JP4028913B2 - N−シクロプロピルアニリン類の製造方法とそのための中間体 - Google Patents
N−シクロプロピルアニリン類の製造方法とそのための中間体 Download PDFInfo
- Publication number
- JP4028913B2 JP4028913B2 JP21251397A JP21251397A JP4028913B2 JP 4028913 B2 JP4028913 B2 JP 4028913B2 JP 21251397 A JP21251397 A JP 21251397A JP 21251397 A JP21251397 A JP 21251397A JP 4028913 B2 JP4028913 B2 JP 4028913B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- difluoro
- general formula
- cyclopropyl
- alkyl group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *c(c(F)c(cc1)F)c1N Chemical compound *c(c(F)c(cc1)F)c1N 0.000 description 6
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/30—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and unsaturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/04—Preparation of compounds containing amino groups bound to a carbon skeleton by substitution of functional groups by amino groups
- C07C209/14—Preparation of compounds containing amino groups bound to a carbon skeleton by substitution of functional groups by amino groups by substitution of hydroxy groups or of etherified or esterified hydroxy groups
- C07C209/18—Preparation of compounds containing amino groups bound to a carbon skeleton by substitution of functional groups by amino groups by substitution of hydroxy groups or of etherified or esterified hydroxy groups with formation of amino groups bound to carbon atoms of six-membered aromatic rings or from amines having nitrogen atoms bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/43—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C211/44—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to only one six-membered aromatic ring
- C07C211/52—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to only one six-membered aromatic ring the carbon skeleton being further substituted by halogen atoms or by nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/52—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups or amino groups bound to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/78—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton
- C07C217/80—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of non-condensed six-membered aromatic rings
- C07C217/82—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of non-condensed six-membered aromatic rings of the same non-condensed six-membered aromatic ring
- C07C217/84—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of non-condensed six-membered aromatic rings of the same non-condensed six-membered aromatic ring the oxygen atom of at least one of the etherified hydroxy groups being further bound to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
【発明の属する技術分野】
本発明は、キノロンカルボン酸系合成抗菌剤の製造に有用なN−シクロプロピルアニリン類の製造方法とそのための中間体に関するものである。
【0002】
【従来の技術】
キノロンカルボン酸類、中でも1位にシクロプロピル基を、6位にフッ素原子を、又、8位にアルキル基(特開昭62−215572号公報、特開昭63−264461号公報を参照)、アルコキシ基(特開昭63−198664号公報、特開昭62−252772号公報を参照)又はフッ素置換メトキシ基(特開平5−255183号公報)をそれぞれ有するキノロンカルボン酸類は、優れた抗菌活性を有することが知られている。
【0003】
このようなキノロンカルボン酸類の製法としては、従来より種々の方法が提案されているが、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有するキノロンカルボン酸類を、N−シクロプロピル−2−置換−3,4−ジフルオロアニリン類、更にはこれから誘導されるアニリノメチレンマロン酸類を経て製造する方法は知られていない。
【0004】
一方、これまでに窒素原子にシクロプロピル基が結合したアニリン類の製造方法として、シアノ化水素ホウ素ナトリウムの存在下、1−エトキシ−1−トリメチルシリロキシシクロプロパンとアニリンを反応させる方法が報告されている〔Tetrahedron Letters, Vol. 36, 7399 (1995)〕。しかしながら、この方法には、N,N−ジシクロプロピル体が多量に副生してしまい、目的物のN−シクロプロピル体の収率が低いという問題があった。
【0005】
又、1−エトキシ−1−トリメチルシリロキシシクロプロパンに対し、三臭化リンを反応させ、まず1−エトキシシクロプロパンの反応性誘導体である1−ブロモ−1−エトキシシクロプロパンを調製し、これを更にアニリン類と反応させてN−(1−エトキシシクロプロパン)体を得る方法、及び、N−(1−エトキシシクロプロパン)体を更に水素化ホウ素ナトリウム及び三フッ化ホウ素エーテル錯体と反応させて、次の目的物を得る方法が報告されている〔J.Chem.Soc.Chem.Comm., 897, (1987)〕。しかし、この方法には、1−ブロモ−1−エトキシシクロプロパンの調製工程が必要な上、1−ブロモ−1−エトキシシクロプロパンは熱的に不安定であり、工業的使用には不都合であるという問題があった。
【0006】
従って、窒素原子にシクロプロピル基が結合したアニリン類のこれまでの製造方法を、前述のキノロンカルボン酸類の中間体として有用なN−シクロプロピル−2−置換−3,4−ジフルオロアニリン類の製造に適用することは、収率や操作性から工業的に見て不利であり、決して満足できるものとは言い難かった。
【0007】
【発明が解決しようとする課題】
本発明は、工業的に安価で入手の容易な原料から、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有し、合成抗菌剤として有用なキノロンカルボン酸類の製造において重要な中間体となる、N−シクロプロピルアニリン類の製造方法とそのための中間体を提供することを課題としてなされたものである。
【0008】
【課題を解決するための手段】
本発明の発明者等は、合成抗菌剤として優れた生物活性を有する、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有するキノロンカルボン酸類の製造のための原料の、工業的に有利な製造方法及びその中間体について種々検討した結果、工業的に安価で入手の容易な3,4−ジフルオロ−2−置換アニリン類を出発原料として用い、これに1−アルコキシ−1−トリアルキルシリロキシシクロプロパンを、従来行われていたブロム化のような活性化をすることなく直接作用させることによって、上記キノロンカルボン酸類の製造における重要な中間体であるN−アルコキシシクロプロピルアニリン類を収率よく製造できること、この方法で得られたN−アルコキシシクロプロピルアニリン類は、これを更に還元した後にアルコキシメチレンマロン酸ジアルキルエステルと反応させることによりアニリノメチレンマロン酸類に変換でき、このアニリノメチレンマロン酸類が、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有するキノロンカルボン酸類の製造において有用であるばかりでなく、その中間体を含めて新規化合物であることを見い出し、本発明を完成した。
【0009】
即ち、本発明は、下記〔1〕〜〔5〕項に記載の発明を提供することによって上記課題を解決したものである。
【0010】
〔1〕アルコール系溶媒中、酸の存在下、一般式(1)
【化16】
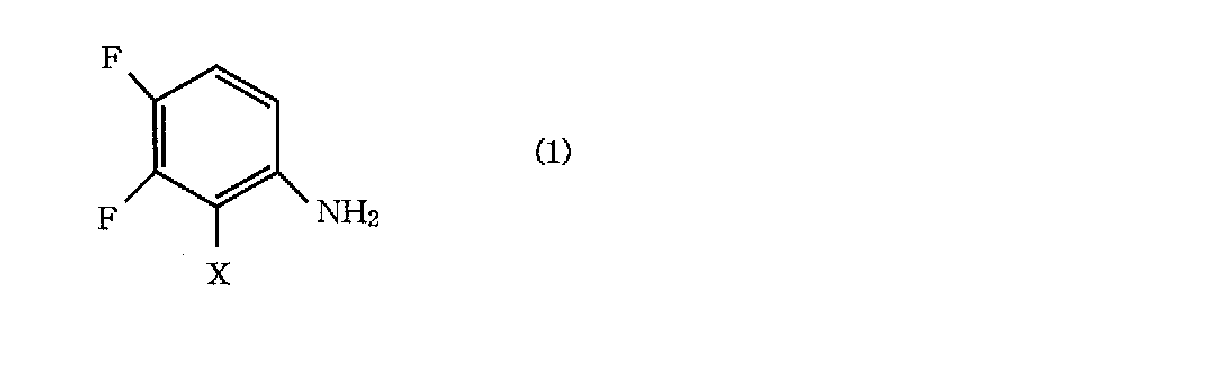
【化17】
【化18】
【0011】
〔2〕アルコール系溶媒中、酸の存在下、一般式(1)
【化19】
【化20】
【化21】
【化22】
【0012】
〔3〕アルコール系溶媒中、酸の存在下、一般式(1)
【化23】
【化24】
【化25】
【化26】
【化27】
【0013】
〔4〕一般式(3)
【化28】
【0014】
〔5〕一般式(6)
【化29】
【化30】
【0015】
【発明の実施の形態】
以下に、本発明を詳細に説明する。
【0016】
本発明の第1の方法においては、一般式(1)で表される3,4−ジフルオロ−2−置換アニリン類と、一般式(2)で表される1−アルコキシ−1−トリアルキルシリロキシシクロプロパンを、アルコール系溶媒中、酸の存在下で反応させることにより、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類を得る。
【0017】
上記反応で原料として使用する3,4−ジフルオロ−2−置換アニリン類は、一般式(1)で示される化合物であればよい。式中のXはアルキル基、アルコキシ基又はフッ素置換メトキシ基を示しており、これらのうちのアルキル基としては、炭素数1〜6(以下、このような置換基等の炭素数については必要に応じ「C1 〜 6」のように略記する。)の直鎖又は分岐鎖のアルキル基であればよく、具体的にはメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、n−ペンチル基及びn−へキシル基等を例示することができ、一般式(1)で表され、Xとしてアルキル基を有するこのような3,4−ジフルオロ−2−アルキルアニリン類としては、例えば3,4−ジフルオロ−2−メチルアニリン及び3,4−ジフルオロ−2−エチルアニリンを挙げることができる。
【0018】
尚、上記3,4−ジフルオロ−2−アルキルアニリン類は、テトラヘドロン(Tetrahedron)、第48巻、第7373頁(1992)に記載の方法等により、3,4−ジフルオロアニリンから容易に製造することができる。
【0019】
式中のXで示されるアルコキシ基としては、炭素数1〜6の直鎖又は分岐鎖のアルコキシ基であればよく、具体的にはメトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、n−ペンチルオキシ基、n−へキシルオキシ基等を例示することができ、一般式(1)で表され、Xとしてアルコキシ基を有するこのような3,4−ジフルオロ−2−アルコキシアニリン類としては、例えば3,4−ジフルオロ−2−メトキシアニリン、3,4−ジフルオロ−2−エトキシアニリン等を挙げることができる。
【0020】
尚、上記3,4−ジフルオロ−2−アルコキシアニリン類は、特開平8−208617号公報記載の方法等により、2,3−ジフルオロ−6−ニトロフェノールから容易に製造することができる。
【0021】
更に、式中のXで示されるフッ素置換メトキシ基としては、フルオロメトキシ基、ジフルオロメトキシ基、トリフルオロメトキシ基であればよく、一般式(1)で表され、Xとしてフッ素置換メトキシ基を有するこのような3,4−ジフルオロ−2−フッ素置換メトキシアニリン類としては、3,4−ジフルオロ−2−フルオロメトキシアニリン、3,4−ジフルオロ−2−ジフルオロメトキシアニリン、3,4−ジフルオロ−2−トリフルオロメトキシアニリンを挙げることができる。
【0022】
上記の3,4−ジフルオロ−2−フッ素置換メトキシアニリン類は、新規化合物であり、このものは、2,3−ジフルオロ−6−ニトロフェノールと、例えばクロロジフルオロ酢酸ナトリウムとを、N,N−ジメチルホルムアミド中、ヨウ化ナトリウム及び炭酸ナトリウムの存在下で反応させる等して、やはり新規化合物である3,4−ジフルオロ−2−フッ素置換メトキシ−ニトロベンゼンとした後、更にこの3,4−ジフルオロ−2−フッ素置換メトキシ−ニトロベンゼンを硫酸存在下で鉄粉等を用いて還元することにより製造できる(後述する参考例3参照)。
【0023】
上記反応におけるもう一方の原料である、一般式(2)で表される1−アルコキシ−1−トリアルキルシリロキシシクロプロパンとしては、1位に〔(C1 〜 6)アルキル〕オキシ基、例えばフェニル基等のアリール基で置換されていても良いアルケニルオキシ基或いはアラルキルオキシ基を(式中のOR1に相当する。)有し、更に1位にトリ〔(C1 〜 6)アルキル〕シリルオキシ基を(式中のOSiR2R3R4に相当する。)有するシクロプロパンであればよい。
【0024】
上記〔(C1 〜 6)アルキル〕オキシ基としては、例えばメトキシ基、エトキシ基、イソプロポキシ基等を、又、上記フェニル基等のアリール基で置換されていても良いアルケニルオキシ基としては、例えば(3−フェニルアリル)オキシ基を、上記アラルキルオキシ基としては、例えばベンジルオキシ基等それぞれ例示することができ、更に、上記トリ〔(C1 〜 6)アルキル〕シリルオキシ基としては、例えばトリメチルシリルオキシ基やジメチル−t−ブチルオキシ基を例示することができる。
【0025】
このような一般式(2)で表される1−アルコキシ−1−トリアルキルシリロキシシクロプロパンとしては、具体的には1−エトキシ−1−トリメチルシリロキシシクロプロパン、1−メトキシ−1−トリメチルシリロキシシクロプロパン、1−イソプロポキシ−1−トリメチルシリロキシシクロプロパン、1−イソプロポキシ−1−ジメチル−t−ブチルシリロキシシクロプロパン、1−(3−フェニルアリル)オキシ−1−トリメチルシリロキシシクロプロパン、1−(3−フェニルアリル)オキシ−1−ジメチル−t−ブチルシリロキシシクロプロパン、1−ベンジルオキシ−1−トリメチルシリロキシシクロプロパン等を挙げることができる。
【0026】
上記反応における一般式(2)で表される1−アルコキシ−1−トリアルキルシリロキシシクロプロパンの使用量としては、一般式(1)で表される3,4−ジフルオロ−2−置換アニリン類に対し、1.0〜1.5倍モル、好ましくは1.0〜1.3倍モルの範囲を例示することができる。
【0027】
尚、上記1−アルコキシ−1−トリアルキルシリロキシシクロプロパンは、オーガニックシンセシス(Organic Synthesis)、第63巻、147頁(1985)に記載されているように、入手の容易なβ−クロロプロピオン酸エチルに対し、クロロトリメチルシランの存在下、金属ナトリウムを反応させることにより、或いは、テトラヘドロンレター(Tetrahedron Letter)、第33巻、785頁(1992)やテトラヘドロンレター(Tetrahedron Letter)、第24巻、1251頁(1983)、シンセシス(Synthesis)、第1巻、第58頁(1982)等に記載の方法により製造することができる。
【0028】
又、上記反応に用いるアルコール系溶媒としては、C1〜6の直鎖、分岐鎖又は脂環構造を有するアルコールであればよく、具体的にはメタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、t−ブタノール、n−へキサノール、シクロペンタノール、シクロヘキサノール等を例示することができる。
【0029】
上記アルコール系溶媒の使用量としては、一般式(1)で表される3,4−ジフルオロ−2−置換アニリン類1モルに対し、50ml〜4000ml、好ましくは200ml〜3000mlという範囲を例示することができる。
【0030】
更に、上記反応に用いる酸としては、有機酸又は無機酸を用いることができ、有機酸としては、例えば蟻酸、酢酸、プロピオン酸、酪酸等を包含する脂肪族カルボン酸;安息香酸、トルイル酸、フタル酸等を包含する芳香族カルボン酸;メタンスルホン酸等を包含する脂肪族スルホン酸;ベンゼンスルホン酸、トルエンスルホン酸等を包含する芳香族スルホン酸等を例示することができる。
【0031】
又、無機酸としては、塩酸、硫酸、燐酸等を例示でき、中でも好ましい酸として、脂肪族カルボン酸、特に酢酸及び蟻酸を挙げることができ、特に酢酸が好ましい。
【0032】
上記酸の使用量としては、一般式(1)で表される3,4−ジフルオロ−2−置換アニリン類に対し、0.005〜50倍モル、好ましくは0.1〜20倍モルという範囲を例示することができる。
【0033】
上記反応の反応温度としては、通常は室温から溶媒の沸点までの間で設定すればよいが、好ましくは20〜90℃であり、当該反応は、常圧下で原料、溶媒及び酸を混合し、所定の温度で撹拌するのみで良く、通常加圧する必要はない。尚、反応時間は通常0.5〜10時間の範囲である。
【0034】
上記反応では、生成したN−アルコキシシクロプロピルアニリン類のシクロプロピル環の1位のアルコキシ基が、用いたアルコール系溶媒との交換反応によって、原料として用いた1−アルコキシ−1−トリアルキルシリロキシシクロプロパン由来のアルコキシ基から、アルコール系溶媒由来のアルコキシ基に置き換わっている化合物が生成する場合があり、そして、このようなアルコキシ基の交換反応の進行の度合いによっては、反応系内で生成する一般式(3)で表されるN−アルコキシシクロプロピルアニリン類が複数種となる場合があるが、これらは混合物のまま次の第2の方法又は第3の方法に使用してもよく、精留する等の適当な方法で各々単離することもできる。
【0035】
次いで第2の方法及び第3の方法について記載する。
【0036】
本発明の第2の方法及び第3の方法では、まず前記本発明の第1の方法により得られた一般式(3)で表されるN−アルコキシシクロプロピルアニリン類を還元し、脱アルコキシ化して一般式(4)で表されるN−シクロプロピル−3,4−ジフルオロアニリン類を得る(本発明の第2の方法は、この工程までで構成される。)。
【0037】
この還元反応では、上述したように、本発明の第1の方法により一般式(3)で表されるN−アルコキシシクロプロピルアニリン類の複数種が生成した場合でも、各々のN−アルコキシシクロプロピルアニリン類を単離することなく、そのまま原料として用いて何ら差し支えない。
【0038】
上記還元の方法としては、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類に対し、溶媒及び三フッ化ホウ素又は金属ハロゲン化物の存在下、水素化ホウ素ナトリウムを作用させる方法を例示することができる。
【0039】
上記還元反応に用いる三フッ化ホウ素は、気体のまま用いることもできるが、三フッ化ホウ素エーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体等の三フッ化ホウ素を錯化した化合物を用いることが、実用上の問題もなく操作上も便利である。
【0040】
上記還元反応に用いる金属ハロゲン化物としては、塩化アルミニウム(AlCl3)、塩化亜鉛(ZnCl2)、塩化鉄(III)(FeCl3)、塩化コバルト(II)(CoCl2)、塩化白金(II)(PtCl2)、塩化ルテニウム(III)(RuCl3)、塩化ロジウム(III)(RhCl3)、塩化パラジウム(II)(PdCl2)、塩化ジルコニウム(IV)(ZrCl4)、塩化カルシウム(CaCl2)及び塩化リチウム(LiCl)等を例示することができる。
【0041】
尚、上記還元反応では、特に塩化アルミニウム、三フッ化ホウ素エーテル錯体又は三フッ化ホウ素テトラヒドロフラン錯体の使用が好ましい。
【0042】
上記還元反応における水素化ホウ素ナトリウムの使用量としては、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類に対し1.0〜4.0倍モル、好ましくは1.0〜2.5倍モルという範囲を、又、併せて使用する金属ハロゲン化物の使用量としては、塩化アルミニウムの場合で一般式(3)で表されるN−アルコキシシクロプロピルアニリン類に対し0.1〜1.0倍モル、好ましくは0.3〜0.7倍モルという範囲を、更に、三フッ化ホウ素エーテル錯体又は三フッ化ホウ素テトラヒドロフラン錯体を用いる場合の使用量としては、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類に対し1.0〜5.5倍モル、好ましくは1.3〜3.3倍モルという範囲を、それぞれ例示することができる。
【0043】
上記還元反応に用いる溶媒としては、例えばテトラヒドロフラン、ジエチルエーテル、ジメトキシエタン、ジエチレングリコールジメチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル等のエーテル系溶媒や、ベンゼン、トルエン、キシレン等の炭化水素系溶媒等、水素化ホウ素ナトリウム、金属ハロゲン化物及び三フッ化ホウ素エーテル錯体或いは三フッ化ホウ素テトラヒドロフラン錯体に対し不活性なものであれば特に制限されることなく使用することができ、又、そのような溶媒の中から適宜に選択し、混合して使用することもできるが、中でもエーテル系溶媒の使用が好ましく、三フッ化ホウ素エーテル錯体又は三フッ化ホウ素テトラヒドロフラン錯体を用いる場合は、生成するボランの安定化が期待できる点で特にテトラヒドロフランが好ましい。
【0044】
上記溶媒の使用量としては、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類1モルに対し50ml〜3000ml、好ましくは200ml〜2000mlの範囲を例示することができる。
【0045】
上記還元反応の反応温度としては、−20℃〜溶媒の沸点、好ましくは−5〜80℃の範囲が好ましく、反応は常圧下で撹拌するのみで進行するので、加圧装置は必要としない。尚、反応時間は通常2〜30時間、好ましくは4〜10時間である。
【0046】
又、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類のその他の還元方法として、水素化アルミニウムリチウムを用いる方法や、接触還元による方法等も適用することが可能である。
【0047】
本発明の第2の方法は、上記本発明の第1の方法及び上記還元反応工程によって構成されるものである。
【0048】
又、本発明の第3の方法は、上記本発明の第1の方法及び第2の方法を経て得られ、一般式(4)で表されるN−シクロプロピル−3,4−ジフルオロアニリン類に、更にアルコキシメチレンマロン酸ジアルキルエステルを反応させることにより、一般式(5)で表されるアニリノメチレンマロン酸類を得るものである。
【0049】
このアルコキシメチレンマロン酸ジアルキルエステルのメチレン炭素に結合するアルコキシ部位のアルコキシ基としては、〔(C1 〜 6)アルキル〕オキシ基、具体的には例えばメトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、イソブトキシ基、ヘキシルオキシ基等を挙げることができ、ジアルキルエステル部位のアルキル基としては、(C1 〜 6)アルキル基、具体的には例えばメチル基、エチル基、プロピル基、イソプロピル基、ブチル基、sec−ブチル基、t−ブチル基等を挙げることができる。
【0050】
このようなアルコキシメチレンマロン酸ジアルキルエステルとしては、例えばエトキシメチレンマロン酸ジエチル、メトキシメチレンマロン酸ジメチル、メトキシメチレンマロン酸ジエチル、プロポキシメチレンマロン酸ジエチル、ブトキシメチレンマロン酸ジエチル、エトキシメチレンマロン酸ジメチル、メトキシメチレンマロン酸ジプロピル、プロポキシメチレンマロン酸ジメチル、ブトキシメチレンマロン酸ジブチル等を挙げることができる。
【0051】
上記反応は、一般式(4)で表されるN−シクロプロピル−3,4−ジフルオロアニリン類に対し、アルコキシメチレンマロン酸ジアルキルエステルを1.0〜1.5倍モル、好ましくは1.0〜1.1倍モルの範囲で混合し、単に加熱撹拌して脱エタノールするのみで進行し、容易に一般式(5)で表されるアニリノメチレンマロン酸類を製造することができる。
【0052】
上記反応においては、特に溶媒は必要無く、通常は無溶媒で実施できるが、これは反応に不活性な溶媒の使用を制限するものではなく、この反応において使用し得る不活性な溶媒としては、例えば芳香族炭化水素溶媒、具体的にはクロロベンゼン、o−ジクロロベンゼン、トルエン、キシレン等を例示することができ、その使用量は一般式(4)で表されるN−シクロプロピル−3,4−ジフルオロアニリン類1モルに対し2000ml以下、好ましくは500ml以下という範囲を例示することができる。
【0053】
上記反応の反応温度としては100〜200℃、好ましくは150〜170℃という範囲が好ましく、又、反応時間は通常2〜20時間である。
【0054】
尚、上記本発明の第3の方法により得られたアニリノメチレンマロン酸類(5)は、例えばポリリン酸、ポリリン酸エステル等の脱アルコール剤と反応させて分子内脱アルコール反応をさせる等、例えば特開平2−45469号公報記載の方法等を利用することにより、容易に目的とするキノロンカルボン酸の製造中間体に導くことができる(後述する参考例1、同2参照)。
【0055】
又、本発明の第1の方法及び第3の方法によって得られる、一般式(3)で表されるN−アルコキシシクロプロピルアニリン類及び一般式(6)で表されるN−シクロプロピル−2−置換アニリン類は、いずれも文献未記載の新規化合物であり、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有する、合成抗菌剤として有用なキノロンカルボン酸の製造中間体として、極めて有用である。
【0056】
【発明の効果】
本発明により、原料として工業的に安価で入手の容易な3,4−ジフルオロ−2−置換アニリン類を出発原料として用いる、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有する、合成抗菌剤として有用なキノロンカルボン酸類の製造原料であるN−アルコキシシクロプロピルアニリン類の高収率で工業的な製造方法、及び、該方法を経由して製造される、1位にシクロプロピル基を、6位にフッ素原子を、8位にアルキル基、アルコキシ基又はフッ素置換メトキシ基をそれぞれ有するキノロンカルボン酸類の重要中間体並びにその製造方法が提供される。
【0057】
【実施例】
次に、本発明化合物及びその製造方法について、実施例により具体的に説明する。
【0058】
実施例1(第1の方法)
還流冷却器、温度計及び磁気撹拌子を備えた50mlの三ツ口フラスコに、3,4−ジフルオロ−2−メトキシアニリン1.59g(10mmol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン1.96g(11.2mmol)、酢酸8.0g(133mmol)及びメタノール20mlを仕込み、窒素気流下、65℃で4時間加熱還流した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータにより濃縮し、更に残渣をクーゲルロールを用いて減圧蒸留することにより、N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン2.0gを得た(収率87.2%)。
[N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンの物性]
沸点:175〜185℃(クーゲルロール外温)/13mmHg
確認データ
GC−MS(m/e):229[M+]
60MHz 1H−NMR(CDCl3)δ値:0.8〜1.2(4H,m,CH2CH2)、3.25(3H,s,OCH3)、3.92(3H,d,J=2.0Hz,芳香核OCH3)、4.9〜5.3(1H,brs,NH)、6.5〜7.4(2H,m,芳香核水素)
【0059】
実施例2(第1の方法)
還流冷却器、温度計及び撹拌機を備えた200mlの四ツ口フラスコに、3,4−ジフルオロ−2−メトキシアニリン7.96g(0.05mol)、酢酸40g(0.67mol)及びエタノール100mlを仕込み、室温で撹拌しながら1−エトキシ−1−トリメチルシリロキシシクロプロパン9.24g(0.053mol)を滴下した。その後、オイルバスで昇温し、窒素気流下、80℃で加熱還流しながら、3時間撹拌した。得られた反応液をアスピレーターで減圧下、ロータリエバポレータにより濃縮し、最後に真空ポンプで残存する酢酸を除去し、油状物質11.5gを得た。次いでこのものを減圧下で蒸留して、N−(1−エトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンを10.1g得た(収率83.0%)。
[N−(1−エトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンの物性]
沸点:92〜96℃/3mmHg
確認データ
GC−MS(m/e):243[M+]
60MHz 1H−NMR(CDCl3)δ値:0.8〜1.1(4H,m,CH2CH2)、1.11(3H,t,J=7Hz,CH3)、3.53(2H,q,J=7Hz,CH2)、3.92(3H,d,J=2Hz,OCH3)、4.9〜5.4(1H,brs,NH)、6.7〜6.9(2H,m,芳香核水素)
【0060】
実施例3(第1の方法)
還流冷却器、温度計及び撹拌機を備えた200mlの四ツ口フラスコに、3,4−ジフルオロ−2−メトキシアニリン7.96g(0.05mol)、酢酸40g(0.67mol)及びイソプロピルアルコール100mlを仕込み、室温で撹拌しながら、1−エトキシ−1−トリメチルシリロキシシクロプロパン9.24g(0.053mol)を滴下した。その後オイルバスにより昇温し、窒素気流下、85℃で4時間、還流下で加熱撹拌した。得られた反応液をアスピレータで減圧下、ロータリーエバポレータにより濃縮し、最後に真空ポンプで残存する酢酸を除去し、褐色の油状物質11.3gを得た。更に減圧下で蒸留し、7.4gの無色の液体を得た(沸点93〜102℃/3mmHg)。この液体をガスクロマトグラフィで分析したところ、全面積%で10.4%のN−(1−エトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンと85.7%のN−〔1−(2−プロピル)オキシ〕シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンが含まれていた。
確認データ
GC−MS(m/e)
1−エトキシ体:243[M+]、212[M+−(OCH3)],184[M+−(OCH3)−(CH2=CH2)]
1−(2−プロピル)オキシ体:257[M+],226[M+−(OCH3)],184(M+−(OCH3)−(CH2=CHCH3)]
【0061】
実施例4(第1の方法)
還流冷却器、温度計及び撹拌機を備えた200mlの四ツ口フラスコに3,4−ジフルオロ−2−メトキシアニリン7.96g(0.05mol)、蟻酸20g(0.43mol)及びメタノール100mlを仕込み、室温で撹拝しながら、1−エトキシ−1−トリメチルシリロキシシクロプロパン9.24g(0.053mol)を滴下した。その後、オイルバスで昇温し、窒素気流下、55℃で4.5時間、還流下で加熱撹拌した。反応液をガスクロマトグラフィーで分析したところ、N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンが84.5%生成していた。
【0062】
実施例5(第2の方法)
還流冷却器、温度計及び撹拌機を備えた200mlの四ツ口フラスコに、3,4−ジフルオロ−2−メトキシアニリン7.96g(0.05mol)、酢酸12g(0.2mol)及びメタノール50mlを仕込み、室温で撹拌しながら、1−エトキシ−1−トリメチルシリロキシシクロプロパン10.0g(0.0574mol)を滴下した。その後、オイルバスにより昇温し、窒素気流下、67℃で3時間、加熱還流下で撹拌した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータで濃縮し、最後に真空ポンプにより残存する酢酸を除去し、粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンを11.52g得た(粗収率100%、純度90%)。
【0063】
次いで、還流冷却器、温度計及び撹拌機を備えた200mlの四ツ口フラスコに、水素化ホウ素ナトリウム3.78g(0.10mol)、無水テトラヒドロフラン50mlを仕込み、窒素気流下で撹拌しながら氷冷し、5℃まで冷却した。その後、三フッ化ホウ素エーテル錯体14.19g(0.10mol)を同温度で滴下し、滴下終了後、5℃で45分間撹拌した。先に得た粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン11.52gを、5〜6℃で40分間にわたって滴下した後、同温度で1.5時間撹拌し、更に20℃に昇温して5時間撹拌した。その後、オイルバス上で加熱し、常圧下でテトラヒドロフランを留出させ、105mlを回収した。蒸留残渣を水200ml中に滴下した後、エーテル200mlで2回抽出した。エーテル層を水で洗浄した後、無水硫酸ナトリウムで乾燥し、ロータリーエバポレータでエーテルを留去し、更に残渣を減圧蒸留して、N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンを8.83g得た(収率88.6%、純度96.3%)。
[N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンの物性]
沸点:78〜79℃/3mmHg
確認データ
GC−MS(m/e):199[M+]、168[M+−(OCH3)]
60MHz 1H−NMR(CDCl3)δ値:0.5〜0.8(4H,m,CH2CH2)、2.1〜2.6(1H,m,CH)、4.3〜4.7(1H,brs,NH)、6.4〜7.3(2H,m,芳香核水素)
【0064】
尚、上記に記載した還元工程における原料として、実施例1乃至4で得られた化合物を(必要に応じ単離して)使用しても、この実施例5の還元工程で得られたN−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンと同一の化合物を得ることができる。
【0065】
実施例6(第3の方法)
還流冷却器、温度計及び撹拌機を備えた500mlの四ツ口フラスコに、3,4−ジフルオロ−2−メトキシアニリン19.1g(0.12mol)、酢酸96g(1.6mol)及びメタノール240mlを仕込み、室温で撹拌しながら、1−エトキシ−1−トリメチルシリロキシシクロプロパン25.0g(0.14mol)を滴下した。その後オイルバスにより昇温し、窒素気流下、67℃で4時間、加熱還流下で撹拌した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータで濃縮し、最後に真空ポンプを用いて残存する酢酸を除去し、粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンを27.3g得た(粗収率99.2%、純度92%)。
【0066】
次いで、還流冷却器、温度計及び撹拌機を備えた500mlの四ツ口フラスコに、水素化ホウ素ナトリウム6.81g(0.18mol)、無水テトラヒドロフラン90mlを仕込み、窒素気流下で撹拌しながら氷冷し、5℃まで冷却した。その後、塩化アルミニウム8.0g(0.06mol)を投入したところ、反応液の温度が15℃まで上昇した。発熱が終了した後、5℃で1.5時間撹拌した。塩化アルミニウムの固体が完全に溶解したのを確認した後、先に得たN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン27.3gをテトラヒドロフラン30mlに溶解し、5℃で滴下し、同温度で3時間撹拌し、更に室温で4時間撹拌した。その後、オイルバス上で昇温し、常圧下でテトラヒドロフランを留出させ、105mlを回収した。500mlのビーカー内に、48%水酸化ナトリウム水溶液45gと水200mlを仕込み、水浴上で冷却し、撹拌しながら先の蒸留残渣を滴下した。このものをエーテル200mlで2回抽出し、有機層を水300mlで2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレータで溶媒を留去し、21.0g(粗収率87.8%)のN−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンを得た(純度90.8%)。更にこのものを減圧下で精留し、N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン(沸点:78〜79℃/3mmHg)を17.9g得た[収率75%(3,4−ジフルオロ−2−メトキシアニリン基準)、純度98.6%]。
【0067】
尚、上記に記載した還元工程における原料として、実施例1乃至4で得られた化合物を(必要に応じ単離して)使用しても、この実施例6の還元工程で得られたN−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリンと同一の化合物を得ることができる。
【0068】
更に、還流冷却器、温度計及び撹拌機を備えた100mlの四ツ口フラスコに、N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン14.9g(0.075mol)及びエトキシメチレンマロン酸ジエチル16.2g(0.075mol)を仕込み、オイルバス上、160℃で6時間加熱撹拌し、N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリノメチレンマロン酸ジエチルを27.3g得た[収率98.5%(N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン基準)、純度95%]。
確認データ
GC−MS(m/e):369[[M+]
60MHz 1H−NMR(CDCl3)δ値:0.6〜0.9(4H,m,CH2CH2),1.1〜1.5(6H,m,2CH3)、3.0〜3.5(1H,m,CH),3.7〜4.4(7H,m,OCH3及び2OCH2)、6.6〜7.3(2H,m,芳香核水素),7.66(1H,s,C=CH)
【0069】
参考例1
還流冷却器、温度計及び撹拌機を備えた300mlの四ツ口フラスコに、N−シクロプロピル−3,4−ジフルオロ−2−メトキシアニリノメチレンマロン酸ジエチル27.3g(0.0739mol)及びポリリン酸142gを仕込み、オイルバス上で加熱し、85〜90℃で30分間撹拌した。反応混合物を40℃まで冷却し、水250mlを滴下した。析出した結晶を濾別し、水洗を繰り返した後に乾燥し、粗の1−シクロプロピル−4−オキソ−6,7−ジフルオロ−8−メトキシ−キノリン−3−カルボン酸エステルを得た。粗収量は19.7gであった(粗収率82.4%)。このものをエタノールで再結晶し、15.3gの精製品を得(収率64%)、得られた生成物を機器分析(融点測定、NMR分析、質量分析)したところ、その分析データは文献値(特公平8−9597号公報、特開昭62−252772号公報参照)と一致した。
【0070】
実施例7(第1の方法)
還流冷却器(無水塩化カルシウム管付き)、温度計及び磁気撹拌子を備えた500mlの四ツ口フラスコに、3,4−ジフルオロ−2−メチルアニリン31.0g(0.217mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン46.9g(0.269mol)、酢酸53.8g(0.896mol)及びメタノール230mlを仕込み、4時間加熱還流した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータにより濃縮し、更に残渣を真空ポンプで濃縮し、固形物質47.6gを得た。得られた結晶をn−ヘキサンで再結晶後、乾燥し、白色結晶のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリン42.0gを得た(収率90.8%)。
[N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリンの物性]
融点:81.0〜82.3℃
確認データ
GC−MS(m/e):213[[M+]
60MHz 1H−NMR(CDCl3)δ値:0.7〜1.3(4H,m,CH2CH2)、2.05(3H,d,J=2.22Hz,芳香核CH3),3.26(3H,s,OCH3)、4.56(1H,brs,NH),6.8〜7.5(2H,m,芳香核水素)
【0071】
実施例8(第1の方法)
還流冷却器(無水塩化カルシウム管付き)、温度計及び磁気撹拌子を備えた200mlの四ツ口フラスコに、3,4−ジフルオロ−2−メチルアニリン4.29g(0.03mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン6.54g(0.0375mol)、酢酸7.21g(0.12mol)及びエタノール60mlを仕込み、5時間加熱還流した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータにより濃縮し、更に残渣を真空ポンプで濃縮し、固形物質7.02gを得た。得られた結晶をn−へキサンで再結晶後、乾燥し、白色結晶のN−(1−エトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリン6.02gを得た(収率88.3%)。
N−(1−エトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリンの物性]
融点:74.2〜75.0℃
確認データ
GC−MS(m/e):227[M+]
60MHz 1H−NMR(CDCl3)δ値:0.8〜1.4(4H,m,CH2CH2)、1.12(3H,t,J=7.02Hz,OCH2CH3のCH3)、2.02(3H,d,J=2.04Hz,芳香核CH3)、3.52(2H、q,J=7.05Hz,OCH2CH3のCH2)、4.50(1H,brs,NH)、6.7〜7.1(2H,m,芳香核水素)
【0072】
実施例9(第1の方法)
還流冷却器(無水塩化カルシウム管付き)、温度計及び磁気撹拌子を備えた四つ口500mlのフラスコに、3,4−ジフルオロ−2−メチルアニリン28.6g(0.2mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン45.32g(0.26mol)、酢酸48.0g(0.8mol)及びイソプロピルアルコール400mlを仕込み、オイルバス温度90℃にて4.5時間反応させた。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータにより濃縮し、精留によりN−〔1−(2−プロピル)オキシ〕シクロプロピル−3,4−ジフルオロ−2−メチルアニリン30.0gを得た(収率62.2%)。
[N−〔1−(2−プロピル)オキシ〕シクロプロピル−3,4−ジフルオロ−2−メチルアニリンの物性]
沸点:92〜95℃/1.5mmHg
確認データ
GC−MS(m/e):241[M+]
60MHz 1H−NMR(CDCl3)δ値:0.7〜1.4(4H,m,CH2CH2)、1.08〔6H,d,J=6.30Hz,CH(CH3)2の(CH3)2〕、2.02(3H,d,J=2.04Hz,芳香核CH3)、3.7〜4.2〔1H,m,CH(CH3)2のCH〕、4.64(1H,brs、NH)、6.7〜7.0(2H,m,芳香核水素)
【0073】
実施例10(第1の方法)
還流冷却器(無水塩化カルシウム管付き)及び磁気撹拌子を備えた50mlのフラスコに、3,4−ジフルオロ−2−メチルアニリン1.43g(0.01mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン2.27g(0.013mol)、蟻酸1.84g(0.04mol)及びメタノール20mlを仕込み、オイルバス温度90℃にて4.5時間反応させた。得られた反応液をガスクロマトグラフィ一で分析したところ、N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリンの生成率は84.7%であった。
【0074】
実施例11(第2の方法)
還流冷却器(無水塩化カルシウム管付き)、温度計及び磁気撹拌子を備えた500mlの四ツ口フラスコに、3,4−ジフルオロ−2−メチルアニリン28.6g(0.2mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン43.6g(0.25mol)、酢酸48.0g(0.8mol)及びメタノール200mlを仕込み、4時間加熱還流した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータにより濃縮し、更に残渣を真空ポンプで濃縮し、粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリンを42.6g得た。
【0075】
次いで、還流冷却器、温度計及び撹拌機を備えた1Lの四ツ口フラスコに、水素化ホウ素ナトリウム9.1g(0.24mol)、無水テトラヒドロフラン300mlを仕込み、氷冷下、5℃まで冷却した。その後、無水塩化アルミニウム16.0g(0.12mol)を同温度で投入したところ、反応液の温度が15℃まで上昇した。発熱が終了した後、5℃で1.5時間撹拌した。先に得た粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリン42.6gを無水テトラヒドロフラン100mlに溶かした溶液を、5℃で1時間かけて滴下した後、5℃にて30分間撹拌し、室温にて1時間撹拌し、更に50℃に昇温し1時間撹拌した。反応液をアスピレーターで減圧下、ロータリーエバポレーターを用いて一部濃縮し、残渣を水800ml中に滴下した後、酢酸エチルで2回抽出した。得られた酢酸エチル層を水で洗浄した後、無水硫酸ナトリウムで乾燥した。溶媒をロータリーエバポレーター用いて留去後、残渣をガスクロマトグラフィ一で分析したところ、N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリンの収率は71.3%であった(3,4−ジフルオロ−2−メチルアニリン基準)。
[N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリンの物性]
沸点:106〜107℃/7mmHg
確認データ
GC−MS(m/e):183[M+]
60MHz 1H−NMR(CDCl3)δ値:0.3〜1.1(4H,m,CH2CH2)、2.00(3H,d,J=2.04Hz,芳香核CH3)、2.2〜2.6(1H,m,CH),3.89(1H,brs,NH)、6.5〜7.3(2H,m,芳香核水素)
【0076】
尚、上記に記載した還元工程における原料として、実施例7乃至10で得られた化合物を(必要に応じ単離して)使用しても、この実施例11の還元工程で得られたN−シクロプロピル−3,4−ジフルオロ−2−メチルアニリンと同一の化合物を得ることができる。
【0077】
実施例12(第3の方法〉
還流冷却器(無水塩化カルシウム管付き)、温度計及び磁気撹拌子を備えた四ツ口500mlフラスコに、3,4−ジフルオロ−2−メチルアニリン28.6g(0.2mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン43.6g(0.25mol)、酢酸48.0g(0.8mol)及びメタノール200mlを仕込み、4時間加熱還流した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレータにより濃縮し、更に残渣を真空ポンプで濃縮し、粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メチルアニリンを43.8g得た。
【0078】
次いで、還流冷却器、温度計及び撹拌機を備えた1Lの四ツ口フラスコに、水素化ホウ素ナトリウム9.1g(0.24mol)、無水テトラヒドロフラン300mlを仕込み、氷冷下、5℃まで冷却した。その後、三フッ化ホウ素エーテル錯体34.1g(0.24mol)を同温度で滴下し、滴下終了後、5℃で1時間撹拌した。先に得た粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−メトキシアニリン43.8gを無水テトラヒドロフラン100mlに溶かした溶液を、5℃で1時間かけて滴下した後、20〜30℃で1時間撹拌し、更に50℃に昇温して3時間撹拌した。反応液をアスピレーターで減圧下、ロータリーエバポレーターを用いて一部濃縮し、残渣を水800ml中に滴下した後、酢酸エチルで2回抽出した。得られた酢酸エチル層を水で洗浄した後、無水硫酸ナトリウムで乾燥した。溶媒をロータリーエバポレーター用いて留去し、最後に減圧蒸留して、N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリンを31.3g得た[収率85.5%(3,4−ジフルオロ−2−メチルアニリン基準)]。
【0079】
尚、上記に記載した還元工程における原料として、実施例7乃至10で得られた化合物を(必要に応じ単離して)使用しても、この実施例12の還元工程で得られたN−シクロプロピル−3,4−ジフルオロ−2−メチルアニリンと同一の化合物を得ることができる。
【0080】
更に、還流冷却器及び磁気撹拌子を備えた50mlのフラスコに、先に得られたN−シクロプロピル−3,4−ジフルオロ−2−メチルアニリン2.92g(0.0159mol)及びエトキシメチレンマロン酸ジエチル3.44g(0.0159mol)を仕込み、オイルバス温度140℃で3時間加熱し、N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリノメチレンマロン酸ジエチル5.53gを得た[収率98.5%(N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリン基準)]。
[N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリノメチレンマロン酸ジエチルの物性]
確認データ
GC−MS(m/e):353[M+]
60MHz 1H−NMR(CDCl3)δ値:0.6〜1.5(10H,m,CH2CH2+2×(CH2CH3のCH3))、2.19(3H,d,J=1.74Hz,芳香核CH3)、2.9〜3.4(1H,m,CH)、3.6〜4.5(4H,m,2×(OCH2CH3のCH2))、6.47〜7.33(2H,m,芳香核水素)、7.6(1H,s,C=CH)
【0081】
参考例2
還流冷却器、温度計、及び撹拌機を備えた50mlの三ツ口フラスコにN−シクロプロピル−3,4−ジフルオロ−2−メチルアニリノメチレンマロン酸ジエチル1.77g(5mmol)及びポリリン酸9.5gを仕込み、80℃で1時間撹拌した。反応混合物を水200mlに滴下した。析出した結晶を濾別し、水洗を繰り返した後、エタノールで再結晶、乾燥し、1−シクロプロピル−6,7−ジフルオロ−8−メチル−4−オキソキノリン−3−カルボン酸エチル1.17gを得た(収率76.0%)。このようにして得られた生成物を機器分析(融点測定、NMR分析、質量分析)したところ、その分析データは文献値(特開昭62−215572号公報、特開昭63−264461号公報)と一致した。
【0082】
参考例3(3,4−ジフルオロ−2−フッ素置換メトキシニアニリンの合成)
(3,4−ジフルオロ−2−ジフルオロメトキシ−ニトロベンゼンの製造)
還流冷却器、温度計及び撹拌機を備えた1lの四ツ口フラスコに、2,3−ジフルオロ−6−ニトロフェノール26.3g(0.15mol)、クロロジフルオロ酢酸ナトリウム塩29.6g(0.2mol)、無水炭酸ナトリウム21.2g(0.2mol)、ヨウ化ナトリウム30.0g(0.2mol)及びN,N−ジメチルホルムアミド200mlを仕込んだ後、オイルバス上で100℃にて1.5時間撹拌した。その後120℃に昇温し、更に2.5時間加熱撹拌した。反応液をガスクロマトグラフィーで分析したところ、目的物の3,4−ジフルオロ−2−ジフルオロメトキシニトロベンゼンが69.6%生成し、原料の2,3−ジフルオロ−6−ニトロフェノールが28.0%残っていた。反応混合物を冷却し、水500mlを加え、エーテル200mlで抽出した。エーテル層を水500mlで2回洗浄し、無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮し、21.6gの褐色の油状物を得た。このものを減圧下で蒸留し、19.1gの3,4−ジフルオロ−2−ジフルオロメトキシ−ニトロベンゼンを得た(収率56.5%、純度97.1%、沸点98〜100℃/6mmHg)。
確認データ
GC−MS(m/e):225[M+]、175[M+−CF2]、158[M+−OCHF2]
60MHz 1H−NMR(CDCl3)δ値:6.73(1H,dd,J=73Hz,OCHF2)、7.1〜8.2(3H,m,芳香核水素)
【0083】
(3,4−ジフルオロ−2−ジフルオロメトキシニアニリンの製造)
還流冷却器、温度計及び撹拌機を備えた200mlの四ツ口フラスコに、鉄粉7.0g(0.125mol)、水50ml、98%硫酸0.1mlを入れ、オイルバス上で95℃に昇温した。次いで先に合成した3,4−ジフルオロ−2−ジフルオロメトキシ−ニトロベンゼン11.3g(0.05mol)を、95〜100℃で10分間に亘って滴下した。更にその後、同温度で2.5時間加熱撹拌した後、ディーン・スターク水分離器を装着して加熱還流を行い、水との共沸で留出した油状物質を分離した。更に水層を塩化メチレン100mlで抽出した。油状物と塩化メチレン層を混合し、濃縮残渣を減圧蒸留することにより、3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン7.1gを得た(収率72.8%、純度99.1%、沸点76〜78℃/6mmHg)。
確認データ
GC−MS(m/e):195[M+]、155[M+−CF2]、128[M+−OCHF2]
60MHz 1H−NMR(CDCl3)δ値:3.5〜4.5(1H,brs,NH)、6.54(1H,dd,J=74Hz,OCHF2)、6.3〜7.2(3H,m,芳香核水素)
【0084】
実施例13(第1の方法)
還流冷却器、温度計、及び撹拌機を備えた50mlの三ツ口フラスコに3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン2.54g(0.013mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン2.62g(0.015mol)、酢酸3.90g(0.065mol)、及びメタノール25mlを仕込み、窒素気流下、オイルバスで昇温し70℃で15時間加熱還流下で撹拌した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレーターにより濃縮し、残渣を減圧下で蒸留してN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン3.16g(収率91.1%、3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン基準)を得た。
[N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリンの物性]
沸点:73〜77℃/0.2mmHg
確認データ
GC−MS(m/e):265[M+]、234[M+−OCH3]、198[M+−OCHF2]
60MHz 1H−NMR(CDCl3)δ値:0.8〜1.4(4H,m,CH2×2)、3.27(3H,s,OCH3)、5.0〜5.4(1H,brs,NH)、6.54(1H,dd,J=74Hz,OCHF2)、6.8〜7.2(2H,m,芳香核水素)
【0085】
実施例14(第3の方法)
還流冷却器、温度計及び磁気撹拌子を備えた50mlの三ツ口フラスコに、3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン3.90g(0.02mol)、1−エトキシ−1−トリメチルシリロキシシクロプロパン4.01g(0.0223mol)、酢酸3.6g(0.06mol)及びメタノール20mlを仕込み、窒素気流下、70℃で12時間、加熱還流した。得られた反応液をアスピレーターで減圧下、ロータリーエバポレーターにより濃縮し、淡黄褐色の油状物5.32gを得た。このものは、機器分析データに基づいて、N−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリンを主生成物として含むことが確認された(純度89.4%)。
【0086】
ついで、還流冷却器、温度計及び撹拌機を備えた100mlの四ツ口フラスコに、水素化ホウ素ナトリウム1.14g(0.03mol)、無水テトラヒドロフラン20mlを仕込み、窒素気流下で撹拌しながら氷冷し、5℃まで冷却した。その後、三フッ化ホウ素テトラヒドロフラン錯体4.33g(0.03mol)を同温度で滴下し、滴下終了後、5℃で1時間撹拌した。先に得た粗のN−(1−メトキシ)シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン5.32gを5〜10℃で滴下した後、徐々60℃に昇温した後、同温度で12時間撹拌した。反応終了後、冷却し、反応混合物を水200ml中に滴下して、室温で30分間撹拌後、エーテル150mlで抽出した。得られたエーテル層を水200mlで洗浄した後、無水硫酸ナトリウムで乾燥し、ロータリーエバポレーター用いてエーテルを留去し、油状物質を4.45得た。このものを減圧蒸留して、N−シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシアニリンを4.17g得た(収率88.6%[3,4−ジフルオロ−2−ジフルオロメトキシアニリン基準)]。純度は91.3%であった。
[N−シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリンの物性]
沸点:95〜100℃/6mmHg
確認データ
GC−MS(m/e):235[M+]、168[M+−OCHF2]
60MHz 1H−NMR(CDCl3)δ値:0.4〜1.0(4H,m,CH2CH2)、2.1〜2.6(1H,m,CH)、4.2〜4.7(1H,brs,NH)、6.4(1H,dd,J=74Hz,OCHF2)、6.6〜7.3(2H,m,芳香核水素)
【0087】
尚、上記に記載した還元工程における原料として、実施例13で得られた化合物を使用しても、この実施例14の還元工程で得られたN−シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリンと同一の化合物を得ることができる。
【0088】
更に、還流冷却器、温度計及び撹拌機を備えた50mlの三ツ口フラスコに、先に得たN−シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン1.18g(0.005mol)及びエトキシメチレンマロン酸ジエチル2.16g(0.01mol)を仕込み、オイルバス上で窒素気流下、170℃で2.5時間加熱撹拌した。反応混合物をミクロ蒸留装置に移し、減圧下で蒸留したところ、まず、初留分としてエトキシメチレンマロン酸ジエチル1.19gが回収された(沸点160〜165℃/0.2mmHg)。次の留分としてN−シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリノメチレンマロン酸ジエチル1.60gを得た[収率78.9%(N−シクロプロピル−3,4−ジフルオロ−2−ジフルオロメトキシ−アニリン基準)]。
[N−シクロプロピル−3,4−ジフルオロ−2−メチルアニリノメチレンマロン酸ジェチルの物性]
沸点:160〜165℃/0.2mmHg
確認データ
GC−MS(m/e):405[M+]、369[M+−C2H5OH]、332[M+−COOC2H5]
60MHz 1H−NMR(CDCl3)δ値:0.6〜0.9(4H,m,CH2CH2)、1.1〜1.7(6H,m,2×(CH2CH3のCH3))、3.1〜3.6(1H,m,CH)、3.85(2H,q,J=7.0Hz,OCH2)、4.18(2H,q,J=7.0Hz,OCH2)、6.77(1H,t,J=73Hz,CHF2)、6.6〜7.4(2H,m,芳香核水素)、7.69(1H,s,NC=CH−)
Claims (5)
- アルコール系溶媒中、酸の存在下、一般式(1)
- アルコール系溶媒中、酸の存在下、一般式(1)
- アルコール系溶媒中、酸の存在下、一般式(1)
- 一般式(3)
- 一般式(6)
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21251397A JP4028913B2 (ja) | 1996-07-22 | 1997-07-22 | N−シクロプロピルアニリン類の製造方法とそのための中間体 |
| ES98911057T ES2216278T3 (es) | 1996-07-22 | 1998-03-27 | Procedimiento para producir n-ciclopropilanilinas y productos intermedios usados para el mismo. |
| US09/423,339 US6162944A (en) | 1996-07-22 | 1998-03-27 | Process for production of N-cyclopropylanilines and intermediates therefor |
| EP98911057A EP0990639B1 (en) | 1996-07-22 | 1998-03-27 | Process for producing n-cyclopropylanilines and intermediates used therefor |
| DE69821934T DE69821934T2 (de) | 1996-07-22 | 1998-03-27 | Verfahren zur herstellung von n-cyclopropylanilinen und zwischenverbindungen zu ihrer herstellung |
| PCT/JP1998/001395 WO1999050226A1 (en) | 1996-07-22 | 1998-03-27 | Process for producing n-cyclopropylanilines and intermediates used therefor |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21060396 | 1996-07-22 | ||
| JP8-210603 | 1996-07-22 | ||
| JP21251397A JP4028913B2 (ja) | 1996-07-22 | 1997-07-22 | N−シクロプロピルアニリン類の製造方法とそのための中間体 |
| PCT/JP1998/001395 WO1999050226A1 (en) | 1996-07-22 | 1998-03-27 | Process for producing n-cyclopropylanilines and intermediates used therefor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH1087584A JPH1087584A (ja) | 1998-04-07 |
| JP4028913B2 true JP4028913B2 (ja) | 2008-01-09 |
Family
ID=27308547
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP21251397A Expired - Fee Related JP4028913B2 (ja) | 1996-07-22 | 1997-07-22 | N−シクロプロピルアニリン類の製造方法とそのための中間体 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0990639B1 (ja) |
| JP (1) | JP4028913B2 (ja) |
| DE (1) | DE69821934T2 (ja) |
| ES (1) | ES2216278T3 (ja) |
| WO (1) | WO1999050226A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6162944A (en) * | 1996-07-22 | 2000-12-19 | Ihara Chemical Industry Co., Ltd. | Process for production of N-cyclopropylanilines and intermediates therefor |
| JP4028913B2 (ja) * | 1996-07-22 | 2008-01-09 | イハラケミカル工業株式会社 | N−シクロプロピルアニリン類の製造方法とそのための中間体 |
| EP1362843B1 (en) * | 1998-08-12 | 2006-06-14 | Ihara Chemical Industry Co., Ltd. | N-cyclopropyl-2-difluoromethoxy-3-bromoaniline and intermediate used for production thereof |
| JP5204438B2 (ja) * | 2007-07-31 | 2013-06-05 | 東ソ−・エフテック株式会社 | 含フッ素アミン化合物の製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0228582B2 (ja) * | 1985-02-06 | 1990-06-25 | Tokuyama Soda Kk | Dai2kyuaminkagobutsunoseizohoho |
| ES2014560A6 (es) * | 1988-12-30 | 1990-07-16 | Marga Investigacion | Proceso para la preparacion de un nuevo reactivo organo-sililpolifosforico y su aplicacion al proceso de sontesis de 3-carboxi-quinolonas o azaquinolonas y sus sales. |
| JP4028913B2 (ja) * | 1996-07-22 | 2008-01-09 | イハラケミカル工業株式会社 | N−シクロプロピルアニリン類の製造方法とそのための中間体 |
-
1997
- 1997-07-22 JP JP21251397A patent/JP4028913B2/ja not_active Expired - Fee Related
-
1998
- 1998-03-27 ES ES98911057T patent/ES2216278T3/es not_active Expired - Lifetime
- 1998-03-27 DE DE69821934T patent/DE69821934T2/de not_active Expired - Fee Related
- 1998-03-27 EP EP98911057A patent/EP0990639B1/en not_active Expired - Lifetime
- 1998-03-27 WO PCT/JP1998/001395 patent/WO1999050226A1/ja not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| EP0990639A4 (en) | 2002-08-14 |
| ES2216278T3 (es) | 2004-10-16 |
| EP0990639A1 (en) | 2000-04-05 |
| JPH1087584A (ja) | 1998-04-07 |
| WO1999050226A1 (en) | 1999-10-07 |
| DE69821934D1 (de) | 2004-04-01 |
| EP0990639B1 (en) | 2004-02-25 |
| DE69821934T2 (de) | 2004-07-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5254974B2 (ja) | レスベラトロールおよびピセアタンノールを得ることを可能にする(e)−スチルベン誘導体を合成するための新規な方法 | |
| CN107417505B (zh) | α-卤代四甲基环己酮及其与(2,3,4,4-四甲基环戊基)甲基羧酸酯的制备方法 | |
| JP2015532309A (ja) | オスペミフェンの製造方法 | |
| JP4028913B2 (ja) | N−シクロプロピルアニリン類の製造方法とそのための中間体 | |
| US6699992B2 (en) | Process for preparing quinolonecarboxylic acids | |
| JP4083842B2 (ja) | N−シクロプロピルアニリン類の製造方法 | |
| US20050261513A1 (en) | Process for producing indenol esters or ethers | |
| EP0184035B1 (en) | Quinolonecarboxylic acid derivatives and process for their preparation | |
| TWI500596B (zh) | 合成3,4-二甲氧基雙環[4.2.0]辛-1,3,5-三烯-7-甲腈之方法,及於合成依伐布雷定(ivabradine)及其與醫藥上可接受酸之加成鹽之應用 | |
| JP4321065B2 (ja) | キノリンカルボキシアルデヒド誘導体の製造法及びその中間体 | |
| JP4540197B2 (ja) | (e)−3−メチル−2−シクロペンタデセノンの製造法 | |
| US6162944A (en) | Process for production of N-cyclopropylanilines and intermediates therefor | |
| JP4667589B2 (ja) | 2,4−ジヒドロキシピリジンの製造方法 | |
| JP2000119221A (ja) | (2,4,5−トリフルオロ−3−メトキシベンゾイル)酢酸エステル誘導体の製造方法及びその製造中間体 | |
| JP2001302658A (ja) | 3−イソクロマノン類の製造方法 | |
| JP7714554B2 (ja) | 2-ベンゾイル安息香酸アルキルの合成のための効率的かつ選択的な経路 | |
| JP4148550B2 (ja) | 5−アミノ−1−シクロプロピル−4−オキソキノリン−3−カルボン酸誘導体とその製造方法 | |
| JP4605766B2 (ja) | アルキリデンシクロペンタノン誘導体の製造法 | |
| US7649107B2 (en) | Process for the preparation of cyclopentanone derivatives | |
| JP4505876B2 (ja) | 4−ハロベンゾフラン誘導体及びその製造方法 | |
| JP3144921B2 (ja) | ベンジルエステル誘導体及びその製造方法 | |
| JP2000344722A (ja) | 4−ヒドロキシメチル−1−アミノシクロペント−2−エン誘導体の製造方法 | |
| JP3144920B2 (ja) | α−アシルアミノケトン誘導体、その製造方法及びその利用 | |
| JP3749564B2 (ja) | テトラリンカルボン酸エステルの製造方法 | |
| JP4039026B2 (ja) | 3−アミノ−2−チオフェンカルボン酸エステルの製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20040628 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20070918 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20070920 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20071015 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20101019 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20111019 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121019 Year of fee payment: 5 |
|
| LAPS | Cancellation because of no payment of annual fees |