CN111116552B - 一种喹唑啉酮类化合物及其制备方法 - Google Patents
一种喹唑啉酮类化合物及其制备方法 Download PDFInfo
- Publication number
- CN111116552B CN111116552B CN202010055413.5A CN202010055413A CN111116552B CN 111116552 B CN111116552 B CN 111116552B CN 202010055413 A CN202010055413 A CN 202010055413A CN 111116552 B CN111116552 B CN 111116552B
- Authority
- CN
- China
- Prior art keywords
- compound
- reaction
- quinazolinone
- hydroxyquinazoline
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 Quinazolinone compound Chemical class 0.000 title claims abstract description 84
- 238000002360 preparation method Methods 0.000 title claims abstract description 34
- AVRPFRMDMNDIDH-UHFFFAOYSA-N 1h-quinazolin-2-one Chemical class C1=CC=CC2=NC(O)=NC=C21 AVRPFRMDMNDIDH-UHFFFAOYSA-N 0.000 claims abstract description 17
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 6
- 150000002367 halogens Chemical class 0.000 claims abstract description 6
- 239000001257 hydrogen Substances 0.000 claims abstract description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 6
- 150000002431 hydrogen Chemical class 0.000 claims abstract description 6
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims abstract description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 3
- 238000006243 chemical reaction Methods 0.000 claims description 86
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Natural products CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 78
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 75
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 75
- HZXJVDYQRYYYOR-UHFFFAOYSA-K scandium(iii) trifluoromethanesulfonate Chemical group [Sc+3].[O-]S(=O)(=O)C(F)(F)F.[O-]S(=O)(=O)C(F)(F)F.[O-]S(=O)(=O)C(F)(F)F HZXJVDYQRYYYOR-UHFFFAOYSA-K 0.000 claims description 48
- QMNUDYFKZYBWQX-UHFFFAOYSA-N 3H-quinazolinyl-4-one Natural products C1=CC=C2C(=O)N=CNC2=C1 QMNUDYFKZYBWQX-UHFFFAOYSA-N 0.000 claims description 46
- 239000007795 chemical reaction product Substances 0.000 claims description 25
- 239000002904 solvent Substances 0.000 claims description 25
- 238000004809 thin layer chromatography Methods 0.000 claims description 25
- 239000003054 catalyst Substances 0.000 claims description 14
- 239000003960 organic solvent Substances 0.000 claims description 13
- 229910052723 transition metal Inorganic materials 0.000 claims description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- 239000000126 substance Substances 0.000 claims description 7
- 239000002994 raw material Substances 0.000 claims description 5
- 238000010898 silica gel chromatography Methods 0.000 claims description 5
- 238000007259 addition reaction Methods 0.000 claims description 4
- 239000000463 material Substances 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- 239000012298 atmosphere Substances 0.000 claims description 3
- 125000003944 tolyl group Chemical group 0.000 claims description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 claims description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 2
- 238000000605 extraction Methods 0.000 claims description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 claims 1
- 239000003795 chemical substances by application Substances 0.000 claims 1
- 238000007670 refining Methods 0.000 claims 1
- 150000001875 compounds Chemical class 0.000 abstract description 6
- 239000003814 drug Substances 0.000 abstract description 6
- 230000000694 effects Effects 0.000 abstract description 6
- 238000009776 industrial production Methods 0.000 abstract description 6
- 230000001773 anti-convulsant effect Effects 0.000 abstract description 5
- 229940079593 drug Drugs 0.000 abstract description 5
- 230000000843 anti-fungal effect Effects 0.000 abstract description 4
- 230000003110 anti-inflammatory effect Effects 0.000 abstract description 4
- 230000000259 anti-tumor effect Effects 0.000 abstract description 4
- 239000001961 anticonvulsive agent Substances 0.000 abstract description 4
- 229960003965 antiepileptics Drugs 0.000 abstract description 4
- 238000003786 synthesis reaction Methods 0.000 abstract description 4
- 229940121375 antifungal agent Drugs 0.000 abstract description 3
- 239000002547 new drug Substances 0.000 abstract description 2
- 230000001766 physiological effect Effects 0.000 abstract description 2
- 239000000047 product Substances 0.000 description 54
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 48
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Natural products CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 44
- 238000001514 detection method Methods 0.000 description 25
- 229910052786 argon Inorganic materials 0.000 description 24
- 239000000706 filtrate Substances 0.000 description 23
- 239000011734 sodium Substances 0.000 description 23
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 22
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 22
- 238000013375 chromatographic separation Methods 0.000 description 22
- 125000004494 ethyl ester group Chemical group 0.000 description 22
- 239000000741 silica gel Substances 0.000 description 22
- 229910002027 silica gel Inorganic materials 0.000 description 22
- KFDVPJUYSDEJTH-UHFFFAOYSA-N 4-ethenylpyridine Chemical compound C=CC1=CC=NC=C1 KFDVPJUYSDEJTH-UHFFFAOYSA-N 0.000 description 13
- 238000000034 method Methods 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000007086 side reaction Methods 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- SGKYOBOFCJAJDT-UHFFFAOYSA-N 3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound C1=NC2=CC=CC=C2C(=O)N1CCC1=CC=NC=C1 SGKYOBOFCJAJDT-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- HIYUMYXSGIKHHE-UHFFFAOYSA-M bismuth trifluoromethanesulfonate Chemical compound [Bi+3].[O-]S(=O)(=O)C(F)(F)F HIYUMYXSGIKHHE-UHFFFAOYSA-M 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- OHAOVNXYYOWZJK-UHFFFAOYSA-N 2-bromo-4-ethenylpyridine Chemical compound BrC1=CC(C=C)=CC=N1 OHAOVNXYYOWZJK-UHFFFAOYSA-N 0.000 description 1
- OXEIJLUTWDDJCK-UHFFFAOYSA-N 2-chloro-4-ethenylpyridine Chemical compound ClC1=CC(C=C)=CC=N1 OXEIJLUTWDDJCK-UHFFFAOYSA-N 0.000 description 1
- VBXYZOUPVPNERZ-UHFFFAOYSA-N 2-ethenyl-3-methylpyridine Chemical compound CC1=CC=CN=C1C=C VBXYZOUPVPNERZ-UHFFFAOYSA-N 0.000 description 1
- WVNIWWGCVMYYJZ-UHFFFAOYSA-N 2-ethenyl-4-methylpyridine Chemical compound CC1=CC=NC(C=C)=C1 WVNIWWGCVMYYJZ-UHFFFAOYSA-N 0.000 description 1
- XUGNJOCQALIQFG-UHFFFAOYSA-N 2-ethenylquinoline Chemical compound C1=CC=CC2=NC(C=C)=CC=C21 XUGNJOCQALIQFG-UHFFFAOYSA-N 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- MYGZNGAHFWOCLI-BUHFOSPRSA-N 3-(2-methylphenyl)-2-[(e)-2-pyridin-2-ylethenyl]quinazolin-4-one Chemical compound CC1=CC=CC=C1N1C(=O)C2=CC=CC=C2N=C1\C=C\C1=CC=CC=N1 MYGZNGAHFWOCLI-BUHFOSPRSA-N 0.000 description 1
- KNSAKZNPHHYWBM-UHFFFAOYSA-N 3-(2-pyridin-2-ylethyl)quinazolin-4-one Chemical compound C1=NC2=CC=CC=C2C(=O)N1CCC1=CC=CC=N1 KNSAKZNPHHYWBM-UHFFFAOYSA-N 0.000 description 1
- MJSBGEIQXDWFPE-UHFFFAOYSA-N 3-(2-pyridin-4-ylethyl)-8-(trifluoromethyl)quinazolin-4-one Chemical compound N1=CC=C(C=C1)CCN1C=NC2=C(C=CC=C2C1=O)C(F)(F)F MJSBGEIQXDWFPE-UHFFFAOYSA-N 0.000 description 1
- SZQJJLNXXLHPMH-UHFFFAOYSA-N 3-(2-quinolin-2-ylethyl)quinazolin-4-one Chemical compound N1=C(C=CC2=CC=CC=C12)CCN1C=NC2=CC=CC=C2C1=O SZQJJLNXXLHPMH-UHFFFAOYSA-N 0.000 description 1
- GORQHKYWDRLFFD-UHFFFAOYSA-N 3-[2-(2-bromopyridin-4-yl)ethyl]quinazolin-4-one Chemical compound BrC1=NC=CC(=C1)CCN1C=NC2=CC=CC=C2C1=O GORQHKYWDRLFFD-UHFFFAOYSA-N 0.000 description 1
- SNXFSZJYCZQFCV-UHFFFAOYSA-N 3-[2-(2-chloropyridin-4-yl)ethyl]quinazolin-4-one Chemical compound ClC1=NC=CC(=C1)CCN1C=NC2=CC=CC=C2C1=O SNXFSZJYCZQFCV-UHFFFAOYSA-N 0.000 description 1
- GBHMPQBICYPOJP-UHFFFAOYSA-N 3-[2-(2-fluoropyridin-4-yl)ethyl]quinazolin-4-one Chemical compound FC1=NC=CC(=C1)CCN1C=NC2=CC=CC=C2C1=O GBHMPQBICYPOJP-UHFFFAOYSA-N 0.000 description 1
- IPQLLDDOBZTJKL-UHFFFAOYSA-N 3-[2-(2-methoxypyridin-4-yl)ethyl]quinazolin-4-one Chemical compound COC1=NC=CC(=C1)CCN1C=NC2=CC=CC=C2C1=O IPQLLDDOBZTJKL-UHFFFAOYSA-N 0.000 description 1
- ZWNRVPGUWWQKTF-UHFFFAOYSA-N 3-[2-(2-methylpyridin-4-yl)ethyl]quinazolin-4-one Chemical compound CC1=NC=CC(=C1)CCN1C=NC2=CC=CC=C2C1=O ZWNRVPGUWWQKTF-UHFFFAOYSA-N 0.000 description 1
- SOOZSTBIPWIUFM-UHFFFAOYSA-N 3-[2-(3-chloropyridin-4-yl)ethyl]quinazolin-4-one Chemical compound ClC=1C=NC=CC1CCN1C=NC2=CC=CC=C2C1=O SOOZSTBIPWIUFM-UHFFFAOYSA-N 0.000 description 1
- HECMKEBONRVDFC-UHFFFAOYSA-N 3-[2-(3-methylpyridin-2-yl)ethyl]quinazolin-4-one Chemical compound CC=1C(=NC=CC1)CCN1C=NC2=CC=CC=C2C1=O HECMKEBONRVDFC-UHFFFAOYSA-N 0.000 description 1
- WBDKJOFPIJIBBF-UHFFFAOYSA-N 3-[2-(4-methylpyridin-2-yl)ethyl]quinazolin-4-one Chemical compound CC1=CC(=NC=C1)CCN1C=NC2=CC=CC=C2C1=O WBDKJOFPIJIBBF-UHFFFAOYSA-N 0.000 description 1
- VFUTUYDMZQPKAW-UHFFFAOYSA-N 3-chloro-4-ethenylpyridine Chemical compound ClC1=CN=CC=C1C=C VFUTUYDMZQPKAW-UHFFFAOYSA-N 0.000 description 1
- WGVZHCAJTSCUEO-UHFFFAOYSA-N 4-ethenyl-2-fluoropyridine Chemical compound FC1=CC(C=C)=CC=N1 WGVZHCAJTSCUEO-UHFFFAOYSA-N 0.000 description 1
- VGJKSVULOADJLV-UHFFFAOYSA-N 4-ethenyl-2-methoxypyridine Chemical compound COC1=CC(C=C)=CC=N1 VGJKSVULOADJLV-UHFFFAOYSA-N 0.000 description 1
- FFOLJFOMOFZLPG-UHFFFAOYSA-N 4-ethenyl-2-methylpyridine Chemical compound CC1=CC(C=C)=CC=N1 FFOLJFOMOFZLPG-UHFFFAOYSA-N 0.000 description 1
- ZDPCOXNOQQRKCN-UHFFFAOYSA-N 5-chloro-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=C1C=CC=C2Cl ZDPCOXNOQQRKCN-UHFFFAOYSA-N 0.000 description 1
- CRJRYFJSKNCLJS-UHFFFAOYSA-N 5-chloro-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound ClC1=C2C(N(C=NC2=CC=C1)CCC1=CC=NC=C1)=O CRJRYFJSKNCLJS-UHFFFAOYSA-N 0.000 description 1
- OVEISJPVPHWEHR-UHFFFAOYSA-N 6-bromo-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC(Br)=CC=C21 OVEISJPVPHWEHR-UHFFFAOYSA-N 0.000 description 1
- ZFHLOJWSTGAWPZ-UHFFFAOYSA-N 6-bromo-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound O=C1C2=CC(Br)=CC=C2N=CN1CCC1=CC=NC=C1 ZFHLOJWSTGAWPZ-UHFFFAOYSA-N 0.000 description 1
- GOBVWEUSCRFCPB-UHFFFAOYSA-N 6-chloro-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC(Cl)=CC=C21 GOBVWEUSCRFCPB-UHFFFAOYSA-N 0.000 description 1
- KVXRKCQDSODBTL-UHFFFAOYSA-N 6-chloro-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound O=C1C2=CC(Cl)=CC=C2N=CN1CCC1=CC=NC=C1 KVXRKCQDSODBTL-UHFFFAOYSA-N 0.000 description 1
- WCSMZAHKVXOYLH-UHFFFAOYSA-N 6-fluoro-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC(F)=CC=C21 WCSMZAHKVXOYLH-UHFFFAOYSA-N 0.000 description 1
- RJTXSWIQJVRUMY-UHFFFAOYSA-N 6-fluoro-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound FC=1C=C2C(N(C=NC2=CC1)CCC1=CC=NC=C1)=O RJTXSWIQJVRUMY-UHFFFAOYSA-N 0.000 description 1
- PUGXMZKDRVGIHC-UHFFFAOYSA-N 6-iodo-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC(I)=CC=C21 PUGXMZKDRVGIHC-UHFFFAOYSA-N 0.000 description 1
- FENWYJKRNHKKES-UHFFFAOYSA-N 6-iodo-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound IC=1C=C2C(N(C=NC2=CC1)CCC1=CC=NC=C1)=O FENWYJKRNHKKES-UHFFFAOYSA-N 0.000 description 1
- NOFVNLZQAOGUIT-UHFFFAOYSA-N 6-methoxy-1h-quinazolin-4-one Chemical compound N1=CNC(=O)C2=CC(OC)=CC=C21 NOFVNLZQAOGUIT-UHFFFAOYSA-N 0.000 description 1
- DABSHWBBROGMPQ-UHFFFAOYSA-N 6-methoxy-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound COC=1C=C2C(N(C=NC2=CC1)CCC1=CC=NC=C1)=O DABSHWBBROGMPQ-UHFFFAOYSA-N 0.000 description 1
- JUCDXPIFJIVICL-UHFFFAOYSA-N 6-methyl-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC(C)=CC=C21 JUCDXPIFJIVICL-UHFFFAOYSA-N 0.000 description 1
- BICQCYLJQASQKJ-UHFFFAOYSA-N 6-methyl-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound CC=1C=C2C(N(C=NC2=CC1)CCC1=CC=NC=C1)=O BICQCYLJQASQKJ-UHFFFAOYSA-N 0.000 description 1
- MOBNCKURXDGQCB-UHFFFAOYSA-N 6-nitro-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC([N+](=O)[O-])=CC=C21 MOBNCKURXDGQCB-UHFFFAOYSA-N 0.000 description 1
- JFQVGCCDEOVWDW-UHFFFAOYSA-N 6-nitro-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound O=C1C2=CC([N+](=O)[O-])=CC=C2N=CN1CCC1=CC=NC=C1 JFQVGCCDEOVWDW-UHFFFAOYSA-N 0.000 description 1
- PMLONMIODRHERC-UHFFFAOYSA-N 7-chloro-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C=2C1=CC(Cl)=CC=2 PMLONMIODRHERC-UHFFFAOYSA-N 0.000 description 1
- LDHOZJUMOHVRNV-UHFFFAOYSA-N 7-chloro-3-(2-pyridin-4-ylethyl)quinazolin-4-one Chemical compound C=1C(Cl)=CC=C(C2=O)C=1N=CN2CCC1=CC=NC=C1 LDHOZJUMOHVRNV-UHFFFAOYSA-N 0.000 description 1
- YVUGNQDFJMZZGV-UHFFFAOYSA-N 8-(trifluoromethyl)-1h-quinazolin-4-one Chemical compound N1=CNC(=O)C2=C1C(C(F)(F)F)=CC=C2 YVUGNQDFJMZZGV-UHFFFAOYSA-N 0.000 description 1
- JEYCTXHKTXCGPB-UHFFFAOYSA-N Methaqualone Chemical compound CC1=CC=CC=C1N1C(=O)C2=CC=CC=C2N=C1C JEYCTXHKTXCGPB-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000003266 anti-allergic effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 230000000078 anti-malarial effect Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 239000003430 antimalarial agent Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003434 antitussive agent Substances 0.000 description 1
- 229940124584 antitussives Drugs 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 230000000147 hypnotic effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000000749 insecticidal effect Effects 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 229960002803 methaqualone Drugs 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 229950011379 piriqualone Drugs 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000036632 reaction speed Effects 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 230000001624 sedative effect Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及化学合成技术领域,具体公开一种喹唑啉酮类化合物及其制备方法。所述喹唑啉酮类化合物,其结构如式(Ⅰ)所示,其中,R1为氢、卤素、甲氧基、甲基、硝基或三氟甲基;R2为 或
或 R3为氢、卤素、甲氧基或甲基。本发明提供了一种新型结构的喹唑啉酮类化合物,丰富了喹唑啉酮类化合物的种类,为发展抗炎、抗肿瘤、抗惊厥或抗真菌药物提供了一种新化合物,对于研究该类化合物的活性以及扩大该类化合物在医学领域和工业生产上的应用具有十分重要的意义,为研究具有独特生理活性的新型药物提供了基础。
R3为氢、卤素、甲氧基或甲基。本发明提供了一种新型结构的喹唑啉酮类化合物,丰富了喹唑啉酮类化合物的种类,为发展抗炎、抗肿瘤、抗惊厥或抗真菌药物提供了一种新化合物,对于研究该类化合物的活性以及扩大该类化合物在医学领域和工业生产上的应用具有十分重要的意义,为研究具有独特生理活性的新型药物提供了基础。
Description
技术领域
本发明涉及化学合成技术领域,尤其涉及一种喹唑啉酮类化合物及其制备方法。
背景技术
喹唑啉酮类化合物代表一类独特的杂环化合物,由于其广泛存在于天然产物和具有生物活性的化合物中,且具有广泛的生物学和药理学活性,在抗过敏、抗癌、抗肿瘤、抗炎、抗高血压、抗微生物和抗真菌、抗疟疾和杀虫灭菌等方面显示出良好的活性;此外,含喹唑啉酮结构的衍生物还表现出细胞毒活性、心血管系统活性、利尿活性,例如具有镇静催眠作用的药物安眠酮(methaqualone),拥有抗痉挛和抗惊厥作用的药物苄啶喹酮(piriqualone),以及镇咳剂chloroqualone均是喹唑啉酮的衍生物。因其在医学领域和工业生产上都具有广泛的应用前景,喹唑啉酮类化合物的设计与合成受到越来越多科研工作者的关注,因此,合成具有新型结构的喹唑啉酮类化合物对于发展抗炎、抗肿瘤、抗惊厥或抗真菌药物具有十分重要的意义。但是,现有合成喹唑啉酮类化合物的方法较为繁琐,无法实现工业化生产。
发明内容
为了丰富喹唑啉酮类化合物的种类以及解决现有技术中合成喹唑啉酮类化合物的方法较为复杂的问题,本发明提供一种喹唑啉酮类化合物及其制备方法。
为解决上述技术问题,本发明提供的技术方案是:
一种喹唑啉酮类化合物,其结构如式(Ⅰ)所示:

其中,R1为氢、卤素、甲氧基、甲基、硝基或三氟甲基;
R2为
R3为氢、卤素、甲氧基或甲基。
相对于现有技术,本发明提供了一种新型结构的喹唑啉酮类化合物,丰富了喹唑啉酮类化合物的种类,为发展抗炎、抗肿瘤、抗惊厥或抗真菌药物提供了一种新化合物,对于研究该类化合物的活性以及扩大该类化合物在医学领域和工业生产上的应用具有十分重要的意义,为研究具有独特生理活性的新型药物提供了基础。
本发明还提供了一种上述喹唑啉酮类化合物的制备方法,以4-羟基喹唑啉化合物和乙烯基氮杂环化合物为原料,以可溶性过渡金属盐为催化剂,通过加成反应制得所述的喹唑啉酮类化合物;其中,所述乙烯基氮杂环化合物为4-乙烯基吡啶化合物、2-乙烯基吡啶化合物或2-乙烯基喹啉化合物;
所述4-乙烯基吡啶化合物的结构式为:

所述2-乙烯基吡啶化合物的结构式为:

所述2-乙烯基喹啉化合物的结构式为:

所述4-羟基喹唑啉化合物的结构式为:

优选的,所述喹唑啉酮类化合物的制备方法包括以下步骤:
将所述4-羟基喹唑啉化合物、乙烯基氮杂环化合物和可溶性过渡金属盐催化剂加入有机溶剂中,在惰性气氛中,于60-110℃反应,反应过程用薄层色谱法监测,反应结束后,淬灭反应,得所述喹唑啉酮类化合物。
目前已经有很多关于喹唑啉酮类化合物合成方法的报道,但是,以4-羟基喹唑啉化合物和乙烯基氮杂环化合物为原料通过一步法合成喹唑啉酮类化合物的方法还未见报道。本发明首次利用加成反应合成了一系列的未见文献报道的喹唑啉酮类化合物,实现了目标化合物的结构多样化,为设计新型结构的喹唑啉酮类化合物及构效关系的分析提供了一定的参考,对于扩展喹唑啉酮类化合物在医药和工业生产领域的应用具有十分重要的价值。
本发明提供的喹唑啉酮类化合物的制备方法是在可溶性过渡金属催化剂的催化作用下,直接利用4-羟基喹唑啉化合物和乙烯基氮杂环化合物进行加成反应,仅一步反应即可制得所需化合物,减少了中间体分离纯化的步骤,操作方便,且原子经济效益高,底物适用范围广,生成成本低,对环境不产生污染,易于实现工业化生产。
本发明中所述惰性气氛可由本领域常规的惰性气体提供,如氮气、氩气等。
优选的,所述可溶性过渡金属盐催化剂为三氟甲磺酸钪、三氟甲磺酸铋或醋酸铜。
更优选的,所述可溶性过渡金属盐催化剂为三氟甲磺酸钪。
优选的催化剂可加快反应的进行,缩短反应时间,降低副反应的发生,提高产品的纯度。
优选的,所述有机溶剂为甲苯、二甲基亚砜、四氢呋喃、乙腈、丙酮、二氯乙烷或1,4-二氧六环中至少一种。
更优选的,所述有机溶剂为甲苯。
优选的有机溶剂可以提高反应速率,使反应原料能够充分反应,并减少副反应的发生。
优选的,所述4-羟基喹唑啉化合物与乙烯基氮杂环化合物的摩尔比为1:1-3。
更优选的,所述4-羟基喹唑啉化合物与乙烯基氮杂环化合物的摩尔比为1:1.5。
优选的反应物质的比可以保证在用量较小的条件下,促进反应正向进行,提高目标产物的产率。
优选的,所述可溶性过渡金属盐催化剂与所述4-羟基喹唑啉化合物的物质的量比为0.1-0.2:1。
更优选的,所述可溶性过渡金属盐催化剂与所述4-羟基喹唑啉化合物的物质的量比为0.1:1。
优选的催化剂用量可以保证在催化剂用量最少的前提,最大限度的提高反应活性,提高反应速率以及产物收率;用量过少,催化效果不明显;用量过多,产率提高不明显,且造成催化剂的浪费以及产物纯度的降低。
优选的,所述有机溶剂的体积与所述4-羟基喹唑啉化合物的物质的量比为1-4:1,其中体积的单位为毫升,物质的量单位为毫摩尔。
更优选的,所述有机溶剂的体积与所述4-羟基喹唑啉化合物的物质的量比为4:1,其中体积的单位为毫升,物质的量单位为毫摩尔。
优选的有机溶剂的用量,可以保证反应原料的充分溶解,使其充分反应,提高目标产物的收率。
优选的,反应温度为110℃。
降低反应温度,反应速度也会随之降低,需要更长的反应时间;提高反应温度,由于副产物的生成,目标产物的收率也会降低。优选的反应温度,可以在最大限度降低副反应的前提下,保证目标产物的收率。
优选的,所述喹唑啉酮类化合物的制备方法还包括精制步骤:向反应产物中加入二氯甲烷萃取,向萃取液中加入饱和氯化钠溶液洗涤,无水硫酸钠脱水,浓缩,硅胶柱层析分离,得精制后的喹唑啉酮类化合物。
优选的,所述硅胶柱层析中的展开剂为体积比为200-500:1的乙酸乙酯和无水甲醇的混合溶剂。
优选的精制方法可提高目标产物的收率。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
为了更好的说明本发明,下面通过实施例做进一步的举例说明。
实施例1
3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮化合物的制备:
将4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)和三氟甲磺酸钪(0.1mmol)加入含有4mL甲苯的单口瓶中,将该烧瓶中的空气置换成氩气,然后在110℃搅拌条件下,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=500:1)层析分离,得到产物228.7mg,产率为91%。
反应方程式如下:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.50–8.43(m,2H),8.23(s,1H),8.17(dd,J=8.0,1.5Hz,1H),7.82(ddd,J=8.5,7.1,1.6Hz,1H),7.65(dd,J=8.2,1.1Hz,1H),7.55(ddd,J=8.2,7.1,1.2Hz,1H),7.30–7.24(m,2H),4.31–4.22(t,J=7.3Hz,2H),3.07(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,150.1,148.3,148.2,147.3,134.8,127.7,127.5,126.5,124.8,121.9,46.7,34.0。
实施例2
6-氟-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-氟-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1mmol)和三氟甲磺酸钪(0.15mmol)依次加入含有1mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在60℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=200:1)层析分离,得到产物237mg,产率为88%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.52–8.44(m,2H),8.24(s,1H),7.87–7.79(m,1H),7.79–7.67(m,2H),7.31–7.25(m,2H),4.28(dd,J=8.1,6.6Hz,2H),3.08(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.0,159.9,150.1,147.7,147.7,147.3,145.2,145.2,130.6,130.5,124.8,123.4,123.2,123.1,111.2,111.0,46.8,33.9。
实施例3
6-氯-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-氯-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(2mmol)、三氟甲磺酸钪(0.2mmol)依次加入含有3mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在80℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=300:1)层析分离,得到产物271.4mg,产率为95%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.51–8.44(m,2H),8.28(s,1H),8.09(d,J=2.5Hz,1H),7.85(dd,J=8.8,2.5Hz,1H),7.68(d,J=8.7Hz,1H),7.31–7.24(m,2H),4.27(t,J=7.3Hz,2H),3.07(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ159.6,150.2,148.7,147.2,147.0,134.9,131.8,130.0,125.4,124.8,123.1,46.8,33.8。
实施例4
6-溴-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-溴-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(3mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在100℃下搅拌,用薄层色谱法监测反应过程。反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=400:1)层析分离,得到产物241.0mg,产率为73%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6):δ8.47(d,J=5.0Hz,2H),8.30–8.20(m,2H),7.96(dd,J=8.7,2.4Hz,1H),7.61(d,J=8.7Hz,1H),7.26(d,J=5.1Hz,2H),4.26(t,J=7.3Hz,2H),3.06(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ159.5,150.2,148.8,147.3,147.2,137.7,130.1,128.6,124.8,123.5,120.0,46.9,33.8。
实施例5
6-碘-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-碘-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=400:1)层析分离,得到产物199.9mg,产率为53%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.51–8.40(m,3H),8.26(s,1H),8.10(dd,J=8.5,2.1Hz,1H),7.44(d,J=8.5Hz,1H),7.30–7.23(m,2H),4.26(t,J=7.3Hz,2H),3.06(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ159.3,150.1,148.9,147.6,147.2,143.1,134.7,129.8,124.8,123.7,92.7,46.8,33.8。
实施例6
6-硝基-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-硝基-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=350:1)层析分离,得到产物213.3mg,产率为72%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.83(s,3H),8.58–8.43(m,12H),7.86(d,J=8.9Hz,3H),7.29(d,J=4.8Hz,6H),4.30(t,J=7.3Hz,6H),3.10(t,J=7.4Hz,6H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ159.9,152.4,151.5,150.2,147.2,145.7,129.6,128.7,124.9,122.6,122.0,47.1,33.7。
实施例7
6-甲基-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-甲基-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=450:1)层析分离,得到产物196.3mg,产率为74%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.50–8.42(m,2H),8.16(s,1H),7.98–7.92(m,1H),7.63(dd,J=8.3,2.1Hz,1H),7.54(d,J=8.3Hz,1H),7.28–7.22(m,2H),4.25(dd,J=7.9,6.6Hz,2H),3.06(t,J=7.3Hz,2H),2.44(s,3H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.5,150.1,147.4,147.4,146.3,137.3,136.0,127.5,125.8,124.8,121.7,46.6,34.0,21.3。
实施例8
6-甲氧基-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将6-甲氧基-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=250:1)层析分离,得到产物154.7mg,产率为55%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.50–8.43(m,2H),8.11(s,1H),7.64–7.49(m,2H),7.42(dd,J=8.9,3.0Hz,1H),7.29–7.23(m,2H),4.27(dd,J=7.9,6.6Hz,2H),3.88(s,3H),3.07(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.3,158.5,150.1,147.4,146.0,142.8,129.4,124.8,124.3,122.8,106.5,56.1,46.6,33.9。
实施例9
7-氯-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将7-氯-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=500:1)层析分离,得到产物208.6mg,产率为73%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.50–8.44(m,2H),8.28(s,1H),8.15(d,J=8.6Hz,1H),7.72(d,J=2.1Hz,1H),7.58(dd,J=8.6,2.1Hz,1H),7.30–7.23(m,2H),4.25(dd,J=8.1,6.6Hz,2H),3.06(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.0,150.2,149.7,149.4,147.3,139.4,128.6,127.8,126.8,124.8,120.8,46.8,33.9。
实施例10
5-氯-3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将5-氯-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=200:1)层析分离,得到产物208.6mg,产率为73%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.52–8.45(m,2H),8.26(s,1H),7.73(t,J=8.0Hz,1H),7.57(ddd,J=10.7,8.0,1.2Hz,2H),7.32–7.26(m,2H),4.21(dd,J=8.3,6.6Hz,2H),3.05(t,J=7.4Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ158.6,150.8,150.2,149.0,147.4,134.6,132.9,130.0,127.3,124.8,118.9,47.0,33.7。
实施例11
3-(2-(吡啶-4-基)乙基)-8-(三氟甲基)喹唑啉-4(3H)-酮的制备:
将8-三氟甲基-4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=300:1)层析分离,得到产物210.7mg,产率为66%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.53–8.41(m,4H),8.21(d,J=7.5Hz,1H),7.69(t,J=7.8Hz,1H),7.30(d,J=5.4Hz,2H),4.29(t,J=7.4Hz,2H),3.10(t,J=7.4Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ159.8,150.2,149.6,147.2,145.7,132.4,132.4,131.3,126.9,125.5,125.4,125.2,124.7,123.2,46.9,33.8。
实施例12
3-(2-(3-氯吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、3-氯-4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=400:1)层析分离,得到产物194.3mg,产率为68%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.57(s,2H),8.44(d,J=4.9Hz,2H),8.23–8.10(m,4H),7.83(ddd,J=8.4,7.1,1.6Hz,2H),7.67(d,J=8.1Hz,2H),7.55(dd,J=15.1,1.2Hz,1H),7.55(s,1H),7.42(d,J=4.9Hz,2H),4.30(t,J=7.0Hz,4H),3.22(t,J=7.0Hz,4H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,149.3,148.6,148.3,148.1,145.0,134.8,132.1,127.7,127.5,126.5,126.4,121.9,45.4,31.7。
实施例13
3-(2-(2-氯吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、2-氯-4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=400:1)层析分离,得到产物191.4mg,产率为67%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.35–8.25(m,2H),8.17(dd,J=8.0,1.5Hz,1H),7.84(ddd,J=8.5,7.2,1.6Hz,1H),7.67(dd,J=8.2,1.1Hz,1H),7.56(ddd,J=8.2,7.1,1.2Hz,1H),7.48(d,J=1.3Hz,1H),7.30(dd,J=5.1,1.4Hz,1H),4.28(dd,J=8.0,6.6Hz,2H),3.11(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,151.7,151.0,150.3,148.3,148.2,134.8,127.7,127.6,126.5,125.0,124.2,121.9,46.3,33.7。
实施例14
3-(2-(2-氟吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、2-氟-4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=350:1)层析分离,得到产物110.4mg,产率为41%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.27(s,1H),8.17(dd,J=8.3,6.3Hz,2H),7.88–7.79(m,1H),7.67(d,J=8.1Hz,1H),7.56(t,J=7.6Hz,1H),7.24(d,J=5.0Hz,1H),7.13(s,1H),4.30(t,J=7.3Hz,2H),3.15(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ164.5,162.6,160.6,154.2,154.2,148.3,148.2,148.1,147.9,134.8,127.7,127.6,126.5,123.1,123.1,121.9,110.3,109.9,46.4,33.8,33.8。
实施例15
3-(2-(2-溴吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、2-溴-4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=450:1)层析分离,得到产物231.1mg,产率为70%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.30(t,J=2.6Hz,2H),8.17(dd,J=7.9,1.5Hz,1H),7.84(s,1H),7.68(d,J=8.1Hz,1H),7.61(s,1H),7.56(t,J=7.5Hz,1H),7.39–7.30(m,1H),4.28(t,J=7.3Hz,2H),3.09(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,151.4,150.8,148.2,142.1,134.8,128.8,127.7,127.6,126.5,124.5,121.9,46.3,33.6。
实施例16
3-(2-(2-甲基吡啶-4-基)乙基)喹唑啉-4(3H)-酮:
将4-羟基喹唑啉(1mmol)、2-甲基-4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=500:1)层析分离,得到产物175.1mg,产率为66%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.34(d,J=5.1Hz,2H),8.26(s,2H),8.18(dd,J=8.0,1.5Hz,2H),7.83(ddd,J=8.5,7.2,1.5Hz,2H),7.66(d,J=8.0Hz,2H),7.56(dd,J=15.2,1.1Hz,1H),7.56(s,1H),7.14(s,2H),7.06(dd,J=5.1,1.6Hz,2H),4.25(dd,J=8.2,6.7Hz,4H),3.02(t,J=7.4Hz,4H),2.41(s,6H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,158.4,149.4,148.3,148.3,147.5,134.8,127.6,127.5,126.5,124.0,121.9,121.8,46.7,33.9,24.4。
实施例17
3-(2-(2-甲氧基吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、2-甲氧基-4-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程。反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=400:1)层析分离,得到产物30.9mg,产率为11%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.24(s,1H),8.18(dd,J=8.0,1.6Hz,1H),8.07(d,J=5.2Hz,1H),7.83(ddd,J=8.4,7.1,1.6Hz,1H),7.69–7.63(m,1H),7.61–7.52(m,1H),6.87(dd,J=5.2,1.4Hz,1H),6.70(s,1H),4.25(t,J=7.3Hz,2H),3.82(s,3H),3.03(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ164.4,160.6,150.5,148.2,147.3,134.8,127.7,127.5,126.5,121.9,118.3,110.9,53.5,46.6,33.8。
实施例18
3-(2-(吡啶-2-基)乙基)喹唑啉-4(3H)-酮:
将4-羟基喹唑啉(1mmol)、2-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=500:1)层析分离,得到产物201.0mg,产率为80%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.59(ddd,J=4.9,1.9,0.9Hz,1H),8.32(dd,J=8.0,1.5Hz,1H),7.89(s,1H),7.73(ddd,J=8.5,7.1,1.6Hz,1H),7.64(dd,J=8.2,1.2Hz,1H),7.60–7.45(m,2H),7.15(ddd,J=7.6,4.9,1.1Hz,1H),7.07(dd,J=7.7,1.1Hz,1H),4.48(t,J=6.7Hz,2H),3.32(t,J=6.7Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ161.2,157.4,149.6,148.1,147.1,136.8,134.1,127.4,127.1,126.6,124.0,122.1,122.0,46.6,36.6。
实施例19
3-(2-(3-甲基吡啶-2-基)乙基)喹唑啉-4(3H)-酮:
将4-羟基喹唑啉(1mmol)、3-甲基-2-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=400:1)层析分离,得到产物220.2mg,产率为83%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.33(dd,J=4.8,1.7Hz,1H),8.30(s,1H),8.18(dd,J=8.0,1.5Hz,1H),7.82(ddd,J=8.5,7.1,1.6Hz,1H),7.66(dd,J=8.3,1.2Hz,1H),7.60–7.50(m,2H),7.16(dd,J=7.6,4.8Hz,1H),4.41(t,J=7.3Hz,2H),3.21(t,J=7.2Hz,2H),2.25(s,3H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.8,156.7,148.7,148.4,146.7,137.9,134.7,132.0,127.59,127.4,126.4,122.2,122.0,45.3,33.6,18.4。
实施例20
3-(2-(喹啉-2-基)乙基)喹唑啉-4(3H)-酮:
将4-羟基喹唑啉(1mmol)、2-乙烯基喹啉(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=350:1)层析分离,得到产物60.3mg,产率20%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.36(s,1H),8.27(d,J=8.4Hz,1H),8.16(dd,J=8.0,1.6Hz,1H),7.94–7.90(m,2H),7.79(ddd,J=8.5,7.1,1.6Hz,1H),7.72(ddd,J=8.3,6.9,1.5Hz,1H),7.62(d,J=8.1Hz,1H),7.57–7.49(m,2H),7.45(d,J=8.4Hz,1H),4.51(t,J=7.0Hz,2H),3.43(t,J=7.1Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.9,159.3,148.9,148.5,147.8,137.1,134.8,130.2,129.0,128.4,127.7,127.5,127.1,126.7,126.6,122.6,122.1,45.9,37.1。
实施例21
3-(2-(4-甲基吡啶-2-基)乙基)喹唑啉-4(3H)-酮:
将4-羟基喹唑啉(1mmol)、4-甲基-2-乙烯基吡啶(1.5mmol)、三氟甲磺酸钪(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=450:1)层析分离,得到产物209.6mg,产率79%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.34(d,J=5.1Hz,1H),8.26(s,1H),8.18(dd,J=8.0,1.5Hz,1H),7.83(ddd,J=8.5,7.2,1.5Hz,1H),7.66(d,J=8.0Hz,1H),7.61–7.52(m,1H),7.06(dd,J=5.1,1.6Hz,1H),4.25(dd,J=8.2,6.7Hz,2H),3.02(t,J=7.4Hz,2H),2.41(s,3H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,158.4,149.4,148.3,148.3,147.5,134.8,127.6,127.5,126.5,124.0,121.9,121.8,46.7,33.9,24.4。
实施例22
3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、三氟甲烷磺酸铋(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱(300~400目,展开剂为乙酸乙酯/甲醇=300:1)层析分离,得到产物163mg,产率为65%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.50–8.43(m,2H),8.23(s,1H),8.17(dd,J=8.0,1.5Hz,1H),7.82(ddd,J=8.5,7.1,1.6Hz,1H),7.65(dd,J=8.2,1.1Hz,1H),7.55(ddd,J=8.2,7.1,1.2Hz,1H),7.30–7.24(m,2H),4.31–4.22(t,J=7.3Hz,2H),3.07(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,150.1,148.3,148.2,147.3,134.8,127.7,127.5,126.5,124.8,121.9,46.7,34.0。
实施例23
3-(2-(吡啶-4-基)乙基)喹唑啉-4(3H)-酮的制备:
将4-羟基喹唑啉(1mmol)、4-乙烯基吡啶(1.5mmol)、乙酸铜(0.1mmol)依次加入含有4mL甲苯的单口烧瓶中,将该烧瓶中的空气置换成氩气,然后在110℃下搅拌,用薄层色谱法监测反应过程,反应完成后,用水(15mL)进行淬灭。
用二氯甲烷(3×20mL)提取反应产物,加入饱和盐水(20mL)洗涤,用无水Na2SO4脱水,过滤,将所得滤液浓缩,通过硅胶柱层析(300~400目,展开剂为乙酸乙酯/甲醇=400:1)得到产物193mg,产率为77%。
反应方程式为:

将所得产物进行核磁检测,1H NMR(400MHz,DMSO-d6)检测结果为:δ8.50–8.43(m,2H),8.23(s,1H),8.17(dd,J=8.0,1.5Hz,1H),7.82(ddd,J=8.5,7.1,1.6Hz,1H),7.65(dd,J=8.2,1.1Hz,1H),7.55(ddd,J=8.2,7.1,1.2Hz,1H),7.30–7.24(m,2H),4.31–4.22(t,J=7.3Hz,2H),3.07(t,J=7.3Hz,2H)。
13C NMR(100MHz,DMSO-d6)检测结果为:δ160.6,150.1,148.3,148.2,147.3,134.8,127.7,127.5,126.5,124.8,121.9,46.7,34.0。
将上述实施例1-23中的有机溶剂替换为二甲基亚砜(DMSO)、四氢呋喃(THF)、乙腈、丙酮、二氯乙烷或1,4-二氧六环中的一种或多种,均可达到与实施例1-23基本相当的效果。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
Claims (9)
1.一种式(Ⅰ)所示的喹唑啉酮类化合物的制备方法,其特征在于,以4-羟基喹唑啉化合物和乙烯基氮杂环化合物为原料,以可溶性过渡金属盐为催化剂,通过加成反应制得所述的喹唑啉酮类化合物;其中,所述乙烯基氮杂环化合物为4-乙烯基吡啶化合物、2-乙烯基吡啶化合物或2-乙烯基喹啉化合物;所述可溶性过渡金属盐催化剂为三氟甲磺酸钪;

其中,R1为氢、卤素、甲氧基、甲基、硝基或三氟甲基;
R2为
R3为氢、卤素、甲氧基或甲基;
所述4-乙烯基吡啶化合物的结构式为:

所述2-乙烯基吡啶化合物的结构式为:
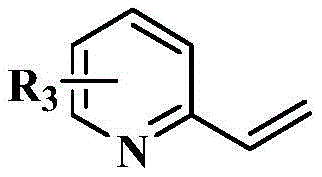
所述2-乙烯基喹啉化合物的结构式为:

所述4-羟基喹唑啉化合物的结构式为:

2.如权利要求1所述的喹唑啉酮类化合物的制备方法,其特征在于,包括如下步骤:
将所述4-羟基喹唑啉化合物、乙烯基氮杂环化合物和可溶性过渡金属盐催化剂加入有机溶剂中,在惰性气氛中,于60-110℃反应,反应过程用薄层色谱法监测,反应结束后,淬灭反应,得所述喹唑啉酮类化合物。
3.如权利要求2所述的喹唑啉酮类化合物的制备方法,其特征在于,所述有机溶剂为甲苯、二甲基亚砜、四氢呋喃、乙腈、丙酮、二氯乙烷或1,4-二氧六环中至少一种。
4.如权利要求3所述的喹唑啉酮类化合物的制备方法,其特征在于,所述有机溶剂为甲苯。
5.如权利要求2-4任一项所述的喹唑啉酮类化合物的制备方法,其特征在于,所述4-羟基喹唑啉化合物与乙烯基氮杂环化合物的摩尔比为1:1-3;和/或
所述可溶性过渡金属盐催化剂与所述4-羟基喹唑啉化合物的物质的量比为0.1-0.2:1。
6.如权利要求5所述的喹唑啉酮类化合物的制备方法,其特征在于,所述有机溶剂的体积与所述4-羟基喹唑啉化合物的物质的量比为1-4:1,其中体积的单位为毫升,物质的量单位为毫摩尔。
7.如权利要求6所述的喹唑啉酮类化合物的制备方法,其特征在于,所述4-羟基喹唑啉化合物与乙烯基氮杂环化合物的摩尔比为1:1.5;和/或
所述可溶性过渡金属盐催化剂与所述4-羟基喹唑啉化合物的物质的量比为0.1:1;和/或
所述有机溶剂的体积与所述4-羟基喹唑啉化合物的物质的量比为4:1,其中体积的单位为毫升,物质的量单位为毫摩尔。
8.如权利要求2所述的喹唑啉酮类化合物的制备方法,其特征在于,还包括精制步骤:向反应产物中加入二氯甲烷萃取,向萃取液中加入饱和氯化钠溶液洗涤,无水硫酸钠脱水,浓缩,硅胶柱层析分离,得精制后的喹唑啉酮类化合物。
9.如权利要求8的喹唑啉酮类化合物的制备方法,其特征在于,所述硅胶柱层析中的展开剂为体积比为200-500:1的乙酸乙酯和无水甲醇的混合溶剂。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010055413.5A CN111116552B (zh) | 2020-01-17 | 2020-01-17 | 一种喹唑啉酮类化合物及其制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010055413.5A CN111116552B (zh) | 2020-01-17 | 2020-01-17 | 一种喹唑啉酮类化合物及其制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111116552A CN111116552A (zh) | 2020-05-08 |
| CN111116552B true CN111116552B (zh) | 2022-10-11 |
Family
ID=70491015
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010055413.5A Active CN111116552B (zh) | 2020-01-17 | 2020-01-17 | 一种喹唑啉酮类化合物及其制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111116552B (zh) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1201393A (zh) * | 1995-10-23 | 1998-12-09 | 酶遗传学股份有限公司 | 治疗骨缺陷疾病的组合物及方法 |
| US6274383B1 (en) * | 1997-12-15 | 2001-08-14 | Sepracor Inc. | Methods for synthesizing libraries of dihydro-quinazolinones |
| CN1421440A (zh) * | 2002-12-06 | 2003-06-04 | 华东理工大学 | 一种喹唑啉-4(3h)衍生物的合成方法 |
| CN101429165A (zh) * | 2007-11-09 | 2009-05-13 | 温州大学 | 一种喹唑啉酮类化合物的合成方法 |
| CN101445487A (zh) * | 2009-01-04 | 2009-06-03 | 上海大学 | 3h-喹唑啉-4-酮类化合物的制备方法 |
-
2020
- 2020-01-17 CN CN202010055413.5A patent/CN111116552B/zh active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1201393A (zh) * | 1995-10-23 | 1998-12-09 | 酶遗传学股份有限公司 | 治疗骨缺陷疾病的组合物及方法 |
| US6274383B1 (en) * | 1997-12-15 | 2001-08-14 | Sepracor Inc. | Methods for synthesizing libraries of dihydro-quinazolinones |
| CN1421440A (zh) * | 2002-12-06 | 2003-06-04 | 华东理工大学 | 一种喹唑啉-4(3h)衍生物的合成方法 |
| CN101429165A (zh) * | 2007-11-09 | 2009-05-13 | 温州大学 | 一种喹唑啉酮类化合物的合成方法 |
| CN101445487A (zh) * | 2009-01-04 | 2009-06-03 | 上海大学 | 3h-喹唑啉-4-酮类化合物的制备方法 |
Non-Patent Citations (1)
| Title |
|---|
| RN: 930554-30-6,930060-11-0,930060-10-9,930056-39-6,858712-88-6,190437-51-5;STN REGISTRY DATABASE;《STN REGISTRY DATABASE》;20070417 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111116552A (zh) | 2020-05-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107698586A (zh) | 一种由铜化合物催化制备吡啶并喹唑啉酮类化合物的方法 | |
| CN110183379B (zh) | 一种铜催化一锅法制备c-4位砜基取代异喹啉酮类化合物的合成方法及应用 | |
| CN107973779A (zh) | 一种n-(2-吡啶/嘧啶基)吲哚衍生物的制备方法 | |
| CN114989094B (zh) | 一种可见光催化合成苯并咪唑衍生物的方法 | |
| CN111116552B (zh) | 一种喹唑啉酮类化合物及其制备方法 | |
| CN117384157B (zh) | 骆驼宁碱b及其衍生物的制备方法和应用 | |
| CN107778240A (zh) | 一种6‑苯基菲啶类化合物的制备方法 | |
| CN109535086B (zh) | 一种喹喔啉-2(1h)-酮c-3位羧酸酯化合物的合成方法 | |
| CN108084076A (zh) | 一种5-溴-7-氮杂吲哚的合成方法 | |
| CN112142796A (zh) | 可见光促进铜催化制备c-4位磷酸酯取代2-氨基异喹啉酮类衍生物的合成方法及应用 | |
| CN114685439B (zh) | 一种拉司米地坦的制备方法 | |
| CN104016917A (zh) | 5,6-二氢啡啶类化合物及其制备方法 | |
| CN106986810B (zh) | 手性3-取代异吲哚啉酮类化合物及其制备方法与应用 | |
| CN116120313A (zh) | 一种吡啶并咪唑氮稠环化合物及其制备方法和应用 | |
| CN110294757B (zh) | 一种铜催化一锅法制备c-3位硫代咪唑并吡啶类衍生物的合成方法及应用 | |
| CN115772171A (zh) | 一价金催化的螺[吲哚啉-3,3’-哌啶]骨架的合成方法 | |
| CN116768788B (zh) | 一种采用微通道反应装置光诱导连续合成吡啶类化合物的方法 | |
| CN112300157B (zh) | 一种新型具有抗肿瘤活性的吡唑并吡啶类化合物及其制备方法 | |
| CN108129402A (zh) | 以二苯乙炔类化合物为原料合成2-苯基喹唑啉酮类化合物的方法 | |
| CN108033919A (zh) | 以苯乙烯类化合物为原料合成2-苯基喹唑啉酮类化合物的方法 | |
| CN106866681A (zh) | 一种合成6,6a‑二氢异吲哚[2,1‑a]喹唑啉‑5,11‑二酮类化合物的方法 | |
| WO2024183094A1 (zh) | 一种配体催化剂在合成吲哚类生物碱的应用 | |
| CN115521307A (zh) | 一种5-卤代-7-氮杂吲哚的制备方法 | |
| CN114920693A (zh) | 一种制备2-氨基喹啉类化合物的方法 | |
| CN106478679B (zh) | 1‑(2‑吡啶)‑9‑环丙基甲基‑β‑咔啉的硝酸铜配合物及其合成方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20231005 Address after: 050000 House A (A6) 301, Building 201 #, Jingshi Collaborative Innovation Demonstration Park, No. 769, Taihang South Street, High tech Zone, Shijiazhuang, Hebei Patentee after: Andixsen medical technology Hebei Co.,Ltd. Address before: 050018 No.26 Yuxiang street, Yuhua District, Shijiazhuang City, Hebei Province Patentee before: HEBEI University OF SCIENCE AND TECHNOLOGY |
|
| TR01 | Transfer of patent right |