JP4245490B2 - 2- (Dichlorophenyl) -4-phenylimidazole compound - Google Patents
2- (Dichlorophenyl) -4-phenylimidazole compound Download PDFInfo
- Publication number
- JP4245490B2 JP4245490B2 JP2004022241A JP2004022241A JP4245490B2 JP 4245490 B2 JP4245490 B2 JP 4245490B2 JP 2004022241 A JP2004022241 A JP 2004022241A JP 2004022241 A JP2004022241 A JP 2004022241A JP 4245490 B2 JP4245490 B2 JP 4245490B2
- Authority
- JP
- Japan
- Prior art keywords
- dichlorophenyl
- mol
- phenyl
- phenylimidazole
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- -1 2- (Dichlorophenyl) -4-phenylimidazole compound Chemical class 0.000 title claims description 19
- 239000000126 substance Substances 0.000 claims description 15
- CLUAAGKBBZBESA-UHFFFAOYSA-N 2-(2,4-dichlorophenyl)-5-methyl-4-phenyl-1h-imidazole Chemical compound CC=1NC(C=2C(=CC(Cl)=CC=2)Cl)=NC=1C1=CC=CC=C1 CLUAAGKBBZBESA-UHFFFAOYSA-N 0.000 claims description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- 239000013078 crystal Substances 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- BVQVLAIMHVDZEL-UHFFFAOYSA-N 1-phenyl-1,2-propanedione Chemical compound CC(=O)C(=O)C1=CC=CC=C1 BVQVLAIMHVDZEL-UHFFFAOYSA-N 0.000 description 10
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 238000004809 thin layer chromatography Methods 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- BGAXCPSNMHVHJC-UHFFFAOYSA-N phenacyl acetate Chemical compound CC(=O)OCC(=O)C1=CC=CC=C1 BGAXCPSNMHVHJC-UHFFFAOYSA-N 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 5
- 239000005695 Ammonium acetate Substances 0.000 description 5
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 235000019257 ammonium acetate Nutrition 0.000 description 5
- 229940043376 ammonium acetate Drugs 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 238000001819 mass spectrum Methods 0.000 description 5
- 238000002844 melting Methods 0.000 description 5
- 230000008018 melting Effects 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- HHOSUMHKSIZYOV-UHFFFAOYSA-N 2-(2,3-dichlorophenyl)-5-phenyl-1h-imidazole Chemical compound ClC1=CC=CC(C=2NC(=CN=2)C=2C=CC=CC=2)=C1Cl HHOSUMHKSIZYOV-UHFFFAOYSA-N 0.000 description 4
- GGKUNTBLCJXNLV-UHFFFAOYSA-N 2-(2,4-dichlorophenyl)-5-phenyl-1h-imidazole Chemical compound ClC1=CC(Cl)=CC=C1C1=NC(C=2C=CC=CC=2)=CN1 GGKUNTBLCJXNLV-UHFFFAOYSA-N 0.000 description 4
- UPAKQFPBLGINHI-UHFFFAOYSA-N 2-(3,4-dichlorophenyl)-5-methyl-4-phenyl-1h-imidazole Chemical compound CC=1NC(C=2C=C(Cl)C(Cl)=CC=2)=NC=1C1=CC=CC=C1 UPAKQFPBLGINHI-UHFFFAOYSA-N 0.000 description 4
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 4
- LLMLNAVBOAMOEE-UHFFFAOYSA-N 2,3-dichlorobenzaldehyde Chemical compound ClC1=CC=CC(C=O)=C1Cl LLMLNAVBOAMOEE-UHFFFAOYSA-N 0.000 description 3
- CDICNGYKVLYRGI-UHFFFAOYSA-N 2-(2,3-dichlorophenyl)-5-methyl-4-phenyl-1h-imidazole Chemical compound CC=1NC(C=2C(=C(Cl)C=CC=2)Cl)=NC=1C1=CC=CC=C1 CDICNGYKVLYRGI-UHFFFAOYSA-N 0.000 description 3
- RSGAJBKILULMBT-UHFFFAOYSA-N 2-(2,6-dichlorophenyl)-5-methyl-4-phenyl-1h-imidazole Chemical compound CC=1NC(C=2C(=CC=CC=2Cl)Cl)=NC=1C1=CC=CC=C1 RSGAJBKILULMBT-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- HYHCSLBZRBJJCH-UHFFFAOYSA-M sodium hydrosulfide Chemical compound [Na+].[SH-] HYHCSLBZRBJJCH-UHFFFAOYSA-M 0.000 description 3
- 230000003595 spectral effect Effects 0.000 description 3
- YSFBEAASFUWWHU-UHFFFAOYSA-N 2,4-dichlorobenzaldehyde Chemical compound ClC1=CC=C(C=O)C(Cl)=C1 YSFBEAASFUWWHU-UHFFFAOYSA-N 0.000 description 2
- DMIYKWPEFRFTPY-UHFFFAOYSA-N 2,6-dichlorobenzaldehyde Chemical compound ClC1=CC=CC(Cl)=C1C=O DMIYKWPEFRFTPY-UHFFFAOYSA-N 0.000 description 2
- RXWOHFUULDINMC-UHFFFAOYSA-N 2-(3-nitrothiophen-2-yl)acetic acid Chemical compound OC(=O)CC=1SC=CC=1[N+]([O-])=O RXWOHFUULDINMC-UHFFFAOYSA-N 0.000 description 2
- ZWUSBSHBFFPRNE-UHFFFAOYSA-N 3,4-dichlorobenzaldehyde Chemical compound ClC1=CC=C(C=O)C=C1Cl ZWUSBSHBFFPRNE-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000003822 epoxy resin Substances 0.000 description 2
- 238000000034 method Methods 0.000 description 2
- 239000012450 pharmaceutical intermediate Substances 0.000 description 2
- 229920000647 polyepoxide Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 0 *c1c(-c2ccccc2)nc(-c2ccccc2)[n]1 Chemical compound *c1c(-c2ccccc2)nc(-c2ccccc2)[n]1 0.000 description 1
- FSAKYMVUZFUEDX-UHFFFAOYSA-N 2-(2,5-dichlorophenyl)-5-methyl-4-phenyl-1h-imidazole Chemical compound CC=1NC(C=2C(=CC=C(Cl)C=2)Cl)=NC=1C1=CC=CC=C1 FSAKYMVUZFUEDX-UHFFFAOYSA-N 0.000 description 1
- OAOJBIRIWDNQLC-UHFFFAOYSA-N 2-(2,5-dichlorophenyl)-5-phenyl-1h-imidazole Chemical compound ClC1=CC=C(Cl)C(C=2NC(=CN=2)C=2C=CC=CC=2)=C1 OAOJBIRIWDNQLC-UHFFFAOYSA-N 0.000 description 1
- IWEYWOGNSYHPRX-UHFFFAOYSA-N 2-(2,6-dichlorophenyl)-5-phenyl-1h-imidazole Chemical compound ClC1=CC=CC(Cl)=C1C1=NC=C(C=2C=CC=CC=2)N1 IWEYWOGNSYHPRX-UHFFFAOYSA-N 0.000 description 1
- GNFKFXXJWPRDJK-UHFFFAOYSA-N 2-(3,4-dichlorophenyl)-5-phenyl-1h-imidazole Chemical compound C1=C(Cl)C(Cl)=CC=C1C1=NC=C(C=2C=CC=CC=2)N1 GNFKFXXJWPRDJK-UHFFFAOYSA-N 0.000 description 1
- ZQKYGKCLSAXVEA-UHFFFAOYSA-N 2-(3,5-dichlorophenyl)-5-methyl-4-phenyl-1h-imidazole Chemical compound CC=1NC(C=2C=C(Cl)C=C(Cl)C=2)=NC=1C1=CC=CC=C1 ZQKYGKCLSAXVEA-UHFFFAOYSA-N 0.000 description 1
- ARQLFXXVTVPIPX-UHFFFAOYSA-N 2-(3,5-dichlorophenyl)-5-phenyl-1h-imidazole Chemical compound ClC1=CC(Cl)=CC(C=2NC(=CN=2)C=2C=CC=CC=2)=C1 ARQLFXXVTVPIPX-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- OMZSGWSJDCOLKM-UHFFFAOYSA-N copper(II) sulfide Chemical compound [S-2].[Cu+2] OMZSGWSJDCOLKM-UHFFFAOYSA-N 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000004188 dichlorophenyl group Chemical group 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- IMACFCSSMIZSPP-UHFFFAOYSA-N phenacyl chloride Chemical compound ClCC(=O)C1=CC=CC=C1 IMACFCSSMIZSPP-UHFFFAOYSA-N 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- SUBJHSREKVAVAR-UHFFFAOYSA-N sodium;methanol;methanolate Chemical compound [Na+].OC.[O-]C SUBJHSREKVAVAR-UHFFFAOYSA-N 0.000 description 1
- 239000012756 surface treatment agent Substances 0.000 description 1
Description
本発明は、化1の一般式で示される新規な2−(ジクロロフェニル)−4−フェニルイミダゾール化合物(但し、2−(2,4−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールを除く)に関するものである。このイミダゾール化合物は、エポキシ樹脂硬化剤や医薬品中間体として有用なものである。 The present invention relates to a novel 2- (dichlorophenyl) -4-phenylimidazole compound represented by the general formula of Chemical Formula 1 (excluding 2- (2,4-dichlorophenyl) -4-phenyl-5-methylimidazole). Is. This imidazole compound is useful as an epoxy resin curing agent or a pharmaceutical intermediate.
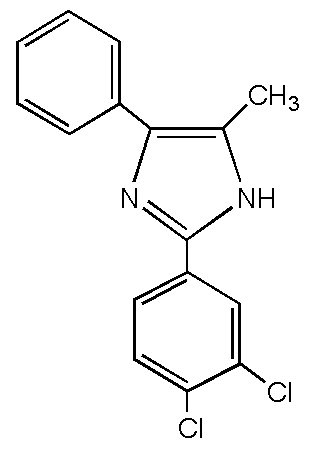
本発明に類似のイミダゾール化合物としては、特許文献1〜3に、2−(2,4−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールが、銅の表面処理剤として使用された例が記載されている。
しかしながら、前記以外の2位のフェニル基が2個の塩素原子に置換されたイミダゾール化合物は知られていない。
As imidazole compounds similar to the present invention, Patent Documents 1 to 3 describe examples in which 2- (2,4-dichlorophenyl) -4-phenyl-5-methylimidazole is used as a copper surface treatment agent. ing.
However, there is no known imidazole compound in which the 2-position phenyl group other than the above is substituted with two chlorine atoms.
本発明は、新規な2−(ジクロロフェニル)−4−フェニルイミダゾール化合物を提供することを目的とする。 An object of the present invention is to provide a novel 2- (dichlorophenyl) -4-phenylimidazole compound.
化1の一般式で示される2−(ジクロロフェニル)−4−フェニルイミダゾール化合物(但し、2−(2,4−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールを除く)を提供する。 A 2- (dichlorophenyl) -4-phenylimidazole compound represented by the general formula of Chemical Formula 1 (excluding 2- (2,4-dichlorophenyl) -4-phenyl-5-methylimidazole) is provided.
本発明の2−(ジクロロフェニル)−4−フェニルイミダゾール化合物は、エポキシ樹脂硬化剤や医薬品中間体として有用なものである。 The 2- (dichlorophenyl) -4-phenylimidazole compound of the present invention is useful as an epoxy resin curing agent or a pharmaceutical intermediate.
本発明の2−(ジクロロフェニル)−4−フェニルイミダゾール化合物は、
2−(2,3−ジクロロフェニル)−4−フェニルイミダゾール、
2−(2,4−ジクロロフェニル)−4−フェニルイミダゾール、
2−(2,5−ジクロロフェニル)−4−フェニルイミダゾール、
2−(2,6−ジクロロフェニル)−4−フェニルイミダゾール、
2−(3,4−ジクロロフェニル)−4−フェニルイミダゾール、
2−(3,5−ジクロロフェニル)−4−フェニルイミダゾール、
2−(2,3−ジクロロフェニル)−4−フェニル−5−メチルイミダゾール、
2−(2,5−ジクロロフェニル)−4−フェニル−5−メチルイミダゾール、
2−(2,6−ジクロロフェニル)−4−フェニル−5−メチルイミダゾール、
2−(3,4−ジクロロフェニル)−4−フェニル−5−メチルイミダゾール及び
2−(3,5−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールである。
The 2- (dichlorophenyl) -4-phenylimidazole compound of the present invention is
2- (2,3-dichlorophenyl) -4-phenylimidazole,
2- (2,4-dichlorophenyl) -4-phenylimidazole,
2- (2,5-dichlorophenyl) -4-phenylimidazole,
2- (2,6-dichlorophenyl) -4-phenylimidazole,
2- (3,4-dichlorophenyl) -4-phenylimidazole,
2- (3,5-dichlorophenyl) -4-phenylimidazole,
2- (2,3-dichlorophenyl) -4-phenyl-5-methylimidazole,
2- (2,5-dichlorophenyl) -4-phenyl-5-methylimidazole,
2- (2,6-dichlorophenyl) -4-phenyl-5-methylimidazole,
2- (3,4-dichlorophenyl) -4-phenyl-5-methylimidazole and 2- (3,5-dichlorophenyl) -4-phenyl-5-methylimidazole.
本発明の2−(ジクロロフェニル)−4−フェニルイミダゾール化合物の内、イミダゾール環の5位が未置換である2−(ジクロロフェニル)−4−フェニルイミダゾール化合物は、化2の反応式に示されるように、2−アセトキシアセトフェノン、ジクロロベンズアルデヒド化合物、アンモニア及び酢酸銅(II)をアルコール中で加熱反応させることにより得られる。 Among the 2- (dichlorophenyl) -4-phenylimidazole compounds of the present invention, the 2- (dichlorophenyl) -4-phenylimidazole compound in which the 5-position of the imidazole ring is unsubstituted is represented by the reaction formula of Chemical Formula 2. , 2-acetoxyacetophenone, dichlorobenzaldehyde compound, ammonia and copper (II) acetate are heated and reacted in alcohol.
前記の反応において、2−アセトキシアセトフェノンの使用量は、ジクロロベンズアルデヒド化合物に対して、0.8〜1.5倍モル、好ましくは0.9〜1.1倍モルである。 In the above reaction, the amount of 2-acetoxyacetophenone used is 0.8 to 1.5 times mol, preferably 0.9 to 1.1 times mol for the dichlorobenzaldehyde compound.
アンモニアの使用量は、ジクロロベンズアルデヒド化合物に対して、10〜50倍モル、好ましくは20〜30倍モルである。 The usage-amount of ammonia is 10-50 times mole with respect to a dichlorobenzaldehyde compound, Preferably it is 20-30 times mole.
酢酸銅(II)の使用量は、ジクロロベンズアルデヒド化合物に対して、1〜5倍モル、好ましくは2〜3倍モルである。 The amount of copper (II) acetate used is 1 to 5 times mol, preferably 2 to 3 times mol, of the dichlorobenzaldehyde compound.
反応温度は、50〜80℃であり、反応時間は1〜10時間である。 The reaction temperature is 50 to 80 ° C., and the reaction time is 1 to 10 hours.
加熱終了後、析出物を濾取しこの析出物をメタノールに懸濁させる。次いで、このメタノール溶液に水硫化ナトリウムを加え、析出した硫化銅を濾別し、メタノールを減圧留去し、残留物を水で洗浄して粗製のイミダゾール環5位未置換の2−(ジクロロフェニル)−4−フェニルイミダゾール化合物を固体として得ることができる。本粗製物は再結晶法により精製することができる。 After heating, the precipitate is collected by filtration and suspended in methanol. Next, sodium hydrosulfide is added to the methanol solution, the precipitated copper sulfide is filtered off, the methanol is distilled off under reduced pressure, and the residue is washed with water to give a crude imidazole ring 5-position unsubstituted 2- (dichlorophenyl). A -4-phenylimidazole compound can be obtained as a solid. This crude product can be purified by a recrystallization method.
また、イミダゾール環の5位にメチル基が置換した2−(ジクロロフェニル)−4−フェニルイミダゾール化合物は、化3の反応式に示されるように、ジクロロベンズアルデヒド化合物、1−フェニル−1,2−プロパンジオン及び酢酸アンモニウムを酢酸中で加熱反応させることにより得られる。 Further, a 2- (dichlorophenyl) -4-phenylimidazole compound in which a methyl group is substituted at the 5-position of the imidazole ring is a dichlorobenzaldehyde compound, 1-phenyl-1,2-propane, as shown in the reaction formula It can be obtained by heating and reacting dione and ammonium acetate in acetic acid.
前記の反応において、1−フェニル−1,2−プロパンジオンの使用量は、ジクロロベンズアルデヒド化合物に対して、0.8〜1.5倍モル、好ましくは0.9〜1.1倍モルである。 In the above reaction, the amount of 1-phenyl-1,2-propanedione to be used is 0.8 to 1.5 times mol, preferably 0.9 to 1.1 times mol, of the dichlorobenzaldehyde compound. .
酢酸アンモニウムの使用量は、ジクロロベンズアルデヒド化合物に対して、2〜10倍モル、好ましくは4〜6倍モルである。 The usage-amount of ammonium acetate is 2-10 times mole with respect to a dichlorobenzaldehyde compound, Preferably it is 4-6 times mole.
反応温度は、80℃以上、好ましくは還流温度であり、反応時間は1〜10時間である。 The reaction temperature is 80 ° C. or higher, preferably the reflux temperature, and the reaction time is 1 to 10 hours.
加熱終了後、反応液または該反応液から酢酸を留去して得た残留物と、水酸化ナトリウム、炭酸ナトリウム、アンモニア等のアルカリ剤を過剰に含む水溶液を混合することにより、粗製の2−(ジクロロフェニル)−4−フェニル−5−メチルイミダゾール化合物を固体の析出物として得ることができる。取り出した析出物は、再結晶操作により精製することができる。 After completion of the heating, the reaction solution or the residue obtained by distilling off acetic acid from the reaction solution is mixed with an aqueous solution containing an excessive amount of an alkali agent such as sodium hydroxide, sodium carbonate, ammonia, etc. The (dichlorophenyl) -4-phenyl-5-methylimidazole compound can be obtained as a solid precipitate. The extracted precipitate can be purified by a recrystallization operation.
以下、本発明を実施例によって具体的に説明するが、本発明はこれらに限定されるものではない。
なお、実施例で使用した主原料は次のとおりである。
EXAMPLES The present invention will be specifically described below with reference to examples, but the present invention is not limited to these examples.
The main raw materials used in the examples are as follows.
[原料]
・2−アセトキシアセトフェノン(参考例に記載した方法で合成した)
・1−フェニル−1,2−プロパンジオン(東京化成工業社製、試薬)
・2,3−ジクロロベンズアルデヒド(東京化成工業社製、試薬)
・2,4−ジクロロベンズアルデヒド(東京化成工業社製、試薬)
・2,6−ジクロロベンズアルデヒド(東京化成工業社製、試薬)
・3,4−ジクロロベンズアルデヒド(東京化成工業社製、試薬)
[material]
2-acetoxyacetophenone (synthesized by the method described in Reference Example)
・ 1-Phenyl-1,2-propanedione (manufactured by Tokyo Chemical Industry Co., Ltd., reagent)
・ 2,3-dichlorobenzaldehyde (manufactured by Tokyo Chemical Industry Co., Ltd., reagent)
・ 2,4-Dichlorobenzaldehyde (manufactured by Tokyo Chemical Industry Co., Ltd., reagent)
2,6-dichlorobenzaldehyde (manufactured by Tokyo Chemical Industry Co., Ltd., reagent)
・ 3,4-dichlorobenzaldehyde (manufactured by Tokyo Chemical Industry Co., Ltd., reagent)
〔参考例〕
<2−アセトキシアセトフェノンの合成>
酢酸カリウム78.5g(0.80mol)、酢酸5.0g(0.08mol)及び2−クロロアセトフェノン123.7g(0.80mol)を、500mlのエタノール中で6時間加熱還流した。加熱終了後、反応液を室温まで冷却し、析出した塩化カリウムを濾去して、エタノールを減圧留去し淡褐色油状物を得た。1lの水に、この油状物を注ぎ入れ析出させた黄褐色の結晶性固体を濾取後、メタノールを使用して再結晶操作を行い、2−アセトキシアセトフェノン113.1g(0.635mol、収率79.3%)を得た。
[Reference example]
<Synthesis of 2-acetoxyacetophenone>
78.5 g (0.80 mol) of potassium acetate, 5.0 g (0.08 mol) of acetic acid and 123.7 g (0.80 mol) of 2-chloroacetophenone were heated to reflux in 500 ml of ethanol for 6 hours. After completion of the heating, the reaction solution was cooled to room temperature, the precipitated potassium chloride was filtered off, and ethanol was distilled off under reduced pressure to obtain a light brown oil. The oily substance was poured into 1 l of water and the precipitated yellow-brown crystalline solid was collected by filtration and then recrystallized using methanol to give 113.1 g (0.635 mol, yield) of 2-acetoxyacetophenone. 79.3%).
〔実施例1〕
<2−(2,3−ジクロロフェニル)−4−フェニルイミダゾールの合成>
2−アセトキシアセトフェノン17.8g(0.1mol)と2,3−ジクロロベンズアルデヒド17.5g(0.1mol)を、120mlのイソプロピルアルコールに溶解させた溶液に、酢酸銅(II)一水和物43.9g(0.22mol)を25%アンモニア水150mlに溶解させた溶液を水冷下で少量ずつ加え、次いで60℃まで1時間、更に78℃まで3時間かけて昇温した。
反応終了後、反応液を5℃まで冷却し、析出物を濾取して水洗後乾燥して暗緑色粉末状物31.1gを得た。この粉末状物をメタノール160mlに懸濁させ、70%水硫化ナトリウム4.9g(0.06mol)を加え1時間加熱還流した。その後、メタノール溶液を冷却して黒色不溶物を濾去した。引き続き、メタノール溶液からメタノールを減圧下で留去し、得られた残留物をクロロホルムに溶解し、水で洗浄後、クロロホルムを減圧留去し、得られた残留物をアセトニトリルを使用して再結晶操作を行い、灰白色粉末状の結晶12.5gを得た(収率43%)。
[Example 1]
<Synthesis of 2- (2,3-dichlorophenyl) -4-phenylimidazole>
To a solution of 17.8 g (0.1 mol) of 2-acetoxyacetophenone and 17.5 g (0.1 mol) of 2,3-dichlorobenzaldehyde in 120 ml of isopropyl alcohol, copper (II) acetate monohydrate 43 A solution prepared by dissolving .9 g (0.22 mol) in 150 ml of 25% aqueous ammonia was added little by little under water cooling, and then the temperature was raised to 60 ° C. for 1 hour and further to 78 ° C. over 3 hours.
After completion of the reaction, the reaction solution was cooled to 5 ° C., the precipitate was collected by filtration, washed with water and dried to obtain 31.1 g of a dark green powder. This powder was suspended in 160 ml of methanol, 4.9 g (0.06 mol) of 70% sodium hydrosulfide was added, and the mixture was heated to reflux for 1 hour. Thereafter, the methanol solution was cooled and black insolubles were removed by filtration. Subsequently, methanol was distilled off from the methanol solution under reduced pressure, the obtained residue was dissolved in chloroform, washed with water, chloroform was distilled off under reduced pressure, and the obtained residue was recrystallized using acetonitrile. The operation was performed to obtain 12.5 g of crystals in the form of grayish white powder (43% yield).
得られた結晶の融点、薄層クロマトグラフィーのRf値、NMR及びマススペクトルデータは、以下のとおりであった。
・mp.144-146℃
・TLC(シリカゲル,クロロホルム/酢酸エチル=9/1):Rf=0.51
・NMR(CDCl3):δ7.2-8.4(m)
・MS
m/z(%):290(70),288(M+,100),253(7),123(4),117(9),89(19)
これらのスペクトルデータから、得られた結晶は、化4で示される2−(2,3−ジクロロフェニル)−4−フェニルイミダゾールであるものと同定した。
The melting point of the obtained crystal, Rf value of thin layer chromatography, NMR and mass spectrum data were as follows.
・ Mp.144-146 ℃
TLC (silica gel, chloroform / ethyl acetate = 9/1): Rf = 0.51
・ NMR (CDCl 3 ): δ7.2-8.4 (m)
・ MS
m / z (%): 290 (70), 288 (M +, 100), 253 (7), 123 (4), 117 (9), 89 (19)
From these spectrum data, the obtained crystal was identified as 2- (2,3-dichlorophenyl) -4-phenylimidazole represented by Chemical formula 4.
〔実施例2〕
<2−(2,4−ジクロロフェニル)−4−フェニルイミダゾールの合成>
2−アセトキシアセトフェノン17.8g(0.1mol)と2,4−ジクロロベンズアルデヒド17.5g(0.1mol)を、150mlのイソプロピルアルコールに溶解させた溶液に、酢酸銅(II)一水和物43.9g(0.22mol)を25%アンモニア水160mlに溶解させた溶液を水冷下で少量ずつ加え、次いで60℃まで1時間、更に80℃まで2.5時間かけて昇温した。
反応終了後、反応液を10℃まで冷却し、析出物を濾取して、水洗後乾燥して暗緑色粉末状物27.3gを得た。この粉末状物をメタノール150mlに懸濁させ、70%水硫化ナトリウム4.3g(0.054mol)を加え1時間加熱還流した。その後、メタノール溶液を冷却して、黒色不溶物を濾去した。引き続き、メタノール溶液からメタノールを減圧下で留去し、得られた残留物をクロロホルムに溶解し、水で洗浄後、クロロホルムを減圧留去し、得られた残留物をアセトニトリルを使用して再結晶操作を行い、黄色粉末状の結晶11.0gを得た(収率38%)。
[Example 2]
<Synthesis of 2- (2,4-dichlorophenyl) -4-phenylimidazole>
To a solution of 17.8 g (0.1 mol) of 2-acetoxyacetophenone and 17.5 g (0.1 mol) of 2,4-dichlorobenzaldehyde in 150 ml of isopropyl alcohol, copper (II) acetate monohydrate 43 A solution of 9.9 g (0.22 mol) dissolved in 160 ml of 25% aqueous ammonia was added little by little under water cooling, and then the temperature was raised to 60 ° C. for 1 hour and further to 80 ° C. over 2.5 hours.
After completion of the reaction, the reaction solution was cooled to 10 ° C., the precipitate was collected by filtration, washed with water and dried to obtain 27.3 g of a dark green powder. This powder was suspended in 150 ml of methanol, 4.3 g (0.054 mol) of 70% sodium hydrosulfide was added, and the mixture was heated to reflux for 1 hour. Thereafter, the methanol solution was cooled, and the black insoluble material was removed by filtration. Subsequently, methanol was distilled off from the methanol solution under reduced pressure, the obtained residue was dissolved in chloroform, washed with water, chloroform was distilled off under reduced pressure, and the obtained residue was recrystallized using acetonitrile. Operation was performed to obtain 11.0 g of yellow powdery crystals (yield 38%).
得られた結晶の融点、薄層クロマトグラフィーのRf値、NMR及びマススペクトルデータは、以下のとおりであった。
・mp.153-156℃
・TLC(シリカゲル,クロロホルム/酢酸エチル=9/1):Rf=0.56
・NMR(CDCl3):δ7.0-8.5(m)
・MS
m/z(%):290(66),288(M+,100),253(5),226(4),185(5),171(4),144(4),
123(7),117(15),100(4),89(19)
これらのスペクトルデータから、得られた結晶は、化5で示される2−(2,4−ジクロロフェニル)−4−フェニルイミダゾールであるものと同定した。
The melting point of the obtained crystal, Rf value of thin layer chromatography, NMR and mass spectrum data were as follows.
・ Mp.153-156 ℃
TLC (silica gel, chloroform / ethyl acetate = 9/1): Rf = 0.56
・ NMR (CDCl 3 ): δ7.0-8.5 (m)
・ MS
m / z (%): 290 (66), 288 (M +, 100), 253 (5), 226 (4), 185 (5), 171 (4), 144 (4),
123 (7), 117 (15), 100 (4), 89 (19)
From these spectral data, the obtained crystal was identified as 2- (2,4-dichlorophenyl) -4-phenylimidazole represented by Chemical formula 5.
〔実施例3〕
<2−(2,3−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールの合成>
1−フェニル−1,2−プロパンジオン14.8g(0.1mol)、2,3−ジクロロベンズアルデヒド17.5g(0.1mol)、酢酸アンモニウム46.2g(0.6mol)を、酢酸100ml中で5時間加熱還流した。反応終了後、得られた反応液を室温まで放冷して、大量の希アンモニア水に注ぎ、析出した固形物を濾取し水洗後、アセトニトリルを使用して再結晶操作を行い、淡緑色粉末状の結晶17.3gを得た(収率57%)。
Example 3
<Synthesis of 2- (2,3-dichlorophenyl) -4-phenyl-5-methylimidazole>
14.8 g (0.1 mol) of 1-phenyl-1,2-propanedione, 17.5 g (0.1 mol) of 2,3-dichlorobenzaldehyde, and 46.2 g (0.6 mol) of ammonium acetate in 100 ml of acetic acid The mixture was heated to reflux for 5 hours. After completion of the reaction, the resulting reaction solution is allowed to cool to room temperature, poured into a large amount of dilute aqueous ammonia, the precipitated solid is collected by filtration, washed with water, and recrystallized using acetonitrile to obtain a pale green powder. 17.3 g of a crystal was obtained (57% yield).
得られた結晶の融点、薄層クロマトグラフィーのRf値、NMR及びマススペクトルデータは、以下のとおりであった。
・mp.183-185℃
・TLC(シリカゲル,クロロホルム/酢酸エチル=9/1):Rf=0.60
・NMR(CDCl3):δ2.5(s,3H),7.2-8.3(m,8H)
・MS
m/z(%):304(69),302(M+,100),267(3),225(2),172(6),151(5),130(35),
103(30),89(20),77(19)
これらのスペクトルデータから、得られた結晶は、化6で示される2−(2,3−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールであるものと同定した。
The melting point of the obtained crystal, Rf value of thin layer chromatography, NMR and mass spectrum data were as follows.
・ Mp.183-185 ℃
・ TLC (silica gel, chloroform / ethyl acetate = 9/1): Rf = 0.60
NMR (CDCl 3 ): δ2.5 (s, 3H), 7.2-8.3 (m, 8H)
・ MS
m / z (%): 304 (69), 302 (M +, 100), 267 (3), 225 (2), 172 (6), 151 (5), 130 (35),
103 (30), 89 (20), 77 (19)
From these spectral data, the obtained crystal was identified as 2- (2,3-dichlorophenyl) -4-phenyl-5-methylimidazole represented by Chemical formula 6.
〔実施例4〕
<2−(2,6−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールの合成>
1−フェニル−1,2−プロパンジオン14.8g(0.1mol)、2,6−ジクロロベンズアルデヒド17.5g(0.1mol)及び酢酸アンモニウム46.2g(0.6mol)を、酢酸100ml中で5時間加熱還流した。反応終了後、得られた反応液を室温まで放冷して、大量の希アンモニア水に注ぎ、析出した固形物を濾取し水洗後乾燥して、30.4gの褐色固形物を得た。この固形物をクロロホルム150mlに溶解し、濃塩酸を加えて析出させた塩酸塩を濾取し、アセトン洗浄後メタノールに溶解し、ソジウムメチラートメタノール溶液を加え脱塩酸し、次いで減圧下でメタノールを留去し、得られた固形物を水洗後乾燥して、乳白色粉末状の結晶18.5gを得た(収率61%)。
Example 4
<Synthesis of 2- (2,6-dichlorophenyl) -4-phenyl-5-methylimidazole>
14.8 g (0.1 mol) of 1-phenyl-1,2-propanedione, 17.5 g (0.1 mol) of 2,6-dichlorobenzaldehyde and 46.2 g (0.6 mol) of ammonium acetate in 100 ml of acetic acid The mixture was heated to reflux for 5 hours. After completion of the reaction, the resulting reaction solution was allowed to cool to room temperature, poured into a large amount of diluted ammonia water, the precipitated solid was collected by filtration, washed with water and dried to obtain 30.4 g of a brown solid. This solid was dissolved in 150 ml of chloroform, the concentrated hydrochloric acid was added to precipitate the hydrochloride, which was collected by filtration, washed with acetone, dissolved in methanol, added with sodium methylate methanol solution to remove hydrochloric acid, and then methanol under reduced pressure. The solid obtained was washed with water and dried to obtain 18.5 g of milky white crystals (yield 61%).
得られた結晶の融点、薄層クロマトグラフィーのRf値、NMR及びマススペクトルデータは、以下のとおりであった。
・mp.185-189℃
・TLC(シリカゲル,クロロホルム/酢酸エチル=9/1):Rf=0.49
・NMR(CDCl3):δ2.5(s,3H),7.2-7.7(m,8H)
・MS
m/z(%):304(66),302(M+,100),267(2),225(2),199(2),172(6),151(4),
130(39),103(29),89(31),77(19)
これらのスペクトルデータから、得られた結晶は、化7で示される2−(2,6−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールであるものと同定した。
The melting point of the obtained crystal, Rf value of thin layer chromatography, NMR and mass spectrum data were as follows.
・ Mp.185-189 ℃
TLC (silica gel, chloroform / ethyl acetate = 9/1): Rf = 0.49
NMR (CDCl 3 ): δ2.5 (s, 3H), 7.2-7.7 (m, 8H)
・ MS
m / z (%): 304 (66), 302 (M +, 100), 267 (2), 225 (2), 199 (2), 172 (6), 151 (4),
130 (39), 103 (29), 89 (31), 77 (19)
From these spectrum data, the obtained crystal was identified as 2- (2,6-dichlorophenyl) -4-phenyl-5-methylimidazole represented by Chemical formula 7.
〔実施例5〕
<2−(3,4−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールの合成>
1−フェニル−1,2−プロパンジオン14.8g(0.1mol)、3,4−ジクロロベンズアルデヒド17.5g(0.1mol)及び酢酸アンモニウム46.2g(0.6mol)を、酢酸100ml中で5時間加熱還流した。反応終了後、得られた反応液を室温まで放冷して、大量の希アンモニア水に注ぎ、析出した固形物を濾取し水洗後、メタノールを使用して再結晶操作を行い、淡黄色粉末状の結晶18.8gを得た(収率62%)。
Example 5
<Synthesis of 2- (3,4-dichlorophenyl) -4-phenyl-5-methylimidazole>
14.8 g (0.1 mol) of 1-phenyl-1,2-propanedione, 17.5 g (0.1 mol) of 3,4-dichlorobenzaldehyde and 46.2 g (0.6 mol) of ammonium acetate in 100 ml of acetic acid The mixture was heated to reflux for 5 hours. After completion of the reaction, the resulting reaction solution is allowed to cool to room temperature, poured into a large amount of dilute aqueous ammonia, the precipitated solid is collected by filtration, washed with water, and recrystallized using methanol to obtain a pale yellow powder. 18.8 g of crystals were obtained (yield 62%).
得られた結晶の融点、薄層クロマトグラフィーのRf値、NMR及びマススペクトルデータは、以下のとおりであった。
・mp.172-175℃
・TLC(シリカゲル,クロロホルム/酢酸エチル=9/1):Rf=0.60
・NMR(CDCl3):δ2.5(s,3H),7.3-7.9(m,8H)
・MS
m/z(%):304(63),302(M+,100),267(3),225(3),199(2),172(7),151(4),
130(25),103(20),89(12),77(15)
これらのスペクトルデータから、得られた結晶は、化8で示される2−(3,4−ジクロロフェニル)−4−フェニル−5−メチルイミダゾールであるものと同定した。
The melting point of the obtained crystal, Rf value of thin layer chromatography, NMR and mass spectrum data were as follows.
・ Mp.172-175 ℃
・ TLC (silica gel, chloroform / ethyl acetate = 9/1): Rf = 0.60
NMR (CDCl 3 ): δ2.5 (s, 3H), 7.3-7.9 (m, 8H)
・ MS
m / z (%): 304 (63), 302 (M +, 100), 267 (3), 225 (3), 199 (2), 172 (7), 151 (4),
130 (25), 103 (20), 89 (12), 77 (15)
From these spectral data, the obtained crystal was identified as 2- (3,4-dichlorophenyl) -4-phenyl-5-methylimidazole represented by Chemical Formula 8.
Claims (1)
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004022241A JP4245490B2 (en) | 2003-03-19 | 2004-01-29 | 2- (Dichlorophenyl) -4-phenylimidazole compound |
| EP04721686A EP1605078B1 (en) | 2003-03-19 | 2004-03-18 | Soldering process using imidazole compound |
| AT04721686T ATE474944T1 (en) | 2003-03-19 | 2004-03-18 | SOLDERING PROCESS USING AN IMIDAZOLE COMPOUND |
| US10/548,544 US7661577B2 (en) | 2003-03-19 | 2004-03-18 | Imidazole compound and use thereof |
| TW093107315A TW200512196A (en) | 2003-03-19 | 2004-03-18 | Novel imidazole compound and usage thereof |
| CN200480007175XA CN1761773B (en) | 2003-03-19 | 2004-03-18 | Novel imidazole compound and usage thereof |
| PCT/JP2004/003658 WO2004083487A1 (en) | 2003-03-19 | 2004-03-18 | Novel imidazole compound and usage thereof |
| DE602004028223T DE602004028223D1 (en) | 2003-03-19 | 2004-03-18 | SOLDERING PROCEDURE USING AN IMIDAZOL CONNECTION |
| KR1020057017354A KR101098506B1 (en) | 2003-03-19 | 2005-09-15 | Novel imidazole compound and usage thereof |
| HK06110659.5A HK1090098A1 (en) | 2003-03-19 | 2006-09-25 | Novel imidazole compound and usage thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003075030 | 2003-03-19 | ||
| JP2004022241A JP4245490B2 (en) | 2003-03-19 | 2004-01-29 | 2- (Dichlorophenyl) -4-phenylimidazole compound |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004300137A JP2004300137A (en) | 2004-10-28 |
| JP4245490B2 true JP4245490B2 (en) | 2009-03-25 |
Family
ID=33421842
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004022241A Expired - Fee Related JP4245490B2 (en) | 2003-03-19 | 2004-01-29 | 2- (Dichlorophenyl) -4-phenylimidazole compound |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4245490B2 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150039551A (en) * | 2013-10-02 | 2015-04-10 | 제이에스알 가부시끼가이샤 | Curable composition, cured film and process for forming the same, and compound |
| JP2016029153A (en) * | 2014-07-24 | 2016-03-03 | 日本合成化学工業株式会社 | Curing agent for anion curable compound, curable composition, and cured product |
| JP2016029152A (en) * | 2014-07-24 | 2016-03-03 | 日本合成化学工業株式会社 | Curing agent for anion curable compound, curable composition, and cured product |
-
2004
- 2004-01-29 JP JP2004022241A patent/JP4245490B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004300137A (en) | 2004-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5016575B2 (en) | A novel synthesis of strontium ranelate and its hydrates | |
| JPH0460990B2 (en) | ||
| JP4245490B2 (en) | 2- (Dichlorophenyl) -4-phenylimidazole compound | |
| EP1440970B1 (en) | Benzenesulfonic acid salt of 1-(6-halogeno-2-benzothiazolyl)ethylamine | |
| EP2118058B9 (en) | Method for the preparation of 5-benzyloxy-2-(4-benzyloxphenyl)-3-methyl-1h-indole | |
| JP4770826B2 (en) | Method for producing 2-oxindole derivatives | |
| JPWO2006001537A1 (en) | Method for producing hydrazone compound | |
| US6790977B2 (en) | Process for preparing 3,4-dihydroxy-benzonitrile | |
| JPH0458468B2 (en) | ||
| JP2004277386A (en) | 2-(dichlorophenyl)-4-methyl-5-phenylimidazole compound | |
| JPH10195319A (en) | Production of polymethinecyanine compound | |
| JP4305747B2 (en) | 2-Phenyl-4- (dichlorophenyl) imidazole compound | |
| JPH1160552A (en) | Production of thiobenzamide derivative | |
| JP4194984B2 (en) | Phenylnaphthylimidazole compound | |
| EP1188752B1 (en) | Process for producing substituted alkylamine derivative | |
| US6372913B1 (en) | Process for preparing 2-substituted 5-formylthiazoles | |
| JP2515122B2 (en) | Method for producing anthranilic acid ester | |
| US6433176B1 (en) | Method for making 8-hydroxyjulolidine compound | |
| JP4925518B2 (en) | Method for producing substituted alkylamine derivative | |
| KR100408431B1 (en) | Process for the preparation of 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1h-imidazol-1-yl)methyl]-4h-carbazol-4-one or pharmaceutically acceptable salts thereof | |
| JP4973210B2 (en) | New synthesis method | |
| JP4299895B2 (en) | Benzo [b] thiophene-2,3-dione-2-oxime derivative and process for producing the same, and process for producing 1,2,3-benzothiadiazole-7-carboxylic acid using the same | |
| JPH07252234A (en) | Production of 2-cyanoimidazole type compound | |
| WO2010074110A1 (en) | Method for producing cyclic aminobenzoic acid derivative | |
| KR100229175B1 (en) | Process for preparation of cephem derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050303 |
|
| A25B | Request for examination refused [due to the absence of examination request for another application deemed to be identical] |
Free format text: JAPANESE INTERMEDIATE CODE: A2522 Effective date: 20081201 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20081215 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090106 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4245490 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120116 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120116 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130116 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130116 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140116 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |