WO1999018124A1 - Assays for nuclear receptor ligands using fret - Google Patents
Assays for nuclear receptor ligands using fret Download PDFInfo
- Publication number
- WO1999018124A1 WO1999018124A1 PCT/US1998/021049 US9821049W WO9918124A1 WO 1999018124 A1 WO1999018124 A1 WO 1999018124A1 US 9821049 W US9821049 W US 9821049W WO 9918124 A1 WO9918124 A1 WO 9918124A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nuclear receptor
- binding domain
- ligand binding
- cbp
- human
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/536—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase
- G01N33/542—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase with steric inhibition or signal modification, e.g. fluorescent quenching
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/566—Immunoassay; Biospecific binding assay; Materials therefor using specific carrier or receptor proteins as ligand binding reagents where possible specific carrier or receptor proteins are classified with their target compounds
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6875—Nucleoproteins
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/70567—Nuclear receptors, e.g. retinoic acid receptor [RAR], RXR, nuclear orphan receptors
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
Definitions
- This invention relates to methods of identifying novel agonists and antagonists of nuclear receptors utilizing the agonist- dependent interaction of such receptors with CREB-binding protein
- CBP nuclear receptor co-activators
- Nuclear receptors are a superfamily of ligand-activated transcription factors that bind as homodimers or heterodimers to their cognate DNA elements in gene promoters.
- the superfamily with more than 150 members, can be divided into subfamilies (e.g. the steroid, retinoid, thyroid hormone, and peroxisome proliferator-activated [PPAR] subfamilies).
- Each subfamily may consist of several members which are encoded by individual genes (e.g. PPAR ⁇ , PPAR ⁇ , and PPAR ⁇ ).
- alternative mRNA splicing can result in more than one isoform of these genes as in the case of specific PPARs (e.g.
- the nuclear receptor superfamily is involved in a wide variety of physiological functions in mammalian cells: e.g., differentiation, proliferation, and metabolic homeostasis. Dysfunction or altered expression of specific nuclear receptors has been found to be involved in disease pathogenesis.
- the PPAR subfamily of nuclear receptors consists of three members: PPAR ⁇ , PPAR ⁇ , and PPAR ⁇ .
- PP.AR ⁇ is highly expressed in liver and kidney.
- Activation of PPAR ⁇ by peroxisome proliferators (including hypolipidimic reagents such as fibrates) or medium and long-chain fatty acids is responsible for the induction of acyl-CoA oxidase and hydratase-dehydrogenase (enzymes required for peroxisomal ⁇ -oxidation), as well as cytochrome P450 4A6 (an enzyme required for fatty acid ⁇ -hydroxylase).
- PPAR ⁇ has an important role in the regulation of lipid metabolism and is part of the mechanism through which hypolipidimic compounds such as fibrates exert their effects. PPAR ⁇ is predominantly expressed in adipose tissue. Recently, a prostaglandin J2 metabolite, 15-Deoxy-D12,14-prostaglandin J2, has been identified as a potential physiological ligand of PPAR ⁇ . Both 15- Deoxy-D12,14-prostaglandin J2 treatment of preadipocytes or retroviral expression of PPAR ⁇ 2 in fibroblasts induced adipocyte differentiation, demonstrating the role of PPAR ⁇ in adipocyte differentiation and lipid storage.
- thiazolidinediones are high affinity ligands for PPAR ⁇ suggesting a broad therapeutic role for PPAR ⁇ ligands in the treatment of diabetes and disorders associated with insulin resistance (e.g. obesity and cardiovascular disease).
- Nuclear receptor proteins contain a central DNA binding domain (DBD) and a COOH-terminal ligand binding domain (LBD).
- DBD central DNA binding domain
- LBD COOH-terminal ligand binding domain
- the DBD is composed of two highly conserved zinc fingers that target the receptor to specific promoter/enhancer DNA sequences known as hormone response elements (HREs).
- HREs hormone response elements
- the LBD is about 200-300 amino acids in length and is less well conserved than the DBD.
- the transactivation function can be viewed as a molecular switch between a transcriptionally inactive and a transcriptionally active state of the receptor. Binding of a ligand which is an agonist flips the switch from the inactive state to the active state.
- the COOH-terminal portion of the LBD contains an activation function domain (AF2) that is required for the switch.
- AF2 activation function domain
- the ligand-induced nuclear receptor molecular switch is mediated through interactions with members of a family of nuclear receptor co-activators (e.g., CBP/p300, SRC-l/NcoA-1, TIF2/GRIP- l/NcoA-2, and p/CIP).
- CBP/p300 nuclear receptor co-activators
- SRC-l/NcoA-1 e.g., SRC-l/NcoA-1
- TIF2/GRIP- l/NcoA-2 e.g., TIF2/GRIP- l/NcoA-2
- p/CIP nuclear receptor co-activators
- CBP co-activators CREB-binding protein
- p300 are two closely related proteins that were originally discovered by virtue of their ability to interact with the transcription factor CREB. These two proteins share extensive amino acid sequence homology. CBP can form a bridge between nuclear receptors and the basic transcriptional machinery (Kamei et al., 1996, Cell 85:403-414; Chakravarti et al., 1996, Nature 383:99-103; Hanstein et al., 1996, Proc. Natl. Acad. Sci.
- CBP also contains intrinsic histone acetyltransferase activity which could result in local chromatin rearrangement and further activation of transcription.
- Ligand- and AF2-dependent interaction between certain nuclear receptors and CBP has been demonstrated in in vitro pull down assays and far- western assays. This interaction is both necessary and sufficient for the transcriptional activation that is mediated by these nuclear receptors.
- an AF2 mutant of the estrogen receptor (ER) which abolishes the transcriptonal function of the receptor is incapable of interacting with CBP.
- Kamei produced GST fusion proteins of the first 100 amino acids of the N-terminus of CBP. These fusion proteins were run out on a polyacrylamide gel, transferred to a membrane, and the membrane was exposed to 32p.l a beled ligand-binding domains of nuclear receptors. In the presence of ligand, a specific binding interaction between the CBP and nuclear receptor fragments was detected in that the 32p-l a beled ligand-binding domains were observed to bind to the bands on the membrane containing the GST-CBP fusion proteins.
- Kamei found that a fragment of CBP (the N- terminal 100 amino acids) was capable of binding to the nuclear receptor-ligand-oligonucleotide complex and shifting the complex's position on the gel to an even higher molecular weight. (4) Kamei was able to co-immunoprecipitate CBP using antibodies to nuclear receptors in extracts from a variety of cells in the presence of ligand.
- Kamei was able to demonstrate specific binding interactions between CBP and the retinoic acid receptor (RAR), glucocorticoid receptor (GR), thyroid hormone receptor (T3R), and retinoid X receptor (RXR). Kamei also demonstrated specific binding between the N-terminus of p300 and RAR. However, Kamei did not demonstrate specific binding between CBP, p300, or any other nuclear receptor co-activators and PPARs. What is striking about the methods used by Kamei is their extremely laborious and time consuming nature.
- Such methods involve, among other things, the construction of fusion proteins, the preparation of 32p-l a beled proteins, the construction of specialized expression vectors for the yeast two-hybrid assay and the transcriptional activation assays, the running of many gels, and the raising of antibodies. Most of these assays take days to carry out and preparing the reagents needed to carry them out may take weeks. Because of the complicated reagents that are involved in these assays and the time needed to prepare and run the assays, these assays tend to be costly.
- Kamei did not use the above-described methods to identify novel agonists or antagonists of nuclear receptors. The focus of Kamei was not on agonists or antagonists, but rather on the interaction between nuclear receptors and CBP.
- the present invention provides novel methods of identifying agonists and antagonists of nuclear receptors.
- the methods take advantage of the agonist-dependent binding of nuclear receptors and CBP, p300, or other nuclear receptor co-activators. In the absence of agonist, binding between the nuclear receptor and CBP, p300, or other nuclear receptor co-activators does not occur. If agonist is present, however, such binding occurs and can be detected by fluorescence resonance energy transfer (FRET) between a fluorescently-labeled nuclear receptor and fluorescently-labeled CBP, p300, or other nuclear receptor co-activator.
- FRET fluorescence resonance energy transfer
- Antagonists can be identified by virtue of their ability to prevent or disrupt the agonist-induced interaction of nuclear receptors and CBP, p300, or other nuclear receptor co-activators.
- the methods of the present invention are simple, rapid, and less costly.
- the present invention provides a nuclear receptor or ligand binding domain thereof labeled with a fluorescent reagent for use in the above-described methods of identifying agonists and antagonists of nuclear receptors.
- the present invention also provides CBP, p300, or other nuclear receptor co-activator, or a binding portion thereof, labeled with a fluorescent reagent.
- Figure 1 illustrates a method of fluorescently labelling a protein or polypeptide with Europium cryptate (Eu3+K).
- FIG. 1 illustrates the format for experiments 1 and 2 of
- FIG. 3 illustrates the format for experiment 3 of
- Figure 5 shows the results of studies using the methods of the present invention with four known PPAR ⁇ agonists.
- -o ⁇ AD5075;
- -D-- Pioglitazone;
- -X- Troglitazone;
- -0-- BRL49653.
- Figure 6 shows a measurement of the binding constant for the interaction between hCBP and PPAR ⁇ lLBD.
- Figure 7A shows the amino acid sequence of human CBP (SEQ.ID.NO.:l).
- Figure 7B shows the nucleotide sequence of a cDNA encoding human CBP (SEQ.ID.NO.:2).
- the open reading frame is at positions 76-1290.
- Figure 8A shows the amino acid sequence of human PPAR ⁇ (SEQ.ID.NO.:3).
- Figure 8B shows the nucleotide sequence of a cDNA encoding human PPAR ⁇ (SEQ.ID.NO.:4).
- the open reading frame is at positions 217-1623.
- Figure 9A shows the amino acid sequence of human PPAR ⁇ l (SEQ.ID.NO.:5).
- Figure 9B shows the nucleotide sequence of a cDNA encoding human PPAR ⁇ l (SEQ.ID.NO.:6).
- the open reading frame is at positions 173-1609.
- Figure 10A shows the amino acid sequence of human PPAR ⁇ (SEQ.ID.NO.:7).
- Figure 10B-C shows the nucleotide sequence of a cDNA encoding human PPAR ⁇ (SEQ.ID.NO.:8).
- the open reading frame is at positions 338-1663.
- an "agonist” is a substance that binds to nuclear receptors in such a way that a specific binding interaction between the nuclear receptor and CBP or other nuclear receptor co-activator can occur.
- an "antagonist” is a substance that is capable of preventing or disrupting the agonist-induced specific binding interaction between a nuclear receptor and CBP, p300, or another nuclear receptor co- activator.
- a "ligand" of a nuclear receptor is an agonist or an antagonist of the nuclear receptor.
- a “specific binding interaction,” “specific binding,” and the like refers to binding between a nuclear receptor and CBP, p300, or other nuclear receptor co-activator which results in the occurrence of fluorescence resonance energy transfer between a fluorescent reagent bound to the nuclear receptor and a fluorescent reagent bound to CBP, p300, or other nuclear receptor co-activator.
- a "binding portion” is that portion of CBP, p300, or other nuclear receptor co-activators that is sufficient for specific binding interactions with nuclear receptors.
- a "ligand binding domain" is that portion of a nuclear receptor that is sufficient to bind an agonist or antagonist of the nuclear receptor.
- the present invention provides a high throughput, time and labor-saving, non-radioactive, inexpensive, and very reliable assay for the identification and characterization of both agonists and antagonists of nuclear receptors.
- the present invention provides methods of identifying agonists and antagonists for any nuclear receptor for which CBP, p300, or another nuclear receptor binding protein is a co-activator. Such agonists and antagonists are identified by virtue of their ability to induce or prevent binding between the ligand binding domain of a nuclear receptor and CBP, p300, or other nuclear receptor co-activator.
- the interaction between the nuclear receptor and CBP, p300, or other nuclear receptor co-activator is monitored by observing the occurrence of fluorescence resonance energy transfer (FRET) between two fluorescent reagents.
- FRET fluorescence resonance energy transfer
- One fluorescent reagent is bound to the nuclear receptor; the other fluorescent reagent is bound to CBP, p300, or other nuclear receptor co-activator.
- the binding of fluorescent reagent to nuclear receptor, CBP, p300, or other nuclear receptor co-activator can be by a covalent linkage or a non-covalent linkage.
- FRET fluorescence resonance energy transfer
- FRET is a process in which energy is transferred from an excited donor fluorescent reagent to an acceptor fluorescent reagent by means of intermolecular long-range dipole-dipole coupling.
- FRET typically occurs over distances of about lOe to lOOe and requires that the emission spectrum of the donor reagent and the absorbance spectrum of the acceptor reagent overlap adequately and that the quantum yield of the donor and the absorption coefficient of the acceptor be sufficiently high.
- the transition dipoles of the donor and acceptor fluorescent reagents must be properly oriented relative to one another.
- the present invention makes use of a nuclear receptor or ligand binding domain thereof labeled with a first fluorescent reagent and CBP, p300, or other nuclear receptor co-activator, or a binding portion thereof, labeled with a second fluorescent reagent.
- the second fluorescent reagent comprises a fluorophore capable of undergoing energy transfer by either (a) donating excited state energy to the first fluorescent reagent, or (b) accepting excited state energy from the first fluorescent reagent.
- either the first or the second fluorescent reagents can be the donor or the acceptor during FRET.
- the first and second fluorescent reagents are spectropscopically complementary to each other. This means that their spectral characteristics are such that excited state energy transfer can occur between them.
- FRET is highly sensitive to the distance between the first and second fluorescent reagents.
- FRET varies inversely with the sixth power of the d stance between the first and second fluorescent reagents.
- the first fluorescent reagent, bound to the nuclear receptor or ligand binding domain thereof will not be near the second fluorescent reagent, bound to CBP, p300, or other nuclear receptor co-activator, or binding portion thereof.
- no FRET, or very little FRET will be observed.
- interaction between the nuclear receptor and CBP, p300, or other nuclear receptor co-activator will occur, thus bringing close together the first and the second fluorescent reagents, allowing FRET to occur and be observed.
- the present invention provides a method of identifying an agonist of a nuclear receptor that comprises providing:
- the nuclear receptor is selected from the group consisting of steroid receptors, thyroid hormone receptors, retinoic acid receptors, peroxisome proliferator-activated receptors, retinoid X receptors, glucocorticoid receptors, vitamin D receptors, and "orphan nuclear receptors" such as LXR, FXR, etc.
- the nuclear receptor or ligand binding domain thereof is a full-length nuclear receptor.
- the nuclear receptor or ligand binding domain thereof is a ligand binding domain of a nuclear receptor.
- the nuclear receptor or ligand binding domain thereof comprises an AF- 2 site of a nuclear receptor.
- the nuclear receptor or ligand binding domain thereof is a full-length PPAR.
- the nuclear receptor or ligand binding domain thereof is the ligand binding domain of a PPAR.
- the PPAR is selected from the group consisting of PPAR ⁇ , PPAR ⁇ l, PPAR ⁇ 2, and PPAR ⁇ .
- the ligand binding domain of the PPAR contains amino acid residues 176-478 of human PPAR ⁇ l.
- the nuclear receptor or ligand binding domain thereof contains amino acids 143-462 of human RAR ⁇ . In another embodiment, the nuclear receptor or ligand binding domain thereof contains amino acids 122-410 of rat T/3R ⁇ l. In another embodiment, the nuclear receptor or ligand binding domain thereof contains amino acids 227-463 of mouse RXR ⁇ . In another embodiment, the nuclear receptor or ligand binding domain thereof contains amino acids 251-595 of human ER.
- the above-described methods utilize full-length CBP, either mouse or human.
- the methods utilize amino acid residues 1-113 of human CBP.
- the methods utilize amino acid residues 1-453 of human CBP.
- the conditions under which the methods described above are carried out are conditions that are typically used in the art for the study of protein-protein interactions: e.g., physiological pH; salt conditions such as those represented by such commonly used buffers as PBS; a temperature of about 4°C to about 55°C.
- the presence of commonly used non-ionic detergents, e.g., NP-40®, sarcosyl, Triton X- 100®, is optional.
- reactions should contain KF at a concentration of at least 200 mM.
- the present invention includes a method of identifying an agonist of a nuclear receptor that comprises providing:
- a nuclear receptor or ligand binding domain thereof labeled with a first fluorescent reagent (a) a nuclear receptor or ligand binding domain thereof labeled with a first fluorescent reagent; (b) a binding portion of a nuclear receptor co-activator, where the binding portion contains the amino acid sequence LXXLL, and where the binding portion is labeled with a second fluorescent reagent; and
- FRET FRET between the first and second fluorescent reagents; where the occurrence of FRET indicates that the substance is an agonist of the nuclear receptor.
- the nuclear receptor co- activator is selected from the group consisting of: human RIP- 140, human SRC-1, mouse TIF-2, human or mouse CBP, human or mouse p300, mouse TIF-1, and human TRIP proteins.
- the nuclear receptor co- activator is human RIP- 140 and the binding portion includes a contiguous stretch of amino acids of human RIP- 140 selected from the group consisting of: positions 20-29, 132-139, 184-192, 266-273, 379-387, 496-506, 712-719, 818-825, 935-944, and 935-942.
- the nuclear receptor co-activator is human SRC-1 and the binding portion includes a contiguous stretch of amino acids of human SRC-1 selected from the group consisting of: positions 45-53, 632-640, 689-696, 748-755, and 1434-1441.
- the nuclear receptor co-activator is mouse TIF-2 and the binding portion includes a contiguous stretch of amino acids of mouse TIF-2 selected from the group consisting of: positions 640-650, 689-699, and 744-754.
- the nuclear receptor co-activator is human or mouse CBP and the binding portion includes a contiguous stretch of amino acids of human or mouse CBP selected from the group consisting of: positions 68-78 and 356-366.
- the nuclear receptor co-activator is human or mouse p300 and the binding portion includes a contiguous stretch of amino acids of human or mouse p300 selected from the group consisting of: positions 80-90 and 341-351.
- the nuclear receptor co-activator is mouse TIF-1 and the binding portion includes a contiguous stretch of amino acids of mouse TIF-1 containing positions 722-732.
- the nuclear receptor co-activator is human TRIP2 and the binding portion includes a contiguous stretch of amino acids of human TRIP2 containing positions 23-33. In another embodiment, the nuclear receptor co-activator is human TRIP3 and the binding portion includes a contiguous stretch of amino acids of human TRIP3 containing positions 97-107.
