CN111315378A - 包含lsz102和瑞博西尼的药物组合 - Google Patents
包含lsz102和瑞博西尼的药物组合 Download PDFInfo
- Publication number
- CN111315378A CN111315378A CN201880072600.5A CN201880072600A CN111315378A CN 111315378 A CN111315378 A CN 111315378A CN 201880072600 A CN201880072600 A CN 201880072600A CN 111315378 A CN111315378 A CN 111315378A
- Authority
- CN
- China
- Prior art keywords
- day
- lsz102
- pharmaceutically acceptable
- treatment
- breast cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- SJXNPGGVGZXKKI-NYYWCZLTSA-N (E)-3-[4-[[2-[2-(1,1-difluoroethyl)-4-fluorophenyl]-6-hydroxy-1-benzothiophen-3-yl]oxy]phenyl]prop-2-enoic acid Chemical compound FC(C)(F)C1=C(C=CC(=C1)F)C1=C(C2=C(S1)C=C(C=C2)O)OC1=CC=C(C=C1)/C=C/C(=O)O SJXNPGGVGZXKKI-NYYWCZLTSA-N 0.000 title claims abstract description 105
- RHXHGRAEPCAFML-UHFFFAOYSA-N 7-cyclopentyl-n,n-dimethyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=C2N(C3CCCC3)C(C(=O)N(C)C)=CC2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 RHXHGRAEPCAFML-UHFFFAOYSA-N 0.000 title claims abstract description 51
- 229950003687 ribociclib Drugs 0.000 title abstract description 29
- 238000011282 treatment Methods 0.000 claims abstract description 60
- 238000000034 method Methods 0.000 claims abstract description 24
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 23
- 150000003839 salts Chemical class 0.000 claims description 53
- 102100038595 Estrogen receptor Human genes 0.000 claims description 24
- 230000035772 mutation Effects 0.000 claims description 24
- 239000003814 drug Substances 0.000 claims description 18
- 201000007281 estrogen-receptor positive breast cancer Diseases 0.000 claims description 18
- 239000003937 drug carrier Substances 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 7
- 101000882584 Homo sapiens Estrogen receptor Proteins 0.000 claims description 5
- 239000006186 oral dosage form Substances 0.000 claims description 4
- 206010028980 Neoplasm Diseases 0.000 abstract description 50
- 239000000203 mixture Substances 0.000 abstract description 31
- 201000011510 cancer Diseases 0.000 abstract description 16
- 230000005764 inhibitory process Effects 0.000 abstract description 15
- 238000006731 degradation reaction Methods 0.000 abstract description 12
- 230000015556 catabolic process Effects 0.000 abstract description 11
- 108010038795 estrogen receptors Proteins 0.000 abstract description 8
- 101000715943 Caenorhabditis elegans Cyclin-dependent kinase 4 homolog Proteins 0.000 abstract description 6
- 230000002265 prevention Effects 0.000 abstract description 4
- 102000015694 estrogen receptors Human genes 0.000 abstract description 3
- 230000009286 beneficial effect Effects 0.000 abstract description 2
- 150000001875 compounds Chemical class 0.000 description 48
- 210000004027 cell Anatomy 0.000 description 46
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 34
- 206010006187 Breast cancer Diseases 0.000 description 33
- 230000000694 effects Effects 0.000 description 32
- 208000026310 Breast neoplasm Diseases 0.000 description 31
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 30
- 229960002258 fulvestrant Drugs 0.000 description 30
- 241000699670 Mus sp. Species 0.000 description 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 17
- 229960001603 tamoxifen Drugs 0.000 description 17
- 229910052805 deuterium Inorganic materials 0.000 description 16
- 239000003795 chemical substances by application Substances 0.000 description 15
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 14
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 12
- 201000010099 disease Diseases 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 108020004999 messenger RNA Proteins 0.000 description 11
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- 230000000259 anti-tumor effect Effects 0.000 description 9
- 230000004663 cell proliferation Effects 0.000 description 9
- 229960005309 estradiol Drugs 0.000 description 9
- 229930182833 estradiol Natural products 0.000 description 9
- 230000014509 gene expression Effects 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 241000282414 Homo sapiens Species 0.000 description 7
- 239000002552 dosage form Substances 0.000 description 7
- 231100000371 dose-limiting toxicity Toxicity 0.000 description 7
- 229940011871 estrogen Drugs 0.000 description 7
- 239000000262 estrogen Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 6
- -1 alkaline earth metal salts Chemical class 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 102000003998 progesterone receptors Human genes 0.000 description 6
- 108090000468 progesterone receptors Proteins 0.000 description 6
- 229940125944 selective estrogen receptor degrader Drugs 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 5
- 108050004787 GREB1 Proteins 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 206010028813 Nausea Diseases 0.000 description 5
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- 206010047700 Vomiting Diseases 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 238000012054 celltiter-glo Methods 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- 230000001394 metastastic effect Effects 0.000 description 5
- 206010061289 metastatic neoplasm Diseases 0.000 description 5
- 230000008693 nausea Effects 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 235000018102 proteins Nutrition 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 4
- 206010012735 Diarrhoea Diseases 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- 206010014759 Endometrial neoplasm Diseases 0.000 description 4
- 108010007005 Estrogen Receptor alpha Proteins 0.000 description 4
- 102000016251 GREB1 Human genes 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 4
- 206010061535 Ovarian neoplasm Diseases 0.000 description 4
- 241000282320 Panthera leo Species 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 4
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 4
- 102100038042 Retinoblastoma-associated protein Human genes 0.000 description 4
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 4
- 208000002495 Uterine Neoplasms Diseases 0.000 description 4
- 210000000577 adipose tissue Anatomy 0.000 description 4
- 230000001833 anti-estrogenic effect Effects 0.000 description 4
- 239000003963 antioxidant agent Substances 0.000 description 4
- 235000006708 antioxidants Nutrition 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 238000001516 cell proliferation assay Methods 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 239000003995 emulsifying agent Substances 0.000 description 4
- 238000009261 endocrine therapy Methods 0.000 description 4
- 239000000328 estrogen antagonist Substances 0.000 description 4
- 235000019441 ethanol Nutrition 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 239000006166 lysate Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 150000007522 mineralic acids Chemical class 0.000 description 4
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 239000008188 pellet Substances 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 4
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 230000008673 vomiting Effects 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- 206010014733 Endometrial cancer Diseases 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 3
- 240000007472 Leucaena leucocephala Species 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 206010033128 Ovarian cancer Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 238000010222 PCR analysis Methods 0.000 description 3
- 206010060862 Prostate cancer Diseases 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 235000010419 agar Nutrition 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 229940046836 anti-estrogen Drugs 0.000 description 3
- 230000001028 anti-proliverative effect Effects 0.000 description 3
- 239000003886 aromatase inhibitor Substances 0.000 description 3
- 229940046844 aromatase inhibitors Drugs 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 229960003722 doxycycline Drugs 0.000 description 3
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 3
- 229940034984 endocrine therapy antineoplastic and immunomodulating agent Drugs 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 239000003701 inert diluent Substances 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 230000003285 pharmacodynamic effect Effects 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- 206010046766 uterine cancer Diseases 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 102100027314 Beta-2-microglobulin Human genes 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 102000003910 Cyclin D Human genes 0.000 description 2
- 108090000259 Cyclin D Proteins 0.000 description 2
- 102000013701 Cyclin-Dependent Kinase 4 Human genes 0.000 description 2
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 2
- 102000013698 Cyclin-Dependent Kinase 6 Human genes 0.000 description 2
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229920002732 Polyanhydride Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- 239000012979 RPMI medium Substances 0.000 description 2
- 108050002653 Retinoblastoma protein Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000001064 anti-interferon Effects 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 238000003149 assay kit Methods 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000012876 carrier material Substances 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- FPAFDBFIGPHWGO-UHFFFAOYSA-N dioxosilane;oxomagnesium;hydrate Chemical compound O.[Mg]=O.[Mg]=O.[Mg]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O FPAFDBFIGPHWGO-UHFFFAOYSA-N 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 230000001804 emulsifying effect Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000002124 endocrine Effects 0.000 description 2
- 230000002357 endometrial effect Effects 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 2
- 229940093471 ethyl oleate Drugs 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 230000009036 growth inhibition Effects 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 230000006951 hyperphosphorylation Effects 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 208000026535 luminal A breast carcinoma Diseases 0.000 description 2
- 208000026534 luminal B breast carcinoma Diseases 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 239000002304 perfume Substances 0.000 description 2
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 238000013517 stratification Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 2
- QIJRTFXNRTXDIP-UHFFFAOYSA-N (1-carboxy-2-sulfanylethyl)azanium;chloride;hydrate Chemical compound O.Cl.SCC(N)C(O)=O QIJRTFXNRTXDIP-UHFFFAOYSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N (R)-alpha-Tocopherol Natural products OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- JNODDICFTDYODH-UHFFFAOYSA-N 2-hydroxytetrahydrofuran Chemical compound OC1CCCO1 JNODDICFTDYODH-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- DODQJNMQWMSYGS-QPLCGJKRSA-N 4-[(z)-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-phenylbut-1-en-2-yl]phenol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 DODQJNMQWMSYGS-QPLCGJKRSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 235000003276 Apios tuberosa Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000010744 Arachis villosulicarpa Nutrition 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 238000009020 BCA Protein Assay Kit Methods 0.000 description 1
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- 108091033409 CRISPR Proteins 0.000 description 1
- 238000010354 CRISPR gene editing Methods 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 201000009273 Endometriosis Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 244000239659 Eucalyptus pulverulenta Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101000882586 Mus musculus Estrogen receptor Proteins 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 1
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 1
- 206010067572 Oestrogenic effect Diseases 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 239000004141 Sodium laurylsulphate Substances 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 108010081355 beta 2-Microglobulin Proteins 0.000 description 1
- 239000003613 bile acid Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 210000001217 buttock Anatomy 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000008004 cell lysis buffer Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 229960001305 cysteine hydrochloride Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003413 degradative effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 208000010643 digestive system disease Diseases 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000000459 effect on growth Effects 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- 239000002834 estrogen receptor modulator Substances 0.000 description 1
- 230000001076 estrogenic effect Effects 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 235000020937 fasting conditions Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 208000018685 gastrointestinal system disease Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 239000000185 hemagglutinin Substances 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000006303 immediate early viral mRNA transcription Effects 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000000266 injurious effect Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 238000010983 kinetics study Methods 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 108020001756 ligand binding domains Proteins 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- 125000005609 naphthenate group Chemical group 0.000 description 1
- 230000035407 negative regulation of cell proliferation Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 230000016087 ovulation Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 1
- 229960004390 palbociclib Drugs 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000000291 postprandial effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 231100000279 safety data Toxicity 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 229940100996 sodium bisulfate Drugs 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940001482 sodium sulfite Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000007962 solid dispersion Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229950000244 sulfanilic acid Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229940087765 tamoxifen 40 mg Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 238000012447 xenograft mouse model Methods 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Images












Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Abstract
本发明涉及包含LSZ102和瑞博西尼的药物组合;包含所述药物组合的药物组合物;以及在治疗或预防其中雌激素受体的降解与CDK4/6抑制组合是有益的病症中、例如在治疗癌症中使用此类组合和组合物的方法。
Description
技术领域
本发明涉及包含LSZ102和瑞博西尼的药物组合;包含所述药物组合的药物组合物;以及在治疗或预防其中雌激素受体的降解与CDK4/6抑制组合是有益的病症中、例如在治疗癌症中使用此类组合和组合物的方法。
背景技术
雌激素在女性和男性生殖组织的发育中起关键作用且促成雌激素受体疾病或障碍(例如乳腺癌、卵巢癌、结肠癌、前列腺癌、子宫内膜癌和子宫癌)的发生和进展。
雌激素受体(ERα)阳性疾病如乳腺癌通常用选择性雌激素受体调节剂(SERM)或芳香酶抑制剂(AI)治疗。尽管这些疗法已经被证实有效地减少乳腺癌进展的发生率,但是一些患者显示出治疗抗性且发展成晚期转移性乳腺癌。
治疗抗性部分因肿瘤演变成对低雌激素水平的超敏状态(AI治疗)或发展对用于激活转录的抗雌激素药的依赖性(SERM治疗)所导致。SERD降解所述受体,从而有效地消除ERα表达且由此规避了针对抗内分泌单一疗法发生的抗性潜在机制。此外,临床和临床前数据显示,可以通过使用显示SERD活性的抗雌激素药来避免大量的抗性途经。
细胞周期蛋白D蛋白在癌细胞分裂中起关键作用并且与CDK4和CDK6蛋白激酶复合以通过高磷酸化和激活视网膜母细胞瘤蛋白(pRb)促进G1进展。导致CDK激活的异常现象在管腔A型和管腔B型乳腺癌亚型中高度富集,其中约85%是ER+/HER2-。所述管腔亚型还维持pRb的表达,这对于从用CDK4/6抑制剂的治疗中获益至关重要。ER+乳腺癌细胞系是对单一药剂CDK4/6抑制以及对内分泌治疗和CDK4/6抑制的组合最敏感的癌症模型。
本发明的组合(LSZ102和瑞博西尼)可以用作治疗雌激素受体疾病或障碍的疗法,所述雌激素受体疾病或障碍例如是排卵机能障碍、子宫癌、子宫内膜癌、卵巢癌、子宫内膜异位症、骨质疏松症、前列腺癌、良性前列腺肥大、雌激素受体α(ERα)阳性乳腺癌,特别是对现存的抗雌激素药和芳香酶抑制剂显示重新产生(de novo)抗性的ERα阳性乳腺癌。
发明内容
本发明提供了药物组合,所述药物组合包含:
(a)(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸(LSZ102)或其药学上可接受的盐,其具有以下结构:

(b)7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺(瑞博西尼)或其药学上可接受的盐,其具有以下结构:

LSZ102或其药学上可接受的盐和瑞博西尼或其药学上可接受的盐的组合在本文中也称为“本发明的组合”。
在本发明的组合的另一个实施例中,LSZ102或其药学上可接受的盐和瑞博西尼或其药学上可接受的盐是在同一配制品中。
在本发明的组合的另一个实施例中,LSZ102或其药学上可接受的盐和瑞博西尼或其药学上可接受的盐是在分开的配制品中。
在另一个实施例中,本发明的组合用于同时或依序(以任何顺序)施用。
在另一个实施例中是用于治疗或预防有需要的受试者中的癌症的方法,所述方法包括向所述受试者施用治疗有效量的本发明的组合。
在所述方法的另一个实施例中,所述癌症是雌激素受体α(ERα)阳性乳腺癌。
在所述方法的另一个实施例中,所述癌症选自卵巢癌、子宫内膜癌、前列腺癌、子宫癌、子宫颈癌和肺癌。
在另一个实施例中,本发明的组合提供了用于在制造治疗雌激素受体α(ERα)阳性乳腺癌的药物中的用途。
