JP6460575B2 - Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery - Google Patents
Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery Download PDFInfo
- Publication number
- JP6460575B2 JP6460575B2 JP2015000652A JP2015000652A JP6460575B2 JP 6460575 B2 JP6460575 B2 JP 6460575B2 JP 2015000652 A JP2015000652 A JP 2015000652A JP 2015000652 A JP2015000652 A JP 2015000652A JP 6460575 B2 JP6460575 B2 JP 6460575B2
- Authority
- JP
- Japan
- Prior art keywords
- active material
- lithium
- secondary battery
- positive electrode
- lithium secondary
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images

Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Landscapes
- Inorganic Compounds Of Heavy Metals (AREA)
- Catalysts (AREA)
- Battery Electrode And Active Subsutance (AREA)
Description
本発明は、新規なリチウム遷移金属複合酸化物を含むリチウム二次電池用正極活物質、その正極活物質を含有するリチウム二次電池用電極、その電極を備えたリチウム二次電池及びその電池を集合した蓄電装置に関する。 The present invention relates to a positive electrode active material for a lithium secondary battery containing a novel lithium transition metal composite oxide, an electrode for a lithium secondary battery containing the positive electrode active material, a lithium secondary battery including the electrode, and a battery thereof. The present invention relates to an assembled power storage device.
従来、リチウム二次電池用正極活物質として、α−NaFeO2型結晶構造を有する「LiMeO2型」活物質(Meは遷移金属)が検討され、LiCoO2を用いたリチウム二次電池が広く実用化されていた。しかし、LiCoO2の放電容量は120〜130mAh/g程度であった。前記Meとして、地球資源として豊富なMnを用いることが望まれてきた。しかし、MeとしてMnを含有させた「LiMeO2型」活物質は、Meに対するMnのモル比Mn/Meが0.5を超える場合には、充電をするとスピネル型へと構造変化が起こり、結晶構造が維持できないため、充放電サイクル性能が著しく劣るという問題があった。 Conventionally, as a positive electrode active material for a lithium secondary battery, a “LiMeO 2 type” active material (Me is a transition metal) having an α-NaFeO 2 type crystal structure has been studied, and lithium secondary batteries using LiCoO 2 have been widely put into practical use. It was converted. However, the discharge capacity of LiCoO 2 was about 120 to 130 mAh / g. As Me, it has been desired to use abundant Mn as a global resource. However, the “LiMeO 2 type” active material containing Mn as Me, when the molar ratio of Mn to Me, Mn / Me exceeds 0.5, the structure changes to spinel type when charged, Since the structure could not be maintained, there was a problem that the charge / discharge cycle performance was extremely inferior.
そこで、Meに対するMnのモル比Mn/Meが0.5以下であり、充放電サイクル性能の点でも優れる「LiMeO2型」活物質が種々提案され、一部実用化されている。例えば、LiNi1/2Mn1/2O2やLiCo1/3Ni1/3Mn1/3O2は、150〜180mAh/gの放電容量を有する。 Accordingly, various “LiMeO 2 type” active materials having a Mn to Me molar ratio Mn / Me of 0.5 or less and excellent in charge / discharge cycle performance have been proposed and partially put into practical use. For example, LiNi 1/2 Mn 1/2 O 2 and LiCo 1/3 Ni 1/3 Mn 1/3 O 2 have a discharge capacity of 150 to 180 mAh / g.
近年、MeとしてNi,Co及びMnを含み、Meに対するMnのモル比Mn/Meが0.5以上であり、充電をしてもα−NaFeO2構造を維持できる活物質材料が提案された。 In recent years, there has been proposed an active material that contains Ni, Co, and Mn as Me, has a molar ratio of Mn to Me, Mn / Me of 0.5 or more, and can maintain an α-NaFeO 2 structure even when charged.
特許文献1及び2には、MeとしてNi,Co及びMnを含み、Meに対するMnのモル比Mn/Meが0.5以上である活物質を用いた電池の製造方法として、4.3V(vs.Li/Li+)を超え4.8V以下(vs.Li/Li+)の正極電位範囲に出現する、電位変化が比較的平坦な領域に少なくとも至る充電を行う製造工程を設けることにより、使用時において、充電時の正極の最大到達電位が4.3V(vs.Li/Li+)以下又は4.4V(vs.Li/Li+)未満である充電方法が採用された場合であっても、200mAh/g以上の放電容量が得られる電池を製造できることが記載されている。
このように、従来の「LiMeO2型」正極活物質の場合とは異なり、MeとしてNi,Co及びMnを含み、Meに対するMnのモル比Mn/Meが0.5以上である正極活物質では、少なくとも最初の充電において4.3Vを超える比較的高い電位、特に4.4V以上の電位に至って行うことにより、高い放電容量が得られるという特徴がある。 Thus, unlike the case of the conventional “LiMeO 2 type” positive electrode active material, the positive electrode active material containing Ni, Co and Mn as Me, and the molar ratio of Mn to Me, Mn / Me is 0.5 or more. In at least the first charge, a relatively high potential exceeding 4.3 V, in particular, a potential of 4.4 V or higher is achieved, whereby a high discharge capacity can be obtained.
なお、この材料は、遷移金属(Me)の比率に対するリチウム(Li)の組成比率Li/Meが1より大きく、例えばLi/Meが1.25〜1.6であるように原料を混合して合成されることから、「リチウム過剰型」活物質とも呼ばれ、合成後の組成はLi1+αMe1−αO2(α>0)と表記できる。ここで、遷移金属(Me)の比率に対するリチウム(Li)の組成比率Li/Meをβとすると、β=(1+α)/(1−α)であるから、例えば、Li/Meが1.5のとき、α=0.2である。 In this material, the raw materials are mixed so that the composition ratio Li / Me of lithium (Li) to the ratio of transition metal (Me) is greater than 1, for example, Li / Me is 1.25 to 1.6. Since it is synthesized, it is also called a “lithium-excess type” active material, and the composition after synthesis can be expressed as Li 1 + α Me 1-α O 2 (α> 0). Here, when the composition ratio Li / Me of lithium (Li) with respect to the ratio of the transition metal (Me) is β, β = (1 + α) / (1-α), and thus, for example, Li / Me is 1.5. In this case, α = 0.2.
特許文献3には、「リチウム含有酸化物から正極活物質を得る正極活物質の製造方法であって、前記リチウム含有酸化物を酸性水溶液で処理する工程を備え、前記リチウム含有酸化物は、Li1+x(MnyM1−y)1−xO2(0<x<0.4、0<y≦1)を含み、前記Mはマンガンを除く少なくとも1種の遷移金属を含み、前記酸性水溶液中の水素イオン量は、前記リチウム含有酸化物1molに対してxmol以上5xmol未満であることを特徴とする正極活物質の製造方法。」(請求項5)の発明が記載され、優れた負荷特性及び高い初期充放電効率を有するリチウム二次電池を製造することができることが記載されている。
In
特許文献4には、Li(LixMnyMez)OpFq(MeはCo、Ni、Cr、Fe、Al、Ti、Zr、Mgから選ばれる少なくとも一種の元素、0.09<x<0.3、0.4≦y/(y+z)≦0.8、x+y+z=1、1.9<p<2.1、0≦q≦0.1)であるリチウム含有複合酸化物の表面にZr、Ti、Sn、Mg、Ba、Pb、Bi、Nb、Ta、Zn、Y、La、Sr、Ce、InおよびAlから選ばれる少なくとも一種の金属元素の酸化物の微粒子を付着する粒子からなるリチウムイオン二次電池用の正極活物質が記載されている。 Patent Document 4 discloses Li (Li x Mn y Me z ) O p F q (Me is at least one element selected from Co, Ni, Cr, Fe, Al, Ti, Zr, and Mg, 0.09 <x <0.3, lithium in which 0.4 ≦ y / (y + z) ≦ 0.8, x + y + z = 1, 1.9 <p <2.1, 0 ≦ q ≦ 0.1) An oxide of at least one metal element selected from Zr, Ti, Sn, Mg, Ba, Pb, Bi, Nb, Ta, Zn, Y, La, Sr, Ce, In and Al on the surface of the composite oxide containing A positive electrode active material for a lithium ion secondary battery comprising particles to which fine particles adhere is described.
特許文献5には、Li(1+x)Co(1−y)MyO(2−z)(MはMg、Al、B、Ti、V、Cr、Mn、Fe、Ni、Cu、Zn、Mo、Sn、Ca、Sr、W、Y、Zrよりなる群から選ばれた少なくとも1種の元素、−0.10≦x≦0.10、0≦y<0.50、z=−0.10≦z≦0.20)で表される複合酸化物粒子表面に設けられ、Liと、Ni及び/又はMnを含む酸化物被覆層と、被覆層に設けられた、ランタノイド元素を含む酸化物表面層を備える非水電解質二次電池用正極活物質であって、粒子同士の結着を抑制し、被覆層の破壊や被覆層の破壊による活性な複合酸化物粒子表面の露呈を防止することが記載されている。
In
特許文献6には、α−NaFeO2型結晶構造を有し、組成式LixMnaNibCocOd(0≦x≦1.3、a+b+c=1、|a−b|≦0.03、0≦c<1、1.7≦d≦2.3)で表されるリチウム遷移金属複合酸化物である母材の電解質と接触する表面に、Ce等の3族の元素が存在する正極活物質であって、充電状態での保存性と充放電サイクル性能に優れたリチウム二次電池を提供できることが記載されている。 Patent Document 6 has an α-NaFeO 2 type crystal structure and a composition formula Li x Mn a Ni b Co c O d (0 ≦ x ≦ 1.3, a + b + c = 1, | a−b | ≦ 0.03, 0 ≦ c <1, 1.7 ≦ d ≦ 2.3) on the surface in contact with the electrolyte of the base metal which is a lithium transition metal composite oxide. It is described that it is a positive electrode active material in which an element is present, and can provide a lithium secondary battery excellent in storage stability and charge / discharge cycle performance in a charged state.
「Li過剰型」の活物質の放電容量は、概して、「LiMeO2型」活物質よりも大きいものの、初期充放電効率(以下「初期効率」という。)が低いことが知られている。このうち、負荷特性及び初期効率については、特許文献3に記載されているように、活物質を酸処理することで向上することが公知である。しかし、近年、電気自動車、ハイブリッド自動車、プラグインハイブリッド自動車といった自動車分野に使用されるリチウム二次電池には、初期効率とともに、放電容量維持率の高い正極活物質が求められている。
Although the discharge capacity of the “Li-excess type” active material is generally larger than that of the “LiMeO 2 type” active material, it is known that the initial charge / discharge efficiency (hereinafter referred to as “initial efficiency”) is low. Among these, as described in
特許文献4には、「Li過剰型」の活物質について、金属元素の酸化物の微粒子を付着させることにより、サイクル特性に効果を上げることが記載されているが、実施例に記載され、効果が確認されているのは、Zrの場合のみである。 Patent Document 4 describes that, for an “Li-excess type” active material, by attaching fine particles of an oxide of a metal element, the effect on cycle characteristics is improved. Is confirmed only in the case of Zr.
特許文献5には、Mn、Niを含み得るリチウムコバルト複合酸化物粒子表面にセリウムを含む表面層を形成した活物質の実施例が記載されているが、この表面層は、粒子同士の結着防止を目的とするものであって、「リチウム過剰型」の活物質において、初期効率と放電容量維持率を高めるという課題を示すものでない。
特許文献6に記載の活物質も、「リチウム過剰型」を想定したものでなく、初期効率と放電容量維持率を高めるという課題を示すものでない。 The active material described in Patent Document 6 is not supposed to be “lithium-excess type”, and does not indicate the problem of increasing the initial efficiency and the discharge capacity retention rate.
本発明は、上記課題に鑑み、初期効率が優れ、かつ放電容量維持率の高いリチウム二次電池用正極活物質、その正極活物質を含有するリチウム二次電池用電極、及びその電極を備えたリチウム二次電池を提供することを目的とする。 In view of the above problems, the present invention includes a positive electrode active material for a lithium secondary battery having excellent initial efficiency and a high discharge capacity retention rate, an electrode for a lithium secondary battery containing the positive electrode active material, and the electrode. An object is to provide a lithium secondary battery.
本発明者らは、上記目的を達成するために、以下の手段を採用する。
(1)α−NaFeO2構造を有するリチウム遷移金属複合酸化物を含む正極活物質であって、前記リチウム遷移金属複合酸化物は、遷移金属(Me)がCo、Ni及びMnを含み、Liと遷移金属(Me)のモル比(Li/Me)が1<Li/Meであり、Mnと遷移金属(Me)のモル比(Mn/Me)が0.5<Mn/Meであり、Ceを含有するリチウム二次電池用正極活物質。
(2)前記リチウム遷移金属複合酸化物は、金属換算したCeの含有比率が0.15〜3.37質量%である前記(1)の正極活物質。
(3)CuKα管球を用いたX線回折パターン解析において、(104)面に帰属される回折ピークの半値幅(FWHM)が0.269≦FWHM≦0.273であることを特徴とする前記(1)又は(2)の正極活物質。
(4)前記(1)〜(3)のいずれかのリチウム二次電池用正極活物質を含有するリチウム二次電池用電極。
(5)前記(4)のリチウム二次電池用電極を備えたリチウム二次電池。
In order to achieve the above object, the present inventors adopt the following means.
(1) A positive electrode active material including a lithium transition metal composite oxide having an α-NaFeO 2 structure, wherein the transition metal (Me) includes Co, Ni, and Mn, Li and The molar ratio of transition metal (Me) (Li / Me) is 1 <Li / Me, the molar ratio of Mn to transition metal (Me) (Mn / Me) is 0.5 <Mn / Me, and Ce is A positive electrode active material for a lithium secondary battery.
(2) The positive electrode active material according to (1), wherein the lithium-transition metal composite oxide has a metal-converted Ce content ratio of 0.15 to 3.37% by mass.
(3) In the X-ray diffraction pattern analysis using a CuKα tube, the half width (FWHM) of the diffraction peak attributed to the (104) plane is 0.269 ≦ FWHM ≦ 0.273 The positive electrode active material according to (1) or (2).
(4) The electrode for lithium secondary batteries containing the positive electrode active material for lithium secondary batteries in any one of said (1)-(3).
(5) A lithium secondary battery comprising the lithium secondary battery electrode of (4).
本発明により、初期効率が優れ、かつ放電容量維持率の高いリチウム二次電池用正極活物質、その正極活物質を含有するリチウム二次電池用電極、及びその電極を備えたリチウム二次電池を提供することができる。 According to the present invention, a positive electrode active material for a lithium secondary battery having excellent initial efficiency and a high discharge capacity maintenance rate, an electrode for a lithium secondary battery containing the positive electrode active material, and a lithium secondary battery including the electrode Can be provided.
本発明者らは、「Li過剰型」の活物質を酸処理することにより、初期効率を向上させることができるものの、充放電サイクル後の放電容量維持率を高くすることができない原因は、酸処理によって、充放電サイクルにともなう活物質からのMnの溶出が促進されるためと推察した。 The present inventors can improve the initial efficiency by acid-treating the “Li-excess type” active material, but the reason why the discharge capacity retention rate after the charge / discharge cycle cannot be increased is as follows. It was inferred that the treatment promoted the elution of Mn from the active material accompanying the charge / discharge cycle.
そこで、「Li過剰型」の活物質の粒子表面を保護することで、充放電中の活物質からのMnの溶出を緩和し、充放電サイクルに伴う放電容量の低下を抑制することができると考え、種々の金属による処理を試みたところ、酸処理水溶液にCeを添加して酸処理した後、加熱することにより、活物質にCeを付与することで効果があることを見出した。 Therefore, by protecting the particle surface of the “Li-excess type” active material, the elution of Mn from the active material during charge / discharge can be relaxed, and the decrease in discharge capacity associated with the charge / discharge cycle can be suppressed. As a result, when treatments with various metals were attempted, it was found that Ce was added to the acid treatment aqueous solution, acid treatment was performed, and then heating was performed to impart Ce to the active material.
以下、本発明に好適な正極活物質及びその製造方法について詳述するが、本発明はこれに限定されるものではない。 Hereinafter, although the positive electrode active material suitable for this invention and its manufacturing method are explained in full detail, this invention is not limited to this.
(正極活物質)
本発明の正極活物質は、典型的には、Li1+α(CoaNibMnc)1−αO2、但し、α>1、a+b+c=1、a>0、b>0、c>0.5で表わされるものであり、Li、Co、Ni及びMnからなる複合酸化物であるが、初期効率及び高率放電性能が優れたリチウム二次電池を得るために、遷移金属元素Meに対するLiのモル比Li/Meは、1.2より大きく且つ1.6より小さいこと、すなわち、組成式Li1+αMe1−αO2において1.2<(1+α)/(1−α)<1.6とすることが好ましい。なかでも、放電容量が特に大きく、初期効率及び高率放電性能が優れたリチウム二次電池を得ることができるという観点から、前記Li/Meが1.25〜1.5のものを選択することが好ましい。なお、本発明において、モル比Li/Meは、酸処理後のものであり、酸処理前の出発物質ではこれよりもやや高くなる。
(Positive electrode active material)
The positive electrode active material of the present invention is typically Li 1 + α (Co a Ni b Mn c ) 1-α O 2 , where α> 1, a + b + c = 1, a> 0, b> 0, c> 0. In order to obtain a lithium secondary battery excellent in initial efficiency and high-rate discharge performance, it is a composite oxide composed of Li, Co, Ni, and Mn. The molar ratio Li / Me is larger than 1.2 and smaller than 1.6, that is, 1.2 <(1 + α) / (1−α) <1 in the composition formula Li 1 + α Me 1-α O 2 . 6 is preferable. Among these, from the viewpoint that a lithium secondary battery having a particularly large discharge capacity and excellent initial efficiency and high rate discharge performance can be obtained, the one having the Li / Me of 1.25 to 1.5 should be selected. Is preferred. In the present invention, the molar ratio Li / Me is after acid treatment, and is slightly higher in the starting material before acid treatment.
また、本発明の正極活物質は、リチウム二次電池の初期効率及び高率放電性能を向上させるために、遷移金属元素Meに対するMnのモル比Mn/Meは0.5以上であることが好ましい。「LiMeO2型」活物質では、モル比Mn/Meが0.5を超える場合、充電をするとスピネル型へと構造変化が起こり、α−NaFeO2構造に帰属される構造を有さないものとなり、リチウム二次電池用活物質として問題があったのに対し、「リチウム過剰型」活物質では、モル比Mn/Meが0.5を超えても、充電をした場合にα−NaFeO2構造を維持できる。したがって、モル比Mn/Meが0.5を超えるという構成は、「リチウム過剰型」活物質を特徴付けるものである。モル比Mn/Meは0.51〜0.75とすることがより好ましい。 In addition, in the positive electrode active material of the present invention, the molar ratio Mn / Me of the transition metal element Me is preferably 0.5 or more in order to improve the initial efficiency and high rate discharge performance of the lithium secondary battery. . In the “LiMeO 2 type” active material, when the molar ratio Mn / Me exceeds 0.5, the structure changes to the spinel type when charged and does not have a structure attributed to the α-NaFeO 2 structure. However, the “lithium-excess type” active material has a problem as an active material for a lithium secondary battery, and the α-NaFeO 2 structure when charged even when the molar ratio Mn / Me exceeds 0.5. Can be maintained. Therefore, the configuration where the molar ratio Mn / Me exceeds 0.5 characterizes the “lithium-rich” active material. The molar ratio Mn / Me is more preferably 0.51 to 0.75.
また、リチウム二次電池の初期効率及び高率放電性能を向上させるために、遷移金属元素Meに対するCoのモル比Co/Meは、0.05〜0.40とすることが好ましく、0.10〜0.30とすることがより好ましい。 In order to improve the initial efficiency and high rate discharge performance of the lithium secondary battery, the molar ratio Co / Me of the Co to the transition metal element Me is preferably 0.05 to 0.40, and 0.10 More preferably, it is set to ˜0.30.
本発明に係るリチウム遷移金属複合酸化物は、本質的に、Li、Co、Ni及びMnからなる複合酸化物であるが、放電容量を向上させるために、Naを1000ppm以上含ませることが好ましい。Naの含有量は、2000〜10000ppmがより好ましい。 The lithium transition metal composite oxide according to the present invention is essentially a composite oxide composed of Li, Co, Ni, and Mn, but it is preferable to contain 1000 ppm or more of Na in order to improve the discharge capacity. As for content of Na, 2000-10000 ppm is more preferable.
Naを含有させるために、後述する炭酸塩前駆体を作製する工程において、炭酸ナトリウム等のナトリウム化合物を中和剤として使用し、洗浄工程でNaを残存させるか、及び、その後の焼成工程において炭酸ナトリウム等のナトリウム化合物を添加する方法を採用することができる。 In order to contain Na, in the step of preparing a carbonate precursor, which will be described later, a sodium compound such as sodium carbonate is used as a neutralizing agent, and Na is left in the washing step. A method of adding a sodium compound such as sodium can be employed.
また、本発明の効果を損なわない範囲で、Na以外のアルカリ金属、Mg,Ca等のアルカリ土類金属、Fe,Zn等の3d遷移金属に代表される遷移金属など少量の他の金属を含有することを排除するものではない。 In addition, a small amount of other metals such as alkali metals other than Na, alkaline earth metals such as Mg and Ca, transition metals typified by 3d transition metals such as Fe and Zn, and the like are included as long as the effects of the present invention are not impaired. It does not exclude doing.
本発明に係るリチウム遷移金属複合酸化物は、α−NaFeO2構造を有している。合成後(充放電を行う前)の上記リチウム遷移金属複合酸化物は、空間群P3112あるいはR3−mに帰属される。このうち、空間群P3112に帰属されるものには、CuKα管球を用いたX線回折図上、2θ=21°付近に超格子ピーク(Li[Li1/3Mn2/3]O2型の単斜晶に見られるピーク)が確認される。ところが、一度でも充電を行い、結晶中のLiが脱離すると結晶の対称性が変化することにより、上記超格子ピークが消滅して、上記リチウム遷移金属複合酸化物は空間群R3−mに帰属されるようになる。ここで、P3112は、R3−mにおける3a、3b、6cサイトの原子位置を細分化した結晶構造モデルであり、R3−mにおける原子配置に秩序性が認められるときに該P3112モデルが採用される。なお、「R3−m」は本来「R3m」の「3」の上にバー「−」を施して表記すべきものである。 The lithium transition metal composite oxide according to the present invention has an α-NaFeO 2 structure. The lithium transition metal composite oxide after synthesis (before charging and discharging) is attributed to the space group P3 1 12 or R3-m. Among these, those belonging to the space group P3 1 12 include a superlattice peak (Li [Li 1/3 Mn 2/3 ] O around 2θ = 21 ° on an X-ray diffraction diagram using a CuKα tube. A peak observed in type 2 monoclinic crystal) is confirmed. However, when charging is performed once and Li in the crystal is desorbed, the symmetry of the crystal changes, whereby the superlattice peak disappears and the lithium transition metal composite oxide belongs to the space group R3-m. Will come to be. Here, P3 1 12 is a crystal structure model in which the atomic positions of the 3a, 3b, and 6c sites in R3-m are subdivided, and when ordering is recognized in the atomic arrangement in R3-m, the P3 1 12 model Is adopted. Note that “R3-m” should be represented by adding a bar “-” on “3” of “R3m”.
本発明に係るリチウム遷移金属複合酸化物は、X線回折パターンを元に空間群R3−mを結晶構造モデルに用いたときに(003)面に帰属される回折ピークの半値幅を0.202°〜0.265°の範囲内とすることが好ましい。また、(104)面に帰属される回折ピークの半値幅を0.265°〜0.285°の範囲内とすることが好ましい。こうすることにより、正極活物質の初期効率を高めることが可能となる。なお、X線回折図上2θ=44°±1°、及び18°±1°に存在する回折ピークが、空間群P3112ではミラー指数hklにおける(114)面、及び(003)面にそれぞれ指数付けされ、空間群R3−mではミラー指数hklにおける(104)面、及び(003)面にそれぞれ指数付けされる。 The lithium transition metal composite oxide according to the present invention has a half width of the diffraction peak attributed to the (003) plane of 0.202 when the space group R3-m is used as a crystal structure model based on the X-ray diffraction pattern. It is preferable to be within the range of ° to 0.265 °. Moreover, it is preferable that the half width of the diffraction peak attributed to the (104) plane is in the range of 0.265 ° to 0.285 °. By doing so, it is possible to increase the initial efficiency of the positive electrode active material. Note that diffraction peaks existing at 2θ = 44 ° ± 1 ° and 18 ° ± 1 ° on the X-ray diffraction diagram are respectively present on the (114) plane and the (003) plane in the Miller index hkl in the space group P3 1 12 respectively. The space group R3-m is indexed on the (104) plane and the (003) plane in the Miller index hkl.
また、本発明に係るリチウム遷移金属複合酸化物は、1000℃で熱処理を行ったときに結晶構造が変化しないものであることが好ましい。これは、1000℃で熱処理を行ったときに、X線回折図上空間群R3−mに帰属される単一相(α−NaFeO2構造)として観察されることにより確認できる。これにより、高率放電性能が優れたリチウム二次電池を得ることができる。 In addition, the lithium transition metal composite oxide according to the present invention preferably has a crystal structure that does not change when heat treatment is performed at 1000 ° C. This can be confirmed by observation as a single phase (α-NaFeO 2 structure) belonging to the space group R3-m on the X-ray diffraction diagram when heat treatment is performed at 1000 ° C. Thereby, a lithium secondary battery excellent in high rate discharge performance can be obtained.
さらに、リチウム遷移金属複合酸化物は、X線回折パターンを基にリートベルト法による結晶構造解析から求められる酸素位置パラメータが、放電末において0.262以下、充電末において0.267以上であることが好ましい。これにより、高率放電性能が優れたリチウム二次電池を得ることができる。なお、酸素位置パラメータとは、空間群R3−mに帰属されるリチウム遷移金属複合酸化物のα―NaFeO2型結晶構造について、Me(遷移金属)の空間座標を(0,0,0)、Li(リチウム)の空間座標を(0,0,1/2)、O(酸素)の空間座標を(0,0,z)と定義したときの、zの値をいう。即ち、酸素位置パラメータは、O(酸素)位置がMe(遷移金属)位置からどれだけ離れているかを示す相対的な指標となる(特許文献1及び2参照)。
Further, in the lithium transition metal composite oxide, the oxygen position parameter obtained from the crystal structure analysis by the Rietveld method based on the X-ray diffraction pattern is 0.262 or less at the end of discharge and 0.267 or more at the end of charge. Is preferred. Thereby, a lithium secondary battery excellent in high rate discharge performance can be obtained. Note that the oxygen positional parameter is Me (transition metal) spatial coordinates (0, 0, 0) for the α-NaFeO 2 type crystal structure of the lithium transition metal composite oxide belonging to the space group R3-m, This is the value of z when the spatial coordinates of Li (lithium) are defined as (0, 0, 1/2) and the spatial coordinates of O (oxygen) are defined as (0, 0, z). In other words, the oxygen position parameter is a relative index indicating how far the O (oxygen) position is from the Me (transition metal) position (see
本発明に係るリチウム遷移金属複合酸化物及びその炭酸塩前駆体は、粒度分布測定における50%粒子径(D50)が5〜18μmであることが好ましい。リチウム遷移金属複合酸化物を水酸化物前駆体から作製する場合はもっと小粒径に制御しないと優れた性能が得られないが、炭酸塩前駆体から作製することにより、粒度分布測定における50%粒子径(D50)が5〜18μm程度であっても、放電容量が大きい正極活物質が得られる。 The lithium transition metal composite oxide and its carbonate precursor according to the present invention preferably have a 50% particle diameter (D50) of 5 to 18 μm in the particle size distribution measurement. When producing a lithium transition metal composite oxide from a hydroxide precursor, excellent performance cannot be obtained unless the particle size is controlled to be smaller, but by producing from a carbonate precursor, 50% in the particle size distribution measurement. Even when the particle diameter (D50) is about 5 to 18 μm, a positive electrode active material having a large discharge capacity can be obtained.
本発明に係る正極活物質のBET比表面積は、初期効率、高率放電性能が優れたリチウム二次電池を得るために、1m2/g以上が好ましく、2〜7m2/gがより好ましい。
また、タップ密度は、高率放電性能が優れたリチウム二次電池を得るために、1.25g/cc以上が好ましく、1.7g/cc以上がより好ましい。
BET specific surface area of the positive electrode active material according to the present invention, the initial efficiency, in order to obtain a lithium secondary battery having excellent high rate discharge performance, preferably at least 1m 2 / g, 2~7m 2 / g is more preferable.
Further, the tap density is preferably 1.25 g / cc or more, and more preferably 1.7 g / cc or more in order to obtain a lithium secondary battery excellent in high rate discharge performance.
本発明に係るリチウム遷移金属複合酸化物は、窒素ガス吸着法を用いた吸着等温線からBJH法で求めた微分細孔容積の最大値を示す細孔径が60nmまでの範囲内の細孔領域(60nmまでの細孔領域)にて0.055cc/g以上の細孔容積とすることが好ましい。これにより、初期効率及び高率放電性能が優れたリチウム二次電池を得ることができる。また、上記細孔容積を0.08cc/g以下とすることにより、高率放電性能が特に優れたリチウム二次電池を得ることができるから、上記細孔容積は0.055〜0.08cc/gであることが好ましい。 The lithium transition metal composite oxide according to the present invention has a pore region having a pore diameter within a range of up to 60 nm, which indicates the maximum value of the differential pore volume determined by the BJH method from the adsorption isotherm using the nitrogen gas adsorption method ( It is preferable that the pore volume be 0.055 cc / g or more in the pore region (up to 60 nm). Thereby, a lithium secondary battery excellent in initial efficiency and high rate discharge performance can be obtained. In addition, by setting the pore volume to 0.08 cc / g or less, a lithium secondary battery having particularly excellent high rate discharge performance can be obtained. Therefore, the pore volume is 0.055 to 0.08 cc / g. It is preferable that it is g.
(正極活物質の製造方法)
次に、本発明のリチウム二次電池用活物質を製造する方法について説明する。
本発明のリチウム二次電池用活物質は、基本的に、活物質を構成する金属元素(Li,Mn,Co,Ni)を目的とする活物質(酸化物)の組成通りに含有する原料を調製し、これを焼成することによって得ることができる。但し、Li原料の量については、焼成中にLi原料の一部が消失することを見込んで、1〜5%程度過剰に仕込むことが好ましい。
目的とする組成の酸化物を作製するにあたり、Li,Co,Ni,Mnのそれぞれの塩を混合・焼成するいわゆる「固相法」や、あらかじめCo,Ni,Mnを一粒子中に存在させた共沈前駆体を作製しておき、これにLi塩を混合・焼成する「共沈法」が知られている。「固相法」による合成過程では、特にMnはCo,Niに対して均一に固溶しにくいため、各元素が一粒子中に均一に分布した試料を得ることは困難である。これまで文献などにおいては固相法によってNiやCoの一部にMnを固溶(LiNi1−xMnxO2など)しようという試みが多数なされているが、「共沈法」を選択する方が原子レベルで均一相を得ることが容易である。そこで、後述する実施例においては、「共沈法」を採用した。
(Method for producing positive electrode active material)
Next, a method for producing the active material for a lithium secondary battery of the present invention will be described.
The active material for a lithium secondary battery of the present invention basically includes a raw material containing a metal element (Li, Mn, Co, Ni) constituting the active material according to the composition of the active material (oxide). It can be obtained by preparing and baking this. However, with respect to the amount of the Li raw material, it is preferable to add an excess of about 1 to 5% in view of the disappearance of a part of the Li raw material during firing.
In producing an oxide having a desired composition, a so-called “solid phase method” in which salts of Li, Co, Ni, and Mn are mixed and fired, or Co, Ni, and Mn were previously present in one particle. A “coprecipitation method” is known in which a coprecipitation precursor is prepared, and a Li salt is mixed and fired therein. In the synthesis process by the “solid phase method”, especially Mn is difficult to uniformly dissolve in Co and Ni, so it is difficult to obtain a sample in which each element is uniformly distributed in one particle. In literatures and the like, many attempts have been made to dissolve Mn in a part of Ni or Co (LiNi 1-x Mn x O 2 etc.) by solid phase method, but the “coprecipitation method” is selected. It is easier to obtain a homogeneous phase at the atomic level. Therefore, the “coprecipitation method” is employed in the examples described later.
共沈前駆体を作製するにあたって、Co,Ni,MnのうちMnは酸化されやすく、Co,Ni,Mnが2価の状態で均一に分布した共沈前駆体を作製することが容易ではないため、Co,Ni,Mnの原子レベルでの均一な混合は不十分なものとなりやすい。特に本発明の組成範囲においては、Mn比率がCo,Ni比率に比べて高いので、水溶液中の溶存酸素を除去することが特に重要である。溶存酸素を除去する方法としては、酸素を含まないガスをバブリングする方法が挙げられる。酸素を含まないガスとしては、限定されるものではないが、窒素ガス、アルゴンガス、二酸化炭素(CO2)等を用いることができる。なかでも、後述する実施例のように、共沈炭酸塩前駆体を作製する場合には、酸素を含まないガスとして二酸化炭素を採用すると、炭酸塩がより生成しやすい環境が与えられるため、好ましい。 When preparing a coprecipitation precursor, Mn is easily oxidized among Co, Ni and Mn, and it is not easy to prepare a coprecipitation precursor in which Co, Ni and Mn are uniformly distributed in a divalent state. Uniform mixing at the atomic level of Co, Ni and Mn tends to be insufficient. In particular, in the composition range of the present invention, since the Mn ratio is higher than the Co and Ni ratios, it is particularly important to remove dissolved oxygen in the aqueous solution. Examples of the method for removing dissolved oxygen include a method of bubbling a gas not containing oxygen. The gas not containing oxygen is not limited, but nitrogen gas, argon gas, carbon dioxide (CO 2 ), or the like can be used. Among these, when preparing a coprecipitated carbonate precursor as in the examples described later, it is preferable to employ carbon dioxide as a gas not containing oxygen because an environment in which carbonate is more easily generated is provided. .
溶液中でCo、Ni及びMnを含有する化合物を共沈させて前駆体を製造する工程におけるpHは、限定されるものではないが、前記共沈前駆体を共沈炭酸塩前駆体として作製しようとする場合には、7.5〜11とすることができる。タップ密度を大きくするためには、pHを制御することが好ましい。pHを9.4以下とすることにより、タップ密度を1.25g/cc以上とすることができ、高率放電性能を向上させることができる。さらに、pHを8.0以下とすることにより、粒子成長速度を促進できるので、原料水溶液滴下終了後の撹拌継続時間を短縮できる。 The pH in the step of producing a precursor by co-precipitation of a compound containing Co, Ni and Mn in a solution is not limited, but let's make the co-precipitation precursor as a co-precipitation carbonate precursor. In this case, it can be set to 7.5 to 11. In order to increase the tap density, it is preferable to control the pH. By setting the pH to 9.4 or less, the tap density can be set to 1.25 g / cc or more, and the high rate discharge performance can be improved. Furthermore, since the particle growth rate can be accelerated by adjusting the pH to 8.0 or less, the stirring continuation time after the raw material aqueous solution dropping is completed can be shortened.
前記共沈前駆体は、MnとNiとCoとが均一に混合された化合物であることが好ましい。本発明においては、放電容量が大きいリチウム二次電池用活物質を得るために、共沈前駆体を炭酸塩とすることが好ましい。また、錯化剤を用いた晶析反応等を用いることによって、より嵩密度の大きな前駆体を作製することもできる。その際、Li源と混合・焼成することでより高密度の活物質を得ることができるので電極面積あたりのエネルギー密度を向上させることができる。 The coprecipitation precursor is preferably a compound in which Mn, Ni, and Co are uniformly mixed. In the present invention, in order to obtain an active material for a lithium secondary battery having a large discharge capacity, the coprecipitation precursor is preferably a carbonate. In addition, a precursor having a larger bulk density can be produced by using a crystallization reaction using a complexing agent. At that time, a higher density active material can be obtained by mixing and firing with a Li source, so that the energy density per electrode area can be improved.
前記共沈前駆体の原料は、Mn化合物としては酸化マンガン、炭酸マンガン、硫酸マンガン、硝酸マンガン、酢酸マンガン等を、Ni化合物としては、水酸化ニッケル、炭酸ニッケル、硫酸ニッケル、硝酸ニッケル、酢酸ニッケル等を、Co化合物としては、硫酸コバルト、硝酸コバルト、酢酸コバルト等を一例として挙げることができる。 The raw material of the coprecipitation precursor is manganese oxide, manganese carbonate, manganese sulfate, manganese nitrate, manganese acetate, etc. as the Mn compound, and nickel hydroxide, nickel carbonate, nickel sulfate, nickel nitrate, nickel acetate as the Ni compound. As examples of the Co compound, cobalt sulfate, cobalt nitrate, cobalt acetate, and the like can be given as examples.
本発明においては、アルカリ性を保った反応槽に前記共沈前駆体の原料水溶液を滴下供給して共沈炭酸塩前駆体を得る反応晶析法を採用する。ここで、中和剤として、リチウム化合物、ナトリウム化合物、カリウム化合物等を使用することができるが、炭酸ナトリウム、炭酸ナトリウムと炭酸リチウム、又は、炭酸ナトリウムと炭酸カリウムの混合物を使用することが好ましい。Naを1000ppm以上残存させるために、炭酸ナトリウムと炭酸リチウムのモル比であるNa/Li、又は、炭酸ナトリウムと炭酸カリウムのモル比であるNa/Kは、1/1[M]以上とすることが好ましい。Na/Li又はNa/Kを1/1[M]以上とすることにより、引き続く洗浄工程でNaが除去されすぎて1000ppm未満となってしまう虞を低減できる。 In the present invention, a reaction crystallization method is employed in which a raw material aqueous solution of the coprecipitation precursor is supplied dropwise to a reaction tank maintaining alkalinity to obtain a coprecipitation carbonate precursor. Here, lithium compounds, sodium compounds, potassium compounds, and the like can be used as the neutralizing agent, but it is preferable to use sodium carbonate, sodium carbonate and lithium carbonate, or a mixture of sodium carbonate and potassium carbonate. In order to leave Na at least 1000 ppm, Na / Li, which is the molar ratio of sodium carbonate to lithium carbonate, or Na / K, which is the molar ratio of sodium carbonate to potassium carbonate, is 1/1 [M] or more. Is preferred. By setting Na / Li or Na / K to 1/1 [M] or more, it is possible to reduce the possibility that Na will be excessively removed in the subsequent washing step and become less than 1000 ppm.
前記原料水溶液の滴下速度は、生成する共沈前駆体の1粒子内における元素分布の均一性に大きく影響を与える。特にMnは、CoやNiと均一な元素分布を形成しにくいので注意が必要である。好ましい滴下速度については、反応槽の大きさ、攪拌条件、pH、反応温度等にも影響されるが、30ml/min以下が好ましい。放電容量を向上させるためには、滴下速度は10ml/min以下がより好ましく、5ml/min以下が最も好ましい。 The dropping speed of the raw material aqueous solution greatly affects the uniformity of element distribution in one particle of the coprecipitation precursor to be generated. In particular, Mn is difficult to form a uniform element distribution with Co and Ni, so care must be taken. The preferred dropping rate is influenced by the reaction vessel size, stirring conditions, pH, reaction temperature, etc., but is preferably 30 ml / min or less. In order to improve the discharge capacity, the dropping rate is more preferably 10 ml / min or less, and most preferably 5 ml / min or less.
また、反応槽内に錯化剤が存在し、かつ一定の対流条件を適用した場合、前記原料水溶液の滴下終了後、さらに攪拌を続けることにより、粒子の自転および攪拌槽内における公転が促進され、この過程で、粒子同士が衝突しつつ、粒子が段階的に同心円球状に成長する。即ち、共沈前駆体は、反応槽内に原料水溶液が滴下された際の金属錯体形成反応、及び、前記金属錯体が反応槽内の滞留中に生じる沈殿形成反応という2段階での反応を経て形成される。従って、前記原料水溶液の滴下終了後、さらに攪拌を続ける時間を適切に選択することにより、目的とする粒子径を備えた共沈前駆体を得ることができる。 In addition, when a complexing agent is present in the reaction tank and a certain convection condition is applied, the particle rotation and revolution in the stirring tank are promoted by continuing the stirring after the dropwise addition of the raw material aqueous solution. In this process, the particles grow concentrically in stages while colliding with each other. That is, the coprecipitation precursor undergoes a reaction in two stages: a metal complex formation reaction when the raw material aqueous solution is dropped into the reaction tank, and a precipitation formation reaction that occurs while the metal complex is retained in the reaction tank. It is formed. Therefore, a coprecipitation precursor having a target particle size can be obtained by appropriately selecting a time for continuing stirring after the dropping of the raw material aqueous solution.
原料水溶液滴下終了後の好ましい攪拌継続時間については、反応槽の大きさ、攪拌条件、pH、反応温度等にも影響されるが、粒子を均一な球状粒子として成長させるために0.5h以上が好ましく、1h以上がより好ましい。また、粒子径が大きくなりすぎることで電池の低SOC領域における出力性能が充分でないものとなる虞を低減させるため、30h以下が好ましく、25h以下がより好ましく、20h以下が最も好ましい。 The preferable stirring duration after completion of dropping of the raw material aqueous solution is influenced by the size of the reaction vessel, stirring conditions, pH, reaction temperature, etc., but 0.5 h or more is required to grow the particles as uniform spherical particles. Preferably, 1 h or more is more preferable. Moreover, in order to reduce the possibility that the output performance in the low SOC region of the battery is not sufficient due to the particle size becoming too large, it is preferably 30 h or less, more preferably 25 h or less, and most preferably 20 h or less.
また、炭酸塩前駆体及びリチウム遷移金属複合酸化物の2次粒子の粒度分布における累積体積が50%となる粒子径であるD50を5〜18μmとするための好ましい攪拌継続時間は、制御するpHによって異なる。例えば、pHを7.5〜8.2に制御した場合には、撹拌継続時間は1〜15hが好ましく、pHを8.3〜9.4に制御した場合には、撹拌継続時間は3〜20hが好ましい。 In addition, the preferable stirring duration for setting D50, which is a particle diameter at which the cumulative volume in the particle size distribution of the secondary particles of the carbonate precursor and the lithium transition metal composite oxide is 50%, to 5 to 18 μm is controlled by pH It depends on. For example, when the pH is controlled to 7.5 to 8.2, the stirring duration is preferably 1 to 15 h, and when the pH is controlled to 8.3 to 9.4, the stirring duration is 3 to 3. 20h is preferred.
炭酸塩前駆体の粒子を、中和剤として炭酸ナトリウム等のナトリウム化合物を使用して作製した場合、その後の洗浄工程において粒子に付着しているナトリウムイオンを洗浄除去するが、本発明においては、Naが1000ppm以上残存するような条件で洗浄除去することが好ましい。例えば、作製した炭酸塩前駆体を吸引ろ過して取り出す際に、イオン交換水200mlによる洗浄回数を5回とするような条件を採用することができる。 When the carbonate precursor particles are prepared using a sodium compound such as sodium carbonate as a neutralizing agent, sodium ions adhering to the particles are washed away in the subsequent washing step. It is preferable to wash and remove under conditions such that Na remains at 1000 ppm or more. For example, when the produced carbonate precursor is removed by suction filtration, a condition that the number of washings with 200 ml of ion-exchanged water is 5 times can be employed.
炭酸塩前駆体は、80℃〜100℃未満で、空気雰囲気中、常圧下で乾燥させることが好ましい。100℃以上にて乾燥を行うことで短時間でより多くの水分を除去できるが、80℃にて長時間かけて乾燥させることで、より優れた電極特性を示す活物質とすることができる。その理由は必ずしも明らかではないが、炭酸塩前駆体は比表面積が50〜100m2/gの多孔体であるため、水分を吸着しやすい構造となっている。そこで、低い温度で乾燥させることによって、前駆体の状態において細孔にある程度の吸着水が残っている状態とした方が、Li塩と混合して焼成する焼成工程において、細孔から除去される吸着水と入れ替わるように、その細孔に溶融したLiが入り込むことができ、これによって、100℃で乾燥を行った場合と比べて、より均一な組成の活物質が得られるためではないかと発明者は推察している。なお、100℃にて乾燥を行って得られた炭酸塩前駆体は黒茶色を呈するが、80℃にて乾燥を行って得られた炭酸塩前駆体は肌色を呈するので、前駆体の色によって区別ができる。 The carbonate precursor is preferably dried at 80 ° C. to less than 100 ° C. in an air atmosphere under normal pressure. By drying at 100 ° C. or higher, more water can be removed in a short time, but by drying at 80 ° C. for a long time, an active material having more excellent electrode characteristics can be obtained. Although the reason is not necessarily clear, since the carbonate precursor is a porous body having a specific surface area of 50 to 100 m 2 / g, it has a structure that easily adsorbs moisture. Therefore, by drying at a low temperature, a state in which a certain amount of adsorbed water remains in the pores in the state of the precursor is removed from the pores in the firing step of mixing with the Li salt and firing. The invention may be because molten Li can enter the pores so as to replace the adsorbed water, and thereby, an active material having a more uniform composition can be obtained compared with the case of drying at 100 ° C. Have guessed. In addition, although the carbonate precursor obtained by drying at 100 ° C. exhibits a black brown color, the carbonate precursor obtained by drying at 80 ° C. exhibits a skin color, so depending on the color of the precursor Can be distinguished.
