(V.詳細な説明)
別段の定義がない限り、本明細書で使用する技術用語及び科学用語はすべて、当業者によって通常理解されるのと同じ意味を有する。本明細書において参照される公開物及び特許はすべて、それらの全体が参照により本明細書に組み込まれる。
(A.定義)
本明細書及び添付の特許請求の範囲において使用する場合、不定冠詞「a」及び「an」ならびに定冠詞「the」には、本文脈が別段に明確に記載しない限り、複数及び単数の指示対象を含む。
「約」または「およそ」という用語は、当業者によって決定される特定の値についての許容され得る誤差を意味し、当該値がどのように測定または決定されるのかに一部依存する。ある実施形態において、「約」または「およそ」という用語は、1標準偏差、2標準偏差、3標準偏差、または4標準偏差内を意味する。ある実施形態において、「約」または「およそ」という用語は、所与の値または範囲の30%以内、25%以内、20%以内、15%以内、10%以内、9%以内、8%以内、7%以内、6%以内、5%以内、4%以内、3%以内、2%以内、1%以内、0.5%以内、0.1%以内、または0.05%以内を意味する。
本明細書で使用する場合、別段の指定がない限り、「治療する」、「治療すること」及び「治療」という用語は、疾患若しくは障害の、または当該疾患若しくは障害と関係する1つ以上の症状の根絶若しくは緩解を指す。ある実施形態において、当該用語は、このような疾患または障害に罹患している対象への1つ以上の予防薬または治療薬の投与から結果的に生じる当該疾患または障害の伝播若しくは悪化を最小限にすることを指す。いくつかの実施形態において、当該用語は、当該特定の疾患の症状の発症後の、本明細書に提供する化合物または剤形の、1つ以上の追加の作用薬(複数可)を伴うまたは伴わない投与を指す。
本明細書で使用する場合、別段の指定がない限り、「予防する」、「予防すること」及び「予防」という用語は、疾患若しくは障害の、またはそれらの1つ以上の症状の発症、再発または伝播の予防を指す。ある実施形態において、当該用語は、症状の発症前に、1つ以上の他の追加の作用薬(複数可)を伴うまたは伴わない、本明細書に提供する化合物若しくは剤形を用いた処置または当該化合物若しくは剤形の投与を指す。当該用語は、当該特定の疾患の症状の阻害または低下を包含する。特に疾患の家族歴のある対象は、ある実施形態における予防的治療計画についての候補である。加えて、再発性症状歴を有する対象も予防のための潜在的な候補である。この点において、「予防」という用語は、「予防的処置」という用語と相互交換可能に使用することがある。
本明細書で使用する場合、別段の指定がない限り、「管理する」、「管理すること」及び「管理」という用語は、疾患若しくは障害の、またはそれらの1つ以上の進行、伝播または悪化を予防または遅延させることを指す。しばしば、対象が予防薬及び/または治療薬から得る有益な効果は、当該疾患または障害の治癒を結果的に生じない。この点において、「管理すること」という用語は、当該特定の疾患の再発を予防または最小限にする試行において当該疾患に罹患した対象を治療することを包含する。
本明細書で使用する場合、特定の医薬組成物の投与による特定の障害の症状の緩解は、永久的であろうと一時的であろうと、持続性であろうと一過性であろうと、当該組成物の投与に属することのできるまたは当該投与と関係することのできる任意の弱化を指す。
本明細書で使用する場合、別段の指定がない限り、化合物の「治療有効量」及び「有効量」という用語は、疾患若しくは障害の治療若しくは管理において治療上の有益性を提供するのに、または当該疾患若しくは障害と関係する1つ以上の症状を遅延若しくは最小限にするのに十分な量を意味する。化合物の「治療有効量」及び「有効量」は、単独での、または当該疾患若しくは障害の治療若しくは管理において治療上の有益性を提供する1つ以上の他の薬剤(複数可)との組み合わせにおける、治療薬の量を意味する。「治療有効量」及び「有効量」という用語は、全体的な療法を改善する、当該疾患若しくは障害の症状若しくは原因を低下若しくは回避する、または別の治療薬の治療有効性を亢進する量を包含することができる。
本明細書で使用する場合、別段の指定がない限り、化合物の「予防有効量」は、疾患若しくは障害を予防するのに、またはその再発を予防するのに十分な量である。化合物の予防有効量は、単独での、または当該疾患の予防において予防上の有益性を提供する1つ以上の他の薬剤(複数可)との組み合わせにおける、治療薬の量を意味する。「予防有効量」という用語は、全体的な予防を改善する量、または別の予防薬の予防有効性を亢進する量を包含することができる。
「腫瘍」は、本明細書で使用する場合、悪性であろうと良性であろうと、新生物の細胞の成長及び増殖を全部、ならびに前癌状態の及び癌性の細胞及び組織を全部指す。「新生物の」は、本明細書で使用する場合、悪性であろうと良性であろうと、異常な組織の成長を結果的に生じる、調節不全のまたは調節されていない細胞の成長の任意の形態を指す。したがって、「新生物細胞」には、調節不全のまたは調節されていない細胞の成長を有する悪性細胞及び良性細胞を含む。
「癌」及び「癌性の」という用語は、調節されていない細胞の成長を典型的に特徴とする哺乳類動物における生理学的容態を指すまたは説明する。癌の例としては、血液由来の腫瘍(例えば、リンパ腫、白血病)及び固形腫瘍が挙げられるが、これらに限定しない。
本明細書で使用する場合、別段の指定がない限り、「増殖性の」障害または疾患という用語は、多細胞生物に対して結果的に有害である、当該多細胞生物における細胞の1つ以上のサブセットの望ましくない細胞増殖(すなわち、不安なまたは短縮した余命)を指す。例えば、本明細書で使用する場合、増殖性の障害または疾患には、新生物性障害または他の増殖性障害を含む。
本明細書で使用する場合、別段の指定がない限り、「再発性の」という用語は、療法後に癌の緩解を有していた対象が、癌細胞の回復を有する状態を指す。
本明細書で使用する場合、別段の指定がない限り、「難治性の」または「耐性の」という用語は、集中治療後でさえ、対象が体内に残余の癌細胞を有している状況を指す。
「組成物」、「製剤」、及び「剤形」という用語は、本明細書で使用する場合、指定された成分(複数可)を(示す場合、指定された量で)含む組成物、及び当該指定された量(複数可)における指定された成分(複数可)の組み合わせから、直接的にまたは間接的に結果として生じる任意の製品(複数可)を包含するよう企図される。「医薬の」または「医薬として許容され得る」は、当該組成物、製剤、または剤形中の任意の希釈剤(複数可)、賦形剤(複数可)または担体(複数可)は、当該他の成分(複数可)と適合性があり、その受け手に対して有害ではないことを意味する。別段の記載がない限り、「組成物」、「製剤」、及び「剤形」という用語は本明細書において、相互交換可能に使用する。
「即時放出」という用語は、本明細書に提供する組成物、製剤または剤形に対する参照において本明細書で使用する場合、当該組成物、製剤、または剤形が、経口投与後に当該組成物、製剤、または剤形から胃の向こうにAPIの一部または全部の空間的及び/または時間的放出を遅延させるよう機能する構成要素(例えば、皮膜)を含んでいないことを意味する。ある実施形態において、即時放出の組成物、製剤、または剤形は、経口投与後に実質的に胃の中でAPIを放出するものである。具体的な実施形態において、即時放出の組成物、製剤、または剤形は、遅延放出ではないものである。具体的な実施形態において、即時放出の組成物、製剤、または剤形は、腸溶コーティングを含んでいないものである。
「腸溶コーティングされていない」及び「非腸溶コーティング」という用語は、本明細書で使用する場合、胃の向こうで(すなわち、腸において)有効成分(複数可)を放出するよう企図された皮膜を含んでいない医薬組成物、製剤、または剤形を指す。ある実施形態において、腸溶コーティングされていない組成物、製剤、または剤形は、実質的に胃の中で当該有効成分(複数可)を放出するよう設計されている。
「実質的に胃の中で」という用語は、本明細書に提供する組成物、製剤、または剤形に対する参照において本明細書で使用する場合、当該シチジン類似体の少なくとも約99%、少なくとも約95%、少なくとも約90%、少なくとも約85%、少なくとも約80%、少なくとも約75%、少なくとも約70%、少なくとも約65%、少なくとも約60%、少なくとも約55%、少なくとも約50%、少なくとも約45%、少なくとも約40%、少なくとも約35%、少なくとも約30%、少なくとも約25%、少なくとも約20%、少なくとも約15%、または少なくとも約10%が胃の中で放出されることを意味する。本明細書で使用する「胃の中で放出される」という用語及び関連用語は、当該シチジン類似体が、胃を裏打ちする細胞による取り込みまたは当該細胞を通過した輸送に利用可能となり、次いで、身体に対して利用可能となる、過程を指す。
「対象」という用語は、霊長類動物(例えば、ヒト)、ウシ、ヒツジ、ヤギ、ウマ、イヌ、ネコ、ウサギ、ラット、マウス及びこれらに類するものを含むがそれらに限定しない哺乳類動物のような動物を含むよう、本明細書において定義される。具体的な実施形態において、当該対象はヒトである。
「併用投与」及び「との組み合わせにおける」という用語には、具体的な時限のない間で同時に、共にまたは連続的に、のいずれかでの2つ以上の治療薬の投与を含む。一実施形態において、当該薬剤は、細胞中に若しくは対象の体内に同時に存在し、または当該薬剤の生物学的効果若しくは治療効果を同時に発揮する。一実施形態において、当該治療薬は、同じ組成物または単位剤形の中にある。他の実施形態において、当該治療薬は、別個の組成物または単位剤形の中にある。ある実施形態において、第一の薬剤は、第二の治療薬の投与の前に(例えば、5分前、15分前、30分前、45分前、1時間前、2時間前、4時間前、6時間前、12時間前、24時間前、48時間前、72時間前、96時間前、1週間前、2週間前、3週間前、4週間前、5週間前、6週間前、8週間前、または12週間前)、当該投与と共に、または当該投与の後に(例えば、5分後、15分後、30分後、45分後、1時間後、2時間後、4時間後、6時間後、12時間後、24時間後、48時間後、72時間後、96時間後、1週間後、2週間後、3週間後、4週間後、5週間後、6週間後、8週間後、または12週間後)に投与することができる。
「溶媒和物」という用語は、化学量論量または非化学量論量で存在する溶質、例えば、本明細書に提供する化合物の1つ以上の分子と、溶媒の1つ以上の分子とによって形成される複合体または凝集体を指す。適切な溶媒には、水、メタノール、エタノール、n−プロパノール、イソプロパノール、及び酢酸を含むが、これらに限定しない。ある実施形態において、当該溶媒は、医薬として許容され得る。一実施形態において、当該複合体または凝集体は、結晶形態にある。別の実施形態において、当該複合体または凝集体は、非晶質形態にある。当該溶媒が水である場合、当該溶媒和物は、水和物である。水和物の例としては、半水和物、一水和物、二水和物、三水和物、四水和物、及び五水和物が挙げられるが、これらに限定しない。
「同位体組成」という用語は、所与の原子配置において存在する各同位体の量を指し、「天然同位体組成」は、所与の原子配置に対する天然の同位体の組成または存在量を指す。これらの天然の同位体の組成を含有する原子配置は、「濃縮されていない」と本明細書で指すこともある。別段の指定がない限り、本明細書に列挙する化合物の原子配置は、当該原子の任意の安定した同位体を表すよう意味する。例えば、別段の記載がない限り、配置が「H」または「水素」と具体的に指定されている場合、当該配置は、その天然同位体組成において水素を有していると理解される。
「同位体濃縮された」という用語は、当該原子の天然同位体組成以外の同位体組成を有する原子配置を指す。「同位体濃縮された」は、当該原子の天然同位体組成以外の同位体組成を有する少なくとも1つの原子配置を含有する化合物を指すこともあり得る。本明細書で使用する場合、「同位体種」とは、同位体濃縮された化合物である。
「同位体濃縮」という用語は、当該原子の天然同位体組成の代わりに、1分子における所与の原子配置における具体的な同位体の量の組み込みの百分率を指す。例えば、所与の配置における1%の重水素濃縮は、所与の試料中の分子の1%が指定配置において重水素を含有することを意味する。重水素の天然の分布は、約0.0156%であるので、濃縮されていない出発材料を用いて合成した化合物における任意の配置における重水素濃縮は、約0.0156%である。
「同位体濃縮因子」という用語は、指定の同位体の同位体組成と天然同位体組成の間の比を指す。
本明細書に提供する化合物に関して、特定の原子配置が重水素または「D」を有するものと指定される場合、当該配置における重水素の存在量は、重水素の天然の存在量よりも実質的に大きく、約0.015%であることは理解される。重水素を有するものと指定される配置は典型的には、特定の実施形態において、各指定の重水素配置における少なくとも1000(15%の重水素組み込み)、少なくとも2000(30%の重水素組み込み)、少なくとも3000(45%の重水素組み込み)、少なくとも3500(52.5%の重水素組み込み)、少なくとも4000(60%の重水素組み込み)、少なくとも4500(67.5%の重水素組み込み)、少なくとも5000(75%の重水素組み込み)、少なくとも5500(82.5%の重水素組み込み)、少なくとも6000(90%の重水素組み込み)、少なくとも6333.3(95%の重水素組み込み)、少なくとも6466.7(97%の重水素組み込み)、少なくとも6600(99%の重水素組み込み)、または少なくとも6633.3(99.5%の重水素組み込み)の最小同位体濃縮因子を典型的に有する。
本明細書に提供する化合物の同位体濃縮及び同位体濃縮因子は、例えば、質量分析、核磁気共鳴分光法、及び結晶解析法を含む、当業者に公知の従来の分析方法を用いて判定することができる。
(B.シチジン類似体)
(1.概観)
本明細書に提供するのは、経口投与の際に実質的に胃の中でAPIを放出するシチジン類似体を含む剤形、医薬製剤及び組成物である。ある実施形態において、当該シチジン類似体は、5−アザシチジンである。ある実施形態において、当該シチジン類似体は、5−アザ−2’−デオキシシチジン(デシタビンまたは5−アザ−CdR)である。ある実施形態において、当該シチジン類似体は、例えば、1−β−D−アラビノフラノシルシトシン(シタラビンまたはAra−C)、プソイドイソ−シチジン(psi ICR)、5−フルオロ−2’−デオキシシチジン(FCdR)、2’−デオキシ−2’,2’−ジフルオロシチジン(ゲムシタビン)、5−アザ−2’−デオキシ‐2’,2’−ジフルオロシチジン、5−アザ−2’−デオキシ−2’−フルオロシチジン、l−β−D−リボフラノシル−2(1H)−ピリミジノン(ゼブラリン)、2’,3’−ジデオキシ−5−フルオロ−3’−チアシチジン(エムトリバ)、2’−シクロシチジン(アンシタビン)、1−β−D−アラビノフラノシル−5−アザシトシン(ファザラビンまたはAra−AC)、6−アザシチジン(6−アザ−CR)、5,6−ジヒドロ−5−アザシチジン(dH−アザ−CR)、N4−ペンチルオキシ−カルボニル−5’−デオキシ−5−フルオロシチジン(カペシタビン)、N4−オクタデシル−シタラビン、エライジン酸シタラビン、あるいはCP−4200(Clavis Pharma ASA)または、アザ−C−5’−ペトロセリン酸エステル若しくはアザ−C−5’−ペトロセライジン酸エステルのような、WO2009/042767に開示された化合物を含むがこれに限定しない、シチジン類似体と脂肪酸とを含む共役化合物(例えば、アザシチジン−脂肪酸共役体)である。
ある実施形態において、本明細書に提供するシチジン類似体には、例えば、5−アザシチジンのエステル化誘導体のような、シチジン類似体のエステル化誘導体を含む。特定の実施形態において、エステル化誘導体は、当該シチジン類似体分子上の1つ以上の位置にエステル部分(例えば、アセチル基)を含有するシチジン類似体である。エステル化誘導体は、当該技術分野で公知の任意の方法によって調製され得る。ある実施形態において、シチジン類似体のエステル化誘導体は、当該シチジン類似体のプロドラッグとして機能し、それにより、例えば、エステル化誘導体の投与後に、当該誘導体は、インビボで脱アセチル化して、当該シチジン類似体を生じる。本明細書の特定の実施形態は、好ましい物理化学的特性及び治療特性を有する2’,3’,5’−トリアセチル−5−アザシチジン(TAC)を提供する。例えば、国際公開第WO2008/092127号(国際出願第PCT/US2008/052124号)、Ziemba,A.J.,ら,「Development of Oral Demethylating Agents for the Treatment of Myelodysplastic Syndrome」(要約番号第3369号),出典:Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; 2009年4月号 18〜22;Denver,Co.フィラデルフィア州(PA):AACR;2009を参照されたい(これらはいずれも、それらの全体が参照により本明細書に組み込まれる)。
ある実施形態において、本明細書に提供するシチジン類似体には、シチジンまたはデオキシシチジンと構造上関連する、ならびにシチジンまたはデオキシシチジンの作用を機能的に模倣及び/または当該作用に拮抗する、任意の化合物を含む。本明細書のある実施形態は、本明細書に提供するシチジン類似体の塩、共結晶、溶媒和物(例えば、水和物)、複合体、プロドラッグ、前駆体、代謝産物、及び/または他の誘導体を提供する。例えば、特定の実施形態は、5−アザシチジンの塩、共結晶、溶媒和物(例えば、水和物)、複合体、前駆体、代謝産物、及び/または他の誘導体を提供する。ある実施形態は、本明細書に提供するシチジン類似体の塩、共結晶、溶媒和物(例えば、水和物)、または複合体ではないシチジン類似体を提供する。例えば、特定の実施形態は、非イオン化、非溶媒和(例えば、無水)、非複合体形成の形態で5−アザシチジンを提供する。本明細書のある実施形態は、本明細書に提供する2つ以上のシチジン類似体からなる混合物を提供する。
本明細書に提供するシチジン類似体は、本明細書で参照するまたはさもなくば当該文献中で入手可能な合成方法及び合成手順を用いて調製してもよい。例えば、5−アザシチジンを合成するための特定の方法は、例えば、それらの各々が参照により本明細書に組み込まれる米国特許第7,038,038号及びその中で考察されている参考文献の中で示されている。5−アザシチジンは、Celgene Corporation,ニュージャージー州ワーレン郡区からも入手可能である。本明細書に提供する他のシチジン類似体は、当業者に利用可能な既に開示されている合成手順を用いて調製してもよい。
ある実施形態において、例示的なシチジン類似体は、以下に提供する構造を有する。
(2.同位体濃縮したシチジン類似体)
本明細書の特定の実施形態は、同位体濃縮したシチジン類似体、そのプロドラッグ、その合成中間体、及びその代謝産物を提供する。例えば、本明細書の具体的な実施形態は、同位体濃縮した5−アザシチジンを提供する。
薬物動態(「PK」)、薬力学(「PD」)、及び毒性特性を改善するための、医薬の同位体濃縮(例えば、重水素化)は、一部のクラスの薬剤を用いてすでに実証されている。例えば、Lijinskyら,Food Cosmet.Toxicol.,20:393(1982)、Lijinskyら,J.Nat.Cancer Inst.,69:1127(1982)、Mangoldら,Mutation Res.308:33(1994)、Gordonら,Drug Metab.Dispos.,15:589(1987)、Zelloら,Metabolism,43:487(1994)、Gatelyら,J.Nucl.Med.,27:388(1986)、Wade,D.,Chem.Biol.Interact.117:191(1999)を参照されたい。
いかなる特定の理論によっても制限されることなく、薬剤の同位体濃縮は、例えば、(1)望ましくない代謝産物を減少若しくは除去する、(2)親薬剤の半減期を延長する、(3)所望の効果を達成するのに必要とされる投薬回数を減少させる、(4)所望の効果を達成するのに必要な投薬量を減少させる、(5)もし形成される場合、作用代謝産物の形成を亢進する、及び/あるいは(6)併用療法が意図的であろうとなかろうと、具体的な組織における有害な代謝産物の産生を低下させ、及び/または当該併用療法のためにより有効な薬剤及び/若しくはより安全な薬剤を作製する、ために使用することができる。
原子とその同位体のうちの1つとの置き換えはしばしば、化学反応の反応速度の変化を結果的に生じることがある。この現象は、動的同位体効果(「KIE」)として公知である。例えば、C−H結合が化学反応における律速段階(すなわち、最大遷移状態エネルギーを有する段階)の間に破壊された場合、重水素と当該水素との置換は、反応速度の低下を生じるであろうし、当該過程は遅延するであろう。この現象は、重水素動的同位体効果(「DKIE」)として公知である。例えば、Fosterら,Adv.Drug Res.,第14巻,1〜36頁(1985)、Kushnerら,Can.J.Physiol.Pharmacol.,第77巻,79〜88頁(1999)を参照されたい。
DKIEの程度は、C−H結合が破壊される所与の反応と、重水素が水素と置換される同じ反応との速度比として表すことができる。DKIEは、約1(同位体効果なし)から50以上など非常に大きな数まで及ぶことができ、重水素が水素と置換される場合、当該反応が50倍以上緩徐であることができることを意味している。特定の理論によって制限されることなく、高いDKIE値は、不確定な原子の結果であるトンネリングとして公知の現象に一部因るのかもしれない。トンネリングは、少量の水素原子に起因しており、プロトンを包含する遷移状態が、必要とされる活性化エネルギーの非存在下で時々形成することができるので、生じる。重水素が水素よりも大きな質量を有しているので、この現象を受けている確率は統計的に非常に低い。
トリチウム(「T」)は、研究、核融合炉、中性子発生装置及び放射性薬剤において使用する水素の放射性同位体である。トリチウムは、核の中に2個の中性子を有しかつ3に近い原子量を有する水素原子である。トリチウムは、非常に低濃度の環境において天然に生じ、最も普遍的にはT2Oとして認められる。トリチウムは緩徐に壊変し(半減期=12.3年)、ヒトの皮膚の外層を貫通できない低エネルギーベータ粒子を放出する。内部被曝は、この同位体と関係した主要なハザードであるが、それでも、有意な健康危険度を引き出すためには多量に摂取しなければならない。重水素と比較した場合、トリチウムが危険レベルに到達する前に、より少量のトリチウムを消費しなくてはならない。トリチウム(「T」)の水素との置換は、重水素よりもそれでもより強い結合を結果的に生じ、数の上ではより大きな同位体効果を付与する。
同様に、炭素に対する13Cまたは14C、硫黄に対する33S、34S、または36S、窒素に対する15N、及び酸素に対する17Oまたは18Oを含むがこれらに限定しない、他の元素に対する同位体置換は、類似の動的同位体効果をもたらすことがある。
動物の身体は、治療薬のような外来物質を当該身体の循環器系から除去する目的のために、種々の酵素を発現している。このような酵素の例としては、チトクロムP450酵素(「CYP」)、エステラーゼ、プロテアーゼ、還元酵素、脱水素酵素、及びモノアミンオキシダーゼが挙げられ、これらの外来物質と反応して、腎排泄のためのより極性のある中間体または代謝産物へと転換する。医薬化合物の最も共通した代謝反応の一部は、炭素−水素(C−H)結合から炭素−酸素(C−O)パイ結合または炭素間(C−C)パイ結合のいずれかへの酸化を包含する。結果として生じる代謝産物は、生理学的条件下で安定または不安定であり得、親化合物に対して実質的に異なる薬物動態特性、薬力学的特性、ならびに急性及び長期の毒性特性を有することができる。多くの薬剤について、このような酸化は迅速である。結果的に、これらの薬剤はしばしば、複数用量または高い日用量の投与を必要とする。
本明細書に提供する化合物のある位置における同位体濃縮は、天然の同位体組成を有する類似の化合物との比較において、本明細書に提供する化合物の薬物動態特性、薬理学的特性及び/または毒性特性に影響する検出可能なKIEを生じ得る。一実施形態において、重水素濃縮は、代謝中のC−H結合切断部位において実施される。
本明細書のある実施形態は、重水素濃縮された5−アザシチジン類似体を提供し、この中で、当該5−アザシチジン分子中の1つ以上の水素(複数可)は、重水素が同位体濃縮されている。ある実施形態において、本明細書に提供されるのは、式(I)の化合物
であり、式中、1つ以上のY原子(複数可)(すなわち、Y
1、Y
2、Y
3、Y
4、Y
5、Y
6、及びY
7)は、重水素が同位体濃縮された水素(複数可)であり、かつ、任意の残余のY原子(複数可)は、濃縮されていない水素原子(複数可)である。特定の実施形態において、示しているY原子(複数可)のうちの1個、2個、3個、4個、5個、6個、または7個は、重水素が同位体濃縮されており、かつ、任意の残余のY原子(複数可)は、濃縮されていない水素(複数可)である。
ある実施形態において、化合物(I)のリボース部分上の1つ以上のY原子は、重水素濃縮されている。特定の例としては、標識「D」が重水素濃縮した原子配置を示す以下の化合物
が挙げられるがこれらに限定せず、すなわち、所与の化合物を含んでいる試料は、示された位置(複数可)において天然の重水素存在量を上回る重水素濃縮を有する。
ある実施形態において、化合物(I)の5−アザシトシン部分上のY原子は、重水素濃縮されている。特定の例としては、標識「D」が重水素濃縮された原子配置を示す以下の化合物
が挙げられ、すなわち、所与の化合物を含んでいる試料は、示される位置(複数可)において天然の重水素存在量を上回る重水素濃縮を有する。
ある実施形態において、リボース部分上の1つ以上のY原子及び化合物(I)の5−アザシトシン部分上のY原子は、重水素濃縮されている。特定の例としては、標識「D」が重水素濃縮された原子配置を示す以下の化合物
が挙げられるが、これらに限定せず、所与の化合物を含んでいる試料は、示される位置(複数可)において天然の重水素存在量を上回る重水素濃縮を有する。
1つ以上の重水素が生理学的条件下で水素と交換され得ることは理解される。
本明細書のある実施形態は、5−アザシチジンの炭素−13濃縮した類似体を提供し、この中で当該5−アザシチジン分子中の1個以上の炭素は、炭素−13で同位体濃縮されている。ある実施形態において、本明細書に提供されるのは、式(II)
の化合物であり、式中、1、2、3、4、5、6、7、または8のうちの1つ以上は、炭素−13で同位体濃縮した炭素原子であり、かつ、1、2、3、4、5、6、7、または8のうちの任意の残余の原子は、濃縮されていない炭素原子(複数可)である。特定の実施形態において、1個、2個、3個、4個、5個、6個、7個、または8個の炭素原子(すなわち、原子1、2、3、4、5、6、7、及び8)は炭素−13で同位体濃縮されており、かつ任意の残余の炭素原子(複数可)は濃縮されていない。
ある実施形態において、化合物(II)のリボース部分の1つ以上の炭素原子は、炭素−13で濃縮されている。特定の例としては、以下の化合物
が挙げられるが、これらに限定せず、すなわち、所与の化合物を含んでいる試料は、示される位置(複数可)において炭素−13の天然の存在量を上回る炭素−13濃縮を有する。
ある実施形態において、化合物(II)の5−アザシトシン部分の1個以上の炭素原子は、炭素−13で濃縮されている。特例の例としては、星印「
*」が炭素−13濃縮した原子配置を示す以下の化合物
が挙げられるが、これらに限定せず、すなわち、所与の化合物を含んでいる試料は、示される位置(複数可)において炭素−13の天然の存在量を上回る炭素−13濃縮を有する。
ある実施形態において、化合物(II)のリボース部分上の1個以上の炭素原子及び5−アザシトシン部分上の1個以上の炭素原子は、炭素−13が濃縮されており、すなわち、リボース部分についての炭素−13濃縮とアザシトシン(azacitosine)部分についての炭素−13濃縮とからなる任意の組み合わせは、本明細書に包含される。
ある実施形態において、1個以上の水素は、重水素で濃縮され、かつ、1個以上の炭素は、炭素−13で濃縮され、すなわち、5−アザシチジンの重水素濃縮と炭素−13濃縮との任意の組み合わせは、本明細書に包含される。
(3.同位体濃縮したシチジン類似体の合成)
本明細書に説明する化合物は、当業者に公知の任意の方法を用いて合成され得る。例えば、本明細書に説明する特定の化合物は、当業者に公知の標準的な有機化学技術を用いて合成される。いくつかの実施形態において、5−アザシチジンの合成についての公知の手順が採用され、この中で、試薬、出発材料、前駆体、または中間体のうちの1つ以上は、1つ以上の重水素濃縮した試薬、出発材料、前駆体、または中間体、及び/あるいは1つ以上の炭素−13濃縮した試薬、出発材料、前駆体、または中間体を含むがこれらに限定しない1つ以上の同位体濃縮された試薬、出発材料、前駆体、または中間体によって置き換えられる。同位体濃縮した試薬、出発材料、前駆体、または中間体は市販されており、あるいは当業者に公知の通例の化学反応によって調製され得る。いくつかの実施形態において、当該経路は、その全体が参照により本明細書に組み込まれる米国特許第7,038,038号において開示された経路を基にしている。
ある実施形態において、重水素濃縮したリボース、重水素濃縮した5−アザシトシン、炭素−13濃縮したリボース、及び/または炭素−13濃縮した5−アザシトシンのような、適切な同位体濃縮した出発材料は、以下の一般的なスキームにおける出発材料として採用して、対応する重水素及び/または炭素−13濃縮した5−アザシチジンを調製し得る(スキーム1を参照されたい)。米国特許第7,038,038号における手順の後、5−アザシトシンをヘキサメチルジシラザン(HMDS)で処理して、シリル化5−アザシトシンにする。テトラアセチル−D−リボースは、Brownら,Biochemical Preparations,1955,4,70〜76における手順に従って、D−リボースを無水酢酸中の酢酸ナトリウムと反応させることによって調製する。シリル化5−アザシトシンをTMS−トリフラートの存在下でテトラアセチル−D−リボースとカップリングさせ、結果として生じる保護された5−アザシチジンをメタノール中のナトリウムメトキシドで処理して、5−アザシチジンを生じる。米国特許第7,038,038号を参照されたい。
スキーム1
いくつかの実施形態において、5−アザシチジンのリボース部分における1つ以上の水素の位置は、重水素で濃縮されている。このような5−アザシチジン類似体は、スキーム1に従って、市販の源から購入したまたは文献手順に従って調製した適切な重水素濃縮したリボースから調製され得る。重水素濃縮したリボース出発材料の具体的な例としては、対応する重水素濃縮した5−アザシチジン類似体へ転換され得る、表1に列挙する以下の化合物が挙げられるが、これらに限定しない。
表1
他の実施形態において、5−アザシチジンの5−アザシトシン環上の水素の位置は、重水素で濃縮される。このような5−アザシチジン類似体は、例えば、重水素化5−アザシトシンからスキーム1に従って調製され得る。重水素化5−アザシトシンは、例えば、スキーム2に示すような適切な重水素化試薬から調製され得る。例えば、Grundmannら,Chem.Ber.1954,87,19〜24、Piskalaら,出典Zorbach及びTipson(編)Synthetic Procedures in Nucleic Acid Chemistry,第1巻,Wiley Interscience,ニューヨーク,1968,107〜108、Piskala,Collect.Czech.Chem.Comm.1967,32,3966〜3976を参照されたい。
スキーム2
他の実施形態において、5−アザシトシン環上の水素の位置と5−アザシチジンのリボース部分における1つ以上の水素の位置とは両方、重水素で濃縮される。このような5−アザシチジン類似体は、例えば、適切な重水素化リボース出発材料を重水素化5−アザシトシンとカップリングさせるスキーム1に従って調製され得る。例えば、化合物I−9、I−10、I−11、I−12、I−13、及びI−14は、表1に列挙する対応する重水素化リボース出発材料、及びスキーム2によって調製される重水素化5−アザシトシンから調製され得る。
いくつかの実施形態において、5−アザシチジンのリボース部分における1つ以上の炭素原子は、炭素−13で濃縮されている。このような5−アザシチジン類似体は、スキーム1に従って、市販の源から購入したまたは文献手順に従って調製した適切な炭素−13濃縮したリボースから調製され得る。炭素−13濃縮したリボース出発材料の具体的な例としては、対応する炭素−13濃縮した5−アザシチジン類似体へ転換され得る、表2に列挙する以下の化合物が挙げられるが、これらに限定しない(星印「
*」は、炭素−13濃縮した原子配置を示す)。
表2
他の実施形態において、5−アザシトシン環における1つ以上の炭素原子は、炭素−13で濃縮されている。このような5−アザシチジン類似体は、炭素−13で濃縮された5−アザシトシンからスキーム1に従って調製され得る。炭素−13濃縮した5−アザシトシン中間体は、スキーム3に示すような適切な炭素−13濃縮した試薬から調製され得る。例えば、Grundmannら,Chem.Ber.1954,87,19〜24、Piskalaら,出典Zorbach及びTipson(編)Synthetic Procedures in Nucleic Acid Chemistry,第1巻,Wiley Interscience,New York,1968,107〜108、Piskala,Collect.Czech.Chem.Comm.1967,32,3966〜3976を参照されたい。
スキーム3
他の実施形態において、5−アザシトシン環上の1つ以上の炭素の位置及び5−アザシチジンのリボース部分における1つ以上の炭素の位置は、炭素−13で濃縮されている。このような5−アザシチジン類似体は、適切な炭素−13濃縮したリボース出発材料を適切な炭素−13濃縮した5−アザシトシンとカップリングさせるスキーム1に従って調製され得る。例えば、化合物は、表2に列挙する炭素−13濃縮したリボース出発材料、及びスキーム3によって調製した炭素−13濃縮した5−アザシトシンから調製され得る。
先に説明した経路及び方法は、重水素濃縮と炭素−13濃縮との両方を有する5−アザシチジンの同位体種(isotopolougue)を提供するよう改変され得る。
(C.医薬製剤)
(1.概観)
本明細書の実施形態は、1つ以上のシチジン類似体、例えば、5−アザシチジン、及び任意に浸透亢進剤を含む医薬製剤及び組成物を包含し、この中で、当該製剤及び組成物は、経口投与のために調製される。特定の実施形態において、当該製剤及び組成物は、実質的に胃の中でのシチジン類似体の放出のために調製される。具体的な実施形態において、当該シチジン類似体、例えば、5−アザシチジンならびに医薬製剤及び組成物は、異常な細胞増殖と関係する疾患及び障害の治療のために使用され、この中で当該シチジン類似体、製剤及び組成物は、経口投与のために、好ましくは実質的に胃の中でのシチジン類似体の放出に調製される。特定の実施形態は、本明細書に提供するような、特定の医学的徴候を治療するための医薬製剤及び組成物の調製のための、1つ以上のシチジン類似体、例えば、5−アザシチジンの使用に関する。本明細書に提供するシチジン類似体を含む医薬製剤及び組成物は、当該シチジン類似体の経口送達を必要とする対象における当該経口送達のために企図される。経口送達フォーマットには、錠剤、カプセル剤、カプレット剤、液剤、懸濁剤、及びシロップ剤を含むが、これらに限定せず、封入されていてもされていなくてもよい複数の顆粒剤、ビーズ剤、散剤またはペレット剤も含んでもよい。このようなフォーマットは、当該シチジン類似体を含有する「薬剤コア」とも本明細書で呼ぶことがある。
本明細書の特定の実施形態は、錠剤またはカプセル剤である固体経口剤形を提供する。ある実施形態において、当該製剤は、シチジン類似体を含む錠剤である。ある実施形態において、当該製剤は、シチジン類似体を含んでいるカプセル剤である。ある実施形態において、本明細書に提供する錠剤またはカプセル剤は任意に、例えば、滑剤、希釈剤、潤滑剤、着色料、崩壊剤、造粒剤、結合剤、ポリマー、及び被覆剤のような1つ以上の賦形剤を含む。ある実施形態において、当該製剤は、即時放出錠剤である。ある実施形態において、当該製剤は、例えば、実質的に胃の中でAPIを放出する徐放性錠剤である。ある実施形態において、当該製剤は、硬質ゼラチンカプセル剤である。ある実施形態において、当該製剤は、軟質ゼラチンカプセル剤である。ある実施形態において、当該カプセル剤は、ヒドロキシプロピルメチルセルロース(HPMC)カプセル剤である。ある実施形態において、当該製剤は、即時放出カプセルである。ある実施形態において、当該製剤は、例えば、実質的に胃の中でAPIを放出する即時放出カプセル剤または徐放性カプセル剤である。ある実施形態において、当該製剤は、投与後に実質的に口腔内で溶解する迅速に崩壊する錠剤である。ある実施形態において、本明細書の実施形態は、異常な細胞増殖と関係する疾患を治療するための医薬組成物の調製のための、シチジン類似体、例えば、5−アザシチジンの使用を包含し、この中で、当該組成物は、経口投与のために調製される。
(2.本明細書に提供するある剤形の性能)
ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体を含む製剤は、経口投与の際にAPIの即時放出をもたらす。特定の実施形態において、例えば、5−アザシチジンのようなシチジン類似体を含んでいる製剤は、治療有効量または予防有効量のシチジン類似体(及び、任意に、1つ以上の賦形剤)を含み、経口投与の際にAPIの即時放出をもたらす。
ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体を含んでいる製剤は、経口投与の際に実質的に胃の中でのAPIの徐放をもたらす。ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体を含んでいる製剤は、治療有効量または予防有効量のシチジン類似体、及び実質的に胃の中でシチジン類似体を放出することのできる薬剤放出制御成分を含む。ある実施形態において、マトリックス(例えば、ポリマーマトリックス)は、シチジン類似体の放出を制御するために当該製剤中に採用され得る。ある実施形態において、皮膜及び/またはシェルは、実質的に胃の中でのシチジン類似体の放出を制御するために、当該製剤中に採用され得る。
ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体を含んでいる製剤は、経口投与の際に実質的に胃の中でAPIを放出する。ある実施形態において、当該製剤は、経口投与の際にシチジン類似体の即時放出をもたらす。ある実施形態において、当該製剤は任意に、薬剤放出制御成分をさらに含み、この中で、当該薬剤放出制御成分は、当該シチジン類似体の放出が実質的に胃の中で生じるよう調整される。特定の実施形態において、当該薬剤放出制御成分は、当該シチジン類似体の放出が即時でありかつ実質的に胃の中で生じるよう調整される。特定の実施形態において、当該薬剤放出制御成分は、当該シチジン類似体の放出が持続的でありかつ実質的に胃の中で生じるよう調整される。ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体の製剤は、実質的に胃の中でAPIを放出し、その後、経口投与の際に腸で残余のAPIを放出する。
薬剤が対象の消化管中のどこで放出されたのかを当業者が評価することのできる方法は、当該技術分野で公知であり、とりわけ、消化管の関連部分における流体を模倣する生体関連媒体中で試験するシンチグラフィー試験を含む。
本明細書に提供する特定の実施形態は、同じシチジン類似体の皮下用量と比較して、当該製剤が経口投与される対象における特定の曝露を達成するシチジン類似体(例えば、5−アザシチジン)を含む医薬製剤(例えば、即時放出経口製剤及び/または実質的に胃の中でAPIを放出する製剤)を提供する。特定の実施形態は、皮下用量と比較して、少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、少なくとも約95%、または約100%の曝露を達成する経口製剤を提供する。
ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体を含んでいる製剤(例えば、即時放出経口製剤及び/または実質的に胃の中でAPIを放出する製剤)は、経口投与の際に、当該製剤中のある特定の百分率のシチジン類似体を全身で生体利用可能にする。ある実施形態において、対象が当該製剤を経口投与された後、当該製剤中のシチジン類似体は、実質的に胃の中で吸収され、全身性曝露を通じて身体に対して利用可能となる。特定の実施形態において、本明細書に提供するシチジン類似体を含んでいる製剤の経口生体利用能は、例えば、当該製剤中のシチジン類似体の総量の約1%超、約5%超、約10%超、約15%超、約20%超、約25%超、約30%超、約35%超、約40%超、約45%超、約50%超、約55%超、約60%超、約65%超、約70%超、約75%超、約80%超、約85%超、約90%超、約95%超、または約100%である。
当業者が、対象における薬剤製剤の経口生体利用能を評価することのできる方法は、当該技術分野で公知である。このような方法には、例えば、最大血漿濃度(「Cmax」)、最大血漿濃度到達時間(「Tmax」)、または曲線下面積(「AUC」)の測定のような、しかしこれらに限定しない、ある投与関連パラメータを比較することを含む。
本明細書の特定の実施形態は、製剤が経口投与される対象(例えば、ヒト)における特定のAUC値(例えば、AUC(0〜t)またはAUC(0〜∞))に到達するシチジン類似体(例えば、5−アザシチジン)を含む医薬製剤(例えば、即時放出経口製剤及び/または実質的に胃の中でAPIを放出する製剤)を提供する。特定の実施形態は、少なくとも約25ng・時/mL、少なくとも約50ng・時/mL、少なくとも約75ng・時/mL、少なくとも約100ng・時/mL、少なくとも約150ng・時/mL、少なくとも約200ng・時/mL、少なくとも約250ng・時/mL、少なくとも約300ng・時/mL、少なくとも約350ng・時/mL、少なくとも約400ng・時/mL、少なくとも約450ng・時/mL、少なくとも約500ng・時/mL、少なくとも約550ng・時/mL、少なくとも約600ng・時/mL、少なくとも約650ng・時/mL、少なくとも約700ng・時/mL、少なくとも約750ng・時/mL、少なくとも約800ng・時/mL、少なくとも約850ng・時/mL、少なくとも約900ng・時/mL、少なくとも約950ng・時/mL、少なくとも約1000ng・時/mL、少なくとも約1100ng・時/mL、少なくとも約1200ng・時/mL、少なくとも約1300ng・時/mL、少なくとも約1400ng・時/mL、少なくとも約1500ng・時/mL、少なくとも約1600ng・時/mL、少なくとも約1700ng・時/mL、少なくとも約1800ng・時/mL、少なくとも約1900ng・時/mL、少なくとも約2000ng・時/mL、少なくとも約2250ng・時/mL、または少なくとも約2500ng・時/mLのAUC値に到達する経口製剤を提供する。特定の実施形態において、AUCの測定は、投与後の動物またはヒトボランティアの血液試料から得た時間−濃度薬物動態特性から得る。
本明細書の特定の実施形態は、当該製剤を経口投与する対象における特定の最大血漿濃度(「Cmax」)に到達するシチジン類似体(例えば、5−アザシチジン)を含んでいる医薬製剤(例えば、即時放出経口製剤及び/または実質的に胃の中でAPIを放出する製剤)を提供する。特定の実施形態は、少なくとも約25ng/mL、少なくとも約50ng/mL、少なくとも約75ng/mL、少なくとも約100ng/mL、少なくとも約150ng/mL、少なくとも約200ng/mL、少なくとも約250ng/mL、少なくとも約300ng/mL、少なくとも約350ng/mL、少なくとも約400ng/mL、少なくとも約450ng/mL、少なくとも約500ng/mL、少なくとも約550ng/mL、少なくとも約600ng/mL、少なくとも約650ng/mL、少なくとも約700ng/mL、少なくとも約750ng/mL、少なくとも約800ng/mL、少なくとも約850ng/mL、少なくとも約900ng/mL、少なくとも約950ng/mL、少なくとも約1000ng/mL、少なくとも約1100ng/mL、少なくとも約1200ng/mL、少なくとも約1300ng/mL、少なくとも約1400ng/mL、少なくとも約1500ng/mL、少なくとも約1600ng/mL、少なくとも約1700ng/mL、少なくとも約1800ng/mL、少なくとも約1900ng/mL、少なくとも約2000ng/mL、少なくとも約2250ng/mL、または少なくとも約2500ng/mLのシチジン類似体のCmaxに到達する経口製剤を提供する。
本明細書の特定の実施形態は、製剤を経口投与する対象における特定の最大血漿濃度到達時間(「Tmax」)に到達するシチジン類似体(例えば、5−アザシチジン)を含んでいる医薬製剤(例えば、即時放出経口製剤及び/または実質的に胃の中でAPIを放出する製剤)を提供する。特定の実施形態は、約10分未満、約15分未満、約20分未満、約25分未満、約30分未満、約35分未満、約40分未満、約45分未満、約50分未満、約55分未満、約60分未満、約65分未満、約70分未満、約75分未満、約80分未満、約85分未満、約90分未満、約95分未満、約100分未満、約105分未満、約110分未満、約115分未満、約120分未満、約130分未満、約140分未満、約150分未満、約160分未満、約170分未満、約180分未満、約190分未満、約200分未満、約210分未満、約220分未満、約230分未満、または約240分未満のシチジン類似体のTmaxに到達する経口製剤を提供する。特定の実施形態において、Tmax値は、製剤を経口投与する時間から測定される。
本明細書の特定の実施形態は、シチジン類似体を含んでいる経口剤形を提供し、この中で、経口剤形は腸溶コーティングを有している。特定の実施形態は、多孔性の透過性または部分的に透過性の(例えば、「漏出性の」)腸溶コーティングを提供する。特定の実施形態において、透過性または部分的に透過性の腸溶コーティングした錠剤は、実質的に胃の中で、即時放出様式で5−アザシチジンを放出する。
(3.本明細書に提供するある剤形の設計)
本明細書に提供するのは、経口投与の際に、例えば、実質的に胃の中で放出するために、経口投与の際のあるシチジン類似体、例えば5−アザシチジン、の吸収及び/または有効な送達を最大限にするよう設計された剤形である。したがって、本明細書のある実施形態は、経口投与の際に、例えば、実質的に胃の中で、APIの即時放出のために設計された医薬賦形剤を用いて、例えば、5−アザシチジンのようなシチジン類似体の固体経口剤形を提供する。特定の即時放出製剤は、特定量のシチジン類似体及び任意に1つ以上の賦形剤を含む。ある実施形態において、当該製剤は、即時放出錠剤または(例えば、HPMCカプセルのような)即時放出カプセルであることがある。
本明細書に提供するのは、本明細書に提供するシチジン類似体を含む、本明細書に提供する製剤(例えば、即時放出経口製剤及び/または実質的に胃の中でAPIを放出する製剤)の作製方法である。特定の実施形態において、本明細書に提供する製剤は、例えば、関連する教科書において説明されるような、医薬製剤の分野の当業者に公知の従来の方法を用いて調製され得る。例えば、Remington,The Science and Practice of Pharmacy,第20版,Lippincott Williams & Wilkins,(2000)、Anselら,Pharmaceutical Dosage Forms and Drug Delivery Systems,第7版,Lippincott Williams & Wilkins,(1999)、Gibson,Pharmaceutical Preformulation and Formulation,CRC Press(2001)を参照されたい。
特定の実施形態において、本明細書に提供する製剤(例えば、即時放出経口製剤、実質的に胃の中でAPIを放出する製剤、または実質的に口腔内で溶解する迅速に崩壊する製剤)は、例えば、5−アザシチジンのようなシチジン類似体を特定量で含む。特定の実施形態において、当該製剤中のシチジン類似体の特定量は、例えば、約10mg、約20mg、約40mg、約60mg、約80mg、約100mg、約120mg、約140mg、約160mg、約180mg、約200mg、約220mg、約240mg、約260mg、約280mg、約300mg、約320mg、約340mg、約360mg、約380mg、約400mg、約420mg、約440mg、約460mg、約480mg、約500mg、約600mg、約700mg、約800mg、約900mg、約1000mg、約1100mg、約1200mg、約1300mg、約1400mg、約1500mg、約1600mg、約1700mg、約1800mg、約1900mg、約2000mg、約2100mg、約2200mg、約2300mg、約2400mg、約2500mg、約3000mg、約4000mg、または約5000mgである。特定の実施形態において、当該製剤中のシチジン類似体の特定量は、例えば、少なくとも約10mg、少なくとも約20mg、少なくとも約40mg、少なくとも約60mg、少なくとも約80mg、少なくとも約100mg、少なくとも約120mg、少なくとも約140mg、少なくとも約160mg、少なくとも約180mg、少なくとも約200mg、少なくとも約220mg、少なくとも約240mg、少なくとも約260mg、少なくとも約280mg、少なくとも約300mg、少なくとも約320mg、少なくとも約340mg、少なくとも約360mg、少なくとも約380mg、少なくとも約400mg、少なくとも約420mg、少なくとも約440mg、少なくとも約460mg、少なくとも約480mg、少なくとも約500mg、少なくとも約600mg、少なくとも約700mg、少なくとも約800mg、少なくとも約900mg、少なくとも約1000mg、少なくとも約1100mg、少なくとも約1200mg、少なくとも約1300mg、少なくとも約1400mg、少なくとも約1500mg、少なくとも約1600mg、少なくとも約1700mg、少なくとも約1800mg、少なくとも約1900mg、少なくとも約2000mg、少なくとも約2100mg、少なくとも約2200mg、少なくとも約2300mg、少なくとも約2400mg、少なくとも約2500mg、少なくとも約3000mg、少なくとも約4000mg、または少なくとも約5000mgである。
ある実施形態において、当該製剤は錠剤であり、この中で当該錠剤は、標準的な当該技術分野で認識される錠剤加工手順及び装置を用いて製造される。ある実施形態において、錠剤の形成方法は、シチジン類似体を単独で、または例えば担体、添加剤、ポリマー、若しくはこれらに類するもののような1つ以上の賦形剤との組み合わせで含んでいる粉状の、結晶の、及び/または顆粒状の組成物の直接圧縮である。ある実施形態において、直接圧縮に対する代替法として、錠剤は、湿式造粒または乾式造粒加工を用いて調製してもよい。ある実施形態において、錠剤は、圧縮されるよりもむしろ鋳造され、湿性のまたはさもなくば扱いやすい材料を用いて出発する。ある実施形態において、圧縮及び造粒技術が使用される。
ある実施形態において、製剤はカプセル剤であり、この中で当該カプセル剤は、標準的な当該技術分野で認識されるカプセル加工手順及び装置を用いて製造され得る。ある実施形態において、当該カプセル剤が、シチジン類似体と、植物油または、例えばポリエチレングリコール及びこれに類するもののような非水性の水混和性材料とからなる混合物を含有する軟質ゼラチンカプセル剤が調製され得る。ある実施形態において、硬質ゼラチンカプセル剤は、例えば、ラクトース、サッカロース、ソルビトール、マンニトール、ジャガイモでんぷん、トウモロコシでんぷん、アミロペクチン、セルロース誘導体、またはゼラチンのような固体粉末状担体との組み合わせでシチジン類似体の顆粒を含有して調製され得る。ある実施形態において、硬質ゼラチンカプセル剤シェルは、ゼラチンと少量のグリセロールのような可塑剤とを含むカプセル組成物から調製され得る。ある実施形態において、ゼラチンに対する代替物として、カプセル剤シェルは、炭水化物材料から製造され得る。ある実施形態において、当該カプセル剤組成物は、ポリマー、着色料、着香料及び乳白剤を必要に応じて追加的に含んでもよい。ある実施形態において、当該カプセル剤はHPMCを含む。
ある実施形態において、例えば、5−アザシチジンのようなシチジン類似体の製剤は、当該シチジン類似体の有意な加水分解性劣化を生じることなく、水性溶媒を用いて調製される。特定の実施形態において、例えば、5−アザシチジンのようなシチジン類似体の製剤は、当該製剤中に当該シチジン類似体の有意な加水分解性劣化を生じることなく水性溶媒を用いて薬剤コアへ適用される皮膜を含有する錠剤である。ある実施形態において、水は、当該薬剤コアを被覆するための溶媒として採用される。ある実施形態において、当該シチジン類似体の経口剤形は、水性溶媒を用いて薬剤コアへ適用されるフィルムコートを含有する錠剤である。特定の実施形態において、水は、フィルムコーティングのための溶媒として採用される。特定の実施形態において、シチジン類似体を含有する錠剤は、医薬組成物の劣化をもたらすことなく水性溶媒を用いてフィルムコーティングされる。特定の実施形態において、水は、当該医薬組成物の劣化をもたらすことなく、フィルムコーティング溶媒として使用される。ある実施形態において、5−アザシチジンと水性薄膜コーティングとを含む経口剤形は、経口送達の際に即時薬剤放出をもたらす。ある実施形態において、5−アザシチジンと水性薄膜コーティングとを含む経口剤形は、経口投与の際に、上部消化管、例えば、胃への薬剤徐放をもたらす。特定の実施形態において、水性薄膜コーティングを用いた錠剤は、5−アザシチジンをAPIとして含む。
ある実施形態において、本明細書に提供するのは、実質的に胃の中でシチジン類似体を放出するシチジン類似体の経口投与のための徐放性医薬製剤であり、a)特定量のシチジン類似体、b)実質的に上部消化管、例えば、胃の中でシチジン類似体の放出を制御するための薬剤放出制御成分、及びc)任意に1つ以上の賦形剤を含む。ある実施形態において、シチジン類似体を含む経口剤形は、当該薬組成物及び任意の賦形剤を含む薬剤コアを含む徐放性錠剤またはカプセル剤として調製される。任意に、「シールコート」または「シェル」が適用される。ある実施形態において、本明細書に提供するシチジン類似体を含む本明細書に提供する製剤は、治療有効量のシチジン類似体、経口投与の際に実質的に胃の中でシチジン類似体の放出を制御する薬剤放出制御成分、及び任意に1つ以上の賦形剤を含む、徐放性錠剤またはカプセル剤である。
特定の実施形態は、胃液への曝露の際に膨潤して、製剤の胃での保持及び実質的に胃の中でポリマーマトリックスからのシチジン類似体の持続性放出をもたらすポリマーマトリックスである薬剤放出制御成分を提供する。ある実施形態において、このような製剤は、製剤中にシチジン類似体を適切なポリマーマトリックスへと組み込むことによって調製され得る。このような製剤の例は、当該技術分野で公知である。例えば、それらの各々が全体として参照により本明細書に組み込まれるShellら,米国特許公開第2002/0051820号(出願番号第09/990,061号)、Shellら,米国特許公開第2003/0039688号(出願番号第10/045,823号)、Guslerら,米国特許公開第2003/0104053号(出願番号第10/029,134号)を参照されたい。
ある実施形態において、薬剤放出制御成分は、薬剤含有コアを取り囲んでいるシェルを含み得、この中で当該シェルは、例えば、コアからのシチジン類似体の拡散を許容して、胃液への曝露の際の胃の中で保持される大きさへの膨潤による製剤の胃での保持を促進することによって、当該コアからシチジン類似体を放出する。ある実施形態において、このような製剤は、薬剤コアを形成するためにシチジン類似体と1つ以上の賦形剤とからなる混合物をまず圧縮してから、シェルを形成するために別の粉状混合物を薬剤コア上で圧縮することによって、または適切な材料でできたカプセルシェルで薬剤コアを封入することによって調製してもよい。このような製剤の例は、当該技術分野で公知である。例えば、全体として参照により本明細書に組み込まれるBernerら,米国特許公開第2003/0104062出願番号第10/213,823号を参照されたい。
本明細書のある実施形態は、シチジン類似体を含む経口剤形を提供し、この中で、当該剤形は、従来の腸溶コーティングにおいて孔を含有している。特定の実施形態において、シチジン類似体の経口剤形は、孔を有している透過性のまたは部分的に透過性の(例えば、「漏出性の」)腸溶コーティングを含有する錠剤である。特定の実施形態において、透過性のまたは部分的に透過性の腸溶コーティング錠剤は、当該錠剤から主として上部消化管、例えば、胃までのシチジン類似体の放出を制御する。特定の実施形態において、透過性または部分的に透過性の腸溶コーティング錠剤は、5−アザシチジンを含む。特定の実施形態において、当該シチジン類似体の残余は、その後、胃を越えて(例えば、腸において)放出される。
ある実施形態において、本明細書に提供する医薬製剤は、シチジン類似体を含む圧縮錠剤である。シチジン類似体に加えて、当該錠剤は任意に、(a)所望の大きさの錠剤を調製するために製剤へ必要なかさを付加し得る希釈剤または充填剤、(b)製剤の粒子の接着を促進し得、造粒を調製することが可能であり得、及び最終的な錠剤の完全性を維持し得る結合剤または接着剤、(c)投与後、改善された薬剤利用可能性のために当該錠剤をより小さな粒子へと崩壊するのを促進し得る崩壊剤(disintegrant)または崩壊剤(disintegrating agent)、(d)錠剤化材料が錠剤ダイスへと流れるのを亢進し得、パンチ及びダイスの摩耗を最小限にし得、充填材料がパンチ及びダイスへ固着するのを防止し得、ならびに光沢を有する錠剤を生成する接着防止剤、滑剤、潤滑剤(lubricant)または潤滑剤(lubricating agent)、ならびに(e)着色料及び着香料のような種々の補助剤を含む、1つ以上の賦形剤を含む。圧縮後、本明細書に提供する錠剤は、本明細書に説明する種々の材料で被覆してもよい。
ある実施形態において、本明細書に提供する医薬製剤は、シチジン類似体の多重圧縮錠剤である。多重圧縮錠剤は、単回圧縮よりも充填材料を多く供することによって調製する。この結果は、多層錠剤または錠剤内錠剤であり得、内部錠剤は、シチジン類似体及び任意に1つ以上の賦形剤を含むコアであり、外部錠剤はシェルであり、当該シェルは、1つ以上の賦形剤を含み、シチジン類似体を含有していても含有していなくてもよい。層状錠剤は、ダイスにおける充填材料の一部の、次いで追加の充填材料の初回圧縮、及び別個の充填剤の数に応じて、2層または3層の錠剤を形成するための圧縮によって調製され得る。各層は、化学的または物理的配合禁忌の理由から相互に別個である異なる治療薬、または段階分類された薬剤放出のための、若しくは単に多層錠剤の独特の様相のための同じ治療薬を含有し得る。充填剤の各部分は、異なる見かけの錠剤を調製するために異なった色付けをしてもよい。内部コアとして圧縮された錠剤を有する錠剤の調製において、特別な機械を用いて、事前形成した錠剤をダイス内に正確に置いて、周辺の充填材料のその後の圧縮に用いてもよい。
ある実施形態において、シチジン類似体の圧縮錠剤は、着色したまたは無着色の糖層で被覆してもよい。このコーティングは、水溶性であり得、かつ口腔内消化の後迅速に溶解し得る。糖衣は、封入した薬剤を環境から保護し、及び不快な味または嗅いに対する遮蔽を提供する目的を果たし得る。糖衣はまた、圧縮錠剤の様相を高め得、製造元の情報を識別する刻印を可能にし得る。ある実施形態において、糖衣錠は、元の被覆していない錠剤よりも50%大きく及び重くあり得る。錠剤の糖衣は、次の任意の工程、すなわち(1)防水加工及び密封(必要な場合)、(2)下がけ、(3)平滑化及び最終丸み付け、(4)仕上げ加工及び着色(必要な場合)、(5)刻印(必要な場合)、ならびに(6)研磨へと分割し得る。
ある実施形態において、シチジン類似体の圧縮された錠剤は、薄膜コーティングされていてもよい。薄膜コーティングされた錠剤は、当該錠剤の上に薄皮のような薄膜を形成することのできるポリマーの薄い層で被覆された圧縮された錠剤であり得る。当該薄膜は通常着色されており、より耐久性があり、あまりかさばらず、かつ適用するのにあまり時間がかからない利点を有する。当該薄膜の組成により、コーティングは、消化管内の所望の位置でコア錠剤を破断及び露出させるよう設計してもよい。圧縮錠剤の上にプラスチック様材料の薄い皮が密着したコーティングを配置する薄膜コーティング法は、元の圧縮錠剤と本質的に同じ重量、形状、及び大きさを有する被覆した錠剤を生成し得る。薄膜コーティングは、錠剤が人目を引いて弁別されるよう着色してもよい。薄膜コーティング溶液は、非水性または水性であり得る。特定の実施形態において、非水性溶液は任意に、錠剤へ所望のコーティングを提供するために次のタイプ、すなわち(1)従来のコーティング条件下で再生可能な、及び例えば、酢酸フタル酸セルロースのような、種々の錠剤形状へ適用可能な滑らかな薄い薄膜を製造することのできるフィルム形成剤、(2)体液による浸透及び例えば、ポリエチレングリコールのような薬剤の治療上の利用可能性を確実にするために水の可溶性または当該薄膜に対する透過性を提供する合金物質、(3)コーティングの可撓性及び弾性を生じ、したがって耐久性を提供する、例えばひまし油のような可塑剤、(4)例えば、ポリオキシエチレンソルビタン誘導体のような、適用中に薄膜の延展性を高めるような界面活性剤、(5)被覆した錠剤の様相が人目を引いて弁別されるような、例えば、不透明体としては二酸化チタン、着色料としては、FD&CまたはD&C染色料のような、不透明体及び着色料、(6)例えば、甘味料としてのサッカリン、ならびに着香料及び芳香料としてのバニリンのような、対象に対する錠剤の許容性を高めるための甘味料、着香料、または芳香料、(7)別個の研磨操作なしで錠剤へ光沢を提供する、例えば、蜜蝋のような光沢剤、ならびに(8)迅速な蒸発を許容して有効な、しかもなお、素早い取り扱いを可能にしながら、当該錠剤上にその他の成分の延展を許容する、例えばアルコール−アセトン混合物のような揮発性溶媒のうちの1つ以上を含有してもよい。ある実施形態において、水性薄膜コーティング製剤は、以下、すなわち、(1)例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、及びメチルセルロースのような、セルロースエーテルポリマーのような、薄膜形成ポリマー、(2)例えば、グリセリン、プロピレングリコール、ポリエチレングリコール、フタル酸ジエチル、及び塩基性酢酸ジブチルのような可塑剤、(3)例えば、FD&CレーキまたはD&Cレーキ及び酸化鉄顔料のような、着色料及び不透明体、または(4)例えば、水のようなビヒクル、のうちの1つ以上を含有してもよい。
ある実施形態において、シチジン類似体の圧縮錠剤は、圧縮被覆され得る。顆粒または粉体の形状にある被覆材料は、特殊な錠剤圧縮で薬剤の錠剤コアへと圧縮され得る。
ある実施形態において、当該医薬製剤は、シチジン類似体のゼラチン被覆錠剤である。ゼラチン被覆錠剤は、被覆した生成物が等量の粉体を充填したカプセルよりも小さくなれるカプセル形状の圧縮錠剤である。ゼラチンコーティングは、嚥下が容易であり、密封していないカプセルと比較して、ゼラチン被覆錠剤は、開封証明性が高くあり得る。
ある実施形態において、当該医薬製剤は、シチジン類似体の舌下錠剤であり得る。舌下錠剤は、口腔粘膜を通じての吸収のために舌の真下で溶解するよう企図される。舌下錠剤は、即座に溶解し得、当該薬剤の迅速な放出を提供し得る。
ある実施形態において、当該医薬製剤は、シチジン類似体の即時放出錠剤である。ある実施形態において、即時放出錠剤は、例えば、特殊コーティング及び他の技術のような、任意の特殊な律速特徴のないAPIを崩壊及び放出するよう設計される。ある実施形態において、当該製剤は、例えば、投与後に実質的に口腔内で溶解する迅速に崩壊する錠剤である。ある実施形態において、当該医薬製剤は、シチジン類似体の放出延長錠剤である。ある実施形態において、当該放出延長錠剤は、例えば、延長した期間にわたって実質的に胃の中でAPIを放出するよう設計される。
ある実施形態において、圧縮錠剤は、湿式造粒によって調製され得る。湿式造粒は、圧縮錠剤の製造のために広範に採用される方法であり、特に1つ以上の以下の工程、すなわち、(1)成分を秤量及び配合する工程、(2)ダンプ塊を調製する工程、(3)ダンプ塊をペレット若しくは顆粒へとふるい分けする工程、(4)造粒物を乾燥させる工程、(5)造粒物を乾燥ふるい分けによってサイズ分けする工程、(6)潤滑剤及び配合剤を添加する工程、ならびに(7)圧縮によって錠剤化する工程を必要とする。
ある実施形態において、圧縮錠剤は、乾式造粒によって調製され得る。乾式造粒法によって、粉体混合物は、大きな複数の片に圧縮され、その後顆粒へと破壊またはサイズ分けされる。しかし、本方法は、有効成分または希釈剤のいずれかが粘着特性を有している。当該成分を秤量及び混合した後、当該粉体混合物は、大きな平らな錠剤またはペレットへとスラギングまたは圧縮され得る。当該スラッグは次に、手によってまたはミルによって粉砕された後、サイズ分けのために所望のメッシュのふるいを通過させる。潤滑剤を通常の様式で添加し、錠剤を圧縮により調製する。あるいは、スラギングの代わりに、粉体圧縮装置を用いて、粉体を高圧ローラの間で圧搾することによって、粉体の密度を高めてもよい。圧縮された材料は次に、破壊され、サイズ分けされ、潤滑化され、錠剤は、通常の様式で圧縮によって調製される。ローラ圧縮法はしばしば、スラギングよりも好ましい。ローラ圧縮製剤において使用する結合剤には、メチルセルロースまたはヒドロキシメチルセルロースを含み、良好な錠剤硬度及び破砕性を生み出すことができる。
ある実施形態において、圧縮錠剤は、直接圧縮によって調製され得る。いくつかの顆粒状化学物質は、湿式造粒及び乾式造粒の必要性なく、錠剤機械において直接圧縮することができる自由流動特性及び接着特性を有する。この性質を有しない化学物質については、直接圧縮によって錠剤の製造に必要な質を与える特殊医薬賦形剤が使用され得る。特定の錠剤化賦形剤には、例えば、噴霧乾燥したラクトース、アルファ一水和物ラクトースの微結晶、スクロース−転化糖−トウモロコシでんぷん混合物、微結晶性セルロース、結晶性マルトース、及びリン酸二カルシウムのような充填剤、直接圧縮でんぷん、でんぷんグリコール酸ナトリウム、架橋したカルボキシメチルセルロース繊維、及び架橋したポリビニルピロリドンのような崩壊剤、ステアリン酸(searate)マグネシウム及びタルクのような潤滑剤、ならびに焙焼した(fumed)二酸化ケイ素のような滑剤を含む。
ある実施形態において、本明細書に提供する錠剤は、鋳造によって調製され得る。鋳造した錠剤のための基剤は概して、粉体化したスクロースの一部を有するまたは有さない細かく粉体化したラクトースからなる混合物である。充填物を調製する上で、当該薬剤は、幾何学的希釈によって基剤とともに均一に混合される。当該粉体混合物は、当該粉体を湿らせるためにのみ十分な水とアルコールとからなる混合物を用いて湿潤化し得、それにより当該粉体を圧縮し得る。ラクトース/スクロース基剤の一部に及ぼす水の溶媒作用は、乾燥の際に粉体混合物の待つことをもたらす。当該アルコール部分は、乾燥加工を促進する。
ある実施形態において、本明細書に提供する医薬製剤は、シチジン類似体及び任意に1つ以上の賦形剤を含有し、「薬剤コア」を形成する。任意の賦形剤には、例えば、当該技術分野で公知のように、例えば、希釈剤(増量剤)、潤滑剤、崩壊剤、充填剤、安定化剤、界面活性剤、保存料、着色料、着香料、結合剤、賦形支持体、滑剤、浸透亢進賦形剤、可塑剤及びこれらに類するものを含む。いくつかの物質が、医薬組成物における1つを超える目的を果たすことは、当業者によって理解されるであろう。例えば、いくつかの物質は、圧縮後に互いに錠剤を保持するのを支援する結合剤であり、いったん標的送達部位に到達すると、錠剤を崩壊させるのを支援する崩壊剤でもある。賦形剤の選択及び使用する量は、当該技術分野で利用可能な標準的な手順及び参考文献の実験及び考慮を基に製剤科学者によって容易に判断され得る。
ある実施形態において、本明細書に提供する製剤は、1つ以上の結合剤を含む。結合剤は、例えば、錠剤へ粘着性を付与するために、したがって、錠剤が圧縮後に未変化のままであることを確実にするために使用され得る。適切な結合剤にはとりわけ、でんぷん(トウモロコシでんぷん及びアルファ化でんぷんを含む)、ゼラチン、糖類(スクロース、グルコース、デキストロース及びラクトースを含む)、ポリエチレングリコール、プロピレングリコール、蝋、ならびに天然ゴム及び合成ゴム、例えば、アカシアアルギン酸ナトリウム、ポリビニルピロリドン、セルロース性ポリマー(ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、カルボキシメチルセルロース及びこれらに類するものを含む)、ビーガム、カルボマー(例えば、カルボポール)、ナトリウム、デキストリン、グアーガム、水素化植物油、マグネシウムアルミニウムシリカート、マルトデキストリン、ポリメタクリラート、ポビドン(例えば、コリドン、プラスドン)、微結晶性セルロースを含むが、これらに限定しない。結合剤には、例えば、アカシア、アガー、アルギン酸、カルボマー(cabomer)、カラギーナン、酢酸フタル酸セルロース、セラトニア、キトサン、アイシング用粉糖、コポビドン、デキストラート、デキストリン、デキストロース、エチルセルロース、ゼラチン、ベヘン酸グリセリル、グアーガム、ヒドロキシエチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルでんぷん、ヒプロメロース、イヌリン、ラクトース、マグネシウムアルミニウムシリカート、マルトデキストリン、マルトース、メチルセルロース、ポロキサマー、ポリカルボフィル、ポリデキストロース、酸化ポリエチレン、ポリメチルアクリラート、ポビドン、アルギン酸ナトリウム、カルボキシメチルセルロースナトリウム、でんぷん、アルファ化でんぷん、ステアリン酸、スクロース、及びゼインも含む。当該結合剤は、適切であると判断された場合、薬剤コアに対して、当該薬剤コアの約2重量%、当該薬剤コアの約4重量%、当該薬剤コアの約6重量%、当該薬剤コアの約8重量%、当該薬剤コアの約10重量%、当該薬剤コアの約12重量%、当該薬剤コアの約14重量%、当該薬剤コアの約16重量%、当該薬剤コアの約18重量%、当該薬剤コアの約20重量%、当該薬剤コアの約22重量%、当該薬剤コアの約24重量%、当該薬剤コアの約26重量%、当該薬剤コアの約28重量%、当該薬剤コアの約30重量%、当該薬剤コアの約32重量%、当該薬剤コアの約34重量%、当該薬剤コアの約36重量%、当該薬剤コアの約38重量%、当該薬剤コアの約40重量%、当該薬剤コアの約42重量%、当該薬剤コアの約44重量%、当該薬剤コアの約46重量%、当該薬剤コアの約48重量%、当該薬剤コアの約50重量%、当該薬剤コアの約52重量%、当該薬剤コアの約54重量%、当該薬剤コアの約56重量%、当該薬剤コアの約58重量%、当該薬剤コアの約60重量%、当該薬剤コアの約62重量%、当該薬剤コアの約64重量%、当該薬剤コアの約66重量%、当該薬剤コアの約68重量%、当該薬剤コアの約70重量%、当該薬剤コアの約72重量%、当該薬剤コアの約74重量%、当該薬剤コアの約76重量%、当該薬剤コアの約78重量%、当該薬剤コアの約80重量%、当該薬剤コアの約82重量%、当該薬剤コアの約84重量%、当該薬剤コアの約86重量%、当該薬剤コアの約88重量%、当該薬剤コアの約90重量%、当該薬剤コアの約92重量%、当該薬剤コアの約94重量%、当該薬剤コアの約96重量%、当該薬剤コアの約98重量%の量、またはそれより多量であることができる。ある実施形態において、適切な量の特定の結合剤は、当業者によって判断される。
ある実施形態において、本明細書に提供する製剤は、1つ以上の希釈剤を含む。希釈剤は、例えば、実用的な大きさの錠剤が最終的に提供されるよう、かさを高めるために使用され得る。適切な希釈剤にはとりわけ、リン酸二カルシウム、硫酸カルシウム、ラクトース、セルロース、カオリン、マンニトール、塩化ナトリウム、乾燥でんぷん、微結晶性セルロース(例えば、AVICEL)、微細セルロース、アルファ化でんぷん、炭酸カルシウム、硫酸カルシウム、糖、デキストラート、デキストリン、デキストロース、二塩基性リン酸カルシウム二水和物、三塩基性リン酸カルシウム、カオリン、炭酸マグネシウム、酸化マグネシウム、マルトデキストリン、マンニトール、ポリメタクリラート(例えば、EUDRAGIT)、塩化カリウム、塩化ナトリウム、ソルビトール及びタルクを含む。希釈剤には、例えば、アルギン酸アンモニウム、炭酸カルシウム、リン酸カルシウム、硫酸カルシウム、酢酸セルロース、圧縮性糖、アイシング用粉糖、デキストラート、デキストリン、デキストロース、エリスリトール、エチルセルロース、フルクトース、フマル酸、グリセリルパルミトステアラート、イソマルト、カオリン、ラシトール(lacitol)、ラクトース、マンニトール、炭酸マグネシウム、酸化マグネシウム、マルトデキストリン、マルトース、中鎖トリグリセリド、微結晶性セルロース、微結晶性ケイ素化セルロース、動力セルロース、ポリデキストロース、ポリメチルアクリラート、シメチコン、アルギン酸ナトリウム、塩化ナトリウム、ソルビトール、でんぷん、アルファ化でんぷん、スクロース、スルホブチルエーテル−β−シクロデキストリン、タルク、トラガカント、トレハロース、及びキシリトールも含む。希釈剤は、錠剤またはカプセル剤のために所望の容積を得るよう算出された量で使用され得、ある実施形態において、希釈剤は、薬剤コアの約5重量%以上、約10重量%以上、約15重量%以上、約20重量%以上、約22重量%以上、約24重量%以上、約26重量%以上、約28重量%以上、約30重量%以上、約32重量%以上、約34重量%以上、約36重量%以上、約38重量%以上、約40重量%以上、約42重量%以上、約44重量%以上、約46重量%以上、約48重量%以上、約50重量%以上、約52重量%以上、約54重量%以上、約56重量%以上、約58重量%以上、約60重量%以上、約62重量%以上、約64重量%以上、約68重量%以上、約70重量%以上、約72重量%以上、約74重量%以上、約76重量%以上、約78重量%以上、約80重量%以上、約85重量%以上、約90重量%以上、または約95重量%以上、薬剤コアの約10重量%〜約90重量%、薬剤コアの約20重量%〜約80重量%、薬剤コアの約30重量%〜約70重量%、薬剤コアの約40重量%〜約60重量%の量で使用される。ある実施形態において、適切な量の特定の希釈剤は、当業者によって判断される。
ある実施形態において、本明細書に提供する製剤は、1つ以上の潤滑剤を含む。潤滑剤は、例えば、錠剤製造を容易にするために使用され得、適切な潤滑剤の例としては、例えば、ピーナッツ油、綿実油、ゴマ油、オリーブ油、トウモロコシ油のような植物油、ならびにテオブロマ、グリセリン、ステアリン酸マグネシウム、ステアリン酸カルシウム、及びステアリン酸の油が挙げられる。ある実施形態において、ステアラートは、存在する場合、薬剤含有コアのおよそ2重量%以下を表す。潤滑剤のさらなる例としては、例えば、ステアリン酸カルシウム、モノステアリン酸グリセリン、ベヘン酸グリセリル、パルミトステアリン酸グリセリル、ラウリル硫酸マグネシウム、ステアリン酸マグネシウム、ミリスチン酸、パルミチン酸、ポロキサマー、ポリエチレングリコール、安息香酸カリウム、安息香酸ナトリウム、塩化ナトリウム、ラウリル硫酸ナトリウム、フマル酸ステアリルナトリウム、ステアリン酸、タルク、及びステアリン酸亜鉛が挙げられる。特定の実施形態において、潤滑剤は、ステアリン酸マグネシウムである。ある実施形態において、潤滑剤は、薬剤コアに対して、薬剤コアの約0.2重量%、薬剤コアの約0.4重量%、薬剤コアの約0.6重量%、薬剤コアの約0.8重量%、薬剤コアの約1.0重量%、薬剤コアの約1.2重量%、薬剤コアの約1.4重量%、薬剤コアの約1.6重量%、薬剤コアの約1.8重量%、薬剤コアの約2.0重量%、薬剤コアの約2.2重量%、薬剤コアの約2.4重量%、薬剤コアの約2.6重量%、薬剤コアの約2.8重量%、薬剤コアの約3.0重量%、薬剤コアの約3.5重量%、薬剤コアの約4重量%、薬剤コアの約4.5重量%、薬剤コアの約5重量%、薬剤コアの約6重量%、薬剤コアの約7重量%、薬剤コアの約8重量%、薬剤コアの約10重量%、薬剤コアの約12重量%、薬剤コアの約14重量%、薬剤コアの約16重量%、薬剤コアの約18重量%、薬剤コアの約20重量%、薬剤コアの約25重量%、薬剤コアの約30重量%、薬剤コアの約35重量%、薬剤コアの約40重量%、薬剤コアの約0.2重量%〜約10重量%、薬剤コアの約0.5重量%〜約5重量%、または薬剤コアの約1重量%〜約3重量%の量で存在する。ある実施形態において、適切な量の特定の潤滑剤は、当業者によって判断される。
ある実施形態において、本明細書に提供する製剤は、1つ以上の崩壊剤を含む。崩壊剤は、例えば錠剤の崩壊を促進するために使用され得、例えば、でんぷん、クレイ、セルロース、アルギン、ガムまたは架橋したポリマーであり得る。崩壊剤には、例えば、アルギン酸、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム(例えば、AC−DI−SOL、PRIMELLOSE)、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、クロスポビドン(例えば、KOLLIDON、POLYPLASDONE)、グアーガム、マグネシウムアルミニウムシリカート、メチルセルロース、微結晶性セルロース、ポラクリンカリウム、粉状セルロース、アルファ化でんぷん、アルギン酸ナトリウム、グリコール酸でんぷんナトリウム(例えば、EXPLOTAB)及びでんぷんも含む。追加の崩壊剤には、例えば、アルギン酸カルシウム、キトサン、ドクサートナトリウム、ヒドロキシプロピルセルロース、及びポビドンを含む。ある実施形態において、崩壊剤は、薬剤コアに対して、当該薬剤コアの約1重量%、当該薬剤コアの約2重量%、当該薬剤コアの約3重量%、当該薬剤コアの約4重量%、当該薬剤コアの約5重量%、当該薬剤コアの約6重量%、当該薬剤コアの約7重量%、当該薬剤コアの約8重量%、当該薬剤コアの約9重量%、当該薬剤コアの約10重量%、当該薬剤コアの約12重量%、当該薬剤コアの約14重量%、当該薬剤コアの約16重量%、当該薬剤コアの約18重量%、当該薬剤コアの約20重量%、当該薬剤コアの約22重量%、当該薬剤コアの約24重量%、当該薬剤コアの約26重量%、当該薬剤コアの約28重量%、当該薬剤コアの約30重量%、当該薬剤コアの約32重量%、当該薬剤コアの約32重量%超、当該薬剤コアの約1重量%〜約10重量%、当該薬剤コアの約2重量%〜約8重量%、当該薬剤コアの約3重量%〜約7重量%、または当該薬剤コアの約4重量%〜約6重量%の量で存在する。ある実施形態において、適切な量の特定の崩壊剤は、当業者によって判断される。
ある実施形態において、本明細書に提供する製剤は、1つ以上の安定化剤を含む。安定化剤(吸収亢進剤ともいう)は、例えば、例として酸化反応を含む薬剤分解反応を阻害または遅延させるために使用され得る。安定化剤には、例えば、d−アルファ−トコフェリルポリエチレングリコール1000スクシナート(ビタミンE TPGS)、アカシア、アルブミン、アルギン酸、ステアリン酸アルミニウム、アルギン酸アンモニウム、アスコルビン酸、パルミチン酸アスコルビル、ベントナイト、酪酸ヒドロキシトルエン、アルギン酸カルシウム、ステアリン酸カルシウム、カルボキシメチルセルロースカルシウム、カラギーナン、セラトニア、コロイド状二酸化ケイ素、シクロデキストリン、ジエタノールアミン、エデト酸塩、エチルセルロース、パルミトステアリン酸エチレングリコール、グリセリンモノステアラート、グアーガム、ヒドロキシプロピルセルロース、ヒプロメロース、転化糖、レシチン、マグネシウムアルミニウムシリカート、モノエタノールアミン、ペクチン、ポロキサマー、ポリビニルアルコール、アルギン酸カリウム、ポラクリンカリウム、ポビドン、没食子酸プロピル、プロピレングリコール、プロピレングリコールアルギナート、ラフィノース、酢酸ナトリウム、アルギン酸ナトリウム、ホウ酸ナトリウム、カルボキシメチルセルロースナトリウム、フマル酸ステアリルナトリウム、ソルビトール、ステアリルアルコール、スルホブチル(sufobutyl)−β−シクロデキストリン、トレハロース、白蝋、キサンタンガム、キシリトール、黄蝋、及び酢酸亜鉛を含む。ある実施形態において、当該安定化剤は、薬剤コアに対して、当該薬剤コアの約1重量%、当該薬剤コアの約2重量%、当該薬剤コアの約3重量%、当該薬剤コアの約4重量%、当該薬剤コアの約5重量%、当該薬剤コアの約6重量%、当該薬剤コアの約7重量%、当該薬剤コアの約8重量%、当該薬剤コアの約9重量%、当該薬剤コアの約10重量%、当該薬剤コアの約12重量%、当該薬剤コアの約14重量%、当該薬剤コアの約16重量%、当該薬剤コアの約18重量%、当該薬剤コアの約20重量%、当該薬剤コアの約22重量%、当該薬剤コアの約24重量%、当該薬剤コアの約26重量%、当該薬剤コアの約28重量%、当該薬剤コアの約30重量%、当該薬剤コアの約32重量%、当該薬剤コアの約1重量%〜約10重量%、当該薬剤コアの約2重量%〜約8重量%、当該薬剤コアの約3重量%〜約7重量%、または当該薬剤コアの約4重量%〜約6重量%の量で存在する。ある実施形態において、適切な量の特定の安定化剤は、当業者によって判断される。
ある実施形態において、本明細書に提供する製剤は、1つ以上の滑剤を含む。滑剤は、例えば、粉体組成物若しくは顆粒の流動特性を改善するために、または投薬の正確性を改善するために使用され得る。滑剤として機能し得る賦形剤には、例えば、コロイド状二酸化ケイ素、三ケイ酸マグネシウム、粉状セルロース、でんぷん、三塩基性リン酸カルシウム、ケイ酸カルシウム、粉状セルロース、コロイド状二酸化ケイ素、ケイ酸マグネシウム、三ケイ酸マグネシウム、二酸化ケイ素、でんぷん、三塩基性リン酸カルシウム、及びタルクを含む。ある実施形態において、当該滑剤は、当該薬剤コアに対して、当該薬剤コアの約1重量%未満、当該薬剤コアの約1重量%、当該薬剤コアの約2重量%、当該薬剤コアの約3重量%、当該薬剤コアの約4重量%、当該薬剤コアの約5重量%、当該薬剤コアの約6重量%、当該薬剤コアの約7重量%、当該薬剤コアの約8重量%、当該薬剤コアの約9重量%、当該薬剤コアの約10重量%、当該薬剤コアの約12重量%、当該薬剤コアの約14重量%、当該薬剤コアの約16重量%、当該薬剤コアの約18重量%、当該薬剤コアの約20重量%、当該薬剤コアの約22重量%、当該薬剤コアの約24重量%、当該薬剤コアの約26重量%、当該薬剤コアの約28重量%、当該薬剤コアの約30重量%、当該薬剤コアの約32重量%、当該薬剤コアの約1重量%〜約10重量%、当該薬剤コアの約2重量%〜約8重量%、当該薬剤コアの約3重量%〜約7重量%、または当該薬剤コアの約4重量%〜約6重量%の量で存在する。ある実施形態において、適切な量の特定の滑剤は、当業者によって判断される。
ある実施形態において、本明細書に提供する製剤は、1つ以上の浸透亢進剤(例えば、透過性亢進剤ともいう)を含む。ある実施形態において、浸透亢進剤は、消化管壁(例えば、胃)を通じてのシチジン類似体の取り込みを亢進させる。ある実施形態において、浸透亢進剤は、血流に入るシチジン類似体の速度及び/または量を変化させる。特定の実施形態において、d−アルファ−トコフェリルポリエチレングリコール−1000スクシナート(ビタミンE TPGS)が浸透亢進剤として使用される。特定の実施形態において、例えば、当該技術分野で公知の任意の浸透亢進剤を含む1つ以上の他の適切な浸透亢進剤が使用される。適切な浸透亢進剤の具体的な例としては、例えば、以下に列挙するものが挙げられる。
他の有望な浸透亢進剤には、例えば、アルコール、ジメチルスルホキシド、グリセリルモノオレアート、グリコフロール、イソプロピルミリスタート、イソプロピルパルミタート、ラノリン、リノール酸、ミリスチン酸、オレイン酸、オレイルアルコール、パルミチン酸、ポリオキシエチレンアルキルエーテル、2−ピロリドン、ラウリル硫酸ナトリウム、及びチモールを含む。
ある実施形態において、浸透亢進剤は、当該製剤の総重量に対する、約0.1%、約0.2%、約0.3%、約0.4%、約0.5%、約0.6%、約0.7%、約0.8%、約0.9%、約1%、約1.1%、約1.2%、約1.3%、約1.4%、約1.5%、約1.6%、約1.7%、約1.8%、約1.9%、約2%、約2.1%、約2.2%、約2.3%、約2.4%、約2.5%、約2.6%、約2.7%、約2.8%、約2.9%、約3%、約3.1%、約3.2%、約3.3%、約3.4%、約3.5%、約3.6%、約3.7%、約3.8%、約3.9%、約4%、約4.1%、約4.2%、約4.3%、約4.4%、約4.5%、約4.6%、約4.7%、約4.8%、約4.9%、約5%、約5.1%、約5.2%、約5.3%、約5.4%、約5.5%、約5.6%、約5.7%、約5.8%、約5.9%、約6%、約6.1%、約6.2%、約6.3%、約6.4%、約6.5%、約6.6%、約6.7%、約6.8%、約6.9%、約7%、約7.1%、約7.2%、約7.3%、約7.4%、約7.5%、約7.6%、約7.7%、約7.8%、約7.9%、約8%、約8.1%、約8.2%、約8.3%、約8.4%、約8.5%、約8.6%、約8.7%、約8.8%、約8.9%、約9%、約9.1%、約9.2%、約9.3%、約9.4%、約9.5%、約9.6%、約9.7%、約9.8%、約9.9%、約10%、約10%超、約12%超、約14%超、約16%超、約18%超、約20%超、約25%超、約30%超、約35%超、約40%超、約45%超、または約50%超の重量による量で当該製剤中に存在する。ある実施形態において、適切な量の本明細書に提供する適切な浸透亢進剤は、当業者によって判断される。
任意の特定の理論に制限されるよう企図することなく、本明細書に提供する浸透亢進剤は、とりわけ、消化管壁を通じてのシチジン類似体の輸送を促進すること(例えば、当該輸送の速度または程度を高めること)によって機能し得る。概して、消化管壁を通じての移動は、例えば、単に濃度勾配によって駆動される様式で膜を貫通する薬剤の移動のような受動拡散、細胞膜中に植え込まれた特化した輸送系を介して細胞膜を貫通する薬剤の移動のような担体仲介性拡散、2つの細胞を通過するよりもむしろ当該細胞間を通過することによって膜を貫通する薬剤の移動のような傍細胞拡散、及び細胞を貫通する薬剤の移動のような細胞貫通拡散によって生じ得る。追加的に、細胞に入る薬剤を汲み出すことによって、薬剤の細胞内蓄積を防止することのできる数多くの細胞タンパク質がある。これらは時として排出ポンプと呼ばれる。あるこのような排出ポンプは、体内の多くの異なる組織(例えば、腸、胎盤膜、血液脳関門)中に存在するp−糖タンパク質を包含するものである。浸透亢進剤は、とりわけ、上記の過程のうちのいずれかを促進することによって(例えば、とりわけ、膜の流動性を高める、細胞間密着結合を開く、及び/または流出を阻害することによって)機能することができる。
ある実施形態において、シチジン類似体、例えば、5−アザシチジンを含む本明細書に提供する組成物は、シチジンデアミナーゼ阻害薬を本質的に含んでいない(例えば、シチジンデアミナーゼ阻害薬を含まない)。ある実施形態において、本明細書に提供する組成物は、シチジンデアミナーゼ阻害薬であるテトラヒドロウリジン(THU)を本質的に含んでいない(例えば、含まない)。本明細書のある実施形態は、治療有効量のシチジン類似体(例えば、5−アザシチジン)を含む医薬組成物を提供し、この中で、当該組成物は、対象への経口投与後に実質的に胃の中でシチジン類似体を放出し、当該組成物は、シチジンデアミナーゼ阻害薬(例えば、THU)を本質的に含んでいない(例えば、含まない)。本明細書のある実施形態は、治療有効量のシチジン類似体(例えば、5−アザシチジン)を含む医薬組成物を提供し、この中で、当該組成物は、対象への経口投与後に実質的に胃の中でシチジン類似体を放出し、当該組成物は、シチジンデアミナーゼ阻害薬(例えば、THU)を本質的に含んでおらず(例えば、含まず)、及び当該組成物は、本明細書に提供する特定の生物学的パラメータ(例えば、本明細書に提供する特定のCmax値、Tmax値、及び/またはAUC値)に到達する。特定の実施形態において、シチジンデアミナーゼ阻害薬(例えば、THU)を本質的に含んでいない本明細書に提供する組成物は、例えば、200mg未満、150mg未満、100mg未満、50mg未満、25mg未満、10mg未満、5mg未満、1mg未満、または0.1mg未満のシチジンデアミナーゼ阻害薬を含む。
(4.追加の治療薬)
特定の実施形態において、本明細書に提供するシチジン類似体経口製剤はさらに、1、2、3、またはより多数の他の薬理学的活性物質(本明細書では「追加の治療薬」、「第二の作用因子」、またはこれらに類するものともいう)を含む。特定の実施形態において、本明細書に提供する経口製剤は、追加の治療薬(複数可)を治療有効量で含む。特定の実施形態において、シチジン類似体(例えば、5−アザシチジン)及び追加の治療薬(複数可)は、本明細書に開示する方法及び当該技術分野で公知の方法を含む有効医薬成分の合剤方法を用いて同じ剤形中にまとめて合剤される。他の実施形態において、シチジン類似体及び追加の治療薬(複数可)は、別個の剤形において併用投与される。ある組み合わせは、例えば、望ましくない血管新生または異常な細胞増殖と関係したまたはこれらを特徴とするタイプの癌ならびにある疾患及び障害を含む特定の疾患または障害の治療において相乗的に作用すると考えられている。本明細書に提供するシチジン類似体経口剤形は、ある第二の作用因子と関係した有害作用を緩和するよう作用することもでき、いくつかの第二の作用因子は、本明細書に提供するシチジン類似体経口剤形と関係した有害作用を緩和するために使用することができる。ある実施形態において、本明細書に提供する経口製剤は、再感作効果を必要とする対象における再感作効果を提供するために1つ以上の治療薬と併用投与される。追加の治療薬は、例えば、大分子(例えば、タンパク質)または小分子(例えば、合成無機分子、有機金属分子、または有機分子)であることができる。特定の実施形態において、当該1つ以上の追加の治療薬(複数可)には、ペンブロリズマブ及びMEDI4736(デュルバルマブ)のような抗PD1/抗PDL1モノクローナル抗体を含むがこれらに限定しない。
本明細書に開示する組成物及び方法において有用な特定の追加の治療薬の例としては、例えば、細胞毒性薬、代謝拮抗薬、葉酸代謝拮抗薬、HDAC阻害薬(例えば、SNDX−275若しくはMS−275としても公知のエンチノスタット、またはスベロイルアニリドヒドロキサム酸(SAHA)若しくはN−ヒドロキシ−N’−フェニル−オクタンジアミドとしても公知のボリノスタット)、DNA挿入剤、DNA架橋剤、DNAアルキル化剤、DNA切断剤、トポイソメラーゼ阻害薬、CDK阻害薬、JAK阻害薬、抗血管新生薬、Bcr−Abl阻害薬、HER2阻害薬、EGFR阻害薬、VEGFR阻害薬、PDGFR阻害薬、HGFR阻害薬、IGFR阻害薬、c−Kit阻害薬、Ras経路阻害薬、PI3K阻害薬、多重標的化キナーゼ阻害薬、mTOR阻害薬、抗エストロゲン薬、抗アンドロゲン薬、アロマターゼ阻害薬、ソマトスタチン類似体、ER調節薬、抗チューブリン薬、ビンカアルカロイド、タキサン、HSP阻害薬、平滑化アンタゴニスト、テロメラーゼ阻害薬、COX−2阻害薬、静止期阻害薬、免疫抑制剤、抗体のような生物製剤、ならびにホルモン療法が挙げられるが、これらに限定しない。特定の実施形態において、併用投与する治療薬は、免疫調節化合物、例えば、サリドマイド、レナリドマイド、またはポマリドマイドである。当該併用投与薬は、例えば、経口でまたは注射によって投与され得る。
追加の治療薬に関する他の例としては、造血性増殖因子、サイトカイン、抗癌薬、顆粒球コロニー刺激因子(G−CSF)、顆粒球−マクロファージコロニー刺激因子(GM−CSF)、エリスロポエチン(EPO)、インターロイキン(IL)、インターフェロン(IFN)、オブリメルセン、メルファラン、トポテカン、ペントキシフィリン、タキソテレ、イリノテカン、シプロフロキサシン、ドキソルビシン、ビンクリスチン、ダカルバジン、Ara−C、ビノレルビン、プレドニゾン、シクロホスファミド、ボルテゾミブ、三酸化ヒ素が挙げられるが、これらに限定しない。このような追加の治療薬は、多発性骨髄腫の治療と関連するものを含むがこれに限定しない、本明細書に開示する方法及び組成物において特に有用である。
追加の治療薬に関する他の例としては、抗体(例えば、リツキシマブ、抗CD33)、造血性増殖因子、サイトカイン、抗癌薬、抗生物質、cox−2阻害薬、免疫調節薬、免疫抑制薬、コルチコステロイド、またはこれらの薬理学邸に活性のある突然変異体若しくは誘導体が挙げられるが、これらに限定しない。例えば、S. Nand et al.、Leukemia and Lymphoma,2008,49(11):2141〜2147を参照されたい(AML及び高リスクのMDSに罹患している高齢患者へのヒドロキシ尿素と低用量ゲムツズマブオゾガマイシンとの組み合わせの投与を包含する第II相試験を説明し、この組み合わせがこの群の患者におけるAML及び高リスクのMDSの治療における安全かつ有効な投与計画のようであると結論付けている)。このような追加の治療薬は、本明細書に開示する疾患及び障害の治療と関連するものを含むがこれに限定しない本明細書に開示する方法及び組成物において特に有用である。
大分子作用薬の例としては、造血性増殖因子、サイトカイン、ならびにモノクローナル抗体及びポリクローナル抗体が挙げられるが、これらに限定しない。典型的な大分子作用薬は、天然のまたは人工的に生成されたタンパク質のような生体分子である。特に有用であるタンパク質には、造血性前駆細胞及び免疫学的に活性のある造血細胞の生存及び/または増殖をインビトロまたはインビボで刺激するタンパク質を含む。その他は、細胞における関与する赤血球系前駆細胞の分割及び分化をインビトロまたはインビボで刺激する。特定のタンパク質には、IL−2(組換えIL−II(「rIL2」)及びカナリア痘IL−2を含む)、IL−10、IL−12、及びIL−18のようなインターロイキン、インターフェロンアルファ−2a、インターフェロンアルファ−2b、インターフェロンアルファ−n1、インターフェロンアルファ−n3、インターフェロンβ−Ia、及びインターフェロンガンマ−Ibのようなインターフェロン、GM−CF及びGM−CSF、ならびにEPOを含むがこれらに限定しない。
本明細書に提供する方法及び組成物において使用することのできる特定のタンパク質には、商標名Neupogen(登録商標)(Amgen,カリフォルニア州サウザンドオークス市)の下で、米国で販売されているフィルグラスチム、商標名Leukine(登録商標)(Immunex,ワシントン州シアトル市)の下で、米国で販売されているサルグラモスチム、及び商標名Epogen(登録商標)(Amgen,カリフォルニア州サウザンドオークス市)の下で、米国で販売されている組換えEPOを含むがこれらに限定しない。
組換え型及び突然変異型のGM−CSFは、それらのすべてが参照により本明細書に組み込まれる米国特許第5,391,485号、第5,393,870号、及び第5,229,496号において説明される通り調製することができる。組換え型及び突然変異型のG−CSFは、それらのすべてが参照により本明細書に組み込まれる米国特許第4,810,643号、第4,999,291号、第5,528,823号、及び第5,580,755号において説明される通り調製することができる。
本明細書の実施形態は、未処置のタンパク質、天然タンパク質、及び組換えタンパク質の使用を包含する。特定の実施形態は、天然タンパク質の突然変異体及び誘導体(例えば、修飾された形態)が、元となるタンパク質の薬理学的活性の少なくとも一部をインビボで呈する当該タンパク質の突然変異体及び誘導体を包含する。突然変異体の例としては、天然形態のタンパク質における対応する残基とは異なる1つ以上のアミノ酸残基を有するタンパク質が挙げられるが、これに限定しない。また「突然変異体」という用語によって包含されるのは、天然形態(例えば、非グリコシル化形態)で通常存在する炭水化物部分を欠失しているタンパク質である。誘導体の例としては、ペグ化された誘導体及び、IgG1またはIgG3を関心対象のタンパク質または当該タンパク質の活性のある部分へ融合することによって形成されるタンパク質のような融合タンパク質が挙げられるが、これらに限定しない。例えば、Penichet,M.L.and Morrison,S.L.,J.Immunol.Methods 248:91〜101(2001)を参照されたい。
本明細書に開示する経口製剤と併用することのできる抗体には、モノクローナル抗体及びポリクローナル抗体を含む。抗体の例としては、トラスツズマブ(Herceptin(登録商標))、リツキシマブ(Rituxan(登録商標))、ベバシズマブ(Avastin(商標))、ペルツズマブ(Omnitarg(商標))、トシツモマブ(Bexxar(登録商標))、エドレコロマブ(Panorex(登録商標))、及びG250が挙げられるが、これらに限定しない。本明細書に開示する経口製剤はまた、抗TNF−α抗体を含むことができ、当該抗体と組み合わせることができ、または当該抗体と併用することができる。好ましい実施形態において、当該抗体は、ペンブロリズマブ及びMEDI4736(デュルバルマブ)のような抗PD1/抗PDL1モノクローナル抗体である。
大分子作用薬は、抗癌ワクチンの形態で投与され得る。例えば、IL−2、G−CSF、及びGM−CSFのようなサイトカインを分泌するまたは当該サイトカインの分泌を生じるワクチンは、本明細書に提供する方法、医薬組成物、及びキットにおいて使用することができる。例えば、Emens,L.A.,et al.,Curr.Opinion Mol.Ther.3(1):77〜84(2001)を参照されたい。
一実施形態において、追加の治療薬(例えば、大分子化合物または小分子化合物)は、本明細書に提供するシチジン類似体の投与(例えば、経口投与)と関係する有害作用を低下、除去、または防止する。特定のシチジン類似体及び疾患または障害に依存することが治療され始め、有害作用には、特定のシチジン類似体と関係することになっている当該技術分野で公知のもののうちでとりわけ、貧血、好中球減少症、発熱性好中球減少症、血小板減少症、肝毒性(例えば、既往症の肝障害に罹患している患者における肝毒性を含むがこれに限定しない)、高血清クレアチン、腎不全、腎尿細管性アシドーシス、低カリウム血症、肝性昏睡、吐き気、嘔吐、消化不良、腹痛、発熱、白血球減少、下痢、便秘、斑状出血、点状出血、固縮、脱力、肺炎、不安、不眠症、嗜眠、及び体重減少を含むことができるが、これらに限定しない。
一部の大分子のように、多くの小分子化合物は、本明細書に開示するシチジン類似体経口製剤と共に投与した場合(例えば、前に、後にまたは同時に)、相乗効果を提供することができると考えられている。小分子の第二の作用薬の例としては、抗癌薬、抗生物質、免疫抑制薬、及びステロイドが挙げられるが、これらに限定しない。
抗癌薬の例としては、アシビシン、アクラルビシン、アコダゾールヒドロクロリド、アクロニン、アドゼレシン、アルデスロイキン、アルトレタミン、アンボマイシン、酢酸アメタントロン、アムサクリン、アナストロゾール、アントラマイシン、アスパラギナーゼ、アスペルリン、アザシチジン、アゼテパ、アゾトマイシン、バチマスタット、ベンゾデパ、ビカルタミド、ビサントレンヒドロクロリド、ビスナフィドジメシラート、ビゼレシン、硫酸ブレオマイシン、ブレキナルナトリウム、ブロピリミン、ブスルファン、カクチノマイシン、カルステロン、カラセミド、カルベチメル(carbetimer)、カルボプラチン、カルムスチン、カルビシンヒドロクロリド、カルゼレシン、セデフィンゴール、セレコキシブ(COX−2阻害薬)、クロラムブシル、シロレマイシン(cirolemycin)、シスプラチン、クラドリビン、クリスナトールメシラート、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシンヒドロクロリド、デシタビン、デキソルマプラチン(dexormaplatin)、デザグアニン、デザグアニンメシラート、ジアジクオン、ドセタキセル、ドキソルビシン、ドキソルビシンヒドロクロリド、ドロロキシフェン、ドロロキシフェンシトラート、ドロモスタノロンプロピオナート、デュアゾマイシン、エダトレキサート、エフロルニチンヒドロクロリド、エルサミトルシン、エンロプラチン、エンプロマート、エピプロピジン、エピルビシンヒドロクロリド、エルブロゾール、エソルビシンヒドロクロリド、エストラムスチン、エストラムスチンリン酸ナトリウム、エタニダゾール、エトポシド、リン酸エトポシド、エトプリン、塩酸ファドロゾール、ファザラビン、フェンレチニド、フロクスウリジン、リン酸フルダラビン、フルオロウラシル、フルオロシタビン、ホスキドン、ホストリエシンナトリウム、ゲムシタビン、ゲムシタビンヒドロクロリド、ヒドロキシ尿素、イダルビシンヒドロクロリド、イフォスファミド、イルモフォシン、イプロプラチン、イリノテカン、イリノテカンヒドロクロリド、酢酸ランレオチド、レトロゾール、酢酸ロイプロリド、リアロゾールヒドロクロリド、ロメトレキソールナトリウム、ロムスチン、ロソキサントロンヒドロクロリド、マソプロコール、マイタンシン、メクロレタミンヒドロクロリド、酢酸メゲストロール、酢酸メレンゲストロール、メルファラン、メノガリル、メルカプトプリン、メトトレキサート、メトトレキサートナトリウム、メトプリン、メツレデパ、ミチンドミド、ミトカルシン、ミトクロミン、ミトギリン、ミトマルシン、ミトマイシン、ミトスペル、ミトタン、ミトキサントロンヒドロクロリド、ミコフェノール酸、ノコダゾール、ノガラマイシン、オルマプラチン、オキシスラン、パクリタキセル、ペグアスパルガーゼ、ペリオマイシン、ペンタムスチン、ペプロマイシンスルファート、ペルホスファミド、ピポブロマン、ピポスルファン、ピロキサントロンヒドロクロリド、プリカマイシン、プロメスタン、ポルフィメルナトリウム、ポルフィロマイシン、プレドニムスチン、プロカルバジンヒドロクロリド、ピューロマイシン、ピューロマイシンヒドロクロリド、ピラゾフリン、リボプリン、サフィンゴール、サフィンゴールヒドロクロリド、セムスチン、シムトラゼン、スパルフォサートナトリウム、スパルソマイシン、スピロゲルマニウムヒドロクロリド、スピロムスチン、スピロプラチン、ストレプトニグリン、ストレプトゾシン、スロフェヌル、タリソマイシン、テコガランナトリウム、タキソテレ、テガフル、テロキサントロンヒドロクロリド、テモポルフィン、テニポシド、テロキシロン、テストラクトン、チアミプリン、チオグアニン、チオテパ、チアゾフリン、チラパザミン、トレミフェンシトラート、酢酸トレストロン、リン酸トリシリビン、トリメトレキサート、トリメトレキサートグルクロナート、トリプトレリン、ツブロゾールヒドロクロリド、ウラシルマスタード、ウレデパ、バプレオチド、ベルテポルフィン、ビンブラスチンスルファート、ビンクリスチンスルファート、ビンデシン、ビンデシンスルファート、ビネピジンスルファート、ビングリシナートスルファート、ビンロイロシンスルファート、ビノレルビンタートラート、ビンロシジンスルファート、ビンゾリジンスルファート、ボロゾール、ゼニプラチン、ジノスタチン、及びゾルビシンヒドロクロリドが挙げられるが、これらに限定しない。
他の抗癌薬には、20−エピ−1,25ジヒドロキシビタミンD3、5−エチニルウラシル、アビラテロン、アクラルビシン、アシルフルベン、アデシペノール、アドゼレシン、アルデスロイキン、ALL−TKアンタゴニスト、アルトレタミン、アンバムスチン、アミドックス、アミホスチン、アミノレブリン酸、アムルビシン、アムサクリン、アナグレリド、アナストロゾール、アンドログラホリド、血管新生阻害薬、アンタゴニストD、アンタゴニストG、アンタレリクス、抗背側化形成タンパク質1、抗アンドロゲン薬、前立腺癌、抗エストロゲン薬、アンチネオプラストン、アンチセンスオリゴヌクレオチド、アフィジコリングリシナート、アポトーシス遺伝子調節薬、アポトーシス調節薬、アプリン酸、ara−CDP−DL−PTBA、アルギニンデアミナーゼ、アスラクリン、アタメスタン、アトリムスチン、アクシナスタチン1、アクシナスタチン2、アクシナスタチン3、アザセトロン、アザトキシン、アザチロシン、バッカチンIII誘導体、バラノール、バチマスタット、BCR/ABLアンタゴニスト、ベンゾクロリン、ベンゾイルスタウロスポリン、ベータラクタム誘導体、ベータ−アレチン、ベタクラマイシンB、ベツリン酸、bFGF阻害薬、ビカルタミド、ビサントレン、ビサジリジニルスペルミン、ビスナフィド、ビストラテンA、ビゼレシン、ブレフラート、ブロピリミン、ブドチタン、ブチオニンスルホキシミン、カルシポトリオール、カルホスチンC、カンプトテシン誘導体、カペシタビン、カルボキサミド−アミノ−トリアゾール、カルボキシアミドトリアゾール、CaRestM3、CARN700、軟骨由来阻害薬、カルゼレシン、カゼインキナーゼ阻害薬(ICOS)、カスタノスペルミン、セクロピンB、セトロレリクス、クロリン(chlorlns)、クロロキノキサリンスルホンアミド、シカプロスト、シス−ポルフィリン、クラドリビン、クロミフェン類似体、クロトリマゾール、コリスマイシンA、コリスマイシンB、コンブレタスタチンA4、コンブレタスタチン類似体、コナゲニン、クランベシジン816、クリスナトール、クリプトフィシン、クリプトフィシンA誘導体、クラシンA、シクロペンタントラキノン、シクロプラタム、シペマイシン、シタラビンオクホスファート、細胞溶解因子、シトスタチン、ダクリキシマブ、デシタビン、デヒドロジデムニンB、デスロレリン、デキサメタゾン、デキシホスファミド、デクスラゾキサン、デクスベラパミル、ジアジクオン、ジデムニンB、ジドックス、ジエチルノルスペルミン、ジヒドロ−5−アザシチジン、ジヒドロタキソール、9−、ジオキサマイシン、ジフェニルスピロムスチン、ドセタキセル、ドコサノール、ドラセトロン、ドキシフルリジン、ドキソルビシン、ドロロキシフェン、ドロナビノール、デュオカルマイシンSA、エブセレン、エコムスチン、エデルホシン、エドレコロマブ、エフロルニチン、エレメン、エミテフル、エピルビシン、エプリステリド、エストラムスチン類似体、エストロゲンアゴニスト、エストロゲンアンタゴニスト、エタニダゾール、リン酸エトポシド、エキセメスタン、ファドロゾール、ファザラビン、フェンレチニド、フィルグラスチム、フィナステリド、フラボピリドール、フレゼラスチン、フルアステロン、フルダラビン、フルオロダウノルビシンヒドロクロリド、フォルフェニメクス、フォルメスタン、フォストリエシン、フォテムスチン、ガドリニウムテキサフィリン、硝酸ガリウム、ガロシタビン、ガニレリクス、ゼラチナーゼ阻害薬、ゲムシタビン、グルタチオン阻害薬、ヘプスルファム、ヘレグリン、ヘキサメチレンビスアセトアミド、ハイペリシン、イバンドロン酸、イダルビシン、イドキシフェン、イドラマントン、イルモフォシン、イロマスタット、イマチニブ(例えば、Gleevec(登録商標))、イミキモド、免疫刺激ペプチド、インスリン様増殖因子1受容体阻害薬、インターフェロンアゴニスト、インターフェロン、インターロイキン、ヨーベングアン、ヨードドキソルビシン、イポメアノール、4−、イロプラクト、イルソグラジン、イソベンガゾール、イソホモハリコンドリンB、イタセトロン、ジャスプラキノリド、カハラリドF、ラメラリン−Nトリアセタート、ランレオチド、レイナマイシン、レノグラスチム、レンチナンスルファート、レプトルスタチン、レトロゾール、白血病抑制因子、白血球アルファインターフェロン、ロイプロリド+エストロゲン+プロゲステロン、ロイプロレリン、レバミソール、リアロゾール、直鎖ポリアミン類似体、親油性二糖ペプチド、親油性白金化合物、リッソクリナミド7、ロバプラチン、ロンブリシン、ロメトレキソール、ロニダミン、ロソキサントロン、ロソキソリビン、ルルトテカン、ルテチウムテキサフィリン、リソフィリン、溶解性ペプチド、マイタンシン、マンノスタチンA、マリマスタット、マソプロコール、マスピン、マトリリジン阻害薬、マトリックスメタロプロテイナーゼ阻害薬、メノガリル、メルバロン、メテレリン、メチオニナーゼ、メトクロプラミド、MIF阻害薬、ミフェプリストン、ミルテホシン、ミリモスチム、ミトグアゾン、ミトラクトール、ミトマイシン類似体、ミトナフィド、ミトトキシン線維芽細胞増殖因子−サポリン、ミトキサントロン、モファロテン、モルグラモスチム、アービタックス、ヒト絨毛性性腺刺激ホルモン、モノホスホリルリピドA+ミコバクテリウム(myobacterium)細胞壁ストレプトキナーゼ(sk)、モピダモール、マスタード抗癌薬、ミカペルオキシドB、ミコバクテリウム細胞壁抽出物、ミリアポロン、N−アセチルジナリン、N置換ベンズアミド、ナファレリン、ナグレスチプ、ナロキソン+ペンタゾシン、ナパビン、ナフテルピン、ナルトグラスチム、ネダプラチン、ネモルビシン、ネリドロン酸、ニルタミド、ニサマイシン、一酸化窒素調節因子、一酸化窒素抗酸化剤、ニトルリン、オブリメルセン(Genasense(登録商標))、O6−ベンジルグアニン、オクトレオチド、オキセノン、オリゴヌクレオチド、オナプリストン、オンダンセトロン、オンダンセトロン、オラシン、口腔サイトカイン誘導因子、オルマプラチン、オサテロン、オキサリプラチン、オキサウノマイシン、パクリタキセル、パクリタキセル類似体、パクリタキセル誘導体、パラウアミン、パルミトイルリゾキシン、パミドロン酸、パナキシトリオール、パノミフェン、パラバクチン、パゼリプチン、ペグアスパルガーゼ、ペルデシン、ペントサンポリスルファートナトリウム、ペントスタチン、ペントロゾール、ペルフルブロン、ペルホスファミド、ペリリルアルコール、フェナジノマイシン、酢酸フェニル、ホスファターゼ阻害薬、ピシバニル、ピロカルピンヒドロクロリド、ピラルビシン、ピリトレキシム、プラセチンA、プラセチンB、プラスミノーゲン活性化因子阻害薬、白金錯体、白金化合物、白金−トリアミン錯体、ポルフィルナーナトリウム、ポルフィマーナトリウム、ポルフィロマイシン、プレドニゾン、プロピルビス−アクリドン、プロスタグランジンJ2、プロテアソーム阻害薬、プロテインA系免疫調節薬、プロテインキナーゼC阻害薬、プロテインキナーゼC阻害薬、微細藻類、プロテインチロシンホスファターゼ阻害薬、プリンヌクレオシドホスホリラーゼ阻害薬、プルプリン、ピラゾロアクリジン、ピリドキシル化ヘモグロビンポリオキシエチレン共役体、rafアンタゴニスト、ラルチトレキセド、ラモセトロン、rasファルネシルタンパク質転移酵素阻害薬、ras阻害薬、ras−GAP阻害薬、脱メチル化レテリプチン、レニウムRe186エチドロナート、リゾキシン、リボザイム、RIIレチナミド、ロヒツキン、ロムルチド、ロキニメクス、ルビギノンB1、ルボキシル、サフィンゴール、サイントピン、SarCNU、サルコフィトールA、サルグラモスチム、Sdi1模倣薬、セムスチン、老化由来阻害因子1、センスオリゴヌクレオチド、シグナル伝達阻害因子、シゾフィラン、ソブゾキサン、ボロカプタートナトリウム、フェニル酢酸ナトリウム、ソルベロール、ソマトメジン結合タンパク質、ソネルミン、スパルホス酸、スピカマイシンD、スピロムスチン、スプレノペンチン、スポンギスタチン1、スクアラミン、スチピアミド、ストロメリシン阻害薬、スルフィノシン、超作用性血管作動性腸ペプチドアンタゴニスト、スラジスタ、スラミン、スワインソニン、タリムスチン、タモキシフェンメチオヂド、タウロムスチン、タザロテン、テコガランナトリウム、テガフル、テルラピリリウム、テロメラーゼ阻害薬、テモポルフィン、テニポシド、テトラクロロデカオキシド、テトラゾミン、サリブラスチン、チオコラリン、トロンボポエチン、トロンボポエチン模倣薬、チマルファシン、チモポエチン受容体アゴニスト、チモトリナン、甲状腺刺激ホルモン、エチルエチオプルプリンすず、チラパザミン、二塩化チタノセン、トプセンチン、トレミフェン、翻訳阻害薬、トレチノイン、トリアセチルウリジン、トリシリビン、トリメトレキサート、トリプトレリン、トロピセトロン、ツロステリド、チロシンキナーゼ阻害薬、チルホスチン、UBC阻害薬、ウベニメクス、泌尿生殖洞由来増殖阻害因子、ウロキナーゼ受容体アンタゴニスト、バプレオチド、バリオリンB、ベラレソール、ベラミン、ベルジン、ベルテポルフィン、ビノレルビン、ビンキサルチン、ビタキシン、ボロゾール、ザノテロン、ゼニプラチン、ジラスコルブ、及びジノスタチンスチマラマーを含むが、これらに限定されない。
具体的な追加の治療薬には、オブリメルセン(Genasense(登録商標))、レミカド、ドセタキセル、セレコキシブ、メルファラン、デキサメタゾン(Decadron(登録商標))、ステロイド、ゲムシタビン、シスプラチン(cisplatinum)、テモゾロミド、エトポシド、シクロホスファミド、テモダール、カルボプラチン、プロカルバジン、グリアデル、タモキシフェン、トポテカン、メトトレキサート、Arisa(登録商標)、タキソール、タキソテレ、フルオロウラシル、ロイコボリン、イリノテカン、キセロダ、CPT−11、インターフェロンアルファ、ペグ化インターフェロンアルファ(例えば、PEG INTRON−A)、カペシタビン、シスプラチン、チオテパ、フルダラビン、カルボプラチン、リポソームダウノルビシン、シタラビン、ドキセタキソール、パクリタキセル、ビンブラスチン、IL−2、GM−CSF、ダカルバジン、ビノレルビン、ゾレドロン酸、パルミトロナート、ビアキシン、ブスルファン、プレドニゾン、ビスホスホナート、三酸化ヒ素、ビンクリスチン、ドキソルビシン(Doxil(登録商標))、パクリタキセル、ガンシクロビル、アドリアマイシン、エストラムスチンリン酸ナトリウム(Emcyt(登録商標))、スリンダク、及びエトポシドを含むが、これらに限定しない。
(D.使用方法)
5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物と、本明細書に提供する抗PD1または抗PDL1モノクローナル抗体を含む1つ以上の追加の治療薬(複数可)との組み合わせは、本明細書に提供するような方法すべてにおいて使用することができる。特に、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物と、本明細書に提供する抗PD1または抗PDL1モノクローナル抗体を含む1つ以上の追加の治療薬(複数可)との組み合わせは、本明細書に提供するすべての疾患障害、または容態の治療、予防または改善において使用することができる。
一実施形態において、疾患または障害に罹患している対象の治療方法であって、当該対象へ治療有効量のシチジン類似体(例えば、5−アザシチジン)、またはその医薬として許容され得る塩、溶媒和物若しくは水和物と抗PD1/抗PDL1モノクローナル抗体を含む治療有効量の1つ以上の治療薬(複数可)とを周期的に投与することを含み、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は経口投与される、当該方法。
一実施形態において、当該疾患または障害は、固形腫瘍である。
一実施形態において、当該疾患または障害は、血液学的障害である。
一実施形態において、当該疾患または障害は、骨髄異形成症候群、急性骨髄性白血病、卵巣癌、または非小細胞肺癌である。
一実施形態において、当該疾患または障害は、再発性または難治性である。
一実施形態において、疾患または障害に罹患している対象は、先行治療に応答しなかった。
一実施形態において、当該先行治療は、注射可能な低メチル化薬を含む。
一実施形態において、当該先行治療は、白金を基にした治療計画を含む。
一実施形態において、当該卵巣癌は、上皮性卵巣癌である。
一実施形態において、当該上皮性卵巣癌は、再発性上皮性卵巣癌である。
一実施形態において、当該再発性または難治性上皮性卵巣癌は、白金を基にした治療計画を含む先行治療に次いで生じた。
一実施形態において、当該再発性または難治性非小細胞肺癌は、白金を基にした治療計画を含む先行治療に次いで生じた。
一実施形態において、当該再発性または難治性上皮性卵巣癌は、注射可能な低メチル化薬を含む先行治療に次いで生じた。
一実施形態において、当該再発性または難治性非小細胞肺癌は、注射可能な低メチル化薬を含む先行治療に次いで生じた。
一実施形態において、当該再発性または難治性骨髄異形成症候群は、注射可能な低メチル化薬を含む先行治療に次いで生じた。
一実施形態において、再発性または難治性急性骨髄性白血病は、注射可能な低メチル化薬を含む先行治療に次いで生じた。
一実施形態において、当該再発性または難治性骨髄異形成症候群は、白金を基にした治療計画を含む先行治療に次いで生じた。
一実施形態において、再発性または難治性急性骨髄性白血病は、白金を基にした治療計画を含む先行治療に次いで生じた。
一実施形態において、当該抗PD1モノクローナル抗体は、ヒト化モノクローナルIgG4抗体である。
一実施形態において、当該抗PDL1モノクローナル抗体は、ヒト化モノクローナルIgG1抗体である。
一実施形態において、当該ヒト化モノクローナルIgG4抗体は、ペンブロリズマブ、MK−3475、ピディリズマブ、ニボルマブ(BMS−936558、MDX−1106、またはONO−4538)である。
一実施形態において、当該ヒト化モノクローナルIgG4抗体は、ペンブロリズマブである。
一実施形態において、当該ヒト化モノクローナルIgG1抗体は、BMS−936559、アテゾリズマブ(MPDL3280A)、またはデュルバルマブ(MEDI4736)である。
一実施形態において、当該ヒト化モノクローナルIgG1抗体は、デュルバルマブ(MEDI4736)である。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で投与されるのに次いで、7日間の連続した休薬日が続く。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、21日周期における14日間の連続した日で投与されるのに次いで、7日間の連続した休薬日が続く。
ある実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日目〜7日目に4週間ごとに(Q4W)投与される。5−アザシチジンは、75mg/m2/日で、1日目〜7日目に、4週間ごとに(Q4W)投与されるであろう。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、28日周期における7日目及び21日目に投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、28日周期における1日目に投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、28日周期における8日目及び21日目に投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、21日周期における1日目に投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、14日周期における1日目に投与される。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で投与されるのに次いで、7日間の連続した休薬日が続き、当該抗PD1/抗PDL1モノクローナル抗体は、当該28日周期の1日目に投与される。当該疾患または障害は、MDSまたはAML(例えば、再発性または難治性のMDSまたはAML、及びより特別には、注射可能な低メチル化薬を用いた治療に応答中ではないMDSまたはAML)である。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で投与されるのに次いで、7日間の連続した休薬日が続き、当該抗PD1/抗PDL1モノクローナル抗体は、当該28日周期の7日目及び21日目に投与される。当該疾患または障害は、MDSまたはAML(例えば、再発性または難治性のMDSまたはAML、及びより特別には、注射可能な低メチル化薬を用いた治療に応答中ではないMDSまたはAML)である。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で投与されるのに次いで、7日間の連続した休薬日が続き、当該抗PD1/抗PDL1モノクローナル抗体は、当該28日周期の8日目及び21日目に投与される。当該疾患または障害は、MDSまたはAML(例えば、再発性または難治性のMDSまたはAML、及びより特別には、注射可能な低メチル化薬を用いた治療に応答中ではないMDSまたはAML)である。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、21日周期における14日間の連続した日で投与されるのに次いで、7日間の連続した休薬日が続き、当該抗PD1/抗PDL1モノクローナル抗体は、当該21日周期の1日目に投与される。当該疾患または障害は、卵巣癌または肺癌(例えば、上皮性卵巣癌または非小細胞肺癌、特に再発性または難治性の上皮性卵巣癌または非小細胞肺癌、及びより特別には、白金を基にした療法の後に再発した上皮性卵巣癌または非小細胞肺癌)である。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日目〜7日目に、4週間ごとに(Q4W)投与され、当該抗PDL1モノクローナル抗体は、2週間ごとに投与される。当該疾患または障害は、MDSまたはAML(例えば、再発性または難治性のMDSまたはAML、及びより特別には、注射可能な低メチル化薬を用いた治療に応答中ではないMDSまたはAML、あるいは未処置の(第一線の)より高いリスクのMDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、21日周期における14日間の連続した日で1日当たり約300mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、ペンブロリズマブは、当該21日周期における1日目に10mg/kgの用量で投与され、当該疾患または障害は、再発性上皮性卵巣癌である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で1日当たり2回、約100mgの、1日当たり2回、約150mgの、1日当たり2回、約200mgのまたは1日当たり2回、約300mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、ペンブロリズマブは、当該28日周期における7日目及び21日目に10mg/kgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)、または骨髄異形成症候群(MDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で1日当たり約200mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、ペンブロリズマブは、当該28日周期の7日目及び21日目に10mg/kgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)、または骨髄異形成症候群(MDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で1日当たり約200mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、ペンブロリズマブは、当該28日周期の7日目及び21日目に5mg/kgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)、または骨髄異形成症候群(MDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における14日間の連続した日で1日当たり約200mgの量で投与されるのに次いで、14日間の連続した休薬日が続き、ペンブロリズマブは、当該28日周期の7日目及び21日目に5mg/kgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)、または骨髄異形成症候群(MDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で1日当たり2回約300mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、ペンブロリズマブは、当該28日周期の8日目及び21日目に10mg/kgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)、または骨髄異形成症候群(MDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、21日周期における14日間の連続した日で1日当たり約300mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、ペンブロリズマブは、当該21日周期の1日目に投与され、当該疾患または障害は、非小細胞肺癌である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、28日周期における21日間の連続した日で1日当たり2回約200mgの量で投与されるのに次いで、7日間の連続した休薬日が続き、デュルバルマブは、当該28日周期の1日目に1日当たり1500mgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)または骨髄異形成症候群(MDS)である。
一実施形態において、当該5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、7日間の連続した日で4週間ごとに(Q4W)約75mg/m2/日の量で投与され、デュルバルマブは、2週間ごとに(Q2W)10mg/体重kgの用量で投与され、当該疾患または障害は、注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない急性骨髄性白血病(AML)または骨髄異形成症候群(MDS)である。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり約50mg、約100mg、約200mg、約300mg、約400mg、約500mg、または約600mgの量で投与される。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり約300mgの量で投与される。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり約200mgの量で投与される。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり1回投与される。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり2回投与される。
一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり2回約200mg、約150mg、または約100mgの量で投与される。一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり2回約200mgの量で投与される。一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり2回約150mgの量で投与される。一実施形態において、5−アザシチジン、またはその医薬として許容され得る塩、溶媒和物若しくは水和物は、1日当たり2回約100mgの量で投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、非経口投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、対象の質量キログラムあたり約0.5mg、約1mg/Kg、約2mg/Kg、約3mg/Kg、約4mg/Kg、約5mg/Kg、約6mg/Kg、約7mg/Kg、約8mg/Kg、約9mg/Kg、約10mg/Kg、約11mg/Kg、約12mg/Kg、約13mg/Kg、約14mg/Kg、約15mg/Kg、約16mg/Kg、約17mg/Kg、約18mg/Kg、約19mg/Kg、または約20mg/Kgの抗PD1/抗PDL1モノクローナル抗体の量で投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、1日当たり約10mg/Kgの量で静脈内投与される。
一実施形態において、当該抗PD1/抗PDL1モノクローナル抗体は、28日周期における7日目及び21日目に、または28日周期における8日目及び21日目に、1日当たり約10mg/Kgの量で静脈内投与される。
一実施形態において、当該抗PD1モノクローナル抗体は、ペンブロリズマブ、MK−3475、ピディリズマブ、ニボルマブ(BMS−936558、MDX−1106、またはONO−4538)であり、30分間の静脈内注入として投与される。
一実施形態において、当該抗PDL1モノクローナル抗体は、BMS−936559、アテゾリズマブ(MPDL3280A)、またはデュルバルマブ(MEDI4736)であり、30分間の静脈内注入として投与される。一実施形態において、1,500mgのデュルバルマブ(MEDI4736)は、1時間の静脈内(IV)注入によって各28日間の治療周期の1日目に投与される。
一実施形態において、5−アザシチジンまたはその医薬として許容され得る塩、溶媒和物若しくは水和物は、カプセル剤、錠剤またはカプレット剤の形態にある。
一実施形態において、当該方法はさらに、治療有効量の追加の作用薬を投与することを含む。
一実施形態において、当該対象はヒトである。
本明細書で説明する場合、本明細書のある実施形態は、例えば、異なる投薬量及び/若しくは投薬期間を可能にすること、代替的な薬物動態特性、薬力学的特性、及び/若しくは安全特性を提供すること、長期療法及び/若しくは維持療法の評価を可能にすること、脱メチル化及び/若しくは遺伝子再発現を最大限にする治療計画を提供すること、持続的な脱メチル化を長期化する治療計画を提供すること、シチジン類似体についての新たな効能を提供すること、ならびに/または他の有望な有利な有益性を提供することと関連する方法において有用なシチジン類似体の経口製剤を提供する。
本明細書に提供するのは、例えば、5−アザシチジンのようなシチジン類似体を含む医薬製剤を経口投与することによる、例えば、血液学的障害及び固形腫瘍を含む癌のような異常な細胞増殖によって顕著化する病理生理学的容態の治療方法であって、当該製剤は、実質的に胃の中で当該シチジン類似体を放出する。本明細書の他の実施形態は、免疫障害の治療方法を提供する。特定の実施形態において、本明細書に提供する方法は、当該シチジン類似体の即時放出をもたらす製剤を経口投与することを包含する。ある実施形態において、当該シチジン類似体及び1つ以上の治療薬は、相乗的な治療効果を生じるよう対象へ併用投与される。当該併用投与される薬剤は、経口でまたは注射によって投与される癌治療薬であり得る。
ある実施形態において、異常な細胞増殖と関連した障害を治療するための、本明細書に提供する方法は、治療有効量のシチジン類似体を含む製剤を経口投与することを含む。本明細書に提供する方法と関連する特定の治療効能を、本明細書に開示する。ある実施形態において、当該医薬製剤中の当該治療有効量のシチジン類似体は、本明細書に開示するような量である。ある実施形態において、当該医薬製剤中の正確な治療有効量のシチジン類似体は、例えば、対象の年齢、体重、疾患及び/または容態に応じて変化するであろう。
特定の実施形態において、異常な細胞増殖と関連した障害には、MDS、AML、ALL、CML、白血病、慢性リンパ性白血病(CLL)、リンパ腫(非ホジキンリンパ腫(NHL)及びホジキンリンパ腫を含む)、多発性骨髄腫(MM)、肉腫、黒色腫、癌腫、腺癌、脊索腫、乳癌、大腸癌、卵巣癌、肺癌(例えば、非小細胞肺癌及び小細胞肺癌)、精巣癌、腎癌、膵癌、骨癌、胃癌、頭頚部癌、及び前立腺癌を含むが、これらに限定しない。特定の実施形態において、異常な細胞増殖と関連した障害は、MDSである。特定の実施形態において、異常な細胞増殖と関連した障害は、AMLである。
ある実施形態において、異常な細胞増殖の障害を治療するための本明細書に提供する方法は、静脈内投与法、皮下投与法及び経口投与法のうちの少なくとも2つを用いてシチジン類似体を投与することを含む。例えば、本明細書の特定の実施形態は、例えば、皮下または静脈内のいずれかで投与する5−アザシチジンのようなシチジン類似体の初回治療周期に次いで続く、当該シチジン類似体のその後の経口投与する治療周期を投与することを提供する。ある実施形態において、治療周期は、複数回用量を必要とする対象へ複数日(例えば、1日間、2日間、3日間、4日間、5日間、6日間、7日間、8日間、9日間、10日間、11日間、12日間、13日間、14日間、または14日間超)にわたって投与する複数回用量に任意に次いで続く治療休薬日(例えば、1日間、2日間、3日間、4日間、5日間、6日間、7日間、8日間、9日間、10日間、11日間、12日間、13日間、14日間、または14日間超)を含む。本明細書の特定の実施形態は、1、2、3、4、5、またはそれより多数の初回周期のための皮下投与及び/または静脈内投与に次いで続くその後の周期のための経口投与を含む治療スケジュールを提供する。例えば、本明細書の特定の実施形態は、1、2、3、4、5、またはそれより多数の初回周期のための皮下投与及び/または静脈内投与に次いで続くその後の周期のための経口投与を含む治療スケジュールを提供する。本明細書に提供する方法に対して適切な薬用量範囲及び量は、本明細書のいたるところで提供される。例えば、ある実施形態において、皮下用量は、約75mg/m2である。ある実施形態において、当該経口用量は、約60mg、約80mg、約120mg、約180mg、約240mg、約300mg、約360mg、約480mg、または約480mg超である。ある実施形態において、経口用量は、皮下曲線下面積の80%、100%、または120%に到達するよう算出される。
ある実施形態において、異常な細胞増殖に関する障害の治療方法は、シチジン類似体(例えば、5−アザシチジン)を含む製剤を単回日用量または複数回日用量として経口投与することを含む。特定の実施形態において、当該シチジン類似体を含む製剤(複数可)は、1日1回、1日2回、1日3回、1日4回、または1日4回超経口投与する。例えば、ある実施形態において、当該シチジン類似体を含む製剤は、約200mg、約300mg、約400mg、約500mg、約600mg、約700mg、約800mg、約900mg、または約1,000mgのシチジン類似体の1日当たり1回、2回、3回、または4回の7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、または30日間の投与を含む治療周期を用いて投与する。ある実施形態において、当該治療方法は、持続的な低用量投与を含む。ある実施形態において、当該シチジン類似体を含む製剤は、1日あたり2回の7日間の当該シチジン類似体約300mgの投与を含む治療周期を用いて投与する。ある実施形態において、当該シチジン類似体を含む製剤は、1日当たり2回の14日間の当該シチジン類似体約300mgの投与を含む治療周期を用いて投与する。ある実施形態において、当該シチジン類似体を含む製剤は、1日当たり3回の7日間の当該シチジン類似体約300mgの投与を含む治療周期を用いて投与する。ある実施形態において、当該シチジン類似体を含む製剤は、1日当たり3回の14日間の当該シチジン類似体約300mgの投与を含む治療周期を用いて投与する。ある実施形態において、本明細書に提供する方法は、本明細書に提供する周期のうちの1つ以上を用いてシチジン類似体を含む製剤を投与すること、及び例えば、1か月間、2か月間、3か月間、4か月間、5か月間、6か月間、7か月間、8か月間、9か月間、10か月間、11か月間、12か月間、または12か月間超の期間、当該周期のうちの1つ以上を反復することを含む。
ある実施形態において、本明細書の方法は、例えば、シチジン類似体の静脈内投与または皮下投与と関係した制限を克服するために本明細書に提供する特定の経口製剤を投与することを含む。例えば、静脈内投与または皮下投与は、規則的な基礎でより長期間、シチジン類似体を送達する能力を制限し、それにより当該シチジン類似体の最大効能を潜在的に制限し得る。長期の静脈内投与スケジュールまたは皮下投与スケジュールの厳密さに従うことが困難であるので、シチジン類似体への長期の皮下曝露または静脈内曝露は、対象(例えば、多発性血球減少に罹患している対象)に当該投与計画を中断させることがある。例えば、その全体が参照により本明細書に組み込まれるLyons,R.M.,et al.,Hematologic Response to Three Alternative Dosing Schedules of Azacitidine in Patients With Myelodysplastic Syndromes,J.Clin.Oncol.(2009)(DOI:10.1200/ JCO.2008.17.1058)を参照されたい。したがって、ある実施形態において、本明細書に提供する方法は、皮下または静脈内でのシチジン類似体投与と関係したこれらのまたは他の制限を克服するために、本明細書に提供する経口製剤を投与することを含む。例えば、ある実施形態において、本明細書に提供する方法は、対象へ本明細書に提供する経口製剤を7日間以上、8日間以上、9日間以上、10日間以上、11日間以上、12日間以上、13日間以上、14日間以上、15日間以上、16日間以上、17日間以上、18日間以上、19日間以上、20日間以上、または21日間以上、毎日投与することを含む。
本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、アザシチジン)を、静脈内投与または皮下投与と比較してより低用量でより長時間にわたって送達することを含む、当該シチジン類似体の経口製剤を投与することを含む方法を提供する。特定の実施形態において、このような方法は、本明細書に提供する経口製剤を投与することによって、用量関連血球減少(例えば、アザシチジンと関係した用量関連血球減少を含む)を管理することを含む。ある実施形態において、本明細書に提供する方法は、同じシチジン類似体を含む静脈内用量または皮下用量と比較して改善された安全性特性を達成するよう、本明細書に提供する経口製剤を投与することを含む。
本明細書に説明する場合、ある実施形態は、本明細書に提供する経口製剤を投与することによって、当該シチジン類似体の静脈内投与または皮下投与と比較した場合、特定の疾患または障害の改善された治療(例えば、固形腫瘍の治療)方法を提供する。特定の実施形態において、本明細書のある方法は、本明細書に提供する経口製剤をより低用量でより長時間投与して、改善された脱メチル化をもたらすことを提供する。例えば、本明細書に提供するある方法は、本明細書に提供する経口製剤を投与して、皮下投与または静脈内投与を介して当該シチジン類似体を投薬することと関係したある用量制限毒性関連副作用を回避しながら固形腫瘍を治療することを含む。シチジン類似体の投与と関係したある毒性関連欠点の例は、例えば、その全体が参照により本明細書に組み込まれるK.Appleton et al.,J.Clin.Oncol.,Vol.25(29):4603〜4609(2007)において説明されている。
本明細書の特定の実施形態は、本明細書に提供する医薬組成物を経口投与することによって、本明細書に提供する疾患または障害に罹患している対象の治療方法を提供し、この中で当該治療は、当該対象の改善された生存を結果として生じる。ある実施形態において、改善された生存は、1つ以上の従来のケア計画と比較して測定される。本明細書の特定の実施形態は、本明細書に提供する医薬組成物を経口投与することによる、本明細書に提供する疾患または障害に罹患している対象の治療方法を提供し、この中で、当該治療は、改善された有効性を提供する。特定の実施形態において、改善された有効性は、米国食品医薬品局(FDA)によって推奨されるように、癌の臨床試験についての1つ以上の評価項目を用いて測定する。例えば、FDAは、癌の薬剤及び生物製剤の認可についての臨床試験評価項目に関する産業についての指針(Guidance for Industry on Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics(http://www.fda.gov/CbER/gdlns/clintrialend.htm))を提供している。FDAの評価項目には、(i)疾患のない生存、(ii)客観的な応答率、(iii)進行に至る時間及び進行のない生存ならびに(iv)治療不全に至る時間、のような全体的な生存、腫瘍評価を基にした評価項目を含むが、これらに限定しない。症状の評価項目を包含する評価項目には、(i)癌の症状の進行に至る時間及び(ii)混合症状の評価項目、のような具体的な症状の評価項目を含み得る。血液または体液から分析した生体マーカーも、当該疾患の管理を判断するために有用であり得る。
ある実施形態において、異常な細胞増殖に関する障害の治療方法は、シチジン類似体の製剤を食物とともに経口投与することを含む。ある実施形態において、異常な細胞増殖に関する障害の治療方法は、シチジン類似体の製剤を食物なしで経口投与することを含む。ある実施形態において、薬理学的パラメータ(例えば、Cmax、Tmax)は、対象の摂食状態に依存する。ある実施形態において、シチジン類似体の製剤は、舌下投与される。
ある実施形態において、シチジン類似体、例えば、5−アザシチジンは、シチジンデアミナーゼ阻害薬と併用投与されない。ある実施形態において、本明細書に提供するシチジン類似体を含む経口製剤は、THUと併用投与されない。本明細書のある実施形態は、実質的に胃の中で放出するために本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を経口投与することを含む、本明細書に提供する疾患または障害(例えば、異常な細胞増殖と関係した疾患)の治療方法を提供し、この中で、当該方法は、本明細書に提供する特定の生物学的パラメータ(例えば、本明細書に提供する特定のCmax値、Tmax値、及び/または曲線下面積値)に到達し、当該方法は、シチジンデアミナーゼ阻害薬を当該シチジン類似体と併用投与しないことを含む。本明細書のある実施形態は、実質的に胃の中での放出のために、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を経口投与することを含む、本明細書に提供する疾患または障害(例えば、異常な細胞増殖と関係した疾患)の治療方法を提供し、この中で、当該方法は、シチジンデアミナーゼ阻害薬を当該シチジン類似体と併用投与しないことによって、当該シチジンデアミナーゼ阻害薬(例えば、THU)を投与することと関係した有害作用を回避する。特定の実施形態において、シチジンデアミナーゼ阻害薬(例えば、THU)は、例えば、約500mg/日未満、約200mg/日未満、約150mg/日未満、約100mg/日未満、約50mg/日未満、約25mg/日未満、約10mg/日未満、約5mg/日未満、約1mg/日未満、または約0.1mg/日未満の量で、当該シチジン類似体と併用投与する。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することによって、血液学的障害を含む本明細書に提供する障害を治療することを含む。特定の実施形態において、5−アザシチジンを含む本明細書に提供する経口剤形は、血液学的障害に罹患している対象を治療するために使用する。血液学的障害には、例えば、血球の形成異常変化及び種々の白血病のような血液学的悪性病変をもたらすことのできる血球の異常な増殖を含む。血液学的障害の例としては、とりわけ、急性骨髄性白血病(AML)、急性前骨髄球性白血病(APML)、急性リンパ芽球性白血病(ALL)、慢性骨髄性白血病(CML)、慢性リンパ性白血病(CLL)、骨髄異形成症候群(MDS)、及び鎌状赤血球貧血が挙げられるが、これらに限定しない。本明細書に提供する方法を用いて治療することのできる他の障害には、例えば、多発性骨髄腫(MM)及び非ホジキンリンパ腫(NHL)を含む。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ当該経口剤形を投与することによって、AMLを治療することを含む。AMLは、成人で生じる最も普遍的なタイプの急性白血病である。いくつかの遺伝的障害及び免疫不全状態は、AMLのリスクの上昇と関係している。これらには、ブルーム症候群、ファンコニー貧血、リ−フラウメニ近親、毛細血管拡張性運動失調症、及び無ガンマグロブリン血症のような、無作為染色体切断をもたらすDNA安定性異常を有している障害を含む。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ当該経口剤形を投与することによって、APMLを治療することを含む。APMLは、AMLの異なる下位群を表す。この下位群は、15;17染色体転座を含有する前骨髄球性芽球を特徴とする。この転座は、レチノイン酸受容体と配列PMLとから構成される融合転写産物の生成をもたらす。
ある実施形態において、本明細書に提供する方法は、シチジンを含む経口剤形を投与することを必要とする対象へ当該経口剤形を投与することによってALLを治療することを含む。ALLは、種々のサブタイプによって表される異なる臨床的特徴を有する異種性疾患である。再発性細胞遺伝性異常は、ALLにおいて実証されてきた。最も普遍的な細胞遺伝性異常は、9;22転座である。結果として生じるフィラデルフィア染色体は、対象の予後不良を表す。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することによってCMLを治療することを含む。CMLとは、多能性幹細胞のクローン性骨髄増殖性疾患である。CMLは、フィラデルフィア染色体を生じる染色体第9番及び第22番の転座を包含する特異的な染色体異常を特徴とする。電離放射線は、CMLの発達と関係している。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することによってMDSを治療することを含む。ある実施形態において、MDSには、以下の骨髄異形成症候群サブタイプ、すなわち、不応性貧血、環状鉄芽球性不応性貧血(好中球減少症若しくは血小板減少症を伴う場合または輸血を必要とする場合)、芽球増加性不応性貧血、形質転換における芽球増加性不応性貧血、及び慢性骨髄単球性白血病のうちの1つ以上を含む。ある実施形態において、当該MDSは、より高いリスクのMDSである。ある実施形態において、本明細書に提供する方法は、MDSに罹患している対象の生存を高める(例えば、寿命を延ばす)ために、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することを含む。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することによってNHLを治療することを含む。非ホジキンリンパ腫(NHL)は、リンパ系の悪性病変の異種性群を表す。血液学的腫瘍及びリンパ腫のWHO分類によると、これらの疾患は、B細胞新生物及びT細胞新生物と分類される。B細胞リンパ腫は、リンパ腫全体の約90%を占め、この2つの最も普遍的な組織学的疾患実体は、濾胞性リンパ腫及びびまん性大細胞型B細胞性リンパ腫である。年間およそ55,000〜60,000の新しい症例のNHLが米国において診断されている。例えば、Ansell,S.M.,et al.,Mayo Clin.Proc.,2005,80(8):1087〜1097を参照されたい。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することによって、MMを治療することを含む。多発性骨髄腫は、最も普遍的に診断された血液学的悪性病変のうちの1つである。2007年に、米国単独において、およそ20,000の新たなMM症例及び10,000名のMMによる死亡があった。当該疾患はとりわけ、免疫グロブリン、例えば、モノクローナル免疫グロブリンGまたはAの過剰産生をもたらすことのできる骨髄中の悪性形質細胞の蓄積を特徴とする。傍タンパク質としても公知のこれらの免疫グロブリンは、MM患者の尿及び血液中において検出することができる。MMの結果には、貧血、破壊性身体病変の発達、及び腎不全を含む。例えば、Rao,K.V.,American Journal of Health−System Pharmacy,2007,64(17):1799〜1807を参照されたい。
ある実施形態において、本明細書に提供する方法は、シチジン類似体を含む経口剤形を投与することを必要とする対象へ、当該経口剤形を投与することによってCLLを治療することを含む。慢性リンパ性白血病(CLL)とは、成熟Bリンパ球の悪性病変であり、米国における最も多いリンパ系悪性病変である。Bリンパ球新生物B細胞悪性病変のWHOによる分類は、悪性細胞の推定標準対応物によりB細胞悪性病変を群分けする。CLLは、血液、骨髄、またはリンパ節に由来するリンパ球の免疫表現型分析によって診断される。例えば、Zent,C.S.,et al.,Current Oncology Reports,2007,9:345〜352を参照されたい。
本明細書のある実施形態は、シチジン類似体を送達する必要のある対象へ、シチジン類似体を含む経口剤形を投与することを含む、当該対象へシチジン類似体を送達するための方法を提供する。特定の実施形態において、経口製剤は、(1)治療有効量のシチジン類似体、及び(2)対象がシチジン類似体を含む経口製剤を摂取した後で実質的に胃の中でシチジン類似体を放出することのできる任意の薬剤放出制御成分を含む。本明細書のある実施形態は、対象におけるシチジン類似体の経口生物学的利用能の亢進方法を提供する。本明細書のある実施形態は、本明細書に提供する医薬組成物を経口投与することを含む、シチジン類似体の経口生物学的利用能の増大方法を提供する。本明細書に提供するある方法において、本明細書に提供する医薬組成物は、対象へ経口投与され、対象の身体の生物学的流体と接触し、例えば実質的に胃の中のような上部消化管において吸収される。
本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を含む経口製剤を投与することによる、本明細書に提供する特定の曝露値への到達方法を提供する。本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を含む経口剤形を投与することによる、本明細書に提供する特定の経口生物学的利用能への到達方法を提供する。本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を含む経口製剤を投与することによる、本明細書に提供する特定の曲線下面積値への到達方法を提供する。本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を含む経口製剤を投与することによる、本明細書に提供する特定のCmax値への到達方法を提供する。本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を含む経口製剤を投与することによる、本明細書に提供する特定のTmax値への到達方法を提供する。
本明細書のある実施形態は、本明細書に提供するシチジン類似体(例えば、5−アザシチジン)を含む経口製剤を投与することによる、望ましくないまたは制御されていない細胞増殖を包含する容態の治療方法を提供する。このような容態には、例えば、良性腫瘍、原発腫瘍及び腫瘍転移のような種々のタイプの癌、血液学的障害(例えば、白血病、骨髄異形成症候群及び鎌状赤血球貧血)、再狭窄(例えば、冠状病変、頚動脈病変、及び大脳病変)、内皮細胞の異常な刺激(動脈硬化)、手術による身体組織への傷害、異常な創傷治癒、異常な血管新生、組織の線維化を生じる疾患、反復運動障害、高度に血管新生していない組織の障害、ならびに臓器移植と関係した増殖応答を含む。
ある実施形態において、良性腫瘍中の細胞は、これらの分化した特徴を保有しており、完全に制御されていない様式では分割しない。良性腫瘍は、局在性及び/または非転移性であり得る。本明細書に提供する方法、組成物、及び製剤を用いて治療することのできる良性腫瘍の具体的なタイプには、例えば、血管腫、肝細胞腺腫、海綿状血管腫、限局性結節性過形成、聴神経腫瘍、神経線維腫、胆管腺腫、胆管シスタノーマ(cystanoma)、線維腫、脂肪腫、平滑筋腫、中皮腫、奇形腫、粘液腫、結節性再生性過形成、トラコーマ及び化膿性肉芽腫を含む。
ある実施形態において、悪性腫瘍における細胞は、未分化となり、身体の成長の制御シグナルに応答せず、及び/または制御されていない様式で増殖する。悪性腫瘍は、侵襲的であり得、遠隔部位へ伝播すること(転移すること)ができる。悪性腫瘍は、2つの区分、すなわち原発及び二次性へと分けることがある。原発腫瘍は、当該腫瘍が見つかった組織から直接生じている。二次性腫瘍、または転移は、体内の他所に由来するが、いまや遠隔器官へと伝播した腫瘍である。転移のための共通経路は、隣接する構造への直接的な増殖であり、血管系またはリンパ系を通じて伝播し、組織面及び体腔(腹水、脊髄液など)に沿って移動する。
メチル化は、細胞制御に決定的な遺伝子のサイレンシング(すなわち、エピジェネティックな遺伝子サイレンシング)をもたらすことができ、大腸癌または肺癌を含む悪性腫瘍の発達における初期の事象であり得る。例えば、M.V.Brock et al.,N.Engl.J.Med.,2008,358(11):1118〜28、P.M. Das et al.,Mol.Cancer,2006,5(28)、G.Gifford et al.,Clin.Cancer Res.,2004,10:4420〜26、J.G.Herman et al.,N.Engl.J.Med.,2003,349:2042〜54、A.M.Jubb et al.,J.Pathology,2001,195:111〜34を参照されたい。したがって、ある実施形態において、本明細書の方法は、例えば、異常なDNAメチル化を反転させることによって、エピジェネティックな遺伝子サイレンシングを防止または反転させるために、本明細書に提供する経口製剤を用いることを提供する。具体的な実施形態において、本明細書に提供する経口製剤は、癌、例えば、家族性ポリポーシスまたは肺癌を発達させる危険にある患者における癌の発達を防止するための早期介入に使用し、この中で、当該癌の原因は、エピジェネティックな遺伝子サイレンシングである。特定の実施形態において、このような早期の介入は、経口投与以外の手段(例えば、静脈内投与または皮下投与)によって実行することは不可能であろう。具体的な実施形態において、本明細書に提供する経口製剤は、早期再発についての危険にある患者における癌、例えば、大腸癌または非小細胞肺癌の再発を防止するための早期介入に使用する。ある実施形態において、当該早期介入は、本明細書に説明する製剤及び/または方法を用いて、長期経口投薬スケジュールを介して達成される。ある実施形態は、例えば、エピジェネティックな変化による遺伝子サイレンシングの危険にある患者における遺伝子サイレンシングの効果を反転させるための、本明細書に提供する経口製剤の投与方法を提供する。特定の実施形態において、本明細書に提供する方法はさらに、HDAC阻害薬化合物を(例えば、異常なDNAメチル化を反転させた後に転写活性のある立体配置へ染色質を回復させるために)投与することを含む。特定の実施形態において、HDAC阻害薬化合物は、標的化療法と相乗的に作用して癌関連HDACアイソフォーム1、2、及び3に対して選択的である経口HDAC阻害薬であるエンチノスタット(SNDX−275、旧MS−275)である。特定の実施形態において、相乗効果は、固形腫瘍(例えば、NSCLC)または血液学的悪性病変(例えば、MDS、CMMoL、またはAML)の治療のために5−アザシチジン及びHDAC阻害薬(例えば、エンチノスタット(etinostat))を併用投与することによって達成される。
ある実施形態において、本明細書に提供する方法、組成物、及び製剤を用いて治療することのできる原発または二次性のいずれかの癌または悪性腫瘍の具体的なタイプには、例えば、白血病、乳癌、皮膚癌、骨癌、前立腺癌、肝癌、肺癌(例えば、非小細胞肺癌及び小細胞肺癌)、脳癌、喉頭、胆嚢、膵臓、直腸、副甲状腺、甲状腺、副腎、神経組織、頭頚部、結腸、胃、気管支、腎臓の癌、基底細胞癌、潰瘍型及び乳頭状の両タイプの扁平上皮癌、転移性皮膚癌、骨肉腫、ユーイング肉腫、細網肉腫(veticulum cell sarcoma)、骨髄腫、巨細胞腫、胆石、膵島細胞腫瘍、原発脳腫瘍、急性及び慢性のリンパ球性腫瘍及び顆粒球性腫瘍、有毛細胞腫瘍、腺腫、過形成、髄様癌、褐色細胞腫、粘膜神経腫(mucosal neuronma)、 腸神経節神経腫、過形成性角膜神経腫瘍、マルファノイド体質腫瘍、ウィルムス腫瘍(Wilm’s tumor)、精上皮腫、卵巣腫瘍、平滑筋腫、頚部異形成及び上皮内癌、神経芽腫、網膜芽細胞腫、髄芽腫、軟部組織肉腫、悪性カルチノイド、局所皮膚病変、菌状息肉症、横紋筋肉腫、カポジ肉腫、骨肉腫及び他の肉腫、悪性高カルシウム血症、腎細胞腫瘍、真性多血症(polycythermia vera)、腺癌、多形膠芽腫(glioblastoma multiforma)、白血病、リンパ腫、悪性黒色腫、類上皮癌、ならびに他の癌腫及び肉腫を含む。
本明細書の特定の実施形態は、例えば、関節手術、腸手術、及びケロイド瘢痕化を含む種々の手術手技のための手術中に身体組織に対する損傷による異常な細胞増殖を治療するために、本明細書に提供する方法、組成物、及び製剤を使用することを提供する。本明細書に提供する方法、組成物、及び製剤を使用して治療され得る臓器移植関連増殖応答には、潜在的な臓器拒絶または関連する合併症に寄与する増殖性応答を含む。具体的には、これらの増殖性応答は、心臓、肺(例えば、非小細胞肺癌及び小細胞肺癌)、肝臓、腎臓、及び他の身体器官または器官系の移植中に生じ得る。
ある実施形態において、本明細書に提供する製剤、その投与方法、または本明細書で明らかにしたような治療方法におけるシチジン類似体の量は、本明細書に提供する具体的な薬用量である。ある実施形態において、経口アザシチジン薬用量、その投与方法、またはMDS及びAMLを含むがこれらに限定しない少なくとも1つの容態の治療方法は、例えば、約50mg/m2/日〜約2,000mg/m2/日、約100mg/m2/日〜約1,000mg/m2/日、約100mg/m2/日〜約500mg/m2/日、または約120mg/m2/日〜約250mg/m2/日に及び得る。ある実施形態において、特定の薬用量は、例えば、約120mg/m2/日、約140mg/m2/日、約150mg/m2/日、約180mg/m2/日、約200mg/m2/日、約220mg/m2/日、約240mg/m2/日、約250mg/m2/日、約260mg/m2/日、約280mg/m2/日、約300mg/m2/日、約320mg/m2/日、約350mg/m2/日、約380mg/m2/日、約400mg/m2/日、約450mg/m2/日、または約500mg/m2/日である。
ある実施形態において、適切な生体マーカーは、疾患状態に及ぼすシチジン類似体を含む医薬組成物の効果を判定または予測して、投薬スケジュールに対する指針を提供する。例えば、本明細書の特定の実施形態は、MDSと診断された患者が、患者の核酸のメチル化状態を評価することによって、シチジン類似体を含む医薬組成物を用いた治療からより大きな利益を得る高い確率を有するかどうかの判断方法を提供する。特定の実施形態において、当該シチジン類似体はアザシチジンである。特定の実施形態において、当該核酸はDNAまたはRNAである。特定の実施形態において、より大きな利益は、全体的な生存利益である。特定の実施形態において、当該メチル化状態は、1つ以上の遺伝子、例えば、MDSまたはAMLと関係する遺伝子において検討される。具体的な実施形態は、基線DNAメチル化レベルが、アザシチジンを用いて治療したMDS(例えば、より高い危険性のMDS)患者における全体的な生存に影響するかどうかの判断方法を包含する。具体的な実施形態は、遺伝子プロモーターメチル化レベルが、MDS(例えば、より高い危険性のMDS)患者における全体的な生存に影響するかどうかの判断方法を提供する。
例えば、本明細書の具体的な実施形態は、MDS(例えば、より高い危険性のMDS)患者における長期化した生存に及ぼす遺伝子のメチル化の影響の評価方法を提供する。特定の実施形態において、このような評価は、例えば、本明細書に提供するようなシチジン類似体を含む医薬組成物を用いた治療の際に、MDS(例えば、より高い危険性のあるMDS)患者における全体的な生存を予測するために使用する。特定の実施形態において、このような評価は、治療上の意思決定に使用する。具体的な実施形態において、このような治療上の意思決定には、患者の治療、例えば、シチジン類似体の投薬計画、量、及び/または持続期間を計画または調整することを含む。
ある実施形態は、例えば特定の遺伝子におけるメチル化レベル分析を用いて全体的な生存利益をシチジン類似体治療から得る高い確率を有する、MDSと診断された個々の患者の識別方法を提供する。具体的な実施形態において、より低レベルの核酸メチル化は、アザシチジン治療後の改善された全体的な生存を得る高い確率と関係している。特定の実施形態において、治療後の改善された全体的な生存を得る高い確率は、例えば、本明細書に提供するようなシチジン類似体を含む医薬組成物を用いた治療後の改善された全体的な生存を得る少なくとも5%高い確率、少なくとも10%高い確率、少なくとも20%高い確率、少なくとも30%高い確率、少なくとも40%高い確率、少なくとも50%高い確率、少なくとも60%高い確率、少なくとも70%高い確率、少なくとも80%高い確率、少なくとも90%高い確率、少なくとも100%高い確率、少なくとも125%高い確率、少なくとも150%高い確率、少なくとも175%高い確率、少なくとも200%高い確率、少なくとも250%高い確率、少なくとも300%高い確率、少なくとも400%高い確率、または少なくとも500%高い確率である。特定の実施形態において、治療後の改善された全体的な生存を得るより高い確率は、MDSと診断された患者の特定の比較集団の平均確率と比較してより高い確率である。具体的な実施形態において、比較集団は、本明細書に説明するような、特定の骨髄異形成サブタイプと分類された一群の患者である。一実施形態において、比較集団は、より高い危険性のMDSに罹患している患者からなる。特定の実施形態において、比較集団は、特定のIPSS細胞遺伝学的下位群からなる。
特定の実施形態において、核酸(例えば、DNAまたはRNA)の過剰メチル化状態は、当該技術分野で公知の任意の方法によって判定され得る。ある実施形態において、DNAの過剰メチル化状態は、例えば、定量的リアルタイムメチル化特異的PCR(「qMSP」)を用いることによって、MDSと診断された患者の骨髄液を用いて判定され得る。ある実施形態において、当該メチル化分析は、ゲノムDNAの亜硫酸水素転化を包含し得る。例えば、ある実施形態において、DNAの亜硫酸水素処理は、メチル化したCpG部位を未処置にしておいたまま、メチル化されていないCpGをUpGへ転化するために使用する。例えば、Frommer,M.,et al.,Proc.Nat’l Acad.Sci.USA 1992,89:1827〜31を参照されたい。市販のキットは、このような亜硫酸水素処理に使用され得る。ある実施形態において、メチル化PCRを容易にするために、プライマー、例えば、メチル化状態にかかわらずDNAを増幅する外側プライマー、及び第一のPCRによって増幅した領域内のメチル化された配列またはメチル化されていない配列へ結合する入れ子式プライマーが、当該技術分野で公知の通り設計される。例えば、Li et al.,Bioinformatics 2002,18:1427〜31を参照されたい。ある実施形態において、プローブ、例えば、メチル化状態にかかわらず亜硫酸水素処理したDNAへ結合するプローブが設計される。ある実施形態において、CpGメチル化は、例えば、外側プライマーを用いて亜硫酸水素処理したDNAのPCR増幅後に検出される。ある実施形態において、初回PCR反応から増幅した生成物は、メチル化特異的プライマーまたは非メチル化特異的プライマーを用いた入れ子式PCR反応のためのテンプレートとして機能する。ある実施形態において、標準曲線は、特定の試料中のメチル化した分子の百分率を測定するために確立される。核酸のメチル化(例えば、RNAまたはDNAのメチル化)の検出方法は、当該技術分野で公知である。例えば、Laird,P.W.,Nature Rev.Cancer 2003,3:253〜66、Belinsky,S.A.,Nature Rev.Cancer 2004,4:1〜11を参照されたい。
ある実施形態において、統計分析は、シチジン類似体を含む特定の医薬組成物を用いた治療の潜在的な利益による特定のメチル化レベルの影響を評価するために実施する。ある実施形態において、全体的な生存に及ぼすメチル化の影響は、例えば、コックス比例ハザードモデル及びカプラン・マイヤー(KM)法を用いて評価する。
ある実施形態において、MDS及び/またはAMLと関係する任意の遺伝子は、患者における当該遺伝子のメチル化状態について検討され得る。特定の遺伝子には、CKDN2B(p15)、SOCS1、CDH1(E−カドヘリン)、TP73、及びCTNNA1(アルファ−カテニン)を含むが、これらに限定しない。本明細書に開示する方法における使用に適しているであろうMDS及び/またはAMLと関係する特定の遺伝子は、当該技術分野で公知である。
(1.1つ以上の追加の治療薬を本明細書に開示する経口製剤と併用投与することを含む方法)
本明細書のある実施形態は、本明細書に開示する疾患または障害(例えば、異常な細胞増殖を包含する疾患または障害)の治療方法を提供し、この中で、当該方法は、相乗的な治療効果を生じるために、本明細書に開示する経口製剤(例えば、5−アザシチジンを含む経口製剤のような)を、1つ以上の追加の治療薬(例えば、抗PD1/抗PDL1モノクローナル抗体、例えば、ペンブロリズマブ、MK−3475、ピディリズマブ、ニボルマブ(BMS−936558、MDX−1106、またはONO−4538)、BMS−936559、アテゾリズマブ(MPDL3280A)、またはデュルバルマブ(MEDI4736)のような)と併用投与することを含む。本明細書に開示する方法において有用な特定の併用投与する治療薬は、本明細書のいたるところで開示する。特定の実施形態において、追加の治療薬は、治療有効量である量で併用投与する。特定の実施形態において、追加の治療薬は、併用投与されるシチジン類似体剤形とは別個の剤形において併用投与する。特定の実施形態において、追加の治療薬は、併用投与するシチジン類似体と一緒の剤形(例えば、単一単位剤形)において併用投与する。このような場合、シチジン類似体(例えば、5−アザシチジン)及び追加の治療薬は、本明細書に開示する方法及び当該技術分野で公知の方法を含む有効医薬成分の同時製剤方法を用いて同じ剤形において一緒に同時製剤され得る。
(参照による組み込み)
本明細書のいたるところで参照する開示(例えば、特許、刊行物、及びウェブページ)はすべて、それらの全体が参照により組み込まれる。加えて、以下の開示、すなわち(1)B.S.Skikne,M.R.Ward,A.Nasser,L.Aukerman,G.Garcia−Maneroによる2008年ASCOポスター要約、(2)G.Garcia−Manero,M.L.Stoltz,M.R.Ward,H.Kantarjian,及びS.Sharma,Leukemia,2008,22,1680−84、ならびに(3)WO2009/139888も、それらの全体が参照により本明細書に組み込まれる。
(VI.実施例)
(A.実施例1)
再発性上皮性卵巣癌に罹患している女性における経口5−アザシチジンを用いたエピジェネティックな刺激を用いたまたは用いないペンブロリズマブによる免疫チェックポイント阻害の第2相多施設無作為化二重盲検偽薬対照試験を実施する。
本試験は、AZAの経口で生体利用可能な製剤である経口5−アザシチジンがEOC患者の腫瘍におけるAIM発現を誘導でき、それによりモノクローナル抗体ペンブロリズマブを用いたPD−1阻害に対するこれらの腫瘍の応答を亢進できるという仮説を検査する。
本試験の目的は、上皮性卵巣癌患者におけるペンブロリズマブの活性及び安全性を単独で及び経口5−アザシチジンとの組み合わせで評価することとする。
(目的)
第一の目的は、両処置群における進行のない生存(PFS)を概算すること、及びペンブロリズマブ単一療法群に対する併用群についてPFSハザード比を概算することとする。第二の目的は、全体的な生存(OS)、客観的応答率(ORR)、臨床上の利益率(CBR)、及び両処置群における臨床上の利益の持続時間を概算し、安全性を評価することとする。探索目的は、ペンブロリズマブ単独または経口5−アザシチジンとの組み合わせに対する応答に及ぼす基線におけるAIM遺伝子発現の影響を評価することとする。
(評価項目)
主要評価項目は、PFSの判定である。副次的評価項目は、OS、ORR、CBR、臨床上の利益の持続期間、及び安全性の判定である。探索的評価項目は、AIM遺伝子発現、循環腫瘍DNA中の経口5−アザシチジン投薬による遺伝子座特異的メチル化の変化、腫瘍浸潤リンパ球の定量及び特徴づけの判定である。
(試験設計)
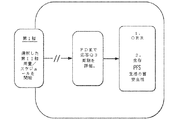
本試験は、無作為化した偽薬対照の並行群の多施設二重盲検第2相試験である。被験者に、2つの処置群のうちの1つに対して1:1の比で無作為に割り当てる。すなわち、1日目にペンブロリズマブを30分間静脈内注入するとともに1〜14日目に経口5−アザシチジン(300mg)または偽薬を21日ごとに経口投与する。本試験は、運営委員会及び独立したデータモニタリング委員会の指針の下で実施する。
(被験者)
およそ120名の患者を2つの処置群間で、1:1ベースでおよそ20か月にわたって無作為に割り当てる。主要分析は、合計およそ80PFSの事象が観察されたときに実施する。この事象数は、最後の患者の無作為化の後に観察されると期待される。PFS分析時に副次的評価項目もすべて分析する。OSの経過観察分析は、80例のOS事象が最後の患者の無作為化後に観察された時に実施する。PFS、ORR、CBR、臨床上の利益の持続時間についての概算値は、OS分析時に更新する。
(組み入れ基準)
本試験についての組み入れ基準は次のとおりである。
1.被験者は、インフォームドコンセント用紙に署名した時点で18歳以上である
2.組織学的に記述された乳頭状血清上皮性卵巣癌
3.白金二重線化学療法を完了した6か月以内に記述された再発
4.0〜1の皮質脳波成績状態
5.RECIST1.1により測定可能な疾患
6.適切な器官機能:
a.2.5×正常範囲上限(ULN)以下の、または肝臓転移がある場合、5×ULN範囲以下のAST(SGOT)、ALT(SGPT)
b.1.5×ULN以下の総ビリルビン
c.1.5×ULN以下のクレアチニン
d.正常範囲内の、または栄養補助食品で修正可能なカリウム
7.適切な骨髄機能:
e.1.5×109個/L以上の絶対的好中球計数
f.100×109個/L以上の血小板
g.9g/dL以上のヘモグロビン
妊娠能力のある女性は、スクリーニング時に陰性の血清妊娠検査結果を有していなければならず、妊娠防止必要条件を遵守しなければならない。
(除外基準)
除外基準は次のとおりである。
1.3g/dL未満の血清アルブミン
2.中枢神経系への転移または癌性髄膜炎に関する既往歴
3.自己免疫障害に関する既往歴
4.肺臓炎若しくは間質性肺疾患またはステロイドの使用を必要とする何らかの他の医学的容態に関する既往歴
5.臨床上有意な心不全または血栓塞栓事象に関する既往歴
6.炎症性腸疾患(例えば、クローン病、潰瘍性大腸炎)、セリアック病、先行胃切除若しくは腸上部除去、あるいは本試験薬の吸収、分布、代謝若しくは排泄に干渉するであろう、または被験者を消化管毒性の高い危険に対して易罹患性にするであろういずれかの他の消化管障害に関する病歴
7.無作為化前2週間以内の大手術または被験者が手術の副作用から回復していない
(治療投与計画)
単一療法群:ペンブロリズマブ:静脈内注入として、30分間にわたって1日目に10mg/kgの用量で21日ごとに、対応する経口5−アザシチジン偽薬とともに投与した。併用群:ペンブロリズマブ:静脈内注入として、30分間にわたって1日目に10mg/kgの用量で21日周期ごとに、1〜14日目に各21日周期で毎日300mgの用量で経口投与する経口5−アザシチジンとともに投与した。
(薬剤供給の詳細)
ペンブロリズマブ:Merck Corporationは、静脈内投与用ペンブロリズマブを供給する。ペンブロリズマブは、単回使用のみのために企図したI型ガラスバイアル中の白色〜オフホワイト色の凍結乾燥した粉末として提供する。注射用ペンブロリズマブ粉末50mg/バイアルは、使用前に注射用滅菌水で元に戻す。薬剤製品は、冷蔵条件(2℃〜8℃)下で安定した凍結乾燥粉末として保管する。5−アザシチジン:Celgene Corporationは、経口投与用の5−アザシチジン(または対応する偽薬)100mg錠を供給する。錠剤はすべて、ブリスターパックに包装されている。
(治療期間)
被験者は、放射線学的疾患の進行(RECIST1.1による)まで、被験者が新たな抗癌治療を開始するまで、同意の撤回、被験者の拒絶、医師の判断、用量遅延若しくは用量減少によって管理できない毒性、死亡、または何らかの理由による試験終了まで、治療され得る。
(治療後の観察期間)
患者は、12週間ごとに、または要求のある場合はより頻繁に生存について追跡される(電話連絡で十分である)。この期間中、さらなる抗癌療法情報(投与計画、開始日及び終了日)が収集される。シスプラチン二重線化学療法後の6か月以内に再発を経験するEOC患者についてのOS中央値はおよそ12.0か月である。
(評価)
効能:RECIST1.1による腫瘍評価は、記述された疾患進行まで、被験者が新たな抗癌治療を開始するまで、同意の撤回、被験者の拒絶、医師の判断、用量遅延若しくは用量減少によって管理できない毒性、死亡、または何らかの理由による試験終了まで、無作為化から6週間ごとに(±5日)実施する。安全性:被験者は全員、被験者がインフォームドコンセントに署名した時点から始まり、IPの最終投与または治療終了(EOT)来院の28日後までのいずれか遅い方まで、有害事象についてモニターされる。医学的容態の完全な評価は、適格性についてのスクリーニング中に実施する。身体的検査(書面記入のみ)、バイタルサイン、実験的評価(例えば、血清化学、血液学)、ECG、及びECOG成績状態は、規則的にモニターする。治験の被験者またはそのパートナーにおける妊娠を回避するための予防措置が取られ、妊娠能力のある女性は、規則的な妊娠検査を受ける。薬物動態:薬物動態パラメータは試験する。生体マーカー:新鮮生検は、同意した被験者から無作為化前の21日間のスクリーニング期間中に収集しなければならない。血液検体及び血漿検体も収集する。
(統計方法)
本試験の第一の目的は、仮説を形式的に検査する代わりに概算することであるので、120の検体サイズを実現可能性に基づいて判定し、形式的な検定力計算を通じては判定しなかった。第一の分析は、合計およそ80のPFS事象を記述した時に実施する。このことは、最後の被験者の無作為化後に生じると見込まれる。併用群とペンブロリズマブ単一療法群とのハザード比についての信頼区間は、数個の仮定上のハザード比を基に算出することができる。PFSは、カプラン・マイヤー法を用いて中央値によって集約する。コックス比例ハザードモデルは、併用群とペンブロリズマブ群とのハザード比(両側の95%CIを含む)を概算するのに使用する。RR、CBR及び臨床上の利益の持続時間といった副次的評価項目は、PFS分析時に評価する。OSは、合計80例の死亡が報告されたときに報告する。他の効能の評価項目(PFS、RR、CBR、臨床上の利益の持続時間)に関する更新した分析もOS分析時に表す。
(B.実施例2)
注射可能な低メチル化薬(HMA)を用いた治療に応答中ではない骨髄異形成症候群(MDS)または急性骨髄性白血病(AML)の治療のためのペンブロリズマブと併用した経口5−アザシチジンの安全性及び忍容性に関する第1/2相国際多施設単一群研究を実施する。試験の流れ図については図1を参照されたい。
(目的)
第I相における第一の目的は、投与計画の安全性及び忍容性を評価し、さらなる評価のために推奨される第II相用量(RP2D)を定義することとする。第II相における第一の目的は、注射可能なHMAに応答しなかったMDS患者またはAML患者を治療するのに使用する場合、この投与計画と関係した効能基準を評価することとする。第I相における第二の目的は、これらの初回の被験者において観察される効能シグナルを評価することとする。第II相における第二の目的は、連続した治療周期において付与する場合の投与計画の安全性及び忍容性を評価することとする。探索目的は、当該投与計画の効能または当該投与計画に対する耐性と相関し得る骨髄及び/または末梢血における分子マーカー及び細胞マーカーを評価することとする。
(評価項目)
第I相における主要評価項目は、用量制限毒性ならびに報告する他の有害事象(AE)の数、タイプ及び重症度の判定である。第II相における主要評価項目は、全体的な応答率の判定である。第I相における副次的評価項目は、客観的応答の数、タイプ、及び臨床上の関連性の判定である。第II相における副次的評価項目は、生存、進行のない生存、応答の持続時間、有害事象の数、タイプ、及び重症度の判定である。探索評価項目は、生体マーカーの評価である。
(試験設計)
これは、注射可能な低メチル化薬(HMA)を用いた治療に応答しなかったMDS患者またはAML患者の治療のための、経口5−アザシチジンとペンブロリズマブとの併用に関する位相I/IIの制御されていない試験である。第I相は、併用投与計画のためにRP2Dを定義して、治療に対する生物学的及び臨床上の応答を探索するよう尽力する。第II相において、検体サイズは、HMA難治性のMDSまたはAMLの治療のための概念証明を得ようとするために、ならびに当該投与計画の安全性及び忍容性と関連した知識体系を増やすために増大させる。本試験は、投与コホート0(経口5−アザシチジン、200mgBID21/28日及びペンブロリズマブ10mg/Kgを周期7日目及び21日目(または8日目及び21日目))において3名の被験者を初回登録する。図2、3、及び4を参照されたい。当該初回の3名の被験者のうちの1名が第一の治療周期において事前に定義しておいた用量制限毒性(DLT)を受ける場合、追加の3名の被験者を当該コホートに登録する。第一の周期のDLTが任意の投与コホートにおいて2以上みられる場合、当該用量レベルは、不耐性であるといえ、3名の被験者は、次のより低い投与コホート、例えば、投与コホート−1に登録される。さもなくば、当該投与コホートは、耐性があるといえ、第II相におけるさらなる評価についての開始用量となる。第I相におけるRP2Dを受容する3〜6名の被験者は、本試験の第II相部分に含まれ、第II相について説明したような効能及び安全性について評価される。十分な人数の被験者が第II相に含まれる。被験者は、第I相からRP2Dにおける治療を開始し、毒性を対処するよう調整した当該RP2Dの用量またはスケジュールを有し得る。被験者は、疾患の応答または進行の徴候を得ようと求めるために、3回の治療周期ごとの後に骨髄評価を受ける。各周期で血液学的パラメータが評価される。被験者が、完全な応答(CR)を経験する場合、ペンブロリズマブを用いた投薬は中断されるが、経口5−アザシチジンを用いた投薬は続行する。治療はすべて、記述された客観的疾患進行を基に中断される。主要評価項目、全体的な応答率の分析は、最後の有効な被験者が6周期の治療を完了した場合に実施する。
(組み入れ基準)
本試験についての組み入れ基準は次のとおりである。
1.MDS(中間1、中間2、高)またはAML
2.先行注射可能HMA(4周期対6か月間)
3.iHMAに対する最良の応答としての進行性疾患または安定疾患
4.記述された客観的PD/SD対「悪化する血球減少、芽細胞の増加、またはFABサブタイプの進行」
5.12週以内(C1D1に対して3週間以上)のiHMAの最終用量
6.ECOG0、1、2
(除外基準)
本試験についての除外基準は次のとおりである。
1.迅速に進行するMDS(客観的基準対侵襲的判断(Inv.Judgment))
2.先行経口デシタビン
3.iHMAに対する先行応答または進行中の応答(再発を除外する場合)
4.ESA、TSAなど、ヒドロキシ尿素、他のモノクローナル抗体、または28日以内の生ワクチン
5.14日以内の他の疾患療法
6.7日以内の全身性副腎皮質ステロイド
7.消化管障害
8.活発な中枢神経系の関与
9.自己免疫障害または他の必要とする免疫抑制
10.間質性肺疾患
11.実験結果
・骨髄芽球30(33)%超
・白血球20K/μL(30K)超
・血清クレアチニン2.5×ULN(2×)超
・血清総ビリルビン1.5×ULN超
・血清アスパラギン酸アミノトランスフェラーゼ(AST)またはアラニンアミノトランスフェラーゼ(ALT)2.5×ULN超
治療投与計画ペンブロリズマブ:静脈内注入として30分間かけて、7日目及び21日目(または8日目及び21日目)に28日周期で10mg/kgの用量で、各28日周期の1〜21日目における200mgBIDの用量で経口投与する経口5−アザシチジンと併用投与した。さらなる第I相投与コホートは、以下の表3に提供する。
表3:第I相投与コホート
(治療持続時間)
被験者は、CR、PD、同意の撤回、被験者の拒絶、医師の判断、用量遅延若しくは用量減少によって管理できない毒性、死亡、または何らかの理由のための試験終了まで治療され得る。CRからの再発の際に再治療する。
(治療後観察期間)
PDまたは新たな療法までQ6週間(来院)、次いで、死亡までQ12週間(電話)。iHMA療法後の疾患進行後の生存は4〜6か月間である。このことは、いずれかの単一の薬剤を用いて延長され得、本発明者らは、併用からの相乗性を期待する。
(評価)
効能:IWG2006応答評価(骨髄生検/骨髄液)は、2周期目、4周期目、及び6周期目における投与の完了後に、次いで、疑いのある疾患の応答または進行を確認するために3回の治療周期ごと(9、12、15、など)における投与の完了後に、及び治療中断の際に実施する。血液学及びヘモグロビンの評価は、毎周期の完了後に実施する。生活の質(QoL)の評価は、毎周期後に測定する。抗ペンブロリズマブ抗体の評価は、毎周期後に測定する。安全性:被験者を全員、疾患の進行/転換についてモニターする。身体的検査(ソースの書面のみ)、バイタルサイン、実験的評価(例えば、血清化学、血液学)、ECG、及びECOG成績状態は、規則的にモニターする。薬物動態:薬物動態パラメータを試験する。図5を参照されたい。
(実施例3)
非小細胞肺癌の治療のためのペンブロリズマブと併用する経口5−アザシチジンに関する第2相多施設無作為化二重盲検偽薬対照試験を実施する。
(目的)
主要目的は、両治療群における進行のない生存(PFS)を概算すること、及びペンブロリズマブ単一療法群と比較した併用群についてのPFSハザード比を概算することとする。第二の目的は、全体的な生存(OS)、客観的応答率(ORR)、臨床上の利益率(CBR)、ならびに両治療群における臨床上の利益の持続時間を概算し、安全性を評価することとする。
(評価項目)
主要評価項目は、PFSの判定である。副次的評価項目は、OS、ORR、CBR、臨床上の利益の持続時間、及び安全性の判断である。
(試験設計)
本試験は、無作為化偽薬対照並行群多施設二重盲検第2相試験である。被験者は、2つの治療群、すなわち21日ごとに1〜14日目に経口投与する経口5−アザシチジン(300mg)または偽薬を伴う、1日目に30分間の静脈内注入としてのペンブロリズマブ、のうちの1つに対して1:1の比において無作為に割り当てる。本試験は、運営委員会及び独立したデータモニタリング委員会の指針の下で実施する
(被験者)
およそ120名の患者をおよそ20か月にわたる2つの治療群間で、1:1ベースで無作為に割り当てる。主要な分析は、合計およそ80例のPFS事象が観察された場合に実施する。この事象数は、最後の患者の無作為化後に観察されると期待される。PFS分析時に、副次的評価項目もすべて分析する。OSについての経過観察分析は、80例のOS事象が最後の患者の無作為後に観察された場合に実施する。PFS、ORR、CBR、及び臨床上の利益の持続時間についての概算値は、OS分析の時点で更新する。
(D.実施例4)
骨髄異形成症候群(MDS)または急性骨髄性白血病(AML)の治療のための5−アザシチジン単独またはデュルバルマブ(MEDI4736)との併用での安全性及び忍容性に関する試験を実施する。経口5−アザシチジンは、低メチル化薬(HMA)を用いた治療に応答しなかったMDS患者に対して単独でまたはデュルバルマブ(MEDI4736)との組み合わせで使用する。注射可能な5−アザシチジンは、未治療(第一の線)のより高い危険にあるMDSに罹患している患者に対して単独でまたはデュルバルマブ(MEDI4736)との組み合わせで使用する。
第一の線の治験はまた、第III相試験のための調製物における同種幹細胞移植を受けるのに適格ではない未治療のAMLに罹患している高齢患者におけるアザシチジン及びデュルバルマブ(MEDI4736)の組み合わせについて、または顕著な効能がある場合、米国における認可の加速のために、無作為化第II相コホートからデータを生じる。
これら2つの適応症は、満たされていない高い医学的需要を実証する。入手可能な科学的データの分析は、注射可能な5−アザシチジンまたは経口5−アザシチジンのいずれかとデュルバルマブとの組み合わせが、これらの疾患に罹患している患者の寿命を優位に改善するために十分相乗的であるという仮説を支持している。現に入手可能な制限された知識を基に、免疫療法及びエピジェネティックな療法の組み合わせの探索は、MDSの管理を改善するためには優先性が高い。
(目的)
本試験は、安全性に関する初回組み合わせデータ、予備的効能シグナル及びおそらく関連性のある生体指標データを得る。
(試験設計)
本試験の第一の相は、3つの適応症における3つの異なる臨床試験を含む。患者は、治験に含まれる進行性の(pregressive)疾患(PD)または安定疾患(SD)により分類され、両タイプについて別個に応答を評価する(evaluate)。
1つの治験において、注射可能な5−アザシチジンは、デュルバルマブ(MEDI4736)単一薬を用いた全体的な応答(OR)に到達しない患者へ、デュルバルマブとの組み合わせで投与する。このことは、他の試験との競合なしで、早期の安全性及び効能シグナルの発生を可能にするであろう。
1つの治験において、経口5−アザシチジンは、単独でまたはデュルバルマブとの組み合わせで、より高い危険性のMDS及びHMA不全に罹患している患者において投与する。この治験は、第一の効能シグナルを生じるべきであり、進む判断/進まない判断のための十分に定義したパラメータを介して、無作為化第II相拡大相において併用療法対経口5−アザシチジン単独の効能を実証する。この治験はまた、その後の第III相治験の計画を支援する。
1つの治験において、安全性慣らしのある第II相治験は、デュルバルマブと注射可能な5−アザシチジンとの組み合わせを評価するために、先行では未治療のより高い危険性にあるMDSに罹患している患者を分ける(part in)。この治験は、より良好な免疫機能を有するかつあまり耐性のない疾患に罹患している集団におけるデータを生じるべきである。同じCelgene依頼の治験において、注射可能なアザシチジンとデュルバルマブの組み合わせ対注射可能な5−アザシチジンは、異種性幹細胞移植のための候補ではないAML高齢患者のコホート(WHOで定義)における無作為化第II相設計において評価する。
5−アザシチジンは、IPSSにより、より低い危険性及びより高い危険性の現行コホートにおけるデュルバルマブに対していかなる時も疾患の進行に応答しない(少なくとも4周期後にCR、PR及びHIなし)または当該疾患の進行を発達させない患者へ投与される。デュルバルマブは、10mg/体重kgで2週間ごとに(Q2W)投与され、5−アザシチジンは、75mg/m2/日で1日目から7日目まで4週間ごと(Q4W)に投与される。あるいは、一部において、経口5−アザシチジンは、200mgBIDを28日における1〜21に投与され(21/28日、Q4W)、患者群における経口5−アザシチジン200mgBIDの安全性を判定する。別の部分において、経口5−アザシチジンは、200mgBIDを28日における1〜21で投与され(21/28日、Q4W、血液学的毒性または消化管の毒性の場合、段階的に減少)、デュルバルマブ(1.5g)は、4週間ごとに投与し、必要な場合、段階的に減少させる。
DLTの場合、用量を段階的に減少させる。
(治療持続時間)
5−アザシチジンは、進行または忍容できない毒性まで投与する。デュルバルマブについて、12か月間の治療期間の終了を通じて完全な応答(CR)、骨髄の完全な緩解(mCR)、部分的応答(PR)、SD、または血液学的改善(HI)を達成及び維持する被験者は、経過観察に入る。経過観察の初期の12か月間に、被験者がPDを有している場合、被験者の試験治療は、さらに最長12か月間再投与され得るとともに、被験者がPDの設定における再治療についての基準を満たし、当該疾患についての他の抗癌治療を受けたことがなく、試験実施計画書についての治験生成物中断基準のうちのいずれも満たさない場合、同じ治療指針に最初の12か月間準拠する。1回の再治療のみが許容される。
(E.実施例5)
注射用アザシチジンまたはデシタビンを用いた治療に対する客観的な応答に到達し損なっている、骨髄異形成症候群に罹患している被験者における経口アザシチジンの効能及び安全性を単独で及びMEDI4736(デュルバルマブ)との組み合わせで評価するための第2相国際多施設無作為化非盲検並行群試験。
(目的)
本試験の第一の目的は、注射可能な低メチル化薬(HMA−注射用アザシチジンまたはデシタビン)を用いた最も直近の治療に応答しなかった、または注射可能なHMAを用いた治療に忍容できなかったMDSに罹患している被験者における経口5−アザシチジンの効能を、単一療法として、及び抗PD−L1モノクローナル抗体であるデュルバルマブとの組み合わせにおいて調査することとする。さらなる目的は、MDSについての治療としての経口5−アザシチジンの単独での及びデュルバルマブとの組み合わせにおける安全性及び忍容性を評価すること、経口5−アザシチジンを単独で及びデュルバルマブとの組み合わせで使用する治療と関係した客観的な血液学的応答及び/または生物学的応答に関する臨床関連性を説明すること、MDSに罹患している被験者における経口5−アザシチジンとの組み合わせで付与する場合のデュルバルマブの免疫原性を判定すること、ならびにMDSに罹患している被験者におけるデュルバルマブ及び経口5−アザシチジンの薬物動態を評価することとする。
(試験設計)
これは、3つの試験相、すなわちスクリーニング、無作為化した治療、および経過観察からなる第2相国際多施設無作為化並行群非盲検試験である。安全性挿入は、経口5−アザシチジン単独及びデュルバルマブとの組み合わせにおける安全性及び忍容性を探索して、組み合わせで送達することになっている2つの薬剤の能力を制限する重複する毒性または相乗的な毒性がないことを確認する(図6を参照されたい)。組み合わせ投与計画の許容性がいったん実証されたら、無作為化治療相への登録を開始してもよい。
本試験のいたるところで、被験者は、MDSのための最も直近の治療として付与したiHMA療法に対する応答により、進行性疾患(PD)または安定疾患(SD)に罹患しているものとして分類する。各治療群に割り当てられたPD及びSDに罹患している被験者の数は、各亜集団について計画された分析を可能にするようモニターする。したがって、本試験において評価する4つのコホートがある。
単一療法、進行性疾患
単一療法、安定疾患
併用療法、進行性疾患
併用療法、安定疾患
無作為化した治療相において、適格性のある被験者は、経口5−アザシチジンを単独でまたはデュルバルマブとの組み合わせで服用する。治療相は、2つの病期で実施し、当該4つの試験コホートの各々についての無益評価は、当該コホートが第2期へ進行するかどうかを判定する(図7を参照されたい)。第一の分析は、第2期の完了を追随し、追加の分析は、最後の被験者が登録したおよそ12か月後に実施する。
本試験は、世界中の最多およそ75の研究施設でおよそ69〜130名の被験者を登録する。少なくとも12名の被験者は、本試験の安全性挿入相に含まれ、6名は経口5−アザシチジン単一療法を受け、6名は経口5−アザシチジン+デュルバルマブ併用療法を受ける(図6を参照されたい)。固定用量のデュルバルマブと組み合わせた経口5−アザシチジンの用量及びスケジュールを決定する。
およそ57名の被験者は、本試験の無作為化した治療相の第1期において登録する。試験登録時に進行性疾患に罹患しているおよそ16名の被験者及び試験登録時に安定疾患に罹患している18名は、経口5−アザシチジン単一療法を受けるよう無作為化する。試験登録時に進行性疾患に罹患しているおよそ9名の被験者及び試験登録時に安定疾患に罹患している14名は、経口5−アザシチジン+デュルバルマブ併用療法を受けるよう無作為化する。十分な客観的応答が1つ以上のコホートで観察された場合、適用可能なコホート(複数可)は、最多およそ130名の被験者について、単一療法群における進行性疾患に罹患している追加のおよそ15名の被験者及び/または安定疾患に罹患している19名の被験者を、ならびに併用療法群において進行性疾患に罹患しているおよそ8名の被験者及び/または安定疾患に罹患している7名の被験者を含むよう、第2期において拡張する(図7を参照されたい)。
無作為化した治療相の間に、PKサンプリングは、必要とする能力を有する選択した施設で併用療法群へ無作為化したおよそ10〜12名の被験者において実施する。このことは、潜在的な薬剤間相互作用及びPKパラメータに及ぼす免疫原性の効果についての評価を可能にする。試験登録時にPDに罹患している少なくとも5名の被験者及びSDに罹患している少なくとも5名の被験者を含むよう尽力し尽くす。
(試験集団)
本試験は、MDSのための最後の治療として注射可能な低メチル化薬(注射用アザシチジンまたはデシタビン)を用いた適切な療法コースを用いた治療に応答しなかった、または少なくとも3か月間の試行した治療後に注射可能なHMAを用いた治療に忍容できなかったMDSに罹患しているおよそ69〜130名の被験者を登録する。少なくとも6名の被験者は、本試験の安全性挿入相の間に2つの治療群、すなわち単一療法(経口5−アザシチジン)または併用療法(経口5−アザシチジン+デュルバルマブ)の各々に登録する。各治療群における進行性疾患(PD)に罹患している少なくとも3名の被験者及び安定疾患(SD)に罹患している3名の被験者を含むようあらゆる試行をする。およそ57名の被験者は、治療相の第1期に登録する。登録は、iHMA療法後のPD及びSDに罹患している適切な数の被験者が、第1期の間のこれらの亜集団における無益を評価するよう含まれていることを確実にするためにモニターする。十分な客観的応答が1つ以上のコホート(SD単一療法、PD単一療法、SD併用療法、PD併用療法)において観察される場合、適用可能なコホート(複数可)は、最多およそ49名の追加の被験者を含むよう、第二の試験期において拡張する。また、登録は、計画した分析を可能にするよう、各コホートにおけるPD及びSDに罹患している適切な数の被験者を確実にするようモニターする。
(試験の長さ)
本試験の総持続時間は、およそ36か月であると期待する。被験者は、インフォームドコンセント文書(ICD)の署名後に最長28日間の期間にわたってスクリーニング手順を受ける。適格性のある被験者は、最長6回の28日治療周期のためにIPを受ける本試験の安全性挿入相または無作為化した治療相へと続く。治療から利益を得る被験者は、当該利益の喪失までIPを続行することがある。治療の中断後、被験者は、28日間の経過観察来院を行った後、本試験の経過観察相において4か月ごとに電話連絡を受ける。本試験の登録期間は、およそ24か月間続くと期待する。試験の治療相および経過観察相は、最後の被験者が登録したおよそ12か月後に結論付けると期待する。それゆえ、本試験の総持続時間は、およそ36か月間であると期待する。治験終了は、本試験を完了するための最後の被験者の最後の来院日、または当該実施計画書及び/または統計分析計画(SAP)において事前に指定したような、一次分析、二次分析及び/または探索分析に必要とする最後の被験者からの最後のデータ点の受理日のいずれかのより遅い日付と定義する。
試験治療(調査製品−IP)(経口アザシチジン)
単一療法治療群へ割り当てられる適格性のある被験者は、本試験の安全性挿入相の間に同定した用量及びスケジュールで経口5−アザシチジンを服用する。用量及びスケジュールは、毒性を管理するために調整し得る。少なくとも2回の十分に忍容した治療周期(cyle)後に経口5−アザシチジンを用いた治療に忍容するが疾患を悪化させる徴候を示すまたは血液学的改善若しくはより良好な(HI)を経験していない被験者は、当該用量が増加することがある。また、用量及びスケジュールは減じることができるが、300mgBID、21/28日を超える用量上昇は許容され得ない。被験者は、実施計画書に指定の再治療基準がすべて適合し続ける限り、経口5−アザシチジンを用いた治療を続行することができる。
(MEDI4736(デュルバルマブ))
併用療法治療群へ割り当てられる適格性のある被験者は、先に説明したような経口5−アザシチジン、及び各28日治療周期の1日目に1時間の静脈内(IV)注入のデュルバルマブ1500mgを受ける。免疫関連AE(irAE)または注入タイプの反応を含む、デュルバルマブを用いた治療と関連すると考えられる毒性については、デュルバルマブの注入は遅延または中断され得る。デュルバルマブについての用量減少は許容され得ない。被験者は、実施計画書の指定する再治療基準がすべて満たされ続ける限り、経口5−アザシチジンと組み合わせたデュルバルマブを用いた治療を続行することができる。経口5−アザシチジンを用いた治療が何らかの理由で中断される場合、デュルバルマブを用いた投与は同様に中断され、被験者は、本試験の経過観察相に入る。
(効能評価の概観)
本治験の主要効能評価項目は、経口5−アザシチジンを単独で用いた及びデュルバルマブとの組み合わせにおける治療に対する、治療客観的応答(HI、PR、CR、または骨髄CR−IWG2006基準から修正)に到達する被験者の比率である。この評価項目を評価するために、骨髄検査は、JPを開始することに続いて最初の6回の治療周期の間の2周期ごとの治療が行われる前に必要とする。第6周期を超えて続行する被験者は、3治療周期ごとの後に、または血液学的応答若しくは疾患進行が疑われるのを確認する必要がある場合、骨髄検査を受ける。骨髄検体(吸引液及び/または生検)は、末梢血塗抹及び関連する臨床情報とともに、疾患の分類、応答、及び/または進行の判断における一貫性を提供するために、独立した病理学者による概観のために提出する。白血球(WBC)が差次的な完全な血球数(CBC)及び血小板を含む血液学的パラメータは、中央実験施設が評価する。本治療の効能に及ぼすデュルバルマブに対する何らかの免疫原性応答の潜在的な影響を理解するために、免疫原性評価を実施する。
(探索評価の概観)
骨髄検体及び末梢血検体は、スクリーニング中に、本試験の治療相のいたるところで、及び治療の中断の際に収集する。これらの検体は、アザシチジン効能(注射用アザシチジンに対する耐性及び/または経口5−アザシチジンの耐性若しくは感受性)に影響し得る生物学的マーカーの潜在的な将来的な探索評価のために保管する。治療の応答または不全を予測する生物学的マーカーを識別することは、MDS患者の個々の疾患特徴を基にしたMDS患者の治療のための低メチル化薬療法の使用における、より標的化したアプローチを可能にし得る。可溶性PD−L1及び血漿サイトカイン/ケモカインのレベルのようなデュルバルマブ療法の薬力学的生体マーカーは、末梢血検体において調査する。PD−1/PD−L1タンパク質発現、遺伝子発現署名、循環中の可溶性タンパク質、遺伝的突然変異及び染色体異常、腫瘍浸潤リンパ球の存在、TCRクローン性、ならびに耐性の潜在的な機序としての他のチェックポイント分子発現(PD−L2、Tim−3、Lag−3、CTLA−4)を含むがこれらに限定しない、経口5−アザシチジン/デュルバルマブ併用療法に対する応答の探索的機械論的な及び予測する生体マーカーを評価する。
本明細書に引用する参考文献の全部は、それらの全体が参照により組み込まれる。本明細書に提供する方法は、特定の実施形態に関して説明してきたが、種々の変更及び改変が添付の特許請求の範囲によって列挙されるような精神及び範囲から逸脱することなく行えることは、当業者に明白であろう。
先に説明した実施形態は、単に例示的であるよう企図されており、当業者は、常用の実験法のみ、具体的な化合物、材料及び手順に関する数多くの等価物を用いて認識するであろうし、または確認することができるであろう。このような等価物はすべて当該範囲内であるとみなし、添付の特許請求の範囲によって包含する。