在另一个实施例中,本发明的组合提供了用于在制造治疗癌症的药物中的用途,所述癌症选自:卵巢癌、子宫内膜癌、前列腺癌、子宫癌、子宫颈癌和肺癌。
在另一个实施例中是药物组合物,所述药物组合物包含本发明的组合。
在另一个实施例中,所述药物组合物进一步包含一种或多种药学上可接受的赋形剂。
附图说明
图1:LSZ102、氟维司群(fulvestrant)和他莫昔芬(tamoxifen)对促进MCF-7细胞内ER降解的比较。
图2A:对在LSZ102处理的MCF-7亲本(WT)细胞中的mRNA的PCR分析。
图2B:对在LSZ102-处理的MCF-7Y537S突变细胞中的mRNA的PCR分析。
图3:LSZ102、氟维司群和他莫昔芬在MCF-7亲本(WT)和Y537S突变细胞中的ERα降解活性的比较。
图4:LSZ102、氟维司群和他莫昔芬在原位人乳腺癌MCF-7异种移植模型中的抗肿瘤功效。
图5:LSZ102、氟维司群和他莫昔芬在原发性人乳腺癌HBRX1298异种移植模型中的抗肿瘤功效。
图6:LSZ102和氟维司群在Y537S ER突变MCF-7乳腺癌异种移植模型中的功效。
图7A:LSZ102和氟维司群在D538G ER突变MCF-7乳腺癌异种移植模型中的功效。
图7B:LSZ102和氟维司群在D538G ER突变MCF-7细胞中的ERα降解活性。
图8A:LSZ102剂量分级对MCF-7异种移植模型中功效的影响。
图8B:LSZ102剂量分级对ER调节的转录物GREB1和PGR mRNA水平的影响。
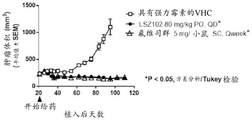
图9:LSZ102与瑞博西尼(LEE011)组合在原位人乳腺癌MCF-7异种移植模型中的抗肿瘤功效。
图10:表明LSZ102与瑞博西尼组合对MCF-7细胞增殖的影响的剂量矩阵和等效线图。
定义
除非另外说明,否则上文和下文中使用的通用术语优选在本披露的上下文中具有以下含义,其中无论在什么情况下使用的更通用的术语可以彼此独立地由更具体的定义代替或保留,从而定义本发明的更详细实施例。
“ESR1突变”是雌激素受体基因(ESR1)突变。突变导致不依赖配体的ER活性。已经鉴定出数种修饰ER的配体结合结构域的突变。这些突变包括但不限于D538G、E380Q和Y537S/N/C,代表80%以上的ESR1突变。由于这些突变在原发性BC肿瘤中几乎不存在(<2%),所以它们是获得性分子事件。ESR1突变在转移环境中已接受芳香酶抑制剂的患者中很常见。突变发生于9%的早期转移性ER+疾病(Y537N/S和D538G)和20%的晚期转移性ER+乳腺癌(Y537C/N/S和D538G)中。与野生型相比,具有D538G和Y537S突变的肿瘤生长更快。
如本文所用,术语“受试者”或“患者”旨在包括易于患有癌症或任何障碍(直接或间接涉及癌症)或受其折磨的动物。受试者的实例包括哺乳动物,例如人、猿、猴、狗、乳牛、马、猪、绵羊、山羊、猫、小鼠、兔、大鼠和转基因非人动物。在一个实施例中,受试者是人,例如患有癌症、具有患癌症的风险或可能易于患有癌症的人。
如本文所用,术语“治疗(treating)”或“治疗(treatment)”包括解除、减轻或缓解受试者的至少一种症状或者实现疾病进展延迟的治疗。例如,治疗可以是减弱障碍的一种或几种症状或者完全根除障碍(如癌症)。在本披露的含义范围内,术语“治疗”还表示阻止、延迟发作(即在疾病的临床表现之前的时间段)和/或降低疾病发展或疾病恶化的风险。
除非另外指明,否则术语“包含”和“包括”在本文中以其开放式和非限制性的含义使用。
除非本文另外指示或与上下文明显矛盾,否则在描述本发明的上下文中(尤其是下文权利要求的上下文中),术语“一个/一种(a)”和“一个/一种(an)”和“所述(the)”以及相似的指示语应解释为包括单数和复数两者。当复数形式用于化合物、盐等时,这也意指单数的化合物、盐等。
如本文所用,术语“协同效应”是指两种治疗剂(例如像作为选择性雌激素受体调节剂的化合物LSZ102、和作为CDK4/6抑制剂的瑞博西尼)的作用,以产生如下效果,例如减缓增殖性疾病(特别是癌症)或其症状的症状进展,这比自身施用的每种药物的效果的简单加和要大。可以例如使用合适的方法(如Sigmoid-Emax方程式(Holford,N.H.G.和Scheiner,L.B.,Clin.Pharmacokinet[临床药代动力学].6:429-453(1981))、Loewe加和性方程式(Loewe,S.和Muischnek,H.,Arch.Exp.Pathol Pharmacol.[实验病理学和药理学的档案]114:313-326(1926))和中值效应方程(Chou,T.C.和Talalay,P.,Adv.Enzyme Regul[酶调节进展].22:27-55(1984))计算协同效应。上面所指的每个方程式都可以应用于实验数据以生成相应的图以帮助评估药物组合的效果。与上文提到的方程相关的相应的图分别是浓度-效果曲线、等效线图曲线和组合指数曲线。
本发明的组合(LSZ102和瑞博西尼)还旨在表示化合物的未经标记的形式以及化合物的同位素标记形式。同位素标记的化合物的一个或多个原子被具有选定原子质量或质量数的原子取代。可以掺入LSZ102和瑞博西尼的同位素的实例包括氢、碳、氮、氧、磷、氟和氯的同位素,例如,分别是2H、3H、11C、13C、14C、15N、18F、31P、32P、35S、36Cl、123I、124I、125I。本发明包括同位素标记的LSZ102和瑞博西尼,例如其中存在放射性同位素(如3H和14C)或非放射性同位素(如2H和13C)。同位素标记的LSZ102和瑞博西尼可用于代谢研究(用14C)、反应动力学研究(例如用2H或3H)、检测或成像技术,例如正电子发射断层扫描(PET)或单光子发射计算机断层扫描(SPECT),包括药物或底物组织分布测定,或用于患者的放射治疗。特别地,用18F标记的LSZ102对于PET或SPECT研究可能是特别理想的。本发明的同位素标记的化合物通常可通过本领域技术人员已知的常规技术或与在使用适当的同位素标记的试剂的所附实例中所述的那些类似的方法来制备。
此外,用较重的同位素,特别是氘(即,2H或D)取代可以提供来源于更大的代谢稳定性(例如,体内半衰期延长或剂量需求减少或治疗指数改进)的某些治疗优点。应当理解,在此上下文中,氘可以被认为是LSZ102或瑞博西尼的取代基。这种较重的同位素(特别是氘)的浓度可以由同位素富集因子来定义。如本文所用,术语“同位素富集因子”意指同位素丰度与特定同位素的天然丰度之间的比率。如果LSZ102或瑞博西尼中的取代基指示氘,这种化合物具有针对每个指定的氘原子的同位素富集因子为至少3500(在每个指定的氘原子上52.5%氘掺入)、至少4000(60%氘掺入)、至少4500(67.5%氘掺入)、至少5000(75%氘掺入)、至少5500(82.5%氘掺入)、至少6000(90%氘掺入)、至少6333.3(95%氘掺入)、至少6466.7(97%氘掺入)、至少6600(99%氘掺入)、或至少6633.3(99.5%氘掺入)。
具体实施方式
LSZ102是一种研究药剂,是一种口服生物可利用的具有混合的SERD和SERM活性的小分子,其在动物中具有抗雌激素作用和促雌激素作用。在体外乳腺癌细胞系中,LSZ102已显示出有效的ER拮抗作用和降解活性。
相对于本发明的药物组合,在一个实施例中是一种药物组合,所述药物组合包含(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐、以及7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐。
在另一个实施例中,(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐、以及7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐分开地、同时地或以任何顺序依序地施用。
在另一个实施例中,所述药物组合用于口服施用。
在另一个实施例中,(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸是口服剂型。
在另一个实施例中,7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺是口服剂型。
在另一个实施例中是药物组合物,所述药物组合物包含(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合以及至少一种药学上可接受的载体。
在另一个实施例中是(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合,用于在治疗野生型ER+乳腺癌中使用。
在另一个实施例中是(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合,用于在治疗ESR1突变型ER+乳腺癌中使用。
在另一个实施例中,所述ESR1突变是表达MCR7的ESR1突变。
在另一个实施例中,所述ESR1突变选自由以下组成的组:D538G、E380Q、Y537S、Y537N和Y537C。
在另一个实施例中,所述ESR1突变选自由以下组成的组:D538G和Y537S。
在另一个实施例中是(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合用于制造治疗ER+乳腺癌的药物的用途。
在另一个实施例中是治疗野生型ER+乳腺癌的方法,所述方法包括向有需要的患者施用(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合、或包含(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合以及至少一种药学上可接受的载体的药物组合物。
在另一个实施例中是治疗ER+乳腺癌的方法,其中所述ER+乳腺癌含有ESR1突变,所述方法包括向有需要的患者施用(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合、或包含(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐的药物组合以及至少一种药学上可接受的载体的药物组合物。
在另一个实施例中,所述突变选自由以下组成的组:D538G、E380Q、Y537S、Y537N和Y537C。
在另一个实施例中,所述突变选自D538G和Y537S。
在另一个实施例中,将(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸以约100mg/天、或200mg/天、或300mg/天、或400mg/天、或500mg/天、或600mg/天的剂量口服施用。
在另一个实施例中,将7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺以约100mg/天、或200mg/天、或300mg/天、或400mg/天、或500mg/天、或600mg/天的剂量口服施用。
在另一个实施例中,将7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺以约100mg/天、或200mg/天、或300mg/天、或400mg/天、或500mg/天、或600mg/天的剂量连续地口服施用。
在另一个实施例中,将7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺以约100mg/天、或200mg/天、或300mg/天、或400mg/天、或500mg/天、或600mg的剂量口服施用,持续21天,随后中断治疗7天。
在另一个实施例中,将7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺以600mg口服施用,持续21天,随后中断治疗7天。
药理学及效用
乳腺癌是女性中癌症死亡的主要原因。尽管经常被泛化为单一疾病,但由于三种关键生物标记物的特征,乳腺癌在临床环境中更通常按其分子亚型进行分类。雌激素和孕酮受体的存在或缺失导致激素受体分类(HR+/HR-),而人表皮生长因子受体2(HER2)的水平升高或降低则导致HER2蛋白分类(HER2+/HER2-)。近74%的乳腺癌表现出高表达的雌激素受体α(ERα),这是一种直接与HR+癌症的进展有关的核激素受体。此配体诱导的转录因子结合激素雌激素以激活并促进致癌基因的表达。
在ERα阳性乳腺癌患者中,治疗长期依赖于内分泌疗法,如他莫昔芬(及其活性代谢物4-羟基他莫昔芬)和阿那曲唑,这两种药物均会阻止配体激活并最终阻止基因表达。他莫昔芬(这类患者的基本护理标准)起雌激素受体调节剂的作用,可有效阻断雌激素与受体的结合并阻断其在乳腺组织中的作用。用这种一线疗法治疗的女性通常会产生积极响应并在临床环境中显示出增加的存活率,但是这些患者获得的抗性最终导致疾病复发,这仍然是一项重大的医学挑战。尽管尚未完全了解ERα阳性肿瘤对他莫昔芬产生抗药性的具体原理,但芳香酶抑制剂(如来曲唑)已在此类难治性癌症中显示出临床疗效。与他莫昔芬相比,芳香酶抑制剂因其活性能降低雌激素生成,更具体地讲,是通过抑制负责形成雌激素过程中关键生物合成步骤的酶。不幸的是,与他莫昔芬一样,芳香酶抑制剂也可导致抗性癌症。
氟维司群是一种批准用于治疗内分泌抗性癌症的选择性雌激素受体降解剂(SERD)。这种基于类固醇的抗雌激素药结合雌激素受体并加速其降解,并且在疾病已发展的经内分泌治疗的患者中是临床有效的。然而,氟维司群的临床效用受到限制,这在很大程度上是由于其不良物理化学性质。所述药物不能口服施用,而以每月两次5mL注射液施用到臀部的500mg的批准临床剂量似乎不足以完全占据所述受体。LSZ102被开发为口服药物,其具有改善的生物利用度,同时保留了所需的雌激素受体降解特性。
有力的临床前数据和基本原理支持将CDK4/6抑制剂引入ER+乳腺癌的治疗中。细胞周期蛋白D蛋白在癌细胞分裂中起关键作用并且与CDK4和CDK6蛋白激酶复合以通过高磷酸化和激活视网膜母细胞瘤蛋白(pRb)促进G1进展。导致CDK激活的异常现象在管腔A型和管腔B型乳腺癌亚型中高度富集,其中约85%是ER+/HER2-。所述管腔亚型还维持pRb的表达,这对于从用CDK4/6抑制剂的治疗中获益至关重要。ER+乳腺癌细胞系是对单一药剂CDK4/6抑制以及对内分泌治疗和CDK4/6抑制的组合最敏感的癌症模型。
基于以下“实例”部分所述的抑制研究,LSZ102和瑞博西尼的组合显示出治疗功效。实例8详细说明了在小鼠原位MCF-7乳腺癌模型中测试的LSZ102和瑞博西尼的组合的功效。以10mg/kg QD的LSZ102和以75mg/kg QD的瑞博西尼的单一药剂治疗导致肿瘤生长抑制(%ΔT/ΔC分别为10%和19%)。出人意料地,两者的组合诱导了28%的肿瘤消退(表7)。
药物组合物
在另一个方面,本发明提供了药学上可接受的组合物,所述药学上可接受的组合物包含治疗有效量的LSZ102和瑞博西尼,其与一种或多种药学上可接受的载体(添加剂)和/或稀释剂配制在一起。如下面详细描述的,本发明的药物组合物可特别配制用于固体或液体形式施用(包括适合口服施用的那些,例如浸液(水性或非水性溶液或悬浮液)、片剂(例如,针对口腔、舌下和全身吸收的那些)、大丸剂、粉剂、颗粒剂、糊剂(应用于舌))。
如本文所用的短语“治疗有效量”是指包含本发明化合物的化合物、材料或组合物的量,其对于在动物中的至少一个细胞亚群中以适用于任何医学治疗的合理的受益/风险比产生一些所期望的治疗效果是有效的。
本文使用的短语“药学上可接受的”是指在合理的医学判断的范围,适合用于与人和动物的组织接触而不产生过度毒性、刺激、过敏反应、或其他问题或并发症,同时具有相称的合理受益/风险比的那些化合物、材料、组合物、和/或剂型。
如本文所用的短语“药学上可接受的载体”是指药学上可接受的材料、组合物或媒剂,如液体或固体填充剂、稀释剂、赋形剂、制造助剂(例如润滑剂、滑石镁、硬脂酸钙或硬脂酸锌或硬脂酸)或溶剂包封材料,参与将主题化合物从一个器官或身体的一部分运送或运输到另一个器官或身体的一部分。在与配制品的其他成分相容并且对患者无害的意义上,每种载体必须是“可接受的”。可用作药学上可接受的载体的材料的一些实例包括:(1)糖,如乳糖、葡萄糖和蔗糖;(2)淀粉,如玉米淀粉和马铃薯淀粉;(3)纤维素以及其衍生物,如羧甲基纤维素钠、乙基纤维素和醋酸纤维素;(4)粉状黄蓍胶;(5)麦芽;(6)明胶;(7)滑石粉;(8)赋形剂,如可可脂和栓剂蜡类;(9)油,如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和豆油;(10)二醇,如丙二醇;(11)多元醇,如甘油、山梨醇、甘露醇和聚乙二醇;(12)酯,如油酸乙酯和月桂酸乙酯;(13)琼脂;(14)缓冲剂,如氢氧化镁和氢氧化铝;(15)藻酸;(16)无热原的水;(17)等渗盐水;(18)林格氏溶液;(19)乙醇;(20)pH缓冲液;(21)聚酯,聚碳酸酯和/或聚酸酐;以及(22)药物配制品中所用的其他无毒可相容物质。
如上文所述,本发明化合物的某些实施例可含有碱性官能团,例如氨基或烷基氨基,并且由此能够与药学上可接受的酸形成药学上可接受的盐。在这方面,术语“药学上可接受的盐”是指本发明的化合物的相对无毒的无机和有机酸加成盐。这些盐可以在施用媒剂或剂型制造过程中原位制备,或者通过使纯化的游离碱形式的本发明化合物与合适的有机或无机酸分别反应,并在随后的纯化期间分离由此形成的盐来制备。代表性盐包括氢溴酸盐、盐酸盐、硫酸盐、硫酸氢盐、磷酸盐、硝酸盐、乙酸盐、戊酸盐、油酸盐、棕榈酸盐、硬脂酸盐、月桂酸盐、苯甲酸盐、乳酸盐、磷酸盐、甲苯磺酸盐、柠檬酸盐、马来酸盐、富马酸盐、琥珀酸盐、酒石酸盐、萘酸盐(napthylate)、甲磺酸盐、葡庚糖酸盐、乳糖醛酸盐和月桂基磺酸盐等。(参见例如Berge等人(1977)“Pharmaceutical Salts[药用盐]”,J.Pharm.Sci.[药物科学杂志]66:1-19)。
主题化合物的药学上可接受的盐包括化合物的常规的无毒盐或季铵盐,例如来自无毒的有机酸或无机酸。例如,此类常规无毒盐包括衍生自无机酸的那些,所述无机酸例如为盐酸、氢溴酸、硫酸、胺磺酸、磷酸、硝酸等;以及从有机酸制备的盐,所述有机酸例如为乙酸、丙酸、琥珀酸、乙醇酸、硬脂酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、棕榈酸、马来酸、羟基马来酸、苯乙酸、谷氨酸、苯甲酸、水杨酸、磺胺酸、2-乙酰氧基苯甲酸、富马酸、甲苯磺酸、甲磺酸、乙二磺酸、草酸、异硫磺酸等。例如,瑞博西尼的药学上可接受的盐是琥珀酸盐。
在其他情况下,本发明的化合物可以含有一个或多个酸性官能团,并且因此能够与药学上可接受的碱形成药学上可接受的盐。在这些例子中,术语“药学上可接受的盐”是指本发明化合物的相对无毒的无机和有机碱加成盐。这些盐也可以在施用媒剂或剂型制造过程中原位制备,或者通过使纯化的游离酸形式的化合物与合适的碱(例如,药学上可接受的金属阳离子的氢氧化物、碳酸盐或碳酸氢盐)与氨、或与药学上可接受的有机伯、仲或叔胺分别反应来制备。代表性的碱或碱土金属盐包括锂盐、钠盐、钾盐、钙盐、镁盐和铝盐等。可用于形成碱加成盐的代表性有机胺包括乙胺、二乙胺、乙二胺、乙醇胺、二乙醇胺、哌嗪等。(参见例如Berge等人,同上)
在组合物中还可以存在润湿剂、乳化剂和润滑剂,如十二烷基硫酸钠和硬脂酸镁,以及着色剂、释放剂、包衣剂、甜味剂、调味剂和芳香剂、防腐剂和抗氧化剂。
药学上可接受的抗氧化剂的实例包括:(1)水溶性抗氧化剂,如抗坏血酸、盐酸半胱氨酸、硫酸氢钠、焦亚硫酸钠、亚硫酸钠等;(2)油溶性抗氧化剂,如抗坏血酸棕榈酸酯、丁基羟基苯甲醚(BHA)、丁基羟基甲苯(BHT)、卵磷脂、没食子酸丙酯、α-生育酚等;以及(3)金属螯合剂,如柠檬酸、乙二胺四乙酸(EDTA)、山梨醇、酒石酸、磷酸等。
本发明的配制品包括适合于口服、鼻、局部(包括口腔和舌下)、直肠、阴道和/或肠胃外施用的那些。配制品可以方便地以单位剂型存在并且可以通过药学领域熟知的任何方法制备。可以与载体材料组合以产生单一剂型的活性成分的量将根据所治疗的宿主和特定的施用方式而变化。可以与载体材料组合以产生单一剂型的活性成分的量通常为产生治疗效果的化合物的量。通常,在百分之百的范围内,量的范围为从约0.1%至约99%的活性成分,优选从约5%至约70%,最优选从约10%至约30%。
在某些实施例中,本发明的配制品包含选自由以下组成的组的赋形剂:环糊精、纤维素、脂质体、胶束形成剂(例如胆汁酸)和聚合物载体(例如聚酯和聚酸酐);和本发明的化合物。在某些实施例中,前述配制品使得本发明化合物在口服方面是生物可利用的。
制备这些配制品或组合物的方法包括将本发明的化合物与载体和任选的一种或多种辅助成分结合的步骤。通常,通过将本发明化合物与液体载体或细碎固体载体或两者均匀且紧密地结合,并且然后如果需要,使产物成形来制备配制品。
适用于口服施用的本发明的配制品可以呈胶囊、扁囊剂、丸剂、片剂、锭剂(使用调味基质,通常为蔗糖和阿拉伯胶或黄芪胶)、粉剂、颗粒剂的形式,或作为在水性或非水性液体中的溶液、悬浮液或固体分散体,或作为水包油或油包水液体乳剂,或作为酏剂或糖浆,或作为软锭剂(使用惰性基质,例如明胶和甘油,或蔗糖和阿拉伯胶),和/或作为漱口水等等,每一形式含有预定量的本发明的化合物作为活性成分。本发明化合物还能以大丸剂、药糖剂或糊剂形式施用。
在用于口服施用的本发明的固体剂型(胶囊、片剂、丸剂、糖衣丸、粉剂、颗粒记、糖锭等)中,将活性成分与一种或多种药学上可接受的载体如柠檬酸钠或磷酸二钙和/或任何下列物质混合到一起:(1)填充剂或增充剂,如淀粉类、乳糖、蔗糖、葡萄糖、甘露醇和/或硅酸;(2)粘合剂,例如像羧甲基纤维素、藻酸盐类、明胶、聚乙烯吡咯烷酮、蔗糖和/或阿拉伯胶;(3)保湿剂,如甘油;(4)崩解剂,如琼脂、碳酸钙、马铃薯或木薯淀粉、藻酸、某些硅酸盐和碳酸钠;(5)溶液阻滞剂,如石蜡;(6)吸收促进剂,如季铵化合物和表面活性剂,例如泊洛沙姆和十二烷基硫酸钠;(7)润湿剂,例如像鲸蜡醇、单硬脂酸甘油酯和非阴离子表面活性剂;(8)吸收剂,如高岭土和膨润土;(9)润滑剂,如滑石粉、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠、硬脂酸锌、硬脂酸钠、硬脂酸、及其混合物;(10)着色剂;以及(11)控制释放剂,如交聚维酮或乙基纤维素。在胶囊、片剂和丸剂的情况下,药物组合物还可以包含缓冲剂。还可以使用此类赋形剂,如乳糖(lactose或milk sugar)以及高分子量聚乙二醇等,将相似类型的固体组合物用作软壳和硬壳明胶胶囊中的填充剂。
片剂可以通过压缩或模制(任选地与一种或多种辅助成分一起)来制备。可以使用粘合剂(例如,明胶或羟丙基甲基纤维素)、润滑剂、惰性稀释剂、防腐剂、崩解剂(例如,羟基乙酸淀粉钠或交联羧甲基纤维素钠)、表面活性剂或分散剂来制备压缩片剂。模制片剂可以通过在合适的机器中模制用惰性液体稀释剂润湿的粉末化合物的混合物来制备。
片剂和本发明的药物组合物的其他固体剂型(如糖衣丸、胶囊、丸剂和颗粒剂)可任选地进行刻痕或制备有包衣和外壳,例如肠溶包衣和药物配制领域熟知的其他包衣。也可以使用例如不同比例的羟丙基甲基纤维素将它们配制成使其中的活性成分缓慢或受控释放以提供所需的释放曲线、其他聚合物基质、脂质体和/或微球。可以将它们配制用于快速释放,例如冷冻干燥的。它们可以例如通过细菌截留过滤器滤过,或通过并入呈无菌固体组合物形式的灭菌剂来灭菌,这些无菌固体组合物可以在使用前立即溶解于无菌水或一些其他无菌可注射介质中。这些组合物也可以任选地包含遮光剂并且可以是如下组合物,所述组合物在胃肠道的某一部分中,任选地以一种延迟的方式仅或者优先释放一种或多种活性成分。可利用的嵌入式组合物的实例包括聚合物质以及蜡。活性成分还可以是微囊化形式,如果适当的话,可包含一种或多种上述赋形剂。
用于口服施用本发明化合物的液体剂型包括药学上可接受的乳剂、微乳剂、溶液、悬浮液、糖浆和酏剂。除了活性成分之外,液体剂型可以含有本领域常用的惰性稀释剂(例如像水或其他溶剂)、增溶剂和乳化剂,例如乙醇、异丙醇、碳酸乙酯、醋酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、油类(特别是棉籽油、花生油、玉米油、胚芽油、橄榄油、蓖麻油和芝麻油)、甘油、四氢呋喃甲醇、聚乙二醇和脱水山梨醇脂肪酸酯,及其混合物。
除了惰性稀释剂之外,口服组合物还可包括辅助剂,例如润湿剂、乳化剂和悬浮剂、甜味剂、调味剂、着色剂、芳香剂以及防腐剂。
除了含有活性化合物外,悬浮液还可以含有悬浮剂如,例如乙氧化异硬脂醇、聚氧乙烯山梨醇和山梨醇酯、微晶纤维素、偏氢氧化铝(aluminum metahydroxide)、膨润土、琼脂和黄芪胶,及其混合物。
可以在本发明的药物组合物中使用的合适的水性和非水性载体的实例包括水、乙醇、多元醇(如,甘油、丙二醇、聚乙二醇等)及其合适的混合物、植物油(如橄榄油)和可注射的有机酯(如油酸乙酯)。适当流动性可以例如通过以下方式来维持:通过使用包衣材料(如卵磷脂),在分散体的情况下通过维持所需粒度,以及通过使用表面活性剂。
这些组合物也可以含有辅助剂,如防腐剂、润湿剂、乳化剂和分散剂。可以通过包括各种抗细菌剂和抗真菌剂(例如,对羟苯甲酸酯、三氯叔丁醇、山梨酸苯酚等)确保阻止微生物对主题化合物起作用。还令人希望的是在组合物中包括等渗剂,如糖、氯化钠等。
当将本发明的化合物作为药物向人和动物施用时,它们可以本身或作为药物组合物施用,所述药物组合物含有例如与药学上可接受的载体组合的0.1%至99%(更优选10%至30%)的活性成分。
通过本领域技术人员已知的常规方法将本发明的化合物(其能以合适的水合形式使用)和/或本发明的药物组合物配制成药学上可接受的剂型。
可以改变本发明药物组合物中活性成分的实际剂量水平,以便获得一定量的活性成分,所述活性成分的量有效地实现对于特定的患者、组合物和施用方式的所期望的治疗反应,而对患者没有毒性。
选择的剂量水平将取决于各种因素,包括所使用的本发明的具体化合物或其酯、盐或酰胺的活性,施用途径,施用时间,所采用的具体化合物的排泄率或代谢率,吸收的速度和程度,治疗的持续时间,与所采用的具体化合物组合使用的其他药物、化合物和/或材料,正被治疗的患者的年龄、性别、体重、病症、一般健康状况和先前病史,以及在医学领域中熟知的类似因素。
具有本领域普通技术的医师或兽医可以容易地确定并开出所需的药物组合物的有效量。例如,医生或兽医能以低于实现所期望的治疗效果所需的水平开始给药于药物组合物中使用的本发明化合物,并逐渐增加剂量直至达到所期望的效果。
通常,本发明的组合的合适日剂量将是每种化合物有效产生治疗效果的最低剂量的量。这种有效剂量通常取决于上述因素。
在另一个方面,本发明提供了药学上可接受的组合物,所述药学上可接受的组合物包含治疗有效量的一种或多种上述主题化合物,其与一种或多种药学上可接受的载体(添加剂)和/或稀释剂配制在一起。
实例
LSZ102和瑞博西尼
根据WO 2014/130310的实例139合成(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸(LSZ102)。根据WO 2010/020675的实例74合成7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺(瑞博西尼)。
本文所述的LSZ102和瑞博西尼的效用可以通过以下实例中的测试来证明。
实例1
LSZ102促进MCF-7细胞中的ER降解
蛋白质印迹。为了在功效研究结束时分析LSZ102、氟维司群和他莫昔芬对MCF-7肿瘤中ERα蛋白水平的影响,将速冻的肿瘤粉碎成粉末,然后转移至与冰冷裂解缓冲液(1X细胞裂解缓冲液;细胞信号传导公司(Cell Signaling),目录号9803S)混合的裂解介质管(Lysing Matrix Tubes)(MP生物化学公司(MP Biomedicals),目录号6913-500)中,所述冰冷裂解缓冲液含有通过Fast Prep 24组织裂解器(MP生物化学公司)均质化的CompleteMini(1片对10mL)、PhosStop(1片对10mL和1M尿素)。根据制造商的说明,通过BCA测定法(Pierce BCA蛋白质测定试剂盒,产品编号23225,赛默科技公司(Thermo Scientific))测试了裂解物的总蛋白浓度。通过SDS-PAGE将裂解物分离,将其转移至膜上,并然后使用抗ERα抗体(圣克鲁斯生物技术公司(Santa Cruz Biotechnology),HC-20)以及抗微管蛋白抗体作为上样对照进行免疫印迹。将蛋白质印迹扫描用于定量免疫印迹带。剩余ERα的百分比是通过比较治疗小鼠的肿瘤与媒剂对照组的肿瘤确定的。图1表明,与氟维司群和他莫昔芬相比,LSZ102促进MCF-7细胞中的ER降解,在同等浓度下,LSZ102引起的ER降解与氟维司群相似,而未观察到在MCF-7细胞中他莫昔芬对ER降解的影响。
MCF-7增殖测定。将耗尽生长因子的MCF-7细胞接种(10,000个细胞/孔)到CSS培养基的96孔板中。过夜孵育后,在雌二醇(0.1nM)存在下,将细胞用化合物处理6天。然后通过CellTiterGlo测定法(普洛麦格公司(Promega))测量细胞活力。
在细胞增殖测定中,LSZ102抑制MCF-7细胞的生长,其中IC50为0.7nM,并且在10μM下对ER-MDA-MB468细胞的生长没有影响,这表明即使在高剂量下,LSZ102也不影响缺乏ER的细胞。LSZ102是有效的SERD,如使用生长因子驱动的增殖测定(其中胰岛素而非雌激素驱动细胞增殖)所证实的。此测定不受SERM他莫昔芬的影响,但受SERD氟维司群的抑制。在使用MCF-7细胞系进行胰岛素驱动的增殖测定中,LSZ102阻断了细胞生长,其中IC50为6nM,优于氟维司群。
实例2
在MCF-7亲本细胞和Y537S细胞中的LSZ102抗增殖和ER降解活性
在MCF-7亲本(野生型或WT)细胞和Y537S突变细胞中研究了LSZ102、氟维司群和他莫昔芬作为单一药剂的作用。将MCF-7WT细胞和Y537S突变细胞在RPMI(无酚红)加10%的活性炭/葡聚糖处理的血清中孵育,并在0.1nM雌二醇(WT)或没有雌二醇(Y537S)的存在下用递增浓度的化合物处理。化合物处理7天后,通过CellTiter-Glo(CTG)测定法确定细胞活力。对于ERE萤光素酶测定,在24小时后使用Bright-Glo测定法测量细胞萤光素酶信号。IC50是抑制50%CTG信号的50%的化合物浓度。使用XLfit软件计算IC50纳摩尔(nM)值,并将其定义为拟合抑制曲线的拐点。LSZ102、氟维司群和他莫昔芬在MCF-7WT和Y537S突变细胞中的抗增殖活性的结果呈现于表1中。
表1

在MCF-7WT细胞中,LSZ102、氟维司群和他莫昔芬对细胞增殖的抑制作用相似。发现在MCF-7WT细胞中,LSZ102在IC50为5.2nM时抑制细胞增殖,而氟维司群在IC50为2.6nM时抑制细胞增殖,而他莫昔芬在IC50为4.5nM时抑制细胞增殖。在MCF-7Y537S突变细胞中,用LSZ102抑制细胞增殖是这三种药物中最有效的。发现在MCF-7Y537S突变细胞中,LSZ102在IC50为27.0nM时抑制细胞增殖,而氟维司群在IC50为53.0nM时抑制细胞增殖,而他莫昔芬在IC50为60.1nM时抑制细胞增殖。尽管所有三种化合物的IC50功效从MCF-7WT到Y537S突变体均有改变,但LSZ102的改变最少,并保留了所测试的三种化合物中最有效的活性。
PCR分析。将来自LSZ102处理的MCF-7WT和Y537S突变细胞的mRNA分离,并使其经受qRT-PCR分析用于ER靶基因表达(图2A和2B)。针对ER(ESR1)的mRNA表达,测量典型ER靶基因GREB1和PGR的mRNA水平,以此作为LSZ102对ER自身影响的对照,以确保所述影响不是由于对ER mRNA水平的影响,而是由于对ER蛋白水平的影响。对于MCF-7WT和Y537S突变细胞中GREB1和PGR的mRNA表达,LSZ102均表现出剂量反应抑制。在WT细胞中此作用更明显,但在高浓度下,GREB1和PGR表达均显著降低。相比之下,在任一细胞系中的ESR1 mRNA水平均未显著降低。
免疫印迹分析。使MCF-7亲本和Y537S突变细胞在不含酚红的、具有10%活性炭/葡聚糖处理的血清的RPMI培养基连续生长(Y537S突变体)或生长3天(亲本),然后用LSZ102、氟维司群和他莫昔芬作为单一药剂处理24hr。对提取的细胞裂解物进行免疫印迹分析(图3)用于ERα蛋白定量。在MCF-7亲本细胞中,LSZ102和氟维司群在剂量反应中均显著降低ER蛋白水平。在MCF-7Y537S突变细胞中,LSZ102跨各剂量地明显降低ER蛋白水平,但氟维司群似乎没有任何作用。
实例3
NSG小鼠中的MCF-7异种移植模型
雌激素反应ER阳性(ER+)MCF-7细胞系显示出在体外对LSZ102敏感。为了证明在NOD scidγ(NSG)小鼠的原位MCF-7异种移植模型中的靶向性抗肿瘤活性,每天一次(QD)口服(PO)施用1、3、10和20mg/kg的LSZ102,以及每只小鼠每周一次(Qweek)皮下(SC)施用5mg氟维司群,且每天一次口服(PO)施用60mg/kg他莫昔芬持续5天/周作为阳性对照。向小鼠补充雌二醇(0.72mg雌二醇/90天释放丸剂),以在细胞植入前数天进一步支持MCF-7肿瘤的生长。通过将50% 中的10x 106个细胞注射到每只小鼠的腋窝乳腺脂肪垫区域中,在雌性NSG小鼠中建立MCF-7肿瘤。当肿瘤达到平均200mm3时,根据肿瘤体积将小鼠随机分入治疗组(n=8)。在第48天,所述治疗对MCF-7乳腺癌异种移植模型中的肿瘤反应的作用呈现于表2中。
中的10x 106个细胞注射到每只小鼠的腋窝乳腺脂肪垫区域中,在雌性NSG小鼠中建立MCF-7肿瘤。当肿瘤达到平均200mm3时,根据肿瘤体积将小鼠随机分入治疗组(n=8)。在第48天,所述治疗对MCF-7乳腺癌异种移植模型中的肿瘤反应的作用呈现于表2中。
表2


LSZ102治疗导致剂量依赖性抗肿瘤功效(图4),并在用20mg/kg QD的剂量治疗的小鼠中观察到最大活性,对应于肿瘤体积对比对照(ΔT/ΔC)的平均变化百分数为2.4%(第48天,p<0.05)。在20mg/kg QD的剂量下,达到了肿瘤停滞并维持48天。10mg/kg QD剂量也显著有效(ΔT/ΔC=25%,p<0.05),而1和3mg/kg QD剂量无显著疗效(%ΔT/ΔC分别为51%和56%)。用作对照的他莫昔芬和氟维司群分别诱导肿瘤停滞和生长抑制。
实例4
NSG小鼠中的ER+原发性乳腺癌模型HBRX1298
在以下条件下在NSG小鼠中测试了对雌激素敏感的ER+原发性乳腺癌模型HBRX1298:LSZ102 20mg/kg PO QD,LSZ102 80mg/kg PO QD,他莫昔芬40mg/kg PO QD,氟维司群5mg/小鼠SC每周一次,媒剂对照和去除雌二醇定时释放丸剂的对照(图5)。在第63天,所述治疗对HBRX1298乳腺癌异种移植模型中的肿瘤反应的作用呈现于表3中。
表3

通过将肿瘤浆(tumor brei)注射到腹股沟乳腺脂肪垫区域中,在NSG雌性小鼠中建立HBRX1298肿瘤。在细胞植入前数天,将0.72mg雌二醇/90天释放丸剂植入小鼠。当肿瘤达到约250mm3时,在第38天,根据肿瘤体积将小鼠随机分入治疗组(所有组n=6,除雌二醇停药组n=4外)。在LSZ102的剂量为20和80mg/kg QD时有疗效优势。LSZ102的80mg/kg QD剂量显示出比媒剂处理的对照在统计学上的显著功效,并且与在去除雌二醇丸剂的小鼠中所见的功效相当。80mg/kg QD剂量抑制的肿瘤体积接近于在去除雌二醇丸剂的小鼠中观察到的程度(%ΔT/ΔC分别为32%和21%;p<0.05)。
实例5
NSG小鼠中的Y537S
ER突变MCF-7乳腺癌模型
在以下条件下在NSG小鼠中测试了Y537S ER突变MCF-7乳腺癌模型:LSZ102 20mg/kg PO QD,LSZ102 80mg/kg PO QD,氟维司群5mg/小鼠SC每周一次,以及媒剂对照(图6)。在第70天,所述治疗对Y537S ER突变MCF-7乳腺癌模型中的肿瘤反应的作用呈现于表4中。
表4

使用CRISPR技术将MCF-7细胞系进行工程改造,以敲除先天性野生型功能性ER,并敲入突变Y537S ER。将50% 中的10x 106个细胞植入到每只切除卵巢的雌性NSG小鼠的腋窝乳腺脂肪垫区域中。当肿瘤达到平均250mm3时,根据肿瘤体积将小鼠随机分入治疗组。
中的10x 106个细胞植入到每只切除卵巢的雌性NSG小鼠的腋窝乳腺脂肪垫区域中。当肿瘤达到平均250mm3时,根据肿瘤体积将小鼠随机分入治疗组。
每天以20和80mg/kg的LSZ102的治疗使表达Y537S ER的MCF-7异种移植物消退,这证明了其在表达这种突变形式的ER的乳腺癌中的活性,而氟维司群未达到统计学上显著的功效。
实例6
NSG小鼠中的D538G
ER突变MCF-7乳腺癌模型
在以下条件下在NSG小鼠中测试了D538G强力霉素(doxycycline)诱导的ER突变MCF-7乳腺癌模型:LSZ102 80mg/kg PO QD,氟维司群5mg/小鼠SC每周一次,以及媒剂对照(图7A)。在第74天,所述治疗对D538G ER突变MCF-7乳腺癌模型中的肿瘤反应的作用呈现于表5中。
表5

用强力霉素诱导的启动子将MCF-7细胞系进行工程改造,以表达D538G突变型ER。将50% 中的10x 106个细胞植入到每只切除卵巢的雌性NSG小鼠的腋窝乳腺脂肪垫区域中。细胞植入后八天,小鼠通过其食物接受了强力霉素。当肿瘤达到平均250mm3时,根据肿瘤体积将小鼠随机分入治疗组。
中的10x 106个细胞植入到每只切除卵巢的雌性NSG小鼠的腋窝乳腺脂肪垫区域中。细胞植入后八天,小鼠通过其食物接受了强力霉素。当肿瘤达到平均250mm3时,根据肿瘤体积将小鼠随机分入治疗组。
每天80mg/kg的LSZ102使表达D538G ER的MCF-7异种移植物消退,这证明了其在表达这种突变形式的ER的乳腺癌中的活性,而氟维司群也具有活性。
在功效结束时从肿瘤中分离出蛋白质,用于蛋白质印迹分析(图7B)。由于突变型ER蛋白被HA标记,因此用抗ER或抗血凝素(HA)抗体将膜进行免疫染色。也针对β-肌动蛋白对膜进行染色以作为上样对照。还包括来自用LSZ102给药两周后收集的肿瘤中的一组蛋白质样品。所述样品显示了此D538G突变型ER蛋白的降解。
实例7
在具有MCF-7异种移植物的小鼠中的LSZ102的剂量分级研究
在具有MCF-7异种移植物的小鼠中的LSZ102的剂量分级研究评估QD给药对比每日两次分次剂量(BID)给药的影响,显示出同等功效,这表明LSZ102通过总暴露量驱动(图8A)。在第61天,所述治疗对MCF-7乳腺癌异种移植模型中的肿瘤反应的作用呈现于表6中。
表6

在第61天最终治疗后,在24小时后收集的肿瘤显示,在24小时剂量间隔内,较高剂量水平对ER调节的转录物GREB1 mRNA和PGR mRNA水平提供了略微更强的抑制作用,但在QD给药和BID分次剂量之间的PD相同(图8B)。绘制各个动物的数据,并将其归一化为β-2微球蛋白(B2M)的表达。
实例8
在小鼠ER+MCF-7乳腺癌模型中,LSZ102与瑞博西尼组合
在小鼠原位MCF-7乳腺癌模型中测试了LSZ102和瑞博西尼的组合的功效(图9)。分别在10mg/kg QD的LSZ102和75mg/kg QD的瑞博西尼的单一药剂治疗中观察到肿瘤生长抑制(%ΔT/ΔC为10%和19%)。出人意料地,两者的组合诱导了28%的肿瘤消退。在第70天,所述治疗对MCF-7乳腺癌模型中的肿瘤反应的作用呈现于表7中。
表7

实例9
在MCF-7细胞中的LSZ102与瑞博西尼的组合
增殖测定。将MCF-7细胞在RPMI培养基加10%的全血清培养基中培养,并以棋盘状设计用递增浓度的组合的化合物进行处理。在化合物处理7天后,通过CellTiter-Glo测定法确定细胞活力,并将其归一化为二甲亚砜(DMSO)对照。使用Chalice软件(马萨诸塞州坎布里奇的CombinatoRx公司(CombinatoRx,Cambridge MA))分析生长抑制百分比和过量抑制百分比。使用Loewe算法获得数据,所述算法计算剂量矩阵的加权“协同作用得分”,所述得分对剂量采样和覆盖范围进行了调整,并且针对高抑制水平下的有利组合效应进行权重(Lehar等人2009)。生成协同作用得分和等效线图,以量化组合强度(图10)。与自身交互(药物与其自身交互;理论协同作用得分为0)内所看到的协同作用得分的变化相比,高于2的协同作用得分被认为是有意义的(Lehar等人2009)。用Loewe协同模型计算过量抑制,所述模型相对于两种药物以剂量叠加方式作用的预期情况来测量对生长的影响。正数代表增强协同作用的区域。LSZ102和瑞博西尼的组合在体外MCF-7细胞中观察到协同抗增殖作用(协同作用得分=3.8)。这些结果有力地支持了LSZ102和瑞博西尼的组合在ER阳性乳腺癌治疗中的潜在作用。
实例10
在晚期或转移性ER+乳腺癌患者中的LSZ102±瑞博西尼的I/Ib期研究
主要目的是:用主要终点表征安全性和耐受性,所述主要终点包括剂量限制性毒性(DLT)和不良事件(AE);并确定单独和/或与瑞博西尼组合的LSZ102的推荐剂量和方案。次要目的是评估初步抗肿瘤活性并研究总反应率(ORR)、反应持续时间(DOR)、无进展存活期(PFS)、疾病控制率(DCR)、药代动力学(PK)和药效动力学(PD)。
合格的患者(≥18岁)患有组织学确认的ER+乳腺癌,所述乳腺癌在内分泌治疗后进展为转移性或局部晚期疾病,或在用芳香酶抑制剂辅助治疗结束时或结束后12个月内复发。
所述研究中的全部患者均在禁食条件下用LSZ102进行治疗。在所述研究的剂量递增部分中,单一药剂剂量递增组中的患者接受起始剂量为200mg、后续剂量为400mg、450mg、600mg和900mg的每日一次(QD)口服LSZ102。截至2017年8月28日,已按以下剂量组治疗了45例患者:LSZ102 200mg QD(n=4)、400mg QD(n=6)、450mg QD(n=13)、600mg QD(n=16)和900mg QD(n=6)。中值年龄是60.0岁,其中67%的ECOG体能状态为零,并且60%先前已接受氟维司群治疗。截至2017年8月28日,有34/45(76%)患者已停止治疗,主要(31/34患者)是由于进行性疾病。这些患者的基线特征和分布情况分别在以下表8和表9中进行详述。
表8

表9

剂量限制性毒性(DLT)是发生在LSZ102 600mg QD(呕吐;n/N=1/16[6%])和900mg QD(腹泻;n/N=2/6[33%])组中的胃肠道障碍。在200-450mg QD剂量组的23例患者中未报告DLT。
最常见(≥25%)的药物相关性不良事件(AE)是腹泻(62%)、恶心(56%)和呕吐(27%;表3)。很少报告药物相关性等级(Gr)3AE:腹泻(3例患者[7%])、恶心(2例患者[4%])、呕吐和贫血(1例患者[2%]),并且没有报告药物相关性Gr 4AE(表10)。
共有6/45患者(13%)需要降低剂量;其中包括4例接受LSZ102 900mg QD的患者(2例患者Gr 3腹泻、1例患者Gr 2恶心、以及1例出现Gr 3恶心),1例接受LSZ102 600mg QD的患者(Gr 3呕吐)和1例接受LSZ102 450mg QD的患者(Gr 2恶心和Gr 2呕吐)。
表10

*基于相似的PK曲线,组合了400和450mg剂量的安全性数据。对于一种治疗,在这种治疗下多次出现DLT的患者在不良事件类别中仅被计数一次。在主要系统器官类别中具有多个DLT的患者在总计行中仅被计数一次。
共有6例患者维持>6个月的研究治疗;其中,有5例患者先前已接受氟维司群治疗。一例其肿瘤含有ESR1 D538G突变的患者,先前已接受9个治疗线,包括在进展前接受氟维司群治疗120天,以及在进展前接受来曲唑+帕博西尼治疗94天。
用单一药剂LSZ102观察到了抗肿瘤活性的初步证据,疾病控制率为33%,表11。
表11

初步PK参数表明LSZ102被快速吸收(中值Tmax 2-3h)。观察到几何平均积累半衰期为5-11h。单一药剂LSZ102暴露似乎按剂量比例增加。第1天和稳定状态的AUC和Cmax通常相当,这表明没有药物积累。在MCF-7小鼠异种移植模型中,在LSZ102 20mg/kg剂量下的暴露水平远超过为3510h*ng/mL的临床前有效暴露。在所有剂量水平下获得的LSZ102暴露均超过临床前模型中的有效暴露。已鉴定出600mg QD的候选禁食推荐扩展剂量(RDE),并且正在进行的研究正在进一步探索食物对LSZ102 PK的影响。临床数据(n=7对)已显示,进食后暴露增加,其中中值AUC增加1.7倍。
口服单一药剂LSZ102呈现良好耐受性,其具有可控安全性曲线。在经大量预治疗的ER+乳腺癌患者中(包括先前用氟维司群和CDK4/6抑制剂治疗的患者)观察到抗肿瘤活性的初步证据。六例患者维持>6个月的研究治疗,所有患者均接受了≥6个先前治疗线(范围6-11线)。
对LSZ102和瑞博西尼的组合正在进行临床试验,其中与200mg(n=5)或400mg(n=5)或600mg(n=4)的LSZ102组合,以300mg口服施用(禁食)瑞博西尼21天,然后停药7天。与400mg(n=4)的LSZ102组合,口服施用(禁食)较高剂量(400mg)的瑞博西尼21天,然后停药7天。数据截至2017年10月09日。另外,与450mg的LSZ102组合,正在以300mg(n=6)口服施用(禁食)瑞博西尼。基于临床数据,所述组合具有良好的耐受性和活性。
应理解,本文所述的实例和实施例仅用于举例说明目的,其各种修饰或改变对于本领域技术人员将是明了的,并包括在本申请的精神和范围内和所附权利要求书的范围内。
Claims (16)
1.一种药物组合,所述药物组合包含(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐。
2.根据权利要求1所述的组合,其中将(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸或其药学上可接受的盐和7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受的盐分开地、同时地或以任何顺序依序地施用。
3.根据权利要求1或2所述的药物组合,将其用于口服施用。
4.根据权利要求1或2所述的药物组合,其中(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸是口服剂型。
5.根据权利要求1或2所述的药物组合,其中7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺是口服剂型。
6.一种药物组合物,所述药物组合物包含根据前述权利要求中任一项所述的药物组合和至少一种药学上可接受的载体。
7.根据权利要求1至5中任一项所述的药物组合或根据权利要求6所述的药物组合物,用于在ER+乳腺癌的治疗中使用。
8.根据权利要求1至5中任一项所述的药物组合或根据权利要求6所述的药物组合物用于制造治疗ER+乳腺癌的药物的用途。
9.一种治疗野生型ER+乳腺癌的方法,所述方法包括向有需要的患者施用根据权利要求1至5中任一项所述的药物组合或根据权利要求6所述的药物组合物。
10.一种治疗ER+乳腺癌的方法,其中所述ER+乳腺癌含有ESR1突变,所述方法包括向有需要的患者施用根据权利要求1至5中任一项所述的药物组合或根据权利要求6所述的药物组合物。
11.根据权利要求10所述的方法,其中所述ESR1突变是表达MCR7的ESR1突变。
12.根据权利要求11所述的方法,其中所述突变选自由以下组成的组:D538G、E380Q、Y537S、Y537N和Y537C。
13.根据权利要求12所述的方法,其中所述突变选自D538G和Y537S。
14.根据权利要求9或10所述的方法,其中将(E)-3-(4-((2-(2-(1,1-二氟乙基)-4-氟苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯基)丙烯酸以约100mg/天、或200mg/天、或300mg/天、或400mg/天、或450mg/天、或500mg/天、或600mg/天、或900mg/天的剂量口服施用。
15.根据权利要求14所述的方法,其中将7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺以约100mg/天、或200mg/天、或300mg/天、或400mg/天、或500mg/天、或600mg/天的剂量口服施用。
16.根据权利要求15所述的方法,其中将7-环戊基-N,N-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7H-吡咯并[2,3-d]嘧啶-6-甲酰胺以600mg口服施用,持续21天,随后中断治疗7天。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762587239P | 2017-11-16 | 2017-11-16 | |
| US62/587,239 | 2017-11-16 | ||
| US201862758835P | 2018-11-12 | 2018-11-12 | |
| US62/758,835 | 2018-11-12 | ||
| PCT/IB2018/058963 WO2019097426A1 (en) | 2017-11-16 | 2018-11-14 | Pharmaceutical combination comprising lsz102 and ribociclib |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111315378A true CN111315378A (zh) | 2020-06-19 |
Family
ID=64572406
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201880072600.5A Pending CN111315378A (zh) | 2017-11-16 | 2018-11-14 | 包含lsz102和瑞博西尼的药物组合 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US11179365B2 (zh) |
| EP (1) | EP3709997A1 (zh) |
| JP (1) | JP2021503448A (zh) |
| CN (1) | CN111315378A (zh) |
| TW (1) | TW201922238A (zh) |
| WO (1) | WO2019097426A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023284790A1 (zh) * | 2021-07-13 | 2023-01-19 | 江苏恒瑞医药股份有限公司 | 选择性雌激素受体共价拮抗剂与cdk4/6抑制剂联合在制备治疗乳腺癌药物中的用途 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019106604A1 (en) * | 2017-12-01 | 2019-06-06 | Novartis Ag | Pharmaceutical combination comprising lsz102 and alpelisib |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017172957A1 (en) * | 2016-04-01 | 2017-10-05 | Kalyra Pharmaceuticals, Inc. | Estrogen receptor modulators |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA200800766A1 (ru) | 2005-09-07 | 2008-06-30 | Лаборатуар Сероно С.А. | Ингибиторы pi3k для лечения эндометриоза |
| PL2331547T3 (pl) | 2008-08-22 | 2015-01-30 | Novartis Ag | Związki pirolopirymidynowe jako inhibitory CDK |
| EP2650401A1 (de) | 2012-04-10 | 2013-10-16 | Siemens Aktiengesellschaft | Kraftwerk basiertes Methanisierungssystem |
| HUE039052T2 (hu) | 2013-02-19 | 2018-12-28 | Novartis Ag | Benzotiofénszármazékok és azok készítményei szelektív ösztrogén receptor lebontóként |
| EP3696276A1 (en) * | 2013-02-25 | 2020-08-19 | Novartis AG | Novel androgen receptor mutation |
| WO2014205138A1 (en) | 2013-06-19 | 2014-12-24 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulator and uses thereof |
| WO2014203129A1 (en) * | 2013-06-19 | 2014-12-24 | Olema Pharmaceuticals, Inc. | Combinations of benzopyran compounds, compositions and uses thereof |
| PT3033086T (pt) | 2013-08-14 | 2021-12-15 | Novartis Ag | Terapia de combinação para o tratamento de cancro |
| WO2015028409A1 (de) | 2013-08-27 | 2015-03-05 | Bayer Pharma Aktiengesellschaft | 6,7-dihydro-5h-benzo[7]annulen-derivate, verfahren zu ihrer herstellung, pharmazeutische präparate die diese enthalten, sowie deren verwendung zur herstellung von arzneimitteln |
| JP6223154B2 (ja) | 2013-11-29 | 2017-11-01 | ロッテ アドバンスト マテリアルズ カンパニー リミテッド | ポリアミド樹脂およびその製造方法 |
| WO2015136016A2 (en) | 2014-03-13 | 2015-09-17 | F. Hoffmann-La Roche Ag | Therapeutic combinations with estrogen receptor modulators |
| IL307981A (en) | 2015-04-29 | 2023-12-01 | Radius Pharmaceuticals Inc | Cancer treatment methods |
| CA2981159A1 (en) | 2015-06-29 | 2017-01-05 | F. Hoffmann-La Roche Ag | Methods of treatment with taselisib |
| ES2917191T3 (es) | 2016-03-29 | 2022-07-07 | Novartis Ag | Medio de reacción que contiene una mezcla de agua-tensioactivo |
| EP3440067B1 (en) * | 2016-04-08 | 2021-05-26 | F. Hoffmann-La Roche AG | Tetrahydroisoquinoline estrogen receptor modulators and uses thereof |
| MX2019008158A (es) | 2017-01-06 | 2019-12-09 | G1 Therapeutics Inc | Terapia de combinacion para el tratamiento del cancer. |
| WO2019106604A1 (en) | 2017-12-01 | 2019-06-06 | Novartis Ag | Pharmaceutical combination comprising lsz102 and alpelisib |
-
2018
- 2018-11-14 US US16/190,298 patent/US11179365B2/en active Active
- 2018-11-14 CN CN201880072600.5A patent/CN111315378A/zh active Pending
- 2018-11-14 EP EP18812283.2A patent/EP3709997A1/en not_active Withdrawn
- 2018-11-14 JP JP2020526417A patent/JP2021503448A/ja active Pending
- 2018-11-14 WO PCT/IB2018/058963 patent/WO2019097426A1/en unknown
- 2018-11-16 TW TW107140852A patent/TW201922238A/zh unknown
-
2021
- 2021-10-15 US US17/502,986 patent/US20220031657A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017172957A1 (en) * | 2016-04-01 | 2017-10-05 | Kalyra Pharmaceuticals, Inc. | Estrogen receptor modulators |
Non-Patent Citations (4)
| Title |
|---|
| NCT02734615: "NCT02734615", 《NCT02734615》 * |
| S. P. CORONA ET AL.,: "Advances in systemic therapy for metastatic breast cancer: future perspectives", 《MEDICAL ONCOLOGY》 * |
| SUZANNE E. WARDELL ET AL.,: "Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy–Resistant Breast Cancer", 《CLINICAL CANCER RESEARCH》 * |
| 陈本川: "治疗晚期乳腺癌新药——瑞博西尼(ribociclib)", 《医药导报》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023284790A1 (zh) * | 2021-07-13 | 2023-01-19 | 江苏恒瑞医药股份有限公司 | 选择性雌激素受体共价拮抗剂与cdk4/6抑制剂联合在制备治疗乳腺癌药物中的用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2021503448A (ja) | 2021-02-12 |
| TW201922238A (zh) | 2019-06-16 |
| US20220031657A1 (en) | 2022-02-03 |
| WO2019097426A1 (en) | 2019-05-23 |
| EP3709997A1 (en) | 2020-09-23 |
| US20190142796A1 (en) | 2019-05-16 |
| US11179365B2 (en) | 2021-11-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2020222296B2 (en) | Pharmaceutical combination comprising TNO155 and ribociclib | |
| CN113382774A (zh) | 包含tno155和krasg12c抑制剂的药物组合 | |
| US20210000810A1 (en) | Pharmaceutical combination comprising lsz102 and alpelisib | |
| US20220031657A1 (en) | Pharmaceutical combination comprising lsz102 and ribociclib | |
| US11083722B2 (en) | Combination therapies for the treatment of breast cancer | |
| EP3890834A1 (en) | Methods for treating cancer resistant to cdk4/6 inhibitors | |
| AU2019246719B2 (en) | A triple pharmaceutical combination comprising dabrafenib, trametinib and an Erk inhibitor | |
| TW202339726A (zh) | 用於治療癌症之與畢尼替尼(Binimetinib)併用之HDAC抑制劑OKI-179 | |
| CN117177752A (zh) | 用于治疗mpnst的化合物和组合物 | |
| RU2813111C2 (ru) | Фармацевтическая комбинация, содержащая tno155 и рибоциклиб | |
| TW202143973A (zh) | 用於治療PRC2介導的疾病或障礙的N—((5—氟—2,3—二氫苯并呋喃—4—基)甲基)—8—(2—甲基吡啶—3—基)—[1,2,4]三唑并[4,3—c]嘧啶—5—胺或其藥學上可接受的鹽之給藥方案 | |
| JP2022511497A (ja) | Esr1変異を含むモデルにおいて癌を治療するための方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20200619 |