本発明のリチウム二次電池用活物質は、前記炭酸塩前駆体とLi化合物とを混合した後、熱処理することで好適に作製することができる。Li化合物としては、水酸化リチウム、炭酸リチウム、硝酸リチウム、酢酸リチウム等を用いることで好適に製造することができる。但し、Li化合物の量については、焼成中にLi化合物の一部が消失することを見込んで、1〜5%程度過剰に仕込むことが好ましい。 The active material for a lithium secondary battery of the present invention can be suitably prepared by mixing the carbonate precursor and the Li compound and then heat-treating them. As a Li compound, it can manufacture suitably by using lithium hydroxide, lithium carbonate, lithium nitrate, lithium acetate, etc. However, with respect to the amount of the Li compound, it is preferable to add an excess of about 1 to 5% in view of the disappearance of a part of the Li compound during firing.
本発明においては、リチウム遷移金属複合酸化物中のNaの含有量を1000ppm以上とするために、炭酸塩前駆体に含まれるNaが1000ppm以下であっても、焼成工程においてLi化合物と共にNa化合物を、前記炭酸塩前駆体と混合することで活物質中に含まれるNa量を1000ppm以上とすることができる。Na化合物としては炭酸ナトリウムが好ましい。 In the present invention, in order to make the content of Na in the lithium transition metal composite oxide 1000 ppm or more, even if Na contained in the carbonate precursor is 1000 ppm or less, the Na compound is added together with the Li compound in the firing step. By mixing with the carbonate precursor, the amount of Na contained in the active material can be 1000 ppm or more. As the Na compound, sodium carbonate is preferable.
焼成温度は、活物質の可逆容量に影響を与える。
焼成温度が高すぎると、得られた活物質が酸素放出反応を伴って崩壊すると共に、主相の六方晶に加えて単斜晶のLi[Li1/3Mn2/3]O2型に規定される相が、固溶相としてではなく、分相して観察される傾向がある。このような分相が多く含まれすぎると、活物質の可逆容量の減少を導くので好ましくない。このような材料では、X線回折図上35°付近及び45°付近に不純物ピークが観察される。従って、焼成温度は、活物質の酸素放出反応の影響する温度未満とすることが好ましい。活物質の酸素放出温度は、本発明に係る組成範囲においては、概ね1000℃以上であるが、活物質の組成によって酸素放出温度に若干の差があるので、あらかじめ活物質の酸素放出温度を確認しておくことが好ましい。特に試料に含まれるCo量が多いほど前駆体の酸素放出温度は低温側にシフトすることが確認されているので注意が必要である。活物質の酸素放出温度を確認する方法としては、焼成反応過程をシミュレートするために、共沈前駆体とリチウム化合物を混合したものを熱重量分析(DTA−TG測定)に供してもよいが、この方法では測定機器の試料室に用いている白金が揮発したLi成分により腐食されて機器を痛めるおそれがあるので、あらかじめ500℃程度の焼成温度を採用してある程度結晶化を進行させた組成物を熱重量分析に供するのが良い。
The firing temperature affects the reversible capacity of the active material.
When the firing temperature is too high, the obtained active material collapses with an oxygen releasing reaction, and in addition to the hexagonal crystal of the main phase, the monoclinic Li [Li 1/3 Mn 2/3 ] O 2 type is obtained. The defined phase tends to be observed as a phase separation rather than as a solid solution phase. If too many such phase separations are contained, it is not preferable because it leads to a reduction in the reversible capacity of the active material. In such materials, impurity peaks are observed around 35 ° and 45 ° on the X-ray diffraction pattern. Therefore, the firing temperature is preferably less than the temperature at which the oxygen release reaction of the active material affects. The oxygen release temperature of the active material is approximately 1000 ° C. or higher in the composition range according to the present invention. However, there is a slight difference in the oxygen release temperature depending on the composition of the active material. It is preferable to keep it. In particular, it is confirmed that the oxygen release temperature of the precursor shifts to the lower temperature side as the amount of Co contained in the sample increases. As a method for confirming the oxygen release temperature of the active material, a mixture of a coprecipitation precursor and a lithium compound may be subjected to thermogravimetric analysis (DTA-TG measurement) in order to simulate the firing reaction process. In this method, the platinum used in the sample chamber of the measuring instrument may be corroded by the Li component volatilized, and the instrument may be damaged. Therefore, a composition in which crystallization is advanced to some extent by adopting a firing temperature of about 500 ° C. in advance. Goods should be subjected to thermogravimetric analysis.
一方、焼成温度が低すぎると、結晶化が十分に進まず、電極特性が低下する傾向がある。本発明においては、焼成温度は少なくとも800℃以上とすることが好ましい。十分に結晶化させることにより、結晶粒界の抵抗を軽減し、円滑なリチウムイオン輸送を促すことができる。
また、発明者らは、本発明活物質の回折ピークの半値幅を詳細に解析することで750℃までの温度で合成した試料においては格子内にひずみが残存しており、それ以上の温度で合成することでほとんどひずみを除去することができることを確認した。また、結晶子のサイズは合成温度が上昇するに比例して大きくなるものであった。よって、本発明活物質の組成においても、系内に格子のひずみがほとんどなく、かつ結晶子サイズが十分成長した粒子を志向することで良好な放電容量を得られるものであった。具体的には、格子定数に及ぼすひずみ量が2%以下、かつ結晶子サイズが50nm以上に成長しているような合成温度(焼成温度)及びLi/Me比組成を採用することが好ましいことがわかった。これらを電極として成型して充放電を行うことで膨張収縮による変化も見られるが、充放電過程においても結晶子サイズは30nm以上を保っていることが得られる効果として好ましい。
On the other hand, if the firing temperature is too low, crystallization does not proceed sufficiently and the electrode characteristics tend to deteriorate. In the present invention, the firing temperature is preferably at least 800 ° C. or higher. By sufficiently crystallizing, the resistance of the crystal grain boundary can be reduced and smooth lithium ion transport can be promoted.
In addition, the inventors have analyzed the half width of the diffraction peak of the active material of the present invention in detail, and in the sample synthesized at a temperature up to 750 ° C., strain remains in the lattice, and at a temperature higher than that, It was confirmed that almost all strains could be removed by synthesis. The crystallite size was increased in proportion to the increase in the synthesis temperature. Therefore, even in the composition of the active material of the present invention, a favorable discharge capacity can be obtained by aiming at a particle having almost no lattice distortion in the system and having a sufficiently grown crystallite size. Specifically, it is preferable to employ a synthesis temperature (firing temperature) and a Li / Me ratio composition in which the strain amount affecting the lattice constant is 2% or less and the crystallite size is grown to 50 nm or more. all right. Although changes due to expansion and contraction are observed by charging and discharging by molding these as electrodes, it is preferable as an effect that the crystallite size is maintained at 30 nm or more in the charging and discharging process.
上記のように、焼成温度は、活物質の酸素放出温度に関係するが、活物質から酸素が放出される焼成温度に至らずとも、900℃を超えると1次粒子が大きく成長することによる結晶化現象が見られる。これは、焼成後の活物質を走査型電子顕微鏡(SEM)で観察することにより確認できる。900℃を超える合成温度を経て合成した活物質は1次粒子が0.5μm以上に成長しており、充放電反応中における活物質中のLi+移動に不利な状態となり、高率放電性能が低下する。1次粒子の大きさは0.5μm未満であることが好ましく、0.3μm以下であることがより好ましい。また、900℃を超える合成温度では、活物質の細孔容積が、60nmまでの細孔領域にて0.055cc/g未満となり、初期効率、高率放電性能が低下する。 As described above, the firing temperature is related to the oxygen release temperature of the active material. However, even if the firing temperature does not reach the firing temperature at which oxygen is released from the active material, the crystal due to large growth of primary particles above 900 ° C. A phenomenological phenomenon is observed. This can be confirmed by observing the fired active material with a scanning electron microscope (SEM). The active material synthesized through a synthesis temperature exceeding 900 ° C. has primary particles grown to 0.5 μm or more, which is disadvantageous for Li + movement in the active material during the charge / discharge reaction, and has a high rate discharge performance. descend. The size of the primary particles is preferably less than 0.5 μm, and more preferably 0.3 μm or less. Further, at a synthesis temperature exceeding 900 ° C., the pore volume of the active material is less than 0.055 cc / g in the pore region up to 60 nm, and the initial efficiency and high rate discharge performance are lowered.
したがって、初期効率、高率放電性能を向上させるために、1.2<モル比Li/Me<1.6の本発明に係るリチウム遷移金属複合酸化物を正極活物質とする場合、焼成温度は800〜900℃とすることが好ましい。 Therefore, in order to improve initial efficiency and high rate discharge performance, when the lithium transition metal composite oxide according to the present invention of 1.2 <molar ratio Li / Me <1.6 is used as the positive electrode active material, the firing temperature is It is preferable to set it as 800-900 degreeC.
焼成後の活物質は、平均粒子サイズ100μm以下であることが望ましく、高出力特性を向上する目的で10μm以下であることが特に望ましい。粉体を所定の形状で得るためには粉砕機や分級機が用いられる。例えば乳鉢、ボールミル、サンドミル、振動ボールミル、遊星ボールミル、ジェットミル、カウンタージェトミル、旋回気流型ジェットミルや篩等が用いられる。粉砕時には水、あるいはヘキサン等の有機溶剤を共存させた湿式粉砕を用いることもできる。分級方法としては、特に限定はなく、篩や風力分級機などが、乾式、湿式ともに必要に応じて用いられる。 The active material after firing is desirably an average particle size of 100 μm or less, and particularly desirably 10 μm or less for the purpose of improving high output characteristics. In order to obtain the powder in a predetermined shape, a pulverizer or a classifier is used. For example, a mortar, a ball mill, a sand mill, a vibrating ball mill, a planetary ball mill, a jet mill, a counter jet mill, a swirling air flow type jet mill or a sieve is used. At the time of pulverization, wet pulverization in the presence of water or an organic solvent such as hexane may be used. There is no particular limitation on the classification method, and a sieve, an air classifier, or the like is used as needed for both dry and wet methods.
(セリウム添加酸処理)
本発明において、セリウムを適用する方法については限定されない。後述する実施例においては、以上のようにして作製したセリウムを含まないリチウム遷移金属複合酸化物に、酸処理を行ってCeを添加する方法を採用した。
この酸処理は、例えば、硫酸セリウム等のセリウム化合物の水溶液に硫酸を加えた酸溶液に、リチウム遷移金属複合酸化物を投入して行うことができる。酸としては硫酸又はリン酸が好ましい。酸として塩酸の使用を避けることが好ましく、これにより、酸処理前の結晶構造が崩れる虞を低減することができる。また、酸としてホウ酸を用いてもよいが、ホウ酸などの弱酸の使用を避けることが好ましく、これにより、活物質からのリチウムの脱離量が少なすぎて初期効率を向上させる効果が充分に奏されない虞を低減することができる。
酸処理時間を長くしすぎないこと、また、pHの値を小さくしすぎないことが好ましく、これにより、活物質からのリチウムの脱離量が多くなりすぎて可逆容量が充分とならない虞を低減できる。酸処理の時間を短くしすぎないこと、また、pHの値を大きくしすぎないことが好ましく、これにより、活物質からのリチウムの離脱量が少なすぎて初期効率の効果が充分に奏されない虞を低減することができる。上記の観点から、酸処理時間は、30秒〜10800秒が好ましく、酸溶液のpHは0.5〜3が好ましい。
酸処理後の溶液は、吸引ろ過等により、ろ紙上のCeを含むリチウム遷移金属複合酸化物を回収した後、乾燥、熱処理することによりCeが添加された活物質を得ることができる。
(Cerium-added acid treatment)
In the present invention, the method for applying cerium is not limited. In the examples described later, a method was adopted in which Ce was added by performing an acid treatment on the lithium transition metal composite oxide not containing cerium prepared as described above.
This acid treatment can be performed, for example, by introducing a lithium transition metal composite oxide into an acid solution obtained by adding sulfuric acid to an aqueous solution of a cerium compound such as cerium sulfate. As the acid, sulfuric acid or phosphoric acid is preferable. It is preferable to avoid the use of hydrochloric acid as the acid, and this can reduce the risk of the crystal structure before the acid treatment being broken. Further, although boric acid may be used as the acid, it is preferable to avoid the use of a weak acid such as boric acid, so that the amount of lithium desorbed from the active material is too small to sufficiently improve the initial efficiency. It is possible to reduce the possibility of being not played.
It is preferable not to make the acid treatment time too long, and it is preferable not to make the pH value too small, thereby reducing the possibility that the amount of lithium desorbed from the active material becomes too large and the reversible capacity is not sufficient. it can. It is preferable not to make the acid treatment time too short, and it is preferable not to make the pH value too large, and thereby the amount of lithium released from the active material is so small that the effect of the initial efficiency may not be sufficiently achieved. Can be reduced. From the above viewpoint, the acid treatment time is preferably from 30 seconds to 10800 seconds, and the pH of the acid solution is preferably from 0.5 to 3.
After the acid treatment, the lithium transition metal composite oxide containing Ce on the filter paper is recovered by suction filtration or the like, and then dried and heat-treated to obtain an active material to which Ce is added.
酸処理後、熱処理を行うことが好ましい。熱処理温度は300〜600℃が好ましい。保持時間は2〜10hが好ましい。熱処理により、活物質表面におけるCeの担持が安定化し、充放電に伴う局所的なMnの溶出を防止し、放電容量維持率を向上させることができる。処理温度を高くしすぎないことにより、結晶構造が変化して正極活物質の特性を低下させる虞を低減できる。また、処理温度を低くしすぎないことにより、Ceの担持が充分とならないものとなる虞を低減できる。保持時間が長すぎても、本発明の効果に影響を与えないが、エネルギーを無駄に消費して生産性が低下することを避けるため、10h以下が好ましい。 It is preferable to perform heat treatment after the acid treatment. The heat treatment temperature is preferably 300 to 600 ° C. The holding time is preferably 2 to 10 hours. The heat treatment stabilizes the support of Ce on the active material surface, prevents local Mn elution associated with charge and discharge, and improves the discharge capacity retention rate. By not setting the treatment temperature too high, the possibility that the crystal structure changes and the characteristics of the positive electrode active material are deteriorated can be reduced. Moreover, the possibility that the loading of Ce will not be sufficient can be reduced by not setting the treatment temperature too low. Even if the holding time is too long, the effect of the present invention is not affected, but 10 h or less is preferable in order to avoid wasteful consumption of energy and decrease in productivity.
本発明に係るリチウム遷移金属複合酸化物が含有するCeの量は、前記酸溶液中のセリウムイオンの濃度により調整することができる。ここで、セリウムイオンの濃度は0.0005〜0.025Mが好ましい。本発明に係るリチウム遷移金属複合酸化物が含有するCeの量は、多い方が、初期効率は向上するため、好ましい。また、Ceの量を多くしすぎないことにより、活物質表面に付着したCeがリチウムイオンの挿入離脱を阻害して活物質高率放電性能が低下する虞を低減できるため、好ましい。
なお、Ceの添加量は、本発明に係る活物質について、ICP分析で求めることができる。また、本発明に係る活物質がCeを含有することについては、CuKα線源を用いたXRDパターンにおいて、CeO2に帰属される最強ピークが2θ=29±1°付近に観測されることにより推定することができる。さらに、本発明に係る活物質を空気中で1000℃で焼成すると、CeO2の結晶性が高まるため、2θ=29±1°付近の回折ピークに加えて、2θ=33±1°付近、47±1°付近及び56±1°付近の回折ピークが顕在化するので、CeO2の存在をより確実に推定できる。
The amount of Ce contained in the lithium transition metal composite oxide according to the present invention can be adjusted by the concentration of cerium ions in the acid solution. Here, the concentration of cerium ions is preferably 0.0005 to 0.025M. A larger amount of Ce contained in the lithium transition metal composite oxide according to the present invention is preferable because the initial efficiency is improved. In addition, it is preferable not to increase the amount of Ce excessively, because Ce adhering to the active material surface can inhibit the insertion and release of lithium ions and reduce the active material high rate discharge performance.
In addition, the addition amount of Ce can be calculated | required by the ICP analysis about the active material which concerns on this invention. In addition, the fact that the active material according to the present invention contains Ce is estimated by observing the strongest peak attributed to CeO 2 in the vicinity of 2θ = 29 ± 1 ° in the XRD pattern using the CuKα radiation source. can do. Further, when the active material according to the present invention is fired at 1000 ° C. in air, the crystallinity of CeO 2 is increased. Therefore, in addition to the diffraction peak around 2θ = 29 ± 1 °, 2θ = around 33 ± 1 °, 47 Since diffraction peaks near ± 1 ° and 56 ± 1 ° are manifested, the presence of CeO 2 can be estimated more reliably.
(負極活物質)
負極材料としては、限定されるものではなく、リチウムイオンを放出あるいは吸蔵することのできる形態のものであればどれを選択してもよい。例えば、Li[Li1/3Ti5/3]O4に代表されるスピネル型結晶構造を有するチタン酸リチウム等のチタン系材料、SiやSb,Sn系などの合金系材料リチウム金属、リチウム合金(リチウム−シリコン、リチウム−アルミニウム,リチウム−鉛,リチウム−スズ,リチウム−アルミニウム−スズ,リチウム−ガリウム,及びウッド合金等のリチウム金属含有合金)、リチウム複合酸化物(リチウム−チタン)、酸化珪素の他、リチウムを吸蔵・放出可能な合金、炭素材料(例えばグラファイト、ハードカーボン、低温焼成炭素、非晶質カーボン等)等が挙げられる。
(Negative electrode active material)
The negative electrode material is not limited, and any negative electrode material may be selected as long as it can release or occlude lithium ions. For example, titanium-based materials such as lithium titanate having a spinel crystal structure represented by Li [Li 1/3 Ti 5/3 ] O 4 , alloy-based materials such as Si, Sb, and Sn-based lithium metal, lithium alloys (Lithium metal-containing alloys such as lithium-silicon, lithium-aluminum, lithium-lead, lithium-tin, lithium-aluminum-tin, lithium-gallium, and wood alloys), lithium composite oxide (lithium-titanium), silicon oxide In addition, an alloy capable of inserting and extracting lithium, a carbon material (for example, graphite, hard carbon, low-temperature fired carbon, amorphous carbon, etc.) can be used.
(正極・負極)
以上、正極及び負極の主要構成成分である正極活物質及び負極材料について詳述したが、前記正極及び負極には、前記主要構成成分の他に、導電剤、結着剤、増粘剤、フィラー等が、他の構成成分として含有されてもよい。
(Positive electrode / Negative electrode)
The positive electrode active material and the negative electrode material, which are the main components of the positive electrode and the negative electrode, have been described in detail above. In addition to the main components, the positive electrode and the negative electrode include a conductive agent, a binder, a thickener, and a filler. Etc. may be contained as other constituents.
導電剤としては、電池性能に悪影響を及ぼさない電子伝導性材料であれば限定されないが、通常、天然黒鉛(鱗状黒鉛,鱗片状黒鉛,土状黒鉛等)、人造黒鉛、カーボンブラック、アセチレンブラック、ケッチェンブラック、カーボンウイスカー、炭素繊維、金属(銅,ニッケル,アルミニウム,銀,金等)粉、金属繊維、導電性セラミックス材料等の導電性材料を1種またはそれらの混合物として含ませることができる。 The conductive agent is not limited as long as it is an electron conductive material that does not adversely affect the battery performance. Usually, natural graphite (such as scaly graphite, scaly graphite, earthy graphite), artificial graphite, carbon black, acetylene black, Conductive materials such as ketjen black, carbon whisker, carbon fiber, metal (copper, nickel, aluminum, silver, gold, etc.) powder, metal fiber, and conductive ceramic material can be included as one kind or a mixture thereof. .
これらの中で、導電剤としては、電子伝導性及び塗工性の観点よりアセチレンブラックが望ましい。導電剤の添加量は、正極または負極の総重量に対して0.1重量%〜50重量%が好ましく、特に0.5重量%〜30重量%が好ましい。特にアセチレンブラックを0.1〜0.5μmの超微粒子に粉砕して用いると必要炭素量を削減できるため望ましい。これらの混合方法は、物理的な混合であり、その理想とするところは均一混合である。そのため、V型混合機、S型混合機、擂かい機、ボールミル、遊星ボールミルといったような粉体混合機を乾式、あるいは湿式で混合することが可能である。 Among these, as the conductive agent, acetylene black is desirable from the viewpoints of electron conductivity and coatability. The addition amount of the conductive agent is preferably 0.1% by weight to 50% by weight, and particularly preferably 0.5% by weight to 30% by weight with respect to the total weight of the positive electrode or the negative electrode. In particular, it is desirable to use acetylene black by pulverizing into ultrafine particles of 0.1 to 0.5 μm because the required carbon amount can be reduced. These mixing methods are physical mixing, and the ideal is uniform mixing. Therefore, powder mixers such as V-type mixers, S-type mixers, crackers, ball mills, and planetary ball mills can be mixed dry or wet.
前記結着剤としては、通常、ポリテトラフルオロエチレン(PTFE),ポリフッ化ビニリデン(PVDF),ポリエチレン,ポリプロピレン等の熱可塑性樹脂、エチレン−プロピレン−ジエンターポリマー(EPDM),スルホン化EPDM,スチレンブタジエンゴム(SBR)、フッ素ゴム等のゴム弾性を有するポリマーを1種または2種以上の混合物として用いることができる。結着剤の添加量は、正極または負極の総重量に対して1〜50重量%が好ましく、特に2〜30重量%が好ましい。 The binder is usually a thermoplastic resin such as polytetrafluoroethylene (PTFE), polyvinylidene fluoride (PVDF), polyethylene, polypropylene, ethylene-propylene-diene terpolymer (EPDM), sulfonated EPDM, styrene butadiene. Polymers having rubber elasticity such as rubber (SBR) and fluororubber can be used as one kind or a mixture of two or more kinds. The addition amount of the binder is preferably 1 to 50% by weight, particularly preferably 2 to 30% by weight, based on the total weight of the positive electrode or the negative electrode.
フィラーとしては、電池性能に悪影響を及ぼさない材料であれば何でも良い。通常、ポリプロピレン,ポリエチレン等のオレフィン系ポリマー、無定形シリカ、アルミナ、ゼオライト、ガラス、炭素等が用いられる。フィラーの添加量は、正極または負極の総重量に対して添加量は30重量%以下が好ましい。 As the filler, any material that does not adversely affect the battery performance may be used. Usually, olefin polymers such as polypropylene and polyethylene, amorphous silica, alumina, zeolite, glass, carbon and the like are used. The addition amount of the filler is preferably 30% by weight or less with respect to the total weight of the positive electrode or the negative electrode.
正極及び負極は、前記主要構成成分(正極においては正極活物質、負極においては負極材料)、およびその他の材料を混練し合剤とし、N−メチルピロリドン,トルエン等の有機溶媒又は水に混合させた後、得られた混合液をアルミニウム箔等の集電体の上に塗布し、または圧着して50℃〜250℃程度の温度で、2時間程度加熱処理することにより好適に作製される。前記塗布方法については、例えば、アプリケーターロールなどのローラーコーティング、スクリーンコーティング、ドクターブレード方式、スピンコーティング、バーコータ等の手段を用いて任意の厚さ及び任意の形状に塗布することが望ましいが、これらに限定されるものではない。 The positive electrode and the negative electrode are prepared by mixing the main constituents (positive electrode active material in the positive electrode, negative electrode material in the negative electrode) and other materials into a mixture and mixing with an organic solvent such as N-methylpyrrolidone or toluene or water. After that, the obtained mixed solution is applied on a current collector such as an aluminum foil, or is pressure-bonded and heat-treated at a temperature of about 50 ° C. to 250 ° C. for about 2 hours. About the application method, for example, it is desirable to apply to any thickness and any shape using means such as roller coating such as applicator roll, screen coating, doctor blade method, spin coating, bar coater, etc. It is not limited.
(非水電解質)
本発明に係るリチウム二次電池に用いる非水電解質は、限定されるものではなく、一般にリチウム電池等への使用が提案されているものが使用可能である。非水電解質に用いる非水溶媒としては、プロピレンカーボネート、エチレンカーボネート、ブチレンカーボネート、クロロエチレンカーボネート、ビニレンカーボネート等の環状炭酸エステル類;γ−ブチロラクトン、γ−バレロラクトン等の環状エステル類;ジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート等の鎖状カーボネート類;ギ酸メチル、酢酸メチル、酪酸メチル等の鎖状エステル類;テトラヒドロフランまたはその誘導体;1,3−ジオキサン、1,4−ジオキサン、1,2−ジメトキシエタン、1,4−ジブトキシエタン、メチルジグライム等のエーテル類;アセトニトリル、ベンゾニトリル等のニトリル類;ジオキソランまたはその誘導体;エチレンスルフィド、スルホラン、スルトンまたはその誘導体等の単独またはそれら2種以上の混合物等を挙げることができるが、これらに限定されるものではない。
(Nonaqueous electrolyte)
The nonaqueous electrolyte used for the lithium secondary battery according to the present invention is not limited, and those generally proposed for use in lithium batteries and the like can be used. Examples of the nonaqueous solvent used for the nonaqueous electrolyte include cyclic carbonates such as propylene carbonate, ethylene carbonate, butylene carbonate, chloroethylene carbonate, and vinylene carbonate; cyclic esters such as γ-butyrolactone and γ-valerolactone; dimethyl carbonate, Chain carbonates such as diethyl carbonate and ethyl methyl carbonate; chain esters such as methyl formate, methyl acetate and methyl butyrate; tetrahydrofuran or derivatives thereof; 1,3-dioxane, 1,4-dioxane, 1,2-dimethoxy Ethers such as ethane, 1,4-dibutoxyethane and methyldiglyme; Nitriles such as acetonitrile and benzonitrile; Dioxolane or derivatives thereof; Ethylene sulfide, sulfolane, sultone or derivatives thereof Examples thereof include a conductor alone or a mixture of two or more thereof, but are not limited thereto.
非水電解質に用いる電解質塩としては、例えば、LiClO4,LiBF4,LiAsF6,LiPF6,LiSCN,LiBr,LiI,Li2SO4,Li2B10Cl10,NaClO4,NaI,NaSCN,NaBr,KClO4,KSCN等のリチウム(Li)、ナトリウム(Na)またはカリウム(K)の1種を含む無機イオン塩、LiCF3SO3,LiN(CF3SO2)2,LiN(C2F5SO2)2,LiN(CF3SO2)(C4F9SO2),LiC(CF3SO2)3,LiC(C2F5SO2)3,(CH3)4NBF4,(CH3)4NBr,(C2H5)4NClO4,(C2H5)4NI,(C3H7)4NBr,(n−C4H9)4NClO4,(n−C4H9)4NI,(C2H5)4N−maleate,(C2H5)4N−benzoate,(C2H5)4N−phthalate、ステアリルスルホン酸リチウム、オクチルスルホン酸リチウム、ドデシルベンゼンスルホン酸リチウム等の有機イオン塩等が挙げられ、これらのイオン性化合物を単独、あるいは2種類以上混合して用いることが可能である。 Examples of the electrolyte salt used for the nonaqueous electrolyte include LiClO 4 , LiBF 4 , LiAsF 6 , LiPF 6 , LiSCN, LiBr, LiI, Li 2 SO 4 , Li 2 B 10 Cl 10 , NaClO 4 , NaI, NaSCN, NaBr. , KClO 4 , KSCN, and other inorganic ion salts containing one of lithium (Li), sodium (Na), or potassium (K), LiCF 3 SO 3 , LiN (CF 3 SO 2 ) 2 , LiN (C 2 F 5 SO 2 ) 2 , LiN (CF 3 SO 2 ) (C 4 F 9 SO 2 ), LiC (CF 3 SO 2 ) 3 , LiC (C 2 F 5 SO 2 ) 3 , (CH 3 ) 4 NBF 4 , ( CH 3 ) 4 NBr, (C 2 H 5 ) 4 NClO 4 , (C 2 H 5 ) 4 NI, (C 3 H 7 ) 4 NBr, (n-C 4 H 9 ) 4 NClO 4 , (n-C 4 H 9) 4 NI, ( C 2 H 5) 4 N-mal ate, (C 2 H 5) 4 N-benzoate, (C 2 H 5) 4 N-phthalate, lithium stearyl sulfonate, lithium octyl sulfonate, organic ion salts of lithium dodecyl benzene sulfonate, and the like. These These ionic compounds can be used alone or in admixture of two or more.
さらに、LiPF6又はLiBF4と、LiN(C2F5SO2)2のようなパーフルオロアルキル基を有するリチウム塩とを混合して用いることにより、さらに電解質の粘度を下げることができるので、低温特性をさらに高めることができ、また、自己放電を抑制することができ、より望ましい。 Furthermore, by mixing and using LiPF 6 or LiBF 4 and a lithium salt having a perfluoroalkyl group such as LiN (C 2 F 5 SO 2 ) 2 , the viscosity of the electrolyte can be further reduced. The low temperature characteristics can be further improved, and self-discharge can be suppressed, which is more desirable.
また、非水電解質として常温溶融塩やイオン液体を用いてもよい。 Moreover, you may use normal temperature molten salt and an ionic liquid as a nonaqueous electrolyte.
非水電解質における電解質塩の濃度としては、高い電池特性を有する非水電解質電池を確実に得るために、0.1mol/l〜5mol/lが好ましく、さらに好ましくは、0.5mol/l〜2.5mol/lである。 The concentration of the electrolyte salt in the non-aqueous electrolyte is preferably 0.1 mol / l to 5 mol / l, more preferably 0.5 mol / l to 2 in order to reliably obtain a non-aqueous electrolyte battery having high battery characteristics. .5 mol / l.
(セパレータ)
セパレータとしては、優れた高率放電性能を示す多孔膜や不織布等を、単独あるいは併用することが好ましい。非水電解質電池用セパレータを構成する材料としては、例えばポリエチレン,ポリプロピレン等に代表されるポリオレフィン系樹脂、ポリエチレンテレフタレート,ポリブチレンテレフタレート等に代表されるポリエステル系樹脂、ポリフッ化ビニリデン、フッ化ビニリデン−ヘキサフルオロプロピレン共重合体、フッ化ビニリデン−パーフルオロビニルエーテル共重合体、フッ化ビニリデン−テトラフルオロエチレン共重合体、フッ化ビニリデン−トリフルオロエチレン共重合体、フッ化ビニリデン−フルオロエチレン共重合体、フッ化ビニリデン−ヘキサフルオロアセトン共重合体、フッ化ビニリデン−エチレン共重合体、フッ化ビニリデン−プロピレン共重合体、フッ化ビニリデン−トリフルオロプロピレン共重合体、フッ化ビニリデン−テトラフルオロエチレン−ヘキサフルオロプロピレン共重合体、フッ化ビニリデン−エチレン−テトラフルオロエチレン共重合体等を挙げることができる。
(Separator)
As the separator, it is preferable to use a porous film or a non-woven fabric exhibiting excellent high rate discharge performance alone or in combination. Examples of the material constituting the separator for a nonaqueous electrolyte battery include polyolefin resins typified by polyethylene and polypropylene, polyester resins typified by polyethylene terephthalate and polybutylene terephthalate, polyvinylidene fluoride, and vinylidene fluoride-hexa. Fluoropropylene copolymer, vinylidene fluoride-perfluorovinyl ether copolymer, vinylidene fluoride-tetrafluoroethylene copolymer, vinylidene fluoride-trifluoroethylene copolymer, vinylidene fluoride-fluoroethylene copolymer, fluorine Vinylidene fluoride-hexafluoroacetone copolymer, vinylidene fluoride-ethylene copolymer, vinylidene fluoride-propylene copolymer, vinylidene fluoride-trifluoropropylene copolymer, vinylidene fluoride - tetrafluoroethylene - hexafluoropropylene copolymer, vinylidene fluoride - ethylene - can be mentioned tetrafluoroethylene copolymer.
セパレータの空孔率は強度の観点から98体積%以下が好ましい。また、充放電特性の観点から空孔率は20体積%以上が好ましい。 The porosity of the separator is preferably 98% by volume or less from the viewpoint of strength. Further, the porosity is preferably 20% by volume or more from the viewpoint of charge / discharge characteristics.
また、セパレータは、例えばアクリロニトリル、エチレンオキシド、プロピレンオキシド、メチルメタアクリレート、ビニルアセテート、ビニルピロリドン、ポリフッ化ビニリデン等のポリマーと電解質とで構成されるポリマーゲルを用いてもよい。非水電解質を上記のようにゲル状態で用いると、漏液を防止する効果がある点で好ましい。 The separator may be a polymer gel composed of a polymer such as acrylonitrile, ethylene oxide, propylene oxide, methyl methacrylate, vinyl acetate, vinyl pyrrolidone, polyvinylidene fluoride, and an electrolyte. Use of the non-aqueous electrolyte in the gel state as described above is preferable in that it has an effect of preventing leakage.
さらに、セパレータは、上述したような多孔膜や不織布等とポリマーゲルを併用して用いると、電解質の保液性が向上するため望ましい。即ち、ポリエチレン微孔膜の表面及び微孔壁面に厚さ数μm以下の親溶媒性ポリマーを被覆したフィルムを形成し、前記フィルムの微孔内に電解質を保持させることで、前記親溶媒性ポリマーがゲル化する。 Furthermore, it is desirable that the separator be used in combination with the above-described porous film, non-woven fabric, or the like and a polymer gel because the liquid retention of the electrolyte is improved. That is, by forming a film in which the surface of the polyethylene microporous membrane and the microporous wall are coated with a solvophilic polymer having a thickness of several μm or less, and holding the electrolyte in the micropores of the film, Gels.
前記親溶媒性ポリマーとしては、ポリフッ化ビニリデンの他、エチレンオキシド基やエステル基等を有するアクリレートモノマー、エポキシモノマー、イソシアナート基を有するモノマー等が架橋したポリマー等が挙げられる。該モノマーは、ラジカル開始剤を併用して加熱や紫外線(UV)を用いたり、電子線(EB)等の活性光線等を用いて架橋反応を行わせることが可能である。 Examples of the solvophilic polymer include polyvinylidene fluoride, an acrylate monomer having an ethylene oxide group or an ester group, an epoxy monomer, a polymer having a monomer having an isocyanate group, and the like crosslinked. The monomer can be subjected to a crosslinking reaction using a radical initiator in combination with heating or ultraviolet rays (UV), or using an actinic ray such as an electron beam (EB).
(リチウム二次電池の構成)
本発明のリチウム二次電池の構成については特に限定されるものではなく、正極、負極及びロール状のセパレータを有する円筒型電池、角型電池、扁平型電池等が一例として挙げられる。図1に角型電池の一例を示す。セパレータを挟んで巻回された正極及び負極よりなる電極群2が角型の電池容器3に収納され、正極リード4‘を介して正極端子4が、負極リード5’を介して負極端子5が電池容器外に導出されている。
(Configuration of lithium secondary battery)
The configuration of the lithium secondary battery of the present invention is not particularly limited, and examples thereof include a cylindrical battery having a positive electrode, a negative electrode, and a roll separator, a square battery, and a flat battery. FIG. 1 shows an example of a square battery. An
(蓄電装置の構成)
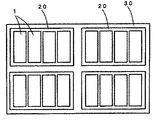
本発明のリチウム二次電池は、特に電気自動車(EV)、ハイブリッド自動車(HEV)、プラグインハイブリッド自動車(PHEV)などの自動車用電源として用いる場合に、複数のリチウム二次電池を集合して構成した蓄電装置(バッテリーモジュール)として搭載することができる。
図2に、リチウム二次電池1が集合した蓄電ユニット20をさらに集合した蓄電装置30の一例を示す。
(Configuration of power storage device)
The lithium secondary battery of the present invention is configured by assembling a plurality of lithium secondary batteries, particularly when used as a power source for an automobile such as an electric vehicle (EV), a hybrid vehicle (HEV), and a plug-in hybrid vehicle (PHEV). It can be mounted as a power storage device (battery module).
FIG. 2 shows an example of a
(充電電位)
従来の正極活物質も、本発明の活物質も、正極電位が4.5V(vs.Li/Li+)付近に至って充放電が可能である。しかしながら、使用する非水電解質の種類によっては、充電時の正極電位が高すぎると、非水電解質が酸化分解され電池性能の低下を引き起こす虞がある。したがって、使用時において、充電時の正極の最大到達電位が4.3V(vs.Li/Li+)以下となるような充電方法を採用しても、充分な放電容量が得られるリチウム二次電池が求められる場合がある。本発明の活物質を用いると、使用時において、充電時の正極の最大到達電位が4.5V(vs.Li/Li+)より低くなるような、例えば、4.4V(vs.Li/Li+)以下や4.3V(vs.Li/Li+)以下となるような充電方法を採用しても、約200mAh/g以上という従来の正極活物質の容量を超える放電電気量を取り出すことが可能である。
(Charge potential)
Both the conventional positive electrode active material and the active material of the present invention can be charged / discharged when the positive electrode potential reaches around 4.5 V (vs. Li / Li + ). However, depending on the type of nonaqueous electrolyte used, if the positive electrode potential during charging is too high, the nonaqueous electrolyte may be oxidized and decomposed, resulting in a decrease in battery performance. Therefore, in use, a lithium secondary battery capable of obtaining a sufficient discharge capacity even when a charging method is adopted in which the maximum potential of the positive electrode during charging is 4.3 V (vs. Li / Li + ) or less. May be required. When the active material of the present invention is used, for example, 4.4 V (vs. Li / Li) such that the maximum potential of the positive electrode during charging is lower than 4.5 V (vs. Li / Li + ) during use. + ) Or less and 4.3 V (vs. Li / Li + ) or less, even if a charging method is adopted, it is possible to take out a discharge electric quantity exceeding the capacity of the conventional positive electrode active material of about 200 mAh / g or more. Is possible.
(実施例1)
<リチウム遷移金属複合酸化物の合成>
硫酸コバルト7水和物14.08g、硫酸ニッケル6水和物21.00g及び硫酸マンガン5水和物65.27gを秤量し、これらの全量をイオン交換水200mlに溶解させ、Co:Ni:Mnのモル比が12.50:19.94:67.56となる2.0Mの硫酸塩水溶液を作製した。一方、2Lの反応槽に750mlのイオン交換水を注ぎ、CO2ガスを30minバブリングさせることにより、イオン交換水中にCO2を溶解させた。反応槽の温度を50℃(±2℃)に設定し、攪拌モーターを備えたパドル翼を用いて反応槽内を700rpmの回転速度で攪拌しながら、前記硫酸塩水溶液を3ml/minの速度で滴下した。ここで、滴下の開始から終了までの間、2.0Mの炭酸ナトリウム、及び0.4Mのアンモニアを含有する水溶液を適宜滴下することにより、反応槽中のpHが常に7.9(±0.05)を保つように制御した。滴下終了後、反応槽内の攪拌をさらに5h継続した。攪拌の停止後、12h以上静置した。
次に、吸引ろ過装置を用いて、反応槽内に生成した共沈炭酸塩の粒子を分離し、さらにイオン交換水を用いて粒子に付着しているナトリウムイオンを洗浄除去し、電気炉を用いて、空気雰囲気中、常圧下、100℃にて20h乾燥させた。その後、粒径を揃えるために、瑪瑙製自動乳鉢で数分間粉砕した。このようにして、共沈炭酸塩前駆体を作製した。
前記共沈炭酸塩前駆体2.278gに、炭酸リチウムをLi:(Co,Ni,Mn)のモル比が1.44となるように加え、瑪瑙製自動乳鉢を用いてよく混合し、混合粉体を調製した。ペレット成型機を用いて、6MPaの圧力で成型し、直径25mmのペレットとした。ペレット成型に供した混合粉体の量は、想定する最終生成物の質量が2gとなるように換算して決定した。前記ペレット1個を全長約100mmのアルミナ製ボートに載置し、箱型電気炉(型番:AMF20)に設置し、空気雰囲気中、常圧下、常温から900℃の温度まで10時間かけて昇温し、昇温後温度で10h焼成した。前記箱型電気炉の内部寸法は、縦10cm、幅20cm、奥行き30cmであり、幅方向20cm間隔に電熱線が入っている。焼成後、ヒーターのスイッチを切り、アルミナ製ボートを炉内に置いたまま自然放冷した。この結果、炉の温度は5時間後には約200℃程度にまで低下するが、その後の降温速度はやや緩やかである。一昼夜経過後、炉の温度が100℃以下となっていることを確認してから、ペレットを取り出し、粒径を揃えるために、瑪瑙製自動乳鉢で数分間粉砕した。このようにして、Naを2100ppm含み、D50が13μmである出発物質のリチウム遷移金属複合酸化物Li1.18Co0.10Ni0.17Mn0.55O2を作製した。
Example 1
<Synthesis of lithium transition metal composite oxide>
Cobalt sulfate heptahydrate (14.08 g), nickel sulfate hexahydrate (21.00 g) and manganese sulfate pentahydrate (65.27 g) were weighed, and all of these were dissolved in 200 ml of ion-exchanged water, and Co: Ni: Mn A 2.0 M aqueous sulfate solution having a molar ratio of 12.50: 19.94: 67.56 was prepared. On the other hand, 750 ml of ion exchange water was poured into a 2 L reaction tank, and CO 2 gas was bubbled for 30 minutes to dissolve CO 2 in the ion exchange water. The temperature of the reaction vessel was set to 50 ° C. (± 2 ° C.), and the aqueous sulfate solution was stirred at a rate of 3 ml / min while stirring the inside of the reaction vessel at a rotational speed of 700 rpm using a paddle blade equipped with a stirring motor. It was dripped. Here, during the period from the start to the end of the dropwise addition, an aqueous solution containing 2.0 M sodium carbonate and 0.4 M ammonia is appropriately dropped, so that the pH in the reaction tank is always 7.9 (± 0. 05). After completion of the dropping, stirring in the reaction vessel was further continued for 5 hours. After the stirring was stopped, the mixture was allowed to stand for 12 hours or more.
Next, using a suction filtration device, the coprecipitated carbonate particles produced in the reaction vessel are separated, and sodium ions adhering to the particles are washed away using ion-exchanged water, and an electric furnace is used. Then, it was dried at 100 ° C. for 20 hours in an air atmosphere under normal pressure. Then, in order to arrange | equalize a particle size, it grind | pulverized for several minutes with the smoked automatic mortar. In this way, a coprecipitated carbonate precursor was produced.
Lithium carbonate is added to 2.278 g of the coprecipitated carbonate precursor so that the molar ratio of Li: (Co, Ni, Mn) is 1.44 and mixed well using a smoked automatic mortar. The body was prepared. Using a pellet molding machine, molding was performed at a pressure of 6 MPa to obtain pellets having a diameter of 25 mm. The amount of the mixed powder subjected to pellet molding was determined by conversion so that the mass of the assumed final product was 2 g. One pellet was placed on an alumina boat having a total length of about 100 mm, placed in a box-type electric furnace (model number: AMF20), and heated in an air atmosphere at normal pressure to a temperature of 900 ° C. over 10 hours. And calcined for 10 hours at the temperature after the temperature elevation. The box-type electric furnace has internal dimensions of 10 cm in length, 20 cm in width, and 30 cm in depth, and heating wires are inserted at intervals of 20 cm in the width direction. After firing, the heater was turned off and allowed to cool naturally with the alumina boat placed in the furnace. As a result, the temperature of the furnace decreases to about 200 ° C. after 5 hours, but the subsequent temperature decrease rate is somewhat moderate. After the passage of day and night, it was confirmed that the furnace temperature was 100 ° C. or lower, and then the pellets were taken out and pulverized for several minutes in a smoked automatic mortar in order to make the particle diameter uniform. In this manner, a starting lithium transition metal composite oxide Li 1.18 Co 0.10 Ni 0.17 Mn 0.55 O 2 containing 2100 ppm Na and having a D50 of 13 μm was prepared.
<セリウム添加酸処理>
300ml三角フラスコに硫酸セリウム四水和物0.0406gと、イオン交換水を加えて溶解し0.0005Mの硫酸セリウム水溶液を調製した。上記水溶液に、pHが1.6となるまで98wt%の硫酸を加えて混合し、酸溶液を調製した。なお、pH調整のために加える硫酸量は0.3ml以下とごく微量であるから、上記酸溶液中のセリウムイオン濃度は、上記水溶液中のセリウムイオン濃度と有効数字の範囲内で等しい。上記の酸溶液をスターラーを用いて、25℃、400rpmで撹拌しているところに、上記のリチウム遷移金属複合酸化物5.0g投入した。リチウム遷移金属複合酸化物を投入してから、30sec後に吸引ろ過することによって、ろ紙上にリチウム遷移金属複合酸化物を回収した。
このリチウム遷移金属複合酸化物を乾燥機(ヤマト科学社製)を用いて、空気中、常圧、80℃で16から18h乾燥させることによって、硫酸セリウム水溶液処理したリチウム遷移金属複合酸化物を得た。
<Cerium-added acid treatment>
To a 300 ml Erlenmeyer flask, 0.0406 g of cerium sulfate tetrahydrate and ion exchange water were added and dissolved to prepare a 0.0005 M aqueous solution of cerium sulfate. To the above aqueous solution, 98 wt% sulfuric acid was added and mixed until the pH reached 1.6 to prepare an acid solution. Since the amount of sulfuric acid added for adjusting the pH is very small, 0.3 ml or less, the cerium ion concentration in the acid solution is equal to the cerium ion concentration in the aqueous solution within the range of significant figures. While the acid solution was being stirred at 25 ° C. and 400 rpm using a stirrer, 5.0 g of the lithium transition metal composite oxide was added. The lithium transition metal composite oxide was collected on the filter paper by suction filtration after 30 seconds from the introduction of the lithium transition metal composite oxide.
This lithium transition metal composite oxide is dried in air at atmospheric pressure and 80 ° C. for 16 to 18 hours using a dryer (manufactured by Yamato Kagaku Co., Ltd.) to obtain a lithium transition metal composite oxide treated with a cerium sulfate aqueous solution. It was.
上記の酸溶液で処理したリチウム遷移金属複合酸化物5.0gをアルミナルツボのフタに加え、箱型電気炉(型番:AMF20)に設置し、空気中、昇温速度が5℃/min、熱処理温度が400℃、保持時間が4hの条件で熱処理することによって、Ceを含むリチウム遷移金属複合酸化物を作製した。 Add 5.0 g of the lithium transition metal composite oxide treated with the above acid solution to the lid of the alumina crucible and place it in a box-type electric furnace (model number: AMF20). In the air, the heating rate is 5 ° C./min. A lithium transition metal composite oxide containing Ce was prepared by heat treatment under conditions of a temperature of 400 ° C. and a holding time of 4 hours.
(実施例2〜5)
酸溶液を調製する際に用いる硫酸セリウム水溶液として、0.001M、0.005M、0.01M、及び0.025Mの水溶液を使用した以外は、実施例1と同様にして、実施例2〜5に係るリチウム遷移金属複合酸化物を作製した。
(Examples 2 to 5)
Examples 2 to 5 are the same as Example 1 except that 0.001M, 0.005M, 0.01M, and 0.025M aqueous solutions were used as the cerium sulfate aqueous solution used in preparing the acid solution. A lithium transition metal composite oxide according to the present invention was prepared.
(比較例1)
酸処理を施さない以外は、実施例1と同様にして、比較例1に係るリチウム遷移金属複合酸化物を作製した。
(Comparative Example 1)
A lithium transition metal composite oxide according to Comparative Example 1 was produced in the same manner as in Example 1 except that the acid treatment was not performed.
(比較例2)
酸溶液として、水素イオン濃度が0.05Mの硫酸水溶液を使用した以外は実施例1と同様にして、比較例2に係るリチウム遷移金属複合酸化物を作製した。
(Comparative Example 2)
A lithium transition metal composite oxide according to Comparative Example 2 was prepared in the same manner as in Example 1 except that a sulfuric acid aqueous solution having a hydrogen ion concentration of 0.05 M was used as the acid solution.
(比較例3〜8)
硫酸セリウム水溶液に代えて、0.0005M、0.001M、0.005M、0.01M、0.025M及び0.05Mの硫酸スズ水溶液をそれぞれ使用した以外は実施例1と同様にして、比較例3〜8に係るリチウム遷移金属複合酸化物を作製した。
(Comparative Examples 3 to 8)
Comparative Example as in Example 1 except that 0.0005M, 0.001M, 0.005M, 0.01M, 0.025M and 0.05M tin sulfate aqueous solutions were used instead of the cerium sulfate aqueous solution, respectively. Lithium transition metal composite oxides according to 3 to 8 were produced.
(比較例9)
硫酸セリウム水溶液に代えて、0.1Mの硫酸鉄水溶液を使用した以外は実施例1と同様にして、比較例9に係るリチウム遷移金属複合酸化物を作製した。
(Comparative Example 9)
A lithium transition metal composite oxide according to Comparative Example 9 was produced in the same manner as in Example 1 except that a 0.1 M aqueous iron sulfate solution was used instead of the cerium sulfate aqueous solution.
(比較例10)
硫酸セリウム水溶液に代えて、0.01Mの硫酸ジルコニウム水溶液を使用した以外は実施例1と同様にして、比較例10に係るリチウム遷移金属複合酸化物を作製した。
(Comparative Example 10)
A lithium transition metal composite oxide according to Comparative Example 10 was produced in the same manner as in Example 1 except that a 0.01 M zirconium sulfate aqueous solution was used instead of the cerium sulfate aqueous solution.
<半値幅の測定>
全ての実施例および比較例に係るリチウム遷移金属複合酸化物は、次の条件及び手順に沿ってX線回折測定を行い、半値幅を決定した。X線回折装置(Rigaku社製、型名:MiniFlexII)を用いて粉末X線回折測定を行った。線源はCuKα、加速電圧及び電流はそれぞれ30kV及び15mAとした。サンプリング幅は0.01deg、走査時間は14分(スキャンスピードは5.0)、発散スリット幅は0.625deg、受光スリット幅は開放、散乱スリットは8.0mmとした。得られたX線回折データについて、前記X線回折装置の付属ソフトである「PDXL」を用いて、X線回折図上2θ=44°±1°に存在する回折ピークについて半値幅を決定した。
<Measurement of half width>
The lithium transition metal composite oxides according to all Examples and Comparative Examples were subjected to X-ray diffraction measurement according to the following conditions and procedures, and the half width was determined. Powder X-ray diffraction measurement was performed using an X-ray diffractometer (manufactured by Rigaku, model name: MiniFlex II). The radiation source was CuKα, and the acceleration voltage and current were 30 kV and 15 mA, respectively. The sampling width was 0.01 deg, the scanning time was 14 minutes (scanning speed was 5.0), the divergence slit width was 0.625 deg, the light receiving slit width was open, and the scattering slit was 8.0 mm. For the obtained X-ray diffraction data, the half width of the diffraction peak at 2θ = 44 ° ± 1 ° on the X-ray diffraction diagram was determined using “PDXL” which is software attached to the X-ray diffractometer.
<金属量の確認試験>
酸処理後の正極活物質に含まれる金属量をICP発光分光分析により定量した。なお、実施例1〜5に係る正極活物質粉末を水中に分散し、超音波洗浄機を用いて2分間超音波振動を与えた。この分散液を試料として粒度分布測定を行ったところ、析出していると考えられるCeO2の粒径に相当する粒子は観測されなかった。したがって、Ce化合物は正極活物質表面に安定的に担持されていることがわかった。
<Metal content confirmation test>
The amount of metal contained in the positive electrode active material after acid treatment was quantified by ICP emission spectroscopic analysis. In addition, the positive electrode active material powder which concerns on Examples 1-5 was disperse | distributed in water, and ultrasonic vibration was given for 2 minutes using the ultrasonic cleaner. When the particle size distribution measurement was performed using this dispersion as a sample, particles corresponding to the particle diameter of CeO 2 considered to be precipitated were not observed. Therefore, it was found that the Ce compound was stably supported on the surface of the positive electrode active material.
<リチウム二次電池の作製>
全ての実施例および比較例に係るリチウム遷移金属複合酸化物をそれぞれリチウム二次電池用正極活物質として用いて、以下の手順でリチウム二次電池を作製した。
<Production of lithium secondary battery>
Using the lithium transition metal composite oxides according to all of the examples and comparative examples as positive electrode active materials for lithium secondary batteries, lithium secondary batteries were fabricated according to the following procedure.
N−メチルピロリドンを分散媒とし、活物質、アセチレンブラック(AB)及びポリフッ化ビニリデン(PVdF)が質量比90:4:6の割合で混練分散されている塗布用ペーストを作製した。該塗布ペーストを厚さ20μmのアルミニウム箔集電体の片方の面に塗布し、正極板を作製した。なお、全ての実施例及び比較例に係るリチウム二次電池同士で試験条件が同一になるように、一定面積当たりに塗布されている活物質の質量及び塗布厚みを統一した。 Using N-methylpyrrolidone as a dispersion medium, an active material, acetylene black (AB), and polyvinylidene fluoride (PVdF) were kneaded and dispersed at a mass ratio of 90: 4: 6. The coating paste was applied to one side of an aluminum foil current collector having a thickness of 20 μm to produce a positive electrode plate. In addition, the mass and coating thickness of the active material applied per fixed area were standardized so that the test conditions were the same among the lithium secondary batteries according to all the examples and comparative examples.
正極の単独挙動を正確に観察する目的のため、対極、即ち負極には金属リチウムをニッケル箔集電体に密着させて用いた。ここで、リチウム二次電池の容量が負極によって制限されないよう、負極には十分な量の金属リチウムを配置した。 For the purpose of accurately observing the single behavior of the positive electrode, metallic lithium was used in close contact with the nickel foil current collector for the counter electrode, that is, the negative electrode. Here, a sufficient amount of metallic lithium was disposed on the negative electrode so that the capacity of the lithium secondary battery was not limited by the negative electrode.
電解液として、エチレンカーボネート(EC)/エチルメチルカーボネート(EMC)/ジメチルカーボネート(DMC)が体積比6:7:7である混合溶媒に濃度が1mol/lとなるようにLiPF6を溶解させた溶液を用いた。セパレータとして、ポリアクリレートで表面改質したポリプロピレン製の微孔膜を用いた。外装体には、ポリエチレンテレフタレート(15μm)/アルミニウム箔(50μm)/金属接着性ポリプロピレンフィルム(50μm)からなる金属樹脂複合フィルムを用い、正極端子及び負極端子の開放端部が外部露出するように電極を収納し、前記金属樹脂複合フィルムの内面同士が向かい合った融着代を注液孔となる部分を除いて気密封止した。前記電解液を注液後、注液孔を封止してリチウム二次電池を作製した。 As an electrolytic solution, LiPF 6 was dissolved in a mixed solvent in which ethylene carbonate (EC) / ethyl methyl carbonate (EMC) / dimethyl carbonate (DMC) had a volume ratio of 6: 7: 7 so that the concentration was 1 mol / l. The solution was used. As the separator, a polypropylene microporous film whose surface was modified with polyacrylate was used. A metal resin composite film made of polyethylene terephthalate (15 μm) / aluminum foil (50 μm) / metal-adhesive polypropylene film (50 μm) is used for the outer package, and the electrodes are exposed so that the open ends of the positive and negative terminals are exposed to the outside And a hermetic seal in which the inner surfaces of the metal resin composite film face each other are hermetically sealed except for a portion serving as a liquid injection hole. After injecting the electrolytic solution, the injection hole was sealed to prepare a lithium secondary battery.
<リチウム二次電池の評価>
(初期効率)
以上の手順にて作製されたリチウム二次電池を、25℃の下、初期充放電工程に供した。充電は、電流0.1CA、電圧4.6Vの定電流定電圧充電とし、充電終止条件は電流値が1/5に減衰した時点とした。放電は、電流0.1CA、終止電圧2.0Vの定電流放電とした。この充放電を1サイクル行った。ここで、充電後及び放電後にそれぞれ10分の休止過程を設けた。この充放電試験における放電容量を充電電気量で割った値(%)を初期充放電効率(表1では、「初期効率」)として記録した。
<Evaluation of lithium secondary battery>
(Initial efficiency)
The lithium secondary battery produced by the above procedure was subjected to an initial charge / discharge process at 25 ° C. Charging was performed at a constant current and a constant voltage with a current of 0.1 CA and a voltage of 4.6 V, and the charge termination condition was the time when the current value was attenuated to 1/5. The discharge was a constant current discharge with a current of 0.1 CA and a final voltage of 2.0 V. This charge / discharge was performed for one cycle. Here, a pause process of 10 minutes was provided after charging and discharging, respectively. A value (%) obtained by dividing the discharge capacity in the charge / discharge test by the amount of charge was recorded as the initial charge / discharge efficiency (“initial efficiency” in Table 1).
(放電容量維持率)
次に、30サイクルの充放電試験を行った。この充放電試験の条件は、充電は、電流0.2CA、電圧4.45Vの定電流定電圧充電とし、充電終止条件は電流値が1/10に減衰した時点とした。放電は、電流0.5CA、終止電圧2.0Vの定電流放電とした。この1サイクル目の放電容量に対する30サイクル目の放電容量の比(%)を放電容量維持率として記録した。
(Discharge capacity maintenance rate)
Next, a 30-cycle charge / discharge test was performed. The charging / discharging test was performed at a constant current / constant voltage with a current of 0.2 CA and a voltage of 4.45 V. The charge termination condition was when the current value was attenuated to 1/10. The discharge was a constant current discharge with a current of 0.5 CA and a final voltage of 2.0 V. The ratio (%) of the discharge capacity at the 30th cycle to the discharge capacity at the 1st cycle was recorded as the discharge capacity retention rate.
実施例及び比較例に係るリチウム遷移金属複合酸化物、及びそれぞれをリチ
ウム二次電池用正極活物質として用いたリチウム二次電池の試験結果を表1に示す。なお、酸処理に用いた酸溶液中のCe、Sn、Fe又はZrイオン濃度を「金属イオン濃度(M)」として表1に示した。
Lithium transition metal composite oxides according to Examples and Comparative Examples, and
Table 1 shows the test results of the lithium secondary battery used as the positive electrode active material for the lithium secondary battery. The Ce, Sn, Fe, or Zr ion concentration in the acid solution used for the acid treatment is shown in Table 1 as “metal ion concentration (M)”.

表1によると、「リチウム過剰型」の活物質に硫酸処理を施した比較例2は、酸処理を施さない比較例1より初期充放電効率が向上しているが、放電容量維持率は低下している。
金属イオン添加の酸処理により、活物質にSn添加(比較例3〜8)、又はFe添加(比較例9)を行った場合も、硫酸処理を施した比較例2の放電容量維持率を上回ることはない。Zr添加を行った比較例10は、硫酸処理を施した比較例2より放電容量維持率は向上しているが、初期効率が低下している。
According to Table 1, Comparative Example 2, in which the “lithium-excess type” active material was subjected to sulfuric acid treatment, had improved initial charge / discharge efficiency than Comparative Example 1, in which acid treatment was not performed, but the discharge capacity retention rate was decreased. doing.
Even when Sn addition (Comparative Examples 3 to 8) or Fe addition (Comparative Example 9) is performed on the active material by acid treatment with addition of metal ions, the discharge capacity maintenance rate of Comparative Example 2 subjected to sulfuric acid treatment is exceeded. There is nothing. In Comparative Example 10 in which Zr was added, the discharge capacity retention rate was improved as compared with Comparative Example 2 in which sulfuric acid treatment was performed, but the initial efficiency was lowered.
これに対して、Ceイオンを添加して酸処理を行った実施例1〜5は、初期効率、放電容量維持率が、いずれも酸処理を施さない比較例1、硫酸処理を施した比較例2、及びSnイオンやFeイオンを添加して酸処理を行った比較例3〜9を上回っている。また、Zrを添加して酸処理を行った比較例10と比べると、放電容量維持率に特段の差異が見られないものの、初期効率が優れている。
実施例1〜5では、X線回折データにおける2θ=44°±1°に存在する(104)面に帰属されるピークの半値幅が0.269〜0.273°であり、酸処理により適度な結晶性を有する活物質が得られ、初期効率が向上したことが確認できる。また、Ceを0.15〜3.37質量%含むことにより、初期効率が優れ、かつ放電容量維持率が高い活物質が得られることも確認できた。正極活物質がCeを含むことによって、正極活物質からのMnの溶出が抑制されたものと考えられる。
On the other hand, Examples 1 to 5 in which Ce ions were added and subjected to acid treatment were comparative examples 1 in which initial efficiency and discharge capacity maintenance rate were not subjected to acid treatment, and comparative examples in which sulfuric acid treatment was performed. 2 and Comparative Examples 3 to 9 where acid treatment was performed by adding Sn ions or Fe ions. Further, compared with Comparative Example 10 in which Zr was added and acid treatment was performed, the initial efficiency was excellent, although no particular difference was observed in the discharge capacity retention rate.
In Examples 1 to 5, the full width at half maximum of the peak attributed to the (104) plane existing at 2θ = 44 ° ± 1 ° in the X-ray diffraction data is 0.269 to 0.273 °, and is moderate by acid treatment. It can be confirmed that an active material having excellent crystallinity was obtained and the initial efficiency was improved. It was also confirmed that an active material having excellent initial efficiency and a high discharge capacity retention rate can be obtained by containing 0.15 to 3.37% by mass of Ce. It is considered that the elution of Mn from the positive electrode active material was suppressed when the positive electrode active material contained Ce.
なお、充放電サイクル後の正極活物質中のモル比Li/Meを次の手順によって確認した。上記充放電試験後、放電末状態の電池を解体して正極を単独で取り出し、金属リチウムを対極にしてセルを組み立て、充電電圧を4.3Vとして電流0.1CmAでの定電流充電を行った後、30分の休止をはさんで0.1CmAにて2.0Vに至るまで定電流放電を行い、放電末状態とした。再び取り出した正極板をジメチルカーボネート(DMC)で洗浄し、室温で30分真空乾燥した。乾燥後の正極から、正極活物質とABとPVdFとが混合している正極合剤50
mgを剥がし取り、35wt%塩酸中に加え、150℃で10分間煮沸することによって正極活物質のみを溶解した。この溶液をろ過することによってABとPVdFを取り除いた。得られたろ液についてICP発光分光分析をおこなった。その結果、モル比Li/Meは、合成後(充放電を行う前)のリチウム遷移金属複合酸化物のモル比Li/Meに対して97%であった。
したがって、原料の仕込み量によって定まるリチウム遷移金属複合酸化物のLi/Meは、活物質として電池電極に用いると、充放電状態によってLi量が変化してしまうが、電池を解体して上記の処理を経て測定されるLi量に、3%分加味することにより、正極活物質の合成後(充放電を行う前)のLi/Me量を推定することができる。
The molar ratio Li / Me in the positive electrode active material after the charge / discharge cycle was confirmed by the following procedure. After the charge / discharge test, the battery in the end-discharge state was disassembled, the positive electrode was taken out alone, a cell was assembled using metallic lithium as a counter electrode, and the constant voltage was charged at a current of 0.1 CmA at a charge voltage of 4.3V. Thereafter, constant current discharge was performed at 0.1 CmA up to 2.0 V with a pause of 30 minutes, and a final discharge state was obtained. The positive electrode plate taken out again was washed with dimethyl carbonate (DMC) and vacuum-dried at room temperature for 30 minutes. Positive electrode mixture 50 in which positive electrode active material, AB, and PVdF are mixed from the positive electrode after drying
The mg was peeled off, added to 35 wt% hydrochloric acid, and boiled at 150 ° C. for 10 minutes to dissolve only the positive electrode active material. AB and PVdF were removed by filtering this solution. The obtained filtrate was subjected to ICP emission spectroscopic analysis. As a result, the molar ratio Li / Me was 97% with respect to the molar ratio Li / Me of the lithium transition metal composite oxide after synthesis (before charging and discharging).
Therefore, when Li / Me of the lithium transition metal composite oxide determined by the amount of raw material charged is used for the battery electrode as an active material, the amount of Li changes depending on the charge / discharge state. The amount of Li / Me after synthesis of the positive electrode active material (before charge / discharge) can be estimated by adding 3% to the amount of Li measured through the above.
また、充放電サイクル後の正極及び負極について、ICP発光分光分析によりCe量を求めたところ、ほぼ100%が正極側から検出された。したがって、充放電サイクル後の電池においても、Ceは正極活物質から消失することがなく、安定的に存在していることがわかる。 Moreover, when the amount of Ce was calculated | required by the ICP emission-spectral-analysis about the positive electrode and negative electrode after a charging / discharging cycle, almost 100% was detected from the positive electrode side. Therefore, it can be seen that Ce does not disappear from the positive electrode active material and exists stably even in the battery after the charge / discharge cycle.
以上のとおり、本発明は、初期効率が優れ、かつ放電容量維持率の高いリチウム二次電池用正極活物質、その正極活物質を含有するリチウム二次電池用電極、及びその電極を備えたリチウム二次電池を提供することができるから、ハイブリッド自動車用、電気自動車用電池としての利用が期待できる。 As described above, the present invention provides a positive electrode active material for a lithium secondary battery that has excellent initial efficiency and a high discharge capacity retention rate, an electrode for a lithium secondary battery containing the positive electrode active material, and lithium including the electrode. Since a secondary battery can be provided, it can be expected to be used as a battery for hybrid vehicles and electric vehicles.
1 リチウム二次電池
2 電極群
3 電池容器
4 正極端子
4’ 正極リード
5 負極端子
5’ 負極リード
20 蓄電ユニット
30 蓄電装置
DESCRIPTION OF
Claims (4)
前記リチウム遷移金属複合酸化物は、遷移金属(Me)がCo、Ni及びMnを含み、
Liと遷移金属(Me)のモル比(Li/Me)が1<Li/Meであり、
Mnと遷移金属(Me)のモル比(Mn/Me)が0.5<Mn/Meであり、
Ceを含有し、
CuKα管球を用いたX線回折パターン解析において、(104)面に帰属される回折ピークの半値幅(FWHM)が0.269≦FWHM≦0.273であることを特徴とするリチウム二次電池用正極活物質。 A positive electrode active material comprising a lithium transition metal composite oxide having an α-NaFeO 2 structure,
In the lithium transition metal composite oxide, the transition metal (Me) includes Co, Ni and Mn,
The molar ratio (Li / Me) between Li and the transition metal (Me) is 1 <Li / Me,
Mn and transition metal (Me) molar ratio (Mn / Me) is 0.5 <Mn / Me,
Containing Ce ,
In the X-ray diffraction pattern analysis using a CuKα tube, the half width (FWHM) of the diffraction peak attributed to the (104) plane is 0.269 ≦ FWHM ≦ 0.273, Positive electrode active material.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015000652A JP6460575B2 (en) | 2015-01-06 | 2015-01-06 | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015000652A JP6460575B2 (en) | 2015-01-06 | 2015-01-06 | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2016126935A JP2016126935A (en) | 2016-07-11 |
| JP2016126935A5 JP2016126935A5 (en) | 2017-11-30 |
| JP6460575B2 true JP6460575B2 (en) | 2019-01-30 |
Family
ID=56359618
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015000652A Active JP6460575B2 (en) | 2015-01-06 | 2015-01-06 | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6460575B2 (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6834363B2 (en) * | 2016-11-02 | 2021-02-24 | 株式会社Gsユアサ | Positive electrode active material for non-aqueous electrolyte secondary batteries, electrodes for non-aqueous electrolyte secondary batteries and non-aqueous electrolyte secondary batteries |
| CN112005410B (en) * | 2018-04-02 | 2024-10-18 | 松下知识产权经营株式会社 | Positive electrode active material for nonaqueous electrolyte secondary battery and nonaqueous electrolyte secondary battery |
| WO2019244955A1 (en) | 2018-06-21 | 2019-12-26 | 株式会社Gsユアサ | Positive electrode active material for non-aqueous electrolyte secondary battery, method for producing positive electrode active material for non-aqueous electrolyte secondary battery, positive electrode for non-aqueous electrolyte secondary battery, non-aqueous electrolyte secondary battery, method for producing non-aqueous electrolyte secondary battery, and method for use of non-aqueous electrolyte secondary battery |
| EP3910707A4 (en) | 2019-03-15 | 2022-03-23 | Basf Toda Battery Materials LLC | Positive electrode active material for non-aqueous electrolyte secondary battery, positive electrode for non-aqueous electrolyte secondary battery, and non-aqueous electrolyte secondary battery |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5055702B2 (en) * | 2005-03-09 | 2012-10-24 | 株式会社Gsユアサ | Positive electrode active material and manufacturing method thereof |
| JP2012138197A (en) * | 2010-12-24 | 2012-07-19 | Asahi Glass Co Ltd | Positive electrode active material for lithium ion secondary battery, positive electrode, lithium ion secondary battery, and method for manufacturing positive electrode active material for lithium ion secondary battery |
| JP6253408B2 (en) * | 2011-06-24 | 2017-12-27 | 住友化学株式会社 | Method for producing positive electrode active material for lithium ion secondary battery |
| CN104247101A (en) * | 2012-04-11 | 2014-12-24 | 旭硝子株式会社 | Positive electrode active material for lithium-ion secondary cell |
| JP6231775B2 (en) * | 2013-05-24 | 2017-11-15 | 旭化成株式会社 | Positive electrode active material and method for producing the same, positive electrode, and non-aqueous secondary battery |
-
2015
- 2015-01-06 JP JP2015000652A patent/JP6460575B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016126935A (en) | 2016-07-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6825559B2 (en) | Positive electrode active material for non-aqueous electrolyte secondary battery, its manufacturing method, electrode for non-aqueous electrolyte secondary battery and non-aqueous electrolyte secondary battery | |
| JP6197939B2 (en) | Non-aqueous electrolyte secondary battery active material, non-aqueous electrolyte secondary battery active material manufacturing method, non-aqueous electrolyte secondary battery electrode, and non-aqueous electrolyte secondary battery | |
| JP6428996B2 (en) | Mixed active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery | |
| JP6497537B2 (en) | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, lithium secondary battery | |
| JP6600944B2 (en) | Lithium secondary battery | |
| JP6044809B2 (en) | Non-aqueous electrolyte secondary battery active material, non-aqueous electrolyte secondary battery electrode, and non-aqueous electrolyte secondary battery | |
| JP6315404B2 (en) | Non-aqueous electrolyte secondary battery positive electrode active material, method for producing the positive electrode active material, non-aqueous electrolyte secondary battery electrode, and non-aqueous electrolyte secondary battery | |
| JP6471693B2 (en) | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery | |
| JP6090661B2 (en) | Positive electrode active material for lithium secondary battery, precursor of the positive electrode active material, electrode for lithium secondary battery, lithium secondary battery | |
| JP6175763B2 (en) | Positive electrode active material for lithium secondary battery, method for producing the positive electrode active material, electrode for lithium secondary battery, and lithium secondary battery | |
| JP5871187B2 (en) | Non-aqueous electrolyte secondary battery active material, method for producing the active material, non-aqueous electrolyte secondary battery electrode, and non-aqueous electrolyte secondary battery | |
| JP6083505B2 (en) | Positive electrode active material for lithium secondary battery, method for producing the positive electrode active material, electrode for lithium secondary battery, and lithium secondary battery | |
| JP5946011B2 (en) | Non-aqueous electrolyte secondary battery active material, non-aqueous electrolyte secondary battery electrode, and non-aqueous electrolyte secondary battery | |
| WO2018012385A1 (en) | Positive electrode active material for nonaqueous electrolyte secondary batteries, transition metal hydroxide precursor, method for producing transition metal hydroxide precursor, method for producing positive electrode active material for nonaqueous electrolyte secondary batteries, electrode for nonaqueous electrolyte secondary batteries, nonaqueous electrolyte secondary battery and electricity storage device | |
| JP2018073481A (en) | Positive electrode active material for nonaqueous electrolyte secondary battery, method for manufacturing the same, positive electrode for nonaqueous electrolyte secondary battery, and nonaqueous electrolyte secondary battery | |
| JP6583662B2 (en) | Positive electrode active material for non-aqueous electrolyte secondary battery and non-aqueous electrolyte secondary battery | |
| JP5757139B2 (en) | Positive electrode active material for non-aqueous electrolyte secondary battery, lithium transition metal composite oxide, method for producing positive electrode active material for non-aqueous electrolyte secondary battery, and non-aqueous electrolyte secondary battery | |
| JP6274536B2 (en) | Method for producing mixed active material for lithium secondary battery, method for producing electrode for lithium secondary battery, and method for producing lithium secondary battery | |
| JP6611074B2 (en) | Mixed active material for lithium secondary battery, positive electrode for lithium secondary battery, and lithium secondary battery | |
| WO2018012384A1 (en) | Lithium-transition-metal composite oxide, transition metal hydroxide precursor, method for manufacturing transition metal hydroxide precursor, method for manufacturing lithium-transition-metal composite oxide, positive-electrode active material for nonaqueous-electrolyte secondary cell, electrode for nonaqueous-electrolyte secondary cell, nonaqueous-electrolyte secondary cell, and power storage device | |
| JP6460575B2 (en) | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery | |
| JP2016015298A (en) | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, lithium secondary battery and power storage device | |
| JP5757140B2 (en) | Non-aqueous electrolyte secondary battery positive electrode active material, lithium transition metal composite oxide, method for producing the positive electrode active material, etc., and non-aqueous electrolyte secondary battery | |
| JP2018073751A (en) | Positive electrode active material for nonaqueous electrolyte secondary battery, electrode for nonaqueous electrolyte secondary battery, and nonaqueous electrolyte secondary battery | |
| JP6474033B2 (en) | Positive electrode active material for lithium secondary battery, electrode for lithium secondary battery, and lithium secondary battery |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171013 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20171017 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180724 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20180725 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180802 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20181210 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20181223 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6460575 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |