CN1177832C - Novel sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion - Google Patents
Novel sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion Download PDFInfo
- Publication number
- CN1177832C CN1177832C CNB998040894A CN99804089A CN1177832C CN 1177832 C CN1177832 C CN 1177832C CN B998040894 A CNB998040894 A CN B998040894A CN 99804089 A CN99804089 A CN 99804089A CN 1177832 C CN1177832 C CN 1177832C
- Authority
- CN
- China
- Prior art keywords
- alkyl
- group
- aryl
- formula
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
- C07C311/19—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/36—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/06—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D239/08—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms directly attached in position 2
- C07D239/12—Nitrogen atoms not forming part of a nitro radical
- C07D239/16—Nitrogen atoms not forming part of a nitro radical acylated on said nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Abstract
The present invention relates to sulfonamide derivatives of formula (I), in which R<1>, R<2>, R<4>, R<5> and R<6> have the meanings indicated in the claims, their physiologically tolerable salts and their prodrugs. The compounds of the formula (I) are valuable pharmaceutical active compounds. They are vitronectin receptor antagonists and inhibitors of cell adhesion and inhibit bone resorption by osteoclasts. They are suitable, for example, for the therapy and prophylaxis of diseases which are caused at least partially by an undesired extent of bone resorption, for example of osteoporosis. The invention furthermore relates to processes for the preparation of compounds of the formula (I), their use, in particular as pharmaceutical active ingredients, and pharmaceutical preparations comprising them.
Description
The present invention relates to the salt that can tolerate on formula I sulfone amide derivative, their physiology and their prodrug,

R wherein
1, R
2, R
4, R
5And R
6Has the following implication that indicates.Described formula I compound is the compound of valuable pharmaceutical active.They are Vitronectic receptor antagonist and cell adhension inhibitors and suppress heavily to absorb by the bone of osteoclast.For example they are suitable for treating and prevent and heavily absorb the disease that causes to the bone that small part is dredged property by for example sclerotin that does not conform with desirability.The invention still further relates to the method for the described formula I compound of preparation, particularly they are as the purposes of active pharmaceutical ingredients with contain their medicinal preparations.
People's bone stands constantly dynamic recovery process, comprises that bone heavily absorbs and bone forming.These processes are controlled by the broad variety cell that is used in particular for these purposes.Bone heavily absorbs on the destructive basis that is based upon the ground substance of bone by osteoclast.Balance disorder between most of bone disorders heavily absorb based on bone forming and bone.Osteoporosis is to cause increasing the disease of the bone fragility of risk of bone fracture for its feature with sclerotin reduction and enhanced.This is because in described process of carrying out refigure, bone is absorbed heavily deficiency in new bone forming causes.Conventional osteoporosis treatment comprises and for example gives diphosphonate, oestrogenic hormon, estrogenic/progestogenic (Hormone Replacement Therapy or HRT), estrogen agonist/antagonist (selective estrogen receptor modulators or SERMs), thyrocalcitonin, novel vitamin D analogues, Rat parathyroid hormone 1-34, growth hormone cinogenic agent or Sodium Fluoride (Jardine etc., Annual Reports in MedicinalChemistry 1996,31,211).
The activatory osteoclast is to have the syncyte of removing ground substance of bone of diameter up to 400 μ m.The activatory osteoclast is attached to described ground substance of bone surface justacrine proteolytic ferment and acid enters what is called " seal area ", and the sealing district is their cytolemma and the zone between the ground substance of bone.Sour environment and proteolytic enzyme cause osteoclasia.Formula I compound suppresses heavily to absorb through the bone of osteoclast.
Multiple research has shown that osteoclast is attached to described bone and is subjected to control at the integrin receptor on described osteoclast surface.Integrin is the superfamily of acceptor, its fibrin original receptor α on the thrombocyte
IIbβ
3With Vitronectic receptor α
vβ
3Described Vitronectic receptor α
vβ
3Be membrane glycoprotein, it is at the various kinds of cell cell surface expression of endotheliocyte, vascular smooth muscle cell, osteoclast and tumour cell for example.The Vitronectic receptor α that on the osteoclast film, expresses
vβ
3Control is attached to bone and the re-absorbed process of bone and therefore helps osteoporosis.In this case, α
vβ
3Be bonded in bone matrix protein for example osteopontin, bone sialoprotein and thrombospondin (thrombospontin), it contains three peptide motif Arg-Gly-Asp (perhaps RGD).
Horton and colleague describe RGD peptide and anti-Vitronectic receptor antibody (23C6), and this antibody suppresses to destroy (Horton etc., Exp.Cell.Res.1991,195,368) by the tooth of osteoclast and osteoclast migration.At J.Cell Biol.1990, in 111,1713, Sato etc. have described echiststin (echistatin) (a kind of RGD peptide from snake venom) as effective inhibitors of bone resorption and adhere to the inhibitor of bone as osteoclast in tissue culture medium (TCM).Fisher etc. (Endocrinology 1993,132,1411) can show in the rat body that echiststin also suppresses bone and heavily absorbs.
Vitronectin α on this further demonstration people's the aorta vessel smooth muscle cell
vβ
3Stimulate these cell migration to enter the neovascularity inner membrance, cause arteriosclerosis and postangioplasty restenosis (Brown etc., Cardiovascular Res.1994,28,1815) at last.
Brooks etc. (Cell 1994,79,1157) show at α
vβ
3Or α
vβ
3The antibody of antagonist can cause tumor shrinkage by the apoptosis of mediation vascular cell during angioplasty.Described Vitronectic receptor α
vβ
3Also participate in the development of multiple other types of cancer and at malignant melanoma cell overexpression (Engleman etc., Annual Reports inMedicinal Chemistry 1996,31,191).Described melanoma cells invasion and attack relevant (Stracke etc., Encylopedia of Cancer, III volume, 1855, AcademicPress, 1997 with this overexpression; Hillis etc., Clinical Science 1996,91,639).Carron etc. (CancerRes.1998,58,1930) describe and use α
vβ
3Antagonist suppresses described tumor growth and suppresses the hypercalcemia of malignant tumour.
Cherest etc. (Science 1995,270,1500) describe its anti-α that suppresses the big rathole medium vessels of described bFGF inductive plasty process
vβ
3Antibody or α
vβ
3Antagonist, this is that it can be used to treat the character in the treatment retinopathy.The influence of described Vitronectic receptor or wherein its related interactional influence therefore provide the possibility that influences the various disease state, this its treatment and prevention need to be continued the appropriate drug activeconstituents.
WO-A-94/12181 describes fragrance or the non-aromatic ring system that replaces, and WO-A-94/08577 describes the substituted heterocycle as fibrinogen receptor antagonist and anticoagulant.EP-A-528586 and EP-A-528587 disclose phenylalanine derivative aminoalkyl group-replacement or heterocyclic radical-replacement, and WO-A-95/32710 discloses the aryl derivatives as the inhibitors of bone resorption that passes through osteoclast.WO-A-96/00574 has described benzodiazepine class and WO-A-96/00730 has described fibrinogen receptor antagonist template, and especially it is connected in the benzodiazepine class as Vitronectic receptor antagonist of the first ring of nitrogenous 5-.WO-A-98/00395 (DE-A-19654483) has described the Vitronectic receptor antagonist that is derived from the tyrosine skeleton.EP-A-820991 (German patent application 19629816.4) has described cycloalkyl derivatives and european patent application 97122520.6 and has described it and be the carbamate derivatives of Vitronectic receptor antagonist.Further study display type I sulfone amide derivative for strong especially Vitronectic receptor with by the re-absorbed inhibitor of the bone of osteoclast.
The present invention relates to formula I compound, salt that can tolerate on the stereoisomer form that they are all and the form of mixtures of any ratio and their physiology and their prodrug,

Wherein
R
1And R
2Independently of one another be hydrogen or be unsubstituted or by R
3(the C that replaces
1-C
6)-alkyl, perhaps radicals R wherein
1-and R
2-be saturated or unsaturated divalence (C together
2-C
9)-alkylidene group is group-(CH of 2,3,4,5,6,7,8 or 9 as p wherein
2)
p-.It is for unsubstituted or by one or more halogen, (C of being selected from
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, (C
3-C
12)-cycloalkyl, (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, wherein by being unsubstituted or by R
3Particularly by one or two radicals R
3The 5-unit that replaces to 7-unit saturated or unsaturated ring and be that carbocyclic ring or the heterocycle that contains one or two theheterocyclic nitrogen atom can condense in (C
2-C
9On the C-C in the)-alkylidene group; R
3Be (C
1-C
10)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
1-C
8)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, hydroxyl, oxo, nitro, amino ,-NH-(C
1-C
4)-alkyl ,-N ((C
1-C
4)-alkyl)
2,-NH-CO-(C
1-C
4)-alkyl ,-CO-(C
1-C
4)-alkyl; R
4Be hydrogen, (C
1-C
6)-alkyl-CO-O-(C
1-C
4)-alkyl-or for unsubstituted or by being selected from hydroxyl, (C
1-C
4)-alkoxyl group, (C
1-C
4)-alkyl-S (O)
2-,-NR
7R
7 'With-N
+R
7R
7 'R
7 "Q
-(the C that replaces of group
1-C
6)-alkyl, wherein R
7, R
7 'And R
7 "Independently of one another is hydrogen, (C
1-C
6)-alkyl, (C
5-C
14)-aryl or (C
5-C
14)-aryl-(C
1-C
6)-alkyl-and Q
-Be the negatively charged ion that can tolerate on the physiology, perhaps R wherein
4Be one of following group
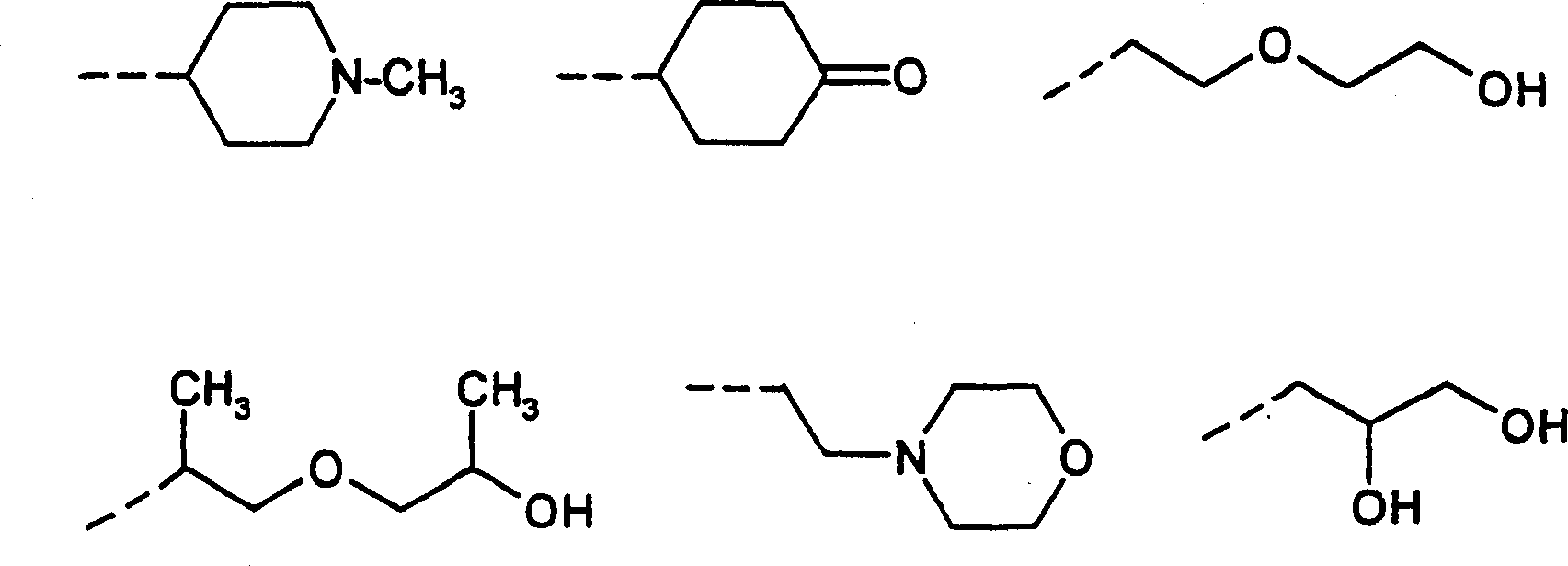
Wherein said group indicates by a dotted line so as to the key that connects;
R
5Be (C
1-C
20)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, wherein each in aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl is unsubstituted or by one, two or three radicals R
3Replace, and wherein in described alkyl, described monocycle alkyl, described bicyclic alkyl and described tricyclic alkyl, one or more carbon atoms, particularly one, two, three or four carbon atom can be substituted by the identical or different atoms that are selected from nitrogen, oxygen and sulphur;
R
6Be hydrogen, (C
1-C
6)-alkyl-O-CO-, hydroxyl, (C
1-C
6)-alkyl-O-CO-O-or nitro.
Several times all groups such as radicals R can appear in formula I compound
3Can have specified implication independently of one another, and can be identical or different in each case.In each case, the group that has the implication of indicating independently of one another can be identical or different.
Preferred formula I compound of the present invention is (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido) propionic acid or its (C
1-C
4The salt that can tolerate on)-alkyl ester and its physiology.
Alkyl can be straight chain or branched and can be saturated monounsaturated or polyunsaturated.If they have substituting group or occur as the substituting group of other group, this also is applied to for example alkoxyl group, alkoxy carbonyl or arylalkyl.This is equally applicable to alkylidene group (=divalent alkyl=saturated or unsaturated alkane 2 basis).The example of the suitable alkyl that contains 1 to 20 carbon atom is methyl, ethyl, propyl group, butyl, amyl group, hexyl, heptyl, octyl group, nonyl, decyl, undecyl, dodecyl, tetradecyl, hexadecyl, octadecyl and eicosyl, the positive isomer of all these groups, sec.-propyl, isobutyl-, isopentyl, neo-pentyl, isohexyl, isodecyl, 3-methyl amyl, 2,3,4-trimethylammonium hexyl, sec-butyl, the tertiary butyl, tert-pentyl.One group of preferred alkyl is formed by methyl, ethyl, n-propyl, sec.-propyl, normal-butyl, isobutyl-, sec-butyl and the tertiary butyl.Corresponding to the divalent group of above-mentioned univalent perssad methylene radical, 1 for example, 1-ethylidene (=methyl methylene radical), 1,2-ethylidene, 1,3-propylidene, propylene (=1-methyl ethylidene and 2-methyl ethylidene), 2,3-butylidene (=1,2-dimethyl-1, the 2-ethylidene), tetramethylene, hexamethylene are the example of alkylidene group.