- the nuclear receptor co-activator is human TRIP4 and the binding portion includes a contiguous stretch of amino acids of human TRIP4 containing positions 36-46.
- the nuclear receptor co-activator is human TRIP5 and the binding portion includes a contiguous stretch of amino acids of human TRIP5 containing positions 26-36.
- the nuclear receptor co-activator is human TRIP8 and the binding portion includes a contiguous stretch of amino acids of human TRIP8 containing positions 36-46.
- the nuclear receptor co-activator is human TRIP9 and the binding portion includes a contiguous stretch of amino acids of human TRIP9 selected from the group consisting of: positions 73-83, 256-266 and 288-298.
- That general method is a method of identifying an agonist of a nuclear receptor that comprises providing:
- FRET fluorescence resonance energy transfer
- the amino acid sequence LXXLL is present in an ⁇ helical portion of the polypeptide. In another embodiment, the amino acid sequence LXXLL is present in an ⁇ helical portion of the polypeptide and the leucines form a hydrophobic face.
- the present invention provides methods for identifying antagonists of a nuclear receptor. Such methods are based on the ability of the antagonist to prevent the occurrence of agonist-induced binding between a nuclear receptor and CBP, p300, or other nuclear receptor co- activator, or to disrupt such binding after it has occurred. Thus, the present invention provides a method for identifying antagonists of nuclear receptors that comprises providing:
- a nuclear receptor or ligand binding domain thereof labeled with a first fluorescent reagent (a) a nuclear receptor or ligand binding domain thereof labeled with a first fluorescent reagent; (b) CBP, p300, or other nuclear receptor co-activator, or a binding portion thereof, labeled with a second fluorescent reagent; (c) an agonist of the nuclear receptor; and
- the nuclear receptor is selected from the group consisting of steroid receptors, thyroid hormone receptors, retinoic acid receptors, peroxisome proliferator-activated receptors, retinoid X receptors, glucocorticoid receptors, vitamin D receptors, and "orphan nuclear receptors" such as LXR, FXR, etc.
- the nuclear receptor or ligand binding domain thereof is a full-length nuclear receptor.
- the nuclear receptor or ligand binding domain thereof is a ligand binding domain of a nuclear receptor.
- the nuclear receptor or ligand binding domain thereof is an AF-2 site of a nuclear receptor.
- the nuclear receptor or ligand binding domain thereof is a full-length PPAR.
- the nuclear receptor or ligand binding domain thereof is the ligand binding domain of a PPAR.
- the PPAR is selected from the group consisting of PPAR ⁇ , PPAR ⁇ , and PPAR ⁇ .
- the ligand binding domain of the PPAR contains amino acid residues 176-478 of human PPAR ⁇ l.
- the nuclear receptor or ligand binding domain thereof contains amino acids 143-462 of human RAR ⁇ . In another embodiment, the nuclear receptor or ligand binding domain thereof contains amino acids 122-410 of rat T 3 R ⁇ l. In another embodiment, the nuclear receptor or ligand binding domain thereof contains amino acids 227-463 of mouse RXR ⁇ . In another embodiment, the nuclear receptor or ligand binding domain thereof contains amino acids 251-595 of human ER.
- the above-described methods utilize full-length CBP, either mouse or human. In other embodiments, the methods utilize amino acid residues 1-113 of human CBP. In another embodiment, the methods utilize amino acid residues 1-453 of human CBP.
- the conditions under which the methods described above are carried out are conditions that are typically used in the art for the study of protein-protein interactions: e.g., physiological pH; salt conditions such as those represented by such commonly used buffers as PBS; a temperature of about 4°C to about 55°C.
- the presence of commonly used non-ionic detergents, e.g., NP-40®, sarcosyl, Triton X- 100®, is optional.
- reactions should contain KF at a concentration of at least 200 mM.
- FRET Fluorescence Reduction Activated FRET
- Emission ratioing monitors the change in the ratio of emission by the acceptor over emission by the donor. An increase in this ratio signifies that energy is being transferred from donor to acceptor and thus that FRET is occurring. Emission ratioing can be measured by employing a laser-scanning confocal microscope.
- Emission ratioing is preferably done by splitting the emitted light from a sample with a dichroic mirror and measuring two wavelength bands (corresponding to the donor and the acceptor emission wavelengths) simultaneously with two detectors. Alternatively, the emitted light can be sampled consecutively at each wavelength (by using appropriate filters) with a single detector. In any case, these and other methods of measuring FRET are well known in the art. Although a variety of donor and acceptor fluorescent reagents can be used in the practice of the present invention, preferred embodiments of the present invention make use of cryptates of fluorescent reagents as donor reagents. Inclusion of a substrate into the intramolecular cavity of a macropolycyclic ligand results in the formation of a cryptate.
- the macropolycyclic ligand shields the substrate from interaction with solvent and other solute molecules. If the substrate is a fluororescent reagent, formation of a cryptate may result in markedly different spectroscopic characteristics for the reagent as compared to the spectroscopic characteristics of the free reagent.
- the present invention includes the use of europium (EuHI) or terbium (TbM) cryptates as donor fluorescent reagents.
- EuHI europium
- TbM terbium
- EuIH or TblH cryptates are well known in the art. For example, see Alpha et al., 1987, Angew. Chem. Int. Ed. Engl. 26:266-267; Mathis, 1995, CHn. Chem. 41:1391-1397.
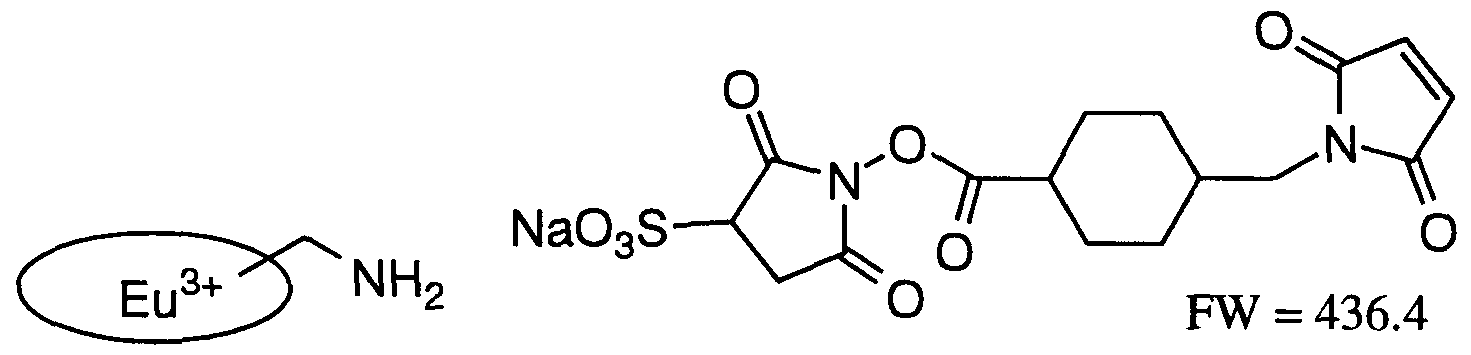
- a europium cryptate is formed by the inclusion of a europium ion into the intramolecular cavity of a macropolycyclic ligand which contains bipyridine groups as light absorbers. When europium cryptates are present in solution together with fluoride ions, a total shielding of the europium cryptate fluorescence is occurs. The molecular structure of a europium cryptate is shown below.
- Europium cryptates can be conjugated to proteins by the use of well-known heterobifunctional reagents (see, e.g. , International Patent Application WO 89/05813; Prat et al., 1991, Anal. Biochem. 195:283-289; Lopez et al., 1993, Clin. Chem. 39:196-201).
- the present invention includes the use of XL665 as the acceptor fluorescent reagent.
- XL665 is a crosslinked derivative of allophycocyanin (APC).
- APC is a porphyrin containing protein which is derived from the light harvesting system of algae (Kronick, 1986, M. Immunol. Meth. 92:1-13).
- XL665 is labeled with streptavidin in order to effect the binding of the streptavidin-labled XL665 to a biotin-labeled substance, e.g. , CBP or the ligand binding domain of a nuclear receptor.
- Streptavidin labeling of XL655 and biotin labeling of CBP, or the ligand binding domain of a nuclear receptor can be performed by well known methods.
- XL665 as the acceptor fluorescent reagent is combined with Europium cryptate (Eu3+K) as the donor fluorescent reagent.
- Europium cryptate (Eu3+K) has a large Stokes shift, absorbing light at 337 nm and emitting at 620 nm.
- the emission maximum of Europium cryptate (Eu3+K) overlaps the absorption maximum of XL665.
- Europium cryptate (Eu3+K) has a large temporal shift; the time between absorption and emission of a photon is about 1 millisecond. This is advantageous because most background fluorescence signals in biological samples are short-lived.
- a fluorescent reagent such as europium cryptate, with a long fluorescent lifetime, permits time-resolved detection resulting in the reduction of background interference.
- the spectral and temporal properties of europium cryptate (Eu3+K) result in essentially no fluorescence background and thus assays using this fluorescent reagent can be carried out in a "mix and read" mode, greatly facilitating its use as a high throughput screening tool.
- the measuring instrument irradiates the sample at 337 nm and measures the fluorescence output at two wavelengths, 620 nm (B counts, europium fluorescence) and 665 nm (A counts, XL665 fluorescence).
- the extent of flurorescent resonance energy transfer is measured as the ratio between these two values. Typically this ratio is multiplied by 10,000 to give whole numbers.
- FRET donor-acceptor pairs are suitable for the practice of the present invention.
- the following donor- acceptor pairs can be used: dansyl/fluorescein; fluorescein/rhodamine; tryptophan/aminocoumarin.
- the present invention provides a nuclear receptor or ligand binding domain thereof labeled with a fluorescent reagent for use in the above-described methods of identifying agonists and antagonists of nuclear receptors.
- the present invention also provides CBP, p300, or other nuclear receptor co-activator, or a binding portion thereof, labeled with a fluorescent reagent.
- the nuclear receptor or ligand binding domain thereof is selected from the group consisting of PPAR ⁇ , PPAR ⁇ , PPAR ⁇ , a ligand binding domain of PPAR ⁇ , PPAR ⁇ , or PPAR ⁇ , and amino acid residues 176-478 of human PPAR ⁇ l and the fluorescent reagent is selected from the group consisting of XL665 and Europium cryptate (Eu3+K).
- the fluorescent reagent is selected from the group consisting of XL665 and Europium cryptate (Eu3+K).
- CBP, p300, or other nuclear receptor co-activator is labeled with a fluorescent reagent selected from the group consisting of XL665 and Europium cryptate (Eu3+K).
- the primers used for hCBPl-113 were:
- the primers for hCBPl-453 were: 5'-ACTCGGATCCAAGCCATGGCTGAGAACTTGCTGGACGG-3'
- the primers were expected to amplify a 1.5 kb DNA fragment.
- the template for the PCR reaction was a human fetal brain cDNA library (Stratagene, Catalogue #IS 937227). Of course, any human cDNA library from a tissue expressing CBP could have been used.
- the PCR amplified 0.9 kb and 1.5 kp DNA fragments which were digested with restriction endonucleases and ligated into pBluescript II vector. DNA sequencing analysis confirmed that the amplified fragments were identical to the corresponding published nucleic acid sequences of human CBP.
- vectors encoding fusion proteins of the polypeptides and glutathione S- transferase were constructed and expressed in E. coli.
- the PCR fragments were subcloned into the expression vector pGEX (Pharmacia).
- IPTG isopropylthio- ⁇ -galactoside
- the cells were harvested by centrifugation for 10 min at 5000g.
- the cell pellet was used for GST-CBP fusion protein purification by following the procedure from Pharmacia Biotech using Glutathione Sepharose beads.
- hCBPl-113 and hCBPl-453 proteins were generated by cleaving the corresponding GST fusion proteins with thrombin.
- SDS- polyacrylamide gel electrophoresis analysis showed that the preparation from pGEXhCBPl-113 gave two polypeptide bands, with apparent molecular weight of 12 kd and 10 kd.
- the 12 kd band is the expected size of hCBPl-113 and the 10 kd band is most likely a premature translational termination product.
- a cDNA encoding the human PPAR ⁇ l ligand binding domain (PPAR ⁇ lLBD; amino acids 176-478 of PPAR ⁇ l) was subcloned from a modified pSG5 vector as a Xho I (site located in the N- terminus of the LBD)/ Xba I (site located in the pSG5 vector) fragment.
- the Xba I site was blunt-ended with T4 DNA polymerase.
- the 1.1 kb fragment containing the LBD was purified from an agarose gel and ligated into pGEX-KG (see Guan & Dixon, 1991, Anal. Biochem.
- Human PPAR ⁇ 2 contains the same amino acid sequence as human PPAR ⁇ l except for an amino terminal addition of 24 amino acids (see Elbrecht et al., 1996, Biochem. Biophys. Res. Comm. 224:431-437).
- the amino acid sequence of the ligand binding domain of human PPAR ⁇ 2 is the same as the amino acid sequence of the ligand binding domain of human PPAR ⁇ l, although the numbering of the amino acids differs (176-478 for human PPAR ⁇ l and 200-502 for human PPAR ⁇ 2).
- the DNA and amino acid sequences of human PPAR ⁇ are disclosed in Sher et al., 1993, Biochemistry 32:5598-5604 and in GenBank, accession no. L02932. See Figure lOA-C.
- hCBPl-113 was biotinylated with Sulfo-NHS- LC-Biotin (PIERCE) to a biotin:hPPAR ⁇ lLBD ratio of 3:1 according to the procedure provided by PIERCE.
- Purified hCBPl-113 was directly labeled with europium cryptate (Eu3+K) by the method illustrated in Figure 1.
- Biotin-labeled hPPAR ⁇ lLBD, Eu3+K-labeled hCBPl-113, and streptavidin-labeled XL665 were incubated together in the presence or absence of 1 ⁇ M of known PPAR ⁇ agonist (BRL49653 or AD5075).
- SA-XL665 streptavidin-labeled XL665; from PACKARD
- this experimental format made use of the fluorescent reagent pair europium cryptate (Eu3+K), which acted as donor, and
- hCBPl-113 was directly labeled with europium cryptate (Eu3+K); hPPAR ⁇ lLBD was indirectly labeled with
- Reactions were carried out in microtiter plates. Reaction conditions were: appropriate volume (total 250 ⁇ l) of the reaction buffer (either PBS or HEPES, see below, containing 500 mM KF, 0.1% bovine serum albumin, BSA) was added to each well, followed by addition of ligands (BRL49653 or AD5075 at a final concentration of 1 ⁇ M and 0.1% dimethylsulfoxide (DMSO) or vehicle control (0.1% DMSO), Eu3+K labeled hCBP (100 nM), biotin-hPPAR ⁇ lLBD (100 nM), and streptavidin- labeled XL665 (100 nM) to appropriate wells.
- DMSO dimethylsulfoxide
- DMSO dimethylsulfoxide
- Eu3+K labeled hCBP 100 nM
- biotin-hPPAR ⁇ lLBD 100 nM
- streptavidin- labeled XL665 100
- HEPES buffer N-2-hydroxyethylpiperazine-N'- 2-ethane sulfonic acid, 100 mM, pH 7.0
- NP40 Nonidet P-40
- the anti-GST antibody was a goat antibody to GST from Pharmacia (catalogue number 27-4577-01) that was labeled with Eu3+K according to the procedure summarized below.
- the detergent was changed from 0.05% NP40 to 0.5% CHAPS (3- ⁇ [3- cholamidopropyl]dimethyl-ammoniol ⁇ -l-propanesulfonate). A three- to four-fold signal-noise ratio was obtained.
- Figure 4 shows the strategy used for experiment 4 and similar experiments.
- Pioglitazone 890 nM.
- the above-described assay can also be used to characterize the interaction between nuclear receptors with co-activators as, e.g. , by determining the binding constant for that interaction.
- Figure 6 shows an example of such an application. Saturating amounts of PPAR ⁇ agonist (10 ⁇ M BRL49653) were used. Increasing concentrations of non- biotinylated hCBPl-453 were used to titrate away biotin-hCBP- PPAR ⁇ lLBD complex and decrease the fluorescence energy transfer.
- a Kd of 300 nM for the interaction between hCBPl-453 and PPAR ⁇ lLBD can be derived from the results illustrated in Figure 6 and this Kd (300 nM) is a measurement ofthe affinity between CBP and PPAR ⁇ .
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Food Science & Technology (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Physics & Mathematics (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description



Claims
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000514931A JP2001519525A (en) | 1997-10-07 | 1998-10-06 | Assay for nuclear receptor ligands using FRET |
| EP98956094A EP1021462A4 (en) | 1997-10-07 | 1998-10-06 | Assays for nuclear receptor ligands using fret |
| CA002305711A CA2305711A1 (en) | 1997-10-07 | 1998-10-06 | Assays for nuclear receptor ligands using fret |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6138597P | 1997-10-07 | 1997-10-07 | |
| US60/061,385 | 1997-10-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999018124A1 true WO1999018124A1 (en) | 1999-04-15 |
Family
ID=22035450
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/021049 WO1999018124A1 (en) | 1997-10-07 | 1998-10-06 | Assays for nuclear receptor ligands using fret |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1021462A4 (en) |
| JP (1) | JP2001519525A (en) |
| CA (1) | CA2305711A1 (en) |
| WO (1) | WO1999018124A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001020326A2 (en) * | 1999-09-13 | 2001-03-22 | Equitech Laboratories, Inc. | Materials and methods for the determination of an analyte |
| WO2001040805A1 (en) * | 1999-11-30 | 2001-06-07 | Akzo Nobel N.V. | Steroid compounds for steroid receptor binding assays |
| WO2001071352A2 (en) * | 2000-03-17 | 2001-09-27 | The Salk Institute For Biological Studies | Compositions associated with complex formation |
| EP1137940A1 (en) * | 1998-10-23 | 2001-10-04 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| EP1140079A1 (en) * | 1998-12-23 | 2001-10-10 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| WO2004042404A1 (en) * | 2002-11-07 | 2004-05-21 | Erasmus Universiteit Rotterdam | Fret probes and methods for detecting interacting molecules |
| WO2005021025A2 (en) * | 2003-08-28 | 2005-03-10 | Choongwae Pharma Corporation | MODULATION OF β-CATENIN/TCF ACTIVATED TRANSCRIPTION |
| EP1546718A2 (en) * | 2002-07-24 | 2005-06-29 | 3-Dimensional Pharmaceuticals, Inc. | Method for determining the regulation of xenobiotic removal |
| US6924311B2 (en) | 2001-10-17 | 2005-08-02 | X-Ceptor Therapeutics, Inc. | Methods for affecting various diseases utilizing LXR compounds |
| WO2005093423A2 (en) * | 2004-03-26 | 2005-10-06 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with peroxisome proliferative activated receptor alpha (ppara) |
| US7166438B2 (en) | 2001-11-07 | 2007-01-23 | Schering Ag | In Vitro screening for ligands of the estrogen receptor |
| KR100704011B1 (en) | 2005-02-16 | 2007-04-04 | 한국과학기술원 | Detection method for specific biomolecular interactions using FRET between metal nanoparticle and quantum dot |
| EP1907564A2 (en) * | 2005-06-28 | 2008-04-09 | Daiichi Sankyo Company, Limited | Lxr ligand testing method |
| US8945884B2 (en) | 2000-12-11 | 2015-02-03 | Life Technologies Corporation | Methods and compositions for synthesis of nucleic acid molecules using multiplerecognition sites |
| WO2015159292A3 (en) * | 2014-04-14 | 2015-12-10 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | A method and kit for determining the tissue or cell origin of dna |
| US9534252B2 (en) | 2003-12-01 | 2017-01-03 | Life Technologies Corporation | Nucleic acid molecules containing recombination sites and methods of using the same |
| EP3464644A4 (en) * | 2016-06-07 | 2020-07-15 | The Regents of The University of California | Cell-free dna methylation patterns for disease and condition analysis |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4803976B2 (en) * | 2003-07-09 | 2011-10-26 | 独立行政法人科学技術振興機構 | Molecular sensor for intracellular IP3 measurement |
| JP5264421B2 (en) | 2007-11-22 | 2013-08-14 | 富士フイルム株式会社 | Test substance measuring carrier and method for producing the same |
| WO2009141926A1 (en) * | 2008-05-23 | 2009-11-26 | 国立大学法人東京大学 | Method for acquisition of compound capable of acting on glucose metabolism/lipid metabolism |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9608143D0 (en) * | 1996-04-19 | 1996-06-26 | Ver Nl Kanker Inst | Assay |
| WO1998056806A1 (en) * | 1997-06-12 | 1998-12-17 | The Regents Of The University Of California | A TRANSCRIPTION FACTOR COACTIVATOR PROTEIN, p/CIP |
| WO1999041608A2 (en) * | 1998-02-12 | 1999-08-19 | Prolifix Limited | Interaction between cyclin d1 and steroid receptor co-activators |
-
1998
- 1998-10-06 EP EP98956094A patent/EP1021462A4/en not_active Withdrawn
- 1998-10-06 CA CA002305711A patent/CA2305711A1/en not_active Abandoned
- 1998-10-06 WO PCT/US1998/021049 patent/WO1999018124A1/en active Application Filing
- 1998-10-06 JP JP2000514931A patent/JP2001519525A/en active Pending
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP1021462A4 * |
| ZHOU G., ET AL.: "NUCLEAR RECEPTORS HAVE DISTINCT AFFINITIES FOR COACTIVATORS: CHARACTERIZATION BY FLUORESCENCE RESONANCE ENERGY TRANSFER.", MOLECULAR ENDOCRINOLOGY, THE ENDOCRINE SOCIETY, US, vol. 12., no. 10., 1 October 1998 (1998-10-01), US, pages 1594 - 1604., XP002915572, ISSN: 0888-8809, DOI: 10.1210/me.12.10.1594 * |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1137940A1 (en) * | 1998-10-23 | 2001-10-04 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| EP1137940A4 (en) * | 1998-10-23 | 2004-06-02 | Glaxo Group Ltd | Assays for ligands for nuclear receptors |
| US6639078B1 (en) | 1998-12-23 | 2003-10-28 | Smithkline Beecham Corporation | Assays for ligands for nuclear receptors |
| US6984650B2 (en) | 1998-12-23 | 2006-01-10 | Smithkline Beecham Corporation | Use of FXR ligands |
| EP1140079A4 (en) * | 1998-12-23 | 2004-06-09 | Glaxo Group Ltd | Assays for ligands for nuclear receptors |
| EP1140079A1 (en) * | 1998-12-23 | 2001-10-10 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| WO2001020326A3 (en) * | 1999-09-13 | 2002-02-21 | Equitech Lab Inc | Materials and methods for the determination of an analyte |
| AU780935B2 (en) * | 1999-09-13 | 2005-04-28 | Equitech Laboratories, Inc. | Materials and methods for the determination of an analyte |
| US6500629B1 (en) | 1999-09-13 | 2002-12-31 | Equitech Laboratories, Inc. | Materials and methods for detection and quantitation of an analyte |
| WO2001020326A2 (en) * | 1999-09-13 | 2001-03-22 | Equitech Laboratories, Inc. | Materials and methods for the determination of an analyte |
| US7030210B2 (en) | 1999-09-13 | 2006-04-18 | Equitech Laboratories, Inc. | Materials and methods for detection and quantitation of an analyte |
| WO2001040805A1 (en) * | 1999-11-30 | 2001-06-07 | Akzo Nobel N.V. | Steroid compounds for steroid receptor binding assays |
| WO2001071352A2 (en) * | 2000-03-17 | 2001-09-27 | The Salk Institute For Biological Studies | Compositions associated with complex formation |
| WO2001071352A3 (en) * | 2000-03-17 | 2002-11-07 | Salk Inst For Biological Studi | Compositions associated with complex formation |
| US9309520B2 (en) | 2000-08-21 | 2016-04-12 | Life Technologies Corporation | Methods and compositions for synthesis of nucleic acid molecules using multiple recognition sites |
| US8945884B2 (en) | 2000-12-11 | 2015-02-03 | Life Technologies Corporation | Methods and compositions for synthesis of nucleic acid molecules using multiplerecognition sites |
| US6924311B2 (en) | 2001-10-17 | 2005-08-02 | X-Ceptor Therapeutics, Inc. | Methods for affecting various diseases utilizing LXR compounds |
| US7166438B2 (en) | 2001-11-07 | 2007-01-23 | Schering Ag | In Vitro screening for ligands of the estrogen receptor |
| EP1546718A2 (en) * | 2002-07-24 | 2005-06-29 | 3-Dimensional Pharmaceuticals, Inc. | Method for determining the regulation of xenobiotic removal |
| EP1546718A4 (en) * | 2002-07-24 | 2006-09-06 | Johnson & Johnson Pharm Res | Method for determining the regulation of xenobiotic removal |
| WO2004042404A1 (en) * | 2002-11-07 | 2004-05-21 | Erasmus Universiteit Rotterdam | Fret probes and methods for detecting interacting molecules |
| US7413862B2 (en) | 2002-11-07 | 2008-08-19 | Erasmus Universteit Rotterdam | FRET probes and methods for detecting interacting molecules |
| WO2005021025A3 (en) * | 2003-08-28 | 2005-07-07 | Choongwae Pharma Corp | MODULATION OF β-CATENIN/TCF ACTIVATED TRANSCRIPTION |
| WO2005021025A2 (en) * | 2003-08-28 | 2005-03-10 | Choongwae Pharma Corporation | MODULATION OF β-CATENIN/TCF ACTIVATED TRANSCRIPTION |
| US7531320B2 (en) | 2003-08-28 | 2009-05-12 | Choongwae Pharma Corporation | Modulation of β-catenin/TCF-activated transcription |
| US9534252B2 (en) | 2003-12-01 | 2017-01-03 | Life Technologies Corporation | Nucleic acid molecules containing recombination sites and methods of using the same |
| WO2005093423A3 (en) * | 2004-03-26 | 2006-02-09 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with peroxisome proliferative activated receptor alpha (ppara) |
| WO2005093423A2 (en) * | 2004-03-26 | 2005-10-06 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with peroxisome proliferative activated receptor alpha (ppara) |
| KR100704011B1 (en) | 2005-02-16 | 2007-04-04 | 한국과학기술원 | Detection method for specific biomolecular interactions using FRET between metal nanoparticle and quantum dot |
| US7989179B2 (en) | 2005-06-28 | 2011-08-02 | Daiichi Sankyo Company, Limited | LXR ligand testing method |
| EP1907564A4 (en) * | 2005-06-28 | 2008-11-19 | Daiichi Sankyo Co Ltd | Lxr ligand testing method |
| EP1907564A2 (en) * | 2005-06-28 | 2008-04-09 | Daiichi Sankyo Company, Limited | Lxr ligand testing method |
| WO2015159292A3 (en) * | 2014-04-14 | 2015-12-10 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | A method and kit for determining the tissue or cell origin of dna |
| CN106574296A (en) * | 2014-04-14 | 2017-04-19 | 耶路撒冷希伯来大学伊森姆研究发展公司 | A method and kit for determining the tissue or cell origin of DNA |
| CN106574296B (en) * | 2014-04-14 | 2021-03-02 | 耶路撒冷希伯来大学伊森姆研究发展公司 | Method and kit for determining tissue or cell origin of DNA |
| US11203784B2 (en) | 2014-04-14 | 2021-12-21 | Hadasit Medical Research Services And Development Ltd. | Method and kit for determining the tissue or cell origin of DNA |
| EP3464644A4 (en) * | 2016-06-07 | 2020-07-15 | The Regents of The University of California | Cell-free dna methylation patterns for disease and condition analysis |
| US11499196B2 (en) | 2016-06-07 | 2022-11-15 | The Regents Of The University Of California | Cell-free DNA methylation patterns for disease and condition analysis |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1021462A4 (en) | 2005-04-13 |
| CA2305711A1 (en) | 1999-04-15 |
| EP1021462A1 (en) | 2000-07-26 |
| JP2001519525A (en) | 2001-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1021462A1 (en) | Assays for nuclear receptor ligands using fret | |
| McFedries et al. | Methods for the elucidation of protein-small molecule interactions | |
| Mao et al. | A novel method for the study of molecular interaction by using microscale thermophoresis | |
| US6689574B1 (en) | Assays for nuclear receptor agonists and antagonists using fluorescence resonance energy transfer | |
| KR20090083925A (en) | Detection system and uses therefor | |
| EP1057896B1 (en) | Methods for identifying or screening agonists for and antagonists to ppar | |
| JP4324474B2 (en) | Novel cell-based assay for G protein-coupled receptor-mediated activity | |
| Vollmer et al. | Subcellular compartmentalization of activation and desensitization of responses mediated by NK2 neurokinin receptors | |
| Bröer | Xenopus laevis oocytes | |
| WO2006122077A2 (en) | Method of screening for drugs that block ligand binding to a lipid binding protein | |
| Graham et al. | Ligand binding by recombinant domains from insect ecdysone receptors | |
| Zhou et al. | Use of homogeneous time-resolved fluorescence energy transfer in the measurement of nuclear receptor activation | |
| Gidon et al. | Studying the regulation of endosomal cAMP production in GPCR signaling | |
| US7238213B2 (en) | Cell-based assays employing voltage and calcium dyes | |
| Crouch et al. | New strategies in drug discovery for GPCRs: high throughput detection of cellular ERK phosphorylation | |
| Fay et al. | Purification of functional CB1 and analysis by site-directed fluorescence labeling methods | |
| US7767409B2 (en) | Method for detection of substance bound to nuclear receptor | |
| US20060110732A1 (en) | Method for the identification of ligands | |
| Zhang et al. | Microscale thermophoresis and fluorescence polarization assays of calcineurin-peptide interactions | |
| US20030039980A1 (en) | Assays for determination of functional binding of compounds to receptors | |
| JP4848282B2 (en) | A novel cell-based assay using potential and calcium dyes | |
| Mancini et al. | Exploring the Technology Landscape of 7TMR Drug Signaling Profiling | |
| Chen et al. | Application of large-scale transient transfection to cell-based functional assays for ion channels and GPCRs | |
| US20070218475A1 (en) | Efficient drug screening for protein targets | |
| WO2004010108A2 (en) | Method for the identification of ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 514931 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998956094 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2305711 Country of ref document: CA Ref country code: CA Ref document number: 2305711 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998956094 Country of ref document: EP |