CN103641725A - Preparation method of phenylethylamine - Google Patents
Preparation method of phenylethylamine Download PDFInfo
- Publication number
- CN103641725A CN103641725A CN201310630723.5A CN201310630723A CN103641725A CN 103641725 A CN103641725 A CN 103641725A CN 201310630723 A CN201310630723 A CN 201310630723A CN 103641725 A CN103641725 A CN 103641725A
- Authority
- CN
- China
- Prior art keywords
- phenylethylamine
- chloroform
- preparation
- toluene
- extraction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention belongs to the technical field of chemical synthesis, and particularly relates to a preparation method of phenylethylamine. The preparation method of phenylethylamine comprises the following steps: adding phenylacetamide and methylbenzene in a tetrahydrofuran solution of zinc borohydride, heating slowly until the inner temperature reaches 90-96 DEG C, preserving heat and stirring for 3.5-4.5 hours, naturally cooling the reaction liquid to the room temperature, pouring 10% hydrochloric acid, filtering, extracting the filter liquor with chloroform, alkalifying with 20% sodium hydroxide until the pH value reaches 11-12, continuously extracting with chloroform, combining the extracting solutions, drying with anhydrous MgSO4, and carrying out reduced pressure distillation after chloroform is recycled to obtain the phenylethylamine. According to the preparation method of phenylethylamine, zinc borohydride is used as a reducing agent to reduce phenylacetamide, the side reactions are few, the yield is high and the cost is low.
Description
Technical field
The invention belongs to chemosynthesis technical field, be specifically related to a kind of preparation method of phenylethylamine.
Background technology
Phenylethylamine (Phenethylamine, PEA) is a kind of alkaloid and monoamine neurotransmitter.Its another name β-phenylethylamine, 2-phenylethylamine, be for No. CAS 618-36-0, molecular formula C
8h
11n, structural formula is:
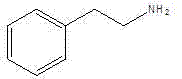
Phenylethylamine is important medicine and dyestuff intermediate, pharmaceutically be mainly used in synthetic following medicine energizer: (Stimulant), as plant alkaloid ephedrine (Ephedrine) and cathinone (Cathinone) and synthetic drugs dextroamphetamine (Dextroamphetamine) with send vinegar methyl esters (Methylphenidate).Fantasy: (Hallucinogen), (Mescaline, beauty card, Mescaline) with synthetic drugs 2C-B as plant alkaloid mescaline.Appetite agent is fallen: (Anorectic), as Phentermine (Phentermine), Phenfluoramine (S-768, Fenfluramine) and amphetamine.Bronchodilator: (Bronchodilator), as salbutamol (Salbutamol) and ephedrine (Ephedrine).Thymoleptic: (Antidepressant), as speed happy (Venlafaxine), Bupropion (Bupropion) and monoamine oxidation ferment inhibitor (Monoamine oxidase inhibitor, MAOI) Phenelzine (Phenelzine) and anti-ring propylamine (Tranylcypromine).
In prior art, multiplex phenylacetamide is the synthetic phenylethylamine of starting raw material, reductive agent is selected respectively sodium borohydride, iron powder, Lithium Aluminium Hydride, although sodium borohydride is gentleer, but long reaction time, consumption is large, and iron powder is low as reductive agent yield, raw material reaction is incomplete, Lithium Aluminium Hydride reaction is very fast, but expensive, side reaction is many.
Summary of the invention
In order to solve above-mentioned technical problem, the invention provides a kind of preparation method of phenylethylamine.
The present invention realizes by following technical scheme:
A preparation method for phenylethylamine, comprises following step:
In the tetrahydrofuran solution of zinc borohydride, add phenylacetamide and toluene, slowly heating makes 90~96 ℃ of Nei Wenda, insulated and stirred 3.5-4.5h, and reaction solution naturally cools to after room temperature, pour in 10% hydrochloric acid, filter, filtrate, after chloroform extraction, is alkalized to pH value 11~12 with 2O% sodium hydroxide, continuation chloroform extraction, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine.
In the preparation method of above-mentioned phenylethylamine, the mass percent concentration of the tetrahydrofuran solution of described zinc borohydride is 5%.
In the preparation method of above-mentioned phenylethylamine, the ratio of described phenylethylamine and toluene is 10.8:70, and its ratio unit is g:ml.
Preferably, in the preparation method of above-mentioned phenylethylamine, described temperature of reaction is 93 ℃.
Preferably, in the preparation method of above-mentioned phenylethylamine, the described reaction times is 4h.
In the preparation method of above-mentioned phenylethylamine, described filtrate is 2 times through chloroform extraction number of times, and the volume ratio of each chloroform consumption and toluene is 2.5:1.
In the preparation method of above-mentioned phenylethylamine, it is 5 times that described alkalization continues with the extraction time of chloroform extraction afterwards, each chloroform consumption be toluene use 1/2.
In the preparation method of phenylethylamine of the present invention, reaction temperature ℃ affects larger on yield, and when being less than 40 ℃, seldom, 50 ℃-70 ℃, yield increases product gradually, and when being greater than 90 ℃, yield can reach 80%.
Science of law reaction formula of the present invention is as follows:

Beneficial effect of the present invention is, adopts zinc borohydride to reduce phenylacetamide as reductive agent, and side reaction is few, and yield is high, and cost is low.
Accompanying drawing explanation
Fig. 1 is the graph of a relation of embodiment of the present invention temperature of reaction and yield.
Embodiment
Below in conjunction with specific embodiment, the present invention is further described, so that those skilled in the art more understands the present invention, but does not therefore limit the present invention.
Embodiment 1
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 93 ℃ of Nei Wenda, insulated and stirred 4h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 8.2g, yield 84.0%.
Embodiment 2
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 90 ℃ of Nei Wenda, insulated and stirred 3.5h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 7.9g, yield 80.8%.
Embodiment 3
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 96 ℃ of Nei Wenda, insulated and stirred 4.5h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 8.1g, yield 83.4%.
Comparative example
Embodiment 4
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 45 ℃ of Nei Wenda, insulated and stirred 4h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 1.5g, yield 15.4%.
Embodiment 5
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 50 ℃ of Nei Wenda, insulated and stirred 4h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 2.8g, yield 28.6%.
Embodiment 6
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 60 ℃ of Nei Wenda, insulated and stirred 4h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 4.4g, yield 45.2%.
Embodiment 7
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 70 ℃ of Nei Wenda, insulated and stirred 4h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 5.7g, yield 58.9%.
Embodiment 8
In the tetrahydrofuran solution of the zinc borohydride 5%, add phenylacetamide 1O.8g and toluene 70mL, slowly add and make 80 ℃ of Nei Wenda, insulated and stirred 4h, reaction solution naturally cools to after room temperature, pours in 150 mL 10% hydrochloric acid, filters, filtrate is used chloroform extraction 2 times, after each chloroform consumption 205ml extracts, with 2O% sodium hydroxide, alkalize to pH value 11~12, continuation chloroform extraction, use respectively 35 mL chloroform extraction 5 times, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine 6.9g, yield 80.5%.
Be below the graph of a relation between each embodiment temperature of reaction and yield, as seen from Figure 1, when temperature of reaction is greater than 90 ℃, yield is higher, surpasses 80%.
Fig. 1 is the graph of a relation of embodiment of the present invention temperature of reaction and yield.As shown in Figure 1, when temperature of reaction is greater than 90 ℃, it is the highest that yield is greater than 80%, 93 ℃ of yield.
Claims (7)
1. a preparation method for phenylethylamine, comprises following step:
In the tetrahydrofuran solution of zinc borohydride, add phenylacetamide and toluene, slowly heating makes 90~96 ℃ of Nei Wenda, insulated and stirred 3.5-4.5h, and reaction solution naturally cools to after room temperature, pour in 10% hydrochloric acid, filter, filtrate, after chloroform extraction, is alkalized to pH value 11~12 with 2O% sodium hydroxide, continuation chloroform extraction, united extraction liquid is with anhydrous MgSO
4dry, to reclaim after chloroform, underpressure distillation, obtains phenylethylamine.
2. the preparation method of phenylethylamine according to claim 1, is characterized in that, the mass percent concentration of the tetrahydrofuran solution of described zinc borohydride is 5%.
3. the preparation method of phenylethylamine according to claim 1, is characterized in that, the ratio of described phenylethylamine and toluene is 10.8:70, and its ratio unit is g:ml.
4. the preparation method of phenylethylamine according to claim 1, is characterized in that, described temperature of reaction is 93 ℃.
5. the preparation method of phenylethylamine according to claim 1, is characterized in that, the described reaction times is 4h.
6. the preparation method of phenylethylamine according to claim 1, is characterized in that, described filtrate is 2 times through chloroform extraction number of times, and the volume ratio of each chloroform consumption and toluene is 2.5:1.
7. the preparation method of phenylethylamine according to claim 1, is characterized in that, it is 5 times that described alkalization continues with the extraction time of chloroform extraction afterwards, each chloroform consumption be toluene use 1/2.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310630723.5A CN103641725A (en) | 2013-12-02 | 2013-12-02 | Preparation method of phenylethylamine |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310630723.5A CN103641725A (en) | 2013-12-02 | 2013-12-02 | Preparation method of phenylethylamine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103641725A true CN103641725A (en) | 2014-03-19 |
Family
ID=50247057
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201310630723.5A Pending CN103641725A (en) | 2013-12-02 | 2013-12-02 | Preparation method of phenylethylamine |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN103641725A (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111500651A (en) * | 2020-04-10 | 2020-08-07 | 宁波酶赛生物工程有限公司 | Preparation method of phenylethylamine |
-
2013
- 2013-12-02 CN CN201310630723.5A patent/CN103641725A/en active Pending
Non-Patent Citations (1)
| Title |
|---|
| 何嘉俊等: "硼氢化锌还原苯乙酰胺的研究", 《化学与生物工程》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111500651A (en) * | 2020-04-10 | 2020-08-07 | 宁波酶赛生物工程有限公司 | Preparation method of phenylethylamine |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105061224B (en) | Synthetic method of L-2-aminobutanol | |
| CN110616236B (en) | Preparation method of (R) -N1, N1-diethyl-1, 4-pentanediamine | |
| CN104263796A (en) | Preparation method of R-1-aminotetralin | |
| CN1990455B (en) | Simple and novel process for preparing indenes derivatives | |
| CN102746161A (en) | Method for synthesizing 1,8-terpene diamine | |
| CN108276296B (en) | Synthesis method of cyanide antidote | |
| CN103641725A (en) | Preparation method of phenylethylamine | |
| CN103641724A (en) | Synthetic method of phenylethylamine | |
| CN102229538A (en) | Method for synthesizing dapoxetine | |
| CN101525293B (en) | Production method of optically active N-benzyl-1-phenylethylamine | |
| CN109134215A (en) | A kind of liquid metals sodium slag method prepares the production method of trimethyl orthoformate | |
| CN113620855B (en) | Isomakava intermediate II and synthesis method thereof | |
| CN1285567C (en) | Process for synthesizing paracetamol | |
| CN104263801B (en) | A kind of preparation method of R-2- tetrahydronaphthalene amines | |
| CN101967103A (en) | Novel preparation method of itopride intermediate body | |
| CN105294449B (en) | Preparation method for (R)-(+)-1-(1-naphthyl)ethylamine and (S)-(-)-1-(1-naphthyl)ethylamine | |
| CN101328129B (en) | Preparation of 3-methoxy propanamine | |
| CN102408329A (en) | Preparation method of 2,4-dihydroxy benzoic acid | |
| CN103319358B (en) | Preparation method of 7-amino heptanoic acid | |
| CN101328130B (en) | Preparation of 2-ethoxy ethyl amine | |
| CN110818548A (en) | Method for preparing benzylidene acetone | |
| CN105523984B (en) | (1R, 2S)- 1- phenyl -2-(1- pyrrolidinyls)The preparation method of -1- propyl alcohol | |
| CN103896784A (en) | Method for reducing nitro of Fingolimod intermediate to amino | |
| CN102442958B (en) | Preparation method of isomer-removed tebuconazole | |
| CN102863354B (en) | A kind of method of the amino-butanamide of dynamic resolution 2 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20140319 |