WO2015083748A1 - フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物 - Google Patents
フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物 Download PDFInfo
- Publication number
- WO2015083748A1 WO2015083748A1 PCT/JP2014/082023 JP2014082023W WO2015083748A1 WO 2015083748 A1 WO2015083748 A1 WO 2015083748A1 JP 2014082023 W JP2014082023 W JP 2014082023W WO 2015083748 A1 WO2015083748 A1 WO 2015083748A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- epoxy resin
- resin composition
- present
- curable resin
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/02—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/02—Polycondensates containing more than one epoxy group per molecule
- C08G59/04—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof
- C08G59/06—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof of polyhydric phenols
- C08G59/08—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof of polyhydric phenols from phenol-aldehyde condensates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/62—Alcohols or phenols
- C08G59/621—Phenols
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G8/00—Condensation polymers of aldehydes or ketones with phenols only
- C08G8/04—Condensation polymers of aldehydes or ketones with phenols only of aldehydes
- C08G8/08—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ
- C08G8/12—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ with monohydric phenols having only one hydrocarbon substituent ortho on para to the OH group, e.g. p-tert.-butyl phenol
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G8/00—Condensation polymers of aldehydes or ketones with phenols only
- C08G8/04—Condensation polymers of aldehydes or ketones with phenols only of aldehydes
- C08G8/08—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ
- C08G8/20—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ with polyhydric phenols
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G8/00—Condensation polymers of aldehydes or ketones with phenols only
- C08G8/04—Condensation polymers of aldehydes or ketones with phenols only of aldehydes
- C08G8/08—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ
- C08G8/24—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ with mixtures of two or more phenols which are not covered by only one of the groups C08G8/10 - C08G8/20
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/34—Monomer units or repeat units incorporating structural elements in the main chain incorporating partially-aromatic structural elements in the main chain
- C08G2261/342—Monomer units or repeat units incorporating structural elements in the main chain incorporating partially-aromatic structural elements in the main chain containing only carbon atoms
- C08G2261/3424—Monomer units or repeat units incorporating structural elements in the main chain incorporating partially-aromatic structural elements in the main chain containing only carbon atoms non-conjugated, e.g. paracyclophanes or xylenes
Definitions
- the present invention relates to a phenol resin, an epoxy resin, an epoxy resin composition, and a cured product thereof that give a cured product having excellent heat resistance.
- the present invention relates to a phenol resin, an epoxy resin, an epoxy resin composition, and a cured product thereof suitable for electrical and electronic material applications requiring heat resistance.
- Curable resin compositions are widely used in the fields of electrical and electronic parts, structural materials, adhesives, paints, etc. due to their workability and excellent electrical properties, heat resistance, adhesion, moisture resistance (water resistance), etc. It is used.
- Non-Patent Document 1 Required characteristics such as heat resistance and high fluidity are required.
- the demand for improved heat resistance has become more severe, and there is a need for a resin with a high Tg and low linear expansion coefficient and of course capable of handling solder reflow. It is done.
- An object of the present invention is to provide a novel highly heat-resistant phenol resin, epoxy resin, epoxy resin composition, and cured product thereof, which are used for electrical / electronic materials such as semiconductor encapsulants.
- each R is independently a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, a hydroxyl group, a methoxy group, an ethoxy group, a nitrile group, an amino group, or a substituted or unsubstituted group.
- the phenol resin according to [1] which is obtained by a reaction between binol and a phenyl compound represented by the following general formula (2).
- each X independently represents a hydroxyl group, an alkoxy group having 1 to 6 carbon atoms or a halogen atom
- each R independently represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or 1 to 6 carbon atoms.
- 6 represents an alkoxy group, a hydroxyl group, a methoxy group, an ethoxy group, a nitrile group, an amino group, or a substituted or unsubstituted phenyl group or naphthyl group, and t represents an integer of 0 to 3.
- [4] An epoxy resin composition comprising at least one phenol resin as described in [1] or [2] above and an epoxy resin.
- An epoxy resin composition comprising the epoxy resin according to the above item [3], a curing agent, and optionally a curing accelerator.
- the curable resin composition using the phenol resin or epoxy resin of the present invention has high heat resistance, insulating materials for electrical and electronic parts, laminated boards (printed wiring boards, build-up boards, etc.), CFRP, etc. It is useful for various composite materials, adhesives, paints and the like. In particular, it is extremely useful for a semiconductor sealing material for protecting a semiconductor element.
- FIG. 2 is a GPC analysis chart of the phenol resin obtained in Example 1.
- the phenol resin (A) of the present invention is a phenol resin represented by the following general formula (1).
- each R is independently a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, a hydroxyl group, a methoxy group, an ethoxy group, a nitrile group, an amino group, or a substituted or unsubstituted group.
- R is preferably a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, and is more preferably water-absorbing, and a substituted or unsubstituted phenyl group or naphthyl group is more excellent in heat resistance.
- t is an integer of 0 to 3, preferably an integer of 0 to 2.
- the softening point is preferably 45 to 190 ° C, more preferably 50 to 180 ° C, and particularly preferably 50 to 150 ° C. If the softening point is too low, the handling property tends to be poor, and if the softening point exceeds 190 ° C, it tends to be difficult to take out as a resin, and the viscosity tends to be very high, and the handling property is poor. There is a fear.
- the hydroxyl equivalent is 100 to 300 g / eq. And more preferably 100 to 250 g / eq. It is.
- the number of repetitions n is preferably 1-15, more preferably 1-10.
- the arrangement of the R group and the hydroxyl group substituted on the benzene skeleton is not particularly specified, but is preferably para-oriented from the balance of heat resistance and mechanical properties.
- the phenolic resin of the present invention may be used alone, but usually the raw material, binol, was obtained by the reaction at a GPC area ratio (area ratio of each peak detected in the chromatogram obtained by GPC analysis).
- the content is preferably 1 to 95 area%, more preferably 5 to 80 area%, and particularly preferably 20 to 70 area% with respect to the phenol resin. If the content of the binol compound is less than 1% by area, the softening point of the resulting resin may become very high, and it tends to be difficult to take out, handling during composition, and composition There is a tendency to greatly influence the moldability after the molding, and there is a possibility that molding becomes difficult.
- binol content of less than 1 area% it is preferable to coexist other phenol resin instead, and specific examples thereof are exemplified as a curing agent that can be used later.
- binol exceeds 95 area%, since the high heat resistance which is the characteristics of this invention may be impaired, it is unpreferable.
- the phenol resin of the present invention can be used as it is as a thermoplastic (or its raw material), or as an epoxy resin raw material and its curing agent as described later.
- the binol used in the present invention is a compound having the structure of the following formula (A).
- Binol has stereoisomers, but either chiral or racemic forms may be used.
- the purity is preferably 90 area% or more, more preferably 93 area% or more, and particularly preferably 98 area% or more by gel permeation chromatography (GPC).
- Impurities include those having a quinone structure and raw material naphthol compounds, and the content of these is preferably 2 area% or less, and more preferably 1 area% or less. Purity can be controlled by crystallization or washing. When the said purity is low, the characteristic of the phenol resin of this reaction may fall and it is unpreferable. Further, the loss on drying is preferably 0.2 area% or less, more preferably 0.1 area% or less.
- the melting point is preferably 200 to 220 ° C, more preferably 212 to 219 ° C. These are commercially available from Aldrich, SR-CHEM.
- the method for synthesizing the phenol resin of the present invention is not particularly limited.
- the phenol resin can be synthesized by a reaction between binol and a phenyl compound represented by the general formula (2).
- each X independently represents a hydroxyl group, an alkoxy group having 1 to 6 carbon atoms or a halogen atom
- each R independently represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or 1 to 6 carbon atoms.
- 6 represents an alkoxy group, a hydroxyl group, a methoxy group, an ethoxy group, a nitrile group, an amino group, or a substituted or unsubstituted phenyl group or naphthyl group, and t represents an integer of 0 to 3.
- a known compound can be used as the phenyl compound represented by the general formula (2), and is not particularly limited.
- bis (hydroxyalkyl) alkylphenol more specifically, bis (hydroxymethyl) cresol, bis ( Hydroxyethyl) cresol, bis (hydroxypropyl) cresol, bis (hydroxybutyl) cresol, etc.
- bis (haloalkyl) alkylphenol halogen includes chlorine, bromine, etc.
- bisalkoxymethylalkylphenol More specific examples include bismethoxymethyl cresol, bisethoxymethyl cresol, bispropoxymethyl cresol, bisbutoxymethyl cresol, and the like.
- bis (hydroxymethyl) cresol, bis (chloromethyl) cresol, and bismethoxymethylcresol are particularly preferable.
- the phenolic resin of the present invention can be obtained by heating binol and a phenyl compound represented by the general formula (2) in the presence of an acid catalyst.
- the order of mixing the binol and the phenyl compound represented by the formula (2) is not particularly specified, and it may be added in portions during mixing.
- the phenol resin of the present invention can be obtained by adding a catalyst to a mixed solution of a binol, a phenyl compound represented by the general formula (2) and a solvent, if necessary, and heating. Further, the phenyl compound represented by the general formula (2) may be gradually added to a solution in which binol is dissolved. Usually, the reaction time is 5 to 150 hours, and the reaction temperature is 40 to 150 ° C.
- the phenolic resin thus obtained can be used without purification depending on the use, but usually, after completion of the reaction, the reaction mixture is subjected to treatment such as neutralization as necessary before crystallization or heating under reduced pressure. It is purified by removing the solvents below and used for various purposes.
- the reaction molar ratio of the binol and the phenyl compound represented by the general formula (2) is preferably 1.2: 1 to 50: 1, more preferably 1.5: 1 to 30: 1, particularly preferably 2: 1 to 20: 1.
- the reaction molar ratio is less than 1.2: 1, that is, when the binol is less than 1.2 with respect to the phenyl compound 1 represented by the general formula (2), the softening point of the resulting phenol resin tends to be too high. There is a risk that it will be difficult to remove.
- the ratio exceeds 50: 1 that is, when the binol exceeds 50 with respect to the phenyl compound 1 represented by the general formula (2), the structure of the formula (1) is relatively small and the heat resistance is poor. There is a fear.
- Solvents that can be used in the synthesis of the phenolic resin of the present invention include, but are not limited to, methanol, ethanol, propanol, isopropanol, toluene, xylene, methyl isobutyl ketone, anone, cyclopentanone, methyl ethyl ketone, and the like. These may be used alone or in combination of two or more.
- the amount of the solvent used is usually in the range of 5 to 500 parts by weight, preferably 10 to 400 parts by weight with respect to 100 parts by weight of binol.
- the catalyst it is basically preferable to use an acidic catalyst.
- the phenyl compound represented by the formula (2) is bis (haloalkyl) cresol, the reaction can proceed smoothly without the addition of a catalyst, and the reaction without a catalyst is performed from the viewpoint of ease of subsequent purification. preferable.
- the acidic catalyst When using the catalyst, specific examples of the acidic catalyst include mineral acids such as hydrochloric acid, sulfuric acid and phosphoric acid; organic acids such as oxalic acid, toluenesulfonic acid and acetic acid; heteropolyacids such as tungstic acid, activated clay, inorganic acids, Examples include stannic chloride, zinc chloride, ferric chloride, and other acidic catalysts usually used for the production of novolak resins such as organic and inorganic acid salts showing acidity. These catalysts are not limited to those described above, and may be used alone or in combination of two or more.
- the amount of the catalyst used is usually in the range of 0.005 to 2.0 times mol, preferably 0.01 to 1.1 times mol of the binol, or usually 0.1 to 10 g, preferably 0 to 100 g of the binol. .3-7g.
- the amount of the catalyst is small, the progress of the reaction tends to be slow. In addition, problems such as the need for a reaction at a high temperature and the reaction not proceeding to the end may occur, which is not preferable.
- the amount of the catalyst is too large, it is not preferable because much labor is required in post-treatment such as neutralization and purification.
- purifies by reaction it is preferable to make it exhaust from the inside of a system by sending inactive gas, such as drawing pressure or nitrogen.
- the epoxy resin of the present invention can be obtained by reacting the phenol resin of the present invention with epihalohydrin. Although it does not specifically limit as a method of reaction, An example of the synthesis
- the specific structural formula of the epoxy resin of the present invention is represented by the following formula (3).
- each R is independently a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, a hydroxyl group, a methoxy group, an ethoxy group, a nitrile group, an amino group, or a substituted or unsubstituted group.
- an epoxy equivalent is 150 to 450 g / eq. And more preferably 200 to 300 g / eq. It is. It becomes easy to obtain the epoxy resin excellent in the heat resistance of a hardened
- Epoxy equivalent is 450 g / eq. In the case of exceeding the range, the epoxy ring is not completely closed, and many compounds having no functional group may be contained. In addition, many of these compounds that could not be ring-closed often contain chlorine, and as an electronic material application, there is concern about the release of chlorine ions under high-temperature and high-humidity conditions and the resulting corrosion of wiring. Absent.
- the epoxy resin of the present invention may be used alone, but usually the glycidylated product of binol, which is a raw material, is 1 to 95% by area, more preferably 5 to 80% with respect to the epoxy resin obtained by the reaction in the GPC area ratio. It is contained in an area%, particularly preferably 20 to 70 area%. When the content of binol glycidyl compound exceeds 95 area%, the high heat resistance, which is a feature of the present invention, may be impaired, which is not preferable.
- the total chlorine remaining in the epoxy resin is preferably 5000 ppm or less, more preferably 3000 ppm or less, and particularly preferably 2000 ppm or less. The adverse effects of chlorine are the same as described above.
- a chlorine ion and a sodium ion 5 ppm or less is respectively preferable, More preferably, it is 3 ppm or less.
- the adverse effects of chlorine ions are described earlier, and it goes without saying that cations such as sodium ions are also a very important factor, especially in power device applications, and contribute to the failure mode when high voltage is applied.
- the arrangement of the R group and glycidyl group substituted on the benzene skeleton is not particularly specified, but is preferably para-oriented from the balance of heat resistance and mechanical properties.
- the epoxy resin of the present invention has a resinous form having a softening point.
- the softening point is preferably 55 to 130 ° C, more preferably 60 to 120 ° C. If the softening point is too low, blocking during storage may be a problem, and problems such as handling at low temperatures may occur. On the other hand, if the softening point is too high, problems such as poor handling may occur during kneading with other resins.
- the melt viscosity is preferably 2 Pa ⁇ s (ICI melt viscosity 150 ° C. cone plate method) or less.
- the epihalohydrin is preferably epichlorohydrin which is easily available industrially.
- the amount of epihalohydrin used is usually 3.0 to 15 mol, preferably 3.0 to 10 mol, more preferably 3.5 to 8.5 mol, particularly preferably 1 mol per mol of the hydroxyl group of the epoxy resin of the present invention. 5.5 to 8.5 moles. If the amount is less than 3.0 mol, the epoxy equivalent may be increased, and the workability of the resulting epoxy resin is likely to deteriorate. Not preferable.
- alkali metal hydroxide examples include sodium hydroxide, potassium hydroxide and the like, and a solid substance may be used, or an aqueous solution thereof may be used. From the viewpoint of moisture, solubility, and handling, it is preferable to use a solid material molded into a flake shape.
- the amount of the alkali metal hydroxide used is usually 0.90 to 1.5 mol, preferably 0.95 to 1.25 mol, more preferably 0.99, based on 1 mol of the hydroxyl group of the phenol resin of the present invention. ⁇ 1.15 moles.
- quaternary ammonium salt such as tetramethylammonium chloride, tetramethylammonium bromide or trimethylbenzylammonium chloride may be added as a catalyst.
- the amount of the quaternary ammonium salt used is usually 0.1 to 15 g, preferably 0.2 to 10 g, per 1 mol of the hydroxyl group of the phenol resin of the present invention.
- a nonpolar proton solvent such as dimethyl sulfoxide, dioxane, dimethylimidazolidinone
- an alcohol having 1 to 5 carbon atoms examples include alcohols such as methanol, ethanol and isopropyl alcohol.
- the amount of the nonpolar protic solvent or alcohol having 1 to 5 carbon atoms is usually 2 to 50% by weight, preferably 4 to 25% by weight, based on the amount of epihalohydrin used.
- epoxidation may be performed while controlling the moisture in the system by a technique such as azeotropic dehydration.
- the obtained epoxy resin tends to deteriorate the electrical reliability, which is not preferred, and it is preferable to synthesize by controlling the moisture to 5% or less.
- an epoxy resin is obtained using a nonpolar proton solvent, an epoxy resin that is more excellent in electrical reliability can be obtained, and therefore a nonpolar proton solvent can be suitably used.
- the reaction temperature is usually 30 to 90 ° C, preferably 35 to 80 ° C. In particular, in the present invention, 60 ° C. or higher is preferable for higher-purity epoxidation, and reaction under conditions close to reflux conditions is particularly preferable.
- the reaction time is usually 0.5 to 10 hours, preferably 1 to 8 hours, particularly preferably 1 to 3 hours. If the reaction time is short, the reaction may not proceed, and if the reaction time is long, a by-product may be formed. After the reaction product of these epoxidation reactions is washed with water or without washing with water, the epihalohydrin, the solvent and the like are removed under heating and reduced pressure.
- the recovered epoxy resin is a solvent of a ketone compound having 4 to 7 carbon atoms (for example, methyl isobutyl ketone, methyl ethyl ketone, cyclopentanone, cyclohexanone, etc.). It can also be dissolved, and an aqueous solution of an alkali metal hydroxide such as sodium hydroxide or potassium hydroxide can be added to react to ensure ring closure.
- a ketone compound having 4 to 7 carbon atoms for example, methyl isobutyl ketone, methyl ethyl ketone, cyclopentanone, cyclohexanone, etc.
- the amount of the alkali metal hydroxide used is usually 0.01 to 0.3 mol, preferably 0.05 to 0.2 mol, based on 1 mol of the hydroxyl group of the phenol resin of the present invention used for epoxidation.
- the reaction temperature is usually 50 to 120 ° C., and the reaction time is usually 0.5 to 2 hours.
- the reaction with epihalohydrin it is preferably substituted with an inert gas such as nitrogen from the beginning of the reaction, and the oxygen concentration in the cavity is preferably 10% or less. Residual oxygen may affect the coloration.
- an inert gas such as nitrogen is blown in the atmosphere (in the air or in the liquid) before the phenol resin of the present invention is charged, or after being evacuated under reduced pressure, the inert gas is replaced. If there is no substitution with an inert gas, the resulting resin may be colored.
- the inert gas is blown, the amount varies depending on the volume of the kettle, but it is preferred that the inert gas be blown in an amount that can replace 1 to 3 times the kettle volume in 0.5 to 10 hours. .
- the produced salt is removed by filtration, washing with water, etc., and the solvent is distilled off under heating and reduced pressure to obtain the epoxy resin of the present invention.
- curable resin composition the epoxy resin composition of the present invention (hereinafter also referred to as “curable resin composition”) will be described.
- the phenol resin of the present invention a curing agent or a polymerization catalyst is used.
- the curable resin composition of this invention can classify
- the curable resin composition A necessarily contains at least one of the epoxy resin of the present invention and the phenol resin of the present invention. That is, a composition comprising an epoxy resin-curing agent as an essential component. Moreover, a hardening accelerator is contained as needed.
- the curable resin composition B is a composition containing the epoxy resin of the present invention and a polymerization catalyst as essential components.
- the amount of the curing agent (which may contain the phenol resin of the present invention) is preferably 0.7 to 1.2 equivalents relative to 1 equivalent of the epoxy groups of all epoxy resins.
- the polymerization catalyst contained in the curable resin composition B of the present invention can be used without limitation as long as it is a catalyst that initiates polymerization by heat or light. Specifically, a curing accelerator or an acidic curing catalyst is preferably used. Can be used.
- curing accelerators that can be used include imidazoles such as 2-methylimidazole, 2-ethylimidazole, 2-ethyl-4-methylimidazole, 2- (dimethylaminomethyl) phenol, 1,8-diaza.
- -Tertiary amines such as bicyclo [5.4.0] undecene-7
- phosphines such as triphenylphosphine, tetrabutylammonium salt, triisopropylmethylammonium salt, trimethyldecanylammonium salt, cetyltrimethylammonium salt, etc.
- quaternary ammonium salts triphenylbenzylphosphonium salts, triphenylethylphosphonium salts, tetrabutylphosphonium salts, and the like.
- the counter ion of the quaternary salt is not particularly specified as halogen, organic acid ion, hydroxide ion, etc., but organic acid ion and hydroxide ion are particularly preferable.
- metal compounds such as tin octylate, etc. Can be mentioned.
- 0.01 to 5.0 parts by weight is used as necessary with respect to 100 parts by weight of the epoxy resin.
- a cationic polymerization initiator is preferable, and a light or thermal cationic polymerization initiator is particularly preferable.
- a cationic polymerization initiator that is activated by active energy rays and / or a cationic polymerization initiator that is activated by heat it can be used as the curable resin composition B described later.
- Cationic polymerization initiators that initiate cationic polymerization of the curable resin composition B of the present invention by irradiation with active energy rays include diazonium salts, iodonium salts, sulfonium salts, selenium salts, pyridinium salts, ferrocenium salts, phosphonium salts. And iodonium salts and sulfonium salts, more preferably diaryl iodonium salts and dialkylphenacyl sulfonium salts, and diaryl iodonium salts can be preferably used.
- a photocationic polymerization initiator such as an iodonium salt or a sulfonium salt is used in the cationic curable resin composition of the present invention
- a photocationic polymerization initiator such as an iodonium salt or a sulfonium salt
- BF 4 ⁇ , AsF 6 ⁇ , SbF 6 ⁇ , PF 6 ⁇ , and B (C 6 F 5 ) 4 — and the like preferably SbF 6 ⁇ , PF 6 ⁇ , or B (C 6 F 5 ) 4 — , particularly preferably SbF 6 — or B (C 6 F 5 ) 4 —. It is.
- a compound that is activated by heat to initiate cationic polymerization that is, a thermal cationic polymerization initiator, can also be used in the curable resin composition B of the present invention.
- a thermal cationic polymerization initiator can also be used in the curable resin composition B of the present invention.
- onium salts such as quaternary ammonium salts, phosphonium salts and sulfonium salts, combinations of alkoxysilanes and aluminum complexes, and the like.
- Adeka Opton CP-66 and Adeka Opton CP-77 both trade names, manufactured by Asahi Denka Kogyo Co., Ltd.
- Sun-Aid SI-60L, Sun-Aid SI-80L and Sun-Aid SI-100L both trade names
- Sanshin Chemical Industry Co., Ltd. and CI series (Nihon Soda Co., Ltd.).
- the epoxy resin of the present invention may be used in addition to the epoxy resin of the present invention, or the epoxy resin of the present invention can be used alone.
- the proportion of the epoxy resin of the present invention in the total epoxy resin is preferably 30% by weight or more, particularly preferably 40% by weight or more.
- the epoxy resin of the present invention is used as a modifier of the curable resin composition, it is added in a proportion of 1 to 30% by weight.
- curable resin composition A when using the phenol resin of this invention as a hardening
- epoxy resins include novolac type epoxy resins, bisphenol A type epoxy resins, biphenyl type epoxy resins, triphenylmethane type epoxy resins, phenol aralkyl type epoxy resins, and the like.
- bisphenol A bisphenol S, thiodiphenol, fluorene bisphenol, terpene diphenol, 4,4′-biphenol, 2,2′-biphenol, 3,3 ′, 5,5′-tetramethyl- [ 1,1′-biphenyl] -4,4′-diol, hydroquinone, resorcin, naphthalenediol, tris- (4-hydroxyphenyl) methane, 1,1,2,2-tetrakis (4-hydroxyphenyl) ethane, phenol (Phenol, alkyl-substituted phenol, naphthol, alkyl-substituted naphthol, dihydroxybenzene, dihydroxynaphthalene, etc
- Curable resin composition A thermal curing with curing agent
- other curing agents may be used in addition to the phenol resin of the present invention, or the phenol resin of the present invention can be used alone.
- the proportion of the phenolic resin of the present invention in the total epoxy resin is preferably 30% by weight or more, particularly preferably 40% by weight or more.
- the epoxy resin of the present invention is used as a modifier of the curable resin composition, it is added in a proportion of 1 to 30% by weight.
- curing agents other than the phenol resin of this invention can be used independently.
- the curing agent examples include a phenol resin, a phenol compound, an amine compound, an acid anhydride compound, an amide compound, a carboxylic acid compound, and the like.
- Specific examples of the curing agent that can be used include phenol resin, phenol compound; bisphenol A, bisphenol F, bisphenol S, fluorene bisphenol, terpene diphenol, 4,4'-biphenol, 2,2'-biphenol, 3,3 '.
- phenol resins include phenol aralkyl resins (resins having an aromatic alkylene structure), particularly preferably a structure having at least one selected from phenol, naphthol, and cresol, and the alkylene portion serving as the linker is benzene.
- a resin characterized by at least one selected from a structure, a biphenyl structure, and a naphthalene structure (specific examples include zylock, naphthol zylock, phenol biphenylene novolak resin, cresol-biphenylene novolak resin, phenol-naphthalene novolak resin, etc.)
- Amine compounds, amide compounds; nitrogen-containing compounds such as diaminodiphenylmethane, diethylenetriamine, triethylenetetramine, diaminodiphenylsulfone, isophoronediamine, dicyandiamide, linolenic acid and polyamide resins synthesized from ethylenediamine; Acid anhydride compounds, carboxylic acid compounds; phthalic anhydride, trimellitic anhydride, pyromellitic anhydride, maleic anhydride, tetrahydrophthalic anhydride, methyltetrahydrophthalic anhydride, methyl nadic anhydride
- the amount of the curing agent used is preferably 0.7 to 1.2 equivalents relative to 1 equivalent of the epoxy groups of all epoxy resins.
- curing may be incomplete and good cured properties may not be obtained.
- a curing accelerator may be used in combination with the curing agent.
- Specific examples of the curing accelerator include those described above.
- the amount is preferably 0.01 to 5.0 parts by weight with respect to 100 parts by weight of the epoxy resin.
- the curable resin composition A of the present invention may contain a phosphorus-containing compound as a flame retardant imparting component.
- the phosphorus-containing compound may be a reactive type or an additive type.
- Specific examples of phosphorus-containing compounds include trimethyl phosphate, triethyl phosphate, tricresyl phosphate, trixylylenyl phosphate, cresyl diphenyl phosphate, cresyl-2,6-dixylylenyl phosphate, 1,3-phenylenebis ( Phosphoric esters such as dixylylenyl phosphate), 1,4-phenylenebis (dixylylenyl phosphate), 4,4′-biphenyl (dixylylenyl phosphate); 9,10-dihydro-9-oxa Phosphanes such as -10-phosphaphenanthrene-10-oxide, 10 (2,5-dihydroxyphenyl) -10H-9-oxa
- Phosphate esters, phosphanes or phosphorus-containing epoxy compounds are preferable, and 1,3-phenylenebis (dixylylenyl phosphate), 1,4-phenylenebis (dixylylene). Nyl phosphate), 4,4′-biphenyl (dixylylenyl phosphate) or phosphorus-containing epoxy compounds are particularly preferred.
- antioxidant to the curable resin composition A of this invention as needed.
- Antioxidants that can be used include phenol-based, sulfur-based, and phosphorus-based antioxidants. Antioxidants can be used alone or in combination of two or more.
- the amount of the antioxidant used is usually 0.008 to 1 part by weight, preferably 0.01 to 0.5 part by weight, based on 100 parts by weight of the resin component in the curable resin composition of the present invention. It is.
- antioxidants examples include a phenol-based antioxidant, a sulfur-based antioxidant, and a phosphorus-based antioxidant.
- phenolic antioxidants include 2,6-di-t-butyl-p-cresol, butylated hydroxyanisole, 2,6-di-t-butyl-p-ethylphenol, stearyl- ⁇ - (3 , 5-di-t-butyl-4-hydroxyphenyl) propionate, isooctyl-3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate, 2,4-bis- (n-octylthio)- Monophenols such as 6- (4-hydroxy-3,5-di-t-butylanilino) -1,3,5-triazine, 2,4-bis [(octylthio) methyl] -o-cresol; 2'-methylenebis (4-methyl-6-t-butylphenol), 2,2'-methylenebis (4-ethyl
- sulfur antioxidant examples include dilauryl-3,3′-thiodipropionate, dimyristyl-3,3′-thiodipropionate, distearyll-3,3′-thiodipropionate, and the like. .
- phosphorus antioxidants include triphenyl phosphite, diphenylisodecyl phosphite, phenyl diisodecyl phosphite, tris (nonylphenyl) phosphite, diisodecylpentaerythritol phosphite, tris (2,4-di-t- Butylphenyl) phosphite, cyclic neopentanetetraylbis (octadecyl) phosphite, cyclic neopentanetetraylbi (2,4-di-t-butylphenyl) phosphite, cyclic neopentanetetraylbi (2,4 -Phosphites such as -di-t-butyl-4-methylphenyl) phosphite, bis [2-tert-butyl-6-methyl
- antioxidants can be used alone, but two or more kinds may be used in combination.
- a phosphorus-based antioxidant is particularly preferable.
- HALS hindered amine-based light stabilizers
- HALS is not particularly limited, but typical examples include dibutylamine, 1,3,5-triazine, N, N′-bis (2,2,6,6-tetramethyl-4- Polycondensate of piperidyl-1,6-hexamethylenediamine and N- (2,2,6,6-tetramethyl-4-piperidyl) butylamine, dimethyl-1- (2-hydroxyethyl) -4-hydroxy succinate -2,2,6,6-tetramethylpiperidine polycondensate, poly [ ⁇ 6- (1,1,3,3-tetramethylbutyl) amino-1,3,5-triazine-2,4-diyl ⁇ ⁇ (2,2,6,6-tetramethyl-4-piperidyl) imino ⁇ hexamethylene ⁇ (2,2,6,6-tetra
- the curable resin composition A of the present invention can be blended with a binder resin as necessary.
- the binder resin include butyral resins, acetal resins, acrylic resins, epoxy-nylon resins, NBR-phenol resins, epoxy-NBR resins, polyamide resins, polyimide resins, and silicone resins.
- the blending amount of the binder resin is preferably within a range that does not impair the flame retardancy and heat resistance of the cured product, and is usually 0.05 to 50 parts by weight, preferably 0.05 to 20 parts per 100 parts by weight of the resin component. Part by weight is used as needed.
- An inorganic filler can be added to the curable resin composition A of the present invention as necessary.
- inorganic fillers include crystalline silica, fused silica, alumina, zircon, calcium silicate, calcium carbonate, silicon carbide, silicon nitride, boron nitride, zirconia, fosterite, steatite, spinel, titania, talc, and the like.
- the present invention is not limited to these. These may be used alone or in combination of two or more.
- the content of these inorganic fillers is used in an amount of 0 to 95% by weight in the curable resin composition of the present invention.
- the curable resin composition of the present invention includes various agents such as silane coupling agents, mold release agents such as stearic acid, palmitic acid, zinc stearate, and calcium stearate, surfactants, dyes, pigments, and ultraviolet absorbers.
- agents such as silane coupling agents, mold release agents such as stearic acid, palmitic acid, zinc stearate, and calcium stearate, surfactants, dyes, pigments, and ultraviolet absorbers.
- a compounding agent and various thermosetting resins can be added.
- the curable resin composition A of the present invention can be obtained by uniformly mixing each component.
- the curable resin composition A of the present invention can be easily made into a cured product by a method similar to a conventionally known method.
- the epoxy resin of the present invention, a curing agent and, if necessary, a curing accelerator, a phosphorus-containing compound, a binder resin, an inorganic filler, and a compounding agent are sufficient until uniform using an extruder, kneader, roll, etc. as necessary.
- potting the curable resin composition melting it (without melting in the case of a liquid), molding using a casting or transfer molding machine, and further 80 to 200 ° C.
- the cured product of the present invention can be obtained by heating for 2 to 10 hours.
- the curable resin composition A of the present invention is dissolved in a solvent such as toluene, xylene, acetone, methyl ethyl ketone, methyl isobutyl ketone, dimethylformamide, dimethylacetamide, N-methylpyrrolidone as necessary, and the curable resin composition varnish.
- the curable resin composition of the present invention is obtained by hot press-molding a prepreg obtained by impregnating a base material such as glass fiber, carbon fiber, polyester fiber, polyamide fiber, alumina fiber, paper, etc. and drying by heating. It can be set as the hardened
- the solvent is used in an amount usually accounting for 10 to 70% by weight, preferably 15 to 70% by weight in the mixture of the curable resin composition of the present invention and the solvent. Moreover, if it is a liquid composition, the epoxy resin hardened
- the curable resin composition A of the present invention can also be used as a film type composition modifier. Specifically, it can be used to improve the flexibility of the B-stage.
- the curable resin composition A of the present invention is applied onto a release film as the curable resin composition varnish, the solvent is removed under heating, and then B-stage is performed. Thus, it is obtained as a sheet-like adhesive.
- This sheet-like adhesive can be used as an interlayer insulating layer in a multilayer substrate or the like.
- Curable resin composition B (cationic curing with acidic curing catalyst (polymerization catalyst))
- the curable resin composition B of the present invention to be cured using an acidic curing catalyst (polymerization catalyst) contains a photopolymerization initiator or a thermal polymerization initiator as an acidic curing catalyst (polymerization catalyst).
- a photopolymerization initiator or a thermal polymerization initiator as an acidic curing catalyst (polymerization catalyst).
- a cationic polymerization initiator is preferable, and a light or thermal cationic polymerization initiator is particularly preferable. It can be used as the curable resin composition B by blending a cationic polymerization initiator activated by active energy rays and / or a cationic polymerization initiator activated by heat.
- Examples of the cationic polymerization initiator for initiating cationic polymerization of the curable resin composition B of the present invention by irradiation with active energy rays include those described above.
- the active energy rays for curing the curable resin composition B of the present invention X-rays, electron beams, ultraviolet rays, visible light, and the like can be used, preferably ultraviolet rays or visible rays, particularly preferably. Is ultraviolet light.
- the wavelength range is not particularly limited, but is preferably 150 to 400 nm, more preferably 200 to 380 nm.
- ultraviolet rays are used, cationic polymerization can be efficiently started.
- the curable resin composition B of the present invention can be used in combination with a sensitizer in order to further increase the activity of the photocationic polymerization initiator, if necessary.
- a sensitizer that can be used in the present invention, for example, a compound disclosed in Krivelloga Advanced in Polymer Science (Adv. In Polymer Sci., 62, 1 (1984)) can be used. Specific examples include pyrene, perylene, acridine orange, thioxanthone, 2-chlorothioxanthone, and benzoflavine.
- thioxanthones such as benzophenone, 2,4-diethylthioxanthone, 2-isopropylthioxanthone, and 2,4-dichlorothioxanthone.
- Benzoin ethers such as benzoin methyl ether, benzoin ethyl ether, benzoin isopropyl ether, benzyl dimethyl ketals such as 2,2-dimethoxy-1,2-diphenylethane-1-one, 2-hydroxy-2-methyl-1 ⁇ -hydroxyalkylphenones such as phenylpropan-1-one, 1- (4-isopropylphenyl) -2-hydroxy-2-methylpropan-1-one, 1-hydroxycyclohexyl phenyl ketone, ⁇ such as camphorquinone -Dicarbo Nil compounds and the like.
- thioxanthones and ⁇ -hydroxyalkylphenones can be particularly preferably used.
- the blending amount of the cationic photopolymerization initiator in the curable resin composition B of the present invention can be appropriately adjusted according to the type of active energy ray and the irradiation amount.
- the amount is preferably 0.1 to 10 parts by mass, more preferably 0.5 to 5 parts by mass, and still more preferably 1 to 1 part by mass with respect to 100 parts by mass in total of the cation curable resin composition. 3 parts by mass.
- the amount of the cationic polymerization initiator is less than 0.1 parts by mass, the curability may be inferior.
- it is more than 10 parts by mass the amount of components that are truly necessary for the cured product is reduced to reduce the amount of the cured product. This is not preferable because the physical properties may deteriorate or the cured product may become more intensely colored.
- the blending amount when a sensitizer is added to the curable resin composition B of the present invention can be appropriately adjusted according to the type of active energy ray and the irradiation amount.
- the amount is preferably 5 parts by mass or less, more preferably 0.2 to 2 parts with respect to 100 parts by mass in total of the curable resin composition B.
- the blending amount of the sensitizer is more than 5 parts by mass, the components that are truly necessary for the cured product may be reduced to deteriorate the physical properties of the cured product, or the cured product may be intensely colored.
- the active energy ray is ultraviolet light or visible light
- the cation curable resin composition is exposed to air.
- the humidity of the atmosphere is preferably low, preferably 80% humidity. H. 70% R.V. H. More preferably, it is as follows.
- a method of sending dry air in front of the light irradiation device or a method of reducing the humidity by attaching a heating device can be employed.
- a compound that is activated by heat to initiate cationic polymerization that is, a thermal cationic polymerization initiator, can also be used in the curable resin composition B of the present invention.
- a thermal cationic polymerization initiator can also be used in the curable resin composition B of the present invention.
- onium salts such as quaternary ammonium salts, phosphonium salts and sulfonium salts, combinations of alkoxysilanes and aluminum complexes, and the like.
- Adeka Opton CP-66 and Adeka Opton CP-77 both trade names, manufactured by Asahi Denka Kogyo Co., Ltd.
- Sun-Aid SI-60L, Sun-Aid SI-80L and Sun-Aid SI-100L both trade names
- Sanshin Chemical Industry Co., Ltd. and CI series (Nihon Soda Co., Ltd.).
- the blending ratio of the thermal cationic polymerization initiator to the curable resin composition B of the present invention is preferably in the range of 0.01 to 10 parts by mass, more preferably 100 parts by mass with respect to the cationic curable resin composition. Is 0.1 to 5 parts by mass, more preferably 0.5 to 3 parts by mass.
- the blending ratio is less than 0.01 parts by mass, the ring-opening reaction of the ring-opening polymerizable group may not be allowed to proceed sufficiently even if it is activated by the action of heat.
- even if it mixes exceeding 10 mass parts since the effect
- the curable resin composition B of the present invention is added with various compounding agents such as the above inorganic fillers, silane coupling materials, mold release agents, pigments, and various thermosetting resins as necessary. can do.
- the curable resin composition B of the present invention can be obtained by uniformly mixing each component. It is also possible to dissolve in an organic solvent such as polyethylene glycol monoethyl ether, cyclohexanone, or ⁇ -butyrolactone, make it uniform, and then use it after removing the solvent by drying. In this case, the solvent is used in an amount of 10 to 70% by weight, preferably 15 to 70% by weight, in the mixture of the curable resin composition B of the present invention and the solvent.
- the curable resin composition B of the present invention can be cured by irradiating with ultraviolet rays, but the amount of ultraviolet irradiation varies depending on the curable resin composition, and therefore is determined by the respective curing conditions.
- the heating after the light irradiation may be performed in the normal curing temperature range of the curable resin composition B.
- the temperature is preferably from room temperature to 150 ° C. for 30 minutes to 7 days.
- the shape of the cured product obtained by curing these curable resin compositions B can be variously selected depending on the application, it is not particularly limited. For example, it may be a film shape, a sheet shape, a bulk shape, or the like. .
- the molding method varies depending on the applicable part and member. For example, a casting method, a casting method, a screen printing method, a spin coating method, a spray method, a transfer method, a dispenser method, or the like can be applied. Although it is mentioned, it is not limited to these.
- polishing glass, hard stainless steel polishing plate, polycarbonate plate, polyethylene terephthalate plate, polymethyl methacrylate plate, or the like can be applied.
- a polyethylene terephthalate film, a polycarbonate film, a polyvinyl chloride film, a polyethylene film, a polytetrafluoroethylene film, a polypropylene film, a polyimide film, or the like can be applied in order to improve releasability from the mold.
- the photocation curable resin composition B of the present invention dissolved in an organic solvent such as polyethylene glycol monoethyl ether, cyclohexanone, or ⁇ -butyrolactone is first coated with copper.
- the composition of the present invention is applied to a film thickness of 5 to 160 ⁇ m on a substrate such as a ceramic substrate or a glass substrate by a method such as screen printing or spin coating to form a coating film.
- the coating film is preliminarily dried at 60 to 110 ° C., and then irradiated with ultraviolet rays (for example, a low pressure mercury lamp, a high pressure mercury lamp, an ultrahigh pressure mercury lamp, a xenon lamp, a laser beam, etc.) through a negative film having a desired pattern.
- ultraviolet rays for example, a low pressure mercury lamp, a high pressure mercury lamp, an ultrahigh pressure mercury lamp, a xenon lamp, a laser beam, etc.
- post-exposure baking is performed at 70 to 120 ° C.
- the unexposed portion is dissolved and removed (developed) with a solvent such as polyethylene glycol monoethyl ether, and if necessary, sufficient by irradiation with ultraviolet rays and / or heating (for example, at 100 to 200 ° C. for 0.5 to 3 hours). Curing is performed to obtain a cured product. In this way, it is also possible to obtain a printed wiring board.
- the cured product obtained by curing the curable resin composition A and the curable resin composition B of the present invention can be used for various applications.
- Typical applications in which the curable resin composition A or the curable resin composition B is used include, for example, adhesives, paints, coating agents, molding materials (including sheets, films, FRP, etc.), insulating materials (prints) In addition to a sealing agent, an additive to other resins and the like are included.
- the adhesive include civil engineering, architectural, automotive, general office, and medical adhesives, and electronic material adhesives.
- adhesives for electronic materials include interlayer adhesives for multilayer substrates such as build-up substrates, die bonding agents, semiconductor adhesives such as underfills, BGA reinforcing underfills, anisotropic conductive films ( ACF) and an adhesive for mounting such as anisotropic conductive paste (ACP).
- Sealing agents and substrates include capacitor, transistor, diode, light emitting diode, IC, LSI potting, dipping, transfer mold sealing, IC, LSI COB, COF, TAB potting sealing, flip chip, etc.
- Underfill, QFP, BGA, CSP and other IC packages such as sealing (including reinforcing underfill) and package substrates.
- substrate use as which a functionality, such as a network board
- Epoxy equivalent compliant with JIS K 7236 (ISO 3001) ICI melt viscosity: compliant with JIS K 7117-2 (ISO 3219) Softening point: compliant with JIS K 7234 GPC: Column (Shodex (trademark) KF-603, KF-602.5, KF-602, KF-601x2) connection Eluent is tetrahydrofuran Flow rate is 0.5 ml / min. Column temperature is 40 ° C Detection: RI (differential refraction detector)
- Example 1 A flask equipped with a stirrer, a reflux condenser, and a stirrer was purged with nitrogen, while binol (GPC purity> 99%, Aldrich reagent melting point 216-218 ° C.) 215 parts, 2,6-bis (hydroxymethyl) -p -Cresol (Asahi Organic Materials Reagents) 41.5 parts, methyl isobutyl ketone (Pure Chemicals reagent) 384 parts, p-toluenesulfonic acid monohydrate (Tokyo Chemicals reagent) 2.9 parts, methanol 20 parts In addition, while removing methanol and water produced, the reaction was carried out at 70 ° C. for 1 hour, 80 ° C.
- Example 2 In a flask equipped with a stirrer, a reflux condenser, and a stirrer, while purging with nitrogen, 145 parts of the phenol resin of the present invention (P-1 hydroxyl equivalent 147 g / eq.), 638 parts epichlorohydrin (7 molar equivalents) Phenol resin) and 191 parts of methanol were added, dissolved under stirring, and the temperature was raised to 70 to 75 ° C. Next, 44 parts of flaky sodium hydroxide was added in portions over 90 minutes, and the reaction was further carried out at 75 ° C. for 75 minutes.
- Examples 3, 4 and Comparative Examples 1, 2, 3, 4 ⁇ Heat resistance test and flame retardancy test> The epoxy resin and phenol resin obtained above were blended in the proportions (parts by weight) shown in Table 1, and mixed and kneaded uniformly using a mixing roll to obtain a curable resin composition for sealing.
- the curable resin composition was pulverized with a mixer and further tableted with a tablet machine. This tableted curable resin composition was transfer molded (175 ° C. ⁇ 60 seconds), and after demolding, cured under the conditions of 160 ° C. ⁇ 2 hours + 180 ° C. ⁇ 6 hours to obtain a test piece for evaluation.
- cured material was measured in the following ways.
- the epoxy resin of the present invention is superior in heat resistance compared to EP-2 and EP-3 having similar structures, and can provide a cured product having high heat resistance. Recognize.
- the curable resin composition using the phenol resin or epoxy resin of the present invention has a high heat resistance
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Epoxy Resins (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
Abstract
本発明は耐熱性に優れた硬化物を与えるフェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物を提供することを目的とする。本発明のフェノール樹脂は、下記一般式(1)で示される。(式中、Rはそれぞれ独立して、水素原子、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、ヒドロキシル基、メトキシ基、エトキシ基、ニトリル基、アミノ基または置換もしくは無置換のフェニル基もしくはナフチル基を表し、tは0~3の整数を示し、nは繰り返し数を意味し、1~20の数を表す。)
Description
本発明は耐熱性に優れた硬化物を与えるフェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物に関する。
本発明は耐熱性が要求される電気電子材料用途に好適なフェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物に関する。
硬化性樹脂組成物は作業性及びその硬化物の優れた電気特性、耐熱性、接着性、耐湿性(耐水性)等により電気・電子部品、構造用材料、接着剤、塗料等の分野で幅広く用いられている。
しかし近年、電気・電子分野においてはその発展に伴い、樹脂組成物の高純度化をはじめ耐湿性、密着性、誘電特性、フィラー(無機または有機充填剤)を高充填させるための低粘度化、成型サイクルを短くするための反応性のアップ等の諸特性の一層の向上が求められている。又、構造材としては航空宇宙材料、レジャー・スポーツ器具用途などにおいて軽量で機械物性の優れた材料が求められている。特に半導体封止分野、基板(基板自体、もしくはその周辺材料)においては、その半導体の変遷に従い、薄層化、スタック化、システム化、三次元化と複雑になっていき、非常に高いレベルの耐熱性や高流動性といった要求特性が求められる(非特許文献1)。なお、特にプラスチックパッケージの車載用途への拡大に伴い、耐熱性の向上要求がいっそう厳しくなっており、高Tgで低線膨張率の樹脂で、かつ当然ながら半田リフローへの対応ができる樹脂が求められる。
"2008年 STRJ報告 半導体ロードマップ専門委員会 平成20年度報告"、第8章、p1-17、[online]、平成21年3月、JEITA(社)電子情報技術産業協会 半導体技術ロードマップ専門委員会、[平成24年5月30日検索]、インターネット<URL:http://strj-jeita.elisasp.net/strj/nenjihoukoku-2008.cfm>
高機能化で特に要求される特性のひとつとして耐熱性が挙げられる。本発明の目的は半導体封止材などの電気・電子材料用途として利用される高耐熱性の新規のフェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物を提供することにある。
本発明者らは前記したような実状に鑑み、鋭意検討した結果、本発明を完成させるに至った。
すなわち本発明は、下記[1]~[7]を提供する。
[1]下記一般式(1)で示されるフェノール樹脂。
すなわち本発明は、下記[1]~[7]を提供する。
[1]下記一般式(1)で示されるフェノール樹脂。

(式中、Rはそれぞれ独立して、水素原子、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、ヒドロキシル基、メトキシ基、エトキシ基、ニトリル基、アミノ基または置換もしくは無置換のフェニル基もしくはナフチル基を表し、tは0~3の整数を示し、nは繰り返し数を意味し、1~20の数を表す。)
[2]バイノールと下記一般式(2)で示されるフェニル化合物との反応により得られる前項[1]に記載のフェノール樹脂。
[2]バイノールと下記一般式(2)で示されるフェニル化合物との反応により得られる前項[1]に記載のフェノール樹脂。

(式中、Xはそれぞれ独立して、水酸基、炭素数1~6のアルコキシ基またはハロゲン原子を示し、Rはそれぞれ独立して、水素原子、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、ヒドロキシル基、メトキシ基、エトキシ基、ニトリル基、アミノ基または置換もしくは無置換のフェニル基もしくはナフチル基を表し、tは0~3の整数を表す。)
[3]前項[1]または[2]に記載のフェノール樹脂とエピハロヒドリンとの反応により得られるエポキシ樹脂。
[4]前項[1]または[2]に記載のフェノール樹脂を少なくとも1種とエポキシ樹脂を含有するエポキシ樹脂組成物。
[5]前項[3]に記載のエポキシ樹脂と硬化剤及び任意に硬化促進剤を含有するエポキシ樹脂組成物。
[6]前項[3]に記載のエポキシ樹脂と重合触媒とを含有するエポキシ樹脂組成物。
[7]前項[4]~[6]のいずれか一項に記載のエポキシ樹脂組成物を硬化して得られる硬化物。
[3]前項[1]または[2]に記載のフェノール樹脂とエピハロヒドリンとの反応により得られるエポキシ樹脂。
[4]前項[1]または[2]に記載のフェノール樹脂を少なくとも1種とエポキシ樹脂を含有するエポキシ樹脂組成物。
[5]前項[3]に記載のエポキシ樹脂と硬化剤及び任意に硬化促進剤を含有するエポキシ樹脂組成物。
[6]前項[3]に記載のエポキシ樹脂と重合触媒とを含有するエポキシ樹脂組成物。
[7]前項[4]~[6]のいずれか一項に記載のエポキシ樹脂組成物を硬化して得られる硬化物。
本発明のフェノール樹脂やエポキシ樹脂を使用する硬化性樹脂組成物は高度な耐熱性を有するため、電気・電子部品用絶縁材料及び積層板(プリント配線板、ビルドアップ基板など)やCFRPを始めとする各種複合材料、接着剤、塗料等に有用である。特に半導体素子を保護する半導体封止材料にきわめて有用である。
本発明のフェノール樹脂(A)は下記一般式(1)で示されるフェノール樹脂である。

(式中、Rはそれぞれ独立して、水素原子、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、ヒドロキシル基、メトキシ基、エトキシ基、ニトリル基、アミノ基または置換もしくは無置換のフェニル基もしくはナフチル基を表し、tは0~3の整数を示し、nは繰り返し数を意味し、1~20の数を表す。)
Rは水素原子または炭素数1~6のアルキル基であると更に吸水性に優れ、置換又は無置換のフェニル基又はナフチル基であると更に耐熱性に優れるため好ましい。tは0~3の整数であり、好ましくは0~2の整数である。
また、本発明のフェノール樹脂において、軟化点は45~190℃が好ましく、より好ましくは50~180℃、特に好ましくは50~150℃である。軟化点が低すぎるとハンドリング性に乏しくなる傾向にあり、軟化点が190℃を越えると樹脂としての取出しが困難となる傾向にある他、粘度が非常に高くなる傾向にあり、取り扱い性が悪くなる恐れがある。また、水酸基当量は100~300g/eq.が好ましく、より好ましくは100~250g/eq.である。
繰り返し数nとしては、好ましくは1~15であり、より好ましくは1~10である。
ここで、上記式(1)において、ベンゼン骨格に置換しているR基と水酸基の配置は特に指定はないが、耐熱性、機械特性のバランスからパラ配向であることが好ましい。
繰り返し数nとしては、好ましくは1~15であり、より好ましくは1~10である。
ここで、上記式(1)において、ベンゼン骨格に置換しているR基と水酸基の配置は特に指定はないが、耐熱性、機械特性のバランスからパラ配向であることが好ましい。
本発明のフェノール樹脂は単独でもかまわないが、通常、原料であるバイノールをGPC面積比率(GPC分析で得られたクロマトグラム中に検出された各ピークの面積の比率)で、反応により得られたフェノール樹脂に対して、1~95面積%含有することが好ましく、より好ましくは5~80面積%含有し、特に好ましくは20~70面積%含有する。バイノール化合物の含有量が1面積%未満である場合、得られる樹脂の軟化点が非常に高くなってしまう恐れがあり、取出しが困難となる傾向にある他、組成物化時のハンドリング、および組成物化した後の成形性に大きく影響する傾向にあり、成型が困難となる恐れがある。1面積%未満のバイノール含有量である場合、代わりに他のフェノール樹脂を共存させることが好ましく、その具体例については後の併用できる硬化剤に例示する。
またバイノールが95面積%を超える場合、本発明の特徴である高耐熱性が損なわれる恐れがあるため、好ましくない。
またバイノールが95面積%を超える場合、本発明の特徴である高耐熱性が損なわれる恐れがあるため、好ましくない。
本発明のフェノール樹脂は、そのままで熱可塑性プラスチック(もしくはその原料)としての使用や、後述するようなエポキシ樹脂の原料やその硬化剤として使用することもできる。
本発明で用いられるバイノールとは、下記式(A)の構造を有する化合物である。

バイノールには立体異性体が存在するがキラル・ラセミ体どちらを用いてもかまわない。なお、その純度はゲルパーミエーションクロマトグラフィー(GPC)で90面積%以上が好ましく、より好ましくは93面積%以上、特に好ましくは98面積%以上である。不純物としてはキノン構造を有するものや、原料のナフトール化合物が挙げられるが、これらの含有量は各々2面積%以下が好ましく、1面積%以下であることがより好ましい。純度は晶析や洗浄によってコントロールできる。当該純度が低いと本反応のフェノール樹脂の特性が低下する可能性があり、好ましくない。また乾燥減量が0.2面積%以下であることが好ましく、より好ましくは0.1面積%以下である。乾燥減量が多い場合、製造工程において製造ラインを汚す等の問題が生じる恐れがある。融点は200~220℃が好ましく、より好ましくは212~219℃である。これらは市販品として、Aldrich、SR-CHEMから入手可能である。
本発明のフェノール樹脂の合成法は特に限定されないが、例えばバイノールと一般式(2)で示されるフェニル化合物との反応によって合成できる。

(式中、Xはそれぞれ独立して、水酸基、炭素数1~6のアルコキシ基またはハロゲン原子を示し、Rはそれぞれ独立して、水素原子、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、ヒドロキシル基、メトキシ基、エトキシ基、ニトリル基、アミノ基または置換もしくは無置換のフェニル基もしくはナフチル基を表し、tは0~3の整数を表す。)
一般式(2)で示されるフェニル化合物は公知のものが使用でき、特に限定されないが、具体的には、ビス(ヒドロキシアルキル)アルキルフェノール(より具体的には、ビス(ヒドロキシメチル)クレゾール、ビス(ヒドロキシエチル)クレゾール、ビス(ヒドロキシプロピル)クレゾール、ビス(ヒドロキシブチル)クレゾールなどが挙げられる。)、ビス(ハロアルキル)アルキルフェノール(ハロゲンとしては、塩素、臭素などが挙げられる。)、ビスアルコキシメチルアルキルフェノール(より具体的には、ビスメトキシメチルクレゾール、ビスエトキシメチルクレゾール、ビスプロポキシメチルクレゾール、ビスブトキシメチルクレゾールなどが挙げられる。)などが挙げられる。本発明においては特にビス(ヒドロキシメチル)クレゾール、ビス(クロロメチル)クレゾール、ビスメトキシメチルクレゾールが好ましい。
具体的な製法としては、バイノールと一般式(2)で示されるフェニル化合物を酸触媒の存在下、加熱することで本発明のフェノール樹脂を得ることができる。バイノール、前記式(2)で示されるフェニル化合物の混合の順番は特に指定はなく、また混合時に分割添加しても構わない。
本発明のフェノール樹脂は、バイノールと一般式(2)で示されるフェニル化合物と溶剤の混合液に、必要に応じて触媒を加えて加熱することにより得られる。また、バイノールを溶解させた溶液に一般式(2)で示されるフェニル化合物を徐々に添加してもよい。通常、反応時間は5~150時間、反応温度は40~150℃である。このようにして得られたフェノール樹脂は用途によって、精製せずに用いることもできるが、通常、反応終了後に反応混合物を必要に応じて中和等の処理をしてから、晶析あるいは加熱減圧下において溶媒類を除去する事で精製して各種用途に使用する。またバイノールと一般式(2)で示されるフェニル化合物の反応モル比は1.2:1~50:1が好ましく、より好ましくは1.5:1~30:1、特に好ましくは2:1~20:1である。反応モル比が1.2:1を下回ると、すなわち一般式(2)で示されるフェニル化合物1に対し、バイノールが1.2未満である場合、生成するフェノール樹脂の軟化点が高くなりすぎる傾向にあり、取り出すことが困難となる恐れがある。また、50:1を上回ると、すなわち、一般式(2)で示されるフェニル化合物1に対し、バイノールが50を越える場合、前記式(1)の構造が比較的少なくなり、耐熱性に乏しくなる恐れがある。
本発明のフェノール樹脂の合成において使用できる溶媒としては、メタノール、エタノール、プロパノール、イソプロパノール、トルエン、キシレン、メチルイソブチルケトン、アノン、シクロペンタノン、メチルエチルケトンなどが挙げられるがこれらに限定されるものではなく、単独で使用しても2種以上を併用してもよい。溶媒の使用量はバイノール100重量部に対し、通常5~500重量部、好ましくは10~400重量部の範囲である。
触媒としては、基本的には酸性触媒を用いることが好ましい。前記式(2)で示されるフェニル化合物がビス(ハロアルキル)クレゾールの場合は触媒の添加がなくともスムーズに反応を進めることができ、後の精製のしやすさの観点から触媒無しでの反応は好ましい。触媒を使用する場合、酸性触媒の具体例としては塩酸、硫酸、リン酸等の鉱酸類;シュウ酸、トルエンスルホン酸、酢酸等の有機酸類;タングステン酸等のヘテロポリ酸、活性白土、無機酸、塩化第二錫、塩化亜鉛、塩化第二鉄等、その他酸性を示す有機、無機酸塩類、等のノボラック樹脂製造用に通常使用される酸性触媒などが挙げられる。これら触媒は、前述したものに限定されるものではなく、単独で使用しても2種以上を併用してもよい。触媒の使用量は、バイノールに対し、通常0.005~2.0倍モル、好ましくは0.01~1.1倍モルの範囲、もしくはバイノール100gに対し通常0.1~10g、好ましくは0.3~7gである。触媒量が少ないと反応の進行が遅くなる傾向にある。また高温での反応が必要になる、反応が最後まで進まない等の課題が生じる場合があり、好ましく無い。また、触媒量が多すぎる場合、中和・精製等の後処理において多大な労力がかかる傾向にあり好ましく無い。
なお、反応により腐食性ガスが精製する場合は、引圧、もしくは窒素等の不活性ガスを送り込むことにより、系内から排出させることが好ましい。
なお、反応により腐食性ガスが精製する場合は、引圧、もしくは窒素等の不活性ガスを送り込むことにより、系内から排出させることが好ましい。
本発明のエポキシ樹脂は本発明のフェノール樹脂とエピハロヒドリンを反応させることで得られる。反応の手法としては特に限定しないが、以下に本発明のエポキシ樹脂の合成方法の一例を記載する。
本発明のエポキシ樹脂の具体的な構造式は下記式(3)で示される。

(式中、Rはそれぞれ独立して、水素原子、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、ヒドロキシル基、メトキシ基、エトキシ基、ニトリル基、アミノ基または置換もしくは無置換のフェニル基もしくはナフチル基を表し、tは0~3の整数を示し、nは繰り返し数を意味し、1~20の数を表す。)
本発明のエポキシ樹脂の好ましい樹脂特性としてはエポキシ当量が150~450g/eq.であることが好ましく、より好ましくは200~300g/eq.である。エポキシ当量が上記範囲内にあることで、硬化物の耐熱性、電気信頼性に優れたエポキシ樹脂を得ることが容易となる。エポキシ当量が450g/eq.を越えている場合、エポキシの環が閉環しきらず、官能基を有さない化合物が多く含まれる恐れがあるため、好ましくない。またこれら閉環しきらなかった化合物の多くには塩素が含有されている場合が多く、電子材料用途としては高温多湿条件での塩素イオンの遊離、およびそれによる配線の腐食が懸念されることから好ましくない。
本発明のエポキシ樹脂は単独でもかまわないが、通常、原料であるバイノールのグリシジル化物をGPC面積比率で、反応により得られたエポキシ樹脂に対して、1~95面積%、より好ましくは5~80面積%含有し、特に好ましくは20~70面積%含有する。バイノールのグリシジル化物の含有量が95面積%を超える場合、本発明の特徴である高耐熱性が損なわれる恐れがあるため、好ましくない。
また、エポキシ樹脂に残存している全塩素としては好ましくは5000ppm以下、より好ましくは3000ppm以下、特に2000ppm以下であることが好ましい。塩素による悪影響については前述同様である。なお、塩素イオン、ナトリウムイオンの含有量については各々5ppm以下が好ましく、より好ましくは3ppm以下である。塩素イオンによる悪影響については先に記載し、いうまでも無いが、ナトリウムイオン等のカチオンも、特にパワーデバイス用途においては非常に重要なファクターとなり、高電圧がかかった際の不良モードの一因となる。
ここで、上記式(3)において、ベンゼン骨格に置換しているR基とグリシジル基の配置は特に指定はないが、耐熱性、機械特性のバランスからパラ配向であることが好ましい。
また、エポキシ樹脂に残存している全塩素としては好ましくは5000ppm以下、より好ましくは3000ppm以下、特に2000ppm以下であることが好ましい。塩素による悪影響については前述同様である。なお、塩素イオン、ナトリウムイオンの含有量については各々5ppm以下が好ましく、より好ましくは3ppm以下である。塩素イオンによる悪影響については先に記載し、いうまでも無いが、ナトリウムイオン等のカチオンも、特にパワーデバイス用途においては非常に重要なファクターとなり、高電圧がかかった際の不良モードの一因となる。
ここで、上記式(3)において、ベンゼン骨格に置換しているR基とグリシジル基の配置は特に指定はないが、耐熱性、機械特性のバランスからパラ配向であることが好ましい。
本発明のエポキシ樹脂は軟化点を有する樹脂状の形態を有する。ここで、軟化点としては55~130℃が好ましく、より好ましくは60~120℃である。軟化点が低すぎると保管時のブロッキングが問題となる恐れがあり、低温で取り扱いをしないといけない等の課題が生じる場合がある。逆に軟化点が高すぎる場合、他の樹脂との混練の際に、ハンドリングが悪くなる等の問題が生じる恐れがある。また、溶融粘度は2Pa・s(ICI 溶融粘度 150℃ コーンプレート法)以下であることが好ましい。無機材料(フィラー等)を混合して用いる場合、流動性が悪い、また、ガラスクロス等もその網目がより微細になっており、含浸性に劣る等の課題が生じる恐れがある。
本発明のエポキシ樹脂を得る反応において、エピハロヒドリンとしては工業的に入手が容易なエピクロルヒドリンが好ましい。エピハロヒドリンの使用量は本発明のエポキシ樹脂の水酸基1モルに対し通常3.0~15モル、好ましくは3.0~10モル、より好ましくは3.5~8.5モルであり、特に好ましくは5.5~8.5モルである。
3.0モルを下回るとエポキシ当量が大きくなる恐れがあり、また、できたエポキシ樹脂の作業性が悪くなる可能性が高いため好ましくなく、15モルを超えると溶剤量が比較的多量となり、産業上好ましくない。
3.0モルを下回るとエポキシ当量が大きくなる恐れがあり、また、できたエポキシ樹脂の作業性が悪くなる可能性が高いため好ましくなく、15モルを超えると溶剤量が比較的多量となり、産業上好ましくない。
上記反応において使用しうるアルカリ金属水酸化物としては水酸化ナトリウム、水酸化カリウム等が挙げられ、固形物を利用してもよく、その水溶液を使用してもよいが、本発明においては特に、水分、溶解性、ハンドリングの面からフレーク状に成型された固形物の使用が好ましい。
アルカリ金属水酸化物の使用量は本発明のフェノール樹脂の水酸基1モルに対して通常0.90~1.5モルであり、好ましくは0.95~1.25モル、より好ましくは0.99~1.15モルである。
アルカリ金属水酸化物の使用量は本発明のフェノール樹脂の水酸基1モルに対して通常0.90~1.5モルであり、好ましくは0.95~1.25モル、より好ましくは0.99~1.15モルである。
反応を促進するためにテトラメチルアンモニウムクロライド、テトラメチルアンモニウムブロマイド、トリメチルベンジルアンモニウムクロライド等の4級アンモニウム塩を触媒として添加してもかまわない。4級アンモニウム塩の使用量としては本発明のフェノール樹脂の水酸基1モルに対し通常0.1~15gであり、好ましくは0.2~10gである。
本反応においては上記エピハロヒドリンに加え、非極性プロトン溶媒(ジメチルスルホキシド、ジオキサン、ジメチルイミダゾリジノン等)や、炭素数1~5のアルコールを併用することが好ましい。炭素数1~5のアルコールとしてはメタノール、エタノール、イソプロピルアルコールなどのアルコール類である。非極性プロトン溶媒もしくは炭素数1~5のアルコールの使用量はエピハロヒドリンの使用量に対し通常2~50重量%、好ましくは4~25重量%である。また、共沸脱水等の手法により、系内の水分をコントロールしながらエポキシ化を行ってもかまわない。
系中の水分が多い場合には、得られたエポキシ樹脂において電気信頼性が悪くなる傾向にあり好ましくなく、水分は5%以下にコントロールして合成することが好ましい。また、非極性プロトン溶媒を使用してエポキシ樹脂を得た際には、電気信頼性により優れるエポキシ樹脂が得られるため、非極性プロトン溶媒は好適に使用できる。
系中の水分が多い場合には、得られたエポキシ樹脂において電気信頼性が悪くなる傾向にあり好ましくなく、水分は5%以下にコントロールして合成することが好ましい。また、非極性プロトン溶媒を使用してエポキシ樹脂を得た際には、電気信頼性により優れるエポキシ樹脂が得られるため、非極性プロトン溶媒は好適に使用できる。
反応温度は通常30~90℃であり、好ましくは35~80℃である。特に本発明においては、より高純度なエポキシ化のために60℃以上が好ましく、還流条件に近い条件での反応が特に好ましい。反応時間は通常0.5~10時間であり、好ましくは1~8時間、特に好ましくは1~3時間である。反応時間が短いと反応が進みきらない場合があり、反応時間が長くなると副生成物ができる場合があることから好ましく無い。
これらのエポキシ化反応の反応物を水洗後、または水洗無しに加熱減圧下でエピハロヒドリンや溶媒等を除去する。また更に加水分解性ハロゲンの少ないエポキシ樹脂とするために、回収したエポキシ樹脂を炭素数4~7のケトン化合物(たとえば、メチルイソブチルケトン、メチルエチルケトン、シクロペンタノン、シクロヘキサノン等が挙げられる。)を溶剤として溶解し、水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物の水溶液を加えて反応を行い、閉環を確実なものにすることも出来る。この場合アルカリ金属水酸化物の使用量はエポキシ化に使用した本発明のフェノール樹脂の水酸基1モルに対して通常0.01~0.3モル、好ましくは0.05~0.2モルである。反応温度は通常50~120℃、反応時間は通常0.5~2時間である。
これらのエポキシ化反応の反応物を水洗後、または水洗無しに加熱減圧下でエピハロヒドリンや溶媒等を除去する。また更に加水分解性ハロゲンの少ないエポキシ樹脂とするために、回収したエポキシ樹脂を炭素数4~7のケトン化合物(たとえば、メチルイソブチルケトン、メチルエチルケトン、シクロペンタノン、シクロヘキサノン等が挙げられる。)を溶剤として溶解し、水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物の水溶液を加えて反応を行い、閉環を確実なものにすることも出来る。この場合アルカリ金属水酸化物の使用量はエポキシ化に使用した本発明のフェノール樹脂の水酸基1モルに対して通常0.01~0.3モル、好ましくは0.05~0.2モルである。反応温度は通常50~120℃、反応時間は通常0.5~2時間である。
またエピハロヒドリンとの反応においては反応初期から窒素等の不活性ガスで置換されていることが好ましく、空腔内の酸素濃度は10%以下であることが好ましい。酸素の残留は着色に影響をする場合がある。手法としては本発明のフェノール樹脂を仕込む前に窒素等不活性ガスを吹き込み(気中、もしくは液中)、もしくは、いったん減圧で真空にした後、不活性ガスで置換する方法が挙げられる。不活性ガスでの置換が無い場合、得られる樹脂に着色が生じる場合がある。不活性ガスの吹き込みを行う場合、その量はその釜の容積によっても異なるが、0.5~10時間でその釜の容積の1~3倍量が置換できる量の不活性ガスの吹き込みが好ましい。
反応終了後、生成した塩を濾過、水洗などにより除去し、更に加熱減圧下溶剤を留去することにより本発明のエポキシ樹脂が得られる。
以下、本発明のエポキシ樹脂組成物(以下、「硬化性樹脂組成物」とも表す。)について説明する。本発明の硬化性樹脂組成物においては、本発明のフェノール樹脂または硬化剤もしくは重合触媒を使用する。
本発明の硬化性樹脂組成物においては大きく二種に分類でき、以下、硬化性樹脂組成物A、硬化性樹脂組成物Bと表現する。
硬化性樹脂組成物Aにおいては、本発明のエポキシ樹脂及び本発明のフェノール樹脂の少なくとも一種を必ず含有する。すなわちエポキシ樹脂-硬化剤を必須成分とする組成物である。また、必要に応じて硬化促進剤を含有する。
硬化性樹脂組成物Bにおいては本発明のエポキシ樹脂と重合触媒を必須成分とする組成物である。
本発明の硬化性樹脂組成物Aにおいて硬化剤(本発明のフェノール樹脂を含んでもよい)の使用量は、全エポキシ樹脂のエポキシ基1当量に対して0.7~1.2当量が好ましい。エポキシ基1当量に対して、0.7当量に満たない場合、あるいは1.2当量を超える場合、いずれも硬化が不完全となり良好な硬化物性が得られない恐れがある。
本発明の硬化性樹脂組成物Bが含有する重合触媒としては、熱または光により重合を開始させる触媒であれば限定なく使用できるが、具体的には、硬化促進剤または酸性硬化触媒が好適に使用できる。
硬化性樹脂組成物Aにおいては、本発明のエポキシ樹脂及び本発明のフェノール樹脂の少なくとも一種を必ず含有する。すなわちエポキシ樹脂-硬化剤を必須成分とする組成物である。また、必要に応じて硬化促進剤を含有する。
硬化性樹脂組成物Bにおいては本発明のエポキシ樹脂と重合触媒を必須成分とする組成物である。
本発明の硬化性樹脂組成物Aにおいて硬化剤(本発明のフェノール樹脂を含んでもよい)の使用量は、全エポキシ樹脂のエポキシ基1当量に対して0.7~1.2当量が好ましい。エポキシ基1当量に対して、0.7当量に満たない場合、あるいは1.2当量を超える場合、いずれも硬化が不完全となり良好な硬化物性が得られない恐れがある。
本発明の硬化性樹脂組成物Bが含有する重合触媒としては、熱または光により重合を開始させる触媒であれば限定なく使用できるが、具体的には、硬化促進剤または酸性硬化触媒が好適に使用できる。
用い得る硬化促進剤の具体例としては、2-メチルイミダゾール、2-エチルイミダゾール、2-エチル-4-メチルイミダゾール等のイミダゾ-ル類、2-(ジメチルアミノメチル)フェノール、1,8-ジアザ-ビシクロ[5.4.0]ウンデセン-7等の第3級アミン類、トリフェニルホスフィン等のホスフィン類、テトラブチルアンモニウム塩、トリイソプロピルメチルアンモニウム塩、トリメチルデカニルアンモニウム塩、セチルトリメチルアンモニウム塩などの4級アンモニウム塩、トリフェニルベンジルフォスフォニウム塩、トリフェニルエチルフォスフォニウム塩、テトラブチルフォスフォニウム塩などの4級フォスフォニウム塩が挙げられる。(4級塩のカウンターイオンはハロゲン、有機酸イオン、水酸化物イオンなど、特に指定は無いが、特に有機酸イオン、水酸化物イオンが好ましい。)また、オクチル酸スズ等の金属化合物等が挙げられる。硬化促進剤を用いる場合は、エポキシ樹脂100重量部に対して0.01~5.0重量部が必要に応じ用いられる。
酸性硬化触媒としてはカチオン重合開始剤が好ましく、光もしくは熱カチオン重合開始剤が特に好ましい。活性エネルギー線によって活性化するカチオン重合開始剤及び/又は熱によって活性化するカチオン重合開始剤を配合することで、後述する硬化性樹脂組成物Bとして使用することができる。
活性エネルギー線の照射により本発明の硬化性樹脂組成物Bのカチオン重合を開始させるカチオン重合開始剤としては、ジアゾニウム塩、ヨードニウム塩、スルフォニウム塩、セレニウム塩、ピリジニウム塩、フェロセニウム塩、フォスフォニウム塩、及びチオピリリニウム塩等が挙げられ、好ましくはヨードニウム塩及びスルフォニウム塩であり、さらに好ましくはジアリールヨードニウム塩及びジアルキルフェナシルスルホニウム塩であり、特にジアリールヨードニウム塩が好適に使用できる。
ヨードニウム塩及びスルフォニウム塩等の光カチオン重合開始剤を本発明のカチオン硬化性樹脂組成物に使用する場合、アニオンとしてはBF4
-、AsF6
-、SbF6
-、PF6
-、及びB(C6F5)4
-等が挙げられ、好ましくはSbF6
-、PF6
-、又はB(C6F5)4
-であり、特に好ましくはSbF6
-又はB(C6F5)4
-である。
光カチオン重合開始剤の具体例を挙げると、ビス(ドデシルフェニル)ヨードニウムヘキサフルオロアンチモネート(GE東芝シリコーン社製、UV-9380Cの主成分)、トリルクミルヨードニウムテトラキス(ペンタフルオロフェニル)ボレート(ローディア社製、PHOTOINITIATOR(商品名)2074)、ビス(アルキル(C=10~14)フェニルヨードニウム)ヘキサフルオロアンチモネート(和光純薬製光カチオン重合開始剤WPI-016)等が挙げられる。
熱により活性化してカチオン重合を開始させる化合物、すなわち熱カチオン重合開始剤を本発明の硬化性樹脂組成物Bに用いることもできる。このものとしては、第四級アンモニウム塩、ホスホニウム塩及びスルホニウム塩等の各種オニウム塩類や、アルコキシシランとアルミニウム錯体の組み合わせ等が例示できる。入手可能な製品としては、アデカオプトンCP-66及びアデカオプトンCP-77(いずれも商品名、旭電化工業(株)製)、サンエイドSI-60L、サンエイドSI-80L及びサンエイドSI-100L(いずれも商品名、三新化学工業(株)製)、及びCIシリーズ(日本曹達(株)製)等が挙げられる。
以下、本発明の硬化性樹脂組成物A、Bについてそれぞれ説明する。
硬化性樹脂組成物Aと硬化性組樹脂成物Bにおいて本発明のエポキシ樹脂以外に他のエポキシ樹脂を併用してもよいし、もしくは本発明のエポキシ樹脂を単独で使用することも出来る。併用する場合、本発明のエポキシ樹脂の全エポキシ樹脂中に占める割合は30重量%以上が好ましく、特に40重量%以上が好ましい。ただし、本発明のエポキシ樹脂を硬化性樹脂組成物の改質剤として使用する場合は、1~30重量%の割合で添加する。なお、硬化性樹脂組成物Aにおいて、本発明のフェノール樹脂を硬化剤として使用する場合は、本発明のエポキシ樹脂以外の他のエポキシ樹脂を単独で使用できる。
他のエポキシ樹脂の具体例としては、ノボラック型エポキシ樹脂、ビスフェノールA型エポキシ樹脂、ビフェニル型エポキシ樹脂、トリフェニルメタン型エポキシ樹脂、フェノールアラルキル型エポキシ樹脂などが挙げられる。具体的には、ビスフェノールA、ビスフェノールS、チオジフェノール、フルオレンビスフェノール、テルペンジフェノール、4,4’-ビフェノール、2,2’-ビフェノール、3,3’,5,5’-テトラメチル-[1,1’-ビフェニル]-4,4’-ジオール、ハイドロキノン、レゾルシン、ナフタレンジオール、トリス-(4-ヒドロキシフェニル)メタン、1,1,2,2-テトラキス(4-ヒドロキシフェニル)エタン、フェノール類(フェノール、アルキル置換フェノール、ナフトール、アルキル置換ナフトール、ジヒドロキシベンゼン、ジヒドロキシナフタレン等)とホルムアルデヒド、アセトアルデヒド、ベンズアルデヒド、p-ヒドロキシベンズアルデヒド、o-ヒドロキシベンズアルデヒド、p-ヒドロキシアセトフェノン、o-ヒドロキシアセトフェノン、ジシクロペンタジエン、フルフラール、4,4’-ビス(クロルメチル)-1,1’-ビフェニル、4,4’-ビス(メトキシメチル)-1,1’-ビフェニル、1,4-ビス(クロロメチル)ベンゼン、1,4-ビス(メトキシメチル)ベンゼン等との重縮合物及びこれらの変性物、テトラブロモビスフェノールA等のハロゲン化ビスフェノール類、アルコール類から誘導されるグリシジルエーテル化物、脂環式エポキシ樹脂、グリシジルアミン系エポキシ樹脂、グリシジルエステル系エポキシ樹脂、等シルセスキオキサン系のエポキシ樹脂(鎖状、環状、ラダー状、あるいはそれら少なくとも2種以上の混合構造のシロキサン構造にグリシジル基、および/またはエポキシシクロヘキサン構造を有するエポキシ樹脂)等の固形または液状エポキシ樹脂が挙げられるが、これらに限定されるものではない。
以下それぞれの硬化性樹脂組成物について言及する。
硬化性樹脂組成物A(硬化剤による熱硬化)
本発明の硬化性樹脂組成物Aが含有する硬化剤としては、本発明のフェノール樹脂以外に他の硬化剤を併用してもよいし、もしくは本発明のフェノール樹脂を単独で使用することも出来る。併用する場合、本発明のフェノール樹脂の全エポキシ樹脂中に占める割合は30重量%以上が好ましく、特に40重量%以上が好ましい。ただし、本発明のエポキシ樹脂を硬化性樹脂組成物の改質剤として使用する場合は、1~30重量%の割合で添加する。なお、硬化性樹脂組成物Aにおいて、本発明のエポキシ樹脂を使用する場合は、本発明のフェノール樹脂以外の他の硬化剤を単独で使用できる。
硬化性樹脂組成物A(硬化剤による熱硬化)
本発明の硬化性樹脂組成物Aが含有する硬化剤としては、本発明のフェノール樹脂以外に他の硬化剤を併用してもよいし、もしくは本発明のフェノール樹脂を単独で使用することも出来る。併用する場合、本発明のフェノール樹脂の全エポキシ樹脂中に占める割合は30重量%以上が好ましく、特に40重量%以上が好ましい。ただし、本発明のエポキシ樹脂を硬化性樹脂組成物の改質剤として使用する場合は、1~30重量%の割合で添加する。なお、硬化性樹脂組成物Aにおいて、本発明のエポキシ樹脂を使用する場合は、本発明のフェノール樹脂以外の他の硬化剤を単独で使用できる。
硬化剤の具体例としては例えばフェノール樹脂、フェノール系化合物、アミン系化合物、酸無水物系化合物、アミド系化合物、カルボン酸系化合物などが挙げられる。用いうる硬化剤の具体例としては、フェノール樹脂、フェノール化合物;ビスフェノールA、ビスフェノールF、ビスフェノールS、フルオレンビスフェノール、テルペンジフェノール、4,4’-ビフェノール、2,2’-ビフェノール、3,3’,5,5’-テトラメチル-[1,1’-ビフェニル]-4,4’-ジオール、ハイドロキノン、レゾルシン、ナフタレンジオール、トリス-(4-ヒドロキシフェニル)メタン、1,1,2,2-テトラキス(4-ヒドロキシフェニル)エタン、フェノール類(フェノール、アルキル置換フェノール、ナフトール、アルキル置換ナフトール、ジヒドロキシベンゼン、ジヒドロキシナフタレン等)とホルムアルデヒド、アセトアルデヒド、ベンズアルデヒド、p-ヒドロキシベンズアルデヒド、o-ヒドロキシベンズアルデヒド、p-ヒドロキシアセトフェノン、o-ヒドロキシアセトフェノン、ジシクロペンタジエン、フルフラール、4,4’-ビス(クロロメチル)-1,1’-ビフェニル、4,4’-ビス(メトキシメチル)-1,1’-ビフェニル、1,4’-ビス(クロロメチル)ベンゼン、1,4’-ビス(メトキシメチル)ベンゼン等との重縮合物及びこれらの変性物、テトラブロモビスフェノールA等のハロゲン化ビスフェノール類、テルペンとフェノール類の縮合物などのポリフェノール類が挙げられるが、これらに限定されるものではない。これらは単独で用いてもよく、2種以上を用いてもよい。
好ましいフェノール樹脂としては、フェノールアラルキル樹脂(芳香族アルキレン構造を有する樹脂)が挙げられ、特に好ましくはフェノール、ナフトール、クレゾールから選ばれる少なくとも一種を有する構造であり、そのリンカーとなるアルキレン部が、ベンゼン構造、ビフェニル構造、ナフタレン構造から選ばれる少なくとも一種であることを特徴とする樹脂(具体的にはザイロック、ナフトールザイロック、フェノールビフェニレンノボラック樹脂、クレゾール-ビフェニレンノボラック樹脂、フェノール-ナフタレンノボラック樹脂などが挙げられる。)である。
アミン系化合物、アミド系化合物;ジアミノジフェニルメタン、ジエチレントリアミン、トリエチレンテトラミン、ジアミノジフェニルスルホン、イソホロンジアミン、ジシアンジアミド、リノレン酸の2量体とエチレンジアミンより合成されるポリアミド樹脂などの含窒素化合物;
酸無水物系化合物、カルボン酸系化合物;無水フタル酸、無水トリメリット酸、無水ピロメリット酸、無水マレイン酸、テトラヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、無水メチルナジック酸、無水ナジック酸、ヘキサヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、ブタンテトラカルボン酸無水物、ビシクロ[2,2,1]ヘプタン-2,3-ジカルボン酸無水物、メチルビシクロ[2,2,1]ヘプタン-2,3-ジカルボン酸無水物、シクロヘキサン-1,3,4-トリカルボン酸-3,4-無水物、などの酸無水物;各種アルコール、カルビノール変性シリコーン、と前述の酸無水物との付加反応により得られるカルボン酸樹脂;
その他;イミダゾール、トリフルオロボラン-アミン錯体、グアニジン誘導体の化合物などが挙げられるが、これらに限定されるものではない。これらは単独で用いてもよく、2種以上を用いてもよい。
アミン系化合物、アミド系化合物;ジアミノジフェニルメタン、ジエチレントリアミン、トリエチレンテトラミン、ジアミノジフェニルスルホン、イソホロンジアミン、ジシアンジアミド、リノレン酸の2量体とエチレンジアミンより合成されるポリアミド樹脂などの含窒素化合物;
酸無水物系化合物、カルボン酸系化合物;無水フタル酸、無水トリメリット酸、無水ピロメリット酸、無水マレイン酸、テトラヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、無水メチルナジック酸、無水ナジック酸、ヘキサヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、ブタンテトラカルボン酸無水物、ビシクロ[2,2,1]ヘプタン-2,3-ジカルボン酸無水物、メチルビシクロ[2,2,1]ヘプタン-2,3-ジカルボン酸無水物、シクロヘキサン-1,3,4-トリカルボン酸-3,4-無水物、などの酸無水物;各種アルコール、カルビノール変性シリコーン、と前述の酸無水物との付加反応により得られるカルボン酸樹脂;
その他;イミダゾール、トリフルオロボラン-アミン錯体、グアニジン誘導体の化合物などが挙げられるが、これらに限定されるものではない。これらは単独で用いてもよく、2種以上を用いてもよい。
本発明の硬化性樹脂組成物Aにおいて硬化剤の使用量は、全エポキシ樹脂のエポキシ基1当量に対して0.7~1.2当量が好ましい。エポキシ基1当量に対して、0.7当量に満たない場合、あるいは1.2当量を超える場合、いずれも硬化が不完全となり良好な硬化物性が得られない恐れがある。
本発明の硬化性樹脂組成物Aにおいては、硬化剤とともに硬化促進剤を併用しても差し支えない。硬化促進剤の具体例としては前記のものが挙げられる。硬化促進剤を用いる場合は、エポキシ樹脂100重量部に対して0.01~5.0重量部が好ましい。
本発明の硬化性樹脂組成物Aには、リン含有化合物を難燃性付与成分として含有させることもできる。リン含有化合物としては反応型のものでも添加型のものでもよい。リン含有化合物の具体例としては、トリメチルホスフェート、トリエチルホスフェート、トリクレジルホスフェート、トリキシリレニルホスフェート、クレジルジフェニルホスフェート、クレジル-2,6-ジキシリレニルホスフェート、1,3-フェニレンビス(ジキシリレニルホスフェート)、1,4-フェニレンビス(ジキシリレニルホスフェート)、4,4’-ビフェニル(ジキシリレニルホスフェート)等のリン酸エステル類;9,10-ジヒドロ-9-オキサ-10-ホスファフェナントレン-10-オキサイド、10(2,5-ジヒドロキシフェニル)-10H-9-オキサ-10-ホスファフェナントレン-10-オキサイド等のホスファン類;エポキシ樹脂と前記ホスファン類の活性水素とを反応させて得られるリン含有エポキシ化合物、赤リン等が挙げられるが、リン酸エステル類、ホスファン類またはリン含有エポキシ化合物が好ましく、1,3-フェニレンビス(ジキシリレニルホスフェート)、1,4-フェニレンビス(ジキシリレニルホスフェート)、4,4’-ビフェニル(ジキシリレニルホスフェート)またはリン含有エポキシ化合物が特に好ましい。リン含有化合物の含有量はリン含有化合物/全エポキシ樹脂=0.1~0.6(重量比)が好ましい。0.1以下では難燃性が不十分であり、0.6以上では硬化物の吸湿性、誘電特性に悪影響を及ぼす懸念がある。
さらに本発明の硬化性樹脂組成物Aには、必要に応じて酸化防止剤を添加しても構わない。使用できる酸化防止剤としては、フェノール系、イオウ系、リン系酸化防止剤が挙げられる。酸化防止剤は単独で又は2種以上を組み合わせて使用できる。酸化防止剤の使用量は、本発明の硬化性樹脂組成物中の樹脂成分に対して100重量部に対して、通常0.008~1重量部、好ましくは0.01~0.5重量部である。
酸化防止剤としては、例えば、フェノール系酸化防止剤、イオウ系酸化防止剤、リン系酸化防止剤などが挙げられる。フェノール系酸化防止剤の具体例として、2,6-ジ-t-ブチル-p-クレゾール、ブチル化ヒドロキシアニソール、2,6-ジ-t-ブチル-p-エチルフェノール、ステアリル-β-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート、イソオクチル-3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート、2,4-ビス-(n-オクチルチオ)-6-(4-ヒドロキシ-3,5-ジ-t-ブチルアニリノ)-1,3,5-トリアジン、2,4-ビス[(オクチルチオ)メチル]-o-クレゾール、等のモノフェノール類;2,2’-メチレンビス(4-メチル-6-t-ブチルフェノール)、2,2’-メチレンビス(4-エチル-6-t-ブチルフェノール)、4,4’-チオビス(3-メチル-6-t-ブチルフェノール)、4,4’-ブチリデンビス(3-メチル-6-t-ブチルフェノール)、トリエチレングリコール-ビス[3-(3-t-ブチル-5-メチル-4-ヒドロキシフェニル)プロピオネート]、1,6-ヘキサンジオール-ビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]、N,N’-ヘキサメチレンビス(3,5-ジ-t-ブチル-4-ヒドロキシ-ヒドロシンナマミド)、2,2-チオ-ジエチレンビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]、3,5-ジ-t-ブチル-4-ヒドロキシベンジルフォスフォネート-ジエチルエステル、3,9-ビス[1,1-ジメチル-2-{β-(3-t-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオニルオキシ}エチル]2,4,8,10-テトラオキサスピロ[5,5]ウンデカン、ビス(3,5-ジ-t-ブチル-4-ヒドロキシベンジルスルホン酸エチル)カルシウム等のビスフェノール類;1,1,3-トリス(2-メチル-4-ヒドロキシ-5-t-ブチルフェニル)ブタン、1,3,5-トリメチル-2,4,6-トリス(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)ベンゼン、テトラキス-[メチレン-3-(3’,5’-ジ-t-ブチル-4’-ヒドロキシフェニル)プロピオネート]メタン、ビス[3,3’-ビス-(4’-ヒドロキシ-3’-t-ブチルフェニル)ブチリックアシッド]グリコールエステル、トリス-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)-イソシアヌレイト、1,3,5-トリス(3’,5’-ジ-t-ブチル-4’-ヒドロキシベンジル)-S-トリアジン-2,4,6-(1H,3H,5H)トリオン、トコフェノール等の高分子型フェノール類が例示される。
イオウ系酸化防止剤の具体例として、ジラウリル-3,3’-チオジプロピオネート、ジミリスチル-3,3’-チオジプロピオネート、ジステアリルル-3,3’-チオジプロピオネート等が例示される。
リン系酸化防止剤の具体例として、トリフェニルホスファイト、ジフェニルイソデシルホスファイト、フェニルジイソデシルホスファイト、トリス(ノニルフェニル)ホスファイト、ジイソデシルペンタエリスリトールホスファイト、トリス(2,4-ジ-t-ブチルフェニル)ホスファイト、サイクリックネオペンタンテトライルビス(オクタデシル)ホスファイト、サイクリックネオペンタンテトライルビ(2,4-ジ-t-ブチルフェニル)ホスファイト、サイクリックネオペンタンテトライルビ(2,4-ジ-t-ブチル-4-メチルフェニル)ホスファイト、ビス[2-t-ブチル-6-メチル-4-{2-(オクタデシルオキシカルボニル)エチル}フェニル]ヒドロゲンホスファイト等のホスファイト類;9,10-ジヒドロ-9-オキサ-10-ホスファフェナントレン-10-オキサイド、10-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)-9,10-ジヒドロ-9-オキサ-10-ホスファフェナントレン-10-オキサイド、10-デシロキシ-9,10-ジヒドロ-9-オキサ-10-ホスファフェナントレン-10-オキサイド等のオキサホスファフェナントレンオキサイド類などが例示される。
これらの酸化防止剤はそれぞれ単独で使用できるが、2種以上を組み合わせて併用しても構わない。特に本発明においてはリン系の酸化防止剤が好ましい。
さらに本発明の硬化性樹脂組成物Aには、必要に応じて光安定剤を添加しても構わない。
光安定剤としては、ヒンダートアミン系の光安定剤、特にHALS等が好適である。HALSとしては特に限定されるものではないが、代表的なものとしては、ジブチルアミン・1,3,5-トリアジン・N,N’―ビス(2,2,6,6-テトラメチル-4-ピペリジル-1,6-ヘキサメチレンジアミンとN-(2,2,6,6-テトラメチル-4-ピペリジル)ブチルアミンの重縮合物、コハク酸ジメチル-1-(2-ヒドロキシエチル)-4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン重縮合物、ポリ〔{6-(1,1,3,3-テトラメチルブチル)アミノ-1,3,5-トリアジン-2,4-ジイル}{(2,2,6,6-テトラメチル-4-ピペリジル)イミノ}ヘキサメチレン{(2,2,6,6-テトラメチル-4-ピペリジル)イミノ}〕、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)〔〔3,5-ビス(1,1-ジメチルエチル)-4-ヒドリキシフェニル〕メチル〕ブチルマロネート、ビス(2,2,6,6-テトラメチル-4-ピペリジル)セバケート、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)セバケート、ビス(1-オクチロキシ-2,2,6,6-テトラメチル-4-ピペリジル)セバケート、2-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)-2-n-ブチルマロン酸ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)、等が挙げられる。HALSは1種のみが用いられても良いし、2種類以上が併用されても良い。
光安定剤としては、ヒンダートアミン系の光安定剤、特にHALS等が好適である。HALSとしては特に限定されるものではないが、代表的なものとしては、ジブチルアミン・1,3,5-トリアジン・N,N’―ビス(2,2,6,6-テトラメチル-4-ピペリジル-1,6-ヘキサメチレンジアミンとN-(2,2,6,6-テトラメチル-4-ピペリジル)ブチルアミンの重縮合物、コハク酸ジメチル-1-(2-ヒドロキシエチル)-4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン重縮合物、ポリ〔{6-(1,1,3,3-テトラメチルブチル)アミノ-1,3,5-トリアジン-2,4-ジイル}{(2,2,6,6-テトラメチル-4-ピペリジル)イミノ}ヘキサメチレン{(2,2,6,6-テトラメチル-4-ピペリジル)イミノ}〕、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)〔〔3,5-ビス(1,1-ジメチルエチル)-4-ヒドリキシフェニル〕メチル〕ブチルマロネート、ビス(2,2,6,6-テトラメチル-4-ピペリジル)セバケート、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)セバケート、ビス(1-オクチロキシ-2,2,6,6-テトラメチル-4-ピペリジル)セバケート、2-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)-2-n-ブチルマロン酸ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)、等が挙げられる。HALSは1種のみが用いられても良いし、2種類以上が併用されても良い。
さらに本発明の硬化性樹脂組成物Aには、必要に応じてバインダー樹脂を配合することも出来る。バインダー樹脂としてはブチラール系樹脂、アセタール系樹脂、アクリル系樹脂、エポキシ-ナイロン系樹脂、NBR-フェノール系樹脂、エポキシ-NBR系樹脂、ポリアミド系樹脂、ポリイミド系樹脂、シリコーン系樹脂などが挙げられるが、これらに限定されるものではない。バインダー樹脂の配合量は、硬化物の難燃性、耐熱性を損なわない範囲であることが好ましく、樹脂成分100重量部に対して通常0.05~50重量部、好ましくは0.05~20重量部が必要に応じて用いられる。
本発明の硬化性樹脂組成物Aには、必要に応じて無機充填剤を添加することができる。無機充填剤としては、結晶シリカ、溶融シリカ、アルミナ、ジルコン、珪酸カルシウム、炭酸カルシウム、炭化ケイ素、窒化ケイ素、窒化ホウ素、ジルコニア、フォステライト、ステアタイト、スピネル、チタニア、タルク等の粉体またはこれらを球形化したビーズ等が挙げられるが、これらに限定されるものではない。これらは単独で用いてもよく、2種以上を用いてもよい。これら無機充填剤の含有量は、本発明の硬化性樹脂組成物中において0~95重量%を占める量が用いられる。更に本発明の硬化性樹脂組成物には、シランカップリング剤、ステアリン酸、パルミチン酸、ステアリン酸亜鉛、ステアリン酸カルシウム等の離型剤、界面活性剤、染料、顔料、紫外線吸収剤等の種々の配合剤、各種熱硬化性樹脂を添加することができる。
本発明の硬化性樹脂組成物Aは、各成分を均一に混合することにより得られる。本発明の硬化性樹脂組成物Aは従来知られている方法と同様の方法で容易にその硬化物とすることができる。例えば本発明のエポキシ樹脂と硬化剤並びに必要により硬化促進剤、リン含有化合物、バインダー樹脂、無機充填材及び配合剤とを必要に応じて押出機、ニーダ、ロール等を用いて均一になるまで充分に混合して硬化性樹脂組成物を得、その硬化性樹脂組成物をポッティング、溶融後(液状の場合は溶融無しに)注型あるいはトランスファー成型機などを用いて成型し、さらに80~200℃で2~10時間加熱することにより本発明の硬化物を得ることができる。
また本発明の硬化性樹脂組成物Aを必要に応じてトルエン、キシレン、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジメチルホルムアミド、ジメチルアセトアミド、N-メチルピロリドン等の溶剤に溶解させ、硬化性樹脂組成物ワニスとし、ガラス繊維、カ-ボン繊維、ポリエステル繊維、ポリアミド繊維、アルミナ繊維、紙などの基材に含浸させて加熱乾燥して得たプリプレグを熱プレス成形することにより、本発明の硬化性樹脂組成物Aの硬化物とすることができる。この際の溶剤は、本発明の硬化性樹脂組成物と該溶剤の混合物中で通常10~70重量%、好ましくは15~70重量%を占める量を用いる。また液状組成物であれば、そのまま例えば、RTM方式でカーボン繊維を含有するエポキシ樹脂硬化物を得ることもできる。
また本発明の硬化性樹脂組成物Aをフィルム型組成物の改質剤としても使用できる。具体的にはB-ステージにおけるフレキ性等を向上させる場合に用いることができる。このようなフィルム型の樹脂組成物は、本発明の硬化性樹脂組成物Aを前記硬化性樹脂組成物ワニスとして剥離フィルム上に塗布し、加熱下で溶剤を除去した後、Bステージ化を行うことによりシート状の接着剤として得られる。このシート状接着剤は多層基板などにおける層間絶縁層として使用することが出来る。
硬化性樹脂組成物B(酸性硬化触媒(重合触媒)によるカチオン硬化)
酸性硬化触媒(重合触媒)を用いて硬化させる本発明の硬化性樹脂組成物Bは、酸性硬化触媒(重合触媒)として光重合開始剤あるいは熱重合開始剤を含有する。さらに、希釈剤、重合性モノマー、重合性オリゴマー、重合開始補助剤、光増感剤等の各種公知の化合物、材料等を含有していてもよい。また、所望に応じて無機充填材、着色顔料、紫外線吸収剤、酸化防止剤、安定剤等、各種公知の添加剤を含有してもよい。
酸性硬化触媒(重合触媒)を用いて硬化させる本発明の硬化性樹脂組成物Bは、酸性硬化触媒(重合触媒)として光重合開始剤あるいは熱重合開始剤を含有する。さらに、希釈剤、重合性モノマー、重合性オリゴマー、重合開始補助剤、光増感剤等の各種公知の化合物、材料等を含有していてもよい。また、所望に応じて無機充填材、着色顔料、紫外線吸収剤、酸化防止剤、安定剤等、各種公知の添加剤を含有してもよい。
酸性硬化触媒(重合触媒)としてはカチオン重合開始剤が好ましく、光もしくは熱カチオン重合開始剤が特に好ましい。活性エネルギー線によって活性化するカチオン重合開始剤及び/又は熱によって活性化するカチオン重合開始剤を配合することで、硬化性樹脂組成物Bとして使用することができる。
活性エネルギー線の照射により本発明の硬化性樹脂組成物Bのカチオン重合を開始させるカチオン重合開始剤としては、前記のものが挙げられる。
本発明の硬化性樹脂組成物Bを硬化させるときの活性エネルギー線としては、X線、電子線、紫外線及び可視光等を使用することもできるが、好ましくは紫外線又は可視光であり、特に好ましくは紫外線である。紫外線を使用する場合、その波長範囲は特に限定されないが、好ましくは150~400nm、さらに好ましくは200~380nmである。紫外線を用いる場合、カチオン重合を効率よく開始できる。
また、本発明の硬化性樹脂組成物Bには、必要に応じてさらに光カチオン重合開始剤の活性を高めるため、増感剤を併用することもできる。本発明で用いることができる増感剤として、例えばクリベロガアドバンスドイン ポリマーサイエンス(Adv.in Plymer Sci.,62,1(1984))で開示している化合物を用いることが可能である。具体的には、ピレン、ペリレン、アクリジンオレンジ、チオキサントン、2-クロロチオキサントン及びペンゾフラビン等がある。また、光ラジカル重合開始剤として広く使用されている化合物も使用することができ、具体的には、ベンゾフェノン、2,4-ジエチルチオキサントン、2-イソプロピルチオキサントン、2,4-ジクロロチオキサントン等のチオキサントン類、ベンゾインメチルエーテル、ベンゾインエチルエーテル、ベンゾインイソプロピルエーテル等のベンゾインエーテル類、2,2-ジメトキシ-1,2-ジフェニルエタン-1-オン等のベンジルジメチルケタール類、2-ヒドロキシ-2-メチル-1-フェニルプロパン-1-オン、1-(4-イソプロピルフェニル)-2-ヒドロキシ-2-メチルプロパン-1-オン、1-ヒドロキシシクロヘキシルフェニルケトン等のα-ヒドロキシアルキルフェノン類、カンファーキノン等のα-ジカルボニル化合物等が挙げられる。本発明においては、チオキサントン類やα-ヒドロキシアルキルフェノン類が特に好適に使用できる。
本発明の硬化性樹脂組成物Bへの光カチオン重合開始剤の配合量は、活性エネルギー線の種類や照射量に応じて適宜に調整できる。例えば紫外線の場合、カチオン硬化性樹脂組成物の合計100質量部に対し、0.1~10質量部とすることが好ましく、より好ましくは0.5~5質量部であり、さらに好ましくは1~3質量部である。カチオン重合開始剤の配合量が0.1質量部よりも少ない場合は硬化性に劣ることがあり、逆に10質量部より多い場合は硬化物に真に必要な成分を減少させて硬化物の物性が低下する場合や、硬化物の着色が激しくなる場合があり好ましくない。
本発明の硬化性樹脂組成物Bに増感剤を添加する場合の配合量は、活性エネルギー線の種類や照射量に応じて適宜に調整できる。例えば紫外線の場合、硬化性樹脂組成物Bの合計100質量部に対し、5質量部以下とすることが好ましく、さらに好ましくは0.2~2部である。増感剤の配合量が5質量部より多い場合は硬化物に真に必要な成分を減少させて硬化物の物性が低下する場合や、硬化物の着色が激しくなる場合がある。
活性エネルギー線が紫外線や可視光である場合、カチオン硬化性樹脂組成物が空気にさらされるが、このとき雰囲気の湿度は低いことが好ましく、好ましくは湿度80%R.H.以下であり、70%R.H.以下であることがさらに好ましい。ここで、紫外線や可視光を生産ラインの中に設置する場合、光照射装置の手前に乾燥空気を送る方法や、加熱装置を取り付けて湿度を下げる方法も採用できる。
熱により活性化してカチオン重合を開始させる化合物、すなわち熱カチオン重合開始剤を本発明の硬化性樹脂組成物Bに用いることもできる。このものとしては、第四級アンモニウム塩、ホスホニウム塩及びスルホニウム塩等の各種オニウム塩類や、アルコキシシランとアルミニウム錯体の組み合わせ等が例示できる。入手可能な製品としては、アデカオプトンCP-66及びアデカオプトンCP-77(いずれも商品名、旭電化工業(株)製)、サンエイドSI-60L、サンエイドSI-80L及びサンエイドSI-100L(いずれも商品名、三新化学工業(株)製)、及びCIシリーズ(日本曹達(株)製)等が挙げられる。
本発明の硬化性樹脂組成物Bへの熱カチオン重合開始剤の配合割合は、カチオン硬化性樹脂組成物100質量部に対し、0.01~10質量部の範囲とすることが好ましく、より好ましくは0.1~5質量部であり、さらに好ましくは0.5~3質量部である。この配合割合が0.01質量部未満の場合には、熱の作用によりこれが活性化しても、開環重合性基の開環反応を十分に進行させることができないことが有る。また、これを10質量部超えて配合したとしても、重合を進行させる作用はそれ以上高まらず、また硬化物の物性が低下する事があるので好ましくない。
更に、本発明の硬化性樹脂組成物Bには、必要に応じて前記のような無機充填剤やシランカップリング材、離型剤、顔料等の種々の配合剤、各種熱硬化性樹脂を添加することができる。
本発明の硬化性樹脂組成物Bは、各成分を均一に混合することにより得られる。またポリエチレングリコールモノエチルエーテルやシクロヘキサノン、γ-ブチロラクトン等の有機溶剤に溶解させ、均一とした後、乾燥により溶剤を除去して使用することも可能である。この際の溶剤は、本発明の硬化性樹脂組成物Bと該溶剤の混合物中で通常10~70重量%、好ましくは15~70重量%を占める量を用いる。本発明の硬化性樹脂組成物Bは紫外線照射することにより硬化できるが、その紫外線照射量については、硬化性樹脂組成物により変化するため、それぞれの硬化条件によって、決定される。光硬化型硬化性樹脂組成物が硬化する照射量であれば良く、硬化物の接着強度が良好である硬化条件を満たしていれば良い。この硬化の際、光が細部まで透過することが必要であることから本発明のエポキシ樹脂、および硬化性樹脂組成物Bにおいては透明性の高いものが望まれる。また、これらエポキシ樹脂系の光硬化では光照射のみでは完全に硬化することが難しく、耐熱性が求められる用途においては光照射後に加熱により完全に反応硬化を終了させる必要がある。
前記、光照射後の加熱は通常の硬化性樹脂組成物Bの硬化温度域で良い。例えば常温~150℃で30分~7日間の範囲が好適である。硬化性樹脂組成物Bの配合により変化するが、特に高い温度域であればあるほど光照射後の硬化促進に効果があり、短時間の熱処理で効果がある。また、低温であればあるほど長時間の熱処理を要する。このような熱アフターキュアすることで、エージング処理になるという効果も出る。
また、これら硬化性樹脂組成物Bを硬化させて得られる硬化物の形状も用途に応じて種々とりうるので特に限定されないが、例えばフィルム状、シート状、バルク状などの形状とすることもできる。成形する方法は適応する部位、部材によって異なるが、例えば、キャスト法、注型法、スクリーン印刷法、スピンコート法、スプレー法、転写法、ディスペンサー方式などの成形方法を適用することができるなどが挙げられるが、これらに限定されるものではない。成形型は研磨ガラス、硬質ステンレス研磨板、ポリカーボネート板、ポリエチレンテレフタレート板、ポリメチルメタクリレート板等を適用することができる。また、成形型との離型性を向上させるためポリエチレンテレフタレートフィルム、ポリカーボネートフィルム、ポリ塩化ビニルフィルム、ポリエチレンフィルム、ポリテトラフルオロエチレンフィルム、ポリプロピレンフィルム、ポリイミドフィルム等を適用することができる。
例えばカチオン硬化性のレジストに使用する際においては、まず、ポリエチレングリコールモノエチルエーテルやシクロヘキサノンあるいはγ-ブチロラクトン等の有機溶剤に溶解させた本発明の光カチオン硬化性樹脂組成物Bを銅張積層板やセラミック基板またはガラス基板等の基板上に、スクリーン印刷、スピンコート法などの手法によって、5~160μmの膜厚で本発明の組成物を塗布し、塗膜を形成する。そして、該塗膜を60~110℃で予備乾燥させた後、所望のパターンの描かれたネガフィルムを通して紫外線(例えば低圧水銀灯、高圧水銀灯、超高圧水銀灯、キセノン灯、レーザー光等)を照射し、ついで、70~120℃で露光後ベーク処理を行う。その後ポリエチレングリコールモノエチルエーテル等の溶剤で未露光部分を溶解除去(現像)した後、さらに必要があれば紫外線の照射及び/または加熱(例えば100~200℃で0.5~3時間)によって十分な硬化を行い、硬化物を得る。このようにしてプリント配線板を得ることも可能である。
本発明の硬化性樹脂組成物Aおよび硬化性樹脂組成物Bを硬化してなる硬化物は各種用途に使用できる。
硬化性樹脂組成物Aまたは硬化性樹脂組成物Bが使用される一般の用途としては、例えば、接着剤、塗料、コーティング剤、成形材料(シート、フィルム、FRP等を含む)、絶縁材料(プリント基板、電線被覆等を含む)、封止剤の他、他樹脂等への添加剤等が挙げられる。接着剤としては、土木用、建築用、自動車用、一般事務用、医療用の接着剤の他、電子材料用の接着剤が挙げられる。これらのうち電子材料用の接着剤としては、ビルドアップ基板等の多層基板の層間接着剤、ダイボンディング剤、アンダーフィル等の半導体用接着剤、BGA補強用アンダーフィル、異方性導電性フィルム(ACF)、異方性導電性ペースト(ACP)等の実装用接着剤等が挙げられる。
封止剤、基板としては、コンデンサ、トランジスタ、ダイオード、発光ダイオード、IC、LSIなどのポッティング、ディッピング、トランスファーモールド封止、IC、LSI類のCOB、COF、TABなどのポッティング封止、フリップチップなどのアンダーフィル、QFP、BGA、CSPなどのICパッケージ類実装時の封止(補強用アンダーフィルを含む)およびパッケージ基板などを挙げることができる。またネットワーク基板や、モジュール基板といった機能性が求められる基板用途へも好適である。
次に本発明を実施例により更に具体的に説明するが、以下において部は特に断わりのない限り重量部である。尚、本発明はこれら実施例に限定されるものではない。
以下に実施例で用いた各種分析方法について記載する。
以下に実施例で用いた各種分析方法について記載する。
エポキシ当量:JIS K 7236(ISO 3001)に準拠
ICI溶融粘度:JIS K 7117-2(ISO 3219)に準拠
軟化点:JIS K 7234に準拠
GPC:
カラム(Shodex(商標)KF-603、KF-602.5、KF-602、KF-601x2)連結
溶離液はテトラヒドロフラン
流速は0.5ml/min.
カラム温度は40℃
検出:RI(示差屈折検出器)
ICI溶融粘度:JIS K 7117-2(ISO 3219)に準拠
軟化点:JIS K 7234に準拠
GPC:
カラム(Shodex(商標)KF-603、KF-602.5、KF-602、KF-601x2)連結
溶離液はテトラヒドロフラン
流速は0.5ml/min.
カラム温度は40℃
検出:RI(示差屈折検出器)
(実施例1)
撹拌機、還流冷却管、撹拌装置を備えたフラスコに、窒素パージを施しながらバイノール(GPC純度>99% Aldrich製 試薬融点216-218℃)215部、2,6-ビス(ヒドロキシメチル)-p-クレゾール(旭有機材料工業製 試薬)41.5部、メチルイソブチルケトン(純正化学製 試薬)384部、パラトルエンスルホン酸・一水和物(東京化成製 試薬)2.9部、メタノール20部加え、メタノールと生成する水を抜きながら、70℃1時間、80℃1時間、100℃2時間反応させ、その後、110-120℃で還流状態とし、そのまま3時間反応を行った。
反応終了後、50℃まで冷却、水洗を繰り返し、水層が中性になったことを確認した後、油層からロータリーエバポレーターを用いて減圧下、溶剤類を留去することで本発明のフェノール樹脂(P-1)を245部を得た。得られたフェノール樹脂の軟化点は132℃であった。なお、GPCのチャートを図1に示す。本GPCの結果から残留バイノール量はフェノール樹脂(P-1)に対する面積比率で50.8面積%である事が確認された。
撹拌機、還流冷却管、撹拌装置を備えたフラスコに、窒素パージを施しながらバイノール(GPC純度>99% Aldrich製 試薬融点216-218℃)215部、2,6-ビス(ヒドロキシメチル)-p-クレゾール(旭有機材料工業製 試薬)41.5部、メチルイソブチルケトン(純正化学製 試薬)384部、パラトルエンスルホン酸・一水和物(東京化成製 試薬)2.9部、メタノール20部加え、メタノールと生成する水を抜きながら、70℃1時間、80℃1時間、100℃2時間反応させ、その後、110-120℃で還流状態とし、そのまま3時間反応を行った。
反応終了後、50℃まで冷却、水洗を繰り返し、水層が中性になったことを確認した後、油層からロータリーエバポレーターを用いて減圧下、溶剤類を留去することで本発明のフェノール樹脂(P-1)を245部を得た。得られたフェノール樹脂の軟化点は132℃であった。なお、GPCのチャートを図1に示す。本GPCの結果から残留バイノール量はフェノール樹脂(P-1)に対する面積比率で50.8面積%である事が確認された。
(実施例2)
撹拌機、還流冷却管、撹拌装置を備えたフラスコに、窒素パージを施しながら本発明のフェノール樹脂(P-1 水酸基当量147g/eq.)145部、エピクロロヒドリン638部(7モル当量 対 フェノール樹脂)、メタノール191部を加え、撹拌下で溶解し、70~75℃にまで昇温した。次いでフレーク状の水酸化ナトリウム44部を90分かけて分割添加した後、更に75℃で75分反応を行った。反応終了後,水132部で水洗を行い、油層からロータリーエバポレーターを用いて減圧下、過剰のエピクロルヒドリン等の溶剤類を留去した。残留物にメチルイソブチルケトン380部を加え溶解し、75℃にまで昇温した。撹拌下で30重量%の水酸化ナトリウム水溶液16部を加え、1時間反応を行った後、油層の洗浄水が中性になるまで水洗を行った。得られた溶液をロータリーエバポレーターを用いて減圧下、メチルイソブチルケトン等を留去することで本発明のエポキシ樹脂(EP-1)200部を得た。得られたエポキシ樹脂のエポキシ当量は226g/eq.、軟化点92.4℃、ICI粘度1.19Pa・sであった。
撹拌機、還流冷却管、撹拌装置を備えたフラスコに、窒素パージを施しながら本発明のフェノール樹脂(P-1 水酸基当量147g/eq.)145部、エピクロロヒドリン638部(7モル当量 対 フェノール樹脂)、メタノール191部を加え、撹拌下で溶解し、70~75℃にまで昇温した。次いでフレーク状の水酸化ナトリウム44部を90分かけて分割添加した後、更に75℃で75分反応を行った。反応終了後,水132部で水洗を行い、油層からロータリーエバポレーターを用いて減圧下、過剰のエピクロルヒドリン等の溶剤類を留去した。残留物にメチルイソブチルケトン380部を加え溶解し、75℃にまで昇温した。撹拌下で30重量%の水酸化ナトリウム水溶液16部を加え、1時間反応を行った後、油層の洗浄水が中性になるまで水洗を行った。得られた溶液をロータリーエバポレーターを用いて減圧下、メチルイソブチルケトン等を留去することで本発明のエポキシ樹脂(EP-1)200部を得た。得られたエポキシ樹脂のエポキシ当量は226g/eq.、軟化点92.4℃、ICI粘度1.19Pa・sであった。
(合成例1)
撹拌機、還流冷却管、撹拌装置を備えたフラスコに、窒素パージを施しながらバイノール214部、エピクロロヒドリン555部(4モル当量 対 フェノール樹脂)、メタノール55部を加え、撹拌下で溶解し、70~75℃にまで昇温した。次いでフレーク状の水酸化ナトリウム60部を90分かけて分割添加した後、更に75℃で75分反応を行った。反応終了後,水400部で水洗を行い、油層からロータリーエバポレーターを用いて減圧下、過剰のエピクロルヒドリン等の溶剤類を留去した。残留物にメチルイソブチルケトン500部を加え溶解し、75℃にまで昇温した。撹拌下で30重量%の水酸化ナトリウム水溶液10部、メタノール10部を加え、1時間反応を行った後、油層の洗浄水が中性になるまで水洗を行い、得られた溶液から、ロータリーエバポレーターを用いて減圧下にメチルイソブチルケトン等を留去することでエポキシ樹脂(EP-2)403部を得た。得られたエポキシ樹脂のエポキシ当量は230g/eq.、軟化点58℃であった。
撹拌機、還流冷却管、撹拌装置を備えたフラスコに、窒素パージを施しながらバイノール214部、エピクロロヒドリン555部(4モル当量 対 フェノール樹脂)、メタノール55部を加え、撹拌下で溶解し、70~75℃にまで昇温した。次いでフレーク状の水酸化ナトリウム60部を90分かけて分割添加した後、更に75℃で75分反応を行った。反応終了後,水400部で水洗を行い、油層からロータリーエバポレーターを用いて減圧下、過剰のエピクロルヒドリン等の溶剤類を留去した。残留物にメチルイソブチルケトン500部を加え溶解し、75℃にまで昇温した。撹拌下で30重量%の水酸化ナトリウム水溶液10部、メタノール10部を加え、1時間反応を行った後、油層の洗浄水が中性になるまで水洗を行い、得られた溶液から、ロータリーエバポレーターを用いて減圧下にメチルイソブチルケトン等を留去することでエポキシ樹脂(EP-2)403部を得た。得られたエポキシ樹脂のエポキシ当量は230g/eq.、軟化点58℃であった。
実施例3、4および比較例1、2、3、4
<耐熱性試験・難燃性試験>
前記で得られたエポキシ樹脂、フェノール樹脂を表1の割合(重量部)で配合し、ミキシングロールを用いて均一に混合・混練し、封止用硬化性樹脂組成物を得た。この硬化性樹脂組成物をミキサーにて粉砕し、更にタブレットマシーンにてタブレット化した。このタブレット化された硬化性樹脂組成物をトランスファー成型(175℃×60秒)し、更に脱型後160℃×2時間+180℃×6時間の条件で硬化、評価用試験片を得た。なお、硬化物の物性は以下の要領で測定した。
<耐熱性試験・難燃性試験>
前記で得られたエポキシ樹脂、フェノール樹脂を表1の割合(重量部)で配合し、ミキシングロールを用いて均一に混合・混練し、封止用硬化性樹脂組成物を得た。この硬化性樹脂組成物をミキサーにて粉砕し、更にタブレットマシーンにてタブレット化した。このタブレット化された硬化性樹脂組成物をトランスファー成型(175℃×60秒)し、更に脱型後160℃×2時間+180℃×6時間の条件で硬化、評価用試験片を得た。なお、硬化物の物性は以下の要領で測定した。
・耐熱性(DMA)
動的粘弾性測定器:TA-instruments、DMA-2980
測定温度範囲:-30~280℃
温速度:2℃/分
試験片サイズ:5mm×50mmに切り出した物を使用した(厚みは約800μm)
Tg:Tan-δのピーク点をTgとした
動的粘弾性測定器:TA-instruments、DMA-2980
測定温度範囲:-30~280℃
温速度:2℃/分
試験片サイズ:5mm×50mmに切り出した物を使用した(厚みは約800μm)
Tg:Tan-δのピーク点をTgとした
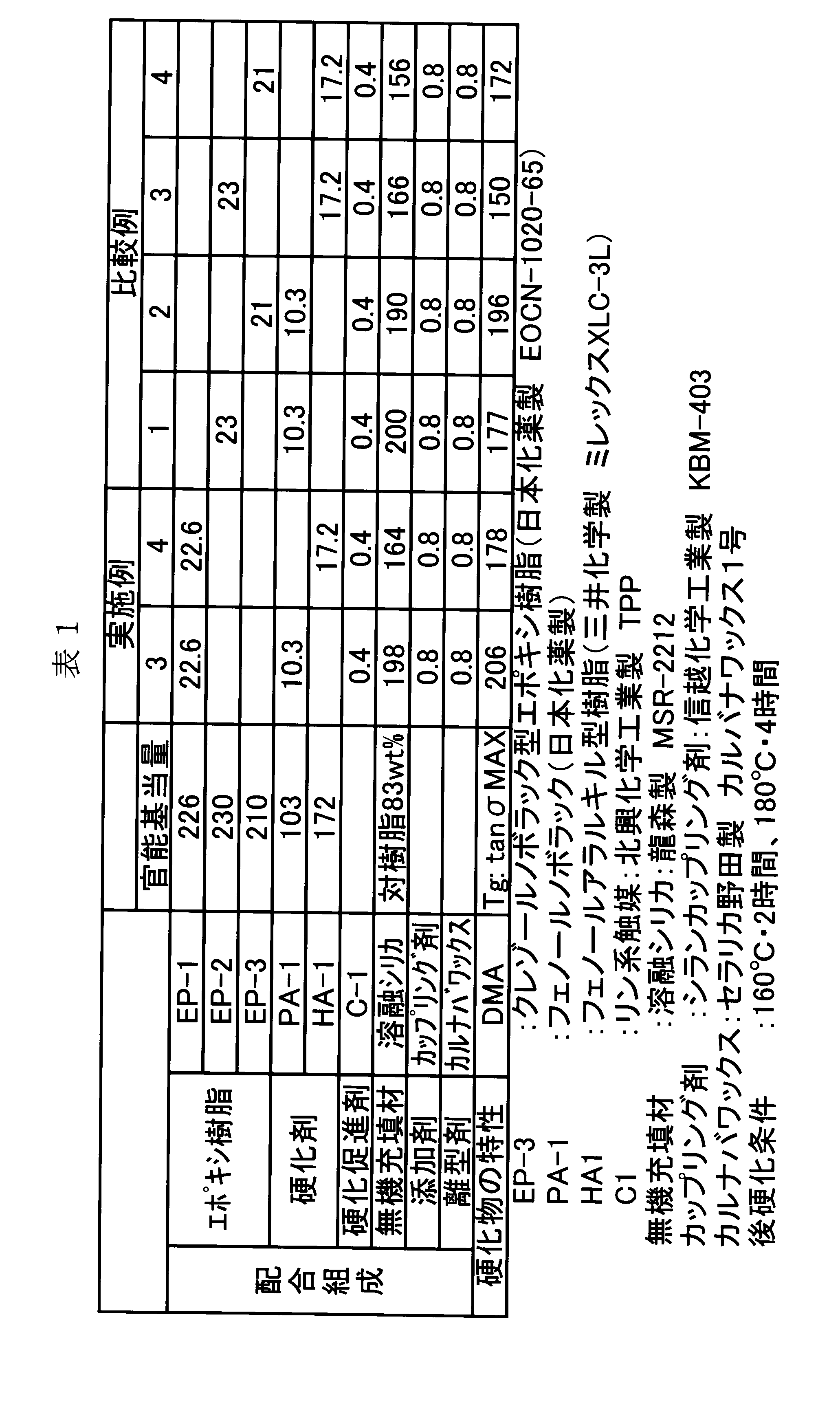
本発明のエポキシ樹脂(実施例3、4)はバイノールエポキシ樹脂(比較例1、3)およびクレゾールノボラック型エポキシ樹脂(比較例2、4)と比較して耐熱性が向上することが確認できる。
以上の結果から、本発明のエポキシ樹脂は、類似構造を有するEP-2、EP-3と比較し、耐熱性に優れることが明らかであり、高い耐熱性を有する硬化物を与えることができることがわかる。
本発明を特定の態様を参照して詳細に説明したが、本発明の精神と範囲を離れることなく様々な変更および修正が可能であることは、当業者にとって明らかである。
なお、本出願は、2013年12月4日付で出願された日本国特許出願(特願2013-250733)に基づいており、その全体が引用により援用される。また、ここに引用されるすべての参照は全体として取り込まれる。
なお、本出願は、2013年12月4日付で出願された日本国特許出願(特願2013-250733)に基づいており、その全体が引用により援用される。また、ここに引用されるすべての参照は全体として取り込まれる。
本発明のフェノール樹脂やエポキシ樹脂を使用する硬化性樹脂組成物は、高度な耐熱性を有するため、電気・電子部品用絶縁材料及びプリント配線板、ビルドアップ基板などの積層板や、CFRPを始めとする各種複合材料、接着剤、塗料等に有用である。特に半導体素子を保護する半導体封止材料にきわめて有用である。
Claims (7)
- 下記一般式(1)で示されるフェノール樹脂。

- バイノールと下記一般式(2)で示されるフェニル化合物との反応により得られる請求項1に記載のフェノール樹脂。

- 請求項1または請求項2に記載のフェノール樹脂とエピハロヒドリンとの反応により得られるエポキシ樹脂。
- 請求項1または請求項2記載のフェノール樹脂を少なくとも1種とエポキシ樹脂を含有するエポキシ樹脂組成物。
- 請求項3に記載のエポキシ樹脂と硬化剤及び任意に硬化促進剤を含有するエポキシ樹脂組成物。
- 請求項3に記載のエポキシ樹脂と重合触媒とを含有するエポキシ樹脂組成物。
- 請求項4及至請求項6のいずれか一項に記載のエポキシ樹脂組成物を硬化して得られる硬化物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015551547A JP6504709B2 (ja) | 2013-12-04 | 2014-12-03 | フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013250733 | 2013-12-04 | ||
| JP2013-250733 | 2013-12-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015083748A1 true WO2015083748A1 (ja) | 2015-06-11 |
Family
ID=53273509
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/082023 WO2015083748A1 (ja) | 2013-12-04 | 2014-12-03 | フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP6504709B2 (ja) |
| TW (1) | TWI642700B (ja) |
| WO (1) | WO2015083748A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6071120B2 (ja) * | 2014-12-18 | 2017-02-01 | 三菱瓦斯化学株式会社 | シアン酸エステル化合物及びその製造方法、樹脂組成物、並びに、硬化物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04275317A (ja) * | 1991-02-28 | 1992-09-30 | Nippon Kayaku Co Ltd | 新規エポキシ樹脂の製法 |
| JP2003201324A (ja) * | 2001-12-28 | 2003-07-18 | Hitachi Ltd | 水性アルカリ可溶性樹脂、感光性樹脂組成物、フォトマスクおよび電子デバイスの製造方法 |
| JP2006248912A (ja) * | 2005-03-08 | 2006-09-21 | Dainippon Ink & Chem Inc | 多価ヒドロキシ化合物、エポキシ樹脂、それらの製造方法、エポキシ樹脂組成物及び硬化物 |
| JP2009203427A (ja) * | 2008-02-29 | 2009-09-10 | Dic Corp | エポキシ樹脂組成物、半導体封止材料及び半導体装置 |
| JP2014009336A (ja) * | 2012-07-02 | 2014-01-20 | Nippon Kayaku Co Ltd | フェノール樹脂、エポキシ樹脂、および硬化性樹脂組成物 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1601515A2 (en) * | 2003-03-13 | 2005-12-07 | 3M Innovative Properties Company | Composite webs and closure systems |
-
2014
- 2014-12-03 JP JP2015551547A patent/JP6504709B2/ja active Active
- 2014-12-03 WO PCT/JP2014/082023 patent/WO2015083748A1/ja active Application Filing
- 2014-12-04 TW TW103142126A patent/TWI642700B/zh active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04275317A (ja) * | 1991-02-28 | 1992-09-30 | Nippon Kayaku Co Ltd | 新規エポキシ樹脂の製法 |
| JP2003201324A (ja) * | 2001-12-28 | 2003-07-18 | Hitachi Ltd | 水性アルカリ可溶性樹脂、感光性樹脂組成物、フォトマスクおよび電子デバイスの製造方法 |
| JP2006248912A (ja) * | 2005-03-08 | 2006-09-21 | Dainippon Ink & Chem Inc | 多価ヒドロキシ化合物、エポキシ樹脂、それらの製造方法、エポキシ樹脂組成物及び硬化物 |
| JP2009203427A (ja) * | 2008-02-29 | 2009-09-10 | Dic Corp | エポキシ樹脂組成物、半導体封止材料及び半導体装置 |
| JP2014009336A (ja) * | 2012-07-02 | 2014-01-20 | Nippon Kayaku Co Ltd | フェノール樹脂、エポキシ樹脂、および硬化性樹脂組成物 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6071120B2 (ja) * | 2014-12-18 | 2017-02-01 | 三菱瓦斯化学株式会社 | シアン酸エステル化合物及びその製造方法、樹脂組成物、並びに、硬化物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2015083748A1 (ja) | 2017-03-16 |
| TWI642700B (zh) | 2018-12-01 |
| TW201527360A (zh) | 2015-07-16 |
| JP6504709B2 (ja) | 2019-04-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6240069B2 (ja) | エポキシ樹脂組成物、およびその硬化物、並びに、硬化性樹脂組成物 | |
| JP5559154B2 (ja) | オレフィン樹脂、エポキシ樹脂、硬化性樹脂組成物及びその硬化物 | |
| JP6090765B2 (ja) | フェノール樹脂、エポキシ樹脂、および硬化性樹脂組成物 | |
| JP5469078B2 (ja) | ジオレフィン化合物、エポキシ樹脂、硬化性樹脂組成物、および硬化物 | |
| JP5430337B2 (ja) | ジオレフィン化合物、エポキシ樹脂、硬化性樹脂組成物及びその硬化物 | |
| KR102047682B1 (ko) | 에폭시 수지 혼합물, 에폭시 수지 조성물 및 그 경화물 | |
| JP6284732B2 (ja) | エポキシ樹脂混合物、および硬化性樹脂組成物 | |
| JP6403554B2 (ja) | フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物 | |
| JP5505960B2 (ja) | ジオレフィン化合物、エポキシ樹脂、硬化性樹脂組成物及びその硬化物 | |
| KR102226437B1 (ko) | 에폭시 수지, 경화성 수지 조성물 및 그 경화물 | |
| WO2015083748A1 (ja) | フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびその硬化物 | |
| JP6016647B2 (ja) | エポキシ樹脂、および硬化性樹脂組成物 | |
| JP2015067615A (ja) | エポキシ樹脂混合物、硬化性樹脂組成物、およびその硬化物 | |
| JP5878865B2 (ja) | ジオレフィン化合物、エポキシ樹脂、硬化性樹脂組成物及びその硬化物 | |
| JP5196663B2 (ja) | ジオレフィン化合物、エポキシ樹脂、硬化性樹脂組成物及びその硬化物 | |
| JP6414845B2 (ja) | フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、およびそれらの硬化物 | |
| JP2010254628A (ja) | ジオレフィン化合物、エポキシ化合物、硬化性樹脂組成物及びその硬化物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14867988 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015551547 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14867988 Country of ref document: EP Kind code of ref document: A1 |