JP6481171B2 - 6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンから調製した(6r,10r)−6,10,14−トリメチルペンタデカン−2−オン - Google Patents
6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンから調製した(6r,10r)−6,10,14−トリメチルペンタデカン−2−オン Download PDFInfo
- Publication number
- JP6481171B2 JP6481171B2 JP2015547083A JP2015547083A JP6481171B2 JP 6481171 B2 JP6481171 B2 JP 6481171B2 JP 2015547083 A JP2015547083 A JP 2015547083A JP 2015547083 A JP2015547083 A JP 2015547083A JP 6481171 B2 JP6481171 B2 JP 6481171B2
- Authority
- JP
- Japan
- Prior art keywords
- dimethylundec
- formula
- group
- ketal
- carbon
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- COFFUYZBRGBAQS-FMIVXFBMSA-N (e)-6,10-dimethylundec-5-en-2-one Chemical compound CC(C)CCC\C(C)=C\CCC(C)=O COFFUYZBRGBAQS-FMIVXFBMSA-N 0.000 title claims description 48
- HNZUNIKWNYHEJJ-UHFFFAOYSA-N 6,10-dimethylundeca-5,9-dien-2-one Chemical compound CC(C)=CCCC(C)=CCCC(C)=O HNZUNIKWNYHEJJ-UHFFFAOYSA-N 0.000 title claims description 26
- LBWPYRZGHYVSEL-QZTJIDSGSA-N (6R,10R)-2,6,10-trimethylpentadecane Chemical compound C[C@H](CCCCC)CCC[C@@H](CCCC(C)C)C LBWPYRZGHYVSEL-QZTJIDSGSA-N 0.000 title 1
- 239000000203 mixture Substances 0.000 claims description 77
- 238000006243 chemical reaction Methods 0.000 claims description 65
- 238000000034 method Methods 0.000 claims description 56
- 238000009876 asymmetric hydrogenation reaction Methods 0.000 claims description 53
- 238000006317 isomerization reaction Methods 0.000 claims description 50
- WHWDWIHXSPCOKZ-UHFFFAOYSA-N hexahydrofarnesyl acetone Natural products CC(C)CCCC(C)CCCC(C)CCCC(C)=O WHWDWIHXSPCOKZ-UHFFFAOYSA-N 0.000 claims description 47
- -1 steps a) to f) is a Chemical compound 0.000 claims description 46
- WHWDWIHXSPCOKZ-IAGOWNOFSA-N (6r,10r)-6,10,14-trimethylpentadecan-2-one Chemical compound CC(C)CCC[C@@H](C)CCC[C@@H](C)CCCC(C)=O WHWDWIHXSPCOKZ-IAGOWNOFSA-N 0.000 claims description 45
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 44
- 150000002576 ketones Chemical class 0.000 claims description 43
- 229910052741 iridium Inorganic materials 0.000 claims description 41
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 claims description 39
- KEVYVLWNCKMXJX-UHFFFAOYSA-N 3,7,11,15-tetramethylhexadec-1-en-3-ol Chemical compound CC(C)CCCC(C)CCCC(C)CCCC(C)(O)C=C KEVYVLWNCKMXJX-UHFFFAOYSA-N 0.000 claims description 38
- RBGLEUBCAJNCTR-GFCCVEGCSA-N (6r)-6,10-dimethylundecan-2-one Chemical compound CC(C)CCC[C@@H](C)CCCC(C)=O RBGLEUBCAJNCTR-GFCCVEGCSA-N 0.000 claims description 32
- 239000003054 catalyst Substances 0.000 claims description 32
- CKJHJTOHWZLGFY-VAFHRPCXSA-N (e,10r)-6,10,14-trimethylpentadec-5-en-2-one Chemical group CC(C)CCC[C@@H](C)CCC\C(C)=C\CCC(C)=O CKJHJTOHWZLGFY-VAFHRPCXSA-N 0.000 claims description 28
- 239000011203 carbon fibre reinforced carbon Substances 0.000 claims description 27
- 238000005984 hydrogenation reaction Methods 0.000 claims description 27
- HSFWRNGVRCDJHI-UHFFFAOYSA-N Acetylene Chemical compound C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 26
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 25
- 238000004821 distillation Methods 0.000 claims description 25
- 125000000217 alkyl group Chemical group 0.000 claims description 24
- 150000001875 compounds Chemical class 0.000 claims description 23
- 230000008569 process Effects 0.000 claims description 23
- CKJHJTOHWZLGFY-UDLNKVAWSA-N (z,10r)-6,10,14-trimethylpentadec-5-en-2-one Chemical compound CC(C)CCC[C@@H](C)CCC\C(C)=C/CCC(C)=O CKJHJTOHWZLGFY-UDLNKVAWSA-N 0.000 claims description 22
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 22
- 238000000926 separation method Methods 0.000 claims description 21
- 239000000126 substance Substances 0.000 claims description 21
- 125000003118 aryl group Chemical group 0.000 claims description 19
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 claims description 18
- XLIJOVLDKFATNT-GICMACPYSA-N (7r)-3,7,11-trimethyldodec-1-en-3-ol Chemical compound CC(C)CCC[C@@H](C)CCCC(C)(O)C=C XLIJOVLDKFATNT-GICMACPYSA-N 0.000 claims description 17
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 17
- 239000000654 additive Substances 0.000 claims description 16
- COFFUYZBRGBAQS-XFXZXTDPSA-N (z)-6,10-dimethylundec-5-en-2-one Chemical compound CC(C)CCC\C(C)=C/CCC(C)=O COFFUYZBRGBAQS-XFXZXTDPSA-N 0.000 claims description 14
- 239000007818 Grignard reagent Substances 0.000 claims description 14
- 229910052799 carbon Inorganic materials 0.000 claims description 14
- 239000011981 lindlar catalyst Substances 0.000 claims description 14
- 125000005843 halogen group Chemical group 0.000 claims description 13
- 229910052731 fluorine Inorganic materials 0.000 claims description 12
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 12
- 229910052751 metal Inorganic materials 0.000 claims description 12
- 229910052757 nitrogen Inorganic materials 0.000 claims description 12
- CKJHJTOHWZLGFY-MRXNPFEDSA-N (10r)-6,10,14-trimethylpentadec-5-en-2-one Chemical class CC(C)CCC[C@@H](C)CCCC(C)=CCCC(C)=O CKJHJTOHWZLGFY-MRXNPFEDSA-N 0.000 claims description 11
- 125000004432 carbon atom Chemical group C* 0.000 claims description 11
- 239000002184 metal Substances 0.000 claims description 11
- 150000004703 alkoxides Chemical class 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- 238000006886 vinylation reaction Methods 0.000 claims description 10
- 239000002253 acid Substances 0.000 claims description 9
- HNZUNIKWNYHEJJ-FMIVXFBMSA-N geranyl acetone Chemical compound CC(C)=CCC\C(C)=C\CCC(C)=O HNZUNIKWNYHEJJ-FMIVXFBMSA-N 0.000 claims description 9
- HNZUNIKWNYHEJJ-XFXZXTDPSA-N geranylacetone Chemical compound CC(C)=CCC\C(C)=C/CCC(C)=O HNZUNIKWNYHEJJ-XFXZXTDPSA-N 0.000 claims description 9
- 230000007062 hydrolysis Effects 0.000 claims description 9
- 238000006460 hydrolysis reaction Methods 0.000 claims description 9
- 229920002554 vinyl polymer Polymers 0.000 claims description 8
- OWRXWSVBJIIORE-GICMACPYSA-N (7r)-3,7,11-trimethyldodec-1-yn-3-ol Chemical compound CC(C)CCC[C@@H](C)CCCC(C)(O)C#C OWRXWSVBJIIORE-GICMACPYSA-N 0.000 claims description 7
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 7
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 claims description 7
- 125000002723 alicyclic group Chemical group 0.000 claims description 7
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 claims description 7
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 claims description 6
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 claims description 6
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 claims description 6
- 230000000996 additive effect Effects 0.000 claims description 6
- 150000001450 anions Chemical class 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 239000013110 organic ligand Substances 0.000 claims description 6
- 125000006705 (C5-C7) cycloalkyl group Chemical group 0.000 claims description 5
- YOWQWFMSQCOSBA-UHFFFAOYSA-N 2-methoxypropene Chemical compound COC(C)=C YOWQWFMSQCOSBA-UHFFFAOYSA-N 0.000 claims description 5
- CKJHJTOHWZLGFY-UHFFFAOYSA-N 6,10,14-trimethylpentadec-5-en-2-one Chemical compound CC(C)CCCC(C)CCCC(C)=CCCC(C)=O CKJHJTOHWZLGFY-UHFFFAOYSA-N 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 claims description 5
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 4
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 claims description 4
- QYPLUMNWWBFAJS-UHFFFAOYSA-N dec-5-en-2-one Chemical compound CCCCC=CCCC(C)=O QYPLUMNWWBFAJS-UHFFFAOYSA-N 0.000 claims description 4
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 claims description 4
- WASQWSOJHCZDFK-UHFFFAOYSA-N diketene Chemical compound C=C1CC(=O)O1 WASQWSOJHCZDFK-UHFFFAOYSA-N 0.000 claims description 3
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 claims description 3
- 125000004437 phosphorous atom Chemical group 0.000 claims description 3
- 229910052723 transition metal Inorganic materials 0.000 claims description 3
- 125000006713 (C5-C10) cycloalkyl group Chemical group 0.000 claims description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 2
- 125000002993 cycloalkylene group Chemical group 0.000 claims description 2
- 238000007040 multi-step synthesis reaction Methods 0.000 claims description 2
- KHQOKXNPFOPAFV-UHFFFAOYSA-N 2,2-dimethylhexadecan-3-one Chemical compound CCCCCCCCCCCCCC(=O)C(C)(C)C KHQOKXNPFOPAFV-UHFFFAOYSA-N 0.000 claims 1
- 238000005481 NMR spectroscopy Methods 0.000 description 42
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 37
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- 238000002474 experimental method Methods 0.000 description 34
- 239000000243 solution Substances 0.000 description 32
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 28
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 27
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- 239000000047 product Substances 0.000 description 20
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 229940087168 alpha tocopherol Drugs 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 17
- 229960000984 tocofersolan Drugs 0.000 description 17
- 239000002076 α-tocopherol Substances 0.000 description 17
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 16
- 229920006295 polythiol Polymers 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000011541 reaction mixture Substances 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- 150000001298 alcohols Chemical class 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- 150000002503 iridium Chemical class 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 239000011732 tocopherol Substances 0.000 description 10
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 9
- 238000009835 boiling Methods 0.000 description 9
- 150000002009 diols Chemical class 0.000 description 9
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- 229960001295 tocopherol Drugs 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 238000004817 gas chromatography Methods 0.000 description 8
- 125000001424 substituent group Chemical group 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 7
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 150000001336 alkenes Chemical class 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 6
- 238000000354 decomposition reaction Methods 0.000 description 6
- 125000005677 ethinylene group Chemical group [*:2]C#C[*:1] 0.000 description 6
- 239000010408 film Substances 0.000 description 6
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 6
- 150000004795 grignard reagents Chemical class 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 229930003799 tocopherol Natural products 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 5
- 239000012159 carrier gas Substances 0.000 description 5
- 238000012512 characterization method Methods 0.000 description 5
- 238000006757 chemical reactions by type Methods 0.000 description 5
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 5
- 150000001993 dienes Chemical class 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 239000002798 polar solvent Substances 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 239000010936 titanium Substances 0.000 description 5
- 235000010384 tocopherol Nutrition 0.000 description 5
- LALRXNPLTWZJIJ-UHFFFAOYSA-N triethylborane Chemical compound CCB(CC)CC LALRXNPLTWZJIJ-UHFFFAOYSA-N 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- GVJHHUAWPYXKBD-QLVXXPONSA-N (S,R,R)-alpha-tocopherol Chemical compound [H][C@@](C)(CCCC(C)C)CCC[C@@]([H])(C)CCC[C@@]1(C)CCC2=C(O1)C(C)=C(C)C(O)=C2C GVJHHUAWPYXKBD-QLVXXPONSA-N 0.000 description 4
- YVSMQHYREUQGRX-UHFFFAOYSA-N 2-ethyloxaluminane Chemical compound CC[Al]1CCCCO1 YVSMQHYREUQGRX-UHFFFAOYSA-N 0.000 description 4
- BTVWZWFKMIUSGS-UHFFFAOYSA-N 2-methylpropane-1,2-diol Chemical compound CC(C)(O)CO BTVWZWFKMIUSGS-UHFFFAOYSA-N 0.000 description 4
- RBGLEUBCAJNCTR-UHFFFAOYSA-N 6,10-dimethylundecan-2-one Chemical compound CC(C)CCCC(C)CCCC(C)=O RBGLEUBCAJNCTR-UHFFFAOYSA-N 0.000 description 4
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 4
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- OWBTYPJTUOEWEK-UHFFFAOYSA-N butane-2,3-diol Chemical compound CC(O)C(C)O OWBTYPJTUOEWEK-UHFFFAOYSA-N 0.000 description 4
- 238000013375 chromatographic separation Methods 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 4
- IJMWREDHKRHWQI-UHFFFAOYSA-M magnesium;ethene;chloride Chemical compound [Mg+2].[Cl-].[CH-]=C IJMWREDHKRHWQI-UHFFFAOYSA-M 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- CPOFMOWDMVWCLF-UHFFFAOYSA-N methyl(oxo)alumane Chemical compound C[Al]=O CPOFMOWDMVWCLF-UHFFFAOYSA-N 0.000 description 4
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 4
- BOTWFXYSPFMFNR-PYDDKJGSSA-N phytol Chemical compound CC(C)CCC[C@@H](C)CCC[C@@H](C)CCC\C(C)=C\CO BOTWFXYSPFMFNR-PYDDKJGSSA-N 0.000 description 4
- YPFDHNVEDLHUCE-UHFFFAOYSA-N propane-1,3-diol Chemical compound OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- NHDIQVFFNDKAQU-UHFFFAOYSA-N tripropan-2-yl borate Chemical compound CC(C)OB(OC(C)C)OC(C)C NHDIQVFFNDKAQU-UHFFFAOYSA-N 0.000 description 4
- MYYZRSFBZQLURU-RTBURBONSA-N (6r,10r)-6,10,14-trimethyl-2,2-bis(2,2,2-trifluoroethoxy)pentadecane Chemical compound CC(C)CCC[C@@H](C)CCC[C@@H](C)CCCC(C)(OCC(F)(F)F)OCC(F)(F)F MYYZRSFBZQLURU-RTBURBONSA-N 0.000 description 3
- 239000001707 (E,7R,11R)-3,7,11,15-tetramethylhexadec-2-en-1-ol Substances 0.000 description 3
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 3
- KYCQOKLOSUBEJK-UHFFFAOYSA-M 1-butyl-3-methylimidazol-3-ium;bromide Chemical compound [Br-].CCCCN1C=C[N+](C)=C1 KYCQOKLOSUBEJK-UHFFFAOYSA-M 0.000 description 3
- AUFZRCJENRSRLY-UHFFFAOYSA-N 2,3,5-trimethylhydroquinone Chemical compound CC1=CC(O)=C(C)C(C)=C1O AUFZRCJENRSRLY-UHFFFAOYSA-N 0.000 description 3
- HBYIIHBZRWCZPX-NHCUHLMSSA-N 2,5,5-trimethyl-2-[(4r,8r)-4,8,12-trimethyltridecyl]-1,3-dioxane Chemical compound CC(C)CCC[C@@H](C)CCC[C@@H](C)CCCC1(C)OCC(C)(C)CO1 HBYIIHBZRWCZPX-NHCUHLMSSA-N 0.000 description 3
- HXLIOPFBVCGHSF-UHFFFAOYSA-N 2,6-dimethylundeca-2,6-diene Chemical compound CCCCC=C(C)CCC=C(C)C HXLIOPFBVCGHSF-UHFFFAOYSA-N 0.000 description 3
- QPRQEDXDYOZYLA-UHFFFAOYSA-N 2-methylbutan-1-ol Chemical compound CCC(C)CO QPRQEDXDYOZYLA-UHFFFAOYSA-N 0.000 description 3
- GNBPEYCZELNJMS-UHFFFAOYSA-N 2-methylbutane-1,3-diol Chemical compound CC(O)C(C)CO GNBPEYCZELNJMS-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 3
- CQTNEUQSGKLZQC-NTEUORMPSA-N CC(C)CCC\C(C)=C\CCC(C)(OCC(F)(F)F)OCC(F)(F)F Chemical compound CC(C)CCC\C(C)=C\CCC(C)(OCC(F)(F)F)OCC(F)(F)F CQTNEUQSGKLZQC-NTEUORMPSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 150000008282 halocarbons Chemical class 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 239000002608 ionic liquid Substances 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000005580 one pot reaction Methods 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 238000012856 packing Methods 0.000 description 3
- 238000002953 preparative HPLC Methods 0.000 description 3
- 150000003138 primary alcohols Chemical class 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 3
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 3
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 3
- VYXHVRARDIDEHS-QGTKBVGQSA-N (1z,5z)-cycloocta-1,5-diene Chemical compound C\1C\C=C/CC\C=C/1 VYXHVRARDIDEHS-QGTKBVGQSA-N 0.000 description 2
- ZHGXLFJFEQDEFC-NTEUORMPSA-N (6e)-2,6-dimethyl-10,10-bis(2,2,2-trifluoroethoxy)undeca-2,6-diene Chemical compound CC(C)=CCC\C(C)=C\CCC(C)(OCC(F)(F)F)OCC(F)(F)F ZHGXLFJFEQDEFC-NTEUORMPSA-N 0.000 description 2
- KQWCXLSGBMBWAD-XUDLPTGHSA-N (Z,10R)-6,10,14-trimethylpentadec-5-ene Chemical compound C/C(=C/CCCC)/CCC[C@@H](CCCC(C)C)C KQWCXLSGBMBWAD-XUDLPTGHSA-N 0.000 description 2
- OADXFGREZMDGOJ-SDNWHVSQSA-N (e)-2,2-dimethoxy-6,10-dimethylundec-5-ene Chemical compound COC(C)(OC)CC\C=C(/C)CCCC(C)C OADXFGREZMDGOJ-SDNWHVSQSA-N 0.000 description 2
- SHNUYFZWJWMWPH-CPNJWEJPSA-N (e)-6,10,14-trimethyl-2,2-bis(2,2,2-trifluoroethoxy)pentadec-5-ene Chemical compound CC(C)CCCC(C)CCC\C(C)=C\CCC(C)(OCC(F)(F)F)OCC(F)(F)F SHNUYFZWJWMWPH-CPNJWEJPSA-N 0.000 description 2
- SHNUYFZWJWMWPH-UIMCMNAESA-N (e,10r)-6,10,14-trimethyl-2,2-bis(2,2,2-trifluoroethoxy)pentadec-5-ene Chemical compound CC(C)CCC[C@@H](C)CCC\C(C)=C\CCC(C)(OCC(F)(F)F)OCC(F)(F)F SHNUYFZWJWMWPH-UIMCMNAESA-N 0.000 description 2
- SHNUYFZWJWMWPH-UYRXBGFRSA-N (z)-6,10,14-trimethyl-2,2-bis(2,2,2-trifluoroethoxy)pentadec-5-ene Chemical compound CC(C)CCCC(C)CCC\C(C)=C/CCC(C)(OCC(F)(F)F)OCC(F)(F)F SHNUYFZWJWMWPH-UYRXBGFRSA-N 0.000 description 2
- CQTNEUQSGKLZQC-ZROIWOOFSA-N (z)-6,10-dimethyl-2,2-bis(2,2,2-trifluoroethoxy)undec-5-ene Chemical compound CC(C)CCC\C(C)=C/CCC(C)(OCC(F)(F)F)OCC(F)(F)F CQTNEUQSGKLZQC-ZROIWOOFSA-N 0.000 description 2
- SHNUYFZWJWMWPH-ZJYZBMPFSA-N (z,10r)-6,10,14-trimethyl-2,2-bis(2,2,2-trifluoroethoxy)pentadec-5-ene Chemical compound CC(C)CCC[C@@H](C)CCC\C(C)=C/CCC(C)(OCC(F)(F)F)OCC(F)(F)F SHNUYFZWJWMWPH-ZJYZBMPFSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- MNCMBBIFTVWHIP-UHFFFAOYSA-N 1-anthracen-9-yl-2,2,2-trifluoroethanone Chemical group C1=CC=C2C(C(=O)C(F)(F)F)=C(C=CC=C3)C3=CC2=C1 MNCMBBIFTVWHIP-UHFFFAOYSA-N 0.000 description 2
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical compound CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 2
- WGLLSSPDPJPLOR-UHFFFAOYSA-N 2,3-dimethylbut-2-ene Chemical compound CC(C)=C(C)C WGLLSSPDPJPLOR-UHFFFAOYSA-N 0.000 description 2
- HBYIIHBZRWCZPX-UHFFFAOYSA-N 2,5,5-trimethyl-2-(4,8,12-trimethyltridecyl)-1,3-dioxane Chemical compound CC(C)CCCC(C)CCCC(C)CCCC1(C)OCC(C)(C)CO1 HBYIIHBZRWCZPX-UHFFFAOYSA-N 0.000 description 2
- HALGESSBEJXZMN-RCCKNPSSSA-N 2,5,5-trimethyl-2-[(e)-4,8,12-trimethyltridec-3-enyl]-1,3-dioxane Chemical compound CC(C)CCCC(C)CCC\C(C)=C\CCC1(C)OCC(C)(C)CO1 HALGESSBEJXZMN-RCCKNPSSSA-N 0.000 description 2
- XJUXWWIQZMRNLI-LFIBNONCSA-N 2-[(3e)-4,8-dimethylnona-3,7-dienyl]-2,5,5-trimethyl-1,3-dioxane Chemical compound CC(C)=CCC\C(C)=C\CCC1(C)OCC(C)(C)CO1 XJUXWWIQZMRNLI-LFIBNONCSA-N 0.000 description 2
- XJUXWWIQZMRNLI-WJDWOHSUSA-N 2-[(3z)-4,8-dimethylnona-3,7-dienyl]-2,5,5-trimethyl-1,3-dioxane Chemical compound CC(C)=CCC\C(C)=C/CCC1(C)OCC(C)(C)CO1 XJUXWWIQZMRNLI-WJDWOHSUSA-N 0.000 description 2
- OODATBVLFDZMTM-LFIBNONCSA-N 2-[(e)-4,8-dimethylnon-3-enyl]-2,5,5-trimethyl-1,3-dioxane Chemical compound CC(C)CCC\C(C)=C\CCC1(C)OCC(C)(C)CO1 OODATBVLFDZMTM-LFIBNONCSA-N 0.000 description 2
- OODATBVLFDZMTM-WJDWOHSUSA-N 2-[(z)-4,8-dimethylnon-3-enyl]-2,5,5-trimethyl-1,3-dioxane Chemical compound CC(C)CCC\C(C)=C/CCC1(C)OCC(C)(C)CO1 OODATBVLFDZMTM-WJDWOHSUSA-N 0.000 description 2
- PFNHSEQQEPMLNI-UHFFFAOYSA-N 2-methyl-1-pentanol Chemical compound CCCC(C)CO PFNHSEQQEPMLNI-UHFFFAOYSA-N 0.000 description 2
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical compound CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 2
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 2
- MXLMTQWGSQIYOW-UHFFFAOYSA-N 3-methyl-2-butanol Chemical compound CC(C)C(C)O MXLMTQWGSQIYOW-UHFFFAOYSA-N 0.000 description 2
- IWTBVKIGCDZRPL-UHFFFAOYSA-N 3-methylpentanol Chemical compound CCC(C)CCO IWTBVKIGCDZRPL-UHFFFAOYSA-N 0.000 description 2
- MYYZRSFBZQLURU-UHFFFAOYSA-N 6,10,14-trimethyl-2,2-bis(2,2,2-trifluoroethoxy)pentadecane Chemical compound CC(C)CCCC(C)CCCC(C)CCCC(C)(OCC(F)(F)F)OCC(F)(F)F MYYZRSFBZQLURU-UHFFFAOYSA-N 0.000 description 2
- LVIPBQZRBBDBOK-UHFFFAOYSA-N 6,10-dimethyl-2,2-bis(2,2,2-trifluoroethoxy)undecane Chemical compound CC(C)CCCC(C)CCCC(C)(OCC(F)(F)F)OCC(F)(F)F LVIPBQZRBBDBOK-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- 239000004358 Butane-1, 3-diol Substances 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229910020366 ClO 4 Inorganic materials 0.000 description 2
- KEVYVLWNCKMXJX-ZCNNSNEGSA-N Isophytol Natural products CC(C)CCC[C@H](C)CCC[C@@H](C)CCC[C@@](C)(O)C=C KEVYVLWNCKMXJX-ZCNNSNEGSA-N 0.000 description 2
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- BLUHKGOSFDHHGX-UHFFFAOYSA-N Phytol Natural products CC(C)CCCC(C)CCCC(C)CCCC(C)C=CO BLUHKGOSFDHHGX-UHFFFAOYSA-N 0.000 description 2
- 239000004146 Propane-1,2-diol Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 229910018286 SbF 6 Inorganic materials 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- HNZBNQYXWOLKBA-UHFFFAOYSA-N Tetrahydrofarnesol Natural products CC(C)CCCC(C)CCCC(C)=CCO HNZBNQYXWOLKBA-UHFFFAOYSA-N 0.000 description 2
- HFKJQIJFRMRSKM-UHFFFAOYSA-N [3,5-bis(trifluoromethyl)phenoxy]boronic acid Chemical compound OB(O)OC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 HFKJQIJFRMRSKM-UHFFFAOYSA-N 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- 150000001241 acetals Chemical class 0.000 description 2
- 238000005903 acid hydrolysis reaction Methods 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 2
- BOTWFXYSPFMFNR-OALUTQOASA-N all-rac-phytol Natural products CC(C)CCC[C@H](C)CCC[C@H](C)CCCC(C)=CCO BOTWFXYSPFMFNR-OALUTQOASA-N 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 229940063013 borate ion Drugs 0.000 description 2
- BMRWNKZVCUKKSR-UHFFFAOYSA-N butane-1,2-diol Chemical compound CCC(O)CO BMRWNKZVCUKKSR-UHFFFAOYSA-N 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 2
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 description 2
- 150000001983 dialkylethers Chemical group 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000011552 falling film Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 description 2
- 239000003205 fragrance Substances 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 2
- 238000005907 ketalization reaction Methods 0.000 description 2
- CDOSHBSSFJOMGT-UHFFFAOYSA-N linalool Chemical compound CC(C)=CCCC(C)(O)C=C CDOSHBSSFJOMGT-UHFFFAOYSA-N 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- ZOCHHNOQQHDWHG-UHFFFAOYSA-N n-hexan-3-ol Natural products CCCC(O)CC ZOCHHNOQQHDWHG-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000004237 preparative chromatography Methods 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 2
- 150000003460 sulfonic acids Chemical class 0.000 description 2
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- VOITXYVAKOUIBA-UHFFFAOYSA-N triethylaluminium Chemical compound CC[Al](CC)CC VOITXYVAKOUIBA-UHFFFAOYSA-N 0.000 description 2
- RSJKGSCJYJTIGS-UHFFFAOYSA-N undecane Chemical compound CCCCCCCCCCC RSJKGSCJYJTIGS-UHFFFAOYSA-N 0.000 description 2
- 238000005292 vacuum distillation Methods 0.000 description 2
- 239000001490 (3R)-3,7-dimethylocta-1,6-dien-3-ol Substances 0.000 description 1
- 239000001618 (3R)-3-methylpentan-1-ol Substances 0.000 description 1
- DNUUVMYAGFEOOX-SDNWHVSQSA-N (6e)-10,10-dimethoxy-2,6-dimethylundeca-2,6-diene Chemical compound COC(C)(OC)CC\C=C(/C)CCC=C(C)C DNUUVMYAGFEOOX-SDNWHVSQSA-N 0.000 description 1
- LVIPBQZRBBDBOK-CQSZACIVSA-N (6r)-6,10-dimethyl-2,2-bis(2,2,2-trifluoroethoxy)undecane Chemical compound CC(C)CCC[C@@H](C)CCCC(C)(OCC(F)(F)F)OCC(F)(F)F LVIPBQZRBBDBOK-CQSZACIVSA-N 0.000 description 1
- OINAUZAZTSKBGK-RTBURBONSA-N (6r,10r)-2,2-dimethoxy-6,10,14-trimethylpentadecane Chemical compound COC(C)(OC)CCC[C@H](C)CCC[C@H](C)CCCC(C)C OINAUZAZTSKBGK-RTBURBONSA-N 0.000 description 1
- RBGLEUBCAJNCTR-LBPRGKRZSA-N (6s)-6,10-dimethylundecan-2-one Chemical compound CC(C)CCC[C@H](C)CCCC(C)=O RBGLEUBCAJNCTR-LBPRGKRZSA-N 0.000 description 1
- ZHGXLFJFEQDEFC-ZROIWOOFSA-N (6z)-2,6-dimethyl-10,10-bis(2,2,2-trifluoroethoxy)undeca-2,6-diene Chemical compound CC(C)=CCC\C(C)=C/CCC(C)(OCC(F)(F)F)OCC(F)(F)F ZHGXLFJFEQDEFC-ZROIWOOFSA-N 0.000 description 1
- YRTZHYFUDNIOGV-YSSOQSIOSA-N (7R)-3,7,11-trimethyldodec-1-ene Chemical compound CC(C=C)CCC[C@@H](CCCC(C)C)C YRTZHYFUDNIOGV-YSSOQSIOSA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- CDOSHBSSFJOMGT-JTQLQIEISA-N (R)-linalool Natural products CC(C)=CCC[C@@](C)(O)C=C CDOSHBSSFJOMGT-JTQLQIEISA-N 0.000 description 1
- GAZJVINYNDPROS-BQYQJAHWSA-N (e)-undec-5-en-2-one Chemical compound CCCCC\C=C\CCC(C)=O GAZJVINYNDPROS-BQYQJAHWSA-N 0.000 description 1
- OADXFGREZMDGOJ-KAMYIIQDSA-N (z)-2,2-dimethoxy-6,10-dimethylundec-5-ene Chemical compound COC(C)(OC)CC\C=C(\C)CCCC(C)C OADXFGREZMDGOJ-KAMYIIQDSA-N 0.000 description 1
- UOCLXMDMGBRAIB-UHFFFAOYSA-N 1,1,1-trichloroethane Chemical compound CC(Cl)(Cl)Cl UOCLXMDMGBRAIB-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- ARKHCBWJALMQJO-UHFFFAOYSA-N 1,2-bis(ethenyl)cyclohexane Chemical compound C=CC1CCCCC1C=C ARKHCBWJALMQJO-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- DUJLUWPMQYPQCD-UHFFFAOYSA-N 1-ethoxypentadecane Chemical compound CCCCCCCCCCCCCCCOCC DUJLUWPMQYPQCD-UHFFFAOYSA-N 0.000 description 1
- FLVFPAIGVBQGET-UHFFFAOYSA-N 1-methylpyrrolidin-3-ol Chemical compound CN1CCC(O)C1 FLVFPAIGVBQGET-UHFFFAOYSA-N 0.000 description 1
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical compound C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- NCUBXPKFLQTRCW-UHFFFAOYSA-N 2,2-dimethylhexadec-6-en-3-one Chemical compound CCCCCCCCCC=CCCC(=O)C(C)(C)C NCUBXPKFLQTRCW-UHFFFAOYSA-N 0.000 description 1
- OLCFWQNMCDUEMF-UHFFFAOYSA-N 2,3-dihydroxy-6-methyl-4-propan-2-ylnaphthalene-1-carboxylic acid Chemical compound C1=C(C)C=C2C(C(C)C)=C(O)C(O)=C(C(O)=O)C2=C1 OLCFWQNMCDUEMF-UHFFFAOYSA-N 0.000 description 1
- SXSWMAUXEHKFGX-UHFFFAOYSA-N 2,3-dimethylbutan-1-ol Chemical compound CC(C)C(C)CO SXSWMAUXEHKFGX-UHFFFAOYSA-N 0.000 description 1
- HALGESSBEJXZMN-LNKKFFRNSA-N 2,5,5-trimethyl-2-[(e,8r)-4,8,12-trimethyltridec-3-enyl]-1,3-dioxane Chemical compound CC(C)CCC[C@@H](C)CCC\C(C)=C\CCC1(C)OCC(C)(C)CO1 HALGESSBEJXZMN-LNKKFFRNSA-N 0.000 description 1
- HALGESSBEJXZMN-QNGOZBTKSA-N 2,5,5-trimethyl-2-[(z)-4,8,12-trimethyltridec-3-enyl]-1,3-dioxane Chemical compound CC(C)CCCC(C)CCC\C(C)=C/CCC1(C)OCC(C)(C)CO1 HALGESSBEJXZMN-QNGOZBTKSA-N 0.000 description 1
- HALGESSBEJXZMN-YLCCHKQJSA-N 2,5,5-trimethyl-2-[(z,8r)-4,8,12-trimethyltridec-3-enyl]-1,3-dioxane Chemical compound CC(C)CCC[C@@H](C)CCC\C(C)=C/CCC1(C)OCC(C)(C)CO1 HALGESSBEJXZMN-YLCCHKQJSA-N 0.000 description 1
- PXSCEAJMCLQOBY-UHFFFAOYSA-N 2,6,6-trimethyloxaluminane Chemical compound C[Al]1CCCC(C)(C)O1 PXSCEAJMCLQOBY-UHFFFAOYSA-N 0.000 description 1
- HBERPQFEJXRHBX-UHFFFAOYSA-N 2,6-bis(sulfanylmethyl)heptanedioic acid Chemical compound C(CCC(C(=O)O)CS)C(C(=O)O)CS HBERPQFEJXRHBX-UHFFFAOYSA-N 0.000 description 1
- LAGGTOBQMQHXON-UHFFFAOYSA-N 2,6-octadiene Chemical compound CC=CCCC=CC LAGGTOBQMQHXON-UHFFFAOYSA-N 0.000 description 1
- PSYGHMBJXWRQFD-UHFFFAOYSA-N 2-(2-sulfanylacetyl)oxyethyl 2-sulfanylacetate Chemical compound SCC(=O)OCCOC(=O)CS PSYGHMBJXWRQFD-UHFFFAOYSA-N 0.000 description 1
- RKRMEYGGSCPEMK-UHFFFAOYSA-N 2-(4,8-dimethylnonyl)-2,5,5-trimethyl-1,3-dioxane Chemical compound CC(C)CCCC(C)CCCC1(C)OCC(C)(C)CO1 RKRMEYGGSCPEMK-UHFFFAOYSA-N 0.000 description 1
- KHSSGPGBNFORJH-UHFFFAOYSA-N 2-(4,8-dimethylnonyl)-2,5-dimethyl-1,3-dioxane Chemical compound CC(C)CCCC(C)CCCC1(C)OCC(C)CO1 KHSSGPGBNFORJH-UHFFFAOYSA-N 0.000 description 1
- OHWMJHMSUDKMGL-UHFFFAOYSA-N 2-(hydroxymethyl)cyclohexan-1-ol Chemical compound OCC1CCCCC1O OHWMJHMSUDKMGL-UHFFFAOYSA-N 0.000 description 1
- SMCMMJBLYPTGIT-NTEUORMPSA-N 2-[(3e)-4,8-dimethylnona-3,7-dienyl]-2-methyl-1,3-dioxolane Chemical compound CC(C)=CCC\C(C)=C\CCC1(C)OCCO1 SMCMMJBLYPTGIT-NTEUORMPSA-N 0.000 description 1
- SMCMMJBLYPTGIT-ZROIWOOFSA-N 2-[(3z)-4,8-dimethylnona-3,7-dienyl]-2-methyl-1,3-dioxolane Chemical compound CC(C)=CCC\C(C)=C/CCC1(C)OCCO1 SMCMMJBLYPTGIT-ZROIWOOFSA-N 0.000 description 1
- RKRMEYGGSCPEMK-MRXNPFEDSA-N 2-[(4r)-4,8-dimethylnonyl]-2,5,5-trimethyl-1,3-dioxane Chemical compound CC(C)CCC[C@@H](C)CCCC1(C)OCC(C)(C)CO1 RKRMEYGGSCPEMK-MRXNPFEDSA-N 0.000 description 1
- KHSSGPGBNFORJH-KLAILNCOSA-N 2-[(4r)-4,8-dimethylnonyl]-2,5-dimethyl-1,3-dioxane Chemical compound CC(C)CCC[C@@H](C)CCCC1(C)OCC(C)CO1 KHSSGPGBNFORJH-KLAILNCOSA-N 0.000 description 1
- WOGUFXCDRXLGSK-CQSZACIVSA-N 2-[(4r)-4,8-dimethylnonyl]-2-methyl-1,3-dioxolane Chemical compound CC(C)CCC[C@@H](C)CCCC1(C)OCCO1 WOGUFXCDRXLGSK-CQSZACIVSA-N 0.000 description 1
- RMZFBVITHXNIPQ-NTEUORMPSA-N 2-[(e)-4,8-dimethylnon-3-enyl]-2-methyl-1,3-dioxolane Chemical compound CC(C)CCC\C(C)=C\CCC1(C)OCCO1 RMZFBVITHXNIPQ-NTEUORMPSA-N 0.000 description 1
- RMZFBVITHXNIPQ-ZROIWOOFSA-N 2-[(z)-4,8-dimethylnon-3-enyl]-2-methyl-1,3-dioxolane Chemical compound CC(C)CCC\C(C)=C/CCC1(C)OCCO1 RMZFBVITHXNIPQ-ZROIWOOFSA-N 0.000 description 1
- TZYRSLHNPKPEFV-UHFFFAOYSA-N 2-ethyl-1-butanol Chemical compound CCC(CC)CO TZYRSLHNPKPEFV-UHFFFAOYSA-N 0.000 description 1
- JJSYPAGPNHFLML-UHFFFAOYSA-N 2-ethyl-2-(hydroxymethyl)propane-1,3-diol;3-sulfanylpropanoic acid Chemical compound OC(=O)CCS.OC(=O)CCS.OC(=O)CCS.CCC(CO)(CO)CO JJSYPAGPNHFLML-UHFFFAOYSA-N 0.000 description 1
- HXPYRCAWQDGUNN-RTBURBONSA-N 2-methyl-2-[(4r,8r)-4,8,12-trimethyltridecyl]-1,3-dioxolane Chemical compound CC(C)CCC[C@@H](C)CCC[C@@H](C)CCCC1(C)OCCO1 HXPYRCAWQDGUNN-RTBURBONSA-N 0.000 description 1
- FYLFGBUXVQLLQH-UIMCMNAESA-N 2-methyl-2-[(e,8r)-4,8,12-trimethyltridec-3-enyl]-1,3-dioxolane Chemical compound CC(C)CCC[C@@H](C)CCC\C(C)=C\CCC1(C)OCCO1 FYLFGBUXVQLLQH-UIMCMNAESA-N 0.000 description 1
- ISTJMQSHILQAEC-UHFFFAOYSA-N 2-methyl-3-pentanol Chemical compound CCC(O)C(C)C ISTJMQSHILQAEC-UHFFFAOYSA-N 0.000 description 1
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 1
- NBWLKCHXMMMNLN-UHFFFAOYSA-N 2-methyldodecan-3-one Chemical compound CCCCCCCCCC(=O)C(C)C NBWLKCHXMMMNLN-UHFFFAOYSA-N 0.000 description 1
- QWGRWMMWNDWRQN-UHFFFAOYSA-N 2-methylpropane-1,3-diol Chemical compound OCC(C)CO QWGRWMMWNDWRQN-UHFFFAOYSA-N 0.000 description 1
- DUXCSEISVMREAX-UHFFFAOYSA-N 3,3-dimethylbutan-1-ol Chemical compound CC(C)(C)CCO DUXCSEISVMREAX-UHFFFAOYSA-N 0.000 description 1
- IUDWWFNDSJRYRV-UHFFFAOYSA-N 3,7-dimethyloct-1-en-3-ol Chemical compound CC(C)CCCC(C)(O)C=C IUDWWFNDSJRYRV-UHFFFAOYSA-N 0.000 description 1
- KSXTZYRIJKDCEA-UHFFFAOYSA-N 3,7-dimethyloct-1-ene Chemical compound CC(C)CCCC(C)C=C KSXTZYRIJKDCEA-UHFFFAOYSA-N 0.000 description 1
- DKIDEFUBRARXTE-UHFFFAOYSA-M 3-mercaptopropionate Chemical compound [O-]C(=O)CCS DKIDEFUBRARXTE-UHFFFAOYSA-M 0.000 description 1
- ZXNBBWHRUSXUFZ-UHFFFAOYSA-N 3-methyl-2-pentanol Chemical compound CCC(C)C(C)O ZXNBBWHRUSXUFZ-UHFFFAOYSA-N 0.000 description 1
- RBQLGIKHSXQZTB-UHFFFAOYSA-N 3-methylpentane-2,4-diol Chemical compound CC(O)C(C)C(C)O RBQLGIKHSXQZTB-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- WVYWICLMDOOCFB-UHFFFAOYSA-N 4-methyl-2-pentanol Chemical compound CC(C)CC(C)O WVYWICLMDOOCFB-UHFFFAOYSA-N 0.000 description 1
- PCWGTDULNUVNBN-UHFFFAOYSA-N 4-methylpentan-1-ol Chemical compound CC(C)CCCO PCWGTDULNUVNBN-UHFFFAOYSA-N 0.000 description 1
- 125000004199 4-trifluoromethylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C(F)(F)F 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000016444 Benign adult familial myoclonic epilepsy Diseases 0.000 description 1
- JLLMOYPIVVKFHY-UHFFFAOYSA-N Benzenethiol, 4,4'-thiobis- Chemical compound C1=CC(S)=CC=C1SC1=CC=C(S)C=C1 JLLMOYPIVVKFHY-UHFFFAOYSA-N 0.000 description 1
- XDKUJXKPZMCOKN-CLZZGJSISA-N C[Si](C)(C)[C@](C(=O)O)(O)[C@H](O)C(=O)O Chemical compound C[Si](C)(C)[C@](C(=O)O)(O)[C@H](O)C(=O)O XDKUJXKPZMCOKN-CLZZGJSISA-N 0.000 description 1
- 238000006563 Carroll rearrangement reaction Methods 0.000 description 1
- 238000005821 Claisen rearrangement reaction Methods 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical group CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 238000007698 E/Z-isomerization reaction Methods 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- ABSPRNADVQNDOU-UHFFFAOYSA-N Menaquinone 1 Natural products C1=CC=C2C(=O)C(CC=C(C)C)=C(C)C(=O)C2=C1 ABSPRNADVQNDOU-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- JOBBTVPTPXRUBP-UHFFFAOYSA-N [3-(3-sulfanylpropanoyloxy)-2,2-bis(3-sulfanylpropanoyloxymethyl)propyl] 3-sulfanylpropanoate Chemical compound SCCC(=O)OCC(COC(=O)CCS)(COC(=O)CCS)COC(=O)CCS JOBBTVPTPXRUBP-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000006307 alkoxy benzyl group Chemical group 0.000 description 1
- 125000005233 alkylalcohol group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- VRPKUXAKHIINGG-UHFFFAOYSA-N biphenyl-4,4'-dithiol Chemical group C1=CC(S)=CC=C1C1=CC=C(S)C=C1 VRPKUXAKHIINGG-UHFFFAOYSA-N 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- PFURGBBHAOXLIO-OLQVQODUSA-N cis-cyclohexane-1,2-diol Chemical compound O[C@H]1CCCC[C@H]1O PFURGBBHAOXLIO-OLQVQODUSA-N 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- ZXIJMRYMVAMXQP-UHFFFAOYSA-N cycloheptene Chemical compound C1CCC=CCC1 ZXIJMRYMVAMXQP-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- UVJHQYIOXKWHFD-UHFFFAOYSA-N cyclohexa-1,4-diene Chemical compound C1C=CCC=C1 UVJHQYIOXKWHFD-UHFFFAOYSA-N 0.000 description 1
- PFURGBBHAOXLIO-UHFFFAOYSA-N cyclohexane-1,2-diol Chemical class OC1CCCCC1O PFURGBBHAOXLIO-UHFFFAOYSA-N 0.000 description 1
- PFURGBBHAOXLIO-WDSKDSINSA-N cyclohexane-1,2-diol Chemical compound O[C@H]1CCCC[C@@H]1O PFURGBBHAOXLIO-WDSKDSINSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- URYYVOIYTNXXBN-UPHRSURJSA-N cyclooctene Chemical compound C1CCC\C=C/CC1 URYYVOIYTNXXBN-UPHRSURJSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- FJBFPHVGVWTDIP-UHFFFAOYSA-N dibromomethane Chemical compound BrCBr FJBFPHVGVWTDIP-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- WTAZEIUCUCCYIA-ZIAGYGMSSA-N dipropan-2-yl (2r,3r)-2,3-bis(trimethylsilyloxy)butanedioate Chemical compound CC(C)OC(=O)[C@H](O[Si](C)(C)C)[C@@H](O[Si](C)(C)C)C(=O)OC(C)C WTAZEIUCUCCYIA-ZIAGYGMSSA-N 0.000 description 1
- XEBCWEDRGPSHQH-YUMQZZPRSA-N dipropan-2-yl (2s,3s)-2,3-dihydroxybutanedioate Chemical compound CC(C)OC(=O)[C@@H](O)[C@H](O)C(=O)OC(C)C XEBCWEDRGPSHQH-YUMQZZPRSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 208000016427 familial adult myoclonic epilepsy Diseases 0.000 description 1
- 235000019387 fatty acid methyl ester Nutrition 0.000 description 1
- ZGNITFSDLCMLGI-UHFFFAOYSA-N flubendiamide Chemical compound CC1=CC(C(F)(C(F)(F)F)C(F)(F)F)=CC=C1NC(=O)C1=CC=CC(I)=C1C(=O)NC(C)(C)CS(C)(=O)=O ZGNITFSDLCMLGI-UHFFFAOYSA-N 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- ZGXMNEKDFYUNDQ-UHFFFAOYSA-N hepta-1,5-diene Chemical compound CC=CCCC=C ZGXMNEKDFYUNDQ-UHFFFAOYSA-N 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical compound C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 description 1
- QNVRIHYSUZMSGM-UHFFFAOYSA-N hexan-2-ol Chemical compound CCCCC(C)O QNVRIHYSUZMSGM-UHFFFAOYSA-N 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- RJMMFJHMVBOLGY-UHFFFAOYSA-N indium(3+) Chemical compound [In+3] RJMMFJHMVBOLGY-UHFFFAOYSA-N 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229930007744 linalool Natural products 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- KAVKNHPXAMTURG-UHFFFAOYSA-N n-(4-bromonaphthalen-1-yl)acetamide Chemical compound C1=CC=C2C(NC(=O)C)=CC=C(Br)C2=C1 KAVKNHPXAMTURG-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- SJYNFBVQFBRSIB-UHFFFAOYSA-N norbornadiene Chemical compound C1=CC2C=CC1C2 SJYNFBVQFBRSIB-UHFFFAOYSA-N 0.000 description 1
- JFNLZVQOOSMTJK-KNVOCYPGSA-N norbornene Chemical compound C1[C@@H]2CC[C@H]1C=C2 JFNLZVQOOSMTJK-KNVOCYPGSA-N 0.000 description 1
- HITROERJXNWVOI-UHFFFAOYSA-N octa-1,5-diene Chemical compound CCC=CCCC=C HITROERJXNWVOI-UHFFFAOYSA-N 0.000 description 1
- 150000002898 organic sulfur compounds Chemical class 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 150000002905 orthoesters Chemical class 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- NWNISXPJONCMSX-UHFFFAOYSA-N pentadec-5-en-2-one Chemical compound CCCCCCCCCC=CCCC(C)=O NWNISXPJONCMSX-UHFFFAOYSA-N 0.000 description 1
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- BCTWNMTZAXVEJL-UHFFFAOYSA-N phosphane;tungsten;tetracontahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.P.[W].[W].[W].[W].[W].[W].[W].[W].[W].[W].[W].[W] BCTWNMTZAXVEJL-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- MBWXNTAXLNYFJB-NKFFZRIASA-N phylloquinone Chemical compound C1=CC=C2C(=O)C(C/C=C(C)/CCC[C@H](C)CCC[C@H](C)CCCC(C)C)=C(C)C(=O)C2=C1 MBWXNTAXLNYFJB-NKFFZRIASA-N 0.000 description 1
- 235000019175 phylloquinone Nutrition 0.000 description 1
- 239000011772 phylloquinone Substances 0.000 description 1
- SHUZOJHMOBOZST-UHFFFAOYSA-N phylloquinone Natural products CC(C)CCCCC(C)CCC(C)CCCC(=CCC1=C(C)C(=O)c2ccccc2C1=O)C SHUZOJHMOBOZST-UHFFFAOYSA-N 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 229960001898 phytomenadione Drugs 0.000 description 1
- DFOXKPDFWGNLJU-UHFFFAOYSA-N pinacolyl alcohol Chemical compound CC(O)C(C)(C)C DFOXKPDFWGNLJU-UHFFFAOYSA-N 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 229940074439 potassium sodium tartrate Drugs 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000004040 pyrrolidinones Chemical class 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- SIXSYDAISGFNSX-UHFFFAOYSA-N scandium atom Chemical compound [Sc] SIXSYDAISGFNSX-UHFFFAOYSA-N 0.000 description 1
- CFPVQGQJRGIWSP-UHFFFAOYSA-N scandium(3+) Chemical class [Sc+3] CFPVQGQJRGIWSP-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 238000009751 slip forming Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 238000011924 stereoselective hydrogenation Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CWERGRDVMFNCDR-UHFFFAOYSA-M thioglycolate(1-) Chemical compound [O-]C(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-M 0.000 description 1
- 150000003573 thiols Chemical group 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 235000019149 tocopherols Nutrition 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Substances COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 1
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 1
- NQVBDTYIJUEQLV-UHFFFAOYSA-N undeca-5,9-dien-2-one Chemical compound CC=CCCC=CCCC(C)=O NQVBDTYIJUEQLV-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- 150000003754 zirconium Chemical class 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
- QUEDXNHFTDJVIY-UHFFFAOYSA-N γ-tocopherol Chemical class OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1 QUEDXNHFTDJVIY-UHFFFAOYSA-N 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/17—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrogenation of carbon-to-carbon double or triple bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/44—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring increasing the number of carbon atoms by addition reactions, i.e. reactions involving at least one carbon-to-carbon double or triple bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/56—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds from heterocyclic compounds
- C07C45/57—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds from heterocyclic compounds with oxygen as the only heteroatom
- C07C45/59—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds from heterocyclic compounds with oxygen as the only heteroatom in five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/62—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by hydrogenation of carbon-to-carbon double or triple bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/70—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form
- C07C45/71—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form being hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/87—Preparation of ketenes or dimeric ketenes
- C07C45/90—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/04—1,3-Dioxanes; Hydrogenated 1,3-dioxanes
- C07D319/06—1,3-Dioxanes; Hydrogenated 1,3-dioxanes not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table compounds of the platinum group
- C07F15/0033—Iridium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/50—Organo-phosphines
- C07F9/5045—Complexes or chelates of phosphines with metallic compounds or metals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Seasonings (AREA)
- Fodder In General (AREA)
Description
本発明は、(6R,10R)−6,10,14−トリメチル−ペンタデカン−2−オンおよびその反応生成物に係る分野に関する。
(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンは、特に(R,R)−イソフィトール[=(3RS,7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−エン−3−オール]、(R,R)−フィトールおよびトコフェロールの合成に係る重要な中間体である。
従って、本発明により解決されるべき課題は、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンの製造プロセスの提供である。
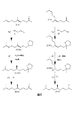
第1の態様において、本発明は、6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンからの多段合成において(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを製造するプロセスに関し、以下のステップを含む。
a)(E)−6,10−ジメチルウンデカ−5−エン−2−オンおよび(Z)−6,10−ジメチルウンデカ−5−エン−2−オンの混合物、または、(E)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンおよび(Z)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンの混合物を提供するステップ、
b)6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンの1種の異性体を、ステップa)の混合物から分離するステップ、
c)キラルイリジウム錯体の存在下において分子水素を用いて不斉水素化を行い、(R)−6,10−ジメチルウンデカン−2−オンを得るステップ、
d)(R)−6,10−ジメチルウンデカン−2−オンを、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチル−ペンタデカ−5−エン−2−オンの混合物に化学転換するステップ、
e)(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの1種の異性体を、ステップd)において得られた混合物から分離するステップ、
f)キラルイリジウム錯体の存在下において分子水素を用いて不斉水素化を行い、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを得るステップ。
ここで、ステップa)〜f)は、a、b、c、d、e、fの順番である。
ステップb)およびe)は、それぞれ、6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンの異性体の1種をステップa)の混合物から分離するステップ、および、(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの異性体の1種をステップd)において得られる混合物から分離するステップに関する。

シス/トランス異性化触媒は、炭素−炭素二重結合を異性化させる触媒である。本発明の目的について、5位および9位における二重結合のシス/トランス異性化を触媒する前記シス/トランス異性化は、特に一酸化窒素(NO)または有機硫黄化合物、特にポリチオールであることが見出された。
(式中、n1は1〜4の整数、特に2を表し、
m1は、2〜8の整数、特に3または4、好ましくは4を表し、
ならびに、Aは、28g/mol〜400g/mol、特に90〜150g/molの分子量のm1価の脂肪族炭化水素基を表す)
さらなる実施形態においては、ステップc)に先立って、ステップc0)が行われる。
c0)ステップb)において分離された6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンの異性体のケタールを形成するステップ。
f0)ステップe)において分離した(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの異性体のケタールを形成するステップ。
(式中、波線は、隣接する炭素−炭素二重結合に結合して、前記炭素−炭素二重結合をZ配置またはE配置のいずれかで有する、炭素−炭素結合を表し、
および、式中における点線を伴う二重結合
は、炭素−炭素単結合または炭素−炭素二重結合のいずれかを表し、
および、
は不斉中心を表し、
ならびに、式中、
Q1およびQ2は、
各々、もしくは、共に、C1〜C10アルキル基もしくはハロゲン化C1〜C10アルキル基を指し、
または、一緒になって、C2〜C6アルキレン基もしくはC6〜C8シクロアルキレン基を形成する。
Q1およびQ2は、特に、
直鎖C1〜C10アルキル基もしくはフッ素化直鎖C1〜C10アルキル基のいずれか、好ましくは直鎖C1〜C4アルキル基もしくは−CH2CF3基
または、Q3、Q4、Q5およびQ6が、相互に独立して、水素原子もしくはメチルもしくはエチル基である、式
の基を指す。)
ステップc)およびf)は、キラルイリジウム錯体の存在下における、分子水素による不斉水素化を含む。
(式中、
P−Q−Nは、不斉中心を含むか、または、面性もしくは軸キラリティーを有し、かつ、前記錯体のイリジウム中心に対する結合部位として窒素およびリン原子を有するキレート化有機リガンドを指し、
Y1、Y2、Y3およびY4は、相互に独立して、水素原子、C1〜12アルキル、C5〜10−シクロアルキルもしくは芳香族基であり;または、これらの少なくとも2つが一緒になって、少なくとも2個の炭素原子を有する少なくとも2価の架橋された基を形成し;ただし、Y1、Y2、Y3およびY4のすべてが水素原子とされることはなく;ならびに
は、アニオンであり、特に、ハライド、PF6 −、SbF6 −、テトラ(3,5−ビス(トリフルオロメチル)フェニル)ホウ酸イオン(BArF −)、BF4 −、過フッ素化スルホン酸イオン、好ましくはF3C−SO3 −またはF9C4−SO3 −;ClO4 −、Al(OC6F5)4 −、Al(OC(CF3)3)4 −、N(SO2CF3)2 − 、N(SO2C4F9)2 −およびB(C6F5)4 −からなる群から選択される)
(式中、G1は、C1〜C4アルキル、C5〜7−シクロアルキル、アダマンチル、フェニル(1〜3個のC1〜5アルキル、C1〜4アルコキシ、C1〜4−パーフルオロアルキル基および/または1〜5個のハロゲン原子により任意選択により置換されている)、ベンジル、1−ナフチル、2−ナフチル、2−フリル基のいずれかを表し、
G2、G3およびG4は、相互に独立して、水素原子またはC1〜C4アルキル、C5〜7−シクロアルキル、アダマンチル、フェニル(1〜3個のC1〜5−、C1〜4アルコキシ、C1〜4−パーフルオロアルキル基および/または1〜5個のハロゲン原子で任意選択により置換されている)、ベンジル、1−ナフチル、2−ナフチル、2−フリル基を表し、
X1およびX2は、相互に独立して、水素原子、C1〜4アルキル、C5〜7−シクロアルキル、アダマンチル、フェニル(1〜3個のC1〜5アルキル、C1〜4アルコキシ、C1〜4−パーフルオロアルキル基および/または1〜5個のハロゲン原子により任意選択により置換されている)、ベンジル、1−ナフチル、2−ナフチル、2−フリルまたはフェロセニルであり、
Phはフェニルを指し、
nは、1または2または3、好ましくは1または2であり、
およびR1、Z1およびZ2は、式(III)について以下に定義されているとおりである)
(式中、
nは、1または2または3、好ましくは1または2であり、
X1およびX2は、相互に独立して、水素原子、C1〜4アルキル、C5〜7−シクロアルキル、アダマンチル、フェニル(1〜3個のC1〜5−、C1〜4アルコキシ、C1〜4−パーフルオロアルキル基および/または1〜5個のハロゲン原子で任意選択により置換されている)、ベンジル、1−ナフチル、2−ナフチル、2−フリルまたはフェロセニルであり、
Z1およびZ2は、相互に独立して、水素原子、C1〜5アルキルまたはC1〜5アルコキシ基であり、
または、Z1およびZ2は、一緒になって、5〜6員環を形成する架橋基を指し、
は、アニオンであり、特に、ハライド、PF6 −、SbF6 −、テトラ(3,5−ビス(トリフルオロメチル)フェニル)ホウ酸イオン(BArF −)、BF4 −、過フッ素化スルホン酸イオン、好ましくはF3C−SO3 −またはF9C4−SO3 −;ClO4 −、Al(OC6F5)4 −、Al(OC(CF3)3)4 −、N(SO2CF3)2 − 、N(SO2C4F9)2 −およびB(C6F5)4 −からなる群から選択され、
R1は、フェニルもしくはo−トリルもしくはm−トリルもしくはp−トリル、または、式(IVa)もしくは(IVb)もしくは(IVc)の基のいずれかを表し、
(式中、R2およびR3は、共にHもしくはC1〜C4アルキル基もしくはハロゲン化C1〜C4アルキル基を表すか、または、ハロゲン原子もしくはC1〜C4アルキル基もしくはC1〜C4アルコキシ基により任意選択により置換されている6員脂環式もしくは芳香族環を一緒になって形成する二価の基を表し、
R4およびR5は、共にHもしくはC1〜C4アルキル基もしくはハロゲン化C1〜C4アルキル基、または、ハロゲン原子もしくはC1〜C4アルキル基もしくはC1〜C4アルコキシ基により任意選択により置換されている6員脂環式もしくは芳香族環を一緒になって形成する二価の基を表し、
R6およびR7およびR8は、各々、C1〜C4アルキル基またはハロゲン化C1〜C4アルキル基を表し、
R9およびR10は、共にHもしくはC1〜C4アルキル基もしくはハロゲン化C1〜C4アルキル基、または、ハロゲン原子もしくはC1〜C4アルキル基もしくはC1〜C4アルコキシ基により任意選択により置換されている6員脂環式もしくは芳香族環を一緒になって形成する二価の基を表す)
ならびに、*は、式(III)の錯体の不斉中心を表す)
(式中、
は不斉中心を表し、
ならびに、Q1およびQ2は式(XI)および(XII)について定義されているとおりである)
(R)−2−(4,8−ジメチルノニル)−2,5,5−トリメチル−1,3−ジオキサン、(R)−6,10−ジメチル−2,2−ビス(2,2,2−トリフルオロエトキシ)ウンデカン、2,5,5−トリメチル−2−((4R,8R)−4,8,12−トリメチル−トリデシル)−1,3−ジオキサンおよび(6R,10R)−6,10,14−トリメチル−2,2−ビス(2,2,2−トリフルオロエトキシ)−ペンタデカン
からなる群から選択されることが好ましい。
(E)−6,10−ジメチルウンデカ−5−エン−2−オン、(E)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンもしくはそのケタール、または、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンもしくはそのケタールが水素化される事例においては、*で示す不斉中心でS配置を有し、
または、
(Z)−6,10−ジメチルウンデカ−5−エン−2−オン、(Z)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンもしくはそのケタール、または、(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンもしくはそのケタールが水素化される事例においては、*で示す不斉中心でR配置を有する。
本発明好ましい実施形態において、ステップc)および/またはステップf)における不斉水素化は、有機スルホン酸、有機スルホン酸の遷移金属塩、金属アルコキシド、アルミノキサン、アルキルアルミノキサンおよびB(R)(3−v)(OZ)vからなる群から選択される添加剤の存在下で行われ、式中、vは0、1、2または3を指し、Rは、F、C1〜6アルキル、ハロゲン化C1〜6アルキル、アリールまたはハロゲン化アリール基を指し;および、Zは、C1〜6アルキル、ハロゲン化C1〜6アルキル、アリールまたはハロゲン化アリール基を指す。
ステップd)において、(R)−6,10−ジメチルウンデカン−2−オンは、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物に化学転換される。
d1)塩基性物質の存在下においてエチンを用いて、(7R)−3,7,11−トリメチルドデカ−1−イン−3−オールを得る(R)−6,10−ジメチルウンデカン−2−オンのエチニル化、
d2)リンドラー触媒の存在下において、(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを得る、分子水素による(7R)−3,7,11−トリメチルドデカ−1−イン−3−オールの水素化、
または
d1’)ビニルグリニャール試薬の添加により(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを得る(R)−6,10−ジメチルウンデカン−2−オンのビニル化、
のいずれかのステップ、これに続いて、
d3)(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールと2−メトキシプロプ−1−エンとを反応させて、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物を得るステップ、
または
d3’)(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを、塩基および/または酸の存在下でアルキルアセト酢酸またはジケテンと反応させて、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物を得るステップ
のいずれかによって行われる。
上記において詳述されているとおり、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを製造するプロセス、
これに続いて、
g)塩基の存在下においてエチンを用いて、(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエチニル化、
h)リンドラー触媒の存在下において、(R,R)−イソフィトールを得る、分子水素による(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールの水素化、
または
h’)ビニルグリニャール試薬の添加により(R,R)−イソフィトールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのビニル化
のいずれかのステップを含む。
上記において詳述されているとおり、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを製造するプロセス、
これに続いて、
g)塩基性物質の存在下においてエチンを用いて、(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエチニル化、
h)リンドラー触媒の存在下において、(R,R)−イソフィトールを得る、分子水素による(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールの水素化、
または
h’)ビニルグリニャール試薬の添加により(R,R)−イソフィトールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのビニル化、
のいずれかのステップ、これに続いて、
m)(R,R)−イソフィトールと式(VI)の化合物とを縮合して、#によって示される中心に係るキラリティーについて異性体混合物である式(V)の化合物を得るステップ
(式中、#は不斉中心を表す)
を含む。
上記において詳述されているとおり、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを製造するプロセス、
これに続いて、
g)塩基性物質の存在下においてエチンを用いて、(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエチニル化、
h)リンドラー触媒の存在下において、(R,R)−イソフィトールを得る、分子水素による(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールの水素化、
または
h’)ビニルグリニャール試薬の添加により(R,R)−イソフィトールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのビニル化、
のいずれかのステップ、これに続いて、
m)(R,R)−イソフィトールと式(VI)の化合物とを縮合して、#によって示される中心に係るキラリティーについて異性体混合物である式(V)の化合物を得るステップ、
(式中、#は不斉中心を表す)、
および
n)式(V−A)の化合物を式(V)の異性体混合物から単離するステップ
を含む。
(E)−6,10−ジメチルウンデカ−5−エン−2−オンのケタール、(Z)−6,10−ジメチル−ウンデカ−5−エン−2−オンのケタール、(E)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンのケタール、(Z)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンのケタール;(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オン、(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オン、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンのケタール、(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンのケタール、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オン、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのケタール、(7R)−3,7,11−トリメチルドデカ−1−イン−3−オール、(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールおよび(R,R)−イソフィトールから選択される物質が、トコフェロール、ビタミンK1、ならびに、香味料および芳香剤または薬学生成物の合成に係る重要な中間体である。これらの大半は特徴的な香りを有するため、これらは、香味料、および、香水などの芳香剤に係る産業の製品における処方成分としての使用にきわめて魅力を有するものである。
− 式(XI)または(XII)の少なくとも1種のケタール、および
− 少なくとも1種のキラルイリジウム錯体
を含む組成物に関する。
(式中、上記式中の点線を伴う二重結合
は炭素−炭素単結合または炭素−炭素二重結合のいずれかを表し;および
波線は、隣接する炭素単結合(
が
を表す)または隣接する炭素−炭素二重結合(
が
を表す)と結合して、前記炭素−炭素二重結合をZ配置またはE配置のいずれかで有する、炭素−炭素結合を表す)
以下の段落において、いくつかの本発明の好ましい実施形態は、概略図1〜6によりさらに考察されている。しかしながら、これは、ここでこれらの図面に記載されている実施形態に本発明が限定されるものと理解されるべきではない。
本発明を、以下の実験によりさらに例示する。
[6,10−ジメチルウンデカ−5−エン−2−オン(DHGA)、(R)−6,10−ジメチルウンデカン−2−オン(THGA)および(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オン(R−THFA)のE/Z比および/または純度のGC定量:]
Agilent 6850、カラムDB−5HT(30m、0.25mm直径、0.10μmフィルム厚)、107kPaヘリウムキャリアガス)。サンプルを、ヘキサン中の溶液として、分割比300:1、インジェクタ温度200℃、検出器温度350℃で注入した。オーブン温度プログラム:100℃(8分間)、200℃まで10℃/分(1分間)、220℃まで20℃/分(4分間)、ランタイム24分間。
Agilent 6850、カラムDB−5HT(30m、0.25mm直径、0.10μmフィルム厚)、115kPaヘリウムキャリアガス)。サンプルを、ヘキサン中の溶液として、分割比300:1、インジェクタ温度200℃、検出器温度350℃で注入した。オーブン温度プログラム:120℃(5分間)、260℃まで14℃/分(2分間)、280℃まで20℃/分(4分間)、ランタイム22分間。
FID.Agilent DB−5カラム(30m、0.32mm直径、0.25μmフィルム厚)を備えるAgilent 6850機器、25psi 分子水素キャリアガス。サンプルを、アセトニトリル中の溶液として、50:1の分割比で注入した。インジェクタ温度:250℃、検出器温度:350℃。オーブン温度プログラム:100℃、250℃まで4℃/分。
Agilent 6850機器、カラムAgilent DB−5(123−5032E、30m×0.32mm、フィルム0.25μm)、サンプルを、アセトニトリル中の溶液として、分割比50:1、インジェクタ250℃、検出器350℃で注入した。オーブン温度プログラム:100℃、250℃まで4℃/分、合計ランタイム37.5分間。
対応するジメチル、エチレングリコール、ネオペンチルおよびビス(トリフルオロエチル)ケタールを水性酸の存在下でケトンに加水分解し、ケトンに対して以下の方法を用いて転換率およびその立体異性体比を分析した。
FID.Agilent HP−5カラム(30m、0.32mm直径、0.25μmフィルム厚)を備えるAgilent 7890A GC、25psi分子水素キャリアガス。サンプルを、ジクロロメタン中の溶液として、10:1の分割比で注入した。インジェクタ温度:250℃、検出器温度:300℃。オーブン温度プログラム:50℃(2分間)、次いで、300℃まで15℃/分、5分間保持。
FAMEカラム用FID.Agilent CP−Sil88(60m、0.25mm直径、0.20μmフィルム厚)を備えるAgilent 6890N GC、16psi分子水素キャリアガス。サンプルを、酢酸エチル中の溶液として、5:1の分割比で注入した。インジェクタ温度:250℃、FID検出器温度:250℃。オーブン温度プログラム:165℃(恒温、240分間)。
7.02kgの6,10−ジメチルウンデカ−5−エン−2−オンを独国特許第1 193 490号明細書の実施例10に従って調製し、上記のGC法により、(E)−6,10−ジメチルウンデカ−5−エン−2−オンおよび(Z)−6,10−ジメチルウンデカ−5−エン−2−オンの57%/43%混合物(99%純度)であると分析した。
異性体(E)−6,10−ジメチルウンデカ−5−エン−2−オン(E−DHGA)(E/Z=99.5/0.5)および(Z)−6,10−ジメチルウンデカ−5−エン−2−オン(Z−DHGA)(Z/E=99/1)の両方を不斉水素化し、以下の方法で相互を分離した。
[a)ジメチルケタールの調製]
6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オン(170.5mmol)をトリメチルオルトギ酸エステル(50.8ml、49.2g、451mmol、2.65当量)に添加し、5℃に冷却した。MeOH(16ml)中の硫酸(96%、32.3mg、0.29mmol、0.2mol%)を5分間以内に添加した。その後、この反応を還流(65℃ IT)に3時間加熱した。冷却した後、薄層クロマトグラフィ(TLC)による分析は完全な転換を示した。NaOMe(0.24mLのMeOH中の25%溶液)を添加して酸を中和した。混合物を減圧中で濃縮し、その後、ヘキサン(50mL)で希釈した。現れた沈殿物をろ出し、濾液を濃縮した。粗生成物を蒸留により精製して、所望のジメチルケタールを得た。
[(E)−10,10−ジメトキシ−2,6−ジメチルウンデカ−2,6−ジエン(E−GA−DM)]
1H NMR(300MHz,CDCl3):δ1.26(s,3H),1.58(s,3H),1.60(s,3H),1.60〜1.65(m,2H)と重畳,1.66(br s,3H),1.92〜2.09(m,6H),3.17(s,6H),5.02〜5.14(m,2H)ppm。
13C NMR(75MHz,CDCl3):δ15.9(1C),17.6(1C),20.8(1C),22.8(1C),25.6(1C),26.6(1C),36.4(1C),39.6(1C),47.9(2C),101.4(1C),123.8(1C),124.2(1C),131.2(1C),135.1(1C)ppm。
MS(El,m/z):240(M+,<1),225.3[(M−CH3)+,1],209.3[(M−CH3O)+,20],193.3(8),176.2(18),161.2(16),139.2(20),123.2(14),107.2(75),89.2(100),69.2(65),41.1(56)。
IR(cm−1):2928(m),2857(w),2828(w),1670(w),1452(m),1376(s),1345(w),1302(w),1262(w),1222(w),1196(m),1172(m),1123(s),1102(s),1053(s),985(w),929(w),854(s),744(w),619(w)。
1H NMR(300MHz,CDCl3):δ1.27(s,3H),1.56〜1.65(m,5H),1.68(br s,6H),1.96〜2.09(m,6H),3.17(s,6H),5.11(t,J=7.2Hz,2H)ppm。
13C NMR(75MHz,CDCl3):δ17.6(1C),20.9(1C),22.7(1C),23.3(1C),25.7(1C),26.6(1C),31.9(1C),36.7(1C),48.0(2C),101.4(1C),124.2(1C),124.6(1C),131.5(1C),135.4(1C)ppm。
MS(El,m/z):カラムにおける分解のためにGC−MSは入手しなかった。
IR(cm−1):2943(m),2858(w),2828(w),1451(m),1376(m),1348(w),1301(w),1261(w),1197(m),1172(m),1153(w),1120(s),1098(m),1053(s),929(w),854(m),833(m),745(w),622(w)。
1H NMR(300MHz,CDCl3):δ0.83(d,J=6.6Hz,6H),1.02〜1.13(m,2H),1.24(s,3H),1.27〜1.39(m,2H),1.49(tqq,J=6.4,6.4,6,4Hz,1H),1.53〜1.63(m,2H)と重畳,1.56(s,3H)と重畳,1.87〜2.03(m,4H),3.13(s,6H),5.07(tq,J=7.0,1.4Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ16.1(1C),21.2(1C),23.0(2C),23.2(1C),26.0(1C),28.2(1C),36.9(1C),39.0(1C),40.2(1C),48.3(2C),101.8(1C),124.0(1C),135.9(1C)ppm。
MS(El,m/z):カラムにおける分解のためにGC−MSは入手しなかった。
IR(cm−1):2953(s),2931(s),2870(m),2828(m),2108(w),1668(w),1460(m),1377(s),1367(m),1345(w),1301(w),1262(m),1221(m),1198(m),1172(s),1119(s),1100(s),1077(s),1053(s),967(w),927(w),854(w),796(w),739(w),620(w)。
1H NMR(300MHz,CDCl3):δ0.88(d,J=6.6Hz,6H),1.12〜1.21(m,2H),1.28(s,3H),1.32〜1.43(m,2H),1.53(dspt,J=6.6,6.6Hz,1H),1.57〜1.66(m,2H),1.68(q,J=1.1Hz,3H),1.94〜2.06(m,4H),3.18(s,6H),5.10(t,J=6.8Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ20.9(1C),22.6(2C),22.7(1C),23.3(1C),25.8(1C),27.9(1C),31.9(1C),36.8(1C),38.9(1C),48.0(2C),101.5(1C),124.3(1C),135.9(1C)ppm。
MS(El,m/z):カラムにおける分解のためにGC−MSは得られなかった。
IR(cm−1):2953(s),2870(w),2828(w),1461(w),1376(m),1301(w),1261(w),1205(m),1172(m),1119(m),1097(m),1074(m),1053(s),1022(w),927(w),854(m),738(w),621(w)。
窒素下で、反応容器に、グリコール(112mL、125g、2.1mol)、p−トルエンスルホン酸一水和物(0.150g、0.5774mmol)、および、0.5molの(E)−6,10−ジメチルウンデカ−5−エン−2−オンまたは(Z)−6,10−ジメチルウンデカ−5−エン−2−オンのいずれかを仕込んだ。この混合物を、周囲温度で、5時間、減圧下(0.39mbar)で撹拌した。減圧を維持しながら、温度をゆっくりと40℃に昇温した。ケトンの95%を超える転換率では、温度をさらに高くして、グリコールを穏やかに蒸留し、99%を超える転換率が達成されるまで継続した。
[(E)−2−(4,8−ジメチルノン−3−エン−1−イル)−2−メチル−1,3−ジオキソラン(E−DHGA−エン)]
1H NMR(300MHz,CDCl3)δ5.12(t,1H),3.95(m,4H),2.2〜2(m,2H),1.94(t,2H),1.8〜1.3(m,11H),1.2〜1.0(m,2H),0.87(d,6H)ppm。
1H NMR(300MHz,CDCl3)δ5.12(t,1H),3.94(m,4H),2.15〜1.9(m,4H),1.7〜1.45(m,6H),1.44〜1.27(m,5H),1.23〜1.08(m,2H),0.88(d,6H)ppm。
表2d(iii)中に示すケトン(90.7mmol)、2,2−ジメチル−1,3−プロパンジオール(ネオペンチルグリコール、32.4g、283mmol、3.4当量)およびp−トルエンスルホン酸一水和物(60mg、0.31mmol、0.3mol%)をトルエン(300mL)中に懸濁させた。この反応を90℃に加熱したところ、均質な溶液が形成された。その後、75℃で、トルエンをゆっくりと蒸発させる(4時間でおよそ100mL)ために注意深く減圧(先ず63mbarで、次いで24mbar)した。4時間後、薄層クロマトグラフィ(TLC)による分析はケトンの完全な転換を示した。この反応を室温に冷却し、ヘプタン(300mL)で希釈したところ過剰量のネオペンチルグリコールが析出した。沈殿物をろ出した(17.4g wet)。濾液をEt3N(1mL)で処理し、続いて、NaHCO3水溶液(2.4%w/w、2×300mL)で洗浄し、MgSO4で乾燥させ、減圧中で濃縮した。粗生成物を蒸留により精製し、所望のネオペンチルケタールを得た。ケタールの特性付けを以下に詳細に示す。
[(E)−2−(4,8−ジメチルノナ−3,7−ジエン−1−イル)−2,5,5−トリメチル−1,3−ジオキサン(E−GA−neo)]
1H NMR(300MHz,CDCl3):δ0.92(s,3H),0.99(s,3H),1.37(s,3H),1.59(s,3H),1.61(s,3H),1.67(s,3H),1.68〜1.75(m,2H),1.94〜2.15(m,6H),ABシグナル(δA=3.46,δB=3.52,JAB=11.3Hz,4H),5.05〜5.17(m,2H)ppm。
13C NMR(75MHz,CDCl3):δ15.9(1C),17.6(1C),20.8(1C),22.0(1C),22.6(1C),22.7(1C),25.6(1C),26.7(1C),29.9(1C),37.3(1C),39.6(1C),70.3(2C),98.8(1C),124.1(1C),124.3(1C),131.2(1C),135.1(1C)ppm。
MS(El,m/z):280(M+,3),265[(M−CH3)+,14],176(21),129[(C7H13O2)+,100],69(63),43(43)。
IR(cm−1):2954(m),2925(m),2858(m),2731(w),1720(w),1669(w),1473(w),1450(m),1394(m),1372(m),1349(w),1306(w),1271(w),1249(m),1211(m),1186(m),1123(s),1088(s),1043(m),1021(m),984(w),950(w),925(w),907(w),862(m),837(w),792(w),742(w),677(w),667(w)。
1H NMR(300MHz,CDCl3):δ0.91(s,3H),0.97(s,3H),1.35(s,3H),1.60(s,3H),1.67(br s,3H)が重畳した1.64〜1.74(m,5H),1.99〜2.18(m,6H),ABシグナル(δA=3.44,δB=3.51,JAB=11.3Hz,4H),5.07〜5.16(m,2H)ppm。
13C NMR(75MHz,CDCl3):δ17.5(1C),20.9(1C),21.3(1C),21.9(1C),22.5(1C),22.6(1C),23.3(1C),25.7(1C),26.6(1C),29.9(1C),31.8(1C),37.5(1C),70.3(1C),98.7(1C),124.3(1C),124.9(1C),131.4(1C),135.2(1C)ppm。
MS(El,m/z):280(M+,3),265[(M−CH3)+,13],176(19),129[(C7H13O2)+,100],107()15,69(62),43(39)。
IR(cm−1):2954(m),2927(m),2858(m),2729(w),1721(w),1671(w),1473(m),1450(m),1394(m),1374(m),1349(w),1315(w),1271(m),1249(m),1211(m),1187(m),1149(w),1120(s),1086(s),1043(m),1021(m),985(w),951(m),925(w),907(m),857(m),833(m),792(w),743(w),677(w),667(w)。
1H NMR(300MHz,CDCl3):δ0.87(d,J=6.6Hz,6H),0.93(s,3H),1.00(s,3H),1.06〜1.22(m,2H),1.38(s,3H)が重畳した1.31〜1.43(m,2H),1.53(tqq,J=6.6,6.6,6.6Hz,1H),1.61(br s,3H),1.65〜1.77(m,2H),1.94(t,J=7.5Hz,2H),2.05〜2.17(m,2H),ABシグナル(δA=3.46,δB=3.54,JAB=11.4Hz,4H),5.13(tq,J=7.1,1.1Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ15.8(1C),20.9(1C),22.0(1C),22.59(1C),22.63(2C),22.7(1C),25.7(1C),27.9(1C),29.9(1C),37.3(1C),38.6(1C),39.9(1C),70.3(2C),98.8(1C),123.8(1C),135.6(1C)ppm。
MS(El,m/z):282(M+,5),267[(M−CH3)+,10),129(100),95(14),69(36),43(32)。
IR(cm−1):2953(s),2929(m),2868(m),1720(w),1468(m),1394(m),1381(m),1368(m),1349(w),1306(w),1270(w),1250(m),1211(m),1187(w),1118(s),1087(s),1066(m),1044(m),1022(m),950(m),925(w),907(m),862(m),791(w),739(w),677(w),666(w)。
1H NMR(300MHz,CDCl3):δ0.87(d,J=6.6Hz,6H),0.93(s,3H),0.97(s,3H),1.10〜1.20(m,2H),1.36(s,3H)が重畳した1.34〜1.41(m,3H),1.53(tqq,J=6.6,6.6,6.6Hz,1H),1.67(q,J=1.5Hz,3H)が重畳した1.64〜1.75(m,2H),1.95〜2.15(m,4H),ABシグナル(δA=3.46,δB=3.51,JAB=11.1Hz,4H),5.12(br t,J=7.2Hz,1H)。
13C NMR(75MHz,CDCl3):δ21.1(1C),22.0(1C),22.61(3C),22.65(1C),23.4(1C),25.7(1C),27.9(1C),29.9(1C),31.9(1C),37.2(1C),38.8(1C),70.3(2C),98.8(1C),124.6(1C),135.8(1C)ppm。
MS(El,m/z):282(M+,6),267[(M−CH3)+11),129(100),95(14),69(35),43(32)。
IR(cm−1):2953(s),2867(m),1722(w),1468(m),1394(m),1368(m),1349(w),1306(w),1270(w),1250(m),1211(m),1189(w),1116(s),1086(s),1043(m),1022(m),951(m),925(w),907(m),856(m),792(w),739(w),677(w),667(w)。
攪拌棒を伴う250mL三首フラスコを高真空で乾燥させ(250℃でヒートガン使用)、次いで、冷却させ、アルゴンでフラッシュし、1,1,1トリフルオロエタノール(TFE)(40mL)をアルゴン下で仕込んだ。このフラスコを、トリメチルアルミニウム(ヘプタン中に2M、20.0mL、40.0mmol、1.95当量)を60分間以内に滴下しながら氷浴で冷却して温度を22℃未満に維持した。数分後に二相(TFE/ヘプタン)混合物は再び清透になり、室温でさらに20分間撹拌した。20.7mmolの(E)−10,10−ジメトキシ−2,6−ジメチルウンデカ−2,6−ジエン(E−GA−DM)または(E)−2,2−ジメトキシ−6,10−ジメチルウンデカ−5−エン(E−DHGA−DM)(上記のとおり調製したもの)を、室温で5分間に滴下した。1.5時間後、GC分析は、出発材料の完全な転換を示した。反応を酒石酸ナトリウムカリウムの水(100mL)中の半飽和溶液で失活させ、2時間室温で撹拌し、最後に、n−ヘキサン(200mL)で希釈した。有機相を分離し、n−ヘキサンで抽出し(2×100mL)、MgSO4で乾燥させ、濃縮した。粗生成物を、カラムクロマトグラフィにより精製した(中性アルミニウム酸化物、溶離液:n−ヘキサン)。ケタールの特性付けを以下に詳細に示す。
[(E)−2,6−ジメチル−10,10−ビス(2,2,2−トリフルオロエトキシ)ウンデカ−2,6−ジエン(E−GA−tfe)]
1H NMR(300MHz,CDCl3):δ1.41(s,3H),1.62(br s,6H),1.67〜1.76(m,2H),1.69(q,J=0.9Hz,3H)と重畳,1.93〜2.15(m,6H),3.73〜3.97(m,4H),5.02〜5.18(m,2H)ppm。
13C NMR(150MHz,CDCl3):δ15.9(1C),17.6(1C),21.3(1C),22.6(1C),25.7(1C),26.6(1C),36.9(1C),39.6(1C),59.3(q,JC,F=35.0Hz,2C),103.4(1C),124.0(q,JC,F=275.0Hz,2C),122.7(1C),124.1(1C),131.5(1C),136.2(1C)ppm。
MS(El,m/z):361[(M−CH3)+,1],276[(M−TFE)+,15],225[(CF3CH2O)2C−CH3)+,86],207(20),153(18),136(58),107(80),69(100),41(40)。
IR(cm−1):2927(w),2859(w),1459(w),1419(w),1385(w),1281(s),1223(w),1156(s),1133(s),1081(s),971(s),889(m),860(w),845(w),678(w),663(w)。
1H NMR(600MHz,CDCl3):δ0.88(d,J=6.8Hz,6H),1.11〜1.17(m,2H),1.35〜1.40(m,2H),1.41(s,3H),1.54(qqt,J=6.7,6.7,6.7Hz,1H),1.61(br s,3H),1.69〜1.73(m,2H),1.95(t,J=7.7Hz,2H),2.03〜2.09(m,2H),3.78〜3.91(m,4H),5.09(tq,J=7.1,1.3Hz,1H)ppm。
13C NMR(151MHz,CDCl3):δ14.1(1C),15.8(1C),21.3(1C),22.56(1C),22.61(1C),25.6(1C),27.9(1C),37.0(1C),38.6(1C),39.8(1C),59.2(q,JC,F=35.0Hz,2C),103.4(1C),124.0(q,JC,F=277.0Hz,2C),122.4(1C),136.7(1C)ppm。
MS(El,m/z):363[(M−CH3)+,1],278[(M−TFE)+,22],225[(CF3CH2O)2C−CH3)+,60],193(100),153(13),127(11),83(CF3CH2 +,25),69(13),43(17)。
IR(cm−1):2956(w),2933(w),2872(w),1462(w),1419(w),1385(w),1368(w),1281(s),1223(w),1156(s),1134(s),1081(s),971(s),889(m),860(w),845(w),679(w),663(m)。
オートクレーブに、表2e〜表2hに示されている6,10−ジメチルウンデカ−5,9−ジエン−2−オンまたは6,10−ジメチルウンデカ−5−エン−2−オンの0.5mmolのケタール、表2e〜表2hに示されている4gの溶剤、および、表2e〜表2hに示されている、前記式において*で示される中心でキラリティーを有する式(III−F)のキラルイリジウム錯体の溶液を、表2e〜表2hに示されている量で仕込んだ。オートクレーブを閉め、分子水素を30barの圧力で印加した。反応混合物を室温で16時間撹拌した。その後、圧力を解放し、溶剤を除去した。
[(R)−2,2−ジメトキシ−6,10−ジメチルウンデカン(R−THGA−DM)]
1H NMR(300MHz,CDCl3):0.852(d,J=6.6Hz,6H)と重畳したδ0.848(d,J=6.6Hz,3H),1.25(s,3H)と重畳した1.01〜1.41(m,11H),1.44〜1.61(m,3H),3.16(s,6H)ppm。
13C NMR(75MHz,CDCl3):δ14.1(1C),19.6(1C),20.9(1C),21.7(1C),22.6(1C),22.7(1C),24.8(1C),27.9(1C),32.7(1C),36.8(1C),37.2(1C),37.4(1C),39.3(1C),47.9(1C),101.7(1C)ppm。
MS(El,m/z):カラムにおける分解のためにGC−MSは入手しなかった。
IR(cm−1):2951(s),2927(m),2870(m),2828(m),1723(w),1462(m),1377(m),1309(w),1256(m),1215(m),1194(m),1172(m),1111(m),1089(m),1053(s),972(w),934(w),920(w),855(m),815(m),736(w),618(w)。
1H NMR(300MHz,CDCl3):δ0.87(d,J=6.6Hz,9H),0.91(s,3H),1.01(s,3H),1.36(s,3H)と重畳した1.04〜1.61(m,12H),1.61〜1.74(m,2H),ABシグナル(δA=3.44,δB=3.54,JAB=11.7Hz,4H)ppm。
13C NMR(75MHz,CDCl3):δ19.7(1C),20.4(1C),21.0(1C),22.56(1C),22.61(1C),22.71(1C),22.77(1C),24.8(1C),28.0(1C),30.0(1C),32.8(1C),37.3(1C),37.4(1C),38.2(1C),39.3(1C),70.3(2C),99.1(1C)ppm。
MS(El,m/z):269[(M−CH3)+,65),199(8),129(100),109(8),69(32),55(10),43(25)。
IR(cm−1):2953(s),2925(s),2868(m),1722(w),1464(m),1394(m),1371(m),1316(w),1258(m),1212(m),1161(m),1141(m),1111(s),1095(s),1043(m),1020(m),951(m),925(m),907(m),870(m),855(m),801(m),792(m),737(m),677(w),667(w)。
1H NMR(300MHz,CDCl3):δ0.88(d,J=6.6Hz,6H),0.87(d,J=6.4Hz,3H),1.03〜1.23(m,5H),1.39(s,3H),1.38〜1.40(m,6H),1.46〜1.71(m,3H),3.73〜3.94(m,4H)。
13C NMR(75MHz,CDCl3):δ19.5(1C),21.39(1C),21.47(1C),22.58(1C),22.68(1C),24.7(1C),28.0(1C),32.6(1C),37.0(1C),37.19(1C),37.23(1C),39.3(1C),59.2(q,2JC,F=32.5Hz,2C),124.1(q,1JC,F=279.0Hz,2C)。
MS(El,m/z):365[(M−CH3)+,1],281(2),225[(CF3CH2O)2C−CH3)+,100],153(8),140(6),83(CF3CH2 +,6),43(7)。
IR(cm−1):2955(w),2929(w),2872(w),1463(w),1419(w),1385(w),1281(s),1216(w),1156(s),1122(m),1082(s),972(m),892(m),861(w),737(w),679(w),663(m)。
(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのケタールの不斉水素化の後、得られた水素化ケタールをケトンに加水分解して、それぞれ、(R)−6,10−ジメチルウンデカン−2−オンまたは(S)−6,10−ジメチルウンデカン−2−オンを得た。
不斉水素化反応からの反応混合物のサンプル(1〜2ml)を、等しい体積の塩酸の1M水溶液で、室温で、1時間撹拌した。ジクロロメタン(2ml)を添加し、層を分離した。水性層をジクロロメタン(2ml)で2回洗浄した。組み合わせた有機層を減圧下で蒸発させて、無色〜薄い黄色の油としてケトンを得た。次いで、粗ケトンを純度および異性体比について分析した。
不斉水素化反応からの反応混合物のサンプル(1〜2ml)を、9:1:0.2(体積)メタノール:水:トリフルオロ酢酸の0.5mlの溶液で、40℃で、1時間撹拌した。ジクロロメタン(2ml)および水(2ml)を添加し、層を分離した。水性層をジクロロメタン(2ml)で2回洗浄した。組み合わせた有機層を減圧下で蒸発させて、無色〜薄い黄色の油としてケトンを得た。次いで、粗ケトンを純度および異性体比について分析した。
オートクレーブ容器に、窒素下で、*で示したキラル中心でR配置の式(III−F)のキラルイリジウム錯体、表2hまたは表2iに示されているケトンまたはケタール(conc.)、表2hまたは表2i)に示されている溶剤を仕込んだ。反応容器を閉め、分子水素で表2hまたは表2iに示す圧力(pH2)に加圧した。反応混合物を、室温で、表2hまたは表2i中に示されている時間(t)、水素下に撹拌した。次いで、圧力を解放し、完全水素化生成物のアッセイ収率および立体異性体分布を判定した。ケタールの場合、アッセイ収率および立体異性体分布は、実験E2cに示されている酸によるケタールの加水分解後に判定した。触媒添加量(S/C)は、ケトンまたはケタール(「基材」)のmmol数/キラルイリジウム錯体のmmol数として定義される。
水素化に添加剤を用いたことを除き、実験E2dと同様に不斉水素化を行った。用いた添加剤および量は、ケタールの水素化については表2j、表2k、ならびに、ケトンの水素化については表2lおよび表2mに示されている。
− MAO/TFE:1.6M MAO(MAO:トルエン(0.64mL)中のメチルアルミノキサン溶液を2,2,2−トリフルオロエタノール(TFE)(3.1mmol)で失活させて、わずかに過剰量の遊離TFEをもたらした。
− EAO/TFE:10重量%EAO(EAO:トルエン(1mmol)中のエチルアルミノキサン溶液をTFE(3.2mmol)で失活させて、わずかに過剰量の遊離TFEをもたらした。
− TMA/TFE:2M TMA(TMA:ヘプタン(1mmol)中のトリメチルアルミニウム(Al(CH3)3))溶液をTFE(3.1mmol)で失活させて、わずかに過剰量の遊離TFEをもたらした。
− TEA/TFE:2M TEA(TEA:ヘプタン(1mmol)中のトリエチルアルミニウム(Al(CH2CH3)3))溶液をTFE(3.1mmol)で失活させて、わずかに過剰量の遊離TFEをもたらした。
− TMA/BHT/TFE:ヘプタン(1mmol)中の2M TMA溶液を2,6−ジ−tert−ブチル−4−メチルフェノール(BHT)(2mmol)、続いて、TFE(3.1mmol)で失活させて、わずかに過剰量の遊離TFEをもたらした。
− Ti(OCH2CF3)4:オルトチタン酸テトライソプロピル(8.1mmol)を2,2,2−トリフルオロエタノール中に50℃で溶解させた。溶剤を除去して、Ti(OCH2CF3)4を白色の残渣として得、これを単離し、Ti(OCH2CF3)4として同定した。
[実験E3a:(R)−6,10−ジメチルウンデカン−2−オンのビニル化(ステップd1’)]
オーバーヘッド攪拌機、温度計、凝縮器およびアルゴンインレットを備える乾燥させた100ml四首フラスコを排気し、アルゴンでパージした。ビニル塩化マグネシウム(23.63mLのTHF中の1.6M溶液、37.8mmol、1.56当量)を室温で加えた。乾燥THF(20mL)中の溶液(R)−6,10−ジメチルウンデカン−2−オン(5.01g、24.24mmol、96.0%、1.0当量)を20分間以内にゆっくりと添加した。氷浴で冷却することで、発熱反応を25〜30℃の内部温度に維持した。添加が完了した後、反応を室温で1時間撹拌した。飽和NH4Cl溶液(10mL)を注意深く添加して過剰量のグリニャール試薬を失活させた。ペンタン(150mL)、水(150mL)および塩水(150mL)を添加した。有機相を塩水(2×150mL)で抽出し、水性相をペンタン(2×150mL)で逆抽出した。組み合わせた有機相を乾燥させ(MgSO4)、減圧中で濃縮し、無色の油を得た(5.42g)。粗生成物をクーゲルロール装置における減圧蒸留により精製した。143℃/3.8−2mbarで主な画分が蒸留され、(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを、94.0%の純度(5.15g、21.38mmol、88%収率)で無色の油として得た。
(R)−6,10−ジメチルウンデカン−2−オン(340g、1.70mol、1.0当量、99.0%)を、サーモスタット、計量供給ポンプ、アセチレンインレットおよびアンモニアインレットを備えるオートクレーブに加えた。反応器をシールし、排気し、窒素でフラッシュし、15℃に冷却した。アンモニア(632g、37.2mol、22.0当量、99.8%)を反応器内に縮合させ、15℃に冷却したところ、8〜9barの圧力が得られた。12barの圧力に達するまでアセチレンを導入し、続いて、KOH(水中に45重量%、6.6g、52.9mmol、3.1mol%)を15℃で計量して添加した。反応の進行をGCにより監視した。90分後、反応混合物を酢酸で中和し、その後、反応器を25℃でベントした。反応混合物を洗浄し、減圧中で濃縮し、減圧中の蒸留により精製して、325gの(7R)−3,7,11−トリメチルドデカ−1−イン−3−オールを95%の純度(81%収率)で得た。
(7R)−3,7,11−トリメチルドデカ−1−イン−3−オール(910g、4.06mol、1.0当量、95%)、リンドラー触媒触媒(850mg)をオートクレーブに入れた。反応器をシールし、排気し、窒素でフラッシュし、その後、45℃に加熱した。反応器をもう一度排気し、水素でフラッシュし、2barに加圧した。この反応を、理論量の分子水素が消費されるまで、45℃でおよそ2〜3時間撹拌した。ろ過の後、884gの(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを91.5%の純度(88%収率)で得た。
(7R)−3,7,11−トリメチルドデカ−1−エン−3−オール(640g、2.59mol、1.0当量、91.5%)、H3PO4(水中に20重量%、4.5g、8.2mmol、0.32mol%)およびイソプロペニルメチルエーテル(600g、8.17mol、3.2当量、98.0%)を、サーモスタット、オーバーヘッド攪拌機および1.1m蒸留カラム(Sulzer充填物)を備える2Lのオートクレーブに入れた。反応器をシールし、排気し、窒素でフラッシュし、その後、80℃に加熱した。蓄積した圧力をゆっくりとベントした。次いで、内部温度を1時間以内で160℃にゆっくりと昇温させた。反応を160℃で3時間撹拌した。反応混合物を30℃に冷却し、減圧中で濃縮し、水およびNaHCO3溶液で洗浄した。次いで、低沸点溶剤を蒸留(ジャケット温度150℃、1mbar)により除去し、690gの(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物を、85%の純度(85%収率)で残渣として得た。混合物は、49%E異性体および36%Z異性体の混合物であるとGCにより分析された。
(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物1.94kg、36%(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび49%(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンを、流下薄膜型エバポレータを備える蒸留器(体積:9リットル)、精留カラム(70mm内径、高さ5m)から構成される分離器具を用いて精留した。カラムは、高効率に構造化された充填物を備えるものであった(Sulzer)。精留プロセスは、およそ2mbarの頂部圧力、および、95〜122℃の範囲内で変化するカラム頂部の温度で行われ、蒸留器中の下部温度は165℃であった。還流比を20に調節した。留出物流の精留により、(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンを含有する画分を得た(Z異性体の含有量=97%)。最後に、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンが蒸留器に残留していた(E異性体の含有量=94%)。両方の異性体を精留によりさらに精製したところ、E異性体およびZ異性体の両方の得られた画分は、それぞれ、99.5%の純度であった。
両方の異性体(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オン)(R,E−THFA)(E/Z=99.5/0.5、R/S=92/8)および(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オン(R,Z−THFA)(Z/E=99.5/0.5、R/S=92/8)を、以下の方法で別々に不斉水素化した。
それぞれ、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンのジメチルケタール、ネオペンチルグリコールケタールまたはビス(トリフルオロエチル)ケタールを、6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンについて上記した実験E2aと同様に得た。
[(R,E)−2,2−ジメトキシ−6,10,14−トリメチルペンタデカ−5−エン(R−E−THFA−DM)]
1H NMR(300MHz,CDCl3):δ0.84(d,J=6.6Hz,3H),0.86(d,J=6.6Hz,6H)と重畳,0.99〜1.44(m,11H),1.28(s,3H)と重畳,1.52(tqq,J=6.6,6.6,6.6Hz,1H),1.60(s,3H),1.60〜1.66(m,2H),1.90〜2.05(m,4H),3.18(s,6H),5.10(tq,J=7.1,1.1Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ16.3(1C),20.1(1C),21.3(1C),23.0(1C),23.1(1C),23.2(1C),25.2(1C),25.7(1C),28.4(1C),33.1(1C),36.9(1C),37.1(1C),37.7(1C),39.8(1C),40.3(1C),48.4(2C),101.9(1C),124.0(1C),136.0(1C)ppm。
MS(El,m/z):カラムにおける分解のためにGC−MSは入手しなかった。
IR(cm−1):2952(m),2927(s),2869(m),2828(w),1461(m),1377(m),1301(w),1262(m),1222(m),1197(m),1172(m),1120(m),1101(m),1076(m),1054(s),930(w),854(m),737(w),620(w)。
1H NMR(300MHz,CDCl3):δ0.85(d,J=6.4Hz,3H),0.87(d,J=6.4Hz,6H)と重畳,1.01〜1.27(m,7H),1.28(s,3H),1.29〜1.44(m,4H),1.53(dqq,J=6.5,6.5Hz,6.5Hz,1H),1.58〜1.66(m,2H),1.68(q,J=1.1Hz,3H),1.91〜2.08(m,4H),3.18(s,6H),5.11(t,J=6.8Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ19.7(1C),20.9(1C),22.60(1C),22.69(1C),22.71(1C),23.4(1C),24.8(1C),25.5(1C),28.0(1C),32.0(1C),32.7(1C),36.8(1C),37.0(1C),37.3(1C),39.3(1C),48.0(2C),101.5(1C),124.3(1C),135.9(1C)ppm。
MS(El,m/z):カラムにおける分解のためにGC−MSは入手しなかった。
IR(cm−1):2952(m),2927(m),2869(m),2828(w),1462(m),1376(m),1301(w),1261(w),1197(w),1172(m),1119(m),1098(m),1074(m),1054(s),1022(w),854(m),736(w),622(w)。
1H NMR(300MHz,CDCl3):δ0.84(d,J=6.4Hz,3H),0.86(d,J=6.6Hz,6H),0.92(s,3H),0.99(s,3H),0.97〜1.44(m,11H)と重畳,1.37(s,3H)と重畳,1.52(qqt,J=6.9,6.9,6.9Hz,1H),1.60(s,3H),1.67〜1.76(m,2H),1.93(t,J=7.4Hz,2H),2.03〜2.18(m,2H),ABシグナル(δA=3.45,δB=3.52,JAB=11.4Hz,4H),5.12(tq,J=7.2,1.0Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ15.8(1C),19.7(1C),20.9(1C),22.0(1C),22.6(2C),22.7(2C),24.8(1C),25.3(1C),27.9(1C),29.9(1C),32.6(1C),36.6(1C),37.3(1C),37.4(1C),39.3(1C),39.9(1C),70.3(2C),98.8(1C),123.8(1C),135.5(1C)ppm。
MS(El,m/z):352(M+,4),337[(M−CH3)+,8),265(6),129(100),95(10),69(25),43(25)。
IR(cm−1):2953(s),2926(s),2867(m),1462(m),1394(w),1369(m),1270(w),1249(m),1211(m),1187(w),1119(s),1088(s),1043(m),1021(m),951(w),925(w),907(w),862(m),791(w),738(w),678(w)。
1H NMR(300MHz,CDCl3):δ0.84(d,J=6.4Hz,3H),0.86(d,J=6.6Hz,6H)と重畳,0.93(s,3H),0.97(s,3H),1.00〜1.42(m,11H),1.36(s,3H)と重畳,1.52(qqt,J=6.7,6.7,6.7Hz,1H),1.63〜1.76(m,2H),1.67(s,3H),1.94〜2.15(m,4H),ABシグナル(δA=3.45,δB=3.51,JAB=11.1Hz,4H),5.12(t,J=7.1Hz,1H)ppm。
13C NMR(75MHz,CDCl3):δ19.6(1C),21.1(1C),21.9(1C),22.60(2C),22.67(2C),22.69(1C),23.4(1C),24.8(1C),25.4(1C),27.9(1C),29.9(1C),32.0(1C),32.7(1C),36.9(1C),37.3(1C),39.3(1C),70.3(2C),98.8(1C),124.6(1C),135.7(1C)ppm。
MS(El,m/z):352(M+,3),337[(M−CH3)+,9),265(6),129(100),95(10),69(24),43(25)。
IR(cm−1):2953(s),2926(s),2860(m),1463(m),1394(w),1371(m),1270(w),1250(w),1211(m),1188(w),1117(s),1086(s),1043(m),1022(w),951(w),925(w),907(w),855(m),792(w),737(w),667(w)。
オートクレーブに、表3bおよび3cに示されている、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンのケタール0.5mmol、表3bおよび3cに示されている溶剤4g、ならびに、表3bおよび表3cに示す、前記式における*で示される中心でキラリティーを有する式(III−F)のキラルイリジウム錯体の溶液を、表3bおよび表3cに記載の量で仕込んだ。オートクレーブを閉め、分子水素を30barの圧力で印加した。反応混合物を室温で16時間撹拌した。その後、圧力を解放し、溶剤を除去した。
[(6R,10R)−2,2−ジメトキシ−6,10,14−トリメチルペンタデカン(RR18−DM)]
1H NMR(300MHz,CDCl3):δ0.83〜0.89(m,12H),0.98〜1.45(m,21H),1.46〜1.65(m,3H),3.18(s,6H)。
13C NMR(75MHz,CDCl3):δ19.68(1C),19.73(1C),21.0(1C),21.7(1C),22.6(1C),22.7(1C),24.5(1C),24.8(1C),28.0(1C),32.72(1C),32.78(1C),36.8(1C),37.28(1C),37.33(1C),37.36(1C),37.41(1C),39.4(1C),48.0(2C),101.7(1C)ppm。
IR(cm−1):2951(s),2926(s),2869(s),2828(m),1734(w),1723(w),1216(w),1463(s),1377(s),1308(w),1255(m),1215(m),1172(s),1105(s),1090(s),1054(s),971(w),933(w),860(s),815(m),736(w)618(w)。
1H NMR(300MHz,CDCl3):δ0.78〜0.95(m,15H),0.95〜1.61(m,19H),1.01(s,3H)と重畳,1.36(s,3H),1.63〜1.74(m,2H),ABシグナル(δA=3.44,δB=3.55,JAB=11.7Hz,4H)ppm。
13C NMR(75MHz,CDCl3):δ19.72(1C),19.74(1C),20.4(1C),20.9(1C),22.56(1C),22.62(1C),22.72(1C),22.77(1C),24.5(1C),24.8(1C),28.0(1C),30.0(1C),32.8(1C),32.8(1C),37.28(1C),37.35(1C),37.42(2C),38.2(1C),39.4(1C),70.3(2C),99.1(1C)ppm。
MS(El,m/z):339[(M−CH3)+,83],269(5),129(100),69(21),43(18)。
IR(cm−1):2952(s),2925(s),2867(m),1463(m),1394(m),1372(m),1258(m),1211(m),1189(w),1141(w),1100(s),1043(m),1020(m),951(w),925(w),907(m),858(m),792(w),737(w),677(w)。
1H NMR(600MHz,CDCl3):δ0.86(d,J=6.6Hz,3H),0.879(d,J=6.6Hz,3H),0.882(d,J=6.6Hz,3H),0.884(d,J=6.6Hz,3H),1.03〜1.46(m,18H),1.40(s,3H)と重畳,1.54(qqt,J=6.6,6.6,6.6Hz,1H),1.60〜1.70(m,2H),3.77〜3.90(m,4H)ppm。
13C NMR(151MHz,CDCl3):δ19.6(1C),19.7(1C),21.4(1C),21.5(1C),22.6(1C),22.7(1C),24.5(1C),24.8(1C),28.0(1C),32.6(1C),32.8(1C),37.0(1C),37.24(1C),37.30(1C),37.34(1C),37.43(1C),39.4(1C),59.2(q,2JC,F=35.0Hz,2C),103.6(1C),124.0(q,1JC,F=277.0Hz,2C)ppm。
MS(El,m/z):435[(M−CH3)+,1],351(1),250(1),225[(CF3CH2O)2C−CH3)+,100],153(7),140(5),83(CF3CH2 +,3),43(6)。
IR(cm−1):2954(m),2927(m),2871(w),1463(w),1419(w),1384(w),1281(s),1215(w),1157(s),1123(m),1082(s),972(s),892(m),861(w),737(w),679(w),663(m)。
(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの対応するケタールの不斉水素化の後、得られた水素化ケタールをケトンに加水分解し、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを得た。
[実験E6−I:(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエテニル化(ステップg)]
(6R,10R)−6,10,14−トリメチルペンタデカン−2−オン(35.0g、129mmol、1.0当量、98.8%)を、サーモスタット、計量供給ポンプ、アセチレンインレットおよびアンモニアインレットを備えるオートクレーブに加えた。反応器をシールし、排気し、次いで窒素でフラッシュし、15℃に冷却した。アンモニア(715g、45.0mol、326当量、99.8%)を反応器内に縮合させ、15℃に冷却したところ、8〜9barの圧力が得られた。12barに達するまでアセチレンを導入し、続いて、KOH(水中に40重量%、5.0g、35.6mmol、28mol%)を15℃で計量して添加した。反応の進行をGCにより監視した。所望の転換率(およそ2時間後)で、反応混合物を酢酸で中和し、その後、反応器を25℃でベントした。反応混合物を洗浄し、減圧中で濃縮し、減圧中の蒸留により精製して、26.9g(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを98.8面積%の純度(70%収率)で得た。
ヘプタン(40g)に溶解した(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オール(10g、33.4mmol、98.4%純度)、および、リンドラー触媒(850mg)をオートクレーブに入れた。反応器をシールし、窒素でフラッシュし、その後、85℃に加熱した。所望の温度に達したら、反応を2bar水素に加圧した。反応を、およそ22時間、この温度で必要量の水素ガスが消費されるまで撹拌した。ろ過の後、粗生成物を第2の反応バッチと組み合わせた。11.9gの粗材料を蒸留により精製し、11.1gの(R,R)−イソフィトール(GCにより97.6%純度、88%全収率)を得た。
オーバーヘッド攪拌機、温度計、凝縮器およびアルゴンインレットを備える乾燥させた100mL四首フラスコを排気し、アルゴンでパージした。ビニル塩化マグネシウム(18.3mLのTHF中の1.6M溶液、29.0mmol、1.59当量)を室温で加えた。(6R,10R)−6,10,14−トリメチルペンタデカン−2−オン(5.00g、18.3mmol、98.2%、1.0当量)の乾燥THF(20mL)中の溶液を25分間以内にゆっくりと添加した。氷浴で冷却することで、発熱反応を25〜30℃の内部温度に維持した。添加が完了した後、反応を室温で1時間撹拌した。飽和NH4Cl溶液(10mL)を注意深く添加して過剰量のグリニャール試薬を失活させた。ペンタン(150mL)、水(150mL)および塩水(150mL)を添加した。有機相を塩水(2×150mL)で抽出し、水性相をペンタン(2×150mL)で逆抽出した。組み合わせた有機相を乾燥させ(MgSO4)、減圧中で濃縮し、無色の油を得た(5.58g)。粗生成物をクーゲルロール装置における減圧蒸留により精製した。143℃/3.5×10−2mbarで主な画分が蒸留され、(R,R)−イソフィトール(=(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−エン−3−オール)を99.3%の純度(5.271g、96%収率)で無色の油として得た。
国際公開第2005/121115 A1号パンフレットに開示の手法に従って、(R,R)−イソフィトール(=(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−エン−3−オール)を2,3,5−トリメチルベンゼン−1,4−ジオール(=2,3,5−トリメチルヒドロキノン)と、縮合触媒の存在下において、(2−ambo)−α−トコフェロールに縮合させた。
(2−ambo)−α−トコフェロールを、キラル相を用いるクロマトグラフィ分離により分離した。分取クロマトグラフィにより(2R,4’R,8’R)−α−トコフェロールおよび(2S,4’R,8’R)−α−トコフェロールを得た。
分取分離を、Agilent 1100デガッサ、Agilent 1100分取ポンプ、Agilent 1100ダイオードアレイ検出器、chemstation/CC−modeソフトウェアパッケージにより制御されるAgilent 1100MPS G2250Aオートサンプラー/画分コレクタから構成されるAgilent 1100シリーズHPLCシステムで行った。
カラム:Daicel Chiracel(登録商標)OD−H、250mm×20mm;溶離液0.5%イソプロパノール、n−ヘプタン中の0.2%酢酸;流量13ml/分;検出220nm、400μl注入。
Claims (16)
- 6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンからの多段合成における(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンの製造方法であって、
a)(E)−6,10−ジメチルウンデカ−5−エン−2−オンおよび(Z)−6,10−ジメチルウンデカ−5−エン−2−オンの混合物、または、(E)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンおよび(Z)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンの混合物を提供するステップ、
b)6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンのEおよびZ異性体のいずれか1種をステップa)の前記混合物から分離するステップ、
c)キラルイリジウム錯体の存在下において分子水素を用いて不斉水素化を行い、(R)−6,10−ジメチルウンデカン−2−オンを得るステップ、
d)(R)−6,10−ジメチルウンデカン−2−オンを、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物に化学転換するステップ、
e)(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オンのEおよびZ異性体のいずれか1種をステップd)において得られた前記混合物から分離するステップ、
f)キラルイリジウム錯体の存在下において分子水素を用いて不斉水素化を行い、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを得るステップを含み、前記ステップa)〜f)がa、b、c、d、e、fの順番である方法。 - 前記ステップc)に先だって、ステップc0)
c0)ステップb)において分離された6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンのEおよびZ異性体のいずれか1種のケタールを形成するステップ
を行うことと、
ステップc)において、6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンの前記ケタールを不斉水素化し、前記不斉水素化の後に、前記水素化ケタールをケトンに加水分解し、(R)−6,10−ジメチルウンデカン−2−オンを得ることと
を特徴とする、請求項1に記載の方法。 - 前記ステップf)に先だって、ステップf0)
f0)ステップe)において分離した(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オンのEおよびZ異性体のいずれか1種のケタールを形成するステップ
を行うことと、
ステップf)において、(R)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの前記ケタールを不斉水素化し、前記不斉水素化の後に、前記水素化ケタールを前記ケトンに加水分解し、(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンを得ることと
を特徴とする、請求項1または2に記載の方法。 - ステップc)および/またはステップf)における前記不斉水素化が、有機スルホン酸、有機スルホン酸の遷移金属塩、金属アルコキシド、アルミノキサン、アルキルアルミノキサンおよびB(R)(3−v)(OZ)vからなる群から選択される添加剤の存在下で行われ、
vが0、1、2または3を指し、
Rが、F、C1〜6アルキル、ハロゲン化C1〜6アルキル、アリールまたはハロゲン化アリール基を指し;ならびに
Zが、C1〜6アルキル、ハロゲン化C1〜6アルキル、アリールまたはハロゲン化アリール基を指す
ことを特徴とする、請求項1〜3のいずれか一項に記載の方法。 - ステップd)において、(R)−6,10−ジメチルウンデカン−2−オンの(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物への前記化学的転換が、
d1)塩基性物質の存在下においてエチンを用いて、(7R)−3,7,11−トリメチルドデカ−1−イン−3−オールを得る(R)−6,10−ジメチルウンデカン−2−オンのエチニル化、
d2)リンドラー触媒の存在下において、(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを得る、分子水素による(7R)−3,7,11−トリメチルドデカ−1−イン−3−オールの水素化、
または
d1’)ビニルグリニャール試薬の添加により(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを得る(R)−6,10−ジメチルウンデカン−2−オンのビニル化、
のいずれかのステップ、これに続いて、
d3)(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールと2−メトキシプロプ−1−エンとを反応させて、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物を得るステップ、
または
d3’)(7R)−3,7,11−トリメチルドデカ−1−エン−3−オールを、塩基または酸の存在下でアルキルアセト酢酸またはジケテンと反応させて、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンおよび(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンの混合物を得るステップ
のいずれかによって行われることを特徴とする、請求項1〜4のいずれか一項に記載の方法。 - ステップb)および/またはe)におけるEおよびZ異性体のいずれか1種の前記分離が、蒸留によって行われることを特徴とする、請求項1〜5のいずれか一項に記載の方法。
- 前記蒸留が、シス/トランス異性化触媒の存在下において行われることを特徴とする、請求項6に記載の方法。
- 前記残存するEまたはZ異性体がシス/トランス異性化触媒により異性化され、ステップa)およびd)のそれぞれによりもたらされた前記対応する異性体の混合物に加えられることを特徴とする、請求項1〜6のいずれか一項に記載の方法。
- ステップc)および/またはf)における前記キラルイリジウム錯体が、式(III−0)
(式中、
P−Q−Nは、不斉中心を含むか、または、面性または軸キラリティーを有し、かつ、前記錯体のイリジウム中心に対する結合部位として窒素およびリン原子を有するキレート化有機リガンドを指し、
Y1、Y2、Y3およびY4は、相互に独立して、水素原子、C1〜12アルキル、C5〜10−シクロアルキルもしくは芳香族基であり;または、これらの少なくとも2つが一緒になって、少なくとも2個の炭素原子を有する少なくとも2価の架橋された基を形成し;ただし、Y1、Y2、Y3およびY4のすべてが水素原子とされることはなく、ならびに
は、アニオンである)
のキラルイリジウム錯体であることを特徴とする、請求項1〜8のいずれか一項に記載の方法。 - ステップc)および/またはf)における前記キラルイリジウム錯体が、式(III)
(式中、
nは、1または2または3であり、
X1およびX2は、相互に独立して、水素原子、C1〜4アルキル、C5〜7−シクロアルキル、アダマンチル、フェニル(1〜3個のC1〜5−、C1〜4−アルコキシ、C1〜4−パーフルオロアルキル基および/または1〜5個のハロゲン原子で任意選択により置換されている))、ベンジル、1−ナフチル、2−ナフチル、2−フリルまたはフェロセニルであり、
Z1およびZ2は、相互に独立して、水素原子、C1〜5アルキルまたはC1〜5アルコキシ基であり、
または、Z1およびZ2は、一緒になって、5〜6員環を形成する架橋基を指し、
は、アニオンであり、
R1は、フェニルもしくはo−トリルもしくはm−トリルもしくはp−トリル、または、式(IVa)もしくは(IVb)もしくは(IVc)の基を表し、
(式中、R2およびR3は、共にHもしくはC1〜C4アルキル基もしくはハロゲン化C1〜C4アルキル基を表すか、または、ハロゲン原子もしくはC1〜C4アルキル基もしくはC1〜C4アルコキシ基により任意選択により置換されている6員脂環式もしくは芳香族環を一緒になって形成する二価の基を表し、
R4およびR5は、共にHもしくはC1〜C4アルキル基もしくはハロゲン化C1〜C4アルキル基、または、ハロゲン原子もしくはC1〜C4アルキル基もしくはC1〜C4アルコキシ基により任意選択により置換されている6員脂環式もしくは芳香族環を一緒になって形成する二価の基を表し、
R6およびR7およびR8は、各々、C1〜C4アルキル基またはハロゲン化C1〜C4アルキル基を表し、
R9およびR10は、共にHもしくはC1〜C4アルキル基もしくはハロゲン化C1〜C4アルキル基、または、ハロゲン原子もしくはC1〜C4アルキル基もしくはC1〜C4アルコキシ基により任意選択により置換されている6員脂環式もしくは芳香族環を一緒になって形成する二価の基を表す)
ならびに、
式中、*は、式(III)の前記錯体の不斉中心を表す)
のキラルイリジウム錯体であることを特徴とする、請求項1〜8のいずれか一項に記載の方法。 - 前記不斉水素化に利用されるステップc)および/またはf)において用いられる式(III)の前記キラルイリジウム錯体が、
(E)−6,10−ジメチルウンデカ−5−エン−2−オンもしくは(E)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンもしくはそのケタール、または、(R,E)−6,10,14−トリメチルペンタデカ−5−エン−2−オンもしくはそのケタールが水素化される事例においては、*で示す前記不斉中心でS配置を有し、
または、
(Z)−6,10−ジメチルウンデカ−5−エン−2−オンもしくは(Z)−6,10−ジメチルウンデカ−5,9−ジエン−2−オンもしくはそのケタール、または、(R,Z)−6,10,14−トリメチルペンタデカ−5−エン−2−オンもしくはそのケタールが水素化される事例においては、*で示す前記不斉中心でR配置を有する
ことを特徴とする、請求項10に記載の方法。 - (R,R)−イソフィトール((3RS,7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−エン−3−オール)の製造方法であって、
請求項1〜11のいずれか一項に記載の(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンの製造方法、
これに続いて、
g)塩基性物質の存在下においてエチンを用いて、(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエチニル化、
h)リンドラー触媒の存在下において、(R,R)−イソフィトールを得る、分子水素による(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールの水素化、
または
h’)ビニルグリニャール試薬の添加により(R,R)−イソフィトールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのビニル化
のいずれかのステップを含む、製造方法。 - 式(V)の化合物の製造方法であって、
請求項1〜11のいずれか一項に記載の(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンの製造方法、
これに続いて、
g)塩基性物質の存在下においてエチンを用いて、(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエチニル化、
h)リンドラー触媒の存在下において、(R,R)−イソフィトールを得る、分子水素による(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールの水素化、
または
h’)ビニルグリニャール試薬の添加により(R,R)−イソフィトールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのビニル化、
のいずれかのステップ、これに続いて、
m)(R,R)−イソフィトールと式(VI)の化合物とを縮合して、#によって示される前記中心に係るキラリティーについて異性体混合物である式(V)の化合物を得るステップ
(式中、#は不斉中心を表す)
を含む製造方法。 - 式(V−A)の化合物の製造方法であって、
請求項1〜11のいずれか一項に記載の(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンの製造方法、
これに続いて、
g)塩基性物質の存在下においてエチンを用いて、(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのエチニル化、
h)リンドラー触媒の存在下において、(R,R)−イソフィトールを得る、分子水素による(7R,11R)−3,7,11,15−テトラメチルヘキサデカ−1−イン−3−オールの水素化、
または
h’)ビニルグリニャール試薬の添加により(R,R)−イソフィトールを得る(6R,10R)−6,10,14−トリメチルペンタデカン−2−オンのビニル化、
のいずれかのステップ、これに続いて、
m)(R,R)−イソフィトールと式(VI)の化合物とを縮合して、#によって示される前記中心に係るキラリティーについて異性体混合物である式(V)の化合物を得るステップ、
(式中、R11、R13およびR14は、相互に独立して、水素またはメチル基であり、ならびに、#は不斉中心を表す)
および
n)式(V−A)の化合物を式(V)の前記異性体混合物から単離するステップ
を含む製造方法。 - − 式(XI)または(XII)の少なくとも1種のケタールおよび
− 少なくとも1種のキラルイリジウム錯体
(式中、波線は、前記隣接する炭素−炭素二重結合に結合して、前記炭素−炭素二重結合を前記Z配置または前記E配置のいずれかで有する、炭素−炭素結合を表し、
および、前記式中の点線を伴う前記二重結合
は、炭素−炭素単結合または炭素−炭素二重結合のいずれかを表し、
ならびに、
は不斉中心を表し、
ならびに、
Q1およびQ2は、
各々、または、共に、C1〜C10アルキル基もしくはハロゲン化C1〜C10アルキル基を指し、
または、一緒になって、C2〜C6アルキレン基またはC6〜C8シクロアルキレン基を形成する)
を含む組成物。 - 式(XX−A)または(XX−B)
(式中、上記式中の点線を伴う二重結合
は、炭素−炭素単結合または炭素−炭素二重結合のいずれかを表し;および
波線は、隣接する炭素単結合(
が
を表す)または隣接する炭素−炭素二重結合(
が
を表す)と結合して、前記炭素−炭素二重結合を前記Z配置または前記E配置のいずれかで有する、炭素−炭素結合を表す)のケタール。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12197835.7 | 2012-12-18 | ||
| EP12197835 | 2012-12-18 | ||
| PCT/EP2013/077187 WO2014096065A1 (en) | 2012-12-18 | 2013-12-18 | (6r,10r)-6,10,14-trimetylpentadecan-2-one prepared from 6,10-dimetylundec-5-en-2-one or 6,10-dimetylundeca-5,9-dien-2-one |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2015537054A JP2015537054A (ja) | 2015-12-24 |
| JP2015537054A5 JP2015537054A5 (ja) | 2019-01-10 |
| JP6481171B2 true JP6481171B2 (ja) | 2019-03-13 |
Family
ID=47458695
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015547083A Active JP6481171B2 (ja) | 2012-12-18 | 2013-12-18 | 6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンから調製した(6r,10r)−6,10,14−トリメチルペンタデカン−2−オン |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9452961B2 (ja) |
| EP (1) | EP2935177B1 (ja) |
| JP (1) | JP6481171B2 (ja) |
| CN (1) | CN104854069B (ja) |
| BR (1) | BR112015014335B1 (ja) |
| EA (1) | EA028852B1 (ja) |
| ES (1) | ES2746749T3 (ja) |
| MY (1) | MY195300A (ja) |
| WO (1) | WO2014096065A1 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104918909B (zh) * | 2012-12-18 | 2018-05-11 | 帝斯曼知识产权资产管理有限公司 | 使用e/z异构体的混合物通过组合的不对称氢化定量获得特定产物 |
| CN104884420A (zh) * | 2012-12-18 | 2015-09-02 | 帝斯曼知识产权资产管理有限公司 | 使用e/z异构体的混合物通过联合不对称氢化和异构化定量获得特定产物 |
| BR112015014284B1 (pt) * | 2012-12-18 | 2020-01-21 | Dsm Ip Assets Bv | processo de hidrogenação assimétrica de cetais e acetais, processo para a fabricação de aldeídos ou cetonas, composição e uso da mesma |
| WO2014096098A1 (en) * | 2012-12-18 | 2014-06-26 | Dsm Ip Assets B.V. | (6r,10r)-6,10,14-trimetylpentadecan-2-one prepared from 6,10,14-trimetylpentadeca-5,9,13-trien-2-one or 6,10,14-trimetylpentadeca-5,9-dien-2-one |
| WO2018108606A1 (en) * | 2016-12-12 | 2018-06-21 | Dsm Ip Assets B.V. | Process for the manufacture of 6,10-dimethylundecan-2-one, isophytol, alpha-tocopherol (acetate) and further intermediates thereof |
| EP3470388A1 (en) * | 2017-10-13 | 2019-04-17 | Basf Se | Synthesis of aliphatic alcohols as aroma chemicals |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4028385A (en) * | 1974-09-02 | 1977-06-07 | Kuraray Co., Ltd. | Process for preparing stereospecific farnesylacetic acid and ester thereof |
| US4292459A (en) | 1975-07-16 | 1981-09-29 | Scm Corporation | Coupling reaction involving a Grignard and allylic halide |
| EP0565975B1 (de) | 1992-04-16 | 1996-09-04 | F. Hoffmann-La Roche Ag | Verfahren zur Herstellung von Isoprenderivaten |
| US5374727A (en) * | 1992-06-19 | 1994-12-20 | Hoffmann-La Roche Inc. | Asymmetric hydrogenation of dihydro-pyrido [1,2-a]indoles |
| US5955636A (en) | 1996-07-05 | 1999-09-21 | Kuraray Co., Ltd. | Process for producing 6-methyl-3-hepten-2-one and 6-methyl-2-heptanone analogues, and process for producing phyton or isophytol |
| US8426617B2 (en) | 2004-12-22 | 2013-04-23 | Dsm Ip Assets B.V. | Asymmetric hydrogenation of alkenes using chiral iridium complexes |
| CN104918909B (zh) * | 2012-12-18 | 2018-05-11 | 帝斯曼知识产权资产管理有限公司 | 使用e/z异构体的混合物通过组合的不对称氢化定量获得特定产物 |
| CN104884420A (zh) * | 2012-12-18 | 2015-09-02 | 帝斯曼知识产权资产管理有限公司 | 使用e/z异构体的混合物通过联合不对称氢化和异构化定量获得特定产物 |
| WO2014096098A1 (en) * | 2012-12-18 | 2014-06-26 | Dsm Ip Assets B.V. | (6r,10r)-6,10,14-trimetylpentadecan-2-one prepared from 6,10,14-trimetylpentadeca-5,9,13-trien-2-one or 6,10,14-trimetylpentadeca-5,9-dien-2-one |
-
2013
- 2013-12-18 US US14/652,303 patent/US9452961B2/en active Active
- 2013-12-18 WO PCT/EP2013/077187 patent/WO2014096065A1/en active Application Filing
- 2013-12-18 EP EP13815472.9A patent/EP2935177B1/en active Active
- 2013-12-18 BR BR112015014335-0A patent/BR112015014335B1/pt active IP Right Grant
- 2013-12-18 MY MYPI2015701882A patent/MY195300A/en unknown
- 2013-12-18 CN CN201380065785.4A patent/CN104854069B/zh active Active
- 2013-12-18 ES ES13815472T patent/ES2746749T3/es active Active
- 2013-12-18 JP JP2015547083A patent/JP6481171B2/ja active Active
- 2013-12-18 EA EA201500648A patent/EA028852B1/ru not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| JP2015537054A (ja) | 2015-12-24 |
| EA028852B1 (ru) | 2018-01-31 |
| BR112015014335B1 (pt) | 2020-12-08 |
| CN104854069B (zh) | 2017-06-20 |
| ES2746749T3 (es) | 2020-03-06 |
| EP2935177B1 (en) | 2019-07-24 |
| BR112015014335A2 (pt) | 2017-07-11 |
| EA201500648A1 (ru) | 2015-12-30 |
| US9452961B2 (en) | 2016-09-27 |
| CN104854069A (zh) | 2015-08-19 |
| US20150329457A1 (en) | 2015-11-19 |
| MY195300A (en) | 2023-01-12 |
| EP2935177A1 (en) | 2015-10-28 |
| WO2014096065A1 (en) | 2014-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6481171B2 (ja) | 6,10−ジメチルウンデカ−5−エン−2−オンまたは6,10−ジメチルウンデカ−5,9−ジエン−2−オンから調製した(6r,10r)−6,10,14−トリメチルペンタデカン−2−オン | |
| JP6406676B2 (ja) | 不斉水素化および異性化を組み合わせることによって特定の生成物を定量的に得るためのe/z異性体の混合物の使用 | |
| JP6390048B2 (ja) | 組み合わされた不斉水素化によって特定の生成物を定量的に得るためのe/z異性体の混合物の使用 | |
| JP6582324B2 (ja) | 6,10,14−トリメチルペンタデカ−5,9,13−トリエン−2−オンまたは6,10,14−トリメチルペンタデカ−5,9−ジエン−2−オンから調製した(6r,10r)−6,10,14−トリメチルペンタデカン−2−オン | |
| EP2935191B1 (en) | Preparation of (6r,10r)-6,10,14-trimetylpentadecan-2-one from 3,7-dimetyloct-2-enal or 3,7-dimetylocta-2,6-dienal | |
| JP6476496B2 (ja) | ケタールおよびアセタールの不斉水素化の方法 | |
| EP2935190B1 (en) | (6r,10r)-6,10,14-trimetylpentadecan-2-one prepared from 6,10-dimetylundeca-3,5,9-trien-2-one | |
| EP2935176B1 (en) | (6r,10r)-6,10,14-trimetylpentadecan-2-one prepared from (r)-3,7-dimetyloct-6-enal | |
| EP2935175B1 (en) | (6r,10r)-6,10,14-trimetylpentadecan-2-one prepared from (r)-6,10,14-trimetylpentadec-5-en-2-one |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161205 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20171012 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A132 Effective date: 20171017 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20171212 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20180322 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180821 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20181120 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190115 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190117 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6481171 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |