JP4355142B2 - 組換え法 - Google Patents
組換え法 Download PDFInfo
- Publication number
- JP4355142B2 JP4355142B2 JP2002563325A JP2002563325A JP4355142B2 JP 4355142 B2 JP4355142 B2 JP 4355142B2 JP 2002563325 A JP2002563325 A JP 2002563325A JP 2002563325 A JP2002563325 A JP 2002563325A JP 4355142 B2 JP4355142 B2 JP 4355142B2
- Authority
- JP
- Japan
- Prior art keywords
- nucleic acid
- acid molecule
- recombination
- protein
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 230000006798 recombination Effects 0.000 title claims description 211
- 238000005215 recombination Methods 0.000 title claims description 211
- 238000000034 method Methods 0.000 title claims description 144
- 150000007523 nucleic acids Chemical class 0.000 claims description 210
- 102000039446 nucleic acids Human genes 0.000 claims description 205
- 108020004707 nucleic acids Proteins 0.000 claims description 205
- 108090000623 proteins and genes Proteins 0.000 claims description 197
- 102000004169 proteins and genes Human genes 0.000 claims description 152
- 108091034117 Oligonucleotide Proteins 0.000 claims description 141
- 210000004027 cell Anatomy 0.000 claims description 141
- 238000000137 annealing Methods 0.000 claims description 112
- 108020004414 DNA Proteins 0.000 claims description 101
- 239000013612 plasmid Substances 0.000 claims description 83
- 230000008439 repair process Effects 0.000 claims description 62
- 102000053602 DNA Human genes 0.000 claims description 56
- 101100316841 Escherichia phage lambda bet gene Proteins 0.000 claims description 43
- 125000003729 nucleotide group Chemical group 0.000 claims description 41
- 239000002773 nucleotide Substances 0.000 claims description 40
- 210000000349 chromosome Anatomy 0.000 claims description 30
- 238000006243 chemical reaction Methods 0.000 claims description 26
- 241000894007 species Species 0.000 claims description 23
- 238000004520 electroporation Methods 0.000 claims description 22
- 241000588724 Escherichia coli Species 0.000 claims description 20
- 238000000338 in vitro Methods 0.000 claims description 19
- 241000700605 Viruses Species 0.000 claims description 18
- 101100226347 Escherichia phage lambda exo gene Proteins 0.000 claims description 17
- 102000001218 Rec A Recombinases Human genes 0.000 claims description 16
- 108010055016 Rec A Recombinases Proteins 0.000 claims description 16
- 238000012217 deletion Methods 0.000 claims description 15
- 230000037430 deletion Effects 0.000 claims description 15
- 210000003527 eukaryotic cell Anatomy 0.000 claims description 13
- 230000000295 complement effect Effects 0.000 claims description 12
- 238000006467 substitution reaction Methods 0.000 claims description 12
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 10
- 230000015572 biosynthetic process Effects 0.000 claims description 10
- 238000003780 insertion Methods 0.000 claims description 10
- 230000037431 insertion Effects 0.000 claims description 10
- 239000000203 mixture Substances 0.000 claims description 10
- 239000013598 vector Substances 0.000 claims description 10
- 238000010367 cloning Methods 0.000 claims description 9
- 240000004808 Saccharomyces cerevisiae Species 0.000 claims description 7
- 230000003321 amplification Effects 0.000 claims description 7
- 230000001580 bacterial effect Effects 0.000 claims description 7
- 238000001727 in vivo Methods 0.000 claims description 7
- 238000006011 modification reaction Methods 0.000 claims description 7
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 7
- 210000001236 prokaryotic cell Anatomy 0.000 claims description 7
- 230000010076 replication Effects 0.000 claims description 7
- 241000701959 Escherichia virus Lambda Species 0.000 claims description 6
- 108020004682 Single-Stranded DNA Proteins 0.000 claims description 6
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 claims description 5
- 230000027455 binding Effects 0.000 claims description 5
- 244000045947 parasite Species 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 230000009466 transformation Effects 0.000 claims description 4
- 230000000692 anti-sense effect Effects 0.000 claims description 3
- 108020004999 messenger RNA Proteins 0.000 claims description 3
- 238000000926 separation method Methods 0.000 claims description 3
- 238000011426 transformation method Methods 0.000 claims description 3
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 2
- 241000219194 Arabidopsis Species 0.000 claims description 2
- 241000203069 Archaea Species 0.000 claims description 2
- 241000193830 Bacillus <bacterium> Species 0.000 claims description 2
- 241000588748 Klebsiella Species 0.000 claims description 2
- 241000588653 Neisseria Species 0.000 claims description 2
- 102000035118 modified proteins Human genes 0.000 claims description 2
- 108091005573 modified proteins Proteins 0.000 claims description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 claims 1
- 241001646716 Escherichia coli K-12 Species 0.000 claims 1
- 241000233866 Fungi Species 0.000 claims 1
- 241000018427 Iphisa elegans Species 0.000 claims 1
- -1 RecT (rac prophage) Proteins 0.000 claims 1
- 230000013120 recombinational repair Effects 0.000 claims 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 53
- 241001045988 Neogene Species 0.000 description 46
- 101150091879 neo gene Proteins 0.000 description 46
- 230000000694 effects Effects 0.000 description 38
- 238000002474 experimental method Methods 0.000 description 38
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 31
- 239000012634 fragment Substances 0.000 description 29
- 229960000318 kanamycin Drugs 0.000 description 28
- 229930027917 kanamycin Natural products 0.000 description 27
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 27
- 229930182823 kanamycin A Natural products 0.000 description 27
- 230000037361 pathway Effects 0.000 description 20
- 238000002744 homologous recombination Methods 0.000 description 19
- 230000006801 homologous recombination Effects 0.000 description 19
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 18
- 238000003556 assay Methods 0.000 description 16
- 230000002950 deficient Effects 0.000 description 16
- 230000004048 modification Effects 0.000 description 16
- 238000012986 modification Methods 0.000 description 16
- 229960000723 ampicillin Drugs 0.000 description 15
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 15
- 230000035772 mutation Effects 0.000 description 14
- 230000006870 function Effects 0.000 description 13
- 229960005091 chloramphenicol Drugs 0.000 description 12
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 12
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 12
- 108700010839 phage proteins Proteins 0.000 description 11
- 230000008685 targeting Effects 0.000 description 11
- 241000699666 Mus <mouse, genus> Species 0.000 description 10
- 238000012552 review Methods 0.000 description 10
- 238000005516 engineering process Methods 0.000 description 8
- 238000010606 normalization Methods 0.000 description 8
- 241000206602 Eukaryota Species 0.000 description 7
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 238000010187 selection method Methods 0.000 description 7
- 239000006228 supernatant Substances 0.000 description 7
- SRBFZHDQGSBBOR-HWQSCIPKSA-N L-arabinopyranose Chemical compound O[C@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-HWQSCIPKSA-N 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- 230000003115 biocidal effect Effects 0.000 description 6
- 239000013613 expression plasmid Substances 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 101150098466 rpsL gene Proteins 0.000 description 6
- 229960005322 streptomycin Drugs 0.000 description 6
- 241000894006 Bacteria Species 0.000 description 5
- 108060002716 Exonuclease Proteins 0.000 description 5
- 108010086093 Mung Bean Nuclease Proteins 0.000 description 5
- 108010084455 Zeocin Proteins 0.000 description 5
- 230000002759 chromosomal effect Effects 0.000 description 5
- 102000013165 exonuclease Human genes 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 101150066555 lacZ gene Proteins 0.000 description 5
- 239000003550 marker Substances 0.000 description 5
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 238000001890 transfection Methods 0.000 description 5
- 241000196324 Embryophyta Species 0.000 description 4
- 241000282414 Homo sapiens Species 0.000 description 4
- 101100342379 Mus musculus Kmt2a gene Proteins 0.000 description 4
- 230000000903 blocking effect Effects 0.000 description 4
- 230000029087 digestion Effects 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 238000009396 hybridization Methods 0.000 description 4
- 230000001939 inductive effect Effects 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 108091008146 restriction endonucleases Proteins 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000010353 genetic engineering Methods 0.000 description 3
- 210000000688 human artificial chromosome Anatomy 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 241000711573 Coronaviridae Species 0.000 description 2
- 230000008836 DNA modification Effects 0.000 description 2
- 230000033616 DNA repair Effects 0.000 description 2
- 238000001712 DNA sequencing Methods 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000709661 Enterovirus Species 0.000 description 2
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 2
- 108091092566 Extrachromosomal DNA Proteins 0.000 description 2
- 108010025076 Holoenzymes Proteins 0.000 description 2
- 241001631646 Papillomaviridae Species 0.000 description 2
- 102000018120 Recombinases Human genes 0.000 description 2
- 108010091086 Recombinases Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 206010042602 Supraventricular extrasystoles Diseases 0.000 description 2
- 239000004098 Tetracycline Substances 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 2
- 101150049515 bla gene Proteins 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 238000009585 enzyme analysis Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 238000010363 gene targeting Methods 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000009545 invasion Effects 0.000 description 2
- 230000003071 parasitic effect Effects 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 101150087770 recT gene Proteins 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- 235000019364 tetracycline Nutrition 0.000 description 2
- 150000003522 tetracyclines Chemical class 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 230000001131 transforming effect Effects 0.000 description 2
- 241001529453 unidentified herpesvirus Species 0.000 description 2
- 241001515965 unidentified phage Species 0.000 description 2
- 230000006269 (delayed) early viral mRNA transcription Effects 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 101150033922 ABCA2 gene Proteins 0.000 description 1
- 241000701242 Adenoviridae Species 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- 241000712892 Arenaviridae Species 0.000 description 1
- 241000712891 Arenavirus Species 0.000 description 1
- 241001533362 Astroviridae Species 0.000 description 1
- 101100355997 Bacillus subtilis (strain 168) recA gene Proteins 0.000 description 1
- 108010077805 Bacterial Proteins Proteins 0.000 description 1
- 241000709687 Coxsackievirus Species 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 241001466953 Echovirus Species 0.000 description 1
- 108010067770 Endopeptidase K Proteins 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 101150094177 Erf gene Proteins 0.000 description 1
- 101100301301 Escherichia coli (strain K12) recE gene Proteins 0.000 description 1
- 101100356230 Escherichia coli (strain K12) recT gene Proteins 0.000 description 1
- 241000711950 Filoviridae Species 0.000 description 1
- 241000710781 Flaviviridae Species 0.000 description 1
- 241000710831 Flavivirus Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 241000700586 Herpesviridae Species 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 102000003855 L-lactate dehydrogenase Human genes 0.000 description 1
- 108700023483 L-lactate dehydrogenases Proteins 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 1
- 241001115401 Marburgvirus Species 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 241000711386 Mumps virus Species 0.000 description 1
- 101100446513 Mus musculus Fgf4 gene Proteins 0.000 description 1
- 101100355599 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) mus-11 gene Proteins 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 241000714209 Norwalk virus Species 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 241000702259 Orbivirus Species 0.000 description 1
- 241000712464 Orthomyxoviridae Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 241000711504 Paramyxoviridae Species 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 241000150350 Peribunyaviridae Species 0.000 description 1
- 241000710778 Pestivirus Species 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 241001631648 Polyomaviridae Species 0.000 description 1
- 241001505332 Polyomavirus sp. Species 0.000 description 1
- 241001272996 Polyphylla fullo Species 0.000 description 1
- 241000700625 Poxviridae Species 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 101150006234 RAD52 gene Proteins 0.000 description 1
- 230000004570 RNA-binding Effects 0.000 description 1
- 102000053062 Rad52 DNA Repair and Recombination Human genes 0.000 description 1
- 108700031762 Rad52 DNA Repair and Recombination Proteins 0.000 description 1
- 101100437728 Rattus norvegicus Bloc1s2 gene Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 241000702247 Reoviridae Species 0.000 description 1
- 241000702263 Reovirus sp. Species 0.000 description 1
- 241000711931 Rhabdoviridae Species 0.000 description 1
- 241000702670 Rotavirus Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000701835 Salmonella virus P22 Species 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 241000338168 Tringa Species 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 240000004922 Vigna radiata Species 0.000 description 1
- 235000010721 Vigna radiata var radiata Nutrition 0.000 description 1
- 235000011469 Vigna radiata var sublobata Nutrition 0.000 description 1
- 241000269368 Xenopus laevis Species 0.000 description 1
- 108700010877 adenoviridae proteins Proteins 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 238000000246 agarose gel electrophoresis Methods 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- 244000309743 astrovirus Species 0.000 description 1
- 230000003416 augmentation Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000012411 cloning technique Methods 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 238000011259 co-electroporation Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000013601 cosmid vector Substances 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 230000003544 deproteinization Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000012361 double-strand break repair Effects 0.000 description 1
- 238000011143 downstream manufacturing Methods 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000012224 gene deletion Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 238000012872 hydroxylapatite chromatography Methods 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000010399 physical interaction Effects 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 230000037432 silent mutation Effects 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 108010068698 spleen exonuclease Proteins 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
Landscapes
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Plant Pathology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Peptides Or Proteins (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Description
a)第1の核酸分子を、この第1の核酸分子とこの第2の核酸分子との間に修復組換えが生じるに適切な条件下で、ファージアニーリングタンパク質、またはその機能的等価物もしくはフラグメントの存在下で第2の核酸分子と接触させる工程であって、この第1の核酸分子は、この第2の核酸分子と共有された配列相同性の少なくとも2つの領域を含む、工程;ならびに
b)配列が第2の核酸分子由来の配列を含むように改変された核酸分子を選択する工程。
a)ファージアニーリングタンパク質またはその機能的等価物もしくはフラグメントを含有する宿主を提供する工程;
b)この宿主中で、第1の核酸分子を、少なくとも2つの、この第1の核酸分子上の領域との配列相同性領域を含む第2の核酸分子と、この第1の核酸分子と第2の核酸分子との間で修復組換えが生じるに適切な条件下で、接触させる工程;
c)この第1の核酸分子と第2の核酸分子との間で修復組換えが生じた宿主を選択する工程、
を包含する。
a)第2の核酸分子の存在下で、第1の核酸分子を、ファージアニーリングタンパク質またはその機能的等価物もしくはフラグメントに曝露して、連結分子を生成する工程であって、ここで、この第1および第2の核酸分子は、少なくとも2つの配列相同性領域を共有する、工程;
b)この第1の核酸分子と第2の核酸分子との間で修復組換えが生じるに適切な条件下で、この連結分子をインキュベートする工程;および
c)その配列が、この第2の核酸分子由来の配列を含むように変更されている核酸分子を選択する工程、
を包含する。
a)目的の核酸分子の配列に相補的な配列を有するオリゴヌクレオチド分子を、コートされた分子または連結分子複合体の形成に適切な条件下で、ファージアニーリングタンパク質、またはそれらの機能的な等価物もしくはフラグメントに曝露する工程
b)このコートした分子または連結分子複合体を、核酸分子の混合物と共にインキュベートする工程;および
c)ファージアニーリングタンパク質またはそれらの機能的な等価物またはフラグメントに結合する核酸分子を選択する工程。
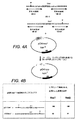
2つのDNA分子の間の修復組換えを媒介するファージアニーリングタンパク質の組換え活性を、図1の実験を用いて、初めに見出した。
pGKneo*およびpGKneoΔを、以下の手順によりpGKneoから作製した:pGKneoを、NcoI制限酵素(これは、neo遺伝子中に固有の認識部位を有する)を用いて線状化した。pGKneo*を作製するために、NcoIで消化したpGKneoの5’オーバーハングを、製造業者(New England Biolabs)の指示書に従ってKlenowおよびヌクレオチドを用いて補充し、次いで、連結により、インタクトな環状プラスミドを作製した。pGKneoΔを作製するために、NcoIで消化したpGKneoの5’オーバーハングを、製造業者(New England Biolabs)の指示書に従ってMung Beanヌクレアーゼを用いて取り除き、次いで、連結により、インタクトな環状プラスミドを作製した。
ファージアニーリングタンパク質の活性を調査するために、さらに、相同性領域の長さと組換えの効率との間の関連性を試験した。
この実験の詳細は、以下のとおりであった。4つの型のpGKneo由来プラスミドを、各々可変長のneo遺伝子中に配列欠損を含み、これらの各々のプラスミドの中にneo遺伝子欠損を与えるように構築した。pGKneoΔにおいて、4個のヌクレオチドを、neo遺伝子内から欠失させ(図1Bを参照のこと)、pGKneoΔ15において、15個のヌクレオチドを欠失させ、pGKneoΔ33において、33個のヌクレオチドを欠失させ;そしてpGKneoΔ60において、60個のヌクレオチドを欠失させた。これらのプラスミドの各々を、25個のヌクレオチドの相同性領域に隣接する損失配列を含むオリゴヌクレオチドと組換えた。従って、全てのpGKneoΔプラスミド改変体を、それ自体に特異的なオリゴヌクレオチドと共に、同時エレクトロポレーションした。この特異的なオリゴヌクレオチドは、25ntの相同性領域(用いられる全てのプラスミドについて同じ)に隣接する、これらのプラスミド型の欠損配列(用いられるプラスミドに依存して4、15、33または60個のヌクレオチド)を含んでいた。全ての組換え反応について、相同性領域の長さは、同じ(すなわち、25nt)であった。pGKneoΔ15、pGKneoΔ33およびpGKneoΔ60を作製するために、pGKneoを、neo遺伝子を独自に切断するNcoIを用いて消化した。引き続いて、製造業者(New England Biolabs)の指示書に従ってBal31と共にインキュベーションすることにより、配列欠損を作製した。Bal31消化の後、得られた分子を連結して、インタクトな環状プラスミドを作製した。作製した配列の長さを、DNA配列決定により決定した。作製した配列の長さを、DNA配列決定により決定した。示したファージタンパク質を、JC5519細胞において、それぞれの遺伝子を含むpBAD24ベースのプラスミドから誘導性発現させた。示したタンパク質を誘導性発現したエレクトロコンピテント細胞を、上記のように(エレクトロポレーション、選択、および得られた組換え効率の標準化で既に述べたように)調製した。
図4Aは、組換えアッセイにおいて用いられるオリゴヌクレオチドおよびプラスミドの図を示す。オリゴヌクレオチドの両方の配向を試験した(上の鎖または下の鎖に相補的、表3を参照のこと)。用いられるオリゴヌクレオチドに依存して、点変異は、(下の鎖に相補的であるオリゴヌクレオチドの場合)欠損neo遺伝子を修復し得る配列に対して5’側にあるか、または(上の鎖に相補的であるオリゴヌクレオチドの場合)欠損neo遺伝子を修復し得る配列に対して3’側にある。導入した点変異は、neo遺伝子に組換えられる場合、neo遺伝子中にサイレントな変異を導入し、それにより、タンパク質配列および遺伝子の機能を変化しない。これらのオリゴヌクレオチドを、pGKneo*プラスミドを用いて、ファージアニーリングタンパク質を発現するJC5519遺伝子へと同時エレクトロポレーションした。
図5Aは、組換えアッセイに用いられるオリゴヌクレオチドおよびプラスミドをの図を示す。オリゴヌクレオチドの両方の配向を試験した(上の鎖または下の鎖に相補的、表3を参照のこと)。両方の配向において、ssDNAオリゴヌクレオチドの3’末端は、ジデオキシ残基を含む。これらのオリゴヌクレオチドを、pGKneo*プラスミドを用いて、ファージアニーリングタンパク質を発現するJC5519遺伝子へと同時エレクトロポレーションした。
E.coli染色体が、一本鎖オリゴヌクレオチドを用いるファージアニーリングタンパク質を媒介した修復組換えを介して、標的化され得るか否かを決定すために、このアッセイを、図6に記載したように行った。
上に示されるデータを得るために、neo遺伝子の修復を、組換え事象についてのスコア付けするための便利な系として利用した。しかし、殆どの実験的適用において、修復組換えにより改変される配列は、抗生物質を用いて選択することができない。従って、記載した活性の絶対的な効率を決定することが望ましい。
ファージアニーリングタンパク質の所望の組換え活性をまた、線状化プラスミドの再環化するために(図7に示すように、この実施例では、ssDNAオリゴヌクレオチドの相同性領域間に既に存在する配列を含むために)適用し得る。ここで、線状化プラスミドを、ssDNAオリゴヌクレオチドと共に、ファージアニーリングタンパク質を発現した宿主株へと同時エレクトロポレーションした。淘汰圧を、線状化プラスミドに存在する選択可能なマーカー遺伝子blaの発現についてのみ用いた。従って、組換え領域については淘汰圧を適用しなかった。組換えの後、ssDNAオリゴヌクレオチドの相同性領域の間に本来存在する配列を含んでいた、インタクトな環状プラスミドを得た。
(プラスミド)
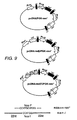
PKGプロモーターおよびneo*からなるDNAフラグメントを、pcDNA3/hyg(−)(Invitrogen)のBst1107I部位に挿入して、pcDNA/PGK−neo*を作製した。redβ遺伝子およびrecE/IRES/recTフラグメントを、pcDNA/PGK−neo*中にCMVプロモーターの制御下で挿入して、pcDNA−redβ/PGK−neo*およびpcDNA−recET/PGK−neo*を作製した(図9を参照のこと)。
50ヌクレオチド(nt)のオリゴヌクレオチドを、NcoI部位の領域中のneo遺伝子の配列に従って合成した。このオリゴヌクレオチドは、2つの22ntの相同領域からなり、各々が、正確なNcoI配列に隣接する(図9の下を参照のこと)。このオリゴヌクレオチドの配列は、以下の通りである:
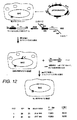
マウスES細胞を、4%のグルコース(Gibco&BRL)、15%のFCS(PAA)、100μm/mlのペニシリン/ストレプトマイシン(Gibco&BRL)、100μMの非必須アミノ酸(Seromed)、1mMのピルビン酸ナトリウム(Gibco&BRL)、1μMのβ−メルカプトエタノール(Sigma)、2mMのL−グルタミン(Gibco&BRL)および500U/mlのLIF「ESGROTM」(Gibco&BRL)を含むDMEM(Gibco&BRL)中で培養した。
発現プラスミド(図1)を、Qiagen Maxi−prepキットを使用して単離し、そしてAhd I(New England Biolabs)で消化して、線状DNAを作製した。沈殿後、DNAを0.5mg/mlでPBS(Gibco&BRL)中に再懸濁した。
10cmディッシュ上のES細胞を、コンフルエントになった後に、10mlのPBSで1回リンスした。1mlのトリプシン/EDTA(Gibco&BRL)溶液を、このディッシュに添加した。このディッシュをインキュベータ中で3〜5分間インキュベートした。10mlのES培養培地を、このディッシュに添加し、そしてES細胞を、上下にピペッティングすることによって単一の細胞に分離した。ES細胞を、1,000rpmで5分間スピンダウンした。上清を除去し、そして細胞ペレットを0.8mlのPBSに再懸濁した。20μgのプラスミドDNAまたは5μgのオリゴヌクレオチドを、ES細胞と混合し、そして4mmのエレクトロポレーションキュベット中に配置した。DNAとES細胞との混合物を、240Vでエレクトロポレーションした。エレクトロポレーションした細胞を、ゼラチンコーティングしたディッシュに移し、そして10mlの培養培地を添加した(図10を参照のこと)。
トランスフェクトした細胞の培地を、毎日交換し、そして選択抗生物質をトランスフェクションの48時間後に添加した。コロニーを、選択のほぼ10日後に観察した。使用した抗生物質の濃度は、以下であった:
G418−200μg/ml(Gibco&BRL)
ハイグロマイシンB−400μg/ml(Boehringer Mannheim)。
1.ES細胞を培養し、そして発現プラスミド(pcDNA/PGK−neo*、pcDNA−redβ/PGK−neo*およびpcDNA−recET/PGK−neo*)でトランスフェクトした(従って、3つの別個のトランスフェクションを実行した)。
本発明の組換え方法はまた、細菌人工染色体(BAC)(これは、挿入物の長さに対する大きな収容能力に起因して、第一のクローニングベクターとなっている)に対しても実行され得る。
a.BAC由来の領域(ここでは、rpsL/neoカセット)を欠失し得る;
b.特定の部位に新規の配列を導入するために使用され得る。ここで、この配列は、短かった。なぜなら、100nt長までの短い配列領域は、オリゴヌクレオチド合成の間に容易に含まれ得るからである。しかし、ssDNAがより長いDNA供給源から他の方法によって調製される場合、より長い配列が含まれ得る。
c.単純で強力かつ効率的である。
従来の形質転換方法に記載されるように、pSC101/γβαA発現プラスミドを、マウスMLL BAC宿主細胞(HS996)中に形質転換する。温度を2℃に設定した冷却Eppendorf遠心分離を使用して、細胞を冷却する。実験前に、dH2Oを少なくとも3時間氷上で冷却するか、または冷蔵庫から冷却dH2Oを取り出して、それを氷上に置く。エレクトロポレーションキュベットもまた、氷上に置くべきである。
1.rpsL−neoカセットおよびpSC101/BAD/γβαと共にMll BACを含むシングルコロニーを、蓋に穴を有するEppendorfチューブ中に、テトラサイクリン(5μg/ml)、カナマイシン(15μg/ml)およびクロラムフェニコール(15μg/ml)を含むLB培地1.4ml中に接種する。加熱ブロックにおいて30℃で攪拌しながら、OD600が約0.15〜0.2になるまで、4〜5時間このチューブをインキュベートする。
または
蓋に穴を有するEppendorfチューブ中に、テトラサイクリン(5μg/ml)、カナマイシン(15μg/ml)およびクロラムフェニコール(15μg/ml)を含むLB培地1.4ml中に30μlの一晩培養物を添加する。加熱ブロックにおいて30℃で攪拌しながら、OD600が約0.2になるまで、約2時間このチューブをインキュベートする。
次いで
2.L−アラビノースを0.1%〜0.2%(最終)まで添加して、リコンビナーゼの発現を誘導する。
3.37℃の加熱ブロック中に移し、そしてOD600が約0.35〜0.4になるまで、45〜60分間37℃でインキュベートする。
4.室温にて、Eppendorf遠心分離において、最高速度を30秒間使用して、細胞をスピンダウンする。
5.上清を廃棄し、そしてチューブを氷上に置く。
6.細胞を、1.0mlの氷冷dH2Oまたは10%の氷冷グリセロール中に、氷上で再懸濁する。
7.最高速度を30秒間使用して、細胞をスピンダウンし、上清を廃棄する。
8.細胞を、1.0mlの氷冷dH2Oまたは10%の氷冷グリセロール中に、氷上で再び再懸濁する。
9.最高速度を30秒間使用して、細胞をスピンダウンする。
10.1mlのピペットを使用して上清を廃棄し、約20〜30μlの溶液を残す。
11.dH2O中のオリゴヌクレオチド(50μM)1μlを添加し、そして氷冷エレクトロポレータキュベット(1mm)中に取り出す。
12.細胞を、Eppendorfエレクトロポレータを使用して1,350Vでエレクトロポレーションする。
13.1mlのLB培地を添加し、37℃で75分間インキュベートする。
14.細胞を、クロラムフェニコール(15μg/ml)およびストレプトマイシン(15μg/ml)または他の抗生物質を含むプレート上に移す。
15.このプレートを、37℃で一晩インキュベートする。ETプラスミド(pSC101/BAD/γβα)は、37℃で失われる。
BACは、しばしば、改変のためには、最も厳しいテンプレートであることが示されてきた。本発明の組換え技術が、BACに対して機能し得ることを実証することによって、E.coli中のすべての他のテンプレート(E.coli染色体、PAC、および他の低コピーテンプレートを含む)ならびに培地および高コピープラスミドがまた、ssオリゴヌクレオチド組換えによって改変され得る。
この実施例は、さらに、本発明の組換え技術がBACと共に機能する能力を実証する。
従来の形質転換方法に記載されるように、pBAD/βγα発現プラスミドを、マウスM11 BAC宿主細胞(HS996)中に形質転換する。−5℃の温度に設定した冷却遠心分離を使用して、E.coli細胞を冷却する。実験前に、dH2Oを少なくとも3時間氷上で冷却するか、または冷蔵庫から冷却dH2Oを取り出して、それを氷上に置く。エレクトロポレーションキュベットもまた、氷上に置くべきである。
実施例10と同じ結論を確立することに加えて、異なるストラテジーおよび異なる発現プラスミドを使用することによって、この実験は、ssオリゴヌクレオチド組換えの絶対的な効率を測定する。相同性アーム中の50ヌクレオチド長の見かけ上最適の長さで、総コロニー数の3%が、正確に組換えられた。この顕著な有効性を考慮すると、抗生物質耐性についての選択が必要なく、そして単純な物理的方法論(例えば、制限分析、PCR、コロニーPCRまたはコロニーハイブリダイゼーション)を使用して、正確な組換え体を同定し得ることが明らかである。
本発明者らは、共有された相同配列を介した修復組換えによって、単一のファージアニーリングタンパク質が2つの分子の組換えを媒介する、新規の組換え活性を本明細書中に記載する。この活性を使用して、いくつかの操作ストラテジー(例えば、欠失(図1A)、挿入(ヌクレオチドおよび短い操作配列(例えば、タンパク質タグ(図1B))から、数キロ塩基対までの)、または配列の置換)は、一定範囲の分子に適切である。この活性のいくつかの重要な特徴を、以下に要約する。これらの特徴の多くは、本明細書に記載される活性を、ET組換え(上記を参照のこと)(これはまた、ファージアニーリングタンパク質の発現を必要とする)から識別する。適切な場合、これらの差異は、強調される。
・記載された活性は、一定範囲の分子中の1個または多数のヌクレオチドの、欠失、挿入および置換を可能にする。
・相同性領域の配列の設計により、記載された活性は、所望の任意の改変可能な部分での分子のDNA操作に適用され得る。
・記載された活性は、外因性(例えば、プラスミド)DNAおよび内因性(染色体)DNAの改変に適切である。
・記載された活性は、ファージアニーリングタンパク質の発現を必要とする。しかし、オルソロガスなエキソヌクレアーゼパートナーの発現は、必要ではない。このことは、アニーリングタンパク質の発現およびそのオルソロガスなエキソヌクレアーゼパートナーの発現が厳密に必要なET組換えとは対照的である(Muyrersら、Genes Dev 14(2000)、1971−1982)。さらに、E.coliにおける他の組換え経路が、記載される活性を媒介できないことが見出された。
・必要な共有された必要とされる相同性の長さは、非常に短い(ET組換え(Zhangら、Nature Genet 20(1998)、123−128;Muyrersら、Genes Dev 14(2000)、1971−1982)よりも短い)。
・これまでに試験した長さまでは、組換え効率は、共有された相同性領域の長さの増加と伴に増加し続ける。
・インビトロで、ファージアニーリングタンパク質は、分子をコーティングし得、そして相同性領域共有分子間で分子の連結を形成し得る。
・記載される活性において、相同性領域における点変異は、組換え体に導入されない(図4)。
・記載される活性が機能する機構において、第二の分子(表5)単独では、複製のためのプライマーとしても岡崎フラグメントとしてもおそらく機能しない。なぜなら、ジデオキシ残基を含む第二の分子は、記載される活性について使用され得(図5)、そしてプラスミドおよび染色体の操作は、リーディング鎖およびラギング鎖の両方由来の一本鎖分子を使用して行われ得るからである(表3、図6)。
・RecBCDの発現は、記載される活性を阻害しないが、一方で、ET組換えを阻害する(Zhangら、Nature Genet 20(1998)、123−128)。
・Erfのみ、またはArf、Erf、Abc1およびAbc2の任意の組み合わせを構成するP22組換え系は、記載される活性について熟練しているが、ET組換えについては熟練していない(おそらく、Erfに対するオルソロガスなエキソヌクレアーゼパートナーの非存在に起因する)。
・記載される活性において、オリゴヌクレオチドから環状プラスミド中に組換える必要のあるヌクレオチドの量の増加は、組換え効率における減少と相関する。このような影響は、ET組換えについては、観察されない。
・RNA分子が、記載される活性において使用され得る。

Claims (50)
- 核酸分子の配列を改変するための方法であって、該方法は、以下の工程:
a)第1の核酸分子を、ファージアニーリングタンパク質の存在下で第2の核酸分子に、該第1の核酸分子と該第2の核酸分子との間で修復組換えが生じるに適切な条件下で接触させる工程であって、該第1の核酸分子は、該第2の核酸分子と共有された配列相同性の少なくとも2つの領域を含む、工程;ならびに
b)該第2の核酸分子由来の配列を含むように配列が改変された核酸分子を選択する工程、
を包含し、
RecEタンパク質もRedαタンパク質も、原核生物細胞において行われるいずれの配列改変反応の間にも存在しないことを条件とする、方法。 - 請求項1に記載の方法であって、前記ファージアニーリングタンパク質が、宿主種内に含まれるか、または宿主種によりコードされる、方法。
- 請求項2に記載の方法であって、前記宿主種が、ウイルス、寄生生物、原核生物または真核生物細胞である、方法。
- 請求項3に記載の方法であって、前記宿主種が、グラム陰性細菌細胞である、方法。
- 請求項4に記載の方法であって、前記細菌細胞が、Escherichia coli細胞である、方法。
- 請求項5に記載の方法であって、前記Escherichia coli細胞が、JC5519株、JC8679株、またはJC9604株のようなEscherichia coli K12株の細胞である、方法。
- 請求項3に記載の方法であって、前記宿主種が、Klebsiella、Bacillus、Neisseria、真菌、およびS.cerevisiaeからなる群より選択される、方法。
- 請求項3に記載の方法であって、前記宿主種がES細胞である、方法。
- 請求項8に記載の方法であって、前記宿主種がマウスES細胞である、方法。
- 請求項2〜9のいずれか1項に記載の方法であって、前記宿主種が、ファージアニーリングタンパク質をコードする遺伝子を発現し得る少なくとも1つのベクターで形質転換される、方法。
- 請求項10に記載の方法であって、前記ファージアニーリングタンパク質をコードする遺伝子の発現が、調節可能なプロモーターの制御下にある、方法。
- 請求項2〜9のいずれか1項に記載の方法であって、前記ファージアニーリングタンパク質が、前記宿主種に導入されたメッセンジャーRNA分子から発現される、方法。
- 請求項1〜12のいずれか1項に記載の方法であって、前記ファージアニーリングタンパク質が、RecT(racプロファージ)、Redβ(ファージλ)、およびErf(p22)からなる群より選択される、方法。
- 請求項13に記載の方法であって、前記ファージアニーリングタンパク質が、RecTであり、該ファージアニーリングタンパク質は、配列番号1に示される配列を有する、方法。
- 請求項13に記載の方法であって、前記ファージアニーリングタンパク質が、Redβであり、該ファージアニーリングタンパク質は、配列番号2に示される配列を有する、方法。
- 請求項13に記載の方法であって、前記ファージアニーリングタンパク質が、Erfであり、該ファージアニーリングタンパク質は、Genbank ID X05268(V01152)に示される配列を有する、方法。
- 請求項1〜16のうちのいずれか1項に記載の方法であって、前記相同性の領域のうちの一方または両方が、前記第1の核酸分子の内部に位置する、方法。
- 請求項1〜17のいずれか1項に記載の方法であって、前記第1の核酸分子が直鎖状である、方法。
- 請求項18に記載の方法であって、前記第1の核酸分子は、一本鎖DNA分子、一本鎖RNA分子、RNA分子、二本鎖DNA分子、5’オーバーハングを有する二本鎖DNA分子、および3’オーバーハングを有する二本鎖DNA分子からなる群より選択される、方法。
- 請求項19に記載の方法であって、前記第1の核酸分子が、一本鎖核酸分子である、方法。
- 請求項18〜20のいずれか1項に記載の方法であって、前記第1のDNA分子が、増幅反応により得られる、方法。
- 請求項1〜21のうちのいずれか1項に記載の方法であって、核酸分子の異種性集団が、前記第1の核酸分子として使用される、方法。
- 請求項1〜22のいずれか1項に記載の方法であって、前記第2の核酸分子が、環状である、方法。
- 請求項23に記載の方法であって、前記第2の核酸分子が、宿主細胞において作動する複製起点を含む染色体外核酸分子である、方法。
- 請求項23または請求項24に記載の方法であって、前記第2の核酸分子が、プラスミド、コスミド、P1ベクター、BACベクターおよびPACベクターからなる群より選択される、方法。
- 請求項23〜25のいずれか1項に記載の方法であって、前記第2の核酸分子が宿主細胞染色体である、方法。
- 請求項26に記載の方法であって、前記宿主細胞染色体が、C.elegans染色体、Arabidopsis染色体、またはDrosophila染色体である、方法。
- 請求項23に記載の方法であって、前記第2の核酸分子が、二本鎖RNAである、方法。
- 請求項1〜28のいずれか1項に記載の方法であって、前記第1の核酸分子と前記第2の核酸分子との間で共有される配列相同性の領域が、各々少なくとも9ヌクレオチドである、方法。
- 請求項2〜29のいずれか1項に記載の方法であって、前記第1および/または前記第2の核酸分子が、形質転換により前記宿主種に導入される、方法。
- 請求項30に記載の方法であって、前記形質転換法がエレクトロポレーションである、方法。
- 請求項2〜31のいずれか1項に記載の方法であって、前記第1および前記第2の核酸分子が、同時形質転換により前記宿主種に導入される、方法。
- 請求項2〜32のいずれか1項に記載の方法であって、前記第1の核酸分子が、前記第2の核酸分子が既に存在する宿主細胞に導入される、方法。
- 請求項1〜33のいずれか1項に記載の方法であって、前記組換え事象がインビトロで生じる、方法。
- 請求項1〜34のいずれか1項に記載の方法であって、前記組換え事象がインビボで生じる、方法。
- 請求項1〜35のうちのいずれか1項に記載の方法であって、recBCDが不活化されておらず、従って、前記組換え事象が、recBCD+バックグラウンドにおいて生じる、方法。
- 請求項1〜36のうちのいずれか1項に記載の方法であって、RecAが発現しない宿主細胞において実施される、方法。
- 多キロ塩基対の長さの核酸分子の欠失、挿入、または置換のための、請求項1〜37のうちのいずれか1項に記載の方法。
- ファージアニーリングタンパク質をコードする遺伝子を発現し得る細胞の、修復組換えを含むクローニング法のための宿主としての使用であって、
該細胞が原核生物である場合に該細胞がRecEもRedαも含まないことを条件とする、使用。 - ファージアニーリングタンパク質をコードする遺伝子を発現し得るベクターの、修復組換えを含むクローニング法のための宿主種における使用であって、
該宿主種が原核生物である場合に該宿主種がRecEもRedαも含まないことを条件とする、使用。 - 核酸分子の配列を改変するための方法であって、該方法は、以下の工程:
a)第1の核酸分子を、第2の核酸分子の存在下で、ファージアニーリングタンパク質に曝して、結合分子を生成する工程であって、該第1および該第2の核酸分子は、配列相同性の少なくとも2つの領域を共有する、工程;
b)該結合分子を、該第1の核酸分子と該第2の核酸分子との間に組換え修復が生じるに適切な条件下でインキュベートする工程;ならびに
c)該第2の核酸分子由来の配列を含むように配列が改変された核酸分子を選択する工程、
を包含し、
RecEタンパク質もRedαタンパク質も、原核生物細胞において行われる配列改変反応の過程の間に存在しないことを条件とする、方法。 - 核酸分子の配列を改変するための方法であって、該方法は、以下の工程:
a)第1の核酸分子を、ファージアニーリングタンパク質に曝して、コートされた核酸分子を生成する工程;
b)該コートされた分子を、第2の核酸分子に、該第1の核酸分子と該第2の核酸分子との間に修復組換えが生じるに適切な条件下で接触させる工程であって、該第1および該第2の核酸分子は、配列相同性の少なくとも2つの領域を共有する、工程;ならびに
c)該第2の核酸分子由来の配列を含むように配列が改変された核酸分子を選択する工程、
を包含し、
RecEタンパク質もRedαタンパク質も、原核生物細胞において行われる配列改変反応の過程の間に存在しないことを条件とする、方法。 - 酵母、古細菌、C.elegans、Drosophila、X.laevis、ウイルス、および寄生生物から選択される宿主において外因性核酸分子および内因性核酸分子を操作するための、請求項42に記載の方法。
- 核酸をクローニングするための方法であって、
請求項1〜38または請求項41〜43のいずれか1項に記載の核酸分子の配列を改変する方法を利用する、
方法。 - 核酸分子の配列を操作するための方法であって、
請求項1〜38または請求項41〜43のいずれか1項に記載の方法の工程
を包含する、方法。 - 改変されたタンパク質リーディングフレームの作製のための、請求項1〜45のうちのいずれか1項に記載の方法または使用。
- 核酸分子の混合物から所望の核酸分子を選択する方法であって、該方法は、以下の工程:
a)該所望の核酸分子の配列に相補的な配列を有するオリゴヌクレオチド分子を、コートされた分子または結合分子複合体の形成に適切な条件下で、ファージアニーリングタンパク質に曝す工程;
b)該コートされた分子または結合分子の複合体を、該核酸分子の混合物とともにインキュベートする工程;ならびに
c)ファージアニーリングタンパク質に結合された核酸分子を選択する工程、
を包含し、
RecEタンパク質もRedαタンパク質も、原核生物細胞において行われるいずれの配列改変反応の間にも存在しないことを条件とする、方法。 - 請求項47に記載の方法であって、前記オリゴヌクレオチドがタグを含む、方法。
- 請求項47または請求項48に記載の方法であって、前記工程c)において選択された核酸分子は、タグ化されたオリゴヌクレオチドを単離するためにアフィニティー分離方法を使用して選択される、方法。
- アンチセンス戦略のための、請求項42に記載のコートされた核酸分子の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0103276A GB0103276D0 (en) | 2001-02-09 | 2001-02-09 | Recombination method |
| GB0120312A GB0120312D0 (en) | 2001-08-21 | 2001-08-21 | Recombination method |
| PCT/IB2002/001415 WO2002062988A2 (en) | 2001-02-09 | 2002-02-08 | Recombination method |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2004535773A JP2004535773A (ja) | 2004-12-02 |
| JP2004535773A5 JP2004535773A5 (ja) | 2008-06-05 |
| JP4355142B2 true JP4355142B2 (ja) | 2009-10-28 |
Family
ID=26245704
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002563325A Expired - Fee Related JP4355142B2 (ja) | 2001-02-09 | 2002-02-08 | 組換え法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20040101520A1 (ja) |
| EP (1) | EP1399546A2 (ja) |
| JP (1) | JP4355142B2 (ja) |
| CA (1) | CA2436743A1 (ja) |
| WO (1) | WO2002062988A2 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2419322C (en) | 2000-08-14 | 2012-10-23 | Donald L. Court | Enhanced homologous recombination mediated by lambda recombination proteins |
| US7521242B2 (en) | 2003-05-09 | 2009-04-21 | The United States Of America As Represented By The Department Of Health And Human Services | Host cells deficient for mismatch repair and their use in methods for inducing homologous recombination using single-stranded nucleic acids |
| US7674621B2 (en) | 2004-05-21 | 2010-03-09 | The United States Of America As Represented By The Department Of Health And Human Services | Plasmids and phages for homologous recombination and methods of use |
| US20070281309A1 (en) * | 2006-05-19 | 2007-12-06 | Massachusetts Institute Of Technology | Microfluidic-based Gene Synthesis |
| GB0909660D0 (en) | 2009-06-04 | 2009-07-22 | Gene Bridges Gmbh | Method of altering nucleic acids |
| GB0922108D0 (en) | 2009-12-17 | 2010-02-03 | Gene Bridges Gmbh | Heterologous hosts |
| GB201009732D0 (en) | 2010-06-10 | 2010-07-21 | Gene Bridges Gmbh | Direct cloning |
| CN103667331B (zh) * | 2013-12-10 | 2015-09-30 | 南京师范大学 | 重组酶基因bet作为一种大肠杆菌异源蛋白表达融合标签的应用 |
| EP4056701A1 (en) * | 2016-08-12 | 2022-09-14 | Nexuspiral Inc. | Genome editing method |
| CN106995813B (zh) | 2017-03-23 | 2020-06-16 | 山东大学 | 基因组大片段直接克隆和dna多分子组装新技术 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1034260B1 (en) * | 1997-12-05 | 2003-06-04 | Europäisches Laboratorium Für Molekularbiologie (Embl) | Novel dna cloning method relying on the e. coli rece/rect recombination system |
| CA2419322C (en) * | 2000-08-14 | 2012-10-23 | Donald L. Court | Enhanced homologous recombination mediated by lambda recombination proteins |
-
2002
- 2002-02-08 US US10/470,679 patent/US20040101520A1/en not_active Abandoned
- 2002-02-08 EP EP02722618A patent/EP1399546A2/en not_active Withdrawn
- 2002-02-08 JP JP2002563325A patent/JP4355142B2/ja not_active Expired - Fee Related
- 2002-02-08 CA CA002436743A patent/CA2436743A1/en not_active Abandoned
- 2002-02-08 WO PCT/IB2002/001415 patent/WO2002062988A2/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| US20040101520A1 (en) | 2004-05-27 |
| CA2436743A1 (en) | 2002-08-15 |
| WO2002062988A3 (en) | 2004-01-15 |
| WO2002062988A2 (en) | 2002-08-15 |
| JP2004535773A (ja) | 2004-12-02 |
| EP1399546A2 (en) | 2004-03-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20250034562A1 (en) | Compositions and methods for improving the efficacy of cas9-based knock-in strategies | |
| CN111373041B (zh) | 用于基因组编辑和调节转录的crispr/cas系统和方法 | |
| Murphy | λ recombination and recombineering | |
| JP7301332B2 (ja) | Dnaが編集された真核細胞を製造する方法、および当該方法に用いられるキット | |
| CN106995813B (zh) | 基因组大片段直接克隆和dna多分子组装新技术 | |
| CN103068995B (zh) | 直接克隆 | |
| JP2019526248A (ja) | プログラム可能cas9−リコンビナーゼ融合タンパク質およびその使用 | |
| JPH11507236A (ja) | 操作された組換え部位を使用する組換えクローニング | |
| JP5703031B2 (ja) | 核酸組換え法 | |
| JP4355142B2 (ja) | 組換え法 | |
| Narayanan et al. | Bacterial artificial chromosome mutagenesis using recombineering | |
| JP2004535773A5 (ja) | ||
| Penewit et al. | Genome editing in staphylococcus aureus by conditional recombineering and CRISPR/Cas9-mediated counterselection | |
| Penewit et al. | Recombineering in Staphylococcus aureus | |
| AU2002253479A1 (en) | Recombination method | |
| AU2008202476A1 (en) | Recombination method | |
| WO2010140066A2 (en) | Method of altering nucleic acids | |
| Brumos et al. | An improved plant toolset for high-throughput recombineering | |
| Casini | Better safe than sorry: new CRISPR/Cas9 tools for improved genome engineering | |
| Linares et al. | A CRISPR-associated transposase presents null cargo integration efficiency when targeting a transcriptionally highly active region |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050127 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20071109 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080129 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080205 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080307 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080314 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20080409 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090323 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090622 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090715 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090731 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120807 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |