day.
- a further aspect of the invention is a patient pack comprising at least one active ingredient: tenofovir disoproxil fumarate, emtricitabine, or a physiologically functional derivative of either of the combination and an information package or product insert containing directions on the use of the combination of the invention.
- Segregation of active ingredients in pharmaceutical powders and granulations is a widely recognized problem that can result in inconsistent dispersions of the active ingredients in final dosage forms.
- Some of the main factors contributing to segregation are particle size, shape and density. Segregation is particularly troublesome when attempting to formulate a single homogenous tablet containing multiple active ingredients having different densities and different particle sizes.
- Glidants are substances that have traditionally been used to improve the flow characteristics of granulations and powders by reducing interparticulate friction. See Lieberman, Lachman , & Schwartz, Pharmaceutical Dosage Forms: Tablets, Volume 1, p. 177-178 (1989), incorporated herein by reference.
- Glidants are typically added to pharmaceutical compositions immediately prior to tablet compression to facilitate the flow of granular material into the die cavities of tablet presses.
- Glidants include: colloidal silicon dioxide, asbestos free talc, sodium aluminosilicate, calcium silicate, powdered cellulose, microcrystalline cellulose, corn starch, sodium benzoate, calcium carbonate, magnesium carbonate, metallic stearates, calcium stearate, magnesium stearate, zinc stearate, stearowet C, starch, starch 1500, magnesium lauryl sulfate, and magnesium oxide.
- Exemplary Tablet Formulation A has colloidal silicon dioxide (Examples).
- Glidants can be used to increase and aid blend composition homogeneity in formulations of anti-HIV drugs (U.S. Pat. No. 6,113,920).
- the novel compositions of the present invention may contain glidants to effect and maintain homogeneity of active ingredients during handling prior to tablet compression.
- the present invention provides pharmaceutical formulations combining the active ingredients tenofovir DF and emtricitabine, or physiologically functional derivatives thereof, in a sufficiently homogenized form, and a method for using this pharmaceutical formulation.
- An object of the present invention is to utilize glidants to reduce the segregation of active ingredients in pharmaceutical compositions during pre-compression material handling.
- Another object of the present invention is to provide a pharmaceutical formulation combining the active ingredients tenofovir DF and emtricitabine, or physiologically functional derivatives thereof, with a pharmaceutically acceptable glidant, resulting in a mixture characterized by a pharmaceutically acceptable measure of homogeneity.
- Formulations include those suitable for oral, rectal, nasal, topical (including transdermal, buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) administration.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. Such methods represent a further feature of the present invention and include the step of bringing into association the active ingredients with the carrier, which constitutes one or more accessory ingredients, and maintaining chemical stability.
- the formulations are prepared by uniformly and intimately bringing into association the active ingredients with liquid carriers or finely divided solid carriers or both, and then if necessary shaping the product.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, caplets, cachets or tablets each containing a predetermined amount of the active ingredients; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be presented as a bolus, electuary or paste.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredients in a free-flowing form such as a powder or granules, optionally mixed with a binder (e.g. povidone, gelatin, hydroxypropyl methylcellulose), lubricant, inert diluent, preservative, disintegrant (e.g. sodium starch glycollate, cross-linked povidone, cross-linked sodium carboxymethyl cellulose) surface-active or dispersing agent.
- Molded tablets may be made by molding a mixture of the powdered compound moistened with an inert liquid diluent in a suitable machine.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredients therein using, for example, cellulose ether derivatives (e.g., hydroxypropyl methylcellulose) or methacrylate derivatives in varying proportions to provide the desired release profile. Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach.
- cellulose ether derivatives e.g., hydroxypropyl methylcellulose
- methacrylate derivatives in varying proportions to provide the desired release profile.
- Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach.
- Formulations suitable for topical administration in the mouth include lozenges comprising the active ingredients in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Formulations for rectal administration may be presented as a suppository with a suitable base comprising, for example, cocoa butter or a salicylates.
- Topical administration may also be by means of a transdermal iontophoretic device.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- Formulations suitable for penile administration for prophylactic or therapeutic use may be presented in condoms, creams, gels, pastes, foams or spray formulations containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- compositions suitable for rectal administration wherein the carrier is a solid are most preferably presented as unit dose suppositories.
- Suitable carriers include cocoa butter and other materials commonly used in the art.
- the suppositories may be conveniently formed by admixture of the active combination with the softened or melted carrier(s) followed by chilling and shaping in moulds.
- Formulations suitable for parenteral administration include aqueous and nonaqueous isotonic sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents; and liposomes or other microparticulate systems which are designed to target the compound to blood components or one or more organs.
- the formulations may be presented in unit-dose or multi-dose sealed containers, for example, ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injection, immediately prior to use.
- sterile liquid carrier for example water for injection
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Exemplary unit dosage formulations are those containing a daily dose or daily subdose of the active ingredients, as hereinbefore recited, or an appropriate fraction thereof. It should be understood that in addition to the ingredients particularly mentioned above the formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question, for example, those suitable for oral administration may include such further agents as sweeteners, thickeners and flavoring agents.
- the compounds of the combination of the present invention may be obtained in a conventional manner, known to those skilled in the art.
- Tenofovir disoproxil fumarate can be prepared, for example, as described in U.S. Pat. No. 5,977,089. Methods for the preparation of FTC are described in WO 92/14743, incorporated herein by reference.
- compositions of the present invention are administered to a human or other mammal in a safe and effective amount as described herein. These safe and effective amounts will vary according to the type and size of mammal being treated and the desired results of the treatment. Any of the various methods known by persons skilled in the art for packaging tablets, caplets, or other solid dosage forms suitable for oral administration, that will not degrade the components of the present invention, are suitable for use in packaging. The combinations may be packaged in glass and plastic bottles. Tablets, caplets, or other solid dosage forms suitable for oral administration may be packaged and contained in various packaging materials optionally including a dessicant, e.g. silica gel. Packaging may be in the form of unit dose blister packaging.
- a package may contain one blister tray of tenofovir DF and another blister tray of emtricitabine pills, tablets, caplets, or capsule.
- a patient would take one dose, e.g. a pill, from one tray and one from the other.
- the package may contain a blister tray of the co-formulated combination of tenofovir DF and emtricitabine in a single pill, tablet, caplet or capsule.
- the combinations of the invention include physiological functional derivatives of tenofovir DF and FTC.
- the packaging material may also have labeling and information related to the pharmaceutical composition printed thereon.
- an article of manufacture may contain a brochure, report, notice, pamphlet, or leaflet containing product information. This form of pharmaceutical information is referred to in the pharmaceutical industry as a “package insert.”
- a package insert may be attached to or included with a pharmaceutical article of manufacture.
- the package insert and any article of manufacture labeling provides information relating to the pharmaceutical composition.
- the information and labeling provides various forms of information utilized by health-care professionals and patients, describing the composition, its dosage and various other parameters required by regulatory agencies such as the United States Food and Drug Agency.
- the combinations of the inventions may be tested for in vitro activity against HIV and sensitivity, and for cytotoxicity in laboratory adapted cell lines, e.g. MT2 and in peripheral blood mononuclear cells (PBMC) according to standard assays developed for testing anti-HIV compounds, such as WO 02/068058 and U.S. Pat. No. 6,475,491.
- Combination assays may be performed at varying concentrations of the compounds of the combinations to determine EC 50 by serial dilutions.
- compositions A, B, C, D, E, and F are prepared by wet granulation of the ingredients with an aqueous solution, addition of extragranular components and then followed by addition of magnesium stearate and compression.
- Formulation A mg/tablet Tenofovir Disoproxil Fumarate 300 emtricitabine 200 Microcrystalline Cellulose 200 Lactose Monohydrate 175 Croscarmellose Sodium 60 Pregelatinized Starch 50 Colloidal silicon dioxide 5 Magnesium Stearate 10 total: 1000
- Formulation B mg/tablet Tenofovir Disoproxil fumarate 300 emtricitabine 100 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 900
- Formulation C mg/tablet Tenofovir Disoproxil fumarate 200 emtricitabine 200 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 900
- Formulation D mg/tablet Tenofovir Disoproxil fumarate 300 emtricitabine 25 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 825
- Formulation E mg/tablet Tenofovir Disoproxil fumarate 200 emtricitabine 25 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 725
- Formulation F mg/tablet Tenofovir Disoproxil fumarate 100 emtricitabine 100 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 700
- This formulation is prepared by wet granulation of the ingredients with an aqueous solution, followed by the addition of magnesium stearate and compression.
- Drug release takes place over a period of about 6-8 hours and is complete after 12 hours.
- a capsule formulation is prepared by admixing the ingredients and filling into a two-part hard gelatin or hydroxypropyl methylcellulose capsule.
- mg/capsule Active Ingredient 500 Microcrystalline Cellulose 143 Sodium Starch Glycollate 25 Magnesium Stearate 2 total: 670
- the following controlled release capsule formulation is prepared by extruding ingredients a, b, and c using an extruder, followed by spheronization of the extrudate and drying. The dried pellets are then coated with release-controlling membrane (d) and filled into a two-piece, hard gelatin or hydroxypropyl methylcellulose capsule.
- mg/capsule (a) Active Ingredient 500 (b) Microcrystalline Cellulose 125 (c) Lactose B.P. 125 (d) Ethyl Cellulose 13 total: 763
- active ingredients are admixed with the ingredients and filling them as dry powder. Purified water is added and mixed well before use. Active Ingredient 500 mg Confectioner's Sugar 2000 mg Simethicone 300 mg Methylparaben 30 mg Propylparaben 10 mg Flavor, Peach 500 mg Purified Water q.s. to 5.00 ml
- Witepsol H15 is melted in a steam-jacketed pan at 45° C. maximum.
- the active ingredients are sifted through a 200 micron sieve and added to the molten base with mixing, using a Silverson fitted with a cutting head, until a smooth dispersion is achieved. Maintaining the mixture at 45° C., the remaining Witepsol H15 is added to the suspension and stirred to ensure a homogenous mix.
- the entire suspension is passed through a 250 micron stainless steel screen and, with continuous stirring, is allowed to cool to 40° C. At a temperature of 38° C. to 40° C., 2.02 g of the mixture is filled into suitable, 2 ml plastic molds. The suppositories are allowed to cool to room temperature.
- TDF tenofovir disoproxil fumarate
- Equipment included a high shear mixer equipped with a pressure tank and spray nozzle tip to add the granulating water, a fluid-bed dryer, a mill, a tumble blender, a rotary tablet press, and a tablet deduster.
- the dried, milled powder was blended with the extragranular microcrystalline cellulose and croscarmellose sodium and then blended with magnesium stearate. Powder samples were removed after mixing with the magnesium stearate. The blend samples were evaluated for, bulk density, mesh analysis and compressibility. The powder blend mixed with the magnesium stearate was compressed into tablets on a press setup.
- TDF/emtricitabine tablet formulation The following Table 1 lists the quantitative composition of the TDF/emtricitabine tablet formulation.
- Magnesium Stearate NF/EP 1.0 10.0 0.12 Purified Water, USP/EP b b b Totals 100.0 1000.0 12.00
- Moisture content was measured by loss on drying using a heat lamp/balance system.
- the powder blend was sampled with a sampling thief fitted with chambers to determine powder blend uniformity. Duplicate samples were removed from each of several locations in the blender. Blend uniformity analysis was performed on one sample from each location.
- Particle size analysis of the final powder blend was determined by sifting a multi-gram sample through a screen using a sonic sifter. The quantity of final powder blend retained on each sieve and the fines collector was determined by calculating the difference in weight between the sieves and fines collector before and after the test. The geometric mean diameter particle size was calculated by logarithmic weighting of the sieved distribution.
- Tablets were characterized for friability using a friabilator, a hardness tester, a thickness micrometer equipped with a printer, and a weighing balance.
- Compression characteristics were determined using a rotary tablet press equipped with a flat-faced, beveled edged punch to a target weight of 400 mg.
- the powder blends were compressed using target upper punch pressures ranging from approximately 100 to 250 MPa.
- the apparent normalized ejection force was determined and normalized for tablet thickness and diameter.
- Tablet hardness was determined using a hardness tester. Tablet thickness was determined using a micrometer, and tablet weights were determined using a top loading balance.
- each wet granulation was deagglomerated with a mill fitted with a screen and an impeller. The milled wet granules were charged into a fluid-bed dryer immediately following wet milling.
- Milled wet granules were dried using an inlet air setpoint temperature of about 70° C. and airflow of approximately 100 cfm.
- the target LOD was about 1.0% with a range of not more than (NMT) 1.5%.
- the total fluid-bed drying time ranged from 53 to 75 minutes.
- Final LOD ranged from 0.4% to 0.7% for all of the batches dried.
- the final exhaust temperatures for all the batches ranged from 47° C. to 50° C.
- the final powder blend moisture level ranged from 0.8% to 1.1% LOD.
- Table 2 Three 300 gm formulations (Table 2) were granulated in a granulator equipped with a 1-L bowl. The quantities of intragranular components were based on a 300 g total batch size. The formulations in lots 1 and 2 differed in the amount of microcrystalline cellulose 30% vs. 20% w/w, respectively. Lots 2 and 3 were identical except for the type of binder. Lot 2 contained 5% w/w of pregelatinized starch and lot 3 contained 5% w/w povidone as binder.
- the final blend batch size was adjusted to 100 g.
- a portion, 81 g of the resulting blend from Lot 1 was blended with 15 g microcrystalline cellulose, 3 g croscarmellose sodium and 1 g magnesium stearate.
- 86 g of the resulting granulation from Lot 2 and Lot 3 were each blended with 10 g microcrystalline cellulose, 3 g croscarmellose sodium and 1 g magnesium stearate.
- Impurities related to tenofovir disoproxil fumarate and emtricitabine were characterized and measured in the bulk API (active pharmaceutical ingredient) before formulation in the three lots of Table 2, and again after formulation in the resulting tablets.
- the impurities include by-products from hydrolysis of the exocyclic amino groups of tenofovir disoproxil fumarate and emtricitabine, and the hydrolysis of the disoproxil (POC) esters of tenofovir disoproxil fumarate. In each lot, the sum total of impurities related to tenofovir disoproxil fumarate and emtricitabine was less than 1% after formulation and tablet manufacture.
- a pharmaceutical composition comprising an effective amount of a compound of the formula:
- R 1 and R are independently selected from H, C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl, C 6 -C 20 substituted aryl, C 6 -C 20 arylalkyl, C 6 -C 20 substituted arylalkyl, acyloxymethyl esters —CH 2 OC( ⁇ O)R 9 and acyloxymethyl carbonates —CH 2 OC( ⁇ O)OR 9 where R 9 is C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl and C 6 -C 20 substituted aryl;
- R 3 is selected from H, C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, or CH 2 OR 8 where R 8 is C 1 -C 6 alkyl, C 1 -C 6 hydroxyalkyl and C 1 -C 6 haloalkyl;
- R 4 and R 5 are independently selected from H, NH 2 , NHR and NR 2 where R is C 1 -C 6 alkyl;
- R 6 and R 7 are independently selected from H and C 1 -C 6 alkyl; or a physiologically functional derivative thereof;
- B is selected from adenine, guanine, cytosine, uracil, thymine, 7-deazaadenine, 7-deazaguanine, 7-deaza-8-azaguanine, 7-deaza-8-azaadenine, inosine, nebularine, nitropyrrole, nitroindole, 2-aminopurine, 2-amino-6-chloropurine, 2,6-diaminopurine, hypoxanthine, pseudouridine, 5-fluorocytosine, 5-chlorocytosine, 5-bromocytosine, 5-iodocytosine, pseudocytosine, pseudoisocytosine, 5-propynylcytosine, isocytosine, isoguanine, 7-deazaguanine, 2-thiopyrimidine, 6-thioguanine, 4-thiothymine, 4-thiouracil, O 6 -methylguanine, N 6 -methyla
- R is selected from H, C 1 -C 18 alkyl, C 1 -C 18 substituted alkyl, C 2 -C 18 alkenyl, C 2 -C 18 substituted alkenyl, C 2 -C 18 alkynyl, C 2 -C 18 substituted alkynyl, C 6 -C 20 aryl, C 6 -C 20 substituted aryl, C 2 -C 20 heterocycle, C 2 -C 20 substituted heterocycle, phosphonate, phosphophosphonate, diphosphophosphonate, phosphate, diphosphate, triphosphate, polyethyleneoxy or a physiologically functional derivative thereof; and
- a pharmaceutically acceptable carrier [0179] a pharmaceutically acceptable carrier.
- R 1 and R 2 are independently selected from H, C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl, C 6 -C 20 substituted aryl, C 6 -C 20 arylalkyl, C 6 -C 20 substituted arylalkyl, acyloxymethyl esters —CH 2 OC( ⁇ O)R 9 and acyloxymethyl carbonates —CH 2 OC( ⁇ O)OR 9 where R 9 is C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl and C 6 -C 20 substituted aryl; and R 3 R 4 , R 5 , R 6 and R 7 are independently H or C 1 -C 6 alkyl.
- C3. A composition of embodiment A1 wherein, in formula 2, B is cytosine or a 5-halocytosine.
- R 1 and R 2 are independently selected from H, C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl, C 6 -C 20 substituted aryl, C 6 -C 20 arylalkyl, C 6 -C 20 substituted arylalkyl, acyloxymethyl esters —CH 2 OC( ⁇ O)R 9 and acyloxymethyl carbonates —CH 2 OC( ⁇ O)OR 9 where R 9 is C 1 -C 6 alkyl, C 1 -C 6 substituted alkyl, C 6 -C 20 aryl and C 6 -C 20 substituted aryl; and R 3 , R 4 , R 5 , R 6 and R 7 are independently H or C 1 -C 6 alkyl; and, in formula 2, B is cytosine or a 5-halocytosine.
- R 1 and R 2 are independently selected from H, acyloxymethyl esters —CH 2 OC( ⁇ O)R 9 and acyloxymethyl carbonates —CH 2 OC( ⁇ O)OR 9 where R 9 is C 1 -C 6 alkyl; and R 3 , R 4 , R 5 , R 6 and R 7 are independently H or C 1 -C 6 alkyl; and, in formula 2, B is cytosine or a 5-halocytosine and R is H.
- F6 A composition of embodiment E5 wherein, in formula 1, R 1 and R 2 are independently selected from H and —CH 2 OC( ⁇ O)OCH(CH 3 ) 2 ; R 3 is —CH 3 ; and R 4 , R 5 , R 6 and R 7 are H; and, in formula 2, B is 5-fluorocytosine and R is H.
- a pharmaceutical composition comprising a pharmaceutically effective amount of [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) or a physiologically functional derivative thereof and a pharmaceutically effective amount of (2R, 5S-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof; and a pharmaceutically acceptable carrier.
- a pharmaceutical formulation of embodiment A1 to G7 further comprising a third active ingredient selected from the group consisting of a protease inhibitor, a nucleoside or nucleotide reverse transcriptase inhibitor, a non-nucleoside reverse transcriptase inhibitor, and an integrase inhibitor.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Biophysics (AREA)
- Inorganic Chemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
The present invention relates to therapeutic combinations of [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester (tenofovir disoproxil fumarate, Viread®) and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine, Emtriva™, (−)-cis FTC) and their physiologically functional derivatives. The combinations may be useful in the treatment of HIV infections, including infections with HIV mutants bearing resistance to nucleoside and/or non-nucleoside inhibitors. The present invention is also concerned with pharmaceutical compositions and formulations of said combinations of tenofovir disoproxil fumarate and emtricitabine, and their physiologically functional derivatives, as well as therapeutic methods of use of those compositions and formulations.
Description
- This non-provisional application claims the benefit of Provisional Application Nos. 60/440,246 and 60/440,308, both filed Jan. 14, 2003, which is incorporated herein by reference.
- The invention relates generally to combinations of compounds with antiviral activity and more specifically with anti-HIV properties. In particular, it relates to chemically stable combinations of structurally diverse anti-viral agents.
- Human immunodeficiency virus (HIV) infection and related diseases are a major public health problem worldwide. Human immunodeficiency virus type 1 (HIV-1) encodes at least three enzymes which are required for viral replication: reverse transcriptase (RT), protease (Prt), and integrase (Int). Although drugs targeting reverse transcriptase and protease are in wide use and have shown effectiveness, particularly when employed in combination, toxicity and development of resistant strains have limited their usefulness (Palella, et al N. Engl. J. Med. (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001). Human immunodeficiency virus type 1 (HIV-1) protease (Prt) is essential for viral replication and is an effective target for approved antiviral drugs. The HIV Prt cleaves the viral Gag and Gag-Pol polyproteins to produce viral structural proteins (p17, p24, p7 and p6) and the three viral enzymes. Combination therapy with RT inhibitors has proven to be highly effective in suppressing viral replication to unquantifiable levels for a sustained period of time. Also, combination therapy with RT and Prt inhibitors (PI) have shown synergistic effects in suppressing HIV replication. Unfortunately, a high percentage, typically 30 to 50% of patients currently fail combination therapy due to the development of drug resistance, non-compliance with complicated dosing regimens, pharmacokinetic interactions, toxicity, and lack of potency. Therefore, there is a need for new HIV-1 inhibitors that are active against mutant HIV strains, have distinct resistance profiles, fewer side effects, less complicated dosing schedules, and are orally active. In particular, there is a need for a less onerous dosage regimen, such as once per day oral dosing, optimally with as few pills as possible.
- The use of combinations of compounds can yield an equivalent antiviral effect with reduced toxicity, or an increase in drug efficacy. Lower overall drug doses can reduce the frequency of occurrence of drug-resistant variants of HIV. Many different methods have been used to examine the effects of combinations of compounds acting together in different assay systems (Furman WO 02/068058). Lower doses predict better patient compliance when pill burden decreases, dosing schedules are simplified and, optionally, if synergy between compounds occurs (Loveday, C. “Nucleoside reverse transcriptase inhibitor resistance” (2001) JAIDS Journal of Acquired Immune Deficiency Syndromes 26:S10-S24). AZT (zidovudine™, 3′-azido, 3′-deoxythymidine) demonstrates synergistic antiviral activity in vitro in combination with agents that act at HIV-1 replicative steps other than reverse transcription, including recombinant soluble CD4 castanospermine and recombinant interferon-α. However, it must be noted that combinations of compounds can give rise to increased cytotoxicity. For example, AZT and recombinant interferon-α have an increased cytotoxic effect on normal human bone marrow progenitor cells.
- Chemical stability of combinations of antiviral agents is an important aspect of co-formulation success and the present invention provides examples of such combinations.
- There is a need for new combinations of orally-active drugs for the treatment of patients infected with certain viruses, e.g. HIV, that provide enhanced therapeutic safety and efficacy, impart lower resistance, and predict higher patient compliance.
- The present invention provides combinations of antiviral compounds, in particular compositions and methods for inhibition of HIV. In an exemplary aspect, the invention includes a composition including tenofovir disoproxil fumarate and emtricitabine which has anti-HIV activity. The composition of tenofovir DF and emtricitabine is both chemically stable and either synergistic and/or reduces the side effects of one or both of tenofovir DF and emtricitabine. Increased patient compliance is likely in view of the lower pill burden and simplified dosing schedule.
- The present invention relates to therapeutic combinations of [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate, tenofovir DF, TDF, Viread®) and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine, Emtriva™, (−)-cis FTC) and their use in the treatment of HIV infections including infections with HIV mutants bearing resistance to nucleoside and/or non-nucleoside inhibitors. The present invention is also concerned with pharmaceutical compositions and formulations of said combinations of tenofovir disoproxil fumarate and emtricitabine. Another aspect of the invention is a pharmaceutical formulation comprising a physiologically functional derivative of tenofovir disoproxil fumarate or a physiologically functional derivative of emtricitabine.
- Therapeutic combinations and pharmaceutical compositions and formulations of the invention include the combination of PMEA or PMPA (tenofovir) compounds with emtricitabine or (2R, 5S, cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (3TC, lamivudine, Epivir™), and their use in the treatment of HIV infections.
- One aspect of the invention is a method for the treatment or prevention of the symptoms or effects of an HIV infection in an infected animal which comprises administering to, i.e. treating, said animal with a therapeutically effective amount of a combination comprising [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir DF, TDF) or a physiologically functional derivative thereof, and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof.
- Another aspect of the invention is a unit dosage form of a therapeutic combination comprising tenofovir disoproxil fumarate and emtricitabine, or physiological functional derivatives thereof. The unit dosage form may be formulated for administration by oral or other routes and is unexpectedly chemically stable in view of the properties of the structurally diverse components.
- Another aspect of the invention is directed to chemically stable combination antiviral compositions comprising tenofovir disoproxil fumarate and emtricitabine. In a further aspect of the invention, the chemically stable combinations of tenofovir disoproxil fumarate and emtricitabine further comprise a third antiviral agent. In this three-component mixture, the unique chemical stability of tenofovir disoproxil fumarate and emtricitabine is taken advantage of in order to enable the combination with the third antiviral agent. Particularly useful third agents include, by way of example and not limitation, those of Table A. Preferably, the third component is an agent approved for antiviral use in humans, more preferably, it is an NNRTI or a protease inhibitor (PI), more preferably yet, it is an NNRTI. In a particularly preferred embodiment, the invention is directed to a combination of the chemically stable mixture of tenofovir disoproxil fumarate and emtricitabine together with efavirenz.
- Another aspect of the invention is a patient pack comprising at least one, typically two, and optionally, three active ingredients and other antiviral agents selected from tenofovir disoproxil fumarate and emtricitabine, and an information insert containing directions on the use of tenofovir disoproxil fumarate and emtricitabine together in combination.
- Another aspect of the invention is a process for preparing the combinations hereinbefore described, which comprises bringing into association tenofovir DF and emtricitabine of the combination in a medicament to provide an antiviral effect. In a further aspect of the present invention, there is provided the use of a combination of the present invention in the manufacture of a medicament for the treatment of any of the aforementioned viral infections or conditions.
- While the invention will be described in conjunction with the enumerated claims, it will be understood that they are not intended to limit the invention to those claims. On the contrary, the invention is intended to cover all alternatives, modifications, and equivalents, which may be included within the scope of the present invention as defined by the claims.
- Definitions
- Unless stated otherwise, the following terms and phrases as used herein are intended to have the following meanings:
- When tradenames are used herein, applicants intend to independently include the tradename product and the active pharmaceutical ingredient(s) of the tradename product.
- The term “chemical stability” means that the two primary antiviral agents in combination are substantially stable to chemical degradation. Preferably, they are sufficiently stable in physical combination to permit commercially useful shelf life of the combination product. Typically, “chemically stable” means that a first component of the mixture does not act to degrade a second component when the two are brought into physical combination to form a pharmaceutical dosage form. More typically, “chemically stable” means that the acidity of a first component does not catalyzes or otherwise accelerate the acid decomposition of a second component. By way of example and not limitation, in one aspect of the invention, “chemically stable” means that tenofovir disoproxil fumarate is not substantially degraded by the acidity of emtricitabine. “Substantially” in this context means at least about less than 10%, preferably less than 1%, more preferably less than 0.1%, more preferably yet, less than 0.01% acid degradation of tenofovir disoproxil fumarate over a 24-hour period when the products are in a pharmaceutical dosage form.
- The terms “synergy” and “synergistic” mean that the effect achieved with the compounds used together is greater than the sum of the effects that results from using the compounds separately, i.e. greater than what would be predicted based on the two active ingredients administered separately. A synergistic effect may be attained when the compounds are: (1) co-formulated and administered or delivered simultaneously in a combined formulation; (2) delivered by alternation or in parallel as separate formulations; or (3) by some other regimen. When delivered in alternation therapy, a synergistic effect may be attained when the compounds are administered or delivered sequentially, e.g. in separate tablets, pills or capsules, or by different injections in separate syringes. In general, during alternation therapy, an effective dosage of each active ingredient is administered sequentially, i.e. serially, whereas in combination therapy, effective dosages of two or more active ingredients are administered together. A synergistic antiviral effect denotes an antiviral effect which is greater than the predicted purely additive effects of the individual compounds of the combination.
- The term “physiologically functional derivative” means a pharmaceutically active compound with equivalent or near equivalent physiological functionality to tenofovir DF or emtricitabine when administered in combination with another pharmaceutically active compound in a combination of the invention. As used herein, the term “physiologically functional derivative” includes any: physiologically acceptable salt, ether, ester, prodrug, solvate, stereoisomer including enantiomer, diastereomer or stereoisomerically enriched or racemic mixture, and any other compound which upon administration to the recipient, is capable of providing (directly or indirectly) such a compound or an antivirally active metabolite or residue thereof.
- “Bioavailability” is the degree to which the pharmaceutically active agent becomes available to the target tissue after the agent's introduction into the body. Enhancement of the bioavailability of a pharmaceutically active agent can provide a more efficient and effective treatment for patients because, for a given dose, more of the pharmaceutically active agent will be available at the targeted tissue sites.
- The compounds of the combinations of the invention may be referred to as “active ingredients” or “pharmaceutically active agents.”
- The term “prodrug” as used herein refers to any compound that when administered to a biological system generates the drug substance, i.e. active ingredient, as a result of spontaneous chemical reaction(s), enzyme catalyzed chemical reaction(s), and/or metabolic chemical reaction(s).
- “Prodrug moiety” means a labile functional group which separates from the active inhibitory compound during metabolism, systemically, inside a cell, by hydrolysis, enzymatic cleavage, or by some other process (Bundgaard, Hans, “Design and Application of Prodrugs” in Textbook of Drug Design and Development (1991), P. Krogsgaard-Larsen and H. Bundgaard, Eds. Harwood Academic Publishers, pp. 113-191). Prodrug moieties can serve to enhance solubility, absorption and lipophilicity to optimize drug delivery, bioavailability and efficacy. A “prodrug” is thus a covalently modified analog of a therapeutically-active compound.
- “Alkyl” means a saturated or unsaturated, branched, straight-chain, branched, or cyclic hydrocarbon radical derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane, alkene, or alkyne. Typical alkyl groups consist of 1-18 saturated and/or unsaturated carbons, such as normal, secondary, tertiary or cyclic carbon atoms. Examples include, but are not limited to: methyl, Me (—CH 3), ethyl, Et (—CH2CH3), acetylenic (—C≡CH), ethylene, vinyl (—CH═CH2), 1-propyl, n-Pr, n-propyl (—CH2CH2CH3), 2-propyl, i-Pr, i-propyl (—CH(CH3)2), allyl (—CH2CH═CH2), propargyl (—CH2C≡CH), cyclopropyl (—C3H5), 1-butyl, n-Bu, n-butyl (—CH2CH2CH2CH3), 2-methyl-1-propyl, i-Bu, i-butyl (—CH2CH(CH3)2), 2-butyl, s-Bu, s-butyl (—CH(CH3)CH2CH3), 2-methyl-2-propyl, t-Bu, t-butyl (—C(CH3)3), 1-pentyl, n-pentyl, (—CH2CH2CH2CH2CH3), 2-pentyl (—CH(CH3)CH2CH2CH3), 3-pentyl (—CH(CH2CH3)2), 2-methyl-2-butyl (—C(CH3)2CH2CH3), cyclopentyl (—C5H9), 3-methyl-2-butyl (—CH(CH3)CH(CH3)2), 3-methyl-1-butyl (—CH2CH2CH(CH3)2), 2-methyl-1-butyl (—CH2CH(CH3)CH2CH3), 1-hexyl (—CH2CH2CH2CH2CH2CH3), 5-hexenyl (—CH2 CH2CH2CH2CH═CH2) 1-hexyl (—CH(CH3)CH2CH2CH2CH3), 3-hexyl (—CH(CH2CH3)(CH2CH2CH3) ), cyclohexyl (—C6H11), 2-methyl-2-pentyl (—C(CH3)2CH2CH2CH3), 3-methyl-2-pentyl (—CH(CH3)CH(CH3)CH2CH3), 4-methyl-2-pentyl (—CH(CH3)CH2CH(CH3)2), 3-methyl-3-pentyl (—C(CH3)(CH2CH3)2), 2-methyl-3-pentyl (—CH(CH2CH3)CH(CH3)2), 2,3-dimethyl-2-butyl (—C(CH3)2CH(CH3)2), and 3,3-dimethyl-2-butyl (—CH(CH3)C(CH3)3.
- “Aryl” means a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system. Typical aryl groups include, but are not limited to, radicals derived from benzene, substituted benzene, naphthalene, anthracene, biphenyl, and the like.
- “Arylalkyl” refers to an acyclic alkyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp 3 carbon atom, is replaced with an aryl radical. Typical arylalkyl groups include, but are not limited to, benzyl, 2-phenylethan-1-yl, 2-phenylethen-1-yl, naphthylmethyl, 2-naphthylethan-1-yl, 2-naphthylethen-1-yl, naphthobenzyl, 2-naphthophenylethan-1-yl and the like. The arylalkyl group 6 to 20 carbon atoms e.g., the alkyl moiety, including alkanyl, alkenyl or alkynyl groups, of the arylalkyl group is 1 to 6 carbon atoms and the aryl moiety is 5 to 14 carbon atoms.
- “Substituted alkyl”, “substituted aryl”, and “substituted arylalkyl” mean alkyl, aryl, and arylalkyl respectively, in which one or more hydrogen atoms are each independently replaced with a substituent. Typical substituents include, but are not limited to, —X, —R, —O −, —OR, —SR, —S−, —NR2, —NR3, ═NR, —CX3, —CN, —OCN, —SCN, —N═C═O, —NCS, —NO, —NO2, ═N2, —N3, NC(═O)R, —C(═O)R, —C(═O)NRR —S(═O)2O−, —S(═O)2OH, —S(═O)2R, —OS(═O)2OR, —S(═O)2NR, —S(═O)R, —OP(═O)O2RR, —P(═O)O2RR —P(═O)(O−)2, —P(═O)(OH)2, —C(═O)R, —C(═O)X, —C(S)R, —C(O)OR, —C(O)O−, —C(S)OR, —C(O)SR, —C(S)SR, —C(O)NRR, —C(S)NRR, —C(NR)NRR, where each X is independently a halogen: F, Cl, Br, or I; and each R is independently —H, alkyl, aryl, heterocycle, or prodrug moiety.
- “Heteroaryl” and “Heterocycle” refer to a ring system in which one or more ring atoms is a heteroatom, e.g. nitrogen, oxygen, and sulfur. Heterocycles are described in: Katritzky, Alan R., Rees, C. W., and Scriven, E. Comprehensive Heterocyclic Chemistry (1996) Pergamon Press; Paquette, Leo A Principles of Modern Heterocyclic Chemistry W. A. Benjamin, New York, (1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28. Exemplary heterocycles include but are not limited to substituents, i.e. radicals, derived from pyrrole, indole, furan, benzofuran, thiophene, benzothiophene, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 2-imidazole, 4-imidazole, 3-pyrazole, 4-pyrazole, pyridazine, pyrimidine, pyrazine, purine, cinnoline, pthalazine, quinazoline, quinoxaline, 3-(1,2,4-N)-triazolyl, 5-(1,2,4-N)-triazolyl, 5-tetrazolyl, 4-(1-0,3-N)-oxazole, 5-(1-O, 3-N)-oxazole, 4-(1-S, 3-N)-thiazole, 5-(1-S, 3-N)-thiazole, 2-benzoxazole, 2-benzothiazole, 4-(1,2,3-N)-benzotriazole, and benzimidazole.
- Stereochemical definitions and conventions used herein generally follow S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; and Eliel, E. and Wilen, S., Stereochemistry of Organic Compounds (1994) John Wiley & Sons, Inc., New York. Many organic compounds exist in optically active forms, i.e., they have the ability to rotate the plane of plane-polarized light. In describing an optically active compound, the prefixes D and L or R and S are used to denote the absolute configuration of the molecule about its chiral center(s). The prefixes d and 1 or (+) and (−) are employed to designate the sign of rotation of plane-polarized light by the compound, with (−) or 1 meaning that the compound is levorotatory. A compound prefixed with (+) or d is dextrorotatory. For a given chemical structure, these compounds, called stereoisomers, are identical except that they are mirror images of one another. A specific stereoisomer is also referred to as an enantiomer, and a mixture of such isomers is often called an enantiomeric mixture. A 50:50 mixture of enantiomers is referred to as a racemic mixture or a racemate. The terms “racemic mixture” and “racemate” refer to an equimolar mixture of two enantiomeric species, devoid of optical activity.
- The term “chiral” refers to molecules which have the property of non-superimposability of the mirror image partner, while the term “achiral” refers to molecules which are superimposable on their mirror image partner.
- The term “stereoisomers” refers to compounds which have identical chemical constitution, but differ with regard to the arrangement of the atoms or groups in space.
- “Diastereomer” refers to a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities. Mixtures of diastereomers may separate under high resolution analytical procedures such as electrophoresis and chromatography.
- “Enantiomers” refer to two stereoisomers of a compound which are non-superimposable mirror images of one another.
- Active Ingredients of the Combinations
- The present invention provides novel combinations of two or more active ingredients being employed together. In some embodiments, a synergistic antiviral effect is achieved. In other embodiments, a chemically stable combination is obtained. The combinations include at least one active ingredient selected from (1) tenofovir disoproxil fumarate and physiologically functional derivatives, and at least one active ingredient selected from (2) emtricitabine and physiologically functional derivatives. The term “synergistic antiviral effect” is used herein to denote an antiviral effect which is greater than the predicted purely additive effects of the individual components (a) and (b) of the combination.
- Tenofovir disoproxil fumarate (also known as Viread®, Tenofovir DF, Tenofovir disoproxil, TDF, Bis-POC-PMPA (U.S. Pat. Nos. 5,935,946, 5,922,695, 5,977,089, 6,043,230, 6,069,249) is a prodrug of tenofovir, and has the structure:

- and including fumarate salt (HO 2CCH2CH2CO2 −).
- The chemical names for Tenofovir disoproxil include: [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester; 9-[(R)-2-[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine; and 2,4,6,8-tetraoxa-5-phosphanonanedioic acid, 5-[[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]-, bis(1-methylethyl)ester, 5-oxide. The CAS Registry numbers include: 201341-05-1; 202138-50-9; 206184-49-8. It should be noted that the ethoxymethyl unit of tenofovir has a chiral center. The R (rectus, right handed configuration) enantiomer is shown. However, the invention also includes the S isomer. The invention includes all enantiomers, diastereomers, racemates, and enriched stereoisomer mixtures of tenofovir (PMPA) and physiologically functional derivatives thereof.
- PMPA or tenofovir (U.S. Pat. Nos. 4,808,716, 5,733,788, 6,057,305) has the structure:

- The chemical names of PMPA, tenofovir include: (R)-9-(2-phosphonylmethoxypropyl)adenine; and phosphonic acid, [[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]. The CAS Registry number is 147127-20-6.
- Tenofovir disoproxil fumarate (DF) is a nucleotide reverse transcriptase inhibitor approved in the United States in 2001 for the treatment of HIV-1 infection in combination with other antiretroviral agents. Tenofovir disoproxil fumarate or Viread® (Gilead Science, Inc.) is the fumarate salt of tenofovir disoproxil. Viread® may be named as: 9-[(R)-2-[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1); or 2,4,6,8-tetraoxa-5-phosphanonanedioic acid, 5-[[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]-, bis(1-methylethyl)ester, 5-oxide, (2E)-2-butenedioate (1:1). The CAS Registry number is 202138-50-9.
- Physiologically functional derivatives of tenofovir disoproxil fumarate include PMEA (adefovir, 9-((R)-2-(phosphonomethoxy)ethyl)adenine) and PMPA compounds. Exemplary combinations include a PMEA or PMPA compound in combination with emtricitabine or 3TC. PMEA and PMPA compounds have the structures:

- where PMEA (R 3 is H) and PMPA (R3 is C1-C6 alkyl, C1-C6 substituted alkyl, or CH2OR8 where R8 is C1-C6 alkyl, C1-C6 hydroxyalkyl or C1-C6 haloalkyl. R6 and R7 are independently H or C1-C6 alkyl. R4 and R5 are independently H, NH2, NHR or NR2 where R is C1-C6 alkyl. R1 and R2 are independently H, C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl, C6-C20 substituted aryl, C6-C20 arylalkyl, C6-C20 substituted arylalkyl, acyloxymethyl esters —CH2OC(═O)R9 (e.g. POM) or acyloxymethyl carbonates —CH2OC(═O)OR9 (e.g. POC) where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl or C6-C20 substituted aryl. For example, R1 and R2 may be pivaloyloxymethoxy, POM, —CH2OC(═O)C(CH3)3; —CH2OC(═O)OC(CH3)3; or POC, —CH2OC(═O)OCH(CH3)2. Also for example, tenofovir has the structure where R3 is CH3, and R1, R2, R4, R5, R6 and R7 are H. Dialkyl phosphonates may be prepared according to the methods of: Quast et al (1974) Synthesis 490; Stowell et al (1990) Tetrahedron Lett. 3261; U.S. Pat. No. 5,663,159.
- The PMPA compound may be enantiomerically-enriched or purified (single stereoisomer) where the carbon atom bearing R 3 may be the R or S enantiomer. The PMPA compound may be a racemate, i.e. a mixture of R and S stereoisomers.
- Adefovir (9-(2-phosphonomethoxyethyl)adenine where R 1—R7═H) is an exemplary PMEA compound (U.S. Pat. Nos. 4,808,716, 4,724,233). As the bis-pivalate prodrug, Adefovir dipivoxil, also known as bis-POM PMEA, (R3—R7═H, R1 and R2═—CH2OC(═O)C(CH3)3, pivoxil, POM, pivaloyloxymethoxy), is effective against HIV and Hepatitis B infections (U.S. Pat. Nos. 5,663,159, 6,451,340). Adefovir dipivoxil has demonstrated minor to moderate synergistic inhibition of HIV replication in combination with other compounds with anti-HIV activity including PMPA, d4T, ddC, nelfinavir, ritonavir, and saquinavir (Mulato et al (1997) Antiviral Research 36:91-97).
- The invention includes all enantiomers, diastereomers, racemates, and enriched stereoisomer mixtures of PMEA and PMPA, and physiologically functional derivatives thereof.
- Emtricitabine ((−)-cis-FTC, Emtriva™), a single enantiomer of FTC, is a potent nucleoside reverse transcriptase inhibitor approved for the treatment of HIV (U.S. Pat. Nos. 5,047,407, 5,179,104, 5,204,466, 5,210,085, 5,486,520, 5,538,975, 5,587,480, 5618820, 5,763,606, 5,814,639, 5,914,331, 6,114,343, 6180639, 6215004; WO 02/070518). The single enantiomer emtricitabine has the structure:

- The chemical names for emtricitabine include: (−)-cis-FTC; β-L-hydroxymethyl-5-(5-fluorocytosin-1-yl)-1,3-oxathiolane; (2R,5S)-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine; and 4-amino-5-fluoro-1-(2-hydroxymethyl-[1,3]-(2R,5S)-oxathiolan-5-yl)-1H-pyrimidin-2-one. The CAS Registry numbers include: 143491-57-0; 143491-54-7. It should be noted that FTC contains two chiral centers, at the 2 and 5 positions of the oxathiolane ring, and therefore can exist in the form of two pairs of optical isomers (i.e. enantiomers) and mixtures thereof including racemic mixtures. Thus, FTC may be either a cis or a trans isomer or mixtures thereof. Mixtures of cis and trans isomers are diastereomers with different physical properties. Each cis and trans isomer can exist as one of two enantiomers or mixtures thereof including racemic mixtures. The invention includes all enantiomers, diastereomers, racemates, and enriched stereoisomer mixtures of emtricitabine and physiologically functional derivatives thereof. For example, the invention includes physiological functional derivatives such as the 1:1 racemic mixture of the enantiomers (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) and its mirror image (2S, 5R, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one, or mixtures of the two enantiomers in any relative amount. The invention also includes mixtures of cis and trans forms of FTC.
- Physiologically functional derivatives of emtricitabine include 1,3 oxathiolane nucleosides having the structure:

- In the 1,3 oxathiolane nucleoside structure above, B is a nucleobase including any nitrogen-containing heterocyclic moiety capable of forming Watson-Crick hydrogen bonds in pairing with a complementary nucleobase or nucleobase analog, e.g. a purine, a 7-deazapurine, or a pyrimidine. Examples of B include the naturally occurring nucleobases: adenine, guanine, cytosine, uracil, thymine, and minor constituents and analogs of the naturally occurring nucleobases, e.g. 7-deazaadenine, 7-deazaguanine, 7-deaza-8-azaguanine, 7-deaza-8-azaadenine, inosine, nebularine, nitropyrrole, nitroindole, 2-aminopurine, 2-amino-6-chloropurine, 2,6-diaminopurine, hypoxanthine, pseudouridine, 5-fluorocytosine, 5-chlorocytosine, 5-bromocytosine, 5-iodocytosine, pseudocytosine, pseudoisocytosine, 5-propynylcytosine, isocytosine, isoguanine, 7-deazaguanine, 2-thiopyrimidine, 6-thioguanine, 4-thiothymine, 4-thiouracil, O 6-methylguanine, N6-methyladenine, O4-methylthymine, 5,6-dihydrothymine, 5,6-dihydrouracil, 4-methylindole, pyrazolo[3,4-D]pyrimidines (U.S. Pat. Nos. 6,143,877 and 6,127,121; WO 01/38584), and ethenoadenine (Fasman (1989) in Practical Handbook of Biochemistry and Molecular Biology, pp. 385-394, CRC Press, Boca Raton, Fl).
- Nucleobases B may be attached in the configurations of naturally-occurring nucleic acids to the 1,3 oxathiolane moiety through a covalent bond between the N-9 of purines, e.g. adenin-9-yl and guanin-9-yl, or N-1 of pyrimidines, e.g. thymin-1-yl and cytosin-1-yl (Blackburn, G. and Gait, M. Eds. “DNA and RNA structure” in Nucleic Acids in Chemistry and Biology, 2nd Edition, (1996) Oxford University Press, pp. 15-81).
- Also in the 1,3 oxathiolane nucleoside structure above, R is H, C 1-C18 alkyl, C1-C18 substituted alkyl, C2-C18 alkenyl, C2-C18 substituted alkenyl, C2-C18 alkynyl, C2-C18 substituted alkynyl, C6-C20 aryl, C6-C20 substituted aryl, C2-C20 heterocycle, C2-C20 substituted heterocycle, phosphonate, phosphophosphonate, diphosphophosphonate, phosphate, diphosphate, triphosphate, polyethyleneoxy, or a prodrug moiety.
- Physiologically functional derivatives of emtricitabine also include 3TC (lamivudine, Epivir®), a reverse transcriptase inhibitor approved in the United States for the treatment of HIV-1 infection in combination with AZT as Combivir® (GlaxoSmithKline). U.S. Pat. Nos. 5,859,021; 5,905,082; 6,177,435; 5,627,186; 6,417,191. Lamivudine (U.S. Pat. Nos. 5,587,480, 5,696,254, 5,618,820, 5,756,706, 5,744,596, 568,164, 5,466,806, 5,151,426) has the structure:

- For example and for some therapeutic uses, 3TC may be a physiologically functional derivative of emtricitabine in combination with tenofovir DF or a physiologically functional derivative of tenofovir DF.
- It will be appreciated that tenofovir DF and emtricitabine, and their physiologically functional derivatives may exist in keto or enol tautomeric forms and the use of any tautomeric form thereof is within the scope of this invention. Tenofovir DF and emtricitabine will normally be utilized in the combinations of the invention substantially free of the corresponding enantiomer, that is to say no more than about 5% w/w of the corresponding enantiomer will be present.
- Prodrugs
- The invention includes all prodrugs of tenofovir and emtricitabine. An exemplary prodrug of tenofovir is tenofovir disoproxil fumarate (TDF, Viread®). A large number of structurally-diverse prodrugs have been described for phosphonic acids (Freeman and Ross in Progress in Medicinal Chemistry 34: 112-147 (1997). A commonly used prodrug class is the acyloxyalkyl ester, which was first used as a prodrug strategy for carboxylic acids and then applied to phosphates and phosphonates by Farquhar et al (1983) J. Pharm. Sci. 72: 324; also U.S. Pat. Nos. 4,816,570, 4,968,788, 5,663,159 and 5,792,756. Subsequently, the acyloxyalkyl ester was used to deliver phosphonic acids across cell membranes and to enhance oral bioavailability. A close variant of the acyloxyalkyl ester strategy, the alkoxycarbonyloxyalkyl ester, may also enhance oral bioavailability as a prodrug moiety in the compounds of the combinations of the invention. Aryl esters of phosphorus groups, especially phenyl esters, are reported to enhance oral bioavailability (DeLambert et al (1994) J. Med. Chem. 37: 498). Phenyl esters containing a carboxylic ester ortho to the phosphate have also been described (Khamnei and Torrence, (1996) J. Med. Chem. 39:4109-4115). Benzyl esters are reported to generate the parent phosphonic acid. In some cases, substituents at the ortho- or para-position may accelerate the hydrolysis. Benzyl analogs with an acylated phenol or an alkylated phenol may generate the phenolic compound through the action of enzymes, e.g. esterases, oxidases, etc., which in turn undergoes cleavage at the benzylic C—O bond to generate the phosphoric acid and the quinone methide intermediate. Examples of this class of prodrugs are described by Mitchell et al (1992) J. Chem. Soc. Perkin Trans. I 2345; Brook et al WO 91/19721. Still other benzylic prodrugs have been described containing a carboxylic ester-containing group attached to the benzylic methylene (Glazier et al WO 91/19721). Thio-containing prodrugs are reported to be useful for the intracellular delivery of phosphonate drugs. These proesters contain an ethylthio group in which the thiol group is either esterified with an acyl group or combined with another thiol group to form a disulfide. Deesterification or reduction of the disulfide generates the free thio intermediate which subsequently breaks down to the phosphoric acid and episulfide (Puech et al (1993) Antiviral Res., 22: 155-174; Benzaria et al (1996) J. Med. Chem. 39: 4958). Cyclic phosphonate esters have also been described as prodrugs of phosphorus-containing compounds.
- Prodrug esters in accordance with the invention are independently selected from the following groups: (1) mono-, di-, and tri-phosphate esters of tenofovir or emtricitabine or any other compound which upon administration to a human subject is capable of providing (directly or indirectly) said mono-, di, or triphosphate ester; (2) carboxylic acid esters (3) sulphonate esters, such as alkyl- or aralkylsulphonyl (for example, methanesulphonyl); (4) amino acid esters (for example, alanine, L-valyl or L-isoleucyl); (5) phosphonate; and (6) phosphonamidate esters.
- Ester groups (1)-(6) may be substituted with; straight or branched chain C 1-C18 alkyl (for example, methyl, n-propyl, t-butyl, or n-butyl); C3-C12 cycloalkyl; alkoxyalkyl (for example, methoxymethyl); arylalkyl (for example, benzyl); aryloxyalkyl (for example, phenoxymethyl); C5-C20 aryl (for example, phenyl optionally substituted by, for example, halogen, C1-C4 alkyl, C1-C4 alkoxy, or amino; acyloxymethyl esters —CH2OC(═O)R9 (e.g. POM) or acyloxymethyl carbonates —CH2OC(═O)OR9 (e.g. POC) where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl or C6-C20 substituted aryl. For example, ester groups may be: —CH2OC(═O)C(CH3)3, —CH2OC(═O)OC(CH3)3 or —CH2OC(═O)OCH(CH3)2.
- An exemplary aryl moiety present in such esters comprises a phenyl or substituted phenyl group. Many phosphate prodrug moieties are described in U.S. Pat. No. 6,312,662; Jones et al (1995) Antiviral Research 27:1-17; Kucera et al (1990) AIDS Res. Hum. Retro Viruses 6:491-501; Piantadosi et al (1991) J. Med. Chem. 34:1408-14; Hosteller et al (1992) Antimicrob. Agents Chemother. 36:2025-29; Hostetler et al (1990) J. Biol. Chem. 265:611127; and Siddiqui et al (1999) J. Med. Chem. 42:4122-28.
- Pharmaceutically acceptable prodrugs refer to a compound that is metabolized in the host, for example hydrolyzed or oxidized, by either enzymatic action or by general acid or base solvolysis, to form an active ingredient. Typical examples of prodrugs of the active ingredients of the combinations of the invention have biologically labile protecting groups on a functional moiety of the active compound. Prodrugs include compounds that can be oxidized, reduced, aminated, deaminated, esterified, deesterified, alkylated, dealkylated, acylated, deacylated, phosphorylated, dephosphorylated, or other functional group change or conversion involving forming or breaking chemical bonds on the prodrug.
- Chemical Stability of a Pharmaceutical Formulation
- The chemical stability of the active ingredients in a pharmaceutical formulation is of concern to minimize the generation of impurities and ensure adequate shelf-life. The active ingredients, tenofovir disoproxil fumarate and emtricitabine, in the pharmaceutical formulations of the invention have relatively low pKa values, indicative of the potential to cause acidic hydrolysis of the active ingredients. Emtricitabine, with a pKa of 2.65 (Emtriva™ Product Insert, Gilead Sciences, Inc. 2003, available at gilead.com) is subject to hydrolytic deamination of the 5-fluoro cytosine nucleobase to form the 5-fluoro uridine nucleobase. Tenofovir disoproxil fumarate, with a pKa of 3.75 (Yuan L. et al “Degradation Kinetics of Oxycarbonyloxymethyl Prodrugs of Phosphonates in Solution”, Pharmaceutical Research (2001) Vol. 18, No. 2, 234-237), is subject also to hydrolytic deamination of the exocyclic amine of the adenine nucleobase, and to hydrolysis of one or both of the POC ester groups (U.S. Pat. No. 5,922,695). It is desirable to formulate a therapeutic combination of tenofovir disoproxil fumarate and emtricitabine, and the physiological functional derivatives thereof, with a minimum of impurities and adequate stability.
- The combinations of the present invention provide combination pharmaceutical dosage forms which are chemically stable to acid degradation of: (1) a first component (such as tenofovir disoproxil fumarate, and physiological functional derivatives; (2) a second component (such as emtricitabine, and physiological functional derivatives; and (3) optionally a third component having antiviral activity. The third component includes anti-HIV agents and include: protease inhibitors (PI), nucleoside reverse transcriptase inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI), and integrase inhibitors. Exemplary third active ingredients to be administered in combination with first and second components are shown in Table A. First and second components are as defined in the above section entitled: ACTIVE INGREDIENTS OF THE COMBINATIONS.
- Salts
- Any reference to any of the compounds in the compositions of the invention also includes any physiologically acceptable salt thereof. Examples of physiologically acceptable salts of tenofovir DF, emtricitabine and their physiologically functional derivatives include salts derived from an appropriate base, such as an alkali metal (for example, sodium), an alkaline earth (for example, magnesium), ammonium and NX 4 + (wherein X is C1-C4 alkyl), or an organic acid such as fumaric acid, acetic acid, succinic acid. Physiologically acceptable salts of an hydrogen atom or an amino group include salts of organic carboxylic acids such as acetic, benzoic, lactic, fumaric, tartaric, maleic, malonic, malic, isethionic, lactobionic and succinic acids; organic sulfonic acids, such as methanesulfonic, ethanesulfonic, benzenesulfonic and p-toluenesulfonic acids; and inorganic acids, such as hydrochloric, sulfuric, phosphoric and sulfamic acids. Physiologically acceptable salts of a compound of an hydroxy group include the anion of said compound in combination with a suitable cation such as Na+ and NX4 + (wherein X is independently selected from H or a C1-C4 alkyl group).
- For therapeutic use, salts of active ingredients of the combinations of the invention will be physiologically acceptable, i.e. they will be salts derived from a physiologically acceptable acid or base. However, salts of acids or bases which are not physiologically acceptable may also find use, for example, in the preparation or purification of a physiologically acceptable compound. All salts, whether or not derived from a physiologically acceptable acid or base, are within the scope of the present invention.
- Administration of the Formulations
- While it is possible for the active ingredients of the combination to be administered alone and separately as monotherapies, it is preferable to administer them as a pharmaceutical co-formulation. A two-part or three-part combination may be administered simultaneously or sequentially. When administered sequentially, the combination may be administered in one, two, or three administrations.
- Preferably, two-part or three-part combinations are administered in a single pharmaceutical dosage form. More preferably, a two-part combination is administered as a single oral dosage form and a three-part combination is administered as two identical oral dosage forms. Examples include a single tablet of tenofovir disoproxil fumarate and emtricitabine, or two tablets of tenofovir disoproxil fumarate, emtricitabine, and efavirenz.
- It will be appreciated that the compounds of the combination may be administered: (1) simultaneously by combination of the compounds in a co-formulation or (2) by alternation, i.e. delivering the compounds serially, sequentially, in parallel or simultaneously in separate pharmaceutical formulations. In alternation therapy, the delay in administering the second, and optionally a third active ingredient, should not be such as to lose the benefit of a synergistic therapeutic effect of the combination of the active ingredients. By either method of administration (1) or (2), ideally the combination should be administered to achieve peak plasma concentrations of each of the active ingredients. A one pill once-per-day regimen by administration of a combination co-formulation may be feasible for some HIV-positive patients. Effective peak plasma concentrations of the active ingredients of the combination will be in the range of approximately 0.001 to 100 μM. Optimal peak plasma concentrations may be achieved by a formulation and dosing regimen prescribed for a particular patient. It will also be understood that tenofovir DF and emtricitabine, or the physiologically functional derivatives of either thereof, whether presented simultaneously or sequentially, may be administered individually, in multiples, or in any combination thereof. In general, during alternation therapy (2), an effective dosage of each compound is administered serially, where in co-formulation therapy (1), effective dosages of two or more compounds are administered together.
- Formulation of the Combinations
- When the individual components of the combination are administered separately they are generally each presented as a pharmaceutical formulation. The references hereinafter to formulations refer unless otherwise stated to formulations containing either the combination or a component compound thereof. It will be understood that the administration of the combination of the invention by means of a single patient pack, or patient packs of each formulation, within a package insert diverting the patient to the correct use of the invention is a desirable additional feature of this invention. The invention also includes a double pack comprising in association for separate administration, formulations of tenofovir disoproxil fumarate and emtricitabine, or a physiologically functional derivative of either or both thereof.
- The combination therapies of the invention include: (1) a combination of tenofovir DF and emtricitabine or (2) a combination containing a physiologically functional derivative of either or both thereof.
- The combination may be formulated in a unit dosage formulation comprising a fixed amount of each active pharmaceutical ingredient for a periodic, e.g. daily, dose or subdose of the active ingredients.
- Pharmaceutical formulations according to the present invention comprise a combination according to the invention together with one or more pharmaceutically acceptable carriers or excipients and optionally other therapeutic agents. Pharmaceutical formulations containing the active ingredient may be in any form suitable for the intended method of administration. When used for oral use for example, tablets, troches, lozenges, aqueous or oil suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups or elixirs may be prepared ( Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, Pa.). Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents including antioxidants, sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation. Tablets containing the active ingredient in admixture with non-toxic pharmaceutically acceptable excipient which are suitable for manufacture of tablets are acceptable. These excipients may be, for example, inert diluents, such as calcium or sodium carbonate, lactose, lactose monohydrate, croscarmellose sodium, povidone, calcium or sodium phosphate; granulating and disintegrating agents, such as maize starch, or alginic acid; binding agents, such as cellulose, microcrystalline cellulose, starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- Formulations for oral use may be also presented as hard gelatin capsules where the active ingredient is mixed with an inert solid diluent, for example pregelatinized starch, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions of the invention contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients include a suspending agent, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia, and dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadecaethyleneoxycetanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene sorbitan monooleate). The aqueous suspension may also contain one or more preservatives such as ethyl or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents and one or more sweetening agents, such as sucrose, sucralose or saccharin.
- Oil suspensions may be formulated by suspending the active ingredient in a vegetable oil, such as arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. The oral suspensions may contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents, such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an antioxidant such as ascorbic acid, BHT, etc.
- Dispersible powders and granules of the invention suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, a suspending agent, and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those disclosed above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
- The pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions or liposome formulations. The oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these. Suitable emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth, naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids and hexitol anhydrides, such as sorbitan monooleate, and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan monooleate. The emulsion may also contain sweetening and flavoring agents. Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- The pharmaceutical compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1,3-butane-diol or prepared as a lyophilized powder. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile fixed oils may conventionally be employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- The pharmaceutical compositions of the invention may be injected parenterally, for example, intravenously, intraperitoneally, intrathecally, intraventricularly, intrastemally, intracranially, intramuscularly or subcutaneously, or they may be administered by infusion techniques. They are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood. The aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary. The preparation of suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
- The pharmaceutical compositions of the invention may also be administered intranasally or by inhalation and are conveniently delivered in the form of a dry powder inhaler or an aerosol spray presentation from a pressurized container or a nebuliser with the use of a suitable propellant, e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoroalkane such as 1,1,1,2-tetrafluoroethane (HFC 134a), carbon dioxide or other suitable gas. In the case of a pressurized aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount. The pressurized container or nebuliser may contain a solution or suspension of the composition, e.g. using a mixture of ethanol and the propellant as the solvent, which may additional contain a lubricant, e.g. sorbitan trioleate. Capsules and cartridges (made, for example, from gelatin) for use in an inhaler or insufflator may be formulated to contain a powder mix of a compound of the formula (I) and a suitable powder base such as lactose or starch. Aerosol or dry powder formulations are preferably arranged so that each metered dose or “puff” contains from 20 μg to 20 mg of a composition for delivery to the patient. The overall daily dose with an aerosol will be in the range of from 20 μg to 20 mg which may be administered in a single dose or, more usually, in divided doses throughout the day.
- The amount of active ingredient that may be combined with the carrier material to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. For example, a time-release formulation intended for oral administration to humans may contain approximately 1 to 1000 mg of active material compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total compositions (weight:weight). The pharmaceutical composition can be prepared to provide easily measurable amounts for administration. For example, an aqueous solution intended for intravenous infusion may contain from about 3 to 500 μg of the active ingredient per milliliter of solution in order that infusion of a suitable volume at a rate of about 30 mL/hr can occur. As noted above, formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion. The active ingredient may also be administered as a bolus, electuary or paste.
- The combinations of the invention may conveniently be presented as a pharmaceutical formulation in a unitary dosage form. A convenient unitary dosage formulation contains the active ingredients in any amount from 1 mg to 1 g each, for example but not limited to, 10 mg to 300 mg. The synergistic effects of tenofovir DF in combination with emtricitabine may be realized over a wide ratio, for example 1:50 to 50:1 (tenofovir DF:emtricitabine). In one embodiment, the ratio may range from about 1:10 to 10:1. In another embodiment, the weight/weight ratio of tenofovir to emtricitabine in a co-formulated combination dosage form, such as a pill, tablet, caplet or capsule will be about 1, i.e. an approximately equal amount of tenofovir DF and emtricitabine. In other exemplary co-formulations, there may be more or less tenofovir than FTC. For example, 300 mg tenofovir DF and 200 mg emtricitabine can be co-formulated in a ratio of 1.5:1 (tenofovir DF: emtricitabine). In one embodiment, each compound will be employed in the combination in an amount at which it exhibits antiviral activity when used alone. Exemplary Formulations A, B, C, D, E, and F (Examples) have ratios of 12:1 to 1:1 (tenofovir DF: emtricitabine). Exemplary Formulations A, B, C, D, E, and F use amounts of tenofovir DF and emtricitabine ranging from 25 mg to 300 mg. Other ratios and amounts of the compounds of said combinations are contemplated within the scope of the invention.
- A unitary dosage form may further comprise tenofovir DF and emtricitabine, or physiologically functional derivatives of either thereof, and a pharmaceutically acceptable carrier.
- It will be appreciated by those skilled in the art that the amount of active ingredients in the combinations of the invention required for use in treatment will vary according to a variety of factors, including the nature of the condition being treated and the age and condition of the patient, and will ultimately be at the discretion of the attending physician or health care practitioner. The factors to be considered include the route of administration and nature of the formulation, the animal's body weight, age and general condition and the nature and severity of the disease to be treated. For example, in a Phase I/II monotherapy study of emtricitabine, patients received doses ranging from 25 mg to 200 mg twice-a-day for two weeks. At each dose regimen greater or equal to 200 mg, a 98-percent (1.75 log 10) or greater viral suppression was observed. A once-a-day dose of 200 mg of emtricitabine reduced the viral load by an average of 99 percent (1.92 log 10). Viread® (tenofovir DF) has been approved by the FDA for the treatment and prophylaxis of HIV infection as a 300 mg oral tablet. Emtriva™ (emtricitabine) has been approved by the FDA for the treatment of HIV as a 200 mg oral tablet.
- It is also possible to combine any two of the active ingredients in a unitary dosage form for simultaneous or sequential administration with a third active ingredient. The three-part combination may be administered simultaneously or sequentially. When administered sequentially, the combination may be administered in two or three administrations. Third active ingredients have anti-HIV activity and include protease inhibitors (PI), nucleoside reverse transcriptase inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI), and integrase inhibitors. Exemplary third active ingredients to be administered in combination with tenofovir DF, emtricitabine, and their physiological functional derivatives, are shown in Table A.
TABLE A 5,6 dihydro-5-azacytidine 5-aza 2′deoxycytidine 5-azacytidine 5-yl-carbocyclic 2′-deoxyguanosine (BMS200,475) 9 (arabinofuranosyl)guanine; 9-(2′ deoxyribofuranosyl)guanine 9-(2′-deoxy 2′fluororibofuranosyl)-2,6-diaminopurine 9-(2′-deoxy 2′fluororibofuranosyl)guanine 9-(2′-deoxyribofuranosyl)-2,6 diaminopurine 9-(arabinofuranosyl)-2,6 diaminopurine Abacavir, Ziagen ® Acyclovir, ACV; 9-(2-hydroxyethoxylmethyl)guanine Adefovir dipivoxil, Hepsera ® amdoxivir, DAPD Amprenavir, Agenerase ® araA; 9-β-D-arabinofuranosyladenine (Vidarabine) atazanivir sulfate (Reyataz ®) AZT; 3′-azido-2′,3′-dideoxythymdine, Zidovudine, (Retrovir ®) BHCG; (.+−.)-(1a,2b,3a)-9-[2,3-bis(hydroxymethyl)cyclobutyl]guanine BMS200,475; 5-yl-carbocyclic 2′-deoxyguanosine Buciclovir; (R) 9-(3,4-dihydroxybutyl)guanine BvaraU; 1-β-D-arabinofuranosyl-E-5-(2-bromovinyl)uracil (Sorivudine) Calanolide A Capravirine CDG; carbocyclic 2′-deoxyguanosine Cidofovir, HPMPC; (S)-9-(3-hydroxy-2- phosphonylmethoxypropyl)cytosine Clevudine, L-FMAU; 2′-Fluoro-5-methyl-β-L-arabino-furanosyluracil Combivir ® (lamivudine/zidovudine) Cytallene; [1-(4′-hydroxy-1′,2′-butadienyl)cytosine] d4C; 3′-deoxy-2′,3′-didehydrocytidine DAPD; (−)-β-D-2,6-diaminopurine dioxolane ddA; 2′,3′-dideoxyadenosine ddAPR; 2,6-diaminopurine-2′,3′-dideoxyriboside ddC; 2′,3′-dideoxycytidine (Zalcitabine) ddI; 2′,3′-dideoxyinosine, didanosine, (Videx ®, Videx ® EC) Delavirdine, Rescriptor ® Didanosine, ddI, Videx ®; 2′,3′-dideoxyinosine DXG; dioxolane guanosine E-5-(2-bromovinyl)-2′-deoxyuridine Efavirenz, Sustiva ® Enfuvirtide, Fuzeon ® F-ara-A; fluoroarabinosyladenosine (Fludarabine) FDOC; (−)-β-D-5-fluoro-1-[2-(hydroxymethyl)-1,3- dioxolane]cytosine FEAU; 2′-deoxy-2′-fluoro-1-β-D-arabinofuranosyl-5-ethyluracil FIAC; 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-5-iodocytosine FIAU; 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-5-iodouridine FLG; 2′,3′-dideoxy-3′-fluoroguanosine FLT; 3′-deoxy-3′-fluorothymidine Fludarabine; F-ara-A; fluoroarabinosyladenosine FMAU; 2′-Fluoro-5-methyl-β-L-arabino-furanosyluracil FMdC Foscarnet; phosphonoformic acid, PFA FPMPA; 9-(3-fluoro-2-phosphonylmethoxypropyl)adenine Gancyclovir, GCV; 9-(1,3-dihydroxy-2-propoxymethyl)guanine GS-7340; 9-[R-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]- phenoxyphosphinyl]methoxy]propyl]adenine HPMPA; (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine HPMPC; (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (Cidofovir) Hydroxyurea, Droxia ® Indinavir, Crixivan ® Kaletra ® (lopinavir/ritonavir) Lamivudine, 3TC, Epivir ™; (2R, 5S, cis)-4-amino-1-(2- hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one L-d4C; L-3′-deoxy-2′,3′-didehydrocytidine L-ddC; L-2′,3′-dideoxycytidine L-Fd4C; L-3′-deoxy-2′,3′-didehydro-5-fluorocytidine L-FddC; L-2′,3′-dideoxy-5-fluorocytidine Lopinavir Nelfinavir, Viracept ® Nevirapine, Viramune ® Oxetanocin A; 9-(2-deoxy-2-hydroxymethyl-β-D-erythro- oxetanosyl)adenine Oxetanocin G; 9-(2-deoxy-2-hydroxymethyl-β-D-erythro- oxetanosyl)guanine Penciclovir PMEDAP; 9-(2-phosphonylmethoxyethyl)-2,6-diaminopurine PMPA, tenofovir; (R)-9-(2-phosphonylmethoxypropyl)adenine PPA; phosphonoacetic acid Ribavirin; 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide Ritonavir, Norvir ® Saquinavir, Invirase ®, Fortovase ® Sorivudine, BvaraU; 1-β-D-arabinofuranosyl-E-5-(2-bromovinyl)uracil Stavudine, d4T, Zerit ®; 2′,3′-didehydro-3′-deoxythymidine Trifluorothymidine, TFT; Trifluorothymidine Trizivir ® (abacavir sulfate/lamivudine/zidovudine) Vidarabine, araA; 9-β-D-arabinofuranosyladenine Zalcitabine, Hivid ®, ddC; 2′,3′-dideoxycytidine Zidovudine, AZT, Retrovir ®; 3′-azido-2′,3′-dideoxythymdine Zonavir; 5-propynyl-1-arabinosyluracil - Another aspect of the present invention is a three-part combination comprising tenofovir DF, FTC, and 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine, also designated herein as GS-7340, which has the structure:

- GS-7340 is a prodrug of tenofovir and the subject of commonly owned, pending application, U.S. Ser. No. 09/909,560, filed Jul. 20, 2001 and Becker et al WO 02/08241.
- For example, a ternary unitary dosage may contain 1 mg to 1000 mg of tenofovir disoproxil fumarate, 1 mg to 1000 mg of emtricitabine, and 1 mg to 1000 mg of the third active ingredient. As a further feature of the present invention, a unitary dosage form may further comprise tenofovir DF, emtricitabine, the third active ingredient, or physiologically functional derivatives of the three active ingredients thereof, and a pharmaceutically acceptable carrier.
- Combinations of the present invention enable patients greater freedom from multiple dosage medication regimens and ease the needed diligence required in remembering and complying with complex daily dosing times and schedules. By combining tenofovir disoproxil fumarate and emtricitabine into a single dosage form, the desired daily regimen may be presented in a single dose or as two or more sub-doses per day. The combination of co-formulated tenofovir DF and emtricitabine may be administered as a single pill, once per day.
- A further aspect of the invention is a patient pack comprising at least one active ingredient: tenofovir disoproxil fumarate, emtricitabine, or a physiologically functional derivative of either of the combination and an information package or product insert containing directions on the use of the combination of the invention.
- Segregation of active ingredients in pharmaceutical powders and granulations is a widely recognized problem that can result in inconsistent dispersions of the active ingredients in final dosage forms. Some of the main factors contributing to segregation are particle size, shape and density. Segregation is particularly troublesome when attempting to formulate a single homogenous tablet containing multiple active ingredients having different densities and different particle sizes. Glidants are substances that have traditionally been used to improve the flow characteristics of granulations and powders by reducing interparticulate friction. See Lieberman, Lachman, & Schwartz, Pharmaceutical Dosage Forms: Tablets, Volume 1, p. 177-178 (1989), incorporated herein by reference. Glidants are typically added to pharmaceutical compositions immediately prior to tablet compression to facilitate the flow of granular material into the die cavities of tablet presses. Glidants include: colloidal silicon dioxide, asbestos free talc, sodium aluminosilicate, calcium silicate, powdered cellulose, microcrystalline cellulose, corn starch, sodium benzoate, calcium carbonate, magnesium carbonate, metallic stearates, calcium stearate, magnesium stearate, zinc stearate, stearowet C, starch, starch 1500, magnesium lauryl sulfate, and magnesium oxide. Exemplary Tablet Formulation A has colloidal silicon dioxide (Examples). Glidants can be used to increase and aid blend composition homogeneity in formulations of anti-HIV drugs (U.S. Pat. No. 6,113,920). The novel compositions of the present invention may contain glidants to effect and maintain homogeneity of active ingredients during handling prior to tablet compression.
- The present invention provides pharmaceutical formulations combining the active ingredients tenofovir DF and emtricitabine, or physiologically functional derivatives thereof, in a sufficiently homogenized form, and a method for using this pharmaceutical formulation. An object of the present invention is to utilize glidants to reduce the segregation of active ingredients in pharmaceutical compositions during pre-compression material handling. Another object of the present invention is to provide a pharmaceutical formulation combining the active ingredients tenofovir DF and emtricitabine, or physiologically functional derivatives thereof, with a pharmaceutically acceptable glidant, resulting in a mixture characterized by a pharmaceutically acceptable measure of homogeneity.
- Formulations include those suitable for oral, rectal, nasal, topical (including transdermal, buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) administration. The formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. Such methods represent a further feature of the present invention and include the step of bringing into association the active ingredients with the carrier, which constitutes one or more accessory ingredients, and maintaining chemical stability. In general, the formulations are prepared by uniformly and intimately bringing into association the active ingredients with liquid carriers or finely divided solid carriers or both, and then if necessary shaping the product.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, caplets, cachets or tablets each containing a predetermined amount of the active ingredients; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion. The active ingredient may also be presented as a bolus, electuary or paste.
- A tablet may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredients in a free-flowing form such as a powder or granules, optionally mixed with a binder (e.g. povidone, gelatin, hydroxypropyl methylcellulose), lubricant, inert diluent, preservative, disintegrant (e.g. sodium starch glycollate, cross-linked povidone, cross-linked sodium carboxymethyl cellulose) surface-active or dispersing agent. Molded tablets may be made by molding a mixture of the powdered compound moistened with an inert liquid diluent in a suitable machine. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredients therein using, for example, cellulose ether derivatives (e.g., hydroxypropyl methylcellulose) or methacrylate derivatives in varying proportions to provide the desired release profile. Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach.
- Formulations suitable for topical administration in the mouth include lozenges comprising the active ingredients in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier. Formulations for rectal administration may be presented as a suppository with a suitable base comprising, for example, cocoa butter or a salicylates. Topical administration may also be by means of a transdermal iontophoretic device.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- Formulations suitable for penile administration for prophylactic or therapeutic use may be presented in condoms, creams, gels, pastes, foams or spray formulations containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- Pharmaceutical formulations suitable for rectal administration wherein the carrier is a solid are most preferably presented as unit dose suppositories. Suitable carriers include cocoa butter and other materials commonly used in the art. The suppositories may be conveniently formed by admixture of the active combination with the softened or melted carrier(s) followed by chilling and shaping in moulds.
- Formulations suitable for parenteral administration include aqueous and nonaqueous isotonic sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents; and liposomes or other microparticulate systems which are designed to target the compound to blood components or one or more organs. The formulations may be presented in unit-dose or multi-dose sealed containers, for example, ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injection, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Exemplary unit dosage formulations are those containing a daily dose or daily subdose of the active ingredients, as hereinbefore recited, or an appropriate fraction thereof. It should be understood that in addition to the ingredients particularly mentioned above the formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question, for example, those suitable for oral administration may include such further agents as sweeteners, thickeners and flavoring agents.
- The compounds of the combination of the present invention may be obtained in a conventional manner, known to those skilled in the art. Tenofovir disoproxil fumarate can be prepared, for example, as described in U.S. Pat. No. 5,977,089. Methods for the preparation of FTC are described in WO 92/14743, incorporated herein by reference.
- Composition Use
- Compositions of the present invention are administered to a human or other mammal in a safe and effective amount as described herein. These safe and effective amounts will vary according to the type and size of mammal being treated and the desired results of the treatment. Any of the various methods known by persons skilled in the art for packaging tablets, caplets, or other solid dosage forms suitable for oral administration, that will not degrade the components of the present invention, are suitable for use in packaging. The combinations may be packaged in glass and plastic bottles. Tablets, caplets, or other solid dosage forms suitable for oral administration may be packaged and contained in various packaging materials optionally including a dessicant, e.g. silica gel. Packaging may be in the form of unit dose blister packaging. For example, a package may contain one blister tray of tenofovir DF and another blister tray of emtricitabine pills, tablets, caplets, or capsule. A patient would take one dose, e.g. a pill, from one tray and one from the other. Alternatively, the package may contain a blister tray of the co-formulated combination of tenofovir DF and emtricitabine in a single pill, tablet, caplet or capsule. As in other combinations and packaging thereof, the combinations of the invention include physiological functional derivatives of tenofovir DF and FTC.
- The packaging material may also have labeling and information related to the pharmaceutical composition printed thereon. Additionally, an article of manufacture may contain a brochure, report, notice, pamphlet, or leaflet containing product information. This form of pharmaceutical information is referred to in the pharmaceutical industry as a “package insert.” A package insert may be attached to or included with a pharmaceutical article of manufacture. The package insert and any article of manufacture labeling provides information relating to the pharmaceutical composition. The information and labeling provides various forms of information utilized by health-care professionals and patients, describing the composition, its dosage and various other parameters required by regulatory agencies such as the United States Food and Drug Agency.
- Assays of the Combinations
- The combinations of the inventions may be tested for in vitro activity against HIV and sensitivity, and for cytotoxicity in laboratory adapted cell lines, e.g. MT2 and in peripheral blood mononuclear cells (PBMC) according to standard assays developed for testing anti-HIV compounds, such as WO 02/068058 and U.S. Pat. No. 6,475,491. Combination assays may be performed at varying concentrations of the compounds of the combinations to determine EC 50 by serial dilutions.
- Exemplary Formulations
- The following examples further describe and demonstrate particular embodiments within the scope of the present invention. Techniques and formulations generally are found in Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, Pa.). The examples are given solely for illustration and are not to be construed as limitations as many variations are possible without departing from spirit and scope of the Invention. The following examples are intended for illustration only and are not intended to limit the scope of the invention in any way. “Active ingredient” denotes tenofovir disoproxil fumarate, emtricitabine, or a physiologically functional derivative of either thereof.
- Tablet Formulation
- The following exemplary formulations A, B, C, D, E, and F are prepared by wet granulation of the ingredients with an aqueous solution, addition of extragranular components and then followed by addition of magnesium stearate and compression.
- Formulation A:
mg/tablet Tenofovir Disoproxil Fumarate 300 emtricitabine 200 Microcrystalline Cellulose 200 Lactose Monohydrate 175 Croscarmellose Sodium 60 Pregelatinized Starch 50 Colloidal silicon dioxide 5 Magnesium Stearate 10 total: 1000 - Formulation B:
mg/tablet Tenofovir Disoproxil fumarate 300 emtricitabine 100 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 900 - Formulation C:
mg/tablet Tenofovir Disoproxil fumarate 200 emtricitabine 200 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 900 - Formulation D:
mg/tablet Tenofovir Disoproxil fumarate 300 emtricitabine 25 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 825 - Formulation E:
mg/tablet Tenofovir Disoproxil fumarate 200 emtricitabine 25 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 725 - Formulation F:
mg/tablet Tenofovir Disoproxil fumarate 100 emtricitabine 100 Microcrystalline Cellulose 200 Lactose Monohydrate 180 Sodium Starch Glycollate 60 Pregelatinized Starch 50 Magnesium Stearate 10 total: 700 - Formulation G (Controlled Release Formulation):
- This formulation is prepared by wet granulation of the ingredients with an aqueous solution, followed by the addition of magnesium stearate and compression.
mg/tablet Tenofovir Disoproxil fumarate 300 emtricitabine 200 Hydroxypropyl Methylcellulose 112 Lactose B.P. 53 Pregelatinized Starch B.P. 28 Magnesium Stearate 7 total: 700 - Drug release takes place over a period of about 6-8 hours and is complete after 12 hours.
- Capsule Formulations
- Formulation H:
- A capsule formulation is prepared by admixing the ingredients and filling into a two-part hard gelatin or hydroxypropyl methylcellulose capsule.
mg/capsule Active Ingredient 500 Microcrystalline Cellulose 143 Sodium Starch Glycollate 25 Magnesium Stearate 2 total: 670 - Formulation I (Controlled Release Capsule):
- The following controlled release capsule formulation is prepared by extruding ingredients a, b, and c using an extruder, followed by spheronization of the extrudate and drying. The dried pellets are then coated with release-controlling membrane (d) and filled into a two-piece, hard gelatin or hydroxypropyl methylcellulose capsule.
mg/capsule (a) Active Ingredient 500 (b) Microcrystalline Cellulose 125 (c) Lactose B.P. 125 (d) Ethyl Cellulose 13 total: 763 - Formulation J (Oral Suspension):
- The active ingredients are admixed with the ingredients and filling them as dry powder. Purified water is added and mixed well before use.
Active Ingredient 500 mg Confectioner's Sugar 2000 mg Simethicone 300 mg Methylparaben 30 mg Propylparaben 10 mg Flavor, Peach 500 mg Purified Water q.s. to 5.00 ml - Formulation K (Suppository):
- One-fifth of the Witepsol H15 is melted in a steam-jacketed pan at 45° C. maximum. The active ingredients are sifted through a 200 micron sieve and added to the molten base with mixing, using a Silverson fitted with a cutting head, until a smooth dispersion is achieved. Maintaining the mixture at 45° C., the remaining Witepsol H15 is added to the suspension and stirred to ensure a homogenous mix. The entire suspension is passed through a 250 micron stainless steel screen and, with continuous stirring, is allowed to cool to 40° C. At a temperature of 38° C. to 40° C., 2.02 g of the mixture is filled into suitable, 2 ml plastic molds. The suppositories are allowed to cool to room temperature.
mg/Suppository Active Ingredient 500 Hard Fat, B.P. (Witepsol H15 - Dynamit Nobel) 1770 total 2270 - Fixed Dose Combination Tablet
- A fixed dose combination tablet of tenofovir disoproxil fumarate (TDF) 300 mg/emtricitabine 200 mg was formulated using a wet granulation/fluid-bed drying process using conventional methods. See: U.S. Pat. No. 5,935,946; L. Young (editor). Tableting Specification Manual 5 th ed., American Pharmaceutical Association, Washington, D.C., (2001); L. Lachman, H. Lieberman (editors). Pharmaceutical Dosage Forms: Tablets (Vol 2), Marcel Dekker Inc., New York, 185-202 (1981); J. T. Fell and J. M. Newton, J. Pharm. Pharmacol. 20, 657-659 (1968); US Pharmacopeia 24-National Formulary 19, “Tablet Friability”, Chapter <1216>, Page 2148 (2000).
- The effects of granulation water level (ranging from 40% to 50% w/w) and wet massing time were studied on the physicochemical properties of the final powder blend and its performance with respect to blend uniformity and compressibility (tablet compactibility). In addition, content uniformity, assay, stability and dissolution performance was evaluated for the TDF/emtricitabine fixed dose combination tablets.
- Formulation Equipment
- Equipment included a high shear mixer equipped with a pressure tank and spray nozzle tip to add the granulating water, a fluid-bed dryer, a mill, a tumble blender, a rotary tablet press, and a tablet deduster.
- Formulation Process
- The dried, milled powder was blended with the extragranular microcrystalline cellulose and croscarmellose sodium and then blended with magnesium stearate. Powder samples were removed after mixing with the magnesium stearate. The blend samples were evaluated for, bulk density, mesh analysis and compressibility. The powder blend mixed with the magnesium stearate was compressed into tablets on a press setup.
- Materials
- The following Table 1 lists the quantitative composition of the TDF/emtricitabine tablet formulation.
TABLE 1 Unit Formula for Quantity per tablet cores 12 kg Batch Ingredient % w/w (mg/tablet) (kg) Tenofovir Disoproxil 30.0 300.0 3.60 Fumaratea Emtricitabinea 20.0 200.0 2.40 Pregelatinized Starch, NF/EP 5.0 50.0 0.60 Croscarmellose Sodium, 6.0 60.0 0.72 NF/EP Lactose Monohydrate, NF/EPa 8.0 80.0 0.96 Microcrystalline Cellulose, 30.0 300.0 3.60 NF/EPc Magnesium Stearate, NF/EP 1.0 10.0 0.12 Purified Water, USP/EP b b b Totals 100.0 1000.0 12.00 - Characterization Equipment
- Moisture content was measured by loss on drying using a heat lamp/balance system. The powder blend was sampled with a sampling thief fitted with chambers to determine powder blend uniformity. Duplicate samples were removed from each of several locations in the blender. Blend uniformity analysis was performed on one sample from each location.
- Particle size analysis of the final powder blend was determined by sifting a multi-gram sample through a screen using a sonic sifter. The quantity of final powder blend retained on each sieve and the fines collector was determined by calculating the difference in weight between the sieves and fines collector before and after the test. The geometric mean diameter particle size was calculated by logarithmic weighting of the sieved distribution.
- Bulk density was determined by filling a graduated cylinder with the final powder blend and measuring the weight differential between the empty and filled graduate cylinder per unit volume.
- Tablets were characterized for friability using a friabilator, a hardness tester, a thickness micrometer equipped with a printer, and a weighing balance.
- Compression characteristics were determined using a rotary tablet press equipped with a flat-faced, beveled edged punch to a target weight of 400 mg. The powder blends were compressed using target upper punch pressures ranging from approximately 100 to 250 MPa. The apparent normalized ejection force was determined and normalized for tablet thickness and diameter.
- Tablet hardness was determined using a hardness tester. Tablet thickness was determined using a micrometer, and tablet weights were determined using a top loading balance.
- Wet Granulation
- The powders were blended in a granulator and then granulated using water. The impeller and chopper speeds were kept constant in the blender at a low setting during the granulation and wet massing operations. After water addition, the impeller and chopper were stopped and the granulator bowl was opened to observe the granulation consistency and texture. The lid was closed and the wet massing phase was performed. Acceptable granules had 40% w/w and 60% w/w water, respectively.
- Wet Milling
- To facilitate a uniform drying process, each wet granulation was deagglomerated with a mill fitted with a screen and an impeller. The milled wet granules were charged into a fluid-bed dryer immediately following wet milling.
- Fluid-Bed Drying
- Milled wet granules were dried using an inlet air setpoint temperature of about 70° C. and airflow of approximately 100 cfm. The target LOD was about 1.0% with a range of not more than (NMT) 1.5%. The total fluid-bed drying time ranged from 53 to 75 minutes. Final LOD ranged from 0.4% to 0.7% for all of the batches dried. The final exhaust temperatures for all the batches ranged from 47° C. to 50° C.
- Dry Milling
- All dried granules were milled through a perforated screen. The mill was equipped with a square impeller and operated. The lots were milled and manually transferred to the V-blender.
- Blending
- Each lot was blended using the V-blender. In one set of three formulations, starting with 12 kg materials, final powder blend yield available for compression after blending ranged from 10.5 kg (87.5%) to 11.1 kg (92.5%). The final powder blend bulk density ranged from 0.48 to 0.58 g/cc and the geometric mean diameter particle size ranged from 112 to 221 μm. Percent water and wet massing time affect final powder blend particle size and bulk density.
- The powder blending for both tenofovir DF and emtricitabine gave a mean (n=10) strength value for tenofovir DF ranged from 100.6% to 102.8% of target strength for the lots and the relative standard deviation (RSD) was from 0.5% to 1.7%. The mean (n=10) strength value for emtricitabine ranged from 101.3% to 104.1% of target strength for the lots with the relative standard deviation (RSD) ranged from 0.6% to 1.7%. The final powder blend moisture level ranged from 0.8% to 1.1% LOD.
- Tablet Compression
- The final blends were compressed using a rotary tablet press and the tablets were film-coated.
- Three 300 gm formulations (Table 2) were granulated in a granulator equipped with a 1-L bowl. The quantities of intragranular components were based on a 300 g total batch size. The formulations in lots 1 and 2 differed in the amount of microcrystalline cellulose 30% vs. 20% w/w, respectively. Lots 2 and 3 were identical except for the type of binder. Lot 2 contained 5% w/w of pregelatinized starch and lot 3 contained 5% w/w povidone as binder.
TABLE 2 Ingredient Lot 1% w/w Lot 2% w/w Lot 3% w/w Tenofovir Disoproxil 30.0 30.0 30.0 Fumarate Emtricitabine 20.0 20.0 20.0 Pregelatinized Starch, 5.0 5.0 N/A NF/EP Povidone, USP/NF (C- N/A N/A 5.0 30) Croscarmellose 6.0 6.0 6.0 Sodium, NF/EP Lactose Monohydrate, 8.0 18.0 18.0 NF/EP Microcrystalline 30.0 20.0 20.0 Cellulose, NF/EPa Magnesium Stearate, 1.0 1.0 1.0 NF/EP Purified Water, a a a USP/EP Total 100.0 100.0 100.0 - After water addition, the impeller and chopper were stopped and the granulator bowl was opened to observe the granulation consistency and texture. To achieve similar granulation consistency, lots 1, 2, and 3 were granulated with 45%, 40%, and 30% w/w water, respectively. The lid was closed and the wet massing phase was performed. All lots had a 30 sec wet massing resulting in acceptable granulations. The wet granulations from all batches were hand screened through a sieve to deagglomerate. The resulting granulations were tray dried in a convection oven set at 60° C. for approximately 20 hours to an LOD <1.0%. The dried granulations from all batches were hand screened through a sieve. In order to fit the granulation into the small scale (300 mL) V-blender, the final blend batch size was adjusted to 100 g. A portion, 81 g of the resulting blend from Lot 1 was blended with 15 g microcrystalline cellulose, 3 g croscarmellose sodium and 1 g magnesium stearate. 86 g of the resulting granulation from Lot 2 and Lot 3 were each blended with 10 g microcrystalline cellulose, 3 g croscarmellose sodium and 1 g magnesium stearate.
- Purity analysis was conducted by reverse-phase HPLC (high performance liquid chromatography). Impurities related to tenofovir disoproxil fumarate and emtricitabine were characterized and measured in the bulk API (active pharmaceutical ingredient) before formulation in the three lots of Table 2, and again after formulation in the resulting tablets. The impurities include by-products from hydrolysis of the exocyclic amino groups of tenofovir disoproxil fumarate and emtricitabine, and the hydrolysis of the disoproxil (POC) esters of tenofovir disoproxil fumarate. In each lot, the sum total of impurities related to tenofovir disoproxil fumarate and emtricitabine was less than 1% after formulation and tablet manufacture.
- The physicochemical properties of tenofovir disoproxil fumarate and emtricitabine tablets were evaluated by visual appearance, water content, label strength, impurity and degradation product contents, and tablet dissolution. Stability studies were conducted on drug product packaged in container-closure systems that are identical to the intended clinical and commercial container-closure system. There was no sign of discoloration or tablet cracking during the course of the stability study. Film-coated tenofovir disoproxil fumarate and emtricitabine tablets exhibited satisfactory stability at 40° C./75% RH (relative humidity) for up to six months when packaged and stored with silica gel desiccant. No significant loss (defined as ≧5% degradation) in % label strength of tenofovir DF or emtricitabine was observed after six months at 40° C./75% RH. when packaged and stored with desiccant. The increase in the total degradation products was 1.5% for tenofovir DF and 0.6-0.7% for emtricitabine after six months at 40° C./75% RH when packaged and stored with 3 grams of desiccant.
- All publications and patent applications cited herein are incorporated by reference to the same extent as if each individual publication or patent application was specifically and individually indicated to be incorporated by reference.
- Although certain embodiments are described in detail above, those having ordinary skill in the art will clearly understand that many modifications are possible in the claims without departing from the teachings thereof. All such modifications are intended to be encompassed within the claims of the invention.
- A1. A pharmaceutical composition comprising an effective amount of a compound of the formula:

- wherein R 1 and R are independently selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl, C6-C20 substituted aryl, C6-C20 arylalkyl, C6-C20 substituted arylalkyl, acyloxymethyl esters —CH2OC(═O)R9 and acyloxymethyl carbonates —CH2OC(═O)OR9 where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl and C6-C20 substituted aryl;
- R 3 is selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, or CH2OR8 where R8 is C1-C6 alkyl, C1-C6 hydroxyalkyl and C1-C6 haloalkyl;
- R 4 and R5 are independently selected from H, NH2, NHR and NR2 where R is C1-C6 alkyl; and
- R 6 and R7 are independently selected from H and C1-C6 alkyl; or a physiologically functional derivative thereof;
- in combination with an effective amount of a compound of the formula

- wherein B is selected from adenine, guanine, cytosine, uracil, thymine, 7-deazaadenine, 7-deazaguanine, 7-deaza-8-azaguanine, 7-deaza-8-azaadenine, inosine, nebularine, nitropyrrole, nitroindole, 2-aminopurine, 2-amino-6-chloropurine, 2,6-diaminopurine, hypoxanthine, pseudouridine, 5-fluorocytosine, 5-chlorocytosine, 5-bromocytosine, 5-iodocytosine, pseudocytosine, pseudoisocytosine, 5-propynylcytosine, isocytosine, isoguanine, 7-deazaguanine, 2-thiopyrimidine, 6-thioguanine, 4-thiothymine, 4-thiouracil, O 6-methylguanine, N6-methyladenine, O4-methylthymine, 5,6-dihydrothymine, 5,6-dihydrouracil, 4-methylindole, and a pyrazolo[3,4-D]pyrimidine; and
- R is selected from H, C 1-C18 alkyl, C1-C18 substituted alkyl, C2-C18 alkenyl, C2-C18 substituted alkenyl, C2-C18 alkynyl, C2-C18 substituted alkynyl, C6-C20 aryl, C6-C20 substituted aryl, C2-C20 heterocycle, C2-C20 substituted heterocycle, phosphonate, phosphophosphonate, diphosphophosphonate, phosphate, diphosphate, triphosphate, polyethyleneoxy or a physiologically functional derivative thereof; and
- a pharmaceutically acceptable carrier.
- B2. A composition of embodiment A1 wherein, in formula 1, R 1 and R2 are independently selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl, C6-C20 substituted aryl, C6-C20 arylalkyl, C6-C20 substituted arylalkyl, acyloxymethyl esters —CH2OC(═O)R9 and acyloxymethyl carbonates —CH2OC(═O)OR9 where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl and C6-C20 substituted aryl; and R3 R4, R5, R6 and R7 are independently H or C1-C6 alkyl.
- C3. A composition of embodiment A1 wherein, in formula 2, B is cytosine or a 5-halocytosine.
- D4. A composition of embodiment A1 wherein, in formula 1, R 1 and R2 are independently selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl, C6-C20 substituted aryl, C6-C20 arylalkyl, C6-C20 substituted arylalkyl, acyloxymethyl esters —CH2OC(═O)R9 and acyloxymethyl carbonates —CH2OC(═O)OR9 where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl and C6-C20 substituted aryl; and R3, R4, R5, R6 and R7 are independently H or C1-C6 alkyl; and, in formula 2, B is cytosine or a 5-halocytosine.
- E5. A composition of embodiment D4 wherein, in formula 1, R 1 and R2 are independently selected from H, acyloxymethyl esters —CH2OC(═O)R9 and acyloxymethyl carbonates —CH2OC(═O)OR9 where R9 is C1-C6 alkyl; and R3, R4, R5, R6 and R7 are independently H or C1-C6 alkyl; and, in formula 2, B is cytosine or a 5-halocytosine and R is H.
- F6. A composition of embodiment E5 wherein, in formula 1, R 1 and R2 are independently selected from H and —CH2OC(═O)OCH(CH3)2; R3 is —CH3; and R4, R5, R6 and R7 are H; and, in formula 2, B is 5-fluorocytosine and R is H.
- G7. A pharmaceutical composition comprising a pharmaceutically effective amount of [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) or a physiologically functional derivative thereof and a pharmaceutically effective amount of (2R, 5S-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof; and a pharmaceutically acceptable carrier.
- H8. A pharmaceutical formulation of embodiment A1 to G7 further comprising a third active ingredient selected from the group consisting of a protease inhibitor, a nucleoside or nucleotide reverse transcriptase inhibitor, a non-nucleoside reverse transcriptase inhibitor, and an integrase inhibitor.
- I9. A pharmaceutical formulation of embodiments A1 to H8 in unit dosage form.
- J10. A method for the treatment or prevention of the symptoms or effects of an HIV infection in an infected animal which comprises administering to said animal a pharmaceutical composition of embodiments claims A1 to I9.
Claims (58)
1. A method for the treatment or prevention of the symptoms or effects of an HIV infection in an infected animal which comprises administering to said animal a therapeutically effective amount of a composition comprising [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) or a physiologically functional derivative thereof, and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof.
2. The method according to claim 1 wherein the composition comprises tenofovir disoproxil fumarate and emtricitabine.
3. The method according to claim 2 wherein the composition comprises about 300 mg of tenofovir disoproxil fumarate and about 200 mg of emtricitabine.
4. The method according to claim 1 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are present in a ratio of about 1:50 to about 50:1 by weight.
5. The method according to claim 1 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are present in a ratio of about 1:10 to about 10:1 by weight.
6. The method according to claim 1 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are each present in an amount from about 1 mg to about 1000 mg per unit dosage form.
7. The method according to claim 1 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are each present in an amount from about 100 mg to about 300 mg per unit dosage form.
8. A method for the treatment or prevention of the symptoms or effects of an HIV infection in an infected animal which comprises administering to said animal a therapeutically effective amount of a composition comprising [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) or a physiologically functional derivative thereof, and a compound of the formula:
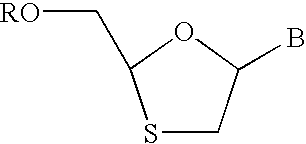
wherein B is selected from adenine, guanine, cytosine, uracil, thymine, 7-deazaadenine, 7-deazaguanine, 7-deaza-8-azaguanine, 7-deaza-8-azaadenine, inosine, nebularine, nitropyrrole, nitroindole, 2-aminopurine, 2-amino-6-chloropurine, 2,6-diaminopurine, hypoxanthine, pseudouridine, 5-fluorocytosine, 5-chlorocytosine, 5-bromocytosine, 5-iodocytosine, pseudocytosine, pseudoisocytosine, 5-propynylcytosine, isocytosine, isoguanine, 7-deazaguanine, 2-thiopyrimidine, 6-thioguanine, 4-thiothymine, 4-thiouracil, 06-methylguanine, N6-methyladenine, 04-methylthymine, 5,6-dihydrothymine, 5,6-dihydrouracil, 4-methylindole, and a pyrazolo[3,4-D]pyrimidine; and
R is selected from H, C1-C18 alkyl, C1-C18 substituted alkyl, C2-C18 alkenyl, C2-C18 substituted alkenyl, C2-C18 alkynyl, C2-C18 substituted alkynyl, C6-C20 aryl, C6-C20 substituted aryl, C2-C20 heterocycle, C2-C20 substituted heterocycle, phosphonate, phosphophosphonate, diphosphophosphonate, phosphate, diphosphate, triphosphate, polyethyleneoxy, and a prodrug moiety.
9. The method according to claim 1 wherein one component of composition is (2R, 5S, cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (3TC).
10. The method according to claim 1 wherein the composition comprises a physiologically functional derivative of emtricitabine which is a racemic mixture of the enantiomers (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one and (2S, 5R, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one.
11. The method according to claim 1 wherein the composition comprises a physiologically functional derivative of tenofovir disoproxil fumarate which has the structure:

wherein R1 and R2 are independently selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl, C6-C20 substituted aryl, C6-C20 arylalkyl, C6-C20 substituted arylalkyl, acyloxymethyl esters —CH2OC(═O)R and acyloxymethyl carbonates —CH2OC(═O)OR9 where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl and C6-C20 substituted aryl;
R3 is selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, or CH2OR8 where R8 is C1-C6 alkyl, C1-C6 hydroxyalkyl and C1-C6 haloalkyl;
R4 and R5 are independently selected from H, NH2, NHR and NR2 where R is C1-C6 alkyl; and
R6 and R7 are independently selected from H and C1-C6 alkyl;
or a pharmaceutically acceptable salt or solvate thereof.
12. The method according to claim 11 wherein at least one of R1 and R2 is —CH2OC(═O)C(CH3)3.
13. The method according to claim 11 wherein at least one of R1 and R2 is —CH2OC(═O)OC(CH3)3.
14. The method according to claim 11 wherein at least one of R1 and R2 is —CH2OC(═O)OCH(CH3)2.
15. A method for the treatment or prevention of the symptoms or effects of an HIV infection in an infected animal comprising administering in combination or alternation a therapeutically effective amount of [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) or a physiologically functional derivative thereof, and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof.
16. The method according to claim 15 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof, and emtricitabine or a physiologically functional derivative thereof, are administered in alternation.
17. The method according to claim 15 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof, and emtricitabine or a physiologically functional derivative thereof, are administered in combination as a single combined formulation.
18. The method according to claim 17 wherein the single combined formulation is administered once per day to an infected human.
19. The method according to claim 1 in which said animal is a human.
20. The method according to claim 1 wherein the composition further comprises a third active ingredient selected from a protease inhibitor (PI), a nucleoside reverse transcriptase inhibitor (NRTI), a non-nucleoside reverse transcriptase inhibitor (NNRTI), and an integrase inhibitor.
21. The method according to claim 20 wherein the third active ingredient is 9-[R-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]-phenoxyphosphinyl]methoxy]propyl]adenine (GS-7340).
22. The method according to claim 1 wherein the composition further comprises a pharmaceutically acceptable glidant.
23. The method according to claim 22 wherein the glidant is selected from silicon dioxide, powdered cellulose, microcrystalline cellulose, metallic stearates, sodium aluminosilicate, sodium benzoate, calcium carbonate, calcium silicate, corn starch, magnesium carbonate, asbestos free talc, stearowet C, starch, starch 1500, magnesium lauryl sulfate, magnesium oxide, and combinations thereof.
24. The method according to claim 23 wherein the metallic stearates are selected from calcium stearate, magnesium stearate, zinc stearate, and combinations thereof.
25. A pharmaceutical formulation comprising [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) or a physiologically functional derivative thereof and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine) or a physiologically functional derivative thereof.
26. The pharmaceutical formulation according to claim 25 further comprising one or more pharmaceutically acceptable carriers or excipients.
27. The pharmaceutical formulation according to claim 26 wherein the pharmaceutically acceptable carriers or excipients are selected from pregelatinized starch, croscarmellose sodium, povidone, lactose monohydrate, microcrystalline cellulose, and magnesium stearate; and combinations thereof.
28. The pharmaceutical formulation according to claim 25 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are present in a ratio of 1:50 to 50:1 by weight.
29. The pharmaceutical formulation according to claim 25 wherein tenofovir or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are present in a ratio of 1:10 to 10:1 by weight.
30. The pharmaceutical formulation according to claim 25 in unit dosage form.
31. The pharmaceutical formulation according to claim 30 wherein tenofovir disoproxil fumarate or a physiologically functional derivative thereof and emtricitabine or a physiologically functional derivative thereof are each and individually present in an amount from 100 mg to 1000 mg per unit dosage form.
32. The pharmaceutical formulation according to claim 31 comprising tenofovir disoproxil fumarate and emtricitabine.
33. The pharmaceutical formulation according to claim 32 comprising about 300 mg of tenofovir disoproxil fumarate and about 200 mg of emtricitabine.
34. The pharmaceutical formulation according to claim 25 suitable for oral administration.
35. The pharmaceutical formulation according to claim 25 in the form of a tablet or capsule.
36. The pharmaceutical formulation according to claim 25 suitable for administration once per day to an infected human.
37. The pharmaceutical formulation according to claim 25 comprising a physiologically functional derivative of emtricitabine which is (2R, 5S, cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (3TC).
38. The pharmaceutical formulation according to claim 25 wherein the combination comprises a physiologically functional derivative of tenofovir disoproxil fumarate which has the structure:

wherein R1 and R2 are independently selected from H, C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl, C6-C20 substituted aryl, C6-C20 arylalkyl, C6-C20 substituted arylalkyl, acyloxymethyl esters —CH2OC(═O)R and acyloxymethyl carbonates —CH2OC(═O)OR9 where R9 is C1-C6 alkyl, C1-C6 substituted alkyl, C6-C20 aryl or C6-C20 substituted aryl;
R3 is H, C1-C6 alkyl, C1-C6 substituted alkyl, or CH2OR8 where R8 is C1-C6 alkyl, C1-C6 hydroxyalkyl or C1-C6 haloalkyl;
R4 and R5 are independently selected from H, NH2, NHR and NR2 where R is C1-C6 alkyl; and
R6 and R7 are independently selected from H and C1-C6 alkyl;
or a pharmaceutically acceptable salt or solvate thereof.
39. The pharmaceutical formulation according to claim 38 wherein at least one of R1 and R2 is —CH2OC(═O)C(CH3)3.
40. The pharmaceutical formulation according to claim 38 wherein at least one of R1 and R2 is —CH2OC(═O)OC(CH3)3.
41. The pharmaceutical formulation according to claim 38 wherein at least one of R1 and R2 is —CH2OC(═O)OCH(CH3)2.
42. A patient pack comprising at least one active ingredient selected from [2-(6-amino-purin-9-yl)-1-methyl-ethoxymethyl]-phosphonic acid diisopropoxycarbonyloxymethyl ester fumarate (tenofovir disoproxil fumarate) and (2R, 5S, cis)-4-amino-5-fluoro-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (emtricitabine), and an information insert containing directions on the use of tenofovir and emtricitabine together in combination.
43. The patient pack according to claim 42 comprising a co-formulated pill, tablet, caplet, or capsule of 100 to 1000 mg of tenofovir disoproxil fumarate and 100 to 1000 mg of emtricitabine.
44. The patient pack according to claim 43 comprising a co-formulated pill, tablet, caplet, or capsule of 300 mg of tenofovir disoproxil fumarate and 200 mg of emtricitabine.
45. The patient pack according to claim 42 comprising a separate pill, tablet, caplet, or capsule of 100 to 1000 mg of tenofovir disoproxil fumarate and 100 to 1000 mg of emtricitabine.
46. The patient pack according to claim 45 comprising a separate pill, tablet, caplet, or capsule of 300 mg of tenofovir disoproxil fumarate and 200 mg of emtricitabine.
47. A chemically stable combination of tenofovir disoproxil fumarate and emtricitabine.
48. The chemically stable combination of claim 47 wherein the combination is a pharmaceutical dosage form.
49. The chemically stable combination of claim 48 wherein the dosage form is oral.
50. The chemically stable combination of any of claims 47, 48, or 49 which further comprises a third antiviral agent.
51. The chemically stable combination of claim 50 where in the third antiviral agent is an NNRTI or PI.
52. The chemically stable combination of claim 51 wherein the third antiviral agent is a PI.
53. The chemically stable combination of Clam 51 wherein the third antiviral agent is an NNRTI.
54. The chemically stable combination of claim 50 wherein the third antiviral agent is selected from Reyataz, Kaletra or Sustiva.
55. A chemically stable oral pharmaceutical dosage form comprising tenofovir disoproxil fumarate and emtricitabine.
56. A chemically stable oral pharmaceutical dosage form comprising tenofovir disoproxil fumarate, emtricitabine and Reyataz.
57. A chemically stable oral pharmaceutical dosage form comprising tenofovir disoproxil fumarate, emtricitabine and Kaletra.
58. A chemically stable oral pharmaceutical dosage form comprising tenofovir disoproxil fumarate, emtricitabine and Sustiva.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/757,141 US20040224917A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US44024603P | 2003-01-14 | 2003-01-14 | |
| US44030803P | 2003-01-14 | 2003-01-14 | |
| US10/757,141 US20040224917A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20040224917A1 true US20040224917A1 (en) | 2004-11-11 |
Family
ID=32776014
Family Applications (11)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/540,782 Abandoned US20060234982A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
| US10/757,122 Abandoned US20040224916A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
| US10/757,141 Abandoned US20040224917A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
| US10/540,794 Abandoned US20060246130A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
| US12/195,161 Expired - Lifetime US8592397B2 (en) | 2003-01-14 | 2008-08-20 | Compositions and methods for combination antiviral therapy |
| US12/204,174 Expired - Lifetime US8716264B2 (en) | 2003-01-14 | 2008-09-04 | Compositions and methods for combination antiviral therapy |
| US14/227,653 Abandoned US20140213556A1 (en) | 2003-01-14 | 2014-03-27 | Compositions and methods for combination antiviral therapy |
| US14/523,758 Expired - Lifetime US9457036B2 (en) | 2003-01-14 | 2014-10-24 | Compositions and methods for combination antiviral therapy |
| US14/523,783 Expired - Lifetime US9744181B2 (en) | 2003-01-14 | 2014-10-24 | Compositions and methods for combination antiviral therapy |
| US15/586,500 Abandoned US20170232019A1 (en) | 2003-01-14 | 2017-05-04 | Compositions and Methods for Combination Antiviral Therapy |
| US15/622,484 Abandoned US20170273994A1 (en) | 2003-01-14 | 2017-06-14 | Compositions and Methods for Combination Antiviral Therapy |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/540,782 Abandoned US20060234982A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
| US10/757,122 Abandoned US20040224916A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
Family Applications After (8)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/540,794 Abandoned US20060246130A1 (en) | 2003-01-14 | 2004-01-13 | Compositions and methods for combination antiviral therapy |
| US12/195,161 Expired - Lifetime US8592397B2 (en) | 2003-01-14 | 2008-08-20 | Compositions and methods for combination antiviral therapy |
| US12/204,174 Expired - Lifetime US8716264B2 (en) | 2003-01-14 | 2008-09-04 | Compositions and methods for combination antiviral therapy |
| US14/227,653 Abandoned US20140213556A1 (en) | 2003-01-14 | 2014-03-27 | Compositions and methods for combination antiviral therapy |
| US14/523,758 Expired - Lifetime US9457036B2 (en) | 2003-01-14 | 2014-10-24 | Compositions and methods for combination antiviral therapy |
| US14/523,783 Expired - Lifetime US9744181B2 (en) | 2003-01-14 | 2014-10-24 | Compositions and methods for combination antiviral therapy |
| US15/586,500 Abandoned US20170232019A1 (en) | 2003-01-14 | 2017-05-04 | Compositions and Methods for Combination Antiviral Therapy |
| US15/622,484 Abandoned US20170273994A1 (en) | 2003-01-14 | 2017-06-14 | Compositions and Methods for Combination Antiviral Therapy |
Country Status (26)
| Country | Link |
|---|---|
| US (11) | US20060234982A1 (en) |
| EP (4) | EP1923063A3 (en) |
| JP (8) | JP2006515624A (en) |
| KR (3) | KR20090053867A (en) |
| CN (2) | CN102670629B (en) |
| AP (1) | AP2089A (en) |
| AT (1) | ATE398455T1 (en) |
| AU (3) | AU2004206827A1 (en) |
| BR (1) | BRPI0406760A (en) |
| CA (2) | CA2512475C (en) |
| CY (1) | CY1108355T1 (en) |
| DE (1) | DE602004014470D1 (en) |
| DK (1) | DK1583542T3 (en) |
| EA (2) | EA015145B1 (en) |
| ES (1) | ES2308136T3 (en) |
| HK (1) | HK1079122A1 (en) |
| HR (2) | HRP20050619A2 (en) |
| IL (1) | IL169243A (en) |
| IS (1) | IS7977A (en) |
| MX (1) | MXPA05007016A (en) |
| NO (3) | NO337917B1 (en) |
| NZ (1) | NZ540728A (en) |
| PL (2) | PL378368A1 (en) |
| PT (1) | PT1583542E (en) |
| SI (1) | SI1583542T1 (en) |
| WO (2) | WO2004064846A1 (en) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070003608A1 (en) * | 2005-04-08 | 2007-01-04 | Almond Merrick R | Compounds, compositions and methods for the treatment of viral infections and other medical disorders |
| US20070003516A1 (en) * | 2005-04-08 | 2007-01-04 | Almond Merrick R | Compounds, compositions and methods for the treatment of poxvirus infections |
| US20070077295A1 (en) * | 2005-06-13 | 2007-04-05 | Gilead Sciences, Inc. | Method and composition for pharmaceutical product |
| WO2008100848A2 (en) * | 2007-02-12 | 2008-08-21 | Board Of Regents, The University Of Texas System | Novel agent for in vivo pet imaging of tumor proliferation |
| US20080317852A1 (en) * | 2005-12-14 | 2008-12-25 | Amar Lulla | Pharmaceutical Combination |
| US20090036408A1 (en) * | 2003-01-14 | 2009-02-05 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| US20090247749A1 (en) * | 2008-03-31 | 2009-10-01 | Apotex Pharmachem Inc. | Salt form and cocrystals of adefovir dipivoxil and processes for preparation thereof |
| US20100065444A1 (en) * | 2007-01-20 | 2010-03-18 | Stefan Henke | Pack containing soft capsules |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2010135569A1 (en) | 2009-05-20 | 2010-11-25 | Pharmasset, Inc. | N- [ (2 ' r) -2 ' -deoxy-2 ' -fluoro-2 ' -methyl-p-phenyl-5 ' -uridylyl] -l-alanine 1-methylethyl ester and process for its production |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| WO2011123668A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Stereoselective synthesis of phosphorus containing actives |
| WO2011156594A2 (en) | 2010-06-09 | 2011-12-15 | Vaccine Technologies, Incorporated | Therapeutic immunization in hiv infected subjects receiving stable antiretroviral treatment |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| WO2012075140A1 (en) | 2010-11-30 | 2012-06-07 | Pharmasset, Inc. | Compounds |
| WO2013082476A1 (en) | 2011-11-30 | 2013-06-06 | Emory University | Antiviral jak inhibitors useful in treating or preventing retroviral and other viral infections |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| US8598185B2 (en) | 2005-06-13 | 2013-12-03 | Bristol-Myers Squibb & Gilead Sciences, Inc. | Unitary pharmaceutical dosage form |
| US8614200B2 (en) | 2009-07-21 | 2013-12-24 | Chimerix, Inc. | Compounds, compositions and methods for treating ocular conditions |
| US8993542B2 (en) | 2008-01-25 | 2015-03-31 | Chimerix Inc. | Methods of treating viral infections |
| US9006218B2 (en) | 2010-02-12 | 2015-04-14 | Chimerix Inc. | Nucleoside phosphonate salts |
| US20150272972A1 (en) * | 2006-02-03 | 2015-10-01 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Serv | Inhibition of hiv infection through chemoprophyalxis |
| US9278135B2 (en) | 2010-04-26 | 2016-03-08 | Chimerix Inc. | Methods of treating retroviral infections and related dosage regimes |
Families Citing this family (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3165220B1 (en) | 2004-12-03 | 2019-04-03 | Merck Sharp & Dohme Corp. | Pharmaceutical composition containing an anti-nucleating agent |
| US8000981B2 (en) | 2005-11-30 | 2011-08-16 | The Invention Science Fund I, Llc | Methods and systems related to receiving nutraceutical associated information |
| US8340944B2 (en) | 2005-11-30 | 2012-12-25 | The Invention Science Fund I, Llc | Computational and/or control systems and methods related to nutraceutical agent selection and dosing |
| US10296720B2 (en) | 2005-11-30 | 2019-05-21 | Gearbox Llc | Computational systems and methods related to nutraceuticals |
| US8068991B2 (en) | 2005-11-30 | 2011-11-29 | The Invention Science Fund I, Llc | Systems and methods for transmitting pathogen related information and responding |
| US8297028B2 (en) | 2006-06-14 | 2012-10-30 | The Invention Science Fund I, Llc | Individualized pharmaceutical selection and packaging |
| US7927787B2 (en) | 2006-06-28 | 2011-04-19 | The Invention Science Fund I, Llc | Methods and systems for analysis of nutraceutical associated components |
| US7827042B2 (en) | 2005-11-30 | 2010-11-02 | The Invention Science Fund I, Inc | Methods and systems related to transmission of nutraceutical associated information |
| US7974856B2 (en) | 2005-11-30 | 2011-07-05 | The Invention Science Fund I, Llc | Computational systems and methods related to nutraceuticals |
| EP2049506B2 (en) | 2006-07-07 | 2024-05-08 | Gilead Sciences, Inc. | Modulators of pharmacokinetic properties of therapeutics |
| WO2008096369A2 (en) * | 2007-02-05 | 2008-08-14 | Matrix Laboratories Limited | Pharmaceutical formulation for use in hiv therapy |
| PT2487163T (en) | 2007-02-23 | 2016-11-21 | Gilead Sciences Inc | Modulators of pharmacokinetic properties of therapeutics |
| US20100055180A1 (en) * | 2007-10-10 | 2010-03-04 | Mallinckrodt Baker, Inc. | Directly Compressible Granular Microcrystalline Cellulose Based Excipient, Manufacturing Process and Use Thereof |
| NZ584521A (en) * | 2007-10-10 | 2011-03-31 | Avantor Performance Mat Inc | Directly compressible high functionality granular microcrystalline cellulose based excipient, manufacturing process and use thereof |
| CA2713423A1 (en) | 2008-01-18 | 2009-07-23 | North Carolina State University | Peptides and methods of use as therapeutics and screening agents |
| MX2010011963A (en) | 2008-05-02 | 2010-12-06 | Gilead Sciences Inc | The use of solid carrier particles to improve the processability of a pharmaceutical agent. |
| AU2010210598B2 (en) | 2009-02-06 | 2015-03-05 | Gilead Sciences, Inc. | Tablets for combination therapy |
| ES2969969T3 (en) * | 2010-01-27 | 2024-05-23 | Viiv Healthcare Co | Combinations of dolutegravir and lamivudine for the treatment of HIV infection |
| EP2389929A1 (en) * | 2010-05-30 | 2011-11-30 | Abdi Ibrahim Ilac Sanayi ve Ticaret Anonim Sirketi | Pharmaceutical formulations of tenofovir |
| CN103038241B (en) * | 2010-08-01 | 2015-06-17 | 正大天晴药业集团股份有限公司 | Crystals of tenofovir disoproxil fumarate |
| CN106511357A (en) * | 2010-11-19 | 2017-03-22 | 吉利德科学公司 | Therapeutic compositions comprising rilpivirine HCL and tenofovir disoproxil fumarate |
| US9550803B2 (en) | 2011-05-06 | 2017-01-24 | University Of Southern California | Method to improve antiviral activity of nucleotide analogue drugs |
| CA2857490C (en) | 2011-12-22 | 2020-03-31 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| AU2012327170A1 (en) * | 2012-02-03 | 2013-08-22 | Gilead Sciences, Inc. | Therapeutic compounds |
| CN104105484A (en) | 2012-02-03 | 2014-10-15 | 吉联亚科学公司 | Combination therapy comprising tenofovir alafenamide hemifumarate and cobicistat for use in the treatment of viral infections |
| CN103665043B (en) * | 2012-08-30 | 2017-11-10 | 江苏豪森药业集团有限公司 | A kind of tenofovir prodrug and its application in medicine |
| US9227990B2 (en) * | 2012-10-29 | 2016-01-05 | Cipla Limited | Antiviral phosphonate analogues and process for preparation thereof |
| UY35213A (en) | 2012-12-21 | 2014-06-30 | Gilead Sciences Inc | COMPOUNDS OF THE TYPE OF POLYCYCLIC CARBAMYLPIRIDONS AND THEIR PHARMACEUTICAL USE |
| CN103127028A (en) * | 2013-03-14 | 2013-06-05 | 南京恒道医药科技有限公司 | Capsule containing tenofovir disoproxil fumarate |
| NO2865735T3 (en) | 2013-07-12 | 2018-07-21 | ||
| AU2014286993B2 (en) | 2013-07-12 | 2018-10-25 | Gilead Sciences, Inc. | Polycyclic-carbamoylpyridone compounds and their use for the treatment of HIV infections |
| MX2016002560A (en) * | 2013-08-29 | 2016-10-26 | Teva Pharma | Unit dosage form comprising emtricitabine, tenofovir, darunavir and ritonavir and a monolithic tablet comprising darunavir and ritonavir. |
| CZ2013985A3 (en) * | 2013-12-09 | 2015-06-17 | Zentiva, K.S. | Stable pharmaceutical composition containing tenofovir disoproxil fumarate |
| TWI660965B (en) | 2014-01-15 | 2019-06-01 | 美商基利科學股份有限公司 | Solid forms of tenofovir |
| CA2942877A1 (en) | 2014-04-08 | 2015-10-15 | Nitzan SHAHAR | Unit dosage form comprising emtricitabine, tenofovir, darunavir and ritonavir |
| TW201613936A (en) | 2014-06-20 | 2016-04-16 | Gilead Sciences Inc | Crystalline forms of(2R,5S,13aR)-8-hydroxy-7,9-dioxo-n-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| NO2717902T3 (en) | 2014-06-20 | 2018-06-23 | ||
| TWI677489B (en) | 2014-06-20 | 2019-11-21 | 美商基利科學股份有限公司 | Synthesis of polycyclic-carbamoylpyridone compounds |
| CN105399771B (en) * | 2014-07-21 | 2020-11-24 | 江苏豪森药业集团有限公司 | Tenofovir prodrug crystal form and preparation method and application thereof |
| TWI738321B (en) | 2014-12-23 | 2021-09-01 | 美商基利科學股份有限公司 | Polycyclic-carbamoylpyridone compounds and their pharmaceutical use |
| RS62434B1 (en) | 2014-12-26 | 2021-11-30 | Univ Emory | Anti-viral n4-hydroxycytidine derivatives |
| EP3273946A1 (en) * | 2015-03-27 | 2018-01-31 | F. Hoffmann-La Roche AG | Pharmaceutical formulation comprising sembragiline |
| NZ735575A (en) | 2015-04-02 | 2018-11-30 | Gilead Sciences Inc | Polycyclic-carbamoylpyridone compounds and their pharmaceutical use |
| PT4070788T (en) * | 2015-06-30 | 2023-06-06 | Gilead Sciences Inc | Pharmaceutical formulations |
| TWI620754B (en) * | 2015-08-26 | 2018-04-11 | Method for preparing amino phosphate derivative and preparation method thereof | |
| JP6621933B2 (en) | 2015-11-09 | 2019-12-18 | ギリアード サイエンシス インコーポレーテッド | Therapeutic composition for treating human immunodeficiency virus |
| US10450335B2 (en) | 2015-12-15 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral oxime phosphoramide compounds |
| CN109641874A (en) | 2016-05-10 | 2019-04-16 | C4医药公司 | C for target protein degradation3The glutarimide degron body of carbon connection |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| CN109562107A (en) | 2016-05-10 | 2019-04-02 | C4医药公司 | Heterocycle degron body for target protein degradation |
| EP3503895B1 (en) | 2016-08-25 | 2021-09-15 | Merck Sharp & Dohme Corp. | Antiviral prodrugs of tenofovir |
| US10736908B2 (en) | 2016-10-26 | 2020-08-11 | Merck Sharp & Dohme Corp. | Antiviral aryl-amide phosphodiamide compounds |
| TR201617448A2 (en) * | 2016-11-29 | 2018-06-21 | Arven Ilac Sanayi Ve Ticaret Anonim Sirketi | SOLID ORAL PHARMACEUTICAL COMPOSITIONS CONTAINING TENOFOVIR AND EMTRISITAB |
| WO2018118826A1 (en) | 2016-12-22 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antiviral benzyl-amine phosphodiamide compounds |
| CN106749254B (en) * | 2017-01-10 | 2018-05-25 | 青岛科技大学 | A kind of adenine ethylnaphthalene acetate compounds and its purposes as plant growth regulator |
| WO2018153977A1 (en) * | 2017-02-24 | 2018-08-30 | Hexal Ag | Stable composition of tenofovir alafenamide |
| GB201705087D0 (en) * | 2017-03-30 | 2017-05-17 | Univ Liverpool | Method for producing a liquid composition |
| RU2019136678A (en) * | 2017-04-18 | 2021-05-18 | Сипла Лимитед | COMBINATION THERAPY FOR USE IN THE TREATMENT OF RETROVIRAL INFECTIONS |
| RU2662160C9 (en) * | 2017-07-03 | 2018-10-22 | Александрович Иващенко Андрей | Combined drug for viral infection therapy |
| KR102077060B1 (en) * | 2017-07-14 | 2020-02-13 | 주식회사 종근당 | Pharmaceutical composition comprising Tenofovir Disoproxil Aspartate salt |
| RU2666727C1 (en) * | 2017-07-18 | 2018-09-12 | Андрей Александрович Иващенко | Hepatitis b (hbv) inhibitor |
| TR201713954A2 (en) | 2017-09-20 | 2019-04-22 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | PHARMACEUTICAL COMBINATIONS OF TENOFOVIR, EMTRISITABIN AND EAVIRENZINE |
| KR102626210B1 (en) | 2017-12-07 | 2024-01-18 | 에모리 유니버시티 | N4-hydroxycytidine and derivatives and anti-viral uses related thereto |
| US11826375B2 (en) | 2018-07-19 | 2023-11-28 | Merck Sharp & Dohme Llc | Phosphinic amide prodrugs of tenofovir |
| RU2726210C2 (en) * | 2018-12-27 | 2020-07-09 | Общество С Ограниченной Ответственностью "Пролонгированные Лекарства" | Combination of antiviral agents, kit and method of treatment based thereon |
| CN110261631A (en) * | 2019-07-26 | 2019-09-20 | 重庆德方信息技术有限公司 | Liquid dropping mechanism for health test apparatus |
| CN110251476B (en) * | 2019-08-01 | 2022-08-09 | 海思科制药(眉山)有限公司 | Emtricitabine tenofovir pharmaceutical composition |
| CN113880898B (en) * | 2020-10-30 | 2023-07-25 | 杭州拉林智能科技有限公司 | Flavonoid glycoside-organic amine antimicrobial compound salt compound and preparation method and application thereof |
Family Cites Families (133)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US568164A (en) | 1896-09-22 | Motor attachment for bicycles | ||
| CH389608A (en) | 1960-01-19 | 1965-03-31 | Sandoz Ag | Process for the production of new ethers |
| US3524846A (en) * | 1967-06-02 | 1970-08-18 | Syntex Corp | Process for the didealkylation of phosphonate esters |
| US3622677A (en) | 1969-07-07 | 1971-11-23 | Staley Mfg Co A E | Compressed tablets containing compacted starch as binder-disintegrant ingredient |
| CH531000A (en) * | 1970-03-11 | 1972-11-30 | Sandoz Ag | Process for the preparation of new benzocycloheptathiophenes |
| US3994974A (en) * | 1972-02-05 | 1976-11-30 | Yamanouchi Pharmaceutical Co., Ltd. | α-Aminomethylbenzyl alcohol derivatives |
| US4003878A (en) * | 1972-12-07 | 1977-01-18 | Avtex Fibers Inc. | Method of preparing an alkali-metal salt of an alkoxysulfonated benzoic acid glycol ester |
| GB1523865A (en) | 1974-09-02 | 1978-09-06 | Wellcome Found | Purine compunds and salts thereof |
| DE2645710C2 (en) * | 1976-10-09 | 1985-06-27 | Merck Patent Gmbh, 6100 Darmstadt | Phenoxy-aminopropanols, process for their manufacture and pharmaceutical preparation |
| US4384005A (en) | 1980-09-26 | 1983-05-17 | General Foods Corporation | Non-friable, readily-soluble, compressed tablets and process for preparing same |
| GB2111043B (en) | 1980-12-12 | 1985-02-06 | Ciba Geigy Ag | Novel cephalosporin esters |
| US4355032B2 (en) * | 1981-05-21 | 1990-10-30 | 9-(1,3-dihydroxy-2-propoxymethyl)guanine as antiviral agent | |
| JPS5879983A (en) * | 1981-11-06 | 1983-05-13 | Kanebo Ltd | Novel benzimidazole derivative, its preparation and pharmaceutical composition thereof |
| US4816570A (en) | 1982-11-30 | 1989-03-28 | The Board Of Regents Of The University Of Texas System | Biologically reversible phosphate and phosphonate protective groups |
| US4476248A (en) * | 1983-02-28 | 1984-10-09 | The Upjohn Company | Crystallization of ibuprofen |
| EP0141927B1 (en) * | 1983-08-18 | 1991-10-30 | Beecham Group Plc | Antiviral guanine derivatives |
| US5155268A (en) * | 1984-05-04 | 1992-10-13 | The Upjohn Company | Antiarrhythmic N-aminoalkylene alkyl and aryl sulfonamides |
| EP0182024B1 (en) | 1984-09-20 | 1991-04-03 | Beecham Group Plc | Purine derivatives and their pharmaceutical use |
| CS263951B1 (en) | 1985-04-25 | 1989-05-12 | Antonin Holy | 9-(phosponylmethoxyalkyl)adenines and method of their preparation |
| CS263952B1 (en) | 1985-04-25 | 1989-05-12 | Holy Antonin | Remedy with antiviral effect |
| GB8607684D0 (en) * | 1986-03-27 | 1986-04-30 | Ici America Inc | Thiazepine compounds |
| US4968788A (en) | 1986-04-04 | 1990-11-06 | Board Of Regents, The University Of Texas System | Biologically reversible phosphate and phosphonate protective gruops |
| CS264222B1 (en) * | 1986-07-18 | 1989-06-13 | Holy Antonin | N-phosphonylmethoxyalkylderivatives of bases of pytimidine and purine and method of use them |
| NZ222553A (en) | 1986-11-18 | 1991-07-26 | Bristol Myers Co | Phosphonomethoxyalkylene purine and pyrimidine derivatives and pharmaceutical compositions |
| DE3790883T1 (en) | 1987-01-20 | 1988-12-08 | ||
| GB8719367D0 (en) * | 1987-08-15 | 1987-09-23 | Wellcome Found | Therapeutic compounds |
| ZA885709B (en) | 1987-08-19 | 1989-04-26 | Fujisawa Pharmaceutical Co | Novel crystalline 7-(2-(2-aminothiazol-4-yl)-2-hydroxyiminoacetamido)-3-vinyl-3-cephem-4-carboxylic acid(syn isomer) |
| US5047407A (en) | 1989-02-08 | 1991-09-10 | Iaf Biochem International, Inc. | 2-substituted-5-substituted-1,3-oxathiolanes with antiviral properties |
| US6175008B1 (en) | 1988-04-11 | 2001-01-16 | Biochem Pharma Inc. | Processes for preparing substituted 1,3-oxathiolanes with antiviral properties |
| US5466806A (en) | 1989-02-08 | 1995-11-14 | Biochem Pharma Inc. | Processes for preparing substituted 1,3-oxathiolanes with antiviral properties |
| US5453503A (en) | 1988-10-11 | 1995-09-26 | Eli Lilly And Company | Azetidinone intermediates to carbacephalosporins and process |
| CA2001715C (en) | 1988-11-14 | 1999-12-28 | Muzammil M. Mansuri | Carbocyclic nucleosides and nucleotides |
| UA45942A (en) * | 1989-02-08 | 2002-05-15 | Біокем Фарма, Інк. | 1,3-OXATHYOLANE, ITS DERIVATIVES, METHOD (OPTIONS) OF ITS PREPARATION AND PHARMACEUTICAL COMPOSITION |
| US6642245B1 (en) | 1990-02-01 | 2003-11-04 | Emory University | Antiviral activity and resolution of 2-hydroxymethyl-5-(5-fluorocytosin-1-yl)-1,3-oxathiolane |
| US6703396B1 (en) | 1990-02-01 | 2004-03-09 | Emory University | Method of resolution and antiviral activity of 1,3-oxathiolane nuclesoside enantiomers |
| US5728575A (en) | 1990-02-01 | 1998-03-17 | Emory University | Method of resolution of 1,3-oxathiolane nucleoside enantiomers |
| US5204466A (en) * | 1990-02-01 | 1993-04-20 | Emory University | Method and compositions for the synthesis of bch-189 and related compounds |
| GB9009861D0 (en) | 1990-05-02 | 1990-06-27 | Glaxo Group Ltd | Chemical compounds |
| WO1991019721A1 (en) | 1990-06-13 | 1991-12-26 | Arnold Glazier | Phosphorous produgs |
| US5177064A (en) * | 1990-07-13 | 1993-01-05 | University Of Florida | Targeted drug delivery via phosphonate derivatives |
| HU9300132D0 (en) | 1990-07-19 | 1993-04-28 | Beecham Group Plc | Method for producing phosphono-alkene-purine derivatives and medical preparatives of anti-viral effect containing them |
| KR100221981B1 (en) * | 1990-08-10 | 1999-09-15 | 안토닌 포레이트 | Novel process for the preparation of nucleotides |
| EP0481214B1 (en) * | 1990-09-14 | 1998-06-24 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Prodrugs of phosphonates |
| CA2054126A1 (en) | 1990-10-26 | 1992-04-27 | Michiyuki Sendai | Cephem compounds, their production and use |
| US5587480A (en) | 1990-11-13 | 1996-12-24 | Biochem Pharma, Inc. | Substituted 1,3-oxathiolanes and substituted 1,3-dithiolanes with antiviral properties |
| US5208221A (en) * | 1990-11-29 | 1993-05-04 | Bristol-Myers Squibb Company | Antiviral (phosphonomethoxy) methoxy purine/pyrimidine derivatives |
| GB9026164D0 (en) | 1990-12-01 | 1991-01-16 | Beecham Group Plc | Pharmaceuticals |
| US5179104A (en) | 1990-12-05 | 1993-01-12 | University Of Georgia Research Foundation, Inc. | Process for the preparation of enantiomerically pure β-D-(-)-dioxolane-nucleosides |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| NZ250842A (en) | 1991-02-22 | 1996-03-26 | Univ Emory | Resolution of a racemic mixture of nucleoside enantiomers such as 2-hydroxymethyl-5-(5-fluorocytosin-1-yl)-1,3-oxathiolane (ftc) |
| US6812233B1 (en) | 1991-03-06 | 2004-11-02 | Emory University | Therapeutic nucleosides |
| CZ285232B6 (en) * | 1991-05-16 | 1999-06-16 | Glaxo Group Limited | Antiviral mixtures and pharmaceutical preparation containing thereof |
| GB9110874D0 (en) | 1991-05-20 | 1991-07-10 | Iaf Biochem Int | Medicaments |
| ZA923641B (en) * | 1991-05-21 | 1993-02-24 | Iaf Biochem Int | Processes for the diastereoselective synthesis of nucleosides |
| GB9111902D0 (en) | 1991-06-03 | 1991-07-24 | Glaxo Group Ltd | Chemical compounds |
| GB9116601D0 (en) | 1991-08-01 | 1991-09-18 | Iaf Biochem Int | 1,3-oxathiolane nucleoside analogues |
| US6177435B1 (en) | 1992-05-13 | 2001-01-23 | Glaxo Wellcome Inc. | Therapeutic combinations |
| ATE173267T1 (en) | 1992-06-23 | 1998-11-15 | Yamanouchi Pharma Co Ltd | NEW CRYSTAL OF HETEROCYCLIC BIS(PHOSPHONIC ACID) MONOHYDRATE DERIVATIVE |
| US5532225A (en) | 1992-07-31 | 1996-07-02 | Sri International | Acyclic purine phosphonate nucleotide analogs as antiviral agents, and related synthetic methods |
| US5432172A (en) | 1992-08-03 | 1995-07-11 | The Research Foundation Of State University Of New York | Biological applications of alkaloids derived from the tunicate Eudistoma sp. |
| US6057305A (en) | 1992-08-05 | 2000-05-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Antiretroviral enantiomeric nucleotide analogs |
| US5665720A (en) | 1992-08-07 | 1997-09-09 | Merck & Co., Inc. | Benzoxazinones as inhibitors of HIV reverse transcriptase |
| NZ248852A (en) | 1992-10-28 | 1997-06-24 | Squibb & Sons Inc | Alpha-phosphono and phosphinosulphonates and medicaments thereof |
| EP0684953A4 (en) * | 1993-02-03 | 1999-12-22 | Gensia Inc | Adenosine kinase inhibitors comprising lyxofuranosyl derivatives. |
| US5514798A (en) * | 1993-06-02 | 1996-05-07 | Gilead Sciences, Inc. | Method and cyclic carbonates for nucleotide analogues |
| GB9311709D0 (en) | 1993-06-07 | 1993-07-21 | Iaf Biochem Int | Stereoselective synthesis of nucleoside analogues using bicycle intermediate |
| CA2126601A1 (en) | 1993-06-29 | 1994-12-30 | Mitsubishi Chemical Corporation | Phosphonate-nucleotide ester derivatives |
| US5798340A (en) * | 1993-09-17 | 1998-08-25 | Gilead Sciences, Inc. | Nucleotide analogs |
| WO1995007920A1 (en) | 1993-09-17 | 1995-03-23 | Gilead Sciences, Inc. | Nucleotide analogs |
| KR100386685B1 (en) | 1993-09-17 | 2003-12-31 | 길리애드 사이언시즈, 인코포레이티드 | Nucleotide Homolog |
| WO1995032957A1 (en) | 1994-05-27 | 1995-12-07 | Astra Aktiebolag | Novel ethoxycarbonyloxymethyl derivatives of substituted benzimidazoles |
| US5514557A (en) | 1994-06-06 | 1996-05-07 | Genetic Testing Institute Inc. | Method and kit for detecting antibodies specific for HLA and/or platelet glycoproteins |
| PE32296A1 (en) | 1994-07-28 | 1996-08-07 | Hoffmann La Roche | L-MONOVALINE ESTER DERIVED FROM 2- (2-AMINO-1,6-DIHYDRO-6-OXO-PURIN-9-IL) METOXI-1,3-PROPANDIOL AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS |
| US5512596A (en) * | 1994-09-02 | 1996-04-30 | Gilead Sciences, Inc. | Aromatic compounds |
| US5486806A (en) * | 1994-11-09 | 1996-01-23 | Firari; Harold A. | Anti-hijacking and theft prevention device for motor vehicles |
| US5684018A (en) | 1994-12-13 | 1997-11-04 | Merck & Co., Inc. | Acyloxyisopropyl carbamates as prodrugs for amine drugs |
| MY115461A (en) | 1995-03-30 | 2003-06-30 | Wellcome Found | Synergistic combinations of zidovudine, 1592u89 and 3tc |
| JPH11507632A (en) * | 1995-06-07 | 1999-07-06 | トリメリス,インコーポレーテッド | Treatment of HIV and other viral infections using combination therapy |
| US5618964A (en) * | 1995-06-07 | 1997-04-08 | Bristol-Myers Squibb Company | Prodrug esters of phosphonosulfonate squalene synthetase inhibitors and method |
| CA2261619C (en) | 1996-07-26 | 2006-05-23 | Gilead Sciences, Inc. | Nucleotide analogs |
| US5922695A (en) * | 1996-07-26 | 1999-07-13 | Gilead Sciences, Inc. | Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavarilability |
| US5733788A (en) | 1996-07-26 | 1998-03-31 | Gilead Sciences, Inc. | PMPA preparation |
| US6121315A (en) * | 1996-09-20 | 2000-09-19 | Warner-Lambert Company | Oral compositions containing a zinc compound |
| GB9622681D0 (en) | 1996-10-31 | 1997-01-08 | Glaxo Group Ltd | Pharmaceutical compositions |
| US6113920A (en) * | 1996-10-31 | 2000-09-05 | Glaxo Wellcome Inc. | Pharmaceutical compositions |
| US5965729A (en) | 1997-02-05 | 1999-10-12 | Merck & Co., Inc. | Process for the crystallization of a reverse transcriptase inhibitor using an anti-solvent |
| US6143877A (en) | 1997-04-30 | 2000-11-07 | Epoch Pharmaceuticals, Inc. | Oligonucleotides including pyrazolo[3,4-D]pyrimidine bases, bound in double stranded nucleic acids |
| ZA986614B (en) | 1997-07-25 | 1999-01-27 | Gilead Sciences | Nucleotide analog composition |
| DK1256584T3 (en) * | 1997-07-25 | 2004-12-06 | Gilead Sciences Inc | Process for preparing adefovir dipivoxil |
| US5935946A (en) | 1997-07-25 | 1999-08-10 | Gilead Sciences, Inc. | Nucleotide analog composition and synthesis method |
| US6270957B1 (en) * | 1997-08-26 | 2001-08-07 | Wisconsin Alumni Research Foundation | Non-Imuunosuppressive cyclosporins and their use in the prevention and treatment of HIV infection |
| CO4970782A1 (en) | 1997-11-13 | 2000-11-07 | Merck & Co Inc | COMBINED THERAPY FOR THE TREATMENT OF AIDS |
| US6087383A (en) * | 1998-01-20 | 2000-07-11 | Bristol-Myers Squibb Company | Bisulfate salt of HIV protease inhibitor |
| US6312662B1 (en) | 1998-03-06 | 2001-11-06 | Metabasis Therapeutics, Inc. | Prodrugs phosphorus-containing compounds |
| US6127121A (en) | 1998-04-03 | 2000-10-03 | Epoch Pharmaceuticals, Inc. | Oligonucleotides containing pyrazolo[3,4-D]pyrimidines for hybridization and mismatch discrimination |
| UA72207C2 (en) * | 1998-04-07 | 2005-02-15 | Брістол- Майєрс Сквібб Фарма Компані | Pharmaceutical formulations of efavirenz and disintegrants providing for increasing dissolution rate and process of manufacturing such tablets or capsules |
| US20020072493A1 (en) * | 1998-05-19 | 2002-06-13 | Yeda Research And Development Co. Ltd. | Activated T cells, nervous system-specific antigens and their uses |
| EP1332757B1 (en) | 1998-05-27 | 2012-06-13 | Merck Sharp & Dohme Corp. | Efavirenz compressed tablet formulation |
| US20010014352A1 (en) * | 1998-05-27 | 2001-08-16 | Udit Batra | Compressed tablet formulation |
| CO5070643A1 (en) | 1998-05-27 | 2001-08-28 | Merck & Co Inc | FORMULATION IN COMPRESSED TABLETS |
| AU4716299A (en) | 1998-06-24 | 2000-01-10 | Emory University | Use of 3'-azido-2',3'-dideoxyuridine in combination with further anti-hiv drugs for the manufacture of a medicament for the treatment of hiv |
| EP1535921A1 (en) | 1998-08-12 | 2005-06-01 | Gilead Sciences, Inc. | Method of manufacture of 1,3-oxathiolane nucleosides |
| GB9820420D0 (en) | 1998-09-18 | 1998-11-11 | Glaxo Group Ltd | Antiviral combinations |
| DK1380303T3 (en) | 1998-11-02 | 2008-12-01 | Gilead Sciences Inc | Combination therapy to treat hepatitis B virus |
| GB9909154D0 (en) | 1999-04-22 | 1999-06-16 | Nippon Glaxo Limited | Pharmaceutical formulation |
| US6660845B1 (en) | 1999-11-23 | 2003-12-09 | Epoch Biosciences, Inc. | Non-aggregating, non-quenching oligomers comprising nucleotide analogues; methods of synthesis and use thereof |
| DE60114772T2 (en) | 2000-02-02 | 2006-07-20 | Dorian Bevec | AROMATIC GUANYLHYDRAZONE FOR THE TREATMENT OF RESISTANT VIRAL INFECTIONS |
| RS51561B (en) * | 2000-02-29 | 2011-08-31 | Bristol-Myers Squibb Co. | Low dose entecavir formulation and use |
| CN1291994C (en) * | 2000-07-21 | 2006-12-27 | 吉里德科学公司 | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| JP2005500252A (en) | 2000-12-15 | 2005-01-06 | トライアングル・フアーマシユーテイカルズ・インコーポレイテツド | Combination therapy with IMDPH inhibitors such as ribavirin or mycophenolic acid and DAPD |
| US6900315B2 (en) | 2001-02-06 | 2005-05-31 | Yale University | 2-amino-9H-purin-9-yl compounds and methods for inhibiting/treating HIV infections and AIDS related symptoms |
| DK1389207T3 (en) | 2001-03-01 | 2006-04-18 | Gilead Sciences Inc | Polymorphic and other crystalline forms of cis-FTC |
| MY169670A (en) | 2003-09-03 | 2019-05-08 | Tibotec Pharm Ltd | Combinations of a pyrimidine containing nnrti with rt inhibitors |
| US20030124186A1 (en) | 2001-11-27 | 2003-07-03 | Hussain Munir A. | Efavirenz tablet formulation having unique biopharmaceutical characteristics |
| PT2260833E (en) | 2002-01-16 | 2012-12-26 | Boehringer Ingelheim Pharma | Bilayer pharmaceutical tablet comprising telmisartan and a diuretic |
| CA2474056A1 (en) * | 2002-01-24 | 2003-07-31 | Sangstat Medical Corporation | Combination therapy for treatment of hiv infection |
| CN1656109A (en) * | 2002-04-26 | 2005-08-17 | 吉里德科学公司 | Non nucleoside reverse transcriptase inhibitors |
| CA2502625A1 (en) | 2002-12-09 | 2004-06-24 | The University Of Georgia Research Foundation, Inc. | Dioxolane thymine and combinations for use against resistant strains of hiv |
| MXPA05006954A (en) * | 2002-12-26 | 2005-09-22 | Pozen Inc | Multilayer Dosage Forms Containing NSAIDs and Triptans. |
| KR20090053867A (en) | 2003-01-14 | 2009-05-27 | 길리애드 사이언시즈, 인코포레이티드 | Compositions and methods for combination antiviral therapy |
| JP2006527184A (en) * | 2003-06-06 | 2006-11-30 | エティファーム | Orally dispersible multilayer tablets |
| CA2598607A1 (en) | 2005-01-18 | 2006-07-27 | Adra N. Chaker | Methods and compositions for cell-cycle regulation |
| TWI471145B (en) | 2005-06-13 | 2015-02-01 | Bristol Myers Squibb & Gilead Sciences Llc | Unitary pharmaceutical dosage form |
| TWI375560B (en) | 2005-06-13 | 2012-11-01 | Gilead Sciences Inc | Composition comprising dry granulated emtricitabine and tenofovir df and method for making the same |
| CA2612788A1 (en) | 2005-06-24 | 2006-12-28 | Steven Cesar Alfons De Jonghe | Pyrido(3,2-d)pyrimidines and pharmaceutical compositions useful for treating hepatitis c |
| US20070026441A1 (en) * | 2005-07-22 | 2007-02-01 | Olson William C | Methods for reducing viral load in HIV-1-infected patients |
| US7448494B2 (en) * | 2005-08-10 | 2008-11-11 | Certain Teed Corporation | Loose fill insulation packaged with additive |
| AP2008004533A0 (en) | 2005-12-14 | 2008-08-31 | Cipla Ltd | Pharmaceutical combination |
| GB0525898D0 (en) | 2005-12-20 | 2006-02-01 | Pharmo Bioscience As | Screening compounds for activity in modulating chloride ion transport |
| AU2008206695A1 (en) | 2007-01-16 | 2008-07-24 | Proteologics Ltd | Methods for enhancing the therapeutic efficacy of topoisomerase inhibitors |
| AU2009279604A1 (en) | 2008-08-06 | 2010-02-11 | Bionevia Pharmaceuticals, Inc. | Flupirtine hydrochloride maleic acid cocrystal |
| WO2012003413A1 (en) | 2010-06-30 | 2012-01-05 | The Broad Institute, Inc. | Novel solid forms of tacedinaline |
| CN106511357A (en) | 2010-11-19 | 2017-03-22 | 吉利德科学公司 | Therapeutic compositions comprising rilpivirine HCL and tenofovir disoproxil fumarate |
-
2004
- 2004-01-13 KR KR1020097009376A patent/KR20090053867A/en not_active Application Discontinuation
- 2004-01-13 US US10/540,782 patent/US20060234982A1/en not_active Abandoned
- 2004-01-13 DK DK04701819T patent/DK1583542T3/en active
- 2004-01-13 CN CN201210094391.9A patent/CN102670629B/en not_active Expired - Lifetime
- 2004-01-13 PT PT04701819T patent/PT1583542E/en unknown
- 2004-01-13 SI SI200430842T patent/SI1583542T1/en unknown
- 2004-01-13 EP EP20080152527 patent/EP1923063A3/en not_active Ceased
- 2004-01-13 EA EA200501134A patent/EA015145B1/en not_active IP Right Cessation
- 2004-01-13 AT AT04701819T patent/ATE398455T1/en active
- 2004-01-13 EA EA201100293A patent/EA201100293A1/en unknown
- 2004-01-13 KR KR20057013069A patent/KR100860136B1/en active Protection Beyond IP Right Term
- 2004-01-13 NZ NZ540728A patent/NZ540728A/en not_active IP Right Cessation
- 2004-01-13 AP AP2005003348A patent/AP2089A/en active
- 2004-01-13 WO PCT/US2004/000868 patent/WO2004064846A1/en active Application Filing
- 2004-01-13 PL PL37836804A patent/PL378368A1/en unknown
- 2004-01-13 US US10/757,122 patent/US20040224916A1/en not_active Abandoned
- 2004-01-13 MX MXPA05007016A patent/MXPA05007016A/en active IP Right Grant
- 2004-01-13 WO PCT/US2004/000832 patent/WO2004064845A1/en active Application Filing
- 2004-01-13 JP JP2006500943A patent/JP2006515624A/en active Pending
- 2004-01-13 US US10/757,141 patent/US20040224917A1/en not_active Abandoned
- 2004-01-13 DE DE200460014470 patent/DE602004014470D1/en not_active Expired - Lifetime
- 2004-01-13 CA CA 2512475 patent/CA2512475C/en not_active Expired - Lifetime
- 2004-01-13 EP EP15190214.5A patent/EP3025718A1/en not_active Withdrawn
- 2004-01-13 CN CN201510697340.9A patent/CN105596356A/en active Pending
- 2004-01-13 CA CA 2512319 patent/CA2512319A1/en not_active Abandoned
- 2004-01-13 JP JP2006500939A patent/JP4996241B2/en not_active Expired - Lifetime
- 2004-01-13 PL PL40825404A patent/PL408254A1/en unknown
- 2004-01-13 BR BRPI0406760 patent/BRPI0406760A/en not_active Application Discontinuation
- 2004-01-13 EP EP20040701819 patent/EP1583542B9/en not_active Revoked
- 2004-01-13 KR KR1020087007002A patent/KR20080032014A/en not_active Application Discontinuation
- 2004-01-13 EP EP20040701840 patent/EP1585527A1/en not_active Ceased
- 2004-01-13 AU AU2004206827A patent/AU2004206827A1/en not_active Abandoned
- 2004-01-13 AU AU2004206821A patent/AU2004206821C1/en not_active Expired
- 2004-01-13 ES ES04701819T patent/ES2308136T3/en not_active Expired - Lifetime
- 2004-01-13 US US10/540,794 patent/US20060246130A1/en not_active Abandoned
-
2005
- 2005-06-16 IL IL169243A patent/IL169243A/en active IP Right Grant
- 2005-07-06 HR HR20050619A patent/HRP20050619A2/en not_active Application Discontinuation
- 2005-08-12 NO NO20053817A patent/NO337917B1/en not_active IP Right Cessation
- 2005-08-12 IS IS7977A patent/IS7977A/en unknown
-
2006
- 2006-02-10 HK HK06101818A patent/HK1079122A1/en not_active IP Right Cessation
-
2008
- 2008-08-20 US US12/195,161 patent/US8592397B2/en not_active Expired - Lifetime
- 2008-09-04 US US12/204,174 patent/US8716264B2/en not_active Expired - Lifetime
- 2008-09-17 CY CY081101015T patent/CY1108355T1/en unknown
-
2009
- 2009-02-04 AU AU2009200414A patent/AU2009200414B2/en not_active Expired
-
2010
- 2010-02-15 JP JP2010030680A patent/JP2010120957A/en not_active Withdrawn
-
2013
- 2013-10-25 JP JP2013221825A patent/JP2014037430A/en not_active Withdrawn
-
2014
- 2014-03-27 US US14/227,653 patent/US20140213556A1/en not_active Abandoned
- 2014-04-25 HR HRP20140379AA patent/HRP20140379A2/en not_active Application Discontinuation
- 2014-10-24 US US14/523,758 patent/US9457036B2/en not_active Expired - Lifetime
- 2014-10-24 US US14/523,783 patent/US9744181B2/en not_active Expired - Lifetime
-
2015
- 2015-02-25 JP JP2015034888A patent/JP2015098488A/en not_active Withdrawn
- 2015-05-22 NO NO20150656A patent/NO340951B1/en not_active IP Right Cessation
-
2017
- 2017-01-06 JP JP2017000869A patent/JP2017057232A/en not_active Withdrawn
- 2017-05-04 US US15/586,500 patent/US20170232019A1/en not_active Abandoned
- 2017-06-14 US US15/622,484 patent/US20170273994A1/en not_active Abandoned
- 2017-07-17 NO NO20171193A patent/NO20171193A1/en not_active Application Discontinuation
-
2018
- 2018-12-03 JP JP2018226434A patent/JP2019052174A/en not_active Withdrawn
-
2020
- 2020-10-16 JP JP2020174621A patent/JP2021004264A/en active Pending
Cited By (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8716264B2 (en) | 2003-01-14 | 2014-05-06 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| US8592397B2 (en) | 2003-01-14 | 2013-11-26 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| US9457036B2 (en) | 2003-01-14 | 2016-10-04 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| US20090036408A1 (en) * | 2003-01-14 | 2009-02-05 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| US9744181B2 (en) | 2003-01-14 | 2017-08-29 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| US20070003608A1 (en) * | 2005-04-08 | 2007-01-04 | Almond Merrick R | Compounds, compositions and methods for the treatment of viral infections and other medical disorders |
| US8642577B2 (en) | 2005-04-08 | 2014-02-04 | Chimerix, Inc. | Compounds, compositions and methods for the treatment of poxvirus infections |
| US20070003516A1 (en) * | 2005-04-08 | 2007-01-04 | Almond Merrick R | Compounds, compositions and methods for the treatment of poxvirus infections |
| US8598185B2 (en) | 2005-06-13 | 2013-12-03 | Bristol-Myers Squibb & Gilead Sciences, Inc. | Unitary pharmaceutical dosage form |
| US8871271B2 (en) | 2005-06-13 | 2014-10-28 | Gilead Sciences, Inc. | Method and composition for pharmaceutical product |
| US9545414B2 (en) | 2005-06-13 | 2017-01-17 | Bristol-Myers Squibb & Gilead Sciences, Llc | Unitary pharmaceutical dosage form |
| US20070077295A1 (en) * | 2005-06-13 | 2007-04-05 | Gilead Sciences, Inc. | Method and composition for pharmaceutical product |
| US9018192B2 (en) | 2005-06-13 | 2015-04-28 | Bristol-Myers Squibb & Gilead Sciences, Inc. | Unitary pharmaceutical dosage form |
| US20080317852A1 (en) * | 2005-12-14 | 2008-12-25 | Amar Lulla | Pharmaceutical Combination |
| US10335423B2 (en) | 2006-02-03 | 2019-07-02 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Inhibition of HIV infection through chemoprophylaxis |
| US9937191B2 (en) | 2006-02-03 | 2018-04-10 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Inhibition of HIV infection through chemoprophylaxis |
| US9579333B2 (en) * | 2006-02-03 | 2017-02-28 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Inhibition of HIV infection through chemoprophyalxis |
| US20150272972A1 (en) * | 2006-02-03 | 2015-10-01 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Serv | Inhibition of hiv infection through chemoprophyalxis |
| US20100065444A1 (en) * | 2007-01-20 | 2010-03-18 | Stefan Henke | Pack containing soft capsules |
| WO2008100848A3 (en) * | 2007-02-12 | 2010-03-25 | Board Of Regents, The University Of Texas System | Novel agent for in vivo pet imaging of tumor proliferation |
| WO2008100848A2 (en) * | 2007-02-12 | 2008-08-21 | Board Of Regents, The University Of Texas System | Novel agent for in vivo pet imaging of tumor proliferation |
| US8993542B2 (en) | 2008-01-25 | 2015-03-31 | Chimerix Inc. | Methods of treating viral infections |
| US20090247749A1 (en) * | 2008-03-31 | 2009-10-01 | Apotex Pharmachem Inc. | Salt form and cocrystals of adefovir dipivoxil and processes for preparation thereof |
| US7935817B2 (en) * | 2008-03-31 | 2011-05-03 | Apotex Pharmachem Inc. | Salt form and cocrystals of adefovir dipivoxil and processes for preparation thereof |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| US8759510B2 (en) | 2008-06-11 | 2014-06-24 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| EP2671888A1 (en) | 2008-12-23 | 2013-12-11 | Gilead Pharmasset LLC | 3',5'-cyclic nucleoside phosphate analogues |
| US9045520B2 (en) | 2008-12-23 | 2015-06-02 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| EP3222628A1 (en) | 2008-12-23 | 2017-09-27 | Gilead Pharmasset LLC | Nucleoside phosphoramidates |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8716263B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| US8957045B2 (en) | 2008-12-23 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP2913337A1 (en) | 2009-05-20 | 2015-09-02 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2'-fluoro-2'-methyl-p-phenyl-5'-uridylyl]-l-alanine 1-methylethyl ester and process for its production |
| EP2610264A2 (en) | 2009-05-20 | 2013-07-03 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2'-fluoro-2'-methyl-p-phenyl-5'-uridylyl]-l-alanine 1-methylethyl ester and process for its production |
| WO2010135569A1 (en) | 2009-05-20 | 2010-11-25 | Pharmasset, Inc. | N- [ (2 ' r) -2 ' -deoxy-2 ' -fluoro-2 ' -methyl-p-phenyl-5 ' -uridylyl] -l-alanine 1-methylethyl ester and process for its production |
| EP3321275A1 (en) | 2009-05-20 | 2018-05-16 | Gilead Pharmasset LLC | Crystalline form of sofosbuvir |
| EP2910562A1 (en) | 2009-05-20 | 2015-08-26 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2 '-fluoro-2'-methyl-p-phenyl-5 '-uridylyl]-l-alanine 1-methylethyl ester in crystalline form |
| US8614200B2 (en) | 2009-07-21 | 2013-12-24 | Chimerix, Inc. | Compounds, compositions and methods for treating ocular conditions |
| US9006218B2 (en) | 2010-02-12 | 2015-04-14 | Chimerix Inc. | Nucleoside phosphonate salts |
| US9765100B2 (en) | 2010-02-12 | 2017-09-19 | Chimerix, Inc. | Nucleoside phosphonate salts |
| WO2011123668A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Stereoselective synthesis of phosphorus containing actives |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| EP2752422A1 (en) | 2010-03-31 | 2014-07-09 | Gilead Pharmasset LLC | Stereoselective synthesis of phosphorus containing actives |
| EP2609923A2 (en) | 2010-03-31 | 2013-07-03 | Gilead Pharmasset LLC | Nucleoside Phosphoramidates |
| EP3290428A1 (en) | 2010-03-31 | 2018-03-07 | Gilead Pharmasset LLC | Tablet comprising crystalline (s)-isopropyl 2-(((s)-(((2r,3r,4r,5r)-5-(2,4-dioxo-3,4-dihydropyrimidin-1 (2h)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| WO2011123645A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| US9694024B2 (en) | 2010-04-26 | 2017-07-04 | Chimerix, Inc. | Methods of treating retroviral infections and related dosage regimes |
| US9956239B2 (en) | 2010-04-26 | 2018-05-01 | Chimerix, Inc. | Methods of treating retroviral infections and related dosage regimes |
| US9278135B2 (en) | 2010-04-26 | 2016-03-08 | Chimerix Inc. | Methods of treating retroviral infections and related dosage regimes |
| WO2011156594A2 (en) | 2010-06-09 | 2011-12-15 | Vaccine Technologies, Incorporated | Therapeutic immunization in hiv infected subjects receiving stable antiretroviral treatment |
| WO2012075140A1 (en) | 2010-11-30 | 2012-06-07 | Pharmasset, Inc. | Compounds |
| US9394331B2 (en) | 2010-11-30 | 2016-07-19 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| EP3042910A2 (en) | 2010-11-30 | 2016-07-13 | Gilead Pharmasset LLC | 2'-spiro-nucleosides for use in the therapy of hepatitis c |
| US8841275B2 (en) | 2010-11-30 | 2014-09-23 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| WO2013082476A1 (en) | 2011-11-30 | 2013-06-06 | Emory University | Antiviral jak inhibitors useful in treating or preventing retroviral and other viral infections |
| EP3750544A2 (en) | 2011-11-30 | 2020-12-16 | Emory University | Jak inhibitors for use in the prevention or treatment of viral infection |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9744181B2 (en) | Compositions and methods for combination antiviral therapy | |
| AU2011253996B2 (en) | Compositions and methods for combination antiviral therapy | |
| DAHL et al. | Patent 2512475 Summary | |
| AU2014216017A1 (en) | Compositions and methods for combination antiviral therapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: GILEAD SCIENCES, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:DAHL, TERRENCE C.;MENNING, MARK M.;OLIYAI, REZA;REEL/FRAME:014636/0215;SIGNING DATES FROM 20040426 TO 20040429 |
|
| STCB | Information on status: application discontinuation |
Free format text: EXPRESSLY ABANDONED -- DURING EXAMINATION |