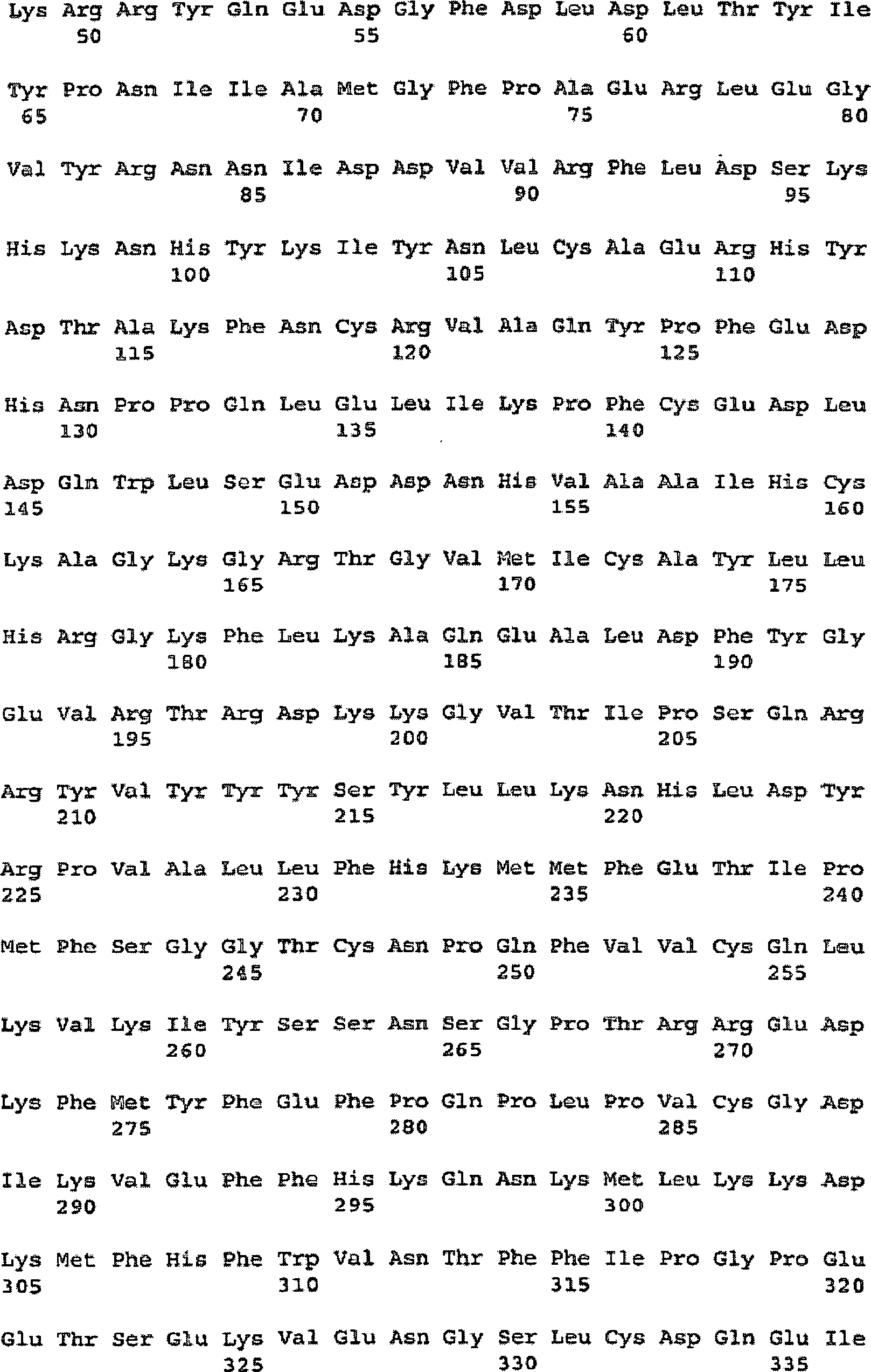-
STAND DER TECHNIK
-
I. Gebiet der Erfindung
-
Die
vorliegende Erfindung betrifft die Gebiete der Onkologie, Genetik
und Molekularbiologie. Insbesondere betrifft die Erfindung die Identifizierung
eines Tumorsuppressorgens auf dem menschlichen Chromosom 10. Defekte
in diesem Gen sind mit der Entwicklung von Krebserkrankungen, wie
beispielsweise Gliomen, verknüpft.
-
II. Stand der Technik
-
Die
Onkogenese wurde von Foulds (1958) als ein aus mehreren Schritten
bestehender, biologischer Prozess beschrieben, von dem inzwischen
bekannt ist, dass er durch eine Ansammlung genetischer Schäden auftritt.
Auf molekularer Ebene umfasst der aus mehreren Schritten bestehende
Prozess der Tumorgenese die Störung
sowohl positiver als auch negativer regulatorischer Effektoren (Weinberg,
1989). Von Vogelstein und Mitarbeiter (1990) haben postuliert, dass
die molekulare Basis menschlicher Colonkarzinome eine Reihe von Onkogenen,
Tumorsuppressorgenen und Reparaturgenen einschließt. In ähnlicher
Weise sind Defekte, die zur Entwicklung des Retinoblastoms führen, mit
einem anderen Tumorsuppressorgen verknüpft worden (Lee et al., 1987).
Bei einer Reihe anderer Malignome wurden wiederum andere Onkogene
und Tumorsuppressorgene identifiziert. Leider ist die Anzahl behandelbarer
Krebserkrankungen noch immer gering und die Auswirkungen einer Krebserkrankung
sind katastrophal – allein
in den USA sind jährlich über eine
halbe Million Todesfälle
zu verzeichnen.
-
Ein
Beispiel für
die verheerende Natur einer Krebserkrankung sind Tumore, die aus
Zellen der Abstammungslinie der Astrozyten entstehen und die zu
den am häufigsten
vorkommenden Primärtumoren
des Zentralnervensystems zählen
(Russell und Rubinstein, 1989). Die meisten dieser Tumoren treten
bei Erwachsenen auf. Primäre
Gehirntumoren sind darüber
hinaus die häufigsten
soliden Tumoren bei pädiatrischen
Patienten und die zweithäufigste
Todesursache bei Kindern unter 15 Jahren. 1994 wurden schätzungsweise 18.500
neue Fälle
von primären
Gehirntumoren diagnostiziert (Boring et al. 1994). Epidemiologische
Studien zeigen, dass die Inzidenz von Gehirntumoren zunimmt und
die dritthäufigste
Ursache von Krebstod bei 18- bis 35-jährigen Patienten darstellt.
Wegen ihrer Position innerhalb des Gehirns und der typischen Infiltration
von Tumorzellen in das umliegende Gewebe ist ein erfolgreiches therapeutisches
Eingreifen bei primären
Gehirntumoren häufig
beschränkt.
Leider erliegen rund zwei Drittel dieser betroffenen Personen der
Krankheit innerhalb von zwei Jahren. Die häufigsten Intrakranialtumoren
bei Erwachsenen entstehen aus Zellen glialer Abstammung und treten
mit einer ungefähren
Frequenz von 48% als Glioblastoma multiforme (GBM), zu 21% als Astrozytome
(A) (anaplastisch (AA) und geringen Grades) und zu 9% als Ependymome
und Oligodendrogliome auf (Levin et al., 1993).
-
Genetische
Studien haben mehrere Gene und ihre entsprechenden Protein-Produkte
mit der Onkogenese primärer
Gehirntumore in Verbindung gebracht. Zu den verschiedenen berichteten
Veränderungen
gehören:
Amplifikation des Rezeptors für
den epidermalen Wachstumsfaktor und eines seiner Liganden, des transformierenden
Wachstumsfaktors alpha (Transforming Growth Factor alpha), N-myc;
gli, verändertes
Splicing und Expression von Rezeptoren für den Fibroblasten-Wachstumsfaktor
und der Verlust der Funktion von p53, p16, Rb, der Neurofibromatosegene
1 und 2, DCC und putativer Tumorsuppressorgene auf Chromosom 4,
10, 17 (non-p53), 19, 22 und X (Wong et al. 1987; EI-Azouzi-et al.,
1989; Nishi et al. 1991; James et al., 1988; Kamb et al. 1984; Henson
et al. 1994; Yamaguchi et al. 1994; Bianchi et al. 1994; Ransom
et al. 1992; Rasheed et al. 1992; Scheck und Coons, 1993; Von Demling
et al. 1994; Rubio et al. 1994; Ritland et al., 1995).
-
Die
häufigsten
Veränderungen
umfassten eine Amplifikation des EGF-Rezeptors (~40%), einen Verlust
der Funktion von p53 (~50%), p16 (~50%), Rb(~30%) und Deletionen
auf Chromosom 10 (> 90%).
Außerdem
korrelieren der Grad oder das Ausmaß der histologischen Malignität von Astrozytentumoren
mit einer verstärkten
Ansammlung genetischer Schäden, ähnlich wie
beim Kolonkarzinom. Überdies
scheinen einige Veränderungen
relativ abstammungs- oder gradspezifisch zu sein. Zum Beispiel treten
Verluste auf Chromosom 19q überwiegend
bei Oligodendrogliomen auf, während
Deletionen auf Chromosom 10 sowie eine Amplifikation und Mutation
des EGF-Rezeptors hauptsächlich
bei GBM vorkommen. Die Deletion einer vollständigen Kopie oder von Abschnitten
von Chromosom 10 ist stark indiziert als häufigstes genetisches Ereignis
in Verbindung mit der häufigsten
Form eines primären
Gehirntumors, GBM.
-
Zytogenetische
Studien und später
durchgeführte
Alleldeletionsstudien bei GBM haben eindeutig häufige und weit reichende molekulargenetische
Veränderungen
in Verbindung mit Chromosom 10 aufgezeigt (Bigner et al., 1988;
Ransom et al., 1992; Rasheed et al. 1992; James et al., 1988: Fujimoto
et al. 1989; Fults et al., 1990, 1993; Karlbom et al., 1993; Rasheed
et al., 1995; Sonoda et al., 1996; Albarosa et al., 1996). Zytogenetische
Analysen haben die Veränderung
von Chromosom 10 eindeutig als häufiges
Ereignis bei GBM demonstriert, wobei die Veränderung bei 60% der Tumoren
vorkam. Alleldeletionsstudien bei GBM haben ebenfalls sehr häufig auftretende
Allel-Ungleichgewichte in Verbindung mit Chromosom 10 (90%) aufgezeigt. Die
Verluste sind jedoch derart weitreichend und häufig, dass in diesen Analysen
keine chromosomale Sublokalisation eines einheitlichen Verlustes
zweifelsfrei definiert werden konnte.
-
Mehrere,
kürzlich
durchgeführte
Studien haben die Region 10q25-26, insbesondere eine 17-cM-Region von D10S190
bis D10S216 impliziert. Durch Alleldeletionsanalyse anhand einer
Reihe von Gliomen niedrigen und hohen Grades, die nur einen partiellen
Verlust von Chromosom 10 aufwiesen, wurde eine telomere Region von
D10S587 bis D10S216 impliziert (Rasheed et al., 1995). Diese Region
(~ 1 cM) war bei 11 GMBs, 4 AAs, 1 A und 1 Oligodendrogliom nicht
mehr vorhanden oder nicht-informativ, was die Lokalisierung einer Kandidatenregion
andeutet. Die Studie zeigte außerdem,
dass Deletionen auf Chromosom 10 in Gliomen niedrigen Grades vorkommen.
Albarosa et al. (1996) legen auf der Basis einer kleinen Alleldeletion
bei einem pädiatrischen
Gehirntumor eine zentromere Kandidatenregion von Marker D10S221
bis D10S209 nahe. Steck und Saya haben anhand einer Reihe von GBM
zwei häufige
Deletionsbereiche, 10q26 und 10q24 (D10S192) vorgeschlagen.
-
Es
ist außerdem
impliziert worden, dass der kurze Arm von Chromosom 10 ein weiteres
Tumorsuppressorgen enthält.
Studien lieferten anfangs funktionelle Hinweise auf ein Tumorsuppressorgen
auf 10p bei Gliomen (Steck et al., 1995), welches später für Prostatakrebs
gezeigt wurde (Sanchez et al., 1995; Murakami et al., 1996). Letztere
Studie implizierte eine Region von 11 cM zwischen D10S1172 und D10S527.
Alleldeletionsstudien an Gliomen haben eine weit reichende Deletion
auf 10p gezeigt, aber auch war keine feste Lokalisierung möglich (Karlbom
et al., 1993; Kimmelman et al., 1996; diese Regionen auf Chromosom
10 sind in 1 unten gezeigt). Darüber hinaus
ist gezeigt worden, dass auch die Amplifikation des EGF-Rezeptors
fast ausschließlich
bei Tumoren vorkommt, die Deletionen auf Chromosom 10 aufwesen,
was auf eine mögliche Verknüpfung zwischen
diesen genetischen Veränderungen
hinweist (Von Deimling et al., 1992).
-
Deletionen
auf dem langen Arm, vor allem 10q24, sind auch für Prostata-, Nieren- und Uteruskarzinome,
kleinzelliges Bronchialkarzinom und endometriale Karzinome, Meningiome
und bei akuten T-Zell-Leukämien
festgestellt worden (Carter et al., 1990; Morita et al., 1991; Herbst
et al., 1984; Jones et al., 1994; Rempel et al., 1993; Peiffer et
al., 1995; Petersen et al., 1997). Kürzlich haben ausführliche
Studien über
Prostatakarzinome gezeigt, dass (1) der kurze und der lange Arm von
Chromosom 10 Tumorsuppressorgene aufzuweisen scheinen und (2) sich
das Suppressorgen des langen Arms im Übergangsbereich 10q23-24 befindet
(Gray et al., 1995; Ittmann, 1996, Trybus et al., 1996). Der von
diesen drei Gruppen identifizierte Bereich der häufigen Deletion befindet sich
im Bereich von D10S215 als Zentrum und ist etwa 10 cM groß (1).
Der Bereich überlappt
mit unserer Kandidatenregion, aber innerhalb der Region ist für das Prostatakarzinom
keine weitere Lokalisierung berichtet worden. Die mit Prostatakarzinom
verknüpften
Allelverluste scheinen auch nur bei etwa 30–40% der untersuchten Tumore
vorzukommen. Darüber
hinaus werden Deletionen bei Tumoren in einem fortgeschrittenerem
Stadium beobachtet, ähnlich
wie bei GBM, und könnten
mit der Metastasierungsfähigkeit verknüpft sein
(Nihei et al., 1995; Komiya et al., 1996). Die Kombination dieser
Ergebnisse lässt
den Schluss zu, dass die Region 10a23-24 bei mehreren Krebserkrankungen
des Menschen eine Rolle spielt.
-
Die
Unterschiede in der Lokalisierung der Kandidatenregionen lassen
mehrere Möglichkeiten
zu. Erstens ist es möglich,
dass zwei oder mehr Tumorsuppressorgene auf 10q vorhanden sind.
Zweitens beeinflussen möglicherweise
nicht alle Deletionen den Tumorsuppressorgenlocus. Diese Alternativen
schließen
sich gegenseitig nicht aus. Letztere Möglichkeit wird dadurch gestützt, dass
ein potenzielles latentes Zentromer auf 10q25 vorhanden sein könnte, was
genetische Veränderungen,
insbesondere Brüche,
hervorrufen könnte (Vouillaire
et al., 1993).
-
Trotz
dieser Informationen bleibt die Identität des Gens (oder der Gene),
die an der Tumorsuppression in Verbindung mit 10q23-24 beteiligt
sind, unklar. Ohne Identifikation eines spezifischen Gens und der
Ableitung des Proteins, für
das es kodiert, ist es unmöglich,
eine wirksame Therapie gegen dieses Produkt zu entwickeln. Es ist
daher ein wichtiges Ziel, den Tumorsuppressor/die Tumorsuppressoren,
die sich in dieser Region befinden, zu isolieren und ihre Struktur
und Funktion zu bestimmen.
-
In
einer weiteren Ausführungsform
stellt die vorliegende Erfindung ein Verfahren des Diagnostizierens des
Cowden-Syndroms bereit, welches die Schritte des Erhaltens einer
Probe von einer Testperson und das Bestimmen der Expression eines
funktionellen TS10q23.3-Genproduktes
in Zellen der Probe umfasst. In besonders bevorzugten Ausführungsformen
können
die Zellen aus der Gruppe ausgewählt
sein, die aus Brust-, Ovarial-, Schilddrüsen- und Endometriumzellen
besteht. In anderen Ausführungsformen
kann die Probe eine Gewebe- oder eine Flüssigkeitsprobe sein. In anderen
Aspekten der Erfindung umfasst das Bestimmen das Testen hinsichtlich
einer Nukleinsäure
aus der Probe. In mehr bevorzugten Aspekten kann das Verfahren des Weiteren
umfassen, die Probe Bedingungen auszusetzen, die geeignet sind,
um die Nukleinsäure
zu amplifizieren. In anderen Ausführungsformen kann das Verfahren
des Weiteren den Schritt des Vergleichens der Expression von TS10q23.3
mit der Expression von TS10q23.3 bei Proben, die nicht vom Cowden-Syndrom
stammen, umfassen. In speziellen Ausführungsformen besteht der Vergleich
im Evaluieren des Levels der Expression von TS10q23.3. In spezielleren
Ausführungsformen
umfasst die Probe von Patienten mit Cowden-Syndrom eine Mutation in der Kodierungssequenz
von TS10q23.3. Bei der Mutation kann es sich um eine Frameshift-Mutation,
eine Deletionsmutation, eine Insertionsmutation oder eine Missense-Mutation
handeln. In spezielleren Ausführungsformen
befindet sich die Mutation in Exon 7. In anderen besonderen Ausführungsformen bewirkt
die Mutation einen vorzeitigen Abbruch des TS10q23.3-Genproduktes.
In anderen Ausführungsformen befindet
sich die Deletionsmutation in Exon 8. In bestimmten Ausführungsformen
befindet sich die Insertion in Exon 2. In besonders bevorzugten
Ausführungsformen
ist die Mutation eine Insertion von AT an Base 791 von Exon 7. In
anderen besonders bevorzugten Ausführungsformen ist die Mutation
eine Insertion von drei Basenpaaren an Base 137 von Exon 2. Spezifischer
kodiert die Insertion von drei Basenpaaren für ein ASN im TS10q23.3-Genprodukt.
-
In
einem weiteren Aspekt ist außerdem
ein Verfahren des Diagnostizierens eines Individuums mit einer Prädisposition
für Brustkrebs
vorgesehen, das die Schritte des Erhaltens einer Probe von einem
Individuum und des Bestimmens der Expression eines funktionellen
TS10q23.3-Genproduktes in Zellen der Probe umfasst. In besonderen
Ausführungsformen
können
die Zellen aus der Gruppe ausgewählt
sein, die aus Brustzellen, Ovarialzellen, Schilddrüsenzellen
und Endometrialzellen besteht. In anderen Ausführungsformen ist die Probe
eine Gewebeprobe oder eine Flüssigkeitsprobe.
In einem besonders bevorzugten Aspekt umfasst das Verfahren des
Weiteren den Schritt des Vergleichs der Expression von TS10q23.3
mit der Expression von TS10q23.3 in normalen Proben. In definierteren
Aspekten umfasst die Probe eine Mutation in der Kodierungssequenz
von TS10q23.3. Bei der Mutation kann es sich um eine Frameshift-Mutation,
eine Deletionsmutation, eine Insertionsmutation oder eine Missense-Mutation handeln.
In spezielleren Ausführungsformen
befindet sich die Mutation in Exon 7. In anderen besonderen Ausführungsformen
bewirkt die Mutation einen vorzeitigen Abbruch des TS10q23.3-Genproduktes.
In anderen Ausführungsformen
befindet sich die Deletionsmutation in Exon 8. In bestimmten Ausführungsformen
befindet sich die Insertion in Exon 2. In besonders bevorzugten Ausführungsformen
ist die Mutation eine Insertion von AT an Base 791 von Exon 7.
-
In
anderen besonders bevorzugten Ausführungsformen ist die Mutation
eine Deletion von dreizehn Basenpaaren an Base 915 von Exon 8. In
einer anderen besonders bevorzugten Ausführungsform ist die Mutation
eine Insertion von drei Basenpaaren an Base 137 von Exon 2. Spezifischer
kodiert die Insertion von drei Basenpaaren für ein ASN im TS10q23.3-Genprodukt.
-
KURZE BESCHREIBUNG DER
ZEICHNUNGEN
-
Die
folgenden Zeichnungen bilden einen Teil der vorliegenden Patentschrift
und sind enthalten, um bestimmte Aspekte der vorliegenden Erfindung
ausführlicher
aufzuzeigen. Die Erfindung wird durch Bezugnahme auf eine oder mehrere
der Zeichnungen in Kombination mit der ausführlichen Beschreibung spezifischer hierin
vorgestellter Ausführungsformen
besser verständlich:
-
1 – Lokalisierung
von Tumorsuppressor-Kandidatenloci auf dem menschlichen Chromosom
10. Verschiedene Loci auf dem menschlichen Chromosom 10 sind als
mögliche
Stellen mit Tumorsuppressoraktivität vorgeschlagen worden. Diese
Stellen und die Reportergruppe sind dargestellt.
-
2 – Darstellung
homozygoter Deletionen in Gliom-Zelllinien. Es wurden verschiedene
Gliom-Zelllinien hinsichtlich des Vorhandenseins von Deletionen
in beiden Kopien von Loci auf Chromosom 10 untersucht. Die Loci
sind auf der senkrechten Achse und die Zelllinien auf der waagrechten
Achse dargestellt. Die Gliom-Zelllinien D54, EFC-2, A172 und LG11
wurden auf Vorhandensein von Marker AFMA086WG9 (AFM086) untersucht.
In Multiplex-Polymerasekettenreaktionsuntersuchungen wurde anhand
einiger weiterer polymorpher Allele auf Chromosom 10 in unabhängigen Reaktionen
gezeigt, dass der Marker deletiert war. Allel D1OS196 ist als Kontrolle
für die
PCRTM-Reaktionen gezeigt. EFC-2-Zellen zeigten
eine homozygote Deletion von 4 aufeinander folgenden Markern (siehe 2).
-
3 – Darstellung
von Regionen auf Chromosom 10: Vorhandensein oder Abwesenheit von DNA-Mikrosatellitenmarkern
in einem Hybridklon. Dargestellt sind Regionen des Vorhandenseins
(gefüllte Kreise)
oder der Abwesenheit (leere Kreise) von DNA auf Chromosom 10, die
den für
Chromosom 10 spezifischen, in A9-Zellen der Maus übertragenen
Mikrosatellitenmarkern aus elf Subklonen des somatischen Zellhybridklons
U251.N10.7 entsprechen. Die somatischen Zellhybride U251.N10.6 und
U251.N10.8 sind vollständig
supprimierte Klone, die in Weichagarose kein oder wenig Wachstum
zeigen, während
die Subklone U251.10.5A und C partiell supprimiert sind (Steck et
al., 1995). Der Unterschied zwischen den vollständig supprimierten Klonen und
den partiell supprimierten Klonen liefert eine funktionelle Lokalisierung
des Tumorsuppressorgens. Die Regionen, die möglicherweise das Tumorsuppressorgen
enthalten, sind durch die schraffierten Kästchen angezeigt. Das schraffierte
Kästchen
an 10q12.2 überlappt
mit den homozygoten Deletionen und der durch Alleldeletionsanalyse
implizierten Region (siehe 2 und 4).
-
4 – Deletionskarte
von Chromosom 10 bei humanen Gliomen. Die von den Markern D10S551
bis D10S583 begrenzte Region befindet sich in einer 10-cM-Region.
Die Mikrosatelliten sind in der Reihenfolge der wahrscheinlichsten
Verknüpfung
gezeigt und ihre ungefähre
chromosomale Lage ist ausgehend von der Radiation-Hybrid-Karte von
Gyapay et al., 1994 kartiert. In den durch Kästchen eingegrenzten Regionen
des Tumors ist die Region auf Chromosom 10 dargestellt, die nicht
an anaplastischen Astrozytomen und einem Gliom beteiligt ist. Die
kritische Region, die von der homozygoten Deletionsanalyse definiert
und von dieser Analyse nicht ausgeschlossen ist, wird durch den
ausgefüllten
Balken auf der rechten Seite gezeigt.
-
5 – Kartierung
von BAC 106d16. Dargestellt ist die Kartierung des BAC mit der Bezeichnung 106d16
sowie der Nachweis der homozygoten Deletion durch Southern-Blotting.
Gezeigt ist die partielle Restriktionskarte von 106d16. Die Darstellung
des Blots zeigt die homozygote Deletion der Eco-Bande Nr. 14 (Mr etwa
11 kb) in EFC-2-Zellen.
-
6 – Kodierungssequenz
und 5'- und 3'-flankierende Regionen
von TS10q23.3. Die Kodierungsregion ist fett gedruckt, ebenso wie
das erste im Leseraster befindliche Stop-Codon.
-
7 – Vorhergesagte
Aminosäuresequenz
des TS10q23.3-Produktes. Abkürzungen
sind A, Alanin; C, Cystein; D, Asparaginsäure; E, Glutaminsäure; F;
Phenylalanin; G, Glycin; H, Histidin; I, Isoleucin; K, Lysin; L,
Leucin; M, Methionin; N, Asparagin; P; Prolin; Q, Glutamin; R, Arginin;
S, Serin; T, Threonin; V, Valin; W, Tryptophan; Y, Tyrosin. Die
Phosphatase-Konsensusstelle
ist fett gedruckt, Tyrosinphosphorylierungsstellen sind kursiv gedruckt
und unterstrichen.
-
8 – Deletionsanalyse
von 10q12.2. Die Gliom-Zelllinie, bei der zu Anfang homozygote Deletionen in
10q23.3 gezeigt worden sind, wurde erneut hinsichtlich des Vorhandenseins
des TS10g23.3-Gens analysiert. Das schattierte Oval zeigt an, dass
die Genregion vorhanden ist; das leere Oval zeigt eine homozygote Deletion
in der Genregion an. *zeigt eingeschlossene Exons an.
-
9 – Homologievergleich
des humanen TS10q23.3 mit Homologen von Maus und Hund. Das ATG-Startcodon
und die Aminosäure
Methionin sind als die Start (1)-Position bezeichnet. Das Terminationscodon
ist TGA (1210). Abweichungen zwischen den Sequenzen von Mensch und
Maus oder Hund auf Genom- oder Aminosäureebene sind durch einen Stern
in der verglichenen Sequenz dargestellt. Die Aminosäuresequenzen
von Hund und Mensch sind identisch; die Maussequenz unterschied
sich an Position 398, wobei die Maus ein Serin aufweist, Hund und
Mensch dagegen ein Threonin.
-
10 – Sequenzen
von Exons und umliegenden Intronregionen von TS10q23.3. Die Exons
sind durch Großbuchstaben
ab Position eins und die Introns mit Kleinbuchstaben angegeben:
eine Ausnahme ist das erste Exon, bei dem das Startcodon bei Position
eins beginnt und sich der Exon/Intron-3'-Übergang
an Position 79/80 befindet. Die Kleinbuchstabenbezeichnung (Tabelle
5) entspricht der Nummerierung der in dieser Figur dargestellten
Sequenz, ausgenommen das erste Exon. Die Mutationen bei U87 und
U138 befinden sich am G-Rest des ersten Introns [G+1>T] nach dem Exon (respektive
Exon 7 und 8). Die Punktmutation bei T98G befindet sich an Position
46 und die Punktmutation bei KE an Position 28 von Exon 2. Bei LnCap-Zellen handelt
es sich bei der Mutation um eine Deletion von Base 16 und 17 im
ersten Intron.
-
11A–G. – Analyse
von Sekundärstrukturen
in TS1023.3.- 11A: Hydrophilie-Plot; 11B: Oberflächenwahrscheinlichkeits-Plot; 11C: Flexibilitäts-Plot; 11D: Antigenitätsindex-Plot; 11E: Plot der amphiphilen Helix; 11F: Plot des amphiphilen Blattes; 11G: Sekundärstruktur-Plot.
-
12A–I. – Vergleich
prognostizierter Merkmale in TS10q23.3 und den Punktmutanten T98G
und KE. 12A: Hydrophilie-Plot von Rest
1–60 des
Wildtyp-Polypeptids; 12B:
Oberflächenwahrscheinlichkeits-Plot
von Rest 1–60
des Wildtyp-Polypeptids; 12C:
Sekundärstruktur-Plot
von Rest 1–60
des Wildtyp-Polypeptids; 12D:
Hydrophilie-Plot von Rest 1–60
der KE-Mutante; 12E: Oberflächenwahrscheinlichkeits-Plot
von Rest 1–60
der KE-Mutante; 12F: Sekundärstruktur-Plot von Rest 1–60 der
KE-Mutante; 12G: Hydrophilie-Plot von Rest
1–60 der
T98G-Mutante; 12H: Oberflächenwahrscheinlichkeits-Plot von Rest 1–60 der
T98G-Mutante; 12I: Sekundärstruktur-Plot von Rest 1–60 der
T98G-Mutante. Die T98G-Mutation
(Leu → Arg)
an Rest 42 bewirkt den Verlust der vorgeschlagenen Helixsekundärstruktur
von TS10q23.3. Die Mutation in KE an Rest 36 (Gly → Glu) würde die
Länge der
vorgeschlagenen Helixstruktur in dem Bereich signifikant verlängern. Beide
Mutationen würden
dieselbe Helixstruktur beeinflussen. Es würden auch kleinere Veränderungen der
Hydrophilie und der Oberflächenwahrscheinlichkeit
entstehen.
-
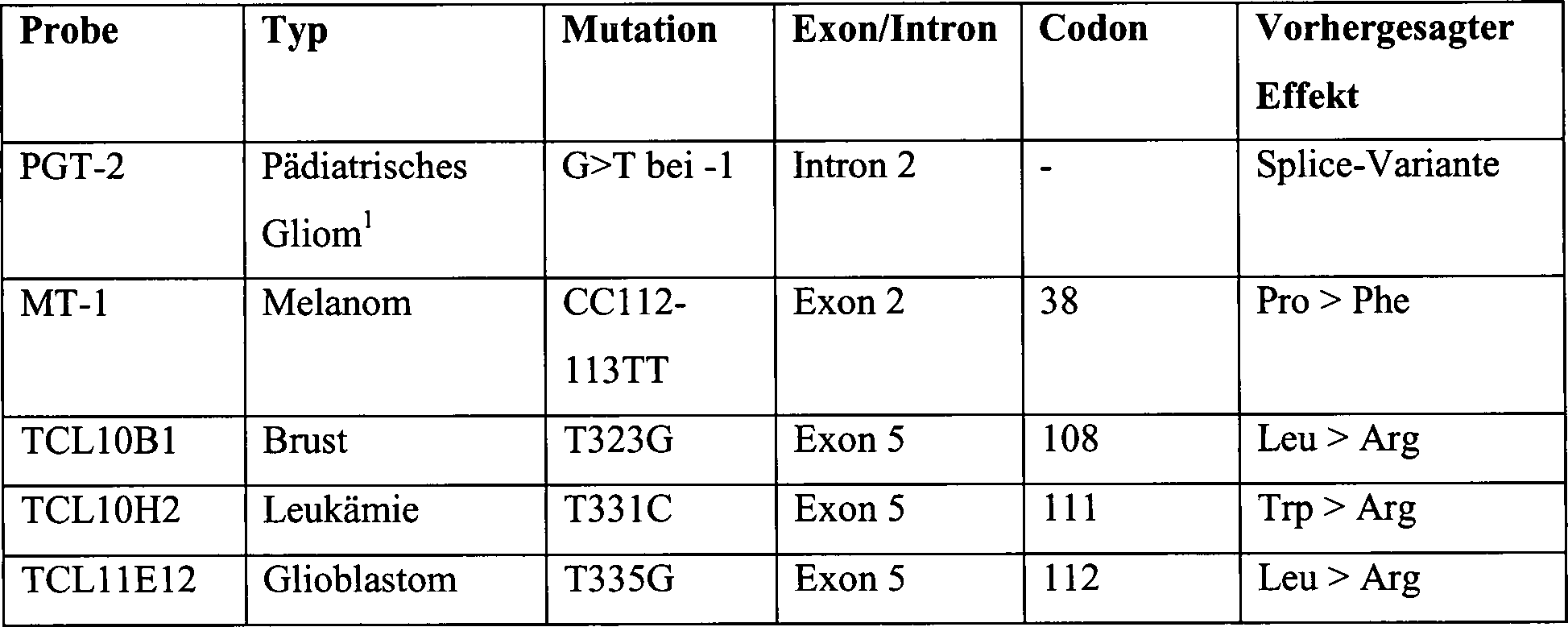
13A. Homozygote Deletion des TS10Q3.3-Gens in
humanen Tumorzelllinien und TS10q23.3-mRNA-Expressionsniveaus in
primären
Glioblastomen. Gezeigt sind vier Zelllinien, Brustkarzinom TCL11A11,
Melanom TCL11D7, Melanom TCL11D9 und Leukämie TCL10G9 (Kontrollprobe
ohne homozygot deletiertes TS10Q3.3), jeweils untersucht durch PCRTM-Amplifikation
anhand der folgenden Muster (Sequence Tagged Sites): (1) TS10Q3.3
Exon1, (2) TS10Q23.3 Exon 2, (3) TS10Q23.3 Exon 3, (4) TS10Q23.3
Exon 4, (5) TS10Q23.3 Exon 5, (6) TS10Q23.3 Exon 6, (7) TS10Q23.3
Exon 7, (8) TS10Q23.3 Exon 8, (9) TS10Q23.3 Exon 9, (10) Kontroll-MKK4-Exon
8.
-
13B. Homozygote Deletion des TS10Q23.3-Gens in
humanen Tumorzelllinien und TS10q23.3-mRNA-Expressionsniveaus in
primären
Glioblastomen. Schema der im TS10Q3.3-Gen der untersuchten TZL beobachteten
homozygoten Deletionen. Ausgefüllte
Kreise geben Exons wieder, die nicht homozygot deletiert sind, während unausgefüllte Kreise
verlorene Exons kennzeichnen.
-
13C. Homozygote Deletion des TS10Q23.3-Gens in
humanen Tumorzelllinien und TS10Q23.3-mRNA-Expressionsniveaus in
primären
Glioblastomen. Expression der TS10q23.3-Boten-RNA in humanem gesundem Gehirn
und GBM-Proben, nachgewiesen durch RT-PCRTM-Analyse. Gezeigt
ist das 5'-terminale
Amplikon von TS10Q23.3. Die gezeigten Bahnen umfassen ein Kontroll-Amplikon
(C) aus cDNA des niedriggradigen Glioms PL-1 und sieben normale
Präparate
und Tumorpräparate.
Sechs der 10 untersuchten GBM wurden auf Heterozygositätsverlust
(LOH) in der Umgebung des TS10Q23.3-Locus und Veränderungen
des TS10Q23.3-Gens untersucht. Alle sechs Proben zeigten Heterozygositätsverlust
(LOH), aber es wurden keine Mutationen entdeckt, wenn die Erfinder
ihre DNAs durch Sequenzierung untersuchten. Die Expressionsniveaus
der GADPH-mRNA wurde als Kontrolle für äquivalente Template-Mengen
und -Qualitäten
verwendet.
-
13D. Homozygote Deletion des TS10Q23.3-Gens in
humanen Tumorzelllinien und TS10Q23.3-mRNA-Expressionsniveaus in
primären
Glioblastomen. Verhältnis
der RT-PCRTM-Amplikon-Intensitäten von TS10Q23.3 zu GAPDH
für jedes
normale Präparat
und GBM-Präparat.
-
14. Darstellung der putativen funkionellen Domänen von
TS10Q23.3 und die Position identifizierter Veränderungen. Die N-terminale
Hälfte
von TS10Q23.3 ist homolog zu Phosphatasen sowie zu den Zytoskelettproteinen
Tensin und Auxilin (brauner Kasten). Gezeigt sind auch die Positionen
der Phosphatase-Kerndomäne
(roter Kasten), drei potenzielle Tyrosinphosphorylierungsstellen
(blaue Kästen)
und zwei potenzielle Serinphosphorylierungsstellen (gelbe Kästen). Das
PDZ-Motiv, ITKV, befindet sich am C-Terminus des Proteins. Gezeigt sind
TS10Q23.3-Varianten, die von Steck et al., (1997), Li et al., (1997)
und Liaw et al., (1997) identifiziert worden sind, sowie in dieser
Studie festgestellte Veränderungen;
Blaue Pfeile markieren Misssense-Substitutionen; schwarze Pfeile
kennzeichnen Insertionen oder Deletion innerhalb des Leserasters;
grüne Pfeile
markieren potenzielle Splicing-Varianten
und rote Pfeile geben Frameshift- oder Nonsense-Mutationen wieder,
die zu Verkürzungen
von TS10Q23.3 führen.
Sternchen zeigen Keimbahnmutationen an, die bei Patienten mit Cowden-Syndrom
festgestellt worden sind (Liaw et al., 1997), während die ausgefüllten Kreise
Läsionen
anzeigen, die bei zwei mutmaßlich
unabhängigen
DNA-Proben beobachtet worden sind.
-
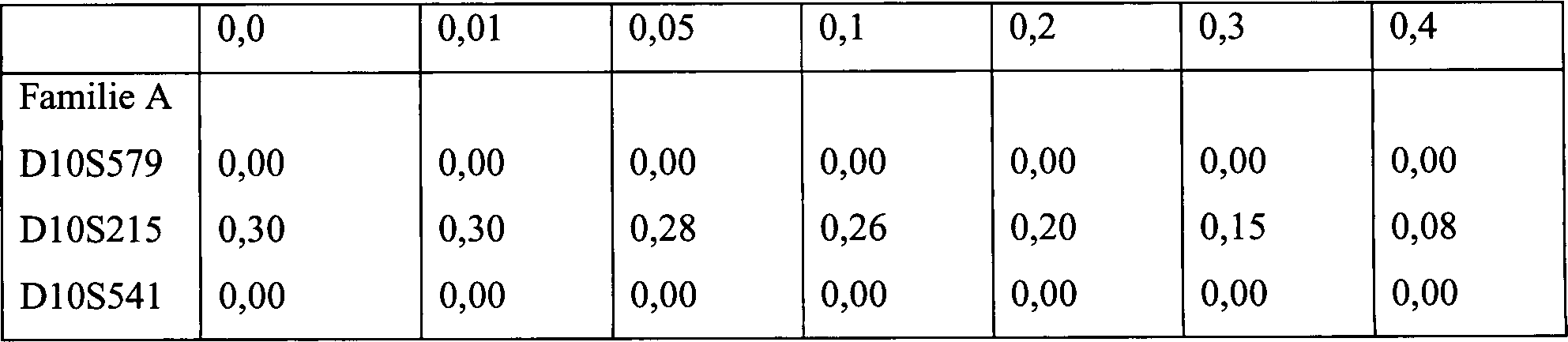
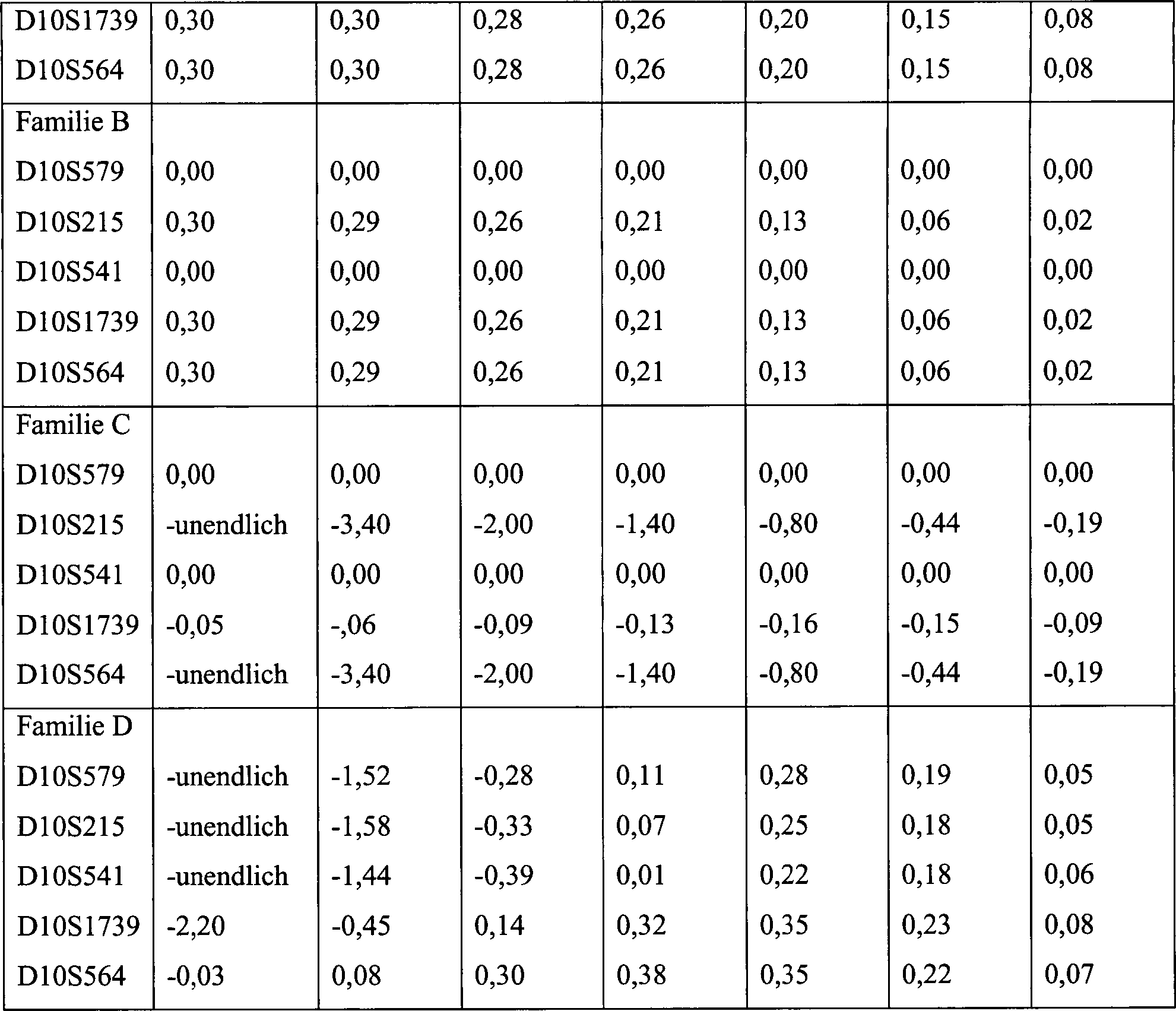
15. Haplotyp-Konstruktion mit Markern auf Chromosom
10 bei vier Familien mit CS.
-
16. DNA-Sequenzierung von TS10Q23.3 bei einer
Familie mit CS und Brustkrebs mit frühzeitigem Auftreten. Die betroffene
Mutter (schwarzer Kreis) wies eine Insertion aus 2 Basenpaaren (AT)
in Exon 5 auf, die bei ihrem nicht betroffenen Bruder (nicht ausgefülltes Quadrat)
nicht vorhanden war. Ihre betroffene Tochter hat die AT-Insertion
geerbt.
-
17. Exogene MMAC1-Proteinexpression. U87MG-Zellen
wurden mit MMCB oder GFCB in den angezeigten Konzentrationen (Partikelzahl/ml)
24 Stunden lang infiziert, anschließend wurden sofort (nach 24 Std.)
oder nach 24 Std. (48 Std.) Lysate hergestellt. Wie bei den Verfahren
beschrieben wurden Western Blots durchgeführt. Auf der linken Seite sind
Proteingrößenmarker
gezeigt. Das MMAC1-Protein wanderte mit etwa 55 kD, was mit den
Angaben von Li et al., 1997 übereinstimmt.
-
18D. FACS-Infektiositäts-Test. U87MG-Zellen wurden
mit GFCB in den angegebenen Konzentrationen 24 Std. lang infiziert.
Der Anteil an Zellen, die grün
fluoreszierendes Protein exprimierten, wurde durchflusszytometrisch
bestimmt. pn/ml: Adenoviruspartikelanzahl pro ml.
-
19A und 19B.
Hemmung der in-vitro-Proliferation durch MMCB; 19A. 3H-Thymidin-Aufnahme. 19B. Untersuchung der Anzahl lebender Zellen.
Fehlerbalken stellen Standardabweichungen (S.D.) dar (3 Wiederholungen).
pn/ml: Adenoviruspartikelanzahl pro ml.
-
20. Bildung von Kolonien in Weichagar. U87MG-Zellen
wurden mit GFCB, MMCB oder FTCB in den angegebenen Konzentrationen
24 Std. lang infiziert. Aufgetragen ist die durchschnittliche Anzahl
an Kolonien ± S.D.
pn/ml: Adenoviruspartikelanzahl pro ml.
-
SEQUENZÜBERBLICK
-
SEQ
ID-Nr.: 1 = TS10q23.3-Gensequenz des Menschen (6 und 9); SEQ ID-Nr.: 2 = Humane TS10q23.3-Peptidsequenz
der CDS von SEQ ID-Nr.: 1; SEQ ID-Nr.: 3 = Translation der Basen
3-119 von SEQ ID-Nr.: 1; SEQ ID-Nr.: 4 = Translation der Basen 123–242 of
SEQ ID-Nr.:1; SEQ ID-Nr.: 5 = Translation der Basen 246–272 von
SEQ ID-Nr.: 1; SEQ ID-Nr.: 6 = Translation der Basen 276–317 von
SEQ ID-Nr.: 1; SEQ ID-Nr.: 7 = Translation der Basen 321–449 von
SEQ ID-Nr.: 1; SEQ ID-Nr.: 8 = Translation der Basen 453–2243 of
SEQ ID-Nr.: 1; SEQ ID-Nr.: 9 = TS10q23.3-Gensequenz der Maus (9); SEQ ID-Nr.: 10 = TS10q23.3-Peptidsequenz der
CDS von SEQ ID-Nr.: 9; SEQ ID-Nr.: 11 = Translation der Basen 14–55 von SEQ
ID-Nr.: 9; SEQ ID-Nr.:12 = Translation der Basen 59–166 von
SEQ ID-Nr.: 9; SEQ ID-Nr.:13
= Translation der Basen 172–222
von SEQ ID-Nr.: 9; SEQ ID-Nr.:14 = Translation der Basen 223–273 von
SEQ ID-Nr.: 9; SEQ ID-Nr.: 15 = Translation der Basen 283–1959 von
SEQ ID Nr.: 9; SEQ ID-Nr.: 16 = TS10q23.3-Gensequenz des Hundes
(9); SEQ ID-Nr.: 17 = TS10q23.3-Peptidsequenz
des Hundes der CDS von SEQ ID-Nr.: 16; SEQ ID-Nr.: 18 = Translation
der Basen 1–1290
von SEQ ID-Nr.: 16; SEQ ID Nr.:19 = Exon 1 (10);
SEQ ID Nr.: 20 = Exon 2 (10); SEQ
ID Nr.: 21 = Exon 3 (10); SEQ ID Nr.:
22 = Exon 4 (10); SEQ ID Nr.: 23 =
Exon 5 (10); SEQ ID Nr.: 24 = Exon
6 (10); SEQ ID Nr.: 25 = Exon 7 (10); SEQ ID Nr.: 26 = Exon 8 (10); SEQ ID Nr: 27 = Exon 9 (10); SEQ ID Nr.: 28 = Ein Motiv aus Rest
88 bis 98; SEQ ID Nr: 29 = konservierte katalytische Domäne einer
Proteintyrosinphosphatase (Denu et al., 1996); SEQ ID Nr.: 30 =
Rest 1–60
des TS10q23.3-Wildtyp-Polypeptids (12A–12C); SEQ ID Nr: 31 = Rest 1–60 der T98G-Mutante des TS10q23.3-Polypeptids
(12D–12F); SEQ ID Nr.: 32 = Rest 1–60 der KE-Mutante des TS10q23.3-Polypeptids
(12G–12I); SEQ ID Nr.: 33 = CA6.ex8.FB-Primer; SEQ ID Nr.:
34 = CA6.ex8.RQ-Primer; SEQ ID Nr.: 35: = CA6.ex8.FC-Primer; SEQ
ID Nr.: 36 = CA6.ex8.RR-Primer; SEQ ID Nr.: 37 = Geschachtelter
(nested) Primer zum Erhalt von Exon 8 des sekundären Amplikons FB-RQ; SEQ ID
Nr.: 38 = = Geschachtelter (nested) Primer zum Erhalt von Exon 9
des sekundären Amplikons
FB-RR; SEQ ID Nr.: 39 = M5'F-Primer; SEQ ID Nr.:
40 = M5' R-Primer;
SEQ ID Nr.: 41 = M3'F-Primer;
SEQ ID Nr.: 42: = F3'R-Primer;
SEQ ID Nr.: 43 = Primer im ersten Durchgang der PCRTM mit
humanem fetalen Gehirn; SEQ ID Nr.: 44 = Primer im ersten Durchgang
der PCRTM mit humanem fetalen Gehirn; SEQ ID
Nr.: 45 = Primer im zweiten Durchgang der PCRTM mit
humanem fetalen Gehirn; SEQ ID Nr.: 46 = Primer im zweiten Durchgang
der PCRTM mit humanem fetalen Gehirn; SEQ
ID Nr.: 47 = Primer zur Herstellung eines spezifischen 303-bp-Produktes
aus dem Pseudogen und nicht aus TS10q23; SEQ ID Nr.: 48 = Primer
zur Herstellung eines spezifischen 303-bp-Produktes aus dem Pseudogen und nicht
aus TS10q23; SEQ ID Nr.: 49 = MMACI-Proteinsequenz der Maus; SEQ ID Nr.:
50 = Peptidsequenz; SEQ ID Nr.: 51 = Translation der Basen 321–1034 von
SEQ ID Nr.: 1; SEQ ID Nr.: 52 = Translation der Basen 169–750 von
SEQ ID Nr.: 9; SEQ ID Nr.: 53 = Translation der Basen 1–108 von
SEQ ID Nr.: 16; SEQ ID Nr.: 54 = MMACI-Gensequenz des Hundes; SEQ
ID Nr.: 55 = MMAC 1-Proteinsequenz des Hundes der CDS von SEQ ID
Nr.: 54; SEQ ID Nr.: 56 = MMAC-Gensequenz der Maus; SEQ ID Nr.:
57 = MMAC1-Proteinsequenz der Maus der CDS von SEQ ID Nr.: 56; SEQ
ID Nr.: 58 = Primer MAC1.6f mit Übereinstimmung
mit Sequenzen in MMAC1-Exon 2; SEQ ID-Nr.: 59 = Primer MAC1,6r mit Übereinstimmung
mit Sequenzen in MMAC1-Exon 5; SEQ ID Nr.: 60 = Translation der Basen
1–54 von
SEQ ID Nr.: 56; SEQ ID Nr.: 61 = Translation der Basen 58–96 von
SEQ ID Nr.: 56; SEQ ID Nr.: 62 = Translation der Basen 98–178 von
SEQ ID Nr.: 56; SEQ ID Nr.: 63 = Translation der Basen 182–208 von
SEQ ID Nr.: 56; SEQ ID Nr.:64 = Sequenz des humanen TS10q23.3-Pseudogens.
-
AUSFÜHRLICHE
BESCHREIBUNG DER BEVORZUGTEN AUSFÜHRUNGSFORMEN
-
I. Die vorliegende Erfindung
-
Wie
oben angegeben, hat eine Reihe verschiedener Gruppen Hinweise auf
eine Tumor supprimierende Aktivität in Verbindung mit der Region
10q des menschlichen Chromosoms 10 gezeigt. Trotz dieser in beträchtlichem
Umfang vorliegenden Arbeiten ist die Identität des Gens oder der Gene, die
für diese
Aktivität
verantwortlich sind, nicht bestimmt worden. Frühere Untersuchungen verwendeten
einen funktionellen Ansatz, der den Transfer von Chromosomen oder
chromosomaler Fragmente, die möglicherweise
ein Tumorsuppressorgen oder Tumorsuppressorgene enthalten, in tumorigene
Gliomzellen umfasst. Diese Arbeiten erlaubten die Definition der
biologischen Aktivität
eines putativen Tumorsuppressorgens oder putativer Tumorsuppressorgene
und trugen zur Lokalisierung einer solchen Aktivität bei. Chromosom
2 und 10 wurden in U251-Gliomzellen und Chromosom 2 und 10 in LG-11-Zellen
eingeführt.
Die LG-11-Zellen haben erwiesenermaßen keine intakten Kopien von
Chromosom 10 und später
stellte sich heraus, dass sich die Bruchstelle an Position 10q24 befindet.
Der Transfer von Chromosom 10 brachte Hybridzellen hervor, die einen
Suppressor-Phänotyp
hatten, einen Verlust der Tumorigenität aufwiesen (keine Tumorbildung)
und die Fähigkeit
verloren hatten, in Weichagarose zu wachsen (50- bis 100-facher
Rückgang;
Pershouse et al., 1993). Die exponentielle Wachstumsrate des Hybrids
war ähnlich
wie die der Herkunftszellen, obgleich die Sättigungsdichte der Hybridzellen
signifikant (10- bis 20-fach) niedriger war als die der Herkunftszellen.
Der Transfer von Chromosom 2 ergab Hybridzellen, die ähnlich wie
die Herkunftszellen agierten.
-
Ein
Ziel dieser Studien war es, das Suppressorgen auf Chromosom 10 durch
Fragmentierung des Neomycin-markierten Chromosoms 10 zu lokalisieren
und anschließend
das fragmentierte Chromosom in Gliomzellen zu übertragen. Die Erfinder haben
jedoch beobachtet, dass einige Hybridzellen spontan Chromosomen-Umordnungen
unterliefen, um Hybridzellen zu ergeben, die nur verschiedene Regionen
des eingeführten Chromosoms
10 zurückbehielten
(Pershouse et al., 1993). Anstatt Fragmentierungsstudien durchzuführen, stellten
die Erfinder Subklone der Hybride her und analysierten diese (Steck
et al., 1995). Die Retention des eingeführten Chromosoms 10 oder seiner
Fragmente wurde mittels informativer RFLP-Marker und FISH-Analyse
verfolgt. Interessanterweise wurde nur das eingeführte Chromosom
einer Umordnung unterzogen. Die Einführung einer vollständigen Kopie
von Chromosom 10 bewirkte eine Hemmung der Transformationseigenschaft
der Hybridzellen, in Weichagarose zu wachsen und in Nacktmäusen Tumore
zu bilden.
-
Durch
sofortige Analyse scheinen diese beiden Phänotypen nun partiell trennbar
zu sein. Manche Subklone (U251.N10.5a-j), die einen Verlust eines überwiegenden
Teils des langen Arms von Chromosom 10 zeigten, wuchsen in Weichagarose,
bildeten jedoch keine Tumore in Nacktmäusen, was anzeigt, dass sich
im verbleibenden Anteil des Chromosoms (10pter bis 10q11) ein Tumorsuppressorlocus
befindet. Klone, die eine distale Region des langen Arms, 10q24
bis 10q26, behalten hatten, wuchsen dagegen weder in Weichagarose noch
in Nacktmäusen
(siehe 4). Dies deutet auf eine weitere
phänotypische
Suppressorregion in der distalen Region des Chromosoms hin. Der
Mangel an weiterem Material in Verbindung mit Chromosom 10 würde außerdem nahe
legen, dass das verbleibende Material von Chromosom 10 für den veränderten
biologischen Phänotyp
verantwortlich ist. Diese Ergebnisse implizieren das Vorhandensein
zweier phänotypisch
unabhängiger
suppressiver Regionen auf Chromosom 10, die am Fortschreiten eines
Glioms beteiligt sind (Steck et al., 1995).
-
Gemäß der vorliegenden
Erfindung haben die Erfinder nun mehrere unabhängige Strategien verfolgt, um
ein Tumorsuppressorgen mit der Bezeichnung TS10q23.3 zu lokalisieren,
das bei Gliomen, Brustkrebs, Prostatakrebs und anderen Krebsarten
eine Rolle spielt. Diese Ansätze,
die ausführlicher
in den folgenden Beispielen beschrieben sind, umfassen (i) Identifizierung
einer homozygoten Deletion in einer Reihe von humanen Gliomzelllinien;
(ii) Feststellen einer einheitlichen Region oder einheitlicher Regionen,
die in Klonen mit unterdrückter
Tumorigenität
beibehalten wurde oder wurden; und (iii) Alleldeletionsstudien mit
Gliomen verschiedenen Grades und entsprechenden Normalproben. Da
das Gen zur Verfügung
steht, ist es nun möglich, die
von dem Gen kodierte Information zu untersuchen, um neuartige diagnostische
und therapeutische Ansätze
in Bezug auf menschliche Krebserkrankungen zu entwickeln.
-
II. Der Tumorsuppressor
auf 10q23.3
-
Gemäß der vorliegenden
Erfindung ist ein Tumorsuppressor identifiert worden, der von einem
Gen auf dem Locus 10q23.3 kodiert wird und hier als TS10q23.3 bezeichnet
wird. Dieses Molekül
kann bei verschiedenen Krebsarten Tumorphänotypen unterdrücken. Beispiele
anderer Tumorsuppressorgene sind p53, Rb und p16, um nur Einige
zu nennen. Obwohl diese Moleküle
strukturell unterschiedlich sind, bilden sie eine Gruppe funktionell
verwandter Moleküle,
zu denen auch TS10q23.3 gehört.
Diese anderen Tumorsuppressoren, die nun untersucht werden, können in
gleicher Weise verwendet werden.
-
Die
vorliegende Erfindung betrifft nicht nur das gesamte TS10q23.3-Molekül, sondern
auch Fragmente des Polypeptids, das die tumorsupprimierende (oder
andere) Aktivität
beibehält
oder nicht. Fragmente einschließlich
des N-Terminus des Moleküls
können
durch gentechnologische Bearbeitung der Translations-Stopp-Stellen
innerhalb der kodierenden Region erzeugt werden (wird nachfolgend
erläutert).
Alternativ kann die Behandlung des TS10q23.3-Moleküls mit proteolytischen Enzymen,
bekannt als Proteasen, eine Vielzahl unterschiedlicher N-terminaler,
C-terminaler und interner Fragmente hervorbringen. Beispiele von
Fragmenten können
aufeinander folgende Reste der in 7 und 9 gezeigten TS10q23.3-Sequenz mit einer Länge von
6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 12, 22,
23, 24, 25, 30, 35, 40, 45, 50, 55, 60, 65, 75, 80, 85, 90, 95,
100, 200, 300, 400 oder mehr Aminosäuren umfassen. Diese Fragmente
können
nach bekannten Verfahren gereinigt werden, beispielsweise durch
Präzipitation
(z. B. Ammoniumsulfat), HPLC, Ionenaustauschchromatographie, Affinitätschromatographie
(einschließlich
Immunaffinitätschromatographie) oder
verschiedene Größenauftrennungen
(Sedimentation, Gelelektrophorese, Gelfiltration).
-
A. Strukturelle Merkmale
des Polypeptids
-
Das
Gen für
TS10q23.3 kodiert für
ein Polypeptid mit 403 Aminosäuren.
Das vorausgesagte Molekulargewicht dieses Moleküls ist 47.122, was einen pI
von 5,86 ergibt. Dieses Molekül
kann daher mindestens als Standard in Assays verwendet werden, in
denen Molekulargewicht und pI-Wert untersucht werden.
-
Eine
Phosphatase-Konsensusstelle befindet sich an den Resten 122–131 und
entspricht vollständig der
Tyrosinphosphatase (PTP)-Konsensussequenz: [I/V]HCxAGxxR[S/T]G.
Außerhalb
der aktiven Domänen unterscheiden
sich die Sequenzen erheblich. PTPs entstehen durch Phosphoenzym-Zwischenstufen.
Die enzymatische Reaktion umfasst die Bildung von Phosphoryl-Cystein-Zwischenstufen
nach einem nukleophilen Angriff des Thiolatanions von Cystein auf
das Phosphoratom des Substrats. Die Reaktion kann als zweistufiger chemischer
Prozess dargestellt werden: Phosphoryltransfer auf das Enzym, begleitet
von einer schnellen Freisetzung des dephosphorylierten Produktes
und Hydrolyse der Thiol-Phosphat-Zwischenstufe, begleitet von einer
schnellen Freisetzung von Phosphat. Das Enzym bindet das Dianion
des phosphathaltigen Substrats und reagiert damit, um den katalytisch
kompetenten Komponentenkomplex zu bilden. Für einen Phosphoryltransfer auf
das Enzym müssen
am Enzym eine Asparaginsäure
protoniert und das nukleophile Cystein deprotoniert werden (Thiolatanion).
Außerdem
befinden sich potenzielle Tyrisonphosphorylierungsstellen an Rest
233–240 und
308–315
und cAMR-Phosphorylierungsstellen befinden sich an Rest 128, 164,
223 und 335. Phosphatasen haben bekannterweise Kinasestellen und
die Phosphataseaktivität
dieser Enzyme kann durch Phosphorylierung an diesen Stellen moduliert
werden, Proteinphosphatasen werden generell in zwei Kategorien unterteilt – Serin-/Threoninphosphatasen
und Tyrosinphosphatasen. Bestimmte Tyrosinphosphatasen sind auch gegen
Phosphoserin und Phosphothreonin aktiv.
-
Die
Wechselwirkung zwischen Kinasen und Phosphatasen und die verschiedenen
Phosphorylierungszustände
von Polypeptiden sind wichtige Merkmale der Zellphysiologie. Kinasen
und Phosphatasen arbeiten durch verschiedenartige Mechanismen bei
unterschiedlichen Wegen in Zellen zusammen, die an der Signal-, Energiespeicherung-
und Zellregulation beteiligt sind. Seit der Identifizierung einer
intrinsischen Tyrosinkinasefunktion in dem transformierenden Protein
src (Collett & Erickson,
1978) erwies sich die Rolle der Phosphorylierung, insbesondere an
Tyrosinresten, als zentraler Vorgang bei der Kontrolle zellulärer Proliferation
und der Induktion von Krebs (Hunter, 1991; Bishop, 1991). Die Rolle,
die Proteinphosphatasen bei der Wachstumsregulation sowie bei vielen
anderen biologischen und biochemischen Aktivitäten spielen, wurde mit dem
Phosphorylierungsstatus biologisch wichtiger Moleküle in Zusammenhang
gebracht (Cohen, 1994).
-
Ausgehend
von seiner Sequenz scheint TS10q23.3 für eine Tyrosinphosphatase oder
eine Phosphatase mit zweifacher Spezifität zu kodieren, die Homologie
zu Zytoskelett-assoziierten Proteinen, dem Tensin des Huhns und
bovinem Auxilin aufweist (Steck et al., 1997; Li et al., 1997).
Die N-terminate Hälfte
von TS10q23.3 ist daher zu mehreren Phosphatasen homolog und sein
putatives Phosphatase-Kernmotiv befindet sich an den Resten 122–134 (Denu
et al., 1996; Tonks und Neel, 1996). Die N-terminale Region von TS10q23.3
könnte
daher enzymatische Aktivität
und zelluläre
Lokalisationsaktivität
aufweisen. Der C-terminale Anteil von TS10q23.3 enthält drei
potenzielle Tyrosinphosphatasestellen an Rest 240, 315 und 336.
Ein gegebenenfalls phosphoryliertes Tyrosin 315 würde eine
potenzielle SH2-Bindungsstelle darstellen, da sich drei Reste vom
Tyrosin entfernt in C-terminaler Richtung ein Leucinrest befindet
(Songyang et al., 1995). In der C-terminalen Hälfte von TS10q23.3 befinden
sich zwei potenzielle Serinphosphorylierungsstellen. Serinrest 338
repräsentiert
eine potenzielle Cal2+/Calmodulinabhängige Proteinkinase-II-Stelle,
während
Serin 355 eine potenzielle Caseinkinase-II-Stelle darstellt (Hardie
und Hanks, 1995). Die letzten vier C-terminalen Aminosäuren, ITKV,
repräsentieren
eine potenzielle PDZ-Bindungsdomäne
(Fanning und Anderson, 1996; Saras und Heldin, 1996). PDZ-Domänen finden
sich in vielen verschiedenen intrazellulären Proteinen und es wird angenommen,
dass sie Protein-Protein-Wechselwirkungen vermitteln, indem sie
direkt an das C-terminale Ende von Zielproteinen binden.
-
Es
sollte ferner erwähnt
werden, dass die etwa 60 Aminosäuren
des N-Terminus des Moleküls
eine gewisse Homologie zu Tensin aufweisen, einem Zytoskelettprotein,
das möglicherweise
an Adhäsionsplaques beteiligt
ist. Dies deutet darauf hin, dass TS10q23.3 an Zelloberflächenphänomenen
beteiligt ist, beispielsweise an der Kontakthemmung, Invasion, Migration
oder bei der Signalübertragung
zwischen Zellen. TS10q23.3-Punktmutationen, die in bestimmten Tumorzelllinien
identifiziert worden sind, betreffen vorgeschlagene Sekundärstrukturen
in dieser Region.
-
B. Funktionelle Aspekte
-
Wenn
die vorliegende Anmeldung die Funktion von TS10q23.3 oder „Wildtyp"-Aktivität betrifft,
ist damit gemeint, dass das fragliche Molekül die Fähigkeit besitzt, die Transformation
einer Zelle von einem normal regulierten Proliferationszustand zu
einem malignen Zustand zu hemmen, d. h. einen Zustand, der mit jeglicher Art
einer anomalen Wachstumsregulierung verbunden ist, oder die Transformation
einer Zelle von einem anomalen Zustand zu einem hoch malignen Zustand
zu hemmen, z. B., Metastasierung oder invasives Tumorwachstum zu
verhindern. Andere Phänotypen,
die als von dem normalen TS10q23.3-Genprodukt reguliert angesehen
werden können,
sind Angiogenese, Adhäsion,
Migration, Signalübertragung
zwischen Zellen, Zellwachstum, Zellproliferation, dichteabhängiges Wachstum,
verankerungsabhängiges
Wachstums und Andere. Anhand von Assays, die einem Fachmann bekannt
sind, kann bestimmt werden, welche Moleküle diese Aktivität besitzen.
Beispielsweise können
durch den Transfer von Genen, die TS10q23.3 oder Varianten davon
kodieren, in Zellen, die kein funktionelles TS10q23.3-Produkt haben
und daher eine beeinträchtigte
Wachstumskontrolle aufweisen, mittels Wachstumsunterdrückung solche
Moleküle
mit TS10q23.3-Funktion identifiziert werden.
-
Wie
oben festgestellt, liegen Hinweise vor, dass es sich bei TS10q23.3
um eine Phosphatase handelt. Der Anteil des Proteins im Bereich
der Reste 88–98
stimmt genau mit der konservierten katalytischen Domäne einer
Proteintyrosinphosphatase überein.
In dem Molekül
befinden sich außerdem
putative Kinaseziele, was ein weiteres Merkmal von Phosphatasen
darstellt. Da andere Tumorsuppressoren mit dieser Art von Aktivität identifiziert
worden sind, ist es wünschenswert,
die Phosphatasefunktion der tumorsupprimierenden Rolle von TS10q23.3
zu bestimmen. Dies könnte
sich auch als erfolgreicher Ansatz zur Entwicklung von Screening-Assays für die Abwesenheit
einer TS10q23.3-Funktion oder bei der Entwicklung von Krebstherapien
erweisen, beispielsweise durch gezielten Angriff der Phosphatasefunktion
von TS10q23.3, durch gezieltes Targeting des Substrats, mit dem
TS10q23.3 reagiert und/oder durch durch gezieltes Targeting der
Kinase oder der Kinasen, die mit TS10q23.3 reagieren.
-
C. Varianten von TS10q23.3
-
Bei
Aminosäuresequenzvarianten
des Polypeptids kann es sich um Substitutions-, Insertions- oder Deletionsvarianten
handeln. In Deletionsvarianten fehlen ein oder mehrere Reste des
nativen Proteins, die für die
Funktion oder immunogene Aktivität
nicht essenziell sind; ein Beispiel dafür sind die oben beschriebenen Varianten,
denen eine Transmembransequenz fehlt. Ein anderer, häufiger Typ
einer Deletionsvariante ist eine Variante, der sekretorische Signalsequenzen
fehlen oder Signalsequenzen, die ein Protein zur Bindung an einen
bestimmten Teil einer Zelle steuern. Insertionsmutanten umfassen
typischerweise die Addition von Material an einer nicht-terminalen
Stelle im Polypeptid. Dazu gehört
beispielsweise die Insertion eines immunreaktiven Epitops oder einfach
eines einzelnen Restes. Terminale Additionen, die Fusionsproteine
genannt werden, sind nachfolgend erläutert.
-
Substitutive
Varianten enthalten typischerweise den Austausch einer Aminosäure durch
eine Andere an einer oder mehreren Stellen innerhalb des Proteins
und können
so konzipiert sein, dass eine oder mehrere Eigenschaften des Polypeptids,
beispielsweise Stabilität
gegenüber
proteolytischer Spaltung, ohne Verlust anderer Funktionen oder Eigenschaften
moduliert werden. Substitutionen dieser Art sind vorzugsweise konservativ,
das heißt,
eine Aminosäure
wird durch eine Andere mit ähnlicher
Form und Ladung ersetzt. Konservative Substitutionen sind aus dem
Stand der Technik gut bekannt und umfassen beispielsweise den Austausch
von: Alanin durch Serin, Arginin durch Lysin, Asparagin durch Glutamin
oder Histidin, Asparaginsäure
durch Glutaminsäure,
Cystein durch Serin, Glutamin durch Asparagin, Glutaminsäure durch
Asparaginsäure,
Glycin durch Prolin, Histidin durch Asparagin oder Glutamin, Isoleucin
durch Leucin oder Valin, Leucin oder Valin oder Isoleucin, Lysin
durch Arginin, Methionin durch Leucin oder Isoleucin, Phenylalanin
durch Tyrosin, Leucin oder Methionin, Serin durch Threonin, Threonin
durch Serin, Tryptophan durch Tyrosin, Tyrosin durch Tryptophan oder
Phenylalanin und Valin durch Isoleucin oder Leucin.
-
In
besonderen Aspekten wird erwogen, dass ein Fachmann Standardtechniken,
die einem Fachmann gut bekannt sind, anwendet, um die Mutanten herzustellen.
Speziell in Betracht gezogen sind N-terminale Deletionen, C-terminale
Deletionen, interne Deletionen sowie zufällige (Random) Mutagenese und
Punktmutagenese.
-
N-terminale
und C-terminale Deletionen sind Formen einer Deletionsmutagenese,
die beispielsweise das Vorhandensein einer geeigneten einzelnen
Restriktionsschnittstelle in der Nähe der C- oder N-terminalen Region
ausnutzt. Die DNA wird an dieser Stelle gespalten und die Schnittenden
werden mit Nukleasen wie BAL31, Exonuklease III, DNase I und S1-Nuklease
verdaut. Durch Wiedervereinigung der beiden Enden entsteht eine
Reihe von DNAs mit Deletionen verschiedener Größe in der Umgebung der Restriktionsschnittstelle. Die
von solchen Mutanten exprimierten Proteine können hinsichtlich ihrer Funktion
bei der Apoptosehemmung und/oder als Chaperon getestet werden, wie
im Verlauf der Patentbeschreibung beschrieben ist. Bei Mutanten mit
internen Deletionen werden ähnliche
Techniken angewandt, aber Mutanten mit interner Deletion werden hergestellt,
indem zwei geeignet platzierte Restriktionsschnittstellen verwendet
werden, was somit die Herstellung einer präzise definierten Deletion und
die Wiedervereinigung der Enden wie oben erlaubt.
-
In
Betracht gezogen sind auch Mutanten mit partieller Deletion. In
solchen Fällen
verwendet ein Fachmann ein Restriktionsenzym, das die DNA je nach
Länge der
Reaktionsdauer an zahlreichen Stellen schneidet („Frequent
Cutter"). Durch
Variation der Reaktionsbedingungen ist es daher möglich, eine
Reihe von Mutanten verschiedener Größe herzustellen, die dann hinsichtlich
ihrer Aktivität
untersucht werden können.
-
Eine
zufällige
Insertionsmutation kann auch durchgeführt werden, indem die DNA-Sequenz beispielsweise
mit einer DNase I geschnitten und eine Abfolge von Nukleotiden eingesetzt
wird, die 3, 6, 9, 12. etc. Aminosäuren kodieren, und die Enden
neu ligiert werden. Nachdem eine solche Mutation hergestellt worden
ist, können
die Mutanten hinsichtlich verschiedener Aktivitäten, die dem Wildtypprotein
eigen sind, untersucht werden.
-
Sobald
allgemeine Bereiche des Gens als für bestimmte Proteindomänen kodierend
festgestellt worden sind, kann eine Punktmutagenese durchgeführt werden,
um genau zu identifizieren, welche Aminosäurereste für bestimmte, mit TS10q23.3
verbundenen Aktivitäten
wichtig sind. Ein Fachmann ist daher in der Lage, Einzelbasenveränderungen
des DNA-Strangs herzustellen, die ein verändertes Codon und eine Missense-Mutation
ergeben.
-
Es
folgt eine Erörterung
der Veränderung
der Aminosäuren
eines Proteins zur Erzeugung eines äquivalenten oder sogar verbesserten
Moleküls
der zweiten Generation. Beispielsweise können in einer Proteinstruktur
bestimmte Aminosäuren
durch andere Aminosäuren
ersetzt werden, ohne dass interaktive Bindungskapazitäten an Strukturen,
wie beispielsweise Antigenbindungsregionen von Antikörpern oder
Bindungsstellen an Substratmolekülen,
wesentlich verloren gehen. Da die biologische funktionelle Aktivität eines
Proteins durch seine interaktiven Fähigkeiten und seine Art bestimmt
wird, können
in einer Proteinsequenz und in ihrer zugrunde liegenden DNA-Kodierungssequenz
bestimmte Aminosäuresubstitutionen
durchgeführt
und dennoch ein Protein mit ähnlichen
Eigenschaften erhalten werden. Es wird von den Erfindern daher in
Betracht gezogen, dass in der DNA-Sequenz von Genen ohne wesentlichen
Verlust ihrer biologischen Nützlichkeit
oder Aktivität
verschiedene Änderungen
durchgeführt
werden können,
wie unten erläutert.
Tabelle 1 zeigt die Codons, die bestimmte Aminosäuren kodieren.
-
Bei
der Durchführung
solcher Veränderungen
kann der hydropathische Index von Aminosäuren berücksichtigt werden. Die Bedeutung
des hydropathischen Aminosäureindex
bei der Übertragung
interaktiver biologischer Funktion auf ein Protein ist im Stand
der Technik im Allgemeinen anerkannt (Kyte & Doolittle, 1982). Es ist anerkannt,
dass die relative hydropathische Eigenschaft der Aminosäure zur
Sekundärstruktur des
resultierenden Proteins beiträgt,
welche wiederum die Interaktion des Proteins mit anderen Molekülen wie beispielsweise
Enzymen, Substraten, Rezeptoren, DNA, Antikörpern, Antigenen und dergleichen
definiert.
-
Jeder
Aminosäure
ist wie folgt auf der Basis ihrer Hydrophobie und Ladungseigenschaften
ein hydropathischer Index zugeordnet worden (Kyte & Doolittle, 1982):
Isoleucin (+4,5), Valin (+4,2), Leucin (+3,8), Phenylalanin (+2,8),
Cystein(Cystin (+2,5), Methionin (+1,9), Alanin (+1,8), Glycin (–0,4), Threonin
(–0,7),
Serin (–0,8),
Tryptophan (–0,9),
Tyrosin (–1,3),
Prolin (–1,6),
Histidin (–3,2),
Glutamat (–3,5),
Glutamin (–3,5),
Aspartat (–3,5),
Asparagin (–3,5),
Lysin (–3,9)
und Arginin (–4,5).
-
Es
ist aus dem Stand der Technik bekannt, dass bestimmte Aminosäuren durch
andere Aminosäuren mit ähnlichen
hydropathischen Indizes oder Werten substituiert werden können und
dennoch ein Protein mit ähnlicher
biologischer Funktion ergeben, d. h. dennoch ein in seiner biologischen
Funktion äquivalentes
Protein. Bei der Durchführung
solcher Veränderungen
ist die Substitution von Aminosäuren,
deren hydropathische Indizes im Bereich von ±2 liegen, bevorzugt, wobei
solche, deren hydropathischer Index im Bereich von ±1 liegt,
besonders bevorzugt sind und solche mit einem hydropathischen Index
im Bereich von ±0,5
noch mehr bevorzugt sind.
-
Es
ist aus dem Stand der Technik klar, dass ähnliche Aminosäuren wirksam
auf der Basis ihrer Hydrophilie substituiert werden können. Das
hierin durch Bezugnahme enthaltene US-Patent 4,554,101 stellt fest, dass die
höchste
lokale durchschnittliche Hydrophilie eines Proteins, bestimmt durch
die Hydrophilie seiner benachbarten Aminosäuren, mit einer biologischen
Eigenschaft des Proteins korreliert. Wie in US-Patent 4,554,101
ausführlich
aufgeführt,
sind Aminosäureresten
folgende Hydrophiliewerte zugeordnet worden: Arginin (+3,0), Lysin
(+3,0), Aspartat (+3,0 ± 1),
Glutamat (+3,0 ± 1),
Serin (+0,3), Asparagin (+0,2), Glutamin (+0,2), Glycin (0), Threonin
(–0,4),
Prolin (–0,5 ± 1), Alanin
(–0,5),
Histidin (–0,5),
Cystein (–1,0),
Methionin (–1,3),
Valin (–1,5),
Leucin (–1,8),
Isoleucin (–1,8),
Tyrosin (–2,3),
Phenylalanin (–2,5)
, Tryptophan (–3,4).
-
Eine
Aminosäure
kann durch eine andere mit ähnlichem
Hydrophiliewert substituiert werden und dennoch ein biologisch äquivalentes
und immunologisch äquivalentes
Protein erhalten werden. Bei solchen Veränderungen ist die Substitution
von Aminosäuren,
deren Hydrophiliewerte im Bereich von +2 liegen, bevorzugt, wobei
solche, deren Hydrophiliewert im Bereich von ±1 liegt, besonders bevorzugt
sind und solche mit einem Hydrophiliewert im Bereich von ±0,5 noch
mehr bevorzugt sind.
-
Wie
oben ausgeführt,
beruhen Aminosäuresubstitutionen
im Allgemeinen auf der relativen Ähnlichkeit der Aminosäureseitenkettensubstituenten,
beispielsweise ihrer Hydrophobie, Hydrophilie, Ladung, Größe und Dergleichen.
Einem Fachmann sind beispielhafte Substitutionen, die verschiedene
der vorherigen Merkmale in Betracht ziehen, gut bekannt und umfassen:
Arginin und Lysin, Glutamat und Aspartat, Serin und Threonin, Glutamin
und Asparagin sowie Valin, Leucin und Isoleucin.
-
Eine
weitere Ausführungsform
für die
erfindungsgemäße Herstellung
von Polypeptiden ist die Verwendung von Peptidmimetika. Mimetika
sind peptidhaltige Moleküle,
die Elemente einer Proteinsekundärstruktur nachahmen.
Siehe beispielsweise Johnson et al., "Peptide Turn Mimetics" in BIOTECHNOLOGY
AND PHARMACY, Pezzuto et al., Hrsg., Chapman und Hall, New York
(1993). Die Begründung
der Verwendung von Peptidmimetika ist, dass das Peptidgerüst von Proteinen
vorwiegend dazu dient, Aminosäureseitenketten
so auszurichten, dass molekulare Wechselwirkungen, wie die von Antikörper und
Antigen, erleichtert sind. Ein Peptidmimetikum soll molekulare Wechselwirkungen
ermöglichen,
die ähnlich
sind wie beim natürlichen
Molekül. Diese
Prinzipien können
in Verbindung mit den oben ausgeführten Prinzipien verwendet
werden, um Moleküle der
zweiten Generation technisch herzustellen, die viele der natürlichen
Eigenschaften von TS10q23.3, jedoch mit veränderten und sogar verbesserten
Merkmalen aufweisen.
-
D. Domänenaustausch
-
Wie
in den Beispielen beschrieben, haben die vorliegenden Erfinder putative
Homologe des humanen TS10q23.3-Gens in Maus und Hund identifiziert.
Darüber
hinaus sind in TS10q23.3 Mutationen identifiziert worden, von denen
angenommen wird, dass sie seine Funktion verändern. Diese Studien sind aus
mindestens zwei Gründen
wichtig. Erstens begründen
sie die Erwartung, dass in verwandten Arten wie der Ratte, dem Kaninchen,
dem Affen, dem Gibbon, dem Schimpansen, dem Menschenaffen, dem Pavian,
der Kuh, dem Schwein, dem Pferd, dem Schaf und der Katze noch weitere
Homologe, Allelvarianten und Mutanten dieses Gens existieren. Nach
Isolierung dieser Homologe, Varianten und Mutanten und in Verbindung
mit anderen Analysen können
bestimmte aktive oder funktionelle Domänen identifiziert werden. Zweitens
wird dies einen Ausgangspunkt für
weitere Mutationsanalysen des Moleküls liefern. Diese Informationen
können
beispielsweise durch Domänenaustausch
untersucht werden.
-
Domänenaustausch
umfasst die Erzeugung chimärer
Moleküle
anhand von unterschiedlichen, aber in diesem Fall verwandten Polypeptiden.
Durch Vergleich der TS10q23.3-Sequenzen von Maus, Hund und Mensch
mit der TS10q23.3-Sequenz anderer Arten und mit Mutanten und Allelvarianten
dieser Polypeptide ist eine Vorhersage in Bezug auf die funktionell
signifikanten Regionen dieser Moleküle möglich. Es ist damit möglich, anschließend verwandte
Domänen
dieser Moleküle
auszutauschen, um die Bedeutung dieser Regionen für die Funktion
von TS10q23.3 zu bestimmen. Diese Moleküle können insofern zusätzlichen
Wert haben, als diese „Chimären" von natürlichen
Molekülen
unterscheidbar sind, obgleich sie dieselbe Funktion vermitteln.
-
Ausgehend
von der Sequenzidentität
der Sequenzen von Maus, Hund und Mensch auf Aminosäureebene
kann abgeleitet werden, dass selbst kleine Veränderungen der Primärsequenz
des Moleküls
seine Funktion beeinflussen. Eine weitergehende Analyse von Mutationen
und ihrem prognostizierten Effekt auf die Sekundärstruktur wird dieses Verständnis erweitern.
-
Ein
weiterer struktureller Aspekt von TS10q23.3, der eine Begründung für die Domänenaustauschstudien
liefert, sind die Tyrosinphosphatase-ähnliche Domäne und die putativen Tyrosinphosphorylierungsstellen. Diese
Domäne
kann durch andere Phosphatasedomänen
substituiert werden, um die Spezifität dieser Funktion zu verändern. Diese
Beobachtung rechtfertigt eine weitere Untersuchung der Homologie
zwischen TS10q23.3 und anderen Phosphatasen.
-
E. Fusionsproteine
-
Eine
spezielle Art einer Insertionsvariante ist das Fusionsprotein. Dieses
Molekül
weist in der Regel alle Abschnitte oder einen wesentlichen Abschnitt
des nativen Moleküls
auf, die bzw. der am N- oder C-Terminus mit einem vollständigen oder
einem Abschnitt eines zweiten Polypeptids verknüpft ist oder die am N- oder C-Terminus
mit einem vollständigen
oder einem Abschnitt eines zweiten Polypeptids verknüpft sind.
Fusionen weisen typischerweise zum Beispiel Führungssequenzen anderer Arten
auf, um die rekombinante Expression eines Proteins in einem heterologen
Wirt zu erlauben. Eine andere nützliche
Fusion umfasst die Addition einer immunologisch aktiven Domäne wie beispielsweise
eines Antikörperepitops,
um die Reinigung des Fusionsproteins zu erleichtern. Der Einschluss
einer Schnittstelle an oder in der Nähe der Verbindungsstelle erleichtert das
Entfernen des externen Polypeptids nach der Reinigung. Weitere nützliche
Funktionen umfassen das Verknüpfen
funktioneller Domänen,
wie beispielsweise aktive Zentren von Enzymen, Glykosylierungsdomänen, Signale
für zelluläres Targeting
oder Transmembranregionen.
-
Eine
bestimmte Fusion, die von Interesse ist, würde ein Deletionskonstrukt
enthalten, dem die Phosphatasebindungsstelle von TS10q23.3 fehlt,
die aber andere Regionen enthält,
die das Substratmolekül
binden könnten.
Eine Fusion an ein Polypeptid, das zur Reinigung des Substrat-TS10q23.3-Komplexes
verwendet werden kann, hätte
den Zweck, das Substrat zur Identifikation und Analyse zu isolieren.
-
Beispiele
von Fusionsproteinexpressionssystemen umfassen das Glutathion-S-Transferase (GST)-System
(Pharmacia, Piscataway, NJ, USA), das Maltosebindungsproteinsystem
(NEB, Beverley, MA, USA), das FLAG-System (IBI, New Haven, CT, USA)
und das 6×His-System
(Qiagen, Chatsworth, CA, USA).
-
Manche
dieser Systeme produzieren rekombinante Polypeptide, die nur eine
kleine Anzahl zusätzlicher
Aminosäuren
aufweisen, welche die antigene Eigenschaft des rekombinanten Polypeptids
mit hoher Wahrscheinlichkeit nicht beeinflussen. Sowohl das FLAG-System
als auch das 6×His-System
fügen nur
kurze Sequenzen hinzu, von denen bekannt ist, dass sie kaum antigen
sind und die Faltung des Polypeptids zu seiner nativen Konformation
nicht negativ beeinflussen.
-
In
anderen Systemen ist es möglich,
Fusionsproteinkonstrukte herzustellen, um die Immunogenität eines
TS10q23.3-Fusionskonstruktes zu steigern. Fusionskonstrukte zur
Steigerung der Immunogenität
sind einem Fachmann gut bekannt, beispielsweise ist eine Fusion
von TS10q23.3 mit einem Helferanigen wie beispielsweise hsp70 oder
Peptidsequenzen wie der der Diphterietoxinkette oder einem Zytokin
wie IL-2 für
die Induktion einer Immunreaktion hilfreich. In anderen Ausführungsformen
können
Fusionskonstrukte hergestellt werden, welche das Targeting der mit
TS10q23.3 verwandten Zusammensetzungen zu einem speziellen Ort oder
einer bestimmten Stelle leiten. Die Fusion von TS10q23.3 oder eines
Proteins des Typs TS10q23.3 mit einem Liganden ist beispielsweise
ein effektives Mittel, um die Zusammensetzung an einen Ort zu leiten,
an dem der Rezeptor für
einen solchen Liganden exprimiert wird. So kann das TS10q23.3 oder
die mit TS10q23.3 verwandte Zusammensetzung über eine rezeptorvermittelte
Aufnahme in eine Zelle gelangen. Das TS10q23.3-Protein kann kovalent
an einen Liganden angebracht oder mit ihm fusioniert sein. Dies
kann als Mechanismus zur Abgabe in eine Zelle verwendet werden.
Der Ligand, an dem das Protein angebracht ist, kann dann von einer
rezeptortragenden Zelle aufgenommen werden.
-
Andere
Fusionssysteme produzieren Polypeptidhybride, wenn es wünschenswert
ist, den Fusionspartner von dem gewünschten Polypeptid abzuschneiden.
In einer Ausführungsform
ist der Fusionspartner mit dem rekombinanten TS10q23.3-Polypeptid
durch eine Peptidsequenz verknüpft,
die eine spezifische Erkennungsstelle für eine Protease enthält. Beispiele
geeigneter Sequenzen sind solche, die von der Tabakätzvirusprotease
(Life Technologies, Gaithersburg, MD, USA) oder Faktor Xa (New England
Biolabs, Beverly, MA, USA) erkannt werden.
-
F. Reinigung
von Proteinen
-
Es
ist wünschenswert,
TS10q23.3 oder Varianten davon zu reinigen. Einem Fachmann sind
Proteinreinigungstechniken gut bekannt. Diese Techniken umfassen
in einer Stufe die grobe Fraktionierung des zellulären Milieus
in Polypeptid- und Nicht-Polypeptidfraktionen. Nachdem das Polypeptid
von anderen Proteinen getrennt ist, kann das Polypeptid von Interesse
anhand chromatographischer und elektrophoretischer Techniken weiter
gereinigt werden, um eine partielle oder vollständige Reinigung zu erzielen
(bzw. eine Reinigung bis zur Homogenität). Analytische Verfahren,
die sich besonders gut für
die Herstellung eines reinen Peptids eignen, sind die Ionenaustauschchromatographie,
die Ausschlusschromatographie, die Polyacrylamidgelelektrophorese
und die isoelektrische Fokussierung. Ein besonders effizientes Verfahren
der Reinigung von Peptiden ist die schnelle Proteinflüssigchromatographie
oder sogar die HPLC.
-
Bestimmte
Aspekte der vorliegenden Erfindung betreffen die Reinigung und in
bestimmten Ausführungsformen
die wesentliche Reinigung eines kodierten Proteins oder Peptids.
Der Begriff „gereinigtes
Protein oder Peptid",
wie hierin verwendet, soll sich auf eine Zusammensetzung beziehen,
die von anderen Bestandteilen isolierbar ist, wobei das Protein
oder Peptid bis zu einer beliebigen Stufe relativ zu seinem natürlicherweise
erzielbaren Zustand gereinigt wird. Ein gereinigtes Protein oder
Peptid bezieht sich daher auch auf ein Protein oder Peptid außerhalb
der Umgebung, in der es natürlicherweise
vorkommt.
-
„Gereinigt" bezieht sich im
Allgemeinen auf eine Protein- oder Peptidzusammensetzung, die einer Fraktionierung
unterzogen worden ist, um verschiedene andere Bestandteile zu entfernen,
und bei der diese Zusammensetzung im Wesentlichen ihre ausgedrückte biologische
Aktivität
beibehält.
Wenn der Begriff „im Wesentlichen
gereinigt" verwendet
wird, bezieht sich diese Bezeichnung auf eine Zusammensetzung, in
der das Protein oder Peptid den Hauptbestandteil der Zusammensetzung
ausmacht, beispielsweise etwa 50%, etwa 60%, etwa 70%, etwa 80%,
etwa 90%, etwa 95% oder mehr der Proteine in der Zusammensetzung
darstellt.
-
Einem
Fachmann sind angesichts der vorliegenden Offenbarung verschiedene
Verfahren zur Quantifizierung des Grads der Reinigung des Proteins
oder Peptids bekannt. Dazu gehört
beispielsweise das Bestimmen der spezifischen Aktivität einer
aktiven Fraktion oder die Ermittlung der Menge von Polypeptiden
in einer Fraktion mittels SDS/PAGE-Analyse. Ein bevorzugtes Verfahren
zum Ermitteln der Reinheit einer Fraktion ist es, die spezifische
Aktivität
der Fraktion zu berechnen, um sie mit der spezifischen Aktivität des ursprünglichen Extraktes
zu vergleichen und so den Reinheitsgrad zu berechnen, der hierin
durch eine „x-fache
Reinigungszahl" ermittelt
wird. Welche tatsächlichen
Einheiten verwendet werden, um das Maß der Aktivität darzustellen, richtet
sich natürlich
nach der jeweiligen Assaytechnik, die nach der Reinigung ausgewählt wird,
und danach, ob oder ob nicht das exprimierte Protein oder Polypeptid
eine nachweisbare Aktivität
aufweist.
-
Einem
Fachmann sind verschiedene Techniken bekannt, die sich zur Anwendung
bei der Proteinreinigung eignen. Dazu gehören beispielsweise das Ausfällen mit
Ammoniumsulfat, PEG, Antikörpern
und dergleichen oder durch Hitzedenaturierung, gefolgt von Zentrifugation,
Chromatographieschritte wie beispielsweise Innenaustausch-, Gelfiltrations-,
Umkehrphasen-, Hydroxyapatit- und Affinitätschromatographie, Isoelektrische
Fokussierung, Gelelektrophorese und Kombinationen solcher und anderer
Techniken. Wie aus dem Stand der Technik im Allgemeinen bekannt
ist, wird angenommen, dass die Reihenfolge der Durchführung der verschiedenen
Reinigungsschritte verändert
werden kann oder dass bestimmte Schritte weggelassen werden können und
dies dennoch ein geeignetes Verfahren für die Reinigung eines im Wesentlichen
gereinigten Proteins oder Peptids hervorbringt.
-
Es
besteht keine allgemeine Voraussetzung, dass das Protein oder Peptid
immer in seinem am stärksten
gereinigten Zustand bereit gestellt werden muss. Tatsächlich besteht
die Überlegung,
dass weniger im Wesentlichen gereinigte Produkte in bestimmten Ausführungsformen
von Nutzen sind. Eine partielle Reinigung kann erreicht werden,
indem weniger Reinigungsschritte in Kombination oder indem verschiedene
Formen desselben allgemeinen Reinigungsschemas verwendet werden.
Beispielsweise bewirkt eine Kationenaustausch-Säulenchromatographie mittels
eines HPLC-Apparates im Allgemeinen eine stärkere „x-fache" Reinigung als die gleiche Technik,
die ein Niedrigdruck-Chromatographiesystem
verwendet. Verfahren, die einen niedrigeren Grad einer relativen
Reinigung aufweisen, können
im Hinblick auf die Gesamtwiedergewinnung des Proteinproduktes oder
der Erhaltung der Aktivität
eines Expressionsproteins Vorteile haben.
-
Es
ist bekannt, dass die Migration eines Polypeptids bei unterschiedlichen
Bedingungen der SDS/PAGE variieren kann, in einigen Fällen signifikant
(Capaldi et al., 1977). Es wird daher anerkannt, dass die scheinbaren
Molekulargewichte gereinigter oder partiell gereinigter Expressionsprodukte
unter unterschiedlichen Elektrophoresebedingungen variieren können.
-
HPLC
(High Performance Liquid Chromatography) zeichnet sich durch eine
sehr schnelle Auftrennung mit außerordentlicher Peakauflösung aus.
Dies wird erreicht, indem sehr feine Teilchen und hoher Druck verwendet
werden, um eine angemessene Durchflussrate aufrecht zu erhalten.
Die Auftrennung kann innerhalb von Minuten oder höchstens
einer Stunde erzielt werden. Darüber
hinaus wird nur ein sehr kleines Probenvolumen benötigt, da
die Teilchen so klein und dicht gepackt sind, dass das Totvolumen
einen sehr geringen Anteil des Bettvolumens ausmacht. Auch die Konzentration
der Probe braucht nicht sehr groß zu sein, da die Banden so
schmal sind, dass die Probe nur sehr wenig verdünnt wird.
-
Die
Gelchromatographie oder Molekularsiebchromatographie ist eine spezielle
Art von Auftrennungschromatographie, die auf der Molekülgröße beruht.
Die Gelchromatographie basiert auf der Theorie, dass die Säule, die
mit winzigen Teilchen einer inerten Substanz hergestellt ist, die
kleine Poren enthalten, größere Moleküle je nach
Größe von kleineren
Molekülen
trennt, wenn diese durch oder um die Poren herum wandern. So lange
wie das Material, aus dem die Teilchen bestehen, die Moleküle nicht
adsorbiert, wird der Durchfluss ausschließlich von der Größe bestimmt.
Die Elution von Molekülen
aus der Säule
erfolgt daher nach abnehmender Größe, so lange die Form relativ
konstant ist. Die Gelchromatographie ist bei der Auftrennung von
Molekülen
unterschiedlicher Größe unübertroffen,
da die Auftrennung von allen anderen Faktoren, wie pH-Wert, Ionenstärke, Temperatur,
etc. unabhängig
ist. Es gibt außerdem
praktisch keine Adsorption, weniger Zonenverbreiterung, und das
Elutionsvolumen ist auf einfache Weise mit dem Molekulargewicht
verbunden.
-
Die
Affinitätschromatographie
ist ein chromatographisches Verfahren, das auf der spezifischen
Affinität
zwischen einer zu isolierenden Substanz und einem spezifisch an
ihr bindenden Molekül
beruht. Es handelt sich dabei um eine Wechselwirkung vom Typ Rezeptor-Ligand. Das Säulenmaterial
wird synthetisiert, indem einer der Bindungspartner kovalent an
eine unlösliche
Matrix gekoppelt wird. Das Säulenmaterial
ist anschließend
in der Lage, die Substanz spezifisch aus der Lösung zu adsorbieren. Zu einer
Elution kommt es, indem die Bedingungen so geändert werden, dass keine Bindung
erfolgt (Änderung
des pH-Wertes, der Ionenstärke, der
Temperatur, etc.).
-
Die
Lektin-Affinitätschromatographie
ist eine besondere Art von Affinitätschromatographie, die bei
der Reinigung von kohlenhydrathaltigen Verbindungen nützlich ist.
Lektine sind eine Klasse von Substanzen, die an eine Vielzahl von
Polysacchariden und Glykoproteine binden. Lektine sind in der Regel
durch Zyanogenbromid an Agarose gekoppelt. An Sepharose gekoppeltes
Concanavalin A war das erste verwendete Material dieser Art und
wurde häufig
bei der Isolierung von Polysacchariden und Glykoproteinen verwendet.
Andere Lektine, die verwendet worden sind, sind Lektin aus Linsen,
Weizenkeimagglutinin, das bei der Reinigung von N-Acetylglucosaminylresten
benutzt wurde, sowie Lektin aus Helix pomatia. Lektine selbst werden
durch Affinitätschromatographie
mit Kohlenhydratliganden gereinigt. Laktose wurde verwendet, um
Lektine aus Rizinussamen und Erdnüssen zu reinigen; Maltose diente
der Extraktion von Lektinen aus Linsen und Jackbohnen; N-Acetyl-D-Galaktosamin wird
zur Reinigung von Lektinen aus Sojabohnen benutzt; N-Acetylglucosaminyl
bindet an Lektine aus Weizenkeimen; D-Galaktosamin wurde zur Gewinnung
von Lektinen aus Muscheln verwendet und L-Fukose bindet an Lektine
aus Lotus.
-
Die
Matrix sollte eine Substanz sein, die selbst Moleküle nicht
wesentlich bindet und die eine sehr weit gefasste chemische, physikalische
und thermale Stabilität
besitzt. Der Ligand sollte so gekoppelt sein, dass seine Bindungseigenschaften
nicht beeinträchtigt
werden. Der Ligand sollte außerdem
eine relativ feste Bindung eingehen. Und es sollte möglich sein,
die Substanz zu eluieren, ohne die Probe oder den Liganden zu zerstören. Eine
der häufigsten
Formen der Affinitätschromatographie
ist die Immunaffinitätschromatographie. Die
Herstellung von Antikörpern,
die zur Verwendung in Übereinstimmung
mit der vorliegenden Erfindung geeignet sind, ist unten erläutert.
-
G. Synthetische Peptide
-
Die
vorliegende Erfindung beschreibt außerdem kleinere mit TS10q23.3
verwandte Peptide zur Verwendung in verschiedenen Ausführungsformen
der vorliegenden Erfindung. Aufgrund ihrer relativ kleinen Größe, können die
Peptide der Erfindung in Übereinstimmung
mit herkömmlichen
Techniken auch in Lösung
oder auf einem festen Träger
synthetisiert werden. Es sind verschiedene automatische Synthesegeräte kommerziell erhältlich und
können
in Übereinstimmung
mit bekannten Protokollen verwendet werden. Siehe beispielsweise Steward
und Young (1984), Tam et al., (1983), Merrifield (1986) und Barany
und Merrifield (1979), jeweils hierin durch Bezugnahme enthalten.
Kurze Peptidsequenzen oder Bibliotheken überlappender Peptide, in der
Regel von etwa 6 bis zu etwa 35 bis 50 Aminosäuren, die den hierin beschriebenen
ausgewählten
Regionen entsprechen, können
leicht synthetisiert und dann in Screening-Assays untersucht werden,
die zur Identifizierung reaktiver Peptide konzipiert sind. Alternativ
kann rekombinante DNA-Technologie angewandt werden, wobei eine Nukleotidsequenz,
die ein Peptid der Erfindung kodiert, in einen Expressionsvektor
eingesetzt wird, in eine geeignete Wirtszelle transformiert oder
transfiziert und unter Bedingungen kultiviert wird, die für eine Expression geeignet
sind.
-
US-Patent
4,554,101 (hierin durch Bezugnahme enthalten) lehrt auch die Identifizierung
und Herstellung von Epitopen aus primären Aminosäuresequenzen auf der Basis
der Hydrophilie. Durch die von Hopp offenbarten Verfahren wäre ein Fachmann
in der Lage, Epitope aus der von einer beliebigen der hierin offenbarten
DNA-Sequenzen kodierten Aminosäure
zu identifizieren.
-
H. Antigenzusammensetzungen
-
Die
vorliegende Erfindung sieht außerdem
die Verwendung von TS10q23.3-Proteinen oder -Peptiden als Antigene
für die
Immunisierung von Tieren in Zusammenhang mit der Herstellung von
Antikörpern
vor. Es ist vorgesehen, dass entweder TS10q23.3 oder Abschnitte
davon über
Linker, Polylinker oder derivatisierte Aminosäuren an ein oder mehrere Mittel
gekoppelt, bondiert, gebunden, konjugiert oder chemisch verknüpft werden.
Dies kann derart durchgeführt
werden, dass eine bispezifische oder multivalente Zusammensetzung oder
Vakzine hergestellt wird. Es ist weiter vorgesehen, dass die bei
der Herstellung dieser Zusammensetzungen verwendeten Verfahren einem
Fachmann bekannt sind und für
die Verabreichung an Tiere geeignet sind, d. h. pharmazeutisch annehmbar
sind. Bevorzugte Mittel sind die Trägerstoffe Keyhole-Limpet-Hämocyanin (KLH)
oder Rinderserumalbumin (bovines Serumalbumin, BSA).
-
III. Nukleinsäuren
-
Die
vorliegende Erfindung sieht in einer anderen Ausführungsform
Gene vor, die TS10q23.3 kodieren. Es sind Gene für das TS10q23.3-Molekül des Menschen,
des Hundes und der Maus identifiziert worden. Die vorliegende Erfindung
ist in ihrem Umfang nicht auf diese Gene beschränkt, aber, ein durchschnittlicher
Fachmann könnte
anhand dieser Nukleinsäuren
verwandte Homologe in verschiedenen anderen Arten (z. B. Ratte, Kaninchen,
Affe, Gibbon, Schimpanse, Menschenaffe, Pavian, Kuh, Schwein, Pferd,
Schaf, Katze und andere Arten) leicht identifizieren. Das Auffinden
von Maus- und Hundhomologen dieses Gens macht es wahrscheinlich,
dass andere Arten, die näher
mit dem Menschen verwandt sind, ebenfalls ein Homolog aufweisen.
-
Darüber hinaus
sollte klar sein, dass die vorliegende Erfindung nicht auf die hierin
offenbarten spezifischen Nukleinsäuren beschränkt ist. Wie unten erläutert, kann
ein „TS10q23.3-Gen" eine Vielzahl unterschiedlicher
Basen enthalten und dennoch ein entsprechendes Polypeptid hervorbringen,
das funktionell, und in manchen Fällen strukturell, von den hierin
offenbarten Genen von Mensch und Maus nicht unterscheidbar ist.
-
Gleichermaßen ist
bei jeder Bezugnahme auf eine Nukleinsäure auch eine Wirtszelle gemeint,
die diese Nukleinsäure
enthält,
und in einigen Fällen
in der Lage ist, das Produkt dieser Nukleinsäure zu exprimieren. Abgesehen
von therapeutischen Überlegungen
könnten
sich Zellen, die Nukleinsäuren
der vorliegenden Erfindung exprimieren, in Zusammenhang mit dem
Screening auf Mittel, die die Funktion von TS10q23.3 induzieren,
zurückhalten,
inhibieren, unterstützen,
damit in Wechselwirkung treten, blockieren, aufheben, stimulieren
oder verstärken,
als nützlich
erweisen.
-
A. Für 10q23.3 kodierende Nukleinsäuren
-
Das
in 6 und 9 offenbarte
humane Gen und das in 9 offenbarte
Maus-Gen sind TS10q23.3-Gene der vorliegenden Erfindung. Nukleinsäuren gemäß der vorliegenden
Erfindung können
ein vollständiges
TS10q23.3-Gen, eine Domäne
von TS10q23.3, die eine tumorsupprimierende Funktion oder Phosphatasefunktion
exprimiert, oder ein beliebiges anderes Fragment der hierin ausgeführten TS10q23.3-Sequenzen
kodieren. Die Nukleinsäure
kann aus genomischer DNA abgeleitet sein, d. h. direkt aus dem Genom
eines bestimmten Organismus kloniert sein. In bevorzugten Ausführungsformen
würde die Nukleinsäure jedoch
komplementäre
DNA (cDNA) umfassen. In Betracht gezogen ist auch eine cDNA plus
ein natürliches
Intron oder ein von einem anderen Gen abgeleitetes Intron; solche
technisch hergestellten Moleküle
werden manchmal als „Minigene" bezeichnet. Diese
und andere Nukleinsäuren
der vorliegenden Erfindung können
mindestens als Molekulargewichtsstandards beispielsweise bei der
Gelelektrophorese verwendet werden.
-
Der
Begriff „cDNA" soll sich auf DNA
beziehen, die anhand von Boten-RNA (Messenger RNA oder mRNA) als
Vorlage hergestellt wird. Die Verwendung einer cDNA hat gegenüber genomischer
DNA oder DNA, die aus einer genomischen, nicht oder teilweise prozessierten
RNA-Vorlage polymerisiert wird, den Vorteil, dass die cDNA überwiegend
kodierende Sequenzen des entsprechenden Proteins enthält. Bisweilen
ist die vollständige
oder partielle genomische Sequenz bevorzugt, beispielsweise dann,
wenn die nicht kodierenden Regionen für eine optimale Expression
erforderlich sind oder wenn auf nicht kodierende Regionen, wie beispielsweise
Introns, in einer Antisense-Strategie abgezielt wird.
-
Es
wird auch in Betracht gezogen, dass ein gegebenes TS10q23.3 von
einer bestimmten Art durch natürliche
Varianten repräsentiert
werden kann, die leicht unterschiedliche Nukleinsäuresequenzen
aufweisen, aber dennoch dasselbe Protein kodieren (siehe Tabelle
1 unten).
-
Wie
in dieser Anmeldung verwendet, bezieht sich der Begriff „eine Nukleinsäure, die
für ein
TS10q23.3 kodiert",
auf ein Nukleinsäuremolekül, das ohne
zelluläre
Gesamtnukleinsäure
isoliert worden ist. In bevorzugten Ausführungsformen betrifft die Erfindung
eine Nukleinsäuresequenz,
im Wesentlichen wie in 6 und 9 ausgeführt. Der Begriff „wie in 6 oder 9 ausgeführt" bedeutet, dass die
Nukleinsäuresequenz
im Wesentlichen einem Abschnitt von 6 oder 9 entspricht. Der Begriff „funktionell äquivalentes
Codon" wird hierin verwendet,
um sich auf Codons zu beziehen, die dieselbe Aminosäure kodieren,
beispielsweise die sechs Codons für Arginin oder Serin (Tabelle
1 unten), und bezieht sich außerdem
auf Codons, die biologisch äquivalente
Aminosäuren
kodieren, wie auf den folgenden Seiten erläutert ist.
-
-
Unter
Berücksichtigung
der Degeneration des genetischen Codes sind Sequenzen, deren Nukleinsäuren zu
mindestens 50%, in der Regel zu etwa 60%, noch häufiger zu etwa 70%, am häufigsten
zu etwa 80%, vorzugsweise zu mindestens etwa 90% und am meisten
bevorzugt zu etwa 95% mit den Nukleotiden von 9 identisch
sind, „wie
in 9 ausgeführt". Sequenzen, die im Wesentlichen gleich
sind wie die in 9 ausgeführten, können funktionell
auch als Sequenzen definiert sein, die unter Standardbedingungen
ein Nukleinsäuresegment
hybridisieren können,
welches das Komplement von 9 enthält.
-
Die
DNA-Segmente der vorliegenden Erfindung umfassen solche, die biologisch
funktionelle äquivalente
TS10q23.3-Proteine und -Peptide enthalten, wie oben beschrieben.
Solche Sequenzen können
als Folge einer Codonredundanz und einer funktionellen Äquivalenz
der Aminosäuren
entstehen, von denen bekannt ist, dass sie in Nukleinsäuresequenzen
und den davon kodierten Proteinen natürlicherweise auftreten. Alternativ können durch
Anwendung rekombinanter DNA-Technologie funktionell äquivalente
Proteine oder Peptide erzeugt werden, in denen die Proteinstruktur
je nach Berücksichtigung
der Eigenschaften der ausgetauschten Aminosäuren technisch geändert ist.
Durch den Menschen konzipierte Änderungen
können
durch Anwendung von Techniken einer positionsgichteten (Site-Directed)
Mutagenese eingeführt
werden oder können
zufällig eingeführt werden
und später
hinsichtlich der gewünschten
Funktion untersucht werden, wie unten beschrieben ist.
-
B. Oligonukleotidsonden
und -primer
-
Natürlich umfasst
die vorliegende Erfindung auch DNA-Segmente, die zu der in 6 und 9 ausgeführten Sequenz
komplementär
oder im Wesentlichen komplementär
sind. Nukleinsäuresequenzen,
die „komplementär" sind, sind solche,
die gemäß den Komplementaritäts-Standardregeln
von Watson-Crick zu einer Basenpaarung fähig sind. Wie hierin verwendet,
bedeutet der Begriff „komplementäre Sequenzen" Nukleinsäuresequenzen,
die im Wesentlichen komplementär
sind, wie durch denselben Nukleinsäurevergleich wie oben ausgeführt bestimmt
wird, oder die so definiert sind, dass sie unter relativ stringenten
Bedingungen, wie beispielsweise den hierin Beschriebenen, an die
Nukleinsäuresegmente
von 6 und 9 hybridisieren
können.
Solche Sequenzen können
das vollständige
TS10q23.3-Protein
oder funktionelle oder nicht-funktionelle Fragmente davon kodieren.
-
Alternativ
kann es sich bei den Hybridisierungssegmenten um kürzere Oligonukleotide
handeln. Sequenzen mit einer Länge
von 17 Basen sollten im Genom des Menschen nur einmal auftreten
und sind daher ausreichend, um eine einmalige Zielsequenz zu spezifizieren.
Obgleich kürzere
Oligomere einfacher herzustellen sind und die Zugänglichkeit
in vivo erhöhen,
sind zahlreiche andere Faktoren an der Festlegung der Spezifität einer
Hybridisierung beteiligt. Sowohl die Bindungsaffinität als auch
die Sequenzspezifität
eines Oligonukleotids zu seinem komplementären Ziel erhöhen sich
mit zunehmender Länge.
Es wird in Betracht gezogen, beispielhafte Oligonukleotide mit 8,
9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45,
50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100 oder mehr Basenpaaren
zu verwenden, obgleich Andere in Betracht gezogen werden. Längere Polynukleotide,
die 250, 500, 1000, 1212, 1500, 2000, 2500, 3000 oder 3431 Basen und
länger
kodieren, werden ebenfalls in Betracht gezogen. Solche Oligonukleotide
können
beispielsweise in Southern oder Northern Blots und als Primer in
Amplifizierungsreaktionen verwendet werden.
-
Einem
Fachmann sind geeignete Hybridisierungsbedingungen gut bekannt.
In bestimmten Anwendungen, beispielsweise bei der Substitution von
Aminosäuren
durch positionsgerichtete Mutagenese, sind Bedingungen mit geringerer
Stringenz erforderlich. Unter diesen Bedingungen kann auch dann
eine Hybridisierung ablaufen, wenn die Sequenzen von Sonde und Zielstrang
nicht perfekt komplementär
sind, sondern an einer Position oder an mehreren Positionen nicht übereinstimmen.
Die Stringenz der Bedingungen kann durch Erhöhung der Salzkonzentration
und Verringerung der Temperatur verringert werden. Beispielsweise
können Bedingungen
mit mittlerer Stringenz durch etwa 0,1 bis 0,25 M NaCl bei Temperaturen
von etwa 37°C
bis etwa 55°C
bereit gestellt werden, während
Bedingungen mit niedriger Stringenz durch etwa 0,15 M bis etwa 0,9
M Salz bei Temperaturen zwischen etwa 20°C und etwa 55°C bereit
gestellt werden kann. Hybridisierungsbedingungen können also
einfach verändert
werden und sind daher im Allgemeinen je nach den gewünschten
Ergebnissen ein Verfahren der Wahl.
-
In
anderen Ausführungsformen
kann eine Hybridisierung unter Bedingungen von beispielsweise 50 mM
Tris-HCl (pH 8,3), 75 mM KCl, 3 mM MgCl2,
10 mM Dithiothreitol, bei Temperaturen von etwa 20°C bis etwa
37°C erreicht
werden. Andere verwendete Hybridisierungsbedingungen umfassen etwa
10 mM Tris-HCl (pH 8,3), 50 mM KCl, 1,5 μM MgCl2 bei
Temperaturen im Bereich von etwa 40°C bis etwa 72°C. Es können auch
Formamid und SDS verwendet werden, um die Hybridisierungsbedingungen
zu verändern.
-
Sonden
und Primer der vorliegenden Erfindung können auch bei der Suche nach
Genen, die mit TS10q23.3 verwandt sind, oder spezieller nach Homologen
von TS10q23.3 anderer Arten verwendet werden. Das Vorhandensein
eines Maushomologs lässt
den Schluss zu, dass in Arten, die näher und vielleicht entfernter
verwandt sind als die Maus, andere Homologe des humanen TS10q23.3
entdeckt werden. Normalerweise ist die Ziel-DNA eine genomische
oder cDNA-Bibliothek,
obgleich ein Screening die Analyse von RNA-Molekülen beinhalten kann. Durch
Variation der Stringenz der Hybridisierung und der Region der Sonde
können unterschiedliche
Homologiegrade entdeckt werden.
-
Ein
weiterer Weg der Anwendung von Sonden und Primern der vorliegenden
Erfindung ist die ortsgerichtete oder ortsspezifische Mutagenese.
Ortsspezifische Mutagenese ist eine Technik, die sich für die Herstellung
individueller Peptide oder biologisch funktioneller äquivalenter
Proteine oder Peptide durch spezifische Mutagenese der zugrunde
liegenden DNA eignet. Die Technik bietet darüber hinaus eine einfache Möglichkeit
zur Herstellung und Testung von Sequenzvarianten, wobei eine oder
mehrere der vorangehenden Überlegungen
miteinbezogen werden, indem eine oder mehrere Nukleotidsequenzen
in die DNA eingeführt werden.
Ortsspezifische Mutagenese ermöglicht
die Herstellung von Mutanten durch Verwendung spezifischer Oligonukleotidsequenzen,
die die DNA-Sequenz der gewünschten
Mutation kodieren, sowie eine ausreichende Zahl benachbarter Nukleotide,
um eine Primersequenz ausreichender Größe und Sequenzkomplexität bereit
zu stellen, um auf beiden Seiten der überbrückten Deletionsübergangsstelle
einen stabilen Duplex zu bilden. Typischerweise ist ein Primer mit
etwa 17 bis 25 Nukleotiden bevorzugt, wobei auf jeder Seite der Übergangsstelle
der Sequenz etwa 5 bis 10 Reste verändert werden.
-
Bei
der Technik wird typischerweise ein Bakteriophagenvektor verwendet,
der sowohl in Einzelstrang- als auch in Doppelstrangform vorkommt.
Zu den typischen Vektoren, die sich für die ortsspezifische Mutagenese
eignen, gehören
Vektoren wie der Phage M13. Diese Phagenvektoren sind kommerziell
verfügbar
und ihre Verwendung ist einem Fachmann im Allgemeinen gut bekannt.
Auch doppelsträngige
Plasmide werden bei der ortsspezifischen Mutagenese routinemäßig verwendet,
was den Schritt der Übertragung
des Gens von Interesse von einem Phagen in ein Plasmid beseitigt.
-
Im
Allgemeinen wird eine ortsspezifische Mutagenese durchgeführt, indem
zunächst
ein einzelsträngiger
Vektor erhalten oder die beiden Stränge eines doppelsträngigen Vektors
durch Schmelzen getrennt werden, wobei der Vektor in seiner Sequenz
eine DNA-Sequenz aufweist, die das gewünschte Protein kodiert. Es wird
ein Oligonukleotidprimer synthetisch hergestellt, der die gewünschte mutierte
Sequenz trägt.
Der Primer wird dann an das einzelsträngige DNA-Präparat
angeheftet, wobei der Grad der Nichtübereinstimmungen bei der Auswahl
der Hybridisierungsbedingungen berücksichtigt wird, und DNA-polymerisierenden
Enzymen, wie dem Klenow-Fragment der Polymerase I von E.coli unterzogen,
um die Synthese des mutationstragenden Stranges abzuschließen. So
bildet sich ein Heteroduplex, in dem ein Strang die ursprüngliche,
nicht mutierte Sequenz kodiert und der zweite Strang die gewünschte Mutation
trägt.
Der Heteroduplexvektor wird dann verwendet, um geeignete Zellen
wie beispielsweise E. coli-Zellen zu transformieren, und es werden
Klone selektiert, die rekombinante Vektoren enthalten, welche die
mutierte Sequenzanordnung tragen.
-
Die
Herstellung von Sequenzvarianten des ausgewählten Gens anhand einer ortsspezifischen
Mutagenese ist als ein Mittel zur Herstellung potenziell nützlicher
Arten vorgesehen und soll nicht einschränkend sein, da Sequenzvarianten
von Genen noch auf andere Art und Weise erhalten werden können. Beispielsweise können rekombinante
Vektoren, die das gewünschte
Gen kodieren, mit mutagenen Mitteln wie Hydroxylamin behandelt werden,
um Sequenzvarianten zu erhalten.
-
C. Antisense-Konstrukte
-
In
manchen Fällen
können
mutante Tumorsuppressoren nicht nicht-funktionell sein. Sie können stattdessen
abweichende Funktionen haben, die durch die Genersatztherapie nicht
ausgeglichen werden können, selbst
dann nicht, wenn das „Wildtyp"-Molekül in viel
höheren
Mengen exprimiert wird als das mutante Polypeptid. Antisense-Behandlungen
sind eine Möglichkeit,
dieser Situation zu begegnen. Antisense-Technologie kann auch verwendet
werden, um die Funktion von TS10q23.3 während der Entwicklung von Zelllinien
oder transgenen Mäusen
für Forschungs-,
Diagnose- und Analysezwecke auszuschalten („knock-out").
-
Bei
der Antisense-Methodik wird die Tatsache genutzt, dass Nukleinsäuren dazu
neigen, mit „komplementären" Sequenzen Paare
zu bilden. Mit komplementär
ist gemeint, dass Polynukleotide diejenigen sind, die entsprechend
den Komplementaritäts-Standardregeln
von Watson-Crick zu einer Basenpaarung fähig sind. Das heißt, dass
die größeren Purine
ein Paar mit den kleineren Pyrimidinen eingehen, um Kombinationen von
Paaren wie Guanin und Cytosin (G:C) und Adenin und Thymin (A:T)
im Falle von DNA oder Adenin und Uracil (A:U) im Falle von RNA zu
bilden. Das Vorhandensein weniger häufiger Basen wie Inosin, 5-Methylcytosin,
6-Methyladenin,
Hypoxanthin und Anderen in Hybridisierungssequenzen beeinflusst
die Paarbildung nicht.
-
Das
Targeting doppelsträngiger
(ds) DNA mit Polynukleotiden führt
zur Bildung einer Dreifachhelix; das Targeting von RNA führt zur
Bildung einer Doppelhelix. Wenn Antisense-Polynukleotide in eine Zielzelle eingebracht
werden, binden sie spezifisch an ihr Zielnukleotid und beeinflussen
die Transkription sowie die Prozessierung, den Transport, die Translation
und/oder die Stabilität
von RNA. Antisense-RNA-Konstrukte oder DNA, die solche Antisense-RNAs kodiert, können verwendet
werden, um Gentranskription oder Translation oder beides entweder
in vitro oder in vivo in einer Wirtszelle zu hemmen, wie beispielsweise
bei einem Wirtstier, einschließlich
einem Menschen.
-
Es
können
Antisense-Konstrukte konzipiert werden, um an den Promotor und andere
Steuerregionen, Exons, Introns oder selbst Exon-Intron-Übergangsstellen
eines Gens zu binden. Es wird in Betracht gezogen, dass die effektivsten
Antisense-Konstrukte Regionen umfassen, die komplementär zu Intron/Exon-Splice-Übergängen sind.
Es wird daher vorgeschlagen, dass eine bevorzugte Ausführungsform
ein Antisense-Konstrukt mit Komplementarität zu Regionen innerhalb von
50–200
Basen eines Intron/Exon-Splice-Übergangs
enthält.
Es wurde beobachtet, dass manche Exonsequenzen in dem Konstrukt
enthalten sein können, ohne
dessen Zielselektivität
ernsthaft zu beeinflussen. Die Menge an enthaltenem Exonmaterial
variiert je nach den jeweils verwendeten Exon- und Intronsequenzen.
Es kann einfach getestet werden, ob zu viel Exon-DNA vorhanden ist,
indem einfach die Konstrukte in vivo getestet werden um zu bestimmen,
ob die normale zelluläre
Funktion beeinflusst ist oder ob die Expression verwandter Gene
mit komplementären
Sequenzen beeinflusst ist.
-
Wie
oben festgelegt, bedeuten „komplementäre" oder „antisense" Polynukleotidsequenzen
solche, die im Wesentlichen über
ihre gesamte Länge
komplementär
sind und sehr wenige Basennichtübereinstimmungen
aufweisen. Sequenzen mit einer Länge
von fünfzehn
Basenpaaren können
als komplementär
bezeichnet werden, wenn sie an dreizehn oder vierzehn Positionen
komplementäre
Nukleotide aufweisen. Natürlich
sind Sequenzen, die vollständig
komplementär
sind, Sequenzen, die über
ihre gesamte Länge
vollkommen komplementär
sind und keine Basennichtübereinstimmungen
aufweisen. Andere Sequenzen mit einem geringeren Homologiegrad werden
ebenfalls in Betracht gezogen. Beispielsweise könnte ein Antisense-Konstrukt,
das eingeschränkte
Regionen hoher Homologie aufweist, aber auch eine nicht-homologe
Region enthält
(z. B. Ribozym, siehe unten), konzipiert werden. Diese Moleküle würden unter
geeigneten Bedingungen an Zielsequenzen binden, obwohl sie eine
Homologie von unter 50% haben.
-
Es
könnte
von Vorteil sein, Abschnitte genomischer DNA mit cDNA oder synthetischen
Sequenzen zu kombinieren, um spezifische Konstrukte zu erzeugen.
Beispielsweise muss ein genomischer Klon verwendet werden, wenn
im fertigen Konstrukt ein Intron gewünscht ist. Die cDNA oder ein
synthetisches Polynukleotid könnte
praktischere Restriktionsschnittstellen für den verbleibenden Abschnitt
des Konstruktes liefern und würde
daher für
den Rest der Sequenz verwendet werden.
-
D. Ribozyme
-
Ein
weiterer Ansatz, der auf den „dominant
negativen" mutanten
Tumorsuppressor abzielt, erfolgt mittels Verwendung von Ribozymen.
Obgleich Proteine traditionell zur Katalyse von Nukleinsäuren verwendet wurden,
hat sich eine andere Klasse von Makromolekülen bei diesem Vorhaben als
nützlich
erwiesen. Ribozyme sind RNA-Protein-Komplexe, die Nukleinsäuren in
positionsspezifischer Art schneiden. Ribozyme haben spezifische
katalytische Domänen,
die Endonukleaseaktivität
aufweisen (Kim und Cook, 1987; Gerlach et al., 1987; Forster und
Symons, 1987). Beispielsweise beschleunigt eine große Anzahl
von Ribozymen Phosphoesterübertragungsreaktionen
mit einem hohen Grad an Spezifität,
wobei häufig
nur einer von mehreren Phosphoestern in einem Oligonukleotidsubstrat
gespalten wird (Cook et al., 1981; Michel und Westhof, 1990; Reinhold-Hurek
und Shub, 1992). Diese Spezifität
wurde der Voraussetzung zugeschrieben, dass das Substrat vor der
chemischen Reaktion über
spezifische Basenpaarungswechselwirkungen an die interne Führungssequenz
(„IGS") des Ribozyms bindet.
-
Ribozymkatalyse
wurde überwiegend
als Teil sequenzspezifischer Schnitt-/Ligationsreaktionen beobachtet, bei
denen Nukleinsäuren
beteiligt waren (Cook et al., 1981). Beispielsweise berichtet US-Patent
Nr. 5.354.855, dass bestimmte Ribozyme als Endonukleasen wirken
können
und eine Sequenzspezifität
aufweisen, die höher
als die bekannter Ribonukleasen ist und fast der der DNA-Restriktionsenzyme
entspricht. Sequenzspezifische, ribozymvermittelte Genexpressionshemmung
kann daher für
therapeutische Anwendungen besonders geeignet sein (Scanlon et al.,
1991; Sarver et al., 1990). Kürzlich
ist berichtet worden, dass Ribozyme in einigen Zelllinien, zu denen
sie gegeben wurden, genetische Veränderungen ausgelöst haben;
zu den veränderten
Genen gehörten
die Onkogene H-ras, c-fos und Gene von HIV. Die meisten dieser Arbeiten
beinhalten die Modifikation einer Ziel-mRNA ausgehend von einem
spezifischen mutierten Codon, das von einem spezifischen Ribozym
geschnitten wird.
-
E. Vektoren für die Klovierung,
Genübertragung
und -expression
-
In
bestimmten Ausführungsformen
werden Expressionsvektoren verwendet, um das TS10q23.3-Polypeptidprodukt
zu exprimieren, welches anschließend gereinigt und beispielsweise
verwendet werden kann, um Tiere zu immunisieren, um Antiseren oder
einen monoklonalen Antikörper
zu erzeugen, mit denen weitere Studien durchgeführt werden können. In
anderen Ausführungsformen
werden die Expressionsvektoren bei einer Gentherapie verwendet.
Expression erfordert, dass geeignete Signal in den Vektoren bereit
gestellt werden, die zudem verschiedene regulatorische Elemente,
wie beispielsweise Verstärker
(Enhancer)/Promotoren aus den viralen und säugetierspezifischen Quellen
umfassen, welche die Expression des Gens von Interesse in Wirtszellen
antreiben. Es sind auch Elemente definiert, die zur Optimierung
der Stabilität
und Translationsfähigkeit
in Wirtszellen konzipiert sind. Es sind auch die Bedingungen für die Verwendung
einer Reihe dominanter Wirkstoffselektionsmarker zur Etablierung
permanenter, stabiler, die Produkte exprimierender Zellklone vorgesehen,
wie beispielsweise ein Element, der die Expression des Wirkstoffselektionsmarkers
mit der Expression des Polypeptids verknüpft.
-
(i) Regulatorische Elemente
-
Promotoren.
-
In
dieser gesamten Anmeldung soll der Begriff „Expressionskonstrukt" jede Art von genetischem
Konstrukt einschließen,
das eine Nukleinsäure
enthält,
die Genprodukte kodiert, in denen ein Teil oder die gesamte Nukleinsäure mit
der Kodierungssequenz transkribiert werden kann. Das Transkript
kann, muss aber nicht, zu einem Protein translatiert werden. In
bestimmten Ausführungsformen
umfasst Expression sowohl die Transkription eines Gens als auch
die Translation von mRNA in ein Genprodukt. In anderen Ausführungsformen
umfasst Expression nur die Transkription der Nukleinsäure, welche
die Gene von Interesse kodiert.
-
Die
Nukleinsäure,
die ein Genprodukt kodiert, steht unter der Transkriptionskontrolle
eines Promotors. Ein „Promotor" bezieht sich auf
eine DNA-Sequenz, die vom Syntheseapparat der Zelle oder einem eingeführten Syntheseapparat,
der erforderlich ist, um die spezifische Transkription eines Gens
einzuleiten, erkannt wird. Der Begriff „unter Transkriptionskontrolle" bedeutet, dass sich
der Promotor in der korrekten Position und Ausrichtung in Bezug
auf die Nukleinsäure
befindet, um die Initiation der RNA-Polymerase und die Expression des
Gens zu kontrollieren.
-
Der
Begriff „Promotor" wird hier verwendet,
um sich auf eine Gruppe von Transkriptionskontrollmodulen zu beziehen,
die um die Initiationsstelle für
die RNA-Polymerase II gruppiert sind. Viele Vorstellungen darüber, wie
Promotoren organisiert sind, entstammt Analysen mehrerer viraler
Promotoren, einschließlich
denen für
die HSV-Thymidinkinase (tk) und der frühen Transkriptionseinheiten
(Early Transcription Units) von SV40. Diese Studien, die von neueren
Arbeiten gestützt
werden, haben gezeigt, dass Promotoren aus diskreten funktionellen
Modulen zusammengesetzt sind, von denen ein Jedes aus etwa 7–20 bp DNA
besteht und eine oder mehrere Erkennungsstellen für transkriptionelle
Aktivator- und Repressorproteine enthalten.
-
Mindestens
ein Modul in jedem Promotor dient dazu, die Startstelle zur RNA-Synthese
festzulegen. Das beste bekannte Beispiel dafür ist die TATA-Box, aber in
einigen Promotoren, denen eine TATA-Box fehlt, wie beispielsweise
dem Promotor für
das terminale Desoxynukleotidyltransferasegen der Säugetiere
und dem Promotor für
die späten
(Late) Gene von SV40, trägt
ein diskretes Element, das die Startstelle überlappt, dazu bei, den Ort
der Initiation festzulegen.
-
Weitere
Promotorelemente regeln die Häufigkeit
der Transkriptionsinitiation. Typischweise befinden sich diese in
der Region 30–110
bp stromaufwärts
(upstream) der Startstelle, obgleich für eine Reihe von Promotoren
kürzlich
gezeigt wurde, dass sie auch funktionelle Elemente stromabwärts (downstream)
der Startstelle aufweisen. Der Abstand zwischen Promotorelementen
ist häufig
flexibel, so dass die Promotorfunktion erhalten bleibt, wenn Elemente
eingefügt
oder relativ zueinander bewegt werden. Im tk-Promotor kann der Abstand
zwischen Promotorelementen auf 50 bp erhöht werden, bevor die Aktivität beginnt
abzunehmen. Es scheint, dass einzelne Elemente je nach Promotor
entweder kooperativ oder unabhängig
funktionieren können,
um die Transkription zu aktivieren.
-
Es
wird angenommen, dass es nicht wichtig ist, welcher Promotor jeweils
zur Regelung der Expression einer Nukleinsäuresequenz von Interesse verwendet
wird, so lange er in der Lage ist, die Expression der Nukleinsäure in der
Zielzelle zu steuern. Handelt es sich bei der Zielzelle also um
eine humane Zelle, ist es vorzuziehen, die kodierende Nukleinsäureregion
neben einen und unter die Kontrolle eines Promotors zu setzen, der
in einer humanen Zelle exprimiert werden kann. Allgemein ausgedrückt, kann
ein solcher Promotor entweder einen humanen oder einen viralen Promotor
umfassen.
-
In
verschiedenen Ausführungsformen
können
der Promotor der Immediate-Early-Gene des humanen Zytomegalievirus
(CMV), der SV40-Early-Promotor, der Long-Terminal-Repeat des Rous-Sarkom-Virus,
der β-Actinpromotor,
der Ratteninsulinpromotor und der Promotor der Glyceraldehyd-3-Phosphatdehydrogenase verwendet
werden, um eine starke Expression der kodierenden Sequenzen von
Interesse zu erreichen. Die Verwendung anderer viraler oder von
Säugetieren
stammenden zellulärer
Promotoren oder bakterieller Phagenpromotoren, die aus dem Stand
der Technik bekannt sind, um eine Expression einer Kodierungssequenz von
Interesse zu erreichen, wird ebenfalls in Betracht gezogen, vorausgesetzt,
dass der Grad der Expression jeweils für einen gegebenen Zweck ausreichend
ist. Durch Verwendung eines Promotors mit gut bekannten Eigenschaften
können
Grad und Muster der Expression des Proteins von Interesse nach Transfektion
oder Transformation optimiert werden.
-
Die
Auswahl eines Promotors, der als Reaktion auf bestimmte physiologische
oder synthetische Signale reguliert wird, kann eine induzierbare
Expression des Genproduktes erlauben. Wenn beispielsweise die Expression
eines Transgens oder von Transgenen, wenn ein multizistronischer
Vektor verwendet wird, für
die Zellen, in denen der Vektor produziert wird, toxisch ist, kann
es wünschenswert
sein, die Expression eines oder mehrerer Transgene zu verhindern
oder zu reduzieren. Beispiele von Transgenen, die für die Produzentenzelllinien
toxisch sein können,
sind proapoptotische Gene und Zytokingene. Wenn das transgene Produkt
toxisch ist, stehen für
die Produktion viraler Vektoren mehrere induzierbare Promotorsysteme
zu Verfügung.
-
Eines
dieser Systeme ist das Ecdysonsystem (Invitrogen, Carlsbad, CA,
USA). Dieses System ist so konzipiert, dass es eine regulierte Expression
eines Gens von Interesse in Säugetierzellen
erlaubt. Es besteht aus einem eng regulierten Expressionsmechanismus,
der praktisch keine Basisexpression des Transgens, aber eine über 200-fache
Induzierbarkeit zulässt.
Das System beruht auf dem heterodimeren Ecdysonrezeptor von Drosophila,
und wenn Ecdyson oder ein Analog wie beispielsweise Muristeron A
an den Rezeptor bindet, aktiviert der Rezeptor einen Promotor, um
die Expression des sich stromabwärts
(downstream) befindenden Transgens einzuschalten. So werden hohe
Mengen an mRNA-Transkripten erhalten. In diesem System werden beide
Monomere des heterodimeren Rezeptors von einem Vektor konstitutiv
exprimiert, wobei der Ecdyson-reaktive Promotor, der die Expression
des Gens von Interesse antreibt, sich auf einem anderen Plasmid befindet.
Die technische Kombination dieser Art von System mit dem Gentransfervektor
von Interesse wäre daher
sinnvoll. Cotransfektion von Plasmiden, die das Gen von Interesse
enthalten, und der Rezeptormonomere in die Produzentenzelllinie
würde dann
die Produktion des Gentransfervektors ohne Expression eines potenziell
toxischen Transgens erlauben. Die Expression des Transgens könnte zu
einem geeigneten Zeitpunkt mit Ecdyson oder Muristeron A aktiviert
werden.
-
Ein
weiteres geeignetes, induzierbares System ist das Tet-OffTM- oder Tet-OnTM-System
(Clontech, Palo Alto, CA, USA), das ursprünglich von Gossen und Bujard
entwickelt wurde (Gossen und Bujard, 1992; Gossen et al., 1995).
Dieses System erlaubt ebenfalls einen hohen Grad an Genexpression,
die als Reaktion auf Tetracyclin oder Tetracyclinderivate wie Doxycyclin
reguliert werden kann. Im Tet-OnTM-System
wird die Genexpression in Gegenwart von Doxycyclin eingeschaltet,
während
die Genexpression im Tet-OffTM-System in Abwesenheit
von Doxycyclin eingeschaltet wird. Diese Systeme beruhen auf zwei
regulatorischen Elementen, die aus dem Tetracyclinresistenzoperon
von E.coli abgeleitet sind, der Tetracyclinoperatorsequenz, an die
der Tetracyclinrepressor bindet, und dem Tetracyclinrepressorprotein.
Das Gen von Interesse wird in ein Plasmid hinter einen Promotor
kloniert, in dem sich Tetracyclin-reaktive Elemente befinden. Ein
zweites Plasmid enthält ein
regulatorisches Element mit der Bezeichnung Tetracyclin-kontrollierter
Transaktivator, der im Tet-OffTM-System
aus der VP16-Domäne
des Herpes-Simplex-Virus und dem Wildtyp des Tetracyclinrepressors
zusammengesetzt ist. So wird in Abwesenheit von Doxycyclin die Transkription
konstitutiv eingeschaltet. Im Tet-OnTM-System
ist der Tetracyclinrepressor nicht vom Wildtyp und aktiviert in
Gegenwart von Doxycyclin die Transkription. Für die Produktion eines Gentherapievektors
wäre das
Tet-OffTM-System vorzuziehen, so dass die
Produzentenzelle in Gegenwart von Tetracyclin oder Doxycyclin gezüchtet und
die Expression eines potenziell toxischen Transgens verhindert werden
kann, aber die Genexpression konstitutiv eingeschaltet werden würde, wenn
der Vektor in den Patienten eingeführt wird.
-
In
manchen Fällen
kann es wünschenswert
sein, die Expression eines Transgens in einem Gentherapievektor
zu regulieren. Beispielsweise können
je nach dem gewünschten
Grad der Expression unterschiedliche virale Promotoren mit verschieden
starker Aktivität
verwendet werden. In Säugetierzellen
wird häufig
der CMV-Immediate-Early-Promotor verwendet, um eine starke Aktivierung
der Transkription zu vermitteln. Es sind auch modifizierte Versionen
des CMV-Promotors, die weniger potent sind, verwendet worden, wenn
ein geringerer Grad der Expression des Transgens erwünscht ist.
Wenn die Expression eines Transgens in hämatopoietischen Zellen erwünscht ist,
werden häufig
retrovirale Promotoren wie die LTR-Sequenzen von MLV oder MMTV verwendet.
Weitere virale Promotoren, die je nach dem gewünschten Effekt verwendet werden
können, sind
SV40, RSV, LTR, HIV-1- und HIV2-LTR, Adenoviruspromotoren wie beispielsweise
aus der E1A-, E2A- oder MLP-Region, AAV LTR, Blumenkohlmosaikvirus,
HSV-TK und Vogelsarkomvirus.
-
Gleichermaßen können gewebespezifische
Promotoren verwendet werden, um eine Transkription in bestimmten
Geweben oder Zellen zu bewirken, um damit eine potenzielle Toxizität oder unerwünschte Effekte auf
Nicht-Zielgewebe zu reduzieren. Promotoren wie PSA, Probasin, Prostata-spezifische
Phosphatase oder Prostata-spezifisches, glanduläres Kallikrein (hK2) können beispielsweise
verwendet werden, um eine gezielte Genexpression in der Prostata
zu bewirken. Gleichermaßen
können
für eine
gezielte Genexpression in anderen Geweben die folgenden Promotoren
verwendet werden (Tabelle 2).
-
-
-
Bei
bestimmten Indikationen kann es wünschenswert sein, die Transkription
zu bestimmten Zeitpunkten nach der Verabreichung des Gentherapievektors
zu aktivieren. Dies kann mithilfe solcher Promotoren erfolgen, die
durch ein Hormon oder Zytokin regulierbar sind. Bei Gentherapieanwendungen,
in denen die Indikation ein Gonadengewebe ist, in dem spezifische
Steroide produziert oder in das spezifische Steroide geleitet werden,
kann beispielsweise die Verwendung von Promotoren vorteilhaft sein,
die durch Androgene oder Estrogen regulierbar sind. Solche hormonregulierten
Promotoren umfassen MMTV, MT-1, Ecdyson und RuBisco. Es wird erwartet,
dass andere hormonregulierte Promotoren, wie die, welche auf Schilddrüsen-, Hypophysen- und
Nebennierenhormone ansprechen, in der vorliegenden Erfindung nützlich sind.
Zu den Promotoren, die auf Zytokine und Entzündungsproteine ansprechen und
verwendet werden können,
gehören
K- und T-Kininogen (Kageyama et al., 1987), c-fos, TNF-alpha, C-reaktives
Protein (Arcone et al., 1988), Haptoglobin (Oliviero et al., 1987),
Serumamyloid A2, C/EBP-alpha, IL-1, IL-6 (Poli und Cortese, 1989),
Komplement C3 (Wilson et al., 1990), IL-8, Alpha-1-Säureglykoprotein
(Prowse und Baumann, 1988), Alpha-1-Antitrypsin, Lipoproteinlipase
(Zechner et al., 1988), Angiotensinogen (Ron et al., 1991), Fibrinogen,
c-jun (induzierbar durch Phorbolester und Retinolsäure), Metallothionein
(induzierbar durch Schwermetall und Glucocorticoide), Stromelysin (induzierbar
durch Phorbolester, Interleukin-1 und EGF), Alpha-2-Makroglobulin
und Alpha-1-Antichymotrypsin.
-
Es
ist vorgesehen, dass zellzyklusregulierbare Promotoren in der vorliegenden
Erfindung nützlich
sein könnten.
Beispielsweise könnte
in einem bizistronischen Gentherapievektor die Verwendung eines
starken CMV-Promotors zur Aktivierung der Expression eines ersten
Gens wie p16, das die Zellen in der G1-Phase anhält, von der Expression eines
zweiten Gens wie p53 gefolgt sein unter der Kontrolle eines Promotors,
der in der G1-Phase des Zellzyklus aktiv ist, wobei dadurch ein „zweiter
Treffer" erzielt
wird, welcher die Zelle in die Apotose treibt. Es können auch
andere Promotoren, wie die verschiedener Cycline, PCNA, Galectin-3, E2F1,
p53 und BRCA1 verwendet werden.
-
Tumorspezifische
Promotoren, wie Osteocalcin, Hypoxie-responsives Element (HRE),
MAGE-4, CEA, Alpha-Fetoprotein, GRP78/BiP und Tyrosinase können ebenfalls
verwendet werden, um Genexpression in Tumorzellen zu regulieren.
Andere Promotoren, die gemäß der vorliegenden
Erfindung benutzt werden können, umfassen
Lac-regulierbare, chemotherapieinduzierbare (z. B. MDR) und Hitze
(Hyperthermie)-induzierbare Promotoren, strahlungsinduzierbare (z.
B. EGR (Joki et al., 1995)), Alpha-Inhibin, RNA pol III tRNA met
und andere Aminosäurepromotoren,
U1 snRNA (Bartlett et al., 1996), MC-1, PGK, β-Aktin und α-Globin. In Walther und Stein (1996)
sind viele andere Promotoren aufgeführt, die nützlich sein könnten.
-
Es
ist geplant, dass jeder der obigen Promotoren alleine oder in Kombination
mit einem Anderen je nach der erwünschten Wirkung gemäß der vorliegenden
Erfindung nützlich
sein kann. Darüber
soll diese Liste von Promotoren nicht erschöpfend oder einschränkend sein;
ein Fachmann kennt andere Promotoren, die in Verbindung mit den
hierin offenbarten Promotoren und Verfahren verwendet werden können.
-
Verstärker (Enhancer).
Enhancer sind genetische Elemente, welche die Transkription von
einem Promotor verstärken,
der sich an einer entfernten Position auf demselben DNA-Molekül befindet.
Enhancer sind weitgehend wie Promotoren organisiert. Das bedeutet,
dass sie aus vielen einzelnen Elementen zusammengesetzt sind, von
denen jedes an ein oder mehrere transkriptionelle Proteine bindet.
Der grundlegende Unterschied zwischen Enhancern und Promotoren ist
funktionsbedingt. Eine Enhancer-Region als Ganzes muss in der Lage
sein, Transkription auf die Entfernung zu stimulieren; für eine Promotorregion
oder ihre Bestandteilselemente muss dies nicht unbedingt zutreffen.
Andererseits muss ein Promotor ein oder mehrere Elemente aufweisen,
die eine direkte Initiierung der RNA-Synthese an einer bestimmten
Stelle und in einer bestimmten Ausrichtung steuern, wohingegen Enhancer
diese Spezifitäten
nicht haben. Promotoren und Enhancer sind häufig überlappend und kontig, wobei
es häufig
scheint, als ob sie sehr ähnliche
modulare Organisation aufweisen.
-
Unten
befindet sich zusätzlich
zu den oben aufgeführten
gewebespezifischen Promotoren eine Liste von Promotoren, zellulären Promotoren/Enhancern
und induzierbaren Promotoren/Enhancern, die in Kombination mit der
Nukleinsäure
verwendet werden können,
die ein Gen von Interesse in einem Expressionskonstrukt kodieren
(Tabelle 3 und Tabelle 4). Darüber
hinaus könnte
auch jede beliebige Promotor/Enhancer-Kombination (wie beispielsweise
anhand der Datenbank EPDB (Eukaryotic Promotor Data Base)) verwendet
werden, um die Expression des Gens anzutreiben. Eukaryontische Zellen
können
zytoplasmatische Transkription bestimmter bakterieller Promotoren
unterstützen,
wenn die geeignete bakterielle Polymerase bereitgestellt wird, entweder
als Teil des Verabreichungskomplexes oder als ein zusätzliches
genetisches Expressionskonstrukt.
-
-
-
-
-
Polyadenylierungssignale.
-
Wo
ein cDNA-Insert eingesetzt wird, würde man typischerweise ein
Polyadenylierungssignal einsetzen wollen, um eine ordnungsgemäße Polyadenylierung
des Gentranskriptes zu bewirken. Es wird angenommen, dass die Art
des Polyadenylierungssignals für
die erfolgreiche Anwendung der Erfindung nicht von zentraler Bedeutung
ist und dass jede solche Sequenz verwendet werden kann, beispielsweise
das Polyadenylierungssignal des humanen oder bovinen Wachstumshormons
oder von SV40. Als ein Element der Expressionskassette wird auch
ein Terminator in Betracht gezogen. Diese Elemente können dazu
dienen, die mRNA-Expression zu steigern und das Ablesen der genetischen
Information der Kassette bis in andere Sequenzen hinein zu minimieren.
-
IRES.
-
In
bestimmten Ausführungsformen
der Erfindung wird die Verwendung interner Ribosomen-Eintrittsstellenelemente
(internal ribosome entry site; IRES) erwogen, um multigene oder
polyzistronische mRNAs zu erzeugen. IRES-Elemente sind in der Lage,
das Ribosomen- Scanning-Modell
der 5'-methylierten
Cap-abhängigen
Translation zu umgehen und beginnen die Translation an internen
Stellen (Pelletier und Sonenberg, 1988). Es sind IRES-Elemente aus
zwei Mitgliedern der Familie der Picornaviren (Poliovirus und Enzephalomyokarditis)
(Pelletier und Sonenberg, 1988) sowie ein IRES aus einer Säugetier-mRNA
(Macejak und Sarnow, 1991) beschrieben worden. IRES-Elemente können an
heterologe offene Leseraster gekoppelt sein. Multiple offene Leseraster
können
gemeinsam transkribiert werden, wobei jeder von einem IRES getrennt
ist, wodurch polyzistrone mRNA entsteht. Durch das IRES-Elements
ist jedes offene Leseraster für
Ribosomen zugänglich,
um eine effektive Translation zu bewirken. Multiple Gene können mittels
eines einzigen Promotors/Enhancers exprimiert werden, um eine einzelne
mRNA zu transkribieren.
-
Jedes
heterologe offene Leseraster kann mit IRES-Elementen verknüpft werden.
Dazu gehören
Gene für
sezernierte Proteine, Proteine mit mehreren Untereinheiten, die
von unabhängigen
Genen kodiert werden, intrazelluläre oder membrangebundene Proteine
und selektierbare Marker. Auf diese Art kann die simultane Expression
mehrerer Proteine in einer Zelle mit einem einzigen Konstrukt und
einem einzigen selektierbaren Marker technisch erreicht werden.
-
(ii) Selektierbare Marker
-
In
bestimmten Ausführungsformen
der Erfindung enthalten die Zellen Nukleinsäurekonstrukte der vorliegenden
Erfindung. Eine Zelle kann in vitro oder in vivo identifiziert werden,
indem in das Expressionskonstrukt ein Marker eingefügt wird.
Solche Marker würden
eine identifizierbare Veränderung
der Zelle bewirken, was die einfache Identifizierung von Zellen,
die das Expressionskonstrukt enthalten, erlaubt. In der Regel hilft das
Einfügen
eines Wirkstoffselektionsmarkers bei der Klonierung und der Selektion
von Transformanden; beispielsweise sind Gene, die Resistenz gegen
Neomycin, Puromycin, Hygromycin, DHFR, GPT, Zeoxin und Histidinol
verleihen, nützliche
selektierbare Marker. Alternativ können Enzyme wie die Thymidinkinase
des Herpes-Simplex-Virus (tk) oder die Chloramphenicoltransferase
(CAT) verwendet werden. Auch immunologische Marker können eingesetzt
werden. Der verwendete selektierbare Marker ist unwichtig, so lange
er simultan mit der Nukleinsäure,
die ein Genprodukt kodiert, exprimiert werden kann. Weitere Beispiele
selektierbarer Marker sind einem Fachmann gut bekannt.
-
(iii) Einführung von
Expressionsvektoren
-
Es
gibt eine Reihe von Möglichkeiten,
Expressionsvektoren in Zellen einzuführen. In bestimmten Ausführungsformen
der Erfindung umfasst das Expressionskonstrukt ein Virus oder ein
technisch hergestelltes Konstrukt, das aus einem viralen Genom abgeleitet
ist. Die Möglichkeit
bestimmter Viren über
rezeptorvermittelte Endozytose in Zellen zu gelangen, sich in das
Genom der Wirtszelle zu integrieren und virale Gene stabil und effizient
zu exprimieren, machten sie zu attraktiven Kandidaten für den Transfer
fremder Gene in Säugetierzellen
(Ridgeway, 1988; Nicolas und Rubenstein, 1988; Baichwal und Sugden,
1986; Temin, 1986). Die ersten Viren, die als Genvektoren verwendet
worden sind, waren DNA-Viren, darunter die Papovaviren (Simianvirus
40, boviner Papillomavirus und Polyomvirus) (Ridgeway, 1988; Baichwal
und Sugden, 1986) und Adenoviren (Ridgeway, 1988; Baichwal und Sugden,
1986). Diese haben eine vergleichsweise geringe Kapazität für Fremd-DNA-Sequenzen
und ein eingeschränktes
Wirtsspektrum. Darüber
hinaus wecken ihr onkogenes Potenzial und ihre zytopathischen Effekte
in permissiven Zellen Sicherheitsbedenken. Sie können lediglich bis zu 8 kb
an genetischem Fremdmaterial aufnehmen, aber leicht in verschiedene
Zelllinien und Labortiere eingeführt
werden (Nicolas und Rubenstein, 1988; Temin, 1986). Der Vektor kann
in der Lage sein, sich innerhalb der Zellen zu replizieren. Alternativ
kann der Vektor replikationsdefizient sein und wird vor der Einführung in Helferzellen
repliziert. Es sind geeignete Vektoren bekannt, beispielsweise wie
in US-Patent 5.252.479 und der veröffentlichten PCT-Anmeldung
WO 93/07282 und in US-Patent 5.691.198, 5.436.146 und 5,753,500
offenbart.
-
Adenoviren.
-
Eine
der bevorzugten Verfahren für
das Einführen
in vivo umfasst die Verwendung eines Adenovirus-Expressionsvektors. „Adenovirus-Expressionsvektor" soll diejenigen
Konstrukte umfassen, die Adenovirussequenzen enthalten, welche ausreichend
sind, um (a) die Verpackung des Konstruktes zu unterstützen und
(b) ein Antisense-Polynukleotid zu exprimieren, das darin kloniert
ist. In diesem Zusammenhang erfordert die Expression nicht, dass
das Genprodukt synthetisiert wird.
-
Der
Expressionsvektor umfasst eine gentechnologisch hergestellte Form
von Adenovirus. Die Kenntnis der genetischen Organisation von Adenovirus,
einem Virus mit linearer doppelsträngiger DNA einer Länge von
36 kb, erlaubt die Substitution großer Stücke adenoviraler DNA durch
Fremdsequenzen bis zu 7 kb (Grunhaus und Horwitz, 1992). Im Gegensatz
zur retroviralen Infektion führt
die adenovirale Infektion von Wirtszellen nicht zu einer chromosomalen
Integration, da sich adenovirale DNA auf episomale Art ohne potenzielle
Gentoxizität
replizieren kann. Darüber
hinaus sind Adenoviren strukturell stabil und es wurde keine Umorganisation des
Genoms nach ausführlicher
Amplifizierung festgestellt. Adenovirus kann praktisch alle epithelialen
Zellen infizieren, unabhängig
von deren Zellzyklusstadium. Bislang scheint eine adenovirale Infektion
nur mit einer schwach ausgeprägten
Erkrankung verknüpft
zu sein, beispielsweise mit akuter respiratorischer Erkrankung beim
Menschen.
-
Adenovirus
ist aufgrund seines mittelgroßen
Genoms, der Einfachheit der Manipulation, seines hohen Titers, des
weiten Zielzellbereichs und der hohen Infektiosität zur Verwendung
als Gentransfervektor besonders geeignet. Beide Enden des viralen
Genoms enthalten umgekehrte Sequenzwiederholungen oder Inverted Repeats
(ITRs) mit einer Länge
von 100–200
Basenpaaren, bei denen es sich um cis-Elemente handelt, die für die Replikation
und Verpackung der viralen DNA erforderlich sind. Die früh (Early,
E) und spät
(Late, L) exprimierten Regionen des Genoms enthalten verschiedene
Transkriptionseinheiten, die sich durch den Beginn der Replikation
der viralen DNA unterscheiden. Die E1-Region (E1A und E1B) kodiert
Proteine, die für
die Regulierung der Transkription des viralen Genoms und einiger
zellulärer
Gene verantwortlich sind. Die Expression der E2-Region (E2A und
E2B) führt
zur Synthese der Proteine für
die Replikation der viralen DNA: Diese Proteine sind an der DNA-Replikation,
der Expression der Late-Gene und dem Abschalten der Wirtszelle beteiligt
(Renan, 1990). Die Produkte der Late-Gene, einschließlich der
Mehrzahl der viralen Kapsidproteine, werden nur nach signifikanter
Prozessierung eines einzelnen primären Transkriptes exprimiert,
die vom starken späten
(Major Late) Promotor (MLP) veranlasst wird. Der MLP((an Kartierungseinheit
(map unit, m.u.) 16,8) ist während
der späten
Phase der Infektion besonders effizient, und alle mRNAs, deren Transkription
von diesem Promotor veranlasst wird, besitzen eine 5'-dreigeteilte Leadersequenz
(TPL), durch die sie zu bevorzugten mRNAs für die Translation werden.
-
In
einem aktuellen System wird rekombinantes Adenovirus durch homologe
Rekombination zwischen Shuttlevektor und Provirusvektor erzeugt.
Aufgrund der möglichen
Rekombination zwischen zwei proviralen Vektoren, kann bei diesem
Prozess Wildtyp-Adenovirus entstehen. Es ist daher wichtig, einen
einzelnen Virusklon aus einem einzelnen Plaque zu isolieren und
seine Genomstruktur zu untersuchen.
-
Herstellung
und Propagierung der derzeitigen Adenovirusvektoren, die replikationsdefizient
sind, hängen
von einer besonderen Helferzelllinie mit der Bezeichnung 293 ab,
die aus humanen embryonalen Nierenzellen mit Ad5-DNA-Fragmenten
transformiert wurde und E1-Proteine konstitutiv exprimiert (Graham
et al., 1977). Da die E3-Region für das Adenovirusgenom entbehrlich
ist (Jones und Shenk, 1978), tragen die derzeitigen Adenovirusvektoren
mithilfe von 293-Zellen Fremd-DNA entweder in der E1-Region, der
D3-Region oder in
beiden Regionen (Graham und Prevec, 1991). Natürlicherweise kann Adenovirus
rund 105% des Wildtypgenoms verpacken (Ghosh-Choudhury et al., 1987),
was Kapazität
für etwa
2 zusätzliche
kb an DNA bietet. Zusammen mit den ungefähr 5,5 kb an DNA, die in den
Regionen E1 und E3 ersetzt werden können, liegt die maximale Kapazität der derzeitigen
Adenovirusvektoren bei unter 7,5 kb oder bei etwa 15% der Gesamtlänge des
Vektors. Mehr als 80% des viralen Genoms von Adenovirus bleiben
im Vektor-Gerüst
und sind die Ursache vektorbedingter Zytotoxizität. Außerdem ist die Replikationsdefizienz
des E1-deletierten Virus unvollständig. Beispielsweise wurde
bei den derzeit erhältlichen
Vektoren bei hoher MOI (Multiplicity of Infection) durchsickernde
Expression viraler Gene beobachtet (Mulligan, 1993).
-
Helferzelllinien
können
aus humanen Zellen abgeleitet werden, beispielsweise aus humanen
embryonalen Nierenzellen, Muskelzellen, hämatopoietischen Zellen oder
anderen humanen embryonalen mesenchymalen oder epithelialen Zellen.
Alternativ können
die Helferzellen von den Zellen anderer Säugetierarten, die für humanes
Adenovirus permissiv sind, abgeleitet werden. Zu diesen Zellen gehören beispielsweise
Vero-Zellen oder andere embryonale mesenchymale oder epitheliale
Zellen des Affen. Wie oben erwähnt,
ist 293 die bevorzugte Helferzelllinie.
-
Kürzlich haben
Racher et al., (1995) verbesserte Verfahren für die Kultivierung von 293-Zellen
und die Propagierung von Adenovirus offenbart. In einem Format werden
natürliche
Zellaggregate gezüchtet,
indem silikonisierte 1-Liter-Spinner-Flaschen (Techne, Cambridge,
GB), die 100–200
ml Medium enthalten, mit einzelnen Zellen beimpft werden. Nach Rühren bei
40 Umdrehungen pro Minute wird mit Trypanblau ein Näherungswert
der Lebensfähigkeit
der Zellen bestimmt. In einem anderen Format werden Fibra-Cel-Mikroträger (Bibby
Sterlin, Stone, GB) (5 g/l) wie folgt eingesetzt. Dem Träger (50
ml) in einer 250-ml-Erlenmayer-Flasche wird
ein Zellinokulum, das in 5 ml Medium resuspendiert ist, zugegeben
und 1 bis 4 Std. unter gelegentlichem Rühren stehen gelassen. Anschließend wird
das Medium durch 50 ml frisches Medium ersetzt und mit Schütteln begonnen.
Für die
Virusproduktion wird den Zellen gestattet bis zu einer Konfluenz
von 80% zu wachsen, wonach das Medium ausgewechselt (mit 25% des
Endvolumens) und Adenovirus bei einer MOI von 0,05 zugegeben wird.
Die Kulturen werden über
Nacht stehen gelassen, wonach das Volumen auf 100% erhöht und das
Schütteln
weitere 72 Stunden fortgesetzt wird.
-
Abgesehen
von der Maßgabe,
dass der Adenovirusvektor replikationsdefekt oder zumindest konditionell
defekt sein soll, dürfte
die Art des Adenovirusvektors für
die erfolgreiche Anwendung der Erfindung nicht ausschlaggebend sein.
Das Adenovirus kann von einem der 42 verschiedenen bekannten Serotypen
oder aus den Subgruppen A-F sein. Adenovirus Typ 5 aus Subgruppe
C ist das bevorzugte Ausgangsmaterial, um den konditionellen replikationsdefekten
Adenovirusvektor zur Verwendung in der vorliegenden Erfindung zu
erhalten. Dies liegt daran, dass Adenovirus Typ 5 ein humanes Adenovirus
ist, über
das umfangreiche biochemische und genetische Daten vorliegen und
es traditionell für
die meisten Konstruktionen verwendet worden ist, in denen Adenovirus
als Vektor eingesetzt worden ist.
-
Wie
oben ausgeführt,
ist der typische Vektor gemäß der vorliegenden
Erfindung replikationsdefekt und hat keine adenovirale E1-Region.
Es ist daher am praktischsten, das Polynukleotid, welches das Gen
von Interesse kodiert, an der Position einzufügen, aus der die kodierenden
E1-Sequenzen entfernt worden sind. Die Position der Insertion des
Konstruktes in die Adenovirussequenzen ist jedoch für die Erfindung
nicht ausschlaggebend. Das Polynukleotid, welches das Gen von Interesse
kodiert, kann auch anstelle der deletierten E3-Region in E3-Austauschvektoren
eingefügt
werden, wie von Karlsson et al., (1984) beschrieben wurde, oder
in die E4-Region, wobei eine Helferzelllinie oder ein Helfervirus
den E4-Defekt ergänzt.
-
Adenovirus
ist einfach zu züchten
und zu manipulieren und weist in vitro und in vivo einen breiten Wirtsbereich
auf. Diese Virusgruppe kann in hohen Titern erhalten werden, z.
B. 109–1011 Plaque-bildende Einheiten pro ml, und
ist hoch infektiös.
Der Lebenszyklus von Adenovirus erfordert keine Integration in das
Wirtszellgenom. Die von Adenvirusvektoren eingeführten Fremdgene liegen episomal
vor und haben daher auf Wirtszellen geringe gentoxische Wirkung.
In Studien einer Vakzinierung mit Wildtyp-Adenovirus wurden keine Nebenwirkungen
berichtet (Couch et al., 1963; Top et al., 1971), was ihre Sicherheit
und ihr therapeutisches Potenzial als Vektoren für einen Gentransfer in vivo
zeigt.
-
Adenovirusvektoren
sind zur Expression eukaryontischer Gene (Levrero et al., 1991;
Gomez-Foix et al., 1992) und bei der Impfstoffentwicklung (Grunhaus
und Horwitz, 1992; Graham und Prevec, 1991) verwendet worden. Kürzlich deuteten
Tierstudien darauf hin, dass rekombinanter Adenovirus für die Gentherapie
verwendet werden könnte
(Stratfort-Perricaudet und Perricaudet, 1991; Stratfort-Perricaudet
et al., 1990; Rich et al., 1993). Studien über die Verabreichung von rekombinantem
Adenovirus an unterschiedliche Gewebe umfassen Luftröhreninstillation
(Rosenfeld et al., 1991; Rosenfeld et al., 1992), Muskelinjektion
(Ragot et al., 1993), periphere intravenöse Injektionen (Herz und Gerard,
1993) und stereotaktische Inokulation in das Gehirn (Le Gal La Salle
et al., 1993).
-
Retroviren.
-
Die
Retroviren sind eine Gruppe von RNA-Einzelstrang-Viren, die sich
durch eine Fähigkeit
der Konvertierung ihrer RNA zu doppelsträngiger DNA in infizierten Zellen
durch einen Vorgang der reversen Transkription auszeichnen (Coffin,
1990). Die resultierende DNA integriert sich danach als Provirus
stabil in die zellulären
Chromosomen und steuert die Synthese viraler Proteine. Die Integration
resultiert in der Beibehaltung der viralen Gensequenzen in der Empfängerzelle
und deren Nachkommen. Das retrovirale Genom enthält drei Gene, gag, pol und
env, die für
Kapsidproteine, Polymeraseenzym und Hüllbestandteile kodieren. Eine
Sequenz, die sich stromaufwärts
(upstream) vom gag-Gen befindet, enthält ein Signal für die Verpackung
des Genoms in Virionen. An den 5'-
und 3'-Enden des
viralen Genoms befinden sich zwei lange terminale Repeat (LTR)-Sequenzen.
Diese enthalten starke Promotor- und Enhancersequenzen und werden
auch für
die Integration in das Wirtszellgenom benötigt (Coffin, 1990).
-
Zur
Konstruktion eines retroviralen Vektors wird eine Nukleinsäure, die
das Gen von Interesse kodiert, in das virale Genom an die Stelle
bestimmter viraler Sequenzen eingesetzt, um ein replikationsdefektes
Virus herzustellen. Für
die Produktion von Virionen wird eine Verpackungszelllinie konstruiert,
die gag-, pol- und env-Gene, aber keine LTR-Sequenz und Verpackungsbestandteile
aufweist (Mann et al., 1983). Wenn ein rekombinantes Plasmid, das
eine cDNA enthält,
zusammen mit der retroviralen LRT-Sequenz und Verpackungssequenzen
in diese Zelllinie eingeführt
wird (durch Kalziumphosphatpräzipitation
beispielsweise), erlaubt die Verpackungssequenz, dass das RNA-Transkript
des rekombinanten Plasmids in virale Partikel verpackt wird, die
dann in das Kulturmedium sezerniert werden (Nicolas und Rubenstein,
1988; Temin, 1986, Mann et al., 1983). Das Medium, das die rekombinanten
Retroviren enthält,
wird dann aufgefangen, wahlweise eingeengt und für den Gentransfer verwendet.
Retrovirale Vektoren können
sehr viele verschiedene Zelltypen infizieren. Integration und stabile
Expression erfordert jedoch die Teilung von Wirtszellen (Paskind
et al., 1975).
-
Kürzlich wurde
ein neuartiger Ansatz auf der Basis der chemischen Modifikation
eines Retrovirus durch die chemische Addition von Laktoseresten
an die Virushülle
entwickelt, der konzipiert ist, um ein spezifisches Targeting von
Retrovirusvektoren zu ermöglichen.
Diese Modifikation könnte
die spezifische Infektion von Hepatozyten über Sialoglykoproteinrezeptoren
ermöglichen.
-
Es
wurde ein anderer Ansatz des Targeting von rekombinanten Retroviren
konzipiert, bei dem biotinylierte Antikörper gegen ein retrovirales
Hüllprotein
und gegen einen spezifischen Zellrezeptor verwendet wurden. Die
Antikörper
wurden über
die Biotinbestandteile mittels Streptavidin gekoppelt (Roux et al.,
1989). Mit Antikörper
gegen Klasse-I- und Klasse-II-Antigene
des Haupthistokompatibilitätskomplexes
zeigten sie mit einem ökotropen
Virus in vitro die Infektion einer Vielzahl menschlicher Zellen,
die diese Oberflächenantigene trugen
(Roux et al., 1989).
-
Mit
der Verwendung von Retrovirusvektoren in allen Spektren der vorliegenden
Erfindung sind bestimmte Einschränkungen
verbunden. Beispielsweise integrieren Retrovirusvektoren in der
Regel an zufälligen Stellen
im zellulären
Genom. Dies kann zu einer Insertionsmutagenese durch Unterbrechung
von Wirtsgenen oder durch die Insertion viraler Regulatorsequenzen
führen,
die mit der Funktion benachbarter Gene wechselwirken können (Varmus
et al., 1981). Ein anderes Problem bei der Verwendung defekter Retrovirusvektoren ist
das mögliche
Auftreten replikationskompetenter Wildtypviren in den Verpackungszellen.
Dies kann das Ergebnis von Rekombinationsereignissen sein, in denen
die intakte Sequenz des rekombinanten Virus stromaufwärts (upstream)
der in das Wirtszellgenom integrierten gag-, pol, env-Sequenz eingefügt wird.
Es stehen mittlerweile jedoch neue Verpackungszelllinien zur Verfügung, die
die Wahrscheinlichkeit einer Rekombination stark vermindern sollten
(Markowitz et al., 1988; Hersdorffer et al., 1990).
-
Herpesvirus.
-
Da
das Herpes-Simplex-Virus (HSV) neurotrop ist, hat es erhebliches
Interesse an der Behandlung von Erkrankungen des Nervensystems geweckt.
Darüber
hinaus ist HSV durch seine Fähigkeit,
sich nicht-teilende neuronale Zellen latent zu infizieren, ohne
sich in Wirtszellenchromosomen zu integrieren oder den Metabolismus
der Wirtszelle anderweitig zu verändern, sowie das Vorhandensein
eines Promotors, der während der
Latenzphase aktiv ist, ein attraktiver Vektor. Und obwohl sich viel
Aufmerksamkeit auf die neurotropen Anwendungen von HSV konzentriert
hat, lässt
sich dieser Vektor angesichts seines weiten Wirtsbereiches auch für andere
Gewebe anwenden.
-
Ein
weiterer Faktor, der HSV zu einem attraktiven Vektor macht, ist
die Größe und Organisation
des Genoms. Da HSV groß ist,
ist der Einbau mehrerer Gene oder Expressionskassetten weniger problematisch als
in anderen kleineren viralen Systemen. Darüber hinaus ermöglicht die
Verfügbarkeit
unterschiedlicher viraler Kontrollsequenzen mit unterschiedlicher
Leistung (in Bezug auf Zeit, Stärke,
etc.) die Expression mehr als in anderen Systemen zu kontrollieren.
Es ist außerdem
von Vorteil, dass das Virus relativ wenig gesplicte mRNA aufweist,
was die genetische Manipulation weiter vereinfacht.
-
HSV
ist außerdem
relativ einfach zu manipulieren und kann bis zu hohen Titern angezüchtet werden. Das
Einführen
(Delivery) ist daher ein geringeres Problem, sowohl in Bezug auf
die Volumen, die erforderlich sind, um eine ausreichende MOI zu
erzielen, als auch in Bezug auf einen verminderten Bedarf für wiederholte Dosierungen.
Eine Übersicht über HSV
als Gentherapievektor geben Glorioso et al., (1995).
-
HSV,
die in die Subtypen 1 und 2 eingeteilt werden, sind Hüllviren,
die zu den häufigsten
Infektionserregern des Menschen gehören und die Millionen von Menschen
weltweit infizieren. Das große
komplexe doppelsträngige
DNA-Genom kodiert für
Dutzende unterschiedlicher Genprodukte, von denen einige von gesplicten
Transkripten abstammen. Neben Virion- und Hüllstrukturbestandteilen kodiert
das Virus für
zahlreiche andere Proteine, darunter eine Protease, eine Ribonukleotidreduktase,
eine DNA-Polymerase, ein ssDNA-Bindungsprotein, eine Helikase/Primase
, eine DNA-abhängige
ATPase, eine dUTPase und andere.
-
HSV-Gene
bilden mehrere Gruppen, deren Expression koordiniert reguliert und
in der Art einer Kaskade sequenziell geordnet ist (Honess und Roizman,
1974; Honess und Roizman, 1975, Roizman und Sears, 1995). Die Expression
von α-Genen,
dem ersten Satz von Genen, die nach einer Infektion exprimiert werden, wird
durch das Virionprotein Nummer 16 oder α-transduzierender Faktor verstärkt (Post
et al., 1981; Batterson und Roizman, 1983; Campbell et al., 1983).
Die Expression von β-Genen
erfordert funktionelle α-Genprodukte, vor
allem ICP4, das vom α4-Gen
kodiert wird (DeLuca et al., 1985). Y Gene, eine heterogene Gruppe
von Genen, die überwiegend
strukturelle Virionproteine kodieren, erfordern für eine optimale
Expression den Beginn viraler DNA-Synthese (Holland et al., 1980).
-
Entsprechend
der Komplexität
des Genoms ist der Lebenszyklus von HSV erheblich kompliziert. Außer dem
lytischen Zyklus, der zur Synthese von Viruspartikeln und schließlich zum
Zelltod führt,
hat das Virus die Möglichkeit,
in ein latentes Stadium einzutreten, in dem das Genom in neuralen
Ganglien erhalten bleibt, bis einige bislang undefinierte Signale
ein Wiedereintreten in den lytischen Zyklus auslösen. Es sind avirulente Varianten
von HSV entwickelt worden und sind zur Verwendung in Verbindung
mit Gentherapie einfach verfügbar
(US-Patent 5.672.344).
-
Adeno-assoziiertes
Virus. Seit kurzem hat sich das Adeno-assoziierte Virus (AAV) als
eine mögliche Alternative
zu den üblicherweise
verwendeten retroviralen und adenoviralen Vektoren entwickelt. Während Studien
mit Retrovirus- und Adenovirus-vermitteltem Gentransfer Bedenken
hinsichtlich potenzieller onkogener Eigenschaften des Ersteren und
immunogener Probleme in Verbindung mit Letzterem auslösen, ist
AAV nicht mit solchen pathologischen Indikationen in Verbindung
gebracht worden.
