EP0000108B1 - Benzo(b)thiéno-pyridines, leur procédé de préparation et composition thérapeutique les contenant - Google Patents
Benzo(b)thiéno-pyridines, leur procédé de préparation et composition thérapeutique les contenant Download PDFInfo
- Publication number
- EP0000108B1 EP0000108B1 EP78400006A EP78400006A EP0000108B1 EP 0000108 B1 EP0000108 B1 EP 0000108B1 EP 78400006 A EP78400006 A EP 78400006A EP 78400006 A EP78400006 A EP 78400006A EP 0000108 B1 EP0000108 B1 EP 0000108B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- formula
- benzo
- thieno
- phenyl
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D333/58—Radicals substituted by nitrogen atoms
Definitions
- the present invention relates to new benzo [b] thienopyridine derivatives, to a process for their preparation and to their applications in human and veterinary medicine.
- lower alkyl or “lower alkoxy” is meant groups having from 1 to 6 carbon atoms.
- the invention also includes the addition salts of the derivatives of formula I with mineral or organic acids.
- these derivatives are described as having tranquilizing properties, while the derivatives of the present invention exhibit sedative and inhibitory properties of platelet aggregation, which is completely different.
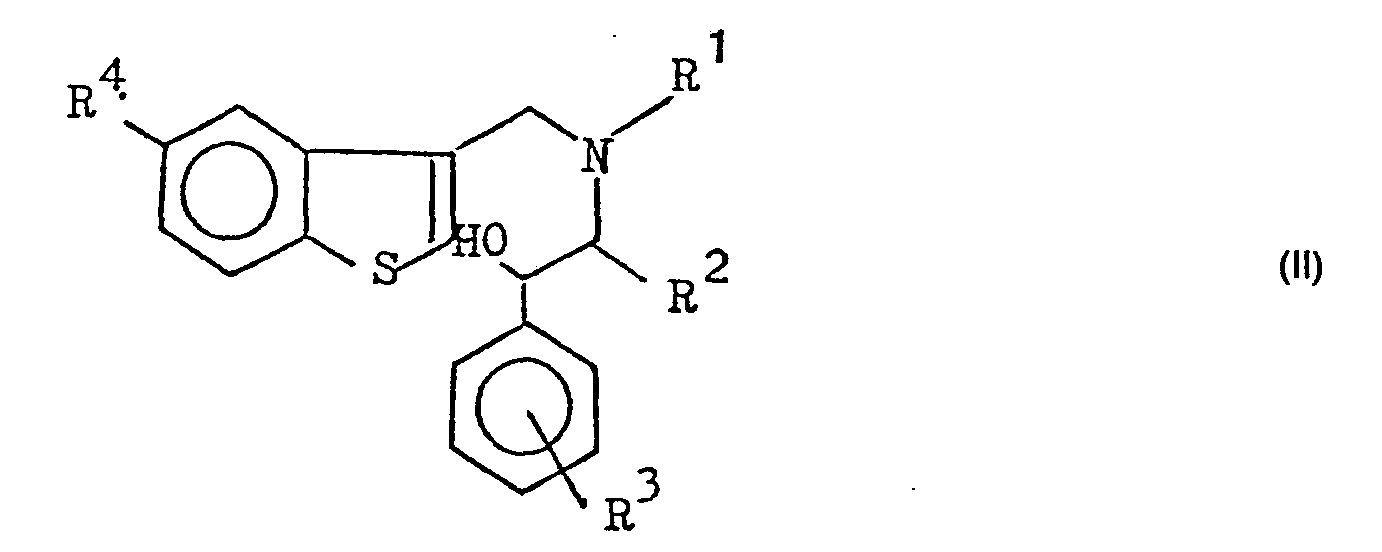
- the subject of the invention is also a process for preparing the compounds of formula (I), characterized in that compounds of formula (II) are cyclized in which R 1 , R 2 , R 3 and R 4 have the meanings given for formula (I), by heating in polyphosphoric acid at temperatures between 60 and 80 ° C.
- the cyclization is preferably carried out in the presence of an inert gas, in particular nitrogen.
- the compounds of formulas (I) for which R 1 is different from hydrogen can also be obtained by condensation of the corresponding compounds of formula (I) in which R 1 is hydrogen, with a compound of formula R 1 X in which X is a halogen atom.
- the reaction is normally carried out in an inert solvent such as ethanol or dimethylformamide, in the presence of a base such as an alkali metal carbonate, for example potassium carbonate.
- a base such as an alkali metal carbonate, for example potassium carbonate.
- X is chlorine or bromine
- the halogenomethyl-2 or -3 benzo (b) thiophenes can be prepared according to: S. AVAKIAN, J. MOSS and G.J. MARTIN, J. amer. chem. Soc., 1948, 70, 3,075; N.B. CHAPMAN, K.CLARKE; B. GORE and S.N. SAWHNEY, J. chem. Soc. (C), 1968, 514; N.B. CHAPMAN, K. CLARKE and B. IDDON, J. chem. Soc. (C), 1965, 774.
- Formyl-2 and -3 benzo (b) thiophenes can be prepared according to: D.A. SHIRLEY and M.J. DANZIG, J.
- the addition salts are prepared with mineral acids (for example hydrochloric, sulfuric acid, etc.) or organic acids (for example methane sulfonic, maleic, tartaric, citric acid, etc.) by usual conventional methods.
- mineral acids for example hydrochloric, sulfuric acid, etc.
- organic acids for example methane sulfonic, maleic, tartaric, citric acid, etc.
- N-methylation of 8-chloro-3-methyl-4-phenyl-1,2,3,4-benzo (b) thieno [3,2-c] pyridine (example 1) is carried out by condensation with methyl iodide, in ethanol, in the presence of potassium carbonate, or by Leuckart reaction (heating in the presence of formaldehyde and formic acid).
- Benzyl bromide is condensed with 8-chloro-3-methyl-4-phenyl-1,2,3,4-benzo (b) thieno [3,2-c] pyridine (Example 1) according to the process described in example 6.
- Methanesulfonate white crystals, F> 260 ° C (acetonitrile), yield: 63%.
- O-nitrobenzyl chloride is condensed with 8-chloro-4-phenyl-1,2,3,4-tetrahydro benzo (b) thieno [3,2-c] pyridine (example 3), according to the process described in l 'example 6.
- O-cyanobenzyl bromide is condensed with 4-phenyl-1,2,3,4-tetrahydro benzo (b) thieno [3,2-c] pyridine (Example 4), according to the method of Example 6.
- O-methoxycarbonylbenzyl bromide is condensed with 8-chloro-3-methylphenyl-1,2,3,4 tetrahydro-1,2,3,4 benzo (b) thieno [3,2-c] pyridine (Example 1), according to the method described in Example 6.
- Butyl bromide is condensed with 8-chloro-3-methyl-4-phenyl-1,2,3,4-benzo (b) thieno [3,2-c] pyridine (example 1), according to the process described in 1 'example 6.
- Phenethyl bromide is condensed with 3-methyl-4-phenyl-1,2,3,4-tetrahydro benzo (b) thieno [3,2-c] pyridine (Example 2), according to the process described in Example 6 .
- a further subject of the invention is therefore a therapeutic composition having in particular activities which inhibit platelet aggregation and sedation, characterized in that it contains, as active principle, a derivative of formula (I) or a salt thereof. addition with a pharmaceutically acceptable acid thereof, in combination with a therapeutically administrable vehicle.
- the compounds of the invention benefit from excellent tolerance and low toxicity.
- the LD 50/24 h / kg of animal, determined in mice according to the Miller and Tainter method, for the oral route, is greater than 400 mg for all the derivatives.
- a blood sample is taken from the jugular vein. From this citrated blood and after centrifugation, a plasma containing 600,000 ⁇ 20,000 platelets per mm 3 is reconstituted, which will be used in all aggregation measurements.
- 0.4 ml of plasma is placed in a silicone tube provided with a magnetic bar itself silicone.
- the tube is introduced into an aggregometer coupled to a device making it possible to record the variations in optical density.
- 0.5 ml of a solution containing 10 ⁇ of ADP is introduced into the tube. (Adenosine-Di Phosphate).
- the aggregation of platelets then causes an increase in light transmission followed a decrease following the disaggregation phase.
- the maximum variation in optical density thus determined characterizes the intensity of the aggregation.
- A.D.P.'s solution is replaced by a collagen solution (bovine tendon extract).
- test products are administered to mice by the oral route at a dose of 100 mg / kg, thirty minutes before the intraperitoneal injection of a solution of 300 mg of chloral in 20 ml of physiological saline.
- a solution of 300 mg of chloral in 20 ml of physiological saline was administered to mice by the oral route at a dose of 100 mg / kg, thirty minutes before the intraperitoneal injection of a solution of 300 mg of chloral in 20 ml of physiological saline.
- the derivatives of the invention considerably potentiate the action of chloral, in particular with regard to the duration of the induced sleep and the number of sleeping mice.
- mice which have received 100 mg of the derivative to be tested by the oral route. It is considered that the mice have undergone a sedative action when they do not manage, within thirty seconds, to effect a recovery which brings at least one of their hind legs on the wire.
- the animals are tested before the test and those which fail to recover within thirty seconds are eliminated. We see during the tests that only 10%. 100 of the tested animals manage to recover.
- a mouse placed in an enclosure containing 4 electrified plates, receives, with each passage of a plate towards another, an electrical stimulus causing a disorderly leak. After n electric shocks, the mouse no longer moves. It is considered that the degree of sedation obtained is proportional to the number n of electric shocks that the treated mouse will have received before it stops in a corner.
- the derivatives of the invention produce an average percentage increase in the number of electric shocks n of the order of 60% after 15 minutes, of 62% after 30 minutes and 51% after ninety minutes.
- the medicament of the invention can be presented, for oral administration, in the form of tablets, coated tablets, capsules, drops and syrup. It can also be presented, for rectal administration, in the form of suppositories and for parenteral administration, in the form of an injectable solution.
- Each unit dose advantageously contains from 0.010 g to 0.500 g of active principle, the doses administered daily being able to vary from 0.010 g to 1.00 g of active principle depending on the age of the patient and the condition treated.
- the medicament of the invention can thus be advantageously administered, as a preventive or curative treatment, in the treatment of diseases causing a pathological change in platelet aggregation such as diseases causing a pathological change in platelet aggregation such as diseases thromboembolic.
Description
- La présente invention est relative à de nouveaux dérivés de benzo[b] thiénopyridines, à un procédé de préparation de ceux-ci et à leurs applications en médecine humaine et vétérinaire.
- Les dérivés de l'invention répondent à la formule

- R1 représente l'hydrogène ou un groupe alcoyle inférieur ; phényl-C1-2 alcoyle éventuellement substitué sur le noyau phényle par au moins un atome d'halogène ou un groupe hydroxy, nitro, amino, cyano, carboxy, carboxamido, alcoxycarbonyle, alcoyle inférieur, alcoxy inférieur ou trifluorométhyle ; (pyridyl-2)méthyle ; (pyridyl-3)méthyle ou (pyridyl-4)méthyle ;
- R2 représente l'hydrogène ou un radical alcoyle inférieur ; et
- R 3 et R4 représentent chacun l'hydrogène, un atome d'halogène ou un groupe hydroxy, alcoyle inférieur ou alcoxy inférieur.
- Par « alcoyle inférieur » ou « alcoxy inférieur » on veut désigner des groupes ayant de 1 à 6 atomes de carbone.
- L'invention comprend aussi les sels d'addition des dérivés de formule I avec des acides minéraux ou organiques.
- On connaît déjà par le brevet US 3 752 820 des dérivés de benzo[b] thiéno pyridine du type (3,2-c), mais ces dérivés ne portent pas, comme les dérivés de la présente invention, de substituant phényle sur les atomes de carbone du cycle pyridinique.
- De plus, ces dérivés sont décrits comme possédant des propriétés tranquillisantes, alors que les dérivés de la présente invention présentent des propriétés sédative et inhibitrice de l'agrégation plaquettaire, ce qui est complètement différent.
- Quant à la demande FR 2 358 150 déposée le 13 juillet 1976 mais publiée le 10 février 1978, elle décrit des dérivés de thiéno (2,3-c) et (3,2-c) pyridines et non pas des benzo(b) thiénopyridines.
- L'invention a également pour objet un procédé de préparation des composés de formule (I), caractérisé en ce qu'on cyclise des composés de formule (II)

- La cyclisation est effectuée de préférence en présence d'un gaz inerte, notamment l'azote.
- Les composés (II) utilisés à titre d'intermédiaires sont des composés nouveaux qui peuvent être préparés par des procédés classiques. On peut obtenir, par exemple, les composés de formule (II) comme suit :
- On fait réagir un phényl-1 éthanolamine de formule (III) ci-dessous dans laquelle R2 et R3 ont les mêmes significations que ci-dessus, soit avec un formyl-3 benzo(b) thiophène et on fait suivre d'une réduction, soit avec un halogénométhyl-3 benzo(b)thiophène, pour obtenir le composé de formule (Ila) ci-dessous, qui est ensuite le cas échéant condensé avec un halogénure de formule R1X dans lequel R1 a la même signification que précédemment et X est un atome d'halogène (chlore, brome ou iode), si on désire que R1 soit autre qu'un atome d'hydrogène.
- Le schéma réactionnel est le suivant :

- Les composés de formules (I) pour lesquels R1 est différent de l'hydrogène peuvent aussi être obtenus par condensation des composés de formule (I) correspondants dans lesquels R1 est de l'hydrogène, avec un composé de formule R1X dans lequel X est un atome d'halogène. La réaction est effectuée normalement dans un solvant inerte tel que l'éthanol ou le diméthylformamide, en présence d'une base telle qu'un carbonate de métal alcalin, par exemple le carbonate de potassium. Lorsque X est le chlore ou le brome, on peut avantageusement ajouter une quantité catalytique d'un iodure minéral tel que l'iodure de potassium.
- Les halogénométhyl-2 ou -3 benzo(b)thiophènes peuvent être préparés selon : S. AVAKIAN, J. MOSS et G.J. MARTIN, J. amer. chem. Soc., 1948, 70, 3 075 ; N.B. CHAPMAN, K.CLARKE ; B. GORE et S.N. SAWHNEY, J. chem. Soc. (C), 1968, 514 ; N.B. CHAPMAN, K. CLARKE et B. IDDON, J. chem. Soc. (C), 1965, 774. Les formyl-2 et -3 benzo(b)thiophènes peuvent être préparés selon : D.A. SHIRLEY et M.J. DANZIG, J. amer. chem. Soc., 1952, 74, 2 935 ; K. CLARKE, C.G. HUGUES, A.J. HUMPHRIES et R.M. SCROWSTON, J. chem. Soc. (C), 1970, 1 013 ; M.S. EL SHANTA et R.M. SCROWSTON, J. chem. Soc. (C), 1967, 2 085 ; E. CAMPAIGNE et E.S. NEISS, J. HET. Chem., 1966, 3, 46.
- Les produits de départ de formule (III) sont des produits disponibles dans le commerce ou bien sont décrits dans la littérature.
- On prépare les sels d'addition avec des acides minéraux (par exemple l'acide chlorhydrique, sulfurique, etc...) ou organiques (par exemple l'acide méthane sulfonique, maléique, tartrique, citrique, etc...) par les procédés classiques usuels.
- Les exemples non limitatifs suivants illustrent l'invention:
- Chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = R3 = H, R2 = CH3, R4 = CI).
- On chauffe à 70 °C, pendant 14 heures, sous atmosphère d'azote, un mélange de 40 g (0,153 mole) de bromométhyl-3 chloro-5 benzo(b)thiophène, 28,7 g (0,153 mole) de chlorhydrate de noréphédrine, 42 g (0,306 mole) de carbonate de potassium sec et 400 cm3 de diméthylformamide. On filtre les sels minéraux et on évapore le solvant à sec sous bon vide. L'huile résiduelle est reprise par du chlorure de méthylène. Les extraits organiques sont lavés à l'eau, séchés sur sulfate de sodium sec et filtrés sur lit de silice. L'évaporation laisse des cristaux que l'on recristallise dans le cyclohexane : cristaux blanchâtres, F = 83 °C, rdt : 71 %.
- On chauffe à 70 °C pendant 1 heure 30, sous atmosphère d'azote, un mélange agité mécaniquement de 16,7 g (0,05 mole) de l'aminoalcool précédent dans 55 g d'acide polyphosphorique commercial. Après refroidissement le milieu réactionnel est versé sur de la glace, rendu basique avec de l'ammoniaque concentrée et extrait au chlorure de méthylène. Les extraits organiques sont séchés sur sulfate de sodium, filtrés sur lit de silice et évaporés à sec.
- Les cristaux obtenus sont recristallisés dans un mélange isopropanol-éthanol : cristaux blancs, F = 174 °C, rdt : 97%.
- Méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = R3 = R4 = H, R2 = CH3).
- Elle est réalisée à partir du chlorométhyl-3 benzo(b) thiophène et de noréphédine selon le mode opératoire de l'exemple 1 (partie a). Base : cristaux blancs, F = 104 °C (cyclohexane) ; rdt : 67 %.
- Elle est réalisée selon le mode opératoire décrit à l'exemple 1 (partie b).
- Base : cristaux blancs rosés, F = 164 °C (isopropanol) ; rdt : 72,5 %.
- Chloro-8 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = R2 = R3 = H, R4 = CI).
- Elle est réalisée à partir du bromométhyl-3 chloro-5 benzo(b) thiophène et de l'amino-2 phényl-1 éthanol, selon le mode opératoire décrit à l'exemple 1 (partie a).
- Base : cristaux blanchâtres, F = 95 °C (cyclohexane) ; rdt : 40 %.
- Elle est réalisée selon le mode opératoire décrit à l'exemple 1 (partie b).
- Méthanesulfonate : cristaux blancs, F = 234 °C (éthanol), rdt : 77 %.
- Phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = R2 = R3 = R4 = H)
- Elle est réalisée à partir du chlorométhyl-3 benzo(b) thiophène et de l'amino-2 phényl-1 éthanol, selon le mode opératoire décrit à l'exemple 1 (partie a). Base : cristaux blancs, F = 108 °C (cyclohexane) ; rdt : 34%
- Elle est réalisée selon le mode opératoire décrit à l'exemple 1 (partie b).
- Méthanesulfonate : cristaux beiges, F = 215 °C (éthanol), rdt : 69%.
- Chloro-8 diméthyl-2,3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = R2 = méthyl ; R 3 = H, R4 = CI)
- On effectue la N-méthylation de la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno [3,2-c]pyridine (exemple 1), par condensation avec de l'iodure de méthyle, dans l'éthanol, en présence de carbonate de potassium, ou par réaction de Leuckart (chauffage en présence de formol et d'acide formique).
- Chlorhydrate : cristaux blancs, F = 229 °C (isopropanol), rdt : 100 % (réaction de Leuckart).
- Chloro-8 o-chlorobenzyl-2 méthyl-3 phényi-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = o.chlorobenzyl ; R2 = méthyl ; R3 = H ; R4 = CI)
- On chauffe à 70 °C, pendant 12 heures, un mélange de 6 g (0,019 mole) de chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1), 3,1 g (0,019 mole) de chlorure d'o-chlorobenzyle et 2,6 g (0,019 mole) de carbonate de potassium sec dans 80 cm3 de diméthylformamide. Après refroidissement les sels minéraux sont filtrés et le solvant est évaporé sous pression réduite. Le résidu est repris par de l'eau et extrait au chlorure de méthylène. Les extraits organiques sont lavés à l'eau, séchés sur sulfate de sodium, filtrés sur lit de silice et évaporés à sec. Le résidu est transformé en chlorhydrate que l'on recristallise dans le méthanol : cristaux blancs, F = 175 °C ; rdt : 55%.
- Benzyl-2 chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = benzyl ; R2 = méthyl ; R3 = H ; R4 = CI)
- On condense du bromure de benzyle avec la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1) selon le procédé décrit à l'exemple 6.
- Méthanesulfonate : cristaux blancs, F > 260 °C (acétonitrile), rdt : 63%.
- Phényl-4 p-tolyl-2 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = p-tolyle ; R2 = R3 = R4 = H) On condense du bromure de. p-tolyle avec la phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c] pyridine (exemple 4) selon le procédé à l'exemple 6.
- Chlorhydrate : cristaux blancs, F = 200 °C (acétate d'éthyle-méthanol), rdt : 99 %.
- Méthyl-3 m-méthoxybenzyl-2 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = m-méthoxybenzyle : R3 = R4 = H ; R2 = CH3)
- On condense du bromure de m-méthoxybenzyle avec la méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 2), selon le procédé décrit à l'exemple 6.
- Chlorhydrate : cristaux blancs, F = 160 °C (acétonitrile-méthanol), rdt : 92 %.
- Chloro-8 (triméthoxy-3,4,5 benzyl)-2 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c] pyridine (R1 = triméthoxy-3,4,4 benzyle ; R3 = R4 = H ; R2 = méthyl)
- On condense le chlorure de triméthoxy-3,4,5 benzyle avec la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1), selon le procédé décrit à l'exemple 6.
- Base : cristaux blancs, F = 172 °C, rdt : 78,5 %.
- Chloro-8 o-nitrobenzyl-2 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = o-nitrobenzyl; R2 = R3 = H; R4 = Cl)
- On condense du chlorure d'o-nitrobenzyle avec la chloro-8 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 3), selon le procédé décrit à l'exemple 6.
- Chlorhydrate : cristaux jaunes, F = 150 °C (acétonitrile) ; rdt : 45 %.
- o-cyanobenzyl-2 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = o-cyanobenzyl ; R2 = R3 = R4 = H)
- On condense du bromure d'o-cyanobenzyle avec la phényl-4 tétrahydro-1,2,3,4 benzo (b) thiéno [3,2-c] pyridine (exemple 4), selon le procédé de l'exemple 6.
- Base : cristaux blanchâtres, F = 143 °C, rdt : 69 %.
- chloro-8 méthyl-3 o-méthoxycarbonylbenzyl-2 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c] pyridine (R1 = o-méthoxycarbonylbenzyl ; R2 = CH3 ; R3 = H ; R4 = CI)
- On condense du bromure d'o-méthoxycarbonylbenzyle avec la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1), selon le procédé décrit à l'exemple 6.
- Chlorhydrate : cristaux blanchâtres, F = 155 °C (acétonitriie), rdt : 86 %.
- o-carboxybenzyl-2 chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = o-carboxybenzyl ; R2 = CH3 : R3 = H ; R4 = CI.
- Ce dérivé est obtenu par hydrolyse basique du composé décrit à l'exemple 13.
- Base : cristaux blancs, F = 258 °C (méthanol-diméthylformamide), rdt : 100 %.
- .Butyl-2 chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = butyl ; R2 = CH3 ; R3 = H ; R4 = Cl)
- On condense du bromure de butyle avec la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1), selon le procédé décrit à l'exemple 6.
- Méthanesulfonate : cristaux blancs, F = 210 °C (acétonitrile), rdt = 58 %.
- Méthyl-3 phénéthyl-2 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = phénéthyl ; R2 = CH3; R3 = R4 = H)
- On condense du bromure de phénéthyle avec la méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 2), selon le procédé décrit à l'exemple 6.
- Chlorhydrate : cristaux blancs, F = 210 °C (isopropanol-méthanol), rdt : 72 %.
- Chloro-8 méthyl-3 phényl-4(pyridyl-3) méthyl-2 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = (pyridyl-3) méthyl ; R2 = CH3 ; R3 = H ; R4 = CI)
- On condense du chlorhydrate de chlorométhyl-3 pyridine avec la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1) selon le procédé décrit à l'exemple 6.
- Dichlorhydrate : cristaux roses, F = 260 °C (méthanoldiméthylformamide), rdt : 69 %.
- Méthyl-3 phényi-4 (pyridyl-2) méthyl-2 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = (pyridyl-2) méthyl ; R2 = CH3 ; R3 = R 4 = H).
- On condense du chlorhydrate de chlorométhyl-2 pyridine avec la méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 2), selon le procédé décrit à l'exemple 6.
- Dichlorhydrate : cristaux blanchâtres ; F = 155 °C (acétate d'éthyle-méthanol) ; rdt : 63 %.
- Chloro-8 méthyl-3 phényl-4 (pyridyl-4) méthyl-2-tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (R1 = (pyridyl-4) méthyl ; R2 = CH3 ;R3 = H ; R4 = CI)
- On condense du chlorhydrate de chlorométhyl-4 pyridine avec la chloro-8 méthyl-3 phényl-4 tétrahydro-1,2,3,4 benzo(b) thiéno[3,2-c]pyridine (exemple 1), selon le procédé décrit à l'exemple 6.
- Dichlorhydrate : cristaux marron clair, F = 258 °C, rdt : 41 %.
- Les résultats des essais toxicologiques et pharmacologiques qui sont rapportés ci-après, ont mis en évidence les intéressantes activités des dérivés de l'invention, notamment inhibitrice de l'agrégation plaquettaire et sédative.
- L'invention a donc encore pour objet une composition thérapeutique présentant en particulier des activités inhibitrice de l'agrégation plaquettaire et sédative, caractérisé en ce qu'elle contient, à titre de principe actif, un dérivé de formule (I) ou un sel d'addition avec un acide pharmaceutiquement acceptable de celui-ci, en association avec un véhicule thérapeutiquement administrable.
- Les composés de l'invention bénéficient d'une excellente tolérance et d'une faible toxicité. Ainsi, la DL50/24 h/kg d'animal, déterminée chez la souris selon la méthode de Miller et Tainter, pour la voie orale, est supérieure à 400 mg pour tous les dérivés.
- En outre, les essais effectués sur la toxicité aiguë, chronique, sub-chronique et retardée, chez diverses espèces animales, n'ont mis en évidence aucune réaction locale ou générale, aucune perturbation dans les contrôles biologiques régulièrement effectués, aucune anomalie dans les examens microscopiques et macroscopiques chez les animaux sacrifiés et autopsiés en fin d'expérimentation.
- Chez des rats de souche Wistar, on effectue un prélèvement sanguin dans la veine jugulaire. A partir de ce sang citraté et après centrifugation, on reconstitue un plasma contenant 600 000 ± 20 000 plaquettes par mm3, qui servira dans toutes les mesures d'agrégation.
- On place 0,4 ml de plasma dans un tube siliconé pourvu d'une barre aimantée elle-même siliconée. Le tube est introduit dans un agrégomètre couplé à un appareil permettant d'enregistrer les variations de densité optique. Lorsque la transmission de la lumière a atteint une valeur stable, on introduit dans le tube 0,5 ml d'une solution contenant 10 µ d'A.D.P. (Adénosine-Di Phosphate).
- L'agrégation des plaquettes provoque alors une augmentation de la transmission lumineuse suivie d'une diminution consécutive à la phase de désagrégation.
- La variation maximale de densité optique ainsi déterminée caractérise l'intensité de l'agrégation.
- La solution d'A.D.P. est remplacée par une solution de collagène (extrait de tendons bovins).
- Différents lots de 20 rats sont utilisés, chaque lot recevant un dérivé à tester par la voie orale, à la dose de 100 mg/kg. Les résultats obtenus au cours de ces 2 essais sont rapportés dans le tableau I suivant qui indique le pourcentage d'inhibition de l'agrégation plaquettaire obtenu, par rapport au témoin, 3 heures après le traitement par le médicament de l'invention, dans le test de l'A.D.P. et au collagène.

- L'action sédative des composés de l'invention a été étudiée selon plusieurs méthodes.
- Cette étude a été effectuée selon la méthode de SAMUEL IRWIN (Ph. D. animal and clinical Pharmacology Technics in drug evaluation). Les dérivés de l'invention sont administrés par la voie orale à des souris à la dose de 100 mg/Kg. L'étude du comportement des animaux traités, pendant les 4 heures suivantes, ainsi que la mesure des différents paramètres physiologiques, température, vitesse cardiaque et respiratoire, met en évidence la nette action sédative des dérivés de l'invention.
- Les produits à tester sont administrés à des souris par la voie orale à la dose de 100 mg/kg, trente minutes avant l'injection intrapéritonéale d'une solution de 300 mg de chloral dans 20 ml de sérum physiologique. On note le nombre de souris endormies, le temps d'endormissement et la durée du sommeil, par rapport aux souris témoins qui n'ont reçu que l'injection du chloral. On constate que les dérivés de l'invention potentialisent considérablement l'action du chloral, notamment en ce qui concerne la durée du sommeil induit et le nombre de souris endormies.
- Ce test consiste à suspendre sur un fil, par les pattes antérieures, des souris qui ont reçu 100 mg du dérivé à tester par la voie orale. On considère que les souris ont subi une action sédative quand elles ne parviennent pas, en trente secondes, à effectuer un rétablissement qui amène au moins une de leurs pattes postérieures sur le fil.
- Les animaux sont testés avant l'essai et ceux qui n'arrivent pas à se rétablir dans le temps de trente secondes sont éliminés. On constate au cours des essais que seulement 10 p. 100 des animaux testés réussissent à se rétablir.
- Une souris, placée dans une enceinte contenant 4 plaques électrifiées, reçoit, à chaque passage d'une plaque vers une autre, un stimulus électrique provoquant une fuite désordonnée. Au bout de n chocs électriques, la souris ne bouge plus. On considère que le degré de sédation obtenu est proportionnel au nombre n de secousses électriques que la souris traitée aura reçu avant qu'elle ne s'immobilise dans un coin.
- On détermine ainsi qu'administrés par la voie orale à la dose de 100 mg/Kg, les dérivés de l'invention produisent un pourcentage moyen d'accroissement du nombre de secousses électriques n de l'ordre de 60 % après 15 minutes, de 62 % après 30 minutes et de 51 % après quatre vingt dix minutes.
- Les résultats de ces études mettent en évidence la bonne tolérance et les intéressantes propriétés inhibitrices de l'agrégation plaquettaire et sédatives des dérivés de l'invention qui les rendent très utiles en médecine humaine et vétérinaire.
- Le médicament de l'invention peut être présenté, pour l'administration orale, sous forme de comprimés, comprimés dragéifiés, capsules, gouttes et sirop. Il peut aussi être présenté, pour l'administration rectale, sous forme de suppositoires et pour l'administration parentérale, sous forme de soluté injectable.
- Chaque dose unitaire contient avantageusement de 0,010 g à 0,500 g de principe actif, les doses administrabes journellement pouvant varier de 0,010 g à 1,00 g de principe actif selon l'âge du patient et l'affection traitée.
- On donnera ci-après, à titre d'exemples non limitatifs, quelques formulations pharmaceutiques du médicament de l'invention.
- 1) Comprimés
- dérivé de l'Ex. 2............0,050 g
- excipient : amidon de maïs, stéarate de magnésium, aérosil, talc, amaranthe, tartrazine.
- 2) Comprimés dragéifiés
- dérivé de l'Ex. 6............0,075 g
- excipient : talc, amidon de maïs, gomme arabique, gomme laque, sucre, glucose, cire blanché, cire de carnauba, blanc de baleine, lactose, jaune orangé S, oxyde de titane.
- 3) Capsules
- dérivé de l'Ex. 9............0,100 g
- excipient : stéarate de magnésium, amidon de maïs, saccharose.
- 4) Ampoules injectables
- dérivé de l'Ex. 14...........0,050 g
- excipient : solvant isotonique q.s.p...5 ml
- 5) Suppositoires
- dérivé de l'Ex. 18...........0,100 g
- excipient : triglycérides semi-synthétiques
- 6) Sirop
- dérivé de l'Ex: 2............1,00 g
- excipient aromatisé q.s.p. 100 ml
- Les études toxicologiques et pharmacologiques qui viennent d'être rapportées ont mis en évidence la bonne tolérance des dérivés de l'invention ainsi que leurs activités inhibitrice de l'agrégation plaquettaire et sédative.
- Le médicament de l'invention peut ainsi être administré avec profit, à titre préventif ou curatif, dans le traitement des maladies provoquant une modification pathologique de l'agrégation plaquettaire telles que les maladies provoquant une modification pathologique de l'agrégation plaquettaire telles que les maladies thromboembolliques.
- Il peut aussi être administré en tant que sédatif et régulateur du système nerveux, dans l'éréthisme nerveux, la neurotonie, les états d'excitation avec insomnie et les troubles de la dentition.
Claims (7)

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR7716878A FR2423494A1 (fr) | 1977-06-02 | 1977-06-02 | Benzo (b) thieno pyridines, leur procede de preparation et leurs applications therapeutiques |
| FR7716878 | 1977-06-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0000108A1 EP0000108A1 (fr) | 1978-12-20 |
| EP0000108B1 true EP0000108B1 (fr) | 1981-08-19 |
Family
ID=9191587
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78400006A Expired EP0000108B1 (fr) | 1977-06-02 | 1978-06-01 | Benzo(b)thiéno-pyridines, leur procédé de préparation et composition thérapeutique les contenant |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US4172134A (fr) |
| EP (1) | EP0000108B1 (fr) |
| JP (1) | JPS543098A (fr) |
| DE (1) | DE2860971D1 (fr) |
| DK (1) | DK152130C (fr) |
| ES (1) | ES470274A1 (fr) |
| FR (1) | FR2423494A1 (fr) |
| GB (1) | GB1572690A (fr) |
| IE (1) | IE46848B1 (fr) |
| LU (1) | LU79763A1 (fr) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4282227A (en) * | 1980-05-22 | 1981-08-04 | Smithkline Corporation | Renal vasodilating 3,4-dihydroxyphenyltetrahydrothienopyridines |
| JPS5939349A (ja) * | 1982-08-31 | 1984-03-03 | 杉 晤夫 | 米の搗精方法と精米機 |
| JP2570692B2 (ja) * | 1986-06-20 | 1997-01-08 | 株式会社豊田自動織機製作所 | 可変容量式回転型圧縮機 |
| JPH0771637B2 (ja) * | 1986-06-30 | 1995-08-02 | 株式会社佐竹製作所 | 竪軸精米装置 |
| JPS63182041A (ja) * | 1987-01-21 | 1988-07-27 | 株式会社 サタケ | 竪軸型精穀機 |
| JPS63218258A (ja) * | 1987-03-06 | 1988-09-12 | 株式会社 サタケ | 竪軸型精穀機 |
| JPH0822389B2 (ja) * | 1987-07-27 | 1996-03-06 | 株式会社佐竹製作所 | 竪軸型摩擦切削式精米機 |
| JPH01262949A (ja) * | 1988-04-14 | 1989-10-19 | Satake Eng Co Ltd | 竪軸型摩擦切削式精穀機 |
| JP3266167B2 (ja) * | 1993-08-06 | 2002-03-18 | 株式会社サタケ | 竪型研削式精穀機の抵抗体調節装置 |
| JPH0775741A (ja) * | 1993-09-07 | 1995-03-20 | Satake Eng Co Ltd | 研削式竪型精穀機の除糠用多孔性筒状体 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1210106A (en) * | 1967-03-08 | 1970-10-28 | Colgate Palmolive Co | Derivatives of 1,2,3,4-tetrahydrobenzothieno [2,3-c]pyridine and of 1,2,3,4-tetrahydro-5h-benzothieno[2,3-c]azepine |
| US3704237A (en) * | 1971-04-29 | 1972-11-28 | Colgate Palmolive Co | Certain 2-amidino-1,2,3,4-tetrahydrobenzothiene(2,3-c)pyridines |
| US3752820A (en) * | 1972-03-20 | 1973-08-14 | Colgate Palmolive Co | Substituted-1,2,3,4-tetrahydrobenzothieno(3,2-c)pyridine derivatives |
| FR2358150A1 (fr) * | 1976-07-13 | 1978-02-10 | Parcor | Nouvelles thieno (2,3-c) et (3,2-c) pyridines, leur procede de preparation et leur application |
-
1977
- 1977-06-02 FR FR7716878A patent/FR2423494A1/fr active Granted
-
1978
- 1978-05-23 US US05/908,856 patent/US4172134A/en not_active Expired - Lifetime
- 1978-05-23 IE IE1022/78A patent/IE46848B1/en unknown
- 1978-05-29 ES ES470274A patent/ES470274A1/es not_active Expired
- 1978-05-30 GB GB24218/78A patent/GB1572690A/en not_active Expired
- 1978-06-01 DE DE7878400006T patent/DE2860971D1/de not_active Expired
- 1978-06-01 EP EP78400006A patent/EP0000108B1/fr not_active Expired
- 1978-06-01 DK DK244878A patent/DK152130C/da not_active IP Right Cessation
- 1978-06-02 LU LU79763A patent/LU79763A1/fr unknown
- 1978-06-02 JP JP6660378A patent/JPS543098A/ja active Granted
Also Published As
| Publication number | Publication date |
|---|---|
| FR2423494B1 (fr) | 1980-10-17 |
| GB1572690A (en) | 1980-07-30 |
| ES470274A1 (es) | 1979-09-16 |
| DE2860971D1 (en) | 1981-11-12 |
| DK152130B (da) | 1988-02-01 |
| IE46848B1 (en) | 1983-10-05 |
| IE781022L (en) | 1978-12-02 |
| DK244878A (da) | 1978-12-03 |
| US4172134A (en) | 1979-10-23 |
| DK152130C (da) | 1988-08-15 |
| EP0000108A1 (fr) | 1978-12-20 |
| LU79763A1 (fr) | 1978-11-28 |
| FR2423494A1 (fr) | 1979-11-16 |
| JPS543098A (en) | 1979-01-11 |
| JPS6230191B2 (fr) | 1987-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0527687B1 (fr) | Nouveaux dérivés d'aryléthylamines, leurs procédés de préparation et les compositions pharmaceutiques qui les contiennent | |
| EP0478446B1 (fr) | Nouvelles amines alkyl hétérocycliques, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| FR2663633A1 (fr) | Nouvelles chalcones, leur procede de preparation et les compositions pharmaceutiques qui les contiennent. | |
| EP0000108B1 (fr) | Benzo(b)thiéno-pyridines, leur procédé de préparation et composition thérapeutique les contenant | |
| FR2663935A1 (fr) | Nouveaux 1,2,3,4,5,6-hexahydroazepino [4,5-b] indoles et 1,2,3,4-tetrahydrobethacarbolines, leurs procedes de preparation et les compositions pharmaceutiques qui les contiennent. | |
| FR2495156A1 (fr) | Derives de la thieno-pyridinone, leur procede de preparation et leur application therapeutique | |
| CH631179A5 (fr) | Tetrahydro-4,5,6,7 thieno (2,3-c) et (3,2-c) pyridines et leur procede de preparation. | |
| EP0366511A1 (fr) | Nouveaux dérivés benzoxazolinoniques, leurs procédés de préparation et les compositions pharmaceutiques qui les contiennent | |
| CH624409A5 (fr) | ||
| EP0114850B1 (fr) | Nouveaux derives substitues du 2,5-diamino 1,4-diazole, leurs procedes de preparation et les compositions pharmaceutiques en renfermant | |
| EP0286515A1 (fr) | Nouvelles amines tricycliques dérivées du tétrahydro-5,6,7,8 naphto [2,3b] dihydro-2,3 furanne, et du tétrahydro-6,7,8,9 5H-benzocyclohepta [2,3b] dihydro-2,3 furanne, leurs procédés de préparation et les compositions pharmaceutiques qui les contiennent | |
| FR2536398A1 (fr) | Nouveaux composes heterocycliques | |
| CH629810A5 (fr) | Derives de thieno (2,3-c) et (3,2-c) pyridines et procede de preparation de ces derives. | |
| EP0239436A1 (fr) | Nouveau dérivé tricyclique dénommé acide (chloro-3 méthyl-6 dioxo-5,5 dihydro-6, 11 dibenzo (c,f) thiazépine (1,2) yl-11 amino) -5 pentanoique, son procédé de préparation et les compositions pharmaceutiques qui le contiennent | |
| EP0385848A1 (fr) | Nouveaux dérivés benzoxazolinoniques, leurs procédés de préparation et les compositions pharmaceutiques qui les contiennent | |
| FR2663634A1 (fr) | Nouvelles acyl benzoxazolinones, leur procede de preparation et les compositions pharmaceutiques qui les contiennent. | |
| FR2637596A1 (fr) | Methyl-4 ((aryl-4 piperazinyl-1)-2 ethyl)-5 thiazole et ses derives, leur procede de preparation et les medicaments en contenant | |
| EP0117171B1 (fr) | Dérivés aminés de la pyridazine substitués en position 6 par un hétérocycle ou un alicycle, procédé d'obtention et médicaments les contenant | |
| EP0136198B1 (fr) | Dérivés de triazolo pyrimidine, leur procédé de préparation et leur application thérapeutique en tant que toni-cardiaques | |
| EP0006772B1 (fr) | Dérivés de la thiéno et furopyridone, procédé pour leur préparation et médicaments les contenant | |
| EP0003920B1 (fr) | Thiéno (3,2-c) et thiéno (2,3-c)pyridines, leur procédé de préparation et leur application en thérapeutique | |
| BE898151A (fr) | Nouveaux dérivés de la quinazoline, leur préparation et leur utilisation comme médicaments. | |
| EP2497774B1 (fr) | Dérivés de dihydro-oxazolobenzodiazépinone, procédés de leur préparation et compositions pharmaceutiques contenant ces composés | |
| EP1256583B1 (fr) | Dérivés de pyrimidin-4-ones, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| CH618173A5 (fr) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): DE NL |
|
| 17P | Request for examination filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): DE NL |
|
| REF | Corresponds to: |
Ref document number: 2860971 Country of ref document: DE Date of ref document: 19811112 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19890616 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19890630 Year of fee payment: 12 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19910101 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19910301 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |