JP5572935B2 - Metal cladding - Google Patents
Metal cladding Download PDFInfo
- Publication number
- JP5572935B2 JP5572935B2 JP2008244724A JP2008244724A JP5572935B2 JP 5572935 B2 JP5572935 B2 JP 5572935B2 JP 2008244724 A JP2008244724 A JP 2008244724A JP 2008244724 A JP2008244724 A JP 2008244724A JP 5572935 B2 JP5572935 B2 JP 5572935B2
- Authority
- JP
- Japan
- Prior art keywords
- diamine
- temperature
- polyamide resin
- acid
- nonanediamine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Description
本発明は、例えば一般工業用の流体金属配管の防錆コーティング、自動車用の燃料・オイル・ブレーキ液などの鋼管・アルミ配管といった金属管の防錆用コーティング、金属ワイヤーのコーティング、水槽タンクなど水回りプレートのコーティングなどの金属被覆用途に使用できる、新規なポリアミド樹脂を含む金属被覆材に関する。詳しくは、ジカルボン酸成分が蓚酸であるポリアミド樹脂を含み、低吸水性で、溶融重合による高分子量化が可能であり、耐薬品性、耐加水分解性に優れ、成形可能温度幅が広く成形加工性に優れる金属被覆材に関するものである。 The present invention includes, for example, rust-proof coatings for fluid metal pipes for general industrial use, rust-proof coatings for metal pipes such as steel pipes and aluminum pipes for fuel, oil, and brake fluids for automobiles, coatings for metal wires, water tank tanks, etc. The present invention relates to a metal coating material containing a novel polyamide resin, which can be used for metal coating applications such as coating of a rotating plate. Specifically, it contains polyamide resin whose dicarboxylic acid component is oxalic acid, has low water absorption, can be made high molecular weight by melt polymerization, has excellent chemical resistance and hydrolysis resistance, and has a wide molding temperature range. The present invention relates to a metal coating material having excellent properties.
ナイロン6、ナイロン66などの結晶性ポリアミドに代表されるポリアミド樹脂は、その優れた特性と溶融成形の容易さから、衣料用、産業資材用繊維、あるいは汎用のエンジニアリングプラスチックとして広く用いられている。特に、金属基材の被覆用途においては、ポリアミド樹脂の金属基材への接着性が求められている。また、これらのポリアミド樹脂においては吸水による物性変化、酸、高温のアルコール、熱水中での劣化などの問題点も指摘されており、より寸法安定性及び耐薬品性にも優れたポリアミドへの要求が高まっている。 Polyamide resins typified by crystalline polyamides such as nylon 6 and nylon 66 are widely used as clothing, industrial material fibers, or general-purpose engineering plastics because of their excellent characteristics and ease of melt molding. In particular, in the coating application of a metal substrate, the adhesiveness of the polyamide resin to the metal substrate is required. In addition, these polyamide resins have been pointed out to have problems such as changes in physical properties due to water absorption, deterioration in acid, high-temperature alcohol, and hot water, resulting in a polyamide with superior dimensional stability and chemical resistance. The demand is growing.
金属基材への接着性に優れるポリアミド樹脂を提供するための技術が例えば特許文献1に開示されている。しかしこの技術では、ポリアミド樹脂の吸水によって、物性変化、金属基材の錆発生、長期使用における金属基材と金属被覆材との間の接着性低下などが生じやすいという問題がある。 For example, Patent Document 1 discloses a technique for providing a polyamide resin having excellent adhesion to a metal substrate. However, in this technique, there is a problem that the property of the polyamide resin is changed, the rust of the metal base material is generated, and the adhesiveness between the metal base material and the metal coating material is likely to be lowered during long-term use due to the water absorption of the polyamide resin.
一方、ジカルボン酸成分として蓚酸を用いるポリアミド樹脂はポリオキサミド樹脂と呼ばれ、同じアミノ基濃度の他のポリアミド樹脂と比較して融点が高いこと、吸水率が低いことが知られ(特許文献2)、吸水による物性変化が問題となっていた従来のポリアミドが使用困難な分野での活用が期待される。 On the other hand, a polyamide resin using oxalic acid as a dicarboxylic acid component is called a polyoxamide resin, and is known to have a higher melting point and lower water absorption than other polyamide resins having the same amino group concentration (Patent Document 2). It is expected to be used in fields where the use of conventional polyamide, where the change in physical properties due to water absorption has been a problem, is difficult.
これまでに、ジアミン成分として種々の脂肪族直鎖ジアミンを用いたポリオキサミド樹脂が提案されている。しかしながら、例えば、ジアミン成分として1,6−ヘキサンジアミンを用いたポリオキサミド樹脂は融点(約320℃)が熱分解温度(窒素中の1%重量減少温度;約310℃)より高いため(非特許文献1)、溶融重合、溶融成形が困難であり実用に耐えうるものではなかった。 So far, polyoxamide resins using various aliphatic linear diamines as diamine components have been proposed. However, for example, a polyoxamide resin using 1,6-hexanediamine as a diamine component has a melting point (about 320 ° C.) higher than the thermal decomposition temperature (1% weight loss temperature in nitrogen; about 310 ° C.) (non-patent document). 1) Melt polymerization and melt molding were difficult and could not withstand practical use.
ジアミン成分が1,9−ノナンジアミンであるポリオキサミド樹脂(以後、PA92と略称する)については、L. Francoらが蓚酸源として蓚酸ジエチルを用いた場合の製造法とその結晶構造を開示している(非特許文献2)。ここで得られるPA92は固有粘度が0.97dL/g、融点が246℃のポリマーであるが、実用に耐える強靭な成形体が成形できない程度の低分子量体しか得られていない。また、特許文献3には、ジカルボン酸エステルとして蓚酸ジブチルを用いた場合について、固有粘度が0.99dL/g、融点が248℃のPA92を製造したことが示されている。この場合も強靭な成形体が成形できない程度の低分子量体しか得られていないという問題点がある。 For a polyoxamide resin whose diamine component is 1,9-nonanediamine (hereinafter abbreviated as PA92), L. Franco et al. Discloses a production method and its crystal structure when diethyl oxalate is used as the oxalic acid source ( Non-patent document 2). The PA 92 obtained here is a polymer having an intrinsic viscosity of 0.97 dL / g and a melting point of 246 ° C., but only a low molecular weight product that can not be molded into a tough molded body that can withstand practical use is obtained. Patent Document 3 shows that PA92 having an intrinsic viscosity of 0.99 dL / g and a melting point of 248 ° C. was produced when dibutyl oxalate was used as the dicarboxylic acid ester. In this case as well, there is a problem that only a low molecular weight body that cannot form a tough molded body is obtained.
本発明者らは、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンを特定の比率で用いたポリアミド樹脂が低吸水性でありながら、溶融成形温度幅が広く、しかも諸特性に優れるポリアミド樹脂(PA92C)であることを開示した(特許文献4)。
しかしながら、このポリアミド樹脂は、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンの3種のジアミンを特定の比率で用いたポリオキサミド樹脂ではない。
The present inventors use oxalic acid as a dicarboxylic acid component, and a polyamide resin using 1,9-nonanediamine and 2-methyl-1,8-octanediamine in a specific ratio as a diamine component has low water absorption, It was disclosed that it is a polyamide resin (PA92C) having a wide melt molding temperature range and excellent properties (Patent Document 4).
However, this polyamide resin uses oxalic acid as the dicarboxylic acid component, and has a specific ratio of three diamines of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine as the diamine component. It is not the polyoxamide resin used in 1.
本発明が解決しようとする課題は、十分な高分子量化が達成され、融点と熱分解温度の差から見積もられる成形可能温度幅がより広く、溶融成形性により優れ、さらに、脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、耐薬品性、柔軟性、耐加水分解性などに優れた金属被覆材を提供することにある。 The problem to be solved by the present invention is that a sufficiently high molecular weight is achieved, the moldable temperature range estimated from the difference between the melting point and the thermal decomposition temperature is wider, the melt moldability is excellent, and the aliphatic linear polyoxamide An object of the present invention is to provide a metal coating material excellent in chemical resistance, flexibility, hydrolysis resistance, etc. without impairing the low water absorption observed in resins.
本発明者らは、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンからなるポリアミド樹脂が、低吸水性でありながら、溶融重合による高分子量化が可能で、融点と熱分解温度の差から見積もられる成形可能温度幅が広く溶融成形性に優れ、更に、耐薬品性、耐加水分解性などにも優れることを既に見出した。そして上記の課題を解決するために鋭意検討を重ねた結果、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下において「C9ジアミン混合物」ともいう。)及び1,6−ヘキサンジアミン(以下において「C6ジアミン」ともいう。)からなり、かつC9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂(以下において「PA92/62T」ともいう)を用いることにより、低吸水性で金属基材の錆の発生及び長期使用時の金属基材と金属被覆材との間の接着性低下を抑制でき、溶融重合による高分子量化が可能で、融点と熱分解温度の差で見積もられる成形可能温度幅が例えば50℃以上と広く溶融成形性に優れ、耐薬品性及び耐加水分解性に優れる金属被覆材が得られることを見出し、本発明を完成した。すなわち本発明は以下の通りである。 The inventors of the present invention have a low water-absorbing property when the dicarboxylic acid component is composed of oxalic acid and the diamine component is composed of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine. However, it can be polymerized by melt polymerization, has a wide moldable temperature range estimated from the difference between the melting point and the thermal decomposition temperature, has excellent melt moldability, and has excellent chemical resistance, hydrolysis resistance, etc. Has already been found. As a result of extensive studies to solve the above problems, the dicarboxylic acid component is composed of oxalic acid, and the diamine component is a mixture of 1,9-nonanediamine and 2-methyl-1,8-octanediamine (hereinafter referred to as “C9”). A polyamide resin consisting of 1,6-hexanediamine (hereinafter also referred to as “C6 diamine”), and having a molar ratio of C9 diamine mixture to C6 diamine of 1:99 to 99: 1. By using (hereinafter also referred to as “PA92 / 62T”), it is possible to suppress the occurrence of rust of the metal base material with low water absorption and the decrease in adhesion between the metal base material and the metal coating material during long-term use, High molecular weight by melt polymerization is possible, and the moldable temperature range estimated by the difference between the melting point and the thermal decomposition temperature is as wide as, for example, 50 ° C. Found that metal coating material having excellent sex and hydrolysis resistance can be obtained, and have completed the present invention. That is, the present invention is as follows.
[1] ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下において「C9ジアミン混合物」ともいう。)及び1,6−ヘキサンジアミン(以下において「C6ジアミン」ともいう。)からなり、かつC9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂を含む、金属被覆材。 [1] The dicarboxylic acid component is composed of oxalic acid, and the diamine component is a mixture of 1,9-nonanediamine and 2-methyl-1,8-octanediamine (hereinafter also referred to as “C9 diamine mixture”) and 1,6-hexane. A metal coating material comprising a polyamide resin comprising a diamine (hereinafter also referred to as “C6 diamine”) and having a molar ratio of the C9 diamine mixture to the C6 diamine of 1:99 to 99: 1.
[2] 前記ポリアミド樹脂の、96%硫酸を溶媒とし、濃度1.0g/dlのポリアミド樹脂溶液を用いて25℃で測定した場合の相対粘度(ηr)が1.8〜6.0である、上記[1]に記載の金属被覆材。 [2] The polyamide resin has a relative viscosity (ηr) of 1.8 to 6.0 when measured at 25 ° C. using a polyamide resin solution having a concentration of 1.0 g / dl using 96% sulfuric acid as a solvent. The metal coating material according to [1] above.
[3] 前記ポリアミド樹脂の、窒素雰囲気下、10℃/分の昇温速度で測定した熱重量分析における1%重量減少温度と窒素雰囲気下、10℃/分の昇温速度で測定した示差走査熱量法により測定した融点との温度差が50℃以上である、上記[1]又は[2]に記載の金属被覆材。 [3] 1% weight loss temperature in thermogravimetric analysis of polyamide resin measured at 10 ° C./min in nitrogen atmosphere and differential scanning measured at 10 ° C./min in nitrogen atmosphere The metal coating material according to the above [1] or [2], wherein the temperature difference from the melting point measured by a calorimetric method is 50 ° C. or more.
[4] 前記ジアミン成分の、1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンとのモル比が5:95〜95:5である、上記[1]〜[3]のいずれかに記載の金属被覆材。 [4] Any of [1] to [3] above, wherein the diamine component has a molar ratio of 1,9-nonanediamine to 2-methyl-1,8-octanediamine of 5:95 to 95: 5. The metal coating | covering material as described in.
[5] 熱可塑性エラストマーを更に含む、上記[1]〜[4]のいずれかに記載の金属被覆材。 [5] The metal coating material according to any one of [1] to [4], further including a thermoplastic elastomer.
[6] スチレン系化合物を主体とする重合体ブロックと共役ジエン化合物又はその部分水添物を主体とする重合体ブロックとからなるブロック共重合体の、共役ジエン化合物に由来する二重結合をエポキシ化したエポキシ化スチレン系熱可塑性エラストマーを更に含む、上記[1]〜[5]のいずれかに記載の金属被覆材。 [6] A double bond derived from a conjugated diene compound of a block copolymer consisting of a polymer block mainly composed of a styrene compound and a polymer block mainly composed of a conjugated diene compound or a partially hydrogenated product thereof is epoxy-bonded. The metal coating material according to any one of the above [1] to [5], further comprising a epoxidized styrenic thermoplastic elastomer.
[7] シランカップリング剤を更に含む、上記[1]〜[6]のいずれかに記載の金属被覆材。 [7] The metal coating material according to any one of [1] to [6], further including a silane coupling agent.
[8] 自動車用金属管を被覆する、上記[1]〜[6]のいずれかに記載の金属被覆材。 [8] The metal coating material according to any one of [1] to [6], which covers a metal pipe for automobiles.
本発明の金属被覆材は、低吸水性でありながら、溶融重合による高分子量化が可能で、融点と熱分解温度の差から見積もられる成形可能温度幅が広く溶融成形性に優れ、耐薬品性及び耐加水分解性に優れるため、一般工業用の流体金属配管の防錆コーティング、自動車用の燃料・オイル・ブレーキ液などの鋼管・アルミ配管といった金属管の防錆用コーティング、金属ワイヤーのコーティング、水槽タンクなど水回りプレートのコーティングなどの金属被覆用途における金属被覆材として広範に使用することができる。 The metal coating material of the present invention is capable of increasing the molecular weight by melt polymerization while having low water absorption, has a wide moldable temperature range estimated from the difference between the melting point and the thermal decomposition temperature, and has excellent melt moldability and chemical resistance. And because of its excellent resistance to hydrolysis, anti-corrosion coating for fluid metal pipes for general industrial use, anti-corrosion coating for metal pipes such as steel pipes and aluminum pipes for fuel, oil and brake fluids for automobiles, metal wire coating, It can be widely used as a metal coating material in metal coating applications such as coating of water-circulating plates such as water tanks.
(I)ポリアミド樹脂
(1)ポリアミド樹脂の構成成分
本発明において用いるポリアミド樹脂は、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下において「C9ジアミン混合物」ともいう。)及び1,6−ヘキサンジアミン(以下において「C6ジアミン」ともいう。)からなり、かつC9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂である。
(I) Polyamide resin (1) Constituent component of polyamide resin The polyamide resin used in the present invention is a mixture of diamine component of 1,9-nonanediamine and 2-methyl-1,8-octanediamine (hereinafter referred to as “C9 diamine mixture”). And a 6,9-hexanediamine (hereinafter also referred to as “C6 diamine”), and a molar ratio of C9 diamine mixture to C6 diamine is 1:99 to 99: 1.
上記ポリアミド樹脂の製造に用いられる蓚酸源としては、蓚酸ジエステルを採用でき、これらはアミノ基との反応性を有するものであれば特に制限はなく、蓚酸ジメチル、蓚酸ジエチル、蓚酸ジn−(又はi−)プロピル、蓚酸ジn−(又はi−、又はt−)ブチル等の脂肪族1価アルコールの蓚酸ジエステル、蓚酸ジシクロヘキシル等の脂環式アルコールの蓚酸ジエステル、蓚酸ジフェニル等の芳香族アルコールの蓚酸ジエステル等が挙げられる。 As the oxalic acid source used in the production of the polyamide resin, oxalic acid diesters can be employed, and there is no particular limitation as long as they have reactivity with amino groups. Dimethyl oxalate, diethyl oxalate, di-n-oxalate (or i-) oxalic acid diesters of aliphatic monohydric alcohols such as propyl, di-n- (or i-, or t-) butyl oxalate, oxalic acid diesters of alicyclic alcohols such as dicyclohexyl oxalate, and aromatic alcohols such as diphenyl oxalate Examples include oxalic acid diesters.
上記の蓚酸ジエステルの中でも炭素原子数が3を超える脂肪族1価アルコールの蓚酸ジエステル、脂環式アルコールの蓚酸ジエステル、芳香族アルコールの蓚酸ジエステルが好ましく、その中でも蓚酸ジブチル及び蓚酸ジフェニルが特に好ましい。 Among the above oxalic acid diesters, oxalic acid diesters of aliphatic monohydric alcohols having more than 3 carbon atoms, oxalic acid diesters of alicyclic alcohols, and oxalic acid diesters of aromatic alcohols are preferred, and among them, dibutyl oxalate and diphenyl oxalate are particularly preferred.
本発明のポリアミド樹脂に用いるC9ジアミン混合物における1,9−ノナンジアミン成分と2−メチル−1,8−オクタンジアミン成分のモル比は、一般的には1:99〜99:1であり、好ましくは5:95〜95:5、より好ましくは5:95〜40:60又は60:40〜95:5、特に5:95〜30:70又は70:30〜90:10である。1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンを上記の特定量共重合することにより、成形可能温度幅が広く、溶融成形性に優れ、かつ低吸水性、耐薬品性、耐加水分解性、透明性などにも優れたポリアミドが得られる。 The molar ratio of the 1,9-nonanediamine component to the 2-methyl-1,8-octanediamine component in the C9 diamine mixture used in the polyamide resin of the present invention is generally 1:99 to 99: 1, preferably 5:95 to 95: 5, more preferably 5:95 to 40:60 or 60:40 to 95: 5, especially 5:95 to 30:70 or 70:30 to 90:10. By copolymerizing 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine in the above specific amounts, the moldable temperature range is wide, the melt moldability is excellent, and the water absorption is low. Polyamide having excellent properties, chemical resistance, hydrolysis resistance, transparency and the like can be obtained.
本発明のポリアミド樹脂においては、ジアミン成分として、上記C9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)に1,6−ヘキサンジアミンを混合したものを用いる。C9ジアミン混合物と1,6−ヘキサンジアミンのモル比は、1:99〜99:1である。C9ジアミン混合物に対して1,6−ヘキサンジアミンをモル比で1/99以上混合することにより、ジカルボン酸成分として蓚酸、ジアミン成分としてC9ジアミン混合物からなるポリアミド樹脂(PA92C)の上記の優れた効果を実質的に保持しながら(特に溶融成形性、低吸水性を損なうことなく)、ポリアミド樹脂の融点が上昇し特に力学的物性を向上させることができる。C9ジアミン混合物と1,6−ヘキサンジアミンのモル比は、好ましくは5.1:94.9〜99:1、より好ましくは10:90〜99:1、さらに好ましくは20:80〜99:1である。特に30:70〜98:2、さらに30:70〜90:10(さらに30:70〜70:30)であることが好ましい。本ポリアミド樹脂においては、C9ジアミン混合物に対して1,6−ヘキサンジアミンをモル比で1/99以上共重合することによって融点への変化は明瞭に現れ、樹脂の融点は上昇するが、1,6−ヘキサンジアミンがモル比で99/1以内であれば溶融成形性は許容できるものが得られる。また、1,6−ヘキサンジアミンがモル比で80/20以内であればポリアミド樹脂の融点は300℃以下となり、重合及び成形加工(溶融成形性)がより容易であり、70/30以内であれば融点が280℃以下になって、溶融成形性がより容易となるのでより好ましい。 In the polyamide resin of the present invention, as the diamine component, the C9 diamine mixture (1,9-nonanediamine and 2-methyl-1,8-octanediamine) mixed with 1,6-hexanediamine is used. The molar ratio of C9 diamine mixture to 1,6-hexanediamine is 1:99 to 99: 1. By mixing 1,6-hexanediamine in a molar ratio of 1/99 or more with respect to the C9 diamine mixture, the above excellent effect of the polyamide resin (PA92C) comprising oxalic acid as the dicarboxylic acid component and C9 diamine mixture as the diamine component Can be maintained (particularly without impairing melt moldability and low water absorption), the melting point of the polyamide resin can be increased, and particularly the mechanical properties can be improved. The molar ratio of the C9 diamine mixture to 1,6-hexanediamine is preferably 5.1: 94.9 to 99: 1, more preferably 10:90 to 99: 1, and even more preferably 20:80 to 99: 1. It is. In particular, it is preferably 30:70 to 98: 2, and more preferably 30:70 to 90:10 (further 30:70 to 70:30). In this polyamide resin, the change to the melting point appears clearly by copolymerizing 1,6-hexanediamine in a molar ratio of 1/99 or more with respect to the C9 diamine mixture, and the melting point of the resin rises. If 6-hexanediamine is within a molar ratio of 99/1, an acceptable melt moldability can be obtained. If the molar ratio of 1,6-hexanediamine is within 80/20, the melting point of the polyamide resin will be 300 ° C. or lower, and polymerization and molding (melt moldability) will be easier, and within 70/30. It is more preferable because the melting point becomes 280 ° C. or lower and the melt moldability becomes easier.
(2)ポリアミド樹脂の製造において配合できる成分
本発明において用いるポリアミド樹脂を製造する際には、本発明の効果を損なわない範囲で他のジカルボン酸成分を混合する事ができる。蓚酸以外の他のジカルボン酸成分としては、マロン酸、ジメチルマロン酸、コハク酸、グルタル酸、アジピン酸、2−メチルアジピン酸、トリメチルアジピン酸、ピメリン酸、2,2−ジメチルグルタル酸、3,3−ジエチルコハク酸、アゼライン酸、セバシン酸、スベリン酸などの脂肪族ジカルボン酸、また、1,3−シクロペンタンジカルボン酸、1,4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、更にテレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、2,7−ナフタレンジカルボン酸、1,4−ナフタレンジカルボン酸、1,4−フェニレンジオキシジ酢酸、1,3−フェニレンジオキシジ酢酸、ジ安息香酸、4,4’−オキシジ安息香酸、ジフェニルメタン−4,4’−ジカルボン酸、ジフェニルスルホン−4,4’−ジカルボン酸、4,4’−ビフェニルジカルボン酸などの芳香族ジカルボン酸などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。更に、トリメリット酸、トリメシン酸、ピロメリット酸などの多価カルボン酸を溶融成形が可能な範囲内で用いることもできる。他のジカルボン酸成分の使用量は、ジカルボン酸成分全体の5モル%以下であることが好ましい。
(2) Components that can be blended in the production of polyamide resin When producing the polyamide resin used in the present invention, other dicarboxylic acid components can be mixed within a range not impairing the effects of the present invention. Examples of dicarboxylic acid components other than succinic acid include malonic acid, dimethylmalonic acid, succinic acid, glutaric acid, adipic acid, 2-methyladipic acid, trimethyladipic acid, pimelic acid, 2,2-dimethylglutaric acid, 3, Aliphatic dicarboxylic acids such as 3-diethylsuccinic acid, azelaic acid, sebacic acid and suberic acid, alicyclic dicarboxylic acids such as 1,3-cyclopentanedicarboxylic acid and 1,4-cyclohexanedicarboxylic acid, and terephthalic acid , Isophthalic acid, 2,6-naphthalenedicarboxylic acid, 2,7-naphthalenedicarboxylic acid, 1,4-naphthalenedicarboxylic acid, 1,4-phenylenedioxydiacetic acid, 1,3-phenylenedioxydiacetic acid, dibenzoic acid Acid, 4,4′-oxydibenzoic acid, diphenylmethane-4,4′-dicarboxylic acid, diphenylsulfuric acid Down-4,4'-dicarboxylic acid, 4,4'-biphenyl and the like alone aromatic dicarboxylic acids such as dicarboxylic acids, or may be added to any mixture thereof during the polycondensation reaction. Furthermore, polyvalent carboxylic acids such as trimellitic acid, trimesic acid and pyromellitic acid can be used as long as melt molding is possible. It is preferable that the usage-amount of another dicarboxylic acid component is 5 mol% or less of the whole dicarboxylic acid component.
また、本発明において用いるポリアミド樹脂を製造する際には、本発明の効果を損なわない範囲で、他のジアミン成分を混合する事ができる。1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミン以外の他のジアミン成分としては、エチレンジアミン、プロピレンジアミン、1,4−ブタンジアミン、1,8−オクタンジアミン、1,10−デカンジアミン、1,12−ドデカンジアミン、3−メチル−1,5−ペンタンジアミン、2,2,4−トリメチル−1,6−ヘキサンジアミン、2,4,4−トリメチル−1,6−ヘキサンジアミン、5−メチル−1,9−ノナンジアミンなどの脂肪族ジアミン、更にシクロヘキサンジアミン、メチルシクロヘキサンジアミン、イソホロンジアミンなどの脂環式ジアミン、更にp−フェニレンジアミン、m−フェニレンジアミン、p−キシレンジアミン、m−キシレンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルエーテルなどの芳香族ジアミンなどを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。他のジアミン成分の使用量は、ジアミン成分全体の5モル%以下であることが好ましい。 Moreover, when manufacturing the polyamide resin used in this invention, another diamine component can be mixed in the range which does not impair the effect of this invention. Other diamine components other than 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine include ethylenediamine, propylenediamine, 1,4-butanediamine, and 1,8-octanediamine. 1,10-decanediamine, 1,12-dodecanediamine, 3-methyl-1,5-pentanediamine, 2,2,4-trimethyl-1,6-hexanediamine, 2,4,4-trimethyl-1 , 6-hexanediamine, aliphatic diamines such as 5-methyl-1,9-nonanediamine, cyclohexanediamine, methylcyclohexanediamine, isophoronediamine and other alicyclic diamines, p-phenylenediamine, m-phenylenediamine, p -Xylenediamine, m-xylenediamine, 4,4'-diamy Diphenylmethane, 4,4'-diaminodiphenyl sulfone, 4,4'-and aromatic diamines, such as diaminodiphenyl ether by itself, or may be added to any mixture thereof during the polycondensation reaction. It is preferable that the usage-amount of another diamine component is 5 mol% or less of the whole diamine component.
本発明で用いるポリアミド樹脂は、本発明の効果を損なわない範囲で、一部が他のポリマー成分で置き換えられたものであってもよい。他のポリマー成分としては、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンからなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミン)と1,6−ヘキサンジアミンのモル比が1:99〜99:1であるポリアミド以外のポリアミドとしての、ポリオキサミド、芳香族ポリアミド、脂肪族ポリアミド、脂環式ポリアミドなどのポリアミド類や、ポリアミド以外の熱可塑性ポリマーなどが挙げられる。本発明において用いるポリアミド樹脂中の、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンからなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミン)と1,6−ヘキサンジアミンのモル比が1:99〜99:1であるポリアミドの割合は、50質量%超、更に70質量%以上が好ましい。 The polyamide resin used in the present invention may be one in which a part thereof is replaced with another polymer component as long as the effects of the present invention are not impaired. As other polymer components, the dicarboxylic acid component is composed of oxalic acid, the diamine component is composed of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine, and a C9 diamine mixture (1 , 9-nonanediamine and 2-methyl-1,8-octanediamine) and 1,6-hexanediamine in a molar ratio of 1:99 to 99: 1 as polyamides other than polyamides, polyoxamides, aromatic polyamides, fats And polyamides such as aromatic polyamides and alicyclic polyamides, and thermoplastic polymers other than polyamides. In the polyamide resin used in the present invention, the dicarboxylic acid component is composed of oxalic acid, the diamine component is composed of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine, and a C9 diamine mixture. The proportion of polyamide in which the molar ratio of (1,9-nonanediamine and 2-methyl-1,8-octanediamine) to 1,6-hexanediamine is from 1:99 to 99: 1 is more than 50% by mass, and more than 70%. The mass% or more is preferable.
(3)ポリアミド樹脂の性状及び物性
本発明において用いるポリアミド樹脂の分子量に特別の制限はないが、ポリアミド樹脂濃度が1.0g/dlの96%濃硫酸溶液を用い、25℃で測定した相対粘度ηrが1.8〜6.0の範囲内であることが好ましく、より好ましくは2.0〜5.5であり、2.5〜4.5が特に好ましい。ηrが1.8より低いと成形物が脆くなり物性が低下する傾向がある。一方、ηrが6.0より高いと溶融粘度が高くなり、成形加工性が悪くなる傾向がある。
(3) Properties and properties of polyamide resin The molecular weight of the polyamide resin used in the present invention is not particularly limited, but the relative viscosity measured at 25 ° C. using a 96% concentrated sulfuric acid solution with a polyamide resin concentration of 1.0 g / dl. ηr is preferably in the range of 1.8 to 6.0, more preferably 2.0 to 5.5, and particularly preferably 2.5 to 4.5. If ηr is lower than 1.8, the molded product tends to be brittle and the physical properties tend to decrease. On the other hand, when ηr is higher than 6.0, the melt viscosity becomes high and the molding processability tends to deteriorate.
本発明において用いるポリアミド樹脂は、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンを共重合することで、蓚酸と1,9−ノナンジアミンからなるポリアミドと比べて、上記相対粘度を増加させること、すなわち分子量を増加させることが可能である。また、実質的な熱分解の指標である1%重量減少温度(以下、Tdと略す)と融点(以下、Tmと略す)の差(Td−Tm)で表される成形可能温度範囲が、蓚酸と1,9−ノナンジアミンからなるポリアミドと比べて拡大し、成形可能温度範囲が好ましくは50℃以上、より好ましくは60℃以上であることができ、更には90℃以上も可能である。本発明において用いるポリアミド樹脂は、Tdが好ましくは280℃以上、より好ましくは300℃以上、更に好ましくは320℃以上であり、高い耐熱性を有することを特徴とする。 The polyamide resin used in the present invention uses oxalic acid as the dicarboxylic acid component and oxalic acid by copolymerizing 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine as the diamine component. It is possible to increase the relative viscosity, that is, to increase the molecular weight, as compared with a polyamide comprising 1,9-nonanediamine. The moldable temperature range represented by the difference (Td−Tm) between the 1% weight loss temperature (hereinafter abbreviated as Td) and the melting point (hereinafter abbreviated as Tm), which is a substantial thermal decomposition index, is oxalic acid. And a moldable temperature range can be preferably 50 ° C. or higher, more preferably 60 ° C. or higher, and even 90 ° C. or higher. The polyamide resin used in the present invention has a Td of preferably 280 ° C. or higher, more preferably 300 ° C. or higher, still more preferably 320 ° C. or higher, and has high heat resistance.
(4)ポリアミド樹脂の製造
本発明において用いるポリアミド樹脂は、ポリアミドを製造する方法として知られている任意の方法を用いて製造することができる。本発明者らの研究によれば、ジアミン及び蓚酸ジエステルをバッチ式又は連続式で重縮合反応させることによりポリアミド樹脂を得ることができる。具体的には、以下の操作で示されるような、(i)前重縮合工程、(ii)後重縮合工程の順で行うのが好ましい。
(4) Manufacture of polyamide resin The polyamide resin used in the present invention can be manufactured using any method known as a method of manufacturing polyamide. According to the study by the present inventors, a polyamide resin can be obtained by polycondensation reaction of diamine and oxalic acid diester in a batch or continuous manner. Specifically, it is preferable to carry out in the order of (i) pre-polycondensation step and (ii) post-polycondensation step as shown by the following operations.
(i)前重縮合工程:まず反応器内を窒素置換した後、ジアミン(ジアミン成分)及び蓚酸ジエステル(蓚酸源)を混合する。混合する場合にジアミン及び蓚酸ジエステルが共に可溶な溶媒を用いても良い。ジアミン成分及び蓚酸源が共に可溶な溶媒としては、特に制限されないが、トルエン、キシレン、トリクロロベンゼン、フェノール、トリフルオロエタノールなどを用いることができ、特にトルエンを好ましく用いることができる。例えば、ジアミンを溶解したトルエン溶液を50℃に加熱した後、これに対して蓚酸ジエステルを加える。このとき、蓚酸ジエステルと上記ジアミンの仕込み比は、蓚酸ジエステル/上記ジアミンで、0.8〜1.5(モル比)、好ましくは0.91〜1.1(モル比)、更に好ましくは0.99〜1.01(モル比)であることができる。 (I) Pre-polycondensation step: First, the inside of the reactor is purged with nitrogen, and then diamine (diamine component) and oxalic acid diester (oxalic acid source) are mixed. When mixing, a solvent in which both the diamine and the oxalic acid diester are soluble may be used. The solvent in which both the diamine component and the oxalic acid source are soluble is not particularly limited, but toluene, xylene, trichlorobenzene, phenol, trifluoroethanol, and the like can be used, and particularly, toluene can be preferably used. For example, after heating the toluene solution which melt | dissolved diamine to 50 degreeC, oxalic acid diester is added with respect to this. At this time, the charging ratio of the oxalic acid diester and the diamine is oxalic acid diester / the diamine, 0.8 to 1.5 (molar ratio), preferably 0.91 to 1.1 (molar ratio), more preferably 0. .99 to 1.01 (molar ratio).
このように仕込んだ反応器内を攪拌及び/又は窒素バブリングしながら、常圧下で昇温する。反応温度は、最終到達温度が80〜150℃、好ましくは100〜140℃の範囲になるように制御するのが好ましい。最終到達温度での反応時間は例えば3時間〜6時間である。 The temperature in the reactor charged in this way is increased under normal pressure while stirring and / or nitrogen bubbling. The reaction temperature is preferably controlled so that the final temperature reaches 80 to 150 ° C., preferably 100 to 140 ° C. The reaction time at the final temperature is, for example, 3 to 6 hours.
(ii)後重縮合工程:更に高分子量化を図るために、前重縮合工程で生成した重合物を常圧下において反応器内で徐々に昇温する。昇温過程において前重縮合工程の最終到達温度、すなわち80〜150℃から、最終的に220℃以上300℃以下、好ましくは230℃以上280℃以下、更に好ましくは240℃以上270℃以下の温度範囲にまで到達させる。昇温時間を含めて1〜8時間、好ましくは2〜6時間保持して反応を行うことが好ましい。更に後重合工程において、必要に応じて減圧下での重合を行うこともできる。減圧重合を行う場合の好ましい最終到達圧力は0.1MPa未満〜13.3Paである。 (Ii) Post-polycondensation step: In order to further increase the molecular weight, the polymer produced in the pre-polycondensation step is gradually heated in the reactor under normal pressure. In the temperature rising process, the final temperature of the prepolycondensation step, that is, from 80 to 150 ° C, is finally 220 ° C to 300 ° C, preferably 230 ° C to 280 ° C, more preferably 240 ° C to 270 ° C. Let reach the range. It is preferable to carry out the reaction for 1 to 8 hours including the temperature raising time, preferably 2 to 6 hours. Furthermore, in the post-polymerization step, polymerization can be performed under reduced pressure as necessary. The preferable final ultimate pressure in the case of performing the vacuum polymerization is less than 0.1 MPa to 13.3 Pa.
本発明に用いるポリアミド樹脂の製造方法の具体的例を説明する。
まず原料の蓚酸ジエステルを容器内に仕込む。容器は、後に行う重縮合反応の温度および圧力に耐え得るものであれば、特に制限されない。その後、容器を原料のジアミンと混合する温度まで昇温させ、次いでジアミンを注入し重縮合反応を開始させる。原料を混合する温度は、原料の蓚酸ジエステルおよびジアミンの融点以上、沸点未満の温度であり、かつ蓚酸ジエステルとジアミンの重縮合反応によって生じるポリオキサミドが熱分解しない温度であれば特に制限されない。例えば、1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン、1,6−ヘキサンジアミンの混合物からなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比が1:99〜99:1であるジアミンと蓚酸ジブチルを原料とするポリオキサミド樹脂の場合、上記混合温度は15℃から300℃が好ましい。また、C9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比は、5:95〜90:10の場合、常温で液状か又は50℃程度に加温するだけで液化するので取り扱いやすいためより好ましい。混合温度が縮合反応によって生成するアルコールの沸点以上の場合、アルコールを留去、凝縮する装置を備えた容器を用いるのが望ましい。また、縮合反応によって生成するアルコール存在下で加圧重合する場合には、耐圧容器を用いる。蓚酸ジエステルとジアミンの仕込み比は、蓚酸ジエステル/上記ジアミンで、0.8〜1.2(モル比)、好ましくは0.91〜1.09(モル比)、更に好ましくは0.98〜1.02(モル比)である。
The specific example of the manufacturing method of the polyamide resin used for this invention is demonstrated.
First, the raw oxalic acid diester is charged into the container. The container is not particularly limited as long as it can withstand the temperature and pressure of the polycondensation reaction to be performed later. Thereafter, the container is heated to a temperature at which it is mixed with the raw material diamine, and then the diamine is injected to start the polycondensation reaction. The temperature at which the raw materials are mixed is not particularly limited as long as it is a temperature not lower than the boiling point and lower than the boiling point of the oxalic acid diester and diamine, and the polyoxamide generated by the polycondensation reaction of the oxalic acid diester and diamine is not thermally decomposed. For example, a mixture of 1,9-nonanediamine, 2-methyl-1,8-octanediamine, 1,6-hexanediamine, and a C9 diamine mixture (1,9-nonanediamine and 2-methyl-1,8-octane) In the case of a polyoxamide resin made from diamine and dibutyl oxalate having a molar ratio of a mixture of diamines) and 1,6-hexanediamine of 1:99 to 99: 1, the mixing temperature is preferably 15 ° C to 300 ° C. When the molar ratio of C9 diamine mixture (1,9-nonanediamine and 2-methyl-1,8-octanediamine mixture) and 1,6-hexanediamine is 5:95 to 90:10, it is liquid at room temperature. Or it is more preferable because it is easy to handle because it liquefies only by heating to about 50 ° C. When the mixing temperature is equal to or higher than the boiling point of the alcohol produced by the condensation reaction, it is desirable to use a container equipped with a device for distilling and condensing the alcohol. In addition, when pressure polymerization is performed in the presence of an alcohol generated by a condensation reaction, a pressure vessel is used. The charging ratio of oxalic acid diester to diamine is oxalic acid diester / the above diamine, 0.8 to 1.2 (molar ratio), preferably 0.91 to 1.09 (molar ratio), more preferably 0.98 to 1. 0.02 (molar ratio).
次に、容器内をポリオキサミド樹脂の融点以上かつ熱分解しない温度以下に昇温する。例えば、1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンからなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比が50:50であり、さらに1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンのモル比が50:50であるジアミンと蓚酸ジブチルを原料とするポリオキサミド樹脂の場合、融点は261℃であることから270℃から300℃に昇温するのが好ましい(圧力は、2MPa〜4MPa)。生成したアルコールを留去しながら、必要に応じて常圧窒素気流下もしくは減圧下において継続して重縮合反応を行う。耐圧容器内で原料を混合し、縮合反応によって生成するアルコール存在下で加圧重合する場合は、まず生成したアルコールを留去しながら放圧する。その後、必要に応じて常圧窒素気流下もしくは減圧下において継続して重縮合反応を行う。減圧重合を行う場合の好ましい最終到達圧力は760〜0.1Torrである。温度は、270〜300℃が好ましい。また、アルコールは水冷コンデンサで冷却して液化し、回収する。 Next, the inside of the container is heated to a temperature not lower than the melting point of the polyoxamide resin and not higher than the temperature at which it does not decompose. For example, it consists of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine, and a C9 diamine mixture (of 1,9-nonanediamine and 2-methyl-1,8-octanediamine). A mixture) and a 1,6-hexanediamine molar ratio of 50:50, and a 1,9-nonanediamine and 2-methyl-1,8-octanediamine molar ratio of 50:50 diamine and dibutyl oxalate. In the case of the polyoxamide resin used as a raw material, the melting point is 261 ° C., so it is preferable to raise the temperature from 270 ° C. to 300 ° C. (pressure is 2 MPa to 4 MPa). While distilling off the produced alcohol, the polycondensation reaction is continued under an atmospheric pressure of nitrogen or reduced pressure as necessary. When the raw materials are mixed in a pressure vessel and subjected to pressure polymerization in the presence of an alcohol produced by a condensation reaction, the pressure is first released while the produced alcohol is distilled off. Thereafter, the polycondensation reaction is continued under an atmospheric pressure of nitrogen or reduced pressure as necessary. The preferable final pressure in the case of carrying out the vacuum polymerization is 760 to 0.1 Torr. The temperature is preferably 270 to 300 ° C. The alcohol is cooled and liquefied by a water-cooled condenser and recovered.
(II)その他の含有成分
本発明においては、上述のポリアミド樹脂に加えて、必要に応じて各種添加剤を組合せることができ、これらはポリアミド重縮合反応時、又はその後に組合せることができる。
(II) Other components In the present invention, various additives can be combined as necessary in addition to the above-mentioned polyamide resin, and these can be combined during or after the polyamide polycondensation reaction. .
各種添加剤としては、接着性改良剤、フィラー、補強繊維、銅化合物などの安定剤、着色剤、紫外線吸収剤、光安定化剤、酸化防止剤、帯電防止剤、難燃剤、結晶化促進剤、ガラス繊維、可塑剤、潤滑剤、耐熱剤などが挙げられる。 Various additives include adhesion improvers, fillers, reinforcing fibers, stabilizers such as copper compounds, colorants, UV absorbers, light stabilizers, antioxidants, antistatic agents, flame retardants, and crystallization accelerators. , Glass fiber, plasticizer, lubricant, heat-resistant agent and the like.
本発明の金属被覆材を金属基材上に例えばプライマーを介さずに形成する場合、金属被覆材は接着性改良剤を更に含むことが好ましい。接着性改良剤としては従来公知のものを使用でき、例えば熱可塑性エラストマー、特にエポキシ化スチレン系エラストマー、変性ポリオレフィンなど、更にシランカップリング剤などが挙げられる。 When forming the metal coating material of this invention on a metal base material, for example without a primer, it is preferable that a metal coating material further contains an adhesive improvement agent. As the adhesion improver, conventionally known ones can be used, and examples thereof include thermoplastic elastomers, particularly epoxidized styrene elastomers, modified polyolefins, and further silane coupling agents.
上記のエポキシ化スチレン系エラストマーとしては、例えば前述の特開2004−346255号公報に記載されるような、スチレン系化合物を主体とする重合体ブロックと、共役ジエン化合物又はその部分水添物を主体とする重合体ブロックとからなるブロック共重合体の、共役ジエン化合物に由来する二重結合をエポキシ化したエポキシ化スチレン系熱可塑性エラストマー(以下、単にエポキシ化スチレン系熱可塑性エラストマーというときは特記しない限り上述のものを指す)などが挙げられる。 Examples of the epoxidized styrene elastomer include a polymer block mainly composed of a styrene compound and a conjugated diene compound or a partially hydrogenated product thereof as described in, for example, the above-mentioned JP-A No. 2004-346255. An epoxidized styrene thermoplastic elastomer obtained by epoxidizing a double bond derived from a conjugated diene compound of a block copolymer comprising a polymer block (hereinafter referred to simply as an epoxidized styrene thermoplastic elastomer) As long as it refers to the above).
上記スチレン系化合物としては、例えば、スチレン、α−メチルスチレン、ビニルトルエン、p−第3級ブチルスチレン、ジビニルベンゼン、p−メチルスチレン、1,1−ジフェニルスチレン、ビニルナフタレン、ビニルアントラセンなどから選択される1種又は2種以上を例示でき、中でもスチレンが好ましい。 The styrene compound is selected from, for example, styrene, α-methylstyrene, vinyltoluene, p-tertiary butylstyrene, divinylbenzene, p-methylstyrene, 1,1-diphenylstyrene, vinylnaphthalene, vinylanthracene and the like. 1 type or 2 types or more can be illustrated, and styrene is preferable among them.
また上記共役ジエン化合物としては、例えば、ブタジエン、イソプレン、1,3−ペンタジエン、2,3−ジメチル−1,3−ブタジエン、ピペリレン、3−ブチル−1,3−オクタジエン、フェニル−1,3−ブタジエンなどから選択される1種又は2種以上を例示でき、中でもブタジエン、イソプレン及びこれらの組み合わせが好ましい。 Examples of the conjugated diene compound include butadiene, isoprene, 1,3-pentadiene, 2,3-dimethyl-1,3-butadiene, piperylene, 3-butyl-1,3-octadiene, phenyl-1,3- One or more types selected from butadiene and the like can be exemplified, and butadiene, isoprene and a combination thereof are particularly preferable.
上記ブロック共重合体における上記スチレン系化合物由来の構成単位の含有量は、5〜70質量%であることが好ましく、より好ましくは10〜60質量%である。また、上記ブロック共重合体の重量平均分子量は、5,000〜600,000であることが好ましく、より好ましくは10,000〜500,000の範囲であり、分子量分布[重量平均分子量(Mw)と数平均分子量(Mn)との比(Mw/Mn)]は、10以下であることが好ましい。 The content of the structural unit derived from the styrenic compound in the block copolymer is preferably 5 to 70% by mass, and more preferably 10 to 60% by mass. The block copolymer preferably has a weight average molecular weight of 5,000 to 600,000, more preferably 10,000 to 500,000, and a molecular weight distribution [weight average molecular weight (Mw). And the ratio of the number average molecular weight (Mn) (Mw / Mn)] is preferably 10 or less.
上記ブロック共重合体の分子構造は、直鎖状であることが好ましい。また上記スチレン系化合物(A)と上記共役ジエン化合物(B)とが、例えばA−B−A、B−A−B−A、A−B−A−B−Aなどの構造をとるスチレン系化合物−共役ジエン化合物ブロック共重合体が好ましい。また上記ブロック共重合体は、分子末端に多官能のカップリング剤残基を有していてもよい。 The molecular structure of the block copolymer is preferably linear. In addition, the styrene compound (A) and the conjugated diene compound (B) have a structure such as ABA, BABA, ABBABA, or the like. A compound-conjugated diene compound block copolymer is preferred. The block copolymer may have a polyfunctional coupling agent residue at the molecular end.
上記ブロック共重合体の製造方法は、上述のような構造を有するものが得られればどのような製造方法でもよい。例えば、特公昭40−23798号、特公昭43−17979号、特公昭46−32415号、特公昭56−28925号公報に記載された方法により、リチウム触媒等を用いて不活性溶媒中でスチレン系化合物−共役ジエン化合物ブロック共重合体を製造することができる。更に、特公昭42−8704号、特公昭43−6636号、又は特開昭59−133203号公報に記載された方法により、不活性溶媒中で水素添加触媒の存在下に水素添加して、本発明に供するエポキシ変性ブロック共重合体の原料である部分的に水素添加したブロック共重合体を製造することができる。なお、水添の程度は、水添前及び水添後のブロック共重合体をNMR分析することによって知ることができる。水添率は、未水添・未エポキシ化の原料ブロック共重合体の共役ジエン化合物に由来する二重結合のうち、水添されたものの百分率として定義する。本発明においては、水添率0〜80%の範囲であることが好ましく、特には10〜70%の範囲であることが好ましい。この範囲で耐熱性、凝集性に優れたエポキシ化スチレン系熱可塑性エラストマーが得られる。 The manufacturing method of the block copolymer may be any manufacturing method as long as the block copolymer having the above-described structure is obtained. For example, a styrenic resin in an inert solvent using a lithium catalyst or the like by the method described in JP-B-40-23798, JP-B-43-17799, JP-B-46-32415, and JP-B-56-28925. A compound-conjugated diene compound block copolymer can be produced. Further, hydrogenation is carried out in the presence of a hydrogenation catalyst in an inert solvent by the method described in JP-B-42-8704, JP-B-43-6636, or JP-A-59-133203. A partially hydrogenated block copolymer which is a raw material for the epoxy-modified block copolymer used in the invention can be produced. The degree of hydrogenation can be determined by NMR analysis of the block copolymer before and after hydrogenation. The hydrogenation rate is defined as the percentage of hydrogenated double bonds derived from the conjugated diene compound of the unhydrogenated / epoxidized raw material block copolymer. In the present invention, the hydrogenation rate is preferably in the range of 0 to 80%, particularly preferably in the range of 10 to 70%. Within this range, an epoxidized styrene thermoplastic elastomer excellent in heat resistance and cohesiveness can be obtained.
上記ブロック共重合体をエポキシ化することにより、エポキシ化スチレン系熱可塑性エラストマーを得ることができる。例えば、上記ブロック共重合体を不活性溶媒中でハイドロパーオキサイド類、過酸類等のエポキシ化剤と反応させることにより得ることができる。 Epoxidized styrene thermoplastic elastomer can be obtained by epoxidizing the block copolymer. For example, it can be obtained by reacting the block copolymer with an epoxidizing agent such as hydroperoxides and peracids in an inert solvent.
不活性溶媒は、原料粘度の低下、エポキシ化剤の希釈による安定化等の目的で使用し、例えばヘキサン、シクロヘキサン、トルエン、ベンゼン、酢酸エチル、四塩化炭素、クロロホルム等を用いることができる。 The inert solvent is used for the purpose of lowering the raw material viscosity and stabilizing by dilution of the epoxidizing agent, and for example, hexane, cyclohexane, toluene, benzene, ethyl acetate, carbon tetrachloride, chloroform and the like can be used.
エポキシ化剤の内、ハイドロパーオキサイド類として、過酸化水素、ターシャリブチルハイドロパーオキサイド、クメンハイドロパーオキサイド等が例示できる。また、「過酸類」として、過ギ酸、過酢酸、過安息香酸、トリフルオロ過酢酸等が例示できる。中でも、工業的に大量に製造され、安価に入手でき、安定度も高い点で過酢酸が好ましい。エポキシ化剤の使用量には厳密な制限がなく、使用する個々のエポキシ化剤、所望されるエポキシ化度、使用する個々のブロック共重合体の性状の違いによって変更することができる。 Among the epoxidizing agents, examples of hydroperoxides include hydrogen peroxide, tertiary butyl hydroperoxide, cumene hydroperoxide, and the like. Examples of “peracids” include performic acid, peracetic acid, perbenzoic acid, trifluoroperacetic acid and the like. Among them, peracetic acid is preferred because it is produced industrially in large quantities, can be obtained at low cost, and has high stability. The amount of the epoxidizing agent is not strictly limited, and can be changed depending on the individual epoxidizing agent used, the desired degree of epoxidation, and the difference in the properties of the individual block copolymers used.
エポキシ化の際には必要に応じて触媒を用いることができる。例えば過酸類の場合、炭酸ソーダ等のアルカリや硫酸等の酸を触媒として用いることができる。一方、ハイドロパーオキサイド類の場合、タングステン酸と苛性ソーダの混合物を過酸化水素と、あるいは有機酸を過酸化水素と、あるいはモリブデンヘキサカルボニルをターシャリブチルハイドロパーオキサイドとそれぞれ併用して触媒効果を得ることができる。 In the epoxidation, a catalyst can be used as necessary. For example, in the case of peracids, an alkali such as sodium carbonate or an acid such as sulfuric acid can be used as a catalyst. On the other hand, in the case of hydroperoxides, a catalytic effect is obtained by using a mixture of tungstic acid and caustic soda with hydrogen peroxide, organic acid with hydrogen peroxide, or molybdenum hexacarbonyl with tertiary butyl hydroperoxide. be able to.
エポキシ化反応の条件には厳密な制限はないが、例えば、過酢酸についていえば0〜70℃が好ましい。70℃を越えると過酢酸の分解が起こるからである。反応混合物の特別な操作は必要なく、例えば原料の混合物を2〜10時間攪拌すればよい。エポキシ化の反応温度は、常法に従い、用いるエポキシ化剤の反応性によって変更することができる。 The epoxidation reaction conditions are not strictly limited, but, for example, peracetic acid is preferably 0 to 70 ° C. This is because decomposition of peracetic acid occurs when the temperature exceeds 70 ° C. No special operation is required for the reaction mixture. For example, the mixture of raw materials may be stirred for 2 to 10 hours. The reaction temperature of epoxidation can be changed according to the reactivity of the epoxidizing agent used in accordance with a conventional method.
得られたエポキシ化スチレン系熱可塑性エラストマーの単離は、例えば貧溶媒で沈殿させる方法、エポキシ化スチレン系熱可塑性エラストマーを熱水中に攪拌の下で投入し溶媒を蒸留除去する方法、加熱及び/又は減圧操作によって溶媒を直接乾燥させる方法等で行うことができる。また、最終的に溶液形態で利用する場合には、単離せずに用いることもできる。 Isolation of the obtained epoxidized styrenic thermoplastic elastomer includes, for example, a method of precipitating with a poor solvent, a method of adding the epoxidized styrenic thermoplastic elastomer into hot water with stirring and distilling off the solvent, heating and The solvent can be directly dried by a decompression operation or the like. Moreover, when finally utilizing by a solution form, it can also be used without isolating.
エポキシ化スチレン系熱可塑性エラストマーのエポキシ化率は、10〜40%であること、特には15〜35%であることが好ましい。10%よりエポキシ基の量が少ないと本発明の効果が小さくなる傾向があり、その反面、40%を越えると、エポキシ基の反応活性が高くなりすぎてゲル化し易くなり、熱安定性が低下する傾向がある。また、特に熱安定性が要求される場合には、水素添加もエポキシ化もされずに不飽和のまま残存する共役ジエン化合物に由来する二重結合が全体の90%未満であることが好ましく、特には40%以下のものが好ましい。 The epoxidation rate of the epoxidized styrene thermoplastic elastomer is preferably 10 to 40%, particularly preferably 15 to 35%. If the amount of the epoxy group is less than 10%, the effect of the present invention tends to be reduced. On the other hand, if the amount exceeds 40%, the reaction activity of the epoxy group becomes too high and gelation tends to occur, and the thermal stability decreases. Tend to. In addition, particularly when thermal stability is required, it is preferable that the double bond derived from the conjugated diene compound remaining unsaturated without being hydrogenated or epoxidized is less than 90% of the total, Particularly preferred is 40% or less.
エポキシ化スチレン系熱可塑性エラストマーのエポキシ化率は、未水素添加・未エポキシ化の原料ブロック共重合体の共役ジエン化合物に由来する二重結合のうち、エポキシ化されたものの百分率であり、エポキシ当量(N)から、式:エポキシ化率={10000×D+2×H×(100−S)}/{(N−16)×(100−S)}で示すことができる(Dは共役ジエン化合物の分子量、Hは水添率(%)、Sはスチレン系化合物の含有量(重量%)を示す)。本発明において好ましく使用できるエポキシ化スチレン系熱可塑性エラストマーのエポキシ当量(N)は、0.1規定の臭化水素酸で滴定し、式:エポキシ当量(N)=10000×W/(f×V)(Wは、滴定に用いたエポキシ化スチレン系熱可塑性エラストマーの重量(g)、Vは、臭化水素酸の滴定量(ml)、fは、臭化水素酸のファクターを示す)で示すことができる。 The epoxidation rate of the epoxidized styrene thermoplastic elastomer is the percentage of epoxidized double bonds derived from the conjugated diene compound of the raw hydrogenated / non-epoxidized raw material block copolymer, and the epoxy equivalent (N) can be represented by the formula: epoxidation rate = {10000 × D + 2 × H × (100−S)} / {(N−16) × (100−S)} (D is a conjugated diene compound) The molecular weight, H is the hydrogenation rate (%), and S is the content (% by weight) of the styrene compound. The epoxy equivalent (N) of the epoxidized styrenic thermoplastic elastomer that can be preferably used in the present invention is titrated with 0.1 N hydrobromic acid, and the formula: epoxy equivalent (N) = 10000 × W / (f × V ) (W is the weight (g) of the epoxidized styrenic thermoplastic elastomer used for the titration, V is the titration amount of hydrobromic acid (ml), and f is the hydrobromic acid factor) be able to.
エポキシ化スチレン系熱可塑性エラストマーの配合量は、ポリアミド樹脂100質量部に対して3〜30質量部が好ましく、3〜28質量部がより好ましく、3〜25質量部が特に好ましい。3質量部以上の場合には金属基材と金属被覆材との間の接着性が特に良好であり、30質量部以下の場合、ポリアミド樹脂が有している機械的特性や表面特性が損なわれ難い。 3-30 mass parts is preferable with respect to 100 mass parts of polyamide resins, as for the compounding quantity of an epoxidized styrene-type thermoplastic elastomer, 3-28 mass parts is more preferable, and its 3-25 mass parts is especially preferable. In the case of 3 parts by mass or more, the adhesion between the metal base material and the metal coating material is particularly good, and in the case of 30 parts by mass or less, the mechanical characteristics and surface characteristics of the polyamide resin are impaired. hard.
一方、シランカップリング剤は、無機材料に対して親和性又は反応性を有する加水分解性のシリル基に、有機樹脂に対して親和性又は反応性を有する有機官能性基を化学的に結合させた構造を持つシラン化合物である。ケイ素に結合した加水分解性基としては、アルコキシ基、ハロゲン、アセトキシ基が挙げられるが、通常、アルコキシ基、特にメトキシ基、エトキシ基が好ましく用いられる。1個のケイ素原子につく加水分解性基の数は、1〜3個の間で選択される。有機官能性基としては、アミノ基、エポキシ基、ビニル基、カルボキシル基、メルカプト基、ハロゲン基、メタクリロキシ基、イソシアネート基等を挙げることができ、好ましくは、アミノ基又はエポキシ基である。 On the other hand, a silane coupling agent chemically bonds an organic functional group having affinity or reactivity to an organic resin to a hydrolyzable silyl group having affinity or reactivity to an inorganic material. It is a silane compound having a different structure. Examples of the hydrolyzable group bonded to silicon include an alkoxy group, a halogen, and an acetoxy group. Usually, an alkoxy group, particularly a methoxy group and an ethoxy group are preferably used. The number of hydrolyzable groups attached to one silicon atom is selected between 1 and 3. Examples of the organic functional group include an amino group, an epoxy group, a vinyl group, a carboxyl group, a mercapto group, a halogen group, a methacryloxy group, and an isocyanate group, and preferably an amino group or an epoxy group.
シランカップリング剤の具体例としては、α−アミノエチルトリエトキシラン、α−アミノプロピルトリエトキシシラン、α−アミノブチルトリエトキシシラン、γ−アミノプロピルトリメトキシシラン、γ−アミノプロピルトリエトキシシラン、γ−アミノプロピルメチルジメトキシシラン、γ−アミノプロピルメチルジエトキシシラン、N−β−(アミノエチル)−γ−アミノプロピルトリメトキシシラン、N−β−(アミノエチル)−γ−アミノプロピルメチルジメトキシシラン、N−β−(アミノエチル)−γ−アミノプロピルトリエトキシシラン、γ−ウレイドプロピルトリメトキシシラン、γ−ウレイドプロピルトリエトキシシラン、N−フェニル−γ−アミノプロピルトリメトキシシラン、N−ベンジル−γ−アミノプロピルトリメトキシシラン、N−ビニルベンジル−γ−アミノプロピルトリエトキシシラン等のアミノ基含有シラン類;γ−グリシドキシプロピルトリメトキシシラン、γ−グリシドキシプロピルメチルジメトキシシラン、γ−グリソドキシプロピルトリエトキシシラン、γ−グリシドキシプロピルメチルジエトキシシラン、β−(3,4エポキシシクロヘキシル)エチルトリメトキシシラン、β−(3,4−エポキシシクロヘキシル)エチルトリエトキシシラン等のエポキシ基含有シラン類;ビニルトリメトキシシラン、ビニルトリエトキシシラン、ビニルメチルジメトキシシラン等のビニル基含有シラン類;β−カルボキシエチルトリエトキシシラン、β−カルボキシエチルフェニルビス(2−メトキシエトキシ)シラン、N−β−(N−カルボキシルメチルアミノエチル)−γ−アミノプロピルトリメトキシシランなどのカルボキシル基含有シラン類;γ−メルカプトプロピルトリメトキシシラン、γ−メルカプトプロピルトリエトキシシラン、γ−メルカプトプロピルメチルジメトキシシラン、γ−メルカプトプロピルメチルジエトキシシラン等のメルカプト基含有シラン類;γ−クロロプロピルトリメトキシシラン等のハロゲン含有シラン類;γ−メタクリロキシプロピルトリメトキシシラン、γ−メタクリロキシプロピルトリエトキシシラン、γ−アクリロキシプロピルトリメトキシシラン、γ−メタクリロキシプロピルメチルジメトキシシラン、γ−メタクリロキシプロピルメチルジエトキシシラン等の(メタ)アクリル基含有シラン類;γ−イソシアネートプロピルトリメトキシシラン、γ−イソシアネートプロピルトリエトキシシラン、γ−イソシアネートプロピルメチルジエトキシシラン、γ−イソシアネートプロピルメチルジメトキシシラン等のイソシアネート基含有シラン類等が挙げられる。 Specific examples of the silane coupling agent include α-aminoethyltriethoxysilane, α-aminopropyltriethoxysilane, α-aminobutyltriethoxysilane, γ-aminopropyltrimethoxysilane, γ-aminopropyltriethoxysilane, γ-aminopropylmethyldimethoxysilane, γ-aminopropylmethyldiethoxysilane, N-β- (aminoethyl) -γ-aminopropyltrimethoxysilane, N-β- (aminoethyl) -γ-aminopropylmethyldimethoxysilane N-β- (aminoethyl) -γ-aminopropyltriethoxysilane, γ-ureidopropyltrimethoxysilane, γ-ureidopropyltriethoxysilane, N-phenyl-γ-aminopropyltrimethoxysilane, N-benzyl- γ-aminopropyltrimethoxy Amino group-containing silanes such as silane and N-vinylbenzyl-γ-aminopropyltriethoxysilane; γ-glycidoxypropyltrimethoxysilane, γ-glycidoxypropylmethyldimethoxysilane, γ-glycosoxypropyltriethoxy Epoxy group-containing silanes such as silane, γ-glycidoxypropylmethyldiethoxysilane, β- (3,4-epoxycyclohexyl) ethyltrimethoxysilane, β- (3,4-epoxycyclohexyl) ethyltriethoxysilane; vinyl Vinyl group-containing silanes such as trimethoxysilane, vinyltriethoxysilane and vinylmethyldimethoxysilane; β-carboxyethyltriethoxysilane, β-carboxyethylphenylbis (2-methoxyethoxy) silane, N-β- (N- Carboxymethyl amino Carboxyl group-containing silanes such as ethyl) -γ-aminopropyltrimethoxysilane; γ-mercaptopropyltrimethoxysilane, γ-mercaptopropyltriethoxysilane, γ-mercaptopropylmethyldimethoxysilane, γ-mercaptopropylmethyldiethoxysilane Mercapto group-containing silanes such as γ-halogen-containing silanes such as γ-chloropropyltrimethoxysilane; γ-methacryloxypropyltrimethoxysilane, γ-methacryloxypropyltriethoxysilane, γ-acryloxypropyltrimethoxysilane, γ -(Meth) acrylic group-containing silanes such as methacryloxypropylmethyldimethoxysilane and γ-methacryloxypropylmethyldiethoxysilane; γ-isocyanatopropyltrimethoxysilane, γ-isocyanate Over preparative triethoxysilane, .gamma. isocyanate propyl methyl diethoxy silane, isocyanate group-containing silanes such as .gamma. isocyanate propyl methyl dimethoxysilane and the like.
シランカップリング剤の量は、上述のポリアミド樹脂100質量部に対し、0.01〜0.5質量部が好ましく、0.01〜0.3質量部がより好ましい。0.01質量部以上の場合には金属被覆材の金属基材に対する接着性が特に良好であり、0.5質量部以下の場合にはポリアミド樹脂が有している流動性や表面特性が損なわれ難い。 The amount of the silane coupling agent is preferably 0.01 to 0.5 parts by mass and more preferably 0.01 to 0.3 parts by mass with respect to 100 parts by mass of the polyamide resin described above. When the amount is 0.01 parts by mass or more, the adhesion of the metal coating material to the metal substrate is particularly good, and when it is 0.5 parts by mass or less, the fluidity and surface characteristics of the polyamide resin are impaired. It's difficult.
上述のエポキシ化スチレン系熱可塑性エラストマーと上述のシランカップリング剤とを併用する場合、金属基材と本発明の金属被覆材との接着性が特に良好であり好ましい。 When the above-mentioned epoxidized styrenic thermoplastic elastomer and the above-mentioned silane coupling agent are used in combination, the adhesion between the metal substrate and the metal coating material of the present invention is particularly good and preferable.
(III)金属被覆材の形成
本発明の金属被覆材は、アルミニウムなどの非鉄金属及び鉄といった幅広い金属の基材に対して適用できる。金属被覆材の用途としては、一般工業用の流体金属配管の防錆コーティング、自動車用の燃料・オイル・ブレーキ液などの鋼管・アルミ配管といった金属管に対する防錆用コーティング、金属ワイヤーのコーティング、水槽タンクなど水回りプレートのコーティングなどが挙げられ、特に自動車用金属管に対して好ましく適用できる。
(III) Formation of metal coating material The metal coating material of the present invention can be applied to a wide range of metal substrates such as non-ferrous metals such as aluminum and iron. Applications of metal coating materials include rust-proof coating for fluid metal pipes for general industrial use, rust-proof coating for metal pipes such as steel pipes and aluminum pipes for automobile fuel, oil, brake fluid, etc., metal wire coating, water tank Examples thereof include coating of a water plate such as a tank, and can be preferably applied particularly to a metal pipe for automobiles.
本発明の金属被覆材で金属基材を被覆する方法としては、これらに限定するものではないが、例えば押出しによる鋼管被覆などのように、既に溶融状態にあるポリアミド樹脂組成物で被着物である金属基材を被覆する方法、粉体塗装のように、被着物である金属基材を加熱しておき、その熱により固体状のポリアミド樹脂組成物を溶融させて金属基材を被覆する方法、及び、金属基材と固体状態のポリアミド樹脂組成物を接触させたものを共に加熱して被覆する方法などが挙げられる。金属基材には本発明の金属被覆材による被覆に先立って金属用の従来公知のプライマーを用いたプライマー処理を施してもよい。 The method of coating the metal substrate with the metal coating material of the present invention is not limited to these methods. For example, it is an adherend with a polyamide resin composition that is already in a molten state, such as a steel pipe coating by extrusion. A method of coating a metal substrate, a method of coating a metal substrate by heating a metal substrate that is an adherend, such as powder coating, and melting a solid polyamide resin composition by the heat, And the method etc. which heat and coat | cover the thing which made the metal base material and the polyamide resin composition of a solid state contact are mentioned. Prior to coating with the metal coating material of the present invention, the metal substrate may be subjected to primer treatment using a conventionally known primer for metal.
なお、金属被覆材の形成時において、ポリアミド樹脂組成物の温度は、該ポリアミド樹脂組成物を変質させない温度に維持することが好ましい。 In forming the metal coating material, the temperature of the polyamide resin composition is preferably maintained at a temperature that does not alter the polyamide resin composition.
以下、実施例及び比較例を挙げて本発明を説明するが、本発明はこれらの実施例に限定されるものではない。 EXAMPLES Hereinafter, although an Example and a comparative example are given and this invention is demonstrated, this invention is not limited to these Examples.
[物性測定、成形、評価方法]
特性値を、以下の方法により測定した。
[Physical property measurement, molding, evaluation method]
The characteristic value was measured by the following method.
(1)相対粘度(ηr)
ηrは、ポリアミドの96%硫酸溶液(濃度:1.0g/dl)を用いて、オストワルド型粘度計により25℃で測定した。
(1) Relative viscosity (ηr)
ηr was measured at 25 ° C. with an Ostwald viscometer using a 96% sulfuric acid solution of polyamide (concentration: 1.0 g / dl).
(2)融点(Tm)及び結晶化温度(Tc)
Tm及びTcは、PerkinELmer社製PYRIS Diamond DSC用いて窒素雰囲気下で測定した。30℃から300℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、300℃で3分保持したのち、−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、次に300℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。得られたDSCチャートから降温ファーストランの発熱ピーク温度をTc、昇温セカンドランの吸熱ピーク温度をTmとした。
(2) Melting point (Tm) and crystallization temperature (Tc)
Tm and Tc were measured under a nitrogen atmosphere using a PYRIS Diamond DSC manufactured by PerkinELmer. The temperature was raised from 30 ° C. to 300 ° C. at a rate of 10 ° C./min (referred to as a temperature rise first run), held at 300 ° C. for 3 minutes, and then lowered to −100 ° C. at a rate of 10 ° C./min (temperature fall first). Then, the temperature was raised to 300 ° C. at a rate of 10 ° C./min (called a temperature rising second run). From the obtained DSC chart, the exothermic peak temperature of the temperature decrease first run was Tc, and the endothermic peak temperature of the temperature increase second run was Tm.
(3)1%重量減少温度(Td)
Tdは島津製作所社製THERMOGRAVIMETRIC ANALYZER TGA−50を用い、熱重量分析(TGA)により測定した。20ml/分の窒素気流下室温から500℃まで10℃/分の昇温速度で昇温し、Tdを測定した。
(3) 1% weight loss temperature (Td)
Td was measured by thermogravimetric analysis (TGA) using THERMOGRAVIMETRIC ANALYZER TGA-50 manufactured by Shimadzu Corporation. The temperature was raised from room temperature to 500 ° C. at a rate of 10 ° C./min under a nitrogen stream of 20 ml / min, and Td was measured.
(4)溶融粘度
溶融粘度はティー・エイ・インスツルメント・ジャパン社製溶融粘弾性測定装置ARESに25mmのコーン・プレートを装着して、窒素中、290℃、せん断速度0.1s-1の条件で測定した。
(4) Melt viscosity Melt viscosity was measured by attaching a 25mm cone plate to a melt viscoelasticity measuring device ARES manufactured by TA Instruments Japan Co., Ltd. Measured under conditions.
(5)フィルム成形
東邦マシナリー社製真空プレス機TMB−10を用いて、ペレットからフィルムを成形した。500〜700Paの減圧雰囲気下において290℃(PA6を用いた場合は260℃、PA66を用いた場合は290℃、PA12を用いた場合は230℃)で5分間加熱溶融させた後、5MPaで1分間プレスを行いフィルム成形した。次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却結晶化させてフィルムを得た。
(5) Film formation A film was formed from the pellets using a vacuum press TMB-10 manufactured by Toho Machinery Co., Ltd. After heating and melting at 290 ° C. in a reduced pressure atmosphere of 500 to 700 Pa (260 ° C. when using PA6, 290 ° C. when using PA66, 230 ° C. when using PA12) for 5 minutes, 1 at 5 MPa The film was formed by pressing for a minute. Next, the reduced-pressure atmosphere was returned to normal pressure, and then cooled and crystallized at room temperature of 5 MPa for 1 minute to obtain a film.
(6)飽和吸水率
上記(5)の条件で成形したフィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を23℃のイオン交換水に浸漬し、所定時間ごとにフィルムを取り出し、フィルムの質量を測定した。フィルム質量の増加率が0.2%の範囲内で3回続いた場合にポリアミド樹脂フィルムへの水分の吸収が飽和に達したと判断して、水に浸漬する前のフィルムの質量(Xg)と飽和に達した時のフィルムの質量(Yg)から次の式(1)により飽和吸水率(%)を算出した。
(6) Saturated water absorption rate A film (dimensions: 20 mm × 10 mm, thickness 0.25 mm; mass of about 0.05 g) formed under the conditions of (5) above is immersed in ion-exchanged water at 23 ° C., and every predetermined time. The film was taken out and the mass of the film was measured. When the rate of increase in the film mass lasts 3 times within the range of 0.2%, it is judged that the absorption of moisture into the polyamide resin film has reached saturation, and the mass (Xg) of the film before being immersed in water The saturated water absorption (%) was calculated from the mass (Yg) of the film when reaching saturation with the following equation (1).
飽和吸水率(%)=(Y−X)/X×100 (1) Saturated water absorption (%) = (Y−X) / X × 100 (1)
(7)耐薬品性
本発明によって得られるポリアミドの熱プレスフィルムを以下の薬品中に7日間浸漬した後に、フィルムの質量残存率(%)及び外観の変化を観測した。濃塩酸、64%硫酸、氷酢酸のそれぞれの溶液において23℃下で浸漬した試料について試験を行った。
(7) Chemical Resistance After the polyamide hot press film obtained according to the present invention was immersed in the following chemicals for 7 days, changes in mass residual rate (%) and appearance of the film were observed. Tests were conducted on samples immersed in concentrated hydrochloric acid, 64% sulfuric acid, and glacial acetic acid at 23 ° C.
(8)耐加水分解性
上記(5)の条件で成形したフィルムをオートクレーブに入れ、水、0.5mol/l硫酸、1mol/l水酸化ナトリウム水溶液中(すなわち、順に、pH=7、pH=1、pH=14)でそれぞれ121℃、60分間処理した後の質量残存率(%)を調べた。
(8) Hydrolysis resistance The film molded under the condition (5) above is put in an autoclave, and in water, 0.5 mol / l sulfuric acid, 1 mol / l sodium hydroxide aqueous solution (that is, pH = 7, pH = 1, pH = 14), and the mass residual ratio (%) after treatment at 121 ° C. for 60 minutes was examined.
(9)機械的物性
以下に示す測定は、下記の試験片を樹脂温度290℃(PA6を用いた場合は260℃、PA66を用いた場合は290℃、PA12を用いた場合は230℃)、金型温度80℃の射出成形により成形し、これを用いて行った。
(9) Mechanical properties The following test specimens were measured at a resin temperature of 290 ° C (260 ° C when using PA6, 290 ° C when using PA66, 230 ° C when using PA12), Molding was carried out by injection molding at a mold temperature of 80 ° C.
〔1〕引張降伏点強度:ASTM D638に記載のTypeIの試験片を用いてASTM D638に準拠して測定した。 [1] Tensile yield strength: Measured according to ASTM D638 using a Type I test piece described in ASTM D638.
〔2〕曲げ弾性率:試験片寸法3.2mm×12.7mm×127mmの試験片を用いてASTM D790に準拠し、23℃で測定した。成形後に調湿せずに評価したものをdry、成形後に23℃湿度65%で調湿した後に評価したものをwetとして表中に記載した。 [2] Flexural modulus: Measured at 23 ° C. in accordance with ASTM D790 using a test piece with a test piece size of 3.2 mm × 12.7 mm × 127 mm. What was evaluated without humidity adjustment after molding was described in the table as dry, and what was evaluated after conditioning at 23 ° C. and 65% humidity after molding was shown in the table.
〔3〕アイゾット衝撃強度:試験片寸法3.2mm×12.7mm×127mmの試験片を用いてASTM D256に準拠し、23℃で測定した。 [3] Izod impact strength: Measured at 23 ° C. in accordance with ASTM D256 using a test piece with a test piece size of 3.2 mm × 12.7 mm × 127 mm.
〔4〕荷重たわみ温度(熱変形温度):試験片寸法3.2mm×12.7mm×127mmの試験片を用いてASTM D648に準拠し、荷重1.82MPaで測定した。 [4] Deflection temperature under load (thermal deformation temperature): Measured at a load of 1.82 MPa in accordance with ASTM D648 using a test piece having a test piece size of 3.2 mm × 12.7 mm × 127 mm.
(10)吸水率
上記(5)の条件で成形したフィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を23℃65%RH条件下におき、所定時間ごとにフィルムを取り出し、フィルムの質量を測定した。フィルム質量の増加率が0.2%の範囲内で3回続いた場合にポリアミド樹脂フィルムへの水分の吸収が飽和に達したと判断して、上記23℃65%RH条件下におく前のフィルムの質量(Xg)と飽和に達した時のフィルムの質量(Yg)から次の式(2)により吸水率(%)を算出した。
(10) Water absorption rate A film (dimensions: 20 mm × 10 mm, thickness 0.25 mm; mass of about 0.05 g) formed under the condition (5) above is placed under conditions of 23 ° C. and 65% RH, and the film is taken every predetermined time Was taken out and the mass of the film was measured. When the rate of increase in the film mass continues three times within the range of 0.2%, it is determined that the absorption of moisture into the polyamide resin film has reached saturation, and before the 23 ° C. and 65% RH conditions are satisfied. The water absorption rate (%) was calculated from the mass (Xg) of the film and the mass (Yg) of the film when saturation was reached by the following formula (2).
吸水率(%)=(Y−X)/X×100 (2) Water absorption (%) = (Y−X) / X × 100 (2)
(11)接着力
上記(5)の条件で調製した150mm×100mm、厚み0.1mmのフィルムを、1辺150mm、厚み0.5mmの金属板(亜鉛メッキ鋼板:JIS G3302 SPGC Z22、アルミプレート:JIS1100番)2枚ではさみ、東邦マシナリー社製真空プレス機TMB−10を用いて、500〜700Paの減圧雰囲気下において260℃で5分間加熱溶融させた後、10MPaで1分間プレスを行いフィルム成形した。次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却させて試料を得た。その試料を25mm幅で切り、JIS K−6853に準じてT型剥離試験を行った。接着力はピーク強度と幅25mm×100mmを完全に剥離するまでに要したエネルギーで評価した。
(11) Adhesive force A 150 mm × 100 mm film with a thickness of 0.1 mm prepared under the conditions in (5) above is a metal plate with a side of 150 mm and a thickness of 0.5 mm (galvanized steel sheet: JIS G3302 SPGC Z22, aluminum plate: JIS No. 1100) Scissors with two sheets, heat-melted at 260 ° C. for 5 minutes in a reduced pressure atmosphere of 500 to 700 Pa using a vacuum press machine TMB-10 manufactured by Toho Machinery Co., Ltd., then pressed at 10 MPa for 1 minute to form a film did. Next, after returning the reduced pressure atmosphere to normal pressure, the sample was cooled at room temperature of 5 MPa for 1 minute to obtain a sample. The sample was cut at a width of 25 mm, and a T-type peel test was performed according to JIS K-6683. The adhesive strength was evaluated by the energy required to completely peel the peak strength and the width of 25 mm × 100 mm.
[製造例1:PA92/62T−1]
攪拌機、温度計、トルクメーター、圧力計、ダイアフラムポンプを直結した原料投入口、窒素ガス導入口、放圧口、圧力調節装置及びポリマー抜出し口を備えた内容積が約150リットルの圧力容器に蓚酸ジブチル28.230kg(139.56モル)を仕込み、圧力容器の内部を純度が99.9999%の窒素ガスで0.5MPaに加圧した後、次に常圧まで窒素ガスを放出する操作を5回繰り返し、窒素置換を行った後、封圧下、攪拌しながら系内を昇温した。約30分間かけて蓚酸ジブチルの温度を100℃にした後、1,9−ノナンジアミン1.241kg(7.84モル)と2−メチル−1,8−オクタンジアミン19.639kg(124.04モル)と1,6−ヘキサンジアミン0.893kg(7.68モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が5.62:88.88:5.50)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.13であった。
[Production Example 1: PA92 / 62T-1]
Oxalic acid in a pressure vessel with an internal volume of about 150 liters equipped with a stirrer, thermometer, torque meter, pressure gauge, raw material inlet directly connected to the diaphragm pump, nitrogen gas inlet, pressure outlet, pressure regulator and polymer outlet The operation of charging 28.230 kg (139.56 mol) of dibutyl, pressurizing the inside of the pressure vessel to 0.5 MPa with nitrogen gas having a purity of 99.9999%, and then releasing nitrogen gas to normal pressure is performed 5 times. After repeated nitrogen substitution, the system was heated while stirring under a sealing pressure. After the temperature of dibutyl oxalate was raised to 100 ° C. over about 30 minutes, 1.241 kg (7.84 mol) of 1,9-nonanediamine and 19.639 kg (124.04 mol) of 2-methyl-1,8-octanediamine were obtained. And a mixture of 0.893 kg (7.68 mol) of 1,6-hexanediamine (molar ratio of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine is 5.62: 88.88: 5.50) was supplied into the reaction vessel by a diaphragm pump at a flow rate of 1.49 liters / minute for about 17 minutes, and at the same time the temperature was raised. The internal pressure in the pressure vessel immediately after the supply increased to 0.35 MPa by butanol generated by the polycondensation reaction, and the temperature of the polycondensate increased to about 170 ° C. Thereafter, the temperature was raised to 235 ° C. over 1 hour. Meanwhile, the internal pressure was adjusted to 0.75 MPa while extracting the generated butanol from the pressure relief port. Immediately after the temperature of the polycondensate reached 235 ° C., butanol was extracted from the pressure release port over about 20 minutes, and the internal pressure was brought to normal pressure. From the normal pressure, the temperature was raised while flowing nitrogen gas at 1.5 liters / minute, the temperature of the polycondensate was brought to 260 ° C. over about 1 hour, and the reaction was carried out at 260 ° C. for 4.5 hours. . Thereafter, stirring was stopped, the inside of the system was pressurized to 1 MPa with nitrogen and allowed to stand for about 10 minutes, then released to an internal pressure of 0.5 MPa, and the polycondensate was extracted in a string form from the lower outlet of the pressure vessel. The string-like polymer was immediately cooled with water, and the water-cooled string-like resin was pelletized with a pelletizer. The obtained polyamide was a white tough polymer, and ηr = 3.13.
[製造例2:PA92/62T−2]
蓚酸ジブチル28.462kg(140.71モル)を仕込み、1,9−ノナンジアミン16.448kg(103.88モル)と2−メチル−1,8−オクタンジアミン2.903kg(18.34モル)と1,6−ヘキサンジアミン2.150kg(18.50モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が73.83:13.03:13.14)を仕込んだほかは、製造例1と同様に反応を行ってポリアミドを得た。得られたポリアミドは白色の強靭なポリマーで、ηr=2.97であった。
[Production Example 2: PA92 / 62T-2]
28.462 kg (140.71 mol) of dibutyl oxalate was charged, 16.448 kg (103.88 mol) of 1,9-nonanediamine, 2.903 kg (18.34 mol) of 2-methyl-1,8-octanediamine and 1 , 6-hexanediamine 2.150 kg (18.50 mol) (mixture ratio of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine is 73.83: 13.13. 03: 13.14) was reacted in the same manner as in Production Example 1 to obtain polyamide. The obtained polyamide was a white tough polymer with ηr = 2.97.
[製造例3:PA92/62T−3]
蓚酸ジブチル30.238kg(149.49モル)を仕込み、1,9−ノナンジアミン4.486kg(28.33モル)と2−メチル−1,8−オクタンジアミン4.486kg(28.33モル)と1,6−ヘキサンジアミン10.79kg(92.85モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が18.95:18.95:62.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1.5時間かけて温度を270℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が270℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を285℃にし、285℃において1.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.88であった。
[Production Example 3: PA92 / 62T-3]
30.238 kg (149.49 mol) of dibutyl oxalate was charged, 4.486 kg (28.33 mol) of 1,9-nonanediamine, 4.486 kg (28.33 mol) of 2-methyl-1,8-octanediamine and 1 , 6-hexanediamine 10.79 kg (92.85 mol) (1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine molar ratio of 18.95: 18. 95: 62.10) was supplied into the reaction vessel with a diaphragm pump at a flow rate of 1.49 liters / minute over about 17 minutes, and at the same time the temperature was raised. The internal pressure in the pressure vessel immediately after the supply increased to 0.35 MPa by butanol generated by the polycondensation reaction, and the temperature of the polycondensate increased to about 170 ° C. Thereafter, the temperature was raised to 270 ° C. over 1.5 hours. Meanwhile, the internal pressure was adjusted to 1.00 MPa while extracting the generated butanol from the pressure relief port. Immediately after the temperature of the polycondensate reached 270 ° C., butanol was extracted from the pressure release port over about 20 minutes, and the internal pressure was brought to normal pressure. From the normal pressure, the temperature was raised while flowing nitrogen gas at 1.5 liters / minute, the temperature of the polycondensate was changed to 285 ° C. over about 1 hour, and the reaction was carried out at 285 ° C. for 1.5 hours. . Thereafter, stirring was stopped, the inside of the system was pressurized to 1 MPa with nitrogen and allowed to stand for about 10 minutes, then released to an internal pressure of 0.5 MPa, and the polycondensate was extracted in a string form from the lower outlet of the pressure vessel. The string-like polymer was immediately cooled with water, and the water-cooled string-like resin was pelletized with a pelletizer. The obtained polyamide was a white tough polymer, and ηr = 2.88.
[製造例4:PA92/62T−4]
蓚酸ジブチル29.864kg(147.64モル)を仕込み、1,9−ノナンジアミン5.598kg(35.36モル)と2−メチル−1,8−オクタンジアミン5.598kg(35.36モル)と1,6−ヘキサンジアミン8.941kg(76.92モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が23.95:23.95:52.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が250℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を270℃にし、270℃において2時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.83であった。
[Production Example 4: PA92 / 62T-4]
29.864 kg (147.64 mol) of dibutyl oxalate was charged, and 5.598 kg (35.36 mol) of 1,9-nonanediamine, 5.598 kg (35.36 mol) of 2-methyl-1,8-octanediamine and 1 , 6-hexanediamine 8.941 kg (76.92 mol) of a mixture (the molar ratio of 1,9-nonanediamine, 2-methyl-1,8-octanediamine and 1,6-hexanediamine was 23.95: 23. 95: 52.10) was supplied to the reaction vessel by a diaphragm pump at a flow rate of 1.49 liters / minute over about 17 minutes, and at the same time the temperature was raised. The internal pressure in the pressure vessel immediately after the supply increased to 0.35 MPa by butanol generated by the polycondensation reaction, and the temperature of the polycondensate increased to about 170 ° C. Thereafter, the temperature was raised to 250 ° C. over 1 hour. Meanwhile, the internal pressure was adjusted to 1.00 MPa while extracting the generated butanol from the pressure relief port. Immediately after the temperature of the polycondensate reached 250 ° C., butanol was extracted from the pressure release port over about 20 minutes, and the internal pressure was brought to normal pressure. The temperature was raised from normal pressure while flowing nitrogen gas at 1.5 liters / minute, and the temperature of the polycondensate was increased to 270 ° C. over about 1 hour and reacted at 270 ° C. for 2 hours. Thereafter, stirring was stopped, the inside of the system was pressurized to 1 MPa with nitrogen and allowed to stand for about 10 minutes, then released to an internal pressure of 0.5 MPa, and the polycondensate was extracted in a string form from the lower outlet of the pressure vessel. The string-like polymer was immediately cooled with water, and the water-cooled string-like resin was pelletized with a pelletizer. The obtained polyamide was a white tough polymer with ηr = 2.83.
[製造例5:PA92/62T−5]
蓚酸ジブチル29.107kg(143.89モル)を仕込み、1,9−ノナンジアミン5.641kg(35.63モル)と2−メチル−1,8−オクタンジアミン10.028kg(63.34モル)と1,6−ヘキサンジアミン5.223kg(44.93モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が24.76:44.02:31.22)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が240℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を265℃にし、265℃において3時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.11であった。
[Production Example 5: PA92 / 62T-5]
29.107 kg (143.89 mol) of dibutyl oxalate was charged, 5.641 kg (35.63 mol) of 1,9-nonanediamine, 10.028 kg (63.34 mol) of 2-methyl-1,8-octanediamine and 1 , 6-hexanediamine 5.223 kg (44.93 mol) (1,9-nonanediamine, 2-methyl-1,8-octanediamine, 1,6-hexanediamine molar ratio 24.76: 44. 02: 31.22) was supplied into the reaction vessel over about 17 minutes at a flow rate of 1.49 liters / minute by means of a diaphragm pump, and the temperature was raised. The internal pressure in the pressure vessel immediately after the supply increased to 0.35 MPa by butanol generated by the polycondensation reaction, and the temperature of the polycondensate increased to about 170 ° C. Thereafter, the temperature was raised to 250 ° C. over 1 hour. Meanwhile, the internal pressure was adjusted to 0.75 MPa while extracting the generated butanol from the pressure relief port. Immediately after the temperature of the polycondensate reached 240 ° C., butanol was extracted from the pressure release port over about 20 minutes, and the internal pressure was brought to normal pressure. The temperature was raised from normal pressure while flowing nitrogen gas at 1.5 liters / minute, and the temperature of the polycondensate was adjusted to 265 ° C. over about 1 hour, and the reaction was carried out at 265 ° C. for 3 hours. Thereafter, stirring was stopped, the inside of the system was pressurized to 1 MPa with nitrogen and allowed to stand for about 10 minutes, then released to an internal pressure of 0.5 MPa, and the polycondensate was extracted in a string form from the lower outlet of the pressure vessel. The string-like polymer was immediately cooled with water, and the water-cooled string-like resin was pelletized with a pelletizer. The obtained polyamide was a white tough polymer with ηr = 3.11.
[参考製造例1:PA92C]
蓚酸ジブチル28.40kg(140.4モル)を仕込み、1,9−ノナンジアミン11.11kg(70.2モル)と2−メチル−1,8−オクタンジアミン11.11kg(70.2モル)の混合物をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.35であった。
[Reference Production Example 1: PA92C]
A mixture of 11.11 kg (70.2 mol) of 1,9-nonanediamine and 11.11 kg (70.2 mol) of 2-methyl-1,8-octanediamine was charged with 28.40 kg (140.4 mol) of dibutyl oxalate. Was supplied into the reaction vessel over about 17 minutes at a flow rate of 1.49 liters / minute by means of a diaphragm pump, and at the same time the temperature was raised. The internal pressure in the pressure vessel immediately after the supply increased to 0.35 MPa by butanol generated by the polycondensation reaction, and the temperature of the polycondensate increased to about 170 ° C. Thereafter, the temperature was raised to 235 ° C. over 1 hour. Meanwhile, the internal pressure was adjusted to 0.5 MPa while extracting the generated butanol from the pressure relief port. Immediately after the temperature of the polycondensate reached 235 ° C., butanol was extracted from the pressure release port over about 20 minutes, and the internal pressure was brought to normal pressure. From the normal pressure, the temperature was raised while flowing nitrogen gas at 1.5 liters / minute, the temperature of the polycondensate was brought to 260 ° C. over about 1 hour, and the reaction was carried out at 260 ° C. for 4.5 hours. . Thereafter, stirring was stopped, the inside of the system was pressurized to 1 MPa with nitrogen and allowed to stand for about 10 minutes, then released to an internal pressure of 0.5 MPa, and the polycondensate was extracted in a string form from the lower outlet of the pressure vessel. The string-like polymer was immediately cooled with water, and the water-cooled string-like resin was pelletized with a pelletizer. The obtained polyamide was a white tough polymer, and ηr = 3.35.
[比較製造例1:PA92の製造]
ジアミン原料として1,9−ノナンジアミン22.25kg(140.4モル)だけを用いて、製造例1と同様に反応を行ってポリアミドを得た。得られた重合物は黄白色のポリマーであり、ηr=2.78であった。
[Comparative Production Example 1: Production of PA92]
Using only 22.25 kg (140.4 mol) of 1,9-nonanediamine as a diamine raw material, a reaction was carried out in the same manner as in Production Example 1 to obtain a polyamide. The obtained polymer was a yellowish white polymer, and ηr = 2.78.
製造例1〜5、参考製造例1及び比較製造例1で製造したポリアミドPA92/62T−1〜PA92/62T−5、PA92C、PA92、並びにナイロン6(宇部興産製、UBEナイロン1015B:PA6)、ナイロン66(宇部興産製、UBEナイロン2020B:PA66)及びナイロン12(宇部興産製、UBESTA3020U:PA12)について、相対粘度、融点、結晶化温度、1%重量減少温度、溶融粘度、飽和吸水率、耐薬品性、耐加水分解性、ドライ及びウェットにおける機械的特性を測定した。結果を表1に示す。 Polyamide PA92 / 62T-1 to PA92 / 62T-5, PA92C, PA92, and nylon 6 (manufactured by Ube Industries, UBE nylon 1015B: PA6) produced in Production Examples 1-5, Reference Production Example 1 and Comparative Production Example 1. For nylon 66 (Ube Industries, UBE nylon 2020B: PA66) and nylon 12 (Ube Industries, UBESTA3020U: PA12), relative viscosity, melting point, crystallization temperature, 1% weight loss temperature, melt viscosity, saturated water absorption, resistance to water Chemical properties, hydrolysis resistance, mechanical properties in dry and wet conditions were measured. The results are shown in Table 1.
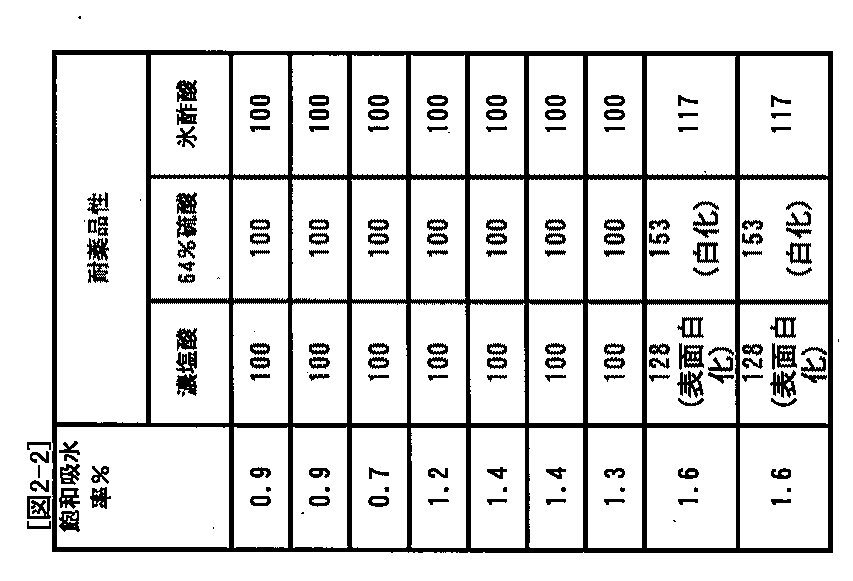
[実施例1〜7、比較例1,2]
エポキシ化スチレン系熱可塑性エラストマーとして、ダイセル化学製エポフレンドA1010を用い、表2に示す組成で、池貝鉄工(株)製2軸混練機PCM−45にて、シリンダー設定温度290℃(PA12を用いた場合は230℃)、回転速度150rpmで溶融混練して金属被覆材を調製した。上記溶融混練した試料を金属基材(亜鉛メッキ鋼板及びアルミプレート)間に挟んだ複合体を作製し、接着力の評価を行った。また、上記溶融混練した試料から上記(5)の条件で成形したフィルムを用いて、それらの飽和吸水率および耐薬品性を、前述した方法により評価した。
[Examples 1 to 7, Comparative Examples 1 and 2]
As the epoxidized styrene-based thermoplastic elastomer, Daicel Chemical's Epofriend A1010 was used, and the composition shown in Table 2 was used. In this case, the metal coating material was prepared by melt-kneading at a rotation speed of 150 rpm. A composite was prepared by sandwiching the melt-kneaded sample between metal substrates (galvanized steel plate and aluminum plate), and the adhesive strength was evaluated. Moreover, the saturated water absorption rate and chemical resistance were evaluated by the above-described methods using a film molded from the melt-kneaded sample under the condition (5).
本発明の金属被覆材は、低吸水性でありながら、溶融重合による高分子量化が可能で、融点と熱分解温度の差から見積もられる成形可能温度幅が広く溶融成形性に優れ、耐薬品性、耐加水分解性に優れるため、例えば一般工業用の流体金属配管の防錆コーティング、自動車用の燃料・オイル・ブレーキ液などの鋼管・アルミ配管といった金属管の防錆用コーティング、金属ワイヤーのコーティング、水槽タンクなど水回りプレートのコーティングなどの金属被覆材として好適に使用できる。 The metal coating material of the present invention is capable of increasing the molecular weight by melt polymerization while having low water absorption, has a wide moldable temperature range estimated from the difference between the melting point and the thermal decomposition temperature, and has excellent melt moldability and chemical resistance. Because of its excellent hydrolysis resistance, for example, anti-corrosion coating for fluid metal pipes for general industrial use, anti-corrosion coating for metal pipes such as steel pipes and aluminum pipes for fuel, oil and brake fluids for automobiles, and coating of metal wires It can be suitably used as a metal coating material such as a coating of a water plate such as a water tank.
Claims (4)
ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、
前記ジアミン成分の、1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンとのモル比が5:95〜95:5であり、
C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂を含み、
エポキシ化スチレン系熱可塑性エラストマーを更に含む、金属被覆材。 The dicarboxylic acid component consists of oxalic acid,
The diamine component is from a mixture of 1,9-nonanediamine and 2-methyl-1,8-octanediamine (hereinafter referred to as “C9 diamine mixture”) and 1,6-hexanediamine (hereinafter referred to as “C6 diamine”). Become
The diamine component has a molar ratio of 1,9-nonanediamine to 2-methyl-1,8-octanediamine of 5:95 to 95: 5,
Including a polyamide resin in which the molar ratio of C9 diamine mixture to C6 diamine is from 1:99 to 99: 1;
A metal coating material further comprising an epoxidized styrenic thermoplastic elastomer .
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008244724A JP5572935B2 (en) | 2008-09-24 | 2008-09-24 | Metal cladding |
| PCT/JP2009/060979 WO2009151145A1 (en) | 2008-06-10 | 2009-06-10 | Novel polyamide resin composition and polyamide resin-containing product |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008244724A JP5572935B2 (en) | 2008-09-24 | 2008-09-24 | Metal cladding |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010077212A JP2010077212A (en) | 2010-04-08 |
| JP5572935B2 true JP5572935B2 (en) | 2014-08-20 |
Family
ID=42208029
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008244724A Expired - Fee Related JP5572935B2 (en) | 2008-06-10 | 2008-09-24 | Metal cladding |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5572935B2 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5379743B2 (en) * | 2010-05-19 | 2013-12-25 | 大成プラス株式会社 | Laminate and manufacturing method thereof |
| US20160046836A1 (en) | 2013-04-09 | 2016-02-18 | Mitsubishi Gas Chemical Company, Inc. | Metal-coating material |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5093466A (en) * | 1990-02-20 | 1992-03-03 | Exxon Chemical Patents Inc. | Polyoxamide oxygen barrier |
| JP3242781B2 (en) * | 1994-02-16 | 2001-12-25 | 株式会社クラレ | Polyamide resin |
| JP2000191771A (en) * | 1998-12-25 | 2000-07-11 | Kuraray Co Ltd | Polyamide and its composition |
| JP2002036382A (en) * | 2000-07-21 | 2002-02-05 | Tokai Rubber Ind Ltd | Method for manufacturing metal composite hose and metal composite hose |
| JP2002286175A (en) * | 2001-03-27 | 2002-10-03 | Tokai Rubber Ind Ltd | Metallic composite hose, insert and hose fastening method |
| JP4891497B2 (en) * | 2001-08-22 | 2012-03-07 | 日鉄防蝕株式会社 | Method of coating polyamide resin-coated steel material and steel material coated by this method |
| JP2004083817A (en) * | 2002-08-29 | 2004-03-18 | Kuraray Co Ltd | Polyamide |
| JP2005273776A (en) * | 2004-03-24 | 2005-10-06 | Tokai Rubber Ind Ltd | Hose for liquid transportation |
| JP4487687B2 (en) * | 2004-08-23 | 2010-06-23 | 宇部興産株式会社 | Low water absorption member |
-
2008
- 2008-09-24 JP JP2008244724A patent/JP5572935B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2010077212A (en) | 2010-04-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2746315B1 (en) | Method for manufacturing crystalline polyamide resin | |
| JP5120518B2 (en) | Polyamide resin | |
| KR101677884B1 (en) | Polyamide resin composition, manufacturing method, and molded article | |
| JP6492199B2 (en) | Semi-aromatic copolyamide resin and polyamide molding composition produced therewith | |
| JP5376112B2 (en) | Multilayer structure | |
| WO2000078838A1 (en) | Epoxy resin composition and process for producing silane-modified epoxy resin | |
| WO2001005862A1 (en) | Glycidyl ether group-containing partial alkoxysilane condensate, silane-modified resin, compositions of these, and processes for producing these | |
| JP2011225830A (en) | Process for producing polyamide resin | |
| WO2002034800A1 (en) | Hydrogenated polymer | |
| JP5217660B2 (en) | Metal cladding | |
| WO2020238440A1 (en) | Semi-aromatic polyamide, method for synthesis thereof, and polyamide molding composition consisting thereof | |
| JP5572935B2 (en) | Metal cladding | |
| WO2018049808A1 (en) | Semi-aromatic copolyamide resin and polyamide molding composition consisting of semi-aromatic copolyamide resin | |
| JP5572922B2 (en) | Polyamide resin composition for engine cooling water system parts, and engine cooling water system parts molded from the composition | |
| JP5430817B2 (en) | Thermoplastic resin composition | |
| JP5321434B2 (en) | Polyamide resin composition for SMT connectors | |
| WO1996016091A1 (en) | Curable resin and composition | |
| JP2009298853A (en) | Polyamide resin composition | |
| JP2004346255A (en) | Polyamide resin composition for coating metal | |
| JP2013095798A (en) | Polyamide resin composition for metal coating, metal coating material obtained by molding the same, and metallic article coated with the same | |
| JP3642362B2 (en) | Phenolic resin-cured epoxy resin and its production method | |
| JP3985316B2 (en) | Polyamide resin composition and parts for parts used in contact with engine coolant | |
| JP3464759B2 (en) | Polyamide resin, polyamide resin composition and molded article | |
| JP5630286B2 (en) | Method for producing polyamide resin | |
| JP5584963B2 (en) | Polyamide resin composition |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110830 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130828 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131017 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140603 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140616 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5572935 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |