JP3584457B2 - Diamine compound, polyamic acid, polyimide, liquid crystal aligning agent and liquid crystal display device - Google Patents
Diamine compound, polyamic acid, polyimide, liquid crystal aligning agent and liquid crystal display device Download PDFInfo
- Publication number
- JP3584457B2 JP3584457B2 JP1880597A JP1880597A JP3584457B2 JP 3584457 B2 JP3584457 B2 JP 3584457B2 JP 1880597 A JP1880597 A JP 1880597A JP 1880597 A JP1880597 A JP 1880597A JP 3584457 B2 JP3584457 B2 JP 3584457B2
- Authority
- JP
- Japan
- Prior art keywords
- liquid crystal
- diamine compound
- dianhydride
- mmol
- polyimide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 CCCC(CC(CCC)(**(*[Tl+]=C)(C1OC1)*(C)=C)N1CCNCC1)*C1CC*(CC2CCC(C)(*)CCC2)CCC1 Chemical compound CCCC(CC(CCC)(**(*[Tl+]=C)(C1OC1)*(C)=C)N1CCNCC1)*C1CC*(CC2CCC(C)(*)CCC2)CCC1 0.000 description 3
Images














Landscapes
- Liquid Crystal (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Liquid Crystal Substances (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Description
【0001】
【発明の属する技術分野】
本発明は、新規なポリアミック酸およびポリイミド並びにこれらの少なくとも一方を含有する液晶配向剤に関する。
【0002】
【従来の技術】
従来、正の誘電異方性を有するネマチック型液晶を、ポリイミドなどからなる液晶配向膜を有する透明電極付き基板でサンドイッチ構造にし、液晶分子の長軸が基板間で90度連続的に捻れるようにしてなるTN(Twisted Nematic)型液晶セルを有する液晶表示素子(TN型液晶表示素子)が知られている。このTN型液晶表示素子における液晶の配向は、ラビング処理が施された液晶配向膜により形成されている。
【0003】
また最近では、コントラストおよび視角依存性に優れた液晶表示素子であるSTN(Super Twisted Nematic)型液晶表示素子や、SH(Super Homeotropic)型液晶表示素子が開発されている。STN型液晶表示素子は、液晶としてネマチック型液晶に光学活性物質であるカイラル剤をブレンドしたものを用い、液晶分子の長軸を基板間で180度以上連続的に捻ることにより生じる複屈折効果を利用するものである。また、SH型液晶表示素子は、液晶分子の誘電異方性が負の液晶を垂直配向させ、電圧印加により分子を倒して単純マトリックス駆動で動作させるものである。
【0004】
【発明が解決しようとする課題】
しかしながら、従来知られているポリイミドなどからなる液晶配向膜を用いてSTN型液晶表示素子を作製した場合、液晶配向膜のプレチルト角が小さいため、液晶を基板間で180度以上捻ることができず、所要の表示機能を得ることは困難である。このため、STN型液晶表示素子においては、液晶を配向させるために、二酸化ケイ素を斜方蒸着して形成した液晶配向膜を用いる必要があるが、この配向膜は製造工程が煩雑で大量生産には適さないという問題がある。また、SH型液晶表示素子は、液晶を垂直配向させるために、二酸化ケイ素を斜方蒸着して形成した基板を用いたり、基板をフッ素系の界面活性剤や長鎖アルキル基を有するカップリング剤で処理することが必要であるが、界面活性剤やカップリング剤を用いる場合には信頼性が乏しくなるという問題がある。
また、TN型液晶表示素子においても、液晶セル駆動時のリバースチルト現象による表示不良を抑制するために、高いプレチルト角を有する液晶配向膜が望まれるようになってきた。
【0005】
従来、高いプレチルト角を発現するためにポリイミド末端部、側鎖部への長鎖アルキル基の導入が一般的に行われてきた。しかし、この長鎖アルキル基で修飾したポリイミド配向膜は、プレチルト角の工程安定性に欠けるといった問題がある。特に、STN型液晶表示素子はその原理上、膜厚ムラ、ラビングムラなどから生じるプレチルト角のバラツキが表示ムラとなって現れるために、プレチルト角発現の工程マージンの広い材料が必要である。
【0006】
本発明の第1の目的は、液晶配向剤として有用である新規なポリアミック酸を提供することにある。
本発明の第2の目的は、液晶配向剤として有用である新規なポリイミドを提供することにある。
本発明の第3の目的は、液晶配向膜としたとき、液晶の配向性が良好で、プレチルト角が大きく、かつ膜厚、ラビング条件などの液晶表示素子の製造工程条件に対するプレチルト角の依存性が小さい液晶配向剤を提供することにある。
本発明のさらに他の目的および利点は、以下の説明から明らかになろう。
【0007】
【課題を解決するための手段】
本発明のポリアミック酸(以下、「特定重合体I」という)は、
下記式(1)
【0008】
【化3】
(式中、R1は炭素数1〜12のアルキル基、炭素数1〜12のハロアルキル基またはハロゲン原子であり、X、Yは互いに独立に下記式(a)〜(d)
【0010】
【化4】
で表わされる2価の結合基であり、aは0〜5の整数である。)
で表わされるジアミン化合物(以下、「特定ジアミン化合物」という)およびテトラカルボン酸二無水物とを反応させて得られるものである。
【0012】
また、本発明のポリイミド(以下、「特定重合体II」という)は、本発明の上記ポリアミック酸を脱水閉環させて得られるものである。
また、本発明の液晶配向剤は、本発明の上記ポリアミック酸および/または上記ポリイミドを含有するものである。
【0013】
【発明の実施の形態】
以下、本発明について詳細に説明する。
特定ジアミン化合物
本発明で用いられる特定ジアミン化合物は、上記式(1)で表わされる化合物である。
式(1)中、R1は炭素数1〜12のアルキル基、炭素数1〜12のハロアルキル基またはハロゲン原子であり、XおよびYは互いに独立に上記式(a)〜(d)のエステル基またはアミド基であり、そしてaは0〜5の整数である。
XとYは同一でも異なっていてもよいが、製造工程上、同一である方がより好ましい。
また、aが2〜5の整数のとき、複数のR1は同一でも異なっていてもよい。
【0014】
R1を表わす炭素数1〜12のアルキル基としては、炭素数1〜4の低級アルキル基が好ましく、その例としては、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、tert−ブチル基などを挙げることができる。
R1を表わす炭素数1〜12のハロアルキル基としては、例えばトリフルオロメチル基を好ましいものとして挙げることができる。
また、R1を表すハロゲン原子としては、例えばフッ素原子を好ましいものとして挙げることができる。
【0015】
上記式(1)で示される特定ジアミン化合物は、例えば次に示す方法によって得られる。
[Xが上記式(a)、Yが上記式(b)で表わされる場合]
工程(i).下記式(2)
【0016】
【化5】
ここでAはハロゲン原子である、
で表わされるジニトロ安息香酸ハライド化合物に、ジヒドロキシシクロヘキサンを反応させて、下記式(3)
【0018】
【化6】
で表わされる水酸基を含有する化合物を得る。
工程(ii).工程(i)で生成した水酸基を有する化合物に、下記式(4)
【0020】
【化7】
ここで、R1、Aおよびaの定義は上記に同じである、
で表わされる化合物を反応させて、下記式(5)
【0022】
【化8】
ここで、R1およびaの定義は上記に同じである、
で表わされるジニトロ化合物を得る。
工程(iii).工程(ii)で得られたジニトロ化合物を還元し、下記式(6)
【0024】
【化9】
ここで、R1およびaの定義は上記に同じである、
で表わされる特定ジアミン化合物を得る。
【0026】
[Xが上記式(c)、Yが上記式(d)で表わされる場合]
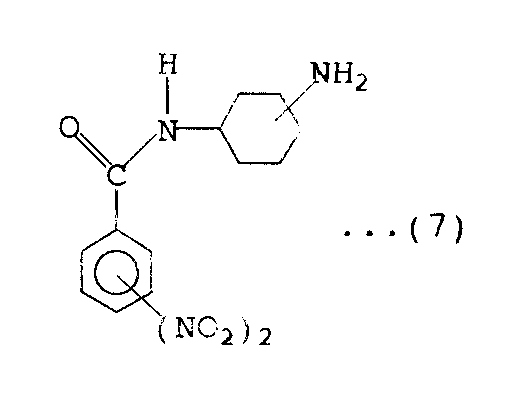
工程(i).上記式(2)で表わされるジニトロ安息香酸ハライドにジアミノシクロヘキサンを反応させて、下記式(7)
【0027】
【化10】
で表わされるアミノ基を有する化合物を得る。
工程(ii).工程(i)で生成したアミノ基を有する化合物に、上記式(4)で表わされる化合物を反応させて、下記式(8)
【0029】
【化11】
ここで、R1およびaの定義は上記に同じである、
で表わされるジニトロ化合物を得る。
工程(iii).工程(ii)で得られたジニトロ化合物を還元し、下記式(9)
【0031】
【化12】
で表わされる特定ジアミン化合物を得る。
【0033】
特定ジアミン化合物の合成反応には、必要に応じて溶媒を用いることができる。上記溶媒としては、特定ジアミン化合物を溶解させることができ、かつ反応を阻害しないものであれば特に限定されず、例えばベンゼン、トルエンなどの芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサンなどのエーテル類;アセトン、メチルエチルケトン、メチルイソブチルケトンなどのケトン類;その他ジメチルスルフォキシド、ジメチルホルムアミド、ジメチルアセトアミドなどを挙げることができる。
上記反応に用いられる塩基性触媒としては、水酸化ナトリウム、水酸化カリウム、ピリジン、トリエチルアミンなどを挙げることができる。
【0034】
上記工程(i)における上記式(2)で表わされるジニトロ安息香酸ハライド化合物とジヒドロキシシクロヘキサン、ジアミノシクロヘキサンなどのシクロヘキサン構造を有する化合物との使用割合は、ジニトロ安息香酸ハライド化合物1モルに対し、シクロヘキサン構造を有する化合物が2〜20モルが好ましい。
上記工程(ii)における工程(i)で得られた化合物1モルに対する上記式(4)または(8)で表わされる化合物の使用割合は1〜1.2モルが好ましい。
【0035】
上記工程(iii)におけるジニトロ化合物の還元には、水素ガス、ヒドラジン、塩酸などの還元剤を周知の触媒の存在化に行うことができる。
上記触媒としては、例えばVIII族金属、すなわち鉄、コバルト、ニッケル、ルテニウム、ロジウム、パラジウム、オスミニウム、インジウム、白金などの金属を活性主体とする金属触媒、具体的には担体に上記金属が担持された触媒、上記金属の錯体触媒を挙げることができ、上記還元反応において均一系でも不均一系であってもよい。
【0036】
上記触媒の使用量は、適宜の割合で使用でき、例えば触媒が前記VIII族の金属を活性主体とする場合、ジニトロ化合物100重量部に対して0.0001〜100重量部、特に0.001〜20重量部の範囲で用いることが好ましい。また、上記還元反応として亜鉛、スズ、炭化スズ(II)、硫化ナトリウム、ナトリウムヒドロスルフィド、亜二チオン酸ナトリウム、硫化アンモニウムなどの還元剤を用いる方法も使用できる。この場合、還元剤は、ジニトロ化合物のニトロ基1モルに対して0.001〜10モルの範囲で使用することが好ましい。
【0037】
上記還元反応に用いる溶媒としては、ジニトロ化合物とジアミン化合物とをともに溶解し、かつ還元反応によって変質しないものが好ましく、例えばメタノール、エタノール、プロパノール、ブタノールなどのアルコール類;ジエチルエーテル、1,2−ジメトキシエタン、テトラヒドロフラン、ジオキサン、アニソールなどのエーテル類を挙げることができる。
【0038】
本発明で使用される特定ジアミン化合物は、単独でまたは2種以上を組合せて使用でき、下記式(10)〜(17)の化合物が好ましいものとして例示される。
【0039】
【化13】
【化14】
また、本発明に使用されるジアミン化合物には、特定ジアミン化合物の他に、本発明の効果を損なわない範囲で他のジアミン化合物を併用することができる。
【0042】
他のジアミン化合物
本発明に使用される他のジアミン化合物としては、以下に示す化合物が例示される。
p−フェニレンジアミン、m−フェニレンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルエタン、4,4’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルホン、3,3’−ジメチル−4,4’−ジアミノビフェニル、4,4’−ジアミノベンズアニリド、4,4’−ジアミノジフェニルエーテル、ビス(4−アミノフェニル)−1,4−ジイソプロピルベンゼン、1,5−ジアミノナフタレン、3,3−ジメチル−4,4’−ジアミノビフェニル、5−アミノ−1−(4’−アミノフェニル)−1,3,3−トリメチルインダン、6−アミノ−1−(4’−アミノフェニル)−1,3,3−トリメチルインダン、3,4’−ジアミノジフェニルエーテル、3,3’−ジアミノベンゾフェノン、3,4’−ジアミノベンゾフェノン、4,4’−ジアミノベンゾフェノン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]プロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]ヘキサフルオロプロパン、2,2−ビス(4−アミノフェニル)ヘキサフルオロプロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]スルホン、1,4−ビス(4−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、1,3−ビス(3−アミノフェノキシ)ベンゼン、9,9−ビス(4−アミノフェニル)−10−ヒドロアントラセン、2,7−ジアミノフルオレン、9,9−ビス(4−アミノフェニル)フルオレン、4,4’−メチレン−ビス(2−クロロアニリン)、2,2’,5,5’−テトラクロロ−4,4’−ジアミノビフェニル、2,2’−ジクロロ−4,4’−ジアミノ−5,5’−ジメトキシビフェニル、3,3’−ジメトキシ−4,4’−ジアミノビフェニル、1,4,4’−(p−フェニレンイソプロピリデン)ビスアニリン、4,4’−(m−フェニレンイソプロピリデン)ビスアニリン、2,2’−ビス[4−(4−アミノ−2−トリフルオロメチルフェノキシ)フェニル]ヘキサフルオロプロパン、4,4’−ジアミノ−2,2’−ビス(トリフルオロメチル)ビフェニル、4,4’−ビス[(4−アミノ−2−トリフルオロメチル)フェノキシ]−オクタフルオロビフェニル、
などの芳香族ジアミン化合物;1,1−メタキシリレンジアミン、1,3−プロパンジアミン、テトラメチレンジアミン、ペンタメチレンジアミン、ヘキサメチレンジアミン、ヘプタメチレンジアミン、オクタメチレンジアミン、ノナメチレンジアミン、4,4−ジアミノヘプタメチレンジアミン、1,4−ジアミノシクロヘキサン、イソホロンジアミン、テトラヒドロジシクロペンタジエニレンジアミン、ヘキサヒドロ−4,7−メタノインダニレンジメチレンジアミン、トリシクロ[6,2,1,02.7]−ウンデシレンジメチルジアミン、4,4’−メチレンビス(シクロヘキシルアミン)などの脂肪族または脂環式ジアミン化合物;
下記式(18)〜(21)
【0043】
【化15】
で表わされるジアミン化合物。
これらの中でp−フェニレンジアミン、4,4’−ジアミノジフェニルメタン、1,5−ジアミノナフタレン、ビス(4−アミノフェニル)−1.4−ジイソプロピルベンゼン、4,4’−ジアミノジフェニルエーテルおよび4,4’−(p−フェニレンイソプロピリデン)ビスアニリンが好ましい。
これらは単独でまたは2種以上を組み合わせて使用できる。
【0045】
本発明に用いられる他のジアミン化合物の使用割合は、全ジアミン化合物中、0〜99.9モル%であり、好ましくは、TN型およびSTN型液晶表示素子用としては、70〜95モル%、SH型液晶表示素子用としては0〜50モル%である。
【0046】
テトラカルボン酸二無水物
本発明に使用されるテトラカルボン酸二無水物としては、以下に示す化合物が例示される。
ブタンテトラカルボン酸二無水物、1,2,3,4−シクロブタンテトラカルボン酸二無水物、1,2,3,4−シクロペンタンテトラカルボン酸二無水物、2,3,5−トリカルボキシシクロペンチル酢酸二無水物、3,5,6−トリカルボキシノルボルナン−2−酢酸二無水物、2,3,4,5−テトラヒドロフランテトラカルボン酸二無水物、1,3,3a,4,5,9b−ヘキサヒドロ−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオン、1,3,3a,4,5,9b−ヘキサヒドロ−5−メチル−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオン、1,3,3a,4,5,9b−ヘキサヒドロ−8−メチル−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオン、5−(2,5−ジオキソテトラヒドロフラル)−3−メチル−3−シクロヘキセン−1,2−ジカルボン酸二無水物、ビシクロ[2,2,2]−オクト−7−エン−2,3,5,6−テトラカルボン酸二無水物などの脂肪族または脂環式テトラカルボン酸二無水物;ピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルスルホンテトラカルボン酸二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物、2,3,6,7−ナフタレンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルエーテルテトラカルボン酸二無水物、3,3’,4,4’−ジメチルジフェニルシランテトラカルボン酸二無水物、3,3’,4,4’−テトラフェニルシランテトラカルボン酸二無水物、1,2,3,4−フランテトラカルボン酸二無水物、4,4’−ビス(3,4−ジカルボキシフェノキシ)ジフェニルスルフィド二無水物、4,4’−ビス(3,4−ジカルボキシフェノキシ)ジフェニルスルホン二無水物、4,4’−ビス(3,4−ジカルボキシフェノキシ)ジフェニルプロパン二無水物、3,3’,4,4’−パーフルオロイソプロピリデンジフタル酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、ビス(フタル酸)フェニルホスフィンオキサイド二無水物、p−フェニレン−ビス(トリフェニルフタル酸)二無水物、m−フェニレン−ビス(トリフェニルフタル酸)二無水物、ビス(トリフェニルフタル酸)−4,4’−ジフェニルエーテル二無水物、ビス(トリフェニルフタル酸)−4,4’−ジフェニルメタン二無水物などの芳香族テトラカルボン酸二無水物。
【0047】
これらのうちではピロメリット酸二無水物、1,2,3,4−シクロブタンテトラカルボン酸二無水物、1,2,3,4−シクロペンタンテトラカルボン酸二無水物、2,3,5−トリカルボキシシクロペンチル酢酸二無水物、1,3,3a,4,5,9b−ヘキサヒドロ−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオン、1,3,3a,4,5,9b−ヘキサヒドロ−5−メチル−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオン、1,3,3a,4,5,9b−ヘキサヒドロ−8−メチル−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオン、5−(2,5−ジオキソテトラヒドロフラル)−3−メチル−3−シクロヘキセン−1,2−ジカルボン酸二無水物およびビシクロ[2,2,2]−オクト−7−エン−2,3,5,6−テトラカルボン酸二無水物が好ましく、ピロメリット酸二無水物、1,2,3,4−シクロブタンテトラカルボン酸二無水物、2,3,5−トリカルボキシシクロペンチル酢酸二無水物、1,3,3a,4,5,9b−ヘキサヒドロ−8−メチル−5−(テトラヒドロ−2,5−ジオキソ−3−フラニル)−ナフト[1,2−c]−フラン−1,3−ジオンが特に好ましい。これらは1種単独でまたは2種以上組み合わせて用いられる。
【0048】
ポリアミック酸
本発明に用いられる特定重合体I(ポリアミック酸)は、特定ジアミン化合物を含有するジアミン化合物とテトラカルボン酸二無水物とを反応させて得られる。かかる反応は有機溶媒中で、通常0〜150℃、好ましくは0〜100℃の反応温度で行われる。
テトラカルボン酸二無水物とジアミン化合物の使用割合は、ジアミン化合物中のアミノ基1当量に対してテトラカルボン酸二無水物の酸無水物基を0.2〜2当量とするのが好ましく、より好ましくは0.3〜1.2当量である。
【0049】
上記有機溶媒としては、反応で生成する特定重合体Iを溶解しうるものであれば特に制限はない。例えば、N−メチル−2−ピロリドン、N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、ジメチルスルホキシド、γ−ブチロラクトン、プロピレンカーボネート、テトラメチル尿素、ヘキサメチルホスホルトリアミドなどの非プロトン系極性溶媒;m−クレゾール、キシレノール、フェノール、ハロゲン化フェノールなどのフェノール系溶媒を挙げることができる。有機溶媒の使用量は、通常、テトラカルボン酸二無水物およびジアミン化合物の総量が、反応溶液の全量に対して0.1〜30重量%になるようにするのが好ましい。
【0050】
なお、上記有機溶媒には、貧溶媒であるアルコール類、ケトン類、エステル類、エーテル類、ハロゲン化炭化水素類、炭化水素類を生成する重合体が析出しない程度に併用することができる。かかる貧溶媒としては、例えばメチルアルコール、エチルアルコール、テトラヒドロフルフリルアルコール、ジアセトンアルコール、イソプロピルアルコール、シクロヘキサノール、エチレングリコール、プロピレングリコール、1,4−ブタンジオール、トリエチレングリコール、エチレングリコールモノメチルエーテル、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン、乳酸メチル、乳酸エチル、乳酸ブチル、酢酸メチル、酢酸エチル、酢酸ブチル、シュウ酸ジエチル、マロン酸ジエチル、ジエチルエーテル、エチレングリコールメチルエーテル、エチレングリコールエチルエーテル、エチレングリコール−n−プロピルエーテル、エチレングリコール−i−プロピルエーテル、エチレングリコール−n−ブチルエーテル、エチレングリコールジメチルエーテル、エチレングリコールエチルエーテルアセテート、ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノメチルエーテルアセテート、ジエチレングリコールモノエチルエーテルアセテート、テトラヒドロフラン、ジクロロメタン、1,2−ジクロロエタン、1,4−ジクロロブタン、トリクロロエタン、クロルベンゼン、o−ジクロルベンゼン、ヘキサン、ヘプタン、オクタン、ベンゼン、トルエン、キシレンなどを挙げることができる。
【0051】
かくして得られる本発明の特定重合体Iは、下記式(22)
【0052】
【化16】
ここで、R1、X、Yおよびaの定義は上記に同じであり、Zはテトラカルボン酸二無水物から2つの酸無水物基を除いた残基である、
で表わされる繰返し単位を有する。
【0054】
ポリイミド
本発明に用いられる特定重合体II(ポリイミド)は、上記の特定重合体Iを、加熱して、または脱水剤およびイミド化触媒の存在下でイミド化することにより得られる。加熱によりイミド化する場合の反応温度は、通常60〜250℃、好ましくは100〜170℃である。反応温度が60℃未満では反応の進行が遅れ、また250℃を越えると特定重合体IIの分子量が大きく低下することがある。また、脱水剤およびイミド化触媒の存在下でイミド化する場合の反応は、前記した有機溶媒中で行うことができる。反応温度は、通常0〜180℃、好ましくは60〜150℃である。脱水剤としては、無水酢酸、無水プロピオン酸、無水トリフルオロ酢酸などの酸無水物を用いることができる。また、イミド化触媒としては、例えばピリジン、コリジン、ルチジン、トリエチルアミンなどの3級アミンを用いることができるが、これらに限定されるものではない。脱水剤の使用量は、相当する特定重合体Iの繰り返し単位1モルに対して1.6〜20モルとするのが好ましい。また、イミド化触媒の使用量は使用する脱水剤1モルに対し、0.5〜10モルとするのが好ましい。また、イミド化反応の条件を適宜調節することにより、イミド化率を調節することができる。
【0055】
かくして得られる本発明の特定重合体IIは、下記式(23)
【0056】
【化17】
ここで、R1、X、Y、aおよびZの定義は上記に同じである、
で表わされる繰返し単位を有する。
【0058】
ポリアミック酸またはポリイミドの固有粘度
このようにして得られる特定重合体IまたはIIの固有粘度[η]inh=(ln ηrel/C、C=0.5g/dl、30℃、N−メチル−2−ピロリドン中、以下同条件にて固有粘度を測定)は、好ましくは0.05〜10dl/g、より好ましくは0.05〜5dl/gである。
【0059】
液晶配向剤
本発明の液晶配向剤は、本発明のポリアミック酸および/またはポリイミドよりなる有効成分が有機溶媒中に溶解含有されて構成される。液晶配向剤を構成する有機溶媒としては、ポリアミック酸の合成反応やイミド化反応に用いられるものとして例示した非プロトン系極性溶媒やフェノール系溶媒を挙げることができる。またポリアミック酸合成の際に併用することができるものとして例示した貧溶媒も適宜選択して併用することができる。
【0060】
本発明の液晶配向剤における有効成分(ポリアミック酸および/またはポリイミド)の濃度は、粘性、揮発性などを考慮して選択されるが、好ましくは1〜10重量%の範囲とされる。溶液からなる液晶配向剤は、印刷法、スピンコート法などにより基板表面に塗布され、次いで、これを乾燥することにより、配向膜材料である塗膜が形成される。有効成分の濃度が1重量%未満である場合には、この塗膜の膜厚が過少となって良好な液晶配向膜を得ることができない。一方、有効成分の濃度が10重量%を越える場合には、塗膜の膜厚が過大となって良好な液晶配向膜を得ることができず、また、液晶配向剤の粘度が増大して塗布特性に劣るものとなる。
本発明の液晶配向剤は、特定重合体Iおよび/または特定重合体IIと基板との接着性のさらなる改善を目的として、官能性シラン含有化合物や、エポキシ系架橋剤を含有することもできる。
【0061】
このような官能性シラン含有化合物としては、例えば3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、2−アミノプロピルトリメトキシシラン、2−アミノプロピルトリエトキシシラン、N−(2−アミノエチル)−3−アミノプロピルトリメトキシシラン、N−(2−アミノエチル)−3−アミノプロピルメチルジメトキシシラン、3−ウレイドプロピルトリメトキシシラン、3−ウレイドプロピルトリエトキシシラン、N−エトシキカルボニル−3−アミノプロピルトリメトキシシラン、N−エトシキカルボニル−3−アミノプロピルトリエトキシシラン、N−トリエトキシシリルプロピルトリエチレントリアミン、N−トリメトキシシリルプロピルトリエチレントリアミン、10−トリメトキシシリル−1,4,7−トリアザデカン、10−トリエトキシシリル−1,4,7−トリアザデカン、9−トリメトキシシリル−3,6−ジアザノニルアセテート、9−トリエトキシシリル−3,6−ジアザノニルアセテート、N−ベンジル−3−アミノプロピルトリメトキシシラン、N−ベンジル−3−アミノプロピルトリエトキシシラン、N−フェニル−3−アミノプロピルトリメトキシシラン、N−フェニル−3−アミノプロピルトリエトキシシラン、N−ビス(オキシエチレン)−3−アミノプロピルトリメトキシシラン、N−ビス(オキシエチレン)−3−アミノプロピルトリエトキシシランなどを挙げることができる。また、上記エポキシ系架橋剤としては、ポリエチレングリコールジグリシジルエーテル、ジグリシジルオルトトルイジン、テトラグリシジルアミノジフェニルメタン、1,3−ビス(N,N’−ジグリシジルアミノメチル)シクロヘキサンなどを好ましいものとして挙げることができる。
【0062】
液晶表示素子
本発明の液晶配向剤を用いて得られる液晶表示素子は、例えば次の方法によって製造することができる。
【0063】
(1)透明導電膜が設けられた基板の透明導電膜側に、本発明の液晶配向剤をロールコーター法、スピンナー法、印刷法などで塗布し、次いで塗布面を加熱することにより塗膜を形成する。
ここに基板としては、例えばフロートガラス、ソーダガラスなどのガラス、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエーテルスルホン、ポリカーボネートなどのプラスチックフィルムなどからなる透明基板を用いることができる。基板の一面に設けられた透明導電膜としては、SnO2からなるNESA膜、In2O3−SnO2からなるITO膜などを用いることができ、これらの透明導電膜のパターニングには、フォト・エッチング法、予めマスクを用いる方法などが用いられる。
【0064】
液晶配向剤の塗布に際しては、基板および透明導電膜と塗膜との接着性をさらに良好にするために、基板および透明導電膜上に、予め官能性シラン含有化合物、チタネートなどを塗布することもできる。加熱温度は、好ましくは80〜250℃とされ、より好ましくは120〜250℃とされる。形成される塗膜の膜厚は、通常、0.001〜1μm、好ましくは0.005〜0.5μmである。
【0065】
(2)形成された塗膜は、ナイロンなどの合成繊維からなる布を巻き付けたロールでラビング処理を行うことにより、液晶配向膜とされる。
【0066】
(3)上記のようにして液晶配向膜が形成された基板は、その2枚を液晶配向膜をラビング方向が直交または逆平行となるよう対向させ、基板の間の周辺部をシール剤でシールし、液晶を充填し、充填孔を封止して液晶セルとし、その両面に偏光方向がそれぞれ基板の液晶配向膜のラビング方向と一致または直交するように張り合わせることにより液晶表示素子とされる。
上記シール剤としては、例えば硬化剤およびスペーサーとしての酸化アルミニウム球を含有したエポキシ樹脂などを用いることができる。
【0067】
上記液晶としては、ネマティック型液晶、スメクティック型液晶、その中でもネマティック型液晶を形成させるものが好ましく、例えばシッフベース系液晶、アゾキシ系液晶、ビフェニル系液晶、フェニルシクロヘキサン系液晶、エステル系液晶、ターフェニル系液晶、ビフェニルシクロヘキサン系液晶、ピリミジン系液晶、ジオキサン系液晶、ビシクロオクタン系液晶、キュバン系液晶などが用いられる。また、これらの液晶に、例えばコレスチルクロライド、コレステリルノナエート、コレステリルカーボネートなどのコレステリック液晶や商品名C−15,CB−15(Merck Ltd.)として販売されているようなカイラル剤などを添加して使用することもできる。さらに、p−デシロキシベンジリデン−p−アミノ−2−メチルブチルシンナメートなどの強誘電性液晶も使用することができる。
【0068】
液晶セルの外側に使用される偏光板としては、ポリビニルアルコールを延伸配向させながら、ヨウ素を吸収させたH膜と呼ばれる偏光膜を酢酸セルロース保護膜で挟んだ偏光板、またはH膜そのものからなる偏光板などを挙げることができる。
【0069】
なお、本発明の液晶配向剤により形成された液晶配向膜に、例えば特開平6−222366号公報や特開平6−281937号公報に示されているような紫外線を照射することによってプレチルト角を変化させるような処理、あるいは特開平5−107544号公報に示されているような、ラビング処理を施した液晶配向膜上にレジストを部分的に形成し、先のラビング処理と異なる方向にラビング処理を行った後にレジスト膜を除去して、液晶配向膜の配向能を変化させるような処理を行うことによって、液晶表示素子の視界特性を改善することが可能である。
【0070】
【実施例】
以下、本発明を実施例により、さらに具体的に説明するが、本発明はこれらの実施例により何ら制限されるものではない。
なお、実施例中におけるプレチルト角の測定は、[T.J. Schffer, et al., J. Appl. Phys., 19, 2013 (1980)]に記載の方法に準拠し、He−Neレーザー光を用いる結晶回転法により行った。
また、液晶セルの配向性評価は、電圧をオン・オフさせた時の液晶セル中の異常ドメインの有無を偏光顕微鏡で観察し、異常ドメインのない場合良好と判断した。
【0071】
特定ジアミン化合物の合成例1
工程(i)
3,5−ジニトロ安息香酸クロリド23.06g(100ミリモル)および1,4−ジヒドロキシシクロヘキサン34.85g(300ミリモル)を、テトラヒドロフラン400gに溶解した。次いで、攪拌氷冷を行いながら、ピリジン7.91g(100ミリモル)を徐々に滴下した。6時間反応を行った後反応液を大過剰の水中にあけ、析出物を濾別し、メチルエチルケトンより再結晶を行い、白色のジニトロ化合物を得た。
【0072】
工程(ii)
工程(i)で得られたジニトロ化合物24.82g(80ミリモル)およびp−トリフルオロメチル安息香酸クロリド18.32g(80ミリモル)を、テトラヒドロフラン400gに溶解した。次いで、攪拌氷冷を行いながら、ピリジン6.33g(80ミリモル)を徐々に滴下した。6時間反応を行った後反応液を大過剰の水中にあけ、析出物を濾別し、メチルエチルケトンより再結晶を行い、白色のジニトロ化合物を得た。
【0073】
工程(iii)
工程(ii)で得られたジニトロ化合物36.20g(72ミリモル)をエタノール400gに溶解させて、5重量%パラジウム−炭素触媒0.30gおよびヒドラジン一水和物5.6g(100ミリモル)を加え、6時間還流反応した。反応液を大過剰の水中にあけ、析出物を濾別し、エタノールより再結晶を行い、上記式(10)で表わされる白色の特定ジアミン化合物(i)24.33gを得た。
得られた特定ジアミン化合物(i)の赤外吸収スペクトルを図1に、NMRスペクトルを図2に示す。
【0074】
特定ジアミン化合物の合成例2
工程(i)
3,5−ジニトロ安息香酸クロリド23.06g(100ミリモル)および1,4−ジヒドロキシシクロヘキサン34.85g(300ミリモル)を、テトラヒドロフラン400gに溶解した。次いで、攪拌氷冷を行いながら、ピリジン7.91g(100ミリモル)を徐々に滴下した。6時間反応を行った後反応液を大過剰の水中にあけ、析出物を濾別し、メチルエチルケトンより再結晶を行い、白色のジニトロ化合物を得た。
【0075】
工程(ii)
工程(i)で得られたジニトロ化合物24.82g(80ミリモル)およびp−フルオロ安息香酸クロリド12.68g(80ミリモル)を、テトラヒドロフラン400gに溶解した。次いで、攪拌氷冷を行いながら、ピリジン6.33g(80ミリモル)を徐々に滴下した。6時間反応を行った後反応液を大過剰の水中にあけ、析出物を濾別し、メチルエチルケトンより再結晶を行い、白色のジニトロ化合物を得た。
【0076】
工程(iii)
工程(ii)で得られたジニトロ化合物31.12g(72ミリモル)をエタノール400gに溶解させて、5重量%パラジウム−炭素触媒0.30gおよびヒドラジン一水和物5.6g(100ミリモル)を加え、6時間還流反応した。反応液を大過剰の水中にあけ、析出物を濾別し、エタノールより再結晶を行い、上記式(10)で表される融点196℃の白色の特定ジアミン化合物(ii)24.33gを得た。
得られた特定ジアミン化合物(ii)の赤外吸収スペクトルを図3に、NMRスペクトルを図4に示す。
【0077】
実施例1(特定重合体Iaの合成)
テトラカルボン酸二無水物として2,3,5−トリカルボキシシクロペンチル酢酸二無水物22.42g(100ミリモル)、ジアミン化合物として上記式(10)で表わされる特定ジアミン化合物4.22g(10ミリモル)およびp−フェニレンジアミン9.73g(90ミリモル)をN−メチル−2−ピロリドン327.33gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.20dl/gの特定重合体Iaを得た。得られた特定重合体Iaの赤外吸収スペクトルを図5に、NMRスペクトルを図6に示す。
【0078】
実施例2(特定重合体IIaの合成)
合成例1で得られた特定重合体Ia36.37gを631.03gのN−メチル−2−ピロリドンに溶解し、ピリジン15.82gおよび無水酢酸20.42gを添加し、115℃で4時間イミド化反応をさせた。
次いで、反応生成液を合成例1と同様に沈澱させ、対数粘度1.20dl/gの特定重合体IIaを得た。得られた特定重合体IIaの赤外吸収スペクトルを図7に、NMRスペクトルを図8に示す。
【0079】
実施例3(特定重合体Ibの合成)
テトラカルボン酸二無水物として2,3,5−トリカルボキシシクロペンチル酢酸二無水物22.42g(100ミリモル)、ジアミン化合物として上記式(10)で表わされる特定ジアミン化合物21.12g(50ミリモル)およびp−フェニレンジアミン5.41g(50ミリモル)をN−メチル−2−ピロリドン440.55gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.00dl/gの特定重合体Ibを得た。
【0080】
実施例4(特定重合体IIbの合成)
合成例3で得られた特定重合体Ib48.95gを930.05gのN−メチル−2−ピロリドンに溶解し、ピリジン15.82gおよび無水酢酸20.42gを添加し、115℃で4時間イミド化反応をさせた。
次いで、反応生成液を合成例1と同様に沈澱させ、対数粘度1.00dl/gの特定重合体IIbを得た。
【0081】
実施例5(特定重合体Icの合成)
テトラカルボン酸二無水物として2,3,5−トリカルボキシシクロペンチル酢酸二無水物22.42g(100ミリモル)およびジアミン化合物として上記式(10)で表わされる特定ジアミン化合物44.24g(100ミリモル)をN−メチル−2−ピロリドン581.94gに溶解させ、60℃で6時間反応させた。 次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度0.80dl/gの特定重合体Icを得た。得られた特定重合体Icの赤外吸収スペクトルを図9に、NMRスペクトルを図10に示す。
【0082】
実施例6(特定重合体IIcの合成)
合成例5で得られた特定重合体Ic64.66gを1228.54gのN−メチル−2−ピロリドンに溶解し、15.82gのピリジンと20.42gの無水酢酸を添加し、115℃で4時間イミド化反応をさせた。
次いで、反応生成液を合成例1と同様に沈澱させ、対数粘度0.80dl/gの特定重合体IIcを得た。得られた特定重合体IIcの赤外吸収スペクトルを図11に、NMRスペクトルを図12に示す。
【0083】
実施例7(特定重合体Idの合成)
テトラカルボン酸二無水物としてピロメリット酸二無水物21.81g(100ミリモル)、ジアミン化合物として上記式(10)で表わされる特定ジアミン化合物21.11g(50ミリモル)およびp−フェニレンジアミン5.41g(50ミリモル)をN−メチル−2−ピロリドン434.97gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.35dl/gの特定重合体Idを得た。
【0084】
実施例8(特定重合体Ieの合成)
テトラカルボン酸二無水物としてピロメリット酸二無水物21.81g(100ミリモル)、ジアミン化合物として上記式(11)で表わされる特定ジアミン化合物21.01g(50ミリモル)およびp−フェニレンジアミン5.41g(50ミリモル)をN−メチル−2−ピロリドン434.07gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.40dl/gの特定重合体Ieを得た。
【0085】
実施例9(特定重合体Ifの合成)
テトラカルボン酸二無水物としてピロメリット酸二無水物21.81g(100ミリモル)、ジアミン化合物として上記式(12)で表わされる特定ジアミン化合物18.41g(50ミリモル)およびp−フェニレンジアミン5.41g(50ミリモル)をN−メチル−2−ピロリドン410.67gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.30dl/gの特定重合体Ifを得た。
【0086】
実施例10(特定重合体Igの合成)
テトラカルボン酸二無水物として1,3,3a,4,5,9b−ヘキサヒドロ−8−メチル−5(テトラヒドロ−2,5−ジオキソ−3−フラニル)ナフト[1,2−c]フラン−1,3−ジオン26.72g(85ミリモル)および1,3−ジメチルシクロブタンテトラカルボン酸二無水物3.36g(15ミリモル)、ジアミン化合物として1,4−フェニレンジアミン8.11g(75ミリモル)、4,4’−ジアミノジフェニルメタン2.97g(15ミリモル)および上記式(10)で表わされる特定化合物4.22g(10ミリモル)をN−メチル−2−ピロリドン408.42gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.10dl/gの特定重合体Igを得た。
【0087】
実施例11(特定重合体IIgの合成)
合成例10で得られた特定重合体Ig45.38を862.22gのN−メチル−2−ピロリドンに溶解し、ピリジン15.82gおよび無水酢酸20.42gを添加し、115℃で4時間イミド化反応させた。
次いで、反応生成物を合成例1と同様に沈澱させ、対数粘度1.10dl/gの特定重合体IIgを得た。
【0088】
実施例12(特定重合体IIhの合成)
テトラカルボン酸二無水物として2,3,5−トリカルボキシシクロペンチル酢酸二無水物22.42g(100ミリモル)、ジアミン化合物として特定ジアミン化合物(ii)3.72g(10ミリモル)およびp−フェニレンジアミン9.73g(90ミリモル)をN−メチル−2−ピロリドン327.33gに溶解させ、60℃で6時間反応させた。次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.10dl/gの特定重合体Ihを得た。得られた特定重合体Ih36.37gを631.03gのN−メチル−2−ピロリドンに溶解し、ピリジン15.82gおよび無水酢酸20.42gを添加し、115℃で4時間イミド化反応をさせた。
次いで、反応生成液を合成例1と同様に沈澱させ、対数粘度1.0dl/gの特定重合対IIhを得た。得られた特定重合体IIhの赤外吸収スペクトルを図13に、NMRスペクトルを図14に示す。
【0089】
比較例1(重合体Iαの合成)
テトラカルボン酸二無水物として2,3,5−トリカルボキシシクロペンチル酢酸二無水物22.42g(100ミリモル)およびジアミン化合物として、p−フェニレンジアミン10.81g(100.00ミリモル)をN−メチル−2−ピロリドン299.07gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.25dl/gの重合体Iαを得た。
【0090】
比較例2(重合体IIαの合成)
比較例1で得られた重合体Ia33.23gを631.37gのN−メチル−2−ピロリドンに溶解し、ピリジン15.82gおよび無水酢酸20.42gを添加し、115℃で4時間イミド化反応をさせた。
次いで、反応生成液を合成例1と同様に沈澱させ、対数粘度1.25dl/gの重合体IIαを得た。
【0091】
比較例3(重合体Iβの合成)
テトラカルボン酸二無水物としてピロメリット酸二無水物21.81g(100ミリモル)およびジアミン化合物としてp−フェニレンジアミン10.81g(100ミリモル)をN−メチル−2−ピロリドン293.58gに溶解させ、60℃で6時間反応させた。
次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.40dl/gの重合体Iβを得た。
【0092】
比較例4(重合体Iγの合成)
テトラカルボン酸二無水物としてピロメリット酸二無水物21.81g(100ミリモル)およびジアミン化合物としてp−フェニレンジアミン5.4g(50ミリモル)、下記式(24)
【0093】
【化18】
で表されるジアミン化合物16.2g(50ミリモル)をN−メチル−2−ピロリドン293.58gに溶解させ、60℃で6時間反応させた。次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.20dl/gの重合体Iγを得た。
【0095】
比較例5(重合体Iδの合成)
テトラカルボン酸二無水物としてピロメリット酸二無水物21.81g(100ミリモル)およびジアミン化合物としてp−フェニレンジアミン5.4g(50ミリモル)、上記式(21)で表されるジアミン化合物14.76g(50ミリモル)をN−メチル−2−ピロリドン293.58gに溶解させ、60℃で6時間反応させた。次いで、反応混合物を大過剰のメタノールに注ぎ、反応生成物を沈澱させた。その後、メタノールで洗浄し、減圧下40℃で15時間乾燥させて、対数粘度1.60dl/gの重合体Iδを得た。
【0096】
実施例13(液晶配向剤の調製・評価)
実施例2で得られた特定重合体IIaをγ−ブチロラクトンに溶解させて、固形分濃度4重量%の溶液とし、この溶液を孔径1μmのフィルターで濾過し、液晶配向剤を調製した。
この液晶配向剤を、ITO膜からなる透明電極付きガラス基板の上に透明電極面に、膜厚が800オングストロームになるようにスピンナーを用いて塗布し、180℃で1時間焼成し塗膜を形成した。
この塗膜にナイロン製の布を巻き付けたロールを有するラビングマシーンにより、ロール毛足押し込み長0.6mm、ロールの回転数500rpm、ステージの移動速度1cm/秒でラビング処理を2回行った。このとき、配向膜と基板との接着性は良好であり、ラビングによる剥がれは観察されなかった。
【0097】
次に、一対のラビング処理された基板の液晶配向膜を有するそれぞれの外縁に、直径17μmの酸化アルミニウム球入りエポキシ樹脂接着剤をスクリーン印刷塗布した後、一対の基板を液晶配向膜面が相対するように、しかもラビング方向が逆平行になるように重ね合わせて圧着し、接着剤を硬化させた。
次いで、液晶注入口より一対の基板間に、ネマティック型液晶(メルク社製、MLC−2001)を充填した後、エポキシ系接着剤で液晶注入口を封止し、基板の外側の両面に偏光板を、偏光板の偏光方向がそれぞれの基板の液晶配向膜のラビング方向と一致するように張り合わせ、液晶表示素子を作製した。
得られた液晶表示素子の配向性は良好であり、プレチルト角を測定したところ、3.5゜であった。これらの結果を表1に示す。
【0098】
実施例14〜45(液晶配向剤の調製・評価)
実施例13において、実施例2、4、6、7、8、9、11および12で得られた特定重合体IIb、IIc、Id、Ie、If、IIgおよびIIhを用いて液晶配向剤を調製し、配向膜の加熱温度を180〜250℃、膜厚を200〜1500オングストローム、ラビング回数を1〜5回とした以外は、実施例13と同様にして液晶表示素子を作製し、その液晶表示素子の配向性およびプレチルト角を測定し、結果を表1および表2に示す。
【0099】
【表1】
【表2】
比較例6〜25(液晶配向剤の調製・評価)
実施例13において、比較例1〜5で得られた重合体を用いて液晶配向剤を調製し、配向膜の加熱温度を180〜250℃、膜厚を200〜1500オングストローム、ラビング回数を1〜5回とした以外は、実施例13と同様にして液晶表示素子を作製し、その液晶表示素子の配向性およびプレチルト角を測定し、結果を表3に示す。
【0102】
【表3】
【発明の効果】
本発明の液晶配向剤によれば、液晶配向膜としたとき、液晶の配向性が良好で、膜厚、ラビング条件などの工程条件に依らない高いプレチルト角を発現できる、TN型、STN型またはSH型表示素子用として好適な液晶配向膜が得られる。
さらに、本発明の液晶配向剤を用いて形成した配向膜を有する液晶表示素子は、液晶の配向性および信頼性に優れ、種々の装置に有効に使用でき、例えば卓上計算機、腕時計、置時計、係数表示板、ワードプロセッサ、パーソナルコンピューター、液晶テレビなどの表示装置に用いられる。
【図面の簡単な説明】
【図1】本発明で用いられるジアミン化合物の具体例の赤外吸収スペクトル図である。
【図2】図1の具体例と同じジアミン化合物のNMRスペクトル図である。
【図3】本発明で用いられるジアミン化合物の他の具体例の赤外吸収スペクトル図である。
【図4】図3の具体例と同じジアミン化合物のNMRスペクトル図である。
【図5】本発明のポリアミック酸の具体例の赤外吸収スペクトル図である。
【図6】図5の具体例と同じポリアミック酸のNMRスペクトル図である。
【図7】本発明のポリイミドの具体例の赤外吸収スペクトル図である。
【図8】図7の具体例と同じポリイミドのNMRスペクトル図である。
【図9】本発明のポリアミック酸の他の具体例の赤外吸収スペクトル図である。
【図10】図9の具体例と同じポリアミック酸のNMRスペクトル図である。
【図11】本発明のポリイミドの他の具体例の赤外吸収スペクトル図である。
【図12】図11の具体例と同じポリイミドのNMRスペクトル図である。
【図13】本発明のポリイミドの他の具体例の赤外吸収スペクトル図である。
【図14】図13の具体例と同じポリイミドのNMRスペクトル図である。[0001]
TECHNICAL FIELD OF THE INVENTION
The present invention relates to a novel polyamic acid and polyimide, and a liquid crystal aligning agent containing at least one of these.
[0002]
[Prior art]
Conventionally, a nematic liquid crystal having a positive dielectric anisotropy is formed into a sandwich structure with a substrate having a transparent electrode having a liquid crystal alignment film made of polyimide or the like so that the major axis of the liquid crystal molecules is continuously twisted by 90 degrees between the substrates. There is known a liquid crystal display element (TN type liquid crystal display element) having a TN (Twisted Nematic) type liquid crystal cell. The alignment of the liquid crystal in this TN type liquid crystal display element is formed by a liquid crystal alignment film subjected to a rubbing treatment.
[0003]
Recently, an STN (Super Twisted Nematic) type liquid crystal display device and an SH (Super Homeotropic) type liquid crystal display device which are liquid crystal display devices excellent in contrast and viewing angle dependence have been developed. The STN-type liquid crystal display device uses a nematic type liquid crystal as a liquid crystal blended with a chiral agent, which is an optically active substance. To use. In addition, the SH type liquid crystal display element vertically operates liquid crystal having negative dielectric anisotropy of liquid crystal molecules, and inverts the molecules by applying a voltage to operate by simple matrix driving.
[0004]
[Problems to be solved by the invention]
However, when an STN-type liquid crystal display device is manufactured using a conventionally known liquid crystal alignment film made of polyimide or the like, the liquid crystal cannot be twisted by 180 degrees or more between substrates because the pretilt angle of the liquid crystal alignment film is small. It is difficult to obtain a required display function. For this reason, in the STN-type liquid crystal display element, it is necessary to use a liquid crystal alignment film formed by obliquely depositing silicon dioxide in order to align the liquid crystal. Is not suitable. In addition, the SH-type liquid crystal display element uses a substrate formed by obliquely depositing silicon dioxide in order to vertically align the liquid crystal, or uses a fluorine-based surfactant or a coupling agent having a long-chain alkyl group for the substrate. However, when a surfactant or a coupling agent is used, there is a problem that reliability is poor.
Also in a TN-type liquid crystal display element, a liquid crystal alignment film having a high pretilt angle has been desired in order to suppress a display defect due to a reverse tilt phenomenon when driving a liquid crystal cell.
[0005]
Conventionally, long-chain alkyl groups have been generally introduced into the terminal and side chains of a polyimide in order to develop a high pretilt angle. However, the polyimide alignment film modified with the long-chain alkyl group has a problem that the process stability of the pretilt angle is lacking. In particular, the STN-type liquid crystal display element requires a material having a wide process margin for expressing the pretilt angle because, in principle, the unevenness of the pretilt angle caused by the unevenness of the film thickness, the rubbing and the like appears as the unevenness of the display.
[0006]
A first object of the present invention is to provide a novel polyamic acid useful as a liquid crystal aligning agent.
A second object of the present invention is to provide a novel polyimide useful as a liquid crystal aligning agent.
A third object of the present invention is to provide a liquid crystal alignment film having good liquid crystal alignment, a large pretilt angle, and dependence of the pretilt angle on the liquid crystal display element manufacturing process conditions such as film thickness and rubbing conditions. Is to provide a liquid crystal aligning agent having a small particle size.
Still other objects and advantages of the present invention will become apparent from the following description.
[0007]
[Means for Solving the Problems]
The polyamic acid of the present invention (hereinafter referred to as “specific polymer I”)
The following equation (1)
[0008]
Embedded image
(Where R1Is an alkyl group having 1 to 12 carbon atoms, a haloalkyl group having 1 to 12 carbon atoms or a halogen atom, and X and Y are each independently the following formulas (a) to (d)
[0010]
Embedded image
And a is an integer of 0 to 5. )
(Hereinafter referred to as “specific diamine compound”) and a tetracarboxylic dianhydride.
[0012]
Further, the polyimide of the present invention (hereinafter, referred to as “specific polymer II”) is obtained by subjecting the above polyamic acid of the present invention to dehydration and ring closure.
Further, the liquid crystal aligning agent of the present invention contains the above polyamic acid and / or the above polyimide of the present invention.
[0013]
BEST MODE FOR CARRYING OUT THE INVENTION
Hereinafter, the present invention will be described in detail.
Specific diamine compound
The specific diamine compound used in the present invention is a compound represented by the above formula (1).
In the formula (1), R1Is an alkyl group having 1 to 12 carbon atoms, a haloalkyl group having 1 to 12 carbon atoms or a halogen atom, X and Y are each independently an ester group or an amide group of the above formulas (a) to (d); a is an integer of 0 to 5.
X and Y may be the same or different, but it is more preferable that they are the same in the manufacturing process.
When a is an integer of 2 to 5, a plurality of R1May be the same or different.
[0014]
R1As the alkyl group having 1 to 12 carbon atoms, a lower alkyl group having 1 to 4 carbon atoms is preferable, and examples thereof include a methyl group, an ethyl group, an n-propyl group, an iso-propyl group and an n-butyl group. , Tert-butyl group and the like.
R1As the haloalkyl group having 1 to 12 carbon atoms which represents, for example, a trifluoromethyl group can be mentioned as a preferable example.
Also, R1As the halogen atom representing, for example, a fluorine atom can be mentioned as a preferable example.
[0015]
The specific diamine compound represented by the above formula (1) is obtained, for example, by the following method.
[When X is represented by the above formula (a) and Y is represented by the above formula (b)]
Step (i). The following equation (2)
[0016]
Embedded image
Where A is a halogen atom,
Is reacted with dihydroxycyclohexane to give a dinitrobenzoic acid halide compound represented by the following formula (3)
[0018]
Embedded image
To obtain a compound containing a hydroxyl group represented by the formula:
Step (ii). The compound having a hydroxyl group generated in the step (i) is added with the following formula (4)
[0020]
Embedded image
Where R1, A and a are as defined above,
And reacting the compound represented by the following formula (5)
[0022]
Embedded image
Where R1And the definitions of a are the same as above,
To obtain a dinitro compound represented by the formula:
Step (iii). The dinitro compound obtained in the step (ii) is reduced to give the following formula (6)
[0024]
Embedded image
Where R1And the definitions of a are the same as above,
Is obtained.
[0026]
[When X is represented by the above formula (c) and Y is represented by the above formula (d)]
Step (i). By reacting dinitrocyclohexane with the dinitrobenzoic halide represented by the above formula (2), the following formula (7)
[0027]
Embedded image
To obtain a compound having an amino group represented by the formula:
Step (ii). The compound having the amino group generated in the step (i) is reacted with the compound represented by the above formula (4) to give the following formula (8)
[0029]
Embedded image
Where R1And the definitions of a are the same as above,
To obtain a dinitro compound represented by the formula:
Step (iii). The dinitro compound obtained in the step (ii) is reduced to give the following formula (9)
[0031]
Embedded image
Is obtained.
[0033]
In the synthesis reaction of the specific diamine compound, a solvent can be used as necessary. The solvent is not particularly limited as long as it can dissolve the specific diamine compound and does not inhibit the reaction. For example, aromatic hydrocarbons such as benzene and toluene; ethers such as diethyl ether, tetrahydrofuran and dioxane And ketones such as acetone, methyl ethyl ketone and methyl isobutyl ketone; and dimethyl sulfoxide, dimethyl formamide, dimethyl acetamide and the like.
Examples of the basic catalyst used in the above reaction include sodium hydroxide, potassium hydroxide, pyridine, triethylamine and the like.
[0034]
In the step (i), the ratio of the dinitrobenzoic acid halide compound represented by the above formula (2) to the compound having a cyclohexane structure such as dihydroxycyclohexane or diaminocyclohexane is such that the cyclohexane structure is used per mole of the dinitrobenzoic acid halide compound. Is preferably 2 to 20 mol.
The use ratio of the compound represented by the formula (4) or (8) to 1 mol of the compound obtained in the step (i) in the step (ii) is preferably 1 to 1.2 mol.
[0035]
The reduction of the dinitro compound in the above step (iii) can be carried out using a reducing agent such as hydrogen gas, hydrazine or hydrochloric acid in the presence of a known catalyst.
Examples of the catalyst include Group VIII metals, that is, metal catalysts mainly composed of metals such as iron, cobalt, nickel, ruthenium, rhodium, palladium, osmium, indium, and platinum. And a complex catalyst of the above-mentioned metals. The reduction reaction may be a homogeneous or heterogeneous system.
[0036]
The catalyst may be used in an appropriate ratio. For example, when the catalyst is mainly composed of the Group VIII metal, 0.0001 to 100 parts by weight, particularly 0.001 to 100 parts by weight, per 100 parts by weight of the dinitro compound is used. Preferably, it is used in the range of 20 parts by weight. In addition, a method using a reducing agent such as zinc, tin, tin (II) carbide, sodium sulfide, sodium hydrosulfide, sodium dithionite, and ammonium sulfide can also be used as the reduction reaction. In this case, the reducing agent is preferably used in the range of 0.001 to 10 mol per 1 mol of the nitro group of the dinitro compound.
[0037]
As the solvent used in the above reduction reaction, those which dissolve both the dinitro compound and the diamine compound and do not deteriorate by the reduction reaction are preferable. For example, alcohols such as methanol, ethanol, propanol and butanol; diethyl ether; Examples thereof include ethers such as dimethoxyethane, tetrahydrofuran, dioxane, and anisole.
[0038]
The specific diamine compound used in the present invention can be used alone or in combination of two or more, and the compounds of the following formulas (10) to (17) are exemplified as preferable ones.
[0039]
Embedded image
Embedded image
Further, in addition to the specific diamine compound, other diamine compounds can be used in combination with the diamine compound used in the present invention as long as the effects of the present invention are not impaired.
[0042]
Other diamine compounds
As other diamine compounds used in the present invention, the following compounds are exemplified.
p-phenylenediamine, m-phenylenediamine, 4,4'-diaminodiphenylmethane, 4,4'-diaminodiphenylethane, 4,4'-diaminodiphenylsulfide, 4,4'-diaminodiphenylsulfone, 3,3'- Dimethyl-4,4'-diaminobiphenyl, 4,4'-diaminobenzanilide, 4,4'-diaminodiphenyl ether, bis (4-aminophenyl) -1,4-diisopropylbenzene, 1,5-diaminonaphthalene, 3 , 3-Dimethyl-4,4'-diaminobiphenyl, 5-amino-1- (4'-aminophenyl) -1,3,3-trimethylindane, 6-amino-1- (4'-aminophenyl)- 1,3,3-trimethylindane, 3,4'-diaminodiphenyl ether, 3,3'-diaminobenzo Enone, 3,4'-diaminobenzophenone, 4,4'-diaminobenzophenone, 2,2-bis [4- (4-aminophenoxy) phenyl] propane, 2,2-bis [4- (4-aminophenoxy) Phenyl] hexafluoropropane, 2,2-bis (4-aminophenyl) hexafluoropropane, 2,2-bis [4- (4-aminophenoxy) phenyl] sulfone, 1,4-bis (4-aminophenoxy) Benzene, 1,3-bis (4-aminophenoxy) benzene, 1,3-bis (3-aminophenoxy) benzene, 9,9-bis (4-aminophenyl) -10-hydroanthracene, 2,7-diamino Fluorene, 9,9-bis (4-aminophenyl) fluorene, 4,4′-methylene-bis (2-chloroaniline), 2,2 ′ 5,5'-tetrachloro-4,4'-diaminobiphenyl, 2,2'-dichloro-4,4'-diamino-5,5'-dimethoxybiphenyl, 3,3'-dimethoxy-4,4'- Diaminobiphenyl, 1,4,4 '-(p-phenyleneisopropylidene) bisaniline, 4,4'-(m-phenyleneisopropylidene) bisaniline, 2,2'-bis [4- (4-amino-2-tri Fluoromethylphenoxy) phenyl] hexafluoropropane, 4,4'-diamino-2,2'-bis (trifluoromethyl) biphenyl, 4,4'-bis [(4-amino-2-trifluoromethyl) phenoxy] -Octafluorobiphenyl,
Aromatic diamine compounds such as 1,1-methaxylylenediamine, 1,3-propanediamine, tetramethylenediamine, pentamethylenediamine, hexamethylenediamine, heptamethylenediamine, octamethylenediamine, nonamethylenediamine, 4,4 -Diaminoheptamethylenediamine, 1,4-diaminocyclohexane, isophoronediamine, tetrahydrodicyclopentadienylenediamine, hexahydro-4,7-methanoindanylenediethylenediamine, tricyclo [6,2,1,02.7An aliphatic or alicyclic diamine compound such as -undecylenedimethyldiamine, 4,4'-methylenebis (cyclohexylamine);
The following equations (18) to (21)
[0043]
Embedded image
A diamine compound represented by the formula:
Among them, p-phenylenediamine, 4,4'-diaminodiphenylmethane, 1,5-diaminonaphthalene, bis (4-aminophenyl) -1.4-diisopropylbenzene, 4,4'-diaminodiphenyl ether and 4,4 '-(P-phenyleneisopropylidene) bisaniline is preferred.
These can be used alone or in combination of two or more.
[0045]
The use ratio of the other diamine compound used in the present invention is 0 to 99.9 mol% in all the diamine compounds, and preferably 70 to 95 mol% for TN type and STN type liquid crystal display elements. It is 0 to 50 mol% for SH type liquid crystal display devices.
[0046]
Tetracarboxylic dianhydride
Examples of the tetracarboxylic dianhydride used in the present invention include the following compounds.
Butanetetracarboxylic dianhydride, 1,2,3,4-cyclobutanetetracarboxylic dianhydride, 1,2,3,4-cyclopentanetetracarboxylic dianhydride, 2,3,5-tricarboxycyclopentyl Acetic acid dianhydride, 3,5,6-tricarboxynorbornane-2-acetic acid dianhydride, 2,3,4,5-tetrahydrofurantetracarboxylic dianhydride, 1,3,3a, 4,5,9b- Hexahydro-5- (tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan-1,3-dione, 1,3,3a, 4,5,9b-hexahydro-5 -Methyl-5- (tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan-1,3-dione, 1,3,3a, 4,5,9b-hexahydro- 8-methyl-5 (Tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan-1,3-dione, 5- (2,5-dioxotetrahydrofural) -3-methyl-3- Aliphatic or alicyclic tetra such as cyclohexene-1,2-dicarboxylic dianhydride, bicyclo [2,2,2] -oct-7-ene-2,3,5,6-tetracarboxylic dianhydride Carboxylic dianhydride; pyromellitic dianhydride, 3,3 ′, 4,4′-benzophenonetetracarboxylic dianhydride, 3,3 ′, 4,4′-biphenylsulfonetetracarboxylic dianhydride, 1,4,5,8-naphthalenetetracarboxylic dianhydride, 2,3,6,7-naphthalenetetracarboxylic dianhydride, 3,3 ′, 4,4′-biphenylethertetracarboxylic dianhydride , 3,3 ', 4, '-Dimethyldiphenylsilanetetracarboxylic dianhydride, 3,3', 4,4'-tetraphenylsilanetetracarboxylic dianhydride, 1,2,3,4-furantetracarboxylic dianhydride, 4, 4′-bis (3,4-dicarboxyphenoxy) diphenylsulfide dianhydride, 4,4′-bis (3,4-dicarboxyphenoxy) diphenylsulfone dianhydride, 4,4′-bis (3,4 -Dicarboxyphenoxy) diphenylpropane dianhydride, 3,3 ', 4,4'-perfluoroisopropylidene diphthalic dianhydride, 3,3', 4,4'-biphenyltetracarboxylic dianhydride, Bis (phthalic acid) phenyl phosphine oxide dianhydride, p-phenylene-bis (triphenylphthalic acid) dianhydride, m-phenylene-bis (triphenyl phthalate) Aromatic tetracarboxylic dianhydrides such as dianhydride, bis (triphenylphthalic acid) -4,4′-diphenyl ether dianhydride and bis (triphenylphthalic acid) -4,4′-diphenylmethane dianhydride Anhydrous.
[0047]
Among these, pyromellitic dianhydride, 1,2,3,4-cyclobutanetetracarboxylic dianhydride, 1,2,3,4-cyclopentanetetracarboxylic dianhydride, 2,3,5- Tricarboxycyclopentylacetic acid dianhydride, 1,3,3a, 4,5,9b-hexahydro-5- (tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan-1 , 3-Dione, 1,3,3a, 4,5,9b-Hexahydro-5-methyl-5- (tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan- 1,3-dione, 1,3,3a, 4,5,9b-hexahydro-8-methyl-5- (tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan -1,3-dione, 5- (2,5 Dioxotetrahydrofural) -3-methyl-3-cyclohexene-1,2-dicarboxylic dianhydride and bicyclo [2,2,2] -oct-7-ene-2,3,5,6-tetracarboxylic acid Preferred are dianhydrides, pyromellitic dianhydride, 1,2,3,4-cyclobutanetetracarboxylic dianhydride, 2,3,5-tricarboxycyclopentylacetic dianhydride, 1,3,3a, 4 , 5,9b-Hexahydro-8-methyl-5- (tetrahydro-2,5-dioxo-3-furanyl) -naphtho [1,2-c] -furan-1,3-dione is particularly preferred. These may be used alone or in combination of two or more.
[0048]
Polyamic acid
The specific polymer I (polyamic acid) used in the present invention is obtained by reacting a diamine compound containing a specific diamine compound with tetracarboxylic dianhydride. This reaction is carried out in an organic solvent at a reaction temperature of usually 0 to 150 ° C, preferably 0 to 100 ° C.
The use ratio of the tetracarboxylic dianhydride to the diamine compound is preferably such that the acid anhydride group of the tetracarboxylic dianhydride is 0.2 to 2 equivalents to 1 equivalent of the amino group in the diamine compound, Preferably it is 0.3 to 1.2 equivalents.
[0049]
The organic solvent is not particularly limited as long as it can dissolve the specific polymer I generated by the reaction. For example, aprotic polar solvents such as N-methyl-2-pyrrolidone, N, N-dimethylacetamide, N, N-dimethylformamide, dimethylsulfoxide, γ-butyrolactone, propylene carbonate, tetramethylurea, and hexamethylphosphortriamide. Phenolic solvents such as m-cresol, xylenol, phenol and halogenated phenol; Usually, the amount of the organic solvent used is preferably such that the total amount of the tetracarboxylic dianhydride and the diamine compound is 0.1 to 30% by weight based on the total amount of the reaction solution.
[0050]
The organic solvent can be used in combination with the organic solvent to such an extent that a polymer which produces a poor solvent such as alcohols, ketones, esters, ethers, halogenated hydrocarbons and hydrocarbons is not precipitated. Examples of such poor solvents include methyl alcohol, ethyl alcohol, tetrahydrofurfuryl alcohol, diacetone alcohol, isopropyl alcohol, cyclohexanol, ethylene glycol, propylene glycol, 1,4-butanediol, triethylene glycol, ethylene glycol monomethyl ether, Acetone, methyl ethyl ketone, methyl isobutyl ketone, cyclohexanone, methyl lactate, ethyl lactate, butyl lactate, methyl acetate, ethyl acetate, butyl acetate, diethyl oxalate, diethyl malonate, diethyl ether, ethylene glycol methyl ether, ethylene glycol ethyl ether, ethylene Glycol-n-propyl ether, ethylene glycol-i-propyl ether, ethylene glycol-n- Butyl ether, ethylene glycol dimethyl ether, ethylene glycol ethyl ether acetate, diethylene glycol dimethyl ether, diethylene glycol diethyl ether, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monomethyl ether acetate, diethylene glycol monoethyl ether acetate, tetrahydrofuran, dichloromethane, 1,2-dichloroethane, , 4-dichlorobutane, trichloroethane, chlorobenzene, o-dichlorobenzene, hexane, heptane, octane, benzene, toluene, xylene and the like.
[0051]
The specific polymer I of the present invention thus obtained is represented by the following formula (22)
[0052]
Embedded image
Where R1, X, Y and a are the same as defined above, and Z is a residue obtained by removing two acid anhydride groups from tetracarboxylic dianhydride.
Having a repeating unit represented by
[0054]
Polyimide
The specific polymer II (polyimide) used in the present invention can be obtained by imidizing the above specific polymer I by heating or in the presence of a dehydrating agent and an imidization catalyst. The reaction temperature when imidizing by heating is usually 60 to 250 ° C, preferably 100 to 170 ° C. If the reaction temperature is lower than 60 ° C., the progress of the reaction is delayed, and if it exceeds 250 ° C., the molecular weight of the specific polymer II may be greatly reduced. The reaction for imidization in the presence of a dehydrating agent and an imidization catalyst can be performed in the above-mentioned organic solvent. The reaction temperature is generally 0-180 ° C, preferably 60-150 ° C. Acid anhydrides such as acetic anhydride, propionic anhydride, and trifluoroacetic anhydride can be used as the dehydrating agent. As the imidation catalyst, for example, tertiary amines such as pyridine, collidine, lutidine, and triethylamine can be used, but the catalyst is not limited to these. The amount of the dehydrating agent used is preferably 1.6 to 20 mol per 1 mol of the corresponding repeating unit of the specific polymer I. The amount of the imidation catalyst used is preferably 0.5 to 10 mol per 1 mol of the dehydrating agent used. The imidation ratio can be adjusted by appropriately adjusting the conditions of the imidization reaction.
[0055]
The specific polymer II of the present invention thus obtained is represented by the following formula (23)
[0056]
Embedded image
Where R1, X, Y, a and Z are as defined above,
Having a repeating unit represented by
[0058]
Intrinsic viscosity of polyamic acid or polyimide
Intrinsic viscosity [η] inh of specific polymer I or II obtained in this manner is (lnηrel / C, C = 0.5 g / dl, 30 ° C., in N-methyl-2-pyrrolidone, under the same conditions) (Measured intrinsic viscosity) is preferably 0.05 to 10 dl / g, more preferably 0.05 to 5 dl / g.
[0059]
Liquid crystal alignment agent
The liquid crystal aligning agent of the present invention is constituted by dissolving and containing an active ingredient comprising the polyamic acid and / or polyimide of the present invention in an organic solvent. Examples of the organic solvent constituting the liquid crystal aligning agent include aprotic polar solvents and phenol solvents exemplified as those used in the synthesis reaction and the imidization reaction of polyamic acid. Poor solvents exemplified as those which can be used together in the synthesis of polyamic acid can also be appropriately selected and used in combination.
[0060]
The concentration of the active ingredient (polyamic acid and / or polyimide) in the liquid crystal aligning agent of the present invention is selected in consideration of viscosity, volatility and the like, but is preferably in the range of 1 to 10% by weight. The liquid crystal alignment agent composed of a solution is applied to the substrate surface by a printing method, a spin coating method or the like, and then dried to form a coating film as an alignment film material. If the concentration of the active ingredient is less than 1% by weight, the thickness of this coating film is too small to obtain a good liquid crystal alignment film. On the other hand, when the concentration of the active ingredient exceeds 10% by weight, the film thickness of the coating film becomes too large to obtain a good liquid crystal aligning film, and the viscosity of the liquid crystal aligning agent increases so that The properties are inferior.
The liquid crystal aligning agent of the present invention can also contain a functional silane-containing compound or an epoxy-based crosslinking agent for the purpose of further improving the adhesiveness between the specific polymer I and / or the specific polymer II and the substrate.
[0061]
Examples of such a functional silane-containing compound include 3-aminopropyltrimethoxysilane, 3-aminopropyltriethoxysilane, 2-aminopropyltrimethoxysilane, 2-aminopropyltriethoxysilane, and N- (2-amino Ethyl) -3-aminopropyltrimethoxysilane, N- (2-aminoethyl) -3-aminopropylmethyldimethoxysilane, 3-ureidopropyltrimethoxysilane, 3-ureidopropyltriethoxysilane, N-ethoxycarbonyl- 3-aminopropyltrimethoxysilane, N-ethoxycarbonyl-3-aminopropyltriethoxysilane, N-triethoxysilylpropyltriethylenetriamine, N-trimethoxysilylpropyltriethylenetriamine, 10-trimethoxysilyl -1,4,7-triazadecane, 10-triethoxysilyl-1,4,7-triazadecane, 9-trimethoxysilyl-3,6-diazanonyl acetate, 9-triethoxysilyl-3,6-diaza Nonyl acetate, N-benzyl-3-aminopropyltrimethoxysilane, N-benzyl-3-aminopropyltriethoxysilane, N-phenyl-3-aminopropyltrimethoxysilane, N-phenyl-3-aminopropyltriethoxysilane , N-bis (oxyethylene) -3-aminopropyltrimethoxysilane, N-bis (oxyethylene) -3-aminopropyltriethoxysilane, and the like. Preferred examples of the epoxy crosslinking agent include polyethylene glycol diglycidyl ether, diglycidyl orthotoluidine, tetraglycidylaminodiphenylmethane, 1,3-bis (N, N'-diglycidylaminomethyl) cyclohexane, and the like. Can be.
[0062]
Liquid crystal display device
The liquid crystal display device obtained by using the liquid crystal alignment agent of the present invention can be manufactured by, for example, the following method.
[0063]
(1) The liquid crystal alignment agent of the present invention is applied to the transparent conductive film side of the substrate provided with the transparent conductive film by a roll coater method, a spinner method, a printing method, or the like, and then the coated surface is heated to form a coating film. Form.
Here, as the substrate, for example, a transparent substrate made of glass such as float glass or soda glass, or a plastic film such as polyethylene terephthalate, polybutylene terephthalate, polyether sulfone, or polycarbonate can be used. As the transparent conductive film provided on one surface of the substrate,
[0064]
In applying the liquid crystal alignment agent, a functional silane-containing compound, titanate, or the like may be applied on the substrate and the transparent conductive film in advance to further improve the adhesiveness between the substrate and the transparent conductive film and the coating film. it can. The heating temperature is preferably from 80 to 250 ° C, more preferably from 120 to 250 ° C. The film thickness of the formed coating film is usually 0.001 to 1 μm, preferably 0.005 to 0.5 μm.
[0065]
(2) The formed coating film is subjected to a rubbing treatment with a roll around which a cloth made of a synthetic fiber such as nylon is wound, thereby forming a liquid crystal alignment film.
[0066]
(3) The two substrates having the liquid crystal alignment films formed thereon are opposed to each other so that the rubbing directions are orthogonal or antiparallel, and the peripheral portion between the substrates is sealed with a sealant. Then, the liquid crystal is filled, the filling hole is sealed to form a liquid crystal cell, and the liquid crystal display element is formed by laminating both surfaces of the liquid crystal cell so that the polarization directions thereof coincide with or perpendicular to the rubbing direction of the liquid crystal alignment film of the substrate. .
As the sealing agent, for example, an epoxy resin containing a hardening agent and aluminum oxide spheres as a spacer can be used.
[0067]
As the liquid crystal, a nematic liquid crystal, a smectic liquid crystal, and among them, a liquid crystal that forms a nematic liquid crystal are preferable. Liquid crystals, biphenylcyclohexane-based liquid crystals, pyrimidine-based liquid crystals, dioxane-based liquid crystals, bicyclooctane-based liquid crystals, cubane-based liquid crystals, and the like are used. Further, to these liquid crystals, for example, cholesteric liquid crystals such as cholesteryl chloride, cholesteryl nonaate, and cholesteryl carbonate, and a chiral agent sold under the trade name C-15, CB-15 (Merck Ltd.) are added. Can also be used. Furthermore, ferroelectric liquid crystals such as p-decyloxybenzylidene-p-amino-2-methylbutylcinnamate can be used.
[0068]
As a polarizing plate used outside the liquid crystal cell, a polarizing plate in which a polarizing film called an H film in which polyvinyl alcohol is stretched and oriented and iodine is absorbed by a cellulose acetate protective film, or a polarizing film composed of the H film itself is used. A plate can be used.
[0069]
The pretilt angle is changed by irradiating the liquid crystal alignment film formed by the liquid crystal alignment agent of the present invention with ultraviolet rays as disclosed in, for example, JP-A-6-222366 and JP-A-6-281937. Or a resist is partially formed on a rubbed liquid crystal alignment film as described in JP-A-5-107544, and a rubbing process is performed in a direction different from the previous rubbing process. The visibility characteristics of the liquid crystal display element can be improved by removing the resist film after the process and performing a process for changing the alignment ability of the liquid crystal alignment film.
[0070]
【Example】
Hereinafter, the present invention will be described more specifically with reference to Examples, but the present invention is not limited to these Examples.
Note that the measurement of the pretilt angle in the examples is described in [T. J. Schffer, et al. , J. et al. Appl. Phys. , 19, 2013 (1980)] according to a crystal rotation method using a He-Ne laser beam.
In the evaluation of the orientation of the liquid crystal cell, the presence or absence of abnormal domains in the liquid crystal cell when the voltage was turned on / off was observed with a polarizing microscope.
[0071]
Synthesis example 1 of specific diamine compound
Step (i)
23.06 g (100 mmol) of 3,5-dinitrobenzoic acid chloride and 34.85 g (300 mmol) of 1,4-dihydroxycyclohexane were dissolved in 400 g of tetrahydrofuran. Then, while stirring and cooling with ice, 7.91 g (100 mmol) of pyridine was gradually added dropwise. After performing the reaction for 6 hours, the reaction solution was poured into a large excess of water, the precipitate was separated by filtration, and recrystallized from methyl ethyl ketone to obtain a white dinitro compound.
[0072]
Step (ii)
24.82 g (80 mmol) of the dinitro compound obtained in the step (i) and 18.32 g (80 mmol) of p-trifluoromethylbenzoic acid chloride were dissolved in 400 g of tetrahydrofuran. Then, while stirring and cooling with ice, 6.33 g (80 mmol) of pyridine was gradually added dropwise. After performing the reaction for 6 hours, the reaction solution was poured into a large excess of water, the precipitate was separated by filtration, and recrystallized from methyl ethyl ketone to obtain a white dinitro compound.
[0073]
Step (iii)
36.20 g (72 mmol) of the dinitro compound obtained in the step (ii) is dissolved in 400 g of ethanol, and 0.30 g of a 5% by weight palladium-carbon catalyst and 5.6 g (100 mmol) of hydrazine monohydrate are added. The mixture was refluxed for 6 hours. The reaction solution was poured into a large excess of water, and the precipitate was separated by filtration and recrystallized from ethanol to obtain 24.33 g of a white specific diamine compound (i) represented by the above formula (10).
FIG. 1 shows an infrared absorption spectrum and FIG. 2 shows an NMR spectrum of the obtained specific diamine compound (i).
[0074]
Synthesis example 2 of specific diamine compound
Step (i)
23.06 g (100 mmol) of 3,5-dinitrobenzoic acid chloride and 34.85 g (300 mmol) of 1,4-dihydroxycyclohexane were dissolved in 400 g of tetrahydrofuran. Then, while stirring and cooling with ice, 7.91 g (100 mmol) of pyridine was gradually added dropwise. After performing the reaction for 6 hours, the reaction solution was poured into a large excess of water, the precipitate was separated by filtration, and recrystallized from methyl ethyl ketone to obtain a white dinitro compound.
[0075]
Step (ii)
24.82 g (80 mmol) of the dinitro compound obtained in the step (i) and 12.68 g (80 mmol) of p-fluorobenzoic acid chloride were dissolved in 400 g of tetrahydrofuran. Then, while stirring and cooling with ice, 6.33 g (80 mmol) of pyridine was gradually added dropwise. After performing the reaction for 6 hours, the reaction solution was poured into a large excess of water, the precipitate was separated by filtration, and recrystallized from methyl ethyl ketone to obtain a white dinitro compound.
[0076]
Step (iii)
31.12 g (72 mmol) of the dinitro compound obtained in the step (ii) is dissolved in 400 g of ethanol, and 0.30 g of a 5% by weight palladium-carbon catalyst and 5.6 g (100 mmol) of hydrazine monohydrate are added. The mixture was refluxed for 6 hours. The reaction solution was poured into a large excess of water, and the precipitate was separated by filtration and recrystallized from ethanol to obtain 24.33 g of a white specific diamine compound (ii) having a melting point of 196 ° C. represented by the above formula (10). Was.
FIG. 3 shows an infrared absorption spectrum and FIG. 4 shows an NMR spectrum of the obtained specific diamine compound (ii).
[0077]
Example 1 (Synthesis of specific polymer Ia)
22.42 g (100 mmol) of 2,3,5-tricarboxycyclopentylacetic dianhydride as a tetracarboxylic dianhydride, 4.22 g (10 mmol) of a specific diamine compound represented by the above formula (10) as a diamine compound and 9.73 g (90 mmol) of p-phenylenediamine was dissolved in 327.33 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Ia having an intrinsic viscosity of 1.20 dl / g. FIG. 5 shows an infrared absorption spectrum and FIG. 6 shows an NMR spectrum of the obtained specific polymer Ia.
[0078]
Example 2 (Synthesis of specific polymer IIa)
36.37 g of the specific polymer Ia obtained in Synthesis Example 1 was dissolved in 631.03 g of N-methyl-2-pyrrolidone, 15.82 g of pyridine and 20.42 g of acetic anhydride were added, and imidization was performed at 115 ° C. for 4 hours. The reaction was allowed.
Next, the reaction product solution was precipitated in the same manner as in Synthesis Example 1 to obtain a specific polymer IIa having an intrinsic viscosity of 1.20 dl / g. FIG. 7 shows an infrared absorption spectrum and FIG. 8 shows an NMR spectrum of the obtained specific polymer IIa.
[0079]
Example 3 (Synthesis of specific polymer Ib)
22.42 g (100 mmol) of 2,3,5-tricarboxycyclopentylacetic acid dianhydride as a tetracarboxylic dianhydride, 21.12 g (50 mmol) of a specific diamine compound represented by the above formula (10) as a diamine compound and 5.41 g (50 mmol) of p-phenylenediamine was dissolved in 440.55 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Ib having a logarithmic viscosity of 1.00 dl / g.
[0080]
Example 4 (Synthesis of specific polymer IIb)
48.95 g of the specific polymer Ib obtained in Synthesis Example 3 was dissolved in 930.05 g of N-methyl-2-pyrrolidone, 15.82 g of pyridine and 20.42 g of acetic anhydride were added, and imidization was performed at 115 ° C. for 4 hours. The reaction was allowed.
Next, the reaction product liquid was precipitated in the same manner as in Synthesis Example 1 to obtain a specific polymer IIb having an logarithmic viscosity of 1.00 dl / g.
[0081]
Example 5 (Synthesis of specific polymer Ic)
22.42 g (100 mmol) of 2,3,5-tricarboxycyclopentylacetic dianhydride as tetracarboxylic dianhydride and 44.24 g (100 mmol) of specific diamine compound represented by the above formula (10) as diamine compound It was dissolved in 581.94 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours. The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Ic having an logarithmic viscosity of 0.80 dl / g. FIG. 9 shows an infrared absorption spectrum and FIG. 10 shows an NMR spectrum of the obtained specific polymer Ic.
[0082]
Example 6 (Synthesis of specific polymer IIc)
64.66 g of the specific polymer Ic obtained in Synthesis Example 5 was dissolved in 1228.54 g of N-methyl-2-pyrrolidone, 15.82 g of pyridine and 20.42 g of acetic anhydride were added, and the mixture was heated at 115 ° C. for 4 hours. An imidation reaction was performed.
Next, the reaction product solution was precipitated in the same manner as in Synthesis Example 1 to obtain a specific polymer IIc having a logarithmic viscosity of 0.80 dl / g. FIG. 11 shows an infrared absorption spectrum and FIG. 12 shows an NMR spectrum of the obtained specific polymer IIc.
[0083]
Example 7 (Synthesis of specific polymer Id)
21.81 g (100 mmol) of pyromellitic dianhydride as a tetracarboxylic dianhydride, 21.11 g (50 mmol) of a specific diamine compound represented by the above formula (10) as a diamine compound and 5.41 g of p-phenylenediamine (50 mmol) was dissolved in 434.97 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Id having an intrinsic viscosity of 1.35 dl / g.
[0084]
Example 8 (Synthesis of specific polymer Ie)
As a tetracarboxylic dianhydride, 21.81 g (100 mmol) of pyromellitic dianhydride, as a diamine compound, 21.01 g (50 mmol) of the specific diamine compound represented by the above formula (11) and 5.41 g of p-phenylenediamine (50 mmol) was dissolved in 434.07 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Ie having an intrinsic viscosity of 1.40 dl / g.
[0085]
Example 9 (Synthesis of specific polymer If)
21.81 g (100 mmol) of pyromellitic dianhydride as a tetracarboxylic dianhydride, 18.41 g (50 mmol) of a specific diamine compound represented by the above formula (12) as a diamine compound and 5.41 g of p-phenylenediamine (50 mmol) was dissolved in 410.67 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer If having an intrinsic viscosity of 1.30 dl / g.
[0086]
Example 10 (Synthesis of specific polymer Ig)
1,3,3a, 4,5,9b-Hexahydro-8-methyl-5 (tetrahydro-2,5-dioxo-3-furanyl) naphtho [1,2-c] furan-1 as tetracarboxylic dianhydride 26.72 g (85 mmol) of 1,3-dione, 3.36 g (15 mmol) of 1,3-dimethylcyclobutanetetracarboxylic dianhydride, 8.11 g (75 mmol) of 1,4-phenylenediamine as a diamine compound, 4 2.97 g (15 mmol) of 4,4'-diaminodiphenylmethane and 4.22 g (10 mmol) of the specific compound represented by the above formula (10) were dissolved in 408.42 g of N-methyl-2-pyrrolidone. Allowed to react for hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Ig having an logarithmic viscosity of 1.10 dl / g.
[0087]
Example 11 (Synthesis of specific polymer IIg)
The specific polymer Ig45.38 obtained in Synthesis Example 10 was dissolved in 862.22 g of N-methyl-2-pyrrolidone, 15.82 g of pyridine and 20.42 g of acetic anhydride were added, and imidization was performed at 115 ° C. for 4 hours. Reacted.
Next, the reaction product was precipitated in the same manner as in Synthesis Example 1 to obtain a specific polymer IIg having an intrinsic viscosity of 1.10 dl / g.
[0088]
Example 12 (Synthesis of specific polymer IIh)
22.42 g (100 mmol) of 2,3,5-tricarboxycyclopentylacetic dianhydride as a tetracarboxylic dianhydride, 3.72 g (10 mmol) of a specific diamine compound (ii) and p-phenylenediamine 9 as a diamine compound 0.73 g (90 mmol) was dissolved in 327.33 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours. The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a specific polymer Ih having an intrinsic viscosity of 1.10 dl / g. 36.37 g of the obtained specific polymer Ih was dissolved in 631.03 g of N-methyl-2-pyrrolidone, 15.82 g of pyridine and 20.42 g of acetic anhydride were added, and an imidization reaction was performed at 115 ° C. for 4 hours. .
Next, the reaction product solution was precipitated in the same manner as in Synthesis Example 1 to obtain a specific polymerization couple IIh having an logarithmic viscosity of 1.0 dl / g. FIG. 13 shows the infrared absorption spectrum and FIG. 14 shows the NMR spectrum of the obtained specific polymer IIh.
[0089]
Comparative Example 1 (Synthesis of Polymer Iα)
22.42 g (100 mmol) of 2,3,5-tricarboxycyclopentylacetic dianhydride as tetracarboxylic dianhydride and 10.81 g (100.00 mmol) of p-phenylenediamine as N-methyl-diamine compound It was dissolved in 299.07 g of 2-pyrrolidone and reacted at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a polymer Iα having a logarithmic viscosity of 1.25 dl / g.
[0090]
Comparative Example 2 (Synthesis of Polymer IIα)
33.23 g of the polymer Ia obtained in Comparative Example 1 was dissolved in 631.37 g of N-methyl-2-pyrrolidone, 15.82 g of pyridine and 20.42 g of acetic anhydride were added, and an imidization reaction was performed at 115 ° C. for 4 hours. Was made.
Next, the reaction product solution was precipitated in the same manner as in Synthesis Example 1 to obtain a polymer IIα having an logarithmic viscosity of 1.25 dl / g.
[0091]
Comparative Example 3 (Synthesis of Polymer Iβ)
21.81 g (100 mmol) of pyromellitic dianhydride as a tetracarboxylic dianhydride and 10.81 g (100 mmol) of p-phenylenediamine as a diamine compound were dissolved in 293.58 g of N-methyl-2-pyrrolidone, The reaction was performed at 60 ° C. for 6 hours.
The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a polymer Iβ having an intrinsic viscosity of 1.40 dl / g.
[0092]
Comparative Example 4 (Synthesis of Polymer Iγ)
Pyromellitic dianhydride 21.81 g (100 mmol) as tetracarboxylic dianhydride and p-phenylenediamine 5.4 g (50 mmol) as diamine compound, the following formula (24)
[0093]
Embedded image
Was dissolved in 293.58 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours. The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a polymer Iγ having an logarithmic viscosity of 1.20 dl / g.
[0095]
Comparative Example 5 (Synthesis of Polymer Iδ)
21.81 g (100 mmol) of pyromellitic dianhydride as a tetracarboxylic dianhydride, 5.4 g (50 mmol) of p-phenylenediamine as a diamine compound, and 14.76 g of a diamine compound represented by the above formula (21) (50 mmol) was dissolved in 293.58 g of N-methyl-2-pyrrolidone and reacted at 60 ° C. for 6 hours. The reaction mixture was then poured into a large excess of methanol, causing the reaction product to precipitate. Thereafter, the polymer was washed with methanol and dried under reduced pressure at 40 ° C. for 15 hours to obtain a polymer Iδ having a logarithmic viscosity of 1.60 dl / g.
[0096]
Example 13 (Preparation and evaluation of liquid crystal aligning agent)
The specific polymer IIa obtained in Example 2 was dissolved in γ-butyrolactone to obtain a solution having a solid content of 4% by weight, and this solution was filtered through a filter having a pore size of 1 μm to prepare a liquid crystal aligning agent.
This liquid crystal aligning agent is applied to the transparent electrode surface using a spinner on a glass substrate with a transparent electrode made of an ITO film so that the film thickness becomes 800 Å, and baked at 180 ° C. for 1 hour to form a coating film. did.
A rubbing machine having a roll in which a nylon cloth was wound around the coating film was subjected to two rubbing treatments at a roll hair indentation length of 0.6 mm, a roll rotation speed of 500 rpm, and a stage moving speed of 1 cm / sec. At this time, the adhesion between the alignment film and the substrate was good, and no peeling due to rubbing was observed.
[0097]
Next, an epoxy resin adhesive containing aluminum oxide spheres having a diameter of 17 μm is screen-printed and applied to the outer edge of each of the pair of rubbed substrates having the liquid crystal alignment film, and then the liquid crystal alignment film faces the pair of substrates. As described above, the rubbing directions were superposed so that the rubbing directions were antiparallel, and the pressure was applied to cure the adhesive.
Next, after filling a nematic liquid crystal (MLC-2001, manufactured by Merck) between the pair of substrates from the liquid crystal injection port, the liquid crystal injection port is sealed with an epoxy-based adhesive, and polarizing plates are provided on both outer surfaces of the substrate. Were bonded so that the polarization direction of the polarizing plate coincided with the rubbing direction of the liquid crystal alignment film of each substrate, to produce a liquid crystal display device.
The orientation of the obtained liquid crystal display device was good, and the pretilt angle was measured and found to be 3.5 °. Table 1 shows the results.
[0098]
Examples 14 to 45 (Preparation and evaluation of liquid crystal aligning agent)
In Example 13, a liquid crystal alignment agent was prepared using the specific polymers IIb, IIc, Id, Ie, If, IIg, and IIh obtained in Examples 2, 4, 6, 7, 8, 9, 11, and 12. A liquid crystal display element was manufactured in the same manner as in Example 13 except that the heating temperature of the alignment film was 180 to 250 ° C., the film thickness was 200 to 1500 Å, and the number of rubbing was 1 to 5 times. The orientation and pretilt angle of the device were measured, and the results are shown in Tables 1 and 2.
[0099]
[Table 1]
[Table 2]
Comparative Examples 6 to 25 (Preparation and Evaluation of Liquid Crystal Alignment Agent)
In Example 13, a liquid crystal aligning agent was prepared using the polymers obtained in Comparative Examples 1 to 5, the heating temperature of the alignment film was 180 to 250 ° C., the film thickness was 200 to 1500 Å, and the number of rubbing was 1 to 1. A liquid crystal display element was manufactured in the same manner as in Example 13 except that the measurement was performed five times, and the orientation and pretilt angle of the liquid crystal display element were measured. The results are shown in Table 3.
[0102]
[Table 3]
【The invention's effect】
According to the liquid crystal aligning agent of the present invention, when a liquid crystal alignment film is formed, the alignment of the liquid crystal is good, and a high pretilt angle that can exhibit a high pretilt angle independent of process conditions such as film thickness and rubbing conditions can be obtained. A liquid crystal alignment film suitable for use in an SH type display element is obtained.
Further, a liquid crystal display device having an alignment film formed using the liquid crystal alignment agent of the present invention has excellent liquid crystal alignment and reliability, and can be effectively used for various devices, such as a desk calculator, a wrist watch, a table clock, and a coefficient. It is used for display devices such as display boards, word processors, personal computers, and liquid crystal televisions.
[Brief description of the drawings]
FIG. 1 is an infrared absorption spectrum diagram of a specific example of a diamine compound used in the present invention.
FIG. 2 is an NMR spectrum diagram of the same diamine compound as the specific example of FIG.
FIG. 3 is an infrared absorption spectrum diagram of another specific example of the diamine compound used in the present invention.
4 is an NMR spectrum diagram of the same diamine compound as the specific example of FIG.
FIG. 5 is an infrared absorption spectrum diagram of a specific example of the polyamic acid of the present invention.
6 is an NMR spectrum diagram of the same polyamic acid as the specific example of FIG.
FIG. 7 is an infrared absorption spectrum diagram of a specific example of the polyimide of the present invention.
FIG. 8 is an NMR spectrum diagram of the same polyimide as the specific example of FIG.
FIG. 9 is an infrared absorption spectrum of another specific example of the polyamic acid of the present invention.
10 is an NMR spectrum diagram of the same polyamic acid as the specific example of FIG.
FIG. 11 is an infrared absorption spectrum diagram of another specific example of the polyimide of the present invention.
12 is an NMR spectrum diagram of the same polyimide as the specific example of FIG.
FIG. 13 is an infrared absorption spectrum of another specific example of the polyimide of the present invention.
14 is an NMR spectrum diagram of the same polyimide as the specific example of FIG.
Claims (5)
で表わされるジアミン化合物。A diamine compound represented by the formula:
テトラカルボン酸二無水物とを反応させて得られるポリアミック酸。 A polyamic acid obtained by reacting the diamine compound according to claim 1 with a tetracarboxylic dianhydride.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP1880597A JP3584457B2 (en) | 1996-02-06 | 1997-01-31 | Diamine compound, polyamic acid, polyimide, liquid crystal aligning agent and liquid crystal display device |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP1956296 | 1996-02-06 | ||
| JP8-19562 | 1996-02-06 | ||
| JP1880597A JP3584457B2 (en) | 1996-02-06 | 1997-01-31 | Diamine compound, polyamic acid, polyimide, liquid crystal aligning agent and liquid crystal display device |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH09272740A JPH09272740A (en) | 1997-10-21 |
| JP3584457B2 true JP3584457B2 (en) | 2004-11-04 |
Family
ID=26355541
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP1880597A Expired - Lifetime JP3584457B2 (en) | 1996-02-06 | 1997-01-31 | Diamine compound, polyamic acid, polyimide, liquid crystal aligning agent and liquid crystal display device |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP3584457B2 (en) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4514247B2 (en) * | 1998-11-12 | 2010-07-28 | Jsr株式会社 | Liquid crystal aligning agent and liquid crystal display element |
| KR100565739B1 (en) * | 2000-10-28 | 2006-03-29 | 엘지.필립스 엘시디 주식회사 | Photo-alignment Characteristic Material and Liquid Crystal Display Device fabricated with it |
| KR100599859B1 (en) * | 2003-10-09 | 2006-07-12 | 제일모직주식회사 | Diamine Compound having Side Chain in Dendron Structure and LC Alignment Material Prepared by the Same |
| JP2005189270A (en) * | 2003-12-24 | 2005-07-14 | Chi Mei Electronics Corp | Alignment layer and liquid crystal display using same |
| JP4569145B2 (en) * | 2004-03-25 | 2010-10-27 | チッソ株式会社 | Polyimide copolymer, liquid crystal aligning agent and liquid crystal display element using the same |
| JP4595417B2 (en) * | 2004-07-16 | 2010-12-08 | チッソ株式会社 | Phenylenediamine, alignment film formed using the same, and liquid crystal display device including the alignment film |
| CN1762978B (en) * | 2004-10-05 | 2010-12-15 | Jsr株式会社 | Novel diamine compound,polymer and liquid crystal tropism agent |
| JP5048742B2 (en) * | 2009-11-06 | 2012-10-17 | 株式会社ジャパンディスプレイイースト | Liquid crystal display |
| CN102102019A (en) | 2009-12-16 | 2011-06-22 | 第一毛织株式会社 | Liquid crystal photo-alignment agent, liquid crystal photo-alignment layer, and liquid crystal display device |
| CN102559205B (en) | 2010-12-29 | 2014-07-30 | 第一毛织株式会社 | Liquid crystal alignment agent, liquid crystal alignment film manufactured using the same, and liquid crystal display device |
| JP5292438B2 (en) * | 2011-05-23 | 2013-09-18 | 株式会社ジャパンディスプレイ | Liquid crystal display |
| KR101444190B1 (en) | 2011-12-19 | 2014-09-26 | 제일모직 주식회사 | Liquid crystal alignment agent, liquid crystal alignment film using the same, and liquid crystal display device including the liquid crystal alignment film |
| JP5537698B2 (en) * | 2013-04-25 | 2014-07-02 | 株式会社ジャパンディスプレイ | Liquid crystal alignment film material |
| CN112375010B (en) * | 2020-09-27 | 2023-03-10 | 浙江中科玖源新材料有限公司 | Novel diamine, polyimide and polyimide film |
| CN115745831B (en) * | 2022-11-21 | 2024-07-05 | 东华大学 | Indenyl-containing diamine, preparation method and application thereof, polyimide film and application thereof |
-
1997
- 1997-01-31 JP JP1880597A patent/JP3584457B2/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JPH09272740A (en) | 1997-10-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3206401B2 (en) | Liquid crystal alignment agent and liquid crystal display device | |
| JP5120047B2 (en) | Vertical alignment type liquid crystal aligning agent and liquid crystal display element | |
| JPH10104633A (en) | Liquid crystal alignment agent | |
| EP0789046B1 (en) | Polyamic acid, polyimide and liquid crystal aligning agent | |
| KR101161447B1 (en) | Liquid Crystal Aligning Agent and Liquid Crystal Display Device | |
| JP3584457B2 (en) | Diamine compound, polyamic acid, polyimide, liquid crystal aligning agent and liquid crystal display device | |
| JP5041169B2 (en) | Liquid crystal aligning agent and liquid crystal display element | |
| JP2001228481A (en) | Liquid crystal alignment film and liquid crystal display device | |
| KR101502628B1 (en) | Liquid crystal aligning agent, liquid crystal alignment film and liquid crystal display device | |
| JP4716061B2 (en) | Liquid crystal aligning agent and liquid crystal display element | |
| JP4788898B2 (en) | Liquid crystal aligning agent and liquid crystal display element | |
| JPH09176315A (en) | Polyimide-based block copolymer | |
| JP2006199751A (en) | Liquid crystal aligning agent | |
| JPH08262450A (en) | Liquid crystal orienting agent | |
| JP3593684B2 (en) | Liquid crystal alignment agent and liquid crystal display device | |
| JP3267347B2 (en) | Liquid crystal alignment agent | |
| JP5041124B2 (en) | Liquid crystal aligning agent and liquid crystal display element | |
| JP4003592B2 (en) | Liquid crystal aligning agent and liquid crystal display element | |
| JP2002338686A (en) | Polyamic acid and polyimide | |
| JP4858686B2 (en) | Liquid crystal alignment agent, liquid crystal alignment film, and liquid crystal display element | |
| JP3279757B2 (en) | Liquid crystal alignment agent and liquid crystal display device | |
| JPH07305065A (en) | Orientation agent for liquid crystal | |
| JP3603491B2 (en) | Liquid crystal sandwich substrate and liquid crystal display element | |
| JP3550672B2 (en) | Liquid crystal alignment agent and liquid crystal display device | |
| JP2005179429A (en) | Liquid crystal-aligning agent and liquid crystal display element |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20040423 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20040712 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20040725 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20070813 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20080813 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090813 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090813 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090813 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100813 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100813 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110813 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110813 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120813 Year of fee payment: 8 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120813 Year of fee payment: 8 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130813 Year of fee payment: 9 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |