-
HINTERGRUND DER ERFINDUNG
-
Gebiet der Erfindung
-
Die vorliegende Erfindung betrifft
die Gebiete der Retinoid-Rezeptor-Biologie und Therapien von Säugetierkrankheiten.
Die vorliegende Erfindung stellt insbesondere Zusammensetzungen
und die Verwendung dieser Zusammensetzungen bei der Herstellung
eines Medikaments zur Verwendung bei der Behandlung eines Tiers,
vorzugsweise eines Menschen, bereit, der an Krebs, einem Hautleiden,
rheumatischer Arthritis oder einer prämalignen Läsion leidet oder dazu prädisponiert
ist, wobei bei der Behandlung dem Tier eine wirksame Menge einer
Zusammensetzung verabreicht wird, welche mindestens einen RAR-Antagonisten,
vorzugsweise einen RARα-Antagonisten,
und mindestens einen RXR-Agonisten
enthält.
-
Verwandte Technik
-
Retinoide
-
Eine Reihe von Studien hat belegt,
dass Retinoide (Derivate von Vitamin A) für normales Wachstum, Sehvermögen, Gewebe-Homöostasie,
Fortpflanzung und allgemeines Überleben
wesentlich sind (zwecks Überblick
und Verweise siehe Sporn et al., The Retinoids, Band 1 und 2, Sporn
et al., Hrsg. Academic Press, Orlando, Florida (1984)). So wurde
zum Beispiel gezeigt, dass Retinoide für die Erhaltung der Haut-Homöostasie
und Sperrfunktion in Säugetieren
von vitaler Bedeutung sind (Fisher, G. J., und Voorhees, J. J.,
FASEB J. 10: 1002–1013
(1996)). Retinoide sind anscheinend auch während der Embryogenese von
entscheidender Bedeutung, da die Nachkommen von Muttertieren mit
Vitamin-A-Mangel (VAD) eine Reihe von Entwicklungsstörungen aufweisen
(Wilson, J. G., et al., Am. J. Anat. 92: 189–217 (1953); Morriss-Kay, G.
M. und Sokolova, N., FASEB J. 10: 961–968 (1996)). Mit Ausnahme
der Auswirkungen auf das Sehvermögen
(Wald, G., et al., Science 162: 230–239 (1968)) und die Spermatogenese
bei Säugetieren
(van Pelt, H. M. M. und De Rooij, D. G., Endocrinology 128: 697–704 (1991)),
können
die meisten Auswirkungen, die durch VAD bei Tieren und deren Föten hervorgerufen
werden, durch die Verabreichung von Retinolsäure (RA) vermieden und/oder
umgekehrt werden (Wilson, J. G., et al., Am. J. Anat. 92: 189–217 (1953);
Thompson et al., Proc. Royal Soc. 159: 510–535 (1964); Morriss-Kay, G.
M. und Sokolova, N., FASEB J. 10: 961–968 (1996)). Die dramatischen
teratogenen Auswirkungen maternaler RA-Verabreichung bei Säugetierembryonen
(Shenefelt, R. E., Teratology 5, 103–108 (1972); Kessel, M., Development
115; 487–501
(1992); Creech Kraft, J., in Retinoids in Normal Development and
Teratogenesis, G. M. Morriss-Kay, Hrsg. Oxford University Press,
Oxford, UK, Seiten 267–280 (1992))
und die spürbaren
Auswirkungen lokaler Verabreichung von Retinoiden auf die embryonale
Entwicklung von Wirbeltieren und die Regeneration von Gliedmaßen bei
Amphibien (Mohanty-Hejmadi et al., Nature 355: 352–353 (1992);
Tabin, C. J., Cell 66: 199– 217
(1991), haben zu der Auffassung beigetragen, dass RA eine entscheidende
Rolle bei der Morphogenese und Organogenese spielen könnte.
-
Retinoid-Rezeptoren
-
Abgesehen von den molekularen Mechanismen,
die beim Sehen (Wald, G. et al., Science 162: 230–239 (1968))
involviert sind, sind die molekularen Mechanismen, die den sehr
unterschiedlichen Auswirkungen von Retinoiden zugrunde liegen, bis
vor kurzem unklar geblieben. Die Entdeckung von nuklearen Rezeptoren
für RA
(Petkavich et al., Nature 330: 444–450 (1987); Giguère et
al., Nature 330: 624–629
(1987)) hat das Verständnis
dafür,
wie die Retinoide ihre pleiotropen Auswirkungen ausüben, entscheidend
verbessert (Leid et al., TIBS 17: 427–433 (1992); Linney, E., Current
Topics in Dev. Biol. 27: 309–350
(1992)). Seit dieser Entdeckung wurde deutlich, dass die genetischen
Aktivitäten
des RA-Signals durch
zwei Rezeptorfamilien – die
RAR-Familie und die RXR-Familie – herbeigeführt werden, die zur Superfamilie
der Ligand-induzierbaren transkriptionalen regulatorischen Faktoren
zählen,
welche Steroid-/Schilddrüsen-Hormon-
und Vitamin-D3-Rezeptoren
einschließen
(Rezensionen siehe Leid et al., TIBS 17: 427–433 (1992); Chambon, P., Semin.
Cell Biol. 5: 115–125
(1994); Chambon, P., FASEB J. 10: 940–954 (1996); Giguère, V.,
Endocrinol. Rev. 15: 61–79
(1994); Mangelsdorf, D. J., und Evans, R. M., Cell 83: 841–850 (1995);
Gronemeyer, H., und Laudet, V., Protein Profile 2: 1173–1236 (1995)).
RAR-Rezeptoren Rezeptoren der RAR-Familie (RARα, β und γ und ihre Isoformen) werden
sowohl von all-trans- als auch 9-Cis-RA aktiviert (Leid et al.,
TIBS 17: 427–433
(1992 ); Chambon, P., Semin. Cell Biol. 5: 115–125 (1994); Dollé, P.,
et al., Mech. Dev. 45: 91–104
(1994); Chambon, P., FASEB J. 10: 940–954 (1996)). Innerhalb einer
bestimmten Spezies sind die DNA-bindenden (C) und die Ligand-bindenden
(E) Domänen
der drei RAR-Typen sehr ähnlich,
während
die C-terminale Domäne
F und die Mitteldomäne
D keine oder nur geringe Ähnlichkeit
aufweisen. Die Aminosäuresequenzen
der drei RAR-Typen unterscheiden sich ebenfalls in ihren B-Regionen
deutlich voneinander, und ihre Haupt-Isoformen (α1 und α2, β1 bis β4 und γ1 und γ2) unterscheiden sich ferner
in ihren N-terminalen A-Regionen (Leid et al., TIBS 17: 427–433 (1992)).
Aminosäuresequenz-Vergleiche
haben ergeben, dass die Interspezies-Erhaltung eines bestimmten
RAR-Typs größer ist
als die Ähnlichkeit,
die zwischen den drei RAR-Typen innerhalb einer bestimmten Spezies
festzustellen ist (Leid et al., TIBS 17: 427–433 (1992)). Diese Interspezies-Erhaltung
ist insbesondere in den Nterminalen A-Regionen der verschiedenen
RARα-, β- und γ-Isoformen
auffällig,
deren A-Regionen-Aminosäuresequenzen
ziemlich abweichend sind. Gemeinsam mit den ausgeprägten spatio-temporalen Expressionsmustern,
die für
die Transkripte jedes RAR- und RXR-Typs im sich entwickelnden Embryo
und in verschiedenen Geweben erwachsener Mäuse festgestellt wurden (Zelent,
A., et al., Nature 339: 714–717 (1989);
Dollé,
P., et al., Nature 342: 702–705
(1989); Dollé et
al., Development 110: 1133–1151
(1990); Ruberte et al., Development 108: 213–222 . (1990); Ruberte et al.,
Development 111: 45–60
(1991); Mangelsdorf et al., Genes & Dev. 6: 329–344 (1992)), hat diese Interspezies-Erhaltung
zur Annahme geführt,
dass jeder RAR-Typ (und Isoform) einzigartige Funktionen erfüllt. Diese
Hypothese wird ferner durch die Erkenntnis untermauert, dass die
verschiedenen RAR-Isoformen
zwei transkriptionale Aktivierungsfunktionen (AFs) enthalten, die
in der Nterminalen A/B-Region (AF-1) und in der C-terminalen E-Region
(AF-2) angeordnet sind, die synergistisch – und bis zu einem gewissen
Grad differential – verschiedene
RAansprechende Promotoren aktivieren können (Leid et al., TIBS 17:
427–433
(1952); Nagpal, S., et al., Cell 70: 1007–1019 (1992); Nagpal, S., et
al., EMBO J. 12: 2349–2360
(1993)).
-
RXR-Rezeptoren
-
Im Gegensatz zu den RARs werden Mitglieder
der Retinoid-X-Rezeptor-Familie (RXRα, β und γ) ausschließlich durch 9-cis-RA aktiviert
(Chambon, P., FASEB J. 10: 940– 954
(1996); Chambon, P., Semin. Cell Biol. 5: 115–125 (1994); Dollé, P.,
et al., Mech. Dev. 45: 91–104
(1994); Linney, E., Current Topics in Dev. Biol. 27: 309–350 (1992);
Leid et al., TIBS 17: 427–433
(1992); Kastner et al., in Vitamin A in Health and Disease, R. Blomhoff,
ed., Marvel Dekker, New York (1993)). Die bis dato charakterisierten
RXRs sind jedoch den RARs insofern ähnlich, als sich die unterschiedlichen
RXR-Typen auch in ihren Nterminalen A/B-Regionen deutlich unterscheiden
(Leid et al., TIBS 17: 427–433
(1992); Leid et al., Cell 68: 377–395 (1992); Mangelsdorf et
al., Genes and Dev: 6: 329–344
(1992)) und sie dieselben transkriptionalen Aktivierungsfunktionen
in ihrer N-terminalen A/B-Region und C-terminalen E-Region enthalten
(Leid et al., TIBS 17: 427–433
(1992); Nagpal, S., et al., Cell 70: 1007–1019 (1992); Nagpal, S., et
al., EMBO J. 12: 2349–2360
(1993)).
-
RXRα und RXRβ weisen ein weit verbreitetes
(möglicherweise
allgegenwärtiges)
Expressionsmuster während
der Mausentwicklung und im erwachsenen Tier auf, das bei allen fötalen und
Erwachsenengeweben festzustellen ist, die bisher überpüft wurden
(Mangelsdorf D. J., et al., Genes & Devel. 6: 329–344 (1992); Dollé, P.,
et al., Mech. Devel. 45: 91–104
(1994); Nagata, T., et al., Gene 142: 183–189 (1994)). RXRγ-Tanskripte scheinen
aber eine eingeschränktere
Verbreitung aufzuweisen, wobei sie in sich entwickelnden Skelettmuskeln
im Embryo (wo ihre Expression das ganze Leben hindurch aufrecht
bleibt), im Herzen (nach der Geburt), in Sinnesepithelen des Seh-
und des Hörsystems,
in spezifischen Strukturen des zentralen Nervensystems und in Geweben
exprimiert sind, die in Schilddrüsenhormon-Homöostasie
involviert sind, z. B. die Schilddrüsenzellen und thyreotropen
Zellen in der Pituitaria (Mangelsdorf, D. J., et al., Genes & Devel. 6: 329–344 (1992);
Dollé,
P., et al., Mech. Devel. 45: 91–104
(1994); Sugawara, A., et al., Endocrinology 136: 1766–1774 (1995);
Liu, Q., und Linney, E., Mol. Endocrinol. 7: 651–658 (1993)).
-
Zur Zeit ist nicht klar, ob alle
molekularen Eigenschaften von RXRs, die in vitro charakterisiert
wurden, für
deren physiologischen Funktionen in vivo relevant sind. Insbesondere
ist nicht bekannt, unter welchen Bedingungen diese Rezeptoren als
9-cis-RAabhängige
transkriptionale Regulatoren agieren (Chambon, P., Semin. Cell Biol.
5: 115–125
(1994)). Die Knockouts von RXRα und
RXRβ in
der Maus haben einen gewissen Einblick in die physiologischen Funktionen
dieser Rezeptoren ermöglicht.
So sind zum Beispiel die okularen und kardialen Fehlbildungen, die
bei RXRα–/– Föten festgestellt
wurden (Kastner, P., et al., Cell 78: 987–1003 (1994); Sucov, H. M.,
et al., Genes & Devel.
8: 1007–1018
(1994)) ähnlich
wie jene; die beim fötalen
VAD-Syndrom festgestellt wurden, was eine wichtige Funktion von
RXRα bei
der Transduktion eines Retinoidsignals während der Entwicklung nahe
legt. Die Einbindung von RXRs bei der Retinoidsignalisierung wird
ferner durch Studien von zusammengesetzten RXRα/RAR-Mutanten unterstützt, welche
Defekte aufweisen, die bei den einzelnen Mutanten entweder fehlen
oder weniger stark: ausgeprägt
sind (Kastner, P., et al., Cell 78: 987–1003 (1994); Kastner, P.,
et al., Cell 83: 859–869
(1995)). Interessanterweise führt
aber das Knockout von RXRγ in der
Maus zu keinen offensichtlichen schädlichen Auswirkungen, und RXRγ–/– Homozygoten,
die auch RXRα–/– oder
RXRβ–/– sind,
weisen abgesehen von jenen Anomalitäten, die bei RXRα–/–,
RXRβ–/– und
fötalen
VAD-Syndrom-Föten
festgestellt wurden (Krezel, W., et al., Proc. Natl. Acad. Sci.
USA 93(17): 9010–9014
(1996)), keine zusätzlichen
Anomalitäten
auf, was die Vermutung nahe legt, dass RXRγ – trotz seines stark gewebespezifischen
Expressionsmusters im sich entwickelnden Embryo – für die embryonale Entwicklung
und das postnatale beben in der Maus entbehrlich ist. Die Beobachtung,
dass lebendgeborene RXRγ–/–/RXRβ–/–/RXRα+/– Mutanten
bis zum Erwachsenenalter heranwachsen können (Krezel et al., Proc.
Natl. Acad. Sci. USA 93(17): 9010–9014 (1996)), zeigt, dass
ein einzelnes RXRα-Allel
ausreicht, um alle vitalen Entwicklungs- und postnatalen Funktionen
der RXR-Rezeptorfamilie auszuführen,
insbesondere alle Entwicklungsfunktionen, die von RARs abhängen und
eine RXR-Partnerschaft
erfordern können
(Dollé,
P., et al., Mech. Dev. 45: 91–104
(1994); Kastner, P., et al., Cell 83: 859–869 (1995)). Ferner zeigt
die Erkenntnis, dass RXRα–/–/RXRγ–/– Doppelmutant-Embryonen
nicht stärker
betroffen sind als einzelne RXRα–/– Mutanten
(Krezel et al., Proc. Natl. Acad. Sci. USA 93(17): 9010–9014 (1996))
deutlich, dass RXRβ alleine
auch einige dieser Funktionen ausführen kann. Daher belegt die
Tatsache, dass RXRα alleine
und – bis
zu einem gewissen Grad – RXRβ alleine
für die
Durchführung
einer Reihe von Entwicklungs-RXR-Funktionen ausreichen, deutlich
die Existenz eines großen
Ausmaßes
von funktioneller Redundanz unter RXRs. In dieser Hinsicht unterscheidet
sich die RXR-Situation von jener der RARs, da alle Arten von RAR-Doppelmutanten
wesentlich breitere Gruppen von Defekten aufwiesen als einzelne
Mutanten (Rowe, A., et al., Develop. 111: 771–778 (1991); Lohnes, D., et
al., Develop. 120: 2723–2748
(1994); Mendelsohn C., Develop. 120: 2749–2771 (1994)).
-
Retinoid-Bindung an RAR-
und RXR-Rezeptoren
-
Die Kristallstrukturen der Ligand-bindenden
Domänen
(LBDs) der RARs und RXRs wurden vor kurzem geklärt (Bourget, W., et al., Nature
375: 377–382
(1995); Renaud, J. P., et al., Nature 378: 681–689 (1995); Wurtz, J. M. et
al., Nature struct. Biol. 3: 87–94
(1996)). Unter den verschiedenen RAR-Typen wird eine wesentliche
Aminosäuresequenzidentität in diesen
Domänen
festgestellt: Ein Vergleich der LBDS von RARα, RARβ und RARγ zeigt, dass nur drei Aminosäurerückstände in der
Ligand-bindenden Tasche dieser Rezeptoren variabel sind. Diese Rückstände sind
anscheinend dafür
verantwortlich, dass die verschiedenen RAR-Typen eine gewisse Selektivität beim Binden
bestimmter synthetischer Retinoide aufweisen (Chen, J.-Y., et al., EMBO
J. 14(6): 1187–1197
(1995); Renaud, J. P., et al., Nature 378: 681–689 (1995)), wobei die Berücksichtigung
dieser divergierenden Rückstände dazu
verwendet werden kann, RAR typenspezifische synthetische Retinoide
zu konstruieren, die agonistisch oder antagonistisch sind (Chambon,
P., FASEB J. 10: 940–954 (1996).
Dieser Konstruktionsansatz kann im Allgemeinen auf andere nukleare
Rezeptoren ausgeweitet werden, wie den Schilddrüsenrezeptor α (Wagner,
R. L., et al., Nature 378: 690– 697
(1995)), deren Ligand-bindenden Taschen chemisch und strukturell
jenen der RARs ähneln
können
(Chambon, P., FASEB J. 10: 940–954
(1996). Umgekehrt zeigt die Molekularmodellierung der Ligand-bindenden
Tasche der RXRs, dass es keine offensichtlichen Unterschiede bei
der Aminosäurezusammensetzung
zwischen RXRα,
RXRβ und
RXRγ gibt
(Bourguet, W., et al., Nature 375: 377–382 (1995); Wurtz, J. M.,
et al., Nature Struct. Biol. 3: 87–94 (1996)), was vermuten lässt, dass
die Konstruktion von typenspezifischen synthetischen Liganden für die RXRs schwieriger
sein kann als für
die RARs (Chambon , P., FASEB J. 10: 940–954 (1996)).
-
Retinoidsignalisierung durch
RAR : RXR Heterodimere
-
Nukleare Rezeptoren (NRs) sind Mitglieder
einer Superfamilie von Ligandinduzierbaren transkriptionalen regulatorischen
Faktoren, die Rezeptoren für
Steroid-Hormone,
Schilddrüsenhormone,
Vitamin D3 und Retinoide einschließen (Leid, M., et al., Trends
Biochem. Sci. 17: 427–433
(1992); Leid, M., et al., Cell 68: 377–395 (1992); und Linney, E.
Curr. Top. Dev. Biol., 27: 309–350
(1992)). NRs weisen eine modulare Struktur auf, welche die Existenz
von mehreren autonomen funktionellen Domänen wiederspiegelt. Basierend
auf der Ähnlichkeit
der Aminosäuresequenz
zwischen dem Huhn-Estrogenrezeptor,
dem menschlichen Estrogen-Rezeptor und dem Glucocorticoid-Rezeptor
und dem v-erb-A-Onkogen haben (Krust, A., et al., EMBO J. 5: 891–897 (1986))
sechs Regionen – A,
B, C, D, E und F – definiert,
die unterschiedliche Grade evolutionärer Konservierung unter verschiedenen
Mitgliedern der Superfamilie der nuklearen Rezeptoren aufweisen.
Die hoch konservierte Region C enthält zwei Zinkfinger und entspricht
dem Kern der DNA-bindenden Domäne (DBD),
die für
die spezifische Erkennung der verwandtern Ansprechelemente verantwortlich
ist. Region E ist funktionell komplex, da sie zusätzlich zur
Ligand-bindenden Domäne
(LBD) eine Ligand-abhängige
Aktivierungsfunktion (AF-2) und eine Dimerisierungsschnittstelle
enthält.
Eine autonome transkriptionale Aktivierungsfunktion (AF-1) ist in
den nicht-erhaltenen N-terminalen A/B-Regionen der Steroid-Rezeptoren
anwesend. Interessanterweise weisen sowohl AF-1 als auch AF-2 der
Steroid-Rezeptoren unterschiedliche transkriptionale Aktivierungseigenschaften
auf, welche sowohl zelltypenspezifisch als auch promotorkontextspezifisch
erscheinen (Gronemeyer, H. Annu. Rev. Genet. 25: 89–123 (1991)).
-
Wie oben beschrieben, werden die
all-trans (T-RA) und 9-cis (9C-RA) Retinolsäuresignale durch zwei Familien
von nuklearen Rezeptoren transduziert, RARα, β und γ (und deren Isoforme) werden
sowohl durch T-RA als auch 9C-RA aktiviert, wobei RXRα, β und γ ausschließlich durch
9C-RA aktiviert werden (Allenby, G. et al., Proc. Natl. Acad. Sci.
USA 90: 30–34
(1993)). Die drei RAR-Typen unterscheiden sich in ihren B- Regionen, und ihre
Haupt-Isoformen (α1
und α2, β1–4 und γ1 und γ2) weisen
unterschiedliche N-terminale A-Regionen auf (Leid, M. et al., Trends
Biochem. Sci. 17: 427– 433
(1992)). Ebenso unterscheiden sich die RXR-Typen in deren A/B-Regionen
(Mangelsdorf, D. J. et al, Genes Dev. 6: 329–344 (1992)).
-
Es wurde auch gezeigt, dass die E-Region
von RARs und RXRs eine Dimerisierungsschnittstelle enthält (Yu,
V. C. et al., Curr. Opin. Biotechnol. 3: 597–602 (1992)). Am interessantesten
ist, dass demonstriert wurde, dass RAR/RXR-Heterodimere in vitro
wesentlich effizienter an eine Reihe von RA-Ansprechelementen (RAREs)
binden als Homodimere von einem der Rezeptoren ((Yu, V. C. et al.,
Cell 67: 1251–1266
(1991); Berrodin, T. J. et al., Mol. Endocrinol 6: 1468–1478 (1992);
Bugge, T. H. et al., EMBO J. 11: 1409–1418 (1992); Hall, R. K. et
al., Mol. Cell. Biol. 12: 5527–5535
(1992); Hallenbeck, P. L. et al., Proc. Natl. Acad. Sci. USA 89: 5572–576 (1992);
Husmann, M. et al., Biochem. Biophys. Res. Commun. 187: 1558–1564 (1992);
Kliewer, S. A. et al., Nature 355: 446–449 (1992); Leid, M. et al.,
Cell 68: 377–395
(1992); Marks, M. S. et al., EMBO J. 11: 1419–1435 (1992); Zhang, X. K.
et al., Nature 355: 441–446
(1992)). RAR- und RXR-Heterodimere werden auch bevorzugt in Lösung in
vitro gebildet (Yu, V. C. et al., Cell 67: 1251–1266 (1991); Leid, M. et al.,
Cell 68: 377–395
(1992); Marks, M. S. et al., EMBO J. 11: 1419–1435 (1992)), obwohl die Zugabe
von 9C-RA die Bildung von RXR-Homodimeren in vitro zu verstärken erscheint
(Lehman, J. M. et al., Science 258: 1944–1946 (1992); Zhang, X. K.
et al., Nature 358: 587–591
(1992b)).
-
Es wurde gezeigt, dass die Aktivierung
von RA-ansprechenden Promotern wahrscheinlich durch RAR-RXR-Heterodimere
anstatt durch Homodimere auftritt (Yu, V. C. et al., Cell 67: 1251–1266 (1991);
Leid et al., Cell 68: 377–395
(1992b); Durand et al., Cell 71: 73–85 (1992); Nagpal et al.,
Cell 70: 1007–1019
(1992); Zhang, X. K., et al., Nature 355, 441-446 (1992); Kliewer
et al., Nature 355: 446–449
(1992); Bugge et al., EMBO J. 11: 1409-1418 (1992); Marks et al., EMBO J. 11:
1419–1435
(1992); You, V. C. et al., Cur. Op. Biotech. 3: 597–602 (1992);
Leid et al., TIBS 17: 427–433
(1992); Laudet und Stehelin, Curr. Biol. 2: 293–295 (1992); Green, S., Nature
361: 590–591
(1993)): Es wurde vorgeschlagen, dass der RXR-Abschnitt dieser Heterodimere
bei der Retinoid-induzierten Signalisierung stumm ist (Kurokawa,
R., et al., Nature 371: 528–531 (1994);
Forman, B. J., et al., Cell 81: 541–550 (1995); Mangelsdorf, D.
J., und Evans, R. M., Cell 83: 835–850 (1995)), obwohl zu diesem
Thema einander widersprechende Ergebnisse berichtet wurden (Apfel,
C. M., et al., J. Biol. Chem. 270(51): 30765–30772 (1995); siehe Chambon,
P. FASEB J. 10: 940–954 (1996)
zwecks Rezension). Obwohl die Ergebnisse dieser Studien die Vermutung
deutlich nahe legen, dass RAR/RXR-Heterodimere tatsächlich funktionelle
Einheiten sind, welche das RA-Signal in vivo transduzieren, ist
unklar, ob alle der vorgeschlagenen heterodimeren Kombinationen
in vivo auftreten (Chambon, P., Semin. Cell Biol. 5: 115–125 (1994)).
Daher kann die Basis für
die hoch pleiotrope Wirkung von Retinoiden, zumindest teilweise, in
der Kontrolle von unterschiedlichen Untergruppen an Retinoid-ansprechenden
Promotern durch zellspezifisch exprimierte heterodimere Kombinationen
von RAR : RXR-Typen (und Isoformen) liegen, deren Aktivität wiederum
durch zellspezifische Pegel von all-traps- und 9-cis-RA geregelt
werden kann (Leid et al., TIBS 17: 427–433 (1992)).
-
Die RXR-Rezeptoren können auch
in RA-unabhängige
Signalisierung involviert sein. So lässt zum Beispiel die Beobachtung
von abweichendem Lipid-Metabolismus in den Sertoli-Zellen von RXRβ–/– Mutanten-Tieren
vermuten, dass es auch zwischen RXRβ und dem Peroxisom-Proliferator-aktiviertem
Rezeptorsignalpfad zu funktionellen Interaktionen kommen kann (WO
94/26100; Kastner, P., et al., Genes & Devel. 10: 80–92 (1996)).
-
Therapeutische Verwendungen
von Retinoiden
-
Überblick
-
Da bekannt ist, dass Retinolsäure die
proliferierenden und differenzierenden Fähigkeiten von einigen Säugetierzelltypen
reguliert (Gudas, L. J., et al., in The Retinoids, zweite Ausgabe,
Sporn, M. B., et al., Hrsg., New York: Raven Press, Seiten 443–520 (1994));
werden Retinoide in verschiedenen chemopräventiven und chemotherapeutischen
Situationen eingesetzt. Es wurde über die Prävention von Mundhöhlenkrebs,
Hautkrebs und Hals-Nasen-Ohrenkrebs bei Patienten berichtet, bei
denen ein Risiko bestand, an diesen Tumoren zu erkranken (Hong,
W. K. et al., N. Engl. J. Md. 315: 1501–1505 (1986); Hong, W. K. et
al., N. Engl. J. Med. 323: 795–801
(1990); Kraemer, K. H. et al., N. Engl. J. Med. 318: 1633–1637 (1988);
Bollag, W. et al., Ann. Oncol. 3: 513–526 (1992); Chiesa, F. et
al., Eur. J. Cancer B. Oral Oncol. 28: 97–102 (1992); Costa, A et al., Cancer
Res. 54: Suppl. 7, 2032–2037
(1994)). Retinoide wurden auch verwendet, um das Plattenepithelkarzinom
des Nackens und der Haut (Verma, A: K. Cancer Res. 47: 5097–5101 (1987);
Lippman S. M. et al., J. Natl Cancer Inst. 84: 235–241 (1992);
Lippman S. M. et al., J. Natl Cancer Inst. 84: 241-245 (1992)) und Kaposi-Sarkom
zu behandeln (Bonhomme, L. et al., Ann. Oncol. 2: 234–235 (1991))
und wurden in beträchtlichem Ausmaß bei der
Therapie von akuter Promyelozytenleukämie eingesetzt (Huang, M. E.
et al., Blood 72: 567–572
(1988); Castaigne, S. et al., Blood 76: 1704–1709 (1990); Chomienne, C.
et al., Blood 76: 1710–1717 (1990);
Chomienne C. et al., J. Clin. Invest. 88: 2150–2154 (1991); Chen Z. et al.,
Leukemia 5: 288–292
(1991); Lo Coco, F. et al., Blood 77: 1657–1659 (1991); Warrell, R. P.,
et al., N. Engl. J. Med. 324: 1385–1393 (1991); Chomienne, C.,
et al., FASEB J. 10: 1025–1030
(1996)).
-
Akute Promyelozytenleukämie (APL)
-
Eine ausgeglichene chromosomale Translokation
t (15; 17) wurde in den meisten Zellen akuter Promyelozytenleukämie (APL)
festgestellt (Larson, A. R., et al., Am. J. Med. 76: 827–841 (1984)).
Der Bruchpunkt für
diese Translokation tritt innerhalb des zweiten Introns des RARα-Gens (Alcalay,
M. D., et al., Proc. Natl. Acad. Sci. USA 88: 1977–1981 (1991);
Chang, K. S., et al., Leukemia 5: 200–204 (1991); Chen, S., et al.,
Blood 78: 2696-2701
(1991) und innerhalb zweier Stellen des Gens ein, das den putativen
Zinkfinger-Transkriptionsfaktor
PML codiert (Goddard, A., et al., Science 254: 1371–1374 (1991)).
Diese reziproke t(15; 17) Translokation führt zur Erzeugung eines PML-RARα- Fusionsproteins,
das mit PML und RARα in
APL-Zellen co-exprimiert wird (zwecks Überblick und Verweisen siehe
Warrell, R. P., et al., N. Engl. J. Med. 329: 177–189 (1993); Grignáni, F.,
et al., Blood 83: 10–25
(1994); Lavau, C., und Dejean, A., Leukemia 8: 1615-1621 (1994); de Thé, H.,
FASEB J. 10: 955–960
(1996)). Die PML-RARα-Fusion
ist augenscheinlich für
den Differenzierungsblock im promyelozytischen Stadium verantwortlich,
da (i) sie bei beinahe allen APL-Patienten festgestellt wird (Warrell,
R. P., et al., N. Engl: J. Med. 329: 177–189 (1993); Grignáni, F.,
et al.; Blood 83: 10–25
(1994); Lavau, C., und Dejean, A., Leukemia 8: 1615–1621 (1994)),
(ii) sie bei Überexprimierung
in U937 oder HL60 myeloblastischen Leukämiezellen Myeloiddifferenzierung
hemmt ((Grignáni,
F., et al., Cell 74: 423–431
(1993)) und (iii) vollständige
klinische Remission auf Grund von . Differenzierung der Leukämiezellen
zu ausgereiften Granulozyten bei Behandlung mit all-trans-Retinolsäure (T-RA)
eng mit der PML-RARα-Expression
verbunden ist (Warrell, R. P., et al., N. Engl. J. Med. 324: 1385–1393 (1991);
Lo Coco, R., et al., Blood 77: 1657–1659 (1991); Chomienne, C.,
et al., FASEB J. 10: 1025–1030
(1996)). Mehrere Studien haben sich mit der möglichen Auswirkung von PML-RARα-Fusionsproteinbildung
auf die Zellenproliferation (Mu, X. M., et al., Mol. Cell. Biol.
14: 6858–6867
(1994)) und Apoptose (Grignani, F., et al., Cell 74: 423–431 (1993)),
AP1-Transrepression (Doucas, V., et al., Proc. Natl. Acad. Sci.
USA 90: 9345–9349
(1993)) und Vitamin-D3-Signalisierung (Perez, A., et al., EMBO J.
12: 3171–3182
(1993)) beschäftigt,
aber der Mechanismus/die Mechanismen, mit dessen/deren Hilfe PML-RARα die Myeloidzellenausreifung
blockiert, ist weiterhin schwer zu bestimmen. In Übereinstimmung
mit der abweichenden nuklearen Kompartimentierung von PML-RARα, der den „PML-Typ“-Standort
bei RA-Behandlung einnimmt (Dyck, J. A., et al., Cell 76: 333–343 (1994);
Weis, K., et al., Cell 76: 345-358
(1994); Koken, M. H., et al., EMBO J. 13: 1073–1083 (1994)), lautet die zur
Zeit vorherrschende Hypothese, dass PML-RARα veränderte transkriptionale Eigenschaften
im Vergleich zu PML oder RARα besitzt
und/oder auf dominant-negative Weise agieren kann (Perez, A., et
al., EMBO J. 12: 3171–3182
(1993); de Thé,
H., et al., Cell 66: 675–684
(1991); Kastner, P., et al, EMBO J. 11: 629–642 (1992)).
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Die Erfindung stellt pharmazeutische
Zusammensetzungen bereit, die mindestens einen RAR-Antagonisten,
der vorzugsweise ein RARα-Antagonist
und am meisten bevorzugt Verbindung A oder Verbindung B ist, mindestens
einen RXR-Agonisten, der am meisten bevorzugt SR11237 ist, und einen
pharmazeutisch annehmbaren Träger
oder ein Arzneibindemittel dafür
umfassen. Die Erfindung betrifft ferner die Verwendung dieser pharmazeutischen
Zusammensetzungen bei der Herstellung eines Medikamentes zur Verwendung
bei der Behandlung eines Tieres, vorzugsweise eines Menschen, das
an Krebserkrankungen (vorzugsweise an Hautkrebs, Mundhöhlenkrebs,
Lungenkrebs, Brustdrüsenkrebs,
Prostatakrebs, Blasenkrebs, Leberkrebs, Bauchspeicheldrüsenkrebs,
Gebärmutterhalskrebs,
Eierstockkrebs, Krebs des Kopf- und Halsbereiches, Darmkrebs, Keimzellenkrebs,
wie Teratokarzinom oder einer Leukämie, und am meisten bevorzugt
akuter Promyelozytenleukämie),
einer Hauterkrankung (vorzugsweise Psoriasis, aktinische Keratose,
Akne, Ichthyosis, Lichtalterung oder kortikoid-induzierte Hautatrophie),
rheumatischer Arthritis und einer prämalignen Läsion leidet oder dazu prädisponiert
ist.
-
KURZE BESCHREIBUNG DER ZEICHNUNGEN
-
1:
Zusammenfassung agonistischer und antagonistischer Aktivitäten der
verschiedenen BMS synthetischen Retinoide. "+" steht beim jeweiligen
Rezeptor für
agonistisch, "(+)" für
schwach agonistisch, "–" für antagonistisch
und "0" für
keine Aktivität.
-
2:
Falschfarbendarstellung einer einzelnen Photon-Kameraanalyse von
Retinoidinduzierter Luciferase-Aktivität, die aus HeLa-Reporterzellen
hervorgeht, die nur mit RARspezifischen Liganden (Lane a) oder mit
10 nM all-trans-RA (Lane b) behandelt wurden.
-
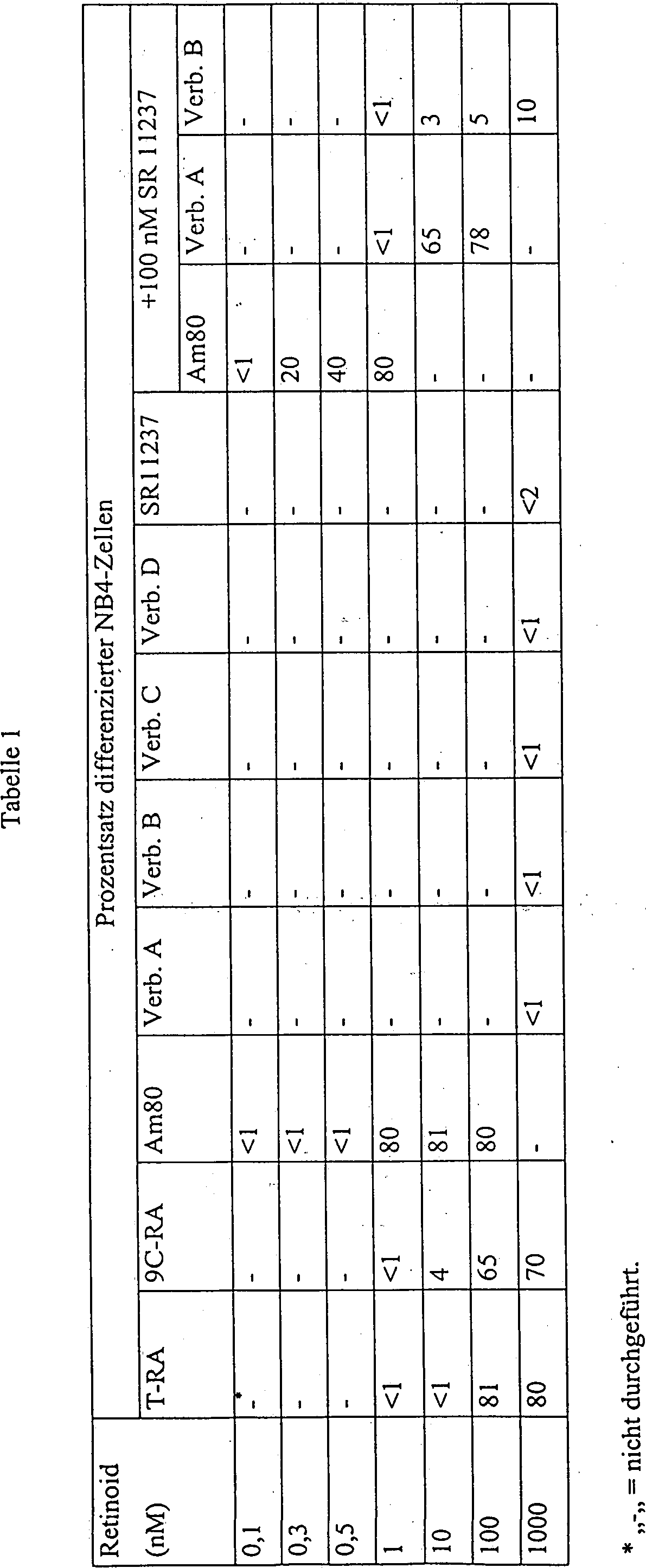
3:
Synergie zwischen RARα-Agonisten
oder Antagonisten und RXR-Agonisten für die Induktion von NB4-Zelldifferenzierung.
Mikrofotografien von Zellen, die mit Nitroblau-Tetrazolium (a–d) gefärbt wurden oder
Immunofluoreszenz-Mikrofotografien von Zellen, die nach 4-tägiger Behandlung
mit Ethanol oder den/dem angeführten
Retinoid(en) mit Anti-PML-Antisera (e–h) gefärbt wurden.
-
4:
Synergie zwischen RARα-Agonisten
oder Antagonisten und RXR-Agonisten für die Induktion von NB4-Zellenapoptose.
Auswirkungen von Retinoiden auf die Zellzyklusverteilung und Auftreten
von „sub-2N“ apoptopischen
Zellen und Partikeln, so wie durch Flusszytometrie-Analyse gezeigt.
Die Horizontalachse bei den Histogrammen zeigt die integrierte Fluoreszenzintensität und die
vertikale Achse die Anzahl an Partikeln an. In jedem Histogramm
werden ungefähr
20.000 Partikel dargestellt; Histogramme, welche die Anzahl der
Zellen anzeigen, die eine 2N, 4N oder Zwischenmenge von DNA für unbehandelte
NB4-Zellen (a) enthalten, Zellen, die mit 1 mM T-RA über einen
Zeitraum von 4, 6 oder 8 Tagen (b–d) behandelt werden, 1 nM Am80 über einen
Zeitraum von 4 oder 8 Tagen (e, f) oder die Kombination aus 100
nM von Verbindung A und 100 nM von SR11237 über einen Zeitraum von 4 oder
8 Tagen (g, h).
-
5:
Retinoid-Induktion von Apoptose in NB4-Zellen. Mikrofotografie eines
Ethidiumbromid-gefärbten
Agarose-Gels in einem DNA-Fragmentations-Assay, ausgeführt mit
Zellen, die in Abwesenheit (Lanes 2–4) oder Gegenwart (Lanes 5–13) der
angeführten
Retinoide in folgenden Konzentrationen gezüchtet werden: T-RA; 100 nM;
Am80, 1 nM; SR11237, 100 nM; Verbindung A, 10 nM (Lanes 9 und 10)
und 100 nM (Lanes 12 und 13).
-
6:
Fotografien von sequenzierenden Gels, welche Synergismus von RARα und RXR
zeigen. Zellen wurden mit Agonisten, so wie angeführt, behandelt,
dann wie unten beschrieben für
Ligations-herbeigeführte
PCR (a, b) oder Umkehr-Transkriptase PCR (c) verarbeitet und auf
einem Sequenzierungsgel aufgelöst. (a)
NB4-Zellen, Autoradiogramm; (b) P19-Zellen, Autoradiogramm; (c)
NB4-Zellen, Ethidiumbromid-gefärbtes Gel
(RNA-Transkripte
des β-Aktin-Gens
wurden als Kontrolle verwendet).
-
DETAILLIERTE BESCHREIBUNG
DER ERFINDUNG
-
Synthese von RAR und RXR-Agonisten
und Antagonisten
-
Die in der vorliegenden Erfindung
zu verwendenden Wirkstoffe können – ohne darauf
beschränkt
zu sein – natürliche oder
synthetische Peptide, Kohlenhydrate, Steroide und Vitaminderivate
(sein (deren Herstellung zum Beispiel unter Anwendung von Verfahren der
synthetischen, organischen und anorganischen Chemie erfolgt, die
auf dem Fachgebiet weithin bekannt sind). Gemäß der Erfindung können die
Wirkstoffe nach dem Zufallsprinzip ausgewählt und gescreent werden. Für Zufalls-Screening
werden Wirkstoffe wie Peptide, Kohlenhydrate, Steroide oder Vitaminderivate
(z. B. RA-Derivate) zufällig
ausgewählt
und untersucht, unter Verwendung direkter oder indirekter Verfahren,
die auf dem Fachgebiet als Routinevorgang gelten, um ihre Fähigkeit,
an einen RAR- oder RXR-Rezeptor oder ein funktionelles Retinoid-RAR
: RXR-Rezeptorheterodimer zu binden, zu untersuchen. So schließen zum
Beispiel mögliche
RAR-Agonisten gemäß der vorliegenden
Erfindung synthetische Retinoide wie Am580, Verbindung 1 und Verbindung
2 (deren Strukturen in Ostrowski et al., Proc. Natl. Acad. Sci.
USA 92: 1812–1816
(1995) offenbart sind) und Am80 (Roy et al., Mol. Cell. Biol. 15(12):
6481–6487
(1995) ein. Mögliche
RXR-Agonisten gemäß der vorliegenden
Erfindung schließen
synthetische Retinoide wie SR11237 (deren Struktur in Lehman, J.
M., et al., Science 258: 1944–1946
(1992) offenbart ist) ein. Mögliche
RAR-Antagonisten
schließen – ohne darauf
beschränkt
zu sein – die
zuvor beschriebenen (Chen et al., EMBO J. 14: 1187–1197 (1995);
Roy et al., Mol. Cell. Biol. 15(12): 6481–6487 (1995); Chen et al.,
EMBO J. 14: 1187–1197
(1995) und Verbindung A und Verbindung B, wie unten im Detail beschrieben, ein.
-
Somit sind auf dem Fachgebiet Verfahren
bekannt, um mögliche
RAR-Antagonisten und RXR-Agonisten für das Screening, so wie unten
beschrieben, zur Verwendung gemäß der vorliegenden
Erfindung zu entwickeln. Die Erfindung kann insbesondere mit den
RAR-Antagonisten-Verbindungen
ausgeführt
werden, die hierin als „Verbindung
A“ und „Verbindung
B“ bezeichnet
werden und folgende Strukturen aufweisen: Verbindung
A:
Verbindung
B:
-
Diese Verbindungen können, so
wie in der US-Patentschrift Nr. 5,559,248 beschrieben, hergestellt werden.
Andere nützliche
RAR-Antagonisten werden zum Beispiel in Eyrolles et al., Med. Chem.
Res. 2: 361–367
(1992) und Apfel et al., Proc. Natl. Acad. Sci. USA 89: 7129–7133 (1992)
beschrieben. Die Erfindung kann auch mit dem RXR-Agonisten LG1069
ausgeführt
werden, dessen Struktur und Herstellung in Boehm et al., J. Med.
Chem. 37: 2930–2941
(1994) beschrieben sind. Andere nützliche RXR-Agonisten sind
zum Beispiel in Lehman et al., Science 258: 1944–1946 (1992) beschrieben. Andere
RAR-Antagonisten und RXR-Agonisten, die sich zur Verwendung in der
vorliegenden Erfindung eignen, können
durch die oben genannten Verfahren und andere Verfahren hergestellt
werden, die für
einen Durchschnittsfachmann als Routine gelten.
-
Screening-Verfahren
Eine Reihe von Verfahren für
das Screening von möglichen
RAR-Antagonisten und
-
RXR-Agonisten, die durch rationales
Design oder Computermodellierung wie oben beschrieben erzeugt werden,
sind auf dem Fachgebiet weithin bekannt und ermöglichen es einem Durchschnittsfachmann, festzustellen,
ob eine Verbindung im Rahmen der vorliegenden Erfindung nützlich ist.
Zum Beispiel wurden in Chen et al., EMBO J. 14(6): 1187–1197 (1995)
drei „Reporter“-Zelllinien
verwendet, um eine Reihe von RARα-, RARβ- oder RARγ-spezifischen
dissoziierenden synthetischen Retinoiden zu charakterisieren, die
selektiv die AF-2-Aktivierungsfunktion induzieren, welche in der
LBD von RARβ (βAF-2) vorhanden
ist (Chen, J.-Y., et al., EMBO J. 14(6): 1187–1197 (1995)). Diese Zelllinien
exprimieren auf stabile Weise chimär Proteine, welche die DNA-Bindungsdomäne des Hefe-Transaktivators
GAL4 enthalten, der an die EF-Regionen (welche die LBD und die AF-2-Aktivierungsfunktion
enthalten) von RARα (GAL-RARα), RARβ (GAL-RARβ) oder RARγ(GAL-RARγ) fusioniert
ist, und ein Luciferase-Reportergen,
das durch ein Pentamer der GAL4-Erkennungssequenz ('17m') vor dem β-Globinpromotor (17m)5-GAL-Luc)
getrieben wird. In diesen Zelllinien induzieren die RAR- Liganden
somit Luciferase-Aktivität,
die in den intakten Zellen unter Verwendung einer Einzel-Photon-Zählungs-Kamera
gemessen werden kann. Dieses Reportersystem ist endogenen Rezeptoren
gegenüber,
welche die GAL4-Bindungstelle nicht erkennen können, unempfindlich. Bei Verwendung
von analogen Screening-Assays wurde festgestellt, dass diese synthetischen
Retinoide, wie RA, das verankerungsunabhängige Wachstum von Onkogen-transformierten
3T3-Zellen hemmen, während
in Bezug auf den Promotor des menschlichen Interleukin-6 (IL-6)
Gens, dessen Produkt in die Regulierung der Hämatopoese, Immunreaktionen
und Entzündung
(Kishimoto, T. et al., Science 258: 593–597 (1992)) involviert ist,
festgestellt wurde, dass er durch RA induziert wird und nicht durch
die synthetischen dissoziierenden Retinoide, welche seine Aktivität unterdrückten. Auf ähnliche
Weise wurden RXR-Agonisten identifiziert, unter Verwendung von Zelllinien, die
einen RXR-Rezeptor exprimieren, der mit einem TREpal-tk-Reportergen
verbunden ist, das sowohl durch RAR-RXR-Heterodimere als auch RXR-Homodimere
aktiviert wird (Lehmann, J. M., et al., Science 258: 1944–1946 (1992)).
Somit können
Reporterzelllinien, die durch Verfahren, die für einen Durchschnittsfachmann
Routine sind, leicht herzustellen sind, nicht nur dazu verwendet
werden, die spezifischen RAR- oder RXR-Typen, an die ein möglicher
Ligand binden wird, zu unterscheiden, sondern auch um festzustellen,
ob dieses Binden einen aktivierenden (d. h. agonistischen) oder
repressiven (d. h. antagonistischen) Effekt induziert. Obwohl die
oben angeführten
Reporterzelllinien die Luciferase- oder Thymidinkinase-Gene als
Reporter umfassten, sind andere Reporter wie Neo, CAT, β-Glactosidase oder
Grünes
Fluoreszierendes Protein auf dem Fachgebiet weithin bekannt und
können
auf ähnliche
Weise verwendet werden, um die vorliegende Erfindung auszuführen. Zu
den Referenzen, die Reporterplasmide offenbaren, welche ein Reportergen
und Expressionsvektoren enthalten, die eine LBD eines nuklearen
Rezeptors codieren, gehören
zum Beispiel Meyer et al., Cell 57: 433–442 (1989); Meyer et al.,
EMBO J. 9(12): 3923–932
(1990); Tasset et al., Cell 62: 1177–1187 (1990); Gronemeyer, H.,
und Laudet, V., Protein Profile 2: 1173–1308 (1995); Webster et al.,
Cell 54: 199–207 (1988);
Strähle
et al., EMBO J. 7: 3389–3395
(1988); Seipel et al., EMBO J. 11: 4961–4968 (1992); und Nagpal et;
al., EMBO J. 12: 2349–2360
(1993).
-
Andere routinemäßige Untersuchungen wurden
verwendet, um Verbindungen auf deren agonistische oder antagonistische
Auswirkungen auf Funktionen von anderen nuklearen Rezeptoren, wie
Steroid-Rezeptoren, zu untersuchen. So wurde zum Beispiel ein transientes
Expressions-/Gelverzögerungssystem
verwendet, um die Auswirkungen der synthetischen Steroide RU486
und R5020 auf die Glucocorticoid- und Progesteronrezeptorfunktion
zu untersuchen (Meyer, M-E., et al., EMBO J. 9(12): 3923–3932 (1990)). Ähnliche
Untersuchungen wurden verwendet, um zu zeigen, dass Tamoxifen Estradiol-induziertes
ERAP160-Binden an den Estrogenrezeptor kompetitiv hemmt; was einen
Mechanismus für
seine wachstumshemmenden Wirkungen bei Brustkrebs vermuten lässt (Halachimi,
S., et al., Science 264: 1455–1458
(1994)). Da die RAR- und RXR-Rezeptoren
anscheinend anderen nuklearen Rezeptoren wie den Steroidrezeptoren
strukturell ähnlich
sind (wie in Chambon, P., FASEB J. 10: 940–954 (1996) besprochen), können routinemäßige Untersuchungen
dieser Art bei der Bewertung von Verbindungen im Hinblick auf deren
agonistisches oder antagonistisches Wirken auf RAR- oder RXR-Rezeptoren
nützlich
sein.
-
Als alternatives, routinemäßiges Verfahren
kann die Auswirkung eines möglichen
Agonisten oder Antagonisten auf das Binden des Ligand-abhängigen AF-2
Modulators TIF1 an eine RAR oder RXR LBD unter Verwendung von Gluathion-S-Transferase
(GST)-Irteraktionsassays
durch Markieren der LBDs mit GST untersucht werden, wie im Detail
in Le Douarin et al., EMBO J. 14: 2020–2033 (1995) beschrieben.
-
In einer anderen Screening-Untersuchung
können
transgene Mäuse
und Zelllinien, die in ihrer Expression eines oder mehrerer RAR-
oder RXR-Rezeptoren verändert
werden, so wie zuvor beschrieben hergestellt (Krezel, W., et al.,
Proc. Natl. Acad. Sci. USA 93(17): 9010–9014 (1996) und verwendet
werden, um Agonisten und Antagonisten spezifischer Mitglieder der
RAR/RXR-Klasse von Rezeptoren unter Anwendung der zuvor beschriebenen
Verfahren (WO 94/26100) zu identifizieren. Bei einer solchen Untersuchung
wird der Wirkstoff, der zu testen ist, mit einer oder mehreren der
transgenen Zelllinien oder Mäuse
oder Geweben, die davon entnommen sind, inkubiert. Danach wird der
Grad der Bindung des Wirkstoffes bestimmt, oder die Auswirkung, welche
der Wirksttoff auf die Entwicklung oder Genexpression hat, wird
mit Hilfe von Techniken überwacht,
die für
einen Durchschnittsfachmann als Routine gelten. So wie hierin verwendet,
ist der Begriff „inkubieren“ so zu verstehen,
dass die Verbindung oder der Wirkstoff, die untersucht werden, mit
der geeigneten Zelle oder dem geeigneten Gewebe in Kontakt gebracht
werden, oder dass der Wirkstoff oder die Verbindung der geeigneten Maus über einen
der weithin bekannten Verabreichungswege, einschließlich enterale,
intravenöse,
subkutane und intramuskuläre
Verabreichung, verabreicht wird.
-
Es können auch andere Untersuchungen,
so wie jene, die im Detail unten in Beispiel 1 und 2 beschrieben
werden, verwendet verwenden, um die agonistischen oder antagonistischen
Auswirkungen von RAR- und RXR-Liganden zu bestimmen. Zum Beispiel
werden bestimmte agonistische Retinoide die Assoziation von endogenem
PML/PML-RARα-Fusionsprotein
mit nuklearen Körpern
in Zellen von APL-Patienten (Dyck, J. A., et al., Cell 76: 333–343 (1994);
Weis, K., et al., Cell 76: 345–356
(1994); Koken, M. H. M., et al., EMBO J. 13(5): 1073–1083 (1994))
oder in verwandten etablierten Zelllinien wie NB4 (Lanotte, M.,
et al., Blood 77(5): 1080–1086
(1991)) induzieren. Diese Auswirkungen von RAR- oder RXR-Agonisten
oder Antagonisten können
zum Beispiel durch verschiedene immunologische Techniken wie Immunofluoreszenz-
oder Immunoelektronmikroskopie unter Verwendung von Antikörpern bestimmt
werden, die für
PML, RAR und/oder PML-RARα-Fusionsproteine
spezifisch sind. RAR- oder RXR-Agonisten oder Antagonisten können auch
durch ihre Fähigkeit,
die in vitro Differenzierung (Ausreifung) von bestimmten etablierten
Zelllinien wie HL-60 myeloblastische Leukämiezellen (Nagy, L., et al.,
Mol. Cell. Biol. 15(7): 3540–3551
(1995)), NB4 promyelozytische Zellen (Lanotte, M., et al., Blood
77(5): 1080–1086
(1991), P19 oder P9 embryonale Karzinomzellen (Roy, B., et al., Mol.
Cell. Biol. 15(12): 6481–6487
(1995); Horn, V., et al., FASEB J. 10: 1071–1077 (1996)) oder ras-transformierte
3T3-Zellen (Chen et al., EMBO J. 14(6): 1187–1197 (1995)) zu induzieren,
identifiziert werden. Ligand-induzierte Differenzierung in diesen
und anderen Zelllinien kann bestimmt werden, indem Ligand-behandelte
oder Ligand-unbehandelte Zellen für die Expression einer Vielzahl
von weithin bekannten Differenzierungsmarkern einer Untersuchung
unterzogen werden, wie in den oben genannten Referenzen im Allgemeinen
beschrieben.
-
Ebenso können die möglichen Antagonisten oder Agonisten
durch Messen ihrer Fähigkeit,
Apoptose (programmierter Zelltod) in zum Beispiel HL-60 Zellen Nagy,
L., et al., Mol. Cell. Biol. 15(7): 3540–3551 (1995)) oder P19-Zellen
(Horn, V., et al., FASEB J. 10: 1071–1077 (1996)) oder in anderen
primären
Zellen oder etablierten Zelllinien zu induzieren, gescreent werden.
Apoptose wird typischerweise durch Messung von Ligandinduzierter
DNA-Fragmentation bewertet, die durch Verfahren wie Gel-Elektrophorese
(Auftreten von kleineren Molekulargewichtbanden), Mikroskopie (Veränderungen
bei der Plasmamembranmorphologie, wie z. B. Bildung von Oberflächenaussstülpungen
(„Blasen“), oder
bei der nuklearen Morphologie, wie z. B. Pyknose oder Fragmentation)
oder Expression des putativen Apoptose-Suppressiv-Proteins BCL-2
(in apoptotischen Zellen verringert) durchgeführt wird; zwecks allgemeiner
Verfahren und Besprechungen dieser Assays, soweit diese sich auf
die RAR- und RXR-Biologie beziehen, siehe Nagy, L., et al., Mol.
Cell. Biol. 15(7): 3540–3551 (1995);
Horn, V., et al., FASEB J. 10: 1071–1077 (1995)). Andere Verfahren
für eine
Untersuchung von Ligand-induzierter Apoptose in primären Zellen
und etablierten Zelllinien, wie Flusszytometrie oder Partikelanalyse
(Auftreten von kleineren Partikeln mit unterschiedlichem Lichtstreuungs-
und/oder DNA-Inhaltsprofil) sind auf dem Fachgebiet weithin bekannt
(Telford; W. G., et al., J. Immunol. Meth. 172(1): 1–16 (1994);
Campana, D. et al., Cytometry 18(2): 68–74 (1994); Sgonc, R., und
Wick, G., Int. Arch. Allergy Immunol. 105(4): 327–332 (1994);
Fraker, P. J., et al., Meth. Cell. Biol. 46: 57–76 (1995); Sherwood, S. W.,
und Schimke, R. T., Meth. Cell. Biol. 46: 77–97 (1995), Carbonari, M, et
al., Cytometry 22(3): 161–167
(1995); Mastrangelo, A. J. und Betenbaugh, M. J., Curr. Opin. Biotechnol.
6(2): 198–202
(1995)). Schließlich
kann das Screening von Agonisten oder Antagonisten durch eine Untersuchung
erfolgen, die als „in
vivo Footprinting“ bekannt
ist (Mueller, P. R., und Wold, B., Science 246: 780–786 (1989);
Garrity, P. A., und Wold, B. J., Proc. Natl. Acad. Sci. USA 89: 1021–1025 (1992)),
wie unten detaillierter in Beispiel 1 und 2 beschrieben, wobei sich
diese Untersuchung für die
Analyse von RA-induzierter Transkription von RARβ2 als nützlich erwiesen hat (Dey, A.,
et al., Mol. Cell. Biol. 14(12): 8191–8201 (1994)).
-
Andere Verfahren zur Bestimmung der
agonistischen oder antagonistischen Aktivitäten eines möglichen Liganden, die auf dem
Fachgebiet als Routine gelten, können
auch bei der Durchführung
der vorliegenden Erfindung verwendet werden. Bei der Durchführung solcher
Untersuchungen wird ein Fachmann in der Lage sein, zu bestimmen,
an welchen RAR- oder RXR-Rezeptortyp ein Wirkstoff bindet, welcher)
spezifische(n) Rezeptoren) von einer bestimmten Verbindung verwendet
wird (werden) und ob der Wirkstoff ein Agonist oder Antagonist des
(der) bestimmten Rezeptors (Rezeptoren) ist.
-
Klinische Indikationen
-
Somit sind Verfahren zur Identifikation,
Synthetisierung und zum Screening von RAR-Antagonisten und RXR-Agonisten
auf dem Fachgebiet weithin bekannt. Diese Liganden können dann
gemäß der vorliegenden
Erfindung bei der Behandlung einer Reihe von physischen Beschwerden
bei Tieren, insbesondere Säugetieren – einschließlich Menschen – eingesetzt
werden. Wie oben beschrieben, ist bekannt, dass Retinolsäure die
proliferierenden und differenzierenden Fähigkeiten von mehreren Säugetierzelltypen
reguliert (Gudas, L. J. et al., (1994), In Sporn, M. B., Roberts,
A. B. und Goodman, D. S. (Hrsg), The Retinoids. 2. Ausgabe, Raven
Press, New York, Seiten 443–520).
Retinoide wurden somit in einer Reihe von chemopräventiven
und chemotherapeutischen Anwendungen eingesetzt. Zu diesen klinischen
Ansätzen
gehören
die Prävention und/oder
Behandlung einer Reihe von Krebsleiden und prämalignen Läsionen davon, wie zum Beispiel
jene der Mundhöhle,
Haut (einschließlich
Plattenepithelkarzinom, Melanom und Kaposi-Sarkom), Kopf- und Halsbereich,
Gebärmutterhals,
Eierstock, Lunge, Brustdrüse,
Blase, Prostata, Leber und Bauchspeicheldrüse (Hong, W. K., et al., N.
Engl. J. Med. 315: 1501–1505
(1986); Verma, A. K., Cancer Res. 47: 5097–5101 (1987); Hong. W. K.,
et al., N. Engl. J. Med. 323: 795–801 (1990); Kraemer, K. H.,
et al., N. Engl. J. Med. 318: 1633–1637 (1988); Bonhomme, L.,
et al., Ann. Oncol. 2: 234–235
(1991); Bollag, W., et al., Ann. Oncol. 3: 513–526 (1992); Chiesa, F., et
al., Eur. J. Cancer B. Oral Oncol. 28: 97–102 (1992); Lippman, S. M.,
et al., J. Natl. Cancer Inst. 84: 235–241 (1992), Lippman, S. M.,
et al., J. Natl. Cancer Inst. 84: 241–245 (1992); Costa, A., et
al., Cancer Res. 54(Suppl. 7): 2032–2037 (1994); de Thé, H.,
FASEB J. 10: 955–960
(1996); Lotan, R., FASEB J. 11): 1031–1039 (1996); Bérard,
J., et al., FASEB J. 10: 1091–1097
(1996)). Retinoide wurden insbesondere dazu verwendet, Patienten
zu behandeln, die unter bestimmten Leukämiearten litten, insbesondere unter
akuter Promyelozytenleukämie
(Huang, M. E., et al., Blood 72: 567–572 (1988); Castaigne, S.,
et al., Blood 76: 1704–1709
(1990); Chomienne, C., et ., Blood 76: 1710–1717 (1990); Chomienne, C.,
et al., J. Clin. Invest. 88: 2150–2154 (1991); Chen. Z., et
al., Leukemia 5: 288–292
(1991); Lo Coco, F., et al., Blood 77: 165701659 (1991); Warrell,
R. P., et al., N. Engl. J. Med. 324: 1385–1393 (1991); Chomienne, C.,
et al., FASEB J. 10: 1025–1030
(1996)). Retinoide haben sich auch bei der Behandlung von bestimmten
Hautleiden wie Psoriasis, Akne, Ichthyosis, Lichtalterung und Corticoid-induzierter
Hautatropie wie jener, die bei lokaler Verwendung von Kortikosteroiden
im Rahmen der Behandlung von Hautentzündungen auftritt, als wirksam
erwiesen (Fisher, G. J. und Voorhees, J. J., FASEB J. 10: 1002–1013 (1996)).
-
Somit können die Kombinationen von
RAR-Antagonisten und RXR-Agonisten der vorliegenden Erfindung bei
der Herstellung eines Medikamentes zur Verwendung bei der Behandlung
eines Tieres, vorzugsweise eines Säugetieres – einschließlich eines Menschen – verwendet
werden, das an einer Reihe von physischen Beschwerden leidet oder
dafür prädisponiert
ist. So wie hierin verwendet, wird ein Tier, das für ein physisches Leiden „prädisponiert“ ist, als
ein Tier definiert, das nicht eine Vielzahl an offensichtlichen
physischen Beschwerdesymptomen aufweist, das aber genetisch, physiologisch
oder aus anderen Gründen
Gefahr läuft,
ein Leiden zu entwickeln. Die Kombinationen aus RAR-Antagonisten und
RXR-Agonisten können
somit prophylaktisch als chemopräventive
Wirkstoffe für
sölche
Leiden eingesetzt werden. Bei der Behandlung des Tieres mit den
Kombinationen der vorliegenden Erfindung kann der RXR-Agonist dem
Tier vor, gleichzeitig mit oder nach der Verabreichung des RAR-Antagonisten
verabreicht werden.
-
Zu den physischen Leiden, die mit
den Kombinationen und Verfahren der vorliegenden Erfindung behandelt
werden können,
zählen
eine Reihe von Krebserkrankungen wie Hautkrebs (einschließlich Melanom und
Kaposi-Sarkom), Mundhöhlenkrebs,
Lungenkrebs, Brustdrüsenkrebs,
Prostatakrebs, Blasenkrebs, Leberkrebs, Bauchspeicheldrüsenkrebs,
Gebärmutterhalskrebs,
Eierstockkrebs, Krebs des Kopf- und Halsbereiches, Darmkrebs, Keimzellenkrebs
(einschließlich
Teratokarzinom) und Leukämie,
am meisten bevorzugt akute Promyelozytenleukämie. Andere physische Leiden,
die mit den Kombinationen und Verfahren der vorliegenden Erfindung
behandelt werden können,
schließen
Hautleiden wie Psoriasis, aktinische Keratose, Akne, Ichthyosis,
Lichtalterung und corticoid-induzierte Hautathropie und rheumatische
Arthritis ein. Die Zusammensetzungen und Verfahren der vorliegenden
Erfindung können
auch bei der Prävention
des Krankheitsfortschrittes eingesetzt werden, wie zum Beispiel
in der Chemoprävention
des Fortschritts einer prämalignen
Läsion
zu einer malignen Läsion.
Die Zusammensetzungen und Verfahren der vorliegenden Erfindung können auch
verwendet werden, um ein Tier zu behandeln, das an anderen physischen
Beschwerden leidet oder für
solche prädisponiert
ist, die auf Behandlung mit Retinoiden ansprechen.
-
Formulierung und Verfahren
zur Verabreichung
-
Wie oben angeführt, ist bekannt, dass RAR-
und RXR-selektive Liganden eine große Bandbreite an Zellreaktionen
hervorrufen, von denen einige in Form klinischer Anwendungen bei
der Behandlung eines Patienten eingesetzt werden. Der Begriff „Patient"
steht im vorliegenden Dokument für
ein Tier, vorzugsweise ein Säugetier,
einschließlich
eines Menschen. Durch die Erfindung kann die Dosis von einem oder
mehreren RAR-Antagonisten
deutlich reduziert werden, wenn sie gemeinsam mit mindestens einem
RXR-Agonisten verabreicht
wird. Wie hierin verwendet, wird „eine wirksame Menge eines
RAR(oder RXR-)Antagonisten“ als
eine Menge definiert, die so wirkt, dass sie eine Zellreaktion in
Zellen hervorruft, die einen RAR- (oder RXR)-Rezeptor exprimieren.
Weiter unten werden beispielhafte klinische Therapien, bei denen
einem Patienten Zusammensetzungen verabreicht werden, die mindestens
einen RAR-Antagonisten und mindestens einen RXR-Agonisten umfassen, näher beschrieben.
-
Kombinationen aus RAR-Agonisten und
RXR-Agonisten mit potentieller Verwendung bei der Therapie von Menschen
sind auf dem Fachgebiet bekannt (Lehmann, J. M., et al., Science
258: 1944–1946
(1992); Durand, B., et al., EMBO J. 13: 5370–5382 (1994); Lotan, R., et
al., Cancer Res. 55: 232–236
(1995); Roy, B., et al., Mol. Cell. Biol. 15(12): 6481–6487 (1995);
Horn, V., et al., FASEB J. 10: 1071–1077 (1996)). Keine dieser vorhergehenden
Offenbarungen hat jedoch die unerwartete Erkenntnis aus der vorliegenden
Erfindung beschrieben oder vorhergesehen, die darin besteht, dass
Kombinationen aus RAR- Antagonisten und RXR-Agonisten bei der Behandlung
einer Reihe von physischen Leiden nützlich sind.
-
Somit werden pharmazeutische Zusammensetzungen
bereitgestellt, die mindestens einen RAR-Antagonisten (wie jene,
die oben beschrieben wurden), mindestens einen RXR-Agonisten (wie jene,
die oben beschrieben wurden) und einen pharmazeutisch annehmbaren
Träger
oder ein Arzneibindemittel umfassen und die oral, rektal, parenteral,
intrasystemisch, intravaginal, intraperitoneal, lokal (beispielsweise
in Form von Pulvern, Salben, Tropfen oder transdermalen Kissen oder
Pflaster), bukkal oder als Mund- oder Nasenspray verabreicht werden
können.
Wichtig ist dabei, dass durch gemeinsames Verabreichen eines RAR-Antagonisten und
eines RXR-Agonisten die klinischen Nebenwirkungen durch Verwendung
geringerer Dosen des RAR-Antagonisten und des RXR-Agonisten reduziert
werden können.
Wie angeführt,
wird erkannt werden, dass der RXR-Agonist entweder vor, nach oder
gleichzeitig mit dem RAR-Antagonisten „gemeinsam verabreicht“ werden
kann, je nach Anforderungen einer speziellen therapeutischen Anwendung.
Unter „pharmazeutisch
annehmbarem Träger“ ist ein
nicht-toxisches, festes, halbfestes oder flüssiges Füllmaterial, Verdünnungsmittel, Einkapselungsmaterial
oder Formulierungshilfsmittel jeglicher Art zu verstehen. Der Begriff „parenteral“ bezieht
sich in der hierin verwendeten Weise auf die Arten der Verabreichung,
zu denen intravenöse,
intramuskuläre,
intraperitoneale, intrasternale, subkutane und intraartikulare Injektion
und Infusion gehören.
-
Pharmazeutische Zusammensetzungen
der vorliegenden Erfindung zur Verwendung bei parenteraler Injektion
können
pharmazeutisch annehmbare, sterile, wässerige: oder nichtwässerige
Lösungen,
Dispersionen, Suspensionen oder Emulsionen sowie sterile Pulver
für die
Rekonstitution in sterile injizierbare Lösungen oder Dispersionen unmittellbar
vor der Verwendung umfassen. Beispiele geeigneter wasseriger und
nicht-wässeriger
Träger,
Verdünnungsmittel,
Lösemittel
oder Vehikel schließen
Wasser, Ethanol, Polyole (wie Glycerin, Propylenglycol, Polyethylenglycol
und dergleichen), Carboxymethylcellulose und geeignete Mischungen
davon, pflanzliche Öle
(wie Olivenöl)
und injizierbare organische Ester wie Ethyloleat ein. Eine geeignete
Fließfähigkeit
kann zum Beispiel durch die Verwendung von Beschichtungsmaterial
wie Lecithin, durch Aufrechterhaltung der erforderlichen Partikelgröße im Falle
von Dispersionen und durch die Verwendung von oberflächenaktiven
Stoffen gewährleistet
werden.
-
Die Zusammensetzungen der vorliegenden
Erfindung können
auch Hilfsstoffe wie Konservierungsstoffe; Benetzungsmittel, Emulgiermittel
und Dispersionsmittel enthalten. Die Prävention der Wirkung von Mikroorganismen
kann durch das Einschließen
verschiedener antibakterieller Wirkstoffe und Fungizide wie zum Beispiel
Paraben, Chlorobutanol, Phenolsorbinsäure und dergleichen gewährleistet
werden. Es kann auch wünschenswert
sein, isotonische Wirkstoffe wie Zucker, Natriumchlorid und dergleichen
einzuschließen.
Eine verlängerte
Absorption der injizierbaren pharmazeutischen Form kann durch das
Einschließen
von Wirkstoffen erfolgen, welche die Absorption verzögern, wie
Aluminiummonostearat und Gelatine.
-
In einigen Fällen ist es wünschenswert,
die Absorption von subkutaner oder intramuskulärer Injektion zu verlangsamen,
um die Wirkung der Arzneimittel zu verlängern. Dies kann durch die
Verwendung einer flüssigen
Suspension von kristallinem oder amorphem Material mit geringer
Wasserlöslichkeit
erfolgen. Die Absorptionsrate des Arzneimittels hängt dann
von seiner Auflösungsrate
ab, die wiederum von der Kristallgröße und der kristallinen Form
abhängen
kann. Alternativ dazu wird eine verzögerte Absorption einer parenteral
verabreichten Arzneimittelform durch Auflösen oder Suspendieren des Arzneimittels
in einem Ölvehikel
erzielt.
-
Injizierbare Depotformen werden durch
Bilden von Mikroeinkapselungsmatrizenn des Arzneimittels in biologisch
abbaubaren Polymeren wie Polylactid-Polyglycolid. hergestellt. In
Abhängigkeit
vom Verhältnis
des Arzneimittels zum Polymer und der Natur des jeweiligen Polymers,
das eingesetzt wird, kann die Rate der Arzneimittelfreigabe gesteuert
werden. Beispiele anderer. biologisch abbaubarer Polymere schließen Poly(orthoester)
und Poly(anhydride) ein. Depot-injizierbare Formulierungen werden
auch durch Einschluss des Arzneimittels in Liposome oder Mikroemulsionen
hergestellt, die mit Körpergeweben
kompatibel sind.
-
Die injizierbaren Formulierungen
können
zum Beispiel mittels Filtrierung durch. einen Bakterienrückhaltefilter
oder durch Aufnahme von sterilisierenden Wirkstoffen in der Form
von sterilen, festen Zusammensetzungen sterilisiert werden, die
in sterilem Wasser oder in einem anderen sterilen, injizierbaren
Medium unmittelbar vor der Werwendung aufgelöst oder dispergiert werden
können.
-
Feste Dosierungsformen für die orale
Verabreichung schließen
Kapseln, Tabletten, Pillen, Pulver und Granulate ein. Bei solchen
festen Dosierungsformen werden die aktiven Verbindungen mit mindestens
einem pharmazeutisch annehmbaren Arzneibindemittel oder Träger wie
Natriumcitrat oder Dicalciumphosphat und/oder a) Füllmitteln
oder Streckmitteln wie Stärken,
Lactose, Sucrose, Glucose, Manitol und Kieselsäure (b) Bindemitteln wie zum
Beispiel Carboxymethylcellulose, Alginaten, Gelatine, Polyvinylpyrrolidon,
Sucrose und Acacia, c) Anfeuchtern wie Glycerin, d) Aufbrechmitteln
wie Agar-Agar, Calciumcarbonat, Kartoffel- oder Tapioka-Stärke, Alginsäure, bestimmte
Silikate und Natriumcarbonat, (e) Lösungsverzögerungsmitteln wie Paraffin,
(f) Absorptionsbeschleunigern wie quaternäre Ammoniumverbindungen, (g)
Benetzungsmitteln wie zum Beispiel Cetylalkohol und Glycerinmonostearat,
(h) Absorptionsmitteln wie Kaolin und Bentonit-Ton und i) Schmiermitteln
wie Talk, Calciumstearat, Magnesiumstearat, festen Polyethylenglycolen, Natriumlaurylsulfat und
Mischungen davon gemischt. Im Falle von Kapseln, Tabletten und Pillen
können
die Dosierformen auch Pufferungsmittel umfassen.
-
Feste Zusammensetzungen eines ähnlichen
Typs können
auch als Füllmittel
in Weich- und Hart-Gelatinekapseln unter Verwendung von Arzneibindemitteln
wie Lactose oder Milchzucker als auch Polyethylenglycolen mit hohem
Molekulargewicht und dergleichen eingesetzt werden.
-
Die festen Dosierungsformen von Tabletten,
Dragees, Kapseln, Pillen und Granulaten können mit Beschichtungen und
Hüllen
wie enterischen Beschichtungen und anderen auf dem Fachgebiet der
pharmazeutischen Formulierung weithin bekannten Beschichtungen hergestellt
werden. Sie können
gegebenenfalls undurchsichtig machende Mittel enthalten und auch
aus einer Zusammensetzung bestehen, bei der sie nur oder bevorzugterweise
die aktiven Inhaltsstoffe/den aktiven Inhaltsstoff in einen bestimmten
Teil des Darmtraktes, optional in verzögerter Weise, freisetzen. Beispiele
einbettender Zusammensetzungen, die verwendet werden können, schließen polymere
Substanzen und Wachse ein.
-
Die aktiven Zusammensetzungen können auch
in mikroeingekapselter Form vorkommen, falls geeignet, mit einem
oder mehreren der oben angeführten
Arzneibindemittel.
-
Flüssige Dosierungsformen für die orale
Verabreichung schließen
pharmazeutisch annehmbare Emulsionen, Lösungen, Suspensionen, Sirups
und Elixiere ein. Zusätzlich
zu den aktiven Verbindungen können
die flüssigen
Dosierungsformen inerte Verdünnungsmittel
enthalten, die auf dem Fachgebiet. häufig verwendet werden, zum
Beispiel Wasser oder andere Lösemittel,
löslich
machende Mittel und Emulgiermittel wie Ethylalkohol, Isopropylalkohol,
Ethylcarbonat, Ethylacetat, Benzylalkohol, Benzylbenzoat, Propylenglycol,
-1,3-Butylenglycol, Dimethylformamid, Öle (insbesondere Baumwollkern-,
Erdnuss-, Mais-, Keim-, Oliven-, Rizinus- und Sesamöl), Glycerin,
Tetrahydrofurfurylalkohol, Polyethylenglycole und Sorbitanfettsäureester
und Mischungen davon.
-
Abgesehen von inerten Verdünnungsmitteln
können
die oralen Zusammensetzungen auch Hilfsstoffe wie Benetzungsmittel,
Emulgier- und Suspendiermittel, Süßstoffe, Geschmacksmittel und
Parfümierungsmittel einschließen.
-
Zusätzlich zu den aktiven Verbindungen
können
Suspensionen Suspensionsmittel wie zum Beispiel ethoxylierte Isostearylalkohole,
Polyoxyethylensorbitol und Sorbitanester, Mikrokristallincellulose,
Aluminummetahydroxid, Bentonit, Agar-Agar und Tragacanth und Mischungen
davon enthalten.
-
Lokale Verabreichung schließt die Verabreichung
an die Haut oder Schleimhaut, einschließlich Oberflächen der
Lunge und des Auges ein. Zusammensetzungen für die lokale Verabreichung,
einschließlich
jener für
Inhalation, können
als ein Trockenpulver hergestellt werden, das unter Druck stehend
oder nicht unter Druck stehend sein kann. In nicht unter Druck stehenden
Pulverzusammensetzungen können
die aktiven Inhaltsstoffe in fein zerteilter Form in Beimengung
mit einem größeren pharmazeutisch
annehmbaren inerten Träger
verwendet werden, der Partikel mit einer Größe von zum Beispiel bis zu
100 μm im
Durchmesser umfasst. Geeignete inerte Träger schließen Zucker wie zum Beispiel
Lactose ein. Wünschenswerterweise
weisen mindestens 95 Gewichtsprozent der Partikel des aktiven Inhaltsstoffes
eine effektive Partikelgröße im Bereich von
0,01 bis 10 μm
auf.
-
Alternativ dazu kann die Komposition
unter Druck stehen und ein Druckgas enthalten, wie Stickstoff oder
ein verflüssigtes
Gastreibmittel. Das verflüssigte
Treibmittelmedium, und in der Tat die gesamte Zusammensetzung, ist
vorzugsweise so gestaltet, dass die aktiven Inhaltsstoffe sich darin
in keinem wesentlichen Ausmaß auflösen. Die
unter Druck stehende Zusammensetzung kann auch einen oberflächenaktiven
Stoff enthalten. Der oberflächenaktive
Stoff kann ein flüssiger
oder fester nicht-ionischer oberflächenaktiver Stoff oder ein
fester anionischer oberflächenaktiver
Stoff sein. Es wird bevorzugt, den festen anionischen oberflächenaktiven
Stoff in Form eines Natriumsalzes zu verwenden.
-
Eine weitere Form der lokalen Verabreichung
ist die Verabreichung ins Auge. Die RAR-Antagonisten/Der RAR-Antagonist
und die RXR-Agonisten/der RXR-Agonist werden in einem pharmazeutisch
annehmbaren ophthalmischen Vehikel verabreicht, so dass die Verbindungen
mit der okularen Oberfläche über einen Zeitraum
in Kontakt gehalten werden, der ausreicht, damit die Verbindungen
die kornealen und internen Regionen des Auges durchdringen, wie
zum Beispiel die vordere Augenkammer, hintere Augenkammer, den Glaskörper, das
Kammerwasser, Humor vitreus, die Hornhaut, Iris/Ziliar, Linsen,
Aderhaut/Netzhaut und Lederhaut. Das pharmazeutisch annehmbare ophthalmische
Vehikel kann zum Beispiel eine Salbe, ein pflanzliches Öl oder ein
einkapselndes Material sein.
-
Zusammensetzungen für rektale
oder vaginale Verabreichung sind vorzugsweise Zäpfchen, die durch Mischen des/der
RAR-Antagonisten und RXR-Agonisten mit geeigneten nicht-reizenden
Arzneibindemitteln oder Trägern
wie Kakaobutter, Polyethylenglycol oder einem Zäpfchenwachs hergestellt werden
können,
die bei Raumtemperatur fest, aber bei Körpertemperatur flüssig sind
und daher im Rektum oder im Scheidenhohlraum schmelzen und die Arzneimittel
freisetzen.
-
Die Zusammensetzungen der vorliegenden
Erfindung können
auch in Form von Liposomen verabreicht werden. Wie auf dem Fachgebiet
bekannt ist, werden Liposome im Allgemeinen von Phospholipiden oder
anderen lipiden Substanzen abgeleitet. Liposome werden durch mono-
oder multi-lamelläre
hydrierte Flüssigkristalle
gebildet, die in einem wässerigen
Medium dispergiert sind. Es kann jedes nicht-toxische, physiologisch
annehmbare und metabolisierbare Lipid verwendet werden, das imstande
ist, Liposome zu bilden. Die vorliegenden Zusammensetzungen in Liposomform
können – zusätzlich zum/zu
den RAR-Antagonisten und dem/den RXR-Agonisten Stabilisatoren, Konservierungsstoffe,
Arzneibindemittel und dergleichen enthalten. Die bevorzugten Lipide
sind Phospholipide und Phosphatidylcholine(Lecithine), sowohl natürliche als
auch synthetische. Verfahren zur Bildung von Liposomen sind auf
dem Fachgebiet bekannt (siehe zum Beispiel Prescott, Ed., Meth.
Cell Bio. 14: Seiten 33 ff (1976)). Dosierung Durch die Erfindung
kann ein RXR-Agonist in vitro, ex vivo oder in vivo an Zellen verabreicht
werden, um die zelluläre
Reaktion auf einen RAR-Antagonisten zu verstärken. Ein Durchschnittsfachmann
wird anerkennen, dass wirksame Mengen eines RAR-Antagonisten und eines RXR-Agonisten
empirisch ermittelt und in purer Form oder – wo solche Formen existieren – in Form eines
pharmazeutisch annehmbaren Salzes, Esters oder als Pro-Arzneimittel
eingesetzt werden können. Der/die
RAR-Antagonisten) und der/die RXR-Agonisten) kann können einem
Patienten, der diese benötigt,
als pharmazeutische Zusammensetzungen) in Kombination mit einem
oder mehreren pharmazeutisch annehmbaren Arzneibindemitteln verabreicht
werden. Es wird verstanden werden, dass bei Verabreichung an einen menschlichen
Patienten die tägliche
Gesamtmenge der Verbindungen und Zusammensetzungen der vorliegenden
Erfindung vom behandelnden Arzt auf der Basis eines fundierten medizinischen
Urteils festgelegt wird. Die spezifische, therapeutisch wirksame
Dosis für
einen bestimmten Patienten hängt
von einer- Reihe von Faktoren ab, einschließlich des Typs und Grads der
zellulären
Reaktion, die es zu erreichen gilt; der Aktivität des jeweiligen RAR-Antagonisten
und RXR-Agonisten, die eingesetzt werden; der jeweiligen, eingesetzten Verbindung;
des Alters, Körpergewichts,
des allgemeinen Gesundheitszustands, des Geschlechts und der Ernährung des
Patienten; der Zeit der Verabreichung, des Verabreichungsweges und
der Ausscheidungsrate des RAR- Antagonisten
und/oder des RXR-Agonisten; der Dauer der Behandlung; der in Kombination
oder gleichzeitig mit dem jeweiligen RAR-Antagonisten und/oder dem
RXF-Agonisten verwendeten Arzneimittel; und ähnlicher Faktoren, die auf
dem Fachgebiet der Medizin weithin bekannt sind. Zum Beispiel ist
es auf dem Fachgebiet üblich,
die Dosen von RAR-Antagonisten
und/oder RXR-Agonisten bei Mengen zu beginnen, die unter jenen liegen,
die erforderlich sind, um die gewünschte therapeutische Wirkung
zu erzielen, um dann die Dosen allmählich zu steigern, bis die
gewünschte
Wirkung erzielt wird.
-
Zum Beispiel werden zufriedenstellende
Ergebnisse durch orale Verabreichung eines RAR-Antagonisten und
eines RXR-Agonisten bei Dosierungen in der Größenordnung von 0,05 bis 10
mg/kg/Tag, bevorzugt 0,1 bis 7,5 mg/kg/Tag, mehr bevorzugt 0,1 bis
2 mg/kg/Tag erzielt, bei einmaliger Verabreichung oder – in aufgeteilten
Dosierungen – 2
bis 4 Mal pro Tag. Bei parenteraler Verabreichung, zum Beispiel
durch intravenöses Tropfen
oder Infusion, können
Dosierungen in der Größenordnung
von 0,01 bis 5 mg/kg/Tag, bevorzugt 0,05 bis 1,0 mg/kg/Tag und mehr
bevorzugt 0,1 bis 1,0 mg/kg/Tag; verwendet werden. Geeignete Tagesdosierungen
für Patienten
liegen somit in der Größenordnung
von 2,5 bis 500 mg p. o., vorzugsweise 5 bis 250 mg p. o, bevorzugter
5 bis 100 mg p. o. oder in der Größenordnung von 0,5 bis 250
mg intravenös,
bevorzugt 2,5 bis 125 mg intravenös und mehr bevorzugt 2,5 bis
50 mg intravenös.
Die Dosierung des RAR-Antagonisten kann so erfolgen wie in
EP 0 661 259 A1 beschrieben.
-
Die Dosierung kann auch in einer
patientenspezifischen Weise erfolgen, um eine vorbestimmte Konzentration
eines RAR-Antagonisten und/oder eines RXR-Agonisten im Blut bereitzustellen,
wie durch auf dem Fachgebiet akzeptierte und als Routine geltende
Techniken bestimmt (HPLC wird bevorzugt). Somit kann die Dosierung
bei Patienten eingestellt werden, um reguläre fortgehende Blutwerte zu
erhalten, wie durch HPLC gemessen und in der Größenordnung von 50 bis 1000
ng/ml, vorzugsweise 1.50 bis 500 .
-
ng/ml.
-
Beispiele
-
Materialien und Verfahren
-
Folgende Materialien und Verfahren
wurden im Allgemeinen in allen Beispielen verwendet, sofern nicht
anders angeführt.
-
Bestimmung agonistischer/antagonistischer
Aktivitäten
von RAR-spezifischen Liganden. HeLa-Reporterzelllinien, die ein
Retinoid-induzierbares Luciferase-Reportergen (17m)5-globin-Luc
in den stabil transfektierten Reporterkonstrukten GAL-RARα, GAL-RARβ oder GAL-RARγ enthalten,
wurden wie zuvor beschrieben konstruiert und verwendet (Chen, J.
Y., et al., EMBO J. 14: 1187–1197
(1995)). Zellen wurden mit RAR-spezifischen Liganden behandelt und
Luciferase-induzierte Biolumineszenz wurde in vivo unter Verwendung
einer Einzel-Photon-Zählungs-Kamera
(Hamamatsu) durch Pflanzen von gleichen Mengen an Zellen in 24-Mulden-Gewebekulturplatten
und deren Inkubieren mit steigenden Konzentrationen an Retinoiden
alleine oder in Gegenwart von T-RA überwacht, um das antagonistische
Potential der Liganden zu bestimmen.
-
Bestimmung der NB4-Zelldifferenzierung.
NB4-Zellen wurden aus ATCC erhalten und in RPMI-1640 (plus 2 mM
L-Glutamin) gezüchtet,
das 100 Einheiten/Mikroliter Penicillin und Streptomycin und 8%
fötales Rinderserum(FBS)
enthält.
Zellen wurden über
einen Zeitraum von vier Tagen mit Retinoid(en) oder Ethanolvehikel
behandelt, danach gewaschen und erneut bei einer Dichte von 5 × 105 Zellen/ml suspendiert. 50 Mikroliter Aliquote
dieser Suspension wurden über
poly-L-lysin-beschichtete Slides gestrichen (Sigma, St. Louis, Missouri).
Nach dem Waschen mit PBS (137 mM NaCI, 2,7 mM KCl, 4,3 mM N2HPO4, 1,4 mM KHP2O4) wurden die Zellen
10 Minuten lang in 2% Formalin (37% Formaldehyd in 15% Methanol
in Wasser) bei Raumtemperatur fixiert.
-
Bestimmung von Nitroblau-Tetrazolium:
Nach dem Fixieren wurden die Zellen drei Mal in PBS gewaschen und
durch TPA(12-O-tetradecanoyl-phorbol-l3-acetat, 200 ng/ml)induzierte
Nitroblau-Tetrazolium-Reduktion (Sigma Fast BCIP/NBT) 30 Minuten
lang bei 37°C
bewertet. Proben wurden auf zelluläre und nukleare Morphologie
hin mit einem optischen Mikroskop (Diavert Leitz) untersucht. Der
Prozentsatz an differenzierten Zellen wurde bestimmt, indem mindestens
300 Zellen für
jede Behandlung gezählt
wurden.
-
Bestimmung der Anti-PML-Immunofluoreszenz:
Nach dem Fixieren wurden die Zellen mit 0,1% Triton X-100 in PBS
(Puffer A) permeabilisiert. Nach dem Waschen in PBS wurden die Zellen
in 0,5 mg/ml normalem Ziegen-IgG (Jackson Laboratories, Bar Harbor,
Maine) in PBS 30 Minuten lang bei Raumtemperatur blockiert und danach über Nacht
bei 4°C
mit einem polyklonalen Anti-PML-Antikörper (freundlicherweise zur
Verfügung gestellt
von Dr. Anne Dejean) inkubiert, verdünnt in Puffer A, der 0,5 mg/ml
normales Ziegen-IgG (Puffer B) enthält, bei 4°C. Nach drei Waschungen in Puffer
A wurden die Zellen mit Esel-Anti-Kaninchen-IgG (H + L) inkubiert,
konjugiert mit Cyanin 3 (Jackson Laboratories, Bar Harbor, Maine)
in Puffer B über
einen Zeitraum von 1 Stunde bei Raumtemperatur. Nach den Waschungen
in PBS wurden die zusammengesetzten Proben mit einem konfokalen
Lasermikroskop (TCS, Leica) untersucht, das mit Cyaninoptik für PML-Lokalisierung ausgestattet
war:
-
Bestimmung der NB4-Zellapoptose
-
Flusszytometrische Analyse. Die Zellzyklusverteilung
und die Gegenwart von „sub-2N“-Zellen
und Partikeln in Kontrolle und Retinoid-behandelten NB4-Zellen wurden
durch Zellzyklusflusszytometrie basierend auf dem DNA-Gehalt unter
Verwendung eines EPICS Profile II Zell-Sortierers (Coulter Electronics,
Inc., Hialeah, Florida) bestimmt, der mit einem 15-Watt-Argonlaser,
der auf eine Anregungswellenlänge
von 488 nm eingestellt war, und Filtersets ausgestattet gewesen
ist, die eine Emissionswellenlänge
von 575 nm bereitstellten. Kulturen von unbehandelten oder Retinoid-behandelten
Zellen wurden zentrifugiert (250 × g) und in 70% Ethanol fixiert
und bei –20°C gespeichert.
Nach zwei Waschungen in PBS wurden die Zellen und subzellulären Partikel
in 1 mg/ml RNase A (59 Kunitz Einheiten/mg, Sigma Chemical Co.,
St. Louis, Missouri) 30 Minuten lang bei 37°C inkubiert, zentrifugiert und
in PBS mit einer Konzentration von ungefähr 106 Zellen
pro ml resuspendiert. Ethidiumbromid wurde zu einer endgültigen Konzentration
von 50 Mikrogramm/ml unmittelbar vor der Probenanalyse hinzugefügt.
-
DNA-Fragmentationsanalyse. Die Induktion
von Apoptose durch Retinoide wurde auch durch das Auftreten einer „Leiter"
von fragmentierter DNA bestimmt, wie zuvor beschrieben (Nagy, L.,
et al., Mol. Cell. Bio1. 15: 3540–3551 (1995)). Die Zellen wurden
alle zwei Tage neu mit frischen Medien und Retinoiden versorgt, und
die Zelldichte wurde deutlich unter der Sättigung gehalten, um Zelltod
auf Grund von Nährstoff
und/oder Mitogenerschöpfung
zu vermeiden. Pro DNA-Spur wurden fünf Mikrogramm in einem 1,5
% Agarosegel elektrophoresiert, danach mit Ethidiumbromid gefärbt, und
DNA-Banden wurden visualisiert und via Ultraviolettbeleuchtung von
Ethidiumbromidfluoreszenz fotografiert.
-
In vivo RARβ2 Footprinting
und RARβ, γ Expression
-
Ligation-vermittelte PCR. NB4-Zellen
wurden wie oben beschrieben kultiviert. P19,6-Zellen wurden aus ATCC erhalten und
in Dulbeccos modifiziertem Eagles Medium kultiviert, das 5% normales
FBS und 5% delipidiertes FBS enthält. NB4-Zellen wurden 24 Stunden
lang entweder mit Ethanol oder den angeführten Retinoiden (
6) behandelt. Nach dem Waschen
in PBS wurden die Zellen 5 Minuten lang bei Raumtemperatur mit 0,1
% Dmethylsulfat (DMS; Aldrich) behandelt. DNA mit hohem Molekulargewicht
wurde extrahiert und mit Piperidin gespalten. DMS-Behandlung in
vitro von nackter DNA erfolgte wie beschrieben (Mueller, P. R.,
und Wold, B., Science 246: 780–786
(1989)). Ligationvermittelte Polymerasekettenreaktion (LM-PCR) wurde
wie beschrieben ausgeführt
(Garrity, P. A., und Wold, B.J., Proc. Natl. Acad. Sci. USA 89:
1021–1025
(1992)), außer,
dass DEEPVENT
TM (exo) DNA-Polymerase (New
England BioLabs, Beverly, Massachusetts) verwendet wurde. Oligonukleotide,
die in der LM-PCR verwendet wurden, um Interaktionen auf dem kodierenden
Strang zu erfassen, waren: a)
menschlicher RARβ2-Promotor:
b)
Maus-RARβ2-Promotor:
-
Jeder Primer 3 (SEQ-ID. Nr. 3 und
5) wurde bei dem 5' Ende
mit [γ-32P]ATP unter Verwendung von T4 Polynukleotidkinase
markiert. Nach der PCR wurden die markierten Produkte in einem 4,8%
Sequenziergel aufgelöst,
und die Gels wurden getrocknet und einem Röntgenfilm zur Produktion von
Autoradiogrammen ausgesetzt.
-
Umkehr-Transkriptase-PCR: Die gesamte
RNA wurde isoliert und umgekehrt in eine Pufferlösung transkribiert, die 10
mM Tris-HCl (pH 8,3), 50 mM KCI, 2 mM MgCl
2 und
1 mM von allen vier Desoxynukleosidtriphosphaten umfasst, unter
Verwendung von 25 Einheiten von AMV Umkehr-Transkriptase in der
Gegenwart von 50 Einheiten von RNAsin 60 Minuten lang bei 42°C. Zur Umkehr-Transkription
wurden 25 Picomol pro Reaktion des allgemeinen Antisense-Primers
5'-GACATGCCCACTTCAAAGCACTTC-3' (SEQ-ID. Nr. 6) für RARα, β oder γ verwendet. Für jede PCR-Reaktion
wurden der allgemeine Antisense-Primer,
ein Viertel des Umkehr-Transkriptionsproduktes und folgende spezifischen
Sense-Primer verwendet:
-
Die Amplifikation wurde in einem
endgültigen
Volumen von 0,1 ml unter Verwendung von Taq DNA Polymerase (Perkin
Elmer Cetus) gemäß den Herstelleranweisungen
durchgeführt.
Achtzehn PCR-Zyklen (30 Sekunden bei 94°C, 30 Sekunden bei 65°C und 30
Sekunden bei 72°C)
wurden ausgeführt,
und ein Aliquot von 10 Mikrolitern jeder Reaktion wurde durch Elektrophorese
auf einem 6% nativen Polyacrylamidgel analysiert, welches dann mit
Ethidiumbromid gefärbt
und durch Ultraviolettbeleuchtung untersucht wurde.
-
Beispiel 1: RAR- und RXR-induzierte
Aktivierung
-
Rezeptor-selektive synthetische Retinoide
(Chen, J.-Y., et al., EMBO J. 14: 1187-1197 (1995)) (
1) wurden verwendet, um die Beiträge von RARα/PML-RARα, RARβ, RARγ und RXRs
zu den molekularen und zellulären
Ereignissen zu untersuchen, die bei T-RA-Behandlung (Lanotte, M., et al.,
Blood 77: 1080–1086 (1991))
zu NB4-Zelldifferenzierung
führen.
Wie in Tabelle 1 dargestellt, hat der Agonist Am80, der bei ≤ 1nM (Chen,
J.-Y., et al., EMBO J. 14: 1187–1197
(1995)) RARα-spezifisch
ist, auf effiziente Weise eine Differenzierung induziert. Retinoide
ohne RARα-agonistische
Aktivität
(Verbindung A und Verbindung C; Chen, J.-Y., et al., EMBO J. 14:
1187–1197
(1995)), Verbindung B und Verbindung D,
1, SR11237 (Lehmann, J. M., et al., Science
258: 1944–1946
(1992)) waren alleine ineffizient (Tabelle 1,
3b,
c). Verbindung C ist die in Beispiel 23
der Europäischen
Patentanmeldung Nr.
EP 0 661
259 beschriebene Verbindung. Verbindung D ist die in Beispiel
1 der Europäischen
Patentanmeldung Nr.
EP 0 747
347 beschriebene Verbindung. Darüber hinaus wurde die durch
1 nM Am80 induzierte Differenzierung durch einen Überschuss
an Verbindung B, einem reinen RARα-Antagonisten (nicht
dargestellt), vollständig
blockiert. Diese Ergebnisse zeigen, dass das Induzieren der transkriptionalen
Aktivität
von AF-2 des nicht-fusionierten RARα-Allels und/oder des RARα-Anteils
von PML-RARα ausreichend
ist, um den Differenzierungsblock zu entlasten, der aus der Bildung
eines PML-RARα-Fusionsproteins
resultiert. Dabei ist zu beachten, dass es nicht wahrscheinlich
ist, dass die AP1-Transrepression durch RARα eine entscheidende Rolle bei
RA-induzierter NB4-Zelldifferenzierung spielt, da „dissoziierte“ Retinoide,
wie Verbindung A, obwohl sie die AP1-Aktivität über alle drei RARs effizient
unterdrücken (Chen,
J-Y, et al., EMBO J. 14: 1187–1197
(1995)), selbst keine Differenzierung induzierten (
2b und
nicht dargestellt).
-
Nach viertägiger Behandlung mit T-RA oder
RARα-Agonisten
hat eine FACS-Analyse
gezeigt, dass NB4-Zellen die mitotische Aktivität reduziert und sich in G1/G0( 4b und e)
akkumuliert hatten. Am Tag 6 haben sich Sub-2N-Partikel in T-RA-behandelten
Kulturen (4c) akkumuliert und am Tag
8 zeigte massive DNA-Fragmentation extensive Apoptose an (4d, f und 5, Lanes 6, 7). Dabei ist
zu beachten, dass Vorwärts-
und Seitwärtsstreuungsanalyse
derselben Zellpräparationen
diese Ergebnisse bestätigten
(nicht dargestellt). Wie im Fall der Differenzierung konnten nur
RARα (aber
nicht RARβ oder
RARγ)-Agonisten
diese Ereignissequenz induzieren (Tabelle 1 und nicht dargestellt).
-
Vor der Differenzierung und wie zuvor
(Dyck, J. A., et al., Cell 76: 333–343 (1994); Weis, K., et al.,
Cell 76: 345–358
(1994); Koken, M. H., et al., EMBO J. 13: 1073–1083 (1994)) für T-RA berichtet,
induzierten RARα-Agonisten
die Assoziation von endogenen PML/PML-RARα mit nuklearen Körpern (3e), während
RARα-Antagonisten
dies nicht taten (3f, und nicht dargestellte
Daten). Darüber
hinaus zeigte in vivo DNA-Footprinting,
dass die Belegung der DR-5-Typen Retinolsäurereaktionselemente (DR5-RAREs), die im RARβ2-Promotor
anwesend waren, und die Rekrutierung anderer promotorbindender Faktoren
durch RARα-Agonisten,
aber nicht Antagonisten (5,
Lanes 3, 5 und 6), induziert wurden. Abgesehen von geringen Unterschieden
waren diese Footprints beinahe nicht von jenen zu unterscheiden,
die ursprünglich
für Maus P19
embryonale Karzinomzellen berichtet wurden (Dey, A., et al., Mol.
Cell: Biol. 14: 8191–8201
(1994)) (6b, Lanes 3, 6, 7). Die Am80-induzierte
Expression von RARβ2
(und RARγ)
wurde durch RT-PCR (nicht dargestellt) bestätigt; bei RARα-Antagonisten
alleine (Verbindung A in 6c und nicht
dargestellt) wurde keine Induktion festgestellt.
-
-
Beispiel 2: Synergismus
zwischen RAR- und RXR-spezifischen Liganden
-
Die mögliche Rolle von RXR in den
oben genannten Ereignissen, abgesehen von jener eines bloßen Partners
für die
Heterodimerisierung, wurde untersucht, indem analysiert wurde, ob
RAR- und RXR-spezifische Liganden synergisieren konnten. In der
Tat war Am80 in der Gegenwart des RXR-spezifischen Agonisten SR11237
beim Induzieren von Differenzierung bei geringen Konzentrationen
deutlich effizienter (Tabelle 1; dabei gilt es zu beachten, dass
SR11237 alleine keine Differenzierung induzierte; 3c). Überraschender
und unerwarteter Weise hat in Kombination mit SR11237 sogar der
RARα-Antagonist
Verbindung A eine Differenzierung mit höherer Effizienz induziert als
T-RA oder 9C-RA (Tabelle 1, 3d).
Es ist unwahrscheinlich, dass diese Differenzierung auf eine Autoinduktion
von RARβ2
durch die RARβ-agonistische
Aktivität
(1) von Verbindung A
in der Gegenwart von SR11237 zurückzuführen ist,
da Verbindung C (1),
ein stärkerer RARβ-Agonist
als Verbindung A (Chen, J-Y., et al., EMBO J. 14: 1187–1197 (1995)),
nicht mit SR11237 synergisierte, um RARβ2-Promotorbelegung (6a, b), RARβ2 mRNA-Produktion
oder Differenzierung zu induzieren (nicht dargestellt). Außerdem hat
Verbindung B, ein „reiner"
RARα-Antagonist,
der kein RARβ oder RARγ bindet (1), auch Differenzierung
in Gegenwart von SR11237 induziert, während Verbindung D, der RARγ(β) Agonist,
der kein RARα binden
kann, das nicht getan hat (Tabelle 1 und nicht dargestellt). Die
geringere Effizienz von Verbindung B im Vergleich zu Verbindung
A kann auf ihre geringere Bindungsaffinität für RARα zurückzuführen sein (2 und
nicht dargestellt).
-
Gemeinsam zeigen die oben erwähnten Ergebnisse
deutlich, dass RARα (oder
PML-RARα) insbesondere-
in die synergistische Induktion von Differenzierung in Gegenwart
des RXR-Agonisten involviert ist. Die Synergie zwischen RARα- und RXR-Liganden
wurde nicht nur für
die Differenzierung, sondern auch für nachfolgende Apoptose beobachtet.
Dies wurde durch die starke antiproliferierende und apoptotische
Wirkung einer Kombination aus dem RARα-Antagonisten Verbindung A und
dem RXR-Agonisten SR11237 demonstriert, von denen jeder alleine
ineffizient gewesen ist (4g und h, 5;
und Vorwärts-
und Seitwärtsstreuungsanalyse,
nicht dargestellt). Vor der Verbindung A/SR11237-induzierten Differenzierung
und Apoptose kam es zu einer Assoziation von PML-RARα mit nuklearen
Körpern,
die von jener, die in Gegenwart-von RARα-Agonisten (3g und h) beobachtet wurde, nicht zu unterscheiden
war. Darüber
hinaus wurden die Belegung von RARβ2 RARE und die Faktorrekrutierung
(Dey, A., et al., Mol. Cell. Biol. 14: 8191–8201 (1994)) zum RARβ2-Promotor
in vivo auch induziert, wenn Zellen mit Verbindung A und SR11237
induziert wurden (6a). Diese Behandlung
führte
zu einer effizienten Akkumulierung von RARβ und RARγ mRNA (6c). Gemäß ihrer
Unfähigkeit,
mit SR11237 zwecks Differenzierung zu synergisieren, war der andere
RARα-Antagonist/RARβ-Agonist
Verbindung C weder imstande, eine nukleare Körperassoziation von PML-RARα (nicht dargestellt),
noch eine RARβ2-Promotorbelegung
(6a) zu induzieren.
-
Allgemeine Besprechung
-
Es wurde früher darauf hingewiesen, dass
die RXR Ligand-abhängige
Transkriptionsaktivierungsfunktion AF-2 in RAR-RXR-Rezeptor heterodimerischen
Signalisierungskomplexen stumm ist (Kurokawa, R., et al., Nature
371: 528–531
(1994); Forman, B. M., et al., Cell 81: 541–550 (1995); Mangelsdorf, D.
J. und Evans; R. M., Cell 83: 835–850 (1995)). Im Gegensatz
dazu zeigen die vorliegenden Ergebnisse, dass in kultivierten menschlichen
NB4-Zellen akuter Promyelozytenleukämie – (APL) in vivo Footprints
des RARβ2-Promotor-RA-Reaktionselements
und RARβ2
mRNA-Expression nicht nur durch Behandlung mit RARα- (und nicht RARβ-, RARγ- oder RXR-)spezifischen
Retinoiden alleine, sondern auch durch die Kombination aus bestimmten
RARα-Antagonisten mit
einem reinen RXR-Agonisten induziert werden können. Außerdem induzieren solche Kombinationen
die Relokation von PML zu nuklearen Körpern und NB4-Zelldifferenzierung
vor der Apoptose. Diese Ergebnisse zeigen, dass die Auswikungen,
die durch die Retinoide auf die APL-Zellen ausgeübt werden, durch Heterodimere
zwischen RARα (oder
PML-RARα und
RXRs) vermittelt werden, wobei die AF-2 wobei nur einem Partner
transkriptional kompetent sein. muss. Somit induzieren RARα-Agonisten
zwei trennbare Ereignisse. Ein Ereignis, das für das Binden von RXR-RARα-Heterodimeren
an DNA in vivo erforderlich ist und es ermöglicht, dass die RXR AF-2 durch
Agonisten aktiviert wird, kann entweder durch einen RARα-Agonisten
oder durch bestimmte RARα-Antagonisten induziert
werden, wobei das andere Ereignis, das RARα AF-2-Aktivität induziert,
nur durch RARα-Agonisten
ausgeübt
werden kann.
-
Der Synergismus zwischen RAR- und
RXR-Liganden ist mit der Schlussfolgerung vorheriger Berichte schwer
in Einklang zu bringen (Kurokawa, R., et al., Nature 371: 528-531 (1994); Forman,
B. M., et al., Cell 81: 541–550
(1995)), die da lautet, dass RXR ein transkriptional stummer Partner
in RXR-RAR-Heterodimeren sowohl bei DR1 als auch DR5 RAREs ist (besprochen
in Mangelsdorf, D. J. und Evans, R. M., Cell 83: 835–850 (1995)).
Im Besonderen stellt sich die Frage, wie ein Heterodimer, das einen
Antagonist-gebundenen RARα und
einen Agonist-gebundenen RXR umfasst, an den RARβ2-Promotor binden und Transkription
vom RARβ2-Promotor
aktivieren konnte, wenn RXR nicht imstande wäre, seinen Liganden bei einer
Assoziierung mit DNA zu binden? In der Tat unterscheidet einer der
obigen Berichte (Forman, B. M., et al., Cell 81: 541–550 (1995))
zwischen den hemmenden Wirkungen von Ligand-freiem und Ligand-gebundenem
RAR auf RXR-Aktivität. Außerdem haben,
im Gegensatz zu obigen Berichten (Kurokawa, R., et al., Nature 371:
528–531
(1994); Forman, B. M., et al., Cell 81: 541–550 (1995)), neuere Studien
(Kersten, S., et al., Biochemistry 35: 3816–3824 (1996); Apfel, R., et
al., J. Biol. Chem. 270: 30765–30772
(1995)) ergeben, dass beide Partner von RXR-RAR-Heterodimeren ihren
verwandten Liganden binden, unabhängig von ihrer Bindung an DNA,
wodurch die frühere
Schlussfolgerung, die aus Transfektionsexperimenten gezogen wurde
und der zufolge beide Partner transkriptional aktiv sein können, unterstützt wird
(Durand, B., et al., Cell 71: 73–85 (1992)). Ungeachtet der
Grundlage für
die einander widersprechenden in vitro Errgebnisse ist klar, dass
bei in vivo der RXR-Partner von RXR-RAR-Dimeren auf einen agonistischen
Liganden ansprechen kann und daher diesen binden muss. Gemäß dieser
Schlussfolgerung wurde in vivo Synergismus zwischen RAR- und RXR-Agonisten zuvor
berichtet (Roy, B., et al., Mol: Cell. Biol. 15: 6481–6487 (1995);
Apfel, R., et al., J. Biol. Chem: 270: 30765–30772 (1995)).
-
Die vorliegenden Studien zeigen,
dass bei hohen Pegeln RAR-Agonisten alleine ausreichen, um die genetischen
Programme, welche zu Differenzierung und Apoptose von NB4-Zellen
führen,
sowie Transkription vom RARβ2-Promotor
zu induzieren. Im Gegensatz dazu war ein RXR-Agonist inaktiv, es
sei denn, er wird mit einem RAR-Agonisten oder bestimmten RAR-Antagonisten
assoziiert. Es erscheint daher, dass (i) die apo-RAR-Struktur im
RXR-RAR-Heterodimer mit Ereignissen inkompatibel ist, die eine Voraussetzung
für die Transaktivierung
in vivo darstellen; und dass (ii) die Transaktivierung durch ein
RXR-RAR-Heterodimer
zwei getrennte RAR-vermittelte Ereignisse umfasst. Ein Ereignis,
das für
das Binden von RXR-RARα-Heterodimeren
an DNA in vivo erforderlich ist und es ermöglicht, dass die RXR AF-2 durch
Agonisten aktiviert wird kann entweder durch einen RARα-Agonisten oder bestimmte
RARα-Antagonisten
induziert werden. Das andere Ereignis induziert RARα AF-2 Aktivität und kann
nur durch RARα-Agonisten
ausgeübt
werden.
-
Bei Ausweitung der vorliegenden Ergebnisse
auf andere RA-Zielgene hat die Schlussfolgerung, dass eine der zwei
Funktionen, die von RAR-Liganden in RXR-RAR- Heterodimeren ausgeübt wird, eine Voraussetzung
dafür ist,
dass RXR-Liganden die Zielgen-Transkription in vivo aktivieren,
wichtige Implikationen. RARs werden ihre RXR-Partner „dominieren" und die Gegenwart
von RXR-Agonisten würde
die Auswirkung von RAR-Liganden verstärken, aber diese nicht ersetzen.
In diesem Zusammenhang ist zu beachten, dass der RXR-spezifische
Ligand SR11237 alleine nicht imstande war, die Differenzierung und
Expression von RA-Zielgenen in F9 und P 19 Zellen zu. induzieren
(Roy, B., et al., Mol. Cell. Biol. 15: 6481–6487 (1995)). Dasselbe mag
auf die RXR heterodimer-vermittelten Signalisierungspfade zutreffen,
die Schilddrüsenhormone
und Vitamin D3 involvieren, während
für andere
RXR-Heterodimere (Mangelsdorf, D. J. und Evans, R. M., Cell 83: 835–850 (1995))
RXR-Liganden als unabhängige
Signalisierungsmoleküle
agieren können.
Die Konstruktion und die Untersuchung von zusätzlichen RAR- und RXR-Agonisten
und Antagonisten durch die hierin offenbarten Verfahren werden zeigen,
ob dieses Konzept der „Dominanz“ von RAR
gegenüber
RXR verallgemeinert werden kann und in welchem Ausmaß andere
synthetische Liganden diese „Dominanz" überwinden
oder ändern
könnten.
-
Weder die RARα-Spezifizität noch der Synergismus zwischen
RARα/Verbindung
A und RXR/SR11237 sind NB4-Zell- oder PML-RARα-spezifisch. Menschliche HL-60
myeloblastische Leukämiezellen,
die kein PML-RARα-Fusionsprotein
aufwiesen, sprachen wie NB4-Zellen auf die verschiedenen Ligandkombinationen in
Bezug auf die RARβ2-Promotor-Belegung,
RARβ2 mRNA-Akkumulierung
und Differenzierung (nicht dargestellt) an. Es wurde jedoch berichtet,
dass – im
Gegensatz zu NB4-Zellen – die
Apoptose von HL-60
Zellen nicht mit einem RAR-Agonisten alleine erzielt werden kann
und insbesondere die Gegenwart eines RXR-Agonisten erfordert (Nagy,
L., et al., Mol. Cell. Biol. 15: 3540–3551 (1995)). RARβ2-Promotorbelegung
und RARβ2 mRNA-Akkumulierung
wurden auch in P19 embryonalen Karzinom (EC)-Zellen beobachtet,
die mit einer Kombination aus einem' RARα-Antagonisten und einem RXR-Agonisten
behandelt wurden (Roy, B., et al., Mol. Cell. Biol. 15: 6481–6487 (1995))
(3b). In starkem Gegensatz dazu steht,
dass dieselbe Liganden-Kombination in F9 EC-Zellen weder RARβ2-Expression
noch Differenzierung (Daten nicht dargestellt) induzierte. Somit
legen die vorliegenden Ergebnisse – unter Berücksichtigung der Fähigkeit „dissoziierter“ Retinoide
(Chen, J.-Y., et al., EMBO J. 14: 1187–1197 (1995)) AP1-Aktivität zu unterdrücken – nahe,
dass es möglich
sein kann, komplexe Genprogramme in einer zellspezifischen Weise
durch entsprechende Auswahl von synthetischen Retinoiden (öder einer
Kombination davon) mit vorbestimmten Eigenschaften zu initiieren.
Die Selektivität
und das synergistische Potential von RAR- und RXR-Liganden, welche
drastisch reduzierte Konzentrationen der individuellen Verbindungen
ermöglichen,
um biologische Aktivität
zu erreichen, sind in Bezug auf die Ausweitung der therapeutischen
Anwendungen von natürlichen
und synthetischen Retinoiden und Retinoidanaloga vielversprechend.
-
-
-
-
-
-