ES2811347T3 - Método de formación de imágenes con un compuesto quelante - Google Patents
Método de formación de imágenes con un compuesto quelante Download PDFInfo
- Publication number
- ES2811347T3 ES2811347T3 ES16837542T ES16837542T ES2811347T3 ES 2811347 T3 ES2811347 T3 ES 2811347T3 ES 16837542 T ES16837542 T ES 16837542T ES 16837542 T ES16837542 T ES 16837542T ES 2811347 T3 ES2811347 T3 ES 2811347T3
- Authority
- ES
- Spain
- Prior art keywords
- acceptable salt
- pharmaceutically acceptable
- cancer
- patient
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000003384 imaging method Methods 0.000 title claims abstract description 68
- 239000002738 chelating agent Substances 0.000 title description 8
- 150000001875 compounds Chemical class 0.000 claims abstract description 245
- 150000003839 salts Chemical class 0.000 claims abstract description 210
- 238000000034 method Methods 0.000 claims abstract description 185
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 147
- 201000011510 cancer Diseases 0.000 claims abstract description 92
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 32
- 125000003118 aryl group Chemical group 0.000 claims abstract description 23
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 15
- 125000004404 heteroalkyl group Chemical group 0.000 claims abstract description 15
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims abstract description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 15
- 239000001257 hydrogen Substances 0.000 claims abstract description 15
- 125000004103 aminoalkyl group Chemical group 0.000 claims abstract description 13
- 125000004181 carboxyalkyl group Chemical group 0.000 claims abstract description 13
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims abstract description 13
- 150000001768 cations Chemical class 0.000 claims abstract description 10
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 claims description 43
- 210000001519 tissue Anatomy 0.000 claims description 41
- 239000011724 folic acid Substances 0.000 claims description 26
- 235000019152 folic acid Nutrition 0.000 claims description 26
- 230000037396 body weight Effects 0.000 claims description 19
- 238000013170 computed tomography imaging Methods 0.000 claims description 19
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 claims description 17
- 229960000304 folic acid Drugs 0.000 claims description 17
- 206010014733 Endometrial cancer Diseases 0.000 claims description 13
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- 210000004369 blood Anatomy 0.000 claims description 10
- 239000008280 blood Substances 0.000 claims description 10
- 210000004072 lung Anatomy 0.000 claims description 10
- 210000003205 muscle Anatomy 0.000 claims description 10
- 102000006815 folate receptor Human genes 0.000 claims description 9
- 108020005243 folate receptor Proteins 0.000 claims description 9
- 210000003734 kidney Anatomy 0.000 claims description 9
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 9
- 206010033128 Ovarian cancer Diseases 0.000 claims description 8
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 8
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 claims description 8
- 210000003169 central nervous system Anatomy 0.000 claims description 8
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 8
- 239000013589 supplement Substances 0.000 claims description 8
- 208000022679 triple-negative breast carcinoma Diseases 0.000 claims description 8
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 8
- 230000003211 malignant effect Effects 0.000 claims description 7
- 229910052713 technetium Inorganic materials 0.000 claims description 7
- 201000009030 Carcinoma Diseases 0.000 claims description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 6
- 210000002376 aorta thoracic Anatomy 0.000 claims description 6
- 229910052702 rhenium Inorganic materials 0.000 claims description 6
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical compound [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 claims description 6
- GKLVYJBZJHMRIY-UHFFFAOYSA-N technetium atom Chemical compound [Tc] GKLVYJBZJHMRIY-UHFFFAOYSA-N 0.000 claims description 6
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 5
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 claims description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 5
- 206010038389 Renal cancer Diseases 0.000 claims description 5
- 206010039491 Sarcoma Diseases 0.000 claims description 5
- GKLVYJBZJHMRIY-OUBTZVSYSA-N Technetium-99 Chemical compound [99Tc] GKLVYJBZJHMRIY-OUBTZVSYSA-N 0.000 claims description 5
- 229910052802 copper Inorganic materials 0.000 claims description 5
- 239000010949 copper Substances 0.000 claims description 5
- 229910052733 gallium Inorganic materials 0.000 claims description 5
- 210000002216 heart Anatomy 0.000 claims description 5
- 229910052738 indium Inorganic materials 0.000 claims description 5
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 claims description 5
- 210000000936 intestine Anatomy 0.000 claims description 5
- 201000010982 kidney cancer Diseases 0.000 claims description 5
- 229940056501 technetium 99m Drugs 0.000 claims description 5
- 206010000830 Acute leukaemia Diseases 0.000 claims description 4
- 206010005003 Bladder cancer Diseases 0.000 claims description 4
- 206010005949 Bone cancer Diseases 0.000 claims description 4
- 208000018084 Bone neoplasm Diseases 0.000 claims description 4
- 206010006143 Brain stem glioma Diseases 0.000 claims description 4
- 206010006187 Breast cancer Diseases 0.000 claims description 4
- 208000026310 Breast neoplasm Diseases 0.000 claims description 4
- 208000011691 Burkitt lymphomas Diseases 0.000 claims description 4
- 208000017897 Carcinoma of esophagus Diseases 0.000 claims description 4
- 206010007953 Central nervous system lymphoma Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 4
- 208000017604 Hodgkin disease Diseases 0.000 claims description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 4
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 4
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 4
- 206010052178 Lymphocytic lymphoma Diseases 0.000 claims description 4
- 238000012879 PET imaging Methods 0.000 claims description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 4
- 208000000821 Parathyroid Neoplasms Diseases 0.000 claims description 4
- 208000007913 Pituitary Neoplasms Diseases 0.000 claims description 4
- 201000005746 Pituitary adenoma Diseases 0.000 claims description 4
- 206010061538 Pituitary tumour benign Diseases 0.000 claims description 4
- 206010035603 Pleural mesothelioma Diseases 0.000 claims description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 4
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 4
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 4
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 4
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 4
- 206010046458 Urethral neoplasms Diseases 0.000 claims description 4
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 4
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 4
- 201000003761 Vaginal carcinoma Diseases 0.000 claims description 4
- 208000019065 cervical carcinoma Diseases 0.000 claims description 4
- 208000024207 chronic leukemia Diseases 0.000 claims description 4
- 208000029742 colonic neoplasm Diseases 0.000 claims description 4
- 208000030381 cutaneous melanoma Diseases 0.000 claims description 4
- 210000000750 endocrine system Anatomy 0.000 claims description 4
- 201000003914 endometrial carcinoma Diseases 0.000 claims description 4
- 201000001343 fallopian tube carcinoma Diseases 0.000 claims description 4
- 206010017758 gastric cancer Diseases 0.000 claims description 4
- 201000007492 gastroesophageal junction adenocarcinoma Diseases 0.000 claims description 4
- 210000004185 liver Anatomy 0.000 claims description 4
- 201000005202 lung cancer Diseases 0.000 claims description 4
- 208000020816 lung neoplasm Diseases 0.000 claims description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 4
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 201000002528 pancreatic cancer Diseases 0.000 claims description 4
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 4
- 210000002990 parathyroid gland Anatomy 0.000 claims description 4
- 208000021310 pituitary gland adenoma Diseases 0.000 claims description 4
- 208000016800 primary central nervous system lymphoma Diseases 0.000 claims description 4
- 201000000849 skin cancer Diseases 0.000 claims description 4
- 201000003708 skin melanoma Diseases 0.000 claims description 4
- 210000000813 small intestine Anatomy 0.000 claims description 4
- 239000011780 sodium chloride Substances 0.000 claims description 4
- 210000000952 spleen Anatomy 0.000 claims description 4
- 201000011549 stomach cancer Diseases 0.000 claims description 4
- 210000001685 thyroid gland Anatomy 0.000 claims description 4
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 4
- 206010046766 uterine cancer Diseases 0.000 claims description 4
- 208000013013 vulvar carcinoma Diseases 0.000 claims description 4
- 206010060862 Prostate cancer Diseases 0.000 claims description 3
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 3
- 206010038038 rectal cancer Diseases 0.000 claims description 3
- 201000001275 rectum cancer Diseases 0.000 claims description 3
- 208000023915 Ureteral Neoplasms Diseases 0.000 claims description 2
- 206010046431 Urethral cancer Diseases 0.000 claims 1
- 230000001919 adrenal effect Effects 0.000 claims 1
- 210000004907 gland Anatomy 0.000 claims 1
- 210000005084 renal tissue Anatomy 0.000 claims 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 abstract description 3
- 238000002347 injection Methods 0.000 description 42
- 239000007924 injection Substances 0.000 description 42
- 150000001413 amino acids Chemical class 0.000 description 23
- 229940024606 amino acid Drugs 0.000 description 22
- 235000001014 amino acid Nutrition 0.000 description 22
- -1 tere-butyl Chemical group 0.000 description 22
- YZSIZVRFVQKMJU-RDGPPVDQSA-N (2r)-5-[[(2s)-2-amino-3-[[(2s)-3-carboxy-1-[[(1r)-1-carboxy-2-sulfanylethyl]amino]-1-oxopropan-2-yl]amino]-3-oxopropyl]amino]-5-oxo-2-[[4-[(4-oxo-1h-pteridin-6-yl)methylamino]benzoyl]amino]pentanoic acid Chemical compound C1=CC(C(=O)N[C@H](CCC(=O)NC[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CS)C(O)=O)C(O)=O)=CC=C1NCC1=CN=C(NC=NC2=O)C2=N1 YZSIZVRFVQKMJU-RDGPPVDQSA-N 0.000 description 16
- 230000000694 effects Effects 0.000 description 11
- 230000003902 lesion Effects 0.000 description 11
- 239000002253 acid Chemical group 0.000 description 9
- 229940014144 folate Drugs 0.000 description 9
- 125000001072 heteroaryl group Chemical group 0.000 description 9
- WHUUTDBJXJRKMK-GSVOUGTGSA-N D-glutamic acid Chemical compound OC(=O)[C@H](N)CCC(O)=O WHUUTDBJXJRKMK-GSVOUGTGSA-N 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 7
- CKLJMWTZIZZHCS-UWTATZPHSA-N D-aspartic acid Chemical compound OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 7
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 7
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000004475 Arginine Substances 0.000 description 6
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 6
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 6
- 239000004472 Lysine Substances 0.000 description 6
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 6
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 6
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 6
- 235000009697 arginine Nutrition 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 6
- 235000013922 glutamic acid Nutrition 0.000 description 6
- 239000004220 glutamic acid Substances 0.000 description 6
- 235000018977 lysine Nutrition 0.000 description 6
- 229960003104 ornithine Drugs 0.000 description 6
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 5
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 5
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 5
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 235000003704 aspartic acid Nutrition 0.000 description 5
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 5
- 230000027455 binding Effects 0.000 description 5
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 5
- 235000018417 cysteine Nutrition 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 230000002055 immunohistochemical effect Effects 0.000 description 5
- 230000002285 radioactive effect Effects 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- QCHPKSFMDHPSNR-UHFFFAOYSA-N 3-aminoisobutyric acid Chemical compound NCC(C)C(O)=O QCHPKSFMDHPSNR-UHFFFAOYSA-N 0.000 description 4
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 4
- 239000004473 Threonine Substances 0.000 description 4
- 235000009582 asparagine Nutrition 0.000 description 4
- 229960001230 asparagine Drugs 0.000 description 4
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 description 4
- 238000001574 biopsy Methods 0.000 description 4
- 210000000038 chest Anatomy 0.000 description 4
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 4
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 4
- 235000004554 glutamine Nutrition 0.000 description 4
- ZTVZLYBCZNMWCF-UHFFFAOYSA-N homocystine Chemical compound [O-]C(=O)C([NH3+])CCSSCCC([NH3+])C([O-])=O ZTVZLYBCZNMWCF-UHFFFAOYSA-N 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000012217 radiopharmaceutical Substances 0.000 description 4
- 229940121896 radiopharmaceutical Drugs 0.000 description 4
- 230000002799 radiopharmaceutical effect Effects 0.000 description 4
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 4
- 235000004400 serine Nutrition 0.000 description 4
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 4
- YSMODUONRAFBET-UHFFFAOYSA-N 5-hydroxylysine Chemical compound NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 206010061252 Intraocular melanoma Diseases 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 206010030137 Oesophageal adenocarcinoma Diseases 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 201000005969 Uveal melanoma Diseases 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 3
- 230000002411 adverse Effects 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 229940044199 carnosine Drugs 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940109239 creatinine Drugs 0.000 description 3
- 125000004093 cyano group Chemical group *C#N 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 125000001475 halogen functional group Chemical group 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 210000004969 inflammatory cell Anatomy 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 201000002575 ocular melanoma Diseases 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 238000011158 quantitative evaluation Methods 0.000 description 3
- 238000012764 semi-quantitative analysis Methods 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 150000003573 thiols Chemical class 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 2
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 2
- 108010082126 Alanine transaminase Proteins 0.000 description 2
- 108010085443 Anserine Proteins 0.000 description 2
- KDZOASGQNOPSCU-WDSKDSINSA-N Argininosuccinic acid Chemical compound OC(=O)[C@@H](N)CCC\N=C(/N)N[C@H](C(O)=O)CC(O)=O KDZOASGQNOPSCU-WDSKDSINSA-N 0.000 description 2
- 108010003415 Aspartate Aminotransferases Proteins 0.000 description 2
- 102000004625 Aspartate Aminotransferases Human genes 0.000 description 2
- QRYRORQUOLYVBU-VBKZILBWSA-N Carnosic acid Natural products CC([C@@H]1CC2)(C)CCC[C@]1(C(O)=O)C1=C2C=C(C(C)C)C(O)=C1O QRYRORQUOLYVBU-VBKZILBWSA-N 0.000 description 2
- 108010087806 Carnosine Proteins 0.000 description 2
- DCXYFEDJOCDNAF-UWTATZPHSA-N D-Asparagine Chemical compound OC(=O)[C@H](N)CC(N)=O DCXYFEDJOCDNAF-UWTATZPHSA-N 0.000 description 2
- XUJNEKJLAYXESH-UWTATZPHSA-N D-Cysteine Chemical compound SC[C@@H](N)C(O)=O XUJNEKJLAYXESH-UWTATZPHSA-N 0.000 description 2
- AGPKZVBTJJNPAG-RFZPGFLSSA-N D-Isoleucine Chemical compound CC[C@@H](C)[C@@H](N)C(O)=O AGPKZVBTJJNPAG-RFZPGFLSSA-N 0.000 description 2
- ONIBWKKTOPOVIA-SCSAIBSYSA-N D-Proline Chemical compound OC(=O)[C@H]1CCCN1 ONIBWKKTOPOVIA-SCSAIBSYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-UWTATZPHSA-N D-Serine Chemical compound OC[C@@H](N)C(O)=O MTCFGRXMJLQNBG-UWTATZPHSA-N 0.000 description 2
- 229930195711 D-Serine Natural products 0.000 description 2
- QNAYBMKLOCPYGJ-UWTATZPHSA-N D-alanine Chemical compound C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 2
- ODKSFYDXXFIFQN-SCSAIBSYSA-N D-arginine Chemical compound OC(=O)[C@H](N)CCCNC(N)=N ODKSFYDXXFIFQN-SCSAIBSYSA-N 0.000 description 2
- 229930028154 D-arginine Natural products 0.000 description 2
- 229930182846 D-asparagine Natural products 0.000 description 2
- RHGKLRLOHDJJDR-SCSAIBSYSA-N D-citrulline Chemical compound OC(=O)[C@H](N)CCCNC(N)=O RHGKLRLOHDJJDR-SCSAIBSYSA-N 0.000 description 2
- 229930182847 D-glutamic acid Natural products 0.000 description 2
- ZDXPYRJPNDTMRX-GSVOUGTGSA-N D-glutamine Chemical compound OC(=O)[C@H](N)CCC(N)=O ZDXPYRJPNDTMRX-GSVOUGTGSA-N 0.000 description 2
- 229930195715 D-glutamine Natural products 0.000 description 2
- HNDVDQJCIGZPNO-RXMQYKEDSA-N D-histidine Chemical compound OC(=O)[C@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-RXMQYKEDSA-N 0.000 description 2
- 229930195721 D-histidine Natural products 0.000 description 2
- 229930182845 D-isoleucine Natural products 0.000 description 2
- ROHFNLRQFUQHCH-RXMQYKEDSA-N D-leucine Chemical compound CC(C)C[C@@H](N)C(O)=O ROHFNLRQFUQHCH-RXMQYKEDSA-N 0.000 description 2
- 229930182819 D-leucine Natural products 0.000 description 2
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 2
- FFEARJCKVFRZRR-SCSAIBSYSA-N D-methionine Chemical compound CSCC[C@@H](N)C(O)=O FFEARJCKVFRZRR-SCSAIBSYSA-N 0.000 description 2
- 229930182818 D-methionine Natural products 0.000 description 2
- COLNVLDHVKWLRT-MRVPVSSYSA-N D-phenylalanine Chemical compound OC(=O)[C@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-MRVPVSSYSA-N 0.000 description 2
- 229930182832 D-phenylalanine Natural products 0.000 description 2
- 229930182820 D-proline Natural products 0.000 description 2
- AYFVYJQAPQTCCC-STHAYSLISA-N D-threonine Chemical compound C[C@H](O)[C@@H](N)C(O)=O AYFVYJQAPQTCCC-STHAYSLISA-N 0.000 description 2
- 229930182822 D-threonine Natural products 0.000 description 2
- 229930182827 D-tryptophan Natural products 0.000 description 2
- QIVBCDIJIAJPQS-SECBINFHSA-N D-tryptophane Chemical compound C1=CC=C2C(C[C@@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-SECBINFHSA-N 0.000 description 2
- OUYCCCASQSFEME-MRVPVSSYSA-N D-tyrosine Chemical compound OC(=O)[C@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-MRVPVSSYSA-N 0.000 description 2
- 229930195709 D-tyrosine Natural products 0.000 description 2
- KZSNJWFQEVHDMF-SCSAIBSYSA-N D-valine Chemical compound CC(C)[C@@H](N)C(O)=O KZSNJWFQEVHDMF-SCSAIBSYSA-N 0.000 description 2
- 229930182831 D-valine Natural products 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 229930195710 D‐cysteine Natural products 0.000 description 2
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 2
- AGPKZVBTJJNPAG-UHNVWZDZSA-N L-allo-Isoleucine Chemical compound CC[C@@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-UHNVWZDZSA-N 0.000 description 2
- SLRNWACWRVGMKD-UHFFFAOYSA-N L-anserine Natural products CN1C=NC(CC(NC(=O)CCN)C(O)=O)=C1 SLRNWACWRVGMKD-UHFFFAOYSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- CQOVPNPJLQNMDC-UHFFFAOYSA-N N-beta-alanyl-L-histidine Natural products NCCC(=O)NC(C(O)=O)CC1=CN=CN1 CQOVPNPJLQNMDC-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 2
- SUHOOTKUPISOBE-UHFFFAOYSA-N O-phosphoethanolamine Chemical compound NCCOP(O)(O)=O SUHOOTKUPISOBE-UHFFFAOYSA-N 0.000 description 2
- BZQFBWGGLXLEPQ-UHFFFAOYSA-N O-phosphoryl-L-serine Natural products OC(=O)C(N)COP(O)(O)=O BZQFBWGGLXLEPQ-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000210053 Potentilla elegans Species 0.000 description 2
- 108010077895 Sarcosine Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 210000001015 abdomen Anatomy 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- MYYIAHXIVFADCU-QMMMGPOBSA-N anserine Chemical compound CN1C=NC=C1C[C@H](NC(=O)CC[NH3+])C([O-])=O MYYIAHXIVFADCU-QMMMGPOBSA-N 0.000 description 2
- 125000005110 aryl thio group Chemical group 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- 229940000635 beta-alanine Drugs 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- CQOVPNPJLQNMDC-ZETCQYMHSA-N carnosine Chemical compound [NH3+]CCC(=O)N[C@H](C([O-])=O)CC1=CNC=N1 CQOVPNPJLQNMDC-ZETCQYMHSA-N 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229960002173 citrulline Drugs 0.000 description 2
- 235000013477 citrulline Nutrition 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 229950006137 dexfosfoserine Drugs 0.000 description 2
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 2
- 239000012216 imaging agent Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000008176 lyophilized powder Substances 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 230000001394 metastastic effect Effects 0.000 description 2
- 206010061289 metastatic neoplasm Diseases 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 210000004197 pelvis Anatomy 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229940043230 sarcosine Drugs 0.000 description 2
- 229940033078 sodium pertechnetate tc 99m Drugs 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 230000001150 spermicidal effect Effects 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 229960003080 taurine Drugs 0.000 description 2
- IUTCEZPPWBHGIX-UHFFFAOYSA-N tin(2+) Chemical compound [Sn+2] IUTCEZPPWBHGIX-UHFFFAOYSA-N 0.000 description 2
- TZHBAZVSQWVUOB-REOHCLBHSA-N (2r)-3-sulfanyl-2-(sulfoamino)propanoic acid Chemical compound OC(=O)[C@H](CS)NS(O)(=O)=O TZHBAZVSQWVUOB-REOHCLBHSA-N 0.000 description 1
- ILRYLPWNYFXEMH-CRCLSJGQSA-N (2s)-2-azaniumyl-4-[(2s)-2-azaniumyl-2-carboxylatoethyl]sulfanylbutanoate Chemical compound OC(=O)[C@@H](N)CCSC[C@@H](N)C(O)=O ILRYLPWNYFXEMH-CRCLSJGQSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- QWCKQJZIFLGMSD-UHFFFAOYSA-N 2-Aminobutanoic acid Natural products CCC(N)C(O)=O QWCKQJZIFLGMSD-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- BRMWTNUJHUMWMS-UHFFFAOYSA-N 3-Methylhistidine Natural products CN1C=NC(CC(N)C(O)=O)=C1 BRMWTNUJHUMWMS-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical class CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010002388 Angina unstable Diseases 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 0 CCC(C(O)=O)NC(C(C)*NC(C(*)CN[C@](C)(*)*(C)[C@@](CCC(*)NC(c(cc1)ccc1NCC(C=N1)NC2=C1N=C(*)NC2=O)=*)OC)O)=O Chemical compound CCC(C(O)=O)NC(C(C)*NC(C(*)CN[C@](C)(*)*(C)[C@@](CCC(*)NC(c(cc1)ccc1NCC(C=N1)NC2=C1N=C(*)NC2=O)=*)OC)O)=O 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000283153 Cetacea Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241001481833 Coryphaena hippurus Species 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- QWCKQJZIFLGMSD-GSVOUGTGSA-N D-alpha-aminobutyric acid Chemical compound CC[C@@H](N)C(O)=O QWCKQJZIFLGMSD-GSVOUGTGSA-N 0.000 description 1
- ILRYLPWNYFXEMH-UHFFFAOYSA-N D-cystathionine Natural products OC(=O)C(N)CCSCC(N)C(O)=O ILRYLPWNYFXEMH-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 241000283070 Equus zebra Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 241000282816 Giraffa camelopardalis Species 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 241000282575 Gorilla Species 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ILRYLPWNYFXEMH-WHFBIAKZSA-N L-cystathionine Chemical compound [O-]C(=O)[C@@H]([NH3+])CCSC[C@H]([NH3+])C([O-])=O ILRYLPWNYFXEMH-WHFBIAKZSA-N 0.000 description 1
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 241000406668 Loxodonta cyclotis Species 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 206010027457 Metastases to liver Diseases 0.000 description 1
- JDHILDINMRGULE-LURJTMIESA-N N(pros)-methyl-L-histidine Chemical compound CN1C=NC=C1C[C@H](N)C(O)=O JDHILDINMRGULE-LURJTMIESA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 208000025174 PANDAS Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000021155 Paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection Diseases 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 240000000220 Panda oleosa Species 0.000 description 1
- 235000016496 Panda oleosa Nutrition 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- 241000282373 Panthera pardus Species 0.000 description 1
- 241000282376 Panthera tigris Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100029753 Reduced folate transporter Human genes 0.000 description 1
- 101710182657 Reduced folate transporter Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 125000000066 S-methyl group Chemical group [H]C([H])([H])S* 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 238000008050 Total Bilirubin Reagent Methods 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 208000007814 Unstable Angina Diseases 0.000 description 1
- 206010046392 Ureteric cancer Diseases 0.000 description 1
- 206010047741 Vulval cancer Diseases 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical class [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 210000002987 choroid plexus Anatomy 0.000 description 1
- 238000002591 computed tomography Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 229940124558 contraceptive agent Drugs 0.000 description 1
- 239000003433 contraceptive agent Substances 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 229950010286 diolamine Drugs 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000005251 gamma ray Effects 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 150000002431 hydrogen Chemical group 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000002991 immunohistochemical analysis Methods 0.000 description 1
- 238000013388 immunohistochemistry analysis Methods 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000009114 investigational therapy Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 238000009533 lab test Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 235000020772 multivitamin supplement Nutrition 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005487 naphthalate group Chemical group 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 201000011216 nasopharynx carcinoma Diseases 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- IQZPDFORWZTSKT-UHFFFAOYSA-N nitrosulphonic acid Chemical class OS(=O)(=O)[N+]([O-])=O IQZPDFORWZTSKT-UHFFFAOYSA-N 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 238000012633 nuclear imaging Methods 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229950004864 olamine Drugs 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical compound OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Chemical group 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 238000009597 pregnancy test Methods 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000036387 respiratory rate Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical compound [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- 229910052717 sulfur Chemical group 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 238000010809 targeting technique Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- YJBKVPRVZAQTPY-UHFFFAOYSA-J tetrachlorostannane;dihydrate Chemical compound O.O.Cl[Sn](Cl)(Cl)Cl YJBKVPRVZAQTPY-UHFFFAOYSA-J 0.000 description 1
- 125000005247 tetrazinyl group Chemical group N1=NN=NC(=C1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 125000003396 thiol group Chemical class [H]S* 0.000 description 1
- 230000009772 tissue formation Effects 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 238000013413 tumor xenograft mouse model Methods 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 201000011294 ureter cancer Diseases 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 201000004916 vulva carcinoma Diseases 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0474—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group
- A61K51/0478—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group complexes from non-cyclic ligands, e.g. EDTA, MAG3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0497—Organic compounds conjugates with a carrier being an organic compounds
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Un método de obtención de una imagen de un cáncer, comprendiendo el método administrar a un paciente un compuesto sin marcar de acuerdo con la Fórmula I **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona del grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, y en donde n es 0 o 1, y administrar al paciente un compuesto marcado de acuerdo con la Fórmula II **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona del grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, en donde n es 0 o 1 y en donde M es un catión de un radionúclido.
Description
DESCRIPCIÓN
Método de formación de imágenes con un compuesto quelante
REFERENCIA CRUZADA A SOLICITUDES RELACIONADAS
La presente solicitud reivindica la prioridad a tenor de 35 U.S.C. § 119(e) de la Solicitud de Patente Provisional de los EE.UU. N.° de serie 62/205.621, presentada el 14 de agosto de 2015.
CAMPO TÉCNICO
La presente divulgación se refiere al campo de la formación de imágenes, por ejemplo, a un método de formación de imágenes usando un compuesto quelante marcado y un compuesto quelante sin marcar.
ANTECEDENTES
El receptor de folato (RF) es una glucoproteína que se sobreexpresa en muchos tipos de células cancerosas y células inflamatorias, pero se distribuye mínimamente en tejidos normales. Por ejemplo, los receptores de folato se sobreexpresan en varios cánceres epiteliales humanos, incluyendo el cáncer de ovario, pulmón, cerebro (primario y metastásico), endometrio y riñón, y en células inflamatorias del sistema inmunitario, tales como macrófagos y monocitos. Mientras que el ácido fólico entra en la mayoría de las células normales a través del transportador de folato reducido, se sabe que el receptor de folato, a través de endocitosis mediada por receptor, es capaz de internalizar los conjugados de folato, ofreciendo de este modo una vía para dirigirse a las células cancerosas o inflamatorias, por ejemplo.
La tecnología dirigida al folato puede ser clínicamente útil, en que pueden usarse agentes de formación de imágenes unidos a folato para identificar la expresión de receptor de folato (RF) en células cancerosas, por ejemplo. Para los cánceres, la expresión de RF se ha determinado tradicionalmente a través del análisis inmunohistoquímico (IHQ) de muestras de ensayo de tejido archivadas obtenidas en el momento de la resección primaria/caracterización histológica del cáncer. Puesto que la expresión de RF puede cambiar durante el curso de la enfermedad, el análisis IHQ puede realizarse en tejido que no esté temporalmente relacionado con el estado actual de la expresión de RF en los pacientes. Las técnicas de formación de imágenes TEP, RM y TCEMF/TC pueden formar imágenes de tejidos casi en tiempo real sin la invasividad de las biopsias y los agentes de formación de imágenes no invasivos dirigidos al folato son avances importantes en el campo. El documento US2014/0065068 desvela 99mTc-etarfolatida en un método de formación de imágenes de cáncer y enseña la preinyección de ácido fólico libre (sin marcar) con el fin de disminuir la actividad de los órganos de fondo.
SUMARIO DE LA INVENCIÓN
La invención se refiere a un método de formación de imágenes usando un compuesto quelante marcado y un compuesto quelante sin marcar. En una realización, se proporciona un método de formación de imágenes de un cáncer. El método comprende administrar a un paciente un compuesto sin marcar de acuerdo con la Fórmula I

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente y en donde n es 0 o 1, y administrar al paciente un compuesto marcado de acuerdo con la Fórmula II

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, en donde n es 0 o 1 y en donde M es un catión de un radionúclido.
Las siguientes cláusulas enumeradas también describen varias realizaciones:
1. Un método de formación de imágenes de un cáncer, comprendiendo el método administrar a un paciente un compuesto sin marcar de acuerdo con la Fórmula I

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente y en donde n es 0 o 1, y administrar al paciente un compuesto marcado de acuerdo con la Fórmula II

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, en donde n es 0 o 1 y en donde M es un catión de un radionúclido.
2. El método de la cláusula 1, en donde el compuesto sin marcar es de fórmula
o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado es de fórmula

o una sal farmacéuticamente aceptable del mismo, y en donde M es un catión de un radionúclido.
3. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, antes de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
4. El método de cualquiera de las cláusulas anteriores, en donde el cáncer se selecciona entre el grupo que consiste en cáncer de pulmón, cáncer de huesos, cáncer de páncreas, cáncer de piel, cáncer de cabeza, cáncer de cuello, melanoma cutáneo, melanoma intraocular, cáncer uterino, cáncer de ovario, cáncer de endometrio, cáncer rectal, cáncer de estómago, cáncer de colon, cáncer de mama, cáncer de mama triple negativo, carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, cáncer del esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de pulmón no microcítico, cáncer de la glándula suprarrenal, sarcoma de tejido blando, cáncer de la uretra, cáncer de próstata, leucemia crónica, leucemia aguda, linfoma linfocítico, mesotelioma pleural, cáncer de la vejiga, linfoma de Burkitt, cáncer del uréter, cáncer de riñón, carcinoma de células renales, carcinoma de la pelvis renal, neoplasias del sistema nervioso central (SNC), linfoma primario del SNC, tumores del eje espinal, glioma del tronco cerebral, adenoma hipofisario y adenocarcinoma de la unión gastroesofágica.
5. El método de cualquiera de las cláusulas anteriores, en donde el cáncer del que se están formando imágenes es un tumor.
6. El método de cualquiera de las cláusulas anteriores, en donde el cáncer es maligno.
7. El método de cualquiera de las cláusulas anteriores, en donde el cáncer es un cáncer que expresa receptor de folato.
8. El método de cualquiera de las cláusulas anteriores, en donde el cáncer es un cáncer de endometrio. 9. El método de cualquiera de las cláusulas 1-7, en donde el cáncer es un cáncer de pulmón no microcítico.
10. El método de cualquiera de las cláusulas 1-7, en donde el cáncer es un cáncer de ovario.
11. El método de cualquiera de las cláusulas 1-7, en donde el cáncer es un cáncer de mama triple negativo. 12. El método de cualquiera de las cláusulas anteriores, en donde el radionúclido se selecciona entre el grupo que consiste en un isótopo de galio, un isótopo de indio, un isótopo de cobre, un isótopo de tecnecio y un isótopo de renio.
13. El método de cualquiera de las cláusulas 1-12, en donde el radionúclido es un isótopo de galio.
14. El método de cualquiera de las cláusulas 1-12, en donde el radionúclido es un isótopo de indio.
15. El método de cualquiera de las cláusulas 1-12, en donde el radionúclido es un isótopo de cobre.
16. El método de cualquiera de las cláusulas 1-12, en donde el radionúclido es un isótopo de tecnecio.
17. El método de cualquiera de las cláusulas 1-12, en donde el radionúclido es un isótopo de renio.
18. El método de cualquiera de las cláusulas anteriores, en donde se forman imágenes del cáncer de aproximadamente 0,5 horas a aproximadamente 8 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
19. El método de cualquiera de las cláusulas 1-17, en donde se forman imágenes del cáncer de aproximadamente 0,5 horas a aproximadamente 6 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
20. El método de cualquiera de las cláusulas 1-17, en donde se forman imágenes del cáncer de aproximadamente 0,5 horas a aproximadamente 4 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
21. El método de cualquiera de las cláusulas 1-17, en donde se forman imágenes del cáncer de aproximadamente 1 hora a aproximadamente 3 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
22. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 1000 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
23. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 100 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo. 24. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 30 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo. 25. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 10 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo. 26. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra de aproximadamente 0,1 mg a aproximadamente 20 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le administra de aproximadamente 0,01 mg a aproximadamente 2 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
27. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le administra de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
28. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 3000 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
29. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 500 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
30. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 400 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
31. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 100 nmol/kg a aproximadamente 300 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
32. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 100 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal
farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
33. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 15 segundos a aproximadamente 2 minutos.
34. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 30 segundos a 90 segundos.
35. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 15 segundos a aproximadamente 2 minutos.
36. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, de aproximadamente 30 segundos a aproximadamente 5 minutos antes de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
37. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, de aproximadamente 1 minuto a aproximadamente 2 minutos antes de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
38. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra solución salina después de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
39. El método de cualquiera de las cláusulas anteriores, en donde se forman imágenes del cáncer mediante formación de imágenes por TEP.
40. El método de cualquiera de las cláusulas 1-38, en donde se forman imágenes del cáncer mediante formación de imágenes por RM.
41. El método de cualquiera de las cláusulas 1-38, en donde se forman imágenes del cáncer mediante formación de imágenes por TCEMF/TC.
42. El método de cualquiera de las cláusulas anteriores que comprende además la etapa de medir una cantidad de radiactividad del cáncer y una cantidad de radiactividad de un tejido de control.
43. El método de la cláusula 42, en donde el tejido de control se selecciona entre el grupo que consiste en sangre, hígado, pulmón, bazo, intestino, corazón, riñón y músculo.
44. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad de sangre del arco aórtico.
45. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad de músculo.
46. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad de pulmón.
47. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad del riñón.
48. El método de cualquiera de las cláusulas 42-47 que comprende además la etapa de calcular una relación de tumor con respecto a fondo como un cociente de la cantidad de radiactividad del cáncer en comparación con la cantidad de radiactividad del tejido de control.
49. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es superior a aproximadamente 2.
50. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de aproximadamente 2 a aproximadamente 150.
51. El método de cualquiera de las cláusulas anteriores, en donde el compuesto marcado comprende de aproximadamente 20 mCi a aproximadamente 25 mCi de tecnecio-99m.
52. El método de cualquiera de las cláusulas anteriores, en donde el paciente no ha tomado un suplemento de ácido fólico en las 3 semanas siguientes a la administración del compuesto marcado.
53. El método de cualquiera de las cláusulas anteriores, en donde el cáncer del que se forman imágenes se evalúa visualmente.
54. El método de cualquiera de las cláusulas anteriores, en donde se administran dosis múltiples del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
55. El método de cualquiera de las cláusulas anteriores, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente, el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administra a continuación al paciente, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente una segunda vez después de que el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administre al paciente.
56. El método de la cláusula 55, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administran a continuación al paciente del día 4 al día 10.
57. El método de la cláusula 55, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administran a continuación al paciente del día 5 al día 8.
58. El método de cualquiera de las cláusulas 55 a 57, en donde al paciente se le administra de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le administra de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo, cada vez que el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente.
59. El método de cualquiera de las cláusulas 55 a 58, en donde se forman imágenes del paciente después de cada administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
60. El método d
61. El método d
66. El método d
67. El método d
68. El método d
69. El método d
70. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de entre aproximadamente 2 y aproximadamente 10.
71. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de entre aproximadamente 2 y aproximadamente 5.
72. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de entre aproximadamente 2 y aproximadamente 4.
BREVE DESCRIPCION DE LOS DIBUJOS
La FIGURA 1 muestra el efecto de la dosis en masa de etarfolatida sin marcar frente al ácido fólico sobre la captación de 99mTc-etarfolatida basado en las relaciones de tumor con respecto a fondo (T:NT).
La FIGURA 2 muestra el efecto del aumento de la dosis en masa de etarfolatida sin marcar y 99mTc-etarfolatida sobre la captación del tumor M109 de 99mTc-etarfolatida.
DEFINICIONES
Como se usa en el presente documento, el término "alquilo" incluye una cadena de átomos de carbono, que está opcionalmente ramificada y contiene de 1 a 20 átomos de carbono. Se comprenderá además que en determinadas realizaciones, el alquilo puede tener ventajosamente una longitud limitada, incluyendo C1-C12, C1-C10, C1-C9, C1-C8, C1-C7, C1-C6 y C1-C4. De manera ilustrativa, dichos grupos alquilo de longitud particularmente limitada, incluyendo C1-C8, C1-C7, C1-C6 y C1-C4, y similares, pueden denominarse "alquilo inferior". Los grupos alquilo ilustrativos incluyen, pero sin limitación, metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, tere-butilo, pentilo, 2-pentilo, 3-pentilo, neopentilo, hexilo, heptilo, octilo y similares. El alquilo puede estar sustituido o sin sustituir. Los grupos sustituyentes típicos incluyen cicloalquilo, arilo, heteroarilo, heteroalicíclico, hidroxi, alcoxi, ariloxi, mercapto, alquiltio, ariltio, ciano, halo, carbonilo, oxo, (=O), tiocarbonilo, O-carbamilo, N-carbamilo, O-tiocarbamilo, N-tiocarbamilo, C-amido, N-amido, C-carboxi, O-carboxi, nitro y amino, o como se describe en las diversas realizaciones que se proporcionan en el presente documento. Se comprenderá que el "alquilo" puede combinarse con otros grupos, tales como los que se han proporcionado anteriormente, para formar un alquilo funcionalizado. A modo de ejemplo, la combinación de un grupo "alquilo", como se describe en el presente documento, con un grupo "carboxi" puede denominarse grupo "carboxialquilo". Otros ejemplos no limitantes incluyen hidroxialquilo, aminoalquilo, heteroalquilo, arilalquilo, heteroarilalquilo y similares.
Como se usa en el presente documento, el término "arilo" se refiere a un grupo monocíclico todo de carbono o policíclico de anillos condensados de 6 a 12 átomos de carbono que tiene un sistema de electrones pi totalmente conjugado. Se comprenderá que, en determinadas realizaciones, el arilo puede tener ventajosamente un tamaño limitado, tal como arilo C6-C10. Los grupos arilo ilustrativos incluyen, pero sin limitación, fenilo, naftalenilo y antracenilo. El grupo arilo puede estar sin sustituir o sustituido como se describe para el alquilo o como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, el término "heteroarilo" se refiere a un grupo monocíclico o de anillos condensados de 5 a 12 átomos de anillo que contiene uno, dos, tres o cuatro heteroátomos de anillo seleccionados entre nitrógeno, oxígeno y azufre, siendo los átomos restantes del anillo átomos de carbono, y que también tiene un sistema de electrones pi totalmente conjugado. Se comprenderá que, en determinadas realizaciones, El heteroarilo puede tener ventajosamente un tamaño limitado, tal como heteroarilo de 3 a 7 miembros, heteroarilo de 5 a 7 miembros y similares. El heteroarilo puede estar sin sustituir o sustituido como se describe para el alquilo o como se describe en las diversas realizaciones que se proporcionan en el presente documento. Los grupos heteroarilo ilustrativos incluyen, pero sin limitación, pirrolilo, furanilo, tiofenilo, imidazolilo, oxazolilo, tiazolilo, pirazolilo, piridinilo, pirimidinilo, quinolinilo, isoquinolinilo, purinilo, tetrazolilo, triazinilo, pirazinilo, tetrazinilo, quinazolinilo, quinoxalinilo, tienilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, triazolilo, bencimidazolilo, benzoxazolilo, benzotiazolilo, bencisoxazolilo, bencisotiazolilo y carbazoloílo, y similares.
Como se usa en el presente documento, "hidroxi" o "hidroxilo" se refieren a un grupo -OH.
Como se usa en el presente documento, "alcoxi" se refiere a un grupo -O-(alquilo) o a un grupo -O-(cicloalquilo sin sustituir). Los ejemplos representativos incluyen, pero sin limitación, metoxi, etoxi, propoxi, butoxi, ciclopropiloxi, ciclobutiloxi, ciclopentiloxi, ciclohexiloxi y similares.
Como se usa en el presente documento, "ariloxi" se refiere a un grupo -O-arilo o a un grupo -O-heteroarilo. Los ejemplos representativos incluyen, pero sin limitación, fenoxi, piridiniloxi, furaniloxi, tieniloxi, pirimidiniloxi, piraziniloxi y similares, y similares.
Como se usa en el presente documento, "mercapto" o "tiol" se refieren a un grupo -SH.
Como se usa en el presente documento, "alquiltio" se refiere a un grupo -S-(alquilo) o a un grupo -S-(cicloalquilo sin sustituir). Los ejemplos representativos incluyen, pero sin limitación, metiltio, etiltio, propiltio, butiltio, ciclopropiltio, ciclobutiltio, ciclopentiltio, ciclohexiltio y similares.
Como se usa en el presente documento, "ariltio" se refiere a un grupo -S-arilo o a un grupo -S-heteroarilo. Los ejemplos representativos incluyen, pero sin limitación, feniltio, piridiniltio, furaniltio, tieniltio, pirimidiniltio y similares. Como se usa en el presente documento, "halo" o "halógeno" se refieren a flúor, cloro, bromo o yodo.
Como se usa en el presente documento, "ciano" se refiere a un grupo -CN.
Como se usa en el presente documento, "O-carbamilo" se refiere a un grupo -OC(O)NR"R", donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "N-carbamilo" se refiere a un grupo R"OC(O)NR"-, donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "O-tiocarbamilo" se refiere a un grupo -OC(S)NR"R", donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "N-tiocarbamilo" se refiere a un grupo R"OC(S)NR"-, donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "amino" se refiere a un grupo -NR"R", donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "C-amido" se refiere a un grupo -C(O)NR"R", donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "N-amido" se refiere a un grupo R"C(O)NR"-, donde R" es cualquier grupo R como se describe en las diversas realizaciones que se proporcionan en el presente documento.
Como se usa en el presente documento, "enlace" se refiere a un enlace covalente.
Como se usa en el presente documento, "aminoácido" significa cualquier molécula que incluye un átomo de carbono alfa unido covalentemente a un grupo amino y a un grupo ácido. El grupo ácido puede incluir un grupo carboxilo. "Aminoácido" puede incluir moléculas que tengan una de las fórmulas:

en donde Ra es un grupo lateral y O incluye al menos 3 átomos de carbono. El "aminoácido" incluye estereoisómeros tales como las formas de aminoácido D y aminoácido L. Los grupos de aminoácidos ilustrativos incluyen, pero sin limitación, los veinte aminoácidos humanos endógenos y sus derivados, tales como lisina (Lys), asparagina (Asn), treonina (Thr), serina (Ser), isoleucina (Ile), metionina (Met), prolina (Pro), histidina (His), glutamina (Gln), arginina (Arg), glicina (Gly), ácido aspártico (Asp), ácido glutámico (Glu), alanina (Ala), valina (Val), fenilalanina (Phe), leucina (Leu), tirosina (Tyr), cisteína (Cys), triptófano (Trp), fosfoserina (PSER), sulfocisteína, ácido arginosuccínico (ASA), hidroxiprolina, fosfoetanolamina (PEA), sarcosina (SARC), taurina (TAU), carnosina (CARN), citrulina (CIT), anserina (ANS), 1,3-metil-histidina (ME-HIS), ácido alfa-amino-adípico ( a A a ) , beta-alanina (BALA), etanolamina (ETN), ácido gamma-amino-butírico (GABA), ácido beta-amino-isobutírico (BAIA), ácido alfaamino-butírico (BABA), L-alo-cistationina (cistationina-A; CYSTA-A), L-cistationina (cistationina-B; CYSTA-B), cistina, alo-isoleucina (ALO-ILE), DL-hidroxilisina (hidroxilisina (I)), DL-alo-hidroxilisina (hidroxilisina (2)), ornitina (ORN), homocistina (HCY) y derivados de los mismos. Se apreciará que cada uno de estos ejemplos también se contempla en relación con la presente divulgación en la configuración D como se ha indicado anteriormente. Específicamente, por ejemplo, D-lisina (D-Lys), D-asparagina (D-Asn), D-treonina (D-Thr), D-serina (D-Ser), D-isoleucina (D-Ile), D-metionina (D-Met), D-prolina (D-Pro), D-histidina (D-His), D-glutamina (D-Gln), D-arginina (D-Arg), D-glicina (D-Gly), Ácido D-aspártico (D-Asp), Ácido D-glutámico (D-Glu), D-alanina (D-Ala), D-valina (D-Val), D-fenilalanina (D-Phe), D-leucina (D-Leu), D-tirosina (D-Tyr), D-cisteína (D-Cys), D-triptófano (D-Trp), D-citrulina (D-CIT), D-carnosina (D-CARN) y similares. En relación con las realizaciones que se describen en el presente documento, los aminoácidos pueden unirse covalentemente a otras porciones de los compuestos que se describen en el presente documento a través de sus grupos funcionales alfa-amino y carboxi (es decir, en una configuración de enlace peptídico) o a través de sus grupos funcionales de cadena lateral (tales como el grupo carboxi de cadena lateral en el ácido glutámico) y sus grupos funcionales alfa-amino o carboxi. Se comprenderá que los aminoácidos, cuando se usan en relación con los compuestos que se describen en el presente documento, pueden existir como zwitteriones en un compuesto en el que se incorporan.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Como se usa en el presente documento "etarfolatida" es el compuesto de fórmula:

Las siguientes cláusulas enumeradas describen varias realizaciones:
1. Un método de formación de imágenes de un cáncer, comprendiendo el método administrar a un paciente un compuesto sin marcar de acuerdo con la Fórmula I

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente y en donde n es 0 o 1, y administrar al paciente un compuesto marcado de acuerdo con la Fórmula II

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, en donde n es 0 o 1 y en donde M es un catión de un radionúclido.
2. El método de la cláusula 1, en donde el compuesto sin marcar es de fórmula

o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado es de fórmula

o una sal farmacéuticamente aceptable del mismo, y en donde M es un catión de un radionúclido.
3. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, antes de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
4. El método de cualquiera de las cláusulas anteriores, en donde el cáncer se selecciona entre el grupo que consiste en cáncer de pulmón, cáncer de huesos, cáncer de páncreas, cáncer de piel, cáncer de cabeza, cáncer de cuello, melanoma cutáneo, melanoma intraocular, cáncer uterino, cáncer de ovario, cáncer de endometrio, cáncer rectal, cáncer de estómago, cáncer de colon, cáncer de mama, cáncer de mama triple negativo, carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, cáncer del esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de pulmón no microcítico, cáncer de la glándula suprarrenal, sarcoma de tejido blando, cáncer de la uretra, cáncer de próstata, leucemia crónica, leucemia aguda, linfoma linfocítico, mesotelioma pleural, cáncer de la vejiga, linfoma de Burkitt, cáncer del uréter, cáncer de riñón, carcinoma de células renales, carcinoma de la pelvis renal, neoplasias del sistema nervioso central (SNC), linfoma primario del SNC, tumores del eje espinal, glioma del tronco cerebral, adenoma hipofisario y adenocarcinoma de la unión gastroesofágica.
5. El método de cualquiera de las cláusulas anteriores, en donde el cáncer del que se están formando imágenes es un tumor.
6. El método de cualquiera de las cláusulas anteriores, en donde el cáncer es maligno.
7. El método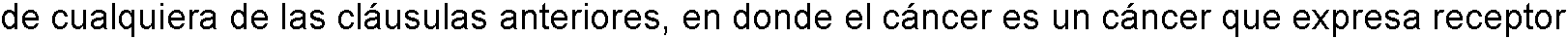
de folato
8. El método
9. El método
10. El método
11. El método
12. El método
que consiste en un
renio.
13. El método
14. El método
15. El método
16. El método
17. El método
18. El método
aproximadamente 0,5 horas a aproximadamente 8 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
19. El método de cualquiera de las cláusulas 1-17, en donde se forman imágenes del cáncer de aproximadamente 0,5 horas a aproximadamente 6 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
20. El método de cualquiera de las cláusulas 1-17, en donde se forman imágenes del cáncer de aproximadamente 0,5 horas a aproximadamente 4 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
21. El método de cualquiera de las cláusulas 1-17, en donde se forman imágenes del cáncer de aproximadamente 1 hora a aproximadamente 3 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
22. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 1000 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
23. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 100 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo. 24. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 30 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo. 25. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra un exceso de aproximadamente 2 a veces a aproximadamente 10 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo. 26. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra de aproximadamente 0,1 mg a aproximadamente 20 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le administra de aproximadamente 0,01 mg a aproximadamente 2 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
27. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le administra de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
28. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 3000 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
29. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 500 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
30. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 400 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
31. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 100 nmol/kg a aproximadamente 300 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
32. El método de cualquiera de las cláusulas 1-21, en donde al paciente se le administra una dosis en masa de aproximadamente 100 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
33. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 15 segundos a aproximadamente 2 minutos.
34. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 30 segundos a 90 segundos.
35. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 15 segundos a aproximadamente 2 minutos.
36. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, de aproximadamente 30 segundos a aproximadamente 5 minutos antes de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
37. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, de aproximadamente 1 minuto a aproximadamente 2 minutos antes de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
38. El método de cualquiera de las cláusulas anteriores, en donde al paciente se le administra solución salina después de que al paciente se le administre el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
39. El método de cualquiera de las cláusulas anteriores, en donde se forman imágenes del cáncer mediante formación de imágenes por TEP.
40. El método de cualquiera de las cláusulas 1-38, en donde se forman imágenes del cáncer mediante formación de imágenes por RM.
41. El método de cualquiera de las cláusulas 1-38, en donde se forman imágenes del cáncer mediante formación de imágenes por TCEMF/TC.
42. El método de cualquiera de las cláusulas anteriores que comprende además la etapa de medir una cantidad de radiactividad del cáncer y una cantidad de radiactividad de un tejido de control.
43. El método de la cláusula 42, en donde el tejido de control se selecciona entre el grupo que consiste en sangre, hígado, pulmón, bazo, intestino, corazón, riñón y músculo.
44. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad de sangre del arco aórtico.
45. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad de músculo.
46. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad de pulmón.
47. El método de la cláusula 42 o la cláusula 43, en donde la cantidad de radiactividad del cáncer se compara con la cantidad de radiactividad del riñón.
48. El método de cualquiera de las cláusulas 42-47 que comprende además la etapa de calcular una relación de tumor con respecto a fondo como un cociente de la cantidad de radiactividad del cáncer en comparación con la cantidad de radiactividad del tejido de control.
49. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es superior a aproximadamente 2.
50. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de aproximadamente 2 a aproximadamente 150.
51. El método de cualquiera de las cláusulas anteriores, en donde el compuesto marcado comprende de aproximadamente 20 mCi a aproximadamente 25 mCi de tecnecio-99m.
52. El método de cualquiera de las cláusulas anteriores, en donde el paciente no ha tomado un suplemento de ácido fólico en las 3 semanas siguientes a la administración del compuesto marcado.
53. El método de cualquiera de las cláusulas anteriores, en donde el cáncer del que se forman imágenes se evalúa visualmente.
54. El método de cualquiera de las cláusulas anteriores, en donde se administran dosis múltiples del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
55. El método de cualquiera de las cláusulas anteriores, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente, el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administra a continuación al paciente, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente una segunda vez después de que el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administre al paciente.
56. El método de la cláusula 55, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administran a continuación al paciente del día 4 al día 10.
57. El método de la cláusula 55, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administran a continuación al paciente del día 5 al día 8.
58. El método de cualquiera de las cláusulas 55 a 57, en donde al paciente se le administra de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le administra de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo, cada vez que el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente.
59. El método de cualquiera de las cláusulas 55 a 58, en donde se forman imágenes del paciente después de cada administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
60. El método d
61. El método
66. El método d
67. El método d
68. El método
69. El método d
70. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de entre aproximadamente 2 y aproximadamente 10.
71. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de entre aproximadamente 2 y aproximadamente 5.
72. El método de la cláusula 48, en donde la relación de tumor con respecto a fondo es de entre aproximadamente 2 y aproximadamente 4.
Como se describe en el presente documento, a un "paciente" se le puede administrar el compuesto sin marcar o el compuesto marcado que se describe en el presente documento, y el paciente puede ser un se humano o, en el caso de las aplicaciones veterinarias, el paciente puede ser un animal de laboratorio, agrícola, doméstico o salvaje. En un aspecto, el paciente puede ser un animal de laboratorio tal como un roedor (por ejemplo, ratón, rata, hámster, etc.), un conejo, un mono, un chimpancé, un animal doméstico tal como un perro, un gato o un conejo, un animal agrícola tal como una vaca, un caballo, un cerdo, una oveja, una cabra, o un animal salvaje en cautiverio tal como un oso, un panda, un león, un tigre, un leopardo, un elefante, una cebra, una jirafa, un gorila, un delfín o una ballena.
En una realización, el compuesto sin marcar puede ser de Fórmula I

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente y en donde n es 0 o 1. En otra realización, el compuesto sin marcar puede ser de fórmula

o una sal farmacéuticamente aceptable del mismo.
En una realización, el compuesto marcado puede ser de Fórmula II

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona entre el grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, en donde n es 0 o 1 y en donde M es un catión de un radionúclido. En otra realización más, el compuesto marcado puede ser de fórmula

o una sal farmacéuticamente aceptable del mismo, y en donde M es un catión de un radionúclido.
En una realización, el radionúclido se selecciona entre el grupo que consiste en un isótopo de galio, un isótopo de indio, un isótopo de cobre, un isótopo de tecnecio y un isótopo de renio. En otras realizaciones, el radionúclido es un isótopo de tecnecio (por ejemplo, 99m-tecnecio).
Como se usa en el presente documento, la expresión "sal farmacéuticamente aceptable" se refiere a las sales cuyos contraiones pueden usarse en productos farmacéuticos. Dichas sales incluyen:
(1) sales de adición de ácido, que puede obtenerse por reacción de la base libre del compuesto parental con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido fosfórico, ácido sulfúrico y ácido perclórico y similares, o con ácidos orgánicos tales como ácido acético, ácido oxálico, ácido (D) o (L) málico, ácido maleico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico y similares; o
(2) sales formadas cuando un protón ácido presente en el compuesto parental o bien se reemplaza por un ion metálico, por ejemplo, un ion de metal alcalino, un ion alcalinotérreo o un ion de aluminio; o se coordina con una base orgánica tal como etanolamina, dietanolamina, trietanolamina, trimetamina, N-metilglucamina y similares. Las sales farmacéuticamente aceptables son bien conocidas por los expertos en la materia y puede contemplarse cualquier sal farmacéuticamente aceptable de este tipo en relación con las realizaciones que se describen en el presente documento.
En diversas realizaciones, se forman sales de adición de ácido adecuadas a partir de ácidos que forman sales atóxicas. Los ejemplos ilustrativos incluyen las sales de acetato, aspartato, benzoato, besilato, bicarbonato/carbonato, bisulfato/sulfato, borato, camsilato, citrato, edisilato, esilato, formiato, fumarato, gluceptato, gluconato, glucuronato, hexafluorofosfato, hibenzato, clorhidrato/cloruro, bromhidrato/bromuro, yodhidrato/yoduro, isetionato, lactato, malato, maleato, malonato, mesilato, metilsulfato, naftilato, 2-napsilato, nicotinato, nitrato, orotato, oxalato, palmitato, pamoato, fosfato/hidrogenofosfato/dihidrogenofosfato, sacarato, estearato, succinato, tartrato, tosilato y trifluoroacetato.
En diversas realizaciones, se forman sales de base adecuadas de los compuestos sin marcar y marcados que se describen en el presente documento a partir de bases que forman sales atóxicas. Los ejemplos ilustrativos incluyen las sales de arginina, benzatina, calcio, colina, dietilamina, diolamina, glicina, lisina, magnesio, meglumina, olamina, potasio, sodio, trometamina y cinc. También pueden formarse hemisales de ácidos y bases, por ejemplo, sales de hemisulfato y hemicalcio.
Como se usa en el presente documento, "opcionalmente" significa que el evento o la circunstancia que se describe posteriormente puede producirse, aunque no necesariamente, y que la descripción incluye casos en los que se produce el evento o la circunstancia y casos en los que no se produce. Por ejemplo, "grupo arilo sustituido opcionalmente con un grupo alquilo" significa que el alquilo puede estar presente, pero no necesariamente, y la descripción incluye situaciones en las que el grupo arilo está sustituido con un grupo alquilo y situaciones en las que el grupo arilo no está sustituido con el grupo alquilo.
La expresión "opcionalmente sustituido" como se usa en el presente documento incluye el reemplazo de átomos de hidrógeno por otros grupos funcionales en el radical que está opcionalmente sustituido. Dichos otros grupos funcionales incluyen de manera ilustrativa, pero sin limitación, amino, hidroxilo, halo, tiol, alquilo, haloalquilo, heteroalquilo, arilo, arilalquilo, arilheteroalquilo, heteroarilo, heteroarilalquilo, heteroarilheteroalquilo, nitro, ácidos sulfónicos y derivados de los mismos, ácidos carboxílicos y derivados de los mismos, y similares. De manera ilustrativa, cualquiera de entre amino, hidroxilo, tiol, alquilo, haloalquilo, heteroalquilo, arilo, arilalquilo, arilheteroalquilo, heteroarilo, heteroarilalquilo, heteroarilheteroalquilo y/o ácido sulfónico está opcionalmente sustituido.
En un aspecto ilustrativo, los compuestos sin marcar o los compuestos marcados que se describen en el presente documento pueden contener uno o más centros quirales, o pueden ser de otro modo capaces de existir como estereoisómeros múltiples. En consecuencia, diversas realizaciones pueden incluir estereoisómeros puros así como mezclas de estereoisómeros, tales como enantiómeros, diastereómeros y mezclas enriquecidas enantioméricamente o diastereoméricamente. En un aspecto, los compuestos sin marcar y los compuestos marcados que se describen en el presente documento pueden ser capaces de existir como isómeros geométricos. En consecuencia, diversas realizaciones pueden incluir isómeros geométricos puros o mezclas de isómeros geométricos.
En algunos aspectos, los compuestos sin marcar o marcados que se describen en el presente documento pueden existir tanto en formas no solvatadas como en formas solvatadas, incluyendo formas hidratadas. En general, las formas solvatadas son equivalentes a las formas no solvatadas y están incluidas dentro del alcance de la presente invención.
En un aspecto ilustrativo, el enlace químico (por ejemplo, "D" o "engarce divalente") en el compuesto sin marcar o marcado que se describe en el presente documento puede ser un enlace directo o el enlace puede ser a través de un engarce intermedio. En una realización, si está presente, un engarce intermedio puede ser cualquier engarce biocompatible conocido en la técnica. En una realización ilustrativa, el engarce divalente comprende de aproximadamente 1 a aproximadamente 30 átomos de carbono. En otra realización ilustrativa, el engarce divalente
comprende de aproximadamente 2 a aproximadamente 20 átomos de carbono. En otras realizaciones, se emplean engarcees divalentes de menor peso molecular (es decir, los que tienen un peso molecular aproximado de aproximadamente 30 a aproximadamente 300).
En una realización, el engarce divalente comprende un heteroátomo unido directamente al folato o al quelante. En una realización, el heteroátomo es nitrógeno. En otra realización, el engarce divalente comprende un diaminoalquileno opcionalmente sustituido. En una realización, el diaminoalquileno opcionalmente sustituido es un diaminoácido. En otra realización, el engarce divalente comprende uno o más restos de diaminoalquileno opcionalmente sustituido y uno o más aminoácidos opcionalmente sustituidos.
En otra realización ilustrativa, el engarce divalente incluye uno o más aminoácidos. En una variación, el engarce divalente incluye un solo aminoácido. En otra variación, el engarce divalente incluye un péptido que tiene de 2 a aproximadamente 50, de 2 a aproximadamente 30 o de 2 a aproximadamente 20 aminoácidos. En otra variación, el engarce divalente incluye un péptido que tiene de aproximadamente 4 a aproximadamente 8 aminoácidos. Dichos aminoácidos se seleccionan ilustrativamente entre los aminoácidos naturales o estereoisómeros de los mismos. En otra realización, el aminoácido también puede ser cualquier otro aminoácido, tal como cualquier aminoácido que tenga la fórmula general:
-N(R1)-(CR2R3)q-C(O)-donde R1 es hidrógeno, alquilo, acilo o un grupo protector de nitrógeno adecuado, R2 y R3 en el aminoácido son hidrógeno o un sustituyente, cada uno de los cuales se selecciona independientemente en cada aparición y q es un número entero tal como 1, 2, 3, 4 o 5. De manera ilustrativa, R y/o R en el aminoácido corresponden independientemente, pero sin limitación, a hidrógeno o las cadenas laterales presentes en aminoácidos de origen natural, tales como metilo, bencilo, hidroximetilo, tiometilo, carboxilo, carboximetilo, guanidinopropilo y similares, y derivados y derivados protegidos de los mismos. La fórmula descrita anteriormente incluye todas las variaciones estereoisoméricas. Por ejemplo, el aminoácido puede ser seleccionarse entre asparagina, ácido aspártico, cisteína, ácido glutámico, lisina, glutamina, arginina, serina, ornitina, treonina y similares. En una variación, el engarce divalente incluye al menos 2 aminoácidos seleccionados entre asparagina, ácido aspártico, cisteína, ácido glutámico, lisina, glutamina, arginina, serina, ornitina y treonina. En otra variación, el engarce divalente incluye entre 2 y aproximadamente 5 aminoácidos seleccionados entre asparagina, ácido aspártico, cisteína, ácido glutámico, lisina, glutamina, arginina, serina, ornitina y treonina. En otra variación, el engarce divalente incluye un tripéptido, tetrapéptido, pentapéptido o hexapéptido que consisten en aminoácidos seleccionados entre ácido aspártico, cisteína, ácido glutámico, lisina, arginina y ornitina, y combinaciones de los mismos.
En otra realización, el engarce divalente también puede incluir uno o más engarcees espaciadores. En la siguiente tabla se muestran engarcees espaciadores ilustrativos. Los siguientes engarcees espaciadores ilustrativos no limitantes se describen donde * indica el punto de unión al folato o al quelante en el compuesto sin marcar o en el compuesto marcado.


En realizaciones ilustrativas, el compuesto marcado puede marcarse con 99mTc. Los métodos típicos conocidos en la técnica para marcar con 99mTc incluyen, pero sin limitación, la reducción de iones pertecnetato en presencia de un precursor quelante para formar el complejo lábil 99mTc-precursor, que, a su vez, reacciona con un grupo de unión metálico. El agente reductor puede ser, por ejemplo, SnCh. El ión estanoso está disponible fácilmente como su dihidrato (tal como dihidrato de cloruro de estaño, SnCl2-2 H2O) o puede generarse in situ a partir de estaño metal (tal
como lámina), gránulos, polvo, limaduras y similares) por contacto con ácido acuoso (tal como HCl). La solución de iones estanosos puede prepararse disolviendo S nC h^^O en HCl acuoso a una concentración preferida para una aplicación particular.
En diversas realizaciones, el compuesto marcado puede prepararse en una farmacia nuclear designada mediante la reconstitución con inyección de Pertecnetato de Sodio Tc-99m, U.S.P., según el Manual de Farmacia Nuclear. En otra realización, el compuesto marcado puede prepararse mediante un método descrito en la Publicación de Solicitud de los EE.UU. N.° 2004/0033195.
En diversas realizaciones, el compuesto marcado comprende de aproximadamente 5 mCi a aproximadamente 100 mCi, de aproximadamente 5 mCi a aproximadamente 80 mCi, de aproximadamente 5 mCi a aproximadamente 50 mCi, de aproximadamente 10 mCi a aproximadamente 30 mCi o de aproximadamente 20 mCi a aproximadamente 25 mCi del radioisótopo.
En diversas realizaciones, el cáncer es un carcinoma, un sarcoma, un linfoma, un melanoma, un mesotelioma, un carcinoma nasofaríngeo, una leucemia, un adenocarcinoma o un mieloma. En otras realizaciones, el cáncer puede ser cáncer de pulmón, cáncer de huesos, cáncer de páncreas, cáncer de piel, cáncer de cabeza, cáncer de cuello, melanoma cutáneo, melanoma intraocular, cáncer uterino, cáncer de ovario, cáncer de endometrio, cáncer rectal, cáncer de estómago, cáncer de colon, cáncer de mama, cáncer de mama triple negativo, carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, cáncer del esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de pulmón no microcítico, cáncer de la glándula suprarrenal, sarcoma de tejido blando, cáncer de la uretra, cáncer de próstata, leucemia crónica, leucemia aguda, un linfoma linfocítico, mesotelioma pleural, cáncer de la vejiga, linfoma de Burkitt, cáncer del uréter, cáncer de riñón, carcinoma de células renales, carcinoma de la pelvis renal, una neoplasia del sistema nervioso central (SNC), linfoma primario del SNC, un tumor del eje espinal, un glioma del tronco encefálico, un adenoma hipofisario o un adenocarcinoma de la unión gastroesofágica.
En algunos aspectos de estas realizaciones, el cáncer es un cáncer que expresa receptor de folato. En algunos aspectos de estas realizaciones, el cáncer es un cáncer de endometrio, un cáncer de pulmón no microcítico, un cáncer de ovario o un cáncer de mama triple negativo. En otra realización, el cáncer del que se están formando imágenes es un tumor. En otra realización, el cáncer es maligno.
En algunos aspectos de estas realizaciones, los métodos que se describen en el presente documento pueden usarse para formar imágenes de un sitio de inflamación en un paciente.
Como se describe en el presente documento, el término "administrar" incluye todos los medios para introducir en el paciente los compuestos sin marcar o marcados que se describen en el presente documento, incluyendo, pero sin limitación, oral (po), intravenoso (iv), intramuscular (im), subcutáneo (sc), transdérmico y similares. Los compuestos sin marcar y marcados que se describen en el presente documento pueden administrarse en formas farmacéuticas unitarias y/o en formulaciones que contengan excipientes, adyuvantes y vehículos farmacéuticamente aceptables convencionales atóxicos.
En un aspecto, un compuesto sin marcar o marcado como se describe en el presente documento puede administrarse directamente en el torrente sanguíneo, en el músculo o en un órgano interno. Las vías adecuadas para dicha administración parenteral incluyen la entrega intravenosa, intraarterial, intraperitoneal, intratecal, epidural, intracerebroventricular, intrauretral, intraesternal, intracraneal, intratumoral, intramuscular y subcutánea. En una realización, los medios para la administración parenteral incluyen inyectores de aguja (incluyendo microagujas), inyectores sin aguja y técnicas de infusión.
En un aspecto ilustrativo, las formulaciones parenterales son normalmente soluciones acuosas que pueden contener vehículos o excipientes tales como sales, hidratos de carbono y agentes tamponantes (preferentemente a un pH de 3 a 9), pero pueden formularse más adecuadamente como una solución no acuosa estéril o como una forma seca para usarse junto con un vehículo adecuado tal como agua estéril apirógena o solución salina estéril. En otras realizaciones, puede adaptarse cualquiera de las formulaciones líquidas que se describen en el presente documento para la administración parenteral de los compuestos sin marcar o marcados como se describe en el presente documento. La preparación en condiciones estériles, mediante liofilización para producir un polvo liofilizado estéril para una formulación parenteral, puede lograrse fácilmente usando técnicas farmacéuticas convencionales bien conocidas por los expertos en la materia. En una realización, la solubilidad de un compuesto sin marcar o marcado utilizado en la preparación de una formulación parenteral puede aumentarse mediante el uso de técnicas de formulación adecuadas, tales como la incorporación de agentes potenciadores de la solubilidad.
En realizaciones ilustrativas, puede administrarse al paciente el compuesto sin marcar o marcado durante un período de tiempo. En diversas realizaciones, la duración del tiempo puede ser de aproximadamente 15 segundos a
aproximadamente 10 minutos, de aproximadamente 15 segundos a aproximadamente 9 minutos, de aproximadamente 15 segundos a aproximadamente 8 minutos, de aproximadamente 15 segundos a aproximadamente 7 minutos, de aproximadamente 15 segundos a aproximadamente 6 minutos, de aproximadamente 15 segundos a aproximadamente 5 minutos, de aproximadamente 15 segundos a aproximadamente 4 minutos, de aproximadamente 15 segundos a aproximadamente 3 minutos, de aproximadamente 15 segundos a aproximadamente 2 minutos, de aproximadamente 15 segundos a aproximadamente 1 minuto, de aproximadamente 15 segundos a aproximadamente 30 segundos o de aproximadamente 30 segundos a 90 segundos. En otras realizaciones, la duración del tiempo puede ser cualquier tiempo entre aproximadamente 15 segundos y aproximadamente 10 minutos, incluyendo, pero sin limitación, aproximadamente 15, 30, 45 o 90 segundos o aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9 o 10 minutos. En un aspecto, el compuesto sin marcar o marcado puede administrarse en una sola dosis o en múltiples dosis.
En diversas realizaciones ilustrativas se administra al paciente un exceso del compuesto sin marcar en comparación con el compuesto marcado. Por ejemplo, el exceso puede ser de aproximadamente 2 veces a aproximadamente 1000 veces, de aproximadamente 2 veces a aproximadamente 500 veces, de aproximadamente 2 veces a aproximadamente 400 veces, de aproximadamente 2 veces a aproximadamente 300 veces, de aproximadamente 2 veces a aproximadamente 200 veces, de aproximadamente 2 veces a aproximadamente 100 veces, de aproximadamente 2 veces a aproximadamente 90 veces, de aproximadamente 2 veces a aproximadamente 80 veces, de aproximadamente 2 veces a aproximadamente 70 veces, de aproximadamente 2 veces a aproximadamente 60 veces, de aproximadamente 2 veces a aproximadamente 50 veces, de aproximadamente 2 veces a aproximadamente 40 veces, de aproximadamente 2 veces a aproximadamente 30 veces, de aproximadamente 2 veces a aproximadamente 20 veces, de aproximadamente 2 veces a aproximadamente 10 veces, de aproximadamente 2 veces a aproximadamente 5 veces, de aproximadamente 2 veces a aproximadamente 4 veces o de aproximadamente 2 veces a aproximadamente 3 veces.
La cantidad del compuesto sin marcar o el compuesto marcado, o el total del compuesto sin marcar y el compuesto marcado juntos, que ha de administrarse puede variar significativamente dependiendo del cáncer del que se están formando imágenes, de la vía de administración del compuesto sin marcar o marcado y de la distribución tisular. La cantidad que ha de administrarse a un paciente puede basarse en el área de superficie corporal, la masa y la evaluación del médico. En diversas realizaciones, las cantidades que han de administrarse pueden variar, por ejemplo, de aproximadamente 0,05 mg a aproximadamente 30 mg, de 0,05 mg a aproximadamente 25,0 mg, de aproximadamente 0,05 mg a aproximadamente 20,0 mg, de aproximadamente 0,05 mg a aproximadamente 15,0 mg, de aproximadamente 0,05 mg a aproximadamente 10,0 mg, de aproximadamente 0,05 mg a aproximadamente 9,0 mg, de aproximadamente 0,05 mg a aproximadamente 8,0 mg, de aproximadamente 0,05 mg a aproximadamente 7,0 mg, de aproximadamente 0,05 mg a aproximadamente 6,0 mg, de aproximadamente 0,05 mg a aproximadamente 5,0 mg, de aproximadamente 0,05 mg a aproximadamente 4,0 mg, de aproximadamente 0,05 mg a aproximadamente 3,0 mg, de aproximadamente 0,05 mg a aproximadamente 2,0 mg, de aproximadamente 0,05 mg a aproximadamente 1,0 mg, de aproximadamente 0,05 mg a aproximadamente 0,5 mg, de aproximadamente 0,05 mg a aproximadamente 0,4 mg, de aproximadamente 0,05 mg a aproximadamente 0,3 mg, de aproximadamente 0,05 mg a aproximadamente 0,2 mg, de aproximadamente 0,05 mg a aproximadamente 0,1 mg, de aproximadamente 0,01 mg a aproximadamente 2 mg, de aproximadamente 0,3 mg a aproximadamente 10 mg, de aproximadamente 0,1 mg a aproximadamente 20 mg o de aproximadamente 0,8 a aproximadamente 3 mg. Un experto en la materia apreciará fácilmente que la dosis puede variar dentro de los diversos intervalos proporcionados anteriormente basándose en los factores indicados anteriormente y puede ser a criterio del médico.
Como se describe en el presente documento, la expresión "dosis en masa" significa la cantidad de compuesto sin marcar y compuesto marcado que se administra al paciente. La dosis en masa puede variar significativamente dependiendo del cáncer del que se están formando imágenes, la vía de administración del compuesto sin marcar y del compuesto marcado y la distribución tisular. En diversas realizaciones, la dosis en masa puede variar, por ejemplo, de aproximadamente 50 nmol/kg a aproximadamente 3000 nmol/kg de peso corporal del paciente, de aproximadamente 50 nmol/kg a aproximadamente 2000 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 1000 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 900 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 800 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 700 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 600 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 500 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 400 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 300 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 200 nmol/kg, de aproximadamente 50 nmol/kg a aproximadamente 100 nmol/kg, de aproximadamente 100 nmol/kg a aproximadamente 300 nmol/kg, de aproximadamente 100 nmol/kg a aproximadamente 500 nmol/kg, de aproximadamente 100 nmol/kg a aproximadamente 1000 nmol/kg, de aproximadamente 100 nmol/kg a aproximadamente 2000 nmol/kg de peso corporal del paciente o cualesquier intervalos de cantidades en el párrafo anterior. En otras realizaciones, la dosis en masa puede ser de aproximadamente 100 nmol/kg, aproximadamente 150 nmol/kg, aproximadamente 200 nmol/kg, aproximadamente 250 nmol/kg, aproximadamente 300 nmol/kg, aproximadamente 350 nmol/kg, aproximadamente 400 nmol/kg, aproximadamente 450 nmol/kg, aproximadamente 500 nmol/kg, aproximadamente 600 nmol/kg,
aproximadamente 700 nmol/kg, aproximadamente 800 nmol/kg, aproximadamente 900 nmol/kg, aproximadamente 1000 nmol/kg, aproximadamente 2000 nmol/kg o aproximadamente 3000 nmol/kg de peso corporal del paciente. En estas realizaciones, "kg" es kilogramos de peso corporal del paciente. En un aspecto, la dosis en masa del compuesto sin marcar y el compuesto marcado puede administrarse en múltiples dosis del compuesto sin marcar y el compuesto marcado.
En una realización ilustrativa, al paciente se le administra el compuesto sin marcar antes de que al paciente se le administre el compuesto marcado. La duración del tiempo que transcurre entre la administración al paciente del compuesto sin marcar y la administración al paciente del compuesto marcado puede variar. Por ejemplo, la duración del tiempo que transcurre entre la administración al paciente del compuesto sin marcar y la administración al paciente del compuesto marcado puede variar de aproximadamente 15 segundos a aproximadamente 10 minutos, de aproximadamente 15 segundos a aproximadamente 9 minutos, de aproximadamente 15 segundos a aproximadamente 8 minutos, de aproximadamente 15 segundos a aproximadamente 7 minutos, de aproximadamente 15 segundos a aproximadamente 6 minutos, de aproximadamente 15 segundos a aproximadamente 5 minutos, de aproximadamente 15 segundos a aproximadamente 4 minutos, de aproximadamente 15 segundos a aproximadamente 3 minutos, de aproximadamente 15 segundos a aproximadamente 2 minutos, de aproximadamente 15 segundos a aproximadamente 1 minuto, de aproximadamente 15 segundos a aproximadamente 30 segundos o de aproximadamente 1 a aproximadamente 2 minutos.
En una realización ilustrativa, al paciente se le administra el compuesto marcado y se forman imágenes después de un período de tiempo. En diversos aspectos, la duración del tiempo que transcurre entre la administración del compuesto marcado y la formación de imágenes del paciente puede variar, por ejemplo, de aproximadamente 30 minutos a aproximadamente 8 horas, de aproximadamente 30 minutos a aproximadamente 7 horas, de aproximadamente 30 minutos a aproximadamente 6 horas, de aproximadamente 30 minutos a aproximadamente 5 horas, de aproximadamente 30 minutos a aproximadamente 4 horas, de aproximadamente 30 minutos a aproximadamente 3 horas, de aproximadamente 30 minutos a aproximadamente 2 horas, de aproximadamente 30 minutos a aproximadamente 1 hora, de aproximadamente 1 hora a aproximadamente 8 horas, de aproximadamente 1 hora a aproximadamente 7 horas, de aproximadamente 1 hora a aproximadamente 6 horas, de aproximadamente 1 hora a aproximadamente 5 horas, de aproximadamente 1 hora a aproximadamente 4 horas, de aproximadamente 1 hora a aproximadamente 3 horas, de aproximadamente 4 horas a aproximadamente 6 horas o de aproximadamente 1 hora a aproximadamente 2 horas. La duración del tiempo puede variar a criterio del médico. En otras realizaciones ilustrativas, se forman imágenes del paciente múltiples veces durante cualquiera de estos períodos de tiempo.
En un aspecto ilustrativo, el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente, el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administra a continuación al paciente, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente una segunda vez después de que el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administre al paciente. En este aspecto, el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, puede administrarse por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, pueden administrarse a continuación al paciente del día 4 al día 10. En otro aspecto, el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, puede administrarse por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, pueden administrarse a continuación al paciente del día 5 al día 8. En estos aspectos ilustrativos, al paciente se le puede administrar de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y al paciente se le puede administrar de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo, cada vez que el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente. En estas realizaciones ilustrativas, pueden formarse imágenes del paciente después de cada administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
En una realización del método que se describe en el presente documento, se forman imágenes del cáncer. En otra realización ilustrativa más, también se forman imágenes de un tejido de control. En una realización ilustrativa, la formación de imágenes se produce mediante formación de imágenes por TEP. En otras realizaciones ilustrativas la formación de imágenes se produce por medio de formación de imágenes por RM o formación de imágenes por TCEMF/TC. Un experto en la materia aprecia que el método de formación de imágenes puede ser cualquier método de formación de imágenes adecuado conocido en la técnica.
En una realización ilustrativa, las imágenes producidas mediante los métodos que se describen en el presente documento pueden cuantificarse. En un aspecto, pueden identificarse y cuantificarse regiones de interés dentro de las imágenes, como se describe en el presente documento, por ejemplo, en la sección "Ejemplos" de la presente solicitud de patente. En una realización ilustrativa, la densidad de píxeles de una imagen se mide en una región de interés. En realizaciones ilustrativas, la densidad de píxeles está relacionada con la cantidad de radiactividad en el
área de la que se están formando imágenes. En diversos aspectos, las imágenes que se describen en el presente documento pueden cuantificarse mediante muchos tipos diferentes de métodos. En una realización, el cáncer del que se forman imágenes se evalúa visualmente.
En realizaciones ilustrativas, el método comprende medir la radiactividad del cáncer y un tejido de control. En una realización, un "tejido de control" puede ser un fluido corporal o un tejido y la cantidad de radiactividad del tejido de control puede compararse con la cantidad de radiactividad del cáncer. En realizaciones ilustrativas, el tejido de control puede ser sangre, tal como sangre del arco aórtico, hígado, pulmón, bazo, intestino, corazón, riñón o músculo.
En realizaciones ilustrativas, el método puede comprender calcular una relación de tumor con respecto a fondo como un cociente de la cantidad de radiactividad del cáncer en comparación con la cantidad de radiactividad del tejido de control. Como se describe en el presente documento, la relación de tumor con respecto a fondo puede calcularse usando las imágenes obtenidas como se describe en el presente documento. En ejemplos ilustrativos, puede calcularse más de una relación de tumor con respecto a fondo.
En realizaciones ilustrativas, la relación de tumor con respecto a fondo puede ser de al menos aproximadamente 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 150, 200, 400, 600 o 800. En realizaciones ilustrativas adicionales, la relación de tumor con respecto a fondo puede ser de aproximadamente 2 a aproximadamente 800, de aproximadamente 2 a aproximadamente 600, de aproximadamente 2 a aproximadamente 400, de aproximadamente 2 a aproximadamente 200, de aproximadamente 2 a aproximadamente 150, de aproximadamente 2 a aproximadamente 100, de aproximadamente 2 a aproximadamente 90, de aproximadamente 2 a aproximadamente 80, de aproximadamente 2 a aproximadamente 70, de aproximadamente 2 a aproximadamente 60, de aproximadamente 2 a aproximadamente 50, de aproximadamente 2 a aproximadamente 40, de aproximadamente 2 a aproximadamente 30, de aproximadamente 2 a aproximadamente 20, de aproximadamente 2 a aproximadamente 10, de aproximadamente 2 a aproximadamente 9, de aproximadamente 2 a aproximadamente 8, de aproximadamente 2 a aproximadamente 7, de aproximadamente 2 a aproximadamente 6, de aproximadamente 2 a aproximadamente 5, de aproximadamente 2 a aproximadamente 4 o de aproximadamente 2 a aproximadamente 3. En una realización ilustrativa, el paciente orina antes de que se obtenga una imagen. En otra realización ilustrativa, el paciente no ha tomado un suplemento de ácido fólico en las 3 semanas siguientes a la administración del compuesto marcado.
En otra realización ilustrativa, pueden usarse escalas relativas para cuantificar la intensidad de captación del compuesto marcado, tal como 99mTc-etarfolatida sobre una base tumor por tumor. En esta realización, la intensidad de captación puede ser, por ejemplo, fuerte si es aproximadamente equivalente a la de un tejido de control (por ejemplo, riñón), moderada si es aproximadamente el 30% de la del tejido de control o débil si aproximadamente el 10% de la del tejido de control. En esta realización, un paciente puede ser considerado como positivo para receptores de folato, lo que indica el tratamiento con fármaco dirigido al folato, si la captación es fuerte o moderada. En otra realización, positividad para receptores de folato puede determinarse, en primer lugar, en ausencia de administración del compuesto sin marcar, y puede demostrar que se indica la formación de imágenes usando los métodos que se describen en el presente documento, si la captación es fuerte o moderada.
Ejemplos
EJEMPLO 1
Administración de 99mTc-etarfolatida y etarfolatida sin marcar
En los modelos de tumores que expresan RF, la etarfolatida muestra una saturación dependiente de la dosis con una constante de disociación alta (Kd = 0,1 nM). Los conjugados de folato son rápidamente captados por tejidos de unión a RF de alta afinidad (por ejemplo, tumores, riñón), así como por tejidos no de unión a RF de baja afinidad (por ejemplo, intestinos, plexo coroideo) con constantes de disociación bajas (Kd~1-10 j M). El aclaramiento de la 99mTcetarfolatida es más lento de los sitios de unión malignos que de los sitios de unión no específicos, como se ha demostrado anteriormente. Dada la semivida relativamente corta de 99mTc (~6 horas), se necesitan medios alternativos para disminuir la captación inespecífica de 99mTc-etarfolatida.
A través de la unión competitiva a RF, se ha demostrado que la captación de fondo de 99mTc-etarfolatida puede bloquearse mediante etarfolatida sin marcar (es decir, etarfolatida "transportadora" o "fría") en exceso. Se ha evaluado el impacto del exceso de ácido fólico o etarfolatida transportadora en la captación tisular de 99mTcetarfolatida en un modelo en ratón de xenoinjerto de tumor KB. Cuando se coinyectó 99mTc-etarfolatida 23 30 nmol/kg con etarfolatida sin marcar en exceso 8,5 molar, las relaciones tumor:fondo (RTF) 4 horas después de la inyección fueron uniformemente más altas con la coinyección de etarfolatida sin marcar que con la coinyección de ácido fólico (Figura 1, Tabla 1). Las líneas punteadas verticales de la Figura 1 representan las estimaciones del valor alométrico de 0,5 mg de ácido fólico (el nivel de dosis utilizado actualmente en desarrollo clínico) o 0,8 mg de etarfolatida, dosis en masa.
Tabla 1: Impacto de etarfolatida o ácido fólico en la relación tumonfondo

EJEMPLO 2
Estudio clínico - Elegibilidad del paciente
Se realizará un estudio clínico para evaluar el impacto de la preinyección de etarfolatida sin marcar en imágenes de TCEMF/TC de 99mTc-etarfolatida en pacientes con cáncer avanzado con malignidades que expresan RF. Pueden ser elegibles pacientes con cáncer avanzado y un historial de >1 lesiones malignas que sean >15°mm en un estudio por TC o RM o >20°mm en una radiografía de tórax a los 2 meses de la entrada en el estudio.
Después de la confirmación de elegibilidad, la expresión de RF se determinará mediante captación por la lesión de 99mTc-etarfolatida en exploraciones por TCEMF/TC de la Sesión de Formación de Imágenes n.°1. Solo los participantes con una captación de 99mTc-etarfolatida en >1 lesiones que miden >15°mm en imágenes de TCEMF/TC del momento basal, volverán para la segunda sesión de formación de imágenes. El estudio inscribirá hasta 18 participantes evaluables. Se consideran participantes evaluables aquellos que completan ambas sesiones de formación de imágenes según el protocolo.
EJEMPLO 3
Variabilidad intraparticipante en la RTF en la formación de imágenes por TCEMF/TC en serie (Cohorte 0)
Se han descrito variaciones intrapaciente con la formación de imágenes por TCEMF y TCEMF/TC con el registro, la normalización en escala de grises (es decir, la alteración del intervalo de intensidad de los píxeles) y la elección de la región de referencia. Como la variabilidad intraparticipante significativa en la RTF podría enturbiar o confundir la capacidad para detectar los cambios en la rTf que podrían atribuirse a la etarfolatida preinyectada, hasta 6 participantes (Cohorte 0) recibirán 99mTc-etarfolatida patrón (0,1 mg de etarfolatida marcada con 20-25 mCi de tecnecio-99m) para ambas sesiones de formación de imágenes para evaluar la variabilidad intraparticipante en la RTF en 3-6 participantes (Cohorte 0). Si la variabilidad intraparticipante en las RTF es inferior al 25 % en 3 participantes, esta cohorte se cerrará. Si la variabilidad intraparticipante en las RTF es >25 % en los primeros 3 participantes, se estudiarán hasta 3 participantes adicionales para la caracterización adicional de la variabilidad. Los participantes pueden proporcionar muestras de ensayo archivadas (por ejemplo, tejido canceroso de biopsia o de aspiración con aguja fina) para la determinación del estado de RF mediante ensayos inmunohistoquímicos (IHQ). EJEMPLO 4
Captación de 99mTc-etarfolatida de fondo en tejidos normales
La captación de 99mTc-etarfolatida se evaluará con respecto a varios patrones de fondo (por ejemplo, actividad de la agrupación de sangre del arco aórtico, pulmón y músculo) para proporcionar a los radiólogos directrices para la identificación de lesiones que expresen RF mediante la captación de 99mTc-etarfolatida.
EJEMPLO 5
Punto temporal formación de imágenes óptima basado en la RTF
Las imágenes tomadas 1 hora después de la inyección frente a 3 horas después de la inyección se compararán en lo que respecta a la retención y el aclaramiento de radionúclidos en tejidos diana y no diana, así como las RTF, para evaluar el efecto del momento de la formación de imágenes después de la inyección.
EJEMPLO 6
Seguridad y tolerabilidad de la inyección secuencial de hasta 4 niveles de dosis de etarfolatida sin marcar y dosis patrón de 99mTc-etarfolatida
La seguridad se evaluará mediante el control de los signos vitales, incluyendo el ritmo cardíaco, la frecuencia respiratoria, la temperatura corporal y la presión sanguínea, antes y después de la inyección de 99mTc-etarfolatida en la primera sesión de formación de imágenes y antes y después de la inyección de etarfolatida sin marcar y marcada en la segunda sesión de formación de imágenes. Se realizarán ensayos de laboratorio clínico basales en el momento de la exploración y la preinyección en la segunda sesión de formación de imágenes. Los participantes se controlarán para determinar eventos adversos durante cuatro días después de cada una de las inyecciones de 99mTc-etarfolatida.
EJEMPLO 7
Diseño del estudio
Se realizará un estudio multicéntrico, sin enmascaramiento y no aleatorizado de múltiples niveles de dosis inyectadas secuencialmente de etarfolatida sin marcar con una dosis patrón de 99mTc-etarfolatida en pacientes con cáncer avanzado. Se inscribirán 18 participantes evaluables. Se consideran participantes evaluables aquellos que completan ambas evaluaciones de sesión de formación de imágenes según el protocolo. Se inscribirán participantes elegibles secuencialmente en cohortes de 3 según el esquema de dosificación comenzando por la Cohorte 0 (véanse las Tablas 2 y 3). Las cohortes se inscribirán de forma escalonada. La cohorte 4 no puede explorarse si no parece haber diferencias sustanciales en las RTF y la calidad de la imagen entre las cohortes 2 y 3.
A la luz del perfil de seguridad bien definido de la etarfolatida, se inscribirán hasta 3 participantes por cohorte simultáneamente. La seguridad se evaluará mediante el historial y la evaluación de laboratorio antes de la inyección de etarfolatida, los signos vitales antes y después de la inyección de etarfolatida y el período de seguimiento de eventos adversos de 4 días después de la última inyección de 99mTc-etarfolatida. Una vez que tres participantes de cada cohorte completen las dos Sesiones de Formación de Imágenes, se completará una revisión de la información de seguridad antes de que comience el tratamiento en la siguiente cohorte.
Tabla 2: Márgenes calculados y dosis propuestas

a DSEAO (todos los demás son DSEO)
b Ensayo de micronúcleos in vivo - etarfolatida no quelada
Tabla 3: Esquema de dosificación de etarfolatida


* indica etarfolatida radiomarcada con 20-25 mCi de Tecnecio-90m
Sesión de Formación de Imágenes n.° 1 - Todos los participantes inscritos de la Cohorte 0-4 se someterán al menos a 1 sesión de formación de imágenes. Durante la Sesión de Formación de Imágenes n.° 1, los participantes recibirán 0,1 mg de etarfolatida radiomarcada con 20-25 mCi de Tecnecio-99m (es decir, la dosis patrón de 99mTcetarfolatida). La formación de imágenes por TCEMF/TC se producirá aproximadamente entre 1 y 3 horas después de la inyección. La ventana para las imágenes de TCEMF/TC de 1 hora es de 0,25 horas; la ventana para las imágenes de TCEMF/TC de 3 horas es de -0,25 horas a 0,50 horas. La expresión de RF se determinará mediante la captación de 90mTc-etarfolatida y el tamaño de la lesión o las lesiones observadas en estas imágenes.
Aproximadamente 4-8 días (pero no menos de 4 días) después de la Sesión de Formación de Imágenes n.° 1, los participantes que tengan una captación de 99mTc-etarfolatida en >1 lesiones que midan >15°mm en las imágenes de TCEMF/TC basales, volverán para la segunda sesión de formación de imágenes.
Sesión de Formación de Imágenes n.° 2 Aproximadamente 4-8 días (pero no menos de 4 días) después de la Sesión de Formación de Imágenes n.°1, los participantes de la Cohorte 0 recibirán de nuevo la dosis patrón de 99mTcetarfolatida y los participantes de las Cohortes 1-4 recibirán, en primer lugar, una inyección de 1 de 4 dosis de etarfolatida sin marcar, seguida de una inyección de dosis patrón de 99mTc-etarfolatida. La formación de imágenes por TCEMF/TC se producirá aproximadamente entre 1 y 3 horas después de las inyecciones. Los participantes de la Cohorte 0 recibirán la misma dosis de 99mTc-etarfolatida para ambas sesiones de formación de imágenes.
El estudio inscribirá hasta 18 participantes evaluables. Son participantes evaluables aquellos que completan ambas sesiones de formación de imágenes según el protocolo. Se inscribirán participantes elegibles secuencialmente en cohortes de 3 según el esquema de dosificación comenzando por la cohorte 0 (véanse la tabla anterior). Las cohortes se inscribirán de forma escalonada. La cohorte 4 no puede explorarse si no parece haber diferencias sustanciales en las RTF y la calidad de la imagen entre las cohortes 2 y 3. A la luz del perfil de seguridad bien definido de la 99mTc-etarfolatida, pueden inscribirse hasta 3 participantes por cohorte simultáneamente. La seguridad se evaluará mediante el historial y la evaluación de laboratorio antes de la inyección de etarfolatida, los signos vitales antes y después de la inyección de etarfolatida y el período de seguimiento de eventos adversos de 4 días después de la inyección de 99mTc-etarfolatida. Una vez que una cohorte de 3 participantes haya completado las dos Sesiones de Formación de Imágenes, se completará una revisión de la información de seguridad antes de que comience la dosificación de la siguiente cohorte.
EJEMPLO 8
Objetivos del estudio
Objetivos primarios:
Identificar la dosis de etarfolatida sin marcar seguida de 99mTc-etarfolatida, que produce la mayor RTF (% de actividad inyectada) en la formación de imágenes por TCEMF/TC.
Objetivos secundarios:
1. Evaluar la variabilidad intraparticipante en RTF en formación de imágenes por TCEMF/TC en serie (Cohorte 0) 2. Explorar la captación de 99mTc-etarfolatida de fondo en diversos tejidos normales.
3. Identificar el punto temporal de formación de imágenes óptima (1 hora después de la inyección frente a 3 horas después de la inyección) basándose en la RTF.
4. Evaluar la seguridad y la tolerabilidad de la inyección secuencial de hasta 4 niveles de dosis de 99mTc-etarfolatida sin marcar y de dosis patrón.
EJEMPLO 9
Criterios de elegibilidad de pacientes
Criterios de inclusión:
1. El participante debe tener 18 años de edad o más.
2. El participante debe proporcionar un consentimiento informado por escrito antes de la inscripción.
3. El participante debe tener un cáncer histológicamente confirmado (biopsia o aspiración con aguja fina) que sea avanzado localmente o metastásico. Nota para los investigadores: Se prefieren cánceres con expresión frecuente de
RF pero no son un requisito para la inclusión en el estudio. Incluyen (pero sin limitación): cáncer de endometrio, cáncer de pulmón no microcítico (CPNM), cáncer de ovario, cáncer de mama triple negativo.
4. El participante debe tener un historial clínico o radiológico de >1 lesión maligna que mida >15°mm o CT o RM, o >20°mm en radiografía de tórax, a las 8 semanas de la entrada en el estudio.
5. El participante debe tener un estado de desempeño de 0 o 1 en el Grupo de Oncología Cooperativa Oriental (ECOG).
6. Los participantes deben tener una función orgánica adecuada:
a) Reserva de médula ósea: Recuento absoluto de neutrófilos (RAN) > 1,5 x 109/l.
Plaquetas > 100 x 109/l. Hemoglobina > 9 g/dl.
b) Hepática: Bilirrubina total < 1,5 x el límite superior de lo normal (LSN).
Alanina aminotransferasa (ALT), aspartato aminotransferasa (AST) <3,0 x LSN O < 5,0 x LSN para participantes con metástasis en el hígado.
c) Renal: Creatinina sérica < 1,5 x LSN o, para participantes con creatinina sérica >1,5 LSN, aclaramiento de creatinina > 50 ml/min.
7. Participante con posibilidad de quedar embarazada:
a) . Todas las mujeres con posibilidad de quedar embarazada deben tener una prueba de embarazo en suero negativa 1 semana antes de cada exposición a etarfolatida o 99mTc-etarfolatida.
b) . Las mujeres con posibilidad de quedar embarazada deben poner en práctica un método anticonceptivo eficaz (por ejemplo, anticonceptivos orales, transdérmicos o inyectables, dispositivo intrauterino [DIU] o anticoncepción de doble barrera, tal como diafragma/preservativo y gel espermicida) durante su participación en el ensayo. c) . Los participantes varones que sean sexualmente activos deben poner en práctica un método anticonceptivo eficaz (por ejemplo, preservativo y gel espermicida). Deben usarse métodos anticonceptivos eficaces durante toda la participación en el estudio.
Criterios de exclusión:
I. El participante está en terapia activa contra el cáncer, distinta de tratamiento hormonal.
2. El participante tiene hipersensibilidad conocida a los componentes del agente de ensayo o sus análogos.
3. La participante está embarazada o amamantando.
4. Enfermedades cardíacas graves o afecciones médicas tales como angina inestable, embolia pulmonar o hipertensión no controlada.
5. El participante participa simultáneamente en otro estudio de investigación de fármacos o dispositivos. El participante debe haber completado la fase de seguimiento de cualquier estudio anterior al menos 30 días antes de la inscripción en este estudio.
6. El participante está tomando actualmente suplementos de ácido fólico y no puede dejar de tomar los suplementos durante un período de aproximadamente 14-21 días (7 días antes de la primera inyección de 99mTc-etarfolatida y un día después del último procedimiento de formación de imágenes).
7. El participante está actualmente en tratamiento con antifolatos tales como metotrexato para la artritis reumatoide en los 28 días siguientes a la primera inyección de 99mTc-etarfolatida.
8. La condición física del participante no es adecuada para la formación de imágenes con radionúclidos.
9. Al participante se le ha administrado otro radiofármaco en los 6 meses anteriores a la inscripción en el estudio que interferiría con la evaluación de las imágenes de 99mTc-etarfolatida.
10. El participante está recibiendo otra quimioterapia, inmunoterapia, radioterapia o terapia de investigación simultánea.
I I . El participante tiene hepatitis B o hepatitis C activas conocidas.
EJEMPLO 10
Procedimientos de estudio
1. Los participantes deben suspender las multivitaminas o los suplementos que contengan ácido fólico al menos 1 semana antes de la primera dosis de 99mTc-etarfolatida.
2. Todos los participantes inscritos se someterán a una primera sesión de formación de imágenes que consistirá en una sesión de formación de imágenes por TCEMF/TC aproximadamente entre 1 y 3 horas después de la inyección de 99mTc-etarfolatida.
3. Los participantes que tengan una captación de 99mTc-etarfolatida en >1 lesiones que midan >15°mm en la primera sesión de formación de imágenes por TCEMF/TC, volverán para la segunda sesión de formación de imágenes que consiste en una sesión de formación de imágenes por TCEMf/TC aproximadamente entre 1 y 3 horas después de las inyecciones de 99mTc-etarfolatida (con o sin una preinyección de etarfolatida sin marcar, dependiendo de la cohorte).
EJEMPLO 11
Procedimientos de TCEMF/TC
1. Instrucciones para el participante
• El participante debe suspender los suplementos de ácido fólico o los suplementos multivitamínicos que contengan ácido fólico, al menos una semana antes de la primera administración de etarfolatida.
• Se debe instruir al participante que beba mucho líquido durante al menos 24 horas después de la administración del radiofármaco.
2. Obtención de imágenes de TCEMF/TC (en ambas sesiones de tiempo de formación de imágenes)
• Obtener imágenes de TCEMF/TC corregistradas aproximadamente entre 1 hora y 3 horas después de la inyección.
• Pedir al participante que orine inmediatamente antes de la obtención de imágenes. Poner al participante en la posición supina para su máximo confort.
• Todos los participantes se someterán a formación de imágenes por TCEMF/TC corregistradas de las regiones de interés. Puede ser el pecho y/o el abdomen y/o la pelvis, dependiendo de la ubicación o ubicaciones de las lesiones tumorales.
• Para una formación de imágenes óptima del cuerpo, los brazos deben elevarse sobre la cabeza si el participante lo tolera.
• Obtener imágenes de acuerdo con la Tabla 4:
Tabla 4: Parámetros de formación de imágenes

3. Transmisión de datos y archivo de imágenes
• Las imágenes de exploración con 99mTc-etarfolatida deben archivarse en medios especificados para, y transmitirse a, el patrocinador o su designado, a intervalos especificados por el patrocinador.
• Las instrucciones detalladas para la transmisión de datos y el archivo de imágenes se detallarán en el Manual de Operaciones del Sitio de formación de imágenes.
Evaluación semicuantitativa de imágenes de TCEMF/TC de 99mTc-etarfolatida
El análisis semicuantitativo se determinará mediante el cálculo de la medición de la radiactividad de una relación tumorfondo (RTF). Se usarán diferentes regiones de fondo (por ejemplo, actividad de la agrupación de sangre del arco aórtico, pulmón o músculo) para determinar qué fondo produce la RTF óptima y reproducible. Las relaciones se expresarán como el cociente de la actividad en las regiones de interés (RDI): recuentos medios por píxel en el tumor y recuentos medios en el fondo.
Métodos estadísticos
1. Evaluación cuantitativa de imágenes de TCEMF/TC
• Los estadísticos descriptivos (media, mediana, desviación típica e intervalo) se calcularán para determinar el %ID en cada región de interés en cada sesión de formación de imágenes.
• El análisis semicuantitativo se determinará mediante el cálculo de la medición de la radiactividad de una relación tumor:fondo (RTF). Las relaciones se expresarán como el cociente de la actividad en las regiones de interés (RDI): recuentos medios por píxel en el tumor y recuentos medios en el fondo.
• Entre los participantes se harán comparaciones de las RTF de las Sesiones de Formación de Imágenes n.° 1 y n.° 2.
• Se harán comparaciones entre las cohortes de las RTF medias.
• También se harán comparaciones entre los puntos temporales de las RTF medias.
2. Seguridad
Los participantes se evaluarán para determinar eventos adversos durante 4 días después de cada inyección de 99mTc-etarfolatida.
Todos los eventos adversos se enumerarán individualmente y se tabularán por la categoría MedDRA.
Estadísticos descriptivos de cada signo vital desde el momento basal hasta después de la inyección.
Estadísticos descriptivos para los resultados de laboratorio clínico desde el momento basal hasta después de la inyección.
EJEMPLO 12
Preparación de etarfolatida
El agente de diagnóstico radiofarmacéutico está compuesto por un ligando dirigido al folato (etarfolatida) que quela Tc-99m. El vial de reactivo no radiactivo (producto farmacológico) se acondiciona como un polvo liofilizado estéril. El principio activo radiactivo se preparará en una farmacia nuclear designada mediante la reconstitución con inyección de Pertecnetato de Sodio Tc-99m, U.S.P., según el Manual de Farmacia Nuclear.
EJEMPLO 13
Administración de etarfolatida
Antes de colocarlo en la mesa de formación de imágenes, se le pedirá al participante que vacíe su vejiga. Durante la Sesión de Formación de Imágenes n.°1: Todos los participantes recibirán una inyección de 99mTc-etarfolatida administrada a través de una sonda permanente IV de flujo libre. Debe administrarse 99mTc-etarfolatida durante un período de tiempo de aproximadamente 30 segundos, seguida de 5-10 ml de solución salina normal. La dosis radiactiva inyectada debería ser de entre 20-25 mCi.
Durante la Sesión de Formación de Imágenes n.° 2: Los participantes de la Cohorte 0 recibirán una inyección de 99mTc-etarfolatida como lo hicieron en la Sesión de Formación de Imágenes n.° 1. Los participantes de las Cohortes 1-4 recibirán dos inyecciones: una inyección de etarfolatida sin marcar seguida de una inyección de etarfolatida radiomarcada, 99mTc-etarfolatida. Para los participantes de las Cohortes 1-4, se administrará etarfolatida sin marcar a la dosis especificada para la cohorte. La etarfolatida sin marcar debe administrarse a través de una sonda permanente IV de flujo libre.
• Para los participantes de la Cohorte 1 y 2, la etarfolatida sin marcar debe administrarse durante un período de tiempo de aproximadamente 30 segundos.
• Para los participantes de la Cohorte 3, la etarfolatida sin marcar debe administrarse durante un período de tiempo de aproximadamente 60 segundos.
• Para los participantes de la Cohorte 4, la etarfolatida sin marcar debe administrarse durante un período de tiempo de aproximadamente 90 segundos.
Aproximadamente 1-2 minutos después de la inyección de etarfolatida sin marcar, se administrará la inyección de
99mTc-etarfolatida (etarfolatida radiomarcada). Debe administrarse 99mTc-etarfolatida durante un período de tiempo de aproximadamente 30 segundos, seguida de 5-10 ml de solución salina normal. La dosis radiactiva inyectada debería ser de entre 20-25 mCi.
Se pedirá a los participantes que proporcionen muestras de ensayo archivadas (por ejemplo, tejido canceroso de biopsia o de aspiración con aguja fina) para la determinación del estado de RF mediante ensayos inmunohistoquímicos (IHQ). La donación de tejido es opcional y no determinará la elegibilidad de los participantes para este estudio.
EJEMPLO 14
Formación de imágenes nuclear
El participante debe suspender los suplementos de ácido fólico por lo menos 1 semana antes de la primera administración de 99mTc-etarfolatida. Se debe instruir al participante que beba mucho líquido durante al menos 24 horas después de la administración del radiofármaco.
La formación de imágenes consistirá en formación de imágenes por TCEMF/TC corregistradas como se detalla a continuación 1 hora y 3 horas después de la inyección de 99mTc-etarfolatida en ambas sesiones de formación de imágenes.
Para la formación de imágenes por TCEMF/TC:
• Todos los participantes se someterán a formación de imágenes por TCEMF/TC corregistradas de las regiones de interés. Puede ser del pecho y/o el abdomen y/o la pelvis, dependiendo de la ubicación o ubicaciones de las lesiones tumorales.
• Para una formación de imágenes óptima por TCEMF/TC corregistradas del cuerpo, los brazos deben elevarse sobre la cabeza, si el participante lo tolera.
• Reconstruir los datos a la máxima resolución de píxeles usando reconstrucción iterativa (se recomienda un mínimo de 6 iteraciones).
• Reconstruir el TCEMF en 3 planos ortogonales: transversal, sagital y coronal.
• Obtener imágenes de acuerdo con la Tabla 4:
Ejemplo 15
Consideraciones estadísticas
Evaluación cuantitativa de imágenes de TCEMF/TC Estadísticos descriptivos (media, mediana, desviación típica e intervalo) se calcularán para determinar el %ID en cada región de interés en cada punto temporal de formación de imágenes. El análisis semicuantitativo se determinará mediante el cálculo de la medición de la radiactividad de una relación tumor:fondo (RTF). Las relaciones se expresarán como el cociente de la actividad en las regiones de interés (RDI): recuentos medios por píxel en el tumor y recuentos medios en el fondo.
Se harán comparaciones entre los participantes de las RTF de la Sesión de Formación de Imágenes n.° 1 y la Sesión de Formación de Imágenes n.°2 TCEMF/TC, así como se harán comparaciones entre las cohortes de las RTF medias.
Ejemplo 16
Formación de imágenes de ratón
Se usaron kits de etarfolatida para la preparación del principio activo radiactivo quelado con 99mTc. Se determinó la pureza radioquímica de la 99mTc-etarfolatida mediante HPLC acoplada a un radiodetector. La 99mTc-etarfolatida se investigó in vivo usando ratones que portaban modelos de xenoinjerto humano positivo para RF. Los artículos de ensayo se administraron por vía intravenosa en ratones a través de la vena lateral de la cola. La biodistribución de 99mTc se determinó extirpando tejidos seleccionados y midiendo su contenido de radiactividad en un contador automático de rayos gamma. Se obtuvieron imágenes tridimensionales de alta resolución del radiomarcador en un sistema dual de microTCEMF/TC.
Después de la administración intravenosa, la 99mTc-etarfolatida se capturó rápidamente y quedó retenida dentro del tumor que expresaba RF y el riñón. Los niveles tisulares máximos se produjeron 1 h después de la inyección. La retención por el tumor se mantuvo cerca del máximo durante aproximadamente 18 horas, mientras que los niveles de 99mTc-etarfolatida en tejidos normales negativos para RF se habían aclarado rápidamente a niveles cercanos a los basales después de 4-8 horas. La formación de imágenes de TCEMF/TC basada en el tiempo corroboró estos hallazgos basados en tejido. Mientras se mantenía constante la dosis radioquímica, se descubrió que el aumento de la dosis total de etarfolatida aumenta sorprendentemente la captación radioquímica en el tumor más de 4 veces,
alcanzando la "cima" con una dosis en el ratón de ~100 nmol/kg. En consecuencia, las relaciones tumor-a-no tumor aumentaron, mejorando de este modo la calidad de la formación de imágenes de tumores. La sustitución de la masa de etarfolatida sin marcar por ácido fólico no mostró estos efectos. De forma importante, las relaciones tumor-a-no tumor disminuyeron a medida que la dosis total de etarfolatida se aumentó a escala adicionalmente hasta el punto de que se observó un bloqueo del tumor competitivo completo a 10.000 nmol/kg.
La biodistribución y la calidad de imagen de TCEMF/TC de la 99mTc-etarfolatida puede mejorarse significativamente i) dejando tiempo suficiente para que se despeje la captación de tejido no afín, y/o ii) administrando una dosis adecuada de etarfolatida. Basándose en estos hallazgos, ambos parámetros experimentales (tiempo de formación de imágenes y dosis de etarfolatida) se están sometiendo a ensayo actualmente a nivel clínico en pacientes con cáncer.
Claims (26)
1. Un método de obtención de una imagen de un cáncer, comprendiendo el método administrar a un paciente un compuesto sin marcar de acuerdo con la Fórmula I

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona del grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, y en donde n es 0 o 1, y administrar al paciente un compuesto marcado de acuerdo con la Fórmula II

o una sal farmacéuticamente aceptable del mismo, en donde R' es hidrógeno o R' se selecciona del grupo que consiste en alquilo, aminoalquilo, carboxialquilo, hidroxialquilo, heteroalquilo, arilo, arilalquilo y heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido, en donde D es un engarce divalente, en donde n es 0 o 1 y en donde M es un catión de un radionúclido.
2. El método de la reivindicación 1, en donde el compuesto sin marcar es de fórmula

o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado es de fórmula

o una sal farmacéuticamente aceptable del mismo, y en donde M es un catión de un radionúclido, en donde se le administra al paciente preferentemente el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, antes de que se le administre al paciente el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
3. El método de cualquiera de las reivindicaciones anteriores, en donde el cáncer se selecciona del grupo que consiste en cáncer de pulmón, cáncer de huesos, cáncer de páncreas, cáncer de piel, cáncer de cabeza, cáncer de cuello, melanoma cutáneo, melanoma intraocular, cáncer uterino, cáncer de ovario, cáncer de endometrio, cáncer rectal, cáncer de estómago, cáncer de colon, cáncer de mama, cáncer de mama triple negativo, carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, cáncer del esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de pulmón no microcítico, cáncer de la glándula suprarrenal, sarcoma de tejido blando, cáncer de la uretra, cáncer de próstata, leucemia crónica, leucemia aguda, linfoma linfocítico, mesotelioma pleural, cáncer de la vejiga, linfoma de Burkitt, cáncer del uréter, cáncer de riñón, carcinoma de células renales, carcinoma de la pelvis renal, neoplasias del sistema nervioso central (SNC), linfoma primario del SNC, tumores del eje espinal, glioma del tronco cerebral, adenoma hipofisario y adenocarcinoma de la unión gastroesofágica.
4. El método de cualquiera de las reivindicaciones anteriores, en donde el cáncer del que se están obteniendo imágenes es un tumor, o, en donde el cáncer es maligno, o, en donde el cáncer es un cáncer que expresa receptores de folato.
5. El método de cualquiera de las reivindicaciones anteriores, en donde el cáncer es un cáncer de endometrio; o cáncer de pulmón no microcítico; o cáncer de ovario; o cáncer de mama triple negativo.
6. El método de cualquiera de las reivindicaciones anteriores, en donde el radionúclido se selecciona del grupo que consiste en un isótopo de galio, un isótopo de indio, un isótopo de cobre, un isótopo de tecnecio, un isótopo de renio, un isótopo de galio, un isótopo de indio, un isótopo de cobre, un isótopo de tecnecio y un isótopo de renio.
7. El método de cualquiera de las reivindicaciones anteriores, en donde se obtienen imágenes del cáncer de aproximadamente 0,5 horas a aproximadamente 8 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 0,5 horas a aproximadamente 6 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 0,5 horas a aproximadamente 4 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 1 hora a aproximadamente 3 horas después de la administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
8. El método de cualquiera de las reivindicaciones 1-7, en donde se le administra al paciente un exceso de aproximadamente 2 veces a aproximadamente 1000 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o un exceso de aproximadamente 2 veces a aproximadamente 100 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o un exceso de aproximadamente 2 veces a aproximadamente 30 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o un exceso de aproximadamente 2 veces a aproximadamente 10 veces del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, con respecto al compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
9. El método de cualquiera de las reivindicaciones 1-7, en donde se le administra al paciente de aproximadamente 0,1 mg a aproximadamente 20 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y se le administra al paciente de aproximadamente 0,01 mg a aproximadamente 2 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y se le administra al paciente de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
10. El método de cualquiera de las reivindicaciones 1-7, en donde se administra al paciente una dosis en masa de aproximadamente 50 nmol/kg a aproximadamente 3000 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 50 nmol/kg a aproximadamente 500 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 50 nmol/kg a aproximadamente 400 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del
mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 100 nmol/kg a aproximadamente 300 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o aproximadamente 100 nmol/kg de peso corporal del paciente del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
11. El método de cualquiera de las reivindicaciones anteriores, en donde se administra al paciente el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 15 segundos a aproximadamente 2 minutos; o de aproximadamente 30 segundos a aproximadamente 90 segundos.
12. El método de cualquiera de las reivindicaciones anteriores, en donde se administra al paciente el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, durante un período de aproximadamente 15 segundos a aproximadamente 2 minutos.
13. El método de cualquiera de las reivindicaciones anteriores, en donde se administra al paciente el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, de aproximadamente 30 segundos a aproximadamente 5 minutos antes de que se le administre al paciente el compuesto marcado, o una sal farmacéuticamente aceptable del mismo; o de aproximadamente 1 minuto a aproximadamente 2 minutos antes de que se le administre al paciente el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
14. El método de cualquiera de las reivindicaciones anteriores, en donde se le administra al paciente solución salina después de que se le administre al paciente el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
15. El método de cualquiera de las reivindicaciones anteriores, en donde se toman imágenes del cáncer mediante formación de imágenes por TEP; o formación de imágenes por RM; o formación de imágenes por TCEMF/TC.
16. El método de cualquiera de las reivindicaciones anteriores que comprende además la etapa de medir una cantidad de radiactividad del cáncer y una cantidad de radiactividad de un tejido de control.
17. El método de la reivindicación 16, en donde el tejido de control se selecciona del grupo que consiste en sangre, hígado, pulmón, bazo, intestino, corazón, riñón y músculo; preferentemente, en donde el tejido de control se selecciona del grupo que consiste en sangre del arco aórtico, tejido muscular, tejido pulmonar y tejido renal.
18. El método de cualquiera de las reivindicaciones 16 o 17 que comprende además la etapa de calcular una relación de tumor con respecto a fondo como un cociente de la cantidad de radiactividad del cáncer en comparación con la cantidad de radiactividad del tejido de control.
19. El método de la reivindicación 18, en donde la relación de tumor con respecto a fondo es superior a aproximadamente 2; o es de aproximadamente 2 a aproximadamente 150.
20. El método de cualquiera de las reivindicaciones anteriores, en donde el compuesto marcado comprende de aproximadamente 20 mCi a aproximadamente 25 mCi de tecnecio-99m.
21. El método de cualquiera de las reivindicaciones anteriores, en donde el paciente no ha tomado un suplemento de ácido fólico en las 3 semanas siguientes a la administración del compuesto marcado.
22. El método de cualquiera de las reivindicaciones anteriores, en donde se administran dosis múltiples del compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
23. El método de cualquiera de las reivindicaciones anteriores, en donde se administra al paciente el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, a continuación se administra al paciente el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, se administra al paciente una segunda vez el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, después de que se administre al paciente el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo.
24. El método de la reivindicación 23, en donde el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra por primera vez al paciente el día 1 y el compuesto sin marcar, o una sal farmacéuticamente aceptable del mismo, y el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administran al paciente a continuación del día 4 al día 10; o del día 5 al día 8.
25. El método de cualquiera de las reivindicaciones 23 o 24, en donde se le administra al paciente de aproximadamente 0,3 mg a aproximadamente 10 mg del compuesto sin marcar, o una sal farmacéuticamente
aceptable del mismo, y se le administra al paciente de aproximadamente 0,05 mg a aproximadamente 0,5 mg del compuesto marcado, o una sal farmacéuticamente aceptable del mismo, cada vez que el compuesto marcado, o una sal farmacéuticamente aceptable del mismo, se administra al paciente.
26. El método de cualquiera de las reivindicaciones 23 a 25, en donde se obtienen imágenes del paciente después de cada administración del compuesto marcado, o una sal farmacéuticamente aceptable del mismo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562205621P | 2015-08-14 | 2015-08-14 | |
| PCT/US2016/046527 WO2017030893A1 (en) | 2015-08-14 | 2016-08-11 | Method of imaging with a chelating compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2811347T3 true ES2811347T3 (es) | 2021-03-11 |
Family
ID=58051942
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16837542T Active ES2811347T3 (es) | 2015-08-14 | 2016-08-11 | Método de formación de imágenes con un compuesto quelante |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10420850B2 (es) |
| EP (1) | EP3334466B1 (es) |
| ES (1) | ES2811347T3 (es) |
| WO (1) | WO2017030893A1 (es) |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2816110A (en) | 1956-11-23 | 1957-12-10 | Merck & Co Inc | Methods for the production of substituted pteridines |
| US5140104A (en) | 1982-03-09 | 1992-08-18 | Cytogen Corporation | Amine derivatives of folic acid analogs |
| US5175343A (en) | 1985-01-14 | 1992-12-29 | Neorx Corporation | Metal radionuclide labeled proteins for diagnosis and therapy |
| US5242679A (en) | 1985-01-14 | 1993-09-07 | Neorx Corporation | Metal radionuclide labeled proteins for diagnosis and therapy |
| NZ217821A (en) | 1985-10-10 | 1989-07-27 | Biotech Australia Pty Ltd | Oral delivery system; complex of active agent and vitamin b12 or analogue thereof |
| GB8608335D0 (en) | 1986-04-04 | 1986-05-08 | Pfizer Ltd | Pharmaceutically acceptable salts |
| EP0273085A1 (en) | 1986-12-29 | 1988-07-06 | IntraCel Corporation | A method for internalizing nucleic acids into eukaryotic cells |
| US5053493A (en) | 1987-04-02 | 1991-10-01 | Centocor Cardiovascular Imaging Partners, L.P. | Method for labeling antibodies with a metal ion |
| US5688488A (en) | 1989-04-03 | 1997-11-18 | Purdue Research Foundation | Composition and method for tumor imaging |
| EP0570493B1 (en) | 1991-02-08 | 2000-01-05 | Diatide, Inc. | TECHNETIUM-99m LABELED POLYPEPTIDES FOR IMAGING |
| US6335434B1 (en) | 1998-06-16 | 2002-01-01 | Isis Pharmaceuticals, Inc., | Nucleosidic and non-nucleosidic folate conjugates |
| US5159079A (en) | 1991-12-20 | 1992-10-27 | Eli Lilly And Company | 2-piperidones as intermediates for 5-deaza-10-oxo- and 5-deaza-10-thio-5,6,7,8-tetrahydrofolic acids |
| US6022966A (en) | 1993-11-22 | 2000-02-08 | Neorx Corporation | Pretargeting methods and compounds |
| JP3538221B2 (ja) | 1993-11-19 | 2004-06-14 | 富士写真フイルム株式会社 | 定着濃厚液およびそれを用いたハロゲン化銀写真感光材料の処理方法 |
| US6083480A (en) | 1997-05-01 | 2000-07-04 | Diatide, Inc. | Calcitonin receptor binding reagents |
| US6093382A (en) | 1998-05-16 | 2000-07-25 | Bracco Research Usa Inc. | Metal complexes derivatized with folate for use in diagnostic and therapeutic applications |
| US6323313B1 (en) | 1999-06-01 | 2001-11-27 | The University Of Washington | Annexin derivative with endogenous chelation sites |
| JP5448284B2 (ja) | 2000-06-02 | 2014-03-19 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | エチレンジシステイン(ec)−薬物結合体 |
| CN1635910A (zh) | 2001-05-02 | 2005-07-06 | 普渡研究基金会 | 巨噬细胞介导的疾病的治疗和诊断 |
| ATE448799T1 (de) | 2002-05-06 | 2009-12-15 | Endocyte Inc | Folatrezeptor gerichtete bildgebende konjugate |
| EP2517730A3 (en) | 2003-01-27 | 2013-01-02 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| WO2004100983A2 (en) | 2003-05-06 | 2004-11-25 | Purdue Research Foundation, Inc. | Treatment of lupus targeting the macrophages or the folate receptor |
| PL2187965T3 (pl) | 2007-08-17 | 2020-05-18 | Purdue Research Foundation | Koniugaty wiążący psma ligand-łącznik i sposoby ich zastosowania |
| US20120322741A1 (en) | 2010-02-25 | 2012-12-20 | Purdue Research Foundation | Psma binding ligand-linker conjugates and methods for using |
-
2016
- 2016-08-11 ES ES16837542T patent/ES2811347T3/es active Active
- 2016-08-11 US US15/751,299 patent/US10420850B2/en active Active
- 2016-08-11 EP EP16837542.6A patent/EP3334466B1/en active Active
- 2016-08-11 WO PCT/US2016/046527 patent/WO2017030893A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| US20190111162A1 (en) | 2019-04-18 |
| US10420850B2 (en) | 2019-09-24 |
| EP3334466B1 (en) | 2020-05-13 |
| EP3334466A4 (en) | 2019-03-20 |
| EP3334466A1 (en) | 2018-06-20 |
| WO2017030893A1 (en) | 2017-02-23 |
| HK1257228A1 (en) | 2019-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4801348B2 (ja) | ビタミンを標的とした造影剤 | |
| ES2972577T3 (es) | Formación de imágenes y terapia dirigidas a la proteína de activación de fibroblastos (FAP) | |
| ES2844586T3 (es) | Inhibidores del antígeno prostático específico de membrana (PSMA) etiquetados con 18F y su uso como agentes de obtención de imágenes para el cáncer de próstata | |
| JP2020165975A (ja) | 陽電子放出断層撮影用の化合物 | |
| ES2736134T3 (es) | Derivado peptídico de tiourea, compuesto marcado con radioisótopos que contiene el mismo y composición farmacéutica que contiene el mismo como ingrediente activo para tratar o diagnosticar el cáncer de próstata | |
| ligan et al. | 99mTc-ethylenedicysteine-folate: a new tumor imaging agent. Synthesis, labeling and evaluation in animals | |
| US12508319B2 (en) | Luteinizing hormone-releasing hormone receptor (LHRH-R) conjugates and uses thereof | |
| US20160303258A1 (en) | Compounds and compositions for imaging gcc-expressing cells | |
| EP4474379A1 (en) | Peptide urea derivative, pharmaceutical composition containing peptide urea derivative, and application of peptide urea derivative | |
| US9365599B2 (en) | N3S1 chelator-folate derivatives, preparation method thereof and composition for diagnosis or treatment of cancer containing the same as an active ingredient | |
| ES2811347T3 (es) | Método de formación de imágenes con un compuesto quelante | |
| CN116472068A (zh) | 治疗性放射性标记的缀合物及其在疗法中的用途 | |
| HK1257228B (en) | Method of imaging with a chelating compound | |
| KR101551232B1 (ko) | N3s1형의 새로운 킬레이터가 접합된 폴레이트 유도체, 이의 제조방법 및 이를 유효성분으로 함유하는 암 진단 또는 치료용 조성물 | |
| JP4801786B6 (ja) | ビタミンを標的とした造影剤 |