CN102531940B - Preparation method for high purity etofenamate - Google Patents
Preparation method for high purity etofenamate Download PDFInfo
- Publication number
- CN102531940B CN102531940B CN201110437413.2A CN201110437413A CN102531940B CN 102531940 B CN102531940 B CN 102531940B CN 201110437413 A CN201110437413 A CN 201110437413A CN 102531940 B CN102531940 B CN 102531940B
- Authority
- CN
- China
- Prior art keywords
- reaction
- preparation
- etofenamate
- organic solvent
- tecramine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention relates to a preparation method for high purity etofenamate. Flufenamic acid reacts with diethylene glycol in non-protic organic solvent in the presence of an organic carboxylic acid activating agent (R*-Cl) such as chlorosilane and sulfonyl chloride, reactant is hydrolyzed under the acidic condition and then extracted and separated simply to obtain high purity etofenamate. Compared with the prior art, the preparation method for the high purity etofenamate has the advantages of being less in equipment investment and reaction steps, easy to operate, high in productivity and product purity and the like and has wide application prospects.
Description
Technical field
The preparation method who the present invention relates to a kind of etofenamate compound, belongs to technical field of medicine synthesis.
Background technology
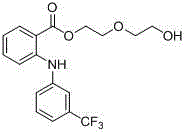
Etofenamate, its English name is Etofenamate, chemical name is 2-(2-hydroxyl-oxethyl)-ethyl-2-[3-(trifluoromethyl) anilino] benzoic ether, No. CAS: [30544-47-9], molecular formula: C
18h
18f
3nO
4, molecular weight: 369.33.Structural formula:

Etofenamate is a kind of fenamic acids NSAID (non-steroidal anti-inflammatory drug), research and develop and went on the market in Germany in 1977 by Beyer Co., Ltd, after the countries and regions listing such as in Australia, Switzerland, Spain, Taiwan in succession.Beyer Co., Ltd released creme in 1999 in China, within 2002, sold emulsifiable paste, within 2005, sold sprays and bulk drug.Bayer (China) got permission to produce emulsifiable paste in 2002, registration goods " excellent stepping " by name (Traumon).This products material medicine is recorded by European Pharmacopoeia.
Etofenamate has analgesia, rheumatism, antipyretic, antiinflammatory property.It has extremely strong close ester and certain wetting ability, and this makes it have the performance of good transdermal.Can suppress release and the effect of bradykinin, cyclooxygenase, ester oxidase, histamine, serotonin, hyaluronic acid and total complement, stablize lysosome membrane, reduce the reaction to foreign matters such as prostaglandin(PG) and other inflammatory mediators, thereby bring into play anti-inflammatory, analgesic activity.Etofenamate can be treated: the various soft tissue injuries in soft tissue rheumatism, joint etc.; The soft tissue rheumatism disease of skeletal musculature, as muscle rheumatism, shoulder pain and stiff, muscle spasm (scapulohumeral periarthritis joint surrounding pain), pain in the back, sciatica, tenosynovitis, bursitis; Rachiopathy and joint disease due to overworked or degeneration, wound are as dampened, sprain and strain etc.
The current disclosed preparation method of etofenamate mainly contains following several:
Patent GB1285400(DE1939112, US3692818) preparation method of etofenamate is disclosed the earliest, use Tecramine sylvite and 2-(2-chloroethoxy) ethanol reacting by heating in DMF, then separate and obtain etofenamate through silicagel column.Because the hydroxyl of 2-(2-chloroethoxy) ethanol is more active, make that this byproduct of reaction is many and concentration is large, be difficult to obtain sterling with general separation purification method, restrict the use of the method.
For the problems referred to above; patent DE2735569 has announced one and has improved one's methods; first by a hydroxyl benzyl protection of glycol ether; then react with sulfur oxychloride; generate chloro diglycol monotertiary benzyl oxide; and then react with Tecramine sylvite, obtaining the etofenamate of benzyl protection, last hydrogenation and removing benzyl protection obtains finished product.This route need be through four-step reaction, complicated operation, and total recovery is low.
Patent ES471375 has announced an other route, use an alkali metal salt of o-iodobenzoic acid, elder generation and 2-(2-chloroethoxy) ethanol synthesis, and then and a trifluoromethyl aniline carry out aminating reaction, generate etofenamate structure, raw materials used expensive and product composition is very complicated, separate that to obtain the yield of sterling very low.
Patent DE2834169 first utilizes Tecramine and PCl
5reaction obtains acyl chlorides, and then carries out esterification with glycol ether, generate etofenamate target product, but reaction product is also very complicated, separation and purification difficulty, and yield is lower than 50%.We attempt using sulfur oxychloride as acylating agent with reference to the method, first obtain the acyl chlorides compound of Tecramine, then react with glycol ether, have obtained and the similar result of patent, and reaction product is very complicated, is difficult to purifying.
What patent DE2834268 and DE4436269 announced is ester exchange method, first use Tecramine and propyl carbinol to carry out esterification, obtain the positive butyl ester of Tecramine, then under sodium methylate catalysis, carry out transesterification reaction with glycol ether, can obtain the etofenamate of 90% purity, then could separating-purifying product (170 ~ 175 ℃/0.001mmHg) by the rectifying of high vacuum high temperature.High-vacuum apparatus investment when reaction is amplified is huge, and we are through finding the THERMAL STABILITY of etofenamate product, it just obviously produces dimerization impurity after 1 hour 170 ℃ of placements, be the impurity D(2 that European Pharmacopoeia EP6.0 describes, 2 '-oxygen-bis-ethyl-bis--[2-[[3-(trifluoromethyl) phenyl] amino] benzoic ether]), the limit of this impurity in pharmacopeia is for being not more than 0.5%.This also means that rectifying need to be used special equipment could obtain the effect of separation and purification.ES512031 adopts similar method, carries out transesterify after first forming methyl esters, and result roughly the same.
Also have patent report to use conventional Lewis acid catalysis, directly make Tecramine and glycol ether carry out the method for esterification, such as DE3811119 and ES2003782 report, yield is also on the low side, purifying products difficulty.
Patent ES534122 has announced a more atypical method, first with formaldehyde and Tecramine reaction, generate the Er hydrogen benzoxazinone structure of 75% yield, then under existing, catalytic amount sodium hydroxide reacts in 175 ℃ with glycol ether, exchange and discharge etofenamate product, the high energy of exchange step yield reaches 78%, but needs high vacuum rectification purifying, and total recovery is also lower.Patent ES548225 has continued to use this thinking, with the first condensation of Vinyl chloroformate replacement formaldehyde, then exist stream next time to carry out cyclization at sulfur oxychloride etc., generate Er hydrogen benzoxazinone structure, react with glycol ether again, high energy separates the etofenamate that obtains 98.5% purity, but total recovery is lower, and the index of quality product and European Pharmacopoeia differs larger.
In the method for the synthetic etofenamate of above bibliographical information, all have that yield is on the low side, separation difficulty, need the shortcomings such as the poor and content of special high vacuum high temperature rectifying device, product appearance is low.At present, domestic also do not have producer to produce this product to mass-producing, is all to rely on import to produce preparation.Therefore, novel method for synthesizing and the route of development this product have very high Social benefit and economic benefit.
Summary of the invention
The present invention aims to provide a kind of high purity etofenamate preparation method, solve that the yield existing in prior art is on the low side, separation difficulty, need the shortcomings such as the poor and content of special high vacuum high temperature rectifying device, product appearance is low, produce the high purity product that meets European Pharmacopoeia, ensure clinical application and security requirement.
Technical scheme provided by the invention is as follows:
The new preparation process of a kind of high purity etofenamate (formula (I)),
Described method comprises reacts with glycol ether Tecramine under the organic carboxyl acid such as chlorosilane, SULPHURYL CHLORIDE activator (R*-Cl) exists in non-proton organic solvent, after reactant acidic hydrolysis, pass through again follow-up lock out operation, can obtain highly purified etofenamate.Reaction formula is as follows:

Wherein, formula (II) compound is Tecramine, and formula (III) compound is glycol ether, and R*-Cl compound is the organic carboxyl acid activators such as chlorosilane or SULPHURYL CHLORIDE.
The preparation method of the etofenamate of said structure provided by the invention, comprises the steps:
Tecramine is joined in non-proton organic solvent, add the organic carboxyl acid such as chlorosilane, SULPHURYL CHLORIDE activator (R*-Cl), stir, then add glycol ether to continue reaction.After reaction finishes, according to the polarity difference of reaction solvent, reaction solution directly can be added in weak acid water and is hydrolyzed, or add in weak acid water and be hydrolyzed after concentrated, generate etofenamate structure.Then regulate pH to alkalescence, stratification or with water-insoluble organic solvent extract, organic layer can obtain highly purified etofenamate again after several washing, activated carbon decolorizing, filtration, concentrating under reduced pressure.
The organic carboxyl acid activator (R*-Cl) using can be the chlorosilane such as trimethylchlorosilane (TMSCl), TERT-BUTYL DIMETHYL CHLORO SILANE (TBDMSCl), or Tosyl chloride (TsCl), Methanesulfonyl chloride (MsCl), trimethyl fluoride sulfonyl chlorine, 4-Nitrobenzenesulfonyl chloride (Nosyl Cl), ortho-nitrophenyl SULPHURYL CHLORIDE (Nps Cl), benzene sulfonyl chloride (C
6h
5sO
2the SULPHURYL CHLORIDE such as Cl), is preferably trimethylchlorosilane, TERT-BUTYL DIMETHYL CHLORO SILANE, Tosyl chloride or Methanesulfonyl chloride.
Non-proton organic solvent used can be nonpolar organic solvent, also can be the organic solvent of polarity, such as toluene, benzene, methylene dichloride, chloroform, 1,2-ethylene dichloride, chlorobenzene, normal hexane, sherwood oil, isopropyl ether, methyl tertiary butyl ether (MTBE), methyl iso-butyl ketone (MIBK) (MIBK), tetrahydrofuran (THF), acetonitrile, DMF etc. one or several, be preferably toluene, methylene dichloride, acetonitrile or DMF.
In above-mentioned synthetic method, the mol ratio of " Tecramine: R*-Cl: glycol ether " is " 1.0: 1.8 ~ 2.5: 2.0 ~ 30.0 ", and preferred mol ratio is " 1.0: 2.0 ~ 2.2: 10.0 ~ 20.0 ".
In above-mentioned synthetic method, the temperature of Tecramine and organic carboxyl acid activator (R*-Cl) reaction can be-20 ℃ ~ 80 ℃, and preferred range is 0 ℃ ~ 40 ℃.The temperature that adds again glycol ether to continue reaction can be 0 ℃ ~ 100 ℃, and preferred range is 20 ℃ ~ 60 ℃.
In above-mentioned synthetic method, when reaction is used non-proton property non-polar organic solvent, as toluene, benzene, methylene dichloride, chloroform, 1, when 2-ethylene dichloride, chlorobenzene, normal hexane, sherwood oil, isopropyl ether, methyl tertiary butyl ether (MTBE), methyl iso-butyl ketone (MIBK) (MIBK) etc., reaction product aftertreatment can directly be added in weak acid water and be hydrolyzed, regulate again pH to the rear directly stratification of alkalescence, do not need to extract with water-insoluble organic solvent again.When reaction is used non-proton property polar organic solvent, during as tetrahydrofuran (THF), acetonitrile, DMF etc., reaction product aftertreatment can first concentrate, and then adds in weak acid water to be hydrolyzed, then regulates pH to extracting with water-insoluble organic solvent after alkalescence.Water-insoluble organic solvent used can be toluene, benzene, methylene dichloride, chloroform, 1, one or several of 2-ethylene dichloride, chlorobenzene, normal hexane, sherwood oil, isopropyl ether, methyl tertiary butyl ether (MTBE), methyl iso-butyl ketone (MIBK) (MIBK) etc. mix, and are preferably toluene, methylene dichloride or MTBE.
In above-mentioned synthetic method, adding weak acid for being hydrolyzed can be one or several of dilute hydrochloric acid, dilute sulphuric acid, glacial acetic acid aqueous solution or saturated aqueous ammonium chloride etc.Regulating pH alkali used can be sodium hydroxide, potassium hydroxide, calcium hydroxide, sodium carbonate, salt of wormwood, sodium bicarbonate, saleratus etc., is preferably sodium carbonate, salt of wormwood, sodium bicarbonate, saleratus.PH can be adjusted to 7 ~ 14, is preferably 8 ~ 10.
As a particularly preferred embodiment according to the invention, the preparation method of etofenamate provided by the invention comprises the steps:
Tecramine is joined in non-proton organic solvent, be cooled to 5 ~ 10 ℃, add the organic carboxyl acid such as chlorosilane, SULPHURYL CHLORIDE activator (R*-Cl), stirring reaction at this temperature, then adds glycol ether, is warming up to 30 ~ 40 ℃ and continues reaction.If the solvent that reaction is used is toluene or methylene dichloride, reaction finishes the rear dilute hydrochloric acid that directly adds in 30 ~ 40 ℃ of reactions that are hydrolyzed; If the solvent that reaction is used is acetonitrile or DMF, reaction finishes rear first concentrated, then adds dilute hydrochloric acid in 30 ~ 40 ℃ of reactions that are hydrolyzed.After hydrolysis reaction finishes, be cooled to 10 ℃ of left and right, then regulate pH to 9 left and right of reaction system with sodium carbonate, stratification or extract with water-insoluble organic solvent, organic layer can obtain highly purified etofenamate again after several washing, activated carbon decolorizing, filtration, concentrating under reduced pressure.The HPLC condition of measuring according to European Pharmacopoeia (EP 6.0) correlative is analyzed, and we all meet the requirements by the correlative impurity of product.
The preparation method of high purity etofenamate provided by the invention compared to the prior art, do not need column purification, do not need special high vacuum high temperature rectifying device to carry out separation and purification product yet, there is the advantages such as reactions steps is few, productive rate is high, separation and purification is simple, product purity is high, outward appearance is good, there is prospect widely.
Accompanying drawing explanation
Fig. 1: the collection of illustrative plates of etofenamate reference substance under EP6.0 correlative HPLC analysis condition.
Fig. 2: the etofenamate typical sample collection of illustrative plates preparing according to this patent method under EP6.0 correlative HPLC analysis condition.
Specific embodiment
Further explain and describe by the following examples content of the present invention.But the embodiment providing should not be understood to protection domain of the present invention to be construed as limiting.
Embodiment 1: the preparation of etofenamate.
281.5 grams of Tecramine are joined in 1000 grams of toluene, stir and be cooled to 10 ℃, add 217.5 grams of trimethylchlorosilanes (R*=TMS), stirring reaction 1 hour at this temperature.Then add 1062 grams of glycol ethers, be warming up to 30 ℃ and continue reaction 5 hours.Reaction process is followed the tracks of and is detected to Tecramine raw material and intermediate disappearance with TLC or HPLC, then adds 800 grams, 10% dilute hydrochloric acid to stir and be hydrolyzed 1 hour in 30 ~ 40 ℃, and HPLC follows the tracks of to detect and is extremely all converted into etofenamate.Reaction solution is cooled to 10 ℃ of left and right, then regulates pH to 9 left and right of reaction system with sodium carbonate, stratification.1000 grams of points of five washings of pure water for organic layer, then add proper amount of active carbon decolouring, filter to obtain colorless clear liquid.50 ℃ are evaporated to solvent-free steaming, and then strengthen vacuum tightness to be less than-0.095MPa, are controlled at 70 ~ 80 ℃ and continue to take out 1 hour, and remaining solvent is all removed, and obtain 329.5 grams of etofenamate finished products, and purity is greater than 99%, yield 89.2%.ESI-MS m/z 370.1 [M+H]
+, HPLC relative retention time is consistent with reference substance, and color is not deeper than GY1, and correlative impurity all meets European Pharmacopoeia requirement.
Embodiment 2: the preparation of etofenamate.
281.5 grams of Tecramine are joined in 1000 grams of toluene, stir and be cooled to 10 ℃, add 331.6 grams of TERT-BUTYL DIMETHYL CHLORO SILANE (R*=TBDMS), and be controlled at 20 ℃ of continuation stirring reactions 1 hour.Then add 1592 grams of glycol ethers, be warming up to 40 ℃ and continue reaction 5 hours.Reaction process is followed the tracks of and is detected to Tecramine raw material and intermediate disappearance with TLC or HPLC, then adds 900 grams, 10% dilute hydrochloric acid to stir and be hydrolyzed 1 hour in 30 ~ 40 ℃, and HPLC follows the tracks of to detect and is extremely all converted into etofenamate.Reaction solution is cooled to 10 ℃ of left and right, then regulates the pH=9 left and right of reaction system with salt of wormwood, stratification.Organic layer with 1200 grams of points of six washings of pure water, then adds proper amount of active carbon decolouring, the clear liquid that filters colourlessly again.50 ℃ are evaporated to solvent-free steaming, and then strengthen vacuum tightness to be less than-0.095MPa, being controlled at 70 ~ 80 ℃ continues to take out 1 hour, remaining solvent is all removed, obtain 321.8 grams of etofenamate finished products, purity is greater than 99%, yield 87.1%, color is not deeper than GY1, and correlative impurity all meets European Pharmacopoeia requirement.
Embodiment 3: the preparation of etofenamate.
281.5 grams of Tecramine are joined in the DMF of 1200 grams and stir and be cooled to 10 ℃, add 381.5 grams of Tosyl chlorides (R*=Ts), and be controlled at 20 ℃ and continue stirring reactions 1 hour.Then add 1062 grams of glycol ethers, be warming up to 40 ℃ and continue reaction 5 hours.Reaction is followed the tracks of and is detected to Tecramine raw material and intermediate disappearance with TLC or HPLC, in the extremely about 1700ml of 50 ~ 60 ℃ of concentrating under reduced pressure reaction solutions left and right, then add 800 grams, 10% dilute hydrochloric acid, stir and be hydrolyzed 2 hours in 30 ~ 40 ℃, HPLC follows the tracks of and detects to being all converted into etofenamate.Reaction solution is cooled to 10 ℃ of left and right, then regulates pH to 10 left and right of reaction system with sodium carbonate, and reaction solution extracts at twice with 1000 grams of toluene.Merge organic layer, with 1000 grams of points of five washings of pure water, then add proper amount of active carbon decolouring, filter to obtain colorless clear liquid.50 ℃ are evaporated to solvent-free steaming, and then strengthen vacuum tightness to be less than-0.095MPa, be controlled at 70 ~ 80 ℃ and continue distillation 1 hour, remaining solvent is all removed, obtain 305.1 grams of etofenamate finished products, purity is greater than 99%, yield 82.6%, color is not deeper than GY1, and correlative impurity all meets European Pharmacopoeia requirement.
Embodiment 4: the preparation of etofenamate.
281.5 grams of Tecramine are joined in 1000 grams of acetonitriles, stir and be cooled to 5 ℃, add 252.1 grams of Methanesulfonyl chlorides (R*=Ms), and be controlled at 5 ℃ of left and right stirring reactions 1 hour.Then add 1592 grams of glycol ethers, be warming up to 30 ℃ and continue reaction 5 hours.Reaction is followed the tracks of and is detected to Tecramine raw material and intermediate disappearance with TLC or HPLC, in the extremely about 2300ml of 40 ℃ of concentrating under reduced pressure reaction solutions left and right, then add 900 grams, 10% dilute hydrochloric acid, stir and be hydrolyzed 2 hours in 30 ~ 40 ℃, HPLC follows the tracks of and detects to being all converted into etofenamate.Reaction solution is cooled to 10 ℃ of left and right, then regulates pH to 10 left and right of reaction system with sodium carbonate, and reaction solution extracts for three times with 1200 grams points of MTBE.Merge organic layer, then with 1200 grams of points of six washings of pure water, then add proper amount of active carbon decolouring, filter to obtain colorless clear liquid.40 ℃ are evaporated to solvent-free steaming, and then strengthen vacuum tightness to be less than-0.095MPa, be controlled at 60 ~ 70 ℃ and continue distillation 1 hour, remaining solvent is all removed, obtain 312.4 grams of etofenamate finished products, purity is greater than 99%, yield 84.6%, color is not deeper than GY1, and correlative impurity all meets European Pharmacopoeia requirement.
Claims (14)
1. a preparation method for high purity etofenamate (formula (I)),

Described method comprises Tecramine under organic carboxyl acid activator R*-Cl exists, react in non-proton organic solvent with glycol ether, reactant passes through follow-up lock out operation after being hydrolyzed under acidic conditions again, can obtain highly purified etofenamate, and reaction formula is as follows:

Wherein, formula (II) compound is Tecramine, and formula (III) compound is glycol ether, and R*-Cl compound is chlorosilane or SULPHURYL CHLORIDE.
2. preparation method according to claim 1, it comprises the steps:
Tecramine is joined in non-proton organic solvent, add organic carboxyl acid activator R*-Cl, stir, then add glycol ether to continue reaction;
After reaction finishes, according to the polarity difference of reaction solvent, reaction solution is directly added in weak acid water and is hydrolyzed, or add in weak acid water and be hydrolyzed after concentrated, generate etofenamate structure, then regulate pH to alkalescence, stratification or with water-insoluble organic solvent extract, organic layer can obtain highly purified etofenamate again after several washing, activated carbon decolorizing, filtration, concentrating under reduced pressure.
3. preparation method according to claim 2, is characterized in that organic carboxyl acid activator R*-Cl is trimethylchlorosilane (TMSCl), TERT-BUTYL DIMETHYL CHLORO SILANE (TBDMSCl); Or Tosyl chloride (TsCl), Methanesulfonyl chloride (MsCl), trimethyl fluoride sulfonyl chlorine, 4-Nitrobenzenesulfonyl chloride (Nosyl Cl), ortho-nitrophenyl SULPHURYL CHLORIDE (Nps Cl), benzene sulfonyl chloride (C
6h
5sO
2cl).
4. preparation method according to claim 3, is characterized in that organic carboxyl acid activator R*-Cl is trimethylchlorosilane, TERT-BUTYL DIMETHYL CHLORO SILANE, Tosyl chloride or Methanesulfonyl chloride.
5. preparation method according to claim 2, it is characterized in that non-proton organic solvent used is toluene, benzene, methylene dichloride, chloroform, 1, one or several mixing of 2-ethylene dichloride, chlorobenzene, normal hexane, sherwood oil, isopropyl ether, methyl tertiary butyl ether (MTBE), methyl iso-butyl ketone (MIBK) (MIBK), tetrahydrofuran (THF), acetonitrile, DMF; The mol ratio of " Tecramine: R*-Cl: glycol ether " is " 1.0: 1.8~2.5: 2.0~30.0 ".
6. preparation method according to claim 5, is characterized in that non-proton organic solvent used is one or several the mixing of toluene, methylene dichloride, acetonitrile or DMF; The mol ratio of " Tecramine: R*-Cl: glycol ether " is " 1.0: 2.0~2.2: 10.0~20.0 ".
7. preparation method according to claim 2, is characterized in that the temperature of Tecramine and organic carboxyl acid activator R*-Cl reaction is-20 ℃~80 ℃; The temperature that adds again glycol ether to continue reaction is 0 ℃~100 ℃.
8. preparation method according to claim 7, is characterized in that the temperature of Tecramine and organic carboxyl acid activator R*-Cl reaction is 0 ℃~40 ℃; The temperature that adds again glycol ether to continue reaction is 20 ℃~60 ℃.
9. preparation method according to claim 2, it is characterized in that reaction is used non-proton property non-polar organic solvent, be selected from toluene, benzene, methylene dichloride, chloroform, 1,2-ethylene dichloride, chlorobenzene, normal hexane, sherwood oil, isopropyl ether, methyl tertiary butyl ether (MTBE), methyl iso-butyl ketone (MIBK) (MIBK), post-reaction treatment directly adds in weak acid water and is hydrolyzed, regulate pH to alkalescence, directly stratification, does not need to extract with water-insoluble organic solvent again.
10. preparation method according to claim 2, is characterized in that reaction is used non-proton property polar organic solvent, is selected from tetrahydrofuran (THF), acetonitrile, DMF, post-reaction treatment is first concentrated, then add in weak acid water and be hydrolyzed, regulate pH to alkalescence, then extract with water-insoluble organic solvent.
11. preparation methods according to claim 10, the water-insoluble organic solvent that it is characterized in that extracting use is toluene, benzene, methylene dichloride, chloroform, 1, one or several of 2-ethylene dichloride, chlorobenzene, normal hexane, sherwood oil, isopropyl ether, methyl tertiary butyl ether (MTBE), methyl iso-butyl ketone (MIBK) (MIBK).
12. preparation methods according to claim 1, is characterized in that acidic conditions used is one or several of dilute hydrochloric acid or dilute sulphuric acid.
13. preparation methods according to claim 2, is characterized in that the weak acid adding is one or several of glacial acetic acid aqueous solution or saturated aqueous ammonium chloride.
14. preparation methods according to claim 2, it is characterized in that after the acidified generation etofenamate of reaction solution structure, regulate pH to alkalescence, alkali used is one or several of sodium hydroxide, potassium hydroxide, calcium hydroxide, sodium carbonate, salt of wormwood, sodium bicarbonate, saleratus.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110437413.2A CN102531940B (en) | 2011-12-23 | 2011-12-23 | Preparation method for high purity etofenamate |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110437413.2A CN102531940B (en) | 2011-12-23 | 2011-12-23 | Preparation method for high purity etofenamate |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102531940A CN102531940A (en) | 2012-07-04 |
| CN102531940B true CN102531940B (en) | 2014-05-28 |
Family
ID=46340124
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201110437413.2A Active CN102531940B (en) | 2011-12-23 | 2011-12-23 | Preparation method for high purity etofenamate |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102531940B (en) |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1939112C3 (en) * | 1969-08-01 | 1975-03-06 | Troponwerke Dinklage & Co, 5000 Koeln | Esters of N- (3-trifluoromethylphenyl) anthranilic acid, process for their preparation and pharmacologically active preparations thereof |
| ES2003782A6 (en) * | 1987-02-06 | 1988-11-16 | Union Quimico Farma | Fluophenamic acid deriv. prepn. |
-
2011
- 2011-12-23 CN CN201110437413.2A patent/CN102531940B/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN102531940A (en) | 2012-07-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114105859B (en) | Synthetic method of 6, 6-dimethyl-3-azabicyclo [3.1.0] hexane | |
| CN105330586B (en) | A kind of preparation method of Apremilast | |
| CN104387320B (en) | A kind of preparation method of high-purity milrinone | |
| US20140370555A1 (en) | Method for preparing (r)-praziquantel | |
| CN106146379A (en) | A kind of synthetic method of oxiracetam | |
| CN106279047A (en) | A kind of preparation method of prostacyclin receptor agonist | |
| WO2015055127A1 (en) | (r)-praziquantel preparation method | |
| CN102531940B (en) | Preparation method for high purity etofenamate | |
| CN108947919A (en) | A kind of novel processing step and its key intermediate of gout suppressant Lesinurad | |
| CN103012350A (en) | Synthetic method of benzopyran chiral compound | |
| CN108239089A (en) | A kind of synthetic method of AVM hereinafter Batan sodium | |
| CN101948455A (en) | Preparation method of 2-n-butyl-3-(4-hydroxybenzoyl)-5-nitrobenzofuran | |
| CN105294556A (en) | Method for preparing montelukast acid | |
| CN114702425A (en) | Preparation method of (S) -2-amino- (S) -3- [ pyrrolidone-2' ] alanine derivative and intermediate | |
| EP3141544B1 (en) | Preparation method for chiral intermediate for use in statins | |
| CN103865976B (en) | A kind of biological chemistry splits the method for 8-benzyl-7,9-dioxo-2,8-diazabicyclo [4.3.0] nonane | |
| CN105541786B (en) | A kind of Montelukast side-chain intermediate and preparation method thereof | |
| CN108129414B (en) | Preparation method of mosapride citrate intermediate | |
| CN104926704A (en) | Nitrogen heterocyclic propane compound and preparation method thereof | |
| CN103965104A (en) | Preparation methods of tyrosine kinase inhibitor and intermediates thereof | |
| US9290428B2 (en) | Process for the preparation of derivatives of 1-(2-fluoro[1,1'-biphenyl]-4-yl)-cyclopropanecarboxylic acid | |
| CN110818679B (en) | Synthetic method of 4-bromobenzo [ b ] thiophene | |
| CN110437063B (en) | Preparation method of ambrisentan key intermediate | |
| CN102690211A (en) | Method for preparing tolvaptan intermediate | |
| CN103102260B (en) | Novel technology for synthesizing S-mandelic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |