EP2155789B1 - Immunoglobulin fc libraries - Google Patents
Immunoglobulin fc libraries Download PDFInfo
- Publication number
- EP2155789B1 EP2155789B1 EP08747239.5A EP08747239A EP2155789B1 EP 2155789 B1 EP2155789 B1 EP 2155789B1 EP 08747239 A EP08747239 A EP 08747239A EP 2155789 B1 EP2155789 B1 EP 2155789B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- domain
- polypeptide
- fcr
- antibody
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Not-in-force
Links
Images


















































Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56911—Bacteria
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/72—Increased effector function due to an Fc-modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/035—Fusion polypeptide containing a localisation/targetting motif containing a signal for targeting to the external surface of a cell, e.g. to the outer membrane of Gram negative bacteria, GPI- anchored eukaryote proteins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
Definitions
- the present invention relates generally to the field of protein engineering. More particularly, it concerns improved methods and compositions for the screening of combinatorial antibody Fc libraries expressed in bacteria.
- mAbs Monoclonal antibodies
- Monoclonal antibodies comprise the majority of recombinant proteins currently in the clinic, with more than 150 products in studies sponsored by companies located worldwide (Pavlou and Belsey, 2005).
- the mAb market is heavily focused on oncology and arthritis, immune and inflammatory disorders, and products within these therapeutic areas are set to continue to be the key growth drivers over the forecast period.
- genetically engineered mAbs generally have higher probability of FDA approval success than small-molecule drugs. At least 50 biotechnology companies and all the major pharmaceutical companies have active antibody discovery programs in place.
- PCR Polymerase Chain Reaction
- Anchored Periplasmic Expression is based on anchoring the antibody fragment on the periplasmic face of the inner membrane of E.coli followed by disruption of the outer membrane, incubation with fluorescently labeled target and sorting of the spheroplasts ( U.S. Patent 7,094,571 ).
- APEx was used for the affinity maturation of antibody fragments (Harvey et al., 2004; Harvey et al., 2006). In one study over 200-fold affinity improvement was obtained after only two rounds of screening.
- ADCC antibody dependent cytotoxicity
- FcRs Fc receptors
- humans contain five different classes of Fc receptors.
- haplotypes, or genetic variants of different FcRs belonging to a particular class are known.
- the binding of an antibody to FcRs determines its ability to recruit other immunological cells and the type of cell recruited. Hence, the ability to engineer antibodies that can recruit only certain kinds of cells can be critically important for therapy.
- mammalian antibodies with engineered Fc regions display increased binding to a particular FcR of interest but in addition they are still capable of binding to other FcRs with normal affinity.
- Such antibodies are more selective than the molecules naturally produced by the immune system they can nonetheless still mediate undesirable immunological responses.
- E.coli possesses a reducing cytoplasm that is unsuitable for the folding of proteins with disulfide bonds which accumulate in an unfolded or incorrectly folded state (Baneyx and Mujacic, 2004).
- the periplasm of E . coli is maintained in an oxidized state that allows the formation of protein disulfide bonds.
- periplasmic expression has been employed successfully for the expression of antibody fragments such as Fvs, scFvs, Fabs or F(ab')2s (Kipriyanov and Little, 1999). These fragments can be made relatively quickly in large quantities with the retention of antigen binding activity.
- antibody fragments lack the Fc domain, they do not bind the FcRn receptor and are cleared quickly; thus, they are only occasionally suitable as therapeutic proteins (Knight et al., 1995).
- full-length antibodies could only be expressed in E. coli as insoluble aggregates and then refolded in vitro (Boss et al., 1984; Cabilly et al., 1984).
- This approach is not amenable to the high throughput screening of antibody libraries since with the current technology it is not possible to refold millions or tens of millions of antibodies individually.
- a further problem is that since E.
- coli expressed antibodies are not glycosylated, they fail to bind to complement factor 1q (C1q) or Fc and many other Fc receptors.
- C1q complement factor 1q
- FcRn neonatal Fc receptor efficiently
- bacterially expressed aglycosylated antibodies do exhibit serum persistence and pharmacokinetics similar to those of fully glycosylated IgGs produced in human cells. Nonetheless, since the aglycosylated antibodies fail to elicit complement activation and can not mediate the recruitment of immune cells such as macrophages, they have previously been ineffective for many therapeutic applications.
- the present invention overcomes a major deficiency in the art in providing aglycosylated antibody Fc domains that bind to Fc receptors and providing methods for the screening and production thereof.
- a method of selecting a bacterial cell comprising an aglycosylated antibody Fc domain having specific affinity for an Fc receptor (FcR) polypeptide comprising the steps of: (a) obtaining a population of Gram negative bacterial cells, cells of which population express an aglycosylated antibody Fc domain in their periplasm, wherein the population expresses a plurality of different Fc domains; (b) contacting the bacterial cells with an FcR polypeptide under conditions wherein the FcR polypeptide contacts the aglycosylated Fc domains; and (c) selecting at least one bacterial cell based on binding of the aglycosylated Fc domain to the FcR polypeptide.
- FcR Fc receptor
- a gram negative bacterial cell of the invention may be defined as an E . coli cell.
- a Gram negative bacterial cell in the context of the invention may be defined as a genetically engineered bacterial cell such as a Jude-1 strain of E . coli.
- Gram negative bacterial cells in the context of the invention are viable bacterial cells.
- the invention involves disrupting, permeablizing or removing the outer membrane of bacteria are well known in the art, for example, see U.S. Patent 7,094,571 .
- the outer membrane of the bacterial cell may be treated with hyperosmotic conditions, physical stress, lysozyme, EDTA, a digestive enzyme, a chemical that disrupts the outer membrane, or by infecting the bacterium with a phage or a combination of the foregoing methods.
- the outer membrane may be disrupted by lysozyme and EDTA treatment.
- the bacterial outer membrane may be removed entirely.
- an antibody Fc domain that is comprised in the bacterial periplasm may be defined as comprising a hinge, CH2 and CH3 region.
- Fc domains of the invention comprise a functional domain fragment.
- the term functional domain fragment means that antibody Fc domain that comprises amino acid deletions relative to wild-type sequence but nonetheless is able to bind to an FcR polypeptide.
- an antibody Fc domain for use in the invention may be an IgA, IgM, IgE, IgD or IgG antibody Fc domain or a variant thereof.
- an antibody of the invention is an IgG antibody Fc domain such as an IgG1, IgG2a, IgG2b, IgG3 or IgG4 antibody Fc domain.
- the antibody Fc domain may be defined as a human Fc domain.
- the Fc domain may be an IgG1 Fc domain, specifically, the Fc domain of an anti-HER2 antibody, more specifically, the Fc domain of trastuzumab.
- a Gram negative bacterial cell in the context of the invention further comprises a nucleic acid sequence encoding an antibody Fc domain.
- the encoded antibody may be any of the antibody Fc domains defined herein.
- a nucleic acid of the invention comprises sequences that facilitate Fc export into the periplasmic space. Such sequences are well known in the art and may comprise a secretion signal fused to the Ig chain ( U.S. Patent Publ. 20030180937 and 20030219870 ).
- an antibody Fc domain encoding nucleic acid may comprise additional elements such as an origin of replication or a selectable marker gene.
- the Fc domain encoding sequences are flanked by known sequences such that the Ig sequence may be amplified by PCR using primers that anneal to the known sequence.
- a nucleic acid sequence encoding an Fc domain of the invention will comprise sequences that mediate periplasmic expression, such as a secretion signal.
- a secretion signal such as a secretion signal.
- a dual arginine secretion signal may be used.
- the secretion signal is from PelB.
- the dsbA secretion signal or any other signal peptide capable of co-translational secretion may be used in order to achieve higher expression.
- Gram negative bacterial cells for use in the invention comprise a plurality of distinct Fc domain sequences.
- a "distinct Fc domain” may be defined as a domain that differs from another Fc by as little as one amino acid.
- Methods for making a library of distinct antibody Fc domains or nucleic acids that encode antibodies are well known in the art and exemplified herein.
- Fc domains may be amplified by error prone PCR as exemplified herein.
- a plurality of antibody Fc domains may comprise a stretch (1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) amino acids that have been randomized.
- residues that are normally glycosylated in an antibody Fc domain may be mutated.
- residues that are normally glycosylated (or adjacent residues) may be used as a site for an insertion of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more amino acids.
- an amino acid insertion may be made at, or adjacent to, a residue corresponding to amino acid 384 of the IgG1 Fc (SEQ ID NO:1).
- a population of gram negative bacteria in the context of the invention may be defined as comprising at least about 1x10 3 , 1x10 4 , 1x10 5 , 1x10 6 , 1x10 7 , 1x10 8 , or more distinct antibodies Fc domains.
- a population of Gram negative bacterial cells may be produced by a method comprising the steps of: (a) preparing a plurality of nucleic acid sequences encoding a plurality of distinct antibody Fc domains; and (b) transforming a population of Gram negative bacteria with said nucleic acids wherein the Gram negative bacteria comprise a plurality of antibody Fc domains expressed in the periplasm.
- an FcR may have specificity for a particular type or subtype of Ig, such as IgA, IgM, IgE or IgG (e.g ., IgG1, IgG2a, IgG2b, IgG3 or IgG4).
- IgA IgA
- IgM IgM
- IgE IgG
- IgG1 IgG2a
- IgG2b IgG3 or IgG4
- an antibody Fc-binding domain may be defined as a mammalian, bacterial or synthetic binding domain.
- Some Fc-binding domains for use in the invention include but are not limited to a binding domain from one of the polypeptides of Table 1.
- an Fc-binding polypeptide may be encoded by an FCGR2A, FCGR2B, FCGR2C, FCGR3A, FCGR3B, FCGR1A, Fcgr1, FCGR2, FCGR2, Fcgr2, Fcgr2, FCGR3, FCGR3, Fcgr3, FCGR3, Fcgr3, FCGRT, mrp4, spa or spg gene.
- an FcR polypeptide for use according to the invention may be an Fc binding region from human Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIb, Fc ⁇ RIIc, Fc ⁇ RIIIa, Fc ⁇ RIIIb, Fc ⁇ RI or C1q.
- an Fc polypeptide may be anchored to the inner membrane of a Gram negative bacteria.
- Methods and compositions for the anchoring of polypeptides to the inner membrane of Gram negative bacterial have previously been described ( U.S. Patent 7,094,571 and U.S. Patent Publ. 20050260736 ).
- an Fc domain may be fused to a polypeptide that is associated with or integrated in a bacterial inner membrane.
- Such a fusion protein may comprise an N terminal or C terminal fusion with an Fc domain and in some case may comprise additional linker amino acids between the membrane anchoring polypeptide and the Fc domain.
- a membrane anchoring polypeptide may be the first six amino acids encoded by the E .
- a membrane anchoring polypeptide may be an inner membrane lipoprotein or fragment thereof such as from AraH, Mglc, MalF, MalG, MalC, MalD, RbsC, RbsC, ArtM, ArtQ, GlnP, ProW, HisM, HisQ, LivH, LivM, LivA, LivE, DppB, DppC, OppB, AmiC, AmiD, BtuC, ThuD, FecC, FecD, FecR, FepD, NikB, NikC, CysT, CysW, UgpA, UgpE, PstA, PstC, PotB, PotC, PotH, Pod, ModB, NosY, Ph
- an FcR may be immobilized on a column or bead (e.g ., a magnetic bead) and the bacterial cell binding to the FcR separated by repeated washing of the bead ( e.g ., magnetic separation) or column.
- a target ligand may be labeled such as with a fluorophor, a radioisotope or an enzyme.
- bacterial cells may, in some cases, be selected by detecting a label on a bound FcR.
- a fluorophore may be used to select cells using fluorescence activated cell sorting (FACS).
- FACS fluorescence activated cell sorting
- bacterial cells may be selected based on binding or lack of binding two or more FcR polypeptides.
- bacteria may be selected that display antibodies that bind to two FcR polypeptides, wherein each FcR is used to select the bacterial sequentially.
- bacteria may be selected that display antibody Fc domains that bind to one FcR (such as an FcR comprising a first label) but not to a second FcR (e.g ., comprising a second label).
- the foregoing method maybe used, for example, to identify antibody Fc domains that bind to a specific FcR but not a second specific FcR.
- methods for producing bacteria of the invention may comprise at least two rounds of selection (step c) wherein the sub-population of bacterial cells obtained in the first round of selection is subjected to at least a second round of selection based on the binding of the candidate antibody Fc domain to an FcR.
- the sub-population of bacterial cells obtained in the first round of selection may be grown under permissive conditions prior to a second selection (to expand the total number of cells).
- methods of the invention may comprise 2, 3, 4, 5, 6, 7, 8, 9, 10 or more rounds of selection.
- a sub-population of bacterial cells obtained from each round of selection will be grown under permissive conditions before a subsequent round of selection.
- Cells isolated following one or more such rounds of selection may be subjected to additional rounds of mutagenesis.
- selection will be performed after removing FcR polypeptide that is not bound to the antibody.
- the stringency of selection may be modified by adjusting the pH, salt concentration, or temperature of a solution comprising bacteria that display antibodies.
- a bacterial cell in the context of the invention is grown at a sub-physiological temperature such as at about 25 °C.
- a method of producing a bacterial cell according to the invention may be further defined as a method of producing a nucleic acid sequence encoding an Fc domain that binds to at least a first FcR.
- a bacterial cell produced by the methods herein may be used to clone a nucleic acid sequence encoding the Fc domain having a specific affinity for an FcR polypeptide.
- Methods for isolating and amplifying such a nucleic acid from a cell for example by PCR are well known in the art and further described below.
- a nucleic acid sequence produced by the forgoing methods is included as part of the instant invention.
- the invention involves a method for producing an Fc domain having a specific affinity for an FcR.
- the invention includes antibody Fc domains produced by the methods of the invention. It will be understood however that the antibody Fc domains produced by such a screen may be combine with antibody variable regions that have an affinity for a particular target ligand and these antibodies are also included as part of the invention.
- the invention provides a polypeptide comprising an aglycosylated antibody Fc domain capable of binding an FcR polypeptide as defined in the claims.
- the aglycosylated Fc domain may be further defined as having a specific affinity for an FcR polypeptide under physiological conditions. For instance an Fc domain may have an equilibrium dissociation constant between about 10 -6 M to about 10 -9 M under physiological conditions.
- an aglycosylated Fc domain may be defined as comprising one or more amino acid substitution or insertion relative to a wild type human sequence.
- a preferred means of preparing such a polypeptide is through the practice of the methods discussed above.
- one can alternatively prepare such polypeptides directly by genetic engineering techniques such as, for example, by introducing selected amino acid substitutions or insertions into a known Fc background, wherein the insertion or substitution provides an improved FcR binding capability to aglycosylated Fc regions.
- the inventors have identified as particularly preferred substitutions for achieving such improved FcR binding as those at positions 331, 382 and/or 428 of the Fc domain (for example, see Nagaoka and Akaike 2003; such as P331, E382 and/or M428 of the human IgG Fc domain sequence as shown in FIG. 46 and also in, e.g ., U.S. Patent Publ. US20060173170 ), and still more preferred are one or more substations defined by P331L, E382V, M428I or M428L.
- Preferred substitutions may further include one or more of 426, 229, 322, 350, 361, 372, 442, 402, 224, 430, 238, 436, 310, 313, 384, 372, 380 or 331 of the Fc domain, such as S426, C229, K322, T350, N361, F372, S442, G402, H224, E430, P238, Y436, H310, W313, N384, F372, E380 or P331 of the human IgG Fc domain, with the specific preferred examples being a) E382 and M428; b) N361, E382 and M428; c) N361, F372, E382 and M428; d) H310, K322, T350, E382, S426 and S442; e) C229R, E382 and M428; f) W313 and M428; g) E382, N384 and M428; h) E380, E382 and N384
- E382V and M428I include a) E382V and M428I; b) E382V; c) N361D, E382V and M428I; d) N361D, F372L, E382V and M428I; e) H310Y, K322R, T350A, E382V, S426T and S442P; f) C229R, E382V and M428I; g) W313R and M428I; h) E382T, N384D and M428I; i) E380R, E382M and N384E; j) N361S, E382V and M428I; k) E382V, M428I and Y436A; 1) P238S, E382V, S426V, M428L and E430H; m) E380D, E382V, N384R, S426V, M
- the inventors have also identified various insertion points that upon insertion of additional amino acids, provide improved FcR binding capability. Most preferred in this regard are insertions of 5 to 15 amino acids, and preferably 10 amino acids, between amino acids N297 and S298 of an Fc domain, such as a human IgG Fc domain.
- Particularly preferred insertions at this position include a) RTETPVYMVM (SEQ ID NO:60); b) WQVFNKYTKP (SEQ ID NO:61); c) LGDGSPCKAN (SEQ ID NO:62); d) EVPLVWMWVS (SEQ ID NO:63) together with F241L and K326E; and e) EQWGSQFGCG (SEQ ID NO:64) together with V282A.
- the Fc domain of the invention may be a human IgG Fc that comprises an amino acid substitution at an amino acid residue corresponding to E382 of the IgG Fc domain.
- an aglycosylated Fc domain may comprise an amino acid sequence insertion ( e.g. , about 1 to 5 amino acids) adjacent to an amino acid residue corresponding to E382 of the IgG Fc domain.
- an Fc domain may comprise a hydrophobic amino acid substitution at E382 such as an E to V substitution.
- an Fc domain of the invention may comprise an amino acid substitution at a residue corresponding to M428 (e.g., M428 to I), S426, C229, H310, K322, T350, N361, F372 or S442 of the human IgG Fc.
- an aglycosylated Fc domain may comprise an amino acid substitution corresponding to those found in the Fc11 (SEQ ID NO:2), Fc5 (SEQ ID NO:3), Fc12 (SEQ ID NO:4), Fc 20 (SEQ ID NO:5), Fc49 (SEQ ID NO:6) or Fc23 Fc (SEQ ID NO:7) domains described herein (see FIG. 14 ).
- an aglycosylated Fc domain may comprise the amino acid sequence of Fc11 (SEQ ID NO:2), Fc5 (SEQ ID NO:3), Fc12 (SEQ ID NO:4), Fc 20 (SEQ ID NO:5), Fc49 (SEQ ID NO:6), Fc23 (SEQ ID NO:7), Fc104 (SEQ ID NO:65), Fc106 (SEQ ID NO:66), Fc110 (SEQ ID NO:67), Fc114 (SEQ ID NO:68), Fc117 (SEQ ID NO:69), Fc143 (SEQ ID NO:70), Fc149 (SEQ ID NO:71), Fc151 (SEQ ID NO:72), Fc152 (SEQ ID NO:73), Fc207 (SEQ ID NO:74), Fc209 (SEQ ID NO:75), Fc216 (SEQ ID NO:76), Fc217 (SEQ ID NO:77), Fc236 (SEQ ID NO:78), Fc331 (SEQ ID NO:2), F
- polypeptides described herein may comprise an Ig variable domain and may be further defined as a full length antibody.
- an aglycosylated Fc domain of the invention comprises a specific binding affinity for an FcR such as human Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIb, Fc ⁇ RIIc, Fc ⁇ RIIIa, Fc ⁇ RIIIb, Fc ⁇ RI or C1q.
- an aglycosylated Fc domain of the invention is defined as an Fc domain with a specific affinity for Fc ⁇ RIa.
- such an Fc domain may be defined as having an equilibrium dissociation constant, with respect to Fc ⁇ RIa binding, of about 10 -6 M to about 10 -9 M under physiological conditions.
- a still further aspect of the invention includes isolated DNA segments encoding a polypeptide in accordance with any one of the foregoing modified Fc regions as well as antibodies, etc., incorporating such a polypeptide.
- DNA segments may preferably be positioned in an expression vector, which is preferably a bacterial expression vector.
- a bacterial growth media that comprises trehalose.
- such a media may be used in a method A method of identifying a bacteria cell comprising a first binding partner associated with an inner membrane comprised in the bacteria cell, wherein the binding partner having specific affinity for a second binding partner, comprising the steps of: a) obtaining a population of bacteria cells, cells of which population comprise the first binding partner associated with the inner membrane in the periplasm of the bacteria cells, wherein the population comprises a plurality of different such first binding partners; b) contacting the bacteria cells with the second binding partner, wherein the first binding partner or the second binding partner comprises a label, wherein a signal is elicited when the first binding partner binds to the second binding partner; and c) selecting at least one bacterial cell by detecting such a signal from at least such a first binding partner binding to at least such second binding partner.
- the signal may be a fluorescent signal.
- a media comprising trehalose as demonstrated herein, provides enhanced fluorescence signal and greatly improves the screening process.
- methods for the used of the trehalose bacterial media in screening such binding partners are included as part of the instant disclosure. Any of the fluorescence screening methods known in the art or described herein may be used in combination with a trehalose bacterial media of the disclosure.
- a fluorescence signal may be detected by flow cytometry.
- bacteria comprising binding partners for detection may have their outer r membrane disrupted or partially disrupted.
- the one of the binding partners for use in the instant methods may be defined as an antibody or an antibody domain.
- a bacterial growth media comprising trehalose may be further defined based upon the trehalose concentration in the media.
- a media comprising about between about 0.05 and 1.5M trehalose or preferably between about 0.1 and 1.0 M trehalose is specifically contemplated herein.
- bacterial media comprising about 0.5 M trehalose is provided.
- Embodiments discussed in the context of a methods and/or composition of the invention may be employed with respect to any other method or composition described herein. Thus, an embodiment pertaining to one method or composition may be applied to other methods and compositions of the invention as well.
- FIG. 1 Two plasmids system for the periplasmic display of Fc using cJun-cFos or cJun(Cys)-cFos(Cys) interaction.
- FIG. 2a -b FACS analysis results of periplasmic displayed Fc homodimer using cJun-cFos and cJun(Cys)-cFos(Cys) interaction pairs.
- FIG. 2a FACS signals of periplasmic displayed Fc using cJun-cFos and cJun(Cys)-cFos(Cys) were compared with a positive and a negative controls.
- FACS signals of periplasmic displayed Fc using cJun-cFos and cJun(Cys)-cFos(Cys) were compared with one plasmid systems not co-expressing NlpA and 6 amino acid residues (CDQSSS (SEQ ID N:84)) fused cJun or cJun(Cys).
- Spheroplasts were incubated with Protein A-FITC probe for detection.
- Mn Mean fluorescence intensity.
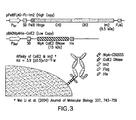
- FIG. 3 Two plasmids system for the periplasmic display of Fc using ColE2-Im2 interaction.
- FIG. 4a -b FACS analysis results for the periplasmic display of Fc homodimer using ColE2-Im2 interaction pairs.
- FIG. 4a Display of Im2 fused M18 scFv or 26-10 scFv co-expressed with APEx displayed ColE2(H578A) and incubated with PA-FITC.
- FIG. 4b Display of Im2 fused M18 scFv or 26-10 scFv co-expressed with APEx displayed ColE2(H578A) and incubated with digoxin-BODIPY.
- Mn Mean fluorescence intensity.
- FIG. 5 Effect of ColE2 for the expression of target proteins, M18 scFv (Lane 1-3), 26-10 scFv (Lane 4-6), and Fc (Lane 7-9).
- M18 scFv Lane 1-3
- 26-10 scFv Lane 4-6
- Fc Fc 7-9.
- lane 1 4 and 7, Im2 fused proteins were co-expressed with APEx displayed ColE2(H578A).
- lane 2 5 and 7, Im2 fused proteins were expressed without APEx displayed ColE2(H578A).
- lane 3 6 and 9, proteins without Im2 fusions were co-expressed APEx displayed ColE2(H578A).
- Anti-ECS antibody peroxidase conjugated was used as a detection antibody for Western blot.
- FIG. 6 Effect of sugars (sorbitol and trehalose) on the FACS analysis for periplasmic displayed Fc or APEx displayed Fc. Spheroplasts were incubated with Protein A-FITC probe for detection. Mn: Mean fluorescence intensity.
- FIG. 7 Effect of trehalose on the periplasmic display of Fc.
- M18 scFv was used as a negative control. Spheroplasts were incubated with Protein A-FITC probe for detection. Mn: Mean fluorescence intensity.
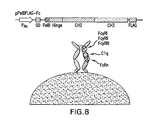
- FIG. 8 One plasmid system for the periplasmic display of trapped Fc with trehalose.
- FIG. 9a -b Effect of trehalose on the expression level and the rentention after spheroplasting for homodimeric Fc.
- FIG.9a Western blot result from reduced gel for the periplasmic expressed Fc and M18 scFv cultured in the media with or without trehalose.
- FIG.9b Western blot result from reduced or non-reduced gel for the periplasmic expressed Fc cultured in the media with or without trehalose.
- Anti-ECS antibody peroxidase conjugated was used as a detection antibody for Western blot.
- FIG. 10a -b Effect of signal leader peptides (PelB and dsbA) on the periplasmic display of Fc.
- FIG. 10a Comparison of FACS signals between PelB and dsbA fused proteins. PelB or dsbA signal peptide fused proteins were cultured with 0.5M trehalose.
- FIG. 10b Comparison of FACS signals between with and without trehalose in the media. DsbA signal peptide fused proteins were cultured with or without 0.5M trehalose.
- Mn Mean fluorescence intensity. Spheroplasts were incubated with Protein A-FITC probe for detection.
- FIG. 11 FACS analysis for the periplasmic displayed antibodies.
- FIG. 12a -b Fluorescence ELISA to detect affinity of FITC labeled Fc ⁇ RIa for IgG-Fc.
- FIG. 12a IgG-Fc was coated onto fluorescence ELISA plate. The fluorescence of serially diluted and bound Fc ⁇ RIa-FITC was detected at excitation 485 nm and emission 528 nm.
- FIG. 12b Fluorescence signals of serially diluted Fc ⁇ RIa-FITC in the IgG-Fc coated wells compared to the signals in the BSA coated wells.
- FIG. 13a -b Fc library screening using FACS sorting.
- FIG. 13a Histogram showing enrichment of high affinity clones sorted by Fc ⁇ RIa-FITC.
- FIG. 13b Histogram showing fluorescence signals of Fc mutants comparing with wild type Fc. Spheroplasts were incubated with Fc ⁇ RIa-FITC for detection.
- Mn Mean fluorescence intensity.
- FIG. 14 Sequences of isolated Fc mutant clones exhibiting high affinity to Fc ⁇ RIa. Depicted sequences are as follows used in the experiment with a FLAG tag attached to the C-terminal end, wt-IgG1 Fc, SEQ ID NO:1, Fc11, SEQ ID NO:2; Fc5, SEQ ID NO:3; Fc12, SEQ ID NO:4; Fc20, SEQ ID NO:5; Fc49, SEQ ID NO:6; and Fc23, SEQ ID NO:7;
- FIG. 15a -b Mutation points of isolated aglycosylated Fcs in 3D structure of glycosylated IgG (PBD Code: 1FC1).
- FIG. 15a Major mutation points in full glycosylated IgG.
- FIG. 15b Interaction of two beta sheets including 382E and 428M in the CH3 region.
- FIG. 16 Fc library comprising 3 kinds of sub-libraries randomized and inserted around 382E and 428M.
- FIG. 17 Histogram showing enrichment of clones showing high affinity to Fc ⁇ RIa by FACS sorting from the library randomized around 382E and 428M in FIG. 16 .
- FIG. 18 Sequence of isolated Fc mutant clones exhibiting high affinity to Fc ⁇ RIa. Spheroplasts were incubated with Fc ⁇ RIa-FITC for detection. FACS mean values are indicated in the parenthesis.
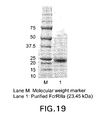
- FIG. 19 SDS-PAGE of purified and refoled Fc ⁇ RIIIa from E. coli inclusion bodies.
- FIG. 20 Histogram showing enrichment of high affinity clones sorted by Fc ⁇ RIIIa-FITC.
- FIG. 21 Histogram showing fluorescence signals of Fc mutants comparing with wild type Fc. Spheroplasts were incubated with Fc ⁇ RIIIa-FITC for detection. M: Mean fluorescence intensity.
- FIG. 22 Sequences of isolated Fc mutant clones exhibiting high affinity to Fc ⁇ RIIIa.
- FIG. 23 Histogram showing enrichment of high affinity clones sorted by Fc ⁇ RIIa-FITC
- FIG. 24 Histogram showing fluorescence signals of Fc mutants comparing with wild type Fc. Spheroplasts were incubated with Fc ⁇ RIIa-FITC for detection. M: Mean fluorescence intensity.
- FIG. 25 Sequences of isolated Fc mutant clones exhibiting high affinity to Fc ⁇ RIIIa.
- FIG. 26 SDS-PAGE showing the expression of wild type Fc ⁇ RIIa and codon optimized Fc ⁇ RIIa, Lane 1: Wild type Fc ⁇ RIIa; Lane 2: codon optimized Fc ⁇ RIIa.
- FIG. 27 SDS-PAGE showing the localization of codon optimized Fc ⁇ RIIa, Lane 1: Total fraction; Lane 2: soluble fraction; Lane 3: insoluble fraction.
- FIG. 28 SDS-PAGE showing the purified Fc ⁇ RIIa. Lane 1: purified Fc ⁇ RIIa.
- FIG. 29 ELISA result of Fc mutants to Fc ⁇ RIIa from the media fraction of cultured Jude-1 cells harboring pDsbAFLAG-Fc mutant plasmids.
- FIG. 30 Soluble expression ofhomodimeric wild type Fc and Fc mutants (5 ml tube culture). Wild type Fc with two different signal peptides (PelB and DsbA) was expressed at different culture temperatures after induction and was harvested at different times. The localization of the protein was also analyzed.
- FIG. 31 Soluble expression of homodimeric wild type Fc and Fc mutants (500 ml flask culture). DsbA leader peptide fused wild type Fc was expressed at different culture temperatures and culture time after induction. The localization of the protein was also analyzed.
- FIG. 32 SDS-PAGE of wild type Fc and Fc mutants purified with Protein A affinity chromatography.
- FIG. 33a -d Chromatogram of wild type Fc ( FIG. 33a ) and Fc mutants using Supedex 200 gel filtration chromatography, including Fc5 ( FIG. 33b ), Fc11 ( FIG. 33c ) and Fc49 ( FIG. 33d ).
- FIG. 34 SDS-PAGE of wild type Fc and Fc mutants purified with Superdex 200 gel filtration chromatography.
- FIG. 35 Direct coating ELISA for the detection of affinity of Fc mutants to Fc ⁇ Rs
- FIG. 36 ELISA result of Fc mutants to Fc ⁇ RI.
- FIG. 37 SPR Sensorgrams of Fc protein binding onto immobilized Fc ⁇ RI.
- FIG. 38 Map of plasmid pSTJ4-HerceptinTM IgG1.
- FIG. 39 Fed batch fermentation for the production of aglycosylated trastuzumab or trastuzumab-Fc5 in a 3.3 L fermenter with 1.2 liter working volume.
- the OD 600 is shown as a function of time after inoculation during the expression of trastuzumab in E . coli
- FIG. 40 Fully assembled IgG as detected by non-denaturing gel electrophoresis and Western bloting with goat anti-human IgG (H+L) antibodies. Results are shown for cells expressing wild type trasuzumab; similar results were obtained for cells expressing trastuzumab-Fc5.
- FIG. 41 Expression of aglycosylated trastuzumab and trastuzumab-Fc5, Lane 1: IgG1 standard; Lane 2: Before induction; Lane 3: aglycosylated trastuzumab; Lane 4: trastuzumab-Fc5.
- FIG. 42 SDS-PAGE showing the purified aglycosylated trastuzumab and trastuzumab-Fc5, Lane 1, 3: Wild type Fc aglycosylated trastuzumab; Lane 2, 4: trastuzumab-Fc5.
- FIG. 43 ELISA assays for binding to Fc ⁇ RIIa. Plates were coated with purified trastuzumab or trastuzumab-Fc5 and the binding of Fc ⁇ R was detected using anti-GST-HRP.
- FIG. 44 ELISA assays for binding to Fc ⁇ RIIb. Plates were coated with purified trastuzumab or trastuzumab-Fc5 and the binding of Fc ⁇ R was detected using either anti-polyhistidine-HRP or anti-GST-HRP.
- FIG. 45 ELISA assays for binding to FcRn at pH 7.4 and 5.5. Plates were coated with purified trastuzumab or trastuzumab-Fc5 and the binding of Fc ⁇ R was detected using anti-GST-HRP.
- FIG. 46 Alignment of sequences for human IgG subclassses (SEQ ID NOS: 110, 111, 112 and 113).
- the instant invention overcomes several major problems with current immunotherapeutic technologies in providing aglycosylated antibody Fc domains that are able to bind to Fc receptor polypeptides. Furthermore, now methods for identifying aglycosylated Fc domains capable of binding to Fc receptors are described. These methods enable isolation of antibody Fc domains that preferentially or selectively bind to specific Fc receptors. Thus, the new compositions and methods will enable manufacture of antibody therapeutics that may be produced in bacteria while retaining their ability to interact with FcR polypeptides and thereby recruit immune affecter cells. Furthermore, Fc receptors may be selected for a particular FcR binding affinity thereby allowing therapeutics to be tailored for recruitment or targeting of specific cell types. Finally, the instant invention provided new media and methods that may be used to enhance prokaryotic interaction screening techniques. Further embodiments and advantages of the invention are described below.
- a polypeptide comprising an antibody Fc domain is expressed in the periplasmic space of a gram negative bacteria.
- an antibody Fc domain may be anchored to the periplasmic face of the inner membrane.
- an Fc domain may be directly fused to a membrane spanning or membrane bound polypeptide or may interact (e.g., via protein-protein interactions) with a membrane spanning or membrane bound polypeptide.
- Such a technique may be termed "Anchored Periplasmic Expression" or "APEx”.
- the periplasmic compartment is contained between the inner and outer membranes of Gram negative cells (see, e.g ., Oliver, 1996). As a subcellular compartment, it is subject to variations in size, shape and content that accompany the growth and division of the cell.
- Within a framework of peptidoglycan heteroploymer is a dense mileau of periplasmic proteins and little water, lending a gel-like consistency to the compartment (Hobot et al., 1984; van Wielink and Duine, 1990).
- the peptidoglycan is polymerized to different extents depending on the proximity to the outer membrane, close-up it forms the murein sacculus that affords cell shape and resistance to osmotic lysis.
- the outer membrane (see Nikaido, 1996) is composed of phospholipids, porin proteins and, extending into the medium, lipopolysaccharide (LPS).
- LPS lipopolysaccharide
- the molecular basis of outer membrane integrity resides with LPS ability to bind divalent cations (Mg 2+ and Ca 2 ) and link each other electrostatically to form a highly ordered quasi-crystalline ordered "tiled roof" on the surface (Labischinski et al., 1985).
- the membrane forms a very strict permeability barrier allowing passage of molecules no greater than around 650 Da (Burman et al., 1972; Decad and Nikaido, 1976) via the porins.
- the large water filled porin channels are primarily responsible for allowing free passage of mono and disaccharides, ions and amino acids in to the periplasm compartment (Nikaido and Nakae, 1979; Nikaido and Vaara, 1985). With such strict physiological regulation of access by molecules to the periplasm it may appear, at first glance, inconceivable that large ligands (i.e ., larger than the 650 Da exclusion limit) could be employed in screening methods. However, the inventors have shown that ligands greater than 2000 Da in size can diffuse into the periplasm without disruption of the periplasmic membrane. Such diffusion can be aided by one or more treatments of a bacterial cell, thereby rendering the outer membrane more permeable, as is described herein below.
- methods are employed for increasing the permeability of the outer membrane to one or more labeled ligand.
- This can allow screening access of labeled ligands otherwise unable to cross the outer membrane.
- certain classes of molecules for example, hydrophobic antibiotics larger than the 650 Da exclusion limit, can diffuse through the bacterial outer membrane itself, independent of membrane porins (Farmer et al., 1999). The process may actually permeabilize the membrane on so doing (Jouenne and Junter, 1990).
- Such a mechanism has been adopted to selectively label the periplasmic loops of a cytoplasmic membrane protein in vivo with a polymyxin B nonapeptide (Wada et al., 1999).
- certain long chain phosphate polymers 100 Pi
- appear to bypass the normal molecular sieving activity of the outer membrane altogether Rao and Torriani, 1988).
- the permeability of the outer membrane of different strains of bacterial hosts can vary widely. It has been shown previously that increased permeability due to OmpF overexpression was caused by the absence of a histone like protein resulting in a decrease in the amount of a negative regulatory mRNA for OmpF translation (Painbeni et al., 1997). Also, DNA replication and chromosomal segregation is known to rely on intimate contact of the replisome with the inner membrane, which itself contacts the outer membrane at numerous points.
- a preferred host for library screening applications is E . coli ABLEC strain, which additionally has mutations that reduce plasmid copy number.
- Treatments such as hyperosmotic shock can improve labeling significantly. It is known that many agents including, calcium ions (Bukau et al., 1985) and even Tris buffer (Irvin et al., 1981) alter the permeability of the outer-membrane. Further, phage infection stimulates the labeling process. Both the filamentous phage inner membrane protein pIII and the large multimeric outer membrane protein pIV can alter membrane permeability (Boeke et al., 1982) with mutants in pIV known to improve access to maltodextrins normally excluded (Marciano et al., 1999).
- a high degree of permeability may be achieved (Daugherty et al., 1999).
- Cells comprising anchored or periplasm-associated polypeptides bound to fluorescently labeled ligands can then be easily isolated from cells that express binding proteins without affinity for the labeled ligand using flow cytometry or other related techniques.
- flow cytometry or other related techniques it will be desired to use less disruptive techniques in order to maintain the viability of cells.
- EDTA and Lysozyme treatments may also be useful in this regard.
- the invention concerns methods for identifying antibody Fc domains with a specific affinity for antibody-binding polypeptide such as an Fc receptor.
- Fc receptors are well known in the art and some examples of receptors are listed below in Table 1.
- Table 1 Selected FcR Polypeptides Protein name Gene name Description Organisms Length (aa) Reference Fc-gamma RII-a (CD32) FCGR2A Low affinity immunoglobulin gamma Fc region receptor II-a precursor Homo sapiens (Human) 317 (Stuart et al., 1987) Fc-gamma RII-a FCGR2A Low affinity immunoglobulin n gamma Fc region receptor II-a precursor Pan troglodytes (Chimpanzee) 316 Fc-gamma RII-b FCGR2B Low affinity immunoglobuli n gamma Fc region receptor II-b precursor Homo sapiens (Human) 310 (Stuart et al.
- Fc-gamma RII-c FCGR2C Low affinity immunoglobuli n gamma Fc region receptor II-c precursor Homo sapiens (Human) 323 (Stuart et al., 1989) Fc-gamma RIIIa FCGR3A Low affinity immunoglobuli n gamma Fc region receptor III-A precursor Homo sapiens (Human) 254 (Ravetch and Perussia, 1989) Fc-gamma RIIIb FCGR3B Low affinity immunoglobuli n gamma Fc region receptor III-B precursor Homo sapiens (Human) 233 (Ravetch and Perussia, 1989) Fc-gamma RI (CD64) FCGR1A High affinity immunoglobuli n gamma Fc receptor I precursor Homo sapiens (Human) 374 (Allen and Seed, 1988) Fc-gamma RI Fcgr1 High affinity immunoglobuli n gamma Fc receptor I precursor Mus
- group G 448 (Fahnestoc k et al., 1986) protein G spg Immunoglobuli n G-binding protein G precursor Streptococcus sp. group G 593 (Olsson et al., 1987) protein H Immunoglobuli n G-binding protein H precursor Streptococcus pyogenes serotype M1 376 (Gomi et al., 1990) Protein sbi sbi Immunoglobuli n G-binding protein sbi precursor Staphylococcu s aureus (strain NCTC 8325-4) 436 (Zhang et al., 1998) Allerge n Asp fl 1 Allergen Asp fl 1 causes an allergic reaction in human.
- Examples of techniques that could be employed in conjunction with the invention for creation of diverse antibody Fc domains and/or antibodies comprising such domains may employ techniques similar to those for expression of immunoglobulin heavy chain libraries described in U.S. Patent 5,824,520 .
- the present invention provides methods for identifying molecules capable of binding to a particular FcR.
- the binding polypeptides screened may comprise a large library of diverse candidate Fc domains, or, alternatively, may comprise particular classes of Fc domains (e.g ., engineered point mutations or amino acid insertions) selected with an eye towards structural attributes that are believed to make them more likely to bind the target ligand.
- the candidate binding protein is an intact antibody, or a fragment or portion thereof comprising an Fc domain.
- a candidate Fc domain capable of binding a target ligand in accordance with the invention, one may carry out the steps of: providing a population of Gram negative bacterial cells that express a distinct antibody Fc domain; admixing the bacteria or phages and at least a first labeled or immobilized target ligand (FcR polypeptide) capable of contacting the antibody and identifying at least a first bacterium expressing a molecule capable of binding the target ligand.
- FcR polypeptide first labeled or immobilized target ligand
- the binding between antibody Fc domain and a labeled FcR polypeptide will prevent diffusing out of a bacterial cell.
- molecules of the labeled ligand can be retained in the periplasm of the bacterium comprising a permeablized outer membrane.
- the periplasm can be removed, whereby the Fc domain will cause retention of the bound candidate molecule since Fc domains are shown to associate with the inner membrane.
- the labeling may then be used to isolate the cell expressing a binding polypeptide capable of binding the FcR polypeptide, and in this way, the gene encoding the Fc domain polypeptide isolated.
- the molecule capable of binding the target ligand may then be produced in large quantities using in vivo or ex vivo expression methods, and then used for any desired application, for example, for diagnostic or therapeutic applications, as described below.
- isolated antibody Fc domains identified may be used to construct an antibody fragment or full-length antibody comprising an antigen binding domain.
- binding affinity of an antibody Fc or other binding protein can, for example, be determined by the Scatchard analysis of Munson & Pollard (1980). Alternatively, binding affinity can be determined by surface plasmon resonance or any other well known method for determining the kinetics and equilibrium constants for protein:protein interactions. After a bacterial cell is identified that produces molecules of the desired specificity, affinity, and/or activity, the corresponding coding sequence may be cloned. In this manner, DNA encoding the molecule can be isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the antibody or binding protein).
- the antibody Fc domain DNA may be placed into expression vectors, which can then transfected into host cells such as bacteria.
- the DNA also may be modified, for example, by the addition of sequence for human heavy and light chain variable domains, or by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non-immunoglobulin polypeptide.
- "chimeric" or “hybrid” binding proteins are prepared to have the desired binding specificity.
- an identified antibody Fc domain may be fused to a therapeutic polypeptide or a toxin and used to target cells ( in vitro or in vivo ) that express a particular FcR.
- Chimeric or hybrid Fc domains also may be prepared in vitro using known methods in synthetic protein chemistry, including those involving crosslinking agents.
- targeted-toxins may be constructed using a disulfide exchange reaction or by forming a thioether bond.
- suitable reagents for this purpose include iminothiolate and methyl-4-mercaptobutyrimidate.
- nucleic acids may be cloned from viable or inviable cells.
- inviable cells for example, it may be desired to use amplification of the cloned DNA, for example, using PCR. This may also be carried out using viable cells either with or without further growth of cells.
- an Fc domain is isolated which has affinity for a labeled FcR polypeptide.
- labeled ligands of potentially any size may be screened. In the absence of removal of the periplasmic membrane, it will typically be preferable that the labeled ligand is less that 50,000 Da in size in order to allow efficient diffusion of the ligand across the bacterial periplasmic membrane.
- an FcR polypeptide which has been labeled with one or more detectable agent(s).
- This can be carried out, for example, by linking the ligand to at least one detectable agent to form a conjugate.
- a "label” or “detectable label” is a compound and/or element that can be detected due to specific functional properties, and/or chemical characteristics, the use of which allows the ligand to which it is attached to be detected, and/or further quantified if desired.
- labels which could be used with the invention include, but are not limited to, enzymes, radiolabels, haptens, fluorescent labels, phosphorescent molecules, chemiluminescent molecules, chromophores, luminescent molecules, photoaffinity molecules, colored particles or ligands, such as biotin.
- a visually-detectable marker is used such that automated screening of cells for the label can be carried out.
- fluorescent labels are beneficial in that they allow use of flow cytometry for isolation of cells expressing a desired binding protein or antibody.
- agents that may be detected by visualization with an appropriate instrument are known in the art, as are methods for their attachment to a desired ligand (see, e.g., U.S. Patents 5,021,236 ; 4,938,948 ; and 4,472,509 ).
- agents can include paramagnetic ions; radioactive isotopes; fluorochromes; NMR-detectable substances and substances for X-ray imaging.
- Types of fluorescent labels that may be used with the invention will be well known to those of skill in the art and include, for example, Alexa 350, Alexa 430, AMCA, BODIPY 630/650, BODIPY 650/665, BODIPY-FL, BODIPY-R6G, BODIPY-TMR, BODIPY-TRX, Cascade Blue, Cy3, Cy5,6-FAM, Fluorescein Isothiocyanate, HEX, 6-JOE, Oregon Green 488, Oregon Green 500, Oregon Green 514, Pacific Blue, REG, Rhodamine Green, Rhodamine Red, Renographin, ROX, TAMRA, TET, Tetramethylrhodamine, and/or Texas Red.
- Magnetic screening techniques are well known to those of skill in the art (see, for example, U.S. Pat. No. 4,988,618 , U.S. Pat. No. 5,567,326 and U.S. Pat. No. 5,779,907 ).
- paramagnetic ions that could be used as labels in accordance with such techniques include ions such as chromium (III), manganese (II), iron (III), iron (II), cobalt (II), nickel (II), copper (II), neodymium (III), samarium (III), ytterbium (III), gadolinium (III), vanadium (II), terbium (III), dysprosium (III), holmium (III) and/or erbium (III).
- Ions useful in other contexts include but are not limited to lanthanum (III), gold (III), lead (II), and especially bismuth (III).
- FcR conjugate contemplated in the present invention are those where the ligand is linked to a secondary binding molecule and/or to an enzyme (an enzyme tag) that will generate a colored product upon contact with a chromogenic substrate.
- enzymes include urease, alkaline phosphatase, (horseradish) hydrogen peroxidase or glucose oxidase. I n such instances, it will be desired that cells selected remain viable.
- Preferred secondary binding ligands are biotin and/or avidin and streptavidin compounds. The use of such labels is well known to those of skill in the art and are described, for example, in U.S. Patents 3,817,837 ; 3,850,752 ; 3,939,350 ; 3,996,345 ; 4,277,437 ; 4,275,149 and 4,366,241 .
- Molecules containing azido groups also may be used to form covalent bonds to proteins through reactive nitrene intermediates that are generated by low intensity ultraviolet light (Potter & Haley, 1983).
- 2- and 8-azido analogues of purine nucleotides have been used as site-directed photoprobes to identify nucleotide-binding proteins in crude cell extracts (Owens & Haley, 1987; Atherton et al., 1985).
- the 2- and 8-azido nucleotides have also been used to map nucleotide-binding domains of purified proteins (Khatoon et al., 1989; King et al., 1989; and Dholakia et al., 1989) and may be used as ligand binding agents.
- Labeling can be carried out by any of the techniques well known to those of skill in the art.
- FcR polypeptides can be labeled by contacting the ligand with the desired label and a chemical oxidizing agent such as sodium hypochlorite, or an enzymatic oxidizing agent, such as lactoperoxidase.
- a ligand exchange process could be used.
- direct labeling techniques may be used, e.g ., by incubating the label, a reducing agent such as SNCl 2 , a buffer solution such as sodium-potassium phthalate solution, and the ligand.
- Intermediary functional groups on the ligand could also be used, for example, to bind labels to a ligand in the presence of diethylenetriaminepentaacetic acid (DTPA) or ethylene diaminetetracetic acid (EDTA).
- DTPA diethylenetriaminepentaacetic acid
- EDTA ethylene diaminetetracetic acid
- Some attachment methods involve the use of an organic chelating agent such as diethylenetriaminepentaacetic acid anhydride (DTPA); ethylenetriaminetetraacetic acid; N-chloro-p-toluenesulfonamide; and/or tetrachloro-3 ⁇ -6 ⁇ -diphenylglycouril-3 attached to the ligand ( U.S. Patents 4,472,509 and 4,938,948 .).
- FcR polypeptides also may be reacted with an enzyme in the presence of a coupling agent such as glutaraldehyde or periodate.
- Conjugates with fluorescein markers can be prepared in the presence of these coupling agents or by reaction with an isothiocyanate.
- imaging of breast tumors is achieved using monoclonal antibodies and the detectable imaging moieties are bound to the antibody using linkers such as methyl-p-hydroxybenzimidate or N-succinimidyl-3-(4-hydroxyphenyl)propionate.
- linkers such as methyl-p-hydroxybenzimidate or N-succinimidyl-3-(4-hydroxyphenyl)propionate.
- an FcR polypeptide may be fused to a reporter protein such as an enzyme as described supra or a fluorescence protein.
- the ability to specifically label periplasmic expressed proteins with appropriate fluorescent ligands also has applications other than library screening. Specifically labeling with fluorescent ligands and flow cytometry can be used for monitoring production of Fc domains during protein manufacturing.
- an Fc domain may be desired to link the molecule to at least one agent to form a conjugate to enhance the utility of that molecule.
- it is conventional to link or covalently bind or complex at least one desired molecule or moiety.
- a molecule or moiety may be, but is not limited to, at least one effector or reporter molecule.
- Effecter molecules comprise molecules having a desired activity, e.g ., cytotoxic activity.
- Non-limiting examples of effector molecules which have been attached to antibodies include toxins, anti-tumor agents, therapeutic enzymes, radio-labeled nucleotides, antiviral agents, chelating agents, cytokines, growth factors, and oligo- or poly-nucleotides.
- a reporter molecule is defined as any moiety which may be detected using an assay. Techniques for labeling such a molecule are known to those of skill in the art and have been described herein above.
- Labeled binding proteins such as Fc domains which have been prepared in accordance with the invention may also then be employed, for example, in immunodetection methods for binding, purifying, removing, quantifying and/or otherwise generally detecting biological components such as protein(s), polypeptide(s) or peptide(s).
- immunodetection methods include enzyme linked immunosorbent assay (ELISA), radioimmunoassay (RIA), immunoradiometric assay, fluoroimmunoassay, chemiluminescent assay, bioluminescent assay, and Western blot to mention a few.
- the Fc domain molecules, including antibodies, prepared in accordance with the present invention may also, for example, in conjunction with both fresh-frozen and/or formalin-fixed, paraffin-embedded tissue blocks prepared for study by immunohistochemistry (IHC).
- IHC immunohistochemistry
- the method of preparing tissue blocks from these particulate specimens has been successfully used in previous IHC studies of various prognostic factors, and/or is well known to those of skill in the art (Abbondanzo et al., 1990).
- FACS fluorescence activated cell sorting
- Instruments for carrying out flow cytometry are known to those of skill in the art and are commercially available to the public. Examples of such instruments include FACS Star Plus, FACScan and FACSort instruments from Becton Dickinson (Foster City, Calif.) Epics C from Coulter Epics Division (Hialeah, Fla.) and MOFLO TM from Cytomation (Colorado Springs, Co).
- Flow cytometric techniques in general involve the separation of cells or other particles in a liquid sample.
- the purpose of flow cytometry is to analyze the separated particles for one or more characteristics thereof, for example, presence of a labeled ligand or other molecule.
- the basis steps of flow cytometry involve the direction of a fluid sample through an apparatus such that a liquid stream passes through a sensing region.
- the particles should pass one at a time by the sensor and are categorized base on size, refraction, light scattering, opacity, roughness, shape, fluorescence, etc .
- Apparati permit quantitative multiparameter analysis of cellular properties at rates of several thousand cells per second. These instruments provide the ability to differentiate among cell types. Data are often displayed in one-dimensional (histogram) or two-dimensional (contour plot, scatter plot) frequency distributions of measured variables.
- the partitioning of multiparameter data files involves consecutive use of the interactive one- or two-dimensional graphics programs.
- Quantitative analysis of multiparameter flow cytometric data for rapid cell detection consists of two stages: cell class characterization and sample processing.
- cell class characterization partitions the cell feature into cells of interest and not of interest.
- sample processing each cell is classified in one of the two categories according to the region in which it falls.
- Analysis of the class of cells is very important, as high detection performance may be expected only if an appropriate characteristic of the cells is obtained.
- U.S. Patent 3,826,364 an apparatus which physically separates particles, such as functionally different cell types.
- a laser provides illumination which is focused on the stream of particles by a suitable lens or lens system so that there is highly localized scatter from the particles therein.
- high intensity source illumination is directed onto the stream of particles for the excitation of fluorescent particles in the stream.
- Certain particles in the stream may be selectively charged and then separated by deflecting them into designated receptacles.
- a classic form of this separation is via fluorescent-tagged antibodies, which are used to mark one or more cell types for separation.
- an important aspect of flow cytometry is that multiple rounds of screening can be carried out sequentially.
- Cells may be isolated from an initial round of sorting and immediately reintroduced into the flow cytometer and screened again to improve the stringency of the screen.
- Another advantage known to those of skill in the art is that nonviable cells can be recovered using flow cytometry. Since flow cytometry is essentially a particle sorting technology, the ability of a cell to grow or propagate is not necessary. Techniques for the recovery of nucleic acids from such non-viable cells are well known in the art and may include, for example, use of template-dependent amplification techniques including PCR.
- Nucleic acid-based expression systems may find use, in certain embodiments of the invention, for the expression of recombinant proteins.
- one embodiment of the invention involves transformation of Gram negative bacteria with the coding sequences for an antibody Fc domain, or preferably a plurality of distinct Fc domains.
- Certain aspects of the invention may comprise delivery of nucleic acids to target cells (e.g ., gram negative bacteria).
- target cells e.g ., gram negative bacteria
- bacterial host cells may be transformed with nucleic acids encoding candidate Fc domains potentially capable binding an FcR.
- it may be desired to target the expression to the periplasm of the bacteria. Transformation of eukaryotic host cells may similarly find use in the expression of various candidate molecules identified as capable of binding a target ligand.
- Suitable methods for nucleic acid delivery for transformation of a cell are believed to include virtually any method by which a nucleic acid (e.g., DNA) can be introduced into such a cell, or even an organelle thereof.
- a nucleic acid e.g., DNA
- Such methods include, but are not limited to, direct delivery of DNA such as by injection ( U.S. Patents 5,994,624 , 5,981,274 , 5,945,100 , 5,780,448 , 5,736,524 , 5,702,932 , 5,656,610 , 5,589,466 and 5,580,859 ), including microinjection (Harland and Weintraub, 1985; U.Sf. Patent 5,789,215 , incorporated herein by reference); by electroporation ( U.S.
- Patent 5,384,253 by calcium phosphate precipitation (Graham and Van Der Eb, 1973; Chen and Okayama, 1987; Rippe et al., 1990); by using DEAE-dextran followed by polyethylene glycol (Gopal, 1985); by direct sonic loading (Fechheimer et al., 1987); by liposome mediated transfection (Nicolau and Sene, 1982; Fraley et al., 1979; Nicolau et al., 1987; Wong et al., 1980; Kaneda et al., 1989; Kato et al., 1991); by microprojectile bombardment ( PCT Application Nos. WO 94/09699 and 95/06128 ; U.S.
- a nucleic acid is introduced into a cell via electroporation.
- Electroporation involves the exposure of a suspension of cells and DNA to a high-voltage electric discharge.
- certain cell wall-degrading enzymes such as pectin-degrading enzymes, are employed to render the target recipient cells more susceptible to transformation by electroporation than untreated cells ( U.S. Patent 5,384,253 ).
- recipient cells can be made more susceptible to transformation by mechanical wounding.
- a nucleic acid is introduced to the cells using calcium phosphate precipitation.
- Vectors may find use with the current invention, for example, in the transformation of a Gram negative bacterium with a nucleic acid sequence encoding a candidate Fc domain which one wishes to screen for ability to bind a target FcR.
- an entire heterogeneous "library" of nucleic acid sequences encoding target polypeptides may be introduced into a population of bacteria, thereby allowing screening of the entire library.
- vector is used to refer to a carrier nucleic acid molecule into which a nucleic acid sequence can be inserted for introduction into a cell where it can be replicated.
- a nucleic acid sequence can be "exogenous,” or “heterologous”, which means that it is foreign to the cell into which the vector is being introduced or that the sequence is homologous to a sequence in the cell but in a position within the host cell nucleic acid in which the sequence is ordinarily not found.
- Vectors include plasmids, cosmids and viruses ( e.g ., bacteriophage).
- viruses e.g ., bacteriophage.
- One of skill in the art may construct a vector through standard recombinant techniques, which are described in Maniatis et al., 1988 and Ausubel et al., 1994.
- expression vector refers to a vector containing a nucleic acid sequence coding for at least part of a gene product capable of being transcribed. In some cases, RNA molecules are then translated into a protein, polypeptide, or peptide.
- Expression vectors can contain a variety of "control sequences,” which refer to nucleic acid sequences necessary for the transcription and possibly translation of an operably linked coding sequence in a particular host organism. In addition to control sequences that govern transcription and translation, vectors and expression vectors may contain nucleic acid sequences that serve other functions as well and are described infra .
- a “promoter” is a control sequence that is a region of a nucleic acid sequence at which initiation and rate of transcription are controlled. It may contain genetic elements at which regulatory proteins and molecules may bind such as RNA polymerase and other transcription factors.
- the phrases "operatively positioned,” “operatively linked,” “under control,” and “under transcriptional control” mean that a promoter is in a correct functional location and/or orientation in relation to a nucleic acid sequence to control transcriptional initiation and/or expression of that sequence.
- a promoter may or may not be used in conjunction with an “enhancer,” which refers to a cis-acting regulatory sequence involved in the transcriptional activation of a nucleic acid sequence.
- a promoter may be one naturally associated with a gene or sequence, as may be obtained by isolating the 5' non-coding sequences located upstream of the coding segment and/or exon. Such a promoter can be referred to as "endogenous.”
- an enhancer may be one naturally associated with a nucleic acid sequence, located either downstream or upstream of that sequence.
- certain advantages will be gained by positioning the coding nucleic acid segment under the control of a recombinant or heterologous promoter, which refers to a promoter that is not normally associated with a nucleic acid sequence in its natural environment.
- a recombinant or heterologous enhancer refers also to an enhancer not normally associated with a nucleic acid sequence in its natural environment.
- Such promoters or enhancers may include promoters or enhancers of other genes, and promoters or enhancers isolated from any other prokaryotic cell, and promoters or enhancers not "naturally occurring," i.e ., containing different elements of different transcriptional regulatory regions, and/or mutations that alter expression.
- sequences may be produced using recombinant cloning and/or nucleic acid amplification technology, including PCRTM, in connection with the compositions disclosed herein (see U.S. Patent 4,683,202 , U.S. Patent 5,928,906 ).
- promoter and/or enhancer that effectively directs the expression of the DNA segment in the cell type chosen for expression.
- promoter that may be used with the invention is the E. coli arabinose or T7 promoter.
- the promoters employed may be constitutive, tissue-specific, inducible, and/or useful under the appropriate conditions to direct high level expression of the introduced DNA segment, such as is advantageous in the large-scale production of recombinant proteins and/or peptides.
- the promoter may be heterologous or endogenous.
- a specific initiation signal also may be required for efficient translation of coding sequences. These signals include the ATG initiation codon or adjacent sequences. Exogenous translational control signals, including the ATG initiation codon, may need to be provided. One of ordinary skill in the art would readily be capable of determining this and providing the necessary signals. It is well known that the initiation codon must be "in-frame" with the reading frame of the desired coding sequence to ensure translation of the entire insert. The exogenous translational control signals and initiation codons can be either natural or synthetic. The efficiency of expression may be enhanced by the inclusion of appropriate transcription enhancer elements.
- Vectors can include a multiple cloning site (MCS), which is a nucleic acid region that contains multiple restriction enzyme sites, any of which can be used in conjunction with standard recombinant technology to digest the vector (see Carbonelli et al., 1999, Levenson et al., 1998, and Cocea, 1997 ) "Restriction enzyme digestion” refers to catalytic cleavage of a nucleic acid molecule with an enzyme that functions only at specific locations in a nucleic acid molecule. Many of these restriction enzymes are commercially available. Use of such enzymes is understood by those of skill in the art.
- MCS multiple cloning site
- a vector is linearized or fragmented using a restriction enzyme that cuts within the MCS to enable exogenous sequences to be ligated to the vector.
- "Ligation” refers to the process of forming phosphodiester bonds between two nucleic acid fragments, which may or may not be contiguous with each other. Techniques involving restriction enzymes and ligation reactions are well known to those of skill in the art of recombinant technology.
- the vectors or constructs prepared in accordance with the present invention will generally comprise at least one termination signal.
- a “termination signal” or “terminator” is comprised of the DNA sequences involved in specific termination of an RNA transcript by an RNA polymerase.
- a termination signal that ends the production of an RNA transcript is contemplated.
- a terminator may be necessary in vivo to achieve desirable message levels.
- Terminators contemplated for use in the invention include any known terminator of transcription described herein or known to one of ordinary skill in the art, including but not limited to, for example, rhp dependent or rho independent terminators.
- the termination signal may be a lack of transcribable or translatable sequence, such as due to a sequence truncation.
- a vector in a host cell may contain one or more origins of replication sites (often termed "ori"), which is a specific nucleic acid sequence at which replication is initiated.
- ori origins of replication sites
- cells containing a nucleic acid construct of the present invention may be identified in vitro or in vivo by including a marker in the expression vector.
- markers would confer an identifiable change to the cell permitting easy identification of cells containing the expression vector.
- a selectable marker is one that confers a property that allows for selection.
- a positive selectable marker is one in which the presence of the marker allows for its selection, while a negative selectable marker is one in which its presence prevents its selection.
- An example of a positive selectable marker is a drug resistance marker.
- a drug selection marker aids in the cloning and identification of transformants, for example, genes that confer resistance to neomycin, puromycin, hygromycin, DHFR, GPT, zeocin and histidinol are useful selectable markers.
- markers conferring a phenotype that allows for the discrimination of transformants based on the implementation of conditions other types of markers including screenable markers such as GFP, whose basis is colorimetric analysis, are also contemplated.
- screenable enzymes such as chloramphenicol acetyltransferase (CAT) may be utilized.
- CAT chloramphenicol acetyltransferase
- One of skill in the art would also know how to employ immunologic markers, possibly in conjunction with FACS analysis. The marker used is not believed to be important, so long as it is capable of being expressed simultaneously with the nucleic acid encoding a gene product. Further examples of selectable and screenable markers are well known to one of skill in the art.
- host cell refers to a prokaryotic cell, and it includes any transformable organism that is capable of replicating a vector and/or expressing a heterologous gene encoded by a vector.
- a host cell can, and has been, used as a recipient for vectors.
- a host cell may be "transfected” or “transformed,” which refers to a process by which exogenous nucleic acid is transferred or introduced into the host cell.
- a transformed cell includes the primary subject cell and its progeny.
- a host cell is a Gram negative bacterial cell.
- Gram negative bacteria are suited for use with the invention in that they posses a periplasmic space between the inner and outer membrane and, particularly, the aforementioned inner membrane between the periplasm and cytoplasm, which is also known as the cytoplasmic membrane.
- any other cell with such a periplasmic space could be used in accordance with the invention.
- Gram negative bacteria that may find use with the invention may include, but are not limited to, E .
- the Gram negative bacterial cell may be still further defined as bacterial cell which has been transformed with the coding sequence of a fusion polypeptide comprising a candidate binding polypeptide capable of binding a selected ligand.
- the polypeptide is anchored to the outer face of the cytoplasmic membrane, facing the periplasmic space, and may comprise an antibody coding sequence or another sequence.
- One means for expression of the polypeptide is by attaching a leader sequence to the polypeptide capable of causing such directing.
- ATCC American Type Culture Collection
- a plasmid or cosmid for example, can be introduced into a prokaryote host cell for replication of many vectors.
- Bacterial cells used as host cells for vector replication and/or expression include DH5 ⁇ , JM109, and KC8, as well as a number of commercially available bacterial hosts such as SURE ® Competent Cells and SOLOPACK TM Gold Cells (STRATAGENE ® , La Jolla).
- bacterial cells such as E. coli LE392 could be used as host cells for bacteriophage.
- a viral vector may be used in conjunction with a prokaryotic host cell, particularly one that is permissive for replication or expression of the vector.
- Some vectors may employ control sequences that allow it to be replicated and/or expressed in both prokaryotic and eukaryotic cells.
- One of skill in the art would further understand the conditions under which to incubate all of the above described host cells to maintain them and to permit replication of a vector. Also understood and known are techniques and conditions that would allow large-scale production of vectors, as well as production of the nucleic acids encoded by vectors and their cognate polypeptides, proteins, or peptides.
- Prokaryote-based systems can be employed for use with the present invention to produce nucleic acid sequences, or their cognate polypeptides, proteins and peptides. Many such systems are commercially and widely available.
- Other examples of expression systems comprise of vectors containing a strong prokaryotic promoter such as T7, Tac, Trc, BAD, lambda pL, Tetracycline or Lac promoters, the pET Expression System and an E . coli expression system.
- antibody Fc domains are expressed on the cytoplasmic or in the periplasmic space membrane of a host bacterial cell.
- FcR target ligand
- Fc domain is intended to refer broadly to any immunoglobulin Fc region such as an IgG, IgM, IgA, IgD or IgE Fc.
- the techniques for preparing and using various antibody-based constructs and fragments are well known in the art. Means for preparing and characterizing antibodies are also well known in the art (See, e.g., Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988 ).
- the Fc domain may be purified, if desired, using filtration, centrifugation and various chromatographic methods such as HPLC or affinity chromatography.
- Fc domains encompassed by the present invention can be synthesized using an automated peptide synthesizer.
- nucleic acids may include, for example, the preparation of vectors for transformation of host cells as well as methods for cloning selected nucleic acid segments from a transgenic cell. Methodology for carrying out such manipulations will be well known to those of skill in the art in light of the instant disclosure.
- Nucleic acids used as a template for amplification may be isolated from cells, tissues or other samples according to standard methodologies (Sambrook et al., 1989). In certain embodiments, analysis may be performed on whole cell or tissue homogenates or biological fluid samples without substantial purification of the template nucleic acid.
- the nucleic acid may be genomic DNA or fractionated or whole cell RNA. Where RNA is used, it may be desired to first convert the RNA to a complementary DNA.
- primer is meant to encompass any nucleic acid that is capable of priming the synthesis of a nascent nucleic acid in a template-dependent process.
- primers are oligonucleotides from ten to twenty and/or thirty base pairs in length, but longer sequences can be employed.
- Primers may be provided in double-stranded and/or single-stranded form, although the single-stranded form is preferred.
- Pairs of primers designed to selectively hybridize to nucleic acids corresponding to a selected nucleic acid sequence are contacted with the template nucleic acid under conditions that permit selective hybridization.
- high stringency hybridization conditions may be selected that will only allow hybridization to sequences that are completely complementary to the primers.
- hybridization may occur under reduced stringency to allow for amplification of nucleic acids comprising one or more mismatches with the primer sequences.
- the template-primer complex is contacted with one or more enzymes that facilitate template-dependent nucleic acid synthesis. Multiple rounds of amplification, also referred to as "cycles," are conducted until a sufficient amount of amplification product is produced.
- the amplification product may be detected or quantified.
- the detection may be performed by visual means.
- the detection may involve indirect identification of the product via chemiluminescence, radioactive scintigraphy of incorporated radiolabel or fluorescent label or even via a system using electrical and/or thermal impulse signals (Affymax technology; Bellus, 1994).
- PCR TM polymerase chain reaction
- a reverse transcriptase PCR TM amplification procedure may be performed to quantify the amount of mRNA amplified.
- Methods of reverse transcribing RNA into cDNA are well known (see Sambrook et al., 1989).
- Alternative methods for reverse transcription utilize thermostable DNA polymerases. These methods are described in WO 90/07641 .
- Polymerase chain reaction methodologies are well known in the art. Representative methods of RT-PCR are described in U.S. Patent 5,882,864 .
- LCR ligase chain reaction
- European Application 320 308 European Application 320 308 .
- U.S. Patent 4,883,750 describes a method similar to LCR for binding probe pairs to a target sequence.
- a method based on PCR TM and oligonucleotide ligase assay (OLA), disclosed in U.S. Patent 5,912,148 may also be used.
- Qbeta Replicase described in PCT Application No. PCT/US87/00880 , may also be used as an amplification method in the present invention.
- a replicative sequence of RNA that has a region complementary to that of a target is added to a sample in the presence of an RNA polymerase.
- the polymerase will copy the replicative sequence which may then be detected.
- An isothermal amplification method in which restriction endonucleases and ligases are used to achieve the amplification of target molecules that contain nucleotide 5'-[alpha-thio]-triphosphates in one strand of a restriction site may also be useful in the amplification of nucleic acids in the present invention (Walker et al., 1992).
- Strand Displacement Amplification (SDA) disclosed in U.S. Patent 5,916,779 , is another method of carrying out isothermal amplification of nucleic acids which involves multiple rounds of strand displacement and synthesis, i.e., nick translation.
- nucleic acid amplification procedures include transcription-based amplification systems (TAS), including nucleic acid sequence based amplification (NASBA) and 3SR (Kwoh et al., 1989; Gingeras et al., PCT Application WO 88/10315 ).
- TAS transcription-based amplification systems
- NASBA nucleic acid sequence based amplification
- 3SR Zaoh et al., 1989; Gingeras et al., PCT Application WO 88/10315 .
- European Application No. 329 822 disclose a nucleic acid amplification process involving cyclically synthesizing single-stranded RNA ("ssRNA”), ssDNA, and double-stranded DNA (dsDNA), which may be used in accordance with the present invention.
- ssRNA single-stranded RNA
- dsDNA double-stranded DNA
- PCT Application WO 89/06700 discloses a nucleic acid sequence amplification scheme based on the hybridization of a promoter region/primer sequence to a target single-stranded DNA ("ssDNA”) followed by transcription of many RNA copies of the sequence. This scheme is not cyclic, i.e ., new templates are not produced from the resultant RNA transcripts.
- Other amplification methods include "race” and "one-sided PCR” (Frohman, 1990; Ohara et al., 1989).
- Oligonucleotides primers (Table 2) and restriction endonucleases used to construct plasmids to display homodimeric protein IgG-Fc were obtained from Integrated DNA Technologies (Coralville, IA) and New England Biolabs (Ipswich, MA), respectively.
- Taq Polymerase and FITC protein labeling kit were from Invitrogen (Carlsbad, CA).
- Recombinant human Fc ⁇ RI/CD64 was purchased from R&D Systems (Minneapolis, MN). Trehalose was obtained from Fisher Scientific (Fair Lawn, NJ).
- Human IgG-Fc and Rabbit anti-ECS antibody peroxidase conjugated were from Bethyl Laboratories (Montgometry, TX).
- Digoxigenin-BODIPY (Digoxigenin-4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl ethtylenediamine) was synthesized as described previously (Harvey et al., 2004).
- PA-FITC was obtained from List Biological Laboratories (Campbell, CA). Protein A-FITC and analytical grades of all other chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise.
- Plasmid pPelBHis was generated by ligating BamHI-HindIII digested skp gene from pMopac12 into pMopac1 digested with the same restriction endonucleases.
- pPelBFLAG was derived from pPelBHis in which polyhistidine tag and c-myc tag were replaced by FLAG tag (DYKDDDDK; SEQ ID NO:114).
- Subcloning of PCR amplified and SfiI digested Fc gene encoding human IgG1-Fc fragment, hinge, CH2 and CH3 region of human IgG1 heavy chain GeneBank Accession No.
- pPelBHis-beta 2 microglobulin was constructed by subcloning soluble mature human beta 2 microglobulin gene synthesized from overlap PCR amplification using 10 primers including 2 external primers (STJ#78 and STJ#84; SEQ ID NOS:14 and 16) and 8 internal primers (STJ#86-93; SEQ ID NOS:17-24) into pPelBHis using SfiI restriction endonuclease site.
- pPelBHis-Fc ⁇ RIIa was generated by introducing SfiI digested soluble mature human Fc ⁇ RIIa gene (GeneBank Accession No. P12318) (Stengelin et al., 1988) amplified from pDNR-LIB-Fc ⁇ RIIa (ATCC: MGC-23887) using primers STJ#74 and STJ#80 (SEQ ID NOS:13 and 15) into pPelBHis.
- Plasmids Used in The Present Examples Plasmids Relevant characteristics Reference or source pMoPac1 Cm r , lac promoter, tetA gene, C-terminal polyhistidine tag and c-myc tag Hayhurst et al., 2003 pMoPac12 Ap r , lac promoter, tetA gene, skp gene, C- terminal polyhistidine tag and c-myc tag Hayhurst et al., 2003 pMoPac16 Ap r , lac promoter, tetA gene, HuCk gene, skp gene, C-terminal polyhistidine tag and c-myc tag Hayhurst et al., 2003 pMoPac1-FLAG-M 18 NlpA fused M18 scFv gene, C-terminal FLAG tag in pMoPac1 Jung et al., 2007 pMoPac1-FLAG-2610 NlpA fuse
- pNlpAFLAG-M18 was constructed by ligating XbaI-HindIII digested fragments for NlpA and 6 amino acid residues (CDQSSS; SEQ ID NO:84) fused M18 scFv gene from pMopac1-FLAG-M18 into pPelBFLAG-M18.
- pNlpAHis-Fc was generated by subcloning SfiI digested Fc gene into pNIpAFLAG-M18 and by replacing FLAG with polyhistidine tag and c-myc tag.
- pBADNlpAFLAG-M18 and pBADNIpAHis-Fc were generated by ligating XbaI-HindIII digested M18 scFv gene and Fc gene from pNlpAFLAG-M18 and pNlpAHis-Fc, respectively, into pBAD30 digested with same restriction endonucleases.
- NotI-SalI digested cFos fragments amplified using two primers STJ#68 and STJ#69; SEQs ID NO:11 and 12
- the cFos(Cys) fragments encoding additional three amino acids including internal two Gly residues and external Cys residue at both ends of C-terminus and N-terminus amplified using two primers STJ#96 and STJ#97; SEQ ID NOS:27 and 28
- STJ#96 and STJ#97 SEQ ID NOS:27 and 28
- pBADNIpAFLAG-cJun and pBADNlpAFLAG-cJun(Cys) were generated by subcloning SfiI digested cJun fragments amplified with primers (STJ#58 and STJ#59; SEQ ID NOS:11 and12) and cJun(Cys) fragments amplified with primers (STJ#94 and STJ#95; SEQ ID NOS:25 and 26) into SfiI digested pBADNlpAFLAG-M18.
- Im2 gene that is PCR amplified using two primers (ST#116 and STJ#117; SEQ ID NOS:31 and 32) and template E. coli WTZ1011 ColE2 harboring plasmid ColE2-P9 (The Coli Genetic Stock Center, Yale Univ. CGSC No.
- the amplified PCR products were SfiI digested and introduced into pBADNlpAHis to generate pBADNlpAHis-ColE2(H574A), pBADNlpAHis-ColE2(H578A), and pBADNlpAHis-ColE2(H574A/H578A).
- Fc and Fc ⁇ RIIa gene fragments were PCR amplified using primers (STJ#144 and STJ#139; SEQ ID NOS:41 and 40) with the templates pPelBHis-Fc for Fc gene and primers (STJ# 136 and STJ#139; SEQ ID NOS:39 and 40) with the template pPelB-Fc ⁇ RIIa for Fc ⁇ RIIa gene, respectively, SalI-HindIII digested, and ligated into dsbA signal sequence (Schierle et al., 2003) inserted pTrc99A.
- E. coli Jude-1 F' [Tn10(Tetr) proAB+ lacIq ⁇ (lacZ)M15] mcrA ⁇ (mrr-hsdRMS-mcrBC) ⁇ 80dlacZAM15 ⁇ lacX74 deoR recA1 araD139 ⁇ (ara leu)7697 galU galK rpsL endA1 nupG) (Kawarasaki et al., 2003).
- cJun-cFos, pPelBHis-Fc-cFos and pPelBHis-Fc-cFos(Cys) were co-transformed with pBADNlpAFLAG-cJun or pBADNlpAFLAG-cJun(Cys) into E. coli Jude-1.
- pPelBFLAG-Fc-Im2 To display Fc using the interaction of ColE2-Im2, pPelBFLAG-Fc-Im2, pPelB-M18 scFv-Im2, and pPeIBFLAG-2610 scFv-Im2 were co-transformed with pBADNlpAHis-ColE2(H574A), pBADNlpAHis-ColE2(H578A), or pBADNlpAHis-ColE2(H574/578) containing single or double mutations at C-terminal ColE2 DNase catalytic domain.
- the transformants harboring two plasmids were grown overnight at 37°C with 250 rpm shaking in Terrific Broth (TB) (Becton Dickinson Diagnostic Systems DIFCOTM, Sparks, MD) supplemented with 2% (wt/vol) glucose, chloramphenicol (40 ⁇ g/ml) and ampicillin (50 ⁇ g/ml). After overnight culture, the cells were diluted 1:100 in fresh TB medium without glucose, incubated at 37°C for 2 h and then cooled at 25°C for 20 min.
- TB Terrific Broth
- PelB signal sequence fused proteins were induced with 1 mM of isopropyl-1-thio- ⁇ -D-galactopyranoside (IPTG) to allow time for correct folding in periplasmic space prior to binding to inner membrane anchored ColE2 mutants.
- IPTG isopropyl-1-thio- ⁇ -D-galactopyranoside
- 0.2% (wt/vol) arabinose was added to induce expression of inner membrane anchored cJun, cJun(Cys), ColE2(H574A), ColE2(H578A), or ColE2(H574A/H578A).
- E. coli transformed with various plasmids pPelBHis-Fc, pPelBHis-beta 2 microglobulin, pPelBFLAG-M18 scFv, pMopac12-M18.1 hum scFv, pMopac12-2610 scFv, pMopac16-M18.1 hum scAb, pMopac16-2610 scAb, pMAZ360-M18.1-Hum-IgG, pMAZ360-26.10 IgG, pNlpAHis-Fc, pTrcdsbAHis-Fc, and pTrcdsbAHis-Fc ⁇ RIIa were cultured overnight at 37 °C with 250 rpm shaking in Terrific Broth (TB) with 2% (wt/vol) glucose.
- Antibiotics chloramphenicol (40 ⁇ g/ml) or ampicillin (50 ⁇ g/ml) appropriate for antibiotic resistance gene of each plasmid, were added for overnight culture.
- the overnight cultured cells were diluted 1:50 in fresh TB medium with 0.5M trehalose and the supplement of appropriate antibiotics, chloramphenicol (40 ⁇ g/ml) or ampicillin (50 ⁇ g/ml).
- the protein expression was induced with 1 mM of isopropyl-1-thio- ⁇ -D-galactopyranoside (IPTG).
- the washed cells were incubated in 1 ml of Solution A with 1 mg/ml of hen egg lysozyme at 37°C for 15 min. After centrifugation at 12,000 x g for 1 min and the resulting spheroplasts pellet were resuspended in 1 ml of cold PBS. 200 ⁇ l of the spheroplasts further diluted in 800 ⁇ l of PBS was mixed with each fluorescent labeled probes, 0.5 ul of Protein A-FITC (5 mg/ml), 2 ul of PA-FITC (0.25 mg/ml), 2.5 ul of Fc ⁇ RIa-FITC (0.6 mg/ml), or 200 nM Digoxin-BODIPY.
- the mixture was pelleted by centrifugation at centrifuged at 12,000 x g for 1 min and resuspended in 1 ml of PBS.
- the 100 ⁇ l of the resuspension was diluted in 1 ml of PBS and analyzed on BD FACSort (BD Bioscience, San Jose, CA).
- Fc partial gene fragments amplified using primers STJ#194 and STJ195 (SEQ ID NOS:43 and 44) were digested with SacII and EcoRI restriction endonucleases and subcloned into SacII-EcoRI digested pPelBFLAG-Fc to generate random peptide loop inserted Fc library.
- the Fc genes in the spheroplasts were rescued by PCR amplification using two specific primers (STJ#16 and STJ#220; SEQ ID NOS:8 and 47), ligated into pPelBFLAG-Fc using SfiI restriction enzyme site, and transformed in electrocompetent E. coli Jude-1cells. The resulting transformants were employed for the next round sorting.
- the multimeric protein should be encoded by single gene to make the homomultimer. Fc encoded by two separate genes generates heterodimeric Fc.
- the homomultimeric proteins should be expressed in the space enabling correct folding and assembly. In bacterial expression system, E . coli periplasmic space provides oxidative environment for disulfide bonds and is suitable for the production of correct folded heterologous protein with the use of cellular folding machinery (Georgiou and Segatori, 2005).
- the expressed and folded multimeric proteins should be tightly anchored to bacterial cells during high throughput Fluorescent Activated Cell Sorting (FACS).
- the first attempted system was leucine zippers cJun-cFos interaction pair.
- the repetitive leucine residues at every seven amino acid of cJun and cFos allow strong non-covalent interaction (Landschulz et al., 1988; Kouzarides and Ziff, 1988).
- Expression of cFos fused Fc from pPelBHis-Fc-cFos was firstly induced for periplasmic expression and then pBADNlpAFLAG-cJun for cJun fused to NlpA leader sequence and six amino acid residues (CDQSSS; SEQ ID NO:84) for inner membrane anchoring was induced for binding to the periplasmic assembled Fc homodimer ( FIG. 1 ).
- Im2 fused Fc was firstly induced for periplasmic Fc assembly and then the expression of ColE2 mutants fused to NlpA leader sequence and six amino acids (CDQSSS; SEQ ID NO:84) was induced for inner membrane anchoring ( FIG. 3 ).
- M18 scFv, 26-10 scFv, and homodimeric Fc were further investigated with M18 scFv, 26-10 scFv, and homodimeric Fc.
- M18 scFv-Im2 and 26-10 scFv-Im2 showed selectively higher fluorescence signal comparing negative controls, M18 scFv and 26-10 scFv not fused to Im2, respectively ( FIG. 4 ), this selective high signals were derived from the deviation in expression levels.
- M18 scFv, 26.10 scFv, and Fc were well expressed.
- the expression of ColE2 with Im2 or without Im2 inhibited the expression of M18 scFv, 26-10 scFv and Fc partially or completely, respectively ( FIG. 5 ).
- the fluorescence signal for the PelB fused Fc was tested when cultured in media comprising sorbitol or trehalose. Surprisingly, 0.5M trehalose greatly increased fluorescence signal intensity in the FACS analysis for both the APEx displayed Fc and the PelB leader peptide fused Fc ( FIG. 6 ). Also, in comparison with other negative controls, PelB fused M18 scFv cultured in the media with or without 0.5M trehalose, the PelB fused Fc clearly exhibited dramatically improved signal intensity and CV value ( FIG. 7 ) providing a selective display system for real affinity maturation of homodimeric Fc ( FIG. 8 ).
- the native human IgG has two N-linked biantennary complex type oligosaccharide chains at the Asn297 amino acid residue of each CH2 domain.
- the two chains are located between the CH2 domains and interact with hydrophobic parts of the domains.
- Effector functions are largely dependent on the presence of the oligosaccharide chains (Wright and Mornson, 1997; Jefferis, 2005) to keep open structure of heavy chains for immune ligands binding (Sondermann et al., 2001). Aglycosylation causes great reduction or complete loss of effector functions (Jefferis, 2005).
- insertion library was constructed by PCR amplification using forward primer STJ#194 (SEQ ID NO:43) containing 10 degenerate codons encoded by the NNS randomization scheme and reverse primer STJ#195 (SEQ ID NO:44) with the same template.
- the amplified PCR fragments were ligated into pPelBFLAG cut with Sfi I restriction sites for the error prone PCR library and with SacII / EcoRI for random 10 a.a. insertion library, respectively.
- the transformation of the resulting library generated 2.8 ⁇ 10 7 transformants.
- error prone PCR was used to generate random mutation for full Fc region.
- the resulting library was 9.2 ⁇ 10 8 individual transformants with 0.49% error rate per gene based on the sequence of 20 library clones randomly selected.
- the Fc5 showing the highest affinity to Fc ⁇ RIa had two mutations E382V and M428I in CH3 region.
- the other five clones contained consensus mutations in E382V as well as M428I or S426T ( FIG. 14 ) suggesting a critical role of two interacting beta sheets including the major mutation points in CH3 region for the binding of aglycosylated Fc to Fc ⁇ RIa ( FIGs. 15a and 15b ).
- Each of the three sublibraries was subcloned into Sex AI / Sap I digested pPelBFLAG.-Fc.
- the resulting plasmids were transformed into E.
- Table 4 shows the sequencing results of 10 randomly picked clones indicating that the expected sequence diversity had been obtained.
- the library cells were cultured in media containing trehalose, protein synthesis was induced with 1 mM IPTG and after 5 hours the cells were harvested and converted into spheroplasts as described in Example 4. Following labeling, spheroplasts were sorted by FACS. In each round the top 3% of the population showing the highest fluorescence due to Fc ⁇ RIa-FITC binding labeling was isolated (-1 ⁇ 10 8 spheroplasts were sorted in each round of sorting).
- the Fc encoding genes were recovered by PCR ligated into vector and the ligation mix was transformed into E. coli Jude-1. Transformants were selected on chloramphenicol containing media and then grown, spheroplasted as above in preparation for the next round of sorting ( FIG. 17 ). After the 4 th round of sorting, 14 individual clones exhibiting high fluorescence were isolated ( FIG. 18 ). However, the parental Fc5 clone (E382V/M428I) showed the highest fluorescence; importantly most of the selected mutants contained the mutations E382V and/or M428I or M428L, again suggesting the importance of these two amino acid substitutions.
- Table 4 Sequence of randomly picked up 10 clones from library randomized around 382E and 428M Underlining indicates mutated or inserted amino acids; * : Stop codon; ⁇ : Blank Wild type 378-AVEWESNG-385 425-CSVMHE ⁇ AL-432 1 AV A W D S R G (SEQ ID NO:85) CSV AL HE ⁇ AL (SEQ ID NO:95) 2 AV Y W S S L G (SEQ ID NO:86) C L V C HS ⁇ AL (SEQ ID NO:96) 3 AV L W G S L G (SEQ ID NO:87) C L V L H G ⁇ AL (SEQ ID NO:97) 4 AV V C Y S Y G (SEQ ID NO:88) C R V * H P ⁇ AL (SEQ ID NO:98) 5 AV S W I S Q G (SEQ ID NO:89) CSV GG HE ⁇ AL (SEQ ID NO:99) 6 AV N W E S K G (SEQ ID NO:90
- the extracellular domain of aglycosylated Fc ⁇ RIIIa was first purified from E. coli inclusion bodies.
- First an E. coli codon optimized Fc ⁇ RIIIa synthetic gene (Nucleotide Sequence #1 (SEQ ID NO:105) was subcloned into pET21a (Novagen) and transformed into E. coli BL21(DE3). After 5 hr induction with 1 mM IPTG induction, Western blot analysis revealed that the majority of the Fc ⁇ RIIIa protein was present as inclusion bodies.
- Inclusion bodies were harvested by centrifugation of cell lysates, washed with U2KP buffer (2M urea in 10 mM potassium phosphate buffer, pH 8.2) and solubilized in U8KP buffer (8M urea, 10mM potassium phosphate buffer, pH 8.2).
- the solubilized and denatured Fc ⁇ RIIIa protein was purified using Ni-NTA affinity chromatography and refolded by consecutive dialysis ( FIG. 19 ) (Jung et al., 2003).
- the purified Fc ⁇ RIIIa was labeled with FITC using a commercial FITC labeling kit (Molecular Probes) as described in the manufacurer's instructions. 1.5 ⁇ l of FITC labeled Fc ⁇ RIIIa (0.8 mg/ml) per 1 ml reaction was used for the labeling of spheroplasts.
- binders to Fc ⁇ RIIIa two libraries were constructed: First, the Fc gene was subjected to random mutagenesis by error prone PCR. Second, a 10 random amino acids insertion library was employed. The library cells were cultured in media containing trehalose, protein synthesis was induced with 1 mM IPTG and after 5 hours the cells were harvested and converted into spheroplasts as described in Example 4. Following labeling, spheroplasts were sorted by FACS. In each round the top 3% of the population showing the highest fluorescence due to Fc ⁇ RIIIa-FITC binding labeling was isolated (1 ⁇ 10 8 spheroplasts were sorted in each round of sorting).
- the Fc encoding genes were recovered by PCR ligated into vector and the ligation mix was transformed into E. coli Jude-1. Transformants were selected on chloramphenicol containing media and then grown, spheroplasted as above in preparation for the next round of sorting ( FIG. 20 ). After the 4th round sorting, five individual clones exhibiting high affinity to Fc ⁇ RIIIa were isolated ( FIG. 21 ). All five clones contained 10 random amino acid insertions. Two of these clones had additional mutations that presumably resulted from PCR amplification ( FIG. 22 ). Although these mutant clones are generally considered to be high affinity binders, as noted above they have been found to exhibit certain variability in different tests.
- Fc ⁇ RIIa binders either the same mixture of library including 10 a.a. insertion library and an error prone PCR library described in Example 7 or the Fc5 error prone PCR library using the template Fc5 (Fc: E382V, M428I) are used.
- the library size of Fc5 error prone PCR library was 1.1 ⁇ 10 7 and the error rate was 0.131% as determined by the sequencing of 20 randomly selected clones.
- the extracellular domain of glycosylated Fc ⁇ RIIa R&D systems
- FITC labeled Fc ⁇ RIIa (0.975 mg/ml) per 1 ml reaction was used.
- the library cells were cultured in media containing trehalose, protein synthesis was induced with 1 mM IPTG and after 5 hours the cells were harvested and converted into spheroplasts as described in Example 4.
- spheroplasts were sorted by FACS. In each round the top 3% of the population showing the highest fluorescence due to Fc ⁇ RIIa-FITC binding labeling was isolated (1 ⁇ 10 8 spheroplasts were sorted in each round of sorting) ( FIG. 23 ).
- the Fc encoding genes were recovered by PCR ligated into vector and the ligation mix was transformed into E. coli Jude-1. Transformants were selected on chloramphenicol containing media and then grown, spheroplasted as above in preparation for the next round of sorting After the 6 th round sorting from the Fc5 error prone PCR library, two individual clones showing high affinity to Fc ⁇ RIIa were isolated ( FIG. 24 ). In addition to the two mutations encoded by the Fc5 parental gene (E382V/M428I), the two isolated clones Fc331 and Fc336 had the mutations G402D and P331L, respectively ( FIG. 25 ). Although these mutant clones are generally considered to be high affinity binders, they may exhibit certain variability in different tests.
- Fc fragment genes were subcloned into Sal I / Hind III digested pDsbAFLAG plasmid and 192 individual colony harboring pDsbAFLAG-Fc mutant genes were cultured in 96 well plates with 200 ⁇ l working volume. The culture supernatant from the induced cells was separated by centrifugation at 4000 rpm for 30 min. For ELISA analysis, the extracellular domain of aglycosylated Fc ⁇ RIIa was purified from E. coli inclusion bodies. First an E.
- coli codon optimized Fc ⁇ RIIa synthetic gene (Nucleotide Sequence #2 SEQ ID NO:106) was subcloned into pET21a (Novagen) and transformed into E. coli BL21(DE3). After 5 hr induction with 1 mM IPTG induction, SDS-PAGE analysis revealed that codon optimized Fc ⁇ RIIa synthetic gene shows dramatically increased expression level comparing with wild type Fc ⁇ RIIa gene ( FIG. 26 ) and the majority of the Fc ⁇ RIIa protein was present as inclusion bodies ( FIG. 27 ).
- Inclusion bodies were harvested by centrifugation of cell lysates, washed with U2KP buffer (2M urea in 10mM potassium phosphate buffer, pH 8.2) and solubilized in U8KP buffer (8M urea, 10mM potassium phosphate buffer, pH 8.2).
- the solubilized and denatured Fc ⁇ RIIa protein was purified using Ni-NTA affinity chromatography and refolded by consecutive dialysis ( FIG. 28 ) (Jung et al., 2003). 100 ⁇ l of the culture supernatants were transferred to 96 well ELISA plates and incubated at 4 °C for overnight.
- Isolated Fc mutants have substitution or insertion mutations in the sequence of wild type Fc (Nucleotide Sequence #3 (SEQ ID NO: 107) and Protein Sequence #1 (SEQ ID NO:1)). Mutation points of the isolated clones showing high affinity to Fc ⁇ Rs are summarized in Table 5.
- Fc mutants (Protein Seqeunce #2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, and 17; SEQ ID NOS:3, 2, 4, 5, 7, 6 and SEQ ID NOS:65-73) show high affinity to Fc ⁇ RIa.
- Fc mutants (Protein Sequences #18, 19, 20, 21, and 22; SEQ ID NOS:74-78) show high affinity to Fc ⁇ RIIIa.
- Fc mutants (Protein Sequence #23, 24, 25, 26, 27; SEQ ID NOS:79, 80, 122, 81 and 82) show high affinity to Fc ⁇ RIIa. Although these mutant clones binding to Fc ⁇ RIIIa and Fc ⁇ IIa are generally considered to be high affinity binders, some of them have been found to exhibit certain variability in different tests.
- pDsbALAG-Fc To construct pDsbALAG-Fc first, a synthetic DNA fragment encoding the 53 bp DsbA signal peptide gene (ATGAAAAAGATTTGGCTGGCGCTGGCTGGTTTAGTTTTAGCGTTTAGCGC ATCGGCG (SEQ ID NO:109)) was introduced into pTrc99A following cleavage with Fat I which is compatible with the Nco I in pTrc99A (Amersham Pharmacia) and also with Sal I. The resulting plasmid was named pDsbA.
- the parental Fc or Fc mutant genes were amplified using he primers STJ#144 (TTTTAGGGGTCGACGACAAAACTCACACATGCCCACCGTG (SEQ ID NO:41)) and STJ#145 (TTTAAGGGAAGCTTCTATTAGGCGCGCCCTTTGTCATCG (SEQ ID NO:42), ligated into pDsbA plasmid using Sal I and Hind III restriction enzyme sites giving rise to pDsbAFLAG-Fc.
- E. coli Jude-1 cells harboring pDsbAFLAG-Fc or Fc mutants were cultured in 2L flask with 500 ml working volume.
- the culture supernatant from the induced cells was separated by centrifugation at 7000 rpm for 30 min.
- the supernatant was filtered using 0.22 ⁇ m bottle top filters (Corning) and loaded onto a column packed with 1 ml of Immobilized Protein A agarose (Pierce). After loading of 400 ml of supernatants by gravity flow, the columns were washed with 75 ml of 20 mM sodium phosphate buffer (pH 7.0) and with 50 ml of 40 mM sodium citrate (pH 5.0).
- Wild type Fc and Fc mutants were eluted using 0.1M glycine (pH 2.5) and neutralized immediately with 1M Tris (pH 8.0) solution.
- the eluted wild type Fc and Fc mutants were analyzed by SDS-PAGE ( FIG. 32 ).
- SDS-PAGE SDS-PAGE
- To collect dimeric Fc the eluted samples from Protein A affinity chromatography column were concentrated with an ultrafiltration unit (10 kDa Mw cutoff: Millipore) and purified using Superdex 200 (Amersham Pharmacia) gel filtration chromatography ( FIG. 33 ). Most of the purified wild type Fc and Fc mutants were dimeric forms ( FIG. 34 ).
- the final yield of purified dimeric Fc and Fc mutants was approximately 800 ⁇ g/ml.
- Fc ⁇ RIa, Fc ⁇ RIIa or Fc ⁇ RIIIa The affinity of purified Fc or Fc mutant proteins for Fc ⁇ RIa, Fc ⁇ RIIa or Fc ⁇ RIIIa was analyzed by ELISA ( FIG. 35 ). 50 ⁇ l of 5 ⁇ g/ml purified wild type Fc, Fc mutants (Fc5, Fc11, Fc49), or glycosylated IgG-Fc (Bethyl laboratories) diluted in 0.05 M Na 2 CO3 (pH 9.6) buffer were coated on 96 well polystyrene ELISA plate (Coming) by overnight incubation at 4 °C.
- Binding of IgG1-Fc domains to the human Fc ⁇ RI was analyzed by surface plasmon resonance using a BIAcore 3000 biosensor (BIAcore).
- the soluble monomeric Fc ⁇ RIa was immobilized on the CM-5 sensor chip by the amine coupling kit as recommended by the manufacturer. Binding experiments were performed in HBS-EP buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3.4 mM EDTA, and 0.005% P20 surfactant).
- Aglycosylated IgG1-Fc fragments, aglycosylated Fc mutants (Fc5, Fc49), or glycosylated IgG1 were injected at flow rate of 100 ul/min for 30 s with dissociation time 300 s. Regeneration was performed by a single injection of 100 mM citric acid, pH 3.0. Fc5 and Fc49 were injected in duplicate at concentrations 0, 80, 100, 200, 400, 600 nM and 0, 200, 400, 600, 800, and 1,000 nM. BIAcore analysis revealed that wt Fc does not bind to Fc ⁇ RI (K D >50 ⁇ M). In contrast, Fc5 and Fc49 exhibited K D values of 31 and 92 nM respectively.
- the equilibrium dissociation constant of commercially available, glycosylated IgG1 was 18 nM.
- the aglycosylated Fc5 mutant and the glycosylated human Fc exhibited experimentally indistinguishable dissociation rate constants, k off and a 2-fold lower association rate constant, k on (Table 6) ( FIG. 37 ).
- Table 6 Kinetic rates and equilibrium dissociation constants of isolated Fc mutants determined by BIACore.
- V L and V H domains of humanized 4D5-8 were synthesized by total gene synthesis with overlap extension PCR using 12 oligonucleotides that included 2 external primers (STJ#302 and STJ#313) and 10 internal primers (STJ#303-312) for V L and 14 primers total 2 external primers (STJ#314 and STJ#327) and 12 internal primers (STJ#315-326) for V H , respectively.
- trastuzumab (HerceptinTM) recognizes HER2/neu (Erb2) which is overexpressed in about 30% of breast carcinomas (Sergina and Moasser, 2007). Extensive evidence indicates that recruitment of innate immune cells via interactions with Fc ⁇ receptors plays an important role in the therapeutic action of trastuzumab (Sergina and Moasser, 2007; Lazar et al., 2006).
- the heavy and light chains were fused to the PelB signal peptide and placed downstream from the lac promoter in a dicistronic operon ( FIG. 38 ).
- Preparative expression was performed by fed-batch fermentation using a 3.3 L jar fermentor (New Brunswick Scientific Co., Edison, NJ) with 1.2 L working volume.
- BL21(DE3) cells were grown at 30°C in R/2 medium (Jeong and Lee" 2003) consisting of: 2 g of (NH 4 ) 2 HPO 4 , 6.75 g of KH 2 PO 4 , 0.93 g of citric acid H 2 O, 0.34 g of MgSO 4 , 20 g of glucose, 0.05 g of ampicillin and 5 ml of trace metal solution dissolved in 2 N HCl (10 g of FeSO 4 -7H 2 O, 2.25 g ZnSO 4 -7H 2 O, 1 g of CuSO 4 -5H 2 O, 0.35 g of MnSO 4 -H 2 O, 0.23 g of Na 2 B 4 O 7 -10H 2 O, 1.5 g of CaCl 2 , and 0.1 g of (NH 4 ) 6 Mo 7 O 24 ) per L).
- E. coli BL21(DE3) EMD Chemicals, Gibbstown, NJ harboring pSTJ4-HerceptinTM IgG1 or pSTJ4-HerceptinTM IgG1-Fc5 were cultured in 500 mL baffled-flask with 120 ml R/2 media at 30°C at 250 rpm for 8 h and used to inoculate the fermenter.
- the dissolved oxygen (DO) concentration was maintained at 40% of air saturation using automatic cascade control by increasing agitation speed from 100 rpm to 1000 rpm, air flow rate from 1 to 3 SLPM (Standard liquid per minute) and pure oxygen flow rate from 0 to 1.5 SLPM when required.
- Cells were pelleted by centrifugation at 11,000 ⁇ g for 30 min, suspended in 1.2 L 100 mM Tris, 10 mM EDTA (pH 7.4), 4 mg of lysozyme (per g of dry cell weight), and 1 mM PMSF and were incubated with shaking at 250 rpm at 30°C for 16 h to release periplasmic proteins. After centrifugation at 14,000 ⁇ g for 30 min, the supernatant was mixed with polyethyleneimine (MP Biomedical, Solon, OH) to a final concentration of 0.2% (w/v) recentrifuged at 14,000 ⁇ g for 30 min, and filtered through 0.2 ⁇ m filter.
- polyethyleneimine MP Biomedical, Solon, OH
- Immobilized Protein A agarose resin pre-equilibrated in 20 mM sodium phosphate buffer (pH 7.0) was added to the supernatant and incubated at 4°C for 16 h. After washing with 200 ml of 20 mM sodium phosphate buffer (pH 7.0) and 200 ml of 40 mM sodium citrate (pH 5.0), IgG1 was eluted from the resin using 15 ml of 0.1 M glycine (pH 3.0) and neutralized immediately with 1M Tris (pH 8.0) solution. The eluted samples were concentrated by ultrafiltration through a 10 kDa Mw cutoff membrane and the retentate was applied to a Superdex 200 gel filtration column developed with PBS (pH 7.4) ( FIG. 42 ).
- the affinity of the purified IgGs for the extracellular domain of Fc ⁇ RIIa and Fc ⁇ RIIb expressed as an N-terminal fusion to GST in 293E cells was determined by ELISA. 50 ⁇ l of 4 ⁇ g/ml of wild type Fc, Fc mutants, aglycosylated trastuzumab, or trastuzumab-Fc5 purified from E. coli, glycosylated IgG trastuzumab (Clinical grade, Fox Chase Cancer Center Pharmacy) or glycosylated IgG1 (Sigma-Aldrich, St.
- ELISAs were carried out as above except that the washing buffer and sample dilution buffers were adjusted to pH 5.5.
- the aglycosylated tratuzumab exhibited low affinity to Fc ⁇ RIIa or Fc ⁇ RIIb ( FIG. 43 and FIG. 44 ).
- Trastuzumab-Fc5 antibody exhibited only slightly higher affinity for Fc ⁇ RIIb.
- the neonatal Fc ⁇ Rn receptor binds to the CH3 domain and is responsible for the endosomal recycling of IgG in plasma (Ghetie and Ward, 2000).
- Glycosylated, aglycosylated and trastuzumab-Fc5 exhibited near identical binding to FcRn at pH 5.5 and low binding at pH 7.5 suggesting that the E382V and M42I substitutions are not likely to affect the circulation half-life of this antibody ( FIG. 45 ).
- Binding of Fc ⁇ RI to the full assembled IgG trastuzumab was also analyzed by immobilizing glycosylated trastuzumab, aglycosylated trastuzumab, and aglycosylated trastuzumab-Fc5 individually on the CM-5 sensor chip. Binding experiments were done in the same HBS-EP buffer. For trastuzumab or trastuzumab-Fc5 Fc ⁇ RIa was injected in duplicate at concentrations 0, 10, 20, 30, 50, and 100 nM for 60 s at a flow rate of 10 ⁇ l/min.
- trastuzumab-Fc5 bound strongly to Fc ⁇ RIa. Specifically, the equilibrium dissociation constants for glycosylated trastuzumab from CHO cells, the E. coli expressed trastuzumab and trastuzumab-Fc5 were 1.7 nM, 0.8 ⁇ M and 3.6 nM respectively.
- trastuzumab-Fc5 exhibits selective binding only to the Fc ⁇ RIa receptor.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Oncology (AREA)
- Food Science & Technology (AREA)
- Cell Biology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Virology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US91518307P | 2007-05-01 | 2007-05-01 | |
| US98265207P | 2007-10-25 | 2007-10-25 | |
| PCT/US2008/062090 WO2008137475A2 (en) | 2007-05-01 | 2008-04-30 | Immunoglobulin fc libraries |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2155789A2 EP2155789A2 (en) | 2010-02-24 |
| EP2155789B1 true EP2155789B1 (en) | 2013-07-24 |
Family
ID=39671394
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP08747239.5A Not-in-force EP2155789B1 (en) | 2007-05-01 | 2008-04-30 | Immunoglobulin fc libraries |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8629245B2 (da) |
| EP (1) | EP2155789B1 (da) |
| AU (1) | AU2008247819B2 (da) |
| CA (1) | CA2685675C (da) |
| DK (1) | DK2155789T3 (da) |
| WO (1) | WO2008137475A2 (da) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019078591A1 (ko) * | 2017-10-20 | 2019-04-25 | 국민대학교 산학협력단 | 세포막 유동성을 이용한 다중체 단백질 디스플레이 시스템 |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1495055B1 (en) * | 2002-04-18 | 2013-08-14 | Genencor International, Inc. | Production of functional antibodies in filamentous fungi |
| CN102549016B (zh) * | 2009-06-30 | 2015-05-06 | 研究发展基金会 | 免疫球蛋白fc多肽 |
| DK2516702T3 (da) | 2009-12-23 | 2018-04-30 | Affinity Biosciences Pty Ltd | Proteinvisning |
| CA2826467C (en) * | 2011-02-07 | 2019-11-12 | Research Development Foundation | Engineered immunoglobulin fc polypeptides |
| EP3626739A1 (en) | 2011-06-24 | 2020-03-25 | Stephen D. Gillies | Light chain immunoglobulin fusion proteins and methods of use thereof |
| JP6013470B2 (ja) | 2011-06-29 | 2016-10-25 | アフィニティ バイオサイエンス ピーティーワイ リミテッド | タンパク質ディスプレイの方法 |
| EP2855529A4 (en) * | 2012-05-24 | 2015-12-09 | Alexion Pharma Inc | HUMANIZED ANTI-FACTOR B ANTIBODIES |
| SG11201408646VA (en) * | 2012-07-06 | 2015-01-29 | Genmab Bv | Dimeric protein with triple mutations |
| US20150353940A1 (en) | 2013-08-05 | 2015-12-10 | Absci, Llc | Vectors for use in an inducible coexpression system |
| AU2013299910B2 (en) | 2012-08-05 | 2019-01-24 | Absci Corporation | Inducible coexpression system |
| WO2014065945A1 (en) | 2012-10-23 | 2014-05-01 | The Board Of Regents Of The University Of Texas System | Antibodies with engineered igg fc domains |
| SG10201800492PA (en) | 2013-04-29 | 2018-03-28 | Hoffmann La Roche | Human fcrn-binding modified antibodies and methods of use |
| CN110981957A (zh) * | 2014-01-15 | 2020-04-10 | 豪夫迈·罗氏有限公司 | 具有改善的蛋白A结合作用的Fc区变体 |
| EP3207150B1 (en) | 2014-10-14 | 2021-04-28 | Research Development Foundation | Methods for generating engineered enzymes |
| CA2976236A1 (en) | 2015-02-09 | 2016-08-18 | Research Development Foundation | Engineered immunoglobulin fc polypeptides displaying improved complement activation |
| CN104774844A (zh) * | 2015-04-16 | 2015-07-15 | 哈尔滨博翱生物医药技术开发有限公司 | 一种利用细菌表面展示系统筛选单链抗体的方法 |
| US10526408B2 (en) | 2015-08-28 | 2020-01-07 | Research Development Foundation | Engineered antibody FC variants |
| WO2017070167A1 (en) * | 2015-10-20 | 2017-04-27 | The University Of North Carolina At Chapel Hill | Methods and compositions for modified factor ix fusion proteins |
| IL314725A (en) | 2015-10-23 | 2024-10-01 | Eureka Therapeutics Inc ׂ A Delaware Corp | Antibody/T-cell receptor chimeric structures and their uses |
| AU2017292184A1 (en) | 2016-07-08 | 2019-02-07 | Staten Biotechnology B.V. | Anti-Apoc3 antibodies and methods of use thereof |
| PE20191034A1 (es) | 2016-10-14 | 2019-08-05 | Xencor Inc | Proteinas de fusion heterodimericas biespecificas que contienen proteinas de fusion fc il-15/il-15r y fragmentos de anticuerpo pd-1 |
| US11414493B2 (en) | 2016-10-27 | 2022-08-16 | Kookmin University Industry Academy Cooperation Foundation | Aglycosylated antibody Fc region for treating cancer |
| AU2018255938A1 (en) | 2017-04-21 | 2019-10-31 | Staten Biotechnology B.V. | Anti-ApoC3 antibodies and methods of use thereof |
| SG10201913656TA (en) | 2017-04-26 | 2020-03-30 | Eureka Therapeutics Inc | Cells expressing chimeric activating receptors and chimeric stimulating receptors and uses thereof |
| US11220550B2 (en) | 2017-05-25 | 2022-01-11 | Bristol-Myers Squibb Company | Antagonistic anti-CD40 antibodies and methods of antagonizing CD40 activity |
| AU2018291497A1 (en) | 2017-06-30 | 2020-01-16 | Xencor, Inc. | Targeted heterodimeric Fc fusion proteins containing IL-15/IL-15Ra and antigen binding domains |
| EP3665195A4 (en) | 2017-08-11 | 2021-05-19 | Research Development Foundation | MANIPULATED ANTIBODY FC VARIANTS FOR INCREASED SERUM HALF-VALUE |
| MX2020004512A (es) | 2017-10-31 | 2020-08-13 | Anticuerpos anti apolipoproteina c-iii (anti-apoc3) y metodos de uso de los mismos. | |
| US10538583B2 (en) | 2017-10-31 | 2020-01-21 | Staten Biotechnology B.V. | Anti-APOC3 antibodies and compositions thereof |
| US11795579B2 (en) | 2017-12-11 | 2023-10-24 | Abalone Bio, Inc. | Yeast display of proteins in the periplasmic space |
| US11524991B2 (en) | 2018-04-18 | 2022-12-13 | Xencor, Inc. | PD-1 targeted heterodimeric fusion proteins containing IL-15/IL-15Ra Fc-fusion proteins and PD-1 antigen binding domains and uses thereof |
| CA3097741A1 (en) | 2018-04-18 | 2019-10-24 | Xencor, Inc. | Tim-3 targeted heterodimeric fusion proteins containing il-15/il-15ra fc-fusion proteins and tim-3 antigen binding domains |
| CN109097382A (zh) * | 2018-08-28 | 2018-12-28 | 东北农业大学 | 一种用于筛选特异性抗体可变区的方法 |
| MA53862A (fr) | 2018-10-12 | 2022-01-19 | Xencor Inc | Protéines de fusion fc d'il-15/il-15ralpha ciblant pd-1 et utilisations dans des polythérapies faisant intervenir celles-ci |
| AR117091A1 (es) | 2018-11-19 | 2021-07-07 | Bristol Myers Squibb Co | Anticuerpos monoclonales antagonistas contra cd40 y sus usos |
| CN113438961A (zh) | 2018-12-20 | 2021-09-24 | Xencor股份有限公司 | 含有IL-15/IL-15Rα和NKG2D抗原结合结构域的靶向异二聚体Fc融合蛋白 |
| TW202128757A (zh) | 2019-10-11 | 2021-08-01 | 美商建南德克公司 | 具有改善之特性的 PD-1 標靶 IL-15/IL-15Rα FC 融合蛋白 |
| EP4421091A1 (en) * | 2021-10-18 | 2024-08-28 | Korea University Research and Business Foundation | Human fc variants having improved fcgamma-riia binding selectivity |
| WO2023239940A1 (en) | 2022-06-10 | 2023-12-14 | Research Development Foundation | Engineered fcriib selective igg1 fc variants and uses thereof |
Family Cites Families (122)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL154598B (nl) | 1970-11-10 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van laagmoleculire verbindingen en van eiwitten die deze verbindingen specifiek kunnen binden, alsmede testverpakking. |
| US3817837A (en) | 1971-05-14 | 1974-06-18 | Syva Corp | Enzyme amplification assay |
| US3826364A (en) | 1972-05-22 | 1974-07-30 | Univ Leland Stanford Junior | Particle sorting method and apparatus |
| US3939350A (en) | 1974-04-29 | 1976-02-17 | Board Of Trustees Of The Leland Stanford Junior University | Fluorescent immunoassay employing total reflection for activation |
| US3996345A (en) | 1974-08-12 | 1976-12-07 | Syva Company | Fluorescence quenching with immunological pairs in immunoassays |
| US4277437A (en) | 1978-04-05 | 1981-07-07 | Syva Company | Kit for carrying out chemically induced fluorescence immunoassay |
| US4275149A (en) | 1978-11-24 | 1981-06-23 | Syva Company | Macromolecular environment control in specific receptor assays |
| US4284412A (en) | 1979-07-13 | 1981-08-18 | Ortho Diagnostics, Inc. | Method and apparatus for automated identification and enumeration of specified blood cell subclasses |
| US4366241A (en) | 1980-08-07 | 1982-12-28 | Syva Company | Concentrating zone method in heterogeneous immunoassays |
| US4957939A (en) | 1981-07-24 | 1990-09-18 | Schering Aktiengesellschaft | Sterile pharmaceutical compositions of gadolinium chelates useful enhancing NMR imaging |
| US4498766A (en) | 1982-03-25 | 1985-02-12 | Becton, Dickinson And Company | Light beam focal spot elongation in flow cytometry devices |
| US4472509A (en) | 1982-06-07 | 1984-09-18 | Gansow Otto A | Metal chelate conjugated monoclonal antibodies |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4661913A (en) | 1984-09-11 | 1987-04-28 | Becton, Dickinson And Company | Apparatus and method for the detection and classification of articles using flow cytometry techniques |
| US4883750A (en) | 1984-12-13 | 1989-11-28 | Applied Biosystems, Inc. | Detection of specific sequences in nucleic acids |
| US4767206A (en) | 1984-12-24 | 1988-08-30 | Flow Cytometry Standards Corporation | Calibration method for flow cytometry using fluorescent microbeads and synthesis thereof |
| US4857451A (en) | 1984-12-24 | 1989-08-15 | Flow Cytometry Standards Corporation | Method of compensating and calibrating a flow cytometer, and microbead standards kit therefor |
| US4774189A (en) | 1984-12-24 | 1988-09-27 | Flow Cytometry Standards Corp. | Fluorescent calibration microbeads simulating stained cells |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4989977A (en) | 1985-07-29 | 1991-02-05 | Becton, Dickinson And Company | Flow cytometry apparatus with improved light beam adjustment |
| US4938948A (en) | 1985-10-07 | 1990-07-03 | Cetus Corporation | Method for imaging breast tumors using labeled monoclonal anti-human breast cancer antibodies |
| US5618920A (en) | 1985-11-01 | 1997-04-08 | Xoma Corporation | Modular assembly of antibody genes, antibodies prepared thereby and use |
| US5576195A (en) | 1985-11-01 | 1996-11-19 | Xoma Corporation | Vectors with pectate lyase signal sequence |
| US4714682A (en) | 1985-12-11 | 1987-12-22 | Flow Cytometry Standards Corporation | Fluorescent calibration microbeads simulating stained cells |
| US4800159A (en) | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| AU622104B2 (en) | 1987-03-11 | 1992-04-02 | Sangtec Molecular Diagnostics Ab | Method of assaying of nucleic acids, a reagent combination and kit therefore |
| WO1988007089A1 (en) | 1987-03-18 | 1988-09-22 | Medical Research Council | Altered antibodies |
| IL86724A (en) | 1987-06-19 | 1995-01-24 | Siska Diagnostics Inc | Methods and kits for amplification and testing of nucleic acid sequences |
| US4988618A (en) | 1987-11-16 | 1991-01-29 | Gene-Trak Systems | Magnetic separation device and methods for use in heterogeneous assays |
| CA1323293C (en) | 1987-12-11 | 1993-10-19 | Keith C. Backman | Assay using template-dependent nucleic acid probe reorganization |
| JP2846018B2 (ja) | 1988-01-21 | 1999-01-13 | ジェネンテク,インコーポレイテッド | 核酸配列の増幅および検出 |
| CA1340807C (en) | 1988-02-24 | 1999-11-02 | Lawrence T. Malek | Nucleic acid amplification process |
| US5858652A (en) | 1988-08-30 | 1999-01-12 | Abbott Laboratories | Detection and amplification of target nucleic acid sequences |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| DE68913658T3 (de) | 1988-11-11 | 2005-07-21 | Stratagene, La Jolla | Klonierung von Immunglobulin Sequenzen aus den variablen Domänen |
| US4932207A (en) | 1988-12-28 | 1990-06-12 | Sundstrand Corporation | Segmented seal plate for a turbine engine |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5302523A (en) | 1989-06-21 | 1994-04-12 | Zeneca Limited | Transformation of plant cells |
| US5550318A (en) | 1990-04-17 | 1996-08-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US7705215B1 (en) | 1990-04-17 | 2010-04-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5322783A (en) | 1989-10-17 | 1994-06-21 | Pioneer Hi-Bred International, Inc. | Soybean transformation by microparticle bombardment |
| US5578464A (en) | 1989-10-31 | 1996-11-26 | Schering Corporation | E. coli secretory strains |
| US5484956A (en) | 1990-01-22 | 1996-01-16 | Dekalb Genetics Corporation | Fertile transgenic Zea mays plant comprising heterologous DNA encoding Bacillus thuringiensis endotoxin |
| US5160974A (en) | 1990-06-25 | 1992-11-03 | Flow Science, Inc. | Closed sample cell for use in flow cytometry |
| US6172197B1 (en) | 1991-07-10 | 2001-01-09 | Medical Research Council | Methods for producing members of specific binding pairs |
| US6916605B1 (en) | 1990-07-10 | 2005-07-12 | Medical Research Council | Methods for producing members of specific binding pairs |
| US5384253A (en) | 1990-12-28 | 1995-01-24 | Dekalb Genetics Corporation | Genetic transformation of maize cells by electroporation of cells pretreated with pectin degrading enzymes |
| US5478722A (en) | 1991-02-17 | 1995-12-26 | The Curators Of The University Of Missouri | Preserved cell preparations for flow cytometry and immunology |
| US5858657A (en) | 1992-05-15 | 1999-01-12 | Medical Research Council | Methods for producing members of specific binding pairs |
| US5994514A (en) | 1991-08-14 | 1999-11-30 | Genentech, Inc. | Immunoglobulin variants |
| AU2515992A (en) | 1991-08-20 | 1993-03-16 | Genpharm International, Inc. | Gene targeting in animal cells using isogenic dna constructs |
| US5610042A (en) | 1991-10-07 | 1997-03-11 | Ciba-Geigy Corporation | Methods for stable transformation of wheat |
| PT1024191E (pt) | 1991-12-02 | 2008-12-22 | Medical Res Council | Produção de auto-anticorpos a partir de reportórios de segmentos de anticorpo e exibidos em fagos |
| AU4116793A (en) | 1992-04-24 | 1993-11-29 | Board Of Regents, The University Of Texas System | Recombinant production of immunoglobulin-like domains in prokaryotic cells |
| US5702932A (en) | 1992-07-20 | 1997-12-30 | University Of Florida | Microinjection methods to transform arthropods with exogenous DNA |
| AU670316B2 (en) | 1992-07-27 | 1996-07-11 | Pioneer Hi-Bred International, Inc. | An improved method of (agrobacterium)-mediated transformation of cultured soybean cells |
| GB9222888D0 (en) | 1992-10-30 | 1992-12-16 | British Tech Group | Tomography |
| US5846709A (en) | 1993-06-15 | 1998-12-08 | Imclone Systems Incorporated | Chemical process for amplifying and detecting nucleic acid sequences |
| FR2708288B1 (fr) | 1993-07-26 | 1995-09-01 | Bio Merieux | Procédé d'amplification d'acides nucléiques par transcription utilisant le déplacement, réactifs et nécessaire pour la mise en Óoeuvre de ce procédé. |
| US5928905A (en) | 1995-04-18 | 1999-07-27 | Glaxo Group Limited | End-complementary polymerase reaction |
| DE69527355T2 (de) | 1994-05-28 | 2003-03-06 | Tepnel Medical Ltd., Manchester | Herstellung von nukleinsaeurekopien |
| US5656610A (en) | 1994-06-21 | 1997-08-12 | University Of Southern California | Producing a protein in a mammal by injection of a DNA-sequence into the tongue |
| US5942391A (en) | 1994-06-22 | 1999-08-24 | Mount Sinai School Of Medicine | Nucleic acid amplification method: ramification-extension amplification method (RAM) |
| CA2195562A1 (en) | 1994-08-19 | 1996-02-29 | Pe Corporation (Ny) | Coupled amplification and ligation method |
| US5567326A (en) | 1994-09-19 | 1996-10-22 | Promega Corporation | Multisample magnetic separation device |
| US5736524A (en) | 1994-11-14 | 1998-04-07 | Merck & Co.,. Inc. | Polynucleotide tuberculosis vaccine |
| US5935825A (en) | 1994-11-18 | 1999-08-10 | Shimadzu Corporation | Process and reagent for amplifying nucleic acid sequences |
| US5843650A (en) | 1995-05-01 | 1998-12-01 | Segev; David | Nucleic acid detection and amplification by chemical linkage of oligonucleotides |
| US5702892A (en) | 1995-05-09 | 1997-12-30 | The United States Of America As Represented By The Department Of Health And Human Services | Phage-display of immunoglobulin heavy chain libraries |
| CA2262403C (en) | 1995-07-31 | 2011-09-20 | Urocor, Inc. | Biomarkers and targets for diagnosis, prognosis and management of prostate disease |
| US6696248B1 (en) | 1995-08-18 | 2004-02-24 | Morphosys Ag | Protein/(poly)peptide libraries |
| US5916779A (en) | 1995-09-21 | 1999-06-29 | Becton, Dickinson And Company | Strand displacement amplification of RNA targets |
| FR2739629B1 (fr) | 1995-10-06 | 1997-12-26 | Systems Bio Ind | Utilisation d'un systeme de secretion sec-dependant pour secreter des proteines normalement secretees par un systeme de secretion sec-independant, bacteries les contenant et leur utilisation |
| US5780448A (en) | 1995-11-07 | 1998-07-14 | Ottawa Civic Hospital Loeb Research | DNA-based vaccination of fish |
| US5612473A (en) | 1996-01-16 | 1997-03-18 | Gull Laboratories | Methods, kits and solutions for preparing sample material for nucleic acid amplification |
| US5928906A (en) | 1996-05-09 | 1999-07-27 | Sequenom, Inc. | Process for direct sequencing during template amplification |
| US5939291A (en) | 1996-06-14 | 1999-08-17 | Sarnoff Corporation | Microfluidic method for nucleic acid amplification |
| US5945100A (en) | 1996-07-31 | 1999-08-31 | Fbp Corporation | Tumor delivery vehicles |
| US5849546A (en) | 1996-09-13 | 1998-12-15 | Epicentre Technologies Corporation | Methods for using mutant RNA polymerases with reduced discrimination between non-canonical and canonical nucleoside triphosphates |
| US5981274A (en) | 1996-09-18 | 1999-11-09 | Tyrrell; D. Lorne J. | Recombinant hepatitis virus vectors |
| US5779907A (en) | 1996-12-06 | 1998-07-14 | Systems Research Laboratories, Inc. | Magnetic microplate separator |
| GB9701425D0 (en) | 1997-01-24 | 1997-03-12 | Bioinvent Int Ab | A method for in vitro molecular evolution of protein function |
| US5849497A (en) | 1997-04-03 | 1998-12-15 | The Research Foundation Of State University Of New York | Specific inhibition of the polymerase chain reaction using a non-extendable oligonucleotide blocker |
| US5866366A (en) | 1997-07-01 | 1999-02-02 | Smithkline Beecham Corporation | gidB |
| WO1999006587A2 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| US5916776A (en) | 1997-08-27 | 1999-06-29 | Sarnoff Corporation | Amplification method for a polynucleotide |
| US5994624A (en) | 1997-10-20 | 1999-11-30 | Cotton Incorporated | In planta method for the production of transgenic plants |
| US5932451A (en) | 1997-11-19 | 1999-08-03 | Incyte Pharmaceuticals, Inc. | Method for unbiased mRNA amplification |
| US6528624B1 (en) | 1998-04-02 | 2003-03-04 | Genentech, Inc. | Polypeptide variants |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| KR20060067983A (ko) | 1999-01-15 | 2006-06-20 | 제넨테크, 인크. | 효과기 기능이 변화된 폴리펩티드 변이체 |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US7183387B1 (en) | 1999-01-15 | 2007-02-27 | Genentech, Inc. | Polypeptide variants with altered effector function |
| EP1178785B1 (en) | 1999-05-06 | 2008-12-24 | Wake Forest University | Compositions and methods for identifying antigens which elicit an immune response |
| EP1077263A1 (de) | 1999-07-29 | 2001-02-21 | F.Hoffmann-La Roche Ag | Verfahren zur Herstellung von natürlich gefalteten und sekretierten Proteinen durch Co-Sekretion von Chaperonen |
| DK1354056T3 (da) | 2000-10-10 | 2015-02-23 | Danisco Us Inc | Forbedret sekretion af et polypeptid ved hjælp af en mikroorganisme |
| US7094571B2 (en) | 2000-10-27 | 2006-08-22 | The Board Of Regents Of The University Of Texas System | Combinatorial protein library screening by periplasmic expression |
| US6979556B2 (en) | 2000-12-14 | 2005-12-27 | Genentech, Inc. | Separate-cistron contructs for secretion of aglycosylated antibodies from prokaryotes |
| AU2002238449A1 (en) | 2000-12-20 | 2002-07-01 | Altana Pharma Ag | Process for the production and purification of secreted proteins ans the production of arrays of proteins |
| US7419783B2 (en) | 2001-11-05 | 2008-09-02 | Research Development Foundation | Engineering of leader peptides for the secretion of recombinant proteins in bacteria |
| US20040002587A1 (en) | 2002-02-20 | 2004-01-01 | Watkins Jeffry D. | Fc region variants |
| US20040132101A1 (en) | 2002-09-27 | 2004-07-08 | Xencor | Optimized Fc variants and methods for their generation |
| US7317091B2 (en) | 2002-03-01 | 2008-01-08 | Xencor, Inc. | Optimized Fc variants |
| US8188231B2 (en) | 2002-09-27 | 2012-05-29 | Xencor, Inc. | Optimized FC variants |
| US7662925B2 (en) | 2002-03-01 | 2010-02-16 | Xencor, Inc. | Optimized Fc variants and methods for their generation |
| DE60310385T2 (de) | 2002-03-23 | 2007-09-20 | Research Development Foundation, Carson City | Sekretion von proteinen mit mehreren disulfidbindungen in bakterien und verwendungen davon |
| US20060235208A1 (en) | 2002-09-27 | 2006-10-19 | Xencor, Inc. | Fc variants with optimized properties |
| US7217798B2 (en) * | 2003-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of Fc-fusion protein serum half-lives by mutagenesis |
| EP2368578A1 (en) | 2003-01-09 | 2011-09-28 | Macrogenics, Inc. | Identification and engineering of antibodies with variant Fc regions and methods of using same |
| US7960512B2 (en) | 2003-01-09 | 2011-06-14 | Macrogenics, Inc. | Identification and engineering of antibodies with variant Fc regions and methods of using same |
| US8084582B2 (en) | 2003-03-03 | 2011-12-27 | Xencor, Inc. | Optimized anti-CD20 monoclonal antibodies having Fc variants |
| WO2005018572A2 (en) * | 2003-08-22 | 2005-03-03 | Biogen Idec Ma Inc. | Improved antibodies having altered effector function and methods for making the same |
| US20050249723A1 (en) | 2003-12-22 | 2005-11-10 | Xencor, Inc. | Fc polypeptides with novel Fc ligand binding sites |
| EP1737890A2 (en) | 2004-03-24 | 2007-01-03 | Xencor, Inc. | Immunoglobulin variants outside the fc region |
| US7229792B2 (en) | 2004-05-03 | 2007-06-12 | Vinod Pandiripally | Method of producing recombinant proteins |
| WO2007024249A2 (en) | 2004-11-10 | 2007-03-01 | Macrogenics, Inc. | Engineering fc antibody regions to confer effector function |
| EP2845865A1 (en) | 2004-11-12 | 2015-03-11 | Xencor Inc. | Fc variants with altered binding to FcRn |
| CA2595169A1 (en) * | 2005-01-12 | 2006-07-20 | Xencor, Inc. | Antibodies and fc fusion proteins with altered immunogenicity |
| US8217147B2 (en) | 2005-08-10 | 2012-07-10 | Macrogenics, Inc. | Identification and engineering of antibodies with variant Fc regions and methods of using same |
| US8312249B1 (en) | 2008-10-10 | 2012-11-13 | Apple Inc. | Dynamic trampoline and structured code generation in a signed code environment |
| FR2957821B1 (fr) | 2010-03-24 | 2014-08-29 | Inst Francais Du Petrole | Nouvelle zone de regeneration du catalyseur divisee en secteurs pour unites catalytiques regeneratives |
-
2008
- 2008-04-30 US US12/112,971 patent/US8629245B2/en active Active
- 2008-04-30 CA CA2685675A patent/CA2685675C/en not_active Expired - Fee Related
- 2008-04-30 AU AU2008247819A patent/AU2008247819B2/en not_active Ceased
- 2008-04-30 WO PCT/US2008/062090 patent/WO2008137475A2/en active Application Filing
- 2008-04-30 DK DK08747239.5T patent/DK2155789T3/da active
- 2008-04-30 EP EP08747239.5A patent/EP2155789B1/en not_active Not-in-force
-
2013
- 2013-12-13 US US14/105,642 patent/US10725037B2/en active Active
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019078591A1 (ko) * | 2017-10-20 | 2019-04-25 | 국민대학교 산학협력단 | 세포막 유동성을 이용한 다중체 단백질 디스플레이 시스템 |
| KR20190044347A (ko) * | 2017-10-20 | 2019-04-30 | 국민대학교산학협력단 | 세포막 유동성을 이용한 다중체 단백질 디스플레이 시스템 |
Also Published As
| Publication number | Publication date |
|---|---|
| US8629245B2 (en) | 2014-01-14 |
| CA2685675C (en) | 2016-02-16 |
| EP2155789A2 (en) | 2010-02-24 |
| WO2008137475A3 (en) | 2009-01-08 |
| AU2008247819B2 (en) | 2013-02-14 |
| US20090136936A1 (en) | 2009-05-28 |
| CA2685675A1 (en) | 2008-11-13 |
| WO2008137475A2 (en) | 2008-11-13 |
| DK2155789T3 (da) | 2013-10-21 |
| AU2008247819A1 (en) | 2008-11-13 |
| US10725037B2 (en) | 2020-07-28 |
| US20140179550A1 (en) | 2014-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2155789B1 (en) | Immunoglobulin fc libraries | |
| US9244070B2 (en) | Immunoglobulin libraries | |
| AU2010273763B2 (en) | Immunoglobulin Fc polypeptides | |
| EP2673299B1 (en) | Engineered immunoglobulin fc polypeptides | |
| EP1907610B1 (en) | Secretion of antibodies without signal peptides from bacteria | |
| WO2005005638A2 (en) | High affinity fusion proteins (rc-scfv-fc or scfv-scfv-fc) binding cytokines, particularly il-6 & il-18 | |
| US9890216B2 (en) | Antibodies with engineered IgG Fc domains | |
| KR101763345B1 (ko) | 단백질 조합 기반의 Fv 라이브러리 및 이의 제조 방법 | |
| US20220348654A1 (en) | AGLYCOSYLATED ANTIBODY Fc REGION FOR TREATING CANCER | |
| GEORGIOU et al. | Patent 2766065 Summary |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20091127 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA MK RS |
|
| 17Q | First examination report despatched |
Effective date: 20110516 |
|
| DAX | Request for extension of the european patent (deleted) | ||
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| INTG | Intention to grant announced |
Effective date: 20130326 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 623403 Country of ref document: AT Kind code of ref document: T Effective date: 20130815 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602008026244 Country of ref document: DE Effective date: 20130919 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: PATENTANWAELTE SCHAAD, BALASS, MENZL AND PARTN, CH |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 Effective date: 20131014 Ref country code: DK Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: TRGR |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 623403 Country of ref document: AT Kind code of ref document: T Effective date: 20130724 |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG4D |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20131125 Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20131024 Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130821 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20131124 Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20131025 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20140425 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602008026244 Country of ref document: DE Effective date: 20140425 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 Ref country code: LU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140430 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 9 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT; INVALID AB INITIO Effective date: 20080430 Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130724 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 10 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20210412 Year of fee payment: 14 Ref country code: DE Payment date: 20210408 Year of fee payment: 14 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IE Payment date: 20210409 Year of fee payment: 14 Ref country code: SE Payment date: 20210413 Year of fee payment: 14 Ref country code: GB Payment date: 20210408 Year of fee payment: 14 Ref country code: DK Payment date: 20210412 Year of fee payment: 14 Ref country code: CH Payment date: 20210416 Year of fee payment: 14 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20210415 Year of fee payment: 14 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 602008026244 Country of ref document: DE Representative=s name: SIMMONS & SIMMONS LLP, DE |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 602008026244 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP Effective date: 20220430 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MM Effective date: 20220501 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20220430 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: EUG |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220501 Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220501 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220430 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220430 Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220430 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20221103 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220430 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220430 Ref country code: DK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220430 |