CN112912507A - 甲病毒新抗原载体和干扰素抑制剂 - Google Patents
甲病毒新抗原载体和干扰素抑制剂 Download PDFInfo
- Publication number
- CN112912507A CN112912507A CN201980068204.XA CN201980068204A CN112912507A CN 112912507 A CN112912507 A CN 112912507A CN 201980068204 A CN201980068204 A CN 201980068204A CN 112912507 A CN112912507 A CN 112912507A
- Authority
- CN
- China
- Prior art keywords
- sequence
- nucleic acid
- acid sequence
- epitope
- encoding nucleic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000013598 vector Substances 0.000 title claims abstract description 171
- 241000710929 Alphavirus Species 0.000 title claims abstract description 140
- 239000003112 inhibitor Substances 0.000 title claims abstract description 71
- 239000000427 antigen Substances 0.000 title claims description 299
- 102000036639 antigens Human genes 0.000 title claims description 298
- 108091007433 antigens Proteins 0.000 title claims description 297
- 102000014150 Interferons Human genes 0.000 title description 2
- 108010050904 Interferons Proteins 0.000 title description 2
- 229940079322 interferon Drugs 0.000 title description 2
- 238000000034 method Methods 0.000 claims abstract description 293
- 230000014509 gene expression Effects 0.000 claims abstract description 165
- 108010014726 Interferon Type I Proteins 0.000 claims abstract description 55
- 102000002227 Interferon Type I Human genes 0.000 claims abstract description 55
- 230000011664 signaling Effects 0.000 claims abstract description 51
- 150000007523 nucleic acids Chemical group 0.000 claims description 396
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 344
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 250
- 206010028980 Neoplasm Diseases 0.000 claims description 196
- 210000004027 cell Anatomy 0.000 claims description 194
- 125000003729 nucleotide group Chemical group 0.000 claims description 147
- 239000002773 nucleotide Substances 0.000 claims description 145
- 108090000623 proteins and genes Proteins 0.000 claims description 143
- 239000000203 mixture Substances 0.000 claims description 132
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 115
- 108091034057 RNA (poly(A)) Proteins 0.000 claims description 89
- 150000002632 lipids Chemical class 0.000 claims description 83
- 229960005486 vaccine Drugs 0.000 claims description 81
- 108091054437 MHC class I family Proteins 0.000 claims description 76
- 241000700605 Viruses Species 0.000 claims description 76
- 102000043129 MHC class I family Human genes 0.000 claims description 74
- 230000035772 mutation Effects 0.000 claims description 67
- 210000004881 tumor cell Anatomy 0.000 claims description 65
- 235000001014 amino acid Nutrition 0.000 claims description 61
- 229920001184 polypeptide Polymers 0.000 claims description 60
- 235000018102 proteins Nutrition 0.000 claims description 60
- 102000004169 proteins and genes Human genes 0.000 claims description 60
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 54
- 108700028369 Alleles Proteins 0.000 claims description 52
- 108091054438 MHC class II family Proteins 0.000 claims description 51
- 238000012384 transportation and delivery Methods 0.000 claims description 51
- 238000012163 sequencing technique Methods 0.000 claims description 50
- 102000043131 MHC class II family Human genes 0.000 claims description 49
- 210000001519 tissue Anatomy 0.000 claims description 48
- 150000001413 amino acids Chemical class 0.000 claims description 46
- 241001217856 Chimpanzee adenovirus Species 0.000 claims description 43
- 230000027455 binding Effects 0.000 claims description 42
- 230000028993 immune response Effects 0.000 claims description 41
- 230000004075 alteration Effects 0.000 claims description 38
- 201000011510 cancer Diseases 0.000 claims description 37
- 239000012634 fragment Substances 0.000 claims description 34
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 32
- 230000037430 deletion Effects 0.000 claims description 31
- 238000012217 deletion Methods 0.000 claims description 31
- 241000282414 Homo sapiens Species 0.000 claims description 29
- -1 amino lipid Chemical class 0.000 claims description 29
- 210000000612 antigen-presenting cell Anatomy 0.000 claims description 27
- 239000002671 adjuvant Substances 0.000 claims description 26
- 239000005089 Luciferase Substances 0.000 claims description 24
- 239000005090 green fluorescent protein Substances 0.000 claims description 22
- 239000002105 nanoparticle Substances 0.000 claims description 21
- 230000001225 therapeutic effect Effects 0.000 claims description 21
- 230000001965 increasing effect Effects 0.000 claims description 20
- 239000003981 vehicle Substances 0.000 claims description 18
- 239000008194 pharmaceutical composition Substances 0.000 claims description 17
- 101710172711 Structural protein Proteins 0.000 claims description 16
- 102000004127 Cytokines Human genes 0.000 claims description 15
- 108090000695 Cytokines Proteins 0.000 claims description 15
- 230000001939 inductive effect Effects 0.000 claims description 15
- 230000008685 targeting Effects 0.000 claims description 15
- 241000710959 Venezuelan equine encephalitis virus Species 0.000 claims description 14
- NRLNQCOGCKAESA-KWXKLSQISA-N [(6z,9z,28z,31z)-heptatriaconta-6,9,28,31-tetraen-19-yl] 4-(dimethylamino)butanoate Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCC(OC(=O)CCCN(C)C)CCCCCCCC\C=C/C\C=C/CCCCC NRLNQCOGCKAESA-KWXKLSQISA-N 0.000 claims description 14
- 210000004443 dendritic cell Anatomy 0.000 claims description 14
- 230000001404 mediated effect Effects 0.000 claims description 14
- 108091033319 polynucleotide Proteins 0.000 claims description 14
- 102000040430 polynucleotide Human genes 0.000 claims description 14
- 239000002157 polynucleotide Substances 0.000 claims description 14
- 230000002829 reductive effect Effects 0.000 claims description 14
- 108091008874 T cell receptors Proteins 0.000 claims description 13
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 13
- 230000000903 blocking effect Effects 0.000 claims description 13
- 238000003776 cleavage reaction Methods 0.000 claims description 13
- 230000001105 regulatory effect Effects 0.000 claims description 13
- 230000007017 scission Effects 0.000 claims description 13
- 230000004936 stimulating effect Effects 0.000 claims description 13
- 230000037433 frameshift Effects 0.000 claims description 12
- 230000008488 polyadenylation Effects 0.000 claims description 12
- 108091027544 Subgenomic mRNA Proteins 0.000 claims description 11
- 230000003321 amplification Effects 0.000 claims description 11
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 11
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 11
- 238000013518 transcription Methods 0.000 claims description 11
- 230000035897 transcription Effects 0.000 claims description 11
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 11
- 108010043121 Green Fluorescent Proteins Proteins 0.000 claims description 10
- 102000004144 Green Fluorescent Proteins Human genes 0.000 claims description 10
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 claims description 10
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 claims description 10
- 238000010362 genome editing Methods 0.000 claims description 10
- 210000001165 lymph node Anatomy 0.000 claims description 10
- 210000004962 mammalian cell Anatomy 0.000 claims description 10
- 238000006467 substitution reaction Methods 0.000 claims description 10
- 206010066919 Epidemic polyarthritis Diseases 0.000 claims description 9
- 101000852870 Homo sapiens Interferon alpha/beta receptor 1 Proteins 0.000 claims description 9
- 102100036714 Interferon alpha/beta receptor 1 Human genes 0.000 claims description 9
- 241000710942 Ross River virus Species 0.000 claims description 9
- 241000710961 Semliki Forest virus Species 0.000 claims description 9
- 241000710960 Sindbis virus Species 0.000 claims description 9
- 108090000848 Ubiquitin Proteins 0.000 claims description 9
- 102000044159 Ubiquitin Human genes 0.000 claims description 9
- 238000000338 in vitro Methods 0.000 claims description 9
- 208000020816 lung neoplasm Diseases 0.000 claims description 9
- 201000001441 melanoma Diseases 0.000 claims description 9
- 238000007920 subcutaneous administration Methods 0.000 claims description 9
- 230000037455 tumor specific immune response Effects 0.000 claims description 9
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 8
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 claims description 8
- 230000009368 gene silencing by RNA Effects 0.000 claims description 8
- 238000003780 insertion Methods 0.000 claims description 8
- 230000037431 insertion Effects 0.000 claims description 8
- 201000005202 lung cancer Diseases 0.000 claims description 8
- 238000007481 next generation sequencing Methods 0.000 claims description 8
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 7
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 7
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 7
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 7
- 206010006187 Breast cancer Diseases 0.000 claims description 7
- 208000026310 Breast neoplasm Diseases 0.000 claims description 7
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 7
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 7
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 7
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 7
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 7
- 108700008625 Reporter Genes Proteins 0.000 claims description 7
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 7
- 239000004012 Tofacitinib Substances 0.000 claims description 7
- 125000000539 amino acid group Chemical group 0.000 claims description 7
- 230000006472 autoimmune response Effects 0.000 claims description 7
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 7
- 208000029742 colonic neoplasm Diseases 0.000 claims description 7
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 7
- 230000005847 immunogenicity Effects 0.000 claims description 7
- 239000002955 immunomodulating agent Substances 0.000 claims description 7
- 229940121354 immunomodulator Drugs 0.000 claims description 7
- 230000002584 immunomodulator Effects 0.000 claims description 7
- 201000002528 pancreatic cancer Diseases 0.000 claims description 7
- 201000003120 testicular cancer Diseases 0.000 claims description 7
- 229960001350 tofacitinib Drugs 0.000 claims description 7
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 claims description 7
- 238000013519 translation Methods 0.000 claims description 7
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims description 6
- 206010009944 Colon cancer Diseases 0.000 claims description 6
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 claims description 6
- 102000042838 JAK family Human genes 0.000 claims description 6
- 108091082332 JAK family Proteins 0.000 claims description 6
- 102000014944 Lysosome-Associated Membrane Glycoproteins Human genes 0.000 claims description 6
- 108010064171 Lysosome-Associated Membrane Glycoproteins Proteins 0.000 claims description 6
- 108700026244 Open Reading Frames Proteins 0.000 claims description 6
- 206010033128 Ovarian cancer Diseases 0.000 claims description 6
- 206010060862 Prostate cancer Diseases 0.000 claims description 6
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 6
- 206010038389 Renal cancer Diseases 0.000 claims description 6
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 6
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 6
- 208000000389 T-cell leukemia Diseases 0.000 claims description 6
- 206010057644 Testis cancer Diseases 0.000 claims description 6
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 claims description 6
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 claims description 6
- 230000030741 antigen processing and presentation Effects 0.000 claims description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 6
- 230000002708 enhancing effect Effects 0.000 claims description 6
- 206010017758 gastric cancer Diseases 0.000 claims description 6
- 201000010536 head and neck cancer Diseases 0.000 claims description 6
- 201000010982 kidney cancer Diseases 0.000 claims description 6
- 229940043355 kinase inhibitor Drugs 0.000 claims description 6
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 6
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 6
- 239000003757 phosphotransferase inhibitor Substances 0.000 claims description 6
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 6
- 230000003595 spectral effect Effects 0.000 claims description 6
- 201000011549 stomach cancer Diseases 0.000 claims description 6
- 229960000814 tetanus toxoid Drugs 0.000 claims description 6
- 108020003589 5' Untranslated Regions Proteins 0.000 claims description 5
- 231100000221 frame shift mutation induction Toxicity 0.000 claims description 5
- 108020004999 messenger RNA Proteins 0.000 claims description 5
- 230000002093 peripheral effect Effects 0.000 claims description 5
- 238000012545 processing Methods 0.000 claims description 5
- 230000008707 rearrangement Effects 0.000 claims description 5
- 150000003384 small molecules Chemical class 0.000 claims description 5
- 108020005345 3' Untranslated Regions Proteins 0.000 claims description 4
- 101710094648 Coat protein Proteins 0.000 claims description 4
- 108090001126 Furin Proteins 0.000 claims description 4
- 108060001084 Luciferase Proteins 0.000 claims description 4
- 101710163270 Nuclease Proteins 0.000 claims description 4
- 108010076504 Protein Sorting Signals Proteins 0.000 claims description 4
- 102000012740 beta Adrenergic Receptors Human genes 0.000 claims description 4
- 108010079452 beta Adrenergic Receptors Proteins 0.000 claims description 4
- 210000000234 capsid Anatomy 0.000 claims description 4
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 claims description 4
- 230000019491 signal transduction Effects 0.000 claims description 4
- 239000000126 substance Substances 0.000 claims description 4
- 230000032258 transport Effects 0.000 claims description 4
- OGHAROSJZRTIOK-KQYNXXCUSA-O 7-methylguanosine Chemical compound C1=2N=C(N)NC(=O)C=2[N+](C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OGHAROSJZRTIOK-KQYNXXCUSA-O 0.000 claims description 3
- 108060003951 Immunoglobulin Proteins 0.000 claims description 3
- 101710128560 Initiator protein NS1 Proteins 0.000 claims description 3
- 108010065805 Interleukin-12 Proteins 0.000 claims description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 3
- 101710144127 Non-structural protein 1 Proteins 0.000 claims description 3
- 101800000515 Non-structural protein 3 Proteins 0.000 claims description 3
- 101800000980 Protease nsP2 Proteins 0.000 claims description 3
- 108010029485 Protein Isoforms Proteins 0.000 claims description 3
- 102000001708 Protein Isoforms Human genes 0.000 claims description 3
- 101800001758 RNA-directed RNA polymerase nsP4 Proteins 0.000 claims description 3
- 230000001413 cellular effect Effects 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 102000018358 immunoglobulin Human genes 0.000 claims description 3
- 230000001629 suppression Effects 0.000 claims description 3
- 102000002260 Alkaline Phosphatase Human genes 0.000 claims description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 claims description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims description 2
- 125000001433 C-terminal amino-acid group Chemical group 0.000 claims description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 2
- 108091035707 Consensus sequence Proteins 0.000 claims description 2
- 108091006020 Fc-tagged proteins Proteins 0.000 claims description 2
- HVLSXIKZNLPZJJ-TXZCQADKSA-N HA peptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 HVLSXIKZNLPZJJ-TXZCQADKSA-N 0.000 claims description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 claims description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 claims description 2
- 108090000172 Interleukin-15 Proteins 0.000 claims description 2
- 108010002350 Interleukin-2 Proteins 0.000 claims description 2
- 108010002586 Interleukin-7 Proteins 0.000 claims description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 2
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 claims description 2
- 108091092724 Noncoding DNA Proteins 0.000 claims description 2
- 108010003723 Single-Domain Antibodies Proteins 0.000 claims description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 2
- 241001492404 Woodchuck hepatitis virus Species 0.000 claims description 2
- 208000014619 adult acute lymphoblastic leukemia Diseases 0.000 claims description 2
- 235000004279 alanine Nutrition 0.000 claims description 2
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 claims description 2
- 229950000321 benralizumab Drugs 0.000 claims description 2
- 108010074108 interleukin-21 Proteins 0.000 claims description 2
- 230000030147 nuclear export Effects 0.000 claims description 2
- 230000001124 posttranscriptional effect Effects 0.000 claims description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 2
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 2
- 102100035233 Furin Human genes 0.000 claims 1
- 208000035475 disorder Diseases 0.000 claims 1
- 230000002519 immonomodulatory effect Effects 0.000 claims 1
- 238000011260 co-administration Methods 0.000 abstract 1
- 108020004566 Transfer RNA Proteins 0.000 description 85
- 125000005647 linker group Chemical group 0.000 description 68
- 238000002649 immunization Methods 0.000 description 53
- 230000003053 immunization Effects 0.000 description 53
- 241000699670 Mus sp. Species 0.000 description 49
- 241000701161 unidentified adenovirus Species 0.000 description 36
- 230000003612 virological effect Effects 0.000 description 31
- 108020004414 DNA Proteins 0.000 description 28
- 102000039446 nucleic acids Human genes 0.000 description 26
- 108020004707 nucleic acids Proteins 0.000 description 26
- 230000005867 T cell response Effects 0.000 description 25
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 24
- 238000004519 manufacturing process Methods 0.000 description 23
- 239000013603 viral vector Substances 0.000 description 20
- 229940045513 CTLA4 antagonist Drugs 0.000 description 18
- 230000000890 antigenic effect Effects 0.000 description 18
- 229910052799 carbon Inorganic materials 0.000 description 18
- 241000282577 Pan troglodytes Species 0.000 description 17
- 208000015181 infectious disease Diseases 0.000 description 17
- 239000013604 expression vector Substances 0.000 description 16
- 150000001721 carbon Chemical group 0.000 description 15
- 238000004949 mass spectrometry Methods 0.000 description 15
- 239000000523 sample Substances 0.000 description 15
- 239000002202 Polyethylene glycol Substances 0.000 description 14
- 230000000295 complement effect Effects 0.000 description 14
- 229920001223 polyethylene glycol Polymers 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 230000010076 replication Effects 0.000 description 14
- 230000004044 response Effects 0.000 description 14
- 230000000694 effects Effects 0.000 description 13
- 238000004806 packaging method and process Methods 0.000 description 13
- 238000001890 transfection Methods 0.000 description 13
- 235000012000 cholesterol Nutrition 0.000 description 12
- 238000001727 in vivo Methods 0.000 description 12
- 230000007935 neutral effect Effects 0.000 description 12
- 238000010186 staining Methods 0.000 description 12
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 11
- 238000013461 design Methods 0.000 description 11
- 239000002245 particle Substances 0.000 description 11
- 238000007912 intraperitoneal administration Methods 0.000 description 10
- 150000003904 phospholipids Chemical class 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- 210000002845 virion Anatomy 0.000 description 10
- 238000011746 C57BL/6J (JAX™ mouse strain) Methods 0.000 description 9
- 230000008901 benefit Effects 0.000 description 9
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 9
- 230000036039 immunity Effects 0.000 description 9
- 230000002163 immunogen Effects 0.000 description 9
- 230000002458 infectious effect Effects 0.000 description 9
- 238000003752 polymerase chain reaction Methods 0.000 description 9
- 210000002966 serum Anatomy 0.000 description 9
- 210000000952 spleen Anatomy 0.000 description 9
- 238000012549 training Methods 0.000 description 9
- 238000011282 treatment Methods 0.000 description 9
- 241000282412 Homo Species 0.000 description 8
- 101000883798 Homo sapiens Probable ATP-dependent RNA helicase DDX53 Proteins 0.000 description 8
- 241000282560 Macaca mulatta Species 0.000 description 8
- 102100038236 Probable ATP-dependent RNA helicase DDX53 Human genes 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 230000036755 cellular response Effects 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 8
- 125000006850 spacer group Chemical group 0.000 description 8
- 238000002255 vaccination Methods 0.000 description 8
- 101150029662 E1 gene Proteins 0.000 description 7
- 206010014611 Encephalitis venezuelan equine Diseases 0.000 description 7
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 7
- 238000003559 RNA-seq method Methods 0.000 description 7
- 208000002687 Venezuelan Equine Encephalomyelitis Diseases 0.000 description 7
- 201000009145 Venezuelan equine encephalitis Diseases 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 7
- 230000037452 priming Effects 0.000 description 7
- 210000004988 splenocyte Anatomy 0.000 description 7
- 230000029812 viral genome replication Effects 0.000 description 7
- 108091026890 Coding region Proteins 0.000 description 6
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 102000035195 Peptidases Human genes 0.000 description 6
- 108091005804 Peptidases Proteins 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000012528 membrane Substances 0.000 description 6
- 239000013612 plasmid Substances 0.000 description 6
- 230000014616 translation Effects 0.000 description 6
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 description 5
- 206010069754 Acquired gene mutation Diseases 0.000 description 5
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 5
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 5
- 108060002716 Exonuclease Proteins 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 108091034117 Oligonucleotide Proteins 0.000 description 5
- 239000004365 Protease Substances 0.000 description 5
- 230000024932 T cell mediated immunity Effects 0.000 description 5
- 230000002776 aggregation Effects 0.000 description 5
- 238000004220 aggregation Methods 0.000 description 5
- 125000002091 cationic group Chemical group 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 150000001875 compounds Chemical class 0.000 description 5
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 5
- 230000002950 deficient Effects 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 238000002405 diagnostic procedure Methods 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 102000013165 exonuclease Human genes 0.000 description 5
- 230000004927 fusion Effects 0.000 description 5
- 210000004602 germ cell Anatomy 0.000 description 5
- 230000001976 improved effect Effects 0.000 description 5
- 238000007918 intramuscular administration Methods 0.000 description 5
- 229960005386 ipilimumab Drugs 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 235000002639 sodium chloride Nutrition 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 230000037439 somatic mutation Effects 0.000 description 5
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 4
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 4
- 102100024263 CD160 antigen Human genes 0.000 description 4
- 102100038078 CD276 antigen Human genes 0.000 description 4
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 4
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 4
- 101150066038 E4 gene Proteins 0.000 description 4
- 102100036242 HLA class II histocompatibility antigen, DQ alpha 2 chain Human genes 0.000 description 4
- 108010050568 HLA-DM antigens Proteins 0.000 description 4
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 4
- 101000761938 Homo sapiens CD160 antigen Proteins 0.000 description 4
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 4
- 241000598171 Human adenovirus sp. Species 0.000 description 4
- 101800000324 Immunoglobulin A1 protease translocator Proteins 0.000 description 4
- 108091092195 Intron Proteins 0.000 description 4
- 241001467552 Mycobacterium bovis BCG Species 0.000 description 4
- 108020004485 Nonsense Codon Proteins 0.000 description 4
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 4
- 241000272534 Struthio camelus Species 0.000 description 4
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 125000003275 alpha amino acid group Chemical group 0.000 description 4
- 125000000129 anionic group Chemical group 0.000 description 4
- 229960000190 bacillus calmette–guérin vaccine Drugs 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000009566 cancer vaccine Methods 0.000 description 4
- 229940022399 cancer vaccine Drugs 0.000 description 4
- 238000012258 culturing Methods 0.000 description 4
- 210000000172 cytosol Anatomy 0.000 description 4
- 231100000135 cytotoxicity Toxicity 0.000 description 4
- 230000003013 cytotoxicity Effects 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000003623 enhancer Substances 0.000 description 4
- 102000054766 genetic haplotypes Human genes 0.000 description 4
- 230000036541 health Effects 0.000 description 4
- 210000005260 human cell Anatomy 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 102000054765 polymorphisms of proteins Human genes 0.000 description 4
- 230000004481 post-translational protein modification Effects 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- 238000010187 selection method Methods 0.000 description 4
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 3
- 108090000565 Capsid Proteins Proteins 0.000 description 3
- 201000009182 Chikungunya Diseases 0.000 description 3
- 108020004705 Codon Proteins 0.000 description 3
- 229920002307 Dextran Polymers 0.000 description 3
- 101150005585 E3 gene Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 102000004961 Furin Human genes 0.000 description 3
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 3
- 108020005004 Guide RNA Proteins 0.000 description 3
- 102100028972 HLA class I histocompatibility antigen, A alpha chain Human genes 0.000 description 3
- 102100028976 HLA class I histocompatibility antigen, B alpha chain Human genes 0.000 description 3
- 102100028971 HLA class I histocompatibility antigen, C alpha chain Human genes 0.000 description 3
- 108010075704 HLA-A Antigens Proteins 0.000 description 3
- 108010058607 HLA-B Antigens Proteins 0.000 description 3
- 108010052199 HLA-C Antigens Proteins 0.000 description 3
- 108010010378 HLA-DP Antigens Proteins 0.000 description 3
- 102000015789 HLA-DP Antigens Human genes 0.000 description 3
- 108010062347 HLA-DQ Antigens Proteins 0.000 description 3
- 108010058597 HLA-DR Antigens Proteins 0.000 description 3
- 102000006354 HLA-DR Antigens Human genes 0.000 description 3
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 3
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 3
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 3
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 3
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 3
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 3
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 3
- 241000713666 Lentivirus Species 0.000 description 3
- 108010026552 Proteome Proteins 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 241000710924 Togaviridae Species 0.000 description 3
- 108020000411 Toll-like receptor Proteins 0.000 description 3
- 102000002689 Toll-like receptor Human genes 0.000 description 3
- 108020000999 Viral RNA Proteins 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 239000001506 calcium phosphate Substances 0.000 description 3
- 229910000389 calcium phosphate Inorganic materials 0.000 description 3
- 235000011010 calcium phosphates Nutrition 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 108091036078 conserved sequence Proteins 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 238000013135 deep learning Methods 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 241001493065 dsRNA viruses Species 0.000 description 3
- 238000005538 encapsulation Methods 0.000 description 3
- 210000002443 helper t lymphocyte Anatomy 0.000 description 3
- 230000006801 homologous recombination Effects 0.000 description 3
- 238000002744 homologous recombination Methods 0.000 description 3
- 238000009396 hybridization Methods 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 239000007927 intramuscular injection Substances 0.000 description 3
- 238000010255 intramuscular injection Methods 0.000 description 3
- 239000006166 lysate Substances 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 238000001823 molecular biology technique Methods 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 239000002086 nanomaterial Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 210000003289 regulatory T cell Anatomy 0.000 description 3
- 229950010550 resiquimod Drugs 0.000 description 3
- BXNMTOQRYBFHNZ-UHFFFAOYSA-N resiquimod Chemical compound C1=CC=CC2=C(N(C(COCC)=N3)CC(C)(C)O)C3=C(N)N=C21 BXNMTOQRYBFHNZ-UHFFFAOYSA-N 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000002356 single layer Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- AWNBSWDIOCXWJW-WTOYTKOKSA-N (2r)-n-[(2s)-1-[[(2s)-1-(2-aminoethylamino)-1-oxopropan-2-yl]amino]-3-naphthalen-2-yl-1-oxopropan-2-yl]-n'-hydroxy-2-(2-methylpropyl)butanediamide Chemical compound C1=CC=CC2=CC(C[C@H](NC(=O)[C@@H](CC(=O)NO)CC(C)C)C(=O)N[C@@H](C)C(=O)NCCN)=CC=C21 AWNBSWDIOCXWJW-WTOYTKOKSA-N 0.000 description 2
- 125000002373 5 membered heterocyclic group Chemical group 0.000 description 2
- 125000004070 6 membered heterocyclic group Chemical group 0.000 description 2
- 125000003341 7 membered heterocyclic group Chemical group 0.000 description 2
- 102100026882 Alpha-synuclein Human genes 0.000 description 2
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 2
- 108010074708 B7-H1 Antigen Proteins 0.000 description 2
- 102100027314 Beta-2-microglobulin Human genes 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- 101710185679 CD276 antigen Proteins 0.000 description 2
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 2
- 108091033409 CRISPR Proteins 0.000 description 2
- 238000010354 CRISPR gene editing Methods 0.000 description 2
- 102100021868 Calnexin Human genes 0.000 description 2
- 102100029968 Calreticulin Human genes 0.000 description 2
- 102000014914 Carrier Proteins Human genes 0.000 description 2
- 102000005600 Cathepsins Human genes 0.000 description 2
- 108010084457 Cathepsins Proteins 0.000 description 2
- 102100023321 Ceruloplasmin Human genes 0.000 description 2
- 101150011126 Chad gene Proteins 0.000 description 2
- 208000006825 Eastern Equine Encephalomyelitis Diseases 0.000 description 2
- 201000005804 Eastern equine encephalitis Diseases 0.000 description 2
- 206010014587 Encephalitis eastern equine Diseases 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 102100031351 Galectin-9 Human genes 0.000 description 2
- 101100229077 Gallus gallus GAL9 gene Proteins 0.000 description 2
- 102100033079 HLA class II histocompatibility antigen, DM alpha chain Human genes 0.000 description 2
- 102100031258 HLA class II histocompatibility antigen, DM beta chain Human genes 0.000 description 2
- 102100031547 HLA class II histocompatibility antigen, DO alpha chain Human genes 0.000 description 2
- 102100031546 HLA class II histocompatibility antigen, DO beta chain Human genes 0.000 description 2
- 102100029966 HLA class II histocompatibility antigen, DP alpha 1 chain Human genes 0.000 description 2
- 102100031618 HLA class II histocompatibility antigen, DP beta 1 chain Human genes 0.000 description 2
- 102100036241 HLA class II histocompatibility antigen, DQ beta 1 chain Human genes 0.000 description 2
- 102100036117 HLA class II histocompatibility antigen, DQ beta 2 chain Human genes 0.000 description 2
- 102100040505 HLA class II histocompatibility antigen, DR alpha chain Human genes 0.000 description 2
- 102100028636 HLA class II histocompatibility antigen, DR beta 4 chain Human genes 0.000 description 2
- 102100028640 HLA class II histocompatibility antigen, DR beta 5 chain Human genes 0.000 description 2
- 108010093061 HLA-DPA1 antigen Proteins 0.000 description 2
- 108010045483 HLA-DPB1 antigen Proteins 0.000 description 2
- 108010086786 HLA-DQA1 antigen Proteins 0.000 description 2
- 108010081606 HLA-DQA2 antigen Proteins 0.000 description 2
- 108010065026 HLA-DQB1 antigen Proteins 0.000 description 2
- 108010067802 HLA-DR alpha-Chains Proteins 0.000 description 2
- 108010040960 HLA-DRB4 Chains Proteins 0.000 description 2
- 108010016996 HLA-DRB5 Chains Proteins 0.000 description 2
- 101000834898 Homo sapiens Alpha-synuclein Proteins 0.000 description 2
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 2
- 101000937544 Homo sapiens Beta-2-microglobulin Proteins 0.000 description 2
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 description 2
- 101000898052 Homo sapiens Calnexin Proteins 0.000 description 2
- 101000793651 Homo sapiens Calreticulin Proteins 0.000 description 2
- 101000866278 Homo sapiens HLA class II histocompatibility antigen, DO alpha chain Proteins 0.000 description 2
- 101000866281 Homo sapiens HLA class II histocompatibility antigen, DO beta chain Proteins 0.000 description 2
- 101000930799 Homo sapiens HLA class II histocompatibility antigen, DQ beta 2 chain Proteins 0.000 description 2
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 2
- 101001117317 Homo sapiens Programmed cell death 1 ligand 1 Proteins 0.000 description 2
- 101001117312 Homo sapiens Programmed cell death 1 ligand 2 Proteins 0.000 description 2
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 2
- 101000652359 Homo sapiens Spermatogenesis-associated protein 2 Proteins 0.000 description 2
- 101000649068 Homo sapiens Tapasin Proteins 0.000 description 2
- 101000955999 Homo sapiens V-set domain-containing T-cell activation inhibitor 1 Proteins 0.000 description 2
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 2
- 101150106931 IFNG gene Proteins 0.000 description 2
- 101710125507 Integrase/recombinase Proteins 0.000 description 2
- 102000002698 KIR Receptors Human genes 0.000 description 2
- 108010043610 KIR Receptors Proteins 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 2
- 102000017578 LAG3 Human genes 0.000 description 2
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 description 2
- 102000011202 Member 2 Subfamily B ATP Binding Cassette Transporter Human genes 0.000 description 2
- 108010023335 Member 2 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 101150039088 PDIA3 gene Proteins 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 2
- 102100037097 Protein disulfide-isomerase A3 Human genes 0.000 description 2
- 101710185720 Putative ethidium bromide resistance protein Proteins 0.000 description 2
- 241000293871 Salmonella enterica subsp. enterica serovar Typhi Species 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- 108091081024 Start codon Proteins 0.000 description 2
- 229930182558 Sterol Natural products 0.000 description 2
- 230000006044 T cell activation Effects 0.000 description 2
- 102100028082 Tapasin Human genes 0.000 description 2
- 238000010162 Tukey test Methods 0.000 description 2
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 description 2
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 2
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 2
- 206010046865 Vaccinia virus infection Diseases 0.000 description 2
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 2
- 108700005077 Viral Genes Proteins 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 229940037003 alum Drugs 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 239000008365 aqueous carrier Substances 0.000 description 2
- 108010068991 arginyl-threonyl-prolyl-prolyl-prolyl-seryl-glycine Proteins 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 238000013475 authorization Methods 0.000 description 2
- 230000005784 autoimmunity Effects 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 229960000397 bevacizumab Drugs 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 210000002421 cell wall Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000012761 co-transfection Methods 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 238000002716 delivery method Methods 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229940000406 drug candidate Drugs 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 2
- 239000003925 fat Substances 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000001476 gene delivery Methods 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 238000012165 high-throughput sequencing Methods 0.000 description 2
- 229960002751 imiquimod Drugs 0.000 description 2
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 2
- 229940126546 immune checkpoint molecule Drugs 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 230000006058 immune tolerance Effects 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 239000000568 immunological adjuvant Substances 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 230000002132 lysosomal effect Effects 0.000 description 2
- 238000010801 machine learning Methods 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 229960003301 nivolumab Drugs 0.000 description 2
- 230000037434 nonsense mutation Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 229960000639 pazopanib Drugs 0.000 description 2
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 235000019419 proteases Nutrition 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000003362 replicative effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 229960003310 sildenafil Drugs 0.000 description 2
- 230000000391 smoking effect Effects 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 230000000392 somatic effect Effects 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 150000003432 sterols Chemical class 0.000 description 2
- 235000003702 sterols Nutrition 0.000 description 2
- 229960001796 sunitinib Drugs 0.000 description 2
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 2
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 238000012385 systemic delivery Methods 0.000 description 2
- 229960000835 tadalafil Drugs 0.000 description 2
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000011269 treatment regimen Methods 0.000 description 2
- 229950007217 tremelimumab Drugs 0.000 description 2
- 230000005748 tumor development Effects 0.000 description 2
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 2
- 108010072106 tumstatin (74-98) Proteins 0.000 description 2
- 208000007089 vaccinia Diseases 0.000 description 2
- 229960002381 vardenafil Drugs 0.000 description 2
- 229940023147 viral vector vaccine Drugs 0.000 description 2
- 238000011179 visual inspection Methods 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- FYGDTMLNYKFZSV-URKRLVJHSA-N (2s,3r,4s,5s,6r)-2-[(2r,4r,5r,6s)-4,5-dihydroxy-2-(hydroxymethyl)-6-[(2r,4r,5r,6s)-4,5,6-trihydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1[C@@H](CO)O[C@@H](OC2[C@H](O[C@H](O)[C@H](O)[C@H]2O)CO)[C@H](O)[C@H]1O FYGDTMLNYKFZSV-URKRLVJHSA-N 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- CITHEXJVPOWHKC-UUWRZZSWSA-N 1,2-di-O-myristoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCC CITHEXJVPOWHKC-UUWRZZSWSA-N 0.000 description 1
- SNKAWJBJQDLSFF-NVKMUCNASA-N 1,2-dioleoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC SNKAWJBJQDLSFF-NVKMUCNASA-N 0.000 description 1
- MWRBNPKJOOWZPW-NYVOMTAGSA-N 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCC\C=C/CCCCCCCC MWRBNPKJOOWZPW-NYVOMTAGSA-N 0.000 description 1
- WTJKGGKOPKCXLL-VYOBOKEXSA-N 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC WTJKGGKOPKCXLL-VYOBOKEXSA-N 0.000 description 1
- KGRVJHAUYBGFFP-UHFFFAOYSA-N 2,2'-Methylenebis(4-methyl-6-tert-butylphenol) Chemical compound CC(C)(C)C1=CC(C)=CC(CC=2C(=C(C=C(C)C=2)C(C)(C)C)O)=C1O KGRVJHAUYBGFFP-UHFFFAOYSA-N 0.000 description 1
- IOJUJUOXKXMJNF-UHFFFAOYSA-N 2-acetyloxybenzoic acid [3-(nitrooxymethyl)phenyl] ester Chemical compound CC(=O)OC1=CC=CC=C1C(=O)OC1=CC=CC(CO[N+]([O-])=O)=C1 IOJUJUOXKXMJNF-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- 102000002627 4-1BB Ligand Human genes 0.000 description 1
- 108010082808 4-1BB Ligand Proteins 0.000 description 1
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 1
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 description 1
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 description 1
- 108010024878 Adenovirus E1A Proteins Proteins 0.000 description 1
- 108010087905 Adenovirus E1B Proteins Proteins 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 241000589158 Agrobacterium Species 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 241000178568 Aura virus Species 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 102100035080 BDNF/NT-3 growth factors receptor Human genes 0.000 description 1
- 108020000946 Bacterial DNA Proteins 0.000 description 1
- 108020004513 Bacterial RNA Proteins 0.000 description 1
- 229920002498 Beta-glucan Polymers 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 108700031361 Brachyury Proteins 0.000 description 1
- 108091079001 CRISPR RNA Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 102000009512 Cyclin-Dependent Kinase Inhibitor p15 Human genes 0.000 description 1
- 108010009356 Cyclin-Dependent Kinase Inhibitor p15 Proteins 0.000 description 1
- 108010009392 Cyclin-Dependent Kinase Inhibitor p16 Proteins 0.000 description 1
- 102100024458 Cyclin-dependent kinase inhibitor 2A Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 150000008574 D-amino acids Chemical class 0.000 description 1
- 238000001061 Dunnett's test Methods 0.000 description 1
- 238000011510 Elispot assay Methods 0.000 description 1
- 102100031726 Endoplasmic reticulum junction formation protein lunapark Human genes 0.000 description 1
- 241001125671 Eretmochelys imbricata Species 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 206010015719 Exsanguination Diseases 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000231322 Fort Morgan virus Species 0.000 description 1
- 102100030708 GTPase KRas Human genes 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 206010061968 Gastric neoplasm Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102100040482 HLA class II histocompatibility antigen, DR beta 3 chain Human genes 0.000 description 1
- 102100040485 HLA class II histocompatibility antigen, DRB1 beta chain Human genes 0.000 description 1
- 108010039343 HLA-DRB1 Chains Proteins 0.000 description 1
- 108010061311 HLA-DRB3 Chains Proteins 0.000 description 1
- 102100035108 High affinity nerve growth factor receptor Human genes 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 108010027412 Histocompatibility Antigens Class II Proteins 0.000 description 1
- 101000779641 Homo sapiens ALK tyrosine kinase receptor Proteins 0.000 description 1
- 101000596896 Homo sapiens BDNF/NT-3 growth factors receptor Proteins 0.000 description 1
- 101000941029 Homo sapiens Endoplasmic reticulum junction formation protein lunapark Proteins 0.000 description 1
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 1
- 101000596894 Homo sapiens High affinity nerve growth factor receptor Proteins 0.000 description 1
- 101001043209 Homo sapiens Leukemia NUP98 fusion partner 1 Proteins 0.000 description 1
- 101000686031 Homo sapiens Proto-oncogene tyrosine-protein kinase ROS Proteins 0.000 description 1
- 101000579425 Homo sapiens Proto-oncogene tyrosine-protein kinase receptor Ret Proteins 0.000 description 1
- 101000800133 Homo sapiens Thyroglobulin Proteins 0.000 description 1
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 101150032643 IVa2 gene Proteins 0.000 description 1
- 108700002232 Immediate-Early Genes Proteins 0.000 description 1
- 102100023915 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102100037850 Interferon gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 101150075239 L1 gene Chemical group 0.000 description 1
- 101150027802 L2 gene Chemical group 0.000 description 1
- 101150084684 L3 gene Chemical group 0.000 description 1
- 101150007425 L4 gene Chemical group 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 108091027974 Mature messenger RNA Proteins 0.000 description 1
- 241000608292 Mayaro virus Species 0.000 description 1
- 102000003792 Metallothionein Human genes 0.000 description 1
- 108090000157 Metallothionein Proteins 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- GUVMFDICMFQHSZ-UHFFFAOYSA-N N-(1-aminoethenyl)-1-[4-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[hydroxy-[[3-[hydroxy-[[3-hydroxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy]phosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy]phosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(2-amino-6-oxo-1H-purin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(2-amino-6-oxo-1H-purin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-[[[2-[[[2-[[[5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[5-(4-amino-2-oxopyrimidin-1-yl)-2-[[hydroxy-[2-(hydroxymethyl)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxyphosphinothioyl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]oxolan-2-yl]-5-methylimidazole-4-carboxamide Chemical compound CC1=C(C(=O)NC(N)=C)N=CN1C1OC(COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)CO)C(OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)O)C1 GUVMFDICMFQHSZ-UHFFFAOYSA-N 0.000 description 1
- 108010084333 N-palmitoyl-S-(2,3-bis(palmitoyloxy)propyl)cysteinyl-seryl-lysyl-lysyl-lysyl-lysine Proteins 0.000 description 1
- 102100029166 NT-3 growth factor receptor Human genes 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108010058846 Ovalbumin Proteins 0.000 description 1
- 239000012270 PD-1 inhibitor Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 102100023347 Proto-oncogene tyrosine-protein kinase ROS Human genes 0.000 description 1
- 102100028286 Proto-oncogene tyrosine-protein kinase receptor Ret Human genes 0.000 description 1
- 239000013614 RNA sample Substances 0.000 description 1
- 206010037742 Rabies Diseases 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 208000002847 Surgical Wound Diseases 0.000 description 1
- 230000006052 T cell proliferation Effects 0.000 description 1
- 208000026651 T-cell prolymphocytic leukemia Diseases 0.000 description 1
- 238000010459 TALEN Methods 0.000 description 1
- 108010055044 Tetanus Toxin Proteins 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 102100039390 Toll-like receptor 7 Human genes 0.000 description 1
- 108091028113 Trans-activating crRNA Proteins 0.000 description 1
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 101000756604 Xenopus laevis Actin, cytoplasmic 1 Proteins 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 230000000947 anti-immunosuppressive effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000009464 antigen specific memory response Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000013528 artificial neural network Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000003833 bile salt Substances 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 238000012410 cDNA cloning technique Methods 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 229960002713 calcium chloride Drugs 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 229960002412 cediranib Drugs 0.000 description 1
- 229940047495 celebrex Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 108091092328 cellular RNA Proteins 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940106189 ceramide Drugs 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 230000008711 chromosomal rearrangement Effects 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000007402 cytotoxic response Effects 0.000 description 1
- 230000004041 dendritic cell maturation Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 239000005546 dideoxynucleotide Substances 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 229960003983 diphtheria toxoid Drugs 0.000 description 1
- 230000009266 disease activity Effects 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 230000037437 driver mutation Effects 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- 229940056913 eftilagimod alfa Drugs 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000002121 endocytic effect Effects 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000010195 expression analysis Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 238000000855 fermentation Methods 0.000 description 1
- 230000004151 fermentation Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 238000012224 gene deletion Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000007274 generation of a signal involved in cell-cell signaling Effects 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000003205 genotyping method Methods 0.000 description 1
- 210000002980 germ line cell Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 230000004727 humoral immunity Effects 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000008004 immune attack Effects 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000001571 immunoadjuvant effect Effects 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 1
- 101150063421 l5 gene Chemical group 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 238000012886 linear function Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007108 local immune response Effects 0.000 description 1
- 238000001325 log-rank test Methods 0.000 description 1
- 238000007477 logistic regression Methods 0.000 description 1
- 230000004777 loss-of-function mutation Effects 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 239000012931 lyophilized formulation Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- 239000002679 microRNA Substances 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 238000009126 molecular therapy Methods 0.000 description 1
- 229940035032 monophosphoryl lipid a Drugs 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 230000009437 off-target effect Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 229940092253 ovalbumin Drugs 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 229940121655 pd-1 inhibitor Drugs 0.000 description 1
- 229960002621 pembrolizumab Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000002205 phenol-chloroform extraction Methods 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229940115272 polyinosinic:polycytidylic acid Drugs 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000002516 postimmunization Effects 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 229960002816 potassium chloride Drugs 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012913 prioritisation Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000001902 propagating effect Effects 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000012175 pyrosequencing Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 238000010223 real-time analysis Methods 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 229950010316 rontalizumab Drugs 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 238000007480 sanger sequencing Methods 0.000 description 1
- 108010038379 sargramostim Proteins 0.000 description 1
- 229960002530 sargramostim Drugs 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 239000002924 silencing RNA Substances 0.000 description 1
- 238000004557 single molecule detection Methods 0.000 description 1
- 239000004055 small Interfering RNA Substances 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229960004249 sodium acetate Drugs 0.000 description 1
- 229960002668 sodium chloride Drugs 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- JAJWGJBVLPIOOH-IZYKLYLVSA-M sodium taurocholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 JAJWGJBVLPIOOH-IZYKLYLVSA-M 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 229940035044 sorbitan monolaurate Drugs 0.000 description 1
- 230000037436 splice-site mutation Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000010907 stover Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 229940118376 tetanus toxin Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 229940021747 therapeutic vaccine Drugs 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000003151 transfection method Methods 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 108010064892 trkC Receptor Proteins 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229940055760 yervoy Drugs 0.000 description 1
Images














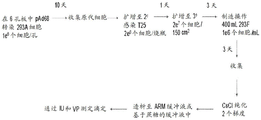













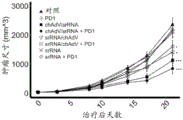
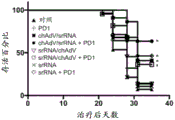
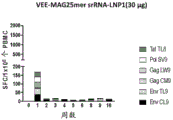
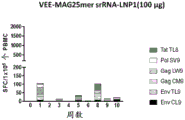
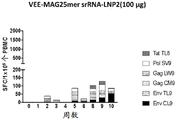

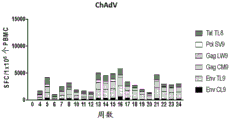
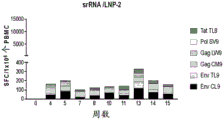



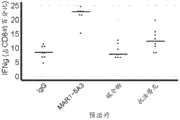

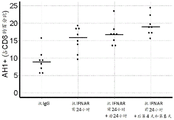

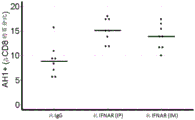
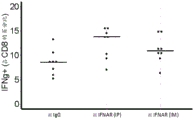










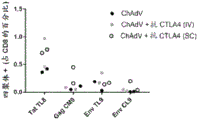


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
- A61K35/761—Adenovirus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/21—Retroviridae, e.g. equine infectious anemia virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4748—Tumour specific antigens; Tumour rejection antigen precursors [TRAP], e.g. MAGE
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2866—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for cytokines, lymphokines, interferons
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/58—Medicinal preparations containing antigens or antibodies raising an immune response against a target which is not the antigen used for immunisation
- A61K2039/585—Medicinal preparations containing antigens or antibodies raising an immune response against a target which is not the antigen used for immunisation wherein the target is cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/60—Medicinal preparations containing antigens or antibodies characteristics by the carrier linked to the antigen
- A61K2039/6031—Proteins
- A61K2039/6037—Bacterial toxins, e.g. diphteria toxoid [DT], tetanus toxoid [TT]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/60—Medicinal preparations containing antigens or antibodies characteristics by the carrier linked to the antigen
- A61K2039/6031—Proteins
- A61K2039/605—MHC molecules or ligands thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/62—Medicinal preparations containing antigens or antibodies characterised by the link between antigen and carrier
- A61K2039/627—Medicinal preparations containing antigens or antibodies characterised by the link between antigen and carrier characterised by the linker
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/70—Multivalent vaccine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/20—Fusion polypeptide containing a tag with affinity for a non-protein ligand
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10341—Use of virus, viral particle or viral elements as a vector
- C12N2710/10343—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16111—Cytomegalovirus, e.g. human herpesvirus 5
- C12N2710/16134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16211—Lymphocryptovirus, e.g. human herpesvirus 4, Epstein-Barr Virus
- C12N2710/16234—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/14011—Deltaretrovirus, e.g. bovine leukeamia virus
- C12N2740/14034—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/36011—Togaviridae
- C12N2770/36111—Alphavirus, e.g. Sindbis virus, VEE, EEE, WEE, Semliki
- C12N2770/36134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/36011—Togaviridae
- C12N2770/36111—Alphavirus, e.g. Sindbis virus, VEE, EEE, WEE, Semliki
- C12N2770/36141—Use of virus, viral particle or viral elements as a vector
- C12N2770/36143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Virology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Epidemiology (AREA)
- Biotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Wood Science & Technology (AREA)
- Oncology (AREA)
- Mycology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Biophysics (AREA)
- Communicable Diseases (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Pulmonology (AREA)
- Analytical Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Toxicology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
Abstract
本文公开了包括基于甲病毒的表达平台的载体。还公开了与基于甲病毒的表达平台和I型干扰素信号传导抑制剂的共同施用相关的方法。
Description
相关申请的交叉引用
本申请要求2018年11月7日提交的美国临时申请号62/756,980的权益,所述临时申请在此出于所有目的以引用的方式整体并入。
序列表
本申请含有序列表,其已经由EFS-Web提交并且在此以引用的方式整体并入本文。创建于2019年11月7日的所述ASCII拷贝,命名为GSO_023WO_sequencelisting.txt,并且大小为422,194个字节。
背景技术
基于肿瘤特异性抗原的治疗性疫苗作为新一代个性化癌症免疫疗法具有广阔的前景。1–3例如,鉴于产生新抗原的可能性相对较高,具有高突变负荷的癌症,诸如非小细胞肺癌(NSCLC)和黑素瘤成为此类疗法的特别值得关注的靶标。4,5早期有证据显示,基于新抗原的疫苗接种可引起T细胞反应6并且靶向新抗原的细胞疗法在某些情况下可在选择的患者中引起肿瘤消退。7
除了当前新抗原预测方法的挑战之外,可用于人中新抗原递送的可用载体系统也存在某些挑战,其中许多衍生自人。例如,许多人由于先前的自然暴露而对人病毒具有预先存在的免疫力,并且这种免疫力可能是使用重组人病毒进行新抗原递送以用于癌症治疗的主要障碍。
基于甲病毒的载体已用于递送平台。然而,对基于甲病毒的载体的免疫应答可能会降低作为递送平台的功效。
仍然需要改进的递送方法,特别是对基于甲病毒的载体而言,诸如对癌症疫苗而言。
发明内容
本文公开了一种用于刺激受试者中的免疫应答的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,其中用于递送所述表达系统的组合物包含所述表达系统,其中所述表达系统包含一个或多个载体,所述一个或多个载体包含:(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(poly(A))序列;以及(b)盒,其中所述盒包含:(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列以及c.任选地3'接头序列;(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
本文还公开了一种用于刺激受试者中的免疫应答的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,其中用于递送所述表达系统的组合物包含所述表达系统,其中所述表达系统包含一个或多个载体,所述一个或多个载体包含:(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含SEQID NO:6中示出的核酸序列,其中所述RNA甲病毒骨架序列包含26S启动子核苷酸序列和poly(A)序列,其中所述26S启动子序列是所述RNA甲病毒骨架的内源序列,并且其中所述poly(A)序列是所述RNA甲病毒骨架的内源序列;以及(b)在所述26S启动子核苷酸序列与所述poly(A)序列之间整合的盒,其中所述盒可操作地连接到所述26S启动子核苷酸序列,并且其中所述盒包含至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列,以及c.任选地3'接头序列;并且其中所述I型干扰素信号传导抑制剂包含抗IFNαβ受体(IFNAR)阻断抗体。
本文还公开了一种用于治疗患有癌症的受试者的方法,所述方法包括向所述受试者施用治疗有效量的用于递送表达系统的组合物以及向所述受试者施用治疗有效量的I型干扰素信号传导抑制剂,其中用于递送所述表达系统的组合物包含所述表达系统,其中所述表达系统包含一个或多个载体,所述一个或多个载体包含:(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(poly(A))序列;以及(b)盒,其中所述盒包含:(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使所述编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列,以及c.任选地3'接头序列;(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
本文还公开了一种用于减小受试者中的肿瘤体积的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,其中用于递送所述表达系统的组合物包含所述表达系统,其中所述表达系统包含一个或多个载体,所述一个或多个载体包含:(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(poly(A))序列;以及(b)盒,其中所述盒包含:(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列以及c.任选地3'接头序列;(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
本文还公开了一种用于刺激受试者中的肿瘤特异性免疫应答的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,其中用于递送所述表达系统的组合物包含所述表达系统,其中所述表达系统包含一个或多个载体,所述一个或多个载体包含:(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(poly(A))序列;以及(b)盒,其中所述盒包含:(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列以及c.任选地3'接头序列;(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
本文还公开了一种增强基于甲病毒的表达系统的递送的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,其中用于递送所述表达系统的组合物包含所述表达系统,其中所述表达系统包含一个或多个载体,所述一个或多个载体包含:(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(poly(A))序列;以及(b)盒,其中所述盒包含:(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使所述编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列以及c.任选地3'接头序列;(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
在一些方面,所述至少一个核酸序列包含所述多肽编码核酸序列。在一些方面,所述多肽编码核酸序列编码所述抗原编码核酸序列。在一些方面,所述抗原编码核酸序列是所述表位编码核酸序列。在一些方面,所述抗原编码核酸序列编码能够经历抗原加工成编码的表位的多肽序列。在一些方面,所述表位编码核酸序列编码已知或怀疑由MHC I类在细胞表面上递呈的表位,任选地其中所述细胞表面是肿瘤细胞表面或感染细胞表面,并且任选地其中所述细胞是所述受试者的细胞。在一些方面,所述细胞是选自由以下组成的组的肿瘤细胞:肺癌、黑素瘤、乳腺癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌,或者其中所述细胞是选自由以下组成的组的感染细胞:病原体感染细胞、病毒感染细胞、细菌感染细胞、真菌感染细胞和寄生虫感染细胞。在一些方面,所述病毒感染细胞是HIV感染细胞。
在一些方面,所述多肽编码核酸序列编码全长蛋白质或其功能部分。在一些方面,所述全长蛋白质或其功能部分选自由以下组成的组:抗体、细胞因子、嵌合抗原受体(CAR)、T细胞受体和基因组编辑系统核酸酶。
在一些方面,所述至少一个核酸序列包含非编码核酸序列。在一些方面,所述非编码核酸序列是RNA干扰(RNAi)多核苷酸或基因组编辑系统多核苷酸。
在一些方面,所述盒包含:i)包含所述多肽编码核酸序列的所述至少一个核酸序列,其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,b.任选地5'接头序列以及c.任选地3'接头序列;(ii)任选地,可操作地连接到所述抗原编码核酸序列的第二启动子核苷酸序列;(iii)任选地,至少一个MHC II类抗原编码核酸序列;(iv)任选地,至少一个编码GPGPG氨基酸接头序列(SEQ ID NO:56)的核酸序列;以及(v)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。在一些方面,所述盒的各元件的有序序列以下式描述,其从5'至3'包含
Pa-(L5b-Nc-L3d)X-(G5e-Uf)Y-G3g
其中P包含所述第二启动子核苷酸序列,其中a=0或1,N包含所述表位编码核酸序列中的一个,其中所述表位编码核酸序列包含MHC I类表位编码核酸序列,其中c=1,L5包含所述5'接头序列,其中b=0或1,L3包含所述3'接头序列,其中d=0或1,G5包含所述至少一个编码GPGPG氨基酸接头的核酸序列中的一个,其中e=0或1,G3包含所述至少一个编码GPGPG氨基酸接头的核酸序列中的一个,其中g=0或1,U包含所述至少一个MHC II类表位编码核酸序列中的一个,其中f=1,X=1至400,其中对于每个X,对应的Nc是表位编码核酸序列,并且Y=0、1或2,其中对于每个Y,对应的Uf是表位编码核酸序列。在一些方面,对于每个X,对应的Nc是不同的MHC I类表位编码核酸序列。在一些方面,对于每个Y,对应的Uf是不同的MHC II类表位编码核酸序列。在一些方面,a=0,b=1,d=1,e=1,g=1,h=1,X=20,Y=2,所述至少一个启动子核苷酸序列是由所述RNA甲病毒骨架提供的单个26S启动子核苷酸序列,所述至少一个聚腺苷酸化poly(A)序列是由所述RNA甲病毒骨架提供的至少100个连续A核苷酸的poly(A)序列,每个N编码长度为7-15个氨基酸的MHC I类表位,L5是编码MHC I表位的原生N末端氨基酸序列的原生5'接头序列,并且其中所述5'接头序列编码长度为至少3个氨基酸的肽,L3是编码MHC I表位的原生C末端氨基酸序列的原生3'接头序列,并且其中所述3'接头序列编码长度为至少3个氨基酸的肽,U是PADRE II类序列和破伤风类毒素MHC II类序列中的一种,所述RNA甲病毒骨架是SEQ ID NO:6中示出的序列,并且所述MHC I类表位编码核酸序列中的一种编码长度为13至25个氨基酸的多肽。
在一些方面,用于递送所述表达系统的所述组合物还包含纳米颗粒递送媒介物。在一些方面,所述纳米颗粒递送媒介物是脂质纳米粒子(LNP)。在一些方面,所述LNP包含可电离的氨基脂质。在一些方面,所述可电离的氨基脂质包含MC3样(二亚油基甲基-4-二甲基氨基丁酸酯)分子。在一些方面,所述纳米颗粒递送媒介物包封所述表达系统。
在一些方面,用于递送所述表达系统的所述组合物还包含多个LNP,其中所述LNP包含:新抗原表达系统;阳离子脂质;非阳离子脂质;以及抑制LNP聚集的缀合脂质,其中所述多个LNP中至少约95%的LNP:具有非层状形态;或者是电子致密的。在一些方面,所述非阳离子脂质是(1)磷脂和(2)胆固醇或胆固醇衍生物的混合物。
在一些方面,所述抑制LNP聚集的缀合脂质是聚乙二醇(PEG)-脂质缀合物。在一些方面,所述PEG-脂质缀合物选自由以下组成的组:PEG-二酰基甘油(PEG-DAG)缀合物、PEG二烷氧基丙基(PEG-DAA)缀合物、PEG-磷脂缀合物、PEG-神经酰胺(PEG-Cer)缀合物及其混合物。在一些方面,所述PEG-DAA缀合物是选自由以下组成的组的成员:PEG-二癸氧基丙基(C10)缀合物、PEG-二月桂氧基丙基(C12)缀合物、PEG-二肉豆蔻酰氧基丙基(C14)缀合物、PEG-二棕榈酰氧基丙基(C16)缀合物、PEG-二硬脂酰氧基丙基(C18)缀合物及其混合物。
在一些方面,用于递送所述表达系统的所述组合物完全包封在LNP中。
在一些方面,LNP的非层状形态包括反六角形(HII)或立方相结构。
在一些方面,所述阳离子脂质占LNP中存在的总脂质的约10mol%至约50mol%。在一些方面,所述阳离子脂质占LNP中存在的总脂质的约20mol%至约50mol%。在一些方面,所述阳离子脂质占LNP中存在的总脂质的约20mol%至约40mol%。
在一些方面,所述非阳离子脂质占LNP中存在的总脂质的约10mol%至约60mol%。在一些方面,所述非阳离子脂质占LNP中存在的总脂质的约20mol%至约55mol%。在一些方面,所述非阳离子脂质占LNP中存在的总脂质的约25mol%至约50mol%。
在一些方面,所述缀合脂质占LNP中存在的总脂质的约0.5mol%至约20mol%。在一些方面,所述缀合脂质占LNP中存在的总脂质的约2mol%至约20mol%。在一些方面,所述缀合脂质占LNP中存在的总脂质的约1.5mol%至约18mol%。
在一些方面,大于95%的LNP具有非层状形态。在一些方面,大于95%的LNP是电子致密的。
在一些方面,用于递送所述表达系统的所述组合物还包含多个LNP,其中所述LNP包含:阳离子脂质,其占LNP中存在的总脂质的50mol%至65mol%;抑制LNP聚集的缀合脂质,其占LNP中存在的总脂质的0.5mol%至2mol%;以及非阳离子脂质,其包含:磷脂和胆固醇或其衍生物的混合物,其中所述磷脂占LNP中存在的总脂质的4mol%至10mol%,并且所述胆固醇或其衍生物占LNP中存在的总脂质的30mol%至40mol%;磷脂和胆固醇或其衍生物的混合物,其中所述磷脂占LNP中存在的总脂质的3mol%至15mol%,并且所述胆固醇或其衍生物占LNP中存在的总脂质的30mol%至40mol%;或LNP中存在的总脂质的高达49.5mol%,并包含磷脂和胆固醇或其衍生物的混合物,其中所述胆固醇或其衍生物占LNP中存在的总脂质的30mol%至40mol%。
在一些方面,用于递送所述表达系统的所述组合物还包含多个LNP,其中所述LNP包含:阳离子脂质,其占LNP中存在的总脂质的50mol%至85mol%;抑制LNP聚集的缀合脂质,其占LNP中存在的总脂质的0.5mol%至2mol%;以及非阳离子脂质,其占LNP中存在的总脂质的13mol%至49.5mol%。
在一些方面,所述磷脂包含二棕榈酰磷脂酰胆碱(DPPC)、二硬脂酰磷脂酰胆碱(DSPC)或其混合物。
在一些方面,所述缀合脂质包含聚乙二醇(PEG)-脂质缀合物。在一些方面,所述PEG-脂质缀合物包含PEG-二酰基甘油(PEG-DAG)缀合物、PEG-二烷氧基丙基(PEG-DAA)缀合物或其混合物。在一些方面,所述PEG-DAA缀合物包含PEG-二肉豆蔻酰氧基丙基(PEG-DMA)缀合物、PEG-二硬脂酰氧基丙基(PEG-DSA)缀合物或其混合物。在一些方面,所述缀合物的PEG部分具有约2,000道尔顿(dalton)的平均分子量。
在一些方面,所述缀合脂质占LNP中存在的总脂质的约1mol%至约2mol%。
在一些方面,所述LNP包含具有式I结构的化合物:

或其药学上可接受的盐、互变异构体、前药或立体异构体,其中:L1和L2各自独立地是-O(C=O)-、-(C=O)O-、-C(=O)-、-O-、-S(O)x-、-S-S-、-C(=O)S-、-SC(=O)-、-RaC(=O)-、-C(=O)Ra-、-RaC(=O)Ra-、-OC(=O)Ra-、-RaC(=O)O-或直接键;G1是Ci-C2亚烷基、-(C=O)-、-O(C=O)-、-SC(=O)-、-RaC(=O)-或直接键;-C(=O)-、-(C=O)O-、-C(=O)S-、-C(=O)Ra-或直接键;G是Ci-C6亚烷基;Ra是H或C1-C12烷基;Rla和R1b在每次出现时独立地是:(a)H或C1-C12烷基;或(b)R1a是H或C1-C12烷基,并且R1b和其所键合的碳原子一起与相邻R1b和其所键合的碳原子一起形成碳-碳双键;R2a和R2b在每次出现时独立地是:(a)H或C1-C12烷基;或(b)R2a是H或C1-C12烷基,并且R2b和其所键合的碳原子一起与相邻R2b和其所键合的碳原子一起形成碳-碳双键;R3a和R3b在每次出现时独立地是:(a)H或C1-C12烷基;或(b)R3a是H或C1-C12烷基,并且R3b和其所键合的碳原子一起与相邻R和其所键合的碳原子一起形成碳-碳双键;R4a和R4b在每次出现时独立地是:(a)H或C1-C12烷基;或(b)R4a是H或C1-C12烷基,并且R4b和其所键合的碳原子一起与相邻R4b和其所键合的碳原子一起形成碳-碳双键;R5和R6各自独立地是H或甲基;R7是C4-C20烷基;R8和R9各自独立地是C1-C12烷基;或者R8和R9与其所连接的氮原子一起形成5元、6元或7元杂环;a、b、c和d各自独立地是1至24的整数;并且x是0、1或2。
在一些方面,所述LNP包含具有式II结构的化合物:

或其药学上可接受的盐、互变异构体、前药或立体异构体,其中:L1和L2各自独立地是-O(C=O)-、-(C=O)O-或碳-碳双键;R1a和R1b在每次出现时独立地是(a)H或C1-C12烷基,或(b)R1a是H或C1-C12烷基,并且R1b和其所键合的碳原子一起与相邻R1b和其所键合的碳原子一起形成碳-碳双键;R2a和R2b在每次出现时独立地是(a)H或C1-C12烷基,或(b)R2a是H或C1-C12烷基,并且R2b和其所键合碳原子一起与相邻R2b和其所键合的碳原子一起形成碳-碳双键;R3a和R3b在每次出现时独立地是(a)H或C1-C12烷基,或(b)R3a是H或C1-C12烷基,并且R3b和其所键合的碳原子一起与相邻R3b和其所键合的碳原子一起形成碳-碳双键;R4a和R4b在每次出现时独立地是(a)H或C1-C12烷基,或(b)R4a是H或C1-C12烷基,并且R4b和其所键合的碳原子一起与相邻R4b和其所键合的碳原子一起形成碳碳双键;R5和R6各自独立地是甲基或环烷基;R7在每次出现时独立地是H或C1-C12烷基;R8和R9各自独立地是未被取代的C1-C12烷基;或者R8和R9与其所连接的氮原子一起形成包含一个氮原子的5元、6元或7元杂环;a和d各自独立地是0至24的整数;b和c各自独立地是1至24的整数;并且e是1或2,其条件是:R1a、R2a、R3a或R4a中的至少一者是C1-C12烷基,或者L1或L2中的至少一者是-O(C=O)-或-(C=O)O-;并且R1a和R1b当a为6时不是异丙基或者当a为8时不是正丁基。
在一些方面,任何上述组合物还包含一种或多种赋形剂,所述赋形剂包含中性脂质、类固醇和聚合物缀合脂质。在一些方面,所述中性脂质包含1,2-二硬脂酰基-sn-甘油-3-磷酸胆碱(DSPC)、1,2-二棕榈酰基-sn-甘油-3-磷酸胆碱(DPPC)、1,2-二肉豆蔻酰基-sn-甘油-3-磷酸胆碱(DMPC)、1-棕榈酰基-2-油酰基-sn-甘油-3-磷酸胆碱(POPC)、1,2-二油酰基-sn-甘油-3-磷酸胆碱(DOPC)和1,2-二油酰基-sn-甘油-3-磷酸乙醇胺(DOPE)中的至少一者。在一些方面,所述中性脂质是DSPC。
在一些方面,所述化合物与所述中性脂质的摩尔比在约2:1至约8:1的范围内。
在一些方面,所述类固醇是胆固醇。在一些方面,所述化合物与胆固醇的摩尔比在约2:1至1:1的范围内。
在一些方面,所述聚合物缀合脂质是聚乙二醇化脂质。在一些方面,所述化合物与所述聚乙二醇化脂质的摩尔比在约100:1至约25:1的范围内。在一些方面,所述聚乙二醇化脂质是PEG-DAG、PEG聚乙烯(PEG-PE)、PEG-琥珀酰基-二酰基甘油(PEG-S-DAG)、PEG-cer或PEG二烷氧基丙基氨基甲酸酯。在一些方面,所述聚乙二醇化脂质具有以下结构III:

或其药学上可接受的盐、互变异构体或立体异构体,其中:R10和R11各自独立地是含有10至30个碳原子的直链或支链的饱和或不饱和的烷基链,其中所述烷基链任选地被一个或多个酯键中断;并且z的平均值在30至60的范围内。在一些方面,R10和R11各自独立地是具有12至16个碳原子的直链的饱和烷基链。在一些方面,平均z是约45。
在一些方面,LNP在与聚阴离子核酸混合时自组装成非双层结构。在一些方面,所述非双层结构的直径在60nm与120nm之间。在一些方面,所述非双层结构的直径为约70nm、约80nm、约90nm或约100nm。在一些方面,其中所述纳米颗粒递送媒介物直径为约100nm。
在一些方面,I型干扰素信号传导抑制剂选自由以下组成的组:IFNα抑制剂、IFNβ抑制剂、IFNAR抑制剂和I型干扰素信号传导途径抑制剂。在一些方面,I型干扰素信号传导抑制剂选自由以下组成的组:抗体或其抗原结合片段、小分子抑制剂、RNAi多核苷酸、基因组编辑系统和Fc融合蛋白。在一些方面,抗体选自由以下组成的组:抗IFNα抗体、抗IFNβ抗体、抗IFNαβ受体(IFNAR)阻断抗体。在一些方面,所述抗IFNα抗体选自由以下组成的组:西法鲁单抗(Sifalumumab)、贝那利珠单抗(Rontalizumab)和ASG-009。在一些方面,所述抗IFNAR阻断抗体选自由以下组成的组:MAR1-5A3、阿尼福鲁单抗(Anifrolumab)、AmS3A5-1、64G12、H2K6、H2K1、H3K6、H3K1 3F11、4G5、11E2和9D4。在一些方面,I型干扰素信号传导途径抑制剂包含JAK激酶抑制剂。在一些方面,所述JAK激酶抑制剂包含小分子。在一些方面,所述JAK激酶抑制剂包含JAK1/2抑制剂或JAK1/3抑制剂。在一些方面,所述JAK1/3抑制剂是托法替尼(Tofacitinib)。
在一些方面,I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物之前、同时或之后施用。在一些方面,I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物前24小时或更短时间施用。在一些方面,I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物后少于12小时施用。在一些方面,I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物后6小时或更短时间施用。在一些方面,I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物前24小时与施用后6小时或更短时间之间施用。
在一些方面,用于递送所述表达系统的所述组合物经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。在一些方面,用于递送所述表达系统的所述组合物经肌肉内(IM)施用。
在一些方面,I型干扰素信号传导抑制剂经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。在一些方面,I型干扰素信号传导抑制剂经肌肉内(IM)施用。在一些方面,I型干扰素信号传导抑制剂经静脉内(IV)施用。
在一些方面,施用I型干扰素信号传导抑制剂的单次施用。
在一些方面,所述盒整合在所述至少一个启动子核苷酸序列与所述至少一个poly(A)序列之间。在一些方面,所述至少一个启动子核苷酸序列可操作地连接到所述盒。
在一些方面,所述一个或多个载体包含一个或多个+-链RNA载体。在一些方面,所述一个或多个+-链RNA载体包含5'7-甲基鸟苷(m7g)帽。在一些方面,所述一个或多个+-链RNA载体通过体外转录来产生。
在一些方面,所述一个或多个载体在哺乳动物细胞内自我复制。
在一些方面,所述RNA甲病毒骨架包含奥拉病毒(Aura virus)、摩根堡病毒(FortMorgan virus)、委内瑞拉马脑炎病毒(Venezuelan equine encephalitis virus)、罗斯河病毒(Ross River virus)、塞姆利基森林病毒(Semliki Forest virus)、辛德毕斯病毒(Sindbis virus)或马雅鲁病毒(Mayaro virus)中的至少一个核苷酸序列。在一些方面,所述RNA甲病毒骨架包含委内瑞拉马脑炎病毒的至少一个核苷酸序列。在一些方面,所述RNA甲病毒骨架包含至少用于非结构蛋白介导扩增的序列,26S启动子序列,poly(A)序列,由奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒的核苷酸序列编码的非结构蛋白1(nsP1)基因、nsP2基因、nsP3基因和nsP4基因。在一些方面,所述RNA甲病毒骨架包含至少用于非结构蛋白介导扩增的序列,26S启动子序列以及由奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒的核苷酸序列编码的poly(A)序列。在一些方面,用于非结构蛋白介导扩增的序列选自由以下组成的组:甲病毒5'UTR、51-nt CSE、24-nt CSE、26S亚基因组启动子序列、19-nt CSE、甲病毒3'UTR或其组合。在一些方面,所述RNA甲病毒骨架不编码结构病毒粒子蛋白衣壳E2和E1。在一些方面,插入所述盒以代替奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒的核苷酸序列内的结构病毒粒子蛋白。
在一些方面,委内瑞拉马脑炎病毒包含SEQ ID NO:3或SEQ ID NO:5的序列。在一些方面,委内瑞拉马脑炎病毒包含SEQ ID NO:3或SEQ ID NO:5的序列,其还包含碱基对7544与11175之间的缺失。在一些方面,所述RNA甲病毒骨架包含SEQ ID NO:6或SEQ ID NO:7中示出的序列。在一些方面,在位置7544处插入所述盒以代替如SEQ ID NO:3或SEQ IDNO:5的序列中所示的碱基对7544与11175之间的缺失。在一些方面,所述盒的插入提供了包含所述nsP1-4基因和所述至少一个核酸序列的多顺反子RNA的转录,其中所述nsP1-4基因和所述至少一个核酸序列在单独的开放阅读框中。
在一些方面,所述至少一个启动子核苷酸序列是由所述RNA甲病毒骨架编码的原生26S启动子核苷酸序列。在一些方面,所述至少一个启动子核苷酸序列是外源RNA启动子。在一些方面,第二启动子核苷酸序列是26S启动子核苷酸序列。在一些方面,第二启动子核苷酸序列包含多个26S启动子核苷酸序列,其中每个26S启动子核苷酸序列提供一个或多个单独的开放阅读框的转录。
在一些方面,所述一个或多个载体的大小各自为至少300nt。在一些方面,所述一个或多个载体的大小各自为至少1kb。在一些方面,所述一个或多个载体的大小各自为2kb。在一些方面,所述一个或多个载体的大小各自小于5kb。
在一些方面,至少一个表位编码核酸序列编码表位,所述表位在表达和翻译时能够由MHC I类在所述受试者的细胞上递呈。在一些方面,至少一个表位编码核酸序列编码表位,所述表位在表达和翻译时能够由MHC II类在所述受试者的细胞上递呈。
在一些方面,所述至少一个核酸序列包含两个或更多个核酸序列。在一些方面,所述至少一个核酸序列包含两个或更多个多肽编码核酸序列。在一些方面,每个多肽编码核酸序列彼此直接连接。
在一些方面,每个多肽编码核酸序列连接到具有编码接头的核酸序列的不同多肽编码核酸序列。在一些方面,所述多肽编码核酸序列是抗原编码核酸序列,并且其中所述接头连接两个MHC I类表位编码核酸序列或将MHC I类表位编码核酸序列连接到MHC II类表位编码核酸序列。在一些方面,所述接头选自由以下组成的组:(1)连续甘氨酸残基,长度为至少2、3、4、5、6、7、8、9或10个残基;(2)连续丙氨酸残基,长度为至少2、3、4、5、6、7、8、9或10个残基;(3)两个精氨酸残基(RR);(4)丙氨酸、丙氨酸、酪氨酸(AAY);(5)长度为至少2、3、4、5、6、7、8、9或10个氨基酸残基的共有序列,其由哺乳动物蛋白酶体有效加工;以及(6)侧接衍生自同源蛋白质来源的抗原并且长度为至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或2-20个氨基酸残基的一个或多个原生序列。在一些方面,所述多肽编码核酸序列是抗原编码核酸序列,并且其中所述接头连接两个MHC II类表位编码核酸序列或将MHC II类表位编码核酸序列连接到MHC I类表位编码核酸序列。在一些方面,所述接头包含序列GPGPG。
在一些方面,所述多肽编码核酸序列是抗原编码核酸序列,并且其中所述抗原编码核酸序列可操作地或直接地连接到单独或连续的序列,所述单独或连续的序列增强所述抗原编码核酸序列的表达、稳定性、细胞运输、加工和递呈和/或免疫原性。在一些方面,所述单独或连续的序列包含以下中的至少一者:泛素序列、经修饰以增加蛋白酶体靶向的泛素序列(例如,泛素序列在位置76处含有Gly至Ala取代)、免疫球蛋白信号序列(例如IgK)、主要组织相容性I类序列、溶酶体相关膜蛋白(LAMP)-1、人树突状细胞溶酶体相关膜蛋白和主要组织相容性II类序列;任选地其中经修饰以增加蛋白酶体靶向的泛素序列是A76。
在一些方面,表位编码核酸序列包含至少一个改变,所述改变使编码的表位相对于翻译的对应野生型核酸序列对其对应MHC等位基因的结合亲和力增加。在一些方面,表位编码核酸序列包含至少一个改变,所述改变使编码的表位相对于翻译的对应野生型核酸序列对其对应MHC等位基因的结合稳定性增加。在一些方面,表位编码核酸序列包含至少一个改变,所述改变使编码的表位相对于翻译的对应野生型核酸序列对其对应MHC等位基因递呈的可能性增加。在一些方面,所述至少一个改变包括点突变、移码突变、非移码突变、缺失突变、插入突变、剪接变体、基因组重排或蛋白酶体产生的剪接抗原。
在一些方面,已知或怀疑所述受试者患有癌症。在一些方面,刺激免疫应答治疗所述癌症。在一些方面,所述癌症选自由以下组成的组:肺癌、黑素瘤、乳腺癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、膀胱癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、成人急性淋巴母细胞性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
在一些方面,所述受试者患有一种或多种肿瘤。在一些方面,刺激免疫应答减小所述一种或多种肿瘤的肿瘤体积。
在一些方面,所述至少一个核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个核酸序列,任选地其中每个核酸序列编码不同的非编码核酸序列、不同的多肽编码核酸序列或其组合。在一些方面,所述至少一个核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个核酸序列,任选地其中每个核酸序列编码不同的非编码核酸序列、不同的多肽编码核酸序列或其组合。在一些方面,在一些方面,所述至少一个核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个多肽编码核酸序列。在一些方面,所述至少一个核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个多肽编码核酸序列。在一些方面,所述至少一个核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个抗原编码核酸序列。在一些方面,所述至少一个核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个抗原编码核酸序列。在一些方面,所述至少一个核酸序列包含至少2-400个抗原编码核酸序列,并且其中所述抗原编码核酸序列中的至少两者编码细胞表面上由MHC I类递呈的多肽序列或其部分。在一些方面,MHC I类表位中的至少两者由MHC I类在肿瘤细胞表面上递呈。
在一些方面,当向所述受试者施用并被翻译时,由所述表位编码核酸序列编码的表位中的至少一者被递呈在抗原递呈细胞上,产生靶向递呈细胞表面上表位中的至少一者的细胞的免疫应答。
在一些方面,所述表位编码核酸序列包含至少一个MHC I类表位编码核酸序列或MHC II类表位编码核酸序列,并且当向所述受试者施用并被翻译时,MHC I类或II类表位中的至少一者被递呈在抗原递呈细胞上,产生靶向递呈细胞表面上表位中的至少一者的细胞的免疫应答,并且任选地其中MHC I类和/或II类表位编码核酸序列中的每一者的表达由所述至少一个启动子核苷酸序列驱动。
在一些方面,所述表位编码核酸序列包含至少一个MHC I类表位编码核酸序列,并且其中每个抗原编码核酸序列编码长度为8至35个氨基酸,任选地长度为9-17、9-25、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35个氨基酸的多肽序列。
在一些方面,存在所述至少一个MHC II类表位编码核酸序列。在一些方面,存在所述至少一个MHC II类表位编码核酸序列并且其包含至少一个MHC II类表位编码核酸序列,所述序列包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变。
在一些方面,所述表位编码核酸序列包含MHC II类表位编码核酸序列,并且其中每个抗原编码核酸序列编码长度为12-20、12、13、14、15、16、17、18、19、20或20-40个氨基酸的多肽序列。
在一些方面,所述表位编码核酸序列包含MHC II类表位编码核酸序列,其中存在所述至少一个MHC II类表位编码核酸序列,并且其中所述至少一个MHC II类表位编码核酸序列包含至少一个通用MHC II类表位编码核酸序列,任选地其中所述至少一个通用序列包含破伤风类毒素和PADRE中的至少一者。
在一些方面,所述至少一个启动子核苷酸序列或第二启动子核苷酸序列是诱导型的。在一些方面,所述至少一个启动子核苷酸序列或第二启动子核苷酸序列是非诱导型的。
在一些方面,至少一个poly(A)序列包含甲病毒原生的poly(A)序列。在一些方面,至少一个poly(A)序列包含甲病毒外源的poly(A)序列。在一些方面,至少一个poly(A)序列可操作地连接到所述至少一个核酸序列中的至少一者。在一些方面,至少一个poly(A)序列是至少20个、至少30个、至少40个、至少50个、至少60个、至少70个、至少80个或至少90个连续A核苷酸。在一些方面,至少一个poly(A)序列是至少100个连续A核苷酸。
在一些方面,所述盒还包含以下中的至少一者:内含子序列、土拨鼠肝炎病毒转录后调控元件(WPRE)序列、内部核糖体进入序列(IRES)序列、编码2A自裂解肽序列的核苷酸序列、编码弗林蛋白酶(Furin)裂解位点的核苷酸序列、或已知增强可操作地连接到所述至少一个核酸序列中的至少一者的mRNA的核输出、稳定性或翻译效率的5'或3'非编码区中的序列。
在一些方面,所述盒还包含报告基因,其包括但不限于绿色荧光蛋白(GFP)、GFP变体、分泌型碱性磷酸酶、荧光素酶、荧光素酶变体或可检测的肽或表位。在一些方面,所述可检测的肽或表位选自由HA标签、Flag标签、His标签或V5标签组成的组。
在一些方面,所述一个或多个载体还包含编码至少一种免疫调节物的一个或多个核酸序列。在一些方面,所述免疫调节物是抗CTLA4抗体或其抗原结合片段、抗PD-1抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。在一些方面,所述抗体或其抗原结合片段是Fab片段、Fab'片段、单链Fv(scFv)、呈单特异性或连接在一起的多特异性的单结构域抗体(sdAb)(例如骆驼科抗体结构域)或全长单链抗体(例如具有通过柔性接头连接的重链和轻链的全长IgG)。在一些方面,所述抗体的重链和轻链序列是由自裂解序列诸如2A或IRES分开的连续序列;或所述抗体的重链和轻链序列由柔性接头诸如连续甘氨酸残基连接。在一些方面,所述免疫调节物是细胞因子。在一些方面,所述细胞因子是IL-2、IL-7、IL-12、IL-15或IL-21或其各自变体中的至少一者。
在一些方面,所述表位编码核酸序列包含MHC I类表位编码核酸序列,并且其中所述MHC I类表位编码核酸序列通过进行以下步骤来选择:(a)获得来自肿瘤的外显子组、转录组或全基因组肿瘤核苷酸测序数据中的至少一者,其中所述肿瘤核苷酸测序数据用于获得代表表位集合中的每一者的肽序列的数据;(b)将每个表位的肽序列输入到递呈模型中,以产生表位中的每一者在肿瘤的肿瘤细胞表面上由一个或多个MHC等位基因递呈的数值可能性集合,所述数值可能性集合已至少基于所接收的质谱数据鉴定;以及(c)基于所述数值可能性集合选择表位集合的子集,以产生经选择的表位集合,其用于产生所述MHC I类表位编码核酸序列。
在一些方面,所述MHC I类表位编码核酸序列中的每一者通过进行以下步骤来选择:(a)获得来自肿瘤的外显子组、转录组或全基因组肿瘤核苷酸测序数据中的至少一者,其中所述肿瘤核苷酸测序数据用于获得代表表位集合中的每一者的肽序列的数据;(b)将每个表位的肽序列输入到递呈模型中,以产生表位中的每一者在肿瘤的肿瘤细胞表面上由一个或多个MHC等位基因递呈的数值可能性集合,所述数值可能性集合已至少基于所接收的质谱数据鉴定;以及(c)基于所述数值可能性集合选择表位集合的子集,以产生经选择的表位集合,其用于产生至少20个MHC I类表位编码核酸序列。在一些方面,经选择的表位集合的数量为2-20个。在一些方面,递呈模型表示以下两者之间的依赖性:(a)MHC等位基因中的一对特定等位基因和在肽序列特定位置处特定氨基酸的存在;以及(b)与在肿瘤细胞表面上由所述对的MHC等位基因中的特定等位基因递呈在特定位置处包含特定氨基酸的此类肽序列的可能性。
在一些方面,选择经选择的表位集合包括基于递呈模型,选择相对于未经选择的表位在肿瘤细胞表面上递呈的可能性增加的表位。在一些方面,选择经选择的表位集合包括基于递呈模型,选择相对于未经选择的表位能够诱导受试者中的肿瘤特异性免疫应答的可能性增加的表位。在一些方面,选择经选择的表位集合包括基于递呈模型,选择相对于未经选择的表位能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加的表位,任选地其中所述APC是树突状细胞(DC)。在一些方面,选择经选择的表位集合包括基于递呈模型,选择相对于未经选择的表位经由中心或外周耐受性受抑制的可能性降低的表位。在一些方面,选择经选择的表位集合包括基于递呈模型,选择相对于未经选择的表位能够诱导受试者中针对正常组织的自身免疫应答的可能性降低的表位。在一些方面,外显子组或转录组核苷酸测序数据通过对肿瘤组织进行测序而获得。在一些方面,测序是下一代测序(NGS)或任何大规模平行测序方法。
在一些方面,所述盒包含由所述盒中的相邻序列形成的连接表位序列。在一些方面,至少一个或每个连接表位序列对MHC的亲和力大于500nM。在一些方面,每个连接表位序列是非自身的。
在一些方面,所述盒不编码包含翻译的野生型核酸序列的非治疗性MHC I类或II类表位核酸序列,其中所述非治疗性表位经预测显示在受试者的MHC等位基因上。在一些方面,经预测的非治疗性MHC I类或II类表位序列是由所述盒中的相邻序列形成的连接表位序列。在一些方面,预测是基于通过将非治疗性表位的序列输入到递呈模型中而产生的递呈可能性。在一些方面,所述盒中抗原编码核酸序列的顺序通过包括以下的一系列步骤来确定:(a)产生对应于所述抗原编码核酸序列的不同顺序的候选盒序列集合;(b)对于每个候选盒序列,基于所述候选盒序列中非治疗性表位的递呈来确定递呈评分;以及(c)选择与低于预定阈值的递呈评分相关的候选盒序列作为用于疫苗的所述盒序列。
在一些方面,用于递送所述表达系统的所述组合物和/或I型干扰素信号传导抑制剂被配制在包含药学上可接受的载剂的药物组合物中。在一些方面,所述方法还包括施用佐剂。
本文还公开了一种试剂盒,其包含本文所述的任何方法的用于递送所述表达系统的所述组合物和I型干扰素信号传导抑制剂,以及使用说明。
在一些方面,衍生的表位编码核酸序列衍生自受试者的肿瘤。在一些方面,所述表位编码核酸序列不衍生自受试者的肿瘤。
在一些方面,所述方法还包括施用一种或多种免疫调节物,任选地其中所述免疫调节物在施用用于递送所述表达系统的所述组合物和/或I型干扰素信号传导抑制剂或其药物组合物之前、同时或之后施用。在一些方面,所述一种或多种免疫调节物选自由以下组成的组:抗CTLA4抗体或其抗原结合片段、抗PD-1抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。在一些方面,所述免疫调节物经静脉内(IV)、肌肉内(IM)、皮内(ID)或皮下(SC)施用。在一些方面,皮下施用是在所述表达系统施用部位的部位附近或者靠近所述表达系统的一个或多个引流淋巴结。
在一些方面,所述方法还包括向所述受试者施用第二疫苗组合物。在一些方面,所述第二疫苗组合物在施用用于递送所述表达系统的所述组合物和/或I型干扰素信号传导抑制剂或其药物组合物之前施用。在一些方面,所述第二疫苗组合物在施用用于递送所述表达系统的所述组合物和/或I型干扰素信号传导抑制剂或其药物组合物之后施用。在一些方面,所述第二疫苗组合物与用于递送所述表达系统的所述组合物或其药物组合物相同。在一些方面,所述第二疫苗组合物不同于用于递送所述表达系统的所述组合物或其药物组合物。在一些方面,所述第二疫苗组合物包含编码至少一个抗原编码核酸序列的黑猩猩腺病毒载体。在一些方面,由黑猩猩腺病毒载体编码的至少一个抗原编码核酸序列与如上述方法权利要求中任一项所述的抗原编码核酸序列相同。在一些方面,I型干扰素信号传导抑制剂或其药物组合物的第二次施用在施用所述第二疫苗组合物之前、同时或之后。
附图说明
参照以下描述和附图将更好地理解本发明的这些和其他特征、方面和优势,在附图中:
图(图)1示出了体外T细胞活化分析的开发。示意性展示所述分析,其中将疫苗盒递送到抗原递呈细胞引起不同肽抗原的表达、表位加工和MHC限制性递呈。经工程改造成具有匹配特定肽-MHC组合的T细胞受体的报告T细胞经活化,引起荧光素酶表达。
图2A示出了对短盒中接头序列的评估并且示出了相对于彼此相同的位置中串接的五个I类MHC限制性表位(表位1至5),接着是两个通用II类MHC表位(MHC-II)。使用不同接头产生各种迭代。在一些情况下,T细胞表位彼此直接连接。在其他情况下,T细胞表位侧接于其天然序列的一侧或两侧上。在其他迭代中,T细胞表位由非天然序列AAY、RR和DPP连接。
图2B示出了对短盒中接头序列的评估并且示出了关于嵌入所述短盒中的T细胞表位的序列信息。
图3示出了对加入到模型疫苗盒中的细胞靶向序列的评估。所述靶向盒用泛素(Ub)、信号肽(SP)和/或跨膜(TM)结构域延伸所述短盒设计,特征在于紧邻五个标志物人T细胞表位(表位1至5)以及两个小鼠T细胞表位SIINFEKL(SII)和SPSYAYHQF(A5),并且使用非天然接头AAY-或天然接头侧接两侧上的T细胞表位(25聚体)。
图4示出了对短盒中接头序列的体内评估。A)使用HLA-A2转基因小鼠进行疫苗盒的体内评估的实验设计。
图5A示出了对21聚体长盒中表位位置的影响的体内评估并且示出了长盒的设计需要用25聚体天然序列中所包含的另外的熟知T细胞I类表位(表位6至21)隔开的包含在25聚体天然序列中的五个标志物I类表位(表位1至5)(接头=天然侧接序列),以及两个通用II类表位(MHC-II0,其中仅I类表位的相对位置改变。
图5B示出了对21聚体长盒中表位位置的影响的体内评估并且示出了关于所用T细胞表位的序列信息。
图6A示出了临床前IND授权研究(IND-enabling study)的最终盒设计并且示出了最终盒的设计包含在25聚体天然序列中所包含的20个MHC I表位(接头=天然侧接序列)以及2个通用MHC II类表位,所述20个MHC I表位由6个非人灵长类动物(NHP)表位、5个人表位、9个鼠类表位构成。
图6B示出了临床前IND授权研究的最终盒设计并且示出了递呈在非人灵长类动物、小鼠和人来源的I类MHC上的所用T细胞表位的序列信息,以及2个通用MHC II类表位PADRE和破伤风类毒素的序列。
图7A示出了在转染之后产生ChAdV68.4WTnt.GFP病毒。使用磷酸钙方案,用ChAdV68.4WTnt.GFP DNA转染HEK293A细胞。在转染之后第10天观察到病毒复制,并且使用光学显微镜(40×放大倍率)观测到ChAdV68.4WTnt.GFP病毒斑块。
图7B示出了在转染之后产生ChAdV68.4WTnt.GFP病毒。使用磷酸钙方案,用ChAdV68.4WTnt.GFP DNA转染HEK293A细胞。在转染之后第10天观察到病毒复制,并且使用荧光显微镜在40×放大倍率下观测到ChAdV68.4WTnt.GFP病毒斑块。
图7C示出了在转染之后产生ChAdV68.4WTnt.GFP病毒。使用磷酸钙方案,用ChAdV68.4WTnt.GFP DNA转染HEK293A细胞。在转染之后第10天观察到病毒复制,并且使用荧光显微镜在100×放大倍率下观测到ChAdV68.4WTnt.GFP病毒斑块。
图8A示出了在转染之后产生ChAdV68.5WTnt.GFP病毒。使用脂染胺(lipofectamine)方案,用ChAdV68.5WTnt.GFP DNA转染HEK293A细胞。在转染之后第10天观察到病毒复制(斑块)。制备溶解产物并用于再感染T25烧瓶中的293A细胞。3天后,使用光学显微镜(40×放大倍率)观测到ChAdV68.5WTnt.GFP病毒斑块并拍照。
图8B示出了在转染之后产生ChAdV68.5WTnt.GFP病毒。使用脂染胺方案,用ChAdV68.5WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制(斑块)。制备溶解产物并用于再感染T25烧瓶中的293A细胞。3天后,使用荧光显微镜在40×放大倍率下观测到ChAdV68.5WTnt.GFP病毒斑块并拍照。
图8C示出了在转染之后产生ChAdV68.5WTnt.GFP病毒。使用脂染胺方案,用ChAdV68.5WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制(斑块)。制备溶解产物并用于再感染T25烧瓶中的293A细胞。3天后,使用荧光显微镜在100×放大倍率下观测到ChAdV68.5WTnt.GFP病毒斑块并拍照。
图9示出了病毒粒子制造方案。
图10示出了甲病毒衍生的VEE自我复制型RNA(srRNA)载体。
图11示出了在用VEE-荧光素酶srRNA接种C57BL/6J小鼠之后的体内报告基因表达。示出了在各种时间点用VEE-荧光素酶srRNA免疫接种C57BL/6J小鼠(每只小鼠10ug,两侧肌肉内注射,MC3包封)之后的代表性荧光素酶信号图像。
图12A示出了在带有B16-OVA肿瘤的小鼠中免疫接种用MC3LNP配制的VEE srRNA之后第14天测量的T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射10ug VEE-荧光素酶srRNA(对照)、VEE-UbAAY srRNA(Vax)、VEE-荧光素酶srRNA和抗CTLA-4(aCTLA-4)或VEE-UbAAY srRNA以及抗CTLA-4(Vax+aCTLA-4)。另外,在第7天开始,用抗PD1 mAb治疗所有小鼠。每组由8只小鼠组成。在免疫接种之后第14天,处死小鼠并收集脾和淋巴结。通过IFN-γELISPOT评定SIINFEKL特异性T细胞反应并且以每106个脾细胞的斑点形成细胞(SFC)报道。线表示中值。
图12B示出了在带有B16-OVA肿瘤的小鼠中免疫接种用MC3LNP配制的VEE srRNA之后第14天测量的T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射10ug VEE-荧光素酶srRNA(对照)、VEE-UbAAY srRNA(Vax)、VEE-荧光素酶srRNA和抗CTLA-4(aCTLA-4)或VEE-UbAAY srRNA以及抗CTLA-4(Vax+aCTLA-4)。此外,在第7天开始,用抗PD1 mAb治疗所有小鼠。每组由8只小鼠组成。在免疫接种之后14天,处死小鼠并收集脾和淋巴结。通过MHCI-五聚体染色评定SIINFEKL特异性T细胞反应,以五聚体阳性细胞占CD8阳性细胞的百分比报道。线表示中值。
图13A示出了在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。另外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过IFN-γELISPOT测量T细胞反应。在用腺病毒免疫接种后第14天,处死小鼠并收集脾和淋巴结。
图13B示出了在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过IFN-γELISPOT测量T细胞反应。在用腺病毒免疫接种后第14天以及在用srRNA加强免疫后第14天(初免之后第28天),处死小鼠并收集脾和淋巴结。
图13C示出了在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过MHC I类五聚体染色测量T细胞反应。在用腺病毒免疫接种后第14天,处死小鼠并收集脾和淋巴结。
图13D示出了在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过MHC I类五聚体染色测量T细胞反应。在用腺病毒免疫接种后第14天以及在用srRNA加强免疫后第14天(初免之后第28天),处死小鼠并收集脾和淋巴结。
图14A示出了在带有CT26(Balb/c)肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。对小鼠免疫接种Ad5-GFP并且在腺病毒初免之后第15天,用经MC3LNP配制的VEE-荧光素酶srRNA(对照)加强免疫,或者用Ad5-UbAAY进行初免并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。向另一个组施用Ad5-GFP/VEE-荧光素酶srRNA初免/加强免疫与抗PD-1的组合(aPD1),而第四组接受Ad5-UbAAY/VEE-UbAAY srRNA初免/加强免疫与抗PD-1mAb的组合(Vax+aPD1)。使用IFN-γELISPOT测量T细胞对AH1肽的反应。在用腺病毒免疫接种后第12天,处死小鼠并收集脾和淋巴结。
图14B示出了在带有CT26(Balb/c)肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。对小鼠免疫接种Ad5-GFP并且在腺病毒初免之后第15天,用经MC3LNP配制的VEE-荧光素酶srRNA(对照)加强免疫,或者用Ad5-UbAAY进行初免并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。向另一个组施用Ad5-GFP/VEE-荧光素酶srRNA初免/加强免疫与抗PD-1的组合(aPD1),而第四组接受Ad5-UbAAY/VEE-UbAAY srRNA初免/加强免疫与抗PD-1mAb的组合(Vax+aPD1)。使用IFN-γELISPOT测量T细胞对AH1肽的反应。在用腺病毒免疫接种后第12天以及在用srRNA加强免疫后第6天(初免之后第21天),处死小鼠并收集脾和淋巴结。
图15示出了ChAdV68引起针对小鼠中小鼠肿瘤抗原的T细胞反应。对小鼠免疫接种ChAdV68.5WTnt.MAG25mer,并且在C57BL/6J雌性小鼠中测量针对MHC I类表位SIINFEKL(OVA)的T细胞反应并在Balb/c小鼠中测量针对MHC I类表位AH1-A5的T细胞反应。呈现在ELISpot分析中测量的每106个脾细胞的平均斑点形成细胞(SFC)。误差条表示标准偏差。
图16示出了在CT26肿瘤模型中单次免疫接种ChAdV6、ChAdV+抗PD-1、srRNA、srRNA+抗PD-1或单独抗PD-1之后的细胞免疫应答。使用ELISpot测量来自每组的6只小鼠的脾细胞中抗原特异性IFN-γ的产生。结果呈现为每106个脾细胞的斑点形成细胞(SFC)。每个组的中值以水平线指示。P值使用Dunnett多重比较检验确定;***P<0.0001,**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mer srRNA。
图17示出了在CT26肿瘤模型中单次免疫接种ChAdV6、ChAdV+抗PD-1、srRNA、srRNA+抗PD-1或单独抗PD-1之后的CD8T细胞反应。使用ICS测量CD8 T细胞中抗原特异性IFN-γ的产生并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。P值使用Dunnett多重比较检验确定;***P<0.0001,**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mer srRNA。
图18示出了在CT26肿瘤模型中用ChAdV/srRNA异源初免/加强免疫、srRNA/ChAdV异源初免/加强免疫或srRNA/srRNA同源初免/加强免疫进行免疫接种之后的肿瘤生长情况。还示出了与在初免和加强免疫期间施用或不施用抗PD1的初免/加强免疫的比较。每周两次测量肿瘤体积并且呈现研究的前21天的平均肿瘤体积。研究起始时每组22-28只小鼠。误差条表示平均值的标准误差(SEM)。P值使用Dunnett检验确定;***P<0.0001,**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mer srRNA。
图19示出了在CT26肿瘤模型中用ChAdV/srRNA异源初免/加强免疫、srRNA/ChAdV异源初免/加强免疫或srRNA/srRNA同源初免/加强免疫进行免疫接种之后的存活情况。还示出了与在初免和加强免疫期间施用或不施用抗PD1的初免/加强免疫的比较。P值使用对数秩检验确定;***P<0.0001,**P<0.001,*P<0.01。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mer srRNA。
图20示出了使用ELISpot测量的抗原特异性细胞免疫应答。在第一次加强免疫之后第1、2、3、4、5、6、8、9或10周,使用ELI Spot测量VEE-MAG25mer srRNA-LNP1(30μg)(图20A)、VEE-MA G25mer srRNA-LNP1(100μg)(图20B)、或VEE-MAG25mer srRNA-LNP2(100μg)(图20C)同源初免/加强免疫或ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA异源初免/加强免疫组(图20D)的PBMC中针对六个不同mamu A01限制性表位的抗原特异性IFN-γ产生情况(每组6只恒河猴)。结果以堆叠条形图格式对于各表位以每106个PBMC的平均斑点形成细胞(SFC)呈现。将每只动物的值相对于放血前(第0周)的水平进行归一化。
图21示出了使用ELISpot测量的抗原特异性细胞免疫应答。在免疫接种之前以及在初始免疫接种之后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周,使用ELISpot测量在用ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA异源初免/加强免疫方案免疫接种后的PBMC中针对六个不同mamu A01限制性表位的抗原特异性IFN-γ产生情况。结果以堆叠条形图格式对于每个表位以每106个PBMC的平均斑点形成细胞(SFC)呈现(每组6只恒河猴)。
图22示出了使用ELISpot测量的抗原特异性细胞免疫应答。在免疫接种之前以及在初始免疫接种之后第4、5、6、7、8、10、11、12、13、14或15周,使用ELISpot测量在用VEE-MAG25mer srRNA LNP2同源初免/加强免疫方案免疫接种后的PBMC中针对六个不同mamuA01限制性表位的抗原特异性IFN-γ产生情况。结果以堆叠条形图格式对于每个表位以每106个PBMC的平均斑点形成细胞(SFC)呈现(每组6只恒河猴)。
图23示出了使用ELISpot测量的抗原特异性细胞免疫应答。在免疫接种之前以及在初始免疫接种之后第4、5、6、7、8、10、11、12、13、14或15周,使用ELISpot测量在用VEE-MAG25mer srRNA LNP1同源初免/加强免疫方案免疫接种后的PBMC中针对六个不同mamuA01限制性表位的抗原特异性IFN-γ产生情况。结果以堆叠条形图格式对于每个表位以每106个PBMC的平均斑点形成细胞(SFC)呈现(每组6只恒河猴)。
图24A和图24B示出了由Promega动态范围标准生成的示例性肽谱。
图25示出了EDGE评分与通过靶向MS检测候选共享新抗原肽的概率之间的相关性。
图26A示出了用VEE-荧光素酶samRNA与抗IFNAR MAb或托法替尼组合免疫接种C57BL/6J小鼠后体内报告蛋白的表达。在用VEE-荧光素酶免疫接种后第1、2和5天对每只小鼠的相对发光(RLU)进行定量(每只小鼠10μg,两侧肌肉内递送)。免疫接种前24小时将抗IFNAR Mab(克隆MAR1-5A3,BioXcell)(2mg)作为单次剂量腹膜内递送。免疫接种前24小时开始口服递送托法替尼,2mg,2次/天,并且持续6天。平均值+/-SEM,每组5只小鼠。
图26B示出了用VEE-MAG samRNA与抗IFNARMAb或托法替尼或对照组合免疫接种Balb/c小鼠后体内抗原特异性T细胞反应。使用细胞内细胞因子染色测量T细胞对AH1-A5肽(SPSYAYHQF)的反应。在用VEE-MAG(10μg,IM)免疫接种后第12天处死小鼠并收集脾。免疫接种前24小时将抗IFNAR Mab(克隆MAR1-5A3,BioXcell)(2mg)作为单次剂量腹膜内递送。免疫接种前24小时开始口服递送托法替尼,2mg,2次/天,并且持续8天。每组8只小鼠,条表示中值。
图27A示出了对于增加的samRNA表达和免疫应答,需要早期抑制IFNa。用1ugsamRNA单次免疫接种后第12天,Balb/c小鼠中的CD8 T细胞反应。在用samRNA免疫接种前或免疫接种后的指定时间,还用抗IgG抗体对照或靶向IFNAR的单克隆阻断抗体治疗小鼠。抗体治疗均为2mg剂量,腹膜内递送。使用细胞内细胞因子染色测量CD8 T细胞中抗原特异性(AH1-A5)IFN-γ的产生并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。
图27B示出了持续的IFNa抑制不降低免疫应答。用1ug samRNA单次免疫后第12天,Balb/c小鼠中的CD8 T细胞反应。在用samRNA免疫接种前和免疫接种后的指定时间,用抗IgG抗体对照或靶向IFNAR的单克隆阻断抗体治疗小鼠。抗体治疗均为2mg剂量,腹膜内递送。使用MHC I类四聚体染色测量抗原特异性(AH1-A5)CD8 T细胞并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。
图27C示出了持续的IFNa抑制不降低免疫应答。用1ug samRNA单次免疫接种后第12天,Balb/c小鼠中的CD8 T细胞反应。在用samRNA免疫接种前和免疫接种后的指定时间,用抗IgG抗体对照或靶向IFNAR的单克隆阻断抗体治疗小鼠。抗体治疗均为2mg剂量,腹膜内递送。使用细胞内细胞因子染色测量CD8 T细胞中抗原特异性(AH1-A5)IFN-γ的产生并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。
图28A示出了IFNa的局部抑制在增加samRNA免疫应答方面与全身递送一样有效。用1ug samRNA单次免疫接种后第12天,Balb/c小鼠中的CD8 T细胞反应。在用samRNA免疫接种前24小时,用腹膜内(IP)递送的抗IgG抗体对照或IP或肌内递送的靶向IFNAR的单克隆阻断抗体治疗小鼠。抗体治疗均为0.5mg剂量。所有肌内注射均两侧递送至胫骨前肌。使用MHCI类四聚体染色测量抗原特异性(AH1-A5)CD8 T细胞并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。
图28B示出了IFNa的局部抑制在增加samRNA免疫应答方面与全身递送一样有效。用1ug samRNA单次免疫接种后第12天,Balb/c小鼠中的CD8 T细胞反应。在用samRNA免疫接种前24小时,用腹膜内(IP)递送的抗IgG抗体对照或IP或肌内递送的靶向IFNAR的单克隆阻断抗体治疗小鼠。抗体治疗均为0.5mg剂量。所有肌内注射均两侧递送至胫骨前肌。使用细胞内细胞因子染色测量CD8 T细胞中抗原特异性(AH1-A5)IFN-γ的产生并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。
图29示出了来自各种物种的具有30(L)、40(XL)或50(XXL)个表位的大抗原盒的模型表位的一般组织。
图30示出了ChAd载体表达长盒,如使用识别所有盒共同序列的抗II类(PADRE)抗体的上文蛋白质印迹所指示。用表达可变大小的大盒(chAd68-50XXL、chAd68-40XL和chAd68-30L)的chAd68载体感染HEK293细胞。感染设置为MOI是0.2。感染后二十四小时,将蛋白酶体抑制剂MG132加入到一组感染孔中(以加号指示)。另一组病毒处理的孔未用MG132处理(以负号指示)。将未感染的HEK293细胞(293F)用作阴性对照。感染后四十八小时收集细胞沉淀物,并通过SDS/PAGE电泳进行分析,并使用兔抗II类PADRE抗体进行免疫印迹。将HRP抗兔抗体和ECL化学发光底物用于检测。
图31示出了在chAd68大盒免疫接种的小鼠中,通过ICS检测到的针对AH1(顶部)和SIINFEKL(底部)的CD8+免疫应答。数据呈现为针对模型表位的IFNg+细胞占总CD8细胞的百分比
图32示出了chAd68大盒疫苗接种后对LD-AH1+(顶部)和Kb-SIINFEKL+(底部)四聚体的CD8+反应。数据呈现为针对模型四聚体肽复合物有反应的总CD8细胞的百分比。通过使用Tukey检验的ANOVA,*p<0.05,**p<0.01。与MAG 20抗原盒比较的所有p值。
图33示出了在甲病毒大盒治疗的小鼠中,通过ICS检测到的针对AH1(顶部)和SIINFEKL(底部)的CD8+免疫应答。数据呈现为针对模型表位的IFNg+细胞占总CD8细胞的百分比。通过使用Tukey检验的ANOVA,*p<0.05,**p<0.01,***p<0.001。与MAG 20抗原盒比较的所有p值。
图34示出了用于评估恒河猴中含有抗原盒的载体的免疫原性的疫苗接种策略。三角形指示在第0周和第32周进行chAd68疫苗接种(1e12 vp/动物)。圆圈表示在第0、4、12、20、28和32周进行甲病毒疫苗接种。正方形表示抗CTLA4抗体的施用。
图35示出了单独用chAd-MAG给药的恒河猴(第4组)中CD8+抗表位反应的时程。示出了平均SFC/1e6个脾细胞。
图36示出了用IV递送的chAd-MAG加抗CTLA4抗体(伊匹单抗(Ipilimumab))给药的恒河猴(第5组)中CD8+抗表位反应的时程。示出了平均SFC/1e6个脾细胞。
图37示出了用SC递送的chAd-MAG加抗CTLA4抗体(伊匹单抗)给药的恒河猴(第6组)中CD8+抗表位反应的时程。示出了平均SFC/1e6个脾细胞。
图38示出了通过ELISpot测量的ChAdV68/samRNA疫苗方案产生的抗原特异性记忆反应。结果以单独的点图呈现,其中每个点表示单只动物。示出了免疫接种前的基线(左图)和初免后第18个月的记忆反应(右图)。
图39示出了通过使用组合四聚体染色和CD45RA/CCR7共染色的流式细胞术的抗原特异性CD8+T细胞的记忆细胞表型分析。
图40示出了在研究第18个月时,四种Mamu-A*01四聚体+CD8+T细胞群体的总和内的记忆细胞类型的分布。记忆细胞的特征如下:CD45RA+CCR7+=初始,CD45RA+CCR7-=效应子(Teff),CD45RA-CCR7+=中枢记忆(Tcm),CD45RA-CCR7-=效应记忆(Tem)。
图41示出了在带有CT26肿瘤的小鼠中CD8+T细胞识别CT26肿瘤抗原AH1的频率。P值使用Tukey多重比较检验的单向ANOVA确定;**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;aCTLA4=抗CTLA4抗体,克隆9D9。
具体实施方式
I.定义
一般说来,权利要求书和说明书中使用的术语意图解释为具有与本领域普通技术人员所理解的普通含义。为清楚起见,以下定义某些术语。如果普通含义与所提供的定义之间存在矛盾,应使用所提供的定义。
如本文所用,术语“抗原”是诱导免疫应答的物质。抗原可以是新抗原。抗原可以是“共享抗原”,其是在特定群体例如癌症患者的特定群体中发现的抗原。
如本文所用,术语“新抗原”是具有至少一个使其不同于对应野生型抗原的改变的抗原,例如经由肿瘤细胞中的突变或特异性针对肿瘤细胞的翻译后修饰。新抗原可以包括多肽序列或核苷酸序列。突变可以包括移码或非移码插入缺失、错义或无义取代、剪接位点改变、基因组重排或基因融合,或产生neoORF的任何基因组或表达改变。突变还可以包括剪接变体。特异性针对肿瘤细胞的翻译后修饰可以包括异常磷酸化。特异性针对肿瘤细胞的翻译后修饰还可以包括蛋白酶体产生的剪接抗原。参见Liepe等,A large fraction ofHLA class I ligands are proteasome-generated spliced peptides;Science.2016年10月21日;354(6310):354-358。此类共享新抗原可用于经由施用诱导受试者中的免疫应答。所述受试者可通过使用各种诊断方法例如下文进一步描述的患者选择方法被鉴定以施用。
如本文所用,术语“肿瘤抗原”是存在于受试者的肿瘤细胞或组织中但不存在于受试者的对应正常细胞或组织中,或衍生自与正常细胞或组织相比已知或已发现在肿瘤细胞或癌组织中改变表达的多肽的抗原。
如本文所用,术语“基于抗原的疫苗”是基于一个或多个抗原(例如多个抗原)的疫苗组合物。疫苗可以是基于核苷酸的(例如基于病毒的、基于RNA的或基于DNA的)、基于蛋白质的(例如基于肽的)或其组合。
如本文所用,术语“候选抗原”是产生可代表抗原的序列的突变或其他畸变。
如本文所用,术语“编码区”是编码蛋白质的基因的部分。
如本文所用,术语“编码突变”是在编码区中出现的突变。
如本文所用,术语“ORF”意指开放阅读框。
如本文所用,术语“NEO-ORF”是由突变或其他畸变诸如剪接而产生的肿瘤特异性ORF。
如本文所用,术语“错义突变”是引起一个氨基酸被另一个氨基酸取代的突变。
如本文所用,术语“无义突变”是引起氨基酸被终止密码子取代或引起典型起始密码子移除的突变。
如本文所用,术语“移码突变”是引起蛋白质框架改变的突变。
如本文所用,术语“插入缺失”是一个或多个核酸的插入或缺失。
如本文所用,在两个或更多个核酸或多肽序列的上下文中,术语“同一性”百分比是指当出于最大对应性比较和比对时,两个或更多个序列或子序列具有指定百分比的核苷酸或氨基酸残基是相同的,如使用下文所述的序列比较算法(例如BLASTP和BLASTN或技术人员可用的其他算法)中的一种测量或通过目测检查所测量。取决于应用,“同一性”百分比可以存在于所比较的序列区域上,例如在功能结构域上,或者存在于待比较的两个序列的全长上。
关于序列比较,通常一个序列充当与测试序列进行比较的参考序列。当使用序列比较算法时,将测试序列和参考序列输入计算机中,必要时指定子序列坐标,并且指定序列算法程序参数。然后,序列比较算法基于指定程序参数计算测试序列相对于参考序列的序列同一性百分比。或者,可以通过组合在所选序列位置(例如序列基序)处特定核苷酸,或对于翻译的序列来说特定氨基酸的存在或不存在来确定序列相似性或不相似性。
用于比较的最佳序列比对可以例如通过Smith和Waterman,Adv.Appl.Math.2:482(1981)的局部同源性算法;Needleman和Wunsch,J.Mol.Biol.48:443(1970)的同源性比对算法;Pearson和Lipman,Proc.Nat'l.Acad.Sci.USA 85:2444(1988)的相似性搜索方法;这些算法的计算机化实施(Wisconsin Genetics软件包中的GAP、BESTFIT、FASTA和TFASTA;Genetics Computer Group,575Science Dr.,Madison,Wis.);或通过目测检查(一般参见Ausubel等,见下文)来进行。
适于测定序列同一性和序列相似性百分比的算法的一个实例是Altschul等,J.Mol.Biol.215:403-410(1990)中描述的BLAST算法。执行BLAST分析的软件可通过国家生物技术信息中心(National Center for Biotechnology Information)公开获得。
如本文所用,术语“无终止或通读”是引起天然终止密码子移除的突变。
如本文所用,术语“表位”是抗原中通常由抗体或T细胞受体结合的特异性部分。
如本文所用,术语“免疫原性”是例如通过T细胞、B细胞或两者引发免疫应答的能力。
如本文所用,术语“HLA结合亲和力”、“MHC结合亲和力”意指特异性抗原与特异性MHC等位基因之间的结合亲和力。
如本文所用,术语“诱饵”是用于从样品富集DNA或RNA的特定序列的核酸探针。
如本文所用,术语“变体”是受试者的核酸与用作对照的参考人基因组之间的差异。
如本文所用,术语“变体识别(variant call)”是对通常由测序确定的变体存在的算法确定。
如本文所用,术语“多态性”是生殖系变体,即在个体的所有携带DNA的细胞中发现的变体。
如本文所用,术语“体细胞变体”是在个体的非生殖系细胞中产生的变体。
如本文所用,术语“等位基因”是基因的一种形式,或基因序列的一种形式,或蛋白质的一种形式。
如本文所用,术语“HLA型”是HLA基因等位基因的补体。
如本文所用,术语“无义介导的衰变”或“NMD”是由过早终止密码子所致的细胞对mRNA的降解。
如本文所用,术语“躯干突变”是起源于肿瘤发展早期且存在于大多数肿瘤细胞中的突变。
如本文所用,术语“亚克隆突变”是起源于肿瘤发展后期且仅存在于肿瘤细胞子集中的突变。
如本文所用,术语“外显子组”是编码蛋白质的基因组的子集。外显子组可以是基因组的集合外显子。
如本文所用,术语“逻辑回归”是来自统计的二进制数据的回归模型,其中因变量等于1的概率的逻辑被建模为因变量的线性函数。
如本文所用,术语“神经网络”是用于分类或回归的机器学习模型,其由多层线性变换,接着是通常经由随机梯度下降和反向传播训练的逐元素非线性组成。
如本文所用,术语“蛋白质组”是由细胞、细胞群或个体表达和/或翻译的全部蛋白质的集合。
如本文所用,术语“肽组”是由MHC-I或MHC-II在细胞表面上递呈的所有肽的集合。肽组可以指细胞或细胞集合(例如肿瘤肽组,意味着构成肿瘤的所有细胞的肽组的联合)的特性。
如本文所用,术语“ELISPOT”意指酶联免疫吸附斑点分析,这是一种用于监测人和动物的免疫应答的常用方法。
如本文所用,术语“葡聚糖肽多聚体”是在流式细胞测量术中用于抗原特异性T细胞染色的基于葡聚糖的肽-MHC多聚体。
如本文所用,术语“耐受性或免疫耐受性”是对一种或多种抗原(例如自身抗原)免疫无反应性的状态。
如本文所用,术语“中心耐受性”是通过缺失自身反应性T细胞克隆或通过促进自身反应性T细胞克隆分化成免疫抑制性调节性T细胞(Treg)而在胸腺中遭受的耐受性。
如本文所用,术语“外周耐受性”是通过下调或不激活经受中心耐受性的自身反应性T细胞或促进这些T细胞分化成Treg而在外周遭受的耐受性。
术语“样品”可以包括借助于包括静脉穿刺、排泄、射精、按摩、活组织检查、针抽吸、灌洗样品、刮取、手术切口或干预在内的手段,或本领域中已知的其他手段从受试者获取单个细胞或多个细胞或细胞碎片或体液等分试样。
术语“受试者”涵盖人或非人,无论体内、离体或体外,雄性或雌性的细胞、组织或生物体。术语受试者包括含人在内的哺乳动物。
术语“哺乳动物”涵盖人和非人两者,并且包括但不限于人、非人灵长类动物、犬科动物、猫科动物、鼠科动物、牛科动物、马科动物和猪科动物。
术语“临床因素”是指受试者状况的量度,例如疾病活动性或严重程度。“临床因素”涵盖受试者健康状况的所有标志物,包括非样品标志物,和/或受试者的其他特征,诸如但不限于年龄和性别。临床因素可以是可在确定条件下评估来自受试者的样品(或样品群体)或受试者而获得的评分、值或值集合。临床因素也可以由标志物和/或其他参数(诸如基因表达替代物)进行预测。临床因素可以包括肿瘤类型、肿瘤亚型和吸烟史。
术语“衍生自肿瘤的抗原编码核酸序列”是指例如经由RT-PCR直接从肿瘤提取的核酸序列;或通过肿瘤测序获得的序列数据,然后使用测序数据例如经由本领域中已知的各种合成或基于PCR的方法合成核酸序列。
术语“甲病毒”是指披膜病毒科(Togaviridae)的成员,并且是正义单链RNA病毒。甲病毒通常分类为旧世界诸如辛德毕斯、罗斯河、马雅鲁、基孔肯尼亚(Chikungunya)和塞姆利基森林病毒,或新世界诸如东部马脑炎、奥拉、摩根堡或委内瑞拉马脑炎及其衍生病毒株TC-83。甲病毒通常是自我复制的RNA病毒。
术语“甲病毒骨架”是指允许病毒基因组自我复制的甲病毒的最小序列。最小序列可包括用于非结构蛋白质介导的扩增的保守序列、非结构蛋白质1(nsP1)基因、nsP2基因、nsP3基因、nsP4基因和polyA序列,以及用于亚基因组病毒RNA表达的序列,包括26S启动子元件。
术语“用于非结构蛋白质介导的扩增的序列”包括本领域技术人员众所周知的甲病毒保守序列元件(CSE)。CSE包括但不限于甲病毒5'UTR、51-nt CSE、24-nt CSE或其他26S亚基因组启动子序列、19-nt CSE和甲病毒3'UTR。
术语“RNA聚合酶”包括催化由DNA模板产生RNA多核苷酸的聚合酶。RNA聚合酶包括但不限于衍生自噬菌体的聚合酶,包括T3、T7和SP6。
术语“脂质”包括疏水性和/或两亲性分子。脂质可以是阳离子、阴离子或中性的。脂质可以是合成或天然来源的,并且在一些情况下是可生物降解的。脂质可包括胆固醇、磷脂、脂质缀合物(包括但不限于聚乙二醇(PEG)缀合物(聚乙二醇化脂质))、蜡、油、甘油酯、脂肪和脂溶性维生素。脂质也可包括二亚油基甲基-4-二甲基胺基丁酸酯(MC3)和MC3样分子。
术语“脂质纳米粒子”或“LNP”包括使用含脂质膜围绕水性内部形成的小泡样结构,也称为脂质体。脂质纳米粒子包括具有通过表面活性剂稳定的固体脂质核心的基于脂质的组合物。核心脂质可以是脂肪酸、酰基甘油、蜡和这些表面活性剂的混合物。生物膜脂质诸如磷脂、鞘磷脂、胆汁盐(牛磺胆酸钠)和固醇(胆固醇),可用作稳定剂。脂质纳米粒子可使用限定比率的不同脂质分子形成,包括但不限于限定比率的一种或多种阳离子脂质、阴离子脂质或中性脂质。脂质纳米粒子可将分子包封在外膜壳内,并且随后可与靶细胞接触以将包封的分子递送到宿主细胞胞溶质。脂质纳米粒子可用非脂质分子修饰或官能化,包括在其表面上。脂质纳米粒子可以是单层的(单层)或多层的(多层)。脂质纳米粒子可与核酸复合。单层脂质纳米粒子可与核酸复合,其中核酸在水性内部。多层脂质纳米粒子可与核酸复合,其中核酸在水性内部,或者形成或包夹在其间。
缩写:MHC:主要组织相容性复合物;HLA:人白细胞抗原或人MHC基因座;NGS:下一代测序;PPV:阳性预测值;TSNA:肿瘤特异性新抗原;FFPE:福尔马林固定、石蜡包埋;NMD:无义介导的衰变;NSCLC:非小细胞肺癌;DC:树突状细胞。
除非上下文另外清楚地规定,否则如本说明书和所附权利要求中所用,单数形式“一个(种)(a/an)”和“所述”包括多个指示物。
除非特别说明或从上下文中可以明显看出,否则如本文所用,术语“约”应理解为在本领域的正常公差范围内,例如在平均值的2个标准偏差内。约可以理解为所述值的10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.1%、0.05%或0.01%内。除非上下文另有明确说明,否则本文提供的所有数值均由术语约修饰。
本文中未直接定义的任何术语应理解为具有与本发明领域内所理解的通常与之相关的含义。本文论述的某些术语是为了向从业人员描述本发明各方面的组合物、装置、方法等以及其制备或使用提供额外的指导。应了解,相同的事物可以按超过一种方式表示。因此,替代性措辞和同义词可以用于本文所论述的任何一个或多个术语。无论本文中是否阐述或论述术语都无关紧要。提供了一些同义词或可取代的方法、材料等。除非明确陈述,否则对一个或数个同义词或等效物的叙述不排除其他同义词或等效物的使用。实例,包括术语实例的使用只是出于说明的目的,且并非在本文中限制本发明各方面的范围和含义。
说明书正文内引用的所有参考文献、颁布的专利和专利申请均出于所有目的以引用的方式整体并入本文。
II.鉴定抗原的方法
用于鉴定共享抗原(例如新抗原)的方法包括鉴定来自受试者肿瘤的抗原,所述抗原可能被递呈于肿瘤或免疫细胞(包括专职抗原递呈细胞诸如树突状细胞)的细胞表面上,和/或可能是免疫原性的。例如,一种此类方法可以包括以下步骤:从受试者的肿瘤细胞获得外显子组、转录组或全基因组肿瘤核苷酸测序和/或表达数据中的至少一者,其中所述肿瘤核苷酸测序和/或表达数据被用于获得代表抗原集合中的每一者的肽序列的数据(例如对于新抗原而言,其中每个新抗原的肽序列包含至少一个使其不同于对应野生型肽序列的改变;或者对于共享抗原而言在不存在突变的情况下,其中肽衍生自与正常细胞或组织相比已知或已发现在肿瘤细胞或癌组织中改变表达的任何多肽);将每个抗原的肽序列输入到一个或多个递呈模型中以产生所述抗原中的每一者被一个或多个MHC等位基因递呈于受试者肿瘤细胞的肿瘤细胞表面或肿瘤中存在的细胞上的数值可能性集合,所述数值可能性集合已被至少基于接收到的质谱数据进行鉴定;以及基于所述数值可能性集合选择所述抗原集合的子集,以产生经选择的抗原的集合。
递呈模型可以包括针对包含对应标记集合的参考数据集合(也称为训练数据集)训练的统计回归或机器学习(例如深度学习)模型,其中所述参考数据集合是从多个不同受试者中的每一者获得,其中任选地,一些受试者可以患有肿瘤,并且其中所述参考数据集合包含以下中的至少一者:代表来自肿瘤组织的外显子组核苷酸序列的数据、代表来自正常组织的外显子组核苷酸序列的数据、代表来自肿瘤组织的转录组核苷酸序列的数据、代表来自肿瘤组织的蛋白质组序列的数据和代表来自肿瘤组织的MHC肽组序列的数据,以及代表来自正常组织的MHC肽组序列的数据。参考数据还可包括工程改造成表达预定MHC等位基因且随后暴露于合成蛋白质的单等位基因细胞系、正常和肿瘤人细胞系,以及新鲜和冷冻原始样品的质谱数据、测序数据、RNA测序数据、表达谱数据和蛋白质组数据以及T细胞分析(例如ELISPOT)。在某些方面,参考数据集合包括每种形式的参考数据。
递呈模型可以包含至少部分从参考数据集合得到的特征集合,并且其中所述特征集合包含等位基因依赖性特征和等位基因非依赖性特征中的至少一种。在某些方面,包括每一特征。
用于鉴定共享抗原的方法还包括通过从受试者的一个或多个肿瘤细胞中鉴定一个或多个可能递呈于肿瘤细胞表面上的抗原,来产生用于构建个性化癌症疫苗的输出。例如,一种此类方法可以包括以下步骤:从受试者的肿瘤细胞和正常细胞获得外显子组、转录组或全基因组核苷酸测序和/或表达数据中的至少一者,其中所述核苷酸测序和/或表达数据用于获得代表通过比较来自肿瘤细胞的核苷酸测序和/或表达数据与来自正常细胞的核苷酸测序和/或表达数据而鉴定的抗原集合中的每一者的肽序列(例如,对于新抗原而言,其中每个新抗原的肽序列包含至少一个使其不同于对应野生型肽序列的改变;或者对于共享抗原而言在不存在突变的情况下,其中肽衍生自与正常细胞或组织相比已知或已发现在肿瘤细胞或癌组织中改变表达的任何多肽)、从受试者正常细胞中鉴定出的肽序列的数据;将每个抗原的肽序列编码成对应的数值向量,每个数值向量包括有关构成肽序列的多个氨基酸和肽序列中的氨基酸位置集合的信息;使用计算机处理器将所述数值向量输入到深度学习递呈模型中,以生成所述抗原集合的递呈可能性集合,所述集合中的每个递呈可能性表示一个或多个II类MHC等位基因在受试者的肿瘤细胞表面上递呈对应抗原的可能性,即深度学习递呈模型;基于所述递呈可能性集合,选择所述抗原集合的子集,以生成经选择的抗原集合;以及基于所述经选择的抗原集合,生成用于构建个性化癌症疫苗的输出。
用于鉴定抗原(包括新抗原)的具体方法是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的方法,所述专利出于所有目的各自以引用的方式整体并入本文。
本文公开了一种治疗患有肿瘤的受试者的方法,其包括进行本文所述的任何抗原鉴定方法的步骤,并且还包括获得包含经选择的抗原集合的肿瘤疫苗,以及向所述受试者施用所述肿瘤疫苗。
本文公开的方法还可以包括鉴定一个或多个T细胞,所述T细胞针对子集中的至少一个抗原具有抗原特异性。在一些实施方案中,鉴定包括在使一个或多个抗原特异性T细胞扩增的条件下,将一个或多个T细胞与子集中的一个或多个抗原共培养。在其他实施方案中,鉴定包括在允许T细胞与四聚体之间结合的条件下,使一个或多个T细胞与包含子集中一个或多个抗原的四聚体接触。在其他实施方案中,本文公开的方法还可以包括鉴定一个或多个已鉴定T细胞的一个或多个T细胞受体(TCR)。在某些实施方案中,鉴定一个或多个T细胞受体包括对一个或多个已鉴定T细胞的T细胞受体序列进行测序。本文公开的方法还可包括对多个T细胞进行基因工程改造以表达一个或多个已鉴定T细胞受体中的至少一者;在使多个T细胞扩增的条件下培养所述多个T细胞;以及将扩增的T细胞注入受试者体内。在一些实施方案中,对多个T细胞进行基因工程改造以表达一个或多个已鉴定T细胞受体中的至少一者包括将一个或多个已鉴定T细胞的T细胞受体序列克隆到表达载体中;以及用所述表达载体转染多个T细胞中的每一者。在一些实施方案中,本文公开的方法还包括在扩增一个或多个已鉴定T细胞的条件下培养所述一个或多个已鉴定T细胞;以及将扩增的T细胞注入受试者体内。
本文还公开了一种分离T细胞,其针对子集中的至少一个经选择的抗原具有抗原特异性。
本文还公开了一种用于制造肿瘤疫苗的方法,其包括以下步骤:从受试者的肿瘤细胞获得外显子组、转录组或全基因组肿瘤核苷酸测序和/或表达数据中的至少一者,其中所述肿瘤核苷酸测序和/或表达数据被用于获得代表抗原集合中的每一者的肽序列的数据(例如对于新抗原而言,其中每个新抗原的肽序列包含至少一个使其不同于对应野生型肽序列的改变;或者对于共享抗原而言在不存在突变的情况下,其中肽衍生自与正常细胞或组织相比已知或已发现在肿瘤细胞或癌组织中改变表达的任何多肽);将每个抗原的肽序列输入到一个或多个递呈模型中以产生所述抗原中的每一者被一个或多个MHC等位基因递呈于受试者肿瘤细胞的肿瘤细胞表面上的数值可能性集合,所述数值可能性集合已被至少基于接收到的质谱数据进行鉴定;以及基于所述数值可能性集合选择所述抗原集合的子集,以产生经选择的抗原的集合;以及产生或已产生包含所述经选择的抗原集合的肿瘤疫苗。
本文还公开了一种包括经选择的抗原集合的肿瘤疫苗,所述经选择的抗原集合通过进行包括以下步骤的方法来选择:从受试者的肿瘤细胞获得外显子组、转录组或全基因组肿瘤核苷酸测序和/或表达数据中的至少一者,其中所述肿瘤核苷酸测序和/或表达数据被用于获得代表抗原集合中的每一者的肽序列以及其中每个抗原的肽序列的数据(例如对于新抗原而言,其中每个新抗原的肽序列包含至少一个使其不同于对应野生型肽序列的改变;或者对于共享抗原的其他而言在不存在突变的情况下,其中肽衍生自与正常细胞或组织相比已知或已发现在肿瘤细胞或癌组织中改变表达的任何多肽);将每个抗原的肽序列输入到一个或多个递呈模型中以产生所述抗原中的每一者被一个或多个MHC等位基因递呈于受试者肿瘤细胞的肿瘤细胞表面上的数值可能性集合,所述数值可能性集合已被至少基于接收到的质谱数据进行鉴定;以及基于所述数值可能性集合选择所述抗原集合的子集,以产生经选择的抗原的集合;以及产生或已产生包含所述经选择的抗原集合的肿瘤疫苗。
肿瘤疫苗可包括核苷酸序列、多肽序列、RNA、DNA、细胞、质粒或载体中的一者或多者。
肿瘤疫苗可包括在肿瘤细胞表面上递呈的一个或多个抗原。
肿瘤疫苗可包括在受试者中具有免疫原性的一个或多个抗原。
肿瘤疫苗可能不包括在受试者中诱导针对正常组织的自身免疫应答的一个或多个抗原。
肿瘤疫苗可包括佐剂。
肿瘤疫苗可包括赋形剂。
本文公开的方法还可包括基于递呈模型,选择相对于未经选择的抗原在肿瘤细胞表面上递呈的可能性增加的抗原。
本文公开的方法还可包括基于递呈模型,选择相对于未经选择的抗原能够在受试者中诱导肿瘤特异性免疫应答的可能性增加的抗原。
本文公开的方法还可包括基于递呈模型,选择相对于未经选择的抗原能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加的抗原,任选地其中所述APC是树突状细胞(DC)。
本文公开的方法还可包括基于递呈模型,选择相对于未经选择的抗原经由中心或外周耐受性受抑制的可能性降低的抗原。
本文公开的方法还可包括基于递呈模型,选择相对于未经选择的抗原能够在受试者中诱导针对正常组织的自身免疫应答的可能性降低的抗原。
外显子组或转录组核苷酸测序和/或表达数据可通过对肿瘤组织进行测序而获得。
测序可以是下一代测序(NGS)或任何大规模平行测序方法。
数值可能性集合可通过至少MHC-等位基因相互作用特征来进一步鉴定,所述特征包含以下中的至少一者:经预测的MHC等位基因与抗原编码肽结合的亲和力;经预测的抗原编码肽-MHC复合物的稳定性;抗原编码肽的序列和长度;如通过质谱蛋白质组学或其他手段所评定,在来自表达特定MHC等位基因的其他个体的细胞中递呈具有类似序列的抗原编码肽的概率;所讨论的受试者中特定MHC等位基因的表达水平(例如,如通过RNA-seq或质谱法所测量);在表达特定MHC等位基因的其他不同受试者中由特定MHC等位基因递呈的总体新抗原编码肽序列独立性概率;在其他不同受试者中由同一家族分子(例如HLA-A、HLA-B、HLA-C、HLA-DQ、HLA-DR、HLA-DP)中的MHC等位基因递呈的总体新抗原编码肽序列独立性概率。
数值可能性集合通过至少MHC-等位基因非相互作用特征来进一步鉴定,所述特征包含以下中的至少一者:在其源蛋白序列内侧接新抗原编码肽的C末端和N末端序列;新抗原编码肽中蛋白酶裂解基序的存在,任选地根据相应蛋白酶在肿瘤细胞中的表达加权(如通过RNA-seq或质谱法所测量);如在适当细胞类型中所测量的源蛋白的周转率;源蛋白的长度,任选地考虑在肿瘤细胞中最高度表达的特异性剪接变体(“同种型”),如通过RNA-seq或蛋白质组质谱法所测量,或如DNA或RNA序列数据中所检测的生殖系或体细胞剪接突变的批注所预测;蛋白酶体、免疫蛋白酶体、胸腺蛋白酶体或其他蛋白酶在肿瘤细胞中的表达水平(其可通过RNA-seq、蛋白质组质谱法或免疫组织化学测量);新抗原编码肽的源基因的表达(例如,如通过RNA-seq或质谱法所测量);新抗原编码肽的源基因在细胞周期的各种阶段期间的典型组织特异性表达;源蛋白和/或其结构域的综合特征目录,如例如uniProt或PDBhttp://www.rcsb.org/pdb/home/home.do中可见;描述含有所述肽的源蛋白的结构域特性的特征,例如:二级或三级结构(例如α螺旋对β折叠);替代性剪接;在其他不同受试者中由所讨论的新抗原编码肽的源蛋白递呈肽的概率;由于技术偏差,肽将不会由质谱法检测到或过量表示的概率;通过RNASeq(其无需含有肽的源蛋白)所测量的提供关于肿瘤细胞、基质或肿瘤浸润淋巴细胞(TIL)状态的信息的各种基因模块/通路的表达;新抗原编码肽的源基因在肿瘤细胞中的拷贝数;肽结合于TAP的概率或经测量或经预测的肽对TAP的结合亲和力;TAP在肿瘤细胞中的表达水平(其可通过RNA-seq、蛋白质组质谱法、免疫组织化学测量);存在或不存在肿瘤突变,包括但不限于:已知癌症驱动基因(诸如EGFR、KRAS、ALK、RET、ROS1、TP53、CDKN2A、CDKN2B、NTRK1、NTRK2、NTRK3)及编码抗原递呈机制中所涉及的蛋白质的基因(例如B2M、HLA-A、HLA-B、HLA-C、TAP-1、TAP-2、TAPBP、CALR、CNX、ERP57、HLA-DM、HLA-DMA、HLA-DMB、HLA-DO、HLA-DOA、HLA-DOB、HLA-DP、HLA-DPA1、HLA-DPB1、HLA-DQ、HLA-DQA1、HLA-DQA2、HLA-DQB1、HLA-DQB2、HLA-DR、HLA-DRA、HLA-DRB1、HLA-DRB3、HLA-DRB4、HLA-DRB5或编码蛋白酶体或免疫蛋白酶体组分的基因中的任一者)中的驱动突变。递呈依赖于肿瘤中经受功能丧失性突变的抗原递呈机制的组分的肽具有降低的递呈概率;存在或不存在功能性生殖系多态性,包括但不限于:在编码抗原递呈机制中所涉及的蛋白质的基因(例如B2M、HLA-A、HLA-B、HLA-C、TAP-1、TAP-2、TAPBP、CALR、CNX、ERP57、HLA-DM、HLA-DMA、HLA-DMB、HLA-DO、HLA-DOA、HLA-DOB、HLA-DP、HLA-DPA1、HLA-DPB1、HLA-DQ、HLA-DQA1、HLA-DQA2、HLA-DQB1、HLA-DQB2、HLA-DR、HLA-DRA、HLA-DRB1、HLA-DRB3、HLA-DRB4、HLA-DRB5或编码蛋白酶体或免疫蛋白酶体组分的基因中的任一者)中;肿瘤类型(例如NSCLC、黑素瘤);临床肿瘤亚型(例如鳞状肺癌对非鳞状肺癌);吸烟史;所述肽的源基因在相关肿瘤类型或临床亚型中的典型表达,任选地通过驱动突变分层。
至少一个改变可以是移码或非移码插入缺失、错义或无义取代、剪接位点改变、基因组重排或基因融合或产生neoORF的任何基因组或表达改变。
肿瘤细胞可选自由以下组成的组:肺癌、黑素瘤、乳癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
本文公开的方法还可包括获得包含经选择的新抗原集合或其子集的肿瘤疫苗,任选地还包括向受试者施用所述肿瘤疫苗。
当呈多肽形式时,经选择的新抗原集合中的至少一个新抗原可包括以下中的至少一者:对于长度为8-15、8、9、10、11、12、13、14或15个氨基酸的MHC I类多肽,对于长度为6-30、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的MHC II类多肽,与MHC的结合亲和力为IC50值小于1000nM,在亲本蛋白质序列中在所述多肽内部或附近存在促进蛋白酶体裂解的序列基序,以及存在促进TAP转运的序列基序。对于MHC II类,在肽内部或附近存在促进通过细胞外或溶酶体蛋白酶(例如组织蛋白酶)裂解或HLA-DM催化的HLA结合的序列基序。
本文公开了用于鉴定可能在肿瘤细胞的肿瘤细胞表面上递呈的一个或多个新抗原的方法,其包括执行以下步骤:接收包含与从衍生自多个新鲜或冷冻肿瘤样品的主要组织相容性复合体(MHC)洗脱的多个分离肽相关的数据的质谱数据;通过至少鉴定肿瘤样品中存在且递呈在与各训练肽序列相关的一个或多个MHC等位基因上的训练肽序列集合来获得训练数据集;基于训练肽序列获得训练蛋白质序列集合;以及使用训练蛋白质序列和训练肽序列训练递呈模型的数值参数集合,所述递呈模型提供来自肿瘤细胞的肽序列在肿瘤细胞表面上由一个或多个MHC等位基因递呈的多个数值可能性。
递呈模型可表示以下两者之间的依赖性:MHC等位基因中的一对特定等位基因和在肽序列特定位置处特定氨基酸的存在;以及与在肿瘤细胞表面上由所述对的MHC等位基因中的特定等位基因递呈在所述特定位置处包含特定氨基酸的此类肽序列的可能性。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自在肿瘤细胞表面上递呈的可能性增加而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自能够在受试者中诱导肿瘤特异性免疫应答的可能性增加而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加而被选择,任选地其中所述APC是树突状细胞(DC)。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自经由中心或外周耐受性受抑制的可能性降低而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自能够在受试者中诱导针对正常组织的自身免疫应答的可能性降低而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于APC各自将在肿瘤细胞中经差异性翻译后修饰的可能性降低而被选择,任选地其中所述APC是树突状细胞(DC)。
除非另外指明,否则本文方法的实践将采用本领域技能范围内的蛋白质化学、生物化学、重组DNA技术和药理学的常规方法。此类技术在文献中被充分解释。参见例如T.E.Creighton,Proteins:Structures and Molecular Properties(W.H.Freeman andCompany,1993);A.L.Lehninger,Biochemistry(Worth Publishers,Inc.,现行版);Sambrook等,Molecular Cloning:A Laboratory Manual(第2版,1989);Methods InEnzymology(S.Colowick和N.Kaplan编,Academic Press,Inc.);Remington'sPharmaceutical Sciences,第18版(Easton,Pennsylvania:Mack Publishing Company,1990);Carey和Sundberg Advanced Organic Chemistry第3版(Plenum Press)A卷和B卷(1992)。
III.鉴定新抗原中的肿瘤特异性突变
本文还公开了用于鉴定某些突变(例如癌细胞中存在的变体或等位基因)的方法。具体来说,这些突变可存在于患有癌症的受试者的癌细胞的基因组、转录组、蛋白质组或外显子组中,而非受试者的正常组织中。用于鉴定对肿瘤具有特异性的新抗原(包括共享新抗原)的具体方法是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的方法,所述专利出于所有目的各自以引用的方式整体并入本文。
如果肿瘤中的基因突变引起肿瘤中特有的蛋白质的氨基酸序列变化,则认为其可用于免疫靶向肿瘤。有用的突变包括:(1)非同义突变,导致蛋白质中的氨基酸不同;(2)通读突变,其中终止密码子被修饰或缺失,导致翻译在C末端具有新颖肿瘤特异性序列的较长蛋白质;(3)剪接位点突变,导致在成熟mRNA中包含内含子且因此导致特有的肿瘤特异性蛋白质序列;(4)染色体重排,在2种蛋白质的接合处产生具有肿瘤特异性序列的嵌合蛋白质(即基因融合);(5)框移突变或缺失,导致具有新颖肿瘤特异性蛋白质序列的新的开放阅读框。突变还可包括非移码插入缺失、错义或无义取代、剪接位点改变、基因组重排或基因融合或产生neoORF的任何基因组或表达改变中的一种或多种。
由例如肿瘤细胞中的剪接位点、移码、通读或基因融合突变产生的具有突变的肽或突变多肽可通过对肿瘤与正常细胞中的DNA、RNA或蛋白质进行测序来鉴定。
突变还可包括先前鉴定的肿瘤特异性突变。已知肿瘤突变可见于癌症体细胞突变目录(COSMIC)数据库。
多种方法可用于检测个体的DNA或RNA中特定突变或等位基因的存在。本领域中的进步已提供精确、容易且便宜的大规模SNP基因分型。例如,已描述若干种技术,包括动态等位基因特异性杂交(DASH)、微孔板阵列对角线凝胶电泳(MADGE)、焦磷酸测序、寡核苷酸特异性连接、TaqMan系统以及各种DNA“芯片”技术,诸如Affymetrix SNP芯片。这些方法利用通常通过PCR扩增靶基因区。仍有其他方法,基于通过侵入性裂解产生小信号分子,接着进行质谱法或固定化挂锁探针和滚环扩增。下文汇总本领域中已知用于检测特异性突变的若干种方法。
基于PCR的检测手段可包括同时多重扩增多个标志物。例如,选择PCR引物以产生大小不重叠且可同时分析的PCR产物是本领域中众所周知的。或者,可用经差异性标记且因此可各自经差异性检测的引物扩增不同的标志物。当然,基于杂交的检测手段允许样品中多个PCR产物的差异检测。本领域中已知其他技术以允许多个标志物的多重分析。
已开发若干种方法以便于基因组DNA或细胞RNA中单核苷酸多态性的分析。例如,单碱基多态性可通过使用特殊化核酸外切酶抗性核苷酸来检测,如例如Mundy,C.R.(美国专利号4,656,127)中所公开。根据所述方法,允许与紧靠着多形位点3'的等位基因序列互补的引物与获自特定动物或人的靶分子杂交。如果靶分子上的多形位点含有与所存在的特定核酸外切酶抗性核苷酸衍生物互补的核苷酸,则所述衍生物将并入到杂交引物的末端上。此类并入使得引物对核酸外切酶具有抗性,从而允许其检测。由于样品的核酸外切酶抗性衍生物的身份是已知的,故引物已对核酸外切酶具有抗性的发现揭露靶分子的多形位点中存在的核苷酸与反应中所用的核苷酸衍生物互补。这种方法的优势在于其不需要确定大量无关序列数据。
可使用基于溶液的方法确定多形位点的核苷酸的身份。Cohen,D.等(法国专利2,650,840;PCT申请号WO91/02087)。如在美国专利号4,656,127的芒迪方法(Mundy method)中,采用与紧靠着多形位点3'的等位基因序列互补的引物。所述方法使用经标记的双脱氧核苷酸衍生物确定所述位点的核苷酸的身份,如果所述核苷酸与多形位点的核苷酸互补,则将并入到引物的末端上。
称为遗传位分析或GBA的替代方法由Goelet,P.等(PCT申请号92/15712)描述。Goelet,P.等的方法使用经标记的终止子和与多形位点3'序列互补的引物的混合物。所并入的经标记的终止子因此通过所评估的靶分子的多形位点中存在的核苷酸确定且与其互补。与Cohen等(法国专利2,650,840;PCT申请号WO91/02087)的方法相比,Goelet,P.等的方法可以是非均相分析,其中引物或靶分子固定于固相。
已描述若干种用于分析DNA中多形位点的引物引导的核苷酸并入程序(Komher,J.S.等,Nucl.Acids.Res.17:7779-7784(1989);Sokolov,B.P.,Nucl.Acids Res.18:3671(1990);Syvanen,A.-C.等,Genomics 8:684-692(1990);Kuppuswamy,M.N.等,Proc.Natl.Acad.Sci.(U.S.A.)88:1143-1147(1991);Prezant,T.R.等,Hum.Mutat.1:159-164(1992);Ugozzoli,L.等,GATA 9:107-112(1992);Nyren,P.等,Anal.Biochem.208:171-175(1993))。这些方法与GBA的不同之处在于其利用并入经标记的脱氧核苷酸来区分多形位点处的碱基。在这种格式中,由于信号与并入的脱氧核苷酸的数量成比例,故在同一核苷酸的操作中发生的多态性可产生与操作的长度成比例的信号(Syvanen,A.-C.等,Amer.J.Hum.Genet.52:46-59(1993))。
许多方案直接从数百万个单独的DNA或RNA分子中并行获取序列信息。实时单分子合成测序技术依赖于荧光核苷酸的检测,因为其并入到与正测序的模板互补的DNA的新生链中。在一种方法中,将长度为30-50个碱基的寡核苷酸在5'端共价锚定于玻璃盖玻片上。这些锚定链执行两种功能。首先,如果模板被配置成具有与表面结合的寡核苷酸互补的捕捉尾部,则其充当靶模板链的捕捉位点。它们还充当模板引导的引物延伸的引物,形成序列阅读的基础。捕捉引物充当固定位点以便使用多个合成、检测及化学裂解染料接头来移除染料的循环进行序列测定。各循环由以下组成:添加聚合酶/经标记的核苷酸混合物、冲洗、成像和染料的裂解。在一个替代方法中,聚合酶被荧光供体分子修饰并固定在载玻片上,而各核苷酸用连接至γ-磷酸的受体荧光部分进行颜色编码。系统检测经荧光标记的聚合酶与经荧光修饰的核苷酸之间的相互作用,因为核苷酸并入到从头链中。也存在其他合成测序技术。
可使用任何适合的合成测序平台鉴定突变。如上文所述,目前可用四种主要合成测序平台:来自Roche/454Life Sciences的基因组测序仪、来自Illumina/Solexa的1G分析仪、来自Applied BioSystems的SOLiD系统以及来自Helicos Biosciences的Heliscope系统。合成测序平台也已由Pacific BioSciences和VisiGen Biotechnologies描述。在一些实施方案中,将要测序的多个核酸分子结合于支撑物(例如固体支撑物)。为将核酸固定于支撑物上,可在模板的3'和/或5'端添加捕捉序列/通用引发位点。核酸可通过将捕捉序列与共价连接于支撑物的互补序列杂交而结合于支撑物。捕捉序列(也称为通用捕捉序列)是与连接至支撑物的序列互补的核酸序列,其可双重充当通用引物。
作为捕捉序列的替代方案,偶联对(例如像抗体/抗原、受体/配体或如例如美国专利申请号2006/0252077中所述的抗生物素蛋白-生物素对)的一个成员可连接于各片段,而被捕捉在用所述偶联对的相应第二成员涂布的表面上。
在捕捉后,可例如通过单分子检测/测序来分析序列,例如,如实施例和美国专利号7,283,337中所述,包括模板依赖性合成测序。在合成测序中,表面结合的分子在聚合酶存在下暴露于多个经标记的三磷酸核苷酸。模板的序列通过并入到生长链的3'端的经标记的核苷酸的顺序来确定。这可实时进行或可以分步重复模式进行。对于实时分析,可将不同的光学标记并入各核苷酸并且可利用多个激光刺激并入的核苷酸。
测序还可包括其他大规模平行测序或下一代测序(NGS)技术和平台。大规模平行测序技术和平台的额外实例是Illumina HiSeq或MiSeq、Thermo PGM或Proton、Pac Bio RSII或Sequel、Qiagen的Gene Reader和Oxford Nanopore MinION。可使用其他类似的当前大规模平行测序技术,以及这些技术的后代。
可利用任何细胞类型或组织来获得用于本文所述的方法的核酸样品。例如,DNA或RNA样品可获自肿瘤或体液,例如通过已知技术(例如静脉穿刺)获得的血液或唾液。或者,可对干燥样品(例如头发或皮肤)进行核酸测试。另外,可从肿瘤获得样品用于测序并且可从正常组织获得另一样品用于测序,其中正常组织具有与肿瘤相同的组织类型。可从肿瘤获得样品用于测序并且可从正常组织获得另一样品用于测序,其中正常组织相对于肿瘤具有不同的组织类型。
肿瘤可包括以下一种或多种:肺癌、黑素瘤、乳腺癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
或者,可使用蛋白质质谱法鉴定或验证与肿瘤细胞上的MHC蛋白质结合的突变肽的存在。肽可从肿瘤细胞或从肿瘤免疫沉淀的HLA分子酸洗脱,然后使用质谱法鉴定。
IV.抗原
抗原可包括核苷酸或多肽。例如,抗原可以是编码多肽序列的RNA序列。因此,可用于疫苗中的抗原可包括核苷酸序列或多肽序列。
本文公开了包含通过本文公开的方法鉴定的肿瘤特异性突变的分离肽、包含已知肿瘤特异性突变的肽以及通过本文公开的方法鉴定的突变多肽或其片段。新抗原肽可描述于其编码序列的上下文中,其中新抗原包括编码相关多肽序列的核苷酸序列(例如DNA或RNA)。
本文还公开了这样的肽,所述肽衍生自与正常细胞或组织相比任何已知或已发现在肿瘤细胞或癌组织中改变表达的多肽,例如与正常细胞或组织相比任何已知或已发现在肿瘤细胞或癌组织中异常表达的多肽。可例如在COSMIC数据库中发现可衍生抗原肽的适合的多肽。COSMIC策划关于人癌症中体细胞突变的综合信息。所述肽包含肿瘤特异性突变。
由抗原核苷酸序列编码的一个或多个多肽可包含以下中的至少一者:对于长度为8-15、8、9、10、11、12、13、14或15个氨基酸的MHC I类肽,与MHC的结合亲和力为IC50值小于1000nM,在肽内部或附近存在促进蛋白酶体裂解的序列基序,以及存在促进TAP转运的序列基序。对于长度为6-30、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的MHC II类肽,在肽内部或附近存在促进通过细胞外或溶酶体蛋白酶(例如组织蛋白酶)裂解或HLA-DM催化的HLA结合的序列基序。
一个或多个抗原可递呈在肿瘤的表面上。
一个或多个抗原在具有肿瘤的受试者中可以是免疫原性的,例如能够在受试者中引发T细胞反应或B细胞反应。
在疫苗生产背景下,对于具有肿瘤的受试者,可不考虑在受试者中诱导自身免疫应答的一个或多个抗原。
至少一个抗原肽分子的大小可包含但不限于约5个、约6个、约7个、约8个、约9个、约10个、约11个、约12个、约13个、约14个、约15个、约16个、约17个、约18个、约19个、约20个、约21个、约22个、约23个、约24个、约25个、约26个、约27个、约28个、约29个、约30个、约31个、约32个、约33个、约34个、约35个、约36个、约37个、约38个、约39个、约40个、约41个、约42个、约43个、约44个、约45个、约46个、约47个、约48个、约49个、约50个、约60个、约70个、约80个、约90个、约100个、约110个、约120个或更多个氨基分子残基,以及其中可导出的任何范围。在具体实施方案中,抗原肽分子等于或小于50个氨基酸。
抗原肽和多肽可以是:对于MHC I类,15个残基或更小的长度且通常由约8至约11个残基、特别是9或10个残基组成;对于MHC II类,6-30个残基(包括端点)。
如果需要,可以若干种方式设计较长的肽。在一种情况下,当HLA等位基因上肽的递呈可能性经预测或已知时,较长的肽可由以下任一者组成:(1)个别递呈的具有朝向各对应基因产物的N末端和C末端延伸2-5个氨基酸的肽;(2)所递呈的肽中的一些或全部与各自的延伸序列的串接。在另一种情况下,当测序揭露肿瘤中所存在的长(>10个残基)新表位序列(例如归因于产生新颖肽序列的移码、通读或内含子包含)时,较长的肽将由以下组成:(3)新颖肿瘤特异性氨基酸的整个延伸段,因此绕过对基于计算或活体外测试选择最强HLA递呈的较短肽的需要。在两种情况下,使用较长的肽允许患者细胞进行内源性加工,并且可引起更有效的抗原递呈和诱导T细胞反应。
抗原肽和多肽可递呈在HLA蛋白质上。在一些方面,抗原肽和多肽以比野生型肽更大的亲和力递呈在HLA蛋白质上。在一些方面,抗原肽或多肽的IC50可至少小于5000nM、至少小于1000nM、至少小于500nM、至少小于250nM、至少小于200nM、至少小于150nM、至少小于100nM、至少小于50nM或更小。
在一些方面,抗原肽和多肽在施用于受试者时不诱导自身免疫应答和/或引起免疫耐受性。
还提供了包含至少两种或更多种抗原肽的组合物。在一些实施方案中,所述组合物含有至少两种不同的肽。至少两种不同的肽可衍生自相同的多肽。不同的多肽意指肽根据长度、氨基酸序列或两者而变化。所述肽衍生自已知或已发现含有肿瘤特异性突变的任何多肽,或者肽衍生自与正常细胞或组织相比任何已知或已发现在肿瘤细胞或癌组织中改变表达的多肽,例如与正常细胞或组织相比任何已知或已发现在肿瘤细胞或癌组织中异常表达的多肽。可例如在COSMIC数据库或AACR基因组证据瘤形成信息交换(GENIE)数据库中发现可衍生抗原肽的适合的多肽。COSMIC策划关于人癌症体细胞突变的综合信息。AACRGENIE汇总并连接了临床级癌症基因组数据与成千上万癌症患者的临床结果。所述肽含有肿瘤特异性突变。在一些方面,肿瘤特异性突变是特定癌症类型的驱动突变。
具有所需活性或特性的抗原肽和多肽可经修饰以提供某些所需属性,例如改进的药理学特征,同时增加或至少保留未修饰肽的实质上所有生物活性以结合所需MHC分子并活化适当T细胞。例如,抗原肽和多肽可进行各种变化,诸如保守或非保守取代,其中此类变化可在其使用中提供某些优势,诸如改进的MHC结合、稳定性或递呈。保守取代意指氨基酸残基用生物学和/或化学上类似的另一个氨基酸残基置换,例如一个疏水性残基置换另一个,或一个极性残基置换另一个。取代包括以下组合,诸如Gly、Ala;Val、Ile、Leu、Met;Asp、Glu;Asn、Gln;Ser、Thr;Lys、Arg;以及Phe、Tyr。单氨基酸取代的效应也可使用D-氨基酸探测。此类修饰可使用众所周知的肽合成程序进行,如例如Merrifield,Science 232:341-347(1986),Barany和Merrifield,The Peptides,Gross和Meienhofer编(N.Y.,AcademicPress),第1-284页(1979);以及Stewart和Young,Solid Phase Peptide Synthesis,(Rockford,Ill.,Pierce),第2版(1984)中所述。
肽和多肽用各种氨基酸模拟物或非天然氨基酸修饰可在提高肽和多肽的体内稳定性方面特别有用。稳定性可以多种方式加以分析。例如,肽酶和各种生物介质诸如人血浆和血清已用于测试稳定性。参见例如Verhoef等,Eur.J.Drug Metab Pharmacokin.11:291-302(1986)。肽的半衰期可使用25%人血清(v/v)分析方便地确定。方案一般如下。将汇集的人血清(AB型,非加热不活化)在使用之前通过离心去脂。然后将血清用RPMI组织培养基稀释至25%并用于测试肽稳定性。在预定时间间隔下,移出少量反应溶液并加入到6%三氯乙酸或乙醇水溶液中。将混浊的反应样品冷却(4℃)15分钟,然后旋转集结沉淀的血清蛋白质。然后使用稳定性特异性色谱条件通过逆相HPLC确定肽的存在。
肽和多肽可经修饰以提供除改进的血清半衰期以外的所需属性。例如,肽诱导CTL活性的能力可通过与含有至少一个能够诱导T辅助细胞反应的表位的序列连接来增强。免疫原性肽/T辅助细胞缀合物可通过间隔分子连接。间隔子通常由相对较小的中性分子诸如氨基酸或氨基酸模拟物构成,其在生理条件下实质上不带电。间隔子通常选自例如Ala、Gly或非极性氨基酸或中性极性氨基酸的其他中性间隔子。应理解,任选地存在的间隔子无需由相同残基组成,并且因此可以是杂寡聚物或均寡聚物。当存在时,间隔子将通常是至少一个或两个残基,更通常三至六个残基。或者,肽可在不存在间隔子的情况下连接到T辅助肽。
抗原肽可直接或经由在肽的氨基或羧基端处的间隔子连接到T辅助肽。抗原肽或T辅助肽的氨基端可经酰化。示例性T辅助肽包括破伤风类毒素830-843、流感307-319、疟疾环子孢子382-398和378-389。
蛋白质或肽可通过本领域技术人员已知的任何技术制造,包括通过标准分子生物学技术表达蛋白质、多肽或肽;从天然来源分离蛋白质或肽;或化学合成蛋白质或肽。先前已公开对应于各种基因的核苷酸和蛋白质、多肽和肽序列,并且可见于本领域一般技术人员已知的计算机化数据库中。一种此类数据库是位于美国国家卫生研究院(NationalInstitutes of Health)网站的国家生物技术信息中心的Genbank和GenPept数据库。已知基因的编码区可使用本文所公开或本领域一般技术人员应知晓的技术扩增和/或表达。或者,蛋白质、多肽和肽的各种市售制剂已为本领域技术人员所知。
在另一方面,抗原包括编码抗原肽或其部分的核酸(例如多核苷酸)。多核苷酸可以是例如DNA、cDNA、PNA、CNA、RNA(例如mRNA)、单链和/或双链、或原生或稳定形式的多核苷酸,例如像具有硫代磷酸骨架的多核苷酸,或其组合,并且其可含有或可不含内含子。另一方面提供一种能够表达多肽或其部分的表达载体。不同细胞类型的表达载体在本领域中众所周知的并且无需过度实验便可选择。一般来说,DNA以适当定向插入到表达载体诸如质粒中且以正确阅读框进行表达。如果需要,DNA可连接到由所需宿主识别的适当转录和翻译调节控制核苷酸序列,而此类控制件一般可用于表达载体中。然后将载体经由标准技术引入到宿主中。指导可见于例如Sambrook等(1989)Molecular Cloning,A Laboratory Manual,Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y.中。
V.疫苗组合物
本文还公开了一种能够引起特异性免疫应答(例如肿瘤特异性免疫应答)的免疫原性组合物,例如疫苗组合物。疫苗组合物通常包含例如使用本文所述的方法选择的一个或多个抗原。疫苗组合物也可称为疫苗。
疫苗可含有1至30个肽;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个不同的肽;6、7、8、9、10、11、12、13或14个不同的肽;或12、13或14个不同的肽。肽可包括翻译后修饰。疫苗可含有1至100或更多个核苷酸序列;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100或更多个不同的核苷酸序列;6、7、8、9、10、11、12、13或14个不同的核苷酸序列;或12、13或14个不同的核苷酸序列。疫苗可含有1至30个抗原序列;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100或更多个不同的抗原序列;6、7、8、9、10、11、12、13或14个不同的抗原序列;或12、13或14个不同的抗原序列。
在一个实施方案中,选择不同的肽和/或多肽或其编码核苷酸序列,以使得肽和/或多肽能够与不同的MHC分子(诸如不同的MHC I类分子和/或不同的MHC II类分子)缔合。在一些方面,一种疫苗组合物包含能够与最常出现的MHC I类分子和/或不同的MHC II类分子缔合的肽和/或多肽的编码序列。因此,疫苗组合物可包含能够与至少2个优选的、至少3个优选的、或至少4个优选的MHC I类分子和/或不同的MHC II类分子缔合的不同片段。
疫苗组合物能够引起特异性细胞毒性T细胞反应和/或特异性辅助T细胞反应。
疫苗组合物还可包含佐剂和/或载剂。有用的佐剂和载剂的实例在下文中给出。组合物可与载剂缔合,例如像蛋白质或抗原递呈细胞,例如像能够将肽递呈于T细胞的树突状细胞(DC)。
佐剂是混合到疫苗组合物中增加或以其他方式修饰对抗原的免疫应答的任何物质。载剂可以是骨架结构,例如能够与抗原缔合的多肽或多糖。任选地,佐剂是共价或非共价结合的。
佐剂提高对抗原的免疫应答的能力通常显现为免疫介导性反应的显著或实质性增加或疾病症状的减少。例如,体液免疫的提高通常显现为针对抗原所产生的抗体的效价显著增加,并且T细胞活性的增加通常显现为细胞增殖或细胞毒性或细胞因子分泌增加。佐剂也可改变免疫应答,例如通过将主要体液或Th反应变为主要细胞或Th反应。
适合的佐剂包括但不限于1018ISS、矾、铝盐、Amplivax、AS15、BCG、CP-870,893、CpG7909、CyaA、dSLIM、GM-CSF、IC30、IC31、咪喹莫特(Imiquimod)、ImuFact IMP321、ISPatch、ISS、ISCOMATRIX、JuvImmune、LipoVac、MF59、单磷酰基脂质A、Montanide IMS 1312、Montanide ISA 206、Montanide ISA 50V、Montanide ISA-51、OK-432、OM-174、OM-197-MP-EC、ONTAK、PepTel载体系统、PLG微粒、雷西莫特(resiquimod)、SRL172、病毒颗粒和其他病毒样颗粒、YF-17D、VEGF捕获剂、R848、β-葡聚糖、Pam3Cys、Aquila的源于皂素的QS21刺激子(Aquila Biotech,Worcester,Mass.,USA)、分支杆菌提取物和合成细菌细胞壁模拟物,以及其他专用佐剂,诸如Ribi的Detox.Quil或Superfos。佐剂诸如不完全弗氏佐剂或GM-CSF是有用的。先前已描述特异性针对树突状细胞的若干种免疫佐剂(例如MF59)及其制备(Dupuis M等,Cell Immunol.1998;186(1):18-27;Allison A C;Dev Biol Stand.1998;92:3-11)。也可使用细胞因子。若干种细胞因子已直接关联于:影响树突状细胞迁移至淋巴组织(例如TNF-α)、加速树突状细胞成熟变为T-淋巴细胞的有效抗原递呈细胞(例如GM-CSF、IL-1和IL-4)(美国专利号5,849,589,其具体地以引用的方式整体并入本文)以及充当免疫佐剂(例如IL-12)(Gabrilovich D I等,J Immunother Emphasis TumorImmunol.1996(6):414-418)。
还已报道CpG免疫刺激性寡核苷酸增强佐剂在疫苗环境中的效应。也可使用其他TLR结合分子,诸如结合RNA的TLR7、TLR 8和/或TLR 9。
有用佐剂的其他实例包括但不限于经化学修饰的CpG(例如CpR、Idera)、聚(I:C)(例如聚i:CI2U)、非CpG细菌DNA或RNA以及免疫活性小分子和抗体,诸如环磷酰胺、舒尼替尼(sunitinib)、贝伐单抗(bevacizumab)、西乐葆(celebrex)、NCX-4016、西地那非(sildenafil)、他达拉非(tadalafil)、伐地那非(vardenafil)、索拉菲尼(sorafinib)、XL-999、CP-547632、帕佐泮尼(pazopanib)、ZD2171、AZD2171、伊匹单抗、曲美木单抗(tremelimumab)和SC58175,其可起治疗作用和/或充当佐剂。佐剂和添加剂的量和浓度可容易由本领域技术人员确定而无需过度实验。额外佐剂包括集落刺激因子,诸如颗粒球巨噬细胞集落刺激因子(GM-CSF,沙格司亭(sargramostim))。
疫苗组合物可包含多于一种不同的佐剂。此外,治疗组合物可包含任何佐剂物质,包括上文中的任一者或其组合。还预期疫苗和佐剂可一起或以任何适当的顺序分开施用。
载剂(或赋形剂)可独立于佐剂存在。载剂的功能可例如为增加特定突变体的分子量以提高活性或免疫原性,赋予稳定性,增加生物活性或增加血清半衰期。此外,载剂可辅助递呈肽至T细胞。载剂可以是本领域技术人员已知的任何适合的载剂,例如蛋白质或抗原递呈细胞。载剂蛋白可以是但不限于匙孔螺血氰蛋白、血清蛋白质(诸如转铁蛋白)、牛血清白蛋白、人血清白蛋白、甲状腺球蛋白或卵白蛋白、免疫球蛋白或激素,诸如胰岛素或棕榈酸。对于人免疫接种,载剂一般为生理学上可接受的载剂,其为人可接受的且为安全的。然而,破伤风类毒素和/或白喉类毒素是适合的载剂。或者,载剂可以是葡聚糖,例如琼脂糖。
细胞毒性T细胞(CTL)识别与MHC分子结合的肽形式的抗原,而非完整外来抗原本身。MHC分子本身位于抗原递呈细胞的细胞表面上。因此,如果存在肽抗原、MHC分子和APC的三聚体复合物,则可能活化CTL。对应地,如果不仅肽用于活化CTL,而且如果另外添加具有相应MHC分子的APC,则可加强免疫应答。因此,在一些实施方案中,疫苗组合物另外含有至少一种抗原递呈细胞。
抗原还可包括在基于病毒载体的疫苗平台中,诸如牛痘、禽痘、自我复制甲病毒、马拉巴病毒(marabavirus)、腺病毒(参见例如Tatsis等,Adenoviruses,MolecularTherapy(2004)10,616-629)或慢病毒,包括但不限于第二、第三或杂交第二/第三代慢病毒以及任一代的重组慢病毒,其经设计以靶向特定细胞类型或受体(参见例如Hu等,Immunization Delivered by Lentiviral Vectors for Cancer and InfectiousDiseases,Immunol Rev.(2011)239(1):45-61;Sakuma等,Lentiviral vectors:basic totranslational,Biochem J.(2012)443(3):603-18;Cooper等,Rescue of splicing-mediated intron loss maximizes expression in lentiviral vectors containingthe human ubiquitin C promoter,Nucl.Acids Res.(2015)43(1):682-690;Zufferey等,Self-Inactivating Lentivirus Vector for Safe and Efficient In Vivo GeneDelivery,J.Virol.(1998)72(12):9873-9880)。取决于上述基于病毒载体的疫苗平台的包装能力,这种方法可递送编码一个或多个新抗原肽的一个或多个核苷酸序列。序列可侧接非突变序列,可由接头分开或可在前面有一个或多个靶向亚细胞区室的序列(参见例如Gros等,Prospective identification of neoantigen-specific lymphocytes in theperipheral blood of melanoma patients,Nat Med.(2016)22(4):433-8;Stronen等,Targeting of cancer neoantigens with donor-derived T cell receptorrepertoires,Science.(2016)352(6291):1337-41;Lu等,Efficient identification ofmutated cancer antigens recognized by T cells associated with durable tumorregressions,Clin Cancer Res.(2014)20(13):3401-10)。在引入到宿主中后,经感染细胞表达抗原,从而引发针对肽的宿主免疫(例如CTL)反应。用于免疫接种方案中的牛痘载体和方法描述于例如美国专利号4,722,848中。另一载体是卡介苗(Bacille Calmette Guerin,BCG)。BCG载体描述于Stover等(Nature 351:456-460(1991))中。根据本文描述,用于抗原的治疗性施用或免疫接种的各种其他疫苗载体,例如伤寒沙门氏菌(Salmonella typhi)载体等对于本领域技术人员将是显而易见的。
V.A.抗原盒
鉴于本文所提供的教导内容,用于选择一个或多个抗原、克隆及构建“盒”及其插入至病毒载体中的方法在本领域的技能内。“抗原盒”意指经选择的抗原或多个抗原与转录抗原且表达转录产物所必需的其他调控元件的组合。抗原或多个抗原可以允许转录的方式可操作地连接到调控组分。此类组分包括可驱动被病毒载体转染的细胞中表达抗原的常规调控元件。因此,抗原盒也可含有经选择的启动子,其连接到抗原并且与其他任选的调控元件一起位于重组载体的经选择的病毒序列内。
有用的启动子可以是组成型启动子或经调控(诱导型)启动子,其将能够控制待表达的抗原的量。例如,合乎需要的启动子是细胞巨大病毒即刻早期启动子/增强子的启动子[参见例如Boshart等,Cell,41:521-530(1985)]。另一种合乎需要的启动子包括劳斯肉瘤(Rous sarcoma)病毒LTR启动子/增强子。另一种启动子/增强子序列是鸡细胞质β-肌动蛋白启动子[T.A.Kost等人,Nucl.Acids Res.,11(23):8287(1983)]。其他适合或合乎需要的启动子可由本领域技术人员选择。
抗原盒还可包括与病毒载体序列异源的核酸序列,包括提供转录本的有效聚腺苷酸化信号(poly(A)、poly-A或pA)的序列及具有功能性剪接供体和受体位点的内含子。本发明的示例性载体中使用的普通poly-A序列衍生自乳多泡病毒SV-40。poly-A序列一般可在基于抗原的序列之后及在病毒载体序列之前插入盒中。普通内含子序列也可衍生自SV-40,并且称为SV-40T内含子序列。抗原盒也可含有位于启动子/增强子序列与抗原之间的此类内含子。这些和其他普通载体元件的选择是常规的[参见例如Sambrook等,“MolecularCloning.A Laboratory Manual.”,第2版,Cold Spring Harbor Laboratory,New York(1989)和其中引用的参考文献]并且许多此类序列可从商业和工业来源以及Genbank获得。
抗原盒可具有一个或多个抗原。例如,给定盒可包含1-10、1-20、1-30、10-20、15-25、15-20、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个抗原。抗原可彼此直接连接。抗原也可用接头彼此连接。抗原可相对于彼此呈任一定向,包括N至C或C至N。
如上文所述,抗原盒可位于病毒载体中任何经选择的缺失位点,诸如E1基因区缺失或E3基因区缺失的位点等可经选择的位点。
可以使用下式描述抗原盒,以从5'到3'描述每个元件的有序序列:
(Pa-(L5b-Nc-L3d)X)Z-(P2h-(G5e-Uf)Y)W-G3g
其中P和P2包含启动子核苷酸序列,N包含MHC I类表位编码核酸序列,L5包含5'接头序列,L3包含3'接头序列,G5包含编码氨基酸接头的核酸序列,G3包含编码氨基酸接头的至少一个核酸序列中的一个,U包含MHC II类抗原编码核酸序列,其中对于每个X,对应的Nc是表位编码核酸序列,其中对于每个Y,对应的Uf是抗原编码核酸序列。可以通过选择存在的元件数来进一步定义组成和有序序列,例如其中a=0或1,其中b=0或1,其中c=1,其中d=0或1,其中e=0或1,其中f=1,其中g=0或1,其中h=0或1,X=1至400,Y=0、1、2、3、4或5,Z=1至400,并且W=0、1、2、3、4或5。
在一个实例中,存在的元件包括其中a=0,b=1,d=1,e=1,g=1,h=0,X=10,Y=2,Z=1和W=1,描述了其中不存在其他启动子(即仅存在由载体骨架(例如病毒骨架诸如甲病毒骨架)提供的启动子核苷酸序列),存在20个MHC I类表位,对于每个N存在5'接头,对于每个N存在3'接头,存在2个MHC II类表位,存在连接两个MHC II类表位的接头,存在将两个MHC II类表位的5'端连接到最后一个MHC I类表位的3'端的接头,以及存在将两个MHCII类表位的3'端连接到载体骨架(例如病毒骨架诸如甲病毒骨架)的接头。将抗原盒的3'端连接到载体骨架(例如病毒骨架诸如甲病毒骨架)的实例包括直接连接到由载体骨架(例如病毒骨架诸如甲病毒骨架)提供的3'UTR元件,诸如3'19-nt CSE。将抗原盒的5'端连接到载体骨架(例如病毒骨架诸如甲病毒骨架)的实例包括直接连接到26S启动子序列、甲病毒5'UTR、51nt CSE或24nt CSE。
其他实例包括:其中a=1描述其中存在不同于载体骨架(例如病毒骨架诸如甲病毒骨架)提供的启动子核苷酸序列的启动子;其中a=1且Z为大于1,其中存在多个除载体骨架(例如病毒骨架诸如甲病毒骨架)提供的启动子核苷酸序列以外的启动子,每个驱动1个或更多个不同的MHC I类表位编码核酸序列的表达;其中h=1描述其中存在一个单独的启动子来驱动MHC II类抗原编码核酸序列的表达;并且其中g=0描述MHC II类抗原编码核酸序列(如果存在)与载体骨架(例如病毒骨架诸如甲病毒骨架)直接连接。
其他实例包括其中存在的每个MHC I类表位可以具有5'接头、3'接头、都不具有或两者都有的情况。在同一抗原盒中存在多于一个MHC I类表位的实例中,一些MHC I类表位可能同时具有5'接头和3'接头,而其他MHC I类表位可能具有5'接头、3'接头或两者都没有。在同一抗原盒中存在多于一个MHC I类表位的其他实例中,一些MHC I类表位可能具有5'接头或3'接头,而其他MHC I类表位可能具有5'接头、3'接头或两者都没有。
在同一抗原盒中存在多于一个MHC II类表位的实例中,一些MHC II类表位可能同时具有5'接头和3'接头,而其他MHC II类表位可能具有5'接头、3'接头或两者都没有。在同一抗原盒中存在多于一个MHC II类表位的其他实例中,一些MHC II类表位可能具有5'接头或3'接头,而其他MHC II类表位可能具有5'接头、3'接头或两者都没有。
启动子核苷酸序列P和/或P2可以与载体骨架(例如病毒骨架诸如甲病毒骨架)提供的启动子核苷酸序列相同。例如,由载体骨架(例如病毒骨架诸如甲病毒骨架)提供的启动子序列Pn和P2可各自包含26S亚基因组启动子。启动子核苷酸序列P和/或P2可以不同于载体骨架(例如病毒骨架诸如甲病毒骨架)提供的启动子核苷酸序列,也可以彼此不同。
5'接头L5可以是原生序列或非天然序列。非天然序列包括但不限于AAY、RR和DPP。3'接头L3也可以是原生序列或非天然序列。另外,L5和L3可以都是原生序列,可以都是非天然序列,或者一个可以是原生的而另一个可以是非天然的。对于每个X,氨基酸接头的长度可以是2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100个或更多个氨基酸。对于每个X,氨基酸接头的长度也可以是至少3、至少4、至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸。
对于每个Y,氨基酸接头G5的长度可以是2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100个或更多个氨基酸。对于每个Y,氨基酸接头的长度也可以是至少3、至少4、至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸。
氨基酸接头G3的长度可以是2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100个或更多个氨基酸。G3的长度也可以是至少3、至少4、至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸。
对于每个X,每个N可以编码长度为7-15个氨基酸的MHC I类表位。对于每个X,每个N也可以编码长度为5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的MHC I类表位。对于每个X,每个N也可以编码长度为至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸的MHC I类表位。
V.B.免疫检查点
本文所述的载体,诸如本文所述的C68载体或本文所述的甲病毒载体,可包含编码至少一种抗原的核酸,并且同一或另一载体可包含编码至少一种免疫调节物(例如抗体诸如scFv)的核酸,其结合于免疫检查点分子且阻断免疫检查点分子的活性。载体可包含抗原盒和一个或多个编码检查点抑制剂的核酸分子。
可靶向用于阻断或抑制的说明性免疫检查点分子包括但不限于CTLA-4、4-1BB(CD137)、4-1BBL(CD137L)、PDL1、PDL2、PD1、B7-H3、B7-H4、BTLA、HVEM、TIM3、GAL9、LAG3、TIM3、B7H3、B7H4、VISTA、KIR、2B4(属于CD2家族的分子并且在所有NK、γδ和记忆CD8+(αβ)T细胞上表达)、CD160(也称为BY55)和CGEN-15049。免疫检查点抑制剂包括抗体或其抗原结合片段或其他结合蛋白,其结合且阻断或抑制以下中的一个或多个的活性:CTLA-4、PDL1、PDL2、PD1、B7-H3、B7-H4、BTLA、HVEM、TIM3、GAL9、LAG3、TIM3、B7H3、B7H4、VISTA、KIR、2B4、CD160和CGEN-15049。说明性免疫检查点抑制剂包括曲美木单抗(CTLA-4阻断抗体)、抗OX40、PD-L1单克隆抗体(抗B7-H1;MEDI4736)、伊匹单抗、MK-3475(PD-1阻断剂)、纳武单抗(Nivolumamb)(抗PD1抗体)、CT-011(抗PD1抗体)、BY55单克隆抗体、AMP224(抗PDL1抗体)、BMS-936559(抗PDL1抗体)、MPLDL3280A(抗PDL1抗体)、MSB0010718C(抗PDL1抗体)和Yervoy/伊匹单抗(抗CTLA-4检查点抑制剂)。抗体编码序列可使用本领域中的普通技能经工程改造至诸如C68的载体中。示例性方法描述于Fang等,Stable antibody expressionat therapeutic levels using the 2A peptide.Nat Biotechnol.2005年5月;23(5):584-90.电子版2005年4月17日中;其出于所有目的以引用的方式并入本文。
也可编码在与抗原编码盒相同的载体系统中编码的免疫调节物(例如检查点抑制剂抗体,诸如抗CTLA4抗体或抗PD1抗体),使得编码免疫调节物的核酸序列被转录为与抗原编码核酸序列相同的转录本的一部分。可将允许抗原和免疫调节物两者翻译的额外的元件并入核酸序列盒中。例如,内部核糖体进入序列(IRES)序列可用于分离编码抗原和免疫调节物的序列,从而允许抗原和免疫调节物的单独翻译。在另一个实例中,可以在抗原与免疫调节物之间并入编码自裂解2A肽的序列,从而允许抗原和免疫调节物两者都翻译为同一蛋白质的一部分,然后裂解2A肽;产生用于抗原和免疫调节物的单独蛋白质。这些实例并不意味着是限制性的,并且还应理解,可以组合多个元件以促进抗原和免疫调节物两者的共表达,诸如同时使用IRES序列和2A肽编码序列。另外,可以将弗林蛋白酶裂解位点编码序列并入2A肽编码序列的5'。弗林蛋白酶裂解位点允许自裂解后移除2A肽残基。
在抗原和免疫调节物在同一转录本上编码的实例中,抗原和免疫调节物的顺序可以是任何顺序。例如,在使用IRES序列分离抗原和免疫调节物的情况下,顺序从5'到3';可以是抗原-IRES-免疫调节物方向,或者免疫调节物-IRES-抗原方向。
另外,也可编码在与抗原编码盒相同的载体系统中编码的免疫调节物,使得编码免疫调节物的核酸序列在与抗原编码核酸序列不同的转录本上转录。例如,可以并入单独的启动子以独立地驱动免疫调节物和抗原编码核酸序列的转录。单独的启动子可以是相同或不同的启动子,并且每个启动子可以是诱导型或组成型启动子。示例性启动子序列包括但不限于CMV、SV40、EF-1、RSV、PGK、MCK、HSA和EBV启动子序列。在另一个实例中,可将抗原编码盒和编码免疫调节物的核酸序列插入同一病毒载体的不同区域(包括缺失区域),使得每个序列独立地被转录。在一个实例中,设计了其中表达盒被引入到缺失的E1区中,并且免疫检查点抑制剂被引入到E1/E3缺失的ChAdV68病毒载体中缺失的E3区中的一种载体。
V.C.疫苗设计和制造的额外考虑
V.C.1.确定覆盖所有肿瘤亚克隆的肽集合
躯干肽,意指由所有或大部分肿瘤亚克隆递呈的那些肽,可优先包括于疫苗中。53任选地,如果不存在经预测以高概率递呈且具有免疫原性的躯干肽,或如果经预测以高概率递呈且具有免疫原性的躯干肽的数量足够小以致额外非躯干肽可包括于疫苗中,则其他肽可通过估计肿瘤亚克隆的数量和身份且选择肽以使疫苗所覆盖的肿瘤亚克隆的数量达到最大而进行优先排序。54
V.C.2.抗原优先排序
在应用所有上述抗原过滤器后,与疫苗技术可支持的相比,更多候选抗原仍可包含于疫苗中。另外,可保留关于抗原分析的各个方面的不确定性,并且候选疫苗抗原的不同特性之间可存在折衷。因此,可考虑整合式多维模型代替选择过程的各步骤中的预定过滤器,将候选抗原置于具有至少以下轴的空间中且使用整合方法优化选择。
1.自身免疫或耐受性的风险(生殖系的风险)(自身免疫的风险较低通常为优选的)
2.测序伪影的概率(伪影的概率较低通常为优选的)
3.免疫原性的概率(免疫原性的概率较高通常为优选的)
4.递呈的概率(递呈的概率较高通常为优选的)
5.基因表达(较高表达通常为优选的)
6.HLA基因的覆盖率(参与抗原集合递呈的HLA分子数量越大可降低肿瘤经由HLA分子的下调或突变逃避免疫攻击的概率)
7.HLA类的覆盖率(同时覆盖HLA-I和HLA-II可能会增加治疗反应的概率并降低肿瘤逃避的概率)
另外,任选地,如果预测抗原是由患者的全部或部分肿瘤中失去或失活的HLA等位基因递呈的,则可以从疫苗接种中除名(例如排除)抗原。HLA等位基因的丢失可能通过体细胞突变、杂合性丢失或基因座的纯合缺失而发生。检测HLA等位基因体细胞突变的方法是本领域中众所周知的,例如(Shukla等,2015)。同样充分描述了检测体细胞LOH和纯合缺失(包括HLA基因座)的方法。(Carter等,2012;McGranahan等,2017;Van Loo等,2010)。如果质谱数据表明预测的抗原未由预测的HLA等位基因递呈,则抗原也可以被除名。
V.D.甲病毒
V.D.1.甲病毒生物学
甲病毒是披膜病毒科的成员,并且为正义单链RNA病毒。成员通常分类为旧世界诸如辛德毕斯、罗斯河、马雅罗、基孔肯尼亚及塞姆利基森林病毒,或新世界诸如东部马脑炎、奥拉、摩根堡、或委内瑞拉马脑炎及其衍生病毒株TC-83(Strauss Microbrial Review1994)。天然甲病毒基因组通常约12kb长,其中前三分之二含有编码非结构蛋白(nsP)的基因,所述非结构蛋白形成用于病毒基因组自我复制的RNA复制复合物,并且最后三分之一含有编码用于病毒粒子产生的结构蛋白的亚基因组表达盒(Frolov RNA 2001)。
甲病毒的模型生命周期涉及若干个不同步骤(Strauss Microbrial Review1994,Jose Future Microbiol 2009)。在病毒附着于宿主细胞后,病毒粒子与内吞区室内的膜融合,导致基因组RNA最终释放至胞溶质中。以正链定向并包含5'甲基鸟苷酸帽和3'polyA尾部的基因组RNA经翻译以产生形成复制复合物的非结构蛋白nsP1-4。在感染早期,正链然后由复合物复制成负链模板。在当前模型中,复制复合物随着感染进展被进一步加工,使得所得经加工的复合物转换成将负链转录成全长正链基因组RNA以及含有结构基因的26S亚基因组正链RNA。甲病毒的若干个保守序列元件(CSE)已鉴定为可能在各种RNA复制步骤中起作用,包括:负链模板的正链RNA复制中的5'UTR的互补序列、基因组模板的负链合成复制中的51-nt CSE、负链的亚基因组RNA转录中的在nsP与26S RNA之间的接合区中的24-nt CSE,以及正链模板的负链合成中的3'19-nt CSE。
在各种RNA物种复制后,病毒粒子然后通常在病毒的天然生命周期中组装。26SRNA经翻译且所得蛋白质经进一步加工以产生结构蛋白,其包括衣壳蛋白、糖蛋白E1和E2以及两个小多肽E3和6K(Strauss 1994)。发生病毒RNA的衣壳化,衣壳蛋白通常仅特异性针对所包装的基因组RNA,接着病毒粒子组装且在膜表面出芽。
V.D.2.甲病毒作为递送载体
甲病毒(包括甲病毒序列、特征和其他元件)可用于生成基于甲病毒的递送载体(也称为甲病毒载体、甲病毒病毒载体、甲病毒疫苗载体、自我复制RNA(srRNA)载体或自我扩增RNA(samRNA)载体)。甲病毒先前已经工程改造以用作表达载体系统(Pushko 1997,Rheme2004)。甲病毒提供若干种优势,特别是在可能需要异源抗原表达的疫苗环境中。由于在宿主胞溶质中自我复制的能力,故甲病毒载体一般能够在细胞内产生高拷贝数的表达盒,从而导致高水平异源抗原产生。另外,载体一般为瞬时的,从而使得生物安全性得以改进以及减少对载体的免疫耐受性的诱导。与其他标准病毒载体诸如人腺病毒相比,公众一般也缺乏对甲病毒载体预先存在的免疫性。基于甲病毒的载体也一般导致对经感染细胞的细胞毒性反应。在一定程度上,细胞毒性在疫苗环境中对于适当违禁引发对所表达的异源抗原的免疫应答可以是重要的。然而,所需细胞毒性的程度可以是平衡作用,并且因此已开发若干种减毒甲病毒,包括VEE的TC-83病毒株。因此,本文所述的抗原表达载体的实例可利用甲病毒骨架,其允许高水平的抗原表达,引发对抗原的稳固免疫应答,不引发对载体本身的免疫应答,并且可以安全方式使用。此外,抗原表达盒可经设计以经由优化载体使用的甲病毒序列(包括但不限于衍生自VEE或其减毒衍生物TC-83的序列)而引发不同水平的免疫应答。
已使用甲病毒序列工程改造若干种表达载体设计策略(Pushko1997)。在一个策略中,甲病毒载体设计包括在结构蛋白基因下游插入26S启动子序列元件的第二拷贝,接着是异源基因(Frolov 1993)。因此,除天然非结构蛋白和结构蛋白之外,也产生表达异源蛋白的额外亚基因组RNA。在此系统中,存在用于产生感染性病毒粒子的所有元件,因此可能发生在未感染细胞中反复轮表达载体的感染。
另一种表达载体设计利用辅助病毒系统(Pushko 1997)。在此策略中,结构蛋白由异源基因替代。因此,在由仍完整的非结构基因介导病毒RNA的自我复制之后,26S亚基因组RNA提供异源蛋白的表达。传统上,表达结构蛋白的额外载体然后诸如通过细胞系的共转染以反式供应,以产生感染性病毒。系统详细描述于USPN 8,093,021中,其出于所有目的以引用的方式整体并入本文。辅助载体系统提供限制形成感染性粒子的可能性的益处,因此提高生物安全性。另外,辅助载体系统减小总载体长度,潜在提高复制和表达效率。因此,本文所述的抗原表达载体的实例可利用结构蛋白由抗原盒替代的甲病毒骨架,所得载体降低生物安全问题,同时由于整体表达载体尺寸减小而促进有效表达。
V.D.3.体外甲病毒产生
甲病毒递送载体一般是正义RNA多核苷酸。本领域中众所周知的用于产生RNA的适宜技术为体外转录IVT。在此技术中,首先通过本领域技术人员众所周知的技术产生所需载体的DNA模板,包括标准分子生物学技术,诸如克隆、限制性消化、连接、基因合成和聚合酶链反应(PCR)。DNA模板在期望转录成RNA的序列的5'端处含有RNA聚合酶启动子。启动子包括但不限于噬菌体聚合酶启动子,诸如T3、T7或SP6。DNA模板然后与适当RNA聚合酶、缓冲剂和核苷酸(NTP)一起温育。所得RNA多核苷酸可任选地经进一步修饰,包括但不限于添加5'帽结构,诸如7-甲基鸟苷或相关结构,以及任选地修饰3'端以包括聚腺苷酸(polyA)尾部。RNA可然后使用本领域中众所周知的技术纯化,诸如苯酚-氯仿提取。
V.D.4.经由脂质纳米粒子递送
在疫苗载体设计中考虑的一个重要方面是针对载体本身的免疫性(Riley 2017)。这可以呈对载体本身(诸如某些人腺病毒系统)预先存在的免疫性形式,或呈在疫苗施用后对载体产生免疫性的形式。如果进行相同疫苗的多次施用(诸如分开的初免和加强剂量),或如果使用相同疫苗载体系统递送不同的抗原盒,则后者为重要的考虑因素。
在甲病毒载体的情况下,标准递送方法是先前论述的辅助病毒系统,其以反式提供衣壳、E1和E2蛋白质以产生感染性病毒粒子。然而,重要的是注意E1和E2蛋白质通常是中和抗体的主要靶标(Strauss 1994)。因此,如果中和抗体靶向感染性粒子,则使用甲病毒载体将所关注的抗原递送到靶细胞的功效可能会降低。
病毒粒子介导的基因递送的替代方案是使用纳米材料递送表达载体(Riley2017)。重要的是,纳米材料媒介物可由非免疫原性材料制成且一般避免引发对递送载体本身的免疫性。这些材料可包括但不限于脂质、无机纳米材料和其他聚合材料。脂质可以是阳离子、阴离子或中性的。材料可以是合成或天然来源的,并且在一些情况下是可生物降解的。脂质可包括脂肪、胆固醇、磷脂、脂质缀合物,包括但不限于聚乙二醇(PEG)缀合物(聚乙二醇化脂质)、蜡、油、甘油酯和脂溶性维生素。
脂质纳米粒子(LNP)是有吸引力的递送系统,因为脂质的两亲性使得能够形成膜和囊泡状结构(Riley 2017)。一般来说,这些囊泡通过吸收至靶细胞的膜中并将核酸释放至胞溶质中来递送表达载体。另外,LNP可经进一步修饰或官能化以有助于靶向特定细胞类型。LNP设计中的另一考虑因素是靶向效率与细胞毒性之间的平衡。脂质组合物一般包括阳离子、中性、阴离子和两性脂质的确定的混合物。在一些情况下,包括特定脂质以防止LNP聚集,防止脂质氧化,或提供有助于额外部分附着的功能性化学基团。脂质组合物可影响整体LNP大小和稳定性。在一个实例中,脂质组合物包含二亚油基甲基-4-二甲基氨基丁酸酯(MC3)或MC3样分子。MC3和MC3样脂质组合物可经配制以包括一种或多种其他脂质,诸如PEG或PEG缀合的脂质、固醇或中性脂质。
直接暴露于血清的核酸载体(诸如表达载体)可具有若干种不期望的结果,包括核酸由血清核酸酶降解或游离核酸对免疫系统的脱靶刺激。因此,包封甲病毒载体可用于避免降解,同时也避免潜在的脱靶影响。在某些实例中,甲病毒载体完全包封在递送媒介物内,诸如在LNP的水性内部。甲病毒载体包封在LNP内可通过本领域技术人员熟知的技术来进行,诸如在微流体液滴生成装置上进行的微流体混合和液滴生成。此类装置包括但不限于标准T形接合装置或流动聚焦装置。在一个实例中,所需脂质制剂(诸如含有MC3或MC3样的组合物)与甲病毒递送载体和其他所需药剂并行提供至液滴生成装置,使得递送载体和所需药剂完全包封在基于MC3或MC3样的LNP内部。在一个实例中,液滴生成装置可控制所产生的LNP的尺寸范围和尺寸分布。例如,LNP的尺寸可在1至1000纳米直径范围内,例如1、10、50、100、500或1000纳米。在液滴生成后,包封表达载体的递送媒介物可经进一步处理或修饰以使其准备用于施用。
V.E.黑猩猩腺病毒(ChAd)
V.E.1.用黑猩猩腺病毒递送病毒
用于递送一个或多个抗原(例如经由抗原盒并包含一个或多个新抗原)的疫苗组合物可通过提供黑猩猩来源的腺病毒核苷酸序列、多种新颖载体和表达黑猩猩腺病毒基因的细胞系来产生。黑猩猩C68腺病毒(在本文中也称为ChAdV68)的核苷酸序列可用于抗原递送的疫苗组合物中(参见SEQ ID NO:1)。衍生自C68腺病毒的载体的使用进一步详细描述于USPN 6,083,716中,其出于所有目的以引用的方式整体并入本文。
在另一方面,本文提供一种重组腺病毒,其包含黑猩猩腺病毒诸如C68的DNA序列和可操作地连接到引导其表达的调控序列的抗原盒。重组病毒能够感染哺乳动物细胞、优选人细胞,并且能够在细胞中表达抗原盒产物。在这种载体中,原生黑猩猩E1基因和/或E3基因和/或E4基因可缺失。抗原盒可插入至这些基因缺失位点中的任一者中。抗原盒可包括抗原,针对其激活的免疫应答是所需的。
在另一方面,本文提供一种被黑猩猩腺病毒诸如C68感染的哺乳动物细胞。
在另一方面,提供一种新颖的哺乳动物细胞系,其表达黑猩猩腺病毒基因(例如来自C68)或其功能片段。
在另一方面,本文提供一种用于将抗原盒递送到哺乳动物细胞中的方法,其包括以下步骤:向细胞中引入有效量的已经工程改造以表达抗原盒的黑猩猩腺病毒,诸如C68。
另一方面提供一种用于引发哺乳动物宿主中的免疫应答以治疗癌症的方法。所述方法可包括以下步骤:向宿主施用有效量的重组黑猩猩腺病毒诸如C68,其包含编码免疫应答所靶向的来自肿瘤的一个或多个抗原的抗原盒。
还公开了一种非猿猴哺乳动物细胞,其表达获自序列SEQ ID NO:1的黑猩猩腺病毒基因。所述基因可选自由以下组成的组:SEQ ID NO:1的腺病毒E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5。
还公开了一种包含黑猩猩腺病毒DNA序列的核酸分子,所述黑猩猩腺病毒DNA序列包含获自序列SEQ ID NO:1序列的基因。所述基因可选自由以下组成的组:SEQ ID NO:1的所述黑猩猩腺病毒E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因。在一些方面,核酸分子包含SEQ ID NO:1。在一些方面,核酸分子包含序列SEQ ID NO:1,其缺少选自由以下组成的组的至少一个基因:SEQ ID NO:1的E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因。
还公开了一种载体,其包含获自SEQ ID NO:1的黑猩猩腺病毒DNA序列以及可操作地连接到一个或多个调控序列的抗原盒,所述一个或多个调控序列引导盒在异源宿主细胞中的表达,任选地其中所述黑猩猩腺病毒DNA序列至少包含用于复制及病毒粒子衣壳化所必需的顺式元件,所述顺式元件侧接抗原盒和调控序列。在一些方面,黑猩猩腺病毒DNA序列包含选自由以下组成的组的基因:SEQ ID NO:1的E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因序列。在一些方面,载体可缺乏E1A和/或E1B基因。
本文还公开经本文所公开的载体(诸如经工程改造以表达抗原盒的C68载体)转染的宿主细胞。本文还公开经由将本文所公开的载体引入细胞中而表达其中引入的经选择的基因的人细胞。
本文还公开了一种用于将抗原盒递送到哺乳动物细胞的方法,其包括向所述细胞中引入有效量的本文所公开的载体,诸如经工程改造以表达抗原盒的C68载体。
本文还公开了一种用于产生抗原的方法,其包括将本文所公开的载体引入哺乳动物细胞中,在适合的条件下培养所述细胞以及产生所述抗原。
V.E.2.表达E1的互补细胞系
为了产生缺失本文所述的基因中的任一者的重组黑猩猩腺病毒(Ad),如果缺失基因区的功能对于病毒的复制和感染性必不可少,则可通过辅助病毒或细胞系(即互补或包装细胞系)供应至重组病毒。例如,为了产生复制缺陷型黑猩猩腺病毒载体,可使用表达人或黑猩猩腺病毒的E1基因产物的细胞系;这种细胞系可包括HEK293或其变体。可遵循产生表达黑猩猩E1基因产物的细胞系的方案(USPN6,083,716的实例3和4),以产生表达任何经选择的黑猩猩腺病毒基因的细胞系。
AAV增强分析可用于鉴定表达黑猩猩腺病毒E1的细胞系。这种分析可用于鉴定通过使用例如来自其他物种的其他未表征的腺病毒的E1基因制备的细胞系中的E1功能。所述分析描述于USPN6,083,716的实例4B中。
经选择的黑猩猩腺病毒基因(例如E1)可在启动子的转录控制下用于在经选择的亲本细胞系中表达。诱导型或组成型启动子可用于此目的。在诱导型启动子中包括可由锌诱导的绵羊金属硫蛋白启动子,或可由糖皮质激素、特别是地塞米松(dexamethasone)诱导的小鼠乳腺肿瘤病毒(MMTV)启动子。其他诱导型启动子,诸如以引用的方式并入本文中的国际专利申请WO95/13392中所鉴定的那些诱导型启动子,也可用于产生包装细胞系。也可采用控制黑猩猩腺病毒基因表达的组成型启动子。
亲本细胞可经选择以产生表达任何所需C68基因的新颖细胞系。没有限制,这种亲本细胞系可以是HeLa[ATCC登录号CCL 2]、A549[ATCC登录号CCL 185]、KB[CCL 17]、Detroit[例如Detroit 510、CCL 72]和WI-38[CCL 75]细胞。其他适合的亲本细胞系可获自其他来源。亲本细胞系可包括CHO、HEK293或其变体、911、HeLa、A549、LP-293、PER.C6或AE1-2a。
表达E1的细胞系可用于产生重组黑猩猩腺病毒E1缺失的载体。使用基本上相同的程序构建的表达一种或多种其他黑猩猩腺病毒基因产物的细胞系可用于产生缺失编码那些产物的基因的重组黑猩猩腺病毒载体。此外,表达其他人Ad E1基因产物的细胞系也可用于产生黑猩猩重组Ad。
V.E.3.作为载体的重组病毒粒子
本文所公开的组合物可包含病毒载体,其将至少一个抗原递送至细胞。此类载体包含黑猩猩腺病毒DNA序列诸如C68和可操作地连接到引导盒表达的调控序列的抗原盒。C68载体能够在经感染的哺乳动物细胞中表达盒。C68载体可能功能性缺失一个或多个病毒基因。抗原盒包含在一个或多个调控序列诸如启动子控制下的至少一个抗原。任选的辅助病毒和/或包装细胞系可向黑猩猩病毒载体供应缺失的腺病毒基因的任何必需产物。
术语“功能性缺失”意指移除或以其他方式改变(例如通过突变或修饰)足够量的基因区,使得基因区不再能够产生一种或多种基因表达的功能性产物。可导致功能性缺失的突变或修饰包括但不限于无义突变,诸如引入过早的终止密码子和去除典型和非典型的起始密码子,改变mRNA剪接或其他转录加工的突变,或其组合。如果需要,可去除整个基因区。
形成本文所公开的载体的核酸序列的修饰,包括序列缺失、插入和其他突变,可使用标准分子生物学技术产生并且在本发明的范围内。
V.E.4.病毒质粒载体的构建
可用于本发明的黑猩猩腺病毒C68载体包括重组缺陷型腺病毒,即在E1a或E1b基因中功能性缺失且任选地携带其他突变(例如其他基因中的温度敏感性突变或缺失)的黑猩猩腺病毒序列。预期这些黑猩猩序列也可用于形成来自其他腺病毒和/或腺相关病毒序列的杂交载体。由人腺病毒制备的同源腺病毒载体描述于公开的文献中[参见例如上文所引用的Kozarsky I和II,以及其中引用的参考文献,美国专利号5,240,846]。
在构建可用于将抗原盒递送至人(或其他哺乳动物)细胞的黑猩猩腺病毒C68载体时,可在载体中采用一系列腺病毒核酸序列。包含最小黑猩猩C68腺病毒序列的载体可与辅助病毒结合使用以产生感染性重组病毒粒子。辅助病毒提供最小黑猩猩腺病毒载体的病毒感染性和繁殖所需的基本基因产物。当在另外的功能性病毒载体中仅产生黑猩猩腺病毒基因的一个或多个经选择的缺失时,可通过在经选择的包装细胞系中繁殖病毒而在病毒载体生产过程中供应缺失的基因产物,所述包装细胞系提供反式缺失的基因功能。
V.E.5.重组最小腺病毒
最小的黑猩猩Ad C68病毒是仅含有复制及病毒粒子衣壳化所必需的腺病毒顺式元件的病毒粒子。也就是说,载体含有腺病毒的顺式作用5'和3'反向末端重复(ITR)序列(其充当复制起点)以及原生5'包装/增强子结构域(其含有用于包装线性Ad基因组所必需的序列和E1启动子的增强子元件)。参见例如在国际专利申请WO96/13597中所描述且以引用的方式并入本文的用于制备“最小”人Ad载体的技术。
V.E.6.其他缺陷型腺病毒
重组复制缺乏型腺病毒也可比最小黑猩猩腺病毒序列含有更多。这些其他Ad载体可通过病毒基因区的各个部分的缺失以及通过任选地使用辅助病毒和/或包装细胞系形成的感染性病毒粒子来表征。
作为一个实例,适合的载体可通过使C68腺病毒立即早期基因E1a和延迟早期基因E1b的全部或足够部分缺失来形成,从而消除其正常的生物功能。当在含有提供对应反式基因产物的功能性腺病毒E1a和E1b基因的黑猩猩腺病毒转化的互补细胞系上生长时,复制缺陷型E1缺失病毒能够复制并产生感染性病毒。基于与已知腺病毒序列的同源性,预期与本领域的人重组E1缺失腺病毒一样,所得重组黑猩猩腺病毒能够感染许多细胞类型并且可表达抗原,但无法在大部分不携带黑猩猩E1区DNA的细胞中复制,除非细胞以极高感染倍率感染。
作为另一个实例,C68腺病毒延迟早期基因E3的全部或一部分可从形成重组病毒的一部分的黑猩猩腺病毒序列消除。
也可构建具有E4基因缺失的黑猩猩腺病毒C68载体。另一个载体可在延迟早期基因E2a中含有缺失。
也可在黑猩猩C68腺病毒基因组的晚期基因L1至L5中的任一者中获得缺失。类似地,中间基因IX和IVa2中的缺失可用于一些目的。可在其他结构性或非结构性腺病毒基因中获得其他缺失。
上文论述的缺失可单独使用,即腺病毒序列可仅含有E1缺失。或者,可以任何组合使用有效破坏或降低其生物活性的完整基因或其部分的缺失。例如,在一个示例性载体中,腺病毒C68序列可缺失E1基因和E4基因,或缺失E1、E2a和E3基因,或缺失E1和E3基因,或在缺失或不缺失E3的情况下缺失E1、E2a和E4基因等。如上文所论述,这些缺失可与其他突变(诸如温度敏感性突变)组合使用,以达成所需结果。
将包含抗原的盒任选地插入至黑猩猩C68 Ad病毒的任何缺失区中。或者,如果需要,则可将盒插入至现有基因区中以破坏所述区的功能。
V.E.7.辅助病毒
取决于用于携带抗原盒的病毒载体的黑猩猩腺病毒基因含量,可使用辅助腺病毒或非复制性病毒片段来提供足够的黑猩猩腺病毒基因序列以产生含有所述盒的感染性重组病毒粒子。
有用的辅助病毒含有经选择的腺病毒基因序列,其不存在于腺病毒载体构建体中和/或不由载体转染的包装细胞系表达。辅助病毒可以是复制缺陷型并且除上述序列之外,还含有多种腺病毒基因。辅助病毒可与本文所述的表达E1的细胞系组合使用。
对于C68,“辅助”病毒可以是通过用SspI剪切C68基因组的C末端形成的片段,其从病毒的左端除去约1300bp。这种经剪切的病毒然后与质粒DNA共转染至表达E1的细胞系中,由此通过与质粒中的C68序列同源重组形成重组病毒。
辅助病毒也可形成聚阳离子缀合物,如Wu等,J.Biol.Chem.,264:16985-16987(1989);K.J.Fisher和J.M.Wilson,Biochem.J.,299:49(1994年4月1日)中所述。辅助病毒可任选地含有报告基因。许多此类报告基因是本领域中已知的。与腺病毒载体上的抗原盒不同,辅助病毒上报告基因的存在允许独立地监测Ad载体和辅助病毒。这种第二报告基因用于纯化后能够将所得重组病毒与辅助病毒分离。
V.E.8.病毒粒子的组装和细胞系的感染
将经选择的腺病毒DNA序列、抗原盒和其他载体元件组装到各种中间质粒和穿梭载体中,以及使用所述质粒和载体产生重组病毒粒子都可使用常规技术实现。此类技术包括常规cDNA克隆技术、体外重组技术(例如吉布森组装(Gibson assembly))、腺病毒基因组的重叠寡核苷酸序列的使用、聚合酶链反应以及提供所需核苷酸序列的任何适合的方法。采用标准转染和共转染技术,例如CaPO4沉淀技术或脂质体介导的转染方法,诸如脂染胺。所采用的其他常规方法包括病毒基因组的同源重组、琼脂覆层中病毒的蚀斑、测量信号产生的方法等。
例如,在构建并组装所需含有抗原盒的病毒载体后,可在辅助病毒存在下将载体体外转染至包装细胞系中。同源重组发生在辅助序列与载体序列之间,其允许载体中的腺病毒抗原序列复制且包装至病毒粒子衣壳中,从而产生重组病毒载体粒子。
所得重组黑猩猩C68腺病毒可用于将抗原盒转移到经选择的细胞中。在使用包装细胞系中生长的重组病毒的体内实验中,E1缺失的重组黑猩猩腺病毒在将盒转移到非黑猩猩(优选地人)细胞中展现效用。
V.E.9.重组病毒载体的用途
所得含有抗原盒的重组黑猩猩C68腺病毒(如上所述,通过腺病毒载体和辅助病毒或腺病毒载体以及包装细胞系的合作产生)因此提供一种有效的基因转移媒介物,其可将抗原体内或离体递送至受试者。
上述重组载体根据公开的基因疗法施用于人。携带抗原盒的黑猩猩病毒载体可施用于患者,优选悬浮于生物相容性溶液或药学上可接受的递送媒介物中。适合的媒介物包括无菌盐水。已知为药学上可接受的载剂并且是本领域技术人员众所周知的其他水性和非水性等张无菌注射溶液以及水性和非水性无菌悬浮液可用于此目的。
黑猩猩腺病毒载体是以足以转导人细胞并提供足够水平的抗原转移和表达的量施用,从而提供治疗益处而无过度不良效应或具有医学上可接受的生理效应,其可由医药领域的技术人员来确定。常规和药学上可接受的施用途径包括但不限于直接递送至肝脏、鼻内、静脉内、肌肉内、皮下、皮内、经口和其他肠胃外施用途径。如果需要,施用途径可以组合。
病毒载体的剂量主要将取决于诸如以下因素:所治疗的病状,患者的年龄、体重和健康状况,并且因此可在不同患者中变化。剂量将经调节以平衡治疗益处与任何副作用,并且这些剂量可根据采用重组载体的治疗应用而变化。可监测抗原表达水平以确定剂量施用频率。
重组复制缺陷型腺病毒可以“药学有效量”施用,也就是说,在施用途径中有效转染所需细胞并提供经选择的基因的足够表达水平以提供疫苗益处(即一些可测量的保护性免疫水平)的重组腺病毒的量。包含抗原盒的C68载体可与佐剂一起共施用。佐剂可与载体(例如矾)分开或在载体内编码,特别是在佐剂为蛋白质时。佐剂是本领域众所周知的。
常规且药学上可接受的施用途径包括但不限于鼻内、肌肉内、气管内、皮下、皮内、经直肠、经口和其他肠胃外施用途径。如果需要,可组合或调整施用途径,视免疫原或疾病而定。例如,在狂犬病预防中,皮下、气管内和鼻内途径是优选的。施用途径主要将取决于所治疗疾病的性质。
可监测对抗原的免疫水平以确定是否需要增强剂。例如,在评定血清中的抗体效价后,可能需要任选的加强免疫。
VI.治疗和制造方法
还提供了一种通过向受试者施用一个或多个抗原诸如多个使用本文公开的方法鉴定的抗原(例如施用包含抗原编码核酸序列的盒,所述盒具有表位编码核酸序列和任选地5'和/或3'接头序列),来刺激受试者中的免疫应答(例如诱导、增加或增强免疫应答,诸如诱导、增加或增强T细胞反应)、刺激受试者中的肿瘤特异性免疫应答、针对肿瘤进行疫苗接种、治疗和/或缓解受试者中的癌症症状的方法。例如,本文所述的方法可诱导、增加或增强表位特异性T细胞反应,诸如诱导、增加或增强表位特异性T细胞增殖、T细胞活化、T细胞细胞因子的产生和/或分泌、T细胞分化、T细胞寿命或其任何组合。本文所述的方法包括刺激受试者中的免疫应答(例如通过与I型干扰素信号传导抑制剂组合,组合施用基于甲病毒的表达系统(例如施用一个或多个基于甲病毒的载体,所述载体编码包含抗原编码核酸序列的盒,所述盒具有表位编码核酸序列和任选地5'和/或3’接头序列),来诱导、增加或增强免疫应答。
还提供了一种通过与I型干扰素信号传导抑制剂组合施用一个或多个具有RNA甲病毒骨架和编码的有效载荷(例如盒)的载体,来增强基于甲病毒的表达系统对有效载荷的递送的方法。有效载荷可以是期望递送至所关注的细胞的任何核苷酸序列。一般来说,所述有效载荷是可操作地连接到启动子以驱动核苷酸序列表达的盒。所述核苷酸序列可以是编码的(即能够被转录并翻译成蛋白质的多肽编码核酸序列)。一般来说,所述多肽编码核酸序列可编码期望在细胞中表达的任何蛋白质。蛋白质的实例包括但不限于抗体、细胞因子、嵌合抗原受体(CAR)、T细胞受体或基因组编辑系统组分(例如基因组编辑系统中使用的核酸酶)。基因组编辑系统包括但不限于CRISPR系统、锌指系统、大范围核酸酶系统或TALEN系统。所述核苷酸序列可以是非编码的(即能够被转录但不被翻译成蛋白质的核酸序列)。一般来说,所述非编码核酸序列可编码期望在细胞中表达的任何非编码多核苷酸。非编码多核苷酸的实例包括但不限于RNA干扰(RNAi)多核苷酸(例如,反义寡核苷酸、shRNA、siRNA、miRNA等)或基因组编辑系统多核苷酸(例如,指导RNA[gRNA]、单指导RNA[sgRNA]、反式激活CRISPR[tracrRNA]和/或CRISPR RNA[crRNA])。
在一些方面,受试者已诊断患有癌症或具有患癌症的风险。受试者可以是人、狗、猫、马或需要肿瘤特异性免疫应答的任何动物。肿瘤可以是任何实体肿瘤诸如乳房肿瘤、卵巢肿瘤、前列腺肿瘤、肺肿瘤、肾脏肿瘤、胃肿瘤、结肠肿瘤、睾丸肿瘤、头颈部肿瘤、胰脏肿瘤、脑肿瘤、黑素瘤和其他组织器官肿瘤,以及血液肿瘤诸如淋巴瘤和白血病,包括急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病和B细胞淋巴瘤。
抗原可以足以诱导CTL反应的量施用。
抗原可以单独或与其他治疗剂组合施用。治疗剂是例如化学治疗剂、辐射或免疫疗法。可针对特定癌症施用任何适合的治疗性治疗。
另外,可向受试者进一步施用抗免疫抑制剂/免疫刺激剂,诸如检查点抑制剂。例如,可向受试者进一步施用抗CTLA抗体或抗PD-1或抗PD-L1。通过抗体阻断CTLA-4或PD-L1可增强患者对癌细胞的免疫应答。具体来说,已显示在按照疫苗接种方案时CTLA-4阻断是有效的。
可以确定包括在疫苗组合物中的每个抗原的最佳量和最佳给药方案。例如,可制备用于静脉内(i.v.)注射、皮下(s.c.)注射、皮内(i.d.)注射、腹膜内(i.p.)注射、肌肉内(i.m.)注射的抗原或其变体。注射方法包括s.c.、i.d.、i.p.、i.m.和i.v.。DNA或RNA注射方法包括i.d.、i.m.、s.c.、i.p.和i.v.。疫苗组合物的其他施用方法是本领域技术人员已知的。
疫苗可经编译以使得组合物中存在的抗原的选择、数量和/或量具有组织、癌症和/或患者特异性。例如,肽的精确选择可通过亲本蛋白质在给定组织中的表达模式来指导,或者通过患者的突变状态来指导。选择可取决于癌症的具体类型、疾病状态、较早的治疗方案、患者免疫状态以及当然患者的HLA单倍型。此外,疫苗可根据特定患者的个人需要而含有个别化组分。实例包括根据抗原在特定患者中的表达改变抗原的选择或在第一轮或治疗方案之后调整二次治疗。
患者可通过使用各种诊断方法例如下文进一步描述的患者选择方法,来鉴定抗原疫苗的施用。患者选择可涉及鉴定一种或多种基因中的突变或表达模式。在一些情况下,患者选择涉及鉴定患者的单倍型。各种患者选择方法可以并行进行,例如测序诊断可以同时鉴定患者的突变和单倍型。各种患者选择方法可以依次进行,例如一个诊断测试鉴定突变,并且单独的诊断测试鉴定患者的单倍型,并且其中每个测试可以是相同的(例如两个高通量测序)或不同的(例如一个高通量测序和另一个Sanger测序)诊断方法。
对于待用作癌症疫苗的组合物,在正常组织中大量表达的具有类似正常自身肽的抗原可以避免或以低量存在于本文所述的组合物中。另一方面,如果已知患者的肿瘤表达大量某一抗原,则用于治疗这种癌症的相应药物组合物可大量存在和/或可包括多于一种特异性针对这种特定抗原或这种抗原的途径的抗原。
可向已患有癌症的个体施用包含抗原的组合物。在治疗应用中,组合物以足以引发对肿瘤抗原的有效CTL反应且治愈或至少部分遏制症状和/或并发症的量施用于患者。足以实现此目标的量定义为“治疗有效剂量”。对这种用途有效的量将取决于例如组合物、施用方式、所治疗疾病的阶段和严重程度、患者的体重和一般健康状况,以及处方医师的判断。应记住,组合物一般可用于严重的疾病状态,也就是说,危及生命或可能危及生命的情形,尤其当癌症已转移时。在此类情况下,鉴于外来物质的最小化和抗原的相对无毒性,主治医师可能且可能感觉需要施用实质性过量的这些组合物。
对于治疗用途,可在检测或手术移除肿瘤时开始施用。此后是加强剂量,直到症状至少实质上减弱且此后持续一段时间。
用于治疗性治疗的药物组合物(例如疫苗组合物)意图用于肠胃外、局部、经鼻、经口或局部施用。药物组合物可以经肠胃外施用,例如静脉内、皮下、皮内或肌肉内施用。组合物可在手术切除部位施用以诱导针对肿瘤的局部免疫应答。本文公开了用于肠胃外施用的组合物,其包含抗原和疫苗组合物溶解或悬浮于可接受的载剂(例如水性载剂)中的溶液。可使用多种水性载剂,例如水、缓冲水、0.9%生理盐水、0.3%甘氨酸、玻尿酸等。这些组合物可以通过常规的众所周知的灭菌技术灭菌,或者可经无菌过滤。所得水性溶液可封装以按原样使用或冻干,冻干制剂在施用之前与无菌溶液组合。组合物可含有接近生理条件所需要的药学上可接受的辅助物质,诸如pH调节剂和缓冲剂、张力调节剂、湿润剂等,例如乙酸钠、乳酸钠、氯化钠、氯化钾、氯化钙、脱水山梨糖醇单月桂酸酯、三乙醇胺油酸酯等。
抗原也可以经由脂质体施用,脂质体使其靶向特定的细胞组织,诸如淋巴组织。脂质体也可用于增加半衰期。脂质体包括乳液、泡沫、胶束、不溶性单层、液晶、磷脂分散体、层状层等。在这些制剂中,有待递送的抗原作为脂质体的一部分单独或与结合于例如淋巴细胞中普遍存在的受体的分子(诸如结合于CD45抗原的单克隆抗体)或与其他治疗性或免疫原性组合物一起并入。因此,用所需抗原填充的脂质体可被引导至淋巴细胞的位点,在此脂质体然后递送经选择的治疗性/免疫原性组合物。脂质体可由标准的形成囊泡的脂质形成,其一般包括中性和带负电荷的磷脂和固醇(诸如胆固醇)。脂质的选择一般通过考虑例如脂质体大小、脂质体在血流中的酸不稳定性和稳定性来指导。多种方法可用于制备脂质体,如例如Szoka等,Ann.Rev.Biophys.Bioeng.9;467(1980),美国专利号4,235,871、4,501,728、4,501,728、4,837,028和5,019,369中所述。
为了靶向免疫细胞,有待并入到脂质体中的配体可包括例如特异性针对所需免疫系统细胞的细胞表面决定子的抗体或其片段。脂质体悬浮液可以一定剂量静脉内、局部、表面等施用,所述剂量尤其根据施用方式、所递送的肽以及所治疗疾病的阶段而变化。
出于治疗或免疫接种目的,编码肽和任选地一种或多种本文所述的肽的核酸也可施用于患者。多种方法方便地用于将核酸递送至患者。例如,核酸可以“裸DNA”形式直接递送。这种方法描述于例如Wolff等,Science 247:1465-1468(1990)以及美国专利号5,580,859和5,589,466中。核酸也可使用弹道式递送施用,如例如美国专利号5,204,253中所述。可施用仅包含DNA的粒子。或者,DNA可黏附于粒子,诸如金粒子。在存在或不存在电穿孔的情况下,用于递送核酸序列的方法可包括病毒载体、mRNA载体和DNA载体。
核酸也可以与阳离子化合物诸如阳离子脂质复合递送。脂质介导的基因递送方法描述于例如9618372WOAWO 96/18372;9324640WOAWO93/24640;Mannino和Gould-Fogerite,BioTechniques 6(7):682-691(1988);美国专利号5,279,833;Rose美国专利号5,279,833;9106309WOAWO 91/06309;以及Felgner等,Proc.Natl.Acad.Sci.US A 84:7413-7414(1987)中。
抗原还可以包括在基于病毒载体的疫苗平台中,诸如牛痘、禽痘、自我复制甲病毒、马拉巴病毒、腺病毒(参见例如Tatsis等,Adenoviruses,Molecular Therapy(2004)10,616-629)或慢病毒,包括但不限于第二、第三或杂交第二/第三代慢病毒以及任一代的重组慢病毒,其经设计以靶向特定细胞类型或受体(参见例如Hu等,Immunization Deliveredby Lentiviral Vectors for Cancer and Infectious Diseases,Immunol Rev.(2011)239(1):45-61;Sakuma等,Lentiviral vectors:basic to translational,Biochem J.(2012)443(3):603-18;Cooper等,Rescue of splicing-mediated intron lossmaximizes expression in lentiviral vectors containing the human ubiquitin Cpromoter,Nucl.Acids Res.(2015)43(1):682-690;Zufferey等,Self-InactivatingLentivirus Vector for Safe and Efficient In Vivo Gene Delivery,J.Virol.(1998)72(12):9873-9880)。取决于上述基于病毒载体的疫苗平台的包装能力,这种方法可递送编码一个或多个抗原肽的一个或多个核苷酸序列。序列可侧接非突变序列,可由接头分开或可在前面有一个或多个靶向亚细胞区室的序列(参见例如Gros等,Prospectiveidentification of neoantigen-specific lymphocytes in the peripheral blood ofmelanoma patients,Nat Med.(2016)22(4):433-8;Stronen等,Targeting of cancerneoantigens with donor-derived T cell receptor repertoires,Science.(2016)352(6291):1337-41;Lu等,Efficient identification of mutated cancer antigensrecognized by T cells associated with durable tumor regressions,Clin CancerRes.(2014)20(13):3401-10)。在引入到宿主中后,经感染细胞表达抗原,从而引发针对肽的宿主免疫(例如CTL)反应。用于免疫接种方案中的牛痘载体和方法描述于例如美国专利号4,722,848中。另一种载体是BCG(卡介苗)。BCG载体描述于Stover等(Nature 351:456-460(1991))中。根据本文描述,可用于抗原的治疗性施用或免疫接种的各种其他疫苗载体,例如伤寒沙门氏菌载体等对于本领域技术人员将是显而易见的。
施用核酸的手段使用编码一个或多个表位的袖珍基因构建体。为产生编码经选择在人细胞中表达的CTL表位(袖珍基因)的DNA序列,逆翻译表位的氨基酸序列。使用人密码子使用表来指导各氨基酸的密码子选择。这些表位编码DNA序列直接邻接,产生连续多肽序列。为了优化表达和/或免疫原性,可将额外元件并入到袖珍基因设计中。可经逆翻译且包括于袖珍基因序列中的氨基酸序列的实例包括:辅助T淋巴细胞、表位、前导(信号)序列和内质网滞留信号。另外,可通过包括相邻于CTL表位的合成(例如聚丙氨酸)或天然存在的侧接序列来改进CTL表位的MHC递呈。通过组装编码袖珍基因的正链和负链的寡核苷酸而将袖珍基因序列转化成DNA。重叠寡核苷酸(30-100个碱基长)是在适当条件下使用众所周知的技术合成、磷酸化、纯化和黏接的。寡核苷酸的末端使用T4 DNA连接酶连接。编码CTL表位多肽的这种合成袖珍基因可以然后克隆到所期望的表达载体中。
可制备经纯化的质粒DNA以便使用多种制剂注射。其中最简单的是冻干DNA在无菌磷酸盐缓冲生理盐水(PBS)中复原。已描述多种方法,并且可使用新技术。如上文所指出,核酸宜用阳离子脂质配制。另外,统称为保护性、相互作用性、非缩合性(PINC)的糖脂、促融脂质体、肽和化合物也可与经纯化的质粒DNA复合以影响诸如以下变量:稳定性、肌肉内分散或运输至特定器官或细胞类型。
还公开了一种制造肿瘤疫苗的方法,其包括进行本文公开的方法的步骤;以及产生包含多个抗原或多个抗原的子集的肿瘤疫苗。
本文公开的抗原可使用本领域中已知的方法制造。例如,产生本文公开的抗原或载体(例如包括编码一个或多个抗原的至少一个序列的载体)的方法可包括在适于表达所述抗原或载体的条件下培养宿主细胞,其中所述宿主细胞包含至少一个编码所述抗原或载体的多核苷酸;以及纯化所述抗原或载体。标准纯化方法包括色谱技术、电泳、免疫、沉淀、透析、过滤、浓缩和色谱聚焦技术。
宿主细胞可包括中国仓鼠卵巢(CHO)细胞、NS0细胞、酵母或HEK293细胞。宿主细胞可用一个或多个包含至少一个编码本文公开的抗原或载体的核酸序列的多核苷酸转化,任选地其中所述分离的多核苷酸还包含可操作地连接到编码抗原或载体的至少一个核酸序列的启动子序列。在某些实施方案中,所述分离的多核苷酸可以是cDNA。
VIIA.抗原和有效载荷的使用和施用
疫苗接种方案可用于向受试者给药一种或多种抗原(例如施用包含抗原编码核酸序列的盒,所述盒具有表位编码核酸序列和任选地5'和/或3'接头序列)。初免疫苗和加强疫苗可用于向受试者给药。初免疫苗可基于C68(例如SEQ ID NO:1或2中所示的序列)或srRNA(例如SEQ ID NO:3或4中所示的序列),并且加强疫苗可基于C68(例如SEQ ID NO:1或2中所示的序列)或srRNA(例如SEQ ID NO:3或4中所示的序列)。各载体通常包括包含抗原的盒。盒可包括约20种抗原,其由间隔子(诸如通常包围各抗原的天然序列)或其他非天然间隔序列(诸如AAY)分开。盒还可包括MHCII抗原,诸如破伤风类毒素抗原和PADRE抗原,其可视为通用II类抗原。盒还可包括靶向序列诸如泛素靶向序列。另外,各疫苗剂量可与检查点抑制剂(CPI)结合(例如同时、之前或之后)施用于受试者。CPI可包括抑制CTLA4、PD1和/或PDL1的那些,诸如抗体或其抗原结合部分。此类抗体可包括曲美木单抗或德瓦鲁单抗(durvalumab)。
初免疫苗可注射(例如肌肉内)于受试者。可使用每一剂量双侧注射。例如,可使用一次或多次ChAdV68(C68)注射(例如总剂量1×1012个病毒粒子);可使用选自0.001至1ugRNA、特别是0.1或1ug范围的低疫苗剂量的一次或多次自我复制RNA(srRNA)注射;或者可使用选自1至100ug RNA、特别是10或100ug范围的高疫苗剂量的一次或多次srRNA注射。
可在初免疫苗接种之后注射(例如肌肉内)疫苗加强剂(加强疫苗)。加强疫苗可在初免后约每1、2、3、4、5、6、7、8、9或10周,例如每4周和/或8周施用。可使用每一剂量双侧注射。例如,可使用一次或多次ChAdV68(C68)注射(例如总剂量1×1012个病毒粒子);可使用选自0.001至1ug RNA、特别是0.1或1ug范围的低疫苗剂量的一次或多次自我复制RNA(srRNA)注射;或者可使用选自1至100ug RNA、特别是10或100ug范围的高疫苗剂量的一次或多次srRNA注射。
也可向受试者施用抗CTLA-4(例如曲美木单抗)。例如,抗CTLA4可在肌肉内疫苗注射(ChAdV68初免或srRNA低剂量)部位附近皮下施用,以确保引流至同一淋巴结。曲美木单抗是CTLA-4的选择性人IgG2 mAb抑制剂。目标抗CTLA-4(曲美木单抗)皮下剂量通常是70-75mg(特别是75mg),其剂量范围为例如1-100mg或5-420mg。
在某些情况下,可使用抗PD-L1抗体诸如德瓦鲁单抗(MEDI4736)。德瓦鲁单抗是一种选择性、高亲和力人IgG1 mAb,其阻断PD-L1结合于PD-1和CD80。德瓦鲁单抗一般每4周以20mg/kg i.v.施用。
可在疫苗施用之前、期间和/或之后进行免疫监测。除其他参数外,此类监测可告知安全性和功效。
为了进行免疫监测,通常使用PBMC。PBMC可在初免疫苗接种之前以及在初免疫苗接种之后(例如4周和8周)分离。PBMC可仅在加强疫苗接种之前以及在每次加强疫苗接种之后(例如4周和8周)收集。
可评定T细胞反应作为免疫监测方案的一部分。可使用本领域中已知的一种或多种方法测量T细胞反应,诸如ELISpot、细胞内细胞因子染色、细胞因子分泌和细胞表面捕捉、T细胞增殖、MHC多聚体染色或通过细胞毒性分析。针对疫苗中编码的表位的T细胞反应可通过使用ELISpot分析测量细胞因子(诸如IFN-γ)的诱导而从PBMC监测。针对疫苗中编码的表位的特异性CD4或CD8 T细胞反应可通过使用流式细胞测量术测量胞内或胞外捕捉的细胞因子(诸如IFN-γ)的诱导而从PBMC监测。针对疫苗中编码的表位的特异性CD4或CD8T细胞反应可通过使用MHC多聚体染色测量表达特异性针对表位/MHC I类复合物的T细胞受体的T细胞群而从PBMC监测。针对疫苗中编码的表位的特异性CD4或CD8 T细胞反应可通过在3H-胸苷、溴脱氧尿苷和羧基荧光素-二乙酸酯-琥珀酰亚胺酯(CFSE)并入后测量T细胞群的离体扩增而从PBMC监测。特异性针对疫苗中编码的表位的PBMC衍生的T细胞的抗原识别能力和溶解活性可通过铬释放分析或替代性比色细胞毒性分析来功能性评定。
方案可用于向受试者给药一种或多种基于甲病毒的表达系统(即施用一个或多个具有RNA甲病毒骨架和编码的有效载荷[例如盒]的载体)。一般来说,可以遵循上文针对疫苗接种的方案来施用本文所述的任何基于甲病毒的表达系统。
VIIB.I型干扰素信号传导抑制剂的使用和施用
本文所述的方法包括一种通过组合施用一个或多个具有RNA甲病毒骨架和编码的有效载荷(例如盒)的载体和施用I型干扰素信号传导抑制剂,来增强基于甲病毒的表达系统对有效载荷的递送的方法。在一些实施方案中,在向受试者施用后,施用I型干扰素信号传导抑制剂改进了基于甲病毒的载体在体内的自我复制/自我扩增。基于甲病毒的载体的自我复制/自我扩增可导致编码的有效载荷(例如盒)的表达提高。可将I型干扰素信号传导抑制剂配制成药物组合物。
I型干扰素信号传导抑制剂可以是IFNα抑制剂、IFNβ抑制剂、IFNAR抑制剂或其他I型干扰素信号传导途径抑制剂。I型干扰素信号传导抑制剂可以是抗体或其抗原结合片段、小分子抑制剂、RNAi多核苷酸、基因组编辑系统、Fc融合蛋白。I型干扰素信号传导抑制剂可以是抗体或其抗原结合片段。I型干扰素信号传导抑制剂可以是抗IFNAR抗体或其抗原结合片段,包括但不限于MAR1-5A3、阿尼福鲁单抗(也称为MEDI546)、AmS3A5-1、64G12、H2K6、H2K1、H3K6、H3K1 3F11、4G5、11E2和9D4,其细节可见于美国专利号7,662,381和7,619,070;所述专利出于所有目的以引用的方式并入本文。I型干扰素信号传导抑制剂可以是抗IFNα抗体或其抗原结合片段,包括但不限于西法鲁单抗、贝那利珠单抗或ASG-009。I型干扰素信号传导途径抑制剂可以是JAK激酶抑制剂,包括JAK激酶的小分子抑制剂。JAK激酶抑制剂包括但不限于JAK1/2抑制剂或JAK1/3抑制剂。JAK1/3抑制剂的实例是托法替尼(商品名Xeljanz、Jakvinus和Tofacinix)或非洛替尼(Filgotinib)(GLPG0634)。JAK1/2抑制剂的实例是鲁索利替尼(Ruxolitinib)、巴瑞克替尼(Baricitinib)或莫洛替尼(Momelotinib)(CYT387)。
I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统(即基于甲病毒的递送平台)的所述组合物之前、同时或之后施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物前24小时或更短时间施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物前24、23、22、21、20、19、18、17、16、15、14、13、12、11、10、9、8、7、6、5、4、3、2或1小时或更短时间施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物后少于12小时施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物后少于12、11、10、9、8、7、6、5、4、3、2或1小时施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物后6小时或更短时间施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物后6、5、4、3、2或1小时或更短时间施用。I型干扰素信号传导抑制剂可以在施用用于递送所述表达系统的所述组合物前24小时与施用后6小时或更短时间之间施用。
I型干扰素信号传导抑制剂可以经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。I型干扰素信号传导抑制剂可以经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用,并且用于递送所述表达系统的所述组合物可以经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。I型干扰素信号传导抑制剂可以经静脉内(IV)施用。I型干扰素信号传导抑制剂可以经静脉内(IV)施用,并且用于递送所述表达系统的所述组合物可以经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。I型干扰素信号传导抑制剂可以经IM施用。I型干扰素信号传导抑制剂可以经IM施用,并且用于递送所述表达系统的所述组合物可以经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。I型干扰素信号传导抑制剂可以经静脉内(IV)施用,并且用于递送所述表达系统的所述组合物可以经肌肉内(IM)施用。I型干扰素信号传导抑制剂可以经IM施用,并且用于递送所述表达系统的所述组合物可以经肌肉内(IM)施用。
可施用I型干扰素信号传导抑制剂的单次施用。可施用I型干扰素信号传导抑制剂的多次施用。I型干扰素信号传导抑制剂的多次施用可与表达递送平台的多次施用(诸如基于甲病毒的递送平台的多次施用)组合施用。I型干扰素信号传导抑制剂的多次施用可以相同的方式或不同的方式施用,诸如通过不同的途径、时间或剂量施用。
VIII.抗原鉴定
VIII.A.抗原候选物鉴定
已描述关于肿瘤和正常外显子组以及转录组的NGS分析的研究方法且将其应用于抗原鉴定空间。6,14,15可考虑在临床环境中对抗原鉴定的灵敏度和特异性更大的某些优化。这些优化可分为两个领域,即与实验室方法相关的那些优化和与NGS数据分析相关的那些优化。优化的实例是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的方法,所述专利出于所有目的各自以引用的方式整体并入本文。
VIII.B.HLA肽的分离和检测
在裂解和溶解组织样品后,使用经典免疫沉淀(IP)方法进行HLA-肽分子的分离(55-58)。澄清的溶解物用于HLA特异性IP。
免疫沉淀使用与珠粒偶联的抗体来进行,其中抗体特异性针对HLA分子。对于泛I类HLA免疫沉淀,使用泛I类CR抗体,对于II类HLA-DR,使用HLA-DR抗体。在过夜温育期间,抗体共价连接到NHS-琼脂糖珠粒。在共价连接后,将珠粒洗涤并等分用于IP。(59,60)免疫沉淀也可以用未共价结合至珠粒的抗体进行。通常使用包被有蛋白A和/或蛋白G的琼脂糖或磁性珠粒将抗体固定在柱上来完成此操作。下面列出了一些可用于选择性富集MHC/肽复合物的抗体。
| 抗体名称 | 特异性 |
| W6/32 | I类HLA-A、B、C |
| L243 | II类–HLA-DR |
| Tu36 | II类–HLA-DR |
| LN3 | II类–HLA-DR |
| Tu39 | II类–HLA-DR、DP、DQ |
将澄清的组织溶解物加入到抗体珠粒中进行免疫沉淀。在免疫沉淀后,从溶解物移除珠粒并且将溶解物储存用于额外实验,包括额外IP。洗涤IP珠粒以移除非特异性结合并且使用标准技术从珠粒洗脱HLA/肽复合物。使用分子量旋转柱或C18分级分离从肽移除蛋白质组分。所得肽通过SpeedVac蒸发变干并且在一些情况下,在MS分析之前储存在-20C下。
干燥的肽在适于反相色谱法的HPLC缓冲液中复原且装载于C-18微毛细管HPLC柱上,以便在Fusion Lumos质谱仪(Thermo)中梯度洗脱。在Orbitrap检测器中以高分辨率收集肽质量/电荷(m/z)的MS1谱,接着在经选择的离子的HCD片段化后,在离子阱检测器中收集MS2低分辨率扫描。另外,可使用CID或ETD片段化方法或三种技术的任何组合来获得MS2谱,以达到肽的更大的氨基酸覆盖率。MS2谱也可在Orbitrap检测器中以高分辨率质量精度测量。
使用Comet(61,62)对来自各分析的MS2谱进行蛋白质数据库搜索,并且使用Percolator(63-65)对肽鉴定进行评分。使用PEAKS studio(Bioinformatics SolutionsInc.)进行其他测序,并且可以使用其他搜索引擎或测序方法,包括光谱匹配和从头测序(97)。
VIII.B.1.支持综合HLA肽测序的MS检测极限研究。
使用肽YVYVADVAAK,使用装载于LC柱上的不同量的肽确定检测极限。所测试的肽的量为1pmol、100fmol、10fmol、1fmol和100amol。(表1)结果示出在图24A和图24B中。这些结果表明,最低检测极限(LoD)在埃摩尔范围(10-18)中,动态范围跨越五个数量级,并且信号噪声比足以在低飞摩尔范围(10-15)测序。质谱可以与本文所述的预测算法结合使用以验证HLA递呈。例如,质谱可用于验证由EDGE预测模型(一种基于通过MS/MS测序的HLA递呈的肽训练的深度学习模型,如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中所述)产生的表位候选物。EDGE评分与通过靶向MS检测候选共享新抗原肽的概率之间的相关性的实例示出在图25中。
表1
| 肽m/z | 装载于柱上 | 1e9个细胞中的拷贝数/细胞 |
| 566.830 | 1pmol | 600 |
| 562.823 | 100fmol | 60 |
| 559.816 | 10fmol | 6 |
| 556.810 | 1fmol | 0.6 |
| 553.802 | 100amol | 0.06 |
IX.递呈模型
递呈模型可用于鉴定患者中肽递呈的可能性。各种递呈模型是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357、WO/2018/208856、WO2016187508和美国专利申请US20110293637中更详细地描述的递呈模型,所述专利出于所有目的各自以引用的方式整体并入本文。
X.训练模块
训练模块可用于基于训练数据集来构建一个或多个递呈模型,所述模型产生肽序列是否会由与所述肽序列相关联的MHC等位基因递呈的可能性。各种训练模块是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的递呈模型,所述专利出于所有目的各自以引用的方式整体并入本文。训练模块可以在独立等位基因基础上构建递呈模型以预测肽的递呈可能性。训练模块还可以在存在两个或更多个MHC等位基因的多等位基因环境中构建递呈模型以预测肽的递呈可能性。
XI.预测模块
预测模块可用于接收序列数据并使用递呈模型选择序列数据中的候选抗原。具体来说,序列数据可以是从患者的肿瘤组织细胞中提取的DNA序列、RNA序列和/或蛋白质序列。预测模块可以通过将从患者的正常组织细胞中提取的序列数据与从患者的肿瘤组织细胞中提取的序列数据相比较以鉴定含有一个或多个突变的部分,由此鉴定出呈突变肽序列的候选新抗原。预测模块可以通过比较从患者的正常组织细胞中提取的序列数据与从患者的肿瘤组织细胞中提取的序列数据,来鉴定与正常细胞或组织相比在肿瘤细胞或癌组织中改变表达的候选抗原,以鉴定不正确表达的候选抗原。
递呈模块可将一个或多个递呈模型应用于经处理的肽序列以估计所述肽序列的递呈可能性。具体来说,预测模块可以通过将递呈模型应用于候选抗原来选择一个或多个可能在肿瘤HLA分子上递呈的候选抗原肽序列。在一个实施方式中,递呈模块选出估计递呈可能性超过预定阈值的候选抗原序列。在另一实施方式中,递呈模型选出N个具有最高估计递呈可能性的候选抗原序列(其中N一般是可以在疫苗中递送的最大表位数量)。包括选择用于给定患者的候选抗原的疫苗可以注射至患者体内以诱导免疫应答。
XI.B.盒设计模块
XI.B.1综述
盒设计模块可用于基于选择用于注射至患者体内的候选肽来产生疫苗盒序列。各种盒设计模块是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的盒设计模块,所述专利出于所有目的各自以引用的方式整体并入本文。
可以基于由预测模块确定的与超过预定阈值的递呈可能性相关联的所选肽产生治疗性表位集合,其中所述递呈可能性由递呈模型测定。然而,应了解,在其他实施方案中,可以基于多种方法中的任一种或多种(单独或组合形式),例如基于针对患者的I类或II类HLA等位基因的结合亲和力或预测的结合亲和力、针对患者的I类或II类HLA等位基因的结合稳定性或预测的结合稳定性、随机取样等产生治疗性表位的集合。
治疗性表位可对应于所选肽本身。除所选肽外,治疗性表位还可包括C末端和/或N末端侧接序列。N末端和C末端侧接序列可以是处于源蛋白质环境中的治疗性疫苗表位的原生N末端和C末端侧接序列。治疗性表位可以表示固定长度的表位。治疗性表位可以表示可变长度的表位,其中表位的长度可以取决于例如C侧接序列或N侧接序列的长度而变化。例如,C末端侧接序列和N末端侧接序列各自可具有2-5个残基的变化的长度,由此产生16种可能的表位选择。
盒设计模块还可通过考虑横跨所述盒中一对治疗性表位之间的接合点的接合点表位的递呈来产生盒序列。接合点表位由于在所述盒中串接治疗性表位和接头序列的过程而在所述盒中产生的新颖非自身但不相关的表位序列。接合点表位的新颖序列不同于所述盒的治疗性表位本身。
盒设计模块可产生降低在患者中递呈接合点表位的可能性的盒序列。具体来说,当将盒注射至患者体内时,接合点表位有可能被患者的HLA I类或HLA II类等位基因递呈,并且分别刺激CD8或CD4T细胞反应。由于T细胞与接合点表位的反应没有治疗益处,且可能因抗原竞争而减弱针对所述盒中所选治疗性表位的免疫应答,故此类反应常常是不合需要的。76
盒设计模块可迭代一个或多个候选盒,且确定与盒序列相关联的接合点表位的递呈评分低于数字阈值的盒序列。接合点表位递呈评分是与所述盒中接合点表位的递呈可能性相关联的量,并且较高的接合点表位递呈评分值指示所述盒的接合点表位会被HLA I类或HLA II类或两者递呈的可能性较高。
在一个实施方案中,盒设计模块可以确定候选盒序列中与最低接合点表位递呈评分相关联的盒序列。
盒设计模块可以迭代一个或多个候选盒序列,确定候选盒的接合点表位递呈评分,且鉴定与低于阈值的接合点表位递呈评分相关联的最佳盒序列。
盒设计模块可以进一步检查所述一个或多个候选盒序列以鉴定候选盒序列中的接合点表位中的任一者是否为设计使用所述疫苗的给定患者的自身表位。为实现此目的,盒设计模块针对已知数据库诸如BLAST检查接合点表位。在一个实施方案中,盒设计模块可以被配置以设计避免接合点自身表位的盒。
盒设计模块可以执行蛮力方法且迭代全部或大部分可能的候选盒序列以选出具有最小接合点表位递呈评分的序列。然而,由于疫苗容量增加,因此此类候选盒的数量可能极大。例如,对于20个表位的疫苗容量,盒设计模块必须迭代约1018个可能的候选盒,才能确定具有最低接合点表位递呈评分的盒。对于盒设计模块在合理的时间量内完成以产生用于患者的疫苗而言,此确定在计算上可能较为繁琐(就所需的计算处理资源而言)且有时难以处理。另外,考虑到每一候选盒的可能接合点表位,甚至可能更为繁琐。因此,盒设计模块可以基于迭代明显少于蛮力方法中的候选盒序列数量的候选盒数量来选择盒序列。
盒设计模块可产生随机产生或至少伪随机产生的候选盒的子集,且选择与低于预定阈值的接合点表位递呈评分相关联的候选盒作为盒序列。另外,盒设计模块可以从所述小组中选择具有最低接合点表位递呈评分的候选盒作为盒序列。例如,盒设计模块可以针对20个所选表位集合产生约1百万个候选盒子集,且选出具有最小接合点表位递呈评分的候选盒。尽管产生随机盒序列子集并从所述子集中选出具有低接合点表位递呈评分的盒序列可能不如蛮力方法好,但其需要明显较少的计算资源,由此使其实施在技术上是可行的。另外,相对于这种更有效的技术,执行蛮力方法可能仅引起接合点表位递呈评分的微小或甚至可忽略的改进,因此从资源分配的观点看,蛮力方法是不值得实施的。盒设计模块可通过将盒的表位序列用非对称旅行商问题(TSP)公式表示来确定改进的盒配置。鉴于节点列表和每对节点之间的距离,TSP确定与最短总距离相关联的节点的序列以访问每个节点恰好一次且返回至原始节点。例如,根据城市A、B和C且已知彼此之间的距离,TSP的解决方案产生一个闭合的城市序列,对于所述序列,访问每个城市恰好一次所行进的总距离是可能途径当中最短的。TSP的非对称形式确定当一对节点之间的距离不对称时节点的最佳序列。例如,从节点A行进至节点B的“距离”可以不同于从节点B行进至节点A的“距离”。通过使用非对称TSP解决改进的最佳盒,盒设计模块可以寻找使所有在所述盒的表位之间的接合点的递呈评分降低的盒序列。非对称TSP解决方案指示对应于应当在盒中串接表位以使所述盒的所有接合点的接合点表位递呈评分减到最小的次序的治疗性表位序列。相较于随机取样方法,经由此方法确定的盒序列可以产生具有明显较少接合点表位递呈的序列,同时可能需要明显较少的计算资源,尤其是在所产生的候选盒序列数量很大时。用于优化盒设计的不同计算方法和比较的说明性实例更详细地描述于国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中,所述专利出于所有目的各自以引用的方式整体并入本文。
XIII.示例计算机
计算机可以用于本文描述的任何计算方法。本领域的技术人员将认识到计算机可以具有不同的架构。计算机的实例是本领域技术人员已知的,例如国际专利申请公布WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的计算机,所述专利出于所有目的各自以引用的方式整体并入本文。
XIV.抗原递送载体实施例
以下是执行本发明的特定实施方案的实施例。所述实施例仅出于说明目的提供,而不旨在以任何方式限制本发明的范围。已作出努力来确保所使用数字(例如量、温度等)的精确性,但一些实验误差和偏差当然应是允许的。
除非另外指明,否则本发明将采用在本领域技能范围内的蛋白质化学、生物化学、重组DNA技术和药理学常规方法实行。所述技术在文献中有充分解释。参见例如T.E.Creighton,Proteins:Structures and Molecular Properties(W.H.Freeman andCompany,1993);A.L.Lehninger,Biochemistry(Worth Publishers,Inc.,现行版);Sambrook等,Molecular Cloning:A Laboratory Manual(第2版,1989);Methods InEnzymology(S.Colowick和N.Kaplan编,Academic Press,Inc.);Remington'sPharmaceutical Sciences,第18版(Easton,Pennsylvania:Mack Publishing Company,1990);Carey和Sundberg Advanced Organic Chemistry第3版(Plenum Press)A卷和B卷(1992)。
XIV.A.新抗原盒设计
可以通过疫苗接种递送刺激相应细胞免疫应答的多个I类MHC限制性肿瘤特异性新抗原(TSNA)。在一个实例中,疫苗盒经工程改造而以单一基因产物形式编码多个表位,其中所述表位系嵌入其天然的周围肽序列内或通过非天然接头序列隔开。鉴定出会潜在地影响抗原加工和递呈且因此影响TSNA特异性CD8 T细胞反应的量值和广度的若干设计参数。在本实例中,设计并构建出若干模型盒以评估:(1)是否可以针对并入单一表达盒中的多个表位产生稳定T细胞反应;(2)什么使得最佳接头置放于表达盒内的TSNA之间,引起所有表位的最佳加工和递呈;(3)所述表位在盒内的相对位置是否影响T细胞反应;(4)盒内表位的数量是否影响针对个别表位的T细胞反应的量值或质量;(5)添加细胞靶向序列是否改善T细胞反应。
产生两个读取结果以评估抗原递呈和对模型盒内的标志物表位具有特异性的T细胞反应:(1)体外基于细胞的筛选,其允许通过专门工程改造的报告T细胞的活化进行衡量,来评定抗原递呈(Aarnoudse等,2002;Nagai等,2012);以及(2)使用HLA-A2转基因小鼠(Vitiello等,1991),通过其对应表位特异性T细胞反应评定盒衍生的人源表位的疫苗接种后免疫原性的体内分析(Cornet等,2006;Depla等,2008;Ishioka等,1999)。
XIV.B.抗原盒设计评估
XIV.B.1.方法与材料
TCR和盒设计以及克隆
当通过A*0201递呈时,所选TCR识别肽NLVPMVATV(PDB#5D2N)、CLGGLLTMV(PDB#3REV)、GILGFVFTL(PDB#1OGA)、LLFGYPVYV(PDB#1AO7)。构建含有2A肽连接的TCR次单元(β然后是α)、EMCV IRES和2A连接的CD8次单元(β然后是α和嘌呤霉素抗性基因)的转移载体。对开放阅读框序列进行密码子优化且由GeneArt合成。
产生用于体外表位加工和递呈研究的细胞系
肽购自ProImmune或Genscript,在含10mM三(2-羧基乙基)膦(TCEP)的水/DMSO(2:8,v/v)中稀释至10mg/mL。除非另外指出,否则细胞培养基和补充剂来自Gibco。热灭活胎牛血清(FBShi)来自Seradigm。QUANTI-Luc底物、吉欧霉素(Zeocin)和嘌呤霉素来自InvivoGen。将Jurkat-Lucia NFAT细胞(InvivoGen)维持在补充有10%FBShi、丙酮酸钠和100μg/mL吉欧霉素的RPMI 1640中。转导后,这些细胞立即另外接受0.3μg/mL嘌呤霉素。在伊氏培养基(Iscove'sMedium,IMDM)加20%FBShi中培养T2细胞(ATCC CRL-1992)。U-87MG(ATCC HTB-14)细胞维持在补充有10%FBShi的MEM伊格尔培养基(MEM Eagles Medium)中。
Jurkat-Lucia NFAT细胞含有NFAT诱导性Lucia报告构建体。Lucia基因在通过接合T细胞受体(TCR)活化时,将利用腔肠素的荧光素酶分泌至培养基中。这种荧光素酶可使用QUANTI-Luc荧光素酶检测试剂测量。用慢病毒转导Jurkat-Lucia细胞以表达抗原特异性TCR。HIV衍生的慢病毒转移载体是从GeneCopoeia获得,并且表达VSV-G(pCMV-VsvG)、Rev(pRSV-Rev)和Gag-pol(pCgpV)的慢病毒支持质粒(support plasmid)是从Cell DesignLabs获得。
通过使用40μl脂染胺和20μg DNA混合物(以重量计4:2:1:1的转移质粒:pCgpV:pRSV-Rev:pCMV-VsvG),用脂染胺2000(Thermo Fisher)转染T75烧瓶中50-80%汇合的HEK293细胞,来制备慢病毒。使用Lenti-X系统(Clontech)浓缩8-10mL含病毒的培养基,且使病毒再悬浮于100-200μl新鲜培养基中。使用此体积覆盖相等体积的Jurkat-Lucia细胞(在不同实验中使用5×10E4-1×10E6个细胞)。在含0.3μg/ml嘌呤霉素的培养基中培养之后,分选细胞以获得克隆性。使用装载肽的T2细胞测试这些Jurkat-Lucia TCR克隆的活性和选择性。
体外表位加工和递呈分析
常规地使用T2细胞,通过TCR检查抗原识别。T2细胞缺乏用于抗原加工的肽转运蛋白(TAP缺陷型)并且不能在内质网中装载内源性肽以在MHC上递呈。然而,T2细胞可以容易地装载外源肽。将五个标志物肽(NLVPMVATV、CLGGLLTMV、GLCTLVAML、LLFGYPVYV、GILGFVFTL)以及两个不相关的肽(WLSLLVPFV、FLLTRICT)装载至T2细胞上。简言之,对T2细胞计数且用IMDM加1%FBShi稀释至1×106个细胞/mL。添加肽以产生10μg肽/1×106个细胞。接着在37℃下温育细胞90分钟。用IMDM加20%FBShi洗涤细胞两次,稀释至5×10E5个细胞/mL并将100μL涂铺至96孔Costar组织培养板中。对Jurkat-Lucia TCR克隆计数并在RPMI1640加10%FBShi中稀释至5×10E5个细胞/毫升,且将100μL添加至T2细胞中。培养板在37℃和5%CO2下温育过夜。然后以400g离心培养板3分钟并将20μL上清液移至白色平底Greiner板中。QUANTI-Luc底物是根据说明书制备且以每孔50μL添加。在MolecularDevices SpectraMax iE3x上读取荧光素酶表达。
为了测试腺病毒盒的标志物表位递呈,使用U-87MG细胞作为替代抗原递呈细胞(APC)且用腺病毒载体转导。收集U-87MG细胞并以5×10E5个细胞/100μl涂铺于96孔Costar组织培养板中的培养基中。在37℃温育培养板约2小时。用MEM加10%FBShi将腺病毒盒稀释至MOI为100、50、10、5、1和0并将其以每孔5μl加入到U-87MG细胞中。再在37℃下温育培养板约2小时。对Jurkat-Lucia TCR克隆计数并在RPMI加10%FBShi中稀释至5×10E5个细胞/毫升,且将其以每孔100μL加入到U-87MG细胞中。接着,在37℃及5%CO2下温育培养板约24小时。以400g离心培养板3分钟并将20μL上清液移至白色平底Greiner板中。QUANTI-Luc底物是根据说明书制备且以每孔50μL添加。在Molecular Devices SpectraMax iE3x上读取荧光素酶表达。
用于免疫原性研究的小鼠品系
转基因HLA-A2.1(HLA-A2 Tg)小鼠是从Taconic Labs,Inc获得。这些小鼠携带由嵌合I类分子组成的转基因,所述嵌合I类分子包含人HLA-A2.1前导序列、α1和α2结构域以及鼠科H2-Kbα3、跨膜和细胞质域(Vitiello等,1991)。用于这些研究的小鼠是基于C57Bl/6背景的野生型BALB/cAnNTac雌性和纯合HLA-A2.1 Tg雄性的第一代后代(F1)。
腺病毒载体(Ad5v)免疫接种
经由两侧肌肉内注射至胫前肌中对HLA-A2 Tg小鼠免疫接种1×1010至1×106个腺病毒载体病毒粒子。在免疫接种后第12天测量免疫应答。
淋巴细胞分离
从新鲜收集的经免疫接种小鼠的脾和淋巴结中分离淋巴细胞。使用GentleMACS组织解离器,根据制造商的说明书,在含有10%胎牛血清与青霉素和链霉素的RPMI(完全RPMI)中解离组织。
离体酶联免疫斑点(enzyme-linked immunospot,ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则(Janetzki等,2015),利用小鼠IFNgELISpotPLUS试剂盒(MABTECH)进行。将1×105个脾细胞与10uM指定肽一起在涂有IFNg抗体的96孔板中温育16小时。使用碱性磷酸酶使斑点显色。对反应定时10分钟并通过用自来水流过板来淬灭反应。使用AID vSpot读取器谱图对斑点计数。对于ELISPOT分析,将饱和度>50%的孔记录为“太多而无法计数”。将复制孔的偏差>10%的样品从分析中排除。然后,使用下式,针对孔汇合度校正斑点计数:斑点计数+2×(斑点计数×汇合度%/[100%-汇合度%])。通过用抗原刺激的孔减去阴性肽刺激孔中的斑点计数来校正阴性背景。最后,将标记为太多而无法计数的孔设定成最高观察校正值,四舍五入至最接近的百分数。
离体细胞内细胞因子染色(ICS)和流式细胞测量术分析
将密度为2-5×106个细胞/mL的新鲜分离的淋巴细胞与10uM指定肽一起温育2小时。两小时之后,添加布雷菲尔德菌素A(brefeldin A)达到5ug/ml浓度且将细胞与刺激剂一起再温育4小时。刺激之后,用可固定的存活力染料eFluor780,根据制造商的方案标记活细胞,并用以1:400稀释的抗CD8 APC(克隆53-6.7,BioLegend)染色。对于细胞内染色,使用1:100稀释的抗IFNg PE(克隆XMG1.2,BioLegend)。将样品收集在Attune NxT流式细胞仪(Thermo Scientific)上。使用FlowJo对流式细胞测量术数据绘图并分析。为了评定抗原特异性反应的程度,计算响应于各肽刺激剂的IFNg+的CD8+细胞百分比以及总IFNg+细胞数量/1×106个活细胞。
XIV.B.2.抗原盒设计的体外评估
作为抗原盒设计评估的实例,开发体外基于细胞的分析以评定在模型疫苗盒内的所选人表位是否被抗原递呈细胞表达、加工和递呈(图1)。在识别后,经工程改造成表达五种对明确表征的肽-HLA组合具有特异性的TCR中的一种的Jurkat-Lucia报告T细胞变得活化且将活化T细胞核因子(NFAT)易位至核中,引起荧光素酶报告基因的转录活化。通过生物发光定量个别报告CD8 T细胞系的抗原刺激。
通过用表达构建体转导慢病毒来改进个别Jurkat-Lucia报告株,所述表达构建体包括通过P2A核糖体跳跃序列(skip sequence)分离以确保等摩尔量翻译产物的抗原特异性TCRβ和TCRα链(Banu等,2014)。将第二CD8β-P2A-CD8α元件添加至慢病毒构建体中提供亲代报告细胞系所缺乏的CD8辅助受体的表达,因为细胞表面上的CD8对于与靶pMHC分子的结合亲和力至关重要并且通过接合其胞质尾区增强信号传导(Lyons等,2006;Yachi等,2006)。
在慢病毒转导之后,使Jurkat-Lucia报告细胞在嘌呤霉素选择下扩增,经历单细胞荧光辅助细胞分选(FACS)且测试单克隆群的荧光素酶表达。由此得到具有功能性细胞反应的针对特定肽抗原1、2、4和5的稳定转导的报告细胞系。(表2)。
表2:体外T细胞活化分析的发展。如通过荧光素酶的诱导所测量的肽特异性T细胞识别指示疫苗盒抗原的有效加工和递呈。
| 短盒设计 | |
| 表位 | AAY |
| 1 | 24.5±0.5 |
| 2 | 11.3±0.4 |
| 3* | n/a |
| 4 | 26.1±3.1 |
| 5 | 46.3±1.9 |
*尚未产生的针对表位3的报告T细胞
在另一实例中,对于一系列短盒,所有标志物表位均并入相同位置中(图2A)且仅分隔HLA-A*0201限制性表位的接头(图2B)是变化的。将报告T细胞个别地与被表达这些短盒的腺病毒构建体感染的U-87抗原递呈细胞(APC)混合,并且相对于未感染对照组测量荧光素酶表达。通过匹配报告T细胞识别模型盒中的全部四个抗原,展示多个抗原的有效加工和递呈。T细胞反应的量值在很大程度上遵循天然和AAY-接头的类似趋势。从基于RR-接头的盒释放的抗原显示较低荧光素酶诱导(表3)。被设计以破坏抗原加工的DPP-接头制造的疫苗盒引起低表位递呈(表3)。
表3:短盒中接头序列的评估。在体外T细胞活化分析中的荧光素酶诱导指示,除基于DPP的盒外,所有接头都有助于盒抗原的有效释放。仅T细胞表位(无接头)=9AA,天然接头一侧=17AA,天然接头两侧=25AA,非天然接头=AAY、RR、DPP

*尚未产生的针对表位3的报告T细胞
在另一实例中,构建另外一系列的短盒,所述盒除人和小鼠表位外,还含有定位于所述盒的N末端或C末端上的靶向序列诸如泛素(Ub)、MHC和Ig-κ信号肽(SP)和/或MHC跨膜(TM)基序。(图3)。当通过腺病毒载体递送至U-87APC时,报告T细胞再次展示多个盒衍生的抗原的有效加工和递呈。然而,各种靶向特征对于T细胞反应的量值无实质影响(表4)。
表4:添加至模型疫苗盒的细胞靶向序列的评估。采用体外活化分析证实,四个HLA-A*0201限制性标志物表位从模型盒有效释放并且靶向序列没有实质改善T细胞识别和活化。


*尚未产生的针对表位3的报告T细胞
XIV.B.3.抗原盒设计的体内评估
作为抗原盒设计评估的另一实例,疫苗盒经设计以含有5个已知以HLA-A*02:01限制性方式刺激CD8 T细胞的明确表征的人I类MHC表位(图2A、图3、图5A)。为了评估体内免疫原性,将含有这些标志物表位的疫苗盒并入腺病毒载体中并用于感染HLA-A2转基因小鼠(图4)。此小鼠模型携带的转基因部分地由人HLA-A*0201和小鼠H2-Kb组成,因此编码由人HLA-A2.1前导序列、连接至鼠类α3的α1和α2域、跨膜和细胞质H2-Kb域组成的嵌合I类MHC分子(Vitiello等,1991)。所述嵌合分子允许HLA-A*02:01限制性抗原递呈,同时维持CD8辅助受体与MHC上的α3域的物种相配的相互作用。
对于短盒,如通过IFN-γELISPOT所测定,所有标志物表位产生T细胞反应,其程度比通常所报道的程度强约10-50倍(Cornet等,2006;Depla等,2008;Ishioka等,1999)。在评估的所有接头中,各自含有通过天然氨基酸序列侧接的极小表位的25聚体序列多联体产生最大且最广泛的T细胞反应(表5)。细胞内细胞因子染色(ICS)和流式细胞测量术分析揭示,抗原特异性T细胞反应衍生自CD8 T细胞。
表5:短盒中接头序列的体内评估。ELISPOT数据指示,HLA-A2转基因小鼠在用1e11腺病毒病毒粒子感染后第17天,针对盒中的所有I类MHC限制性表位产生T细胞反应。

在另一实例中,构建一系列长疫苗盒并将其并入腺病毒载体中,其紧邻着原始的5个标志物表位含有另外16个具有已知CD8 T细胞反应性的HLA-A*02:01、A*03:01和B*44:05表位(图5A、图5B)。这些长盒的尺寸近似地模仿最终临床盒设计,并且只有表位相对于彼此的位置是不同的。对于长疫苗盒与短疫苗盒,CD8 T细胞反应在量值及广度方面是相当的,证实(a)添加更多表位不会实质影响针对原始表位集合的免疫应答的量值,以及(b)表位在盒中的位置不会实质影响随之而来的针对其的T细胞反应(表6)。
表6:有关长盒中表位位置的影响的体内评估。ELISPOT数据指示,对于长疫苗盒与短疫苗盒,HLA-A2转基因小鼠在用5e10腺病毒病毒粒子感染后第17天,产生的T细胞反应的量值相当。

*疑似技术失误引起T细胞反应的缺乏。
XIV.B.4.用于免疫原性和毒理学研究的抗原盒设计
总体而言,有关模型盒评估的研究结果(图2-图5,表2-表6)证实,对于模型疫苗盒,当采用“珠串(string of beads)”法在基于腺病毒的载体的背景下编码约20个表位时,实现强大的免疫原性。所述表位通过串接25聚体序列组装,所述序列各自嵌入在两侧上通过其天然、周围肽序列(例如在每一侧上的8个氨基酸残基)侧接的极小CD8T细胞表位(例如9个氨基酸残基)。如本文所用,“天然”或“原生”侧接序列是指给定表位在所述表位处于其源蛋白质内的天然存在环境中的N末端和/或C末端侧接序列。例如,HCMV pp65 MHC I表位NLVPMVATV通过原生5'序列WQAGILAR侧接于其5'端上且通过原生3'序列QGQNLKYQ侧接于其3'端上,由此产生在HCMV pp65源蛋白质内发现的25聚体肽WQAGILARNLVPMVATVQGQNLKYQ。天然或原生序列也可指编码通过原生侧接序列侧接的表位的核苷酸序列。每个25聚体序列直接连接至后面的25聚体序列。在极小CD8 T细胞表位大于或小于9个氨基酸的实例中,侧接肽长度可以经调整以使得总长度仍为25聚体肽序列。例如,10个氨基酸的CD8T细胞表位可以通过8个氨基酸的序列和7个氨基酸侧接。多联体之后是两个通用的II类MHC表位,包括所述表位是为了刺激CD4 T辅助细胞并改善疫苗盒抗原的总体体内免疫原性。(Alexander等,1994;Panina-Bordignon等,1989)。II类表位通过GPGPG氨基酸接头(SEQ ID NO:56)连接至最终I类表位。所述两个II类表位也通过GPG PG氨基酸接头彼此连接且通过GPGPG氨基酸接头侧接在C末端上。表位的位置和数量似乎都不会实质影响T细胞识别或反应。靶向序列似乎也不会实质影响盒衍生的抗原的免疫原性。
作为另一实例,基于用模型盒获得的体外和体内数据(图2-图5,表2-表6),产生交替已知在非人灵长类动物(NHP)、小鼠和人中具有免疫原性的明确表征的T细胞表位的盒设计。全部嵌入天然25聚体序列中的20个表位之后是存在于所评估的所有模型盒中的两个通用II类MHC表位(图6)。使用这种盒设计在多个物种中研究免疫原性以及药理学和毒理学研究。
XIV.B.5.用于30、40和50种抗原的抗原盒设计和评估
大抗原盒被设计具有30(L)、40(XL)或50(XXL)个表位,每个表位的长度为25个氨基酸。所述表位是人、NHP和小鼠表位的混合物,用于模拟疾病抗原,包括肿瘤抗原。图29示出了来自各种物种的表位的一般组织。表37、表38和表39分别描述了人、灵长类动物和小鼠模型表位使用的模型抗原。表37、表38和表39中的每一者都描述了表位位置、名称、最小表位描述和MHC类别。
这些盒被克隆到所述的chAd68和srRNA疫苗载体中,以评估较长的多表位盒的功效。图30示出了由ChAdV载体表达的每个大抗原盒,如通过蛋白质印迹由预期大小的至少一个主要条带所指示。
将小鼠如所述进行免疫接种以评估大盒的功效。在用chAd68载体(分别为图31/表40和图32/表41)免疫接种后通过ICS和四聚体染色,以及在用srRNA载体(图33/表42)免疫接种后通过ICS分析针对表位AH1(顶部图)和SINNFEKL(底部图)的T细胞反应。使用表达30(L)、40(XL)或50(XXL)表位的chAd68和srRNA疫苗载体进行的免疫接种,可诱导对模型疾病表位的CD8+免疫应答。
表37-大盒中的人表位

表38–大盒中的NHP表位

表39-大盒中的小鼠表位

表40:ChAd大盒治疗的小鼠中平均IFNg+细胞对AH1和SIINFEKL肽的反应。数据呈现为总CD8细胞的百分比。示出了每组的平均值和标准偏差,以及通过使用Tukey检验的ANOVA的p值。与MAG 20抗原盒比较的所有p值。
| #抗原 | 抗原 | 平均值 | 标准偏差 | p值 | N |
| 20 | SIINFEKL | 5.308 | 0.660 | n/a | 8 |
| 30 | SIINFEKL | 4.119 | 1.019 | 0.978 | 8 |
| 40 | SIINFEKL | 6.324 | 0.954 | 0.986 | 8 |
| 50 | SIINFEKL | 8.169 | 1.469 | 0.751 | 8 |
| 20 | AH1 | 6.405 | 2.664 | n/a | 8 |
| 30 | AH1 | 4.373 | 1.442 | 0.093 | 8 |
| 40 | AH1 | 4.126 | 1.135 | 0.050 | 8 |
| 50 | AH1 | 4.216 | 0.808 | 0.063 | 8 |
表41:ChAd大盒治疗的小鼠中AH1和SIINFEKL抗原的平均四聚体+细胞。数据呈现为总CD8细胞的百分比。示出了每组的平均值和标准偏差,以及通过使用Tukey检验的ANOVA的p值。与MAG20抗原盒比较的所有p值。
| #抗原 | 抗原 | 平均值 | 标准偏差 | p值 | N |
| 20 | SIINFEKL | 10.314 | 2.384 | n/a | 8 |
| 30 | SIINFEKL | 4.551 | 2.370 | 0.003 | 8 |
| 40 | SIINFEKL | 5.186 | 3.254 | 0.009 | 8 |
| 50 | SIINFEKL | 14.113 | 3.660 | 0.072 | 8 |
| 20 | AH1 | 6.864 | 2.207 | n/a | 8 |
| 30 | AH1 | 4.713 | 0.922 | 0.036 | 8 |
| 40 | AH1 | 5.393 | 1.452 | 0.223 | 8 |
| 50 | AH1 | 5.860 | 1.041 | 0.543 | 8 |
表42:SAM大盒治疗的小鼠中平均IFNg+细胞对AH1和SIINFEKL肽的反应。数据呈现为总CD8细胞的百分比。示出了每组的平均值和标准偏差,以及通过使用Tukey检验的ANOVA的p值。与MAG 20抗原盒比较的所有p值。


XV.ChAd抗原盒递送载体
XV.A.ChAd抗原盒递送载体构建
在一个实例中,将黑猩猩腺病毒(ChAd)工程改造成用于抗原盒的递送载体。在另一实例中,基于缺失E1(nt 457至3014)和E3(nt 27,816-31,332)序列的AC_000011.1(来自专利US 6083716的序列2)合成全长ChAdV68载体。插入处于CMV启动子/增强子控制下的报告基因代替缺失的E1序列。将此克隆转染至HEK293细胞中不会产生感染性病毒。为了确定野生型C68病毒的序列,从ATCC获得分离株VR-594,传代,并且然后独立测序(SEQ ID NO:10)。当将AC_000011.1序列与野生型ChAdV68病毒的ATCC VR-594序列(SEQ ID NO:10)相比较时,鉴定出6个核苷酸差异。在一个实例中,基于对应ATCC VR-594核苷酸在五个位置被取代的AC_000011.1产生经修饰的ChAdV68载体(ChAdV68.5WTnt SEQ ID NO:1)。
在另一实例中,基于缺失E1(nt 577至3403)和E3(nt 27,816-31,332)序列且对应ATCC VR-594核苷酸在四个位置被取代的AC_000011.1产生经修饰的ChAdV68载体。插入处于CMV启动子/增强子控制下的GFP报告蛋白(ChAdV68.4WTnt.GFP;SEQ ID NO:11)或模型新抗原盒(ChAdV68.4WTnt.MAG25mer;SEQ ID NO:12)代替缺失的E1序列。
在另一实例中,基于缺失E1(nt 577至3403)和E3(nt 27,125-31,825)序列且对应ATCC VR-594核苷酸在五个位置被取代的AC_000011.1产生经修饰的ChAdV68载体。插入处于CMV启动子/增强子控制下的GFP报告蛋白(ChAdV68.5WTnt.GFP;SEQ ID NO:13)或模型新抗原盒(ChAdV68.5WTnt.MAG25mer;SEQ ID NO:2)代替缺失的E1序列。
下文描述了相关载体:
-全长ChAdVC68序列“ChAdV68.5WTnt”(SEQ ID NO:1);相应ATCC VR-594核苷酸在五个位置被取代的AC_000011.1序列。
-ATCC VR-594C68(SEQ ID NO:10);独立测序;全长C68
-ChAdV68.4WTnt.GFP(SEQ ID NO:11);AC_000011.1缺失E1(nt 577至3403)和E3(nt 27,816-31,332)序列;相应ATCC VR-594核苷酸在四个位置被取代;插入处于CMV启动子/增强子控制下的GFP报告蛋白代替缺失的E1
-ChAdV68.4WTnt.MAG25mer(SEQ ID NO:12);AC_000011.1缺失E1(nt 577至3403)和E3(nt 27,816-31,332)序列;相应ATCC VR-594核苷酸在四个位置被取代;插入处于CMV启动子/增强子控制下的模型新抗原盒代替缺失的E1
-ChAdV68.5WTnt.GFP(SEQ ID NO:13);AC_000011.1缺失E1(nt 577至3403)和E3(nt 27,125-31,825)序列;相应ATCC VR-594核苷酸在五个位置被取代;插入处于CMV启动子/增强子控制下的GFP报告蛋白代替缺失的E1
XV.B.ChAd抗原盒递送载体测试
XV.B.1.ChAd载体评估方法和材料
使用脂染胺转染HEK293A细胞
使用以下方案,制备ChAdV68构建体(ChAdV68.4WTnt.GFP、ChAdV68.5WTnt.GFP、ChAdV68.4WTnt.MAG25mer和ChAdV68.5WTnt.MAG25mer)的DNA并将其转染至HEK293A细胞中。
用PacI消化10ug质粒DNA以释放病毒基因组。然后,根据制造商的说明书,对于较长DNA片段,使用GeneJet DNA净化微型柱(DNA cleanup Micro columns;Thermo Fisher)纯化DNA,并且在20ul预热的水中洗脱;在洗脱步骤之前,使柱在37℃下保持0.5-1小时。
在转染之前,将HEK293A以106个细胞/孔的细胞密度引入6孔培养板中,保持14-18小时。用每孔1ml新鲜培养基(含青霉素/链霉素和谷氨酸的DMEM-10%hiFBS)覆盖细胞。在根据制造商的方案,用微升体积(2-4ul)脂染胺2000两次转染中使用每孔1-2ug的纯化DNA。将0.5ml含有转染混合物的OPTI-MEM培养基加入到各孔中的1ml标准生长培养基中并在细胞上保持过夜。
在37℃下温育经转染的细胞培养物至少5-7天。如果在转染后第7天未见到病毒蚀斑,则将细胞以1:4或1:6分离,并在37℃下温育以监测蚀斑的产生。或者,收集经转染的细胞且进行3个循环的冷冻和解冻,并使用细胞溶解产物感染HEK293A细胞且温育细胞直至观察到病毒蚀斑。
使用磷酸钙将ChAdV68转染至HEK293A细胞中并产生第三代病毒原液
使用以下方案,制备ChAdV68构建体(ChAdV68.4WTnt.GFP、ChAdV 68.5WTnt.GFP、ChAdV68.4WTnt.MAG25mer、ChAdV68.5WTnt.MAG25 mer)的DNA并将其转染至HEK293A细胞中。
在转染前一天,将HEK293A细胞以106个细胞/孔接种于6孔板的5%BS/DMEM/1XP/S、1XGlutamax中。每次转染需要两个孔。在转染前二至四小时,将培养基更换成新鲜培养基。用PacI使ChAdV68.4WTnt.GFP质粒线性化。然后,用酚氯仿提取经线性化的DNA并使用十分之一体积的3M乙酸钠pH 5.3和两体积的100%乙醇使其沉淀。通过以12,000xg离心5分钟使沉淀的DNA集结,然后用70%乙醇洗1次。空气干燥集结粒并使其再悬浮于50μL无菌水中。使用NanoDropTM(ThermoFisher)测定DNA浓度并将体积调整至5μgDNA/50μL。
将169μL无菌水加入到微量离心管中。然后将5μL 2M CaCl2加入到水中并通过移液管移液徐缓地混合。将50μL DNA逐滴加入到CaCl2水溶液中。然后添加26微升2M CaCl2并通过微量移液管移液两次徐缓地混合。此最终溶液应由5μg DNA于250μL的0.25M CaCl2中组成。然后制备含有250μL的2XHBS(Hepes缓冲溶液)的第二管。使用连接至Pipet-Aid空气的2mL无菌移液管缓慢鼓泡通过2XHBS溶液。同时,逐滴添加于0.25M CaCl2溶液中的DNA溶液。在添加最终DNA液滴之后,继续鼓泡约5秒。接着在室温温育溶液达20分钟,然后添加至293A细胞中。将250μL DNA/磷酸钙溶液逐滴添加至前一天以106个细胞/孔接种于6孔板中的293A细胞单层中。将细胞放回温育箱中并温育过夜。24小时后更换培养基。72小时后,将细胞以1:6分至6孔板中。每天通过光学显微镜检查监测细胞单层的细胞病变效应(CPE)的迹象。转染后7-10天,观察到病毒蚀斑且通过用移液管吸移孔中的培养基以使细胞升高来收集细胞单层。将收集的细胞和培养基转移至50mL离心管中,接着进行三轮冷冻解冻(在-80℃和37℃下)。随后的溶解产物,称为初代病毒原液,通过在桌上型(bench top)离心机(4300Xg)上全速离心来澄清且使用一部分溶解产物(10-50%)感染T25烧瓶中的293A细胞。将感染的细胞温育48小时,然后在完全CPE下收集细胞和培养基。再次收集细胞,冷冻解冻并澄清,然后使用此第二代病毒原液感染每个烧瓶接种1.5×107个细胞的T150烧瓶。一旦在72小时实现完全CPE,就以先前病毒原液相同的方式收集并处理培养基和细胞以产生第三代原液。
在293F细胞中的制造
在8%CO2的温育箱中,在293FreeStyleTM(ThermoFisher)培养基中生长的293F细胞中进行ChAdV68病毒的制造。感染当天,将细胞稀释至106个细胞/毫升,存活率为98%,并在每个制造操作于1L摇瓶(Corning)中使用400mL。每次感染使用靶MOI>3.3的4mL第三代病毒原液。将细胞温育48-72小时,直至通过台盼蓝测量到存活率<70%。然后,通过全速桌上型离心机离心来收集经感染细胞并在1XPBS中洗涤,再离心,然后使其再悬浮于20mL的10mMTris pH7.4中。通过冷冻解冻3次将细胞集结粒溶解并通过以4,300Xg离心5分钟使其澄清。
通过CsCl离心纯化
通过CsCl离心使病毒DNA纯化。执行两次不连续梯度操作。第一次是从细胞组分中纯化出病毒并且第二次是从细胞组分进一步优化分离并将缺陷性粒子与感染性粒子分离。
将10mL的1.2(26.8g CsCl溶解于92mL的10mM Tris pH 8.0中)CsCl添加至异质同晶聚合物管中。然后,使用移液管递送至管底部,小心地添加8mL的1.4CsCl(53g CsCl溶解于87mL的10mM Tris pH 8.0中)。将澄清的病毒小心地铺在1.2层的顶部上。必要时,再添加10mM Tris以使各管平衡。然后将所述管置放于SW-32Ti旋转器中并在10℃下离心2小时30分钟。然后将所述管移至层流柜中并使用18号针和10mL针筒抽吸病毒带。应避免取出污染性宿主细胞DNA和蛋白质。然后用10mM Tris pH 8.0将所述病毒带稀释至少2倍并如前所述铺在如上文所描述的不连续梯度上。如前所述进行操作,其中例外为此时进行所述操作过夜。次日,小心抽吸病毒带以避免抽吸出任何缺陷性粒子带。然后使用Slide-A-LyzerTM盒(Pierce)针对ARM缓冲液(20mM Tris pH 8.0、25mM NaCl、2.5%丙三醇)透析病毒。进行此操作3次,每次更换缓冲液保持1小时。然后将病毒等分以在-80℃下储存。
病毒分析
基于1.1×1012个病毒粒子(VP)的消光系数相当于在OD260 nm下的吸光度值1,通过使用OD 260分析测定VP浓度。在病毒溶解缓冲液(0.1%SDS、10mM Tris pH 7.4、1mMEDTA)中制备腺病毒的两种稀释液(1:5和1:10)。一式两份测量所述两种稀释液的OD并且通过用OD260值乘以稀释因子乘以1.1×1012VP来测量每毫升VP浓度。
利用病毒原液的限制性稀释分析来计算感染单位(IU)滴度。病毒最初在DMEM/5%NS/1X PS中100倍稀释,随后使用10倍稀释法稀释至1X10-7。然后,将100μL这些稀释液添加至在之前至少一小时以3e5个细胞/孔接种于24孔板中的293A细胞中。此操作一式两份进行。在37℃下,在CO2(5%)温育箱中温育板48小时。然后用1XPBS洗涤细胞,且接着用100%冷甲醇(-20℃)固定。然后在-20℃温育所述板最少20分钟。用1XPBS洗涤各孔,然后在室温下在1×PBS/0.1%BSA中阻断1小时。添加兔抗Ad抗体(Abcam,Cambridge,MA)于阻断缓冲液中的1:8,000稀释液(每孔0.25ml)并在室温下温育1小时。用每孔0.5mL PBS洗涤各孔4次。每孔添加1000倍稀释的HRP结合的山羊抗兔抗体(Bethyl Labs,Montgomery Texas)并温育1小时,然后进行最后一轮洗涤。进行5次PBS洗涤并使用于含0.01%H2O2的Tris缓冲生理盐水中的二氨基联苯胺四盐酸盐(Diaminobenzidine tetrahydrochloride,DAB)底物(0.67mg/mL DAB于50mM Tris pH 7.5、150mM NaCl)使所述板显色。使各孔显色5分钟,然后计数。使用产生每个视野4-40个经染色细胞的稀释液,在10X下对细胞计数。所用视野是0.32mm2栅格,相当于在24孔板上每个视野有625个栅格。可以通过每个栅格中经染色细胞的数量乘以每个视野的栅格数量乘以稀释因子10来测定每毫升中感染性病毒的数量。类似地,当用GFP表达性细胞操作时,可以使用荧光而非衣壳染色来测定每毫升中GFP表达性病毒粒子的数量。
免疫接种
经两侧肌肉内注射向C57BL/6J雌性小鼠和Balb/c雌性小鼠注射1×108个ChAdV68.5WTnt.MAG25mer病毒粒子(VP),体积为100uL(每条腿50uL)。
脾细胞解离
将每只小鼠的脾和淋巴结汇集于3mL完全RPMI(RPMI、10%FBS、青霉素/链霉素)中。使用gentleMACS解离器(Miltenyi Biotec),遵循制造商的方案进行机械解离。通过40微米过滤器过滤解离的细胞并用ACK溶解缓冲液(150mM NH4Cl、10mM KHCO3、0.1mMNa2EDTA)溶解红细胞。再次经由30微米过滤器过滤细胞且然后使其再悬浮于完全RPMI中。在Attune NxT流式细胞仪(Thermo Fisher)上使用碘化丙锭染色对细胞计数以排除死亡和凋亡的细胞。然后将细胞调整至适当活细胞浓度以供后续分析。
离体酶联免疫斑点(ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用小鼠IFNg ELISpotPLUS试剂盒(MABTECH)进行。将5x104个脾细胞与10uM指定肽一起在涂有IFNg抗体的96孔板中温育16小时。使用碱性磷酸酶使斑点显色。对反应定时10分钟并通过用自来水流过板终止反应。使用AID vSpot读取器谱图对斑点计数。对于ELISPOT分析,将饱和度>50%的孔记录为“太多而无法计数”。将复制孔的偏差>10%的样品从分析中排除。然后,使用下式,针对孔汇合度校正斑点计数:斑点计数+2×(斑点计数×汇合度%/[100%-汇合度%])。通过用抗原刺激的孔减去阴性肽刺激孔中的斑点计数来校正阴性背景。最后,将标记为太多而无法计数的孔设定成最高观察校正值,四舍五入至最接近的百分数。
XV.B.2.在DNA转染之后ChAdV68病毒递送粒子的制造
在一个实例中,将ChAdV68.4WTnt.GFP(图7)和ChAdV68.5WT nt.GFP(图8)DNA转染至HEK293A细胞中并在转染之后7-10天观察病毒复制(病毒蚀斑)。使用光学显微镜检查(图7A和8A)和荧光显微镜检查(图7B-图7C和图8B-图8C)观测ChAdV68病毒蚀斑。GFP表示产毒ChAdV68病毒递送粒子的产生。
XV.B.3.ChAdV68病毒递送粒子扩增
在一个实例中,使ChAdV68.4WTnt.GFP、ChAdV68.5WTnt.GFP和ChAdV68.5WTnt.MAG25mer病毒在HEK293F细胞中扩增并在转染之后18天,制备纯化的病毒原液(图9)。定量经纯化ChAdV68病毒原液中的病毒粒子数且与使用相同方案制造的5型腺病毒(Ad5)和ChAdVY25(密切相关的ChAdV;Dicks,2012,PloS ONE 7,e40385)病毒原液相比较。ChAdV68病毒滴度与Ad5和ChAdVY25相当(表7)。
表7.在293F悬浮细胞中产生腺病毒载体
| 构建体 | 平均VP/细胞+/-SD |
| Ad5-载体(多个载体) | 2.96e4+/-2.26e4 |
| Ad5-GFP | 3.89e4 |
| chAdY25-GFP | 1.75e3+/-6.03e1 |
| ChAdV68.4WTnt.GFP | 1.2e4+/-6.5e3 |
| ChAdV68.5WTnt.GFP | 1.8e3 |
| ChAdV68.5WTnt.MAG25mer | 1.39e3+/-1.1e3 |
*SD仅在执行多个制造操作情况下报道
XV.B.4.肿瘤模型中的免疫原性评估
在小鼠免疫原性研究中评估表达小鼠肿瘤抗原的C68载体以证实C68载体引起T细胞反应。在C57BL/6J雌性小鼠中测量针对I类MHC表位SIINFEKL的T细胞反应并在Balb/c小鼠中测量针对I类MHC表位AH1-A5(Slansky等,2000,Immunity13:529-538)的T细胞反应。如图15中所示,在对小鼠免疫接种ChAdV68.5WTnt.MAG25mer之后测量到相对于对照较强的T细胞反应。当对C57BL/6J或Ba lb/c小鼠免疫接种ChAdV68.5WTnt.MAG25mer时,在免疫接种之后10天,在ELISpot分析中分别观察到每106个脾细胞8957个或4019个斑点形成细胞(SFC)的平均细胞免疫应答。
还在评估ChAdV和抗CTLA4抗体的共同施用的CT26肿瘤模型中评估了肿瘤浸润淋巴细胞。对小鼠植入CT26肿瘤细胞,并在植入后第7天免疫接种ChAdV疫苗,并用抗CTLA4抗体(克隆9D9)或IgG作为对照治疗。免疫接种后第12天分析肿瘤浸润淋巴细胞。使用gentleMACS解离器(Miltenyi Biotec)和小鼠肿瘤解离试剂盒(Miltenyi Biotec)解离每只小鼠的肿瘤。通过30微米过滤器过滤解离的细胞并且使其再悬浮于完全RPMI中。在Attune NxT流式细胞仪(Thermo Fisher)上使用碘化丙锭染色对细胞计数以排除死亡和凋亡的细胞。然后将细胞调整至适当活细胞浓度以供后续分析。抗原特异性细胞通过MHC-四聚体复合物鉴定,并与抗CD8和存活力标志物共染色。初免免疫接种后第12天收集肿瘤。
肿瘤内的抗原特异性CD8+T细胞细胞分别占ChAdV、抗CTLA4和ChAdV+抗CTLA4治疗组中总活细胞群体的中值3.3%、2.2%或8.1%(图41和表36)。与单独的ChAdV和单独的抗CTLA4相比,使用抗CTLA与活性ChAdV免疫接种组合治疗导致抗原特异性CD8+T细胞频率有统计学上的显著增加,表明与chAd68疫苗共同施用时,抗CTLA4增加肿瘤内浸润T细胞的数量。
表36-CT26肿瘤中四聚体+浸润CD8 T细胞的频率

XVI.甲病毒抗原盒递送载体
XVI.A.甲病毒递送载体评估材料和方法
体外转录以产生RNA
对于体外测试:通过用PmeI限制性消化使质粒DNA线性化,遵循制造商的方案(GeneJet DNA净化试剂盒,Thermo)进行柱纯化并用作模板。根据制造商的方案,使用RiboMAX大规模RNA生产系统(Promega),利用m7G帽类似物(Promega)进行体外转录。根据制造商的方案,使用RNeasy试剂盒(Qiagen)纯化mRNA。
对于体内研究:产生RNA且由TriLInk Biotechnologies纯化并用Enzymatic Cap1封端。
RNA转染
转染前约16小时,HEK293A细胞对于96孔是以6e4个细胞/孔接种且对于24孔是以2e5个细胞/孔接种。使用MessengerMAX脂染胺(Invitrogen)且遵循制造商的方案,用mRNA转染细胞。对于96孔,每孔使用0.15uL脂染胺和10ng mRNA,且对于24孔,每孔使用0.75uL脂染胺和150ng mRNA。GFP表达mRNA(TriLink Biotechnologies)用作转染对照。
荧光素酶分析
使用ONE-Glo荧光素酶分析(Promega),遵循制造商的方案,在每种条件下于白色壁96孔板中一式三份进行荧光素酶报告蛋白分析。使用SpectraMax测量发光。
qRT-PCR
在转染后2小时,用新鲜培养基冲洗经转染的细胞并更换培养基以移除任何未经转染的mRNA。然后,在多个时间点,将细胞收集于RLT plus溶解缓冲液(Qiagen)中,使用QiaShredder(Qiagen)均质化并使用RNeasy试剂盒(Qiagen)提取RNA,所有操作都遵循制造商的方案。使用Nanodrop(Thermo Scientific)定量总RNA。根据制造商的方案,在qTower3(Analytik Jena)上使用Quantitect探针一步法RT-PCR试剂盒(Probe One-Step RT-PCRkit;Qiagen)进行qRT-PCR,每一反应使用20ng总RNA。对于每个探针,各样品是一式三份地操作。肌动蛋白或GusB用作参考基因。定制引物/探针由IDT产生(表8)。
表8.qPCR引物/探针

B16-OVA肿瘤模型
在C57BL/6J小鼠左下方侧腹部中注射105个B16-OVA细胞/动物。在免疫接种之前,使肿瘤生长3天。
CT26肿瘤模型
在Balb/c小鼠左下方侧腹部中注射106个CT26细胞/动物。在免疫接种之前,使肿瘤生长7天。
免疫接种
对于srRNA疫苗,经两侧肌肉内注射向小鼠注射10ug RNA,体积100uL(每条腿50uL)。对于Ad5疫苗,经两侧肌肉内注射向小鼠注射5×1010个病毒粒子(VP),体积100uL(每条腿50uL)。每周2次,经由腹膜内注射向动物注射250ug剂量的抗CTLA-4(克隆9D9,BioXcell)、抗PD-1(克隆RMP1-14,BioXcell)或抗IgG(克隆MPC-11,BioXcell)。
体内生物发光成像
在每个时间点,经由腹膜内注射向小鼠注射150mg/kg荧光素底物并在注射之后10-15分钟,使用IVIS体内成像系统(PerkinElmer)测量生物发光。
脾细胞解离
将每只小鼠的脾和淋巴结汇集于3mL完全RPMI(RPMI、10%FBS、青霉素/链霉素)中。使用gentleMACS解离器(Miltenyi Biotec),遵循制造商的方案进行机械解离。通过40微米过滤器过滤解离的细胞并用ACK溶解缓冲液(150mM NH4Cl、10mM KHCO3、0.1mMNa2EDTA)溶解红细胞。再次经由30微米过滤器过滤细胞且然后使其再悬浮于完全RPMI中。在Attune NxT流式细胞仪(Thermo Fisher)上使用碘化丙锭染色对细胞计数以排除死亡和凋亡的细胞。然后将细胞调整至适当活细胞浓度以供后续分析。
离体酶联免疫斑点(ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用小鼠IFNg ELISpotPLUS试剂盒(MABTECH)进行。将5x104个脾细胞与10uM指定肽一起在涂有IFNg抗体的96孔板中温育16小时。使用碱性磷酸酶使斑点显色。对反应定时10分钟并通过用自来水流过板终止反应。使用AID vSpot读取器谱图对斑点计数。对于ELISPOT分析,将饱和度>50%的孔记录为“太多而无法计数”。将复制孔的偏差>10%的样品从分析中排除。然后,使用下式,针对孔汇合度校正斑点计数:斑点计数+2×(斑点计数×汇合度%/[100%-汇合度%])。通过用抗原刺激的孔减去阴性肽刺激孔中的斑点计数来校正阴性背景。最后,将标记为太多而无法计数的孔设定成最高观察校正值,四舍五入至最接近的百分数。
XVI.B.甲病毒载体
XVI.B.1.甲病毒载体体外评估
在本发明的一个实施方式中,从基于委内瑞拉马脑炎(VEE)(Kinney,1986,Virology 152:400-413)的自我复制RNA(srRNA)载体产生用于抗原表达系统的RNA甲病毒骨架。在一个实例中,编码位于26S亚基因组启动子3'端的VEE结构蛋白的序列缺失(VEE序列7544至11,175缺失;编号基于Kinney等1986;SEQ ID NO:6)且被抗原序列(SEQ ID NO:14和SEQ ID NO:4)或荧光素酶报告蛋白(例如VEE-荧光素酶,SEQ ID NO:15)替换(图10)。由srRNA DNA载体体外转录RNA,将其转染至HEK293A细胞中并测量荧光素酶报告蛋白表达。此外,用编码荧光素酶的(非复制性)mRNA转染以供比较。当比较23小时测量值与2小时测量值时,对于VEE-荧光素酶srRNA观察到srRNA报告基因信号有约30,000倍增加(表9)。相比之下,在相同时间段内,mRNA报告基因展现小于10倍的信号增加(表9)。
表9.来自VEE自我复制载体的荧光素酶的表达随时间增加。在96孔中用每孔10ngVEE-荧光素酶srRNA或10ng非复制性荧光素酶mRNA(TriLink L-6307)转染HEK293A细胞。在转染后多个时间测量发光。荧光素酶表达以相对发光单位(RLU)报道。每个数据点是3个转染孔的平均值+/-SD。


在另一实例中,通过使用定量逆转录聚合酶链反应(qRT-PCR)测量编码荧光素酶的srRNA(VEE-荧光素酶)或编码多表位盒的srRNA(VEE-MAG25mer)转染之后的RNA含量来直接确定srRNA的复制。对于VEE-荧光素酶srRNA观察到约150倍的RNA增加(表10),而对于VEE-MAG25mer srRNA观察到30-50倍的RNA增加(表11)。这些数据证实,当转染至细胞中时,VEE srRNA载体复制。
表10.VEE-荧光素酶srRNA转染的细胞中RNA复制的直接测量。用VEE-荧光素酶srRNA(150ng/孔,24孔)转染HEK293A细胞并在转染之后多个时间,通过qRT-PCR定量RNA水平。基于肌动蛋白参考基因使各测量值归一化并呈现相对于2小时时间点的倍数变化。

表11.VEE-MAG25mer srRNA转染的细胞中RNA复制的直接测量。用VEE-MAG25mersrRNA(150ng/孔,24孔)转染HEK293细胞并在转染之后多个时间,通过qRT-PCR定量RNA水平。基于GusB参考基因使各测量值归一化并呈现相对于2小时时间点的倍数变化。图上的不同线表示2个不同的qPCR引物/探针集,所述两个集合都检测srRNA的表位盒区。


XVI.B.2.甲病毒载体的体内评估
在另一实例中,在体内评估VEE-荧光素酶报告蛋白表达。对小鼠注射10ug封装于脂质纳米粒子(MC3)中的VEE-荧光素酶srRNA并在注射后24小时和48小时,以及7天和14天使其成像以测定生物发光信号。在注射后24小时检测到荧光素酶信号且其随时间增加,并在srRNA注射之后7天出现峰值(图11)。
XVI.B.3.甲病毒载体肿瘤模型评估
在一个实施方式中,为了确定VEE srRNA载体是否在体内引导抗原特异性免疫应答,产生表达2种不同I类MHC小鼠肿瘤表位SIINFEKL和AH1-A5的VEE srRNA载体(VEE-UbAAY,SEQ ID NO:14)(Slansky等,2000,Immunity 13:529-538)。利用B16-OVA黑素瘤细胞系表达SFL(SIINFEKL)表位,并且AH1-A5(SPSYAYHQF;Slansky等,2000,Immunity)表位诱导T细胞靶向由CT26结肠癌细胞系表达的相关表位(AH1/SPSYVYHQF;Huang等,1996,ProcNatl Acad Sci USA 93:9730-9735)。在一个实例中,对于体内研究,通过使用T7聚合酶(TriLink Biotechnologies)体外转录来产生VEE-UbAAY srRNA并将其包封在脂质纳米粒子(MC3)中。
在用MC3配制的VEE-UbAAY srRNA免疫接种带有B16-OVA肿瘤的小鼠之后两周,观察到相对于对照强烈的靶向SFL的抗原特异性T细胞反应。在一个实例中,在用SFL肽刺激之后,在ELISpot分析中测量每106个脾细胞3835个(中值)斑点形成细胞(SFC)(图12A,表12)并且如通过五聚体染色所测量,1.8%(中值)的CD8 T细胞具有SFL抗原特异性(图12B,表12)。在另一实例中,共同施用抗CTLA-4单克隆抗体(mAb)与VEE srRNA疫苗引起总体T细胞反应的适度增加,并且在ELISpot分析中测量到每106个脾细胞的4794.5个(中值)SFC(图12A,表12)。
表12.在带有B16-OVA肿瘤的C57BL/6J小鼠中VEE srRNA免疫接种后14天ELISPOT和MHCI-五聚体染色分析的结果。

*应注意,从Vax组中小鼠#6得到的结果由于三个重复孔之间变化较大而从分析中排除。
在另一实施方式中,为反映临床方法,在B16-OVA和CT26小鼠肿瘤模型中进行异源初免/加强免疫,其中带有肿瘤的小鼠先用表达相同抗原盒的腺病毒载体(Ad5-UbAAY)免疫接种,接着在Ad5-UbAAY初免之后14天,用VEE-UbAAY srRNA疫苗加强免疫。在一个实例中,通过Ad5-UbAAY疫苗诱导抗原特异性免疫应答,由此在ELISpot分析中测量到每106个脾细胞7330个(中值)SFC(图13A,表13)并且通过五聚体染色测量到2.9%(中值)的CD8 T细胞靶向SFL抗原(图13C,表13)。在另一实例中,在VEE-UbAAY srRNA加强免疫之后2周,B16-OVA模型中仍维持T细胞反应,并且在ELISpot分析中测量到每106个脾细胞3960个(中值)SFL特异性SFC(图13B,表13)并且通过五聚体染色测量到3.1%(中值)的CD8 T细胞靶向SFL抗原(图13D,表13)。
表13.用Ad5疫苗初免和srRNA加强免疫进行异源初免/加强免疫之后B16-OVA小鼠的免疫监测。
第14天

第28天

在另一实施方式中,在Ad5-UbAAY初免和VEE-UbAAY srRNA加强免疫之后,在CT26小鼠模型中观察到类似结果。在一个实例中,在Ad5-UbAAY初免(第14天)之后观察到AH1抗原特异性反应并且在ELISpot分析中测量到每106个脾细胞平均5187个SFC(图14A,表14)并且在VEE-UbAAY srRNA加强免疫(第28天)之后于ELISpot分析中测量到每106个脾细胞平均3799个SFC(图14B,表14)。
表14.在CT26肿瘤小鼠模型中异源初免/加强免疫之后的免疫监测。

XVII.ChAdV/srRNA组合肿瘤模型评估
在鼠类CT26肿瘤模型中评估使用ChAdV68和自我复制RNA(srRNA)的各种给药方案。
XVII.A ChAdV/srRNA组合肿瘤模型评估方法和材料
肿瘤注射
对Balb/c小鼠注射CT26肿瘤细胞系。注射肿瘤细胞之后7天,将小鼠随机分成不同研究组(28-40只小鼠/组)并开始治疗。在Balb/c小鼠左下方侧腹部中注射106个CT26细胞/动物。在免疫接种之前,使肿瘤生长7天。研究组详细地描述于表15中。
表15-ChAdV/srRNA组合肿瘤模型评估研究组

免疫接种
对于srRNA疫苗,经两侧肌肉内注射对小鼠注射10ug VEE-MAG25mer srRNA,体积100uL(每条腿50μL)。对于C68疫苗,经两侧肌肉内注射对小鼠注射1×1011个ChAdV68.5WTnt.MAG25mer病毒粒子(VP),体积100μL(每条腿50μL)。每周2次,经由腹膜内注射对动物注射250ug剂量的抗PD-1(克隆RMP1-14,BioXcell)或抗IgG(克隆MPC-11,BioXcell)。
脾细胞解离
将每只小鼠的脾和淋巴结汇集于3mL完全RPMI(RPMI、10%FBS、青霉素/链霉素)中。使用gentleMACS解离器(Miltenyi Biotec),遵循制造商的方案进行机械解离。通过40微米过滤器过滤解离的细胞并用ACK溶解缓冲液(150mM NH4Cl、10mM KHCO3、0.1mMNa2EDTA)溶解红细胞。再次经由30微米过滤器过滤细胞且然后使其再悬浮于完全RPMI中。在Attune NxT流式细胞仪(Thermo Fisher)上使用碘化丙锭染色对细胞计数以排除死亡和凋亡的细胞。然后将细胞调整至适当活细胞浓度以供后续分析。
离体酶联免疫斑点(ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用小鼠IFNg ELISpotPLUS试剂盒(MABTECH)进行。将5x104个脾细胞与10uM指定肽一起在涂有IFNg抗体的96孔板中温育16小时。使用碱性磷酸酶使斑点显色。对反应定时10分钟并通过用自来水流过板终止反应。使用AID vSpot读取器谱图对斑点计数。对于ELISPOT分析,将饱和度>50%的孔记录为“太多而无法计数”。将复制孔的偏差>10%的样品从分析中排除。然后,使用下式,针对孔汇合度校正斑点计数:斑点计数+2×(斑点计数×汇合度%/[100%-汇合度%])。通过用抗原刺激的孔减去阴性肽刺激孔中的斑点计数来校正阴性背景。最后,将标记为太多而无法计数的孔设定成最高观察校正值,四舍五入至最接近的百分数。
XVII.B CT26肿瘤模型中的ChAdV/srRNA组合评估
在CT26小鼠肿瘤模型中评估ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA异源初免/加强免疫或VEE-MAG25mer srRNA同源初免/加强免疫疫苗的免疫原性和功效。对Balb/c小鼠注射CT26肿瘤细胞系。注射肿瘤细胞之后7天,将小鼠随机分成不同研究组并开始治疗。研究组详细地描述于表15中且较粗略地描述于表16中。
表16-初免/加强免疫研究组


在初免疫苗接种之后14天收集脾进行免疫监测。一周两次获取肿瘤和体重测量值并监测存活情况。在所有活性疫苗组中观察到相对于对照的强烈免疫应答。
在第一次免疫接种之后14天,在ELISpot分析中,在分别免疫接种ChAdV68.5WTnt.MAG25mer(ChAdV/第3组)、ChAdV68.5WTnt.MAG25mer+抗PD-1(ChAdV+PD-1/第4组)、VEE-MAG25mer srRNA(srRNA/第5组与第7组的组合的中值)或VEE-MAG25mer srRNA+抗PD-1(srRNA+PD-1/第6组与第8组的组合的中值)的小鼠中观察到每106个脾细胞10,630个、12,976个、3319个或3745个斑点形成细胞(SFC)的中值细胞免疫应答(图16和表17)。相比之下,疫苗对照(第1组)或疫苗对照与抗PD-1的组合(第2组)分别展现每106个脾细胞296个或285个SFC的中值细胞免疫应答。
表17-CT26肿瘤模型中的细胞免疫应答

与ELISpot数据相符,在第一次免疫接种之后14天,免疫接种ChAdV68.5WTnt.MAG25mer(ChAdV/第3组)、ChAdV68.5WTnt.MAG25mer+抗PD-1(ChAdV+PD-1/第4组)、VEE-MAG25mer srRNA(srRNA/第5组与第7组的组合的中值)或VEE-MAG25mer srRNA+抗PD-1(srRNA+PD-1/第6组与第8组的组合的中值)的小鼠中分别有5.6%、7.8%、1.8%或1.9%的CD8 T细胞(中值)在细胞内细胞因子染色(ICS)分析中展现抗原特异性反应(图17和表18)。免疫接种疫苗对照或疫苗对照与抗PD-1的组合的小鼠分别显示0.2%和0.1%的抗原特异性CD8反应。
表18-CT26肿瘤模型中的CD8 T细胞反应

在CT26结肠肿瘤模型中测量所有组的肿瘤生长情况,且到开始治疗后21天(注射CT-26肿瘤细胞之后28天),出现肿瘤生长。在开始治疗后21天,基于较大肿瘤尺寸(>2500mm3)处死小鼠;因此,仅呈现前21天以避免分析偏差。ChAdV68.5WTnt.MAG25mer初免/VEE-MAG25mer srRNA加强免疫(第3组)、ChAdV68.5WTnt.MAG25mer初免/VEE-MAG25mersrRNA加强免疫+抗PD-1(第4组)、VEE-MAG25mer srRNA初免/ChAdV68.5WTnt.MAG25mer加强免疫(第5组)、VEE-MAG25mer srRNA初免/ChAdV68.5WTnt.MAG25mer加强免疫+抗PD-1(第6组)、VEE-MAG25mer srRNA初免/VEE-MAG25mer srRNA加强免疫(第7组)和VEE-MAG25mersrRNA初免/VEE-MAG25mer srRNA加强免疫+抗PD-1(第8组)在21天时的平均肿瘤体积分别为1129、848、2142、1418、2198和1606mm3(图18和表19)。疫苗对照或疫苗对照与抗PD-1的组合的平均肿瘤体积分别为2361或2067mm3。基于这些数据,用ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA(第3组)、ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA+抗PD-1(第4组)、VEE-MAG25mer srRNA/ChAdV68.5WTnt.MAG25mer+抗PD-1(第6组)和VEE-MAG25mersrRNA/VEE-MAG25mer srRNA+抗PD-1(第8组)的疫苗治疗在21天时引起肿瘤生长减慢,这显著不同于对照(第1组)。
表19-第21天测量的CT26模型中的肿瘤尺寸

在CT-26肿瘤模型中,在开始治疗后,监测存活情况35天(注射CT-26肿瘤细胞之后42天)。在小鼠疫苗接种4个测试组合之后,观察到存活率提高。在疫苗接种之后,利用ChAdV68.5WTnt.MAG25mer初免/VEE-MAG25mer srRNA加强免疫与抗PD-1的组合(第4组;相对于对照组1,P<0.0001)、VEE-MAG25mer srRNA初免/VEE-MAG25mer srRNA加强免疫与抗PD-1的组合(第8组;相对于对照组1,P=0.0006)、ChAdV68.5WTnt.MAG25mer初免/VEE-MAG25mer srRNA加强免疫(第3组;相对于对照组1,P=0.0003)以及VEE-MAG25mer srRNA初免/ChAdV68.5WTnt.MAG25mer加强免疫与抗PD-1的组合(第6组;相对于对照组1,P=0.0016)的小鼠的存活率分别为64%、46%、41%和36%(图19和表20)。其余治疗组[VEE-MAG25mer srRNA初免/ChAdV68.5WTnt.MAG25mer加强免疫(第5组)、VEE-MAG25mer srRNA初免/VEE-MAG25mer srRNA加强免疫(第7组)和单独抗PD-1(第2组)]的存活率与对照组1无显著不同(≤14%)。
表20-CT26模型中的存活率

总之,相对于对照,ChAdV68.5WTnt.MAG25mer和VEE-MAG25mer srRNA引起针对由疫苗编码的小鼠肿瘤抗原的强烈T细胞反应。向带有肿瘤的小鼠施用ChAdV68.5WTnt.MAG25mer初免和VEE-MAG25mer srRNA加强免疫并且共同施用或不共同施用抗PD-1、施用VEE-MAG25mer srRNA初免和ChAdV68.5WTnt.MAG25mer加强免疫与抗PD-1的组合或施用VEE-MAG25mer srRNA同源初免加强免疫与抗PD-1的组合使存活率提高。
XVIII.非人灵长类动物研究
在非人灵长类动物(NHP)中评估使用ChAdV68和自我复制RNA(srRNA)的各种给药方案。
材料和方法
在每个NHP中肌肉注射(IM)初免疫苗以启动研究(疫苗初免)。在每个NHP中也肌肉内注射了一种或多种加强疫苗(疫苗加强)。根据表中概述并在下面总结的组施用每剂量的双侧注射。
免疫接种
用在LNP-1或LNP-2中配制的1×1012个病毒粒子(每次注射5×1011个病毒粒子)的ChAdV68.5WTnt.MAG25mer、30ug VEE-MAG25MER srRNA、100ug VEE-MAG25mer srRNA或300ug VEE-MAG25mer srRNA在双侧免疫接种Mamu-A*01印度恒河猴。在初免疫苗接种之后的指定时间,经肌肉内施用30ug、100ug或300ug VEE-MAG25mer srRNA疫苗加强免疫。
免疫监测
在初免疫苗接种之后的指定时间,使用淋巴细胞分离培养基(LSM,MPBiomedicals)和LeucoSep分离管(Greiner Bio-One)分离PBMC并使其再悬浮于含有10%FBS和青霉素/链霉素的RPMI中。在Attune NxT流式细胞仪(Thermo Fisher)上使用碘化丙锭染色对细胞计数以排除死亡和凋亡的细胞。然后将细胞调整至适当活细胞浓度以供后续分析。对于研究中的每只猴,使用ELISpot或流式细胞测量方法测量T细胞反应。通过使用离体酶联免疫斑点(ELISpot)分析测量诸如IFN-γ的细胞因子的诱导来监测PBMC中针对疫苗中编码的6个不同恒河猴Mamu-A*01的I类表位的T细胞反应。ELISpot分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用猴IFNg ELISpotPLUS试剂盒(MABTECH)进行。将200,000个PBMC与10μM指定肽一起在涂有IFNg抗体的96孔板中温育16小时。使用碱性磷酸酶使斑点显色。对反应定时10分钟并通过用自来水流过板终止反应。使用AID vSpot读取器谱图对斑点计数。对于ELISPOT分析,将饱和度>50%的孔记录为“太多而无法计数”。将复制孔的偏差>10%的样品从分析中排除。然后,使用下式,针对孔汇合度校正斑点计数:斑点计数+2×(斑点计数×汇合度%/[100%-汇合度%])。通过用抗原刺激的孔减去阴性肽刺激孔中的斑点计数来校正阴性背景。最后,将标记为太多而无法计数的孔设定成最高观察校正值,四舍五入至最接近的百分数。
通过使用流式细胞测量术测量诸如IFN-γ的细胞内细胞因子的诱导来监测PBMC中针对疫苗中编码的6个不同恒河猴Mamu-A*01的I类表位的特异性CD4和CD8 T细胞反应。由两种方法得到的结果指示,以抗原特异性方式诱导针对表位的细胞因子。
恒河猴中的免疫原性
本研究被设计用于(a)评估30μg和100μg剂量的VEE-MAG25mer srRNA与ChAdV68.5WTnt.MAG25mer组合作为同源初免/加强免疫或异源初免/加强免疫的免疫原性和初步安全性;(b)使用LNP1与LNP2比较脂质纳米粒子中VEE-MAG25mer srRNA的免疫应答;(c)评估对VEE-MAG25mer srRNA和ChAdV68.5WTnt.MAG25mer免疫接种的T细胞反应的动力学。
研究组在Mamu-A*01印度恒河猴中进行,以证明免疫原性。本研究中使用的所选抗原仅在恒河猴中识别,特别是具有Mamu-A*01MHC I类单倍型的那些。将Mamu-A*01印度恒河猴随机分配到不同的研究组(每组6只恒河猴),并双侧IM注射施用编码包括多个Mamu-A*01限制性表位的模型抗原的ChAdV68.5WTnt.MAG25mer或VEE-MAG25mer srRNA载体。研究组如下所述。
表21:印度恒河猴中的非GLP免疫原性研究

在免疫接种之前以及初始免疫接种之后第1、2、3、4、5、6、8、9和10周收集PBMC,以进行免疫监测。
结果
在免疫接种之前以及初始免疫接种之后第1、2、3、4、5、6、8、9和10周,针对六个不同Mamu-A*01限制性表位测量外周血单核细胞(PBMC)中的抗原特异性细胞免疫应答。如表21所述,动物在第4周和第8周接受剂量为30μg或100μg并用LNP1或LNP2配制的VEE-MAG25mer srRNA的加强免疫。对于每个免疫监测时间点绘制所有六个表位的组合免疫应答(图20A-图20D和表22-表25)。
在初始VEE-MAG25mer srRNA-LNP1(30μg)初免免疫接种后第1、2、3、4、5、6、8、9或10周,分别在每106个PBMC中170、14、15、11、7、8、14、17、12个SFC(六个组合表位)的所有测量结果下观测到组合抗原特异性免疫应答(图20A)。在初始VEE-MAG25mer srRNA-LNP1(100μg)初免免疫接种后第1、2、3、4、5、6、8、9或10周,分别在每106个PBMC中108、-3、14、1、37、4、105、17、25个SFC(六个组合表位)的所有测量结果下观测到组合抗原特异性免疫应答(图20B)。在初始VEE-MAG25mer srRNA-LNP2(100μg)初免免疫接种后第1、2、3、4、5、6、8、9或10周,分别在每106个PBMC中-17、38、14、-2、87、21、104、129、89个SFC(六个组合表位)的所有测量结果下观测到组合抗原特异性免疫应答(图20C)。负值是对每个表位/动物的放血前值进行归一化的结果。
在初始ChAdV68.5WTnt.MAG25mer初免免疫接种后第1、2、3、4、5、6、8、9或10周,分别在每106个PBMC中1218、1784、1866、973、1813、747、797、1249和547个SFC(六个组合表位)的所有测量结果下观测到组合抗原特异性免疫应答(图20D)。免疫应答显示出预期的特征,在初免免疫接种后约2-3周测得免疫应答峰值,接着在4周后免疫应答收缩。在用ChAdV68.5WTnt.MAG25mer初始免疫接种后第5周(即在用VEE-MAG25mer srRNA第一次加强免疫后第1周),测得组合抗原特异性细胞免疫应答为每106个PBMC中1813个SFC(六个组合表位)。用VEE-MAG25mer srRNA第一次加强免疫后第1周(第5周)测得的免疫应答与ChAdV68.5WTnt.MAG25mer初免免疫接种(第3周)测得的峰值免疫应答相当(图20D)。在用ChAdV68.5WTnt.MAG25mer初始免疫接种后第9周(即在用VEE-MAG25mer srRNA进行第二次加强免疫后第1周),测得组合抗原特异性细胞免疫应答为每106个PBMC中1249个SFC(六个组合表位)。在用VEE-MAG25mer srRNA进行第二次加强免疫后第1周(第9周)测得的免疫应答是加强免疫接种之前测得的免疫应答的约2倍(图20D)。
表22:VEE-MAG25mer srRNA-LNP1(30μg)的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM(第1组)

表23:VEE-MAG25mer srRNA-LNP1(100μg)的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM(第2组)


表24:VEE-MAG25mer srRNA-LNP2(100μg)的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM(第3组)

表25:ChAdV68.5WTnt.MAG25mer初免的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM

印度恒河猴中的非GLP RNA剂量范围研究(较高剂量)
本研究被设计用于(a)评估300μg剂量的VEE-MAG25mer srRNA与ChAdV68.5WTnt.MAG25mer组合作为同源初免/加强免疫或异源初免/加强免疫的免疫原性;(b)使用LNP1与LNP2比较脂质纳米粒子中300μg剂量的VEE-MAG25mer srRNA的免疫应答;以及(c)评估针对VEE-MAG25mer srRNA和ChAdV68.5WTnt.MAG25mer免疫接种的T细胞反应的动力学。
研究组在Mamu-A*01印度恒河猴中进行,以证明免疫原性。非人灵长类物种诸如恒河猴中的疫苗免疫原性是人中疫苗效力的最佳预测指标。此外,本研究中使用的所选抗原仅在恒河猴中被识别,特别是具有Mamu-A*01MHC I类单倍型的恒河猴。将Mamu-A*01印度恒河猴随机分配到不同的研究组(每组6只恒河猴),并双侧IM注射施用编码包括多个Mamu-A*01限制性抗原的模型抗原的ChAdV68.5-WTnt.MAG25mer或VEE-MAG25mer srRNA。研究组如下所述。
在免疫接种之前以及初始免疫接种后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周收集PBMC以对第1组进行免疫监测(异源初免/加强免疫)。在免疫接种之前以及初始免疫接种后第4、5、7、8、10、11、12、13、14或15周收集PBMC以对第2组和第3组进行免疫监测(异源初免/加强免疫)。
表26:印度恒河猴中的非GLP免疫原性研究

结果
用ChAdV68.5-WTnt.MAG25mer免疫接种Mamu-A*01印度恒河猴。在免疫接种之前以及初始免疫接种后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周针对六个不同的Mamu-A*01限制性表位测量外周血单核细胞(PBMC)中的抗原特异性细胞免疫应答(图21和表27)。在第4、12和20周时,使用LNP2制剂,使动物接受VEE-MAG25mer srRNA加强免疫。在用ChAdV68.5WTnt.MAG25mer初始免疫接种后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周测得组合抗原特异性免疫应答为每106个PBMC中1750、4225、1100、2529、3218、1915、1708、1561、5077、4543、4920、5820、3395、2728、1996、1465、4730、2984、2828或3043个SFC(六个组合表位)(图21)。用VEE-MAG25mer srRNA第二次加强免疫后第1周(第13周)测得的免疫应答是加强免疫前(第12周)测得的免疫应答的约3倍。用VEE-MAG25mer srRNA进行第三次加强免疫后第1周(第21周)测得的免疫应答是加强免疫前(第20周)测得的免疫应答的约3倍,类似于第二次加强免疫时观测到的应答。
还使用两种不同的LNP制剂(LNP1和LNP2),用VEE-MAG25mer srRNA对Mamu-A*01印度恒河猴进行免疫接种。在免疫接种之前以及在初始免疫接种后第4、5、6、7、8、10、11、12、13、14或15周,针对六个不同Mamu-A*01限制性表位测量外周血单核细胞(PBMC)中的抗原特异性细胞免疫应答(图22和图23,表28和表29)。在第4周和第12周,分别使用LNP1或LNP2制剂,使动物接受使用VEE-MAG25mer srRNA的加强免疫。在用VEE-MAG25mer srRNA-LNP2免疫接种后第4、5、7、8、10、11、13、14、15周测得组合抗原特异性免疫应答为每106个PBMC中168、204、103、126、140、145、330、203和162个SFC(六个组合表位)(图22)。在用VEE-MAG25mersrRNA-LNP1免疫接种后第4、5、7、8、10、11、12、13、14、15周测得组合抗原特异性免疫应答为每106个PBMC中189、185、349、437、492、570、233、886、369和381个SFC(六个组合表位)(图23)。
表27:用ChAdV68.5WTnt.MAG25mer初免疫苗接种的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM(第1组)

表28:用VEE-MAG25mer srRNA-LNP2(300μg)初免疫苗接种的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM(第2组)


表29:用VEE-MAG25mer srRNA-LNP1(300μg)初免疫苗接种的每个表位的每106个PBMC中的平均斑点形成细胞(SFC)±SEM(第3组)

srRNA剂量范围研究
在本发明的一种实施方式中,可以在mamu A01印度恒河猴中进行srRNA剂量范围研究,以鉴定哪个srRNA剂量可以进行NHP免疫原性研究。在一个实例中,可以通过IM注射向Mamu A01印度恒河猴施用包括多个mamu A01限制性表位的编码模型抗原的srRNA载体。在另一个实例中,抗CTLA-4单克隆抗体可以在IM疫苗注射部位附近SC施用,以靶向一组动物中的疫苗引流淋巴结。可在初始疫苗接种后每两周收集PBMC,以进行免疫监测。研究组描述如下(表30)。
表30:印度恒河猴中的非GLP RNA剂量范围研究

*在高剂量≤300μg下测定的srRNA的剂量范围。
印度恒河猴中的免疫原性研究
使用抗原载体在mamu A01印度恒河猴(NHP)中进行疫苗研究以证明免疫原性。图34示出了疫苗接种策略。对三组NHP免疫接种ChAdV68.5-WTnt.MAG25mer和检查点抑制剂抗CTLA-4抗体伊匹单抗(第5组和第6组)或不使用检查点抑制剂(第4组)。抗体经静脉内(第5组)或皮下(第6组)施用。三角形指示在第0周和第32周进行chAd68疫苗接种(1e12 vp/动物)。圆圈表示在第0、4、12、20、28和32周进行甲病毒疫苗接种。
呈现了单独免疫接种chAd-MAG(图35和表31A)、免疫接种chAd-MAG与IV递送的检查点抑制剂(图36和表31B)以及免疫接种chAd-MAG与SC递送的检查点抑制剂(图37和表31C)的免疫接种的NHP中CD8+抗表位反应的时程。结果表明chAd68载体有效地激活灵长类动物中的CD8+反应,甲病毒载体有效地加强chAD68疫苗初免反应,检查点抑制剂无论是通过IV还是SC递送都放大初免反应和加强反应,并且chAd载体在疫苗接种后重新施用以有效加强免疫应答。
表31A:用chAd-MAG给药的恒河猴(第4组)中的CD8+抗表位反应。示出了平均SFC/1e6个脾细胞+/-标准误差



表31B:用IV递送的chAd-MAG加抗CTLA4抗体(伊匹单抗)给药的恒河猴(第5组)中的CD8+抗表位反应。示出了平均SFC/1e6个脾细胞+/-标准误差


表31C:用SC递送的chAd-MAG加抗CTLA4抗体(伊匹单抗)给药的恒河猴(第6组)中的CD8+抗表位反应。示出了平均SFC/1e6个脾细胞+/-标准误差


印度恒河猴中的记忆表型分析
在存在或不存在抗CTLA4的情况下,用ChAdV68.5WTnt.MAG25mer/VEE-MAG25mersrRNA异源初免/加强免疫方案对恒河猴进行免疫接种,并且再用ChAdV68.5WTnt.MAG25mer加强免疫。最终ChAdV68施用后第11个月(研究第18个月)对各组进行评定。如所述通过ELISpot进行。图38和表43示出了免疫接种前(左图)和免疫接种后第18个月(右图)如通过ELISpot测量的对六个不同的Mamu-A*01限制性表位的细胞反应。对限制性表位的反应的检测表明,ChAd V68/samRNA疫苗方案产生了抗原特异性记忆反应。
为了评定记忆,使用双色Mamu-A*01四聚体标记监测识别疫苗中编码的4个不同恒河猴Mamu-A*01I类表位的CD8+T细胞,其中每种抗原均由独特的双阳性组合表示,并允许在单个样品内鉴定所有4种抗原特异性群体。通过与细胞表面标志物CD45RA和CCR7共染色来进行记忆细胞表型分析。图39和表44示出了识别四个不同的Mamu-A*01限制性表位的记忆T细胞的组合四聚体染色和CD45RA/CCR7共染色的结果。T细胞表型也通过流式细胞测量术来评定。图40示出了在研究第18个月时,四种Mamu-A*01四聚体+CD8+T细胞群体的总和内的记忆细胞类型的分布。记忆细胞的特征如下:CD45RA+CCR7+=初始,CD45RA+CCR7-=效应子(Teff),CD45RA-CCR7+=中枢记忆(Tcm),CD45RA-CCR7-=效应记忆(Tem)。总体而言,结果表明,在最后一次加强免疫后至少一年内检测到记忆反应表示免疫力持久,包括效应子、中枢记忆和效应子记忆群。
表43在初免前和记忆评定时间点(第18个月),每只动物的每106个PBMC中的平均斑点形成细胞(SFC)。


ND=由于技术排斥而未确定
表44活CD8+细胞中Mamu-A*01四聚体阳性的百分比

XIX.疫苗接种与干扰素信号传导抑制组合
为了增加基于甲病毒的samRNA疫苗的表达和效力,评估了I型IFN介导的抑制对疫苗的抗原表达和免疫原性的影响。参见例如Pepini等J Immunol.2017年5月15日;198(10):4012-4024。
材料和方法
对C57BL/6J小鼠免疫接种LNP配制的VEE-荧光素酶samRNA(SEQ ID NO:15)(每只小鼠10μg,两侧肌肉内递送)和抗IFNAR MAb或托法替尼。免疫接种前24小时将抗IFNAR Mab(克隆MAR1-5A3,BioXcell)(2mg)作为单次剂量腹膜内递送。免疫接种前24小时开始口服递送托法替尼,2mg,2次/天,并且持续6天。在用VEE-荧光素酶免疫接种后第1、2和5天对每只小鼠的相对发光(RLU)进行定量。
对Balb/c小鼠免疫接种VEE-MAG25mer samRNA(SEQ ID NO:4;每只小鼠10μg,两侧肌肉内递送)和抗IFNAR MAb或托法替尼。免疫接种前24小时将抗IFNAR MAb(克隆MAR1-5A3,BioXcell)(2mg)作为单次剂量腹膜内递送。免疫接种前24小时开始口服递送托法替尼,2mg,2次/天,并且持续8天。在用VEE-MAG免疫接种后第12天处死小鼠并收集脾。使用细胞内细胞因子染色测量T细胞对AH1-A5肽(SPSYAYHQF)的反应。
结果
为了抑制I型IFN,在疫苗接种表达荧光素酶报告基因的VEE-荧光素酶或表达模型抗原盒的VEE-MAG前24小时,将阻断IFNαβ受体(IFNAR)的单克隆抗体(MAR1-5A3)或JAK1和JAK3的小分子抑制剂托法替尼注射到小鼠中。通过体内成像在免疫接种后的各个时间点评估萤光素酶表达。免疫后24小时,用aIFNAR MAb进行预治疗导致荧光素酶表达增加约200倍,并且使用托法替尼进行预治疗导致荧光素酶表达增加约60倍(图26A;表32)。
表32.用VEE-荧光素酶SAM与抗IFNAR MAb或托法替尼组合免疫接种C57BL/6J小鼠后的平均RLU值。

免疫接种后第12天,通过细胞内细胞因子染色测量抗原特异性T细胞反应。与表达增加相一致,用aIFNAR MAb或托法替尼预治疗导致抗原特异性T细胞显著增加(与IgG治疗的对照中的9%相比,aIFNAR:平均22%IFNg+(占CD8+的%),p<0.001;与媒介物治疗的对照中的9%相比,托法替尼:平均13%IFNg+(占CD8+的%),p<0.05)。参见图26B和表33。
表33.用VEE-MAG SAM与抗IFNAR MAb或托法替尼或对照组合免疫接种后第12天的平均抗原特异性T细胞(占CD8的%)。
| 预治疗 | IFNg+(占CD8的%) |
| IgG | 8.6 |
| MAR1-5A3 | 22.2 |
| 媒介物 | 8.9 |
| 托法替尼 | 12.9 |
XX.干扰素信号传导抑制时间的评估
评估了I型IFN介导的抑制的时间和/或组成(contituation)对疫苗的抗原表达和免疫原性的影响。
材料和方法
对Balb/c小鼠免疫接种VEE-MAG25mer samRNA(SEQ ID NO:4;每只小鼠1μg,两侧肌肉内递送)和抗IFNAR MAb或抗IgG抗体对照。如下文所指示,在用samRNA免疫接种前或免疫接种后的指定时间,腹膜内递送抗IFNAR MAb(克隆MAR1-5A3,BioXcell)(2mg)。在用VEE-MAG免疫接种后第12天处死小鼠并收集脾。使用细胞内细胞因子染色测量CD8 T细胞中抗原特异性(AH1-A5)IFN-γ的产生并且使用MHC I类四聚体染色测量AH1-A5特异性CD8T细胞的百分比。
结果
为了进一步评估改进samRNA疫苗接种中的免疫应答中的I型IFN抑制,评估了相对于施用samRNA疫苗,施用抗IFNAR Mab的时间。具体来说,抗IFNAR MAb在施用samRNA疫苗前24小时、同时、施用samRNA疫苗后6小时、施用samRNA疫苗后12小时或施用samRNA疫苗后24小时施用。分析CD8 T细胞中的抗原特异性(AH1-A5)IFN-γ产生来评定免疫应答,并量化为占总CD8 T细胞的百分比。如图27A和表34A所示,与施用对照抗体相比,在施用samRNA疫苗前24小时、同时或施用samRNA疫苗后6小时施用抗IFNAR MAb产生显著改进的抗原特异性免疫应答。相比之下,相对于施用对照抗体,在施用samRNA疫苗后12小时或施用samRNA疫苗后24小时施用抗IFNAR Mab并不会产生改进的免疫应答。因此,结果表明IFNa的早期抑制导致改进的抗原特异性免疫应答。
表34A-AH1-A5特异性IFN-γ产生(占总CD8 T细胞的百分比)
| 组 | Ab治疗 | 中值 | 平均值 | SD | P值* |
| 1 | aIgG | 3.6 | 3.6 | 0.9 | |
| 2 | 前24小时 | 12.3 | 11.7 | 2.3 | 1.8E-05 |
| 3 | 同时 | 12.5 | 11.7 | 5.3 | 2.1E-05 |
| 4 | 后6小时 | 8.8 | 9.1 | 1.5 | 3.5E-03 |
| 5 | 后12小时 | 1.8 | 2.0 | 1.0 | 0.68 |
| 6 | 后24小时 | 1.4 | 1.6 | 0.8 | 0.49 |
*Dunnett检验
为了进一步评估改进samRNA疫苗接种中的免疫应答中的I型IFN抑制,评估了抗IFNAR Mab的持续施用,诸如尽管SAM的表达增加,I型IFN信号传导的持续抑制是否会对总体免疫应答(例如T细胞活化)产生负面影响。为了评估上述内容,在施用samRNA疫苗前24小时、施用samRNA疫苗前24小时和施用samRNA疫苗后24小时,或施用samRNA疫苗前24小时和施用samRNA疫苗后第4和第8天施用抗IFNAR MAb。分析CD8 T细胞中的抗原特异性(AH1-A5)MHC四聚体染色和IFN-γ产生来评定免疫应答,并量化为占总CD8 T细胞的百分比。如图27B和表34B(MHC-四聚体染色)以及图27C和表34C(IFN-γ产生)所示,相对于在samRNA疫苗接种前24小时施用抗IFNAR MAb的单一治疗,在施用samRNA疫苗后额外的抗IFNAR MAb的后续施用不会改变免疫应答,尽管相对于施用对照抗体,所有抗IFNAR MAb的治疗方案确实产生改进的抗原特异性免疫应答。因此,结果表明持续的IFNa抑制不会明显改变(例如钝化)由于抑制I型IFN信号传导而导致的表位特异性免疫应答增加。
表34B-AH1-A5特异性MHC-四聚体染色(占总CD8 T细胞的百分比)

表34C–AH1-A5特异性IFN-γ产生(占总CD8 T细胞的百分比)

XXI.干扰素信号传导抑制施用途径的评估
评估了I型IFN介导的抑制的施用途径对疫苗的抗原表达和免疫原性的影响。
材料和方法
对Balb/c小鼠免疫接种VEE-MAG25mer samRNA(SEQ ID NO:4;每只小鼠1μg,两侧肌肉内递送)和抗IFNAR MAb或抗IgG抗体对照。在用samRNA免疫接种前24小时,IP或肌肉内递送抗IFNAR MAb(克隆MAR1-5A3,BioXcell)(0.5mg)。在用samRNA免疫接种前24小时,IP递送抗IgG抗体对照(0.5mg)。在用VEE-MAG免疫接种后第12天处死小鼠并收集脾。使用细胞内细胞因子染色测量CD8 T细胞中抗原特异性(AH1-A5)IFN-γ的产生并且使用MHC I类四聚体染色测量AH1-A5特异性CD8 T细胞的百分比。
结果
为了进一步评估改进samRNA疫苗接种中的免疫应答中的I型IFN的抑制,评估了抗IFNAR Mab的施用途径。具体来说,在用samRNA免疫接种前24小时,IP或肌内(IM)递送来施用抗-IFNAR MAb(0.5mg)。分析CD8 T细胞中的抗原特异性(AH1-A5)MHC四聚体染色和IFN-γ产生来评定免疫应答,并量化为占总CD8 T细胞的百分比。如图28A和表35A(MHC-四聚体染色)以及图28B和表35B(IFN-γ产生)所示,相对于施用对照抗体,IM(局部)或IP(全身)施用抗IFNAR MAb都会产生显著改进的免疫应答。因此,结果表明在增加对samRNA疫苗的免疫应答方面,局部抑制I型IFN信号传导与全身递送一样有效。
表35A-AH1-A5特异性MHC-四聚体染色(占总CD8 T细胞的百分比)
| 组 | aIFNAR.方案 | 中值 | 平均值 | SD | P值* |
| 1 | 对照 | 8.9 | 9.1 | 3.3 | - |
| 5 | 0.5mg IP | 15.2 | 15.2 | 2.4 | 5.7E-04 |
| 6 | 0.5mg IM | 14.0 | 13.9 | 2.6 | 4.8E-03 |
*Dunnett检验
表35B–AH1-A5特异性IFN-γ产生(占总CD8 T细胞的百分比)
| 组 | aIFNAR.方案 | 中值 | 平均值 | SD | P值* |
| 1 | 对照 | 8.6 | 8.6 | 2.6 | - |
| 5 | 0.5mg IP | 13.8 | 12.5 | 3.2 | 0.02 |
| 6 | 0.5mg IM | 10.9 | 11.2 | 2.8 | 0.14 |
*Dunnett检验
某些序列
本文所提及的载体、盒和抗体在下文中描述,并由SEQ ID NO所提及。



参考文献
1.Desrichard,A.,Snyder,A.&Chan,T.A.Cancer Neoantigens andApplications for Immunotherapy.Clin.Cancer Res.Off.J.Am.Assoc.Cancer Res.(2015).doi:10.1158/1078-0432.CCR-14-3175
2.Schumacher,T.N.&Schreiber,R.D.Neoantigens in cancerimmunotherapy.Science 348,69-74(2015).
3.Gubin,M.M.,Artyomov,M.N.,Mardis,E.R.&Schreiber,R.D.Tumorneoantigens:building a framework for personalized cancerimmunotherapy.J.Clin.Invest.125,3413-3421(2015).
4.Rizvi,N.A.et al.Cancer immunology.Mutational landscape determinessensitivity to PD-1 blockade in non-small cell lung cancer.Science 348,124-128(2015).
5.Snyder,A.et al.Genetic basis for clinical response to CTLA-4blockade in melanoma.N.Engl.J.Med.371,2189-2199(2014).
6.Carreno,B.M.et al.Cancer immunotherapy.A dendritic cell vaccineincreases the breadth and diversity of melanoma neoantigen-specific Tcells.Science348,803-808(2015).
7.Tran,E.et al.Cancer immunotherapy based on mutation-specific CD4+Tcells in a patient with epithelial cancer.Science 344,641-645(2014).
8.Hacohen,N.&Wu,C.J.-Y.United States Patent Application:20110293637-COMPOSITIONS AND METHODS OF IDENTIFYING TUMOR SPECIFIC NEOANTIGENS.(A1).at<http://appft1.uspto.gov/netacgi/nph-Parser?Sect1=PTOl&Sect2=HITOFF&d=PG01&p=1&u=/netahtml/PTO/srchnum.html&r=1&f=G&1=50&s1=20110293637.PGNR.>
9.Lundegaard,C.,Hoof,I.,Lund,O.&Nielsen,M.State of the art andchallenges in sequence based T-cell epitope prediction.Immunome Res.6 Suppl2,S3(2010).
10.Yadav,M.et al.Predicting immunogenic tumour mutations by combiningmass spectrometry and exome sequencing.Nature 515,572-576(2014).
11.Bassani-Stemberg,M.,Pletscher-Frankild,S.,Jensen,L.J.&Mann,M.Massspectrometry of human leukocyte antigen class I peptidomes reveals strongeffects of protein abundance and turnover on antigen presentation.Mol.Cell.Proteomics MCP14,658-673(2015).
12.Van Allen,E.M.et al.Genomic correlates of response to CTLA-4blockade in metastatic melanoma.Science 350,207-211(2015).
13.Yoshida,K.&Ogawa,S.Splicing factor mutations and cancer.WileyInterdiscip.Rev.RNA 5,445-459(2014).
14.Cancer Genome Atlas Research Network.Comprehensive molecularprofiling of lung adenocarcinoma.Nature 511,543-550(2014).
15.Rajasagi,M.et al.Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia.Blood 124,453-462(2014).
16.Downing,S.R.et al.United States Patent Application:0120208706-OPTIMIZATION OF MULTIGENE ANALYSIS OF TUMOR SAMPLES.(A1).at<http://appft1.uspto.gov/netacgi/nph-Parser?Sect1=PTO1&Sect2=HITOFF&d=PG01&p=1&u=/netahtml/PTO/srchnum.html&r=1&f=G&1=50&s1=20120208706.PGNR.>
17.Target Capture for NextGen Sequencing-IDT.at<http://www.idtdna.com/pages/products/nextgen/target-capture>
18.Shukla,S.A.et al.Comprehensive analysis of cancer-associatedsomatic mutations in class I HLA genes.Nat.Biotechnol.33,1152-1158(2015).
19.Cieslik,M.et al.The use of exome capture RNA-seq for highlydegraded RNA with application to clinical cancer sequencing.Genome Res.25,1372-1381(2015).
20.Bodini,M.et al.The hidden genomic landscape of acute myeloidleukemia:subclonal structure revealed by undetected mutations.Blood 125,600-605(2015).
21.Saunders,C.T.et al.Strelka:accurate somatic small-variant callingfrom sequenced tumor-normal sample pairs.Bioinforma.Oxf.Engl.28,1811-1817(2012).
22.Cibulskis,K.et al.Sensitive detection of somatic point mutationsin impure and heterogeneous cancer samples.Nat.Biotechnol.31,213-219(2013).
23.Wilkerson,M.D.et al.Integrated RNA and DNA sequencing improvesmutation detection in low purity tumors.Nucleic Acids Res.42,e107(2014).
24.Mose,L.E.,Wilkerson,M.D.,Hayes,D.N.,Perou,C.M.&Parker,J.S.ABRA:improved coding indel detection via assembly-based realignment.Bioinforma.Oxf.Engl.30,2813-2815(2014).
25.Ye,K.,Schulz,M.H.,Long,Q.,Apweiler,R.&Ning,Z.Pindel:a patterngrowth approach to detect break points of large deletions and medium sizedinsertions from paired-end short reads.Bioinforma.Oxf.Engl.25,2865-2871(2009).
26.Lam,H.Y.K.et al.Nucleotide-resolution analysis of structuralvariants using BreakSeq and a breakpoint library.Nat.Biotechnol.28,4755(2010).
27.Frampton,G.M.et al.Development and validation of a clinical cancergenomic profiling test based on massively parallel DNAsequencing.Nat.Biotechnol.31,1023-1031(2013).
28.Boegel,S.et al.HLA typing from RNA-Seq sequence reads.GenomeMed.4,102(2012).
29.Liu,C.et al.ATHLATES:accurate typing of human leukocyte antigenthrough exome sequencing.Nucleic Acids Res.41,e142(2013).
30.Mayor,N.P.et al.HLA Typing for the Next Generation.PloS One 10,e0127153(2015).
31.Roy,C.K.,Olson,S.,Graveley,B.R.,Zamore,P.D.&Moore,M.J.Assessinglong-distance RNA sequence connectivity via RNA-templated DNA-DNAligation.eLife 4,(2015).
32.Song,L.&Florea,L.CLASS:constrained transcript assembly of RNA-seqreads.BMC Bioinformatics 14 Suppl 5,S14(2013).
33.Maretty,L.,Sibbesen,J.A.&Krogh,A.Bayesian transcriptomeassembly.Genome Biol.15,501(2014).
34.Pertea,M.et al.StringTie enables improved reconstruction of atranscriptome from RNA-seq reads.Nat.Biotechnol.33,290295(2015).
35.Roberts,A.,Pimentel,H.,Trapnell,C.&Pachter,L.Identification ofnovel transcripts in annotated genomes using RNA-Seq.Bioinforma.Oxf.Engl.(2011).doi:10.1093/bioinformatics/btr355
36.Vitting-Seerup,K.,Porse,B.T.,Sandelin,A.&Waage,J.spliceR:an Rpackage for classification of alternative splicing and prediction ofcodingpotential from RNA-seq data.BMC Bioinformatics 15,81(2014).
37.Rivas,M.A.et al.Human genomics.Effect of predicted protein-truncating genetic variants on the human transcriptome.Science 348,666-669(2015).
38.Skelly,D.A.,Johansson,M.,Madeoy,J.,Wakefield,J.&Akey,J.M.Apowerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data.Genome Res.21,1728-1737(2011).
39.Anders,S.,Pyl,P.T.&Huber,W.HTSeq--a Python framework to work withhigh-throughput sequencing data.Bioinforma.Oxf.Engl.31,166-169(2015).
40.Furney,S.J.et al.SF3B1 mutations are associated with alternativesplicing in uveal melanoma.Cancer Discov.(2013).doi:10.1158/2159-8290.CD-13-0330
41.Zhou,Q.et al.A chemical genetics approach for the functionalassessment of novel cancer genes.Cancer Res.(2015).doi:10.1158/0008-5472.CAN-14-2930
42.Maguire,S.L.et al.SF3B1mutations constitute a novel therapeutictarget in breast cancer.J.Pathol.235,571-580(2015).
43.Carithers,L.J.et al.A Novel Approach to High-Quality PostmortemTissue Procurement:The GTEx Project.Biopreservation Biobanking 13,311-319(2015).
44.Xu,G.et al.RNA CoMPASS:a dual approach for pathogen and hosttranscriptome analysis of RNA-seq datasets.PloS One 9,e89445(2014).
45.Andreatta,M.&Nielsen,M.Gapped sequence alignment using artificialneural networks:application to the MHC class I system.Bioinforma.Oxf.Engl.(2015).doi:10.1093/bioinformatics/btv639
46. K.W.,Rasmussen,M.,Buus,S.&Nielsen,M.NetMHCstab-predictingstability of peptide-MHC-I complexes;impacts for cytotoxic T lymphocyteepitope discovery.Immunology 141,18-26(2014).
K.W.,Rasmussen,M.,Buus,S.&Nielsen,M.NetMHCstab-predictingstability of peptide-MHC-I complexes;impacts for cytotoxic T lymphocyteepitope discovery.Immunology 141,18-26(2014).
47.Larsen,M.V.et al.An integrative approach to CTL epitopeprediction:a combined algorithm integrating MHC class I binding,TAP transportefficiency,and proteasomal cleavage predictions.Eur.J.Immunol.35,2295-2303(2005).
48.Nielsen,M.,Lundegaard,C.,Lund,O.& C.The role of the proteasomein generating cytotoxic T-cell epitopes:insights obtained from improvedpredictions of proteasomal cleavage.Immunogenetics 57,33-41(2005).
C.The role of the proteasomein generating cytotoxic T-cell epitopes:insights obtained from improvedpredictions of proteasomal cleavage.Immunogenetics 57,33-41(2005).
49.Boisvert,F.-M.et al.A Quantitative Spatial Proteomics Analysis ofProteome Turnover in Human Cells.Mol.Cell.Proteomics 11,M111.011429-M111.011429(2012).
50.Duan,F.et al.Genomic and bioinformatic profiling of mutationalneoepitopes reveals new rules to predict anticancerimmunogenicity.J.Exp.Med.211,2231-2248(2014).
51.Janeway’s Immunobiology:9780815345312:Medicine&Health ScienceBooks@Amazon.com.at<http://www.amazon.com/Janeways-Immunobiology-Kenneth-Murphy/dp/0815345313>
52.Calis,J.J.A.et al.Properties ofMHC Class I Presented Peptides ThatEnhance Immunogenicity.PLoS Comput.Biol.9,e1003266(2013).
53.Zhang,J.et al.Intratumor heterogeneity in localized lungadenocarcinomas delineated by multiregion sequencing.Science 346,256-259(2014)
54.Walter,M.J.et al.Clonal architecture of secondary acute myeloidleukemia.N.Engl.J.Med.366,1090-1098(2012).
55.Hunt DF,Henderson RA,Shabanowitz J,Sakaguchi K,Michel H,Sevilir N,Cox AL,Appella E,Engelhard VH.Characterization of peptides bound to the classI MHC molecule HLA-A2.1 by mass spectrometry.Science 1992.255:1261-1263.
56.Zarling AL,Polefrone JM,Evans AM,Mikesh LM,Shabanowitz J,Lewis ST,Engelhard VH,Hunt DF.Identification of class I MHC-associated phosphopeptidesas targets for cancer immunotherapy.Proc Natl Acad Sci U S A.2006 Oct 3;103(40)∶14889-94.
57.Bassani-Stemberg M,Pletscher-Frankild S,Jensen LJ,Mann M.Massspectrometry of human leukocyte antigen class I peptidomes reveals strongeffects of protein abundance and turnover on antigen presentation.Mol CellProteomics.2015Mar;14(3):658-73.doi:10.1074/mcp.M114.042812.
58.Abelin JG,Trantham PD,Penny SA,Patterson AM,Ward ST,Hildebrand WH,Cobbold M,Bai DL,Shabanowitz J,Hunt DF.Complementary IMAC enrichment methodsfor HLA-associated phosphopeptide identification by mass spectrometry.NatProtoc.2015 Sep;10(9):1308-18.doi:10.1038/nprot.2015.086.Epub 2015 Aug 6
59.Bamstable C J,Bodmer WF,Brown G,Galfre G,Milstein C,Williams AF,Ziegler A.Production of monoclonal antibodies to group A erythrocytes,HLA andother human cell surface antigens-new tools for genetic analysis.Cell.1978May;14(1):9-20.
60.Goldman JM,Hibbin J,Kearney L,Orchard K,Th′ng KH.HLA-DR monoclonalantibodies inhibit the proliferation of normal and chronic granulocyticleukaemia myeloid progenitor cells.Br J Haematol.1982 Nov;52(3)∶411-20.
61.Eng JK,Jahan TA,Hoopmann MR.Comet:an open-source MS/MS sequencedatabase search tool.Proteomics.2013 Jan;13(1):22-4.doi:10.1002/pmic.201200439.Epub 2012 Dec 4.
62.Eng JK,Hoopmann MR,Jahan TA,Egertson JD,Noble WS,MacCoss MJ.Adeeper look into Comet--implementation and features.J Am Soc MassSpectrom.2015 Nov;26(11):1865-74.doi:10.1007/s13361-015-1179-x.Epub 2015 Jun27.
63.Lukas Jesse Canterbury,Jason Weston,William Stafford Noble andMichael J.MacCoss.Semi-supervised learning for peptide identification fromshotgun proteomics datasets.Nature Methods4:923925,November2007
Jesse Canterbury,Jason Weston,William Stafford Noble andMichael J.MacCoss.Semi-supervised learning for peptide identification fromshotgun proteomics datasets.Nature Methods4:923925,November2007
64.Lukas John D.Storey,Michael J.MacCoss and William StaffordNoble.Assigning confidence measures to peptides identified by tandem massspectrometry.Journal of Proteome Research,7(1):29-34,January 2008
John D.Storey,Michael J.MacCoss and William StaffordNoble.Assigning confidence measures to peptides identified by tandem massspectrometry.Journal of Proteome Research,7(1):29-34,January 2008
65.Lukas John D.Storey and William Stafford Noble.Nonparametricestimation of posterior error probabilities associated with peptidesidentified by tandem mass spectrometry.Bioinformatics,24(16):i42-i48,August2008
John D.Storey and William Stafford Noble.Nonparametricestimation of posterior error probabilities associated with peptidesidentified by tandem mass spectrometry.Bioinformatics,24(16):i42-i48,August2008
66.Kinney RM,BJ Johnson,VL Brown,DW Trent.Nucleotide Sequence of the26 S mRNA of the Virulent Trinidad Donkey Strain of Venezuelan EquineEncephalitis Virus and Deduced Sequence of the Encoded StructuralProteins.Virology 152(2),400-413.1986 Jul 30.
67.Jill E Slansky,Frédérique M Rattis,Lisa F Boyd,Tarek Fahmy,Elizabeth M Jaffee,Jonathan P Schneck,David H Margulies,Drew MPardoll.Enhanced Antigen-Specific Antitumor Immunity with Altered PeptideLigands that Stabilize the MHC-Peptide-TCR Complex.Immunity,Volume 13,Issue4,1 October 2000,Pages 529-538.
68.A Y Huang,P H Gulden,A S Woods,M C Thomas,C D Tong,W Wang,V HEngelhard,G Pasternack,R Cotter,D Hunt,D M Pardoll,and E M Jaffee.Theimmunodominant major histocompatibility complex class I-restricted antigen ofa murine colon tumor derives from an endogenous retroviral gene product.ProcNatl Acad Sci U S A.;93(18):9730-9735,1996 Sep 3.
69.JOHNSON,BARBARA J.B.,RICHARD M.KINNEY,CRYSTLE L.KOST AND DENNISW.TRENT.Molecular Determinants of Alphavirus Neurovirulence:Nucleotide andDeduced Protein Sequence Changes during Attenuation of Venezuelan EquineEncephalitis Virus.J Gen Virol 67∶1951-1960,1986.
70.Aarnoudse,C.A.,Krüse,M.,Konopitzky,R.,Brouwenstijn,N.,and Schrier,P.I.(2002).TCR reconstitution in Jurkat reporter cells facilitates theidentification of novel tumor antigens by cDNA expression cloning.Int JCancer 99,7-13.
71.Alexander,J.,Sidney,J.,Southwood,S.,Ruppert,J.,Oseroff,C.,Maewal,A.,Snoke,K.,Serra,H.M.,Kubo,R.T.,and Sette,A.(1994).Development of highpotency universal DR-restricted helper epitopes by modification of highaffinity DR-blocking peptides.Immunity 1,751-761.
72.Banu,N.,Chia,A.,Ho,Z.Z.,Garcia,A.T.,Paravasivam,K.,Grotenbreg,GM.,Bertoletti,A.,and Gehring,A.J.(2014).Building and optimizing a virus-specificT cell receptor library for targeted immunotherapy in viralinfections.Scientific Reports4,4166.
73.Cornet,S.,Miconnet,I.,Menez,J.,Lemonnier,F.,and Kosmatopoulos,K.(2006).Optimal organization of a polypeptide-based candidate cancer vaccinecomposed of cryptic tumor peptides with enhanced immunogenicity.Vaccine 24,2102-2109.
74.Depla,E.,van der Aa,A.,Livingston,B.D.,Crimi,C.,Allosery,K.,deBrabandere,V.,Krakover,J.,Murthy,S.,Huang,M.,Power,S.,et al.(2008).Rationaldesign of a multiepitope vaccine encoding T-lymphocyte epitopes for treatmentof chronic hepatitis B virus infections.Journal of Virology 82,435-450.
75.Ishioka,G.Y.,Fikes,J.,Hermanson,G.,Livingston,B.,Crimi,C.,Qin,M.,del Guercio,M.F.,Oseroff,C.,Dahlberg,C.,Alexander,J.,et al.(1999).Utilizationof MHC class I transgenic mice for development of minigene DNA vaccinesencoding multiple HLA-restricted CTL epitopes.J Immunol 162,3915-3925.
76.Janetzki,S.,Price,L.,Schroeder,H.,Britten,C.M.,Welters,M.J.P.,andHoos,A.(2015).Guidelines for the automated evaluation of Elispot assays.NatProtoc 10,1098-1115.
77.Lyons,G.E.,Moore,T.,Brasic,N.,Li,M.,Roszkowski,J.J.,and Nishimura,M.I.(2006).Influence of human CD8 on antigen recognition by T-cell receptor-transduced cells.Cancer Res 66,11455-11461.
78.Nagai,K.,Ochi,T.,Fujiwara,H.,An,J.,Shirakata,T.,Mineno,J.,Kuzushima,K.,Shiku,H.,Melenhorst,J.J.,Gostick,E.,et al.(2012).Aurora kinaseA-specific T-cell receptor gene transfer redirects T lymphocytes to displayeffective antileukemia reactivity.Blood 119,368-376.
79.Panina-Bordignon,P.,Tan,A.,Termijtelen,A.,Demotz,S.,Corradin,G.,and Lanzavecchia,A.(1989).Universally immunogenic T cell epitopes:promiscuousbinding to human MHC class II and promiscuous recognition by T cells.Eur JImmunol 19,2237-2242.
80.Vitiello,A.,Marchesini,D.,Furze,J.,Sherman,L.A.,and Chesnut,R.W.(1991).Analysis of the HLA-restricted influenza-specific cytotoxic Tlymphocyte response in transgenic mice carrying a chimeric human-mouse classI major histocompatibility complex.J Exp Med 173,1007-1015.
81.Yachi,P.P.,Ampudia,J.,Zal,T.,and Gascoigne,N.R.J.(2006).Alteredpeptide ligands induce delayed CD8-T cell receptor interaction--a role forCD8 in distinguishing antigen quality.Immunity 25,203-211.
82.Pushko P,Parker M,Ludwig GV,Davis NL,Johnston RE,SmithJF.Replicon-helper systems from attenuated Venezuelan equine encephalitisvirus:expression of heterologous genes in vitro and immunization againstheterologous pathogens in vivo.Virology.1997 Dec 22;239(2):389-401.
83.Strauss,JH and E G Strauss.The alphaviruses:gene expression,replication,and evolution.Microbiol Rev.1994 Sep;58(3):491-562.
84. C,Ehrengruber MU,Grandgirard D.Alphaviral cytotoxicity andits implication in vector development.Exp Physiol.2005 Jan;90(1):45-52.Epub2004 Nov 12.
C,Ehrengruber MU,Grandgirard D.Alphaviral cytotoxicity andits implication in vector development.Exp Physiol.2005 Jan;90(1):45-52.Epub2004 Nov 12.
85.Riley,Michael K.II,and Wilfred Vermerris.Recent Advances inNanomaterials for Gene Delivery-A Review.Nanomaterials 2017,7(5),94.
86.Frolov I,Hardy R,Rice CM.Cis-acting RNA elements at the 5′end ofSindbis virus genome RNA regulate minus-and plus-strand RNAsynthesis.RNA.2001Nov;7(11):1638-51.
87.Jose J,Snyder JE,Kuhn RJ.A structural and functional perspectiveof alphavirus replication and assembly.Future Microbiol.2009 Sep;4(7):837-56.
88.Bo Li and C.olin N.Dewey.RSEM:accurate transcript quantificationfrom RNA-Seq data with or without a referenfe genome.BMC Bioinformatics,12:323,August 2011
89.Hillary Pearson,Tariq Daouda,Diana Paola Granados,Chantal Durette,Eric Bonneil,Mathieu Courcelles,Anja Rodenbrock,Jean-Philippe Laverdure,Caroline Sylvie Mader,Sébastien Lemieux,Pierre Thibault,and ClaudePerreault.MHC class I-associated peptides derive from selective regions ofthe human genome.The Journal of Clinical Investigation,2016,
Sylvie Mader,Sébastien Lemieux,Pierre Thibault,and ClaudePerreault.MHC class I-associated peptides derive from selective regions ofthe human genome.The Journal of Clinical Investigation,2016,
90.Juliane Liepe,Fabio Marino,John Sidney,Anita Jeko,DanielE.Bunting,Alessandro Sette,Peter M.Kloetzel,Michael P.H.Stumpf,AlbertJ.R.Heck,Michele Mishto.A large fraction of HLA class I ligands areproteasome-generated spliced peptides.Science,21,October 2016.
91.Mommen GP.,Marino,F.,Meiring HD.,Poelen,MC.,van Gaans-van denBrink,JA.,Mohammed S.,Heck AJ.,and van Els CA.Sampling From the Proteome tothe Human Leukocyte Antigen-DR(HLA-DR)Ligandome Proceeds Via HighSpecificity.Mol Cell Proteomics 15(4):1412-1423,April 2016.
92.Sebastian Kreiter,Mathias Vormehr,Niels van de Roemer,MustafaDiken,Martin Jan Diekmann,Sebastian Boegel,Barbara
Jan Diekmann,Sebastian Boegel,Barbara Fulvia Vascotto,John C.Castle,Arbel D.Tadmor,Stephen P.Schoenberger,Christoph Huber,
Fulvia Vascotto,John C.Castle,Arbel D.Tadmor,Stephen P.Schoenberger,Christoph Huber, Türeci,and Ugur Sahin.Mutant MHC class II epitopes drive therapeutic immuneresponses to caner.Nature 520,692-696,April 2015.
Türeci,and Ugur Sahin.Mutant MHC class II epitopes drive therapeutic immuneresponses to caner.Nature 520,692-696,April 2015.
93.Tran E.,Turcotte S.,Gros A.,Robbins P.F.,Lu Y.C.,Dudley M.E.,Wunderlich J.R.,Somerville R.P.,Hogan K.,Hinrichs C.S.,Parkhurst M.R.,YangJ.C.,Rosenberg S.A.Cancer immunotherapy based on mutation-specific CD4+Tcells in a patient with epithelial cancer.Science 344(6184)641-645,May 2014.
94.Andreatta M.,Karosiene E.,Rasmussen M.,Stryhn A.,Buus S.,NielsenM.Accurate pan-specific prediction of peptide-MHC class II binding affinitywith improved binding core identification.Immunogenetics 67(11-12)641-650,November 2015.
95.Nielsen,M.,Lund,O.NN-align.An artificial neural network-basedalignment algorithm for MHC class II peptide binding prediction.BMCBioinformatics 10:296,September 2009.
96.Nielsen,M.,Lundegaard,C.,Lund,O.Prediction of MHC class II bindingaffinity using SMM-align,a novel stabilization matrix alignment method.BMCBioinformatics 8:238,July 2007.
97.Zhang,J.,et al.PEAKS DB:de novo sequencing assisted databasesearch for sensitive and accurate peptide identification.Molecular&CellularProteomics.11(4):1-8.1/2/2012.
98.Jensen,Kamilla Kjaergaard,et al.“Improved Methods for PreditingPeptide Binding Affinity to MHC Class II Molecules.”Immunology,2018,doi:10.1111/imm.12889.
99.Carter,S.L.,Cibulskis,K.,Helman,E.,McKenna,A.,Shen,H.,Zack,T.,Laird,P.W.,Onofrio,R.C.,Winckler,W.,Weir,B.A.,et al.(2012).Absolutequantification of somatic DNA alterations in human cancer.Nat.Biotechnol.30,413-421
100.McGranahan,N.,Rosenthal,R.,Hiley,C.T.,Rowan,A.J.,Watkins,T.B.K.,Wilson,G.A.,Birkbak,N.J.,Veeriah,S.,Van Loo,P.,Herrero,J.,et al.(2017).Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution.Cell171,1259-1271.el 1.
101.Shukla,S.A.,Rooney,M.S.,Rajasagi,M.,Tiao,G.,Dixon,P.M.,Lawrence,M.S.,Stevens,J.,Lane,W.J.,Dellagatta,J.L.,Steelman,S.,et al.(2015).Comprehensive analysis of cancer-associated somatic mutations in class I HLAgenes.Nat.Biotechnol.33,1152-1158.
102.Van Loo,P.,Nordgard,S.H.,Lingjaerde,O.C.,Russnes,H.G.,Rye,I.H.,Sun,W.,Weigman,V.J.,Marynen,P.,Zetterberg,A.,Naume,B.,et al.(2010).Allele-specific copy number analysis of tumors.Proc.Natl.Acad.Sci.U.S.A.107,16910-16915.
103.Van Loo,P.,Nordgard,S.H.,Lingjaerde,O.C.,Russnes,H.G.,Rye,I.H.,Sun,W.,Weigman,V.J.,Marynen,P.,Zetterberg,A.,Naume,B.,et al.(2010).Allele-specific copy number analysis of tumors.Proc.Natl.Acad.Sci.U.S.A.107,16910-16915.































































































































































































Claims (161)
1.一种用于刺激受试者中的免疫应答的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,
其中用于递送所述表达系统的所述组合物包含所述表达系统,
其中所述表达系统包含一个或多个载体,
所述一个或多个载体包含:
(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:
(i)至少一个启动子核苷酸序列,和
(ii)至少一个聚腺苷酸化(poly(A))序列;以及
(b)盒,其中所述盒包含:
(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,
b.任选地5'接头序列,以及
c.任选地3'接头序列;
(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及
(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
2.一种用于刺激受试者中的免疫应答的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,
其中用于递送所述表达系统的所述组合物包含所述表达系统,
其中所述表达系统包含一个或多个载体,
所述一个或多个载体包含:
(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含SEQ ID NO:6中示出的核酸序列,其中所述RNA甲病毒骨架序列包含26S启动子核苷酸序列和poly(A)序列,其中所述26S启动子序列是所述RNA甲病毒骨架的内源序列,并且其中所述poly(A)序列是所述RNA甲病毒骨架的内源序列;以及
(b)在所述26S启动子核苷酸序列与所述poly(A)序列之间整合的盒,其中所述盒可操作地连接到所述26S启动子核苷酸序列,并且其中所述盒包含至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,
b.任选地5'接头序列,以及
c.任选地3'接头序列;并且
其中所述I型干扰素信号传导抑制剂包含抗IFNαβ受体(IFNAR)阻断抗体。
3.如权利要求1或2所述的方法,其中所述至少一个核酸序列包含所述多肽编码核酸序列。
4.如权利要求3所述的方法,其中所述多肽编码核酸序列编码所述抗原编码核酸序列。
5.如权利要求4所述的方法,其中所述抗原编码核酸序列是所述表位编码核酸序列。
6.如权利要求4所述的方法,其中所述抗原编码核酸序列编码能够经历抗原加工成编码的表位的多肽序列。
7.如权利要求4-7中任一项所述的方法,其中所述表位编码核酸序列编码已知或怀疑由MHC I类在细胞表面上递呈的表位,任选地其中所述细胞表面是肿瘤细胞表面或感染细胞表面,并且任选地其中所述细胞是所述受试者的细胞。
8.如权利要求7所述的方法,其中所述细胞是选自由以下组成的组的肿瘤细胞:肺癌、黑素瘤、乳癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌,或者
其中所述细胞是选自由以下组成的组的感染细胞:病原体感染细胞、病毒感染细胞、细菌感染细胞、真菌感染细胞和寄生虫感染细胞。
9.如权利要求8所述的方法,其中所述病毒感染细胞是HIV感染细胞。
10.如权利要求3所述的方法,其中所述多肽编码核酸序列编码全长蛋白质或其功能部分。
11.如权利要求10所述的方法,其中所述全长蛋白质或其功能部分选自由以下组成的组:抗体、细胞因子、嵌合抗原受体(CAR)、T细胞受体和基因组编辑系统核酸酶。
12.如权利要求1或2所述的方法,其中所述至少一个核酸序列包含非编码核酸序列。
13.如权利要求12所述的方法,其中所述非编码核酸序列是RNA干扰(RNAi)多核苷酸或基因组编辑系统多核苷酸。
14.如权利要求1所述的方法,其中所述盒包含:
i)所述至少一个核酸序列,其包含所述多肽编码核酸序列,其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变
b.任选地5'接头序列,以及
c.任选地3'接头序列;
(ii)任选地,可操作地连接到所述抗原编码核酸序列的第二启动子核苷酸序列;
(iii)任选地,至少一个MHC II类表位编码核酸序列;
(iv)任选地,至少一个编码GPGPG氨基酸接头序列(SEQ ID NO:56)的核酸序列;以及
(v)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
15.如权利要求14所述的方法,其中所述盒的各元件的有序序列以下式描述,其从5'至3'包含:
Pa-(L5b-Nc-L3d)X-(G5e-Uf)Y-G3g
其中P包含所述第二启动子核苷酸序列,其中a=0或1,
N包含所述表位编码核酸序列中的一个,其中所述表位编码核酸序列包含MHC I类表位编码核酸序列,其中c=1,
L5包含所述5'接头序列,其中b=0或1,
L3包含所述3'接头序列,其中d=0或1,
G5包含所述至少一个编码GPGPG氨基酸接头的核酸序列中的一个,其中e=0或1,
G3包含所述至少一个编码GPGPG氨基酸接头的核酸序列中的一个,其中g=0或1,
U包含所述至少一个MHC II类表位编码核酸序列中的一个,其中f=1,
X=1至400,其中对于每个X,对应的Nc是表位编码核酸序列,并且
Y=0、1或2,其中对于每个Y,对应的Uf是表位编码核酸序列。
16.如权利要求15所述的方法,其中对于每个X,对应的Nc是不同的MHC I类表位编码核酸序列。
17.如权利要求15或16所述的方法,其中对于每个Y,对应的Uf是不同的MHC II类表位编码核酸序列。
18.如权利要求15-17中任一项所述的方法,其中
a=0,b=1,d=1,e=1,g=1,h=1,X=20,Y=2,
所述至少一个启动子核苷酸序列是由所述RNA甲病毒骨架提供的单个26S启动子核苷酸序列,
所述至少一个聚腺苷酸化poly(A)序列是由所述RNA甲病毒骨架提供的至少100个连续A核苷酸的poly(A)序列,
每个N编码长度为7-15个氨基酸的MHC I类表位,
L5是编码所述MHC I表位的原生N末端氨基酸序列的原生5'接头序列,并且其中所述5'接头序列编码长度为至少3个氨基酸的肽,
L3是编码所述MHC I表位的原生C末端氨基酸序列的原生3'接头序列,并且其中所述3'接头序列编码长度为至少3个氨基酸的肽,
U是PADRE II类序列和破伤风类毒素MHC II类序列中的每一者,
所述RNA甲病毒骨架是SEQ ID NO:6中示出的序列,并且
所述MHC I类表位编码核酸序列中的每一者编码长度为13至25个氨基酸的多肽。
19.如上述权利要求中任一项所述的方法,其中用于递送所述表达系统的所述组合物还包含纳米颗粒递送媒介物。
20.如权利要求19所述的方法,其中所述纳米颗粒递送媒介物是脂质纳米粒子(LNP)。
21.如权利要求20所述的方法,其中所述LNP包含可电离的氨基脂质。
22.如权利要求21所述的方法,其中所述可电离的氨基脂质包含MC3样(二亚油基甲基-4-二甲基氨基丁酸酯)分子。
23.如权利要求19-22中任一项所述的方法,其中所述纳米颗粒递送媒介物包封所述表达系统。
24.如权利要求19-23中任一项所述的方法,其中所述纳米颗粒递送媒介物直径为约100nm。
25.如上述权利要求中任一项所述的方法,其中所述I型干扰素信号传导抑制剂选自由以下组成的组:IFNα抑制剂、IFNβ抑制剂、IFNAR抑制剂和I型干扰素信号传导途径抑制剂。
26.如权利要求25所述的方法,其中所述I型干扰素信号传导抑制剂选自由以下组成的组:抗体或其抗原结合片段、小分子抑制剂、RNAi多核苷酸、基因组编辑系统和Fc融合蛋白。
27.如权利要求26所述的方法,其中所述抗体选自由以下组成的组:抗IFNα抗体、抗IFNβ抗体、抗IFNαβ受体(IFNAR)阻断抗体。
28.如权利要求27所述的方法,其中所述抗IFNα抗体选自由以下组成的组:西法鲁单抗、贝那利珠单抗和ASG-009。
29.如权利要求27所述的方法,其中所述抗IFNAR阻断抗体选自由以下组成的组:MAR1-5A3、阿尼福鲁单抗、AmS3A5-1、64G12、H2K6、H2K1、H3K6、H3K1 3F11、4G5、11E2和9D4。
30.如权利要求25所述的方法,其中所述I型干扰素信号传导途径抑制剂包含JAK激酶抑制剂。
31.如权利要求30所述的方法,其中所述JAK激酶抑制剂包含小分子。
32.如权利要求30或31所述的方法,其中所述JAK激酶抑制剂包含JAK1/2抑制剂或JAK1/3抑制剂。
33.如权利要求32所述的方法,其中所述JAK1/3抑制剂是托法替尼。
34.如上述权利要求中任一项所述的方法,其中所述I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物之前、同时或之后施用。
35.如权利要求34所述的方法,其中所述I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物前24小时或更短时间施用。
36.如权利要求34所述的方法,其中所述I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物后少于12小时施用。
37.如权利要求34所述的方法,其中所述I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物后6小时或更短时间施用。
38.如权利要求34所述的方法,其中所述I型干扰素信号传导抑制剂在施用用于递送所述表达系统的所述组合物前24小时与施用后6小时或更短时间之间施用。
39.如上述权利要求中任一项所述的方法,其中用于递送所述表达系统的所述组合物经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。
40.如上述权利要求中任一项所述的方法,其中用于递送所述表达系统的所述组合物经肌肉内(IM)施用。
41.如上述权利要求中任一项所述的方法,其中所述I型干扰素信号传导抑制剂经肌肉内(IM)、皮内(ID)、皮下(SC)或静脉内(IV)施用。
42.如上述权利要求中任一项所述的方法,其中所述I型干扰素信号传导抑制剂经肌肉内(IM)施用。
43.如上述权利要求中任一项所述的方法,其中所述I型干扰素信号传导抑制剂经静脉内(IV)施用。
44.如上述权利要求中任一项所述的方法,其中施用所述I型干扰素信号传导抑制剂的单次施用。
45.如权利要求1、3-17或19-44中任一项所述的方法,其中所述盒整合在所述至少一个启动子核苷酸序列与所述至少一个poly(A)序列之间。
46.如权利要求1、3-17或19-45中任一项所述的方法,其中所述至少一个启动子核苷酸序列可操作地连接到所述盒。
47.如权利要求1、3-17或19-46中任一项所述的方法,其中所述一个或多个载体包含一个或多个+-链RNA载体。
48.如权利要求47所述的方法,其中所述一个或多个+-链RNA载体包含5'7-甲基鸟苷(m7g)帽。
49.如权利要求47或48所述的方法,其中所述一个或多个+-链RNA载体通过体外转录产生。
50.如权利要求1、3-17或19-49中任一项所述的方法,其中所述一个或多个载体在哺乳动物细胞内自我复制。
51.如权利要求1、3-17或19-50中任一项所述的方法,其中所述RNA甲病毒骨架包含奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒中的至少一个核苷酸序列。
52.如权利要求1、3-17或19-50中任一项所述的方法,其中所述RNA甲病毒骨架包含委内瑞拉马脑炎病毒的至少一个核苷酸序列。
53.如权利要求51或52所述的方法,其中所述RNA甲病毒骨架包含至少用于非结构蛋白介导扩增的序列,26S启动子序列,poly(A)序列,由所述奥拉病毒、所述摩根堡病毒、所述委内瑞拉马脑炎病毒、所述罗斯河病毒、所述塞姆利基森林病毒、所述辛德毕斯病毒或所述马雅鲁病毒的核苷酸序列编码的非结构蛋白1(nsP1)基因、nsP2基因、nsP3基因和nsP4基因。
54.如权利要求51或52所述的方法,其中所述RNA甲病毒骨架包含至少用于非结构蛋白介导扩增的序列,26S启动子序列以及由所述奥拉病毒、所述摩根堡病毒、所述委内瑞拉马脑炎病毒、所述罗斯河病毒、所述塞姆利基森林病毒、所述辛德毕斯病毒或所述马雅鲁病毒的核苷酸序列编码的poly(A)序列。
55.如权利要求53或54所述的方法,其中用于非结构蛋白介导扩增的序列选自由以下组成的组:甲病毒5'UTR、51-nt CSE、24-nt CSE、26S亚基因组启动子序列、19-nt CSE、甲病毒3'UTR或其组合。
56.如权利要求53-55中任一项所述的方法,其中所述RNA甲病毒骨架不编码结构病毒粒子蛋白衣壳E2和E1。
57.如权利要求56所述的方法,其中插入所述盒以代替所述奥拉病毒、所述摩根堡病毒、所述委内瑞拉马脑炎病毒、所述罗斯河病毒、所述塞姆利基森林病毒、所述辛德毕斯病毒或所述马雅鲁病毒的核苷酸序列内的结构病毒粒子蛋白。
58.如权利要求51或52所述的方法,其中所述委内瑞拉马脑炎病毒包含SEQ ID NO:3或SEQ ID NO:5的序列。
59.如权利要求51或52所述的方法,其中所述委内瑞拉马脑炎病毒包含SEQ ID NO:3或SEQ ID NO:5的序列,其还包含碱基对7544与11175之间的缺失。
60.如权利要求59所述的方法,其中所述RNA甲病毒骨架包含SEQ ID NO:6或SEQ IDNO:7中示出的序列。
61.如权利要求59或60所述的方法,其中在位置7544处插入所述盒以代替如SEQ IDNO:3或SEQ ID NO:5的序列中所示的碱基对7544与11175之间的缺失。
62.如权利要求57-61所述的方法,其中所述盒的插入提供了包含所述nsP1-4基因和所述至少一个核酸序列的多顺反子RNA的转录,其中所述nsP1-4基因和所述至少一个核酸序列在单独的开放阅读框中。
63.如权利要求1、3-17或19-62中任一项所述的方法,其中所述至少一个启动子核苷酸序列是由所述RNA甲病毒骨架编码的原生26S启动子核苷酸序列。
64.如权利要求1、3-17或19-62中任一项所述的方法,其中所述至少一个启动子核苷酸序列是外源RNA启动子。
65.如权利要求1、3-17或19-64中任一项所述的方法,其中所述第二启动子核苷酸序列是26S启动子核苷酸序列。
66.如权利要求1、3-17或19-64中任一项所述的方法,其中所述第二启动子核苷酸序列包含多个26S启动子核苷酸序列,其中每个26S启动子核苷酸序列提供一个或多个单独的开放阅读框的转录。
67.如上述权利要求中任一项所述的方法,其中所述一个或多个载体的大小各为至少300nt。
68.如上述权利要求中任一项所述的方法,其中所述一个或多个载体的大小各为至少1kb。
69.如上述权利要求中任一项所述的方法,其中所述一个或多个载体的大小各为2kb。
70.如上述权利要求中任一项所述的方法,其中所述一个或多个载体的大小各小于5kb。
71.如上述权利要求中任一项所述的方法,其中至少一个表位编码核酸序列编码表位,所述表位在表达和翻译时能够由MHC I类在所述受试者的细胞上递呈。
72.如上述权利要求中任一项所述的方法,其中至少一个表位编码核酸序列编码表位,所述表位在表达和翻译时能够由MHCII类在所述受试者的细胞上递呈。
73.如权利要求1-17或19-72中任一项所述的方法,其中所述至少一个核酸序列包含两个或更多个核酸序列。
74.如权利要求1-17或19-72中任一项所述的方法,其中所述至少一个核酸序列包含两个或更多个多肽编码核酸序列。
75.如权利要求74所述的方法,其中每个多肽编码核酸序列彼此直接连接。
76.如权利要求1-17或19-75中任一项所述的方法,其中每个多肽编码核酸序列连接到具有编码接头的核酸序列的不同多肽编码核酸序列。
77.如权利要求76所述的方法,其中所述多肽编码核酸序列是抗原编码核酸序列,并且其中所述接头连接两个MHC I类表位编码核酸序列或将MHCI类表位编码核酸序列连接到MHCII类表位编码核酸序列。
78.如权利要求77所述的方法,其中所述接头选自由以下组成的组:(1)连续甘氨酸残基,长度为至少2、3、4、5、6、7、8、9或10个残基;(2)连续丙氨酸残基,长度为至少2、3、4、5、6、7、8、9或10个残基;(3)两个精氨酸残基(RR);(4)丙氨酸、丙氨酸、酪氨酸(AAY);(5)长度为至少2、3、4、5、6、7、8、9或10个氨基酸残基的共有序列,其由哺乳动物蛋白酶体有效加工;以及(6)侧接衍生自同源蛋白质来源的抗原并且长度为至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或2-20个氨基酸残基的一个或多个原生序列。
79.如权利要求76所述的方法,其中所述多肽编码核酸序列是抗原编码核酸序列,并且其中所述接头连接两个MHC II类表位编码核酸序列或将MHC II类序列连接到MHC I类表位编码核酸序列。
80.如权利要求79所述的方法,其中所述接头包含所述序列GPGPG。
81.如权利要求1-17或19-80中任一项所述的方法,其中所述多肽编码核酸序列是抗原编码核酸序列,并且其中所述抗原编码核酸序列可操作地或直接地连接到单独或连续的序列,所述单独或连续的序列增强所述抗原编码核酸序列的表达、稳定性、细胞运输、加工和递呈和/或免疫原性。
82.如权利要求81所述的方法,其中所述单独或连续的序列包含以下中的至少一者:泛素序列、经修饰以增加蛋白酶体靶向的泛素序列(例如,所述泛素序列在位置76处含有Gly至Ala取代)、免疫球蛋白信号序列(例如IgK)、主要组织相容性I类序列、溶酶体相关膜蛋白(LAMP)-1、人树突状细胞溶酶体相关膜蛋白和主要组织相容性II类序列;任选地其中所述经修饰以增加蛋白酶体靶向的泛素序列是A76。
83.如上述权利要求中任一项所述的方法,其中所述表位编码核酸序列包含至少一个改变,所述改变使编码的表位相对于翻译的对应野生型核酸序列对其对应MHC等位基因的结合亲和力增加。
84.如上述权利要求中任一项所述的方法,其中所述表位编码核酸序列包含至少一个改变,所述改变使编码的表位相对于翻译的对应野生型核酸序列对其对应MHC等位基因的结合稳定性增加。
85.如上述权利要求中任一项所述的方法,其中所述表位编码核酸序列包含至少一个改变,所述改变使编码的表位相对于翻译的对应野生型核酸序列在其对应MHC等位基因上递呈的可能性增加。
86.如上述权利要求中任一项所述的方法,其中所述至少一个改变包括点突变、移码突变、非移码突变、缺失突变、插入突变、剪接变体、基因组重排或蛋白酶体产生的剪接抗原。
87.如上述权利要求中任一项所述的方法,其中已知或怀疑所述受试者患有癌症。
88.如权利要求87所述的方法,其中刺激所述免疫应答治疗所述癌症。
89.如权利要求87或88所述的方法,其中所述癌症选自由以下组成的组:肺癌、黑素瘤、乳腺癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、膀胱癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、成人急性淋巴母细胞性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌
90.如上述权利要求中任一项所述的方法,其中所述受试者患有一种或多种肿瘤。
91.如权利要求90所述的方法,其中刺激所述免疫应答减小所述一种或多种肿瘤的肿瘤体积。
92.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个核酸序列,任选地其中每个核酸序列编码不同的非编码核酸序列、不同的多肽编码核酸序列或其组合。
93.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个核酸序列,任选地其中每个核酸序列编码不同的非编码核酸序列、不同的多肽编码核酸序列或其组合。
94.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个多肽编码核酸序列。
95.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个多肽编码核酸序列。
96.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个抗原编码核酸序列。
97.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个抗原编码核酸序列。
98.如权利要求1-17或19-91中任一项所述的方法,其中所述至少一个核酸序列包含至少2-400个抗原编码核酸序列,并且其中所述抗原编码核酸序列中的至少两者编码细胞表面上由MHC I类递呈的多肽序列或其部分。
99.如权利要求18所述的方法,其中所述MHC I类表位中的至少两者由MHC I类在所述肿瘤细胞表面上递呈。
100.如上述权利要求中任一项所述的方法,其中当向所述受试者施用并被翻译时,由所述表位编码核酸序列编码的所述表位中的至少一者被递呈在抗原递呈细胞上,产生靶向递呈所述细胞表面上的所述表位中的至少一者的细胞的免疫应答。
101.如上述权利要求中任一项所述的方法,其中所述表位编码核酸序列包含至少一个MHC I类表位编码核酸序列或MHC II类表位编码核酸序列,并且当向所述受试者施用并被翻译时,所述MHC I类或II类表位中的至少一者被递呈在抗原递呈细胞上,产生靶向递呈所述细胞表面上的所述表位中的至少一者的细胞的免疫应答,并且任选地其中所述MHC I类和/或II类表位编码核酸序列中的每一者的表达由所述至少一个启动子核苷酸序列驱动。
102.如权利要求1-17或19-101中任一项所述的方法,其中所述表位编码核酸序列包含至少一个MHC I类表位编码核酸序列,并且其中每个抗原编码核酸序列编码长度为8至35个氨基酸,任选地长度为9-17、9-25、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35个氨基酸的多肽序列。
103.如权利要求14-17或19-102中任一项所述的方法,其中存在所述至少一个MHC II类表位编码核酸序列。
104.如权利要求14-17或19-102中任一项所述的方法,其中存在所述至少一个MHC II类表位编码核酸序列并且其包含至少一个MHC II类表位编码核酸序列,所述序列包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变。
105.如权利要求1-17或19-104中任一项所述的方法,其中所述表位编码核酸序列包含MHCII类表位编码核酸序列,并且其中每个抗原编码核酸序列编码长度为12-20、12、13、14、15、16、17、18、19、20或20-40个氨基酸的多肽序列。
106.如权利要求1-17或19-105中任一项所述的方法,其中所述表位编码核酸序列包含MHCII类表位编码核酸序列,其中存在所述至少一个MHC II类表位编码核酸序列,并且其中所述至少一个MHC II类表位编码核酸序列包含至少一个通用MHC II类表位编码核酸序列,任选地其中所述至少一个通用序列包含破伤风类毒素和PADRE中的至少一者。
107.如权利要求1、3-17或19-106中任一项所述的方法,其中所述至少一个启动子核苷酸序列或所述第二启动子核苷酸序列是诱导型的。
108.如权利要求1、3-17或19-106中任一项所述的方法,其中所述至少一个启动子核苷酸序列或所述第二启动子核苷酸序列是非诱导型的。
109.如权利要求1、3-17或19-108中任一项所述的方法,其中所述至少一个poly(A)序列包含所述甲病毒的原生poly(A)序列。
110.如权利要求1、3-17或19-108中任一项所述的方法,其中所述至少一个poly(A)序列包含所述甲病毒的外源poly(A)序列。
111.如权利要求1、3-17或19-110中任一项所述的方法,其中所述至少一个poly(A)序列可操作地连接到所述至少一个核酸序列中的至少一者。
112.如权利要求1、3-17或19-111中任一项所述的方法,其中所述至少一个poly(A)序列是至少20个、至少30个、至少40个、至少50个、至少60个、至少70个、至少80个或至少90个连续A核苷酸。
113.如权利要求1、3-17或19-111中任一项所述的方法,其中所述至少一个poly(A)序列是至少100个连续A核苷酸。
114.如上述权利要求中任一项所述的方法,其中所述盒还包含以下中的至少一者:内含子序列、土拨鼠肝炎病毒转录后调控元件(WPRE)序列、内部核糖体进入序列(IRES)序列、编码2A自裂解肽序列的核苷酸序列、编码弗林蛋白酶裂解位点的核苷酸序列、或已知增强可操作地连接到所述至少一个核酸序列中的至少一者的mRNA的核输出、稳定性或翻译效率的5'或3'非编码区中的序列。
115.如上述权利要求中任一项所述的方法,其中所述盒还包含报告基因,其包括但不限于绿色荧光蛋白(GFP)、GFP变体、分泌型碱性磷酸酶、荧光素酶、荧光素酶变体或可检测的肽或表位。
116.如权利要求115所述的方法,其中所述可检测的肽或表位选自由HA标签、Flag标签、His标签或V5标签组成的组。
117.如上述权利要求中任一项所述的方法,其中所述一个或多个载体还包含编码至少一种免疫调节物的一个或多个核酸序列。
118.如权利要求117所述的方法,其中所述免疫调节物是抗CTLA4抗体或其抗原结合片段、抗PD-1抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。
119.如权利要求118所述的方法,其中所述抗体或其抗原结合片段是Fab片段、Fab'片段、单链Fv(scFv)、呈单特异性或连接在一起的多特异性的单结构域抗体(sdAb)(例如骆驼科抗体结构域)或全长单链抗体(例如具有通过柔性接头连接的重链和轻链的全长IgG)。
120.如权利要求118或119所述的方法,其中所述抗体的重链和轻链序列是由自裂解序列诸如2A或IRES分开的连续序列;或所述抗体的重链和轻链序列由柔性接头诸如连续甘氨酸残基连接。
121.如权利要求117所述的方法,其中所述免疫调节物是细胞因子。
122.如权利要求121所述的方法,其中所述细胞因子是IL-2、IL-7、IL-12、IL-15或IL-21或其各自变体中的至少一者。
123.如权利要求1-17或19-122中任一项所述的方法,其中所述表位编码核酸序列包含MHC I类表位编码核酸序列,并且其中所述MHC I类表位编码核酸序列通过进行以下步骤来选择:
(a)获得来自所述肿瘤的外显子组、转录组或全基因组肿瘤核苷酸测序数据中的至少一者,其中所述肿瘤核苷酸测序数据用于获得代表表位集合中的每一者的肽序列的数据;
(b)将每个表位的肽序列输入到递呈模型中,以产生所述表位中的每一者在所述肿瘤的肿瘤细胞表面上由所述MHC等位基因中的一者或多者递呈的数值可能性集合,所述数值可能性集合已至少基于所接收的质谱数据鉴定;以及
(c)基于所述数值可能性集合选择所述表位集合的子集,以产生经选择的表位集合,其用于产生所述MHCI类表位编码核酸序列。
124.如权利要求18所述的方法,其中所述MHC I类表位编码核酸序列中的每一者通过进行以下步骤来选择:
(a)获得来自所述肿瘤的外显子组、转录组或全基因组肿瘤核苷酸测序数据中的至少一者,其中所述肿瘤核苷酸测序数据用于获得代表表位集合中的每一者的肽序列的数据;
(b)将每个表位的肽序列输入到递呈模型中,以产生所述表位中的每一者在所述肿瘤的肿瘤细胞表面上由所述MHC等位基因中的一者或多者递呈的数值可能性集合,所述数值可能性集合已至少基于所接收的质谱数据鉴定;以及
(c)基于所述数值可能性集合选择所述表位集合的子集,以产生经选择的表位集合,其用于产生至少20个MHC I类表位编码核酸序列。
125.如权利要求123所述的方法,其中所述经选择的表位集合的数量为2-20个。
126.如权利要求123-125所述的方法,其中所述递呈模型表示以下两者之间的依赖性:
(a)所述MHC等位基因中的一对特定等位基因和在肽序列特定位置处特定氨基酸的存在;以及
(b)在所述肿瘤细胞表面上由所述对MHC等位基因中的特定等位基因递呈在所述特定位置处包含所述特定氨基酸的此类肽序列的可能性。
127.如权利要求123-126所述的方法,其中选择所述经选择的表位集合包括基于所述递呈模型,选择相对于未经选择的表位在所述肿瘤细胞表面上递呈的可能性增加的表位。
128.如权利要求123-127所述的方法,其中选择所述经选择的表位集合包括基于所述递呈模型,选择相对于未经选择的表位能够诱导所述受试者中的肿瘤特异性免疫应答的可能性增加的表位。
129.如权利要求123-128所述的方法,其中选择所述经选择的表位集合包括基于所述递呈模型,选择相对于未经选择的表位能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加的表位,任选地其中所述APC是树突状细胞(DC)。
130.如权利要求123-129所述的方法,其中选择所述经选择的表位集合包括基于所述递呈模型,选择相对于未经选择的表位经由中心或外周耐受性受抑制的可能性降低的表位。
131.如权利要求123-130所述的方法,其中选择所述经选择的表位集合包括基于所述递呈模型,选择相对于未经选择的表位能够诱导所述受试者中针对正常组织的自身免疫应答的可能性降低的表位。
132.如权利要求123-131所述的方法,其中外显子组或转录组核苷酸测序数据通过对所述肿瘤组织进行测序而获得。
133.如权利要求132所述的方法,其中所述测序是下一代测序(NGS)或任何大规模平行测序方法。
134.如上述权利要求中任一项所述的方法,其中所述盒包含由所述盒中的相邻序列形成的连接表位序列。
135.如权利要求134所述的方法,其中至少一个或每个连接表位序列对MHC的亲和力大于500nM。
136.如权利要求134或135所述的方法,其中每个连接表位序列是非自身的。
137.如上述权利要求中任一项所述的方法,其中所述盒不编码包含翻译的野生型核酸序列的非治疗性MHC I类或II类表位核酸序列,其中所述非治疗性表位经预测显示在所述受试者的MHC等位基因上。
138.如权利要求137所述的方法,其中所述经预测的非治疗性MHC I类或II类表位序列是由所述盒中的相邻序列形成的连接表位序列。
139.如权利要求134-138所述的方法,其中所述预测是基于通过将所述非治疗性表位的序列输入到递呈模型中而产生的递呈可能性。
140.如权利要求134-139中任一项所述的方法,其中所述盒中的所述抗原编码核酸序列的顺序通过包括以下的一系列步骤来确定:
(a)产生对应于所述抗原编码核酸序列的不同顺序的候选抗原盒序列集合;
(b)对于每个候选盒序列,基于所述候选盒序列中非治疗性表位的递呈来确定递呈评分;以及
(c)选择与低于预定阈值的递呈评分相关的候选盒序列作为用于疫苗的所述盒序列。
141.如上述权利要求中任一项所述的方法,其中用于递送所述表达系统的所述组合物和/或所述I型干扰素信号传导抑制剂被配制在包含药学上可接受的载剂的药物组合物中。
142.如上述权利要求中任一项所述的方法,其中所述方法还包括施用佐剂。
143.一种试剂盒,其包含上述方法权利要求中任一项所述的用于递送所述表达系统的所述组合物和所述I型干扰素信号传导抑制剂,以及使用说明。
144.如上述权利要求中任一项所述的方法,其中衍生的所述表位编码核酸序列衍生自所述受试者的肿瘤。
145.如上述权利要求中任一项所述的方法,其中所述表位编码核酸序列不衍生自所述受试者的肿瘤。
146.如上述权利要求中任一项所述的方法,所述方法还包括施用一种或多种免疫调节物,任选地其中所述免疫调节物在施用用于递送所述表达系统的所述组合物和/或所述I型干扰素信号传导抑制剂或其药物组合物之前、同时或之后施用。
147.如权利要求146所述的方法,其中所述一种或多种免疫调节物选自由以下组成的组:抗CTLA4抗体或其抗原结合片段、抗PD-1 抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。
148.如权利要求146或147所述的方法,其中所述免疫调节物经静脉内(IV)、肌肉内(IM)、皮内(ID)或皮下(SC)施用。
149.如权利要求148所述的方法,其中所述皮下施用是在所述表达系统施用部位的部位附近或者靠近所述表达系统的一个或多个引流淋巴结。
150.如上述权利要求中任一项所述的方法,其还包括向所述受试者施用第二疫苗组合物。
151.如权利要求150所述的方法,其中所述第二疫苗组合物在施用用于递送所述表达系统的所述组合物和/或所述I型干扰素信号传导抑制剂或其药物组合物之前施用。
152.如权利要求150所述的方法,其中所述第二疫苗组合物在施用用于递送所述表达系统的所述组合物和/或所述I型干扰素信号传导抑制剂或其药物组合物之后施用。
153.如权利要求150-152中任一项所述的方法,其中所述第二疫苗组合物与用于递送所述表达系统的所述组合物或其药物组合物相同。
154.如权利要求150-152中任一项所述的方法,其中所述第二疫苗组合物不同于用于递送所述表达系统的所述组合物或其药物组合物。
155.如权利要求154所述的方法,其中所述第二疫苗组合物包含编码至少一个抗原编码核酸序列的黑猩猩腺病毒载体。
156.如权利要求155所述的方法,其中由所述黑猩猩腺病毒载体编码的所述至少一个抗原编码核酸序列与如上述方法权利要求中任一项所述的抗原编码核酸序列相同。
157.如权利要求150-156中任一项所述的方法,其中所述I型干扰素信号传导抑制剂或其药物组合物的第二次施用在施用所述第二疫苗组合物之前、同时或之后。
158.一种用于治疗有癌患症的受试者的方法,所述方法包括向所述受试者施用治疗有效量的用于递送表达系统的组合物以及向所述受试者施用治疗有效量的I型干扰素信号传导抑制剂,
其中用于递送所述表达系统的所述组合物包含所述表达系统,
其中所述表达系统包含一个或多个载体,
所述一个或多个载体包含:
(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:
(i)至少一个启动子核苷酸序列,和
(ii)至少一个聚腺苷酸化(poly(A))序列;以及
(b)盒,其中所述盒包含:
(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,
b.任选地5'接头序列,以及
c.任选地3'接头序列;
(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及
(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
159.一种用于减小受试者中的肿瘤体积的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,
其中用于递送所述表达系统的所述组合物包含所述表达系统,
其中所述表达系统包含一个或多个载体,
所述一个或多个载体包含:
(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:
(i)至少一个启动子核苷酸序列,和
(ii)至少一个聚腺苷酸化(poly(A))序列;以及
(b)盒,其中所述盒包含:
(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,
b.任选地5'接头序列,以及
c.任选地3'接头序列;
(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及
(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
160.一种用于刺激受试者中的肿瘤特异性免疫应答的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,
其中用于递送所述表达系统的所述组合物包含所述表达系统,
其中所述表达系统包含一个或多个载体,
所述一个或多个载体包含:
(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:
(i)至少一个启动子核苷酸序列,和
(ii)至少一个聚腺苷酸化(poly(A))序列;以及
(b)盒,其中所述盒包含:
(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,
b.任选地5'接头序列,以及
c.任选地3'接头序列;
(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及
(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
161.一种增强基于甲病毒的表达系统递送的方法,所述方法包括向所述受试者施用用于递送表达系统的组合物以及向所述受试者施用I型干扰素信号传导抑制剂,
其中用于递送所述表达系统的所述组合物包含所述表达系统,
其中所述表达系统包含一个或多个载体,
所述一个或多个载体包含:
(a)RNA甲病毒骨架,其中所述RNA甲病毒骨架包含:
(i)至少一个启动子核苷酸序列,和
(ii)至少一个聚腺苷酸化(poly(A))序列;以及
(b)盒,其中所述盒包含:
(i)至少一个核酸序列,任选地其中所述至少一个核酸序列包含多肽编码核酸序列,任选地其中所述多肽编码核酸序列是抗原编码核酸序列,其包含:
a.表位编码核酸序列,其任选地包含至少一个使编码的表位序列不同于由野生型核酸序列编码的对应肽序列的改变,
b.任选地5'接头序列,以及
c.任选地3'接头序列;
(ii)任选地,可操作地连接到所述至少一个核酸序列的第二启动子核苷酸序列;以及
(iii)任选地,至少一个第二poly(A)序列,其中所述第二poly(A)序列是所述甲病毒的原生poly(A)序列或外源poly(A)序列。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862756980P | 2018-11-07 | 2018-11-07 | |
| US62/756,980 | 2018-11-07 | ||
| PCT/US2019/060355 WO2020097393A1 (en) | 2018-11-07 | 2019-11-07 | Alphavirus neoantigen vectors and interferon inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112912507A true CN112912507A (zh) | 2021-06-04 |
Family
ID=70611178
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980068204.XA Pending CN112912507A (zh) | 2018-11-07 | 2019-11-07 | 甲病毒新抗原载体和干扰素抑制剂 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20220125919A1 (zh) |
| EP (1) | EP3877531A4 (zh) |
| JP (1) | JP2022512912A (zh) |
| KR (1) | KR20210090650A (zh) |
| CN (1) | CN112912507A (zh) |
| AU (1) | AU2019374874A1 (zh) |
| CA (1) | CA3119752A1 (zh) |
| IL (1) | IL282793A (zh) |
| WO (1) | WO2020097393A1 (zh) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018208856A1 (en) | 2017-05-08 | 2018-11-15 | Gritstone Oncology, Inc. | Alphavirus neoantigen vectors |
| TW202043256A (zh) | 2019-01-10 | 2020-12-01 | 美商健生生物科技公司 | 前列腺新抗原及其用途 |
| BR122024002387A2 (pt) | 2019-05-30 | 2024-03-12 | Gritstone Bio, Inc. | Vetores de adenovírus, composição farmacêutica, sequência de nucleotídeo isolada, célula isolada, vetor, kit, usos de um vetor, método para fabricar o vetor, métodos para produzir um vírus e vetor viral |
| WO2022011429A1 (en) * | 2020-07-17 | 2022-01-20 | The University Of Western Australia | Methods to increase response to immune checkpoint modulation |
| CN111991569B (zh) * | 2020-07-22 | 2021-09-28 | 华中科技大学 | 一种双靶向乳腺癌细胞及其淋巴结转移灶的纳米颗粒、制备方法及应用 |
| WO2022032196A2 (en) | 2020-08-06 | 2022-02-10 | Gritstone Bio, Inc. | Multiepitope vaccine cassettes |
| WO2024054882A1 (en) * | 2022-09-09 | 2024-03-14 | Spark Therapeutics, Inc. | Enhancing non-viral dna delivery and expression |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003083065A2 (en) * | 2002-03-25 | 2003-10-09 | University Of North Carolina At Chapel Hill | Improved methods for achieving expression from alphavirus vectors |
| WO2018098362A1 (en) * | 2016-11-23 | 2018-05-31 | Gritstone Oncology, Inc. | Viral delivery of neoantigens |
| WO2018195357A1 (en) * | 2017-04-19 | 2018-10-25 | Gritstone Oncology, Inc. | Neoantigen identification, manufacture, and use |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6451592B1 (en) * | 1996-04-05 | 2002-09-17 | Chiron Corporation | Recombinant alphavirus-based vectors with reduced inhibition of cellular macromolecular synthesis |
| WO2006078294A2 (en) * | 2004-05-21 | 2006-07-27 | Novartis Vaccines And Diagnostics Inc. | Alphavirus vectors for respiratory pathogen vaccines |
| US20210213122A1 (en) * | 2018-05-23 | 2021-07-15 | Gritstone Oncology, Inc. | Immune checkpoint inhibitor co-expression vectors |
-
2019
- 2019-11-07 AU AU2019374874A patent/AU2019374874A1/en not_active Abandoned
- 2019-11-07 US US17/291,984 patent/US20220125919A1/en active Pending
- 2019-11-07 CA CA3119752A patent/CA3119752A1/en active Pending
- 2019-11-07 KR KR1020217016771A patent/KR20210090650A/ko unknown
- 2019-11-07 WO PCT/US2019/060355 patent/WO2020097393A1/en unknown
- 2019-11-07 CN CN201980068204.XA patent/CN112912507A/zh active Pending
- 2019-11-07 EP EP19882269.4A patent/EP3877531A4/en active Pending
- 2019-11-07 JP JP2021524020A patent/JP2022512912A/ja not_active Withdrawn
-
2021
- 2021-04-29 IL IL282793A patent/IL282793A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003083065A2 (en) * | 2002-03-25 | 2003-10-09 | University Of North Carolina At Chapel Hill | Improved methods for achieving expression from alphavirus vectors |
| WO2018098362A1 (en) * | 2016-11-23 | 2018-05-31 | Gritstone Oncology, Inc. | Viral delivery of neoantigens |
| WO2018195357A1 (en) * | 2017-04-19 | 2018-10-25 | Gritstone Oncology, Inc. | Neoantigen identification, manufacture, and use |
Non-Patent Citations (1)
| Title |
|---|
| MIN WANG等: "Alphavirus vector-based replicon particles expressing multivalent cross-protective Lassa virus glycoproteins", VACCINE, vol. 36, no. 5, pages 683 - 690 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20210090650A (ko) | 2021-07-20 |
| US20220125919A1 (en) | 2022-04-28 |
| EP3877531A1 (en) | 2021-09-15 |
| CA3119752A1 (en) | 2020-05-14 |
| IL282793A (en) | 2021-06-30 |
| WO2020097393A1 (en) | 2020-05-14 |
| EP3877531A4 (en) | 2022-08-31 |
| AU2019374874A1 (en) | 2021-06-10 |
| JP2022512912A (ja) | 2022-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11510973B2 (en) | Alphavirus antigen vectors | |
| CN112368386A (zh) | 共有抗原 | |
| WO2018098362A1 (en) | Viral delivery of neoantigens | |
| JP7457733B2 (ja) | 改変アデノウイルス | |
| CN112912507A (zh) | 甲病毒新抗原载体和干扰素抑制剂 | |
| US20210213122A1 (en) | Immune checkpoint inhibitor co-expression vectors | |
| US20230020089A1 (en) | Shared neoantigen vaccines | |
| US20230128001A1 (en) | Hiv antigens and mhc complexes | |
| CN115803049A (zh) | 抗原编码盒 | |
| CN117279660A (zh) | 经修饰的甲病毒载体 | |
| WO2024026329A2 (en) | Egfr vaccine cassettes | |
| CA3231295A1 (en) | Neoantigen adjuvant and maintenance therapy | |
| CN117957015A (zh) | Kras新抗原疗法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| CB02 | Change of applicant information |
Address after: California, USA Applicant after: Millstone biological Co. Address before: California, USA Applicant before: Gritstone Oncology, Inc. |
|
| CB02 | Change of applicant information |