EP0416744B1 - Rückgewinnung von Tertiärbutylhydroperoxid und Tertiärbutylalkohol - Google Patents
Rückgewinnung von Tertiärbutylhydroperoxid und Tertiärbutylalkohol Download PDFInfo
- Publication number
- EP0416744B1 EP0416744B1 EP90308547A EP90308547A EP0416744B1 EP 0416744 B1 EP0416744 B1 EP 0416744B1 EP 90308547 A EP90308547 A EP 90308547A EP 90308547 A EP90308547 A EP 90308547A EP 0416744 B1 EP0416744 B1 EP 0416744B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- tertiary butyl
- fraction
- butyl alcohol
- propylene
- butyl hydroperoxide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 title claims description 95
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 title claims description 84
- 238000011084 recovery Methods 0.000 title description 24
- 238000006735 epoxidation reaction Methods 0.000 claims description 55
- 239000003054 catalyst Substances 0.000 claims description 44
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims description 43
- 229910052750 molybdenum Inorganic materials 0.000 claims description 43
- 239000011733 molybdenum Substances 0.000 claims description 43
- 238000004821 distillation Methods 0.000 claims description 39
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 claims description 28
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 28
- 239000012535 impurity Substances 0.000 claims description 23
- 238000000034 method Methods 0.000 claims description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 20
- 239000007788 liquid Substances 0.000 claims description 20
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 claims description 19
- 239000007795 chemical reaction product Substances 0.000 claims description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 15
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 claims description 14
- 239000002253 acid Substances 0.000 claims description 13
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 12
- 239000000920 calcium hydroxide Substances 0.000 claims description 8
- 229910001861 calcium hydroxide Inorganic materials 0.000 claims description 8
- 239000000292 calcium oxide Substances 0.000 claims description 8
- ODINCKMPIJJUCX-UHFFFAOYSA-N calcium oxide Inorganic materials [Ca]=O ODINCKMPIJJUCX-UHFFFAOYSA-N 0.000 claims description 8
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 claims description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 7
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 claims description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 6
- BRPQOXSCLDDYGP-UHFFFAOYSA-N calcium oxide Chemical compound [O-2].[Ca+2] BRPQOXSCLDDYGP-UHFFFAOYSA-N 0.000 claims description 6
- 235000019253 formic acid Nutrition 0.000 claims description 6
- 150000001735 carboxylic acids Chemical class 0.000 claims description 5
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 claims description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 4
- 239000000706 filtrate Substances 0.000 claims description 4
- 238000007738 vacuum evaporation Methods 0.000 claims description 3
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 claims description 2
- 238000002360 preparation method Methods 0.000 claims description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 claims 2
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical group CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 claims 1
- RUPAXCPQAAOIPB-UHFFFAOYSA-N tert-butyl formate Chemical compound CC(C)(C)OC=O RUPAXCPQAAOIPB-UHFFFAOYSA-N 0.000 claims 1
- 238000001704 evaporation Methods 0.000 description 24
- 230000008020 evaporation Effects 0.000 description 22
- 239000010408 film Substances 0.000 description 16
- 239000000463 material Substances 0.000 description 16
- 239000000047 product Substances 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 11
- 238000000354 decomposition reaction Methods 0.000 description 11
- 239000000243 solution Substances 0.000 description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- 241001279686 Allium moly Species 0.000 description 7
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- 235000011116 calcium hydroxide Nutrition 0.000 description 6
- 239000011552 falling film Substances 0.000 description 6
- 229930195733 hydrocarbon Natural products 0.000 description 6
- 150000002430 hydrocarbons Chemical class 0.000 description 6
- 229940074869 marquis Drugs 0.000 description 5
- 238000009835 boiling Methods 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 230000018044 dehydration Effects 0.000 description 4
- 238000006297 dehydration reaction Methods 0.000 description 4
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- -1 alkyl hydroperoxides Chemical group 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- QMGLMRPHOITLSN-UHFFFAOYSA-N 2,4-dimethyloxolane Chemical compound CC1COC(C)C1 QMGLMRPHOITLSN-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical group CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000000446 fuel Substances 0.000 description 2
- 239000001282 iso-butane Substances 0.000 description 2
- BHIWKHZACMWKOJ-UHFFFAOYSA-N methyl isobutyrate Chemical compound COC(=O)C(C)C BHIWKHZACMWKOJ-UHFFFAOYSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- GQCZPFJGIXHZMB-UHFFFAOYSA-N 1-tert-Butoxy-2-propanol Chemical class CC(O)COC(C)(C)C GQCZPFJGIXHZMB-UHFFFAOYSA-N 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 150000001734 carboxylic acid salts Chemical class 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- SWXVUIWOUIDPGS-UHFFFAOYSA-N diacetone alcohol Natural products CC(=O)CC(C)(C)O SWXVUIWOUIDPGS-UHFFFAOYSA-N 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- WDAXFOBOLVPGLV-UHFFFAOYSA-N isobutyric acid ethyl ester Natural products CCOC(=O)C(C)C WDAXFOBOLVPGLV-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 150000002751 molybdenum Chemical class 0.000 description 1
- 239000005078 molybdenum compound Substances 0.000 description 1
- 150000002752 molybdenum compounds Chemical class 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002924 oxiranes Chemical class 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 238000004457 water analysis Methods 0.000 description 1
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D301/00—Preparation of oxiranes
- C07D301/02—Synthesis of the oxirane ring
- C07D301/03—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds
- C07D301/19—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds with organic hydroperoxides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/74—Separation; Purification; Use of additives, e.g. for stabilisation
- C07C29/76—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment
- C07C29/80—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment by distillation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C407/00—Preparation of peroxy compounds
- C07C407/003—Separation; Purification; Stabilisation; Use of additives
Definitions
- This invention relates to the recovery of tertiary butyl hydroperoxide and tertiary butyl alcohol from a heavy distillation fraction comprising tertiary butyl hydroperoxide, tertiary butyl alcohol and impurities, including dissolved molybdenum catalyst.
- this invention relates to the recovery of tertiary butyl hydroperoxide and tertiary butyl alcohol from a heavy distillation fraction comprising tertiary butyl hydroperoxide, tertiary butyl alcohol and impurities, including dissolved molybdenum catalyst which remains after a distillate propylene fraction, a distillate propylene oxide fraction and a distillate tertiary butyl alcohol fraction are removed, by distillation, from the reaction product formed by the reaction of excess propylene with tertiary butyl hydroperoxide in solution in tertiary butyl alcohol in the presence of a soluble molybdenum catalyst.
- this invention relates to the separation of a a heavy distillation fraction comprising tertiary butyl hydroperoxide, tertiary butyl alcohol and impurities, including dissolved molybdenum catalyst in appropriate vacuum evaporation equipment such as a thin film evaporator, a wiped film evaporator, a forced circulation evaporator, etc., under evaporation conditions including a temperature of 25° to 100°C and a pressure of 0.25 to 27 kPa (2 to 200 mm Hg.) into a lighter condensate fraction comprising 60 to 95 wt.% of the heavy distillation fraction and containing from 70 to 95 wt.% of tertiary butyl alcohol, 1 to 20 wt.% of the tertiary butyl hydroperoxide and from 15 to 3 wt.% of impurites and also into a clear liquid heavier residue fraction comprising tertiary butyl alcohol, tertiary butyl hydroperoxid
- the heavy distillation fraction remaining after removing unreacted propylene, propylene oxide and tertiary butyl alcohol from the reaction product formed by the reaction of excess propylene with tertiary butyl hydroperoxide in solution in tertiary butyl alcohol in the presence of a soluble molybdenum catalyst has generally been considered to be a "waste" product to be treated for the recovery of molybdenum therefrom in order to permit use of the molybdenum-free components as fuel.
- a process for the coproduction of an epoxide such as propylene oxide with an alcohol such as tertiary butyl alcohol is disclosed and described in Kollar U. S.-A-3,350,422 and 3,351,635.
- the olefin is propylene and the hydroperoxide is tertiary butyl hydroperoxide
- the principal reaction products are propylene oxide and tertiary butyl alcohol.
- propylene and tertiary butyl hydroperoxide are reacted in solution in tertiary butyl alcohol in an epoxidation reaction zone in the presence of a soluble molybdenum catalyst under epoxidation reaction conditions to form an epoxidation reaction product which is discharged from the epoxidation reaction zone and charged to a distillation zone containing an appropriate number of distillation columns wherein the epoxidation reaction product is typically resolved into a distillate propylene recycle fraction, a distillate propylene oxide product fraction and a distillate tertiary butyl alcohol fraction.
- a heavy liquid distillation fraction normally a bottoms fraction, will remain after the separation of the unreacted propylene, the propylene oxide and the tertiary butyl alcohol which will be composed of unreacted tertiary butyl hydroperoxide, tertiary butyl alcohol, impurities and the dissolved molybdenum catalyst.
- Levine U. S.-A-3,819,663 is directed to a method for treating a heavy distillation fraction of this nature in order to recover the molybdenum in the concentrated bottoms fraction for recycle to the epoxidation reaction zone as makeup catalyst.
- Levine conducts his wiped-film evaporation process under conditions including a temperature of 273 to 330°C (550-650°F) at atmospheric pressure to obtain his desired residual fraction for recycle as catalyst makeup and a distillate fraction comprising about 85% or more of the heavy distillation fraction.
- the high temperatures used by Levine and the concentration of molybdenum in his wiped film evaporator are such that at least partial dehydration of the tertiary butyl hydroperoxide and tertiary butyl alcohol will occur.
- One of Levine's objectives is the provision of a molybdenum-free overhead that can be burned as a fuel.
- Sweed describes a process for evaporation of the heavy distillation fraction in order to provide a spent catalyst solution containing between about 0.1 and 2.0 wt.% of molybdenum.
- the evaporation is accomplished under special evaporation conditions at a pressure of less than about 52 kPa (400 mm Hg) in a circulation evaporator and heating means designed so that the feed to the evaporator is not preheated under pressure.
- Thornton et al. U. S.-A-4,584,413 discloses a process for removing contaminating quantities of primary and secondary alkyl hydroperoxides from tertiary butyl hydroperoxide formed by the oxidation of isobutane by mixing an isobutane oxidation product such as one comprising tertiary butyl hydroperoxide, tertiary butyl alcohol and oxygenated by-products including primary and secondary alkyl hydroperoxides with an aqueous solution of an alkali metal or alkaline earth metal hydroxide followed by distillation of the mixture to obtain a tertiary butyl alcohol fraction and a two-phase tertiary butyl hydroperoxide azeotrope fraction, the tertiary butyl hydroperoxide phase of which is neutralized in order to provide a more purified tertiary butyl hydroperoxide product.
- an isobutane oxidation product such as one comprising
- a heavy distillation fraction comprising tertiary butyl hydroperoxide, tertiary butyl alcohol, impurities and dissolved molybdenum catalyst resulting from the removal of propylene, propylene oxide and tertiary butyl alcohol from an epoxidation reaction product is further fractionated in a falling film, wiped-film, or forced circulation evaporator under evaporating conditions including a temperature of 25 to 100°C.
- an overhead fraction comprising 60 to 95 wt.% of the charged heavy distillation fraction and where said overhead fraction is composed of from 70 to 96 wt.% of tertiary butyl alcohol and 1 to 20 wt.% of tertiary butyl hydroperoxide.
- an epoxidation reaction mixture which will contain not only unreacted feed components and the desired propylene oxide and tertiary butyl alcohol, but also the soluble molybdenum catalyst and impurities including oxygen-containing impurities and hydrocarbon impurities such as methyl formate, acetone, isobutyraldehyde, methanol, isopropyl alcohol, ditertiary butyl peroxide, formic acid, acetic acid, isobutyric acid, methyl formate, methyl acetate, and methyl isobutyrate, and hydrocarbons resulting from the undesired polymerization of propylene such as hydrocarbons containing 6 or more carbon atoms.
- this heavy distillation fraction is used as a charge stock for a falling film, wiped-film or forced circulation evaporator which is operated under evaporating conditions such as a temperature of 25° to 100°C, such as 50° to 100°C. and a pressure of 0.25 to 27 kPa (2 to 200 mm Hg.)
- the heavy liquid distillation fraction is resolved in the falling film, wiped-film or forced circulation evaporator, into an overhead fraction comprising from 60 to 95 wt.% of the charged heavy liquid residue fraction and a bottoms fraction comprising the balance.
- the heavy liquid distillation bottoms fraction that is obtained by the process of the present invention is a liquid fraction which can be handled with comparative ease insofar as its disposal is concerned. Typically, this heavy residual fraction will be sold to a company that reclaims metals from hydrocarbon fractions in order that the molybdenum contained therein may be recovered for reuse.
- the evaporated overhead fraction obtained in the falling film, wiped-film or forced circulation evaporator will typically contain from 60 to 95 wt.% of tertiary butyl alcohol, 1 to 20 wt.% of tertiary butyl hydroperoxide and, correspondingly, from 15 to 3 wt.% of impurities, principally oxygen-containing impurities having boiling points of less than about 250°C. at atmospheric pressure.
- impurities that will typically be present are impurities such as formic acid, acetic acid and isobutyric acid and propylene glycol esters.
- the lighter evaporated overhead condensate fraction from the vacuum evaporation equipment e.g., a falling film evaporator, a wiped film evaporator, a forced circulation evaporator, etc.
- the vacuum evaporation equipment e.g., a falling film evaporator, a wiped film evaporator, a forced circulation evaporator, etc.
- lighter evaporated condensate fraction has an acid number of more than about 12 meq/g, it is usually unsuitable for direct recycle because of the high content of carboxylic acids, which will normally lead to poor propylene oxide selectivities because the high acidity promotes reaction of propylene oxide with alcohols present in the reaction mixture.
- the lighter evaporated condensate fraction can be treated with calcium oxide and/or hydroxide in the manner disclosed and claimed in EP-A-0415572.
- the lighter evaporated condensate fraction can be charged to a treating zone and treated with 1/2 to 1 equivalents of calcium oxide and/or calcium hydroxide, based on the carboxylic acid content of the condensate fraction to form a slurry of partially precipitated carboxylic acid impurities.
- the precipitate may be separated from the treated product by any suitable means such as centrifugation, filtration, etc., to provide a filtrate that is suitable for reycle to the epoxidation reaction zone, in that precipitation of the molybdenum catalyst will not result from the recycle operation.
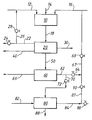
- the figure is a schematic diagram of the reaction and purification sequence that is used in the course of the present invention for the recovery of tertiary butyl alcohol and tertiary butyl hydroxide from a heavy distillation fraction recovered from the reaction product formed by the reaction of propylene with tertiary butyl hydroxide in the presence of a soluble molybdenum catalyst to provide propylene oxide and additional tertiary butyl alcohol.
- An epoxidation reaction zone 10 is provided and propylene is charged thereto by a line 12 together with a soluble molybdenum catalyst charged by a line 14 and a solution of tertiary butyl hydroperoxide and tertiary butyl alcohol charged by a line 16.
- the epoxidation reaction is an epoxidation reaction of the type disclosed by Kollar U. S.-A-3,351,653 as further elaborated upon, for example, in GB-A-1,298,253 wherein propylene is reacted with tertiary butyl hydroperoxide under reaction conditions including a reaction temperature within the range of 82° to 149°C (180° to 300°F.), a pressure of 2.18 to 7 MPa (300 to 1000 psig.) and, more preferably, a temperature of 180°C to 138°C (220°F. to 280°F) and a pressure of 3.5 to 5.6 MPa (500 to 800 psig.)
- the soluble molybdenum catalyst charged to the epoxidation reaction zone by the line 14 may be an epoxidation catalyst of the type known in the art such as those disclosed by the Kollar patent or the British patent or by Marquis et al. U. S.-A-4,626,596, U. S.-A-4,650,886, U. S.-A-4,654,427, or U. S.-A-4,758,681.
- the Marquis et al. patents are directed to molybdenum/alkanol complexes such as solutions of molybdenum compounds in ethylene glycol which contain a high concentration of molybdenum and are particularly useful as catalysts in the epoxidation reaction. Marquis et al.
- the tertiary butyl hydroperoxide that is charged to the epoxidation reaction zone 10 by way of line 16 is a 40 to 75 wt.% solution of tertiary butyl hydroperoxide in tertiary butyl alcohol.
- the catalyst is charged to the epoxidation reaction zone 10 by the charge line 14 in an amount such as to provide from 50 to 1000 ppm of molybdenum, based on the total of the reactants charged and, more preferably, from 200 to 600 ppm.
- the reaction is preferably conducted at superatmospheric pressure such as a pressure of 2.2 to 7.0 MPa (300 to 1000 psig).
- the feed materials are charged to the epoxidation reaction zone 10 through the lines 12, 14 and 16 at rates sufficient to maintain the desired concentration of reactants and an equivalent volume of epoxidation reaction mixture is withdrawn from the epoxidation reaction zone 10 by way of a discharge line 18.
- the reaction product discharged by the line 18 will normally comprise unreacted propylene, a minor amount of unreacted tertiary butyl hydroperoxide, propylene oxide, tertiary butyl alcohol, including tertiary butyl alcohol formed by the reaction of the tertiary butyl hydroperoxide with propylene, the molybdenum catalyst and impurities such as propane, propionaldehyde, acetone, methanol, isopropanol, water, acetaldehyde, methyl formate, acetic acid, formic acid, isobutyric acid, hydrocarbons containing 6 or more carbon atoms and high boiling residue components.
- the reaction product 18 is charged to an epoxidation reaction product distillation zone 20 where it is separated by distillation into desired fractions in accordance with methods known to those skilled in the art.
- the distillation sequence disclosed in GB-A-1,298,253 may be used.
- One of the distillate products that is recovered in the zone 20 is a propylene fraction which is discharged by a line 22 controlled by a valve 24 and provided with a branch line 26 controlled by a valve 28 in order to permit the recycle of unreacted propylene to the epoxidation reaction zone 10 through the propylene charge line 12.
- the propylene oxide product fraction may be purified in the epoxidation reaction distillation zone 20 by known techniques such as, for example, those disclosed in Burnes et al. U. S.-A-3,715,284, Schmidt et al. U. S.-A-3,909,366, Schmidt U. S.-A-3,881,996, Jubin U. S.-A-3,607,669, Schmidt U. S.-A-3,843,488 or Schmidt U. S.-A-4,140,588.
- a tertiary butyl alcohol distillate product 40 which may be further purified, if desired, to remove oxygenated impurities therefrom by catalytic treatment as disclosed, for example, in Sanderson et al. U. S.-A-4,704,482, Sanderson et al. U. S.-A-4,705,903 or Sanderson et al. U. S.-A-4,742,149.
- a heavy distillation fraction 50 is also discharged from the epoxidation reaction zone 20.
- the heavy distillation fraction will contain substantially all of the molybdenum catalyst initially charged to the epoxidation reaction zone 10 by way of the line 14.
- the heavy distillation fraction 50 will contain other products such as tertiary butyl hydroperoxide, tertiary butyl alcohol and impurities including oxygenates lighter than tertiary butyl alcohol such as acetaldehyde, acetone and isopropyl alcohol, oxygenates heavier than tertiary butyl alcohol but lighter than tertiary butyl hydroperoxide, and residue components heavier than tertiary butyl hydroperoxide such as propylene glycol tertiary butyl ethers and hydrocarbons containing 6 or more carbon atoms. As indicated, the heavy distillation fraction 50 will also contain carboxylic acids such as formic acid, acetic acid and isobutyric acid.
- carboxylic acids such as formic acid, acetic acid and isobutyric acid.
- the heavy distillation fraction 50 is charged to a heavy distillation evaporation zone 60 which may comprise, for example, a wiped-film evaporator, a falling film evaporator, a forced circulation evaporator, etc., which is operated at a subatmospheric pressure within the range of 0.25 to 27 kPa (2 to 200 mm Hg.) and at a temperature of 50 to 100°C. with a residence time such that from 60 to 95 wt.% of the heavy distillation fraction charged by the line 50 is taken overhead as condensate by a discharge line 62 controlled by a valve 64.
- the remaining 40-5 wt.% of the material charged to the evaporation zone 60 by the line 50 will be discharged by way of a line 66 and will contain substantially all of the molybdenum initially charged to the epoxidation reaction zone 10.
- the overhead fraction 62 discharged from the evaporator 60 by way of the line 62 may be charged directly to a tertiary butyl hydroperoxide recycle line 66 controlled by a valve 68. If the acid number is higher, the fraction 62 may be routed by a branch line 70 controlled by a valve 72 in order to reduce the acid number to an acceptable level. This is preferably done by treatment of the fraction 70 with calcium oxide and/or calcium hydroxide in accordance with the Marquis et al.
- the reduced-acidity filtrate is discharged from the condensate treating zone 80 by a line 84 controlled by a valve 86 which is provided with a branch line 87 controlled by a valve 90 leading to the tertiary butyl hydroperoxide recycle line 66.
- the heavy liquid distillation fraction 50 discharged from the column 20 is charged by way of line 50 to a wiped film, rotary or forced circulation evaporator 60 wherein the operating conditions include a temperature within the range of 50° to 100°C. and a pressure within the range of 0.25 to 27 kPa (2 to 200 mm Hg) the average residence time of the fraction 50 in the evaporator 60 being such that from about 60 to about 95 wt.% of the material charged by way of the line 50 is taken overhead as a condensate fraction by way of the line 62; the remaining 40-5 wt.% of the material charged by way of the line 50 being discharged from the evaporator 60 by way of a bottoms discharge line 66.
- the fraction 66 will contain substantially all of the molybdenum initially discharged from the epoxidation reaction zone 10.
- the fraction 62 discharged from the evaporator 60 by way of the line 62 may be charged directly to the tertiary butyl hydroperoxide charge line 16 If the acid number is higher, the distillate fraction 62 may be treated with CaO or Ca(OH)2 (see EP-A-0415572) in order to reduce the acid number to an acceptable level after which the thus-treated stream will be recycled to the epoxidation reaction zone by way of the charge line 16.
- the distillate fraction 62 may be treated with CaO or Ca(OH)2 which will react with any acid such as acetic acid, formic acid or isobutyric acid present in the fraction 62 in order to form salts such as the calcium salts which can be removed by filtration and after water wash of this TBHP/TBA stream it can be recycled to the epoxidation reactor.
- any acid such as acetic acid, formic acid or isobutyric acid present in the fraction 62 in order to form salts such as the calcium salts which can be removed by filtration and after water wash of this TBHP/TBA stream it can be recycled to the epoxidation reactor.
- epoxidation catalyst bottoms refers to a stream obtained from an epoxidation reactor effluent after unreacted propylene, low boiling byproducts, propylene oxide, byproducts such as methanol and acetone and most of the tertiary butyl alcohol have been removed by distillation as distillate products.
- Epoxidation catalyst bottoms were the feed used in this wiped-film evaporator run in which conditions were found that result in the quantitative recovery of the unreacted TBHP present in epoxidation catalyst bottoms.
- the epoxidation of propylene was accomplished by reacting t-butyl hydroperoxide (TBA solution) with propylene in the presence of a molybdenum catalyst. Unreacted propylene, low boiling byproducts, propylene oxide and a portion of t-butyl alcohol were recovered in conventional distillation columns affording a stream called "epoxidation catalyst bottoms", containing 9.48 wt.% TBHP (by wet titration), and 677 ppm molybdenum.
- TSA solution t-butyl hydroperoxide
- Unreacted propylene, low boiling byproducts, propylene oxide and a portion of t-butyl alcohol were recovered in conventional distillation columns affording a stream called "epoxidation catalyst bottoms", containing
- the overhead was titrated and found to contain 37.86 grams of TBHP, while the bottoms contained 9.52 grams of TBHP for an overall TBHP recovery or balance of 47.38 grams compared to TBHP feed of 500.0 grams x 9.48% or 47.40 grams, the overall TBHP recovery or balance was essentially quantitative (99.96%).
- the TBHP recovered overhead represents 80% of that recovered with 20% of the TBHP remaining in the bottoms.
- the quantitative recovery of TBHP without decomposition to TBA (and further to isobutylene and water) is remarkable in light of the 5600 ppm molybdenum catalyst level in the bottoms recovered from the wiped-film evaporator.
- Table II is included to show that TBHP when first heated in the presence of molybdenum at 120-125°C. for 1-2 hours without allowing distillation of TBA/TBHP affords substantial decomposition of TBHP.
- TABLE II Ex. Conditions Temp,Time ppm Moly in Feed Wt. % Formic in Feed Wt. % PG in Feed Wt. % TBHP in Feed* Wt. %* TBHP in treated Material % TBHP Decomposition 1 100°C, 1 hr. 1000 0.0 9.2 9.806 8.906 9.18 2 125°C, 1 hr. 1000 0.0 0.0 9.656 8.199 15.09 3 125°C, 1 hr.
- Our invention involves low temperature evaporation in a wiped film, rotary or forced circulation evaporator to avoid TBA dehydration to isobutylene and avoids TBHP decomposition to TBA even in the presence of concentrated moly catalyst.
- the success is evidenced by our essentially quantitative recovery of TBHP (quantitative material balance of TBHP in overheads and bottoms compared to the TBHP present in the feed). Further, we observe an essentially quantitative recovery of TBA during our low temperature evaporation in the WFE. TBA balances (recovery) were generally >90%.
- an advantage of the present invention is the essentially quantitative recovery of unreacted tertiary butyl hydroperoxide from the reactor effluent from the epoxidation reaction product and the recovery of residual quantities of tertiary butyl alcohol with minimal dehydration of the tertiary butyl alcohol to isobutylene.
- epoxidation catalyst bottoms was concentrated with the use of a forced circulation evaporator.
- the catalyst bottoms were evaporated at a temperature and pressure of 77°C. and 15.6 kPa (120 mm Hg,) respectively.
- the overall recovery across the unit was 100.1% while component recoveries of TBA and TBHP were 99.1 and 96.4%, respectively.
- 105kg (232 lb) of material was processed, 94kg (207.5 lb). was taken overhead and 11.2kg (24.8 lb.) remained as bottoms (see Table IV).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Epoxy Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Claims (6)
- Ein Verfahren zur Herstellung von Propylenoxid und tert.-Butylalkohol, in dem Propylen (12) und tert.-Butylhydroperoxid (16) in einer Epoxidationszone (10) in Lösung in tert.-Butylalkohol in der Gegenwart eines löslichen Molybdänkatalysators (14) zur Reaktion gebracht werden, um ein Epoxidationsreaktionsprodukt (18) bereitzustellen, das nichtumgesetztes Propylen, nicht-umgesetztes tert.-Butylhydroperoxid, tert.-Butylalkohol, gelösten Molybdänkatalysator und Verunreinigungen umfaßt, und in dem das Epoxidationsreaktionsprodukt (18) durch Destillation (20) in eine Propylenfraktion (22), eine Propylenoxidfraktion (30), eine tert.-Butylalkoholfraktion (40) und eine hochsiedende flüssige Fraktion (50) die primär aus tert.-Butylhydroperoxid, tert.-Butylalkohol und Verunreinigungen besteht, einschließlich gelöstem Molybdänkatalysator, aufgetrennt wird, gekennzeichnet durch Trennen besagter hochsiedender flüssiger Fraktion (50) durch Vakuumverdampfung bei einer Temperatur von 25 bis 100°C und einem Druck von 0,25 bis 27 kPa (2 bis 200 mm Hg) in eine Überkopffraktion (62), die 60 bis 95 Gew.-% der eingesetzten hochsiedenden flüssigen Fraktion umfaßt, und einen klaren flüssigen Rückstand (66), wobei besagte Überkopffraktion (62) von 70 bis 95 Gew.-% Tert.-Butylalkohol, 1 bis 20 Gew.-% tert.-Butylhydroperoxid und entsprechend 15 bis 3 Gew.-% Verunreinigungen enthält und besagte klare Rückstandsfraktion (66) tert.-Butylhydroperoxid, tert.-Butylalkohol und Verunreinigungen enthält, einschließlich im wesentlichen dem gesamten Molybdän, das in besagter hochsiedender flüssiger Fraktion enthalten ist.
- Ein Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß die hochsiedende flüssige Fraktion (50) bei einer Temperatur von 50 bis 100°C verdampft wird.
- Ein Verfahren nach Anspruch 1 oder 2, dadurch gekennzeichnet, daß die hochsiedende flüssige Fraktion (50) in einem Verdampfer mit Verteilerbürsten, einem Rotationsverdampfer oder einem Zwangsumlaufverdampfer verdampft wird.
- Ein verfahren, wie beansprucht in einem der Ansprüche 1 bis 3, dadurch gekennzeichnet, daß die Verunreinigungen im Epoxidationsreaktionsprodukt (18) eines oder mehrere von Acetaldehyd, Methanol, Methylformiat, Aceton, Di-tert.-butylperoxid, tert.-Butylformiat, Isobutanol, tert.-Butylether von Propylenglykol, Carbonsäuren, einschließlich Armeisensäure, Essigsäure, Isobuttersäure, etc., umfassen.
- Ein Verfahren nach einem der Ansprüche 1 bis 4, dadurch gekennzeichnet, daß die Überkopffraktion (62) eine Säurezahl von weniger als 12 m vol/g besitzt und zu besagter Epoxidationszone (10) rückgeführt wird (64, 67, 68, 16).
- Ein Verfahren nach einem der Ansprüche 1 bis 4, dadurch gekennzeichnet, daß die Überkopffraktion (62) eine Säurezahl von mehr als 12 m vol/g besitzt und zu einer Behandlungszone (80) geleitet wird (70, 72), in der sie mit 1/2 bis 1 Äquivalenten Calciumoxid und/oder Calciumhydroxid (82), bezogen auf ihren Carbonsäuregehalt, behandelt wird, um wenigstens einen Teil (88) der Carbonsäuren auszufällen, und ein Filtrat (84) zu besagter Epoxidationszone (10) rückgeführt wird (87, 90, 67, 68, 16).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US07/401,381 US4992566A (en) | 1989-08-31 | 1989-08-31 | Recovery of tertiary butyl hydroperoxide and tertiary butyl alcohol |
| US401381 | 1989-08-31 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0416744A2 EP0416744A2 (de) | 1991-03-13 |
| EP0416744A3 EP0416744A3 (en) | 1991-08-28 |
| EP0416744B1 true EP0416744B1 (de) | 1994-09-14 |
Family
ID=23587522
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP90308547A Expired - Lifetime EP0416744B1 (de) | 1989-08-31 | 1990-08-02 | Rückgewinnung von Tertiärbutylhydroperoxid und Tertiärbutylalkohol |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US4992566A (de) |
| EP (1) | EP0416744B1 (de) |
| JP (1) | JP2804616B2 (de) |
| DE (1) | DE69012491T2 (de) |
| ES (1) | ES2058805T3 (de) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5104493B1 (en) * | 1991-01-24 | 1995-08-15 | Arco Chem Tech | Tertiary butyl hydroperoxide concentration |
| US5274138A (en) * | 1993-01-21 | 1993-12-28 | Texaco Chemical Company | Epoxidation process for manufacture of olefin oxide and alcohol |
| US5414145A (en) * | 1994-08-11 | 1995-05-09 | Texaco Chemical Inc. | Production of tertiary butyl alcohol from isobutane |
| CA2159280A1 (en) | 1994-11-01 | 1996-05-02 | David Durham Chess | Recovery and purification of tertiary butyl alcohol |
| US5639355A (en) * | 1995-03-13 | 1997-06-17 | Texaco Chemical Inc. | Method for enhancing the yield of tertiary butyl alcohol in a tertiary butyl alcohol recovery process |
| CA2253215A1 (en) * | 1997-11-13 | 1999-05-13 | Shell Internationale Research Maatschappij B.V. | Process for preparing organic hydroperoxides |
| JP2002322167A (ja) * | 2001-04-27 | 2002-11-08 | Sumitomo Chem Co Ltd | プロピレンオキサイドの製造方法 |
| CN112409296B (zh) * | 2020-11-27 | 2022-07-29 | 信汇科技有限公司 | 一种环氧丙烷的制备方法 |
| CN112920144B (zh) * | 2021-01-29 | 2022-06-21 | 北京水木滨华科技有限公司 | 一种环氧丙烷的制备方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3452055A (en) * | 1966-07-29 | 1969-06-24 | Halcon International Inc | Process for the recovery of epoxides wherein alkylaromatic compound is removed in two distillation zones |
| BE790548A (fr) * | 1971-10-27 | 1973-04-25 | Halcon International Inc | Recyclage de catalyseur d'époxydation |
| US4455283A (en) * | 1982-05-24 | 1984-06-19 | Atlantic Richfield Company | Molybdenum epoxidation catalyst recovery |
-
1989
- 1989-08-31 US US07/401,381 patent/US4992566A/en not_active Expired - Lifetime
-
1990
- 1990-08-02 EP EP90308547A patent/EP0416744B1/de not_active Expired - Lifetime
- 1990-08-02 DE DE69012491T patent/DE69012491T2/de not_active Expired - Fee Related
- 1990-08-02 ES ES90308547T patent/ES2058805T3/es not_active Expired - Lifetime
- 1990-08-30 JP JP2226824A patent/JP2804616B2/ja not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| EP0416744A3 (en) | 1991-08-28 |
| US4992566A (en) | 1991-02-12 |
| ES2058805T3 (es) | 1994-11-01 |
| JPH03178944A (ja) | 1991-08-02 |
| EP0416744A2 (de) | 1991-03-13 |
| DE69012491D1 (de) | 1994-10-20 |
| JP2804616B2 (ja) | 1998-09-30 |
| DE69012491T2 (de) | 1995-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0581530B1 (de) | Katalytische Entfernung von Peroxidverunreinigungen aus einem t-Butylalkoholzurückführungsstrom | |
| EP0415572B1 (de) | Abtrennung von sauren Verunreinigungen von Tertiärbutylhydroperoxid | |
| EP0608093A1 (de) | Epoxidierungsverfahren zur Herstellung eines Olefinoxids und eines Alkohols | |
| EP0416744B1 (de) | Rückgewinnung von Tertiärbutylhydroperoxid und Tertiärbutylalkohol | |
| US4977285A (en) | Recovery of tertiary butyl hydroperoxide and tertiary butyl alcohol | |
| EP0540225B1 (de) | Reinigung von Propylenoxid durch Extraktivdestillation | |
| JPH05194454A (ja) | プロピレンオキシドの精製方法 | |
| EP0518499B1 (de) | Entfernung von Spuren von Molybdenum | |
| EP0596718B1 (de) | Wiedergewinnung von Molybdän | |
| US5151530A (en) | Treatment of tertiary butyl hydroperoxide distillation fraction to remove acidic contaminants | |
| US5101052A (en) | Molybdenum recovery | |
| US5128492A (en) | Precipitation of molybdenum | |
| EP0180418B1 (de) | Regenerierung von auflösbaren Molybdänkatalysatoren aus Strömen verwendeter Katalysatoren | |
| EP0404379B1 (de) | Verfahren zur Gewinnung von festen Molybdän-Verbindungen | |
| EP0710640B1 (de) | Gewinnung und Reinigung von tertiärem Butylalkohol | |
| US5216182A (en) | Preparation of mono epoxides and tertiary butyl alcohol using regenerated catalyst | |
| EP0584956B1 (de) | Verfahren zur Herstellung von Monoepoxiden und tert. Butylalkohol mit einem regeneriertem Katalysator | |
| US5736112A (en) | Aqueous oxidation of organic molybdenum compounds | |
| EP0574204B1 (de) | Entfernung von Peroxidverunreinigungen aus tert.-Butylalkohol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): BE DE ES FR GB IT NL |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): BE DE ES FR GB IT NL |
|
| 17P | Request for examination filed |
Effective date: 19920205 |
|
| 17Q | First examination report despatched |
Effective date: 19930625 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): BE DE ES FR GB IT NL |
|
| REF | Corresponds to: |
Ref document number: 69012491 Country of ref document: DE Date of ref document: 19941020 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2058805 Country of ref document: ES Kind code of ref document: T3 |
|
| ITF | It: translation for a ep patent filed | ||
| ET | Fr: translation filed | ||
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E |
|
| NLS | Nl: assignments of ep-patents |
Owner name: TEXACO CHEMICAL INC. |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: TP |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: PC2A |
|
| BECH | Be: change of holder |
Free format text: 970410 *TEXACO CHEMICAL INC. UNE SOC. DE L'ETAT DE DELWARE |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: TP |
|
| NLS | Nl: assignments of ep-patents |
Owner name: HUNTSMAN SPECIALTY CHEMICALS CORPORATION |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: PC2A |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20000620 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20000703 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20000803 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20000816 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20000830 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20000915 Year of fee payment: 11 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010802 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010803 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010831 |
|
| BERE | Be: lapsed |
Owner name: HUNTSMAN SPECIALTY CHEMICALS CORP. Effective date: 20010831 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20020301 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20010802 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20020430 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 20020301 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20020501 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20020911 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20050802 |