JP3857431B2 - Polymer purification method and curable composition - Google Patents
Polymer purification method and curable composition Download PDFInfo
- Publication number
- JP3857431B2 JP3857431B2 JP22141498A JP22141498A JP3857431B2 JP 3857431 B2 JP3857431 B2 JP 3857431B2 JP 22141498 A JP22141498 A JP 22141498A JP 22141498 A JP22141498 A JP 22141498A JP 3857431 B2 JP3857431 B2 JP 3857431B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- polymer
- vinyl polymer
- alkenyl group
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Landscapes
- Processes Of Treating Macromolecular Substances (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polymerization Catalysts (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Description
【0001】
【発明の属する技術分野】
本発明は、ビニル系重合体の精製方法、硬化性組成物に関する。
【0002】
【従来の技術】
ヒドロシリル化反応は官能基変換や、架橋反応等に利用され、工業的に非常に有用な反応の一つである。例えば、分子鎖の末端に官能基としてアルケニル基を有する重合体はヒドロシリル基含有化合物を硬化剤として用いることにより、架橋硬化し、耐熱性、耐久性等の優れた硬化物を与えること、また、末端にアルケニル基を有する重合体に架橋性シリル基を有するヒドロシリル基含有化合物を反応させることにより、架橋性シリル基を末端に有する重合体が製造されることが知られている。これらのヒドロシリル化反応は加熱することにより進行するが、反応をより迅速に進めるために、ヒドロシリル化触媒が添加される。このようなヒドロシリル化触媒としては、有機過酸化物やアゾ化合物等のラジカル開始剤、および遷移金属触媒が挙げられる。特に、遷移金属触媒を用いると触媒量でヒドロシリル化を迅速に進めることができることが知られている。
【0003】
一方、重合体の精密合成法としてリビング重合法が一般的に知られている。リビング重合は分子量、分子量分布のコントロールが可能であるというだけでなく、末端構造が明確な重合体が得られる。従って、リビング重合は重合体末端に官能基を導入する有効な方法の一つとして挙げられる。最近、ラジカル重合においても、リビング重合が可能な重合系が見いだされ、リビングラジカル重合の研究が活発に行われている。特に原子移動ラジカル重合を利用することにより分子量分布の狭いビニル系重合体が得られる。
【0004】
本発明者らは、これらの原子移動ラジカル重合法を用いて、ビニル系モノマーをラジカル重合し、重合体末端をアルケニル基含有置換基に変換することにより、分子鎖の末端にアルケニル基を有するビニル系重合体が得られることを見出した。
【0005】
【発明が解決しようとする課題】
しかし、原子移動ラジカル重合法を利用して製造された末端にアルケニル基を有するビニル系重合体において、硬化剤としてヒドロシリル基含有化合物、ヒドロシリル化触媒として遷移金属触媒を用いて、硬化物の作製を試みたが、硬化しないもしくは硬化が非常に遅いという問題点が生じた。すなわち、ヒドロシリル化反応が阻害されることがわかった。
【0006】
本発明はこの問題点を解決し、ヒドロシリル化反応が阻害されることなく進行するするためのビニル系重合体の精製方法を提供するものである。
【0007】
【課題を解決するための手段】
本発明は、(A)主鎖が原子移動ラジカル重合により製造されるビニル系重合体を、(B)吸着剤に接触させ、引き続き吸着剤を取り除くことによってビニル系重合体を精製する方法である。
また、本発明は、主鎖が原子移動ラジカル重合により製造される末端にアルケニル基を有するビニル系重合体と、ヒドロシリル基含有化合物を反応させるにあたって、予め、該重合体又は該重合体を製造する工程で得られる中間生成物を、吸着剤に接触させ、引き続き吸着剤を取り除くことによってビニル系重合体を精製する方法でもある。
【0008】
また、本発明は、(C)吸着剤処理により精製されたアルケニル基を有するビニル系重合体、(D)ヒドロシリル基含有化合物、を含有する硬化性組成物でもある。
【0009】
【発明の実施の形態】
本発明におけるビニル系重合体とは、主鎖が原子移動ラジカル重合法により製造されるものである。原子移動ラジカル重合とは、リビングラジカル重合の一つであり、有機ハロゲン化物、またはハロゲン化スルホニル化合物等を開始剤、遷移金属を中心金属とする金属錯体を触媒としてビニル系モノマーをラジカル重合する方法である。具体的には、Matyjaszewskiらの文献J.Am.Chem.Soc.1995,117,5614,Macromolecules 1995,28,7901,Science 1996,272,866;あるいはSawamotoらの文献、Macromolecules 1995,28,1721、国際公開特許WO96/30421及びWO97/18247等に記載の方法が挙げられる。
【0010】
リビング重合とは狭義においては、末端が常に活性を持ち続けて分子鎖が生長していく重合のことを示すが、一般には、末端が不活性化されたものと活性化されたものが平衡状態にありながら生長していく擬リビング重合も含まれる。本発明におけるリビング重合の定義も後者である。
本発明におけるビニル系重合体の主鎖を構成するビニル系のモノマ−としては特に限定されず、各種のものを用いることができる。例示するならば、(メタ)アクリル酸、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸−n−プロピル、(メタ)アクリル酸イソプロピル、(メタ)アクリル酸−n−ブチル、(メタ)アクリル酸イソブチル、(メタ)アクリル酸−tert−ブチル、(メタ)アクリル酸−n−ペンチル、(メタ)アクリル酸−n−ヘキシル、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸−n−ヘプチル、(メタ)アクリル酸−n−オクチル、(メタ)アクリル酸−2−エチルヘキシル、(メタ)アクリル酸ノニル、(メタ)アクリル酸デシル、(メタ)アクリル酸ドデシル、(メタ)アクリル酸フェニル、(メタ)アクリル酸トルイル、(メタ)アクリル酸ベンジル、(メタ)アクリル酸−2−メトキシエチル、(メタ)アクリル酸−3−メトキシブチル、(メタ)アクリル酸−2−ヒドロキシエチル、(メタ)アクリル酸−2−ヒドロキシプロピル、(メタ)アクリル酸ステアリル、(メタ)アクリル酸グリシジル、(メタ)アクリル酸2−アミノエチル、γ−(メタクリロイルオキシプロピル)トリメトキシシラン、(メタ)アクリル酸のエチレンオキサイド付加物、(メタ)アクリル酸トリフルオロメチルメチル、(メタ)アクリル酸2−トリフルオロメチルエチル、(メタ)アクリル酸2−パーフルオロエチルエチル、(メタ)アクリル酸2−パーフルオロエチル−2−パーフルオロブチルエチル、(メタ)アクリル酸2−パーフルオロエチル、(メタ)アクリル酸パーフルオロメチル、(メタ)アクリル酸ジパーフルオロメチルメチル、(メタ)アクリル酸2−パーフルオロメチル−2−パーフルオロエチルメチル、(メタ)アクリル酸2−パーフルオロヘキシルエチル、(メタ)アクリル酸2−パーフルオロデシルエチル、(メタ)アクリル酸2−パーフルオロヘキサデシルエチル等の(メタ)アクリル酸系モノマー;スチレン、ビニルトルエン、α−メチルスチレン、クロルスチレン、スチレンスルホン酸及び塩等のスチレン系モノマー;パーフルオロエチレン、パーフルオロプロピレン、フッ化ビニリデン等のフッ素含有ビニルモノマー;ビニルトリメトキシシラン、ビニルトリエトキシシラン等のケイ素含有ビニル系モノマー;無水マレイン酸、マレイン酸、マレイン酸のモノアルキルエステル及びジアルキルエステル;フマル酸、フマル酸のモノアルキルエステル及びジアルキルエステル;マレイミド、メチルマレイミド、エチルマレイミド、プロピルマレイミド、ブチルマレイミド、ヘキシルマレイミド、オクチルマレイミド、ドデシルマレイミド、ステアリルマレイミド、フェニルマレイミド、シクロヘキシルマレイミド等のマレイミド系モノマー;アクリロニトリル、メタクリロニトリル等のニトリル基含有ビニル系モノマー;アクリルアミド、メタクリルアミド等のアミド基含有ビニル系モノマー;酢酸ビニル、プロピオン酸ビニル、ピバリン酸ビニル、安息香酸ビニル、桂皮酸ビニルなどのビニルエステル類;エチレン、プロピレンなどのアルケン類;ブタジエン、イソプレンなどの共役ジエン類;塩化ビニル、塩化ビニリデン、塩化アリル、アリルアルコールなどが挙げられ、これらは単独で用いても良いし、複数を共重合させても構わない。なお上記表現形式で例えば(メタ)アクリル酸とは、アクリル酸および/あるいはメタクリル酸を表す。
これらの内では、生成物の物性等から、スチレン系モノマー及び(メタ)アクリル系モノマーが好ましく、更にアクリル酸エステル系モノマーが好ましく、特にアクリル酸ブチルが好ましい。本発明においては、これらの好ましいモノマーを他のモノマーと共重合させても構わなく、その際は、これらの好ましいモノマーが重量比で40%含まれていることが好ましい。
【0011】
ビニル系重合体の数平均分子量は特に限定されないが、500〜1,000,000の範囲が好ましく、500〜100,000がさらに好ましい。
また、ビニル系重合体の分子量分布は、特に限定されないが、ゲルパーミエーションクロマトグラフィーで測定した重量平均分子量と数平均分子量の比が通常は1.8未満であり、好ましくは1.7以下であり、より好ましくは1.6以下であり、さらに好ましくは1.5以下であり、特に好ましくは1.4以下であり、最も好ましくは1.3以下である。本発明でのGPC測定においては、通常、移動相としてクロロホルムを用い、測定はポリスチレンゲルカラムにておこない、数平均分子量等はポリスチレン換算で求めることができる。
【0012】
本発明におけるアルケニル基とは、一般式(1)に示される基である。
CH2=C(R1)− (1)
(式中、R1は水素又はメチル基を表す。)
本発明におけるアルケニル基を有するビニル系重合体において、アルケニル基は重合体の側鎖または末端のいずれにあってもよく、さらに1分子鎖中に含まれるアルケニル基の数は特に限定されない。しかし、ヒドロシリル基含有化合物を硬化剤として用いて、アルケニル基を有するビニル系重合体の硬化物を得るためには、1分子鎖中に少なくとも1個のアルケニル基が必要であり、さらに、ゴム弾性の優れた硬化物を得るためにはアルケニル基が側鎖よりも末端にあることが好ましい。
【0013】
本発明におけるビニル系重合体を製造する方法として原子移動ラジカル重合が用いられる。重合に用いられる開始剤としては、特に限定されないが、例えば、有機ハロゲン化物、特に、活性化された有機ハロゲン化物(例えば、α位にハロゲンを有するエステル化合物や、ベンジル位にハロゲンを有する化合物)、あるいはハロゲン化スルホニル化合物等が挙げられる。これらの化合物を開始剤として用いた場合には、重合体末端にハロゲンを有するビニル系重合体を得ることができる。この末端ハロゲンを後述の方法により変換することによって末端にアルケニル基を有するビニル系重合体を得ることができる。このような開始剤を具体的に例示すれば、C6H5−CH2X、C6H5−C(H)(X)CH3、C6H5−C(X)(CH3)2(式中、C6H5は、フェニル基を表す。Xは、塩素、臭素又はヨウ素を表す。);R2−C(H)(X)−CO2R3、R2−C(CH3)(X)−CO2R3、R2−C(H)(X)−C(O)R3、R2−C(CH3)(X)−C(O)R3(式中、R2及びR3は、同一若しくは異なって、水素原子、炭素数1〜20のアルキル基、炭素数6〜20のアリール基又は炭素数7〜20のアラルキル基を表す。Xは、塩素、臭素又はヨウ素を表す。);R2−C6H4−SO2X(式中、R2は、水素原子、炭素数1〜20のアルキル基、炭素数6〜20のアリール基又は炭素数7〜20のアラルキル基を表す。Xは、塩素、臭素又はヨウ素を表す。)等が挙げられる。
【0014】
開始剤として、重合を開始する官能基以外の官能基を有する有機ハロゲン化物又はハロゲン化スルホニル化合物を用いることもできる。この場合、一方の主鎖末端には前記開始剤に含まれる官能基に由来する官能基を有し、他方の末端にはハロゲンを有する重合体が製造される。上記の官能基としてアルケニル基、架橋性シリル基、ヒドロキシル基、エポキシ基、アミノ基、アミド基、カルボキシル基等が挙げられる。
【0015】
アルケニル基を有する有機ハロゲン化物としては限定されず、例えば、一般式2に示す構造を有するものが例示される。
R4R5C(X)−R6−R7−C(R8)=CH2 (2)
(式中、R8は水素、またはメチル基、R4、R5は水素、または、炭素数1〜20の1価のアルキル基、炭素数6〜20の1価のアリール基、または炭素数7〜20の1価のアラルキル、または他端において相互に連結したもの、R6は、−C(O)O−(エステル基)、−C(O)−(ケト基)、またはo−,m−,p−フェニレン基、R7は直接結合、または炭素数1〜20の2価の有機基で1個以上のエーテル結合を含んでいても良い、Xは塩素、臭素、またはヨウ素)
置換基R4、R5の具体例としては、水素、メチル基、エチル基、n−プロピル基、イソプロピル基、ブチル基、ペンチル基、ヘキシル基等が挙げられる。R4とR5は他端において連結して環状骨格を形成していてもよい。
【0016】
アルケニル基を有する有機ハロゲン化物としてはさらに一般式(3)で示される化合物が挙げられる。
H2C=C(R8)−R7−C(R4)(X)−R9−R5 (3)
(式中、R4、R5、R7、R8は上記に同じ。R9は、直接結合、−C(O)O−(エステル基)、−C(O)−(ケト基)、または、o−,m−,p−フェニレン基を表す。Xは上記に同じ。)
R7は直接結合、または炭素数1〜20の2価の有機基(1個以上のエーテル結合を含んでいても良い)であるが、直接結合である場合は、ハロゲンの結合している炭素にビニル基が結合しており、ハロゲン化アリル化物である。この場合は、隣接ビニル基によって炭素−ハロゲン結合が活性化されているので、R9としてC(O)O基やフェニレン基等を有する必要は必ずしもなく、直接結合であってもよい。R7が直接結合でない場合は、炭素−ハロゲン結合を活性化するために、R9としてはC(O)O基、C(O)基、フェニレン基が好ましい。
【0017】
アルケニル基を有するハロゲン化スルホニル化合物の具体例を挙げるならば、
o−,m−,p−CH2=CH−(CH2)n−C6H4−SO2X、
o−,m−,p−CH2=CH−(CH2)n−O−C6H4−SO2X、
(上記の各式において、Xは塩素、臭素、またはヨウ素、nは0〜20の整数)
等である。
【0018】
架橋性シリル基を有する有機ハロゲン化物としては特に限定されず、例えば一般式(4)に示す構造を有するものが例示される。
R4R5C(X)−R6−R7−C(H)(R8)CH2−[Si(R10)2-b(Y)bO]m−Si(R11)3-a(Y)a (4)
(式中、R4、R5、R6、R7、R8、Xは上記に同じ、R10、R11は、いずれも炭素数1〜20のアルキル基、炭素数6〜20のアリール基、炭素数7〜20のアラルキル基、または(R’)3SiO−(R’は炭素数1〜20の1価の炭化水素基であって、3個のR’は同一であってもよく、異なっていてもよい)で示されるトリオルガノシロキシ基を示し、R10またはR11が2個以上存在するとき、それらは同一であってもよく、異なっていてもよい。Yは水酸基または加水分解性基を示し、Yが2個以上存在するときそれらは同一であってもよく、異なっていてもよい。aは0,1,2,または3を、また、bは0,1,または2を示す。mは0〜19の整数である。ただし、a+mb≧1であることを満足するものとする)
架橋性シリル基を有する有機ハロゲン化物としてはさらに、一般式(5)で示される構造を有するものが例示される。
【0019】
(R11)3-a(Y)aSi−[OSi(R10)2-b(Y)b]m−CH2−C(H)(R8)−R7−C(R4)(X)−R9−R5 (5)
(式中、R4、R5、R7、R8、R9、R10、R11、a、b、m、X、Yは上記に同じ)
ヒドロキシル基を持つ有機ハロゲン化物、またはハロゲン化スルホニル化合物としては特に限定されず、下記のようなものが例示される。
【0020】
HO−(CH2)n−OC(O)C(H)(R)(X)
(上記の各式において、Xは塩素、臭素、またはヨウ素、Rは水素原子または炭素数1〜20のアルキル基、炭素数6〜20のアリール基、炭素数7〜20のアラルキル基、nは1〜20の整数)
アミノ基を持つ有機ハロゲン化物、またはハロゲン化スルホニル化合物としては特に限定されず、下記のようなものが例示される。
H2N−(CH2)n−OC(O)C(H)(R)(X)
(上記の各式において、Xは塩素、臭素、またはヨウ素、Rは水素原子または炭素数1〜20のアルキル基、炭素数6〜20のアリール基、炭素数7〜20のアラルキル基、nは1〜20の整数)
エポキシ基を持つ有機ハロゲン化物、またはハロゲン化スルホニル化合物としては特に限定されず、下記のようなものが例示される。
【0021】
【化1】
(上記の各式において、Xは塩素、臭素、またはヨウ素、Rは水素原子または炭素数1〜20のアルキル基、炭素数6〜20のアリール基、炭素数7〜20のアラルキル基、nは1〜20の整数)
さらに、開始剤として、2つ以上の開始点を有する有機ハロゲン化物又はハロゲン化スルホニル化合物を用いて重合を行うこともできる。このような場合、ハロゲンを1分子内に2つ以上有するビニル系重合体が製造される。
【0023】
上記の2つの開始点を有する開始剤を具体的に例示すれば、
【0024】
【化2】
(式中、C6H4は、フェニレン基を表す。Xは、塩素、臭素又はヨウ素を表す。Rは、炭素数1〜20のアルキル基、炭素数6〜20のアリール基又は炭素数7〜20のアラルキル基を表す。nは、0〜20の整数を表す。);
【0026】
【化3】
(式中、Xは、塩素、臭素又はヨウ素を表す。nは、0〜20の整数を表す。C6H4は、フェニレン基を表す。)等が挙げられる。
重合触媒として用いられる遷移金属錯体としては特に限定されず、好ましいものとして、0価の銅、1価の銅、2価のルテニウム、2価の鉄又は2価のニッケルの錯体が挙げられる。なかでも、銅の錯体が好ましい。1価の銅化合物を具体的に例示するならば、塩化第一銅、臭化第一銅、ヨウ化第一銅、シアン化第一銅、酸化第一銅、過塩素酸第一銅等である。銅化合物を用いる場合、触媒活性を高めるために2,2′−ビピリジル及びその誘導体、1,10−フェナントロリン及びその誘導体、テトラメチルエチレンジアミン、ペンタメチルジエチレントリアミン、ヘキサメチルトリス(2−アミノエチル)アミン等のポリアミン等の配位子が添加される。また、2価の塩化ルテニウムのトリストリフェニルホスフィン錯体(RuCl2(PPh3)3)も触媒として好適である。ルテニウム化合物を触媒として用いる場合は、活性化剤としてアルミニウムアルコキシド類が添加される。更に、2価の鉄のビストリフェニルホスフィン錯体(FeCl2(PPh3)2)、2価のニッケルのビストリフェニルホスフィン錯体(NiCl2(PPh3)2)、及び、2価のニッケルのビストリブチルホスフィン錯体(NiBr2(PBu3)2)も、触媒として好適である。
【0028】
重合は無溶媒又は各種の溶媒中で行うことができる。溶媒を用いる場合には溶媒としては、例えば、ベンゼン、トルエン等の炭化水素系溶媒;ジエチルエーテル、テトラヒドロフラン、ジフェニルエーテル、アニソール、ジメトキシベンゼン等のエーテル系溶媒;塩化メチレン、クロロホルム、クロロベンゼン等のハロゲン化炭化水素系溶媒;アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン系溶媒;メタノール、エタノール、プロパノール、イソプロパノール、n−ブチルアルコール、tert−ブチルアルコール等のアルコール系溶媒;アセトニトリル、プロピオニトリル、ベンゾニトリル等のニトリル系溶媒;酢酸エチル、酢酸ブチル等のエステル系溶媒;エチレンカーボネート、プロピレンカーボネート等のカーボネート系溶媒等が挙げられる。これらは、単独又は2種以上を混合して用いることができる。また、エマルジョン系もしくは超臨界流体CO2を媒体とする系においても重合を行うことができる。
【0029】
重合は、0〜200℃の範囲で行うことができるが、好ましくは、室温〜150℃の範囲である。
重合に用いられるビニル系モノマーとしては既に例示したもの全てが好適に使用されてよい。
ビニル系モノマーの原子移動ラジカル重合により得られるビニル系重合体の末端ハロゲンをアルケニル基含有置換基に変換することによって末端にアルケニル基を導入することができる。アルケニル基を有するビニル系重合体の合成方法としては特に限定されず、例えば次に述べる(A−a)〜(A−j)の方法などを挙げることができる。
【0030】
(A−a)ラジカル重合によりビニル系重合体を合成する際に、所定のビニル系モノマーとともに、下記一般式(7)等で表される一分子中に重合性のアルケニル基および重合性の低いアルケニル基を併せ持つ化合物をも反応させる方法。
H2C=C(R12)−R13−R14−C(R12)=CH2 (7)
(式中、R12は水素又はメチル基で、2個のR12は互いに同一でも異なっていてもよい。R13は−C(O)O−、またはo−,m−,p−フェニレン基を示し、R14は直接結合、または炭素数1〜20の2価の有機基を示し、1個以上のエーテル結合を含んでいてもよい。)
なお、一分子中に重合性のアルケニル基と重合性の低いアルケニル基を併せ持つ化合物を反応させる時期に制限はないが、ゴム的な性質を期待する場合には重合反応の終期あるいは所定のモノマーの反応終了後に、第2のモノマーとして反応させるのが好ましい。
【0031】
(A−b)原子移動ラジカル重合によりビニル系重合体を合成する際に、重合反応の終期あるいは所定のモノマーの反応終了後に、例えば1,5−ヘキサジエン、1,7−オクタジエン、1,9−デカジエンなどのような重合性の低いアルケニル基を少なくとも2個有する化合物を反応させる方法。
(A−c)原子移動ラジカル重合で得られる末端にハロゲンを有するビニル系重合体に、例えばアリルトリブチル錫、アリルトリオクチル錫などの有機錫のようなアルケニル基を有する各種の有機金属化合物を反応させてハロゲンを置換する方法。
【0032】
(A−d)原子移動ラジカル重合で得られる末端にハロゲンを有するビニル系重合体に、一般式(8)に挙げられるようなアルケニル基を有する安定化カルバニオンを反応させてハロゲンを置換する方法。
M+C-(R15)(R16)−R17−C(R18)=CH2 (8)
(式中、R18は水素又はメチル基、R15、R16はともにカルバニオンC-を安定化する電子吸引基であるか、または一方が前記電子吸引基で他方が水素または炭素数1〜10のアルキル基、またはフェニル基を示す。R17は直接結合、または炭素数1〜10の2価の有機基を示し、1個以上のエーテル結合を含んでいてもよい。M+はアルカリ金属イオン、または4級アンモニウムイオンを示す)
R15、R16の電子吸引基としては、−CO2R、−C(O)Rおよび−CNの構造を有するものが特に好ましい。
【0033】
(A−e)原子移動ラジカル重合で得られる末端にハロゲンを有するビニル系重合体に、例えば亜鉛のような金属単体あるいは有機金属化合物を作用させてエノレートアニオンを調製し、しかる後にハロゲンやアセチル基のような脱離基を有するアルケニル基含有化合物、アルケニル基を有するカルボニル化合物、アルケニル基を有するイソシアネート化合物、アルケニル基を有する酸ハロゲン化物等の、アルケニル基を有する求電子化合物と反応させる方法。
【0034】
(A−f)原子移動ラジカル重合で得られる末端にハロゲンを有するビニル系重合体に、例えば一般式(9)あるいは(10)に示されるようなアルケニル基を有するオキシアニオンあるいはカルボキシレートアニオンを反応させてハロゲンを置換する方法。
H2C=C(R19)−R20−O-M+ (9)
(式中、R19は水素又はメチル基、M+は上記に同じ。R20は炭素数1〜20の2価の有機基で1個以上のエーテル結合を含んでいてもよい)
H2C=C(R19)−R21−C(O)O-M+ (10)
(式中、R19は水素又はメチル基、M+は上記に同じ。R21は直接結合、または炭素数1〜20の2価の有機基で1個以上のエーテル結合を含んでいてもよい)
などが挙げられる。
【0035】
更に、上記アルケニル基を有するビニル系重合体は、水酸基を有するビニル系重合体から得ることもできるが、具体的な方法としては特に限定されず、例えば下記の(A−g)〜(A−j)の方法等を挙げることができる。なお上記水酸基を有するビニル系重合体は後述する(B−a)〜(B−f)の方法により得ることができる。
【0036】
(A−g)ナトリウムメトキシドのような塩基を作用させ、塩化アリルのようなアルケニル基含有ハロゲン化物と反応させる方法。
(A−h)アリルイソシアネート等のアルケニル基含有イソシアネート化合物を反応させる方法。
(A−i)(メタ)アクリル酸クロリドのようなアルケニル基含有酸ハロゲン化物をピリジン等の塩基存在下に反応させる方法。
【0037】
(A−j)アクリル酸等のアルケニル基含有カルボン酸を酸触媒の存在下に反応させる方法;等が挙げられる。
末端に水酸基を有するビニル系重合体の製造方法は以下のような方法が例示されるが、これらの方法に限定されるものではない。
(B−a)原子移動ラジカル重合によりビニル系重合体を合成する際に、例えば下記の一般式(11)に挙げられるような一分子中に重合性のアルケニル基と水酸基を併せ持つ化合物を第2のモノマーとして反応させる方法。
H2C=C(R22)−R23−R24−OH (11)
(式中、R22は水素又はメチル基である。R23は−C(O)O−、またはo−,m−,p−フェニレン基を示し、R24は直接結合、または炭素数1〜20の2価の有機基を示し、1個以上のエーテル結合を含んでいてもよい。)
なお、一分子中に重合性のアルケニル基と水酸基を併せ持つ化合物を反応させる時期に制限はないが、特にリビングラジカル重合で、ゴム的な性質を期待する場合には重合反応の終期あるいは所定のモノマーの反応終了後に、第2のモノマーとして反応させるのが好ましい。
【0038】
(B−b)原子移動ラジカル重合によりビニル系重合体を合成する際に、重合反応の終期あるいは所定のモノマーの反応終了後に、例えば10−ウンデセノール、5−ヘキセノール、アリルアルコールのようなアルケニルアルコールを反応させる方法。
(B−c)原子移動ラジカル重合で得られるビニル系重合体のハロゲンを加水分解あるいは水酸基含有化合物と反応させることにより、末端に水酸基を導入する方法。
(B−d)原子移動ラジカル重合で得られるビニル系重合体の末端ハロゲンに、一般式(12)に挙げられるような水酸基を有する安定化カルバニオンを反応させてハロゲンを置換する方法。
【0039】
M+C-(R25)(R26)−R27−OH (12)
(式中、R25、R26はともにカルバニオンC-を安定化する電子吸引基であるか、または一方が前記電子吸引基で他方が水素または炭素数1〜10のアルキル基、またはフェニル基を示す。R27は直接結合、または炭素数1〜10の2価の有機基を示し、1個以上のエーテル結合を含んでいてもよい。M+はアルカリ金属イオン、または4級アンモニウムイオンを示す)
R25、R26の電子吸引基としては、−CO2R、−C(O)Rおよび−CNの構造を有するものが特に好ましい。
【0040】
(B−e)原子移動ラジカル重合で得られるビニル系重合体の末端ハロゲンに、例えば亜鉛のような金属単体あるいは有機金属化合物を作用させてエノレートアニオンを調製し、しかる後にアルデヒド類、又はケトン類を反応させる方法。
(B−f)原子移動ラジカル重合で得られるビニル系重合体の末端ハロゲンに、例えば一般式(13)あるいは(14)に示されるような水酸基を有するオキシアニオンあるいはカルボキシレートアニオンを反応させてハロゲンを置換する方法。
HO−R28−O-M+ (13)
(式中、M+は上記に同じ。R28は炭素数1〜20の2価の有機基で1個以上のエーテル結合を含んでいてもよい)
HO−R29−C(O)O-M+ (14)
(式中、M+は上記に同じ。R29は直接結合、または炭素数1〜20の2価の有機基で1個以上のエーテル結合を含んでいてもよい)等が挙げられる。
【0041】
さらに、末端にアルケニル基を有するビニル系重合体は、ビニル系重合体同士をカップリングする方法も利用できる。以下に例示する方法が利用できるがこれらに限定されるわけではない。
官能基を有する開始剤を用いてビニル系モノマーの原子移動ラジカル重合を行い、得られた重合体の末端ハロゲンに対して、ハロゲンを置換できる同一又は異なった官能基を合計2個以上有する化合物を用いてカップリングする。上記ハロゲンを置換できる、同一または異なった官能基を合計2個以上有する化合物としては例えば、ポリオール、ポリアミン、ポリカルボン酸、ポリチオール、およびそれらの塩、アルカリ金属硫化物等が挙げられる。
【0042】
次に、吸着剤を用いたビニル系重合体の精製方法について詳述する。
本発明で使用される吸着剤としては特に限定されないが、例えば、活性炭、二酸化ケイ素、固体酸、酸性白土、活性白土、活性アルミナ、シリカゲル、シリカ・アルミナ、アルミニウムシリケート、等が挙げられる。これらのうち、活性炭、活性アルミナ、二酸化ケイ素、アルミニウムシリケートが好ましく、活性炭、アルミニウムシリケートがより好ましい。
【0043】
活性炭とは大部分が炭素質の炭であり、吸着性は高い。製法は、例えば木材、褐炭、泥炭などを活性化剤として塩化亜鉛やリン酸などで処理して乾留するか、あるいは木炭などを水蒸気で活性化する。通常は粉状あるいは粒状であり、いずれも使用することができる。
二酸化ケイ素は、結晶性、無定形、非晶質、ガラス状、合成品、天然品などの種類が知られるが、ここでは、粉体状であれば使用することができる。二酸化ケイ素としては、活性白土を酸処理して得られる粘土鉱物から作られるケイ酸、カープレックスBS304、カープレックスBS304F、カープレックス#67、カープレックス#80(いずれもシオノギ製薬)などの合成ケイ酸が挙げられるが、これらに限定されるわけではない。
【0044】
また、アルミニウムシリケートとはケイ酸のケイ素の一部がアルミニウムに置換されたもので、軽石、フライアッシュ、カオリン、ベントナイト、活性白土、ケイソウ土、ゼオライトなどの天然のアルミニウムシリケートや合成のアルミニウムシリケートが知られている。この中でも、合成のアルミニウムシリケートは比表面積も大きく吸着能力が高い。合成アルミニウムシリケートとしてはキョーワード700PEL、キョーワード700PL、キョーワード700SL(いずれも協和化学製)などが挙げられるが、これらに限定されるわけではない。上記の吸着剤は単独で用いても2種以上を混合して用いてもかまわない。
【0045】
吸着剤による精製は、上述の製造方法により合成されたアルケニル基を有するビニル系重合体と吸着剤を接触させることにより行うことができる。無溶剤でもかまわないが、取り扱いの容易さからベンゼン、トルエン、酢酸エチル等の溶媒中で行うことが好ましい。吸着剤処理の温度や圧力については特に制限はないが、一般に常圧で0℃〜200℃、好ましくは室温〜150℃で行うのがよい。最終的に吸着剤を除去することによって精製されたビニル系重合体が得られる。また、吸着剤の使用量は、ビニル系重合体100重量部に対して0.1〜500重量部の範囲であるが、経済性と操作面から更に好適には5〜200重量部の範囲である。
【0046】
吸着剤と重合体溶液の固液接触には様々な実施態様が可能であるが、撹拌混合と固液分離を回分操作で行う回分式のほか、吸着剤を容器に充填し重合体溶液を通液する固定層方式、吸着剤の移動層に液を通じる移動層式、吸着剤を液で流動化して吸着を行う流動層式等も利用できる。さらに必要に応じて撹拌による混合分散に加えて、容器の振とう、超音波の利用など、分散効率を向上させる諸操作を取り入れることができる。
【0047】
重合体溶液を吸着剤に接触させた後、濾過、遠心分離、沈降分離等の方法で吸着剤を除去し、必要に応じて水洗を加え、目的とする清澄な重合体溶液を得る。
通常、吸着剤による精製はアルケニル基を有するビニル系重合体に対して行えばよいが、前記重合体を製造する工程の途中で吸着剤の精製を行ってもよい。すなわち、アルケニル基を有するビニル系重合体の製造工程で得られる中間生成物、例えば末端にハロゲンを有するビニル系重合体、末端に水酸基を有するビニル系重合体等に対して、予め吸着剤による精製を行ってもよい。
【0048】
ビニル系重合体を吸着剤で精製することによって、ビニル系重合体のアルケニル基にヒドロシリル基含有化合物を速やかに付加させることができる。
例えば、ビニル系重合体の末端アルケニル基に架橋性シリル基を有するヒドロシリル基含有化合物を付加させれば、末端に架橋性シリル基を有するビニル系重合体を得ることができる。架橋性シリル基を有するヒドロシリル基含有化合物としては特に制限はないが、代表的なものを示すと、一般式15で示される化合物が例示される。
H−[Si(R30)2-b(Y)bO]m−Si(R31)3-a(Y)a (15)
(式中、R30およびR31は、いずれも炭素数1〜20のアルキル基、炭素数6〜20のアリール基、または炭素数7〜20のアラルキル基、または(R’)3Si−(R’は炭素数1〜20の1価の炭化水素基であって、3個のR’は同一であってもよく、異なっていてもよい)で示されるトリオルガノシロキシ基を示し、R30またはR31が2個以上存在するとき、それらは同一であってもよく、異なっていてもよい。Yは水酸基または加水分解性基を示し、Yが2個以上存在するとき、それらは同一であってもよく、異なっていてもよい。aは0,1,2,または3を、また、bは0,1,または2を示す。mは0〜19の整数である。ただし、a+mb≧1であることを満足するものとする。)
上記Yで示される加水分解性基としては特に限定されず、従来公知のものを用いることができ、具体的には、水素、ハロゲン原子、アルコキシ基、アシルオキシ基、ケトキシメート基、アミノ基、アミド基、酸アミド基、アミノオキシ基、メルカプト基、アルケニルオキシ基等が挙げられ、加水分解性がマイルドで取り扱いやすいという点から、アルコキシ基が特に好ましい。該加水分解性基や水酸基は1個のケイ素原子に1〜3個の範囲で結合することができ、a+mb、すなわち、加水分解性基の総和は1〜5の範囲が好ましい。加水分解性基や水酸基が架橋性ケイ素基中に2個以上結合するときは、それらは同一であっても、異なっていてもよい。架橋性ケイ素化合物を構成するケイ素原子は1個でもよく、2個以上であってもよいが、シロキサン結合により連結されたケイ素原子の場合は20個程度まであってもよい。
【0049】
一般式15におけるR30およびR31の具体例としては、例えば、メチル基やエチル基などのアルキル基、シクロヘキシル基等のシクロアルキル基、フェニル基などのアリール基、ベンジル基などのアラルキル基、R’がメチル基やフェニル基等である(R’)3Si−で示されるトリオルガノシロキシ基等が挙げられる。
【0050】
これらヒドロシリル基含有化合物の中でも、特に一般式16
H−Si(R31)3-a(Y)a (16)
(式中、R31、Y、aは前記に同じ)
で示される架橋性基を有する化合物が入手容易な点から好ましい。一般式15又は16で示される架橋性基を有するヒドロシリル基含有化合物の具体例としては、
HSiCl3、HSi(CH3)Cl2、HSi(CH3)2Cl、HSi(OCH3)3、HSi(CH3)(OCH3)2、HSi(CH3)2OCH3、HSi(OC2H5)3、HSi(CH3)(OC2H5)2、HSi(CH3)2OC2H5、HSi(OC3H7)3、HSi(C2H5)(OCH3)2、HSi(C2H5)2OCH3、HSi(C6H5)(OCH3)2、HSi(C6H5)2(OCH3)、HSi(CH3)(OC(O)CH3)2、HSi(CH3)2O−[Si(CH3)2O]2-Si(CH3)
(OCH3)2、HSi(CH3)[O−N=C(CH3)2]2
(ただし、上記化学式中、C6H5はフェニル基を示す)
等が挙げられる。
【0051】
架橋性シリル基を有するヒドロシラン化合物をアルケニル基に付加させる際には、通常ヒドロシリル化触媒が添加される。このようなヒドロシリル化触媒として、例えば、白金単体、アルミナ、シリカ、カーボンブラック等の担体に白金固体を分散させたもの、塩化白金酸、塩化白金酸とアルコール、アルデヒド、ケトン等との錯体、白金−オレフィン錯体、白金(0)−ジビニルテトラメチルジシロキサン錯体が挙げられる。白金化合物以外の触媒の例としては、RhCl(PPh3)3、RhCl3、RuCl3、IrCl3、FeCl3、AlCl3、PdCl2・H2O、NiCl2、TiCl4等が挙げられる。これらの触媒は単独で用いてもよく、2種類以上を併用してもかまわない。触媒量としては特に制限はないが、ビニル系重合体のアルケニル基1molに対し、10-1〜10-8molの範囲で用いるのが良く、好ましくは10-3〜10-6molの範囲で用いるのがよい。10-8molより少ないと硬化が十分に進行しない。またヒドロシリル化触媒は高価であるので10-1mol以上用いないのが好ましい。
【0052】
また、上述の方法により精製されたアルケニル基を有するビニル系重合体は、これを含有する硬化性組成物を与える。この硬化性組成物は(C)アルケニル基を有するビニル系重合体、(D)ヒドロシリル基含有化合物、を含有するものである。
(C)成分のアルケニル基を有するビニル系重合体は、主鎖がビニル系モノマーの原子移動ラジカル重合により得られ、且つ分子内に少なくとも1個のアルケニル基を有するものである。アルケニル基は重合体の側鎖、又は末端のいずれに導入されていてもよいが、優れたゴム弾性を有する硬化物を得るためには末端にあることが好ましい。アルケニル基を有するビニル系重合体は、単独で用いても、また、2種類以上を混合して用いても良い。(C)成分の分子量としては特に制限はないが、500〜100000の範囲にあるのが好ましい。500以下であると、ビニル系重合体の本来の特性が発現されにくく、100000以上であると、非常に高粘度あるいは溶解性が低くなり、取り扱いが困難になる。
【0053】
(D)成分であるヒドロシリル基含有化合物としては、少なくとも2個以上のヒドロシリル基を有するものであれば、特に限定されない。すなわち、一般式17または18で表される鎖状ポリシロキサン
R32 3SiO−[Si(R32)2O]a−[Si(H)(R33)O]b−[Si(R33)(R34)O]c−SiR32 3 (17)
HR32 2SiO−[Si(R32)2O]a−[Si(H)(R33)O]b−[Si(R33)(R34)O]c−SiR32 2H (18)
(式中R32およびR33は炭素数1〜6のアルキル基、または、フェニル基、R34は炭素数1〜10のアルキル基またはアラルキル基、aは0≦a≦100、bは2≦b≦100、cは0≦c≦100の整数を示す)、一般式19で表される環状シロキサン
【0054】
【化4】
(式中R32、R33、R34は上記に同じ、dは0≦d≦8、eは2≦e≦10、fは0≦f≦8の整数を示し、かつ3≦d+e+f≦10である)を用いることができる。
これらは単独で用いても2種以上を混合して用いてもかまわない。これらのシロキサンの中でも(メタ)アクリル系重合体との相溶性の観点から、フェニル基を有する、一般式20、21で示される鎖状シロキサンや、一般式22、23で示される環状シロキサンが好ましい。
(CH3)3SiO−[Si(H)(CH3)O]g−[Si(C6H5)2O]h−Si(CH3)3 (20)
(CH3)3SiO−[Si(H)(CH3)O]g−[Si(CH3){CH2C(H)(R35)C6H5}O]h−Si(CH3)3 (21)
(式中、R35は水素またはメチル基、gは2≦g≦100、hは0≦h≦100の整数、C6H5はフェニル基を示す)
【0056】
【化5】
(式中、R36は水素又はメチル基、iは2≦i≦10、jは0≦j≦8、かつ3≦i+j≦10である整数、C6H5はフェニル基)
(D)成分の少なくとも2個以上のヒドロシリル基を有する硬化剤としてはさらに、分子中に2個以上のアルケニル基を有する低分子化合物に対し、式17〜23に示したヒドロシリル基含有化合物を、反応後にも一部のヒドロシリル基が残るようにして付加反応させて得られる化合物を用いることもできる。分子中に2個以上のアルケニル基を有する化合物としては、各種のものを用いることができる。例示するならば、1,4−ペンタジエン、1,5−ヘキサジエン、1,6−ヘプタジエン、1,7−オクタジエン、1,8−ノナジエン、1,9−デカジエン等の炭化水素系化合物、O,O’−ジアリルビスフェノールA、3,3’−ジアリルビスフェノールA等のエーテル系化合物、ジアリルフタレート、ジアリルイソフタレート、トリアリルトリメリテート、テトラアリルピロメリテート等のエステル系化合物、ジエチレングリコールジアリルカーボネート等のカーボネート系化合物が挙げられる。
【0058】
式17〜23に示した過剰量のヒドロシリル基含有化合物に対し、ヒドロシリル化触媒の存在下、上に挙げたアルケニル基含有化合物をゆっくり滴下することにより該化合物を得ることができる。このような化合物のうち、原料の入手容易性、過剰に用いたシロキサンの除去のしやすさ、さらには(C)成分の重合体への相溶性を考慮して、下記のものが好ましい。
【0059】
【化6】
重合体(C)と硬化剤(D)は任意の割合で混合することができるが、硬化性の面から、アルケニル基とヒドロシリル基のモル比が5〜0.2の範囲にあることが好ましく、さらに、2.5〜0.4であることが特に好ましい。モル比が5以上になると硬化が不十分でべとつきのある強度の小さい硬化物しか得られず、また、0.2より小さいと、硬化後も硬化物中に活性なヒドロシリル基が大量に残るので、クラック、ボイドが発生し、均一で強度のある硬化物が得られない。
【0061】
重合体(C)と硬化剤(D)との硬化反応は、2成分を混合して加熱することにより進行するが、反応をより迅速に進めるために、ヒドロシリル化触媒が添加される。このようなヒドロシリル化触媒としては、既に例示したもの全てが上述の条件で好適に使用されてよい。
本発明の2成分(C)、(D)、および必要に応じてヒドロシリル化触媒を混合し硬化させれば、発泡等の現象を伴うことなく、深部硬化性に優れた均一な硬化物が得られる。硬化条件については特に制限はないが、一般に0℃〜200℃、好ましくは30℃〜150℃、特に80℃〜150℃で硬化するのがよい。硬化物の性状は用いる(C)成分の重合体および(D)成分の硬化剤の主鎖骨格や分子量に依存するが、ゴム状のものから樹脂状のものまで幅広く作成することができる。本組成物から得られる硬化物の具体的な用途を挙げるならば、シーリング材、接着剤、粘着材、弾性接着剤、塗料、粉体塗料、発泡体、電気電子用ポッティング材、フィルム、ガスケット、各種成形材料、人工大理石等である。
【0062】
【実施例】
以下に、この反応の具体的な実施例を示すが、この反応は、下記実施例に限定されるものではない。
(製造例1) 2−アリロキシエチルメタクリレートの製造例
撹拌機、温度計、還流冷却管、ディーンスターク管を取り付けた三つ口フラスコに、メタクリル酸(137.7g、1.6mol)、エチレングリコールモノアリルエーテル(80.7g、0.8mol)、p−トルエンスルホン酸(0.76g、4.0mmol)、およびトルエン(650mL)を仕込んだ。120℃で5時間反応させた後、p−トルエンスルホン酸を0.12g追加し、さらに同じ温度で6時間反応させ、p−トルエンスルホン酸を0.1g追加した。同じ温度でさらに9時間反応させて反応を終了した。この間、液体クロマトグラフィーでメタクリル酸とエチレングリコールモノアリルエーテルを追跡し、転化率は最終的に98%に達した。NaHCO3水溶液を加えて中和し、2層を分離した。水層をトルエンで1回抽出し、有機層をCaCl2で乾燥した後、揮発分を減圧下留去した。粗生成物を減圧蒸留する(60℃、2mmHg)ことにより、下式に示す2−アリロキシエチルメタクリレートを98.7g得た(収率73%)。
H2C=C(CH3)CO2(CH2)2OCH2CH=CH2
(製造例2) アルケニル基を有する開始剤の製造例
50mLの2口フラスコを窒素置換し、2−アリルオキシエタノール(2.5mL、23.4mmol)、ピリジン(3mL)、およびTHF(10mL)を仕込んだ。溶液を0℃に冷却し、2−ブロモプロピオン酸クロライド(2mL、19.52mmol)をゆっくり滴下した。そのままの温度で1時間撹拌を続けた後、酢酸エチル(10mL)を加え、生成したピリジンの塩酸塩を濾過により除去した。濾液を希塩酸(10mL)、NaHCO3水溶液(10mL)、さらにブライン(10mL)で洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧化留去した。得られた粗生成物を減圧蒸留することにより、下式に示すアリルオキシエチル−2−ブロモプロピオネートを得た。(78.5〜81℃(1.3mmHg)、2.986g)。
CH3C(H)(Br)C(O)O−CH2CH2−O−CH2CH=CH2(製造例3) 水酸基を有する開始剤の製造例
窒素雰囲気下、エチレングリコール(10.9mL、195mmol)とピリジン(3g、39mmol)のTHF溶液(10mL)に2−ブロモプロピオン酸クロライド(2mL、3.35g、19.5mmol)を0℃でゆっくり滴下した。そのままの温度で溶液を2時間撹拌した。希塩酸(20mL)と酢酸エチル(30mL)を加え、2層を分離した。有機層を希塩酸、およびブラインで洗浄し、Na2SO4で乾燥した後、揮発分を減圧下留去し、粗成生物を得た(3.07g)。この粗生成物を減圧蒸留することにより(70〜73℃、0.5mmHg)、下式に示す、ヒドロキシエチル−2−ブロモプロピオネートを得た(2.14g、56%)。
CH3C(H)(Br)C(O)O(CH2)2−OH
(製造例4) アルケニル基を有するカルボン酸塩の製造例1
水酸化カリウムの1/2Nエタノ−ル溶液(200mL)にウンデシレン酸(18.8g、0.102mol)を撹拌しながら0℃でゆっくり滴下した。揮発分を減圧下留去することにより粗生成物を得た。粗生成物をアセトンで洗浄後、減圧下加熱することにより下式に示すウンデシレン酸カリウム塩の白色固体を得た(8.88g、収率88%)。
CH2=CH−(CH2)8−CO2 -K+
(製造例5) アルケニル基を有するカルボン酸塩の製造例2
メタノ−ル(245mL)に4−ペンテン酸(49g、0.489mol)、カリウム−tert−ブトキシド(54.9g、0.489mol)を仕込み、0℃で撹拌した。揮発分を減圧下留去することにより下式に示すペンテン酸カリウム塩を得た。
CH2=CH−(CH2)2−CO2 -K+
(製造例6)
1Lの耐圧反応容器に、アクリル酸−n−ブチル(112mL、100g、0.78mol)、製造例3で得られた水酸基含有開始剤(3.07g、15.6mmol)、臭化第一銅(2.24g、15.6mmol)、2,2’−ビピリジル(4.87g、31.2mmol)、酢酸エチル(90mL)、アセトニトリル(22mL)を仕込み、窒素バブリングを行って溶存酸素を除去した後、封管した。混合物を130℃に加熱し、2時間反応させた。反応容器を室温にもどし、メタクリル酸−2−ヒドロキシエチル(3.92mL、4.06g、31.2mmol)を加え、110℃で2時間反応させた。混合物を酢酸エチル(200mL)で希釈し、不溶分を濾別した後、濾液を10%塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥した後、溶媒を減圧下留去し、末端に水酸基を有するポリ(アクリル酸−n−ブチル)を82g得た。重合体の数平均分子量はGPC測定(ポリスチレン換算)により、5100、分子量分布は1.29であった。
【0063】
次に、上記のようにして得られた末端に水酸基を有するポリ(アクリル酸−n−ブチル)(50g)およびピリジン(10mL)のトルエン溶液(100mL)に、窒素雰囲気下、75℃で、ウンデセン酸クロリド(7.22mL、6.81g、33.6mmol)をゆっくりと滴下し、75℃で3時間撹拌した。生成した白色固体を濾過し、有機層を希塩酸およびブラインで洗浄した。有機層をNa2SO4で乾燥し、減圧下に濃縮することにより、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)(43g)を得た。
(製造例7)
還流管をつけた100mLの三口丸底フラスコに臭化第一銅(0.625g、15.6mmol)、アセトニトリル(5.0mL)、及び、ペンタメチルジエチレントリアミン(0.91mL)を仕込み、窒素ガスで置換した。アクリル酸−n−ブチル(50mL、44.7g、0.39mol)、及び、ジエチル−2,5−ジブロモアジペート(1.57g、4.36mmol)を添加し、70℃で7時間加熱撹拌した。混合物を酢酸エチルで希釈し、活性アルミナで処理した。揮発分を減圧下留去し、末端にハロゲンを有するポリ(アクリル酸−n−ブチル)を35.0g得た(重合収率87%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により10700、分子量分布は1.15であった。
【0064】
次に、還流管をつけた200mLの三口丸底フラスコに、上記のようにして得られた末端にハロゲンを有するポリ(アクリル酸−n−ブチル)(35.0g)、製造例2で合成された4−ペンテン酸カリウム塩(2.23g、16.1mmol)、及び、ジメチルアセトアミド(35mL)を仕込み、窒素雰囲気下、70℃で4時間反応させた。混合物を酢酸エチルで希釈し、2%塩酸、ブラインで洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去することにより重合体を単離した。
(製造例8)
100mLのガラス反応容器に、アクリル酸ブチル(50.0mL、44.7g、0.349mol)、臭化第一銅(1.25g、8.72mmol)、ペンタメチルジエチレントリアミン(1.82mL、1.51g、8.72mmol)、およびアセトニトリル(5mL)を仕込み、冷却後減圧脱気したのち窒素ガスで置換した。よく撹拌した後、ジエチル2,5−ジブロモアジペート(1.57g、4.36mmol)を添加し、70℃で加熱撹拌した。60分後に1,7−オクタジエン(6.44mL、4.80g、43.6mmol)を添加し、70℃で加熱撹拌を2時間継続した。混合物を活性アルミナで処理した後、揮発分を減圧下加熱して留去した。生成物を酢酸エチルに溶解させ、2%塩酸、ブラインで洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下加熱して留去することにより、末端にアルケニル基を有する重合体を得た。得られた重合体の数平均分子量はGPC測定(ポリスチレン換算)により13100、分子量分布は1.22であった。数平均分子量基準のオレフィン官能基導入率は2.01であった。
(製造例9)
30mLの耐圧ガラス反応容器に、アクリル酸メチル(2.5mL、2.4g、27.8mmol)、α,α’−ジブロモ−p−キシレン(146mg、0.555mmol)、臭化第一銅(79.5mg、4.44mmol)、2,2’−ビピリジル(259mg、1.67mmol)、および酢酸エチル(2.0mL)、アセトニトリル(0.5mL)を仕込み、凍結脱揮により溶存酸素を除去した後、封管した。混合物を130℃に加熱し、2.5時間反応させた。室温に冷却した後、製造例1で得られた2−アリロキシエチルメタクリレート(381mg、2.22mmol)を窒素ガス雰囲気下で添加して封管した。混合物を80℃に加熱し、2時間反応させた。混合物を酢酸エチル(50mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、アルケニル基を有するポリアクリル酸メチルを得た。重合体の数平均分子量はGPC測定(ポリスチレン換算)により6000、分子量分布は1.53であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1HNMR分析より、3.1個であった。
(実施例1)
製造例6で得られた重合体のトルエン溶液にアルミニウムシリケート(協和化学製:キョ−ワ−ド700PEL)を添加して還流温度で撹拌し、アルミニウムシリケートを除去した後、溶媒を留去することによって重合体を得た。
【0065】
次に、30mLの耐圧ガラス反応容器に、上記重合体(1.0g)、ジメトキシメチルヒドロシラン(0.16mL、1.30mmol)、トルエン(1.0mL)、及び、白金触媒を仕込んだ。ただし、白金触媒の使用量は、重合体のアルケニル基に対して、モル比で10-4当量とした。反応混合物を75℃で3時間加熱したところ、アルケニル基の35%が反応して重合体末端にシリル基が導入された。
(実施例2)
製造例7で得られた重合体に重合体と等重量のアルミニウムシリケート(協和化学製:キョ−ワ−ド700PEL)を添加して100℃で4時間撹拌し、末端にアルケニル基を有するポリ(アクリル酸ブチル)を得た。オリゴマ−1分子当たりに導入されたアルケニル基は、1H NMR分析より、1.82個であった。
【0066】
次に、200mLの耐圧ガラス反応容器に、上記重合体(15.0g)、ジメトキシメチルヒドロシラン(1.8mL、14.5mmol)、オルトぎ酸ジメチル(0.26mL、2.42mmol)、及び、白金触媒を仕込んだ。ただし、白金触媒の使用量は、重合体のアルケニル基に対して、モル比で2×10-4当量とした。反応混合物を100℃で4時間加熱した。混合物の揮発分を減圧留去することにより、末端にシリル基を有するポリ(アクリル酸−n−ブチル)を得た。オリゴマ−1分子当たりに導入されたシリル基は、1H NMR分析より、1.46個であった。
(実施例3)
製造例8で得られた重合体(30.5g)、重合体と等重量のアルミニウムシリケート(協和化学製:キョ−ワ−ド700PEL)とをトルエンに混合し、100℃で撹拌した。4時間後、アルミニウムシリケートを濾過し、濾液の揮発分を減圧下加熱して留去することによって重合体を精製した。
【0067】
次に、200mLの耐圧ガラス反応容器に、上記重合体(23.3g)、ジメトキシメチルヒドロシラン(2.55mL、20.7mmol)、オルトぎ酸ジメチル(0.38mL、3.45mmol)、および白金触媒を仕込んだ。ただし、白金触媒の使用量は、重合体のアルケニル基に対して、モル比で2×10-4当量とした。反応混合物を100℃で3時間加熱した。混合物の揮発分を減圧留去することにより、末端にシリル基を有するポリ(アクリル酸−n−ブチル)を得た。オリゴマ−1分子当たりに導入されたシリル基は、1H NMR分析より、1.41個であった。
(比較例1)
製造例9で得られた重合体(0.51g)、ジメトキシメチルヒドロシラン(0.040mL、0.326mmol)、および白金触媒を仕込んだ。ただし、白金触媒の使用量は、重合体のアルケニル基に対して、モル比で10-3当量とした。反応混合物を55℃で1時間加熱したが、シリル基は全く導入されなかった。
(製造例10)
100mLの耐圧ガラス反応容器に、アクリル酸メチル(20.0mL、19.1g、0.222mmol)、α,α’−ジブロモ−p−キシレン(1.168g、4.44mmol)、臭化第一銅(0.636g、4.44mmol)、2,2’−ビピリジル(2.07g、13.3mmol)、および酢酸エチル(16.0mL)、アセトニトリル(4.0mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、1時間反応させた。室温に冷却した後、製造例1で得られた2−アリロキシエチルメタクリレート(3.0g、17.6mmol)を窒素ガス雰囲気下で添加して封管した。混合物を80℃に加熱し、1時間反応させた。混合物を酢酸エチル(50mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、
揮発分を減圧下留去し、末端にアルケニル基を有するポリアクリル酸ブチルを20.5g得た(重合収率93%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により7900、分子量分布は1.99であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1HNMR分析より、3.3個であった。
(製造例11)
30mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(2.5mL、2.24g、17.45mmol)、α,α’−ジブロモ−p−キシレン(92.5mg、0.35mmol)、臭化第一銅(50mg、0.35mmol)、2,2’−ビピリジル(163mg、1.05mmol)、および酢酸エチル(2mL)、アセトニトリル(0.5mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、1時間反応させた。室温に冷却した後、製造例1で得られた2−アリロキシエチルメタクリレート(600mg、3.5mmol)を窒素ガス雰囲気下で添加して封管した。混合物を80℃に加熱し、1時間反応させた。混合物を酢酸エチル(20mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)を1.97g得た(重合収率88%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により6700、分子量分布は1.60であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1H NMR分析より、5.4個であった。
(製造例12)
50mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(10mL、8.94g、69.8mmol)、製造例2で得られたアリルオキシエチル−2−ブロモプロピオネート(332mg、1.4mmol)、臭化第一銅(200mg、1.4mmol)、2,2’−ビピリジル(433mg、2.8mmol)、および酢酸エチル(8mL)、アセトニトリル(2mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、2時間反応させた。室温に冷却した後、製造例1で得られたアリロキシエチルメタクリレート(480mg、2.8mmol)を窒素ガス雰囲気下で添加して封管した。混合物を100℃に加熱し、2時間反応させた。混合物を酢酸エチル(50mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)を8.88g得た(重合収率94%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により6700、分子量分布は1.79であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1H NMR分析より、1.2個であった。
(製造例13)
100mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(10mL、8.94g、69.8mmol)、製造例5で製造したアルケニル基を有する開始剤(332mg、1.40mmol)、臭化第一銅(200mg、1.40mmol)、2,2’−ビピリジル(433mg、2.80mmol)、アセトニトリル(2mL)、および酢酸エチル(8mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、1.5時間反応させた。室温に冷却した後、p−ジビニルベンゼン(364mg、2.80mmol)を窒素ガス雰囲気下で添加して封管した。混合物を100℃に加熱し、2時間反応させた。混合物を酢酸エチル(30mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、下式に示す両末端にアルケニル基を有するポリアクリル酸−n−ブチルを6.43g得た(69%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により3900、分子量分布は5.35であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1H NMR分析より、1.73個であった。
(製造例14)
50mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(10mL、8.94g、69.8mmol)、α,α’−ジブロモ−p−キシレン(370mg、1.4mmol)、臭化第一銅(200mg、1.4mmol)、2,2’−ビピリジル(433mg、2.8mmol)、およびメチルイソブチルケトン(10mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、20分反応させた。室温に冷却した後、混合物を酢酸エチル(20mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、末端に臭素を有するポリ(アクリル酸−n−ブチル)を5.21g得た(58%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により3700、分子量分布は1.41であった。
【0068】
次に、30mLの耐圧反応管に、上記のようにして得られたポリ(アクリル酸−n−ブチル)(2.0g)、p−ジビニルベンゼン(281mg、2.16mmol)、臭化第一銅(77mg、0.54mmol)、2,2’−ビピリジル(167mg、1.08mmol)、およびメチルイソブチルケトン(4mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、30分反応させた。室温に冷却した後、混合物を酢酸エチル(10mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)を2.11g得た。重合体の数平均分子量はGPC測定(ポリスチレン換算)により7300、分子量分布は2.47であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1H NMR分析より、2.1個であった。
(製造例15)
還流管付き500mL三つ口フラスコで、触媒として臭化第一銅(1.50g、10.5mmol)、配位子としてペンタメチルジエチレントリアミン(1.65mL)、開始剤としてジエチル−2,5−ジブロモアジペート(9.42g、26.2mol)、溶媒としてアセトニトリル(30mL)を用いて、アクリル酸−n−ブチル(300mL)を窒素雰囲気下70℃で重合し、アクリル酸−n−ブチルの重合率が93%の時点で、1,7−オクタジエン(38.6mL,0.261mol)を添加し、同温度で加熱した。反応混合物を酢酸エチルで希釈し、活性アルミナのカラムを通して触媒を除き、揮発分を減圧留去することにより、末端にアルケニル基を有する重合体を得た。重合体の数平均分子量はGPC測定(ポリスチレン換算)により13800、分子量分布は1.28であった。オリゴマ−1分子当たりに導入されたアルケニル基は、1H NMR分析より、1.84個であった。
(製造例16)
30mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(7.5mL、6.72g、51.3mmol)、α,α' −ジブロモ−p−キシレン(270mg、1.03mmol)、臭化第一銅(150mg、1.03mmol)、2,2' −ビピリジル(322mg、2.06mmol)、酢酸エチル(6mL)、及び、アセトニトリル(1.5mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、1.5時間反応させた。混合物を酢酸エチル(20mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、末端にハロゲンを有するポリ(アクリル酸−n−ブチル)を5.0g得た(重合収率75%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により5600、分子量分布は1.32であった。
【0069】
上記重合体(5.00g)、製造例4で合成されたウンデシレン酸カリウム塩(476mg、2.14mmol)、及び、ジメチルアセトアミド(10mL)を仕込み、窒素雰囲気下、70℃で6時間反応させた。混合物の揮発分を減圧留去した後、酢酸エチルを加えて不溶分を濾別した。濾液の揮発分を減圧留去することにより、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)4.77gを得た。オリゴマ−1分子当たりに導入されたアルケニル基は、1H NMR分析より、1.70個であった。
(製造例17)
製造例16と同様にして、触媒として臭化第一銅(1.50g、10.5mmol)、配位子としてペンタメチルジエチレントリアミン(0.69mL)、開始剤としてジエチル−2,5−ジブロモアジペート(9.42g、26.2mol)、溶媒としてアセトニトリル(30mL)を用いて、アクリル酸−n−ブチル(300mL)を窒素雰囲気下70℃で重合し、末端にハロゲンを有するポリ(アクリル酸−n−ブチル)を得た。重合体の数平均分子量はGPC測定(ポリスチレン換算)により11300、分子量分布は1.16であった。
【0070】
次に、カルボン酸塩として製造例5で製造されたペンテン酸カリウム塩を用いて製造例10と同様の操作により、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)を得た。オリゴマ−1分子当たりに導入されたアルケニル基は、1H NMR分析より、1.84個であった。
(製造例18)
30mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(5mL、4.47g、34.9mmol)、α,α’−ジブロモ−p−キシレン(180mg、0.69mmol)、臭化第一銅(98mg、0.69mmol)、2,2’−ビピリジル(319g、2.06mmol)、および酢酸エチル(4mL)、アセトニトリル(1ml)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、1時間反応させた。混合物を冷却後、窒素雰囲気下でアリルトリブチル錫(0.51mL、1.64mmol)を添加し、100℃で1時間反応させた。混合物を酢酸エチル(20mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去し、末端にアルケニル基を有するポリアクリル酸−n−ブチルとブロモトリブチル錫の混合物を得た(収量4.48g)。重合体の数平均分子量はGPC測定により(ポリスチレン換算)により6300、分子量分布は1.57であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1HNMR分析より、2.2個であった。
(製造例19)
30mLの耐圧ガラス反応容器に、アクリル酸−n−ブチル(2.5mL、2.24g、17.45mmol)、上で得られたアルケニル基を有する開始剤(165mg、0.698mmol)、臭化第一銅(100mg、0.698mmol)、2,2’−ビピリジル(218mg、1.40mmol)、アセトニトリル(0.5mL)、酢酸エチル(2mL)を仕込み、窒素ガスを10分間吹き込んで溶存酸素を除去した後、封管した。混合物を130℃に加熱し、50分反応させた。室温に冷却した後、混合物を酢酸エチル(20mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去して、片末端にアルケニル基、他の末端には臭素を有するポリ(アクリル酸−n−ブチル)を1.90g得た(79%)。重合体の数平均分子量はGPC測定により(ポリスチレン換算)により3600、分子量分布は1.51であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1H NMR分析より、0.75個であった。
【0071】
次に、撹拌子、還流冷却管を備えた50mLの3つ口フラスコに、上記重合体(1.90g)、Na2S・9H2O(70.2mg、0.293mmol)、およびエタノール(3mL)を仕込み、還流温度で3時間撹拌した。室温に冷却した後、酢酸エチル(10mL)、希塩酸(10mL)を加え、2層を分離した。有機層を希塩酸とブラインで洗浄し、Na2SO4で乾燥した後、揮発分を減圧下留去することにより、末端にアルケニル基を有するポリ(アクリル酸−n−ブチル)を1.69g得た。重合体の数平均分子量はGPC測定により(ポリスチレン換算)により5100、分子量分布は1.73であった。
(製造例20)
30mLの耐圧ガラス反応容器に、アクリル酸メチル(5mL、4.78g、55.5mmol)、2−メチル−2−ブロモプロピオン酸アリル(0.354mL、460mg、2.22mmol)、臭化第一銅(318mg、2.22mmolmmol)、2,2’−ビピリジル(1.04g、6.66mmol)、アセトニトリル(1mL)、酢酸エチル(4mL)を仕込み、真空脱気を3回行って溶存酸素を除去した後、封管した。混合物を80℃に加熱し、3時間反応させた。室温に冷却した後、混合物を酢酸エチル(20mL)で希釈し、生成した不溶固体をろ過した後、濾液を希塩酸で2回、ブラインで1回洗浄した。有機層をNa2SO4で乾燥し、揮発分を減圧下留去して、片末端にアルケニル基、他の末端には臭素を有するポリ(アクリル酸メチル)を3.93g得た(75%)。重合体の数平均分子量はGPC測定(ポリスチレン換算)により2700、分子量分布は1.48であった。また、オリゴマー1分子当たりに導入されたアルケニル基は、1H NMR分析より、0.81個であった。
【0072】
次に、撹拌子、還流冷却管を備えた50mLの3つ口フラスコに、上記のようにして得られた重合体(1.17g)、Na2S・9H2O(57.6mg、0.240mmol)、およびエタノール(2mL)を仕込み、還流温度で3時間撹拌した。室温に冷却した後、酢酸エチル(10mL)、希塩酸(10mL)を加え、2層を分離した。有機層を希塩酸とブラインで洗浄し、Na2SO4で乾燥した後、揮発分を減圧下留去することにより、末端にアルケニル基を有するポリ(アクリル酸メチル)を1.11g得た。重合体の数平均分子量はGPC測定により(ポリスチレン換算)により4200、分子量分布は1.71であった。
(実施例4)
製造例10で製造された、末端にアルケニル基を有する重合体(1.32g)をトルエン(20mL)に溶解し、重合体と等重量の活性炭(和光純薬:活性炭素、粉末)を添加して室温で4時間撹拌した。活性炭を濾別し、濾液の揮発分を減圧留去することによって、重合体0.62gを得た。
【0073】
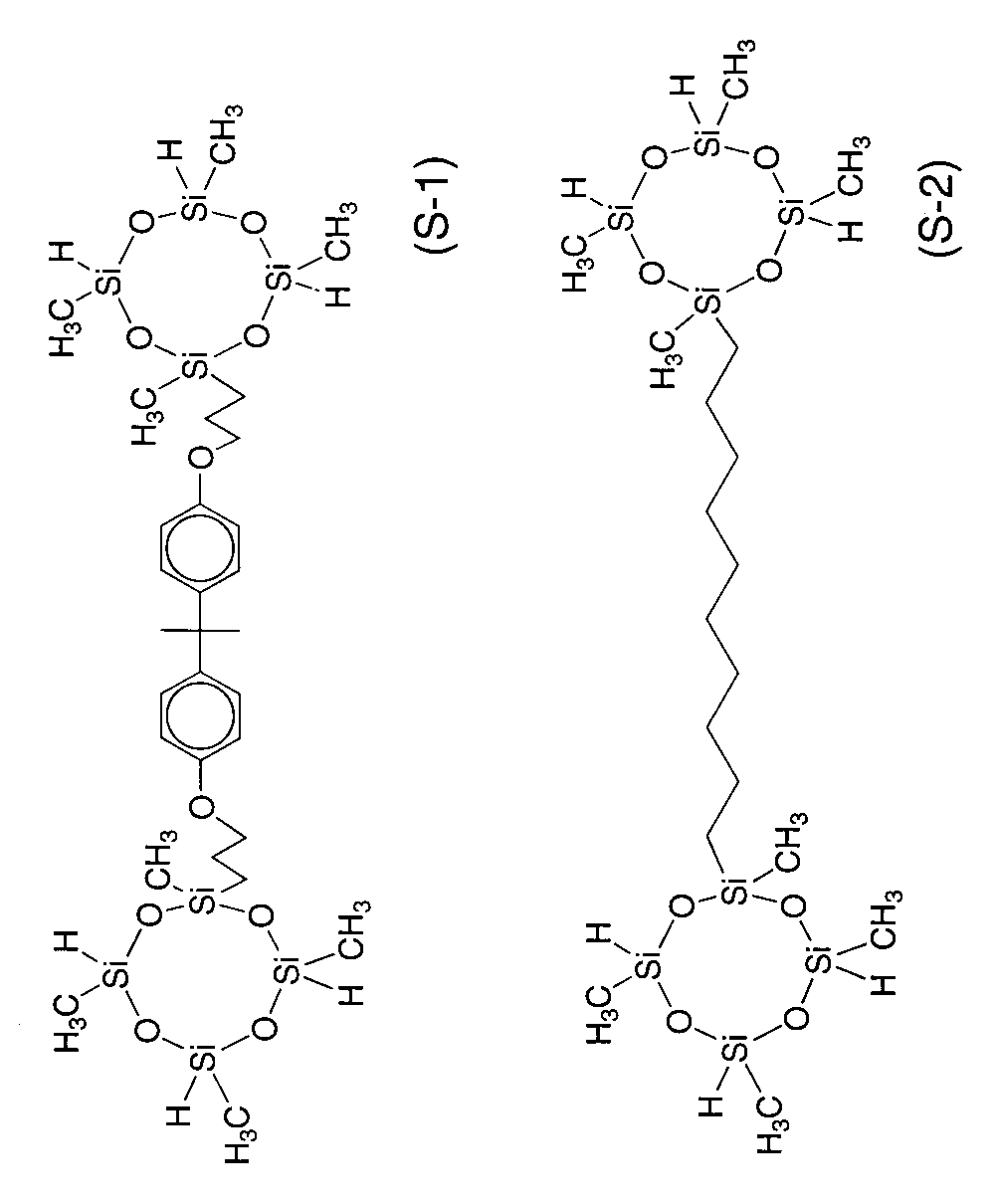
上記重合体と、下式に示す多価ハイドロジェンシリコン化合物(S−1)、および、0価白金の1,1,3,3−テトラメチル−1,3−ジビニルジシロキサン錯体(8.3×10-8molのキシレン溶液)をよく混合した。多価ハイドロジェンシリコン化合物の使用量は、重合体のアルケニル基とハイドロジェンシリコン化合物のヒドロシリル基がモル比で1/1.2となる量、また、白金触媒の使用量は、重合体のアルケニル基に対して、モル比で10-3当量とした。
【0074】
このようにして得られた組成物の一部を130℃のホットプレート上にて硬化試験を行い、ゲル化時間を測定した。また、残りの組成物を減圧下に脱気し、型枠に流し込んで加熱硬化させ、ゴム状の硬化物を得た。硬化物をアセトンに24時間浸漬し、前後の重量変化からそのゲル分率を測定した。結果を表1に示した。
【0075】
【化7】
(実施例5)
製造例10で製造された、末端にアルケニル基を有する重合体を、実施例1において活性炭のかわりにアルミニウムシリケート(協和化学製:キョーワード700PEL)を用いる以外は同様の操作により精製した。
精製された重合体について、実施例1と同様の操作により、硬化試験を行い、ゲル化時間およびゲル分率を測定した。結果を表1に示した。
(実施例6)
製造例11で製造された、末端にアルケニル基を有する重合体(1.70g)をトルエン(20mL)に溶解し、重合体と等重量のアルミニウムシリケート(協和化学製:キョーワード700PEL)を添加して還流温度で2時間撹拌した。アルミニウムシリケートを濾別し、濾液の揮発分を減圧留去することによって、重合体1.50gを得た。
【0077】
精製された重合体について、実施例1と同様の操作により、硬化試験を行い、ゲル化時間およびゲル分率を測定した。結果を表1に示した。
(実施例7〜19)
製造例12〜20、製造例6で得られた、末端にアルケニル基を有する重合体を、アルミニウムシリケート(協和化学製:キョーワード700PEL)を用いて実施例6と同様の操作により精製した。
【0078】
精製された重合体について、実施例1と同様の操作により、硬化試験を行い、ゲル化時間およびゲル分率を測定した。結果を表1に示した。なお、多価ハイドロジェンシリコン化合物として、下記に示す化合物(S−1、S−2)、もしくはα−メチルスチレンで一部変性したメチルハイドロジェンシロキサン(S−3:SiH価7.69mmol/g)のいずれかを用いた。多価ハイドロジェンシリコン化合物の使用量は、重合体のアルケニル基とハイドロジェンシリコン化合物のSiH基がモル比で1/1.2〜1/1.5となる量とした。白金触媒は、重合体のアルケニル基に対して、所定量添加した。
【0079】
【化8】
比較例2
製造例10で得られた末端にアルケニル基を有する重合体について、吸着剤精製を行わずに実施例1と同様の操作により硬化試験を行い、ゲル化時間を測定したところ、1時間経過後においても硬化しなかった。結果を表1に示した。
【0081】
【表1】
【発明の効果】
本発明によれば、原子移動ラジカル重合により製造されたアルケニル基を有するビニル系重合体を吸着剤で精製することにより、未精製では反応が阻害されていたアルケニル基のヒドロシリル化反応を速やかに進行させることができる。また、末端にアルケニル基を有するビニル系重合体、ヒドロシリル基含有化合物を含有する硬化性組成物が得られる。[0001]
BACKGROUND OF THE INVENTION
The present invention relates to a method for purifying a vinyl polymer and a curable composition.
[0002]
[Prior art]
The hydrosilylation reaction is used for functional group conversion, crosslinking reaction, and the like, and is one of industrially very useful reactions. For example, a polymer having an alkenyl group as a functional group at the end of a molecular chain can be crosslinked and cured by using a hydrosilyl group-containing compound as a curing agent to give a cured product having excellent heat resistance and durability. It is known that a polymer having a crosslinkable silyl group at the terminal is produced by reacting a polymer having an alkenyl group at the terminal with a hydrosilyl group-containing compound having a crosslinkable silyl group. These hydrosilylation reactions proceed by heating, but a hydrosilylation catalyst is added to advance the reaction more rapidly. Such hydrosilylation catalysts include radical initiators such as organic peroxides and azo compounds, and transition metal catalysts. In particular, it is known that when a transition metal catalyst is used, hydrosilylation can be rapidly advanced with a catalytic amount.
[0003]
On the other hand, a living polymerization method is generally known as a precision polymer synthesis method. In the living polymerization, not only the molecular weight and molecular weight distribution can be controlled, but also a polymer having a clear terminal structure can be obtained. Therefore, living polymerization can be cited as one effective method for introducing a functional group to the polymer terminal. Recently, also in radical polymerization, a polymerization system capable of living polymerization has been found, and research on living radical polymerization has been actively conducted. In particular, a vinyl polymer having a narrow molecular weight distribution can be obtained by utilizing atom transfer radical polymerization.
[0004]
By using these atom transfer radical polymerization methods, the present inventors radically polymerize vinyl monomers, and convert the terminal of the polymer to an alkenyl group-containing substituent, whereby vinyl having an alkenyl group at the end of the molecular chain. It has been found that a polymer can be obtained.
[0005]
[Problems to be solved by the invention]
However, in a vinyl polymer having an alkenyl group at the terminal produced by using an atom transfer radical polymerization method, a cured product can be prepared using a hydrosilyl group-containing compound as a curing agent and a transition metal catalyst as a hydrosilylation catalyst. Attempts have been made that do not cure or cure very slowly. That is, it was found that the hydrosilylation reaction is inhibited.
[0006]
The present invention solves this problem and provides a method for purifying a vinyl polymer so that the hydrosilylation reaction proceeds without being inhibited.
[0007]
[Means for Solving the Problems]
The present invention is a method for purifying a vinyl polymer by bringing (A) a vinyl polymer having a main chain produced by atom transfer radical polymerization into contact with (B) an adsorbent and subsequently removing the adsorbent. .
In the present invention, when the vinyl polymer having an alkenyl group at the terminal whose main chain is produced by atom transfer radical polymerization is reacted with the hydrosilyl group-containing compound, the polymer or the polymer is produced in advance. It is also a method of purifying the vinyl polymer by bringing the intermediate product obtained in the process into contact with an adsorbent and subsequently removing the adsorbent.
[0008]
The present invention is also a curable composition containing (C) a vinyl polymer having an alkenyl group purified by an adsorbent treatment and (D) a hydrosilyl group-containing compound.
[0009]
DETAILED DESCRIPTION OF THE INVENTION
The vinyl polymer in the present invention is one whose main chain is produced by an atom transfer radical polymerization method. Atom transfer radical polymerization is one of living radical polymerization, and is a method of radical polymerization of vinyl monomers using an organic halide or a sulfonyl halide compound as an initiator and a metal complex having a transition metal as a central metal as a catalyst. It is. Specifically, Matyjazewski et al. Am. Chem. Soc. 1995, 117, 5614, Macromolecules 1995, 28, 7901, Science 1996, 272, 866; or the method described in Sawamoto et al., Macromolecules 1995, 28, 1721, International Publications WO 96/30421, WO 97/18247 It is done.
[0010]
In the narrow sense, living polymerization indicates polymerization in which the terminal always has activity and the molecular chain grows. In general, the terminal is inactivated and the terminal is in an equilibrium state. This includes pseudo-living polymerization that grows in spite of this. The definition of living polymerization in the present invention is also the latter.
The vinyl monomer constituting the main chain of the vinyl polymer in the present invention is not particularly limited, and various types can be used. For example, (meth) acrylic acid, methyl (meth) acrylate, ethyl (meth) acrylate, (meth) acrylic acid-n-propyl, (meth) acrylic acid isopropyl, (meth) acrylic acid-n- Butyl, isobutyl (meth) acrylate, (tert-butyl) (meth) acrylate, n-pentyl (meth) acrylate, n-hexyl (meth) acrylate, cyclohexyl (meth) acrylate, (meth) acryl Acid-n-heptyl, (meth) acrylic acid-n-octyl, (meth) acrylic acid-2-ethylhexyl, (meth) acrylic acid nonyl, (meth) acrylic acid decyl, (meth) acrylic acid dodecyl, (meth) Phenyl acrylate, toluyl (meth) acrylate, benzyl (meth) acrylate, 2-methoxyethyl (meth) acrylate (Meth) acrylic acid-3-methoxybutyl, (meth) acrylic acid-2-hydroxyethyl, (meth) acrylic acid-2-hydroxypropyl, (meth) acrylic acid stearyl, (meth) acrylic acid glycidyl, (meth) 2-aminoethyl acrylate, γ- (methacryloyloxypropyl) trimethoxysilane, ethylene oxide adduct of (meth) acrylic acid, trifluoromethylmethyl (meth) acrylate, 2-trifluoromethylethyl (meth) acrylate , 2-perfluoroethylethyl (meth) acrylate, 2-perfluoroethyl-2-perfluorobutylethyl (meth) acrylate, 2-perfluoroethyl (meth) acrylate, perfluoromethyl (meth) acrylate , (Perfluoromethylmethyl) (meth) acrylate, (me T) 2-perfluoromethyl-2-perfluoroethylmethyl acrylate, 2-perfluorohexylethyl (meth) acrylate, 2-perfluorodecylethyl (meth) acrylate, 2-perfluoro (meth) acrylate (Meth) acrylic acid monomers such as hexadecylethyl; styrene monomers such as styrene, vinyltoluene, α-methylstyrene, chlorostyrene, styrenesulfonic acid and salts; such as perfluoroethylene, perfluoropropylene and vinylidene fluoride Fluorine-containing vinyl monomers; silicon-containing vinyl monomers such as vinyltrimethoxysilane and vinyltriethoxysilane; maleic anhydride, maleic acid, monoalkyl esters and dialkyl esters of maleic acid; fumaric acid, monoalkyl esters of fumaric acid and dialkyl Alkyl esters; maleimide monomers such as maleimide, methylmaleimide, ethylmaleimide, propylmaleimide, butylmaleimide, hexylmaleimide, octylmaleimide, dodecylmaleimide, stearylmaleimide, phenylmaleimide, cyclohexylmaleimide; nitrile groups such as acrylonitrile and methacrylonitrile Vinyl monomers; amide group-containing vinyl monomers such as acrylamide and methacrylamide; vinyl esters such as vinyl acetate, vinyl propionate, vinyl pivalate, vinyl benzoate and vinyl cinnamate; alkenes such as ethylene and propylene; butadiene And conjugated dienes such as isoprene; vinyl chloride, vinylidene chloride, allyl chloride, allyl alcohol, etc., which are used alone Alternatively, a plurality of them may be copolymerized. In the above expression format, for example, (meth) acrylic acid represents acrylic acid and / or methacrylic acid.
Of these, styrene monomers and (meth) acrylic monomers are preferred, acrylate monomers are preferred, and butyl acrylate is particularly preferred, from the physical properties of the product. In the present invention, these preferable monomers may be copolymerized with other monomers, and in this case, it is preferable that these preferable monomers are contained in a weight ratio of 40%.
[0011]
The number average molecular weight of the vinyl polymer is not particularly limited, but is preferably in the range of 500 to 1,000,000, and more preferably 500 to 100,000.
The molecular weight distribution of the vinyl polymer is not particularly limited, but the ratio of the weight average molecular weight to the number average molecular weight measured by gel permeation chromatography is usually less than 1.8, preferably 1.7 or less. More preferably 1.6 or less, even more preferably 1.5 or less, particularly preferably 1.4 or less, and most preferably 1.3 or less. In the GPC measurement in the present invention, chloroform is usually used as the mobile phase, the measurement is performed with a polystyrene gel column, and the number average molecular weight and the like can be determined in terms of polystyrene.
[0012]
The alkenyl group in the present invention is a group represented by the general formula (1).
CH2= C (R1-(1)
(Wherein R1Represents hydrogen or a methyl group. )
In the vinyl polymer having an alkenyl group in the present invention, the alkenyl group may be present at either the side chain or the terminal of the polymer, and the number of alkenyl groups contained in one molecular chain is not particularly limited. However, in order to obtain a cured product of a vinyl polymer having an alkenyl group using a hydrosilyl group-containing compound as a curing agent, at least one alkenyl group is required in one molecular chain, and rubber elasticity In order to obtain an excellent cured product, it is preferred that the alkenyl group is at the terminal rather than the side chain.
[0013]
As a method for producing the vinyl polymer in the present invention, atom transfer radical polymerization is used. The initiator used for the polymerization is not particularly limited. For example, an organic halide, particularly an activated organic halide (for example, an ester compound having a halogen at the α-position or a compound having a halogen at the benzyl-position). Or a halogenated sulfonyl compound. When these compounds are used as an initiator, a vinyl polymer having a halogen at the polymer end can be obtained. A vinyl polymer having an alkenyl group at the terminal can be obtained by converting this terminal halogen by the method described later. A specific example of such an initiator is C6HFive-CH2X, C6HFive-C (H) (X) CHThree, C6HFive-C (X) (CHThree)2(Where C6HFiveRepresents a phenyl group. X represents chlorine, bromine or iodine. ; R2-C (H) (X) -CO2RThree, R2-C (CHThree) (X) -CO2RThree, R2-C (H) (X) -C (O) RThree, R2-C (CHThree) (X) -C (O) RThree(Wherein R2And RThreeAre the same or different and each represents a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, or an aralkyl group having 7 to 20 carbon atoms. X represents chlorine, bromine or iodine. ; R2-C6HFour-SO2X (wherein R2Represents a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, or an aralkyl group having 7 to 20 carbon atoms. X represents chlorine, bromine or iodine. ) And the like.
[0014]
As the initiator, an organic halide or a sulfonyl halide compound having a functional group other than the functional group for initiating polymerization can also be used. In this case, a polymer having a functional group derived from the functional group contained in the initiator at one main chain end and a halogen at the other end is produced. Examples of the functional group include an alkenyl group, a crosslinkable silyl group, a hydroxyl group, an epoxy group, an amino group, an amide group, and a carboxyl group.
[0015]
The organic halide having an alkenyl group is not limited, and examples thereof include those having a structure represented by Formula 2.
RFourRFiveC (X) -R6-R7-C (R8) = CH2(2)
(Wherein R8Is hydrogen or a methyl group, RFour, RFiveIs hydrogen, a monovalent alkyl group having 1 to 20 carbon atoms, a monovalent aryl group having 6 to 20 carbon atoms, or a monovalent aralkyl group having 7 to 20 carbon atoms, or those connected to each other at the other end , R6-C (O) O- (ester group), -C (O)-(keto group), or o-, m-, p-phenylene group, R7Is a direct bond, or a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds, X is chlorine, bromine, or iodine)
Substituent RFour, RFiveSpecific examples thereof include hydrogen, methyl group, ethyl group, n-propyl group, isopropyl group, butyl group, pentyl group, hexyl group and the like. RFourAnd RFiveMay be connected at the other end to form a cyclic skeleton.
[0016]
Examples of the organic halide having an alkenyl group further include a compound represented by the general formula (3).
H2C = C (R8-R7-C (RFour) (X) -R9-RFive(3)
(Wherein RFour, RFive, R7, R8Is the same as above. R9Represents a direct bond, —C (O) O— (ester group), —C (O) — (keto group), or o-, m-, p-phenylene group. X is the same as above. )
R7Is a direct bond or a divalent organic group having 1 to 20 carbon atoms (which may contain one or more ether bonds). The groups are bonded and are allylic halides. In this case, since the carbon-halogen bond is activated by the adjacent vinyl group, R9It is not always necessary to have a C (O) O group or a phenylene group, and a direct bond may be used. R7In order to activate the carbon-halogen bond,9Is preferably a C (O) O group, a C (O) group, or a phenylene group.
[0017]
If specific examples of the sulfonyl halide compound having an alkenyl group are given,
o-, m-, p-CH2= CH- (CH2)n-C6HFour-SO2X,
o-, m-, p-CH2= CH- (CH2)n-OC6HFour-SO2X,
(In the above formulas, X is chlorine, bromine, or iodine, and n is an integer of 0 to 20)
Etc.
[0018]
It does not specifically limit as an organic halide which has a crosslinkable silyl group, For example, what has a structure shown in General formula (4) is illustrated.
RFourRFiveC (X) -R6-R7-C (H) (R8) CH2-[Si (RTen)2-b(Y)bO]m-Si (R11)3-a(Y)a(4)
(Wherein RFour, RFive, R6, R7, R8, X is the same as above, RTen, R11Are all alkyl groups having 1 to 20 carbon atoms, aryl groups having 6 to 20 carbon atoms, aralkyl groups having 7 to 20 carbon atoms, or (R ')ThreeA triorganosiloxy group represented by SiO— (R ′ is a monovalent hydrocarbon group having 1 to 20 carbon atoms, and three R ′ may be the same or different). , RTenOr R11When two or more are present, they may be the same or different. Y represents a hydroxyl group or a hydrolyzable group, and when two or more Y exist, they may be the same or different. a represents 0, 1, 2, or 3, and b represents 0, 1, or 2. m is an integer of 0-19. However, it shall be satisfied that a + mb ≧ 1)
Examples of the organic halide having a crosslinkable silyl group further include those having a structure represented by the general formula (5).
[0019]
(R11)3-a(Y)aSi- [OSi (RTen)2-b(Y)b]m-CH2-C (H) (R8-R7-C (RFour) (X) -R9-RFive(5)
(Wherein RFour, RFive, R7, R8, R9, RTen, R11, A, b, m, X, Y are the same as above)
It does not specifically limit as an organic halide with a hydroxyl group, or a halogenated sulfonyl compound, The following are illustrated.
[0020]
HO- (CH2)n-OC (O) C (H) (R) (X)
(In the above formulas, X is chlorine, bromine, or iodine, R is a hydrogen atom or an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, an aralkyl group having 7 to 20 carbon atoms, and n is 1-20 integer)
It does not specifically limit as an organic halide with an amino group, or a halogenated sulfonyl compound, The following are illustrated.
H2N- (CH2)n-OC (O) C (H) (R) (X)
(In the above formulas, X is chlorine, bromine, or iodine, R is a hydrogen atom or an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, an aralkyl group having 7 to 20 carbon atoms, and n is 1-20 integer)
It does not specifically limit as an organic halide which has an epoxy group, or a halogenated sulfonyl compound, The following are illustrated.
[0021]
[Chemical 1]
(In the above formulas, X is chlorine, bromine, or iodine, R is a hydrogen atom or an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, an aralkyl group having 7 to 20 carbon atoms, and n is 1-20 integer)
Furthermore, it can also superpose | polymerize using the organic halide or sulfonyl halide compound which has a 2 or more start point as an initiator. In such a case, a vinyl polymer having two or more halogen atoms in one molecule is produced.
[0023]
If an initiator having the above two starting points is specifically illustrated,
[0024]
[Chemical 2]
(Where C6HFourRepresents a phenylene group. X represents chlorine, bromine or iodine. R represents an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, or an aralkyl group having 7 to 20 carbon atoms. n represents an integer of 0 to 20. );
[0026]
[Chemical Formula 3]
(In the formula, X represents chlorine, bromine or iodine. N represents an integer of 0 to 20. C.6HFourRepresents a phenylene group. ) And the like.
The transition metal complex used as the polymerization catalyst is not particularly limited, and preferred examples include a complex of zero-valent copper, monovalent copper, divalent ruthenium, divalent iron, or divalent nickel. Of these, a copper complex is preferable. Specific examples of monovalent copper compounds include cuprous chloride, cuprous bromide, cuprous iodide, cuprous cyanide, cuprous oxide, cuprous perchlorate, etc. is there. When a copper compound is used, 2,2′-bipyridyl and its derivatives, 1,10-phenanthroline and its derivatives, tetramethylethylenediamine, pentamethyldiethylenetriamine, hexamethyltris (2-aminoethyl) amine, etc. in order to increase the catalytic activity A ligand such as a polyamine is added. In addition, tristriphenylphosphine complex of divalent ruthenium chloride (RuCl2(PPhThree)Three) Is also suitable as a catalyst. When a ruthenium compound is used as a catalyst, an aluminum alkoxide is added as an activator. Furthermore, a divalent iron bistriphenylphosphine complex (FeCl2(PPhThree)2) Bivalent nickel bistriphenylphosphine complex (NiCl)2(PPhThree)2), And a bivalent nickel bistributylphosphine complex (NiBr)2(PBuThree)2) Is also suitable as a catalyst.
[0028]
The polymerization can be carried out without solvent or in various solvents. When a solvent is used, examples of the solvent include hydrocarbon solvents such as benzene and toluene; ether solvents such as diethyl ether, tetrahydrofuran, diphenyl ether, anisole and dimethoxybenzene; halogenated carbonization such as methylene chloride, chloroform and chlorobenzene. Hydrogen solvents; ketone solvents such as acetone, methyl ethyl ketone, methyl isobutyl ketone; alcohol solvents such as methanol, ethanol, propanol, isopropanol, n-butyl alcohol, tert-butyl alcohol; acetonitrile, propionitrile, benzonitrile, etc. Examples thereof include nitrile solvents; ester solvents such as ethyl acetate and butyl acetate; carbonate solvents such as ethylene carbonate and propylene carbonate. These can be used individually or in mixture of 2 or more types. Also, emulsion system or supercritical fluid CO2Polymerization can also be carried out in a system in which
[0029]
The polymerization can be carried out in the range of 0 to 200 ° C, preferably in the range of room temperature to 150 ° C.
As the vinyl monomers used for the polymerization, all those already exemplified may be suitably used.
An alkenyl group can be introduced at the terminal by converting the terminal halogen of the vinyl polymer obtained by atom transfer radical polymerization of a vinyl monomer into an alkenyl group-containing substituent. The method for synthesizing the vinyl polymer having an alkenyl group is not particularly limited, and examples thereof include the following methods (Aa) to (Aj).
[0030]
(Aa) When synthesizing a vinyl polymer by radical polymerization, together with a predetermined vinyl monomer, a polymerizable alkenyl group and low polymerizability in one molecule represented by the following general formula (7) A method in which a compound having an alkenyl group is also reacted.
H2C = C (R12-R13-R14-C (R12) = CH2(7)
(Wherein R12Is hydrogen or a methyl group and two R12May be the same as or different from each other. R13Represents —C (O) O— or o-, m-, p-phenylene, R14Represents a direct bond or a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds. )
There is no limitation on the timing for reacting a compound having both a polymerizable alkenyl group and a low polymerizable alkenyl group in one molecule. However, when a rubber-like property is expected, the end of the polymerization reaction or a predetermined monomer is used. It is preferable to make it react as a 2nd monomer after completion | finish of reaction.
[0031]
(Ab) When a vinyl polymer is synthesized by atom transfer radical polymerization, for example, 1,5-hexadiene, 1,7-octadiene, 1,9- A method of reacting a compound having at least two alkenyl groups having low polymerizability such as decadiene.
(Ac) Reaction of various organometallic compounds having an alkenyl group such as organotin such as allyltributyltin and allyltrioctyltin with a vinyl polymer having halogen at the terminal obtained by atom transfer radical polymerization To replace the halogen.
[0032]
(Ad) A method of substituting halogen by reacting a vinyl-based polymer having halogen at a terminal obtained by atom transfer radical polymerization with a stabilized carbanion having an alkenyl group as shown in the general formula (8).
M+C-(R15) (R16-R17-C (R18) = CH2(8)
(Wherein R18Is hydrogen or methyl group, R15, R16Are both carbanion C-Are one of the electron-withdrawing groups and the other is hydrogen, an alkyl group having 1 to 10 carbon atoms, or a phenyl group. R17Represents a direct bond or a divalent organic group having 1 to 10 carbon atoms, and may contain one or more ether bonds. M+Represents an alkali metal ion or a quaternary ammonium ion)
R15, R16As an electron withdrawing group,2Those having the structures of R, -C (O) R and -CN are particularly preferred.
[0033]
(Ae) An enolate anion is prepared by allowing a metal polymer such as zinc or an organometallic compound to act on a vinyl polymer having halogen at the terminal obtained by atom transfer radical polymerization, and then halogen or acetyl A method of reacting with an electrophilic compound having an alkenyl group, such as an alkenyl group-containing compound having a leaving group such as a group, a carbonyl compound having an alkenyl group, an isocyanate compound having an alkenyl group, and an acid halide having an alkenyl group.
[0034]
(Af) Reaction of an oxyanion or carboxylate anion having an alkenyl group as shown by, for example, general formula (9) or (10) with a vinyl polymer having halogen at the terminal obtained by atom transfer radical polymerization To replace the halogen.
H2C = C (R19-R20-O-M+(9)
(Wherein R19Is hydrogen or methyl group, M+Is the same as above. R20Is a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds)
H2C = C (R19-Rtwenty one-C (O) O-M+(10)
(Wherein R19Is hydrogen or methyl group, M+Is the same as above. Rtwenty oneMay be a direct bond or a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds)
Etc.
[0035]
Furthermore, the vinyl polymer having an alkenyl group can be obtained from a vinyl polymer having a hydroxyl group, but the specific method is not particularly limited. For example, the following (Ag) to (A- The method of j) can be mentioned. The vinyl polymer having a hydroxyl group can be obtained by the methods (Ba) to (Bf) described later.
[0036]
(Ag) A method of reacting a base such as sodium methoxide with an alkenyl group-containing halide such as allyl chloride.
(Ah) A method of reacting an alkenyl group-containing isocyanate compound such as allyl isocyanate.
(Ai) A method in which an alkenyl group-containing acid halide such as (meth) acrylic acid chloride is reacted in the presence of a base such as pyridine.
[0037]
(Aj) A method in which an alkenyl group-containing carboxylic acid such as acrylic acid is reacted in the presence of an acid catalyst;
Although the following methods are illustrated as a manufacturing method of the vinyl polymer which has a hydroxyl group at the terminal, it is not limited to these methods.
(Ba) When synthesizing a vinyl polymer by atom transfer radical polymerization, for example, a compound having both a polymerizable alkenyl group and a hydroxyl group in one molecule as shown in the following general formula (11) is used. The method of making it react as a monomer.
H2C = C (Rtwenty two-Rtwenty three-Rtwenty four-OH (11)
(Wherein Rtwenty twoIs hydrogen or a methyl group. Rtwenty threeRepresents —C (O) O— or o-, m-, p-phenylene, Rtwenty fourRepresents a direct bond or a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds. )
There is no limitation on the timing of reacting the compound having both a polymerizable alkenyl group and a hydroxyl group in one molecule. However, particularly in the case of living radical polymerization, when the rubber-like properties are expected, the end of the polymerization reaction or a predetermined monomer. After completion of the reaction, it is preferable to react as the second monomer.
[0038]
(Bb) When synthesizing a vinyl polymer by atom transfer radical polymerization, an alkenyl alcohol such as 10-undecenol, 5-hexenol, or allyl alcohol is added at the end of the polymerization reaction or after completion of the reaction of a predetermined monomer. How to react.
(Bc) A method of introducing a hydroxyl group at the terminal by hydrolyzing or reacting a halogen of a vinyl polymer obtained by atom transfer radical polymerization with a hydroxyl group-containing compound.
(Bd) A method in which halogen is substituted by reacting a terminal halogen of a vinyl polymer obtained by atom transfer radical polymerization with a stabilized carbanion having a hydroxyl group as shown in the general formula (12).
[0039]
M+C-(Rtwenty five) (R26-R27-OH (12)
(Wherein Rtwenty five, R26Are both carbanion C-Are one of the electron-withdrawing groups and the other is hydrogen, an alkyl group having 1 to 10 carbon atoms, or a phenyl group. R27Represents a direct bond or a divalent organic group having 1 to 10 carbon atoms, and may contain one or more ether bonds. M+Represents an alkali metal ion or a quaternary ammonium ion)
Rtwenty five, R26As an electron withdrawing group,2Those having the structures of R, -C (O) R and -CN are particularly preferred.
[0040]
(Be) An enolate anion is prepared by allowing a metal simple substance or an organometallic compound such as zinc to act on a terminal halogen of a vinyl polymer obtained by atom transfer radical polymerization, and then an aldehyde or a ketone How to react.
(Bf) A terminal halogen of a vinyl polymer obtained by atom transfer radical polymerization is allowed to react with an oxyanion or carboxylate anion having a hydroxyl group as shown in, for example, the general formula (13) or (14). How to replace
HO-R28-O-M+(13)
(Where M+Is the same as above. R28Is a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds)
HO-R29-C (O) O-M+(14)
(Where M+Is the same as above. R29May be a direct bond or a divalent organic group having 1 to 20 carbon atoms and may contain one or more ether bonds).
[0041]
Furthermore, the vinyl polymer which has an alkenyl group at the terminal can utilize the method of coupling vinyl polymers. Although the method illustrated below can be utilized, it is not necessarily limited to these.
A compound having a total of two or more of the same or different functional groups capable of substituting halogen with respect to the terminal halogen of the polymer obtained by performing atom transfer radical polymerization of a vinyl-based monomer using an initiator having a functional group Use to couple. Examples of the compound having two or more of the same or different functional groups that can substitute for the halogen include polyols, polyamines, polycarboxylic acids, polythiols, and salts thereof, alkali metal sulfides, and the like.
[0042]
Next, a method for purifying a vinyl polymer using an adsorbent will be described in detail.
The adsorbent used in the present invention is not particularly limited, and examples thereof include activated carbon, silicon dioxide, solid acid, acid clay, activated clay, activated alumina, silica gel, silica / alumina, and aluminum silicate. Among these, activated carbon, activated alumina, silicon dioxide, and aluminum silicate are preferable, and activated carbon and aluminum silicate are more preferable.
[0043]
Activated carbon is mostly carbonaceous charcoal and has high adsorptivity. In the production method, for example, wood, lignite, peat or the like is used as an activator to treat with zinc chloride or phosphoric acid and dry distillation, or charcoal or the like is activated with steam. Usually, it is powdery or granular, and both can be used.
Silicon dioxide is known to be crystalline, amorphous, amorphous, glassy, synthetic or natural, but can be used here in the form of powder. As silicon dioxide, synthetic silicic acid such as silicic acid made from clay mineral obtained by acid treatment of activated clay, Carplex BS304, Carplex BS304F, Carplex # 67, Carplex # 80 (all of which are Shionogi) However, it is not necessarily limited to these.
[0044]
Aluminum silicate is a part of silicon silicate that is replaced with aluminum. Natural aluminum silicates such as pumice, fly ash, kaolin, bentonite, activated clay, diatomaceous earth, zeolite, and synthetic aluminum silicate are used. Are known. Among these, synthetic aluminum silicate has a large specific surface area and high adsorption capacity. Examples of the synthetic aluminum silicate include Kyoward 700PEL, Kyoword 700PL, Kyoword 700SL (all manufactured by Kyowa Chemical), and the like, but are not limited thereto. The above adsorbents may be used alone or in admixture of two or more.
[0045]
Purification by the adsorbent can be performed by bringing the adsorbent into contact with a vinyl polymer having an alkenyl group synthesized by the above-described production method. Although no solvent may be used, it is preferably carried out in a solvent such as benzene, toluene, ethyl acetate or the like because of easy handling. Although there is no restriction | limiting in particular about the temperature and pressure of an adsorbent process, Generally it is good to carry out at normal temperature and 0 to 200 degreeC, Preferably it is room temperature to 150 degreeC. Finally, the purified vinyl polymer is obtained by removing the adsorbent. The amount of the adsorbent used is in the range of 0.1 to 500 parts by weight with respect to 100 parts by weight of the vinyl polymer, but more preferably in the range of 5 to 200 parts by weight from the viewpoint of economy and operation. is there.
[0046]
Various embodiments are possible for the solid-liquid contact between the adsorbent and the polymer solution. In addition to the batch method in which stirring and mixing and solid-liquid separation are performed by batch operation, the adsorbent is filled in a container and the polymer solution is passed through. A liquid fixed bed system, a moving bed system in which liquid is passed through the moving bed of adsorbent, a fluidized bed system in which the adsorbent is fluidized with liquid and adsorbed can be used. Furthermore, in addition to mixing and dispersing by stirring, various operations for improving the dispersion efficiency such as shaking of the container and use of ultrasonic waves can be incorporated as necessary.
[0047]
After the polymer solution is brought into contact with the adsorbent, the adsorbent is removed by a method such as filtration, centrifugation, and sedimentation separation, and water washing is performed as necessary to obtain a desired clear polymer solution.
Usually, purification with an adsorbent may be performed on a vinyl polymer having an alkenyl group, but the adsorbent may be purified in the course of the process of producing the polymer. That is, the intermediate product obtained in the production process of a vinyl polymer having an alkenyl group, for example, a vinyl polymer having halogen at the terminal, a vinyl polymer having a hydroxyl group at the terminal, etc. is purified in advance by an adsorbent. May be performed.
[0048]
By purifying the vinyl polymer with an adsorbent, the hydrosilyl group-containing compound can be quickly added to the alkenyl group of the vinyl polymer.
For example, when a hydrosilyl group-containing compound having a crosslinkable silyl group is added to the terminal alkenyl group of the vinyl polymer, a vinyl polymer having a crosslinkable silyl group at the terminal can be obtained. Although there is no restriction | limiting in particular as a hydrosilyl group containing compound which has a crosslinkable silyl group, When a typical thing is shown, the compound shown by General formula 15 will be illustrated.
H- [Si (R30)2-b(Y)bO]m-Si (R31)3-a(Y)a(15)
(Wherein R30And R31Are all alkyl groups having 1 to 20 carbon atoms, aryl groups having 6 to 20 carbon atoms, or aralkyl groups having 7 to 20 carbon atoms, or (R ')ThreeA triorganosiloxy group represented by Si— (R ′ is a monovalent hydrocarbon group having 1 to 20 carbon atoms, and three R ′ may be the same or different). , R30Or R31When two or more are present, they may be the same or different. Y represents a hydroxyl group or a hydrolyzable group, and when two or more Y exist, they may be the same or different. a represents 0, 1, 2, or 3, and b represents 0, 1, or 2. m is an integer of 0-19. However, it shall be satisfied that a + mb ≧ 1. )
The hydrolyzable group represented by Y is not particularly limited, and conventionally known ones can be used. Specifically, hydrogen, halogen atom, alkoxy group, acyloxy group, ketoximate group, amino group, amide group can be used. , An acid amide group, an aminooxy group, a mercapto group, an alkenyloxy group and the like, and an alkoxy group is particularly preferred from the viewpoint that it is mild and easy to handle. The hydrolyzable group or hydroxyl group can be bonded to one silicon atom in the range of 1 to 3, and a + mb, that is, the total sum of hydrolyzable groups is preferably in the range of 1 to 5. When two or more hydrolyzable groups or hydroxyl groups are bonded to the crosslinkable silicon group, they may be the same or different. The number of silicon atoms constituting the crosslinkable silicon compound may be one or two or more. In the case of silicon atoms linked by a siloxane bond, up to about 20 silicon atoms may be used.
[0049]
R in general formula 1530And R31Specific examples of these include, for example, an alkyl group such as a methyl group or an ethyl group, a cycloalkyl group such as a cyclohexyl group, an aryl group such as a phenyl group, an aralkyl group such as a benzyl group, and R ′ is a methyl group or a phenyl group. Yes (R ')ThreeExamples thereof include a triorganosiloxy group represented by Si-.
[0050]
Among these hydrosilyl group-containing compounds, in particular, the general formula 16
H-Si (R31)3-a(Y)a(16)
(Wherein R31Y, a are the same as above)
The compound which has a crosslinkable group shown by is preferable from a point with easy acquisition. Specific examples of the hydrosilyl group-containing compound having a crosslinkable group represented by the general formula 15 or 16 include
HSiClThree, HSi (CHThree) Cl2, HSi (CHThree)2Cl, HSi (OCHThree)Three, HSi (CHThree) (OCHThree)2, HSi (CHThree)2OCHThree, HSi (OC2HFive)Three, HSi (CHThree) (OC2HFive)2, HSi (CHThree)2OC2HFive, HSi (OCThreeH7)Three, HSi (C2HFive) (OCHThree)2, HSi (C2HFive)2OCHThree, HSi (C6HFive) (OCHThree)2, HSi (C6HFive)2(OCHThree), HSi (CHThree) (OC (O) CHThree)2, HSi (CHThree)2O- [Si (CHThree)2O]2-Si (CHThree)
(OCHThree)2, HSi (CHThree) [O-N = C (CHThree)2]2
(However, in the above chemical formula, C6HFiveRepresents a phenyl group)
Etc.
[0051]
When a hydrosilane compound having a crosslinkable silyl group is added to an alkenyl group, a hydrosilylation catalyst is usually added. Examples of such hydrosilylation catalyst include platinum alone, alumina, silica, carbon black and the like in which a platinum solid is dispersed, chloroplatinic acid, a complex of chloroplatinic acid and alcohol, aldehyde, ketone, etc., platinum -An olefin complex and a platinum (0) -divinyltetramethyldisiloxane complex are mentioned. Examples of catalysts other than platinum compounds include RhCl (PPhThree)Three, RhClThree, RuClThree, IrClThree, FeClThreeAlClThree, PdCl2・ H2O, NiCl2TiClFourEtc. These catalysts may be used alone or in combination of two or more. The amount of the catalyst is not particularly limited, but 10% with respect to 1 mol of the alkenyl group of the vinyl polymer.-1-10-8It should be used in the range of mol, preferably 10-3-10-6It is good to use in the range of mol. 10-8If it is less than mol, curing does not proceed sufficiently. Also, since hydrosilylation catalysts are expensive, 10-1It is preferable not to use more than mol.
[0052]
Moreover, the vinyl polymer which has the alkenyl group refine | purified by the above-mentioned method gives the curable composition containing this. This curable composition contains (C) a vinyl polymer having an alkenyl group and (D) a hydrosilyl group-containing compound.
The vinyl polymer having an alkenyl group as component (C) has a main chain obtained by atom transfer radical polymerization of a vinyl monomer and has at least one alkenyl group in the molecule. The alkenyl group may be introduced at either the side chain or the terminal of the polymer, but is preferably at the terminal in order to obtain a cured product having excellent rubber elasticity. The vinyl polymer having an alkenyl group may be used alone or in combination of two or more. Although there is no restriction | limiting in particular as molecular weight of (C) component, It exists in the range of 500-100000. If it is 500 or less, the original characteristics of the vinyl polymer are hardly expressed, and if it is 100,000 or more, the viscosity or solubility becomes very low, and handling becomes difficult.
[0053]
The hydrosilyl group-containing compound as component (D) is not particularly limited as long as it has at least two hydrosilyl groups. That is, a linear polysiloxane represented by the general formula 17 or 18
R32 ThreeSiO- [Si (R32)2O]a-[Si (H) (R33) O]b-[Si (R33) (R34) O]c-SiR32 Three (17)
HR32 2SiO- [Si (R32)2O]a-[Si (H) (R33) O]b-[Si (R33) (R34) O]c-SiR32 2H (18)
(Where R32And R33Is an alkyl group having 1 to 6 carbon atoms, or a phenyl group, R34Is an alkyl group having 1 to 10 carbon atoms or an aralkyl group, a is 0 ≦ a ≦ 100, b is an integer of 2 ≦ b ≦ 100, and c is an integer of 0 ≦ c ≦ 100), cyclic represented by the general formula 19 Siloxane
[0054]
[Formula 4]
(Where R32, R33, R34Is the same as above, d is an integer of 0 ≦ d ≦ 8, e is an integer of 2 ≦ e ≦ 10, f is an integer of 0 ≦ f ≦ 8, and 3 ≦ d + e + f ≦ 10).
These may be used alone or in combination of two or more. Among these siloxanes, chain siloxanes represented by general formulas 20 and 21 and cyclic siloxanes represented by general formulas 22 and 23 having a phenyl group are preferable from the viewpoint of compatibility with (meth) acrylic polymers. .
(CHThree)ThreeSiO- [Si (H) (CHThree) O]g-[Si (C6HFive)2O]h-Si (CHThree)Three(20)
(CHThree)ThreeSiO- [Si (H) (CHThree) O]g-[Si (CHThree) {CH2C (H) (R35) C6HFive} O]h-Si (CHThree)Three(21)
(Wherein R35Is hydrogen or a methyl group, g is 2 ≦ g ≦ 100, h is an integer of 0 ≦ h ≦ 100, C6HFiveRepresents a phenyl group)
[0056]
[Chemical formula 5]
(Wherein R36Is hydrogen or a methyl group, i is 2 ≦ i ≦ 10, j is an integer satisfying 0 ≦ j ≦ 8 and 3 ≦ i + j ≦ 10, C6HFiveIs a phenyl group)
As the curing agent having at least two or more hydrosilyl groups as the component (D), a hydrosilyl group-containing compound represented by formulas 17 to 23 for a low molecular compound having two or more alkenyl groups in the molecule, A compound obtained by addition reaction such that a part of the hydrosilyl group remains after the reaction can also be used. Various compounds can be used as the compound having two or more alkenyl groups in the molecule. For example, hydrocarbon compounds such as 1,4-pentadiene, 1,5-hexadiene, 1,6-heptadiene, 1,7-octadiene, 1,8-nonadiene, 1,9-decadiene, O, O Ether compounds such as' -diallyl bisphenol A, 3,3'-diallyl bisphenol A, ester compounds such as diallyl phthalate, diallyl isophthalate, triallyl trimellitate, tetraallyl pyromellitate, carbonates such as diethylene glycol diallyl carbonate System compounds.
[0058]
The compound can be obtained by slowly dropping the above-mentioned alkenyl group-containing compound in the presence of a hydrosilylation catalyst with respect to an excessive amount of the hydrosilyl group-containing compound represented by Formulas 17 to 23. Of these compounds, the following are preferred in view of the availability of raw materials, the ease of removal of the excessively used siloxane, and the compatibility of the component (C) with the polymer.
[0059]
[Chemical 6]
The polymer (C) and the curing agent (D) can be mixed at an arbitrary ratio, but from the viewpoint of curability, the molar ratio of the alkenyl group to the hydrosilyl group is preferably in the range of 5 to 0.2. Furthermore, it is particularly preferably 2.5 to 0.4. When the molar ratio is 5 or more, curing is insufficient and only a cured product having low tackiness and a low strength can be obtained. When the molar ratio is less than 0.2, a large amount of active hydrosilyl groups remain in the cured product even after curing. Cracks and voids are generated, and a uniform and strong cured product cannot be obtained.
[0061]
The curing reaction between the polymer (C) and the curing agent (D) proceeds by mixing and heating the two components, but a hydrosilylation catalyst is added to advance the reaction more rapidly. As such a hydrosilylation catalyst, all those already exemplified may be suitably used under the above-mentioned conditions.
If the two components (C) and (D) of the present invention and the hydrosilylation catalyst are mixed and cured as necessary, a uniform cured product excellent in deep part curability can be obtained without causing a phenomenon such as foaming. It is done. Although there is no restriction | limiting in particular about hardening conditions, Generally it is good to harden | cure at 0 to 200 degreeC, Preferably it is 30 to 150 degreeC, Especially 80 to 150 degreeC. The properties of the cured product depend on the main chain skeleton and molecular weight of the polymer of component (C) and the curing agent of component (D) to be used, but can be widely prepared from rubbery to resinous. If the specific use of the hardened | cured material obtained from this composition is given, a sealing material, an adhesive agent, an adhesive material, an elastic adhesive agent, a coating material, a powder coating material, a foam, the potting material for electric and electronics, a film, a gasket, Various molding materials, artificial marble, etc.
[0062]
【Example】
Specific examples of this reaction are shown below, but this reaction is not limited to the following examples.
(Production Example 1)Production example of 2-allyloxyethyl methacrylate
In a three-necked flask equipped with a stirrer, thermometer, reflux condenser, and Dean-Stark tube, methacrylic acid (137.7 g, 1.6 mol), ethylene glycol monoallyl ether (80.7 g, 0.8 mol), p -Toluenesulfonic acid (0.76 g, 4.0 mmol) and toluene (650 mL) were charged. After reacting at 120 ° C. for 5 hours, 0.12 g of p-toluenesulfonic acid was added, and further reacted at the same temperature for 6 hours, and 0.1 g of p-toluenesulfonic acid was added. The reaction was terminated at the same temperature for another 9 hours. During this time, methacrylic acid and ethylene glycol monoallyl ether were traced by liquid chromatography, and the conversion finally reached 98%. NaHCOThreeAn aqueous solution was added to neutralize and the two layers were separated. The aqueous layer is extracted once with toluene, and the organic layer is extracted with CaCl.2After drying, the volatiles were removed under reduced pressure. The crude product was distilled under reduced pressure (60 ° C., 2 mmHg) to obtain 98.7 g of 2-allyloxyethyl methacrylate represented by the following formula (yield 73%).
H2C = C (CHThreeCO2(CH2)2OCH2CH = CH2
(Production Example 2)Production example of initiator having alkenyl group
The 50 mL 2-necked flask was purged with nitrogen, and 2-allyloxyethanol (2.5 mL, 23.4 mmol), pyridine (3 mL), and THF (10 mL) were charged. The solution was cooled to 0 ° C. and 2-bromopropionic acid chloride (2 mL, 19.52 mmol) was slowly added dropwise. After stirring at the same temperature for 1 hour, ethyl acetate (10 mL) was added, and the resulting hydrochloride of pyridine was removed by filtration. The filtrate was diluted with dilute hydrochloric acid (10 mL), NaHCO 3ThreeWashed with aqueous solution (10 mL) and then brine (10 mL). The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure. The resulting crude product was distilled under reduced pressure to obtain allyloxyethyl-2-bromopropionate represented by the following formula. (78.5-81 degreeC (1.3mmHg), 2.986g).
CHThreeC (H) (Br) C (O) O-CH2CH2-O-CH2CH = CH2(Production Example 3)Production example of an initiator having a hydroxyl group
2-Bromopropionic acid chloride (2 mL, 3.35 g, 19.5 mmol) was slowly added dropwise at 0 ° C. to a THF solution (10 mL) of ethylene glycol (10.9 mL, 195 mmol) and pyridine (3 g, 39 mmol) in a nitrogen atmosphere. did. The solution was stirred at that temperature for 2 hours. Dilute hydrochloric acid (20 mL) and ethyl acetate (30 mL) were added, and the two layers were separated. The organic layer is washed with dilute hydrochloric acid and brine, and Na2SOFourAfter drying, the volatile matter was distilled off under reduced pressure to obtain a crude product (3.07 g). The crude product was distilled under reduced pressure (70 to 73 ° C., 0.5 mmHg) to obtain hydroxyethyl-2-bromopropionate represented by the following formula (2.14 g, 56%).
CHThreeC (H) (Br) C (O) O (CH2)2-OH
(Production Example 4)Production Example 1 of carboxylate having alkenyl group
Undecylenic acid (18.8 g, 0.102 mol) was slowly added dropwise to a 1 / 2N ethanol solution (200 mL) of potassium hydroxide at 0 ° C. with stirring. Volatile components were distilled off under reduced pressure to obtain a crude product. The crude product was washed with acetone and then heated under reduced pressure to obtain a white solid of potassium undecylenate represented by the following formula (8.88 g, yield 88%).
CH2= CH- (CH2)8-CO2 -K+
(Production Example 5)Production Example 2 of carboxylate having alkenyl group
4-Pentenoic acid (49 g, 0.489 mol) and potassium tert-butoxide (54.9 g, 0.489 mol) were charged in methanol (245 mL), and the mixture was stirred at 0 ° C. Volatile components were distilled off under reduced pressure to obtain potassium pentenoate represented by the following formula.
CH2= CH- (CH2)2-CO2 -K+
(Production Example 6)
In a 1 L pressure-resistant reaction vessel, acrylic acid-n-butyl (112 mL, 100 g, 0.78 mol), the hydroxyl group-containing initiator obtained in Production Example 3 (3.07 g, 15.6 mmol), cuprous bromide ( 2.24 g, 15.6 mmol), 2,2′-bipyridyl (4.87 g, 31.2 mmol), ethyl acetate (90 mL), acetonitrile (22 mL) were charged, and nitrogen bubbling was performed to remove dissolved oxygen. Sealed. The mixture was heated to 130 ° C. and reacted for 2 hours. The reaction vessel was returned to room temperature, 2-hydroxyethyl methacrylate (3.92 mL, 4.06 g, 31.2 mmol) was added, and the mixture was reacted at 110 ° C. for 2 hours. The mixture was diluted with ethyl acetate (200 mL), insolubles were filtered off, and the filtrate was washed twice with 10% hydrochloric acid and once with brine. The organic layer is Na2SOFourAfter drying, the solvent was distilled off under reduced pressure to obtain 82 g of poly (acrylic acid-n-butyl) having a hydroxyl group at the terminal. The number average molecular weight of the polymer was 5100 according to GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.29.
[0063]
Next, poly (acrylic acid-n-butyl) having a hydroxyl group at the terminal obtained as described above (50 g) and a toluene solution (100 mL) of pyridine (10 mL) were added to undecene at 75 ° C. in a nitrogen atmosphere. Acid chloride (7.22 mL, 6.81 g, 33.6 mmol) was slowly added dropwise and stirred at 75 ° C. for 3 hours. The resulting white solid was filtered and the organic layer was washed with dilute hydrochloric acid and brine. The organic layer is Na2SOFourAnd concentrated under reduced pressure to obtain poly (acrylic acid-n-butyl) having an alkenyl group at the terminal (43 g).
(Production Example 7)
A 100 mL three-necked round bottom flask equipped with a reflux tube was charged with cuprous bromide (0.625 g, 15.6 mmol), acetonitrile (5.0 mL), and pentamethyldiethylenetriamine (0.91 mL). Replaced. Acrylic acid-n-butyl (50 mL, 44.7 g, 0.39 mol) and diethyl-2,5-dibromoadipate (1.57 g, 4.36 mmol) were added, and the mixture was heated and stirred at 70 ° C. for 7 hours. The mixture was diluted with ethyl acetate and treated with activated alumina. Volatile components were distilled off under reduced pressure to obtain 35.0 g of poly (acrylic acid-n-butyl) having a halogen at the terminal (polymerization yield: 87%). The number average molecular weight of the polymer was 10700 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.15.
[0064]
Next, in a 200 mL three-necked round bottom flask equipped with a reflux tube, poly (acrylic acid-n-butyl) (35.0 g) having halogen at the terminal obtained as described above was synthesized in Production Example 2. 4-pentenoic acid potassium salt (2.23 g, 16.1 mmol) and dimethylacetamide (35 mL) were charged and reacted at 70 ° C. for 4 hours in a nitrogen atmosphere. The mixture was diluted with ethyl acetate and washed with 2% hydrochloric acid, brine. The organic layer is Na2SOFourAnd the polymer was isolated by removing the volatiles under reduced pressure.
(Production Example 8)
In a 100 mL glass reaction vessel, butyl acrylate (50.0 mL, 44.7 g, 0.349 mol), cuprous bromide (1.25 g, 8.72 mmol), pentamethyldiethylenetriamine (1.82 mL, 1.51 g). , 8.72 mmol), and acetonitrile (5 mL) were charged, and after cooling, degassed under reduced pressure, and then replaced with nitrogen gas. After stirring well, diethyl 2,5-dibromoadipate (1.57 g, 4.36 mmol) was added, and the mixture was heated and stirred at 70 ° C. After 60 minutes, 1,7-octadiene (6.44 mL, 4.80 g, 43.6 mmol) was added, and heating and stirring were continued at 70 ° C. for 2 hours. After the mixture was treated with activated alumina, the volatile components were distilled off by heating under reduced pressure. The product was dissolved in ethyl acetate and washed with 2% hydrochloric acid, brine. The organic layer is Na2SOFourThe polymer having alkenyl groups at the ends was obtained by heating and evaporating volatile components under reduced pressure. The number average molecular weight of the obtained polymer was 13100 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.22. The introduction rate of the olefin functional group based on the number average molecular weight was 2.01.
(Production Example 9)
In a 30 mL pressure-resistant glass reaction vessel, methyl acrylate (2.5 mL, 2.4 g, 27.8 mmol), α, α′-dibromo-p-xylene (146 mg, 0.555 mmol), cuprous bromide (79 0.5 mg, 4.44 mmol), 2,2′-bipyridyl (259 mg, 1.67 mmol), and ethyl acetate (2.0 mL), acetonitrile (0.5 mL), and after removing dissolved oxygen by freeze devolatilization , Sealed. The mixture was heated to 130 ° C. and reacted for 2.5 hours. After cooling to room temperature, 2-allyloxyethyl methacrylate (381 mg, 2.22 mmol) obtained in Production Example 1 was added under a nitrogen gas atmosphere and sealed. The mixture was heated to 80 ° C. and reacted for 2 hours. The mixture was diluted with ethyl acetate (50 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain polymethyl acrylate having an alkenyl group. The number average molecular weight of the polymer was 6000 as measured by GPC (polystyrene conversion), and the molecular weight distribution was 1.53. The alkenyl group introduced per oligomer molecule is1According to HNMR analysis, the number was 3.1.
(Example 1)
Aluminum silicate (manufactured by Kyowa Chemical: Kyowaard 700PEL) is added to the toluene solution of the polymer obtained in Production Example 6 and stirred at reflux temperature to remove the aluminum silicate and then the solvent is distilled off. To obtain a polymer.
[0065]
Next, the polymer (1.0 g), dimethoxymethylhydrosilane (0.16 mL, 1.30 mmol), toluene (1.0 mL), and a platinum catalyst were charged into a 30 mL pressure-resistant glass reaction vessel. However, the amount of platinum catalyst used is 10 by molar ratio with respect to the alkenyl group of the polymer.-FourEquivalent. When the reaction mixture was heated at 75 ° C. for 3 hours, 35% of the alkenyl groups reacted to introduce silyl groups at the ends of the polymer.
(Example 2)
To the polymer obtained in Production Example 7, an aluminum silicate having an equal weight to the polymer (Kyowa Chemical Co., Ltd .: Kyowaard 700 PEL) was added and stirred at 100 ° C. for 4 hours. Butyl acrylate) was obtained. The alkenyl group introduced per oligomer-1 molecule is1According to 1 H NMR analysis, it was 1.82.
[0066]
Next, in a 200 mL pressure-resistant glass reaction vessel, the polymer (15.0 g), dimethoxymethylhydrosilane (1.8 mL, 14.5 mmol), dimethyl orthoformate (0.26 mL, 2.42 mmol), and platinum The catalyst was charged. However, the amount of platinum catalyst used is 2 × 10 in molar ratio to the alkenyl group of the polymer.-FourEquivalent. The reaction mixture was heated at 100 ° C. for 4 hours. The volatile component of the mixture was distilled off under reduced pressure to obtain poly (acrylic acid-n-butyl) having a silyl group at the terminal. The silyl group introduced per oligomer-1 molecule is1From 1 H NMR analysis, it was 1.46.
(Example 3)
The polymer obtained in Production Example 8 (30.5 g), the polymer and an equal weight of aluminum silicate (manufactured by Kyowa Chemical Co., Ltd .: Kyowaard 700PEL) were mixed in toluene and stirred at 100 ° C. After 4 hours, the aluminum silicate was filtered, and the polymer was purified by heating and distilling off the volatiles of the filtrate under reduced pressure.
[0067]
Next, in a 200 mL pressure-resistant glass reactor, the polymer (23.3 g), dimethoxymethylhydrosilane (2.55 mL, 20.7 mmol), dimethyl orthoformate (0.38 mL, 3.45 mmol), and platinum catalyst Was charged. However, the amount of platinum catalyst used is 2 × 10 in molar ratio to the alkenyl group of the polymer.-FourEquivalent. The reaction mixture was heated at 100 ° C. for 3 hours. The volatile component of the mixture was distilled off under reduced pressure to obtain poly (acrylic acid-n-butyl) having a silyl group at the terminal. The silyl group introduced per oligomer-1 molecule is1According to 1 H NMR analysis, the number was 1.41.
(Comparative Example 1)
The polymer (0.51 g) obtained in Production Example 9, dimethoxymethylhydrosilane (0.040 mL, 0.326 mmol), and a platinum catalyst were charged. However, the amount of platinum catalyst used is 10 by molar ratio with respect to the alkenyl group of the polymer.-3Equivalent. The reaction mixture was heated at 55 ° C. for 1 hour, but no silyl groups were introduced.
(Production Example 10)
In a 100 mL pressure-resistant glass reaction vessel, methyl acrylate (20.0 mL, 19.1 g, 0.222 mmol), α, α′-dibromo-p-xylene (1.168 g, 4.44 mmol), cuprous bromide (0.636 g, 4.44 mmol), 2,2′-bipyridyl (2.07 g, 13.3 mmol), ethyl acetate (16.0 mL) and acetonitrile (4.0 mL) were charged, and nitrogen gas was blown in for 10 minutes. After removing the dissolved oxygen, the tube was sealed. The mixture was heated to 130 ° C. and allowed to react for 1 hour. After cooling to room temperature, 2-allyloxyethyl methacrylate (3.0 g, 17.6 mmol) obtained in Production Example 1 was added under a nitrogen gas atmosphere and sealed. The mixture was heated to 80 ° C. and reacted for 1 hour. The mixture was diluted with ethyl acetate (50 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourDried with
Volatiles were distilled off under reduced pressure to obtain 20.5 g of polybutyl acrylate having an alkenyl group at the terminal (polymerization yield: 93%). The number average molecular weight of the polymer was 7900 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.99. The alkenyl group introduced per oligomer molecule is1According to HNMR analysis, the number was 3.3.
(Production Example 11)
In a 30 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (2.5 mL, 2.24 g, 17.45 mmol), α, α′-dibromo-p-xylene (92.5 mg, 0.35 mmol), bromide Cuprous (50 mg, 0.35 mmol), 2,2′-bipyridyl (163 mg, 1.05 mmol), ethyl acetate (2 mL), acetonitrile (0.5 mL) were charged, and nitrogen gas was blown for 10 minutes to dissolve oxygen. Was removed and sealed. The mixture was heated to 130 ° C. and allowed to react for 1 hour. After cooling to room temperature, 2-allyloxyethyl methacrylate (600 mg, 3.5 mmol) obtained in Production Example 1 was added under a nitrogen gas atmosphere and sealed. The mixture was heated to 80 ° C. and reacted for 1 hour. The mixture was diluted with ethyl acetate (20 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 1.97 g of poly (acrylic acid-n-butyl) having an alkenyl group at the terminal (polymerization yield: 88%). The number average molecular weight of the polymer was 6700 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.60. The alkenyl group introduced per oligomer molecule is1From H NMR analysis, it was 5.4.
(Production Example 12)
In a 50 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (10 mL, 8.94 g, 69.8 mmol), allyloxyethyl-2-bromopropionate obtained in Production Example 2 (332 mg, 1.4 mmol) , Cuprous bromide (200 mg, 1.4 mmol), 2,2′-bipyridyl (433 mg, 2.8 mmol), ethyl acetate (8 mL), acetonitrile (2 mL), and nitrogen gas was blown for 10 minutes to dissolve. After removing oxygen, the tube was sealed. The mixture was heated to 130 ° C. and reacted for 2 hours. After cooling to room temperature, allyloxyethyl methacrylate (480 mg, 2.8 mmol) obtained in Production Example 1 was added under a nitrogen gas atmosphere and sealed. The mixture was heated to 100 ° C. and reacted for 2 hours. The mixture was diluted with ethyl acetate (50 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 8.88 g of poly (n-butyl acrylate) having an alkenyl group at the terminal (polymerization yield: 94%). The number average molecular weight of the polymer was 6700 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.79. The alkenyl group introduced per oligomer molecule is1According to 1 H NMR analysis, the number was 1.2.
(Production Example 13)
In a 100 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (10 mL, 8.94 g, 69.8 mmol), an initiator having an alkenyl group prepared in Production Example 5 (332 mg, 1.40 mmol), first bromide Copper (200 mg, 1.40 mmol), 2,2′-bipyridyl (433 mg, 2.80 mmol), acetonitrile (2 mL), and ethyl acetate (8 mL) were charged, and nitrogen gas was blown for 10 minutes to remove dissolved oxygen. , Sealed. The mixture was heated to 130 ° C. and reacted for 1.5 hours. After cooling to room temperature, p-divinylbenzene (364 mg, 2.80 mmol) was added under a nitrogen gas atmosphere and sealed. The mixture was heated to 100 ° C. and reacted for 2 hours. The mixture was diluted with ethyl acetate (30 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 6.43 g of poly (n-butyl acrylate) having an alkenyl group at both ends represented by the following formula (69%). The number average molecular weight of the polymer was 3900 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 5.35. The alkenyl group introduced per oligomer molecule is1From 1 H NMR analysis, it was 1.73.
(Production Example 14)
In a 50 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (10 mL, 8.94 g, 69.8 mmol), α, α′-dibromo-p-xylene (370 mg, 1.4 mmol), cuprous bromide ( 200 mg, 1.4 mmol), 2,2′-bipyridyl (433 mg, 2.8 mmol), and methyl isobutyl ketone (10 mL) were charged, nitrogen gas was blown for 10 minutes to remove dissolved oxygen, and the tube was sealed. The mixture was heated to 130 ° C. and reacted for 20 minutes. After cooling to room temperature, the mixture was diluted with ethyl acetate (20 mL), the insoluble solid produced was filtered, and the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 5.21 g of poly (n-butyl acrylate) having bromine at the terminal (58%). The number average molecular weight of the polymer was 3700 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.41.
[0068]
Next, in a 30 mL pressure-resistant reaction tube, poly (acrylic acid-n-butyl) (2.0 g) obtained as described above, p-divinylbenzene (281 mg, 2.16 mmol), cuprous bromide. (77 mg, 0.54 mmol), 2,2′-bipyridyl (167 mg, 1.08 mmol), and methyl isobutyl ketone (4 mL) were charged, nitrogen gas was blown for 10 minutes to remove dissolved oxygen, and the tube was sealed. The mixture was heated to 130 ° C. and reacted for 30 minutes. After cooling to room temperature, the mixture was diluted with ethyl acetate (10 mL), the insoluble solid formed was filtered, and the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 2.11 g of poly (acrylic acid-n-butyl) having an alkenyl group at the terminal. The number average molecular weight of the polymer was 7300 according to GPC measurement (polystyrene conversion), and the molecular weight distribution was 2.47. The alkenyl group introduced per oligomer molecule is1According to 1 H NMR analysis, the number was 2.1.
(Production Example 15)
In a 500 mL three-necked flask with a reflux tube, cuprous bromide (1.50 g, 10.5 mmol) as a catalyst, pentamethyldiethylenetriamine (1.65 mL) as a ligand, and diethyl-2,5-dibromo as an initiator Using adipate (9.42 g, 26.2 mol) and acetonitrile (30 mL) as a solvent, acrylic acid-n-butyl (300 mL) was polymerized at 70 ° C. in a nitrogen atmosphere, and the polymerization rate of acrylic acid-n-butyl was At 93%, 1,7-octadiene (38.6 mL, 0.261 mol) was added and heated at the same temperature. The reaction mixture was diluted with ethyl acetate, the catalyst was removed through an activated alumina column, and the volatile component was distilled off under reduced pressure to obtain a polymer having an alkenyl group at the terminal. The number average molecular weight of the polymer was 13800 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.28. The alkenyl group introduced per oligomer-1 molecule is1From 1 H NMR analysis, it was 1.84.
(Production Example 16)
In a 30 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (7.5 mL, 6.72 g, 51.3 mmol), α, α′-dibromo-p-xylene (270 mg, 1.03 mmol), first bromide Copper (150 mg, 1.03 mmol), 2,2′-bipyridyl (322 mg, 2.06 mmol), ethyl acetate (6 mL), and acetonitrile (1.5 mL) were charged, and nitrogen gas was blown for 10 minutes to dissolve dissolved oxygen. After removal, the tube was sealed. The mixture was heated to 130 ° C. and reacted for 1.5 hours. The mixture was diluted with ethyl acetate (20 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 5.0 g of poly (acrylic acid-n-butyl) having a halogen at the terminal (polymerization yield: 75%). The number average molecular weight of the polymer was 5600 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.32.
[0069]
The above polymer (5.00 g), undecylenic acid potassium salt synthesized in Production Example 4 (476 mg, 2.14 mmol) and dimethylacetamide (10 mL) were charged and reacted at 70 ° C. for 6 hours in a nitrogen atmosphere. . Volatiles of the mixture were removed under reduced pressure, ethyl acetate was added, and insolubles were filtered off. The filtrate was evaporated under reduced pressure to obtain 4.77 g of poly (n-butyl acrylate) having an alkenyl group at the terminal. The alkenyl group introduced per oligomer-1 molecule is1From 1 H NMR analysis, it was 1.70.
(Production Example 17)
In the same manner as in Production Example 16, cuprous bromide (1.50 g, 10.5 mmol) as a catalyst, pentamethyldiethylenetriamine (0.69 mL) as a ligand, and diethyl-2,5-dibromoadipate (initiator) 9.42 g, 26.2 mol), and acetonitrile (30 mL) as a solvent, acrylate-n-butyl (300 mL) was polymerized at 70 ° C. in a nitrogen atmosphere, and poly (acrylic acid-n-) having a halogen at the terminal was polymerized. Butyl) was obtained. The number average molecular weight of the polymer was 11300 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.16.
[0070]
Next, poly (acrylic acid-n-butyl) having an alkenyl group at the terminal was obtained by the same operation as in Production Example 10 using the pentenoic acid potassium salt produced in Production Example 5 as the carboxylate. The alkenyl group introduced per oligomer-1 molecule is1From 1 H NMR analysis, it was 1.84.
(Production Example 18)
In a 30 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (5 mL, 4.47 g, 34.9 mmol), α, α′-dibromo-p-xylene (180 mg, 0.69 mmol), cuprous bromide ( 98 mg, 0.69 mmol), 2,2′-bipyridyl (319 g, 2.06 mmol), ethyl acetate (4 mL), and acetonitrile (1 ml) were charged, and nitrogen gas was blown for 10 minutes to remove dissolved oxygen. Tubed. The mixture was heated to 130 ° C. and allowed to react for 1 hour. After cooling the mixture, allyltributyltin (0.51 mL, 1.64 mmol) was added under a nitrogen atmosphere and reacted at 100 ° C. for 1 hour. The mixture was diluted with ethyl acetate (20 mL) and the insoluble solid produced was filtered, and then the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain a mixture of poly (n-butyl acrylate) having an alkenyl group at the end and bromotributyltin (yield 4.48 g). The number average molecular weight of the polymer was 6300 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.57. The alkenyl group introduced per oligomer molecule is1According to HNMR analysis, the number was 2.2.
(Production Example 19)
In a 30 mL pressure-resistant glass reaction vessel, acrylic acid-n-butyl (2.5 mL, 2.24 g, 17.45 mmol), an initiator having an alkenyl group obtained above (165 mg, 0.698 mmol), A cuprous (100 mg, 0.698 mmol), 2,2′-bipyridyl (218 mg, 1.40 mmol), acetonitrile (0.5 mL), and ethyl acetate (2 mL) were charged, and dissolved oxygen was removed by blowing nitrogen gas for 10 minutes. And then sealed. The mixture was heated to 130 ° C. and reacted for 50 minutes. After cooling to room temperature, the mixture was diluted with ethyl acetate (20 mL), the insoluble solid produced was filtered, and the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 1.90 g of poly (acrylic acid-n-butyl) having an alkenyl group at one end and bromine at the other end (79%). The number average molecular weight of the polymer was 3600 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.51. The alkenyl group introduced per oligomer molecule is1According to 1 H NMR analysis, it was 0.75.
[0071]
Next, a 50 mL three-necked flask equipped with a stirrer and a reflux condenser was added to the polymer (1.90 g), Na2S ・ 9H2O (70.2 mg, 0.293 mmol) and ethanol (3 mL) were charged, and the mixture was stirred at reflux temperature for 3 hours. After cooling to room temperature, ethyl acetate (10 mL) and dilute hydrochloric acid (10 mL) were added, and the two layers were separated. The organic layer is washed with dilute hydrochloric acid and brine, and Na2SOFourAfter drying, the volatile matter was distilled off under reduced pressure to obtain 1.69 g of poly (acrylic acid-n-butyl) having an alkenyl group at the terminal. The number average molecular weight of the polymer was 5100 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.73.
(Production Example 20)
In a 30 mL pressure-resistant glass reaction vessel, methyl acrylate (5 mL, 4.78 g, 55.5 mmol), allyl 2-methyl-2-bromopropionate (0.354 mL, 460 mg, 2.22 mmol), cuprous bromide (318 mg, 2.22 mmol), 2,2′-bipyridyl (1.04 g, 6.66 mmol), acetonitrile (1 mL), and ethyl acetate (4 mL) were charged, and the vacuum was degassed three times to remove dissolved oxygen. After that, it was sealed. The mixture was heated to 80 ° C. and reacted for 3 hours. After cooling to room temperature, the mixture was diluted with ethyl acetate (20 mL), the insoluble solid produced was filtered, and the filtrate was washed twice with dilute hydrochloric acid and once with brine. The organic layer is Na2SOFourThe volatile matter was distilled off under reduced pressure to obtain 3.93 g of poly (methyl acrylate) having an alkenyl group at one end and bromine at the other end (75%). The number average molecular weight of the polymer was 2700 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.48. The alkenyl group introduced per oligomer molecule is1It was 0.81 from 1 H NMR analysis.
[0072]
Next, a 50 mL three-necked flask equipped with a stirrer and a reflux condenser was added to the polymer (1.17 g) obtained as described above, Na2S ・ 9H2O (57.6 mg, 0.240 mmol) and ethanol (2 mL) were charged, and the mixture was stirred at reflux temperature for 3 hours. After cooling to room temperature, ethyl acetate (10 mL) and dilute hydrochloric acid (10 mL) were added, and the two layers were separated. The organic layer is washed with dilute hydrochloric acid and brine, and Na2SOFourAfter drying, the volatile matter was distilled off under reduced pressure to obtain 1.11 g of poly (methyl acrylate) having an alkenyl group at the terminal. The number average molecular weight of the polymer was 4200 by GPC measurement (polystyrene conversion), and the molecular weight distribution was 1.71.
(Example 4)
The polymer having an alkenyl group at the terminal (1.32 g) produced in Production Example 10 is dissolved in toluene (20 mL), and activated carbon (Wako Pure Chemicals: activated carbon, powder) of the same weight as the polymer is added. And stirred at room temperature for 4 hours. Activated carbon was separated by filtration, and the volatile matter in the filtrate was distilled off under reduced pressure to obtain 0.62 g of a polymer.
[0073]
The above polymer, the polyvalent hydrogen silicon compound (S-1) represented by the following formula, and a 1,1,3,3-tetramethyl-1,3-divinyldisiloxane complex of zero-valent platinum (8.3) × 10-8Mole xylene solution) was mixed well. The amount of polyvalent hydrogen silicon compound used is such that the alkenyl group of the polymer and the hydrosilyl group of the hydrogen silicon compound are 1 / 1.2 in molar ratio, and the amount of platinum catalyst used is the alkenyl group of the polymer. 10 molar ratio to the group-3Equivalent.
[0074]
A part of the composition thus obtained was subjected to a curing test on a hot plate at 130 ° C., and the gelation time was measured. The remaining composition was degassed under reduced pressure, poured into a mold and heat-cured to obtain a rubber-like cured product. The cured product was immersed in acetone for 24 hours, and the gel fraction was measured from the weight change before and after. The results are shown in Table 1.
[0075]
[Chemical 7]
(Example 5)
The polymer having an alkenyl group at the terminal produced in Production Example 10 was purified in the same manner as in Example 1 except that aluminum silicate (Kyowa Chemical Co., Ltd .: Kyoward 700PEL) was used instead of activated carbon.
About the refine | purified polymer, the hardening test was done by operation similar to Example 1, and the gelation time and the gel fraction were measured. The results are shown in Table 1.
(Example 6)
The polymer having an alkenyl group at the terminal (1.70 g) produced in Production Example 11 was dissolved in toluene (20 mL), and an aluminum silicate (Kyowa Chemical: Kyoward 700PEL) having the same weight as the polymer was added. And stirred at reflux temperature for 2 hours. Aluminum silicate was separated by filtration, and volatile matter of the filtrate was distilled off under reduced pressure to obtain 1.50 g of a polymer.
[0077]
About the refine | purified polymer, the hardening test was done by operation similar to Example 1, and the gelation time and the gel fraction were measured. The results are shown in Table 1.
(Examples 7 to 19)
The polymers having an alkenyl group at the terminal obtained in Production Examples 12 to 20 and Production Example 6 were purified by the same operation as in Example 6 using aluminum silicate (manufactured by Kyowa Chemical Co., Ltd .: Kyoward 700PEL).
[0078]
About the refine | purified polymer, the hardening test was done by operation similar to Example 1, and the gelation time and the gel fraction were measured. The results are shown in Table 1. In addition, as a polyvalent hydrogen silicon compound, the following compounds (S-1, S-2) or methyl hydrogen siloxane partially modified with α-methylstyrene (S-3: SiH value 7.69 mmol / g) ) Was used. The amount of the polyvalent hydrogen silicon compound used was such that the molar ratio of the alkenyl group of the polymer to the SiH group of the hydrogen silicon compound was 1 / 1.2-1 / 1.5. A predetermined amount of platinum catalyst was added to the alkenyl group of the polymer.
[0079]
[Chemical 8]
Comparative Example 2
The polymer having an alkenyl group at the terminal obtained in Production Example 10 was subjected to a curing test by the same operation as in Example 1 without purifying the adsorbent, and the gelation time was measured. Also did not cure. The results are shown in Table 1.
[0081]
[Table 1]
【The invention's effect】
According to the present invention, by purifying a vinyl polymer having an alkenyl group produced by atom transfer radical polymerization with an adsorbent, the hydrosilylation reaction of the alkenyl group, which has been inhibited in the unpurified state, proceeds rapidly. Can be made. Moreover, the curable composition containing the vinyl polymer which has an alkenyl group at the terminal, and a hydrosilyl group containing compound is obtained.
Claims (11)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP22141498A JP3857431B2 (en) | 1997-08-06 | 1998-08-05 | Polymer purification method and curable composition |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21167397 | 1997-08-06 | ||
| JP9-211673 | 1997-08-06 | ||
| JP22141498A JP3857431B2 (en) | 1997-08-06 | 1998-08-05 | Polymer purification method and curable composition |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005177865A Division JP4167246B2 (en) | 1997-08-06 | 2005-06-17 | Polymer purification method and curable composition |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH11193307A JPH11193307A (en) | 1999-07-21 |
| JP3857431B2 true JP3857431B2 (en) | 2006-12-13 |
Family
ID=26518771
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP22141498A Expired - Lifetime JP3857431B2 (en) | 1997-08-06 | 1998-08-05 | Polymer purification method and curable composition |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP3857431B2 (en) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4977286B2 (en) | 2000-03-07 | 2012-07-18 | 日東電工株式会社 | Method for producing polymer |
| EP1288230B1 (en) | 2000-05-12 | 2008-10-01 | Kaneka Corporation | Method of purifying vinyl polymer |
| JP2001323011A (en) * | 2000-05-12 | 2001-11-20 | Kanegafuchi Chem Ind Co Ltd | Method for purifying vinyl-based polymer |
| JP4499260B2 (en) * | 2000-08-25 | 2010-07-07 | 株式会社カネカ | Method for purifying vinyl polymer |
| US7276574B2 (en) | 2001-10-17 | 2007-10-02 | Kaneka Corporation | Process for producing vinyl polymer |
| JP4455900B2 (en) * | 2004-02-23 | 2010-04-21 | 株式会社カネカ | Curable composition with improved compatibility with curing agent |
| WO2006093283A1 (en) | 2005-03-03 | 2006-09-08 | Kaneka Corporation | Process for producing vinyl polymer |
| JP2008024833A (en) * | 2006-07-21 | 2008-02-07 | Kaneka Corp | Atom transfer radical polymerization method |
| JP2008081738A (en) * | 2006-09-01 | 2008-04-10 | Kaneka Corp | Method for producing (meth)acrylic acid polymer, polymer and curable composition |
| WO2012020545A1 (en) | 2010-08-10 | 2012-02-16 | 株式会社カネカ | Manufacturing method of (meth) acrylic polymer |
-
1998
- 1998-08-05 JP JP22141498A patent/JP3857431B2/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JPH11193307A (en) | 1999-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6274688B1 (en) | Functional groups-terminated vinyl polymers | |
| EP0976766B1 (en) | Polymers, processes for producing the same, and curable compositions produced therefrom | |
| JP3857431B2 (en) | Polymer purification method and curable composition | |
| JP3895460B2 (en) | POLYMER, PROCESS FOR PRODUCING THE POLYMER, AND CURABLE COMPOSITION USING THE POLYMER | |
| JP3808622B2 (en) | POLYMER, PROCESS FOR PRODUCING THE POLYMER, AND CURABLE COMPOSITION USING THE POLYMER | |
| JP4098896B2 (en) | Polymer, method for producing the polymer, and curable composition using the polymer | |
| WO2000059960A1 (en) | Method of treating polymer | |
| JP2002069121A (en) | Method for purifying vinyl polymer | |
| JPH11116617A (en) | Polymer and its use | |
| JP3751753B2 (en) | Method for producing polymer having alkenyl group at terminal and curable composition using the polymer | |
| JPH1180571A (en) | Curable composition | |
| JP4167246B2 (en) | Polymer purification method and curable composition | |
| JP4393670B2 (en) | Method for purifying vinyl polymer | |
| JP2000344831A (en) | Method for treating polymer | |
| JP4582880B2 (en) | Method for purifying vinyl polymer | |
| WO2001085804A1 (en) | Method of purifying vinyl polymer | |
| JP2003096130A (en) | Purification method for vinyl polymer | |
| JP4750246B2 (en) | Method for purifying vinyl polymer | |
| JP4237483B2 (en) | Method for producing vinyl polymer | |
| JP4745486B2 (en) | Method for purifying vinyl polymer | |
| JP2000128924A (en) | Production of polymer containing terminal alkenyl group and curable composition using the polymer | |
| JP2000119527A (en) | Composition for materials of electric and electronic parts and electric and electronic parts | |
| JP2000143853A (en) | Foamable resin composition and foam prepared therefrom | |
| JP3985890B2 (en) | Method for purifying acrylic polymer | |
| JP2001323011A (en) | Method for purifying vinyl-based polymer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20050412 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20050419 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050617 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20050617 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20051025 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051226 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060822 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060914 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20060922 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100922 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110922 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110922 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120922 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120922 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130922 Year of fee payment: 7 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130922 Year of fee payment: 7 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |