WO2002000653A2 - Fused cyclic compounds as modulators of nuclear hormone receptor function - Google Patents
Fused cyclic compounds as modulators of nuclear hormone receptor function Download PDFInfo
- Publication number
- WO2002000653A2 WO2002000653A2 PCT/US2001/019663 US0119663W WO0200653A2 WO 2002000653 A2 WO2002000653 A2 WO 2002000653A2 US 0119663 W US0119663 W US 0119663W WO 0200653 A2 WO0200653 A2 WO 0200653A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- methanoimidazo
- alkyl
- heterocyclo
- dione
- Prior art date
Links
- 0 CC*(**)N(C(C)(*)C(*)O)C(OC(C)(C)C)=O Chemical compound CC*(**)N(C(C)(*)C(*)O)C(OC(C)(C)C)=O 0.000 description 16
- IFAMSTPTNRJBRG-UHFFFAOYSA-N CC(C)(C)OC(N(C1CC2CC1)C2C(O)=O)=O Chemical compound CC(C)(C)OC(N(C1CC2CC1)C2C(O)=O)=O IFAMSTPTNRJBRG-UHFFFAOYSA-N 0.000 description 1
- WYWWEZFASQNBCU-ZJUUUORDSA-N CC(C)COC(N1[C@H](CO)C[C@@H](C)C1)=O Chemical compound CC(C)COC(N1[C@H](CO)C[C@@H](C)C1)=O WYWWEZFASQNBCU-ZJUUUORDSA-N 0.000 description 1
- RESTWAHJFMZUIZ-UHFFFAOYSA-N CCc(cc1)ccc1[N+]([O-])=O Chemical compound CCc(cc1)ccc1[N+]([O-])=O RESTWAHJFMZUIZ-UHFFFAOYSA-N 0.000 description 1
- ZPTVNYMJQHSSEA-UHFFFAOYSA-N Cc(cc1)ccc1[N+]([O-])=O Chemical compound Cc(cc1)ccc1[N+]([O-])=O ZPTVNYMJQHSSEA-UHFFFAOYSA-N 0.000 description 1
- RCMIVPNJRBRFCM-UHFFFAOYSA-N Cc(cc1C(F)(F)F)ccc1F Chemical compound Cc(cc1C(F)(F)F)ccc1F RCMIVPNJRBRFCM-UHFFFAOYSA-N 0.000 description 1
- HSQGRXTXMXMKPQ-UHFFFAOYSA-N Cc1c(C)[s]nn1 Chemical compound Cc1c(C)[s]nn1 HSQGRXTXMXMKPQ-UHFFFAOYSA-N 0.000 description 1
- AGQOIYCTCOEHGR-UHFFFAOYSA-N Cc1ccn[o]1 Chemical compound Cc1ccn[o]1 AGQOIYCTCOEHGR-UHFFFAOYSA-N 0.000 description 1
- AZEYWBIKLUFKOJ-YPXMJKPUSA-N O=C([C@@H](C1CC2CC1)N2C1=O)N1c1cccc(C(F)(F)F)c1 Chemical compound O=C([C@@H](C1CC2CC1)N2C1=O)N1c1cccc(C(F)(F)F)c1 AZEYWBIKLUFKOJ-YPXMJKPUSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/655—Azo (—N=N—), diazo (=N2), azoxy (>N—O—N< or N(=O)—N<), azido (—N3) or diazoamino (—N=N—N<) compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/04—Drugs for genital or sexual disorders; Contraceptives for inducing labour or abortion; Uterotonics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/12—Drugs for genital or sexual disorders; Contraceptives for climacteric disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/16—Masculine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/18—Feminine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/26—Androgens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/30—Oestrogens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/40—Mineralocorticosteroids, e.g. aldosterone; Drugs increasing or potentiating the activity of mineralocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/42—Drugs for disorders of the endocrine system of the suprarenal hormones for decreasing, blocking or antagonising the activity of mineralocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/44—Glucocorticosteroids; Drugs increasing or potentiating the activity of glucocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/46—Drugs for disorders of the endocrine system of the suprarenal hormones for decreasing, blocking or antagonising the activity of glucocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/08—Plasma substitutes; Perfusion solutions; Dialytics or haemodialytics; Drugs for electrolytic or acid-base disorders, e.g. hypovolemic shock
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/12—Oxygen or sulfur atoms
Definitions
- the present invention relates to fused cyclic compounds, to methods of using such compounds in the treatment of nuclear hormone receptor-associated conditions such as cancer, and to pharmaceutical compositions containing such compounds.
- Nuclear hormone receptors constitute a large super-family of ligand- dependent and sequence-specific transcription factors. Members of this family influence transcription either directly, through specific binding to the promoter target genes (Evans, in Science 240: 889-895 (1988)), or indirectly, via protein-protein interactions with other transcription factors (Jonat et al., Cell 62: 1189-1204 (1990), Schuele et al., Cell 62: 1217-1226 (1990), and Yang-Yen et al., Cell 62: 1205-1215 (1990)).
- the nuclear hormone receptor super-family (also known in the art as the "steroid/thyroid hormone receptor super-family”) includes receptors for a variety of hydrophobic ligands, including cortisol, aldosterone, estrogen, progesterone, testosterone, vitamine D3, thyroid hormone and retinoic acid (Evans, 1988, supra).
- the super-family contains a number of proteins that have no known ligands, termed orphan nuclear hormone receptors (Mangelsdorf et al., Cell 83: 835-839 (1995), O'Malley et al., Mol. Endocrinol 10: 1293 (1996), Enmark et al., Mol. Endocrinol.
- the conventional nuclear hormone receptors are generally transactivators in the presence of ligand, and can either be active repressors or transcriptionally inert in the absence of ligand. Some of the orphan receptors behave as if they are transcriptionally inert in the absence of ligand. Others, however, behave as either constitutive activators or repressors. These orphan nuclear hormone receptors are either under the control of ubiquitous ligands that have not been identified, or do not need to bind ligand to exert these activities.
- the nuclear hormone receptors have a modular structure, being comprised of three distinct domains: an N-terminal domain of variable size containing a transcriptional activation function AF-1, a highly conserved DNA binding domain and a moderately conserved ligand-binding domain.
- the ligand-binding domain is not only responsible for binding the specific ligand but also contains a transcriptional activation function called AF-2 and a dimerisation domain (Wurtz et al., Nature Struc. Biol. 3, 87-94 (1996), Parker et al., Nature Struc. Biol 3, 113-115 (1996) and Kumar et al., Steroids 64, 310-319 (1999)).
- SB -NHR' s The steroid binding nuclear hormone receptors (SB -NHR' s) comprise a subfamily of nuclear hormone receptors. These receptors are related in that they share a stronger sequence homology to one another, particularly in the ligand binding domain (LBD), than to the other members of the NHR super-family (Evans, 1988, supra) and they all utilize steroid based ligands.
- NHR's are the androgen receptor (AR), the estrogen receptor (ER), the progesterone receptor (PR), the glucocorticoid receptor (GR), the mineralocorticoid receptor (MR), the aldosterone receptor (ALDR) and the steroid and xenobiotic receptor (SXR) (Evans et al, WO 99/35246).
- AR androgen receptor
- ER estrogen receptor
- PR progesterone receptor
- GR glucocorticoid receptor
- MR mineralocorticoid receptor
- ADR aldosterone receptor
- SXR steroid and xenobiotic receptor
- the natural ligands for each is derived from a common steroid core.
- examples of some of the steroid based ligands utilized by members of the SB-NHR's include cortisol, aldosterone, estrogen, progesterone, testosterone and dihydrotestosterone. Specificity of a particular steroid based ligand for one SB-NHR versus another is obtained by differential substitution about the steroid core.
- RU486 is an example of a synthetic agonist of the PR, which is utilized as a birth control agent (Vegeto et al., Cell 69: 703-713 (1992)), and Flutamide is an example of an antagonist of the AR, which is utilized for the treatment of prostate cancer (Neri et al, Endo. 91, 427-437 (1972)).
- Tamoxifen is an example of a tissues specific modulator of the ER function, that is used in the treatment of breast cancer (Smigel J. Natl. Cancer Inst. 90, 647-648 (1998)).
- Tamoxifen can function as an antagonist of the ER in breast tissue while acting as an agonist of the ER in bone (Grese et al., Proc. Natl. Acad. Sci. USA 94, 14105-14110 (1997)). Because of the tissue selective effects seen for Tamoxifen, this agent and agents like it are referred to as "partial-agonist" or partial-antagonist". In addition to synthetically derived non-endogenous ligands, non-endogenous ligands for NHR's can be obtained from food sources (Regal et al., Proc. Soc. Exp. Biol. Med. 223, 372- 378 (2000) and Hempstock et al., J. Med. Food!, 267-269 (1999)).
- the flavanoid phytoestrogens are an example of an unnatural ligand for SB-NHR's that are readily obtained from a food source such as soy (Quella et al., J. Clin. Oncol. 18, 1068-1074 (2000) and Banz et al., J. Med. Food 2, 271 -273 (1999)).
- soy Quella et al., J. Clin. Oncol. 18, 1068-1074 (2000) and Banz et al., J. Med. Food 2, 271 -273 (1999)
- the ability to modulate the transcriptional activity of individual NHR by the addition of a small molecule ligand makes them ideal targets for the development of pharmaceutical agents for a variety of disease states.
- non-natural ligands can be synthetically engineered to serve as modulators of the function of NHR's.
- engineering of an unnatural ligand can include the identification of a core structure which mimics the natural steroid core system. This can be achieved by random screening against several SB-NHR's or through directed approaches using the available crystal structures of a variety of NHR ligand binding domains (Bourguet et al., Nature 375, 377-382 (1995), Brzozowski, et al, Nature 389, 753-758 (1997), Shiau et al, Cell 95, 927-937 (1998) and Tanenbaum et al, Proc. Natl. Acad: Sci.
- Differential substitution about such a steroid mimic core can provide agents with selectivity for one receptor versus another. In addition, such modifications can be employed to obtain agents with agonist or antagonist activity for a particular SB- NHR. Differential substitution about the steroid mimic core can result in the formation of a series of high affinity agonists and antagonists with specificity for, for example, ER versus PR versus AR versus GR versus MR. Such an approach of differential substitution has been reported, for example, for quinoline based modulators of steroid NHR inJ Med.
- the compounds of the present invention comprise a core which serves as a steroid mimic, and are useful as modulators of the function of steroid binding nuclear hormone receptors, as well as other NHR as described following.
- the present invention provides fused cyclic compounds of the following formula I and salts thereof, which compounds are especially useful as modulators of nuclear hormone receptor function:
- Z 2 is O, S, NH, or NR 6 ;
- A is CR 7 orN
- a 2 is CR 7 or N
- M is a bond, O, CR 7 R 7' or NR 10
- M' is a bond or NR 10 , with the proviso that at least one of M or M' must be a bond
- R 1 and R 1 are each independently H, alkyl or substituted alkyl, cycloalkyl or substituted cycloalkyl, cycloalkenyl or substituted cycloalkenyl, heterocyclo or substituted heterocyclo, cycloalkylalkyl or substituted cycloalkyalkyl, cycloalkenylalkyl or substituted cycloalkenylalkyl, heterocycloalkyl or substituted heterocycloalkyl, aryl or substituted aryl, arylalkyl or substituted arylalkyl;
- R 2 is alky
- R 2 thiol, alkylthio or substituted alkylthio
- R 7 and R 7 are each independently H, alkyl or substituted alkyl, alkenyl or substituted alkenyl, cycloalkyl or substituted cycloalkyl, cycloalkenyl or substituted cycloalkenyl, heterocyclo or substituted heterocyclo, cycloalkylalkyl or substituted cycloalkylalkyl, cycloalkenylalkyl or substituted cycloalkenylalkyl, heterocycloalkyl or substituted heterocycloalkyl, aryl or substituted aryl, arylalkyl or substituted arylalkyl, halo, CN, OR 1 , nitro, hydroxylamine, hydroxylamide, amino, NHR 4 , NR 2 R 5 , NOR 1 , thiol, alkylthio or substituted alkylthio, R ⁇ O
- G-L- is not phenyl, 4-methoxyphenyl, 4-chlorophenyl, or certain (optionally substituted aryl)-(C r C 3 )-alkyl- groups (e.g., benzyl), when W and Y are -CH 2 -CH 2 -; and
- alkyl and alk refers to a straight or branched chain alkane
- hydrocarbon radical containing from 1 to 12 carbon atoms, preferably 1 to 6 carbon atoms.
- exemplary such groups include methyl, ethyl, propyl, isopropyl, n-butyl, t- butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, 4,4-dimethylpentyl, octyl, 2,2,4- trimethylpentyl, nonyl, decyl, undecyl, dodecyl, and the like.
- “Substituted alkyl” refers to an alkyl group substituted with one or more substituents, preferably 1 to 4 substituents, at any available point of attachment.
- Substituted alkenyl refers to an alkenyl group substituted with one or more substituents, preferably 1 to 4 substituents, at any available point of attachment.
- substituents include, but are not limited to, alkyl or substituted alkyl, as well as those groups recited above as exemplary alkyl substituents.
- alkynyl refers to a straight or branched chain hydrocarbon radical containing from 2 to 12 carbon atoms and at least one carbon to carbon triple bond.
- exemplary such groups include ethynyl
- Substituted alkynyl refers to an alkynyl group substituted with one or more substituents, preferably 1 to 4 substituents, at any available point of attachment.
- substituents include, but are not limited to, alkyl or substituted alkyl, as well as those groups recited above as exemplary alkyl substituents.
- cycloalkyl refers to a fully saturated cyclic hydrocarbon group containing from 1 to 4 rings and 3 to 8 carbons per ring. Exemplary such groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, etc.
- Substituted cycloalkyl refers to a cycloalkyl group substituted with one or more substituents, preferably 1 to 4 substituents, at any available point of attachment.
- substituents include, but are not limited to, nitro, cyano, alkyl or substituted alkyl, as well as those groups recited above as exemplary alkyl substituents, and as previously mentioned as preferred aryl substituents in the definition for G.
- substituents also include spiro-attached or fused cyclic substituents, especially cycloalkenyl or substituted cycloalkenyl.
- cycloalkenyl refers to a partially unsaturated cyclic hydrocarbon group containing 1 to 4 rings and 3 to 8 carbons per ring. Exemplary such groups include cyclobutenyl, cyclopentenyl, cyclohexenyl, etc.
- Substituted cycloalkenyl refers to a cycloalkenyl group substituted with one more substituents, preferably 1 to 4 substituents, at any available point of attachment.
- substituents include but are not limited to nitro, cyano, alkyl or substituted alkyl, as well as those groups recited above as exemplary alkyl substituents, and as previously mentioned as preferred aryl substituents in the definition for G.
- Exemplary substituents also include spiro-attached or fused cyclic substituents, especially cycloalkyl or substituted cycloalkyl.
- alkoxy or “alkylthio” refer to an alkyl group as described above bonded through an oxygen linkage (-O-) or a sulfur linkage (-S-), respectively.
- substituted alkoxy or “substituted alkylthio” refer to a substituted alkyl group as described above bonded through an oxygen or sulfur linkage, respectively.
- alkoxycarbonyl refers to an alkoxy group bonded through a carbonyl group.
- alkylcarbonyl refers to an alkyl group bonded through a carbonyl group.
- alkylcarbonyloxy refers to an alkylcarbonyl group bonded through an oxygen linkage.
- arylalkyl refers to aryl, cycloalkyl, cycloalkenylalkyl, “substituted cycloalkenylalkyl”, “heterocycloalkyl” and “substituted heterocycloalkyl” refer to aryl, cycloalkyl, cycloalkenyl and heterocyclo groups bonded through an alkyl group, substituted on the aryl, cycloalkyl, cycloalkenyl or heterocyclo and/or the alkyl group where indicated as “substituted.”
- aryl refers to cyclic, aromatic hydrocarbon groups which have 1 to 5 aromatic rings, especially monocyclic or bicyclic groups such as phenyl, biphenyl or naphthyl. Where containing two or more aromatic rings (bicyclic, etc.), the aromatic rings of the aryl group may be joined at a single point (e.g., biphenyl), or fused (e.g., naphthyl, phenanthrenyl and the like).
- “Substituted aryl” refers to an aryl group substituted by one or more substituents, preferably 1 to 3 substituents, at any point of attachment. Exemplary substituents include, but are not limited to, nitro, cycloalkyl or substituted cycloalkyl, cycloalkenyl or substituted cycloalkenyl, cyano, alkyl-
- exemplary substituents also include fused cyclic substituents, such as heterocyclo or cycloalkenyl, or substituted heterocyclo or cycloalkenyl, groups.
- Carbamoyl refers to the group -CONH- which is bonded on one end to the remainder of the molecule and on the other to hydrogen or an organic moiety (such as alkyl, substituted alkyl, aryl, substituted aryl, heterocycle, alkylcarbonyl, hydroxyl and substituted nitrogen).
- Carbamoyl refers to the group -CONH- which is bonded on one end to the remainder of the molecule and on the other to hydrogen or an organic moiety (such as alkyl, substituted alkyl, aryl, substituted aryl, heterocycle, alkylcarbonyl, hydroxyl and substituted nitrogen).
- “Carbamate” refers to the group -O-CO-NH- which is bonded on one end to the remainder of the molecule and on the other to hydrogen or an organic moiety (such as those listed above).
- “Urea” refers to the group -NH-CO- NH- which is bonded on one end to the remainder of the molecule and on
- “Substituted carbamoyl,” “substituted carbamate,” “substituted urea” and “substituted amidinyl” refer to carbamoyl, carbamate, urea or amidinyl groups as described above in which one more of the hydrogen groups are replaced by an organic moiety (such as those listed above).
- heterocycle refers to fully saturated, or partially or fully unsaturated, including aromatic (i.e., “heteroaryl”) cyclic groups (for example, 4 to 7 membered monocyclic, 7 to 11 membered bicyclic, or 10 to 16 membered tricyclic ring systems) which have at least one heteroatom in at least one carbon atom-containing ring.
- aromatic i.e., "heteroaryl”
- cyclic groups for example, 4 to 7 membered monocyclic, 7 to 11 membered bicyclic, or 10 to 16 membered tricyclic ring systems
- Each ring of the heterocyclic group containing a heteroatom may have 1, 2, 3, or 4 heteroatoms selected from nitrogen atoms, oxygen atoms and/or sulfur atoms, where the nitrogen and sulfur heteroatoms may optionally be oxidized and the nitrogen heteroatoms may optionally be quaternized.
- heteroarylium refers to a heteroaryl group bearing a quaternary nitrogen atom and thus a positive charge.
- the heterocyclic group may be attached to the remainder of the molecule at any heteroatom or carbon atom of the ring or ring system.
- Exemplary monocyclic heterocyclic groups include azetidinyl, pyrrolidinyl, pyrrolyl, pyrazolyl, oxetanyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, oxazolyl, oxazolidinyl, isoxazolinyl, isoxazolyl, thiazolyl, thiadiazolyl, thiazolidinyl, isothiazolyl, isothiazolidinyl, furyl, tetrahydrofuryl, thienyl, oxadiazolyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolodinyl, 2-oxoazepinyl, azepinyl, hexahydrodiazepinyl, 4-piperidonyl, pyridy
- bicyclic heterocyclic groups include indolyl, isoindolyl, benzothiazolyl, benzoxazolyl, benzoxadiazolyl, benzothienyl, quinuclidinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuryl, benzofurazanyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl (such as furo[2,3-c]pyridinyl, furo[3,2-b]pyridinyl] or furo[2,3- b]pyridinyl), dihydroisoindolyl, dihydroquinazolinyl (such as 3,4-dihydr
- Substituted heterocycle refers to heterocycle, heterocyclic or heterocyclo groups substituted with one or more substituents, preferably 1 to 4 substituents, at any available point of attachment.
- quaternary nitrogen refers to a tetravalent positively charged nitrogen atom including, for example, the positively charged nitrogen in a tetraalkylammonium group (e.g., tetramethylammonium, N-methylpyridinium), the positively charged nitrogen in protonated ammonium species (e.g., trimethyl- hydroammonium, N-hydropyridinium), the positively charged nitrogen in amine N- oxides (e.g., N-methyl-morpholine-N-oxide, pyridine-N-oxide), and the positively charged nitrogen in an N-amino-ammonium group (e.g., N-aminopyridinium).
- a tetraalkylammonium group e.g., tetramethylammonium, N-methylpyridinium
- protonated ammonium species e.g., trimethyl- hydroammonium, N-hydropyridinium
- the positively charged nitrogen in amine N- oxides
- halogen or halo refer to chlorine, bromine, fluorine or iodine.
- hydroxylamine and “hydroxylamide” refer to the groups OH-NH- and OH-NH-CO-, respectively.
- protecting groups for the methods and compounds described herein include, without limitation, those described in standard textbooks, such as Greene, T. W. et al, Protective Groups in Organic Synthesis, Wiley, N.Y. (1991).
- (CRR)n When a term such as “(CRR)n” is used, it denotes an optionally substituted alkyl chain existing between the two fragments to which it is bonded, the length of which chain is defined by the range described for the term n.
- n 0-3, implying from zero to three (CRR) units existing between the two fragments, which are attached to the primary and terminal (CRR) units.
- CRR zero to three
- any heteroatom with unsatisfied valences is assumed to have hydrogen atoms sufficient to satisfy the valences.
- Divalent groups such as those in the definition of W (e.g., NR 9 -CR 7 R 7' ), may be bonded in either direction to the remainder of the molecule (e.g, -A r NR 9 -CR 7 R 7' -A 2 -or, -A r CR 7 R 7' -NR 9 -A 2 - for the aforementioned group within
- Carboxylate anion refers to a negatively charged group -COO-.
- the compounds of formula I form salts which are also within the scope of this invention. Reference to a compound of the formula I herein is understood to include reference to salts thereof, unless otherwise indicated.
- the term "salt(s)", as employed herein, denotes acidic and/or basic salts formed with inorganic and/or organic acids and bases. .
- zwitterions inner salts
- Salts of the compounds of the formula I may be formed, for example, by reacting a compound I with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- the compounds of formula I which contain a basic moiety may form salts with a variety of organic and inorganic acids.
- Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example, trifluoroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclopentanepropionates, digluconates, dodecylsulfates, ethanesulfonates, fumarates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, hydrochlorides, hydrobromides, hydroiodides, hydroxyethane
- the compounds of formula I which contain an acidic moiety may form salts with a variety of organic and inorganic bases.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as benzathines, dicyclohexylamines, hydrabamines (formed with N,N- bis(dehydroabietyl)ethylenediamine), N-methyl-D-glucamines, N-methyl-D- glycamides, t-butyl amines, and salts with amino acids such as arginine, lysine and the like.
- Basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g. methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g. dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g. decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g. benzyl and phenethyl bromides), and others.
- lower alkyl halides e.g. methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfates e.g. dimethyl, diethyl, dibutyl, and diamyl sulfates
- Prodrugs and solvates of the compounds of the invention are also contemplated herein.
- the term "prodrug” as employed herein denotes a compound which, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield a compound of the formula I, or a salt and/or solvate thereof.
- Solvates of the compounds of formula I include, for example, hydrates.
- All stereoisomers of the present compounds are contemplated within the scope of this invention.
- Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers (e.g., as a pure or substantially pure optical isomer having a specified activity), or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers.
- the chiral centers of the present invention may have the S or R configuration as defined by the IUPAC 1974 Recommendations.
- racemic forms can be resolved by physical methods, such as, for example, fractional crystallization, separation or crystallization of diastereomeric derivatives or separation by chiral column chromatography.
- the individual optical isomers can be obtained from the racemates by any suitable method, including without limitation, conventional methods, such as, for example, salt formation with an optically active acid followed by crystallization.
- the compounds of the present invention may be prepared by methods such as those illustrated in the following Schemes I to XV. Solvents, temperatures, pressures, and other reaction conditions may readily be selected by one of ordinary skill in the art. Starting materials are commercially available or readily prepared by one of ordinary skill in the art or prepared by methods illustrated in Figures 1 to 3.
- Combinatorial techniques may be employed in the preparation of compounds, for example, where the intermediates possess groups suitable for these techniques. See the following for alternative methods which may be employed in the preparation of compounds of the present invention: Tetrahedron, 27, 3119 (1971); Tetrahedron, 30, 2977 (1974); Tetrahedron. Let, 31, 2631 (1969); J. Org. Chem., 35, 3097 (1970); Bull. Chem. Soc. Jpn., 67, 3082 (1994); Bull. Chem. Soc. Jpn., 65, 61 (1992); European Patent (EP) No. 406119; U.S. Patent No. 4,397,857; Pons et al, Eur. J. Org.
- Z 2 S, O, NH, NR 6
- compounds of formula I can be obtained from azabicyclo-3-ethylcarboxylate intermediates of formula II.
- Intermediates of formula II can be prepared, for example, from the synthetic approaches described in Bull Chem. Soc. Jpn., 65, 61 (1992), Tetrahedron Let. 31, 2603 (1990), Chem. Commun. 597 (1999), Tetrahedron Lett. 38, 4021, (1997), Tetrahedron Lett. 40, 7929 (1999), Synlett. 1, 29 (1991), J. Chem. Soc, Chem. Commun. 1601 (1988), J. Org. Chem. 31, 1059 (1966), Synthesis 10, 925 (1990), Tetrahedron Lett.
- the individual optical isomers of a compound of Formula IV can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- Scheme II describes a method for preparing compounds of formula I wherein an intermediate of formula II is treated with a phosgene like reagent of formula Cl-E-Cl in the presence of a base, such as NaHCO 3 , to yield an intermediate of formula V.
- a base such as NaHCO 3
- the phosgene like intermediates of formula Cl-E-Cl can be obtained from commercially available sources or can readily be prepared by one skilled in the art.
- the intermediate of formula V can be reacted with an amine of formula H 2 N-L- G in the presence of a base, such as diisopropylamine or triethylamine, with or without a coupling reagent, such as DMAP, to give an intermediate of formula VI.
- a base such as diisopropylamine or triethylamine
- DMAP a coupling reagent
- the amine intermediates of formula H 2 N-L-G can be obtained from commercially available sources or can readily be prepared by one skilled in the art.
- the intermediate of formula VI can be converted to a compound of formula VII by heating with or without the presence of a base, such as DBU or triethylamine.
- the individual antipodes of a compound of formula VII can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula VII can be obtained, for example, by separation of a resulting mixture by standard techniques.
- Scheme III describes a method for preparing compounds of formula I wherein an intermediate of formula II is saponified to an acid of formula VIII by treatment with a base, such as sodium hydroxide.
- the acid can then by coupled to an amine of formula H 2 N-L-G via a variety of coupling reagents, for example, as described in The Practice of Peptide Synthesis, Springer- Verlag, 2 nd Ed., Bodanszy, Miklos, 1993 (incorporated herein by reference in its entirety), to yield an amide intermediate of formula IX.
- the individual antipodes of a compound of formula VII can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula VII can be obtained, for example, by separation of a resulting mixture by standard techniques.
- WSCD water soluble coupling reagent
- the substituted cyano-thioureas of formula NC-NH-C(S)-NH- " L-G can be obtained from commercially available sources or can readily be prepared by one skilled in the art.
- An intermediate of formula X can be heated with or without the presence of a base, such as DBU, to yield a compound of formula XI, which is a compound of formula I where, in addition to E being C-N-CN, M and M' are each a bond.
- a base such as DBU
- the individual antipodes of a compound of formula XI can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula XI can be obtained, for example, by separation of a resulting mixture by standard techniques.
- a compound of formula I where Q is equal to substituents as defined herein other than H
- a base such as LDA and an alkyl halide such as methyl iodide
- a solvent such as tetrahydrofuran at low temperatures (e.g., -78°C)
- the individual antipodes of a compound of formula IV can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula XII or by separation of the racemic mixture by standard techniques.
- the individual or ⁇ isomers of a compound of formula IV can be obtained, for example, by use of the corresponding individual endo or exo isomers of a compound of formula XII or by separation of a resulting mixture by standard techniques.
- Z 2 S, O, NH, NR e
- compounds of formula I can be synthesized by means of a solid support route.
- the above synthetic route allows for the synthesis of combinatorial libraries of compounds of formula I via, for example, standard procedures of automated solid phase synthesis.
- a protecting agent such as di-tertbutylcarbonate
- a base such as sodium hydroxide
- the intermediate of formula XIII can be attached to a solid support, such as a modified Merrifield resin, by treatment with a coupling reagent such as 2,6-dichloro-benzoyl chloride in the presence of pyridine .
- the individual antipodes of a compound of formula IV can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula IV can be obtained, for example, by separation of a resulting mixture by standard techniques.
- Scheme VII shows an alternate approach to the synthesis of compounds of formula I on solid support.
- an intermediate of formula XV can readily be synthesized.
- the intermediate of formula XV can be treated, with or without the presence of a base such as triethylamine or NaHCO 3 , with a phosgene like reagent of formula Cl-E-Cl, to yield an intermediate of formula XVII.
- the intermediate of formula XVII can be reacted with an amine of formula H 2 N-L-G in the presence of a base, such as diisopropylamine, with or without a coupling reagent, such as 4-dimethylamino pyridine, to give an intermediate of formula XVIII.
- the final product VII can be formed and liberated from the solid support by heating the intermediate of formula XVIII with or without a base, such as DBU.
- the individual antipodes of a compound of formula VII can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual or ⁇ isomers of a compound of formula VII can be obtained, for example, by separation of a resulting mixture by standard techniques.
- an intermediate of formula VI can be readily synthesized.
- Scheme VIII treatment of an intermediate of formula VI with a substituted O-diphenylphosphinylhydroxylamine of formula Ph 2 POONH-R 10 , and potassium hydride as described in Synthesis, 7, 592 (1982) and Tetrahedron Let., 29, 1777 (1988) (both incorporated herein by reference in their entirety), yields an intermediate of formula XXII.
- the individual antipodes of a compound of formula XXIII can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula XXIII can be obtained, for example, by separation of a resulting mixture by standard techniques.
- an intermediate of formula XIII can be readily synthesized.
- the acid intermediate of formula XIII can be coupled to an amine of formula H 2 N-L-G via use of a variety of coupling reagents, as described in Scheme III, to yield an amide intermediate of formula XXIV.
- Treatment of the intermediate of formula XXIV with potassium hydride and a substituted O- diphenylphosphinylhydroxylamine of formula Ph 2 POONH-R 10 as described in
- Scheme VIII followed by removal of the BOC protecting group by treatment with an acid, such as trifluoroacetic acid, yields an intermediate of formula XXV.
- the individual antipodes of a compound of formula XXVI can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula XIII or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula XXVI can be obtained, for example, by separation of a resulting mixture by standard techniques.
- an intermediate of formula XXJV can be readily synthesized.
- treatment of an intermediate of formula XXIV with agents suitable for forming a hydroxylamide moiety such as TMS-C1 followed by MoO 5 (DMF) 2 as described inJ. Org. Chem., 54, 5852 (1989) and J. Org. Chem., 59, 8065 (1994) (both incorporated herein by reference in their entirety), and for deprotection of a BOC group, such as ethanol saturated with HCl gas, results in the generation of a hydroxylamide intermediate of formula XXVII.
- the individual antipodes of a compound of formula XXVIII can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula XIII or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula XXVIII can be obtained, for example, by separation of a resulting mixture by standard techniques.
- the individual antipodes of a compound of formula VII can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula VII can be obtained, for example, by use of the corresponding individual endo or exo isomers of a compound of formula II or by separation of a resulting mixture by standard techniques.
- a compound of formula IX can readily be made by the process described.
- XXXIV which is a compound of formula I where M and M' are each a bond and E is CHR 7 .
- the individual antipodes of a compound of formula XXXIV can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula XXXTV can be obtained by separation of a resulting mixture by standard techniques.
- the individual antipodes of a compound of formula XXXV can be obtained, for example, by use of the corresponding individual antipodes of a compound of formula II or by separation of the racemic mixture by standard techniques.
- the individual ⁇ or ⁇ isomers of a compound of formula XXXV can be obtained by separation of a resulting mixture by standard techniques.
- Scheme XIV describes a method for preparing compounds of formula I wherein an intermediate of formula II (where Z, is O) is saponified to an acid of formula VIII by treatment with a base, such as sodium hydroxide.
- the acid can then by coupled to an amine of formula H 2 N-L-G via a variety of coupling reagents, for example, as described in The Practice of Peptide Synthesis, Springer- Verlag, 2 nd Ed., Bodanszy, Miklos, 1993 (incorporated herein by reference in its entirety), to yield an amide intermediate of formula IX.
- the condensation can be conducted, for example, in the presence of a base by heating the mixture of IX and R ⁇ '-C- 'X 2 in an inert solvent.
- Preferred dihalides of formula R 7 R 7 -C-X'X 2 are ethyl bromofluoroacetate and ethyl bromodifluoroacetate.
- suitable bases include alkali salts of carbonate, such as potassium, sodium and lithium, and hydride bases such as sodium hydride.
- inert solvents examples include ethers such as diethyl ether, tetrahydrofuran and dioxane; esters such as ethyl acetate; amides such as dimethylformamide; and acetonitrile.
- ethers such as diethyl ether, tetrahydrofuran and dioxane
- esters such as ethyl acetate
- amides such as dimethylformamide
- acetonitrile examples include ethers such as diethyl ether, tetrahydrofuran and dioxane
- esters such as ethyl acetate
- amides such as dimethylformamide
- acetonitrile examples include acetonitrile.
- compounds of formula I where Z, is O, M and M' are bonds and E is CR 7 R 7' can be prepared by transforming the imidazolinones of formula XXVII.
- the ester of formula XXXVII is hydrolyzed, for example, with sodium hydroxide in a solvent such as methanol or ethanol at about 0°C to 50°C to provide the corresponding carboxylic acid.
- a base such as potassium carbonate
- an inert solvent such as dimethylformamide
- DCC 1,3-dicyclohexylcarbodiimide
- R 13 is alkyl (e.g., C,-C 6 alkyl) or substituted alkyl; alkenyl (e.g., C,-C 6 alkenyl) or substituted alkenyl; cycloalkyl (e.g., C 3 -C 6 cycloalkyl) or substituted cycloalkyl; heterocycloalkyl or substituted heterocycloalkyl; aryl or substituted aryl (e.g., substituted by alkyl and additional substituents); heterocyclo or substituted heterocyclo (e.g., heteroaryl or substituted heteroaryl, such as heteroaryl substituted by alkyl and additional substituents), in the presence of a base such as potassium carbonate, in an inert solvent such as acetonitrile, produces compounds of formula XXXVIII, wherein R 7' CH 2 OR 13 . Other R 7' substitutions are also obtainable from the CO 2 Et
- Scheme XVI describes another approach to incorporating additional substitution onto a compound of formula I.
- a compound of formula XXXIX which can be prepared in accordance with the above Schemes, can be incubated in the presence of a suitable enzyme or microorganism resulting in the formation of a hydroxylated analog of formula XL.
- a suitable enzyme or microorganism resulting in the formation of a hydroxylated analog of formula XL.
- Such a process can be employed to yield regiospecific as well as enantiospecific incorporation of a hydroxyl group into a molecule of formula XXXIX by a specific microorganism or by a series of different microorganisms.
- Compound XL is a compound of formula I where Y is as described above and A, and A 2 are preferably CR 7 .
- Scheme XVII describes another approach to incorporating additional substitution onto a compound of formula I.
- a compound of formula XLI which can be prepared in accordance with the above Schemes, can be incubated in the presence of a suitable enzyme or microorganism resulting in the formation of a diol analog of formula XLII.
- a suitable enzyme or microorganism resulting in the formation of a diol analog of formula XLII.
- Such a process can be employed to yield regiospecific as well as enantiospecific transformation of a compound of formula XLI to a 1-2 diol of formula XLII by a specific microorganism or by a series of different microorganisms.
- Such microorganisms can, for example, be bacterial, yeast or fungal in nature and can be obtained from distributors such as ATCC or identified for use in this method such as by methods known to one skilled in the art.
- Compound XLII is a compound of formula I where Y is as described above and A ! and A 2 are preferably CR 7 .
- the present invention also provides the methods of Schemes XVI and XVII.
- the present invention provides a method for preparation of a compound of the following formula XL, or salt thereof:
- the present invention provides a method for preparation of a compound of the following formula XLII, or salt thereof:
- Conversion of one isomer selectively when contacting an isomeric mixture is a preferred embodiment of the invention.
- Conversion to one isomer selectively e.g., hydroxylation on the exo face "exo isomer” preferentially to the endo face "endo isomer” or regioselective opening of an epoxide to form only one of two possible regioisomers of a trans diol
- Hydroxylation of an achiral intermediate to form a single optical isomer of the hydroxylated product is also a preferred embodiment of the invention.
- Resolution of a recemic mixture of an intermediate by selective hydroxylation, or epoxide ring opening and diol formation, to generate one of the two possible optical isomers is also a preferred embodiment of the invention.
- resolution denotes partial, as well as, preferably, complete resolution.
- enzyme process denotes a process or method of the present invention employing an enzyme or microorganism.
- hydroxylation denotes the addition of a hydroxyl group to a methylene group as described above. Hydroxylation can be achieved, for example, by contact with molecular oxygen according to the methods of the present invention. Diol formation can be achieved, for example, by contact with water according to the methods of the present invention.
- Use of "an enzyme or microorganism” in the present methods includes use of two or more, as well as a single, enzyme or microorganism.
- the enzyme or microorganism employed in the present invention can be any enzyme or microorganism capable of catalyzing the enzymatic conversions described herein.
- the enyzmatic or microbial materials regardless of origin or purity, can be employed in the free state or immobilized on a support such as by physical adsorption or entrapment.
- Microorganisms or enzymes suitable for use in the present invention can be selected by screening for the desired activity, for example, by contacting a candidate microorganism or enzyme with a starting compound XXXIX or XLI or salt thereof, and noting conversion to the corresponding compound XL or XLII or salt thereof.
- the enzyme may, for example, be in the form of animal or plant enzymes or mixtures thereof, cells of microorganisms, cmshed cells, extracts of cells, or of synthetic origin.
- Exemplary microorganisms include those within the genera: Streptomyces or Amycolatopsis. Particularly preferred microorganisms are those within the species Streptomyces griseus, especially Streptomyces griseus ATCC 10137, and Amycolatopsis orientalis such as ATCC 14930, ATCC 21425, ATCC 35165, ATCC 39444, ATCC 43333, ATCC 43490, ATCC 53550, ATCC 53630, and especially ATCC 43491.
- ATCC refers to the accession number of the American Type Culture Collection, 10801 University Boulevard., Manassas Virginia
- mutants of these organisms are also contemplated by the present invention, for use in the methods described herein, such as those modified by the use of chemical, physical (for example, X-rays) or biological means (for example, by molecular biology techniques).
- Preferred enzymes include those derived from microorganisms, particularly those microorganisms described above. Enzymes may be isolated, for example, by extraction and purification methods such as by methods known to those of ordinary skill in the art. An enzyme may, for example, be used in its free state or in immobilized form.
- a suitable carrier e.g., diatomaceous earth (porous Celite Hyflo Supercel), microporous polypropylene (Enka Accurel® polypropylene powder), or a nonionic polymeric adsorbent such as Amberlite® XAD-2 (polystyrene) or XAD-7 (polyacrylate) from Rohm and Haas Co.
- a carrier When employed to immobilize an enzyme, a carrier may control the enzyme particle size and prevent aggregation of the enzyme particles when used in an organic solvent. Immobilization can be accomplished, for example, by precipitating an aqueous solution of the enzyme with cold acetone in the presence of the Celite Hyflo Supercel followed by vacuum drying, or in the case of a nonionic polymeric adsorbent, incubating enzyme solutions with adsorbent on a shaker, removing excess solution and drying enzyme-adsorbent resins under vacuum. While it is desirable to use the least amount of enzyme possible, the amount of enzyme required will vary depending upon the specific activity of the enzyme used. Hydroxylation as described above can occur in vivo.
- liver enzyme can selectively, relative to the endo isomer, hydroxylate the exo isomer of a compound of the present invention.
- liver microsomal hydroxylase can be employed as the enzyme for catalysis.
- These processes may also be carried out using microbial cells containing an enzyme having the ability to catalyze the conversions.
- microbial cells containing an enzyme having the ability to catalyze the conversions.
- these procedures are conveniently carried out by adding the cells and the starting material to the desired reaction medium.
- the cells may be used in the form of intact wet cells or dried cells such as lyophilized, spray-dried or heat-dried cells, or in the form of treated cell material such as ruptured cells or cell extracts.
- the host cell may be any cell, e.g. Escherichia coli, modified to contain a gene or genes for expressing one or more enzymes capable of catalysis as described herein.
- the enzymatic methods of the present invention may be carried out subsequent to the fermentation of the microorganism (two-stage fermentation and conversion), or concurrently therewith, that is, in the latter case, by in situ fermentation and conversion (single-stage fermentation and conversion).
- microorganisms can be achieved by one of ordinary skill in the art by the use of an appropriate medium.
- Appropriate media for growing microorganisms include those which provide nutrients necessary for the growth of the microbial cells.
- a typical medixim for growth includes necessary carbon sources, nitrogen sources, and elements (e.g. in trace amounts). Inducers may also be added.
- the term "inducer”, as used herein, includes any compound enhancing formation of the desired enzymatic activity within the microbial cell.
- Carbon sources can include sugars such as maltose, lactose, glucose, fructose, glycerol, sorbitol, sucrose, starch, mannitol, propylene glycol, and the like; organic acids such as sodium acetate, sodium citrate, and the like; and alcohols such as ethanol, propanol and the like.
- sugars such as maltose, lactose, glucose, fructose, glycerol, sorbitol, sucrose, starch, mannitol, propylene glycol, and the like
- organic acids such as sodium acetate, sodium citrate, and the like
- alcohols such as ethanol, propanol and the like.
- Nitrogen sources can include N-Z amine A, corn steep liquor, soy bean meal, beef extracts, yeast extracts, molasses, baker's yeast, tryptone, nutrisoy, peptone, yeastamin, amino acids such as sodium glutamate and the like, sodixxm nitrate, ammonium sulfate and the like.
- Trace elements can include magnesium, manganese, calcium, cobalt, nickel, iron, sodium and potassium salts. Phosphates may also be added in trace or, preferably, greater than trace amounts.
- the medium employed can include more than one carbon or nitrogen source or other nutrient.
- Preferred media for growth include aqueous media.
- the agitation and aeration of the reaction mixture affects the amount of oxygen available during the conversion process when conducted, for example, in shake-flask cultures or fermentor tanks during growth of microorganisms.
- Incubation of the reaction medium is preferably at a temperature between about 4 and about 60°C.
- the reaction time can be appropriately varied depending upon the amount of enzyme used and its specific activity. Reaction times may be reduced by increasing the reaction temperature and/or increasing the amount of enzyme added to the reaction solution.
- an aqueous liquid as the reaction medium, • although an organic liquid, or a miscible or immiscible (biphasic) organic/aqueous liquid mixture, may also be employed.
- the amount of enzyme or microorganism employed relative to the starting material is selected to allow catalysis of the enzymatic conversions of the present invention.
- Solvents for the organic phase of a biphasic solvent system may be any organic solvent immiscible in water, such as toluene, cyclohexane, xylene, trichlorotrifluoroethane and the like.
- the aqueous phase is conveniently of water, preferably deionized water, or a suitable aqueous buffer solution, especially a phosphate buffer solution.
- the biphasic solvent system preferably comprises between about 10 to 90 percent by volume of organic phase and between about 90 to 10 percent by volume of aqueous phase, and most preferably contains at or about 20 percent by volume of organic phase and at or about 80 percent by volume of the aqueous phase.
- An exemplary embodiment of such processes starts with preparation of an aqueous solution of the enzyme(s) or microbes to be used.
- the preferred enzyme(s) or microbes can be added to a suitable amount of an aqueous solvent, such as phosphate buffer or the like. This mixture is preferably adjusted to and maintained at a desired pH.
- the compounds XL and XLII produced by the processes of the present invention can be isolated and purified, for example, by methods such as extraction, distillation, crystallization, and column chromatography.
- Such a cyclization can be enhanced by the addition of metal salts, such as but not limited to Ytterbium (III) trifluoromethanesulfonate, as described in the documents cited previously.
- An intermediate of formula II can be made where Q ⁇ H, by protection of the secondary nitrogen with a protection group such as a BOC, followed by treatment with reactive intermediates of formula Q-X, where X represents a leaving group or X is an electrophilic center which can react to ultimately make up the definition of Q as described earlier, in the presence of base, such as LDA, or a coupling agent as is readily known by one skilled in the art, followed by deprotection of the BOC group with an acid such as saturated ethanolic HCl.
- a protection group such as a BOC
- reactive intermediates of formula Q-X where X represents a leaving group or X is an electrophilic center which can react to ultimately make up the definition of Q as described earlier, in the presence of base, such as LDA, or a coupling agent as is readily known by one skilled in the art, followed by deprotection of the BOC group with an acid such as saturated ethanolic HCl.
- the commercially available chiral (pure D or L) intermediate N-(tert-butoxycarbonyl)-L- 4-hydroxyproline, B can be treated with a reducing agent, such as BH 3 «THF, to yield a primary alcohol, which can then be selectively protected with an agent such as TBSOTf, in the presence of base (e.g., 2,6-lutidine), to yield the intermediate alcohol C.
- a reducing agent such as BH 3 «THF
- the secondary alcohol of C can then be differentially protected by treatment with an agent such as TsCl, in.the presence of a base (e.g., pyridine), followed by deprotection of the primary alcohol (which can be achieved by treatment with an acid, such as para-toluenesulphonic acid), to yield intermediate alcohol D.
- a base e.g., pyridine
- the resulting alcohol D can be oxidized, such as under standard Swem conditions, to yield the corresponding aldehyde intermediate E.
- the aldehyde intermediate E can be directly treated with benzylamine and diethyl cyanophosphonate to give intermediate F.
- the intermediate of formula H can be treated with reactive intermediates of formula Q-X, where X represents a leaving group or X is an electrophilic center which can react to ultimately make up the definition of Q as described earlier, in the presence of base, such as LDA, or a coupling agent as is readily known by one skilled in the art, which, after treatment with an agent such as palladium on charcoal, yields an intermediate of formula Ila where Q ⁇ H.
- the various intermediates of Figure 2 can be purified, for example, by silica purification, or can, for example, be simply carried forward in situ to the next step (e.g., converting D to F without isolating E).
- the following method is novel as are the individual steps and intermediates produced therein (e.g., E, F, G, H, J and Ila): a method for the preparation of a compound of the following formula Ila:
- BOC is t-butoxycarbonyl
- the method of Figure 2 is especially useful for the preparation of unnatural amino acids Ila which can be employed, by methods analogous to those using compounds of the formula II, in the preparation of the present compounds of formula I.
- the activated i ine intermediate M can be generated by the reactions of an activated sulfonyl isocyanate, such as p-toluenesulfonyl isocyanate, with ethyl glyoxylate and heating.
- Imine M can undergo cyclization with an appropriate diene intermediate of formula A to give an intermediate of formula II'.
- metal salts such as but not limited to Ytterbium (III) trifluoromethanesulfonate, as described in the references cited previously.
- the tosyl protecting group can be removed from intermediate II' by a number of reagents, such as those known to one skilled in the art, such as hydrogen bromide in acetic acid, to yield an intermediate of formula II.
- the intermediate of formula II' can be treated with reactive intermediates of formula Q-X, where X represents a leaving group or X is an electrophilic center which can react to ultimately make up the definition of Q as described earlier, in the presence of base, such as LDA, or a coupling agent as is readily known by one skilled in the art, to yield the intermediate of formula T.
- the tosyl protecting group can be removed from intermediate T by a number of reagents known to one skilled in the art, such as hydrogen bromide in acetic acid, to yield an intermediate of formula II, where Q ⁇ H.
- a preferred subgenus of the compounds of the present invention includes compounds of the formula I or salts thereof wherein one or more, preferably all, of the following substituents are as defined below:
- M is a bond or NR 10
- M' is a bond or NR 10 , with the proviso that at least one of M or M' must be a bond
- R 1 and R 1 are each independently H, alkyl, perfluoroalkyl, cycloalkyl, heterocyclo, cycloalkylalkyl, or heterocycloalkyl;
- R 2 is alkyl, perfluoroalkyl, cycloalkyl, heterocyclo, cycloalkylalkyl, or heterocycloalkyl;
- R 3 and R 3 are each independently H, alkyl, perfluoroalkyl, cycloalkyl, heterocyclo, cycloalkylalkyl, heterocycloalkyl, CI, F, Br, I, CN, alkoxy, amino, NR ] R 2 , thiol, or alkylthio;
- R 1 NHC O, SO 2 OR ⁇ or SO.NR'R 1' ;
- R'OO, R 1 NHC O, SO 2 OR', or SO ⁇ R'R 1' ;
- R 9 and R 9 are each independently H, alkyl, alkenyl, cycloalkyl, heterocyclo, cycloalkylalkyl, heterocycloalkyl
- a more preferred subgenus of the compounds of the invention includes compounds of the formula I or salts thereof wherein one or more, preferably all, of the following substituents are as defined below:
- G is an aryl or heteroaryl group, where said group is mono- or polycyclic, and which is optionally substituted at one or more positions with hydrogen, C r C 3 alkyl, allyl or substituted allyl, alkynyl, CI, F, Br, I, CN, R'C-O, R'HNGO,
- R 1 R 2 NC O, haloalkyl (especially, perfluoroalkyl), C r C 3 hydroxyalkyl, HOCR 3 R 3' , nitro, R ] OCH 2 , R 1 ⁇ , NR 4 R 5 , or SR 1 ;
- Z is O, S, or NCN;
- Z 2 is O, S, or NCN;
- a ! is CR 7 (especially, CH);
- a 2 is CR 7 (especially, CH);
- R 1 and R 1' are each independently H, alkyl, cycloalkyl, heterocycloalkyl, or perfluoroalkyl;
- R 2 is alkyl, cycloalkyl, heterocycloalkyl, or perfluoroalkyl;
- R 3 and R 3' are each independently H, alkyl, perfluoroalkyl, CI, F, Br, I, CN, alkoxy, amino, NR ⁇ R 2 , thiol, or alkylthio;
- R 4 is H, alkyl, cycloalkyl, heterocycloalkyl, R ⁇ O, R'NHC ), SOjOR 1 , or
- Z 2 is O or CN;
- A is CR 7 (especially, CH);
- a 2 is CR 7 (especially, CH);
- M is a bond and M' is a bond;
- L is a bond;
- R 1 and R 1 are each independently H, alkyl, or perfluoroalkyl
- R 2 is alkyl, or perfluoroalkyl
- R 3 and R 3 ' are each independently H, alkyl, perfluoroalkyl, CI, F, Br, I, CN, alkoxy, amino, N ⁇ R 2 , thiol, or alkylthio;
- a particularly preferred subgenus of the compounds of the invention includes compounds of the formula I or salts thereof wherein one or more, preferably all, of the substituents are as defined below:
- G is an aryl (especially, phenyl or naphthyl) or heterocyclo (especially benzo- fused heterocyclic groups such as indole, benzothiophene, benzothiazole, benzothiadiazole, benzisoxazole, benzoxadiazole, oxidobenzothiophene, benzofuran or benzopyran) group, where said group is mono- or polycyclic, and which is optionally substituted at one or more positions, such as 1 to 5 positions (preferably 1 to 2 positions), with substituents selected from one or more of hydrogen, NH 2> alkyl (especially having 1 to 4 carbons) or substituted alkyl (especially having 1 to 4 carbons and substituted with halo, such as the substituted alkyl group CF 3 ), halo (especially F, CI, Br or I), heterocyclo (such as tetrazole or oxazole), CN, nitro, SR 1 or R 1 O (especially where R 1 is alky
- Q is H, alkyl (especially having 1 to 4 carbons), alkenyl (especially having 1 to 4 carbon atoms), arylalkyl (especially benzyl) or substituted arylalkyl (especially substituted benzyl, such as halo-substituted benzyl);
- M is a bond or NH (especially a bond), and M' is a bond;
- L is a bond;
- R 1 and R 1' are each independently alkyl (especially having 1 to 4 carbons) or substituted alkyl (especially having 1 to 4 carbons and substituted with halo), heterocyclo (such as imidazole or isoxazole) or substituted heterocyclo (such as imidazole substituted with methyl), aryl
- aryl especially phenyl or substituted aryl (especially phenyl substituted with one or more of halo, nitro, halo-substituted alkyl such as CF 3 , or alkyl having 1 to 4 carbons), arylalkyl (especially benzyl or phenethyl) or substituted arylalkyl (especially substituted benzyl such as halo- and/or nitro-substituted benzyl); and
- G-L- can be, for example, an optionally substituted naphthyl or optionally substituted fused bicyclic heterocyclic group such as an optionally substituted benzo-fused heterocyclic group (e.g., bonded to the remainder of the molecule through the benzene portion), especially such group wherein the heterocyclic ring bonded to benzene has 5 members exemplified by benzoxazole, benzothiazole, benzothiadiazole, benzoxadiazole or benzothiophene, for example:
- X is halo (especially CI), OH, CN, NO 2 or
- U is O or S (where S can optionally be oxygenated, e.g., to SO);
- U 1 is CH 3 or CF 3 ; each U 2 is independently N, CH or CF;
- U 3 is N, O or S;
- each U 4 and U 5 together with the atoms to which they are bonded, form an optionally substituted 5-membered heterocyclic ring which can be partially unsaturated or aromatic and which contains 1 to 3 ring heteroatoms; each U 6 is independently CH or N; and
- R 9 and U 2 are as defined above, such as optionally substituted arylcarbonyl or optionally substituted aryloxycarbonyl, and X a is an aryl substituent, such as nitro.
- n 1 or 2;
- Q is H, methyl or ethyl
- each G a is independently CN, NO 2 , CF 3 , CI, Br, F, OCH 3 , SCH 3 , 1, CH 3 , C(O)-CH 3 or
- G b is CN, H, F, Br, NO 2 or " ⁇ ⁇
- R 1d is -CH 3 , or ⁇ CF3
- the compounds of the present invention modulate the function of nuclear hormone receptors (NHR), and include compounds which are, for example, agonists, partial agonists, antagonists or partial antagonists of the androgen receptor (AR), the estrogen receptor (ER), the progesterone receptor (PR), the glucocorticoid receptor (GR), the mineralocorticoid receptor (MR), the steroid and xenobiotic receptor (SXR), other steroid binding NHR, the Orphan receptors or other NHR.
- NHR nuclear hormone receptors
- AR androgen receptor
- ER estrogen receptor
- PR progesterone receptor
- GR glucocorticoid receptor
- MR mineralocorticoid receptor
- SXR steroid and xenobiotic receptor
- Modes includes, for example, activation (e.g., agonist activity such as selective androgen receptor agonist activity) or inhibition (e.g., antagonist activity).
- NHR-associated condition denotes a condition or disorder which can be treated by modulating the function of a NHR in a subject, wherein treatment comprises prevention (e.g., prophylatic treatment), partial alleviation or cure of the condition or disorder. Modulation may occur locally, for example, within certain tissues of the subject, or more extensively throughout a subject being treated for such a condition disorder.
- the compounds of the present invention are useful for the treatment of a variety of conditions and disorders including, but not limited to, those described following:
- Compounds of formula I can be applied as agonists, partial agonists, antagonists, or partial antagonists of the estrogen receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the estrogen receptor pathway.
- Applications of said compounds include but are not limited to: osteoporosis, hot flushes, vaginal dryness, prostate cancer, breast cancer, endometrial cancer, cancers expressing the estrogen receptor such as the aforementioned cancers and others, contraception, pregnancy termination, menopause, amennoreahea, and dysmennoreahea.
- Compounds of formula I can be applied as agonists, partial agonists, antagonists or partial antagonists of the progesterone receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the progesterone receptor pathway.
- Applications of said compounds include but are not limited to: breast cancer, other cancers containing the progesterone receptor, endometriosis, cachexia, contraception, menopause, cyclesynchrony, meniginoma, dysmennoreahea, fibroids, pregnancy termination, labor induction and osteoporosis.
- Compounds of formula I can be applied as agonists, partial agonists, antagonists or partial antagonists of the glucocorticoid receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the glucocorticoid receptor pathway.
- Applications of said compounds include but are not limited to: inflammatory diseases, autoimmune diseases, prostate cancer, breast cancer, Alzheimer's disease, psychotic disorders, drug dependence, non-insulin dependent Diabetes Mellitus, and as dopamine receptor blocking agents or otherwise as agents for the treatment of dopamine receptor mediated disorders.
- Compounds of formula I can be applied as agonists, partial agonists, antagonists or partial antagonists of the mineralocorticoid receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the mineralocorticoid receptor pathway.
- Applications of said compounds include but are not limited to: drug withdrawal syndrome and inflammatory diseases.
- Compounds of formula I can be applied as agonists, partial agonists, antagonists or partial antagonists of the aldosterone receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the aldosterone receptor pathway.
- One application of said compounds includes but is not limited to: congestive heart failure.
- Compounds of formula I can be applied as agonists, partial agonists, antagonists or partial antagonists of the androgen receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the androgen receptor pathway.
- Applications of said compounds include but are not limited to: hirsutism, acne, seborrhea, Alzheimer's disease, androgenic alopecia, hypogonadism, hyperpilosity, benign prostate hypertrophia, adenomas and neoplasies of the prostate (such as advanced metastatic prostate cancer), treatment of benign or malignant tumor cells containing the androgen receptor such as is the case for breast, brain, skin, ovarian, bladder, lymphatic, liver and kidney cancers, pancreatic cancers modulation of VCAM expression and applications therein for the treatment of heart disease, inflammation and immune modulations, modulation of VEGF expression and the applications therein for use as antiangiogenic agents, osteoporosis, suppressing spermatogenesis, libid
- Compounds of formula I can be applied as (preferably, selective) antagonists of the mutated androgen receptor found in many tumor lines.
- mutants are those found in representative prostate tumor cell lines such as LNCap, (T877A mutation, Biophys. Acta, 187, 1052 (1990)), PCa2b, (L701H & T877A mutations, J. Urol., 162, 2192 (1999)) and CWR22, (H874Y mutation, Mol. Endo., 11, 450 (1997)).
- Applications of said compounds include but are not limited to: adenomas and neoplasies of the prostate, breast cancer and endometrial cancer.
- Compounds of formula I can be applied as agonists, partial agonists, antagonists or partial antagonists of the steroid and xenobiotic receptor, preferably selectively to that receptor, in an array of medical conditions which involve modulation of the steroid and xenobiotic receptor pathway.
- Applications of said compounds include but are not limited to: treatment of disregulation of cholesterol homeostasis, attenuation of metabolism of pharmaceutical agents by co- administration of an agent (compound of the present invention) which modulates the P450 regulator effects of SXR.
- NHR NHR due to strong sequence homology to other NHR
- Orphan receptors demonstrate strong sequence homology to other NHR
- compounds of formula I include those which serve as modulators of the function of the Orphan NHR.
- Orphan receptors which are modulated by NHR modulators such as compounds within the scope of formula I are exemplified, but not limited to, those listed in Table 1.
- Exemplary therapeutic applications of modulators of said Orphan receptors are also listed in Table 1, but are not limited to the examples therein.
- the present invention thus provides methods for the treatment of NHR- associated conditions, comprising the step of administering to a subject in need thereof at least one compound of formula I in an amount effective therefor.
- Other therapeutic agents such as those described below may be employed with the inventive compounds in the present methods (for example, separately, or formulated together as a fixed dose).
- such other therapeutic agent(s) can be administered prior to, simultaneously with or following the administration of the compound(s) of the present invention.
- the present invention also provides pharmaceutical compositions comprising at least one of the compounds of the formula I capable of treating a NHR-associated condition in an amount effective therefor, and a pharmaceutically acceptable carrier (vehicle or diluent).
- compositions of the present invention can contain other therapeutic agents as described below, and can be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (for example, excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation.
- the compounds of the present invention are, without limitation as to their mechanism of action, useful in treating any of the conditions or disorders listed or described herein such as inflammatory diseases or cancers, or other proliferate diseases, and in compositions for treating such conditions or disorders.
- Such conditions and disorders include, without limitation, any of those described previously, as well as those described following such as: maintenance of muscle strength and function (e.g., in the elderly); reversal or prevention of frailty or age- related functional decline ("ARFD") in the elderly (e.g., sarcopenia); treatment of catabolic side effects of glucocorticoids; prevention and/or treatment of reduced bone mass, density or growth (e.g., osteoporosis and osteopenia); treatment of chronic fatigue syndrome (CFS); chronic malagia; treatment of acute fatigue syndrome and muscle loss following elective surgery (e.g., post-surgical rehabilitation); acceleration of wound healing; accelerating bone fracture repair (such as accelerating the recovery of hip fracture patients); accelerating healing of complicated fractures, e.g.,
- distraction osteogenesis in joint replacement; prevention of post-surgical adhesion formation; acceleration of tooth repair or growth; maintenance of sensory function (e.g., hearing, sight, olefaction and taste); treatment of periodontal disease; treatment of wasting secondary to fractures and wasting in connection with chronic obstructive pulmonary disease (COPD), chronic liver disease, AIDS, weightlessness, cancer cachexia, burn and trauma recovery, chronic catabolic state (e.g., coma), eating disorders (e.g., anorexia) and chemotherapy; treatment of cardiomyopathy; treatment of thrombocytopenia; treatment of growth retardation in connection with Crohn's disease; treatment of short bowel syndrome; treatment of irritable bowel syndrome; treatment of inflammatory bowel disease; treatment of Crohn's disease and ulcerative colits; treatment of complications associated with transplantation; treatment of physiological short stature including growth hormone deficient children and short stature associated with chronic illness; treatment of obesity and growth retardation associated with obesity; treatment of anorexia (e.g., associated with cachexia
- the present compounds have therapeutic utility in the modulation of immune cell activation/proliferation, e.g., as competitive inhibitors of intercellular ligand/receptor binding reactions involving CAMs (Cellular Adhesion Molecules) and Leukointegrins.
- the present compounds modulate LFA-ICAM 1, and are particularly useful as LFA-ICAM 1 antagonists, and in the treatment of all conditions associated with LFA-ICAM 1 such as immunological disorders.
- Preferred utilities for the present compounds include, but are not limited to: inflammatory conditions such as those resulting from a response of the non-specific immune system in a mammal (e.g., adult respiratory distress syndrome, shock, oxygen toxicity, multiple organ injury syndrome secondary to septicemia, multiple organ injury syndrome secondary to trauma, reperfusion injury of tissue due to cardiopulmonary bypass, myocardial infarction or use with thrombolysis agents, acute glomerulonephritis, vasculitis, reactive arthritis, dermatosis with acute inflammatory components, stroke, thermal injury, hemodialysis, leukapheresis, ulcerative colitis, necrotizing enterocolitis and granulocyte transfusion associated syndrome) and conditions resulting from a response of the specific immune system in a mammal (e.g., psoriasis, organ/tissue transplant rejection, graft vs.
- inflammatory conditions such as those resulting from a response of the non-specific immune system in a mammal (e.g., adult
- the present compounds can be used in treating asthma or as an adjunct to minimize toxicity with cytokine therapy in the treatment of cancers.
- the present compounds can be employed in the treatment of all diseases currently treatable through steroid therapy.
- the present compounds may be employed for the treatment of these and other disorders alone or with other immunosuppressive or antiinflammatory agents.
- a compound of the formula I can be administered prior to the onset of inflammation (so as to suppress an anticipated inflammation) or after the initiation of inflammation.
- the immunosupressive compound(s) are preferably provided in advance of any inflammatory response or symptom (for example, prior to, at, or shortly after the time of an organ or tissue transplant but in advance of any symptoms or organ rejection).
- the prophylactic administration of a compound of the formula I prevents or attenuates any subsequent inflammatory response (such as, for example, rejection of a transplanted organ or tissue, etc.)
- Administration of a compound of the formula I attenuates any actual inflammation (such as, for example, the rejection of a transplanted organ or tissue).
- the compounds of the formula I can be administered for any of the uses described herein by any suitable means, for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents.
- suitable means for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.
- the present compounds can, for example, be administered in a form suitable for immediate release or extended release. Immediate release or extended release can be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps.
- the present compounds can also be administered liposomally.
- compositions for oral administration include suspensions which can contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which can contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art.
- the compounds of formula I can also be delivered through the oral cavity by sublingual and/or buccal administration.
- Molded tablets, compressed tablets or freeze-dried tablets are exemplary forms which may be used.
- Exemplary compositions include those formulating the present compound(s) with fast dissolving diluents such as mannitol, lactose, sucrose and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (avicel) or polyethylene glycols (PEG).
- Such formulations can also include an excipient to aid mucosal adhesion such as hydroxy propyl cellulose (HPC), hydroxy propyl methyl cellulose (HPMC), sodium carboxy methyl cellulose (SCMC), maleic anhydride copolymer (e.g., Gantrez), and agents to control release such as polyacrylic copolymer (e.g. Carbopol 934).
- HPC hydroxy propyl cellulose
- HPMC hydroxy propyl methyl cellulose
- SCMC sodium carboxy methyl cellulose
- maleic anhydride copolymer e.g., Gantrez
- agents to control release such as polyacrylic copolymer (e.g. Carbopol 934).
- Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
- compositions for nasal aerosol or inhalation administration include solutions in saline which can contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- exemplary compositions for parenteral administration include injectable solutions or suspensions which can contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
- compositions for rectal administration include suppositories which can contain, for example, a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- exemplary compositions for topical administration include a topical carrier such as Plastibase (mineral oil gelled with polyethylene).
- the effective amount of a compound of the present invention can be determined by one of ordinary skill in the art, and includes exemplary dosage amounts for a adult human of from about 1 to 100 (for example, 15) mg/kg of body weight of active compound per day, which can be administered in a single dose or in the form of individual divided doses, such as from 1 to 4 times per day. It will be understood that the specific dose level and frequency of dosage for any particular subject can be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition.
- Preferred subjects for treatment include animals, most preferably mammalian species such as humans, and domestic animals such as dogs, cats and the like, subject to NHR-associated conditions.
- the compounds of the present invention can be employed alone or in combination with each other and/or other suitable therapeutic agents useful in the treatment of NHR-associated conditions, e.g., an antibiotic or other pharmaceutically active material.
- the compounds of the present invention can be combined with growth promoting agents, such as, but not limited to, TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345, e.g., zeranol, and compounds disclosed in U.S. Patent No. 4,036,979, e.g., sulbenox or peptides disclosed in U.S. Patent No. 4,411,890.
- growth promoting agents such as, but not limited to, TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345, e.g., zeranol, and compounds disclosed in U.S. Patent No. 4,036,979, e.g., sulbenox or peptides disclosed in U.S. Patent No. 4,411,890
- the compounds of the invention can also be used in combination with growth hormone secretagogues such as GHRP-6, GHRP-1 (as described in U.S. Patent No. 4,411,890 and publications WO 89/07110 and WO 89/07111), GHRP-2 (as described in WO 93/04081), NN703 (Novo Nordisk), LY444711 (Lilly), MK-677 (Merck), CP424391 (Pfizer) and B-HT920, or with growth hormone releasing factor and its analogs or growth hormone and its analogs or somatomedins including IGF-1 and IGF-2, or with alpha-adrenergic agonists, such as clonidine or serotinin 5-HT D agonists, such as sumatriptan, or agents which inhibit somatostatin or its release, such as physostigmine and pyridostigmine.
- growth hormone secretagogues such as GHRP-6, GHRP-1 (as described in U.
- a still further use of the compounds of the invention is in combination with estrogen, testosterone, a selective estrogen receptor modulator, such as tamoxifen or raloxifene, or other androgen receptor modulators, such as those disclosed in Edwards, J. P. et al., Bio. Med. Chem. Let., 9, 1003-1008 (1999) and Hamann, L. G. et al., J. Med. Chem., 42, 210-212 (1999).
- a further use of the compounds of this invention is in combination with progesterone receptor agonists ("PRA”), such as levonorgestrel, medroxyprogesterone acetate (MPA).
- PRA progesterone receptor agonists
- the compounds of the present invention can be employed alone or in combination with each other and/or other modulators of nuclear hormone receptors or other suitable therapeutic agents useful in the treatment of the aforementioned disorders including: anti-diabetic agents; anti-osteoporosis agents; anti-obesity agents; anti-inflammatory agents; anti-anxiety agents; anti-depressants; anti- hypertensive agents; anti-platelet agents; anti-thrombotic and thrombolytic agents; cardiac glycosides; cholesterol/lipid lowering agents; mineralocorticoid receptor antagonists; phospodiesterase inhibitors; protein tyrosine kinase inhibitors; thyroid mimetics (including thyroid receptor agonists); anabolic agents; HIV or AIDS therapies; therapies useful in the treatment of Alzheimer's disease and other cognitive disorders; therapies useful in the treatment of sleeping disorders; anti-proliferative agents; and anti-tumor agents.
- suitable therapeutic agents useful in the treatment of the aforementioned disorders including: anti-diabetic agents; anti-o
- Suitable anti-diabetic agents for use in combination with the compounds of the present invention include biguanides (e.g., metformin), glucosidase inhibitors (e.g,. acarbose), insulins (including insulin secretagogues or insulin sensitizers), meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide and glipizide), biguanide/glyburide combinations (e.g., Glucovance®), thiazolidinediones (e.g., troglitazone, rosiglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonists, PPAR alpha/gamma dual agonists, SGLT2 inhibitors, glycogen phosphorylase inhibitors, inhibitors of fatty acid binding protein (aP2) such as those disclosed in U.
- Suitable anti-osteoporosis agents for use in combination with the compounds of the present invention include alendronate, risedronate, PTH, PTH fragment, raloxifene, calcitonin, steroidal or non-steroidal progesterone receptor agonists, RANK ligand antagonists, calcium sensing receptor antagonists, TRAP, inhibitors, selective estrogen receptor modulators (SERM), estrogen and AP-1 inhibitors.
- Suitable anti-obesity agents for use in combination with the compounds of the present invention include aP2 inhibitors, such as those disclosed in U.S. Serial No. 09/519,079 filed March 6, 2000, PPAR gamma antagonists, PPAR delta agonists, beta 3 adrenergic agonists, such as AJ9677 (Takeda/Dainippon), L750355 (Merck), or CP331648 (Pfizer) or other known beta 3 agonists as disclosed in U.S. Patent Nos.
- aP2 inhibitors such as those disclosed in U.S. Serial No. 09/519,079 filed March 6, 2000
- PPAR gamma antagonists such as those disclosed in U.S. Serial No. 09/519,079 filed March 6, 2000
- PPAR gamma antagonists such as those disclosed in U.S. Serial No. 09/519,079 filed March 6, 2000
- PPAR gamma antagonists such as those disclosed in U.S. Serial No
- a lipase inhibitor such as orlistat or ATL-962 (Alizyme)
- a serotonin (and dopamine) reuptake inhibitor such as sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron)
- a thyroid receptor beta drug such as a thyroid receptor ligand as disclosed in WO 97/21993 (U.
- anorectic agent such as dexamphetamine, phentermine, phenylpropanolamine or mazindol.
- Suitable anti-inflammatory agents for use in combination with the compoxinds of the present invention include prednisone, dexamethasone, Enbrel®, cyclooxygenase inhibitors (i.e., COX-1 and/or COX-2 inhibitors such as NSAIDs, aspirin, indomethacin, ibuprofen, piroxicam, Naproxen®, Celebrex®, Vioxx®), CTLA4-Ig agonists/antagonists, CD40 ligand antagonists, IMPDH inhibitors, such as mycophenolate (CellCept®) integrin antagonists, alpha-4 beta-7 integrin antagonists, cell adhesion inhibitors, interferon gamma antagonists, ICAM-1, tumor necrosis factor (TNF) antagonists (e.g., infliximab, OR1384), prostaglandin synthesis inhibitors, budesonide, clofazimine, CNI-1493, CD4 antagonists (e.g.,
- Example of suitable anti-anxiety agents for use in combination with the compounds of the present invention include diazepam, lorazepam, buspirone, oxazepam, and hydroxyzine pamoate.
- Suitable anti-depressants for use in combination with the compounds of the present invention include citalopram, fluoxetine, nefazodone, sertraline, and paroxetine.
- Suitable anti-hypertensive agents for use in combination with the compounds of the present invention include beta adrenergic blockers, calcium channel blockers (L-type and T-type; e.g. diltiazem, verapamil, nifedipine, amlodipine and mybefradil), diuretics (e.g., chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triamtrenene, amiloride, spironolactone), renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril, fosino
- Dual ET/AII antagonist e.g., compounds disclosed in WO 00/01389
- neutral endopeptidase (NEP) inhibitors neutral endopeptidase (NEP) inhibitors
- vasopepsidase inhibitors dual NEP-ACE inhibitors
- omapatrilat and gemopatrilat e.g., omapatrilat and gemopatrilat
- Suitable anti-platelet agents for use in combination with the compounds of the present invention include GPIIb/IIIa blockers (e.g., abciximab, eptifibatide, tirofiban), P2Y12 antagonists (e.g., clopidogrel, ticlopidine, CS-747), thromboxane receptor antagonists (e.g., ifetroban), aspirin, and PDE-III inhibitors (e.g., dipyridamole) with or without aspirin.
- GPIIb/IIIa blockers e.g., abciximab, eptifibatide, tirofiban
- P2Y12 antagonists e.g., clopidogrel, ticlopidine, CS-747
- thromboxane receptor antagonists e.g., ifetroban
- aspirin e.g., ifetroban
- PDE-III inhibitors e.g., dipyridamole
- Suitable cardiac glycosides for use in combination with the compounds of the present invention include digitalis and ouabain.
- suitable cholesterol/lipid lowering agents for use in combination with the compounds of the present invention include HMG-CoA reductase inhibitors (e.g., pravastatin, lovastatin, atorvastatin, simvastatin, NK-104 (a.k.a. itavastatin, or nisvastatin or nisbastatin) and ZD-4522 (a.k.a.
- squalene synthetase inhibitors include rosuvastatin, or atavastatin or visastatin (squalene synthetase inhibitors), fibrates, bile acid sequestrants, ACAT inhibitors, MTP inhibitors, lipooxygenase inhibitors, cholesterol absorption inhibitors, and cholesterol ester transfer protein inhibitors (e.g., CP-529414).
- mineralocorticoid receptor antagonists for use in combination with the compounds of the present invention include spironolactone and eplerinone.
- Suitable phospodiesterase inhibitors for use in combination with the compounds of the present invention include PDEIII inhibitors such as cilostazol, and PDE V inhibitors such as sildenafil.
- thyroid mimetics examples include thyrotropin, polythyroid, KB-130015, and dronedarone.
- Suitable therapies for treatment of Alzheimer's disease and cognitive disorders for use in combination with the compounds of the present invention include donepezil, tacrine, revastigmine, 5HT6, gamma secretase inhibitors, beta secretase inhibitors, SK channel blockers, Maxi-K blockers, and KCNQs blockers.
- Suitable therapies for treatment of sleeping disorders for use in combination with the compounds of the present invention include melatonin analogs, melatonin receptor antagonists, ML IB agonists, and GAB A/NMD A receptor antagonists.
- Suitable anti-proliferative agents for use in combination with the compounds of the present invention include cyclosporin A, paclitaxel, FK 506, and adriamycin.
- Suitable anti-tumor agents for use in combination with the compounds of the present invention include paclitaxel, adriamycin, epothilones, cisplatin and carboplatin.
- Compounds of the present invention can further be used in combination with nutritional supplements such as those described in U.S. 5,179,080, especially in combination with whey protein or casin, amino acids (such as leucine, branched amino acids and hydroxymethylbutyrate), triglycerides, vitamins (e.g., A, B6, B12, folate, C, D and E), minerals (e.g., selenium, magnesium, zinc, chromium, calcium and potassium), carnitine, lipoic acid, creatine, and coenzyme Q-10.
- nutritional supplements such as those described in U.S. 5,179,080, especially in combination with whey protein or casin, amino acids (such as leucine, branched amino acids and hydroxymethylbutyrate), triglycerides, vitamins (e.g., A, B6, B12, folate, C, D and E), minerals (e.g., selenium, magnesium, zinc, chromium, calcium and potassium), carnitine, lipo
- compounds of the present invention can be used in combination with therapeutic agents used in the treatment of sexual dysfunction, including but not limited to PDE5 inhibitors, such as sildenafil or IC-351; with an antiresorptive agent, hormone replacement therapies, vitamin D analogues, calcitonins, elemental calcium and calcium supplements, cathepsin K inhibitors, MMP inhibitors, vitronectin receptor antagonists, Src SH 2 antagonists, vacular -H + - ATPase inhibitors, progesterone receptor agonists, ipriflavone, fluoride, RANK antagonists, PTH and its analogues and fragments, Tibolone, HMG-CoA reductase inhibitors, SERM's, p38 inhibitors, prostanoids, 17-beta hydroxysteroid dehydrogenase inhibitors and Src kinase inhibitors.
- PDE5 inhibitors such as sildenafil or IC-351
- hormone replacement therapies such as sil
- Compounds of the present invention can be used in combination with male contraceptives, such as nonoxynol 9 or therapeutic agents for the treatment of hair loss, such as minoxidil and fmasteride or chemotherapeutic agents, such as with LHRH agonists.
- male contraceptives such as nonoxynol 9 or therapeutic agents for the treatment of hair loss, such as minoxidil and fmasteride or chemotherapeutic agents, such as with LHRH agonists.
- the compounds of the present invention can be administered either alone or in combination with other anti- cancer and cytotoxic agents and treatments useful in the treatment of cancer or other proliferative diseases, for example, where the second drug has the same or different mechanism of action than the present compounds of formula I.
- Examples of classes of anti-cancer and cytotoxic agents useful in combination with the present compounds include but are not limited to: alkylating agents such as nitrogen mustards, alkyl sulfonates, nitrosoureas, ethylenimines, and triazenes; antimetabolites such as folate antagonists, purine analogues, and pyrimidine analogues; antibiotics such as anthracyclines, bleomycins, mitomycin, dactinomycin, and plicamycin; enzymes such as L-asparaginase; famesyl-protein transferase inhibitors; 5 ⁇ reductase inhibitors; inhibitors of 17 ⁇ -hydroxy steroid dehydrogenase type 3; hormonal agents such as glucocorticoids, estrogens/ antiestrogens, androgens/ antiandrogens, progestins, and luteinizing hormone-releasing hormone antagonists, octreotide acetate; microtubule- disruptor
- epothilones such as epothilones A-F and their analogs
- plant-derived products such as vinca alkaloids, epipodophyllotoxins, taxanes; and topiosomerase inhibitors
- prenyl-protein transferase inhibitors prenyl-protein transferase inhibitors
- miscellaneous agents such as hydroxyurea, procarbazine, mitotane, hexamethylmelamine, platinum coordination complexes such as cisplatin and carboplatin
- other agents used as anti-cancer and cytotoxic agents such as biological response modifiers, growth factors; immune modulators and monoclonal antibodies.
- the compounds of the invention may also be used in conjunction with radiation therapy.
- anti-cancer and cytotoxic agents include but are not limited to mechlorethamine hydrochloride, cyclophosphamide, chlorambucil, melphalan, ifosfamide, busulfan, carmustin, lomustine, semustine, streptozocin, thiotepa, dacarbazine, methotrexate, thioguanine, mercaptopurine, fludarabine, pentastatin, cladribin, cytarabine, fluorouracil, doxorubicin hydrochloride, daunorubicin, idarubicin, bleomycin sulfate, mitomycin C, actinomycin D, safracins, saframycins, quinocarcins, discodermolides, vincristine, vinblastine, vinorelbine tartrate, etoposide, etoposide phosphate, teni
- Preferred member of these classes include, but are not limited to, paclitaxel, cisplatin, carboplatin, doxorubicin, carminomycin, daunorubicin, aminopterin, methotrexate, methopterin, mitomycin C, ecteinascidin 743, or porfiromycin, 5- fluorouracil, 6-mercaptopurine, gemcitabine, cytosine arabinoside, podophyllotoxin or podophyllotoxin derivatives such as etoposide, etoposide phosphate or teniposide, melphalan, vinblastine, vincristine, leurosidine, vindesine and leurosine.
- anticancer and other cytotoxic agents include the following: epothilone derivatives as found in German Patent No. 4138042.8; WO 97/19086, WO 98/22461, WO 98/25929, WO 98/38192, WO 99/01124, WO 99/02224, WO 99/02514, WO 99/03848, WO 99/07692, WO 99/27890, WO 99/28324, WO 99/43653, WO 99/54330, WO 99/54318, WO 99/54319, WO 99/65913, WO 99/67252, WO 99/67253 and WO 00/00485; cyclin dependent kinase inhibitors as found in WO 99/24416 (see also U.S.
- NHR modulators including, but not limited to, those of present invention
- the combinations of the present invention can also be formulated or co- administered with other therapeutic agents that are selected for their particular usefulness in administering therapies associated with the aforementioned conditions.
- the compounds of the invention may be formulated with agents to prevent nausea, hypersensitivity and gastric irritation, such as antiemetics, and H, and H 2 antihistaminics.
- the compounds of this invention are most preferably used alone or in combination with anti-cancer treatments such as radiation therapy and/or with cytostatic and/or cytotoxic agents, such as, but not limited to, DNA interactive agents, such as cisplatin or doxorubicin; inhibitors of farnesyl protein transferase, such as those described in U.S. Patent No.
- anti-cancer treatments such as radiation therapy and/or with cytostatic and/or cytotoxic agents, such as, but not limited to, DNA interactive agents, such as cisplatin or doxorubicin; inhibitors of farnesyl protein transferase, such as those described in U.S. Patent No.
- topoisomerase II inhibitors such as etoposide
- topoisomerase I inhibitors such as CPT-11 or topotecan
- tubulin stabilizing agents such as paclitaxel, docetaxel, other taxanes, or epothilones
- hormonal agents such as tamoxifen
- thymidilate synthase inhibitors such as 5-fluorouracil
- antimetabolites such as methoxtrexate
- antiangiogenic agents such as angiostatin, ZD6474, ZD6126 and comberstatin A2
- kinase inhibitors such as her2 specific antibodies, Iressa and CDK inhibitors
- histone deacetylase inhibitors such as CI-994 and MS-27-275.
- Such compounds may also be combined with agents which suppress the production of circulating testosterone such as LHRH agonists or antagonists or with surgical castration.
- known therapies for advanced metastatic prostate cancer include "complete androgen ablation therapy” wherein tumor growth is inhibited by controlling the supply of androgen to the prostate tissues via chemical castration (castration serves to inhibit the production of circulating testosterone (T) and dihydrotestosterone (DHT)) followed by the administration of androgen receptor
- AR anti-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative-proliferative, or Cyproterone acetate.
- Another application of the present compounds is in combination with antibody therapy such as but not limited to antibody therapy against PSCA.
- An additional application is in concert with vaccine / immune modulating agents for the treatment of cancer.
- Compounds of the present invention can be employed in accordance with the methods described in U.S. Provisional Patent Application Serial No.
- the following assays can be employed in ascertaining the activity of a compound as a NHR modulator.
- Various compounds of the present invention were determined to have AR modulator activity utilizing the transactivation assay, and standard AR binding assays as described following. At the concentration tested, certain compounds within formula I showed poor or no in vivo activity in the assay employed (e.g., compounds of Example 97).
- transactivation assays of a transfected reporter construct and using the endogenous androgen receptor of the host cells.
- the transactivation assay provides a method for identifying functional agonists and partial agonists that mimic, or antagonists that inhibit, the effect of native hormones, in this case, dihydrotestosterone (DHT).
- DHT dihydrotestosterone
- This assay can be used to predict in vivo activity as there is a good correlation in both series of data. See, e.g. T. Berger et al., J. Steroid Biochem. Molec. Biol. 773 (1992), the disclosure of which is herein incorporated by reference.
- reporter plasmid For the transactivation assay a reporter plasmid is introduced by transfection (a procedure to induce cells to take foreign genes) into the respective cells.
- This reporter plasmid comprising the cDNA for a reporter protein, such as secreted alkaline phosphatase (SEAP), controlled by prostate specific antigen (PSA) upstream sequences containing androgen response elements (AREs).
- SEAP secreted alkaline phosphatase
- PSA prostate specific antigen
- AREs upstream sequences containing androgen response elements
- This reporter plasmid functions as a reporter for the transcription-modulating activity of the AR.
- the reporter acts as a surrogate for the products (mRNA then protein) normally expressed by a gene under control of the AR and its native hormone.
- the transactivation assay is carried out in the presence of constant concentration of the natural AR hormone (DHT) known to induce a defined reporter signal.
- DHT natural AR hormone
- Increasing concentrations of a suspected antagonist will decrease the reporter signal (e.g., SEAP production).
- exposing the transfected cells to increasing concentrations of a suspected agonist will increase the production of the reporter signal.
- LNCaP and MDA 453 cells were obtained from the American Type Culture Collection (Rockville, MD), and maintained in RPMI 1640 or DMEM medium supplemented with 10% fetal bovine serum (FBS; Gibco) respectively.
- the respective cells were transiently transfected by electroporation according to the optimized procedure described by Heiser, 130 Methods Mol. Biol., 117 (2000), with the pSEAP2/PSA540/Enhancer reporter plasmid.
- the reporter plasmid was constructed as follows: commercial human placental genomic DNA was used to generate by Polymerase Cycle Reaction (PCR) a fragment containing the Bglll site (position 5284) and the Hind III site at position 5831 of the human prostate specific antigen promoter (Accession # U37672), Schuur, et al., J. Biol. Chem., Ill (12): 7043-51 (1996). This fragment was subcloned into the ⁇ SEAP2/basic (Clontech) previously digested with Bglll and Hindlll to generate the ⁇ SEAP2/PSA540 construct.
- PCR Polymerase Cycle Reaction
- Each cell suspension was distributed into two Gene Pulser Cuvetts (Bio-Rad) which then received 8 ⁇ g of the reporter construct, and electoporated using a Bio-Rad Gene Pulser at 210 volts and 960 ⁇ Faraday. Following the transfections the cells were washed and incubated with media containing charcoal stripped fetal bovine serum in the absence (blank) or presence (control) of 1 nM dihydrotestosterone (DHT; Sigma Chemical) and in the presence or absence of the standard anti-androgen bicalutamide or compounds of the present invention in concentrations ranging from 10-10 to 10-5 M (sample). Duplicates were used for each sample.
- the compound dilutions were performed on a Biomek 2000 laboratory workstation. After 48 hours, a fraction of the supernatant was assayed for SEAP activity using the Phospha-Light Chemiluminescent Reporter Gene Assay System (Tropix, Inc). Viability of the remaining cells was determined using the CellTiter 96 Aqueous Non- Radioactive Cell Proliferation Assay (MTS Assay, Promega).
- a mix of a tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)- 2- (4-sulfophenyl)-2H-tetrazolium, inner salt; MTS) and an electron coupling reagent (phenazine methosulfate; PMS) are added to the cells.
- MTS Oleen's reagent
- PMS phenazine methosulfate
- the reporter plasmid utilized was comprised of the cDNA for the reporter SEAP protein, as described for the AR specific transactivation assay. Expression of the reporter SEAP protein was controlled by the mouse mammary tumor virus long terminal repeat (MMTV LTR) sequences that contains three hormone response elements (HREs) that can be regulated by both GR and PR see, e.g. G. Chalepakis et al., Cell, 53(3), 371 (1988). This plasmid was transfected into A549 cells, which expresses endogenous GR, to obtain a GR specific transactivation assay.
- MMTV LTR mouse mammary tumor virus long terminal repeat
- HREs hormone response elements
- A549 cells were obtained from the American Type Culture Collection (Rockville, MD), and maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Gibco). Determination of the GR specific antagonist activity of the compounds of the present invention was identical to that described for the AR specific transactivation assay, except that the DHT was replaced with 5 nM dexamethasone (Sigma Chemicals), a specific agonist for GR. Determination of the GR specific agonist activity of the compounds of the present invention was performed as described for the AR transactivation assay, wherein one measures the activation of the GR specific reporter system by the addition of a test compound, in the absence of a known GR specific agonists ligand.
- the reporter plasmid utilized was comprised of the cDNA for the reporter SEAP protein, as described for the AR specific transactivation assay. Expression of the reporter SEAP protein was controlled by the mouse mammary tumor virus long terminal repeat (MMTV LTR) sequences that contains three hormone response elements (HREs) that can be regulated by both GR and PR. This plasmid was transfected into T47D, which expresses endogenous PR, to obtain a PR specific transactivation assay. T47D cells were obtained from the American Type Culture Collection (Rockville, MD), and maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS; Gibco).
- FBS fetal bovine serum
- Determination of the PR specific antagonist activity of the compounds of the present invention was identical to that described for the AR specific transactivation assay, except that the DHT was replaced with 1 nM Promegastone (NEN), a specific agonist for PR. Determination of the PR specific agonist activity of the compounds of the present invention was performed as described for the AR transactivation assay, wherein one measures the activation of the PR specific reporter system by the addition of a test compound, in the absence of a known PR specific agonists ligand.
- human LNCaP cells T877A mutant AR or MDA 453 (wild type AR) in 96-well microtiter plates containing RPMI 1640 or DMEM supplemented with 10% charcoal stripped CA-FBS (Cocaleco Biologicals) respectively, were incubated at 37°C to remove any endogenous ligand that might be complexed with the receptor in the cells. After 48 hours, either a saturation analysis to determine the K j for tritiated dihydrotestosterone, [ 3 H]-DHT, or a competitive binding assay to evaluate the ability of test compounds to compete with [ 3 H]-DHT were performed.
- media RPMI 1640 or DMEM - 0.2% CA-FBS
- [ 3 H]-DHT in concentrations ranging from 0.1 nM to 16 nM
- an aliquot of the total binding media at each concentration of [ 3 H]-DHT was removed to estimate the amount of free [ 3 H]-DHT.
- IC 50 values were determined.
- the IC 50 is defined as the concentration of competing ligand needed to reduce specific binding by 50%.
- the K d s for [ 3 H]-DHT for MDA 453 and LNCaP were 0.7 and 0.2 nM respectively.
- test compounds were tested ("test compounds") on the proliferation of human prostate cancer cell lines.
- MDA PCa2b cells a cell line derived from the metastasis of a patient that failed castration, Navone et al., Clin. Cancer Res. , 3, 2493-500 (1997), were incubated with or without the test compoxmds for 72 hours and the amount of [ 3 H]-thymidine incorporated into DNA was quantified as a way to assess number of cells and therefore proliferation.
- the MDA PCa2b cell line was maintained in BRFF-HPCl media (Biological Research Faculty & Facility Inc., MD) supplemented with 10% FBS.
- cells were plated in Biocoated 96-well microplates and incubated at 37°C in 10% FBS (charcoal-stri ⁇ ped)/BRFF- BMZERO (without androgens). After 24 hours, the cells were treated in the absence (blank) or presence of 1 nM DHT (control) or with test compounds (sample) of the present invention in concentrations ranging from 10 "10 to 10 "5 M. Duplicates were used for each sample. The compound dilutions were performed on a Biomek 2000 laboratory work station. Seventy two hours later 0.44 uCi.
- % Inhibition 100 x ( 1 - [average control - average bIank / average sample - average bIank ]) Data was plotted and the concentration of compound that inhibited 50% of the [ 3 H]- Thymidine incorporation was quantified (IC 50 ).
- C2C12 Mouse Myoblast Transactivation Assay Two functional transactivation assays were developed to assess the efficacy of androgen agonists in a muscle cell background using a luciferase reporter.
- the first assay (ARTA Stable 1) uses a cell line, Stable 1 (clone #72), which stably expresses the full length rat androgen receptor but requires the transient transfection of an enhancer/reporter. This cell line was derived from C2C12 mouse moyoblast cells.
- the second assay (ARTA Stable 2) uses a cell line, Stable 2 (clone #133), derived from Stable 1 which stably expresses both rAR and the enhancer/luciferase reporter.
- the enhancer/reporter construct used in this system is pGL3/2XDR- 1 /luciferase.
- 2XDR-1 was reported to be an AR specific response element in CV-1 cells, Brown et. al. The Journal of Biological Chemisty 272, 8227-8235, (1997). It was developed by random mutagenesis of an AR/GR consensus enhancer sequence.
- Stable 1 cells are plated in 96 well format at 6,000 cells/well in high glucose DMEM without phenol red (Gibco BRL, Cat. No.: 21063-029) containing 10% charcoal and dextran treated FBS (HyClone Cat. No.: SH30068.02), 50 mM HEPES Buffer (Gibco BRL, Cat. No.: 15630-080), IX MEM Na Pyruvate (Gibco
- sperm DNA (as carrier) are diluted with 5 ⁇ l well Opti-MEMem media (Gibco BRL, Cat. No.: 31985-070). To this, 0.5 ⁇ l/well Plus reagent is added. This mixture is incubated for 15 minutes at room temperature. In a separate vessel, 0.385 ⁇ l/well LipofectAMINE reagent is diluted with 5 ⁇ l/well Opti-MEM. The DNA mixture is then combined with the LipofectAMINE mixture and incubated for an additional 15 minutes at room temperature. During this time, the media from the cells is removed and replaced with 60 ⁇ l/well of Opti-MEM. To this is added 10 ⁇ l/well of the DNA/LipofectAMINE transfection mixture. The cells are incubated for 4 hours. 3. The transfection mixture is removed from the cells and replaced with 90 ⁇ l of media as in #1 above. 4. 10 ⁇ l/well of appropriate drug dilution is placed in each well.
- Stable 2 cells are plated in 96 well format at 6,000 cells/well in high glucose DMEM without phenol red (Gibco BRL, Cat. No.: 21063-029) containing 10% charcoal and dextran treated FBS (HyClone Cat. No.: SH30068.02), 50 mM HEPES Buffer (Gibco BRL, Cat. No.: 15630-080), IX MEM Na Pyruvate (Gibco BRL, Cat. No. : 11360-070), 0.5X Antibiotic-Antimycotic, 800 ⁇ g/ml Geneticin
- the media on the cells is removed and replaced with 90 ⁇ l fresh. 10 ⁇ l/well of appropriate drug dilution is placed in each well. 3. 24 hours later, the Steady-GloTM Luciferase Assay System is used to detect activity according to the manufacturer's instructions (Promega, Cat. No.: E2520).
- test compounds The ability of compoxmds of the present invention to modulate the function of the AR was determined by testing said compounds in a proliferation assay using the androgen responsive murine breast cell line derived from the Shionogi tumor, Hiraoka et al, Cancer Res., 47, 6560-6564 (1987). Stable AR dependent clones of the parental Shionogi line were established by passing tumor fragments under the general procedures originally described in Tetuo, et. al, Cancer Research 25, 1168-1175 (1965). From the above procedure, one stable line, SCI 14, was isolated, characterized and utilized for the testing of example compounds.
- SCI 14 cells were incubated with or without the test compounds for 72 hours and the amount of [3H]-thymidine incorporated into DNA was quantified as a surrogate endpoint to assess the number of cells and therefore the proliferation rate as described in Suzuki et. al, J. Steroid Biochem. Mol. Biol. 37, 559-567 (1990).
- the SCI 14 cell line was maintained in MEM containing IO "8 M testosterone and 2% DCC-treated FCS.
- cells were plated in 96-well microplates in the maintenance media and incubated at 37°C.
- the medium was changed to serum free medium [Ham's F-12:MEM (1 ;1, v/v) containing 0.1% BSA] with (antagonist mode) or without (agonist mode) IO "8 M testosterone and the test compounds of the present invention in concentrations ranging from 10 "10 to IO "5 M.
- Duplicates were used for each sample. The compound dilutions were performed on a Biomek 2000 laboratory work station. Seventy two hours later 0.44uCi of [3H]-Thymidine (Amersham) was added per well and incubated for another 2 hr followed by tripsinization, and harvesting of the cells onto GF/B filters. Micro-scint PS were added to the filters before counting them on a Beckman TopCount. For the antagonist mode, the % Inhibition was calculated as:
- the AP-1 assay is a cell based luciferase reporter assay.
- A549 cells which contain endogenous glucocorticoid receptor, were stably transfected with an AP-1 DNA binding site attached to the luciferase gene. Cells are then grown in RPMI + 10% fetal calf serum (charcoal-treated) + Penicillin/Streptomycin with 0.5mg/ml geneticin. Cells are plated the day before the assay at approximately 40000 cells/well. On assay day, the media is removed by aspiration and 20 ⁇ l assay buffer (RPMI without phenol red + 10% FCS (charcoal-treated) + Pen/Strep) is added to each well.
- test compounds the compounds of the present invention
- dexamethasome 100 nM in DMSO, positive control
- the plates are then pre-incubated for 15 minutes at 37°C, followed by stimulation of the cells with 10 ng/ml PMA.
- the plates are then incubated for 7 hrs at 37°C after which 40 ⁇ l luciferase substrate reagent is added to each well.
- Activity is measured by analysis in a luminometer as compared to control experiments treated with buffer or dexamethasome.
- Activity is designated as % inhibition of the reporter system as compared to the buffer control with 10 ng/ml PMA alone.
- the control, dexamethasone, at a concentration of ⁇ 10 ⁇ M typically suppresses activity by 65%.
- Test compounds which demonstrate an inhibition of PMA induction of 50% or greater at a concentration of test compound of ⁇ 10 ⁇ M are deemed active.
- T serum testosterone
- LH pituitary luteinizing hormone
- FSH follicle stimulating hormone
- DHT dihydrotestosterone
- mice were castrated under metofane ansestesia. Five days after surgery these castrated rats (60-70g, 23-25 day-old) were dosed for 3 days. Animals were dosed sub-cutaneously (s.c.) lmg/kg with Testosterone Proprionate (TP) in arachis oil vehicle and anti-androgen test compounds (compounds of the present invention) were dosed orally by gavage (p.o.) in dissolved/suspensions of 80% PEG 400 and 20% Tween 80 (PEGTW). Animals were dosed (v/w) at 0.5 ml of vehicle /100g body weight. Experimental groups were as follows:
- Testosterone Propionate (3 mg/rat/day, subcutaneous) 3.
- TP plus Casodex (administered p.o. in PEGTW, QD) , a recognized antiandrogen, as a reference compound.
- test compound a compound of the present invention was administered (p.o. in PEGTW, QD) with TP (s.c. as administered in group 2) in a range of doses.
- test compound a compound of the present invention was administered alone (p.o.. in PEGTW, QD) in a range of doses.
- the animals were sacrificed, and the ventral prostate weighed.
- the sexual organs weights were first standardized as mg per 100 g of body weight, and the increase in organ weight induced by TP was considered as the maximum increase (100%).
- ANOVA followed by one-tailed Student or Fischer's exact test was used for statistical analysis.
- the gain and loss of sexual organ weight reflect the changes of the cell number (DNA content) and cell mass (protein content), depending upon the serum androgen concentration. See Y. Okuda et al., J. Urol, 145, 188-191 (1991), the disclosure of which is herein incorporated by reference. Therefore, measurement of organ wet weight is sufficient to indicate the bioactivity of androgens and androgen antagonist.
- SV seminal vesicles
- VP ventral prostate
- the maximum increase in organ weight was 4 to 5-fold when dosing 3 mg/rat/day of testosterone (T) or 1 mg/rat day of testosterone propionate (TP) for 3 days.
- the EC 50 of T and TP were about 1 mg and 0.03 mg, respectively.
- the increase in the weight of the VP and SV also correlated with the increase in the serum T and DHT concentration.
- administration of T showed 5-times higher serum concentrations of T and DHT at 2 hours after subcutaneous injection than that of TP, thereafter, these high levels declined very rapidly.
- the serum concentrations of T and DHT in TP-treated animals were fairly consistent during the 24 hours, and therefore, TP showed about 10-30-fold higher potency than free T.
- a known AR antagonist (Casodex) was also administered simultaneously with 0.1 mg of TP (ED g0 ), inhibiting the testosterone-mediated increase in the weights of the VP and SV in a dose dependent manner.
- the antagonist effects were similar when dosing orally or subcutaneously.
- Compounds of the invention also exhibited AR antagonist activity by suppressing the testosterone-mediated increase in the weights of VP and SV.
- Testosterone Propionate (TP) (3 mg/rat/day, subcutaneous)
- test compound was administered (p.o. in PEGTW, QD) with TP (s.c. as administered in group 2) in a range of doses. 5.
- test compound was administered alone (p.o. in PEGTW, QD) in a range of doses.
- the animals were sacrificed by carbon dioxide, and the levator ani, seminal vesicle and ventral prostate weighed.
- the levator ani muscle and sexual organ weights were first standardized as mg per 100 g of body weight, and the increase in organ weight induced by TP was considered as the maximum increase (100%).
- Super-anova one factor was used for statistical analysis.
- the gain and loss of sexual organ weight reflect the changes of the cell number (DNA content) and cell mass (protein content), depending upon the serum androgen concentration. See Y. Okuda et al, J. Urol, 145, 188-191 (1991), the disclosure of which is herein incorporated by reference. Therefore, measurement of organ wet weight is sufficient to indicate the bioactivity of androgens and androgen antagonist. In immature castrated rats, replacement of exogenous androgens increases levator ani, seminal vesicles (S V) and prostate in a dose dependent manner.
- the maximum increase in organ weight was 4 to 5-fold when dosing 3 mg/rat/day of testosterone (T) or 1 mg/rat/day of testosterone propionate (TP) for 3 days.
- the EC 50 of T and TP were about 1 mg and 0.03 mg, respectively.
- the increase in the weight of the VP and SV also correlated with the increase in the serum T and DHT concentration.
- administration of T showed 5-times higher serum concentrations of T and DHT at 2 hours after subcutaneous injection than that of TP, thereafter, these high levels declined very rapidly.
- the serum concentrations of T and DHT in TP -treated animals were fairly consistent during the 24 hours, and therefore, TP showed about 10-30-fold higher potency than free T.
- MDA-PCa-2b human prostate tumors were maintained in Balb/c nu/nu nude mice. Tumors were propagated as subcutaneous transplants in adult male nude mice (4-6 weeks old) using tumor fragments obtained from donor mice. Tumor passage occurred every 5-6 weeks.
- the required number of animals needed to detect a meaningful response were pooled at the start of the experiment and each was given a subcutaneous implant of a tumor fragment ( ⁇ 50 mg) with a 13 -gauge trocar. Tumors were allowed to grow to approx. 100-200 mg (tumors outside the range were excluded) and animals were evenly distributed to various treatment and control groups. Treatment of each animal was based on individual body weight. Treated animals were checked daily for treatment related toxicity/mortality. Each group of animals was weighed before the initiation of treatment (Wtl) and then again following the last treatment dose (Wt2). The difference in body weight (Wt2-Wtl) provides a measure of treatment-related toxicity.
- Tumor response was determined by measurement of tumors with a caliper twice a week, until the tumors reach a predetermined "target" size of 0.5 gm.
- Tumor response end-point was expressed in terms of tumor growth inhibition (%T/C), defined as the ratio of median tumor weights of the treated tumors (T) to that of the control group (C).
- the tumor volume doubling time was first calculated with the formula:
- Dunning R3327H prostate tumor is a spontaneously derived, well differentiated androgen responsive adenocarcinoma of the prostate (Smolev JK, Heston WD, Scott WW, and Coffey DS, Cancer Treat Rep. 61, 273-287 (1977)).
- the growth of the R3327H subline has been selected for its highly androgen-dependent and reproducible growth in intact male rats. Therefore, this model and other sublines of this tumor have been widely used to evaluate in vivo antitumor activities of antiandrogens such as flutamide and bacilutamide/Casodex (Maucher A., and von Angerer, J. Cancer Res. Clin.
- the Dunning tumor pieces (about 4 x 4 mm) are transplanted subcutaneously to the flank of mature male Copenhagen rats (6-7 weeks old, Harlan-Sprague Dawley, Indianapolis, MD). About 6 weeks after the implantation, the animals with tumors of measurable size (about 80 - 120 mm 2 ) are randomized into treatment groups (8-10 rats/group) and the treatments are initiated. One group of the rats are castrated to serve as the negative control of tumor growth. Animals are treated daily with compounds of the current invention, standard antiandrogens such as bacilutamide or vehicle (control) for an average of 10 to 14 weeks.
- standard antiandrogens such as bacilutamide or vehicle (control) for an average of 10 to 14 weeks.
- Test compounds are dissolved in a vehicle of (2.5 ml/kg of body weight) 10% polyethylene glycol and 0.05% Tween-80 in 1 % carboxymethyl cellulose, PEG/CMC, (Sigma, St Louis, MO).
- Typical therapeutic experiments would include three groups of three escalating doses for each standard or test compound (in a range of 300-3 mg/kg).
- Tumors in the vehicle (control) group reach a size of 1500 to 2500 mm 3 , whereas the castrated animal group typically shows tumor stasis over the 14 weeks of observation.
- Animals treated orally with 20 mg/kg of bicalutamide or flutamide would be expected to show a 40% reduction in tumor volumes compared to control after 14 weeks of treatment.
- the activity of compounds of the present invention were investigated in a mature male rat model, which is a variation of the Levator ani & wet prostate weight assay described above.
- the above in vivo assays are recognized assays for determining the anabolic effects in muscle and sustaining effects in sex organs for a given compound, as described in L. G. Hershberger et al, 83 Proc. Soc. Expt. Biol Med., 175 (1953); B. L. Beyler et al, "Methods for evaluating anabolic and catabolic agents in laboratory animals", 23 J. Amer. Med. Women 's Ass., 708 (1968); H.
- T serum testosterone
- LH pituitary luteinizing hormone
- FSH follicle stimulating hormone
- DHT dihydrotestosterone
- Testosterone production in the Leydig cells of the testis is controlled by the level of circulating LH released from the pituitary gland. LH levels are themselves controlled by the level of LHRH produced in the hypothalmic region. Testosterone levels in the blood serve to inhibit the secretion of LHRH and subsequently reduce levels of LH and ultimately the levels of circulating testosterone levels.
- test compounds By measuring blood levels of LH as they are effected by compounds of the present invention ("test compounds”), it is possible to determine the level of agonist or antagonist activity of said compounds at the hypothalamic axis of this endocrine cycle.
- Control vehicle p.o., PEGTW, QD
- Bicalutamide Casodex, a recognized antiandrogen, as a reference compound
- a compound of the present invention p.o. in PEGTW QD. (in a range of doses).
- the animals were sacrificed, and the ventral prostate, the seminal vesicles, and the levator ani were removed surgically and weighed.
- the organs weights were first standardized as mg per 100 g of body weight, and expressed as a percentage of the value of the respective organ in the intact group.
- Rat luteinizing hormone is quantitatively determined with the Biotrak [125 I] kit (Amersham Pharmacia Biotek), following the manufacturer directions. The assay is based on the competition by the LH present in the serum of the binding of [ 125 I] rLH to an Amerlex-M bead/antibody suspension. The radioactivity that remains after incubation with the serum and subsequent washes is extrapolated into a standard curve to obtain a reading in ng/ml.
- the gain and loss of sexual organ and levator ani weight reflect the changes of the cell number (DNA content) and cell mass (protein content), depending upon the serum androgen concentration, see Y. Okuda et al, J. Urol, 145, 188-191 (1991), the disclosure of which in herein incorporated by reference. Therefore, measurement of organ wet weight is sufficient to indicate the bioactivity of androgens and androgen antagonist.
- active agonist agents will have no effect or will increase the weight of one or more of the androgen responsive organs (levator ani, prostate, seminal vessicle) and will have no effect or a suppressive effect on LH secretion.
- Compounds with antagonist activity will decrease the weight of one or more of the androgen responsive organs (levator ani, prostate, seminal vesicle) and will have no effect or a reduced suppressive effect on LH secretion.
- CWR22 Human Prostate Zenograft Assay In Vivo Antitumor Testing: CWR22 human prostate tumors were maintained in Balb/c nu/nu nude mice. Tumors were propagated as subcutaneous transplants in adult male nude mice (4-6 weeks old) using tumor fragments obtained from donor mice. Tumor passage occurred every 5-6 weeks.
- the required number of animals needed to detect a meaningful response were pooled at the start of the experiment and each was given a subcutaneous implant of a tumor fragment (-50 mg) with a 13-gauge trocar. Tumors were allowed to grow to approx. 100-200 mg (tumors outside the range were excluded) and animals were evenly distributed to various treatment and control groups. Treatment of each animal was based on individual body weight. Treated animals were checked daily for treatment related toxicity/mortality. Each group of animals was weighed before the initiation of treatment (Wt1) and then again following the last treatment dose (Wt2). The difference in body weight (Wt2-Wt1) provides a measure of treatment-related toxicity.
- Tumor response was determined by measurement of tumors with a caliper twice a week, until the tumors reach a predetermined "target" size of 0.5 gm.
- Tumor response end-point was expressed in terms of tumor growth inhibition (%T/C), defined as the ratio of median tumor weights of the treated tumors (T) to that of the control group (C).
- the tumor volume doubling time was first calculated with the formula:
- TVDT Median time (days) for control tumors to reach target size - Median time (days) for control tumors to reach half the target size
- Log cell kill (T-C) ⁇ (3.32 x TVDT)
- TLC thin layer chromatography
- TBSOTf tert-butyldimethylsilyl trifluoromethane sulfonate
- TEA triethylamine
- n-BuLi n-butyllithium
- LC liquid chromatography
- the intermediate compound 4A (180 mg, 0.5 mmol) was dissolved in anhydrous toluene (5 mL) and DBU (0.042 mL) was added. The reaction was heated at 80°C for 1.5 h and then cooled to 25°C. The volatiles were removed in vacuo and the resulting residue was purified by flash chromatography on SiO 2 eluting with a gradient of 0 to 20% acetone/hexane giving pure compound 4B (67 mg) as a yellow oil.
- Resin 9C (1 g) was suspended in 50% TFA/DMF (30 mL) and sonicated at 60°C for 18 h. The reaction was then filtered and washed with DMF (5x20 mL), methanol (2x20 mL), methylene chloride (2x20 mL) and dried under vacuum to give 0.7 g of Resin 9D as a white powder.
- Resin 9D (0.50 g, synthesized as described in Example 9) was suspended in CH 2 C1 2 (10 mL) and phosgene (20% in toluene, 4.5 g) and NaHCO 3 (1.5 g) were added. The resin was shaken for 22 h at 22°C and then filtered rinsing with CH 2 C1 2 (5 50 mL). The resin was then dried in vacuo to give Resin 10A as a yellow resin.
- Resin 10A (0.70 g) was suspended in CH 2 C1 2 (15 mL) and 2-naphthal amine (0.58 g, 4.0 mmol) was added. H ⁇ nig's base (0.88 mL) and catalytic 4-DMAP were added and the mixture was shaken at 70°C for 20 h. After cooling to 22°C, the resin was filtered and washed with CH 2 C1 2 (8x20 mL) and dried in vacuo to give Resin 10B as a yellow solid.
- Example 12 (5 ⁇ .8 ⁇ .8a ⁇ & (5 ⁇ .8 ⁇ ,8a ⁇ >Tetrahydro-2-f3-(trifluoromethyl)phenyll-5,8- methanoimidazo[l,5-a]pyridme-l,3(2H,5H)-dione (12i & 12ii, respectively)
- LDA was prepared by treating diisopropyl amine (0.091 mL, 0.650 mmol) in THF at -78°C with n-BuLi (1.6 M in hexanes, 0.304 mL). After 20 min, Compound 11 (0.100 g, 0.325 mmol) was slowly added to the LDA in THF. The reaction was slowly warmed to -20°C and held for 15 min. The reaction was then cooled to -78°C and quenched by the addition of sat NH 4 C1. The solution was then extracted with CH 2 C1 2 (3 x 30 mL) and the organics were dried over anhydrous sodium sulfate.
- Example 9 was dissolved in CH 2 C1 2 (8.0 mL) and TEA (2.31 mL, 16.6 mmol) and 2,6-dichlorobenzoyl chloride (0.549 mL, 4.15 mmol) were added. The mixture was stirred for 14 h and 2-aminonaphthal (0.593 g, 4.15 mmol) was added in CH 2 C1 2 followed by addition of 4-DMAP (0.010 g). After 3h, the reaction was diluted with CH 2 C1 2 and washed once with IN HCl (40 mL), once with sat aq NaHCO 3 (40 mL) and dried over anhydrous sodium sulfate.
- TEA 2.31 mL, 16.6 mmol
- 2,6-dichlorobenzoyl chloride 0.549 mL, 4.15 mmol
- the crude intermediate (1.00 g, 2.73 mmol) was dissolved in CH 2 C1 2 (2.0 mL) and treated with TFA (2.0 mL) at 20°C. After 3h, the reaction was quenched with saturated aq. NaHCO 3 and extracted with CH 2 C1 2 (3 x 30 mL) and dried over anhydrous sodium sulfate. The crude reaction was purified by preparative reverse phase HPLC to give 0.770 g of Compound 15A as a white solid.
- LC YMC S5 ODS column 4.6 x 50 mm eluting with 10-90% MeOH/H 2 O over 4 minutes containing 0.2% phosphoric acid, 4 mL/min, monitoring at 220 nm
- LCMS YMC S5 ODS column, 4.6 X 50 mm eluting with 10-90% MeOH/H 2 O over 4 minutes containing 0.1% TFA; 4 mL/min, monitoring at 220 nm
- This example illustrates a preferred method for obtaining a compound of formula Ila, which compound is useful as an intermediate in the preparation of compounds of formula I (see, for example, Figure 2 herein).
- N-(ter/-butoxycarbonyl)-L-4-hydroxyproline (10.0 g, 43.3 mmol) was dissolved in THF and cooled to 0°C. Borane/THF (1.0 M solution, 86.6 mL) was then added over a 15 min period. The reaction was then warmed to 25°C followed by heating to reflux for 16 h. The reaction flask was then removed from the heat source and anhydrous methanol (35 mL) was added slowly. After cooling to 25°C, the solvent was removed in vacuo and the resulting crude diol intermediate was taken on directly.
- the crude tosylate intermediate was dissolved in THF (50 mL), to which was added H 2 O (0.5 mL) followed by pTSA-H 2 O (1.03 mmol). Once the reaction was complete as determined by TLC, the mixture was poured into saturated aqueous NaHCO 3 (150 mL) and extracted with methylene chloride (3 x 50 mL). The combined organics were dried over sodium sulfate.
- the crude alcohol was purified by flash chromatography on SiO 2 eluting with acetone/chloroform (0-5-10% acetone) to give 2.71 g (71% for 2-steps) of intermediate Compound 89B as a clear oil.
- the reaction was then quenched with H 2 O (25 mL) and diluted with CH 2 C1 2 (100 mL). The mixture was then washed sequentially with 1 N HCl (1 x 100 mL), saturated aqueous NaHCO 3 (50 mL), and water (2 x 50 mL). The organics were dried over anhydrous sodium sulfate and the volatile organics removed in vacuo.
- the crade aldehyde intermediate (1.60 g, 4.34 mmol) was dissolved in THF (25 mL) and diethyl cyanophosphonate (90%, 0.95 mL, 5.64 mmol) was added followed by benzyl amine (1.23 L, 11.3 mmol).
- HPLC RT 3.133 min (100%) (YMC S5 ODS column, 4.6 X 50 mm; 10-90% MeOH/H 2 O gradient ⁇ 0.1% TFA; 4 mL/min, 220 nM detection); both as white foams.
- This example illustrates a preferred method for obtaining a compound of formula Ila, which compound is useful as an intermediate in the preparation of compounds of formula I (see, for example, Figure 2 herein).
- Cis-4-hydroxy-D-proline (10.0 g, 131.1 mmol) was suspended in absolute EtOH (100 mL) and anhydrous HCl (g) was bubbled through the reaction until a homogenous solution resulted. This was left at 25°C for 1 h and then the volatiles organics were removed in vacuo. The resulting HCl salt was triturated with diethyl ether and filtered to give the crude ethyl ester as a white powder. The ethyl ester salt was used directly in the next reaction.
- the salt (-12 g) was suspended in acetone and cooled to 0°C. 10% aq Na 2 C0 3 (6.0 mL) was then added followed by BOC 2 0 (1.37 g, 6.29 mmol) and then the reaction was slowly warmed to 25°C. After 12 h, the reaction mixture was poured into water and extracted with methylene chloride (3 x 100 mL). The organics were then dried over anhydrous sodium sulfate and concentrated in vacuo to give the crude compound 92A as a white powder. This material was taken on without further purification.
- HPLC RT 3.280 min (100%) (YMC S5 ODS column, 4.6 X 50 mm; 10-90% MeOH/H 2 0 gradient, + 0.1 % TFA; 4 mL/min, 220 nM detection) and Compound 93ii (0.061 g, 16%) MS (ES): m/z 437.09 [M+H] + .
- HPLC RT 3.133 min (100%) (YMC S5 ODS column, 4.6 X 50 mm; 10-90% MeOH/H 2 O gradients 0.1% TFA; 4 mL/min, 220 nM detection); both as white foams.
- N,N-(Diisopropyl)aminomethyl polystyrene (3.49 mmol/g, 60 mg) was then added to each reaction vessel followed by the addition of the desired acid chloride, isocyanate, chloroformate or sulfonyl chloride (0.10 mmol) in 0.5 mL dichloroethane by automated synthesizer.
- the reaction vessels were shaken at 25°C for 24 h and then Tris-(2- Aminoethyl)amine Polystyrene HL (200-400 mesh, 3.3 mmol/g, 75 mg) was added to each reaction vessel and the vessels shaken again for 18 h at 25°C.
- the tube was shaken at 25°C for 24 h and then Tris-(2-Aminoethyl)amine Polystyrene HL (200-400 mesh, 3.3 mmol/g, 75 mg) was added to the reaction vessel and it was shaken again for 18 h at 25°C.
- the liquid was drained into a pretared 2.5 ml STR tube and the resin was rinsed with dichloromethane (3 x 0.25 L).
- N,N- (Diisopropyl)aminomethyl polystyrene (3.49 mmol/g, 65 mg) was then added to each reaction vessel followed by addition of 4-fluorophenylchloro formate (0.033 g, 0.19 mmol).
- the tube was shaken at 25°C for 24 h and then Tris-(2-Aminoethyl)amine Polystyrene HL (200-400 mesh, 3.3 mmol/g, 75 mg) was added to the reaction vessel and it was shaken again for 18 h at 25°C.
- the liquid was drained into a pretared 2.5 ml STR tube and the resin was rinsed with dichloromethane (3 x 0.25 L).
- N,N- (Diisopropyl)aminomethyl polystyrene (3.49 mmol/g, 65 mg) was then added to each reaction vessel followed by addition of imidazolesulfonylchloride (0.034 g, 0.19 mmol).
- the tube was shaken at 25°C for 24 h and then Tris-(2-Aminoethyl)amine Polystyrene HL (200-400 mesh, 3.3 mmol/g, 75 mg) was added to the reaction vessel and it was shaken again for 18 h at 25°C.
- the liquid was drained into pretared 2.5 ml STR tube and the resin was rinsed with dichloromethane (3 x 0.25 mL).
- the TFA salt of the compound 95 (0.010 g, 0.022 mmol) was dissolved in DMF (0.5 mL) followed by addition of K 2 CO 3 (0.009 g, 0.088 mmol) and benzyl bromide (0.005 mL, 0.044 mmol). After 1 h, the DMF was removed in vacuo and the crude product was purified by flash chromatography on silica eluting with 5% acetone in chloroform. This gave 0.008 g of compound 103 as a yellow solid. Proton NMR showed an intact hydantoin ring system.
- LCMS YMC S5 ODS column, 4.6 X 50 mm eluting with 10-90% MeOH/H 2 O over 4 minutes containing 0.1% TFA; 4 mL/min, monitoring at 220 nm.
- LCMS* YMC S5 ODS column, 4.6 X 50 mm eluting with 10-90% MeOH H 2 O over 2 minutes containing 0.1% TFA; 4 mL/min, monitoring at 220 nm.
- LCMS YMC S5 ODS column, 4.6 X 50 mm eluting with 10-90% MeOH/H 2 O over 4 minutes containing 0.1% TFA; 4 mL/min, monitoring at 220 nm.
- LCMS* YMC S5 ODS column, 4.6 X 50 mm eluting with 10-90% MeOH/H 2 O over 2 minutes containing 0.1% TFA; 4 mL/min, monitoring at 220 nm.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Neurology (AREA)
- Reproductive Health (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Physical Education & Sports Medicine (AREA)
- Epidemiology (AREA)
- Gynecology & Obstetrics (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Obesity (AREA)
- Virology (AREA)
- Hospice & Palliative Care (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pregnancy & Childbirth (AREA)
- Urology & Nephrology (AREA)
Abstract
Description
















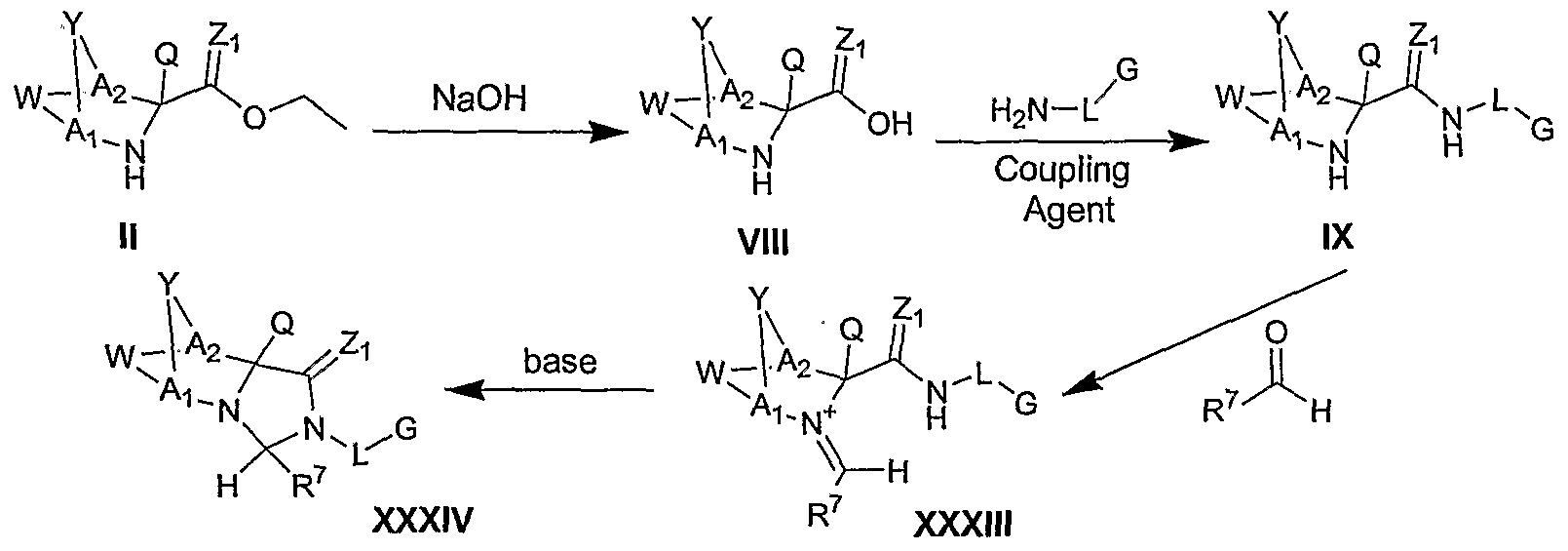







































































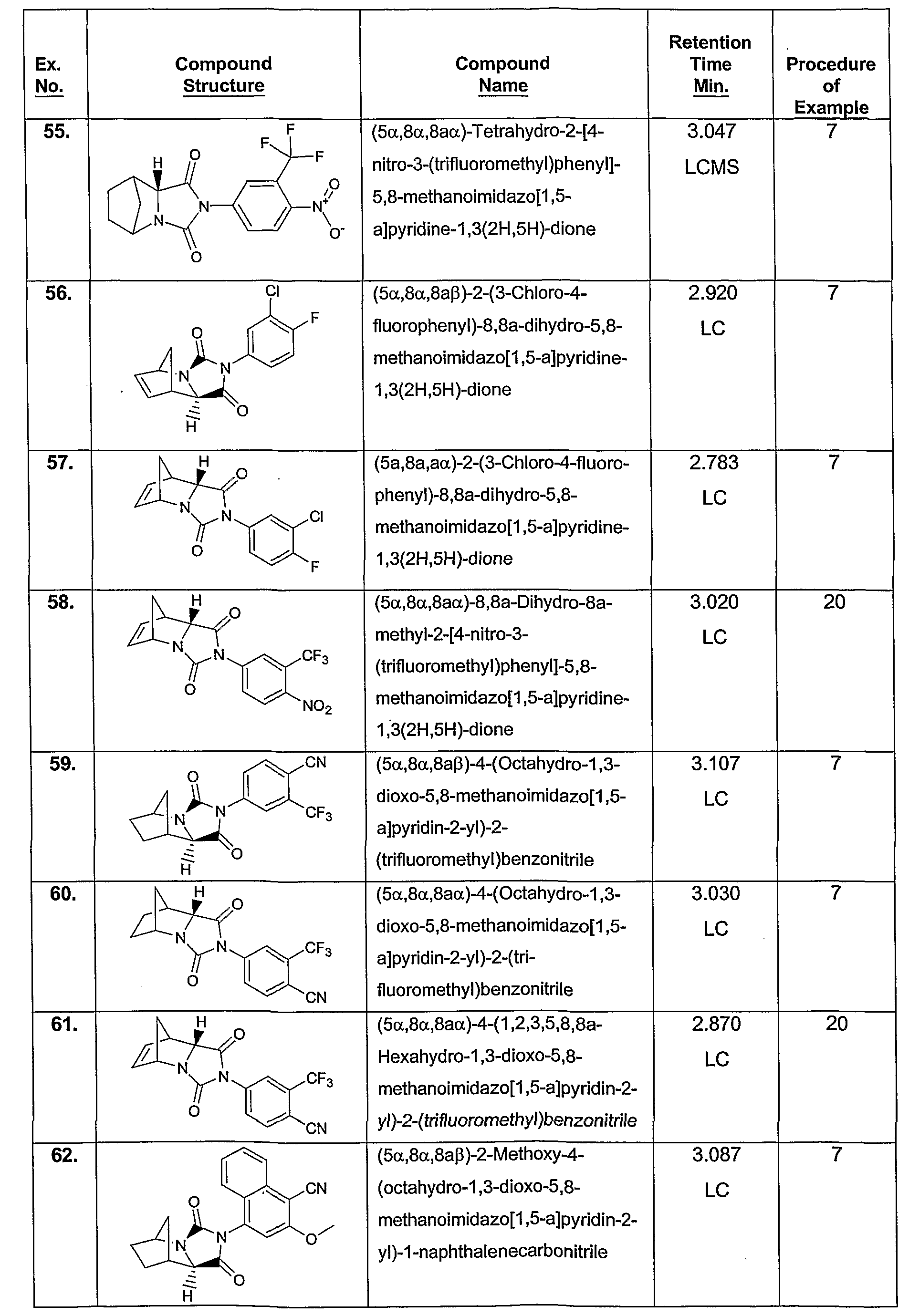




















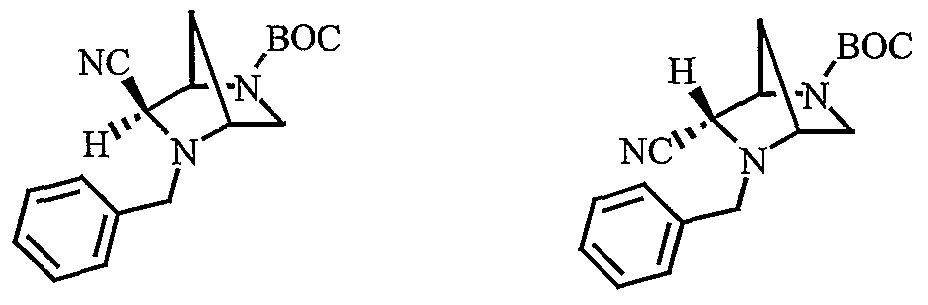
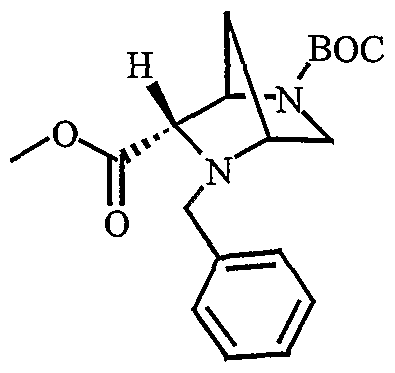






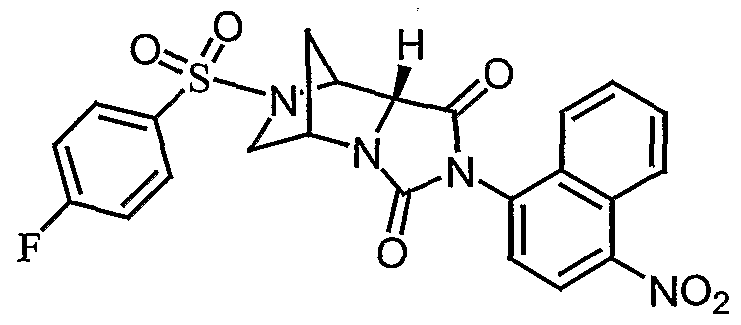





















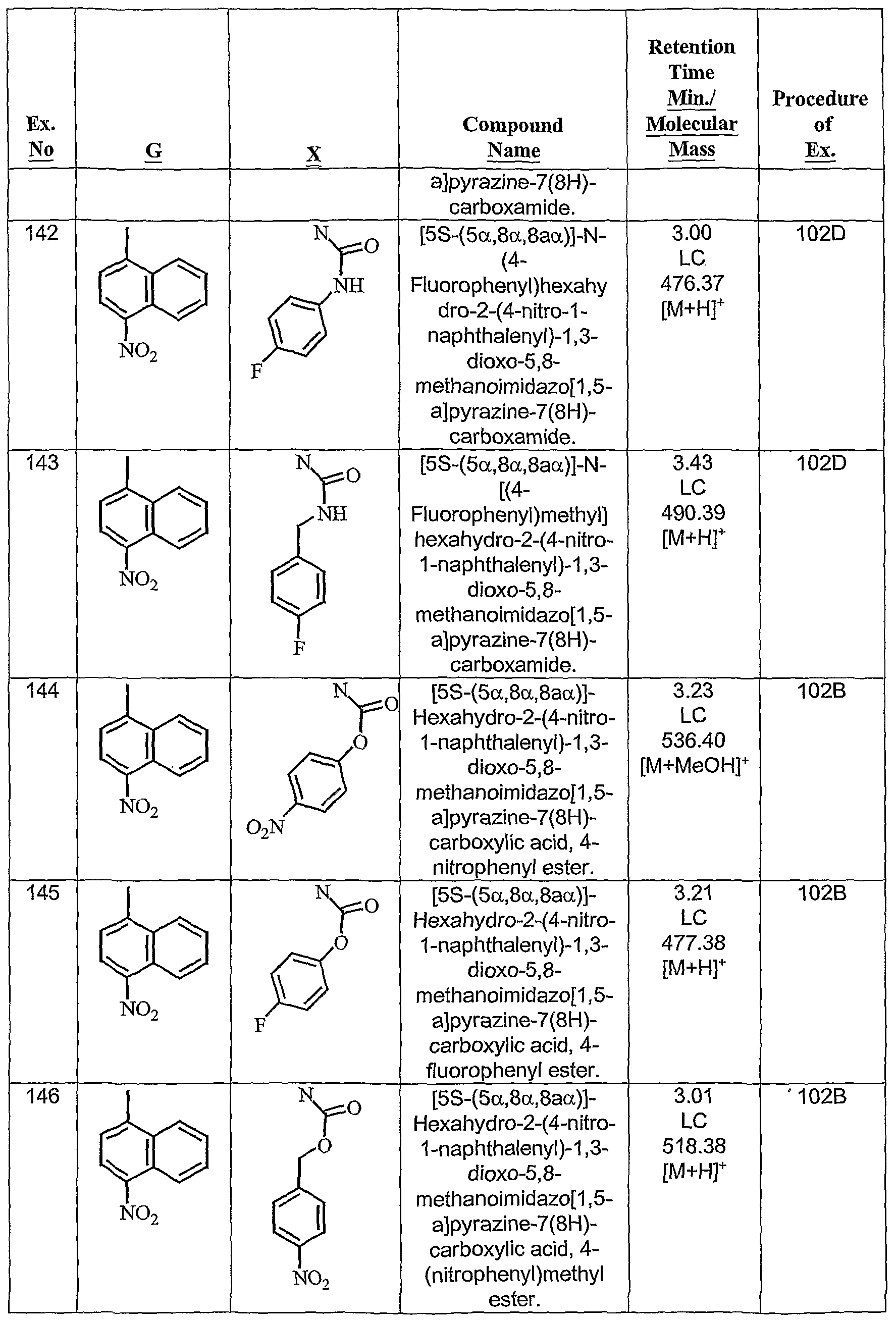








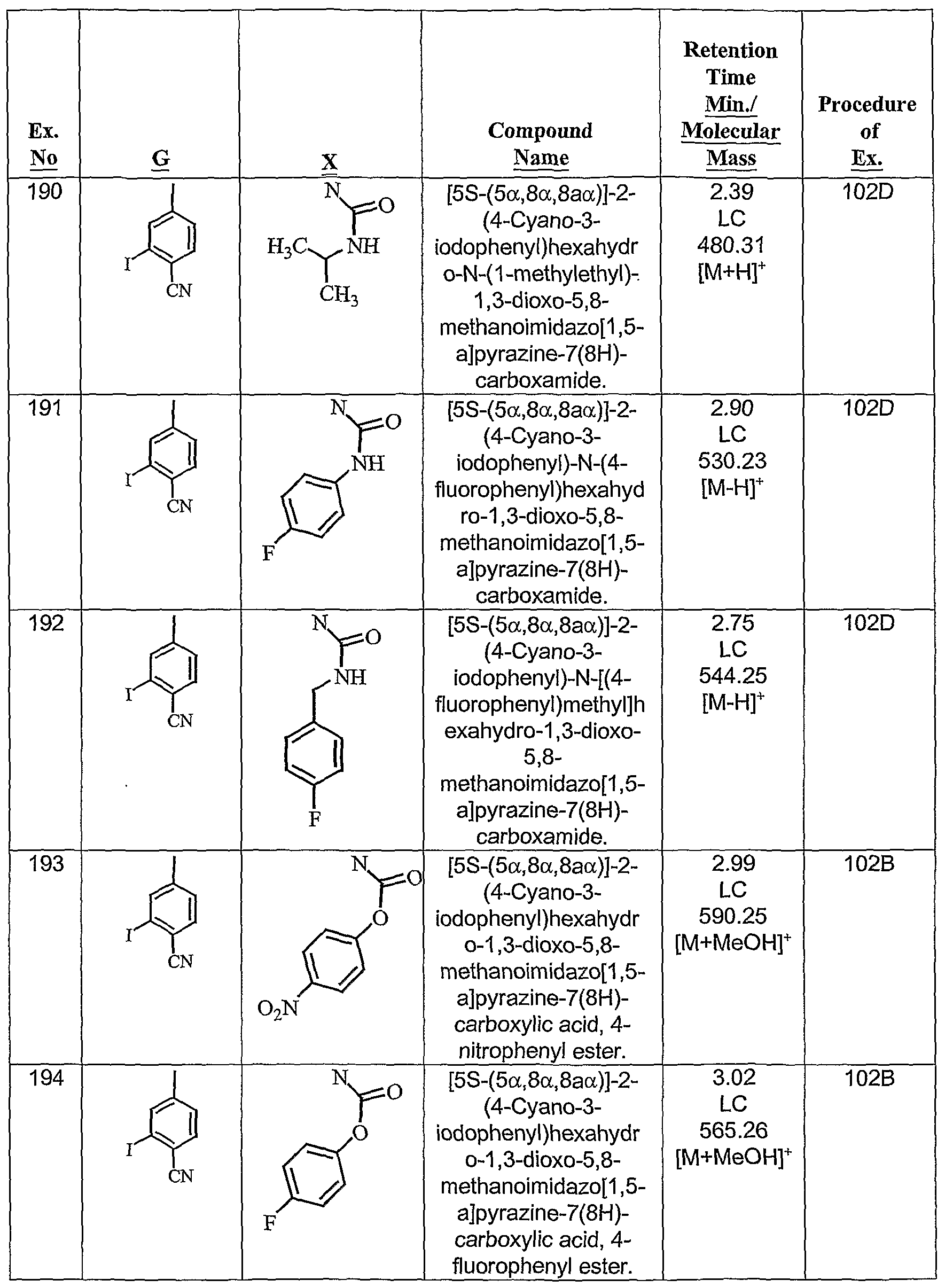

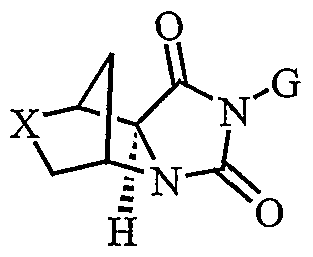


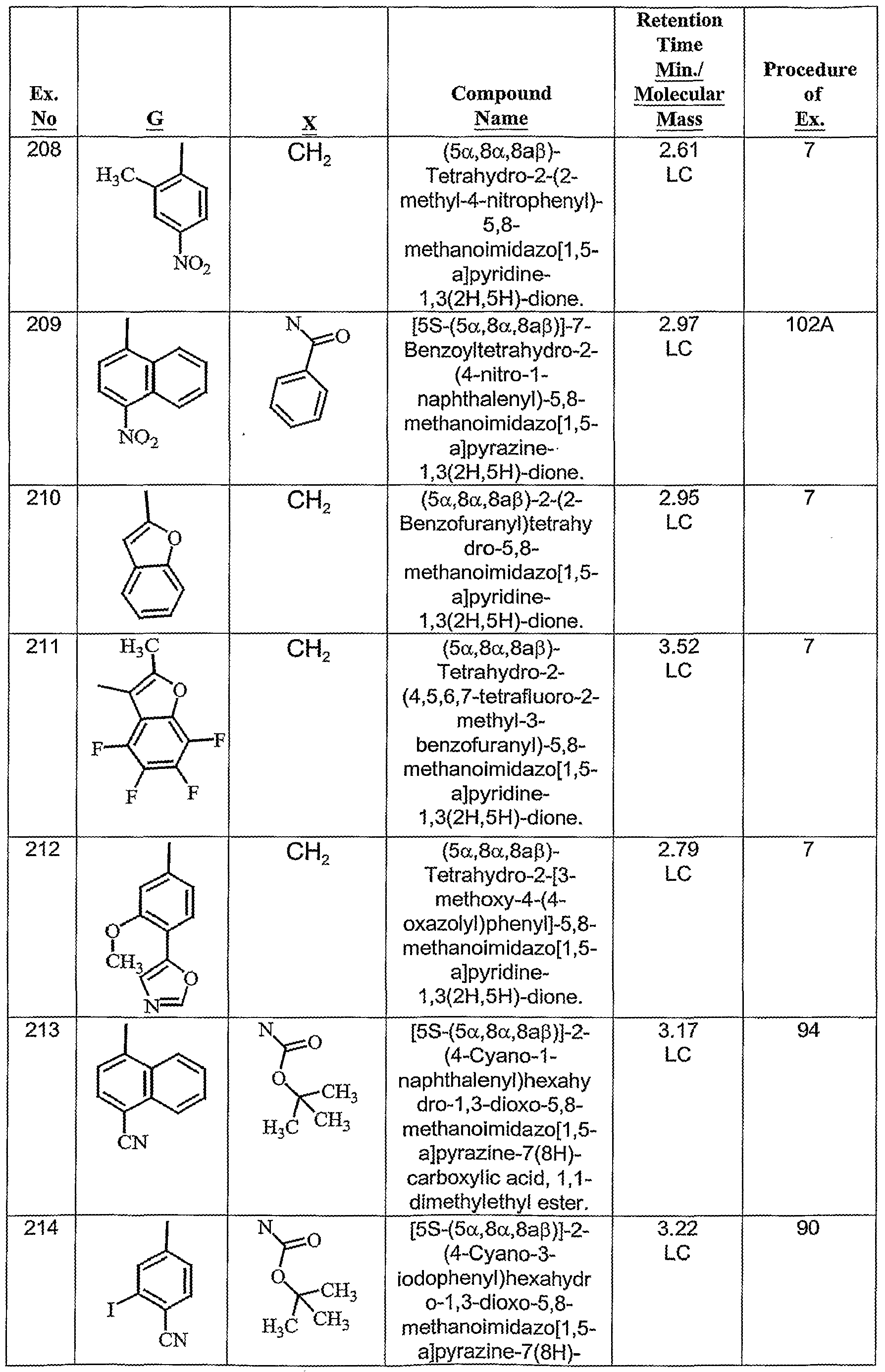

Claims


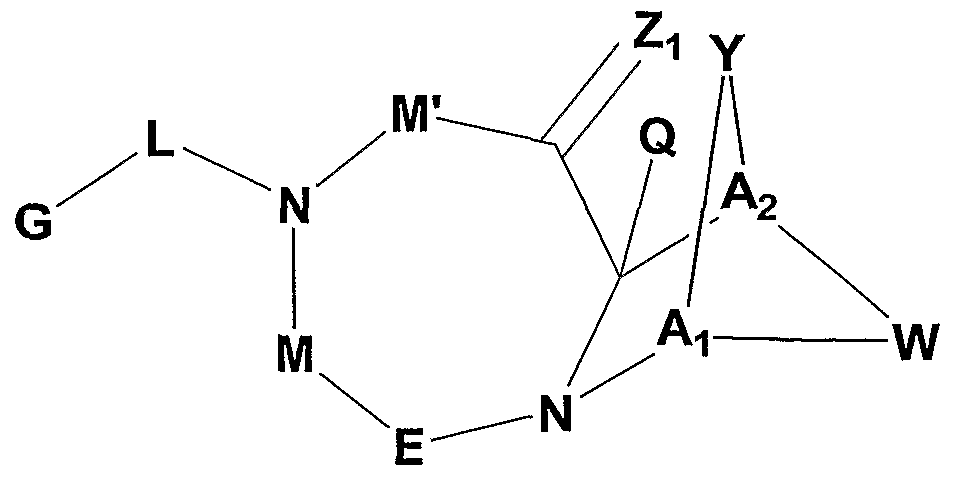















Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IL15289001A IL152890A0 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds and pharmaceutical compositions containing the same |
| JP2002505777A JP2004515462A (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
| HU0303165A HUP0303165A3 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function, process for their preparation and pharmaceutical compositions containing them |
| NZ523802A NZ523802A (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
| EP01984054A EP1299385A2 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic modulators of nuclear hormone receptor function |
| MXPA02012563A MXPA02012563A (en) | 2000-06-28 | 2001-06-20 | Fused cyclic modulators of nuclear hormone receptor function. |
| CA002413683A CA2413683A1 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
| AU2002215609A AU2002215609A1 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
| PCT/US2001/019663 WO2002000653A2 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
| BR0111869-2A BR0111869A (en) | 2000-06-28 | 2001-06-20 | Cyclic compounds fused as modulators of receptor function for nuclear homonium |
| NO20026167A NO20026167D0 (en) | 2000-06-28 | 2002-12-20 | Condensed cyclic compounds as modulators of nuclear hormone receptor function |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21439200P | 2000-06-28 | 2000-06-28 | |
| US60/214,392 | 2000-06-28 | ||
| US28461701P | 2001-04-18 | 2001-04-18 | |
| US28443801P | 2001-04-18 | 2001-04-18 | |
| US60/284,438 | 2001-04-18 | ||
| US60/284,617 | 2001-04-18 | ||
| PCT/US2001/019663 WO2002000653A2 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002000653A2 true WO2002000653A2 (en) | 2002-01-03 |
| WO2002000653A3 WO2002000653A3 (en) | 2002-07-25 |
Family
ID=27485945
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/019663 WO2002000653A2 (en) | 2000-06-28 | 2001-06-20 | Fused cyclic compounds as modulators of nuclear hormone receptor function |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP1299385A2 (en) |
| JP (1) | JP2004515462A (en) |
| AU (1) | AU2002215609A1 (en) |
| BR (1) | BR0111869A (en) |
| CA (1) | CA2413683A1 (en) |
| HU (1) | HUP0303165A3 (en) |
| MX (1) | MXPA02012563A (en) |
| NO (1) | NO20026167D0 (en) |
| NZ (1) | NZ523802A (en) |
| WO (1) | WO2002000653A2 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002000617A2 (en) * | 2000-06-28 | 2002-01-03 | Bristol-Myers Squibb Company | Selective androgen receptor modulators and methods for their identification, design and use |
| WO2002020526A2 (en) * | 2000-09-07 | 2002-03-14 | Bayer Pharmaceuticals Corporation | Cyclic and acyclic amidines and pharmaceutical compositions containing them for use as progesterone receptor binding agents |
| US6586368B1 (en) * | 1998-09-10 | 2003-07-01 | Sumitomo Chemical Company, Limited | Condensed heterocylic compounds and herbicides containing them |
| WO2003080118A2 (en) * | 2002-03-18 | 2003-10-02 | Pharmacia Corporation | Combination of an aldosterone receptor antagonist and a fibric acid derivative |
| EP1463728A2 (en) * | 2001-12-19 | 2004-10-06 | Bristol-Myers Squibb Company | Fused cyclic modulators of nuclear hormone receptor function |
| EP1467979A2 (en) * | 2001-12-19 | 2004-10-20 | Bristol-Myers Squibb Company | FUSED HETEROCYCLIC COMPOUNDS AND ANALOGS THEREOF: MODULATORS OF NUCLEAR HORMONE RECEPTOR FUNCTION |
| EP1722793A1 (en) * | 2004-03-04 | 2006-11-22 | Brystol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US7199125B2 (en) | 2003-10-02 | 2007-04-03 | Bristol-Myers Squibb Company | Spiro-cyclic compounds useful as anti-inflammatory agents |
| US7405234B2 (en) | 2002-05-17 | 2008-07-29 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US7417040B2 (en) | 2004-03-01 | 2008-08-26 | Bristol-Myers Squibb Company | Fused tricyclic compounds as inhibitors of 17β-hydroxysteroid dehydrogenase 3 |
| US7649001B2 (en) | 2002-08-12 | 2010-01-19 | Takeda Pharmaceutical Company Limited | Fused benzene derivative and use |
| US7696241B2 (en) | 2004-03-04 | 2010-04-13 | Bristol-Myers Squibb Company | Bicyclic compounds as modulators of androgen receptor function and method |
| US7820702B2 (en) | 2004-02-04 | 2010-10-26 | Bristol-Myers Squibb Company | Sulfonylpyrrolidine modulators of androgen receptor function and method |
| US8470823B2 (en) | 2008-08-29 | 2013-06-25 | Instituto Di Ricerche Di Biologia Molecolare P. Angeletti S.R.L. | Saturated bicyclic heterocyclic derivatives as SMO antagonists |
| US9051267B2 (en) | 2005-11-28 | 2015-06-09 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
| US9409856B2 (en) | 2005-11-28 | 2016-08-09 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
| US9427418B2 (en) | 2009-02-23 | 2016-08-30 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
| US9624161B2 (en) | 2009-02-23 | 2017-04-18 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10162375A1 (en) * | 2001-12-19 | 2003-07-10 | Bayer Ag | Bicyclic N-aryl amides |
| ES2377667T3 (en) * | 2006-02-10 | 2012-03-29 | Janssen Pharmaceutica N.V. | Heterocycles of bicyclic imidazole or thiadiazole useful as selective androgen receptor modulators |
| WO2008007664A1 (en) | 2006-07-11 | 2008-01-17 | Takeda Pharmaceutical Company Limited | Bicyclic heterocyclic compound and use thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996019458A2 (en) * | 1994-12-22 | 1996-06-27 | Ligand Pharmaceuticals Incorporated | Steroid receptor modulator compounds and methods |
| WO1997049709A1 (en) * | 1996-06-27 | 1997-12-31 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| WO2002000617A2 (en) * | 2000-06-28 | 2002-01-03 | Bristol-Myers Squibb Company | Selective androgen receptor modulators and methods for their identification, design and use |
-
2001
- 2001-06-20 WO PCT/US2001/019663 patent/WO2002000653A2/en active IP Right Grant
- 2001-06-20 JP JP2002505777A patent/JP2004515462A/en active Pending
- 2001-06-20 NZ NZ523802A patent/NZ523802A/en unknown
- 2001-06-20 HU HU0303165A patent/HUP0303165A3/en unknown
- 2001-06-20 BR BR0111869-2A patent/BR0111869A/en not_active IP Right Cessation
- 2001-06-20 EP EP01984054A patent/EP1299385A2/en not_active Withdrawn
- 2001-06-20 MX MXPA02012563A patent/MXPA02012563A/en unknown
- 2001-06-20 CA CA002413683A patent/CA2413683A1/en not_active Abandoned
- 2001-06-20 AU AU2002215609A patent/AU2002215609A1/en not_active Abandoned
-
2002
- 2002-12-20 NO NO20026167A patent/NO20026167D0/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996019458A2 (en) * | 1994-12-22 | 1996-06-27 | Ligand Pharmaceuticals Incorporated | Steroid receptor modulator compounds and methods |
| WO1997049709A1 (en) * | 1996-06-27 | 1997-12-31 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| WO2002000617A2 (en) * | 2000-06-28 | 2002-01-03 | Bristol-Myers Squibb Company | Selective androgen receptor modulators and methods for their identification, design and use |
Non-Patent Citations (7)
| Title |
|---|
| DENISON,M.S. ET AL.: "Xenobiotic-inducible Transcription of Cytochrome P450 Genes" J.BIOL.CHEM., vol. 270, no. 31, 4 August 1995 (1995-08-04), pages 18175-18178, XP002188273 ROCKVILLE PIKE * |
| EVANS R M: "THE STERIOD AND THYRIOD HORMONE RECEPTOR SUPERFAMILY" SCIENCE, AMERICAN ASSOCIATION FOR THE ADVANCEMENT OF SCIENCE, US, vol. 240, no. 4854, 13 May 1988 (1988-05-13), pages 889-895, XP000574469 ISSN: 0036-8075 * |
| REMUZON P ET AL: "FLUORONAPHTHYRIDINES AS ANTIBACTERIAL AGENTS. 6. SYNTHESIS AND STRUCTURE-ACTIVITY RELATIONSHIPS OF NEW CHIRAL 7-(1-,3-,4-, AND 6-METHYL-2,5-DIAZABICLOU2.2.1HEPTAN-2-YL) NAPHTHYRIDINE ANALOGUES OF 7-U(1R,4R)-2,5-DIAZABICYCLOU2.2.1 HEPTAN-2-YL-1-(1,1-DIMETHYLETHYL)-6 -FLUORO-1,4-DIHYDRO-4-OXO- 1,8-NAP" JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 35, no. 15, 1992, pages 2898-2909, XP002034280 ISSN: 0022-2623 * |
| ROSEN,T. ET AL.: "Design, Synthesis and Properties of (4S)-7-(4-Amino-2-substituted-pyrrolidin-1 -yl)quinolinone-3-carboxylic acids" J.MED.CHEM., vol. 31, no. 8, 1988, pages 1598-1611, XP001026364 WASHINGTON * |
| TANAKA K-I ET AL: "Diastereoselective Syntheses of (2R,3R,5R)- and (2S,3S,5S)-3-Hydroxy-5-methyl-2-pyrrolidin ecarboxylic Acid as a Component of Actinomycin Z1" TETRAHEDRON, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 54, no. 34, 20 August 1998 (1998-08-20), pages 10029-10042, XP004129981 ISSN: 0040-4020 * |
| WALTER,W.G.: "Antitumor Imide Derivatives of 7-Oxabicyclo[2.2.1]heptane-2,3-di- methyl-2,3-dicarboxylic acid" J.PHARM.SCI., vol. 78, no. 1, January 1989 (1989-01), pages 66-67, XP002188687 * |
| YU,B. ET AL.: "Pharmacology of some natural products of china" TRENDS IN PHARMACOLOGICAL SCIENCE, vol. 2, no. 10, October 1981 (1981-10), pages 271-272, XP002188686 AMSTERDAM * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6586368B1 (en) * | 1998-09-10 | 2003-07-01 | Sumitomo Chemical Company, Limited | Condensed heterocylic compounds and herbicides containing them |
| WO2002000617A2 (en) * | 2000-06-28 | 2002-01-03 | Bristol-Myers Squibb Company | Selective androgen receptor modulators and methods for their identification, design and use |
| WO2002000617A3 (en) * | 2000-06-28 | 2003-01-30 | Bristol Myers Squibb Co | Selective androgen receptor modulators and methods for their identification, design and use |
| WO2002020526A2 (en) * | 2000-09-07 | 2002-03-14 | Bayer Pharmaceuticals Corporation | Cyclic and acyclic amidines and pharmaceutical compositions containing them for use as progesterone receptor binding agents |
| WO2002020526A3 (en) * | 2000-09-07 | 2002-05-30 | Bayer Ag | Cyclic and acyclic amidines and pharmaceutical compositions containing them for use as progesterone receptor binding agents |
| EP1463728A4 (en) * | 2001-12-19 | 2005-12-21 | Bristol Myers Squibb Co | Fused cyclic modulators of nuclear hormone receptor function |
| EP1463728A2 (en) * | 2001-12-19 | 2004-10-06 | Bristol-Myers Squibb Company | Fused cyclic modulators of nuclear hormone receptor function |
| EP1467979A2 (en) * | 2001-12-19 | 2004-10-20 | Bristol-Myers Squibb Company | FUSED HETEROCYCLIC COMPOUNDS AND ANALOGS THEREOF: MODULATORS OF NUCLEAR HORMONE RECEPTOR FUNCTION |
| EP1467979A4 (en) * | 2001-12-19 | 2006-03-15 | Bristol Myers Squibb Co | Fused heterocyclic compounds and analogs thereof: modulators of nuclear hormone receptor function |
| WO2003080118A3 (en) * | 2002-03-18 | 2004-01-15 | Pharmacia Corp | Combination of an aldosterone receptor antagonist and a fibric acid derivative |
| WO2003080118A2 (en) * | 2002-03-18 | 2003-10-02 | Pharmacia Corporation | Combination of an aldosterone receptor antagonist and a fibric acid derivative |
| US7405234B2 (en) | 2002-05-17 | 2008-07-29 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US7649001B2 (en) | 2002-08-12 | 2010-01-19 | Takeda Pharmaceutical Company Limited | Fused benzene derivative and use |
| US7199125B2 (en) | 2003-10-02 | 2007-04-03 | Bristol-Myers Squibb Company | Spiro-cyclic compounds useful as anti-inflammatory agents |
| US7820702B2 (en) | 2004-02-04 | 2010-10-26 | Bristol-Myers Squibb Company | Sulfonylpyrrolidine modulators of androgen receptor function and method |
| US7989640B2 (en) | 2004-02-04 | 2011-08-02 | Bristol-Myers Squibb Company | Sulfonylpyrrolidine modulators of androgen receptor function and method |
| US7417040B2 (en) | 2004-03-01 | 2008-08-26 | Bristol-Myers Squibb Company | Fused tricyclic compounds as inhibitors of 17β-hydroxysteroid dehydrogenase 3 |
| US7625923B2 (en) | 2004-03-04 | 2009-12-01 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| EP1722793A4 (en) * | 2004-03-04 | 2009-12-16 | Bristol Myers Squibb Co | Bicyclic modulators of androgen receptor function |
| EP1722793A1 (en) * | 2004-03-04 | 2006-11-22 | Brystol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US7696241B2 (en) | 2004-03-04 | 2010-04-13 | Bristol-Myers Squibb Company | Bicyclic compounds as modulators of androgen receptor function and method |
| US9051267B2 (en) | 2005-11-28 | 2015-06-09 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
| US9409856B2 (en) | 2005-11-28 | 2016-08-09 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
| US8470823B2 (en) | 2008-08-29 | 2013-06-25 | Instituto Di Ricerche Di Biologia Molecolare P. Angeletti S.R.L. | Saturated bicyclic heterocyclic derivatives as SMO antagonists |
| US9427418B2 (en) | 2009-02-23 | 2016-08-30 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
| US9624161B2 (en) | 2009-02-23 | 2017-04-18 | Gtx, Inc. | Estrogen receptor ligands and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| NO20026167L (en) | 2002-12-20 |
| EP1299385A2 (en) | 2003-04-09 |
| MXPA02012563A (en) | 2003-05-14 |
| NO20026167D0 (en) | 2002-12-20 |
| BR0111869A (en) | 2003-09-23 |
| CA2413683A1 (en) | 2002-01-03 |
| NZ523802A (en) | 2005-11-25 |
| HUP0303165A3 (en) | 2006-05-29 |
| WO2002000653A3 (en) | 2002-07-25 |
| AU2002215609A1 (en) | 2002-01-08 |
| JP2004515462A (en) | 2004-05-27 |
| HUP0303165A2 (en) | 2003-12-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7001911B2 (en) | Fused cyclic modulators of nuclear hormone receptor function | |
| EP1463728B1 (en) | Fused cyclic modulators of nuclear hormone receptor function | |
| EP1458723B1 (en) | Fused heterocyclic succinimide compounds and analogs thereof, modulators of nuclear hormone receptor function | |
| AU2001269943B2 (en) | Fused heterocyclic succinimide compounds and analogs thereof, modulators of nuclear hormone receptor function | |
| US20080214643A1 (en) | Fused heterocyclic succinimide compounds and analogs thereof, modulators of nuclear hormone receptor function | |
| EP1467979B1 (en) | Fused heterocyclic compounds and analogs thereof modulators of nuclear hormone receptor function | |
| US20050124625A1 (en) | Piperazine derivatives and their use as modulators of nuclear hormone receptor function | |
| EP1299385A2 (en) | Fused cyclic modulators of nuclear hormone receptor function | |
| AU2001269943A1 (en) | Fused heterocyclic succinimide compounds and analogs thereof, modulators of nuclear hormone receptor function | |
| AU2002361785A1 (en) | Fused heterocyclic succinimidecompounds and analogs thereof, modulators of nuclear hormone receptor function | |
| US6953679B2 (en) | Method for the preparation of fused heterocyclic succinimide compounds and analogs thereof | |
| US7655689B2 (en) | Fused heterocyclic succinimide compounds and analogs thereof, modulators of nuclear hormone receptor function | |
| KR20030014407A (en) | Fused Cyclic Modulators of Nuclear Hormone Receptor Function |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 152890 Country of ref document: IL |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/012563 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2413683 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/10313 Country of ref document: ZA Ref document number: 200210313 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2002-4250 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002215609 Country of ref document: AU Ref document number: 2002/02702 Country of ref document: TR |
|
| ENP | Entry into the national phase |
Ref document number: 2002 505777 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 02116482 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 01811976X Country of ref document: CN Ref document number: 1020027017857 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5/MUMNP/2003 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 523802 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2003101918 Country of ref document: RU Kind code of ref document: A Ref country code: RU Ref document number: RU A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001984054 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027017857 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001984054 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-4250 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 523802 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 523802 Country of ref document: NZ |