Unsaturated alkyl perhaps is alkynyl such as ethynyl, 1-proyl or propargyl for for example alkenyl such as vinyl, 1-propenyl, allyl group, butenyl, 3-methyl-2-butene base.Unsaturated alkylene is that alkylene group can be the same as straight chain or branched with alkynylene (=alkene two bases and alkynes two bases).The example of alkylene group is vinylene or propenylidene, and the example of alkynylene is ethynylene or inferior proyl.When they were substituted, alkyl also can be for undersaturated.At described moieties, the example of undersaturated arylalkyl is styryl (=2-phenyl vinyl).
Unless otherwise specifically indicated, cycloalkyl can be monocycle, dicyclo or three rings, and promptly they can be monocycle alkyl, bicyclic alkyl and tricyclic alkyl, and condition is the carbon atom that they have suitable number.The monocycle alkyl is for example cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, suberyl, ring octyl group, ring nonyl, ring decyl, ring undecyl, cyclo-dodecyl, ring tetradecyl, ring octadecyl, and they also can be by for example (C
1-C
4)-alkyl replaces.The example of the cycloalkyl of the replacement that can mention is 4-methylcyclohexyl and 2, the 3-dimethylcyclopentyl.
Bicyclic alkyl and tricyclic alkyl can for unsubstituted or in any required suitable position for example by one or more oxo groups and/or one or more identical or different (C
1-C
4)-alkyl is methyl or sec.-propyl for example, is preferably methyl substituted.Described dicyclo or three cyclic groups enough are positioned on the position of any requirement in the molecule so as to the free bond energy that connects; Therefore described group can connect by bridge end atom or the atom on bridge.Described free key also can be positioned on the stereochemistry position of any requirement, for example outside-position or interior-position on.The example of bicyclic alkyl and tricyclic alkyl be camphyl (camphanyl), bornyl, adamantyl for example 1-adamantyl and 2-adamantyl, carane base, table isobornyl (epiisobornyl), show bornyl, fall bornyl (norbornyl) and fall the pinane base.
Halogen for example is fluorine, chlorine, bromine or iodine.
(C
5-C
14)-aryl comprises heterocycle (C
5-C
14)-aryl (=(C
5-C
14)-heteroaryl), one or more in wherein said 5 to 14 ring carbon atoms by heteroatoms such as nitrogen, oxygen or sulphur and carbocyclic ring (C
6-C
14)-aryl substitutes.Carbocyclic ring (C
6-C
14The example of)-aryl is phenyl, naphthyl, xenyl, anthryl or fluorenyl, and wherein 1-naphthyl, 2-naphthyl and phenyl are preferred.If statement in addition, aryl particularly phenyl can be unsubstituted or by one or more groups, is preferably one, two or three identical or different groups and replaces.Particularly aryl can be by the identical or different (C that is selected from
1-C
8)-alkyl ((C particularly
1-C
4)-alkyl), (C
1-C
8)-alkoxyl group ((C particularly
1-C
4)-alkoxyl group), for example fluorine, chlorine and bromine, nitro, amino, trifluoromethyl, hydroxyl, methylene-dioxy, cyano group, hydroxycarbonyl group, aminocarboxyl, (C of halogen
1-C
4The group of)-alkoxy carbonyl, phenyl, phenoxy group, benzyl and benzyloxy replaces.Generally having only nearly in formula I compound of the present invention, two nitros can take place as substituting group.
In mono-substituted phenyl, described substituting group can be positioned at the 2-position, on 3-position or the 4-position, and preferred 3-and 4-position.If phenyl is replaced by two, substituting group can be in 2,3-position, 2,4-position, 2,5-position, 2,6-position, 3,4-position or 3,5-position.In di-substituted-phenyl, described two substituting group preferred arrangement are with respect to 3 of described link position, the 4-position.In tri-substituted phenyl, described substituting group can be in 2,3,4-position, 2,3,5-position, 2,3,6-position, 2,4,5-position, 2,4,6-position or 3,4,5-position.Similarly, naphthyl and other aryl can be substituted on the position of any requirement, and for example the 1-naphthyl is on 2-, 3-, 4-, 5-, 6-, 7-and 8-position, and the 2-naphthyl is substituted on 1-, 3-, 4-, 5-, 6-, 7-and 8-position.
Except the carbocyclic ring system, (C
5-C
14)-aryl also can or encircle for example dicyclo or three rings for monocycle more, wherein 1,2,3,4 or 5 aromatic nucleus system that ring carbon atom is particularly replaced by the identical or different heteroatomss that are selected from nitrogen, oxygen and sulphur by heteroatoms.Heterocycle (C
5-C
14)-aryl and (C
5-C
14The example of)-heteroaryl is that pyridyl is as the 2-pyridyl, 3-pyridyl and 4-pyridyl, pyrryl is as 2-pyrryl and 3-pyrryl, furyl is as 2-furyl and 3-furyl, thienyl is as 2-thienyl and 3-thienyl, imidazolyl, pyrazolyl oxazolyl isoxazolyl, thiazolyl, isothiazolyl, tetrazyl, pyridazinyl, pyrazinyl, pyrimidyl, indyl, pseudoindoyl, indazolyl, the 2 base, quinolyl, isoquinolyl, quinoxalinyl, quinazolyl, the cinnolines base, β-Ka Lin base or benzo thick and, pentamethylene thick and, hexanaphthene thick and or suberane is thick and the derivative of these groups.Described heterocyclic system can replace by the identical substituting group by the isocyclic aryl system of as above mentioning in all suitable positions.
In these heteroaryls of described series, preferably have 1,2 or 3 heteroatoms particularly 1 or 2 heteroatomic monocycle or the Bicyclic member ring systems that are selected from N, O and S, it can be unsubstituted or be selected from (C by 1,2 or 3
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, fluorine, chlorine, nitro, amino, trifluoromethyl, hydroxyl, (C
1-C
4The substituting group of)-alkoxy carbonyl, phenyl, phenoxy group, benzyloxy and benzyl replaces.Be preferably especially at this and have 1 to 3 heteroatoms that is selected from N, O and S and be in particular and have 1 or 2 heteroatomic monocycle or Bicyclic 5-unit to 10-unit member ring systems, it can be selected from (C by 1 to 2
1-C
4)-alkyl, (C
1-C
4The substituting group of)-alkoxyl group, phenyl, phenoxy group, benzyl and benzyloxy replaces.More special being preferably contained 1 or 2 particularly 1 heteroatomic 5-unit or 6-unit's bicyclic heteroaryl and 9-or 10-unit bicyclic heteroaryl that is selected from N, O and S, and it is unsubstituted or is substituted like that as previously described.
If described two radicals R
1-and R
2-represent the saturated or unsaturated (C of divalence together
2-C
9During)-alkylidene group, it is monocyclic 1 that the described central carbon atom of the guanidine radicals that two nitrogen-atoms of two nitrogen-atoms and these that these two groups connect with their are connected forms, 3-diaza heterocycle, and described heterocycle is at group (CH
2)
2The 2-position of-CO-NH is connected in described nitrogen-atoms.Can be as specified at (C
2-C
9On the)-alkylidene group and also substituted described 1 on the nitrogen-atoms of guanidine radicals, the example of 3-diaza heterocyclic group is a 2-imidazolyl, 4,5-dihydro-2-imidazolyl, 1,4,5,6-tetrahydrochysene-2-pyrimidyl or described 4,5,6,7-tetrahydrochysene-1H-1,3-diaza -2-base.If 5-unit is thick and in (C to 7-unit ring
2-C
9During C-C on the)-alkylidene group, two radicals R so
1And R
2The central carbon atom of stating of the guanidine radicals that two nitrogen-atoms of two nitrogen-atoms and these that connect with them are connected forms bicyclic heterocycle, and it is connected in group (CH
2)
2On the nitrogen-atoms on the-CO-NH and its can be as specified being substituted.It is saturated, monounsaturated diunsaturated or fragrant that the described thick and 5-unit of (or condensation) to 7-unit ring can be.Therefore, for example, pentamethylene ring, cyclohexane ring, cyclohexene ring, cyclohexadiene ring, suberane ring or phenyl ring can be by condensations.Can be connected in group (CH
2)
2The examples of groups of the such bicyclic heterocycle on the nitrogen-atoms of-CO-NH is 1,3a, 4,5,6,6a-six hydrogen-1,3-diaza 2-pentenyl, 1H-benzimidazolyl-2 radicals-Ji, 3a, 4,5,6,7,7a-six hydrogen-1H-benzimidazolyl-2 radicals-Ji, 4,5,6,7-tetrahydrochysene-1H-benzimidazolyl-2 radicals-Ji, 4,7-dihydro-1H-benzimidazolyl-2 radicals-Ji or 1H-imidazo [4,5-b] pyridine-2-base.If if the ring of condensation is substituted and/or (C
2-C
9)-alkylidene group is substituted, and they are preferred independently of one another by identical or different radicals R
3The single replacement or two replacement.If expression R
1And/or R
2Alkyl be substituted, they are preferred independently of one another by identical or different radicals R
3The single replacement or two replacement, be in particular single replacement.
The opticity carbon atom that appears in the formula I compound can have R configuration or S configuration independently of one another.Formula I compound can present with the form of pure enantiomorph or pure diastereomer or present with the form of mixture of enantiomers, for example presents with the form of racemic modification or the form of non-enantiomer mixture.The present invention relates to pure enantiomorph and mixture of enantiomers and relate to pure diastereomer and non-enantiomer mixture.The present invention includes two kinds of formula I or more than the steric isomer of all proportions in two kinds of steric isomers and the mixture.Formula I compound can be chosen wantonly as E isomer or Z isomer and present.The present invention relates to pure E isomer and pure Z isomer and the E/Z mixture that exists with all proportions.The present invention also comprises the formula I compound of all tautomeric forms, for example, the form that shows, also comprises the wherein conduct-CO-N=C of acylguanidines unit (NHR in described formula I
1)-NR
2R
6The form that group presents, and all other the form that comprises that its mobile hydrogen that passes through different positions distinguishes.For example can comprise that the E/Z isomer separation becomes one isomer to diastereomer by chromatography.The chromatography that for example goes up mutually by chirality by ordinary method or by splitting can be separated into racemic modification two enantiomorphs.By starting raw material that uses the stereochemistry homogeneous or the formula I compound that also can access the stereochemistry homogeneous by the reaction of use stereoselectivity.
The salt that can tolerate on the physiology of formula I compound is acceptable on the physiology, particularly pharmaceutically can utilize the atoxic salt of salt.Contain acidic-group for example this type of salt of the formula I compound of carboxyl for example for an alkali metal salt or alkaline earth salt, for example sodium salt, sylvite, magnesium salts and calcium salt, also have with physiology on the quaternary amine ionic salt that can tolerate and with ammonia and physiology on the organic amine that can tolerate, for example acid salt that forms of triethylamine, thanomin or three-(2-hydroxyethyl) amine.The formula I compound that contains basic group for example with mineral acid example hydrochloric acid, sulfuric acid or phosphoric acid or with organic carboxyl acid and sulfonic acid, for example acid salt that forms of acetate, Citric Acid, phenylformic acid, toxilic acid, fumaric acid, tartrate, methylsulfonic acid or tosic acid.Contain basic group and acidic-group for example the formula I compound of guanidine radicals and carboxyl can present as zwitter-ion (betaine), it comprises in the present invention equally.
Work as R
4During for the alkyl that replaces by the positive charge ammonium group, be included in the negatively charged ion Q that can tolerate on the physiology in the formula I compound
-Be in particular on atoxic, the physiology of univalent anion or of equal value multivalent anions available, especially also be pharmaceutically available mineral acid or organic acid, for example be applicable to the negatively charged ion or the negatively charged ion Equivalent of one of above-mentioned acid of forming acid salt.Therefore, Q
-Can be one of described negatively charged ion (perhaps being the negatively charged ion Equivalent) that for example is selected from muriate, vitriol, phosphoric acid salt, acetate, citrate, benzoate, maleate, fumarate, tartrate, mesylate and tosilate.
By ordinary method well known by persons skilled in the art for example by at solvent or disperse to make in the thing formula I compound and mineral acid or organic acid or alkali to combine, perhaps carry out cationic exchange or anionresin, salt that can preparation I compound by salt from other.The present invention also comprises all salt of formula I compound, it still for example is suitable for as the intermediate of other chemically modified of carrying out formula I compound or as the starting raw material that is used to prepare the salt that can tolerate on the physiology owing to tolerability on the low physiology directly is not suitable for using in medicament.
In addition, the present invention includes all solvates of formula I compound, for example hydrate or with the adducts of alcohols, and also comprise the derivative of formula I compound, for example the active metabolite of derivative that can tolerate on ester class, prodrug and other the physiology and formula I compound.The present invention be more particularly directed to the prodrug of formula I compound, they can be converted into formula I compound under physiological condition.It is known for those skilled in the art that the prodrug of suitable formula I compound promptly has the chemically modified derivative that has improved the formula I compound of character in desired mode.The more detailed INFORMATION DISCOVERY that relates to prodrug is at for example Fleisher etc., Advanced Drug DeliveryReviews 19 (1996) 115-130;
The prodrug design, H.Bundgaard, Ed., Elsevier, 1985; H.Bundgaard,
Following medicine16 (1991) 443; Saulnier etc., Bioorg.Med.Chem.Lett.4 (1994) 1985; Safadi etc., among the Pharmaceutical Res.10 (1993) 1350, it is attached to herein by reference.The suitable prodrug of formula I compound is in particular the ester prodrugs of hydroxy-acid group, particularly as group COOR
4In R
4During for hydrogen, there is the COOH group, for example the alkyl ester picture (C of this group
1-C
6)-alkyl ester or (C
1-C
4)-alkyl ester, but and also to can be the acyl group prodrug of nitrogen-containing group of acylations and carbamate prodrugs for example amino and be in particular guanidine radicals.In described acyl group prodrug or carbamate prodrugs, the hydrogen atom that is positioned in these groups on the nitrogen-atoms is replaced one or many, for example twice by carboxyl groups or carbamate groups.Suitable carboxyl groups and the carbamate groups that is used for described acyl group prodrug and carbamate prodrugs has for example radicals R
10-CO and R
11O-CO, wherein R
10Be hydrogen, (C
1-C
18)-alkyl, (C
3-C
14)-cycloalkyl, (C
3-C
14)-cycloalkyl-(C
1-C
8)-alkyl-, (C
5-C
14)-aryl, wherein 1 to 5 carbon atom can by heteroatoms for example N, O or S substitute, perhaps be (C
5-C
14)-aryl-(C
1-C
8)-alkyl-, wherein 1 to 5 carbon atom in described aryl moiety can for example N, O or S substitute and R wherein by heteroatoms
11Have except hydrogen R
10Specified implication.
In formula I compound, radicals R
1And R
2Be preferably hydrogen or be saturated or unsaturated, particularly saturated divalence (C together
2-C
5)-alkylidene group, be in particular (C
2-C
4)-alkylidene group, especially be (C
2-C
3)-alkylidene group, it is for unsubstituted or by one or two identical or different halogen, (C of being selected from
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, (C
3-C
12)-cycloalkyl, (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, be unsubstituted or here by R
3Replace, particularly by one or two radicals R
3Replace, and its for carbocyclic ring or contain the saturated or unsaturated ring of heterocyclic 5-unit to 7-unit of one or two theheterocyclic nitrogen atom can be thick and C-C in described alkylidene group on.In formula I compound, radicals R
1And R
2Be preferably hydrogen especially or, be preferably 2,3 or 4, be preferably group-(CH of 2 or 3 especially for p wherein is a number 2,3,4 or 5
2)
P-, and it is for unsubstituted or by one or two identical or different halogen, (C of being selected from
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, (C
3-C
12)-cycloalkyl, (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, be unsubstituted or here by R
3Particularly by one or two radicals R
3Replace, and its for carbocyclic ring or contain the saturated or unsaturated ring of heterocyclic 5-unit to 7-unit of one or two theheterocyclic nitrogen atom can be thick and in group-(CH
2)
P-in C-C on.Described radicals R
1And R
2More especially preferably be group-(CH together
2)
P-, wherein p is a number 2,3,4 or 5, is preferably 2,3 or 4, is preferably 2 or 3 especially, it is preferably unsubstituted.Described radicals R
1-and R
2-especially preferably be described divalent group-CH together
2-CH
2-CH
2-, i.e. R
1And R
2The nitrogen-atoms that connects with them forms 1,4,5,6-tetrahydrochysene-2-pyrimidyl with the guanidine radicals central carbon atom that is connected with these two nitrogen-atoms;
R
3Be preferably (C
1-C
10)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
1-C
8)-alkoxyl group, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, (C
5-C
14)-heteroaryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl.R
3(C more preferably
1-C
4)-alkyl, (C
3-C
10)-monocycle alkyl, (C
5-C
12)-bicyclic alkyl, (C
5-C
12)-tricyclic alkyl, (C
1-C
4)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl.R
3Be preferably (C especially
1-C
4)-alkyl, (C
3-C
10)-monocycle alkyl, (C
5-C
12)-bicyclic alkyl, (C
5-C
12)-tricyclic alkyl, (C
1-C
4)-alkoxyl group, (C
6-C
14)-aryl, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl.
R
4Be preferably hydrogen or (C unsubstituted or that replace
1-C
6)-alkyl is preferably hydrogen or especially by being selected from (C
1-C
4)-alkoxyl group, (C
1-C
4)-alkyl-S (O)
2-and-NR
7R
7 '(the C that replaces of group
1-C
6)-alkyl, wherein R
7And R
7 'Independently of one another is hydrogen or (C
1-C
4)-alkyl.R
4Very particularly preferably be hydrogen or (C unsubstituted or that replace
1-C
4)-alkyl is preferably hydrogen or as the preceding indicated unsubstituted or (C that replaces in addition
1-C
4)-alkyl.
R
5Be preferably (C
1-C
20)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl is unsubstituted or by one, radicals R that two or three are identical or different
3Replace.R
5(C more preferably
1-C
10)-alkyl, (C
3-C
15)-monocycle alkyl, (C
5-C
15)-bicyclic alkyl, (C
5-C
15)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each in wherein said aryl, described heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl is unsubstituted or by one, radicals R that two or three are identical or different
3Replace.Except these preferred group, the preferred radicals R of a class
5By wherein can substituted or otherwise as above indicated such adorned group (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl and (C
5-C
20)-tricyclic alkyl forms, and more preferably by (C
5-C
15)-monocycle alkyl, (C
5-C
15)-bicyclic alkyl, (C
5-C
15)-tricyclic alkyl forms.Another kind of preferred radicals R
5By group (C
1-C
20)-alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-formation, wherein (C
6-C
14)-aryl and (C
5-C
14)-heteroaryl is preferred, and it can be substituted or otherwise as above indicated the modification.The particularly preferred radicals R of one class
5Form by group phenyl and naphthyl, promptly can be by unsubstituted or form as the above phenyl, 1-naphthyl and the 2-naphthyl that replace of indicating.
R
6Be preferably hydrogen or (C
1-C
6)-alkyl-O-CO-is preferably hydrogen or (C especially
1-C
4)-alkyl-O-CO-is in particular hydrogen.
Preferred formula I compound has preferred implication for those wherein one or more groups or has special or some special their compounds of difference implication, and all of so preferred implication or special implication are combined into purpose of the present invention.Particularly preferred formula I compound is those wherein R
1And R
2For hydrogen or be saturated or unsaturated divalence (C together
2-C
5)-alkylidene group is in particular hydrogen or is the group-(CH of number 2,3,4 or 5 for p wherein together
2)
p-compound, (C here
2-C
5)-alkylidene group and group-(CH
2)
p-for unsubstituted or by being selected from halogen, (C
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, (C
3-C
12)-cycloalkyl, (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, and be unsubstituted or here by R
3Especially by one or two radicals R
3Replace, and be that the saturated or unsaturated ring of carbocyclic ring or the heterocyclic 5-unit of containing one or two theheterocyclic nitrogen atom to 7-unit can be thick and in (C
2-C
5)-alkylidene group and group-(CH
2)
P-in C-C on;
R
3Be (C
1-C
10)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
1-C
8)-alkoxyl group, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, (C
5-C
14)-heteroaryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl;
R
4For hydrogen or its for unsubstituted or by being selected from (C
1-C
4)-alkoxyl group, (C
1-C
4)-alkyl-S (O)
2-and NR
7R
7 '(the C that replaces of group
1-C
6)-alkyl, wherein R
7And R
7 'Independently of one another is hydrogen or (C
1-C
4)-alkyl;
R
5Be (C
1-C
20)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three radicals R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Replace;
R
6Be hydrogen or (C
1-C
6)-alkyl-O-CO-;
Salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and their physiology and their prodrug.
Formula I compound very particularly preferably is those compounds, wherein
R
1And R
2For hydrogen or be saturated or unsaturated divalence (C together
2-C
4)-alkylidene group is in particular hydrogen or is the group-(CH of number 2,3 or 4 for p wherein together
2)
p-, (C here
2-C
4)-alkylidene group and group-(CH
2)
p-for unsubstituted or by being selected from halogen, (C
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, (C
3-C
12)-cycloalkyl, (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, and it is unsubstituted or by R here
3Especially by one or two radicals R
3Replace, and be that the saturated or unsaturated ring of carbocyclic ring or the heterocyclic 5-unit of containing one or two theheterocyclic nitrogen atom to 7-unit can be thick and in (C
2-C
4)-alkylidene group and group-(CH
2)
P-in C-C on;
R
3Be (C
1-C
4)-alkyl, (C
3-C
10)-monocycle alkyl, (C
5-C
12)-bicyclic alkyl, (C
5-C
12)-tricyclic alkyl, (C
1-C
4)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl;
R
4Be hydrogen or (C
1-C
6)-alkyl;
R
5Be (C
1-C
10)-alkyl, (C
3-C
15)-monocycle alkyl, (C
5-C
15)-bicyclic alkyl, (C
5-C
15)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces;
R
6Be preferably hydrogen or (C
1-C
4)-alkyl-O-CO-;
Salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and their physiology and their prodrug.
Particularly preferred formula I compound is such compound, wherein:
R
1And R
2For hydrogen or be saturated or unsaturated divalence (C together
2-C
3)-alkylidene group is in particular hydrogen or is the group-(CH of number 2 or 3 for p wherein together
2)
p-, (C here
2-C
3)-alkylidene group and group-(CH
2)
p-for unsubstituted or by being selected from halogen, (C
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, (C
3-C
12)-cycloalkyl, (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, and be unsubstituted or here by R
3Especially by one or two R
3Group replaces and is that the saturated or unsaturated ring of carbocyclic ring or the heterocyclic 5-unit of containing one or two theheterocyclic nitrogen atom to 7-unit can be thick and in (C
2-C
3)-alkylidene group and group-(CH
2)
P-in C-C on;
R
3Be (C
1-C
4)-alkyl, (C
3-C
10)-monocycle alkyl, (C
5-C
12)-bicyclic alkyl, (C
5-C
12)-tricyclic alkyl, (C
1-C
4)-alkoxyl group, (C
6-C
14)-aryl, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl;
R
4Be hydrogen or (C
1-C
6)-alkyl;
R
5Be (C
1-C
10)-alkyl, (C
3-C
15)-monocycle alkyl, (C
5-C
15)-bicyclic alkyl, (C
5-C
15)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces;
R
6Be hydrogen or (C
1-C
6)-alkyl-O-CO-;
Salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and their physiology and their prodrug.
Preferred in addition formula I compound for those with all they stereoisomer form and exist with their form of mixture of all proportions with their physiology on the compound that exists of the salt that can tolerate and their prodrug, wherein R
5Be (C
6-C
14)-aryl or (C
5-C
14)-heteroaryl is preferably (C
6-C
14)-aryl, wherein aryl and heteroaryl each be unsubstituted or by one, two or three identical or different R
3Group replaces, and is preferably unsubstituted or by one or two identical or different R
3Group replaces.Even more preferably formula I compound for those with all they stereoisomer form and with their form of mixture of all proportions exist with they physiology on the compound that exists of the salt that can tolerate and their prodrug, wherein R
5Be naphthyl for example 1-naphthyl or 2-naphthyl, it is unsubstituted or by one, two or three R
3Group replaces, and it is preferably unsubstituted for example unsubstituted 1-naphthyl or unsubstituted 2-naphthyl.
Preferred formula I compound also is those wherein said two radicals R
4O-CO-and R
5-SO
2The carbon atom that-NH-connects has the compound of S configuration, its with all they stereoisomer form and with all proportions they form of mixtures and their physiology on the compound that exists of the salt that can tolerate and their prodrug.
One group of special formula I compound passes through wherein R
1And R
2Independently of one another be hydrogen or for replacement not or by R
3(the C that replaces
1-C
6The compound formation of)-alkyl,
Perhaps radicals R wherein
1-and R
2-be saturated or unsaturated divalence (C together
2-C
9)-alkylidene group, for example wherein p is group-(CH of 2,3,4,5,6,7,8 or 9
2)
p-, it is for unsubstituted or by one or more halogen, (C of being selected from
1-C
6)-alkyl, (C
1-C
6)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl, (C
3-C
12)-cycloalkyl and (C
3-C
12)-cycloalkyl-(C
1-C
6)-alkyl-and the group of oxo replace, be unsubstituted or here by R
3Especially by one or two R
3Group replaces, and is that the saturated or unsaturated ring of carbocyclic ring or the heterocyclic 5-unit of containing one or two theheterocyclic nitrogen atom to 7-unit can be thick and in (C
2-C
9On the C-C in the)-alkylidene group;
R
3Be (C
1-C
8)-alkyl, (C
1-C
8)-alkoxyl group, (C
5-C
14)-aryl, (C
5-C
14)-aryl-(C
1-C
4)-alkyl, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
4)-alkyl, halogen, trifluoromethyl, hydroxyl, oxo, nitro, amino, NH-(C
1-C
4)-alkyl, N-((C
1-C
4)-alkyl)
2, NH-CO-(C
1-C
4)-alkyl, CO-(C
1-C
4)-alkyl;
R
4Be hydrogen, unsubstituted or by being selected from hydroxyl, (C
1-C
4)-alkoxyl group, (C
1-C
4)-alkyl-SO
2-,-NR
7R
7 'With-N
+R
7R
7 'R
7 "Q
-(the C that replaces of group
1-C
6)-alkyl-CO-O-(C
1-C
4)-alkyl or (C
1-C
6)-alkyl, wherein R
7, R
7 'And R
7 "Independently of one another is hydrogen, (C
1-C
6)-alkyl, (C
5-C
14)-aryl or (C
5-C
14)-aryl-(C
1-C
6)-alkyl and Q
-Be the negatively charged ion that can tolerate on the physiology, perhaps R wherein
4Be one of described group;
Wherein said group shows by a dotted line so as to the free key that connects;
R
5Be (C
1-C
20)-alkyl, (C
5-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces, and wherein in described alkyl, monocycle alkyl, bicyclic alkyl and tricyclic alkyl, one or more carbon atoms, particularly one, two, three or four carbon atom; Can be substituted by the identical or different atoms that are selected from nitrogen, oxygen and sulphur; R
6Be hydrogen, (C
1-C
6)-alkyl-O-CO, hydroxyl, (C
1-C
6)-alkyl-O-CO-O or nitro; Salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and their physiology and their prodrug.
The present invention also relates to be used for the method for preparation I compound.Described compound generally can for example can prepare from two or more fragments of formula I retrosynthesis (retrosynthetically) derivation by connecting in assembling building-up process.In the preparation of formula I compound; when introducing can cause the functional group of undesirable reaction or side reaction in dividing other synthesis step; it with after be converted into desirable functional group precursor forms exist or the described building-up process of blocking group strategy temporary interruption functional group by being suitable for described composition problem in, this is normally favourable or necessary.Such strategy to those skilled in the art be know (referring to for example Greene and Wuts,
Blocking group in the organic synthesis, Wiley, 1991).As the example of precursor group, nitro and cyano group can be mentioned, and can for example be separately converted to amino and amino methyl by catalytic hydrogenation by reduction after them.
Formula I compound can be for example carboxylic acid or carboxylic acid derivative by connection mode II in original known method prepare

R wherein
4And R
5As above specified such definition to formula I; perhaps wherein other functional group existed to be converted into the precursor forms that is present in the described group in the formula I compound afterwards; perhaps wherein functional group exists with the form of protecting; and wherein X is the leavings group of nucleophilic substitution with derivative of the guanidine of formula III or guanidine
R wherein
1, R
2And R
6As above specified such definition to described formula I, perhaps other functional group was converted into the precursor forms that is present in the group in the described formula I compound afterwards with it and existed, and perhaps functional group exists with the form of protection.
In formula II, group COX is preferably hydroxy-acid group COOH or activatory carboxylic acid derivative.X for example for hydroxyl or halogen especially for chlorine or bromine, alkoxyl group, be preferably methoxy or ethoxy, aryloxy for example phenoxy group or penta fluoro benzene oxygen base, thiophenyl, methylthio group, 2-pyridine sulfo-or the Azacyclyl that connects by nitrogen-atoms, be in particular the pyrroles, for example the 1-imidazolyl.X can be for example ((C in addition
1-C
4)-alkyl)-O-CO-O-or tosyloxy, so the activatory acid derivative can be mixed acid anhydride.
If X is a hydroxyl, if the i.e. guanidine of formula III and carboxylic acid reaction, described carboxylic acid at first activates for convenience then.For example, with dicyclohexylcarbodiimide (DCCI) or with O-((cyano group (ethoxy carbonyl)-methylene radical) amino)-1,1,3,3-tetramethyl-urea (uronium) a tetrafluoro borate (TOTU; Konig etc., the 21st Europ.Peptide Symp.1990 of Proc. (Eds.Giralt, Andreu), Escom, 1991, the 143 pages of Leiden) or other activating reagent common in chemistry of peptides can carry out described activation.
Except formula III free guanidine, guanidinesalt also can be used in the reaction with formula II compound, can prepare on the spot thus or prepare the free guanidine by alkali in separating step.With known method itself, the reaction of the guanidine of formula II activatory carboxylic acid derivative and formula III (derivative) is preferably in proton or non-proton polarity but be to carry out in the organic solvent inert.In this case, under boiling temperature from 0 ℃ to these solvents, for example, confirmed that solvent is suitable as methyl alcohol, Virahol, the trimethyl carbinol, dimethyl formamide or tetrahydrofuran (THF) in methyl ester (X=methoxyl group) or in the reaction of ethyl ester (X=oxyethyl group) and guanidine.The reaction of described Type C OX compound and salt-free guanidine is advantageously for example carried out in dimethyl formamide, tetrahydrofuran (THF), glycol dimethyl ether or the dioxane at non-proton inert solvent, if suitably, can add alkali for example potassium tert.-butoxide or sodium methylate.Yet in the reaction of formula II compound and guanidine, for example water also can be used as solvent when using alkali such as sodium hydroxide.If X is for example chlorine, follow to add for example other alkali of acid scavenger or in the presence of the excessive guanidine (derivative) that is used in conjunction with the hydrochloric acid that generates, described reaction is advantageously carried out.Extract described reaction mixture, if requirement, the ordinary method purifying that reaction product is familiar with through those skilled in the art then.
The optional product that obtains from formula II and III compound that still is present in of blocking group is also removed by standard method then.For example, by handling, be described hydroxy-acid group with tert-butyl ester groups converted with trifluoroacetic acid, remove benzyl or remove the fluorenyl methoxy carbonyl by hydrogenation by secondary amine.If requirement is carried out other reaction, for example acylation reaction or esterification then by standard method.In addition, can be converted into the salt or the prodrug that can tolerate on the physiology then by currently known methods.
Formula II relevant and the raw material of III with providing formula I compound be commercially available or can according to or be similar to the method for in described document, describing and be prepared.The preparation of the initial composition of formula II is by the Examples set in the following flow process 1, and the present invention is not limited to this synthetic or their initial composition.To those skilled in the art, synthetic shown in carrying out improves and does not cause any problem, and this preparation to The compounds of this invention is necessary.
Therefore, for example in the presence of pyridine and piperidines, the carboxyl benzaldehyde of formula IV can be for example and the malonic ester reactant salt of formula V, obtains the cinnamic acid derivative of formula VI, and it for example after the hydrogenation, obtains formula VII compound in the presence of palladium carbon.After the activation of described hydroxy-acid group, formula VII compound can with 2 of formula VIII, the condensation of 3-diaminopropionic acid derivative obtains formula IX compound (flow process 1).Described condensation can be carried out in the presence of for example TOTU or the another kind of conventional reagent that is used for the activating carboxy acid.
In formula VIII, Y can be radicals R
5-SO
2-; it is present in the final formula I compound of the present invention and it can be retained in the described molecule then; perhaps Y can for the group of temporary protection 2-bit amino and after step in be removed to obtain the group of free amino, this amino then can be by being used to prepare the standard method of sulphonamide for example by making described unhindered amina and formula R
5-SO
2The SULPHURYL CHLORIDE reaction of-Cl is converted into R
5-SO
2-NH group.The described benzyloxycarbonyl (Z group) of an example of the blocking group of expression Y for removing by catalytic hydrogenation.Be suitable for the introducing radicals R
5-SO
2Described formula R
5-SO
2The sulfonic acid of the SULPHURYL CHLORIDE of-Cl and other be commercially available or can according to or be similar to the method for in described document, describing and be prepared.Other the ester that replaces the tert-butyl ester to appear in formula VIII and the IX compound can exist, and only the described acid groups of temporary protection or its also can appear in the final compound of formula I of the present invention and can be retained in the described molecule for they.The compound that is similar to the VI compound also can transform carbonyl by other to be become alkene and for example obtains by the method for Wittig reaction.
Flow process 1
Formula IX compound is that wherein X is the formula II examples for compounds of methoxyl group.These by above-mentioned synthetic in compound that obtain and that contain activatory carboxylic acid derivative group and analogue can be directly and the formula III compound react.Yet, under standard conditions, also can be at first be present in the relevant locational ester group of formula IX compound at the above compound that obtains in synthetic and transform into corresponding carboxylic acid by the described methyl ester group of cracking or another, it is after with regard to activatable, for example with TOTU or DCCI activation after, perhaps transforming into the activatory carboxylic acid derivative after, react with the guanidine of described formula III.(formula II, X=Cl), this conversion can be for example by using thionyl chloride carry out if the plan preparation example is as the carboxyl acyl chloride derivative as the activatory acid derivative.If plan preparation example as from as described in carboxylic acid as described in methyl ester (formula II, X=methoxyl group), this can handle and carry out by being used in hydrogen chloride gas in the methyl alcohol.With known method itself, other activatory acid derivative can be from described carboxyl acyl chloride or directly (formula II X=OH) prepares from their described carboxylic acids of being alkalized wherein.Example passes through with carbonyl dimidazoles (referring to Staab for it, Angew.Chem.Int.Ed.Engl.1,351-367 (1962)) handles imidazoles (the formula II that described acid obtains, the X=1-imidazolyl), perhaps in inert solvent, at amine for example in the presence of the triethylamine, by with chloro-formic ester for example Vinyl chloroformate reaction or the described mixed acid anhydride that obtains with the toluene sulfonyl chloride reaction.The multiple method that is suitable for preparing the activatory carboxylic acid derivative is at J.March,
Advanced Organic Chemistry, the third edition, John Wiley ﹠amp; Sons indicates in the document in 1985, the 350 pages in detail.
Formula I compound is valuable active pharmaceutical ingredients, and it is fit to for example be used for the treatment of and prevent bone disorders, tumor disease or cardiovascular disorder.Salt that can tolerate on formula I compound and their physiology and their prodrug can give animal as the medicament that is used for the treatment of or prevent, and are preferably Mammals and particularly people.They can be with they self or give with the mixture of another kind of medicine or with the form of medicinal preparations, this medicament allows enteron aisle or parenterai administration and except routine medicinal nontoxic carrier and/or additive, it contains as salt that can tolerate at least a formula I compound of the significant quantity of activeconstituents and/or its physiology and/or its prodrug.
Therefore, the present invention also relates to as salt that can tolerate on the formula I compound of medicament and/or their physiology and/or their prodrug, relate to the salt that can tolerate on formula I compound and/or their physiology and/or their prodrug treatment and prevention is above or below purposes in the medicament production of the disease mentioned, for example be used for the treatment of and prevent osteopathia, and the salt that can tolerate on formula I compound that also relates to and/or their physiology and/or their prodrug are in treatment with prevent purposes in these diseases.The present invention relates in addition except the medicinal harmless carrier of routine, also contains the salt that can tolerate at least a formula I compound of significant quantity and/or their physiology and/or the medicinal preparations or the medicinal compositions of their prodrug.
Described medicament can be for example with the form oral administration of pill, tablet, film tablet, coated tablet, granule, hard and Gelseal, solution, syrup, emulsion, suspensoid or aerosol mixt.Yet, administration also can be for example with the form rectal administration of suppository or for example carry out vein, intramuscular or subcutaneous parenterai administration with the form of injection solution or infusion solution agent, micro-capsule, implant or stylus, perhaps for example carry out through skin or topical, perhaps for example with the otherwise administration of form of aerosol or nasal spray with the form of ointment, solution or tincture.
Medicinal preparations of the present invention is known and are method preparations of being familiar with to those skilled in the art with itself, (they) prodrug of the salt that on (they) physiology of formula I compound and/or it, can tolerate and/or it, use medicinal inert inorganic carrier or organic carrier.For the production of pill, tablet, coated tablet and hard-gelatin capsules, for example using, lactose, W-Gum or their derivative, talcum powder, stearic acid or its salt etc. are possible.The carrier that is used for Gelseal and suppository is for example fat, wax, semisolid and liquid polyol, the natural or oils etc. of hardening.Be used to produce solution for example the suitable carrier of injection liquid or emulsion or syrup be for example water, alcohols, glycerine, many alcohols, sucrose, Nulomoline, glucose, plant wet goods.Be used for the multipolymer of the suitable carrier of microcapsule, implant or stylus for for example glyconic acid and lactic acid.Described medicinal preparations normally contains the salt that can tolerate on the formula I compound of about 0.5 to 90% (weight) and/or their physiology and/or their prodrug.The salt that can tolerate on formula I activeconstituents described in the described medicinal preparations and/or its physiology and/or the amount of its prodrug normally are 0.2 to 500mg, are preferably 1 to 200mg.
Except described activeconstituents and carrier, described medicinal preparations can contain additive in addition, material in weighting agent, disintegrating agent, tackiness agent, lubricant, wetting agent, stablizer, emulsifying agent, sanitas, sweeting agent, tinting material, correctives or perfume compound, thickening material, thinner, the buffering for example, and also contain and be useful on solvent or the solubility promoter that obtains the storage effect, and also contain and be useful on salt, Drug coating or the antioxidant that changes isoosmotic pressure power.They also can contain the salt that can tolerate on two or more formulas I compound and/or their physiology and/or their prodrug.In addition, the salt and/or its prodrug that can tolerate at least a formula I compound and/or its physiology, they also can contain one or more other treatment or preventative activeconstituents.
Formula I compound is Vitronectic receptor antagonist and cell adhension inhibitors.They have and for example suppress osteoclast and be incorporated into bone surface and suppress the re-absorbed ability of bone by osteoclast thus.For example, the effect of formula I compound can be confirmed in a kind of test, has measured vitronectin and be bonded to the cell inhibiting effect that contains described Vitronectic receptor in this test.Below provide the details of such test.As Vitronectic receptor antagonist, salt that can tolerate on formula I compound and their physiology and their prodrug generally are suitable for treating and prevent based on interactional disease between Vitronectic receptor and their part in cell-cell interaction process or the cell-matrix interaction process, perhaps it can perhaps be used for their prevention, alleviation or cure desired such interactional restraining effect by suppressing such interaction.As this paper begin to explain, such interaction for example heavily absorbs, rises at vasculogenesis or in vascular smooth muscle cell proliferation partial action at bone.Therefore, salt that can tolerate on the physiology of formula I compound and they and their prodrug are applicable to and for example alleviate or cure the disease that is caused by heavily absorption of bone, vasculogenesis or the vascular smooth muscle cell proliferation of the degree that do not meet the requirements to small part.
Can use the osteopathia of formula I compounds for treating of the present invention and prevention to be in particular osteoporosis, hypercalcemia, the osteopenia that for example causes, odontopathy, hyperparathyroidism, the circumarticular erosion in rheumatoid arthritis and Pei Jiteshi disease by transfer.In addition, formula I compound can be used in alleviation, avoids or treats the osteopathy that is caused by glucocorticosteroid, steroidal or adrenocortical hormones in treating or do as one likes hormonoprivia.All these diseases to be being its feature based on equilibrated bone loss between bone forming and the osteoclasia, and it can advantageously be suppressed to influence through the bone of osteoclast is re-absorbed.For example the treatment or preventing osteoporosis disease in, with the associating of conventional osteoporosis treatment, for example with the combination therapy of diphosphonate, oestrogenic hormon, estrogenic/progestogenic, estrogen agonist/antagonist, thyrocalcitonin, novel vitamin D analogues, Rat parathyroid hormone 1-34, growth hormone cinogenic agent or Sodium Fluoride in, the salt and/or its prodrug that can tolerate on formula I compound and/or its physiology also can be advantageously used for inhibitors of bone resorption.Salt that can tolerate on formula I compound and/or its physiology and/or its prodrug can with as above list in treatment or preventing osteoporosis disease sample disease effectively other activeconstituents successively simultaneously jointly or dividually give continuously with any order.To the purposes in such combination therapy or prevention, salt that can tolerate on formula I compound and/or their physiology and/or their prodrug and one or more other activeconstituentss as the medicine that those had before been listed can be present in the single medicinal preparations together, for example tablet and granule, perhaps it can be present in and is included in the unitary package or in two or more medicinal preparationss that separate in two or more packings of separating.Salt that can tolerate on formula I compound and/or their physiology and/or their the prodrug purposes in such combination therapy or prevention also is a purpose of the present invention with they are used for the medicament of such combination therapy or prevention in production purposes.The present invention relate in addition the salt that can tolerate at least a formula I compound that comprises significant quantity and/or its physiology and/or its prodrug and at least a medicine of as above listing in treatment or preventing osteoporosis disease or suppress bone heavily absorb in the effective medicinal preparations of other activeconstituents and conventional medicinal harmless carrier.The explanation respective application of described above relevant medicinal preparations is in so medicinal combined preparation.
Except being used as through the inhibitors of bone resorption of osteoclast, salt that can tolerate on formula I compound and their physiology and their prodrug can be as the inhibitor of tumor growth and metastases, as anti-inflammatory drug, be used for the treatment of or preventing cardiovascular disease for example arteriosclerosis or restenosis, perhaps be used for the treatment of or prevent for example diabetic retinopathy of ephrosis change or retinopathy.As the inhibitor of tumor growth or metastases, salt that can tolerate on formula I compound and/or their physiology and/or their prodrug also can be advantageously and conventional cancer therapy drug combination.Bertino (editor), Encyclopedia of Cancer, Academic Press provides the example of conventional cancer therapy in 1997, and it is attached to herein by reference.All statements above-mentioned and use formula I compound and conventional osteoporosis combination therapy, for example possible mode of administration and pharmaceutical combination formulation can correspondingly be applied to the combination therapy of formula I compound and conventional cancer.
When the formula of use I compound, dosage can change in the restriction of broad and be conventional, and it is suitable for the individual condition in each individual case.For example, whether it is according to employed compound, the character of complying with the disease of being treated and seriousness and according to treating or not carrying out prophylaxis of acute or chronic disease and decide.Under the oral administration situation, per daily dose is generally 0.01 to 100mg/kg, be preferably 0.1 to 50mg/kg, be in particular 0.1 to 5mg/kg, for example 0.3 to 0.5mg/kg (in each case in every kg body weight mg) obtains effective result in the adult of the about 75kg of body weight.In the intravenously administrable situation, per daily dose generally also is about 0.01 to 100mg/kg, is preferably 0.05 to 10mg/kg (every in each case kg body weight).Particularly in the situation of a large amount of relatively administrations, described per daily dose can be divided into several parts of administrations, for example 2,3 or 4 parts of administrations.Generally speaking, according to individual behavior, it is necessary that specified per daily dose is floated up or down.
Except being used as active pharmaceutical ingredients, formula I compound also can arrive described site of action (=drug targeting to transport described activeconstituents specificity as the media or the carrier of activeconstituents; Referring to for example
Targeted drug transmits, R.C.Juliano,
The experimental pharmacology handbook, the 100th volume, Ed.Born, G.V.R. etc., Springer Verlag, it is attached to herein by reference).The activeconstituents of being transported is in particular those compositions that can be used in the above-mentioned disease of treatment.
The salt of formula I compound and they can be used for diagnostic purpose in addition, for example is used in in-vitro diagnosis and as the auxiliary means of biochemical research, wherein blocks Vitronectic receptor or influence cell-cell or the cell-matrix interaction meets the requirements.They can be in addition as the synthetic intermediate of preparation other compound, particularly other active pharmaceutical ingredients, this can be for example by modifying or introduce group or functional group obtains from formula I compound.
Embodiment
Described product is identified by mass spectrum (MS) and/or NMR spectrum.According to last synthesis step and/or treatment process (for example carrying out lyophilize) and decide, in some cases, compound partly or completely obtains with the form of salt, and for example the form with acetate, trifluoroacetate or hydrochloride obtains.
Embodiment 1
(2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

A) 4-(2-methoxycarbonyl-vinyl) phenylformic acid
18.74g (0.12mol) malonic acid monomethyl sylvite is suspended in the 18ml pyridine, at room temperature reaches to follow and stir 4-carboxyl benzaldehyde and 0.85g (0.01mol) piperidines that adds 15.01g (0.1mol) down.Described mixture is refluxed up to CO
2Overflow (about 2 hours) fully, add other 60ml pyrido then and stirred other 1 hour at following the described mixture that reflux.Follow and stir down, handle this reaction mixture with 500ml ice and 110ml concentrated hydrochloric acid.Add finish after, reaction mixture was stirred other 20 minutes, and, washes with water this product suction filtration, and from Virahol recrystallization.Output: 12.85g.
1H-NMR (200MHz, d
6-DMSO): δ=3.75 (s, 3H, OCH
3), 6.76 (d, J=15Hz, 1H, CHCOOCH
3), 7.73 (d, J=15Hz, 1H, Ar-CH), 7.84 (d, J=9Hz, 2H, Ar-H), 7.98 (d, J=9Hz, 2H, Ar-H), 13.11 (s, wide, 1H, COOH).MS(Cl
+):m/e=207.2(M+H
+)。
HPLC:RP18, Nucleosil 300-5-C18,250 * 4mm; Buffer A: H
2O, 0.1% trifluoroacetic acid (TFA); Buffer B: acetonitrile (80%v/v)/H
2O (20%v/v), 0.1%TFA; Gradient: first 5 minutes 90% buffer A/10% buffer B, then during 20 minutes to 90% buffer B, 5 minutes 90% buffer B then; Flow velocity 1ml/ minute; R
t=18.05 minutes.
B) 4-(2-methoxycarbonyl-ethyl) phenylformic acid
4-(2-methoxycarbonyl-vinyl) phenylformic acid of 8g (38.8mmol) is suspended in the 250ml dioxane, and at room temperature, clings under the hydrogen-pressure through Pd/C (10% concentration) hydrogenation 7 hours 1.Described mixture is filtered, and under vacuum, remove and desolvate.Output: 8.05g.
1H-NMR (200MHz, d
6-DMSO): δ=2.67 (t, J=8Hz, 2H, CH
2-COOCH
3), 2.93 (t, J=8Hz, 2H, Ar-CH
2), 3.59 (s, 3H, OCH
3), 7.35 (d, 2H, Ar-H), 7.86 (d, J=9Hz, 2H, Ar-H), 12.80 (s, wide, 1H, COOH).
MS(Cl
+):m/e=209.2(M+H
+)。
HPLC:RP18, Nucleosil 300-5-C18,250 * 4mm; Buffer A: H
2O, 0.1%TFA; Buffer B: acetonitrile (80%v/v)/H
2O (20%v/v), 0.1%TFA; Gradient: first 5 minutes 90% buffer A, 10% buffer B, then during 20 minutes to 90% buffer B, 5 minutes 90% buffer B then; Flow velocity 1ml/ minute; R
t=17.03 minutes.
C) (2S)-2-benzyloxycarbonyl amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido) propionic acid tert-butyl ester
4-(2-methoxycarbonyl-ethyl) phenylformic acid of 354mg (1.7mmol) and (2S)-3-amino of 500mg (1.7mmol)-2-benzyloxycarbonyl alanine tert-butyl ester be dissolved in the 3ml dimethyl formamide and with the O-((cyano group-(ethoxy carbonyl)-methylene radical) amino)-1 of 557mg (1.7mmol), 1,3,3-tetramethyl-urea a tetrafluoro borate (TOTU) and 204mg (1.7mmol) diisopropylethylamine are handled, and at room temperature in pH7-8 described mixture are stirred 7 hours.Solvent removed in vacuo, this resistates is dissolved in the ethyl acetate and with this solution at every turn with KHSO
4Solution and NaHCO
3Solution washing 3 times is until neutrality.Separate organic phase and dry also vacuum distilling except that desolvating.Output: 770mg.MS (ES
+): m/e=485.2 (M+H
+).
D) (2S)-2-amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester
Be dissolved in (2S)-2-benzyl oxygen carbonylamino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 6.88g (14.2mmol) in the 200ml methyl alcohol and add 852mg (14.2mmol) acetate.At room temperature, in 1 the crust hydrogen-pressure under through Pd/C (5% concentration) with described mixture hydrogenation 7 hours.Described mixture is filtered and solvent removed in vacuo.Wash described resistates and vacuum-drying with normal heptane.Output: 6.9g.MS(ES
+):m/e=351.2(M+H
+)。
E) (2S)-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-2-(naphthalene-1-sulfuryl amino)-propionic acid tert-butyl ester
(2S)-2-amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 0.123g (0.35mmol) is dissolved in the 2ml dimethyl formamide, and handles with the 1-naphthalic sulfonic chloride of 0.196ml (1.4mmol) triethylamine and 0.159g (0.7mmol).At room temperature with described solution stirring 4 hours.Solvent removed in vacuo, resistates is dissolved in the methylene dichloride and with water with described solution washing three times.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.On silicagel column with described resistates chromatography, with normal heptane/ethyl acetate (1/1) wash-out.Output: 47mg.MS(ES
+):m/e=541.3(M+H
+)。
F) (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester
Be dissolved in (2S)-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-2-(naphthalene-1-sulfuryl amino)-propionic acid tert-butyl ester of 47mg (0.087mmol) in the 2ml dimethyl formamide and add the 2-amino-1 of 86mg (0.87mmol); 4; 5, the 6-tetrahydropyrimidine.At room temperature described reactant was stirred 20 hours.Solvent removed in vacuo, on silicagel column with described resistates chromatography, with methylene chloride (1/1) wash-out, subsequently with methylene chloride/acetic acid/water (85/15/1.5/1.5) wash-out.Output: 46mg.MS(ES
+):m/e=608.3(M+H
+)。
G) (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Be dissolved in (2S)-2-(naphthalene-1-sulfuryl amino)-3-of 45mg (0.074mmol) (4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester in the 2ml methylene dichloride and add the 2ml trifluoroacetic acid.At room temperature with described solution stirring 3 hours.Solvent removed in vacuo and toluene joined in the described resistates and then vacuum remove.Be dissolved in the acetonitrile/water (1/1) described resistates and lyophilize.Output: 50mg.MS(ES
+):m/e=552(M+H
+)。
Embodiment 2
(2S)-2-(quinoline-8-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
A) (2S)-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-2-(quinoline-8-sulfuryl amino)-propionic acid tert-butyl ester
(2S)-2-amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 0.123g (0.35mmol) is dissolved in the 2ml dimethyl formamide, and handles with the 8-quinoline sulfuryl chloride of 0.196ml (1.4mmol) triethylamine and 0.159g (0.7mmol).At room temperature, with described solution stirring 4 hours.Solvent removed in vacuo, described resistates is dissolved in the methylene dichloride and with water described solution washing three times.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.On silicagel column with described resistates chromatography, with normal heptane/ethyl acetate (1/1) wash-out, output: 67mg.MS(ES
+):m/e=542.2(M+H
+)。
B) (2S)-2-(quinoline-8-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester
Be dissolved in (2S)-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-2-(quinoline-8-sulfuryl amino)-propionic acid tert-butyl ester of 65mg (0.12mmol) in the 1ml dimethyl formamide and add the 2-amino-1 of 59mg (0.6mmol); 4; 5, the 6-tetrahydropyrimidine.At room temperature described reactant was stirred 20 hours.Solvent removed in vacuo, on silicagel column with described resistates chromatography, with methylene chloride (1/1) wash-out, subsequently with methylene chloride/acetic acid/water (85/15/1.5/1.5) wash-out.Output: 72mg.MS(ES
+):m/e=609.3(M+H
+)。
C) (2S)-2-(quinoline-8-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Be dissolved in (2S)-2-(quinoline-8-sulfuryl amino)-3-of 72mg (0.11mmol) (4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester in the 2ml methylene dichloride and add the 2ml trifluoroacetic acid.At room temperature with described solution stirring 3 hours.Solvent removed in vacuo and toluene joined in the described resistates and then vacuum remove.Be dissolved in the acetonitrile/water (1/1) described resistates and lyophilize.Output: 54mg.MS(ES
+):m/e=553(M+H
+)。
Embodiment 3
(2S)-2-(4-acetylaminohydroxyphenylarsonic acid benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

A) (2S)-2-(4-acetylaminohydroxyphenylarsonic acid benzenesulfonyl amino)-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester
(2S)-2-amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 0.123g (0.35mmol) is dissolved in the 2ml dimethyl formamide, and handles with the 4-acetylaminohydroxyphenylarsonic acid benzene sulfonyl chloride of 0.196ml (1.4mmol) triethylamine and 0.164g (0.7mmol).At room temperature, with described solution stirring 4 hours.Solvent removed in vacuo, described resistates is dissolved in the methylene dichloride and with water with described solution washing three times.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.On silicagel column with described resistates chromatography, with normal heptane/ethyl acetate (1/2) wash-out, output: 90mg.MS(ES
+):m/e=548(M+H
+)。
B) (2S)-2-(4-acetylaminohydroxyphenylarsonic acid benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester
Be dissolved in (2S)-2-(4-acetylaminohydroxyphenylarsonic acid benzenesulfonyl amino)-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 87mg (0.16mmol) in the 1ml dimethyl formamide and add the 2-amino-1 of 79mg (0.8mmol); 4; 5, the 6-tetrahydropyrimidine.At room temperature described reactant was stirred 20 hours.Solvent removed in vacuo, on silicagel column with described resistates chromatography, with methylene chloride (1/1) wash-out, subsequently with methylene chloride/acetic acid/water (85/15/1.5/1.5) wash-out.Output: 57mg.MS(ES
+):m/e=615.3(M+H
+)。
C) (2S)-2-(4-acetylaminohydroxyphenylarsonic acid benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
(4-acetylaminohydroxyphenylarsonic acid benzenesulfonyl amino)-((2-(1 for 4-for 3-with (2S)-2-of 57mg (0.09mmol); 4; 5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-the propionic acid tert-butyl ester is dissolved in the 2ml methylene dichloride and adds the 2ml trifluoroacetic acid.At room temperature with described solution stirring 3 hours.Solvent removed in vacuo and toluene joined in the described resistates and then vacuum remove.Be dissolved in the acetonitrile/water (1/1) described resistates and lyophilize.Output: 42mg.MS(ES
+):m/e=559.2(M+H
+)。
Embodiment 4
(2S)-2-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
A) 2-((1S)-1-tert-butoxycarbonyl-2-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-ethyl sulfamyl)-methyl benzoate
(2S)-2-amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 0.123g (0.35mmol) is dissolved in the 2ml dimethyl formamide, and handles with 2-(methoxycarbonyl)-benzene sulfonyl chloride of 0.196ml (1.4mmol) triethylamine and 0.164g (0.7mmol).At room temperature, with described solution stirring 4 hours.Solvent removed in vacuo, described resistates is dissolved in the methylene dichloride and with water with described solution washing three times.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.On silicagel column with described resistates chromatography, with normal heptane/ethyl acetate (1/1) wash-out, output: 75mg.MS(ES
+):m/e=549(M+H
+)。
B) (2S)-2-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester
Be dissolved in 2-((1S)-1-tert-butoxycarbonyl-2-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-ethyl sulfamyl)-methyl benzoate of 75mg (0.13mmol) in the 1ml dimethyl formamide and add the 2-amino-1 of 68mg (0.69mmol); 4; 5, the 6-tetrahydropyrimidine.At room temperature described reactant was stirred 20 hours.Solvent removed in vacuo, on silicagel column with described resistates chromatography, with methylene chloride (1/1) wash-out, subsequently with methylene chloride/acetic acid/water (85/15/1.5/1.5) wash-out.Output: 54mg.MS(ES
+):m/e=683.3(M+H
+)。
C) (2S)-2-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
(2-(1 with (2S)-2-of 54mg (0.08mmol); 4; 5; 6-tetrahydropyrimidine-2-base formamyl)-benzenesulfonyl amino)-((2-(1 for 4-for 3-; 4; 5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-the propionic acid tert-butyl ester is dissolved in the 2ml methylene dichloride and adds the 2ml trifluoroacetic acid.At room temperature with described solution stirring 3 hours.Solvent removed in vacuo and toluene joined in the described resistates and then vacuum remove.Be dissolved in the acetonitrile/water (1/1) described resistates and lyophilize.Output: 34mg.MS(ES
+):m/e=627(M+H
+)。
Logical method 1 (synthesizing of the compound of embodiment 5 to 24)
Step a: with the reaction of SULPHURYL CHLORIDE
(2S)-2-amino-3-(4-(2-methoxycarbonyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester of 0.2g is dissolved in the 2ml dimethyl formamide, and handles with 4 molar equivalent triethylamines and the suitable SULPHURYL CHLORIDE of 2 molar equivalents.At room temperature, with described solution stirring 4 hours.Solvent removed in vacuo, described resistates is dissolved in the methylene dichloride and with water with described solution washing three times.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.On silica gel with described resistates chromatography, with normal heptane/ethyl acetate (1/1) wash-out.
Step b: the formation of acylguanidines
Be dissolved in the product of step a in the 1ml dimethyl formamide and add 5 molar equivalent 2-amino-1,4,5,6-tetrahydropyrimidine.At room temperature described reaction mixture was stirred 20 hours.Solvent removed in vacuo, on silica gel with described resistates chromatography, with methylene chloride (1/1) wash-out, subsequently with methylene chloride/acetic acid/water (85/15/1.5/1.5) wash-out.
Step c: the cracking of the tert-butyl ester
Be dissolved in the product of step b in the 2ml methylene dichloride and add the 2ml trifluoroacetic acid.At room temperature with described solution stirring 3 hours.Solvent removed in vacuo and toluene joined in the described resistates and then vacuum remove.Be dissolved in the acetonitrile/water (1/1) described resistates and lyophilize.
Embodiment 5
(2S)-2-(the 4-tertiary butyl-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the 4-tert.-butylbenzene SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 83mg 547.2 (M+H) of step a
+
The product 85mg 614.3 (M+H) of step b
+
The product of step c (title compound) 75mg 558.3 (M+H)
+
Embodiment 6
(2S)-2-(propane-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the propane-1-SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 54mg 457.3 (M+H) of step a
+
The product 55mg 524.3 (M+H) of step b
+
The product of step c (title compound) 45mg 468.3 (M+H)
+
Embodiment 7
(2S)-2-(2-phenyl-ethane sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the trans-β-vinylbenzene SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 49mg 517.1 (M+H) of step a
+
The product 45mg 584.3 (M+H) of step b
+
The product of step c (title compound) 37mg 528.3 (M+H)
+
Embodiment 8
(2S)-2-(4-propyl group-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use 4-(the n-propyl)-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 78mg 533.2 (M+H) of step a
+
The product 73mg 600.4 (M+H) of step b
+
The product of step c (title compound) 55mg 544.3 (M+H)
+
Embodiment 9
(2S)-2-(2-methyl-propane-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

Use the 2-methyl-propane-1-SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 21mg 471.3 (M+H) of step a
+
The product 10mg 538.4 (M+H) of step b
+
The product of step c (title compound) 13mg 482.3 (M+H)
+
Embodiment 10
(2S)-2-(4-butoxy-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

Use 4-(the n-butoxy)-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 83mg 563.2 (M+H) of step a
+
The product 83mg 630.4 (M+H) of step b
+
The product of step c (title compound) 63mg 574.3 (M+H)
+
Embodiment 11
(2S)-2-(2-cyano group-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

Use the 2-cyano group-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 91mg 516.2 (M+H) of step a
+
The product 71mg 583.3 (M+H) of step b
+
The product of step c (title compound) 73mg 527.3 (M+H)
+
Embodiment 12
(2S)-2-(7,7-dimethyl-2-oxo-two ring [2.2.1] heptan-1-base methylsulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the 10-sulphur acyl chloride of camphor among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 85mg 565.3 (M+H) of step a
+
The product 88mg 632.4 (M+H) of step b
+
The product of step c (title compound) 71mg 576.4 (M+H)
+
Embodiment 13
(2S)-2-(4-chloro-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
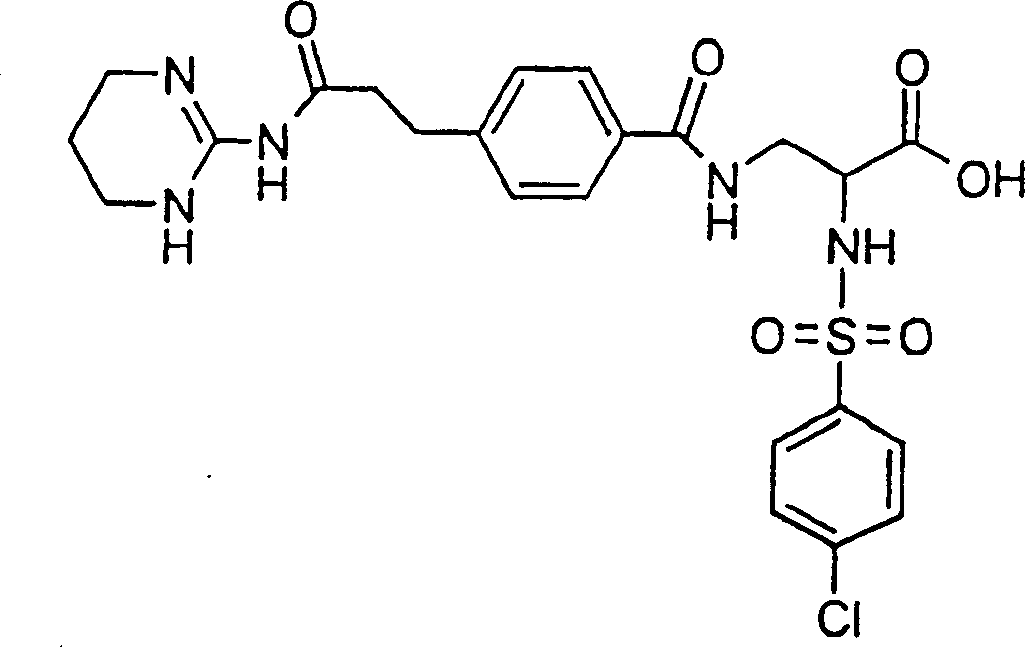
Use the 4-chloro-benzene sulfonyl chloride among the step a, according to logical method 1, synthetic described title compound.
Output MS (ES
+): m/e
The product 91mg 525.1 (M+H) of step a
+
The product 82mg 592.3 (M+H) of step b
+
The product of step c (title compound) 64mg 536.3 (M+H)
+
Embodiment 14
(2S)-2-(3-chloro-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the 3-chloro-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 65mg 525.2 (M+H) of step a
+
The product 58mg 592.3 (M+H) of step b
+
The product of step c (title compound) 52mg 536.3 (M+H)
+
Embodiment 15
(2S)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-2-(3-trifluoromethyl-benzenesulfonyl amino)-propionic acid
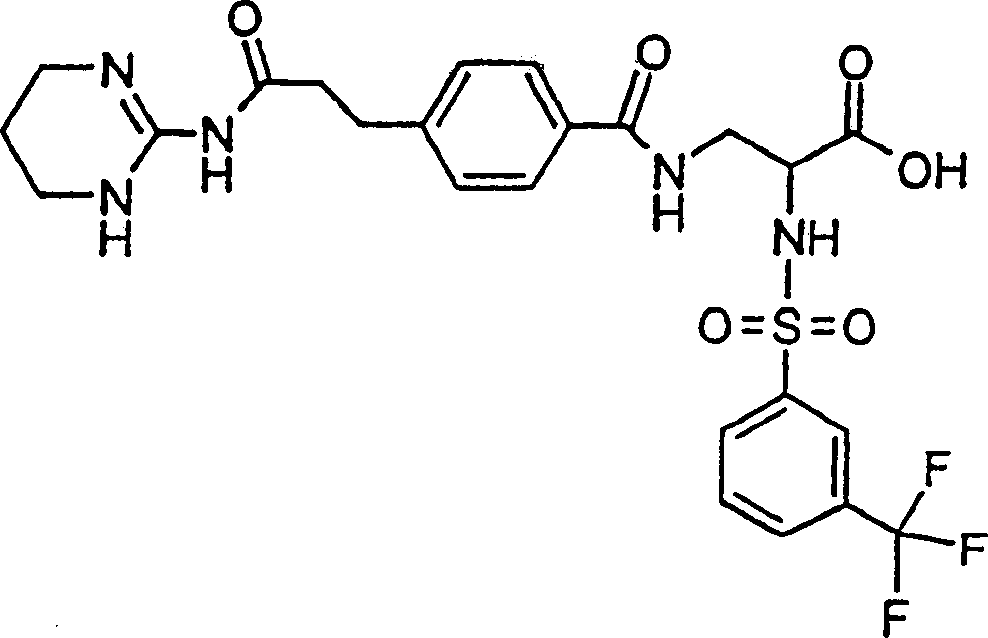
Use the 3-trifluoromethyl-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 86mg 559.2 (M+H) of step a
+
The product 94mg 626.3 (M+H) of step b
+
The product of step c (title compound) 84mg 570.3 (M+H)
+
Embodiment 16
(2S)-2-(4-methoxyl group-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the 4-methoxyl group-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 66mg 521.1 (M+H) of step a
+
The product 71mg 588.3 (M+H) of step b
+
The product of step c (title compound) 49mg 532.3 (M+H)
+
Embodiment 17
(2S)-2-benzenesulfonyl amino-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 76mg 491.2 (M+H) of step a
+
The product 77mg 558.3 (M+H) of step b
+
The product of step c (title compound) 64mg 502.3 (M+H)
+
Embodiment 18
(2S)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-2-(thiophene-2-sulfuryl amino)-propionic acid
Use the 2-thiophene SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 87mg 497.1 (M+H) of step a
+
The product 74mg 564.2 (M+H) of step b
+
The product of step c (title compound) 64mg 508.2 (M+H)
+
Embodiment 19 (2S)-2-(xenyl-4-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

Use the 4-xenyl SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 39mg 567.1 (M+H) of step a
+
The product 40mg 634.4 (M+H) of step b
+
The product of step c (title compound) 33mg 578.3 (M+H)
+
Embodiment 20
(2S)-2-(naphthalene-2-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid

Use the 2-naphthalic sulfonic chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 76mg 541.2 (M+H) of step a
+
The product 74mg 608.3 (M+H) of step b
+
The product of step c (title compound) 62mg 552.3 (M+H)
+
Embodiment 21
(2S)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-2-(2,4,6-trimethylammonium-benzenesulfonyl amino)-propionic acid
Use 2,4 among the step a, the 6-trimethylbenzene chloride is according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 68mg 533.2 (M+H) of step a
+
The product 71mg 600.4 (M+H) of step b
+
The product of step c (title compound) 54mg 544.3 (M+H)
+
Embodiment 22
(2S)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-2-(4-trifluoromethyl-benzenesulfonyl amino)-propionic acid
Use the 4-trifluoromethyl-benzene sulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 105mg 559.3 (M+H) of step a
+
The product 93mg 626.4 (M+H) of step b
+
The product of step c (title compound) 70mg 570.3 (M+H)
+
Embodiment 23
(2S)-2-(butane-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the butane-1-SULPHURYL CHLORIDE among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 87mg 471.4 (M+H) of step a
+
The product 60mg 538.4 (M+H) of step b
+
The product of step c (title compound) 57mg 482.3 (M+H)
+
Embodiment 24
(2S)-2-methylsulfonyl amino-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the methylsulfonyl chloride among the step a, according to logical method 1, synthesising title compound.
Output MS (ES
+): m/e
The product 87.8mg 429.3 (M+H) of step a
+
The product 98mg 496.4 (M+H) of step b
+
The product of step c (title compound) 74mg 440.3 (M+H)
+
Logical method 2 (synthesizing of the compound of embodiment 25 to 27)
Step a: with the reaction of SULPHURYL CHLORIDE
(2S)-2-amino-3-(4-(2-carboxyl-ethyl)-benzoyl-amido)-propionic acid tert-butyl ester hydrochloride of 0.1g is dissolved in the 3ml dimethyl formamide, and under 0 ℃, handles with 3 molar equivalent diisopropyl ethyl amines and the suitable SULPHURYL CHLORIDE of 2 molar equivalents.Under 0 ℃, with described solution stirring 3 hours.By adding the ethyl acetate diluted reaction mixture, solution washs once with the aqueous potassium hydrogen sulfate washed twice with saturated sodium-chloride water solution.With the described organic phase of dried over mgso, filter and solvent removed in vacuo.On silica gel with described resistates chromatography, with methylene chloride/acetic acid/water (97.5/2.5/0.25/0.25) wash-out.
Step b: the formation of acylguanidines
Be dissolved in the product of step a in the 2ml tetrahydrofuran (THF) and add 1.2 molar equivalent 2-amino-1,4,5,6-tetrahydropyrimidine, 4 molar equivalent diisopropyl ethyl amines and 1.1 molar equivalent O-(7-azepine benzo triazol-1-yl)-1,1,3,3-tetramethyl-urea hexafluorophosphate.At room temperature described reactant was stirred 20 hours.Solvent removed in vacuo also is dissolved in described resistates in the ethyl acetate, and described solution with the saturated sodium bicarbonate aqueous solution washed twice and with the saturated sodium-chloride water solution washing once.With the described organic phase of dried over mgso, filter and solvent removed in vacuo.On silica gel with described resistates chromatography, with methylene chloride/acetic acid/water (85/15/1.5/1.5) wash-out, subsequently with methylene chloride/acetic acid/water (9/1/0.1/0.1) wash-out.
Step c: the cracking of tert-butyl ester ester
The product of described step b is dissolved in 1.5ml trifluoroacetic acid/water (95/5).At room temperature with described solution stirring 1 hour.Solvent removed in vacuo and toluene joined in the described resistates and then vacuum remove.Be dissolved in the acetonitrile/water (1/1) described resistates and lyophilize.
Embodiment 25
(2S)-2-(propane-2-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the propane-2-SULPHURYL CHLORIDE among the step a, according to logical method 2, synthesising title compound.
Output MS:m/e
The product 17mg 443.2 (M+H) of step a
+(FAB
+)
The product 7mg 524.2 (M+H) of step b
+(ES
+)
The product of step c (title compound) 7.2mg 468.2 (M+H)
+(ES
+)
Embodiment 26
(2S)-2-chloromethane sulfuryl amino-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid
Use the chloromethane SULPHURYL CHLORIDE among the step a, according to logical method 2, synthesising title compound.
Output MS:m/e
The product 94mg 449.1 (M+H) of step a
+(FAB
+)
The product 28mg 530.2 (M) of step b
+(ES
+)
The product of step c (title compound) 34mg 474.2 (M+H)
+(ES
+)
Embodiment 27
(2S)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-2-(2,2,2-three fluoro-ethylsulfonylamino)-propionic acid
Use 2,2 among the step a, 2-trifluoro ethyl sulfonyl chloride is according to logical method 2, synthesising title compound.
Output MS:m/e
The product 43mg 483.2 (M+H) of step a
+(FAB
+)
The product 24mg 564.2 (M+H) of step b
+(ES
+)
The product of step c (title compound) 22mg 508.2 (M+H)
+(ES
+)
Embodiment 28
(2S)-2-(the 4-tertiary butyl-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-ethyl propionate
Be dissolved in 700mg (2S)-2-(the 4-tertiary butyl-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid in the 100ml ethanol and add 15 vitriol oils.With described reaction soln boiling 3.5 hours.Solvent removed in vacuo is dissolved in described resistates in the methylene dichloride and with saturated sodium bicarbonate aqueous solution washing three times.With methylene dichloride once and wash twice of the organic phase of described merging with saturated sodium-chloride water solution with described aqueous extraction.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.Described resistates is dissolved in the 2N aqueous hydrochloric acid.Vacuum is removed hydrochloric acid, is dissolved in described resistates in the acetonitrile and adds entry.With this mixture lyophilize.Output: 480mg.MS(ES
+):m/e=586.4(M+H)
+。
Embodiment 29
(2S)-2-(the 4-tertiary butyl-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-isopropyl propionate

Be dissolved in 700mg (2S)-2-(the 4-tertiary butyl-benzenesulfonyl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid in the 100ml Virahol and add 15 vitriol oils.With described reaction soln boiling 2.5 days.Solvent removed in vacuo is dissolved in described resistates in the methylene dichloride and with saturated sodium bicarbonate aqueous solution washing three times.With methylene dichloride once and wash twice of the organic phase of described merging with saturated sodium-chloride water solution with described aqueous extraction.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.Described resistates is dissolved in the 2N aqueous hydrochloric acid.Vacuum is removed hydrochloric acid, is dissolved in described resistates in the acetonitrile and adds entry.With this mixture lyophilize.Output: 444mg.MS(ES
+):m/e=600.4(M+H)
+。
Embodiment 30
(2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-ethyl propionate
Be dissolved in 590mg (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid in the 80ml ethanol and add 12 vitriol oils.With described reaction soln boiling 3 hours.Solvent removed in vacuo is dissolved in described resistates in the methylene dichloride and with saturated sodium bicarbonate aqueous solution washing three times.With methylene dichloride once and wash twice of the organic phase of described merging with saturated sodium-chloride water solution with described aqueous extraction.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.Described resistates is dissolved in the 2N aqueous hydrochloric acid.Vacuum is removed hydrochloric acid, is dissolved in described resistates in the acetonitrile and adds entry.With this mixture lyophilize.Output: 381mg.MS(ES
+):m/e=580.3(M+H)
+。
Embodiment 31
(2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-isopropyl propionate
Be dissolved in 1.5g (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid in the 250ml Virahol and add the 1ml vitriol oil.With described reaction soln boiling 3 days.Solvent removed in vacuo is dissolved in described resistates in the methylene dichloride and with saturated sodium bicarbonate aqueous solution washing three times.With methylene dichloride once and wash twice of the organic phase of described merging with saturated sodium-chloride water solution with described aqueous extraction.With the described organic phase of dried over sodium sulfate, filter and solvent removed in vacuo.Described resistates is dissolved in the 2N aqueous hydrochloric acid.Vacuum is removed hydrochloric acid, is dissolved in described resistates in the acetonitrile and adds entry.With this mixture lyophilize.Output: 950mg.MS(ES
+):m/e=594.4(M+H)
+。
Embodiment 32
(2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-isobutyl propionate
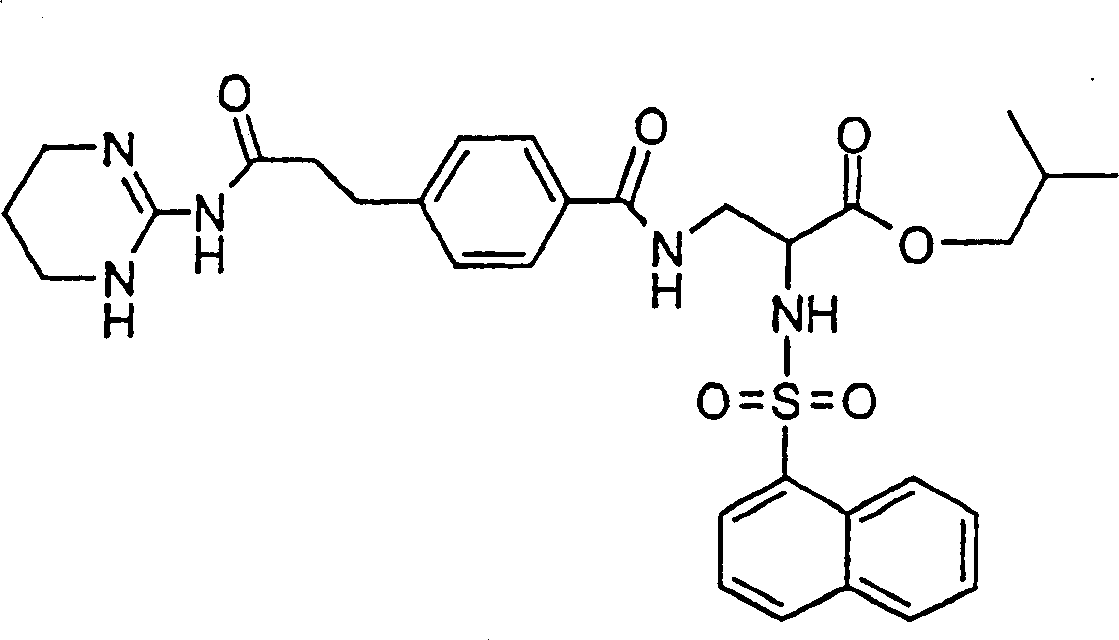
Be dissolved in 600mg (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido)-propionic acid in the 12ml isopropylcarbinol and add the 0.1ml vitriol oil.With described reaction soln boiling 24 hours.Solvent removed in vacuo, and on silica gel with described resistates chromatography, with methylene chloride/acetic acid/water (9/1/0.1/0.1) wash-out.Be dissolved in the acetic acid/water described product and lyophilize.Output: 250mg.MS(ES
+):m/e=608.5(M+H)
+。
Pharmacological testing
For example, by the heavy absorption test (" PIT ASSAY ") of the osteoclast that for example is similar to WO-A-95/32710, can measure The compounds of this invention to the re-absorbed restraining effect of bone, it is attached to herein by reference.
For example, as following described, can measure The compounds of this invention and suppress Vitronectic receptor α
vβ
3Restraining effect.
Be used to measure the 293 cell inhibiting effects tests (Vn/293 test cell line) that are incorporated into people's vitronectin
1. the purifying of people's vitronectin
From people's blood plasma the separation of human vitronectin and according to the affinity stratographic analysis purification in the method for Yatohyo etc. (
Cellularstructure and function, 1988,23,281-292).
2. test cell line
According to described FACS method, select to be used for Vitronectic receptor α with the dna sequence dna cotransfection
vβ
3α
vAnd β
3Being used for of 293 cells (human embryonic kidney cell line) of subunit expressed (>500,000 α at high proportion
vβ
3Acceptor/cell).Cultivate selected cell and, be every cell>1,000, the α of 000 template to obtain the having expression ratio by FACS sorting once more
vβ
3Stable clone (15D).
Under 4 ℃, will have flat Linbro 96 hole tissue culturing plates bag with the people's vitronectin in the salt brine solution (PBS) of phosphate buffered (0.01mg/ml, 0.05ml/ hole) and be spent the night, the BSA (bSA) with 0.5% concentration blocks then.Preparation 10
-10Mol/l to 2 * 10
-3The described of mol/l tried the solution of material in containing the DMEM nutrient solution of glucose, and the 0.05ml/ hole solution is joined on the plate under each situation.To be expressed as high-caliber α
vβ
3The cell suspension of (for example 15D) is in the DMEM nutrient solution that contains glucose, and described suspension is adjusted to content is 25,000 cells/0.05ml nutrient solution.0.05ml joins in every hole with this cell suspending liquid, and under 37 ℃ described plate is hatched 90 minutes.With warm PBS described plate is washed three times, to remove unconjugated cell.The citrate buffer that contains 0.25%Triton X-100 (25mM, pH5.0) in freezing described bonded cell.Add hexoseamidase substrate p-nitrophenyl-N-ethanoyl-β-D-glucosaminide then, and under 37 ℃, described plate was hatched 90 minutes.Stop described reaction with glycine (50mM)/EDTA (5mM) damping fluid (pH10.4), and under 405-650nm, measure the absorption in every hole.Analyze described data according to standard method.
Obtain the following stated test-results (inhibition concentration IC
50).
Embodiment numbers IC
50The Vn/293 test cell line
1 6nM
2 52nM
3 27nM
5 18nM
6 32nM
7 28nM
8 20nM
9 69nM
10 29nM
11 24nM
12 27nM
13 27nM
14 10nM
15 6nM
16 26nM
17 9nM
18 14nM
19 38nM
20 5nM
21 21nM
22 10nM
23 30nM
24 100nM
Claims (16)
1. formula I compound, the salt that can tolerate on the stereoisomer form that they are all and the form of mixtures of all proportions thereof and their physiology
Wherein
Radicals R
1-and R
2-be saturated or unsaturated divalence (C together
2-C
9)-alkylidene group; R
4Be hydrogen, (C
1-C
6)-alkyl-CO-O-(C
1-C
4)-alkyl-or (C
1-C
6)-alkyl, it is for unsubstituted or by being selected from hydroxyl, (C
1-C
4)-alkoxyl group, (C
1-C
4)-alkyl-S (O)
2-,-NR
7R
7 'With-N
+R
7R
7 'R
7 "Q
-Group replace R wherein
7, R
7 'And R
7 "Independently of one another is hydrogen, (C
1-C
6)-alkyl, (C
5-C
14)-aryl or (C
5-C
14)-aryl-(C
1-C
6)-alkyl-and Q
-Be the negatively charged ion that can tolerate on the physiology, perhaps R wherein
4Be one of following groups
Wherein said group indicates by a dotted line so as to the key that connects;
R
5Be (C
1-C
20)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces, and wherein in described alkyl, monocycle alkyl, bicyclic alkyl and tricyclic alkyl, one or more carbon atoms can be replaced by the identical or different atoms that are selected from nitrogen, oxygen and sulphur;
R
3Be (C
1-C
10)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
1-C
8)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, (C
5-C
14)-heteroaryl, (C
5-C
14)-heteroaryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, hydroxyl, oxo, nitro, amino ,-NH-(C
1-C
4)-alkyl ,-N ((C
1-C
4)-alkyl)
2,-NH-CO-(C
1-C
4)-alkyl ,-CO-(C
1-C
4)-alkyl;
R
6Be hydrogen, (C
1-C
6)-alkyl-O-CO-, hydroxyl, (C
1-C
6)-alkyl-O-CO-O-or nitro; Described aryl is selected from phenyl, naphthyl, xenyl, anthryl or fluorenyl; And described heteroaryl is selected from pyridyl, pyrryl, furyl, thienyl, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, tetrazyl, pyridazinyl, pyrazinyl, pyrimidyl, indyl, pseudoindoyl, indazolyl, 2 base, quinolyl, isoquinolyl, quinoxalinyl, quinazolyl, cinnolines base, β-Ka Lin base or benzo is thick and, pentamethylene is thick and, hexanaphthene is thick and or suberane is thick and the derivative of these groups.
2. the claimed formula I compound of claim 1, the salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and their physiology, wherein R
1And R
2For hydrogen or be saturated or unsaturated divalence (C together
2-C
5)-alkylidene group; R
3Be (C
1-C
10)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
1-C
8)-alkoxyl group, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, (C
5-C
14)-heteroaryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl;
R
4For hydrogen or its for unsubstituted or by being selected from (C
1-C
4)-alkoxyl group, (C
1-C
4)-alkyl-S (O)
2-and NR
7R
7 '(the C that replaces of group
1-C
6)-alkyl, wherein R
7And R
7 'Independently of one another is hydrogen or (C
1-C
4)-alkyl;
R
5Be (C
1-C
20)-alkyl, (C
3-C
20)-monocycle alkyl, (C
5-C
20)-bicyclic alkyl, (C
5-C
20)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces;
R
6Be hydrogen or (C
1-C
6)-alkyl-O-CO-.
3. claim 1 or 2 claimed formula I compounds, the salt that can tolerate on the stereoisomer form that they are all and the form of mixtures of all proportions thereof and their physiology, wherein R
1And R
2For hydrogen or be saturated or unsaturated divalence (C together
2-C
4)-alkylidene group; R
3Be (C
1-C
4)-alkyl, (C
3-C
10)-monocycle alkyl, (C
5-C
12)-bicyclic alkyl, (C
5-C
12)-tricyclic alkyl, (C
1-C
4)-alkoxyl group, (C
6-C
14)-aryl, (C
6-C
14)-aryl-(C
1-C
4)-alkyl-, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl;
R
4Be hydrogen or (C
1-C
6)-alkyl;
R
5Be (C
1-C
10)-alkyl, (C
3-C
15)-monocycle alkyl, (C
5-C
15)-bicyclic alkyl, (C
5-C
15)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces;
R
6Be hydrogen or (C
1-C
6)-alkyl-O-CO-.
4. claim 1 or 2 claimed formula I compounds, the salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and their physiology, wherein R
1And R
2For hydrogen or be saturated or unsaturated divalence (C together
2-C
3)-alkylidene group;
R
3Be (C
1-C
4)-alkyl, (C
3-C
10)-monocycle alkyl, (C
5-C
12)-bicyclic alkyl, (C
5-C
12)-tricyclic alkyl, (C
1-C
4)-alkoxyl group, (C
6-C
14)-aryl, halogen, trifluoromethyl, cyano group, oxo ,-N ((C
1-C
4)-alkyl)
2Or-NH-CO-(C
1-C
4)-alkyl;
R
4Be hydrogen or (C
1-C
6)-alkyl;
R
5Be (C
1-C
10)-alkyl, (C
3-C
15)-monocycle alkyl, (C
5-C
15)-bicyclic alkyl, (C
5-C
15)-tricyclic alkyl, (C
6-C
14)-aryl, (C
5-C
14)-heteroaryl, (C
6-C
14)-aryl-(C
1-C
6)-alkyl-or (C
5-C
14)-heteroaryl-(C
1-C
6)-alkyl-, each is unsubstituted or by one, two or three R in wherein said aryl, heteroaryl, alkyl, monocycle alkyl, bicyclic alkyl and the tricyclic alkyl
3Group replaces;
R
6Be hydrogen or (C
1-C
4)-alkyl-O-CO-.
5. claim 1 or 2 claimed formula I compound, wherein R
5Be (C
6-C
14)-aryl or (C
5-C
14Each is unsubstituted or by one, two or three identical or different R in the)-heteroaryl, wherein said aryl and heteroaryl
3Group replaces, the salt that can tolerate on the stereoisomer form that they are all and the form of mixtures of all proportions thereof and their physiology.
6. claim 1 or 2 claimed formula I compounds, the salt that can tolerate on the form of the stereoisomer form that they are all and the mixture of all proportions thereof and its physiology, wherein R
5Be naphthyl.
7. claim 1 or 2 claimed formula I compounds, it is 2-(R
5-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido) propionic acid, wherein R
5-sulfuryl amino substituting group is selected from benzenesulfonyl amino; 4-(n-propyl) benzenesulfonyl amino; 4-tert.-butylbenzene sulfuryl amino; 2; 4; 6-Three methyl Benzene sulfuryl amino; 4-anisole sulfuryl amino; 4-(n-butoxy) benzenesulfonyl amino; 3-chlorobenzene sulfuryl amino; 4-chlorobenzene sulfuryl amino; 3-trifluoromethylbenzene sulfuryl amino; 4-trifluoromethylbenzene sulfuryl amino; 4-P-acetamido benzene sulfonyl base amino; 2-cyano group benzenesulfonyl amino; naphthalene-1-sulfuryl amino; naphthalene-2-sulfuryl amino; xenyl-4-sulfuryl amino; thiophene-2-sulfuryl amino; quinoline-8-sulfuryl amino; methylsulfonyl amino; propane-1-sulfuryl amino; propane-2-sulfuryl amino; butane-1-sulfuryl amino; 2-methylpropane-1-sulfuryl amino; the chloromethane sulfuryl amino; 2; 2; 2-trifluoro ethylsulfonylamino; 7; 7-dimethyl-2-oxo-dicyclo [2.2.1] heptan-the amino and 2-phenyl ethylsulfonylamino of 1-base methylsulfonyl, the salt that can tolerate on the stereoisomer form that they are all and the form of mixtures of all proportions thereof and their physiology.
8. claim 1 or 2 claimed formula I compounds, it is the salt that can tolerate on (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl) benzoyl-amido) propionic acid and its physiology.
9. claim 1 or 2 claimed formula I compounds, it is (2S)-2-(naphthalene-1-sulfuryl amino)-3-(4-(2-(1,4,5,6-tetrahydropyrimidine-2-base formamyl)-ethyl)-benzoyl-amido) propionic acid or its (C
1-C
4The salt that can tolerate on)-alkyl ester and its physiology.
10. claim 1 or 2 claimed formula I compounds; it is that (2S)-2-(naphthalene-1-sulfuryl amino)-((2-(1 for 4-for 3-; 4,5,6-tetrahydropyrimidine-2-base formamyl)-and ethyl) benzoyl-amido) salt that can tolerate on ethyl propionate or its physiology.
11. the method for claimed formula I compound in any one of the claim 1 to 10 of preparation, this method comprises carboxylic acid or the carboxylic acid derivative that makes formula II,

R wherein
4And R
5Such as in any one of claim 1 to 10 definition or other functional group exist with precursor forms or with the form of protection, and X is the commutable leavings group of nucleophilicity, with the guanidine or the guanidine derivative reaction of formula III,

R wherein
1, R
2And R
6Such as in any one of claim 1 to 10 definition, perhaps other functional group exists with precursor forms or with the form of protection.
12. a medicinal preparations, it comprises pharmaceutically harmless carrier, and comprises the salt that can tolerate on any claimed formula I compound of at least a claim 1 to 10 and/or its physiology.
13. the purposes of salt in the preparation Vitronectic receptor antagonist that can tolerate on claimed formula I compound and/or its physiology in any one of the claim 1 to 10.
14. the salt that can tolerate on formula I compound that claim 13 is claimed and/or its physiology the preparation inhibitors of bone resorption or preparation be used for the treatment of or the medicine of preventing osteoporosis disease in purposes.
15. the purposes of the salt that can tolerate on formula I compound that claim 13 is claimed and/or its physiology in the inhibitor of preparation tumor growth or metastases.
16. the salt that can tolerate on formula I compound that claim 13 is claimed and/or its physiology the preparation antiphlogiston or preparation be used for the treatment of preventing cardiovascular disease, restenosis, arteriosclerosis, ephrosis become or the medicine of retinopathy in purposes.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US1248998A | 1998-01-23 | 1998-01-23 | |
| US09/012,489 | 1998-01-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1293662A CN1293662A (en) | 2001-05-02 |
| CN1177832C true CN1177832C (en) | 2004-12-01 |
Family
ID=21755208
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB998040894A Expired - Fee Related CN1177832C (en) | 1998-01-23 | 1999-01-16 | Novel sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion |
Country Status (25)
| Country | Link |
|---|---|
| EP (1) | EP1049677A1 (en) |
| JP (1) | JP2002501054A (en) |
| KR (1) | KR20010034319A (en) |
| CN (1) | CN1177832C (en) |
| AP (1) | AP1269A (en) |
| AR (1) | AR014456A1 (en) |
| AU (1) | AU752882B2 (en) |
| BG (1) | BG104630A (en) |
| BR (1) | BR9907735A (en) |
| CA (1) | CA2318221A1 (en) |
| EA (1) | EA003102B1 (en) |
| HR (1) | HRP20000493A2 (en) |
| HU (1) | HUP0100520A3 (en) |
| ID (1) | ID26219A (en) |
| IL (1) | IL137423A0 (en) |
| NO (1) | NO318795B1 (en) |
| NZ (1) | NZ505613A (en) |
| PL (1) | PL341871A1 (en) |
| SK (1) | SK10632000A3 (en) |
| TR (1) | TR200002160T2 (en) |
| TW (1) | TWI247742B (en) |
| UA (1) | UA63990C2 (en) |
| WO (1) | WO1999037621A1 (en) |
| YU (1) | YU47200A (en) |
| ZA (1) | ZA99476B (en) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2338275A1 (en) * | 1998-07-29 | 2000-02-10 | Merck & Co., Inc. | Integrin receptor antagonists |
| EP1028114A1 (en) | 1999-02-13 | 2000-08-16 | Aventis Pharma Deutschland GmbH | Novel guanidine derivatives as inhibitors of cell adhesion |
| EP1065207A1 (en) * | 1999-07-02 | 2001-01-03 | Aventis Pharma Deutschland GmbH | Naphthyridine derivatives, processes for their preparation, their use, and pharmaceutical compositions comprising them |
| EP1065208A1 (en) * | 1999-07-02 | 2001-01-03 | Aventis Pharma Deutschland GmbH | Substituted purine derivatives as inhibitors of cell adhesion |
| EP1070707A1 (en) * | 1999-07-21 | 2001-01-24 | Aventis Pharma Deutschland GmbH | 1,4,5,6-tetrahydropyrimidine derivative as a vitronectin inhibitor |
| US6849639B2 (en) | 1999-12-14 | 2005-02-01 | Amgen Inc. | Integrin inhibitors and their methods of use |
| EP1108721A1 (en) * | 1999-12-15 | 2001-06-20 | Aventis Pharma Deutschland GmbH | Thienylalanine derivatives as inhibitors of cell adhesion |
| EP1244616A1 (en) * | 1999-12-24 | 2002-10-02 | SmithKline Beecham plc | (hetero)bicyclylmethanesulfonylamino-substituted hydroxamic acid derivatives |
| FR2808798A1 (en) * | 2000-05-09 | 2001-11-16 | Hoechst Marion Roussel Inc | New N-heterocyclyl-aminoacid derivatives and analogs, are vitronectin analogs useful e.g. for treating osteoporosis, tumor growth or metastasis, inflammation or cardiovascular disease |
| FR2847254B1 (en) | 2002-11-19 | 2005-01-28 | Aventis Pharma Sa | NOVEL VITRONECTIN RECEPTOR ANTAGONIST DERIVATIVES, PROCESS FOR PREPARING THEM, THEIR APPLICATION AS MEDICAMENTS AND THE PHARMACEUTICAL COMPOSITIONS REFLECTING THEM |
| EP1727811B1 (en) | 2004-03-24 | 2014-11-12 | Shire Orphan Therapies GmbH | New compounds for the inhibition of angiogenesis and use of thereof |
| FR2870541B1 (en) | 2004-05-18 | 2006-07-14 | Proskelia Sas | ANTIGONISTIC PYRIMIDINE DERIVATIVES OF VITRONECTIN RECEPTOR |
| GB0412553D0 (en) * | 2004-06-04 | 2004-07-07 | Univ Aberdeen | Therapeutic agents for the treatment of bone conditions |
| UA87854C2 (en) | 2004-06-07 | 2009-08-25 | Мерк Энд Ко., Инк. | N-(2-benzyl)-2-phenylbutanamides as androgen receptor modulators |
| FR2873585B1 (en) * | 2004-07-27 | 2006-11-17 | Aventis Pharma Sa | NEW GALENIC FORMULATIONS OF ACTIVE PRINCIPLES |
| GB0705400D0 (en) | 2007-03-21 | 2007-05-02 | Univ Aberdeen | Therapeutic compounds andm their use |
| GB0817208D0 (en) | 2008-09-19 | 2008-10-29 | Pimco 2664 Ltd | Therapeutic apsap compounds and their use |
| GB0817207D0 (en) | 2008-09-19 | 2008-10-29 | Pimco 2664 Ltd | therapeutic apsac compounds and their use |
| GB201311361D0 (en) | 2013-06-26 | 2013-08-14 | Pimco 2664 Ltd | Compounds and their therapeutic use |
| RU2016146826A (en) | 2014-05-30 | 2018-07-04 | Пфайзер Инк. | CARBONITRIL DERIVATIVES AS SELECTIVE ANDROGEN RECEPTOR MODULATORS |
| LT3262028T (en) | 2014-12-17 | 2022-01-10 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamide and n-(-4hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)-benzenesulfonamide compounds and their therapeutic use |
| WO2023275715A1 (en) | 2021-06-30 | 2023-01-05 | Pfizer Inc. | Metabolites of selective androgen receptor modulators |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5741796A (en) * | 1994-05-27 | 1998-04-21 | Merck & Co., Inc. | Pyridyl and naphthyridyl compounds for inhibiting osteoclast-mediated bone resorption |
| JP3895792B2 (en) * | 1995-12-08 | 2007-03-22 | プロスケリア・エス・ア・エス | Bone formation promoter |
-
1999
- 1999-01-16 BR BR9907735-3A patent/BR9907735A/en not_active IP Right Cessation
- 1999-01-16 TR TR2000/02160T patent/TR200002160T2/en unknown
- 1999-01-16 IL IL13742399A patent/IL137423A0/en unknown
- 1999-01-16 UA UA2000084983A patent/UA63990C2/en unknown
- 1999-01-16 WO PCT/EP1999/000242 patent/WO1999037621A1/en not_active Application Discontinuation
- 1999-01-16 HU HU0100520A patent/HUP0100520A3/en unknown
- 1999-01-16 KR KR1020007008041A patent/KR20010034319A/en active IP Right Grant
- 1999-01-16 AU AU25181/99A patent/AU752882B2/en not_active Ceased
- 1999-01-16 AP APAP/P/2000/001863A patent/AP1269A/en active
- 1999-01-16 CA CA002318221A patent/CA2318221A1/en not_active Abandoned
- 1999-01-16 CN CNB998040894A patent/CN1177832C/en not_active Expired - Fee Related
- 1999-01-16 JP JP2000528545A patent/JP2002501054A/en not_active Abandoned
- 1999-01-16 NZ NZ505613A patent/NZ505613A/en unknown
- 1999-01-16 YU YU47200A patent/YU47200A/en unknown
- 1999-01-16 SK SK1063-2000A patent/SK10632000A3/en unknown
- 1999-01-16 PL PL99341871A patent/PL341871A1/en unknown
- 1999-01-16 ID IDW20001407A patent/ID26219A/en unknown
- 1999-01-16 EP EP99904789A patent/EP1049677A1/en not_active Withdrawn
- 1999-01-16 EA EA200000785A patent/EA003102B1/en not_active IP Right Cessation
- 1999-01-22 ZA ZA9900476A patent/ZA99476B/en unknown
- 1999-01-29 AR ARP990100244A patent/AR014456A1/en not_active Application Discontinuation
- 1999-04-30 TW TW088100862A patent/TWI247742B/en active
-
2000
- 2000-07-21 HR HR20000493A patent/HRP20000493A2/en not_active Application Discontinuation
- 2000-07-21 NO NO20003765A patent/NO318795B1/en unknown
- 2000-07-21 BG BG104630A patent/BG104630A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN1293662A (en) | 2001-05-02 |
| HUP0100520A3 (en) | 2002-11-28 |
| HRP20000493A2 (en) | 2001-06-30 |
| UA63990C2 (en) | 2004-02-16 |
| NO20003765D0 (en) | 2000-07-21 |
| YU47200A (en) | 2002-11-15 |
| AP1269A (en) | 2004-04-03 |
| AP2000001863A0 (en) | 2000-09-30 |
| AR014456A1 (en) | 2001-02-28 |
| WO1999037621A1 (en) | 1999-07-29 |
| PL341871A1 (en) | 2001-05-07 |
| AU752882B2 (en) | 2002-10-03 |
| ZA99476B (en) | 1999-08-05 |
| EP1049677A1 (en) | 2000-11-08 |
| KR20010034319A (en) | 2001-04-25 |
| ID26219A (en) | 2000-12-07 |
| CA2318221A1 (en) | 1999-07-29 |
| EA200000785A1 (en) | 2001-02-26 |
| AU2518199A (en) | 1999-08-09 |
| EA003102B1 (en) | 2002-12-26 |
| BR9907735A (en) | 2000-10-17 |
| BG104630A (en) | 2001-04-30 |
| NO20003765L (en) | 2000-09-25 |
| JP2002501054A (en) | 2002-01-15 |
| TR200002160T2 (en) | 2001-07-23 |
| NZ505613A (en) | 2002-11-26 |
| IL137423A0 (en) | 2001-07-24 |
| TWI247742B (en) | 2006-01-21 |
| SK10632000A3 (en) | 2001-02-12 |
| HUP0100520A1 (en) | 2001-07-30 |
| NO318795B1 (en) | 2005-05-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1177832C (en) | Novel sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion | |
| CN1205191C (en) | Novel acylguanidine derivatives as inhibitors of bone resorption and as vitronectin receptor antagonists | |
| CN1158279C (en) | 1-(P-thienylbenzyl)-imidazoles as angiotensin-(1-7) receptor agonists, method for the production and the utilization thereof and pharmaceutical preparations containing said compounds | |
| CN1183111C (en) | Cycloalkano-indote and againdole derivatives, process for their preparation and pharmaceutical composition containing the same | |
| CN1054603C (en) | Tri-substituted phenyl derivatives and processes for their preparation | |
| CN1030251C (en) | Substituted 4-(quinoline-2-yl-methoxy) phenylacetic acid derivatives | |
| CN1273466C (en) | Quinoline derivatives and quinazoline derivatives having azolyl group | |
| CN1186332C (en) | New thryoid receptor ligands and process II | |
| CN1072647C (en) | Novel inhibitors of bone reabsorption and antagonists of vitronectin receptors | |
| CN1158254C (en) | 'Beta'-sulfonyl hydroxamic acids as matrix metalloproteinases inhibitors | |
| CN1046725C (en) | Condensed imidazole compounds, their production and use | |
| CN1101816C (en) | Substituted purine derivatives, processes for their preparation, their use, and compositions comprising them | |
| CN1016778B (en) | Spiro-substituted glutaramida diuretic agents | |
| CN1193276A (en) | Use of oxido-squalene cyclase inhibitors to lower blood cholesterol | |
| CN87101285A (en) | 8-replaces-2-amino-1,2,3, the 4-tetrahydro-bitter edible plant | |
| CN1382144A (en) | Heteroalkylamino-substituted bicyclic nitrogen heterocycles as inhibitors of P38 protein kinase | |
| CN1471515A (en) | Compounds effective as beta-2-adrenoreceptor agonists as well as PDE-4-inhibitors | |
| CN1418194A (en) | Amide compounds and use thereof | |
| CN1036566A (en) | 1,3,4,5-tetrahydro benzo [c, d] diindyl compounds | |
| CN1300281A (en) | Compounds with growth hormone releasing properties | |
| CN1161340C (en) | Benzimidazole compounds that are vitronectin receptor antagonists | |
| CN1107857A (en) | Cyclic amino acid derivativges | |
| CN1051733A (en) | Acyl-CoA: cholesteryl transferase inhibitor | |
| CN1033804A (en) | New anti-arrhythmic agents and preparation method thereof | |
| CN1055920C (en) | Alkyl benzoylguanidines derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |