CN112513071A - Gene therapy constructs and methods of use - Google Patents
Gene therapy constructs and methods of use Download PDFInfo
- Publication number
- CN112513071A CN112513071A CN201980029113.5A CN201980029113A CN112513071A CN 112513071 A CN112513071 A CN 112513071A CN 201980029113 A CN201980029113 A CN 201980029113A CN 112513071 A CN112513071 A CN 112513071A
- Authority
- CN
- China
- Prior art keywords
- disease
- gene therapy
- therapy vector
- type
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000001415 gene therapy Methods 0.000 title claims abstract description 232
- 238000000034 method Methods 0.000 title claims abstract description 50
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 284
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 280
- 239000013598 vector Substances 0.000 claims abstract description 261
- 230000001225 therapeutic effect Effects 0.000 claims abstract description 227
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 375
- 201000010099 disease Diseases 0.000 claims description 353
- 150000007523 nucleic acids Chemical class 0.000 claims description 248
- 108010076504 Protein Sorting Signals Proteins 0.000 claims description 205
- 150000001413 amino acids Chemical group 0.000 claims description 197
- 108020004707 nucleic acids Proteins 0.000 claims description 161
- 102000039446 nucleic acids Human genes 0.000 claims description 161
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 149
- 208000008955 Mucolipidoses Diseases 0.000 claims description 110
- 208000026350 Inborn Genetic disease Diseases 0.000 claims description 100
- 208000016361 genetic disease Diseases 0.000 claims description 100
- 108700041152 Endoplasmic Reticulum Chaperone BiP Proteins 0.000 claims description 93
- 102100021451 Endoplasmic reticulum chaperone BiP Human genes 0.000 claims description 93
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 87
- 108010028144 alpha-Glucosidases Proteins 0.000 claims description 67
- 201000008051 neuronal ceroid lipofuscinosis Diseases 0.000 claims description 67
- 241000710127 Cricket paralysis virus Species 0.000 claims description 64
- 239000012634 fragment Substances 0.000 claims description 64
- 101001076292 Homo sapiens Insulin-like growth factor II Proteins 0.000 claims description 61
- 102000005327 Palmitoyl protein thioesterase Human genes 0.000 claims description 56
- 108020002591 Palmitoyl protein thioesterase Proteins 0.000 claims description 56
- 239000013603 viral vector Substances 0.000 claims description 52
- 206010053185 Glycogen storage disease type II Diseases 0.000 claims description 51
- 208000032007 Glycogen storage disease due to acid maltase deficiency Diseases 0.000 claims description 50
- 201000004502 glycogen storage disease II Diseases 0.000 claims description 50
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 50
- 102100033448 Lysosomal alpha-glucosidase Human genes 0.000 claims description 48
- 208000011045 mucopolysaccharidosis type 3 Diseases 0.000 claims description 47
- 241000589601 Francisella Species 0.000 claims description 44
- 210000004027 cell Anatomy 0.000 claims description 42
- 102000004190 Enzymes Human genes 0.000 claims description 41
- 108090000790 Enzymes Proteins 0.000 claims description 41
- 229940088598 enzyme Drugs 0.000 claims description 41
- 238000006467 substitution reaction Methods 0.000 claims description 37
- 208000024720 Fabry Disease Diseases 0.000 claims description 36
- 230000007812 deficiency Effects 0.000 claims description 36
- 230000002132 lysosomal effect Effects 0.000 claims description 36
- 241000963438 Gaussia <copepod> Species 0.000 claims description 35
- 229920002683 Glycosaminoglycan Polymers 0.000 claims description 35
- 208000002537 Neuronal Ceroid-Lipofuscinoses Diseases 0.000 claims description 35
- 238000012217 deletion Methods 0.000 claims description 35
- 230000037430 deletion Effects 0.000 claims description 35
- 241000702421 Dependoparvovirus Species 0.000 claims description 32
- 102000009133 Arylsulfatases Human genes 0.000 claims description 30
- 201000011442 Metachromatic leukodystrophy Diseases 0.000 claims description 30
- 102000005348 Neuraminidase Human genes 0.000 claims description 30
- 108010006232 Neuraminidase Proteins 0.000 claims description 30
- 239000002773 nucleotide Substances 0.000 claims description 30
- 125000003729 nucleotide group Chemical group 0.000 claims description 30
- 108060007951 sulfatase Proteins 0.000 claims description 30
- 208000015439 Lysosomal storage disease Diseases 0.000 claims description 29
- 206010072928 Mucolipidosis type II Diseases 0.000 claims description 29
- 108010030291 alpha-Galactosidase Proteins 0.000 claims description 29
- -1 batttenin Proteins 0.000 claims description 29
- 102100029199 Iduronate 2-sulfatase Human genes 0.000 claims description 28
- 101710096421 Iduronate 2-sulfatase Proteins 0.000 claims description 28
- 102000005840 alpha-Galactosidase Human genes 0.000 claims description 28
- 102000004627 Iduronidase Human genes 0.000 claims description 27
- 108010003381 Iduronidase Proteins 0.000 claims description 27
- 230000014621 translational initiation Effects 0.000 claims description 27
- ZSLZBFCDCINBPY-ZSJPKINUSA-N Acetyl-CoA Natural products O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 ZSLZBFCDCINBPY-ZSJPKINUSA-N 0.000 claims description 26
- 102100031491 Arylsulfatase B Human genes 0.000 claims description 26
- 102000004547 Glucosylceramidase Human genes 0.000 claims description 26
- 108010017544 Glucosylceramidase Proteins 0.000 claims description 26
- 102000004157 Hydrolases Human genes 0.000 claims description 26
- 108090000604 Hydrolases Proteins 0.000 claims description 26
- 108010027520 N-Acetylgalactosamine-4-Sulfatase Proteins 0.000 claims description 26
- 102100031688 N-acetylgalactosamine-6-sulfatase Human genes 0.000 claims description 26
- 101710099863 N-acetylgalactosamine-6-sulfatase Proteins 0.000 claims description 26
- 108010055297 Sterol Esterase Proteins 0.000 claims description 26
- 102000000019 Sterol Esterase Human genes 0.000 claims description 26
- 239000008194 pharmaceutical composition Substances 0.000 claims description 26
- 108010023546 Aspartylglucosylaminase Proteins 0.000 claims description 25
- 206010011777 Cystinosis Diseases 0.000 claims description 25
- 102100028875 Formylglycine-generating enzyme Human genes 0.000 claims description 25
- 101710192607 Formylglycine-generating enzyme Proteins 0.000 claims description 25
- 102100028496 Galactocerebrosidase Human genes 0.000 claims description 25
- 108010042681 Galactosylceramidase Proteins 0.000 claims description 25
- 102000053187 Glucuronidase Human genes 0.000 claims description 25
- 108010060309 Glucuronidase Proteins 0.000 claims description 25
- 108010003272 Hyaluronate lyase Proteins 0.000 claims description 25
- 206010072929 Mucolipidosis type III Diseases 0.000 claims description 25
- 102100021003 N(4)-(beta-N-acetylglucosaminyl)-L-asparaginase Human genes 0.000 claims description 25
- 108091000080 Phosphotransferase Proteins 0.000 claims description 25
- 101710192761 Serine-type anaerobic sulfatase-maturating enzyme Proteins 0.000 claims description 25
- 108010061312 Sphingomyelin Phosphodiesterase Proteins 0.000 claims description 25
- 102000012086 alpha-L-Fucosidase Human genes 0.000 claims description 25
- 108010061314 alpha-L-Fucosidase Proteins 0.000 claims description 25
- 108010012864 alpha-Mannosidase Proteins 0.000 claims description 25
- 102000019199 alpha-Mannosidase Human genes 0.000 claims description 25
- 108010005774 beta-Galactosidase Proteins 0.000 claims description 25
- 108010055059 beta-Mannosidase Proteins 0.000 claims description 25
- 108010089932 heparan sulfate sulfatase Proteins 0.000 claims description 25
- 229960002773 hyaluronidase Drugs 0.000 claims description 25
- 239000000203 mixture Substances 0.000 claims description 25
- 102000020233 phosphotransferase Human genes 0.000 claims description 25
- 208000020916 Gaucher disease type II Diseases 0.000 claims description 24
- 208000028735 Gaucher disease type III Diseases 0.000 claims description 24
- 101710202061 N-acetyltransferase Proteins 0.000 claims description 24
- 210000002569 neuron Anatomy 0.000 claims description 23
- 206010028095 Mucopolysaccharidosis IV Diseases 0.000 claims description 22
- 230000001684 chronic effect Effects 0.000 claims description 22
- 208000021811 Sandhoff disease Diseases 0.000 claims description 21
- 208000002491 severe combined immunodeficiency Diseases 0.000 claims description 21
- 241001529453 unidentified herpesvirus Species 0.000 claims description 21
- 206010072927 Mucolipidosis type I Diseases 0.000 claims description 20
- 206010072930 Mucolipidosis type IV Diseases 0.000 claims description 20
- 102100026502 Mucolipin-1 Human genes 0.000 claims description 20
- 208000017460 Sialidosis type 2 Diseases 0.000 claims description 20
- 201000008977 glycoproteinosis Diseases 0.000 claims description 20
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 claims description 19
- 102000055025 Adenosine deaminases Human genes 0.000 claims description 19
- 208000020322 Gaucher disease type I Diseases 0.000 claims description 19
- 102220576120 Oligodendrocyte transcription factor 1_Y27W_mutation Human genes 0.000 claims description 19
- 208000016532 chronic granulomatous disease Diseases 0.000 claims description 19
- 238000011282 treatment Methods 0.000 claims description 19
- 102220518120 DNA-directed RNA polymerases I and III subunit RPAC1_E6R_mutation Human genes 0.000 claims description 18
- 230000001965 increasing effect Effects 0.000 claims description 18
- 230000028327 secretion Effects 0.000 claims description 18
- NDYKCIOPBXHJTK-DPNYYRAHSA-N N[C@@H](CC(=O)O)C(=O)NC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO Chemical compound N[C@@H](CC(=O)O)C(=O)NC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO NDYKCIOPBXHJTK-DPNYYRAHSA-N 0.000 claims description 16
- 229920006063 Lamide® Polymers 0.000 claims description 15
- 230000002950 deficient Effects 0.000 claims description 15
- 208000037157 Azotemia Diseases 0.000 claims description 14
- 102100025947 Insulin-like growth factor II Human genes 0.000 claims description 14
- NBSCHQHZLSJFNQ-QTVWNMPRSA-N D-Mannose-6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@@H]1O NBSCHQHZLSJFNQ-QTVWNMPRSA-N 0.000 claims description 13
- 241000700605 Viruses Species 0.000 claims description 13
- 208000009852 uremia Diseases 0.000 claims description 13
- 102220511796 F-actin-capping protein subunit beta_F26S_mutation Human genes 0.000 claims description 12
- 208000015872 Gaucher disease Diseases 0.000 claims description 12
- 102220474905 Insulin-like 3_V43L_mutation Human genes 0.000 claims description 12
- 201000000788 Niemann-Pick disease type C1 Diseases 0.000 claims description 12
- 201000000785 Niemann-Pick disease type C2 Diseases 0.000 claims description 12
- 102220638483 Protein PML_K65R_mutation Human genes 0.000 claims description 12
- 210000003712 lysosome Anatomy 0.000 claims description 12
- 230000001868 lysosomic effect Effects 0.000 claims description 12
- 208000010978 mucopolysaccharidosis type 4 Diseases 0.000 claims description 12
- 102220332512 rs1555032911 Human genes 0.000 claims description 12
- 102220297864 rs761846391 Human genes 0.000 claims description 12
- 208000035374 Chronic visceral acid sphingomyelinase deficiency Diseases 0.000 claims description 11
- 201000000791 Niemann-Pick disease type B Diseases 0.000 claims description 11
- 208000008291 Type B Niemann-Pick Disease Diseases 0.000 claims description 11
- 208000035343 Infantile neurovisceral acid sphingomyelinase deficiency Diseases 0.000 claims description 10
- 201000000794 Niemann-Pick disease type A Diseases 0.000 claims description 10
- 208000007824 Type A Niemann-Pick Disease Diseases 0.000 claims description 10
- 102000005962 receptors Human genes 0.000 claims description 10
- 108020003175 receptors Proteins 0.000 claims description 10
- 102220487360 Guanine nucleotide-binding protein G(i) subunit alpha-1_A54R_mutation Human genes 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 9
- 238000007913 intrathecal administration Methods 0.000 claims description 8
- 102100034746 Cyclin-dependent kinase-like 5 Human genes 0.000 claims description 7
- 239000003814 drug Substances 0.000 claims description 7
- 230000002829 reductive effect Effects 0.000 claims description 7
- 101000945692 Homo sapiens Cyclin-dependent kinase-like 5 Proteins 0.000 claims description 6
- 102000003746 Insulin Receptor Human genes 0.000 claims description 6
- 230000012202 endocytosis Effects 0.000 claims description 6
- 208000017476 juvenile neuronal ceroid lipofuscinosis Diseases 0.000 claims description 6
- 201000007607 neuronal ceroid lipofuscinosis 3 Diseases 0.000 claims description 6
- 230000001737 promoting effect Effects 0.000 claims description 6
- 230000001177 retroviral effect Effects 0.000 claims description 6
- 108010001127 Insulin Receptor Proteins 0.000 claims description 5
- NCYCYZXNIZJOKI-OVSJKPMPSA-N Retinaldehyde Chemical compound O=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-OVSJKPMPSA-N 0.000 claims description 5
- NCYCYZXNIZJOKI-UHFFFAOYSA-N vitamin A aldehyde Natural products O=CC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-UHFFFAOYSA-N 0.000 claims description 5
- 241000710198 Foot-and-mouth disease virus Species 0.000 claims description 4
- 241001502974 Human gammaherpesvirus 8 Species 0.000 claims description 4
- 241000710778 Pestivirus Species 0.000 claims description 4
- 241000709664 Picornaviridae Species 0.000 claims description 4
- 208000022292 Tay-Sachs disease Diseases 0.000 claims description 4
- 208000006454 hepatitis Diseases 0.000 claims description 4
- 231100000283 hepatitis Toxicity 0.000 claims description 4
- 238000007918 intramuscular administration Methods 0.000 claims description 4
- 238000001990 intravenous administration Methods 0.000 claims description 4
- 238000007914 intraventricular administration Methods 0.000 claims description 4
- 238000007920 subcutaneous administration Methods 0.000 claims description 4
- 206010068220 Aspartylglucosaminuria Diseases 0.000 claims description 3
- 108700001567 Type I Schindler Disease Proteins 0.000 claims description 3
- 206010046865 Vaccinia virus infection Diseases 0.000 claims description 3
- 150000001768 cations Chemical class 0.000 claims description 3
- 150000001875 compounds Chemical class 0.000 claims description 3
- 229920000642 polymer Polymers 0.000 claims description 3
- 238000002360 preparation method Methods 0.000 claims description 3
- 150000003839 salts Chemical class 0.000 claims description 3
- 208000007089 vaccinia Diseases 0.000 claims description 3
- 208000029602 Alpha-N-acetylgalactosaminidase deficiency Diseases 0.000 claims description 2
- 208000005176 Hepatitis C Diseases 0.000 claims description 2
- 208000015178 Hurler syndrome Diseases 0.000 claims description 2
- 206010056886 Mucopolysaccharidosis I Diseases 0.000 claims description 2
- 208000025797 Mucopolysaccharidosis type 4A Diseases 0.000 claims description 2
- 241000125162 Rhopalosiphum Species 0.000 claims description 2
- 241000125167 Rhopalosiphum padi Species 0.000 claims description 2
- 208000005252 hepatitis A Diseases 0.000 claims description 2
- 208000026872 Addison Disease Diseases 0.000 claims 3
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 claims 3
- 102000009066 Hyaluronoglucosaminidase Human genes 0.000 claims 2
- 102000005936 beta-Galactosidase Human genes 0.000 claims 2
- 102220274185 rs762526848 Human genes 0.000 claims 2
- 208000031277 Amaurotic familial idiocy Diseases 0.000 claims 1
- 241000175212 Herpesvirales Species 0.000 claims 1
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 claims 1
- 101100066427 Homo sapiens FCGR1A gene Proteins 0.000 claims 1
- YTTRPBWEMMPYSW-HRRFRDKFSA-N N(4)-(beta-N-acetyl-D-glucosaminyl)-L-asparagine Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1NC(=O)C[C@H]([NH3+])C([O-])=O YTTRPBWEMMPYSW-HRRFRDKFSA-N 0.000 claims 1
- 235000011205 Ocimum Nutrition 0.000 claims 1
- 241001529734 Ocimum Species 0.000 claims 1
- 230000008685 targeting Effects 0.000 abstract description 12
- 230000001976 improved effect Effects 0.000 abstract description 5
- 230000014509 gene expression Effects 0.000 abstract description 4
- 108020001507 fusion proteins Proteins 0.000 description 115
- 102000037865 fusion proteins Human genes 0.000 description 115
- 101710132601 Capsid protein Proteins 0.000 description 72
- 101710094648 Coat protein Proteins 0.000 description 72
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 72
- 101710125418 Major capsid protein Proteins 0.000 description 72
- 101710141454 Nucleoprotein Proteins 0.000 description 72
- 101710083689 Probable capsid protein Proteins 0.000 description 72
- 102000016679 alpha-Glucosidases Human genes 0.000 description 64
- 208000011580 syndromic disease Diseases 0.000 description 52
- 208000005340 mucopolysaccharidosis III Diseases 0.000 description 39
- 208000014060 Niemann-Pick disease Diseases 0.000 description 37
- 241000701161 unidentified adenovirus Species 0.000 description 28
- 230000027455 binding Effects 0.000 description 26
- 102100022548 Beta-hexosaminidase subunit alpha Human genes 0.000 description 25
- 208000001905 GM2 Gangliosidoses Diseases 0.000 description 25
- 201000008905 GM2 gangliosidosis Diseases 0.000 description 25
- 101001045440 Homo sapiens Beta-hexosaminidase subunit alpha Proteins 0.000 description 25
- 230000000366 juvenile effect Effects 0.000 description 25
- 108090000565 Capsid Proteins Proteins 0.000 description 24
- 102100026189 Beta-galactosidase Human genes 0.000 description 23
- 102000001974 Hyaluronidases Human genes 0.000 description 23
- 208000022018 mucopolysaccharidosis type 2 Diseases 0.000 description 20
- 102100025621 Cytochrome b-245 heavy chain Human genes 0.000 description 18
- 208000035475 disorder Diseases 0.000 description 18
- 241000699670 Mus sp. Species 0.000 description 17
- 238000011813 knockout mouse model Methods 0.000 description 17
- 241000713666 Lentivirus Species 0.000 description 16
- 238000003860 storage Methods 0.000 description 16
- 241001430294 unidentified retrovirus Species 0.000 description 16
- 239000003981 vehicle Substances 0.000 description 16
- 201000008892 GM1 Gangliosidosis Diseases 0.000 description 15
- 102000004196 processed proteins & peptides Human genes 0.000 description 15
- 150000003408 sphingolipids Chemical class 0.000 description 15
- 241000725303 Human immunodeficiency virus Species 0.000 description 12
- 241000714177 Murine leukemia virus Species 0.000 description 12
- 241000711975 Vesicular stomatitis virus Species 0.000 description 12
- 241000712461 unidentified influenza virus Species 0.000 description 12
- 108010051109 Cell-Penetrating Peptides Proteins 0.000 description 11
- 102000020313 Cell-Penetrating Peptides Human genes 0.000 description 11
- 229940068935 insulin-like growth factor 2 Drugs 0.000 description 11
- 239000002502 liposome Substances 0.000 description 11
- 201000007769 mucolipidosis Diseases 0.000 description 11
- 239000002105 nanoparticle Substances 0.000 description 11
- 102220486129 Alpha-galactosidase A_R49S_mutation Human genes 0.000 description 10
- 108091006088 activator proteins Proteins 0.000 description 10
- 201000002273 mucopolysaccharidosis II Diseases 0.000 description 10
- 239000013607 AAV vector Substances 0.000 description 9
- 108010039203 Tripeptidyl-Peptidase 1 Proteins 0.000 description 9
- 102100034197 Tripeptidyl-peptidase 1 Human genes 0.000 description 9
- 208000020460 mucolipidosis II alpha/beta Diseases 0.000 description 9
- 229920002527 Glycogen Polymers 0.000 description 8
- 101000574223 Homo sapiens Palmitoyl-protein thioesterase 1 Proteins 0.000 description 8
- 229940096919 glycogen Drugs 0.000 description 8
- 230000000694 effects Effects 0.000 description 7
- 208000025974 neutral lipid storage disease Diseases 0.000 description 7
- 101150002416 Igf2 gene Proteins 0.000 description 6
- 230000004700 cellular uptake Effects 0.000 description 6
- 208000025919 mucopolysaccharidosis type 7 Diseases 0.000 description 6
- 201000007642 neuronal ceroid lipofuscinosis 1 Diseases 0.000 description 6
- 208000033436 CLN6 disease Diseases 0.000 description 5
- 208000012239 Developmental disease Diseases 0.000 description 5
- 108090000371 Esterases Proteins 0.000 description 5
- 201000008394 GM1 gangliosidosis type 2 Diseases 0.000 description 5
- 201000008232 GM2 gangliosidosis, AB variant Diseases 0.000 description 5
- 208000010055 Globoid Cell Leukodystrophy Diseases 0.000 description 5
- 101000575454 Homo sapiens Major facilitator superfamily domain-containing protein 8 Proteins 0.000 description 5
- 108700037017 Hyaluronidase Deficiency Proteins 0.000 description 5
- 208000005503 Hyaluronidase deficiency Diseases 0.000 description 5
- 208000028226 Krabbe disease Diseases 0.000 description 5
- 208000003221 Lysosomal acid lipase deficiency Diseases 0.000 description 5
- 102100025613 Major facilitator superfamily domain-containing protein 8 Human genes 0.000 description 5
- 208000002720 Malnutrition Diseases 0.000 description 5
- 208000021642 Muscular disease Diseases 0.000 description 5
- 201000009623 Myopathy Diseases 0.000 description 5
- 206010029148 Nephrolithiasis Diseases 0.000 description 5
- 208000012617 Neutral lipid storage myopathy Diseases 0.000 description 5
- 102100025824 Palmitoyl-protein thioesterase 1 Human genes 0.000 description 5
- 206010033799 Paralysis Diseases 0.000 description 5
- 208000037006 Progressive epilepsy-intellectual disability syndrome, Finnish type Diseases 0.000 description 5
- 201000005660 Protein C Deficiency Diseases 0.000 description 5
- 208000026375 Salivary gland disease Diseases 0.000 description 5
- 241001486234 Sciota Species 0.000 description 5
- 230000003213 activating effect Effects 0.000 description 5
- 239000012190 activator Substances 0.000 description 5
- 208000017478 adult neuronal ceroid lipofuscinosis Diseases 0.000 description 5
- SHZGCJCMOBCMKK-PHYPRBDBSA-N alpha-D-fucose Chemical compound C[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O SHZGCJCMOBCMKK-PHYPRBDBSA-N 0.000 description 5
- 201000008333 alpha-mannosidosis Diseases 0.000 description 5
- 208000016704 atypical Gaucher disease due to saposin C deficiency Diseases 0.000 description 5
- 208000033159 atypical due to saposin A deficiency Krabbe disease Diseases 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000002146 bilateral effect Effects 0.000 description 5
- 210000000988 bone and bone Anatomy 0.000 description 5
- 208000025729 dengue disease Diseases 0.000 description 5
- 206010015037 epilepsy Diseases 0.000 description 5
- 208000013746 hereditary thrombophilia due to congenital protein C deficiency Diseases 0.000 description 5
- 230000000121 hypercalcemic effect Effects 0.000 description 5
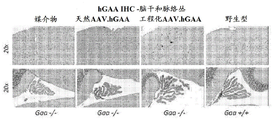
- 238000003364 immunohistochemistry Methods 0.000 description 5
- 208000017169 kidney disease Diseases 0.000 description 5
- 208000035157 late-onset glycogen storage disease due to acid maltase deficiency Diseases 0.000 description 5
- 208000036546 leukodystrophy Diseases 0.000 description 5
- 230000001071 malnutrition Effects 0.000 description 5
- 235000000824 malnutrition Nutrition 0.000 description 5
- 208000012253 mucopolysaccharidosis IVA Diseases 0.000 description 5
- 208000027333 mucopolysaccharidosis type IIID Diseases 0.000 description 5
- 208000012091 mucopolysaccharidosis type IVB Diseases 0.000 description 5
- 230000002580 nephropathic effect Effects 0.000 description 5
- 230000000926 neurological effect Effects 0.000 description 5
- 201000007640 neuronal ceroid lipofuscinosis 7 Diseases 0.000 description 5
- 201000007638 neuronal ceroid lipofuscinosis 8 Diseases 0.000 description 5
- 201000007635 neuronal ceroid lipofuscinosis 8 northern epilepsy variant Diseases 0.000 description 5
- 208000015380 nutritional deficiency disease Diseases 0.000 description 5
- 208000021090 palsy Diseases 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 229920006295 polythiol Polymers 0.000 description 5
- 230000000750 progressive effect Effects 0.000 description 5
- 230000007026 protein scission Effects 0.000 description 5
- 208000002320 spinal muscular atrophy Diseases 0.000 description 5
- 230000002123 temporal effect Effects 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 4
- 102100031675 DnaJ homolog subfamily C member 5 Human genes 0.000 description 4
- 201000008393 GM1 gangliosidosis type 1 Diseases 0.000 description 4
- 101000845893 Homo sapiens DnaJ homolog subfamily C member 5 Proteins 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- 208000031406 ceroid lipofuscinosis, neuronal, 4 (Kufs type) Diseases 0.000 description 4
- 238000002641 enzyme replacement therapy Methods 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 229920001184 polypeptide Polymers 0.000 description 4
- 210000000278 spinal cord Anatomy 0.000 description 4
- 241001655883 Adeno-associated virus - 1 Species 0.000 description 3
- 241000702423 Adeno-associated virus - 2 Species 0.000 description 3
- 241000202702 Adeno-associated virus - 3 Species 0.000 description 3
- 241000580270 Adeno-associated virus - 4 Species 0.000 description 3
- 241001634120 Adeno-associated virus - 5 Species 0.000 description 3
- 241000972680 Adeno-associated virus - 6 Species 0.000 description 3
- 241001164823 Adeno-associated virus - 7 Species 0.000 description 3
- 241001164825 Adeno-associated virus - 8 Species 0.000 description 3
- 201000011240 Frontotemporal dementia Diseases 0.000 description 3
- 101001018026 Homo sapiens Lysosomal alpha-glucosidase Proteins 0.000 description 3
- 208000000609 Pick Disease of the Brain Diseases 0.000 description 3
- 208000024571 Pick disease Diseases 0.000 description 3
- 101150010487 are gene Proteins 0.000 description 3
- 102000045921 human GAA Human genes 0.000 description 3
- 102000051672 human PPT1 Human genes 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 230000001915 proofreading effect Effects 0.000 description 3
- 102100026277 Alpha-galactosidase A Human genes 0.000 description 2
- 102100038238 Aromatic-L-amino-acid decarboxylase Human genes 0.000 description 2
- 102100022440 Battenin Human genes 0.000 description 2
- 101710199232 Battenin Proteins 0.000 description 2
- 102100034480 Ceroid-lipofuscinosis neuronal protein 6 Human genes 0.000 description 2
- 101710163532 Ceroid-lipofuscinosis neuronal protein 6 Proteins 0.000 description 2
- 102100024335 Collagen alpha-1(VII) chain Human genes 0.000 description 2
- 102100029140 Cyclic nucleotide-gated cation channel beta-3 Human genes 0.000 description 2
- 206010013801 Duchenne Muscular Dystrophy Diseases 0.000 description 2
- 102100021519 Hemoglobin subunit beta Human genes 0.000 description 2
- 101000909498 Homo sapiens Collagen alpha-1(VII) chain Proteins 0.000 description 2
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 2
- 208000012860 Horse disease Diseases 0.000 description 2
- 108010031792 IGF Type 2 Receptor Proteins 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- 101710184277 Insulin-like growth factor 1 receptor Proteins 0.000 description 2
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 2
- 206010029748 Noonan syndrome Diseases 0.000 description 2
- 108050000176 Sialidase-3 Proteins 0.000 description 2
- 102000013275 Somatomedins Human genes 0.000 description 2
- 208000027073 Stargardt disease Diseases 0.000 description 2
- 108091081024 Start codon Proteins 0.000 description 2
- 108010035075 Tyrosine decarboxylase Proteins 0.000 description 2
- 102100031835 Unconventional myosin-VIIa Human genes 0.000 description 2
- 102000010126 acid sphingomyelin phosphodiesterase activity proteins Human genes 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 102000028416 insulin-like growth factor binding Human genes 0.000 description 2
- 108091022911 insulin-like growth factor binding Proteins 0.000 description 2
- 108010045758 lysosomal proteins Proteins 0.000 description 2
- 229920001542 oligosaccharide Polymers 0.000 description 2
- 150000002482 oligosaccharides Chemical class 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000002381 plasma Anatomy 0.000 description 2
- 235000008001 rakum palm Nutrition 0.000 description 2
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 102100022712 Alpha-1-antitrypsin Human genes 0.000 description 1
- 101710081722 Antitrypsin Proteins 0.000 description 1
- 101100404726 Arabidopsis thaliana NHX7 gene Proteins 0.000 description 1
- 108700019265 Aromatic amino acid decarboxylase deficiency Proteins 0.000 description 1
- 208000033418 CLN1 disease Diseases 0.000 description 1
- 101100084118 Caenorhabditis elegans ppt-1 gene Proteins 0.000 description 1
- 102100037182 Cation-independent mannose-6-phosphate receptor Human genes 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 208000033810 Choroidal dystrophy Diseases 0.000 description 1
- 201000001873 Christianson syndrome Diseases 0.000 description 1
- 102100022641 Coagulation factor IX Human genes 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 208000006992 Color Vision Defects Diseases 0.000 description 1
- 108010069156 Connexin 26 Proteins 0.000 description 1
- 101710093675 Cyclic nucleotide-gated cation channel beta-3 Proteins 0.000 description 1
- 101710178912 Cyclin-dependent kinase-like 5 Proteins 0.000 description 1
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 208000011518 Danon disease Diseases 0.000 description 1
- 206010011882 Deafness congenital Diseases 0.000 description 1
- 102000001039 Dystrophin Human genes 0.000 description 1
- 108010069091 Dystrophin Proteins 0.000 description 1
- 108010076282 Factor IX Proteins 0.000 description 1
- 208000001948 Farber Lipogranulomatosis Diseases 0.000 description 1
- 208000033149 Farber disease Diseases 0.000 description 1
- 208000001914 Fragile X syndrome Diseases 0.000 description 1
- 102100030708 GTPase KRas Human genes 0.000 description 1
- 102100037156 Gap junction beta-2 protein Human genes 0.000 description 1
- 102000004366 Glucosidases Human genes 0.000 description 1
- 108010056771 Glucosidases Proteins 0.000 description 1
- 208000001500 Glycogen Storage Disease Type IIb Diseases 0.000 description 1
- 208000035148 Glycogen storage disease due to LAMP-2 deficiency Diseases 0.000 description 1
- 208000018565 Hemochromatosis Diseases 0.000 description 1
- 102000048988 Hemochromatosis Human genes 0.000 description 1
- 108700022944 Hemochromatosis Proteins 0.000 description 1
- 108091005904 Hemoglobin subunit beta Proteins 0.000 description 1
- 208000031220 Hemophilia Diseases 0.000 description 1
- 208000009292 Hemophilia A Diseases 0.000 description 1
- 101150065637 Hfe gene Proteins 0.000 description 1
- 101000823116 Homo sapiens Alpha-1-antitrypsin Proteins 0.000 description 1
- 101001028831 Homo sapiens Cation-independent mannose-6-phosphate receptor Proteins 0.000 description 1
- 101000771083 Homo sapiens Cyclic nucleotide-gated cation channel beta-3 Proteins 0.000 description 1
- 101000967216 Homo sapiens Eosinophil cationic protein Proteins 0.000 description 1
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 1
- 101001003584 Homo sapiens Prelamin-A/C Proteins 0.000 description 1
- 101000712530 Homo sapiens RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 description 1
- 101000801643 Homo sapiens Retinal-specific phospholipid-transporting ATPase ABCA4 Proteins 0.000 description 1
- 101000814438 Homo sapiens Retinoschisin Proteins 0.000 description 1
- 101001087416 Homo sapiens Tyrosine-protein phosphatase non-receptor type 11 Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- 208000000563 Hyperlipoproteinemia Type II Diseases 0.000 description 1
- 102000038455 IGF Type 1 Receptor Human genes 0.000 description 1
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 1
- 102000038460 IGF Type 2 Receptor Human genes 0.000 description 1
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 1
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 1
- 108010001831 LDL receptors Proteins 0.000 description 1
- 108010031099 Mannose Receptor Proteins 0.000 description 1
- 102000019218 Mannose-6-phosphate receptors Human genes 0.000 description 1
- 208000001826 Marfan syndrome Diseases 0.000 description 1
- 206010056893 Mucopolysaccharidosis VII Diseases 0.000 description 1
- 208000025915 Mucopolysaccharidosis type 6 Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 108010082739 NADPH Oxidase 2 Proteins 0.000 description 1
- 208000024818 Nipple disease Diseases 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- 101710112083 Para-Rep C1 Proteins 0.000 description 1
- 102100026531 Prelamin-A/C Human genes 0.000 description 1
- 240000003085 Quassia amara Species 0.000 description 1
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 description 1
- 102100022881 Rab proteins geranylgeranyltransferase component A 1 Human genes 0.000 description 1
- 102100033617 Retinal-specific phospholipid-transporting ATPase ABCA4 Human genes 0.000 description 1
- 102100039507 Retinoschisin Human genes 0.000 description 1
- 102000057028 SOS1 Human genes 0.000 description 1
- 108700022176 SOS1 Proteins 0.000 description 1
- 101100197320 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) RPL35A gene Proteins 0.000 description 1
- 229940122055 Serine protease inhibitor Drugs 0.000 description 1
- 101710102218 Serine protease inhibitor Proteins 0.000 description 1
- 201000001828 Sly syndrome Diseases 0.000 description 1
- 101150100839 Sos1 gene Proteins 0.000 description 1
- 101710119887 Trans-acting factor B Proteins 0.000 description 1
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 1
- 102100033019 Tyrosine-protein phosphatase non-receptor type 11 Human genes 0.000 description 1
- 208000026723 Urinary tract disease Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 208000006110 Wiskott-Aldrich syndrome Diseases 0.000 description 1
- 208000023940 X-Linked Combined Immunodeficiency disease Diseases 0.000 description 1
- 201000001408 X-linked juvenile retinoschisis 1 Diseases 0.000 description 1
- 208000017441 X-linked retinoschisis Diseases 0.000 description 1
- 208000035222 X-linked skeletal dysplasia-intellectual disability syndrome Diseases 0.000 description 1
- 201000004852 Yunis-Varon syndrome Diseases 0.000 description 1
- 201000000761 achromatopsia Diseases 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 208000006682 alpha 1-Antitrypsin Deficiency Diseases 0.000 description 1
- 201000006288 alpha thalassemia Diseases 0.000 description 1
- 230000001475 anti-trypsic effect Effects 0.000 description 1
- 201000004001 aromatic L-amino acid decarboxylase deficiency Diseases 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 208000005980 beta thalassemia Diseases 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- CRQQGFGUEAVUIL-UHFFFAOYSA-N chlorothalonil Chemical compound ClC1=C(Cl)C(C#N)=C(Cl)C(C#N)=C1Cl CRQQGFGUEAVUIL-UHFFFAOYSA-N 0.000 description 1
- 210000002987 choroid plexus Anatomy 0.000 description 1
- 208000003571 choroideremia Diseases 0.000 description 1
- 201000007254 color blindness Diseases 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 229960004222 factor ix Drugs 0.000 description 1
- 201000001386 familial hypercholesterolemia Diseases 0.000 description 1
- 108010054451 glucosamine acetyltransferase Proteins 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000004116 glycogenolysis Effects 0.000 description 1
- 150000003278 haem Chemical class 0.000 description 1
- 102000057877 human IGF2 Human genes 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 208000017482 infantile neuronal ceroid lipofuscinosis Diseases 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 108091009639 insulin receptor binding proteins Proteins 0.000 description 1
- 102000008371 intracellularly ATP-gated chloride channel activity proteins Human genes 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 208000025014 late infantile neuronal ceroid lipofuscinosis Diseases 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 125000000311 mannosyl group Chemical group C1([C@@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 208000000690 mucopolysaccharidosis VI Diseases 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 210000001087 myotubule Anatomy 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 201000000744 recessive dystrophic epidermolysis bullosa Diseases 0.000 description 1
- 201000007714 retinoschisis Diseases 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 239000003001 serine protease inhibitor Substances 0.000 description 1
- 239000009874 shenqi Substances 0.000 description 1
- 208000007056 sickle cell anemia Diseases 0.000 description 1
- 208000025869 skeletal dysplasia-intellectual disability syndrome Diseases 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 150000004044 tetrasaccharides Chemical class 0.000 description 1
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
Images
































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A61K48/0066—Manipulation of the nucleic acid to modify its expression pattern, e.g. enhance its duration of expression, achieved by the presence of particular introns in the delivered nucleic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0075—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the delivery route, e.g. oral, subcutaneous
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/65—Insulin-like growth factors, i.e. somatomedins, e.g. IGF-1, IGF-2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/38—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against protease inhibitors of peptide structure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against enzymes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K19/00—Hybrid peptides, i.e. peptides covalently bound to nucleic acids, or non-covalently bound protein-protein complexes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/24—Hydrolases (3) acting on glycosyl compounds (3.2)
- C12N9/2402—Hydrolases (3) acting on glycosyl compounds (3.2) hydrolysing O- and S- glycosyl compounds (3.2.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y302/00—Hydrolases acting on glycosyl compounds, i.e. glycosylases (3.2)
- C12Y302/01—Glycosidases, i.e. enzymes hydrolysing O- and S-glycosyl compounds (3.2.1)
- C12Y302/0102—Alpha-glucosidase (3.2.1.20)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y302/00—Hydrolases acting on glycosyl compounds, i.e. glycosylases (3.2)
- C12Y302/01—Glycosidases, i.e. enzymes hydrolysing O- and S-glycosyl compounds (3.2.1)
- C12Y302/0105—Alpha-N-acetylglucosaminidase (3.2.1.50)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/02—Fusion polypeptide containing a localisation/targetting motif containing a signal sequence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/06—Fusion polypeptide containing a localisation/targetting motif containing a lysosomal/endosomal localisation signal
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/70—Fusion polypeptide containing domain for protein-protein interaction
- C07K2319/74—Fusion polypeptide containing domain for protein-protein interaction containing a fusion for binding to a cell surface receptor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2750/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssDNA viruses
- C12N2750/00011—Details
- C12N2750/14011—Parvoviridae
- C12N2750/14111—Dependovirus, e.g. adenoassociated viruses
- C12N2750/14141—Use of virus, viral particle or viral elements as a vector
- C12N2750/14143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2840/00—Vectors comprising a special translation-regulating system
- C12N2840/10—Vectors comprising a special translation-regulating system regulates levels of translation
- C12N2840/105—Vectors comprising a special translation-regulating system regulates levels of translation enhancing translation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y301/00—Hydrolases acting on ester bonds (3.1)
- C12Y301/02—Thioester hydrolases (3.1.2)
- C12Y301/02022—Palmitoyl-protein hydrolase (3.1.2.22)
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Immunology (AREA)
- Virology (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurology (AREA)
- Physical Education & Sports Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Provided herein are improved gene therapy vectors and methods of use, which in some embodiments comprise sequences for improving expression and cell targeting of therapeutic proteins.
Description
Cross referencing
The present application claims the benefit of U.S. provisional application No. 62/664,741 filed on 30.4.2018, U.S. provisional application No. 62/688,640 filed on 22.6.2018, and U.S. provisional application No. 62/744,068 filed on 10.10.2018, each of which is incorporated herein by reference in its entirety.
Background
Genetic disorders occur through heritable or nascent mutations that occur in the coding regions of genes of the genome. In some cases, such genetic disorders are treated by administering a protein encoded by a gene that is mutated in an individual having the genetic disorder. However, such treatment is challenging as administration of the protein does not always result in the protein reaching the organ, cell or organelle in which it is needed. In addition, such treatments typically require infusion once every two weeks, whereas gene therapy does not, where a single treatment can provide long-lasting relief. Thus, gene therapy is likely to provide improved results compared to currently available treatments for genetic disorders.
Disclosure of Invention
Provided herein are compositions and methods for treating genetic disorders using gene therapy. Also provided herein are gene therapy vector compositions and methods for gene therapy for improving protein expression and increasing cellular uptake or delivery and intracellular or subcellular targeting of therapeutic proteins provided by gene therapy vectors.
In certain aspects, gene therapy vectors are provided, e.g., gene therapy vectors comprising a nucleic acid construct, the nucleusThe acid construct comprises, in 5 'to 3' order: (a) a translation initiation sequence, and (b) a nucleic acid sequence encoding a therapeutic protein. In some embodiments, the translation initiation sequence comprises a Kozak sequence. In some embodiments, the translation initiation sequence and the nucleic acid sequence encoding the therapeutic protein may overlap such that the last three nucleotides of the translation initiation sequence are also the start codon of the therapeutic protein. In some embodiments, the Kozak sequence comprises the sequence AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide. In some embodiments, X1Contains A. In some embodiments, X2Contains G. In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence differs from the sequence of AAGATGA (SEQ ID NO:29) by one or two nucleotides. In some embodiments, the Kozak sequence comprises AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises a nucleic acid sequence at least 85% identical to GCAAGATG (SEQ ID NO:44), wherein the last three nucleotides (ATG) are also the start codon of the therapeutic protein. In some embodiments, the Kozak sequence differs from the sequence of GCAAGATG (SEQ ID NO:44) by one or two nucleotides. In some embodiments, the Kozak sequence comprises GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to CACCATG (SEQ ID NO: 47). In some embodiments, the Kozak sequence differs from the sequence of CACCATG (SEQ ID NO:47) by one or two nucleotides. In some embodiments, the Kozak sequence comprises CACCATG (SEQ ID NO: 47). In some embodiments, the nucleic acid construct further comprises a nucleic acid sequence encoding a signal peptide capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide. In some embodiments, the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide. In some embodiments, the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17. In some embodiments, the signal peptide is identical to a sequence selected from the group consisting of SEQ ID Nos. 13-17 By 5 or less, 4 or less, 3 or less, 2 or less, or 1 amino acid. In some embodiments, the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17. In some embodiments, the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32. In some embodiments, the signal peptide differs from the sequence of SEQ ID No. 32 by 5 or fewer, 4 or fewer, 3 or fewer, 2 or fewer, or 1 amino acid. In some embodiments, the Gaussia signal peptide comprises the amino acid sequence of SEQ ID NO 32. In some embodiments, the nucleic acid construct further comprises an Internal Ribosome Entry Sequence (IRES). In some embodiments, the IRES is a cricket paralysis virus (CrPV) IRES. In some embodiments, the IRES comprises a nucleic acid sequence having at least 90% identity to SEQ ID NO 12. In some embodiments, the IRES comprises SEQ ID NO 12.
In a further aspect, there is provided a gene therapy vector comprising a nucleic acid construct comprising, in 5 'to 3' order: (a) a nucleic acid sequence encoding a signal peptide, and (b) a nucleic acid sequence encoding a therapeutic protein, wherein the signal peptide is capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide. In some embodiments, the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide. In some embodiments, the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17. In some embodiments, the signal peptide differs from a sequence selected from the group consisting of SEQ ID Nos. 13-17 by 5 or less, 4 or less, 3 or less, 2 or less, or 1 amino acid. In some embodiments, the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17. In some embodiments, the signal peptide comprises a Gaussia signal peptide. In some embodiments, the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32. In some embodiments, the signal peptide differs from the sequence of SEQ ID No. 32 by 5 or fewer, 4 or fewer, 3 or fewer, 2 or fewer, or 1 amino acid. In some embodiments In one embodiment, the Gaussia signal peptide comprises SEQ ID NO 32. In some embodiments, the nucleic acid construct further comprises a translation initiation sequence. In some embodiments, the translation initiation sequence comprises a Kozak sequence comprising AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide. In some embodiments, X1Contains A. In some embodiments, X2Contains G. In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence differs from the sequence of AAGATGA (SEQ ID NO:29) by one or two nucleotides. In some embodiments, the Kozak sequence comprises AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence differs from the sequence of GCAAGATG (SEQ ID NO:44) by one or two nucleotides. In some embodiments, the Kozak sequence comprises GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to CACCATG (SEQ ID NO: 47). In some embodiments, the Kozak sequence differs from the sequence of CACCATG (SEQ ID NO:47) by one or two nucleotides. In some embodiments, the Kozak sequence comprises CACCATG (SEQ ID NO: 47). In some embodiments, the nucleic acid construct further comprises an Internal Ribosome Entry Sequence (IRES). In some embodiments, the IRES comprises an IRES selected from the group consisting of: cricket paralysis virus (CrPV) IRES, picornavirus IRES, foot and mouth disease virus IRES, Kaposi's sarcoma-associated herpesvirus IRES, hepatitis a IRES, hepatitis c IRES, pestivirus IRES, cricket paralysis virus (criptavirus) IRES, Rhopalosiphum palustris (Rhopalosiphum padi) virus IRES, and Merek's disease virus IRES. In some embodiments, the IRES comprises a nucleic acid sequence having at least 90% identity to SEQ ID NO 12. In some embodiments, the IRES comprises SEQ ID NO 12.
In other aspects, gene therapy vectors are provided comprising a nucleic acid construct comprising, in 5 'to 3' orderComprises the following components: (a) an Internal Ribosome Entry Sequence (IRES), and (b) a nucleic acid sequence encoding a therapeutic protein. In some embodiments, the IRES comprises an IRES selected from the group consisting of: cricket paralysis virus (CrPV) IRES, picornavirus IRES, foot and mouth disease virus IRES, Kaposi sarcoma-associated herpesvirus IRES, hepatitis A IRES, hepatitis C IRES, pestivirus IRES, cricket paralysis virus IRES, gloomycosis graminearum virus IRES, and Merrex virus IRES. In some embodiments, the IRES is a cricket paralysis virus (CrPV) IRES. In some embodiments, the IRES comprises a nucleic acid sequence having at least 90% identity to SEQ ID NO 12. In some embodiments, the IRES comprises SEQ ID NO 12. In some embodiments, the nucleic acid construct further comprises a translation initiation sequence. In some embodiments, the translation initiation sequence comprises a Kozak sequence comprising AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide. In some embodiments, X1Contains A. In some embodiments, X 2Contains G. In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence differs from the sequence of GCAAGATG (SEQ ID NO:44) by one or two nucleotides. In some embodiments, the Kozak sequence comprises GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to CACCATG (SEQ ID NO: 47). In some embodiments, the Kozak sequence differs from the sequence of CACCATG (SEQ ID NO:47) by one or two nucleotides. In some embodiments, the Kozak sequence comprises CACCATG (SEQ ID NO: 47). In some embodiments, the nucleic acid construct further comprises a signal nucleic acid sequence encoding a signal peptide capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide. In some embodiments, the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide. In some cases In embodiments, the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17. In some embodiments, the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17. In some embodiments, the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32. In some embodiments, the Gaussia signal peptide comprises SEQ ID NO 32.
In some embodiments, any of the nucleic acid constructs provided herein further comprises a nucleic acid sequence encoding a peptide that selectively binds CI-MPR with high affinity, wherein the therapeutic protein and the peptide that selectively binds CI-MPR are expressed as a fusion protein. In some embodiments, the nucleic acid construct further comprises a sequence encoding a linker peptide between the nucleic acid encoding the peptide that selectively binds to the CI-MPR nucleotide sequence and the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the sequence of the linker peptide may overlap with the sequence of the therapeutic peptide or the sequence of the peptide that selectively binds to CI-MPR, or both. In some embodiments, the peptide that binds with high affinity to CI-MPR is a variant IGF2 peptide (vIGF 2). In some embodiments, the vIGF2 peptide facilitates uptake into cells. In some embodiments, the vIGF2 peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 1-11. In some embodiments, the vIGF2 peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOs 2-11. In some embodiments, the vIGF2 nucleotide sequence is 5' to the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the vIGF2 nucleotide sequence is 3' to the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the nucleic acid construct further comprises a sequence encoding a linker peptide between said vIGF2 nucleotide sequence and said nucleic acid sequence encoding a therapeutic protein. In some embodiments, the linker peptide consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO 18-21, SEQ ID NO 33, or SEQ ID NO 37. In some embodiments, the linker peptide comprises SEQ ID NO 18-21, SEQ ID NO 33, or SEQ ID NO 37. In some embodiments, the therapeutic protein is associated with a lysosomal storage disease. In some embodiments, the therapeutic protein is a lysosomal enzyme or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is selected from the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein is an alpha-galactosidase, or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is an alpha-glucosidase, or an enzymatically active fragment thereof. In some embodiments, the therapeutic protein is a Palmitoyl Protein Thioesterase (PPT), including palmitoyl protein thioesterases 1 and 2 (PPT 1 and PPT2, respectively). In some embodiments, the therapeutic protein is palmitoyl protein thioesterase 1. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the therapeutic protein is associated with a genetic disorder selected from the group consisting of: CDKL5 deficiency, cystic fibrosis, alpha-thalassemia and beta-thalassemia, sickle cell anemia, Marfan syndrome, fragile X syndrome, Huntington's disease, hemochromatosis, congenital deafness (non-syndromic), Tay-saxose (Tay-Sachs), familial hypercholesterolemia, Duchenne muscular dystrophy (Duchenne muscular dystrophy), Stargardt disease (Stargardt disease), User syndrome, choroideremia, achromatopsia, X-linked retinoschisis, hemophilia, Weather-Austenitis syndrome (Wiskott-Aldrich syndrome), X-linked chronic granulomatosis, aromatic L-amino acid decarboxylase deficiency, recessive dystrophic epidermolysis bullosa, alpha 1 antitrypsin deficiency, Husky-early syndrome (HG-early-wish syndrome), Gilson disease, Gilson-Harq syndrome, Gilson-Hardy's disease, Gilson-Daphse syndrome, Gilson syndrome, Sjohnson-induced deficiency, Sjohnson syndrome, S, Noonan syndrome (Noonan syndrome), severe combined immunodeficiency X-SCID. In some embodiments, the therapeutic protein is selected from the group consisting of: CDKL5, connexin 26, hexosidase A, LDL receptor, dystrophin, CFTR, β -globin, HFE, huntingtin, ABCA4, myosin VIIA (MYO7A), Rab convoyin-1 (REP1), cyclic nucleotide-gated channel β 3(CNGB3), retinoschisin 1(RS1), heme subunit β (HBB), factor IX, WAS, cytochrome B-245 β chain, Dopa Decarboxylase (DDC), collagen type VII α 1 chain (COL7a1), serine protease inhibitor family a member 1(SERPINA1), LMNA, PTPN11, SOS1, RAF1, KRAS, and IL2 receptor γ genes. In some embodiments, the therapeutic protein is capable of replacing a defective or deficient protein associated with a genetic disorder in a subject having the genetic disorder. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucosaminuria, Barretch disease, cystinosis, Fabry disease (Fabry disease), Gaucher disease type I (Gaucher disease), Gaucher disease type II, Gaucher disease type III, Pompe disease (Pompe disease), Tay Sachs disease (Tay Sachs disease), Mulberry Hoff disease (Sandhoff disease), metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Herle disease (Hurler disease), Hunter disease (Hunter disease), Sannippo disease type A (Sannnippo disease), Sannippo disease type B, Sannippy disease type C, Murphy disease type D, Morquio disease type A (Morquio disease), Moraxe disease type B, Quassie disease (Mardie disease), and Mentius-Pimpire disease (horse-Pick disease), Nippon disease type A (Pimpinesis disease), Moraxe disease type A (horse disease-Pick disease), Moraxe disease type A-Nipple disease (horse disease-Pick disease), Morchelle disease type A, Sandhoff disease, Sandperce disease, Murphy disease, and Morse type D, Niemann-pick disease type C1, niemann-pick disease type C2, sinderler disease type I (Schindler disease), and sinderler disease type II. In some embodiments, the lysosomal storage disease is selected from the group consisting of: deficiency of activating factor, GM 2-gangliosidosis; GM 2-gangliosidosis, AB variant; alpha-mannosidosis (type 2, moderate form; type 3, neonatal, severe); beta-mannoside storage disease; lysosomal acid lipase deficiency; cystinosis (type of late onset juvenile or juvenile nephropathy; nephropathic nature in infants); Chanarin-Dorfman syndrome (Chanarin-Dorfman syndrome); neutral lipid storage disease with myopathy; NLSDM; danong disease (Danon disease); fabry disease; late onset Fabry disease type II; faber disease (Farber disease); farber fatty granulomatosis; fucoside storage disorders; galactosialistorage disorder (combined neuraminidase and beta-galactosidase deficiency); gaucher disease; gaucher disease type II; gaucher disease type III; gaucher disease type IIIC; atypical gaucher disease due to sphingolipid activator protein C deficiency; GM 1-gangliosidosis (late-onset/juvenile GM 1-gangliosidosis; adult/chronic GM 1-gangliosidosis); globular cell leukodystrophy, Krabbe disease (late onset infant; juvenile onset; adult onset); atypical krabbe's disease due to sphingolipid activator protein a deficiency; metachromatic leukodystrophy (juvenile; adult); partial sulfaterebroside deficiency; pseudoarylsulfatase a deficiency; metachromatic leukodystrophy due to sphingolipid activator B deficiency; mucopolysaccharide storage disorder: MPS I, heler syndrome; MPS I, heler-schey syndrome; MPS I, schey syndrome; MPS II, hunter syndrome; MPS II, hunter syndrome; sanfilippo syndrome/MPS IIIA type a; sanfilippo syndrome/MPS IIIB type B; sanfilippo syndrome/MPS IIIC type C; sanfilippo syndrome/MPS IIID type D; morquio syndrome, type a/MPS IVA; morquio syndrome, type B/MPS IVB; MPS IX hyaluronidase deficiency; MPS VI mallotte-Lamy syndrome (Maroteaux-Lamy syndrome); MPS VII stri syndrome (Sly syndrome); salivary gland disease type I, II mucolipidosis; i-cell disease, Leroy disease (Leroy disease), mucolipidosis II; shampooing malnutrition/mucolipidosis type III; mucolipidosis IIIC/ML III GAMMA; type IV mucolipidosis; polythiol esterase deficiency; niemann-pick disease (type B; type C1/chronic neurological form; type C2; type D/Nova scotia); neuronal ceroid lipofuscinosis: CLN6 disease, atypical late infant, late onset variant, early childhood; Batten-Spielmeyer-Vogt/juvenile NCL/CLN3 disease; finnish variant late infant CLN 5; Jansky-Bielschowsky disease/late infant CLN2/TPP 1; kufs (Kufs)/NCL adult onset/CLN 4 disease (type B); northern epilepsy/variant late infant CLN 8; Santavario-Haldiya (Santavuori-Haltia)/infantile CLN1/PPT disease; pompe disease (glycogen storage disease type II); late onset pompe disease; dense bone developmental disorder; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sinderler disease (type III/moderate, variable); shenqi disease (Kanzaki disease); sala disease (sala disease); infantile asialostorage disease (ISSD); spinal muscular atrophy with progressive myospasm epilepsy (SMAPME); Tay-saxophone/GM 2 gangliosidosis; juvenile onset tay-saxophone disease; late onset tay-saxophone; christian syndrome (Christianson syndrome); laowei (Lowe) eye-brain-kidney syndrome; 4J type Charcot-Marie-dus disease (Charcot-Marie-Tooth), CMT 4J; the Yuris-Varon syndrome (Yunis-Varon syndrome); bilateral temporal occipital polycephalic gyra (BTOP); x-linked hypercalcemic nephrolithiasis, dent-1; and dengue disease 2, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID) and neuronal ceroid lipofuscinosis. In some embodiments, the genetic disorder is pompe disease. In some embodiments, the genetic disorder is neuronal ceroid lipofuscinosis. In some embodiments, the neuronal ceroid lipofuscinosis is selected from the group consisting of: infant NCL (Santaovri-Hardy's disease), advanced infant NCL (Janski-Bilski disease), Barteng disease, adult NCL (Kofski disease), Finland advanced infant NCL, variant advanced infant NCL, CLN7, CLN8, Turkey advanced infant NCL, NCL 9, and CLN 10. In some embodiments, the gene therapy vector is a viral vector. In some embodiments, the viral vector is an adeno-associated viral vector, a retroviral vector, a lentiviral vector, a poxviral vector, a vaccinia viral vector, an adenoviral vector, or a herpesvirus vector. In some embodiments, the viral vector is an AAV vector. In some embodiments, the AAV vector comprises an Inverted Terminal Repeat (ITR). In some embodiments, the AAV vector is selected from the group consisting of: AAV1 vector, AAV2 vector, AAV3 vector, AAV4 vector, AAV5 vector, AAV6 vector, AAV7 vector, AAV8 vector, AAV9 vector, AAVrhS vector, AAVrh10 vector, AAVrh33 vector, AAVrh34 vector, AAVrh74 vector, AAV Anc80 vector, aavphp.b vector, AAVhu68 vector and AAV-DJ vector.
In certain aspects, gene therapy vectors are provided, for example, gene therapy vectors comprising: (a) a nucleic acid sequence encoding a therapeutic protein, and (b) a nucleic acid sequence encoding a peptide that binds with high affinity to CI-MPR. In some embodiments, the peptide is a variant IGF2(vIGF2) peptide. In some embodiments, the vIGF2 peptide comprises an amino acid sequence at least 90% identical to SEQ ID No. 1 and having at least one substitution at one or more positions selected from the group consisting of: positions 6, 26, 27, 43, 48, 49, 50, 54, 55 and 65 of SEQ ID NO. 1. In some embodiments, the at least one substitution is selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R, L55R and K65R of SEQ ID NO. 1. In some embodiments, the vIGF2 peptide comprises at least two substitutions at two or more positions selected from the group consisting of: positions 6, 26, 27, 43, 48, 49, 50, 54 and 55 of SEQ ID NO. 1. In some embodiments, the at least two substitutions are selected from the group consisting ofGroup consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R and L55R of SEQ ID NO. 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at position 1 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-2 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-3 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO:1 and substitutions of E6R, Y27L, and K65R. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO:1 and substitutions E6R and Y27L. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-5 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-6 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-7 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide has reduced or no affinity for insulin receptor and IGF1R compared to the native IGF2 peptide. In some embodiments, the vIGF2 peptide is capable of promoting uptake of a therapeutic protein into a cell. In some embodiments, the vIGF2 peptide is capable of promoting uptake of a therapeutic protein into lysosomes. In some embodiments, the therapeutic protein is capable of replacing a defective or deficient protein associated with a genetic disorder in a subject having the genetic disorder. In some embodiments, the therapeutic protein is a lysosomal enzyme or enzymatically active fragment thereof. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uropathy, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, sandhoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Leehler disease, Hunter disease, Sanfilippo disease type A, Sanfilippo disease type B, Sanfilippo disease type C, Sanfilippon disease type C Ribopathy, Sanfilippo disease type D, Moquine disease type A, Moquine disease type B, Manoto-Lamy disease, Spirosis, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Sindler disease type I, and Sindler disease type II. In some embodiments, the lysosomal storage disease is selected from the group consisting of: deficiency of activating factor, GM 2-gangliosidosis; GM 2-gangliosidosis, AB variant; alpha-mannosidosis (type 2, moderate form; type 3, neonatal, severe); beta-mannoside storage disease; lysosomal acid lipase deficiency; cystinosis (type of late onset juvenile or juvenile nephropathy; nephropathic nature in infants); christian-dofmann syndrome; neutral lipid storage disease with myopathy; NLSDM; disease of agricultural crops; fabry disease; late onset Fabry disease type II; fabry disease; farber fatty granulomatosis; fucoside storage disorders; galactosialistorage disorder (combined neuraminidase and beta-galactosidase deficiency); gaucher disease; gaucher disease type II; gaucher disease type III; gaucher disease type IIIC; atypical gaucher disease due to sphingolipid activator protein C deficiency; GM 1-gangliosidosis (advanced infantile/juvenile GM 1-gangliosidosis; adult/chronic GM 1-gangliosidosis); globulocyte leukodystrophy, krabbe's disease (late onset infant type; juvenile onset type; adult onset type); atypical krabbe's disease due to sphingolipid activator protein a deficiency; metachromatic leukodystrophy (juvenile; adult); partial sulfaterebroside deficiency; pseudoarylsulfatase a deficiency; metachromatic leukodystrophy due to sphingolipid activator B deficiency; mucopolysaccharide storage disorder: MPS I, heler syndrome; MPS I, heler-schey syndrome; MPS I, schey syndrome; MPS II, hunter syndrome; MPS II, hunter syndrome; sanfilippo syndrome/MPS IIIA type a; sanfilippo syndrome/MPS IIIB type B; sanfilippo syndrome/MPS IIIC type C; sanfilippo syndrome/MPS IIID type D; morquio syndrome, type a/MPS IVA; morquio syndrome, type B/MPS IVB; MPS IX hyaluronidase deficiency; MPS VI marlotte-lamide syndrome; MPS VII sri syndrome; salivary gland disease type I, II mucolipidosis; i-cell disease, Leluo-lolo disease, mucolipidosis Storage disorder II; shampooing malnutrition/mucolipidosis type III; mucolipidosis IIIC/ML III GAMMA; type IV mucolipidosis; polythiol esterase deficiency; niemann-pick disease (type B; type C1/chronic neurological form; type C2; type D/Nova scotia); neuronal ceroid lipofuscinosis: CLN6 disease, atypical late infant, late onset variant, early childhood; pasteur/juvenile NCL/CLN3 disease; finnish variant late infant CLN 5; jenski-biziksky disease/late infantile CLN2/TPP1 disease; kuves/adult onset NCL/CLN4 disease (type B); northern epilepsy/variant late infant CLN 8; Santaiwaii-Hardiya/infant CLN1/PPT disease; pompe disease (glycogen storage disease type II); late onset pompe disease; dense bone developmental disorder; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sinderler disease (type III/moderate, variable); qi disease of the Shen; sara disease; infantile asialostorage disease (ISSD); spinal muscular atrophy with progressive myospasm epilepsy (SMAPME); Tay-saxophone/GM 2 gangliosidosis; juvenile onset tay-saxophone disease; late onset tay-saxophone; klissinostian syndrome; lowboy eye-brain-kidney syndrome; type 4J chak-mary-dus disease, CMT 4J; ulis-von syndrome; bilateral temporal occipital multicephalic gyrus (BTOP); x-linked hypercalcemic nephrolithiasis, dent-1; and dengue disease 2, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatous Disease (CGD), and neuronal ceroid lipofuscinosis. In some embodiments, the genetic disorder is pompe disease. In some embodiments, the genetic disorder is neuronal ceroid lipofuscinosis. In some embodiments, the neuronal ceroid lipofuscinosis is selected from the group consisting of: infant NCL (Santaovri-Hardy's disease), advanced infant NCL (Janski-Bilski disease), Barteng disease, adult NCL (Kofski disease), Finland advanced infant NCL, variant advanced infant NCL, CLN7, CLN8, Turkey advanced infant NCL, NCL 9, and CLN 10. In some embodiments, the therapeutic protein is a soluble lysosomal enzyme. In some embodiments, the therapeutic protein comprises a peptide selected from the group consisting of An enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-2-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., beta 0-L), acetyl-CoA: beta 1-glucosaminyl N-acetyltransferase, glycosaminoglycan beta 3-L-iduronal hydrolase, heparan N-sulfatase, N-acetyl-beta 4-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, alpha-galactosidase-galactosaminyl-N-glucosaminidase, beta-glucosaminidase, glucosylcerase, glucosylceramide-N-sulfatase, glucosylcerase, glucosylcera, N-acetylgalactosamine-6-sulfatase, glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, β 5-N-acetylneuraminidase (sialidase), ganglioside sialidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, battenin, palmitoyl protein thioesterase, and other battten-related proteins (e.g., ceroid-lipofuscinosis neuronal protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein is an alpha-glucosidase or an enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the palmitoyl protein thioesterase is a palmitoyl protein thioesterase 1(PPT1) or 2(PPT 2). In some embodiments, the palmitoyl protein thioesterase is palmitoyl protein thioesterase 1. In some embodiments, the nucleic acid construct further comprises a translation initiation sequence. In some embodiments, the translation initiation sequence comprises a Kozak sequence. In some embodiments, the Kozak sequence comprises the sequence AX 1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide. In some embodiments, X1Contains A. In some embodiments, X2Contains G. In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises AAGATGA (SEQ ID NO: 29). At one endIn some embodiments, the Kozak sequence comprises a nucleic acid sequence that is at least 85% identical to GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence differs from the sequence of GCAAGATG (SEQ ID NO:44) by one or two nucleotides. In some embodiments, the Kozak sequence comprises GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to CACCATG (SEQ ID NO: 47). In some embodiments, the Kozak sequence differs from the sequence of CACCATG (SEQ ID NO:47) by one or two nucleotides. In some embodiments, the Kozak sequence comprises CACCATG (SEQ ID NO: 47). In some embodiments, the nucleic acid construct further comprises a nucleic acid sequence encoding a signal peptide, wherein the signal peptide is capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide. In some embodiments, the nucleic acid construct further comprises a nucleic acid sequence encoding a signal peptide, wherein the signal peptide is capable of increasing secretion of the therapeutic protein compared to the therapeutic protein with the native signal peptide. In some embodiments, the nucleic acid construct comprises a nucleic acid sequence encoding a non-native signal peptide, wherein the non-native signal peptide is capable of increasing secretion of the therapeutic protein compared to a native signal peptide of the therapeutic protein. In some embodiments, the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide. In some embodiments, the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17. In some embodiments, the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17. In some embodiments, the signal peptide comprises a Gaussia signal peptide. In some embodiments, the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32. In some embodiments, the Gaussia signal peptide comprises SEQ ID NO 32. In some embodiments, the vIGF2 nucleic acid sequence is 5' to the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the vIGF2 nucleic acid sequence is 3' to the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the nucleic acid construct further comprises a linker peptide encoding between said vIGF2 nucleotide sequence and said nucleic acid sequence encoding a therapeutic protein And (4) sequencing. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker peptide comprises SEQ ID NO 18-21 or SEQ ID NO 33. In some embodiments, the gene therapy vector is a viral vector. In some embodiments, the viral vector is an adenoviral vector, an adeno-associated virus (AAV) vector, a retroviral vector, a lentiviral vector, or a herpesvirus vector. In some embodiments, the viral vector is an AAV vector. In some embodiments, the AAV vector comprises an Inverted Terminal Repeat (ITR). In some embodiments, the AAV vector is selected from the group consisting of: AAV1 vector, AAV2 vector, AAV3 vector, AAV4 vector, AAV5 vector, AAV6 vector, AAV7 vector, AAV8 vector, AAV9 vector, AAVrhS vector, AAVrh10 vector, AAVrh33 vector, AAVrh34 vector, AAVrh74 vector, AAV Anc80 vector, aavphp.b vector, AAVhu68 vector and AAV-DJ vector.
A gene therapy vector comprising a nucleic acid construct comprising: (a) a nucleic acid sequence encoding a therapeutic protein, and (b) a nucleic acid sequence encoding a peptide that increases endocytosis of the therapeutic protein. In some embodiments, the peptide that increases endocytosis of the therapeutic protein is a peptide that binds to CI-MPR. In some embodiments, the peptide is a variant IGF2(vIGF2) peptide, HIRMab or TfRMab, or other cell targeting peptide or protein. In some embodiments, the peptide is vIGF 2. In some embodiments, the vIGF2 peptide comprises an amino acid sequence at least 90% identical to SEQ ID No. 1 and having at least one substitution at one or more positions selected from the group consisting of: positions 6, 26, 27, 43, 48, 49, 50, 54, 55 and 65 of SEQ ID NO. 1. In some embodiments, the at least one substitution is selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R, L55R and K65R of SEQ ID NO. 1. In some embodiments, the at least one substitution is selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R and L55R of SEQ ID NO. 1. In some embodiments, the vI The GF2 peptide comprises at least two substitutions at two or more positions selected from the group consisting of: positions 6, 26, 27, 43, 48, 49, 50, 54 and 55 of SEQ ID NO. 1. In some embodiments, the at least two substitutions are selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R and L55R of SEQ ID NO. 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at position 1 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-6 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-2 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-3 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO:1 and substitutions of E6R, Y27L, and K65R. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO:1 and substitutions E6R and Y27L. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-5 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-6 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-7 of SEQ ID NO: 1. In some embodiments, the vIGF2 peptide has increased specificity for the cation-independent M6P receptor (CI-MPR) compared to the native IGF2 peptide. In some embodiments, the vIGF2 peptide is capable of promoting uptake of a therapeutic protein into lysosomes in cells. In some embodiments, the therapeutic protein is capable of replacing a defective or deficient protein associated with a genetic disorder in a subject having the genetic disorder. In some embodiments, the therapeutic protein is a lysosomal enzyme or enzymatically active fragment thereof. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucosaminuria, Barten, cystinosis, Fabry's disease, gaucher type I, gaucher type II, gaucher type III, Pompe disease, Tay-saxophone disease, sandhoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, holler disease, hunter disease, sanfilippo disease type a, sanfilippo disease type B, sanfilippo disease type C, sanfilippo disease type D, morqui disease type a, morqui disease type B, maroto-lamy disease, sri disease, niemann-pick disease type a, niemann-pick disease type B, niemann-pick disease type C1, niemann-pick disease type C2, sindler disease type I, and sindler disease type II. In some embodiments, the lysosomal storage disease is selected from the group consisting of: deficiency of activating factor, GM 2-gangliosidosis; GM 2-gangliosidosis, AB variant; alpha-mannosidosis (type 2, moderate form; type 3, neonatal, severe); beta-mannoside storage disease; lysosomal acid lipase deficiency; cystinosis (type of late onset juvenile or juvenile nephropathy; nephropathic nature in infants); christian-dofmann syndrome; neutral lipid storage disease with myopathy; NLSDM; disease of agricultural crops; fabry disease; late onset Fabry disease type II; fabry disease; farber fatty granulomatosis; fucoside storage disorders; galactosialistorage disorder (combined neuraminidase and beta-galactosidase deficiency); gaucher disease; gaucher disease type II; gaucher disease type III; gaucher disease type IIIC; atypical gaucher disease due to sphingolipid activator protein C deficiency; GM 1-gangliosidosis (advanced infantile/juvenile GM 1-gangliosidosis; adult/chronic GM 1-gangliosidosis); globulocyte leukodystrophy, krabbe's disease (late onset infant type; juvenile onset type; adult onset type); atypical krabbe's disease due to sphingolipid activator protein a deficiency; metachromatic leukodystrophy (juvenile; adult); partial sulfaterebroside deficiency; pseudoarylsulfatase a deficiency; metachromatic leukodystrophy due to sphingolipid activator B deficiency; mucopolysaccharide storage disorder: MPS I, heler syndrome; MPS I, heler-schey syndrome; MPS I, schey syndrome; MPS II, hunter syndrome; MPS II, hunter syndrome; sanfilippo syndrome/MPS IIIA type a; sanfilippo syndrome/MPS IIIB type B; sanfilippo syndrome/MPS IIIC type C; stiff wave syndrome D MPS IIID; morquio syndrome, type a/MPS IVA; morquio syndrome, type B/MPS IVB; MPS IX hyaluronidase deficiency; MPS VI marlotte-lamide syndrome; MPS VII sri syndrome; salivary gland disease type I, II mucolipidosis; i-cell disease, Leluo disease, mucolipidosis II; shampooing malnutrition/mucolipidosis type III; mucolipidosis IIIC/ML III GAMMA; type IV mucolipidosis; polythiol esterase deficiency; niemann-pick disease (type B; type C1/chronic neurological form; type C2; type D/Nova scotia); neuronal ceroid lipofuscinosis: CLN6 disease, atypical late infant, late onset variant, early childhood; pasteur/juvenile NCL/CLN3 disease; finnish variant late infant CLN 5; jenski-biziksky disease/late infantile CLN2/TPP1 disease; kuves/adult onset NCL/CLN4 disease (type B); northern epilepsy/variant late infant CLN 8; Santaiwaii-Hardiya/infant CLN1/PPT disease; pompe disease (glycogen storage disease type II); late onset pompe disease; dense bone developmental disorder; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sinderler disease (type III/moderate, variable); qi disease of the Shen; sara disease; infantile asialostorage disease (ISSD); spinal muscular atrophy with progressive myospasm epilepsy (SMAPME); Tay-saxophone/GM 2 gangliosidosis; juvenile onset tay-saxophone disease; late onset tay-saxophone; klissinostian syndrome; lowboy eye-brain-kidney syndrome; type 4J chak-mary-dus disease, CMT 4J; ulis-von syndrome; bilateral temporal occipital multicephalic gyrus (BTOP); x-linked hypercalcemic nephrolithiasis, dent-1; and dengue disease 2, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatous Disease (CGD), and neuronal ceroid lipofuscinosis. In some embodiments, the genetic disorder is pompe disease. In some embodiments, the genetic disorder is neuronal ceroid lipofuscinosis. In some embodiments, the neuronal ceroid lipofuscinosis is selected from the group consisting of: infantile NCL (Santaooli-Hardy disease), advanced NCL (Janski-Bierwolski disease), Barteng disease, adult NCL (Kofski disease), Finnish late infant NCL, variant late infant NCL, CLN7, CLN8, Turkey late infant NCL, NCL 9 and CLN 10. In some embodiments, the therapeutic protein is a soluble lysosomal enzyme or enzymatically active fragment thereof. In some embodiments, the therapeutic protein comprises an enzyme selected from the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-2-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., beta 0-L), acetyl-CoA: beta 1-glucosaminyl N-acetyltransferase, glycosaminoglycan beta 3-L-iduronal hydrolase, heparan N-sulfatase, N-acetyl-beta 4-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, alpha-galactosidase-galactosaminyl-N-glucosaminidase, beta-glucosaminidase, glucosylcerase, glucosylceramide-N-sulfatase, glucosylcerase, glucosylcera, N-acetylgalactosamine-6-sulfatase, glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, β 5-N-acetylneuraminidase (sialidase), ganglioside sialidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, battenin, palmitoyl protein thioesterase, and other battten-related proteins (e.g., ceroid-lipofuscinosis neuronal protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein is an alpha-glucosidase, or an enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the palmitoyl protein thioesterase is a palmitoyl protein thioesterase 1(PPT1) or 2(PPT 2). In some embodiments, the palmitoyl protein thioesterase is palmitoyl protein thioesterase 1. In some embodiments, the nucleic acid construct further comprises a translation initiation sequence. In some embodiments, the translation initiation sequence comprises a Kozak sequence. In some embodiments, the Kozak sequence comprises the sequence AX 1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide. In some casesIn the embodiment, X1Contains A. In some embodiments, X2Contains G. In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence differs from the sequence of GCAAGATG (SEQ ID NO:44) by one or two nucleotides. In some embodiments, the Kozak sequence comprises GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to CACCATG (SEQ ID NO: 47). In some embodiments, the Kozak sequence differs from the sequence of CACCATG (SEQ ID NO:47) by one or two nucleotides. In some embodiments, the Kozak sequence comprises CACCATG (SEQ ID NO: 47). In some embodiments, the nucleic acid construct further comprises a signal nucleic acid sequence encoding a signal peptide, wherein the signal peptide is capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide. In some embodiments, the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide. In some embodiments, the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17. In some embodiments, the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17. In some embodiments, the signal peptide comprises a Gaussia signal peptide. In some embodiments, the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32. In some embodiments, the Gaussia signal peptide comprises SEQ ID NO 32. In some embodiments, the vIGF2 nucleic acid sequence is 5' to the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the vIGF2 nucleic acid sequence is 3' to the nucleic acid sequence encoding the therapeutic protein. In some embodiments, the nucleic acid construct further comprises a linker sequence encoding a linker peptide between said vIGF2 nucleotide sequence and said nucleic acid sequence encoding a therapeutic protein. In some embodiments, the linker is composed of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids Amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker peptide comprises SEQ ID NO 18-21 or SEQ ID NO 33. In some embodiments, the gene therapy vector is a viral vector. In some embodiments, the viral vector is an adenoviral vector, an adeno-associated virus (AAV) vector, a retroviral vector, a lentiviral vector, or a herpesvirus vector. In some embodiments, the viral vector is an AAV vector. In some embodiments, the AAV vector comprises an Inverted Terminal Repeat (ITR). In some embodiments, the AAV vector is selected from the group consisting of: AAV1 vector, AAV2 vector, AAV3 vector, AAV4 vector, AAV5 vector, AAV6 vector, AAV7 vector, AAV8 vector, AAV9 vector, AAVrhS vector, AAVrh10 vector, AAVrh33 vector, AAVrh34 vector, AAVrh74 vector, AAV Anc80 vector, aavphp.b vector, AAVhu68 vector and AAV-DJ vector.
In a further aspect, there is provided a pharmaceutical composition comprising (i) a therapeutically effective amount of any one of the gene therapy vectors herein, and (ii) a pharmaceutically acceptable carrier or excipient. In some embodiments, the carrier or excipient comprises a non-ionic, low-permeability compound, buffer, polymer, salt, or combination thereof.
In other aspects, methods are provided for treating a genetic disorder, the methods comprising administering to a subject in need thereof any one of the gene therapy vectors provided herein or any one of the pharmaceutical compositions provided herein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine diabetes, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, MuldoHoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Hewler disease, Hunter disease, Sanfilippo disease type A, Sanfilippo disease type B, Sanfilippo disease type C, Sanfilippo disease type D, Moquini disease type A, Moquine disease type B, Maruto-Lami disease, Sprius disease, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Ocimir disease type I and Ocimier disease type II. In some embodiments, the lysosomal storage disease is selected from the group consisting of: deficiency of activating factor, GM 2-gangliosidosis; GM 2-gangliosidosis, AB variant; alpha-mannosidosis (type 2, moderate form; type 3, neonatal, severe); beta-mannoside storage disease; lysosomal acid lipase deficiency; cystinosis (type of late onset juvenile or juvenile nephropathy; nephropathic nature in infants); christian-dofmann syndrome; neutral lipid storage disease with myopathy; NLSDM; disease of agricultural crops; fabry disease; late onset Fabry disease type II; fabry disease; farber fatty granulomatosis; fucoside storage disorders; galactosialistorage disorder (combined neuraminidase and beta-galactosidase deficiency); gaucher disease; gaucher disease type II; gaucher disease type III; gaucher disease type IIIC; atypical gaucher disease due to sphingolipid activator protein C deficiency; GM 1-gangliosidosis (advanced infantile/juvenile GM 1-gangliosidosis; adult/chronic GM 1-gangliosidosis); globulocyte leukodystrophy, krabbe's disease (late onset infant type; juvenile onset type; adult onset type); atypical krabbe's disease due to sphingolipid activator protein a deficiency; metachromatic leukodystrophy (juvenile; adult); partial sulfaterebroside deficiency; pseudoarylsulfatase a deficiency; metachromatic leukodystrophy due to sphingolipid activator B deficiency; mucopolysaccharide storage disorder: MPS I, heler syndrome; MPS I, heler-schey syndrome; MPS I, schey syndrome; MPS II, hunter syndrome; MPS II, hunter syndrome; sanfilippo syndrome/MPS IIIA type a; sanfilippo syndrome/MPS IIIB type B; sanfilippo syndrome/MPS IIIC type C; sanfilippo syndrome/MPS IIID type D; morquio syndrome, type a/MPS IVA; morquio syndrome, type B/MPS IVB; MPS IX hyaluronidase deficiency; MPS VI marlotte-lamide syndrome; MPS VII sri syndrome; salivary gland disease type I, II mucolipidosis; i-cell disease, Leluo disease, mucolipidosis II; shampooing malnutrition/mucolipidosis type III; mucolipidosis IIIC/ML III GAMMA; type IV mucolipidosis; polythiol esterase deficiency; niemann-pick disease (type B; type C1/chronic neurological form; type C2; type D/Nova scotia); neuronal ceroid lipofuscinosis: CLN6 disease, atypical late infant, late onset variant, early childhood; pasteur/juvenile NCL/CLN3 disease; finnish variant late infant CLN 5; jenski-biziksky disease/late infantile CLN2/TPP1 disease; kuves/adult onset NCL/CLN4 disease (type B); northern epilepsy/variant late infant CLN 8; Santaiwaii-Hardiya/infant CLN1/PPT disease; pompe disease (glycogen storage disease type II); late onset pompe disease; dense bone developmental disorder; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sinderler disease (type III/moderate, variable); qi disease of the Shen; sara disease; infantile asialostorage disease (ISSD); spinal muscular atrophy with progressive myospasm epilepsy (SMAPME); Tay-saxophone/GM 2 gangliosidosis; juvenile onset tay-saxophone disease; late onset tay-saxophone; klissinostian syndrome; lowboy eye-brain-kidney syndrome; type 4J chak-mary-dus disease, CMT 4J; ulis-von syndrome; bilateral temporal occipital multicephalic gyrus (BTOP); x-linked hypercalcemic nephrolithiasis, dent-1; and dengue disease 2, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatous Disease (CGD), CDKL5 deficiency, and neuronal ceroid lipofuscinosis. In some embodiments, the genetic disorder is pompe disease. In some embodiments, the genetic disorder is neuronal ceroid lipofuscinosis. In some embodiments, the neuronal ceroid lipofuscinosis is selected from the group consisting of: infant NCL (Santaovri-Hardy's disease), advanced infant NCL (Janski-Bilski disease), Barteng disease, adult NCL (Kofski disease), Finland advanced infant NCL, variant advanced infant NCL, CLN7, CLN8, Turkey advanced infant NCL, NCL 9, and CLN 10. In some embodiments, administration is intrathecal, intraocular, intravitreal, transretinal, intravenous, intramuscular, intraventricular, intracerebral, intracerebellar, intracerebroventricular, intraparenchymal, intracameral, subcutaneous, or a combination thereof. In some embodiments, administration is performed intrathecally. In some embodiments, administration is intraocular, intravitreal, or transretinal.
In a further aspect, there is provided a pharmaceutical composition comprising any one of the gene therapy vectors herein and a pharmaceutically acceptable carrier or excipient for use in the treatment of a genetic disorder. In other aspects, pharmaceutical compositions are provided comprising any one of the gene therapy vectors herein and a pharmaceutically acceptable carrier or excipient for use in the preparation of a medicament for the treatment of a genetic disorder. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine diabetes, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, MuldoHoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Hewler disease, Hunter disease, Sanfilippo disease type A, Sanfilippo disease type B, Sanfilippo disease type C, Sanfilippo disease type D, Moquine disease type A, Moquine disease type B, Maruto-Lami disease, Sprius disease, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Sinidel disease type I and Sindler disease type II. In some embodiments, the lysosomal storage disease is selected from the group consisting of: deficiency of activating factor, GM 2-gangliosidosis; GM 2-gangliosidosis, AB variant; alpha-mannosidosis (type 2, moderate form; type 3, neonatal, severe); beta-mannoside storage disease; lysosomal acid lipase deficiency; cystinosis (type of late onset juvenile or juvenile nephropathy; nephropathic nature in infants); christian-dofmann syndrome; neutral lipid storage disease with myopathy; NLSDM; disease of agricultural crops; fabry disease; late onset Fabry disease type II; fabry disease; farber fatty granulomatosis; fucoside storage disorders; galactosialistorage disorder (combined neuraminidase and beta-galactosidase deficiency); gaucher disease; gaucher disease type II; gaucher disease type III; gaucher disease type IIIC; atypical gaucher disease due to sphingolipid activator protein C deficiency; GM 1-gangliosidosis (advanced infantile/juvenile GM 1-gangliosidosis; adult/chronic GM 1-gangliosidosis); globulocyte leukodystrophy, krabbe's disease (late onset infant type; juvenile onset type; adult onset type); atypical krabbe's disease due to sphingolipid activator protein a deficiency; metachromatic leukodystrophy (juvenile; adult); partial sulfaterebroside deficiency; pseudoarylsulfatase a deficiency; metachromatic leukodystrophy due to sphingolipid activator B deficiency; mucopolysaccharide storage disorder: MPS I, heler syndrome; MPS I, heler-schey syndrome; MPS I, schey syndrome; MPS II, hunter syndrome; MPS II, hunter syndrome; sanfilippo syndrome/MPS IIIA type a; sanfilippo syndrome/MPS IIIB type B; sanfilippo syndrome/MPS IIIC type C; sanfilippo syndrome/MPS IIID type D; morquio syndrome, type a/MPS IVA; morquio syndrome, type B/MPS IVB; MPS IX hyaluronidase deficiency; MPS VI marlotte-lamide syndrome; MPS VII sri syndrome; salivary gland disease type I, II mucolipidosis; i-cell disease, Leluo disease, mucolipidosis II; shampooing malnutrition/mucolipidosis type III; mucolipidosis IIIC/ML III GAMMA; type IV mucolipidosis; polythiol esterase deficiency; niemann-pick disease (type B; type C1/chronic neurological form; type C2; type D/Nova scotia); neuronal ceroid lipofuscinosis: CLN6 disease, atypical late infant, late onset variant, early childhood; pasteur/juvenile NCL/CLN3 disease; finnish variant late infant CLN 5; jenski-biziksky disease/late infantile CLN2/TPP1 disease; kuves/adult onset NCL/CLN4 disease (type B); northern epilepsy/variant late infant CLN 8; Santaiwaii-Hardiya/infant CLN1/PPT disease; pompe disease (glycogen storage disease type II); late onset pompe disease; dense bone developmental disorder; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sinderler disease (type III/moderate, variable); qi disease of the Shen; sara disease; infantile asialostorage disease (ISSD); spinal muscular atrophy with progressive myospasm epilepsy (SMAPME); Tay-saxophone/GM 2 gangliosidosis; juvenile onset tay-saxophone disease; late onset tay-saxophone; klissinostian syndrome; lowboy eye-brain-kidney syndrome; type 4J chak-mary-dus disease, CMT 4J; ulis-von syndrome; bilateral temporal occipital multicephalic gyrus (BTOP); x-linked hypercalcemic nephrolithiasis, dent-1; and dengue disease 2, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatous Disease (CGD), CDKL5 deficiency, and neuronal ceroid lipofuscinosis. In some embodiments, the genetic disorder is pompe disease. In some embodiments, the genetic disorder is neuronal ceroid lipofuscinosis. In some embodiments, the neuronal ceroid lipofuscinosis is selected from the group consisting of: infant NCL (Santaovri-Hardy's disease), advanced infant NCL (Janski-Bilski disease), Barteng disease, adult NCL (Kofski disease), Finland advanced infant NCL, variant advanced infant NCL, CLN7, CLN8, Turkey advanced infant NCL, NCL 9, and CLN 10. In some embodiments, the composition is formulated for intrathecal, intraocular, intravitreal, transretinal, intravenous, intramuscular, intraventricular, intracerebral, intracerebellar, ocular, or subcutaneous administration. In some embodiments, the composition is formulated for intrathecal administration. In some embodiments, the composition is formulated for intrathecal administration for the treatment of a neurodegenerative disorder. In some embodiments, the composition is formulated for ocular, intravitreal, or transretinal administration.
Provided herein are gene therapy vectors comprising a nucleic acid construct encoding a polypeptide comprising: (a) a therapeutic protein; (b) a peptide that binds with high affinity to cation-independent mannose 6-phosphate (M6P) receptor (CI-MPR); and (c) a linker between the therapeutic protein and the CI-MPR binding peptide. In some embodiments, the peptide is a variant IGF2(vIGF2) peptide. In some embodiments, the vIGF2 peptide comprises an amino acid sequence at least 90% identical to SEQ ID No. 1 and having at least one substitution at one or more positions selected from the group consisting of: positions 6, 26, 27, 43, 48, 49, 50, 54, 55 and 65 of SEQ ID NO. 1. In some embodiments, the at least one substitution is selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R, L55R and K65R of SEQ ID NO. 1. In some embodiments, the vIGF2 peptide comprises at least two substitutions at two or more positions selected from the group consisting of: 1, positions 6, 26, 27, 43, 48, 49, 50, 54, 55, 65. In some embodiments, the at least two substitutions are selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R, L55R, K65R of SEQ ID NO. 1. In some embodiments, the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID NO: 1. In some embodiments, wherein the vIGF2 peptide has reduced affinity for insulin receptor and IGF1R compared to the native IGF2 peptide. In some embodiments, the vIGF2 peptide is capable of promoting uptake of the therapeutic protein into a cell. In some embodiments, the vIGF2 peptide is capable of promoting uptake of the therapeutic protein into lysosomes. In some embodiments, the therapeutic protein is capable of replacing a defective or deficient protein associated with a genetic disorder in a subject having the genetic disorder. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Hewlett-packard disease, Hunter disease, sanfilippo disease type A, sanfilippo disease type B, sanfilippo disease type C, sanfilippo disease type D, moquinase type A, moquinase type B, maratoto-lamic disease, sjgren disease, niemann-pick disease type A, niemann-pick disease type B, niemann-pick disease type C1, niemann-pick disease type C2, sinderler disease type I, sinderler disease type II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatosis (CGD), and neuronal ceroid lipofuscinosis. In some embodiments, the genetic disorder is pompe disease. In some embodiments, the genetic disorder is CLN1 disease. In some embodiments, the therapeutic protein comprises a soluble lysosomal enzyme or enzymatically active fragment thereof. In some embodiments, the therapeutic protein comprises a lysosomal enzyme or enzymatically active fragment thereof, wherein the lysosomal enzyme is selected from the group consisting of: alpha-galactosidase A, beta-glucocerebrosidase, lysosomal acid lipase, glycosaminoglycan alpha-L-iduronate hydrolase, iduronate-2-sulfatase, N-acetylgalactosamine-6-sulfatase, glycosaminoglycan N-acetylgalactosamine 4-sulfatase, palmitoyl protein thioesterase, cyclin-dependent kinase-like 5, and alpha-glucosidase. In some embodiments, the therapeutic protein is an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is a palmitoyl protein thioesterase or an enzymatically active fragment thereof. In some embodiments, the therapeutic protein is palmitoyl protein thioesterase-1 or an enzymatically active fragment thereof. In some embodiments, the nucleic acid construct further comprises a translation initiation sequence. In some embodiments, the translation initiation sequence comprises a Kozak sequence. In some embodiments, the nucleic acid construct further comprises a nucleic acid sequence encoding a signal peptide, wherein the signal peptide is capable of increasing secretion of a therapeutic protein compared to the therapeutic protein without the signal peptide. In some embodiments, the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide. In some embodiments, the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17. In some embodiments, the vIGF2 peptide comprises the sequence of SEQ ID No. 31. In some embodiments, the construct comprises SEQ ID NO 36. In some embodiments, the polypeptide comprises SEQ ID NO 23. In some embodiments, the construct comprises SEQ ID NO 38. In some embodiments, vIGF2 is at the N-terminus of the polypeptide. In some embodiments, vIGF2 is C-terminal to the polypeptide. In some embodiments, the linker peptide comprises SEQ ID NO 18-21 or SEQ ID NO 33. In some embodiments, the gene therapy vector is a viral vector selected from the group consisting of: adenoviral vectors, adeno-associated virus (AAV) vectors, retroviral vectors, lentiviral vectors, poxvirus vectors, vaccinia virus vectors, adenoviral vectors, and herpesvirus vectors.
In certain aspects, fusion proteins, e.g., fusion proteins comprising a variant IGF2 peptide and a therapeutic protein, are provided. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37, also referred to herein as "2 GS") or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein is encoded by a nucleic acid comprising a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the fusion protein is encoded by a nucleic acid comprising a Kozak sequence.
In a further aspect, a fusion protein is provided comprising a signal peptide and a therapeutic protein, wherein the signal peptide is post-translationally removed upon secretion from a cell. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the fusion protein is encoded by a nucleic acid comprising a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the fusion protein is encoded by a nucleic acid comprising a Kozak sequence.
In other aspects, a nucleic acid sequence encoding a fusion protein comprising a therapeutic protein is provided, wherein the fusion protein is encoded by a nucleic acid comprising a cricket palsy virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the fusion protein is encoded by a nucleic acid comprising a Kozak sequence.
In a further aspect, a fusion protein comprising a therapeutic protein is provided, wherein the fusion protein is encoded by a nucleic acid comprising a Kozak sequence. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the fusion protein is encoded by a nucleic acid comprising a cricket paralysis virus internal ribosome entry sequence (CrPV IRES).
In a further aspect, there is provided a nucleic acid encoding a fusion protein, for example a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and a therapeutic protein. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus.
In other aspects, nucleic acids encoding fusion proteins comprising a signal peptide and a therapeutic protein are provided. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus.
In a further aspect, there is provided a nucleic acid encoding a fusion protein comprising a therapeutic protein, wherein the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus.
In other aspects, there is provided a nucleic acid encoding a fusion protein comprising a therapeutic protein, wherein the nucleic acid further comprises a Kozak sequence. In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminide N-acetyltransferase, glycosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminide (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, N-acetyl-alpha-D-glucosaminide (NAGLU), N-acetyl-glucosaminyl-2-sulfatase, alpha-L-glucosaminyl-6-sulfatase, N-acetyl-sulfatase, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminide (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus.
In a further aspect, there is provided a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein.
In a further aspect, there is provided a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a signal peptide and a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein.
In other aspects, compositions are provided, comprising: (a) a nucleic acid encoding a fusion protein comprising a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy, wherein the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein.
In other aspects, compositions are provided, comprising: (a) a nucleic acid encoding a fusion protein comprising a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy, wherein the nucleic acid further comprises a Kozak sequence. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein.
In a further aspect, there is provided a method of treating a genetic disorder in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, cells from an individual are treated ex vivo, and the cells are administered to the individual after ex vivo treatment.
In a further aspect, there is provided a method of treating a genetic disorder in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a signal peptide and a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, cells from an individual are treated ex vivo, and the cells are administered to the individual after ex vivo treatment.
In other aspects, there is provided a method of treating a genetic disorder in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a therapeutic protein and a targeting peptide; and (b) a buffer or excipient suitable for gene therapy, wherein the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, cells from an individual are treated ex vivo, and the cells are administered to the individual after ex vivo treatment.
In a further aspect, there is provided a method of treating a genetic disorder in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a therapeutic protein; and (b) a buffer or excipient suitable for gene therapy, wherein the nucleic acid further comprises a Kozak sequence. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, cells from an individual are treated ex vivo, and the cells are administered to the individual after ex vivo treatment.
In other aspects, methods of treating a genetic disorder in an individual are provided, the methods comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and a therapeutic protein. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, the cell is derived from an individual.
In a further aspect, there is provided a method of treating a genetic disorder in an individual, the method comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a signal peptide and a therapeutic protein. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, the cell is derived from an individual.
In a further aspect, there is provided a method of treating a genetic disorder in an individual, the method comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a therapeutic protein, wherein the nucleic acid further comprises a cricket palsy virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the nucleic acid further comprises a Kozak sequence. In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient suitable for gene therapy comprises a liposome, a nanoparticle, or a cell penetrating peptide. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, the cell is derived from an individual.
In other aspects, methods of treating a genetic disorder in an individual are provided, the methods comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a therapeutic protein, wherein the nucleic acid further comprises a Kozak sequence. In some embodiments, the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES). In some embodiments, the fusion protein further comprises a linker. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 7, 8, 9, 10, 11, 12, or 13 amino acids. In some embodiments, the linker comprises the amino acid sequence of GGGGSGGGG (SEQ ID NO:18), GGGGS (SEQ ID NO:19), GGGSGGGGS (SEQ ID NO:20), GGGGSGGGS (SEQ ID NO:21), GGGGSGGGGS (SEQ ID NO:37), or GGSGSGSTS (SEQ ID NO: 33). In some embodiments, the fusion protein further comprises a signal peptide. In some embodiments, the signal peptide comprises a Binding Immunoglobulin Protein (BiP) signal peptide. In some embodiments, the fusion protein further comprises a variant IGF2 peptide. In some embodiments, the therapeutic protein comprises at least one enzyme of the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof. In some embodiments, the therapeutic protein comprises an alpha-glucosidase or enzymatically active fragment thereof. In some embodiments, the therapeutic protein is N-acetyl- α -D-glucosaminidase (NAGLU). In some embodiments, the nucleic acid further comprises a promoter. In some embodiments, the nucleic acid is contained within a viral vector. In some embodiments, the viral vector comprises a retrovirus, adenovirus, adeno-associated virus, lentivirus, or herpes virus. In some embodiments, the buffer or excipient is suitable for gene therapy. In some embodiments, the buffer or excipient suitable for gene therapy comprises a viral coat protein. In some embodiments, the viral coat protein is selected from the group consisting of: vesicular stomatitis virus coat protein, adenovirus coat protein, adeno-associated virus coat protein, murine leukemia virus coat protein, HIV coat protein, and influenza virus coat protein. In some embodiments, the genetic disorder is a lysosomal storage disorder. In some embodiments, the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease A, san Francisella disease B, san Francisella disease C, san Francisella disease D, morqui disease A, morqui disease B, Maruo-lamide disease, Spirosis, Niemann-pick disease A, Niemann-pick disease B, Niemann-pick disease C1, Niemann-pick disease C2, Sindler disease I, Sindler disease II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), and Chronic Granulomatosis (CGD). In some embodiments, the cell is derived from an individual.
Also provided herein is a fusion protein comprising a native signal peptide, an ER protein cleavage domain, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide, wherein the fusion protein is encoded by a nucleic acid comprising a Kozak sequence.
Further provided herein is a fusion protein comprising a Binding Immunoglobulin Protein (BiP) signal peptide, a variant IGF2 peptide, and an alpha-glucosidase, wherein the fusion protein is encoded by a nucleic acid comprising a Kozak sequence.
Further provided herein is a fusion protein comprising an immunoglobulin protein (BiP) binding signal peptide, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide, wherein the fusion protein is encoded by a nucleic acid comprising a cricket paralysis virus internal ribosome entry sequence (CrPV IRES).
Further provided herein is a nucleic acid encoding a fusion protein comprising a native signal peptide, an ER protein cleavage domain, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide.
Further provided herein is a nucleic acid encoding a fusion protein comprising an immunoglobulin protein (BiP) binding signal peptide, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide, wherein the nucleic acid further comprises a Kozak sequence.
Further provided herein is a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and an alpha-glucosidase, wherein the nucleic acid further comprises a cricket paralysis virus internal ribosome entry sequence (CrPV IRES).
Further provided herein is a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a native signal peptide, an ER protein cleavage domain, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide; and (b) a buffer or excipient suitable for gene therapy.
Further provided herein is a composition comprising: (a) a nucleic acid encoding a fusion protein comprising an immunoglobulin protein (BiP) -binding signal peptide, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide, wherein the nucleic acid further comprises a Kozak sequence; and (b) a buffer or excipient suitable for gene therapy.
Further provided herein is a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and an alpha-glucosidase, wherein the nucleic acid further comprises a cricket palsy virus internal ribosome entry sequence (CrPV IRES); and (b) a buffer or excipient suitable for gene therapy.
Further provided herein is a method of treating pompe disease in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a native signal peptide, an ER protein cleavage domain, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide; and (b) a buffer or excipient suitable for gene therapy.
Further provided herein is a method of treating pompe disease in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising an immunoglobulin protein (BiP) -binding signal peptide, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide, wherein the nucleic acid further comprises a Kozak sequence; and (b) a buffer or excipient suitable for gene therapy.
Further provided herein is a method of treating pompe disease in an individual, the method comprising administering a composition comprising: (a) a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and an alpha-glucosidase, wherein the nucleic acid further comprises a cricket palsy virus internal ribosome entry sequence (CrPV IRES); and (b) a buffer or excipient suitable for gene therapy.
Further provided herein is a method of treating pompe disease in an individual comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a native signal peptide, an ER protein cleavage domain, a variant IGF2 peptide, and an alpha-glucosidase lacking its native signal peptide.
Further provided herein is a method of treating pompe disease in an individual, the method comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a Binding Immunoglobulin Protein (BiP) signal peptide, a variant IGF2 peptide, and an alpha-glucosidase or enzymatically active fragment thereof, wherein the nucleic acid further comprises a Kozak sequence.
Further provided herein is a method of treating pompe disease in an individual, the method comprising administering a cell comprising a nucleic acid encoding a fusion protein comprising a variant IGF2 peptide and an alpha-glucosidase, wherein the nucleic acid further comprises a cricket palsy virus internal ribosome entry sequence (CrPV IRES).
Is incorporated by reference
All publications, patents, and patent applications mentioned in this specification are herein incorporated by reference to the same extent as if each individual publication, patent, or patent application was specifically and individually indicated to be incorporated by reference.
Drawings
The patent application document contains at least one drawing executed in color. Copies of this patent application with color drawing(s) will be provided by the office upon request and payment of the necessary fee. An understanding of the features and advantages of the present disclosure will be obtained with reference to the following detailed description that sets forth illustrative embodiments, in which the principles of the disclosure are utilized, and the accompanying drawings of which:
figure 1 shows GAA activity of the arabinosidase-alpha rhGAA with and without M6P. FIG. 1 shows the scale of a commercial ERT that can incorporate CI-MPR. The first peak is rhGAA that lacks any M6P-containing glycans and therefore cannot be taken up and delivered to lysosomes. The second peak is a fraction containing at least one phosphorylated glycan and having the potential to be taken up by cells and delivered to lysosomes for glycogenolysis.
Figure 2 shows the structure of CI-MPR comprising different binding domains of IGF2 and mono-and di-phosphorylated oligosaccharides.
Figure 3 shows the sequence and structure of mature human IGF2 peptide. Site-specific amino acid substitutions are proposed to affect the binding of other receptors.
Figure 4 shows the binding of wild-type IGF2(wtIGF2) peptide to CI-MPR as measured by surface plasmon resonance.
Figure 5 shows the binding of variant IGF2(vIGF2) peptide bound to CI-MPR as measured by surface plasmon resonance.
Figure 6 shows the addition of vIGF2 to glucosidase a to increase the benefit of binding to IGF 2/CI-MPR.
Figure 7 shows the benefit of adding vIGF2 to recombinant human N-acetyl- α -D-glucamine enzyme (rhNAGLU) to increase binding to IGF 2/CI-MPR.
Figure 8 shows the binding of wild-type human IGF2 to the insulin receptor.
Fig. 9 shows no detectable binding of vIGF2 to the insulin receptor.
Figure 10 shows the binding of wild-type IGF2 to insulin-like growth factor 1 receptor.
Figure 11 shows that vIGF2 peptide has reduced binding to insulin-like growth factor 1 receptor compared to wild-type IGF 2.
FIG. 12 shows two examples of gene therapy expression cassettes encoding native hGAA and engineered hGAA. Native hGAA is poorly phosphorylated, resulting in poor CIMPR binding and cellular uptake. The engineered hGAA has the addition of an element for improved cimr binding (vIGF2), a 2GS linker that reduces the steric hindrance of vIGF2-GAA protein to cimr, and a BiP signal peptide for improved secretion.
Figure 13 shows western blots of PPT1 from cells expressing recombinant human PPT1(PPT1-1), recombinant human PPT1 with a vIGF2 targeting domain (PPT1-2), and recombinant human PPT1 with a vIGF2 targeting domain and a BiP signal sequence (PPT 1-29).
Figure 14 shows binding of PPT1 construct to CI-MPR.
FIG. 15 shows GAA activity in conditioned media of CHO cells expressing engineered or native hGAA.
Figure 16 shows the study design of a 4-week gene therapy mouse study in GAA knockout mice.
Figure 17 shows GAA plasma activity in untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles as indicated.
Figure 18 shows GAA levels measured in untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles as indicated.
Figure 19 shows cell surface receptor binding of rhGAA from plasma samples obtained from treated mice as indicated.
Figure 20 shows GAA activity and tetrasaccharide histopathology scores of the tibialis anterior of untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles as indicated.
Figure 21 shows glycogen PAS from the tibialis anterior as indicated from untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 22 shows hGAA immunohistochemistry as indicated for tibialis anterior from untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 23 shows histopathological scores of brain GAA activity, brain glycogen and spinal cord glycogen as indicated from brain and spinal cord of untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 24 shows glycogen PAS from brain of untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles as indicated.
Figure 25 shows hGAA immunohistochemistry for brainstem and choroid plexus as indicated from untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 26 shows glycogen PAS from spinal cord of untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles as indicated.
Figure 27 shows hGAA immunohistochemistry as indicated from spinal cord of untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 28 shows quadriceps GAA activity and glycogen histopathology scores as indicated from untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 29 shows glycogen luxol/PAS as indicated from quadriceps of untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 30 shows hGAA immunohistochemistry for quadriceps as indicated from untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 31 shows triceps GAA activity and histopathological scores as indicated from untreated wild type ("normal") mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 32 shows glycogen luxol/PAS as indicated from triceps of untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles.
Figure 33 shows hGAA immunohistochemistry for triceps from untreated wild type mice or GAA knockout mice treated with gene therapy vectors or vehicles as indicated.
Detailed Description
Gene therapy of single gene genetic disorders provides a potential single treatment for diseases and disorders, some of which have destructive symptoms that may occur early in life and sometimes lead to lifelong disability. Neurogenetic disorders, such as lysosomal storage diseases, are typically treated with enzyme replacement therapy, which administers to the patient a therapeutic protein in the form of an active protein that is deficient or deficient in the disease or disorder state. However, current therapies present challenges, including frequent treatment, development of an immune response to the therapeutic protein, and difficulty in targeting the therapeutic protein to the affected tissue, cellular, or subcellular compartments. Gene therapy offers advantages including reduced treatment times and sustained efficacy.
Provided herein are compositions for gene therapy vectors that provide improvements in gene therapy, such as providing more therapeutic protein where needed, thus improving therapeutic efficacy. Such challenges are addressed herein by improving the expression and cellular uptake or delivery of therapeutic proteins and intracellular or subcellular targeting. Specific tools or components provided herein include (but are not limited to): signal peptides for increased secretion (e.g., Binding Immunoglobulin Protein (BiP) and Gaussia signal peptides); and peptides that increase endocytosis of the therapeutic protein (e.g., peptides that bind with high affinity to CI-MPR to increase cellular uptake and lysosomal delivery). Such peptides are fused to therapeutic proteins encoded by gene therapy vectors. In some embodiments, the peptide is an IGF2 (insulin-like growth factor 2) peptide or a variant thereof. In some embodiments, it is contemplated that the gene therapy vectors provided herein comprise a nucleic acid encoding a therapeutic protein fused to a peptide that binds with high affinity and CI-MPR to optimize the efficacy of gene therapy.
Gene therapy constructs designed for use in enzyme replacement gene therapy. Translation initiation sequences include, but are not limited to, Kozak sequences or IRES sequences located at the 5' end of the construct, such as CrPV IRES; followed by a nucleic acid encoding a signal peptide for one or more of the GAA signal peptides; a nucleic acid encoding an antitrypsin inhibitor; and a nucleic acid encoding a BiP sequence. These are followed by nucleic acids encoding a cell targeting domain, which may be vIGF-2, HIRMab or TfRMab or other cell targeting peptides or proteins. The gene therapy construct further comprises a nucleic acid encoding a linker and a nucleic acid encoding a proofreading enzyme or an enzymatically active fragment thereof, wherein the linker connects the cell-targeting domain to the proofreading enzyme or an enzymatically active fragment thereof. Suitable proofreading enzymes include, but are not limited to, alpha-Glucosidase (GAA), alpha-Galactosidase (GLA), Iduronidase (IDUA), iduronate-2-sulfatase (IDS), PPT1, or enzymatically active fragments thereof, and other enzymes found in deficient amounts in an individual.
Intracellular targeting of therapeutic proteins
The N-linked carbohydrates of most lysosomal proteins are modified to contain a specific carbohydrate structure called mannose 6-phosphate (M6P). M6P is a biological signal that enables transport of lysosomal proteins to lysosomes via membrane-bound M6P receptors. Enzyme replacement therapy for lysosomal storage diseases uses the M6P receptor for uptake and delivery of therapeutic proteins to lysosomes. Certain therapies do not utilize the M6P receptor, including And other recombinant human GCase forms, utilize mannose receptors capable of binding terminal mannose on proteinaceous glycans and delivering to lysosomes. Some enzyme replacement therapies face problems: small amounts of M6P were present on the enzyme therapeutics, which required higher doses to achieve therapeutic efficacy. This results in substantially longer infusion times, higher probability of generating an immune response to the therapeutic agent, and higher drug requirements, requiring increased protein manufacture, resulting in increased costs.
And other recombinant human GCase forms, utilize mannose receptors capable of binding terminal mannose on proteinaceous glycans and delivering to lysosomes. Some enzyme replacement therapies face problems: small amounts of M6P were present on the enzyme therapeutics, which required higher doses to achieve therapeutic efficacy. This results in substantially longer infusion times, higher probability of generating an immune response to the therapeutic agent, and higher drug requirements, requiring increased protein manufacture, resulting in increased costs.
CI-MPR captures the M6P-containing lysosomal enzyme from the circulation. The receptor has different binding domains for M6P and insulin-like growth factor (domains 1-3 and 7-9, see fig. 2), and is therefore also known as IGF 2/mannose-6-phosphate receptor or IGF 2/CI-MPR. Such receptors are useful for enzyme replacement therapy targeting variants containing M6P or IGF2 or IGF 2. The binding affinities of this receptor for these ligands (including insulin-like growth factor) are provided in table 1. Notably, IGF2 peptide has higher binding affinity for CI-MPR than mono-or di-phosphorylated oligosaccharides.

Therapeutic fusion proteins for gene therapy
Provided herein are therapeutic fusion proteins produced from gene therapy vectors. In some embodiments, the fusion protein is secreted by a cell transduced with a gene therapy vector encoding the fusion protein. In some embodiments, the transduced cell is within a tissue or organ (e.g., liver). Once secreted from the cell, the fusion protein is transported through the vascular system of the patient and reaches the relevant tissue. In some embodiments, the therapeutic fusion protein is engineered to have improved secretion. In some embodiments, the fusion protein comprises a signal peptide for increasing secretion levels compared to a corresponding therapeutic protein or a fusion protein comprising a therapeutic protein but lacking the signal peptide.
In some embodiments, the provided gene therapy vectors are engineered to address the problems associated with gene therapy with respect to delivery of therapeutic proteins. For example, in some cases, if an insufficient amount of a therapeutic protein is delivered into a cell in need of the therapeutic protein, gene therapy may not achieve the intended treatment simply by producing a sufficient amount of the therapeutic protein in the patient due to, for example, physical and/or biological barriers that hinder the distribution of the therapeutic protein to the desired site. Thus, even if gene therapy is able to submerge blood or tissue to a saturation point with high concentrations of therapeutic proteins, gene therapy may not be sufficiently therapeutic. In addition, non-productive clearance pathways may remove the vast majority of therapeutic proteins. Even if a therapeutic protein is transported from the vascular system to an interstitial space within a tissue (e.g., a muscle fiber), a sufficient therapeutic effect cannot be ensured. In order to effectively treat lysosomal storage disorders, a therapeutically effective amount of the therapeutic protein must be endocytosed and lysosomal delivered to produce meaningful efficacy. The present disclosure addresses these problems by providing gene therapy vectors encoding fusion proteins comprising peptides capable of endocytosing a therapeutic protein into a therapeutic target cell to produce an effective therapy. In some embodiments, the peptide capable of endocytosis is a CI-MPR binding peptide. In some embodiments, the CI-MPR binding peptide is a vIGF2 peptide.
Provided herein are gene therapy vectors encoding fusion proteins comprising peptides capable of endocytosing a therapeutic protein into a therapeutic target cell. In some embodiments, the gene therapy vector encodes a fusion protein comprising a therapeutic protein and a peptide that binds CI-MPR. These fusion proteins, when expressed from a gene therapy vector, target a therapeutic protein (e.g., an enzyme replacement therapeutic agent) to a cell in need thereof, increase delivery into or cellular uptake by these cells, and target the therapeutic protein to a subcellular location (e.g., a lysosome). In some embodiments, the peptide is an IGF2 peptide or variant thereof, which can target the therapeutic protein to a lysosome. Furthermore, in some embodiments, the fusion proteins herein further comprise a signal peptide that increases secretion, such as a BiP signal peptide or a Gaussia signal peptide. In some embodiments, the fusion protein comprises a linker sequence. In some embodiments, a nucleic acid encoding a fusion protein herein comprises an internal ribosomal entry sequence.
Provided herein are therapeutic proteins for gene therapy comprising a vIGF2 peptide. Exemplary proteins are provided in table 2 below.




The components of the fusion proteins provided herein are further described below.
Peptides that bind CI-MPR (e.g., vIGF2 peptide)
Provided herein are peptides that bind CI-MPR. Fusion proteins comprising these peptides and therapeutic proteins, when expressed from a gene therapy vector, target the therapeutic protein to cells in need thereof, increase cellular uptake of these cells, and target the therapeutic protein to subcellular locations (e.g., lysosomes). In some embodiments, the peptide is fused to the N-terminus of the therapeutic peptide. In some embodiments, the peptide is fused to the C-terminus of the therapeutic protein. In some embodiments, the peptide is a vIGF2 peptide. Some vIGF2 peptides maintained high affinity binding to CI-MPR, while their affinity for IGF1 receptor, insulin receptor, and IGF binding protein (IGFBP) was reduced or eliminated. Thus, some variant IGF2 peptides are substantially more selective and have reduced safety risks as compared to wt IGF 2. The vIGF2 peptides herein include those having the amino acid sequence of SEQ ID No. 31. Variant IGF2 peptides also include those having variant amino acids at positions 6, 26, 27, 43, 48, 49, 50, 54, 55, or 65 as compared to wt IGF2(SEQ ID NO: 1). In some embodiments, the vIGF2 peptide has a sequence with one or more substitutions from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, a54R, L55R, and K65R. In some embodiments, the vIGF2 peptide has a sequence with an E6R substitution. In some embodiments, the vIGF2 peptide has a sequence with an F26S substitution. In some embodiments, the vIGF2 peptide has a sequence with Y27L substitutions. In some embodiments, the vIGF2 peptide has a sequence with a V43L substitution. In some embodiments, the vIGF2 peptide has a sequence with an F48T substitution. In some embodiments, the vIGF2 peptide has a sequence with a R49S substitution. In some embodiments, the vIGF2 peptide has a sequence with an S50I substitution. In some embodiments, the vIGF2 peptide has a sequence with an a54R substitution. In some embodiments, the vIGF2 peptide has a sequence with L55R substitutions. In some embodiments, the vIGF2 peptide has a sequence with a K65R substitution. In some embodiments, vIGF2 peptide has a sequence with E6R, F26S, Y27L, V43L, F48T, R49S, S50I, a54R, and L55R substitutions. In some embodiments, the vIGF2 peptide has an N-terminal deletion. In some embodiments, the vIGF2 peptide has an N-terminal deletion of one amino acid. In some embodiments, the vIGF2 peptide has an N-terminal deletion of two amino acids. In some embodiments, the vIGF2 peptide has an N-terminal deletion of three amino acids. In some embodiments, the vIGF2 peptide has an N-terminal deletion of four amino acids. In some embodiments, the vIGF2 peptide has an N-terminal deletion of four amino acids and substitutions of E6R, Y27L, and K65R. In some embodiments, the vIGF2 peptide has an N-terminal deletion of four amino acids and substitutions of E6R and Y27L. In some embodiments, the vIGF2 peptide has an N-terminal deletion of five amino acids. In some embodiments, the vIGF2 peptide has an N-terminal deletion of six amino acids. In some embodiments, the vIGF2 peptide has an N-terminal deletion of seven amino acids. In some embodiments, the vIGF2 peptide has an N-terminal deletion of seven amino acids and substitutions of Y27L and K65R.



Internal ribosome entry sequence
Provided herein are gene therapy constructs useful for treating disorders, further comprising an Internal Ribosome Entry Sequence (IRES) for increasing gene expression by bypassing the bottleneck of translation initiation. Suitable internal ribosome entry sequences for optimizing gene therapy expression include, but are not limited to, cricket paralysis virus (CrPV) IRES, picornavirus IRES, foot and mouth disease virus IRES, kaposi's sarcoma-associated herpesvirus IRES, hepatitis a IRES, hepatitis c IRES, pestivirus IRES, cricket paralysis virus IRES, gahnia virus IRES, merremik disease virus IRES, and other suitable IRES sequences. In some embodiments, the gene therapy construct comprises a CrPV IRES. In some embodiments, the CrPV IRES has the following nucleic acid sequence: AAAAATGTGATCTTGCTTGTAAATACAATTTTGAGAGGTTAATAAATTACAAGTAGTGCTATTTTTGTATTTAGGTTAGCTATTTAGCTTTACGTTCCAGGATGCCTAGTGGCAGCCCCACAATATCCAGGAAGCCCTCTCTGCGGTTTTTCAGATTAGGTAGTCGAAAAACCTAAGAAATTTACCTGCT (SEQ ID NO: 12). In some embodiments, the CrPV IRES sequence is at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identical to SEQ ID No. 12.
Signal peptide
In some embodiments, the gene therapy constructs provided herein further comprise a signal peptide that improves secretion of the therapeutic protein from a cell transduced with the gene therapy construct. In some embodiments, the signal peptide improves protein processing of the therapeutic protein and facilitates translocation of nascent polypeptide-ribosome complexes to the ER and ensures proper co-and post-translational modification. In some embodiments, the signal peptide is located (i) upstream of the signal translation initiation sequence, (ii) between the translation initiation sequence and the therapeutic protein, or (iii) downstream of the therapeutic protein. Signal peptides useful in gene therapy constructs include, but are not limited to, immunoglobulin protein Binding (BiP) signal peptides and Gaussia signal peptides from the HSP70 protein family (e.g., HSPA5, heat shock protein family a member 5), and variants thereof. These signal peptides have an ultra-high affinity for signal recognition particles. Examples of BiP and Gaussia amino acid sequences are provided in table 5 below. In some embodiments, the signal peptide has an amino acid sequence that is at least 90% identical to a sequence selected from the group consisting of SEQ ID Nos. 13-17. In some embodiments, the signal peptide differs from a sequence selected from the group consisting of SEQ ID Nos. 13-17 by 5 or less, 4 or less, 3 or less, 2 or less, or 1 amino acid.

BiP signal peptide-Signal Recognition Particle (SRP) interactions facilitate translocation to the ER. This interaction is illustrated in fig. 20.
The Gaussia signal peptide is derived from the marine copepod (Gaussia princeps) luciferase and directs increased protein synthesis and secretion of therapeutic proteins fused to this signal peptide. In some embodiments, the Gaussia signal peptide has an amino acid sequence having at least 90% identity to SEQ ID NO: 32. In some embodiments, the signal peptide differs from SEQ ID NO:32 by 5 or fewer, 4 or fewer, 3 or fewer, 2 or fewer, or 1 amino acid.
Joint
In some embodiments, the gene therapy constructs provided herein comprise a linker between the targeting peptide and the therapeutic protein. In some embodiments, these linkers maintain proper spacing and reduce steric clash between the vIGF2 peptide and the therapeutic protein. In some embodiments, the linker comprises repeating glycine residues, repeating glycine-serine residues, and combinations thereof. In some embodiments, the linker consists of 5-20 amino acids, 5-15 amino acids, 5-10 amino acids, 8-12 amino acids, or about 5, 6, 7, 8, 9, 10, 11, 12, or 13 amino acids. Suitable linkers for the gene therapy constructs herein include, but are not limited to, those provided in table 6 below.

Translation initiation sequence
Gene therapy constructs provided herein comprise a vector having a translation initiation sequence (e.g., a Kozak sequence, which facilitatesIn initiating translation of the mRNA). The Kozak sequences encompassed herein have the common sequence of (gcc) RccATGG (SEQ ID NO:27), with lower case letters indicating the most common base at that position and base changes, and upper case letters indicating highly conserved bases with only minor changes. R indicates a purine (adenine or guanine) consistently observed at that position. The sequence in parentheses (gcc) has no significance. In some embodiments, the Kozak sequence comprises the sequence AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide. In some embodiments, X1Contains A. In some embodiments, X2Contains G. In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence differs from the sequence of AAGATGA (SEQ ID NO:29) by one or two nucleotides. In some embodiments, the Kozak sequence provided herein has the sequence of AAGATGA (SEQ ID NO: 29). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence differs from the sequence of GCAAGATG (SEQ ID NO:44) by one or two nucleotides. In some embodiments, the Kozak sequence comprises GCAAGATG (SEQ ID NO: 44). In some embodiments, the Kozak sequence comprises a nucleic acid sequence having at least 85% identity to CACCATG (SEQ ID NO: 47). In some embodiments, the Kozak sequence differs from the sequence of CACCATG (SEQ ID NO:47) by one or two nucleotides. In some embodiments, the Kozak sequence comprises CACCATG (SEQ ID NO: 47).
Therapeutic proteins
Gene therapy constructs provided herein comprise nucleic acids encoding therapeutic proteins for treating a genetic disorder resulting from the absence of a protein or a defective protein due to a genetic defect in an individual. The therapeutic protein expressed from the gene therapy construct replaces the absent protein or the defective protein. Thus, a therapeutic protein is selected based on the genetic defect in an individual in need of treatment. In some embodiments, the therapeutic protein is a structural protein. In some embodiments, the therapeutic protein is an enzyme. In some embodiments, the therapeutic protein is a regulatory protein. In some embodiments, the therapeutic protein is a receptor. In some embodiments, the therapeutic protein is a peptide hormone. In some embodiments, the therapeutic protein is a cytokine or chemokine.
In some embodiments, the gene therapy constructs herein can encode an enzyme, for example an enzyme having a genetic defect in an individual having a lysosomal storage disorder. In some embodiments, the gene therapy construct encodes a lysosomal enzyme, such as a glycosidase, protease, or sulfatase. In some embodiments, the enzymes encoded by the gene therapy constructs provided herein include (but are not limited to): alpha-D-mannosidase; n-aspartyl- β -glucosaminidase; beta-galactosidase; a ceramidase; a fucosidase; galactocerebrosidase; arylsulfatase A; n-acetylglucosamine-1-phosphotransferase; iduronate sulfatase; n-acetylglucosaminidase; acetyl-CoA alpha-glucosaminide acetyltransferase; n-acetylglucosamine 6-sulfatase; beta-glucuronidase; hyaluronic acid enzyme; a sialidase; a sulfatase; sphingomyelinase; acid beta-mannosidase; cathepsin K; 3-hexosidase a; beta-hexosidase B; alpha-N-acetylgalactosaminidase; sialic acid transporter protein (sialin); hexosidase A; a beta-glucosidase; alpha-iduronidase; alpha-galactosidase a; β -glucocerebrosidase; a lysosomal acid lipase; glycosaminoglycan alpha-L-iduronic aldehyde hydrolase; iduronate-2-sulfatase; n-acetylgalactosamine-6-sulfatase; glycosaminoglycan N-acetylgalactosamine 4-sulfatase; an alpha-glucosidase; heparan sulphamidase; gp-91 subunit of NADPH oxidase; adenosine deaminase; cyclin-dependent kinase-like 5; and palmitoyl protein thioesterase 1. In some embodiments, the enzyme encoded by the gene therapy constructs provided herein comprises an alpha-glucosidase. In some embodiments, the therapeutic protein is associated with a genetic disorder selected from the group consisting of: CDKL5 deficiency, cystic fibrosis, alpha-thalassemia and beta-thalassemia, sickle cell anemia, marfan's syndrome, fragile X syndrome, huntington's disease, hemochromatosis, congenital deafness (non-syndromic), tay-saxose disease, familial hypercholesterolemia, duchenne muscular dystrophy, stedt's disease, ewing's syndrome, choroideremia, achromatopsia, X-linked retinoschisis, hemophilia, wir-austenitic syndrome, X-linked chronic granulomatosis, aromatic L-amino acid decarboxylase deficiency, recessive dystrophic epidermolysis bullosa, alpha 1 antitrypsin deficiency, hakinson-gilford progeria syndrome (HGPS), noonan syndrome, X-linked severe combined immunodeficiency syndrome (X-SCID). In some embodiments, the therapeutic protein is selected from the group consisting of: CDKL5, connexin 26, hexosidase A, LDL receptor, dystrophin, CFTR, β -globin, HFE, huntingtin, ABCA4, myosin VIIA (MYO7A), Rab convoyin-1 (REP1), cyclic nucleotide-gated channel β 3(CNGB3), retinoschisin 1(RS1), heme subunit β (HBB), factor IX, WAS, cytochrome B-245 β chain, Dopa Decarboxylase (DDC), collagen type VII α 1 chain (COL7a1), serine protease inhibitor family a member 1(SERPINA1), LMNA, PTPN11, SOS1, RAF1, KRAS, and IL2 receptor γ genes.
Examples of Gene therapy vectors
Gene therapy vectors and compositions
Provided herein are gene therapy vectors in which a nucleic acid (e.g., DNA) encoding a therapeutic fusion protein (e.g., a vIGF2 fusion) optionally has a signal peptide. The gene therapy vector optionally comprises an internal ribosome entry sequence. Retroviral (e.g., lentiviral) derived vectors are suitable tools for achieving long-term gene transfer, as they allow long-term stable integration of a transgene and its propagation in daughter cells. Lentivirus and adeno-associated virus vectors have the additional advantage over vectors derived from oncogenic retroviruses, such as murine leukemia virus, in that they are capable of transducing non-proliferating cells, such as hepatocytes and neurons. They also have the additional advantage of low immunogenicity.
Exemplary gene therapy vectors herein encode therapeutic proteins and therapeutic fusion proteins comprising a vIGF2 peptide. Nucleic acids encoding exemplary fusion protein amino acid sequences are provided in table 7 below.










In some embodiments, the vectors provided herein comprising a nucleic acid encoding a desired therapeutic fusion protein (e.g., a vIGF2 fusion or a signal peptide fusion), optionally with an internal ribosome entry sequence, are adeno-associated viral vectors (a 5/35).
In some embodiments, nucleic acids encoding therapeutic fusion proteins (e.g., vIGF2 fusions) and optionally having internal ribosome entry sequences are cloned into various types of vectors. For example, in some embodiments, the nucleic acid is cloned into a vector, including (but not limited to) plasmids, phagemids, phage derivatives, animal viruses, and cosmids. Vectors of particular interest include expression vectors, replication vectors, probe generation vectors, and sequencing vectors.
Furthermore, in some embodiments, an expression vector encoding a desired therapeutic fusion protein (e.g., a vIGF2 fusion or a signal peptide fusion) and optionally having an internal ribosome entry sequence is provided to a cell in the form of a viral vector. Viral vector technology is described, for example, in Sambrook et al, 2012, Molecular Cloning: A laboratory Manual, volumes 1-4, Cold Spring Harbor Press, NY) and other handbooks for virology and Molecular biology. Viruses suitable for use as vectors include, but are not limited to, retroviruses, adenoviruses, adeno-associated viruses, herpes viruses, and lentiviruses. In general, suitable vectors contain an origin of replication functional in at least one organism, a promoter sequence, a suitable restriction endonuclease site, and one or more selectable markers (e.g., WO 01/96584; WO 01/29058; and U.S. Pat. No. 6,326,193).
Also provided herein are compositions and systems for gene transfer. A number of virus-based systems have been developed for gene transfer into mammalian cells. For example, retroviruses provide a suitable platform for gene delivery systems. In some embodiments, the selected gene is inserted into a vector and packaged in a retroviral particle using a suitable technique. The recombinant virus is then isolated and delivered to cells of the subject in vivo or ex vivo. A variety of retroviral systems are suitable for gene therapy. In some embodiments, an adenoviral vector is used. A variety of adenoviral vectors are suitable for gene therapy. In some embodiments, an adeno-associated viral vector is used. A variety of adeno-associated viruses are suitable for gene therapy. In one embodiment, a lentiviral vector is used.
Gene therapy constructs provided herein comprise a vector (or gene therapy expression vector) into which the relevant gene is cloned or which comprises the relevant gene in a manner such that the nucleotide sequence of the vector allows expression (constitutive expression or regulated in some way) of the relevant gene. Vector constructs provided herein include any suitable gene expression vector capable of delivery to the relevant tissue and which will provide for expression of the relevant gene in the selected relevant tissue.
In some embodiments, the vector is an adeno-associated virus (AAV) vector due to the ability of the AAV vector to cross the blood brain barrier and transduce neuronal tissue. In the methods provided herein, the use of any serotype of AAV is contemplated. In certain embodiments, the viral vector of the serotype used is selected from the group consisting of: AAV1 vector, AAV2 vector, AAV3 vector, AAV4 vector, AAV5 vector, AAV6 vector, AAV7 vector, AAV8 vector, AAV9 vector, AAVrhS vector, AAVrh10 vector, AAVrh33 vector, AAVrh34 vector, AAVrh74 vector, AAV Anc80 vector, aavphp.b vector, AAVhu68 vector, AAV-DJ vector and other vectors suitable for gene therapy.
AAV vectors are DNA parvoviruses that are non-pathogenic to mammals. Briefly, AAV-based vectors remove the rep and cap viral genes that represent 96% of the viral genome, leaving two flanking 145 base pair Inverted Terminal Repeats (ITRs) for initiating viral DNA replication, packaging and integration.
Other embodiments include the use of other serotype capsids to produce: AAV1 vector, AAV2 vector, AAV3 vector, AAV4 vector, AAV5 vector, AAV6 vector, AAV7 vector, AAV8 vector, AAV9 vector, AAVrhS vector, AAVrh10 vector, AAVrh33 vector, AAVrh34 vector, AAVrh74 vector, AAV Anc80 vector, aavphp.b vector, AAV-DJ vector and other vectors suitable for gene therapy. Optionally, the AAV viral capsid is AAV2/9, AAV9, AAVrhS, AAVr h10, AAVAnc80, or AAV php.b.
Additional promoter elements (e.g., enhancers) regulate the transcription initiation frequency. Typically, these are located in the region 30-110bp upstream of the start site, although many promoters have been shown to contain functional elements also downstream of the start site. The spacing between promoter elements is typically flexible such that promoter function is retained when the elements are inverted or moved relative to each other. In the thymidine kinase (tk) promoter, the spacing between promoter elements is typically increased to 50bp apart before activity begins to decline. Depending on the promoter, the individual elements appear to function cooperatively or independently to activate transcription.
An example of a promoter capable of expressing a therapeutic fusion protein (e.g., a vIGF2 fusion or a signal peptide fusion) and optionally having an internal ribosome entry sequence, a transgene in mammalian T cells is the EF1a promoter. The native EF1a promoter drives expression of the alpha subunit of the elongation factor-1 complex, which is responsible for enzymatic delivery of the aminoacyl tRNA to the ribosome. The EF1a promoter has been widely used in mammalian expression and has been shown to be effective in driving expression of transgenes from cloning into lentiviral vectors (see, e.g., Milone et al, mol. Ther.17(8):1453-1464 (2009)). Another example of a promoter is the immediate early Cytomegalovirus (CMV) promoter sequence. Such promoter sequences are strong constitutive promoter sequences capable of driving high levels of expression of any polynucleotide sequence to which they are operably linked. However, other constitutive promoter sequences may sometimes be used, including, but not limited to, the chicken β actin promoter, the P546 promoter, the simian virus 40(SV40) early promoter, the Mouse Mammary Tumor Virus (MMTV), the Human Immunodeficiency Virus (HIV) Long Terminal Repeat (LTR) promoter, the MoMuLV promoter, the avian leukemia virus promoter, the Epstein-Barr virus (Epstein-Barr virus) immediate early promoter, the Rous sarcoma (Rous sarcoma) virus promoter, and human gene promoters such as, but not limited to, the actin promoter, the myosin promoter, the elongation factor-1 a promoter, the heme promoter, and the creatine kinase promoter. Furthermore, it is contemplated that gene therapy vectors are not limited to the use of constitutive promoters. Inducible promoters are also contemplated herein. The use of an inducible promoter provides a molecular switch that is capable of turning on expression of the polynucleotide sequence to which it is operably linked when such expression is desired or turning off expression when expression is not desired. Examples of inducible promoters include, but are not limited to, metallothionein promoters, glucocorticoid promoters, progesterone promoters, and tetracycline regulated promoters.
To assess the expression of a therapeutic fusion protein (e.g., a vIGF fusion or a signal peptide fusion) optionally having an internal ribosome entry sequence or portion thereof, the expression vector to be introduced into a cell typically contains a selectable marker gene or a reporter gene or both to facilitate the identification and selection of expressing cells from a population of cells that are intended to be transfected or infected by a viral vector. In other aspects, the selectable marker is typically carried on a separate DNA segment and used in a co-transfection procedure. Selectable markers and reporter genes are sometimes flanked by appropriate regulatory sequences to achieve expression in a host cell. Useful selectable markers include, for example, antibiotic resistance genes, such as neo and the like.
Methods and compositions for introduction into cells and expression of genes are suitable for the methods herein. In the case of expression vectors, the vectors are readily introduced into host cells (e.g., mammalian, bacterial, yeast, or insect cells) by any method known in the art. For example, the expression vector is transferred into a host cell by physical, chemical or biological means.
Physical methods and compositions for introducing polynucleotides into host cells include calcium phosphate precipitation, lipofection, particle bombardment, microinjection, gene guns, electroporation, and the like. Methods for making cells comprising the vector and/or the exogenous nucleic acid are suitable for the methods herein (see, e.g., Sambrook et al, 2012, Molecular Cloning: A Laboratory Manual, Vol. 1-4, Cold Spring Harbor Press, NY). One method of introducing polynucleotides into host cells is calcium phosphate transfection.
Chemical means and compositions for introducing polynucleotides into host cells include colloidal dispersion systems such as macromolecular complexes, nanocapsules, microspheres, beads, and lipid-based systems including oil-in-water emulsions, micelles, mixed micelles, nucleic acid-lipid particles, and liposomes. An exemplary colloidal system for use as an in vitro and in vivo delivery vehicle is a liposome (e.g., an artificial membrane vesicle). Other methods of targeted delivery of nucleic acids in currently advanced technologies are available, such as delivery of polynucleotides with targeted nanoparticles or other suitable submicron-sized delivery systems.
Where a non-viral delivery system is utilized, an exemplary delivery vehicle is a liposome. The use of lipid formulations for introducing nucleic acids into host cells (in vitro, ex vivo or in vivo) is contemplated. In another aspect, the nucleic acid is associated with a lipid. In some embodiments, the nucleic acid associated with a lipid is encapsulated within the aqueous interior of a liposome, interspersed within the lipid bilayer of a liposome, linked to the liposome via a linker molecule associated with the liposome and an oligonucleotide, encapsulated in the liposome, complexed with the liposome, dispersed in a solution containing the lipid, mixed with the lipid, combined with the lipid, contained in suspension in the lipid, contained in or complexed with a micelle, or otherwise associated with the lipid. The composition associated with the lipid, lipid/DNA or lipid/expression vector is not limited to any particular structure in solution. For example, in some embodiments, they are present in a bilayer structure, in the form of micelles or with a "collapsed" structure. Alternatively, they may simply be interpenetrated in solution, possibly forming aggregates that are not uniform in size or shape. Lipids are fatty substances, which in some embodiments are naturally occurring or synthetic lipids. For example, lipids include droplets of fat naturally occurring in the cytoplasm as well as classes of compounds containing long chain aliphatic hydrocarbons and their derivatives (e.g., fatty acids, alcohols, amines, amino alcohols, and aldehydes).
Suitable lipids are available from commercial sources. For example, in some embodiments, dimyristylphosphatidylcholine ("DMPC") is obtained from Sigma, st.louis, mo.; in some embodiments, dicetyl phosphate ("DCP") is obtained from K & K Laboratories (Plainview, n.y.); in some embodiments, cholesterol ("Choi") is obtained from Calbiochem-Behring; dimyristylphosphatidylglycerol ("DMPG") and other Lipids are commonly available from Avanti Polar Lipids, Inc. Stock solutions of lipids in chloroform or chloroform/methanol are typically stored at about-20 ℃. Chloroform was used as the only solvent because it evaporates more readily than methanol. "liposomes" is a generic term that encompasses a variety of mono-and multilamellar lipid vehicles formed by the production of encapsulated lipid bilayers or aggregates. Liposomes are generally characterized as having a vesicular structure with a phospholipid bilayer membrane and an internal aqueous medium. Multilamellar liposomes have multiple lipid layers separated by an aqueous medium. They form spontaneously when phospholipids are suspended in excess aqueous solution. The lipid components rearrange themselves before forming a closed structure and trap water and dissolved solutes between lipid bilayers (Ghosh et al, 1991Glycobiology 5: 505-10). However, compositions having a structure in solution that is different from the normal vesicle structure are also contemplated. For example, in some embodiments, the lipids present a micellar structure or merely exist as non-uniform aggregates of lipid molecules. Lipid-staining amine-nucleic acid complexes are also contemplated.
Regardless of the method used to introduce the exogenous nucleic acid into the host cell or expose the cell to the therapeutic fusion protein provided herein (e.g., a vIGF2 fusion or a signal peptide fusion), optionally with an internal ribosome entry sequence, various assays are contemplated for confirming the presence of recombinant DNA sequences in the host cell. Such assays include, for example, "molecular biology" assays suitable for the methods herein, such as Southern and Northern blots, RT-PCR and PCR; "biochemical" assays, e.g., detecting the presence or absence of a particular peptide, identify reagents falling within the scope hereof, e.g., by immunological means (ELISA and western blot) or by assays described herein.
The disclosure also provides a vector comprising a therapeutic fusion protein (e.g., a vIGF2 fusion or a signal peptide fusion), optionally with an internal ribosome entry sequence, encoding a nucleic acid molecule. In one aspect, the therapeutic fusion protein vector is capable of being directly transduced into a cell. In one aspect, the vector is a cloning or expression vector, such as, but not limited to, vectors including: one or more plasmids (e.g., expression plasmids, cloning vectors, minicircles, microcarriers, double minichromosomes), retroviral and lentiviral vector constructs. In one aspect, the vector is capable of expressing the vIGF 2-therapeutic fusion protein construct in a mammalian cell. In one aspect, the mammalian cell is a human cell.
Use and method of treatment
Also provided herein are methods of treating a genetic disorder using gene therapy, comprising administering to an individual a nucleic acid encoding a therapeutic fusion protein disclosed herein (e.g., a vIGF2 fusion or a signal peptide-vIGF 2 fusion) and optionally having an internal ribosome entry sequence. Genetic disorders suitable for treatment using the methods herein include disorders in an individual caused by one or more mutations in the genome that result in the absence of an expressed protein or the expression of a dysfunctional protein by a mutant gene.
Also provided herein are pharmaceutical compositions comprising a gene therapy vector, e.g., a gene therapy vector comprising a nucleic acid encoding a therapeutic fusion protein disclosed herein (e.g., a vIGF2 fusion or a signal peptide-vIGF 2 fusion) and optionally having an internal ribosome entry sequence, and a pharmaceutically acceptable carrier or excipient, for use in the preparation of a medicament for the treatment of a genetic disorder.
Genetic disorders suitable for treatment by the methods herein include (but are not limited to): achondroplasia, alpha-1 antitrypsin deficiency, antiphospholipid Syndrome, autosomal dominant polycystic kidney Disease, Chack-Marie-Dus Disease (Charcot-Marie-Tooth), colon cancer, Cat's Syndrome (Cri du chat), Crohn's Disease, cystic fibrosis, Derch's Disease, Dunne Syndrome (Duane Syndrome), Duchenne muscular dystrophy, Factor V Laden Disease (Factor V Leiden Thrombophilia), familial hypercholesterolemia, familial Mediterranean fever, Fragile X Syndrome, gaucher's Disease, hemochromatosis, hemophilia, forebrain non-cracking malformation, Huntington's Disease (Huntington ' Disease), Klinefelter Syndrome (Klefelder Syndrome), Marek's Syndrome, muscular dystrophia, neurofibroma, neurofibromyalgia Syndrome, osteogenesis, Parkinson's Disease (Parkinson's Disease), Phenylketonuria, Poland Anomaly, porphyria, progeria, retinitis pigmentosa, Severe Combined Immunodeficiency (SCID), sickle cell Disease, spinal muscular atrophy, Thai-saxophone Disease, thalassemia, trimethylaminouria, Turner Syndrome, jaw face Syndrome, WAGR Syndrome, or Wilson Disease. In some embodiments, the genetic disorder is selected from the group consisting of: CDKL5 deficiency, cystic fibrosis, alpha-thalassemia and beta-thalassemia, sickle cell anemia, marfan's syndrome, fragile X syndrome, huntington's disease, hemochromatosis, congenital deafness (non-syndromic), tay-saxose disease, familial hypercholesterolemia, duchenne muscular dystrophy, stedt's disease, ewing's syndrome, choroideremia, achromatopsia, X-linked retinoschisis, hemophilia, wir-austenitic syndrome, X-linked chronic granulomatosis, aromatic L-amino acid decarboxylase deficiency, recessive dystrophic epidermolysis bullosa, alpha 1 antitrypsin deficiency, hakinson-gilford progeria syndrome (HGPS), noonan syndrome, X-linked severe combined immunodeficiency syndrome (X-SCID).
In some embodiments, the genetic disorder suitable for treatment using the methods provided herein is a lysosomal storage disorder. In some embodiments, gene therapy is used herein to deliver a deletion or defective enzyme to a patient to treat a lysosomal storage disorder. In some embodiments, the methods herein deliver an enzyme fused to vIGF2 or to a signal peptide to a patient in order to deliver the enzyme into a cell in need thereof. In some embodiments, the lysosomal storage disease is selected from the group consisting of: aspartylglucamine diabetes, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, MuldoHoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Hewler disease, Hunter disease, Sanfilippo disease type A, Sanfilippo disease type B, Sanfilippo disease type C, Sanfilippo disease type D, Moquini disease type A, Moquine disease type B, Maruto-Lami disease, Sprius disease, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Ocimir disease type I and Ocimier disease type II. In some embodiments, the lysosomal storage disease is selected from the group consisting of: deficiency of activating factor, GM 2-gangliosidosis; GM 2-gangliosidosis, AB variant; alpha-mannosidosis (type 2, moderate form; type 3, neonatal, severe); beta-mannoside storage disease; aspartylglucosaminuria; lysosomal acid lipase deficiency; cystinosis (type of late onset juvenile or juvenile nephropathy; nephropathic nature in infants); christian-dofmann syndrome; neutral lipid storage disease with myopathy; NLSDM; danong disease (Danon disease); fabry disease; late onset fabry disease type II; fabry disease; farber fatty granulomatosis; fucoside storage disorders; galactosialistorage disorder (combined neuraminidase and beta-galactosidase deficiency); gaucher disease; gaucher disease type II; gaucher disease type III; gaucher disease type IIIC; atypical gaucher disease due to sphingolipid activator protein C deficiency; GM 1-gangliosidosis (advanced infantile/juvenile GM 1-gangliosidosis; adult/chronic GM 1-gangliosidosis); globulocyte leukodystrophy, krabbe's disease (late onset infant type; juvenile onset type; adult onset type); atypical krabbe's disease due to sphingolipid activator protein a deficiency; metachromatic leukodystrophy (juvenile; adult); partial sulfaterebroside deficiency; pseudoarylsulfatase a deficiency; metachromatic leukodystrophy due to sphingolipid activator B deficiency; mucopolysaccharide storage disorder: MPS I, heler syndrome; MPS I, heler-schey syndrome; MPS I, schey syndrome; MPS II, hunter syndrome; MPS II, hunter syndrome; sanfilippo syndrome/MPS IIIA type a; sanfilippo syndrome/MPS IIIB type B; sanfilippo syndrome/MPS IIIC type C; sanfilippo syndrome/MPS IIID type D; morquio syndrome, type a/MPS IVA; morquio syndrome, type B/MPS IVB; MPS IX hyaluronidase deficiency; MPS VI marlotte-lamide syndrome; MPS VII sri syndrome; salivary gland disease type I, II mucolipidosis; i-cell disease, Leluo disease, mucolipidosis II; shampooing malnutrition/mucolipidosis type III; mucolipidosis IIIC/ML III GAMMA; type IV mucolipidosis; polythiol esterase deficiency; niemann-pick disease (type B; type C1/chronic neurological form; type C2; type D/Nova scotia); neuronal ceroid lipofuscinosis: CLN6 disease, atypical late infant, late onset variant, early childhood; pasteur/juvenile NCL/CLN3 disease; finnish variant late infant CLN 5; jenski-biziksky disease/late infantile CLN2/TPP1 disease; kuves/adult onset NCL/CLN4 disease (type B); northern epilepsy/variant late infant CLN 8; Santaiwaii-Hardiya/infant CLN1/PPT disease; pompe disease (glycogen storage disease type II); late onset pompe disease; dense bone developmental disorder; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sandhoff disease/GM 2 gangliosidosis; sinderler disease (type III/moderate, variable); qi disease of the Shen; sara disease; infantile asialostorage disease (ISSD); spinal muscular atrophy with progressive myospasm epilepsy (SMAPME); Tay-saxophone/GM 2 gangliosidosis; juvenile onset tay-saxophone disease; late onset tay-saxophone; klissinostian syndrome; lowboy eye-brain-kidney syndrome; type 4J chak-mary-dus disease, CMT 4J; ulis-von syndrome; bilateral temporal occipital multicephalic gyrus (BTOP); x-linked hypercalcemic nephrolithiasis, dent-1; and dengue 2. In some embodiments, the therapeutic protein is associated with a lysosomal storage disease, and the therapeutic protein is selected from the group consisting of: GM 2-activator protein; alpha-mannosidase; MAN2B 1; lysosomal β -mannosidase; a glycosylasparaginase enzyme; a lysosomal acid lipase; cystine transporter protein (cystinosin); a CTNS; PNPLA 2; lysosomal associated membrane protein-2; alpha-galactosidase a; GLA; acid ceramidase; an alpha-L-fucosidase; protective protein/cathepsin a; acid beta-glucosidase; GBA; a PSAP; beta-galactosidase-1; GLB 1; galactosylceramide beta-galactosidase; GALC; a PSAP; arylsulfatase A; ARSA; alpha-L-iduronidase; iduronate 2-sulfatase; heparan N-sulfatase; n- α -acetylglucosaminidase; heparan acetyl-CoA alpha-glucosaminide acetyltransferase; n-acetylglucosamine 6-sulfatase; galactosamine-6-sulfatase; beta-galactosidase; hyaluronic acid enzyme; arylsulfatase B; beta-glucuronidase; neuraminidase; NEU 1; gamma subunits of N-acetylglucosamine-1-phosphotransferase; mucrolipin-1; sulfatase modification factor-1; acid sphingomyelinase; SMPD 1; NPC 1; and NPC 2.
In some embodiments, a gene encoding a therapeutic protein is delivered into a cell in need of the therapeutic protein via treatment by the methods herein. In some embodiments, the treatment delivers the gene to all somatic cells in the individual. In some embodiments, the therapeutic replacement targets a defective gene in the cell. In some embodiments, the cells treated ex vivo to express the therapeutic protein are delivered to an individual.
Gene therapy for the conditions disclosed herein provides therapeutic results that are superior to conventional therapies, including enzyme replacement therapy, because it does not require long-term infusion therapy. In addition, it reduces the risk of an individual developing an immune response to the therapeutic protein, which is often experienced in individuals receiving enzyme replacement therapy.
Definition of
As used herein, "ex vivo gene therapy" refers to a method in which patient cells are genetically modified outside of a subject, for example, to express a therapeutic gene. The cells with the new genetic information are then returned to the subject from which they were derived.
As used herein, "in vivo gene therapy" refers to a method in which a vector carrying a therapeutic gene is directly administered to a subject.
As used herein, "fusion protein" and "therapeutic fusion protein" are used interchangeably herein and refer to a therapeutic protein having at least one additional protein, peptide, or polypeptide attached thereto. In some cases, a fusion protein is a single protein molecule containing two or more proteins, or fragments thereof, covalently linked via peptide bonds within their respective peptide chains without chemical linkers. In some embodiments, the fusion protein comprises a therapeutic protein and a signal peptide (a peptide that increases endocytosis of the fusion protein) or both. In some embodiments, the peptide that increases endocytosis is a CI-MPR binding peptide.
As used herein, "vector" or "gene therapy vector" used interchangeably herein refers to a gene therapy delivery vehicle or vector that delivers a therapeutic gene to a cell. A gene therapy vector is any vector suitable for use in gene therapy, such as any vector suitable for the therapeutic delivery of a nucleic acid polymer (encoding a polypeptide or variant thereof) into a target cell (e.g., a sensory neuron) of a patient. In some embodiments, the gene therapy vector delivers a nucleic acid encoding a therapeutic protein or therapeutic fusion protein to a cell that expresses the therapeutic protein or fusion and is secreted from the cell. The vector may be of any type, for example it may be a plasmid vector or a minicircle DNA. Typically, the vector is a viral vector. These include both gene-disabled viral (e.g., adenoviral) vectors and non-viral vectors (e.g., liposomes). The viral vector may, for example, be derived from an adeno-associated virus (AAV), a retrovirus, lentivirus, herpes simplex virus or adenovirus. Vectors of AAV origin. The vector may comprise an AAV genome or derivative thereof.
As used herein, "construct" refers to a nucleic acid molecule or sequence that encodes a therapeutic protein or fusion protein and optionally includes additional sequences (e.g., translation initiation sequences or IRES sequences).
As used herein, "plasmid" refers to a circular double-stranded DNA unit that replicates independently of chromosomal DNA within a cell.
As used herein, "promoter" refers to a site on DNA that binds to the enzyme RNA polymerase and initiates transcription of DNA into RNA.
As used herein, "somatic cell therapy" refers to a method in which gene expression in cells that will be corrective for a patient rather than inherited to the next generation is manipulated. Somatic cells include all non-germ cells in the human body.
As used herein, "somatic cell" refers to all cells of the body except germ cells.
As used herein, "tropism" refers to the preference of a vector (e.g., a virus) for a certain cell or tissue type. Various factors determine the ability of the vector to infect a particular cell. Viruses, for example, must bind to specific cell surface receptors to enter cells. If the cell does not express the essential receptor, the virus is generally unable to infect the cell.
The term "transduction" is used to refer to the in vivo or in vitro administration/delivery of a nucleic acid encoding a therapeutic protein to a target cell via a replication-deficient rAAV of the present disclosure, thereby causing the recipient cell to express a functional polypeptide. Transduction of cells with a gene therapy vector (e.g., rAAV) of the present disclosure results in sustained expression of the polypeptide or RNA encoded by the rAAV. Accordingly, the present disclosure provides methods of administering/delivering a gene therapy vector (e.g., rAAV) encoding a therapeutic protein to a subject by intrathecal, intraretinal, intraocular, intravitreal, intracerebroventricular, intraparenchymal, or intravenous routes, or any combination thereof. By "intrathecal" delivery is meant delivery into the subintimal space of the brain or spinal cord. In some embodiments, intrathecal administration is via intracisternal administration.
The terms "recipient," "individual," "subject," "host," and "patient" are used interchangeably herein, and in some cases refer to any mammalian subject, particularly a human, in need of diagnosis, treatment, or therapy. "mammal" for purposes of treatment refers to any animal classified as a mammal, including humans, domestic animals and farm animals, as well as laboratory, zoo, sports, or pet animals, such as dogs, horses, cats, cows, sheep, goats, pigs, mice, rats, rabbits, guinea pigs, monkeys, and the like. In some embodiments, the mammal is a human.
As used herein, in some instances, the terms "treating", "ameliorating", and the like, refer to administering an agent or performing a procedure for the purpose of obtaining a therapeutic effect (including inhibiting, alleviating, reducing, preventing, or altering at least one aspect or marker of a disorder) in a statistically significant manner or in a clinically significant manner. The terms "improving" or "treating" do not indicate or imply a cure for the underlying condition. As used herein, "treating" or "ameliorating" (etc.) may include treating a mammal, particularly a human, and includes: (a) preventing a disorder or symptoms of a disorder from occurring in a subject who may be predisposed to the disorder but has not yet been diagnosed as diseased (e.g., including disorders that may be associated with or caused by a primary disorder); (b) inhibiting the disorder, i.e., arresting its development; (c) relieving the condition, i.e., causing regression of the condition; and (d) ameliorating at least one symptom of the disorder. Treatment may refer to any indicator of success in treating or ameliorating or preventing a disorder, including any objective or subjective parameter, such as elimination; (iii) alleviating; reducing symptoms or making the condition more tolerable to the patient; slowing the rate of degeneration or decline; or make the final point of degradation less debilitating. Treating or ameliorating the symptoms is based on one or more objective or subjective parameters; including the results of the examination by the physician. Thus, the term "treating" includes administering a compound or agent of the invention to prevent or delay, reduce or arrest or inhibit the development of the symptoms or conditions associated with the disorder. The term "therapeutic effect" refers to a reduction, elimination, or prevention of a disorder, a symptom of a disorder, or a side effect of a disorder in a subject.
The term "affinity" refers to the strength of binding between a molecule and its binding partner or receptor.
As used herein, the phrase "high affinity" refers to, for example, a therapeutic fusion containing such a peptide that binds CI-MPR with an affinity to CI-MPR that is about 100 to 1,000-fold or 500 to 1,000-fold that of a therapeutic protein without the peptide. In some embodiments, the affinity is at least 100, at least 500, or at least 1000-fold greater than without the peptide. For example, when a therapeutic protein and CI-MPR are combined at relatively equal concentrations, a peptide with high affinity will bind to the available CI-MPR in order to shift the equilibrium towards a high concentration of the resulting complex.
As used herein, "secreted" refers to the release of a protein from a cell into the blood stream, e.g., to be carried to the relevant tissue or site of action of a therapeutic protein. When the gene therapy product is secreted into the interstitial space of an organ, secretion may allow cross-correction of neighboring cells.
As used herein, "delivery" means drug delivery. In some embodiments, the delivery process means transporting the drug substance (e.g., a therapeutic protein or fusion protein produced from a gene therapy vector) from outside the cell (e.g., blood, tissue, or interstitial space) into the therapeutically active target cell of the drug substance.
As used herein, "engineering" or "protein engineering" refers to the manipulation of the structure of a protein by providing appropriate nucleic acid sequences encoding the protein in order to produce a protein of a desired property or having a particular structure.
In some instances, a "therapeutically effective amount" means an amount sufficient to effect treatment of a disorder when administered to a subject for treating the disorder.
As used herein, the term "about" a number refers to a range that spans from less than 10% of the number to greater than 10% of the number, and includes values within the range (e.g., the number itself).
As used herein, the term "comprising" or "comprises" an element or elements of a claim means that those elements do not preclude the inclusion of one or more additional elements.
Examples
The following examples are given for the purpose of illustrating various embodiments of the invention and are not intended to limit the invention in any way. The examples, together with the methods described herein, are presently representative of the preferred embodiments, are exemplary, and are not intended as limitations on the scope of the invention. Variations thereof and other uses will occur to those skilled in the art which are encompassed within the spirit of the invention as defined by the scope of the claims.
Example 1: binding of variant IGF2 peptides to CI-MPR receptors
Surface Plasmon Resonance (SPR) experiments were performed using Biacore to measure the binding of wild-type and variant IGF2(vIGF2) to CI-MPR receptors. The wild-type human mature IGF2 peptide (wt IGF2) has the sequence shown in SEQ ID NO: 1. The vIGF2 sequence differs from wt IGF2 in that it lacks residues 1-4 and contains the following mutations: E6R, Y27L and K65R. It has the amino sequence: SRTLCGGELVDTLQFVCGDRGFLFSRPASRVSRRSRGIVEECCFRSCDLALLETYCATPARSE (SEQ ID NO: 31). vIGF2 also has an N-terminal linker with the sequence GGGGSGGGG (SEQ ID NO: 18). The combined sequence was GGGGSGGGGSRTLCGGELVDTLQFVCGDRGFLFSRPASRVSRRSRGIVEECCFRSCDLALLETYCATPARSE (SEQ ID NO: 43). Figure 4 shows that, as expected, wild-type IGF2 peptide binds with high affinity (0.2nM) to CI-MPR receptor. Figure 5 shows that the variant IGF2 peptide (vIGF2) also bound with high affinity (0.5nM) to the CI-MPR receptor. These data indicate that vIGF2 peptide has high affinity for the expected CI-MPR receptor for targeting therapeutic agents to lysosomes.
SPR was used to measure peptides binding to the insulin receptor to assess potential side effects. Insulin binds to the insulin receptor with high affinity (-8 nM; data not shown). Wild-type IGF2 and vIGF2 were tested, wherein vIGF2 has sequence SRTLCGGELVDTLQFVCGDRGFLFSRPASRVSRRSRGIVEECCFRSCDLALLETYCATPARSE (SEQ ID NO:31) with an N-terminal linker having the sequence GGGGSGGGG (SEQ ID NO: 18). Fig. 8 shows that wild-type IGF2 also binds insulin receptor with relatively high affinity (-100 nM). The IGF2 peptide from the Biomarin/zytor IGF2-GAA fusion protein (BMN-701) also binds to the insulin receptor with high affinity and was shown to cause hypoglycemia in clinical trials. Figure 9 shows that there was no measurable binding of vIGF2 peptide to insulin receptor. These data indicate that vIGF2 peptide confers a higher safety profile than the wt IGF2 peptide fusion.
The interaction of vIGF2 peptide with IGF1 receptor was characterized using the same SPR binding assay. Fig. 10 shows that wild-type IGF2 peptide binds IGF1 receptor with relatively high affinity (-100 nM). Figure 11 shows no measurable binding of vIGF2 peptide to IGF1 receptor, indicating an improved safety profile compared to wt IGF 2.

Example 2: vIGF2 converts low affinity ligands for CI-MPR to high affinity ERT
A vIGF2 peptide (SEQ ID NO:31) with an N-terminal linker (SEQ ID NO:18) was chemically coupled to the alpha-glucosidase, referred to herein as vIGF 2-alpha-glucosidase to determine whether the vIGF2 peptide could improve affinity for CI-MPR. As shown in figure 6, the binding affinities of the arabinosidase-alpha and vIGF 2-arabinosidase-alpha were directly compared using a CI-MPR plate binding assay in a CI-MPR coated 96-well ELISA plate. Unbound enzyme was washed away before measuring bound enzyme activity. Varying concentrations of both enzyme preparations were used with or without free WT IGF2 peptide. vIGF2 substantially improved the affinity for CI-MPR. Furthermore, binding of vIGF 2-arabinosidase-a was blocked by free WT IGF2, indicating that binding is IGF2 dependent. (data not shown.) coupling of vIGF2 peptide did not impair GAA enzyme activity.
vIGF2 was coupled to recombinant human N-acetyl- α -D-glucosaminidase (rhNAGLU). Rhnaglu, a lysosomal enzyme lacking M6P, was used to determine whether peptides could convert non-ligands to high affinity ligands for CI-MPR. In this experiment, rhNAGLU and vIGF2-rhNAGLU were compared directly using CI-MPR coated plates in conjunction with the assay. Unbound enzyme was washed away before measuring bound enzyme activity. Varying concentrations of both enzyme preparations were used with or without free vIGF2 peptide. As shown in fig. 7, vIGF2-rhNAGLU had significantly higher affinity for CI-MPR than rhNAGLU lacking vIGF 2. Furthermore, vIGF2-rhNAGLU binding was blocked by the free vIGF2 peptide, indicating that receptor binding is specific for the IGF2 peptide. These results indicate that vIGF2 peptide can be used to improve drug targeting to lysosomes.
Example 3: myoblast uptake of vIGF2-GAA fusion protein
vIGF2-GAA fusion protein (same sequence as in example 1-2) was administered and the L6 myoblast uptake of the enzyme was measured. Figure 6 shows better absorption of vIGF2-rhGAA compared to rhGAA and M6P-GAA. Therefore, vIGF2 was effective in targeting GAA to cells.
Example 4: by passingConstructs for ERT for gene therapy delivery
Two different constructs are shown in figure 12. The top panel is a construct encoding "native hGAA" (SEQ ID NO:45) containing a Kozak sequence and a nucleic acid encoding recombinant human GAA with a native signal peptide (SEQ ID NO: 45). The middle panel is the construct Kozak-BiP-vIGF2-2GS-GAA encoding "engineered hGAA" (SEQ ID NO: 23). This construct is characterized by a Kozak sequence, a nucleic acid encoding a BiP signal peptide, a nucleic acid encoding a vIGF2 peptide (having the sequence shown in SEQ ID NO: 31) and a nucleic acid encoding a 2GS linker (SEQ ID NO:18), followed by a nucleic acid encoding recombinant human GAA in which the N-terminal 60 amino acids (SEQ ID NO:46) are removed to prevent premature processing and removal of vIGF 2.
Example 5: enhanced secretion of gene therapy constructs
Engineered hGAA have higher secretion and are capable of interacting with cell surface receptors suitable for cellular uptake and lysosomal targeting
CHO expressing engineered hGAA (described in more detail below) or native hGAA is cultured and conditioned media is collected for measurement of GAA activity. Fig. 15 shows the relative activities of engineered and native hGAA, indicating that the engineered hGAA has increased activity compared to native hGAA, indicating more efficient secretion of the engineered hGAA.
Example 6: analysis of PPT1 in conditioned Medium
Cloning of the PPT1 construct
The PPT1 construct was cloned into pcdna3.1 expression vector (ThermoFisher, catalog # V79020) containing the CMV promoter. The constructs tested included PPT1-1(WT-PPT1) (SEQ ID NO:24), PPT1-2(WT-vIGF2-PPT1) (SEQ ID NO:25), PPT1-29(BiP2aa-vIGF2-PPT1) (SEQ ID NO: 26).
PPT1 secretion and binding
The PPT1 construct was transiently expressed in HEK293T cells for 3 days and PPT1 was secreted into the culture medium. Secreted PPT1 was quantified by western blotting and CI-MPR binding was analyzed using well-established methods. The secreted PPT1 is shown in figure 13. The CI-MPR combination is shown in FIG. 14.
Example 7: testing gene therapy vectors in Pompe disease animal models
Pompe gene therapy: preclinical proof of concept study design
In GAA knockout (GAA KO) mice, preclinical studies were performed using high doses for initial comparison of constructs. The constructs are shown in figure 12. Mice were treated with vehicle or one of the two constructs (native hGAA or engineered hGAA). Mice were administered 5e11 gc/mouse (approximately 2.5e13 gc/kg). GAA knockout mice of 2 months of age were used. Normal (wild type) mice were used as controls. The study design is summarized in figure 16.
Pompe gene therapy: blood plasma
Plasma was collected from wild type (normal) mice or GAA KO mice treated with vehicle or gene therapy vector as indicated, and GAA activity and cell surface binding were measured. The data are summarized in fig. 17, 27 and 19. Similar high GAA levels were seen in mice treated with gene therapy vectors (fig. 17, fig. 18). However, higher cell-targeted receptor binding was observed with the engineered constructs (fig. 19).
Pompe gene therapy: quadriceps muscle
GAA activity and glycogen storage/cytoplasmic vacuolization were assessed in normal (wild-type) and treated GAA KO mice (fig. 28). GAA activity in quadriceps is about 20 times that of the wild type. Glycogen PAS (fig. 29) and immunohistochemistry (fig. 30) were also evaluated. Immunohistochemistry showed that the engineered hGAA had higher lysosomal targeting compared to the wild type. The glycogen reduction of engineered hGAA was more constant according to PAS staining.
Pompe gene therapy: triceps muscle
GAA activity and glycogen storage/cytoplasmic vacuolation were assessed in normal (wild-type) and treated GAA KO mice (fig. 31). The GAA activity is about 10-15 times that of the wild type. Immunohistochemistry and glycogen PAS were also evaluated (fig. 32 and 33). Immunohistochemistry indicated that the engineered hGAA had higher lysosomal targeting compared to wild-type GAA. The glycogen reduction of the engineered hGAA was more constant as measured by PAS staining.
Pompe gene therapy: tibialis Anterior (TA)
GAA activity and glycogen storage/cytoplasmic vacuolation were assessed in normal (wild-type) and treated GAA KO mice (fig. 20). GAA activity in TA is about 15-20 times that of wild type. Immunohistochemistry and glycogen PAS were also evaluated (fig. 21 and 22). Immunohistochemistry indicated that the engineered hGAA had higher lysosomal targeting compared to wild-type GAA. Glycogen levels are close to wild-type levels. The glycogen reduction of engineered hGAA was more constant according to PAS staining.
Pompe gene therapy: brain and spinal cord
GAA activity, glycogen content and glycogen storage/cytoplasmic vacuolization were assessed in normal (wild-type) and treated GAA KO mice (fig. 23). GAA activity in brain is about 5 times lower than that of wild type. Immunohistochemistry and glycogen PAS were also evaluated (fig. 24, 25, 26, 27). Immunohistochemistry indicates that there may be direct transduction of some cells. However, the native construct achieved little to no glycogen clearance. Glycogen levels of the engineered constructs were close to wild-type levels, even though activity was only 20% of wild-type. PAS staining in the spinal cord showed little to no glycogen clearance of the native construct. In the ventral horn, which included motor neurons, engineered constructs were observed to approximate wild-type glycogen levels. Immunohistochemistry indicated direct transduction in spinal cord neurons. Engineered hGAA produced by choroid plexus and neuronal cells can reduce glycogen by cross-correction in the spinal cord, while little glycogen reduction is observed for native hGAA.
Conclusion
Taken together, the data in this example indicate that the engineered gene therapy constructs have significantly better tissue uptake and glycogen reduction, including effects in the brain and spinal cord, than wild-type GAA used in conventional therapy.
Example 8: animal study protocol
AAVhu68 Vector was generated and titrated by Penn Vector Core as described. (Lock, Alvira et al, 2010, "Rapid, simple, and university influencing of recombinant adono-associated viral vectors at scale," Hum Gene Ther 21(10): 1259-.
Gaa knockout mice (pompe mice) were purchased at Jackson Labs (stock #004154, also known as 6neo mice) in a C57BL/6/129 background creator.
Will receive 5X 10 in 0.1mL via the lateral tail vein11GC (about 2.5X 10)13GC/kg) mice of aavhu68.cag. hGAA (containing either native hGAA (SEQ ID NO:45) or engineered hGAA (SEQ ID NO:38)) were bled for serum separation on days 7 and 21 post vector administration and final bleeds (for plasma separation) were performed 28 days post injection and euthanized by bleeds. Tissue was collected rapidly, starting from the brain.
GAA Activity
Plasma was mixed with 5.6mM 4-MU- α -glucopyranoside pH 4.0 and incubated at 37 ℃ for three hours. The reaction was stopped with 0.4M sodium carbonate pH 11.5. Relative fluorescence units RFU were measured using a Victor3 fluorometer (355nm excitation and 460nm emission). Activity in nmol/mL/hr was calculated by interpolation from a 4-MU standard curve. The activity in individual tissue samples was further normalized based on the total protein content in the homogenate.
GAA signature peptides according to LC/MS
Plasma was precipitated in 100% methanol and centrifuged. The supernatant was discarded. The pellet was spiked with the unique stable isotope labeled peptide of hGAA as an internal standard and resuspended with trypsin and incubated for one hour at 37 ℃. Digestion was stopped with 10% formic acid. Tryptic peptides were separated by C-18 reverse phase chromatography and identified and quantified by ESI-mass spectrometry. The total GAA concentration in plasma was calculated from the tag peptide concentration.
Cell surface receptor binding assays
96-well plates were coated with receptor, washed and blocked with BSA. 28-day plasma from AAV-treated mice was serially diluted to give a series of reduced concentrations and incubated with the coupled receptor. After incubation, the plates were washed to remove any unbound hGAA and 4-MU- α -glucopyranoside was added at 37 ℃ for one hour. The reaction was stopped with 1.0M glycine pH 10.5 and the RFU read by a Spectramax fluorometer (excitation 370, emission 460). RFU was converted to activity (nmol/mL/hr) for each sample by interpolation according to a standard curve of 4-MU. Nonlinear regression was performed using GraphPad Prism.
Histology
Tissues were formalin fixed and paraffin embedded. Muscle sections were stained with PAS; CNS sections were stained with luxol fast blue/periodic acid-Schiff (PAS). Board certified veterinary pathologists (JH) blindly reviewed histological sections. Semi-quantitative estimates of the total percentage of cells with glycogen storage and cytoplasmic vacuolization were performed on the scanned sections. Scores from 0 to 4 are listed as described in the table below.

Immuno-histochemistry (IHC)
We studied transgene expression and cellular localization from sections immunostained with anti-human GAA antibody (Sigma HPA 029126).
Example 9: histology-tissue processing protocol and results in Pompe disease animal model
All tissues were fixed in 10% NBF (neutral buffered formalin). Assays (PAS and IHC) are routinely used in the art.
PAS staining of quadriceps and triceps (FIGS. 29 and 32)Tissues were fixed in 10% NBF and embedded in paraffin. Sections were post-fixed in 1% periodic acid and stained with Schiff reagent. Thereafter, the sections were counterstained with hematoxylin. Glycogen appears as magenta aggregates (lysosome binding) or diffuse pink (cytoplasm); the core is blue. From the images and assuming that each image represents a group, the ranking order with respect to glycogen clearance is: engineered hGAA>Natural hGAA. Throughout the image, the engineered hGAA construct produced more staining than the rest, indicating improved endocytosis of the GAA protein mediated by binding of vIGF2 to CI-MPR.
PAS staining of spinal cord (FIG. 26)Tissues were fixed in 10% NBF. Post-fixation in 1% periodic acid can be performed before or after paraffin embedding. Sections were stained with Schiff reagent and possibly counterstained with methylene blue. Glycogen appears as magenta aggregates (lysosome binding); the nerve fibers appear blue. The images focus on glycogen accumulation in the ventral horn of the spinal cord and motor neurons. In the constructs, the engineered hGAA appeared to be most effective in glycogen reduction.
GAAIHC (FIG. 22, FIG. 25, FIG. 27, FIG. 30 and FIG. 35)Tissues were fixed in 10% NBF and embedded in paraffin. Sections were incubated with anti-GAA primary antibody followed by a secondary antibody that recognized the primary antibody and carried enzyme-labeled HRP. Subsequently, an enzymatic reaction proceeds and a brown precipitated product is formed. The sections were then counterstained with hematoxylin. The construct showed GAA uptake into muscle fibers (fig. 31). Engineered hGAA>Natural hGAA. Throughout the image, the BiP-vIGF2 construct had more diffuse staining than the rest.
Engineered hGAA produced more GAA IHC signal than other vectors, with a spot-like appearance inside the muscle fibers, indicating far more efficient lysosomal targeting (fig. 22).
In summary, engineered hGAA consistently demonstrated superiority in tissue uptake, lysosomal targeting, and glycogen reduction in various tissues in the constructs.
While preferred embodiments of the present invention have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only. Numerous variations, changes, and substitutions will now occur to those skilled in the art without departing from the invention. It should be understood that various alternatives to the embodiments described herein may be employed. It is intended that the following claims define the scope of the invention and that methods and structures within the scope of these claims and their equivalents be covered thereby.
Claims (151)
1. A gene therapy vector comprising a nucleic acid construct comprising:
(a) a nucleic acid sequence encoding a therapeutic protein, and
(b) a nucleic acid sequence encoding a peptide that binds with high affinity to cation-independent mannose 6-phosphate (M6P) receptor (CI-MPR).
2. The gene therapy vector of claim 1, wherein the peptide is a variant IGF2(vIGF2) peptide.
3. The gene therapy vector of claim 2, wherein the vIGF2 peptide comprises an amino acid sequence having at least 90% identity to SEQ ID No. 1 and having at least one substitution at one or more positions selected from the group consisting of: positions 6, 26, 27, 43, 48, 49, 50, 54, 55 and 65 of SEQ ID NO. 1.
4. The gene therapy vector of claim 3, wherein the at least one substitution is selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R, L55R and K65R of SEQ ID NO. 1.
5. The gene therapy vector of claims 3-4, wherein the vIGF2 peptide comprises at least two substitutions at two or more positions selected from the group consisting of: positions 26, 27, 43, 48, 49, 50, 54 and 55 of SEQ ID NO. 1.
6. The gene therapy vector of claim 5, wherein the at least two substitutions are selected from the group consisting of: E6R, F26S, Y27L, V43L, F48T, R49S, S50I, A54R, L55R and K65R of SEQ ID NO. 1.
7. The gene therapy vector of any one of claims 1-6, wherein the vIGF2 peptide comprises an N-terminal deletion at position 1 of SEQ ID No. 1.
8. The gene therapy vector of claim 7, wherein the vIGF2 peptide comprises an N-terminal deletion at positions 1-4 of SEQ ID No. 1.
9. The gene therapy vector of any one of claims 1-8, wherein the vIGF2 peptide has reduced or no affinity for insulin receptor and IGFR1 compared to native IGF2 peptide.
10. The gene therapy vector of claim 9 and wherein the vIGF2 peptide is capable of promoting uptake of the therapeutic protein into lysosomes in cells.
11. The gene therapy vector of any one of claims 1-10, wherein the therapeutic protein is capable of replacing a defective or deficient protein associated with a genetic disorder in a subject having the genetic disorder.
12. The gene therapy vector of claim 11, wherein the genetic disorder is a lysosomal storage disorder.
13. The gene therapy vector of claim 11 or claim 12, wherein the genetic disorder is selected from the group consisting of: aspartyl glucosamine uraemia, Barten disease (Batten disease), cystinosis, Fabry disease (Fabry disease), Gaucher disease type I (Gaucher disease), Gaucher disease type II, Gaucher disease type III, Pompe disease (Pompe disease), Tay Sachs disease (Tay Sachs disease), Sandhoff disease (Sandhoff disease), metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Leehler disease (Hurler disease), Hunter disease (Hunter disease), Sanphyspo disease type A (filappo disease), Sanphyr disease type B, Sanphyrperi disease type C, Pherpheri disease type D, Morquio disease type A (Morquio disease), Morphe disease type B, and Marmy-Nippon disease (Marmy disease), Mary-Nippon disease type A (Mary disease), Mary-type B (Mary-disease, Mary-Nippon disease, Mary-type A, Mary-type I, Mary disease, Mary-type I, and Mary type I, and Mar, Niemann-pick disease type B, niemann-pick disease type C1, niemann-pick disease type C2, sindler disease type I (Schindler disease), sindler disease type II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatosis (CGD), CDKL5 deficiency, and neuronal ceroid lipofuscinosis.
14. The gene therapy vector of claim 11 or claim 12, wherein the genetic disorder is pompe disease.
15. The gene therapy vector of claim 11 or claim 12, wherein the genetic disorder is neuronal ceroid lipofuscinosis.
16. The gene therapy vector of any one of claims 1-15, wherein the therapeutic protein comprises an enzyme selected from the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof.
17. The gene therapy vector of claim 16, wherein the therapeutic protein is an alpha-glucosidase or an enzymatically active fragment thereof.
18. The gene therapy vector of claim 16, wherein the therapeutic protein is a palmitoyl protein thioesterase.
19. The gene therapy vector of claim 1, wherein the nucleic acid construct further comprises a translation initiation sequence.
20. The gene therapy vector of claim 19, wherein the translation initiation sequence comprises a Kozak sequence.
21. The gene therapy vector of claim 20, wherein the Kozak sequence comprises the sequence AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide.
22. The gene therapy vector of claim 21, wherein X1Contains A.
23. The gene therapy vector of claim 21 or claim 22, wherein X2Contains G.
24. The gene therapy vector of claim 21 or claim 22, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29).
25. The gene therapy vector of any one of claims 20-24, wherein the Kozak sequence comprises AAGATGA (SEQ ID NO: 29).
26. The gene therapy vector of claim 20, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
27. The gene therapy vector of claim 20 or claim 26, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
28. The gene therapy vector of claim 1, wherein the nucleic acid construct further comprises a signal nucleic acid sequence encoding a signal peptide, wherein the signal peptide is capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide.
29. The gene therapy vector of claim 28, wherein the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide.
30. The gene therapy vector of claim 29, wherein the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17.
31. The gene therapy vector of claim 29, wherein the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17.
32. The gene therapy vector of claim 28, wherein the signal peptide comprises a Gaussia signal peptide.
33. The gene therapy vector of claim 32, wherein the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32.
34. The gene therapy vector of claim 32, wherein the Gaussia signal peptide comprises SEQ ID NO: 32.
35. The gene therapy vector of any one of claims 1-34, wherein the vIGF2 nucleic acid sequence is 5' to the nucleic acid sequence encoding a therapeutic protein.
36. The gene therapy vector of any one of claims 1-34, wherein the vIGF2 nucleic acid sequence is 3' to the nucleic acid sequence encoding a therapeutic protein.
37. The gene therapy vector of any one of claims 1-36, wherein the nucleic acid construct further comprises a linker sequence encoding a linker peptide between the vIGF2 nucleotide sequence and the nucleic acid sequence encoding a therapeutic protein.
38. The gene therapy vector of claim 37, wherein the linker peptide comprises SEQ ID NOs 18-21, 33, or 37.
39. The gene therapy vector of any one of claims 1-38, wherein the gene therapy vector is a viral vector.
40. The gene therapy vector of claim 39, wherein the viral vector is an adenoviral vector, an adeno-associated virus (AAV) vector, a retroviral vector, a lentiviral vector, a poxviral vector, a vaccinia viral vector, an adenoviral vector, or a herpes viral vector.
41. A pharmaceutical composition comprising a therapeutically effective amount of the gene therapy vector of any one of claims 1 to 40 and a pharmaceutically acceptable carrier or excipient.
42. The pharmaceutical composition of claim 41, wherein the excipient comprises a non-ionic, low-permeability compound, buffer, polymer, salt, or combination thereof.
43. A method for treating a genetic disorder, the method comprising administering to a subject in need thereof a gene therapy vector of any one of claims 1 to 40 or a pharmaceutical composition of claim 41 or claim 42.
44. The method of claim 43, wherein the genetic disorder is a lysosomal storage disorder.
45. The method of claim 43 or claim 44, wherein the genetic disorder is selected from the group consisting of: aspartylglucosaminuria, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Muldoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Hewler disease, Hunter disease, Sanfilippo disease type A, Sanfilippo disease type B, Sanfilippo disease type C, Sanfilippo disease type D, Moquini disease type A, Moquine disease type B, Maruo-Lami disease, Spanish disease, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Singler disease type I, Ocimum disease type II, Addison disease (CGA-severe combined immunodeficiency), and Addison disease (ADD), CDKL5 deficiency and neuronal ceroid lipofuscinosis.
46. The method of claim 43 or claim 44, wherein the genetic disorder is Pompe disease.
47. The method of claim 43 or claim 44, wherein the genetic disorder is neuronal ceroid lipofuscinosis.
48. The method of any one of claims 43-47, wherein the administering is performed intrathecally, intraocularly, intravitreally, transretinally, intravenously, intramuscularly, intraventricularly, intracerebrally, intracerebellarly, intracerebroventricularly, intraparenchymally, subcutaneously, or a combination thereof.
49. The method of any one of claims 43-47, wherein the administering is performed intrathecally.
50. A pharmaceutical composition comprising the gene therapy vector of any one of claims 1 to 40 and a pharmaceutically acceptable carrier or excipient for use in the treatment of a genetic disorder.
51. A pharmaceutical composition comprising the gene therapy vector of any one of claims 1 to 40 and a pharmaceutically acceptable carrier or excipient for use in the preparation of a medicament for the treatment of a genetic disorder.
52. The pharmaceutical composition of claim 50 or claim 51, wherein the genetic disorder is a lysosomal storage disorder.
53. The pharmaceutical composition of any one of claims 50-52, wherein the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Hewlett-packard disease, Hunter disease, sanfilippo disease type A, sanfilippo disease type B, sanfilippo disease type C, sanfilippo disease type D, moquinase type A, moquinase type B, maratoto-lamic disease, sjgren disease, niemann-pick disease type A, niemann-pick disease type B, niemann-pick disease type C1, niemann-pick disease type C2, sinderler disease type I, sinderler disease type II, adenosine deaminase severe combined immunodeficiency disease (ADA-SCID), Chronic Granulomatosis (CGD), and neuronal ceroid lipofuscinosis.
54. The pharmaceutical composition of any one of claims 50-53, wherein the genetic disorder is Pompe disease.
55. The pharmaceutical composition of any one of claims 50-53, wherein the genetic disorder is neuronal ceroid lipofuscinosis.
56. The pharmaceutical composition of any one of claims 50-55, wherein the composition is formulated for intrathecal, intraocular, intravitreal, transretinal, intravenous, intramuscular, intraventricular, intracerebral, intracerebellar, or subcutaneous administration.
57. The pharmaceutical composition of any one of claims 50-56, wherein the composition is formulated for intrathecal administration.
58. A gene therapy vector comprising a nucleic acid construct comprising, in 5 'to 3' order:
(a) a translation initiation sequence, and
(b) a nucleic acid sequence encoding a therapeutic protein.
59. The gene therapy vector of claim 58, wherein the translation initiation sequence comprises a Kozak sequence.
60. The gene therapy vector of claim 58, wherein the Kozak sequence comprises the sequence AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide.
61. The gene therapy vector of claim 59, wherein X1Contains A.
62. The gene therapy vector of claim 59 or claim 60, wherein X2Contains G.
63. The gene therapy vector of any one of claims 58-61, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29).
64. The gene therapy vector of any one of claims 58-62, wherein the Kozak sequence comprises AAGATGA (SEQ ID NO: 29).
65. The gene therapy vector of claim 58, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
66. The gene therapy vector of claim 58 or claim 64, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
67. The gene therapy vector of any one of claims 58-65, wherein the nucleic acid construct further comprises a nucleic acid sequence encoding a signal peptide capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide.
68. The gene therapy vector of claim 66, wherein the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide.
69. The gene therapy vector of claim 67, wherein the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17.
70. The gene therapy vector of claim 67, wherein the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17.
71. The gene therapy vector of claim 67, wherein the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32.
72. The gene therapy vector of claim 67, wherein the Gaussia signal peptide comprises the amino acid sequence of SEQ ID NO. 32.
73. The gene therapy vector of any one of claims 58-71, wherein the nucleic acid construct further comprises an Internal Ribosome Entry Sequence (IRES).
74. The gene therapy vector of claim 72, wherein the IRES is a cricket paralysis virus (CrPV) IRES.
75. The gene therapy vector of claim 72 or claim 73, wherein the IRES comprises a nucleic acid sequence having at least 90% identity to SEQ ID NO 12.
76. The gene therapy vector of claim 72 or claim 73, wherein the IRES comprises SEQ ID NO 12.
77. A gene therapy vector comprising a nucleic acid construct comprising, in 5 'to 3' order:
(a) a nucleic acid sequence encoding a signal peptide, and
(b) a nucleic acid sequence encoding a therapeutic protein, wherein the signal peptide is capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide.
78. The gene therapy vector of claim 76, wherein the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide.
79. The gene therapy vector of claim 77, wherein the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17.
80. The gene therapy vector of claim 77, wherein the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOS 13-17.
81. The gene therapy vector of any one of claims 76-79, wherein the signal peptide comprises a Gaussia signal peptide.
82. The gene therapy vector of claim 80, wherein the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32.
83. The gene therapy vector of claim 80, wherein the Gaussia signal peptide comprises SEQ ID NO 32.
84. The gene therapy vector of any one of claims 76-82, wherein the nucleic acid construct further comprises a translation initiation sequence.
85. The gene therapy vector of claim 83, wherein the translation initiation sequence comprises a Kozak sequence comprising sequence AX 1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of the above.
86. The gene therapy vector of claim 84, wherein X1Contains A.
87. The gene therapy vector of claim 84 or claim 85, wherein X2Contains G.
88. The gene therapy vector of any one of claims 84-86, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29).
89. The gene therapy vector of any one of claims 84-87, wherein the Kozak sequence comprises AAGATGA (SEQ ID NO: 29).
90. The gene therapy vector of claim 83, wherein the translation initiation sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
91. The gene therapy vector of claim 83 or claim 89, wherein the translation initiation sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
92. The gene therapy vector of any one of claims 76-90, wherein the nucleic acid construct further comprises an Internal Ribosome Entry Sequence (IRES).
93. The gene therapy vector of claim 91, wherein the IRES comprises an IRES selected from the group consisting of: cricket paralysis virus (CrPV) IRES, picornavirus IRES, foot and mouth disease virus IRES, Kaposi's sarcoma-associated herpesvirus IRES, hepatitis a IRES, hepatitis c IRES, pestivirus IRES, cricket paralysis virus (criptavirus) IRES, Rhopalosiphum palustris (Rhopalosiphum padi) virus IRES, and Merek's disease virus IRES.
94. The gene therapy vector of claim 91 or claim 92, wherein the IRES comprises a nucleic acid sequence having at least 90% identity to SEQ ID NO 12.
95. The gene therapy vector of any one of claims 91-93, wherein the IRES comprises SEQ ID NO 12.
96. A gene therapy vector comprising a nucleic acid construct comprising, in 5 'to 3' order:
(a) an Internal Ribosome Entry Sequence (IRES), and
(b) a nucleic acid sequence encoding a therapeutic protein.
97. The gene therapy vector of claim 95, wherein the IRES comprises an IRES selected from the group consisting of: cricket paralysis virus (CrPV) IRES, picornavirus IRES, foot and mouth disease virus IRES, Kaposi sarcoma-associated herpesvirus IRES, hepatitis A IRES, hepatitis C IRES, pestivirus IRES, cricket paralysis virus IRES, gloomycosis graminearum virus IRES, and Merrex virus IRES.
98. The gene therapy vector of claim 96, wherein the IRES is a cricket paralysis virus (CrPV) IRES.
99. The gene therapy vector of claim 96 or claim 97, wherein the IRES comprises a nucleic acid sequence having at least 90% identity to SEQ ID NO 12.
100. The gene therapy vector of any one of claims 96-98, wherein the IRES comprises SEQ ID NO 12.
101. The gene therapy vector of any one of claims 95-99, wherein the nucleic acid construct further comprises a translation initiation sequence.
102. The gene therapy vector of claim 100, wherein the translation initiation sequence comprises a Kozak sequence comprising AX1X2ATGA (SEQ ID NO:28), wherein X1And X2Each of which is any nucleotide.
103. The gene therapy vector of claim 101, wherein X1Contains A.
104. The gene therapy vector of claim 101 or claim 102, wherein X2Contains G.
105. The gene therapy vector of any one of claims 101-103, wherein the Kozak sequence comprises a nucleic acid sequence having at least 90% identity to AAGATGA (SEQ ID NO: 29).
106. The gene therapy vector of any one of claims 101-104, wherein the Kozak sequence comprises AAGATGA (SEQ ID NO: 29).
107. The gene therapy vector of claim 100, wherein the translation initiation sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
108. The gene therapy vector of claim 100 or claim 106, wherein the translation initiation sequence comprises a nucleic acid sequence having at least 90% identity to GCAAGATG (SEQ ID NO: 44).
109. The gene therapy vector of any one of claims 95-107, wherein the nucleic acid construct further comprises a nucleic acid sequence encoding a signal peptide capable of increasing secretion of the therapeutic protein compared to the therapeutic protein without the signal peptide.
110. The gene therapy vector of claim 106, wherein the signal peptide is selected from the group consisting of a Binding Immunoglobulin Protein (BiP) signal peptide and a Gaussia signal peptide.
111. The gene therapy vector of claim 109, wherein the BiP signal peptide comprises an amino acid sequence having at least 90% identity to an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17.
112. The gene therapy vector of claim 109, wherein the BiP signal peptide comprises an amino acid sequence selected from the group consisting of SEQ ID NOs 13-17.
113. The gene therapy vector of claim 109, wherein the Gaussia signal peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NO: 32.
114. The gene therapy vector of claim 109, wherein the Gaussia signal peptide comprises SEQ ID NO: 32.
115. The gene therapy vector of any one of claims 58-113, wherein the nucleic acid construct further comprises a nucleic acid sequence encoding an IGF2 peptide.
116. The gene therapy vector of claim 114, wherein the IGF2 peptide is a vIGF2 peptide comprising an amino acid sequence at least 90% identical to an amino acid sequence selected from the group consisting of: 1-11, 30-31, 34 and 35.
117. The gene therapy vector of claim 114, wherein the IGF2 peptide is a vIGF2 peptide comprising an amino acid sequence selected from the group consisting of: 1-11, 30-31, 34 and 35.
118. The gene therapy vector of any one of claims 114-116, wherein the vIGF2 nucleic acid sequence is 5' to a nucleic acid sequence encoding a therapeutic protein.
119. The gene therapy vector of any one of claims 114-116, wherein the vIGF2 nucleic acid sequence is 3' to a nucleic acid sequence encoding a therapeutic protein.
120. The gene therapy vector of any one of claims 114-118, wherein the nucleic acid construct further comprises a linker sequence encoding a linker peptide between the vIGF2 nucleotide sequence and the nucleic acid sequence encoding a therapeutic protein.
121. The gene therapy vector of claim 119, wherein the linker peptide comprises an amino acid sequence having at least 90% identity to SEQ ID NOs 18-21, 33, or 37.
122. The gene therapy vector of claim 119, wherein the linker peptide comprises SEQ ID NOs 18-21, 33, or 37.
123. The gene therapy vector of any one of claims 58-121, wherein the therapeutic protein is selected from the group consisting of: alpha-galactosidase (A or B), beta-galactosidase, beta-hexosidase (A or B), galactosylceramidase, arylsulfatase (A or B), beta-glucocerebrosidase, lysosomal acid lipase, lysosomal enzyme acid sphingomyelinase, formylglycine generating enzyme, iduronidase (e.g., alpha-L), acetyl-CoA alpha-glucosaminyl N-acetyltransferase, glucosaminoglycan alpha-L-iduronate hydrolase, heparan N-sulfatase, N-acetyl-alpha-D-glucosaminidase (NAGLU), iduronate-2-sulfatase, galactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, and the like, Glycosaminoglycan N-acetylgalactosamine 4-sulfatase, β -glucuronidase, hyaluronidase, α -N-acetylneuraminidase (sialidase), gangliosidase, phosphotransferase, α -glucosidase, α -D-mannosidase, β -D-mannosidase, aspartylglucosaminidase, α -L-fucosidase, batttenin, palmitoyl protein thioesterase, and other Batten-related proteins (e.g., ceroid-lipofuscinosis neuron protein 6), or enzymatically active fragments thereof.
124. The gene therapy vector of claim 122, wherein the therapeutic protein is an alpha-galactosidase or enzymatically active fragment thereof.
125. The gene therapy vector of claim 122, wherein the enzyme is a palmitoyl protein thioesterase.
126. The gene therapy vector of claim 122, wherein the therapeutic protein is capable of replacing a defective or deficient protein associated with a genetic disorder in a subject having the genetic disorder.
127. The gene therapy vector of claim 125, wherein the genetic disorder is a lysosomal storage disorder.
128. The gene therapy vector of claim 125, wherein the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease type A, san Francisella disease type B, san Francisella disease type C, san Francisella disease type D, morqui disease type A, morqui disease type B, Maruo-lamide disease, Spirosis, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Sindler disease type I, Sindler disease type II, adenosine deaminase severe combined immunodeficiency (ADA-SCID), and neuronal ceroid lipofuscinosis.
129. The gene therapy vector of claim 125, wherein the genetic disorder is pompe disease.
130. The gene therapy vector of claim 125, wherein the genetic disorder is neuronal ceroid lipofuscinosis.
131. The gene therapy vector of any one of claims 58-129, wherein the gene therapy vector is a viral vector.
132. The gene therapy vector of claim 130, wherein the viral vector is an adeno-associated viral vector, a retroviral vector, a lentiviral vector, a poxviral vector, a vaccinia viral vector, an adenoviral vector, or a herpesvirus vector.
133. A pharmaceutical composition comprising (i) a therapeutically effective amount of the gene therapy vector of any one of claims 58-131 and (ii) a pharmaceutically acceptable carrier or excipient.
134. The pharmaceutical composition of claim 132, wherein said carrier or excipient comprises a non-ionic low-permeability compound, buffer, polymer, salt, or combination thereof.
135. A method for treating a genetic disorder, the method comprising administering to a subject in need thereof a gene therapy vector of any one of claims 58-131 or a pharmaceutical composition of claim 132 or claim 133.
136. The method of claim 134, wherein the genetic disorder is a lysosomal storage disorder.
137. The method of claim 134 or claim 135, wherein the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease type A, san Francisella disease type B, san Francisella disease type C, san Francisella disease type D, morqui disease type A, morqui disease type B, Maruo-lamide disease, Spirosis, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Sindler disease type I, Sindler disease type II, adenosine deaminase severe combined immunodeficiency (ADA-SCID), and neuronal ceroid lipofuscinosis.
138. The method of claim 134 or claim 135, wherein the genetic disorder is pompe disease.
139. The method of claim 134 or claim 135, wherein the genetic disorder is neuronal ceroid lipofuscinosis.
140. The method of any one of claims 134-138, wherein the administering is performed intrathecally, intravenously, intramuscularly, intraventricularly, intracerebrally, intracerebroventricularly, intraparenchymally, ocularly, subcutaneously, or a combination thereof.
141. The method of any one of claims 134-139, wherein the administering is performed intrathecally.
142. A pharmaceutical composition comprising the gene therapy vector of any one of claims 58-131 and a pharmaceutically acceptable carrier or excipient for use in the treatment of a genetic disorder.
143. A pharmaceutical composition comprising the gene therapy vector of any one of claims 58-131 and a pharmaceutically acceptable carrier or excipient for use in the preparation of a medicament for the treatment of a genetic disorder.
144. The pharmaceutical composition of claim 141 or claim 142, wherein the genetic disorder is a lysosomal storage disorder.
145. The pharmaceutical composition of any one of claims 141-143, wherein the genetic disorder is selected from the group consisting of: aspartylglucamine uremia, Barten disease, cystinosis, Fabry disease, gaucher disease type I, gaucher disease type II, gaucher disease type III, Pompe disease, Tay-saxophone disease, Sandohoff disease, metachromatic leukodystrophy, mucolipidosis type I, mucolipidosis type II, mucolipidosis type III, mucolipidosis type IV, Heller disease, hunter's disease, san Francisella disease type A, san Francisella disease type B, san Francisella disease type C, san Francisella disease type D, morqui disease type A, morqui disease type B, Maruo-lamide disease, Spirosis, Niemann-pick disease type A, Niemann-pick disease type B, Niemann-pick disease type C1, Niemann-pick disease type C2, Sindler disease type I, Sindler disease type II, adenosine deaminase severe combined immunodeficiency (ADA-SCID), and neuronal ceroid lipofuscinosis.
146. The pharmaceutical composition of any one of claims 141-144, wherein the genetic disorder is pompe disease.
147. The pharmaceutical composition of any one of claims 141-144, wherein the genetic disorder is neuronal ceroid lipofuscinosis.
148. The pharmaceutical composition of any one of claims 141-146, wherein said composition is formulated for intrathecal, intravenous, intramuscular, intraventricular, intracerebral, intracardial, ocular, or subcutaneous administration.
149. The pharmaceutical composition of any one of claims 141-147, wherein the composition is formulated for intrathecal administration.
150. A gene therapy vector comprising a nucleic acid construct comprising:
(a) a nucleic acid sequence encoding a therapeutic protein, and
(b) a nucleic acid sequence encoding a peptide that increases endocytosis of the therapeutic protein.
151. The gene therapy vector of claim 149, wherein the peptide that increases endocytosis of the therapeutic protein is a peptide that binds CI-MPR.
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862664741P | 2018-04-30 | 2018-04-30 | |
| US62/664,741 | 2018-04-30 | ||
| US201862688640P | 2018-06-22 | 2018-06-22 | |
| US62/688,640 | 2018-06-22 | ||
| US201862744068P | 2018-10-10 | 2018-10-10 | |
| US62/744,068 | 2018-10-10 | ||
| PCT/US2019/030076 WO2019213180A1 (en) | 2018-04-30 | 2019-04-30 | Gene therapy constructs and methods of use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112513071A true CN112513071A (en) | 2021-03-16 |
Family
ID=66669057
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980029113.5A Pending CN112513071A (en) | 2018-04-30 | 2019-04-30 | Gene therapy constructs and methods of use |
Country Status (16)
| Country | Link |
|---|---|
| US (3) | US10874750B2 (en) |
| EP (1) | EP3788064A1 (en) |
| JP (2) | JP2021521851A (en) |
| KR (1) | KR20210005154A (en) |
| CN (1) | CN112513071A (en) |
| AU (1) | AU2019261982A1 (en) |
| BR (1) | BR112020021962A2 (en) |
| CA (1) | CA3098674A1 (en) |
| CL (1) | CL2020002809A1 (en) |
| CO (1) | CO2020014729A2 (en) |
| MX (1) | MX2020011514A (en) |
| PH (1) | PH12020551818A1 (en) |
| SG (1) | SG11202010533WA (en) |
| TW (2) | TWI808169B (en) |
| WO (1) | WO2019213180A1 (en) |
| ZA (1) | ZA202006905B (en) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2019261982A1 (en) | 2018-04-30 | 2020-10-15 | Amicus Therapeutics, Inc. | Gene therapy constructs and methods of use |
| WO2020077114A2 (en) | 2018-10-10 | 2020-04-16 | Amicus Therapeutics, Inc. | Disulfide bond stabilized polypeptide compositions and methods of use |
| EP4041284A4 (en) * | 2019-10-10 | 2023-10-18 | Amicus Therapeutics, Inc. | Variant igf2 constructs |
| IL293068A (en) * | 2019-11-19 | 2022-07-01 | Asklepios Biopharmaceutical Inc | Therapeutic adeno-associated virus comprising liver-specific promoters for treating pompe disease and lysosomal disorders |
| IL298178A (en) * | 2020-05-14 | 2023-01-01 | Univ Pennsylvania | Compositions useful for treatment of pompe disease |
| MX2023000815A (en) | 2020-07-27 | 2023-04-11 | Voyager Therapeutics Inc | Compositions and methods for the treatment of neurological disorders related to glucosylceramidase beta deficiency. |
| WO2022026410A2 (en) | 2020-07-27 | 2022-02-03 | Voyager Therapeutics, Inc | Compositions and methods for the treatment of niemann-pick type c1 disease |
| US20220153869A1 (en) * | 2020-11-16 | 2022-05-19 | Tavotek Biotherapeutics (Hong Kong) Limited | Shielded and homing bispecific antibody that simultaneously inhibits angiogenic pathway targets and her2 family proteins and uses thereof |
| IL303239A (en) | 2020-12-01 | 2023-07-01 | Univ Pennsylvania | Compositions and uses thereof for treatment of angelman syndrome |
| WO2023086939A1 (en) * | 2021-11-12 | 2023-05-19 | Amicus Therapeutics, Inc. | Compositions and methods for treating mucopolysaccharidosis iiia |
| WO2023091949A2 (en) | 2021-11-17 | 2023-05-25 | Voyager Therapeutics, Inc. | Compositions and methods for the treatment of neurological disorders related to glucosylceramidase beta deficiency |
| WO2023240236A1 (en) | 2022-06-10 | 2023-12-14 | Voyager Therapeutics, Inc. | Compositions and methods for the treatment of spinal muscular atrophy related disorders |
| WO2024151982A1 (en) | 2023-01-13 | 2024-07-18 | Amicus Therapeutics, Inc. | Gene therapy constructs for the treatment of pompe disease |
| WO2024163012A1 (en) | 2023-02-02 | 2024-08-08 | Voyager Therapeutics, Inc. | Compositions and methods for the treatment of neurological disorders related to glucosylceramidase beta deficiency |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1280616A (en) * | 1997-10-03 | 2001-01-17 | 耶路撒冷希伯来大学伊森姆研究发展公司 | Method and compositions for inducing tumor-specific cytotoxicity |
| DE10237639A1 (en) * | 2002-08-13 | 2004-02-26 | Martin-Luther-Universität Halle-Wittenberg | Preparing virus-like particles, useful as gene therapy vectors, comprises assembling capsomers, optionally with active molecules attached, in presence of nonionic stabilizers |
| CN1903374A (en) * | 2005-07-27 | 2007-01-31 | 中国人民解放军军事医学科学院生物医学分析中心 | Application of colon bacillus for preparing medicine for diagnosis and treatment of tumor |
| US20070212375A1 (en) * | 2004-04-30 | 2007-09-13 | Caston Jose R | Process For Producing In Yeast Empty Viral Capsids Consisting Of Proteins Derived From Pvp2 Of The Infectious Bursal Disease Virus (Ibdv) |
| CN103338784A (en) * | 2010-11-30 | 2013-10-02 | 奥菲泽米有限公司 | Methods for increasing intracellular activity of Hsp70 |
| CN103796672A (en) * | 2011-06-10 | 2014-05-14 | 普洛特拉有限公司 | Pharmaceutical compositions containing proteases and methods for the treatment of lysosomal storage diseases |
| CN104822701A (en) * | 2012-11-27 | 2015-08-05 | 生物马林药物股份有限公司 | Targeted therapeutic lysosomal enzyme fusion proteins and uses thereof |
| CN105189742A (en) * | 2013-03-15 | 2015-12-23 | 阿米库斯治疗学公司 | Chemical crosslinkers |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001029058A1 (en) | 1999-10-15 | 2001-04-26 | University Of Massachusetts | Rna interference pathway genes as tools for targeted genetic interference |
| US6326193B1 (en) | 1999-11-05 | 2001-12-04 | Cambria Biosciences, Llc | Insect control agent |
| EP1118334A1 (en) | 2000-01-11 | 2001-07-25 | Aventis Behring Gesellschaft mit beschränkter Haftung | Method for the production of conjugates and uses thereof for the prevention and treatment of allergic reactions and autoimmune diseases |
| WO2001096584A2 (en) | 2000-06-12 | 2001-12-20 | Akkadix Corporation | Materials and methods for the control of nematodes |
| US7396811B2 (en) | 2001-04-30 | 2008-07-08 | Zystor Therapeutics, Inc. | Subcellular targeting of therapeutic proteins |
| US7629309B2 (en) | 2002-05-29 | 2009-12-08 | Zystor Therapeutics, Inc. | Targeted therapeutic proteins |
| US7560424B2 (en) | 2001-04-30 | 2009-07-14 | Zystor Therapeutics, Inc. | Targeted therapeutic proteins |
| US20030072761A1 (en) | 2001-10-16 | 2003-04-17 | Lebowitz Jonathan | Methods and compositions for targeting proteins across the blood brain barrier |
| JP2005506340A (en) * | 2001-10-16 | 2005-03-03 | シムバイオンティクス インコーポレイテッド | Methods and compositions for targeting underglycosylated proteins across the blood brain barrier |
| ATE521701T1 (en) | 2003-01-22 | 2011-09-15 | Univ Duke | IMPROVED CONSTRUCTS FOR EXPRESSING LYSOSOMAL POLYPEPTIDES |
| EP3513814A1 (en) | 2003-05-01 | 2019-07-24 | Genzyme Corporation | Gene therapy for neurometabolic disorders |
| GB0314856D0 (en) * | 2003-06-25 | 2003-07-30 | Unitargeting Res As | Protein expression system |
| US7355018B2 (en) | 2003-09-30 | 2008-04-08 | Regeneron Pharmaceuticals, Inc. | Modified IGF1 polypeptides with increased stability and potency |
| EP1716232B9 (en) | 2004-02-10 | 2010-10-13 | ZyStor Therapeutics , Inc. | Acid alpha-glucosidase and fragments thereof |
| WO2006007560A2 (en) | 2004-07-01 | 2006-01-19 | University Of Pennsylvania | Targeted protein replacement for the treatment of lysosomal storage disorders |
| AU2007322123A1 (en) | 2006-11-13 | 2008-05-29 | Biomarin Pharmaceutical Inc. | Methods for treating Pompe disease |
| HUE034850T2 (en) | 2008-05-07 | 2018-03-28 | Biomarin Pharm Inc | Lysosomal targeting peptides and uses thereof |
| LT3912643T (en) | 2009-02-13 | 2023-02-10 | Immunomedics Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| CA2818689C (en) | 2010-11-22 | 2021-09-21 | Callidus Biopharma, Inc. | Novel signal sequences to improve protein expression and secretion of recombinant enzymes and other proteins |
| US8580922B2 (en) | 2011-03-04 | 2013-11-12 | Shire Human Genetic Therapies, Inc. | Peptide linkers for polypeptide compositions and methods for using same |
| CN103443273B (en) | 2011-03-16 | 2016-10-26 | 天野酶制品株式会社 | Modification type alpha-glucosidase and application thereof |
| ES2660185T3 (en) | 2011-05-27 | 2018-03-21 | Amicus Therapeutics, Inc. | Methods for coupling recombinant lysosomal enzyme targeting peptides to optimize the treatments of lysosomal deposition diseases |
| US20160243260A1 (en) | 2013-10-24 | 2016-08-25 | Uniqure Ip B.V. | Treatment of neurological diseases using adeno-associated virus (AAV) comprising AAV-5 capsid proteins |
| WO2015085238A1 (en) | 2013-12-05 | 2015-06-11 | The Regents Of The University Of California, A California Corporation | Inhibitors of lpxc |
| AU2019261982A1 (en) | 2018-04-30 | 2020-10-15 | Amicus Therapeutics, Inc. | Gene therapy constructs and methods of use |
-
2019
- 2019-04-30 AU AU2019261982A patent/AU2019261982A1/en active Pending
- 2019-04-30 TW TW108115137A patent/TWI808169B/en active
- 2019-04-30 BR BR112020021962-2A patent/BR112020021962A2/en unknown
- 2019-04-30 SG SG11202010533WA patent/SG11202010533WA/en unknown
- 2019-04-30 JP JP2020560355A patent/JP2021521851A/en active Pending
- 2019-04-30 CA CA3098674A patent/CA3098674A1/en active Pending
- 2019-04-30 TW TW112122938A patent/TW202344691A/en unknown
- 2019-04-30 MX MX2020011514A patent/MX2020011514A/en unknown
- 2019-04-30 KR KR1020207033891A patent/KR20210005154A/en unknown
- 2019-04-30 US US16/399,979 patent/US10874750B2/en active Active
- 2019-04-30 EP EP19727170.3A patent/EP3788064A1/en active Pending
- 2019-04-30 WO PCT/US2019/030076 patent/WO2019213180A1/en unknown
- 2019-04-30 CN CN201980029113.5A patent/CN112513071A/en active Pending
-
2020
- 2020-10-29 CL CL2020002809A patent/CL2020002809A1/en unknown
- 2020-10-30 PH PH12020551818A patent/PH12020551818A1/en unknown
- 2020-11-05 ZA ZA2020/06905A patent/ZA202006905B/en unknown
- 2020-11-27 CO CONC2020/0014729A patent/CO2020014729A2/en unknown
- 2020-12-18 US US17/127,001 patent/US11491243B2/en active Active
-
2022
- 2022-11-07 US US18/053,160 patent/US20230233711A1/en active Pending
-
2024
- 2024-04-19 JP JP2024068431A patent/JP2024119788A/en active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1280616A (en) * | 1997-10-03 | 2001-01-17 | 耶路撒冷希伯来大学伊森姆研究发展公司 | Method and compositions for inducing tumor-specific cytotoxicity |
| DE10237639A1 (en) * | 2002-08-13 | 2004-02-26 | Martin-Luther-Universität Halle-Wittenberg | Preparing virus-like particles, useful as gene therapy vectors, comprises assembling capsomers, optionally with active molecules attached, in presence of nonionic stabilizers |
| US20070212375A1 (en) * | 2004-04-30 | 2007-09-13 | Caston Jose R | Process For Producing In Yeast Empty Viral Capsids Consisting Of Proteins Derived From Pvp2 Of The Infectious Bursal Disease Virus (Ibdv) |
| CN1903374A (en) * | 2005-07-27 | 2007-01-31 | 中国人民解放军军事医学科学院生物医学分析中心 | Application of colon bacillus for preparing medicine for diagnosis and treatment of tumor |
| CN103338784A (en) * | 2010-11-30 | 2013-10-02 | 奥菲泽米有限公司 | Methods for increasing intracellular activity of Hsp70 |
| CN103796672A (en) * | 2011-06-10 | 2014-05-14 | 普洛特拉有限公司 | Pharmaceutical compositions containing proteases and methods for the treatment of lysosomal storage diseases |
| CN104822701A (en) * | 2012-11-27 | 2015-08-05 | 生物马林药物股份有限公司 | Targeted therapeutic lysosomal enzyme fusion proteins and uses thereof |
| CN105189742A (en) * | 2013-03-15 | 2015-12-23 | 阿米库斯治疗学公司 | Chemical crosslinkers |
Non-Patent Citations (1)
| Title |
|---|
| FRANCESCO PUZZO: "Rescue of pompe disease in mice by AAV-mediated liver delivery of secretable acid a-glucosidase", 《SCIENCE TRANSLATIONAL MEDICINE》, pages 1 - 12 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20230233711A1 (en) | 2023-07-27 |
| PH12020551818A1 (en) | 2021-07-26 |
| ZA202006905B (en) | 2022-03-30 |
| TW202344691A (en) | 2023-11-16 |
| EP3788064A1 (en) | 2021-03-10 |
| WO2019213180A1 (en) | 2019-11-07 |
| JP2024119788A (en) | 2024-09-03 |
| CO2020014729A2 (en) | 2020-12-21 |
| AU2019261982A1 (en) | 2020-10-15 |
| TW202003053A (en) | 2020-01-16 |
| CA3098674A1 (en) | 2019-11-07 |
| MX2020011514A (en) | 2020-12-09 |
| JP2021521851A (en) | 2021-08-30 |
| BR112020021962A2 (en) | 2021-01-26 |
| US11491243B2 (en) | 2022-11-08 |
| CL2020002809A1 (en) | 2021-04-30 |
| KR20210005154A (en) | 2021-01-13 |
| TWI808169B (en) | 2023-07-11 |
| US20190343968A1 (en) | 2019-11-14 |
| US20210162075A1 (en) | 2021-06-03 |
| SG11202010533WA (en) | 2020-11-27 |
| US10874750B2 (en) | 2020-12-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11491243B2 (en) | Gene therapy constructs and methods of use | |
| US20240091321A1 (en) | Variant igf2 constructs | |
| US12064485B2 (en) | Disulfide bond stabilized polypeptide compositions and methods of use | |
| JP2022538497A (en) | Vector compositions and methods of their use for treating lysosomal storage diseases | |
| WO2023150051A1 (en) | Compositions and methods of using two-promoter vector for treatment of lysosomal storage disorders | |
| Ioannou et al. | Gene therapy for lysosomal storage disorders | |
| US20240318157A1 (en) | Neurotensin Variants And Tagged Proteins Comprising Neurotensin Or Sortilin Propeptide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 40039189 Country of ref document: HK |