RU2156458C1 - Способ определения азота в биологических образцах - Google Patents
Способ определения азота в биологических образцах Download PDFInfo
- Publication number
- RU2156458C1 RU2156458C1 RU99111126A RU99111126A RU2156458C1 RU 2156458 C1 RU2156458 C1 RU 2156458C1 RU 99111126 A RU99111126 A RU 99111126A RU 99111126 A RU99111126 A RU 99111126A RU 2156458 C1 RU2156458 C1 RU 2156458C1
- Authority
- RU
- Russia
- Prior art keywords
- nitrogen
- sample
- solution
- volume
- mixture
- Prior art date
Links
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 title claims abstract description 64
- 229910052757 nitrogen Inorganic materials 0.000 title claims abstract description 31
- 238000000034 method Methods 0.000 title claims abstract description 24
- 239000012472 biological sample Substances 0.000 title claims abstract description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims abstract description 24
- 239000000523 sample Substances 0.000 claims abstract description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 20
- 239000012153 distilled water Substances 0.000 claims abstract description 18
- 238000004458 analytical method Methods 0.000 claims abstract description 13
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims abstract description 12
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims abstract description 12
- 238000004448 titration Methods 0.000 claims abstract description 10
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 claims abstract description 8
- 239000000779 smoke Substances 0.000 claims abstract description 8
- 229920002472 Starch Polymers 0.000 claims abstract description 7
- 235000019698 starch Nutrition 0.000 claims abstract description 7
- 239000008107 starch Substances 0.000 claims abstract description 7
- 239000005708 Sodium hypochlorite Substances 0.000 claims abstract description 5
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 claims abstract description 5
- 239000011734 sodium Substances 0.000 claims description 13
- 239000011572 manganese Substances 0.000 claims description 6
- 239000013068 control sample Substances 0.000 claims description 5
- 238000001816 cooling Methods 0.000 claims description 5
- 238000002845 discoloration Methods 0.000 claims description 4
- 238000010438 heat treatment Methods 0.000 claims description 3
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 claims description 2
- 229910052748 manganese Inorganic materials 0.000 claims description 2
- 238000005070 sampling Methods 0.000 claims description 2
- 239000000203 mixture Substances 0.000 abstract description 12
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 abstract description 6
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 abstract description 4
- 235000019345 sodium thiosulphate Nutrition 0.000 abstract description 4
- 239000000126 substance Substances 0.000 abstract description 4
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 abstract description 3
- 235000013305 food Nutrition 0.000 abstract description 2
- 230000001863 plant nutrition Effects 0.000 abstract description 2
- 239000013074 reference sample Substances 0.000 abstract 2
- 230000000694 effects Effects 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 41
- 239000000460 chlorine Substances 0.000 description 39
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Chemical compound Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 description 31
- 238000006243 chemical reaction Methods 0.000 description 27
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 12
- 229910052801 chlorine Inorganic materials 0.000 description 12
- JGJLWPGRMCADHB-UHFFFAOYSA-N hypobromite Chemical compound Br[O-] JGJLWPGRMCADHB-UHFFFAOYSA-N 0.000 description 10
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 9
- QDHHCQZDFGDHMP-UHFFFAOYSA-N Chloramine Chemical class ClN QDHHCQZDFGDHMP-UHFFFAOYSA-N 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- JSYGRUBHOCKMGQ-UHFFFAOYSA-N dichloramine Chemical compound ClNCl JSYGRUBHOCKMGQ-UHFFFAOYSA-N 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 4
- KJFMBFZCATUALV-UHFFFAOYSA-N phenolphthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2C(=O)O1 KJFMBFZCATUALV-UHFFFAOYSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 238000001228 spectrum Methods 0.000 description 4
- 238000007696 Kjeldahl method Methods 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- XKMRRTOUMJRJIA-UHFFFAOYSA-N ammonia nh3 Chemical compound N.N XKMRRTOUMJRJIA-UHFFFAOYSA-N 0.000 description 3
- 239000010425 asbestos Substances 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 229910052895 riebeckite Inorganic materials 0.000 description 3
- DHCDFWKWKRSZHF-UHFFFAOYSA-L thiosulfate(2-) Chemical compound [O-]S([S-])(=O)=O DHCDFWKWKRSZHF-UHFFFAOYSA-L 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 229910021538 borax Inorganic materials 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000004744 fabric Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910001437 manganese ion Inorganic materials 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 235000010339 sodium tetraborate Nutrition 0.000 description 2
- 239000004328 sodium tetraborate Substances 0.000 description 2
- 239000012224 working solution Substances 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- UQGFMSUEHSUPRD-UHFFFAOYSA-N disodium;3,7-dioxido-2,4,6,8,9-pentaoxa-1,3,5,7-tetraborabicyclo[3.3.1]nonane Chemical compound [Na+].[Na+].O1B([O-])OB2OB([O-])OB1O2 UQGFMSUEHSUPRD-UHFFFAOYSA-N 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000021384 green leafy vegetables Nutrition 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- RSAZYXZUJROYKR-UHFFFAOYSA-N indophenol Chemical compound C1=CC(O)=CC=C1N=C1C=CC(=O)C=C1 RSAZYXZUJROYKR-UHFFFAOYSA-N 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 229940099596 manganese sulfate Drugs 0.000 description 1
- 239000011702 manganese sulphate Substances 0.000 description 1
- 235000007079 manganese sulphate Nutrition 0.000 description 1
- SQQMAOCOWKFBNP-UHFFFAOYSA-L manganese(II) sulfate Chemical compound [Mn+2].[O-]S([O-])(=O)=O SQQMAOCOWKFBNP-UHFFFAOYSA-L 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 229910000474 mercury oxide Inorganic materials 0.000 description 1
- UKWHYYKOEPRTIC-UHFFFAOYSA-N mercury(ii) oxide Chemical compound [Hg]=O UKWHYYKOEPRTIC-UHFFFAOYSA-N 0.000 description 1
- QEHKBHWEUPXBCW-UHFFFAOYSA-N nitrogen trichloride Chemical compound ClN(Cl)Cl QEHKBHWEUPXBCW-UHFFFAOYSA-N 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 238000004451 qualitative analysis Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
Landscapes
- Investigating Or Analyzing Non-Biological Materials By The Use Of Chemical Means (AREA)
Abstract
Изобретение относится к пищевой промышленности, сельскому хозяйству, агрохимии и может быть использовано при диагностике питания растений в процессе выращивания, а также при контроле качества сельскохозяйственной продукции, то есть для количественного определения азота в природных и промышленных продуктах. Способ определения азота в биологических образцах включает отбор пробы, внесение перекиси водорода и серной кислоты, нагрев до выделения белого дыма и полного обесцвечивания, охлаждение, добавление дистиллированной воды, далее в смесь дополнительно вносят боратный буфер, нейтрализуют раствором NaOH, затем вносят растворы марганца, гипохлорита натрия, KJ, H2SO4, добавляют крахмал и титруют до обесцвечивания, потом аналогичную процедуру выполняют и для контрольной пробы, после чего определяют содержание азота по формуле Х=(0,1166(V0-V1)•100•100%):m•V2, где V0 - объем Na2S2O3, пошедший на титрование контрольной пробы, мл; V1 - объем Na2S2O3, пошедший на титрование пробы, мл; V2 - объем зольного раствора пробы, взятой на анализ, мл; m - масса навески, мг; 0,1166 - количество мг азота, эквивалентное 1 мл 0,025 н. растворa Na2S2O3; 100 - исходный объем зольного раствора пробы, мл. Данный способ позволяет сократить время проведения анализа, а также улучшить воспроизводимость результата и повысить точность определения аммонийного азота.
Description
Изобретение относится к пищевой промышленности, сельскому хозяйству, агрохимии и может быть использовано при диагностике питания растений в процессе выращивания, а также при контроле качества сельскохозяйственной продукции, то есть для количественного определения азота в природных и промышленных продуктах.
Известен способ определения азота в органических веществах - метод Кьельдаля, включающий отбор пробы, внесение перекиси водорода и серной кислоты, нагрев до выделения белого дыма и полного обесцвечивания, охлаждение, добавление дистиллированной воды (Г.Т.Кларк "Руководство по качественному и количественному органическому анализу". ОНТИ НКТП Государственное научно-техническое издательство Украины, 1934, с. 314-316).
После того, как Кьельдаль предложил метод определения азота в органических веществах, начались поиски способа исключения громоздкой и трудоемкой процедуры отгонки аммиака. К этому времени Гофманом была уже открыта реакция между аммиаком и хлором, в результате которой образуется элементарный азот, составляющий 3 часть объема хлора (1). Позже она была проведена в водном растворе с гипохлоритом вместо хлора (2).
2NH4 + + 3OCl- = N2 + 2H+ + 3Cl- + 3H2 (1)
Однако попытки применить ее для определения аммонийного азота оказались безуспешными, поскольку не удавалось добиться необходимой точности. Когда гипохлорит был заменен на гипобромит (3), благодаря чему получались вполне удовлетворительные результаты, интерес к ней был окончательно потерян. Высказывалось предположение, что в этой реакции параллельно основной протекает какая-то побочная (4).
Однако попытки применить ее для определения аммонийного азота оказались безуспешными, поскольку не удавалось добиться необходимой точности. Когда гипохлорит был заменен на гипобромит (3), благодаря чему получались вполне удовлетворительные результаты, интерес к ней был окончательно потерян. Высказывалось предположение, что в этой реакции параллельно основной протекает какая-то побочная (4).
Таким образом, недостатком известного способа является невозможность определения аммонийного азота в органических веществах и большая продолжительность анализа.
Существующий в настоящее время метод определения протеина (белка) в кормах не позволяет своевременно выполнить анализ, и данные по питательности кормов, например, лаборатория птицефабрики получает после того, как корма скормлены. Практическая ценность такого анализа практически равна 0. Предлагаемый способ определения белка дает возможность получить данные в течение 1 часа и, в случае необходимости, своевременно доработать до нужных показателей. Это позволяет избежать срывов в технологии производства и экономических потерь.
Цель изобретения - сокращение времени проведения анализа, улучшение воспроизводимости результатов, точность определения аммонийного азота.
Поставленная цель достигается тем, что в предлагаемом способе определения азота в биологических образцах, включающем подготовку образца, внесение перекиси водорода, серной кислоты, нагрев до появления белого дыма и полного обесцвечивания, охлаждение, добавление дистиллированной воды, в смесь дополнительно вносят боратный буфер, нейтрализуют раствором NaOH, затем вносят растворы марганца, гипохлорита натрия, KJ, H2SO4, добавляют крахмал и титруют до обесцвечивания, далее аналогичную процедуру выполняют и для контрольной пробы, после чего определяют содержание азота по формуле:
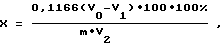
где V0 - объем Na2S2O3, пошедший на титрование контрольной пробы, мл;
V1 - объем Na2S2O3, пошедший на титрование пробы, мл;
V2 - объем зольного раствора пробы, взятой на анализ, мл;
m - масса навески, мг;
0,1166 - количество мг азота, эквивалентное 1 мл 0,025 н. раствору Na2S2O3;
100 - исходный объем зольного раствора пробы, мл.
где V0 - объем Na2S2O3, пошедший на титрование контрольной пробы, мл;
V1 - объем Na2S2O3, пошедший на титрование пробы, мл;
V2 - объем зольного раствора пробы, взятой на анализ, мл;
m - масса навески, мг;
0,1166 - количество мг азота, эквивалентное 1 мл 0,025 н. раствору Na2S2O3;
100 - исходный объем зольного раствора пробы, мл.
Способ осуществляется следующим образом.
Раствор гипохлорита готовят из коммерческого препарата "Белизна" в боратном буфере в концентрации 0,075 н. Нужный pH доводился растворами NaOH или H2SO4. Стандартный раствор аммония готовился из фиксанала хлористого аммония (0,1 г-экв). Активный хлор определяется методом йодометрии. Йод оттитровывался 0,025 н. раствором тиосульфата натрия, приготовленного из фиксанала (0,1 г-экв).
Абсолютные спектры реакционной смеси регистрировались на дифференциальном спектрофотометре SPECORD (ФРГ), pH определялось на ORION RESEARCH (США), азот измерялся на автоанализаторе CONTIFO (Венгрия) (реакция индофенольной зелени) после определения Cl+. Окислительно-восстановительный потенциал измерялся на pH 340.
Определение азота в биологических образцах по методу Кьельдаля проводилось следующим образом. В колбу бралась навеска около 500 мг, затем в нее вносили 5 мл 30% H2O2 и 10 мл концентрированной H2SO4. После бурного кипения колба нагревалась на электроплитке в течение 10-15 минут, затем ставилась на асбестовую ткань для охлаждения. После повторного внесения 5 мл перекиси водорода колба вновь нагревалась до выделения белого дыма и полного обесцвечивания содержимого. При необходимости эта процедура повторялась вновь.
Объем охлажденной пробы доводят до 100 мл дистиллированной водой.
50 мл зольного раствора брались для отгонки аммиака в 0,05 н. раствор H2SO4, приготовленный из фиксанала (0,1 г-экв). Титр раствора щелочи уточнялся 0,05 н. раствором H2SO4. Оставшийся зольный раствор доводился водой до 100 мл и использовался в предлагаемом методе.
Многими авторами было показано, что промежуточными продуктами реакции (1) являются хлорамины, причем какой преимущественно хлорамин образуется зависит от pH смеси. При pH более 7 преобладает NH2Cl, а в пределах от 5 до 4 NHCl2, и менее 2-NCl3 (5, 6). Каждый из них находился в равновесии со своим соседом, и при любом данном pH в системе не может существовать более двух хлораминов (7). В этой работе реакция исследуется только в слабощелочной среде.
Посмотрим, что происходит по мере прибавления гипохлорита к раствору аммония. Для удобства изложения обозначим хлор с валентностью +1, так называемый "активный хлор" через Cl+, мольное отношение исходного Cl+ к исходному аммонию -R и мольное отношение израсходованного в реакции Cl+ к исходному аммонию -r.
Следовательно, идет известная реакция:
NH4 + + OCl- = NH2Cl + H2O. (2)
Дальнейшее увеличение соотношения хлора к азоту до R = 1 вызывает смещение равновесия реакций (2) вправо, о чем свидетельствует повышение максимума поглощения. При эквимолярном соотношении структура спектра резко меняется. Во-первых, снижается содержание NH2Cl в системе и появляются еще два максимума поглощения при 206 нм и 292 нм. Последний соответствует иону гипохлорита (8), а другой обусловлен присутствием дихлорамина. Дело в том, что спектр дихлорамина имеет два максимума поглощения: при 206 нм и 295 нм (9), но поскольку гипохлорит поглощает при 292 нм, их полосы накладываются. В смеси эти соединения можно дифференцировать по более выраженной полосе поглощения дихлорамина (206 нм), так как его коэффициент молярной экстинции при этой длине волны в 7 раз превосходит коэффициент при 295 нм (9). Таким образом, по мере увеличения R до значения, равного 1.5, протекает следующая реакция:
NH2Cl + OCl- = NHCl2 + OH-. (3)
При R = 2 в смеси содержится незначительное количество хлораминов в равновесии с гипохлоритом. Спектр при более высоком значении R (3.0) демонстрирует полное отсутствие хлораминов и преобладание в системе гипохлорита.
NH4 + + OCl- = NH2Cl + H2O. (2)
Дальнейшее увеличение соотношения хлора к азоту до R = 1 вызывает смещение равновесия реакций (2) вправо, о чем свидетельствует повышение максимума поглощения. При эквимолярном соотношении структура спектра резко меняется. Во-первых, снижается содержание NH2Cl в системе и появляются еще два максимума поглощения при 206 нм и 292 нм. Последний соответствует иону гипохлорита (8), а другой обусловлен присутствием дихлорамина. Дело в том, что спектр дихлорамина имеет два максимума поглощения: при 206 нм и 295 нм (9), но поскольку гипохлорит поглощает при 292 нм, их полосы накладываются. В смеси эти соединения можно дифференцировать по более выраженной полосе поглощения дихлорамина (206 нм), так как его коэффициент молярной экстинции при этой длине волны в 7 раз превосходит коэффициент при 295 нм (9). Таким образом, по мере увеличения R до значения, равного 1.5, протекает следующая реакция:
NH2Cl + OCl- = NHCl2 + OH-. (3)
При R = 2 в смеси содержится незначительное количество хлораминов в равновесии с гипохлоритом. Спектр при более высоком значении R (3.0) демонстрирует полное отсутствие хлораминов и преобладание в системе гипохлорита.
Были исследованы результаты количественного контроля за содержанием азота и Cl+, проведенного параллельно качественному анализу. Потеря как азота, так и Cl+, хотя и незначительная, происходит даже тогда, когда в системе преобладает монохлорамин, но наиболее резкая наблюдается при R>1.0. При эквимолярном соотношении реакция (1) прореагировала приблизительно на 80%. Полное завершение ее достигается при исходном мольном соотношении хлора к азоту, равном 2.25.
Фактическое потребление Cl+ на окисление аммония до молекулярного азота на 10% выше теоретически рассчитанного по реакции (1). Причина потери хлора, возможно кроется в механизме последующих реакций дихлорамина. Было показано (10), что стабильность его сохраняется только до тех пор, пока в системе присутствует ион аммония. При исчерпании NH4 +, вызванного избытком гипохлорита идут следующие сопряжения реакции:
NHCl2 + OCl- = NCl3 + OH-, (4)
NHCl2 + NCl3 + 3OH- = N2 + 2HOCl- + 3Cl- + H2O, (5)
HOCl = OCl- + H+. (6)
Образование хлорноватистой кислоты можно наблюдать визуально по желтовато-зеленоватой окраске смеси в период наиболее бурного протекания реакции (1<R<1,5). Обесцвечивание обусловлено диссоциацией HОCl на ионы H+ и OCl-. Гипохлорит включается в реакцию (4), ускоряя ее, поэтому распад дихлорамина является аутокаталитическим процессом.
NHCl2 + OCl- = NCl3 + OH-, (4)
NHCl2 + NCl3 + 3OH- = N2 + 2HOCl- + 3Cl- + H2O, (5)
HOCl = OCl- + H+. (6)
Образование хлорноватистой кислоты можно наблюдать визуально по желтовато-зеленоватой окраске смеси в период наиболее бурного протекания реакции (1<R<1,5). Обесцвечивание обусловлено диссоциацией HОCl на ионы H+ и OCl-. Гипохлорит включается в реакцию (4), ускоряя ее, поэтому распад дихлорамина является аутокаталитическим процессом.
Как показали дальнейшие опыты, потеря хлора предотвращается присутствием в реагирующей смеси иона марганца в боратном буфере при pH 8,5 гипохлорит не окисляет Mn++, но внесение в эту смесь NH4 + вызывает помутнение раствора, обусловленное образованием двуокиси марганца, точнее ее гидрата MnO(OH)2. Такое же помутнение происходит при пропускании через смесь боратного буфера с Mn++ (pH 8,5) как Cl2, так и NCl3. Вероятно реакция (5) протекает через стадию образования Cl2.
NHCl2 + NCl3 + OH- = N2 + 2Cl2 + Cl- + H2O, (7)
Cl2 + OH- = HOCl + Cl-. (8)
Если уравнение реакции (8) умножить на 2 и почленно сложить с уравнением (7), то получиться уравнение (5), выделившаяся двуокись марганца определяется методом йодометрии аналогично определению Cl+.
Cl2 + OH- = HOCl + Cl-. (8)
Если уравнение реакции (8) умножить на 2 и почленно сложить с уравнением (7), то получиться уравнение (5), выделившаяся двуокись марганца определяется методом йодометрии аналогично определению Cl+.
Мольное отношение израсходованного хлора к исходному аммонию (r) в этом случае получается равным 1.5, что соответствует отношению стехиометрических коэффициентов в уравнении (1).
С увеличением pH в системе преобладает NH2Cl, а с уменьшением NHCl2, то максимум скорости реакции, вероятно, достигается при оптимальном соотношении монохлорамина и дихлорамина. В присутствии Mn++ r, принимает значение, равное теоретическому в интервале 7,5-9,5. Резкое снижение r за пределами этого интервала вызвано тем, что реакция не идет до конца, и оставшиеся часть азота содержится в форме NHCl2 в более кислой среде и NH2Cl- в более щелочной.
Полученные данные позволили подобрать оптимальные условия для протекания реакции (1) и позволили применить ее для определения азота в биологических образцах, подвергнутых кислотному разложению по Кьельдалю.
А. Приготовление зольного раствора пробы.
В мерную на 100 мл колбу берут 100-200 мг (в зависимости от содержания азота) тщательно измельченного и перемешанного образца, вносят 5 мл 30% перекиси водорода, 4 мл концентрированной H2SO4. Колбу ставят на электроплитку и нагревают до появления белого дыма (15 минут), а затем снимают и ставят на асбестовую ткань для охлаждения (10 мин). В остывшую колбу вновь вносят 5 мл перекиси и ставят на электроплитку для дальнейшего сжигания до появления белого дыма и обесцвечивания содержимого (25 минут). Объем колбы, остывшей на асбесте, доводят дистиллированной водой до метки и тщательно перемешивают.
Все перечисленные операции параллельно выполняют и для слепой (контрольной) пробы, содержащей только 4 мл концентрированной H2SO4.
Б. Приготовление реактивов.
1. Раствор йодистого калия. Растворяют 50 г KJ в 1 л дистиллированной воды. Раствор хранят в темной посуде.
2. Раствор серной кислоты (2 н.). В мерную колбу на 1 л берут 53,6 мл концентрированной H2SO4, доводят до метки дистиллированной водой.
3. Раствор гипосульфита натрия (0,025 н.). Растворяют фиксанал, содержащий 0,1 г-экв Na2S2O3 в 4 литрах дистиллированной воды.
4. В полученный раствор вносят 400 мг Na2CO3 или 1 г Na2CO3•10H2O.
5. Раствор крахмала. Отвешивают 0,5 г "растворимого крахмала" и тщательно растирают его с несколькими миллилитрами холодной дистиллированной воды. Полученную пасту вливают в 100 мл кипящей дистиллированной воды, кипятят 2 минуты и горячим фильтруют. Хранят в холодильнике.
6. Раствор гидроокиси натрия. Растворяют 100 г NaOH в 1 л дистиллированной воды.
7. Приготовление рабочего раствора гипохлорита. Растворяют в мерной колбе на 1 л 30 г буры в 800 - 900 мл теплой дистиллированной воды и вносят 50 мл 2 н. H2SO4. Затем определяют объем исходного раствора гипохлорита (бытовой препарат "Белизна") для получения 0,075 н. рабочего раствора. Для этого вносят в колбу на 250 мл с магнитной мешалкой 1 мл "Белизны", 50 мл дистиллированной воды, 10 мл раствора KJ и 10 мл 2 н. H2SO4. Выделившийся йод титруют 0,025 н. раствором Na2S2O3 до соломенно-желтой окраски, после чего прибавляют около 0.5 мл раствора крахмала и титруют до обесцвечивания синей окраски от одной капли. Объем (V) определяют по формуле
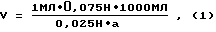
где a - объем 0,025 н. раствора гипосульфита, пошедший на титрование.
где a - объем 0,025 н. раствора гипосульфита, пошедший на титрование.
После внесения найденного объема "Белизны" колбу доводят до метки дистиллированной водой и тщательно перемешивают, pH полученного раствора должен быть в пределах 8.5.
8. Раствор сернокислого марганца. Растворяют 2.5 г MnSO4•5H2O в 100 мл дистиллированной воды.
9. Боратный буфер. В мерную колбу на 1 литр берут навеску фенолфталеина, равную 15 мг, вносят некоторое количество дистиллированной воды и 1 мл раствора NaOH (N 5), перемешивают и растворяют 30 г тетрабората натрия теплой дистиллированной водой.
10. Объем доводят до метки.
В коническую колбу на 250 мл вносят 10-20 мл зольного раствора пробы, 10 мл боратного буфера. Смесь нейтрализуют на магнитной мешалке раствором NaOH до слабо-розовой окраски. Затем последовательно вносят 2 мл раствора марганца, 20 мл гипохлорита натрия, 10 мл KJ и 10 мл 2 н. H2SO4, после чего добавляют около 0,5 мл крахмала и титруют до обесцвечивания синей окраски от одной капли. Аналогичную процедуру выполняют и для слепой (контрольной) пробы. Процентное содержание азота в пробе (X) определяют по формуле:
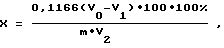
где V0 - объем Na2S2O3, пошедший на титрование контрольной пробы, мл;
V1 - объем Na2S2O3, пошедший на титрование пробы, мл;
V2 - объем зольного раствора пробы, взятой на анализ, мл;
m - масса навески, мг;
0,1166 - количество мг азота, эквивалентное 1 мл 0,025 н. раствору Na2S2O3;
100 - исходный объем зольного раствора пробы, мл.
где V0 - объем Na2S2O3, пошедший на титрование контрольной пробы, мл;
V1 - объем Na2S2O3, пошедший на титрование пробы, мл;
V2 - объем зольного раствора пробы, взятой на анализ, мл;
m - масса навески, мг;
0,1166 - количество мг азота, эквивалентное 1 мл 0,025 н. раствору Na2S2O3;
100 - исходный объем зольного раствора пробы, мл.
Если почленно сложить уравнение (2)-(6), умножив (2), (3), (6) на 2, то получится уравнение (1). Таким образом, конечные продукты этой реакции являются результатом сложной цепи последовательных превращений исходных продуктов. При этом Cl+ с валентностью +1, превращаясь в хлорид с валентностью -1, проходит стадию с нулевой валентностью, и в этом состоянии возможна его потеря.
Определенный вклад в эту потерю может вносить и треххлористый азот потому, что он обладает более выраженным по сравнению с NH2Cl и NHCl2 гидрофобными свойствами. В двухфазной системе, по имеющимся данным (7), отношения распределения между Cl4C и водой равно 0,058, 0,88 и 32,0 соответственно для NH2Cl и NHCl2 и NCl3. Поэтому некоторая часть NCl3 вместе с Cl2, вероятно, увлекается бурно выделяющимся газообразным азотом, приводя к потери "активного хлора". Марганец, очевидно, вызывает резкое смещение реакции (7) вправо, поскольку Cl2 расходуется на образование двуокиси.
Равенство r отношения стехиометрических коэффициентов исходных продуктов реакции (1) в присутствии Mn++ дает возможность рассчитать титр раствора гипосульфита по азоту. Между тем в некоторых предлагаемых гипобромитных методах определения азота это далеко не всегда возможно, и тогда титр гипосульфита уточняется по стандартному раствору аммония (11).
Чаще всего в качестве индикатора при доведении pH до нужного значения в предлагаемых методах с гипобромитом используют окись ртути, так как органические индикаторы окисляются гипогалогенами, искажая результаты анализа. В данном случае для доведения pH реакционной смеси до 8,5 применен фенолфталеин таким образом, что количество его одинаково как в слепой (контрольной), так и в исследуемых пробах ошибка, связанная с расходованием Cl+ на окисление индикатора, уничтожается в формуле (2). Поскольку предел pH, в которых значение r равно 1.5, сравнительно широкий, требования к точности доведения реакционной среды - невысокие.
Нормальность 0,075 рабочего раствора гипохлорита рассчитана на определение азота в пределах от 0 до 3 мг в реакционной смеси, но этот интервал можно расширить, повысив концентрацию Cl+.
Сжигание биологических образцов проводится в концентрированной H2SO4 с окислителем (H2O2) без добавления каких-либо катализаторов, так как они мешают определению азота. Рекомендуемые некоторыми авторами способы удаления катализаторов из минерализата (12) сложны и неоправданно увеличивают время анализа. Перекись значительно эффективнее катализаторов, но и она тоже может вносить помеху в случае неполного испарения, завышая результаты анализа. Однако, как показали исследования, H2O2 в нем отсутствует, если сжигаемая смесь кипит с выделением белого дыма (330oC).
Гипохлорит натрия является более доступным окислителем, чем гипобромид, поэтому иногда в предлагаемых гипобромидных методах применяется KBr, окисляющийся гипохлоритом до гипобромита.
Однако реакция (1), в отличие от гипобромитной, применима только для метода обратного титрования, так как она идет до конца только при значительном избытке гипохлорита.
Выводы
1. Точность предлагаемого метода определения азота в биологических образцах близка к точности метода Кьельдаля.
1. Точность предлагаемого метода определения азота в биологических образцах близка к точности метода Кьельдаля.
2. Участие иона марганца в данной реакции является важным условием достижения этой точности.
3. Для полного завершения реакции исходное мольное отношение хлора к аммонию должно быть не менее 2.25.
4. Оптимальная для протекания реакция pH в пределах 7,5-9.0.
Источники информации
1. Hofmann A. Ber. 1882 15. 2664.
1. Hofmann A. Ber. 1882 15. 2664.
2. Hentschell W. Ber. 1897. 30. 1434.
3. Rapp E., Rossler E. Arch. Farm. 1905. 243. 104.
4. Dowell С. Bray W. Am. Chem. Soc. 1917. 39. 896.
5. Chapin R. J. Am. Chem. Soc. 1931. 53. 912.
6. Metcalf W. J. Chem. Soc. 1932. 148.
7. Czech F., Fuches R., Antezak H. Anatyt. Chem. 1961. 33. 705.
8. Anber M. J. Chem. Soc. 1954. 1105.
9. Soulard M. Bloc F., Hatterer A. J. Chem. Soc. 1981. 2300.
10. Sisler H. Jnorg Chem. 1983, 22. 1449.
11. Belcher R., West Т., Williams M. J. Chem. Soc. 1957. 4323.
12. Hashmi M., Ali E., Umar M. Analyt. Chem. 1962. 34. 988-991.
Claims (1)
- Способ определения азота в биологических образцах, включающий отбор пробы, внесение перекиси водорода и серной кислоты, нагрев до выделения белого дыма и полного обесцвечивания, охлаждение, добавление дистиллированной воды, отличающийся тем, что дополнительно вносят боратный буфер, нейтрализуют раствором NaOH, затем вносят растворы марганца, гипохлорита натрия, KJ, H2SO4, добавляют крахмал и титруют до обесцвечивания, далее аналогичную процедуру выполняют и для контрольной пробы, после чего определяют содержание азота по формуле
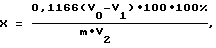
где V0 - объем Na2S2O3, пошедший на титрование контрольной пробы, мл;
V1 - объем Na2S2O3, пошедший на титрование пробы, мл;
V2 - объем зольного раствора пробы, взятой на анализ, мл;
m - масса навески, мг;
0,1166 - количество мг азота, эквивалентное 1 мл 0,025 н раствора Na2S2O3; 100 - исходный объем зольного раствора пробы, мл.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU99111126A RU2156458C1 (ru) | 1999-05-28 | 1999-05-28 | Способ определения азота в биологических образцах |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU99111126A RU2156458C1 (ru) | 1999-05-28 | 1999-05-28 | Способ определения азота в биологических образцах |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2156458C1 true RU2156458C1 (ru) | 2000-09-20 |
Family
ID=20220413
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU99111126A RU2156458C1 (ru) | 1999-05-28 | 1999-05-28 | Способ определения азота в биологических образцах |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2156458C1 (ru) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2216992C2 (ru) * | 2001-07-04 | 2003-11-27 | Всероссийский научно-исследовательский институт мясного скотоводства | Способ определения качества протеина кормов |
| RU2407003C1 (ru) * | 2009-07-20 | 2010-12-20 | Государственное образовательное учреждение высшего профессионального образования "Алтайский государственный университет" | Способ определения содержания азота в нитратах целлюлоз |
| CN104391076A (zh) * | 2014-11-03 | 2015-03-04 | 乌鲁木齐市华泰隆化学助剂有限公司 | 引发剂Tx-36中过氧化双(3,5,5-三甲基己酰)含量检测方法 |
| RU2554799C2 (ru) * | 2013-11-06 | 2015-06-27 | Федеральное государственное бюджетное научное учреждение "Северо-Кавказский зональный научно-исследовательский институт садоводства и виноградарства" | Способ определения общего азота методом капиллярного электрофореза |
| CN111693474A (zh) * | 2020-06-29 | 2020-09-22 | 中国第一重型机械股份公司 | 一种氨基氮/粗蛋白的新型检测方法 |
-
1999
- 1999-05-28 RU RU99111126A patent/RU2156458C1/ru active
Non-Patent Citations (1)
| Title |
|---|
| Кларк Г.Т. Руководство по качественному и количественному органическому анализу. ОНТИ НКТП Государственное научно-техническое издательство Украины, 1934, с.314-316. Колосов С.Г. и др. Физико-химические методы контроля ингредиентов, питательных сред и биопрепаратов./ Методическое пособие для биофабрик и ветеринарных лабораторий. М., 1970, с.4-11. * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2216992C2 (ru) * | 2001-07-04 | 2003-11-27 | Всероссийский научно-исследовательский институт мясного скотоводства | Способ определения качества протеина кормов |
| RU2407003C1 (ru) * | 2009-07-20 | 2010-12-20 | Государственное образовательное учреждение высшего профессионального образования "Алтайский государственный университет" | Способ определения содержания азота в нитратах целлюлоз |
| RU2554799C2 (ru) * | 2013-11-06 | 2015-06-27 | Федеральное государственное бюджетное научное учреждение "Северо-Кавказский зональный научно-исследовательский институт садоводства и виноградарства" | Способ определения общего азота методом капиллярного электрофореза |
| CN104391076A (zh) * | 2014-11-03 | 2015-03-04 | 乌鲁木齐市华泰隆化学助剂有限公司 | 引发剂Tx-36中过氧化双(3,5,5-三甲基己酰)含量检测方法 |
| CN111693474A (zh) * | 2020-06-29 | 2020-09-22 | 中国第一重型机械股份公司 | 一种氨基氮/粗蛋白的新型检测方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Viles Jr et al. | Determination of starch and cellulose with anthrone | |
| US8065906B2 (en) | Detection of free chlorine in water | |
| US3082146A (en) | Process for the treatment of water | |
| JPH069202A (ja) | 非水性媒体における二酸化塩素の製造方法 | |
| RU2156458C1 (ru) | Способ определения азота в биологических образцах | |
| Kolthoff et al. | Calcium hypochlorite as a volumetric oxidizing agent: Stability and standardization of the solution. Determination of ammonia | |
| Beattie et al. | Copper-catalyzed oxidation of cyanide by peroxide in alkaline aqueous solution | |
| Swank et al. | The determination of iron with mercaptoacetic acid | |
| Kleinberg et al. | Absorption spectrum of aqueous monochloramine solutions | |
| Holzbecher et al. | The fluorimetric determination of mercury | |
| Busch et al. | Bleaches and sterilants | |
| Johannesson | The determination of monobromamine and monochloramine in water | |
| EP0985929B1 (fr) | Solution aqueuse à base d'un colorant azoique, son procédé de fabrication et son utilisation | |
| Hosseini et al. | Spectrophotometric determination of chlorate ions in drinking water | |
| Ayres et al. | Catalytic Decomposition of Hypochlorite Solution by Iridium Compounds. I. The pH-Time Relationship1, 2 | |
| JP2006170897A (ja) | 化学的酸素要求量測定方法及び測定装置 | |
| WO2017075342A1 (en) | Storage stable standards for aqueous chlorine analysis | |
| Mary et al. | Spectrophotometric determination of iodine species in table salt and pharmaceutical preparations | |
| JPH11116205A (ja) | 二酸化塩素水製造時の原料溶液供給流量の選定方法及びこれを用いた二酸化塩素水の製造方法 | |
| Szabó et al. | On the iodometric determination of the bromide ion | |
| IT7370738A1 (it) | Procedimento per la produzione di blu di prussia | |
| Grampp et al. | Kinetics of the formation of the blue complex CrO (O2) 2 formed by dichromate and H2O2 in acid solutions. A stopped-flow investigation using rapid-scan UV-VIS detection | |
| SU1508146A1 (ru) | Способ определени белка в хлебопродуктах | |
| Tan et al. | A spectrofluorimetric method for the determination of small amounts of sulphate ion | |
| KR970000300B1 (ko) | 안정화 이산화염소 수용액의 제조방법 |