RU2670767C9 - Method for producing low molecular weight heparin - Google Patents
Method for producing low molecular weight heparin Download PDFInfo
- Publication number
- RU2670767C9 RU2670767C9 RU2017146005A RU2017146005A RU2670767C9 RU 2670767 C9 RU2670767 C9 RU 2670767C9 RU 2017146005 A RU2017146005 A RU 2017146005A RU 2017146005 A RU2017146005 A RU 2017146005A RU 2670767 C9 RU2670767 C9 RU 2670767C9
- Authority
- RU
- Russia
- Prior art keywords
- heparin
- benzethonium
- solution
- benzyl ester
- sodium acetate
- Prior art date
Links
- 239000003055 low molecular weight heparin Substances 0.000 title claims abstract description 7
- 229940127215 low-molecular weight heparin Drugs 0.000 title claims abstract description 7
- 238000004519 manufacturing process Methods 0.000 title abstract description 8
- 229920000669 heparin Polymers 0.000 claims abstract description 67
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 claims abstract description 57
- 229960002897 heparin Drugs 0.000 claims abstract description 49
- 239000000243 solution Substances 0.000 claims abstract description 35
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims abstract description 34
- 238000000034 method Methods 0.000 claims abstract description 31
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 31
- 229960003872 benzethonium Drugs 0.000 claims abstract description 30
- -1 heparin benzyl ester Chemical class 0.000 claims abstract description 29
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 23
- SIYLLGKDQZGJHK-UHFFFAOYSA-N dimethyl-(phenylmethyl)-[2-[2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethoxy]ethyl]ammonium Chemical compound C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 SIYLLGKDQZGJHK-UHFFFAOYSA-N 0.000 claims abstract description 22
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 claims abstract description 21
- 238000005406 washing Methods 0.000 claims abstract description 21
- 239000001632 sodium acetate Substances 0.000 claims abstract description 16
- 235000017281 sodium acetate Nutrition 0.000 claims abstract description 16
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 claims abstract description 16
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 claims abstract description 13
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 13
- 239000008213 purified water Substances 0.000 claims abstract description 13
- 238000002604 ultrasonography Methods 0.000 claims abstract description 11
- 238000001556 precipitation Methods 0.000 claims abstract description 10
- 238000005574 benzylation reaction Methods 0.000 claims abstract description 9
- 238000002955 isolation Methods 0.000 claims abstract description 8
- 239000000010 aprotic solvent Substances 0.000 claims abstract description 6
- 230000005855 radiation Effects 0.000 claims abstract description 6
- 230000003993 interaction Effects 0.000 claims abstract description 5
- 238000007068 beta-elimination reaction Methods 0.000 claims abstract description 4
- 150000003839 salts Chemical class 0.000 claims abstract description 4
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 3
- 239000012047 saturated solution Substances 0.000 claims abstract description 3
- 238000000926 separation method Methods 0.000 claims abstract description 3
- 238000003776 cleavage reaction Methods 0.000 claims abstract 2
- 230000007017 scission Effects 0.000 claims abstract 2
- 238000003756 stirring Methods 0.000 claims description 12
- 239000002244 precipitate Substances 0.000 claims description 11
- 229940040526 anhydrous sodium acetate Drugs 0.000 claims description 5
- 229920002521 macromolecule Polymers 0.000 claims description 4
- 229920006395 saturated elastomer Polymers 0.000 claims description 3
- 239000000126 substance Substances 0.000 abstract description 9
- 229960001950 benzethonium chloride Drugs 0.000 abstract description 4
- 230000032050 esterification Effects 0.000 abstract description 4
- 238000005886 esterification reaction Methods 0.000 abstract description 4
- 230000000694 effects Effects 0.000 abstract description 2
- 238000007493 shaping process Methods 0.000 abstract 1
- 239000002699 waste material Substances 0.000 abstract 1
- 239000000047 product Substances 0.000 description 16
- 238000006243 chemical reaction Methods 0.000 description 15
- 230000008569 process Effects 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 235000019441 ethanol Nutrition 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 238000010511 deprotection reaction Methods 0.000 description 6
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 5
- 229940073608 benzyl chloride Drugs 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 230000002255 enzymatic effect Effects 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 108010022901 Heparin Lyase Proteins 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 238000005904 alkaline hydrolysis reaction Methods 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 230000001590 oxidative effect Effects 0.000 description 3
- BQSMUQUKNCGJCT-SLPGGIOYSA-N 2-N,6-O-disulfo-D-glucosamine Chemical group OS(=O)(=O)OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](NS(O)(=O)=O)C=O BQSMUQUKNCGJCT-SLPGGIOYSA-N 0.000 description 2
- 0 CC*(CC)*C(CC12NC)[C@]1OC(CO*)CC2(CO[C@@](C(C1O)O*)OC[C@@]1C(CC)(CC)O[C@@](C(C1O)NC)OC(C*)[C@]1OCC(*)(COC(*)=C1)C1O)O* Chemical compound CC*(CC)*C(CC12NC)[C@]1OC(CO*)CC2(CO[C@@](C(C1O)O*)OC[C@@]1C(CC)(CC)O[C@@](C(C1O)NC)OC(C*)[C@]1OCC(*)(COC(*)=C1)C1O)O* 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229960004969 dalteparin Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 229960000610 enoxaparin Drugs 0.000 description 2
- ZFGMDIBRIDKWMY-PASTXAENSA-N heparin Chemical compound CC(O)=N[C@@H]1[C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O[C@@H]1O[C@@H]1[C@@H](C(O)=O)O[C@@H](O[C@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@@H](O[C@@H]3[C@@H](OC(O)[C@H](OS(O)(=O)=O)[C@H]3O)C(O)=O)O[C@@H]2O)CS(O)(=O)=O)[C@H](O)[C@H]1O ZFGMDIBRIDKWMY-PASTXAENSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000003301 hydrolyzing effect Effects 0.000 description 2
- 210000004379 membrane Anatomy 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 239000010451 perlite Substances 0.000 description 2
- 235000019362 perlite Nutrition 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- PURMPUDWXOWORS-LECHCGJUSA-N (2s,3r,4r,5s)-2,3,4-trihydroxy-6-oxo-5-sulfooxyhexanoic acid Chemical compound OC(=O)[C@@H](O)[C@H](O)[C@@H](O)[C@H](OS(O)(=O)=O)C=O PURMPUDWXOWORS-LECHCGJUSA-N 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 206010000891 acute myocardial infarction Diseases 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229960003616 bemiparin Drugs 0.000 description 1
- MTDHILKWIRSIHB-QZABAPFNSA-N beta-D-glucosamine 6-sulfate Chemical class N[C@H]1[C@H](O)O[C@H](COS(O)(=O)=O)[C@@H](O)[C@@H]1O MTDHILKWIRSIHB-QZABAPFNSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 208000034158 bleeding Diseases 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000002612 cardiopulmonary effect Effects 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 238000001631 haemodialysis Methods 0.000 description 1
- 230000000322 hemodialysis Effects 0.000 description 1
- 229960001008 heparin sodium Drugs 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 150000002772 monosaccharides Chemical group 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- GKTNLYAAZKKMTQ-UHFFFAOYSA-N n-[bis(dimethylamino)phosphinimyl]-n-methylmethanamine Chemical compound CN(C)P(=N)(N(C)C)N(C)C GKTNLYAAZKKMTQ-UHFFFAOYSA-N 0.000 description 1
- 230000014508 negative regulation of coagulation Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 235000011837 pasties Nutrition 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 239000012716 precipitator Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229910052594 sapphire Inorganic materials 0.000 description 1
- 239000010980 sapphire Substances 0.000 description 1
- 239000010802 sludge Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 238000009210 therapy by ultrasound Methods 0.000 description 1
- 229960005062 tinzaparin Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 239000012224 working solution Substances 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0075—Heparin; Heparan sulfate; Derivatives thereof, e.g. heparosan; Purification or extraction methods thereof
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
Область техники, к которой относится изобретениеFIELD OF THE INVENTION
Изобретение относится к технологии получения фармацевтической субстанции низкомолекулярного гепарина (НМГ) щелочной деполимеризацией бензилового эфира коммерческого высокомолекулярного гепарина натрия.The invention relates to a technology for the production of pharmaceutical substance of low molecular weight heparin (NMH) by alkaline depolymerization of benzyl ether of commercial high molecular weight sodium heparin.
Краткое описание чертежейBrief Description of the Drawings
На Фиг. 1 представлена структура фрагмента молекулы высокомолекулярного гепарина.In FIG. 1 shows the structure of a fragment of a high molecular weight heparin molecule.
На Фиг. 2 изображена схема получения НМГ окислительной деполимеризацией.In FIG. 2 shows a scheme for the production of NMH by oxidative depolymerization.
На Фиг. 3 показана структура дальтепарина.In FIG. 3 shows the structure of dalteparin.
На Фиг. 4 приведена схема ферментативной деполимеризации в синтезе тинза-парина.In FIG. 4 shows the scheme of enzymatic depolymerization in the synthesis of tinza-parine.
На Фиг. 5 показана структура фрагмента макромолекулы НМГ (эноксапарин), полученного щелочным гидролизом.In FIG. 5 shows the structure of a fragment of the NMG macromolecule (enoxaparin) obtained by alkaline hydrolysis.
На Фиг. 6 изображена схема реакций, протекающих при получении гепарината бензетония.In FIG. 6 shows a diagram of the reactions occurring upon receipt of benzethonium heparin.
На Фиг. 7 представлена схема реакций бензилирования гепарината бензетония (ГБ) и удаления защиты сульфогрупп.In FIG. 7 is a diagram of the reactions of benzylation of benzetonium heparin benzylation (GB) and deprotection of sulfo groups.
На Фиг. 8 дана схема реакций, протекающих при эпимеризации и циклизации.In FIG. 8 is a diagram of the reactions occurring during epimerization and cyclization.
Уровень техникиState of the art
Гепарин - органопрепарат, получаемый из легких или мукозы - слизистой оболочки крупного рогатого скота или свиней. Он является прямым антикоагулянтом и используется для приготовления лекарственных форм, применяемых для профилактики и терапии тромбоэмболических заболеваний, тромбообразования при операциях на сердце и кровеносных сосудах, при остром инфаркте миокарда, а также для поддержания жидкого состояния крови в аппаратах искусственного кровообращения и гемодиализа.Heparin is an organ preparation obtained from the lungs or mucose, the mucous membrane of cattle or pigs. It is a direct anticoagulant and is used for the preparation of dosage forms used for the prevention and treatment of thromboembolic diseases, thrombosis during operations on the heart and blood vessels, in acute myocardial infarction, as well as for maintaining the liquid state of the blood in cardiopulmonary bypass and hemodialysis machines.
Коммерческий нефракционированный гепарин (НФГ) представляет собой смесь сульфатированных полисахаридов различной структуры (Фиг. 1) с молекулярной массой от 3000 до 30000 Да. От длины молекулы гепарина и величины ее заряда зависит фармакокинетика и фармакодинамика лекарственного препарата, а также способность гепарина взаимодействовать с белками крови и клетками организма [1]. Наряду с несомненными преимуществами препараты НФГ обладают серьезными недостатками, такими как: возможность возникновения неконтролируемых кровотечений, аллергические реакции, остеопороз, необходимость повторных инъекций и др. [2].Commercial unfractionated heparin (UFH) is a mixture of sulfated polysaccharides of various structures (Fig. 1) with a molecular weight of 3,000 to 30,000 Da. The pharmacokinetics and pharmacodynamics of the drug, as well as the ability of heparin to interact with blood proteins and body cells, depend on the length of the heparin molecule and the magnitude of its charge [1]. Along with the undoubted advantages, UFH preparations have serious disadvantages, such as the possibility of uncontrolled bleeding, allergic reactions, osteoporosis, the need for repeated injections, etc. [2].
Фракционирование природного гепарина известными методами (гельфильтрация, диализ и др.) дает низкий выход целевой фракции с низкой молекулярной массой и высокой антикоагулянтной активностью. Более эффективна в этом отношении так называемая контролируемая деполимеризация гепарина [3] под действием неорганических или органических веществ, ферментов или радиационного излучения. Реализуемые в настоящее время способы контролируемой деполимеризации можно свести к процессам гидролиза, окислительной, радикальной или ферментативной деструкции.Fractionation of natural heparin by known methods (gel filtration, dialysis, etc.) gives a low yield of the target fraction with a low molecular weight and high anticoagulant activity. In this respect, the so-called controlled depolymerization of heparin [3] is more effective under the action of inorganic or organic substances, enzymes, or radiation. Currently implemented methods of controlled depolymerization can be reduced to processes of hydrolysis, oxidative, radical or enzymatic degradation.
Окислительная деполимеризация НФГ заключается в обработке его водных растворов различными окислителями, из которых чаще всего используют нитрит натрия [4]. В результате действия выделяющейся азотистой кислоты на НФГ происходит разрыв гликозидных связей основной цепи [5] с образованием гидроксильных и альдегидных групп которые восстанавливают, например, боргидридом натрия [6] как схематично показано на Фиг. 2. Таким образом получают, например, дальтепарин, который имеет среднюю молекулярную массу около 6,0 кДа и ангидроманнозные концевые фрагменты (Фиг. 3).Oxidative depolymerization of UFH consists in the treatment of its aqueous solutions with various oxidizing agents, of which sodium nitrite is most often used [4]. As a result of the action of nitrous acid released on UFH, the glycosidic bonds of the main chain break [5] to form hydroxyl and aldehyde groups which are reduced, for example, with sodium borohydride [6] as shown schematically in FIG. 2. Thus, for example, dalteparin is obtained, which has an average molecular weight of about 6.0 kDa and anhydromannose terminal fragments (Fig. 3).
Ферментативную деполимеризацию осуществляют при воздействии, например, микробных гепариназ (гепарин-лиазы), специфически разрушающих α-гликозидные связи между N-сульфатированным D-глюкозамин-6-сульфатом и 2-О-сульфатом идуроновой кислоты. Таким образом получают НМГ тинзапарин со средней молекулярной массой около 4,5 кДа и концевыми 2-N,6-O-дисульфо-D-глюкозаминовым и 4,5-ненасыщенным уроновокислым фрагментами [7] (Фиг. 4) В настоящее время известны способы ферментативной деполимеризации НФГ с помощью и других гепариназ [8]. К достоинствам ферментативной деполимеризации относятся мягкие условия синтеза и высокая селективность процесса.Enzymatic depolymerization is carried out under the influence, for example, of microbial heparinases (heparin lyases), which specifically destroy α-glycosidic bonds between N-sulfated D-glucosamine-6-sulfate and iduronic acid 2-O-sulfate. Thus, NMH tinzaparin with an average molecular weight of about 4.5 kDa and terminal 2-N, 6-O-disulfo-D-glucosamine and 4,5-unsaturated uronic acid fragments is obtained [7] (Fig. 4). Currently, methods are known. enzymatic depolymerization of UFH using other heparinases [8]. The advantages of enzymatic depolymerization include mild synthesis conditions and high selectivity of the process.
Гидролитическая деполимеризация представляет собой специфический метод направленной деструкции НФГ, который подразумевает получение производных НФГ с их последующим щелочным гидролизом. Обычно процесс такого рода включает в себя предварительную защиту сульфогрупп НФГ реакцией с хлоридом бензетония и последующее преобразование карбоксильных групп в сложноэфирные за счет этерификации полученного продукта бензилхлоридом. Получаемый в результате бензиловый эфир гепарината бензетония осаждают, например, этанолом [9] с последующим удалением защиты с групп SO3 -, и полученный неполный сложный эфир подвергают щелочному гидролизу [5]. При этом образуются молекулы НМГ с 2-O-сульфо-4-енопиранозуроновым и 2-N, 6-О-дисульфо-D-глюкозаминовым концевыми фрагментами (Фиг. 5). Таким образом получают ультранизкомолекулярный семулопарин, а также такие НМГ, как бемипарин и эноксапарин. Дополнительно деполимеризацию такого рода можно стимулировать микроволновым облучением.Hydrolytic depolymerization is a specific method of directed destruction of UFH, which involves the preparation of derivatives of UFH with their subsequent alkaline hydrolysis. Typically, this kind of process involves the preliminary protection of UFH sulfogroups by reaction with benzetonium chloride and the subsequent conversion of carboxyl groups to ester groups due to the esterification of the resulting product with benzyl chloride. The resulting benzethonium heparinate benzyl ester is precipitated, for example, with ethanol [9] followed by deprotection of the SO 3 - groups, and the resulting partial ester is subjected to alkaline hydrolysis [5]. In this case, NMH molecules are formed with 2-O-sulfo-4-enopyranosuronic and 2-N, 6-O-disulfo-D-glucosamine end fragments (Fig. 5). Thus, ultra-low molecular weight semuloparin is obtained, as well as LMWHs such as bemiparin and enoxaparin. Additionally, depolymerization of this kind can be stimulated by microwave irradiation.
Производное НФГ получают аналогичным способом - реакцией с хлоридом бензетония и последующей этерификации бензилхлоридом. Полученный продукт подвергают гидролитической деполимеризации в органических растворителях таких как: формамид, диметилформамид или метиленхлорид, а в качестве катализатора применяют сильные основания семейства фосфазенов. Считается, что процесс в неводной среде проходит в сравнительно мягких условиях, благодаря чему снижается доля побочных реакций и сохраняется биологическая активность гепарина.The derivative of UFH is obtained in a similar way - by reaction with benzetonium chloride and subsequent esterification with benzyl chloride. The resulting product is subjected to hydrolytic depolymerization in organic solvents such as formamide, dimethylformamide or methylene chloride, and strong bases of the phosphazene family are used as a catalyst. It is believed that the process in a non-aqueous medium takes place under relatively mild conditions, which reduces the proportion of adverse reactions and preserves the biological activity of heparin.
Наиболее близким по технической сущности к заявляемому изобретению является способ получения, раскрытый в описании и формуле изобретения к патенту [9], включающий стадии получения бензетониевой соли гепарина, бензилирования этой соли в неводном растворителе, спиртового осаждения неполного бензилового эфира бензетониевой соли гепарина и щелочной деполимеризации этого продукта, отличающийся тем, что бензетониевую соль нефракционированного гепарина получают в 0,05-0,5 М водном растворе натрия хлорида при температуре 50-60°С, рН в интервале 8,2-8,8 и массовом соотношении гепарин/бензетоний хлорид 1/(2,35-2,70), бензилирование бензетониевой соли гепарина проводят в течение 2-3 часов в среде биполярного апротонного растворителя бензилхлоридом в соотношении гепаринат/бензилхлорид 1/(0,2-1,0), который предварительно подвергают активированию в апротонном растворителе в течение 15-20 минут, осаждение бензилового эфира гепарина проводят методом Спиро этиловым спиртом, предварительно насыщенным безводным натрия ацетатом, с последующим удалением защиты с сульфогрупп, проведение β-элиминирования бензилового эфира со степенью этерификации гепарина 9-13% 1±0,5 N щелочью NaOH при температуре 55±5°С, длительности процесса 40-60 минут и массовым соотношением реагентов бензиловый эфир/щелочь 1/(0,5-2).Closest to the technical nature of the claimed invention is a production method disclosed in the description and claims of the patent [9], which includes the steps of producing the benzethonium salt of heparin, benzylation of this salt in a non-aqueous solvent, the alcohol deposition of a partial benzyl ester of the benzethonium salt of heparin and alkaline depolymerization of this product, characterized in that the benzethonium salt of unfractionated heparin is obtained in a 0.05-0.5 M aqueous solution of sodium chloride at a temperature of 50-60 ° C, pH in the range of 8.2 -8.8 and a mass ratio of heparin /
Стадию бензилирования проводят в смеси апротонных растворителей. Выделение бензилового эфира бензетониевой соли проводят осаждением этанолом в присутствии соосадителя ацетата натрия, удаление защиты сульфогрупп осуществляют действием насыщенного этанольного раствора того же реактива. Процесс отмывки бензетониевой соли от примесей проводят обработкой дистиллированной водой.The benzylation step is carried out in a mixture of aprotic solvents. The isolation of the benzyl ester of the benzethonium salt is carried out by ethanol precipitation in the presence of sodium acetate co-precipitator, the sulfo groups are deprotected by the action of a saturated ethanol solution of the same reagent. The process of washing benzethonium salt from impurities is carried out by treatment with distilled water.
Среди недостатков этого способа следует отметить:Among the disadvantages of this method should be noted:
- большой расход воды для отмывки гепарината бензетония от избытка бензетония хлорида, остаточное количество которого на последующих стадиях синтеза НМГ значительно осложняет технологический процесс и приводит к значительным потерям целевого продукта, поскольку продукты реакции представляют собой липкие тестообразные вещества;- a large flow of water for washing benzetonium heparin from an excess of benzetonium chloride, the residual amount of which in the subsequent stages of the synthesis of NMH significantly complicates the process and leads to significant losses of the target product, since the reaction products are sticky pasty substances;
- проведение процессов осаждения бензилового эфира бензетониевой соли и последующей процедуры удаления защиты сульфогрупп гепарина одним и тем же реагентом без выделения промежуточного продукта не приводит к получению структурированного продукта и увеличивает суммарную продолжительность процесса;- the process of precipitation of benzyl ester of benzethonium salt and the subsequent procedure for removing the protection of heparin sulfogroups by the same reagent without isolation of the intermediate product does not lead to a structured product and increases the total process time;
- выделение готового продукта 96%-м этанолом осложнено высокой гигроскопичностью порошка.- the selection of the finished product with 96% ethanol is complicated by the high hygroscopicity of the powder.
Целью настоящего изобретения является преодоление недостатков ближайшего аналога, в частности - снижение содержания примесей в готовом продукте, увеличение выхода полупродуктов на отдельных стадиях производства, сокращение расхода промывочной воды. Преимуществами предлагаемого технического решения являются снижение трудоемкости способа и затрат на производство готового НМГ.The aim of the present invention is to overcome the disadvantages of the closest analogue, in particular - reducing the content of impurities in the finished product, increasing the yield of intermediates at certain stages of production, reducing the consumption of washing water. The advantages of the proposed technical solution are the reduction of the complexity of the method and the cost of manufacturing the finished NMG.
Раскрытие сущности изобретения.Disclosure of the invention.
Цель настоящего изобретения достигается с помощью способа получения низкомолекулярного гепарина, включающего стадии:The purpose of the present invention is achieved using a method of producing low molecular weight heparin, comprising the steps of:
(а) формирования защиты сульфогрупп взаимодействием высокомолекулярного гепарина с бензетония хлоридом с образованием гепарината бензетония (ГБ),(a) formation of sulfo group protection by the interaction of high molecular weight heparin with benzetonium chloride to form benzetonium hepinate (GB),
(б) этерификации полученной соли бензилированием в апротонном растворителе,(b) esterifying the obtained salt with benzylation in an aprotic solvent,
(в) выделения сложного бензилового эфира гепарина осаждением и удаления защиты сульфогрупп насыщенным раствором ацетата натрия в метиловом спирте,(c) isolating heparin benzyl ester by precipitation and deprotecting the sulfo groups with a saturated solution of sodium acetate in methyl alcohol,
(г) щелочной деполимеризации макромолекулы гепарина и(g) alkaline depolymerization of a heparin macromolecule; and
(д) формирования концевых 1,6-ангидрогрупп β-элиминированием при взаимодействии с сильным восстановителем,(e) the formation of
в котором на стадии (а) отмывку гепарината бензетония от избытка непрореаги-ровавшего бензетония хлорида производят многократной дробной промывкой водой очищенной с применением ультразвука рабочей частоты 30-40 кГц, мощностью излучения 200-400 Вт и на стадии (в) выделение сложного бензилового эфира гепарина осаждением и удаление защиты сульфогрупп проводят как две последовательные операции, а применяемым спиртом является метанол.in which, at stage (a), benzethonium heparin is washed from excess unreacted benzetonium chloride by repeated fractional washing with water purified using ultrasound at an operating frequency of 30-40 kHz, a radiation power of 200-400 W, and at stage (c), heparin benzyl ester is isolated precipitation and deprotection of sulfo groups is carried out as two sequential operations, and the alcohol used is methanol.
Предпочтительно многократную дробную промывку с применением ультразвука осуществляют водой очищенной при массовом соотношении ГБ/вода равном 1/(50-60) и температуре 40-60°С при продолжительности каждого цикла ультразвукового воздействия 10-35 минут.Preferably, multiple fractional washing using ultrasound is carried out with purified water at a mass ratio of GB / water equal to 1 / (50-60) and a temperature of 40-60 ° C with a duration of each cycle of ultrasonic exposure of 10-35 minutes.
Также предпочтительно выделение неполного сложного бензилового эфира гепарината бензетония из раствора производят осаждением 1,5-2,5 объемами 8-10% метанольного раствора натрия ацетата при температуре от -2 до 4°С при перемешивании в течение 4-6 часов.It is also preferable to isolate the benzethonium heparin incomplete benzyl ester from the solution by precipitation of 1.5-2.5 volumes of an 8-10% methanol solution of sodium acetate at a temperature of from -2 to 4 ° C. with stirring for 4-6 hours.
Далее предпочтительно удаление бензетониевой защиты сульфогрупп производят после отделения осадка взаимодействием с 4-6-кратным массовым количеством насыщенного метанольного раствора безводного натрия ацетата при температуре 18-24°С при перемешивании в течение 8-10 часов.It is further preferred that the benzethonium protection of the sulfo groups is removed after separation of the precipitate by reaction with a 4-6-fold mass amount of a saturated methanol solution of anhydrous sodium acetate at a temperature of 18-24 ° C. with stirring for 8-10 hours.
Кроме того, предпочтительно стадию щелочной деполимеризации проводят в присутствии инертного носителя перлита при массовом соотношении бензиловый эфир гепарина/перлит 1/(0,25-0,5).In addition, preferably, the alkaline depolymerization step is carried out in the presence of an inert perlite carrier in a weight ratio of heparin /
В результате проведенных обширных исследований авторы изобретения неожиданно установили, что при проведении отмывки бензетония хлорида с применением ультразвука его частота и мощность имеют решающее значение для качества получаемого полупродукта. При рабочей частоте выше 40 кГЦ и мощности излучения более 400 Вт существует опасность непредсказуемой деструкции не только в полимерной цепи, но также и в отдельных звеньях моносахаридов, что приводит к потере активности готового продукта.As a result of extensive research, the inventors unexpectedly found that when washing benzetonium chloride with ultrasound, its frequency and power are critical to the quality of the resulting intermediate. At an operating frequency above 40 kHz and a radiation power of more than 400 W, there is a danger of unpredictable degradation not only in the polymer chain, but also in individual units of monosaccharides, which leads to a loss of activity of the finished product.
Природа спиртового растворителя также имеет большое значение для достижения технического результата изобретения. В ближайшем аналоге [9] для выделения полупродуктов из рабочих растворов на различных стадиях процесса применяется 96% этанол. При этом первоначально выделяется маслоподобная, липкая и вязкая гигроскопичная субстанция, которую невозможно извлечь из технологического оборудования без значительных потерь. Для получения порошка необходимо многократно перетирать это вещество с осадителем до получения аморфного порошка удовлетворительного качества.The nature of the alcohol solvent is also of great importance for achieving the technical result of the invention. In the closest analogue [9], 96% ethanol is used to isolate intermediates from working solutions at various stages of the process. In this case, an oil-like, sticky and viscous hygroscopic substance that is impossible to extract from technological equipment without significant losses is initially released. To obtain a powder, it is necessary to grind this substance with a precipitant repeatedly until an amorphous powder of satisfactory quality is obtained.
В предлагаемом способе на стадии получения бензилового эфира бензетониевой соли процесс выделения из раствора продукта бензилирования с неудаленной бензетониевой защитой сульфогрупп осаждением метанолом проводят как отдельную операцию, причем в качестве соосадителя используют небольшое количество ацетата натрия.In the proposed method, at the stage of producing benzethonium salt benzyl ester, the process of isolating a benzylation product from the solution with the non-removed benzethonium protection of sulfo groups by methanol precipitation is carried out as a separate operation, and a small amount of sodium acetate is used as a precipitant.
В соответствии с изобретением процесс удаления бензетониевой защиты выделяют в отдельную стадию. Химизм этой реакции заключается во взаимодействии бензетониевого соединения с ацетатом натрия. В аналоге [9] процессы выделения и удаления защиты проводят многократной промывкой этанолом, насыщенным натрия ацетатом (его максимальная концентрация составляет 2,33% масс.). Для этого требуется трех-четырехкратная обработка продукта этим раствором с промежуточным выделением субстанции, т.к. в одной порции раствора реагента недостаточно.In accordance with the invention, the process of removing the benzethonium protection is isolated in a separate stage. The chemistry of this reaction consists in the interaction of the benzethonium compound with sodium acetate. In analogue [9], the processes of isolation and deprotection are carried out by repeated washing with ethanol saturated with sodium acetate (its maximum concentration is 2.33% by weight). This requires three to four times the treatment of the product with this solution with an intermediate allocation of substance, because in one portion of the reagent solution is not enough.
Основомоль гепарина содержит три сульфогруппы и, в соответствии с этим, для удаления бензетониевой защиты с макромолекулы необходимо израсходовать три эквивалента натрия ацетата. В метаноле растворимость натрия ацетата составляет 16,22% масс., что в семь раз больше, чем в этаноле. Поэтому расход осадителя значительно меньший. Кроме того, неожиданно достигается существенно лучшее качество осадка.The basic mole of heparin contains three sulfo groups, and, accordingly, to remove the benzethonium protection from the macromolecule, three equivalents of sodium acetate must be consumed. In methanol, the solubility of sodium acetate is 16.22% by mass, which is seven times more than in ethanol. Therefore, the consumption of precipitant is much smaller. In addition, a significantly better sludge quality is unexpectedly achieved.
Таким образом, техническим результатом предлагаемого изобретения является усовершенствованный способ получения низкомолекулярного гепарина с улучшенными показателями чистоты и общего выхода субстанции при существенном снижении расхода отмывочной воды и образующихся стоков.Thus, the technical result of the invention is an improved method for producing low molecular weight heparin with improved purity and overall yield of the substance with a significant reduction in the consumption of washing water and the resulting effluent.
Осуществление изобретенияThe implementation of the invention
Далее возможность осуществления изобретения с достижением технического результата будет показана на неограничивающих примерах.Further, the possibility of carrying out the invention with the achievement of a technical result will be shown in non-limiting examples.
Пример 1. Синтез гепарината бензетонияExample 1. Synthesis of benzetonium heparin
Схема превращений приведена на Фиг. 6. В стеклянную двугорлую колбу вместимостью 250 мл, помещенную в водяную баню на платформе магнитной термостатируемой мешалки, наливают 50 мл воды очищенной и при перемешивании загружают 8,4 г бензетония хлорида (Hyamine 1622, Lonza Group Ltd.). После полного растворения устанавливают рН в интервале 6,0-6,7.The transformation scheme is shown in FIG. 6. In a 250-ml glass two-necked flask placed in a water bath on the platform of a thermostatically controlled magnetic stirrer, pour 50 ml of purified water and load with stirring 8.4 g of benzethonium chloride (Hyamine 1622, Lonza Group Ltd.). After complete dissolution, the pH is adjusted in the range of 6.0-6.7.
В другом стакане готовят раствор 3,2 г гепарина мукозного в 30 мл 0,2 М раствора NaCl при комнатной температуре. После полного растворения гепарина устанавливают рН в интервале 8,5-8,7.In another glass, a solution of 3.2 g of mucosal heparin in 30 ml of a 0.2 M NaCl solution is prepared at room temperature. After complete dissolution of the heparin, the pH is adjusted in the range of 8.5-8.7.
Температуру водяной бани повышают до 60±1°С и с помощью капельной воронки в реактор при перемешивании, избегая вспенивания содержимого колбы, приливают раствор гепарина в течение 30-40 минут. После прибавления всего количества раствора реакцию продолжают еще 50-60 минут. Образовавшийся осадок горячим отделяют фильтрацией на воронке Бюхнера или центрифугируют. Жидкую фазу отбрасывают, предварительно измерив ее объем и оптическую плотность для количественного определения содержания примесей.The temperature of the water bath is raised to 60 ± 1 ° C and, using a dropping funnel, the heparin solution is poured into the reactor with stirring, avoiding foaming of the contents of the flask, for 30-40 minutes. After adding the entire amount of the solution, the reaction is continued for another 50-60 minutes. The precipitate formed is hot separated by filtration on a Buchner funnel or centrifuged. The liquid phase is discarded, having previously measured its volume and optical density to quantify the content of impurities.
Осадок переносят в стакан вместимостью 100 мл, прибавляют 50 мл воды очищенной и помещают его в ультразвуковую ванну (тип УЗВ, Сапфир). Экстракцию избытка непрореагировавших реагентов (отмывку) производят при температуре 40-60°С и воздействии ультразвуком с рабочей частотой 30-40 кГц и мощностью излучения 200-400 Вт при продолжительности каждого цикла ультразвукового воздействия 10-35 минут. Степень отмывки и содержание примесей в промывочной воде определяют измерением оптической плотности раствора (D270) спектрофотометрически (Cary 60 UV-Vis, Agilent) при длине волны 270 нм по калибровочному графику. Промывку повторяют до достижения оптической плотности промывной воды менее 1,5. Результаты эксперимента с применением ультразвука (УЗ) приведены в таблице 1. Как видно из этих данных удовлетворительный результат достигается уже за 7 циклов промывки, на что расходуется 430 мл воды очищенной. Промытый продукт - гепаринат бензетония (ГБ) лиофильно высушивают (лиофильная сушилка Иней-6, ФГБУН ИБП РАН). Выход ГБ составляет 8,23 г при влагосодержании 5,3%. Расход воды очищенной в расчете на 1 г порошка составляет 52,2 мл.The precipitate is transferred to a glass with a capacity of 100 ml, 50 ml of purified water is added and placed in an ultrasonic bath (ultrasound type, sapphire). Extraction of excess unreacted reagents (washing) is carried out at a temperature of 40-60 ° C and exposure to ultrasound with an operating frequency of 30-40 kHz and a radiation power of 200-400 W with a duration of each cycle of ultrasonic exposure of 10-35 minutes. The degree of washing and the content of impurities in the wash water is determined by measuring the optical density of the solution (D 270 ) spectrophotometrically (Cary 60 UV-Vis, Agilent) at a wavelength of 270 nm according to the calibration graph. Washing is repeated until the optical density of the washing water is less than 1.5. The results of the experiment with the use of ultrasound (US) are shown in Table 1. As can be seen from these data, a satisfactory result is achieved for 7 washing cycles, which consumes 430 ml of purified water. The washed product is benzetonium heparin (GB), freeze-dried (Iney-6 freeze dryer, FSBII IBP RAS). The GB yield is 8.23 g with a moisture content of 5.3%. The consumption of purified water per 1 g of powder is 52.2 ml.
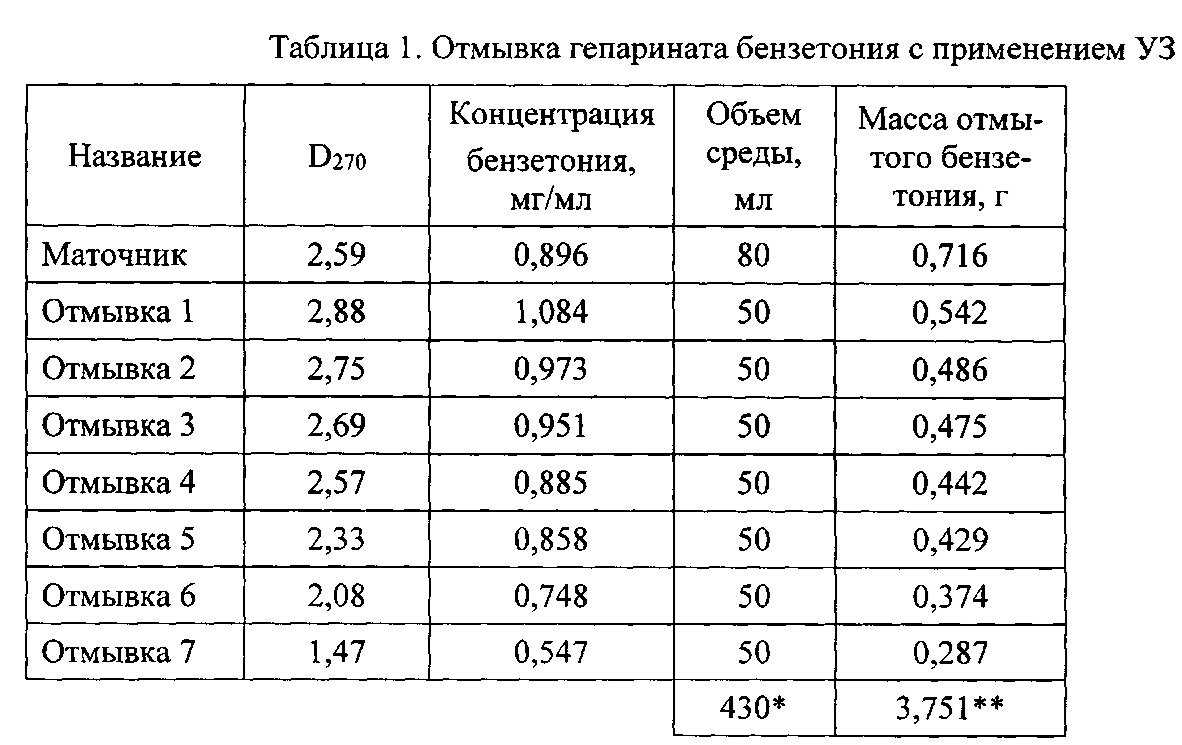
*в том числе промывочных вод 350 мл.* including wash water 350 ml.
**в том числе с промывной водой 3,035 г.** including with wash water 3.035 g.
Параллельно проводят промывку при тех же условиях без применения УЗ (таблица 2). При этом для достижения требуемого результата необходимо провести 14-15 циклов промывки, причем расход воды очищенной составляет 700 мл. Выход порошка ГБ составляет 7,9 г при влагосодержании 5,1%. Расход воды очищенной в расчете на 1 г продукта составляет 88,6 мл.In parallel, washing is carried out under the same conditions without the use of ultrasound (table 2). Moreover, to achieve the desired result, it is necessary to carry out 14-15 washing cycles, and the purified water consumption is 700 ml. The GB powder yield is 7.9 g with a moisture content of 5.1%. The consumption of purified water per 1 g of product is 88.6 ml.


*в том числе промывочных вод 700 мл.* including wash water 700 ml.
**в том числе с промывной водой 3,424 г.** including 3.424 g.
Как видно из приведенных данных, применение отмывки с помощью УЗ обработки приводит к значительной интенсификации процесса, снижению расхода воды очищенной на отмывку ГБ, количества стоков и трудозатрат на проведение процесса в два раза.As can be seen from the above data, the use of washing using ultrasonic treatment leads to a significant intensification of the process, reducing the consumption of purified water for washing GB, the amount of effluent and labor costs for the process by half.
Пример 2. Синтез неполного бензилового эфира гепаринаExample 2. The synthesis of partial benzyl ester of heparin
Схема синтеза приведена на Фиг. 7.The synthesis scheme is shown in FIG. 7.
2.1. Синтез бензилового эфира гепарината бензетония2.1. Synthesis of benzethonium heparin benzyl ester
В трехгорлую колбу вместимостью 1 л, снабженную механической мешалкой и гидрозатвором, помещенную в термостатируемую водяную баню при температуре 37±1°С, наливают 250 мл N,N-диметилформамида (ДМФА) и прибавляют при перемешивании 45,0 г гепарината бензетония, полученного по методике изложенной в примере 1. После полного растворения ГБ в колбу с раствором с помощью капельной воронки прибавляют 40 мл бензилхлорида. Реакцию продолжают при температуре 37±1°С и перемешивании в течение 8 часов после чего содержимое колбы охлаждают до комнатной температуры. Выделение бензилового эфира гепарината бензетония из раствора производят осаждением 525 мл 9% метанольного раствора натрия ацетата при температуре 2-4°С при перемешивании в течение 4-6 часов. Осадок отделяют на воронке Бюхнера и влажным используют на стадии удаления бензетониевой защиты.In a three-necked flask with a capacity of 1 l, equipped with a mechanical stirrer and a water lock, placed in a thermostatic water bath at a temperature of 37 ± 1 ° С, 250 ml of N, N-dimethylformamide (DMF) were poured and 45.0 g of benzetonium heparinate obtained by the method described in example 1. After complete dissolution of the GB in a flask with a solution using a dropping funnel add 40 ml of benzyl chloride. The reaction is continued at a temperature of 37 ± 1 ° C and stirring for 8 hours after which the contents of the flask are cooled to room temperature. Isolation of benzethonium heparinate benzyl ester from solution is carried out by precipitation of 525 ml of a 9% methanol solution of sodium acetate at a temperature of 2-4 ° C with stirring for 4-6 hours. The precipitate is separated on a Buchner funnel and wet used at the stage of removal of the benzethonium protection.
Выход влажного бензилового эфира гепарината бензетония 24,4 г с содержанием бензилового эфира гепарина 16,8 г.The yield of wet benzethonium heparin benzyl ester is 24.4 g with a heparin benzyl ester content of 16.8 g.
2.2. Удаление защиты сульфогрупп бензилового эфира гепарината бензетония2.2. Deprotection of benzogonium hepinate benzyl ester sulfo groups
В коническую плоскодонную колбу вместимостью 500 мл, помещенную на платформу магнитной мешалки, загружают 24.0 г влажного бензилового эфира гепарината бензетония, полученного по примеру 2.1, и приливают 100 мл насыщенного метанольного раствора безводного натрия ацетата. В колбу помещают якорь магнитной мешалки и закрывают ее притертой пробкой. Реакцию проводят при комнатной температуре при перемешивании в течение 8-10 часов. После окончания процесса осадок отделяют на воронке Бюхнера, и высушивают в вакуумном сушильном шкафу при температуре не выше 40С°.A 500 ml conical flat-bottomed flask placed on a magnetic stirrer platform was charged with 24.0 g of wet benzethonium hepinate benzyl ester obtained in Example 2.1, and 100 ml of a saturated methanol solution of anhydrous sodium acetate were added. The anchor of the magnetic stirrer is placed in the flask and closed with a ground stopper. The reaction is carried out at room temperature with stirring for 8-10 hours. After the end of the process, the precipitate is separated on a Buchner funnel, and dried in a vacuum oven at a temperature not exceeding 40 ° C.
Выход неполного бензилового эфира гепарина равен 16,2 г, что составляет 96% от теоретического.The yield of incomplete benzyl ester of heparin is 16.2 g, which is 96% of theoretical.
Пример 3. Гидролиз бензилового эфира гепарина натрияExample 3. Hydrolysis of heparin sodium benzyl ester
Схема синтеза приведена на Фиг. 8. В стакан вместимостью 250 мл, помещенный в водяную баню на платформе магнитной термостатируемой мешалки, наливают 75 мл воды очищенной и при перемешивании загружают 16,0 г бензилового эфира гепарина, полученного как описано в примере 2. Устанавливают температуру 60±1°С и перемешивают до полного растворения порошка. Затем в растворе устанавливают рН равным 11,0-11,1 с помощью свежеприготовленного 1 М раствора NaOH и прибавляют 8 мл 1 М раствора натрия гидроксида. Далее с помощью капельной воронки в течение 30-45 минут дозируют еще 8 мл того же раствора. Реакцию продолжают в течение 45 минут, поддерживая рН не ниже 11,0.The synthesis scheme is shown in FIG. 8. In a 250 ml beaker placed in a water bath on the platform of a thermostatically controlled magnetic stirrer, 75 ml of purified water is poured and 16.0 g of heparin benzyl ester obtained as described in Example 2 are loaded with stirring. The temperature is set to 60 ± 1 ° C and mix until the powder is completely dissolved. Then, the solution is adjusted to pH 11.0-11.1 with a freshly prepared 1 M NaOH solution and 8 ml of 1 M sodium hydroxide solution are added. Then, with the help of a dropping funnel, an additional 8 ml of the same solution is dosed for 30-45 minutes. The reaction is continued for 45 minutes, maintaining the pH not lower than 11.0.
По окончании выдержки охлаждают реакционную массу до комнатной температуры, устанавливают рН равным 6,0±0,2 добавлением соляной кислоты (1:3), и раствор фильтруют. К прозрачному желтоватому фильтрату прибавляют 1,6 г безводного ацетата натрия и при необходимости корректируют рН до 6,0±0,2. Полученный раствор фильтруют через мембрану с размером пор 0,45 мкм, фильтрат охлаждают до температуры 0-4°С в течение 30-60 минут и осаждают продукт охлажденным метанолом (250-300 мл). Раствор помещают в холодильник и оставляют для созревания осадка на 3-4 часа. Надосадочную жидкость декантируют, а образовавшийся осадок дополнительно промывают 50-60 мл метанола. Осадок отделяют на воронке Бюхнера и высушивают в вакуумном сушильном шкафу при температуре не выше 40С°. Выход гидролизованого гепарина составляет 11,3 г, что составляет 84,3% от теоретического.At the end of the exposure, the reaction mixture is cooled to room temperature, the pH is adjusted to 6.0 ± 0.2 by the addition of hydrochloric acid (1: 3), and the solution is filtered. 1.6 g of anhydrous sodium acetate are added to the clear yellowish filtrate and, if necessary, the pH is adjusted to 6.0 ± 0.2. The resulting solution is filtered through a membrane with a pore size of 0.45 μm, the filtrate is cooled to a temperature of 0-4 ° C for 30-60 minutes and the product precipitated with chilled methanol (250-300 ml). The solution is placed in the refrigerator and left to mature the precipitate for 3-4 hours. The supernatant was decanted, and the precipitate formed was further washed with 50-60 ml of methanol. The precipitate is separated on a Buchner funnel and dried in a vacuum oven at a temperature not exceeding 40 ° C. The yield of hydrolyzed heparin is 11.3 g, which is 84.3% of theory.
Пример 4. Боргидрирование сырца низкомолекулярного гепарина натрияExample 4. Borohydrogenation of raw low molecular weight sodium heparin
В стакан вместимостью 100 мл, снабженный мешалкой, наливают 50 мл воды очищенной и при перемешивании прибавляют 11,0 г продукта, полученного в примере 3. После полного растворения порошка устанавливают рН равным 8,4±0,2 и прибавляют 0,20 г боргидрида натрия. Реакцию продолжают в течение 50-60 мин. По окончании реакции устанавливают рН равным 4,0±0,2 добавлением соляной кислоты (1:3) при перемешивании в течение 10 минут, после чего добавлением 1 М раствора гидроксида натрия устанавливают рН равным 6,0±0,2. Полученный раствор фильтруют через мембрану с размером пор 0,45 мкм. Фильтрат охлаждают до температуры 0-4°С в течение 30-60 минут. Охлажденный фильтрат помещают в стакан вместимостью 200 мл и осаждают продукт охлажденным метанолом (150-200 мл). Раствор помещают в холодильник и оставляют для созревания осадка на 3 часа. Надосадочную жидкость декантируют, и образовавшийся осадок дополнительно промывают 30 мл метанола. Осадок отделяют на воронке Бюхнера и высушивают в вакууме при температуре не выше 40С°. Масса готового продукта составляет 9,7 г.50 ml of purified water is poured into a 100 ml beaker equipped with a stirrer and 11.0 g of the product obtained in Example 3 are added with stirring. After complete dissolution of the powder, the pH is adjusted to 8.4 ± 0.2 and 0.20 g of borohydride is added. sodium. The reaction is continued for 50-60 minutes At the end of the reaction, the pH was adjusted to 4.0 ± 0.2 by the addition of hydrochloric acid (1: 3) with stirring for 10 minutes, after which the pH was adjusted to 6.0 ± 0.2 by adding a 1 M sodium hydroxide solution. The resulting solution is filtered through a membrane with a pore size of 0.45 μm. The filtrate is cooled to a temperature of 0-4 ° C for 30-60 minutes. The cooled filtrate is placed in a glass with a capacity of 200 ml and the product is precipitated with chilled methanol (150-200 ml). The solution was refrigerated and left to mature for 3 hours. The supernatant was decanted and the precipitate formed was washed further with 30 ml of methanol. The precipitate is separated on a Buchner funnel and dried in vacuum at a temperature not exceeding 40 ° C. The mass of the finished product is 9.7 g.
Полученная субстанция охарактеризована показателями, приведенными в таблице 3.The resulting substance is characterized by the indicators shown in table 3.


ЛитератураLiterature
1. Hirsh J. et al. Clinicalof Chest Physicians Evidence-Based Parenteral Anticoagulants: American College Practice Guidelines (8th Edition) / Chest, 2008, V. 133, pp. 141S-159S.1. Hirsh J. et al. Clinicalof Chest Physicians Evidence-Based Parenteral Anticoagulants: American College Practice Guidelines (8th Edition) / Chest, 2008, V. 133, pp. 141S-159S.
2. Макаров B.A. и др. Применение гепаринов в клинической практике / РМЖ, 1998, №3, с. 4.2. Makarov B.A. et al. The use of heparins in clinical practice / breast cancer, 1998, No. 3, p. four.
3. Кудрявцев В.Н. / Химия высоких энергий, 1993, Т. 27, №1, С. 41.3. Kudryavtsev V.N. / High Energy Chemistry, 1993, T. 27, No. 1, S. 41.
4. Авторское свидетельство SU 1570651 A3, опубл. 07.06.1990.4. Copyright certificate SU 1570651 A3, publ. 06/07/1990.
5.Linhardt R.J. et. al. Production and chemical processing of low molecular weight heparins / Seminars in Thrombosis and Hemostasi, 1999, V. 25, N. 3, pp. 5-16; патент US 6384021 B1, опубл. 07.05.2002.5.Linhardt R.J. et. al. Production and chemical processing of low molecular weight heparins / Seminars in Thrombosis and Hemostasi, 1999, V. 25, N. 3, pp. 5-16; US patent 6384021 B1, publ. 05/07/2002.
6. Linhardt R.J. et. al. Differential anticoagulant activity of heparin fragments prepared using microbial heparinase / Journal of Biological Chemistry, 1982, V. 257, N. 13, pp. 7310-7313.6. Linhardt R.J. et. al. Differential anticoagulant activity of heparin fragments prepared using microbial heparinase / Journal of Biological Chemistry, 1982, V. 257, N. 13, pp. 7310-7313.
7. Патент RU 2295538 C2, опубл. 20.03.2007.7. Patent RU 2295538 C2, publ. 03/20/2007.
8. Патент RU 2396282 C1, опубл. 10.08.2010.8. Patent RU 2396282 C1, publ. 08/10/2010.
9. Патент RU 2512768 C1, опубл. 10.04.2014.9. Patent RU 2512768 C1, publ. 04/10/2014.
Claims (9)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2017146005A RU2670767C9 (en) | 2017-12-26 | 2017-12-26 | Method for producing low molecular weight heparin |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2017146005A RU2670767C9 (en) | 2017-12-26 | 2017-12-26 | Method for producing low molecular weight heparin |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2670767C1 RU2670767C1 (en) | 2018-10-25 |
| RU2670767C9 true RU2670767C9 (en) | 2018-11-26 |
Family
ID=63923439
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2017146005A RU2670767C9 (en) | 2017-12-26 | 2017-12-26 | Method for producing low molecular weight heparin |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2670767C9 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2725545C1 (en) * | 2020-01-30 | 2020-07-02 | федеральное государственное бюджетное образовательное учреждение высшего образования "МИРЭА-Российский технологический университет" | Method of producing low-molecular heparin |
| CN115043959A (en) * | 2022-05-25 | 2022-09-13 | 湖北亿诺瑞生物制药有限公司 | Preparation method of high-yield enoxaparin sodium |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FI3724235T3 (en) | 2017-12-11 | 2024-09-27 | Biological E Ltd | Process for the preparation of low molecular weight heparin |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2295538C2 (en) * | 2005-03-01 | 2007-03-20 | Гематологический научный центр РАМН | Method for preparing heparins of low molecular mass |
| RU2396282C1 (en) * | 2009-04-02 | 2010-08-10 | Государственное учреждение гематологический научный центр Российской академии медицинских наук (ГУ ГНЦ РАМН) | Method for preparing heparin with low molecular weight and anticoagulant activity |
| RU2512768C1 (en) * | 2012-12-18 | 2014-04-10 | Федеральное бюджетное учреждение "Государственный институт кровезаменителей и медицинских препаратов (ФБУ "ГИКиМП") | Method of obtaining low-molecular heparin |
-
2017
- 2017-12-26 RU RU2017146005A patent/RU2670767C9/en active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2295538C2 (en) * | 2005-03-01 | 2007-03-20 | Гематологический научный центр РАМН | Method for preparing heparins of low molecular mass |
| RU2396282C1 (en) * | 2009-04-02 | 2010-08-10 | Государственное учреждение гематологический научный центр Российской академии медицинских наук (ГУ ГНЦ РАМН) | Method for preparing heparin with low molecular weight and anticoagulant activity |
| RU2512768C1 (en) * | 2012-12-18 | 2014-04-10 | Федеральное бюджетное учреждение "Государственный институт кровезаменителей и медицинских препаратов (ФБУ "ГИКиМП") | Method of obtaining low-molecular heparin |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2725545C1 (en) * | 2020-01-30 | 2020-07-02 | федеральное государственное бюджетное образовательное учреждение высшего образования "МИРЭА-Российский технологический университет" | Method of producing low-molecular heparin |
| CN115043959A (en) * | 2022-05-25 | 2022-09-13 | 湖北亿诺瑞生物制药有限公司 | Preparation method of high-yield enoxaparin sodium |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2670767C1 (en) | 2018-10-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2670767C9 (en) | Method for producing low molecular weight heparin | |
| SU1658820A3 (en) | Method for preparing mucopolysaccharides | |
| US4981955A (en) | Depolymerization method of heparin | |
| CN101735336B (en) | Oligomeric fucosylated glycosaminoglycan and preparation method thereof | |
| JPS63278901A (en) | Normal structure low molecular weight heparin, manufacture and biological application | |
| EP3298047B1 (en) | Process for the preparation of polysaccharides | |
| NO304991B1 (en) | Process for preparing mixtures of sulfated polysaccharides | |
| EA005995B1 (en) | Heparin-derived polysaccharide mixtures, preparation method and pharmaceutical compositions containing same | |
| JPH04502928A (en) | Heparin saccharides and pharmaceutical compositions | |
| RU2512768C1 (en) | Method of obtaining low-molecular heparin | |
| CA2908959C (en) | Low-molecular-weight glycosaminoglycan derivative containing terminal 2, 5-anhydrated talose or derivative thereof | |
| AU2002321225B2 (en) | Polysaccharidic esters of retinoic acid | |
| AU2002321225A1 (en) | Polysaccharidic esters of retinoic acid | |
| Tang et al. | A regular Chlorella mannogalactan and its sulfated derivative as a promising anticoagulant: structural characterization and anticoagulant activity | |
| RU2753678C1 (en) | Method for obtaining nadroparin calcium | |
| CA2907887C (en) | Low-molecular-weight glycosaminoglycan derivative, pharmaceutical composition thereof, preparation method therefor and use thereof | |
| CN109400730B (en) | Lycium barbarum polysaccharide, and preparation method and application thereof | |
| CN116515013A (en) | Ultralow molecular heparin and preparation method and application thereof | |
| EP3724235B1 (en) | Process for the preparation of low molecular weight heparin | |
| CA2914256A1 (en) | New processes for the production of chemically-modified heparins | |
| CN108484792B (en) | Dextran sulfate and method for preparing dextran sulfate | |
| RU2725545C1 (en) | Method of producing low-molecular heparin | |
| CN102585034B (en) | Method for sulfonating pectin | |
| CN104725532A (en) | Method for precisely and quantitatively controlling chondroitin sulfate and dermatan sulfate contents of heparin/heparinoid | |
| RU2441024C1 (en) | Method of producing low-molecular pectin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| TH4A | Reissue of patent specification |