CN102633779A - Fasudil acetate as well as preparation method and application thereof - Google Patents
Fasudil acetate as well as preparation method and application thereof Download PDFInfo
- Publication number
- CN102633779A CN102633779A CN2012101252852A CN201210125285A CN102633779A CN 102633779 A CN102633779 A CN 102633779A CN 2012101252852 A CN2012101252852 A CN 2012101252852A CN 201210125285 A CN201210125285 A CN 201210125285A CN 102633779 A CN102633779 A CN 102633779A
- Authority
- CN
- China
- Prior art keywords
- fasudil
- acetate
- acetic acid
- glacial acetic
- organic solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Abstract
The invention relates to fasudil acetate as well as a preparation method and application thereof, belonging to the technical field of medicines and chemicals. According to the method, fasudil acetate is prepared through reaction between fasudil and acetic acid, and fasudil hydrochloride with high purity is prepared from fasudil acetate, therefore, the column chromatography operation is effectively avoided, the problems of need of column chromatography, high toxicity and long separation period existing in a fasudil hydrochloride preparation process are solved, and therefore, the process is more suitable for industrial production.
Description
Technical field
The present invention relates to a kind of new compound fasudil acetate, belong to pharmaceutical chemistry technical field.
Background technology
Fasudil is an isoquinoline 99.9 sulfamido material; Chemical name is six hydrogen-1-(5-alkylsulfonyl isoquinoline 99.9)-1H-1; The 4-diazepine, English name fasudil is researched and developed by Japanese Asahi Kasei Corporation (Asahi Kasei Pharma); Have the effect of powerful vasodilative effect and protection ischemic tissue of brain, belong to the Rho-SU11752.Fasudil went on the market in Japan in nineteen ninety-five, and at present marketed drug be its mono-hydrochloric salts form, and the cerebral vasospasm that is used to improve after the subarachnoid hemorrhage reaches the symptoms of cerebral ischemia that thereupon causes.New indication-the acute cerebral thrombosis of this medicine also waits for ratification in Japan at present.
In the preparation method of the HA-1077 of having reported; The main bullion that adopts 5-isoquinoline 99.9 SULPHURYL CHLORIDE and the reaction of high piperazine to obtain fasudil; This bullion makes fasudil through column chromatographic isolation and purification, and last and hydrochloric acid salify makes the title product HA-1077.
In the prior art, the major impurity in the fasudil bullion is the two acylates shown in the formula (II), for removing two acylates and other impurity in the fasudil bullion, obtains the fasudil of better purity, the general column chromatography method that adopts in the technology.But the silica gel consumption of this method is big, needs higher chloroform of toxicity or methylene dichloride as elutriant, and solvent is volatile; Loss is big, and the separation and purification cycle is long, and cost is high; The process scale of having the more important thing is the big limitations of this method is not suitable for the industrial production of mass-producing.

Summary of the invention
To the deficiency of prior art, the invention provides a kind of new midbody compound, six hydrogen-1-(5-alkylsulfonyl isoquinoline 99.9)-1H-1,4-diazepine acetate, i.e. fasudil acetate, structural formula is suc as formula shown in (I):
N=1 or 2 wherein.
The preparation method of this fasudil acetate is: in organic solvent, fasudil and glacial acetic acid are made in 0-60 ℃ of reaction, reaction equation is following:

Reaction finishes, and slowly is cooled to 2-8 ℃ of scope interior crystallization 6-8 hour, suction filtration, washing, the dry fasudil acetate that gets.
Wherein, described organic solvent is selected from methyl alcohol, ethanol, Virahol, acetone, methylene dichloride, trichloromethane, acetonitrile, N, dinethylformamide, DMAC N,N, one or more in the THF.
In the preparation process because glacial acetic acid is weak organic acid, can with fasudil free alkali salify, but can not react salifies with the more weak two acylates of alkalescence.Fasudil in the fasudil bullion through with the glacial acetic acid salify after separate out through crystallization; And two acidylate impurity are not because of continuing to stay in the mother liquor with the glacial acetic acid salify; Therefore; This salification process can effectively be removed the two acylates in the fasudil bullion, obtains highly purified fasudil acetate.In order to guarantee that the fasudil bullion can fully dissolve, abundant with glacial acetic acid reaction salify, temperature of reaction is controlled at 0-60 ℃, preferred 40-60 ℃.General exothermic effect in order to prevent that topical agent is excessive and possibly to occur, glacial acetic acid adopts the mode that drips.In addition, in this preparation process, fasudil is different with the mol ratio of glacial acetic acid, and the acetate of the fasudil that obtains is also inequality: when the mol ratio of glacial acetic acid and fasudil is 1-1.1:1, obtain the fasudil Monoacetate; When the mol ratio of glacial acetic acid and fasudil is 2-4:1, obtain the fasudil diacetate.
The molecular formula of fasudil acetate and molecular weight thereof such as table 1 are listed among the present invention:
The molecular formula and the molecular weight thereof of table 1 fasudil acetate
| Title | Molecular formula | Molecular weight |
| Fasudil Monoacetate (n=1) | C 14H 17N 3O 2S·CH 3COOH | 351.42 |
| Fasudil diacetate (n=2) | C 14H 17N 3O 2S·2CH 3COOH | 411.47 |
In addition, the present invention also provides the method for preparing highly purified HA-1077 through the fasudil acetate.Adopt the fasudil acetate to prepare HA-1077 and can avoid the column chromatography operation effectively; Solved the problem that the need column chromatography, the toxicity that exist among the HA-1077 preparation technology are big, separation cycle is long; Make technology be fit to suitability for industrialized production more, comprise the steps:
(1) in the organic solvent of fasudil acetate, add inorganic base aqueous solution, conditioned reaction liquid pH value 9-11 guarantees that fasudil fully dissociates out, tells organic phase, washing, and drying is filtered, and concentrating under reduced pressure obtains highly purified fasudil;
(2) highly purified fasudil is dissolved in the methyl alcohol, adds hydrochloric acid reaction, conditioned reaction liquid pH value 4-5 guarantees the abundant salify of fasudil, is cooled to-5-5 ℃ crystallization, obtains highly purified HA-1077.Wherein, the excessive salify that causes of pH value is insufficient; The pH value is too small, can produce dihydrochloride in the product, causes the yield of title product fasudil mono-hydrochloric salts to reduce.
Organic solvent is selected from methylene dichloride described in the preparation process of above-mentioned high-purity hydrochloric acid fasudil, trichloromethane, one or more in the ETHYLE ACETATE; Said mineral alkali is selected from one or more in sodium hydroxide, Pottasium Hydroxide, yellow soda ash, salt of wormwood, sodium hydrogencarbonate, the saleratus.
In sum; The invention provides a kind of fasudil acetate and preparation method thereof, and utilize this fasudil acetate to make highly purified HA-1077, detect through performance liquid chromatography (HPLC) method; HA-1077 purity is higher than 99.8%; Avoid the column chromatography operation effectively, solved the problem that the need column chromatography, the toxicity that exist among the HA-1077 preparation technology are big, separation cycle is long, make technology be fit to suitability for industrialized production more.
Embodiment
Below, foregoing of the present invention is done further to specify through the embodiment of embodiment form.Protection scope of the present invention includes but not limited to the description of following examples.The fasudil bullion all adopts the preparation of patent CN1183782A reported method in following examples, wherein the two acylate content 3.66% of impurity.
Embodiment 1
Take by weighing fasudil bullion 50.8g (0.174mol), add the 50ml absolute ethyl alcohol,, dropwise in 55-60 ℃ of dropping glacial acetic acid 11ml (0.174mol); Slowly be cooled to 5-8 ℃ of crystallization 7 hours, suction filtration, cold ethanol filter wash cake gets white to the off-white color solid; Drying gets product 41.8g, productive rate 68.4%; Detect through performance liquid chromatography (HPLC) method, purity is 99.92%, and the two acylates of impurity do not detect.Product is the fasudil Monoacetate.
Ultimate analysis: (C
14H
17N
3O
2SCH
3COOH): C, 54.70; H, 5.99; N, 11.97; S, 9.11;
Theoretical value: C, 54.68; H, 6.02; N, 11.96; S, 9.12.
1H-NMR(600?MHz,?DMSO-d6)
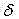 :9.70(s,?2H)?,?9.52(s,?1H),?8.73(d,?J?=?6Hz,?1H),?8.46(d,?J?=?6.0Hz,?1H),?8.35(d,?J?=?7.2Hz,?1H),?8.19(d,?J?=?8.4Hz,?1H),?7.69(t,?J?=?7.8Hz,?1H),?3.51(t,?J?=?6.0Hz,?2H),?3.46(t,?J?=?5.4Hz,?2H),?3.00(t,?J?=?4.8Hz,?2H),?2.95(t,?J?=?6.0Hz,?2H),?2.09(s,?3H)?,?1.85(m,?2H)。
:9.70(s,?2H)?,?9.52(s,?1H),?8.73(d,?J?=?6Hz,?1H),?8.46(d,?J?=?6.0Hz,?1H),?8.35(d,?J?=?7.2Hz,?1H),?8.19(d,?J?=?8.4Hz,?1H),?7.69(t,?J?=?7.8Hz,?1H),?3.51(t,?J?=?6.0Hz,?2H),?3.46(t,?J?=?5.4Hz,?2H),?3.00(t,?J?=?4.8Hz,?2H),?2.95(t,?J?=?6.0Hz,?2H),?2.09(s,?3H)?,?1.85(m,?2H)。
MS(ESI,?m/z?),?292.1[M-CH
3COO]
+?,?314.3[M-CH
3COOH+Na]
+。
IR(KBr)cm
-1:3433,3392,2950,1647,1619,1563,1404,1308,1130,1015,899,812,716,599。
Embodiment 2
Take by weighing fasudil bullion 50.8g (0.174mol), add the 150ml anhydrous methanol,, dropwise in 30-35 ℃ of dropping glacial acetic acid 12ml (0.19mol); Slowly be cooled to 2-5 ℃ of crystallization 6 hours, suction filtration, cold methanol filter wash cake gets white to the off-white color solid; Drying gets product 36.6g, productive rate 59.9%; Detect through performance liquid chromatography (HPLC) method, purity is 99.94%, and the two acylates of impurity do not detect.Product is the Monoacetate of fasudil.
Embodiment 3
Take by weighing fasudil bullion 50.8g (0.174mol), add the 100ml methylene dichloride,, dropwise in 40-45 ℃ of dropping glacial acetic acid 12ml (0.19mol); Slowly be cooled to 4-6 ℃ of crystallization 6 hours, suction filtration, cold methylene dichloride filter wash cake gets white to the off-white color solid; Drying gets product 40.2g, productive rate 65.8%; Detect through performance liquid chromatography (HPLC) method, purity is 99.95%, and the two acylates of impurity do not detect.Product is the fasudil Monoacetate.
Embodiment 4
Take by weighing fasudil bullion 50.8g (0.174mol), add 120ml acetone,, dropwise in 0-20 ℃ of dropping glacial acetic acid 11ml (0.174mol); Slowly be cooled to 6-8 ℃ of crystallization 8 hours, suction filtration, cold acetone filter wash cake gets white to the off-white color solid; Drying gets product 41.8g, productive rate 68.4%; Detect through performance liquid chromatography (HPLC) method, purity is 99.92%, and the two acylates of impurity do not detect.Product is the fasudil Monoacetate.
Embodiment 5
Take by weighing fasudil bullion 200g (0.69mol), add the 300ml absolute ethyl alcohol,, dropwise in 30-40 ℃ of dropping glacial acetic acid 86.4ml (1.51mol); Slowly be cooled to 2-3 ℃ of crystallization 6 hours, suction filtration, cold ethanol filter wash cake; Must be white to the off-white color solid, drying gets product 192.4g; Productive rate 68.2%, (HPLC) purity is 99.97%, the two acylates of impurity do not detect.Product is the diacetate of fasudil.
Ultimate analysis: (C
14H
17N
3O
2S2CH
3COOH): C, 52.58; H, 5.09; N, 10.23; S, 7.78;
Theoretical value: C, 52.54; H, 6.12; N, 10.21; S, 7.79.
1H-NMR(600?MHz,?DMSO-d6)
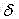 :10.12(s,?1H),?9.65(s,?2H),?9.53(s,?1H),?8.81(d,?J?=?6Hz,?1H),?8.51(d,?J?=?6.0Hz,?1H),?8.37(d,?J?=?7.2Hz,?1H),?8.22(d,?J?=?8.4Hz,?1H),?7.73(t,?J?=?7.8Hz,?1H),?3.51(t,?J?=?6.0Hz,?2H),?3.49(t,?J?=?5.4Hz,?2H),?3.11(t,?J?=?4.8Hz,?2H),?2.97(t,?J?=?6.0Hz,?2H),?2.11(s,?6H)?,?1.87(m,?2H)。
:10.12(s,?1H),?9.65(s,?2H),?9.53(s,?1H),?8.81(d,?J?=?6Hz,?1H),?8.51(d,?J?=?6.0Hz,?1H),?8.37(d,?J?=?7.2Hz,?1H),?8.22(d,?J?=?8.4Hz,?1H),?7.73(t,?J?=?7.8Hz,?1H),?3.51(t,?J?=?6.0Hz,?2H),?3.49(t,?J?=?5.4Hz,?2H),?3.11(t,?J?=?4.8Hz,?2H),?2.97(t,?J?=?6.0Hz,?2H),?2.11(s,?6H)?,?1.87(m,?2H)。
MS(ESI,?m/z?),?292.1[M-2CH
3COOH+H]
+?,?314.3[M-2CH
3COOH+Na]
+。
IR(KBr)cm
-1:3430,3393,2950,1690,1646,1620,1564,1403,1333,1130,1014,894,713,596。
Embodiment 6
Take by weighing fasudil bullion 200g (0.69mol), add the 600ml anhydrous methanol,, dropwise in 10-20 ℃ of dropping glacial acetic acid 158ml (2.76mol);, slowly be cooled to 6-8 ℃ of crystallization 8 hours, suction filtration, small amount of cold methyl alcohol filter wash cake; Must be white to the off-white color solid, drying gets product 172.1g, productive rate 61%; Detect through performance liquid chromatography (HPLC) method, purity is 99.96%, and the two acylates of impurity do not detect.Product is the diacetate of fasudil.
Embodiment 7:
(1) takes by weighing the fasudil Monoacetate 35.1g (0.1mol, purity 99.92%) that embodiment 1 makes, add the 200ml methylene dichloride; The pH that stirs the sodium hydroxide solution adjusting water that adds 2mol/L down is 9, tells organic phase, washing; Drying is filtered, and concentrating under reduced pressure gets white to off-white color solid 28.4g; Be fasudil, detect that purity is 99.7% through performance liquid chromatography (HPLC) method.
(2) the gained fasudil is joined in the 110ml methyl alcohol, add the hydrochloric acid soln of 4mol/L after the stirring and dissolving, the pH to 4 of regulator solution; And-5-0 ℃ following stirring and crystallizing 4 hours; Suction filtration, methyl alcohol filter wash cake, the gained filter cake obtains white solid and is HA-1077 after drying; Amount to 29.4g, yield 89.7%; Detect through performance liquid chromatography (HPLC) method, purity is 99.96%, and the two acylates of impurity do not detect.
Embodiment 8:
(1) takes by weighing embodiment 5 fasudil diacetate 41.1g (0.1mol, purity 99.97%), add the 240ml methylene dichloride; The pH that stirs the sodium hydroxide solution adjusting water that adds 2mol/L down is 11, tells organic phase, washing; Drying is filtered, and concentrating under reduced pressure gets white to off-white color solid 27.2g; Be fasudil, detect that purity is 99.7% through performance liquid chromatography (HPLC) method.
(2) the gained fasudil is joined in the 100ml methyl alcohol, add the hydrochloric acid soln of 4mol/L after the stirring and dissolving, conditioned reaction liquid pH to 5; And at 0-5 ℃ of following stirring and crystallizing 6 hours, suction filtration, small amount of methanol washing leaching cake; The gained filter cake obtains white solid and is HA-1077 after drying; Amount to 27.5g, yield 83.9%; Detect through performance liquid chromatography (HPLC) method, purity is 99.98%, and the two acylates of impurity do not detect.
Embodiment 9:
(1) takes by weighing the fasudil Monoacetate 35.1g (0.1mol, purity 99.95%) that embodiment 3 makes, add 150ml ETHYLE ACETATE; The pH that stirs the potassium hydroxide solution adjusting water that adds 2mol/L down is 9, tells organic phase, washing; Drying is filtered, and concentrating under reduced pressure gets white to off-white color solid 29.2g; Be fasudil, detect that purity is 99.7% through performance liquid chromatography (HPLC) method.
(2) the gained fasudil is joined in the 110ml methyl alcohol, add the hydrochloric acid soln of 4mol/L after the stirring and dissolving, the pH to 5 of regulator solution; And-5-0 ℃ following stirring and crystallizing 4 hours; Suction filtration, methyl alcohol filter wash cake, the gained filter cake obtains white solid and is HA-1077 after drying; Amount to 28.8g, yield 87.9%; Detect through performance liquid chromatography (HPLC) method, purity is 99.98%, and the two acylates of impurity do not detect.
Claims (8)
1. the fasudil acetate has the structure shown in the formula (I), and chemical name is: six hydrogen-1-(5-alkylsulfonyl isoquinoline 99.9)-1H-1, and 4-diazepine acetate,

N=1 or 2 wherein.
2. prepare the method for the described fasudil acetate of claim 1, it is characterized in that: in organic solvent with fasudil and glacial acetic acid behind 0-60 ℃ of reaction response, cooling crystallization, suction filtration, washing, drying make,

Wherein, n=1 or 2.
3. the method for preparing the fasudil acetate according to claim 2 is characterized in that: recrystallization temperature is 2-8 ℃, and the crystallization time is 6-8 hour.
4. the method for preparing the fasudil acetate according to claim 2; It is characterized in that: said organic solvent is selected from methyl alcohol, ethanol, Virahol, acetone, methylene dichloride, trichloromethane, acetonitrile, N; Dinethylformamide, N; The N-N,N-DIMETHYLACETAMIDE, one or more in the THF.
5. the method for preparing the fasudil acetate according to claim 2 is characterized in that: temperature of reaction is 40-60 ℃.
6. the method for preparing the fasudil acetate according to claim 2 is characterized in that: the mol ratio of acetate and fasudil be below arbitrary ratio:
When (1) mol ratio of glacial acetic acid and fasudil is 1-1.1:1, obtain the fasudil Monoacetate, i.e. n=1;
When (2) mol ratio of glacial acetic acid and fasudil is 2-4:1, obtain the fasudil diacetate, i.e. n=2.
7. application rights requires 1 described fasudil acetate to prepare the method for high-purity hydrochloric acid fasudil, it is characterized in that: comprise the steps:
(1) in the organic solvent of fasudil acetate, add inorganic base aqueous solution, conditioned reaction liquid pH value 9-11 tells organic phase, washing, and drying is filtered, and concentrating under reduced pressure obtains highly purified fasudil;
(2) will go up the highly purified fasudil that goes on foot acquisition and be dissolved in the methyl alcohol, and add hydrochloric acid reaction, conditioned reaction liquid pH value 4-5 is cooled to-5-5 ℃ crystallization, obtains highly purified HA-1077.
8. the preparation method of high-purity hydrochloric acid fasudil according to claim 7 is characterized in that: said organic solvent is selected from methylene dichloride, trichloromethane, one or more in the ETHYLE ACETATE; Said mineral alkali is selected from one or more in sodium hydroxide, Pottasium Hydroxide, yellow soda ash, salt of wormwood, sodium hydrogencarbonate, the saleratus.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210125285.2A CN102633779B (en) | 2012-04-26 | 2012-04-26 | Fasudil acetate as well as preparation method and application thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210125285.2A CN102633779B (en) | 2012-04-26 | 2012-04-26 | Fasudil acetate as well as preparation method and application thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102633779A true CN102633779A (en) | 2012-08-15 |
| CN102633779B CN102633779B (en) | 2014-01-22 |
Family
ID=46618372
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210125285.2A Active CN102633779B (en) | 2012-04-26 | 2012-04-26 | Fasudil acetate as well as preparation method and application thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102633779B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109705096A (en) * | 2019-03-07 | 2019-05-03 | 山东新华制药股份有限公司 | A kind of refining methd of Fasudic hydrochloride |
| WO2020177291A1 (en) * | 2019-03-04 | 2020-09-10 | 中国药科大学 | Fasudil compound salt, preparation method therefor and use thereof |
| CN112010805A (en) * | 2020-08-26 | 2020-12-01 | 山东威高药业股份有限公司 | Method for refining fasudil hydrochloride |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0187371A2 (en) * | 1984-12-27 | 1986-07-16 | Asahi Kasei Kogyo Kabushiki Kaisha | Substituted isoquinolinesulfonyl compounds |
| CN101341122A (en) * | 2005-12-20 | 2009-01-07 | 吉瑞工厂 | Novel process for production of highly pure polymorph (I) donepezil hydrochloride |
| CN102120739A (en) * | 2010-01-07 | 2011-07-13 | 成都欣捷高新技术开发有限公司 | Preparation method of fasudil hydrochloride |
-
2012
- 2012-04-26 CN CN201210125285.2A patent/CN102633779B/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0187371A2 (en) * | 1984-12-27 | 1986-07-16 | Asahi Kasei Kogyo Kabushiki Kaisha | Substituted isoquinolinesulfonyl compounds |
| CN101341122A (en) * | 2005-12-20 | 2009-01-07 | 吉瑞工厂 | Novel process for production of highly pure polymorph (I) donepezil hydrochloride |
| CN102120739A (en) * | 2010-01-07 | 2011-07-13 | 成都欣捷高新技术开发有限公司 | Preparation method of fasudil hydrochloride |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020177291A1 (en) * | 2019-03-04 | 2020-09-10 | 中国药科大学 | Fasudil compound salt, preparation method therefor and use thereof |
| CN109705096A (en) * | 2019-03-07 | 2019-05-03 | 山东新华制药股份有限公司 | A kind of refining methd of Fasudic hydrochloride |
| CN109705096B (en) * | 2019-03-07 | 2023-06-09 | 山东新华制药股份有限公司 | Refining method of fasudil hydrochloride |
| CN112010805A (en) * | 2020-08-26 | 2020-12-01 | 山东威高药业股份有限公司 | Method for refining fasudil hydrochloride |
| CN112010805B (en) * | 2020-08-26 | 2023-12-05 | 山东威高药业股份有限公司 | Refining method of fasudil hydrochloride |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102633779B (en) | 2014-01-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105330609B (en) | A kind of method for preparing LCZ696 | |
| CN106349245A (en) | Sitagliptin phosphate impurities, method for preparing same and application of sitagliptin phosphate impurities | |
| CN102395591B (en) | Method for preparing prasugrel | |
| CN106256824A (en) | A kind of preparation method of high-purity De Lasha star meglumine salt | |
| CN102633779B (en) | Fasudil acetate as well as preparation method and application thereof | |
| CN101985442B (en) | Convenient and quick method for preparing high-purity imatinib and mesylate thereof | |
| CN103408548B (en) | The method of synthesis (R)-9-(2-hydroxypropyl) VITAMIN B4 | |
| CN103145636B (en) | 1,4-diacyl-3,6-diphenyl-1,4-dihydrotetrazine compound as well as preparation method and application thereof | |
| CN103787924A (en) | New purification method of antitumor drug Belinostat | |
| CN104557877B (en) | A kind of avanaphil intermediate and its preparation method and application | |
| CN103087090B (en) | Synthetic method of 2-dipalmitoyl-sn-glycero-3-phosphoethanolamine | |
| CN105566260A (en) | Furosemide preparation method | |
| CN102911186A (en) | Ceftizoxime sodium preparation and refining method | |
| CN103396373A (en) | Preparation method of deferasirox and intermediate compound of deferasirox | |
| CN104250251A (en) | Preparation method for ticagrelor | |
| CN104725349A (en) | Polycrystalline A-type crystal of alogliptin polycrystalline, preparation method and production purpose thereof | |
| CN106478624A (en) | A kind of purification process of moxifloxacin hydrochloride | |
| CN102329317B (en) | Method for synthesizing theobromine | |
| CN108069900B (en) | Preparation method and application of aripiprazole hydrochloride | |
| CN106588786A (en) | Preparation method of high purity favipiravir impurity | |
| CN105294620A (en) | Synthetic method for 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid | |
| CN104211705B (en) | A kind of preparation method of sldenafil | |
| CN103880807B (en) | The synthesis technique of high-purity alpha-mangostin | |
| CN104557922A (en) | Synthetic method for 6-bromoimidazo[1,2-a]pyridine-8-carboxylic acid | |
| CN101787035A (en) | Preparation method of 7-aminocephalo-5-mercapto-1-methyltetrazole |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20201026 Address after: 570314 -A, Nanhai Avenue, national hi tech Development Zone, Hainan, Haikou, 273 Patentee after: QILU PHARMACEUTICAL (HAINAN) Co.,Ltd. Patentee after: Qilu Pharmaceutical Co.,Ltd. Address before: 250100 No. 243 industrial North Road, Shandong, Ji'nan Patentee before: Qilu Pharmaceutical Co.,Ltd. |
|
| TR01 | Transfer of patent right |