-
AAV
besitzt darüber
hinaus einige besondere Merkmale, die es wünschenswerter als die anderen
Vektoren macht. Im Gegensatz zu Retroviren kann AAV nicht-teilende
Zellen infizieren; Wildtyp-AAV wurde durch ortsspezifische Integration
in Chromosom 19 menschlicher Zellen charakterisiert (Kotin und Berns,
1989; Kotin et al., 1990; Kotin et al., 1991; Samulski et al., 1991),
und AAV besitzt außerdem
anti-onkogene Eigenschaften (Ostrove et al., 1981; Berns und Giraud,
1996). Rekombinante AAV-Genome werden konstruiert, indem DNA-Sequenzen
von Interesse molekular zwischen die AAV-ITRs kloniert werden, wodurch
die gesamten kodierenden Sequenzen des AAV-Wildtypgenoms beseitigt
werden. Die so produzierten AAV-Vektoren
weisen also keine kodierenden Sequenzen des AAV-Wildtypgenoms auf,
haben aber die Eigenschaft einer stabilen Chromosomenintegration
und Expression der rekombinanten Gene nach Transduktion in vitro
und in vivo beibehalten (Berns, 1990; Berns und Bohensky, 1987;
Bertran et al., 1996; Kearns et al., 1996; Ponnazhagan et al., 1997a).
Bis vor Kurzem wurde angenommen, dass AAV fast alle Zelltypen infiziert,
und selbst Artenbarrieren überschreitet.
Es wurde nun jedoch festgestellt, dass die AAV-Infektion rezeptorvermittelt
ist (Ponnazhagan et al., 1996; Mizukami et al., 1996).
-
AAV
verwendet eine lineare einzelsträngige
DNA von etwa 4700 Basenpaaren. Das Genom wird von umgekehrten terminalen
Sequenzwiederholungen oder Inverted Terminal Repeats (ITRs) flankiert.
In dem Genom sind zwei Gene vorhanden, die eine Anzahl unterschiedlicher
Genprodukte hervorbringen. Das erste, das cap-Gen, produziert drei
verschiedene Virionproteine (VP), die als VP-1, VP-2 und VP-3 bezeichnet
werden. Das zweite, das rep-Gen, kodiert vier nicht strukturelle
Proteine (NP). Eines oder mehrere dieser rep-Genprodukte ist für die Transaktivierung der
AAV-Transkription verantwortlich. Die Sequenz von AAV ist von Srivastava
et al., (1993) und in US-Patent 5.252.479 (dessen gesamter Text
hierin spezifisch durch Bezugnahme enthalten ist) bereitgestellt.
-
Die
drei Promotoren in AAV sind nach ihrer Position in Kartierungseinheiten
(Map Units) im Genom gekennzeichnet. Es sind von links nach rechts
p5, p19 und p40. Durch Transkription entstehen sechs Transkripte,
wobei jeweils zwei an jedem der drei Promotoren initiiert werden
und aus jedem Paar eines gesplict wird. Bei jedem Transkript befindet
sich die Splice-Stelle an der Position der Kartierungseinheiten
42–46.
Die vier nicht strukturellen Proteine entstammen offensichtlich
dem längeren
Transkript, während
die drei Virionproteine alle aus dem kleinsten Transkript abgeleitet
sind.
-
AAV
ist beim Menschen nicht mit einem pathologischen Zustand verbunden.
Für eine
effiziente Replikation erfordert AAV interessanterweise „Helfer"-Funktionen von Viren
wie Herpes-Simplex-Virus I und II, Cytomegalovirus, Pseudorabies-Virus
und natürlich
Adenovirus. Der am besten charakterisierte Helfer ist Adenovirus,
und es ist gezeigt worden, dass viele frühen (early) Funktionen dieses
Virus die AAV-Replikation unterstützen. Es wird angenommen, dass
eine schwache Expression von AAV-rep-Proteinen die Expression struktureller
AAV-Gene in Schach hält,
und dass die Infektion mit Helferviren diese Blockade beseitigt.
-
Vakzinia-Virus.
-
Vakzinia-Virusvektoren
sind aufgrund der Einfachheit ihrer Herstellung, dem relativ hohen
Grad an Expression, den man erhalten kann, dem weiten Wirtsbereich
und der großen
Kapazität
zur Aufnahme von DNA extensiv verwendet worden. Vakzinia enthält ein Genom
aus linearer doppelsträngiger
DNA von etwa 186 kb, die eine ausgeprägte Präferenz von „A-T"-Paaren aufweist. Umgekehrte terminale
Sequenzwiederholungen oder Inverted Terminal Repeats (ITRs) von
etwa 10,5 kb flankieren das Genom. Die Mehrzahl der essenziellen Gene
scheint sich in der Zentralregion zu befinden, welche bei Pockenviren
am stärksten
konserviert ist. Vakzinia-Viren enthalten rund 150 bis 200 offene
Leseraster. Obgleich beide Stränge
kodieren, kommt es selten zu einer weit reichenden Überlappung
der Leseraster.
-
In
das Genom des Vakzinia-Virus können
mindestens 25 kb eingefügt
werden (Smith und Moss, 1983). Prototypische Vakzinia-Vektoren enthalten
Transgene, die über
homologe Rekombination in das virale Thymidinkinasegen eingefügt worden
sind. Vektoren werden auf der Basis eines tk-Phänotyps ausgewählt. Durch den
Einschluss der untranslatierten Leadersequenz des Enzephalomyokarditisvirus
wird die Expression gegenüber
herkömmlichen
Vektoren verstärkt,
wobei das Transgen nach 24 Stunden 10% oder mehr der Proteine der
infizierten Zelle ausmacht (Elroy-Stein et al., 1989).
-
Nicht-viraler Transfer.
-
Damit
Sense- oder Antisense-Genkonstrukte exprimiert werden können, muss
das Expressionskonstrukt in eine Zelle eingeführt werden. Dieses Einführen kann
in vitro erreicht werden wie bei Laborverfahren zur Transformation
von Zelllinien, oder in vivo oder ex vivo wie bei der Behandlung
bestimmter Erkrankungsstadien. Ein Mechanismus für die Einführung besteht aus einer viralen
Infektion, wobei das Expressionskonstrukt in einem infektiösen Viruspartikel
verkapselt ist.
-
In
der vorliegenden Erfindung werden auch einige nicht-virale Verfahren
für den
Transfer von Expressionskonstrukten in kultivierte Säugetierzellen
in Betracht gezogen. Dazu gehören Kalziumphosphatpräzipitation
(Graham und Van Der Eb, 1973; Chen und Okayama, 1987; Rippe et al.,
1990), DEAE-Dextran (Gopal, 1985), Elektroporation (Tur-Kaspa et
al., 1986; Potter et al., 1984), direkte Mikroinjektion (Harland
und Weintraub, 1985), DNA-beladene Liposomen (Nicolau und Sene,
1982; Fraley et al., 1979) und Lipofectamin-DNA-Komplexe, Zellsonifikation
(Fechheimer et al., 1987), Genbeschuss mit Hochgeschwindigkeits-Mikroprojektilen
(Yang et al., 1990) und rezeptorvermittelte Transfektion (Wu und
Wu, 1987; Wu und Wu, 1988). Einige dieser Techniken könnten erfolgreich
für die
Verwendung in vivo oder ex vivo adaptiert werden.
-
Wenn
das Expressionskonstrukt in die Zelle eingeführt worden ist, wird die Nukleinsäure, die
das Gen von Interesse kodiert, unter Umständen an verschiedenen Stellen
positioniert und exprimiert. In bestimmten Ausführungsformen kann die Nukleinsäure, die
das Gen kodiert, stabil in das Genom der Zelle integriert werden.
Diese Integration kann über
homologe Rekombination (Genaustausch) an der verwandten Position
und in verwandter Ausrichtung erfolgen oder die Integration erfolgt
an einer zufälligen,
nicht spezifischen Position (Genaugmentation). In weiteren Ausführungsformen
kann die Nukleinsäure
in der Zelle stabil als ein gesondertes episomales Segment einer
DNA erhalten bleiben. Solche Nukleinsäuresegmente oder „Episomen" kodieren Sequenzen,
die ausreichend sind, um eine Erhaltung und Replikation unabhängig von
oder synchronisiert mit dem Zellzyklus zu erlauben. Wie das Expressionskonstrukt
in eine Zelle eingeführt
wird und wo die Nukleinsäure
in der Zelle bleibt, hängt
von der Art des verwendeten Expressionskonstruktes ab.
-
In
einer weiteren Ausführungsform
der Erfindung kann das Expressionskonstrukt einfach aus nackter rekombinanter
DNA oder nackten rekombinanten Plasmiden bestehen. Der Transfer
des Konstruktes kann durch jedes beliebige der oben erwähnten Verfahren
erfolgen, das die Zellmembran physikalisch oder chemisch durchlässig macht:
Dies eignet sich vor allem für
den Transfer in vitro, lässt
sich aber auch für
den Gebrauch in vivo verwenden. Dubensky et al., (1984) injizierten
Polyomvirus-DNA in Form von Kalziumphosphatpräzipitaten erfolgreich in Leber
und Milz adulter und neugeborener Mäuse, wobei aktive virale Replikation
und akute Infektion gezeigt wurden. Benvenisty und Neshif (1986)
zeigten auch, dass eine direkte intraperitoneale Injektion von Kalziumphosphat-präzipitierten
Plasmiden zu einer Expression der transfizierten Gene führt. Es könnte sein,
dass DNA, die ein Gen von Interesse kodiert, auf ähnliche
Weise in vivo übertragen
werden kann und das Genprodukt exprimiert.
-
Eine
weitere Ausführungsform
der Erfindung zur Übertragung
eines Expressionskonstruktes aus nackter DNA in Zellen kann Teilchenbeschuss
umfassen. Dieses Verfahren beruht auf der Möglichkeit, DNA-beschichtete
Mikroprojektile auf eine hohe Geschwindigkeit zu beschleunigen,
wodurch sie die Zellmembran durchdringen und in die Zellen hinein
gelangen können,
ohne sie zu töten
(Klein et al., 1987). Es sind mehrere Vorrichtungen zum Beschleunigen
kleiner Teilchen entwickelt worden. Eine solche Vorrichtung basiert
auf einer Hochspannungsentladung zur Erzeugung eines elektrischen
Flusses, der wiederum die Antriebskraft liefert (Yang et al., 1990).
Die verwendeten Mikroprojektile bestanden aus biologisch inerten
Substanzen wie beispielsweise Tungsten oder Goldkügelchen.
-
Es
sind ausgewählte
Organe, einschließlich
Leber, Haut und Muskelgewebe von Ratten und Mäusen in vivo bombardiert worden
(Yang et al., 1990; Zelenin et al., 1991). Dies kann die chirurgische
Freilegung des Gewebes oder der Zellen erforderlich machen, um jedes
Gewebe, das zwischen der Pistole und dem Zielorgan liegt, zu beseitigen,
d. h. eine ex vivo-Behandlung. Auch eine DNA, die ein bestimmtes
Gen kodiert, kann durch dieses Verfahren eingeführt und dennoch in die vorliegende
Erfindung eingebaut werden.
-
In
einer weiteren Ausführungsform
der Erfindung kann das Expressionskonstrukt in einem Liposom eingeschlossen
sein. Liposomen sind vesikuläre
Strukturen, die durch eine Phospholipid-Doppelmembran und ein inneres
wässriges
Medium gekennzeichnet sind. Multilamellare Liposomen haben mehrere
Lipidschichten, die durch wässriges
Medium getrennt sind. Sie bilden sich spontan, wenn Phospholipide
in einem Überschuss an
wässriger
Lösung
suspendiert werden. Die Lipidbestandteile durchlaufen vor der Bildung
geschlossener Strukturen eine Neuanordnung und schließen Wasser
und aufgelöste
lösliche
Stoffe zwischen den Lipidschichten ein (Ghosh und Bachhawat, 1991).
In Betracht gezogen werden auch Lipofectam-DNA-Komplexe.
-
Liposomen-vermitteltes
Einführen
von Nukleinsäuren
und Expression von Fremd-DNA in vitro waren sehr erfolgreich. Wong
et al., (1989) zeigten die Durchführbarkeit einer Liposomen-vermittelten
Einführung
und Expression von Fremd-DNA in kultivierte Zellen von Hühnerembryonen
sowie in HeLa- und Hepatomzellen. Nicolau et al., (1987) gelang
ein erfolgreicher Liposomen-vermittelter Gentransfer in Ratten nach
intravenöser Injektion.
-
In
bestimmten Ausführungsformen
der Erfindung können
Komplexe aus Liposom und einem Hämagglutininvirus
(HVJ) gebildet werden. Es wurde gezeigt, dass die Fusion mit der
Zellmembran damit erleichtert und das Eintreten von in Liposomen
verkapselter DNA in die Zelle gefördert wird (Kaneda et al.,
1989). In anderen Ausführungsformen
kann das Liposom komplexiert oder in Verbindung mit nukleären chromosomalen Nicht-Histonproteinen
(HMG-1) verwendet werden (Kato et al., 1991). In weiteren Ausführungsformen
kann das Liposom komplexiert oder in Verbindung mit HVJ und HMG-1
verwendet werden. Insofern, als solche Expressionskonstrukte erfolgreich
beim Transfer und der Expression von Nukleinsäuren in vitro und in vivo eingesetzt
worden sind, sind sie für
die vorliegende Erfindung geeignet. Wenn in dem DNA-Konstrukt ein
bakterieller Promotor verwendet wird, ist es außerdem wünschenswert, eine geeignete
bakterielle Polymerase in das Liposom aufzunehmen.
-
Andere
Expressionskonstrukte, die verwendet werden können, um eine Nukleinsäure, die
ein bestimmtes Gen kodiert, in Zellen einzuführen, sind rezeptorvermittelte
Einführungsvehikel.
Diese nutzen die selektive Aufnahme von Makromolekülen durch
rezeptorvermittelte Endozytose in fast alle eukaryontischen Zellen.
Aufgrund der zelltypspezifischen Verteilung verschiedener Rezeptoren
kann diese Art des Einführens hoch
spezifisch sein (Wu und Wu, 1993).
-
Vehikel
für rezeptorvermitteltes
Gentargeting bestehen im Allgemeinen aus zwei Komponenten: einem Zellrezeptor-spezifischen
Liganden und einem DNA-Bindungsmittel. Es sind mehrere Liganden
für rezeptorvermittelten
Gentransfer verwendet worden. Die am ausführlichsten charakterisierten
Liganden sind Asialoorosomucoid (ASOR) (Wu und Wu, 1987) und Transferrin
(Wagner et al., 1990). Kürzlich
ist ein synthetisches Neoglykoprotein, das denselben Rezeptor wie
ASOR erkennt, als Vehikel zum Einführen von Genen verwendet worden
(Ferkol et al., 1993; Perales et al., 1994), und auch EGF (Epidermaler
Wachstumsfaktor) wurde benutzt, um Gene in Plattenepithelkarzinomzellen
einzuführen
(Myers, EPO 0273085).
-
In
anderen Ausführungsformen
kann das Einführungsvehikel
einen Liganden und ein Liposom umfassen. Nicolau et al., (1987)
beispielsweise setzten Lactosyl-Ceramid ein, ein galaktoseterminales
Asialogangliosid, das in Liposomen eingebaut war, und beobachteten
eine Erhöhung
der Aufnahme des Insulingens von Hepatozyten. Es ist daher möglich, dass
auch eine Nukleinsäure,
die ein bestimmtes Gen kodiert, durch eine beliebige Anzahl an Rezeptor-Ligand-Systemen mit oder
ohne Liposomen spezifisch in einen Zelltyp wie Lungen-, Epithel-
oder Tumorzellen eingeführt
wird. Beispielsweise kann EGF (Epidermaler Wachstumsfaktor) in vielen
Tumorzellen, die eine Hochregulierung des EGF-Rezeptors aufweisen,
als Rezeptor für
die vermittelte Einführung
einer Nukleinsäure,
die ein Gen kodiert, verwendet werden. Mannose kann eingesetzt werden, wenn
das Ziel der Mannoserezeptor auf Leberzellen ist. Auch Antikörper gegen
CDS (CLL), CD22 (Lymphom), CD25 (T-Zellleukämie) und MAA (Melanom) können als
Targeting-Einheiten eingesetzt werden.
-
In
bestimmten Ausführungsformen
könnte
ein Gentransfer einfacher unter ex vivo-Bedingungen durchgeführt werden. Ex-vivo-Gentherapie
bezieht sich auf die Isolierung von Zellen aus einem Tier, das Einführen einer
Nukleinsäure
in die Zellen in vitro, und anschließende Rückführung der modifizierten Zellen
in ein Tier. Dies kann die chirurgische Entnahme von Geweben/Organen
aus einem Tier oder die Primärkultur
von Zellen und Geweben umfassen.
-
Primäre Säugetierzellkulturen
können
auf verschiedene Art und Weise hergestellt werden. Damit die Zellen
in vitro und während
des Kontaktes mit dem Expressionskonstrukt lebensfähig bleiben,
muss sichergestellt werden, dass die Zellen Kontakt mit Sauerstoff
und Kohlendioxid und Nährstoffen
im richtigen Verhältnis aufrecht
erhalten, aber vor mikrobieller Kontamination geschützt sind.
Zellkulturtechniken sind gut dokumentiert und hierin durch Bezugnahme
enthalten (Freshner, 1992).
-
Eine
Ausführungsform
des Vorangehenden umfasst die Anwendung eines Gentransfers immortalisierter
Zellen für
die Produktion von Proteinen. Das Gen für das Protein von Interesse
kann wie oben beschrieben in geeignete Wirtszellen überführt werden,
gefolgt von einer Kultur der Zellen unter geeigneten Bedingungen. Auf
diese Weise können
Gene für
praktisch jedes beliebige Polypeptid eingesetzt werden. Die Herstellung
rekombinanter Expressionsvektoren und der darin enthaltenen Elemente
ist oben erläutert.
Alternativ kann es sich bei dem herzustellenden Protein um ein endogenes
Protein handeln, das normalerweise von der fraglichen Zelle synthetisiert
wird.
-
Beispiele
geeigneter Säugetier-Wirtszelllinien
sind Vero- und HeLa-Zellen und Zelllinien aus dem Eierstock des
chinesischen Hamsters, W138-, BHK-, COS-7-, 293-, HepG2-, NIH3T3-,
RIN- und MDCK-Zellen. Darüber
hinaus kann ein Wirtszellstamm ausgewählt werden, der die Expression
der eingefügten
Sequenzen moduliert oder das Genprodukt auf die gewünschte Art
und Weise modifiziert und prozessiert. Solche Modifikationen (z.
B. Glykosylierung) und Prozessierung (z. B. Spaltung) von Proteinprodukten
können
für die
Funktion des Proteins wichtig sein. Verschiedene Wirtszellen verfügen über charakteristische
und spezifische Mechanismen für
die posttranslationale Prozessierung und Modifikation von Proteinen.
Es können
geeignete Zelllinien oder Wirtssysteme gewählt werden, um die korrekte
Modifikation und Prozessierung des exprimierten Fremdproteins sicherzustellen.
-
Es
kann eine Reihe von Selektionssystemen verwendet werden, einschließlich, aber
nicht beschränkt auf,
das HSV-Thymidinkinasegen in tk-Zellen, das Hpyoxanthin-Guaninphosphoribosyltransferasegen
in hgptr–-Zellen
und das Adeninphosphoribosyltransferasegen in aprt–-Zellen.
Als Basis für
die Selektion kann auch Antimetabolitenresistenz verwendet werden,
beispielsweise das dhfr-Gen, das Resistenz gegen Dehydrofolsäurereduktase
vermittelt, das gpt-Gen, das Resistenz gegen Mycophenolsäure vermittelt,
das neo-Gen, das Resistenz gegen Aminoglykosid G418 vermittelt,
und das hygro-Gen,
das Resistenz gegen Hygromycin vermittelt.
-
Tierische
Zellen lassen sich in vitro auf zweierlei Art und Weise propagieren:
als nicht-ankerabhängige Zellen,
die während
der gesamten Kultur in Suspension wachsen, oder als ankerabhängige Zellen,
welche die Haftung an ein solides Substrat benötigen um zu wachsen (d. h.
ein Zellwachstum als einlagige Zellschicht, d. h. als Monolayertyp).
-
Nicht-ankerabhängige Kulturen
oder Suspensionskulturen aus kontinuierlich etablierten Zelllinien
sind das am häufigsten
verwendete Mittel für
die Produktion von Zellen und Zellprodukten in großem Maßstab. In Suspension
kultivierte Zellen haben jedoch Einschränkungen, beispielsweise ein
tumorigenes Potenzial und geringere Proteinproduktion als adhärente Zellen.
-
Suspensionskulturen
von Säugetierzellen
in großem
Maßstab
in Rührtanks
ist ein gängiges
Verfahren für
die Herstellung rekombinanter Proteine. Weit verbreitet ist die
Verwendung zweier Suspensionskulturreaktordesigns: Der Rührkesselreaktor
und der Airlift-Reaktor.
Die Rührtechnik
wurde erfolgreich für
die Produktion von Interferon im Maßstab von 8000 Liter eingesetzt.
Die Zellen werden in einem Edelstahltank mit einem Verhältnis von
Höhe zu
Durchmesser von 1:1 oder 3:1 gezüchtet.
Die Kultur wird in der Regel mit einem oder mehreren Rührwerken
auf der Basis von Rührblattscheiben
oder Propellern gemischt. Es sind Rührwerke beschrieben worden,
die geringere Scherkräfte
als Rührblätter bieten.
Der Rührvorgang
kann entweder direkt oder indirekt über magnetisch gekoppelte Motoren
angetrieben werden. Indirekte Antriebe reduzieren das Risiko einer
mikrobiellen Kontamination durch Dichtungen an Rührschäften.
-
Der
Airlift-Reaktor, der ursprünglich
ebenfalls für
die mikrobielle Fermentation beschrieben und später für die Kultur von Säugetierzellen
angepasst worden ist, beruht auf einem Gasstrom, um die Kultur zu
mischen und mit Sauerstoff zu versorgen. Der Gasstrom gelangt in
einen Steigrohrabschnitt des Reaktors und treibt die Zirkulation
an. Gas tritt an der Oberfläche
der Kultur aus, was bewirkt, dass dichtere Flüssigkeit ohne Gasbläschen nach
unten in den Fallrohrabschnitt des Reaktors wandert. Der Hauptvorteil
dieses Designs ist seine Einfachheit und das Entfallen der Notwendigkeit
mechanischen Mischens. Das Verhältnis
von Höhe
zu Durchmesser ist typischerweise 10:1. Der Maßstab des Airlift-Reaktors
kann relativ einfach vergrößert werden,
er weist eine gute Masseübernagung
von Gasen und im Allgemeinen relativ niedrige Scherkräfte auf.
-
Die
Antikörper
der vorliegenden Erfindung sind besonders für die Isolierung von Antigenen
durch Immunpräzipitation
geeignet. Immunpräzipitation
umfasst die Trennung des Zielantigenbestandteils von einer komplexen
Mischung und wird verwendet, um kleinste Mengen von Protein abzutrennen
oder zu isolieren. Für die
Isolierung von Membranproteinen müssen Zellen in Detergensmizellen
aufgelöst
werden. Nicht-ionische Salze sind bevorzugt, da andere Mittel wie
Gallensalze bei saurem pH-Wert oder in Gegenwart bivalenter Kationen
ausfallen. Antikörper
und ihr Gebrauch werden unten ausführlicher erläutert.
-
III. Herstellung von Antikörpern mit
Reaktivität
gegen TS10q23.3
-
In
einem weiteren Aspekt betrifft die vorliegende Erfindung einen Antikörper, der
gegen ein TS10q23.3-Molekül
der vorliegenden Erfindung oder einen beliebigen Abschnitt davon
immunreaktiv ist. Ein Antikörper
kann ein polyklonaler oder monoklonaler Antikörper sein. In einer bevorzugten
Ausführungsform
ist ein Antikörper
ein monoklonaler Antikörper.
Mittel zur Herstellung und Charakterisierung von Antikörpern sind aus
dem Stand der Technik gut bekannt (siehe z. B. Howell und Lane,
1988).
-
Kurz
beschrieben, wird ein polyklonaler Antikörper hergestellt, indem ein
Tier mit einem Immunogen immunisiert wird, das ein Polypeptid der
vorliegenden Erfindung umfasst, und als Antiserum von diesem immunisierten
Tier gewonnen wird. Für
die Produktion von Antiserum können
viele verschiedene Tierarten verwendet werden. Ein Tier, das zur
Produktion von Antiseren verwendet wird, ist typischerweise nicht
menschlich, einschließlich
Kaninchen, Mäuse,
Ratten, Hamster, Schweine oder Pferde. Aufgrund des relativ großen Blutvolumens
von Kaninchen, sind Kaninchen eine bevorzugte Wahl für die Produktion
polyklonaler Antikörper.
-
Antikörper, sowohl
polyklonale als auch monoklonale, die für Antigenisoformen spezifisch
sind, können
anhand herkömmlicher
Immunisierungstechniken, die einem Fachmann im Allgemeinen bekannt
sind, hergestellt werden. Eine Zusammensetzung, die antigene Epitope
der Verbindungen der vorliegenden Erfindung enthält, kann verwendet werden,
um eines oder mehrere Versuchstiere zu immunisieren, beispielsweise ein
Kaninchen oder eine Maus, die dann spezifische Antikörper gegen
die Verbindungen der vorliegenden Erfindung produzieren. Nach einem
Zeitraum zur Bildung von Antikörpern
können
polyklonale Antiseren erhalten werden, indem dem Tier einfach Blut
abgenommen und aus dem Vollblut Serumproben gewonnen werden. Die monoklonalen
Antikörper
der vorliegenden Erfindung könnten
sinnvolle Anwendung in immunchemischen Standardverfahren finden,
wie beispielsweise in ELISA- und Western-Blot-Verfahren, und in immunhistochemischen
Verfahren wie beispielsweise der Gewebefärbung, sowie in anderen Verfahren,
die Antikörper
verwenden, die spezifisch gegen in Zusammenhang mit TS10q23.3 stehende
Antigenepitope einsetzen. Darüber hinaus
könnten
monoklonale Antikörper,
die spezifisch gegen das jeweilige TS10q23.3 verschiedener Arten gerichtet
sind, in anderen nützlichen
Anwendungen Einsatz finden.
-
Im
Allgemeinen können
in verschiedenen Ausführungsformen
sowohl polyklonale als auch monoklonale Antikörper gegen TS10q23.3 verwendet
werden. Beispielsweise können
sie in Antikörper-Klonierungsprotokollen
eingesetzt werden, um cDNAs oder Gene, die andere TS10q23.3 kodieren,
zu erhalten. Sie können auch
in Inhibierungsstudien verwendet werden, um die Effekte von in Zusammenhang
mit TS10q23.3 stehenden Peptiden in Zellen oder Tieren zu analysieren.
Anti-TS1Oq23.3-Antikörper
eignen sich auch für
Immunlokalisierungsstudien, um die Verteilung von TS10q23.3 während verschiedener
zellulärer
Ereignisse zu untersuchen, beispielsweise um die zelluläre oder
gewebespezifische Verteilung von TS10q23.3-Polypeptiden in unterschiedlichen
Stadien des Zellzyklus zu bestimmen. Eine besonders nützliche
Anwendung solcher Antikörper
ist bei der Reinigung nativer oder rekombinanter TS10q23.3, beispielsweise
durch Verwendung einer Antikörperaffinitätssäule. Die
Anwendung aller solcher immunologischer Techniken ist einem Fachmann
angesichts der vorliegenden Offenbarung bekannt.
-
Mittel
zum Herstellen und Charakterisieren von Antikörpern sind aus dem Stand der
Technik gut bekannt (siehe z. B. Harlow und Lane, 1988; hierin durch
Bezugnahme enthalten). Spezifischere Beispiele der Herstellung monoklonaler
Antikörper
sind in den Beispielen unten gegeben.
-
Wie
aus dem Stand der Technik bekannt ist, kann eine gegebene Zusammensetzung
in ihrer Immunogenität
variieren. Es ist daher häufig
erforderlich, das Immunsystem des Wirts zu stärken, was beispielsweise erreicht
werden kann, indem ein Peptid- oder Polypeptidimmunogen an einen
Träger
gekoppelt wird. Beispielhafte und bevorzugte Träger sind Keyhole-Limpet-Hämozyanin (KLH) und Rinderserumalbumin
(BSA). Andere Albumine wie beispielsweise Ovalbumin, Mausserumalbumin
oder Kaninchenserumalbumin können auch
als Träger
verwendet werden. Mittel zum Konjugieren eines Polypeptids an ein
Trägerprotein
sind aus dem Stand der Technik bekannt und umfassen Glutaraldehyd,
m-Maleimidobencoyl-N-hydroxysuccinimidester,
Carbodiimid und bis-biazotisiertes Benzidin.
-
Wie
aus dem Stand der Technik ebenso bekannt ist, kann die Immunogenität einer
bestimmten immunogenen Zusammensetzung verstärkt werden, indem nicht-spezifische
Stimulatoren der Immunantwort, die als Adjuvantien bekannt sind,
verwendet werden. Beispielhafte und bevorzugte Adjuvantien umfassen
das Freund-Adjuvans (ein unspezifischer Stimulator der Immunantwort,
der abgetötetes
Mycobacterium tuberculosis enthält),
unvollständiges
Freund-Adjuvans und Aluminiumhydroxydadjuvans.
-
Die
Menge an immunogener Zusammensetzung, die bei der Produktion polyklonaler
Antikörper
verwendet wird, variiert je nach Art des Immunogens sowie des für die Immunisierung
verwendeten Tieres. Die Verabreichung des Immunogens ist auf unterschiedliche
Art und Weise möglich
(subkutan, intramuskulär,
intradermal, intravenös
und intraperitoneal). Die Produktion polyklonaler Antikörper kann
durch Abnahme von Blut von dem immunisierten Tier zu verschiedenen
Zeitpunkten nach der Immunisierung überwacht werden. Es kann auch
eine zweite verstärkende
so genannte „Booster"-Injektion gegeben
werden. Der Vorgang der Verstärkung
(Boosting) und der Titrierung wird wiederholt, bis ein geeigneter
Titer erreicht ist. Wenn ein gewünschter
Grad an Immunität
erreicht ist, kann das immunisierte Tier ausgeblutet und das Serum
isoliert und aufbewahrt werden und/oder das Tier kann verwendet
werden, um mAbs herzustellen.
-
mAbs
können
leicht durch Anwendung gut bekannter Techniken hergestellt werden,
wie beispielsweise die, die in US-Patent 4.196.265, das hierin durch
Bezugnahme enthalten ist, veranschaulicht sind. Typischerweise umfasst
diese Technik das Immunisieren eines geeigneten Tieres mit einer
ausgewählten
Immunogenzusammensetzung, z. B. einem gereinigten oder partiell
gereinigtem TS10q23.3-Protein, -Polypeptid oder -Peptid oder Zelle,
die TS10q23.3 in hohem Maß exprimiert.
Die immunisierende Zusammensetzung wird derart verabreicht, dass
sie Antikörper
produzierende Zellen wirksam stimuliert. Nager wie Mäuse und
Ratten sind bevorzugte Tiere, aber es ist auch die Verwendung von
Kaninchen, und Zellen von Hypopachus variolosus möglich. Die
Verwendung von Ratten könnte
bestimmte Vorteile bieten (Goding, 1986), aber Mäuse sind bevorzugt, wobei die
BALB/c-Maus die am meisten bevorzugte ist, da sie am häufigsten
verwendet und im Allgemeinen einen höheren Prozentanteil stabiler
Fusionen ergibt.
-
Nach
der Immunisierung werden Zellen mit dem Potenzial zur Produktion
von Antikörpern,
speziell B-Lymphozyten (B-Zellen), zur Verwendung in den Protokoll
zur Erzeugung von mAbs ausgewählt,
Diese Zellen können
aus Milz-, Tonsillen- oder Lymphknotenbiopsien oder aus einer peripheren
Blutprobe erhalten werden. Milzzellen und periphere Blutzellen sind
bevorzugt, Erstere, weil sie eine reichhaltige Quelle an Antikörper produzierenden
Zellen sind, die sich im Stadium eines teilenden Plasmablasten befinden,
und Letztere, weil peripheres Blut leicht zugänglich ist. Häufig wird
eine Reihe von Tieren immunisiert und die Milz des Tiers, das den
höchsten
Antikörpertiter
aufweist, wird entnommen und die Milzlymphozyten erhalten, indem
die Milz mit einer Spritze homogenisiert wird. Eine Milz einer immunisierten
Maus enthält
rund 5 × 107 bis 2 × 108 Lymphozyten.
-
Die
Antikörper
produzierenden B-Lymphozyten des immunisierten Tieres werden anschließend mit Zellen
einer unsterblichen Myelomzelle fusioniert, in der Regel von der
gleichen Art wie das immunisierte Tier. Myelomzelllinien, die zur
Verwendung in Hybridom produzierenden Fusionsverfahren geeignet
sind, sind vorzugsweise nicht Antikörper produzierend, haben eine
hohe Fusionseffizienz und Enzymmangelzustände, die es ihnen unmöglich machen,
in bestimmten Selektionsmedien zu wachsen, welche das Wachstum von
lediglich den gewünschten
Fusionszellen (Hybridomen) unterstützen.
-
Es
kann eine Beliebige aus einer Reihe von Hybridomzellen verwendet
werden, wie sie einem Fachmann bekannt sind (Goding, 1986). Wenn
das immunisierte Tier beispielsweise eine Maus ist, kann P3-X63/Ag8,
P3-X63/Ag8.653, NS1/1.Ag41, Sp210-Ag14, FO, NSO/U, MPC-11, MPC11-X45-GTG
1.7 und S194/5XX0 Bul verwendet werden; für Ratten kann R210.RCY3, Y3-Ag
1.2.3, IR983F und 4B210 verwendet werden, und U-266, GM1500-GRG2,
LICR-LON-HMy2 und
UC729-6 sind in Verbindung mit Zellfusionen geeignet.
-
Verfahren
zum Erzeugen von Hybriden Antikörper
produzierender Milz- oder Lymphknotenzellen und Myelomzellen umfassen
in der Regel das Mischen somatischer Zellen mit Myelomzellen im
Verhältnis
von 2:1, obgleich das Verhältnis
in Gegenwart eines Mittels oder von Mitteln (chemisch oder elektrisch),
welche die Fusion von Zellmembranen fördern, von etwa 20:1 bis etwa
1:1 (respektive) variieren kann. Es sind Fusionsverfahren unter
Verwendung von Sendai-Virus beschrieben worden (Köhler und
Milstein, 1975, 1976) und solche, bei denen Polyethylenglycol (PEG),
beispielsweise 37% (Vol./Vol.) verwendet wird (Gefter et al., 1977).
Auch die Verwendung elektrisch induzierter Fusionsverfahren ist
geeignet (Goding, 1986).
-
Fusionsverfahren
bringen in der Regel lebensfähige
Hybride in niedriger Häufigkeit
hervor, d. h. etwa 1 × 10–6 bis
1 × 10–8.
Dies stellt allerdings kein Problem dar, da die lebensfähigen fusionierten
Hybride durch Kultivierung in einem Selektionsmedium von den nicht
fusionierten Elternzellen (insbesondere den nicht fusionierten Myelomzellen,
die sich normalerweise unendlich weiter teilen würden), differenziert sind.
Das Selektionsmedium ist im Allgemeinen eines, das ein Mittel enthält, welches
die de-novo-Synthese von Nukleotiden in dem Gewebekulturmedium blockiert.
Beispielhafte und bevorzugte Mittel sind Aminopterin, Methotrexat
und Azaserin. Aminopterin und Methotrexat blockieren die de-novo-Synthese
von Purinen und Pyrimidinen, während
Azaserin nur die Purinsynthese blockiert. Wenn Aminopterin und Methotrexat
verwendet werden, wird das Medium mit Hypoxanthin und Thymidin als
Nukleotidquelle supplementiert (HAT-Medium). Wenn Azaserin verwendet
wird, wird das Medium mit Hypoxanthin supplementiert.
-
Das
bevorzugte Selektionsmedium ist HAT. In HAT-Medium können nur
Zellen überleben,
die fähig sind,
auf einen Nukleotid-Ausweich-Stoffwechselweg (Salvage Pathway) umzustellen.
Den Myelomzellen fehlen wichtige Enzyme des Ausweich-Stoffwechselwegs,
z. B. Hypoxanthinphosphoribosyltransferase (HPRT), und sie sind
daher nicht überlebensfähig. Bei
den B-Zellen ist dieser Stoffwechselweg aktiv, aber sie haben in Kultur
eine eingeschränkte
Lebensdauer und sterben in der Regel innerhalb von zwei Wochen.
Die einzigen Zellen, die in dem Selektionsmedium überleben
können,
sind daher die aus Myelom- und B-Zellen gebildeten Hybride.
-
Diese
Kultivierung liefert eine Population von Hybridomen, aus denen spezifische
Hybridome ausgewählt
werden. Die Auswahl von Hybridomen erfolgt typischerweise durch
Kultivieren der Zellen mittels Einzelklonverdünnung in Mikrotiterplatten,
gefolgt von einer Testung der einzelnen klonalen Überstände (nach etwa
zwei bis drei Wochen) hinsichtlich der gewünschten Reaktivität. Der Test
sollte empfindlich, einfach und schnell sein, wie beispielsweise
Radioimmunassays, Enzymimmunassays, Zytotoxizitätsassays, Plaque-Assays, Punkt-Immun-Bindungs-Assays
und dergleichen.
-
Die
ausgewählten
Hybridomas würden
dann seriell verdünnt
und zu einzelnen Antikörper
produzierende Zelllinien kloniert werden, wobei die Klone anschließend unbegrenzt
weiter propagiert werden können,
um mAbs zu liefern. Die Zelllinien lassen sich auf zwei Arten für die Produktion
von mAbs nutzen. Eine Probe des Hybridoms kann einem histokompatiblen
Tier der Art, die zur Bereitstellung der somatischen Zellen und
Myelomzellen für
die ursprüngliche
Fusion verwendet worden ist, injiziert werden (häufig in die Peritonealhöhle). Das
injizierte Tier entwickelt Tumoren, welche den von dem fusionierten
Zellhybrid produzierten spezifischen monoklonalen Antikörper sezernieren.
Die Körperflüssigkeiten
des Tiers, beispielsweise Serum oder Aszitesflüssigkeit, können dann entnommen werden,
um mAbs in hoher Konzentration bereit zu stellen. Die einzelnen Zelllinien
können
auch in vitro kultiviert werden, wobei die mAbs natürlicherweise
in das Zellkulturmedium sezerniert werden, aus dem sie in hoher
Konzentration einfach gewonnen werden können. Die durch eines der beiden
Verfahren hergestellten mAbs können
anhand von Filtrations-, Zentrifugations- und verschiedenen Chromatographieverfahren,
wie beispielsweise HPLC oder Affinitätschromatographie, gegebenenfalls
weiter gereinigt werden.
-
Die
einzelnen Zelllinien könnten
auch in vitro kultiviert werden, wobei die mAbs natürlicherweise
in das Zellkulturmedium sezerniert werden, aus dem sie in hoher
Konzentration einfach gewonnen werden können.
-
Die
durch eines der beiden Verfahren hergestellten mAbs können anhand
von Filtrations-, Zentrifugations- und verschiedenen Chromatographieverfahren,
wie beispielsweise HPLC oder Affinitätschromatographie, gegebenenfalls
weiter gereinigt werden. Aus den gereinigten monoklonalen Antikörpern lassen
sich durch Verfahren, die einen Verdau mit Enzymen wie Pepsin oder
Papain und/oder durch Spaltung von Disulfidbrücken durch chemische Reduktion
umfassen, Fragmente der monoklonalen Antikörper der Erfindung erhalten.
Alternativ können
monoklonale Antikörperfragmente,
die von der vorliegenden Erfindung umfasst werden, mit einem automatisierten
Peptidsynthesegerät
synthetisiert werden.
-
Es
wird auch in Betracht gezogen, dass ein molekularer Klonierungsansatz
verwendet werden könnte, um
monoklonale Antikörper
herzustellen. Dazu werden kombinatorische Immunglobulin-Phagemid-Bibliotheken
aus RNA hergestellt, die aus der Milz des immunisierten Tiers isoliert
worden ist, und Phagemide, die geeignete Antikörper exprimieren, werden durch
Panning ausgewählt,
wobei Zellen, die das Antigen exprimieren, und Kontrollzellen verwendet
werden, z. B. Normalzellen gegenüber
Tumorzellen. Die Vorteile dieses Ansatzes gegenüber herkömmlichen Hybridomtechniken
sind, dass etwa 104-mal mehr Antikörper produziert
und auf einmal getestet werden können
und dass durch Kombination der H- und L-Ketten neue Spezifitäten entstehen,
was die Chance zur Auffindung geeigneter Antikörper weiter erhöht.
-
Humanisierte
monoklonale Antikörper
sind Antikörper
tierischer Abstammung, die anhand gentechnischer Verfahren modifiziert
worden sind, um Gerüstsequenzen
der konstanten Region und/oder der variablen Region durch humane
Sequenzen zu ersetzen, während
die ursprüngliche
Antigenspezifität
erhalten bleibt. Solche Antikörper
werden normalerweise von Nagerantikörpern mit Spezifität gegen
humane Antigene abgeleitet. Solche Antikörper eignen sich im Allgemeinen
für therapeutische
Anwendungen in vivo. Diese Strategie reduziert die Reaktion des
Wirts auf den Fremdantikörper
und erlaubt eine Selektion der humanen Effektorfunktionen.
-
Die
Techniken zur Produktion humanisierter Immunglobuline sind einem
Fachmann gut bekannt. US-Patent Nr. 5.693.762 offenbart beispielsweise
Verfahren zur Herstellung und Zusammensetzungen von humanen Immunglobulinen,
die eine oder mehrere Komplementaritätsbestimmende Regionen (CDRs)
aufweisen. Bei Kombination mit einem intakten Antikörper sind
die humanisierten Immunglobuline im Menschen im Wesentlichen nicht
immunogen und behalten im Wesentlichen dieselbe Affinität zu dem
Antigen, beispielsweise einem Protein oder einer anderen, Epitop-haltigen
Verbindung, wie das Spenderimmunglobulin bei.
-
Andere
US-Patente, die hierin durch Bezugnahme enthalten sind und welche
die Produktion von Antikörpern
lehren, die in der vorliegenden Erfindung nützlich sind, umfassen US-Patent
Nr. 5.565.332, das die Produktion chimärer Antikörper mittels eines kombinatorischen
Ansatzes beschreibt; 4.816.567, das rekombinante Immunglobulinpräparationen
beschreibt, und 4.867.973, das Konjugate aus Antikörper und
therapeutischen Mitteln beschreibt.
-
US-Patent
5.565.332 beschreibt Verfahren für
die Produktion von Antikörpern
oder Antikörperfragmenten,
welche dieselbe Bindungsspezifität
wie ein Elternantikörper
besitzen, aber verstärkte
humane Merkmale aufweisen. Humanisierte Antikörper können durch das Chain-Shuffling-Verfahren,
unter Umständen durch
Anwendung von Phagendisplaytechnologie erhalten werden. Insofern
als solche Verfahren in der vorliegenden Erfindung nützlich sind,
ist der gesamte Text von US-Patent Nr. 5.565.332 hierin durch Bezugnahme enthalten.
Humane Antikörper
können
auch hergestellt werden, indem B-Zellen mit EBV transformiert und
sezernierende Transformanden anschließend kloniert werden, wie von
Hoon et al., (1993) beschrieben wurde.
-
Antikörperkonjugate,
bei denen ein TS10q23.3-Antikörper
mit einer nachweisbaren Markierung oder einem zytotoxischen Mittel
verknüpft
ist, stellen weitere Aspekte der Erfindung dar. Diagnostische Antikörperkonjugate
können
sowohl in der in-vitro-Diagnostik als auch in verschiedenen Immunassays
und in der in-vivo-Diagnostik, beispielsweise bei Bildgebungstechnologien,
verwendet werden.
-
Bestimmte
Antikörperkonjugate
umfassen solche, die hauptsächlich
für die
Verwendung in vitro bestimmt sind, wobei der Antikörper mit
einem sekundären
Bindungsliganden oder mit einem Enzym (einem Enzymetikett) verknüpft ist,
der bzw. das bei Kontakt mit einem chromogenen Substrat ein Farbprodukt
erzeugt. Beispiele geeigneter Enzyme umfassen Urease, alkalische
Phosphatase, (Meerrettich)-Hydrogenphosphatase und Glukoseoxidase.
Bevorzugte sekundäre
Bindungsliganden sind Biotin- und Avidin- oder Streptavidinverbindungen.
Der Gebrauch solcher Marker ist einem Fachmann gut bekannt und ist
beispielsweise in US-Patent 3.817.837, 3.850.752, 3.939.350, 4.277.437,
4.275.149 und 4.366.241, die alle hierin durch Bezugnahme enthalten
sind, beschrieben.
-
Radioaktiv
markierte monoklonale Antikörper
der vorliegenden Erfindung können
nach gut bekannten Verfahren aus dem Stand der Technik produziert
werden. Beispielsweise können
monoklonale Antikörper durch
Kontakt mit Natrium- oder Kaliumiodid und einem chemischen Oxidationsmittel
wie Natriumhypochlorit oder einem enzymatischen Oxidationsmittel
wie Lactoperoxidase iodiert werden. Monoklonale Antikörper gemäß der vorliegenden
Erfindung können
durch Ligandenaustausch mit Technetium-99m markiert
werden, indem beispielsweise Pertechnat mit einer Zinnlösung reduziert
wird, das reduzierte Technetium auf einer Sephadex-Säule cheliert wird und der Antikörper auf
diese Säule
aufgetragen wird, oder durch direkte Markierungstechniken, z. B.
durch Inkubieren von Pertechnat, einem Reduktionsmittel wie SNCl2, einer Pufferlösung wie Natriumkaliumphthalat-Lösung und
dem Antikörper.
-
Intermediäre funktionelle
Gruppen, die häufig
verwendet werde, um Radioisotope, die als Metallionen vorliegen,
an Antikörper
zu binden, sind Diethylentetraaminpentaessigsäure (DTPA) und Ehylendiamintetraessigsäure (EDTA).
Fluoreszenzmarker umfassen Rhodamin, Fluoreszeinisothiocyanat und
Renographin.
-
IV. Diagnose von Krebs
in Verbindung mit TS10q23.3
-
Die
vorliegenden Erfinder haben festgestellt, dass Veränderungen
in TS10q23.3 mit Malignomen verbunden sind. Daher können TS10q23.3
und die entsprechenden Gene als ein diagnostischer oder prgnostischer
Indikator für
Krebs verwendet werden. Spezifischer könnten Punktmutationen, Deletionen,
Insertionen oder regulatorische Störungen in Verbindung mit TS10q23.3
Krebs verursachen oder die Entwicklung von Krebs fördern, Tumorprogression
an einer primären
Stelle verursachen oder fördern
und/oder Metastasierung verursachen oder fördern. Andere Phänomene in
Verbindung mit Malignomen, die von der Expression von TS10q23.3
beeinflusst sein könnten,
umfassen Angiogenese und Gewebeinvasion.
-
A. Gendiagnose
-
Eine
Ausführungsform
der vorliegenden Erfindung umfasst ein Verfahren zum Nachweis von
Variationen in der Expression von TS10q23.3 Dies kann auch die Bestimmung
des Expressionsgrades von TS10q23.3 oder die Bestimmung spezifischer
Veränderungen
des exprimierten Produktes umfassen. Diese Art von Assay hat offenkundig
Bedeutung bei der Diagnose damit in Zusammenhang stehender Krebsarten.
Solche Krebsarten können
Karzinome in Gehirn (Glioblastome, Medulloblastome, Astrozytome,
Oligodendrogliome, Ependymome), Lunge, Leber, Milz, Niere, Bauchspeicheldrüse, Dünndarm,
Blutzellen, Lymphknoten, Dickdarm, Brust, Endometrium, Magen, Prostata,
Hoden, Ovar, Haut, Kopf und Hals, Speiseröhre, Knochenmark, Blut oder
anderen- Geweben umfassen. Die vorliegende Erfindung betrifft insbesondere
die Diagnose von Gliomen.
-
Die
biologische Probe kann jedes beliebige Gewebe oder jede beliebige
Flüssigkeit
sein. Verschiedene Ausführungsformen
umfassen Zellen von Haut, Muskeln, Faszie, Gehirn, Prostata, Brust,
Endometrium, Lunge, Kopf & Hals,
Bauchspeicheldrüse,
Dünndarm,
Blutzellen, Leber, Hoden, Eierstock, Dickdarm, Haut, Magen, Speiseröhre, Milz,
Lymphknoten, Knochenmark oder Nieren. Andere Ausführungsformen
umfassen Flüssigproben
wie peripheres Blut, Lymphflüssigkeit,
Aszites, seröse
Flüssigkeit,
pleurale Effusion, Sputum, Gehirn-Rückenmark-Flüssigkeit,
Tränenflüssigkeit,
Stuhl oder Urin.
-
Verwendete
Nukleinsäure
wird in Übereinstimmung
mit Standardmethoden (Sambrook et al., 1989) aus Zellen isoliert,
die in der biologischen Probe enthalten sind. Die Nukleinsäure kann
genomische DNA oder fraktionierte oder zelluläre Gesamt-RNA sein. Wenn RNA
verwendet wird, kann es wünschenswert
sein, die RNA in eine komplementäre
DNA umzuwandeln. In einer Ausführungsform
handelt es sich bei der RNA um zelluläre Gesamt-RNA; in einer anderen
Ausführungsform
handelt es sich dabei um Poly-A-RNA. Die Nukleinsäure wird
normalerweise amplifiziert.
-
Je
nach Format wird die spezifische Nukleinsäure von Interesse in der Probe
mittels Amplifikation direkt oder nach der Amplifikation mit einer
zweiten bekannten Nukleinsäure
identifiziert. Anschließend
wird das identifizierte Produkt nachgewiesen. In bestimmten Anwendungen
kann der Nachweis visuell erfolgen (z. B. durch Ethidiumbromidfärbung eines
Gels). Alternativ kann der Nachweis die indirekte Identifizierung
des Produktes über
Chemilumineszenz, radioaktive Szintigraphie eines radioaktiven Markers
oder eines fluoreszierenden Markers oder sogar über ein System umfassen, das
elektrische oder thermische Impulssignale verwendet (Affymax Technologies;
Bellus, 1994).
-
Nach
dem Nachweis können
die Ergebnisse, die bei einem bestimmten Patienten vorhanden sind,
mit einer statistisch signifikanten Referenzgruppe normaler Patienten
und Patienten mit Pathologien in Zusammenhang mit TS10q23.3 verglichen
werden. Auf diese Art ist es möglich,
die Menge oder Art des nachgewiesenen TS10q23.3 mit verschiedenen
klinischen Stadien zu korrelieren.
-
Von
den vorliegenden Erfindern sind verschiedene Arten von Defekten
identifiziert worden. „(Ver)änderungen" sind daher so zu
verstehen, als sie Deletionen, Insertionen, Punktmutationen und
Duplikationen umfassen. Punktmutationen führen zu Stop-Codons, Frameshift-Mutationen
oder Aminosäuresubstitutionen. Somatische
Mutationen sind solche, die in Nichtkeimbahngeweben auftreten. Keimbahnmutationen
können
in jedem Gewebe auftreten und sind vererbt. Mutationen in und außerhalb
der kodierenden Region können
ebenfalls die Menge an produziertem TS10q23.3 beeinflussen, sowohl
durch Veränderung
der Transkription des Gens oder durch Destabilisierung oder anderweitige
Veränderung
der Prozessierung des Transkripts (mRNA) oder des Proteins.
-
Eine
Zelle unternimmt einen Schritt in Richtung einer onkogenen Transformation,
wenn ein Allel eines Tumorsuppressorgens aufgrund der Vererbung
einer Keimbahnläsion
oder Akquisition einer somatischen Mutation inaktiviert wird. Die
Inaktivierung des anderen Allels des Gens umfasst in der Regel eine
somatische Mikromutation oder eine chromosomale Alleldeletion, die
zu einem Verlust der Heterozygosität (LOH) führt. Alternativ können beide
Kopien eines Tumorsuppressorgens durch homozygote Deletion verloren
gehen.
-
Die
ersten Schritte der Erfinder zur Identifizierung neuer Mutationen
in TS10q23.3 waren, Primärtumoren
und Tumorzelllinien (TZL) hinsichtlich eines Heterozygositätsverlustes
(LOH) innerhalb dieser Region von 10q23 zu analysieren. Präparate von
Primärtumoren
und TZL wurden anhand von polymorphen kurzen Tandem-Sequenzwiederholungen
(Short Tandem Repeats) auf Chromosom 10 in der Nähe des TS10q23.3-Locus untersucht
(Tabelle 6). In dieser Reihe von Proben beobachteten die Erfinder
einen Heterozygositätsverlust (LOH)
in Präparaten
von Primärtumoren
mit Häufigkeiten
zwischen 20% in Colonpräparaten
bis zu 75% in Glioblastoma multiforme-Präparaten (GBM), bei einer LOH-Gesamthäufigkeit
von ~49%. Für
TZL mit einer Stichprobengrößen über neun
variierte die Inzidenz eines Heterozygositätsverlustes (LOH) zwischen
28% (Colon) bis 82% (GBM) bei einer Gesamthäufigkeit von ~46%.
-
In
Primärtumoren,
die einen Heterozygositätsverlust
(LOH) in der Umgebung des TS10q23.3-Locus aufwiesen, entdeckten
die Erfinder eine Frameshift-Mutation bei Brustkarzinom, eine Nonsense-Mutation
bei pädiatrischem
GBM, eine Splice-Variante bei pädiatrischem
GBM und eine Missense-Variante bei Melanom (Tabelle 7). Die Erfinder
untersuchten auch TZL mit Heterozygositätsverlust (LOH) und identifizierten
zehn homozygote Deletionen, die die kodierenden Regionen von TS10q23.3
beeinflussten (13A und 13B). Die
homozygoten Deletionen waren in TZL von Astrozytomen, Blasenkarzinom,
Brustkarzinom, Glioblastom, Lungenkarzinom, Melanom und Prostatakarzinom
vorhanden. Während
zwei der Zelllinien alle neun TS10q23.3-Exons verloren hatten, waren
bei den anderen acht TZL unterschiedliche kodierende Abschnitte des
Gens homozygot deletiert. Analyse der verbleibenden TZL ergab eine
Frameshift-, eine Nonsense- und sieben nicht konservative Missense-Varianten
(Tabelle 7). Diese bestimmten Mutationen könnten das Ziel von Oligonukleotiden
sein, die speziell konzipiert sind, um diese Mutationen zu identifizieren,
oder von Antikörpern, die
diese Marker von Wildtyp-TS10q23.3 unterscheiden.
-
Es
wird in Betracht gezogen, dass gemäß der vorliegenden Erfindung
andere Mutationen in dem TS10q23.3-Gen identifiziert werden können. In
dieser Hinsicht wird eine Vielzahl verschiedener Assays in Betracht
gezogen, einschließlich,
jedoch nicht ausschließlich,
Fluoreszenz-in-situ-Hybridisierung (FISH; US-Patent 5.633.365 und
US-Patent 5.665.549, jeweils hierin durch Bezugnahme enthalten),
direkte DNA-Sequenzierung, PFGE-Analyse, Southern oder Northern
Blotting, Einzelstrang-Konformationsanalyse (SSCA), RNase-Protektionsassay,
allelspezifische Oligonukleotide (ASO), Dotblot-Analyse, denaturierende
Gradienten-Gelelektrophorese (z. B. US-Patent 5.190.856, hierin
durch Bezugnahme enthalten), RFLP (z. B. US-Patent 5.324.631, hierin
durch Bezugnahme enthalten) und PCRTM-SSCP.
-
(i) Primer und Sonden
-
Der
Begriff Primer, wie hierin verwendet, soll jede Nukleinsäure umfassen,
die in der Lage ist, die Synthese einer entstehenden Nukleinsäure in einem
Template (Muster)-abhängigen
Vorgang zu starten (zu primen). Primer sind typischerweise Oligonukleotide
mit einer Länge
zwischen zehn und zwanzig Basenpaaren, aber es können auch längere Sequenzen verwendet werden.
Primer können
in Einzel- oder Doppelstrangform bereit gestellt sein, obgleich
die Einzelstrangform bevorzugt ist. Sonden sind anders definiert,
obgleich ihre Funktion auch die eines Primers sein kann. Sonden
binden an die Ziel-DNA oder -RNA und müssen nicht in einem Amplifikationsverfahren
verwendet werden, sind aber dennoch in der Lage, als Primer zu wirken.
-
In
bevorzugten Ausführungsformen
sind die Sonden oder Primer mit radioaktiven Arten (32P, 14C, 35S, 3H oder anderen Markern), mit einem Fluorophor
(Rhodamin, Fluoreszein) oder einem chemilumineszierenden Marker
(Luziferase) markiert.
-
(ii) Template (Muster)-abhängige Amplifizierungsverfahren
-
Es
ist eine Reihe von Template (Muster)-abhängigen Verfahren verfügbar, um
die in einer gegebenen Template (Muster)-Probe vorhandenen Markersequenzen
zu amplifizieren. Eines der am besten bekannten Amplifizierungsverfahren
ist die Polymerasekettenreaktion (als PCRTM bezeichnet,
die in US-Patent Nr. 4.683.185, 4.683,202 und 4.800.159 und in Innis
et al., 1990, die hierin jeweils in Gänze durch Bezugnahme enthalten
sind, ausführlich
beschrieben ist.
-
Kurz
beschrieben, werden bei der PCRTM zwei Primersequenzen
hergestellt, die zu Regionen auf gegenüber liegenden komplementären Strängen der
Markersequenz komplementär
sind. Zu einer Reaktionsmischung werden Desoxynukleosidtriphosphate
im Überschuss
zugegeben, sowie eine DNA-Polymerase, z. B. Taq-Polymerase. Wenn
die Markersequenz in einer Probe vorhanden ist, binden die Primer
an den Marker und die Polymerase bewirkt anschließend die
Verlängerung
der Primer entlang der Markersequenz durch Hinzufügen von
Nukleotiden. Indem die Temperatur der Reaktionsmischung erhöht und gesenkt
wird, lösen
sich die verlängerten
Primer von dem Marker ab, um Reaktionsprodukte zu bilden. Überschüssige Primer
binden an den Marker und an die Reaktionsprodukte, und der Vorgang
wird wiederholt.
-
Zur
Quantifizierung der Menge an amplifizierter mRNA kann ein Reverse-Transkriptase-PCRTM-Amplifikationsverfahren
durchgeführt
werden. Verfahren der reversen Transkription von RNA in cDNA sind
gut bekannt und in Sambrook et al., 1989 beschrieben. Alternative
Verfahren für
eine reverse Transkription verwenden thermostabile RNA-abhängige DNA-Polymerase.
Diese Verfahren sind in WO 90/07641 beschrieben, das am 21.Dezember
1990 beantragt wurde. Verfahren der Polymerasekettenreaktion sind
aus dem Stand der Technik gut bekannt.
-
Ein
weiteres Verfahren für
die Amplifizierung ist die Ligasekettenreaktion („LCR"), die in EPO Nr.
320 308 offenbart ist, das hierin in Gänze durch Bezugnahme enthalten
ist. Bei der LCR werden zwei komplementäre Sondenpaare hergestellt,
und in Gegenwart der Zielsequenz bindet jedes Paar derart an gegenüber liegende
komplementäre
Stränge
des Ziels, dass sie anliegen. In Gegenwart einer Ligase werden die
beiden Sondenpaare verknüpft,
um eine einzelne Einheit zu bilden. Durch Temperaturzyklen wie bei
der PCRTM lösen sich die gebunden, ligierten
Einheiten vom Ziel und dienen dann als „Zielsequenzen" für die Ligation überschüssiger Sondenpaare.
US-Patent Nr. 4.883.750 beschreibt ein Verfahren zum Binden von
Sondenpaaren an eine Zielsequenz, das der LCR gleicht.
-
Qbeta-Replikase,
beschrieben in PCT-Anmeldung Nr. PCT/US87/00880, kann ebenfalls als
weiteres Amplifikationsverfahren in der vorliegenden Erfindung verwendet
werden. In diesem Verfahren wird eine replikative Sequenz einer
RNA, die eine Region aufweist, welche komplementär zu der eines Ziel ist, in
Gegenwart einer RNA-Polymerase zu einer Probe gegeben. Die Polymerase
kopiert die replikative Sequenz, die anschließend nachgewiesen werden kann.
-
Zur
Amplifizierung von Nukleinsäuren
in der vorliegenden Erfindung eignet sich auch ein isothermisches
Amplifikationsverfahren, bei dem Restriktionsendonukleasen und Ligasen
verwendet werden, um die Amplifizierung von Zielmolekülen zu erreichen,
die Nukleotid-5'-[alpha-thio]-Triphosphate
in einem Strang einer Restriktionsschnittstelle enthalten (Walker
et al., 1992a). SDA (Strand Displacement Amplification) ist ein weiteres
Verfahren zum Durchführen
einer isothermischen Amplifizierung von Nukleinsäuren, das mehrere Zyklen einer
Strangablösung
und -synthese, d. h. Nick-Translation, enthält. Siehe US-Patent 5.270.184
und 5.455.166 und Walker et al. (1992b) hinsichtlich des SDA-Verfahrens
und Spargo et al., (1996) hinsichtlich des thermophilen SDA-Verfahrens.
-
Das
RCR-Verfahren (Repair Chain Reaction) umfasst das Anheften mehrerer
Sonden im Verlauf einer Region, die amplifiziert werden soll, gefolgt
von einer Reparaturreaktion, in der nur zwei der vier Basen vorhanden
sind. Die anderen beiden Basen können
als biotinylierte Derivate zugegeben werden, um den Nachweis zu
erleichtert. Beim SDA-Verfahren wird eine ähnliche Strategie eingeschlagen.
Zielspezifische Sequenzen können
auch anhand einer zyklischen Sondenreaktion (Cyclic Probe Reaction,
CPR) nachgewiesen werden. Beim CPR-Verfahren wird eine Sonde, die
3'- und 5'-Sequenzen unspezifischer
DNA und eine Mittelsequenz spezifischer RNA enthält, an in einer Probe vorhandene
DNA hybridisiert. Nach der Hybridisierung wird die Reaktion mit
RNase H behandelt und die Produkte der Sonde als distinkte Produkte
identifiziert, die nach dem Verdau freigesetzt werden. Das Originaltemplate
(das Originalmuster) wird an eine andere Sonde des Zyklus angeheftet
und die Reaktion wird wiederholt.
-
Gemäß der vorliegenden
Erfindung können
noch andere Amplifizierungsverfahren verwendet werden, die in der
GB-Anmeldung Nr. 2 202 328 und in der PCT-Anmeldung Nr. PCT/US89/01025
beschrieben sind, von denen jede hierin in Gänze durch Bezugnahme enthalten
ist. In ersterer Anmeldung werden „modifizierte" Primer in einer
PCRTM-ähnlichen,
Template (Muster)- und enzymabhängigen
Synthese verwendet. Die Primer können „durch
Markieren mit einer Capture-Einheit (z. B. Biotin) und/oder einer
Detektoreinheit (z. B. Enzym) modifiziert sein. In letzterer Anmeldung
werden markierte Sonden im Überschuss
zu einer Probe gegeben. In Gegenwart der Zielsequenz bindet die
Sonde und wird katalytisch gespalten. Nach dem Spalten wird die
Zielsequenz intakt freigesetzt, um von überschüssiger Sonde gebunden zu werden.
Die Spaltung der markierten Sonde signalisiert das Vorhandensein
der Zielsequenz.
-
Weitere
Nukleinsäure-Amplifizierungsverfahren
umfassen transkriptionsbasierte Amplifizierungssysteme (TAS), einschließlich nukleinsäuresequenzbasierte
Amplifizierung (NASBA) und 3SR (Kwoh et al., 1989; Gingeras et al.,
PCT-Anmeldung WO 88/10315, hierin in Gänze durch Bezugnahme enthalten).
Siehe auch US-Patent 5.409.818, Fahy et al., (1991) und Compton
(1991) hinsichtlich 3SR-Verfahren und NASBA-Verfahren. Bei NASBA-Verfahren
können
die Nukleinsäuren
durch Phenol/Chloroform-Standardextraktion, Hitzedenaturierung einer
klinischen Probe, Behandlung mit Lysepuffer und Minispin-Säulen zur
Isolierung von DNA und RNA oder Guanidiniumchloridextraktion von
RNA für
die Amplifizierung vorbereitet werden. Diese Amplifizierungstechniken
umfassen die Anheftung eines Primers mit zielspezifischen Sequenzen.
Nach der Polymerisierung werden DNA/RNA-Hybride mit RNase H verdaut,
während
doppelsträngige
DNA-Moleküle
erneut hitzedenaturiert werden. In jedem Fall wird aus der einzelsträngigen DNA
durch Addition des zweiten zielspezifischen Primers, gefolgt von
Polymerisierung, ein vollständiger
Doppelstrang. Die doppelsträngigen
DNA-Moleküle
werden dann von einer RNA-Polymerase wie T7 oder SP6 multiplikativ
transkribiert. In einer isothermischen zyklischen Reaktion werden
die RNAs in einzelsträngige
DNA revers transkribiert, welche dann in doppelsträngige DNA
umgewandelt wird und dann noch einmal mit einer RNA-Polymerase wie
T7 oder SP6 transkribiert wird. Die resultierenden Produkte, verkürzt oder
vollständig,
zeigen zielspezifische Sequenzen an.
-
Davy
et al., EPO Nr. 329/822 (hierin Gänze durch Bezugnahme enthalten)
offenbaren ein Nukleinsäureamplifizierungsverfahren,
das das zyklische Synthetisieren einsträngiger RNA („ssRNA"), ssDNA und doppelsträngiger DNA
(„dsDNA") umfasst, welche
gemäß der vorliegenden
Erfindung verwendet werden können. Die
ssRNA ist ein Template (Muster) für ein erstes Primeroligonukleotid,
das durch eine Reverse Transkriptase (RNA-abhängige DNA-Polymerase) verlängert wird. Die RNA wird anschließend aus
dem resultierenden DNA:RNA-Duplex
durch die Wirkung von Ribonuklease H (RNase H, eine RNase mit Spezifität für RNA als Duplex
mit DNA oder RNA) entfernt. Die resultierende ssDNA ist ein Template
(Muster) für
einen zweiten Primer, der 5' zu
seiner zum Template (Muster) homologen Sequenz außerdem die
Sequenzen eines RNA-Polymerase-Promotors (als Beispiel T7-RNA-Polymerase)
enthält.
Dieser Primer wird dann von der DNA-Polymerase (als Beispiel das
große „Klenow"-Fragment der DNA-Polymerase
I von E.coli) verlängert,
woraus ein doppelsträngiges
DNA-Molekül
(„sdDNA") mit einer Sequenz
resultiert, die identisch zu der der ursprünglichen RNA zwischen den Primern
ist und an einem Ende außerdem
eine Promotorsequenz aufweist. Diese Promotorsequenz kann von der
geeigneten RNA-Polymerase verwendet werden, um viele RNA-Kopien der DNA herzustellen.
Diese Kopien können
dann wieder in den Zyklus eintreten, was zu einer sehr schnellen
Amplifizierung führt.
Bei geeigneter Wahl der Enzyme kann diese Amplifizierung isotherm
durchgeführt
werden, ohne dass in jedem Zyklus Enzyme zugegeben werden müssen. Aufgrund
der zyklischen Natur dieses Vorgangs kann die Startsequenz wahlweise
die Form einer DNA oder RNA aufweisen.
-
Miller
et al., PCT-Anmeldung WO 89/06700 (hierin in Gänze durch Bezugnahme enthalten)
offenbaren ein Nukleinsäuresequenzampilfizierungsschema
auf der Basis der Hybridisierung einer Promotor/Primer-Sequenz mit
einer einzelsträngigen
Ziel-DNA („ssDNA"), gefolgt von Transkription
vieler RNA-Kopien der Sequenz. Dieses Schema ist nicht zyklisch,
d. h. neue Templates (Muster) werden nicht aus den resultierenden RNA-Transkripten
hergestellt. Weitere Amplifizierungsverfahren umfassen „RACE" und „einseitige
PCTTM" (Frohman,
1990; Ohara et al., 1989, jeweils hierin in Gänze durch Bezugnahme enthalten).
-
In
dem Amplifizierungsschritt der vorliegenden Erfindung können auch
Verfahren verwendet werden, die auf einer Ligation von zwei (oder
mehreren) Oligonukleotiden in Gegenwart von Nukleinsäure mit
der Sequenz des resultierenden „Di-Oligonukleotids" beruhen, wodurch
das Di-Oligonukleotid amplifiziert wird (Wu et al., 1989; hierin
in Gänze
durch Bezugnahme enthalten).
-
(iii) Southern/Northern
Blotting
-
Blottingtechniken
sind einem Fachmann gut bekannt. Southern Blotting umfasst die Verwendung
von DNA als Ziel, während
beim Northern Blotting RNA als ein Ziel verwendet wird. Jedes Verfahren
liefert unterschiedliche Arten von Information, obgleich das cDNA-Blotting in vielerlei
Hinsicht zum Blotting von RNA-Arten analog ist.
-
Kurz
beschrieben wird eine Sonde verwendet, um an DNA oder RNA-Arten
spezifisch zu binden, die auf einer geeigneten Matrix, häufig ein
Nitrozellulosefilter immobilisiert sind. Die unterschiedlichen Arten
sollten räumlich
getrennt sein, um eine Analyse zu erleichtern. Dies wird häufig durch
Gelelektrophorese von Nukleinsäurearten,
gefolgt vom „Blotting" auf den Filter,
erreicht.
-
Anschließend wird
das geblottete Zielmolekül
mit einer Sonde (üblicherweise
markiert) unter Bedingungen inkubiert, die eine Denaturierung und
Rehybridisierung fördern.
Da die Sonde Basenpaarungen mit dem Ziel bilden kann, bindet sie
unter Renaturierungsbedingungen einen Abschnitt der Zielsequenz.
Anschließend
wird ungebundene Sonde entfernt und der Nachweis wie oben beschrieben
durchgeführt.
-
(iv) Trennverfahren
-
In
dem einen oder anderen Stadium ist es normalerweise wünschenswert,
das Amplifikationsprodukt vom Template (Muster) und dem überschüssigen Primer
zu trennen um zu bestimmen, ob eine spezifische Amplifikation stattgefunden
hat. In einer Ausführungsform
werden Amplifikationsprodukte durch Agarose-, Agaroseacrylamid-
oder Polyacrylamidgelelektrophorese mittels Standardverfahren getrennt.
Siehe Sambrook et al., 1989.
-
Alternativ
können
zur Durchführung
einer Trennung chromatographische Techniken angewandt werden. Es
gibt viele Chromatographiearten, die in der vorliegenden Erfindung
verwendet werden können:
Adsorption, Partition, Ionenaustausch und Molekularsieb, und viele
spezialisierte Techniken zu deren Anwendung, darunter Säulen-, Papier-,
Dünnschicht-
und Gaschromatographie (Freifelder, 1982).
-
(v) Nachweisverfahren
-
Zur
Bestätigung
der Amplifikation der Markersequenzen können Produkte visualisiert
werden. Ein typisches Visualisierungsverfahren umfasst das Färben eines
Gels mit Ethidiumbromid und Visualisierung unter UV-Licht. Wenn
die Amplifizierungsprodukte integral mit radioaktiv oder fluorometrisch
markierten Nukleotiden markiert sind, können die Amplifizierungsprodukte
nach der Trennung alternativ einem Röntgenfilm ausgesetzt oder im
geeigneten anregenden Spektrum visualisiert werden.
-
In
einer Ausführungsform
wird die Visualisierung indirekt erreicht. Nach der Auftrennung
von Amplifizierungsprodukten wird eine markierte Nukleinsäuresonde
in Kontakt mit der amplifizierten Markersequenz gebracht. Die Sonde
ist vorzugsweise mit einem Chromophor konjugiert, kann aber auch
radioaktiv markiert sein. In einer anderen Ausführungsform ist die Sonde mit
einem Bindungspartner konjugiert, beispielsweise einem Antikörper oder
Biotin und der andere Partner des Bindungspaars trägt eine
nachweisbare Einheit.
-
In
einer Ausführungsform
erfolgt der Nachweis durch eine markierte Sonde. Die diesbezüglichen
Techniken sind einem Fachmann gut bekannt und sind in vielen Standardbüchern über molekulare
Protokolle enthalten. Siehe Sambrook et al., 1989.
-
Beispielsweise
identifizieren ein Chromophor oder radioaktiv markierte Sonden oder
Primer das Ziel während
oder nach der Amplifizierung.
-
Ein
Beispiel für
das Vorangehende ist in US-Patent Nr. 5.279.721 beschrieben, das
hierin durch Bezugnahme enthalten ist, welches einen Apparat und
ein Verfahren für
automatisierte Elektrophorese und Transfer von Nukleinsäuren offenbart.
Der Apparat erlaubt Elektrophorese und Blotting ohne externe Manipulation
des Gels und eignet sich ideal, um Verfahren gemäß der vorliegenden Erfindung
durchzuführen.
-
Die
oben beschriebenen Amplifizierungsprodukte können darüber hinaus einer Sequenzanalyse
anhand von Sequenzanalyse-Standardtechniken unterzogen werden, um
spezielle Arten von Variationen zu identifizieren. Mithilfe bestimmter
Verfahren wird eine erschöpfende
Analyse von Genen mittels Sequenzanalyse durchgeführt, bei
der Primersätze
verwendet werden, die für
eine optimale Sequenzierung konzipiert sind (Pignon et al., 1994).
Die vorliegende Erfindung sieht Verfahren vor, durch die beliebige
oder alle dieser Analysearten angewandt werden können. Unter Verwendung der
hierin offenbarten Sequenzen können
Oligonukleotidprimer konzipiert werden, um die Amplifizierung von
Sequenzen im gesamten TS10q23.3-Gen zu ermöglichen, das dann durch direkte
Sequenzierung analysiert werden kann.
-
(vi) Kitbestandteile
-
Alle
wichtigen Materialien und Reagenzien, die für Nachweis und Sequenzierung
von TS10q23.3 und von Varianten davon benötigt werden, können gemeinsam
in einem Kit zusammengestellt werden. Dieses umfasst im Allgemeinen
im Voraus ausgewählte
Primer und Sonden. Es können
auch Enzyme enthalten sein, die zur Amplifizierung von Nukleinsäuren geeignet
sind, einschließlich
verschiedener Polymerasen (RT, Taq, Sequenase TM,
etc.), Desoxynukleotide und Puffer, um die notwendige Reaktionsmischung
für eine
Amplifizierung bereit zu stellen. Solche Kits umfassen im Allgemeinen
und in geeigneten Mitteln getrennte Behälter für jedes einzelne Reagens und
Enzym sowie für
jeden Primer oder für
jede Sonde.
-
(vii) Design und theoretische
Gesichtspunkte der relativen quantitativen RT-PCRTM
-
Reverse
Transkription (RT) von RNA zu cDNA, gefolgt von relativer quantitativer
PCRTM (RT-PCRTM) lässt sich
verwenden, um die relativen Konzentrationen spezifischer, aus Patienten
isolierter mRNA-Arten zu bestimmen. Indem bestimmt wird, dass die
Konzentration einer spezifischen mRNA-Art variiert, wird gezeigt, dass
das Gen, welches die spezifische mRNA kodiert, differenziell exprimiert
wird.
-
Bei
der PCRTM erhöht sich die Anzahl an Molekülen der
amplifizierten Ziel-DNA mit jedem Reaktionszyklus um einen Faktor
von nahezu zwei, bis einige Reagenzien limitierend werden. Danach
geht die Amplifikationsrate immer mehr zurück, bis keine Erhöhung des
amplifizierten Ziels zwischen den Zyklen mehr stattfindet. Wenn
ein Schaubild aufgezeichnet wird, in dem die Zyklusnummer auf der
X-Achse und der Logarithmus der Konzentration der amplifizierten
Ziel-DNA auf der Y-Achse aufgetragen sind, bildet sich bei Verbinden der
aufgetragenen Punkte eine Kurve mit charakteristischer Form. Beginnend
mit dem ersten Zyklus ist die Steigung der Kurve positiv und konstant.
Dies gilt als linearer Abschnitt der Kurve. Nachdem ein Reagens
limitierend wird, beginnt die Steigung der Kurve abzunehmen und
wird schließlich
null. An diesem Punkt nähert sich
die Konzentration der amplifizierten Ziel-DNA asymptotisch einem
festen Wert an. Dies gilt als Plateau-Abschnitt der Kurve.
-
Die
Konzentration der Ziel-DNA im linearen Abschnitt der PCRTM-Amplifizierung ist direkt proportional zu
der Startkonzentration der Ziel-DNA vor Reaktionsbeginn. Indem die
Konzentration der amplifizierten Produkte der Ziel-DNA in PCRTM-Reaktionen, welche dieselbe Anzahl von
Zyklen abgeschlossen haben und sich in ihren linearen Bereichen
befinden, bestimmt wird, ist es möglich, die relativen Konzentrationen
der spezifischen Ziel-Sequenz in der ursprünglichen DNA-Mischung zu bestimmen.
Wenn es sich bei den DNA-Mischungen um cDNAs handelt, die aus RNAs
synthetisiert worden sind, die aus unterschiedlichen Geweben oder
Zellen isoliert wurden, kann für
die jeweiligen Gewebe oder Zellen die relative Häufigkeit der spezifischen mRNA, von
der die Zielsequenz abstammt, bestimmt werden. Diese direkte Proportionalität zwischen
der Konzentration der PCRTM-Produkte und
den relativen mRNA-Häufigkeiten
tritt nur im linearen Bereich der PCRTM-Reaktion
auf.
-
Die
Endkonzentration der Ziel-DNA im Plateau-Abschnitt der Kurve wird
durch die Verfügbarkeit
von Reagenzien in der Reaktionsmischung bestimmt und ist unabhängig von
der ursprünglichen
Konzentration an Ziel-DNA. Die erste Bedingung, die erfüllt sein
muss, bevor die relativen Häufigkeiten
einer mRNA-Art für
eine Sammlung von RNA-Populationen durch RT-PCRTM bestimmt
werden kann, ist, dass die Konzentrationen der amplifizierten PCRTM-Produkte geprüft werden müssen, wenn die PCRTM-Reaktionen
sich im linearen Abschnitt ihrer Kurven befinden.
-
Die
zweite Bedingung, die erfüllt
sein muss, damit in einem RT-PCRTM-Experiment
die relativen Häufigkeiten
einer bestimmten mRNA-Art erfolgreich bestimmt werden kann, ist,
dass relative Konzentrationen der amplifizierbaren cDNAs gegenüber einem
unabhängigen
Standard normalisiert werden müssen.
Das Ziel eines RT-PCRTM-Experimentes ist
es, die Häufigkeit
einer bestimmten mRNA-Art relativ zu der mittleren Häufigkeit
aller in der Probe vorhandenen mRNA-Arten zu bestimmen. In den unten
beschriebenen Experimenten wurden mRNAs für β-Aktin, Asparaginsynthetase und Lipocortin
II als externe und interne Standards verwendet, mit denen die relative
Häufigkeit
anderer mRNAs verglichen werden.
-
Die
meisten Protokolle für
kompetitive PCRTM verwenden interne PCRTM-Standards, die ungefähr genauso häufig sind
wie das Zielmolekül.
Diese Strategien sind effektiv, wenn die Produkte der PCRTM-Amplifizierungen während ihrer linearen Phasen
geprüft
werden. Werden die Produkte geprüft,
wenn die Reaktionen in die Nähe
der Plateau-Phase kommen, ist das weniger häufige Produkt relativ überrepräsentiert.
Vergleiche relativer Häufigkeiten
vieler verschiedener RNA-Proben, wie es beispielsweise bei der Untersuchung
von RNA-Proben auf differenzielle Expression der Fall ist, werden
so verzerrt, dass Unterschiede der relativen Häufigkeiten von RNAs geringer
erscheinen, als sie tatsächlich
sind. Dies ist kein signifikantes Problem, wenn der interne Standard
viel häufiger
ist als das Ziel. Wenn der interne Standard häufiger ist als das Ziel, kann zwischen
RNA-Proben ein direkter linearer Vergleich vorgenommen werden.
-
Die
obige Erläuterung
beschreibt theoretische Gesichtspunkte eines RT-PCRTM-Assays
für Materialien klinischer
Herkunft. Die Probleme, die mit klinischen Proben einhergehen, sind,
dass sie von variabler Menge (was eine Normalisierung problematisch
macht) und von variabler Qualität
sind (was die Co-Amplifizierung einer zuverlässigen internen Kontrolle erforderlich
macht, die vorzugsweise größer ist
als das Ziel). Diese beiden Probleme werden überwunden, wenn die RT-PCRTM als eine relative quantitative RT-PCRTM mit einem internen Standard durchgeführt wird,
bei welcher der interne Standard ein amplifizierbares cDNA-Fragment
ist, das größer als
das cDNA-Zielfragment ist, und bei der die Häufigkeit der mRNA, die für den internen
Standard kodiert, ungefähr
5-100-fach größer ist
als die der mRNA, die für
das Ziel kodiert. Dieser Assay misst relative Häufigkeiten, keine absoluten
Häufigkeiten
der jeweiligen mRNA-Arten.
-
Es
können
andere Studien durchgeführt
werden, in denen ein konventionellerer relativ quantitativer RT-PCRTM-Assay mit einem externen Standard angewandt
wird. Diese Assays prüfen
die PCRTM-Produkte im linearen Abschnitt
ihrer Amplifizierungskurven. Die Anzahl an PCRTM-Zyklen,
die für
die Prüfung
optimal sind, muss für
jedes cDNA-Zielfragment empirisch bestimmt werden. Darüber hinaus
müssen
die Reverse-Transkriptase-Produkte jeder RNA-Population, die aus den verschiedenen
Gewebeproben isoliert worden ist, sorgfältig auf gleiche Konzentrationen
amplifizierbarer cDNAs normalisiert werden. Dieser Gesichtspunkt
ist sehr wichtig, da der Assay absolute mRNA-Häufigkeit misst. Absolute mRNA-Häufigkeit
kann nur in normalisierten Proben als Maß einer differenziellen Genexpression
herangezogen werden. Während
eine empirische Bestimmung des linearen Bereichs der Amplifizierungskurve
und eine Normalisierung von cDNA-Präparationen mühevolle
und zeitaufwändige
Vorgänge
sind, sind die resultierenden RT-PCRTM-Assays
unter Umständen
solchen, die vom relativen quantitativen RT-PCRTM-Assay
mit einem internen Standard abgeleitet sind, überlegen sein.
-
Ein
Grund für
diesen Vorteil ist, dass ohne den internen Standard/Kompetitor alle
Reagenzien im linearen Bereich der Amplifizierungskurve in ein einzelnes
PCRTM-Produkt umgewandelt werden können, was
die Empfindlichkeit des Assays erhöht. Ein anderer Grund ist,
dass bei nur einem PCRTM-Produkt die Darstellung des
Produktes in einem Elektrophoresegel oder einem anderen Anzeigeverfahren
weniger komplex wird, weniger Hintergrund aufweist und einfacher
zu interpretieren ist.
-
(viii) Chip-Technologien
-
Von
den vorliegenden Erfindern werden chipbasierte DNA-Technologien
wie die von Hacia et al., (1996) und Shoemaker et al., (1996) beschriebenen
spezifisch in Betracht gezogen Kurz beschrieben, umfassen diese
Techniken quantitative Verfahren für das schnelle und genaue Analysieren
einer großen
Anzahl von Genen. Indem Gene mit Oligonukleotiden markiert oder
feste Sondenanordnungen verwendet werden, lässt sich die Chip-Technologie
anwenden, um Zielmoleküle
als hochdichte Anordnungen zu isolieren und diese Moleküle auf der
Basis von Hybridisierung zu testen. Siehe auch Pease et al., (1994),
Fodor et al., (1991).
-
B. Immundiagnose
-
Antikörper der
vorliegenden Erfindung können
verwendet werden, um den TS10q23.3-Gehalt gesunder und erkrankter Gewebe
durch Techniken wie ELISA-Verfahren und Western Blotting zu charakterisieren. Dies
könnte
sich als Prüftest
für das
Vorliegen oder Nichtvorliegen einer Malignität oder als Prädiktor einer
zukünftigen
Krebserkrankung erweisen.
-
Es
wird die Verwendung von Antikörpern
der vorliegenden Erfindung in einem ELISA-Assay in Betracht gezogen. Beispielsweise
sind die Anti-TS10q23.3-Antikörper
auf einer ausgewählten
Fläche
immobilisiert, vorzugsweise auf einer Fläche, die eine Proteinaffinität aufweist,
wie beispielsweise die Vertiefungen einer Mikrotiterplatte aus Polystyrol.
Nachdem gewaschen wird, um unvollständig adsorbiertes Material
zu entfernen, ist es wünschenswert,
ein unspezifisches Protein an die Assayplatte zu binden oder die
Assayplatte mit einem unspezifischen Protein zu beschichten, von
dem bekannt ist, dass es im Hinblick auf die Testantiseren neutral
ist, beispielsweise Rinderserumalbumin (BSA), Casein oder Trockenmilchlösungen.
Dies erlaubt eine Blockierung unspezifischer Adsorptionsstellen
auf der immobilisierenden Fläche
und reduziert so den Hintergrund, der durch unspezifische Bindung
von Antigen an die Fläche
hervorgerufen wird.
-
Nach
dem Binden von Antikörper
an die Vertiefung, Beschichten mit einem nicht-reaktiven Material zur Reduzierung von
Hintergrund und Waschen zum Entfernen ungebundenen Materials wird
die immobilisierende Fläche
mit der zu testenden Probe so in Kontakt gebracht, das sich Immunkomplexe
(Antigen/Antikörper) bilden
können.
-
Nach
der Bildung spezifischer Immunkomplexe zwischen der Testprobe und
dem gebundenen Antikörper
und anschließendem
Waschen kann das Vorkommen und selbst die Menge der gebildeten Immunkomplexe
bestimmt werden, indem dieselben einem zweiten Antikörper mit
Spezifität
für TS10q23.3
ausgesetzt werden, der sich vom ersten Antikörper unterscheidet. Geeignete
Bedingungen umfassen vorzugsweise das Verdünnen der Probe mit Verdünnungsmitteln
wie BSA, bovinem Gammaglobulin (BGG) und phosphatgepufferter Salzlösung (PBS)/Tween®.
Diese zugegebenen Mittel unterstützen
außerdem
die Reduzierung des Hintergrunds. Die Antiserumschicht kann anschließend etwa
2 bis 4 Stunden inkubieren, bei Temperaturen, die vorzugsweise im
Berech von etwa 25° bis
etwa 27° C
liegen. Nach der Inkubation wird die mit Antiserum kontaktierte
Fläche
gewaschen, um Material zu entfernen, das keine Immunkomplexe gebildet
hat. Ein bevorzugtes Waschverfahren umfasst das Waschen mit einer
Lösung
wie PBS/Tween® oder
Boratpuffer.
-
Zur
Bereitstellung eines Nachweismittels weist der sekundäre Antikörper vorzugsweise
ein mit ihm verknüpftes
Enzym auf, das bei Inkubation mit einem geeigneten chromogenen Substrat
eine Farbentwicklung erzeugt. Es könnte beispielsweise erwünscht sein,
die an den zweiten Antikörper
gebundene Fläche
eine Zeit lang und unter Bedingungen, welche die Entwicklung der
Bildung von Immunkomplexen begünstigen,
mit einem Unease- oder Peroxidase-konjugierten Anti-Human-IgG zu
inkubieren (z. B. Inkubation für
2 Stunden bei Raumtemperatur in einer PBS-haltigen Lösung wie
beispielsweise PBS/Tween®).
-
Nach
der Inkubation mit dem zweiten enzymmarkierten Antikörper und
nach dem Waschen zum Entfernen von ungebundenem Material wird die
Menge des Markers durch Inkubation mit einem chromogenen Substrat
wie Harnstoff und Bromkresolviolett oder 2,2'-Azino-di-(3-ethyl-benzthiazolin)-sulfonsäure (ABTS) und
H2O2 quantifiziert,
wenn der Enzymmarker Peroxidase ist. Eine Quantifizierung wird anschließend erreicht, indem
der Grad der Farbstoffbildung gemessen wird, z. B. indem ein Spektrophotometer
mit Licht im sichtbaren Spektrum verwendet wird.
-
Das
vorhergehende Format kann geändert
werden, indem zuerst die Probe an die Assayplatte gebunden wird.
Anschließend
wird der Primärantikörper mit
der Assayplatte inkubiert, gefolgt vom Nachweis gebundenen Primärantikörpers durch
Verwendung eines markierten Sekundärantikörpers mit Spezifität für den Primärantikörper.
-
In
der wissenschaftlichen Literatur sind die Schritte verschiedener
anderer geeigneter Immunnachweisverfahren beschrieben worden, beispielsweise
in Nakamura et al., (1987; hierin durch Bezugnahme enthalten). Im
einfachsten und direktesten Sinn sind Immunassays Bindungsassays.
Bestimmte bevorzugte Immunassays sind die verschiedenen Arten von
RAdioimmunassays (RIA) und Immunobead-Capture-Assays. Besonders
geeignet ist auch der immunhistochemische Nachweis anhand von Gewebeschnitten.
Anerkannterweise ist der Nachweis jedoch nicht auf solche Techniken
beschränkt,
und Western Blotting, Dot Blotting, FACS-Analyse und dergleichen
können
in Verbindung mit der vorliegenden Erfindung verwendet werden.
-
Die
Antikörperzusammensetzung
der vorliegenden Erfindung findet umfassende Verwendung bei der Immunblotanalyse
oder Western-Blot-Analyse. Die Antikörper können als hoch affine Primäneagenzien
zur Identifizierung von Proteinen verwendet werden, die auf einer
festen Trägermatrix
immobilisiert sind, beispielsweise auf Nitrozellulose, Nylon oder
Kombinationen davon. In Verbindung mit einer Immunpräzipitation,
gefolgt von einer Gelelektrophorese, können sie als Einzelschrittreagenzien
zur Anwendung beim Nachweis von Antigenen eingesetzt werden, gegen
die beim Nachweis des Antigens verwendete Sekundärreagenzien einen unerwünschten
Hintergrund verursachen. Zu den auf immunologischen Prinzipien basierenden
Nachweismethoden zum Gebrauch in Verbindung mit Western Blotting
gehören
enzymatisch, radioaktiv oder fluoreszent markierte Sekundärantikörper gegen
die Toxinhälfte
gelten in dieser Hinsicht als von besonderem Nutzen. US-Patente,
welche die Verwendung solcher Marker betreffen, umfassen 3.817.837,
3.850.752, 3.939.350, 3.996.345, 4.277.437, 4.275.149 und 4.366.241,
die alle hierin durch Bezugnahme enthalten sind. Selbstverständlich können sich
durch den Gebrauch eines sekundären
Bindungsliganden wie beispielsweise einem Sekundärantikörper oder einer Biotin/Avidin-Ligandenbindungsanordnung,
wie aus dem Stand der Technik bekannt ist, weitere Vorteile ergeben.
-
V. Verfahren zur Testung
aktiver Verbindungen
-
Die
vorliegende Erfindung zieht ferner die Verwendung von TS10q23.3
und aktiver Fragmente und von dafür kodierenden Nukleinsäuren bei
der Testung von Verbindungen hinsichtlich einer Aktivität bei der
Stimulierung der Aktivität
von TS10q23.3, bei der Aufhebung des Mangels an TS10q23.3 oder bei
der Blockierung der Wirkung eines mutanten TS10q23.3-Moleküls in Erwägung. Diese
Assays können
viele verschiedene Formate anwenden und können auf der Art von „Aktivität" beruhen, für welche
der Test durchgeführt
wird. In Betracht gezogene funktionelle „Ausgabeformen" umfassen das Binden
an eine Verbindung, das Hemmen der Bindung an ein Substrat, einen
Liganden oder einen anderen Bindungspartner durch eine Verbindung,
Phosphataseaktivität,
Anti-Phosphatase-Aktivität,
Phosphorylierung von TS10q23.3, Dephosphorylierung von TS10q23.3,
Hemmung oder Stimulation der Signalweiterleitung von Zelle zu Zelle,
Wachstum, Metastasierung, Zellteilung, Zellmigration, Koloniebildung
in Weichagar, Kontakthemmung, Invasivität, Angiogenese, Apoptose, Tumorprogression
oder andere maligne Phänotypen.
-
Das
Polypeptid der Erfindung kann auch zum Testen von Verbindungen verwendet
werden, die als ein Ergebnis einer Technologie unter Verwendung
einer kombinatorischen Bibliothek entwickelt worden sind. Die Technologie
unter Verwendung einer kombinatorischen Bibliothek bietet eine effiziente
Möglichkeit
der Testung einer potenziell großen Anzahl unterschiedlicher
Substanzen hinsichtlich ihrer Fähigkeit
zur Modulierung der Aktivität
eines Polypeptids. Solche Bibliotheken und ihre Verwendung sind
aus dem Stand der Technik bekannt. Die Verwendung von Peptidbibliotheken
ist bevorzugt. Siehe beispielsweise SO97/02048.
-
Kurz
beschrieben, kann ein Verfahren zum Testen auf eine Substanz, welche
die Aktivität
eines Polypeptids moduliert, das Inkontaktbringen einer oder mehrerer
Testsubstanzen mit dem Polypeptid in einem geeigneten Reaktionsmedium,
das Testen der Aktivität
des behandelten Polypeptids und das Vergleichen dieser Aktivität mit der
Aktivität
des Polypeptids in einem vergleichbaren Reaktionsmedium, das nicht
mit der Testsubstanz oder den Testsubstanzen behandelt worden ist,
umfassen. Ein Unterschied der Aktivität zwischen den behandelten
und unbehandelten Polypeptiden zeigt einen modulierenden Effekt
der relevanten Testsubstanz oder der relevanten Testsubstanzen an.
-
Vor
einer Testung hinsichtlich einer Modulierung der Aktivität oder zusätzlich zu
einer solchen Testung können
Testsubstanzen hinsichtlich ihrer Fähigkeit zur Wechselwirkung
mit dem Polypeptid geprüft
werden, z. B. in einem Hefe-Zwei-Hybrid-System (z. B. Bartel et
al., 1993, Fields und Song, 1989), Chevray und Nathans, 1992, Lee
et al., 1995). Dieses System kann als grobes Auswahlwerkzeug vor
der Testung einer Substanz auf ihre eigentliche Fähigkeit
zur Modulierung der Aktivität
des Polypeptids verwendet werden. Alternativ könnte das System verwendet werden,
um Testsubstanzen auf Bindung an einen spezifischen KVLQT1- oder KCNE1-Bindungspartner
zu testen oder um Mimetika des KVLQT1- oder KCNE1-Polypeptids zu
finden.
-
Nach
der Identifizierung einer Substanz, welche die Polypeptidaktivität moduliert
oder beeinflusst, kann die Substanz weiter untersucht werden. Des
Weiteren kann sie hergestellt und/oder bei der Zubereitung, d. h.
Herstellung oder Formulierung, oder einer Zusammensetzung wie beispielsweise
eines Medikamentes, einer pharmazeutischen Zusammensetzung oder
eines Arzneimittels verwendet werden. Diese können an Individuen verabreicht
werden.
-
Die
vorliegende Erfindung betrifft also in verschiedenen Aspekten nicht
nur eine Substanz, die in Übereinstimmung
mit dem hierein Offenbarten anhand eines Nukleinsäuremoleküls als Modulator
einer Polypeptidaktivität
identifiziert worden ist, sondern auch eine pharmazeutische Zusammensetzung,
ein Medikament, ein Arzneimittel oder eine andere Zusammensetzung,
die eine solche Substanz umfasst, ein Verfahren, das die Verabreichung
einer solchen Zusammensetzung, die eine solche Substanz umfasst,
umfasst. ein Verfahren, das die Verabreichung einer solchen Zusammensetzung
an einen Patienten umfasst, z. B. zur Behandlung (die auch eine
präventive
Behandlung einschließen
kann) von LQT, die Verwendung einer solchen Substanz bei der Herstellung
einer Zusammensetzung zur Verabreichung, z. B. zur Behandlung von
LQT, und ein Verfahren zum Herstellen einer pharmazeutischen Zusammensetzung
umfassend das Mischen solch einer Substanz mit einem pharmazeutisch
annehmbaren Exzipienten, Vehikel oder Trägerstoff und wahlweise anderen
Inhaltsstoffen.
-
A. In-Vitro-Assays
-
In
einer Ausführungsform
soll die Erfindung zum Testen von Verbindungen verwendet werden,
die an das TS10q23.3-Molekül
oder Fragmente davon bindet. Das Polypeptid oder Fragment kann entweder
frei in Lösung,
an einen Träger
fixiert, in einer Zelle oder auf der Oberfläche einer Zelle exprimiert
sein. Entweder das Polypeptid oder die Verbindung können markiert
sein, was eine Feststellung der Bindung erlaubt.
-
In
einer anderen Ausführungsform
kann der Assay die Hemmung der Bindung von TS10q23.3 an ein natürliches
oder künstliches
Substrat oder an natürliche
oder künstliche
Bindungspartner messen. Es können kompetitive
Bindungsassays durchgeführt
werden, in denen eines der Mittel (TS10q23.3, Bindungspartner oder
Verbindung) markiert ist. In der Regel ist das Polypeptid markiert.
Mann kann die Menge an freiem Marker gegenüber der Menge an gebundenem
Marker messen, um eine Bindung oder eine Hemmung der Bindung festzustellen.
-
Eine
andere Technik zum Testung von Verbindungen mit hohem Durchsatz
ist in WO 84/03564 beschrieben. Eine große Anzahl von kleinen Peptidtestverbindungen
wird auf einem festen Substrat wie Kunststoffstiften oder einer
anderen Fläche
synthetisiert. Die Peptidtestverbindungen werden mit TS10q23.3 umgesetzt
und gewaschen. Gebundenes Polypeptid wird anhand verschiedener Verfahren
nachgewiesen.
-
Gereinigtes
TS10q23.3 kann direkt auf Platten beschichtet werden, die in den
zuvor erwähnten
Arzneimittelprüftechniken
verwendet werden. Nicht neutralisierende Antikörper gegen das Polypeptid können jedoch
verwendet werden, um das Polypeptid an einer festen Phase zu immobilisieren.
Außerdem
können
Fusionsproteine mit einer reaktiven Region (vorzugsweise einer terminalen
Region) verwendet werden, um die aktive Region von TS10q23.3 mit
einer Festphase zu verknüpfen.
-
Er
können
verschiedene Zelllinien mit Wildtyp oder natürlichen oder gentechnisch eingeführten Mutationen
in TS10q23.3 verwendet werden, um verschiedene funktionelle Attribute
von TS10q23.3 zu untersuchen und wie eine Kandidatenverbindung diese
Attribute beeinflusst. Verfahren zum gentechnischen Einführen von
Mutationen sind an anderer Stelle in diesem Dokument beschrieben,
wie auch natürlich
auftretende Mutationen in TS10q23.3, die zu Malignomen führen, zu
Malignomen beitragen und/oder Malignome auf andere Art und Weise
verursachen. In solchen Assays würde
die Verbindung in Anbetracht ihrer biologischen Natur geeignet formuliert
sein und mit einer Zielzelle in Kontakt gebracht. Je nach Assay
kann eine Kultur erforderlich sein. Die Zelle kann anschließend mittels
einer Reihe unterschiedlicher physiologischer Assays untersucht werden.
Alternativ kann eine molekulare Analyse durchgeführt werden, in welcher die
Funktion von TS10q23.3 oder verwandter Pfade untersucht werden kann.
Dies kann Assays umfassen, wie beispielsweise solche zur Testung
von Proteinexpression, Enzymfunktion, Substratnutzung, Phosphorylierungszustand
verschiedener Moleküle,
einschließlich
TS10q23.3, cAMP-Mengen, mRNA-Expression (einschließlich differentielles
Display oder Gesamt-RNA oder polyA-RNA) und Anderen.
-
B. In-Vivo-Assays
-
Die
vorliegende Erfindung umfasst auch die Verwendung verschiedener
Tiermodelle. Hier bietet die Identität zwischen humanem und murinem
TS10q23.3 eine hervorragende Möglichkeit,
die Funktion von TS10q23.3 in einem vollständigen Tiersystem zu untersuchen,
in dem es normalerweise exprimiert wird. Es können durch Entwicklung oder
Isolierung mutanter Zelllinien, die kein normales TS10q23.3 exprimieren
können,
Krebsmodelle in Mäusen
erzeugt werden, die für
Krebserkrankungen beim Menschen und anderen Säugetieren hoch prädiktiv sind.
Diese Modelle können
die orthotopische oder systemische Verabreichung von Tumorzellen
anwenden, um primäre
und/oder metastatische Krebserkrankungen nachzuahmen. Alternativ
kann Krebs in Tieren ausgelöst
werden, indem Mittel bereitgestellt werden, von denen bekannt ist,
dass sie für
bestimmte Ereignisse in Zusammenhang mit maligner Transformation
und/oder Tumorprogression verantwortlich sind. Schließlich können transgene
Tiere (unten erläutert),
denen Wildtyp-TS10q23.3 fehlt, als Modelle für Krebsentwicklung und -behandlung
verwendet werden.
-
Behandlung
von Tieren mit Testverbindungen umfasst die Verabreichung der Verbindung
in geeigneter Form an das Tier. Die Verabreichung kann auf jedem
Weg erfolgen, der für
klinische oder nicht klinische Zwecke genutzt werden kann, einschließlich, aber
nicht ausschließlich,
oral, nasal, bukkal, rektal, vaginal oder topisch. Alternativ kann
die Verabreichung durch intratracheale Instillation, bronchiale
Instillation, intradermale, subkutane, intramuskuläre, intraperitoneale
oder intravenöse
Injektion erfolgen. Speziell in Erwägung gezogen werden intravenöse Injektion,
regionale Verabreichung über
Blut- und Lymphversorgung und intratumorale Injektion.
-
Die
Bestimmung der Wirksamkeit einer Verbindung in vivo kann viele verschiedene
Kriterien einschließen.
Solche Kriterien umfassen Überleben,
Verminderung der Tumorlast oder Tumormasse, Stillstand oder Verlangsamung
der Tumorprogression, Eliminierung von Tumoren, Inhibition oder
Prävention
von Metastasierung, erhöhter
Aktivitätsgrad,
Verbesserung der Immuneffektorfunktion und verbesserte Nahrungsaufnahme, sind
aber nicht darauf beschränkt.
-
C. Rationales Wirkstoffdesign
-
Das
Ziel des rationalen Wirkstoffdesigns ist es, strukturelle Analoga
biologisch aktiver Polypeptide oder Verbindungen, mit denen sie
wechselwirken (Agonisten, Antagonisten, Inhibitoren, Bindungspartner, etc.),
herzustellen. Durch Erzeugung solcher Analoga ist es möglich, Wirkstoffe
zu schaffen, die aktiver oder stabiler als die natürlichen
Moleküle
sind, die unterschiedliche Empfindlichkeit gegenüber Veränderungen sind oder die die
Funktion verschiedener anderer Moleküle beeinflussen können. In
einem Ansatz würde
eine dreidimensionale Struktur für
TS10q23.3 oder ein Fragment davon erzeugt werden. Dies könnte durch
Röntgenkristallographie,
Computer-Modellierung oder eine Kombination beider Ansätze erreicht
werden. Ein alternativer Ansatz, das „Alanin-Scan"-Verfahren, umfasst
den zufallsbasierten Austausch von Resten im gesamten Molekül durch
Alanin, wobei im Anschluss die resultierende Wirkung auf die Funktion
bestimmt wird.
-
Es
ist außerdem
möglich,
einen TS10q23.3-spezifischen Antikörper zu isolieren, der durch
einen funktionellen Assays ausgewählt worden ist, und dann seine
Kristallstruktur aufzuklären.
Im Prinzip ergibt dieser Ansatz ein Pharmakophor, auf dessen Basis
späteres
Wirkstoffdesign erfolgen kann. Es ist möglich, Proteinkristallographie
ganz zu umgehen, indem Anti-Idiotyp-Antikörper gegen einen funktionellen,
pharmakologisch aktiven Antikörper
erzeugt werden. Als Spiegelbild eines Spiegelbildes wäre die Bindungsstelle
eines Anti-Idiotyp-Antikörpers ein
Analog des ursprünglichen
Antigens. Der Anti-Idiotyp-Antikörper
könnte
dann verwendet werden, um Peptide aus Banken chemisch oder biologisch
produzierter Peptide zu identifizieren und zu isolieren. Ausgewählte Peptide
würden
dann als der Pharmakophor dienen. Anti-Idiotyp-Antikörper können erzeugt werden,
indem die hierin für
die Produktion von Antikörpern
beschriebenen Verfahren verwendet werden, wobei ein Antikörper als
das Antigen eingesetzt wird.
-
Es
können
somit Wirkstoffe konzipiert werden, die verbesserte TS10q23.3-Aktivität aufweisen,
oder die als Stimulatoren, Inhibitoren, Agonisten, Antagonisten
von TS10q23.3 oder von Molekülen,
die von der Funktion von TS10q23.3 beeinflusst werden, wirken. Durch
die Verfügbarkeit
von klonierten TS10q23.3-Sequenzen können ausreichende Mengen von
TS10q23.3 produziert werden, um kristallographische Studien durchzuführen. Darüber hinaus
erlaubt die Kenntnis der Polypeptidsequenzen eine rechnerunterstützte Vorhersage
von Struktur-Funktions-Zusammenhängen.
-
Eine
Substanz, die als ein Modulator einer Polypeptidfunktion identifiziert
wird, kann von ihrer Art her ein Peptid oder ein Nichtpeptid sein.
Für viele
pharmazeutische Anwendungen in vivo sind kleine Nicht-Peptid-Moleküle bevorzugt.
Entsprechend kann ein Mimetikum oder ein Imitator der Substanz (vor
allem, wenn es sich um ein Peptid handelt) für die pharmazeutische Anwendung
konzipiert werden.
-
Das
Design von Mimetika einer bekannten pharmazeutisch aktiven Verbindung
ist ein bekannter Ansatz für
die Entwicklung von Pharmazeutika auf der Basis einer Leitverbindung.
-
Dies
kann wünschenswert
sein, wenn die aktive Verbindung schwierig oder kostspielig zu synthetisieren
ist oder wenn sie für
ein bestimmtes Verabreichungsverfahren ungeeignet ist, z. B. sind
reine Peptidasen ungeeignete aktive Mittel für orale Zusammensetzungen,
da sie dazu neigen, von Proteasen im Verdauungskanal schnell abgebaut
zu werden. Design, Synthese und Testung von Mimetika werden im Allgemeinen
verwendet, um eine zufallsbasierte Testung einer großen Anzahl
von Molekülen
hinsichtlich einer Zieleigenschaft zu vermeiden.
-
Das
Design eines Mimetikums ausgehend von einer Verbindung mit einer
bestimmten Zieleigenschaft erfolgt üblicherweise in mehreren Schritten.
Zunächst
werden die bestimmten Teile der Verbindung bestimmt, die bei der
Festlegung der Zieleigenschaft ausschlaggebend und/oder wichtig
sind. Im Fall eines Peptids kann dies erfolgen, indem systematisch
die Aminosäurereste
in dem Peptid variiert werden, z. B. indem jeder Rest abwechselnd
substituiert wird. Häufig
werden Alanin-Scans von Peptiden verwendet, um solche Peptidmotive zu
redefinieren. Diese Teile oder Reste, welche die aktive Region der
Verbindung ausmachen, sind als ihr „Pharmakophor" bekannt.
-
Wenn
der Pharmakophor gefunden ist, wird anhand seiner physikalischen
Eigenschaften, z. B. Stereochemie, Bindung, Größe und/oder Ladung, unter Verwendung
von Daten aus einer Reihe von Quellen, z. B. spektroskopischen Techniken,
Röntgenbeugungsdaten
und NMR, ein Modell seiner Struktur erstellt. Bei diesem Vorgang
der Modellerstellung können
rechnerunterstützte
Analyse, Kartierung von Ähnlichkeiten
(welche ein Modell der Ladung und/oder des Volumens eines Pharmakophors
anstelle der Bindung zwischen Atomen erstellen) und andere Techniken
verwendet werden.
-
In
einer Variante dieses Ansatzes wird ein Modell der dreidimensionalen
Struktur des Liganden und seines Bindungspartners erstellt. Dies
kann vor allem dann nützlich
sein, wenn der Ligand und/oder sein Bindungspartner bei der Bindung
eine Konformationsänderung
durchlaufen, was es dem Modell ermöglicht, dieses beim Design
des Mimetikums zu berücksichtigen.
-
Anschließend wird
ein Template (Muster)-Molekül
ausgewählt,
auf das chemische Gruppen, die den Pharmakophor nachahmen, aufgesetzt
werden können.
Das Template(Muster)-Molekül und die
darauf aufgesetzten chemischen Gruppen können praktischerweise so gewählt werden,
dass das Mimetikum einfach zu synthetisieren ist, wahrscheinlich
pharmazeutisch akzeptabel ist und in vivo nicht zerfällt, während die
biologische Aktivität
der Leitverbindung erhalten bleibt. Wenn das Mimetikum peptidbasiert
ist, kann alternativ die Stabilität erhöht werden, indem das Peptid
zyklisch gemacht wird, was seine Steifigkeit erhöht. Das Mimetikum oder die
Mimetika, die durch diesen Ansatz gefunden werden, können dann
analysiert werden um zu prüfen, ob
oder in welchem Ausmaß sie
die Zieleigenschaft aufweisen. Danach kann eine weitere Optimierung
oder Modifikation durchgeführt
werden, um schließlich
eines oder mehrere endgültige
Mimetika für
die Testung in vivo oder für
klinische Tests zu erhalten.
-
VI. Verfahren zum Behandeln
von in Zusammenhang mit 10q23.3 stehenden Malignomen
-
Die
vorliegende Erfindung umfasst in einer anderen Ausführungsform
außerdem
die Behandlung von Krebs. Die Arten von Krebserkrankungen, die gemäß der vorliegenden
Erfindung behandelt werden könnten, sind
nur durch die Beteiligung von TS10q23.3 beschränkt. Mit Beteiligung ist gemeint,
dass es nicht einmal eine Voraussetzung ist, dass TS10q23.3 mutiert
oder anomal ist – die Überexpression
dieses Tumorsuppressors kann faktisch andere Läsionen in der Zelle überwinden.
Es wird daher in Betracht gezogen, eine breite Vielzahl von Tumoren
mit einer TS10q23.3-Therapie zu behandeln, einschließlich Krebserkrankungen
in Gehirn (Glioblastom, Astrozytom, Oligodendrogliom, Ependymome),
Lunge, Leber, Milz, Niere, Lymphknoten, Bauchspeicheldrüse, Dünndarm,
Blutzellen, Dickdarm, Magen, Brust, Endometrium, Prostata, Hoden,
Eierstock, Kopf und Hals, Speiseröhre, Knochenmark, Blut oder
andere Gewebe.
-
In
vielen Fällen
ist es nicht erforderlich, dass die Tumorzelle abgetötet wird
oder ein normaler Zelltod oder eine „Apoptose" der Tumorzelle induziert wird. Um eine
sinnvolle Behandlung zu erreichen, ist lediglich erforderlich, dass
das Tumorwachstum bis zu einem gewissen Grad verlangsamt wird. Es
kann jedoch sein, dass das Tumorwachstum vollständig blockiert oder eine gewisse
Tumorregression erreicht wird. Es wird auch klinische Terminologie
wie „Remission" und „Verringerung
der Tumor"-Last
in Betracht gezogen, wenn sie wie üblich gebraucht wird.
-
A. Genbasierte Therapien
-
Eine
der therapeutischen Ausführungsformen,
die von den vorliegenden Erfindern in Betracht gezogen werden, ist
der Eingriff in die Ereignisse, die an der Tumorgenese einiger Krebserkrankungen
beteiligt sind, auf molekularer Ebene. Speziell beabsichtigen die
vorliegenden Erfinder, ein Expressionskonstrukt für eine Krebszelle
bereit zu stellen, das dieser Zelle TS10q23.3 liefern kann. Aufgrund
der Sequenzhomologie zwischen den Genen von Mensch, Maus und Hund
könnte
jede dieser Nukleinsäuren
bei der Therapie von Menschen verwendet werden, genauso wie jede
der oben erläuterten
Gensequenzen, die dasselbe oder ein biologisch äquivalentes Polypeptid kodieren.
Die ausführliche
Diskussion von Expressionsvektoren und der hierin verwendeten genetischen
Elemente ist in diesem Abschnitt durch Bezugnahme enthalten. Besonders
bevorzugte Expressionsvektoren sind virale Vektoren, wie Adenovirus,
Adeno-assoziiertes Virus, Herpesvirus, Vakziniavirus und Retrovirus.
Außerdem
bevorzugt ist ein liposomal verkapselter Expressionsvektor.
-
Einem
Fachmann ist gut bekannt, wie ein Verfahren der Einführung von
Genen (Gen-Delivery)
in vivo und ex vivo angewandt wird. Für virale Vektoren wird man
im Allgemeinen einen viralen Vektorstamm herstellen. Je nach Art
des Virus und des zu erhaltenden Titers werden dem Patienten 1 × 104, 1 × 105, 1 × 106, 1 × 107, 1 × 108, 1 × 109, 1 × 1010, 1 × 1011 oder 1 × 1012 infektiöse Partikel
verabreicht. Ähnliche
Zahlen können für liposomale
oder andere nichtvirale Formulierungen extrapoliert werden, indem
relative Aufnahme-Effizienzen verglichen werden. Eine Formulierung
als eine pharmazeutisch annehmbare Komposition ist unten erläutert.
-
Für verschiedene
Tumortypen werden verschiedene Verabreichungswege in Betracht gezogen.
Der Abschnitt unten, in dem es um Verabreichungswege geht, enthält eine
ausführliche
Liste an möglichen
Verabreichungswegen. Eine systemische Verabreichung wird praktisch
für jeden
Tumor in Betracht gezogen. Dies würde sich vor allem für das Angreifen
von mikroskopischem oder metastatischem Krebs als wichtig erweisen. Wo
eine diskrete Tumormasse identifiziert werden kann, können viele
verschiedene direkte, lokale und regionale Ansätze ergriffen werden. Beispielsweise
kann der Expressionsvektor direkt in den Tumor injiziert werden. Ein
Tumorbett kann vor, während
oder nach einer Resektion behandelt werden. Nach einer Resektion
würde der
Vektor im Allgemeinen über
einen Katheter verabreicht werden, der nach der Operation an Ort
und Stelle verbleibt. Es könnte
die Tumorvaskulatur verwendet werden, um den Vektor in den Tumor
einzuführen,
indem in eine zuführende
Vene oder eine Arterie injiziert wird. Es könnte auch der Weg über eine
distalere Blutzufuhr verwendet werden.
-
In
einer anderen Ausführungsform
wird eine Gentherapie ex vivo in Betracht gezogen. Dieser Ansatz ist
besonders geeignet für
die Behandlung von Krebserkrankungen in Verbindung mit Knochenmark,
ist aber nicht beschränkt
darauf. In einer ex-vivo-Ausführungsform
werden dem Patienten Zellen entnommen und außerhalb des Körpers für mindestens
eine Zeit lang erhalten. Während
dieses Zeitraums wird eine Therapie verabreicht, nach der die Zellen
wieder in den Patienten verbracht werden; Hoffentlich alle Tumorzelle
in der Probe wurden dabei abgetötet.
Ein autologes Knochenmarktransplantat (ABMT) ist ein Beispiel einer
Gentherapie ex vivo. Die Idee hinter einem ABMT ist im Grunde, dass
der Patient als sein eigener Knochenmarkspender fungiert. Damit
kann dem Patienten eine normalerweise tödliche Dosis an Strahlung oder
Chemotherapie verabreicht werden, um die Tumorzellen abzutöten, und
das Knochenmark mit den eigenen Zellen des Patienten neu besiedelt
werden, die ex vivo erhalten (und möglicherweise expandiert) worden
sind. Da Knochenmark häufig
mit Tumorzellen kontaminiert ist, ist es wünschenswert, diese Zellen aus
dem Knochenmark zu beseitigen. Die Verwendung von Gentherapie, um
dieses Ziel zu erreichen, ist eine weitere Art und Weise, wie TS10q23.3
gemäß der vorliegenden
Erfindung genutzt werden könnte.
-
Bei
der Ausübung
der Gentherapieverfahren der vorliegenden Erfindung können Gentransfersysteme nützlich sein,
die aus dem Stand der Technik bekannt sind. Dazu gehören virale
und nicht-virale Transferverfahren. Eine Reihe von Viren ist als
Gentransfervektoren oder als Basis für reparierende Gentransfervektoren verwendet
worden, darunter Papovaviren, (z. B. SV40, Madzak et al.,1992),
Adenovirus (Berkner, 1992; Berkner et al., 1988; Gorziglia und Kapikian,
1992; Quantin et al., 1992; Rosenfeld et al., 1992; Wilkinson und
Akrigg, 1992; Stratford-Perricaudet et al., 1990; Schneider et al.,
1998), Vakziniavirus (Moss, 1992; Moss, 1996), Adenoassoziiertes
Virus (Muzyczka, 1992; Ohi et al., 1990; Russen und Hirata, 1998),
Herpes-Viren, einschließlich
HSV und EBV (Margolskee, 1992; Johnson et al., 1992; Fink et al.,
1992; Breakefield und Geller, 1987; Freese et al., 1990; Fink et
al., 1996), Lentiviren (Naldini et al., 1996), Sindbis- und Semliki-Forest-Virus (Berglund
et al., 1993) und Retroviren des Vogels (Bandyopadhyay und Temin,
1984; Petropoulos et al., 1992), der Maus (Miller, 1992; Miller
et al., 1985; Sorge et al., 1984; Mann und Baltimore, 1985; Miller
et al., 1988) und des Menschen (Shimada et al., 1991; Helseth et
al., 1990; Page et al., 1990; Buchschacher und Panganiban, 1992).
-
Nicht-virale
Gentransferverfahren, die aus dem Stand der Technik bekannt sind,
umfassen chemische Techniken wie Kalziumphosphat-Copräzipitation
(Graham und van der Eb, 1973; Pellicer et al., 1980), mechanische
Techniken, wie beispielsweise Mikroinjektion (Anderson et al., 1980;
Gordon et al., 1980; Brinster et al., 1981; Costantini und Lacy,
1981), Membranfusions-vermittelter Transfer über Liposomen (FeIgner et al.,
1987; Wang und Huang, 1989; Kaneda et al., 1989; Stewart et al.,
1992; Nabel et al., 1990; Lim et al., 1991) und direkte DNA-Aufnahme
und rezeptorvermittelter DNA-Transfer (Wolff et al., 1990; Wu et
al., 1991; Zenke et al., 1990; Wu et al., 1989; Wolff et al., 1991;
Wagner et al., 1990; Wagner et al., 1991; Cotten et al., 1990; Curiel et
al., 1992; Curiel et al., 1991). Virusvermittelter Gentransfer kann
durch Verwendung einer liposomalen Verabreichung mit einem direkten
in-vivo-Gentransfer kombiniert werden, was es ermöglicht,
dass die viralen Vektoren zu den Tumorzellen und nicht in die umliegenden,
sich nicht teilenden Zellen geleitet werden. Alternativ kann die
Zelllinie, die den retroviralen Vektor produziert, in Tumoren injiziert
werden (Culver et al., 1992). Die Injektion von Produzentenzellen
würde dann
eine kontinuierliche Quelle von Vektorpartikeln bereitstellen. Diese
Technik ist für
die Verwendung am Menschen bei inoperablen Gehirntumoren zugelassen.
-
In
einem Ansatz, der biologische und physikalische Gentransferverfahren
kombiniert, wird eine Plasmid-DNA beliebiger Größe mit einem Polylysin-konjugierten
Antikörper
kombiniert, der spezifisch gegen das Adenovirushexonprotein gerichtet
ist, und der resultierende Komplex wird an einen Adenovirusvektor
gebunden. Der trimolekulare Komplex wird dann verwendet, um Zellen
zu infizieren. Der Adenovirusvektor erlaubt eine effiziente Bindung,
Internalisierung und Degradation des Endosoms, bevor die gekoppelte
DNA geschädigt
wird. Zu anderen Techniken für
die Verabreichung von Adenovirus-basierten Vektoren siehe Schneider
et al., (1998) und US-Patent Nr. 5.691.198m 5.747.469, 5.436.146
und 5.753.500.
-
Es
ist gezeigt worden, dass Liposom/DNA-Komplexe in der Lage sind,
einen direkten Gentransfer in vivo zu vermitteln. Während der
Vorgang des Gentransfers in Liposomenstandardpräparationen unspezifisch ist,
wurden eine lokalisierte Aufnahme in vivo und Expression bei Tumorablagerungen
berichtet, beispielsweise direkt nach einer Verabreichung in situ
(Nabel, 1992).
-
Expressionsvektoren
in Zusammenhang mit Gentherapie sollten solche Konstrukte enthalten,
die Sequenzen aufweisen, welche ausreichend sind, um ein Polynukleotid
zu exprimieren, das dort hinein kloniert worden ist. In viralen
Expressionsvektoren enthält
das Konstrukt virale Sequenzen, die ausreichend sind, um eine Verpackung
des Konstruktes zu unterstützen.
Wenn das Polynukleotid ein Antisense-Polynukleotid oder ein Ribozym
kodiert, wird durch die Expression das Antisense-Polynukleotid oder
-Ribozym produziert. In diesem Zusammenhang erfordert eine Expression
daher nicht, dass ein Proteinprodukt synthetisiert wird. Zusätzlich zu
dem in den Expressionsvektor klonierten Polynukleotid enthält der Vektor
auch einen Promotor, der in eukaryontischen Zellen funktional ist.
Die klonierte Polynukleotidsequenz steht unter der Kontrolle dieses
Promotors. Geeignete eukaryontische Promotoren umfassen solche,
die oben beschrieben sind. Der Expressionsvektor kann auch Sequenzen,
wie beispielsweise selektierbare Marker und andere hierin beschriebene
Sequenzen, umfassen.
-
B. Immuntherapien
-
Immuntherapien
beruhen im Allgemeinen auf der Verwendung von Immuneffektorzellen
und -molekülen,
um Krebszellen gezielt anzugreifen und zu zerstören. Der Immuneffektor kann
beispielsweise ein Antikörper
sein, der für
einen Marker auf der Oberfläche
einer Tumorzelle spezifisch ist. Der Antikörper kann alleine als ein Effektor
der Therapie wirken oder er kann andere Zellen hinzuziehen, um die
Zelle zu töten.
Der Antikörper
kann auch mit einem Arzneimittel oder Toxin konjugiert sein (chemotherapeutisch,
Radionuklid, Ricin-A-Kette, Choleratoxin, Pertussis-Toxin, etc.)
und nur als Mittel zum Targeting dienen. Alternativ kann der Effektor
ein Lymphozyt sein, der ein Oberflächenmolekül trägt, welches entweder direkt
oder indirekt mit einem Tumorzellziel interagiert. Die verschiedenen
Effektorzellen umfassen zytotoxische T-Zellen und NK-Zellen.
-
Gemäß der vorliegenden
Erfindung ist es unwahrscheinlich, dass TS10q23.3 ein Ziel für einen
Immuneffektor darstellen könnte,
da es (i) wahrscheinlich nicht an der Oberfläche der Zelle exprimiert ist
und (ii) das Vorhandensein, nicht das Nichtvorhandensein, von TS10q23.3
mit dem normalen Zustand verbunden ist. Es ist aber möglich, dass
bestimmte mutante Formen von TS10q23.3 ein Ziel einer Immuntherapie
darstellen können,
bei der entweder Antikörper,
Antikörperkonjugate
oder Immuneffektorzellen verwendet werden.
-
Ein
wahrscheinlicheres Szenario ist, dass eine Immuntherapie als Teil
einer kombinierten Therapie in Verbindung mit einer auf TS10q23.3
abzielenden Gentherapie verwendet wird. Der allgemeine Ansatz für eine kombinierte
Therapie ist unten erläutert.
Im Allgemeinen muss die Tumorzelle einen Marker tragen, der als
Ziel dienen kann, d. h. auf der Mehrzahl anderer Zellen nicht vorhanden
ist. Es existieren viele Tumormarker und jeder davon könnte sich
für ein
Targeting in Zusammenhang mit der vorliegenden Erfindung eignen.
Gängige Tumormarker
umfassen Karzinoembryonales Antigen, Prostata-spezifisches Antigen,
Tumor-assoziiertes Harnantigen, fetales Antigen, Tyrosinase (p97),
gp68, TAG-72, HMFG, Sialyl-Lewis-Antigen, MucA, MucB, PLAP, Östrogenrezeptor,
Lamininrezeptor, erb B und p155.
-
Immunkonjugate.
-
Die
Erfindung sieht ferner Immuntoxine vor, bei denen ein Antikörper, der
an einen Krebsmarker bindet, beispielsweise an ein mutantes TS10q23.3,
mit einem zytotoxischen Mittel verknüpft ist. Immuntoxintechnologie
ist relativ weit fortgeschritten und einem Fachmann gut bekannt.
Immuntoxine sind Mittel, in denen der Antikörperbestandteil mit einem anderen
Mittel verknüpft
ist, insbesondere mit einem zytotoxischen oder anderweitig antizellulären Mittel,
das die Fähigkeit
besitzt, das Wachstum oder die Zellteilung zu unterdrücken oder
die Zelle zu töten.
-
Wie
hierin verwendet, werden die Begriffe „Toxin" und „Toxineinheit" verwendet, um sich
auf jedes zytotoxische oder anderweitig antizelluläre Mittel
zu beziehen, das eine solche tödliche
oder suppressive Eigenschaft aufweist. Toxine sind also pharmakologische
Mittel, die mit einem Antikörper
konjugiert sein und einer Zelle in aktiver Form verabreicht werden
können,
wo sie eine erhebliche schädigende
Wirkung ausüben.
-
Die
Herstellung von Immuntoxinen ist aus dem Stand der Technik im Allgemeinen
gut bekannt (siehe z.B. US-Patent 4.340.535, das hierin durch Bezugnahme
enthalten ist). Es ist außerdem
bekannt, dass auf Fab'-Fragmenten
basierende Immuntoxine im Allgemeinen bessere Gewebe durchdringende
Eigenschaft haben als IgG-basierte Immuntoxine, während IgG-basierte
Immuntoxine typischerweise bessere Bindungsfähigkeiten aufweisen und aus
dem Blut langsamer entfernt werden als ihre Fab'-Gegenstücke.
-
Beispielhafte
antizelluläre
Mittel umfassen chemotherapeutische Mittel, Radioisotope sowie Zytotoxine.
Beispiele für
chemotherapeutische Mittel sind Hormone wie Steroide, Antimetabolite
wie Cytosinarabinosid, Fluoruracil, Methotrexat oder Aminopterin,
Anthracyclin, Mitomycin C, Vincaalkaloide, Demecolcin, Etoposid,
Mithramycin oder alkylierende Mittel wie Chlorambucil oder Melphalan.
-
Bevorzugte
Immuntoxine enthalten häufig
ein von Pflanzen, Pilzen oder Bakterien abgeleitetes Toxin, wie
beispielsweise ein A-Ketten-Toxin, ein Ribosom inaktivierendes Protein, α-Sarcin,
Aspergillin, Restirictocin, eine Ribonuklease, Diphtherie-Toxin
oder Pseudomonas-Exotoxin,
um nur einige Beispiele zu nennen. Die Verwendung von Toxin-Antikörper-Konstrukten sowie
deren Anbringung an Antikörper
sind aus dem Stand der Technik über
Immuntoxine gut bekannt. Natürlich
können
auch Kombinationen der verschiedenen Toxine an ein Antikörpermolekül gekoppelt
werden, wodurch eine variable oder sogar verstärkte Zytotoxizität ermöglicht wird.
-
Eine
Art von Toxin zur Anbringungen von Antikörpern ist Ricin, wobei die
deglykosylierte Ricin-A-Kette besonders bevorzugt ist. Wie hierin
verwendet, soll ich der Begriff „Ricin" auf Ricin beziehen, das aus natürlichen
Quellen und mit rekombinanten Mitteln hergestellt ist. Einem Fachmann
sind verschiedene „rekombinante" oder „gentechnisch
hergestellte" Formen
des Ricinmoleküls
bekannt, von denen alle gemäß der vorliegenden Erfindung
verwendet werden können.
-
Die
deglykosylierte Ricin-A-Kette (dgA) ist aufgrund ihrer extremen
Potenz und längeren
Halbwertszeit bevorzugt, sowie deshalb, weil sie in klinisch erforderlichem
Reinheitsgrad und erforderlicher Menge kostengünstig hergestellt werden kann
(kommerziell von Inland Laboratories, Austin, TX, USA verfügbar). Auch
eine verkürzte
Ricin-A-Kette, von der die 30 N-terminalen
Aminosäuren
durch Nagase (Sigma) entfernt worden sind, kann verwendet werden.
-
Verknüpfen oder
Koppeln einer oder mehrerer Toxinhälften an einen Antikörper kann
durch verschiedene Mechanismen erreicht werden, beispielsweise durch
kovalente Bindung, Affinitätsbindung,
Interkalierung, koordinative Bindung und Komplexbildung. Bevorzugte
Bindungsverfahren sind solche, die kovalente Bindung umfassen, wie
beispielsweise durch Verwendung von chemischen Vernetzungsmitteln,
natürlichen Peptiden
oder Disulfidbrücken.
-
Die
kovalente Bindung kann entweder durch direkte Kondensation vorhandener
Seitenketten oder durch Einbau externer Überbrückungsmoleküle erreicht werden. Viele bivalente
oder polyvalente Mittel sind beim Koppeln von Proteinmolekülen an andere
Proteine, Peptide oder Aminfunktionen geeignet. Beispiele von Kopplungsmitteln
sind Carbodiimide, Diisocyanate, Glutaraldehyd, Diazobenzole und
Hexamethylendiamine. Diese Liste soll die verschiedenen Kopplungsmittel,
die aus dem Stand der Technik bekannt sind, nicht erschöpfend wiedergeben,
sondern die gängigeren
Kopplungsmittel, die verwendet werden können, exemplarisch wiedergeben.
-
In
bevorzugten Ausführungsformen
wird in Betracht gezogen, dass gewünscht sein könnte, zuerst
den Antikörper
zu derivatisieren und dann den Toxinbestandteil an das derivatisierte
Produkt anzubringen. Wie hierin verwendet, wird der Begriff „derivatisiert" werden, um die chemische
Modifikation des Antikörpersubstrates
mit einem geeigneten Vernetzungsmittel zu beschreiben. Beispiele
für Vernetzungsmittel
zur Verwendung auf diese Art umfassen die Disulfidbrücken enthaltenden
Linker SPDP (N-Succinimidyl-3-(2-pyridyldithio)propionat)
und SMPT (4-Succinimidyl-oxycarbonyl-α-methyl-α(2-pyridyldithio)toluol).
-
Biologisch
lösbare
Bindungen sind bei der Umsetzung eines klinisch aktiven Immuntoxins
in die Praxis insofern wichtig, als es möglich sein muss, dass die Toxineinheit
von dem Antikörper
freigesetzt wird, nachdem er in die Zielzelle gelangt ist. Es sind
zahlreiche Arten von Verknüpfungskonstrukten
bekannt, einschließlich einfach
direkte Disulfidbrückenbildung
zwischen Sulihydrylgruppen, die an Aminosäuren wie Cystein vorhanden
sind oder anderweitig in entsprechende Proteinstrukturen eingeführt wurden,
und Disulfidverknüpfungen anhand
verfügbarer
oder künstlich
hergestellter Linkereinheiten.
-
Es
sind zahlreiche Arten von Disulfidbrücken enthaltenden Linkern bekannt,
die erfolgreich verwendet werden können, um Toxineinheiten an
Antikörper
zu konjugieren, während
bestimmte Linker jedoch bevorzugt sind; so sind beispielsweise sterisch
behinderte Disulfidbrückenlinker
aufgrund ihrer höheren
Stabilität
in vivo bevorzugt, wodurch sie die Freisetzung der Toxineinheit
vor der Bindung an den Wirkort verhindern. Ein besonders bevorzugtes
Vernetzungsreagens ist SMPT, obgleich andere Linker wie SATA, SPDP
und 2-Iminothiolan
ebenfalls verwendet werden können.
-
Nach
der Konjugation ist es wichtig, das Konjugat zu reinigen, um verunreinigende
Stoffe wie beispielsweise unkonjugierte A-Kette oder Antikörper zu
entfernen. Aufgrund der Möglichkeit
einer erhöhten
Toxizität
ist es wichtig, unkonjugierte A-Kette zu entfernen. Darüber hinaus
ist es wichtig, unkonjugierten Antikörper zu entfernen, um die Möglichkeit
einer kompetitiven Bindung des Antigens an konjugierte und unkonjugierte
Arten zu vermeiden. In jedem Fall gibt es eine Reihe von Reinigungstechniken,
um Konjugate in ausreichendem Reinheitsgrad bereit zu stellen, um
sie klinisch verwendet zu können.
-
Die
am meisten bevorzugte Technik beinhaltet im Allgemeinen die Verwendung
von Blue-Sepharose mit einem Gelfiltrations- oder Gelpermeationsschritt.
Blue-Sepharose ist eine Säulenmatrix,
die aus Cibacron Blue 3GA und Agarose zusammengesetzt ist und sich
bei der Reinigung von Immunkonjugaten als nützlich erwiesen hat. Die Verwendung
von Blue-Sepharose
kombiniert die Eigenschaften eines Ionenaustausches mit A-Ketten-Bindung,
um eine gute Trennung konjugierter und unkonjugierter Bindungen
zu liefern. Blue-Sepharose erlaubt die Beseitigung des freien (unkonjugierten)
Antikörpers
aus der Konjugatzubereitung. Es kann ein Chromatographieschritt
des molekularen Ausschlusses verwendet werden, um das freie (unkonjugierte)
Toxin (z. B. dgA) zu entfernen, indem entweder ein herkömmliches
Gelfiltrationsverfahren oder eine HPLC (High Performance Liquid
Chromatograhy) verwendet wird.
-
Nachdem
ein ausreichend gereinigtes Konjugat hergestellt worden ist, wäre es im
Allgemeinen wünschenswert,
dieses in Form einer pharmazeutischen Zusammensetzung zuzubereiten,
die parenteral verabreicht werden kann. Dies wird dadurch erreicht,
indem für
den letzten Reinigungsschritt ein Medium mit einer geeigneten pharmazeutischen
Zusammensetzung verwendet wird. Solche Formulierungen enthalten
typischerweise pharmazeutische Puffer, zusammen mit Exzipienten,
Stabilisatoren und dergleichen. Die pharmazeutischen Zusammensetzungen
sind steril, nicht-immunogen und nicht-pyrogen. Einzelheiten ihrer
Zubereitung sind aus dem Stand der Technik gut bekannt und hierin
weiter beschrieben. Endotoxinverunreinigung sollte minimal auf einem
unbedenklichen Niveau gehalten werden, beispielsweise bei unter
0,5 ng/mg Protein.
-
Erfindungsgemäße geeignete
pharmazeutische Zusammensetzungen umfassen im Allgemeinen von etwa
10 bis etwa 100 mg des gewünschten
Konjugates in einer Mischung mit einem annehmbaren pharmazeutischen
Verdünnungsmittel
oder Exzipienten, wie beispielsweise einer sterilen wässrigen
Lösung,
um in Bezug auf das Konjugat eine Endkonzentration von etwa 0,25
bis etwa 2,5 mg/ml zu erhalten.
-
Wie
oben erwähnt,
können
die Antikörper
der Erfindung mit einem oder mehreren chemotherapeutischen Mittel
verknüpft
werden, wie beispielsweise Anti-Tumor-Wirkstoffen, Zytokinen, Antimetaboliten,
alkylierenden Mitteln, Hormonen, Nukleinsäuren und dergleichen, die so
mittels des Antikörperkonjugates
zielgerichtet zu einer TS10q23.3-exprimierenden Krebszelle geleitet
werden können.
Der Vorteil Antikörper-konjugierter Mittel
gegenüber
ihren nicht-Antikörper-konjugierten
Gegenstücken
ist die durch den Antikörper
vermittelte höhere
Selektivität.
-
Bei
der Analyse der unterschiedlichen chemotherapeutischen und pharmakologischen
Mittel, die für die
Konjugation an einen Antikörper
zur Verfügung
stehen, würde
man insbesondere solche in Betracht ziehen, von denen bereits gezeigt
worden ist, dass sie erfolgreich mit Antikörpern konjugiert werden können und
pharmakologisch wirken. Beispielhafte antineoplastische Mittel,
die verwendet worden sind, umfassen Doxorubicin, Daunomycin, Methotrexat,
Vinblastin. Darüber
hinaus wurde auch die Anbringung anderer Mittel wie Neokarzinostatin,
Macromycin, Trnimon und α-Amantin
beschrieben. Die hierin präsentierten
Listen geeigneter Mittel sind natürlich lediglich beispielhaft
insofern, als die Technologie zum Anbringen pharmazeutischer Mittel
an Antikörper
für die
spezifische Verabreichung an Gewebe gut etabliert ist.
-
Es
wird daher im Allgemeinen angenommen, dass es möglich ist, jedes pharmakologische
Mittel an Antikörper
zu konjugieren, das eine primäre
oder sekundäre
Amingruppe, Hydrazid- oder
Hydrazingruppe, Carboxylalkoholgruppe, Phosphatgruppe oder alkylierende
Gruppe aufweist, die für
die Bindung oder Vernetzung mit den Aminosäuren oder Kohlenhydratgruppen
des Antikörper
verfügbar
ist. In Falle von Proteinstrukturen wird dies am einfachsten mit
einem Vernetzungsmittel erreichet, wie oben für die Immuntoxine beschrieben.
Eine Anbringung kann auch mit einer säurelabilen Acylhydrazon- oder
cis-Aconitrylverknüpfung
zwischen dem Wirkstoff und dem Antikörper erreichet werden oder
indem ein Peptid Abstandshalter wie beispielsweise L-Leu-L-Ala-L-Leu-L-Ala
zwischen der γ-Carbonylgruppe
des Wirkstoffes und einer Aminosäure
des Antikörpers
verwendet wird.
-
C. Proteintherapie
-
Ein
weiterer Therapieansatz ist die Bereitstellung von TS10q23.3-Polypeptid,
aktiven Fragmenten, synthetischen Peptiden, Mimetika oder anderen
Analoga davon an ein Individuum. Das Protein kann durch rekombinante
Expressionsmittel produziert werden oder, falls es klein genug ist,
von einem automatisierten Peptidsynthesegerät erzeugt werden. Formulierungen
würden
je nach Verabreichungsweg und Zweck ausgewählt werden, einschließlich, aber
nicht beschränkt
auf, liposomale Formulierungen und klassische pharmazeutische Zubereitungen.
-
D. Kombinierte Therapie
mit Immuntherapie, klassischer Chemo- und Strahlentherapie
-
Tumorzellresistenz
gegen DNA-schädigende
Mittel stellt ein großes
Problem in der klinischen Onkologie dar. Ein Ziel derzeitiger Krebsforschung
ist es, Wege zu finden, um die Wirksamkeit von Chemo- und Strahlentherapie
zu verbessern. Eine Möglichkeit
ist die Kombination solcher traditioneller Therapien mit Gentherapie.
Beispielsweise induzierte das Herpes-Simplex-Thymidinkinase (HS-tk)-Gen
nach Verabreichung an Gehirntumoren mittels eines retroviralen Vektorsystems
Empfindlichkeit gegenüber
dem Antivirusmittel Ganciclovir (Culver et al., 1992). In Zusammenhang
mit der vorliegenden Erfindung wird in Betracht gezogen, dass TS10q23.3-Substitutionstherapie
auf ähnliche
Weise in Verbindung mit einer chemo- oder strahlentherapeutischen
Intervention verwendet werden könnte.
Es könnte
sich auch als wirksam erweisen, eine TS10q23.3-Gentherapie mit einer
Immuntherapie zu kombinieren, wie oben beschrieben ist.
-
In
der Regel würde
man im Allgemeinen eine „Ziel"-Zelle mit einem
TS10q23.3-Expressionskonstrukt und
mindestens einem anderen Mittel in Kontakt bringen, um Zellen durch
Anwendung der Verfahren und Zusammensetzungen der vorliegenden Erfindung
zu töten,
Zellwachstum zu hemmen, Metastasierung zu hemmen, Angiogenese zu
hemmen oder den malignen Phänotyp
von Tumorzellen anderweitig umzukehren oder zu reduzieren. Diese
Zusammensetzungen würden
in einer kombinierten Menge bereitgestellt werden, die wirksam ist,
um die Zelle zu töten
oder die Proliferation der Zelle zu hemmen. Dieser Vorgang kann
das Inkontaktbringen der Zellen mit dem Expressionskonstrukt und
gleichzeitig mit dem Mittel/den Mitteln oder dem Faktor/den Faktoren
umfassen. Dies kann erreicht werden, indem die Zelle mit einer einzigen
Zusammensetzung oder pharmakologischen Formulierung in Kontakt gebracht
wird, die beide Mittel enthält,
oder indem die Zelle gleichzeitig mit zwei verschiedenen Zusammensetzungen
oder Formulierungen in Kontakt gebracht wird, wobei eine Zusammensetzung
das Expressionskonstrukt und die Andere das Mittel enthält.
-
Alternativ
kann die Gentherapiebehandlung der Behandlung mit einem anderen
Mittel in Abständen, die
von Minuten zu Wochen reichen können,
vorangehen oder folgen. In Ausführungsformen,
in denen das andere Mittel und das Expressionskonstrukt separat
auf die Zelle angewandt werden, würde man im Allgemeinen sicherstellen,
dass zwischen dem Zeitpunkt jeder Verabreichung kein signifikanter
Zeitraum verstreicht, so dass das Mittel und das Expressionskonstrukt
dennoch in der Lage wären,
einen vorteilhaften kombinierten Effekt auf die Zelle auszuüben. In
solchen Fällen
wird in Betracht gezogen, dass man die Zelle mit beiden Modalitäten in Kontakt
bringt, wobei der Abstand zwischen den jeweiligen Modalitäten etwa
12–24
Stunden und mehr bevorzugt etwa 6–12 Stunden beträgt, wobei
eine Verzögerungszeit
von nur etwa 12 Stunden am meisten bevorzugt ist. In manchen Situationen
kann es jedoch wünschenswert
sein, den Zeitraum für
die Behandlung wesentlich auszudehnen, wobei mehrere Tage (2, 3,
4, 5, 6 oder 7) oder mehrere Wochen (1, 2, 3, 4, 5, 6, 7 oder 8)
zwischen den jeweiligen Verabreichungen liegen können.
-
Es
ist auch vorstellbar, dass mehr als eine Verabreichung von entweder
TS10q23.3 oder dem anderen Mittel erwünscht sind. Es können verschiedene
Kombinationen verwendet werden, wobei TS10q23.3 „A" ist und das andere Mittel „B" ist, wie unten beispielhaft
dargestellt.
A/B/A B/A/B B/B/A A/A/B B/A/A A/B/B B/B/B/A B/B/A/B
A/A/B
A/B/A/B A/B/B/A B/B/A/A B/A/B/A B/A/A/B B/B/B/A
A/A/A/B B/A/A/A
A/B/A/A A/A/B/A A/B/B/B B/A/B/B B/B/A/B
-
Andere
Kombinationen werden in Betracht gezogen. Auch hier müssen beide
Mittel in einer kombinierten Menge, die ein Abtöten der Zelle bewirkt, an eine
Zelle verabreicht werden, um ein Abtöten der Zelle zu erreichen.
-
Mittel
oder Faktoren, die zur Verwendung in einer kombinierten Therapie
geeignet sind, sind alle chemischen Verbindungen oder Behandlungsverfahren,
die bei Anwendung auf eine Zelle eine Schädigung der DNA induzieren.
Solche Mittel und Faktoren umfassen Strahlung und Wellen, die eine
DNA-Schädigung
induzieren, wie beispielsweise Gammastrahlung, Röntgenstrahlen, UV-Strahlung,
Mikrowellen, elektronische Emissionen und dergleichen.
-
Verschiedene
chemische Verbindungen, die auch as „chemotherapeutische Mittel" beschrieben sind, bewirken
eine DNA-Schädigung,
von denen alle in den hierin offenbarten Verfahren einer kombinierten
Behandlung verwendet werden könnten.
Chemotherapeutische Mittel, die verwendet werden können, umfassen z.
B. Adriamycin, 5-Fluoruracil (5FU), Etoposid (VP-16), Camptothecin,
Actinomycin-D, Mitomycin C, Cisplatin (CDDP) und sogar Wasserstoffperoxid.
Die Erfindung umfasst auch die Verwendung einer Kombination von einem
oder mehreren DNA-schädigenden
Mitteln, sei es auf der Basis von Strahlung oder tatsächliche
Verbindungen, beispielsweise die Verwendung von Röntgenstrahlen
mit Cisplatin oder die Verwendung von Cisplatin mit Etoposid. In
bestimmten Ausführungsformen
ist die Verwendung von Cisplatin in Kombination mit einem TS10q23.3-Expressionskonstrukt
als diese Verbindung besonders bevorzugt.
-
Bei
der erfindungsgemäßen Behandlung
von Krebs würden
die Tumorzellen außer
mit dem Expressionskonstrukt mit einem Mittel in Kontakt gebracht
werden. Dies kann erreicht werden, indem die lokalisierte Tumorstelle
mit Strahlung wie Röntgenstrahlen;
UV-Licht, Gammastrahlen oder sogar Mikrowellen bestrahlt wird. Alternativ
können
die Tumorzellen mit dem Agent in Kontakt gebracht werden, indem
eine therapeutisch wirksame Menge einer pharmazeutischen Zusammensetzung
an ein Individuum verabreicht wird, die eine Verbindung wie Adriamycin,
5-Fluoruracil, Etoposid, Camptothecin, Actinomycin-D, Mitomycin
C oder mehr bevorzugt Cisplatin umfasst. Das Mittel kann als eine
kombinierte therapeutische Zusammensetzung oder ein Kit hergestellt
und verwendet werden, indem es, wie oben beschrieben, mit einem
TS10q23.3-Expressionskonstrukt kombiniert wird.
-
Es
sind Mittel vorgesehen, die Nukleinsäuren, spezifisch DNA, direkt
vernetzen, um die DNA-Schädigung
zu erleichtern, was eine synergistische, antineoplastische Kombination
mit TS10q23.3 bewirkt. Es können
Mittel wie Cisplatin und andere DNA-alkylierende Mittel verwendet
werden. Cisplatin ist häufig
verwendet worden, um Krebs zu behandeln, wobei in klinischen Anwendungen
wirksame Dosismengen von 20 mg/m2 für 5 Tage
alle drei Wochen für
insgesamt drei Zyklen verwendet worden sind. Cisplatin wird nicht
oral resorbiert und muss daher per Injektion intravenös, subkutan,
intratumoral oder intraperitoneal verabreicht werden.
-
DNA-schädigende
Mittel umfassen auch Verbindungen, die in die DNA-Replikation, Mitose
und chromosomale Segregation eingreifen. Solche chemotherapeutischen
Verbindungen umfassen Adriamycin, auch bekannt als Doxorubicin,
Etoposid, Verapamil, Podophyllotoxin und dergleichen. Diese Verbindungen
werden im klinischen Umfeld häufig
für die
Behandlung von Neoplasmen verwendet und durch Bolusinjektionen verabreicht,
im Fall von Adriamycin im Bereich von 25–75 mg/m2 intravenös in 21-tägigen Intervallen,
bei Etoposid im Bereich von 35–50mg/m2 intravenös oder in doppelter Dosis oral.
-
Mittel,
welche die Synthese und Wiedergabegenauigkeit von Nukleinsäurevorläufern und
Untereinheiten unterbrechen, führen
ebenfalls zu DNA-Schädigung.
Als solche wurde eine Reihe von Nukleinsäurevorläufern entwickelt. Besonders
geeignet sind Mittel, die ausführliche
Tests durchlaufen haben und einfach erhältlich sind. Als solche werden
Mittel wie 5-Fluoruracil (5-FU) von neoplastischem Gewebe bevorzugt
verwendet, was dieses Mittel für
ein Targeting von neoplastischen Zellen besonders geeignet macht.
Obgleich es relativ toxisch ist, ist 5-FU in einem weiten Bereich
von Trägerstoffen
einsetzbar einschließlich
topisch, wobei jedoch die intravenöse Verabreichung in Dosismengen
von 3 bis 15 mg/kg/Tag häufig
verwendet wird.
-
Andere
Faktoren, die eine DNA-Schädigung
verursachen und häufig
verwendet wurde, sind, was im Allgemeinen als Gammastrahlen, Röntgenstrahlen
und/oder direkte Verabreichung von Radioisotopen an Tumorzellen
bekannt ist. Andere Formen DNA-schädigender Faktoren werden ebenfalls
in Betracht gezogen, wie beispielsweise Mikrowellen und UV-Strahlung.
Es ist sehr wahrscheinlich, dass alle dieser Faktoren eine weit gefasste
Schädigung
von DNA bewirken, auf die Vorläufer
von DNA, die Replikation und Reparatur von DNA und dem Zusammenbau
und der Erhaltung von Chromosomen ausüben. Dosisbereiche für Röntgenstrahlen liegen
bei täglichen
Dosismengen von 50 bis 200 Röntgen
für längere Zeiträume (3 bis
4 Wochen) bis hin zu Einzeldosismengen von 2000 bis 6000 Röntgen. Dosisbereiche
für Radioisotope
sind sehr unterschiedlich und richten sich nach der Halbwertszeit
des Isotops, der Stärke
und dem Typ der emittierten Strahlung und der Aufnahme von den neoplastischen
Zellen.
-
Der
erfahrene Fachmann sei auf „Remington's Pharmaceutical
Sciences", 15. Auflage,
Kapitel 33, insbesondere die Seiten 624–652 verwiesen. Je nach Zustand
des behandelten Individuums wird es natürlich zu einer gewissen Variation
der Dosis kommen. Die für
die Verabreichung zuständige
Person wird in jedem Fall die geeignete Dosis für das Individuum bestimmen.
Für die
Verabreichung an Menschen sollten Zubereitungen die Sterilitäts- und
Pyrogenitätsstandards,
die allgemeinen Sicherheits- und Reinheitsstandards erfüllen, wie sie
vom FDA Office of Biologics Standards vorgeschrieben sind.
-
Die
Erfinder schlagen vor, dass die regionale Verabreichung von TS10q23.3-Expressionskonstrukten an
Patienten mit Krebserkrankungen, die in Verbindung mit 10q23.3 stehen,
ein sehr effizientes Verfahren für die
Verabreichung eines therapeutisch effektiven Gens darstellt, um
der klinischen Krankheit entgegen zu wirken. In ähnlicher Weise kann in bestimmten
Umständen,
wenn eine weit reichende Metastasierung aufgetreten ist, die systemische
Verabreichung von Expressionskonstrukt und/oder dem Mittel angemessen
sein.
-
Außer der
Kombination von Therapien, bei denen TS10q23.3 das Ziel ist, mit
Chemo- und Strahlentherapien
wird auch in Betracht gezogen, dass eine Kombination mit anderen
Gentherapien vorteilhaft ist. Beispielsweise kann ein gleichzeitiges
Targeting von TS10q23.3- und
p53- oder p16-Mutationen eine verbesserte Anti-Krebs-Behandlung
bewirken. Jedes andere in Verbindung mit einem Tumor stehende, vorstellbare
Gen kann auf diese Weise angegriffen werden, beispielsweise p21,
Rb, APC, DCC, NF-1, NF-2, BCRA2, p16, FHIT, WT-1, MEN-I, MEN-II,
BRCA1, VHL, FCC, MCC, ras, myc, neu, raf, erb, src, fms, jun, trk,
ret, gsp, hast, bcl und abl.
-
Es
sollte auch darauf hingewiesen werden, dass sich jede der vorhergehenden
Therapien auch für sich
alleine bei der Behandlung eines TS10q23.3. als nützlich erweisen
könnte.
In dieser Hinsicht sollte die Bezugnahme auf Chemotherapien und
Nicht-TS10q23.3-Gentherapie in Kombination als Überlegung verstanden werden,
dass diese Ansätze
auch getrennt angewandt werden können.
-
E. Formulierungen und
Verabreichungswege an Patienten
-
Wenn
klinische Anwendungen in Betracht gezogen werden, ist es erforderlich,
pharmazeutische Zusammensetzungen – Expressionsvektoren, Virusstämme, Proteine,
Antikörper
und Wirkstoffe – in
einer Form herzustellen, die sich für die beabsichtigte Anwendung
eignet. Dies umfasst im Allgemeinen das Herstellen von Zusammensetzungen,
die vollständig
frei von Pyrogenen sind sowie von anderen Unreinheiten, die für Menschen
oder Tiere schädlich
sein könnten.
-
Es
wäre im
Allgemeinen wünschenswert,
geeignete Salze und Puffer zu verwenden, um die Verabreichungsvektoren
zu stabilisieren und eine Aufnahme durch Zielzellen zu ermöglichen.
Puffer werden auch eingesetzt, wenn rekombinante Zellen in einen
Patienten eingeführt
werden. Wässrige
Zusammensetzungen der vorliegenden Erfindung umfassen die Verabreichung
einer wirksamen Menge des Vektors an Zellen, aufgelöst oder
verteilt in einem pharmazeutisch annehmbaren Trägerstoff oder einem wässrigen
Medium. Solche Zusammensetzungen werden auch als Inokula bezeichnet.
Der Begriff „pharmazeutisch
oder pharmakologisch annehmbar" bezieht
sich auf molekulare Einheiten und Zusammensetzungen, die keine nachteiligen,
allergischen oder andere unangemessene Reaktionen hervorbringen,
wenn sie einem Tier oder einem Menschen verabreicht werden. Wie
hierin verwendet, umfasst „pharmazeutisch
annehmbarer Träger" jedes und alle Lösungsmittel,
Dispersionsmedien, Beschichtungen, antibakterielle und fungizide
Mittel, isotonische und absorptionsverzögernde Mittel und dergleichen.
Die Verwendung solcher Medien und Mittel für pharmazeutisch aktive Substanzen
ist aus dem Stand der Technik gut bekannt. Außer insofern, als ein herkömmliches
Medium oder Mittel mit dem Vektor oder den Zellen der vorliegenden
Erfindung nicht verträglich
ist, wird seine Verwendung in therapeutischen Zusammensetzungen
in Betracht gezogen. In die Zusammensetzungen können auch ergänzende aktive
Inhaltsstoffe aufgenommen werden.
-
Die
aktiven Zusammensetzungen der vorliegenden Erfindung können klassische
pharmazeutische Zubereitungen umfassen. Die Verabreichung dieser
Zusammensetzungen gemäß der vorliegenden
Erfindung erfolgt über
einen beliebigen gängigen
Weg, vorausgesetzt, das Zielgewebe ist über diesen Weg zugänglich. Dies
umfasst den oralen, nasalen, bukkalen, rektalen, vaginalen oder
topischen Weg. Alternativ kann die Verabreichung durch eine orthotopische,
intradermale, subkutane, intramuskuläre, intraperitoneale oder intravenöse Injektion
erfolgen. Solche Zusammensetzungen würden normalerweise als pharmazeutisch
annehmbare Zusammensetzungen, die oben beschrieben sind, verabreicht
werden.
-
Die
aktiven Verbindungen können
auch parenteral oder intraperitoneal verabreicht werden. Lösungen der
aktiven Verbindungen als freie Base oder pharmakologisch annehmbaren
Salzen können
in Wasser hergestellt werden, das geeignet mit einem oberflächenaktiven
Stoff wie Hydroxypropylcellulose gemischt ist. Dispersionen können auch
in Glycerol, flüssigen
Polyethylenglykolen und Mischungen davon und in Ölen hergestellt werden. Unter
normalen Bedingungen der Aufbewahrung und Verwendung enthalten diese
Zubereitungen ein Konservierungsmittel, um das Wachstum von Mikroorganismen
zu verhindern.
-
Die
pharmazeutischen Formen, die zur Verwendung als Injektion geeignet
sind, umfassen sterile wässrige
Lösungen
oder Dispersionen und sterile Pulver für die unvorbereitete Herstellung
steriler injizierbarer Lösungen
oder Dispersionen. In allen Fallen muss die Form steril und so flüssig sein,
dass eine einfache Aufnahme in eine Spritze möglich ist. Sie muss unter den
Herstellungs- und Aufbewahrungsbedingungen stabil sein und gegen
die kontaminierende Wirkung von Mikroorganismen, beispielsweise
Bakterien und Pilze konserviert sein. Der Träger kann ein Lösungsmittel
oder ein Dispersionsmedium sein, das beispielsweise Wasser, Ethanol,
Polyol (beispielsweise Glycerol, Propylenglycol und flüssiges Polyethylenglycol
und dergleichen), geeignete Mischungen davon und pflanzliche Öle enthält. Die
richtige Fluidität kann
beispielsweise durch Verwendung einer Beschichtung wie Lecithin,
durch Beibehaltung einer erforderliche Partikelgröße im Falle
einer Dispersion und durch Verwendung von oberflächenaktiven Stoffen aufrechterhalten
werden. Die Prävention der
Wirkung von Mikroorganismen kann durch verschiedene antibakterielle
und fungizide Mittel wie beispielsweise Parabene, Chlorbutanol,
Phenol, Sorbinsäure,
Thimerosal und dergleichen vermittelt werden. In vielen Fällen ist
es bevorzugt, isotope Mittel, beispielsweise Zucker oder Natriumchlorid,
einzuschließen.
Verlängerte Absorption
der injizierbaren Zusammensetzungen kann vermittelt werden, indem
in den Zusammensetzungen Mittel verwendet werden, die eine Absorption
verzögern,
beispielsweise Aluminiummonostearat und Gelatine.
-
Sterile
injizierbare Lösungen
werden hergestellt, indem die aktiven Verbindungen in der erforderlichen Menge
dem geeigneten Lösungsmittel
mit Verschiedenen der anderen Bestandteile, die oben aufgezählt sind, wie
erforderlich hinzugefügt
werden, gefolgt von Sterilfiltration. Dispersionen werden im Allgemeinen
hergestellt, indem die verschiedenen sterilisierten aktiven Inhaltstoffe
einem sterilen Vehikel zugefügt
werden, welches das Dispersionsbasismedium und die erforderlichen
anderen Bestandteile aus den oben Aufgezählten enthält. Im Falle steriler Pulver
für die
Zubereitung steriler injizierbarer Lösungen sind Vakuumtrocknungs-
und Gefriertrocknungstechniken die bevorzugten Verfahren der Zubereitung,
die ein Pulver des aktiven Inhaltsstoffes plus jedes weiteren gewünschten
Inhaltsstoffes aus einer zuvor steril filtrierten Lösung davon
ergibt.
-
Wie
hierin verwendet, umfasst „pharmazeutisch
annehmbarer Träger" jedes und alle Lösungsmittel, Dispersionsmedien,
Beschichtungen, antibakterielle und fungizide Mittel, isotonische
und resorptionsverzögernde
Mittel und dergleichen. Die Verwendung solcher Medien und Mittel
für pharmazeutisch
aktive Substanzen ist aus dem Stand der Technik gut bekannt. Außer insofern,
als ein herkömmliches
Medium oder Mittel mit dem aktiven Inhaltsstoff nicht verträglich ist,
wird seine Verwendung in therapeutischen Zusammensetzungen in Betracht
gezogen. Den Zusammensetzungen können
auch ergänzende
aktive Inhaltsstoffe zugefügt
werden.
-
Zur
oralen Verabreichung können
die Polypeptide der vorliegenden Erfindung mit Exzipienten versehen
sein und in Form nicht einnehmbarer Mundwässer oder Zahnpasten verwendet
werden. Ein Mundwasser kann hergestellt werden, indem der aktive
Inhaltsstoff in der erforderlichen Menge in einem geeigneten Lösungsmittel,
wie beispielsweise eine Natriumboratlösung (Dobells Lösung), hinzugefügt wird.
Alternativ kann der aktive Inhaltsstoff einer antiseptischen Waschlösung zugefügt werden,
die Natriumborat, Glycerin und Kaliumbicarbonat enthält. Der
aktive Inhaltsstoff kann auch in Zahnpasten verteilt sein, einschließlich: Gels,
Pasten, Pulvern und Schlämmen.
Der aktive Inhaltsstoff kann einer Zahnpasta, die Wasser, Bindemittel,
Schleifmittel, Geschmacksstoffe, Schaumbilder und Befeuchtungsmittel
enthalten kann, in einer therapeutisch wirksamen Menge zugegeben
werden.
-
Die
Zusammensetzungen der vorliegenden Erfindung können in einer neutralen Form
oder als Salzform formuliert sein. Pharmazeutisch annehmbare Salze
umfassen die Säureadditionssalze
(gebildet mit freien Aminogruppen des Proteins) und welche, die
mit anorganischen Säuren
wie beispielsweise Salzsäure
oder Phosphorsäure
oder organischen Säuren
wie Essigsäure,
Oxasäure,
Weinsäure,
Mandelsäure
und dergleichen gebildet sind. Salze, die mit den freien Carbonylgruppen
gebildet sind, können
auch von anorganischen Basen abstammen, wie beispielsweise Natrium-,
Kalium-, Ammonium-, Kalzium- oder Eisen(III)hydroxiden und solchen
organischen Basen wie Isopropylamin, Trimethylamin, Histidin, Procain
und dergleichen.
-
Nach
der Formulierung werden Lösungen
auf eine Weise verabreicht, die mit der Dosisformulierung kompatibel
ist, und in einer solchen Menge, wie sie therapeutisch wirksam ist.
Die Formulierungen werden in verschiedenen Dosisformen einfach verabreicht,
beispielsweise als Injektionslösung,
Wirkstofffreisetzungskapseln und dergleichen. Für eine parenterale Verabreichung
in einer wässrigen
Lösung
beispielsweise sollte die Lösung
geeignet gepuffert sein und das flüssige Verdünnungsmittel zunächst mit
ausreichend Salzlösung oder
Glukose isotonisch gemacht werden. Diese speziellen wässrigen
Lösungen
sind besonders für
die intravenöse,
intramuskuläre,
subkutane und intraperitoneale Verabreichung geeignet. In diesem
Zusammenhang sind einem Fachmann angesichts der vorliegenden Offenbarung
sterile wässrige
Medien bekannt. Eine Dosis könnte
beispielsweise in 1 ml isotonischer NaCl-Lösung gelöst und entweder zu 1000 ml
Hypodermoclysis-Flüssigkeit
gegeben oder an der beabsichtigten Infusionsstelle injiziert werden
(siehe beispielsweise „Remington's Pharmaceutical
Sciences", 15. Auflage,
Seite 1035–1038
und 1570–1580).
Je nach Zustand des behandelten Individuums wird natürlich eine
gewisse Variation der Dosismengen erforderlich sein. Die für die Verabreichung
zuständige
Person wird in jedem Fall die geeignete Dosis für das Individuum bestimmen.
Für die Verabreichung
an Menschen sollten Zubereitungen darüber hinaus die Sterilitäts- und
Pyrogenitätsstandards, sowie
die allgemeinen Sicherheits- und Reinheitsstandards erfüllen, wie
sie vom FDA Office of Biologics Standards vorgeschrieben sind.
-
VII. Transgene Tiere/Knockout-Tiere
-
In
einer Ausführungsform
der Erfindung werden transgene Tiere hergestellt, die ein funktionelles Transgen
enthalten, welches ein funktionelles TS10q23.3-Polypeptid oder Varianten
davon kodiert. Transgene Tiere, die TS10q23.3-Transgene exprimieren,
von solchen Tieren stammende rekombinante Zelllinien und transgene
Embryos können
bei Verfahren zum Testen auf und zur Identifizierung von Mitteln,
die die Funktion von TS10q23.3 induzieren oder unterdrücken, nützlich sein.
Transgene Tiere der vorliegenden Erfindung können auch als Modelle für die Untersuchung
von Indikationen wie Krebserkrankungen verwendet werden.
-
In
einer Ausführungsform
der Erfindung wird ein TS10q23.3-Transgen in einen nicht-menschlichen Wirt
eingeführt,
um ein transgenes Tier hervorzubringen, das ein humanes oder murines
TS10q23.3-Gen exprimiert. Das transgene Tier wird hergestellt, indem
das Transgen in das Genom derart integriert wird, dass das Transgen
exprimiert werden kann. Verfahren zum Produzieren von transgenen
Tieren sind im Allgemeinen von Wagner und Hoppe (US-Patent Nr. 4.873.191;
welches hierin durch Bezugnahme enthalten ist), Brinster et al.,
1985; welches hierin in Gänze
durch Bezugnahme enthalten ist, und in „Manipulating the Mouse Embryo; A
Laboratory Manual" 2.
Auflage (Hrsg. Hogan, Beddington, Constantimi und Long, Cold Spring
Harbour Laboratory Press, 1994; welches hierin in Gänze durch
Bezugnahme enthalten ist) beschrieben.
-
Es
kann wünschenswert
sein, das endogene TS10q23.3 durch homologe Rekombination zwischen dem
Transgen und dem endogenen Gen zu ersetzen; oder das endogene Gen
kann durch Deletion eliminiert werden, wie bei der Herstellung von „Knockout"-Tieren. Typischerweise
wird ein TS10q23.3-Gen, das von genomischen Sequenzen flankiert
wird, durch Mikroinjektion in ein befruchtetes Ei überführt. Die
mikroinjizierten Eier werden in ein weibliches Wirtstier implantiert
und die Nachkommen werden auf Expression des Transgens getestet.
Transgene Tiere können
aus den befruchteten Eiern einer Reihe von Tieren hergestellt werden,
darunter, jedoch nicht ausschließlich, aus Reptilien, Amphibien,
Vögeln,
Säugetieren
und Fischen. In einer besonders bevorzugten Ausführungsform werden transgene
Mäuse erzeugt,
die TS10q23.3 überexprimieren
oder eine mutante Form des Polypeptids exprimieren. Alternativ erlaubt
die Abwesenheit von TS10q23.3 in „Knockout"-Mäusen
die Untersuchung der Effekte, die ein Verlust von TS10q23.3-Protein
auf eine Zelle in vivo hat. „Knockout"-Mäuse stellen
außerdem
ein Modell für
die Entwicklung von Krebserkrankungen in Zusammenhang mit TS10q23.3
dar.
-
Verfahren
zum Herstellen von Knockout-Tieren sind im Allgemeinen von Shastry
(1995, 1998) und Osterrieder und Wolf (1998) beschrieben. Die Herstellung
konditionierter Knockout-Tiere,
bei denen das Gen aktiv ist, bis es zum gewünschten Zeitpunkt ausgeschaltet
wird, ist allgemein beschrieben von Feil et al. (1996), Gagneten
et al., (1997) und Lobe und Nagy (1998). Jede dieser Bezugsquellen
ist hierin durch Bezugnahme enthalten.
-
Wie
oben bemerkt, finden transgene Tiere und Zelllinien, die von solchen
Tieren abstammen, Anwendung in bestimmten Testversuchen. Diesbezüglich können transgene
Tiere und Zelllinien, die Wildtyp-TS10q23.3 oder mutantes TS10q23.3
exprimieren können,
Testsubstanzen ausgesetzt werden. Diese Testsubstanzen können hinsichtlich
ihrer Fähigkeit,
die Expression oder Funktion von Wildtyp-TS10q23.3 zu verstärken oder
die Expression oder Funktion von mutantem TS10q23.3 zu vermindern,
getestet werden.
-
BEISPIELE
-
Die
folgenden Beispiele sind enthalten, um bevorzugte Ausführungsformen
der Erfindung aufzuzeigen. Einem Fachmann sollte klar sein, dass
Techniken, die in den folgenden Beispielen offenbart sind, Techniken
darstellen, bei denen die Erfinder entdeckt haben, dass sie in der
praktischen Umsetzung der Erfindung gut funktionieren, und daher
als bevorzugte Wege für
deren praktische Umsetzung gelten können. Ein Fachmann sollte jedoch
angesichts der vorliegenden Offenbarung anerkennen, dass in den
spezifischen offenbarten Ausführungsformen
viele Änderungen
vorgenommen werden können
und dennoch ein gleiches oder ähnliches
Ergebnis erhalten wird, ohne vom Konzept, dem Geist und dem Umfang
der Erfindung abzuweichen.
-
Insbesondere
ist offensichtlich, dass die hierin beschriebenen Mittel durch bestimmte
Mittel, die sowohl chemisch als auch physikalisch verwandt sind,
ersetzt werden können,
während
dieselben oder ähnlichen
Ergebnisse erzielt werden würden.
Alle derartigen Substitutionen und Modifikationen, die einem Fachmann
bekannt sind, sind in Geist, Umfang und Konzept der Erfindung, wie
von den beigefügten
Ansprüchen definiert,
enthalten.
-
BEISPIEL 1
-
Homozygote Deletion in
Gliomzelllinien
-
Die
Erfinder haben DNA aus einer Reihe von 21 Gliomzelllinien und Primärkulturen
sowie normale Zellen untersucht, um homozygote Deletionen von genomischem
Material auf Chromosom 10 zu identifizieren. Die Auswahl der Marker
erfolgte nach nach ihrer ungefähren
Position an oder in der Nähe
der zuvor implizierten Regionen (1). Die
analysierten Zellen wurden in der Abteilung für Neuroonkologie UTMDACC hergestellt
(LG11, EFC-2, PL-1, PC-1, JW, FG-2, FG-0, NG-1, PH-2, KE, PC-3 und
D77), waren kommerziell erhältlich
(U138, A172, U373, U87, U251, U118 und T98G) oder wurden von Mitarbeitern
erhalten (13 Wochen, Astrozytom, D54-MG). Die Marker wurden von
Research Genetics, Huntsville, AL, USA, erhalten, oder nach bekannten
Sequenzen synthetisiert. Bei einer Zelllinie, EFC-2, ergab sich
eine große
homozygote Deletion in Zusammenhang mit vier Markern um D10S215
(2). Diese Deletion wurde auch im FISH-Verfahren
anhand von YAC 746h6 beobachtet, welches in der Region liegt. Drei
andere Zelllinien (D-54, A172 und LG11) zeigten ebenfalls homozygote
Deletionen an AFM086, was stark darauf hinweist, dass die Region
ein putatives Tumorsuppressorgen aufweist (2). Deletionen
in PCRTM-Reaktionen wurden in Gegenwart
zweiter Primerpaare (gestaffelt) durchgeführt, um geeignete Amplifikationsbedingungen
sicherzustellen. Alle Deletionen wurden mindestens in Dreifachreaktionen
bestätigt.
Dieselbe Region wurde auch in Verbindung mit Prostatakarzinom gebracht
(Gray et al., 1995). Homozygote Deletionen in Zelllinien wurden
auch verwendet, um einen Tumorsuppressorgenlocus an 3p21.2 bei kleinzelligem
Lungenkarzinom zu definieren (Daly et al., 1993; Kok et al., 1994,
Wei et al., 1996).
-
BEISPIEL 2
-
Retention von 10q-Loci
in supprimierten Hybridzellen
-
Die
zweite Strategie der Erfinder war es, die Regionen von Chromosom
10 zu untersuchen, die bei supprimierten Hybridklonen erhalten waren,
aber in revertanten Klonen fehlten. Diese Analyse ging über die vorige
Studie der Erfinder hinaus, wobei das Vorhandensein zweier Tumorsuppressorloci
auc Chromosom 10 gezeigt und die erhaltenen Regionen analysiert
wurden. Hybride, bei denen 10q gesamt oder in Abschnitten erhalten
war, zeigten kein Wachstum in Weichagarose und Nacktmäusen („vollständig" supprimierte Klone), während Hybridzellen,
die den größten Teil
des eingesetztes Chromosoms 10q verloren hatten, in Weichagarose
wuchsen, aber nicht-tumorigen waren („partiell" supprimierte Klone; Steck et al., 1995, 3 rechts).
Zuvor wurde gezeigt, dass Originalklone U251N10.6, N10.7 und N10.8
nur Fragmente von 10q erhalten hatten (Pershouse et al., 1993; Steck
et al., 1995). Anhand von weiteren informativen Mikrosatellitenmarkern
wurden in allen drei supprimierten Klonen drei erhaltene Regionen
identifiziert: eine 22-cM-Region von D10S219 bis D10S110, eine 14-cM-Region von D10S192
bis D10S187 und eine 18-cM-Region von D10S169 bis D10S1134 (3).
-
Zur
Umgehung dieser Einschränkung
wurde das ursprünglich übertragene
Neomycinresistenz-markierte Chromosom 10 von Hybrid U251.N10.7 durch
Mikrozellenvermittelten Chromosomentransfer in A9-Mauszellen überführt („rescued"). Damit sind alle
humanen Mikrosatellitenmarker hinsichtlich des Vorhandenseins von
Chromosom 10 informativ. Diese Analyse beruht darauf, dass alle „vollständig" supprimierten Subklone
eine gemeinsame Region erhalten haben sollten und diese Region in
den „partiell" supprimierten Subklonen
deletiert ist. Ein weiterer Anstoß war, dass N10.7 in Bezug
auf die Größe des erhaltenen
Chromosoms 10 beträchtliche
Heterogenität
aufwies, wie anhand von FISH mit CHromosom-10-spezifischen Sonden festgestellt wurde.
Außerdem
wurden die für
diese Übernagung
(„Rescue") verwendeten Hybride
zuerst auf Weichagarosewachstum getestet und zeigten keine Koloniebildung.
Alle Maushybride, die das übertragene humane
Chromosom 10 enthielten, enthielten den kurzen Arm von Chromosom
10. Dieselbe Region war in den „partiell" supprimierten Klonen (N10.5a-j), die
in Weichagarose wuchsen, erhalten (Steck et al., 1995), wodurch
ausgeschlossen werden konnte, dass diese Region (10pter-10q11) das
10q-Tumorsuppressorgen
enthält.
Eine Untersuchung der erhaltenen Regionen von 10q zeigte beträchtliche
Heterogenität
(3). Die Mehrzahl der Klone zeigte entweder partielle
oder extensive Deletionen von 10q23-26. Nur zwei Regionen waren
in allen untersuchten Subklonen erhalten. Die am meisten zentromer
gelegene, erhaltene Region umfasste die Marker D10S210 und D10S219.
Diese Marker waren jedoch in den ursprünglichen Klonen N10.6 und/oder N10.8
nicht vorhanden, weshalb diese Region ausgeschlossen wurde (3).
Die andere Region befand sich zentromer von D4S536, aber telomer
von D10S215 (~4 cM). Die Marker AFM086 und D10S536 waren in allen untersuchten
Klonen erhalten (Kastenbereich in 3). Diese
Marker waren in den partiell supprimierten Klonen (N10.5a-j) nicht
vorhanden. Diese Ergebnisse zeigten, dass eine gemeinsame Region
in der Umgebung von AFM086 in allen Hybridzellen, die phänotypisch
supprimiert sind, erhalten ist. Dieselbe Region ist bei mehreren
Gliomzelllinien deletiert.
-
Diese
Analyse hat mehrere Einschränkungen.
Zunächst
können
die übertragenen
(„rescued") Klone nicht auf
biologische Aktivität
analysiert werden, weshalb alle Veränderungen auf Chromosom 10,
die während oder
nach dem Transfer aufgetreten sein könnten, nicht entdeckt werden
konnten. Um diesem Einwand zu begegnen, wurde die Analyse der Erfinder
durchgeführt,
sobald die Klone geerntet werden konnten. Darüber hinaus könnte die
Retention dieses Abschnitts des Chromosoms nur eine in vitro künstlich
herbeigerufene Deletion „korrigieren". Folglich wurden
Alleldeletionsstudien durchgeführt
um festzustellen, ob diese Region bei Gliomen eine Rolle spielt.
In dieser Analyse deutete sich außerdem eine alternative Region
bei D10S1158 an, die bei allen Klonen außer einem (C7) erhalten war.
Die erhaltene Region bei AFM086 zeigte jedoch auch homozygote Deletionen
und wurde deshalb im Vergleich zu D10S1158 durch zwei alternative
Verfahren impliziert. Interessanterweise ist außerdem anzumerken, dass es
scheint, als bliebe die Tumorsuppressorgenregion vorzugsweise erhalten
während
der Rest von 10q fragmentiert wird.
-
BEISPIEL 3
-
Alleldeletionsanalyse
von 10q
-
Mit
DNA aus einer Reihe von 53 Gliompräparaten und entsprechenden
Patientenlymphozyten wurde anhand von Mikrosatellitenmarkern für Chromosom
10 eine Alleldeletionsstudie durchgeführt. Diese Studie wurde unternommen
um festzustellen, ob unsere kritische Region auch in Gliompräparaten
eine Rolle spielt. Bei der Mehrzahl der aus GBM stammenden Präparate wurden
extensive Deletionen beobachtet, wobei 30 von 38 GBM eine Deletion
der meisten oder aller Chromosom-10-Marker aufwiesen. Wenn die Präparate aus anaplastischen
Astrozytomen stammten, wurden weniger extensive Deletionen beobachtet,
während
in Astrozytomen und den meisten Oligodendrogliomen Deletionen selten
beobachtet wurden (4 und Daten nicht gezeigt).
Die Mehrzahl der in dieser Analyse verwendeten Marker lag in der
Region 10q23-26 (Gyapay et al., 1994). Ähnlich wie bei anderen Studien
war es aufgrund der großen
Deletionen in den meisten GBM-Proben nicht möglich, eine gemeinsame Region überzeugend
aufzuzeigen (Fults et al., 1993; Rasheed et al., 1995).
-
Unter
den untersuchten GBM-Präparaten
ergab sich allerdings bei allen Tumorproben außer einer (Nr. 9; 4)
Deletionen im Bereich der Region von D10S579 bis D10 S541. Darüber hinaus
zeigte lediglich ein AA und keines der Astrozytome eine Deletion
in der kritischen Region der Erfinder. Zwei Oligodendrogliome wiesen
Deletionen in der kritischen Region auf, aber beide wurden als maligne
diagnostiziert. Diese Studie liefert mehrere Möglichkeiten. Erstens treten
die Deletionen, welche die kritische Region der Erfinder umfassten, überwiegend
in GBM und nicht in Tumoren geringeren Grades auf. Dies würde darauf
hinweisen, dass ein Verlust des Tumorsuppressorgens auf Chromosom
10q in der kritischen Region der Erfinder eine genetische Veränderung
darstellt, die mit der Progression zu GBM zusammenhängt. Diese
Hypothese wird dadurch gestützt, dass
in diesen Präparaten
keine gemeinsame Region identifiziert wurde, in der Deletionen an
10q vorhanden waren, obgleich in Tumoren geringeren Grades Deletionen
an 10q auftreten. Diese Beobachtung würde wiederum den vorigen Vorschlag
der Erfinder unterstützen,
dass eine Deletion des Tumorsuppressorgens an 10q überwiegend
mit GBM assoziiert ist und nicht alle Deletionen an 10q das Tumorsuppressorgen
betreffen. Die Region D10S216 bis D10S587 wies extensive Deletionen
auf, aber mehrere GBM hatten in dieser Region eine Heterozygosität bewahrt
(Tumor Nr. 2, Nr. 9, Nr. 13, Nr. 26, 4). Wenn
Tumoren geringen Grades aus der Studie ausgeschlossen werden, spielt
darüber
hinaus die Region der Erfinder in allen GBM eine Rolle. Diese Kombination
von unabhängigen
Ansätzen
deutet stark darauf hin, dass sich in Region D10S512 bis D10S541, insbesondere
bei AFM086, ein Tumorsuppressorgen befindet.
-
BEISPIEL 4
-
Kartierung der Tumorsuppressorgen-Kandidatenregion
-
Die
kritische Region, welche die Erfinder identifiziert haben, befindet
sich zentriert an AFM086 und wird von D10S215 und S10S451 eingegrenzt
(2 und 8). Diese Region ist relativ
klein und ist in mehreren YACs vorhanden (787d7, 746h8, 934d3).
FISH-Farbgebung mit YAC 746h8 auf EFC-2-Metaphaseausstrichen zeigt,
dass die homozygote Deletion in dem YAC vorhanden ist, da das YAC
partiell beobachtet und auf beiden Seiten benachbarte YACs vorhanden
waren. Es sind bakterielle artifizielle Chromosomen (BACs) oder
PACs für
alle Marker in der Region isoliert worden (8). Das
BAC-Kontig der Region wurde aus Endsequenzen von BACs konstruiert,
die in dieser Region kartiert worden sind. Es sind mehrere bemerkenswerte Merkmale
identifiziert worden. Erstens wurden zwei überlappende BACs identifiziert
worden (46b12 und 220) und verifizieren die genomische Integrität von 106d16.
Zweitens wurde an einem Ende der BACs eine NotI-Schnittstelle identifiziert.
Das Vorhandensein der NotI-Schnittstelle und ein paralleler Verdau
mit SacII, EagI und BssIII weist auf das Vorhandensein einer CpG-Insel
in 106d16 hin.
-
Die
EcoRI-Fragmente von BAC 106d16 wurden verwendet, um das Ausmaß der homozygoten
Deletionen in den Gliomzellen, bei denen zuvor gezeigt worden war,
dass sie AFM086 homozygot deletiert haben, durch Southern Blotting
zu untersuchen (2 und 5). Die rechte
Seite (EcoRI-Fragment 14) enthält
die mögliche
CpG-Insel und ist in drei der vier Zelllinien vorhanden. Ein NotI/EcoRI-Fragment
(Nr.3) wurde auf einem Southern Blot, der mehrere BACs und die Gliomzelllinie
enthielt, als Sonde verwendet (2). Wenn Sonden
von 46b12 verwendet wurden, wurden außer in EFC-2-Zellen keine Deletionen
auf der telomeren Seite (rechte Seite) entdeckt. In den Zellen wurden
jedoch in der von 106d16 definierten Region (~65 kb) weitere homozygote
Deletionen beobachtet. Eine homozygote Deletion für Bande
3 wird für
LG11- und EFC-2-Zellen beobachtet, aber nicht in den weiteren Gliomzellen
oder normalen Kontrollen. 106d16 (Bande 12) war in allen Zellen
vorhanden (EFC-2 weist eine Bande mit veränderter Migrationseigenschaft
auf), was darauf hinweist, dass die homozygote Deletion vollständig im
Bereich 106d16 enthalten ist.
-
BEISPIEL 5
-
Identifikation von exprimierten
Genen in der kritischen Region
-
Es
wurden EcoRI-Fragmente von BAC 106d16 hergestellt und durch Agarose-Gelelektrophorese
nach Größe getrennt.
Einzelne Banden oder Pools von Banden ähnlicher Größe wurden in pSPL3 (GIBCO, Gaithersburg,
MD, USA) ligiert. Putative Exons wurden identifiziert, wie vom Hersteller
beschrieben. Zwei Exons wurden korrekt in den Trapping-Vektor gesplict.
Die Exons stammten aus Bandenpool 2, 3, 4, 5 und Bande 7. Die Sequenz
der getrappten Exons wurde bestimmt und nach der bekannten Sequenz
des Trapping-Vektors definiert. Anhand von BLAST-Recherchen in der
Datenbank mit exprimierten Sequenzmarkern (dbEST) wurden fünf potenzielle
exprimierte Sequenzmarker (Expressed Sequence Tags; ESTs) identifiziert. Zwei
ESTs (gb/H92038, AA009519) enthielten entweder ein Exon oder beide
Exons (wenn auch ein EST die falsche Ausrichtung hatte).
-
Aus
den ESTs wurden Sequenzierprimer erstellt und verwendet, um putative
Exon-Intron-Übergänge zu definieren,
wobei BAC46b12 als Template („Muster") diente. Es wurden
neun Exons identifiziert. Sequenzunterschiede zwischen den ESTs
und dem genomischen Template („Muster") wurden korrigiert.
Alle Exons waren in BAC46b12 vorhanden. Aus den Intronsequenzen
neben den Exons wurden Primer erzeugt, um Amplikon-Einheiten für jedes
Exon zu bilden. Zwei der Exons entsprachen dem getrappten Exon aus
den BAC106d16-EcoRI-Sequenzen.
Die Sequenz des Gens ist in 6 gezeigt.
Die vorhergesagte Aminosäure-Sequenz
war definiert durch Vorhandensein einer ATG-Startstelle, TGA- und
TAA-Stopcodons im Leseraster, dem Vorhandensein multipler Stop-Codons
in allen drei Leserastern an anderer Stelle in der Sequenz, neun
Splice-Stellen und das Vorhandensein von Kozak-Signalen in der Nähe der Initiationsstelle.
Die Sequenz von 403 Aminosäuren
ist in 7 und 9 gezeigt.
Das vorhergesagte Molekulargewicht beträgt 47.122 mit einem pI-Wert
von 5,86.
-
Eine
Sequenzhomologie mit mehreren Proteinmotiven zeigt eine mögliche funktionelle
Rolle des Proteinproduktes an. Ein kritisches Motiv von Rest 88
bis 98 [IHCKAGKGRTG](SEQ ID Nr.: 28) stimmt genau mit der konservierten
katalytischen Domäne
einer Proteintyrosinphosphatase [(I/V)HCxAGxxR(S/T)G] (SEQ ID Nr.:
29; Denu et al., 1996) überein.
Mehrere andere Motive wurden identifiziert, die mit der Phosphatasefunktion
des Tumorsuppressorgens übereinstimmen
würden.
-
Aus
im Zufallsverfahren geprimter (Random Priming) DNA wurden Amplikons
(PCRTM-Produkte,
die aus verschiedenen Regionen des Gens hergestellt worden sind)
generiert. Die Sequenz der Amplikons entsprach der DNA-Sequenz.
Nicht-überlappende
Amplikons wurden als Sonden für
Northern Blots mit normalem Gewebe aus verschiedenen Organen (Clontech,
Palo Alto, USA; Multigewebeblots) eingesetzt. Alle Amplikons identifizierten
auf den Northern Blots eine Hauptbande bei 5,5 bis 6 kb und mehrere
kleinere Banden. Die mRNA war in allen untersuchten Geweben exprimiert
(Herz, Gehirn, Plazenta, Lunge, Leber, Skelettmuskel, Niere, Bauchspeicheldrüse, Milz,
Thymus, Prostata, Hoden, Eierstock, Dünndarm, Dickdarm und periphere Blutlymphozyten).
-
BEISPIEL 6
-
Mutationsanalyse
-
Die
Mutationsanalysen wurden anfangs mit zwei Strategien verfolgt. Erstens
wurden die Gliomzelllinien, bei denen anfangs gezeigt wurde, dass
sie homozygote Deletionen aufweisen, auf Vorhandensein des Kandidatengens
analysiert. Wie in 8 gezeigt, wiesen alle Zelllinien
mit einer Deletion von AFM086 homozygote Deletionen multipler Exons
des Kandidatengens auf. Darüber
hinaus traten die Deletionen in der Mitte des Gens auf, wodurch
die Deletionsgrenzen (ähnliche
Deletionen in allen Zelllinien) zwischen Exon 2 und 7 definiert
wurden. Deletionen, die die Mitte des Gens betreffen, zeigen außerdem,
dass das identifizierte Gen das Zielgen der Mutation darstellt.
-
Mit
einer Reihe von Gliomzelllinien wurde außerdem eine vorläufige Analyse
auf Sequenzmutationen durchgeführt.
Mutationen und/oder Deletionen wurden in allen untersuchten Gliomzellinien,
außer
dreien, beobachtet (Tabelle 5). Die Basenzahl in der Tabelle bezieht
sich auf das Exon, nicht die Gesamtsequenzen, d. h. die 98. Base
von Exon 7 bei U251.
-
TABELLE
5 IDENTIFIZIERTE
MUTATIONEN IN DEM KANDIDATENGEN
-
Deletionen
von Exons wurden auch in LnCap, einer Prostatazelllinie, gefunden.
Die Gliomzellen, die keine Mutation/Deletion zeigten, stammten von
Tumoren geringen Grades (PC-3
und PH-2), bei denen keine Alleldeletion von Chromosom 10 erwartet
und für
diese Zellen beobachtet wurde. Bei den anderen Zellen (D77) handelte
es sich um eine Primärzellkultur,
und Chromosom 10 erwies sich als heterozygot zu einem 1-bp-Polyrnorphismus
in dem Gen. Auch eine Brustkrebszelllinie zeigte eine Mutation.
Diese erste Analyse unterstützt die
Schlussfolgerung der Erfinder, dass der Verlust eines 10q-Tumorsuppressorgens
einen kritischen molekularen Marker für Glioblastome und eine Progression
der Krankheit darstellt.
-
BEISPIEL 7
-
Analyse von TS10q23.3-Mutanten
in Krebspräparaten
und Tumorzelllinien
-
In
einer ausführlicheren
Studie berichten die Erfinder das Auftreten von TS10q23.3-Mutationen in 342 Präparaten
von Primärtumoren
und 164 Tumorzelllinien (TZL) verschiedener Krebstypen, die auf
dem gesamten TS10q23.3-Locus einen scheinbaren Verlust der Heterozygosität (LOH)
aufweisen. Unter den 75 TZL, die auf dem gesamten TS10q23.3-Locus
einen scheinbaren Heterozygositätsverlust
(Loss of Heterozygosity, LOH) aufweisen, fanden die Erfinder zehn
homozygote Deletionen, die kodierende Abschnitt von TS10q23.3 entfernten,
sowie eine Frameshift-, eine Nonsense- und sieben Missense-Varianten.
Dagegen entdeckten die Erfinder unter 84 Primärtumoren, die auf Heterozygositätsverlust
(LOH) voranalysiert worden waren, nur eine Frameshift-Läsion, eine
Nonsense-Mutation, eine Splice-Variante und eine Missense-Variante.
Interessanterweise erwies sich die Expression von TS10q23.3 bei
hochgradigen Glioblastomen im Vergleich zu normalem Gehirngewebe
als signifikant reduziert.
-
Verfahren
-
OH-Analyse:
-
Aus
gefrorenen Präparaten
oder entparaffinierten Schnitten wurde genomische Gesamt-DNA gereinigt.
Genomische Gesamt-DNA aus Krebszelllinien wurde mit dem Easy-DNA-Kit
(Invitrogen, San Diego, CA, USA) gereinigt. Die LOH-Analyse wurde
durchgeführt,
wie zuvor beschrieben (Teng et al., 1996; Steck et al., 1995). Folgende
polymorphen STR-Marker (Short Terminal Repeats) wurden in dieser
Studie verwendet: D10S1687 (Heterozygositätsindex, H.I. = 0,81; Ldb (Collins
et al., 1996), Position von p-Telomer
nach Radiation Hybrid Mapping, R. L. = 85 Mb), D10S579 (H.I. = 0,59;
R. L. = 86,4 Mb), D10S541 (H.I. = 0,78; R. L. = 86,5 Mb), AFM280WE1
(H.I. = nicht bestimmt; R. L. = 87 Mb), AFMA114XB1 (H.I. = 0,70;
R. L. = 91,9 Mb) und D10S1753 (H.I. = 0,74; R. L. = 92,48 Mb), TS10q23.3
wie definiert durch AFM086WE1 befindet sich an Position 86,5 Mb.
In Präparaten
von Primärtumoren
wurde der Heterozygositätsverlust
(LOH) in den meisten Fällen bestimmt,
indem STR-Marker-Amplikons, die aus Tumor-DNA und normaler DNA jedes getesteten
Individuums hergestellt worden sind, quantitativ verglichen wurden.
Im Falle von TZL und einigen Primärtumoren wurde der LOG auf
der Basis der kombinierten scheinbaren Hemizygosität von AFMA114XB1,
D10S541 und D10S1753 bestimmt; die Wahrscheinlichkeit, dass alle
drei dieser STR-Marker in einer gegebenen Probe homozygot sind,
ist kleiner als 0,017.
-
Analyse auf homozygote
Deletion:
-
Mit
den genomischen DNAs aus den Zelllinien als Template („Muster") wurden gestaffelte
PCRTM-Amplifikationen entweder mit TaqPlus
(Stratagene, La Jolla, CA, USA) oder AmpliTaqGold (Perkin Elmer,
Foster City, CA, USA) durchgeführt.
Die Primer, die zur Herstellung von TS10q23.3- und MMK4-Amplikons
verwendet wurden, sowie die verwendeten PCRTM-Bedingungen
sind unten beschrieben. Zwanzig Mikroliter der Sekundärreaktionen
wurden auf 2-3%igen Nu Sieve (FMC Bioproducts)-Agarosegelen fraktioniert und danach
visualisiert.
-
Mutationsanalyse:
-
Die
Erfinder führten
gestaffelte PCRTM-Amplifikationen mit genomischer
DNA von Tumorpräparaten oder
TZL durch und testeten die resultierenden Amplikons auf Sequenzvarianten
gemäß der Verfahren
von Steck et al., (1997) mit mehreren Modifikationen. Zuerst wurde
Exon 6 mit einem einzelnen sekundären Amplikon anhand des Exon-6-FB-RR-Primerpaars
getestet. Als Zweites wurde das Exon nach einer primären Amplifikation
von Exon 8 unter Verwendung von FA-RP-Primern als zwei sekundäre Amplikons
mit den folgenden FB-RQ- und FC-RR-Primern getestet:
CA6.ex8.FB
GTTTTCCCAGTCACGACGAGGTGACAGATTTTCTTTTTTA (SEQ ID Nr.: 33)
CA6.ex8.RQ
AGGAAACAGCTATGACCATTCGGTTGGCTTTGTCTTTA (SEQ ID Nr.: 34)
CA6.ex8.FC
GTTTTCCCAGTCACGACGCATTTGCAGTATAGAGCGT (SEQ ID Nr.: 35)
CA6.ex8.RR
AGGAAACAGCTATGACCATAGCTGTACTCCTAGAATTA (SEQ ID Nr.: 36)
-
Drittens,
da Mononukleotiddurchläufe
in bestimmten Introns schlechtes Dye-Primer-Sequencing verursachten, erhielten die
Erfinder Dye-Terminator-Sequenzdaten über die sekundären Amplikons
Exon 8 FB-RQ und Exon 9 FB-RR, indem respektive die gestaffelten
Primer 5'-TTTTTTTTTAGGACAAAATGTTTC-3' (SEQ ID Nr.: 37)
und 5'-AATTCAGACTTTTGTAATTTGTG-3' (SEQ ID Nr.: 38)
verwendet wurden. Die Erfinder erhielten eine mehr als 90%ige Abdeckung
der kodierenden Sequenz von TS10q23.3 für alle analysierten Proben;
alle Mutationen wurde durch Sequenzierung eines neu amplifizierten
Produktes bestätigt.
-
RT-PCRTM-Expression:
-
Aus
Gefrierschnitten von 10 normalen Geweben und 10 hoch normalen Geweben
und 10 Präparaten hochgradiger
Glioblastome wurde Boten-RNA (Messenger RNA) isoliert. Die Gefrierschnitte
(jeweils 20, 5 μm) wurden
geschnitten und verwendet, um mRNA zu isolieren (Micro-Fast Track;
Invitrogen, San Diego, CA, USA). Aufeinander folgende Schnitte wurden
histologisch untersucht und es wurde gezeigt, dass die Schnitte überwiegend
normale Zellen oder Tumorzellen enthielten. Normale Schnitte wurden
aus Regionen erhalten, die im normalen Verlauf therapeutischer Kraniotomien
tumorfrei waren. Komplementäre
DNA wurde durch Verwendung von Superscript II und Primern zur Amplifizierung
von TS10q23.3 von –28
bis 347 oder 345 bis 1232 der kodierenden Region hergestellt. Die
verwendeten Primer waren:
M5'F: TCCTTTTTCTTCAGCCACAG (SEQ ID Nr.:
39)
M5'R: ATTGCTGCAACATGATTGTC
(SEQ ID Nr.: 40);
M3'F:
TGACAATCATGTTGCAGCA (SEQ ID Nr.: 41);
F3'R: TTTATTTTCATGGTGTTTTATCC (SEQ ID Nr.:
42).
-
Die
PCRTM-Bedingungen waren ähnlich wie die zuvor beschriebenen,
außer,
dass der Anheftungsschritt bei 53°C
durchgeführt
wurde.
-
Charakterisierung eines
TS10q23.3-Pseudogens:
-
sAus
einer cDNA-Bibliothek aus humanem fetalem Gehirn wurden DNA-Fragmente
amplifiziert, indem Pfu-Polymerase und eine gestaffelte PCRTM-Strategie verwendet wurden. Die Anfangsreaktion
mit χ μl enthielt 100
ng cDNA. Das in der ersten Runde der Amplifikation verwendete Primerpaar
war CTTCAGCCACAGGCTCCCAGAC (SEQ ID Nr.: 43) und GGTGTTTTATCCCTCTTG
(SEQ ID Nr.: 44), wonach die Reaktion 20-fach verdünnt und
mit den Primern CGGGATCCATGACAGCCATCATCAAAGAGATC (SEQ ID Nr.: 45)
und CGGAATTCTCAGACTTTTGTAATTG (SEQ ID Nr.: 46) reamplifiziert wurde.
Die verwendeten PCRTM-Bedingungen umfassten
einen anfänglichen
Denaturierungsschritt bei 94°C
für 5 Minuten,
gefolgt von 30 Zyklen bei 94°C
für 45
Sekunden, 55°C
für 30
Sekunden und 72°C
für 1 Minute.
Zur Bestimmung der chromosomalen Position dieses Pseudogens führten die
Erfinder ein Radiation Hybrid Mapping anhand des Genebridge 4 Panels
(Genome Systems) und des folgenden Primerpaars durch, das ein spezifisches
303-bp-Produkt des Pseudogens, aber nicht von TS10q23.3 erzeugte:
ATCCTCAGTTTGTGGTCTGC (SEQ ID Nr.: 47) und GAGCGTGCAGATAATGACAA (SEQ
ID Nr.: 48). Anhand dieser STS stellten die Erfinder fest, dass
sich das Pseudogen bei etwa 160 cM auf Chromosom 9 befand. Darüber hinaus isolierten
die Erfinder zwei bakterielle artifizielle Klone (BACs), 145c22
und 188122, die dieses Pseudogen tragen, und bestätigten seine
genomische DNA-Sequenz. Ein Vergleich der Kodierungssequenz von
TS10q23.3 mit der des Pseudogens ergab die folgenden Basenunterschiede:
T2G, C89T, T202C, T242C, G248A, A258G, G397A, A405T, G407A, T531C,
T544G, C556G, A672G, C700T, A705G, C720T, C900T und A942G. Die Nukleotidsequenz
für das
humane TS10q23.3-Pseudogen ist in SEQ ID Nr.: 64 ausgeführt.
-
Da
TS10q23.3 ein Tumorsuppressorgen zu kodieren scheint, war der erste
Schritt der Erfinder zur Identifizierung neuer Mutationen in diesem
Gen die Voranalyse von Primärtumoren
und TZL hinsichtlich eines Heterozygositätsverlustes (LOH) in dieser
Region von 10q23. Insgesamt wurden 342 Präparate von Primärtumoren
und 164 TZL anhand von polymorphen kurzen Tandem-Repeat-Markern
auf Chromosom 10, die sich in der Nähe des TS10q23.3-Locus befinden,
untersucht (Tabelle 6). In dieser Probenreihe beobachteten die Erfinder
in Präparaten
von Primärtumoren
einen Heterozygositätsverlust
(LOH) im Bereich von 20% bei Darmpräparaten bis zu 75% bei Glioblastoma
multiforme (GBM), wobei die LOH-Gesamthäufigkeit
~49% betrug. Bei TZL mit Probengrößen über neun variierte die LOH-Inzidenz von 28%
(Darm) bis 82% (GBM) mit einer Gesamthäufigkeit von ~46%.
-
TABELLE
6 LOH-ANALYSE
VON TUMORPRÄPARATEN
UND TUMORZELLLINIEN
-
-
- 1Der wurde nur für Probengrößen über neun berechnet.
- 2Proben, die erfolgreich amplifiziert
und sequenziert wurden (> 90%
der Kodierungssequenz wurde untersucht).
- 3Die Anzahl an Nicht-LOH-Proben, die
sequenziert wurde, ist in Klammer gezeigt. Bestimmte Primärtumor-DNAs,
besonders aus Pankreaskarzinomen und endometrialen Karzinomen, wurden
aus Mikroparaffinschnitten isoliert und konnten aufgrund schlechter
Template-Qualität
nicht zu > 90% sequenziert
werden.
- 4Alle untersuchten TZL zeigten einen
scheinbaren Heterozygositätsverlust
(LOH). TZL mit homozygoten Deletionen im kodierenden Abschnitt von
TS10q23.3 wurden nicht durch Sequenzierung untersucht.
- 5Diese Gesamtwerte beinhalten Proben,
die zuvor von Steck et al. (1997) berichtet worden sind.
- 6Fünf
dieser Kolonproben bestanden aus Krebserkrankungen, die in die Leber
metastasiert hatten, wenn auch die Lebermetastasen keinen Heterozygositätsverlust
(LOH) zeigten.
- 7Die Prostatazelllinie NCIH660 (TCL10F4)
wurde von Li et al. (1997) charakterisiert und zeigte eine homozygote
Deletion von Exon 2–9
von TS10q23.3.
- 8Diese Präparate metastasierter Tumore
stammten aus Adenokarzinomen, einem Sarkom, einem Nierenzellkarzinom
und einem Melanom. Die Metastaseläsionen befanden sich in der
Lunge, außer
bei dem Melanom, wo sie sich in der Leiste befanden.
- 9Von diesen 137 durch Sequenzierung
analysierten Präparaten
sind 45 bereits bekannt (Steck et al., 1997), 8 zeigten keinen Heterozygositätsverlust
(LOH) und 84 zeigten einen Heterozygositätsverlust (LOH).
-
Für die Suche
nach kodierenden Varianten von TS10q23.3 in Primärtumoren sequenzierten die
Erfinder Amplikons, die aus den Exons und flankierenden Splice-Übergangsstellen dieses Gens
bestanden, das aus Tumor-DNAs mit Heterozygositätsverlust (LOH) amplifiziert
wurde. Ein Vorbehalt dieses Ansatzes ist, dass er keine regulatorischen
Mutationen identifizieren kann, die das Expressionsniveau dieses
Gens beeinflussen. Darüber
hinaus schließt
diese Analyse die Möglichkeit
aus, mutante Homozygote und gemischte Heterozygote zu finden, aber
die Inzidenz dieser Art von Mutanten ist vermutlich gering. Zuvor
hatten die Erfinder berichtet, dass die Inzidenz von TS10q23.3-Kodierungsvarianten
bei Glioblastomen bei 6/26 (23%), bei Brustkarzinomen bei 2/14 (14%)
und bei Nierenkarzinomen bei 1/4 lag (Steck et al., 1997). In dieser
Studie entdeckten die Erfinder unter 84 Primärtumoren mit Heterozygositätsverlust
(LOH) im Bereich des TS10q23.3-Locus eine Frameshift-Mutation (Brustkarzinom),
eine Nonsense-Mutation (pädiatrisches
GBM), eine Splice-Variante (pädiatrisches
GBM) und eine Missense-Variante (Melanom, Tabelle 7).
-
TABELLE
7 TS10q23.3-Varianten
in Primärtumoren
und Tumorzelllinien
-
-
- 1Präparate von Primärtumoren.
- 2Analyse korrespondierender normaler
DNA zeigte, dass die TS10q23.3-Mutation dieser Probe eines primären Brusttumors
somatisch ist. Eine ähnliche
Analyse der TS10q23.3-Veränderungen
in den anderen drei Präparaten
von Primärtumoren
war nicht möglich,
da keine korrespondierenden normalen DNAs verfügbar waren. Die Erfinder stellten
jedoch fest, dass alle neun Mutationen bei Primärtumoren, die zuvor von Steck
et al. (1997) beobachtet worden sind, somatisch entstanden.
-
Außer Primärtumoren
untersuchten die Erfinder einer Reihe von Tumorzelllinien auf Veränderungen im
TS10q23.3-Gen. Diese TZL erlaubten es den Erfindern, Krebstypen
zu untersuchen, die in der Reihe der analysierten Primärtumoren
nicht vertreten waren, einschließlich Leukämie, Lymphom, Neuroblastom,
Retinoblastom und Blasen-, Hoden- und Uteruskarzinome. Unter den
75 TZL mit Heterozygositätsverlust
(LOH) identifizierten die Erfinder zehn homozygote Deletionen, welche
die Kodierungsregionen von TS10q23.3 betrafen (13A und 13B).
Die homozygoten Deletionen waren vorhanden in TZL von Astrozytomen
(1/1), Blasenkarzinom (1/3), Brustkarzinom (1/14), Glioblastom (2/8),
Lungenkarzinom (1/7), Melanom (4/7) und Prostatakarzinom (1/2).
Während
zwei der Zelllinien alle neun TS10q23.3-Exons verloren hatten, zeigten
die übrigen
8 TZL eine homozygote Deletion verschiedener Kodierungsabschnitte
des Gens. Eine Analyse der verbleibenden 65 TZL ergab eine Frameshift-,
eine Nonsense- und sieben nicht konservative Missense-Varianten (Tabelle
7).
-
Aufgrund
der relativ geringen Häufigkeit
beobachteter TS10q23.3-Mutationen in Primärtumoren im Vergleich zu TTL
untersuchten die Erfinder die Expression von TS10q23.3 in einer
Reihe von zehn GBM und zehn normalen Präparaten. Alle Normalproben
zeigten eine Expression von TS10q23.3, während keine der GBM-Proben
eine signifikante Expression dieser mRNA aufwies (13C und 13D).
In bestimmten Proben wurden nach längerer Exposition schwache
Signale beobachtet, wenn auch die Erfinder nicht unterscheiden konnten,
ob dieses Signal auf eine Kontamination der Schnitte durch normale
Zellen oder eine niedrige TS10q23.3-Expression in den Tumorzellen
zurückzuführen war.
Diese Beobachtung deutet allerdings an, dass die veränderte Expression
von TS10q23.3 bei der Tumorentstehung dieser GBM eine Rolle spielen
könnte. Die
Mechanismen oder der Mechanismus der Hemmung der TS10q23.3-Expression
und des TS10q23.3-Expressionsniveaus in anderen Arten von Primärtumoren
wird derzeit untersucht.
-
Die
Erfinder haben eine lange Reihe von Primärtumoren und TZL, die hinsichtlich
eines Heterozygositätsverlustes
(LOH) voranalysiert worden sind, auf Veränderungen von TS10q23.3 untersucht.
In dieser Reihe von 84 Primärtumoren
entdeckten die Erfinder nur vier mögliche inaktivierende TS10q23.3-Mutationen.
Zusammen mit den früheren
Befunden der Erfinder (Steck et al., 1997) zeigten damit 8/31 (26%)
der primären Glioblastome,
3/31 (10%) der primären
Brusttumore, 1/8 (13%) der primären
Nierentumore und 1/11 (9%) der primären Melanome Veränderungen
von TS10q23.3. Interessanterweise wiesen zwei der fünf pädiatrischen GBM
Veränderungen
von TS10q23.3 auf, die zur Expression eines nicht funktionellen
Proteins führen
dürften, was
darauf hinweist, dass eine weitergehende Analyse der Rolle von TS10q23.3
bei dieser Kinderkrankheit angebracht ist. In der Reihe von 75 TZL
beobachteten die Erfinder insgesamt 19 putative inaktivierende TS10q23.3-Mutationen.
-
Die
tatsächliche
Inzidenz von TS10q23.3-Mutationen in den verschiedenen Krebstypen
liegt wahrscheinlich zwischen der bei Primärumoren und der bei TZL beobachteten
Häufigkeit.
Die Befunde der Erfinder zeigen, dass TZL im Vergleich zu Primärtumoren
eine signifikant höhere
Inzidenz von Mutationen in TS10q23.3 aufweisen (Tabelle 6). Eine ähnliche
Beobachtung wurde von Spruck et al. für Mutationen von p16 bei Blasenkrebs
berichtet (Spruck III et al., 1994). Diese Diskrepanz ist vermutlich
auf eine oder mehrere der folgenden Möglichkeit zurückzuführen. Erstens
könnten
Tumorzellen bestimmte Kombinationen genetischer Läsionen,
die in vivo erworben worden sind, benötigen, um in vitro erfolgreich
kultiviert werden zu können. Zweitens
könnten
Mutationsereignisse in TS10q23.3 während der Passagierung von
TZL in vitro einen Wachstumsvorteil mit sich bringen oder klonale
Selektion bewirken. Drittens deutet die wesentlich reduzierte Expression
von TS10q23.3, wie sie bei 10/10 Präparaten primärer GBM beobachtet
wurde, darauf hin, dass bestimmte Tumore möglicherweise keine Kodierungsmutationen
in diesem Gen aufweisen, sondern stattdessen eine kleine Menge an
funktionellem TS10q23.3 exprimieren. Und viertens könnte die
Kontamination mit normalen Zellen und Heterogenität der Präparate von
Primärtumoren
die Feststellung homozygoter Deletionen, einem Mutationsmechanismen,
der bei einer signifikanten Anzahl von TZL am TS10q23.3-Locus beobachtet
wurde, verhindern. In Kontrollexperimenten wurde festgestellt, dass
das Vorhandensein von lediglich 5% kontaminierender DNA aus Normalgewebe
in den Tumorproben die Identifikation homozygoter Deletionen durch
diese Verfahren verhindert. Das Vorhandensein homozygoter Deletionen
mit Einfluss auf T510q23.3 in Primärtumoren könnte daher in der Analyse der
Erfinder leicht unterschätzt
worden sein und erfordert alternative Ansätze zur Evaluierung ihres Auftretens.
Eine weitere Komplikation ist allerdings das Vorhandensein eines
scheinbar ungesplicten TS10q23.3-Pseudogens auf Chromosom 9q; die
Kodierungssequenz von TS10q23.3 weicht an 16/1209 Basen von diesem
putativen Pseudogen ab (siehe „Verfahren").
-
Eine
Zusammenstellung von TS10q23.3-Veränderungen zeigt, dass das Spektrum
der Varianten sehr divers ist (14).
Alle identifizierten nicht-konservativen Missense-Substitutionen befinden
sich im N-terminalen Abschnitt von TS10q23.3 innerhalb seiner putativen
Phosphatasedomäne.
Die Läsionen,
die zu einer Verkürzung
von TS10q23.3 führen,
sind dagegen im gesamten Gen verteilt. Wenn alle verkürzten Formen
von TS10q23.3 nicht-funktional
sind, weisen die Daten darauf hin, dass die carboxyterminale Region
von TS10q23.3 für
die Expression des aktiven Proteins essenziell ist. Dies stimmt
mit der Ansicht überein,
dass die potenziellen Phosphorylierungsstellen und das PDZ-Motiv
für die
Funktion von TS10q23.3 wichtig sind. Alternativ könnten die
Sequenzen der C-terminalen Region dieses Proteins für eine korrekte
Faltung erforderlich sein. Interessanterweise wurden die einzigen,
bislang berichteten Keimbahnmutationen von TS10q23.3 bei Individuen
mit Cowden-Syndrom festgestellt (Liaw et al., 1997); alle anderen
charakterisierten TS10q23.3-Varianten aus Primärtumoren sind somatisch entstanden
(Tabelle 7). Die Diversität
der beobachteten Veränderungen
von TS10q23.3 lassen den Schluss zu, dass in der Bevölkerung
viele verschiedene Läsionen
dieses Gens vorhanden sind. Insgesamt legen die Daten nahe, dass
TS10q23.3 ein Tumorsuppressor ist, der bei der Entstehung vieler
Arten von Krebserkrankungen eine wesentliche Rolle spielt.
-
BEISPIEL 8
-
Rolle von TS10q23.3-Mutationen
bei Brustkrebs mit frühzeitiger
Manifestation; kausativ in Verbindung mit Cowden-Syndrom und ausgeschlossen
in brca1-negativen Fällen
Verfahren
-
Klinisches Material:
-
Nach
Einholung des informierten Einverständnisses wurden Personen mit
Cowden-Syndrom Blutproben abgenommen. Ein Aliquot wurde zur DNA-Extraktion verwendet,
während
aus einer zweiten Probe mononukleäre periphere Blutzellen gereinigt
und verwendet wurden, um eine EBV-transformierte lmyphoblastoide Zelllinie
zu erzeugen. Die Diagnose von CS erfolgte nach den CS-Diagnosekriterien
des International Cowden's
Consortium (Nelen et al., 1996). Bei Personen mit Brustkrebs mit
frühzeitiger
Manifestation bestand die Stichprobe aus 63 Frauen, die mit unter
35 Jahren Brustkrebs entwickelten (Durchschnittsalter bei der Diagnosestellung
ist 27,7 Jahre), bei denen CS klinisch diagnostiziert wurden und
bei denen zuvor gezeigt wurde, dass sie keine eindeutig nachteiligen
BRCA1-Mutationen trugen (Frauen in der Stichprobe trugen Missense-Polymorphismen
unbekannter Signifikanz). Diese Frauen bildeten eine Untergruppe
einer Stichprobe von 798 nicht verwandten Personen aus 20 Partnereinrichtungen,
die aus Familien ausgewählt
wurden, bei denen allgemein ein erhöhtes Risiko für BRCA1-Mutationen
vorlag. Die meisten Familien wurden gewählt, da mehrere Fälle von
Brustkrebs vorlagen, der Brustkrebs im frühen Alter diagnostiziert wurde
und eine Inzidenz von Ovarialkrebs vorhanden war; diese Zustände sind
bereits früher
mit Keimbahnmutationen von BRCA1 in Verbindung gebracht worden.
Einige der Familien erstreckten sich bis zu Verwandten zweiten Grades.
Alle Proben aus Einrichtungen in den Vereinigten Staaten stammten
von Personen, die an Forschungsstudien über die Genetik von Brustkrebs
teilnahmen. Alle Proben aus Einrichtungen außerhalb der Vereinigten Staaten
wurden in Übereinstimmung
mit den geltenden Richtlinien in Bezug auf Forschung am Menschen
erhalten, die von den entsprechenden Behörden, die für die Einrichtungen zuständig waren,
aufgestellt wurden. In die Stichprobe wurde nur ein Vertreter jeder
Familie eingeschlossen, und es wurden keine Familien eingeschlossen,
bei denen bekannterweise eine genetische Verknüpfung zu BRCA1 vorliegt. Es
handelt sich hierbei um eine heterogene Probe, die eine Diversität unter
Patienten wiedergibt, welche in Kliniken als Hochrisikopersonen
vorstellig wurden, im Gegensatz zu einer kontrollierten Stichprobennahme
bei Familien- oder Populationsstudien. Dies veranlasste die Erfinder,
bei ihren Analysen Proben zu verwenden, die nicht erfordern, dass
die Probenhäufigkeit
von Untergruppen die Häufigkeit
in der allgemeinen Population wiedergibt. Die Erfinder können daher
beispielsweise die Wahrscheinlichkeit prüfen, dass eine Frau, bei der
im Alter von 30 Jahren Brustkrebs diagnostiziert wird, eine schädliche Mutation
von BRCA1 oder TS10q23.3 (auch als MMAC1) aufweist, aber die Erfinder
können
keine Schätzung
der Häufigkeit
solche Frauen in der allgemeinen Bevölkerung vornehmen. Von den
Proben, die in der TS10q23.3-Studie verwendet wurden, wurden alle
identifizierenden Merkmale entfernt.
-
DNA-Extraktion:
-
Nach
Erhalt des informierten Einverständnisse
wurde die genomische DNA der Patienten aus Vollblut oder lymphoblastoiden
Zelllinien unter Verwendung des Q1Amp Blut Maxi Kits extrahiert.
Die Konzentration wurde durch OD250 gemessen,
und die Reinheit wurde anhand des Verhältnisses von OD260/OD280 bestimmt.
-
Genotypbestimmung:
-
Primerpaare
für den
Locus auf Chromosom 10 wurden von Research Genetics erhalten. Der
Vorwärtsstrangprimer
wurde in Gegenwart von 32P-γATP und Polynukleotidkinase
endständig
markiert. PCRTM-Reaktionen wurden in einem
Reaktionsgesamtvolumen von 30 Mikrolitern durchgeführt. Die
Reaktionen bestanden aus 10 mM eines jeden Primers, 200 mM Desoxynukleotiden,
1,5 Einheiten Taq-DNA-Polymerase und 50 ng genomischer DNA. Die
durchgeführte
PCRTM bestand aus 35 Zyklen mit 45 Sekunden
Denaturierung bei 94°C, 45
Sekunden Anheftung bei 55°C
und 1 Minute Verlängerung
bei 72°C.
Am Schluss fand eine zusätzliche
Verlängerungsphase
von 10 Minuten statt. Die PCRTM-Reaktionen wurden
durch Zugabe von 20 Mikrolitern Stopp-Lösung (95% Formamid, 1 mM EDTA,
0,25% Bromphenolblau, 0,25% Xylolcyanol) angehalten. Anschließend wurden
die Reaktionen 5 Minuten lang bei 94°C denaturiert und die Produkte
wurden auf einem 8%igen denaturierenden Polyacrylamidgel aufgetrennt.
Die Allelgrößen wurden
durch Vergleich mit dem SequaMark (Research Genomics) bestimmt,
der als Größenstandard
auf den Gelen vorhanden war.
-
Kopplungsanalyse:
-
Anhand
von MLINK wurde eine Zweipunkt-Kopplungsanalyse durchgeführt. Personen
unter 20 Jahren wurden als unbekannt eingestuft. Die Häufigkeit
des Krankheitsgens wurde überall
auf 0,000001 gesetzt und die Markerallelhäufigkeiten wurden mit ILINK
geschätzt.
Sowohl MLINK als auch ILINK stammen aus dem LINKAGE-Paket, Version
5.2 (Lathtop et al., 1984). Die Rekonstruktion der wahrscheinlichsten
Haplotypen in Familie D erfolgte mit GENEHUNTER (Kruglyak et al.,
1996). Stammbäume
wurden mit Cyrillic Version 2.02 gezeichnet.
-
Ergebnisse
-
Beim
Cowden-Syndrom (CS) (Lloyd und Dermis, 1963) oder multiplen Hamartomsyndrom
(Weary et al., 1972) handelt es sich um eine autosomal dominante
Erkrankung, die mit der Entwicklung von Hamartomen und gutartigen
Tumoren in unterschiedlichen Geweben in Verbindung steht, darunter
der Haut, der Schilddrüse,
der Brust, dem Darm und dem Gehirn. Es ist darauf hingewiesen worden,
dass für
Frauen mit CS ein erhöhtes
Brustkrebsrisiko besteht (Brownstein et al., 1978), und, wie bei
anderen Anfälligkeitssyndromen,
scheinen sie Brustkrebs in einem frühen Alter zu entwickeln. CS
ist außerdem
mit einer spezifischen Hautläsion, dem
Trichilemmom (Tumor des follikulären
Infundibulums), verbunden, und deshalb ist dieses Brustkrebsanfälligkeitssyndrom
am Vorhandensein eines kutanen Biomarkers erkennbar (Brownstein
et al., 1977, 1978). Die Erfinder haben die klinischen und pathologischen
Befunde in diesem Syndrom ausführlich
untersucht und gezeigt, dass das Durchschnittsalter der Präsentation
mit malignem Brustkrebs bei CS 46 Jahre beträgt, wobei der Altersbereich
der Präsentation
mit Brustkrebs bei betroffenen Frauen bei 33 bis 73 Jahren liegt.
Darüber hinaus
lag bei nur sehr wenigen der Frauen mit CS eine Familiengeschichte
mit Brustkrebs vor. Interessanterweise scheint es für Männer mit
CS kein erhöhtes
Risiko zur Entwicklung von Brustkrebs zu geben (Brownstein et al.,
1978). Die Erfinder haben auch gezeigt, dass Frauen mit CS sehr
häufige,
gutartige Brusterkrankungen entwickeln und häufig vor der Entwicklung von
Brustkrebs mehrere Brustbiopsien durchgeführt worden sind. Eine Krankengeschichte
mit Hautkrankheit und gutartiger Brusterkrankung kann daher eine
Identifizierung betroffener Personen vor der Entwicklung von Brustkrebs
bei dieser Hochrisikopopulation ermöglichen.
-
Es
ist bereits zuvor gezeigt worden, dass sich auf Chromosom 10 ein
Locus für
CS befindet (Nelen et al., 1996). In dieser Studie wurden insgesamt
12 Familien untersucht, was zur Identifizierung des für das Cowden-Syndrom
kritischen Intervalls zwischen Marker D10S215 und D10S564 führte. Bestimmte
betroffene Personen in diesen Familien hatten CS und Lhermette-Duclos-Krankheit
(LDD) (Nelen et al., 1996; Liaw et al., 1997), eine seltene Gehirnerkrankung,
die durch ein dysplastisches Gangliozytom des Zerebellums gekennzeichnet
ist (Albrecht et al., 1992). Die Feinkartierung dieses Bereichs
führte
zur Verfeinerung dieses ersten Ergebnisses (Liaw et al., 1997),
was darauf hinweist, dass sich das CS-Gen zwischen Marker D10S215
und D10S541 befindet. In der jüngeren
Vergangenheit wurde gezeigt, dass betroffene Personen in vier Familien mit
CS Keimbahnmutationen (Liaw et al., 1997) in einem als PTEN bezeichneten
Gen (Li et al.,1997), TS10q23.3 (Steck et al., 1997) oder TEP1 (Li
et al., 1997) aufweisen, welches sich im für das Cowden-Syndrom kritischen
Intervall auf Chromosom 10 befindet. Interessanterweise enthält das vorhergesagte TS10q23.3-Protein
Sequenzmotive mit signifikanter Homologie zu der katalytischen Domäne von Proteinphosphatasen
und zu den Zytoskelettproteinen Tensin und Auxillin (Li et al.,
1997, Steck et al., 1997). Darüber
hinaus sind in humanen Tumoren oder Tumorzelllinien der Brust, des
Gehirns, der Prostata und der Nieren Mutationen der Kodierungsregion
von TS10q23.3 beobachtet worden (Li et al., 1997, Steck et al.,
1997). Obwohl die Funktion dieses Gens unbekannt ist, ist es wahrscheinlich,
dass TS10q23.3 bei der Regulierung der Zellproliferation eine Rolle
spielt und dass der Verlust seiner Funktion bei der Entwicklung
humaner Tumor von Bedeutung ist.
-
Kopplungsanalyse und Mutationsanalyse
bei CS-Sippen
-
Zur
Erweiterung der Beobachtungen, die auf einen CS-Locus auf Chromosom
10 hinweisen, führten die
Erfinder eine Zweipunkt-Kopplungsanalyse anhand von fünf Markern
im für
das Cowden-Syndrom kritischen Intervall bei vier Familien mit klinisch
erwiesenem CS durch (Nelen et al., 1996). Alle Familien wurden ausführlich untersucht
und die Diagnose dieses Syndroms erfolgte nach den CS-Diagnosekriterien
des International Cowden's
Consortium (Nelen et al., 1996). Zwei kleine Familien zeigten positive
LOD-Werte, was eine Kopplung mit drei Loci auf Chromosom 10 nicht
ausschließen
konnte (siehe Familie A und B, Tabelle 8). Zwei andere Familien
mit klinischen Befunden, die mit den oben beschriebenen identisch
waren, zeigten für
einige der Marker in dieser Region signifikant negative LOD-Werte
(Familie C und D, Tabelle 8). Auch ein Heterogenitätstest wurde
durchgeführt,
der nicht-signifikante Ergebnisse lieferte. Diese Befunde wurden
durch die Konstruktion der Haplotypen unterstützt (15).
Insbesondere gibt Person 2 aus Familie C den vom nicht-betroffenen
Vater geerbten Haplotyp an beide betroffene Kinder weiter. In Familie
D schließlich
haben Person 2 und 20 einen Haplotyp geerbt, der sich von dem ihrer
betroffenen Verwandten unterscheidet.
-
TABELLE
8 Zweipunktanalyse
von CS-Familien mit CA-Wiederholungsmarkern
-
-
-
Anhand
eines auf PCRTM und Sequenzierung basierenden
Ansatzes haben die Erfinder in 16 betroffenen Personen aus diesen
4 Familien die 9 Exons und damit verbundenen Splice-Übergänge von TS10q23.3 untersucht,
wobei die beschriebenen Primer verwendet wurden (Steck et al., 1997).
Interessanterweise hatten 4 dieser 16 Personen Brustkrebs und 2
der 4 hatten Brustkrebs vor dem 40. Lebensjahr. Die Erfinder konnten in
der Kodierungssequenz dieser 16 Personen aus diesen 4 Familien mit
den klassischen Anzeichen und Symptomen von CS keine Mutationen
feststellen.
-
Mutationsanalyse bei Personen
mit CS
-
Die
Erfinder haben einen Satz von 31 Personen aus 23 Familien mit CS
analysiert, deren Verwandte nicht in den Kopplungsstudien der Erfinder
verwendet worden sind. Von den 31 Personen waren 13 verwandte Personen
aus 5 Familien. Insgesamt wurden also 23 nicht verwandte Probanden
getestet. Eine einzelne betroffene weibliche Person (Walton et al.,
1986) zeigte eine Frameshift-Mutation in Exon 7 der Kodierungssequenz
(siehe 16). Spezifischer zeigten die
Erfinder eine AT-Insertion nach Nukleotid 791 (791insAT), was zu
einem Frameshift und zu einem stromabwärts (downstream) liegenden
vorzeitigen Abbruchcodon führte. Interessanterweise
entwickelte diese Frau einen im Mammogramm negativen Brustkrebs
im Alter von 36 Jahren, der zum Zeitpunkt einer prophylaktischen
Mastektomie entdeckt wurde (Walton et al., 1986). Die Probandin
hatte einen nicht betroffenen Bruder und eine betroffene Tochter.
Direkte Sequenzierung von Exon 7 in diesen Personen zeigte das Vorhandensein
der identischen Mutation in der betroffenen Tochter (16) und das Nichtvorhandensein der Mutation in
dem nicht betroffenen Bruder. Bei der Analyse einer zweiten Person
mit CS und Brustkrebs mit frühzeitiger
Manifestation (im Alter von 33 Jahren) zeigten die Erfinder eine
Insertion von drei Basen in Exon 2 (137ins3), die zur Insertion
einer Aminosäure
(Asn) führte.
Bei einer anderen Frau mit beidseitigem Brustkrebs und Endometrialkrebs
identifizierten die Erfinder schließlich eine Frameshift-Deletion
von 13 Basenpaaren in Exon 8 (915del12). Diese Daten zeigen 3 mutante
Allele von TS10q23.3, die mit CS (Liaw et al., 1997) und insbesondere
mit CS und Brustkrebs (Brownstein et al., 1978) verknüpft sind.
Bei 27 Personen aus 20 Familien entdeckten die Erfinder jedoch keine
Mutationen in den Kodierungssequenzen von TS10q23.3. In dieser Population
hatten 7 dieser Personen Brustkrebs, wenn auch jede dieser Frauen Brustkrebs
im Alter über
40 Jahren entwickelte. Durch Kombination der Familiendaten und die
dieser Personen entdeckten die Erfinder daher Kodierungssequenzmutationen
bei 4 Personen aus 3 CS-Familien, entdeckten jedoch keine Veränderungen
der Kodierungssequenz (d. h. Missense- oder Silent-Varianten, d. h.
Varianten ohne Phänotyp)
in 43 weiteren Personen aus 24 Familien mit CS.
-
Mutationsanalyse bei Frauen
mit Brustkrebs mit frühzeitiger
Manifestation
-
Vielen
weist auf die Existenz eines genetischen Mechanismus hin, der die
Bildung eines Brusttumors bei Brustkrebs mit früher Manifestation (Entwicklung
von Brustkrebs vor dem Alter von 40 Jahren) reguliert (Claus et
al., 1990). Da CS auf autosomal dominante Art vererbt wird, könnten die
genetischen Mechanismen, welche die Entwicklung von Brustkrebs bei
dieser Population regulieren, auch eine Rolle bei der Entwicklung von
Brustkrebs mit früher
Manifestation spielen. Da die Erfinder TS10q23.3-Keimbahnmutationen
in CS in Verbindung mit Brustkrebs mit früher Manifestation entdeckt
haben und Mutationen in diesem Gen bei Brusttumoren und Brusttumorzelllinien
mit relativ großer
Häufigkeit
auftreten (Steck et al., 1997, Li et al., 1997), wollten die Erfinder
die Rolle von TS10q23.3-Keimbahnmutationen bei Brustkrebs mit früher Manifestation
weiter untersuchen. Um einen Stichprobensatz zu erhalten, in dem
TS10q23.3-Keimbahnmutationen mit größerer Häufigkeit vorkommen, sequenzierten
die Erfinder das Gen bei 63 Frauen, die Brustkrebs vor dem Alter
von 35 Jahren entwickelt hatten (Durchschnittsalter bei der Diagnosestellung
war 27,7 Jahre), bei denen offensichtlich keine klinische Diagnose
von CS vorlag und bei denen zuvor gezeigt worden war, dass sie eindeutig
keine nachteilige Mutationen in BRCA1 aufweisen (5 Frauen in der
Stichprobe trugen Polymorphismen unbekannter Signifikanz). In den
9 Exons von TS10q23.3 in dieser Stichprobe wurden keine Veränderungen
der Kodierungssequenz entdeckt. Wenn dagegen dieselben Kriterien
zum Mutationsnachweis und zur Mutationsanalyse auf einen in ähnlich definierten
Satz von nicht-Ashkenazi-stämmigen
Personen mit Brustkrebs angewandt werden würden (ohne Ausschluss von BRCA1-Trägern), würden die
Erfinder erwarten, 7 nachteilige Mutationen und 5 Missense-Polymorphismen
unbekannter Signifikant bei BRCA1 zu entdecken. Darüber hinaus
haben die Erfinder abgesehen von den oben beschriebenen 4 CS-Patienten
mit TS10q23.3-Keimbahnmutationen
in über 200
Keimbahnchromosomen keine Sequenzpolymorphismen in der Kodierungssequenz
dieses Gens festgestellt und tatsächlich nur einen Sequenzunterschied
(silent, still) zwischen den Sequenzen von Mensch und Schimpanse
gefunden. Wenn die Häufigkeit
von Varianten der Kodierungssequenz und der proximalen Splice-Übergangssequenzen
bei TS10q23.3 in der Population, aus der diese Stichprobe entnommen
wurde, bei 5% liegen würde,
hätten
die Erfinder eine Chance von 95%, eine oder mehrere solcher Varianten
zu entdecken.
-
Diskussion
-
Das
Cowden-Syndrom nimmt unter den autosomal dominant vererbten genetischen
Syndromen insofern eine Sonderstellung ein, als es die Anfälligkeit
zur Entwicklung von Brustkrebs erhöht und einen besonderen Biomarker
auf der Haut aufweist, das Trichilemmom (Brownstein et al., 1997;
1978). Darüber
hinaus liegt bei Frauen mit CS häufig
eine Krankengeschicht mit mehreren Brustbiopsien aufgrund gutartiger
Brusterkrankung vor der Entwicklung von Brustkrebs vor. Die meisten
dieser Frauen weisen keine Familiengeschichte von Brustkrebs auf.
Bislang ist die Verbindung zwischen CS und weiblichem Brustkrebs
die am besten beschriebene Verbindung zwischen CS und der Anfälligkeit
für eine
organspezifische Krebserkrankung (Brownstein et al., 1977). Andere
Organsysteme, wie beispielsweise die Schilddrüse, scheinen bei solchen Personen
Krebs mit erhöhter
Häufigkeit
zu entwickeln. Im Vergleich zu anderen Syndromen mit Anfälligkeit
für autosomalen Brustkrebs.
wie beispielsweise dem in Verbindung mit Mutationen in BRCA1 (Ford
et al., 1995), ist die Entwicklung von Ovarialkrebs in diesem Syndrom
relativ selten. CS hat mit diesen Syndromen gemeinsam, dass sich der
Brustkrebs in einem frühen
Alter manifestiert und die Wahrscheinlichkeit eines zweiseitigen
Brustkrebses erhöht
ist. Frühere
Beobachtungen zeigten eine Kopplung von CS mit Chromosom 10q22-23
(Nelen et al., 1996). Darüber
hinaus ist inzwischen offenkundig, dass CS bei Personen mit Mutationen
in einem Gen (Liaw et al., 1997), das unter der Bezeichnung PTEN
bekannt ist (Li et al., 1997), TS10q23.3 (Steck et al., 1997) oder TEP1
(Li et al., 1997), die in für
da Cowden-Syndrom kritischen Intervall auf Chromosom 10 gefunden
wurden, verknüpft
ist (Liaw et al., 1997),
-
In
den hier berichteten Beobachtungen identifizierten die Erfinder
drei neue Keimbahnmutationen in der Kodierungssequenz von TS10q23.3
in Verbindung mit CS und vor allem bei Personen mit CS und Brustkrebs.
Bei zwei verwandten Personen mit CS beschrieben die Erfinder eine
Frameshift-Mutation in Exon 7, die zu einem vorzeitigen Terminationscodon
führt und
die bei einer betroffenen Mutter und ihrer betroffenen Tochter identisch
ist. Diese TS10q23.3-Mutation scheint mit Brustkrebs mit frühzeitiger
Manifestation verknüpft
zu sein, da eine der beiden betroffenen Personen Brustkrebs im Alter
von 36 Jahren entwickelte. Bei einer dritten betroffenen Person
identifizierten die Erfinder eine Deletion von 13 Basenpaaren in
Exon 8. Diese Person entwickelte Brustkrebs zwar nicht in einem
frühen
Alter, hatte aber eine Krankengeschichte von beidseitigem Brustkrebs.
Außerdem
entwickelte sie während
der Behandlung mit Tamoxifen Endometrialkrebs. In Anbetracht dessen,
dass Endometrialkrebs mit CS (Starink et al., 1986) und der Verwendung
von Tamoxifen (Fornander et al., 1989) in Verbindung gebracht wurde,
ist der Beitrag beider Risikofaktoren zur Krankheitsentwicklung
bei dieser Frau unbekannt. Dies eröffnet aber die Möglichkeit,
dass die Unterpopulation von Frauen, die während der Behandlung mit Tamoxifen
Endometrialkrebs entwickeln, CS und/oder Mutationen in TS10q23.3 aufweisen
könnten.
Schließlich
haben die Erfinder bei einer anderen Frau, die im Alter von 33 Jahren
Brustkrebs entwickelte, eine Insertion von 3 Basen in Exon 2 identifiziert.
-
In
dem von den Erfindern analysierten Satz von Personen mit CS haben
die Erfinder in 4 Personen aus 3 Familien TS10q23.3-Keimbahnmutationen
entdeckt, aber in den verbleibenden 43 Personen aus 24 nicht verwandten
Familien keine Veränderungen
der Kodierungssequenz festgestellt. Diese Daten unterstützen die
eingeschränkten
Kopplungsdaten der Erfinder und deuten an, dass nicht bei allen
CS-Familien eine Verknüpfung
zu dem auf Chromosom 10 identifizierten Locus vorhanden ist. Während die
von den Erfordern durchgeführten
Studien nicht ausschließen,
dass Mutationen in den 5'-regulatorischen
Regionen oder in der 3'-untranslatierten
Region von TS10q23.3- oder andere Mechanismen, die das Expressionsniveau
verändern, beispielsweise
Silencing durch Methylierung, mit CS in Verbindung stehen, unterstützen sowohl
die Kopplungsdaten als auch die Ergebnisse der DNA-Sequenzierung
die Vorstellung, dass CS genetisch heterogen ist. Tuberöse Sklerose,
eine andere autosomal dominante Erkrankung, die mit der Bildung
von Hamartomen in der Haut und anderen Organen verknüpft ist,
erwies sich als genetisch heterogen mit unterschiedlichen Loci auf
Chromosom 9q34 (Haines et al., 1991) und Chromosom 16p13.3 (Kandt
et al., 1992). Die Ergebnisse der Erfinder deuten an, dass dies
auch für
CS zutreffen könnte.
Warum dies im Rahmen der ersten Beobachtungen nicht gezeigt werden
konnte, ist unklar, könnte
aber auf den ethnischen Hintergrund der ersten untersuchten Familien
zurückzuführen sein
(Nelen et al., 1996, Liaw et al., 1997). Darüber hinaus präsentierten
Bestimmte dieser Personen mit CS und Lhermette-Duclos-Krankheit,
welche die Erfinder nie bei einem CS-Probanden oder in einer CS-Familie
gesehen haben (Nelen et al., 1996, Liaw et al., 1997).
-
Vieles
weist auf das Vorhandensein eines genetischen Mechanismus hin, der
die Bildung eines Brusttumors bei Brustkrebs mit frühzeitiger
Manifestation reguliert (Claus et al., 1990). Tatsächlich wurde
Brustkrebs mit fühzeitiger
Manifestation mit Mutationen in BRCA1 (Miki et al., 1994) und BRCA2
(Wooster et al., 1995) in Verbindung gebracht. Es besteht eine Verknüpfung zwischen
CS und Brustkrebs mit frühzeitiger
Manifestation, und bei dem Krebs handelt es sich in der Regel um
ein duktales Karzinom ( eBrownsteint al., 1977, Brownstein et al.,
1978). Rachel Cowden, nach der dieses Syndrom bekannt ist, verstarb
offensichtlich im Alter von 31 Jahren an Brustkrebs (Lloyd und Dennis,
1963, Brownstein et al., 1978). Wie hierin beschrieben, haben die
Erfinder TS10q23.3-Mutationen bei 2 Personen mit CS mit Brustkrebs
mit frühzeitiger
Manifestation sowie bei 1 Person mit beidseitigem Brustkrebs identifiziert.
Wenn die Erfinder allerdings bei einer Untergruppe von Frauen mit
Brustkrebs mit frühzeitiger
Manifestation ohne Anzeichen von CS und zuvor gezeigten Wildtyp-Sequenzen
von BRCA1 nach TS10q23.3-Keimbahnmutationen suchten, konnten die
Erfinder keine Sequenzvarianten entdecken. Diese Daten weisen darauf
hin, dass Keimbahnmutationen in TS10q23.3 in zumindest dieser Unterpopulation
von Fällen
mit Brustkrebs mit frühzeitiger
Manifestation selten auftreten.
-
BEISPIEL 9
-
Suppression der Tumorigenität von Glioblastomzellen
durch Adenovirus-vermittelten MMAC1/PTEN-Gentransfer
-
Es
wurden weitere Studien konzipiert, um die Funktion von MMAC1/PTEN
als Tumorsuppressor weiter zu untersuchen. Es wurde ein replikationsdefektes
Adenovirus (MMCB) konstruiert, um MMAC1 effizient und transient
in Tumorzellen zu transduzieren. Die in diesem Beispiel vorgestellten
Daten unterstützen
eine Tumorsuppressionsaktivität
von MMAC1/PTEN in vivo und deuten an, dass in-vivo-Gentranser mit
diesem rekombinanten adenoviralen Vektor bei der Krebs-Gentherape
nützlich
sein wird.
-
Material und Verfahren
-
Zelllinien:
-
Die
MMAC1-mutierte Glioblastom-Zelllinie U87MG wurde von der American
Type Culture Collection (ATCC) erhalten. Die Zellen wurden in Kulturmedium
(DME/10% FBS/1% L-Glutamin) in einer befeuchteten Atmosphäre mit 7%
CO2 bei 37°C gehalten. Embryonale Nierenzellen
293 wurden ebenfalls von ATCC erhalten und in DME-Kulturmedium gezüchtet, das
mit 10% FBS angereichert war.
-
RT-PCR Analyse:
-
Aus
U87MG-Zellen (Tri Reagent, Molecular Research Center) wurde nach
den Anleitungen des Herstellers Gesamt-RNA isoliert. RNA wurde unter
Verwendung von MuLV-RNA (RNA PCR Kit, Perkin-Elmer), Random-Hexamer
und anderen Kitreagenzien revers transkribiert, gefolgt von PCR
unter Verwendung der Primer MAC1.6f (5'-CTG CAG AAA GAC TTG AAG GCG TA-3', SEQ ID Nr.: 58)
und MAC1.6r (5'-GCC
CCG ATG TAA TAA ATA TGC AC-3')
(SEQ ID Nr.: 59), die Sequenzen in den MMAC1-Exons 2 und 5 entsprachen.
Die Amplifizierungsbedingungen waren Denaturierung bei 95°C für 1 Minute,
anschließend
(95°C, 15
s; 55°C,
30 s) für
25 Zyklen, dann 72°C
für 5 Minuten.
Die erwartete Größe des Normalproduktes
war 317 bp. Die anomale Bande von U87MG wurde aus einem Agarosegel
ausgeschnitten, gereinigt (UltraClean, Mo Bio Labs) und mit einem
automatischen Sequenziersystem (ABI 373A, Perkin Elmer) direkt sequenziert.
-
Viren:
-
Ein
rekombinantes Adenovirus, das Wildtyp p53 (FTBC) enthielt, wurde
wie zuvor beschrieben (Wills et al., 1994) konstruiert. Das Genom
dieses Vektors weist Deletionen der E1- und E3-Region und des Protein-IX-Gens
auf und exprimiert sein Transgen unter der Kontrolle des Immediate-Early-Promotors/Enhancers des
humanen Zytomegalievirus (CMV). Der MMAC1/PTEN-Vektor MMCB wurde
auf genau dieselbe Art und Weise konstruiert, außer, dass p53 durch eine cDNA,
die MMAC1 in voller Länge
kodiert (Steck et al., 1997), ersetzt wurde. Der Kontrollvektor
GFCB wurde so konstruiert, dass er MMCB entsprach, mit Ausnahme
seines Transgens, verstärktem
grün fluoreszierenden
Protein (Clontech). Es wurde ein weiterer passender Kontrollvektor,
ZZCB, ohne ein Transgen konstruiert. Der BGCA-Kontrollvektor, der
LacZ von E.coli, angetrieben durch den CMV-Promotor, exprimiert,
wurde aufgrund der Einschränkungen
in Bezug auf die Verpackungsgröße in ein
Genom mit partieller E4-Deletion in Ergänzung zur Deletion von E1,
E3 und Protein IX konstruiert (Wang et al., 1997). Alle Viren wurden
in 293-Zellen gezüchtet
und durch DEAE-Säulenchromatographie
wie beschrieben (Huyghe et al., 1995) gereinigt. Viruspartikelkonzentrationen
wurden durch Resource Q HPLC (Shabram et al., 1997) bestimmt und
die Primärstruktur
aller Transgene wurde durch automatisiertes Sequenzieren viraler
DNA verifiziert.
-
Immunnachweis von MMAC1-Protein:
-
Zellmonolayer
wurden 24 Std. lang mit GFCB oder MMCB mit einer unterschiedlichen
Anzahl an Viruspartikeln pro Milliliter Wachstumsmedium (pn/ml)
infiziert. Virushaltige Lösungen
wurden nach 24 Std. entfernt und die Zellen wurden zu diesem Zeitpunkt
entweder geerntet oder mit Wachstumsmedium gefüttert und zu späteren Zeitpunkten
geerntet. Das Ernten der Zellen erfolgte durch Abschaben in kalte
phosphatgepufferte Salzlösung
(PBS), Zentrifugation und erneutes Waschen in kaltem PBS, anschließendes Gefrieren-Auftauen und
Resuspendieren in Lysepuffer [50 mM MOPS, pH 7,0, 150 mM NaCl, 1
% NP-40, 5% Glycerol, 0,4 mM EDTA und ergänzt durch 1 mM DTT und IX Complete
Protease Inhibitor Cocktail (Boehringer Mannheim)]. Zelllysate wurde
durch Zentrifugation bei 10.000 × g für 15 Minuten geklärt und die Überstände auf
Proteingehalt normalisiert. Die Proben wurden durch SDS-PAGE in
vorgegossenen 8%igen TRIS-Glycin-Gelen
(Novex) aufgetrennt, anschließend
auf Poly(vinylidendifluorid)-Membranen (Immobilon-P) für ein Western
Blotting überführt. Die
Membranen wurden mit TBST blockiert, das 5% Magermilch enthielt,
und dann mit einem polyklonalen Anti-MMAC1-Kaninchenantikörper (BL74),
gefolgt von Esel-Anti-Kaninchen-IgG, konjugiert mit Meerrettichperoxidase
(Amersham), geblottet. MMAC1 wurde durch Chemilumineszenz (HCL Kit,
Pierce) anhand von Kodak XAR-5-Film nachgeweisen.
-
FACS-Assay zur Bestimmung
der Infektiosität:
-
U87MG-Zellen
wurden mit 2 × 105 Zellen/Vertiefung in Platten mit 6 Vertiefungen
ausplattiert und über Nacht
inkubiert, anschließend
24 Stunden lang mit GFCB in Konzentrationen zwischen 1 × 105 bis 1 × 109 Partikeln/ml infiziert. Die Zellen wurden
durch Trypsinierung geerntet und durchflusszytometrisch (Becton
Dickinson FACScan) hinsichtlich grüner Fluoreszenz (Peaknachweis
bei 525 nm, Filter FL-I) analysiert. Zellen wurden hinsichtlich
ihrer Grösse
(„forward
scatter", FSC) und
Granularität
(„side
scatter", SSC) eingegrenzt
und ein Schwellenwert für
die Fluoreszenzintensität
wurde derart eingerichtet, dass ~99% der nicht infizierten Zellen
negativ waren. Anschließend
wurde der Prozentanteil von GFCB-infizierten Zellen, deren Fluoreszenz
diesen Schwellenwert übertraf,
ermittelt, was eine Mindestschätzung
des Prozentanteils infizierter Zellen darstellte.
-
3H-Thymidineinbautest:
-
Zellen
wurden mit 5 × 103
Zellen/Vertiefung in Mikrotiterplatten mit 96 Vertiefungen (Costar)
ausplattiert und über
Nacht inkubiert. Verdünnungen
von ZZCB, GFCB und FTCB und MMCB in Medium im Bereich von 5 × 106 bis
1 × 109
Partikel/ml wurden in dreifacher Ausführung zu den Zellmonolayern
gegeben und anschließend
24 Std. inkubiert. Virushaltige Lösungen wurden 24 Stunden nach
der Infektion entfernt und für
weitere 24 Stunden durch neues Gewebekulturmedium ersetzt. 4 Stunden
vor dem Ernten wurden die Zellen mit 1 μCi 3H-Thymidin
pro Vertiefung behandelt. Zellen wurden auf Glasfaserfilter geerntet
und der Einbau von 3H-Thymidin wurde mit
Flüssigszintillation
(Top Count, Packard) bestimmt. Die Ergebnisse sind als Prozentanteil
pufferbehandelter Kontrollen aufgetragen (Mittelwert ± SD).
-
Zellzahl/Testung auf Lebensfähigkeit:
-
Subkonfluente
Monolayer von U87MG-Zellen wurden in dreifacher Ausführung mit
MMCB oder GFCB-Adenovirus in verschiedenen Konzentrationen für 24 Stunden
infiziert, wonach Überstände für weitere
48 Stunden mit frischem Gewebekulturmedium ersetzt wurden. Zellen
wurden anschließend
durch Trypsinierung geerntet und lebensfähige Zellen wurden im Trypanblauausschlussverfahren
mit einem Hämozytometer
gezählt.
-
Testung auf Koloniebildung
in Weichagar:
-
U87MG-Zellen,
die wie oben 24 Stunden lang mit 5 × 106,
5 × 107 oder 5 × 108 Partikeln/Füllung infiziert wurden,
wurden in Gewebekulturmedium, das 0,35% Agar enthielt, suspendiert
und in dreifacher Ausführung auf
0,7%igem Agar in 35-mm-Gewebekulturvertiefungen geschichtet. Die
Kulturen wurden in einer befeuchteten Atmosphäre, die 7% CO2 enthielt,
bei 37°C
mit darüber
liegendem Gewebekulturmedium inkubiert, das alle fünf Tage
ausgetauscht wurde. Das Koloniewachstum wurde 14 Tage nach der Infektion
beurteilt.
-
Tumorigenitäts-Assay:
-
U87MG-Zellen
wurden in einer Dichte von 1 × 107 Zellen je T225-Flasche ausplattiert. Nach
Inkubation über
Nacht wurden die Zellmonolayer 24 Stunden lang mit 5 × 107 oder 5 × 108 Partikeln/ml
der Adenoviren GFCB, FTCB, BGCA oder MMCB infiziert. Infizierte
oder nicht infizierte Zellen wurden durch Trypsinierung geerntet,
in Gegenwart von Trypan Blau gezählt
und subkutan (5 × 106 lebensfähige
Zellen pro Flasche) in athymische weibliche nu/nu-Mäuse (Simonsen
Labs) injiziert. Die Mäuse
wurden nach 21 oder 30 Tagen auf Vorhandensein von Tumoren untersucht;
Die dreidimensionalen Tumordurchmesser wurden mit Vernier-Messschiebern
gemessen und die Tumorvolumen wurden als Produkt davon berechnet.
-
Ergebnisse und Diskussion
-
Die
Auswahl humaner Glioblastomzellen U87MG (Ponten und Macintyre, 1968)
für die
Studie erfolgte auf der Basis ihrer bekannten MMAC1-Mutation (Steck
et al., 1997), ihrer Fähigkeit
zur Bildung von Kolonien in Weichagar und ihrer subkutanen Tumorigenität in Nacktmäusen. Durch
Sequenzierung zeigte sich, dass in einem anomal kleinen RT-PCR-Produkt,
das mit Primern in Exon 2 und 5 (siehe obiger Abschnitt „Verfahren" in Beispiel 9) aus
U87MG-RNA erhalten
wurden, Exon 3 fehlte, was der Mutation der Intron-3-Splice-Donorstelle
(Steck et al., 1997) entsprach. Obgleich Exon 3 45 bp (15 Codons)
enthält
und ein Readthrough-Produkt (Produkt, bei dessen Transkription ein
Stop-Codon überlesen
wird) im Leseraster möglich
ist, kodieren die fehlenden Reste eine konservierte Alphahelix im
nativen Protein und ihr Verlust hob die wachstumshemmende Aktivität auf, wie
von Fumari et al., (1997) gemessen wurden.
-
Das
gereinigte rekombinante MMAC1-enthaltende Adenovirus (MMCB) wurde
durch Western Blotting von Zelllysaten mit einem polyklonalen Kaninchenantikörper hinsichtlich
der Expression des Transgens in U87MG-Zellen charakterisiert (17). In nicht infizierten oder mit Kontrollvirus
infizierten Zellen wurde kein endogenes MMAC1-Protein festgestellt,
wurde aber am Ende des 24-stündigen
Infektionszeitraums sowie nach 48 Stunden, 72 Stunden und 96 Stunden
in dosisabhängiger
Art in MMCB-infizierten Zellen nachgewiesen (17).
Diese Studie verifizierte die effiziente Transduktion und akute
Expression von exogenem MMAC1-Protein
in U87MG-Gliomzellen und validiert seinen Nachweis durch Western
Blotting mit Antikörper BL74.
-
Die
Infektiosität
von U87MG-Zellen wurde anhand eines rekombinanten Adenovirus, das mit
Ausnahme seines Transgens, das grün fluroeszierendes Protein
kodierte, identisch zu MMCB war, quantitativ durch FACS-Analyse
bestimmt, (18). Es wurde die erwartete
sigmoide Infektiositätskurve
erhalten, aus der geschätzt
wurde, dass 85 – 90
% der Zellen 24 Stunden lang mit einer viralen Dosis von 5 × 107 Partikeln/ml infiziert worden waren. Bemerkenswert
ist, dass die hierin verwendeten Dosierungsparameter nicht auf der Plaque-bildenden
Einheit oder ihrer Ableitung, der Multiplicity of Infection, beruhen.
Zuvor ist gezeigt worden, dass die Konzentration und der Infektionszeitpunkt
von Adenovirus die primären
Determinanten einer Transduktion in vitro sind (Nyberg-Hoffman et
al., 1997).
-
In-vitro-Proliferation
von MMCB-infizierten gegenüber
Kontroll-Ad-infizierten U87MG-Zellen
wurde durch Einbau von 3H-Thymidin für eine Reihe
vitaler Konzentrationen gemessen (19).
Im Vergleich zu zwei Kontrolladenoviren (GFCB und ZZCB) wurde U87MG
bei den meisten viralen Dosismengen differenziell inhibiert, wie
bereits zuvor für
einige Zelllinien festgestellt worden ist (Harris et al., 1995).
Die Hemmung der DNA-Synthese durch MMCB war vergleichbar mit der
durch adenoviralen p53-Gentransfer induzierten (FTCB, 19A).
-
Wachstumshemmung
wurde in einem zweiten in-vitro-Assay bestätigt, indem lebensfähige Zellen
72 Stunden nach Start der Infektion gezählt wurden (19B). Zu diesem Zeitpunkt hatte MMCB die Zellzahlen im
Vergleich zu GFCB in gleicher Dosis zu etwa 50% reduziert. Diese
Hemmung war in ihrer Größenordnung mit
der Hemmung vergleichbar, die bei der transienten Plasmidtransfektion
beobachtet wird (Furnari et al., 1997). MMCB- und GFCB-infizierte Kulturen
wiesen nach 72 Stunden ähnliche
Lebensfähigkeitsraten,
und morphologische Hinweise auf Zelltod auf, wie beispielsweise
Zell-Blasenbildung („Blebbing") oder Kernfragmentierung,
wurden bei Behandlung mit MMCB nicht beobachtet.
-
Die
Effekte von MMAC1 auf ankerunabhängiges
Wachstum wurden ebenfalls durch Koloniebildung in Weichagar nach
Transduktion durch MMCB gegenüber
GFCB oder FTCB bestimmt. Letzteres wurde eingeschlossen, um den
Assay mit einem etablierten Tumorsuppressorgen zu validieren. Bei
einer Dosis von 5 × 107 Partikeln/ml für 24 Stunden wurde die Koloniebildung
mit MMCB oder FTCB gegenüber
der GFCB-Kontrolle um etwa 50% gehemmt, wohingegen bei 5 × 108/ml MMCB oder FFCB eine Hemmung von >85% (relativ zu GFCB)
erreicht werden konnte (20).
In diesem in-vitro-Assay trat daher ein dosisabhängiger, genspezifischer Effekt
von MMAC1 hervor.
-
Es
sind zwei Tumorigenitäts-Assays
durchgeführt
worden, in denen 5 × 106 MMCB-infizierte U87MG-Zellen
pro Injektion verwendet und mit derselben Anzahl von Zellen, die
von drei verschiedenen Kontroll-Ad infiziert wurden, verglichen
wurden: GFCB (grün
fluoreszierendes Protein zum Abgleich des ΔE1/ΔE3-Hintergrunds), FTCB (p53
zum Abgleich des ΔE1/ΔE3-Hintergrunds)
und BGCA (LacZ für
den ΔE1/ΔE3/ΔE4-Hintergrund)
(Tabelle 10). Die Unterschiede zwischen Versuch 1 und 2 beinhalteten
die Verwendung von zwei Dosisstufen gegenüber einer und die Beendigung
nach 21 gegenüber
30 Tagen. Nach 21 oder 30 Tagen waren MMCB-infizierte U87-Zellen
vollständig
nicht-tumorigen, mit Ausnahme dreier sehr kleiner Tumore (~10 mm3) in der niedrigeren Dosisstufe in Versuch
1. Bei allen 39 Mäusen,
die mit nicht-infizierten oder Kontroll-Ad-infizierten Zellen injiziert
worden waren, bildeten sich Tumoren. Die Reportergen-enthaltenden
Kontroll-Ads GFCB und BGCA wiesen gegenüber pufferbehandelten Zellen
eine gewisse Aktivität
hinsichtlich einer Reduzierung der durchschnittlichen Tumorgröße auf,
ein unspezifischer „adenoviraler" Effekt, der von
den Erfordern bereits zuvor bemerkt worden war (Will et al., 1994,
Harris et al., 1995). Das p53-enthaltende
Adenovirus hatte einen drastischeren Effekt auf die durchschnittliche
Tumorgröße (~68
mm3), aber dennoch bildeten sich in 6 von
6 Mäusen
Tumoren. Diese Ergebnisse stimmen mit den wachstumshemmenden Effekten
eines p53-Adenovirus-Gentransfers in U87MG-Zellen überein,
von denen an anderer Stelle berichtet wurde, auch wenn diese Zellen
p53-Allele mit der Wildtypsequenz enthalten (Gomez-Manzani et al., 1996,
Kock et al., 1996). Auf jeden Fall deuten diese Daten auf eine genspezifische
Tumorsuppressionsaktivität
von MMCB in U87MG-Zellen
bei mäßigen Virusdosismengen
hin.
-
Durch
Verwendung eines rekombinanten adenoviralen Gentransfersystems wurde
eine wachstumshemmende Wirkung von MMAC1/PTEN auf U87MG-Zellen in
vitro gezeigt. Die Verwendung eines rekombinanten Adenovirus war
hilfreich, um die technische Schwierigkeit der Untersuchung von
Tumorzellen, die potenziell wachstumshemmende Proteine wie MMAC1
exprimieren, zu umgehen. In dem in-vivo-Assay wurde eine spezifische
Tumorsuppressionsfunktion von MMAAC1 am deutlichsten festgestellt,
was die Bedeutung des Tumorigenitätsassays bei der Bestimmung
einer Tumorsuppressionsfunktion hervorhebt. Diese Daten weisen darauf
hin, dass die MMAC1-Inaktivierung bei der Entstehung von Glioblastom-Tumoren eine Rolle spielt,
und lassen weiter den Schluss zu, dass ein MMAC1/PTEN-Gentransfer in vivo
als potenzieller Gentherapieansatz angesehen werden kann.
-
Während die
Zusammensetzungen und Verfahren dieser Erfindung in Form von bevorzugten
Ausführungsformen
beschrieben worden sind, ist es für einen Fachmann offensichtlich,
dass die Zusammensetzungen und/oder Verfahren und die Schritte oder
der Ablauf von Schritten der hierin beschriebenen Verfahren variiert
werden können,
ohne vom Konzept, Geist und Umfang der Erfindung abzuweichen. Insbesondere
ist offensichtlich, dass bestimmte Mittel, die sowohl chemisch als
auch physiologisch verwandt sind, als Ersatz für die hierin beschriebenen
Mittel verwendet werden können,
wobei dieselben oder ähnliche
Ergebnisse erzielt werden würden.
Alle derartigen Substitute und Modifikationen, die für einen
Fachmann offensichtlich sind, gelten als im Geist, Umfang und Konzept
der Erfindung enthalten, wie in den beigefügten Ansprüchen definiert ist.
-
BEZUGSQUELLEN
-
Insofern
als sie exemplarische verfahrensbezogene oder andere Details in
Ergänzung
zu den hierin Ausgeführten
bereitstellen, sind die folgenden Bezugsquellen hierin spezifisch
durch Bezugnahme enthalten:
- Albarosa et al.,
Am. J. Genet, 58:1260–1267,
1996.
- Albrecht, et al., Cancer, 70:869–875, 1992.
- Anderson WF, et al. (1980). Proc. Natl. Acad. Sci. USA 77:5399–5403.
- Arcone et al., Nuc. Acids Res. 16(8):3195–3207, 1988.
- Baichwal und Sugden, In: Gene Transfer, Kucherlapati R, Hrsg.,
New York, Plenum Press, pp. 117–148,
1986.
- Bandyopadhyay PK und Temin HM (1984). Mol. Cell Biol. 4:749–754.
- Barany und Merrifield, The Peptides, Gross und Meienhofer, Hrsg.,
Academic Press, New York, pp. 1–284, 1979.
- Bartel PL, et al. (1993). "Using
the 2-hybrid system to detect protein-protein interactions" In Cellular Interactions
in Development: A Practical Approach, Oxford University Press, pp.
153–179.
- Bartlett et al., Proc. Natl. Acad. Sci. USA, 93:8852–8857, 1996.
- Batterson und Roizman, J. Virol.; 46:371–377, 1983.
- Bellus, J. Macromo.l Sci. Pure Appl. Chem, A311: 1355–1376, 1994.
- Benvenisty und Neshif Proc. Natl. Acad. Sci. USA, 83:9551–555, 1986.
- Berglund P, et al. (1993). Biotechnology 11:916–920.
- Berkner KL, et al. (1988). BioTechniques 6:616–629.
- Berkner KL (1992). Curr. Top. Microbiol. Immunol. 158:39–66.
- Berns und Bohenzky, Adv. Virus Res., 32:243–307, 1987.
- Bems und Giraud, Curr. Top. Microbiol. Immunol. 218:1–23, 1996.
- Berns, Microbiol Rev., 54:316–329, 1990.
- Bertran, et al., J Virol, 70 (10)6759–6766, 1996.
- Bianchi et al., Nature Genetics, 6:185–192, 1994.
- Bigner et al., Cancer Res., 48:405–411, 1988.
- Bishop, J.M., Cell, 64:2351–248,
1991.
- Boring et al., Cancer Statistics, 1994 CA, 43:7–26, 1994.
- Breakefield XO und Geller AI (1987). Mol. Neurobiol. 1:337–371.
- Brinster RL, et al. (1981). Cell 27:223–231.
- Brinster et al., Proc. Natl. Acad. Sci. USA, 82: 4438–4442, 1985.
- Brownstein, et al., JAMA, 238:26, 1977.
- Brownstein, et al., Cancer, 41:2393–2398, 1978.
- Buchschacher GL und Panganiban AT(1992). J. Virol 66:2731–2739.
- Campbell et al., J. Mol. Biol., 180:1–19, 1984.
- Capaldi et al., Biochem. Biophys. Res. Comm., 76:425, 1977
- Carter et al., Proc. Natl. Acad. Sci. USA, 87:8751–8755, 1990.
- Chang et al., Hepatology, 14:124A, 1991.
- Chen und Okayama, Mol. Cell. Biol., 7:2745–2752, 1987.
- Chevray PM und Nathans DN (1992). Proc. Natl. Acad. Sci. USA
89:5789–5793.
- Claus, et al., Am. J. Epidemiol, 131:961–972, 1990.
- Coffin, Retroviridae and Their Replication. In: Virology, Fields
et al., Hrsg., Raven Press, New York, pp. 1437–1500, 1990.
- Cohen, P., Bioessays, 61-583–588, 1994.
- Collet et al., Proc. Natl. Acad. Sci. USA, 75:2021–2024, 1978.
- Compton J(1991). Nature 350:91–92.
- Cooker al., Cell, 27:487–496,
1981.
- Costantini F und Lacy E (1981). Nature 294:92–94.
- Cotten M, et al. (1990). Proc. Natl. Acad. Sci. USA 87:4033–4037.
- Couch et al., Am. Rev. Resp. Dis., 88:394–403, 1963.
- Coupar et al., Gene, 68:1–10,
1988
- Culver et al., Science, 256:1550–1552, 1992.
- Curiel DT, et al. (1991). Proc. Natl. Acad. Sci. USA 88:8850–8854.
- Curiel DT, et al. (1992). Hum. Gene Ther. 3:147–154.
- Daly et al., Oncogene 8:1721–1729, 1993.
- Dani, et al., J. Biol Chem., 264:10119–10125, 1989.
- Davey et al., EPO No. 329 822.
- DeLuca et al., J. Virol, 56:558–570, 1985.
- Denu et al., Ce1187:361–364,
1996.
- Dubensky et al., Proc. Natl. Acad. Sci. USA, 81:7529–7533, 1984.
- El-Azouzi et al., Proc. Natl. Acad. Sci. USA, 86:7186–7190, 1989.
- Elroy-Stein et al., Proc. Natl. Acad. Sci. USA, 1989.
- EP 329 822 , Davey
et al.
- Fahy E, et al. (1991). PCR Methods Appl. 1:25–33.
- Fanning und Anderson, Curr. Biol., 6:11,1385–1388, 1996.
- Fearron und Vogelstein, Cell, 61:759–767, 1990.
- Fechheimer et al., Proc. Natl. Acad. Sci. USA, 84:8463–8467, 1987.
- Feil et al., Proc. Natl. Acad. Sci. USA 93:10887–10890,
1996.
- Feigner PL, et al. (1987). Proc. Natl. Acad. Sci. USA 84:7413–7417.
- Ferkol et al., FASEB J., 7:1081–1091, 1993.
- Fields S und Song OK (1989). Nature 340:245–246.
- Fink DJ, et al. (1992). Hum. Gene Ther. 3:11–19.
- Fink DJ, et al. (1996). Ann. Rev. Neurosci. 19:265–287.
- Fodor et al., Science, 251:767–773, 1991.
- Ford, et al., Am. J. Hum. Genet., 57:1457–1462, 1995.
- Fornander et al., Lancet, 1, 8630:117–120, 1989.
- Forster und Symons, Cell, 49:211–220, 1987.
- Foulds, J. Chronic Dis., 8:2–37, 1958.
- Fraley et al., Proc. Natl. Acad. Sci. USA, 76:3348–3352, 1979.
- Freese A, et al. (1990). Biochem. Pharmacol. 40:2189–2199.
- Freifelder, Physical Biochemistry Applications to Biochemistry
and Molecular Biology, 2. Auflage. Wm. Freeman and Co., New York,
NY, 1982.
- Freshner, Animal Cell Culture: A Practical Approach, 2. Auflage,
Oxford/New York, IRL Press, Oxford University Press, 1992.
- Friedmann, Science, 244:1275–1281, 1989.
- Friedman T (1991). In Therapy for Genetic Diseases. T. Friedman,
Hrsg., Oxford University Press, pp. 105–121.
- Frohman, In: PCR Protocols: A Guide To Methods And Applications,
Academic Press, N.Y. 1990.
- Fujimoto et al., Genomics, 4:210–214, 1989.
- Fults und Pedone, Genes Chromosom. Cancer 7:173–177, 1993.
- Fults et al., Cancer Res., 50:5784–5789, 1990.
- Fumari et al., Proc. Natl. Acad. Sci. USA, 94:12479–12484,
1997
- GB-Anmeldung 2 202 328
- Ganeten et al., Nucl. Acids Res. 25:3326–3331, 1997.
- Gefter et al., Somatic Cell Genet., 3: 231–236, 1977.
- Gerlach et al., Nature London, 328:802–805, 1987.
- Ghosh-Choudhury et al., EMBO J., 6:1733–1739, 1987.
- Ghosh und Bachhawat, In: Liver Diseases, Targeted Diagnosis
and Therapy Using Specific Receptors and Ligands. Wu et al., Hrsg.,
Marcel Dekker, New York, pp. 87–104,
1991.
- Gingeras et al., PCT-Anmeldung WO 88/10315,
- Giorioso et al., Ann. Rev. Microbiol, 49:675–710, 1995.
- Goding, 1986, In: Monoclonal Antibodies: Principles and Practice,
2. Auflage, Academic Press, Orlando, Fla., pp. 60–61, und
71–74,
1986.
- Gomez-Foix et al.,J. Biol. Chem., 267:25129–25134, 1992.
- Gomez-Manzani et al., Cancer Res, 6:694–699, 1996.
- Gopal, Mol. Cell. Biol., 5:1188–1190, 1985.
- Gordon JW, et al. (1980). Proc. Natl. Acad. Sci. USA 77:7380–7384.
- Gorziglia M und Kapikian AZ (1992). J. Virol. 66:4407–4412.
- Gossen und Bujard, Proc. Natl. Acad. Sci. USA, 89:5547–5551, 1992.
- Gossen et al., Science, 268:1766–1769, 1995.
- Graham und Prevec, In: Methods in Molecular Biology: Gene Transfer
and Expression Protocol, E.J. Murray, Hrsg., Humana Press, Clifton,
NJ, 7:109–128,
1991.
- Graham und van der Eb, Virology, 52:456–467, 1973.
- Graham et al., J. Gen. Yirol, 36:59–72, 1977.
- Gray et al., Cancer Res., 55:4800–4803, 1995.
- Grunhaus und Horwitz, Seminar in Virology, 3:237–252, 1992.
- Gyapay et al., Nat. Genet., 7:246–339, 1994.
- Hacia et al., Nature Genetics, 14:441–447, 1996.
- Haines et al., Am. J. Hum. Genet, 49:764–772, 1991.
- Hardie und Hanks, In: The Protein Kinase Facts Book, 1995
- Harland und Weintraub, J. Cell. Biol., 101:1094–1099, 1985.
- Harlow und Lane, Antibodies: A Laboratory manual, Cold Spring
Harbor Laboratory, 1988.
- Harris et al., Cancer Gene Ther., 3:121–130, 1995.
- Helseth E, et al. (1990). J. Virol. 64:2416–2420.
- Henson et al., Ann. Neurol., 36:714–721, 1994.
- Herbst et al., Cancer Res., 54:3111–3114, 1994.
- Hermonat und Muzycska, Proc. Natl. Acad. Sci. USA, 81:6466–6470, 1984.
- Hersdorffer et al., DNA Cell Biol., 9:713–723, 1990.
- Herz und Gerard, Proc. Natl. Acad. Sci. USA, 90:2812–2816, 1993.
- Holland et al., Virology, 101:10–18, 1980.
- Honess und Roizman, J. Yirol, 14:8–19, 1974.
- Honess und Roizman, J. Virol, 16:1308–1326, 1975.
- Hoon et al., J. Urol., 150(6):2013–2018, 1993.
- Horwich et al., J. Yirol, 64:642–650, 1990.
- Hunt et al., Proc. Natl. Acad. Sci. USA, 83:3786–3790, 1986.
- Hunter, Cell, 64-249–270,
1991.
- Huyghe et al., Hum. Gene Ther., 6:1403–1416, 1995.
- Innis et al., PCR Protocols, Academic Press, Inc., San Diego
CA, 1990.
- Ittmann, Cancer Res., 56:2143–2147, 1996.
- James et al., Cancer Res., 48:5546–5551, 1988.
- Johnson PA, et al. (1992). J. Virol., 66:2952–2965.
- Johnson et al., Peptide Turn Mimetics In: Biotechnology And
Pharmacy, Pezzuto et al., Hrsg., Chapman and Hall, New York, 1993.
- Joki et al., Human Gene Ther., 6:1507–1513, 1995.
- Jones und Shenk, Cell, 13:181–188, 1978.
- Jones, et al., Genes Chromosomes Cancer, 9:2, 119–123, 1994.
- Kageyama et al., J. Biol. Chem., 262(5):2345–2351, 1987.
- Kamb et al., Science, 264:436–440, 1984.
- Kaneda Y, et al. (1989). J. Biol. Chem. 264:12126–12129.
- Kandt et al., Nature Genet, 2:37–41,1992.
- Kaneda et al., Science, 243:375–378, 1989.
- Karlbom et al., Hum. Genet, 92:169–174, 1993.
- Karlsson et al., EMBO J, 5:2377–2385, 1986.
- Kato et al., J. Biol. Chem, 266:3361–3364, 1991.
- Kearns et al., Gene Ther., 3:748–755, 1996.
- Kim und Cook, Proc. Natl. Acad. Sci. USA, 84:8788–8792, 1987.
- Kimmelman et al., Genomics 34:250–254, 1996.
- Klein et al., Nature, 327:70–73, 1987.
- Kock et al., Int. J. Cancer, 67:808–815, 1996.
- Köhler
und Milstein, Eur. J. Immunol 6:511–519, 1976.
- Köhler
und Milstein, Nature, 256:495–497,
1975.
- Kok et al., Cancer Res. 54:4183–4187, 1994.
- Komiya et al., Genes Chromo. Cancer 17:245–253, 1996.
- Korhonen, et al., Blood, 86(5):1828–1835, 1995.
- Kotin und Bems, Virol, 170:460–467, 1989.
- Kotin et al., Genomics, 10:831–834, 1991.
- Kotin et al., Proc. Natl. Acad. Sci. USA, 87:2211–2215, 1990.
- Kruglyak, et al., Am. J. Hum. Genet., 58:347–1363, 1996.
- Kwoh et al., Proc. Natl. Acad. Sci. USA, 86: 1173, 1989.
- Kyte und Doolittle, J. Mol. Biol, 157,1: 105–132, 1982.
- Lathrop, et al., Proc. Natl. Acad. Sci. USA, 81:3443–3446, 1984.
- Le Gal La Salle et al., Science, 259:988–990, 1993.
- Lee et al., Science, 235:1394–1399, 1987.
- Lee JE, et al. (1995). Science 268:836–844.
- Levin et al., In: Cancer: Principles & Practice of Oncology, 4. Auflage,
DeVita et al., Hrsg., J.B. Lippincott Co., Philadelphia, 1993.
- Levrero et al., Gene, 101:195–202, 1991.
- Li und Sun, Cancer Res., 57:2124–2129, 1997.
- Li et al., Science, 275:1943–1947, 1997.
- Liaw et al., Nature Genet., 16:64–67, 1997.
- Lim CS, et al. (1991). Circulation 83:2007–2011.
- Lloyd und Dennis, Ann. Intern. Med, 58:136–142, 1963.
- Lobe und Nagy, Bioessays, 20:200–208, 1998.
- Macejak und Sarnow, Nature, 353:90–94, 1991.
- Madzak C, et al. (1992). J. Gen. Virol., 73:1533–1536.
- Manipulating the Mouse Embryo: A Laboratory Manual, 2. Auflage,
Hogan et al., Hrsg., Cold Spring Harbor Laboratory Press, 1994.
- Mann R und Baltimore D (1985). J. Virol 54:401–407.
- Mann et al., Cell, 33:153–159,
1983.
- Margolskee RF (1992). Curr. Top. Microbiol. Immunol., 158:67–95.
- Markowitz et al., J. Virol., 62:1120–1124, 1988.
- Merrifield, Science, 232: 341–347, 1986.
- Michel und Westhof, J. Mol. Biol., 216:585–610, 1990.
- Miki et al., Science, 266:66–71, 1994.
- Miller et al., PCT-Anmeldung WO 89/06700
- Miller AD, et al. (1988). J. Virol., 62:4337–4345.
- Miller AD (1992). Curr. Top. Microbiol. Immunol., 158:1–24.
- Miller AD, et al., (1985). Mol. Cell. Biol 5:431–437.
- Mizukami et al., Virology, 217:124–130, 1996.
- Morita et al., Cancer Res., 51:5817–5820, 1991.
- Moss B (1992). Curr. Top. Microbiol. Immunol., 158:25–38.
- Moss B (1996). Proc. Natl. Acad. Sci. USA 93:11341–11348.
- Mulligan, Science, 260:926–932,
1993.
- Murakami et al., Cancer Res., 56:2157–2160, 1996.
- Muzyczka N (1992). Curr. Top. Microbiol. Immunol., 158:97–129.
- Myers, EP 0273085
- Nabel EG, et al. (1990). Science, 249:1285–1288.
- Nabel (1992). Hum. Gene Ther. 3:399–410.
- Nakamura et al., In: Handbook of Experimental Immunology (4.
Auflage.), Weir, E., Herzenberg, L.A., Blackwell, C, Herzenberg,
L. (Hrsg.). Band 1, Kapitel 27, Blackwell Scientific Publ., Oxford,
1987.
- Naldini L, et al. (1996). Science 272:263–267
- Nelen et al., Nature Genet., 13:114–116, 1996.
- Nicolas und Rubenstein, In: Vectors: A survey of molecular cloning
vectors and their uses, Rodriguez und Denhardt (Hrsg.), Stoneham:
Butterworth, pp. 493–513,
1988.
- Nicolau und Sene, Biochim. Biophys. Acta, 721:185–190, 1982.
- Nicolau et al., Methods Enzymol., 149:157–176, 1987.
- Nihei et al., Genes Chromosom. Cancer, 14:112–119, 1995.
- Nishi et al., Oncogene, 6:1555–1559, 1991.
- Nyberg-Hoffman et al., Nat. Medicine, 7:808–811, 1997.
- Ohara et al., Proc. Natl. Acad. Sci. USA, 86: 5673–5677, 1989.
- Ohi S,et al. (1990). Gene 89:279–282.
- Olivierio et al., EMBO J., 6(7):1905–1912, 1987.
- Osterrieder und Wolf, Rev. Sci. Tech. 17:351–364, 1998.
- Ostrove et al., Virology, 113:532–533, 1981.
- Page KA, et al. (1990). J. Virol., 64:5270–5276.
- Pape und Kim, Mol. Cell. Biol., 974–982, 1989.
- Paskind et al., Virology, 67:242–248, 1975.
- PCT/US87/00880
- PCT/US89/01025
- Veröffentliche
PCT-Anmeldung WO 93/07282
- Veröffentliche
PCT-Anmeldung WO 97/02048
- Pease et al., Proc. Natl. Acad. Sci. USA, 91:5022–5026, 1994.
- Peiffer et al., Cancer Res., 55:1922–1926, 1995.
- Pelletier und Sonenberg, Nature, 334:320–325, 1988.
- Pellicer A, et al. (1980). Science, 209:1414–1422.
- Perales et al., Proc. Natl. Acad. Sci. 91:4086–4090, 1994.
- Pershouse et al., Cancer Res. 53:5043–5050, 1993.
- Petersen et al., Brit. J. Cancer 75:79–86, 1997.
- Petropoulos CJ,et al.(1992). J. Virol., 66:3391–3397.
- Pignon et al., Hum. Mutat., 3: 126–132, 1994.
- Poll und Cortese, Proc. Natl. Acad. Sci. USA, 86:8202–8206, 1989.
- Ponnazhagan et al., Hum. Gene Ther., 8:275–284, 1997a.
- Ponnazhagan et al., J. Gen. Virol., 77:1111–1122, 1996.
- Post et al., Cell, 24:555–565,
1981.
- Potter et al., Proc. Natl. Acad. Sci. USA, 81:7161–7165, 1984.
- Prowse und Baumann, Mol. Cell. Biol., 8(1):42–51, 1988.
- Quantin B, et al. (1992). Proc. Natl. Acad. Sci. USA 89:2581–2584.
- Racher et al., Biotechnology Techniques, 9:169–174, 1995.
- Ragot et al., Nature, 361:647–650, 1993.
- Ransom et al., Genes Chromosom. Cancer 5:357–374, 1992.
- Rasheed et al., Genes Chromo. Cancer, 5:75–82, 1992.
- Rasheed et al., Oncogene, 11:2243–2246, 1995.
- Reinhold-Hurek und Shub, Nature, 357:173–176, 1992.
- Remington's
Pharmaceutical Sciences, 15. Auflage, pp. 1035–1038 und 1570–1580.
- Rempel et al., Cancer Res., 53:2386–2392, 1993.
- Renan, Radiother. Oncol., 19:197–218, 1990.
- Rich et al., Hum. Gene Ther., 4:461–476, 1993.
- Ridgeway, In: Vectors: A survey of molecular cloning vectors
and their uses, Rodriguez RL, Denhardt DT, Hrsg., Stoneham:Butterworth,
pp. 467–492,
1988.
- Rippe ET AL., Mol. Cell. Biol., 10:689–695, 1990.
- Ritland et al., Genes. Chromo. Cancer, 12:277–282, 1995.
- Rosenfeld et al., In vivo transfer of the human cystic fibrosis
transmembrane conductance regulator gene to the airway epithelium.
Cell, 68:143–155,
1992.
- Rosenfeld et al., Science, 252:431–434, 1991.
- Rosenfeld MA, et al. (1992). Cell, 68:143–155.
- Roux et al., Proc. Natl. Acad. Sci. USA, 86:9079–9083, 1989.
- Rubio et al., Cancer Res., 54:45–47, 1994.
- Russell D und Hirata R (1998). Nature Genetics 18:323–328.
- Russell und Rubinstein, In: Pathology of Tumors of the Nervous
System, 5. Auflage, Williams und Wilkins, Hrsg., pp. 82–219, 1989.
- Sambrook et al., In: Molecular Cloning: A Laboratory Manual,
2. Auflage, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
NY, 1989.
- Samutski et al., EMBO J,10:3941–3950, 1991.
- Sanchez et al., Proc. Natl. Acad. Sci. USA, pp. 2551–2556, 1996.
- Sanchez, et al., Mil.Med., 160:8, 416–419, 1995.
- Saras und Heldin CH, Trends Biochem. Sci. 21,12 455–458, 1996.
- Sarver et al., Science, 247:1222–1225, 1990.
- Scanlon et al., Proc. Natl. Acad. Sci. USA, 88:10591–10595,
1991.
- Scheck und Coons, Cancer Res., 53:5605–5609, 1993.
- Schneider G, et al. (1998). Nature Genetics 18:180–183.
- Shabram et al., Hum. Gene Ther., 8:453–465, 1997.
- Shastry, Experientia 51:1028–1039, 1995.
- Shastry, Mol. Cell. Biochem. 181:163–179, 1998.
- Shimada T, et al. (1991). J. Clin. Invest., 88:1043–1047.
- Shoemaker et al., Nature Genetics 14:450–456, 1996.
- Smith und Moss, Gene, 25:21–28,
1983.
- Songyang et al., J. Biol. Chem., 270(44): 26029–26032,
1995
- Sonoda et al., Cancer Res., 55:2166–2168, 1995.
- Sorge J, et al. (1984). Mol. Cell. Biol., 4:1730–1737.
- Spargo CA, et al. (1996). Mol. Cell. Probes, 10:247–256.
- Speigelman, et al.,J. Biol Chem., 264(3), 1811–1815, 1989.
- Spruck, et al., Nature,:370:6486, 183–184, siehe auch Kommentar
in Nature 1994 Jul 21, 370, 6486:180 1994
- Srivastava et al.,J. Yirol., 45:555–564, 1983.
- Starink, et al., Clin. Genet., 29:222–233, 1986.
- Steck und Saya, Curr. Opin. Oncol., 3:3,476–484, 1991.
- Steck et al., Genes Chromosom. Cancer 712:255–261, 1995.
- Steck et al., Nature Genet., 15:356–362, 1997.
- Stewart und Young, Solid Phase Peptide Synthesis, 2. Auflage,
Pierce Chemical Co., 1984.
- Stewart MJ, et al. (1992). Hum. Gene Ther. 3:267–275.
- Stratford-Perricaudet und Perricaudet, Gene transfer into animals:
the promise of adenovirus. In: Human Gene Transfer, O. Cohen-Haguenauer
et al., Hrsg., John Libbey Eurotext, Frankreich, pp. 51–61, 1991.
- Stratford-Perricaudet et al., Hum. Gene. Ther, 1:241–256, 1990.
- Tam et al., J. Am. Chem. Soc., 105:6442, 1983.
- Tavtigian et al., Nature Genet., 12:333–337, 1996.
- Temin, In: Gene Transfer, Kucherlapati (Hrsg.), New York: Plenum
Press, pp. 149–188,
1986.
- Teng et al., Cancer Res., 57:5221–5225, 1997.
- Tonks und Neel, Cell 87:3, 365–368, siehe auch Kommentar
in Cell 1996 Nov 1, 87, 3:361–4,
199<
- Tope/a/., J. Infect. Dis., 124:155–160, 1971.
- Trybus et al., Cancer Res., 56:2263–2267, 1996.
- Tur-Kaspa et al., Mol. Cell. Biol., 6:716–718, 1986.
- US-Patent 4.554.101
- US-Patent 4.683.195
- US-Patent 4.683.202
- US-Patent 4.800.159
- US-Patent 4.873.191, Wagner und Hoppe
- US-Patent 4.883.750
- US-Patent 5.252.479
- US-Patent 5.270.184
- US-Patent 5.279.721
- US-Patent 5.354.855
- US-Patent 5.409.818
- US-Patent 5.436.146
- US-Patent 5.455.166
- US-Patent 5.672.344
- US-Patent 5.691.198
- US-Patent 5.747.469
- US-Patent 5.753.500.
- Varmus et al., Cell, 25:23–36,
1981.
- Vogelstein, et al., Genes Chromosomes Cancer, 2:2,159–162, 1990.
- von Deimling, et al., J Neurosurg, 77:2, 295–301, 1992.
- von Deimling et al., Int. J. Cancer, 57:676–680, 1994.
- Voullaire et al., Am. J Hum. Genet., 52:1153–1163, 1993.
- Wagner und Hoppe, US-Patent Nr. 4.873.191
- Wagner et al., Proc. Natl. Acad. Sci. 87, 9:3410–3414, 1990.
- Wagner E, et al. (1991). Proc. Natl Acad. Set USA, 88:4255–4259.
- Wagner et al., Science, 260:1510–1513, 1993.
- Walker et al., Proc. Natl. Acad. Sci. USA, 89:392–396, 1992a.
- Walker GT, et al., (1992b). Nucl. Acids Res. 20:1691–1696.
- Walther und Stein, J. Mol. Med., 74:379–392, 1996.
- Walton, et al., Surgery, 99:82–86, 1986.
- Wang CY und Huang L (1989). Biochemistr, 28:9508–9514.
- Wang et al., Cancer Res., 57:351–354, 1997.
- Weary, et al., Arch. Dermatol., 106:682–690, 1972.
- Wei et al., Cancer Res., 56:1487–1494, 1996.
- Weinberg, Biochemistry, 28:8263–8269, 1989.
- Wilkinson GW und Akrigg A (1992). Nucleic Acids Res., 20:2233–2239.
- Wills et al., Human Gene Therapy, 5:1079–1088, 1994.
- Wilson et al., Mol. Cell. Biol., 6181–6191, 1990.
- Wolff JA, et al. (1990). Science, 247:1465–1468.
- Wolff JA, et al. (1991). BioTechniques, 11:474–485.
- Wong et al., Gene, 10:87–94,
1980.
- Wong et al., Proc. Natl. Acad. Sci. USA, 84:6899–6903, 1987.
- Wooster et al., Nature, 378:789–792, 1995.
- Wu und Wu, Biochemistry, 27:887–892, 1988.
- Wu und Wu, J. Biol. Chem., 262:4429–4432, 1987.
- Wu und Wu, Adv. Drug Delivery Rev., 12:159–167, 1993.
- Wu et al., Genomics, 4:560, 1989.
- Wu CH, et al. (1989). J. Biol. Chem., 264:16985–16987.
- Wu GY, et al. (1991). J. Biol Chem., 266:14338–14342.
- Yamaguchi et al., Proc. Natl. Acad. Sci. USA, 91:484–488, 1994.
- Yang et al., Proc. Natl. Acad. Sci. USA, 87:9568–9572, 1990.
- Zechner et al., Mol Cell Biol, 2394–2401, 1988.
- Zelenin et al., FEBS Lett., 280:94–96, 1991.
- Zenke M, et al. (1990). Proc. Natl Acad. Sci. USA, 87:3655–3659.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-