CN1294115A - Process for preparing loxoprofen sodium - Google Patents
Process for preparing loxoprofen sodium Download PDFInfo
- Publication number
- CN1294115A CN1294115A CN 00127293 CN00127293A CN1294115A CN 1294115 A CN1294115 A CN 1294115A CN 00127293 CN00127293 CN 00127293 CN 00127293 A CN00127293 A CN 00127293A CN 1294115 A CN1294115 A CN 1294115A
- Authority
- CN
- China
- Prior art keywords
- compound
- reaction
- loxoprofen sodium
- preparation
- under
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Abstract
A process for preparing loxoprofen sodium includes such steps as Friedel-Crafts reaction of 2-chlorpropionyl chlorine as initial raw material to obtain 2-chloro-1-(4-methylphenyl)-1-aceton, reaction on neopentanediol under catalysis of p-toluenesulfonic acid, toluene azeotropic dewatering to obtain ketal, hydrolysis, acidifying to obtain 2-(4-methylphenyl) propionic acid, brominating to obtain 2-(4-bromomethylphenyl) propionic acid, obtaining 2-ethoxycarbonyl cyclopontanone from diethyl adipic acid, reaction of both to obtain ester compound, hydrolysis, decarboxylating, and reaction on sodium hydroxide and alcohol.
Description
The present invention is a kind of preparation method of loxoprofen sodium, and its chemical name is 2-[4-(2-oxo-cyclopentane-1-ylmethyl) phenyl] the Sodium Propionate dihydrate is a kind of medicine.
Loxoprofen sodium (Loxoprofen sodium) is a kind of phenylpropionic acid NSAID (non-steroidal anti-inflammatory drug), and its structural formula is as follows:
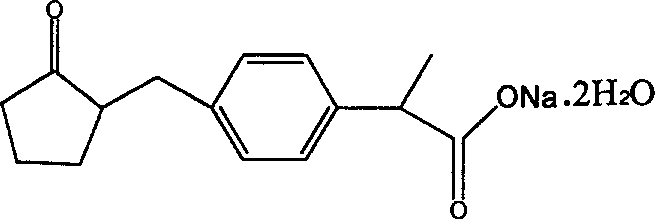 Loxoprofen sodium, the synthetic route of 1 loxoprofen sodium is a lot, and now the synthetic route that will deliver is listed below:
Loxoprofen sodium, the synthetic route of 1 loxoprofen sodium is a lot, and now the synthetic route that will deliver is listed below:
1, be the feedstock production loxoprofen sodium with 2-ethoxycarbonyl cyclopentanone
2-ethoxycarbonyl cyclopentanone (2) can be by diethylene adipate under the sodium ethylate effect, through the Dieckmann prepared in reaction.
2-ethoxycarbonyl cyclopentanone is under the potassium hydroxide effect, with 2-[4-bromine (or chlorine) aminomethyl phenyl] and propionic acid or its ester (3, X=Cl or Br; R=H, CH
3, C
2H
5) carry out hydrocarbonylation, get compound 4,4 behind hydrolysis decarboxylation in the presence of 48% Hydrogen bromide, promptly get object loxoprofen sodium (1) with the sodium hydroxide salify.Reaction formula is as follows:

2, with the cyclopentanone be the feedstock production loxoprofen sodium
(1) by the enamine intermediate
The reaction of cyclopentanone and Pyrrolidine, change enamine (5) into after, with 2-[4-bromine (or chlorine) aminomethyl phenyl] propionic acid or its ester (3) carry out hydrocarbonylation and get compound 6,6 hydrolysis after salify promptly get 1.
(2) by the oxime intermediate
The reaction of cyclopentanone and oxammonium hydrochloride generates cyclopentanone oxime (7), and salify promptly gets 1 after carrying out hydrocarbonylation and get intermediate 8,8 hydrolysis with compound 3 again.
(3) react by stock
Enamine 9 gets alpha, beta-unsaturated ketone 11 with 2-(to the formyl radical phenyl) propionic ester (10) condensation, and compound 11 is after two key reduction, and the hydrolysis salify promptly gets 1.

(4) by intermediate cyano group phosphoric acid ester
Cyclopentanone is that raw material makes 2-benzyl rings pentanone (12), with Acetyl Chloride 98Min., aluminum trichloride (anhydrous) carry out Friedel-Crafts react compound 13,13 and lithium cyanide, the DEPC reaction, generate the cyano group phosphoric acid ester, then catalytic hydrogenation gets nitrile compound 14,14 and makes loxoprofen sodium 1 with sodium hydroxide hydrolysis, salify.

The objective of the invention is to develop that a kind of raw material is easy to get, the preparation method of process stabilizing, yield height, loxoprofen sodium that product price is not high.
The objective of the invention is to develop a kind of preparation method of medicinal loxoprofen sodium.
With 2-chlorpromazine chloride (I) is starting raw material, with toluene under Catalyzed by Anhydrous Aluminium Chloride, carry out Friedel-Crafts reaction, 2-chloro-1-(4-aminomethyl phenyl)-1-acetone (II).Compound ii and neopentyl glycol are under Catalyzed by p-Toluenesulfonic Acid, and the methylbenzene azeotropic dehydration obtains ketal (III); The compound III carries out 1 under the catalysis of zinc oxide and Red copper oxide, the migration of 2-aryl, and then hydrolysis, acidifying obtain 2-(4-aminomethyl phenyl) propionic acid (IV).Bromination reaction takes place and gets 2-(4-2-bromomethylphenyl) propionic acid (V) under illumination in compound IV and bromine, benzoyl peroxide.
Diethylene adipate (VI) carries out the Dieckmann ester condensation under the sodium Metal 99.5 effect, get 2-ethoxycarbonyl cyclopentanone (VII).Alkylation reaction takes place in compound VII and compound V under the effect of sodium hydroxide, get 2-[4-(1-ethoxycarbonyl-2-oxo-cyclopentane-1-ylmethyl) phenyl] propionic acid (VIII).Ester hydrolysis, decarboxylic reaction take place in the compound VIII under sulfuric acid, acetic acid catalysis, get 2-[4-(2-oxo-cyclopentane-1-ylmethyl) phenyl] propionic acid (IX); Compound IX and sodium hydroxide, aqueous ethanol effect promptly get purpose product loxoprofen sodium (1).
Each step prior art personnel of the inventive method all can realize according to this area knowledge, the present invention has taken all factors into consideration various factors, as factors such as cost, raw material, yield and product prices, realized that with the 2-chlorpromazine chloride be the purpose that starting raw material finally obtains the loxoprofen sodium product.
For further understanding content of the present invention, each step process is described below: 1, the preparation of 2-chloro-1-(4-aminomethyl phenyl)-1-acetone (compound ii)
Reaction formula
This step is an acylation reaction, and chemical compounds I is added in well-beaten dry toluene, the aluminum trichloride (anhydrous), and 0~5 ℃ of temperature of reaction dropwises, and continues stirring reaction 8~10 hours.Separate compound ii.
2, the preparation of 2-(4-aminomethyl phenyl) propionic acid (compound IV)
Reaction formula


This step reaction has ketalization, rearrangement, alkaline hydrolysis, acidifying.Add compound ii, neopentyl glycol, tosic acid, toluene, in the reactor of water trap was housed, the heating azeotropic dehydration reacted 18~20 hours approximately.Separate washing.Add zinc oxide and Red copper oxide again, heated and stirred reaction 8~10 hours.Remove zinc oxide and Red copper oxide, washing.In above-mentioned rearrangement liquid, add NaOH, heated and stirred 6~8 hours (NaOH can be mixed with 25~35% solution adding).Hcl acidifying after the hydrolysis, getting white solid is the compound IV.
3, the preparation of 2-(4-2-bromomethylphenyl) propionic acid (compound V)
Reaction formula

In the exsiccant reactor, add compound IV, sherwood oil, benzoyl peroxide, illumination is stirred, and 15~20 ℃ of following dripping bromine, reacts colourless to solution.Separation, purifying.
4, the preparation of 2-ethoxycarbonyl cyclopentanone (compound VII)
Reaction formula

In the exsiccant reactor, add dry toluene, stirring and add sodium Metal 99.5 down, drip diethylene adipate with 1: 1 mol ratio, to react 3~5 hours down at 40~50 ℃, separated and collected gets the compound VII.This step must guarantee that all raw materials are anhydrous.
5,2-[4-(2-oxo-cyclopentane-1-ylmethyl) phenyl] preparation of propionic acid (compound IX)
Reaction formula
In dry reactor, add compound VII, N, dinethylformamide, dehydrated alcohol add sodium hydroxide or potassium hydroxide after the stirring and dissolving, 35~40 ℃ of reactions down, react to yellow solution.The compound V is dissolved in N, and dinethylformamide added to this drips of solution in 15~30 minutes in the above-mentioned reactor of yellow solution, 50~60 ℃ of following stirring reactions 8~10 hours, dewatered, and it is the compound VIII that underpressure distillation gets red liquid.Add Glacial acetic acid, sulfuric acid (25%) in liquid, heated and stirred back flow reaction 10~12 hours is separated, washing, and reclaim under reduced pressure toluene gets the compound IX.
6,2-[4-(2-oxo-cyclopentane-1-ylmethyl) phenyl] preparation of Sodium Propionate dihydrate (loxoprofen sodium 1)
Reaction formula

The compound IX is dissolved in 95% ethanol, and stirring and frozen water cooling drip NaOH down in loxoprofen's sodium ethoxide solution, and temperature is lower than 25 ℃ in the control, and stirring at room was reacted 2~3 hours.Decolouring, separation get the loxoprofen sodium product.
For each step of the present invention was both economized in raw materials, react completely again, make reaction process just right, the feed ratio (mol ratio) of each step reaction compound of the present invention is:
Toluene: chemical compounds I: Alcl
3=(4~6): 1: (1.2~1.3);
Compound ii: neopentyl glycol=1: (1.7~2.0);
Compound IX: bromine=1: (1.1~1.15);
Compound V: compound VII=1: (1.4~1.5);
Compound IX: sodium hydroxide=1: (1.1~1.15).
In above-mentioned feed ratio scope, the reaction yield of each step is higher.
When the present invention prepares the compound ii acylation reaction, be in dry reactor, to add dry toluene, aluminum trichloride (anhydrous), be cooled to 0 ℃ or lower; Chemical compounds I is dropped in the above-mentioned reactor 0~5 ℃ of temperature of reaction, 7~10 hours reaction times.As this method, in chemical compounds I, yield about 80%.
When the compound IV prepared, ketal reaction can be followed the tracks of with tlc, and developping agent is a normal hexane: ether=9: 1, iodine colour developing.
When the compound V prepares, in the dry reactor that the compound IV is arranged, add sherwood oil and benzoyl peroxide, stir down, use UV-irradiation, dripping bromine in 15~20 ℃.Meanwhile, available water pump extracts the bromize hydrogen gas of generation.
When the compound VIII prepares, in dry reactor, add compound VII, dimethyl formamide, dehydrated alcohol, the dissolving back adds NaOH, 35~40 ℃ of controlled temperature, continue to stir yellow solution, reclaim the dimethyl formamide that will be dissolved with the compound V behind the solvent and in 15~30 minutes, be added dropwise to above-mentioned reactor, in 50~55 ℃ of following stirring reactions 8~10 hours.
The hydrolysis of compound VIII ester, decarboxylic reaction, 90~100 ℃ of heated and stirred reflux, and 10~12 hours time, underpressure distillation gets the compound IX.
During refining final product 1 of the present invention, use the dissolve with ethanol product, add gac, reflux, filtered while hot, the filtrate reflux, adding ethyl acetate while hot extremely just has solid to separate out.
Product of the present invention is made tablet or granule, is that a kind of side effect is little, the pharmaceutical chemicals that the anti-inflammatory analgesic effect is good.
The inventive method raw material sources are extensive, technology uniqueness, novelty, stable, and each step products yield is higher; In building-up process, use to such an extent that multiple solvent is all recyclable, reuse, greatly reduce production cost.After testing, reliable product quality that the present invention obtains, stable performance.Product of the present invention is made tablet or granules medicine, has anti-inflammatory, analgesic, analgesic good result, and gastrointestinal side effect is low than other similar medicine, is the kind that China's medical sci-tech development program is recommended.
Embodiment:
Each reactions steps feeds intake and reacts as follows:
1, the preparation raw material specification and the charging capacity of 2-chloro-1-(4-aminomethyl phenyl)-1-acetone (compound ii)
Feed ratio (mole) toluene: raw material I: AlCl
3=5: 1: 1.2
| Raw material (molecular weight) | Specification | Charging capacity |
| Dry toluene (92.142) | GB2284-8.0 colourless liquid proportion 0.867 | (1.1L reaction) 0.9L (extraction) |
| 2-chlorpromazine chloride (126.971) | Colourless or weak yellow liquid has strong tearing property and corrodibility content 〉=98.5% | 254g (1.970 moles) |
| Aluminum trichloride (anhydrous) (133.341) | Industry first grade content 〉=98.5% easy deliquescence | 320g (2.364 moles) |
This step feeds intake as previously mentioned, drips chemical compounds I and needs 4~5 hours approximately, and 0~5 ℃ was stirred 9 hours, and reactant is poured in the trash ice, told organic layer then, was washed to pH=5~6, reclaimed toluene, collected product.
2, raw material specification and charging capacity
Feed ratio (mole) intermediate II: neopentyl glycol=1: 1.7
| Raw material (molecular weight) | Specification | Charging capacity |
| Intermediate II (182.652) | Tearing property is arranged | 200g (1.095 moles) |
| Neopentyl glycol (104.150) | White crystals content 〉=97% | 193g (1.853 moles) |
| Tosic acid (172.204) | White crystals content 〉=95% | (21.8g 0.127 mole) |
| Zinc oxide (81.369) | White powder content 〉=99% | (5.5g 0.068 mole) |
| Red copper oxide (143.091) | Taupe brown content of powder 〉=99.5% | 2g (0.014 mole) |
| Sodium hydroxide | GB209-63 content 〉=98.5% | 54g (1.35 moles) |
| Toluene | GB2284-8.0 colourless liquid proportion 0.867 | (1.3L reaction) 1.5L (extraction) |
| 30% hydrochloric acid | GB320-64 content 〉=30% | ????200ml |
Feed intake as previously mentioned, react and finished in 18 hours, tell organic layer, with warm water washing 5 times, azeotropic dehydration gets the compound III.In the compound III, add zinc oxide and Red copper oxide, reacted 9 hours, be cooled to room temperature.Remove catalyzer, add NaOH54g (be mixed with 30% solution), heated and stirred 6~7 hours, decolouring then separates.Mother liquor transfers to pH=1~2 with 30% hydrochloric acid, divides oil-yielding stratum, reclaims toluene, collects cut, gets the compound IV, is white solid.Calculate with compound ii, three step total recoverys are 70~80%.
3, raw material specification and charging capacity
Feed ratio (mole) intermediate IV: bromine=1: 1.1
| Raw material (molecular weight) | Specification | Charging capacity |
| Intermediate IV (164.206) | Colourless or little yellow liquid content 〉=98% mp 〉=37 ℃ | 50g (0.298 mole) |
| Bromine (159.814) | QB349-63 content 〉=98.5% has strong corrosion to be sure not to contact skin | 55g (0.339 mole) |
| Sherwood oil | 60-90 ℃ of content 〉=99.5% of colourless liquid boiling range | ????200ml |
| Benzoyl peroxide | White powder content 〉=98.5% | ????1g |
Feed intake as previously mentioned, 16 ℃ of following ultraviolet lightings extract the bromize hydrogen gas of generation simultaneously with water pump, 4~5 hours dropping time, continue insulated and stirred and react colourless to solution.Filter, washing, recrystallization gets product V.
4, raw material specification and charging capacity
Feed ratio (mole) diethylene adipate: sodium Metal 99.5=1: 1
| Raw material (molecular weight) | Specification | Charging capacity |
| Diethylene adipate (202.253) | Colourless oil liquid content 〉=98.5% | (2.04Kg 9.935 moles) |
| Toluene | Ditto | 10L (reaction) 5L (extraction) |
| Sodium Metal 99.5 (22.9898) | Meet water burning content 〉=99.5% | 230g (9.954 moles) |
Feed intake as previously mentioned.After adding, sodium Metal 99.5 become microgranular cooling slightly can drip the compound VI, and about 6 hours, difficult as if stirring, can add dry toluene, drip and finish, continuation stirring reaction 3 hours separates, cools off, and regulates Ph=5~6, washing, drying gets the compound VII, yield 65~70%.
5, raw material specification and charging capacity
Feed ratio (mole) intermediate V: intermediate VII=1: 1.4~1.5
| Raw material (molecular weight) | Specification | Charging capacity |
| Intermediate V (243.105) | Faint yellow or white powder mp114-120 ℃ content 〉=95% | 50g (0.206 mole) |
| Intermediate VII (156.183) | Bp86-88 ℃/5mmHg of colorless and odorless liquid content 〉=98.5% | 46g (0.295 mole) |
| N, N-dimethyl formyl | Bp150-155 ℃ of moisture content≤0.2% of industry first grade colourless oil liquid | ????220ml |
| Dehydrated alcohol | GB678-65 content 〉=99.5% | ????100ml |
| Sodium hydroxide | GB209-63 content 〉=98.5% | (11.8g 0.291 mole) |
| The vitriol oil | GB534-65 content 〉=98% | ????25ml |
| Glacial acetic acid | HG2-403-77 content 〉=99% | ????140g |
Feed intake as previously mentioned, add NaOH after, about 37 ℃ of temperature controls, stir yellow solution, reclaim solvent.Again the compound V is dissolved in N, in the dinethylformamide, splashes in the above-mentioned reactor in 15~30 minutes, 50~55 ℃ of stirring reactions 9 hours reclaim N, dinethylformamide, pH=7 is regulated in cooling, reclaims toluene, the compound VIII.Add glacial acetic acid, 25% sulfuric acid 160g in the compound VIII, refluxed 10 hours at 95~100 ℃, separate, washing gets the compound IX behind the recovery toluene.
6, raw material specification and charging capacity
Feed ratio (mole) intermediate IX: sodium hydroxide=1: 1.1
| Raw material (molecular weight) | Specification | Charging capacity |
| Intermediate IX (230.310) | ????230-240℃/1-2mmHg | 50g (0.203 mole) |
| Sodium hydroxide | GB209-63 content 〉=98.5% | (9.5g 0.234 mole) |
| 95% ethanol | GB/T679-94 content 〉=95% | ????400ml |
Feed intake as previously mentioned.The back stirring at room that feeds intake 2 hours refluxes, and decolouring separates, and gets final product of the present invention.Process for purification
Get this product 50g, add 95% ethanol 200ml, reflux to solid all dissolves.Cold slightly, add gac 1g, refluxed filtered while hot 5 minutes.The filtrate reflux adds ethyl acetate while hot, to just having solid to separate out, needs 300ml approximately.Add small amount of ethanol again, reflux clear solution.Cooling, suction filtration gets white crystalline solid.With a small amount of cold washing with alcohol.In 50~60 ℃ of oven dry, get elaboration, yield about 80%.
Claims (9)
1, a kind of preparation method of loxoprofen sodium is characterized in that with chemical compounds I, and the 2-chlorpromazine chloride is a starting raw material, with toluene under Catalyzed by Anhydrous Aluminium Chloride, carry out Friedel-Crafts reaction, compound ii, 2-chloro-1-(4-aminomethyl phenyl)-1-acetone; Compound ii and neopentyl glycol are under Catalyzed by p-Toluenesulfonic Acid, and the methylbenzene azeotropic dehydration obtains compound III, ketal; The compound III carries out 1 under the catalysis of zinc oxide and Red copper oxide, the migration of 2-aryl, and then hydrolysis, acidifying obtain the compound IV, 2-(4-aminomethyl phenyl) propionic acid; Bromination reaction takes place and gets the compound V, 2-(4-2-bromomethylphenyl) propionic acid under illumination in compound IV and bromine, benzoyl peroxide; Compound VI, diethylene adipate are carried out the Dieckmann ester condensation under the sodium Metal 99.5 effect, get the compound VII, 2-ethoxycarbonyl cyclopentanone; Alkylation reaction takes place in compound VII and compound V under the effect of sodium hydroxide, get the compound VIII, 2-[4-(1-ethoxycarbonyl-2-oxo-cyclopentane-1-ylmethyl) phenyl] propionic acid; Ester hydrolysis, decarboxylic reaction take place in the compound VIII under sulfuric acid, acetic acid catalysis, get the compound IX, 2-[4-(2-oxo-cyclopentane-1-ylmethyl) phenyl] propionic acid; Compound IX and sodium hydroxide, aqueous ethanol effect get loxoprofen sodium.
2, the preparation method of loxoprofen sodium according to claim 1 is characterized in that the feed ratio (mol ratio) of compound in each step is:
Toluene: chemical compounds I: AlCl
3=(4~6): 1: (1.2~1.3);
Compound ii: neopentyl glycol=1: (1.7~2.0);
Compound IV: bromine=1: (1.1~1.15);
Compound V: compound VII=1: (1.4~1.5);
Compound IX: sodium hydroxide=1: (1.1~1.15).
3, the preparation method of loxoprofen sodium according to claim 1 is characterized in that chemical compounds I is to be added drop-wise in dry toluene, the aluminum trichloride (anhydrous) to go, 0~5 ℃ of control reaction temperature, 7~10 hours reaction times.
4, the preparation method of loxoprofen sodium according to claim 1 is characterized in that ketal reaction follows the tracks of with tlc.
5, the preparation method of loxoprofen sodium according to claim 1 is characterized in that compound IV and sherwood oil, and benzoyl peroxide stirs down at 15~20 ℃, dripping bromine under the illumination.
6, the preparation method of loxoprofen sodium according to claim 1, earlier the compound VII is dissolved in dimethyl formamide, dehydrated alcohol when it is characterized in that preparing the compound VIII, temperature of reaction is 35~40 ℃, the dimethyl formamide that will be dissolved with the compound V again splashes into, and reacts 8~10 hours down in 50~55 ℃.
7, the preparation method of loxoprofen sodium according to claim 1 is characterized in that the hydrolysis of compound VIII ester, decarboxylic reaction are at 90~100 ℃, 10~20 hours time, gets the compound IX.
8, the preparation method of loxoprofen sodium according to claim 1 is characterized in that it is to use dissolve with ethanol that loxoprofen sodium is made with extra care, and adds gac, and reflux and filter adds ethyl acetate while hot to just having solid to separate out.
9, the preparation method of loxoprofen sodium according to claim 1 is characterized in that product of the present invention makes tablet or granule, with hyoscine.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN00127293A CN1101802C (en) | 2000-11-07 | 2000-11-07 | Process for preparing loxoprofen sodium |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN00127293A CN1101802C (en) | 2000-11-07 | 2000-11-07 | Process for preparing loxoprofen sodium |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1294115A true CN1294115A (en) | 2001-05-09 |
| CN1101802C CN1101802C (en) | 2003-02-19 |
Family
ID=4592309
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN00127293A Expired - Fee Related CN1101802C (en) | 2000-11-07 | 2000-11-07 | Process for preparing loxoprofen sodium |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN1101802C (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101412670B (en) * | 2007-10-19 | 2011-11-09 | 浙江普洛医药科技有限公司 | Method for synthesizing loxoprofen sodium |
| CN102716768A (en) * | 2012-07-03 | 2012-10-10 | 复旦大学 | 2-aryl-zinc-propionate catalyst and preparation method and application thereof |
| CN103193620A (en) * | 2013-04-07 | 2013-07-10 | 天长市禾益化学药品有限公司 | Method for preparing 2-(4-Chloromethylphenyl) propionic acid as loxoprofen key intermediate |
| CN103351318A (en) * | 2013-07-19 | 2013-10-16 | 常州工程职业技术学院 | Preparation method of 2-(3-carboxymethyl-4-thiophenyl-phenyl) propionic acid |
| CN105218351A (en) * | 2015-11-05 | 2016-01-06 | 上海立科化学科技有限公司 | A kind of synthetic method of loxoprofen sodium |
| CN105601500A (en) * | 2016-03-07 | 2016-05-25 | 山东罗欣药业集团股份有限公司 | Loxoprofen-sodium sesquialter hydrate crystal form and preparing method thereof |
| CN107324990A (en) * | 2017-07-12 | 2017-11-07 | 山东省药学科学院 | A kind of preparation method of felbinac |
| CN107353195A (en) * | 2017-06-13 | 2017-11-17 | 威海迪素制药有限公司 | A kind of preparation method of loxoprofen sodium open loop impurity |
| CN109694326A (en) * | 2017-10-24 | 2019-04-30 | 湖北迅达药业股份有限公司 | A kind of preparation method of loxoprofen sodium |
| CN111440059A (en) * | 2020-05-14 | 2020-07-24 | 上海柏狮生物科技有限公司 | Synthetic method of loxoprofen |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62161740A (en) * | 1986-01-09 | 1987-07-17 | Sankyo Yuki Gosei Kk | Production of phenylpropionic acid derivative |
-
2000
- 2000-11-07 CN CN00127293A patent/CN1101802C/en not_active Expired - Fee Related
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101412670B (en) * | 2007-10-19 | 2011-11-09 | 浙江普洛医药科技有限公司 | Method for synthesizing loxoprofen sodium |
| CN102716768A (en) * | 2012-07-03 | 2012-10-10 | 复旦大学 | 2-aryl-zinc-propionate catalyst and preparation method and application thereof |
| CN103193620A (en) * | 2013-04-07 | 2013-07-10 | 天长市禾益化学药品有限公司 | Method for preparing 2-(4-Chloromethylphenyl) propionic acid as loxoprofen key intermediate |
| CN103193620B (en) * | 2013-04-07 | 2015-11-11 | 安徽禾益化学股份有限公司 | A kind of loxoprofen's key intermediate 2-for preparing is to the method for chloromethyl phenyl propionic acid |
| CN103351318A (en) * | 2013-07-19 | 2013-10-16 | 常州工程职业技术学院 | Preparation method of 2-(3-carboxymethyl-4-thiophenyl-phenyl) propionic acid |
| CN105218351A (en) * | 2015-11-05 | 2016-01-06 | 上海立科化学科技有限公司 | A kind of synthetic method of loxoprofen sodium |
| CN105601500A (en) * | 2016-03-07 | 2016-05-25 | 山东罗欣药业集团股份有限公司 | Loxoprofen-sodium sesquialter hydrate crystal form and preparing method thereof |
| CN107353195A (en) * | 2017-06-13 | 2017-11-17 | 威海迪素制药有限公司 | A kind of preparation method of loxoprofen sodium open loop impurity |
| CN107353195B (en) * | 2017-06-13 | 2020-10-13 | 迪嘉药业集团有限公司 | Preparation method of loxoprofen sodium ring-opening impurity |
| CN107324990A (en) * | 2017-07-12 | 2017-11-07 | 山东省药学科学院 | A kind of preparation method of felbinac |
| CN109694326A (en) * | 2017-10-24 | 2019-04-30 | 湖北迅达药业股份有限公司 | A kind of preparation method of loxoprofen sodium |
| CN111440059A (en) * | 2020-05-14 | 2020-07-24 | 上海柏狮生物科技有限公司 | Synthetic method of loxoprofen |
| CN111440059B (en) * | 2020-05-14 | 2022-11-15 | 上海柏狮生物科技有限公司 | Synthetic method of loxoprofen |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1101802C (en) | 2003-02-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1140500C (en) | Certain 5-alkyl-2-arylaminophenylacetic acids and derivatives | |
| CN102241582B (en) | Synthesis technology of sodium valproate | |
| CN1101802C (en) | Process for preparing loxoprofen sodium | |
| CN101412670B (en) | Method for synthesizing loxoprofen sodium | |
| JPS6122046A (en) | Stilbene derivative | |
| WO2019119934A1 (en) | Method for continuous preparation of 2-methyl allyl alcohol | |
| JPS5823683A (en) | Manufacture of substituted benzofuran | |
| CN101172953B (en) | Method of preparing telmisartan midbody of angiotensin medicament for treating hypertension | |
| CA2107150C (en) | Chromenic derivatives having a triene lateral chain, process for the preparation thereof and pharmaceutical compositions containing them | |
| US4057641A (en) | Method of treating inflammation with 2-(2,3-dihydro-2-isopropyl-4-oxo-4H-1-benzopyran-6-yl)propionic acid | |
| CN101434613B (en) | Process for synthesizing Al-salicylic aldehyde acylhydrazone complexes | |
| CN102716768A (en) | 2-aryl-zinc-propionate catalyst and preparation method and application thereof | |
| Daub et al. | The Monocyanoethylation of Anthrone. An Improved Synthesis of β-(9-Anthranyl)-propionic Acid and β-(9, 10-Dihydro-9-anthranyl)-propionic Acid1 | |
| CN101665418B (en) | Methods for preparing E-3,5-dimethoxy-4'-oxhydryl diphenylethene and derivative thereof | |
| JPS59104347A (en) | Manufacture of arylalkanoic acid ester | |
| CN111087357B (en) | Preparation method of Prisamod | |
| Csákÿ et al. | Asymmetric synthesis of cyclopentenones with benzylic α-quaternary carbon stereogenic centres from furans | |
| CN111087356B (en) | Preparation method of Iguratimod | |
| CN102351834B (en) | Economic, practical, and environment friendly method for preparing norathyriol | |
| CN1279035C (en) | Method for synthesizing optical enantiomer 6-fluoro-3,4-dihydro-2H-1-benzopyran-2-carboxylic acid and 6-fluoro-3,4-dihydro-2H-1-benzopyran-2-carboxylate | |
| CN100462362C (en) | Preparing process of 2-(10-oxy-10,11-dihydrodibenz [b,f]-thiotropilium-2-yl) propionic acid | |
| CN1232522C (en) | Synthesis method for preparation of psoralen | |
| JPH01186844A (en) | Production of 3-(4'-bromobiphenyl)-3-hydroxyl- 4-phenylbutyric ester | |
| CN111423319B (en) | Preparation method of loxoprofen | |
| US4013692A (en) | Certain 3-phenyl-benzofuran lower alkanoic acids and esters thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| BB1A | Publication of application | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |