JP2006257580A - Polyacrylonitrile-based polymer for precursor fiber of carbon fiber, precursor fiber of carbon fiber and method for producing carbon fiber - Google Patents
Polyacrylonitrile-based polymer for precursor fiber of carbon fiber, precursor fiber of carbon fiber and method for producing carbon fiber Download PDFInfo
- Publication number
- JP2006257580A JP2006257580A JP2005076621A JP2005076621A JP2006257580A JP 2006257580 A JP2006257580 A JP 2006257580A JP 2005076621 A JP2005076621 A JP 2005076621A JP 2005076621 A JP2005076621 A JP 2005076621A JP 2006257580 A JP2006257580 A JP 2006257580A
- Authority
- JP
- Japan
- Prior art keywords
- carbon fiber
- fiber
- polyacrylonitrile
- polymer
- fiber precursor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Artificial Filaments (AREA)
- Inorganic Fibers (AREA)
Abstract
Description
本発明は、引張強度および引張弾性率に優れた炭素繊維の製造方法に関するものである。更には、前記した高性能な炭素繊維を製造するのに好適な炭素繊維前駆体繊維用ポリアクリロニトリル系重合体および炭素繊維前駆体繊維の製造方法に関するものである。 The present invention relates to a method for producing a carbon fiber excellent in tensile strength and tensile elastic modulus. Further, the present invention relates to a polyacrylonitrile-based polymer for carbon fiber precursor fibers suitable for producing the above-described high-performance carbon fibers and a method for producing carbon fiber precursor fibers.
炭素繊維は、その優れた力学特性および電気特性からさまざまな用途に利用されている。近年では、従来のゴルフクラブや釣竿などのスポーツ用途、航空機用途に加え、自動車部材、CNGタンク、建造物の耐震補強、船舶部材などいわゆる一般産業用途への展開が進み、それに伴い、求められる力学特性のレベルも高まっている。例えば航空機用途では、軽量化のため構造部材の多くが炭素繊維強化プラスチックに置き換えられつつあり、引張強度と引張弾性率が高いレベルで両立した炭素繊維が求められている。 Carbon fibers are used in various applications because of their excellent mechanical and electrical properties. In recent years, in addition to conventional golf clubs and fishing rods such as sports applications and aircraft applications, the development of so-called general industrial applications such as automobile members, CNG tanks, seismic reinforcement of buildings, ship members, etc. has progressed. The level of characteristics is also increasing. For example, in aircraft applications, many structural members are being replaced with carbon fiber reinforced plastics for weight reduction, and carbon fibers that are compatible with high levels of tensile strength and tensile elastic modulus are required.
炭素繊維は、工業的にはポリアクリロニトリルなどの前駆体繊維を200〜300℃の空気中で熱処理する耐炎化工程、300〜3000℃の不活性雰囲気中で熱処理する炭化工程を経て製造される。一般に、炭化時の最高温度を高くするほど引張弾性率は高くできるものの、引張強度は1500℃付近で極大となり、さらに高温の領域では、著しい低下が見られ、いわゆるトレードオフの関係となる。炭化温度の制御以外で、引張強度、引張弾性率の両方を高める技術について、これまでいくつかの提案がなされている。 Industrially, carbon fibers are manufactured through a flameproofing process in which precursor fibers such as polyacrylonitrile are heat-treated in air at 200 to 300 ° C. and a carbonizing process in which heat treatment is performed in an inert atmosphere at 300 to 3000 ° C. In general, the higher the maximum temperature during carbonization, the higher the tensile elastic modulus, but the tensile strength reaches a maximum at around 1500 ° C., and a significant decrease is observed in a higher temperature region, which is a so-called trade-off relationship. Several proposals have been made so far for techniques for increasing both the tensile strength and the tensile modulus other than the control of the carbonization temperature.
例えば、用いる重合体の共重合成分を制御することにより、炭素繊維前駆体繊維の酸素透過性を向上させ、耐炎化繊維内の酸素濃度分布を均一に制御し、得られる炭素繊維の引張強度および引張弾性率を向上させる技術が提案されている(特許文献1)。本技術によれば、確かに相対的には、引張強度、引張弾性率の向上効果が認められるものの、その強度レベルは5.1GPaと低く、現在の要求レベルを満足するものではない。しかも、本技術では、酸素透過性を高めるために1.5%を超える多くの共重合成分を用いており、得られる炭素繊維前駆体繊維の耐熱性が低下してしまうという問題があった。耐熱性の低下は、製糸の乾燥熱処理工程やスチーム延伸工程、耐炎化、炭化といった焼成工程における単繊維同士の接着発生の増加を誘起し、得られる炭素繊維の強度レベルを低下させるのである。
一方、用いる重合体の極限粘度を高め、かつ炭素繊維前駆体繊維の繊度を小さくすることで、緻密性を向上させ、高い引張強度、引張弾性率を持つ炭素繊維を製造する技術が提案されている(特許文献2)。本技術では、引張強度9GPa、引張弾性率360GPaと非常に高い力学物性を実現しているものの、用いる重合体の極限粘度を高めることは、(1)重合体のゲル化が生じやすくなり重合体の安定性低下、(2)製糸における延伸性低下、につながり、また、炭素繊維前駆体繊維の繊度を小さくすることは、(3)可紡性の低下、(4)生産性の低下、につながるという多くの問題点があり、工業的に低コストで実現できる技術とは言えないのである。
On the other hand, by increasing the intrinsic viscosity of the polymer to be used and reducing the fineness of the carbon fiber precursor fiber, a technique for producing carbon fibers having high tensile strength and high tensile elastic modulus has been proposed. (Patent Document 2). Although this technology realizes very high mechanical properties such as a tensile strength of 9 GPa and a tensile elastic modulus of 360 GPa, increasing the intrinsic viscosity of the polymer used is as follows: (1) the polymer is easily gelled. Decrease in stability of the fiber, and (2) decrease in stretchability in yarn production. Also, reducing the fineness of the carbon fiber precursor fiber is (3) decrease in spinnability and (4) decrease in productivity. There are many problems of being connected, and it cannot be said that the technology can be realized at low cost industrially.
本発明の課題は、生産性、プロセス性を損なうことなく、引張強度、引張弾性率が共に優れた炭素繊維を製造する方法を提供することにある。 The subject of this invention is providing the method of manufacturing the carbon fiber which was excellent in both tensile strength and a tensile elasticity modulus, without impairing productivity and process property.
かかる本発明の目的を達成するために、本発明は次の構成を有する。
すなわち、極限粘度が1.2〜2.2、耐炎化処理時の酸化深さDが3.6〜6.0μmかつ示差走査熱量計(以下、DSC)により測定される湿熱下融点Tmが180〜190℃である炭素繊維前駆体繊維用ポリアクリロニトリル系重合体である。
また、前記した炭素繊維前駆体繊維用ポリアクリロニトリル系重合体を湿式または乾湿式法により紡糸し、乾燥熱処理後、スチーム延伸する炭素繊維前駆体繊維を製造する方法において、乾燥熱処理温度Td(℃)スチーム延伸温度Ts(℃)、および該重合体のDSCにより測定される湿熱下融点Tm(℃)が下記式を満たす炭素繊維前駆体繊維の製造方法である。
In order to achieve the object of the present invention, the present invention has the following configuration.
That is, the intrinsic viscosity is 1.2 to 2.2, the oxidation depth D at the time of flameproofing is 3.6 to 6.0 μm, and the melting point Tm under wet heat measured by a differential scanning calorimeter (hereinafter DSC) is 180. It is a polyacrylonitrile polymer for carbon fiber precursor fibers having a temperature of ˜190 ° C.
Further, in the method for producing a carbon fiber precursor fiber that is obtained by spinning the polyacrylonitrile-based polymer for carbon fiber precursor fiber by a wet or dry wet method, followed by a dry heat treatment and then steam drawing, a dry heat treatment temperature Td (° C.) This is a method for producing a carbon fiber precursor fiber in which the steam stretching temperature Ts (° C.) and the melting point Tm (° C.) under wet heat measured by DSC of the polymer satisfy the following formula.
(Tm―30)≦Td≦(Tm−10)
(Tm―50)≦Ts≦(Tm−30)
さらには、前記した方法により製造される炭素繊維前駆体繊維を、200〜300℃の空気中において延伸比0.90〜1.20で延伸しながら耐炎化した後、300〜800℃の不活性雰囲気中において延伸比1.00〜1.30で延伸しながら予備炭化し、1000〜2000℃の不活性雰囲気中において延伸比0.97〜1.10で延伸しながら炭化する炭素繊維の製造方法である。
(Tm-30) ≦ Td ≦ (Tm-10)
(Tm-50) ≦ Ts ≦ (Tm-30)
Further, the carbon fiber precursor fiber produced by the above-described method is flame-resistant while being stretched at a stretch ratio of 0.90 to 1.20 in air at 200 to 300 ° C., and then inert at 300 to 800 ° C. Process for producing carbon fiber that is pre-carbonized while being drawn at a draw ratio of 1.00 to 1.30 in an atmosphere, and carbonized while being drawn at a draw ratio of 0.97 to 1.10 in an inert atmosphere at 1000 to 2000 ° C. It is.
本発明によれば、生産性、プロセス性を損なうことなく、焼成工程での高延伸を実現でき、それにより引張強度、引張弾性率さらには圧縮強度に優れた炭素繊維を低コストで製造できる。 According to the present invention, high stretching in the firing step can be realized without impairing productivity and processability, and thereby carbon fibers excellent in tensile strength, tensile elastic modulus and compressive strength can be produced at low cost.
本発明者らは、用いる重合体の極限粘度、酸素透過性および耐熱性を特定の範囲に制御することで、安定性よく、つづく製糸工程および焼成工程における高い延伸性を同時に実現できることを見出し、本発明に到達した。すなわち、用いる重合体の極限粘度を特定範囲とすることにより、原液安定性に優れた重合体とすることができ、用いる重合体の酸化深さDで表される酸素透過性を特定範囲とすることにより、均一な耐炎化構造が得られ、それにより焼成工程における延伸性を向上でき、用いる重合体の湿熱下融点Tmで表される耐熱性を特定範囲とすることで、製糸工程における乾燥熱処理、スチーム延伸処理を、単繊維間の接着なく効率的に行うことができるのである。
The present inventors have found that by controlling the intrinsic viscosity, oxygen permeability and heat resistance of the polymer to be used within a specific range, it is possible to achieve high stability at the same time in the yarn forming step and the firing step with good stability. The present invention has been reached. That is, by setting the intrinsic viscosity of the polymer to be used in a specific range, it is possible to obtain a polymer excellent in stock solution stability, and the oxygen permeability represented by the oxidation depth D of the polymer to be used is in a specific range. Thus, a uniform flameproof structure can be obtained, whereby the stretchability in the firing process can be improved, and the heat resistance expressed by the melting point Tm under the wet heat of the polymer to be used is in a specific range, thereby drying heat treatment in the yarn making process In addition, the steam stretching process can be efficiently performed without adhesion between single fibers.
まず、本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体について説明する。 First, the polyacrylonitrile polymer for carbon fiber precursor fiber of the present invention will be described.
本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体は、極限粘度が1.2〜2.2、耐炎化処理時の酸化深さDが3.6〜6.0μmかつDSCにより測定される湿熱下融点Tmが180〜190℃である。 The polyacrylonitrile-based polymer for carbon fiber precursor fiber of the present invention has an intrinsic viscosity of 1.2 to 2.2, an oxidation depth D of 3.6 to 6.0 μm during flameproofing treatment, and is measured by DSC. The melting point Tm under wet heat is 180-190 ° C.
本発明における炭素繊維前駆体繊維用ポリアクリロニトリル系重合体の極限粘度は、原液安定性の観点から、1.2〜2.2の範囲とすることが必須である。極限粘度が1.2を下回る様な低分子量になると、可紡性が低下し、また、極限粘度が2.2を超える様な高分子量になるとゲル化し易くなり、安定した紡糸が困難となる。該極限粘度は1.4〜2.0がより好ましく、1.5〜1.9がさらに好ましい。極限粘度は、重合時のモノマー濃度、重合開始剤や連鎖移動剤の量などにより制御することができる。
本発明において、極限粘度とはジメチルフォルムアミドを溶媒とし、オストワルド粘度計を用い、25℃で測定した比粘度をもとに算出した極限粘度のことをいう。具体的には、以下のような手順で測定する。予め120℃で2時間熱処理し絶乾した炭素繊維前駆体繊維用ポリアクリロニトリル系重合体150mgを、25℃において50mlのチオシアン酸ナトリウム0.1mol/リットル添加ジメチルフォルムアミドに溶解する。得られた溶液を25℃に温調し、予め25℃に温調してあるオストワルド粘度計を用いて標線間の落下時間を1/100秒の精度で測定し、その時間をt(秒)とする。同様に、炭素繊維前駆体繊維用ポリアクリロニトリル系重合体を溶解していないチオシアン酸ナトリウム0.1mol/リットル添加ジメチルフォルムアミドについても測定し、その落下時間をt0(秒)とする。次式を用いて極限粘度[η]を算出する。
[η]={(1+1.32×ηsp)^(1/2)―1}/0.198
ηsp=(t/t0)−1
該重合体の、耐炎化処理時の酸化深さDは、焼成工程における延伸性を向上させる観点から、3.6〜6.0μmであることが必須であり、より好ましくは3.8〜5.8μm、さらに好ましくは4.0〜5.5μmである。酸化深さDが3.6μmを下回ると、つづく予備炭化および炭化工程における延伸性を向上させる効果が明確に発揮されず、得られる炭素繊維の引張強度、引張弾性率を向上させることができない。一方、6μmを超えると、耐炎化工程における酸化反応が過剰に進み、つづく予備炭化および炭化工程での延伸性低下、得られる炭素繊維の収率低下のおそれがある。
The intrinsic viscosity of the polyacrylonitrile-based polymer for carbon fiber precursor fibers in the present invention is essential to be in the range of 1.2 to 2.2 from the viewpoint of stock solution stability. When the molecular weight is so low that the intrinsic viscosity is less than 1.2, the spinnability is lowered, and when the molecular weight is such that the intrinsic viscosity is more than 2.2, gelation tends to occur and stable spinning becomes difficult. . The intrinsic viscosity is more preferably 1.4 to 2.0, and further preferably 1.5 to 1.9. The intrinsic viscosity can be controlled by the monomer concentration at the time of polymerization, the amount of polymerization initiator or chain transfer agent, and the like.
In the present invention, the intrinsic viscosity means an intrinsic viscosity calculated based on a specific viscosity measured at 25 ° C. using an Ostwald viscometer using dimethylformamide as a solvent. Specifically, the measurement is performed according to the following procedure. 150 mg of a polyacrylonitrile polymer for carbon fiber precursor fibers, which has been heat-treated at 120 ° C. for 2 hours and dried completely, is dissolved in 50 ml of dimethylformamide containing 0.1 mol / liter of sodium thiocyanate at 25 ° C. The temperature of the obtained solution was adjusted to 25 ° C., and the Ostwald viscometer previously adjusted to 25 ° C. was used to measure the drop time between the marked lines with an accuracy of 1/100 second. ). Similarly, dimethylformamide added with 0.1 mol / liter of sodium thiocyanate in which the polyacrylonitrile-based polymer for carbon fiber precursor fiber is not dissolved is also measured, and the drop time is defined as t0 (seconds). The intrinsic viscosity [η] is calculated using the following formula.
[Η] = {(1 + 1.32 × ηsp) ^ (1/2) −1} /0.198
ηsp = (t / t0) −1
The oxidation depth D of the polymer during the flameproofing treatment is essential to be 3.6 to 6.0 μm, more preferably 3.8 to 5 from the viewpoint of improving stretchability in the firing step. 0.8 μm, more preferably 4.0 to 5.5 μm. When the oxidation depth D is less than 3.6 μm, the effect of improving the stretchability in the subsequent pre-carbonization and carbonization steps is not clearly exhibited, and the tensile strength and tensile elastic modulus of the obtained carbon fiber cannot be improved. On the other hand, if the thickness exceeds 6 μm, the oxidation reaction in the flameproofing process proceeds excessively, and there is a risk of lowering the stretchability in the subsequent pre-carbonization and carbonization processes and the yield of the resulting carbon fibers.
耐炎化処理時の酸化深さDは次のようにして定義、測定される。まず、重合体をジメチルスルホキシド、ジメチルフォルムアミドなどのポリアクリロニトリルが可溶な溶媒に、重量濃度で25%となるよう溶解し、次に該溶液をガラス板上にキャストして、一定の厚みになるように塗布する。次に、重合体溶液を塗布したガラス板を、熱風乾燥機等を用いて、空気中120℃で6時間乾燥し、溶媒を蒸発させて、厚み20〜40μmのフィルムとする。得られたフィルムを、熱風乾燥機等を用いて、空気中240℃で60分、さらに空気中250℃で60分熱処理し、耐炎化処理を行う。得られた耐炎化フィルムを樹脂包埋した上で研磨し、そのフィルム表面に対して垂直な断面を光学顕微鏡を用いて倍率800倍で観察する。断面において酸化が進んだ部分は暗い層として、進んでいない部分は明るい層として観察されるので、フィルム表面から、暗い層と明るい層の境界までの距離を少なくとも5点計測し、その算術平均を酸化深さD(μm)とする。
本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体のDSCにより測定される湿熱下融点Tmは、得られる炭素繊維前駆体繊維の耐熱性、ひいては、製糸工程におけるスチーム延伸性を向上させる目的から、180〜190℃が必須であり、より好ましくは182〜189℃、さらに好ましくは184〜188℃である。湿熱下融点Tmが180℃を下回ると、単繊維間の接着が顕著となり、製糸工程における乾燥およびスチーム延伸処理時の温度を低下させなくてはならず、より長時間の処理が必要となり、結果として生産性の低下、得られる炭素繊維の品位、力学物性の低下が生じる。190℃を超えると、スチーム延伸の際に、より高温すなわちより高圧力のスチームが必要となり、その高圧力による繊維の破断が顕著となるため、結果として生産性の低下、得られる炭素繊維の品位、力学物性の低下が生じる。
The oxidation depth D during the flameproofing treatment is defined and measured as follows. First, the polymer is dissolved in a solvent in which polyacrylonitrile such as dimethyl sulfoxide and dimethylformamide is soluble so that the concentration is 25% by weight, and then the solution is cast on a glass plate to obtain a constant thickness. Apply as follows. Next, the glass plate coated with the polymer solution is dried in air at 120 ° C. for 6 hours using a hot air dryer or the like, and the solvent is evaporated to form a film having a thickness of 20 to 40 μm. The obtained film is heat-treated at 240 ° C. in air for 60 minutes and further in air at 250 ° C. for 60 minutes using a hot air dryer or the like to perform flameproofing treatment. The obtained flame resistant film is polished after being embedded in a resin, and a cross section perpendicular to the film surface is observed with an optical microscope at a magnification of 800 times. In the cross section, the part where oxidation has progressed is observed as a dark layer, and the part which has not progressed is observed as a bright layer. Therefore, measure the distance from the film surface to the boundary between the dark layer and the bright layer at least 5 points, and calculate the arithmetic average. The oxidation depth is D (μm).
The melting point Tm under wet heat as measured by DSC of the polyacrylonitrile-based polymer for carbon fiber precursor fibers of the present invention is for the purpose of improving the heat resistance of the resulting carbon fiber precursor fibers, and hence the steam stretchability in the yarn making process. 180 to 190 ° C is essential, more preferably 182 to 189 ° C, and still more preferably 184 to 188 ° C. When the melting point Tm under wet heat is less than 180 ° C., the adhesion between the single fibers becomes remarkable, the temperature during drying and steam drawing in the yarn forming process must be lowered, and a longer treatment is required. As a result, a decrease in productivity, a quality of the obtained carbon fiber, and a decrease in mechanical properties occur. When the temperature exceeds 190 ° C., steam at a higher temperature, that is, higher pressure, is required for steam drawing, and fiber breakage due to the high pressure becomes remarkable. As a result, productivity is lowered and the quality of the obtained carbon fiber is reduced. , Mechanical properties are degraded.
湿熱下融点Tmは次のように定義、測定する。まず重合体を粉砕し、長径0.5mm以下の粉体とする。該粉体を5mg精秤し、耐圧2MPa以上の密閉可能なDSC用サンプルパンに、5mgの純水とともに密封する。10℃/分の昇温速度で、室温から220℃までDSC測定し、150〜200℃付近に現れる吸熱ピークの頂点に対応する温度を湿熱下融点Tm(℃)とする。 The melting point Tm under wet heat is defined and measured as follows. First, the polymer is pulverized to obtain a powder having a major axis of 0.5 mm or less. 5 mg of the powder is precisely weighed and sealed with 5 mg of pure water in a DSC sample pan capable of sealing with a pressure resistance of 2 MPa or more. DSC measurement is performed from room temperature to 220 ° C. at a rate of temperature increase of 10 ° C./min, and the temperature corresponding to the end of the endothermic peak appearing near 150 to 200 ° C. is defined as the melting point Tm (° C.) under wet heat.
本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体は、動的粘弾性測定により求められるガラス転移点Tgが60〜75℃であることが好ましい。ガラス転移点Tgは、非晶部の運動性を示す指標であり、Tgが高いほど非晶部の運動性が低いことを意味する。該Tgが60℃を下回ると耐熱性が低下し、そのためスチーム延伸性が低下することがあり、75℃を超えると分子間の抵抗が大きくなり、結果としてスチーム延伸性の低下、ひいては生産性の低下および得られる炭素繊維の品位、力学物性低下を招くおそれがあるので、好ましくない。ガラス転移点Tgは、より好ましくは62〜73℃であり、さらに好ましくは64℃〜70℃である。 The polyacrylonitrile-based polymer for carbon fiber precursor fibers of the present invention preferably has a glass transition point Tg determined by dynamic viscoelasticity measurement of 60 to 75 ° C. The glass transition point Tg is an index indicating the mobility of the amorphous part, and the higher the Tg, the lower the mobility of the amorphous part. When the Tg is less than 60 ° C., the heat resistance is lowered, so that the steam stretchability may be lowered. When the Tg is more than 75 ° C., the intermolecular resistance is increased, and as a result, the steam stretchability is lowered. This is not preferable because it may cause a decrease and deterioration of the quality and mechanical properties of the obtained carbon fiber. The glass transition point Tg is more preferably 62 to 73 ° C, and further preferably 64 ° C to 70 ° C.
ガラス転移点Tgは次のように定義、測定する。まず、前記した酸化深さDの測定と同様の方法でフィルムを作製する。該フィルムを用いて、動的粘弾性測定を行う。測定は、0.2Hzの正弦波で引張荷重を加えながら、昇温速度10℃/分で室温から200℃まで行う。荷重および振幅は、可能な限り塑性変形が生じないように調整することが好ましい。得られたデータから損失弾性率E”を求め、そのピークに相当する温度をガラス転移点Tg(℃)とする。
本発明における炭素繊維前駆体繊維用ポリアクリロニトリル系重合体は、得られる炭素繊維前駆体繊維の耐熱性や緻密性の観点からアクリロニトリルが95mol%以上からなることが好ましく、より好ましくは98.5mol%以上、さらに好ましくは99.0mol%以上である。該アクリロニトリル量は、得られる炭素繊維前駆体繊維の耐熱性を高める目的からは高い方が好ましいが、酸化深さDを本発明に規定する特定値とするためには、後述するような共重合成分を加えることが好ましいため、99.9mol%以下とするのが良い。
The glass transition point Tg is defined and measured as follows. First, a film is produced by the same method as the measurement of the oxidation depth D described above. Using this film, dynamic viscoelasticity is measured. The measurement is performed from room temperature to 200 ° C. at a heating rate of 10 ° C./min while applying a tensile load with a sine wave of 0.2 Hz. The load and amplitude are preferably adjusted so that plastic deformation does not occur as much as possible. The loss elastic modulus E ″ is obtained from the obtained data, and the temperature corresponding to the peak is defined as the glass transition point Tg (° C.).
In the polyacrylonitrile-based polymer for carbon fiber precursor fibers in the present invention, acrylonitrile is preferably composed of 95 mol% or more, more preferably 98.5 mol% from the viewpoint of heat resistance and denseness of the obtained carbon fiber precursor fiber. As mentioned above, More preferably, it is 99.0 mol% or more. The amount of the acrylonitrile is preferably higher for the purpose of increasing the heat resistance of the obtained carbon fiber precursor fiber, but in order to set the oxidation depth D to a specific value defined in the present invention, a copolymer as described later is used. Since it is preferable to add a component, it is good to set it as 99.9 mol% or less.
本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体は、前記した極限粘度、酸化深さD、湿熱下融点Tmを満足すれば、共重合成分、分子量分布、立体規則性などに制約は無い。前記した耐炎化時の酸化深さDを向上させるために、大きい側鎖を持つビニルモノマーを共重合することにより、酸素透過性を高めることが有効な手段として挙げられる。湿熱下融点は該ビニルモノマーの共重合量が多くなるほど低下する傾向にあり、共重合量を変えることにより制御することができる。
前記した耐炎化時の酸化深さDと湿熱下融点Tmを同時に満足するためには、酸素透過性向上効果の高い共重合成分を必要最小量共重合することが好ましい。より具体的には、モル体積が100〜400cm3/molであるビニルモノマーを0.1〜1.0mol%共重合することが好ましい。ここで、モル体積とは、分子量を20℃における比重で割った値であり、モノマーのかさ高さに対応するパラメータである。このモル体積が大きいほど、酸素透過性向上効果を大きくできるものの、400cm3/molを超えると、アクリロニトリルに対する重合性の低下や、得られる炭素繊維前駆体繊維および炭素繊維の緻密性の低下、また耐炎化時に酸化が進みすぎることによる収率の低下が顕著となる場合がある。該モル体積は、より好ましくは140〜350cm3/molであり、さらに好ましくは180〜300cm3/molである。
前記したモル体積が100〜400cm3/molであるビニルモノマーの共重合量は、0.1mol%を下回ると、明確な酸素透過性向上効果が得にくくなる。一方、多いほど酸素透過性は高まるものの、湿熱下融点Tmが低下するため、1.0mol%を超えない範囲とすることが好ましい。
前記したモル体積が100〜400cm3/molであるビニルモノマーとしては、アクリロニトリルに対する重合性、工業的な入手のしやすさから、アクリル酸およびメタクリル酸のエステルを好ましく用いることができる。より具体的には、エチルアクリレート、ブチルアクリレート、イソブチルアクリレート、オクチルアクリレート、イソオクチルアクリレート、2−エチルヘキシルアクリレート、ラウリルアクリレート、ステアリルアクリレート、ベヘニルアクリレート、シクロヘキシルアクリレート、2−ヒドロキシエチルアクリレート、2−ヒドロキシプロピルアクリレート、4−ヒドロキシブチルアクリレート、グリセリンモノアクリレート、テトラヒドロフルフリルアクリレート、エチルメタクリレート、ブチルメタクリレート、イソブチルメタクリレート、オクチルメタクリレート、イソオクチルメタクリレート、2−エチルヘキシルメタクリレート、ラウリルメタクリレート、ステアリルメタクリレート、ベヘニルメタクリレート、シクロヘキシルメタクリレート、2−ヒドロキシエチルメタクリレート、2−ヒドロキシプロピルメタクリレート、4−ヒドロキシブチルメタクリレート、グリセリンモノメタクリレート、テトラヒドロフルフリルメタクリレートを好ましく例示できる。製糸の延伸性を向上させる観点からは、これらの化合物には側鎖中に分岐が少ないことが好ましい。その観点からは、エチルアクリレート、ブチルアクリレート、オクチルアクリレート、ラウリルアクリレート、ステアリルアクリレート、ベヘニルアクリレート、2−ヒドロキシエチルアクリレート、2−ヒドロキシプロピルアクリレート、4−ヒドロキシブチルアクリレート、グリセリンモノアクリレート、エチルメタクリレート、ブチルメタクリレート、オクチルメタクリレート、ラウリルメタクリレート、ステアリルメタクリレート、ベヘニルメタクリレート、2−ヒドロキシエチルメタクリレート、2−ヒドロキシプロピルメタクリレート、4−ヒドロキシブチルメタクリレート、グリセリンモノメタクリレートをより好ましく例示できる。さらに好ましくは、エチルアクリレート、ブチルアクリレート、オクチルアクリレート、ラウリルアクリレート、ステアリルアクリレート、ベヘニルアクリレート、エチルメタクリレート、ブチルメタクリレート、オクチルメタクリレート、ラウリルメタクリレート、ステアリルメタクリレート、ベヘニルメタクリレートを例示できる。
本発明において、炭素繊維前駆体繊維用ポリアクリロニトリル系重合体の酸素透過性を効率良く向上させ、湿熱下融点との両立を図るためには、前記したモル体積が100〜400cm3/molであるビニルモノマーが、重合体中に均一に存在していることが好ましい。重合体中における共重合成分の均一性は、重合方法、選択する共重合成分のアクリロニトリルに対する反応性、共重合成分の重合時の添加方法、例えば逐次的添加などにより制御することができる。中でも、アクリロニトリルに対する反応性比r1が0.4〜3であるビニルモノマーを選択し、ラジカル重合により重合体を製造することが、工業的に容易であり好ましい。反応性比r1は、アクリロニトリル同士の連鎖成長反応速度定数をk11、アクリロニトリルラジカル末端と共重合成分との連鎖成長反応速度定数をk12とすると、次式で定義される。
The polyacrylonitrile-based polymer for carbon fiber precursor fiber of the present invention is not limited in copolymerization component, molecular weight distribution, stereoregularity, etc. as long as the above-mentioned intrinsic viscosity, oxidation depth D, and melting point Tm under wet heat are satisfied. . In order to improve the oxidation depth D at the time of flame resistance described above, it is effective means to increase oxygen permeability by copolymerizing a vinyl monomer having a large side chain. The melting point under wet heat tends to decrease as the copolymerization amount of the vinyl monomer increases, and can be controlled by changing the copolymerization amount.
In order to satisfy the oxidation depth D and the melting point Tm under wet heat at the same time, it is preferable to copolymerize the minimum necessary amount of a copolymer component having a high effect of improving oxygen permeability. More specifically, it is preferable to copolymerize 0.1 to 1.0 mol% of a vinyl monomer having a molar volume of 100 to 400 cm 3 / mol. Here, the molar volume is a value obtained by dividing the molecular weight by the specific gravity at 20 ° C., and is a parameter corresponding to the bulkiness of the monomer. The larger the molar volume, the greater the effect of improving oxygen permeability. However, when it exceeds 400 cm 3 / mol, the polymerizability with respect to acrylonitrile decreases, the resulting carbon fiber precursor fiber and carbon fiber become less dense, There may be a significant decrease in yield due to excessive oxidation during flame resistance. The molar volume is more preferably 140 to 350 cm 3 / mol, and still more preferably 180 to 300 cm 3 / mol.
When the amount of copolymerization of the vinyl monomer having a molar volume of 100 to 400 cm 3 / mol is less than 0.1 mol%, it is difficult to obtain a clear oxygen permeability improvement effect. On the other hand, the oxygen permeability increases as the amount increases, but the melting point Tm decreases under wet heat, so that it is preferably within a range not exceeding 1.0 mol%.
As the above-mentioned vinyl monomer having a molar volume of 100 to 400 cm 3 / mol, esters of acrylic acid and methacrylic acid can be preferably used from the viewpoint of polymerizability with respect to acrylonitrile and industrial availability. More specifically, ethyl acrylate, butyl acrylate, isobutyl acrylate, octyl acrylate, isooctyl acrylate, 2-ethylhexyl acrylate, lauryl acrylate, stearyl acrylate, behenyl acrylate, cyclohexyl acrylate, 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate 4-hydroxybutyl acrylate, glycerin monoacrylate, tetrahydrofurfuryl acrylate, ethyl methacrylate, butyl methacrylate, isobutyl methacrylate, octyl methacrylate, isooctyl methacrylate, 2-ethylhexyl methacrylate, lauryl methacrylate, stearyl methacrylate, behenyl methacrylate, cyclohexyl Methacrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, 4-hydroxybutyl methacrylate, glycerol monomethacrylate, tetrahydrofurfuryl methacrylate can be preferably exemplified. From the viewpoint of improving the drawability of yarn production, these compounds preferably have few branches in the side chain. From that viewpoint, ethyl acrylate, butyl acrylate, octyl acrylate, lauryl acrylate, stearyl acrylate, behenyl acrylate, 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate, 4-hydroxybutyl acrylate, glycerin monoacrylate, ethyl methacrylate, butyl methacrylate More preferable examples include octyl methacrylate, lauryl methacrylate, stearyl methacrylate, behenyl methacrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, 4-hydroxybutyl methacrylate, and glycerin monomethacrylate. More preferable examples include ethyl acrylate, butyl acrylate, octyl acrylate, lauryl acrylate, stearyl acrylate, behenyl acrylate, ethyl methacrylate, butyl methacrylate, octyl methacrylate, lauryl methacrylate, stearyl methacrylate, and behenyl methacrylate.
In the present invention, in order to efficiently improve the oxygen permeability of the polyacrylonitrile-based polymer for carbon fiber precursor fibers and to achieve compatibility with the melting point under wet heat, the aforementioned molar volume is 100 to 400 cm 3 / mol. It is preferred that the vinyl monomer is present uniformly in the polymer. The homogeneity of the copolymer component in the polymer can be controlled by the polymerization method, the reactivity of the selected copolymer component with acrylonitrile, the addition method of the copolymer component during polymerization, for example, sequential addition. Among them, it is industrially easy and preferable to select a vinyl monomer having a reactivity ratio r1 to acrylonitrile of 0.4 to 3 and to produce a polymer by radical polymerization. The reactivity ratio r1 is defined by the following equation, where k11 is a chain growth reaction rate constant between acrylonitriles and k12 is a chain growth reaction rate constant between an acrylonitrile radical end and a copolymerization component.
r1=k11/k12
r1は、重合時にアクリロニトリルユニットの次にアクリロニトリルが重合されるか、他の共重合成分が重合されるかの確率を表す指数であり、1に近いほど等確率となり、共重合量を反映した均一な共重合体が得られることを示す。本発明において、反応性比r1が0.4を下回ったり、3を上回ると、共重合成分が共重合体中において偏って重合されるため、共重合量に対して得られる実質的な酸素透過性向上効果が十分ではないことがあり、好ましくない。
r1 = k11 / k12
r1 is an index representing the probability of whether acrylonitrile is polymerized next to the acrylonitrile unit at the time of polymerization or other copolymerization components are polymerized. This shows that a good copolymer can be obtained. In the present invention, when the reactivity ratio r1 is less than 0.4 or exceeds 3, the copolymer component is polymerized in the copolymer in an uneven manner, so that the substantial oxygen permeation obtained with respect to the copolymerization amount is obtained. The effect of improving the properties may not be sufficient, which is not preferable.
アクリロニトリルに対する反応性比r1は、実験的に求める方法、および計算により推算する方法がある。前者については、線形化法、直線交差法、曲線合致法などがあり、たとえば高分子学会編「共重合1反応解析」、初版、株式会社培風館社、1975年6月20日、p.59〜68に記載されている方法に従い求めることができる。後者については、いわゆるアルフレイ−プライス(Alfrey−Price)のQ値、e値を用いて算出する方法であり、共重合成分のQ値、e値をそれぞれQc、ecとすると次式を用いて算出することができる。 The reactivity ratio r1 with respect to acrylonitrile includes a method for experimental determination and a method for estimation by calculation. The former includes a linearization method, a straight line intersection method, a curve matching method, and the like. For example, edited by Polymer Society of Japan, “Copolymerization 1 reaction analysis”, first edition, Baifukan Co., Ltd., June 20, 1975, p. It can be determined according to the method described in 59-68. The latter is a method of calculating by using the so-called Alfrey-Price Q value and e value. When the Q value and e value of the copolymer component are Qc and ec, respectively, the calculation is performed using the following equations. can do.
r1=(0.48/Qc)×EXP(―1.23×(1.23−ec)
また、多くのビニルモノマーについての反応性比r1、Alfrey−PriceのQ値、e値は、J.Brandrup、E.H. Immergut、E.A. Grulke著、「ポリマーハンドブック(Polymer Handbook)」、(米国)、第4版、ジョンワイリーアンドサンズ(John Wiley & Sons Inc)、1999年、p.II/181〜II/319などにまとめられており、参照することもできる。
r1 = (0.48 / Qc) × EXP (−1.23 × (1.23−ec)
In addition, the reactivity ratio r1, the Alfree-Price Q value, and the e value for many vinyl monomers are as described in J. Org. Brandrup, E.I. H. Immergut, E .; A. Grulke, “Polymer Handbook” (USA), 4th edition, John Wiley & Sons Inc., 1999, p. II / 181 to II / 319 and the like can be referred to.
アクリロニトリルに対する反応性比r1が0.4〜3であるビニルモノマーの具体例としては、アクリル酸のエステルを好ましいものとして挙げることができる。より具体的には、エチルアクリレート、ブチルアクリレート、オクチルアクリレート、ラウリルアクリレートなどを挙げることができる。 Specific examples of vinyl monomers having a reactivity ratio r1 to acrylonitrile of 0.4 to 3 include esters of acrylic acid. More specifically, ethyl acrylate, butyl acrylate, octyl acrylate, lauryl acrylate, and the like can be given.
本発明において、炭素繊維前駆体繊維用ポリアクリロニトリル系重合体は、共重合体としての耐炎化促進成分を0.1〜1.0mol%含むことが好ましい。耐炎化促進成分としては、カルボキシル基またはアミド基を一つ以上有するものが、好ましく例示できる。耐炎化促進成分の共重合量を多くするほど耐炎化反応が促進され、短時間で耐炎化処理でき、生産性を高める目的から好ましい。しかし一方で、該共重合量が多くなるほど、湿熱下融点Tmが低下したり、発熱速度が大きくなり暴走反応の危険が生じることがあるため、1.0mol%を超えない範囲とすることが好ましく、より好ましくは0.15〜0.5mol%、さらに好ましくは0.2〜0.4mol%である。 In the present invention, the polyacrylonitrile-based polymer for carbon fiber precursor fibers preferably contains 0.1 to 1.0 mol% of a flameproofing promoting component as a copolymer. Preferred examples of the flame resistance promoting component include those having one or more carboxyl groups or amide groups. Increasing the copolymerization amount of the flameproofing promoting component promotes the flameproofing reaction, can be flameproofed in a short time, and is preferable for the purpose of increasing productivity. However, on the other hand, the higher the amount of copolymerization, the lower the melting point Tm under wet heat, or the higher the rate of heat generation and the risk of a runaway reaction. More preferably, it is 0.15-0.5 mol%, More preferably, it is 0.2-0.4 mol%.
該耐炎化促進成分の具体例としては、アクリル酸、メタクリル酸、イタコン酸、クロトン酸、シトラコン酸、エタクリル酸、マレイン酸、メサコン酸、アクリルアミド、メタクリルアミドなどが好ましく例示できる。湿熱下融点Tmの低下を防止するという目的からは、耐炎化促進効果の高いモノマーを少量用いることが好ましく、アミド基よりもカルボキシル基を有する耐炎化促進成分を用いることが好ましい。また含有されるアミド基、カルボキシル基の数については1つよりも2つ以上であることがより好ましく、その観点からは、アクリル酸、メタクリル酸、イタコン酸、クロトン酸、シトラコン酸、エタクリル酸、マレイン酸、メサコン酸がより好ましく、イタコン酸、マレイン酸、メサコン酸がさらに好ましく挙げられる。
湿熱下融点の低下を防止するという目的からは、アクリロニトリル以外の共重合成分のトータル量は5mol%を超えないことが好ましく、より好ましくは1.5mol%、さらに好ましくは1.0mol%を超えない範囲とすることがよい。
本発明における炭素繊維前駆体繊維用ポリアクリロニトリル系重合体を製造する重合方法としては、溶液重合、懸濁重合、乳化重合など公知の重合方法を選択することができるが、共重合成分を均一に重合する目的からは、溶液重合を用いることが好ましい。
溶液重合で行う場合の溶液としては、ジメチルスルホキシド、ジメチルフォルムアミド、ジメチルアセトアミドなどのポリアクリロニトリルが可溶な溶媒を用いるのが好ましい。中でも、溶解性の観点から、ジメチルスルホキシドがより好ましい。
次に、本発明の炭素繊維前駆体繊維の製造方法について説明する。
本発明の炭素繊維前駆体繊維は、前記した本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体を用いて製造する。該重合体をジメチルスルホキシド、ジメチルフォルムアミド、ジメチルアセトアミドなどのポリアクリロニトリルが可溶な溶媒に溶解し、紡糸原液とする。溶液重合を用いる場合、重合に用いる溶媒と紡糸溶媒を同じものにしておくと、得られた重合体を分離し紡糸溶媒に再溶解する工程が不要となり好ましい。紡糸原液中の該重合体の濃度は、原液安定性の観点から、10〜40重量%であることが好ましい。
かかる紡糸原液を紡糸する前に目開き1μm以下のフィルターに通し、ポリマー原料および各工程において混入した不純物を除去することが高強度な炭素繊維を得るためには好ましい。
Specific examples of the flame resistance promoting component include acrylic acid, methacrylic acid, itaconic acid, crotonic acid, citraconic acid, ethacrylic acid, maleic acid, mesaconic acid, acrylamide, and methacrylamide. For the purpose of preventing the melting point Tm from decreasing under wet heat, it is preferable to use a small amount of a monomer having a high flame resistance promoting effect, and it is preferable to use a flame resistance promoting component having a carboxyl group rather than an amide group. In addition, the number of amide groups and carboxyl groups contained is more preferably two or more than one. From that viewpoint, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, citraconic acid, ethacrylic acid, Maleic acid and mesaconic acid are more preferable, and itaconic acid, maleic acid and mesaconic acid are more preferable.
For the purpose of preventing lowering of the melting point under wet heat, the total amount of copolymerization components other than acrylonitrile is preferably not more than 5 mol%, more preferably 1.5 mol%, still more preferably not more than 1.0 mol%. It is better to be in the range.
As a polymerization method for producing the polyacrylonitrile-based polymer for carbon fiber precursor fiber in the present invention, a known polymerization method such as solution polymerization, suspension polymerization, emulsion polymerization or the like can be selected. For the purpose of polymerization, solution polymerization is preferably used.
As the solution in the case of solution polymerization, it is preferable to use a solvent in which polyacrylonitrile such as dimethyl sulfoxide, dimethylformamide, dimethylacetamide or the like is soluble. Among these, dimethyl sulfoxide is more preferable from the viewpoint of solubility.
Next, the manufacturing method of the carbon fiber precursor fiber of this invention is demonstrated.
The carbon fiber precursor fiber of the present invention is produced using the above-described polyacrylonitrile polymer for carbon fiber precursor fiber of the present invention. The polymer is dissolved in a solvent in which polyacrylonitrile such as dimethyl sulfoxide, dimethylformamide, dimethylacetamide or the like is soluble to obtain a spinning dope. In the case of using solution polymerization, it is preferable that the solvent used for polymerization and the spinning solvent be the same because the step of separating the obtained polymer and re-dissolving in the spinning solvent is unnecessary. The concentration of the polymer in the spinning dope is preferably 10 to 40% by weight from the viewpoint of stock solution stability.
In order to obtain a high-strength carbon fiber, it is preferable to pass through a filter having an opening of 1 μm or less before spinning the spinning solution to remove the polymer raw material and impurities mixed in each step.
紡糸原液を、湿式紡糸法または乾湿式紡糸法により口金から紡出し、凝固浴に導入して繊維を凝固せしめる。得られる炭素繊維前駆体繊維の緻密性を高め、また得られる炭素繊維の力学物性を高める目的からは、乾湿式紡糸法を用いることが、より好ましい。 The spinning solution is spun from the die by a wet spinning method or a dry-wet spinning method, and introduced into a coagulation bath to coagulate the fibers. In order to increase the density of the obtained carbon fiber precursor fiber and to increase the mechanical properties of the obtained carbon fiber, it is more preferable to use a dry and wet spinning method.
本発明において、前記凝固浴には、紡糸原液の溶媒として用いたジメチルスルホキシド、ジメチルホルムアミド、ジメチルアセトアミドなどの溶媒と、いわゆる凝固促進成分を含ませることが好ましい。凝固促進成分としては、前記重合体を溶解せず、かつ紡糸原液に用いる溶媒と相溶性があるものが使用できる。具体的には、水を使用するのが好ましい。 In the present invention, the coagulation bath preferably contains a solvent such as dimethyl sulfoxide, dimethylformamide, or dimethylacetamide used as a solvent for the spinning dope and a so-called coagulation promoting component. As the coagulation accelerating component, a component that does not dissolve the polymer and is compatible with the solvent used in the spinning dope can be used. Specifically, it is preferable to use water.
凝固浴中に導入して糸条を凝固せしめた後、水洗工程、浴中延伸工程、油剤付与工程、乾燥熱処理工程、スチーム延伸工程を経て、炭素繊維前駆体繊維が得られる。 After introducing into a coagulation bath and coagulating the yarn, a carbon fiber precursor fiber is obtained through a washing step, an in-bath drawing step, an oil agent application step, a drying heat treatment step, and a steam drawing step.
ただし、凝固後の糸条は、水洗工程を省略して直接浴中延伸を行っても良いし、溶媒を水洗工程により除去した後に浴中延伸を行っても良い。
かかる浴中延伸は、通常、30〜98℃に温調された単一又は複数の延伸浴中で行うことが好ましい。延伸倍率は、1〜5倍であることが好ましく、2〜4倍であることがより好ましい。
However, the solidified yarn may be directly stretched in the bath without the water washing step, or may be stretched in the bath after the solvent is removed by the water washing step.
Such stretching in a bath is usually preferably performed in a single or a plurality of stretching baths adjusted to a temperature of 30 to 98 ° C. The draw ratio is preferably 1 to 5 times, and more preferably 2 to 4 times.
浴中延伸工程の後、単繊維同士の接着を防止する目的から、糸条にシリコーン等からなる油剤を付与することが好ましい。かかるシリコーン油剤は、変性されたシリコーンを用いることが好ましく、耐熱性の高いアミノ変性シリコーンを含有するものを用いることがより好ましい。 After the stretching process in the bath, it is preferable to apply an oil agent made of silicone or the like to the yarn for the purpose of preventing adhesion between the single fibers. As such a silicone oil agent, it is preferable to use a modified silicone, and it is more preferable to use a material containing an amino-modified silicone having high heat resistance.
前記した、水洗工程、浴中延伸工程、油剤付与工程の後、乾燥熱処理およびスチーム延伸を行うことにより、炭素繊維前駆体繊維を製造する。かかる乾燥熱処理およびスチーム延伸においては、炭素繊維前駆体繊維用ポリアクリロニトリル系重合体のDSCにより測定される湿熱下融点をTm(℃)とすると、乾燥熱処理温度Td(℃)およびスチーム延伸温度Ts(℃)を下記式の範囲内に設定することが好ましい。 A carbon fiber precursor fiber is manufactured by performing a drying heat treatment and steam drawing after the water washing step, the drawing step in bath, and the oil agent application step. In such dry heat treatment and steam drawing, assuming that the melting point under wet heat measured by DSC of the polyacrylonitrile-based polymer for carbon fiber precursor fibers is Tm (° C.), the dry heat treatment temperature Td (° C.) and the steam drawing temperature Ts ( Is preferably set within the range of the following formula.
(Tm―30)≦Td≦(Tm−10)
(Tm―50)≦Ts≦(Tm−30)
また、より好ましくは、
(Tm―20)≦Td≦(Tm−10)
(Tm―40)≦Ts≦(Tm−30)
である。一般に、ポリアクリロニトリルは乾熱下では融点を示さないものの、水が共存していると、その融点が降下することが知られている。乾燥熱処理およびスチーム延伸は、水共存下で行われるため、その処理温度が、用いる炭素繊維前駆体繊維用ポリアクリロニトリル系重合体の湿熱下融点Tmを上回ると融解がおき、プロセス性、得られる炭素繊維前駆体繊維または炭素繊維の力学物性が低下することがあり、好ましくない。また、前記したように、用いる炭素繊維前駆体繊維用ポリアクリロニトリル系重合体の共重合成分やその共重合量によって、湿熱下融点Tmは変化するため、そのTmに合わせて、乾燥熱処理温度Tdおよびスチーム延伸温度Tsを設定することが、高い引張強度、高い引張弾性率を持つ炭素繊維を生産性よく得るために、重要である。
(Tm-30) ≦ Td ≦ (Tm-10)
(Tm-50) ≦ Ts ≦ (Tm-30)
More preferably,
(Tm−20) ≦ Td ≦ (Tm−10)
(Tm-40) ≦ Ts ≦ (Tm-30)
It is. In general, polyacrylonitrile does not show a melting point under dry heat, but it is known that the melting point of the polyacrylonitrile drops when water coexists. Since the drying heat treatment and steam stretching are performed in the presence of water, melting occurs when the treatment temperature exceeds the melting point Tm under wet heat of the polyacrylonitrile polymer for the carbon fiber precursor fiber to be used. The mechanical properties of the fiber precursor fiber or carbon fiber may decrease, which is not preferable. Further, as described above, since the melting point Tm under wet heat changes depending on the copolymerization component of the polyacrylonitrile-based polymer for the carbon fiber precursor fiber to be used and the amount of the copolymerization, the drying heat treatment temperature Td and the temperature are adjusted according to the Tm. Setting the steam stretching temperature Ts is important in order to obtain a carbon fiber having high tensile strength and high tensile elastic modulus with high productivity.
また、一方で乾燥熱処理温度Tdが高いほど、乾燥効率、得られる炭素繊維前駆体繊維の緻密性を高める目的から有利であり、また、スチーム延伸温度Tsが高いほど、可塑化が進みやすく延伸性向上に有利である。このことから、前記範囲でTdおよびTsを制御することにより、本発明の炭素繊維前駆体繊維用ポリアクリロニトリル系重合体のポテンシャルを最大限に引き出すことができるのである。 On the other hand, the higher the drying heat treatment temperature Td, the more advantageous for the purpose of increasing the drying efficiency and the denseness of the resulting carbon fiber precursor fiber, and the higher the steam stretching temperature Ts, the easier the plasticization proceeds and the stretchability. It is advantageous for improvement. Therefore, by controlling Td and Ts within the above range, the potential of the polyacrylonitrile polymer for carbon fiber precursor fiber of the present invention can be maximized.
本発明において、スチーム延伸工程における延伸倍率は、生産性および得られる炭素繊維の力学物性の観点から3倍以上、より好ましくは4倍以上、さらに好ましくは5倍以上であるのがよい。 In the present invention, the draw ratio in the steam drawing step is preferably 3 times or more, more preferably 4 times or more, and still more preferably 5 times or more from the viewpoint of productivity and mechanical properties of the obtained carbon fiber.
本発明において、炭素繊維前駆体繊維の単繊維繊度は、好ましくは0.5〜1.5dtex、より好ましくは0.55〜1.0dtex、さらに好ましくは0.6〜0.8dtexであることが良い。該単繊維繊度が0.5dtexを下回ると、可紡性の低下、ローラー、ガイドとの接触による糸切れ発生などにより、製糸工程および焼成工程のプロセス安定性が低下することがある。一方、1.5dtexを超えると耐炎化後の各単繊維における内外構造差が大きくなり、つづく炭化工程でのプロセス性低下や、得られる炭素繊維の引張強度、引張弾性率が低下することがあり、好ましくない。 In the present invention, the single fiber fineness of the carbon fiber precursor fiber is preferably 0.5 to 1.5 dtex, more preferably 0.55 to 1.0 dtex, and still more preferably 0.6 to 0.8 dtex. good. When the single fiber fineness is less than 0.5 dtex, the process stability of the yarn forming process and the firing process may be reduced due to a decrease in spinnability and the occurrence of yarn breakage due to contact with a roller or a guide. On the other hand, if it exceeds 1.5 dtex, the difference between the inner and outer structures of each single fiber after flame resistance will increase, and the processability in the subsequent carbonization process may decrease, and the tensile strength and tensile modulus of the resulting carbon fiber may decrease. It is not preferable.
また、本発明において、炭素繊維前駆体繊維の1糸条当たりのフィラメント数は、好ましくは1,000〜3,000,000、より好ましくは12,000〜3,000,000、さらに好ましくは24,000〜2,500,000、最も好ましくは36,000〜2,000,000であるのが良い。該フィラメント数は、生産性の向上の目的からは、1,000以上で多い方が好ましいが、3,000,000を超えると束内部まで均一に耐炎化処理できないことがあり、好ましくない。 In the present invention, the number of filaments per yarn of the carbon fiber precursor fiber is preferably 1,000 to 3,000,000, more preferably 12,000 to 3,000,000, still more preferably 24. 3,000 to 2,500,000, most preferably 36,000 to 2,000,000. The number of filaments is preferably greater than 1,000 for the purpose of improving productivity. However, when the number exceeds 3,000,000, the inside of the bundle may not be uniformly flameproofed, which is not preferable.
次に、本発明の炭素繊維の製造方法について説明する。
本発明の炭素繊維は、前記した本発明の炭素繊維前駆体繊維を200〜300℃の空気中において延伸比0.90〜1.20で延伸しながら耐炎化した後、300〜800℃の不活性雰囲気中において延伸比1.00〜1.30で延伸しながら予備炭化し、1000〜2000℃の不活性雰囲気中において延伸比0.97〜1.10で延伸しながら炭化して製造することが好ましい。
Next, the manufacturing method of the carbon fiber of this invention is demonstrated.
The carbon fiber of the present invention is flame resistant while stretching the carbon fiber precursor fiber of the present invention described above in the air at 200 to 300 ° C. at a stretch ratio of 0.90 to 1.20, Pre-carbonize while stretching at a stretch ratio of 1.00 to 1.30 in an active atmosphere, and carbonize while stretching at a stretch ratio of 0.97 to 1.10 in an inert atmosphere at 1000 to 2000 ° C. Is preferred.
本発明において、耐炎化する際の延伸比は、0.90〜1.20が好ましく、より好ましくは0.95〜1.15、さらに好ましくは0.97〜1.10である。該延伸比は0.90を下回ると、得られる耐炎化繊維の配向度が不十分となり、また得られる炭素繊維の力学物性が低下することがある。また、該延伸比が1.20を超えると、毛羽発生、糸切れ発生によりプロセス性が低下することがある。 In the present invention, the stretch ratio when making flame resistant is preferably 0.90 to 1.20, more preferably 0.95 to 1.15, and still more preferably 0.97 to 1.10. If the draw ratio is less than 0.90, the degree of orientation of the resulting flame-resistant fiber may be insufficient, and the mechanical properties of the resulting carbon fiber may be reduced. On the other hand, when the draw ratio exceeds 1.20, processability may be deteriorated due to generation of fluff and yarn breakage.
本発明において、耐炎化の処理時間は、10〜100分の範囲で適宜選択することができるが、得られる耐炎化繊維の比重が1.3〜1.38の範囲となるよう設定することが、つづく予備炭化工程のプロセス性、および得られる炭素繊維の力学物性向上の目的から好ましい。 In the present invention, the flameproofing treatment time can be appropriately selected within the range of 10 to 100 minutes, but the specific gravity of the obtained flameproofed fiber can be set within the range of 1.3 to 1.38. In view of the processability of the subsequent pre-carbonization step and the purpose of improving the mechanical properties of the resulting carbon fiber.
本発明において、予備炭化工程、炭化工程、黒鉛化工程は不活性雰囲気中で行うが、用いるガスとしては、窒素、アルゴン、キセノンなどが好ましく例示でき、経済的な観点からは窒素を好ましく用いることができる。 In the present invention, the preliminary carbonization step, the carbonization step, and the graphitization step are performed in an inert atmosphere. As the gas to be used, nitrogen, argon, xenon and the like can be preferably exemplified, and nitrogen is preferably used from an economical viewpoint. Can do.
本発明において、予備炭化工程における温度は300〜800℃が好ましく、その範囲における昇温速度は500℃/分以下に設定することが好ましい。 In the present invention, the temperature in the preliminary carbonization step is preferably 300 to 800 ° C., and the temperature increase rate in the range is preferably set to 500 ° C./min or less.
本発明において、予備炭化を行う際の延伸比は1.00〜1.30が好ましく、より好ましくは1.05〜1.25、さらに好ましくは1.08〜1.20である。該延伸比は1.00を下回ると、得られる予備炭化繊維の配向度が不十分となり、炭素繊維の力学物性が低下することがある。また、該延伸比が1.30を超えると、毛羽発生、糸切れ発生によりプロセス性が低下することがある。 In the present invention, the draw ratio during preliminary carbonization is preferably 1.00 to 1.30, more preferably 1.05 to 1.25, and still more preferably 1.08 to 1.20. When the draw ratio is less than 1.00, the degree of orientation of the obtained preliminary carbonized fiber becomes insufficient, and the mechanical properties of the carbon fiber may be lowered. On the other hand, when the draw ratio exceeds 1.30, processability may be deteriorated due to occurrence of fluff and yarn breakage.
本発明において、炭化工程における温度は1000〜2000℃が好ましく、その最高温度は、所望する炭素繊維の力学物性に応じて適宜設定するのがよい。一般に該炭化工程の最高温度が高いほど、得られる炭素繊維の引張弾性率が高くなるものの、引張強度は1500℃付近で極大となる。引張強度と引張弾性率の両方を高めるという目的からは、該炭化の最高温度は1200〜1700℃がより好ましく、1300〜1600℃であるのがさらに好ましい。 In the present invention, the temperature in the carbonization step is preferably 1000 to 2000 ° C., and the maximum temperature is appropriately set according to the desired mechanical properties of the carbon fiber. Generally, the higher the maximum temperature of the carbonization step, the higher the tensile elastic modulus of the obtained carbon fiber, but the tensile strength becomes maximum at around 1500 ° C. In order to increase both the tensile strength and the tensile modulus, the maximum carbonization temperature is more preferably 1200 to 1700 ° C, and further preferably 1300 to 1600 ° C.
本発明において、炭化を行う際の延伸比は0.970〜1.100が好ましく、より好ましくは0.975〜1.005、さらに好ましくは0.980〜1.000である。該延伸比は0.970を下回ると、得られる炭素繊維の配向度や緻密性が不十分となり、力学物性が低下することがある。また、該延伸比が1.10を超えると、毛羽発生、糸切れ発生によりプロセス性が低下することがある。
引き続き、上述の方法で得られた炭素繊維を不活性雰囲気中、2,000〜3,000℃で延伸比1.000〜1.200で延伸しながら黒鉛化することによって、より高い弾性率を有した黒鉛化繊維とすることもできる。
In the present invention, the stretching ratio during carbonization is preferably 0.970 to 1.100, more preferably 0.975 to 1.005, and still more preferably 0.980 to 1.000. When the draw ratio is less than 0.970, the degree of orientation and denseness of the obtained carbon fiber may be insufficient, and the mechanical properties may be lowered. On the other hand, when the draw ratio exceeds 1.10, processability may be deteriorated due to generation of fluff and yarn breakage.
Subsequently, the carbon fiber obtained by the above-described method is graphitized while being stretched at a stretch ratio of 1.000 to 1.200 at 2,000 to 3,000 ° C. in an inert atmosphere, thereby obtaining a higher elastic modulus. It can also be a graphitized fiber.
本発明において、黒鉛化を行う際の延伸比は、より好ましくは1.005〜1.150、さらに好ましくは1.010〜0.1.100である。該延伸比は1.000を下回ると、得られる黒鉛化繊維の配向度や緻密性が不十分となり、力学物性が低下することがある。また、該延伸比が1.200を超えると、毛羽発生、糸切れ発生によりプロセス性が低下することがある。 In the present invention, the draw ratio during graphitization is more preferably 1.005 to 1.150, and still more preferably 1.010 to 0.1.100. When the draw ratio is less than 1.000, the degree of orientation and denseness of the resulting graphitized fiber may be insufficient, and the mechanical properties may deteriorate. On the other hand, when the draw ratio exceeds 1.200, the processability may be deteriorated due to the occurrence of fluff and yarn breakage.
得られた炭素繊維、黒鉛化繊維はその表面改質のため、電解処理することができる。電解処理に用いる電解液には、硫酸、硝酸、塩酸等の酸性溶液や、水酸化ナトリウム、水酸化カリウム、テトラエチルアンモニウムヒドロキシド、炭酸アンモニウム、重炭酸アンモニウムといったアルカリ又はそれらの塩を水溶液として使用することができる。ここで、電解処理に要する電気量は、適用する炭素繊維、黒鉛化繊維の炭化度に応じて適宜選択することができる。 The obtained carbon fiber and graphitized fiber can be subjected to electrolytic treatment for surface modification. As an electrolytic solution used for the electrolytic treatment, an acidic solution such as sulfuric acid, nitric acid, hydrochloric acid, an alkali such as sodium hydroxide, potassium hydroxide, tetraethylammonium hydroxide, ammonium carbonate, ammonium bicarbonate or a salt thereof is used as an aqueous solution. be able to. Here, the amount of electricity required for the electrolytic treatment can be appropriately selected according to the carbonization degree of the applied carbon fiber and graphitized fiber.
かかる電解処理により、得られる複合材料において炭素繊維、黒鉛繊維とマトリックスとの接着性が適正化でき、接着が強すぎることによる複合材料のブリトルな破壊や、繊維方向の引張強度が低下する問題や、繊維方向における引張強度は高いものの、樹脂との接着性に劣り、非繊維方向における強度特性が発現しないといった問題が解消され、得られる複合材料において、繊維方向と非繊維方向の両方向にバランスのとれた強度特性が発現されるようになる。 Such electrolytic treatment can optimize the adhesion between the carbon fiber and graphite fiber and the matrix in the composite material obtained, the brittle breakage of the composite material due to too strong adhesion, and the problem that the tensile strength in the fiber direction decreases. Although the tensile strength in the fiber direction is high, the problem of poor adhesion to the resin and the absence of strength properties in the non-fiber direction has been resolved, and the resulting composite material has a balance in both the fiber direction and the non-fiber direction. Excellent strength characteristics are developed.
かかる電解処理の後、得られる炭素繊維、黒鉛化繊維に集束性を付与するため、サイジング処理をすることもできる。サイジング剤には、使用する樹脂の種類に応じて、樹脂との相溶性の良いサイジング剤を適宜選択することができる。 After the electrolytic treatment, a sizing treatment can also be performed in order to impart convergence to the obtained carbon fiber and graphitized fiber. As the sizing agent, a sizing agent having good compatibility with the resin can be appropriately selected according to the type of resin used.
本発明において、適宜条件設定することにより、ストランド引張強度が6.5GPa以上、ストランド弾性率が330GPa以上である力学物性に優れた炭素繊維が得られる。ストランド引張強度が6.7GPa以上、ストランド弾性率が340GPa以上がより好ましく、ストランド引張強度が7.0GPa以上、ストランド弾性率が350GPa以上がさらに好ましい。 In the present invention, by appropriately setting conditions, a carbon fiber excellent in mechanical properties having a strand tensile strength of 6.5 GPa or more and a strand elastic modulus of 330 GPa or more can be obtained. More preferably, the strand tensile strength is 6.7 GPa or more, the strand elastic modulus is 340 GPa or more, the strand tensile strength is 7.0 GPa or more, and the strand elastic modulus is 350 GPa or more.
また、本発明において、適宜条件設定することにより、ストランド引張強度が6GPa以上、ストランド弾性率が400GPa以上である力学物性に優れた黒鉛化繊維が得られる。 In the present invention, by appropriately setting conditions, a graphitized fiber excellent in mechanical properties having a strand tensile strength of 6 GPa or more and a strand elastic modulus of 400 GPa or more is obtained.
本発明により得られる炭素繊維および黒鉛化繊維は、引張強度が高く、引張弾性率が高い(すなわち高伸度)であり、また、相対的に低い焼成温度で高い弾性率が得られるため、同時に高い圧縮強度を発現することができる。従って、プリプレグとしてオートクレーブ成形、織物などのプリフォームとしてレジントランスファーモールディングで成形、フィラメントワインディングで成形するなど種々の成型法により、航空機部材、圧力容器部材、自動車部材、釣り竿、ゴルフシャフトなどのスポーツ部材として、好適に用いることができる。 The carbon fiber and graphitized fiber obtained by the present invention have high tensile strength, high tensile elastic modulus (that is, high elongation), and high elastic modulus can be obtained at a relatively low firing temperature. High compressive strength can be expressed. Therefore, as a prepreg, autoclave molding, as a preform such as fabric, molded by resin transfer molding, molded by filament winding, etc., as a sports member such as aircraft members, pressure vessel members, automobile members, fishing rods, golf shafts, etc. Can be preferably used.
本発明をより具体的に説明する。なお、実施例で用いた各種物性値の測定方法は以下に記載の方法によるものである。
<極限粘度>
予め120℃で2時間熱処理し絶乾した炭素繊維前駆体繊維用ポリアクリロニトリル系重合体150mgを、25℃において50mlのチオシアン酸ナトリウム0.1mol/リットル添加ジメチルフォルムアミド(いずれも和光純薬社製特級)に溶解した。得られた溶液を、25℃に温調し、予め25℃に温調してあるオストワルド粘度計を用いて標線間の落下時間を1/100秒の精度で測定し、その時間をt(秒)とした。同様に、炭素繊維前駆体繊維用ポリアクリロニトリル系重合体を溶解していないチオシアン酸ナトリウム0.1mol/リットル添加ジメチルフォルムアミドについても測定し、その落下時間をt0(秒)とした。次式を用いて極限粘度[η]を算出した。
[η]={(1+1.32×ηsp)^(1/2)―1}/0.198
ηsp=(t/t0)−1
<耐炎化処理時の酸化深さD>
測定に供する重合体を、ジメチルスルホキシドに重量濃度で25%となるよう溶解し、次に該溶液をガラス板上にキャストして、ベーカー式アプリケーターを用いて約80μmの厚みになるように塗布した。次に、重合体溶液を塗布したガラス板を、熱風乾燥機を用いて空気中120℃で6時間乾燥し、ジメチルスルホキシドを蒸発させて、厚み約20μmのフィルムとした。得られたフィルムを、熱風乾燥機を用いて、空気中240℃で60分、さらに空気中250℃で60分熱処理し、耐炎化処理を行った。得られた耐炎化フィルムを樹脂包埋した上で研磨し、そのフィルム表面に対して垂直な断面を光学顕微鏡を用いて倍率800倍で観察し、写真撮影した。断面において酸化が進んだ部分は暗い層として、進んでいない部分は明るい層として観察されるので、フィルム表面から、暗い層と明るい層の境界までの距離を、写真上で5点計測し、その算術平均を酸化深さD(μm)とした。
<湿熱下融点Tm>
測定に供する重合体を、液体窒素中で凍結粉砕した後、目開き0.5mmの篩いを通し粉体を得た。該粉体を5mg精秤し、メトラー社製DSC用中圧パンME29990(耐圧2MPa)に、5mgの純水とともに密封した。ブルカー社製DSC3100SAを用いて、10℃/分の昇温速度で、室温から220℃までDSC測定し、150〜200℃付近に現れる吸熱ピークの頂点に対応する温度を読み取り、湿熱下融点Tm(℃)とした。
<ガラス転移点Tg>
前記した酸化深さDの測定と同様の方法でフィルムを作製した。該フィルムを幅3mmの短冊状に切り、厚みおよび幅を正確に測定した後、試長間が20mmとなるように動的粘弾性測定装置にセットした。測定はブルカー・エイエックスエス社製TMA4010SAの粘弾性測定モードを用いて行った。最低荷重3g、最高荷重6g、周波数0.2Hzの正弦波で引張荷重を加えながら、昇温速度10℃/分で、室温から200℃まで昇温しながら測定を行った。得られたデータから損失弾性率E”を求め、そのピークに相当する温度を読み取り、ガラス転移点Tg(℃)とした。
<耐炎化繊維比重>
JIS R7601(1986)記載の方法に従った。試薬はエタノール(和光純薬社製特級)を精製せずに用いた。1.0〜1.5gの繊維を採取し、熱風乾燥機を用い、空気中120℃で2時間絶乾した。絶乾質量A(g)を測定した後、比重既知(比重ρ)のエタノールに含浸し、エタノール中の繊維質量B(g)を測定し、次式、繊維比重=(A×ρ)/(A−B)により繊維比重Dを求めた。
<炭素繊維のストランド引張強度及びストランド引張弾性率>
JIS R7601(1986)「樹脂含浸ストランド試験法」に従って求めた。
The present invention will be described more specifically. In addition, the measuring method of the various physical-property values used in the Example is based on the method as described below.
<Intrinsic viscosity>
150 mg of polyacrylonitrile polymer for carbon fiber precursor fiber, which was heat-treated at 120 ° C. for 2 hours in advance and completely dried, was added at 25 ° C. with 50 ml of sodium thiocyanate 0.1 mol / liter dimethylformamide (both manufactured by Wako Pure Chemical Industries, Ltd.) It was dissolved in special grade). The temperature of the resulting solution was adjusted to 25 ° C., and the Ostwald viscometer previously adjusted to 25 ° C. was used to measure the drop time between the marked lines with an accuracy of 1/100 second. Seconds). Similarly, 0.1 mol / liter-added dimethylformamide with sodium thiocyanate in which the polyacrylonitrile-based polymer for carbon fiber precursor fibers was not dissolved was also measured, and the drop time was defined as t0 (seconds). The intrinsic viscosity [η] was calculated using the following formula.
[Η] = {(1 + 1.32 × ηsp) ^ (1/2) −1} /0.198
ηsp = (t / t0) −1
<Oxidation depth D during flameproofing treatment>
The polymer used for the measurement was dissolved in dimethyl sulfoxide to a weight concentration of 25%, and then the solution was cast on a glass plate and applied to a thickness of about 80 μm using a Baker type applicator. . Next, the glass plate coated with the polymer solution was dried in air at 120 ° C. for 6 hours using a hot air dryer to evaporate dimethyl sulfoxide to obtain a film having a thickness of about 20 μm. The obtained film was heat-treated at 240 ° C. in air for 60 minutes and further in air at 250 ° C. for 60 minutes using a hot air dryer to perform flameproofing treatment. The obtained flame-resistant film was polished after being embedded in a resin, and a cross section perpendicular to the film surface was observed with an optical microscope at a magnification of 800 times and photographed. In the cross section, the part where oxidation has progressed is observed as a dark layer, and the part which has not progressed is observed as a bright layer. Therefore, the distance from the film surface to the boundary between the dark layer and the bright layer is measured at five points on the photograph The arithmetic average was defined as the oxidation depth D (μm).
<Melting point Tm under wet heat>
The polymer used for the measurement was freeze-ground in liquid nitrogen and then passed through a sieve having an aperture of 0.5 mm to obtain a powder. 5 mg of the powder was precisely weighed and sealed with 5 mg of pure water in a medium pressure pan ME29990 for DSC manufactured by Mettler (withstand pressure of 2 MPa). Using DSC3100SA manufactured by Bruker, DSC measurement was performed from room temperature to 220 ° C. at a rate of temperature increase of 10 ° C./min, and the temperature corresponding to the end of the endothermic peak appearing in the vicinity of 150 to 200 ° C. was read. ° C).
<Glass transition point Tg>
A film was produced in the same manner as the measurement of the oxidation depth D described above. The film was cut into a strip shape having a width of 3 mm, and after measuring the thickness and width accurately, the film was set in a dynamic viscoelasticity measuring apparatus so that the test length was 20 mm. The measurement was performed using the viscoelasticity measurement mode of TMA4010SA manufactured by Bruker AXS. Measurement was performed while increasing the temperature from room temperature to 200 ° C. at a temperature increase rate of 10 ° C./min while applying a tensile load with a sine wave having a minimum load of 3 g, a maximum load of 6 g and a frequency of 0.2 Hz. The loss elastic modulus E ″ was obtained from the obtained data, and the temperature corresponding to the peak was read and used as the glass transition point Tg (° C.).
<Flame resistant fiber specific gravity>
The method described in JIS R7601 (1986) was followed. As a reagent, ethanol (special grade manufactured by Wako Pure Chemical Industries, Ltd.) was used without purification. 1.0 to 1.5 g of fiber was collected and dried in air at 120 ° C. for 2 hours using a hot air dryer. After measuring the absolute dry mass A (g), it was impregnated in ethanol with a known specific gravity (specific gravity ρ), and the fiber mass B (g) in ethanol was measured. The following formula, fiber specific gravity = (A × ρ) / ( The fiber specific gravity D was determined by AB).
<Strand tensile strength and strand tensile modulus of carbon fiber>
It was determined according to JIS R7601 (1986) “Resin-impregnated strand test method”.
ここで、測定する炭素繊維の樹脂含浸ストランドは、ユニオンカーバイド(株)製、”BAKELITE(登録商標)”ERL4221(100重量部)/3フッ化ホウ素モノエチルアミン(3重量部)/アセトン(4重量部)を、炭素繊維に含浸させ、130℃、30分熱処理し硬化させて作製した。また、ストランドの測定本数は6本とし、各測定結果の算術平均値を、その炭素繊維の引張強度、引張弾性率とした。
[実施例1〜8、比較例1〜4]
表1に示した組成からなる共重合体成分をジメチルスルホキシドを溶媒とする溶液重合法により、アゾビスイソブチロニトリルを開始剤としてラジカル重合し、極限粘度1.5〜1.6の炭素繊維前駆体繊維用共重合体を得た。得られた重合体について、酸化深さD(μm)、湿熱下融点Tm(℃)、およびガラス転移点Tg(℃)を測定した。
該重合体の濃度が、ジメチルスルホキシド中、25重量%となるよう調製した後、アンモニアガスをpHが8.5になるまで吹き込むことで、イタコン酸を中和しつつ、アンモニウム基をポリアクリロニトリル系共重合体に導入し、紡糸原液を作製した。得られた紡糸原液を、目開き0.5μmのフィルター通過後、40℃で、単孔の直径0.15mm、孔数6,000の紡糸口金を用い、一旦空気中に吐出し、約4mmの空間を通過させた後、3℃にコントロールした35重量%ジメチルスルホキシドの水溶液からなる凝固浴に導入する乾湿式紡糸法により凝固糸条とした。この凝固糸条を、常法により水洗した後、温水中で3.5倍に延伸し、さらにアミノ変性シリコーン系シリコーン油剤を付与して単繊維繊度2.6dtexの浴中延伸糸を得た。この浴中延伸糸を、165℃に加熱したローラーを用いて乾燥熱処理を行い、次に145℃の加圧スチーム中にて延伸倍率を0.1倍ずつ変えながら糸切れの有無を測定し、糸切れの発生しない最大倍率をスチーム延伸性(倍)とした。併せて前記条件において、加圧スチーム中で3.7倍延伸し、全延伸倍率13倍、単繊維繊度0.7dtex、フィラメント数6,000の炭素繊維前駆体繊維を得た。
得られた炭素繊維前駆体繊維を4本合糸し、トータルフィラメント数24,000とした上で、240〜260℃の空気中において延伸比1.0で延伸しながらで耐炎化処理し、比重1.35の耐炎化繊維を得た。
続いて300〜700℃の窒素雰囲気中において、延伸比1.15で延伸しながら予備炭化処理を行い、さらに最高温度1500℃の窒素雰囲気中において、延伸比を0.99に設定して炭化処理を行い、比重1.80〜1.83の炭素繊維を得た。この際、炭化工程の出側において、走行中の糸条の毛羽数を長さ30mに亘って目視により計測し、その1m当たりの毛羽数を炭化毛羽個数(個/m)とした。また、得られた炭素繊維について、ストランド引張強度およびストランド引張弾性率を測定した。
得られた結果を表2および表3に示す。
酸化深さDが大きい重合体を用いるほど、炭化毛羽個数が少なく、焼成におけるプロセス性が良好で、得られる炭素繊維の物性も良好であった。また、湿熱下融点Tmが低いものは、製糸におけるスチーム延伸性が低下し、炭化毛羽個数、得られる炭素繊維のストランド引張強度、ストランド引張弾性率も低下することがわかった。
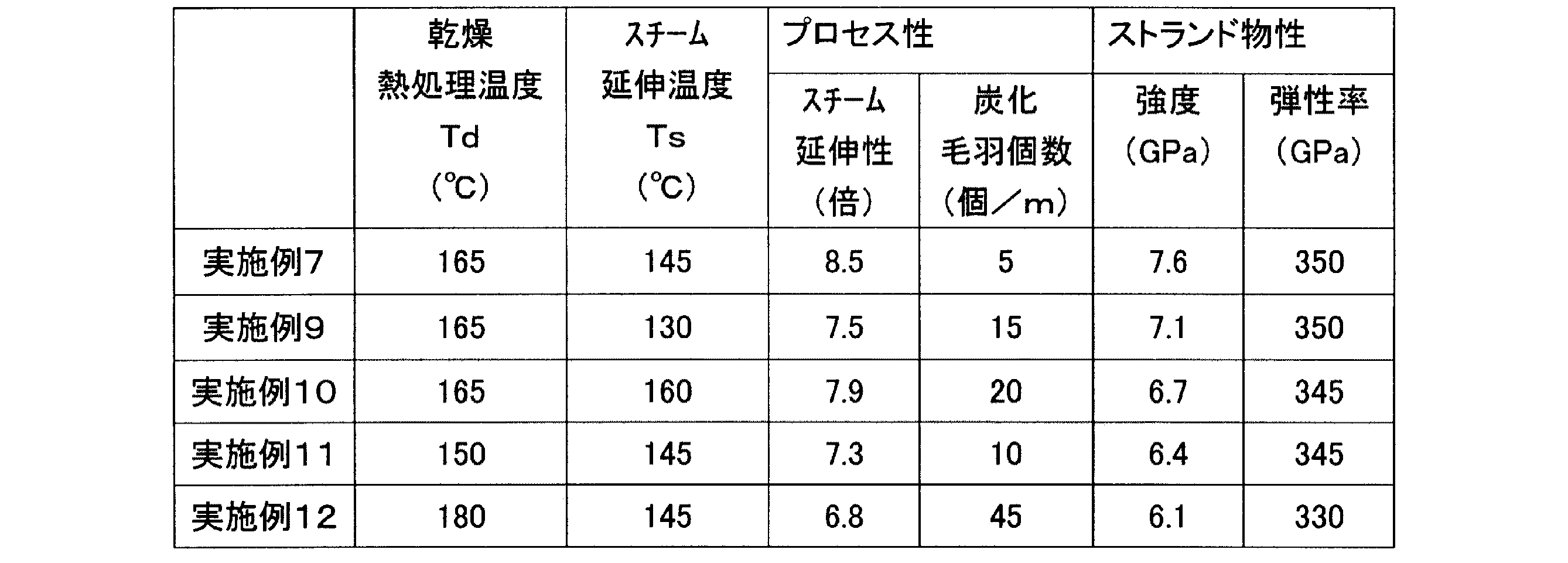
[実施例9〜12]
製糸の乾燥熱処理温度Tdおよびスチーム延伸温度Tsを、表4のように変更した他は、実施例7と同様にして、製糸、焼成、評価を行った。
Here, the resin-impregnated strand of carbon fiber to be measured was “BAKELITE (registered trademark)” ERL 4221 (100 parts by weight) / 3 boron trifluoride monoethylamine (3 parts by weight) / acetone (4 weights) manufactured by Union Carbide Corporation. Part) was impregnated into carbon fiber, and heat treated at 130 ° C. for 30 minutes and cured. Further, the number of strands measured was 6, and the arithmetic average value of each measurement result was taken as the tensile strength and tensile modulus of the carbon fiber.
[Examples 1-8, Comparative Examples 1-4]
A carbon fiber having an intrinsic viscosity of 1.5 to 1.6 is obtained by radical polymerization of a copolymer component having the composition shown in Table 1 using azobisisobutyronitrile as an initiator by a solution polymerization method using dimethyl sulfoxide as a solvent. A copolymer for precursor fibers was obtained. About the obtained polymer, oxidation depth D (micrometer), melting | fusing point Tm (degreeC) under wet heat, and glass transition point Tg (degreeC) were measured.
The polymer concentration was adjusted to 25% by weight in dimethyl sulfoxide, and then ammonia gas was blown until the pH reached 8.5 to neutralize itaconic acid, while the ammonium group was polyacrylonitrile-based. The solution was introduced into the copolymer to prepare a spinning dope. The obtained spinning dope is passed through a filter having a mesh opening of 0.5 μm, and at 40 ° C., once discharged into the air using a spinneret having a single hole diameter of 0.15 mm and a hole number of 6,000, about 4 mm After passing through the space, a coagulated yarn was obtained by a dry and wet spinning method introduced into a coagulation bath composed of an aqueous solution of 35% by weight dimethyl sulfoxide controlled at 3 ° C. The coagulated yarn was washed with water by a conventional method, then stretched 3.5 times in warm water, and further amino-modified silicone-based silicone oil was added to obtain a stretched yarn in a bath having a single fiber fineness of 2.6 dtex. The drawn yarn in the bath is subjected to a drying heat treatment using a roller heated to 165 ° C., and then the presence or absence of yarn breakage is measured while changing the draw ratio by 0.1 times in a pressurized steam at 145 ° C. The maximum magnification at which no yarn breakage occurred was defined as steam stretchability (times). At the same time, the carbon fiber precursor fiber was stretched 3.7 times in pressurized steam to obtain a carbon fiber precursor fiber having a total draw ratio of 13 times, a single fiber fineness of 0.7 dtex, and a filament number of 6,000.
Four carbon fiber precursor fibers obtained were combined to give a total filament number of 24,000, and subjected to a flame resistance treatment while being drawn in air at 240 to 260 ° C. at a draw ratio of 1.0. 1.35 flameproof fiber was obtained.
Subsequently, pre-carbonization is performed while stretching at a stretch ratio of 1.15 in a nitrogen atmosphere at 300 to 700 ° C., and further, carbonization is performed by setting the stretch ratio to 0.99 in a nitrogen atmosphere at a maximum temperature of 1500 ° C. To obtain carbon fibers having a specific gravity of 1.80 to 1.83. At this time, on the exit side of the carbonization step, the number of fluffs of the running yarn was measured visually over a length of 30 m, and the number of fluffs per meter was defined as the number of fluffs (pieces / m). Moreover, strand tensile strength and strand tensile elastic modulus were measured about the obtained carbon fiber.
The obtained results are shown in Tables 2 and 3.
As the polymer having a larger oxidation depth D was used, the number of carbonized fluff was smaller, the processability in firing was better, and the physical properties of the resulting carbon fiber were better. Further, it was found that when the melting point Tm under wet heat is low, the steam stretchability in the yarn production is lowered, and the number of carbonized fuzz, the strand tensile strength of the obtained carbon fiber, and the strand tensile elastic modulus are also lowered.
[Examples 9 to 12]
Yarn-making, firing, and evaluation were performed in the same manner as in Example 7 except that the drying heat treatment temperature Td and the steam drawing temperature Ts of the yarn-making were changed as shown in Table 4.
得られた結果を表4に併せて示す。 The obtained results are also shown in Table 4.
乾燥熱処理温度Tdおよびスチーム延伸温度Tsによって、プロセス性および得られる炭素繊維のストランド引張強度、ストランド引張弾性率が変化し、特定の範囲で最適化できることがわかった。 It was found that the processability and strand tensile strength and strand tensile elastic modulus of the resulting carbon fiber change depending on the drying heat treatment temperature Td and the steam drawing temperature Ts, and can be optimized within a specific range.
Claims (7)
(Tm―30)≦Td≦(Tm−10)
(Tm―50)≦Ts≦(Tm−30) In a method for producing a carbon fiber precursor fiber that is formed by spinning the polyacrylonitrile-based polymer for a carbon fiber precursor fiber according to any one of claims 1 to 4 by a wet or dry-wet spinning method, followed by a dry heat treatment and then steam drawing. A method for producing a carbon fiber precursor fiber, wherein a drying heat treatment temperature Td (° C.), a steam stretching temperature Ts (° C.), and a melting point Tm (° C.) under wet heat measured by a differential scanning calorimeter of the polymer satisfy the following formula.
(Tm-30) ≦ Td ≦ (Tm-10)
(Tm-50) ≦ Ts ≦ (Tm-30)
A method for producing graphitized fiber, wherein the carbon fiber produced by the method according to claim 6 is graphitized while being drawn at a draw ratio of 1.00 to 1.20 in an inert atmosphere at 2000 to 3000 ° C.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005076621A JP4617940B2 (en) | 2005-03-17 | 2005-03-17 | Polyacrylonitrile-based polymer for carbon fiber precursor fiber, carbon fiber precursor fiber, and method for producing carbon fiber |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005076621A JP4617940B2 (en) | 2005-03-17 | 2005-03-17 | Polyacrylonitrile-based polymer for carbon fiber precursor fiber, carbon fiber precursor fiber, and method for producing carbon fiber |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006257580A true JP2006257580A (en) | 2006-09-28 |
| JP4617940B2 JP4617940B2 (en) | 2011-01-26 |
Family
ID=37097149
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005076621A Expired - Fee Related JP4617940B2 (en) | 2005-03-17 | 2005-03-17 | Polyacrylonitrile-based polymer for carbon fiber precursor fiber, carbon fiber precursor fiber, and method for producing carbon fiber |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4617940B2 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008248219A (en) * | 2006-10-18 | 2008-10-16 | Toray Ind Inc | Polyacrylonitrile-based polymer and method for producing the same, and method for producing carbon fiber precursor, and carbon fiber and method for producing the same |
| WO2012050171A1 (en) | 2010-10-13 | 2012-04-19 | 三菱レイヨン株式会社 | Carbon-fiber-precursor fiber bundle, carbon fiber bundle, and uses thereof |
| US20120126442A1 (en) * | 2005-12-13 | 2012-05-24 | Toray Industries, Inc. | Processes for producing polyacrylonitrile-base precursor fibers and carbon fibers |
| KR101219965B1 (en) * | 2010-12-30 | 2013-01-08 | 주식회사 효성 | Preparation method of carbon fiber Precursor |
| KR101276469B1 (en) | 2009-12-31 | 2013-06-19 | 주식회사 효성 | Method of preparing precursors for polyacrylonitrile-based carbon fibers |
| JP2015067910A (en) * | 2013-09-27 | 2015-04-13 | 東レ株式会社 | Carbon fiber and manufacturing method thereof |
| JP2015071722A (en) * | 2013-10-04 | 2015-04-16 | 三菱レイヨン株式会社 | Acrylonitrile-based copolymer, carbon fiber precursor acrylonitrile-based fiber, carbon fiber, and manufacturing method of carbon fiber |
| US10017881B2 (en) | 2011-07-22 | 2018-07-10 | Mitsubishi Chemical Corporation | Polyacrylonitrile-based copolymer, polyacrylonitrile-based precursor fiber for carbon fiber, carbon fiber bundles, process for producing stabilized fiber bundles, and process for producing carbon fiber bundles |
| CN113789607A (en) * | 2021-09-22 | 2021-12-14 | 辽宁兴汇碳材料科技有限公司 | Polyacrylonitrile-based fibrofelt and preparation method and application thereof |
| US11535957B2 (en) | 2016-11-23 | 2022-12-27 | Lg Chem, Ltd. | Method for producing polyacrylonitrile-based fiber and polyacrylonitrile-based copolymer used therein |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1112856A (en) * | 1997-06-23 | 1999-01-19 | Toray Ind Inc | Acrylic precursor fiber for carbon fiber and its production |
| JPH11241230A (en) * | 1997-12-11 | 1999-09-07 | Toray Ind Inc | Carbon fiber, precursor fiber for carbon fiber, composite material and production of carbon fiber |
| JP2004316052A (en) * | 2002-09-30 | 2004-11-11 | Toray Ind Inc | Oil formulation for producing carbon fiber and method for producing the carbon fiber |
-
2005
- 2005-03-17 JP JP2005076621A patent/JP4617940B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1112856A (en) * | 1997-06-23 | 1999-01-19 | Toray Ind Inc | Acrylic precursor fiber for carbon fiber and its production |
| JPH11241230A (en) * | 1997-12-11 | 1999-09-07 | Toray Ind Inc | Carbon fiber, precursor fiber for carbon fiber, composite material and production of carbon fiber |
| JP2004316052A (en) * | 2002-09-30 | 2004-11-11 | Toray Ind Inc | Oil formulation for producing carbon fiber and method for producing the carbon fiber |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120126442A1 (en) * | 2005-12-13 | 2012-05-24 | Toray Industries, Inc. | Processes for producing polyacrylonitrile-base precursor fibers and carbon fibers |
| JP2008248219A (en) * | 2006-10-18 | 2008-10-16 | Toray Ind Inc | Polyacrylonitrile-based polymer and method for producing the same, and method for producing carbon fiber precursor, and carbon fiber and method for producing the same |
| KR101276469B1 (en) | 2009-12-31 | 2013-06-19 | 주식회사 효성 | Method of preparing precursors for polyacrylonitrile-based carbon fibers |
| US10233569B2 (en) | 2010-10-13 | 2019-03-19 | Mitsubishi Chemical Corporation | Carbon-fiber-precursor fiber bundle, carbon fiber bundle, and uses thereof |
| WO2012050171A1 (en) | 2010-10-13 | 2012-04-19 | 三菱レイヨン株式会社 | Carbon-fiber-precursor fiber bundle, carbon fiber bundle, and uses thereof |
| US11332852B2 (en) | 2010-10-13 | 2022-05-17 | Mitsubishi Chemical Corporation | Carbon-fiber-precursor fiber bundle, carbon fiber bundle, and uses thereof |
| US10662556B2 (en) | 2010-10-13 | 2020-05-26 | Mitsubishi Chemical Corporation | Carbon-fiber-precursor fiber bundle, carbon fiber bundle, and uses thereof |
| US9920456B2 (en) | 2010-10-13 | 2018-03-20 | Mitsubishi Chemical Corporation | Carbon-fiber-precursor fiber bundle, carbon fiber bundle, and uses thereof |
| KR101219965B1 (en) * | 2010-12-30 | 2013-01-08 | 주식회사 효성 | Preparation method of carbon fiber Precursor |
| US10017881B2 (en) | 2011-07-22 | 2018-07-10 | Mitsubishi Chemical Corporation | Polyacrylonitrile-based copolymer, polyacrylonitrile-based precursor fiber for carbon fiber, carbon fiber bundles, process for producing stabilized fiber bundles, and process for producing carbon fiber bundles |
| JP2015067910A (en) * | 2013-09-27 | 2015-04-13 | 東レ株式会社 | Carbon fiber and manufacturing method thereof |
| JP2015071722A (en) * | 2013-10-04 | 2015-04-16 | 三菱レイヨン株式会社 | Acrylonitrile-based copolymer, carbon fiber precursor acrylonitrile-based fiber, carbon fiber, and manufacturing method of carbon fiber |
| US11535957B2 (en) | 2016-11-23 | 2022-12-27 | Lg Chem, Ltd. | Method for producing polyacrylonitrile-based fiber and polyacrylonitrile-based copolymer used therein |
| CN113789607A (en) * | 2021-09-22 | 2021-12-14 | 辽宁兴汇碳材料科技有限公司 | Polyacrylonitrile-based fibrofelt and preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4617940B2 (en) | 2011-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4617940B2 (en) | Polyacrylonitrile-based polymer for carbon fiber precursor fiber, carbon fiber precursor fiber, and method for producing carbon fiber | |
| JP4957251B2 (en) | Carbon fiber, method for producing polyacrylonitrile-based precursor fiber for carbon fiber production, and method for producing carbon fiber | |
| JP5691366B2 (en) | Carbon fiber manufacturing method | |
| KR101467620B1 (en) | Manufacturing method of carbon fiber and precursor | |
| JP2008308776A (en) | Method for producing polyacrylonitrile-based precursor fiber, method for producing carbon fiber, and carbon fiber | |
| WO2013015210A1 (en) | Polyacrylonitrile-based copolymer, polyacrylonitrile-based precursor fiber for carbon fiber, carbon fiber bundles, process for producing flameproofed fiber bundles, and process for producing carbon fiber bundles | |
| JP4924469B2 (en) | Carbon fiber precursor fiber and method for producing carbon fiber | |
| JP5434187B2 (en) | Polyacrylonitrile-based continuous carbon fiber bundle and method for producing the same | |
| JP2007162144A (en) | Method for producing carbon fiber bundle | |
| JP2006348462A (en) | Method for producing acrylonitrile-based precursor fiber for carbon fiber | |
| JP2010100970A (en) | Method for producing carbon fiber | |
| JP2011213773A (en) | Polyacrylonitrile-based polymer and carbon fiber | |
| KR101268173B1 (en) | Polyacrylonitrile-based polymer solution, preparing method of the same, Carbon fiber precursor, manufacturing method of the same and manufacturing method of carbon fiber using the same | |
| JP2010053468A (en) | Method for producing carbon fiber precursor fiber | |
| JP5504678B2 (en) | Polyacrylonitrile polymer solution, carbon fiber precursor fiber, and method for producing carbon fiber | |
| JP2011017100A (en) | Method for producing carbon fiber | |
| JP2008308777A (en) | Carbon fiber and method for producing polyacrylonitrile-based precursor fiber for producing carbon fiber | |
| JP5066952B2 (en) | Method for producing polyacrylonitrile-based polymer composition, and method for producing carbon fiber | |
| JP2009079343A (en) | Method for producing precursor fiber for carbon fiber and carbon fiber | |
| JP4983709B2 (en) | Carbon fiber precursor fiber and method for producing carbon fiber | |
| KR20150127870A (en) | The method of producing the polyacrylonitrile precursor for carbon fiber and the method of producing carbon fiber | |
| JP2004060069A (en) | Polyacrylonitrile-based carbon fiber, and method for producing the same | |
| JP5146394B2 (en) | Method for producing carbon fiber precursor fiber and method for producing carbon fiber | |
| JP2007321267A (en) | Method for producing polyacrylonitrile-based fiber and carbon fiber | |
| JP2011213774A (en) | Polyacrylonitrile for producing carbon fiber, polyacrylonitrile-based precursor fiber, and method for producing carbon fiber |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080313 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100713 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100819 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100928 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20101011 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131105 Year of fee payment: 3 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 4617940 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131105 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |