ES2777181T3 - 6-cloro-3-(fenil-d5)-inden-1-ona y su uso - Google Patents
6-cloro-3-(fenil-d5)-inden-1-ona y su uso Download PDFInfo
- Publication number
- ES2777181T3 ES2777181T3 ES13811511T ES13811511T ES2777181T3 ES 2777181 T3 ES2777181 T3 ES 2777181T3 ES 13811511 T ES13811511 T ES 13811511T ES 13811511 T ES13811511 T ES 13811511T ES 2777181 T3 ES2777181 T3 ES 2777181T3
- Authority
- ES
- Spain
- Prior art keywords
- phenyl
- chloro
- indan
- compound
- dichloro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Abstract
Un procedimiento para la producción del compuesto (I) en donde el compuesto (I) **(Ver fórmula)** se obtiene a través de la reacción de 3-bromo-6-cloro-inden-1-ona con 4,4,5,5-tetrametil-2-d5-fenil-[1,3,2]dioxaborolano.
Description
DESCRIPCIÓN
6-cloro-3-(fenil-d5)-inden-1 -ona y su uso
Campo de la invención
La presente invención se refiere a un procedimiento para la preparación de 6-cloro-3-(fenil-d5)-inden-1-ona, así como el uso del compuesto obtenido.
Antecedentes de la invención
Se han descrito 1-piperazin-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia en la solicitud estadounidense n.° 13/527.364. La solicitud estadounidense n.° 13/527.364 también describe cómo pueden obtenerse 1 -piperazin-3-fenil-indanos deuterados específicos a través de 6-cloro-3-(fenil-d5)-indan-1 -ona. Sin embargo, las rutas descritas para la síntesis de 6-cloro-3-(fenil-d5)-indan-1 -ona tanto racémica como enantioméricamente puro se basan en el uso antieconómico de una alta carga de o bien un catalizador de rodio quiral o bien un catalizador de paladio quiral. En consecuencia, son deseables nuevos procedimientos para la síntesis de 6-cloro-3-(fenil-d5)-indan-1-ona racémica y enantioméricamente puro, y en el presente documento se describen procedimientos a través del compuesto 6-cloro-3-(fenil-d5)-inden-1 -ona (I).
Clark, W. M. et al en Organic Letter, 1999, vol. 1, n.° 11, págs. 1839-1842 intentaron preparar 3-arilindenonas, tales como 6-cloro-3-(fenil-d5)-indan-1-ona (I), con grupos aceptores de electrones (Cl, Br, NO2) en la posición C(5) o C(6) del anillo de indenona utilizando una metodología Suzuki, pero no pudieron obtener cantidades apreciables de los productos deseados. Por el contrario, la presente invención describe la preparación exitosa de tales 3-arilindenonas, por ejemplo, 6-cloro-3-(fenil-d5)-indan-1 -ona (I), a través de una metodología Suzuki.
Compendio de la invención
La presente invención describe un procedimiento para la preparación del compuesto 6-cloro-3-(fenil-d5)-inden-1-ona (I).

En un aspecto adicional, la presente invención describe el uso de 6-cloro-3-(fenil-d5)-inden-1-ona (I) para obtener 6 cloro-3-(fenil-d5)-indan-1-ona (VIII) racémica o (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX).
Además, la presente invención describe el uso de 6-cloro-3-(fenil-d5)-inden-1 -ona (I) para obtener 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1 -il)-1 (d3),2,2-trimetilpiperazina y sus sales farmacéuticamente aceptables.
Descripción detallada de la invención
Lista de compuestos
(I): 6-cloro-3-(fenil-d5)-inden-1-ona
(II) : 6-cloro-1-indanona
(III) : 3-bromo-6-cloro-inden-1-ona
(IV) : 6-cloro-3-(fenil-d5)-1 H-indeno
(V) : 5-cloro-1-indanona
(VI): (±)-6-cloro-3-(fenil-d5)-1H-inden-1-ol
(VIa) 6-cloro-3-(fenil-d5)-1 H-inden-1 -ol
(VII) : (S)-6-cloro-3-(fenil-d5)-1 H-inden-1-ol
(VIII) : (±)-6-cloro-3-(fenil-d5)-indan-1-ona
(VIIIa): 6-cloro-3-(fenil-d5)-indan-1-ona
(IX): (S)-6-cloro-3-(fenil-d5)-indan-1-ona
(X): (±)-c/'s-6-cloro-3-(fenil-d5)-indan-1 -ol
(Xa): (1 S,3S)-6-cloro-3-(fenil-d5)-indan-1 -ol
(Xb): 6-cloro-3-(fenil-d5)-indan-1 -ol
(XI) : (±)-c/'s-3,5-dicloro-1-(fenil-d5)-indano
(Xla): (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano
(XIb): 3,5-dicloro-1-(fenil-d5)-indano
(XII) : maleato de (±)-frans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina
(XIIa): maleato de 1 -((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1 -il)-3,3-dimetil-piperazina
(Xllb): sal farmacéuticamente aceptable de 1-(6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina
(XIII) : succinato de (±)-frans-4-(6-cloro-3-(fenil-d5)-indan-1-il)-1 (d3),2,2-trimetil-piperazina
(XIIIa): 4-(6-cloro-3-(fenil-d5)-indan-1-il)-1 (d3),2,2-trimetil-piperazina
(XIV) : L-(+)-tartrato de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1 (d3),2,2-trimetil-piperazina
(XV) : fumarato de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1 -il)-1 (d3),2,2-trimetil-piperazina
(XVa) sal farmacéuticamente aceptable de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina (XVI) : bis-2,2,2-trifluoroacetato de 1(d3),2,2-trimetilpiperazina
(XVII): 2,2-dimetilpiperazina
(XVIII) : hemi-D,L-tartrato de 3,3-dimetilpiperazin-1-carboxilato de ferc-butilo
(XIX) : (E)-1 -(6-cloro-3-fenil(d5)-1 H-inden-1 -ilidenmetil)-A/,W-dimetilamina
La presente invención proporciona procedimientos para la preparación del compuesto (I)

así como el uso del compuesto para la preparación de 4-((1R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetilpiperazina y sales farmacéuticamente aceptables de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetilpiperazina.
La presente invención describe cómo puede lograrse la síntesis del compuesto (I) mediante una primera etapa que comprende la síntesis del compuesto (III), 3-bromo-6-cloro-inden-1-ona, seguido de una segunda etapa donde (III) se hace reaccionar con 4,4,5,5-tetrametil-2-d5-fenil-[1,3,2]dioxaborolano, en presencia de un catalizador, por ejemplo acetato de paladio (II) y base, por ejemplo fosfato de potasio, apropiados.
La presente invención describe además cómo puede lograrse la síntesis del compuesto (I) a partir del compuesto (IV), 6-cloro-3-(fenil-d5)-1 H-indeno, a través de la oxidación de una enamina (XIX), (£)-1-(6-cloro-3-fenil(d5)-1H-inden-1-ilidenmetil)-W,W-dimetilamina, por ejemplo en presencia de una sal de peryodato.
En un aspecto adicional, la presente invención describe el uso del compuesto (I) para la preparación del compuesto (VIII) o (IX) mediante cualquiera de las siguientes rutas.
(1) Reducción seguida de reordenamiento para obtener el compuesto (VIII) (rutas B y D2 en el esquema 1, a continuación)
(2) Reducción enantioselectiva seguida de reordenamiento para obtener el compuesto (IX) (rutas C y E1 en el esquema 1, a continuación)
(3) Hidrogenación para obtener el compuesto (VIII) (ruta D1 en el esquema 1, a continuación)
(4) Hidrogenación por transferencia asimétrica organocatalítica para obtener el compuesto (IX) (ruta E2 en el esquema 1, a continuación)
(5)  Hidrogenación asimétrica para obtener el compuesto (IX) (ruta E3 en el esquema 1, a continuación). Estas rutas sintéticas de la invención pueden resumirse tal como sigue:
Hidrogenación asimétrica para obtener el compuesto (IX) (ruta E3 en el esquema 1, a continuación). Estas rutas sintéticas de la invención pueden resumirse tal como sigue:

Esquema 1: Preparación de los compuestos (I), (VI/VIa), (VII), (VIII)/(VIIIa) y (IX)
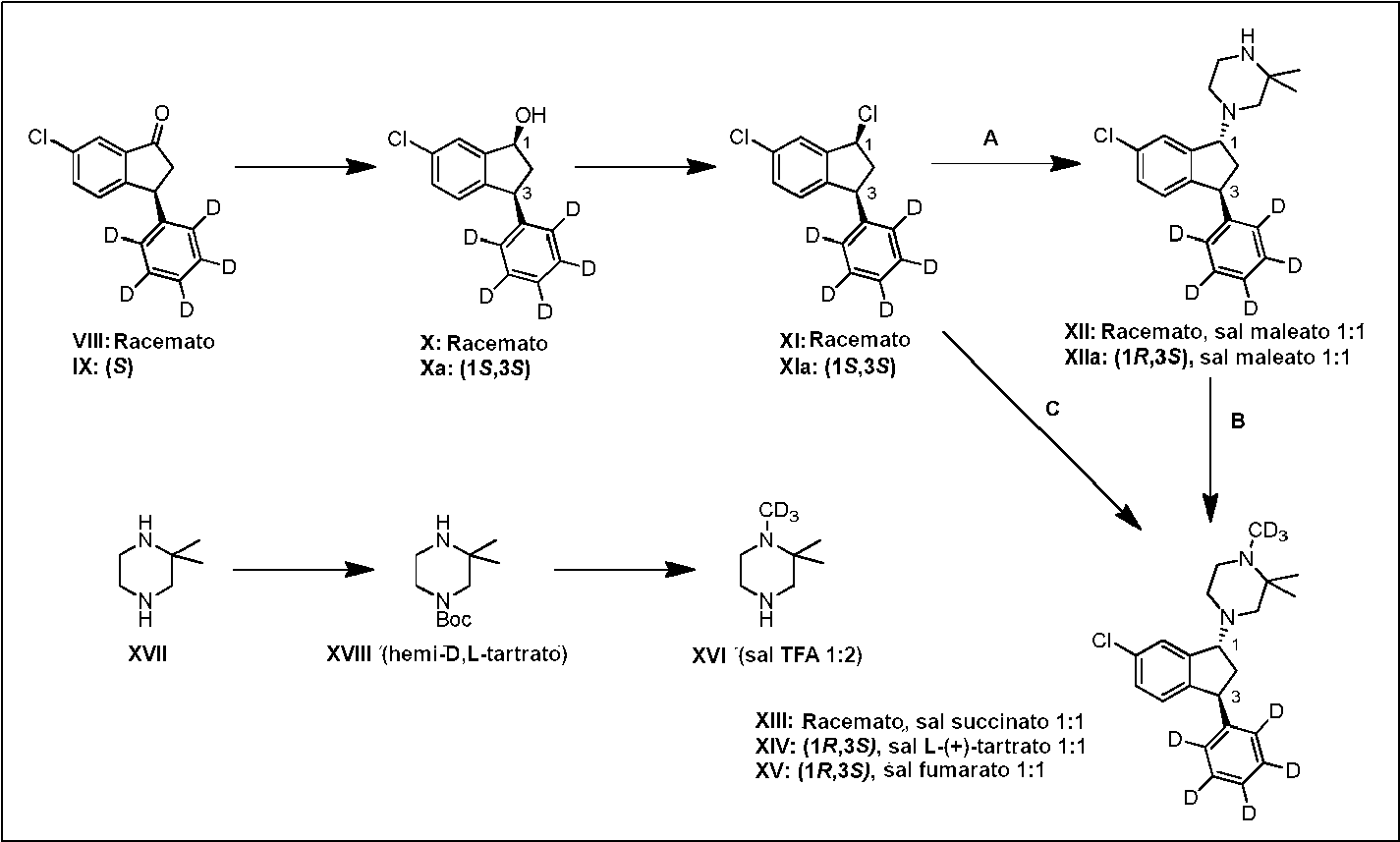
En un aspecto adicional, la presente invención describe el uso del compuesto (VIII) obtenido tal como se describió anteriormente para la preparación del compuesto (XIV) a través de los compuestos (X), (XI), (XII) y (XIII) (ruta A y B en el esquema 2).
En un aspecto adicional, la presente invención describe el uso del compuesto (IX) obtenido tal como se describió anteriormente para la preparación del compuesto (XV) o el compuesto (XIV) a través de los compuestos (Xa) y (Xla) (ruta C en el esquema 2).
En un aspecto adicional, la presente invención describe el uso del compuesto (IX) obtenido tal como se describió anteriormente para la preparación del compuesto (XV) o el compuesto (XIV) a través de los compuestos (Xa), (Xla) y (Xlla) (ruta A y B en el esquema 2).
En otro aspecto, la presente invención describe la preparación del compuesto (XVI) a partir del compuesto (XVII) a través del compuesto (XVIII) (Esquema 2).
Las rutas sintéticas de la invención pueden resumirse tal como sigue:
La invención se ilustrará en los siguientes ejemplos no limitativos.
Realizaciones según la invención
A menos que se especifique de otro modo, la referencia a cualquiera de los compuestos en las realizaciones a continuación cubre el compuesto enantioméricamente puro o mezclas de los enantiómeros en cualquier razón. Por ejemplo, el compuesto (VIIIa) 6-cloro-3-(fenil-d5)-indan-1-ona se refiere a la mezcla racémica de (VIIIa), es decir (±)-6-cloro-3-(fenil-d5)-indan-1-ona, así como los enantiómeros de (VIIIa) en cualquier razón.
En una primera realización (E1), la presente invención se refiere a un procedimiento para la producción del compuesto que tiene la estructura (I) (también denominado compuesto de fórmula (I) o compuesto (I))

obtenido a partir de la reacción de 3-bromo-6-cloro-inden-1-ona (III) con 4,4,5,5-tetrametil-2-afe-fenil-[1,3,2] dioxaborolano.
En una realización adicional (E2) de (E1), el compuesto (III) se obtiene a partir de (II) como compuesto de partida. En una realización adicional (E3) de (E2), la síntesis del compuesto (I) comprende las siguientes etapas:
1. Bromación del compuesto (II), por ejemplo, mediante la adición de 2,2'-azo-bis-isobutironitrilo y N-bromosuccinimida a una disolución que comprende 6-cloro-1-indanona (II).
2. Eliminación inducida por base mediante la adición de una base, por ejemplo, trietilamina, a la disolución de la etapa 1 para obtener 3-bromo-6-cloro-inden-1-ona (III).
3. Se separa opcionalmente 3-bromo-6-cloro-inden-1-ona (III) obtenida en la etapa 2 y se hace reaccionar con 4,4,5,5-tetrametil-2-afe-fenil-[1,3,2]dioxaborolano, en presencia de un catalizador y una base apropiados para obtener el compuesto (I).
En una realización (E4) de (E1), la síntesis del compuesto (I) comprende las siguientes etapas:
1. Síntesis de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) mediante reacción entre una especie organometálica (obtenida a partir de benceno-d5 monohalogenado) y 5-cloro-1-indanona (V) seguido de deshidratación. 2. Reacción de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) a compuesto (XIX) y escisión oxidativa adicional del mismo para obtener el compuesto (I).
En una realización (E5) de (E4), el compuesto (I) se obtiene mediante un procedimiento que comprende:
1. Síntesis de 6-cloro-3-(fenil-d5)-1 H-indeno (IV) mediante reacción de Grignard entre bromobenceno-d5, magnesio y 5-cloro-1-indanona (V) seguido de deshidratación.
2. Hacer reaccionar 6-cloro-3-(fenil-d5)-1 H-indeno (IV) con 1,1-dimetoxi-H,H-dimetilmetanamina seguido de escisión oxidativa del compuesto (XIX) formado para obtener el compuesto (I).
En una realización adicional (E6) de (E4) y (E5), la escisión oxidativa en la síntesis del compuesto (I) se lleva a cabo mediante el uso de un agente oxidante seleccionado del grupo que consiste en metaperyodato de sodio, metaperyodato de potasio, ozono, dicromato de potasio, dicromato de sodio, oxígeno singlete y ácido mcloroperbenzoico.
En una realización particular (E7) de (E6), la escisión oxidativa se lleva a cabo mediante el uso de metaperyodato de sodio.
En una realización (E8), el compuesto (I) obtenido en (E1) se reduce para obtener (VIa), en particular (±)-6-cloro-3-(fenil-d5)-1H-inden-1-ol (VI).
En una realización adicional (E9) de (E8) la reducción tiene lugar en presencia de reductores seleccionados del grupo que consiste en borohidruro de sodio, borohidruro de magnesio, borohidruro de calcio, borohidruro de litio, triacetoxiborohidruro de sodio, triacetoxiborohidruro de litio, hidruro de litio y aluminio, dihidruro de sodio y bis(2-metoxietoxi)aluminio, hidruro de diisobutilaluminio y trietilborohidruro de litio.
En una realización particular (E10) de (E9) la reducción tiene lugar en presencia de hidruro de diisobutilaluminio. En una realización (E11), el compuesto (VIa) obtenido en (E8) se convierte en (VIIIa), en particular (±)-6-cloro-3-(fenild5)-indan-1-ona (VIII), a través de reordenamiento inducido por base.
En una realización (E12), el compuesto (I) obtenido en (E1) se convierte en (S)-6-cloro-3-(fenil-d5)-1H-inden-1-ol (VII) a través de reducción enantioselectiva.
En una realización particular (E13) de (E12) la reducción enantioselectiva tiene lugar en presencia de catalizadores y reductores enantioselectivos seleccionados del grupo que consiste en 2-metil-CBS-oxazaborolidina enantioméricamente pura, o-tolil-CBS-oxazaborolidina, 2-butil-CBS-oxazaborolidina, Alpine-Borane® y B-chlorodiisopinocanfeilborano.
En una realización particular (E14) de (E13), la reducción enantioselectiva tiene lugar en presencia de 2-metil-CBS-oxazaborolidina enantioméricamente pura.
En una realización (E15) de (E12) a (E14), el compuesto (VII) se convierte en (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) a través de reordenamiento inducido por base.
En una realización adicional (E18) de cualquiera de (E11) y (E15), el reordenamiento inducido por base tiene lugar en presencia de una base adecuada seleccionada del grupo que consiste en 1,4-diazabiciclo[2.2.2]octano, bis(trimetilsilil) amida de potasio y bis(trimetilsilil)amida de litio.
En una realización particular (E17) de (E16), el reordenamiento inducido por base tiene lugar en presencia de 1,4-diazabiciclo [2.2.2]octano.
En una realización (E18), el compuesto (I) obtenido en (E1) se convierte para obtener (VIIIa), en particular (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII) a través de hidrogenación en presencia de un catalizador adecuado en un disolvente adecuado.
En una realización específica (E19) de (E18), el compuesto (I) se convierte en el compuesto (VIII) en presencia de cloruro de tris(trifenilfosfina)rodio (I).
En una realización específica (E20) de (E18), el disolvente es acetato de etilo.
En una realización (E21), el compuesto (I) obtenido en (E1) se convierte en (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) a través de hidrogenación asimétrica en presencia de un catalizador adecuado en un disolvente adecuado.
En una realización adicional (E22) de (E21), la hidrogenación asimétrica del compuesto (I) se lleva a cabo en presencia de una sal de rodio.
En una realización adicional (E23) de cualquiera de (E21) y (E22), la hidrogenación asimétrica de (I) se lleva a cabo en presencia de un ligando de fosfina quiral.
En una realización específica (E24) de (E22), la sal de rodio seleccionada es del grupo que consiste en trifluorometanosulfonato de bis(norbornadien)rodio (I), tetrafluoroborato de bis(norbornadien)rodio (I),
trifluorometanosulfonato de bis(1,5-ciclooctadien)rodio (I), tetrafluoroborato de bis(norbornadien)rodio (I) y tetrakis [(bis(3,5-trifluorometil)fenil]borato de bis(1,5-ciclooctadien)rodio (I).
En una realización específica (E25) de (E23), el ligando de fosfina quiral seleccionado del grupo que consiste en (R)-(-)-5,5'-bis[di(3,5-di-ferc-butil-4-metoxifenil)fosfin]-4,4'-bi-1,3-benzodioxol ((R)-DTBM-SEGp Ho S), (S)-(+)-4,12-bis (difenilfosfin)-[2.2]-paraciclofano ((S)-Phanephos) y (s)-(+)-4,12-bis[di(3,5-xilil)fosfin]-[2.2]-paraciclofano ((S)-DM-Phanephos).
En una realización específica (E26) de (E21) el disolvente es acetato de etilo.
En una realización (E27), el compuesto (VIII) de cualquiera de las realizaciones (E11) y (E18) se convierte en (Xb), en particular (±)-c/s-6-cloro-3-(fenil-d5)-indan-1-ol (X).
En una realización (E28) de cualquiera de las realizaciones (E15) a (E17) y (E21) a (E26), (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) se convierte en (1 S,3S)-c/s-6-cloro-3-(fenil-d5)-indan-1-ol (Xa).
En una realización (E29) de (E27), (Xb) se convierte en (XIb) mediante cloración, en particular (±)-c/s-6-cloro-3-(fenild5)-indan-1-ol (X) se convierte en (±)-c/s-3,5-dicloro-1-(fenil-d5)-indano (XI) mediante cloración.
En una realización (E30) de (E28), (1 S,3S)-6-cloro-3-(fenil-d5)-indan-1-ol (Xa) se convierte en (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano (XIa) mediante cloración.
En una realización adicional (E31) de cualquiera de (E29) y (E30) la cloración tiene lugar en presencia de un reactivo seleccionado del grupo que consiste en cloruro de tionilo, oxicloruro de fósforo y pentacloruro de fósforo.
En una realización particular (E32) de (E31), la cloración tiene lugar en presencia de cloruro de tionilo.
En una realización (E33) de (E29), 3,5-dicloro-1-(fenil-d5)-indano (XIb) se convierte en (XIIb) mediante sustitución nucleófila con 2,2-dimetilpiperazina o un compuesto que posteriormente puede transformarse en el resto 3,3-dimetilpiperazina de (XIIb); en particular, (XI) se convierte en (XII) mediante sustitución nucleófila con 2,2-dimetilpiperazina o un compuesto que posteriormente puede transformarse en el resto 3,3-dimetilpiperazina de (XII). En una realización (E34) de (E30), (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano (XIa) se convierte en el compuesto (XIIa) mediante sustitución nucleófila con 2,2-dimetilpiperazina o un compuesto que posteriormente puede transformarse en el resto 3,3-dimetilpiperazina del compuesto (XIIa). En una realización adicional (E35) de cualquiera de (E33) y (E34), la sustitución nucleófila se lleva a cabo con 2,2-dimetilpiperazina en presencia de una base.
En una realización particular (E36) de (E35), la base es un carbonato, por ejemplo carbonato de potasio.
En una realización (E37), el compuesto (XII) se convierte en el compuesto (XIII) mediante alquilación.
En una realización (E40), el compuesto (Xlla) se convierte en el compuesto (XVa), tal como (XIV) o (XV) mediante alquilación.
En una realización adicional (E41) de cualquiera de (E37) y (E38), la alquilación se lleva a cabo en presencia de un donante de metilo-d3 activo y una base.
En una realización específica (E40) de (E39), el donante de metilo activo se elige del grupo que consiste en yoduro de metilo-d3, bromuro de metilo-d3 y sulfato de dimetilo-d6.
En una realización particular (E41) de cualquiera de (E39) y (E40), el donante de metilo activo es yoduro de metilo-d3. En una realización específica (E42) de (E39), la base se elige del grupo que consiste en hidróxido de sodio y potasio, carbonato de sodio y potasio, y ferc-butóxido de sodio y potasio.
En una realización particular (E43) de cualquiera de (E39) y (E42), la base es hidróxido de potasio.
En una realización (E44), (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano (XIa) de (E30) se convierte en el compuesto (XIV) o (XV) mediante sustitución nucleófila con el compuesto (XVI) o un compuesto que posteriormente puede transformarse en resto 1 (d3),2,2-trimetilpiperazina del compuesto (XIV) o compuesto (XV).
En una realización adicional (E45) de (E44), la sustitución nucleófila se lleva a cabo con el compuesto (XVI) en presencia de una base.
En una realización específica (E46) de (E45), la base se elige del grupo que consiste en hidróxido de sodio y potasio, carbonato de sodio y potasio, y terc-butóxido de sodio y potasio.
En una realización particular (E47) de (E46), la base es carbonato de potasio.
En una realización particular (E48) de (E45), el compuesto (XVI) se obtiene a partir del compuesto (XVII) a través del compuesto (XVIII).
Definiciones
Se define exceso enantiomérico como la diferencia absoluta entre la fracción molar de cada enantiómero.
El porcentaje de exceso enantiomérico se calcula como

en donde R y S son las respectivas fracciones molares de enantiómeros en la mezcla de tal manera que R+S = 1. La invención se ilustrará en los siguientes ejemplos no limitativos.
Los compuestos descritos en el presente documento pretenden designar cualquier forma del compuesto, tal como la base libre, sales farmacéuticamente aceptables del mismo, por ejemplo sales de adición de ácido farmacéuticamente aceptables, tales como sal succinato, sal tartrato, en particular sal L-(+)-tartrato, y sales malonato, hidratos o solvatos de la base libre o sales de los mismos, así como formas anhidras, formas amorfas, formas cristalinas y disoluciones. Las sales farmacéuticamente aceptables de compuestos de la presente invención incluyen sales de adición de ácido farmacéuticamente aceptables. Las sales de adición de ácido incluyen sales de ácidos inorgánicos, así como ácidos orgánicos. Los ejemplos representativos de ácidos inorgánicos adecuados incluyen ácidos clorhídrico, bromhídrico, yodhídrico, fosfórico, sulfúrico, sulfámico, nítrico y similares. Los ejemplos representativos de ácidos orgánicos adecuados incluyen ácidos fórmico, acético, tricloroacético, trifluoroacético, propiónico, benzoico, cinámico, cítrico, fumárico, glicólico, itacónico, láctico, metanosulfónico, maleico, málico, malónico, mandélico, oxálico, pícrico, pirúvico, salicílico, succínico, metanosulfónico, etanosulfónico, tartárico, ascórbico, pamoico, bismetilensalicílico, etanodisulfónico, glucónico, citracónico, aspártico, esteárico, palmítico, EDTA, glicólico, p-aminobenzoico, glutámico, bencensulfónico, p-toluensulfónico, ácidos acéticos de teofilina, así como las 8-haloteofilinas, por ejemplo, 8 bromoteofilina y similares.
A menos que se especifique de otro modo, la referencia a cualquiera de los compuestos descritos en esta solicitud cubre el compuesto enantioméricamente puro, así como mezclas de los enantiómeros en cualquier razón.
Experimental
Experimental general
A menos que se indique de otro modo, todas las reacciones se llevaron a cabo bajo nitrógeno. Las reacciones se monitorizaron mediante análisis de cromatografía en capa fina (TLC) y/o CL-EM. Todos los reactivos se compraron y se usaron sin purificación adicional. Las manchas se visualizaron mediante exposición a luz ultravioleta (UV) (254 nm), o mediante tinción con una disolución al 5% p/p de ácido fosfomolibdénico (PMA) en etanol o permanganato de potasio básico acuoso (KMnO4) y luego calentamiento. Se llevó a cabo cromatografía en columna usando gel de sílice Merck C60 (40-63 pm, malla 230-240). Los espectros de RMN se registraron a 250, 500 o 600 MHz (1H RMN), y se calibraron con respecto al pico de disolvente residual. Se usaron las siguientes abreviaturas se usan para los datos de RMN: s, singlete; d, doblete; t, triplete; m, multiplete. Las constantes de acoplamiento están redondeadas al 0,5 Hz más cercano. El exceso enantiomérico se determinó mediante HPLC quiral.
La resolución de compuestos racémicos puede llevarse a cabo tal como se describe en, por ejemplo, los documentos WO12/093165 y WO11/003423.
Método de CL-EM
Columna Acquity UPLC BEH C18 de 1,7 pm; 2,1 x 50 mm funcionando a 60°C con flujo de 1,2 ml/minuto de un gradiente binario que consiste en agua ácido fórmico al 0,1% (A) y acetonitrilo 5% de agua ácido fórmico al 0,1% (B). Detección UV a 254 nm.
Método de HPLC quiral
Columna Phenomenex Lux Celullose-2 de 5p; 250 x 4,6 mm funcionando a 30°C con flujo de 0,5 o 1,0 ml/minuto de n-hexano:isopropanol:dietilamina, 90:10:0,1. Detección UV a 220 nm.
Métodos de HPLC
Método 1: columna Chromolith Performance Rp-18e de 2 p; 100 x 4,6 mm funcionando a 30°C con flujo de 2,0 ml/minuto de agua:trietilamina:acetonitrilo, 1000:5,5:1000, ajustado hasta pH 3 con H3PO4. Detección UV a 254 nm.
Método 2: columna Agilent Zorbax SB-Phenyl de 3,5 p; 150 x 4,6 mm funcionando a 40°C con flujo de 1,0 ml/minuto. Detección UV a 220 nm. Fase móvil A: agua ácido trifluoroacético = 1000 0,5 ml; fase móvil B: acetonitrilo ácido trifluoroacético = 1000 0,5 ml. Gradiente: 0 min: 90% de A, 10% de B; 20 min: 5% de A, 95% de B; 25 min: 5% de A, 95% de B; 25,1 min: 90% de A, 10% de B; 30 min: 90% de A, 10% de B.
Método 3: columna Phenomenex Luna C18 de 3,0 ^ 150 x 4,6 mm funcionando a 40°C con flujo de 1,0 ml/minuto. Detección UV a 220 nm. Fase móvil A: tampón fosfato 25 mM pH 7,4:acetonitrilo = 40:60; fase móvil B: agua:acetonitrilo = 10:90. Gradiente: 0 min: 100% de A, 0% de B; 32 min: 100% de A, 0% de B; 35 min: 50% de A, 50% de B; 37 min: 50% de A, 50% de B; 39 min: 100% de A, 0% de B; 40 min: 100% de A, 0% de B.
Método de GC
Rtx-5 amina de 0,5 ^ 30 m x 0,25 mm con flujo de 1 ml/minuto de helio. Detección FID (250°C). Gradiente: 0 min: 50°C; 9 min: 140°C; 11 min: 140°C; 21 min: 240°C; 23 min 240°C; 26 min: 300°C; 28 min: 300°C.
Síntesis de compuestos de la invención
A. Síntesis de 6-cloro-3-(fenil-d5)-inden-1-ona (I)
Esquema 3:

A1. A través de reacción de Suzuki (Esquema 3, ruta A1):
Síntesis de 3-bromo-6-cloro-inden-1 -ona (III)
A una disolución de 6-cloro-1-indanona (II) (100,0 g, 600,2 mmol) en 1,2-dicloroetano (1,00 l) se le añadió 2,2'-azobis-isobutironitrilo (9,86 g, 60,0 mmol) seguido de W-bromosuccinimida (224,3 g, 1,26 mol). La mezcla de reacción se calentó rápidamente hasta reflujo. Después de 30 minutos a reflujo, se le añadió más 2,2'-azo-bis-isobutironitrilo (9,86 g, 60,0 mmol). La mezcla de reacción se mantuvo durante 4,5 h a reflujo. Posteriormente, la mezcla se agitó a temperatura ambiente durante la noche. La mezcla se enfrió hasta 0°C, y se le añadió trietilamina (126 ml, 904 mmol) gota a gota. La mezcla se agitó durante 1 hora a 0°C y luego se dejó calentar hasta temperatura ambiente. Se añadió agua (1,0 l). La mezcla se agitó vigorosamente durante 15 minutos. Se detuvo la agitación, y la fase acuosa se retiró por succión. Se añadió agua dulce (1,0 l) y la mezcla se agitó durante 15 minutos. Entonces, se separó la fase acuosa por succión. La fase orgánica se agitó adicionalmente con salmuera (500 ml) en un embudo de separación.
Se separó la fase orgánica y se agitó con MgSÜ4 y carbón activo durante 30 min. La mezcla se filtró a través de una capa de Celite. El filtrado se evaporó a sequedad a vacío. Esto produjo 3-bromo-6-cloro-inden-1 -ona (III) bruto (190 g) como sólido, que se usó en la siguiente etapa sin purificación adicional.
Síntesis de 6-cloro-3-(fenil-d5)-inden-1-ona (I)
Al 3-bromo-6-cloro-inden-1 -ona (III) en bruto anterior se le añadió acetato de paladio (5,78 g, 25,8 mmol), trifenilfosfina (13,5 g, 51,5 mmol) y 4,4,5,5-tetrametil-2-d5-fenil-[1,3,2]dioxaborolano (116 g, 566 mmol) seguido de THF (1,50 l) a temperatura ambiente. Se añadió agua (750 ml) y fosfato de potasio (115 g, 541 mmol). La mezcla de reacción se agitó vigorosamente durante 2 h a temperatura ambiente. Se formó una disolución oscura casi negra. Se le añadió heptano (0,70 l). Entonces, se lavó la fase orgánica con agua (1,0 l) y salmuera (0,5 l), se secó sobre MgSÜ4, se filtró y se evaporó a sequedad a vacío. Esto produjo 6-cloro-3-(fenil-d5)-inden-1-ona (I) bruto como un sólido oscuro. El 6-cloro-3-(fenil-d5)-inden-1-ona (I) en bruto se disolvió en una mezcla de heptano-EtÜAc (2:1) y la disolución se filtró a través de gel de sílice. El filtrado se evaporó a sequedad a vacío. El residuo se volvió a precipitar en heptano disolviéndolo en heptano en ebullición, filtrándolo en caliente y dejándolo enfriar lentamente hasta temperatura ambiente para producir 6-cloro-3-(fenil-d5)-inden-1-ona (I) (75,8 g, 52%) como un sólido naranja oscuro, con una pureza del 95% según el análisis de CL-EM.
Datos analíticos para 6-cloro-3-(fenil-d5)-inden-1-ona (I):
1H RMN (600 MHz, CDCh) 5h 6,04 (s, 1H), 7,32 (d, J = 8,0 Hz, 1H), 7,37 (dd, J = 2,0, 8,0 Hz, 1H), 7,49 (dd, J = 0,5, 2,0 Hz, 1H); 13C RMN (150 MHz, CDCh) 5c 122,7, 123,1, 123,5, 127,2 (t, J = 23,5 Hz), 128,7 (t, J = 23,5 Hz), 130,6 (t,
J = 23,5 Hz), 132,2, 132,6, 134,2, 134,4 (t, J = 23,5 Hz), 135,7, 142,1, 162,8; CL-EM (APPI): m/e calc. para C15H5 D5CIO [M+H]+ 246,1, encontrado 246,1.
A2. A través de oxidación (Esquema 3, ruta A2):
Ejemplo 1:
Síntesis de 6-cloro-3-(fenil-d5)-1 H-indeno (IV)
A una suspensión de magnesio (4,43 g, 182 mmol) en THF (15,0 ml) se le añadió Red-AI (0,50 ml, 1,67 mmol, 65% p/p en tolueno). Se añadió una pequeña cantidad (aproximadamente 5 ml) de una disolución de bromobenceno-d5 (29,3 g, 181 mmol) en THF (100 ml) a temperatura ambiente. La mezcla se calentó suavemente (40-50°C), lo que inició la reacción. El inicio de la reacción se detectó mediante una exoterma y se añadió la disolución restante de bromobenceno-d5 gota a gota para mantener un reflujo constante, que tardó 35 minutos en completarse. Después, la mezcla se calentó a reflujo durante 1,5 h. La mezcla resultante se enfrió hasta temperatura ambiente, y la disolución se decantó (usando una cánula) del exceso de magnesio. A la disolución se le añadió una disolución de 5-cloro-1-indanona (V) (20,0 g, 120,0 mmol) en THF (100 ml) a lo largo de un período de 30 min, que mantuvo la temperatura por debajo de 50°C (sin calentamiento o enfriamiento externos). Una vez completada la adición, se dejó agitar la mezcla de reacción durante 1 h (sin calentamiento ni enfriamiento externos). Se le añadió ácido sulfúrico concentrado (13,3 ml, 96% p/p) muy lentamente y con cuidado mientras se mantenía una temperatura por debajo de 50°C en la mezcla de reacción. Una vez que se terminó la adición, se añadió agua (125 ml). La mayor parte del THF se eliminó mediante evaporación a vacío. La mezcla acuosa restante se extrajo con heptano dos veces (2x 100 ml). Los extractos combinados se lavaron con disolución acuosa saturada de NaHCO3 (100 ml), agua (2x 100 ml) y salmuera (100 ml). La fase orgánica se agitó vigorosamente con MgSO4 y carbón activo durante 20 min, y se filtró a través de una capa de Celite. El filtrado se evaporó hasta sequedad. El residuo se evaporó conjuntamente con etanol hasta sequedad a vacío para eliminar la mayor parte del heptano mediante destilación azeotrópica. Esto produjo 6-cloro-3-(fenil-ds)-1 H-indeno (IV) bruto (26,7 g) como un sólido. El producto bruto se volvió a precipitar en etanol disolviéndolo en la mínima cantidad de etanol en ebullición y enfriándolo lentamente hasta 5°C con agitación para proporcionar 6-cloro-3-(fenilds)-1 H-indeno (IV) (20,5 g, 74%) como un sólido de color amarillento, con una pureza del 99% según el análisis de CL-EM.
Datos analíticos para 6-cloro-3-(fenil-ds)-1 H-indeno (IV):
1H RMN (600 MHz, CDCla) 5h 3,49 (d, J = 2,0 Hz, 2 H), 6,57 (t, J = 2,0 Hz, 1H), 7,29 (dd, J = 2,0, 8,0 Hz, 1H), 7,48 (d, J = 8,0 Hz, 1 H), 7,50 (m, 1 H); 13C RMN (150 MHz, CDCla) 5c 38,1, 121,2, 124,6, 126,5, 127,3 (t, J = 24,0 Hz), 127,4 (t, J = 24,0 Hz), 128,3 (t, J = 24,0 Hz), 131,1, 131,2, 135,6, 142,6, 144,7, 146,6; CL-EM (APPI): m/e calc. para C^HaDaCl (M+) 231,1, encontrado 231,1.
Síntesis de 6-cloro-3-(fenil-d5)-inden-1-ona (I)
A una disolución de 6-cloro-3-(fenil-d5)-1 H-indeno (3,00 g, 12,9 mmol) en THF (30,0 ml) se le añadió 1,1-dimetoxi-N,N-dimetilmetanamina (4,30 ml, 32,4 mmol) a temperatura ambiente. La mezcla se calentó a 45°C durante 2,5 h. Se le añadió agua (15,0 ml) seguido de metaperyodato de sodio (8,31 g, 38,8 mmol). La mezcla se calentó adicionalmente a 60°C con agitación vigorosa durante 1,5 h. La mezcla se filtró a través de una capa de Celite. La torta del filtro se lavó a fondo con diclorometano. Los filtrados combinados se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se evaporaron hasta sequedad a vacío. El residuo se purificó por cromatografía en columna eluyendo con una mezcla de heptano-EtOAc (20:1) para proporcionar 6-cloro-3-(fenil-d5)-inden-1-ona (I) (2,86 g, 90%) como un sólido de color amarillo-naranja, con una pureza del 97% según el análisis de CL-EM.
Los datos analíticos (RMN y CL-EM) para el compuesto (I) fueron los mismos que los presentados anteriormente.
Ejemplo 2:
Síntesis de (E)-1 -(6-cloro-3-fenil(d5)-1 H-inden-1 -ilidenmetil)-N,N-dimetilamina (XIX)
Se suspendieron virutas de magnesio (5,60 kg, 230 mol) en 2-MeTHF (21,3 l). A las virutas de magnesio se les añadió cloruro de isopropilmagnesio (25 ml, 50,0 mmol, 2 M) en THF y la suspensión de virutas de magnesio se calentó hasta reflujo con agitación. A las virutas de magnesio se les añadió una disolución de bromobenceno-d5 (34,23 kg, 211 mol) en 2-MeTHF (79,6 l) a lo largo de un período de 1 h 3 min. Se añadió 2-MeTHF (10,5 l) y la reacción se calentó a reflujo durante 38 min. Luego, la reacción se enfrió hasta 22°C antes de añadir una disolución de 5-cloro-1-indanona (V) (32,5 kg, 195 mol) disuelta en 2-MeTHF (198 l) a lo largo de un período de 42 min, con una temperatura máxima de 44°C. Se añadió 2-MeTHF (10,5 l) y la reacción se agitó durante la noche. A la reacción se le añadió una disolución de HCl ac. (80 l, 15% p/p), y la reacción se agitó durante 2 h 46 min. Las fases se separaron y la fase orgánica se lavó con una disolución de NaCl ac. (40 l, 15% p/p). Las fases se separaron y la fase orgánica se redujo en volumen mediante destilación hasta 170 l. La reacción se enfrió hasta 30°C y luego se le añadió 1,1-dimetoxi-N,N-dimetilmetamina (31,0 kg, 260 mol). La reacción se agitó durante la noche y luego se enfrió hasta 6°C. El precipitado formado se filtró y se lavó con heptano dos veces (2x 38 l). El sólido resultante se secó en un horno de vacío a 50°C
durante dos días para producir (£)-1-(6-cloro-3-fenil(d5)-1 H-inden-1-iliden)-W,W-dimetilmetamina (XIX) (48,0 kg, 86%) con una pureza > 99% según el análisis de HPLC (método 1).
Datos analíticos para ('E)-1-(6-cloro-3-fenil(d5)-1H-inden-1-ilidenmetil)-W,W-dimetilamina (XIX):
1H RMN (250 MHz, CDCta) 5h 3,26 (s, 6H), 7,11 (s, 1 H), 7,12 (dd, J = 2,0, 8,5 Hz, 1H), 7,37 (s, 1H), 7,58 (d, J = 2,0 Hz, 1H), 7,66 (d, J = 8,0 Hz, 1 H).
Síntesis de 6-cloro-3-(fenil-d5)-inden-1-ona (I)
Se agitó una mezcla de (£)-1-(6-cloro-3-fenil-1H-inden-1-ilidenmetil)-W,W-dimetilamina (XIX) (803 g, 2,80 mol), metaperyodato de sodio (1,80 kg, 8,40 mol), THF (3,9 l) y agua (3,9 l) a 30°C. Después de 48 minutos, la exoterma de la reacción había calentado la mezcla hasta 36°C, la temperatura máxima alcanzada durante la reacción. La reacción se agitó durante la noche a 30°C y luego se enfrió hasta 21°C. Se añadieron tolueno (280 ml), ácido metanosulfónico (546 ml) y heptano (4,2 l) y la reacción se calentó hasta 29°C. Las fases se separaron y la fase orgánica se lavó con agua (2 x 4 l). A la fase orgánica se añadió heptano (4 l) y el volumen de la fase orgánica se redujo mediante destilación a vacío (máx. 45°C) hasta 3 l. Se añadieron THF (280 ml) y heptano (4 l), y la reacción se agitó durante la noche. La reacción se enfrió hasta 5°C durante 2 h antes de que el precipitado formado se filtrara y se lavara con heptano (2,5 l). El sólido se secó en un horno de vacío a 40°C durante la noche para producir 6-cloro-3-(fenil-d5)-inden-1 -ona (I) (508 g, 74%) con una pureza de > 99% según el análisis de HPLC (método 1).
Los datos analíticos (RMN y CL-EM) para el compuesto (I) fueron los mismos que los presentados anteriormente. B. Síntesis de (±)-6-cloro-3-(fenil-d5)-1 H-inden-1-ol (VI)
Esquema 4:

A una disolución de 6-cloro-3-(fenil-d5)-inden-1-ona (I) (1,00 g, 4,07 mmol) en THF (10,0 ml) se añadió a lo largo de un período de 45 minutos de hidruro de diisobutilaluminio en THF (5,70 ml, 5,70 mmol, 1,0 M) a -10°C con agitación. La mezcla de reacción resultante se agitó durante 30 min a -10°C. Se le añadió metanol (3,0 ml) a -10°C, y se eliminó el enfriamiento. Después de 5 min se añadió disolución de tartrato de sodio y potasio acuosa saturada (10 ml). La mezcla resultante se agitó durante 15 min, y se añadió disolución de NH4Cl acuosa saturada (5 ml) seguido de diclorometano (30 ml). La fase orgánica se separó, y se lavó con salmuera. La fase orgánica se secó sobre MgSÜ4, se filtró y se evaporó hasta sequedad a vacío.
El residuo se purificó por cromatografía en columna eluyendo con heptano-EtÜAc (4:1) para proporcionar (±)-6-cloro-3-(fenil-d5)-1 H-inden-1-ol (VI) (907 mg, 90%) como un sólido de color blanquecino, con una pureza del 98% según el análisis de CL-EM.
Datos analíticos para (±)-6-cloro-3-(fenil-d5)-1H-inden-1-ol (VI):
1H RMN (600 MHz, CDCh) 5h 5,18 (dd, J = 2,0, 7,0 Hz, 1H), 5,76 (d, J = 7,0 Hz, 1H), 6,55 (d, J = 2,0 Hz, 1H), 7,36 (dd, J = 2,0, 8,0 Hz, 1H), 7,42 (d, J = 8,0 Hz, 1H), 7,53 (m, 1H); 13C RMN (150 MHz, CDCh) 5c 74,8, 121,7, 124,3, 127,0 (t, J = 24,0 Hz), 127,8, 128,1 (t, J = 24,0 Hz), 128,6 (t, J = 24,0 Hz), 131,1, 134,1, 136,5, 140,2, 142,4, 150,5; CL-EM (APPI): m/e calc. para C15H7D5GÜ [M+H]+ 248,1, encontrado 248,2.
C. Síntesis de (S)-6-cloro-3-(fenil-d5)-1 H-inden-1-ol (VII)
Esquema 5:

A una disolución de (fi)-(+)-2-metil-CBS-oxazaborolidina en THF (61 pL, 61 pmol, 1,0 M) se le añadió THF (4,0 ml) seguido de una disolución de complejo de borano-THF en THF (1,34 ml, 1,34 mmol, 1,0 M). La disolución resultante se enfrió hasta -10°C, y se añadió lentamente una disolución de 6-cloro-3-(fenil-d5)-inden-1-ona (I) (300 mg, 1,22 mmol) en THF (4,0 ml) a lo largo de un período de 1,5 h. La mezcla de reacción se agitó durante 45 minutos
adicionales a -10°C. Se añadió metanol (5 ml) para extinguir la reacción, y la mezcla se dejó calentar hasta temperatura ambiente. La mezcla se evaporó conjuntamente con gel de sílice. El material obtenido se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (4:1) proporcionó (S)-6-cloro-3-(fenil-d5)-1 H-inden-1-ol (VII) (243 mg, 80%) como un sólido blanco, con el 97% de ee según el análisis de HPLC quiral.
Los datos analíticos (RMN y CL-EM) para el compuesto (VII) fueron los mismos que los presentados anteriormente para el compuesto (VI).
D. Síntesis de (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII)
D1. A través de hidrogenación:
Esquema 6:

Método general:
A una mezcla sólida de 6-cloro-3-(fenil-d5)-inden-1 -ona (I) (200 mg, 0,814 mmol) y cloruro de tris(trifenilfosfina)rodio (I) (7,5 mg, 8,1 pmol) se le añadió disolvente (3,0 ml, véase la tabla 1 para más detalles). La disolución resultante se hidrogenó a 4 bar de gas hidrógeno durante 22 h a temperatura ambiente. La mezcla de reacción se evaporó sobre gel de sílice, se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (20:1) proporcionó (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII). El compuesto obtenido (VIII) se analizó por CL-EM, véase la tabla 1 para más detalles.
Tabla 1. Selección de disolventes:1

1. Condiciones de reacción: (Ph3P)3RhCl (1% en moles), gas hidrógeno (4 bar), temperatura ambiente, 22 h.
2. Porcentaje de área UV en CL-EM.
Ejemplo:
A una mezcla sólida de 6-cloro-3-(fenil-d5)-inden-1 -ona (I) (200 mg, 0,814 mmol) y cloruro de tris(trifenilfosfina)rodio (I) (7,5 mg, 8,1 pmol) se le añadió EtOAc (3,0 ml). La disolución resultante se hidrogenó a 4 bar de gas hidrógeno durante 22 h a temperatura ambiente. La mezcla de reacción se evaporó sobre gel de sílice, se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (20:1) proporcionó (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII) (164 mg, 81%). Datos analíticos para (±)-6-cloro-3-(fenil-d5)-inden-1-ona (VIII):
1H RMN (500 MHz, CDCh) óh 2,72 (dd, 1H, J = 4,0, 19,5 Hz), 3,27 (dd, 1H, J = 8,0, 19,5 Hz), 4,55 (dd, 1H, J = 4,0, 8,0 Hz), 7,21 (d, 1 H; J = 8,0 Hz), 7,52 (dd, 1H, J = 2,0, 8,0 Hz), 7,77 (d, 1H, J = 2,0 Hz); 13C RMN (125 MHz, CDCh) óc 44,0, 47,2, 123,2, 126,8 (t, J = 24,0 Hz), 127,3 (t, J = 24,0 Hz), 128,7 (t, J = 24,0 Hz), 134,4, 135,1, 138,2, 142,9, 156,0, 206,4; CL-EM (APPI): m/e calc. para C15H7D5ClO [M+H]+ 248,1, encontrado 247,6.
D2. A través de reordenamiento:
Esquema 7:

A una disolución de (±)-6-cloro-3-(fenil-d5)-1H-inden-1-ol (VI) (200 mg, 0,807 mmol) y DABCO (1,4-diazabiciclo[2.2.2] octano) (45,3 mg, 0,404 mmol) en THF (3,0 ml) se le añadió trietilamina (281 gL , 2,02 mmol) a temperatura ambiente. La mezcla de reacción se calentó a 60°C durante 1 h. La mezcla de reacción se enfrió y se evaporó conjuntamente con gel de sílice. El material obtenido se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (10:1) proporcionó (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII) (188 mg, 94%).
Los datos analíticos (RMN y CL-EM) para el compuesto (VIII) fueron los mismos que los presentados anteriormente. E. Síntesis de (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX)
E1. A través de reordenamiento:
Esquema 8:

A una disolución de (S)-6-cloro-3-(fenil-d5)-1 H-inden-1 -ol (VII) (200 mg, 0,807 mmol, 97% de ee) y DABCO (45,3 mg, 0,404 mmol) en THF (3,0 ml) se le añadió trietilamina (281 gL, 2,02 mmol) a temperatura ambiente. La mezcla de reacción se calentó a 60°C durante 1 h. La mezcla de reacción se enfrió y se evaporó conjuntamente con gel de sílice. El material obtenido se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (10:1) proporcionó (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) (188 mg, 94%), con el 80% de ee según el análisis de HPLC quiral.
Los datos analíticos (RMN y CL-EM) para el compuesto (IX) fueron los mismos que los presentados anteriormente para el compuesto (VIII).
E2. A través de hidrogenación por transferencia asimétrica organocatalítica:
Esquema 9:

Método general:
A una disolución de 6-cloro-3-(fenil-d5)-inden-1 -ona (I) (300 mg) en un disolvente se le añadió catalizador y reductor a temperatura ambiente o a 60°C (véase la tabla 2 para los detalles). La mezcla de reacción se agitó durante 10-24 h. La mezcla de reacción se evaporó sobre gel de sílice, se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (20:1) proporcionó (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX).
Tabla 2. Selección de condiciones de reacción:

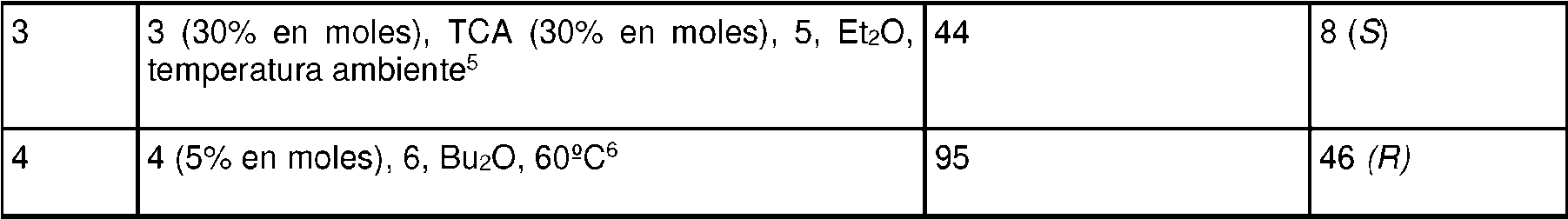
1. TCA = ácido tricloroacético; Et2Ü = éter dietílico; BU2O = óxido de dibutilo
2. Catalizadores:

3. Reductores:

4. Del análisis de la mezcla de  por HPLC quiral.
por HPLC quiral.
5. Jamison B. Tuttle et al., J. Am. Chem Soc. 2006, 128, 12662-12663.
6. Nolwenn J. A. Martin et al., J. Am. Chem Soc. 2006, 128, 13368-13369.
Ejemplo:
El catalizador 4 se elaboró mezclando cantidades equimolares de (R)-TRIP y éster terc-butílico de L-valina en Et2O. El precipitado formado se filtró y se secó a vacío para producir el catalizador 4.
A una mezcla sólida de 6-cloro-3-(fenil-d5)-inden-1-ona (I) (300 mg, 1,22 mmol), reductor 6 (402 mg, 1,59 mmol) y catalizador 4 (57 mg, 0,0610 mmol) se le añadió Bu2O a temperatura ambiente. La mezcla de reacción se calentó a 60°C durante 10 h. La mezcla de reacción se evaporó conjuntamente con gel de sílice, se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (de 20:1 a 10:1) proporcionó (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) (287 mg, 95%), con el 46% de ee según el análisis de HPLC quiral.
Los datos analíticos (RMN y CL-EM) para el compuesto (IX) fueron los mismos que los presentados anteriormente para el compuesto (VIII).
E3. A través de hidrogenación asimétrica (Esquema 9):
Método general:
A una mezcla sólida de precursor metálico y ligando, o catalizador se le añadió disolvente. La mezcla se agitó vigorosamente durante 30 minutos a temperatura ambiente, después de lo cual se le añadió una disolución de 6-cloro-3-(fenil-d5)-inden-1-ona (I) en disolvente (véase la tabla 3-6 para los detalles). La mezcla resultante se hidrogenó a 4 bar de gas hidrógeno con agitación durante 18-70 h a temperatura ambiente. La mezcla de reacción se analizó directamente por CL-EM y HPLC quiral. El producto (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) podría aislarse por evaporación de la mezcla de reacción a vacío y purificación por cromatografía en columna eluyendo con heptano-EtOAc (20:1), o reprecipitación en etanol.
Tabla 3. Selección inicial de condiciones de reacción:1






1. Condiciones de reacción: razón molar sustrato/Rh = 50; razón molar ligando/Rh = 1; 200 mg de 6-cloro-3-(fenil-d5)-inden-1-ona (I); 3 ml de disolvente; 4 bar de gas hidrógeno; temperatura ambiente; 18-70 h.
2. Precursores metálicos y ligandos/catalizadores:
7: (COD)2 lrBArF, cas n.12666826-16-0.
8: (Me-alil)2(COD)Ru, cas n.212289-94-0.
9: Pd(OCOCF3)2, cas n.242196-31-6.
10: (NBD)2RhBF4, cas n.236620-11 -8.
11: (R)-BINAP, cas n.276189-55-4.
12: (S)-T-BINAP, cas n.2100165-88-6.
13: (R)-DM-BINAP, cas n.2137219-86-4.
14: (R)-DTBM-SEGPHOS, cas n.2566940-03-2.
15: (Rj-Monophos, cas n.° 157488-65-8.
16: (S,R,R)-(+)-(3,5-Dioxa-4-fosfaciclohepta[2,1-a:3,4-a']dinaftalen-4-il)bis(1 -feniletil)amina, cas n.° 415918-91 -1.
17: ((1R,1'R,2S,2'S)-Duanphos, cas n.2528814-26-8.
18: (Sj-Phanephos, cas n.° 192463-40-4.
19: [((4R,5R)-Ph2-Ubaphox)Ir(COD)]BArF, cas n.2880262-16-8.
20: (Sj-Me-f-Ketalphos, cas n.2488760-58-3.
21: (R,R)-DIOP, cas n.232305-98-9.
22: (S,S)-Me-Duphos, cas n.° 136735-95-0.
23: (Rj-Prophos, cas n.° 67884-32-6.
24: (S,S)-Chiraphos, cas n.° 64896-28-2.
25: (Rj-C3-TunePhos, cas n.2301847-89-2.
26: RuCl2[(fíj-BINAP][(Sj-DAIPEN]
27: RuCl2[(Sj-(DM-BINAP)][(Sj-DAIPEN] cas n.2220114-01-2.
28: RuCl(p-cimeno)[(S,S)-Ts-DPEN], cas n.2192139-90-5.
29: [('Rj-BINAP]RuCl2p-cimeno, cas n.° 145926-28-9.
30: [(Sj-BINAP]RuCl2, cas n.2134524-84-8.
31: (S,S)-Et-Duphos, cas n.2136779-28-7.
32: (Rj-Me-BoPhoz, cas n.2406680-94-2.
33: (Sj-BINAPINE, cas n.2528854-26-4.
34: (R,R)-Et-BPE, cas n.2136705-62-9.
35: Taniaphos SL-T001 -1, cas n.21003012-96-1.
36: Walphos SL-W001-1, cas n.2387868-06-6.
37: Josiphos SL-J001-1, cas n.° 155806-35-2.
38: (S,S',R,R')-Tangphos, cas n.° 470480-32-1.
39: (Rj-Xilil-Phanephos, cas n.° 325168-89-6.
40: (Rj-DM-SEGPHOS, cas n.2850253-53-1.
41: (Rj-SEGPHOS, cas n.2244261-66-3.
3. Porcentaje de área UV en CL-EM.
4. El producto se aisló por cromatografía en columna sobre gel de sílice. ND = no determinado.
5. Del análisis de la mezcla de reacción por HPLC quiral.
6. Disolventes utilizados:
DCE = 1,2-dicloroetano
MeOH = metanol
TFE = 2,2,2-trifluoroetanol
THF = tetrahidrofurano
EtOAc = acetato de etilo
Optimización con cuatro ligandos principales:
Tabla 4. influencia de la razón ligando/rodio:1

1. Condiciones de reacción: 1% en moles de (NBD)2RhBF4; ligando; 200 mg de 6-cloro-3-(fenil-d5)-inden-1-ona (I); 3 ml de EtOAc; gas hidrógeno (4 bar); temperatura ambiente; 18 h.
2. Los números se refieren a la numeración en la tabla 3.
3. Porcentaje de área UV en CL-EM.
4. Del análisis de la mezcla de reacción mediante HPLC quiral.
5. 2% en moles de (NBD)2RhBF4.
Optimización con (S)-Phanephos:
Tabla 5. Selección de disolvente:1

1. Condiciones de reacción: 1% en moles de (NBD)2RhBF4; 3% en moles de (S)-Phanephos; 200 mg de 6-cloro-3-(fenil-d5)-inden-1-ona (I); 3 ml de disolvente; gas hidrógeno (4 bar); temperatura ambiente; 18 h.
2. Porcentaje de área UV en CL-EM.
3. Del análisis de la mezcla de reacción por HPLC quiral.
4. Disolventes utilizados:
TBME = terc-butil metil éter
i-PrOAc = acetato de isopropilo
Tabla 6. Influencia de la carga del catalizador:1

1. Condiciones de reacción: (NBD)2RhBF4; (S)-Phanephos; Ligando/Rh = 3; 200 mg de 6-cloro-3-(fenil-d5)-inden-1-ona (I); 3 ml de EtOAc; gas hidrógeno (4 bar); temperatura ambiente; 18 h.
2. Porcentaje de área UV en CL-EM.
3. Del análisis de la mezcla de reacción por HPLC quiral.
Ejemplo 3:
A una mezcla sólida de (NBD)2RhBF4 (0,8 mg, 2 |umol) y (S)-Phanephos (3,5 mg, 6,1 |umol) se le añadió EtOAc (libre de oxígeno, 4,0 ml). La mezcla se agitó vigorosamente durante 30 minutos, después de lo cual se añadió una disolución de 6-cloro-3-(fenil-d5)-inden-1 -ona (I) (1,00 g, 4,07 mmol) en EtOAc (libre de oxígeno, 3,0 ml) a la disolución nebulosa. La mezcla resultante se hidrogenó a 4 bar de gas hidrógeno con agitación durante 18 h. El análisis mediante HPLC quiral de la mezcla de reacción mostró la formación de (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) con el 86% de ee. La mezcla de reacción se evaporó hasta sequedad a vacío, y el residuo se volvió a disolver en el mínimo etanol en ebullición, y la disolución se dejó enfriar lentamente hasta temperatura ambiente. El precipitado formado se filtró de la disolución y se secó a vacío para producir (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) (712 mg, 71%) como polvo de color blanquecino, con el 98% de ee según el análisis mediante HPLC quiral. Se pudo obtener una segunda cosecha enfriando el filtrado en el congelador (-5°C) para obtener (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) (64 mg, 6%), con el 93% de ee según el análisis de HPLC quiral.
Los datos analíticos (RMN y CL-EM) para el compuesto (IX) fueron los mismos que los presentados anteriormente para el compuesto (VIII).
Ejemplo 4:
A una mezcla sólida de complejo de [(S)-Phanephos][NBD]RhBF4 (17 mg, 20 |umol) y 6-cloro-3-(fenil-d5)-inden-1-ona (I) (10,0 g, 40,7 mmol) se le añadió EtOAc (libre de oxígeno, 100 ml). La mezcla se hidrogenó a 4 bar de gas hidrógeno con agitación durante 2 h. El análisis mediante HPLC quiral de la mezcla de reacción mostró la formación de (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) con el 89% de ee. La mezcla de reacción se agitó con carbón activo (1 g) durante 1 h, y se filtró a través de Celite. El filtrado se evaporó hasta sequedad a vacío, y el residuo se volvió a disolver en el mínimo etanol en ebullición, y la disolución se dejó enfriar lentamente hasta temperatura ambiente. El precipitado formado se filtró de la disolución y se secó a vacío para producir (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) (7,1 g, 70%), con el 99% de ee según el análisis de HPLC quiral.
Los datos analíticos (RMN y CL-EM) para el compuesto (IX) fueron los mismos que los presentados anteriormente para el compuesto (VIII).
Ejemplo 5:
A una mezcla sólida de (NBD)2RhBF4 (435 mg, 1,16 mmol) y (S)-Phanephos (1,31 g, 2,27 mmol) se le añadió EtOAc (libre de oxígeno, 300 ml). La mezcla se agitó vigorosamente durante 30 min, y se añadió a una suspensión de 6-cloro-3-(fenil-d5)-inden-1-ona (I) (400 g, 1,63 mol) en EtOAc (libre de oxígeno, 2,7 l). La mezcla se transfirió a un autoclave de 25 l y se hidrogenó a 4 bar de gas hidrógeno durante 22 h a t.a. Luego, se mezcló la mezcla de reacción con carbón activo (56 g) y se agitó durante 1 h, y se filtró a través de Arbocel BC 200® usando EtOAc adicional (200 ml). El filtrado se evaporó hasta sequedad a vacío, y se le añadió etanol (1,2 l). La mezcla se calentó hasta 80°C para formar una disolución homogénea, que posteriormente se dejó enfriar lentamente con agitación hasta t.a., y la suspensión resultante se enfrió adicionalmente en un baño de agua con hielo, y se filtró. El precipitado se lavó con etanol enfriado con hielo (200 ml), y se secó a vacío a 50°C durante un día para producir (S)-6-cloro-3-(fenil-d5)-indan
1 -ona (IX) (339 g, 84%) como sólido, con el 99% de ee según el análisis de HPLC quiral y una pureza de > 99% según el análisis de CL-EM.
Los datos analíticos (RMN y CL-EM) para el compuesto (IX) fueron los mismos que los presentados anteriormente para el compuesto (VIII).
F. Síntesis de (±)-c/s-6-cloro-3-(fenil)-d5)-indan-1 -ol (X)
Esquema 10:

A una suspensión de borohidruro de sodio (443 mg, 11,7 mmol) en IPA (10,0 ml) se le añadió una disolución de (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII) (1,45 g, 5,85 mmol) en IPA (10,0 ml) y THF (5,0 ml) a -10°C. La mezcla se dejó calentar hasta t.a. lentamente durante la noche. Se añadió cuidadosamente una disolución de HCl ac. (10 ml, 4 M) mientras la mezcla de reacción se enfriaba en un baño de agua con hielo para mantener la temperatura a o por debajo de temperatura ambiente. La mezcla resultante se concentró mediante evaporación a vacío, y se le añadió agua (20 ml). La mezcla acuosa se extrajo con EtOAc tres veces (3 x 30 ml). Los extractos combinados se lavaron con salmuera (20 ml), se secaron sobre MgSO4 y se filtraron. El filtrado se evaporó conjuntamente con gel de sílice. El material obtenido se cargó en una columna de gel de sílice y la elución con heptano-EtOAc (4:1) proporcionó (±)-c/s-6-cloro-3-(fenil-d5)-indan-1-ol (X) (1,43 g, 98%) como un sólido de color blanquecino, con una razón cis:trans de 97:3 según el análisis de 1H RMN, y pureza del 97% según el análisis de CL-EM.
Datos analíticos para (±)-c/s-6-cloro-3-(fenil-d5)-indan-1-ol (X):
1H RMN (600 MHz, CDCta) 5h 1,96 (ddd, J = 8,0, 13,0 Hz, 1H), 2,06 (d, J = 8,0 Hz, 1H), 3,03 (dt, J = 8,0, 13,0 Hz, 1H), 4,14 (t, J = 8,0 Hz, 1 H), 5,25 (q, J = 8,0 Hz, 1H), 6,86 (dd, J = 1,0, 8,0 Hz, 1H), 7,20 (dd, J = 2,0, 8,0 Hz, 1 H), 7,45 (d, J = 2,0 Hz, 1H); 13C RMN (150 MHz, CDCta) 5c 47,3, 47,8, 74,7, 124,1, 126,3, 126,4, 128,0, 128,5, 129,3, 133,2, 143,6, 144,1, 147,2; CL-EM (APPI): m/e calc. para C15H9D5ClO [M+H]+ 250,1, encontrado 250,0.
G. Síntesis de maleato de (±)-írans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina (XII) a través de (±)-c/s-3,5-dicloro-1 -(fenil)-d5)-indano (XI)
Esquema 11:

XII: Racemato, sal maleato 1:1
Se mezclaron cloruro de tionilo (2,01 kg, 16,9 mol) y THF (7,2 kg) y la mezcla se enfrió hasta 10-15°C. Se añadió lentamente una disolución de (±)-c/s-6-cloro-3-(fenil-d5)-indan-1-ol (X) (2,76 kg, 11,1 mol) en THF (7,2 kg) y, una vez completado, se añadió THF (5,9 kg). La mezcla de reacción se agitó a 15°C durante aproximadamente 90 h. Se enfrió agua (16,7 kg) hasta 11 °C y se añadió lentamente a la mezcla de reacción, luego se añadió lentamente disolución de NaOH ac. (7,8 kg, 27,7% p/p), seguido de EtOAc (10 kg). La mezcla se agitó durante 20-40 min. Las fases se separaron y la fase orgánica se redujo hasta un volumen de aproximadamente 6 l mediante destilación. Se añadió MIBK (16 kg) y el volumen se redujo hasta aproximadamente 8 l mediante destilación para producir una disolución del compuesto (XI). Se añadieron carbonato de potasio (1,58 kg, 11,4 mol), 2,2-dimetilpiperazina (1,69 kg, 14,8 mol) y MIBK (13,6 kg). La mezcla de reacción se agitó 35 h a 90-95°C. Después de enfriar hasta temperatura ambiente, se añadió agua (11 kg) y la mezcla se agitó durante 30-60 min. Se separaron las fases. A la fase orgánica se le añadió agua (13,7 kg) y la mezcla se agitó lentamente durante 30-60 min. Las fases se separaron y la fase orgánica se filtró en blanco. Se añadieron MIBK (5 kg), agua (7,8 kg) y disolución de HCl ac. (5,9 kg, 36% p/p) y la mezcla se agitó a 50°C durante de 30 a 60 min. Se separaron las fases, y a la fase acuosa se le añadió MIBK (8 kg) y la mezcla se enfrió hasta 10-15°C. A la mezcla se le añadió lentamente una mezcla de MIBK (3,5 kg) y disolución de NH3 ac. (7,8 kg, 25% p/p) y la mezcla de reacción se agitó a 20-25°C durante 60-90 min. Se separaron las fases y la fase orgánica se lavó con agua (10,5 kg). La fase orgánica se redujo hasta 8 l mediante destilación. Se añadieron ácido maleico (1,19 kg, 10,3 mol) y MIBK
(9 kg) y la mezcla de reacción se calentó después hasta 75-80°C. Tras enfriar hasta 10-15°C, el precipitado se filtró y se lavó con MIBK (10 kg). El sólido se secó en un horno de vacío a 50°C durante aproximadamente 20 h para proporcionar maleato de (±)-frans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina (XII) (3,47 kg, 68%).
Datos analíticos para maleato de (±)-frans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina (XII):
1H RMN (250 MHz, DMSO-de) óh 1,31 (s, 3H), 1,33 (s, 3H), 2,12 (ddd, J = 6,0, 8,0, 14,0 Hz, 1H), 2,31(d, J = 12,0 Hz, 1H), 2,58-2,50 (m, 3H), 2,77 (sa, 1H), 3,16 (sa, 2H), 3,37 (sa, 1H), 4,48 (dd, J = 6,0, 8,5 Hz, 1 H), 4,56 (dd, J = 5,0, 8,0 Hz, 1H), 6,04 (s, 2H, ácido maleico), 6,98 (d, J = 8,0 Hz, 1 H), 7,29 (dd, J = 2,0, 8,0 Hz, 1H), 7,39 (d, J = 1,5 Hz, 1H), 8,60 (sa, 2H).
H. Síntesis de succinato de (±)-írans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetilpiperazina (XIII)
Esquema 12:

Se agitaron el compuesto (XII) (1,1 kg, 2,38 mol), TBME (11 l), agua (1,8 l) y disolución de NH3 ac. (1 l, 25% p/p) durante 1-2 h. Se separaron las fases y la fase orgánica se lavó con agua (2 x 2 l). A la fase orgánica se le añadieron una disolución de k Oh (254 g, 3,85 mol, 85% p/p) y agua (1,5 l), seguido de la adición de yoduro de metilo-d3 (450 g, 3,11 mol). La mezcla de reacción se agitó a 20-25°C durante 16-24 h. Se le añadió agua (2 l) y se filtró el subproducto precipitaba. Al filtrado se le añadieron agua (0,8 l) y disolución de NH3 ac. (0,2 l, 25% p/p) y la mezcla se agitó durante 20-40 min. Se separaron las fases y la fase orgánica se lavó con agua (2 l). Las fases se separaron y se añadió cloruro de acetilo (38 g, 0,48 mol) a la fase orgánica que se agitó durante 20-40 min. Se añadieron agua (0,8 l) y una disolución de NH3 ac. (0,2 l, 25% p/p) y la mezcla se agitó durante 20-40 min. Se separaron las fases y la fase orgánica se lavó con agua (2 l). La fase orgánica se evaporó hasta sequedad. Se añadieron ácido succínico (225 g, 1,91 mol) y acetona para alcanzar un volumen de reacción total de 6-6,5 l. La mezcla de reacción se calentó hasta reflujo y luego se enfrió hasta 5-10°C. El precipitado se filtró y se lavó con acetona (1 l). El sólido se secó en un horno de vacío a 50°C durante más de 16 h para proporcionar el compuesto (XIII) (630 g, 55%).
Datos analíticos para succinato de (±)-trans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina (XIII):
1H RMN (600 MHz, DMSO-de) óh 1,02 (s, 3H), 1,04 (s, 3H), 2,02 (ddd, J = 6,0, 8,0, 14,0 Hz, 1H), 2,13 (d, J = 11,0 Hz, 1H), 2,31 (sa, 1H), 2,37 (s, 4H, ácido succínico), 2,46-2,41 (m, 1H), 2,65-2,56 (m, 4H), 4,46 (dd, J = 6,0, 9,0 Hz, 1H), 4,46 (dd, J = 5,0, 8,0 Hz, 1H), 6,95 (d, J = 8,0 Hz, 1 H), 7,26 (dd, J = 2,0, 8,0 Hz, 1H), 7,33 (d, J = 2,0 Hz, 1 H).
I. Síntesis de L-(+)-tartrato de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina (XIV) mediante resolución
Esquema 13:

XIII: Racemato, sal succinato 1:1 XIV: (1R,3S), sal L-(+)-tartrato 1:1
Se agitaron el compuesto (XIII) (1,00 kg, 2,08 mol), EtOAc (8 l), agua (2 l) y disolución de NH3 ac. (1 l, 25% p/p) durante 0,5-1 h. Se separaron las fases y la fase orgánica se lavó con agua (2 l). La fase orgánica se redujo hasta aproximadamente 1,5 l mediante destilación. Se añadieron acetona (10 l) y ácido L-(+)-tartárico (312 g, 2,08 mol). La mezcla se calentó hasta reflujo y luego se enfrió hasta 5-10°C. El precipitado se filtró, y se lavó con acetona (1,2 l). La torta del filtro húmeda se mezcló con etanol (11 l). La mezcla se calentó hasta reflujo y luego se enfrió hasta 5-10°C.
El precipitado se filtró y se lavó con etanol absoluto (1,2 l). El sólido se secó en un horno de vacío a 50°C durante más de 16 h para proporcionar el compuesto (XIV) (395 g, 37% de rendimiento).
Datos analíticos para L-(+)-tartrato de 4-((1 fí,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina (XIV):
1H RMN (600 MHz, DMSO-de) 5h 1,18 (s, 3H), 1,21 (s, 3H), 2,04 (ddd, J = 6,0, 8,0, 14,0 Hz, 1H), 2,31(d, J = 12,0 Hz, 1H), 2,61 -2,50 (m, 3H), 2,77 (sa, 1H), 2,95 (sa, 1 H), 4,07 (s, 2H, tartrato), 4,45 (dd, J = 6,0, 8,5 Hz, 1H), 4,50 (dd, J = 5,0, 8,0 Hz, 1 H), 6,96 (d, J = 8,0 Hz, 1 H), 7,27 (d, J = 8,0 Hz, 1H), 7,36 (s, 1H).
J. Síntesis de (7S,3S)-6-cloro-3-(fenil-d3)-indan-1-ol (Xa)
Esquema 14:

Se disolvió borohidruro de sodio (67 g, 1,77 mol) en IPA (2,1 l) y la disolución se enfrió hasta -10°C. Se disolvió (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) (438 g, 1,77 mol) en THF (2,3 l) e IPA (0,4 l). La disolución de (S)-6-cloro-3-(fenild5)-indan-1-ona (IX) se añadió a la disolución de borohidruro de sodio a lo largo de un período de 2 h 24 min con una temperatura máxima de -4°C durante la adición. La reacción se agitó durante la noche mientras la temperatura alcanzaba la temperatura ambiente. La reacción se enfrió hasta -2°C y se añadió disolución de HCl ac. (1,55 l, 4 M) a lo largo de un período de 1 h 35 min. El volumen de la reacción se redujo mediante destilación a vacío hasta aproximadamente 2,5 l. Se añadió agua (2,5 l) y tolueno (4 l) y la reacción se agitó a 45°C durante 15 min. Se separaron las fases y la fase orgánica se lavó con disolución de NaCl ac. (3 l, 5% p/p). Se separaron las fases y la fase orgánica se redujo mediante destilación a vacío (máx. 70°C) hasta aproximadamente 1,4 l. La fase orgánica se añadió a lo largo de un período de 9 min a heptano (12,5 l). La reacción se enfrió hasta -5°C y el precipitado formado se filtró 1 h 20 min más tarde. El precipitado se lavó con heptano (1 l) y luego se secó en un horno de vacío a 40°C durante la noche para producir (1 S,3S>6-cloro-3-(fenil-d5)-indan-1-ol (Xa) (377 g, 86%) como un sólido de color blanquecino, con una pureza del 99,5% según el análisis por HPLC (método 2).
Los datos analíticos (RMN y CL-EM) para el compuesto (Xa) fueron los mismos que los presentados anteriormente para el compuesto (X).
K. Síntesis de L-(+)-tartrato de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1 (d3),2,2-trimetil-piperazina (XIV) a través de (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano (XIa)
Esquema 15:

XIV: (1 R,3S), sal L-(+)-tartrato 1:1
Se añadió una disolución enfriada con hielo del compuesto (Xa) (25 g, 100 mmol) en 2-MeTHF (80 ml) a una disolución enfriada con hielo de cloruro de tionilo (11,0 ml, 152 mmol) en 2-MeTHF (60 ml) a lo largo de un período de 10 min, con una temperatura máxima de 1°C. La reacción se agitó durante la noche a temperatura ambiente, luego se enfrió hasta 2°C, antes de añadirse agua (180 ml) a lo largo de un período de 25 min, manteniendo la temperatura por debajo de 18°C. El pH se ajustó hasta 7 mediante la adición de disolución de NH3 ac. (34 ml, 25% p/p) y luego se separaron las fases. La fase orgánica se evaporó a vacío y el aceite resultante se separó una vez con MIBK (50 ml) para producir el compuesto (Xla) bruto. Se le añadieron MIBK (160 ml), carbonato de potasio (42,8 g, 310 mmol) y el compuesto (XVI) (43,1 g, 120 mmol) y la reacción se calentó a 90°C durante 24 h. La reacción se enfrió hasta temperatura ambiente y luego se le añadió agua (300 ml). La reacción se agitó durante 15 minutos, las fases se separaron y la fase orgánica se lavó con agua (300 ml). Las fases se separaron y se añadió cloruro de acetilo (1,0 ml) a la fase orgánica. La reacción se agitó durante 3 h, luego se añadieron agua (20 ml) y disolución de NH3 ac. (6 ml, 25% p/p). Las fases se separaron y la fase orgánica se lavó con agua (130 ml). La fase orgánica se filtró a través de Arbocel BC-200 y luego se añadió disolución de HCl ac. (240 ml, 1,08 mol, 4,5 M). La reacción se calentó hasta 50°C, las fases se
separaron y a la fase acuosa se le añadieron MIBK (300 ml) y luego disolución de NH3 ac. (180 ml, 25% p/p). Las fases se separaron y la fase orgánica se lavó con agua (300 ml) y luego se redujo en volumen mediante evaporación a vacío. El aceite resultante se separó con acetona (100 ml). El aceite se disolvió luego en etanol (300 ml) y se le añadió ácido L-(+)-tartárico (15,0 g, 100 mmol). La reacción se calentó hasta reflujo y luego se enfrió hasta temperatura ambiente. El precipitado resultante se filtró y el sólido se lavó con acetona (50 ml). El sólido se secó en un horno de vacío a 50°C durante la noche para producir el compuesto (XIV) (29,8 g, 58%), con una pureza del 97,8% según el análisis de HPLC (método 3).
Los datos analíticos (RMN y CL-EM) para el compuesto (XIV) fueron los mismos que los presentados anteriormente.
L. Síntesis de fumarato de 4-((1 fí,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina (XV) a través de (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano (Xla)
Esquema 16:

XV: (1R,3S), sal fumarato 1:1
Se añadió una disolución enfriada con hielo del compuesto (Xa) (23,7 g, 94,9 mmol) en 2-MeTHF (80 ml) a una disolución enfriada con hielo de cloruro de tionilo (10,3 ml, 142 mmol) en 2-MeTHF (60 ml) a lo largo de un período de 10 min, con una temperatura máxima de 1 °C. La reacción se agitó durante la noche a temperatura ambiente, luego se enfrió hasta 3°C, antes de añadirse agua (180 ml) a lo largo de un período de 25 min, manteniendo la temperatura por debajo de 16°C. El pH se ajustó hasta 7 mediante la adición de disolución de NH3 ac. (35 ml, 25% p/p) y luego se separaron las fases. La fase orgánica se evaporó a vacío y el aceite resultante se separó una vez con MIBK (50 ml) para producir el compuesto (Xla) bruto. Se añadieron MIBK (160 ml), carbonato de potasio (40,7 g, 295 mmol) y compuesto (XVI) (40,9 g, 114 mmol) y la reacción se calentó a 80°C durante 68 h. La reacción se enfrió hasta 39°C y luego se le añadió agua (270 ml). La reacción se agitó durante 15 min, se separaron las fases y la fase orgánica se lavó con agua (270 ml). Las fases se separaron y se añadió cloruro de acetilo (0,9 ml) a la fase orgánica. La reacción se agitó durante 72 h, luego se añadieron agua (25 ml) y disolución de NH3 ac. (7 ml, 25% p/p). Las fases se separaron y la fase orgánica se lavó con disolución de NaCl ac. (100 ml, 7,5% p/p) y posteriormente con agua (100 ml). La fase orgánica se filtró a través de Arbocel BC-200 y luego se añadió disolución de HCl ac. (250 ml, 1,0 mol, 4,0 M). La reacción se calentó hasta 55°C, las fases se separaron y a la fase acuosa se le añadió MIBK (300 ml) y luego disolución de NH3 ac. (100 ml, 25% p/p). Las fases se separaron y la fase orgánica se lavó con agua (300 ml) y luego se redujo en volumen mediante evaporación a vacío. El aceite resultante se separó con acetona (200 ml) y luego con etanol (200 ml). Luego, se disolvió el aceite en etanol (200 ml) y se le añadió ácido fumárico (9,75 g, 84,0 mmol). La reacción se calentó hasta 55°C y luego se enfrió hasta temperatura ambiente. El precipitado resultante se filtró y el sólido se lavó con etanol dos veces (2 x 25 ml). El sólido se secó en un horno de vacío a 50°C durante dos días para producir el compuesto (XV) (26,4 g, 58%), con una pureza del 99,2% según el análisis de HPLC (método 3).
Datos analíticos para fumarato de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1 (d3),2,2-trimetil-piperazina (XV):
1H RMN (250 MHz, CDCh) óh 1,14 (s, 3H), 1,16 (s, 3H), 2,04 (ddd, J = 6,0, 8,0, 13,5 Hz, 1H), 2,26 (d, J = 12,0 Hz, 1H), 2,73-2,40 (m, 3H), 2,86-2,75 (m, 1H), 2,92-2,86 (m, 2H), 4,52-4,41 (m, 2H), 6,53 (s, 2H, fumarato), 6,95 (d, J = 8,0 Hz, 1H), 7,26 (dd, J = 2,5, 8,0 Hz, 1 H), 7,34 (d, J = 2,5 Hz, 1 H).
M. Síntesis de hemi-D,L-tartrato de 3,3-dimetilpiperazin-1-carboxilato de terc-butilo (XVIII)
Esquema 17:

XVII XVIII (hemi-D,L-tartrato)
Se disolvió 2,2-dimetilpiperazina (11,5 kg, 101 mol) en etanol (48,5 l) y la disolución se enfrió hasta aproximadamente 9°C. Se disolvió dicarbonato de di-terc-butilo (21,9 kg, 100 mol) en etanol (41,7 l). La disolución de dicarbonato de diterc-butilo se añadió a la disolución de dimetilpiperazina a lo largo de un período de 2 h 30 min, manteniendo la
temperatura de la reacción por debajo de 15°C. Se añadió etanol (12,4 l) y la disolución se agitó durante la noche a una temperatura de entre 12-25°C. La reacción se calentó hasta reflujo y se destilaron 75 l. Se añadió etanol (76 l) a la reacción y la disolución se calentó hasta 52°C y se transfirió a una suspensión de ácido D,L-tartárico (7,5 kg, 50,0 mol) en etanol (25,2 l) y se calentó hasta 51 °C. Se añadió etanol (25,3 l) y la reacción se mantuvo a 20°C durante la noche. El precipitado se filtró y se lavó con etanol (28,1 l). El sólido se secó en un horno de vacío a 50°C durante la noche para producir el compuesto (XVIII) (27,1 kg, 93%) con una pureza del 99% según el análisis de GC.
Datos analíticos para hemi-D,L-tartrato de 3,3-dimetilpiperazin-1-carboxilato de tero-butilo (XVIII):
1H RMN (250 MHz, CDCh) óh 1,35 (s, 6H), 1,46 (s, 9H), 3,10 (sa, 2H), 3,42 (sa, 2H), 3,63 (sa, 2H), 4,29 (s, 1H, tartrato), 7,60 ppm (sa, 3H); 13C RMN (62,5 MHz, CDCh) óc 22,3, 28,3, 39,0, 40,8, 50,2, 51,8, 53,6, 73,6 (tartrato), 80,6, 154,2, 178,3 (tartrato).
N. Síntesis de bis-2,2,2-trifluoroacetato de 1(d3),2,2-trimetilpiperazina (XVI)
Esquema 18:

XVIII (hemi-D,L-tartrato) XVI (sal de TFA 1:2)
Se suspendió en tolueno (133 l) el compuesto XVIII (23,0 kg, 79,5 mol), se añadieron agua (85,2 l) y disolución de NaOH ac. (14,1 kg, 27,7% p/p) y la reacción se agitó durante 1 h. Tras la separación de fases, la fase orgánica se añadió a carbonato de potasio (11,1 kg, 80,3 mol). Se le añadió W-metilpirrolidina (7,0 kg). Se disolvió yodometano-d3 (12,7 kg, 87,6 mol) en tolueno (11,5 l) y luego se añadió a la reacción, seguido de tolueno (11,5 l). La reacción se agitó a 23°C durante la noche. Después de que un control en proceso mostrara que quedaba el 5,7% de compuesto (XVIII), se añadieron yodometano-d3 (0,9 kg, 6,21 mol) y tolueno (12,7 l) y la reacción se agitó durante la noche a 23°C. Se añadieron agua (85 l) y disolución de NH3 ac. (3,5 kg, 25% p/p) y la reacción se agitó durante 40 min. Las fases se separaron y la fase orgánica se redujo mediante destilación a vacío hasta aproximadamente 20 l. La reacción se enfrió hasta 0°C y se añadió ácido trifluoroacético (38,0 kg, 333 mol) a lo largo de un período de 36 min. La reacción se agitó a 39°C durante la noche y luego se enfrió hasta 13°C. Se añadió éter dietílico (77,1 l) y la reacción se agitó a aproximadamente 22°C durante la noche. La reacción se enfrió hasta 8°C, se agitó allí durante 3,5 h y luego se filtró. La torta del filtro se lavó con éter dietílico (44,9 l) y luego con más éter dietílico (30,8 l). El sólido resultante se secó en un horno de vacío a 50°C durante la noche para producir el compuesto (XVI) (23,4 kg, 82%) con una pureza del 93,2% según el análisis de GC.
Datos analíticos para bis-2,2,2-trifluoroacetato de 1 (d3),2,2-trimetilpiperazina (XVI):
1H RMN (250 MHz, D2O) 5h 1,35 (s, 6H), 3,16 (d, J = 14,5 Hz, 1H), 3,31 -3,21 (m, 1H), 3,58-3,35 ppm (m, 4 H); 13C RMN (62,5 MHz, D2O) 5c 17,0, 23,8, 37,0, 41,9, 47,3, 51,7, 60,8, 117,6 (q, J = 36 Hz, TFA), 164,0 (q, J = 291 Hz, TFA).
Claims (14)
- REIVINDICACIONES1 Un procedimiento para la producción del compuesto (n donde el compuesto (I) se obtiene a través de la reacción de 3-bromo-6-cloro-inden-1 -ona con 4,4,5,5-tetrametil-2-a5-fenil-[1,3,2]dioxaborolano.

- 2. El procedimiento de la reivindicación 1 que comprende las siguientes etapasa) se añade 2,2'-azo-bis-isobutironitrilo y W-bromosuccinimida a una disolución que comprende 6-cloro-1-indanonab) se añade trietilamina a la disolución de la etapa a) para obtener 3-bromo-6-cloro-inden-1-onac) se separa 3-bromo-6-cloro-inden-1-ona y se hace reaccionar con 4,4,5,5-tetrametil-2-cfe-fenil-[1,3,2]dioxaborolano en presencia de un catalizador y una base apropiados para obtener el compuesto (I).
- 3. El procedimiento de la reivindicación 2 en donde la etapa c) se lleva a cabo en presencia de acetato de paladio y trifenilfosfina.
- 4. Un procedimiento para la producción del compuestoque comprende las etapas de
 a) síntesis de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) por reacción entre una especie organometálica obtenida a partir de benceno-d5 monohalogenado y 5-cloro-1-indanona (V) seguido de deshidratación,b) reacción de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) al compuesto (£)-1-(6-cloro-3-fenil(d5)-1H-inden-1-ilidenmetil)-W,W-dimetilamina (XIX) y escisión oxidativa adicional de la misma para obtener el compuesto (I).
a) síntesis de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) por reacción entre una especie organometálica obtenida a partir de benceno-d5 monohalogenado y 5-cloro-1-indanona (V) seguido de deshidratación,b) reacción de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) al compuesto (£)-1-(6-cloro-3-fenil(d5)-1H-inden-1-ilidenmetil)-W,W-dimetilamina (XIX) y escisión oxidativa adicional de la misma para obtener el compuesto (I). - 5. El procedimiento según la reivindicación 4 que comprende las etapas dea) síntesis de 6-cloro-3-(fenil-ds)-1 H-indeno (IV) por reacción de Grignard entre bromobenceno-d5, magnesio y 5-cloro-1-indanona seguido de deshidratación,b) hacer reaccionar 6-cloro-3-(fenil-ds)-1 H-indeno de la etapa a) con 1,1-dimetoxi-W,W-dimetilmetanamina seguido de escisión oxidativa del intermedio de enamina formado (XIX) para obtener el compuesto (I).
- 6. El procedimiento de las reivindicaciones 4 o 5 en donde la escisión oxidativa en la etapa b) se lleva a cabo mediante el uso de un agente oxidante seleccionado del grupo que consiste en metaperyodato de sodio, metaperyodato de potasio, ozono, dicromato de potasio, dicromato de sodio, oxígeno singlete y ácido mcloroperbenzoico.
- 7. Un procedimiento según cualquiera de las reivindicaciones 1 o 4 que comprende además las etapasa) el compuesto (I) se reduce para obtener 6-cloro-3-(fenil-ds)-1 H-inden-1 -ol (VIa),b) el compuesto (VIa) se convierte en 6-cloro-3-(fenil-ds)-indan-1-ona (VIIIa) a través de reordenamiento inducido por base.
- 8. Un procedimiento según cualquiera de las reivindicaciones 1 o 4 que comprende además las etapasa) el compuesto (I) se convierte en (S)-6-cloro-3-(fenil-d5)-1H-inden-1-ol (VII) a través de reducción enantioselectiva en presencia de reductor y catalizador enantioselectivob) el compuesto (VII) se convierte en (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) a través de reordenamiento inducido por base.
- 9. Un procedimiento según cualquiera de las reivindicaciones 1 o 4 que comprende además la conversión del compuesto (I) para obtener 6-cloro-3-(fenil-ds)-indan-1-ona (VIIIa) a través de hidrogenación en presencia de un catalizador adecuado, tal como cloruro de tris(trifenilfosfina)rodio (I), en un disolvente adecuado, tal como acetato de etilo.
- 10. Un procedimiento según cualquiera de las reivindicaciones 1 o 4 que comprende además la conversión del compuesto (I) para obtener (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) a través de hidrogenación asimétrica en presencia de un catalizador adecuado, tal como una sal de rodio, y un ligando de fosfina quiral y un disolvente adecuado, tal como acetato de etilo.
- 11. Un procedimiento según cualquiera de las reivindicaciones 7 a 10 en donde los compuestos obtenidos en cualquiera de las reivindicaciones 7 a 10 se convierten en 3,5-dicloro-1 -(fenil-d5)-indano, en particular (±)-c/s-3,5-dicloro-1-(fenil-d5)-indano o (1 S,3S)-3,5-dicloro-1-(fenil-d5)-indano, comprendiendo el procedimiento las siguientes etapasa) reducción de (±)-6-cloro-3-(fenil-d5)-indan-1-ona (VIII), 6-cloro-3-(fenil-d5)-indan-1-ona (VIIIa) o (S)-6-cloro-3-(fenil-d5)-indan-1-ona (IX) para obtener el indanol correspondiente:(±)-cis-6-cloro-3-(fenil-d5)-indan-1-ol (X), 6-cloro-3-(fenil-d5)-indan-1-ol (Xb) o (1S,3S)-6-cloro-3-(fenil-d5)-indan-I -ol (Xa) en presencia de un agente reductor adecuado, tal como NaBH4;b) cloración, por ejemplo en presencia de cloruro de tionilo, de cualquiera de los compuestos obtenidos en la etapa a) para obtener el compuesto de indano clorado correspondiente (±)-cis-3,5-dicloro-1-(fenil-d5)-indano (XI), 3,5-dicloro-1-(fenil-d5)-indano (Xlb) o (1S,3S)-3,5-dicloro-1-(fenil-d5)-indano (Xla).
- 12. Un procedimiento según la reivindicación 11 en donde el (±)-cis-3,5-dicloro-1-(fenil-d5)-indano (XI), 3,5-dicloro-1-(fenil-d5)-indano (XIb) o (1S,3S)-3,5-dicloro-1-(fenil-d5)-indano (XIa) tal como se obtiene en la reivindicación I I se convierten en una sal farmacéuticamente aceptable de (±)-trans-1-(6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina, 1 -(6-cloro-3-(fenil-ds)-indan-1 -il)-3,3-dimetil-piperazina o 1 -((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-3,3-dimetil-piperazina, comprendiendo el procedimiento las siguientes etapasa) reacción con 2,2-dimetilpiperazina o un compuesto que posteriormente pueda transformarse en el resto 3,3-dimetilpiperazina de (±)-trans-1 -(6-cloro-3-(fenil-ds)-indan-1 -il)-3,3-dimetil-piperazina, 1 -(6-cloro-3-(fenil-ds)-indan-1 -il)-3,3-dimetil-piperazina o 1 -((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1 -il)-3,3-dimetil-piperazina;b) formación y opcionalmente precipitación de la sal farmacéuticamente aceptable mediante la adición del ácido correspondiente.
- 13. Un procedimiento según la reivindicación 12 en donde la sal farmacéuticamente aceptable de (±)-trans-1-(6-cloro-3-(fenil-d5)-indan-1 -il)-3,3-dimetil-piperazina, 1 -(6-cloro-3-(fenil-ds)-indan-1 -il)-3,3-dimetil-piperazina o 1 -((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1 -il)-3,3-dimetil-piperazina obtenida en la reivindicación 12 se convierte en una sal farmacéuticamente aceptable de 4-((1R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina, comprendiendo el procedimiento las siguientes etapasa) alquilación en presencia de un donante de metilo-d3 activo, tal como yoduro de metilo-d3 , y una base, tal como hidróxido de potasio, yb) precipitación de la sal farmacéuticamente aceptable mediante la adición del ácido correspondiente.
- 14. Un procedimiento según la reivindicación 11 en donde el 3,5-dicloro-1-(fenil-ds)-indano, en particular (±)-c/s-3,5-dicloro-1 -(fenil-d5)-indano o (1 S,3S)-3,5-dicloro-1 -(fenil-ds)-indano, obtenido en la reivindicación 11, se convierte en una sal farmacéuticamente aceptable de 4-((1R,3S)-6-cloro-3-(fenil-d5)-indan-1-il)-1(d3),2,2-trimetilpiperazina, comprendiendo el procedimiento las siguientes etapasa) sustitución nucleófila de 3,5-dicloro-1-(fenil-ds)-indano con bis-2,2,2-trifluoroacetato de 1(d3),2,2-trimetilpiperazina o un compuesto que posteriormente pueda transformarse en el resto 1(d3),2,2-trimetilpiperazina de 4-((1 R,3S)-6-cloro-3-(fenil-d5)-indan-1 -il)-1 (d3),2,2-trimetilpiperazinab) precipitación de la sal farmacéuticamente aceptable mediante la adición del ácido correspondiente.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261739095P | 2012-12-19 | 2012-12-19 | |
| DKPA201200811 | 2012-12-19 | ||
| PCT/EP2013/077314 WO2014096151A2 (en) | 2012-12-19 | 2013-12-19 | 6-chloro-3-(phenyl-d5)-inden-1-one and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2777181T3 true ES2777181T3 (es) | 2020-08-04 |
Family
ID=71783777
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13811511T Active ES2777181T3 (es) | 2012-12-19 | 2013-12-19 | 6-cloro-3-(fenil-d5)-inden-1-ona y su uso |
Country Status (3)
| Country | Link |
|---|---|
| ES (1) | ES2777181T3 (es) |
| HU (1) | HUE048382T2 (es) |
| ME (1) | ME03635B (es) |
-
2013
- 2013-12-19 ME MEP-2020-38A patent/ME03635B/me unknown
- 2013-12-19 ES ES13811511T patent/ES2777181T3/es active Active
- 2013-12-19 HU HUE13811511A patent/HUE048382T2/hu unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ME03635B (me) | 2020-07-20 |
| HUE048382T2 (hu) | 2020-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2013366693B2 (en) | 6-chloro-3-(phenyl-d5)-inden-1-one and use thereof | |
| JP5671456B2 (ja) | 3座配位子を有する新規ルテニウムカルボニル錯体、並びにその製造法及び用途 | |
| US5066815A (en) | Process for preparing optically active alcohol | |
| US6043387A (en) | Chiral bisphosphines | |
| JPS63316742A (ja) | 光学活性アルコ−ルの製造法 | |
| TW200813079A (en) | Method for making optically active amino phosphinylbutanoic acids | |
| AU2011301115B2 (en) | Biaryl diphosphine ligands, intermediates of the same and their use in asymmetric catalysis | |
| ES2777181T3 (es) | 6-cloro-3-(fenil-d5)-inden-1-ona y su uso | |
| US20020042540A1 (en) | New 6,6'-bis-(1-phosphanorbornadiene) diphosphines, their preparation and their uses | |
| JPH09208558A (ja) | 光学活性4−ヒドロキシ−2−ピロリドンの製造方法 | |
| JP5232374B2 (ja) | 2位に置換基を有する光学活性キヌクリジノール類の製造方法 | |
| JP3431749B2 (ja) | 新規な1−置換−2−ジフェニルホスフィノナフタレンおよびこれを配位子とする遷移金属錯体 | |
| JP2012224600A (ja) | 2−メントキシエタノールの製造方法 | |
| JP4845470B2 (ja) | 光学活性アミノアルコール類の製造方法 | |
| JPH05194568A (ja) | 新規なホモキラルジホスフィン類 | |
| WO2019031232A1 (ja) | 2,3-ビスホスフィノピラジン誘導体の製造方法及びホスフィン遷移金属錯体の製造方法 | |
| JP2004099609A (ja) | 光学活性7−アミノ−5−アザスピロ[2.4]ヘプタンの製造法 | |
| Mokotoff et al. | Potential inhibitors of L-asparagine biosynthesis. I.. beta.-Elimination reactions with. beta.-hydroxyaspartic acid derivatives | |
| Ibn El Alami et al. | α-Amino-Oximes Based on Optically Pure. | |
| Brunner et al. | Stabilization of the labile metal configuration in halfsandwich complexes [CpRh (PN) Hal] X | |
| JPH0670016B2 (ja) | (+)−n−ホルミル−1−(4−メトキシフェニルメチレン)−1,2,3,4,5,6,7,8−オクタヒドロイソキノリンの製造方法 | |
| WO2009044130A1 (en) | Biaryl diphosphines | |
| JP2008505152A (ja) | 光学活性アルキルコハク酸モノアルキルエステルの製造法 |