WO2024219480A1 - Method for producing cyclic peptide compound - Google Patents
Method for producing cyclic peptide compound Download PDFInfo
- Publication number
- WO2024219480A1 WO2024219480A1 PCT/JP2024/015529 JP2024015529W WO2024219480A1 WO 2024219480 A1 WO2024219480 A1 WO 2024219480A1 JP 2024015529 W JP2024015529 W JP 2024015529W WO 2024219480 A1 WO2024219480 A1 WO 2024219480A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal
- group
- peptide compound
- alkyl
- formula
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 483
- 108010069514 Cyclic Peptides Proteins 0.000 title claims abstract description 177
- 102000001189 Cyclic Peptides Human genes 0.000 title claims abstract description 177
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 42
- 239000013078 crystal Substances 0.000 claims abstract description 426
- 238000000034 method Methods 0.000 claims abstract description 304
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 174
- 238000007363 ring formation reaction Methods 0.000 claims abstract description 124
- 239000007791 liquid phase Substances 0.000 claims abstract description 9
- -1 Q 9 Chemical compound 0.000 claims description 229
- 239000002904 solvent Substances 0.000 claims description 174
- 239000012453 solvate Substances 0.000 claims description 128
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 104
- 229910052739 hydrogen Inorganic materials 0.000 claims description 88
- 239000001257 hydrogen Substances 0.000 claims description 88
- 150000003839 salts Chemical class 0.000 claims description 86
- 239000003153 chemical reaction reagent Substances 0.000 claims description 84
- 125000006239 protecting group Chemical group 0.000 claims description 81
- 238000009833 condensation Methods 0.000 claims description 79
- 230000005494 condensation Effects 0.000 claims description 79
- 229910052757 nitrogen Inorganic materials 0.000 claims description 79
- 125000000217 alkyl group Chemical group 0.000 claims description 75
- 229910052799 carbon Inorganic materials 0.000 claims description 72
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 68
- 125000001433 C-terminal amino-acid group Chemical group 0.000 claims description 59
- 125000000729 N-terminal amino-acid group Chemical group 0.000 claims description 58
- 125000000623 heterocyclic group Chemical group 0.000 claims description 57
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 54
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 53
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 50
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 50
- 125000003118 aryl group Chemical group 0.000 claims description 47
- 125000003277 amino group Chemical group 0.000 claims description 42
- 125000004104 aryloxy group Chemical group 0.000 claims description 38
- 229910052736 halogen Inorganic materials 0.000 claims description 37
- 150000002367 halogens Chemical class 0.000 claims description 35
- 125000005415 substituted alkoxy group Chemical group 0.000 claims description 29
- 239000003495 polar organic solvent Substances 0.000 claims description 27
- 125000002723 alicyclic group Chemical group 0.000 claims description 26
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 23
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 22
- 125000004432 carbon atom Chemical group C* 0.000 claims description 16
- 150000002430 hydrocarbons Chemical class 0.000 claims description 15
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 14
- 239000004215 Carbon black (E152) Substances 0.000 claims description 14
- 229930195733 hydrocarbon Natural products 0.000 claims description 14
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 12
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 12
- 150000001408 amides Chemical class 0.000 claims description 11
- 239000004210 ether based solvent Substances 0.000 claims description 10
- 150000002825 nitriles Chemical class 0.000 claims description 10
- 239000003759 ester based solvent Substances 0.000 claims description 9
- 238000001914 filtration Methods 0.000 claims description 8
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 5
- 239000003660 carbonate based solvent Substances 0.000 claims description 5
- 125000005843 halogen group Chemical group 0.000 claims description 5
- 150000001413 amino acids Chemical class 0.000 abstract description 92
- 125000004122 cyclic group Chemical group 0.000 abstract description 48
- 239000006227 byproduct Substances 0.000 abstract description 33
- 238000002425 crystallisation Methods 0.000 abstract description 11
- 230000008025 crystallization Effects 0.000 abstract description 11
- 239000012634 fragment Substances 0.000 abstract description 10
- 230000008878 coupling Effects 0.000 abstract description 4
- 238000010168 coupling process Methods 0.000 abstract description 4
- 238000005859 coupling reaction Methods 0.000 abstract description 4
- 239000002243 precursor Substances 0.000 abstract description 4
- 102000004196 processed proteins & peptides Human genes 0.000 abstract description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 200
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 112
- 238000006243 chemical reaction Methods 0.000 description 108
- 239000000243 solution Substances 0.000 description 98
- 229940024606 amino acid Drugs 0.000 description 86
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 80
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 75
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 74
- 230000002829 reductive effect Effects 0.000 description 68
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 66
- 239000002585 base Substances 0.000 description 66
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 65
- 239000000203 mixture Substances 0.000 description 65
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 61
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 57
- 150000001721 carbon Chemical group 0.000 description 54
- 239000012044 organic layer Substances 0.000 description 54
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 53
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 53
- 239000011347 resin Substances 0.000 description 52
- 229920005989 resin Polymers 0.000 description 52
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 49
- 239000000047 product Substances 0.000 description 46
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 45
- 238000004128 high performance liquid chromatography Methods 0.000 description 39
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 37
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 35
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 34
- GPDHNZNLPKYHCN-DZOOLQPHSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.CCOC(=O)C(\C#N)=N/OC(=[N+](C)C)N1CCOCC1 GPDHNZNLPKYHCN-DZOOLQPHSA-N 0.000 description 33
- 229940125904 compound 1 Drugs 0.000 description 31
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 31
- 238000003786 synthesis reaction Methods 0.000 description 31
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 30
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- 230000015572 biosynthetic process Effects 0.000 description 30
- 239000010410 layer Substances 0.000 description 29
- 230000014759 maintenance of location Effects 0.000 description 28
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 27
- 235000019439 ethyl acetate Nutrition 0.000 description 27
- 239000007788 liquid Substances 0.000 description 26
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 25
- 238000005259 measurement Methods 0.000 description 25
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 24
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 24
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 23
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 21
- 125000003710 aryl alkyl group Chemical group 0.000 description 21
- 125000000753 cycloalkyl group Chemical group 0.000 description 21
- 238000003756 stirring Methods 0.000 description 21
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 20
- 239000011541 reaction mixture Substances 0.000 description 20
- 238000000926 separation method Methods 0.000 description 20
- 125000001424 substituent group Chemical group 0.000 description 20
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 19
- 150000001336 alkenes Chemical class 0.000 description 19
- 125000001072 heteroaryl group Chemical group 0.000 description 19
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 18
- 125000003342 alkenyl group Chemical group 0.000 description 18
- 125000000304 alkynyl group Chemical group 0.000 description 18
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 18
- 208000012839 conversion disease Diseases 0.000 description 18
- 239000012046 mixed solvent Substances 0.000 description 18
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 18
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 17
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 17
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 17
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 16
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 description 16
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 16
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 16
- KPFBUSLHFFWMAI-HYRPPVSQSA-N [(8r,9s,10r,13s,14s,17r)-17-acetyl-6-formyl-3-methoxy-10,13-dimethyl-1,2,7,8,9,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl] acetate Chemical compound C1C[C@@H]2[C@](CCC(OC)=C3)(C)C3=C(C=O)C[C@H]2[C@@H]2CC[C@](OC(C)=O)(C(C)=O)[C@]21C KPFBUSLHFFWMAI-HYRPPVSQSA-N 0.000 description 15
- 239000011259 mixed solution Substances 0.000 description 15
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 15
- 239000011780 sodium chloride Substances 0.000 description 15
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 14
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 14
- 238000004455 differential thermal analysis Methods 0.000 description 14
- 238000002411 thermogravimetry Methods 0.000 description 14
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 13
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 13
- 238000001514 detection method Methods 0.000 description 13
- 239000012071 phase Substances 0.000 description 13
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 13
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 13
- 239000003643 water by type Substances 0.000 description 13
- 125000003963 dichloro group Chemical group Cl* 0.000 description 12
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 12
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 12
- YAYGSLOSTXKUBW-UHFFFAOYSA-N ruthenium(2+) Chemical compound [Ru+2] YAYGSLOSTXKUBW-UHFFFAOYSA-N 0.000 description 12
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 12
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 11
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 11
- RDWDVLFMPFUBDV-PXMDEAMVSA-N [(e)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-tripyrrolidin-1-ylphosphanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.C1CCCN1[P+](N1CCCC1)(O/N=C(C(=O)OCC)\C#N)N1CCCC1 RDWDVLFMPFUBDV-PXMDEAMVSA-N 0.000 description 11
- 239000002253 acid Substances 0.000 description 11
- 125000000539 amino acid group Chemical group 0.000 description 11
- 238000006482 condensation reaction Methods 0.000 description 11
- 229940011051 isopropyl acetate Drugs 0.000 description 11
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 11
- OVRKATYHWPCGPZ-UHFFFAOYSA-N 4-methyloxane Chemical compound CC1CCOCC1 OVRKATYHWPCGPZ-UHFFFAOYSA-N 0.000 description 10
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Chemical compound OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 10
- 150000008065 acid anhydrides Chemical class 0.000 description 10
- 239000012190 activator Substances 0.000 description 10
- 150000004820 halides Chemical class 0.000 description 10
- 125000004430 oxygen atom Chemical group O* 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 102000016914 ras Proteins Human genes 0.000 description 10
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 10
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 9
- 125000003545 alkoxy group Chemical group 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- 238000004364 calculation method Methods 0.000 description 9
- 229940125810 compound 20 Drugs 0.000 description 9
- 239000012141 concentrate Substances 0.000 description 9
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 9
- 238000010828 elution Methods 0.000 description 9
- 125000000524 functional group Chemical group 0.000 description 9
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 9
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 9
- 239000002994 raw material Substances 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 9
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 8
- AUHZEENZYGFFBQ-UHFFFAOYSA-N 1,3,5-Me3C6H3 Natural products CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 8
- RXYPXQSKLGGKOL-UHFFFAOYSA-N 1,4-dimethylpiperazine Chemical compound CN1CCN(C)CC1 RXYPXQSKLGGKOL-UHFFFAOYSA-N 0.000 description 8
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 8
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 8
- YQHJFPFNGVDEDT-UHFFFAOYSA-N 2-tert-butyl-1,1,3,3-tetramethylguanidine Chemical compound CN(C)C(N(C)C)=NC(C)(C)C YQHJFPFNGVDEDT-UHFFFAOYSA-N 0.000 description 8
- BMTZEAOGFDXDAD-UHFFFAOYSA-M 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium;chloride Chemical compound [Cl-].COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 BMTZEAOGFDXDAD-UHFFFAOYSA-M 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- 125000002252 acyl group Chemical group 0.000 description 8
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 8
- 229940125961 compound 24 Drugs 0.000 description 8
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 238000005649 metathesis reaction Methods 0.000 description 8
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 8
- 238000005464 sample preparation method Methods 0.000 description 8
- 229910000029 sodium carbonate Inorganic materials 0.000 description 8
- 238000010532 solid phase synthesis reaction Methods 0.000 description 8
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 8
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 8
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 8
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 7
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 7
- LINDOXZENKYESA-UHFFFAOYSA-N TMG Natural products CNC(N)=NC LINDOXZENKYESA-UHFFFAOYSA-N 0.000 description 7
- 229940125773 compound 10 Drugs 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 7
- 238000010966 qNMR Methods 0.000 description 7
- 125000003003 spiro group Chemical group 0.000 description 7
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 6
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- 229940126657 Compound 17 Drugs 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 6
- 238000002441 X-ray diffraction Methods 0.000 description 6
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 230000008859 change Effects 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 229940126208 compound 22 Drugs 0.000 description 6
- 238000004090 dissolution Methods 0.000 description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 6
- GAEKPEKOJKCEMS-UHFFFAOYSA-N gamma-valerolactone Chemical compound CC1CCC(=O)O1 GAEKPEKOJKCEMS-UHFFFAOYSA-N 0.000 description 6
- 238000007429 general method Methods 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 150000007530 organic bases Chemical class 0.000 description 6
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 6
- PGMYKACGEOXYJE-UHFFFAOYSA-N pentyl acetate Chemical compound CCCCCOC(C)=O PGMYKACGEOXYJE-UHFFFAOYSA-N 0.000 description 6
- 125000004434 sulfur atom Chemical group 0.000 description 6
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 5
- RUKVGXGTVPPWDD-UHFFFAOYSA-N 1,3-bis(2,4,6-trimethylphenyl)imidazolidine Chemical group CC1=CC(C)=CC(C)=C1N1CN(C=2C(=CC(C)=CC=2C)C)CC1 RUKVGXGTVPPWDD-UHFFFAOYSA-N 0.000 description 5
- HVFQJWGYVXKLTE-UHFFFAOYSA-N 3,5-bis(trifluoromethyl)benzoic acid Chemical compound OC(=O)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 HVFQJWGYVXKLTE-UHFFFAOYSA-N 0.000 description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 5
- 125000000649 benzylidene group Chemical group [H]C(=[*])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 5
- 229940125797 compound 12 Drugs 0.000 description 5
- 229940125833 compound 23 Drugs 0.000 description 5
- 125000004663 dialkyl amino group Chemical group 0.000 description 5
- 238000007599 discharging Methods 0.000 description 5
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 5
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 150000004682 monohydrates Chemical class 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 5
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 5
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 5
- 239000007790 solid phase Substances 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 125000000565 sulfonamide group Chemical group 0.000 description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 5
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 4
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 description 4
- TXYQMVROVVJZHJ-UHFFFAOYSA-N 1,1,2,3-tetramethyl-3-[8-[methyl-(n,n,n'-trimethylcarbamimidoyl)amino]naphthalen-1-yl]guanidine Chemical compound C1=CC(N(C)C(=NC)N(C)C)=C2C(N(C)C(N(C)C)=NC)=CC=CC2=C1 TXYQMVROVVJZHJ-UHFFFAOYSA-N 0.000 description 4
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 4
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 4
- OEBXWWBYZJNKRK-UHFFFAOYSA-N 1-methyl-2,3,4,6,7,8-hexahydropyrimido[1,2-a]pyrimidine Chemical compound C1CCN=C2N(C)CCCN21 OEBXWWBYZJNKRK-UHFFFAOYSA-N 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- LJCZNYWLQZZIOS-UHFFFAOYSA-N 2,2,2-trichlorethoxycarbonyl chloride Chemical group ClC(=O)OCC(Cl)(Cl)Cl LJCZNYWLQZZIOS-UHFFFAOYSA-N 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 4
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical group O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 4
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 4
- 125000001318 4-trifluoromethylbenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])*)C(F)(F)F 0.000 description 4
- VARHFUNXFXTFII-UHFFFAOYSA-N 6052-72-8 Chemical compound C1CCC2=CN=CC3=C2N1CCC3 VARHFUNXFXTFII-UHFFFAOYSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- 102100030708 GTPase KRas Human genes 0.000 description 4
- 102100039788 GTPase NRas Human genes 0.000 description 4
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 4
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 4
- 150000001576 beta-amino acids Chemical class 0.000 description 4
- 238000006664 bond formation reaction Methods 0.000 description 4
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- AJDPNPAGZMZOMN-UHFFFAOYSA-N diethyl (4-oxo-1,2,3-benzotriazin-3-yl) phosphate Chemical compound C1=CC=C2C(=O)N(OP(=O)(OCC)OCC)N=NC2=C1 AJDPNPAGZMZOMN-UHFFFAOYSA-N 0.000 description 4
- 239000000539 dimer Substances 0.000 description 4
- 238000011978 dissolution method Methods 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- JBTWLSYIZRCDFO-UHFFFAOYSA-N ethyl methyl carbonate Chemical compound CCOC(=O)OC JBTWLSYIZRCDFO-UHFFFAOYSA-N 0.000 description 4
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 4
- 239000005457 ice water Substances 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 4
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 4
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 3
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 3
- KYVBNYUBXIEUFW-UHFFFAOYSA-N 1,1,3,3-tetramethylguanidine Chemical compound CN(C)C(=N)N(C)C KYVBNYUBXIEUFW-UHFFFAOYSA-N 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- ZRPFJAPZDXQHSM-UHFFFAOYSA-L 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazole;dichloro-[(2-propan-2-yloxyphenyl)methylidene]ruthenium Chemical compound CC(C)OC1=CC=CC=C1C=[Ru](Cl)(Cl)=C1N(C=2C(=CC(C)=CC=2C)C)CCN1C1=C(C)C=C(C)C=C1C ZRPFJAPZDXQHSM-UHFFFAOYSA-L 0.000 description 3
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 3
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 3
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 3
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 3
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 3
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- 102100029974 GTPase HRas Human genes 0.000 description 3
- 101000584633 Homo sapiens GTPase HRas Proteins 0.000 description 3
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 3
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 3
- RJUFJBKOKNCXHH-UHFFFAOYSA-N Methyl propionate Chemical compound CCC(=O)OC RJUFJBKOKNCXHH-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 3
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 229940043232 butyl acetate Drugs 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 229940125846 compound 25 Drugs 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 150000004691 decahydrates Chemical class 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- QLVWOKQMDLQXNN-UHFFFAOYSA-N dibutyl carbonate Chemical compound CCCCOC(=O)OCCCC QLVWOKQMDLQXNN-UHFFFAOYSA-N 0.000 description 3
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 3
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 229940093499 ethyl acetate Drugs 0.000 description 3
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 3
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 3
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 3
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 229940017219 methyl propionate Drugs 0.000 description 3
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 3
- 235000019799 monosodium phosphate Nutrition 0.000 description 3
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 150000004686 pentahydrates Chemical class 0.000 description 3
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 3
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 3
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 3
- 229940090181 propyl acetate Drugs 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 150000003456 sulfonamides Chemical class 0.000 description 3
- 150000003512 tertiary amines Chemical class 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 3
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 3
- 150000004684 trihydrates Chemical class 0.000 description 3
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 3
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 2
- GANZFSNGFPNJGI-VLIAUNLRSA-N (2s,4r)-4-ethoxy-1-(9h-fluoren-9-ylmethoxycarbonyl)pyrrolidine-2-carboxylic acid Chemical compound C1[C@H](OCC)C[C@@H](C(O)=O)N1C(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 GANZFSNGFPNJGI-VLIAUNLRSA-N 0.000 description 2
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- OOWSDKUFKGVADH-UHFFFAOYSA-N 1-diphenylphosphoryloxy-2,3,4,5,6-pentafluorobenzene Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1OP(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 OOWSDKUFKGVADH-UHFFFAOYSA-N 0.000 description 2
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 2
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- DEXFNLNNUZKHNO-UHFFFAOYSA-N 6-[3-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperidin-1-yl]-3-oxopropyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1CCN(CC1)C(CCC1=CC2=C(NC(O2)=O)C=C1)=O DEXFNLNNUZKHNO-UHFFFAOYSA-N 0.000 description 2
- TZCYLJGNWDVJRA-UHFFFAOYSA-N 6-chloro-1-hydroxybenzotriazole Chemical compound C1=C(Cl)C=C2N(O)N=NC2=C1 TZCYLJGNWDVJRA-UHFFFAOYSA-N 0.000 description 2
- 230000035502 ADME Effects 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 102000018898 GTPase-Activating Proteins Human genes 0.000 description 2
- 108091006094 GTPase-accelerating proteins Proteins 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 108010067218 Guanine Nucleotide Exchange Factors Proteins 0.000 description 2
- 102000016285 Guanine Nucleotide Exchange Factors Human genes 0.000 description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 125000006598 aminocarbonylamino group Chemical group 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- IADUEWIQBXOCDZ-UHFFFAOYSA-N azetidine-2-carboxylic acid Chemical compound OC(=O)C1CCN1 IADUEWIQBXOCDZ-UHFFFAOYSA-N 0.000 description 2
- 125000002393 azetidinyl group Chemical group 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- PNPBGYBHLCEVMK-UHFFFAOYSA-L benzylidene(dichloro)ruthenium;tricyclohexylphosphane Chemical compound Cl[Ru](Cl)=CC1=CC=CC=C1.C1CCCCC1P(C1CCCCC1)C1CCCCC1.C1CCCCC1P(C1CCCCC1)C1CCCCC1 PNPBGYBHLCEVMK-UHFFFAOYSA-L 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 210000000692 cap cell Anatomy 0.000 description 2
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 2
- 125000005708 carbonyloxy group Chemical group [*:2]OC([*:1])=O 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940125851 compound 27 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125807 compound 37 Drugs 0.000 description 2
- 230000006552 constitutive activation Effects 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- DIBHLCJAJIKHGB-UHFFFAOYSA-N dec-5-ene Chemical compound [CH2]CCCC=CCCCC DIBHLCJAJIKHGB-UHFFFAOYSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 150000004683 dihydrates Chemical class 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- 125000000532 dioxanyl group Chemical group 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- LCFXLZAXGXOXAP-QPJJXVBHSA-N ethyl (2e)-2-cyano-2-hydroxyiminoacetate Chemical compound CCOC(=O)C(=N\O)\C#N LCFXLZAXGXOXAP-QPJJXVBHSA-N 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 150000004688 heptahydrates Chemical class 0.000 description 2
- 150000004687 hexahydrates Chemical class 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 125000005945 imidazopyridyl group Chemical group 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 150000004689 octahydrates Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 125000005740 oxycarbonyl group Chemical group [*:1]OC([*:2])=O 0.000 description 2
- 238000005897 peptide coupling reaction Methods 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 125000004437 phosphorous atom Chemical group 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-M prolinate Chemical compound [O-]C(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-M 0.000 description 2
- 229960002429 proline Drugs 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000007142 ring opening reaction Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 2
- 150000003377 silicon compounds Chemical class 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- 235000014347 soups Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 2
- 125000006296 sulfonyl amino group Chemical group [H]N(*)S(*)(=O)=O 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 239000013076 target substance Substances 0.000 description 2
- RNLQHMIDSCYLAK-UHFFFAOYSA-N tert-butyl 2-(methylamino)acetate;hydron;chloride Chemical compound Cl.CNCC(=O)OC(C)(C)C RNLQHMIDSCYLAK-UHFFFAOYSA-N 0.000 description 2
- 125000001302 tertiary amino group Chemical group 0.000 description 2
- 150000004685 tetrahydrates Chemical class 0.000 description 2
- 150000003527 tetrahydropyrans Chemical group 0.000 description 2
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000005500 uronium group Chemical group 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- UXOBFFXDCHDYNQ-IBGZPJMESA-N (2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]pent-4-enoic acid Chemical compound C1=C2C3=C(C=CC=C3)C(COC(=O)N(C)[C@@H](CC=C)C(=O)O)C2=CC=C1 UXOBFFXDCHDYNQ-IBGZPJMESA-N 0.000 description 1
- YMEGJWTUWMVZPD-QFIPXVFZSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-[4-(trifluoromethyl)phenyl]propanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)C1=CC=C(C(F)(F)F)C=C1 YMEGJWTUWMVZPD-QFIPXVFZSA-N 0.000 description 1
- HKELUUGCKFRJQM-IBGZPJMESA-N (2s)-2-[9h-fluoren-9-ylmethoxycarbonyl(methyl)amino]pentanoic acid Chemical compound C1=CC=C2C(COC(=O)N(C)[C@@H](CCC)C(O)=O)C3=CC=CC=C3C2=C1 HKELUUGCKFRJQM-IBGZPJMESA-N 0.000 description 1
- CRFFPDBJLGAGQL-QMMMGPOBSA-N (2s)-2-amino-3-[4-(trifluoromethyl)phenyl]propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(C(F)(F)F)C=C1 CRFFPDBJLGAGQL-QMMMGPOBSA-N 0.000 description 1
- JSHXJPFZKBRLFU-JQWIXIFHSA-N (2s,3s)-3-methyl-2-(phenylmethoxycarbonylamino)pentanoic acid Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)OCC1=CC=CC=C1 JSHXJPFZKBRLFU-JQWIXIFHSA-N 0.000 description 1
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- ZCTHVVPOQJDLEW-UHFFFAOYSA-N 1,1,2,3-tetramethyl-3-[1-[methyl-(n,n,n'-trimethylcarbamimidoyl)amino]naphthalen-2-yl]guanidine Chemical compound C1=CC=CC2=C(N(C)C(=NC)N(C)C)C(N(C)C(N(C)C)=NC)=CC=C21 ZCTHVVPOQJDLEW-UHFFFAOYSA-N 0.000 description 1
- 125000005919 1,2,2-trimethylpropyl group Chemical group 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- CZSRXHJVZUBEGW-UHFFFAOYSA-N 1,2-thiazolidine Chemical group C1CNSC1 CZSRXHJVZUBEGW-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- YIWGJFPJRAEKMK-UHFFFAOYSA-N 1-(2H-benzotriazol-5-yl)-3-methyl-8-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carbonyl]-1,3,8-triazaspiro[4.5]decane-2,4-dione Chemical compound CN1C(=O)N(c2ccc3n[nH]nc3c2)C2(CCN(CC2)C(=O)c2cnc(NCc3cccc(OC(F)(F)F)c3)nc2)C1=O YIWGJFPJRAEKMK-UHFFFAOYSA-N 0.000 description 1
- HMUNWXXNJPVALC-UHFFFAOYSA-N 1-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C(CN1CC2=C(CC1)NN=N2)=O HMUNWXXNJPVALC-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- JFLSOKIMYBSASW-UHFFFAOYSA-N 1-chloro-2-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 JFLSOKIMYBSASW-UHFFFAOYSA-N 0.000 description 1
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- JQCSUVJDBHJKNG-UHFFFAOYSA-N 1-methoxy-ethyl Chemical group C[CH]OC JQCSUVJDBHJKNG-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 102000010400 1-phosphatidylinositol-3-kinase activity proteins Human genes 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000004778 2,2-difluoroethyl group Chemical group [H]C([H])(*)C([H])(F)F 0.000 description 1
- PDELQDSYLBLPQO-UHFFFAOYSA-N 2,3,3a,4,5,6,7,7a-octahydro-1h-indole Chemical group C1CCCC2NCCC21 PDELQDSYLBLPQO-UHFFFAOYSA-N 0.000 description 1
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 1
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 1
- JQMFQLVAJGZSQS-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-N-(2-oxo-3H-1,3-benzoxazol-6-yl)acetamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)NC1=CC2=C(NC(O2)=O)C=C1 JQMFQLVAJGZSQS-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- YBDQLHBVNXARAU-UHFFFAOYSA-N 2-methyloxane Chemical compound CC1CCCCO1 YBDQLHBVNXARAU-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000005979 2-naphthyloxy group Chemical group 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000006479 2-pyridyl methyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 1
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- HJBLUNHMOKFZQX-UHFFFAOYSA-N 3-hydroxy-1,2,3-benzotriazin-4-one Chemical compound C1=CC=C2C(=O)N(O)N=NC2=C1 HJBLUNHMOKFZQX-UHFFFAOYSA-N 0.000 description 1
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000006201 3-phenylpropyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005977 3-phenylpropyloxy group Chemical group 0.000 description 1
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 description 1
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- DMAYBPBPEUFIHJ-UHFFFAOYSA-N 4-bromobut-1-ene Chemical compound BrCCC=C DMAYBPBPEUFIHJ-UHFFFAOYSA-N 0.000 description 1
- VRJHQPZVIGNGMX-UHFFFAOYSA-N 4-piperidinone Chemical group O=C1CCNCC1 VRJHQPZVIGNGMX-UHFFFAOYSA-N 0.000 description 1
- IEUNIAZAENZEMT-UHFFFAOYSA-N 5,9-dioxaspiro[3.5]nonane Chemical group C1CCC21OCCCO2 IEUNIAZAENZEMT-UHFFFAOYSA-N 0.000 description 1
- CONKBQPVFMXDOV-QHCPKHFHSA-N 6-[(5S)-5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-2-oxo-1,3-oxazolidin-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C[C@H]1CN(C(O1)=O)C1=CC2=C(NC(O2)=O)C=C1 CONKBQPVFMXDOV-QHCPKHFHSA-N 0.000 description 1
- WTFUTSCZYYCBAY-SXBRIOAWSA-N 6-[(E)-C-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-N-hydroxycarbonimidoyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C/C(=N/O)/C1=CC2=C(NC(O2)=O)C=C1 WTFUTSCZYYCBAY-SXBRIOAWSA-N 0.000 description 1
- DFGKGUXTPFWHIX-UHFFFAOYSA-N 6-[2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]acetyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)C1=CC2=C(NC(O2)=O)C=C1 DFGKGUXTPFWHIX-UHFFFAOYSA-N 0.000 description 1
- LLQHSBBZNDXTIV-UHFFFAOYSA-N 6-[5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-4,5-dihydro-1,2-oxazol-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC1CC(=NO1)C1=CC2=C(NC(O2)=O)C=C1 LLQHSBBZNDXTIV-UHFFFAOYSA-N 0.000 description 1
- PRJHEJGMSOBHTO-UHFFFAOYSA-N 7-[(4-chlorophenyl)methyl]-1-(3-hydroxypropyl)-3-methyl-8-[3-(trifluoromethoxy)phenoxy]purine-2,6-dione Chemical compound C=1C=C(Cl)C=CC=1CN1C=2C(=O)N(CCCO)C(=O)N(C)C=2N=C1OC1=CC=CC(OC(F)(F)F)=C1 PRJHEJGMSOBHTO-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- IADUEWIQBXOCDZ-VKHMYHEASA-N Azetidine-2-carboxylic acid Natural products OC(=O)[C@@H]1CCN1 IADUEWIQBXOCDZ-VKHMYHEASA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical group [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 description 1
- 125000005865 C2-C10alkynyl group Chemical group 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- PUTNDPKAXXVOGH-IBGZPJMESA-N CN(C)C([C@H](CC(O)=O)N(C)C(OCC1C2=CC=CC=C2C2=C1C=CC=C2)=O)=O Chemical compound CN(C)C([C@H](CC(O)=O)N(C)C(OCC1C2=CC=CC=C2C2=C1C=CC=C2)=O)=O PUTNDPKAXXVOGH-IBGZPJMESA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- 238000010485 C−C bond formation reaction Methods 0.000 description 1
- 150000008574 D-amino acids Chemical class 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 238000007341 Heck reaction Methods 0.000 description 1
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical group C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 239000004201 L-cysteine Substances 0.000 description 1
- 235000013878 L-cysteine Nutrition 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 230000037364 MAPK/ERK pathway Effects 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- NEAPKZHDYMQZCB-UHFFFAOYSA-N N-[2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]ethyl]-2-oxo-3H-1,3-benzoxazole-6-carboxamide Chemical compound C1CN(CCN1CCNC(=O)C2=CC3=C(C=C2)NC(=O)O3)C4=CN=C(N=C4)NC5CC6=CC=CC=C6C5 NEAPKZHDYMQZCB-UHFFFAOYSA-N 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- VCUFZILGIRCDQQ-KRWDZBQOSA-N N-[[(5S)-2-oxo-3-(2-oxo-3H-1,3-benzoxazol-6-yl)-1,3-oxazolidin-5-yl]methyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C1O[C@H](CN1C1=CC2=C(NC(O2)=O)C=C1)CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F VCUFZILGIRCDQQ-KRWDZBQOSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 102100031424 Ras-related protein Ral-A Human genes 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 241000289690 Xenarthra Species 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- DGYIJVNZSDYBOE-UHFFFAOYSA-N [CH2]C1=CC=NC=C1 Chemical group [CH2]C1=CC=NC=C1 DGYIJVNZSDYBOE-UHFFFAOYSA-N 0.000 description 1
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 1
- HBQDCKPPSIWZLY-UHFFFAOYSA-M [dimethylamino-(2-oxopyridin-1-yl)oxymethylidene]-dimethylazanium;trifluoroborane;fluoride Chemical compound [F-].FB(F)F.CN(C)C(=[N+](C)C)ON1C=CC=CC1=O HBQDCKPPSIWZLY-UHFFFAOYSA-M 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- XBJFCYDKBDVADW-UHFFFAOYSA-N acetonitrile;formic acid Chemical compound CC#N.OC=O XBJFCYDKBDVADW-UHFFFAOYSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000006323 alkenyl amino group Chemical group 0.000 description 1
- 125000005089 alkenylaminocarbonyl group Chemical group 0.000 description 1
- 125000005090 alkenylcarbonyl group Chemical group 0.000 description 1
- 125000005091 alkenylcarbonylamino group Chemical group 0.000 description 1
- 125000005193 alkenylcarbonyloxy group Chemical group 0.000 description 1
- 125000003302 alkenyloxy group Chemical group 0.000 description 1
- 125000005092 alkenyloxycarbonyl group Chemical group 0.000 description 1
- 125000005137 alkenylsulfonyl group Chemical group 0.000 description 1
- 125000005108 alkenylthio group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 125000005130 alkyl carbonyl thio group Chemical group 0.000 description 1
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 1
- 125000004691 alkyl thio carbonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 125000006319 alkynyl amino group Chemical group 0.000 description 1
- 125000005095 alkynylaminocarbonyl group Chemical group 0.000 description 1
- 125000005087 alkynylcarbonyl group Chemical group 0.000 description 1
- 125000005088 alkynylcarbonylamino group Chemical group 0.000 description 1
- 125000005198 alkynylcarbonyloxy group Chemical group 0.000 description 1
- 125000005133 alkynyloxy group Chemical group 0.000 description 1
- 125000005225 alkynyloxycarbonyl group Chemical group 0.000 description 1
- 125000005139 alkynylsulfonyl group Chemical group 0.000 description 1
- 125000005109 alkynylthio group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000002344 aminooxy group Chemical group [H]N([H])O[*] 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 1
- 125000005140 aralkylsulfonyl group Chemical group 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000005098 aryl alkoxy carbonyl group Chemical group 0.000 description 1
- 125000005125 aryl alkyl amino carbonyl group Chemical group 0.000 description 1
- 125000001691 aryl alkyl amino group Chemical group 0.000 description 1
- 125000005126 aryl alkyl carbonyl amino group Chemical group 0.000 description 1
- 125000005099 aryl alkyl carbonyl group Chemical group 0.000 description 1
- 125000004659 aryl alkyl thio group Chemical group 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 125000005100 aryl amino carbonyl group Chemical group 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 125000005141 aryl amino sulfonyl group Chemical group 0.000 description 1
- 125000004658 aryl carbonyl amino group Chemical group 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- 125000005199 aryl carbonyloxy group Chemical group 0.000 description 1
- 125000005162 aryl oxy carbonyl amino group Chemical group 0.000 description 1
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 1
- 125000004657 aryl sulfonyl amino group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- YHNUDLCUIKMNSN-UHFFFAOYSA-N bis(1,2,4-triazol-1-yl)methanone Chemical compound C1=NC=NN1C(=O)N1C=NC=N1 YHNUDLCUIKMNSN-UHFFFAOYSA-N 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 1
- 125000006254 cycloalkyl carbonyl group Chemical group 0.000 description 1
- 125000005167 cycloalkylaminocarbonyl group Chemical group 0.000 description 1
- 125000005145 cycloalkylaminosulfonyl group Chemical group 0.000 description 1
- 125000005169 cycloalkylcarbonylamino group Chemical group 0.000 description 1
- 125000005201 cycloalkylcarbonyloxy group Chemical group 0.000 description 1
- 125000005170 cycloalkyloxycarbonyl group Chemical group 0.000 description 1
- 125000005144 cycloalkylsulfonyl group Chemical group 0.000 description 1
- 125000005366 cycloalkylthio group Chemical group 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000005131 dialkylammonium group Chemical group 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- HPYNZHMRTTWQTB-UHFFFAOYSA-N dimethylpyridine Natural products CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 1
- HGGNZMUHOHGHBJ-UHFFFAOYSA-N dioxepane Chemical group C1CCOOCC1 HGGNZMUHOHGHBJ-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- 125000006260 ethylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- HQVFCQRVQFYGRJ-UHFFFAOYSA-N formic acid;hydrate Chemical compound O.OC=O HQVFCQRVQFYGRJ-UHFFFAOYSA-N 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 125000005241 heteroarylamino group Chemical group 0.000 description 1
- 125000005222 heteroarylaminocarbonyl group Chemical group 0.000 description 1
- 125000005223 heteroarylcarbonyl group Chemical group 0.000 description 1
- 125000005224 heteroarylcarbonylamino group Chemical group 0.000 description 1
- 125000005204 heteroarylcarbonyloxy group Chemical group 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 125000005226 heteroaryloxycarbonyl group Chemical group 0.000 description 1
- 125000005143 heteroarylsulfonyl group Chemical group 0.000 description 1
- 125000005419 heteroarylsulfonylamino group Chemical group 0.000 description 1
- 125000005368 heteroarylthio group Chemical group 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 239000011987 hoveyda–grubbs catalyst Substances 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- AUONNNVJUCSETH-UHFFFAOYSA-N icosanoyl icosanoate Chemical compound CCCCCCCCCCCCCCCCCCCC(=O)OC(=O)CCCCCCCCCCCCCCCCCCC AUONNNVJUCSETH-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- CMWYAOXYQATXSI-UHFFFAOYSA-N n,n-dimethylformamide;piperidine Chemical compound CN(C)C=O.C1CCNCC1 CMWYAOXYQATXSI-UHFFFAOYSA-N 0.000 description 1
- SFMJNHNUOVADRW-UHFFFAOYSA-N n-[5-[9-[4-(methanesulfonamido)phenyl]-2-oxobenzo[h][1,6]naphthyridin-1-yl]-2-methylphenyl]prop-2-enamide Chemical compound C1=C(NC(=O)C=C)C(C)=CC=C1N1C(=O)C=CC2=C1C1=CC(C=3C=CC(NS(C)(=O)=O)=CC=3)=CC=C1N=C2 SFMJNHNUOVADRW-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- QWMHUFMEYKIYPC-JGAZGGJJSA-N n-cyclohexylcyclohexanamine;(2s,3s)-3-methyl-2-(phenylmethoxycarbonylamino)pentanoic acid Chemical compound C1CCCCC1[NH2+]C1CCCCC1.CC[C@H](C)[C@@H](C([O-])=O)NC(=O)OCC1=CC=CC=C1 QWMHUFMEYKIYPC-JGAZGGJJSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- ZUSSTQCWRDLYJA-UHFFFAOYSA-N n-hydroxy-5-norbornene-2,3-dicarboximide Chemical compound C1=CC2CC1C1C2C(=O)N(O)C1=O ZUSSTQCWRDLYJA-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical group C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- LVSJDHGRKAEGLX-UHFFFAOYSA-N oxolane;2,2,2-trifluoroacetic acid Chemical compound C1CCOC1.OC(=O)C(F)(F)F LVSJDHGRKAEGLX-UHFFFAOYSA-N 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical group O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- QGKLPGKXAVVPOJ-UHFFFAOYSA-N pyrrolidin-3-one Chemical group O=C1CCNC1 QGKLPGKXAVVPOJ-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000006479 redox reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 102000030938 small GTPase Human genes 0.000 description 1
- 108060007624 small GTPase Proteins 0.000 description 1
- XHFLOLLMZOTPSM-UHFFFAOYSA-M sodium;hydrogen carbonate;hydrate Chemical compound [OH-].[Na+].OC(O)=O XHFLOLLMZOTPSM-UHFFFAOYSA-M 0.000 description 1
- LMUMMJCCZMWLEN-UHFFFAOYSA-N spiro[3.3]heptyl Chemical group [CH]1CCC11CCC1 LMUMMJCCZMWLEN-UHFFFAOYSA-N 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000001174 sulfone group Chemical group 0.000 description 1
- 125000003375 sulfoxide group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- XJJBXZIKXFOMLP-ZETCQYMHSA-N tert-butyl (2s)-pyrrolidine-2-carboxylate Chemical compound CC(C)(C)OC(=O)[C@@H]1CCCN1 XJJBXZIKXFOMLP-ZETCQYMHSA-N 0.000 description 1
- IUUYANMOEMBTBV-FJXQXJEOSA-N tert-butyl (2s)-pyrrolidine-2-carboxylate;hydron;chloride Chemical compound Cl.CC(C)(C)OC(=O)[C@@H]1CCCN1 IUUYANMOEMBTBV-FJXQXJEOSA-N 0.000 description 1
- 150000005621 tetraalkylammonium salts Chemical class 0.000 description 1
- YBDQLHBVNXARAU-ZCFIWIBFSA-N tetrahydro-2-methyl-2H-pyran Natural products C[C@@H]1CCCCO1 YBDQLHBVNXARAU-ZCFIWIBFSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- RLTPJVKHGBFGQA-UHFFFAOYSA-N thiadiazolidine Chemical group C1CSNN1 RLTPJVKHGBFGQA-UHFFFAOYSA-N 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical group C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000004950 trifluoroalkyl group Chemical group 0.000 description 1
- HYWCXWRMUZYRPH-UHFFFAOYSA-N trimethyl(prop-2-enyl)silane Chemical compound C[Si](C)(C)CC=C HYWCXWRMUZYRPH-UHFFFAOYSA-N 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/02—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length in solution
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/30—Extraction; Separation; Purification by precipitation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/34—Extraction; Separation; Purification by filtration, ultrafiltration or reverse osmosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/50—Cyclic peptides containing at least one abnormal peptide link
- C07K7/52—Cyclic peptides containing at least one abnormal peptide link with only normal peptide links in the ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
Definitions
- the present invention relates to a method for producing a cyclic peptide compound, in particular a method for producing a cyclic peptide compound that has selective KRAS inhibitory activity against HRAS and NRAS.
- RAS is a protein belonging to the small GTPase family, and KRAS, NRAS, and HRAS are known.
- the activated or inactivated state of RAS is determined by its binding state with GDP or GTP. It is activated by the exchange reaction of GDP to GTP by GEF (guanine nucleotide exchange factor) and inactivated by the hydrolysis of GTP by GAP (GTPase-activating proteins) (Non-patent Document 1).
- GEF renal nucleotide exchange factor
- GAP GTPase-activating proteins
- Activated RAS induces cell proliferation, survival, and differentiation by activating various downstream signals such as the MAPK pathway, the PI3K/Akt pathway, and the RAL pathway, and the constitutive activation of RAS plays an important role in the development and progression of cancer.
- Non-patent Document 2 It is known that the RAS-RAF-MEK-ERK pathway is activated in cancer due to activation of upstream signals of RAS, constitutive activation of RAS, and/or activating mutations of RAS. These activating mutations of RAS have been observed in many types of cancer. G12, G13, and Q61 are known to be hotspots for RAS mutations, with high frequency mutations observed at G12 in KRAS and at Q61 in NRAS. It is also known that these mutations are associated with patient prognosis (Non-Patent Document 3).
- cyclic peptide compound (1) represented by the following formula (1) (hereinafter also referred to as cyclic peptide compound (1)) has been reported, which has selectivity in RAS, specifically, a selective KRAS inhibitory effect over HRAS and NRAS (Patent Document 1).
- Patent Document 1 describes a method for producing a cyclic peptide compound (1) by a cyclization reaction at cyclization position A.
- a cyclic dimer was confirmed as a by-product in addition to the desired cyclic peptide compound (1), and the ratio of the cyclic peptide compound (1) to the cyclic dimer was 75:25, indicating low selectivity.
- the cyclized precursor peptide compound described in Patent Document 1 is produced by sequentially linking amino acids or some tripeptides by the Fmoc method and synthesizing on a solid phase.
- Solid phase synthesis requires excessive amounts of amino acids and reagents, and large amounts of organic solvents are required for washing in each step, so it has been desirable to avoid solid phase synthesis as much as possible when producing large quantities.
- crystals of cyclic peptide compound (1) there have been no reports of crystals of cyclic peptide compound (1).
- the present invention has been made in light of the above circumstances, and an objective of the present invention is to provide an efficient method for producing a cyclic peptide compound (1) having a double bond bridged between amino acids. Another object of the present invention is to provide crystals of the cyclic peptide compound (1) that are highly stable.
- the present invention provides a cyclization method for producing a cyclic peptide compound (1) having a double bond bridged between amino acids, which is a cyclization method based on the cyclization position, capable of reducing the amount of cyclic dimer produced as a by-product.
- the present invention provides an efficient production method for producing a cyclized precursor peptide compound, in which three fragment peptides are each synthesized and synthesized by liquid phase fragment coupling.
- the present invention provides crystals of the cyclic peptide compound (1) and a method for producing the crystals by crystallization.
- the present invention includes, in one non-limiting specific embodiment, the following.
- a method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof comprising a step of cyclizing the compound by reacting an N-terminal amino acid residue with a C-terminal amino acid residue of a peptide compound represented by formula (2) or (3) in a solvent (cyclization step).
- R 1 is C 1 -C 6 alkyl
- P1 is C1 - C6 alkyl
- R2 is C1 - C6 alkyl
- R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring
- P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle
- P4 is C1 - C6 alkyl
- R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl
- P6 is C1 - C6 alkyl
- a method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof comprising the steps of: (a) preparing a peptide compound represented by any one of formulas (4) to (6), or a salt thereof, or a solvate of said peptide compound or salt; (b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4) to (6) in a solvent (linking step); and (c) the step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in the step (b) by reacting them in a solvent (cyclization step).
- R 1 is C 1 -C 6 alkyl
- P1 is C1 - C6 alkyl
- R2 is C1 - C6 alkyl
- R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring
- P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle
- P4 is C1 - C6 alkyl
- R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl
- P6 is C1 - C6 alkyl
- step (b) (b-1)
- the method according to [A2] comprising a step (linking step) of reacting and linking the N-terminal amino acid residue of a peptide compound represented by formula (5) and the C-terminal amino acid residue of a peptide compound represented by formula (6) in a solvent to convert them into a peptide compound represented by formula (7).
- step (b) further (b-2) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4) and the C-terminal amino acid residue of the peptide compound represented by formula (7) in a solvent to convert them into the peptide compound represented by formula (2) (linking step); and
- step (c) (c-1)
- step (b) further (b-3) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7) and the C-terminal amino acid residue of the peptide compound represented by formula (4) in a solvent to convert them into a compound represented by formula (3) (linking step); and
- step (c) (c-2)
- the method according to [A3] which comprises a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3) in a solvent (cyclization step).
- [A6] The method according to any of [A1] to [A5], wherein the linkage between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is a linkage between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue.
- [A7] The method according to any of [A1] to [A6], wherein the link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is an amide bond between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue.
- [A8] The method according to any one of [A1] to [A7], wherein the solvent in the cyclization step comprises one or more selected from the group consisting of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents.
- the nitrile-based solvent is one or more selected from the group consisting of acetonitrile and propionitrile;
- the halogen-based solvent is one or more selected from the group consisting of dichloromethane, chloroform, and 1,2-dichloroethane;
- the ether-based solvent is one or more selected from the group consisting of diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, 4-methyltetrahydropyran, 1,3-dioxolane, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, t-butyl methyl ether, diglyme, triglyme, anisole, and tetraglyme;
- the amide solvent is one or more selected from the group consisting of DMF, NMP, DMA, NEP, NBP, and formamide;
- the ester solvent is one or
- [A12] The method according to [A8], wherein the solvent in the cyclization step is acetonitrile, 2-methyltetrahydrofuran, or ethyl acetate.
- [A13] The method according to any one of [A1] to [A12], wherein the cyclization step is carried out in the presence of a condensation reagent.
- the condensation reagent in the cyclization step is one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop.
- [A15] The method according to [A13], wherein the condensation reagent in the cyclization step is one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P.
- [A16] The method according to [A13], wherein the condensation reagent in the cyclization step is HATU.
- [A17] The method according to [A13], wherein the condensation reagent in the cyclization step is COMU.
- [A18] The method according to [A13], wherein the condensation reagent in the cyclization step is HATU, and the solvent in the cyclization step is acetonitrile.
- the base in the cyclization step is 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN), 7-methyl-1,5
- DBU 1,8-diazabicyclo[5.4.0]-7-undecene
- DBU 1,8-diazabicyclo[5.4.0]-7-undecene
- DBU 1,8-diazabicyclo[5.4.0]-7-undecene
- DBU 1,8-di
- [A30] The method according to [A22], wherein the condensation reagent in the cyclization step is COMU, the solvent in the cyclization step is 2-methyltetrahydrofuran, and the base in the cyclization step is 2,6-lutidine.
- [A31] The method according to any one of [A1] to [A30], wherein the cyclization step is carried out by a liquid phase method.
- [A32] The method according to any of [A1] to [A31], wherein in the cyclization step, the peptide compound and the base are mixed into a mixed solution obtained by mixing a solvent for the cyclization step with the condensation reagent.
- [A33] The method according to any one of [A1] to [A32], characterized in that the content of all by-products produced in the cyclization step is less than 20%, less than 15%, less than 10%, less than 5%, or less than 3%, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
- [A34] The method according to any one of [A1] to [A33], characterized in that the content of each by-product generated in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
- [A35] The method according to any one of [A1] to [A34], characterized in that the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount, based on the total amount of the product, as determined by a UV area value at 220 nm by HPLC analysis, and the by-products include epimers and/or cyclic dimers.
- the solvent in the linking step comprises one or more selected from the group consisting of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents.
- the nitrile solvent is one or more selected from the group consisting of acetonitrile and propionitrile;
- the halogen-based solvent is one or more selected from the group consisting of dichloromethane, chloroform, and 1,2-dichloroethane;
- the ether-based solvent is one or more selected from the group consisting of diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, 4-methyltetrahydropyran, 1,3-dioxolane, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, t-butyl methyl ether, diglyme, triglyme, anisole, and tetraglyme;
- the amide solvent is one or more selected from the group consisting of DMF, NMP, DMA, NEP, NBP, and formamide;
- the ester solvent is one or more selected
- [A40] The method according to [A38], wherein the solvent in the linking step is one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, THF, ethyl acetate, isopropyl acetate, DMF, and anisole.
- the solvent in the linking step is a mixed solvent of one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran P, THF, ethyl acetate, isopropyl acetate, and anisole, and DMF.
- [A42] The method according to [A38], wherein the solvent in the linking step is a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF.
- [A43] The method according to [A38], wherein the solvent in the linking step is a mixed solvent of 2-methyltetrahydrofuran and DMF.
- [A44] The method according to any one of [A1] to [A43], wherein the linking step is carried out in the presence of a condensation reagent.
- [A52] The method according to [A44], wherein the condensation reagent in the linking step is COMU, and the solvent in the linking step is acetonitrile or 2-MeTHF.
- [A53] The method according to [A44], wherein the condensation reagent in the linking step is COMU, and the solvent in the linking step is a mixed solvent of acetonitrile, 2-MeTHF, and DMF.
- [A54] The method according to any one of [A1] to [A53], wherein the linking step is carried out in the presence of a base.
- [A55] The method according to [A54], wherein the base in the linking step is an organic base.
- the base in the linking step is 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN), 7-methyl-1,5
- DBU 1,8-diazabicyclo[5.4.0]-7-undecene
- DBU 1,8-diazabicyclo[5.4.0]-7-undecene
- [A58] The method according to [A54], wherein the base in the linking step is one or more selected from the group consisting of 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, and pyridine.
- the condensation reagent in the linking step is HATU
- the solvent in the linking step is acetonitrile or 2-MeTHF
- the base in the linking step is N,N-diisopropylethylamine (DIPEA).
- [A62-2] The method according to [A54], wherein the condensation reagent in the linking step is HATU, the solvent in the linking step is acetonitrile, and the base in the linking step is N-methylmorpholine.
- [A63] The method according to any one of [A1] to [A62], wherein the linking step is carried out by a liquid phase method.
- [A64] The method according to any one of [A1] to [A63], wherein column chromatography is used for isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof.
- [A65] The method according to any one of [A1] to [A63], wherein column chromatography is not used for isolation and/or purification of the cyclic peptide compound, or a salt thereof, or a solvate thereof.
- [A66] The method according to any one of [A1] to [A65], further comprising a step of isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof by crystallization to obtain the cyclic peptide compound, or a salt thereof, or a crystal thereof.
- [A67] The method according to any one of [A1] to [A66], wherein R 1 is C 3 -C 4 alkyl.
- [A67-1] The method according to any one of [A1] to [A66], wherein R 1 is n-propyl.
- [A67-2] The method according to any one of [A1] to [A66], wherein R 1 is 2-methylpropyl.
- [A68] The method according to any one of [A1] to [A67], wherein P 1 is C 1 -C 4 alkyl.
- [A68-1] The method according to any one of [A1] to [A67], wherein P 1 is methyl.
- [A69] The method according to any one of [A1] to [A68], wherein R 2 is C 3 -C 4 alkyl.
- [A69-1] The method according to any one of [A1] to [A68], wherein R 2 is 1-methylpropyl.
- [A70] The method according to any one of [A1] to [A69], wherein R 3 is hydrogen or R 3 forms a 5-membered saturated heterocycle together with P 3 , the carbon atom to which R 3 is bonded, and the nitrogen atom to which P 3 is bonded.
- [A70-1] The method according to any one of [A1] to [A69], wherein R 3 is hydrogen.
- [A70-2] The method according to any one of [A1] to [A69], wherein R 3 forms a 5-membered saturated heterocycle together with P 3 , the carbon atom to which R 3 is bonded, and the nitrogen atom to which P 3 is bonded.
- [A71] The method according to any one of [A1] to [A70], wherein P3 is C 1 -C 4 alkyl, or P3 forms a 5-membered saturated heterocycle together with R3 , the carbon atom to which R3 is bonded, and the nitrogen atom to which P3 is bonded.
- [A71-1] The method according to any one of [A1] to [A70], wherein P3 is methyl.
- [A71-2] The method according to any one of [A1] to [A70], wherein P3 forms a 5-membered saturated heterocycle together with R3 , the carbon atom to which R3 is bonded, and the nitrogen atom to which P3 is bonded.
- [A72] The method according to any one of [A1] to [A71], wherein P4 is C 1 -C 4 alkyl.
- [A72-1] The method according to any one of [A1] to [A71], wherein P4 is methyl.
- [A73] The method according to any one of [A1] to [A72], wherein R 5 is benzyl optionally substituted by C 1 -C 4 haloalkyl.
- [A73-1] The method according to any one of [A1] to [A72], wherein R 5 is 4-trifluoromethylbenzyl.
- [A74] The method according to any one of [A1] to [A73], wherein P6 is C 1 -C 4 alkyl.
- [A74-1] The method according to any one of [A1] to [A73], wherein P6 is methyl.
- [A75] The method according to any one of [A1] to [A74], wherein R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy.
- [A75-1] The method according to any one of [A1] to [A74], wherein R 7 is 3-methoxy-4-trifluoromethylphenethyl.
- [A75-2] The method according to any one of [A1] to [A74], wherein R 7 is 3,5-difluoro-4-trifluoromethylphenethyl.
- [A76] The method according to any one of [A1] to [A75], wherein R 8 together with P 8 , the carbon atom to which R 8 is bonded, and the nitrogen atom to which P 8 is bonded forms a 5-membered saturated heterocycle, and the 5-membered saturated heterocycle is substituted with C 1 -C 4 alkyl.
- [A76-1] The method according to any one of [A1] to [ A75 ], wherein R 8 forms a 5-membered saturated heterocycle together with P 8 , the carbon atom to which R 8 is bonded, and the nitrogen atom to which P 8 is bonded, and the 5-membered saturated heterocycle is substituted with ethoxy.
- [A77] The method according to any one of [A1] to [A76], wherein R 9 forms a 4- to 6-membered alicyclic ring together with Q 9 and the carbon atom to which R 9 and Q 9 are bonded.
- [A77-1] The method according to any one of [A1] to [A76], wherein R 9 forms a 4-membered alicyclic ring together with Q 9 and the carbon atom to which R 9 and Q 9 are bonded.
- [A77-2] The method according to any one of [A1] to [A76], wherein R 9 forms a 5-membered alicyclic ring together with Q 9 and the carbon atom to which R 9 and Q 9 are bonded.
- [A78] The method according to any one of [A1] to [A77], wherein P 9 is hydrogen or C 1 -C 4 alkyl.
- [A78-1] The method according to any one of [A1] to [A77], wherein P 9 is hydrogen.
- [A81] The method according to any one of [A1] to [A80], wherein R 11 is diC 1 -C 4 alkylaminocarbonyl, or 5- to 6-membered cyclic aminocarbonyl.
- [A81-1] The method according to any one of [A1] to [A80], wherein R 11 is dimethylaminocarbonyl.
- [A82] The method according to any one of [A1] to [A81], wherein P 11 is C 1 -C 4 alkyl.
- [A82-1] The method according to any one of [A1] to [A81], wherein P 11 is methyl.
- the carbamate protecting group is one selected from the group consisting of an Fmoc group, a Cbz group, a Troc group, an Alloc group, a Teoc group, a TSoc group, a BIBSoc group, an IPCSoc group, a BBSoc group, a CHBSoc group, a CDBSoc group, and a Boc group.
- the acyl-based protecting group is one selected from the group consisting of a trifluoroacetyl group, an acetyl group, and a benzoyl group.
- [A86] The method according to [A83], wherein the sulfonamide protecting group is one selected from the group consisting of a 2-nitrobenzenesulfonyl group, a 4-nitrobenzenesulfonyl group, and a 2,4-dinitrobenzenesulfonyl group.
- the silyl protecting group is one selected from the group consisting of a TMS group, a TBDMS group, a TES group, a TIPS group, and a TBDPS group.
- X 1 is hydrogen or a carbamate protecting group.
- [A88-1] The method according to any one of [A1] to [A87], wherein X 1 is hydrogen.
- [A88-2] The method according to any of [A1] to [A87], wherein X 1 is an Fmoc group.
- [A89] The method according to any one of [A1] to [A88], wherein X3 is hydrogen or a carbamate protecting group.
- [A89-1] The method according to any one of [A1] to [A88], wherein X 3 is hydrogen.
- [A89-2] The method according to any of [A1] to [A88], wherein X 3 is a Cbz group.
- [A90] The method according to any one of [A1] to [A89], wherein X 5 is hydrogen or a carbamate protecting group.
- [A90-1] The method according to any one of [A1] to [A89], wherein X 5 is hydrogen.
- [A90-2] The method according to any of [A1] to [A89], wherein X5 is a Cbz group.
- [A91-1] The method according to any one of [A1] to [A90], wherein the halogen is chlorine or bromine.
- [A91-2] The method according to any one of [A1] to [A90], wherein the optionally substituted alkoxy is t-butoxy, methoxy, ethoxy, or isopropoxy.
- [A91-3] The method according to any one of [A1] to [A90], wherein the optionally substituted aryloxy is pentafluorophenyloxy or nitrophenyloxy.
- [A91-4] The method according to any one of [A1] to [A90], wherein the optionally substituted aralkoxy is optionally substituted benzyloxy.
- [A91-5] The method according to any of [A1] to [A90], wherein the optionally substituted cyclic aminooxy is N-hydroxysuccinic acid iminoxy.
- [ A91-6 ] The method according to any one of [A1] to [A90 ] , wherein the group represented by -OSiRxRyRz is trimethylsilyloxy, triethylsilyloxy, triisopropylsilyloxy, triphenylsilyloxy, tri-t-butylsilyloxy, di-t-butylisobutylsilyloxy, or tris(triethylsilyl)silyloxy.
- [A92] The method according to any one of [A1] to [A91], wherein X 2 is a hydroxyl group, t-butoxy, or benzyloxy.
- [A92-1] The method according to any one of [A1] to [A91], wherein X 2 is a hydroxyl group.
- [A92-2] The method according to any of [A1] to [A91], wherein X 2 is t-butoxy.
- [A93] The method according to any one of [A1] to [A92], wherein X 4 is a hydroxyl group, t-butoxy, or benzyloxy.
- [A93-1] The method according to any one of [A1] to [A92], wherein X 4 is a hydroxyl group.
- [A93-2] The method according to any of [A1] to [A92], wherein X 4 is t-butoxy.
- [A94] The method according to any one of [A1] to [A93], wherein X 6 is a hydroxyl group, t-butoxy, or benzyloxy.
- [A94-1] The method according to any one of [A1] to [A93], wherein X 6 is a hydroxyl group.
- [A94-2] The method according to any one of [A1] to [A93], wherein X 6 is t-butoxy.
- the cyclic peptide compound is represented by formula (1a): The method according to any one of [A1] to [A95], [A97] The cyclic peptide compound is represented by formula (1a): The method according to any one of [A1] to [A95], wherein the crystal is a crystal of a cyclic peptide compound represented by the formula: [A97-1] The method according to [A97], wherein the crystal of the cyclic peptide compound is a non-solvate crystal or a solvate crystal. [A97-2] The method according to [A97], wherein the crystal of the cyclic peptide compound is a solvate crystal.
- [A97-3] The method according to [A97-2], wherein the solvate crystal of the cyclic peptide compound is a hydrate crystal.
- [B1] A method for producing a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, comprising a step of cyclization (cyclization step) of reacting an N-terminal amino acid residue with a C-terminal amino acid residue of a peptide compound represented by formula (2a) or (3a) in a solvent.
- X1 and X5 each independently represent a hydrogen atom or a protecting group for an amino group
- X2 and X4 each independently represent a hydroxyl group, an optionally substituted alkoxy group, an optionally substituted aryloxy group, an optionally substituted aralkoxy group, an optionally substituted cyclic aminooxy group, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz each independently represent an alkyl group or an aryl group).
- a method for producing a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof comprising the steps of: (a) providing a peptide compound represented by any one of formulas (4a) to (6a), or a salt thereof, or a solvate of said peptide compound or salt; (b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4a) to (6a) in a solvent (linking step); and (c) the step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in the step (b) by reacting them in a solvent (cyclization step).
- X 1 , X 3 and X 5 each independently represent a hydrogen atom or a protecting group for an amino group
- X 2 , X 4 and X 6 each independently represent a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y and R z each independently represent an alkyl or an aryl).
- step (b) (b-1)
- the method according to [B2] which comprises a step (linking step) of reacting and linking the N-terminal amino acid residue of a peptide compound represented by formula (5a) and the C-terminal amino acid residue of a peptide compound represented by formula (6a) in a solvent to convert them into a peptide compound represented by formula (7a).
- step (b) further (b-2) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4a) and the C-terminal amino acid residue of the peptide compound represented by formula (7a) in a solvent to convert them into the peptide compound represented by formula (2a) (linking step); and
- step (c) (c-1)
- the method according to [B3] which comprises a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2a) in a solvent (cyclization step).
- step (b) further (b-3) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7a) and the C-terminal amino acid residue of the peptide compound represented by formula (4a) in a solvent to convert them into the peptide compound represented by formula (3a) (linking step); and
- step (c) (c-2)
- the method according to [B3] which comprises a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3a) by reacting them in a solvent (cyclization step).
- [B6] The method according to any one of [B1] to [B5], wherein the solvent in the cyclization step is a solvent according to any one of [A8] to [A12].
- [B7] The method according to any one of [B1] to [B6], wherein the cyclization step is carried out in the presence of a condensation reagent.
- [B8] The method according to [B7], wherein the condensation reagent in the cyclization step is a condensation reagent according to any one of [A14] to [A17].
- [B9] The method according to [B7], wherein the solvent and condensation reagent in the cyclization step are the solvent and condensation reagent according to any one of [A18] to [A21].
- [B10] The method according to any one of [B1] to [B9], wherein the cyclization step is carried out in the presence of a base.
- the method according to [B10], wherein the base in the cyclization step is a base according to any one of [A24] to [A26].
- [B12] The method according to [B10], wherein the solvent, condensation reagent, and base in the cyclization step are the solvent, condensation reagent, and base according to any one of [A27] to [A30].
- [B13] The method according to any one of [B1] to [B12], wherein the cyclization step is carried out by a liquid phase method.
- [B14] The method according to any one of [B1] to [B13], wherein in the cyclization step, the peptide compound and the base are mixed into a mixed solution obtained by mixing a solvent for the cyclization step with the condensation reagent.
- [B15] The method according to any one of [B1] to [B14], characterized in that the content of all by-products produced in the cyclization step is less than 20%, less than 15%, less than 10%, less than 5%, or less than 3%, based on the total amount of the product, as determined by the UVarea value at 220 nm by HPLC analysis.
- [B16] The method according to any one of [B1] to [B15], characterized in that the content of each by-product generated in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount, based on the total amount of the product, as determined by the UV area value at 220 nm by HPLC analysis.
- [B17] The method according to any one of [B1] to [B16], characterized in that the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount based on the total amount of the product, as determined by the UV area value at 220 nm by HPLC analysis, and the by-products include epimers and/or cyclic dimers.
- [B21] The method according to any one of [B1] to [B20], wherein the linking step is carried out in the presence of a condensation reagent.
- the condensation reagent in the linking step is a condensation reagent according to any one of [A45] to [A49].
- the solvent and condensation reagent in the linking step are the solvent and condensation reagent according to any one of [A50] to [A53].
- the ligation step is carried out in the presence of a base.
- [B25] The method according to [B24], wherein the base in the linking step is a base according to any one of [A55] to [A59].
- [B26] The method according to [B24], wherein the solvent, condensation reagent and base in the linking step are the solvent, condensation reagent and base according to any one of [A60] to [A62].
- [B27] The method according to any one of [B1] to [B26], wherein the linking step is carried out by a liquid phase method.
- [B28] The method according to any one of [B1] to [B27], wherein column chromatography is used for isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof.
- [B29] The method according to any one of [B1] to [B27], wherein column chromatography is not used for isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof.
- [B30] The method according to any one of [B1] to [B29], further comprising a step of isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof by crystallization to obtain the cyclic peptide compound, or a salt thereof, or a crystal thereof.
- [C1] A compound represented by formula (4a) or a salt thereof.
- X 1 is hydrogen or a protecting group for an amino group
- X2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
- Rx , Ry , and Rz are each independently an alkyl or an aryl.
- [C4] The compound or salt thereof according to any one of [C1] to [C3], wherein X 2 is t-butoxy.
- [C6] A compound represented by formula (5a) or a salt thereof.
- X3 is hydrogen or a protecting group for an amino group
- X4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
- Rx , Ry , and Rz are each independently an alkyl or an aryl.
- [C9] The compound or salt thereof according to any one of [C6] to [C8], wherein X 4 is t-butoxy.
- [C11] A compound represented by formula (6a) or a salt thereof.
- X5 is hydrogen or a protecting group for an amino group
- X6 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
- Rx , Ry , and Rz are each independently an alkyl or an aryl.
- [C14] The compound or a salt thereof according to any one of [C11] to [C13], wherein X 6 is a hydroxyl group or t-butoxy.
- [C16] A compound represented by formula (7a) or a salt thereof.
- X5 is hydrogen or a protecting group for an amino group
- X4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
- [C17] The compound or a salt thereof according to [C16], wherein X 5 is hydrogen.
- X 5 is a Cbz group.
- [C21] A compound represented by formula (2a) or a salt thereof.
- X5 is hydrogen or a protecting group for an amino group
- X2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
- Rx , Ry , and Rz are each independently an alkyl or an aryl.
- [C24] The compound or a salt thereof according to any one of [C21] to [C23], wherein X 2 is a hydroxyl group or t-butoxy.
- [C25] 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino ]-2-Cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]p
- [C26] A compound represented by the formula (3a) or a salt thereof.
- X 1 is hydrogen or a protecting group for an amino group
- X4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
- [C27] The compound or a salt thereof according to [C26], wherein X 1 is hydrogen.
- [C28] The compound or salt thereof according to [C26], wherein X 1 is an Fmoc group.
- [C31] A method for producing the compound according to any one of [C1] to [C30], which does not use solid-phase synthesis.
- [C32] A method for producing a cyclic peptide compound represented by formula (1a), which does not use solid-phase synthesis.
- [D1] A crystal of a cyclic peptide compound represented by formula (1a), a salt thereof, or a solvate thereof.
- [D5-2] The crystal according to [D4], wherein the hydrate crystal is a Form A crystal having peaks at the following diffraction angles (2 ⁇ values) in powder X-ray diffraction: 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° ( ⁇ 0.2°).
- [D5-3] The Form A crystal according to any one of [D5] to [D5-2], wherein the diffraction angle (2 ⁇ value) is a diffraction angle (2 ⁇ value) of a hydrate crystal stored at a relative humidity of 10% or more for 15 minutes or more.
- [D7-2] The crystal according to [D3], wherein the solvate crystal is a Form F crystal comprising peaks at the following diffraction angles (2 ⁇ values) in powder X-ray diffraction: 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° ( ⁇ 0.2°).
- [D7-3] The crystal according to any one of [D7] to [D7-2], wherein the solvate crystal is an acetone/heptane/hydrate.
- [D8-3] The Form J crystal according to any one of [D8] to [D8-2], wherein the diffraction angle (2 ⁇ value) is a diffraction angle (2 ⁇ value) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more.
- [D9-3] The Form Y crystal according to any one of [D9] to [D9-2], wherein the diffraction angle (2 ⁇ value) is a diffraction angle (2 ⁇ value) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes or more.
- [D10-2] The crystal according to [D3], wherein the solvate crystal is a Form K crystal comprising peaks at the following diffraction angles (2 ⁇ values) in powder X-ray diffraction: 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° ( ⁇ 0.2°).
- [D11] The crystal according to [D3], wherein the solvate crystal is a dimethylsulfoxide/heptane/hydrate crystal of Form G having the structure shown in Figure 1 by single crystal X-ray analysis.
- [D11-1] The crystal according to [D3], wherein the solvate crystal is a 2-propanol/heptane/hydrate crystal of Form E having the structure shown in FIG. 3 by single crystal X-ray analysis.
- [D11-2] The crystal according to [D3], wherein the solvate crystal is an ethanol/hydrate crystal of Form H having the structure shown in Figure 8 by single crystal X-ray analysis.
- [D11-3] The crystal according to [D3], wherein the solvate crystal is a 1,4-dioxane/hydrate crystal of Form D having the structure shown in FIG. 26 by single crystal X-ray analysis.
- [D11-7] The crystal according to [D3], wherein the solvate crystal is a propylene glycol/hydrate crystal of Form M, the data of which is shown in FIG. 30(A) by simultaneous thermogravimetry and differential thermal analysis.
- [D11-8] The crystal according to [D3], wherein the solvate crystal is a propylene glycol solvate crystal of Form N having the diffraction angle shown in FIG. 29 by powder X-ray diffraction.
- [D11-9] The crystal according to [D3], wherein the solvate crystal is a propylene glycol solvate crystal of Form N showing the data of simultaneous thermogravimetry and differential thermal analysis shown in FIG. 30 (B).
- [D12] A method for producing a crystal of a cyclic peptide compound according to any one of [D1] to [D10-2], comprising the steps of dissolving the cyclic peptide compound in an amount of a polar organic solvent in which the cyclic peptide compound can be dissolved to obtain a solution, and adding a hydrocarbon solvent or water to the solution to obtain a crystal of the cyclic peptide compound.
- [D12-1] The method according to [D12], wherein the purity of the starting cyclic peptide compound is 85% or more.
- [D13] A method for producing a crystal of a cyclic peptide compound according to any one of [D1] to [D10-2], comprising the step of adding a mixture of a hydrocarbon solvent and a polar organic solvent, or a mixture of water and a polar organic solvent, to the cyclic peptide compound in an amorphous state to obtain a crystal of the cyclic peptide compound.
- [D14] The method according to any one of [D12] to [D13-1], wherein the polar organic solvent is one or more selected from the group consisting of DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, propylene glycol, 1,4-dioxane, and ethyl acetate.
- the polar organic solvent is one or more selected from the group consisting of DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, propylene glycol, 1,4-dioxane, and ethyl acetate.
- the polar organic solvent is acetone.
- [D15-1] The method according to [D14], wherein the polar organic solvent is ethanol.
- [D16] The method according to any one of [D12] to [D13-1], wherein the hydrocarbon solvent is one or more selected from the group consisting of heptane, hexane, pentane, toluene, and xylene.
- the hydrocarbon solvent is heptane.
- [D18] A method for producing a crystal of a cyclic peptide compound according to any one of [D1] to [D10-2], comprising the steps of dissolving the cyclic peptide compound in an amorphous state in DMSO to obtain a solution, freeze-drying the solution to obtain a freeze-dried product of the cyclic peptide compound, and adding a mixture of water and a polar organic solvent to the freeze-dried product to obtain a crystal of the cyclic peptide compound.
- [D19] The method according to [D18], wherein the polar organic solvent is one or more selected from the group consisting of DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, 1,4-dioxane and propylene glycol.
- the polar organic solvent is acetone.
- [D22] The method according to any one of [D12] to [D21], further comprising a step of drying the crystals after the step of obtaining the crystals of the cyclic peptide compound.
- [D23] The method according to any one of [D12] to [D22], wherein the crystal of the cyclic peptide compound is a solvate crystal.
- [D24] The method according to [D23], wherein the solvate crystal of the cyclic peptide compound is a hydrate crystal.
- [D25] The method according to [D21] or [D22], wherein the crystals of the cyclic peptide compound are formed as solvate crystals in a solvent and are obtained as hydrate crystals after a filtering step and/or a drying step.
- [D26] A composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, wherein the compound contains an impurity, the cyclic dimer of formula (1a), at a ratio of 1.5 wt % or less.
- [D27] A composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, wherein the compound contains an impurity, the cyclic dimer of formula (1a), at a ratio of 0.001 w/w% or more.
- [D28] A composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, wherein the compound contains acetone in an amount of 2.0 w/w% or less.
- [D29] A composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, wherein the compound contains acetone in a proportion of 0.001 w/w% or more.
- the numbers referred to in the dependent claims include their subnumbers unless otherwise specified.
- [A67] referred to in the dependent claims includes [A67] as well as its subnumber [A67-1]. The same applies to other numbering schemes.
- the present invention it is possible to efficiently produce a cyclic peptide compound, or a salt or solvate thereof, while suppressing the production of the by-product cyclic dimer.
- the production method of the present invention is particularly useful for the synthesis of peptides on a large scale, since it can reduce the production costs of peptide compounds and reduce the environmental burden.
- Example 3-1 The crystal structure of the crystal (Form G) obtained in Example 3-1 is shown.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form A) obtained in Example 3-2 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the crystal structures of the crystals obtained in Example 3-3 are shown.
- Compound 1 is depicted using the Capped Stick model, and the others are depicted using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form E) obtained in Example 3-3 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of powder X-ray diffraction measurement of the crystal (Form B) obtained in Example 3-4 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of powder X-ray diffraction measurement of the crystal (Form B) obtained in Example 3-5 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the crystal structure of the crystal (Form H) obtained in Example 3-6 is shown.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form B) obtained in Example 3-6 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the crystal structure of the crystal (Form C) obtained in Example 3-7 is shown.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form B) obtained in Example 3-7 are shown.
- the vertical axis is the diffraction intensity
- the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of powder X-ray diffraction measurement of the crystal (Form A) obtained in Example 3-8 are shown.
- the vertical axis is the diffraction intensity
- the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of simultaneous thermogravimetry and differential thermal analysis of the crystal (Form A) obtained in Example 3-8 are shown.
- the horizontal axis is temperature (°C)
- the right vertical axis is the weight change (%) of the sample in the thermogravimetric analysis.
- the left vertical axis represents the heat flow observed in the differential thermal analysis.
- the results of 1 H-NMR measurement of the crystal (Form A) obtained in Example 3-8 are shown below.
- the vertical axis represents signal intensity
- the horizontal axis represents chemical shift ⁇ (ppm).
- the results of powder X-ray diffraction measurement of the crystal (Form A) obtained in Example 3-9 are shown.
- the vertical axis is the diffraction intensity
- the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of simultaneous thermogravimetry and differential thermal analysis of the crystal (Form A) obtained in Example 3-9 are shown.
- the horizontal axis is temperature (°C)
- the right vertical axis is the weight change (%) of the sample in the thermogravimetric analysis.
- the left vertical axis represents the heat flow observed in the differential thermal analysis.
- the results of 1 H-NMR measurement of the crystal (Form A) obtained in Example 3-9 are shown below.
- the vertical axis represents signal intensity
- the horizontal axis represents chemical shift ⁇ (ppm).
- the results of powder X-ray diffraction measurement of the crystal (Form F) obtained in Example 3-10 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the crystal structure of the crystal (Form F) obtained in Example 3-11 is shown.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form A) obtained in Example 3-12 are shown.
- the vertical axis is the diffraction intensity
- the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of simultaneous thermogravimetry and differential thermal analysis of the crystal (Form A) obtained in Example 3-12 are shown.
- the horizontal axis is temperature (°C), and the right vertical axis is the weight change (%) of the sample in the thermogravimetric analysis.
- the left vertical axis represents the heat flow observed in the differential thermal analysis.
- the results of powder X-ray diffraction measurements of the crystals obtained in Example 3-12 at relative humidities of (A) 0% (Form J), (B) 10% (Form A), (C) 20% (Form A), (D) 50% (Form A), and (E) 90% (Form A) are shown.
- the vertical axis represents the diffraction intensity
- the horizontal axis represents the diffraction angle 2 ⁇ (°). The peaks near 4.89° and 6.65° in the figure are peaks derived from the measuring equipment.
- the results of powder X-ray diffraction measurement of the crystal (Form B) obtained in Example 3-13 are shown.
- the vertical axis is the diffraction intensity
- the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of simultaneous thermogravimetry and differential thermal analysis of the crystal (Form B) obtained in Example 3-13 are shown.
- the horizontal axis is temperature (°C)
- the right vertical axis is the weight change (%) of the sample in the thermogravimetric analysis.
- the left vertical axis represents the heat flow observed in the differential thermal analysis.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the crystal structure of the crystal (Form L) obtained in Example 3-16 is shown.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form L) obtained in Example 3-16 (A: wet powder, B: dry powder) are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of powder X-ray diffraction measurement of the crystals obtained in Example 3-17 are shown (A: Form M, B: Form N).
- the vertical axis is the diffraction intensity
- the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of simultaneous thermogravimetric and differential thermal analysis of the crystal (Form N) obtained in Example 3-17 are shown.
- the horizontal axis is temperature (°C)
- the right vertical axis is the weight change (%) of the sample in the thermogravimetric analysis.
- the left vertical axis represents the heat flow observed in the differential thermal analysis.
- the crystal structure of the crystal (Form M) obtained in Example 3-18 is shown.
- Compound 1 is drawn using the Capped Stick model, and the others are drawn using the Ball and Stick model.
- the results of powder X-ray diffraction measurement of the crystal (Form K) obtained in Example 3-19 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- the results of simultaneous thermogravimetry and differential thermal analysis of the crystal (Form K) obtained in Example 3-19 are shown.
- the horizontal axis is temperature (°C), and the right vertical axis is the weight change (%) of the sample in the thermogravimetric analysis.
- the left vertical axis represents the heat flow observed in the differential thermal analysis.
- the results of 1 H-NMR measurement of the crystal (Form K) obtained in Example 3-19 are shown below.
- the vertical axis represents signal intensity, and the horizontal axis represents chemical shift ⁇ (ppm).
- Example 5-9 The results of powder X-ray diffraction measurements of the crystals obtained in (A) Example 5-1, (B) Example 5-2, (C) Example 5-3, (D) Example 5-4, (E) Example 5-5, (F) Example 5-6, (G) Example 5-7, (H) Example 5-8, and (I) Example 5-9 are shown.
- the vertical axis represents the diffraction intensity, and the horizontal axis represents the diffraction angle 2 ⁇ (°).
- the results of powder X-ray diffraction measurement of the crystal (Form B) obtained in Example 3-22 are shown.
- the vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- halogen examples include F, Cl, Br, and I.
- alkyl refers to a monovalent group derived by removing any one hydrogen atom from an aliphatic hydrocarbon, which does not contain heteroatoms (atoms other than carbon and hydrogen atoms) or unsaturated carbon-carbon bonds in the skeleton, and has a subset of hydrocarbyl or hydrocarbon group structures containing hydrogen and carbon atoms. Alkyl includes not only linear but also branched chain alkyls.
- alkyl examples include alkyls having 1 to 20 carbon atoms (C 1 -C 20 , hereinafter "C p -C q " means that the number of carbon atoms is p to q), preferably C 1 -C 10 alkyl, and more preferably C 1 -C 6 alkyl.
- alkyl examples include methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, isobutyl (2-methylpropyl), n-pentyl, s-pentyl (1-methylbutyl), t-pentyl (1,1-dimethylpropyl), neopentyl (2,2-dimethylpropyl), isopentyl (3-methylbutyl), 3-pentyl (1-ethylpropyl), 1,2-dimethylpropyl, 2-methylbutyl, n-hexyl, 1,1,2-trimethylpropyl, 1,2,2-trimethylpropyl, 1,1,2,2-tetramethylpropyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-dimethylmethylprop
- alkenyl refers to a monovalent group having at least one double bond (two adjacent sp2 carbon atoms). Depending on the arrangement of the double bond and the substituents (if present), the geometry of the double bond can be in an
- E E
- Z cis or trans configuration.
- Alkenyl includes not only linear but also branched chains. Preferred examples of alkenyl include C 2 -C 10 alkenyl, more preferably C 2 -C 6 alkenyl.
- alkenyl examples include vinyl, allyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl (including cis and trans), 3-butenyl, pentenyl, 3-methyl-2-butenyl, hexenyl, and the like.
- alkynyl refers to a monovalent group having at least one triple bond (two adjacent sp carbon atoms). Alkynyl includes not only straight chain but also branched chain. Preferred alkynyl groups include C 2 -C 10 alkynyl groups, more preferably C 2 -C 6 alkynyl groups. Specific examples of alkynyl groups include ethynyl, 1-propynyl, propargyl, 3-butynyl, pentynyl, hexynyl, and the like.
- cycloalkyl refers to a saturated or partially saturated cyclic monovalent aliphatic hydrocarbon group, including a monocyclic ring, a bicyclo ring, and a spiro ring.
- Preferred examples of cycloalkyl include C 3 -C 8 cycloalkyl, more preferably C 3 -C 7 cycloalkyl, and even more preferably C 3 -C 6 cycloalkyl.
- cycloalkyl examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, bicyclo[2.2.1]heptyl, and spiro[3.3]heptyl.
- aryl refers to a monovalent aromatic hydrocarbon ring, i.e., an aromatic hydrocarbon ring group.
- Preferred examples of aryl include C 6 -C 10 aryl.
- Specific examples of aryl include phenyl, naphthyl (e.g., 1-naphthyl, 2-naphthyl), and the like.
- heteroaryl refers to an aromatic monovalent cyclic group containing 1 to 5 heteroatoms in addition to carbon atoms, i.e., an aromatic heterocyclic group.
- the ring may be a single ring or a condensed ring with other rings, and may be partially saturated.
- the number of atoms constituting the heteroaryl ring is preferably 5 to 10 (5- to 10-membered heteroaryl), and more preferably 5 to 7 (5- to 7-membered heteroaryl).
- heteroaryl examples include furyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, triazolyl, tetrazolyl, pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl, benzofuranyl, benzothienyl, benzothiadiazolyl, benzothiazolyl, benzoxazolyl, benzoxadiazolyl, benzimidazolyl, benzotriazolyl, indolyl, isoindolyl, indazolyl, azaindolyl, quinolyl, isoquinolyl, cinnolinyl, quinazolinyl, quinoxalinyl, benzodioxolyl,
- heteroarylalkyl refers to a group in which one or more hydrogen atoms of an "alkyl” as defined herein are substituted with a “heteroaryl” as defined herein.
- a 5- to 10-membered heteroaryl C 1 -C 6 alkyl is preferred, and a 5- to 10-membered heteroaryl C 1 -C 2 alkyl is more preferred.
- heteroarylalkyl examples include 3-thienylmethyl, 4-thiazolylmethyl, 2-pyridylmethyl, 3-pyridylmethyl, 4-pyridylmethyl, 2-(2-pyridyl)ethyl, 2-(3-pyridyl)ethyl, 2-(4-pyridyl)ethyl, 2-(6-quinolyl)ethyl, 2-(7-quinolyl)ethyl, 2-(6-indolyl)ethyl, 2-(5-indolyl)ethyl, 2-(5-benzofuranyl)ethyl, and the like.
- alkoxy refers to an oxy group bonded to an "alkyl” as defined herein.
- alkoxy C 1 -C 6 alkoxy is preferred, and C 1 -C 4 alkoxy is more preferred.
- Specific examples of the alkoxy include methoxy, ethoxy, 1-propoxy, 2-propoxy, n-butoxy, i-butoxy, s-butoxy, t-butoxy, pentyloxy, 3-methylbutoxy, and the like.
- alkoxyalkyl refers to a group in which one or more hydrogen atoms of an "alkyl” as defined herein are substituted with an “alkoxy” as defined herein.
- alkoxyalkyl C 1 -C 6 alkoxy C 1 -C 6 alkyl is preferred, and C 1 -C 6 alkoxy C 1 -C 2 alkyl is more preferred.
- alkoxyalkyl examples include methoxymethyl, ethoxymethyl, 1-propoxymethyl, 2-propoxymethyl, n-butoxymethyl, i-butoxymethyl, s-butoxymethyl, t-butoxymethyl, pentyloxymethyl, 3-methylbutoxymethyl, 1-methoxyethyl, 2-methoxyethyl, 2-ethoxyethyl, and the like.
- aryloxy refers to an oxy group bonded to an "aryl” as defined herein.
- Preferred examples of aryloxy include C 6 -C 10 aryloxy.
- Specific examples of aryloxy include phenoxy, 1-naphthyloxy, and 2-naphthyloxy.
- aralkoxy refers to an oxy group bonded to an "aralkyl” as defined herein.
- aralkoxy C 7 -C 14 aralkoxy is preferred, and C 7 -C 10 aralkoxy is more preferred.
- Specific examples of the aralkoxy include benzyloxy, phenethyloxy, and 3-phenylpropoxy.
- amino means -NH2 in a narrow sense, and -NRR' in a broad sense, where R and R' are independently selected from hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl, or heteroaryl, or R and R' mean a group forming a ring together with the nitrogen atom to which they are attached.
- Preferred examples of amino include -NH2 , mono- C1 - C6 alkylamino, di- C1 - C6 alkylamino, and 4- to 8-membered cyclic amino.
- monoalkylamino refers to a group in which R is hydrogen and R' is an "alkyl” as defined herein, among “amino” as defined herein.
- Preferred examples of monoalkylamino include mono-C 1 -C 6 alkylamino.
- Specific examples of monoalkylamino include methylamino, ethylamino, n-propylamino, i-propylamino, n-butylamino, s-butylamino, and t-butylamino.
- dialkylamino refers to an "amino" as defined herein, in which R and R' are independently “alkyl” as defined herein.
- Preferred examples of dialkylamino include diC 1 -C 6 alkylamino.
- Specific examples of dialkylamino include dimethylamino and diethylamino.
- cyclic amino refers to a group in which R and R' form a ring together with the nitrogen atom to which they are attached, as defined in this specification.
- Preferred examples of cyclic amino include 4- to 8-membered cyclic amino.
- Specific examples of cyclic amino include 1-azetidyl, 1-pyrrolidyl, 1-piperidyl, 1-piperazyl, 4-morpholinyl, 3-oxazolidyl, 1,1-dioxidethiomorpholinyl-4-yl, 3-oxa-8-azabicyclo[3.2.1]octan-8-yl, and the like.
- cyclic aminooxy refers to an oxy group to which "cyclic amino” as defined in this specification is bonded.
- cyclic aminooxy preferably, 4- to 8-membered cyclic aminooxy is mentioned.
- Specific examples of the cyclic amino include 1-azetidyloxy, 1-pyrrolidyloxy, 1-piperidyloxy, 1-piperazyloxy, 4-morpholinyloxy, 3-oxazolidyloxy, 1,1-dioxidethiomorpholinyl-4-yloxy, 3-oxa-8-azabicyclo[3.2.1]octan-8-yloxy, etc.
- aminocarbonyl refers to a carbonyl group bonded to "amino" as defined herein.
- Preferred examples of aminocarbonyl include -CONH 2 , mono C 1 -C 6 alkylaminocarbonyl, mono C 3 -C 6 cycloalkylaminocarbonyl, di C 1 -C 6 alkylaminocarbonyl, and 4- to 8-membered cyclic aminocarbonyl.
- aminocarbonyl examples include -CONH 2 , dimethylaminocarbonyl, 1-azetidinylcarbonyl, 1-pyrrolidinylcarbonyl, 1-piperidinylcarbonyl, 1-piperazinylcarbonyl, 4-morpholinylcarbonyl, 3-oxazolidinylcarbonyl, 1,1-dioxidethiomorpholinyl-4-ylcarbonyl, and 3-oxa-8-azabicyclo[3.2.1]octan-8-ylcarbonyl.
- haloalkyl refers to a group in which one or more hydrogen atoms of an "alkyl” as defined herein are substituted with halogen.
- haloalkyl haloC 1 -C 6 alkyl is preferred, and fluoroC 1 -C 6 alkyl is more preferred.
- Specific examples of haloalkyl include difluoromethyl, trifluoromethyl, 2,2-difluoroethyl, 2,2,2-trifluoroethyl, 3,3-difluoropropyl, 4,4-difluorobutyl, 5,5-difluoropentyl, and the like.
- alicyclic ring means a non-aromatic hydrocarbon ring.
- the alicyclic ring may have an unsaturated bond in the ring.
- the carbon atoms constituting the ring may be oxidized to form a carbonyl.
- the alicyclic ring may be a monocyclic ring (referred to as a monocyclic alicyclic ring in this specification) or may form a condensed ring with a saturated alicyclic ring such as a cyclopentane ring or a cyclohexane ring, or an aromatic hydrocarbon ring such as a benzene ring or a naphthalene ring.
- Preferred examples of the alicyclic ring include 3- to 10-membered alicyclic rings, and more preferably 3- to 8-membered alicyclic rings.
- Specific examples of the alicyclic ring include a cyclopropane ring, a cyclobutane ring, a cyclopentane ring, a cyclohexane ring, a cycloheptane ring, a cyclooctane ring, and a bicyclo[2.2.1]heptane ring.
- saturated heterocycle refers to a non-aromatic heterocycle that contains preferably 1 to 5, more preferably 1 to 3, heteroatoms in the atoms that constitute the ring and has no unsaturated bonds in the ring.
- a saturated heterocycle does not have double and/or triple bonds in the ring.
- a saturated heterocycle may be a monocycle, or may form a condensed ring or a spiro ring with another ring, for example, a saturated alicyclic ring such as a cyclopentane ring or a cyclohexane ring, or a saturated heterocycle such as a tetrahydropyran ring, a dioxane ring, or a pyrrolidine ring.
- saturated heterocycles include 4- to 10-membered saturated heterocycles, more preferably 4- to 7-membered saturated heterocycles, and even more preferably 5-membered saturated heterocycles.
- Specific examples of saturated heterocyclic rings include an azetidine ring, an oxoazetidine ring, an oxetane ring, a tetrahydrofuran ring, a tetrahydropyran ring, a morpholine ring, a thiomorpholine ring, a pyrrolidine ring, a 2-oxopyrrolidine ring, a 4-oxopyrrolidine ring, a piperidine ring, a 4-oxopiperidine ring, a piperazine ring, a pyrazolidine ring, an imidazolidine ring, an oxazolidine ring, an isoxazolidine ring, a thiazolidine ring, an isothiazolidine ring,
- amino group protecting group includes carbamate protecting groups, acyl protecting groups, sulfonamide protecting groups, and silyl protecting groups.
- carbamate protecting groups include 9-fluorenylmethyloxycarbonyl group (Fmoc group), benzyloxycarbonyl group (Cbz group), 2,2,2-trichloroethoxycarbonyl group (Troc group), allyloxycarbonyl group (Alloc group), 2-(trimethylsilyl)ethoxycarbonyl group (Teoc group), triisopropylsilyloxycarbonyl group (TSoc group), di-t-butylisobutyl
- the protecting group include a silyloxycarbonyl group (BIBSoC group), a di-i-propyl-t-butylsilyloxycarbonyl group (IPCSoc group), a benzyl-di-t-butylsilyloxycarbonyl group (BBSoC group
- acyl protecting group examples include a trifluoroacetyl group, an acetyl group, a benzoyl group, etc.
- sulfonamide protecting group examples include a 2-nitrobenzenesulfonyl group, a 4-nitrobenzenesulfonyl group, a 2,4-dinitrobenzenesulfonyl group, etc.
- silyl-based protecting groups include trimethylsilyl (TMS), t-butyldimethylsilyl (TBDMS), triethylsilyl (TES), triisopropylsilyl (TIPS), and t-butyldiphenylsilyl (TBDPS).
- one or more means one or more than one.
- the term means a number from one to the maximum number of substituents permitted by that group. Specific examples of "one or more” include 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and/or more.
- peptide compound as used herein means two or more amino acid residues linked together by amide bonds. Peptides having an ester bond in part of the main chain, such as depsipeptides, are also included in the term “peptide compound” as used herein.
- the number of amino acid residues contained in a peptide in this disclosure is not particularly limited, but may be preferably 5 to 30 residues, more preferably 8 to 15 residues, and even more preferably 9 to 13 residues.
- the peptide compound in this disclosure preferably contains at least three N-substituted amino acids, more preferably contains at least five, and even more preferably contains at least six. These N-substituted amino acids may be present consecutively or discontinuously in the peptide compound.
- the peptide compound in this disclosure may be linear or cyclic, with cyclic peptide compounds being preferred.
- cyclic peptide compound refers to a peptide compound having a cyclic structure composed of four or more amino acid residues.
- the cyclic peptide compound may have amino acids not included in the cyclic structure or a chain peptide structure. It may also have a
- Cyclization of a peptide compound means forming a cyclic structure of a cyclic portion containing four or more amino acid residues.
- the number of amino acids contained in the cyclic portion of the cyclic peptide compound in this specification is not particularly limited, but examples include 4 to 20 residues, 5 to 15 residues, and 6 to 13 residues.
- a method for converting a linear peptide compound to a cyclic peptide compound can be carried out by performing a bond formation reaction within the molecule using a method described in Comprehensive Organic Transformations, A Guide to Functional Group Preparations, 3rd Edition (by R. C. Larock) or March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (by M. B. Smith and J.
- Examples of C-C bond formation reactions using transition metals as catalysts include Suzuki reaction, Heck reaction, and Sonogashira reaction.
- Examples of functional group conversion reactions that are performed after bond-forming reactions include oxidation reactions and reduction reactions. Specifically, examples include reactions in which sulfur atoms are oxidized to convert to sulfoxide groups or sulfone groups.
- Another example is a reduction reaction in which a triple bond or a double bond among carbon-carbon bonds is reduced to a double bond or a single bond.
- amino acid includes natural amino acids and non-natural amino acids.
- amino acid may refer to an amino acid residue.
- natural amino acid refers to Gly, Ala, Ser, Thr, Val, Leu, Ile, Phe, Tyr, Trp, His, Glu, Asp, Gln, Asn, Cys, Met, Lys, Arg, and Pro.
- Non-natural amino acids are not particularly limited, but examples thereof include ⁇ -amino acids, D-amino acids, N-substituted amino acids, ⁇ , ⁇ -disubstituted amino acids, amino acids whose side chains are different from those of natural amino acids, and hydroxycarboxylic acids. As used herein, amino acids are allowed to have any configuration.
- the side chain of the amino acid can be freely selected from, in addition to a hydrogen atom, for example, an alkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, a heteroarylalkyl group, a cycloalkyl group, and a spiro-linked cycloalkyl group.
- a hydrogen atom for example, an alkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, a heteroarylalkyl group, a cycloalkyl group, and a spiro-linked cycloalkyl group.
- substituents are not limited, and may be independently selected from any substituents including, for example, a halogen atom, an oxygen atom, a nitrogen atom, a sulfur atom, a boron atom, a silicon atom, or a phosphorus atom. That is, examples include an optionally substituted alkyl group, an alkoxy group, an alkoxyalkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, a cycloalkyl group, or an oxo, an aminocarbonyl, a halogen atom, and the like.
- the amino acid in this specification may be a compound having a carboxyl group and an amino group in the same molecule (even in this case, proline, hydroxyproline, azetidine-2-carboxylic acid, etc., in which the nitrogen atom of the amino group and any atom of the side chain form a ring together, are also included in the amino acid).
- amino acid residues that make up a peptide compound may be simply referred to as "amino acids.”
- N-terminal amino acid residue refers to the amino acid residue located at the N-terminus of a peptide.
- C-terminal amino acid residue refers to the amino acid residue located at the C-terminus of a peptide.
- the "number of amino acids” and “number of amino acid residues” refer to the number of amino acid residues (amino acid units) that make up a peptide compound, and refer to the number of amino acid units that are generated when the amide bonds, ester bonds, and cyclized bonds that link the amino acids are cleaved.
- amino acids constituting the peptide compounds in this specification include all corresponding isotopes.
- An isotope of an "amino acid” is one in which at least one atom is replaced with an atom having the same atomic number (proton number) but a different mass number (sum of the number of protons and neutrons) in a ratio different from that in nature.
- substituents containing a halogen atom include alkyl groups, cycloalkyl groups, alkenyl groups, alkynyl groups, aryl groups, heteroaryl groups, and aralkyl groups each having a halogen as a substituent, and more specifically, examples include fluoroalkyl, difluoroalkyl, and trifluoroalkyl.
- Examples of oxy (-OR) include alkoxy, cycloalkoxy, alkenyloxy, alkynyloxy, aryloxy, heteroaryloxy, aralkyloxy, etc.
- alkoxy C 1 -C 4 alkoxy and C 1 -C 2 alkoxy are preferred, and among these, methoxy and ethoxy are preferred.
- Examples of oxycarbonyl include alkyloxycarbonyl, cycloalkyloxycarbonyl, alkenyloxycarbonyl, alkynyloxycarbonyl, aryloxycarbonyl, heteroaryloxycarbonyl, and aralkyloxycarbonyl.
- Examples of carbonyloxy include alkylcarbonyloxy, cycloalkylcarbonyloxy, alkenylcarbonyloxy, alkynylcarbonyloxy, arylcarbonyloxy, heteroarylcarbonyloxy, and aralkylcarbonyloxy.
- thiocarbonyl examples include alkylthiocarbonyl, cycloalkylthiocarbonyl, alkenylthiocarbonyl, alkynylthiocarbonyl, arylthiocarbonyl, heteroarylthiocarbonyl, and aralkylthiocarbonyl.
- Examples of carbonylthio include alkylcarbonylthio, cycloalkylcarbonylthio, alkenylcarbonylthio, alkynylcarbonylthio, arylcarbonylthio, heteroarylcarbonylthio, and aralkylcarbonylthio.
- aminocarbonyl examples include alkylaminocarbonyl (e.g., C1 - C6 or C1 - C4 alkylaminocarbonyl, particularly ethylaminocarbonyl, methylaminocarbonyl, etc.), cycloalkylaminocarbonyl, alkenylaminocarbonyl, alkynylaminocarbonyl, arylaminocarbonyl, heteroarylaminocarbonyl, aralkylaminocarbonyl, etc.
- alkylaminocarbonyl e.g., C1 - C6 or C1 - C4 alkylaminocarbonyl, particularly ethylaminocarbonyl, methylaminocarbonyl, etc.
- cycloalkylaminocarbonyl alkenylaminocarbonyl, alkynylaminocarbonyl, arylaminocarbonyl, heteroarylaminocarbonyl, aralkylaminocarbonyl,
- Examples of carbonylamino include alkylcarbonylamino, cycloalkylcarbonylamino, alkenylcarbonylamino, alkynylcarbonylamino, arylcarbonylamino, heteroarylcarbonylamino, and aralkylcarbonylamino.
- Examples of oxycarbonylamino include alkoxycarbonylamino, cycloalkoxycarbonylamino, alkenyloxycarbonylamino, alkynyloxycarbonylamino, aryloxycarbonylamino, heteroaryloxycarbonylamino, and aralkyloxycarbonylamino.
- sulfonylamino examples include alkylsulfonylamino, cycloalkylsulfonylamino, alkenylsulfonylamino, alkynylsulfonylamino, arylsulfonylamino, heteroarylsulfonylamino, aralkylsulfonylamino, etc.
- H atom bonded to the N atom in -NH-SO 2 -R is further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
- aminosulfonyl examples include alkylaminosulfonyl, cycloalkylaminosulfonyl, alkenylaminosulfonyl, alkynylaminosulfonyl, arylaminosulfonyl, heteroarylaminosulfonyl, aralkylaminosulfonyl, etc.
- examples include groups in which the H atom bonded to the N atom in -SO 2 -NHR is further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
- sulfamoylamino examples include alkylsulfamoylamino, cycloalkylsulfamoylamino, alkenylsulfamoylamino, alkynylsulfamoylamino, arylsulfamoylamino, heteroarylsulfamoylamino, aralkylsulfamoylamino, etc.
- the two H atoms bonded to the N atom in -NH-SO 2 -NHR may be substituted with substituents independently selected from the group consisting of alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, and these two substituents may form a ring.
- thio examples are selected from alkylthio, cycloalkylthio, alkenylthio, alkynylthio, arylthio, heteroarylthio, aralkylthio, etc.
- sulfonyl examples include alkylsulfonyl, cycloalkylsulfonyl, alkenylsulfonyl, alkynylsulfonyl, arylsulfonyl, heteroarylsulfonyl, aralkylsulfonyl, and the like.
- secondary amino examples include alkylamino, cycloalkylamino, alkenylamino, alkynylamino, arylamino, heteroarylamino, and aralkylamino.
- tertiary amino examples include, for example, alkyl(aralkyl)amino and other amino groups having any two substituents independently selected from alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, and the like, and these any two substituents may form a ring.
- Specific examples include dialkylamino, particularly C 1 -C 6 dialkylamino, C 1 -C 4 dialkylamino, dimethylamino, diethylamino, and the like.
- C p -C q dialkylamino group refers to a group in which an amino group is substituted with two C p -C q alkyl groups, and both C p -C q alkyl groups may be the same or different.
- substituted amidino examples include those in which the three substituents R, R', and R" on the N atom are each independently selected from alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, such as alkyl(aralkyl)(aryl)amidino.
- substituted guanidino examples include groups in which R, R', R", and R''' are each independently selected from alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, or groups in which these groups form a ring.
- aminocarbonylamino examples include groups in which R, R', and R" are each independently selected from a hydrogen atom, alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, or groups in which these form a ring.
- the compound of the present invention may be a salt thereof, preferably a chemically acceptable salt thereof.
- the compound of the present invention or a salt thereof may be a solvate thereof, preferably a chemically acceptable solvate thereof.
- the salt of the compound of the present invention may be, for example, hydrochloride; hydrobromide; hydroiodide; phosphate; phosphonate; sulfate; sulfonate such as methanesulfonate and p-toluenesulfonate; carboxylate such as acetate, citrate, malate, tartrate, succinate, salicylate; or alkali metal salt such as sodium salt and potassium salt; alkaline earth metal salt such as magnesium salt and calcium salt; ammonium salt such as ammonium salt, alkylammonium salt, dialkylammonium salt, trialkylammonium salt, tetraalkylammonium salt, etc.
- a solvate of a compound refers to a compound that forms a molecular group together with a solvent, and is not particularly limited as long as it is a solvate formed by a solvent. If the solvent is water, it is called a hydrate.
- the solvates of the compounds of the present invention are preferably hydrates, and specific examples of such hydrates include monohydrates to decahydrates, preferably monohydrates to pentahydrates, and more preferably monohydrates to trihydrates.
- solvates of the compounds of the present invention include solvates with a single solvent such as water, alcohol (e.g., methanol, ethanol, 1-propanol, 2-propanol, etc.), and dimethylformamide, as well as solvates with multiple solvents.
- a single solvent such as water, alcohol (e.g., methanol, ethanol, 1-propanol, 2-propanol, etc.), and dimethylformamide, as well as solvates with multiple solvents.
- the compound according to the present invention When the compound according to the present invention is obtained as a free form, the compound can be converted into its hydrate or solvate according to a conventional method.
- the compound according to the present invention when the compound according to the present invention is obtained as a free form, the compound can be converted into the salt that the compound may form, or into its hydrate or solvate according to a conventional method.
- the hydrate, ethanol solvate, etc. of the cyclic peptide compound represented by formula (1a) or its salt can be included.
- the cyclic peptide compound represented by formula (1a) may be hemihydrate, monohydrate, dihydrate, trihydrate, tetrahydrate, pentahydrate, hexahydrate, heptahydrate, octahydrate, nodahydrate, decahydrate, or monoethanolate, or the sodium salt of the cyclic peptide compound represented by formula (1a) may be hemihydrate, monohydrate, dihydrate, trihydrate, tetrahydrate, pentahydrate, hexahydrate, heptahydrate, octahydrate, nodahydrate, decahydrate, or monoethanolate, or the hydrochloride salt of the cyclic peptide compound represented by formula (1a) may be hydrated or ethanolate, but is not limited thereto.
- the hydrate or solvate may be produced in a crystalline or non-crystalline form, and in the case of a crystalline form, it may be a crystalline polymorph.
- a solvent such as ethanol and/or water can be added to the cyclic peptide compound represented by formula (1a) or a peptide compound described in the present specification, and the hydrate or solvate can be obtained by a conventional method, such as stirring, cooling, concentrating, and/or drying.
- the compound according to the present invention when obtained as a salt, hydrate, or solvate of the compound, the compound can be converted to its free form by a conventional method.
- solvent A/hydrate crystal means a crystal in which solvent A molecules and water molecules are contained in the crystal lattice of a compound.
- solvent A/solvent B/hydrate crystal means a crystal in which solvent A molecules, solvent B molecules, and water molecules are contained in the crystal lattice of a compound.
- acetone/heptane/hydrate crystal means a crystal in which acetone, heptane, and water are contained in the crystal lattice of a compound.
- A, B, and/or C includes the following seven variations: (i) A, (ii) B, (iii) C, (iv) A and B, (v) A and C, (vi) B and C, (vii) A, B, and C.
- epimer refers to a compound (epimer) in which the configuration of the side chain bonded to the ⁇ -carbon of an amino acid residue constituting a cyclic peptide compound is inverted.
- “Epimer” includes cyclic peptide compounds in which the ⁇ -carbon of the C-terminal amino acid residue of a linear peptide compound is inverted when the linear peptide compound is cyclized to produce the cyclic peptide compound.
- the epimer in the total product containing the cyclic peptide compound produced by the method of the present invention can be determined, for example, by the UVarea value at 210 nm or 220 nm by HPLC analysis.
- cyclic dimer refers to a compound in which peptide compounds, the raw materials of a cyclic peptide compound, are bonded together in a linear chain and then cyclized.
- the amount of cyclic dimers in the total product containing the cyclic peptide compound produced by the method of the present invention can be determined, for example, by the UVarea value at 210 nm or 220 nm by HPLC analysis.
- the present invention relates to a method for producing a cyclic peptide compound represented by formula (1), or a salt or solvate thereof, which comprises a step of cyclizing a peptide compound represented by formula (2) or (3) by reacting the N-terminal amino acid residue with the C-terminal amino acid residue in a solvent (cyclization step) (hereinafter, also referred to as "embodiment 1").
- the present invention relates to a method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof, the method comprising: (a) preparing a peptide compound represented by any one of formulas (4) to (6), or a salt thereof, or a solvate of said peptide compound or salt; (b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4) to (6) in a solvent (linking step); and (c) A step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in step (b) by reacting them in a solvent (cyclization step) (hereinafter, also referred to as "Mode 2").
- the step (b) may include (b-1) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (5) and the C-terminal amino acid residue of the peptide compound represented by formula (6) in a solvent to convert them into the peptide compound represented by formula (7) (linking step).
- the step (b) includes, in addition to the step (b-1), (b-2) a step (linking step) of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4) and the C-terminal amino acid residue of the peptide compound represented by formula (7) in a solvent to convert them into the peptide compound represented by formula (2), and the step (c) may include (c-1)
- the method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2) by reaction in a solvent (cyclization step).
- the step (b) includes, in addition to the step (b-1), (b-3) a step (linking step) of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7) and the C-terminal amino acid residue of the peptide compound represented by formula (4) in a solvent to convert them into the compound represented by formula (3)
- the step (c) may include (c-2)
- the method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3) by reaction in a solvent (cyclization step).
- the link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is preferably a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue, and more preferably a link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue via an amide bond.
- the solvent for the cyclization step preferably includes one or more selected from the group consisting of nitrile solvents, halogen solvents, ether solvents, amide solvents, ester solvents, and carbonate solvents.
- nitrile solvents include acetonitrile and propionitrile.
- halogen solvents include dichloromethane, chloroform, and 1,2-dichloroethane.
- ether solvents include diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, 4-methyltetrahydropyran, 1,3-dioxolane, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, t-butyl methyl ether, diglyme, triglyme, anisole, and tetraglyme.
- amide solvents include DMF, NMP, DMA, NEP, NBP, and formamide.
- ester solvents include methyl acetate, ethyl acetate, methyl propionate, butyl acetate, propyl acetate, isopropyl acetate, isobutyl acetate, pentyl acetate, and ⁇ -valerolactone.
- carbonate ester solvents include dimethyl carbonate, diethyl carbonate, and dibutyl carbonate.
- the solvent for the cyclization step is preferably one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, dichloromethane, DMF, and anisole, more preferably one or more selected from the group consisting of acetonitrile, 2-methyltetrahydrofuran, ethyl acetate, and dichloromethane, and even more preferably acetonitrile, 2-methyltetrahydrofuran, or ethyl acetate.
- the cyclization step can be carried out in a solvent, in the presence or absence of a condensation reagent, in the presence or absence of a base, at a temperature between -20°C and the boiling point of the solvent, preferably between -20°C and 100°C, and preferably between -5°C and 60°C, by stirring the reaction mixture for 10 minutes to 48 hours.
- the condensation reagent, base, and amounts thereof used in the cyclization step are not particularly limited, and condensation reagents, bases, and amounts thereof generally used in peptide synthesis are preferred (e.g., Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.)).
- condensation reagents, bases, and amounts thereof generally used in peptide synthesis are preferred (e.g., Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.)).
- a carboxyl group that has been converted into an active ester in advance may be used.
- Condensation reagents for the cyclization step include, for example, N,N'-dicyclohexylcarbodiimide (DCC), N,N'-diisopropylcarbodiimide (DIC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI), HCl), 1-hydroxy-1H-benzotriazole (HOBt), 1-hydroxy-7-azabenzotriazole (HOAt), ethyl 2-cyano-2-(hydroxyimino)acetate (oxyma), 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HOOBt or HODhbt), N-hydroxy-5-norbornene-2,3-dicarboximide (HONB), 2,3,4,5,6-pentafluorophenol (HOPfp), N-hydroxysuccinimide (HOSu), 6-chloro-1-hydroxy-1H-benzo
- the condensation reagent for the cyclization step is preferably one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop, more preferably one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P, and even more preferably HATU and COMU.
- HATU and acetonitrile or 2-methyltetrahydrofuran are preferred, since by-products can be further suppressed.
- an organic base is preferably used, and among them, an organic base containing a tertiary amine is preferable.
- Specific examples of such bases include 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonane (NDABCO), and 1,5-diazabicyclo[4.3.0]-5-nonane (NDABCO).
- DMAP dimethylaminopyridine
- the base for the cyclization step is preferably one or more selected from the group consisting of 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, and pyridine, and N,N-diisopropylethylamine (DIPEA) and 2,6-lutidine are preferred.
- DIPEA N,N-diisopropylethylamine
- DIPEA N,N-diisopropylethylamine
- the combinations of solvent, condensation reagent, and base are more effective in suppressing by-products, and therefore are preferably HATU, acetonitrile, and N,N-diisopropylethylamine (DIPEA); HATU, 2-methyltetrahydrofuran, and N,N-diisopropylethylamine (DIPEA); COMU, acetonitrile, and 2,6-lutidine; and COMU, 2-methyltetrahydrofuran, and 2,6-lutidine.
- DIPEA N,N-diisopropylethylamine
- DIPEA 2-methyltetrahydrofuran
- DIPEA N,N-diisopropylethylamine
- COMU acetonitrile, and 2,6-lutidine
- COMU 2-methyltetrahydrofuran, and 2,6-lutidine.
- the cyclization step is carried out in a liquid phase process.
- the cyclization step is carried out by mixing the peptide compound and, optionally, a base into a mixture obtained by mixing a solvent and a condensation reagent.
- this operation is sometimes referred to as "reverse dripping.”
- reverse dripping By reverse dripping the peptide compound and base over a long period of time, for example, several hours to several days, preferably 1 to 24 hours, and more preferably 1 to 10 hours, it is possible to suppress the production of by-products without using a large amount of solvent for dilution.
- the cyclic peptide compounds produced by the method of the present invention have a low content of by-products (e.g., epimers, cyclic dimers, etc.) and are highly pure, as described below.
- by-products e.g., epimers, cyclic dimers, etc.
- the content of total by-products produced in the cyclization step is less than 20%, less than 15%, less than 10%, less than 5%, or less than 3% based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
- the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
- the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis, and the by-products include epimers and/or cyclic dimers.
- the by-product produced in the cyclization step includes an epimer, and the epimer content is less than 10%, less than 7.5%, less than 5%, less than 2.5%, or less than 1% based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
- the by-products produced in the cyclization step include cyclic dimers, and the content of the cyclic dimers is less than 15%, less than 10%, less than 5%, less than 2.5%, or less than 1% based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
- the solvent in the linking step preferably includes one or more selected from the group consisting of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents.
- nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents include those exemplified as the solvent in the cyclization step.
- the solvent in the linking step is preferably one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, DMF, and anisole, more preferably a mixed solvent of one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, and anisole and DMF, even more preferably a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF, and particularly preferably a mixed solvent of 2-methyltetrahydrofuran and DMF.
- the linking step can be carried out in a solvent, in the presence or absence of a condensation reagent, in the presence or absence of a base, at a temperature between -20°C and the boiling point of the solvent, preferably between -20°C and 100°C, and preferably between -5°C and 60°C, by stirring the reaction composition for 10 minutes to 48 hours.
- condensation reagent, base, and amounts thereof used in the coupling step are not particularly limited, and condensation reagents, bases, and amounts thereof generally used in peptide synthesis are preferred (e.g., Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.)).
- condensation reagents, bases, and amounts thereof generally used in peptide synthesis are preferred (e.g., Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.)).
- a reagent in which the carboxyl group has been converted into an active ester in advance may be used.
- Condensation reagents for the linking step include those exemplified as condensation reagents for the cyclization step.
- the condensation reagent for the linking step is preferably one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop, more preferably one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P, and even more preferably HATU and COMU.
- solvents and condensation reagents are preferred, since they can further suppress by-products: HATU and acetonitrile or 2-methyltetrahydrofuran; a mixed solvent of HATU and acetonitrile, 2-methyltetrahydrofuran, and DMF; COMU and acetonitrile or 2-methyltetrahydrofuran; and a mixed solvent of COMU and acetonitrile, 2-methyltetrahydrofuran, and DMF.
- the bases in the linking step include those exemplified as the bases in the cyclization step above.
- the bases in the linking step include 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene, 1,1,3,3-tetramethyl
- TMG guanidine
- the combinations of solvent, condensation reagent, and base can further suppress by-products, so the following are preferred: HATU and acetonitrile or 2-methyltetrahydrofuran and N,N-diisopropylethylamine (DIPEA); HATU and a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF and N,N-diisopropylethylamine (DIPEA); COMU and acetonitrile or 2-methyltetrahydrofuran and N-methylmorpholine or 2,6-lutidine; and COMU and a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF and N-methylmorpholine or 2,6-lutidine.
- DIPEA HATU and acetonitrile or 2-methyltetrahydrofuran and N,N-diisopropylethylamine
- DIPEA HATU and a mixed solvent of ace
- the linking step is carried out using a liquid phase method.
- the method of the present invention further comprises the step of preparing a peptide compound represented by formulas (4) to (6), or a salt thereof, or a solvate thereof.
- the peptide compound represented by formulas (4) to (6) can be produced, for example, by the following general method.
- Pg4 and Pg5 represent protecting groups for the amino group
- Xg6 represents an oxygen atom and a protecting group bonded thereto
- R5 represents the side chain of the amino acid
- P4 and P6 represent substituents for the nitrogen atom.
- Peptide compound (4) can be prepared using the following method.
- an aldehyde with the protected amino acid according to the method of Freidinger et al. (J. Org. Chem., 1983, 48(1), 77-81)
- an oxazolidinone body with a cyclic protecting group can be obtained.
- an alkyl group having an olefin on the nitrogen atom can be introduced.
- an amino acid with a protected C-terminus can be condensed to extend the amino acid to the C-terminus.
- the condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a combination of mixed acid anhydrides or acid halides as a carboxyl group activator.
- a combination of DIC and Oxyma a combination of DIC and HOAt
- a combination of HATU and DIPEA a combination of mixed acid anhydrides or acid halides as a carboxyl group activator.
- the protecting group of the amino group is deprotected, and then the protected amino acid having an olefin on the side chain can be extended.
- the olefins in the molecule can then be cyclized by a metathesis reaction.
- catalysts such as dichloro(2-isopropoxybenzylidene)(tricyclohexylphosphine)ruthenium(II): CAS number 203714-71-0, dichloro(3-phenyl-1H-inden-1-ylidene)bis(tricyclohexylphosphine)ruthenium(II): CAS number 250220-36-1, dichloro(benzylidene)bis(tricyclohexylphosphine)ruthenium(II): CAS number 172222-30-9, [1,3-bis(2,4,6-trimethylphenyl)-2-imido]-2,3-dichloro(2,4,6-trimethylphenyl) ...
- the protecting group at the C-terminus is deprotected to produce peptide compound (4) having an unprotected C-terminus, or the protecting group at the N-terminus is deprotected to produce peptide compound (4) having an unprotected N-terminus.
- Peptide compound (4) can also be prepared using the following method.
- An alkyl group having an olefin can be introduced to the nitrogen atom of an amino acid by reacting an alkylating agent having an olefin in the presence of a base. Then, an amino acid with a protected C-terminus can be condensed to extend the amino acid to the C-terminus.
- the condensation reaction can be carried out using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a mixed acid anhydride or acid halide as an activator of the carboxyl group.
- the protected amino acid having an olefin in the side chain can be extended.
- the olefin in the molecule can be cyclized by a metathesis reaction.
- the catalyst used in the metathesis reaction is the same as that described above.
- the protecting group of the C-terminus can be deprotected to produce a peptide compound (4) with an unprotected C-terminus, or the protecting group of the N-terminus can be deprotected to produce a peptide compound (4) with an unprotected N-terminus.
- Peptide compound (5) can be prepared using the following method.
- the amino acid can be extended to the N-terminus by condensing a protected amino acid to an amino acid with a protected C-terminus.
- the condensation reaction can be carried out using the condensation reagent and base used in the linking step described above.
- various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a mixed acid anhydride or acid halide, as an activator of the carboxyl group.
- a fragment consisting of three amino acids can be synthesized by condensing the protected amino acid.
- the protecting group of the C-terminus can be deprotected to produce a peptide compound (5) with an unprotected C-terminus, or the protecting group of the N-terminus can be deprotected to produce a peptide compound (5) with an unprotected N-terminus.
- Peptide compound (5) can also be synthesized by solid-phase synthesis.
- Xg 3 in the above formula is an amino acid bound to a solid-phase support via an oxygen atom and a linker bound thereto, and an amino acid can be extended to the N-terminus by condensing a protected amino acid with this.
- the condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a combination via a mixed acid anhydride or acid halide as an activator of the carboxyl group.
- a fragment consisting of three amino acids can be synthesized by sequentially deprotecting the protecting group of the amino group and condensing the protected amino acid. Then, the peptide compound (5) with an unprotected C-terminus can be produced by cutting out from the solid phase.
- Peptide compound (6) can be prepared using the following method.
- the amino acid can be extended to the N-terminus by condensing a protected amino acid to an amino acid having a ⁇ -amino acid skeleton with a protected carboxyl group.
- the condensation reaction can be carried out using the condensation reagent and base used in the above-mentioned linking step.
- various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a mixed acid anhydride or acid halide, as an activator of the carboxyl group.
- a fragment consisting of five amino acids can be synthesized by sequentially deprotecting the protecting group of the amino group and condensing the protected amino acid. Next, the C-terminal protecting group can be deprotected to produce a peptide compound (6) with an unprotected C-terminus, or the N-terminal protecting group can be deprotected to produce a peptide compound (6) with an unprotected N-terminus.
- Peptide compound (6) can also be synthesized by solid-phase synthesis.
- Xg 11 in the above formula is an amino acid having a ⁇ -amino acid skeleton bound to a solid-phase support via an oxygen atom and a linker bound thereto, and an amino acid can be extended to the N-terminus by condensing a protected amino acid with this.
- the condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a combination via a mixed acid anhydride or acid halide as an activator of the carboxyl group.
- a fragment consisting of five amino acids can be synthesized by sequentially deprotecting the protecting group of the amino group and condensing the protected amino acid.
- the peptide compound (6) with an unprotected C-terminus can be produced by cutting out from the solid phase.
- the cyclic peptide compound, or a salt thereof, or a solvate thereof produced by the method of the present invention may or may not be isolated and/or purified using column chromatography.
- the cyclic peptide compound, or its salt, or its solvate produced by the method of the present invention can be isolated and/or purified by crystallization by crystallization. Specifically, for example, the reaction solution after the condensation reaction is subjected to a liquid separation operation, and the organic layer is concentrated and/or filtered as necessary, and then a solvent suitable for crystallization is added to the obtained residue, and seed crystals are optionally added and stirred as necessary to obtain crystals of the cyclic peptide compound, or its salt, or its solvate.
- the solvent added during crystallization is not particularly limited as long as it is a solvent in which the cyclic peptide compound can form crystals, but a solvent in which the solubility of the cyclic peptide compound can be reduced in the solution in which the cyclic peptide compound is dissolved is preferable.
- a solvent in which the solubility of the cyclic peptide compound can be reduced by adding a poor solvent or cooling the solution examples of solvents that allow such operations are exemplified.
- a solvent that allows such operations can be used for crystallization.
- Specific examples of solvents that can be added during crystallization include acetone, water, DMSO, acetonitrile, ethanol, and mixtures of these solvents.
- R 1 is C 1 -C 6 alkyl.
- R 1 is preferably C 3 -C 4 alkyl, more preferably n-propyl, 2-methylpropyl.
- P1 is C1 - C6 alkyl.
- P1 is preferably C1 - C4 alkyl, more preferably methyl.
- R2 is C1 - C6 alkyl.
- R2 is preferably C3 - C4 alkyl, more preferably 1-methylpropyl.
- R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached forms a 4- to 7-membered saturated heterocyclic ring.
- R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached forms a 5-membered saturated heterocyclic ring.
- R 3 is preferably hydrogen.
- R 3 is preferably together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached forms a 5- membered saturated heterocyclic ring.
- P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached forms a 4-7 membered saturated heterocyclic ring.
- P3 is C1 - C4 alkyl, or P3 together with R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached forms a 5 membered saturated heterocyclic ring.
- P3 is preferably methyl.
- P3 is preferably together with R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached forms a 5 membered saturated heterocyclic ring.
- P4 is C 1 -C 6 alkyl.
- P4 is preferably C 1 -C 4 alkyl, more preferably methyl.
- R 5 is benzyl optionally substituted by one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl.
- R 5 is preferably benzyl optionally substituted by C 1 -C 4 haloalkyl, more preferably 4-trifluoromethylbenzyl.
- P6 is C 1 -C 6 alkyl.
- P6 is preferably C 1 -C 4 alkyl, more preferably methyl.
- R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy.
- R 7 is preferably phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy, and is more preferably 3-methoxy-4-trifluoromethylphenethyl or 3,5-difluoro-4-trifluoromethylphenethyl.
- R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocyclic ring, which may be substituted by C 1 -C 6 alkoxy.
- R 8 preferably together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocyclic ring, which is substituted by C 1 -C 4 alkyl, and more preferably together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocyclic ring, which is substituted by ethoxy.
- R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 3-8 membered alicyclic ring, which may be substituted by one or more C 1 -C 6 alkyls.
- R 9 preferably together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 4-6 membered alicyclic ring.
- R 9 preferably together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 4 membered alicyclic ring.
- R 9 preferably together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 5 membered alicyclic ring.
- P 9 is hydrogen or C 1 -C 6 alkyl.
- P 9 is preferably hydrogen or C 1 -C 4 alkyl. In some embodiments, P 9 is preferably hydrogen. In some embodiments, P 9 is preferably methyl.
- R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl.
- R 10 is preferably C 4 -C 6 cycloalkyl, more preferably cyclopentyl.
- P 10 is C 1 -C 6 alkyl.
- P 10 is preferably C 1 -C 4 alkyl, more preferably methyl.
- R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4- to 8-membered cyclic aminocarbonyl.
- R 11 is preferably diC 1 -C 4 alkylaminocarbonyl, or 5- to 6-membered cyclic aminocarbonyl, more preferably dimethylaminocarbonyl.
- P 11 is C 1 -C 6 alkyl.
- P 11 is preferably C 1 -C 4 alkyl, more preferably methyl.
- X1 , X3 , and X5 are each independently hydrogen or a protecting group of an amino group.
- X1 , X3 , and X5 are preferably each independently one selected from the group consisting of hydrogen, a carbamate-based protecting group, an acyl-based protecting group, a sulfonamide-based protecting group, and a silyl-based protecting group.
- the carbamate-based protecting group is one selected from the group consisting of an Fmoc group, a Cbz group, a Troc group, an Alloc group, a Teoc group, a TSoc group, a BIBSoc group, an IPCSoc group, a BBSoc group, a CHBSoc group, a CDBSoc group, and a Boc group.
- the acyl-based protecting group is one selected from the group consisting of a trifluoroacetyl group, an acetyl group, and a benzoyl group.
- the sulfonamide protecting group is one selected from the group consisting of 2-nitrobenzenesulfonyl, 4-nitrobenzenesulfonyl, and 2,4-dinitrobenzenesulfonyl groups.
- the silyl protecting group is one selected from the group consisting of TMS, TBDMS, TES, TIPS, and TBDPS groups.
- X 1 is hydrogen or a carbamate-based protecting group. In some embodiments, X 1 is preferably hydrogen. In some embodiments, X 1 is preferably an Fmoc group.
- X3 is hydrogen or a carbamate-based protecting group. In some embodiments, X3 is preferably hydrogen. In some embodiments, X3 is preferably a Cbz group.
- X5 is hydrogen or a carbamate protecting group. In some embodiments, X5 is preferably hydrogen. In some embodiments, X5 is preferably a Cbz group.
- X 2 , X 4 , and X 6 are each independently a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently an alkyl or an aryl).
- X 2 , X 4 , and X 6 are preferably each independently a halogen, a hydroxyl group, an optionally substituted C 1 -C 6 alkoxy, an optionally substituted C 6 -C 10 aryloxy, an optionally substituted C 7 -C 14 aralkoxy, an optionally substituted 4- to 8-membered cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently a C 1 -C 6 alkyl, or a C 6 -C 10 aryl).
- the halogen is chlorine or bromine.
- the optionally substituted alkoxy is t-butoxy, methoxy, ethoxy, or isopropoxy.
- the optionally substituted aryloxy is pentafluorophenyloxy, or nitrophenyloxy.
- the optionally substituted aralkoxy is an optionally substituted benzyloxy.
- the optionally substituted cyclic aminooxy is N- hydroxysuccinimoxy .
- the group represented by -OSiRxRyRz is trimethylsilyloxy, triethylsilyloxy, triisopropylsilyloxy, triphenylsilyloxy, tri -t-butylsilyloxy, di-t-butylisobutylsilyloxy, or tris(triethylsilyl)silyloxy.
- X2 is hydroxyl, t-butoxy, or benzyloxy. In some embodiments, X2 is preferably hydroxyl. In some embodiments, X2 is preferably t-butoxy.
- X 4 is hydroxyl, t-butoxy, or benzyloxy. In some embodiments, X 4 is preferably hydroxyl. In some embodiments, X 4 is preferably t-butoxy.
- X 6 is hydroxyl, t-butoxy, or benzyloxy. In some embodiments, X 6 is preferably hydroxyl. In some embodiments, X 6 is preferably t-butoxy.
- R 1 is C 1 -C 6 alkyl
- P1 is C1 - C6 alkyl
- R2 is C1 - C6 alkyl
- R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring
- P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle
- P4 is C1 - C6 alkyl
- R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 halo
- R 1 is C 3 -C 4 alkyl
- P1 is C1 - C4 alkyl
- R2 is C3 - C4 alkyl
- R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring
- P3 is C 1 -C 4 alkyl or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocycle
- P4 is C1 - C4 alkyl
- R 5 is benzyl optionally substituted by C 1 -C 4 haloalkyl
- P6 is C1 - C4 alkyl
- R7 is phenethyl optionally substituted with one or more groups selected from the group consisting of
- R 1 is n-propyl, 2-methylpropyl; P1 is methyl; R2 is 1-methylpropyl; R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring; P3 is methyl or forms together with P3 , R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached a 5-membered saturated heterocycle; P4 is methyl; R5 is 4-trifluoromethylbenzyl; P6 is methyl; R 7 is 3-methoxy-4-trifluoromethylphenethyl, or 3,5-difluoro-4-trifluoromethylphenethyl; R 8 together with P 8 , the carbon atom to which R 8 is attached, and
- R 1 is C 1 -C 6 alkyl
- P1 is C1 - C6 alkyl
- R2 is C1 - C6 alkyl
- R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring
- P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle
- P4 is C1 - C6 alkyl
- R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloal
- R 1 is C 3 -C 4 alkyl
- P1 is C1 - C4 alkyl
- R2 is C3 - C4 alkyl
- R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring
- P3 is C 1 -C 4 alkyl or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocycle
- P4 is C1 - C4 alkyl
- R 5 is benzyl optionally substituted by C 1 -C 4 haloalkyl
- P6 is C1 - C4 alkyl
- R7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy;
- the cyclic peptide compound produced by the method of the present invention has the following formula (1a): or a salt or solvate thereof.
- the present invention relates to a method for producing a cyclic peptide compound represented by formula (1a), or a salt or solvate thereof, the method comprising: (a) providing a peptide compound represented by any one of formulas (4a) to (6a), or a salt thereof, or a solvate of said peptide compound or salt; (b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4a) to (6a) (linking step); and (c) A step of cyclizing the peptide compound obtained in step (b) by reacting the N-terminal amino acid residue with the C-terminal amino acid residue (cyclization step) (hereinafter, also referred to as "Mode 2'").
- the step (b) may include a step (b-1) of converting the N-terminal amino acid residue of the peptide compound represented by formula (5a) and the C-terminal amino acid residue of the peptide compound represented by formula (6a) into the peptide compound represented by formula (7a).
- the step (b) includes, in addition to the step (b-1), (b-2) a step (linking step) of converting a peptide compound represented by formula (4a) to a peptide compound represented by formula (7a) by linking the N-terminal amino acid residue with the C-terminal amino acid residue of the peptide compound represented by formula (7a) to obtain a peptide compound represented by formula (2a), and the step (c) may include (c-1)
- the method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2a) by reaction in a solvent (cyclization step).
- the step (b) includes, in addition to the step (b-1), (b-3) a step (linking step) of converting a peptide compound represented by formula (7a) to a peptide compound represented by formula (4a) by linking the N-terminal amino acid residue with the C-terminal amino acid residue of the peptide compound represented by formula (4a) to obtain a peptide compound represented by formula (3a), and the step (c) may include (c-2)
- the method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3a) by reaction in a solvent (cyclization step).
- the link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is preferably a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue, and more preferably a link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue via an amide bond.
- the solvent in the cyclization step, the condensation reagent in the cyclization step, the base in the cyclization step, the by-products generated in the cyclization step, the solvent in the linking step, the condensation reagent in the linking step, the base in the linking step, etc. are the same as those described in embodiments 1 and 2 above.
- the present invention relates to a compound represented by formula (4a):
- X 1 is hydrogen or a protecting group of an amino group.
- the protecting group in X 1 is the same as that described in the above embodiments 1 and 2.
- X 1 is preferably hydrogen.
- X 1 is preferably an Fmoc group.
- X 2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl).
- R x , R y , and R z are each independently alkyl or aryl.
- the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 2 are the same as those explained in the above embodiments 1 and 2.
- X 2 is preferably t-butoxy.
- the compound represented by formula (4a) is preferably tert-butyl 2-[methyl-[(2S)-2-[(4Z,7S)-7-(methylamino)-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]amino]acetic acid (compound 9).
- the compound represented by formula (4a) (peptide compound (4a)) can be prepared using the following method.
- the protected amino acid (4a-1) can be reacted with an aldehyde according to the method of Freidinger et al. (J. Org. Chem., 1983, 48(1), 77-81) to obtain an oxazolidinone (4a-2) having a cyclic protecting group.
- a ring-opening reaction can be performed using a silicon compound having an olefin according to the method of Nguyen et al. (Synthesis, 2009, 12, 1991) to obtain a compound (4a-3) having an alkyl group having an olefin on the nitrogen atom.
- the amino acid (4a-4) having a protected C-terminus can be condensed to extend the amino acid to the C-terminus.
- the condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a combination of mixed acid anhydrides or acid halides as a carboxyl group activator.
- the protected amino acid (4a-6) having an olefin in the side chain can be elongated.
- the olefin in the molecule can then be cyclized by a metathesis reaction.
- catalysts such as dichloro(2-isopropoxybenzylidene)(tricyclohexylphosphine)ruthenium(II): CAS No. 203714-71-0, dichloro(3-phenyl-1H-inden-1-ylidene)bis(tricyclohexylphosphine)ruthenium(II): CAS No. 250220-36-1, dichloro(benzylidene)bis(tricyclohexylphosphine)ruthenium(II): CAS No.
- Peptide compound (4a) can also be prepared using the following method. By reacting an alkylating agent having an olefin in the presence of a base with the amino acid (4a-9), an amino acid (4a-10) having an olefin-containing alkyl group introduced at the N atom can be obtained. Next, the amino acid (4a-4) with a protected C-terminus can be condensed to extend the amino acid to the C-terminus.
- the condensation reaction can be carried out using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a mixed acid anhydride or acid halide as an activator of the carboxyl group.
- the protecting group of the amino group is deprotected, and then the protected amino acid having an olefin in the side chain can be extended.
- the olefin in the molecule can be cyclized by a metathesis reaction.
- the catalyst used in the metathesis reaction is the same as that described above.
- the protecting group at the C-terminus can be deprotected to produce a peptide compound (4a) with an unprotected C-terminus, or the protecting group at the N-terminus can be deprotected to produce a peptide compound (4a) with an unprotected N-terminus.
- the present invention relates to a compound represented by formula (5a):
- X3 is water or an amino-protecting group.
- the amino-protecting group in X3 is the same as that described in the above embodiments 1 and 2.
- X3 is preferably hydrogen.
- X3 is preferably an Fmoc group or a Cbz group.
- X 4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl).
- R x , R y , and R z are each independently alkyl or aryl.
- the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 2 are the same as those explained in the above embodiments 1 and 2.
- X 4 is preferably t-butoxy.
- the compound represented by formula (5a) is preferably tert-butyl(2S)-1-[(2S,3S)-3-methyl-2-[[(2S)-2-(methylamino)pentanoyl]amino]pentanoyl]pyrrolidine-2-acetic acid (compound 13).
- the compound represented by formula (5a) (peptide compound (5a)) can be prepared using the following method.
- Compound (5a-3) is obtained by condensing protected amino acid (5a-2) to proline (5a-1) whose C-terminus is protected.
- the condensation reaction can be carried out using the condensation reagent and base used in the above-mentioned linking step.
- various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a mixed acid anhydride or acid halide, as an activator of the carboxyl group.
- a fragment (5a-5) consisting of three amino acids can be synthesized by condensing protected amino acid (5a-4).
- the protecting group of the C-terminus can be deprotected to produce peptide compound (5a) with an unprotected C-terminus, or the protecting group of the N-terminus can be deprotected to produce peptide compound (5a) with an unprotected N-terminus.
- the present invention relates to a compound represented by formula (6a):
- X 6 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl).
- R x , R y , and R z are each independently alkyl or aryl.
- the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 6 are the same as those explained in the above embodiments 1 and 2.
- X 6 is preferably t-butoxy.
- the compound represented by formula (6a) is preferably (3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid (compound 20).
- the compound represented by formula (6a) (peptide compound (6a)) can be prepared using the following method.
- Compound (6a-3) is obtained by condensing protected amino acid (6a-2) with amino acid (6a-1) having a ⁇ -amino acid skeleton with a protected carboxyl group.
- the condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step.
- various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a mixed acid anhydride or acid halide as an activator of the carboxyl group.
- the present invention relates to a compound represented by formula (7a):
- X5 is hydrogen or a protecting group for an amino group.
- the protecting group for an amino group in X5 is the same as that described in the above embodiments 1 and 2.
- X5 is preferably hydrogen.
- X5 is preferably a Cbz group.
- X 4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl).
- R x , R y , and R z are each independently alkyl or aryl.
- the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 4 are the same as those explained in the above embodiments 1 and 2.
- X 4 is preferably a hydroxyl group or t-butoxy.
- the compound represented by formula (7a) is preferably (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentylacetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylic acid (compound 22).
- the compound represented by formula (7a) is preferably tert-butyl (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate (compound 37).
- the compound represented by formula (7a) can be produced by reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (5a) and the C-terminal amino acid residue of the peptide compound represented by formula (6a) in a solvent (linking step).
- the solvent in the linking step, the condensation reagent in the linking step, the base in the linking step, etc. are the same as those described in the above embodiments 1 and 2.
- the present invention relates to a compound represented by formula (2a):
- X5 is hydrogen or a protecting group for an amino group.
- the protecting group for an amino group in X5 is the same as that described in the above embodiments 1 and 2.
- X5 is preferably hydrogen.
- X5 is preferably a Cbz group.
- X 2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl).
- R x , R y , and R z are each independently alkyl or aryl.
- the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 2 are the same as those explained in the above embodiments 1 and 2.
- X 2 is preferably a hydroxyl group or t-butoxy.
- the compound represented by formula (2a) is preferably 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbo Nyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carbonyl]-methyl-amino]
- the compound represented by formula (2a) can be produced by reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4a) and the C-terminal amino acid residue of the peptide compound represented by formula (7a) in a solvent (linking step).
- the solvent for the linking step, the condensation reagent for the linking step, the base for the linking step, etc. are the same as those described in the above embodiments 1 and 2.
- the present invention relates to a compound represented by formula (3a):
- X1 is hydrogen or an amino-protecting group.
- the amino-protecting group in X1 is the same as that described in the above embodiments 1 and 2.
- X1 is preferably hydrogen.
- X1 is preferably an Fmoc group.
- X 4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl).
- R x , R y , and R z are each independently alkyl or aryl.
- the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 4 are the same as those explained in the above embodiments 1 and 2.
- X 4 is preferably a hydroxyl group or t-butoxy.
- the compound represented by formula (3a) is preferably (S)-2-[(S)-3-[(S)-2-cyclopentyl-2-[1-[(2S,4R)-4-ethoxy-1-[(S)-4-[3-methoxy-4-(trifluoromethyl)phenyl]-2-[(2-[(S)-N-methyl-2-[(R,Z)-3-(methylamino)-2-oxo-3,4,7,8-tetrahydroazocin-1(2H)-yl]-3-[4-(trifluoromethyl)phenyl]propanamido]acetamido)butanoyl]-N-methylpyrrolidine-2-carboxamido]-N-methylcyclobutane-1-carboxamido]-N-methylacetamido]-4-(dimethylamino)-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleuc
- the compound represented by formula (3a) can be produced by reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7a) and the C-terminal amino acid residue of the peptide compound represented by formula (4a) in a solvent (linking step).
- the solvent in the linking step, the condensation reagent in the linking step, the base in the linking step, etc. are the same as those described in the above embodiments 1 and 2.
- the present invention relates to a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the crystal of this compound include a nonsolvate crystal or a solvate crystal of this compound, or a nonsolvate crystal or a solvate crystal of a salt of this compound.
- a solvate crystal of the cyclic peptide compound represented by formula (1a) is preferred.
- a preferred example of the solvate crystal is a hydrate crystal.
- the diffraction angle 2 ⁇ in powder X-ray diffraction is the diffraction peak measured using CuK ⁇ or CuK ⁇ 1 radiation.
- These solvate crystals further specified by the diffraction angle 2 ⁇ in powder X-ray diffraction are sometimes called "Form A crystals" of the hydrate shown below, for example, but are sometimes simply called "Form A”.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form A crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably those of a hydrate crystal stored at a relative humidity of 10% or more for 15 minutes or more, more preferably those of a hydrate crystal stored at a relative humidity of 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form A crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the hydrate crystal stored at a relative humidity of 10% or more for 15 minutes or more, more preferably the diffraction angles (2 ⁇ values) of the hydrate crystal stored at a relative humidity of 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form A crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably the diffraction angles (2 ⁇ values) of the hydrate crystal stored at a relative humidity of 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form B crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably those of a hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably those of a hydrate crystal stored at a relative humidity of 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form B crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably those of a hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably those of a hydrate crystal stored at a relative humidity of 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form B crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably the diffraction angles (2 ⁇ values) of the hydrate crystal stored at a relative humidity of 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form F crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as a diffraction angle 2 ⁇ in powder X-ray diffraction. 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° ( ⁇ 0.2°)
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form F crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as a diffraction angle 2 ⁇ in powder X-ray diffraction. 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° ( ⁇ 0.2°)
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form F crystal having a powder X-ray diffraction pattern including the following peaks as a diffraction angle 2 ⁇ in powder X-ray diffraction. 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° ( ⁇ 0.2°)
- the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal
- the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal
- the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal
- the crystal is a Form J crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more, more preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form J crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has a diffraction angle 2 ⁇ of 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° ( ⁇ 0.2°) in powder X-ray diffraction at a relative humidity of less than 10%.
- Form J crystals having a powder X-ray diffraction pattern containing at least seven peaks, and at a relative humidity of 10% or greater it includes Form A crystals having a powder X-ray diffraction pattern containing at least seven peaks at the following positions: 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° ( ⁇ 0.2°).
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has a diffraction angle 2 ⁇ of 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° ( ⁇ 0.2°) in powder X-ray diffraction at a relative humidity of less than 10%.
- Form J crystals having a powder X-ray diffraction pattern containing at least eight peaks, and at a relative humidity of 10% or greater it includes Form A crystals having a powder X-ray diffraction pattern containing at least eight peaks at the following positions: 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° ( ⁇ 0.2°).
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has the following diffraction angles 2 ⁇ in powder X-ray diffraction at a relative humidity of less than 10%: 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97°.
- Form A crystals having a powder X-ray diffraction pattern including peaks at 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° ( ⁇ 0.2°) at a relative humidity of 10% or greater.
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction.
- the following diffraction angles (2 ⁇ values) are preferably diffraction angles (2 ⁇ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, more preferably diffraction angles (2 ⁇ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal
- the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, more preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal
- the crystal is a Form Y crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal
- the crystal is a Form Y crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2 ⁇ in powder X-ray diffraction:
- the following diffraction angles (2 ⁇ values) are preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2 ⁇ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes.
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal exhibits, in powder X-ray diffraction, a diffraction angle 2 ⁇ of at least one of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° ( ⁇ 0.2°) at a relative humidity of less than 30%.
- Form Y crystals having a powder X-ray diffraction pattern containing at least seven peaks at 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° ( ⁇ 0.2°).
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal exhibits, in powder X-ray diffraction, a diffraction angle 2 ⁇ of at least one of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° ( ⁇ 0.2°) at a relative humidity of less than 30%.
- Form Y crystals having a powder X-ray diffraction pattern containing at least eight peaks at 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° ( ⁇ 0.2°).
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has diffraction angles 2 ⁇ of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° ( ⁇ 0.05°) in powder X-ray diffraction at a relative humidity of less than 30%.
- Form Y crystals having a powder X-ray diffraction pattern including peaks at 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° ( ⁇ 0.2°).
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a water solvate crystal, the crystal is a Form K crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as a diffraction angle 2 ⁇ in powder X-ray diffraction. 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° ( ⁇ 0.2°)
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a water solvate crystal, the crystal is a Form K crystal having a powder X-ray diffraction pattern including at least eight of the following peaks at a diffraction angle 2 ⁇ in powder X-ray diffraction: 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° ( ⁇ 0.2°)
- the crystal of the cyclic peptide compound of formula (1a) when the crystal of the cyclic peptide compound of formula (1a) is a water solvate crystal, the crystal is a Form K crystal having a powder X-ray diffraction pattern including the following peaks at a diffraction angle 2 ⁇ in powder X-ray diffraction. 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° ( ⁇ 0.2°)
- the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the method includes the steps of dissolving the cyclic peptide compound in an amount of a polar organic solvent in which the cyclic peptide compound can be dissolved to obtain a solution, and adding a hydrocarbon solvent or water to the solution to obtain a crystal of the cyclic peptide compound (hereinafter, also referred to as "embodiment 3").
- the nature of the cyclic peptide compound to be dissolved is not particularly limited, and for example, a cyclic peptide compound in a solid state, an amorphous state, or a crystalline state can be used.
- the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the method includes the step of adding a mixture of a hydrocarbon solvent and a polar organic solvent, or a mixture of water and a polar organic solvent, to the cyclic peptide compound in an amorphous or crystalline state to obtain a crystal of the cyclic peptide compound (hereinafter, also referred to as "embodiment 4").
- the polar organic solvent used in embodiments 3 and 4 include DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, ethyl acetate, propylene glycol, and mixtures thereof, with acetone being a more preferred example.
- the "amount capable of dissolving the cyclic peptide compound” can be in the range of 3 to 10 v/w, preferably 3 to 7 v/w, relative to the cyclic peptide compound of formula (1a).
- hydrocarbon solvent used in embodiments 3 and 4 include heptane, hexane, pentane, toluene, xylene, and mixed solvents thereof, with heptane being a more preferred example.
- the mixture ratio of the hydrocarbon solvent and the polar organic solvent in the mixture of the hydrocarbon solvent and the polar organic solvent can be 0.5 to 10 parts by weight of the hydrocarbon solvent per 1 part by weight of the polar organic solvent, preferably 1 to 7 parts by weight of the hydrocarbon solvent, and more preferably 1 to 5 parts by weight of the hydrocarbon solvent.
- the mixture ratio of the water and the polar organic solvent in the mixture of the water and the polar organic solvent can be 0.5 to 10 parts by weight of water per 1 part by weight of the polar organic solvent, preferably 1 to 7 parts by weight of water, and more preferably 1 to 5 parts by weight of water.
- glass beads e.g., 1 to 5 beads
- the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the method includes the steps of dissolving the cyclic peptide compound in an amorphous state in DMSO to obtain a solution, freeze-drying the solution to obtain a freeze-dried product of the cyclic peptide compound, and adding a mixture of water and a polar organic solvent to the freeze-dried product to obtain a crystal of the cyclic peptide compound (hereinafter, also referred to as "embodiment 5").
- polar organic solvent used in embodiment 5 include DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, propylene glycol, and mixed solvents thereof, with acetone being a more preferred example.
- the mixing ratio of water to polar organic solvent in the mixture of water and polar organic solvent can be 0.5 to 10 parts by weight of water per 1 part by weight of polar organic solvent, preferably 1 to 7 parts by weight of water, and more preferably 1 to 5 parts by weight of water.
- the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the method includes a step of heating the crystal of the cyclic peptide compound to obtain another crystalline polymorph of the cyclic peptide compound (hereinafter, also referred to as "embodiment 6").
- the heating temperature is, for example, 30 to 350°C, and preferably 30 to 120°C.
- a step of filtering the crystals may be further included.
- a step of drying the crystals may be further included.
- the crystals of the cyclic peptide compound produced by the method of the present invention are formed as solvate crystals in a solvent, and are obtained as hydrate crystals after the filtering and/or drying steps. In another embodiment, the crystals of the cyclic peptide compound produced by the method of the present invention are obtained as hydrate crystals by absorbing moisture from the atmosphere after the filtering and/or drying steps.
- compositions containing cyclic peptide compounds in one embodiment, the present invention relates to compositions containing a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, which contains 1.5 w/w% or less of the cyclic dimer of formula (1a) as an impurity.
- the present invention relates to a composition
- a composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the compound contains the cyclic dimer of formula (1a) as an impurity at a ratio of 0.001 w/w% or more.
- the present invention relates to a composition
- a composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof.
- the compound contains acetone in an amount of 0.001 w/w% or more.
- Measuring device Bruker Avance III 400 Internal standard substance: 3,5-bis(trifluoromethyl)benzoic acid Measurement conditions ( 19 F-NMR): CDCl 3 or DMSO-d 6 , 24.8° C., pulse angle 90°, digital resolution 0.24 Hz, relaxation Time: 15 seconds, no spin, 64 times
- Sample preparation method 1 A mixture containing the target compound was diluted with acetonitrile.
- Sample preparation method 2 The mixture containing the target compound was diluted with a mixture of acetonitrile and water in a ratio of 9:1.
- Sample preparation method K A mixture containing the target compound was diluted with a mixture of acetonitrile and n-propylamine in a ratio of 100:1.
- the reaction conversion rate was calculated by one of the following formulas using the area values of the raw materials and the target product, or the area values of the raw materials, the area values of the raw materials and the target product, or the area values of the raw materials before the reaction and the area values of the raw materials after the reaction, all calculated by HPLC analysis.
- Reaction conversion rate (%) 100 - (area value of raw material after reaction / area value of raw material before reaction x 100)
- N-methyl-2-pyrrolidone (17.0 L) and compound 3 (2.44 kg) were added to a nitrogen-purged reaction vessel at room temperature and stirred.
- Sarcosine tert-butyl ester hydrochloride (0.87 kg) and HATU (2.18 kg) were then added at 20°C and stirred for 30 minutes.
- DIPEA (1.85 kg) was added dropwise over 60 minutes at an internal temperature of 15-20°C.
- the reaction mixture was stirred at 20-25°C for 3 hours and then diluted with methyl tert-butyl ether (48.8 L).
- the organic layer was washed with water (48.8 L x 2) and 5% saline (24.4 L), and then concentrated under reduced pressure to obtain a concentrated, dry product containing compound 4.
- the combined organic layer was washed with 5% saline (14.9 L x 2), and the organic layer was concentrated under reduced pressure at 30 ⁇ 5°C.
- 2-Chlorotrityl chloride resin (1.12 mmol/g, 70 g, 78.4 mmol) and DCM (560 mL) were placed in a filter-equipped reaction vessel (1 L) and left to stand at room temperature for 30 minutes. After the DCM was filtered under reduced pressure, (3S)-4-(dimethylamino)-3-[9H-fluoren-9-ylmethoxycarbonyl (methyl)amino]-4-oxy-butanoic acid (23.4 g, 56.0 mmol) dissolved in DCM (140 mL) was added and washed with DCM (140 mL).
- DIPEA (27.4 mL) was added to the reaction vessel, and after stirring for 45 minutes, DCM (140 mL) was added and stirred at room temperature for 105 minutes. The reaction solution was filtered under reduced pressure and washed with DCM (280 mL x 2). A solution of methanol (28.0 mL) and DIPEA (14.0 mL) in DMF (238 mL) was added to a reaction vessel and stirred at room temperature for 120 minutes. The reaction solution was filtered under reduced pressure, and then IPA (280 mL) was added and stirred. After 15 minutes, the reaction solution was filtered under reduced pressure, and DMF (280 mL) was added and stirred for 15 minutes.
- Example 1-14 After the reaction solution was filtered under reduced pressure, the entire amount of Compound 14-resin, excluding the amount used for measuring the loading amount, was used to proceed to Example 1-14.
- the amount of amino acid supported on the resin was calculated as follows. The obtained compound 14-resin (4.76 mg) was placed in a reaction vessel, and 20% Pip/DMF solution (50 mL) was added and shaken at room temperature for 1 hour. The absorbance (301 nm) of the solution was measured (Shimadzu, UV-1600PC (cell length 1.0 cm)), and the amount of compound 14-resin supported was calculated to be 0.632 mmol/g.
- the extension reaction was carried out by adding Fmoc-MeGly(cPent)-OH (Cas number 187475-29-2, 42.5 g), oxyma (7.96 g) and DIC (34.9 mL) in a DMF solution (280 mL) to the resin, stirring for 5 minutes, and leaving it at room temperature for 16 hours.
- the solution of the extension reaction was filtered under reduced pressure, and IPA (280 mL) was added and stirred. After 10 minutes, the reaction solution was filtered under reduced pressure, and DMF (280 mL) was added and stirred for 10 minutes. After the reaction solution was filtered under reduced pressure, the entire amount of compound 15-resin except for the amount loaded for measurement was used to proceed to Example 1-15.
- the amount of amino acid supported on the resin was calculated as follows.
- the obtained compound 15-resin (4.91 mg) was placed in a reaction vessel, and 20% Pip/DMF solution (50 mL) was added and shaken at room temperature for 1 hour.
- the absorbance (301 nm) of the solution was measured (Shimadzu, UV-1600PC (cell length 1.0 cm)), and the amount of compound 15-resin supported was calculated to be 0.635 mmol/g.
- a small amount of compound 15 supported on the resin was used to cleave the compound from the resin with TFE/DCM (1/1), and the structure was confirmed by LC/MS.
- LCMS (ESI) of compound 15: Retention time: 2.53 minutes, m/z 536 [M+H] + (LCMS analysis conditions method 2)
- the extension reaction was carried out by adding Fmoc-MecVal-OH (Cas No. 1700368-07-5, 39.4 g) and oxyma (7.96 g) in DMF solution (280 mL), adding DIC (34.9 mL), stirring for 5 minutes, and then leaving it to stand for 94 hours.
- the solution of the extension reaction was filtered under reduced pressure, and the resin was washed with DMF (280 mL), IPA (280 mL), and DMF (280 mL).
- the washing solution was filtered under reduced pressure, and the obtained compound 16-resin was used in its entirety except for the amount measured for the loading amount, and then proceeded to Example 1-17.
- the amount of amino acid supported on the resin was calculated as follows.
- reaction conversion rate calculation formula 1 The external temperature of the reaction vessel was set to 40°C, and the mixture was concentrated under reduced pressure. After concentration under reduced pressure, DCM (214 mL) was added and the operation of concentrating under reduced pressure was repeated twice. The resulting concentrate was dried under reduced pressure overnight to obtain a concentrated, dried product containing compound 17 (45.5 g, 95% yield).
- the obtained resin was subjected to an elongation reaction of compound 17.
- the elongation reaction was performed by sequentially adding a DCM solution (280 mL) of compound 17 (46.0 g) obtained in Example 1-16 and Collidine (74.0 mL) and stirring for 5 minutes, and leaving it at room temperature for 4 hours. After the extension reaction solution was filtered under reduced pressure, the resin was washed with DCM (280 mL), and the entire amount of the obtained Compound 19-resin, excluding the amount used for measuring the loading amount, was used in Example 1-18.
- the amount of amino acid supported on the resin was calculated as follows.
- the obtained resin was subjected to the extension reaction of Cbz-Hph(4-CF3-3-OMe)-OH.
- the extension reaction was carried out by adding a DMF solution (280 mL) of Cbz-Hph(4-CF3-3-OMe)-OH (46.1 g) and oxyma (7.96 g) and DIC (34.9 mL) to the resin, stirring for 5 minutes, and then leaving it to stand for 3.5 hours. After the extension reaction solution was filtered under reduced pressure, the resin was washed twice with DMF (280 mL). IPA (280 mL) was added and stirred for 15 minutes. After draining the solution, DCM (280 mL) was added and stirred for 15 minutes.
- the concentrated dry matter containing compound 20 (80.7 g) and compound 13 (45.8 g) were added to the reaction vessel. After replacing with nitrogen, 2-MeTHF (484 mL), DIPEA (64.5 mL) and DMF (121 mL) were added at room temperature and stirred. After confirming complete dissolution, HATU (48.0 g) was added at room temperature. After 1 hour, the reaction mixture was sampled and sample preparation (sample preparation method 2) was performed, and it was confirmed that the reaction conversion rate was 98% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). The external temperature of the reaction vessel was set to 5°C, and 2.5% aqueous ammonia solution (484 mL) was added to the reaction vessel.
- the concentrated dry product including the concentrated dry product of compound 19 obtained in Example 1-20 (111 g) and 2-MeTHF (666 mL) were added in sequence and stirred at room temperature. After confirming dissolution, the mixture was cooled to an external temperature of 5°C, hexamethyldisilazane (69.5 mL) was added, and trimethylsilyl trifluoromethanesulfonate (44.9 mL) was added so that the internal temperature did not exceed 15°C. After the addition was completed, the external temperature was raised to 10°C and stirred for 1 hour and 20 minutes.
- reaction conversion rate to compound 22 was 99% (calculation formula 1 for reaction conversion rate).
- sample preparation method 2 a mixed solution of 5% aqueous sodium carbonate solution (555 mL) and 5% saline (333 mL) was added so that the internal temperature did not exceed 25°C.
- aqueous layer was discharged by liquid separation.
- the obtained organic layer was washed with a 10% aqueous sodium hydrogen sulfate solution (555 mL), a 5% aqueous sodium carbonate solution (555 mL), and a 5% saline solution (555 mL).
- Example 1-22 Compound 23: tert-butyl 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanoyl 2-(4-(trifluoromethyl)phenyl)propanoyl)-methyl-amino]-2-cyclopentyl-acetyl-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl-methyl-amino]pentanoyl-amino]-3-methyl-
- reaction conversion rate was confirmed to be 98% or more by HPLC analysis (calculation formula 1 for reaction conversion rate).
- a 2.8% aqueous ammonia solution (564 mL) was added to the reaction vessel at room temperature. After the aqueous layer was discharged by liquid separation, a 10% aqueous sodium hydrogen sulfate solution (564 mL) was added to the organic layer and stirred. After liquid separation, the liquid was separated and the aqueous layer was discharged. The obtained organic layer was washed successively with a 5% aqueous sodium carbonate solution (564 mL) and a 5% saline solution (564 mL).
- the concentrated dry solid containing compound 23 (90.8 g), L-cysteine (6.3 g), and 2-MeTHF (363 mL) were added in sequence to the reaction vessel. After replacing the reaction vessel with nitrogen, the external temperature was set to 20 ° C. Hexamethyldisilazane (119 mL) was added while stirring. Then, trimethylsilyl trifluoromethanesulfonate (93.5 mL) was added over 20 minutes. After stirring for 25 minutes, the temperature was raised to 50 ° C. After stirring for 8 hours at an internal temperature of 50 ° C, the reaction vessel was cooled to room temperature.
- the deprotection reaction of the Cbz group is generally carried out under catalytic hydrogen reduction conditions in the presence of a metal catalyst such as palladium/carbon.
- a metal catalyst such as palladium/carbon.
- the present inventors have discovered a method that makes it possible to deprotect the Cbz group without reducing the olefin in the molecule by using the conditions described in Example 1-23, i.e., the TMSOTf/HMDS conditions.
- Example 1-24-1 Compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.5.1.0 4,8 .0 26,30 Synthesis of ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide (cyclization position B)
- the external temperature of the reaction vessel was set to 40°C, and the mixture was concentrated and dried while stirring.
- the external temperature of the reaction vessel was cooled to 25°C, and isopropyl acetate (627 mL) and 2.5% aqueous ammonia solution (627 mL) were added to the obtained concentrate and stirred.
- Example 1-25 After discharging the aqueous layer by liquid-liquid separation, 10% aqueous sodium hydrogen sulfate solution (627 mL) was added to the obtained organic layer and stirred. After discharging the aqueous layer by liquid-liquid separation, the obtained organic layer was washed successively with 5% aqueous disodium hydrogen phosphate solution (627 mL x 2) and 5% aqueous sodium chloride solution (627 mL). The obtained organic layer was washed with 0.5% aqueous sodium chloride solution (627 mL x 2). The external temperature was set to 40°C, and the obtained organic layer was concentrated under reduced pressure to obtain 92.23 g of a concentrated and dried product containing compound 1. The obtained concentrated and dried product containing compound 1 was used in Example 1-25.
- Example 1-24-2 Compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.5.1.0 4,8 .0 26,30 Synthesis of ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide (cyclization position B)
- HATU (72.6 mg) and acetonitrile (1.80 mL) were added to the reaction vessel.
- a solution of the concentrated dry product containing compound 24 (100 mg) obtained in Example 1-23 and acetonitrile (5.5 mL) containing DIPEA (40 ⁇ L) was added dropwise over 5 hours and 42 minutes.
- the reaction mixture was sampled and prepared as a sample (sample preparation method 2).
- HPLC of compound 1 retention time: 3.99 minutes (HPLC analysis condition method 1) Cyclic dimer HPLC: Retention time: 4.80 minutes (HPLC analysis condition method 1)
- acetone/heptane/hydrate crystals (Form F) (about 1.00 mg) of compound 1 obtained by the same operation as in Example 3-8 were added to the reaction vessel and stirred at 35 ° C. for 23 hours. The mixture was cooled to 25°C and stirred for another 6 hours. Heptane (4.90mL) was added over 1 hour and stirred at 25°C for 14 hours. Heptane (4.90mL) was further added over 1 hour and stirred for 3 hours. Finally, heptane (4.90mL) was added over 2 hours and stirred for 3 hours.
- the reaction mixture was filtered under reduced pressure, and the obtained crystals were washed with a mixture of acetone (7.76mL) and heptane (11.6mL).
- the obtained crystals were dried for 16 hours with the external temperature set to 40°C.
- the dried powder was collected, and a white powder (6.8g, Form A) was obtained.
- Example 1-26 Compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.5.1.0 4,8 .0 26,30 Synthesis of ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide (cyclization position A)
- Example 2-2 Compound 27: tert-Butyl (S)-N-(2-(but-3-en-1-ylamino)-3-(4-(trifluoromethyl)phenyl)propanoyl)-N-methylglycinate hydrochloride
- toluene (751 mL) and 1N NaOH aqueous solution (536 mL) were added to the reaction mixture, and the mixture was stirred for 30 minutes, after which the aqueous layer was discharged by liquid-liquid separation.
- the organic layer was stored overnight at room temperature. After storage, 5% aqueous sodium carbonate solution (536 mL) was added to the organic layer, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation. Next, 5% aqueous sodium dihydrogen phosphate solution (751 mL) was added to the organic layer, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation.
- 5% aqueous sodium dihydrogen phosphate solution (751 mL) was added to the organic layer once more, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation.
- 5% saline solution (751 mL) was added to the organic layer, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation.
- the organic layer was stored overnight at an external temperature of 5° C. After storage, the organic layer was concentrated under reduced pressure at 40° C. until it became about 215 mL. Toluene (215 mL) was added to the concentrated liquid, and concentrated under reduced pressure at 40° C. until it became about 215 mL, and this operation was repeated twice.
- the precipitated inorganic salt was filtered, the target substance and toluene in the obtained filtrate were quantified, and toluene was added so that the amount was 296 mL.
- pyridine hydrochloride 43.28 g
- acetonitrile 148 mL
- Crystal precipitation was confirmed during the dropwise addition.
- toluene (1.7 L) was added, and after stirring for 1 hour, the external temperature was lowered to 0 ° C. and stirred for another 2 hours.
- Example 2-3 Compound 28: 2-[[(2S)-2-[(4Z,7S)-7-[9H-Fluoren-9-ylmethoxycarbonyl (methyl)amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanyl]-methyl-amino]acetic acid
- Toluene (583 g) and 5% aqueous sodium hydrogen sulfate (2066 g) were added to N-cyclohexylcyclohexanaminium (2S,3S)-2- ⁇ [(benzyloxy)carbonyl]amino ⁇ -3-methylpentanoate (135 g), and the mixture was stirred at room temperature for 10 minutes, and the organic layer was separated. The resulting organic layer was washed with 5% aqueous sodium hydrogen sulfate (2066 g) and 5% saline (1397 g) in that order, and the solvent was distilled off under reduced pressure.
- Example 2-7-1 The solvent was distilled off from 9.5346 g of this solution under reduced pressure, and heptane (100 mL) was added to the resulting residue.
- the mixture was dissolved at an external temperature of 50° C., and the seed crystals (11.0 mg) obtained in Example 2-7-1 were added at an internal temperature of 40° C.
- the mixture was stirred at an external temperature of 42° C. for 15 minutes, at an external temperature of 43° C. for 13 minutes, and at an external temperature of 44° C. for 17 minutes, then the external temperature was cooled to 0° C. at a rate of 12° C. per hour, and further stirred for 1.5 hours at an external temperature of 0° C.
- Example 2-7-1 Compound 13: Synthesis of seed crystals of tert-butyl N-methyl-L-norvalyl-L-isoleucyl-L-prolinate A part of the solution containing the title compound obtained in the reaction of Example 2-7 was concentrated under reduced pressure, and heptane (7622 ⁇ L) was added to the resulting residue (0.3811 g). The resulting solid was dissolved at an external temperature of 50° C., cooled to room temperature with stirring, and heptane (3811 ⁇ L) was added to the resulting slurry and stirring was continued.
- 2-Methyltetrahydrofuran (95 g) was added to the obtained residue, and the operation of concentrating under reduced pressure was repeated twice.
- 2-Methyltetrahydrofuran (95 g), acetonitrile (75 g), DIPEA (35.46 g, 274 mmol), and dimethylamine hydrochloride (7.88 g, 96.6 mmol) were added to the obtained residue (47.91 g) at 25° C.
- a solution of propylphosphonic anhydride in 2-methyltetrahydrofuran (50.4 wt %, 61.33 g, 97.1 mmol) was added dropwise over 1 hour and 30 minutes.
- a reaction vessel was charged with 10% palladium carbon (54.33% wet, 3.39 g, 1.45 mmol, 3 mol% on Pd metal basis) and 2-methyltetrahydrofuran (75 g). The atmosphere was replaced with nitrogen and hydrogen at 25°C, and the mixture was stirred under a hydrogen atmosphere (0.40 MPaG) for 2 hours. A solution of compound A11 (42.39 g) obtained in Example 2-8-1 and 2-methyltetrahydrofuran (22 g) were added. After stirring for 1 hour and 30 minutes under a hydrogen atmosphere (0.20 MPaG), a sample was taken, and the completion of the reaction was confirmed by HPLC analysis.
- reaction conversion rate calculation formula 1 The reaction mixture was sampled and sample preparation (sample preparation method K) was performed, and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (reaction conversion rate calculation formula 1).
- 5% potassium carbonate aqueous solution (447 mL) and N,N-dimethyl-4-aminopyridine (90 g) were added, and the mixture was further stirred for 1 hour and 30 minutes.
- the internal temperature was cooled to 25°C, toluene (373mL) was added, and the aqueous layer was discharged by liquid separation.
- the organic layer was washed with 4% aqueous sulfuric acid solution (447mL x 2) and 5% aqueous sodium carbonate solution (447mL x 2).
- a THF solution (163.97 g) of compound 26 obtained in Example 2-8, THF (665 mL), and 4.57 wt. % Pd/C (14.71 g) were placed in a pressure-resistant reaction vessel (5 L), and the external temperature was set to 25 ° C. and stirred. The atmosphere was replaced with 0.20 MPa nitrogen three times, and further replaced with 0.20 MPa hydrogen three times, and stirred for 1 hour under a 0.20 MPa hydrogen atmosphere.
- the reaction mixture was sampled and prepared as a sample (sample preparation method 1), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate).
- the inside of the reaction vessel was replaced with 0.20 MPa nitrogen three times, and Pd/C was removed by filtration.
- reaction mixture was sampled and prepared as a sample (sample preparation method 1), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (calculation formula 1 for reaction conversion rate).
- the inside of the reaction vessel was replaced with nitrogen at 0.20 MPa three times, and Pd/C was removed by filtration. The removed Pd/C was washed three times with 2-methyltetrahydrofuran (232 mL). The filtrate and the washings were mixed and concentrated under reduced pressure to 182 mL at an external temperature of 50° C. 2-Methyltetrahydrofuran (774 mL) was added, and the mixture was concentrated under reduced pressure to 182 mL.
- a part of the obtained concentrated liquid (5.5 g) was placed in a reaction vessel (50 mL), and 2-methyltetrahydrofuran (1.34 mL) was added.
- the obtained solution was heated to an internal temperature of 67.5°C and stirred.
- Heptane (35.6 mL) was added, and the crystals of compound 33 (322 mg) obtained by the same operation as in Example 2-11 were suspended in heptane (0.40 mL) and added to the reaction vessel.
- the mixture was stirred at an internal temperature of 67.5°C for 1 hour or more, cooled to 57.5°C over 2 hours or more, and further stirred for 1 hour or more.
- the internal temperature was further cooled to 47.5°C over 1 hour or more, and further stirred for 1 hour or more.
- the internal temperature was further cooled to below 10°C over 2 hours or more, and further stirred for 1 hour or more.
- the reaction mixture was filtered through a Kiriyama funnel, and the obtained crystals were washed twice with heptane (10.0 mL) and with a heptane/2-methyltetrahydrofuran (6/1) solution (10.0 mL) cooled to 10°C.
- the obtained crystals were dried for 2 hours under reduced pressure with an external temperature set to 40°C. The dried powder was collected, and a white powder (1.39 g) was obtained.
- compound 34 (206.8 g) was obtained from 4-bromo-2-methoxy-1-(trifluoromethyl)benzene (190 g).
- Nickel bromide trihydrate (1.23 kg) and 1,3-dimethyl-2-imidazolidinone (98.8 kg) were mixed as a slurry and charged into a reaction vessel (1000 L), followed by charging 4,4'-di-tert-butyl-2,2-dipyridyl (1.21 kg) and stirring at 20.8-22.7°C for 28 minutes.
- reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate).
- sample preparation method 1 15% aqueous ammonium chloride solution (332 kg) was added dropwise.
- Toluene (135 kg) was added to the resulting mixture, which was stirred for 30 minutes or more, and then filtered using Celite. After washing the Celite with toluene (135 kg), the aqueous layer was discarded.
- reaction conversion rate calculation formula 1 The reaction mixture was sampled and sample preparation (sample preparation method 1), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (reaction conversion rate calculation formula 1).
- the mixture was filtered, and the Pd/C residue was washed with toluene (26.7 kg). The filtrate was obtained again by the same operation, then combined, and concentrated to 155 L at an external temperature setting of 40 ° C., to obtain 155 kg of a solution containing the title compound.
- LCMS (ESI) of compound 34- ⁇ 2: Retention time: 5.72 minutes, m/z 278.37 [M-Boc+H] + (LCMS analysis conditions method H)
- Nickel bromide trihydrate (0.198 g) and 1,3-dimethyl-2-imidazolidinone (15 mL) were mixed and charged as a slurry in a reaction vessel (100 mL), then 4,4'-di-tert-butyl-2,2-dipyridyl (0.195 g) was charged and stirred at 25°C for 30 minutes or more.
- reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate).
- 10% potassium hydroxide aqueous solution 35 mL was added dropwise at an internal temperature of 0 to 25°C.
- the reaction mixture was sampled and sample preparation (sample preparation method H) was performed, and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula H for reaction conversion rate).
- the reaction mixture was cooled, and water (186 kg) was added dropwise over 80 minutes at an internal temperature of 25.9°C to 32.0°C. After removing the organic layer, the mixture was washed three times with toluene (124 kg), and the pH was adjusted to 6.52 with 40% aqueous potassium phosphate tripotassium solution (95.7 kg). Acetonitrile (61.2 kg) was then added, and the pH was adjusted to 7.83 again using 40% tripotassium phosphate aqueous solution (25.4 kg). N-(benzyloxycarbonyloxy)succinimide (16.0 kg) was added to the reaction mixture after pH adjustment to start the reaction.
- reaction mixture was sampled and sample preparation (sample preparation method 1), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (calculation formula H for reaction conversion rate).
- the pH of the obtained reaction mixture was adjusted to 7.73 again using 40% tripotassium phosphate aqueous solution (44.7 kg), and then methyl-tert-butyl ether (46.3 kg) and heptane (42.4 kg) were added, and it was confirmed that the mixture was separated into three layers.
- the obtained organic layer was washed four times with a mixed solution of 24% sodium hydroxide aqueous solution (2.1 kg)-sodium chloride (12.4 kg)-water (110.0 kg), and the organic layer was sampled and sampled (sample preparation method 1), and it was confirmed by NMR analysis that the amount of residual acetonitrile was 0.44 v/w or less (calculation formula H for residual acetonitrile).
- the obtained organic layer was washed with a 0.2 M sodium hydroxide aqueous solution (62.1 kg), and then washed with a mixed solution of 24% sodium hydroxide aqueous solution (2.1 kg)-sodium chloride (3.1 kg)-water (72.3 kg).
- sample preparation method 1 The organic layer after washing was sampled and subjected to sample preparation (sample preparation method 1), and it was confirmed by HPLC analysis that the residual impurities were 0.10% or less.
- the obtained organic layer was washed with 1M hydrochloric acid (310 kg), and then the organic layer after washing was sampled and subjected to sample preparation (sample preparation method H), and it was confirmed by HPLC analysis that the residual impurities were 0.10% or less.
- the obtained organic layer was washed with 10% saline (166 kg), concentrated to 62 L, and then toluene (80.9 kg) was added and concentrated again to 62 L to carry out solvent replacement with toluene.
- ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt) 91.0 g was suspended in a toluene (2.51 kg)-heptane (511 g) mixed solution and charged. After cooling the internal temperature from 67.0 ° C. to 52.1 ° C. over 1 hour 38 minutes, stirring was continued for more than 1 hour, and then the internal temperature was cooled from 50.1 ° C. to 23.0 ° C. over 1 hour 55 minutes.
- sample preparation method H sample preparation method H
- the supernatant concentration was confirmed to be 2 mg / mL or less by HPLC analysis.
- the obtained slurry was filtered and washed with a toluene (47.4 kg) - heptane (37.2 kg) mixed solution to obtain wet powder.
- the obtained wet powder was dried at an external temperature of 38 ° C. for 50 hours to obtain ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt) (25.6 kg).
- LCMS (ESI) of compound 34: Retention time: 5.89 minutes, m/z 368.41 [M-CO2-DCHA+H] + (LCMS analysis conditions method H)
- HMDS HMDS (19.0 g) was added, the external temperature of the reaction vessel was set to 0 ° C, and the mixture was stirred for 15 minutes.
- TMSOTf (15.4 g) was slowly added dropwise so that the internal temperature did not exceed 20 ° C.
- the external temperature of the reaction vessel was set to 20°C, and the reaction mixture was stirred for 1 hour, then diluted with 2-methyltetrahydrofuran (244mL), and the external temperature of the reaction vessel was set to 10°C.
- 5% aqueous solution of dipotassium hydrogen phosphate (478mL) was slowly added dropwise, and stirring was stopped and the aqueous layer was discharged.
- Example 2-16 Compound 21: Synthesis of tert-butyl (2S)-1-[(2s,3s)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate
- the obtained organic layer was washed in the order of 10% aqueous ammonia (280 mL), 4% dilute sulfuric acid (280 mL), 4% dilute sulfuric acid (280 mL), and 5% aqueous sodium carbonate (280 mL).
- the obtained organic layer was concentrated under reduced pressure to obtain a 2-methyltetrahydrofuran solution of compound 21 (132.34 g).
- Example 2-18 Compound 38: Synthesis of tert-butyl [(S)-2-[(S)-3-[(S)-2-(1-[(2S,4R)-1-[(S)-2-amino-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-N-methylpyrrolidine-2-carboxamido]-N-methylcyclobutane-1-carboxamido)-2-cyclopentyl-N-methylacetamido]-4-[dimethylamino]-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleucyl-L-proline tert-butyl
- a 2-MeTHF solution (17.6 kg) of compound 37 (3.41 kg) was added to the reaction vessel at room temperature, followed by the addition of 2-MeTHF (12.3 kg), compound 28 monotoluene solvate (2.38 kg), and acetonitrile (6.20 kg) in that order.
- the external temperature was set to 5°C, and NMM (1.26 kg) and HATU (1.65 kg) were added at an internal temperature of 30°C or less, followed by stirring for 1 hour at an internal temperature of 25°C.
- a 5% aqueous potassium carbonate solution (23.9 kg) and 1-methylimidazole (234 g) were added and stirred at room temperature for 30 minutes, and the aqueous layer was discharged by liquid-liquid separation.
- the resulting organic layer was washed with 10% aqueous ammonia (23.0 kg), 5% aqueous sodium hydrogen sulfate (23.9 kg), and 5% aqueous sodium carbonate (23.9 kg), and then concentrated at an external temperature of 40°C until the volume reached 10 L.
- Acetonitrile (16.2 kg) was added to the concentrated solution, and the concentration was repeated twice until the volume reached 10 L, yielding a solution of Compound 38 in acetonitrile (19.4 kg).
- HATU (949 g) and acetonitrile (25.6 kg) were added to the reaction vessel and dissolved to prepare an acetonitrile solution of HATU.
- Compound 40 (1.31 kg) in acetonitrile (5.52 kg), acetonitrile (19.1 kg), and N-methylmorpholine (504 g) were added to another vessel to prepare an acetonitrile solution of compound 40.
- the acetonitrile solution of compound 40 prepared in the acetonitrile solution of HATU at an external temperature of 25° C. was dropped over 4 hours and 22 minutes, and acetonitrile (1.04 kg) was used to wash it in. After dropping, the mixture was stirred for another hour.
- the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (LC analysis condition method cyc) (calculation formula 1 for reaction conversion rate).
- the external temperature was set to 40°C, and the reaction mixture was concentrated to approximately 12.5 L.
- MTBE (14.5 kg) and 2.5% aqueous ammonia (13.3 kg) were added to the resulting concentrate at room temperature and stirred for 34 minutes.
- cyclization positions B and C in the present invention have a smaller amount of cyclic dimer impurities than the existing cyclization position A, and are therefore efficient cyclization positions for the production of compound 1.
- Example 3 Crystallization Example 3-1 Amorphous compound 1 (77.4 mg) was dissolved in dimethyl sulfoxide (0.387 mL), and the resulting solution (0.015 mL) was freeze-dried at -20°C for 3 days. A 2-butanone-heptane mixture (1:4 (v/v), 0.015 mL) was added to the resulting freeze-dried product, and the mixture was shaken and stirred at room temperature for 7 days to obtain crystals of compound 1. The resulting crystals were confirmed to be dimethyl sulfoxide/heptane/hydrate crystals (Form G) by single crystal X-ray structural analysis (Examples 4-7). The structure is shown in Figure 1.
- Example 3-2 Amorphous Compound 1 (11.4 mg) was dissolved in acetone (34.2 ⁇ L), and about 0.1 mg of the crystals of Example 3-1 was added. Heptane (22.8 ⁇ L) was then added, and the mixture was shaken and stirred at room temperature for 3 hours. Heptane (22.8 ⁇ L) was further added, and the mixture was shaken and stirred at room temperature for 2 hours. Heptane (22.8 ⁇ L) was further added, and the mixture was shaken and stirred at room temperature for 18 hours, after which it was filtered and dried under reduced pressure. The obtained crystals were hydrate crystals of Compound 1 (Form A). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in FIG. 2.
- Example 3-3 Amorphous compound 1 (10.7 mg) was dissolved in 2-propanol (32.1 ⁇ L), and about 0.1 mg of the crystals obtained in Example 3-1 was added. Heptane (21.4 ⁇ L) was then added, and the mixture was shaken and stirred at room temperature for 3 hours. Heptane (21.4 ⁇ L) was further added, and the mixture was shaken at room temperature for 2 hours, after which heptane (21.4 ⁇ L) was added, and the mixture was shaken and stirred at room temperature for 3 hours. Heptane (21.4 ⁇ L) was further added, and the mixture was shaken and stirred at room temperature for 15 hours to obtain a crystal of compound 1 (Form E).
- the obtained crystal was subjected to single crystal X-ray structural analysis (Example 4-8), and it was confirmed that the crystal was 2-propanol/heptane/hydrate.
- the crystal structure is shown in FIG. 3.
- the obtained Form E was dried under reduced pressure, and powder X-ray diffraction measurement (Example 4-3) was performed.
- the results are shown in FIG. 4.
- the diffraction pattern showed Form A, and it was confirmed that 2-propanol/heptane/hydrate (Form E) was transformed into a hydrate crystal (Form A) by drying under reduced pressure.
- Examples 3-4 To the Form A crystals of Compound 1 (5.9 mg), an ethanol-heptane mixture (1:1 (v/v), 0.017 mL) and one glass bead were added. The mixture was stirred at 37° C. and 2000 rpm for 7 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). The powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in FIG. 5.
- Examples 3-5 To the Form A crystals of Compound 1 (5.8 mg), an ethyl acetate-heptane mixture (1:1 (v/v), 0.017 mL) and one glass bead were added. The mixture was stirred at 37° C. and 2000 rpm for 7 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). The powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in FIG. 6.
- Example 3-6 Amorphous compound 1 (9.0 mg) was dissolved in ethanol (27.0 ⁇ L), heptane (27.0 ⁇ L) was added, and the mixture was shaken and stirred at room temperature for 4 minutes. When about 0.1 mg of the crystals obtained in Example 3-4 was added and the mixture was shaken and stirred at room temperature for 1 minute, a crystal of compound 1 was obtained. Single crystal X-ray structure analysis (Example 4-9) was performed, and it was confirmed that the crystal was an ethanol/hydrate (Form H). The crystal structure is shown in FIG. 7. The obtained Form H was dried under reduced pressure, and powder X-ray diffraction measurement (Example 4-3) was performed. The results are shown in FIG. 8. The diffraction pattern shows Form B, and it was confirmed that the ethanol/hydrate (Form H) was transformed into a hydrate crystal (Form B) by drying under reduced pressure.
- Examples 3-7 Amorphous compound 1 (10.9 mg) was dissolved in ethyl acetate (32.7 ⁇ L), heptane (32.7 ⁇ L) was added, and the mixture was shaken and stirred at room temperature for 4 minutes. About 0.1 mg of the crystals of Example 3-5 was added, and the mixture was shaken and stirred at room temperature for 1 minute to obtain crystals of compound 1.
- Single crystal X-ray structure analysis (Example 4-9) was performed, and it was confirmed that the crystals were ethyl acetate/hydrate (Form C). The crystal structure is shown in FIG. 9.
- the obtained Form C was dried under reduced pressure, and powder X-ray diffraction measurement (Example 4-3) was performed. The results are shown in FIG. 10.
- the diffraction pattern shows Form B, and it was confirmed that the ethyl acetate/hydrate (Form C) was transformed into a hydrate crystal (Form B) by drying under reduced pressure.
- Examples 3-8 Amorphous compound 1 (112.3 mg) was dissolved in acetone (337 ⁇ L), and about 0.1 mg of Form A crystals was added as seed crystals. Heptane (112 ⁇ L) was added and stirred at room temperature for 2 minutes using a stirrer. Heptane (112 ⁇ L) was added again and stirred at room temperature for 10 minutes. After confirming the dissolution of the seed crystals, about 0.1 mg of Form A crystals was added again and stirred at room temperature for 1 hour. Heptane (225 ⁇ L) was then added and stirred at room temperature for 20 minutes. About 0.1 mg of Form A crystals were further added and stirred at room temperature for 40 minutes.
- Examples 3-9 Amorphous Compound 1 (61.7 mg) was dissolved in 2-propanol (185 ⁇ L), heptane (123 ⁇ L) was added, and the mixture was stirred at room temperature for 3 minutes using a stirrer. After adding about 0.1 mg of Form A crystals as seed crystals, the mixture was stirred at room temperature for 30 minutes. Thereafter, heptane (123 ⁇ L) was added, and the mixture was stirred at room temperature for 2 hours. Furthermore, heptane (185 ⁇ L) was added, and the mixture was shaken and stirred at room temperature for 18 hours. The reaction product was filtered under reduced pressure and dried in vacuum for 1 day to obtain 23.3 mg of Form A crystals.
- Powder X-ray diffraction measurement (Example 4-3) was performed on Form A. The measurement results are shown in FIG. 14. The results of simultaneous thermogravimetry and differential thermal analysis of Form A are shown in FIG. 15, and the results of 1 H-NMR measurement are shown in FIG. 16. Since the powder X-ray diffraction pattern in FIG. 14 coincided with the pattern of Example 3-8, a weight loss of 2 wt % was observed in FIG. 15, and only 1 wt % of 2-propanol was observed in FIG. 16, it was determined that Form A was a hydrate and that 2-propanol was present as a residual solvent.
- Example 3-10 Amorphous compound 1 (60.1 mg) was dissolved in acetone (180 ⁇ L) and stirred using a stirrer. Heptane (120 ⁇ L) was added and stirred at room temperature for 1 minute. About 0.1 mg of Form A crystals was added and stirred for 30 minutes. Heptane (120 ⁇ L) was added again and stirred at room temperature for 1 minute. About 0.1 mg of Form A crystals was added again and stirred at room temperature for 2.5 hours. Heptane (180 ⁇ L) was further added and stirred at room temperature for 18 hours.
- Form F acetone/heptane/hydrate crystals of compound 1 (Form F) were obtained as wet powder.
- the obtained Form F was enclosed in a glass capillary, and powder X-ray diffraction measurement (Example 4-2) was performed, confirming the following main peaks: 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47°.
- the measurement results are shown in FIG.
- Example 3-11 Amorphous Compound 1 (10.0 mg) was dissolved in acetone (30 ⁇ L) and stirred using a stirrer. Heptane (20 ⁇ L) was added and stirred at room temperature for 1 minute using a stirrer. Approximately 0.1 mg of Form A crystal was added and stirred for 5 minutes. Heptane (20 ⁇ L) was added again and stirred at room temperature for 2 hours, resulting in a high-quality single crystal. The obtained crystal was confirmed to be acetone/heptane/hydrate (Form F) by single crystal structure analysis (Example 4-8). The crystal structure is shown in FIG. 18.
- Example 3-12 When the powder X-ray diffraction measurement (Example 4-1) of the crystal (Form A) obtained in Example 1-25 was carried out, the main peaks were confirmed as 6.93 °, 7.56 °, 8.26 °, 9.00 °, 9.58 °, 10.35 °, 11.35 °, 12.26 °, 12.85 °, 13.51 °, 14.12 °, 14.69 °, 15.46 °, 15.92 °, 17.43 °, and 17.73 °. The measurement results are shown in FIG. 19. The results of simultaneous thermogravimetry and differential thermal analysis are shown in FIG. 20.
- Form B was maintained at a relative humidity of 30% to 90%, but was transformed to Form Y at a relative humidity of 0%.
- Form Y has major peaks at 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24°.
- Example 3-14 The crystal obtained in Example 3-13 was confirmed to be a hydrate crystal (Form B) by single crystal X-ray structure analysis (Example 4-9). The crystal structure is shown in FIG.
- Example 3-15 Amorphous compound 1 (77.4 mg) was dissolved in dimethyl sulfoxide (0.387 mL), and the solution (0.015 mL) was freeze-dried at -20°C for 3 days. A 1,4-dioxane-heptane mixture (1:4 (v/v), 0.015 mL) was added to the freeze-dried product and stirred with shaking at room temperature for 7 days to obtain crystals of compound 1. The obtained crystals were confirmed to be 1,4-dioxane/hydrate crystals (Form D) by single crystal X-ray structural analysis (Example 4-8). The crystal structure is shown in Figure 26.
- Example 3-16 The crystals of Compound 1 obtained in Example 3-8 (Form A, 6.8 mg) were dissolved in dimethyl sulfoxide (0.12 mL) and stirred at room temperature for 13 days by shaking. Water (1 ⁇ L) was then added and stirred at room temperature for 34 days to obtain crystals of Compound 1. The obtained crystals were confirmed to be dimethyl sulfoxide/hydrate crystals (Form L) by single crystal X-ray structure analysis (Example 4-8). The crystal structure is shown in FIG. 27. Powder X-ray diffraction measurement of the crystals obtained by the same procedure (Example 4-3) confirmed that Form L was a solvate crystal that became amorphous by drying under reduced pressure. The powder X-ray diffraction pattern is shown in FIG. 28.
- Example 3-17 Propylene glycol (0.12 mL) was added to the crystals of Compound 1 (Form B, 45.8 mg) and stirred at room temperature for 5 minutes. Propylene glycol (0.020 mL) was further added and stirred at 40° C. for 5 minutes, then propylene glycol (0.070 mL) was added and stirred at 40° C. for 5 minutes, and the mixture was allowed to stand for another 5 hours. Propylene glycol (0.070 mL) was further added and stirred at 35° C. overnight, then propylene glycol (0.070 mL) was added and stirred for about 1 minute to obtain wet powder of Compound 1.
- the obtained wet powder was confirmed to be Form M crystals by powder X-ray diffraction measurement (Example 4-3). It was confirmed by powder X-ray diffraction measurement that Form M crystals were transformed to Form N crystals when dried under reduced pressure at room temperature overnight.
- the powder X-ray diffraction patterns of Form M and Form N are shown in FIG. 29.
- the results of simultaneous thermogravimetry and differential thermal analysis are shown in FIG. 30. A weight loss of about 11% was observed at 157° C., which corresponds to 2.5 molecules of propylene glycol per compound 1. From the above, it was confirmed that Form M and Form N were propylene glycol solvates.
- Example 3-18 The crystals of Compound 1 (Form B, 12.66 mg) were dissolved in propylene glycol (0.10 mL) and stirred at room temperature for 20 days. Furthermore, about 0.1 mg of the wet powder obtained in Example 3-17 was added and stirred at room temperature for 2.5 hours. Propylene glycol (0.10 mL) was added, and the mixture was heated and stirred at 60° C. for 30 minutes on a hot plate, and then shaken and stirred at room temperature for 4.5 hours to obtain crystals of Compound 1. The obtained crystals were confirmed to be propylene glycol/hydrate crystals (Form M) by single crystal X-ray structure analysis (Example 4-8). The crystal structure is shown in FIG. 31.
- Example 3-19 The crystals (Form A) obtained in Example 1-25 were weighed in amounts of 7.773 mg, 7.258 mg, 8.665 mg, and 13.047 mg (total 36.7 mg) in open aluminum pans, and heated using a simultaneous thermogravimetric and differential thermal analyzer (Example 4-11). The obtained crystals were subjected to powder X-ray diffraction measurement (Example 4-2), and the main peaks were confirmed to be 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51°.
- the powder X-ray diffraction pattern of Form K is shown in FIG. 32.
- the results of simultaneous thermogravimetry and differential thermal analysis of the obtained Form K crystals are shown in Figure 33, and the results of 1 H-NMR measurement are shown in Figure 34.
- a weight loss of about 1.8 wt % was observed in Figure 33, while the 1 H-NMR measurement showed no peaks other than those corresponding to the measurement solvent DMSO (containing tetramethylsilane), Compound 1, and water. From these results, it was determined that Form K was a hydrate.
- Example 3-20 The crude solution with low purity was concentrated to dryness and dried under reduced pressure at 40°C to prepare a crude amorphous solid.
- the crude amorphous solid (20 mg) was dissolved in 2-propanol (60 ⁇ L) and stirred using a stirrer. Heptane (60 ⁇ L) was added. Approximately 0.1 mg of Form A seed crystals were added and stirred at room temperature for 1 hour. As no crystal precipitation was observed, further 2-propanol (20 ⁇ L) was added, which resulted in oiling out and no crystals being obtained.
- Example 3-21 The crude solution was concentrated to dryness and dried under reduced pressure at 40°C to prepare a crude amorphous solid.
- the crude amorphous solid (20 mg) was dissolved in ethanol (60 ⁇ L) and stirred using a stirrer. Heptane (60 ⁇ L) was added.
- Two identical solutions were prepared, one of which was added with about 0.1 mg of Form A seed crystals, and the other was added with about 0.1 mg of Form B seed crystals, and the mixture was stirred at room temperature for 1 hour. In both cases, crystals precipitated, and the mixture was filtered to obtain a wet powder.
- Form B was obtained despite the conditions in which Form A seed crystals were added.
- Form B was obtained under the conditions in which Form B seed crystals were added.
- Example 3-22 The crude solution (17.7 kg) was concentrated to 7 L at an external temperature of 50° C. A part of the concentrated solution (4.31 kg) was charged into a reactor, and ethanol (1.16 kg) was added. The external temperature was then set to 42° C. and the mixture was stirred. Purified water (1.01 kg) and seed crystals (Form B, 5.17 g) were added and stirred for 1 hour. Purified water (2.08 kg) was then added dropwise over 80 minutes. Purified water (1.04 kg) was further added dropwise over 40 minutes. The internal temperature was lowered to 25° C. or less over 48 minutes. The mixture was stirred overnight, filtered, and the crystals were washed twice with ethanol/purified water (2.50 L/1.66 L mixture).
- Example 4 Evaluation of crystals
- Example 4-1 Powder X-ray measurements of Examples 3-8, 3-12, 3-13, and 3-19 were carried out under the following conditions.
- Measurement device SmartLab System, D/Tex Ultra detector (manufactured by Rigaku Corporation) Radiation source: CuK ⁇ 1 Tube voltage: 45 kV Tube current: 200mA Scanning range: 4 to 50 degrees Sampling width: 0.005°
- Example 4-2 Powder X-ray measurements of Examples 3 to 10 were carried out under the following conditions to evaluate the crystal forms in the suspensions.
- Measurement device SmartLab System, D/Tex Ultra detector (manufactured by Rigaku Corporation) Radiation source: CuK ⁇ 1 Tube voltage: 45 kV Tube current: 200mA Scanning range: 4 to 50 degrees Sampling width: 0.005° Measurement: The sampled suspension was packed into a capillary for X-ray crystallography and measured.
- Measuring device D8 Discover, 2D VANTEC-500 solid state detector (manufactured by Bruker) Radiation source: CuK ⁇ Tube voltage, tube current: 50 kV, 1000 ⁇ A Measurement range: 5 to 31 degrees Exposure time: 40 seconds
- Example 4-4 Powder X-ray diffraction measurements of Example 3-12 were carried out under the following conditions, and the crystal form at each relative humidity was evaluated. Measurements were taken at 0% (FIG. 21(A)), 10% (FIG. 21(B)), 20% (FIG. 21(C)), 50% (FIG. 21(D)), and 90% (FIG. 21(E)) at points between when the relative humidity was lowered to 0% and when it was raised to 90%.
- Measurement equipment SmartLab System, D/Tex Ultra detector, water vapor generator HUM-SL (manufactured by Rigaku Corporation)
- Anticathode Cu Tube voltage: 45 kV Tube current: 200mA Scanning range: 4 to 35° Scanning speed: 5°/min Sampling width: 0.02° Humidity change conditions:
- Example 3-13 Powder X-ray diffraction measurements of Example 3-13 were carried out under the following conditions, and the crystal form at each relative humidity was evaluated. Measurements were taken at 0% (FIG. 24(A)), 30% (FIG. 24(B)), 50% (FIG. 24(C)), and 90% (FIG. 24(D)) at points between when the relative humidity was lowered to 0% and when it was raised to 90%.
- Measurement equipment SmartLab System, D/Tex Ultra detector, water vapor generator HUM-SL (manufactured by Rigaku Corporation)
- Anticathode Cu Tube voltage: 45 kV Tube current: 200mA Scanning range: 4 to 35° Scanning speed: 5°/min Sampling width: 0.02° Humidity change conditions:
- Examples 4-6 Thermogravimetry and differential thermal analysis (TG-DTA) of Examples 3-8, 3-9, 3-12, 3-13, 3-17, and 3-19 were carried out under the following conditions.
- Measurement device STA7200RV+AS-3T (Hitachi High-Tech Science) Measurement range: 30 to 350°C Heating rate: 10° C./min.
- Atmosphere nitrogen Measurement: A sample was weighed in an open aluminum pan, covered with a mesh and then the measurement was performed.
- Example 4-7 The single crystal X-ray structure analysis of Example 3-1 was carried out under the following conditions. Measurement device: Rigaku XtaLAB Synergy Custom with a VariMax Cu Diffractometer (manufactured by Rigaku Corporation) Anticathode: Cu Tube voltage: 40 kV Tube current: 30mA Temperature: -180°C Measurement: Measurement was performed using a strategy and exposure time that was believed to provide sufficient diffraction spots for structural analysis. Structural analysis: The Olex2 program was used, the initial structure was determined using SIR2008, and the structure was refined using the Full-matrix least-squares method (SHELXL-2018/3).
- Examples 4-8 The single crystal X-ray structure analyses of Examples 3-3, 3-11, 3-15, 3-16, and 3-18 were carried out under the following conditions.
- Measurement device Rigaku XtaLAB Synergy Custom with a VariMax Cu Diffractometer (manufactured by Rigaku Corporation)
- Anticathode Cu Tube voltage: 40 kV Tube current: 30mA
- Temperature: -180°C Measurement Measurement was performed using a strategy and exposure time that was believed to provide sufficient diffraction spots for structural analysis.
- Structural analysis Using the Olex2 program, initial structure determination was performed using the Dual Space method (SHELXD-2008), and structure refinement was performed using the Full-matrix least-squares method (SHELXL-2018/3).
- Examples 4-9 The single crystal X-ray structure analyses of Examples 3-6, 3-7, and 3-14 were carried out under the following conditions.
- Structural analysis Using the Olex2 program, initial structure determination was performed using the Dual Space method (SHELXD-2008), and structure refinement was performed using the Full-matrix least-squares method (SHELXL-2018/3).
- Examples 4-10 The 1 H-NMR measurements of Examples 3-8, 3-9, and 3-19 were carried out under the following conditions.
- Measuring device JNM-ECX500II (manufactured by JEOL)
- Measurement solvent DMSO-d6, contains 0.03% (v/v)
- TMS Measurement temperature: 295K
- Sample preparation A commercially available deuterated solvent was mixed with the compound to be measured to prepare a sample.
- Accumulation number and relaxation waiting time Measurement was performed the number of times (8, 16 or 512) that was considered to provide a sufficient s/n ratio, and for a time (60 seconds) sufficiently longer than the relaxation time.
- the residual solvent ratio (wt %) was calculated based on the integral value: signal area intensity ratio of each signal.
- Example 4-11 The sample preparation for Example 3-19 was carried out under the following conditions. Equipment used: STA7200RV+AS-3T (Hitachi High-Tech Science) Temperature range: 30 to 120°C Heating rate: 20° C./min (30-120° C.), maintained at 120° C. for 60 minutes. Atmosphere: nitrogen. Preparation: A sample was weighed in an open aluminum pan and heated.
- Example 5 Slurry conversion experiment
- Example 5-1 Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.0 mg) of Compound 1 were mixed, and an acetone-heptane mixture (1:1 (v/v), 0.030 mL) and one glass bead were added.
- the mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B).
- Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (A). It was confirmed that Form B crystals were the stable form in the acetone-heptane mixture (1:1 (v/v)).
- Example 5-2 Form A crystals (3.1 mg), Form B crystals (3.1 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and a 2-propanol-heptane mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (B). It was confirmed that Form B crystals were the stable form in the 2-propanol-heptane mixture (1:1 (v/v)).
- Example 5-3 Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and an ethanol-heptane mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred and shaken at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (C). It was confirmed that Form B crystals were the stable form in the ethanol-heptane mixture (1:1 (v/v)).
- Example 5-4 Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.0 mg) of Compound 1 were mixed, and an acetone-water mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (D). It was confirmed that Form B crystals were the stable form in the acetone-water mixture (1:1 (v/v)).
- Example 5-5 Form A crystals (3.0 mg), Form B crystals (3.1 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and a 2-propanol-water mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (E). It was confirmed that Form B crystals were the stable form in the 2-propanol-water mixture (1:1 (v/v)).
- Examples 5-6 Form A crystals (3.1 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and an ethanol-water mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (F). It was confirmed that Form B crystals were the stable form in the ethanol-water mixture (1:1 (v/v)).
- Examples 5-7 Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and an acetone-water mixture (1:4 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (G). It was confirmed that Form B crystals were the stable form in the acetone-water mixture (1:4 (v/v)).
- Examples 5-8 Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and a 2-propanol-water mixture (1:4 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (H). It was confirmed that Form B crystals were the stable form in the 2-propanol-water mixture (1:4 (v/v)).
- Examples 5-9 Form A crystals (3.0 mg), Form B crystals (3.1 mg), and Form K crystals (3.0 mg) of Compound 1 were mixed, and an ethanol-water mixture (1:4 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 7 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (I). It was confirmed that Form B crystals were the stable form in the ethanol-water mixture (1:4 (v/v)).
- the present invention provides a method for producing a cyclic peptide compound, or a salt thereof, or a solvate thereof that is useful as a pharmaceutical, and a method for producing a peptide compound used in the production of the cyclic peptide compound, or a salt thereof, or a solvate thereof.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Life Sciences & Earth Sciences (AREA)
- Analytical Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Water Supply & Treatment (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
本発明は、環状ペプチド化合物の製造方法、特にHRASおよびNRASに対して選択的なKRAS阻害作用を有する環状ペプチド化合物の製造方法に関する。 The present invention relates to a method for producing a cyclic peptide compound, in particular a method for producing a cyclic peptide compound that has selective KRAS inhibitory activity against HRAS and NRAS.
RASはsmall GTPase familyに属するタンパクで、KRAS、NRAS、HRASが知られている。RASはGDPまたはGTPとの結合状態によって活性化状態または不活性化状態が規定されており、GEF (guanine nucleotide exchange factor)によるGDPからGTPへの交換反応によって活性化され、GAP (GTPase-activating proteins)によるGTPの加水分解反応によって不活性化される(非特許文献1)。活性化されたRASはMAPK経路、PI3K/Akt経路、RAL経路など様々な下流のシグナルを活性化することで、細胞の増殖、生存、分化を誘導し、RASの恒常活性化はがんの発生や進展に重要な役割を果たす。がんではRASの上流シグナルの活性化、RASの恒常的活性化、および/またはRASの活性型変異により、RAS-RAF-MEK-ERK経路が活性化していることが知られている(非特許文献2)。これらのRASの活性型変異は数多くのがん種で認められている。RAS変異のホットスポットとしてG12、G13、Q61が知られており、KRASではG12に、NRASではQ61に高頻度に変異が認められる。また、これらの変異は患者の予後に関連していることも知られている(非特許文献3)。 RAS is a protein belonging to the small GTPase family, and KRAS, NRAS, and HRAS are known. The activated or inactivated state of RAS is determined by its binding state with GDP or GTP. It is activated by the exchange reaction of GDP to GTP by GEF (guanine nucleotide exchange factor) and inactivated by the hydrolysis of GTP by GAP (GTPase-activating proteins) (Non-patent Document 1). Activated RAS induces cell proliferation, survival, and differentiation by activating various downstream signals such as the MAPK pathway, the PI3K/Akt pathway, and the RAL pathway, and the constitutive activation of RAS plays an important role in the development and progression of cancer. It is known that the RAS-RAF-MEK-ERK pathway is activated in cancer due to activation of upstream signals of RAS, constitutive activation of RAS, and/or activating mutations of RAS (Non-patent Document 2). These activating mutations of RAS have been observed in many types of cancer. G12, G13, and Q61 are known to be hotspots for RAS mutations, with high frequency mutations observed at G12 in KRAS and at Q61 in NRAS. It is also known that these mutations are associated with patient prognosis (Non-Patent Document 3).
これに関して、RASにおける選択性、具体的にはHRASおよびNRASに対して選択的なKRAS阻害作用を有する、下記式(1)で表される環状ペプチド化合物(以下、環状ペプチド化合物(1)ともいう)が報告されている(特許文献1)。
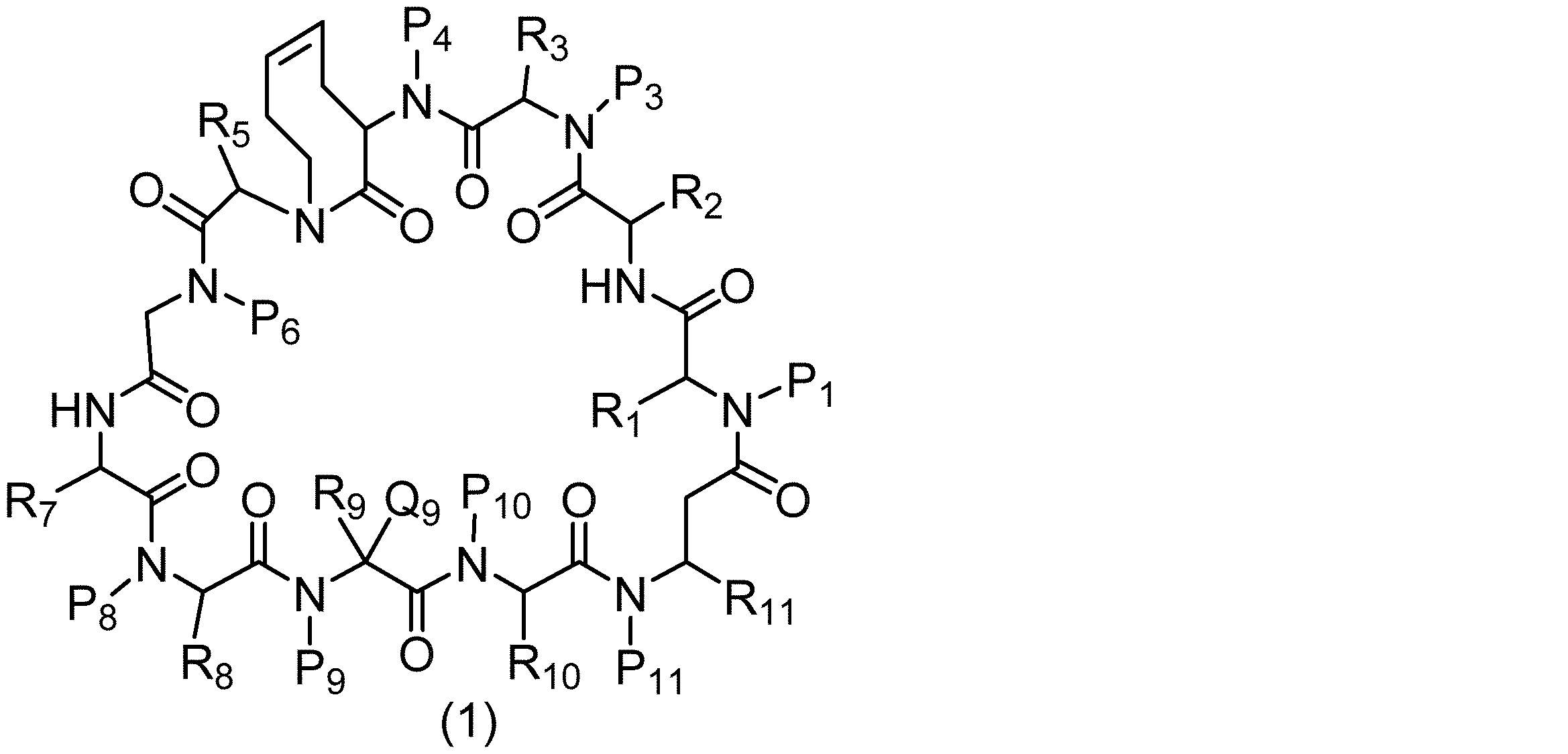
本発明者の知る限り、アミノ酸間に架橋した二重結合を有する環状ペプチド化合物(1)の製造において、環化前駆体ペプチド化合物のどの位置で環化させるのが好ましいのかについての報告例はない。特許文献1には、環化位置Aでの環化反応による環状ペプチド化合物(1)の製造方法が記載されている。しかしながら、後述する実施例1-26に記載されているように、特許文献1に記載されている製造方法では、目的とする環状ペプチド化合物(1)に加えて環状ダイマーが副生成物として確認されており、当該環状ペプチド化合物(1)と環状ダイマーとの比率が75:25と、その選択性は低いものであった。 To the best of the inventor's knowledge, there have been no reports on which position of a cyclization precursor peptide compound is preferred for cyclization in the production of a cyclic peptide compound (1) having a double bond bridged between amino acids. Patent Document 1 describes a method for producing a cyclic peptide compound (1) by a cyclization reaction at cyclization position A. However, as described in Examples 1-26 below, in the production method described in Patent Document 1, a cyclic dimer was confirmed as a by-product in addition to the desired cyclic peptide compound (1), and the ratio of the cyclic peptide compound (1) to the cyclic dimer was 75:25, indicating low selectivity.

また、特許文献1に記載されている環化前駆体ペプチド化合物の製造は、Fmoc法によるアミノ酸または一部トリペプチドを順次連結させて、固相上で合成することにより行われている。固相合成はアミノ酸および試薬が過剰量必要であり、各工程の洗浄には大量の有機溶媒を必要とするため、大量の製造を行う上では可能な限り固相合成を避けることが望まれていた。また、本発明者らの知る限り、環状ペプチド化合物(1)の結晶についての報告例はない。 The cyclized precursor peptide compound described in Patent Document 1 is produced by sequentially linking amino acids or some tripeptides by the Fmoc method and synthesizing on a solid phase. Solid phase synthesis requires excessive amounts of amino acids and reagents, and large amounts of organic solvents are required for washing in each step, so it has been desirable to avoid solid phase synthesis as much as possible when producing large quantities. Furthermore, to the inventors' knowledge, there have been no reports of crystals of cyclic peptide compound (1).
本発明はこのような状況を踏まえてなされたものであり、アミノ酸間に架橋した二重結合を有する環状ペプチド化合物(1)の効率的な製造方法を提供することを課題とする。
また、本発明は環状ペプチド化合物(1)の安定性に優れた結晶を提供とすることを課題とする。
The present invention has been made in light of the above circumstances, and an objective of the present invention is to provide an efficient method for producing a cyclic peptide compound (1) having a double bond bridged between amino acids.
Another object of the present invention is to provide crystals of the cyclic peptide compound (1) that are highly stable.
上記事情に鑑み、本発明者らは鋭意検討の結果、本発明を完成するに至った。すなわち本発明は一局面において、アミノ酸間に架橋した二重結合を有する環状ペプチド化合物(1)の製造における環化方法として、副生成物である環状ダイマーの生成量を低減することができる環化位置による環化方法を提供する。また、本発明は一局面において、環化前駆体ペプチド化合物の製造方法として、3つのフラグメントペプチドをそれぞれ合成し、液相フラグメントカップリングにより合成する、効率的な製造方法を提供する。更に、本発明は一局面において環状ペプチド化合物(1)の結晶と、晶析による該結晶の製造方法を提供する。 In view of the above circumstances, the present inventors have conducted extensive research and have completed the present invention. That is, in one aspect, the present invention provides a cyclization method for producing a cyclic peptide compound (1) having a double bond bridged between amino acids, which is a cyclization method based on the cyclization position, capable of reducing the amount of cyclic dimer produced as a by-product. In another aspect, the present invention provides an efficient production method for producing a cyclized precursor peptide compound, in which three fragment peptides are each synthesized and synthesized by liquid phase fragment coupling. Furthermore, in one aspect, the present invention provides crystals of the cyclic peptide compound (1) and a method for producing the crystals by crystallization.
本発明は、非限定の具体的な一態様において以下を包含する。
〔A1〕式(1)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法であって、溶媒中、式(2)または(3)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを反応させて環化する工程(環化工程)を含む、前記方法。

P1は、C1-C6アルキルであり;
R2は、C1-C6アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P3は、C1-C6アルキル、もしくはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P4は、C1-C6アルキルであり;
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルであり;
P6は、C1-C6アルキルであり;
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1-C6アルコキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよく;
P9は、水素またはC1-C6アルキルであり;
R10は、C1-C6アルキル、またはC3-C8シクロアルキルであり;
P10は、C1-C6アルキルであり;
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルであり;
P11は、C1-C6アルキルであり;
X1およびX5は、それぞれ独立して水素、またはアミノ基の保護基であり;
X2およびX4は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔A2〕式(1)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法であって、
(a) 式(4)~(6)で表されるペプチド化合物、もしくはその塩または前記ペプチド化合物もしくは塩の溶媒和物を用意する工程、
(b) 式(4)~(6)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて連結する工程(連結工程)、および
(c) (b)工程で得られたペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、前記方法。

P1は、C1-C6アルキルであり;
R2は、C1-C6アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P3は、C1-C6アルキル、またはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P4は、C1-C6アルキルであり;
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルであり;
P6は、C1-C6アルキルであり;
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1-C6アルコキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよく;
P9は、水素またはC1-C6アルキルであり;
R10は、C1-C6アルキル、またはC3-C8シクロアルキルであり;
P10は、C1-C6アルキルであり;
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルであり;
P11は、C1-C6アルキルであり;
X1、X3およびX5は、それぞれ独立して水素、またはアミノ基の保護基であり;
X2、X4およびX6は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔A3〕前記(b)の工程において、
(b-1)式(5)で表されるペプチド化合物のN末端のアミノ酸残基と、式(6)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(7)で表されるペプチド化合物に変換する工程(連結工程)を含む、〔A2〕に記載の方法。
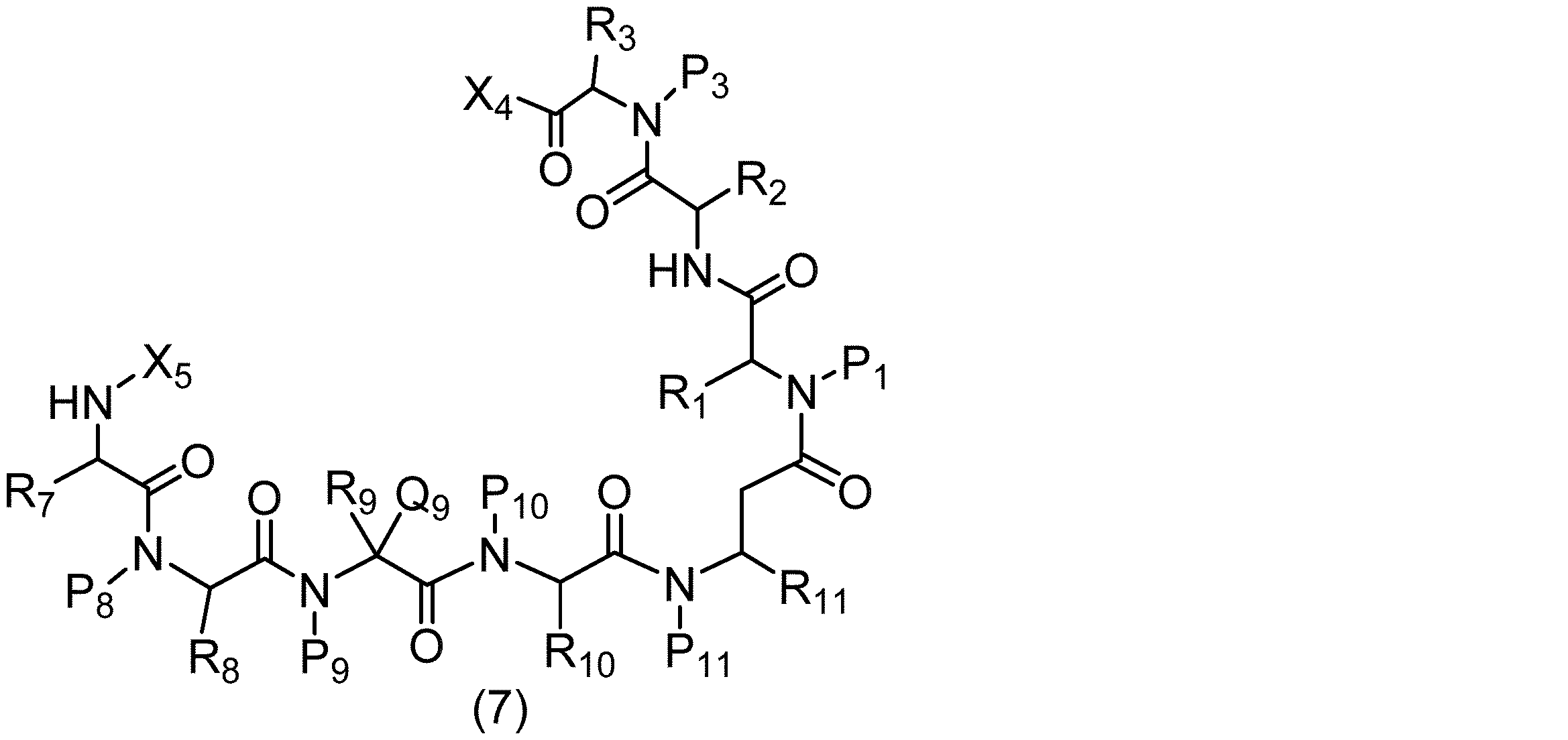
〔A4〕前記(b)の工程において、さらに、
(b-2)式(4)で表されるペプチド化合物のN末端のアミノ酸残基と、式(7)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(2)で表されるペプチド化合物に変換する工程(連結工程)、および、
前記(c)の工程において、
(c-1)式(2)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、〔A3〕に記載の方法。
〔A5〕前記(b)の工程において、さらに、
(b-3)式(7)で表されるペプチド化合物のN末端のアミノ酸残基と、式(4)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(3)で表される化合物に変換する工程(連結工程)、および、
前記(c)の工程において、
(c-2)式(3)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、〔A3〕に記載の方法。
〔A6〕前記ペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基との連結が、N末端のアミノ酸残基のアミノ基とC末端のアミノ酸残基のカルボキシル基との連結である、〔A1〕~〔A5〕のいずれかに記載の方法。
〔A7〕前記ペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基との連結が、N末端のアミノ酸残基のアミノ基とC末端のアミノ酸残基のカルボキシル基とのアミド結合による連結である、〔A1〕~〔A6〕のいずれかに記載の方法。
〔A8〕前記環化工程の溶媒が、ニトリル系溶媒、ハロゲン系溶媒、エーテル系溶媒、アミド系溶媒、エステル系溶媒、および炭酸エステル系溶媒からなる群より選択される1つ以上を含む、〔A1〕~〔A7〕のいずれかに記載の方法。
〔A9〕前記ニトリル系溶媒が、アセトニトリル、およびプロピオニトリルからなる群より選択される1種または複数であり、
前記ハロゲン系溶媒が、ジクロロメタン、クロロホルム、および1,2-ジクロロエタンからなる群より選択される1種または複数であり、
前記エーテル系溶媒が、ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、シクロペンチルメチルエーテル、4-メチルテトラヒドロピラン、1,3-ジオキソラン、1,4-ジオキサン、1,2-ジメトキシエタン、ジイソプロピルエーテル、t-ブチルメチルエーテル、ジグリム、トリグリム、アニソール、およびテトラグリムからなる群より選択される1種または複数であり、
前記アミド系溶媒が、DMF、NMP、DMA、NEP、NBP、およびホルムアミドからなる群より選択される1種または複数であり、
前記エステル系溶媒が、酢酸メチル、酢酸エチル、プロピオン酸メチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、酢酸イソブチル、酢酸ペンチル、およびγ-バレロラクトンからなる群より選択される1種または複数であり、
前記炭酸エステル系溶媒が、炭酸ジメチル、炭酸ジエチル、および炭酸ジブチルからなる群より選択される1種または複数である、〔A8〕に記載の方法。
〔A10〕前記環化工程の溶媒が、アセトニトリル、炭酸ジメチル、2-メチルテトラヒドロピラン、4-メチルテトラヒドロピラン、テトラヒドロフラン、酢酸エチル、酢酸イソプロピル、ジクロロメタン、DMF、およびアニソールからなる群より選択される1種または複数である、〔A8〕に記載の方法。
〔A11〕前記環化工程の溶媒が、アセトニトリル、2-メチルテトラヒドロフラン、酢酸エチル、およびジクロロメタンからなる群より選択される1種または複数である、〔A8〕に記載の方法。
〔A12〕前記環化工程の溶媒が、アセトニトリル、2-メチルテトラヒドロフラン、または酢酸エチルである、〔A8〕に記載の方法。
〔A13〕前記環化工程が、縮合試薬の存在下で行われる、〔A1〕~〔A12〕のいずれかに記載の方法。
〔A14〕前記環化工程の縮合試薬が、HATU、COMU、DMT-MM、PyOxim、PyBOP、HCTU、T3P、EDCI、BEP、およびPyClopからなる群より選択される1種または複数である、〔A13〕に記載の方法。
〔A15〕前記環化工程の縮合試薬が、HATU、COMU、PyOxim、PyBOP、HCTU、およびT3Pからなる群より選択される1つである、〔A13〕に記載の方法。
〔A16〕前記環化工程の縮合試薬が、HATUである、〔A13〕に記載の方法。
〔A17〕前記環化工程の縮合試薬が、COMUである、〔A13〕に記載の方法。
〔A18〕前記環化工程の縮合試薬が、HATUであり、前記環化工程の溶媒が、アセトニトリルである、〔A13〕に記載の方法。
〔A19〕前記環化工程の縮合試薬が、HATUであり、前記環化工程の溶媒が、2-メチルテトラヒドロフランである、〔A13〕に記載の方法。
〔A20〕前記環化工程の縮合試薬が、COMUであり、前記環化工程の溶媒が、アセトニトリルである、〔A13〕に記載の方法。
〔A21〕前記環化工程の縮合試薬が、COMUであり、前記環化工程の溶媒が、2-メチルテトラヒドロフランである、〔A13〕に記載の方法。
〔A22〕前記環化工程が、塩基の存在下で行われる、〔A1〕~〔A21〕のいずれかに記載の方法。
〔A23〕前記環化工程の塩基が、有機塩基である、〔A22〕に記載の方法。
〔A24〕前記環化工程の塩基が、3級アミンを含む有機塩基である、〔A22〕に記載の方法。
〔A25〕前記環化工程の塩基が、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、ピリジン、1,8-ジアザビシクロ〔5.4.0〕-7-ウンデセン(DBU)、2,3,6,7-テトラヒドロ-1H,5H-9-アザベンゾ[ij]キノリジン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,5-ジアザビシクロ[4.3.0]-5-ノネン(DBN)、7-メチル-1,5,7-トリアザビシクロ[4.4.0]デカ-5-エン、1,1,3,3-テトラメチルグアニジン(TMG)、1,8-ビス(テトラメチルグアニジノ)ナフタレン(TMGN)、2-tert-ブチル-1,1,3,3-テトラメチルグアニジン(BTMG)、トリエチルアミン(TEA)、トリメチルアミン、1-メチルピぺリジン、N,N’-ジメチルピペラジン、N-エチルモルホリン、およびp-ジメチルアミノピリジン(DMAP)からなる群より選択される1種または複数である、〔A22〕に記載の方法。
〔A26〕前記環化工程の塩基が、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、およびピリジンからなる群より選択される1種または複数である、である、〔A22〕に記載の方法。
〔A27〕前記環化工程の縮合試薬が、HATUであり、前記環化工程の溶媒が、アセトニトリルであり、前記環化工程の塩基が、N,N-ジイソプロピルエチルアミン(DIPEA)である、〔A22〕に記載の方法。
〔A28〕前記環化工程の縮合試薬が、HATUであり、前記環化工程の溶媒が、2-MeTHFであり、前記環化工程の塩基が、N,N-ジイソプロピルエチルアミン(DIPEA)である、〔A22〕に記載の方法。
〔A29〕前記環化工程の縮合試薬が、COMUであり、前記環化工程の溶媒が、アセトニトリルであり、前記環化工程の塩基が、2,6-ルチジンである、〔A22〕に記載の方法。
〔A30〕前記環化工程の縮合試薬が、COMUであり、前記環化工程の溶媒が、2-メチルテトラヒドロフランであり、前記環化工程の塩基が、2,6-ルチジンである、〔A22〕に記載の方法。
〔A31〕前記環化工程が、液相法で行われる、〔A1〕~〔A30〕のいずれかに記載の方法。
〔A32〕前記環化工程において、前記環化工程の溶媒と前記縮合試薬とを混合して得られた混合液中に、前記ペプチド化合物および前記塩基を混合する、〔A1〕~〔A31〕のいずれかに記載の方法。
〔A33〕前記環化工程において生成する総副生成物の含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、20%未満、15%未満、10%未満、5%未満、または3%未満であることを特徴とする、〔A1〕~〔A32〕のいずれかに記載の方法。
〔A34〕前記環化工程において生成する副生成物各々の含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、3%未満、1%未満、または検出不能な量であることを特徴とする、〔A1〕~〔A33〕のいずれかに記載の方法。
〔A35〕前記環化工程において生成する副生成物各々の含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、3%未満、1%未満、または検出不能な量であり、前記副生成物がエピマーおよび/または環状ダイマーを含むことを特徴とする、〔A1〕~〔A34〕のいずれかに記載の方法。
〔A36〕前記環化工程において生成する副生成物が、エピマーを含み、前記エピマーの含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、10%未満、7.5%未満、5%未満、2.5%未満、または1%未満であることを特徴とする、〔A1〕~〔A35〕のいずれかに記載の方法。
〔A37〕前記環化工程において生成する副生成物が、環状ダイマーを含み、前記環状ダイマーの含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、2.5%未満、または1%未満であることを特徴とする、〔A1〕~〔A36〕のいずれかに記載の方法。
〔A38〕前記連結工程の溶媒が、ニトリル系溶媒、ハロゲン系溶媒、エーテル系溶媒、アミド系溶媒、エステル系溶媒、および炭酸エステル系溶媒からなる群より選択される1つ以上を含む、〔A1〕~〔A37〕のいずれかに記載の方法。
〔A39〕前記ニトリル系溶媒が、アセトニトリル、およびプロピオニトリルからなる群より選択される1種または複数であり、
前記ハロゲン系溶媒が、ジクロロメタン、クロロホルム、および1,2-ジクロロエタンからなる群より選択される1種または複数であり、
前記エーテル系溶媒が、ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、シクロペンチルメチルエーテル、4-メチルテトラヒドロピラン、1,3-ジオキソラン、1,4-ジオキサン、1,2-ジメトキシエタン、ジイソプロピルエーテル、t-ブチルメチルエーテル、ジグリム、トリグリム、アニソール、およびテトラグリムからなる群より選択される1種または複数であり、
前記アミド系溶媒が、DMF、NMP、DMA、NEP、NBP、およびホルムアミドからなる群より選択される1種または複数であり、
前記エステル系溶媒が、酢酸メチル、酢酸エチル、プロピオン酸メチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、酢酸イソブチル、酢酸ペンチル、およびγ-バレロラクトンからなる群より選択される1種または複数であり、
前記炭酸エステル系溶媒が、炭酸ジメチル、炭酸ジエチル、および炭酸ジブチルからなる群より選択される1種または複数である、〔A38〕に記載の方法。
〔A40〕前記連結工程の溶媒が、アセトニトリル、炭酸ジメチル、2-メチルテトラヒドロフラン、4-メチルテトラヒドロピラン、THF、酢酸エチル、酢酸イソプロピル、DMF、およびアニソールからなる群より選択される1種または複数である、〔A38〕に記載の方法。
〔A41〕前記連結工程の溶媒が、アセトニトリル、炭酸ジメチル、2-メチルテトラヒドロフラン、4-メチルテトラヒドロピランP、THF、酢酸エチル、酢酸イソプロピル、およびアニソールからなる群より選択される1種または複数と、DMFとの混合溶媒である、〔A38〕に記載の方法。
〔A42〕前記連結工程の溶媒が、アセトニトリル、2-メチルテトラヒドロフラン、およびDMFの混合溶媒である、〔A38〕に記載の方法。
〔A43〕前記連結工程の溶媒が、2-メチルテトラヒドロフランおよびDMFの混合溶媒である、〔A38〕に記載の方法。
〔A44〕前記連結工程が、縮合試薬の存在下で行われる、〔A1〕~〔A43〕のいずれかに記載の方法。
〔A45〕前記連結工程の縮合試薬が、HATU、COMU、DMT-MM、PyOxim、PyBOP、HCTU、T3P、EDCI、BEP、およびPyClopからなる群より選択される1種または複数である、〔A44〕に記載の方法。
〔A46〕前記連結工程の縮合試薬が、HATU、COMU、PyOxim、PyBOP、HCTU、およびT3Pからなる群より選択される1つである、〔A44〕に記載の方法。
〔A47〕前記連結工程の縮合試薬が、HATU、およびCOMUからなる群より選択される1つである、〔A44〕に記載の方法。
〔A48〕前記連結工程の縮合試薬が、HATUである、〔A44〕に記載の方法。
〔A49〕前記連結工程の縮合試薬が、COMUである、〔A44〕に記載の方法。
〔A50〕前記連結工程の縮合試薬が、HATUであり、前記連結工程の溶媒が、アセトニトリルまたは2-MeTHFである、〔A44〕に記載の方法。
〔A51〕前記連結工程の縮合試薬が、HATUであり、前記連結工程の溶媒が、アセトニトリル、2-MeTHF、およびDMFの混合溶媒である、〔A44〕に記載の方法。
〔A52〕前記連結工程の縮合試薬が、COMUであり、前記連結工程の溶媒が、アセトニトリルまたは2-MeTHFである、〔A44〕に記載の方法。
〔A53〕前記連結工程の縮合試薬が、COMUであり、前記連結工程の溶媒が、アセトニトリル、2-MeTHF、およびDMFの混合溶媒である、〔A44〕に記載の方法。
〔A54〕前記連結工程が、塩基の存在下で行われる、〔A1〕~〔A53〕のいずれかに記載の方法。
〔A55〕前記連結工程の塩基が、有機塩基である、〔A54〕に記載の方法。
〔A56〕前記連結工程の塩基が、3級アミンを含む有機塩基である、〔A54〕に記載の方法。
〔A57〕前記連結工程の塩基が、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、ピリジン、1,8-ジアザビシクロ〔5.4.0〕-7-ウンデセン(DBU)、2,3,6,7-テトラヒドロ-1H,5H-9-アザベンゾ[ij]キノリジン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,5-ジアザビシクロ[4.3.0]-5-ノネン(DBN)、7-メチル-1,5,7-トリアザビシクロ[4.4.0]デカ-5-エン、1,1,3,3-テトラメチルグアニジン(TMG)、1,8-ビス(テトラメチルグアニジノ)ナフタレン(TMGN)、2-tert-ブチル-1,1,3,3-テトラメチルグアニジン(BTMG)、トリエチルアミン(TEA)、トリメチルアミン、1-メチルピぺリジン、N,N’-ジメチルピペラジン、N-エチルモルホリン、およびp-ジメチルアミノピリジン(DMAP)からなる群より選択される1種または複数である、〔A54〕に記載の方法。
〔A58〕前記連結工程の塩基が、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、およびピリジンからなる群より選択される1種または複数である、〔A54〕に記載の方法。
〔A59〕前記連結工程の縮合試薬が、HATUであり、前記連結工程の溶媒が、アセトニトリルまたは2-MeTHFであり、前記連結工程の塩基が、N,N-ジイソプロピルエチルアミン(DIPEA)である、〔A54〕に記載の方法。
〔A60〕前記連結工程の縮合試薬が、HATUであり、前記連結工程の溶媒が、アセトニトリル、2-メチルテトラヒドロフラン、およびDMFの混合溶媒であり、前記連結工程の塩基が、N,N-ジイソプロピルエチルアミン(DIPEA)である、〔A54〕に記載の方法。
〔A61〕前記連結工程の縮合試薬が、COMUであり、前記連結工程の溶媒が、アセトニトリルまたは2-メチルテトラヒドロフランであり、前記連結工程の塩基が、N-メチルモルホリンまたは2,6-ルチジンである、〔A54〕に記載の方法。
〔A62〕前記連結工程の縮合試薬が、COMUであり、前記連結工程の溶媒が、アセトニトリル、2-メチルテトラヒドロフラン、およびDMFの混合溶媒であり、前記連結工程の塩基が、N-メチルモルホリンまたは2,6-ルチジンである、〔A54〕に記載の方法。
〔A62-1〕前記連結工程の縮合試薬が、HATUであり、前記連結工程の溶媒が、アセトニトリルおよび2-メチルテトラヒドロフランの混合溶媒であり、前記連結工程の塩基が、N-メチルモルホリンである、〔A54〕に記載の方法。
〔A62-2〕前記連結工程の縮合試薬が、HATUであり、前記連結工程の溶媒が、アセトニトリルであり、前記連結工程の塩基が、N-メチルモルホリンである、〔A54〕に記載の方法。
〔A63〕前記連結工程が、液相法で行われる、〔A1〕~〔A62〕のいずれかに記載の方法。
〔A64〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の単離および/または精製にカラムクロマトグラフィーを用いる、〔A1〕~〔A63〕のいずれかに記載の方法。
〔A65〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の単離および/または精製にカラムクロマトグラフィーを用いない、〔A1〕~〔A63〕のいずれかに記載の方法。
〔A66〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を晶析により単離および/または精製して、前記環状ペプチド化合物、もしくはその塩またはそれらの結晶を得る工程をさらに含む、〔A1〕~〔A65〕のいずれかに記載の方法。
〔A67〕R1が、C3-C4アルキルである、〔A1〕~〔A66〕のいずれかに記載の方法。
〔A67-1〕R1が、n-プロピルである、〔A1〕~〔A66〕のいずれかに記載の方法。
〔A67-2〕R1が、2-メチルプロピルである、〔A1〕~〔A66〕のいずれかに記載の方法。
〔A68〕P1が、C1-C4アルキルである、〔A1〕~〔A67〕のいずれかに記載の方法。
〔A68-1〕P1が、メチルである、〔A1〕~〔A67〕のいずれかに記載の方法。
〔A69〕R2が、C3-C4アルキルである、〔A1〕~〔A68〕のいずれかに記載の方法。
〔A69-1〕R2が、1-メチルプロピルである、〔A1〕~〔A68〕のいずれかに記載の方法。
〔A70〕R3が、水素であるか、またはR3が、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している、〔A1〕~〔A69〕のいずれかに記載の方法。
〔A70-1〕R3が、水素である、〔A1〕~〔A69〕のいずれかに記載の方法。
〔A70-2〕R3が、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している、〔A1〕~〔A69〕のいずれかに記載の方法。
〔A71〕P3が、C1-C4アルキルであるか、またはP3が、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している、〔A1〕~〔A70〕のいずれかに記載の方法。
〔A71-1〕P3が、メチルである、〔A1〕~〔A70〕のいずれかに記載の方法。
〔A71-2〕P3が、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している、〔A1〕~〔A70〕のいずれかに記載の方法。
〔A72〕P4が、C1-C4アルキルである、〔A1〕~〔A71〕のいずれかに記載の方法。
〔A72-1〕P4が、メチルである、〔A1〕~〔A71〕のいずれかに記載の方法。
〔A73〕R5が、C1-C4ハロアルキルによって置換されていてもよいベンジルである、〔A1〕~〔A72〕のいずれかに記載の方法。
〔A73-1〕R5が、4-トリフルオロメチルベンジルである、〔A1〕~〔A72〕のいずれかに記載の方法。
〔A74〕P6が、C1-C4アルキルである、〔A1〕~〔A73〕のいずれかに記載の方法。
〔A74-1〕P6が、メチルである、〔A1〕~〔A73〕のいずれかに記載の方法。
〔A75〕R7が、ハロゲン、トリフルオロメチル、およびメトキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルである、〔A1〕~〔A74〕のいずれかに記載の方法。
〔A75-1〕R7が、3-メトキシ-4-トリフルオロメチルフェネチルである、〔A1〕~〔A74〕のいずれかに記載の方法。
〔A75-2〕R7が、3,5-ジフルオロ-4-トリフルオロメチルフェネチルである、〔A1〕~〔A74〕のいずれかに記載の方法。
〔A76〕R8が、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はC1-C4アルキルによって置換されている、〔A1〕~〔A75〕のいずれかに記載の方法。
〔A76-1〕R8が、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はエトキシによって置換されている、〔A1〕~〔A75〕のいずれかに記載の方法。
〔A77〕R9が、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって4~6員脂環式環を形成している、〔A1〕~〔A76〕のいずれかに記載の方法。
〔A77-1〕R9が、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって4員脂環式環を形成している、〔A1〕~〔A76〕のいずれかに記載の方法。
〔A77-2〕R9が、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって5員脂環式環を形成している、〔A1〕~〔A76〕のいずれかに記載の方法。
〔A78〕P9が、水素またはC1-C4アルキルである、〔A1〕~〔A77〕のいずれかに記載の方法。
〔A78-1〕P9が、水素である、〔A1〕~〔A77〕のいずれかに記載の方法。
〔A78-2〕P9が、メチルである、〔A1〕~〔A77〕のいずれかに記載の方法。
〔A79〕R10が、C4-C6シクロアルキルである、〔A1〕~〔A78〕のいずれかに記載の方法。
〔A79-1〕R10が、シクロペンチルである、〔A1〕~〔A78〕のいずれかに記載の方法。
〔A80〕P10が、C1-C4アルキルである、〔A1〕~〔A79〕のいずれかに記載の方法。
〔A80-1〕P10が、メチルである、〔A1〕~〔A79〕のいずれかに記載の方法。
〔A81〕R11が、ジC1~C4アルキルアミノカルボニル、または5~6員環状アミノカルボニルである、〔A1〕~〔A80〕のいずれかに記載の方法。
〔A81-1〕R11が、ジメチルアミノカルボニルである、〔A1〕~〔A80〕のいずれかに記載の方法。
〔A82〕P11が、C1-C4アルキルである、〔A1〕~〔A81〕のいずれかに記載の方法。
〔A82-1〕P11が、メチルである、〔A1〕~〔A81〕のいずれかに記載の方法。
〔A83〕X1、X3、およびX5は、それぞれ独立して水素、カルバメート系保護基、アシル系保護基、スルホンアミド系保護基、およびシリル系保護基からなる群より選択される1つである、〔A1〕~〔A82〕のいずれかに記載の方法。
〔A84〕前記カルバメート系保護基は、Fmoc基、Cbz基、Troc基、Alloc基、Teoc基、TSoc基、BIBSoc基、IPCSoc基、BBSoc基、CHBSoc基、CDBSoc基、およびBoc基からなる群より選択される1つである、〔A83〕に記載の方法。
〔A85〕前記アシル系保護基は、トリフルオロアセチル基、アセチル基、およびベンゾイル基からなる群より選択される1つである、〔A83〕に記載の方法。
〔A86〕前記スルホンアミド系保護基は、2-ニトロベンゼンスルホニル基、4-ニトロベンゼンスルホニル基、および2,4-ジニトロベンゼンスルホニル基からなる群より選択される1つである、〔A83〕に記載の方法。
〔A87〕前記シリル系保護基は、TMS基、TBDMS基、TES基、TIPS基、およびTBDPS基からなる群より選択される1つである、〔A83〕に記載の方法。
〔A88〕X1は、水素、またはカルバメート系保護基である、〔A1〕~〔A87〕のいずれかに記載の方法。
〔A88-1〕X1は、水素である、〔A1〕~〔A87〕のいずれかに記載の方法。
〔A88-2〕X1は、Fmoc基である、〔A1〕~〔A87〕のいずれかに記載の方法。
〔A89〕X3は、水素、またはカルバメート系保護基である、〔A1〕~〔A88〕のいずれかに記載の方法。
〔A89-1〕X3は、水素である、〔A1〕~〔A88〕のいずれかに記載の方法。
〔A89-2〕X3は、Cbz基である、〔A1〕~〔A88〕のいずれかに記載の方法。
〔A90〕X5は、水素、またはカルバメート系保護基である、〔A1〕~〔A89〕のいずれかに記載の方法。
〔A90-1〕X5は、水素である、〔A1〕~〔A89〕のいずれかに記載の方法。
〔A90-2〕X5は、Cbz基である、〔A1〕~〔A89〕のいずれかに記載の方法。
〔A91〕X2、X4、およびX6は、それぞれ独立してハロゲン、水酸基、置換されていてもよいC1-C6アルコキシ、置換されていてもよいC6-C10アリールオキシ、置換されていてもよいC7-C14アラルコキシ、置換されていてもよい4~8員環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、C1-C6アルキル、またはC6-C10アリールである)で表される基である、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A91-1〕前記ハロゲンは、塩素、または臭素である、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A91-2〕前記置換されていてもよいアルコキシは、t-ブトキシ、メトキシ、エトキシ、またはイソプロポキシである、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A91-3〕前記置換されていてもよいアリールオキシは、ペンタフルオロフェニルオキシ、またはニトロフェニルオキシである、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A91-4〕前記置換されていてもよいアラルコキシは、置換されていてもよいベンジルオキシである、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A91-5〕前記置換されていてもよい環状アミノオキシは、N-ヒドロキシコハク酸イミノオキシである、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A91-6〕前記-OSiRxRyRzで表される基は、トリメチルシリルオキシ、トリエチルシリルオキシ、トリイソプロピルシリルオキシ、トリフェニルシリルオキシ、トリ-t-ブチルシリルオキシ、ジ-t-ブチルイソブチルシリルオキシ、またはトリス(トリエチルシリル)シリルオキシである、〔A1〕~〔A90〕のいずれかに記載の方法。
〔A92〕X2は、水酸基、t-ブトキシ、またはベンジルオキシである、〔A1〕~〔A91〕のいずれかに記載の方法。
〔A92-1〕X2は、水酸基である、〔A1〕~〔A91〕のいずれかに記載の方法。
〔A92-2〕X2は、t-ブトキシである、〔A1〕~〔A91〕のいずれかに記載の方法。
〔A93〕X4は、水酸基、t-ブトキシ、またはベンジルオキシである、〔A1〕~〔A92〕のいずれかに記載の方法。
〔A93-1〕X4は、水酸基である、〔A1〕~〔A92〕のいずれかに記載の方法。
〔A93-2〕X4は、t-ブトキシである、〔A1〕~〔A92〕のいずれかに記載の方法。
〔A94〕X6は、水酸基、t-ブトキシ、またはベンジルオキシである、〔A1〕~〔A93〕のいずれかに記載の方法。
〔A94-1〕X6は、水酸基である、〔A1〕~〔A93〕のいずれかに記載の方法。
〔A94-2〕X6は、t-ブトキシである、〔A1〕~〔A93〕のいずれかに記載の方法。
〔A95〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物が、前記環状ペプチド化合物の溶媒和物である、〔A1〕~〔A94〕のいずれかに記載の方法。
〔A95-1〕前記環状ペプチド化合物の溶媒和物が、前記環状ペプチド化合物の水和物である、〔A95〕に記載の方法。
〔A96〕前記環状ペプチド化合物が、式(1a):

〔A97〕前記環状ペプチド化合物が、式(1a):
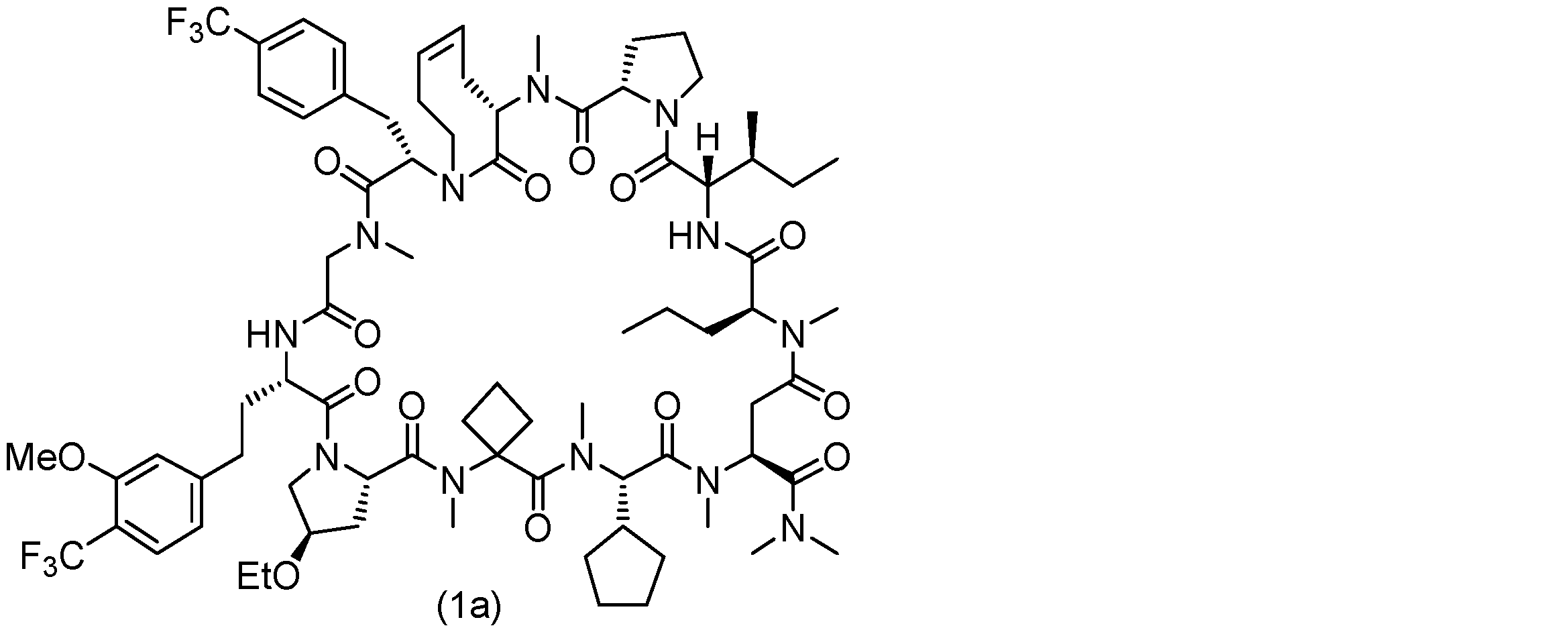
〔A97-1〕前記環状ペプチド化合物の結晶が、非溶媒和物結晶、または溶媒和物結晶である、〔A97〕に記載の方法。
〔A97-2〕前記環状ペプチド化合物の結晶が、溶媒和物結晶である、〔A97〕に記載の方法。
〔A97-3〕前記環状ペプチド化合物の溶媒和物結晶が、水和物結晶である、〔A97-2〕に記載の方法。
〔B1〕式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法であって、溶媒中、式(2a)または(3a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを反応させて環化する工程(環化工程)を含む、前記方法。

X2およびX4は、それぞれ独立して水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔B2〕式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法であって、
(a) 式(4a)~(6a)で表されるペプチド化合物、もしくはその塩または前記ペプチド化合物もしくは塩の溶媒和物を用意する工程、
(b) 式(4a)~(6a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて連結する工程(連結工程)、および
(c) (b)工程で得られたペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、前記方法。
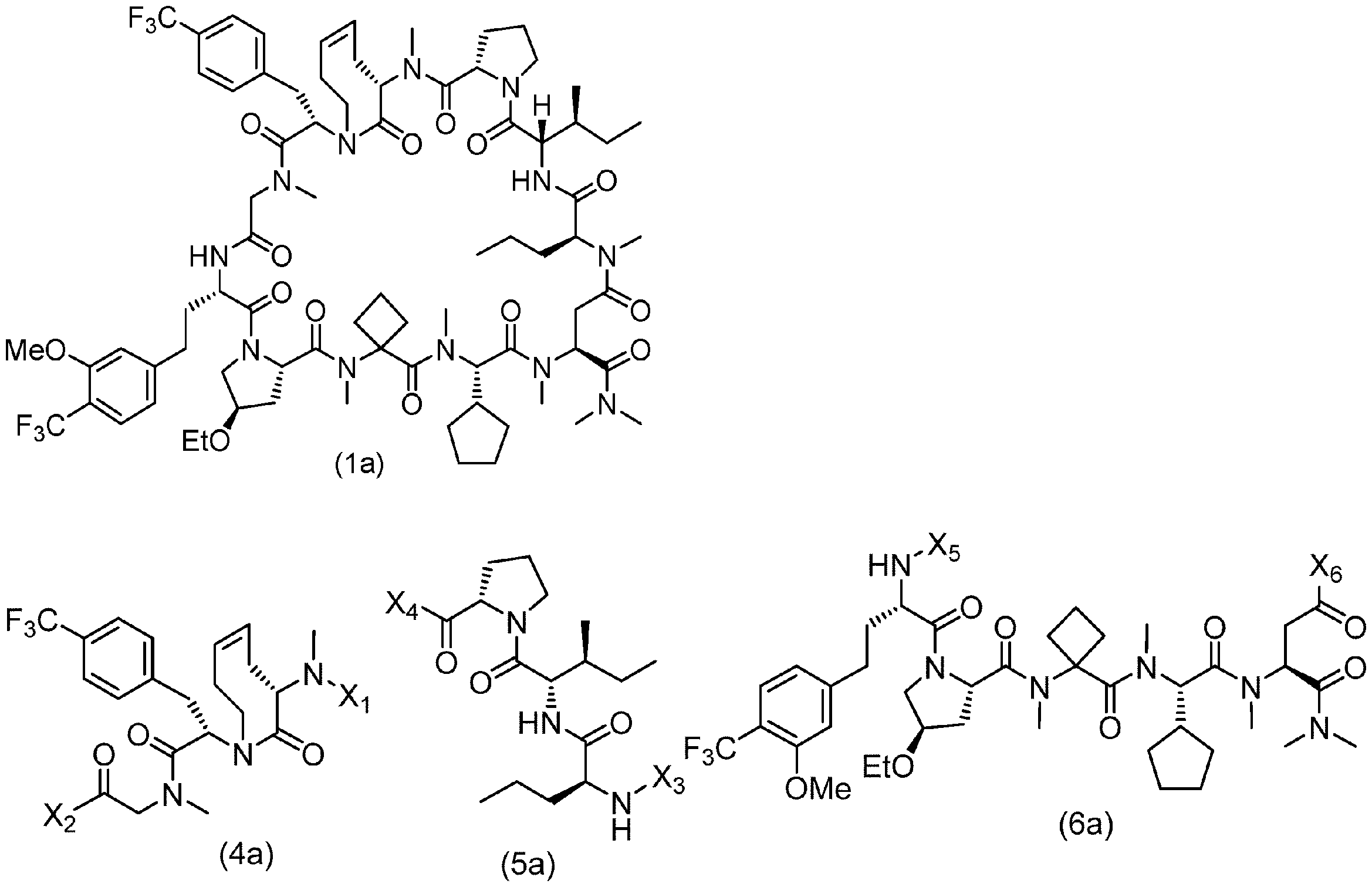
X2、X4およびX6は、それぞれ独立して水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔B3〕前記(b)の工程において、
(b-1)式(5a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(6a)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(7a)で表されるペプチド化合物に変換する工程(連結工程)を含む、〔B2〕に記載の方法。

〔B4〕前記(b)の工程において、さらに、
(b-2)式(4a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(7a)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(2a)で表されるペプチド化合物に変換する工程(連結工程)、および、
前記(c)の工程において、
(c-1)式(2a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、〔B3〕に記載の方法。
〔B5〕前記(b)の工程において、さらに、
(b-3)式(7a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(4a)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(3a)で表されるペプチド化合物に変換する工程(連結工程)、および、
前記(c)の工程において、
(c-2)式(3a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、〔B3〕に記載の方法。
〔B6〕前記環化工程の溶媒が、〔A8〕~〔A12〕のいずれかに記載の溶媒である、〔B1〕~〔B5〕のいずれかに記載の方法。
〔B7〕前記環化工程が、縮合試薬の存在下で行われる、〔B1〕~〔B6〕のいずれかに記載の方法。
〔B8〕前記環化工程の縮合試薬が、〔A14〕~〔A17〕のいずれかに記載の縮合試薬である、〔B7〕に記載の方法。
〔B9〕前記環化工程の溶媒および縮合試薬が、〔A18〕~〔A21〕のいずれかに記載の溶媒および縮合試薬である、〔B7〕に記載の方法。
〔B10〕前記環化工程が、塩基の存在下で行われる、〔B1〕~〔B9〕のいずれかに記載の方法。
〔B11〕前記環化工程の塩基が、〔A24〕~〔A26〕のいずれかに記載の塩基である、〔B10〕に記載の方法。
〔B12〕前記環化工程の溶媒、縮合試薬および塩基が、〔A27〕~〔A30〕のいずれかに記載の溶媒、縮合試薬および塩基である、〔B10〕に記載の方法。
〔B13〕前記環化工程が、液相法で行われる、〔B1〕~〔B12〕のいずれかに記載の方法。
〔B14〕前記環化工程において、前記環化工程の溶媒と前記縮合試薬とを混合して得られた混合液中に、前記ペプチド化合物および前記塩基を混合する、〔B1〕~〔B13〕のいずれかに記載の方法。
〔B15〕前記環化工程において生成する総副生成物の含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、20%未満、15%未満、10%未満、5%未満、または3%未満であることを特徴とする、〔B1〕~〔B14〕のいずれかに記載の方法。
〔B16〕前記環化工程において生成する副生成物各々の含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、3%未満、1%未満、または検出不能な量であることを特徴とする、〔B1〕~〔B15〕のいずれかに記載の方法。
〔B17〕前記環化工程において生成する副生成物各々の含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、3%未満、1%未満、または検出不能な量であり、前記副生成物がエピマーおよび/または環状ダイマーを含むことを特徴とする、〔B1〕~〔B16〕のいずれかに記載の方法。
〔B18〕前記環化工程において生成する副生成物が、前記エピマーを含み、前記エピマーの含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、10%未満、7.5%未満、5%未満、2.5%未満、または1%未満であることを特徴とする、〔B1〕~〔B17〕のいずれかに記載の方法。
〔B19〕前記環化工程において生成する副生成物が、環状ダイマーを含み、前記環状ダイマーの含有率が、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、2.5%未満、または1%未満であることを特徴とする、〔B1〕~〔B18〕のいずれかに記載の方法。
〔B20〕前記連結工程の溶媒が、〔A38〕~〔A43〕のいずれかに記載の溶媒である、〔B1〕~〔B19〕のいずれかに記載の方法。
〔B21〕前記連結工程が、縮合試薬の存在下で行われる、〔B1〕~〔B20〕のいずれかに記載の方法。
〔B22〕前記連結工程の縮合試薬が、〔A45〕~〔A49〕のいずれかに記載の縮合試薬である、〔B21〕に記載の方法。
〔B23〕前記連結工程の溶媒および縮合試薬が、〔A50〕~〔A53〕のいずれかに記載の溶媒および縮合試薬である、〔B21〕に記載の方法。
〔B24〕前記連結工程が、塩基の存在下で行われる、〔B1〕~〔B23〕のいずれかに記載の方法。
〔B25〕前記連結工程の塩基が、〔A55〕~〔A59〕のいずれかに記載の塩基である、〔B24〕に記載の方法。
〔B26〕前記連結工程の溶媒、縮合試薬および塩基が、〔A60〕~〔A62〕のいずれかに記載の溶媒、縮合試薬および塩基である、〔B24〕に記載の方法。
〔B27〕前記連結工程が、液相法で行われる、〔B1〕~〔B26〕のいずれかに記載の方法。
〔B28〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の単離および/または精製にカラムクロマトグラフィーを用いる、〔B1〕~〔B27〕のいずれかに記載の方法。
〔B29〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の単離および/または精製にカラムクロマトグラフィーを用いない、〔B1〕~〔B27〕のいずれかに記載の方法。
〔B30〕前記環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を晶析により単離および/または精製して、前記環状ペプチド化合物、もしくはその塩またはそれらの結晶を得る工程をさらに含む、〔B1〕~〔B29〕のいずれかに記載の方法。
〔C1〕式(4a)で表される化合物またはその塩。

X2は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔C2〕X1が水素である、〔C1〕に記載の化合物またはその塩。
〔C3〕X1がFmoc基である、〔C1〕に記載の化合物またはその塩。
〔C4〕X2がt-ブトキシである、〔C1〕~〔C3〕のいずれかに記載の化合物またはその塩。
〔C5〕tert-ブチル2-[メチル-[(2S)-2-[(4Z,7S)-7-(メチルアミノ)-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]アミノ]酢酸(化合物9)。
〔C6〕式(5a)で表される化合物またはその塩。

X4は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔C7〕X3が水素である、〔C6〕に記載の化合物またはその塩。
〔C8〕X3がFmoc基またはCbz基である、〔C6〕に記載の化合物またはその塩。
〔C9〕X4がt-ブトキシである、〔C6〕~〔C8〕のいずれかに記載の化合物またはその塩。
〔C10〕tert-ブチル(2S)-1-[(2S,3S)-3-メチル-2-[[(2S)-2-(メチルアミノ)ペンタノイル]アミノ]ペンタノイル]ピロリジン-2-酢酸(化合物13)。
〔C11〕式(6a)で表される化合物またはその塩。
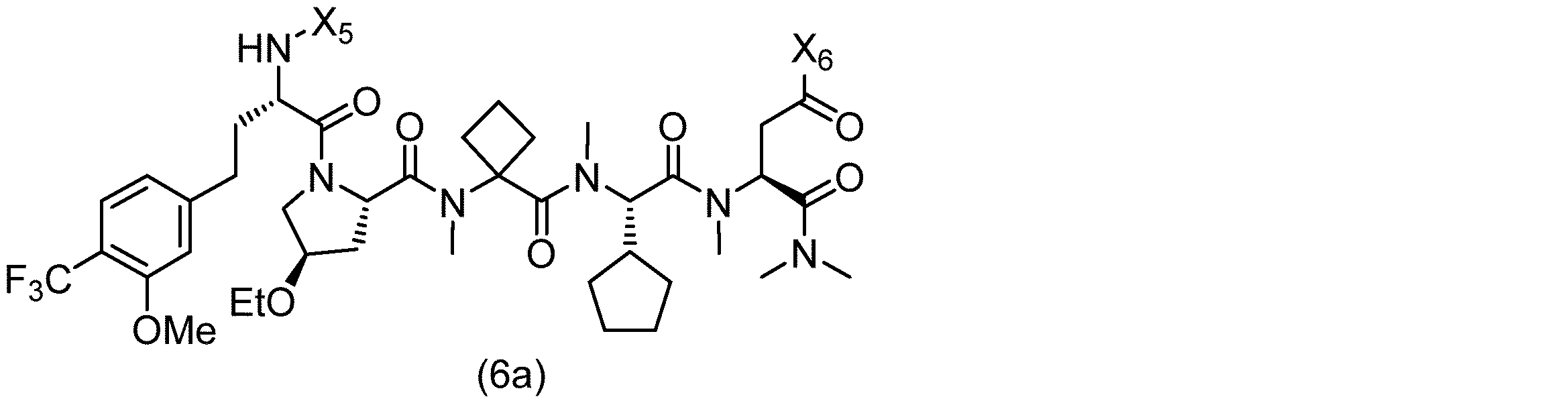
X6は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔C12〕X5が水素である、〔C11〕に記載の化合物またはその塩。
〔C13〕X5がCbz基である、〔C11〕に記載の化合物またはその塩。
〔C14〕X6が水酸基またはt-ブトキシである、〔C11〕~〔C13〕のいずれかに記載の化合物またはその塩。
〔C15〕(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸(化合物20)。
〔C16〕式(7a)で表される化合物またはその塩。

X4は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔C17〕X5が水素である、〔C16〕に記載の化合物またはその塩。
〔C18〕X5がCbz基である、〔C16〕に記載の化合物またはその塩。
〔C19〕X6が水酸基またはt-ブトキシである、〔C16〕~〔C18〕のいずれかに記載の化合物またはその塩。
〔C20〕(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチルアセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸(化合物22)。
〔C20-1〕tert-ブチル (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-アミノ-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボキシラート(化合物37)。
〔C21〕式(2a)で表される化合物またはその塩。
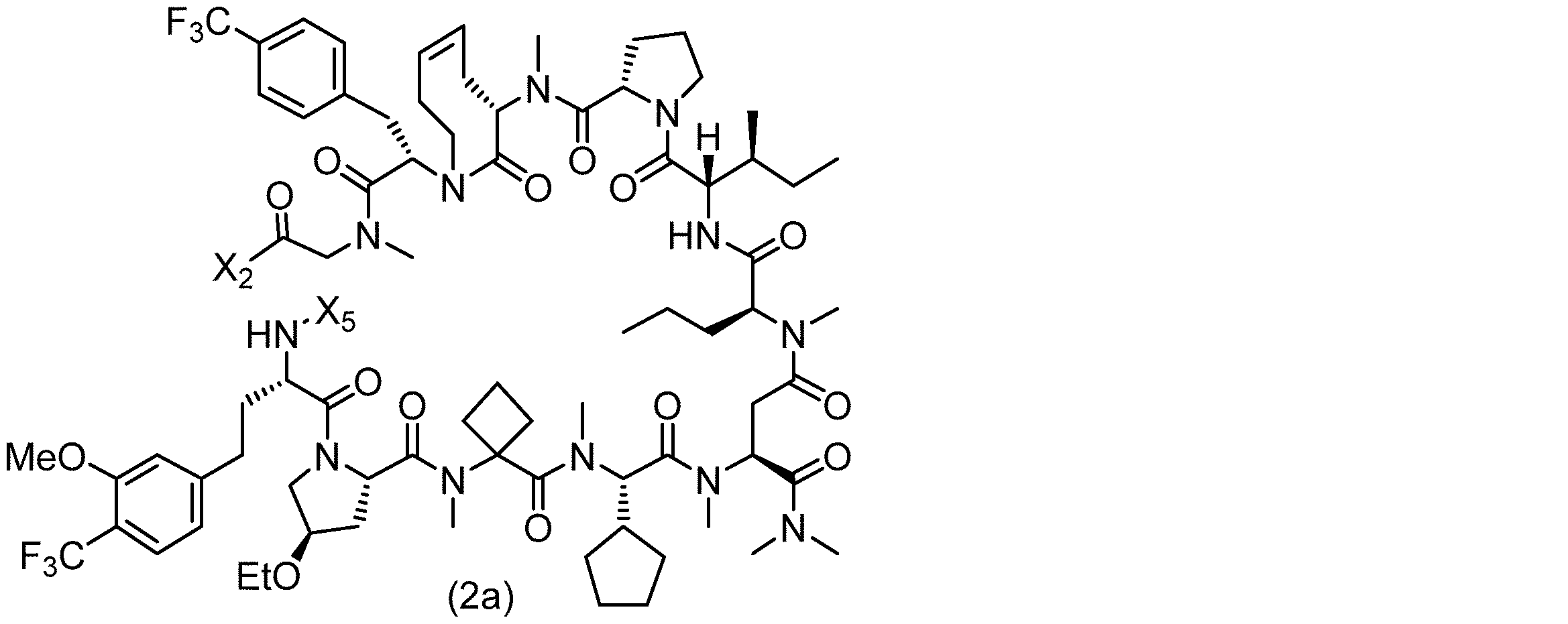
X2は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔C22〕X5が水素である、〔C21〕に記載の化合物またはその塩。
〔C23〕X5がCbz基である、〔C21〕に記載の化合物もしくはその塩。
〔C24〕X2が水酸基またはt-ブトキシである、〔C21〕~〔C23〕のいずれかに記載の化合物またはその塩。
〔C25〕2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-アミノ-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボニル]-メチル-アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸(化合物24)。
〔C26〕式(3a)で表される化合物またはその塩。
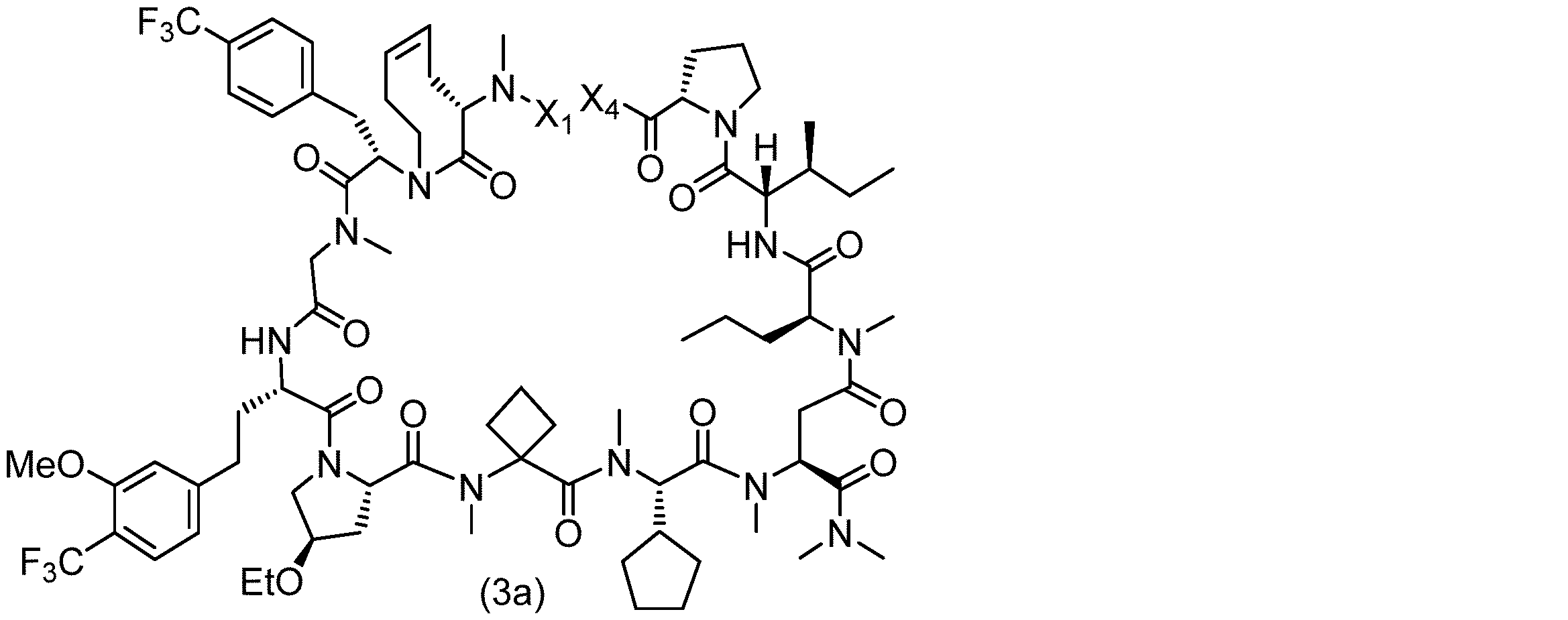
X4は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。]
〔C27〕X1が水素である、〔C26〕に記載の化合物またはその塩。
〔C28〕X1がFmoc基である、〔C26〕に記載の化合物またはその塩。
〔C29〕X4が水酸基またはt-ブトキシである、〔C26〕~〔C28〕のいずれかに記載の化合物またはその塩。
〔C30〕(S)-2-[(S)-3-[(S)-2-シクロペンチル-2-[1-[(2S,4R)-4-エトキシ-1-[(S)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]-2-[(2-[(S)-N-メチル-2-[(R,Z)-3-(メチルアミノ)-2-オキソ-3,4,7,8-テトラヒドロアゾシン-1(2H)-イル]-3-[4-(トリフルオロメチル)フェニル]プロパンアミド]アセトアミド)ブタノイル]-Nメチルピロリジン-2-カルボキシアミド]-N-メチルシクロブタン-1-カルボキシアミド]-N-メチルアセトアミド]-4-(ジメチルアミノ)-N-メチル-4-オキソブタンアミド]ペンタノイル]-L-イソロイシル-L-プロリン(化合物40)。
〔C31〕〔C1〕~〔C30〕のいずれかに記載の化合物の製造方法であって、固相合成を用いない、製造方法。
〔C32〕式(1a)で表される環状ペプチド化合物の製造方法であって、固相合成を用いない、製造方法。
〔D1〕式(1a)で表される環状ペプチド化合物、もしくはその塩、またはそれらの溶媒和物の結晶。

〔D3〕結晶が、環状ペプチド化合物の溶媒和物結晶である、〔D2〕に記載の結晶。
〔D4〕溶媒和物結晶が、環状ペプチド化合物の水和物結晶である、〔D3〕に記載の結晶。
〔D5〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)からなる群から選択される、少なくとも7個のピークを含むForm A結晶である、〔D4〕に記載の結晶。
〔D5-1〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)からなる群から選択される、少なくとも8個のピークを含むForm A結晶である、〔D4〕に記載の結晶。
〔D5-2〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)のピークを含むForm A結晶である、〔D4〕に記載の結晶。
〔D5-3〕前記回折角(2θ値)が、10%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)である、〔D5〕~〔D5-2〕のいずれかに記載のForm A結晶。
〔D6〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)からなる群から選択される、少なくとも7個のピークを含むForm B結晶である、〔D4〕に記載の結晶。
〔D6-1〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)からなる群から選択される、少なくとも8個のピークを含むForm B結晶である、〔D4〕に記載の結晶。
〔D6-2〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)のピークを含むForm B結晶である、〔D4〕に記載の結晶。
〔D6-3〕前記回折角(2θ値)が、30%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)である、〔D6〕~〔D6-2〕のいずれかに記載のForm B結晶。
〔D7〕前記溶媒和物結晶が、粉末X線回折による回折角(2θ値)として、6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°(±0.2°)からなる群から選択される、少なくとも7個のピークを含むForm F結晶である、〔D3〕に記載の結晶。
〔D7-1〕前記溶媒和物結晶が、粉末X線回折による回折角(2θ値)として、6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°(±0.2°)からなる群から選択される、少なくとも8個のピークを含むForm F結晶である、〔D3〕に記載の結晶。
〔D7-2〕前記溶媒和物結晶が、粉末X線回折による回折角(2θ値)として、6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°(±0.2°)のピークを含むForm F結晶である、〔D3〕に記載の結晶。
〔D7-3〕前記溶媒和物結晶が、アセトン/へプタン/水和物である、〔D7〕~〔D7-2〕のいずれかに記載の結晶。
〔D8〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)からなる群から選択される、少なくとも7個のピークを含むForm J結晶である、〔D4〕に記載の結晶。
〔D8-1〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)からなる群から選択される、少なくとも8個のピークを含むForm J結晶である、〔D4〕に記載の結晶。
〔D8-2〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)のピークを含むForm J結晶である、〔D4〕に記載の結晶。
〔D8-3〕前記回折角(2θ値)が、10%未満の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)である、〔D8〕~〔D8-2〕のいずれかに記載のForm J結晶。
〔D9〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)からなる群から選択される、少なくとも7個のピークを含むForm Y結晶である、〔D4〕に記載の結晶。
〔D9-1〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)からなる群から選択される、少なくとも8個のピークを含むForm Y結晶である、〔D4〕に記載の結晶。
〔D9-2〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)のピークを含むForm Y結晶である、〔D4〕に記載の結晶。
〔D9-3〕前記回折角(2θ値)が、30%未満の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)である、〔D9〕~〔D9-2〕のいずれかに記載のForm Y結晶。
〔D10〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°(±0.2°)からなる群から選択される、少なくとも7個のピークを含むForm K結晶である、〔D3〕に記載の結晶。
〔D10-1〕前記水和物結晶が、粉末X線回折による回折角(2θ値)として、7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°(±0.2°)からなる群から選択される、少なくとも8個のピークを含むForm K結晶である、〔D3〕に記載の結晶。
〔D10-2〕前記溶媒和物結晶が、粉末X線回折による回折角(2θ値)として、7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°(±0.2°)のピークを含むForm K結晶である、〔D3〕に記載の結晶。
〔D11〕前記溶媒和物結晶が、図1に示す単結晶X線解析の構造をもつForm Gのジメチルスルホキシド/ヘプタン/水和物結晶である、〔D3〕に記載の結晶。
〔D11-1〕前記溶媒和物結晶が、図3に示す単結晶X線解析の構造をもつForm Eの2-プロパノール/へプタン/水和物結晶である、〔D3〕に記載の結晶。
〔D11-2〕前記溶媒和物結晶が、図8に示す単結晶X線解析の構造をもつForm Hのエタノール/水和物結晶である、〔D3〕に記載の結晶。
〔D11-3〕前記溶媒和物結晶が、図26に示す単結晶X線解析の構造をもつForm Dの1,4-ジオキサン/水和物結晶である、〔D3〕に記載の結晶。
〔D11-4〕前記溶媒和物結晶が、図28に示す単結晶X線解析の構造をもつForm Lのジメチルスルホキシド/水和物結晶である、〔D3〕に記載の結晶。
〔D11-5〕前記溶媒和物結晶が、図31に示す単結晶X線解析の構造をもつForm Mのプロピレングリコール/水和物結晶である、〔D3〕に記載の結晶。
〔D11-6〕前記溶媒和物結晶が、図29に示す粉末X線回折による回折角をもつForm Mのプロピレングリコール/水和物結晶である、〔D3〕に記載の結晶。
〔D11-7〕前記溶媒和物結晶が、図30(A)に示す熱重量・示差熱同時測定のデータを示すForm Mのプロピレングリコール/水和物結晶である、〔D3〕に記載の結晶。
〔D11-8〕前記溶媒和物結晶が、図29に示す粉末X線回折による回折角を持つ、Form Nのプロピレングリコール和物結晶である、〔D3〕に記載の結晶。
〔D11-9〕前記溶媒和物結晶が、図30(B)に示す熱重量・示差熱同時測定のデータを示すForm Nのプロピレングリコール和物結晶である、〔D3〕に記載の結晶。
〔D12〕〔D1〕~〔D10-2〕のいずれかに記載の環状ペプチド化合物の結晶を製造する方法であって、該環状ペプチド化合物を該環状ペプチド化合物が溶解可能な量の極性有機溶媒に溶解させて溶液を得る工程、および該溶液に炭化水素系溶媒または水を加えて、該環状ペプチド化合物の結晶を得る工程を含む、方法。
〔D12-1〕原料の環状ペプチド化合物の純度が85%以上である、〔D12〕に記載の方法。
〔D13〕〔D1〕~〔D10-2〕のいずれかに記載の環状ペプチド化合物の結晶を製造する方法であって、アモルファス状態の該環状ペプチド化合物に、炭化水素系溶媒と極性有機溶媒の混合液、または水と極性有機溶媒の混合液を加えて、該環状ペプチド化合物の結晶を得る工程を含む、方法。
〔D14〕前記極性有機溶媒が、DMSO、アセトン、2-ブタノン、メタノール、エタノール、1-プロパノール、2-プロパノール、プロピレングリコール、1,4-ジオキサン、および酢酸エチルからなる群より選択される1種または複数である、〔D12〕~〔D13-1〕のいずれかに記載の方法。
〔D15〕前記極性有機溶媒が、アセトンである、〔D14〕に記載の方法。
〔D15-1〕前記極性有機溶媒が、エタノールである、〔D14〕に記載の方法。
〔D16〕前記炭化水素系溶媒が、ヘプタン、ヘキサン、ペンタン、トルエン、およびキシレンからなる群より選択される1種または複数である、〔D12〕~〔D13-1〕のいずれかに記載の方法。
〔D17〕前記炭化水素系溶媒が、ヘプタンである、〔D16〕に記載の方法。
〔D18〕〔D1〕~〔D10-2〕のいずれかに記載の環状ペプチド化合物の結晶を製造する方法であって、アモルファス状態の該環状ペプチド化合物をDMSOに溶解させて溶液を得る工程、該溶液を凍結乾燥して該環状ペプチド化合物の凍結乾燥体を得る工程、および、該凍結乾燥体に水と極性有機溶媒の混合液を加えて、該環状ペプチド化合物の結晶を得る工程を含む、方法。
〔D19〕前記極性有機溶媒が、DMSO、アセトン、2-ブタノン、メタノール、エタノール、1-プロパノール、2-プロパノール、1,4-ジオキサンおよびプロピレングリコールからなる群より選択される1種または複数である、〔D18〕に記載の方法。
〔D20〕前記極性有機溶媒が、アセトンである、〔D19〕に記載の方法。
〔D21〕前記環状ペプチド化合物の結晶を得る工程の後に、さらに該結晶をろ過する工程を含む、〔D12〕~〔D20〕のいずれかに記載の方法。
〔D22〕前記環状ペプチド化合物の結晶を得る工程の後に、さらに該結晶を乾燥する工程を含む、〔D12〕~〔D21〕のいずれかに記載の方法。
〔D23〕前記環状ペプチド化合物の結晶が、溶媒和物結晶である、〔D12〕~〔D22〕のいずれかに記載の方法。
〔D24〕前記環状ペプチド化合物の溶媒和物結晶が、水和物結晶である、〔D23〕に記載の方法。
〔D25〕前記環状ペプチド化合物の結晶が、溶媒中で溶媒和物結晶として形成され、ろ過する工程および/または乾燥する工程の後に、水和物結晶として得られる、〔D21〕または〔D22〕に記載の方法。
〔D26〕式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を含む組成物であって、該化合物は不純物である式(1a)の環状ダイマーを1.5w/w%以下の割合で含有する、前記組成物。

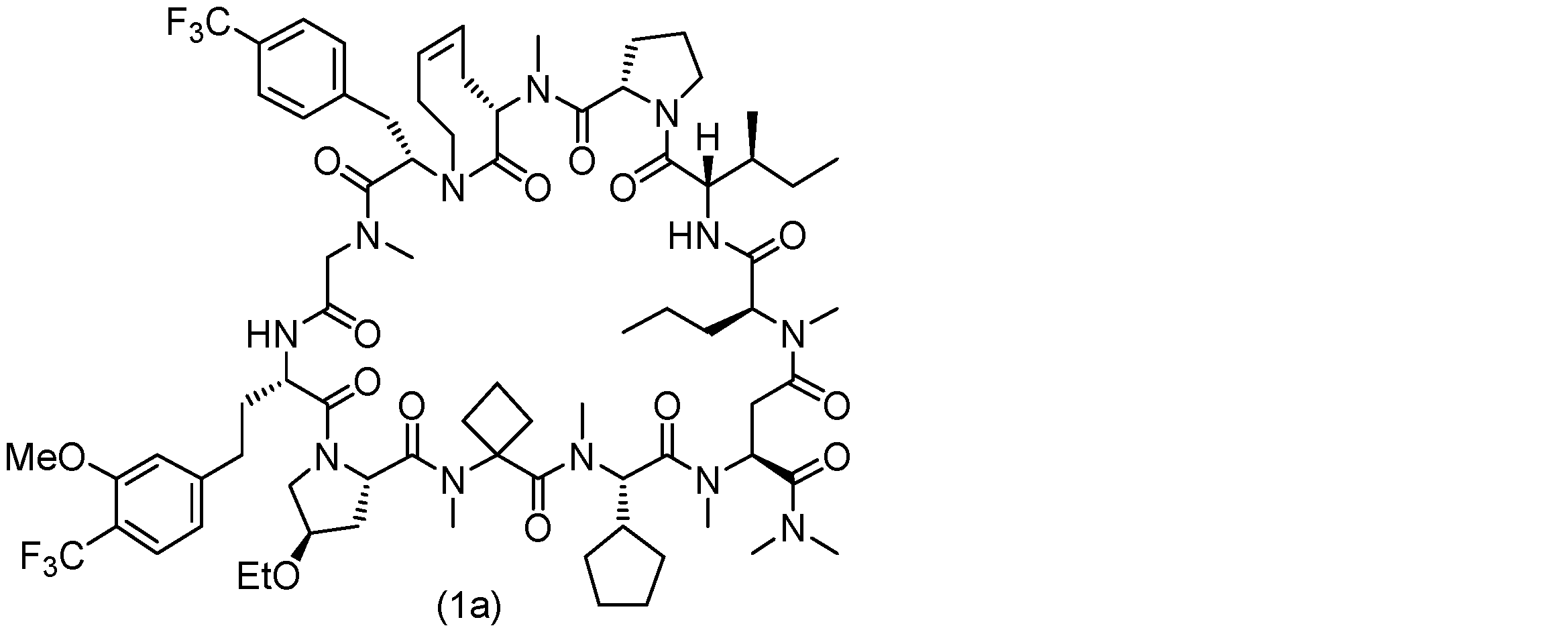
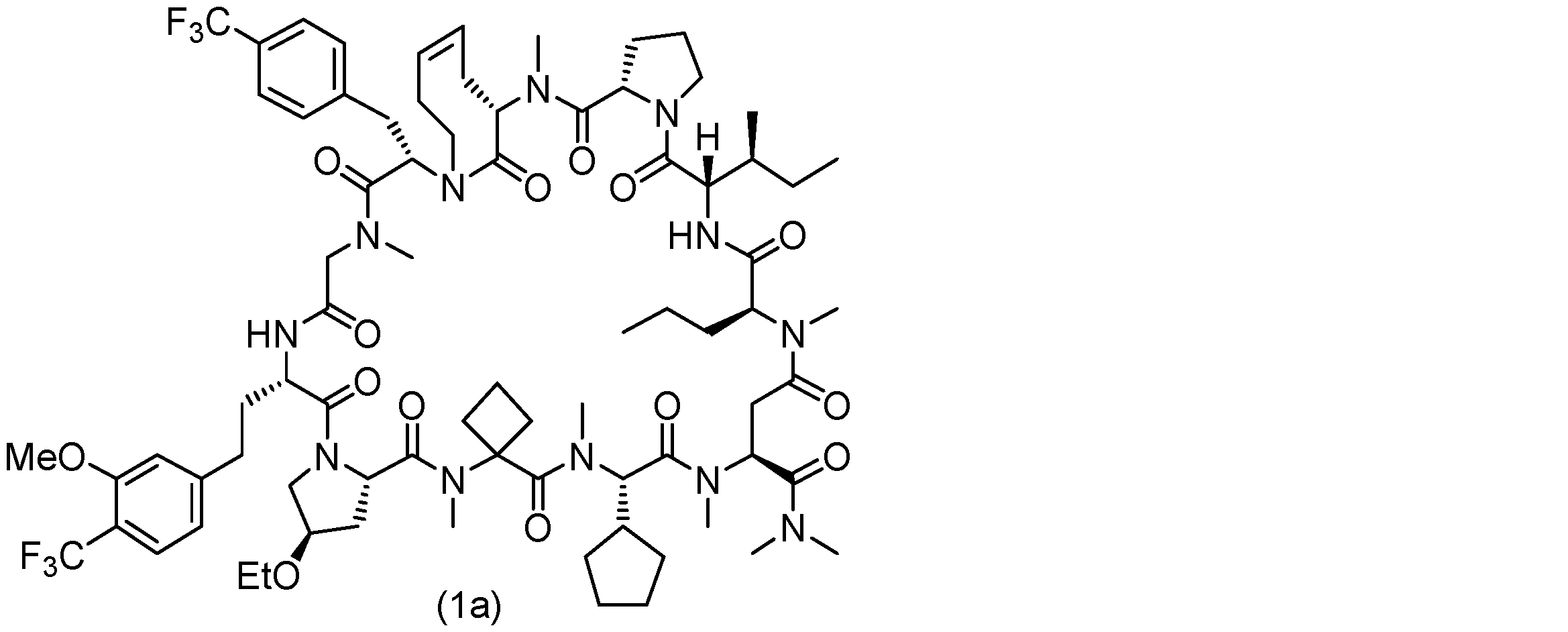
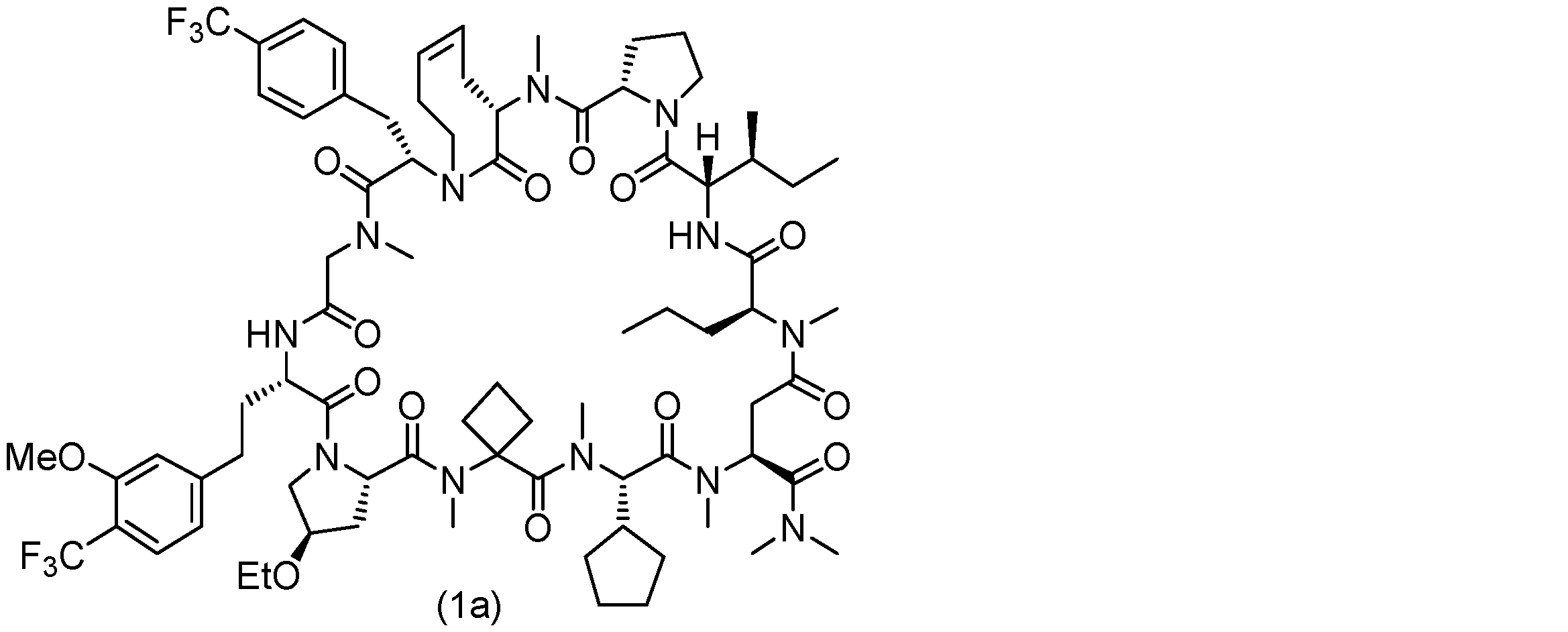
[A1] A method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof, comprising a step of cyclizing the compound by reacting an N-terminal amino acid residue with a C-terminal amino acid residue of a peptide compound represented by formula (2) or (3) in a solvent (cyclization step).
P1 is C1 - C6 alkyl;
R2 is C1 - C6 alkyl;
R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring;
P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle ;
P4 is C1 - C6 alkyl;
R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl;
P6 is C1 - C6 alkyl;
R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocycle, which is optionally substituted by C 1 -C 6 alkoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 3- to 8-membered alicyclic ring, which is optionally substituted by one or more C 1 -C 6 alkyl;
P9 is hydrogen or C1 - C6 alkyl;
R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl;
P10 is C1 - C6 alkyl;
R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4-8 membered cyclic aminocarbonyl;
P11 is C1 - C6 alkyl;
X1 and X5 are each independently hydrogen or an amino-protecting group;
X2 and X4 each independently represent a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz each independently represent an alkyl or an aryl ) .
[A2] A method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof, comprising the steps of:
(a) preparing a peptide compound represented by any one of formulas (4) to (6), or a salt thereof, or a solvate of said peptide compound or salt;
(b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4) to (6) in a solvent (linking step); and
(c) the step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in the step (b) by reacting them in a solvent (cyclization step).
P1 is C1 - C6 alkyl;
R2 is C1 - C6 alkyl;
R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring;
P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle ;
P4 is C1 - C6 alkyl;
R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl;
P6 is C1 - C6 alkyl;
R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocycle, which is optionally substituted by C 1 -C 6 alkoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 3- to 8-membered alicyclic ring, which is optionally substituted by one or more C 1 -C 6 alkyl;
P9 is hydrogen or C1 - C6 alkyl;
R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl;
P10 is C1 - C6 alkyl;
R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4-8 membered cyclic aminocarbonyl;
P11 is C1 - C6 alkyl;
X 1 , X 3 and X 5 are each independently hydrogen or a protecting group for an amino group;
X 2 , X 4 and X 6 each independently represent a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y and R z each independently represent an alkyl or an aryl).
[A3] In the step (b),
(b-1) The method according to [A2], comprising a step (linking step) of reacting and linking the N-terminal amino acid residue of a peptide compound represented by formula (5) and the C-terminal amino acid residue of a peptide compound represented by formula (6) in a solvent to convert them into a peptide compound represented by formula (7).
[A4] In the step (b), further
(b-2) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4) and the C-terminal amino acid residue of the peptide compound represented by formula (7) in a solvent to convert them into the peptide compound represented by formula (2) (linking step); and
In the step (c),
(c-1) The method according to [A3], comprising a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2) in a solvent (cyclization step).
[A5] In the step (b), further
(b-3) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7) and the C-terminal amino acid residue of the peptide compound represented by formula (4) in a solvent to convert them into a compound represented by formula (3) (linking step); and
In the step (c),
(c-2) The method according to [A3], which comprises a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3) in a solvent (cyclization step).
[A6] The method according to any of [A1] to [A5], wherein the linkage between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is a linkage between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue.
[A7] The method according to any of [A1] to [A6], wherein the link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is an amide bond between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue.
[A8] The method according to any one of [A1] to [A7], wherein the solvent in the cyclization step comprises one or more selected from the group consisting of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents.
[A9] The nitrile-based solvent is one or more selected from the group consisting of acetonitrile and propionitrile;
The halogen-based solvent is one or more selected from the group consisting of dichloromethane, chloroform, and 1,2-dichloroethane;
the ether-based solvent is one or more selected from the group consisting of diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, 4-methyltetrahydropyran, 1,3-dioxolane, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, t-butyl methyl ether, diglyme, triglyme, anisole, and tetraglyme;
The amide solvent is one or more selected from the group consisting of DMF, NMP, DMA, NEP, NBP, and formamide;
the ester solvent is one or more selected from the group consisting of methyl acetate, ethyl acetate, methyl propionate, butyl acetate, propyl acetate, isopropyl acetate, isobutyl acetate, pentyl acetate, and γ-valerolactone;
The method according to [A8], wherein the carbonate ester solvent is one or more selected from the group consisting of dimethyl carbonate, diethyl carbonate, and dibutyl carbonate.
[A10] The method according to [A8], wherein the solvent in the cyclization step is one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydropyran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, dichloromethane, DMF, and anisole.
[A11] The method according to [A8], wherein the solvent in the cyclization step is one or more selected from the group consisting of acetonitrile, 2-methyltetrahydrofuran, ethyl acetate, and dichloromethane.
[A12] The method according to [A8], wherein the solvent in the cyclization step is acetonitrile, 2-methyltetrahydrofuran, or ethyl acetate.
[A13] The method according to any one of [A1] to [A12], wherein the cyclization step is carried out in the presence of a condensation reagent.
[A14] The method according to [A13], wherein the condensation reagent in the cyclization step is one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop.
[A15] The method according to [A13], wherein the condensation reagent in the cyclization step is one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P.
[A16] The method according to [A13], wherein the condensation reagent in the cyclization step is HATU.
[A17] The method according to [A13], wherein the condensation reagent in the cyclization step is COMU.
[A18] The method according to [A13], wherein the condensation reagent in the cyclization step is HATU, and the solvent in the cyclization step is acetonitrile.
[A19] The method according to [A13], wherein the condensation reagent in the cyclization step is HATU, and the solvent in the cyclization step is 2-methyltetrahydrofuran.
[A20] The method according to [A13], wherein the condensation reagent in the cyclization step is COMU, and the solvent in the cyclization step is acetonitrile.
[A21] The method according to [A13], wherein the condensation reagent in the cyclization step is COMU, and the solvent in the cyclization step is 2-methyltetrahydrofuran.
[A22] The method according to any one of [A1] to [A21], wherein the cyclization step is carried out in the presence of a base.
[A23] The method according to [A22], wherein the base in the cyclization step is an organic base.
[A24] The method according to [A22], wherein the base in the cyclization step is an organic base containing a tertiary amine.
[A25] The base in the cyclization step is 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN), 7-methyl-1,5 The method according to [A22], wherein the compound is one or more selected from the group consisting of ,7-triazabicyclo[4.4.0]dec-5-ene, 1,1,3,3-tetramethylguanidine (TMG), 1,8-bis(tetramethylguanidino)naphthalene (TMGN), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG), triethylamine (TEA), trimethylamine, 1-methylpiperidine, N,N'-dimethylpiperazine, N-ethylmorpholine, and p-dimethylaminopyridine (DMAP).
[A26] The method according to [A22], wherein the base in the cyclization step is one or more selected from the group consisting of 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, and pyridine.
[A27] The method according to [A22], wherein the condensation reagent in the cyclization step is HATU, the solvent in the cyclization step is acetonitrile, and the base in the cyclization step is N,N-diisopropylethylamine (DIPEA).
[A28] The method according to [A22], wherein the condensation reagent in the cyclization step is HATU, the solvent in the cyclization step is 2-MeTHF, and the base in the cyclization step is N,N-diisopropylethylamine (DIPEA).
[A29] The method according to [A22], wherein the condensation reagent in the cyclization step is COMU, the solvent in the cyclization step is acetonitrile, and the base in the cyclization step is 2,6-lutidine.
[A30] The method according to [A22], wherein the condensation reagent in the cyclization step is COMU, the solvent in the cyclization step is 2-methyltetrahydrofuran, and the base in the cyclization step is 2,6-lutidine.
[A31] The method according to any one of [A1] to [A30], wherein the cyclization step is carried out by a liquid phase method.
[A32] The method according to any of [A1] to [A31], wherein in the cyclization step, the peptide compound and the base are mixed into a mixed solution obtained by mixing a solvent for the cyclization step with the condensation reagent.
[A33] The method according to any one of [A1] to [A32], characterized in that the content of all by-products produced in the cyclization step is less than 20%, less than 15%, less than 10%, less than 5%, or less than 3%, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
[A34] The method according to any one of [A1] to [A33], characterized in that the content of each by-product generated in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
[A35] The method according to any one of [A1] to [A34], characterized in that the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount, based on the total amount of the product, as determined by a UV area value at 220 nm by HPLC analysis, and the by-products include epimers and/or cyclic dimers.
[A36] The method according to any one of [A1] to [A35], wherein a by-product produced in the cyclization step contains an epimer, and the content of the epimer is less than 10%, less than 7.5%, less than 5%, less than 2.5%, or less than 1%, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
[A37] The method according to any one of [A1] to [A36], characterized in that a by-product produced in the cyclization step contains a cyclic dimer, and the content of the cyclic dimer is less than 15%, less than 10%, less than 5%, less than 2.5%, or less than 1%, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
[A38] The method according to any one of [A1] to [A37], wherein the solvent in the linking step comprises one or more selected from the group consisting of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents.
[A39] The nitrile solvent is one or more selected from the group consisting of acetonitrile and propionitrile;
The halogen-based solvent is one or more selected from the group consisting of dichloromethane, chloroform, and 1,2-dichloroethane;
the ether-based solvent is one or more selected from the group consisting of diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, 4-methyltetrahydropyran, 1,3-dioxolane, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, t-butyl methyl ether, diglyme, triglyme, anisole, and tetraglyme;
The amide solvent is one or more selected from the group consisting of DMF, NMP, DMA, NEP, NBP, and formamide;
the ester solvent is one or more selected from the group consisting of methyl acetate, ethyl acetate, methyl propionate, butyl acetate, propyl acetate, isopropyl acetate, isobutyl acetate, pentyl acetate, and γ-valerolactone;
The method according to [A38], wherein the carbonate ester solvent is one or more selected from the group consisting of dimethyl carbonate, diethyl carbonate, and dibutyl carbonate.
[A40] The method according to [A38], wherein the solvent in the linking step is one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, THF, ethyl acetate, isopropyl acetate, DMF, and anisole.
[A41] The method according to [A38], wherein the solvent in the linking step is a mixed solvent of one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran P, THF, ethyl acetate, isopropyl acetate, and anisole, and DMF.
[A42] The method according to [A38], wherein the solvent in the linking step is a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF.
[A43] The method according to [A38], wherein the solvent in the linking step is a mixed solvent of 2-methyltetrahydrofuran and DMF.
[A44] The method according to any one of [A1] to [A43], wherein the linking step is carried out in the presence of a condensation reagent.
[A45] The method according to [A44], wherein the condensation reagent in the linking step is one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop.
[A46] The method according to [A44], wherein the condensation reagent in the linking step is one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P.
[A47] The method according to [A44], wherein the condensation reagent in the linking step is one selected from the group consisting of HATU and COMU.
[A48] The method according to [A44], wherein the condensation reagent in the linking step is HATU.
[A49] The method according to [A44], wherein the condensation reagent in the linking step is COMU.
[A50] The method according to [A44], wherein the condensation reagent in the linking step is HATU, and the solvent in the linking step is acetonitrile or 2-MeTHF.
[A51] The method according to [A44], wherein the condensation reagent in the linking step is HATU, and the solvent in the linking step is a mixed solvent of acetonitrile, 2-MeTHF, and DMF.
[A52] The method according to [A44], wherein the condensation reagent in the linking step is COMU, and the solvent in the linking step is acetonitrile or 2-MeTHF.
[A53] The method according to [A44], wherein the condensation reagent in the linking step is COMU, and the solvent in the linking step is a mixed solvent of acetonitrile, 2-MeTHF, and DMF.
[A54] The method according to any one of [A1] to [A53], wherein the linking step is carried out in the presence of a base.
[A55] The method according to [A54], wherein the base in the linking step is an organic base.
[A56] The method according to [A54], wherein the base in the linking step is an organic base containing a tertiary amine.
[A57] The base in the linking step is 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN), 7-methyl-1,5 The method according to [A54], wherein the compound is one or more selected from the group consisting of ,7-triazabicyclo[4.4.0]dec-5-ene, 1,1,3,3-tetramethylguanidine (TMG), 1,8-bis(tetramethylguanidino)naphthalene (TMGN), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG), triethylamine (TEA), trimethylamine, 1-methylpiperidine, N,N'-dimethylpiperazine, N-ethylmorpholine, and p-dimethylaminopyridine (DMAP).
[A58] The method according to [A54], wherein the base in the linking step is one or more selected from the group consisting of 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, and pyridine.
[A59] The method according to [A54], wherein the condensation reagent in the linking step is HATU, the solvent in the linking step is acetonitrile or 2-MeTHF, and the base in the linking step is N,N-diisopropylethylamine (DIPEA).
[A60] The method according to [A54], wherein the condensation reagent in the linking step is HATU, the solvent in the linking step is a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF, and the base in the linking step is N,N-diisopropylethylamine (DIPEA).
[A61] The method according to [A54], wherein the condensation reagent in the linking step is COMU, the solvent in the linking step is acetonitrile or 2-methyltetrahydrofuran, and the base in the linking step is N-methylmorpholine or 2,6-lutidine.
[A62] The method according to [A54], wherein the condensation reagent in the linking step is COMU, the solvent in the linking step is a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF, and the base in the linking step is N-methylmorpholine or 2,6-lutidine.
[A62-1] The method according to [A54], wherein the condensation reagent in the linking step is HATU, the solvent in the linking step is a mixed solvent of acetonitrile and 2-methyltetrahydrofuran, and the base in the linking step is N-methylmorpholine.
[A62-2] The method according to [A54], wherein the condensation reagent in the linking step is HATU, the solvent in the linking step is acetonitrile, and the base in the linking step is N-methylmorpholine.
[A63] The method according to any one of [A1] to [A62], wherein the linking step is carried out by a liquid phase method.
[A64] The method according to any one of [A1] to [A63], wherein column chromatography is used for isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof.
[A65] The method according to any one of [A1] to [A63], wherein column chromatography is not used for isolation and/or purification of the cyclic peptide compound, or a salt thereof, or a solvate thereof.
[A66] The method according to any one of [A1] to [A65], further comprising a step of isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof by crystallization to obtain the cyclic peptide compound, or a salt thereof, or a crystal thereof.
[A67] The method according to any one of [A1] to [A66], wherein R 1 is C 3 -C 4 alkyl.
[A67-1] The method according to any one of [A1] to [A66], wherein R 1 is n-propyl.
[A67-2] The method according to any one of [A1] to [A66], wherein R 1 is 2-methylpropyl.
[A68] The method according to any one of [A1] to [A67], wherein P 1 is C 1 -C 4 alkyl.
[A68-1] The method according to any one of [A1] to [A67], wherein P 1 is methyl.
[A69] The method according to any one of [A1] to [A68], wherein R 2 is C 3 -C 4 alkyl.
[A69-1] The method according to any one of [A1] to [A68], wherein R 2 is 1-methylpropyl.
[A70] The method according to any one of [A1] to [A69], wherein R 3 is hydrogen or R 3 forms a 5-membered saturated heterocycle together with P 3 , the carbon atom to which R 3 is bonded, and the nitrogen atom to which P 3 is bonded.
[A70-1] The method according to any one of [A1] to [A69], wherein R 3 is hydrogen.
[A70-2] The method according to any one of [A1] to [A69], wherein R 3 forms a 5-membered saturated heterocycle together with P 3 , the carbon atom to which R 3 is bonded, and the nitrogen atom to which P 3 is bonded.
[A71] The method according to any one of [A1] to [A70], wherein P3 is C 1 -C 4 alkyl, or P3 forms a 5-membered saturated heterocycle together with R3 , the carbon atom to which R3 is bonded, and the nitrogen atom to which P3 is bonded.
[A71-1] The method according to any one of [A1] to [A70], wherein P3 is methyl.
[A71-2] The method according to any one of [A1] to [A70], wherein P3 forms a 5-membered saturated heterocycle together with R3 , the carbon atom to which R3 is bonded, and the nitrogen atom to which P3 is bonded.
[A72] The method according to any one of [A1] to [A71], wherein P4 is C 1 -C 4 alkyl.
[A72-1] The method according to any one of [A1] to [A71], wherein P4 is methyl.
[A73] The method according to any one of [A1] to [A72], wherein R 5 is benzyl optionally substituted by C 1 -C 4 haloalkyl.
[A73-1] The method according to any one of [A1] to [A72], wherein R 5 is 4-trifluoromethylbenzyl.
[A74] The method according to any one of [A1] to [A73], wherein P6 is C 1 -C 4 alkyl.
[A74-1] The method according to any one of [A1] to [A73], wherein P6 is methyl.
[A75] The method according to any one of [A1] to [A74], wherein R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy.
[A75-1] The method according to any one of [A1] to [A74], wherein R 7 is 3-methoxy-4-trifluoromethylphenethyl.
[A75-2] The method according to any one of [A1] to [A74], wherein R 7 is 3,5-difluoro-4-trifluoromethylphenethyl.
[A76] The method according to any one of [A1] to [A75], wherein R 8 together with P 8 , the carbon atom to which R 8 is bonded, and the nitrogen atom to which P 8 is bonded forms a 5-membered saturated heterocycle, and the 5-membered saturated heterocycle is substituted with C 1 -C 4 alkyl.
[A76-1] The method according to any one of [A1] to [ A75 ], wherein R 8 forms a 5-membered saturated heterocycle together with P 8 , the carbon atom to which R 8 is bonded, and the nitrogen atom to which P 8 is bonded, and the 5-membered saturated heterocycle is substituted with ethoxy.
[A77] The method according to any one of [A1] to [A76], wherein R 9 forms a 4- to 6-membered alicyclic ring together with Q 9 and the carbon atom to which R 9 and Q 9 are bonded.
[A77-1] The method according to any one of [A1] to [A76], wherein R 9 forms a 4-membered alicyclic ring together with Q 9 and the carbon atom to which R 9 and Q 9 are bonded.
[A77-2] The method according to any one of [A1] to [A76], wherein R 9 forms a 5-membered alicyclic ring together with Q 9 and the carbon atom to which R 9 and Q 9 are bonded.
[A78] The method according to any one of [A1] to [A77], wherein P 9 is hydrogen or C 1 -C 4 alkyl.
[A78-1] The method according to any one of [A1] to [A77], wherein P 9 is hydrogen.
[A78-2] The method according to any one of [A1] to [A77], wherein P 9 is methyl.
[A79] The method according to any one of [A1] to [A78], wherein R 10 is C 4 -C 6 cycloalkyl.
[A79-1] The method according to any one of [A1] to [A78], wherein R 10 is cyclopentyl.
[A80] The method according to any one of [A1] to [A79], wherein P 10 is C 1 -C 4 alkyl.
[A80-1] The method according to any one of [A1] to [A79], wherein P10 is methyl.
[A81] The method according to any one of [A1] to [A80], wherein R 11 is diC 1 -C 4 alkylaminocarbonyl, or 5- to 6-membered cyclic aminocarbonyl.
[A81-1] The method according to any one of [A1] to [A80], wherein R 11 is dimethylaminocarbonyl.
[A82] The method according to any one of [A1] to [A81], wherein P 11 is C 1 -C 4 alkyl.
[A82-1] The method according to any one of [A1] to [A81], wherein P 11 is methyl.
[A83] The method according to any one of [A1] to [A82], wherein X 1 , X 3 , and X 5 are each independently one selected from the group consisting of hydrogen, a carbamate protecting group, an acyl protecting group, a sulfonamide protecting group, and a silyl protecting group.
[A84] The method according to [A83], wherein the carbamate protecting group is one selected from the group consisting of an Fmoc group, a Cbz group, a Troc group, an Alloc group, a Teoc group, a TSoc group, a BIBSoc group, an IPCSoc group, a BBSoc group, a CHBSoc group, a CDBSoc group, and a Boc group.
[A85] The method according to [A83], wherein the acyl-based protecting group is one selected from the group consisting of a trifluoroacetyl group, an acetyl group, and a benzoyl group.
[A86] The method according to [A83], wherein the sulfonamide protecting group is one selected from the group consisting of a 2-nitrobenzenesulfonyl group, a 4-nitrobenzenesulfonyl group, and a 2,4-dinitrobenzenesulfonyl group.
[A87] The method according to [A83], wherein the silyl protecting group is one selected from the group consisting of a TMS group, a TBDMS group, a TES group, a TIPS group, and a TBDPS group.
[A88] The method according to any one of [A1] to [A87], wherein X 1 is hydrogen or a carbamate protecting group.
[A88-1] The method according to any one of [A1] to [A87], wherein X 1 is hydrogen.
[A88-2] The method according to any of [A1] to [A87], wherein X 1 is an Fmoc group.
[A89] The method according to any one of [A1] to [A88], wherein X3 is hydrogen or a carbamate protecting group.
[A89-1] The method according to any one of [A1] to [A88], wherein X 3 is hydrogen.
[A89-2] The method according to any of [A1] to [A88], wherein X 3 is a Cbz group.
[A90] The method according to any one of [A1] to [A89], wherein X 5 is hydrogen or a carbamate protecting group.
[A90-1] The method according to any one of [A1] to [A89], wherein X 5 is hydrogen.
[A90-2] The method according to any of [A1] to [A89], wherein X5 is a Cbz group.
[A91] The method according to any one of [A1] to [A90], wherein X 2 , X 4 , and X 6 are each independently a halogen, a hydroxyl group, an optionally substituted C 1 -C 6 alkoxy, an optionally substituted C 6 -C 10 aryloxy, an optionally substituted C 7 -C 14 aralkoxy, an optionally substituted 4- to 8-membered cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently a C 1 -C 6 alkyl, or a C 6 -C 10 aryl).
[A91-1] The method according to any one of [A1] to [A90], wherein the halogen is chlorine or bromine.
[A91-2] The method according to any one of [A1] to [A90], wherein the optionally substituted alkoxy is t-butoxy, methoxy, ethoxy, or isopropoxy.
[A91-3] The method according to any one of [A1] to [A90], wherein the optionally substituted aryloxy is pentafluorophenyloxy or nitrophenyloxy.
[A91-4] The method according to any one of [A1] to [A90], wherein the optionally substituted aralkoxy is optionally substituted benzyloxy.
[A91-5] The method according to any of [A1] to [A90], wherein the optionally substituted cyclic aminooxy is N-hydroxysuccinic acid iminoxy.
[ A91-6 ] The method according to any one of [A1] to [A90 ] , wherein the group represented by -OSiRxRyRz is trimethylsilyloxy, triethylsilyloxy, triisopropylsilyloxy, triphenylsilyloxy, tri-t-butylsilyloxy, di-t-butylisobutylsilyloxy, or tris(triethylsilyl)silyloxy.
[A92] The method according to any one of [A1] to [A91], wherein X 2 is a hydroxyl group, t-butoxy, or benzyloxy.
[A92-1] The method according to any one of [A1] to [A91], wherein X 2 is a hydroxyl group.
[A92-2] The method according to any of [A1] to [A91], wherein X 2 is t-butoxy.
[A93] The method according to any one of [A1] to [A92], wherein X 4 is a hydroxyl group, t-butoxy, or benzyloxy.
[A93-1] The method according to any one of [A1] to [A92], wherein X 4 is a hydroxyl group.
[A93-2] The method according to any of [A1] to [A92], wherein X 4 is t-butoxy.
[A94] The method according to any one of [A1] to [A93], wherein X 6 is a hydroxyl group, t-butoxy, or benzyloxy.
[A94-1] The method according to any one of [A1] to [A93], wherein X 6 is a hydroxyl group.
[A94-2] The method according to any one of [A1] to [A93], wherein X 6 is t-butoxy.
[A95] The method according to any one of [A1] to [A94], wherein the cyclic peptide compound, or a salt thereof, or a solvate thereof is a solvate of the cyclic peptide compound.
[A95-1] The method according to [A95], wherein the solvate of the cyclic peptide compound is a hydrate of the cyclic peptide compound.
[A96] The cyclic peptide compound is represented by formula (1a):
[A97] The cyclic peptide compound is represented by formula (1a):
[A97-1] The method according to [A97], wherein the crystal of the cyclic peptide compound is a non-solvate crystal or a solvate crystal.
[A97-2] The method according to [A97], wherein the crystal of the cyclic peptide compound is a solvate crystal.
[A97-3] The method according to [A97-2], wherein the solvate crystal of the cyclic peptide compound is a hydrate crystal.
[B1] A method for producing a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, comprising a step of cyclization (cyclization step) of reacting an N-terminal amino acid residue with a C-terminal amino acid residue of a peptide compound represented by formula (2a) or (3a) in a solvent.
X2 and X4 each independently represent a hydroxyl group, an optionally substituted alkoxy group, an optionally substituted aryloxy group, an optionally substituted aralkoxy group, an optionally substituted cyclic aminooxy group, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz each independently represent an alkyl group or an aryl group).
[B2] A method for producing a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, comprising the steps of:
(a) providing a peptide compound represented by any one of formulas (4a) to (6a), or a salt thereof, or a solvate of said peptide compound or salt;
(b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4a) to (6a) in a solvent (linking step); and
(c) the step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in the step (b) by reacting them in a solvent (cyclization step).
X 2 , X 4 and X 6 each independently represent a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y and R z each independently represent an alkyl or an aryl).
[B3] In the step (b),
(b-1) The method according to [B2], which comprises a step (linking step) of reacting and linking the N-terminal amino acid residue of a peptide compound represented by formula (5a) and the C-terminal amino acid residue of a peptide compound represented by formula (6a) in a solvent to convert them into a peptide compound represented by formula (7a).
[B4] In the step (b), further
(b-2) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4a) and the C-terminal amino acid residue of the peptide compound represented by formula (7a) in a solvent to convert them into the peptide compound represented by formula (2a) (linking step); and
In the step (c),
(c-1) The method according to [B3], which comprises a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2a) in a solvent (cyclization step).
[B5] In the step (b), further
(b-3) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7a) and the C-terminal amino acid residue of the peptide compound represented by formula (4a) in a solvent to convert them into the peptide compound represented by formula (3a) (linking step); and
In the step (c),
(c-2) The method according to [B3], which comprises a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3a) by reacting them in a solvent (cyclization step).
[B6] The method according to any one of [B1] to [B5], wherein the solvent in the cyclization step is a solvent according to any one of [A8] to [A12].
[B7] The method according to any one of [B1] to [B6], wherein the cyclization step is carried out in the presence of a condensation reagent.
[B8] The method according to [B7], wherein the condensation reagent in the cyclization step is a condensation reagent according to any one of [A14] to [A17].
[B9] The method according to [B7], wherein the solvent and condensation reagent in the cyclization step are the solvent and condensation reagent according to any one of [A18] to [A21].
[B10] The method according to any one of [B1] to [B9], wherein the cyclization step is carried out in the presence of a base.
[B11] The method according to [B10], wherein the base in the cyclization step is a base according to any one of [A24] to [A26].
[B12] The method according to [B10], wherein the solvent, condensation reagent, and base in the cyclization step are the solvent, condensation reagent, and base according to any one of [A27] to [A30].
[B13] The method according to any one of [B1] to [B12], wherein the cyclization step is carried out by a liquid phase method.
[B14] The method according to any one of [B1] to [B13], wherein in the cyclization step, the peptide compound and the base are mixed into a mixed solution obtained by mixing a solvent for the cyclization step with the condensation reagent.
[B15] The method according to any one of [B1] to [B14], characterized in that the content of all by-products produced in the cyclization step is less than 20%, less than 15%, less than 10%, less than 5%, or less than 3%, based on the total amount of the product, as determined by the UVarea value at 220 nm by HPLC analysis.
[B16] The method according to any one of [B1] to [B15], characterized in that the content of each by-product generated in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount, based on the total amount of the product, as determined by the UV area value at 220 nm by HPLC analysis.
[B17] The method according to any one of [B1] to [B16], characterized in that the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount based on the total amount of the product, as determined by the UV area value at 220 nm by HPLC analysis, and the by-products include epimers and/or cyclic dimers.
[B18] The method according to any one of [B1] to [B17], wherein a by-product produced in the cyclization step contains the epimer, and the content of the epimer is less than 10%, less than 7.5%, less than 5%, less than 2.5%, or less than 1%, based on the total amount of the product, as determined by a UV area value at 220 nm by HPLC analysis.
[B19] The method according to any one of [B1] to [B18], characterized in that a by-product produced in the cyclization step contains a cyclic dimer, and the content of the cyclic dimer is less than 15%, less than 10%, less than 5%, less than 2.5%, or less than 1%, based on the total amount of the product, as determined by a UVarea value at 220 nm by HPLC analysis.
[B20] The method according to any one of [B1] to [B19], wherein the solvent in the linking step is a solvent according to any one of [A38] to [A43].
[B21] The method according to any one of [B1] to [B20], wherein the linking step is carried out in the presence of a condensation reagent.
[B22] The method according to [B21], wherein the condensation reagent in the linking step is a condensation reagent according to any one of [A45] to [A49].
[B23] The method according to [B21], wherein the solvent and condensation reagent in the linking step are the solvent and condensation reagent according to any one of [A50] to [A53].
[B24] The method according to any one of [B1] to [B23], wherein the ligation step is carried out in the presence of a base.
[B25] The method according to [B24], wherein the base in the linking step is a base according to any one of [A55] to [A59].
[B26] The method according to [B24], wherein the solvent, condensation reagent and base in the linking step are the solvent, condensation reagent and base according to any one of [A60] to [A62].
[B27] The method according to any one of [B1] to [B26], wherein the linking step is carried out by a liquid phase method.
[B28] The method according to any one of [B1] to [B27], wherein column chromatography is used for isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof.
[B29] The method according to any one of [B1] to [B27], wherein column chromatography is not used for isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof.
[B30] The method according to any one of [B1] to [B29], further comprising a step of isolating and/or purifying the cyclic peptide compound, or a salt thereof, or a solvate thereof by crystallization to obtain the cyclic peptide compound, or a salt thereof, or a crystal thereof.
[C1] A compound represented by formula (4a) or a salt thereof.
X2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
[C2] The compound or salt thereof according to [C1], wherein X 1 is hydrogen.
[C3] The compound or salt thereof according to [C1], wherein X 1 is an Fmoc group.
[C4] The compound or salt thereof according to any one of [C1] to [C3], wherein X 2 is t-butoxy.
[C5] tert-Butyl 2-[methyl-[(2S)-2-[(4Z,7S)-7-(methylamino)-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]amino]acetic acid (compound 9).
[C6] A compound represented by formula (5a) or a salt thereof.
X4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
[C7] The compound or a salt thereof according to [C6], wherein X 3 is hydrogen.
[C8] The compound or salt thereof according to [C6], wherein X 3 is an Fmoc group or a Cbz group.
[C9] The compound or salt thereof according to any one of [C6] to [C8], wherein X 4 is t-butoxy.
[C10] tert-Butyl (2S)-1-[(2S,3S)-3-methyl-2-[[(2S)-2-(methylamino)pentanoyl]amino]pentanoyl]pyrrolidine-2-acetic acid (compound 13).
[C11] A compound represented by formula (6a) or a salt thereof.
X6 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
[C12] The compound or salt thereof according to [C11], wherein X 5 is hydrogen.
[C13] The compound or salt thereof according to [C11], wherein X 5 is a Cbz group.
[C14] The compound or a salt thereof according to any one of [C11] to [C13], wherein X 6 is a hydroxyl group or t-butoxy.
[C15] (3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid (compound 20).
[C16] A compound represented by formula (7a) or a salt thereof.
X4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
[C17] The compound or a salt thereof according to [C16], wherein X 5 is hydrogen.
[C18] The compound or salt thereof according to [C16], wherein X 5 is a Cbz group.
[C19] The compound or a salt thereof according to any one of [C16] to [C18], wherein X 6 is a hydroxyl group or t-butoxy.
[C20] (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentylacetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylic acid (compound 22).
[C20-1] tert-butyl (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate (compound 37).
[C21] A compound represented by formula (2a) or a salt thereof.
X2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
[C22] The compound or a salt thereof according to [C21], wherein X 5 is hydrogen.
[C23] The compound or a salt thereof according to [C21], wherein X 5 is a Cbz group.
[C24] The compound or a salt thereof according to any one of [C21] to [C23], wherein X 2 is a hydroxyl group or t-butoxy.
[C25] 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino ]-2-Cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carbonyl]-methyl-amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetic acid (compound 24).
[C26] A compound represented by the formula (3a) or a salt thereof.
X4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently an alkyl or an aryl).
[C27] The compound or a salt thereof according to [C26], wherein X 1 is hydrogen.
[C28] The compound or salt thereof according to [C26], wherein X 1 is an Fmoc group.
[C29] The compound or a salt thereof according to any one of [C26] to [C28], wherein X 4 is a hydroxyl group or t-butoxy.
[C30] (S)-2-[(S)-3-[(S)-2-cyclopentyl-2-[1-[(2S,4R)-4-ethoxy-1-[(S)-4-[3-methoxy-4-(trifluoromethyl)phenyl]-2-[(2-[(S)-N-methyl-2-[(R,Z)-3-(methylamino)-2-oxo-3,4,7,8-tetrahydroazocin-1(2H)-yl]-3-[4-(trifluoromethyl)phenyl]propanamido]acetamido)butanoyl]-N-methylpyrrolidine-2-carboxamide]-N-methylcyclobutane-1-carboxamide]-N-methylacetamide]-4-(dimethylamino)-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleucyl-L-proline (compound 40).
[C31] A method for producing the compound according to any one of [C1] to [C30], which does not use solid-phase synthesis.
[C32] A method for producing a cyclic peptide compound represented by formula (1a), which does not use solid-phase synthesis.
[D1] A crystal of a cyclic peptide compound represented by formula (1a), a salt thereof, or a solvate thereof.
[D3] The crystal according to [D2], which is a solvate crystal of a cyclic peptide compound.
[D4] The crystal according to [D3], wherein the solvate crystal is a hydrate crystal of a cyclic peptide compound.
[D5] The crystal according to [D4], wherein the hydrate crystal is a Form A crystal having at least seven peaks selected from the group consisting of 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D5-1] The crystal according to [D4], wherein the hydrate crystal is a Form A crystal comprising at least eight peaks selected from the group consisting of 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D5-2] The crystal according to [D4], wherein the hydrate crystal is a Form A crystal having peaks at the following diffraction angles (2θ values) in powder X-ray diffraction: 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°).
[D5-3] The Form A crystal according to any one of [D5] to [D5-2], wherein the diffraction angle (2θ value) is a diffraction angle (2θ value) of a hydrate crystal stored at a relative humidity of 10% or more for 15 minutes or more.
[D6] The crystal according to [D4], wherein the hydrate crystal is a Form B crystal having at least seven peaks selected from the group consisting of 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D6-1] The crystal according to [D4], wherein the hydrate crystal is a Form B crystal comprising at least eight peaks selected from the group consisting of 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°) as diffraction angles (2θ values) in powder X-ray diffraction.
[D6-2] The crystal according to [D4], wherein the hydrate crystal is a Form B crystal having peaks at the following diffraction angles (2θ values) in powder X-ray diffraction: 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°).
[D6-3] The Form B crystal according to any one of [D6] to [D6-2], wherein the diffraction angle (2θ value) is a diffraction angle (2θ value) of a hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more.
[D7] The crystal according to [D3], wherein the solvate crystal is a Form F crystal comprising at least seven peaks selected from the group consisting of 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° (±0.2°) as diffraction angles (2θ values) in powder X-ray diffraction.
[D7-1] The crystal according to [D3], wherein the solvate crystal is a Form F crystal comprising at least eight peaks selected from the group consisting of 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° (±0.2°) as diffraction angles (2θ values) in powder X-ray diffraction.
[D7-2] The crystal according to [D3], wherein the solvate crystal is a Form F crystal comprising peaks at the following diffraction angles (2θ values) in powder X-ray diffraction: 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° (±0.2°).
[D7-3] The crystal according to any one of [D7] to [D7-2], wherein the solvate crystal is an acetone/heptane/hydrate.
[D8] The crystal according to [D4], wherein the hydrate crystal is a Form J crystal having at least seven peaks selected from the group consisting of 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D8-1] The crystal according to [D4], wherein the hydrate crystal is a Form J crystal comprising at least eight peaks selected from the group consisting of 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D8-2] The crystal according to [D4], wherein the hydrate crystal is a Form J crystal having peaks at the following diffraction angles (2θ values) in powder X-ray diffraction: 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°).
[D8-3] The Form J crystal according to any one of [D8] to [D8-2], wherein the diffraction angle (2θ value) is a diffraction angle (2θ value) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more.
[D9] The crystal according to [D4], wherein the hydrate crystal is a Form Y crystal having at least seven peaks selected from the group consisting of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D9-1] The crystal according to [D4], wherein the hydrate crystal is a Form Y crystal comprising at least eight peaks selected from the group consisting of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D9-2] The crystal according to [D4], wherein the hydrate crystal is a Form Y crystal having peaks at the following diffraction angles (2θ values) in powder X-ray diffraction: 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°).
[D9-3] The Form Y crystal according to any one of [D9] to [D9-2], wherein the diffraction angle (2θ value) is a diffraction angle (2θ value) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes or more.
[D10] The crystal according to [D3], wherein the hydrate crystal is a Form K crystal having at least seven peaks selected from the group consisting of 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° (±0.2°) as diffraction angles (2θ values) by powder X-ray diffraction.
[D10-1] The crystal according to [D3], wherein the hydrate crystal is a Form K crystal comprising at least eight peaks selected from the group consisting of 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° (±0.2°) as diffraction angles (2θ values) in powder X-ray diffraction.
[D10-2] The crystal according to [D3], wherein the solvate crystal is a Form K crystal comprising peaks at the following diffraction angles (2θ values) in powder X-ray diffraction: 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° (±0.2°).
[D11] The crystal according to [D3], wherein the solvate crystal is a dimethylsulfoxide/heptane/hydrate crystal of Form G having the structure shown in Figure 1 by single crystal X-ray analysis.
[D11-1] The crystal according to [D3], wherein the solvate crystal is a 2-propanol/heptane/hydrate crystal of Form E having the structure shown in FIG. 3 by single crystal X-ray analysis.
[D11-2] The crystal according to [D3], wherein the solvate crystal is an ethanol/hydrate crystal of Form H having the structure shown in Figure 8 by single crystal X-ray analysis.
[D11-3] The crystal according to [D3], wherein the solvate crystal is a 1,4-dioxane/hydrate crystal of Form D having the structure shown in FIG. 26 by single crystal X-ray analysis.
[D11-4] The crystal according to [D3], wherein the solvate crystal is a dimethylsulfoxide/hydrate crystal of Form L having the structure shown in FIG. 28 by single crystal X-ray analysis.
[D11-5] The crystal according to [D3], wherein the solvate crystal is a propylene glycol/hydrate crystal of Form M having the structure shown in FIG. 31 by single crystal X-ray analysis.
[D11-6] The crystal according to [D3], wherein the solvate crystal is a propylene glycol/hydrate crystal of Form M having the diffraction angle shown in FIG. 29 by powder X-ray diffraction.
[D11-7] The crystal according to [D3], wherein the solvate crystal is a propylene glycol/hydrate crystal of Form M, the data of which is shown in FIG. 30(A) by simultaneous thermogravimetry and differential thermal analysis.
[D11-8] The crystal according to [D3], wherein the solvate crystal is a propylene glycol solvate crystal of Form N having the diffraction angle shown in FIG. 29 by powder X-ray diffraction.
[D11-9] The crystal according to [D3], wherein the solvate crystal is a propylene glycol solvate crystal of Form N showing the data of simultaneous thermogravimetry and differential thermal analysis shown in FIG. 30 (B).
[D12] A method for producing a crystal of a cyclic peptide compound according to any one of [D1] to [D10-2], comprising the steps of dissolving the cyclic peptide compound in an amount of a polar organic solvent in which the cyclic peptide compound can be dissolved to obtain a solution, and adding a hydrocarbon solvent or water to the solution to obtain a crystal of the cyclic peptide compound.
[D12-1] The method according to [D12], wherein the purity of the starting cyclic peptide compound is 85% or more.
[D13] A method for producing a crystal of a cyclic peptide compound according to any one of [D1] to [D10-2], comprising the step of adding a mixture of a hydrocarbon solvent and a polar organic solvent, or a mixture of water and a polar organic solvent, to the cyclic peptide compound in an amorphous state to obtain a crystal of the cyclic peptide compound.
[D14] The method according to any one of [D12] to [D13-1], wherein the polar organic solvent is one or more selected from the group consisting of DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, propylene glycol, 1,4-dioxane, and ethyl acetate.
[D15] The method according to [D14], wherein the polar organic solvent is acetone.
[D15-1] The method according to [D14], wherein the polar organic solvent is ethanol.
[D16] The method according to any one of [D12] to [D13-1], wherein the hydrocarbon solvent is one or more selected from the group consisting of heptane, hexane, pentane, toluene, and xylene.
[D17] The method according to [D16], wherein the hydrocarbon solvent is heptane.
[D18] A method for producing a crystal of a cyclic peptide compound according to any one of [D1] to [D10-2], comprising the steps of dissolving the cyclic peptide compound in an amorphous state in DMSO to obtain a solution, freeze-drying the solution to obtain a freeze-dried product of the cyclic peptide compound, and adding a mixture of water and a polar organic solvent to the freeze-dried product to obtain a crystal of the cyclic peptide compound.
[D19] The method according to [D18], wherein the polar organic solvent is one or more selected from the group consisting of DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, 1,4-dioxane and propylene glycol.
[D20] The method according to [D19], wherein the polar organic solvent is acetone.
[D21] The method according to any one of [D12] to [D20], further comprising a step of filtering the crystals after the step of obtaining the crystals of the cyclic peptide compound.
[D22] The method according to any one of [D12] to [D21], further comprising a step of drying the crystals after the step of obtaining the crystals of the cyclic peptide compound.
[D23] The method according to any one of [D12] to [D22], wherein the crystal of the cyclic peptide compound is a solvate crystal.
[D24] The method according to [D23], wherein the solvate crystal of the cyclic peptide compound is a hydrate crystal.
[D25] The method according to [D21] or [D22], wherein the crystals of the cyclic peptide compound are formed as solvate crystals in a solvent and are obtained as hydrate crystals after a filtering step and/or a drying step.
[D26] A composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, wherein the compound contains an impurity, the cyclic dimer of formula (1a), at a ratio of 1.5 wt % or less.
本発明によれば、副生成物である環状ダイマーの生成を抑制して、環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を効率的に製造することができる。本発明の製造方法は、ペプチド化合物の製造コストを削減でき、環境負荷も軽減できるため、大規模スケールでのペプチドの合成に特に有用である。 According to the present invention, it is possible to efficiently produce a cyclic peptide compound, or a salt or solvate thereof, while suppressing the production of the by-product cyclic dimer. The production method of the present invention is particularly useful for the synthesis of peptides on a large scale, since it can reduce the production costs of peptide compounds and reduce the environmental burden.
略語
本明細書において使用される略語を以下に記す。
2-MeTHF:2-メチルテトラヒドロフラン
20% Pip/DMF:20%ピぺリジンを含むN,N-ジメチルホルムアミド溶液
EtOAc:酢酸エチル
Alloc:アリルオキシカルボニル
BEP:2-ブロモ-1-エチルピリジニウム テトラフルオロホウ酸塩
BHT:2,6-ジ-tert-ブチル-4-メチルフェノール
Boc:t-ブトキシカルボニル
Cbz:ベンジルオキシカルボニル
COMU:(1-シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ-モルホリノ-カルベニウムヘキサフルオロリン酸塩
CPME:シクロペンチルメチルエーテル
CSA:10-カンファースルホン酸
DCM:ジクロロメタン
DEPBT:りん酸ジエチル3,4-ジヒドロ-4-オキソ-1,2,3-ベンゾトリアジン-3-イル
DIPEA:N,N-ジイソプロピルエチルアミン
DMA:N,N-ジメチルアセトアミド
DMAP:4-ジメチルアミノピリジン
DMF:N,N-ジメチルホルムアミド
DMSO:ジメチルスルホキシド
DMT-MM:4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド
EDCI:1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド
FDPP:ペンタフルオロフェニルジフェニルホスフィナート
FmocまたはFMOC:9-フルオレニルメチルオキシカルボニル
HATU:O-(7-アザベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムヘキサフルオロリン酸塩
HMDS:1,1,1,3,3,3-ヘキサメチルジシラザン
HOAt:1-ヒドロキシ-7-アザベンゾトリアゾール
HOBt:1-ヒドロキシベンゾトリアゾール
IPAc:酢酸イソプロピル
MeCN:アセトニトリル
MTBE:メチルtert-ブチルエーテル
MTHP:4-メチルテトラヒドロピラン
NMM:4-メチルモルホリン
NMP:N-メチルピロリドン
PyBOP:1H-ベンゾトリアゾール-1-イルオキシトリピロリジノホスホ二ウムヘキサフルオロリン酸塩
PyClop:クロロトリピロリジノホスホニウムヘキサフルオロホスファート
PyOxim:(エチルシアノ(ヒドロキシイミノ)アセタト-O2)-トリ-(1-ピロリジニル)-ホスホニウム ヘキサフルオロリン酸塩
T3P:プロピルホスホン酸無水物
TBAF:テトラブチルアンモニウムフルオリド
Teoc:2-(トリメチルシリル)エトキシカルボニル
TFA:トリフルオロ酢酸
THF:テトラヒドロフラン
TMSOTf:トリフルオロメタンスルホン酸トリメチルシリル
Troc:2,2,2-トリクロロエトキシカルボニル
PDA:フォトダイオードアレイ
FA:ギ酸
qNMR:定量核磁気共鳴
UPLC:Ultra-Performance Liquid Chromatography (trademark of the Waters Corporation)
HPLC:高速液体クロマトグラフ
Et:エチル
DBU:1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン
ESI:エレクトロスプレーイオン化
Bu:ブチル
Me:メチル
oxyma:シアノ(ヒドロキシイミノ)酢酸エチル
DIC:N,N'-ジイソプロピルカルボジイミド
TFE:2,2,2-トリフルオロエタノール
IPA:2-プロパノール
NMI:1-メチルイミダゾール
TCFH:クロロ-N,N,N',N'-テトラメチルホルムアミジニウム ヘキサフルオロホスファート
NMR:核磁気共鳴
TMS:テトラメチルシラン
Abbreviations The following abbreviations are used in this specification:
2-MeTHF: 20% 2-methyltetrahydrofuran Pip/DMF: 20% piperidine in N,N-dimethylformamide
EtOAc: ethyl acetate
Alloc: Allyloxycarbonyl
BEP: 2-bromo-1-ethylpyridinium tetrafluoroborate
BHT: 2,6-di-tert-butyl-4-methylphenol
Boc: t-butoxycarbonyl
Cbz: benzyloxycarbonyl
COMU: (1-cyano-2-ethoxy-2-oxoethylideneaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate
CPME: Cyclopentyl methyl ether
CSA: 10-camphorsulfonic acid
DCM: dichloromethane
DEPBT: Diethyl 3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl phosphate
DIPEA: N,N-diisopropylethylamine
DMA: N,N-dimethylacetamide
DMAP: 4-dimethylaminopyridine
DMF: N,N-dimethylformamide
DMSO: Dimethyl sulfoxide
DMT-MM: 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
EDCI: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
FDPP: Pentafluorophenyl diphenyl phosphinate
Fmoc or FMOC: 9-fluorenylmethyloxycarbonyl
HATU: O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate
HMDS: 1,1,1,3,3,3-hexamethyldisilazane
HOAt: 1-hydroxy-7-azabenzotriazole
HOBt: 1-hydroxybenzotriazole
IPAc: Isopropyl acetate
MeCN: Acetonitrile
MTBE: Methyl tert-butyl ether
MTHP: 4-methyltetrahydropyran
NMM: 4-methylmorpholine
NMP: N-methylpyrrolidone
PyBOP: 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
PyClop: Chlorotripyrrolidinophosphonium hexafluorophosphate
PyOxim: (Ethylcyano(hydroxyimino)acetato-O2)-tri-(1-pyrrolidinyl)-phosphonium hexafluorophosphate
T3P: Propylphosphonic anhydride
TBAF: Tetrabutylammonium fluoride
Teoc: 2-(trimethylsilyl)ethoxycarbonyl
TFA: Trifluoroacetic acid
THF: Tetrahydrofuran
TMSOTf: Trimethylsilyl trifluoromethanesulfonate
Troc: 2,2,2-trichloroethoxycarbonyl
PDA: Photodiode array
FA: Formic acid
qNMR: Quantitative Nuclear Magnetic Resonance
UPLC: Ultra-Performance Liquid Chromatography (trademark of the Waters Corporation)
HPLC: High Performance Liquid Chromatograph
Et: Ethyl
DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene
ESI: electrospray ionization
Bu: butyl
Me: Methyl
oxyma: ethyl cyano(hydroxyimino)acetate
DIC: N,N'-diisopropylcarbodiimide
TFE: 2,2,2-trifluoroethanol
IPA: 2-propanol
NMI: 1-methylimidazole
TCFH: Chloro-N,N,N',N'-tetramethylformamidinium hexafluorophosphate
NMR: Nuclear magnetic resonance
TMS: Tetramethylsilane
官能基等の定義(本明細書における全ての用語および語句は、当該技術分野において一般的に理解されているものとして使用される。以下に例示するが、これに限定されない。) Definitions of Functional Groups, etc. (All terms and phrases in this specification are used as they are commonly understood in the art. Examples are given below, but are not limited to these.)
本明細書における「ハロゲン」としては、F、Cl、BrまたはIが例示される。 In this specification, examples of "halogen" include F, Cl, Br, and I.
本明細書において「アルキル」とは、脂肪族炭化水素から任意の水素原子を1個除いて誘導される1価の基であり、骨格中にヘテロ原子(炭素及び水素原子以外の原子をいう。)または不飽和の炭素-炭素結合を含有せず、水素及び炭素原子を含有するヒドロカルビルまたは炭化水素基構造の部分集合を有する。アルキルは直鎖状のものだけでなく、分枝鎖状のものも含む。アルキルとして具体的には、炭素原子数1~20(C1-C20、以下「Cp-Cq」とは炭素原子数がp~q個であることを意味する)のアルキルであり、好ましくはC1-C10アルキル、より好ましくはC1-C6アルキルが挙げられる。アルキルとして、具体的には、メチル、エチル、n-プロピル、i-プロピル、n-ブチル、s-ブチル、t-ブチル、イソブチル(2-メチルプロピル)、n-ペンチル、s-ペンチル(1-メチルブチル)、t-ペンチル(1,1-ジメチルプロピル)、ネオペンチル(2,2-ジメチルプロピル)、イソペンチル(3-メチルブチル)、3-ペンチル(1-エチルプロピル)、1,2-ジメチルプロピル、2-メチルブチル、n-ヘキシル、1,1,2-トリメチルプロピル、1,2,2-トリメチルプロピル、1,1,2,2-テトラメチルプロピル、1,1-ジメチルブチル、1,2-ジメチルブチル、1,3-ジメチルブチル、2,2-ジメチルブチル、2,3-ジメチルブチル、3,3-ジメチルブチル、1-エチルブチル、2-エチルブチル等が挙げられる。 As used herein, "alkyl" refers to a monovalent group derived by removing any one hydrogen atom from an aliphatic hydrocarbon, which does not contain heteroatoms (atoms other than carbon and hydrogen atoms) or unsaturated carbon-carbon bonds in the skeleton, and has a subset of hydrocarbyl or hydrocarbon group structures containing hydrogen and carbon atoms. Alkyl includes not only linear but also branched chain alkyls. Specific examples of alkyl include alkyls having 1 to 20 carbon atoms (C 1 -C 20 , hereinafter "C p -C q " means that the number of carbon atoms is p to q), preferably C 1 -C 10 alkyl, and more preferably C 1 -C 6 alkyl. Specific examples of the alkyl include methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, isobutyl (2-methylpropyl), n-pentyl, s-pentyl (1-methylbutyl), t-pentyl (1,1-dimethylpropyl), neopentyl (2,2-dimethylpropyl), isopentyl (3-methylbutyl), 3-pentyl (1-ethylpropyl), 1,2-dimethylpropyl, 2-methylbutyl, n-hexyl, 1,1,2-trimethylpropyl, 1,2,2-trimethylpropyl, 1,1,2,2-tetramethylpropyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-dimethylbutyl, 1-ethylbutyl, and 2-ethylbutyl.
本明細書において「アルケニル」とは、少なくとも1個の二重結合(2個の隣接sp2炭素原子)を有する1価の基である。二重結合及び置換分(存在する場合)の配置によって、二重結合の幾何学的形態は、エントゲーゲン(E)またはツザンメン(Z)、シスまたはトランス配置をとることができる。アルケニルは、直鎖状のものだけでなく、分枝鎖状ものも含む。アルケニルとして好ましくはC2-C10アルケニル、より好ましくはC2-C6アルケニルが挙げられる。アルケニルとして具体的には、例えば、ビニル、アリル、1-プロペニル、2-プロペニル、1-ブテニル、2-ブテニル(シス、トランスを含む)、3-ブテニル、ペンテニル、3-メチル-2-ブテニル、ヘキセニル等が挙げられる。 As used herein, "alkenyl" refers to a monovalent group having at least one double bond (two adjacent sp2 carbon atoms). Depending on the arrangement of the double bond and the substituents (if present), the geometry of the double bond can be in an entgegen (E) or zusammen (Z), cis or trans configuration. Alkenyl includes not only linear but also branched chains. Preferred examples of alkenyl include C 2 -C 10 alkenyl, more preferably C 2 -C 6 alkenyl. Specific examples of alkenyl include vinyl, allyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl (including cis and trans), 3-butenyl, pentenyl, 3-methyl-2-butenyl, hexenyl, and the like.
本明細書において「アルキニル」とは、少なくとも1個の三重結合(2個の隣接sp炭素原子)を有する、1価の基である。アルキニルは、直鎖状のものだけでなく、分枝鎖状のものも含む。アルキニルとして好ましくはC2-C10アルキニル、より好ましくはC2-C6アルキニルが挙げられる。アルキニルとして具体的には、例えば、エチニル、1-プロピニル、プロパルギル、3-ブチニル、ペンチニル、ヘキシニル等が挙げられる。 As used herein, "alkynyl" refers to a monovalent group having at least one triple bond (two adjacent sp carbon atoms). Alkynyl includes not only straight chain but also branched chain. Preferred alkynyl groups include C 2 -C 10 alkynyl groups, more preferably C 2 -C 6 alkynyl groups. Specific examples of alkynyl groups include ethynyl, 1-propynyl, propargyl, 3-butynyl, pentynyl, hexynyl, and the like.
本明細書において「シクロアルキル」とは、飽和または部分的に飽和した環状の1価の脂肪族炭化水素基を意味し、単環、ビシクロ環、スピロ環を含む。シクロアルキルとして好ましくはC3-C8シクロアルキル、より好ましくはC3-C7シクロアルキル、さらに好ましくはC3-C6シクロアルキルが挙げられる。シクロアルキルとして具体的には、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、ビシクロ[2.2.1]ヘプチル、スピロ[3.3]ヘプチル等が挙げられる。 As used herein, the term "cycloalkyl" refers to a saturated or partially saturated cyclic monovalent aliphatic hydrocarbon group, including a monocyclic ring, a bicyclo ring, and a spiro ring. Preferred examples of cycloalkyl include C 3 -C 8 cycloalkyl, more preferably C 3 -C 7 cycloalkyl, and even more preferably C 3 -C 6 cycloalkyl. Specific examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, bicyclo[2.2.1]heptyl, and spiro[3.3]heptyl.
本明細書において「アリール」とは1価の芳香族炭化水素環、すなわち芳香族炭化水素環基を意味する。アリールとして好ましくはC6-C10アリールが挙げられる。アリールとして具体的には、フェニル、ナフチル(例えば、1-ナフチル、2-ナフチル)等が例示される。 In the present specification, "aryl" refers to a monovalent aromatic hydrocarbon ring, i.e., an aromatic hydrocarbon ring group. Preferred examples of aryl include C 6 -C 10 aryl. Specific examples of aryl include phenyl, naphthyl (e.g., 1-naphthyl, 2-naphthyl), and the like.
本明細書において「ヘテロアリール」とは、炭素原子に加えて1~5個のヘテロ原子を含有する、芳香族性の1価の環式基、すなわち芳香族複素環基を意味する。環は単環でも、他の環との縮合環でもよく、部分的に飽和されていてもよい。ヘテロアリールの環を構成する原子の数は好ましくは5~10(5~10員ヘテロアリール)であり、より好ましくは5~7(5~7員ヘテロアリール)である。ヘテロアリールとして具体的には、フリル、チエニル、ピロリル、イミダゾリル、ピラゾリル、チアゾリル、イソチアゾリル、オキサゾリル、イソオキサゾリル、オキサジアゾリル、チアジアゾリル、トリアゾリル、テトラゾリル、ピリジル、ピリミジル、ピリダジニル、ピラジニル、トリアジニル、ベンゾフラニル、ベンゾチエニル、ベンゾチアジアゾリル、ベンゾチアゾリル、ベンゾオキサゾリル、ベンゾオキサジアゾリル、ベンゾイミダゾリル、ベンゾトリアゾリル、インドリル、イソインドリル、インダゾリル、アザインドリル、キノリル、イソキノリル、シンノリニル、キナゾリニル、キノキサリニル、ベンゾジオキソリル、インドリジニル、イミダゾピリジル、ピラゾロピリジル、イミダゾピリジル、トリアゾロピリジル、ピロロピラジニル、フロピリジル等が例示される。 As used herein, "heteroaryl" refers to an aromatic monovalent cyclic group containing 1 to 5 heteroatoms in addition to carbon atoms, i.e., an aromatic heterocyclic group. The ring may be a single ring or a condensed ring with other rings, and may be partially saturated. The number of atoms constituting the heteroaryl ring is preferably 5 to 10 (5- to 10-membered heteroaryl), and more preferably 5 to 7 (5- to 7-membered heteroaryl). Specific examples of heteroaryl include furyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, triazolyl, tetrazolyl, pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl, benzofuranyl, benzothienyl, benzothiadiazolyl, benzothiazolyl, benzoxazolyl, benzoxadiazolyl, benzimidazolyl, benzotriazolyl, indolyl, isoindolyl, indazolyl, azaindolyl, quinolyl, isoquinolyl, cinnolinyl, quinazolinyl, quinoxalinyl, benzodioxolyl, indolizinyl, imidazopyridyl, pyrazolopyridyl, imidazopyridyl, triazolopyridyl, pyrrolopyrazinyl, and furopyridyl.
本明細書において「アラルキル(アリールアルキル)」とは、本明細書定義の「アルキル」の1つまたは複数の水素原子が本明細書定義の「アリール」で置換された基を意味する。アラルキルとして、C7-C14アラルキルが好ましく、C7-C10アラルキルがより好ましい。アラルキルとして具体的には、ベンジル、フェネチル、3-フェニルプロピル等が例示される。 As used herein, "aralkyl (arylalkyl)" refers to a group in which one or more hydrogen atoms of an "alkyl" as defined herein are substituted with an "aryl" as defined herein. As the aralkyl, a C 7 -C 14 aralkyl is preferred, and a C 7 -C 10 aralkyl is more preferred. Specific examples of the aralkyl include benzyl, phenethyl, and 3-phenylpropyl.
本明細書において「ヘテロアリールアルキル」とは、本明細書定義の「アルキル」の1つまたは複数の水素原子が本明細書定義の「ヘテロアリール」で置換された基を意味する。ヘテロアリールアルキルとして、5~10員ヘテロアリールC1-C6アルキルが好ましく、5~10員ヘテロアリールC1-C2アルキルがより好ましい。ヘテロアリールアルキルとして具体的には、3-チエニルメチル、4-チアゾリルメチル、2-ピリジルメチル、3-ピリジルメチル、4-ピリジルメチル、2-(2-ピリジル)エチル、2-(3-ピリジル)エチル、2-(4-ピリジル)エチル、2-(6-キノリル)エチル、2-(7-キノリル)エチル、2-(6-インドリル)エチル、2-(5-インドリル)エチル、2-(5-ベンゾフラニル)エチル等が例示される。 As used herein, the term "heteroarylalkyl" refers to a group in which one or more hydrogen atoms of an "alkyl" as defined herein are substituted with a "heteroaryl" as defined herein. As the heteroarylalkyl, a 5- to 10-membered heteroaryl C 1 -C 6 alkyl is preferred, and a 5- to 10-membered heteroaryl C 1 -C 2 alkyl is more preferred. Specific examples of the heteroarylalkyl include 3-thienylmethyl, 4-thiazolylmethyl, 2-pyridylmethyl, 3-pyridylmethyl, 4-pyridylmethyl, 2-(2-pyridyl)ethyl, 2-(3-pyridyl)ethyl, 2-(4-pyridyl)ethyl, 2-(6-quinolyl)ethyl, 2-(7-quinolyl)ethyl, 2-(6-indolyl)ethyl, 2-(5-indolyl)ethyl, 2-(5-benzofuranyl)ethyl, and the like.
本明細書において「アルコキシ」とは、本明細書定義の「アルキル」が結合したオキシ基を意味する。アルコキシとして、C1-C6アルコキシが好ましく、C1-C4アルコキシがより好ましい。アルコキシとして具体的には、メトキシ、エトキシ、1-プロポキシ、2-プロポキシ、n-ブトキシ、i-ブトキシ、s-ブトキシ、t-ブトキシ、ペンチルオキシ、3-メチルブトキシ等が例示される。 As used herein, "alkoxy" refers to an oxy group bonded to an "alkyl" as defined herein. As the alkoxy, C 1 -C 6 alkoxy is preferred, and C 1 -C 4 alkoxy is more preferred. Specific examples of the alkoxy include methoxy, ethoxy, 1-propoxy, 2-propoxy, n-butoxy, i-butoxy, s-butoxy, t-butoxy, pentyloxy, 3-methylbutoxy, and the like.
本明細書における「アルコキシアルキル」とは、本明細書定義の「アルキル」の1つまたは複数の水素が本明細書定義の「アルコキシ」で置換された基を意味する。アルコキシアルキルとして、C1-C6アルコキシC1-C6アルキルが好ましく、C1-C6アルコキシC1-C2アルキルがより好ましい。アルコキシアルキルとして具体的には、メトキシメチル、エトキシメチル、1-プロポキシメチル、2-プロポキシメチル、n-ブトキシメチル、i-ブトキシメチル、s-ブトキシメチル、t-ブトキシメチル、ペンチルオキシメチル、3-メチルブトキシメチル、1-メトキシエチル、2-メトキシエチル、2-エトキシエチル等が例示される。 As used herein, "alkoxyalkyl" refers to a group in which one or more hydrogen atoms of an "alkyl" as defined herein are substituted with an "alkoxy" as defined herein. As the alkoxyalkyl, C 1 -C 6 alkoxy C 1 -C 6 alkyl is preferred, and C 1 -C 6 alkoxy C 1 -C 2 alkyl is more preferred. Specific examples of the alkoxyalkyl include methoxymethyl, ethoxymethyl, 1-propoxymethyl, 2-propoxymethyl, n-butoxymethyl, i-butoxymethyl, s-butoxymethyl, t-butoxymethyl, pentyloxymethyl, 3-methylbutoxymethyl, 1-methoxyethyl, 2-methoxyethyl, 2-ethoxyethyl, and the like.
本明細書において「アリールオキシ」とは、本明細書定義の「アリール」が結合したオキシ基を意味する。アリールオキシとして、好ましくはC6-C10アリールオキシが挙げられる。アリールオキシとして具体的には、フェノキシ、1-ナフチルオキシ、2-ナフチルオキシ等が例示される。 As used herein, "aryloxy" refers to an oxy group bonded to an "aryl" as defined herein. Preferred examples of aryloxy include C 6 -C 10 aryloxy. Specific examples of aryloxy include phenoxy, 1-naphthyloxy, and 2-naphthyloxy.
本明細書において「アラルコキシ」とは、本明細書定義の「アラルキル」が結合したオキシ基を意味する。アラルコキシとして、C7-C14アラルコキシが好ましく、C7-C10アラルコキシがより好ましい。アラルコキシとして具体的には、ベンジルオキシ、フェネチルオキシ、3-フェニルプロポキシ等が例示される。 As used herein, "aralkoxy" refers to an oxy group bonded to an "aralkyl" as defined herein. As the aralkoxy, C 7 -C 14 aralkoxy is preferred, and C 7 -C 10 aralkoxy is more preferred. Specific examples of the aralkoxy include benzyloxy, phenethyloxy, and 3-phenylpropoxy.
本明細書において「アミノ」とは、狭義には-NH2を意味し、広義には-NRR’を意味し、ここでR及びR’は独立して、水素、アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクリル、アリール、またはヘテロアリールから選択されるか、あるいはR及びR’はそれらが結合している窒素原子と一緒になって環を形成した基を意味する。アミノとして好ましくは、-NH2、モノC1-C6アルキルアミノ、ジC1-C6アルキルアミノ、4~8員環状アミノなどが挙げられる。 In the present specification, "amino" means -NH2 in a narrow sense, and -NRR' in a broad sense, where R and R' are independently selected from hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl, or heteroaryl, or R and R' mean a group forming a ring together with the nitrogen atom to which they are attached. Preferred examples of amino include -NH2 , mono- C1 - C6 alkylamino, di- C1 - C6 alkylamino, and 4- to 8-membered cyclic amino.
本明細書において「モノアルキルアミノ」とは、本明細書定義の「アミノ」のうち、Rが水素であり、かつR’が本明細書定義の「アルキル」である基を意味する。モノアルキルアミノとして、好ましくは、モノC1-C6アルキルアミノが挙げられる。モノアルキルアミノとして具体的には、メチルアミノ、エチルアミノ、n-プロピルアミノ、i-プロピルアミノ、n-ブチルアミノ、s-ブチルアミノ、t-ブチルアミノ等が例示される。 As used herein, "monoalkylamino" refers to a group in which R is hydrogen and R' is an "alkyl" as defined herein, among "amino" as defined herein. Preferred examples of monoalkylamino include mono-C 1 -C 6 alkylamino. Specific examples of monoalkylamino include methylamino, ethylamino, n-propylamino, i-propylamino, n-butylamino, s-butylamino, and t-butylamino.
本明細書において「ジアルキルアミノ」とは、本明細書定義の「アミノ」のうち、R及びR’が独立して本明細書定義の「アルキル」である基を意味する。ジアルキルアミノとして、好ましくは、ジC1-C6アルキルアミノが挙げられる。ジアルキルアミノとして具体的には、ジメチルアミノ、ジエチルアミノ等が例示される。 As used herein, "dialkylamino" refers to an "amino" as defined herein, in which R and R' are independently "alkyl" as defined herein. Preferred examples of dialkylamino include diC 1 -C 6 alkylamino. Specific examples of dialkylamino include dimethylamino and diethylamino.
本明細書において「環状アミノ」とは、本明細書定義の「アミノ」のうち、R及びR’はそれらが結合している窒素原子と一緒になって環を形成する基を意味する。環状アミノとして、好ましくは、4~8員環状アミノが挙げられる。環状アミノとして具体的には、1-アゼチジル、1-ピロリジル、1-ピペリジル、1-ピペラジル、4-モルホリニル、3-オキサゾリジル、1,1-ジオキシドチオモルホリニル-4-イル、3-オキサ-8-アザビシクロ[3.2.1]オクタン-8-イル等が例示される。 In this specification, "cyclic amino" refers to a group in which R and R' form a ring together with the nitrogen atom to which they are attached, as defined in this specification. Preferred examples of cyclic amino include 4- to 8-membered cyclic amino. Specific examples of cyclic amino include 1-azetidyl, 1-pyrrolidyl, 1-piperidyl, 1-piperazyl, 4-morpholinyl, 3-oxazolidyl, 1,1-dioxidethiomorpholinyl-4-yl, 3-oxa-8-azabicyclo[3.2.1]octan-8-yl, and the like.
本明細書において「環状アミノオキシ」とは、本明細書定義の「環状アミノ」が結合したオキシ基を意味する。環状アミノオキシとして、好ましくは、4~8員環状アミノオキシが挙げられる。環状アミノとして具体的には、1-アゼチジルオキシ、1-ピロリジルオキシ、1-ピペリジルオキシ、1-ピペラジルオキシ、4-モルホリニルオキシ、3-オキサゾリジルオキシ、1,1-ジオキシドチオモルホリニル-4-イルオキシ、3-オキサ-8-アザビシクロ[3.2.1]オクタン-8-イルオキシ等が例示される。 In this specification, "cyclic aminooxy" refers to an oxy group to which "cyclic amino" as defined in this specification is bonded. As the cyclic aminooxy, preferably, 4- to 8-membered cyclic aminooxy is mentioned. Specific examples of the cyclic amino include 1-azetidyloxy, 1-pyrrolidyloxy, 1-piperidyloxy, 1-piperazyloxy, 4-morpholinyloxy, 3-oxazolidyloxy, 1,1-dioxidethiomorpholinyl-4-yloxy, 3-oxa-8-azabicyclo[3.2.1]octan-8-yloxy, etc.
本明細書において「アミノカルボニル」とは、本明細書定義の「アミノ」が結合したカルボニル基を意味する。アミノカルボニルとして、好ましくは、-CONH2、モノC1-C6アルキルアミノカルボニル、モノC3-C6シクロアルキルアミノカルボニル、ジC1-C6アルキルアミノカルボニル、4~8員環状アミノカルボニルが挙げられる。アミノカルボニルとして具体的には、-CONH2、ジメチルアミノカルボニル、1-アゼチジニルカルボニル、1-ピロリジニルカルボニル、1-ピペリジニルカルボニル、1-ピペラジニルカルボニル、4-モルホリニルカルボニル、3-オキサゾリジニルカルボニル、1,1-ジオキシドチオモルホリニル-4-イルカルボニル、3-オキサ-8-アザビシクロ[3.2.1]オクタン-8-イルカルボニル等が例示される。 As used herein, "aminocarbonyl" refers to a carbonyl group bonded to "amino" as defined herein. Preferred examples of aminocarbonyl include -CONH 2 , mono C 1 -C 6 alkylaminocarbonyl, mono C 3 -C 6 cycloalkylaminocarbonyl, di C 1 -C 6 alkylaminocarbonyl, and 4- to 8-membered cyclic aminocarbonyl. Specific examples of aminocarbonyl include -CONH 2 , dimethylaminocarbonyl, 1-azetidinylcarbonyl, 1-pyrrolidinylcarbonyl, 1-piperidinylcarbonyl, 1-piperazinylcarbonyl, 4-morpholinylcarbonyl, 3-oxazolidinylcarbonyl, 1,1-dioxidethiomorpholinyl-4-ylcarbonyl, and 3-oxa-8-azabicyclo[3.2.1]octan-8-ylcarbonyl.
本明細書における「ハロアルキル」とは、本明細書定義の「アルキル」の1つまたは複数の水素がハロゲンで置換された基を意味する。ハロアルキルとして、ハロC1-C6アルキルが好ましく、フルオロC1-C6アルキルがより好ましい。ハロアルキルとして具体的には、ジフルオロメチル、トリフルオロメチル、2,2-ジフルオロエチル、2,2,2-トリフルオロエチル、3,3-ジフルオロプロピル、4,4-ジフルオロブチル、5,5-ジフルオロペンチル等が例示される。 As used herein, "haloalkyl" refers to a group in which one or more hydrogen atoms of an "alkyl" as defined herein are substituted with halogen. As the haloalkyl, haloC 1 -C 6 alkyl is preferred, and fluoroC 1 -C 6 alkyl is more preferred. Specific examples of haloalkyl include difluoromethyl, trifluoromethyl, 2,2-difluoroethyl, 2,2,2-trifluoroethyl, 3,3-difluoropropyl, 4,4-difluorobutyl, 5,5-difluoropentyl, and the like.
本明細書における「脂環式環」は、非芳香族炭化水素環を意味する。脂環式環は、環中に不飽和結合を有してもよい。また環を構成する炭素原子は酸化されてカルボニルを形成してもよい。脂環式環は単環でもよく(本明細書において単環式脂環式環と呼称する)、シクロペンタン環、シクロヘキサン環などの飽和脂環式環、ベンゼン環、ナフタレン環などの芳香族炭化水素環と縮合環を形成してもよい。脂環式環として好ましくは3~10員脂環式環、より好ましくは3~8員脂環式環が挙げられる。脂環式環として具体的には、シクロプロパン環、シクロブタン環、シクロペンタン環、シクロヘキサン環、シクロヘプタン環、シクロオクタン環、ビシクロ[2.2.1]ヘプタン環等が例示される。 In this specification, "alicyclic ring" means a non-aromatic hydrocarbon ring. The alicyclic ring may have an unsaturated bond in the ring. Furthermore, the carbon atoms constituting the ring may be oxidized to form a carbonyl. The alicyclic ring may be a monocyclic ring (referred to as a monocyclic alicyclic ring in this specification) or may form a condensed ring with a saturated alicyclic ring such as a cyclopentane ring or a cyclohexane ring, or an aromatic hydrocarbon ring such as a benzene ring or a naphthalene ring. Preferred examples of the alicyclic ring include 3- to 10-membered alicyclic rings, and more preferably 3- to 8-membered alicyclic rings. Specific examples of the alicyclic ring include a cyclopropane ring, a cyclobutane ring, a cyclopentane ring, a cyclohexane ring, a cycloheptane ring, a cyclooctane ring, and a bicyclo[2.2.1]heptane ring.
本明細書における「飽和複素環」は、環を構成する原子中に好ましくは1~5個、より好ましくは1~3個のヘテロ原子を含有する、環中に不飽和結合を有さない、非芳香族複素環を意味する。飽和複素環は、環中に二重及びまたは三重結合を有さない。飽和複素環は単環でもよく、他の環、たとえばシクロペンタン環、シクロヘキサン環などの飽和脂環式環や、テトラヒドロピラン環、ジオキサン環、ピロリジン環などの飽和複素環と縮合環またはスピロ環を形成してもよい。飽和複素環として好ましくは4~10員飽和複素、より好ましくは4~7員飽和複素環、さらに好ましくは5員飽和複素環が挙げられる。飽和複素環として具体的には、アゼチジン環、オキソアゼチジン環、オキセタン環、テトラヒドロフラン環、テトラヒドロピラン環、モルホリン環、チオモルホリン環、ピロリジン環、2-オキソピロリジン環、4-オキソピロリジン環、ピペリジン環、4-オキソピペリジン環、ピペラジン環、ピラゾリジン環、イミダゾリジン環、オキサゾリジン環、イソオキサゾリジン環、チアゾリジン環、イソチアゾリジン環、チアジアゾリジン環、オキサゾリドン環、ジオキソラン環、ジオキサン環、チエタン環、オクタヒドロインドール環、インドリン環、アゼパン環、ジオキセパン環、5,9-ジオキサスピロ[3.5]ノナン環等が例示される。 In this specification, "saturated heterocycle" refers to a non-aromatic heterocycle that contains preferably 1 to 5, more preferably 1 to 3, heteroatoms in the atoms that constitute the ring and has no unsaturated bonds in the ring. A saturated heterocycle does not have double and/or triple bonds in the ring. A saturated heterocycle may be a monocycle, or may form a condensed ring or a spiro ring with another ring, for example, a saturated alicyclic ring such as a cyclopentane ring or a cyclohexane ring, or a saturated heterocycle such as a tetrahydropyran ring, a dioxane ring, or a pyrrolidine ring. Preferred examples of saturated heterocycles include 4- to 10-membered saturated heterocycles, more preferably 4- to 7-membered saturated heterocycles, and even more preferably 5-membered saturated heterocycles. Specific examples of saturated heterocyclic rings include an azetidine ring, an oxoazetidine ring, an oxetane ring, a tetrahydrofuran ring, a tetrahydropyran ring, a morpholine ring, a thiomorpholine ring, a pyrrolidine ring, a 2-oxopyrrolidine ring, a 4-oxopyrrolidine ring, a piperidine ring, a 4-oxopiperidine ring, a piperazine ring, a pyrazolidine ring, an imidazolidine ring, an oxazolidine ring, an isoxazolidine ring, a thiazolidine ring, an isothiazolidine ring, a thiadiazolidine ring, an oxazolidone ring, a dioxolane ring, a dioxane ring, a thietane ring, an octahydroindole ring, an indoline ring, an azepane ring, a dioxepane ring, and a 5,9-dioxaspiro[3.5]nonane ring.
本明細書における「アミノ基の保護基」として、カルバメート系保護基、アシル系保護基、スルホンアミド系保護基、およびシリル系保護基が挙げられる。カルバメート系保護基として、具体的には9-フルオレニルメチルオキシカルボニル基(Fmoc基)、ベンジルオキシカルボニル基(Cbz基)、2,2,2-トリクロロエトキシカルボニル基(Troc基)、アリルオキシカルボニル基(Alloc基)、2-(トリメチルシリル)エトキシカルボニル基(Teoc基)、トリイソプロピルシリルオキシカルボニル基(TSoc基)、ジ-t-ブチルイソブチルシリルオキシカルボニル基(BIBSoc基)、ジ-i-プロピル-t-ブチルシリルオキシカルボニル基(IPCSoc基)、ベンジル-ジ-t-ブチルシリルオキシカルボニル基(BBSoc基)、ジ-t-ブチルシクロへキシルシリルオキシカルボニル基(CHBSoc基)、ジ-t-ブチルオクタデシルシリルオキシカルボニル基(CDBSoc基)、t-ブトキシカルボニル基(Boc基)等が例示される。アシル系保護基として、具体的にはトリフルオロアセチル基、アセチル基、ベンゾイル基等が例示される。スルホンアミド系保護基として、具体的には2-ニトロベンゼンスルホニル基、4-ニトロベンゼンスルホニル基、2,4-ジニトロベンゼンスルホニル基等が例示される。シリル系保護基として、具体的にはトリメチルシリル基(TMS基)、t-ブチルジメチルシリル基(TBDMS基)、トリエチルシリル基(TES基)、トリイソプロピルシリル基(TIPS基)、t-ブチルジフェニルシリル基(TBDPS基)等が例示される。 In this specification, the term "amino group protecting group" includes carbamate protecting groups, acyl protecting groups, sulfonamide protecting groups, and silyl protecting groups. Specific examples of carbamate protecting groups include 9-fluorenylmethyloxycarbonyl group (Fmoc group), benzyloxycarbonyl group (Cbz group), 2,2,2-trichloroethoxycarbonyl group (Troc group), allyloxycarbonyl group (Alloc group), 2-(trimethylsilyl)ethoxycarbonyl group (Teoc group), triisopropylsilyloxycarbonyl group (TSoc group), di-t-butylisobutyl Examples of the protecting group include a silyloxycarbonyl group (BIBSoC group), a di-i-propyl-t-butylsilyloxycarbonyl group (IPCSoc group), a benzyl-di-t-butylsilyloxycarbonyl group (BBSoC group), a di-t-butylcyclohexylsilyloxycarbonyl group (CHBSoc group), a di-t-butyloctadecylsilyloxycarbonyl group (CDBSoc group), a t-butoxycarbonyl group (Boc group), etc. Specific examples of the acyl protecting group include a trifluoroacetyl group, an acetyl group, a benzoyl group, etc. Specific examples of the sulfonamide protecting group include a 2-nitrobenzenesulfonyl group, a 4-nitrobenzenesulfonyl group, a 2,4-dinitrobenzenesulfonyl group, etc. Specific examples of silyl-based protecting groups include trimethylsilyl (TMS), t-butyldimethylsilyl (TBDMS), triethylsilyl (TES), triisopropylsilyl (TIPS), and t-butyldiphenylsilyl (TBDPS).
本明細書において「置換されていてもよい」とは、ある基が任意の置換基によって置換されていてもよいことを意味する。 In this specification, "optionally substituted" means that a group may be substituted with any substituent.
本明細書において「保護されていてもよい」とは、ある基が任意の保護基によって保護されていてもよいことを意味する。 In this specification, "optionally protected" means that a group may be protected by any protecting group.
本明細書において「1つまたは複数の」とは、1つまたは2つ以上の数を意味する。「1つまたは複数の」が、ある基の置換基に関連する文脈で用いられる場合、この用語は、1つからその基が許容する置換基の最大数までの数を意味する。「1つまたは複数の」として具体的には、たとえば、1、2、3、4、5、6、7、8、9、10、および/またはそれより大きい数が挙げられる。 As used herein, "one or more" means one or more than one. When "one or more" is used in the context of a substituent of a group, the term means a number from one to the maximum number of substituents permitted by that group. Specific examples of "one or more" include 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and/or more.
本明細書における「ペプチド化合物」は、2以上のアミノ酸残基がアミド結合によって連結したものを意味する。デプシペプチドのように主鎖の一部にエステル結合を有するペプチドも、本明細書における「ペプチド化合物」に含まれる。本開示におけるペプチドに含まれるアミノ酸残基数は特に限定されないが、好ましくは5~30残基、より好ましくは8~15残基、さらに好ましくは9~13残基であってよい。本開示におけるペプチド化合物は、N置換アミノ酸を少なくとも3つ含むことが好ましく、少なくとも5つ含むことがより好ましく、少なくとも6つ含むことがさらに好ましい。これらのN置換アミノ酸は、ペプチド化合物中に連続して存在していても、不連続に存在していてもよい。本開示におけるペプチド化合物は、直鎖状でも環状でもよく、環状ペプチド化合物が好ましい。 The term "peptide compound" as used herein means two or more amino acid residues linked together by amide bonds. Peptides having an ester bond in part of the main chain, such as depsipeptides, are also included in the term "peptide compound" as used herein. The number of amino acid residues contained in a peptide in this disclosure is not particularly limited, but may be preferably 5 to 30 residues, more preferably 8 to 15 residues, and even more preferably 9 to 13 residues. The peptide compound in this disclosure preferably contains at least three N-substituted amino acids, more preferably contains at least five, and even more preferably contains at least six. These N-substituted amino acids may be present consecutively or discontinuously in the peptide compound. The peptide compound in this disclosure may be linear or cyclic, with cyclic peptide compounds being preferred.
本明細書において「環状ペプチド化合物」とは、4以上のアミノ酸残基によって構成される環状構造を有するペプチド化合物をいう。環状ペプチド化合物の環状構造にはアミド結合以外の結合が含まれていてもよく、例えば、酸素原子を利用したC-O-C結合、C(O)-O結合、C(S)-O結合、硫黄原子を利用したC(O)-S結合、C(S)-S結合、C-S-S-C結合、C-S-C結合、C-S(O)-C結合、C-S(O2)-C結合、窒素原子を利用した、C-N-C結合、C=N-C結合、N-C(O)-N結合、N-C(S)N結合、C(S)-N結合、及びC-C結合からなる群から選択される結合が含まれていても良い。環状ペプチド化合物は、環状構造以外に、環状構造に含まれないアミノ酸や鎖状ペプチド構造を有していてもよい。また、アミノ酸や鎖状ペプチド構造以外の構造を有していてもよい。 In this specification, the term "cyclic peptide compound" refers to a peptide compound having a cyclic structure composed of four or more amino acid residues. The cyclic structure of the cyclic peptide compound may contain bonds other than amide bonds, for example, bonds selected from the group consisting of C-O-C bonds, C(O)-O bonds, C(S)-O bonds using oxygen atoms, C(O)-S bonds using sulfur atoms, C(S)-S bonds, C-S-S-C bonds, C-S-C bonds, C-S(O)-C bonds, C-S(O2)-C bonds, C-N-C bonds using nitrogen atoms, C=N-C bonds, N-C(O)-N bonds, N-C(S)N bonds, C(S)-N bonds, and C-C bonds. In addition to the cyclic structure, the cyclic peptide compound may have amino acids not included in the cyclic structure or a chain peptide structure. It may also have a structure other than amino acids and chain peptide structures.
ペプチド化合物の「環化」とは、4以上のアミノ酸残基を含む環状部環状構造を形成することを意味する。本明細書における環状ペプチド化合物の環状部に含まれるアミノ酸の数は特に限定されないが、4~20残基、5~15残基、6~13残基が例示される。直鎖状のペプチド化合物を環状ペプチド化合物に変換する方法は、Comprehensive Organic Transformations, A Guide to Functional Group Preparations, 3rd Edition(R. C. Larock著)、またはMarch's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition、(M. B. Smith, J. March著)などに記載の方法により、分子内で結合形成反応を行うことにより実施することができる。結合形成反応の後に、さらに官能基変換反応を行うこともできる。結合形成反応は、カルボン酸とアミンから形成されるC(O)-N結合、酸素原子を利用したC-O-C結合、C(O)-O結合、C(S)-O結合、硫黄原子を利用したC(O)-S結合、C(S)-S結合、C-S-S-C結合、C-S-C結合、C-S(O)-C結合、C-S(O2)-C結合、窒素原子を利用した、C-N-C結合、C=N-C結合、N-C(O)-N結合、N-C(S)N結合、C(S)-N結合などが例示される。さらに、鈴木反応、Heck反応、Sonogashira反応等の遷移金属を触媒としたC-C結合の形成反応などが挙げられる。結合形成反応の後に、さらに行う官能基変換反応として、酸化反応または還元反応が例示される。具体的には硫黄原子を酸化して、スルホキシド基やスルホン基に変換する反応が例示される。また、炭素-炭素結合のうち、三重結合や二重結合を還元して、二重結合または単結合に変換する還元反応が例示される。2つのアミノ酸がアミノ酸の主鎖において結合すると、ペプチド結合により閉環構造が形成されるが、2つのアミノ酸の側鎖同士、側鎖と主鎖の結合等により、2つのアミノ酸間の共有結合が形成されてもよい。 "Cyclization" of a peptide compound means forming a cyclic structure of a cyclic portion containing four or more amino acid residues. The number of amino acids contained in the cyclic portion of the cyclic peptide compound in this specification is not particularly limited, but examples include 4 to 20 residues, 5 to 15 residues, and 6 to 13 residues. A method for converting a linear peptide compound to a cyclic peptide compound can be carried out by performing a bond formation reaction within the molecule using a method described in Comprehensive Organic Transformations, A Guide to Functional Group Preparations, 3rd Edition (by R. C. Larock) or March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (by M. B. Smith and J. March), etc. After the bond formation reaction, a functional group conversion reaction can also be performed. Examples of bond-forming reactions include C(O)-N bonds formed from carboxylic acids and amines, C-O-C bonds using oxygen atoms, C(O)-O bonds, C(S)-O bonds, C(O)-S bonds using sulfur atoms, C(S)-S bonds, C-S-S-C bonds, C-S-C bonds, C-S(O)-C bonds, C-S(O2)-C bonds, C-N-C bonds, C=N-C bonds, N-C(O)-N bonds, N-C(S)N bonds, and C(S)-N bonds using nitrogen atoms. Further examples include C-C bond formation reactions using transition metals as catalysts, such as Suzuki reaction, Heck reaction, and Sonogashira reaction. Examples of functional group conversion reactions that are performed after bond-forming reactions include oxidation reactions and reduction reactions. Specifically, examples include reactions in which sulfur atoms are oxidized to convert to sulfoxide groups or sulfone groups. Another example is a reduction reaction in which a triple bond or a double bond among carbon-carbon bonds is reduced to a double bond or a single bond. When two amino acids are bonded to each other in the main chain of the amino acid, a closed ring structure is formed by a peptide bond, but a covalent bond between two amino acids may also be formed by bonding the side chains of the two amino acids together, or between the side chain and the main chain, etc.
本明細書における「アミノ酸」には、天然アミノ酸、及び非天然アミノ酸が含まれる。また本明細書において「アミノ酸」はアミノ酸残基を意味することがある。本明細書における「天然アミノ酸」とは、Gly、Ala、Ser、Thr、Val、Leu、Ile、Phe、Tyr、Trp、His、Glu、Asp、Gln、Asn、Cys、Met、Lys、Arg、Proを指す。非天然アミノ酸は特に限定されないが、β-アミノ酸、D型アミノ酸、N置換アミノ酸、α,α-ジ置換アミノ酸、側鎖が天然アミノ酸と異なるアミノ酸、ヒドロキシカルボン酸などが例示される。本明細書におけるアミノ酸としては、任意の立体配置が許容される。アミノ酸の側鎖の選択は特に制限を設けないが、水素原子の他にも例えばアルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基、ヘテロアリールアルキル基、シクロアルキル基、スピロ結合したシクロアルキル基から自由に選択される。それぞれには置換基が付与されていてもよく、それら置換基も制限されず、例えば、ハロゲン原子、酸素原子、窒素原子、硫黄原子、ホウ素原子、ケイ素原子、又はリン原子を含む任意の置換基の中から独立して1つ又は2つ以上自由に選択されてよい。すなわち、置換されていてもよいアルキル基、アルコキシ基、アルコキシアルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基、シクロアルキル基など、または、オキソ、アミノカルボニル、ハロゲン原子などが例示される。非限定の一態様において、本明細書におけるアミノ酸は、同一分子内にカルボキシル基とアミノ基を有する化合物であってよい(この場合であっても、アミノ基の窒素原子と側鎖の任意の原子がと一緒になって環を形成したプロリン、ヒドロキシプロリン、アゼチジン-2-カルボン酸などもアミノ酸に含まれる。)。 In the present specification, the term "amino acid" includes natural amino acids and non-natural amino acids. In addition, in the present specification, the term "amino acid" may refer to an amino acid residue. In the present specification, the term "natural amino acid" refers to Gly, Ala, Ser, Thr, Val, Leu, Ile, Phe, Tyr, Trp, His, Glu, Asp, Gln, Asn, Cys, Met, Lys, Arg, and Pro. Non-natural amino acids are not particularly limited, but examples thereof include β-amino acids, D-amino acids, N-substituted amino acids, α,α-disubstituted amino acids, amino acids whose side chains are different from those of natural amino acids, and hydroxycarboxylic acids. As used herein, amino acids are allowed to have any configuration. There are no particular limitations on the selection of the side chain of the amino acid, but it can be freely selected from, in addition to a hydrogen atom, for example, an alkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, a heteroarylalkyl group, a cycloalkyl group, and a spiro-linked cycloalkyl group. Each of them may have a substituent, and the substituents are not limited, and may be independently selected from any substituents including, for example, a halogen atom, an oxygen atom, a nitrogen atom, a sulfur atom, a boron atom, a silicon atom, or a phosphorus atom. That is, examples include an optionally substituted alkyl group, an alkoxy group, an alkoxyalkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, a cycloalkyl group, or an oxo, an aminocarbonyl, a halogen atom, and the like. In a non-limiting embodiment, the amino acid in this specification may be a compound having a carboxyl group and an amino group in the same molecule (even in this case, proline, hydroxyproline, azetidine-2-carboxylic acid, etc., in which the nitrogen atom of the amino group and any atom of the side chain form a ring together, are also included in the amino acid).
本明細書においてペプチド化合物を構成する「アミノ酸残基」を単に「アミノ酸」ということがある。 In this specification, the "amino acid residues" that make up a peptide compound may be simply referred to as "amino acids."
本明細書において「N末端のアミノ酸残基」とは、ペプチドのN末端に位置するアミノ酸残基を意味する。本明細書において「C末端のアミノ酸残基」とは、ペプチドのC末端に位置するアミノ酸残基を意味する。 As used herein, "N-terminal amino acid residue" refers to the amino acid residue located at the N-terminus of a peptide. As used herein, "C-terminal amino acid residue" refers to the amino acid residue located at the C-terminus of a peptide.
本明細書において「アミノ酸の数(アミノ酸数)」および「アミノ酸残基の数(アミノ酸残基数)」とは、ペプチド化合物を構成するアミノ酸残基(アミノ酸ユニット)の数のことであり、アミノ酸を連結しているアミド結合、エステル結合、及び環化部の結合を切断した際に生じるアミノ酸ユニットの数を意味する。 In this specification, the "number of amino acids" and "number of amino acid residues" refer to the number of amino acid residues (amino acid units) that make up a peptide compound, and refer to the number of amino acid units that are generated when the amide bonds, ester bonds, and cyclized bonds that link the amino acids are cleaved.
本明細書におけるペプチド化合物を構成する「アミノ酸」にはそれぞれに対応する全ての同位体を含む。「アミノ酸」の同位体は、少なくとも1つの原子が、原子番号(陽子数)が同じで,質量数(陽子と中性子の数の和)が異なる原子で天然とは異なる存在比で置換されたものである。本発明のペプチド化合物を構成する「アミノ酸」に含まれる同位体の例としては、水素原子、炭素原子、窒素原子、酸素原子、リン原子、硫黄原子、フッ素原子、塩素原子などがあり、それぞれ、2H、3H、13C、14C、15N、17O、18O、31P、32P、35S、18F、36Cl等が含まれる。全ての割合の放射性または非放射性の同位元素を含有する本明細書の化合物は、本発明の範囲に包含される。 The "amino acids" constituting the peptide compounds in this specification include all corresponding isotopes. An isotope of an "amino acid" is one in which at least one atom is replaced with an atom having the same atomic number (proton number) but a different mass number (sum of the number of protons and neutrons) in a ratio different from that in nature. Examples of isotopes contained in the "amino acids" constituting the peptide compounds of the present invention include hydrogen atoms, carbon atoms, nitrogen atoms, oxygen atoms, phosphorus atoms, sulfur atoms, fluorine atoms, and chlorine atoms, each of which includes 2 H, 3 H, 13 C, 14 C, 15 N, 17 O, 18 O, 31 P, 32 P, 35 S, 18 F, and 36 Cl. Compounds of this specification containing radioactive or non-radioactive isotopes in all ratios are within the scope of the present invention.
本明細書におけるハロゲン原子を含む置換基としては、ハロゲンを置換基に有するアルキル基、シクロアルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基などが例示され、より具体的には、フルオロアルキル、ジフルオロアルキル、トリフルオロアルキルなどが例示される。 In this specification, examples of substituents containing a halogen atom include alkyl groups, cycloalkyl groups, alkenyl groups, alkynyl groups, aryl groups, heteroaryl groups, and aralkyl groups each having a halogen as a substituent, and more specifically, examples include fluoroalkyl, difluoroalkyl, and trifluoroalkyl.
O原子を含む置換基としては、ヒドロキシ(-OH)、オキシ(-OR)、カルボニル(-C=O-R)、カルボキシ(-CO2H)、オキシカルボニル(-C=O-OR)、カルボニルオキシ(-O-C=O-R)、チオカルボニル(-C=O-SR)、カルボニルチオ(-S-C=O-R)、アミノカルボニル(-C=O-NHR)、カルボニルアミノ(-NH-C=O-R)、オキシカルボニルアミノ(-NH-C=O-OR)、スルホニルアミノ(-NH-SO2-R)、アミノスルホニル(-SO2-NHR)、スルファモイルアミノ(-NH-SO2-NHR)、チオカルボキシル(-C(=O)-SH)、カルボキシルカルボニル(-C(=O)-CO2H)などの基が挙げられる。 Examples of the substituent containing an O atom include hydroxyl (-OH), oxy (-OR), carbonyl (-C=O-R), carboxy (-CO 2 H), oxycarbonyl (-C=O-OR), carbonyloxy (-O-C=O-R), thiocarbonyl (-C=O-SR), carbonylthio (-S-C=O-R), aminocarbonyl (-C=O-NHR), carbonylamino (-NH-C=O-R), oxycarbonylamino (-NH-C=O-OR), sulfonylamino (-NH-SO 2 -R), aminosulfonyl (-SO 2 -NHR), sulfamoylamino (-NH-SO 2 -NHR), thiocarboxyl (-C(=O)-SH), and carboxylcarbonyl (-C(=O)-CO 2 H).
オキシ(-OR)の例としては、アルコキシ、シクロアルコキシ、アルケニルオキシ、アルキニルオキシ、アリールオキシ、ヘテロアリールオキシ、アラルキルオキシなどが挙げられる。アルコキシとしては、C1-C4アルコキシ、C1-C2アルコキシが好ましく、なかでもメトキシ、又はエトキシが好ましい。 Examples of oxy (-OR) include alkoxy, cycloalkoxy, alkenyloxy, alkynyloxy, aryloxy, heteroaryloxy, aralkyloxy, etc. As the alkoxy, C 1 -C 4 alkoxy and C 1 -C 2 alkoxy are preferred, and among these, methoxy and ethoxy are preferred.
カルボニル(-C=O-R)の例としては、ホルミル(-C=O-H)、アルキルカルボニル、シクロアルキルカルボニル、アルケニルカルボニル、アルキニルカルボニル、アリールカルボニル、ヘテロアリールカルボニル、アラルキルカルボニルなどが挙げられる。 Examples of carbonyl (-C=O-R) include formyl (-C=O-H), alkylcarbonyl, cycloalkylcarbonyl, alkenylcarbonyl, alkynylcarbonyl, arylcarbonyl, heteroarylcarbonyl, and aralkylcarbonyl.
オキシカルボニル(-C=O-OR)の例としては、アルキルオキシカルボニル、シクロアルキルオキシカルボニル、アルケニルオキシカルボニル、アルキニルオキシカルボニル、アリールオキシカルボニル、ヘテロアリールオキシカルボニル、アラルキルオキシカルボニルなどが挙げられる。 Examples of oxycarbonyl (-C=O-OR) include alkyloxycarbonyl, cycloalkyloxycarbonyl, alkenyloxycarbonyl, alkynyloxycarbonyl, aryloxycarbonyl, heteroaryloxycarbonyl, and aralkyloxycarbonyl.
カルボニルオキシ(-O-C=O-R)の例としては、アルキルカルボニルオキシ、シクロアルキルカルボニルオキシ、アルケニルカルボニルオキシ、アルキニルカルボニルオキシ、アリールカルボニルオキシ、ヘテロアリールカルボニルオキシ、アラルキルカルボニルオキシなどが挙げられる。 Examples of carbonyloxy (-O-C=O-R) include alkylcarbonyloxy, cycloalkylcarbonyloxy, alkenylcarbonyloxy, alkynylcarbonyloxy, arylcarbonyloxy, heteroarylcarbonyloxy, and aralkylcarbonyloxy.
チオカルボニル(-C=O-SR)の例としては、アルキルチオカルボニル、シクロアルキルチオカルボニル、アルケニルチオカルボニル、アルキニルチオカルボニル、アリールチオカルボニル、ヘテロアリールチオカルボニル、アラルキルチオカルボニルなどが挙げられる。 Examples of thiocarbonyl (-C=O-SR) include alkylthiocarbonyl, cycloalkylthiocarbonyl, alkenylthiocarbonyl, alkynylthiocarbonyl, arylthiocarbonyl, heteroarylthiocarbonyl, and aralkylthiocarbonyl.
カルボニルチオ(-S-C=O-R)の例としては、アルキルカルボニルチオ、シクロアルキルカルボニルチオ、アルケニルカルボニルチオ、アルキニルカルボニルチオ、アリールカルボニルチオ、ヘテロアリールカルボニルチオ、アラルキルカルボニルチオなどが挙げられる。 Examples of carbonylthio (-S-C=O-R) include alkylcarbonylthio, cycloalkylcarbonylthio, alkenylcarbonylthio, alkynylcarbonylthio, arylcarbonylthio, heteroarylcarbonylthio, and aralkylcarbonylthio.
アミノカルボニル(-C=O-NHR)の例としては、アルキルアミノカルボニル(例えば、C1-C6又はC1-C4アルキルアミノカルボニル、なかでもエチルアミノカルボニル、メチルアミノカルボニルなどが例示される。)、シクロアルキルアミノカルボニル、アルケニルアミノカルボニル、アルキニルアミノカルボニル、アリールアミノカルボニル、ヘテロアリールアミノカルボニル、アラルキルアミノカルボニルなどが挙げられる。これらに加えて、-C=O-NHR中のN原子と結合したH原子が、アルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルでさらに置換された基が挙げられる。 Examples of aminocarbonyl (-C=O-NHR) include alkylaminocarbonyl (e.g., C1 - C6 or C1 - C4 alkylaminocarbonyl, particularly ethylaminocarbonyl, methylaminocarbonyl, etc.), cycloalkylaminocarbonyl, alkenylaminocarbonyl, alkynylaminocarbonyl, arylaminocarbonyl, heteroarylaminocarbonyl, aralkylaminocarbonyl, etc. In addition to these, examples include groups in which the H atom bonded to the N atom in -C=O-NHR is further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
カルボニルアミノ(-NH-C=O-R)の例としては、アルキルカルボニルアミノ、シクロアルキルカルボニルアミノ、アルケニルカルボニルアミノ、アルキニルカルボニルアミノ、アリールカルボニルアミノ、ヘテロアリールカルボニルアミノ、アラルキルカルボニルアミノなどが挙げられる。これらに加えて-NH-C=O-R中のN原子と結合したH原子が、アルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルでさらに置換された基が挙げられる。 Examples of carbonylamino (-NH-C=O-R) include alkylcarbonylamino, cycloalkylcarbonylamino, alkenylcarbonylamino, alkynylcarbonylamino, arylcarbonylamino, heteroarylcarbonylamino, and aralkylcarbonylamino. In addition, the H atom bonded to the N atom in -NH-C=O-R may be further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
オキシカルボニルアミノ(-NH-C=O-OR)の例としては、アルコキシカルボニルアミノ、シクロアルコキシカルボニルアミノ、アルケニルオキシカルボニルアミノ、アルキニルオキシカルボニルアミノ、アリールオキシカルボニルアミノ、ヘテロアリールオキシカルボニルアミノ、アラルキルオキシカルボニルアミノなどが挙げられる。これらに加えて、-NH-C=O-OR中のN原子と結合したH原子がアルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルでさらに置換された基が挙げられる。 Examples of oxycarbonylamino (-NH-C=O-OR) include alkoxycarbonylamino, cycloalkoxycarbonylamino, alkenyloxycarbonylamino, alkynyloxycarbonylamino, aryloxycarbonylamino, heteroaryloxycarbonylamino, and aralkyloxycarbonylamino. In addition, examples include groups in which the H atom bonded to the N atom in -NH-C=O-OR is further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
スルホニルアミノ(-NH-SO2-R)の例としては、アルキルスルホニルアミノ、シクロアルキルスルホニルアミノ、アルケニルスルホニルアミノ、アルキニルスルホニルアミノ、アリールスルホニルアミノ、ヘテロアリールスルホニルアミノ、アラルキルスルホニルアミノなどが挙げられる。これらに加えて、-NH-SO2-R中のN原子と結合したH原子がアルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルでさらに置換された基が挙げられる。 Examples of sulfonylamino (-NH-SO 2 -R) include alkylsulfonylamino, cycloalkylsulfonylamino, alkenylsulfonylamino, alkynylsulfonylamino, arylsulfonylamino, heteroarylsulfonylamino, aralkylsulfonylamino, etc. In addition to these, there are also groups in which the H atom bonded to the N atom in -NH-SO 2 -R is further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
アミノスルホニル(-SO2-NHR)の例としては、アルキルアミノスルホニル、シクロアルキルアミノスルホニル、アルケニルアミノスルホニル、アルキニルアミノスルホニル、アリールアミノスルホニル、ヘテロアリールアミノスルホニル、アラルキルアミノスルホニルなどが挙げられる。これらに加えて、-SO2-NHR中のN原子と結合したH原子がアルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルでさらに置換された基が挙げられる。 Examples of aminosulfonyl (-SO 2 -NHR) include alkylaminosulfonyl, cycloalkylaminosulfonyl, alkenylaminosulfonyl, alkynylaminosulfonyl, arylaminosulfonyl, heteroarylaminosulfonyl, aralkylaminosulfonyl, etc. In addition to these, examples include groups in which the H atom bonded to the N atom in -SO 2 -NHR is further substituted with an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, or aralkyl.
スルファモイルアミノ(-NH-SO2-NHR)の例としては、アルキルスルファモイルアミノ、シクロアルキルスルファモイルアミノ、アルケニルスルファモイルアミノ、アルキニルスルファモイルアミノ、アリールスルファモイルアミノ、ヘテロアリールスルファモイルアミノ、アラルキルスルファモイルアミノなどが挙げられる。さらに、-NH-SO2-NHR中のN原子と結合した2つのH原子はアルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、およびアラルキルからなる群より独立して選択される置換基で置換されていてもよく、またこれらの2つの置換基は環を形成しても良い。 Examples of sulfamoylamino (-NH-SO 2 -NHR) include alkylsulfamoylamino, cycloalkylsulfamoylamino, alkenylsulfamoylamino, alkynylsulfamoylamino, arylsulfamoylamino, heteroarylsulfamoylamino, aralkylsulfamoylamino, etc. Furthermore, the two H atoms bonded to the N atom in -NH-SO 2 -NHR may be substituted with substituents independently selected from the group consisting of alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, and these two substituents may form a ring.
S原子を含む置換基としては、チオール(-SH)、チオ(-S-R)、スルフィニル(-S=O-R)、スルホニル(-SO2-R)、スルホ(-SO3H)などの基が挙げられる。 Examples of the substituent containing an S atom include thiol (-SH), thio (-S-R), sulfinyl (-S=O-R), sulfonyl (-SO 2 -R), and sulfo (-SO 3 H).
チオ(-S-R)の例としては、アルキルチオ、シクロアルキルチオ、アルケニルチオ、アルキニルチオ、アリールチオ、ヘテロアリールチオ、アラルキルチオなどの中から選択される。 Examples of thio (-S-R) are selected from alkylthio, cycloalkylthio, alkenylthio, alkynylthio, arylthio, heteroarylthio, aralkylthio, etc.
スルホニル(-SO2-R)の例としては、アルキルスルホニル、シクロアルキルスルホニル、アルケニルスルホニル、アルキニルスルホニル、アリールスルホニル、ヘテロアリールスルホニル、アラルキルスルホニルなどが挙げられる。 Examples of sulfonyl (—SO 2 —R) include alkylsulfonyl, cycloalkylsulfonyl, alkenylsulfonyl, alkynylsulfonyl, arylsulfonyl, heteroarylsulfonyl, aralkylsulfonyl, and the like.
N原子を含む置換基として、アジド(-N3、「アジド基」ともいう)、シアノ(-CN)、1級アミノ(-NH2)、2級アミノ(-NH-R;モノ置換アミノともいう。)、3級アミノ(-NR(R');ジ置換アミノともいう。)、アミジノ(-C(=NH)-NH2)、置換アミジノ(-C(=NR)-NR'R")、グアニジノ(-NH-C(=NH)-NH2)、置換グアニジノ(-NR-C(=NR''')-NR'R")、アミノカルボニルアミノ(-NR-CO-NR'R")、ピリジル、ピペリジノ、モルホリノ、アゼチジニルなどの基が挙げられる。 Examples of the substituent containing an N atom include azide (-N 3 , also referred to as an "azide group"), cyano (-CN), primary amino (-NH 2 ), secondary amino (-NH-R; also referred to as mono-substituted amino), tertiary amino (-NR(R'); also referred to as di-substituted amino), amidino (-C(=NH)-NH 2 ), substituted amidino (-C(=NR)-NR'R"), guanidino (-NH-C(=NH)-NH 2 ), substituted guanidino (-NR-C(=NR'")-NR'R"), aminocarbonylamino (-NR-CO-NR'R"), pyridyl, piperidino, morpholino, and azetidinyl.
2級アミノ(-NH-R;モノ置換アミノ)の例としては、アルキルアミノ、シクロアルキルアミノ、アルケニルアミノ、アルキニルアミノ、アリールアミノ、ヘテロアリールアミノ、アラルキルアミノなどが挙げられる。 Examples of secondary amino (-NH-R; monosubstituted amino) include alkylamino, cycloalkylamino, alkenylamino, alkynylamino, arylamino, heteroarylamino, and aralkylamino.
3級アミノ(-NR(R');ジ置換アミノ)の例としては、例えばアルキル(アラルキル)アミノなど、アルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルなどの中からそれぞれ独立して選択される、任意の2つの置換基を有するアミノ基が挙げられ、これらの任意の2つの置換基は環を形成しても良い。具体的には、ジアルキルアミノ、なかでもC1-C6ジアルキルアミノ、C1-C4ジアルキルアミノ、ジメチルアミノ、ジエチルアミノなどが例示される。本明細書において「Cp-Cqジアルキルアミノ基」とは、アミノ基にCp-Cqアルキル基が2個置換された基をいい、両Cp-Cqアルキル基は同一であっても異なっていてもよい。 Examples of tertiary amino (-NR(R'); disubstituted amino) include, for example, alkyl(aralkyl)amino and other amino groups having any two substituents independently selected from alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, and the like, and these any two substituents may form a ring. Specific examples include dialkylamino, particularly C 1 -C 6 dialkylamino, C 1 -C 4 dialkylamino, dimethylamino, diethylamino, and the like. In this specification, the term "C p -C q dialkylamino group" refers to a group in which an amino group is substituted with two C p -C q alkyl groups, and both C p -C q alkyl groups may be the same or different.
置換アミジノ(-C(=NR)-NR'R")の例としては、N原子上の3つの置換基R、R'、およびR"が、アルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルの中からそれぞれ独立して選択された基、例えばアルキル(アラルキル)(アリール)アミジノなどが挙げられる。 Examples of substituted amidino (-C(=NR)-NR'R") include those in which the three substituents R, R', and R" on the N atom are each independently selected from alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, such as alkyl(aralkyl)(aryl)amidino.
置換グアニジノ(-NR-C(=NR''')-NR'R")の例としては、R,R'、R"、およびR'''が、アルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルの中からそれぞれ独立して選択された基、あるいはこれらが環を形成した基などが挙げられる。 Examples of substituted guanidino (-NR-C(=NR''')-NR'R") include groups in which R, R', R", and R''' are each independently selected from alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, or groups in which these groups form a ring.
アミノカルボニルアミノ(-NR-CO-NR'R")の例としては、R、R'、およびR"が、水素原子、アルキル、シクロアルキル、アルケニル、アルキニル、アリール、ヘテロアリール、アラルキルの中からそれぞれ独立して選択された基、あるいはこれらは環を形成した基などが挙げられる。 Examples of aminocarbonylamino (-NR-CO-NR'R") include groups in which R, R', and R" are each independently selected from a hydrogen atom, alkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, and aralkyl, or groups in which these form a ring.
本発明の化合物は、その塩、好ましくはその化学的に許容される塩であることができる。また本発明の化合物またはその塩は、それらの溶媒和物、好ましくはその化学的に許容される溶媒和物であることができる。本発明の化合物の塩には、例えば、塩酸塩;臭化水素酸塩;ヨウ化水素酸塩;リン酸塩;ホスホン酸塩;硫酸塩;メタンスルホン酸塩、p-トルエンスルホン酸塩などのスルホン酸塩;酢酸塩、クエン酸塩、リンゴ酸塩、酒石酸塩、コハク酸塩、サリチル酸塩などのカルボン酸塩;または、ナトリウム塩、カリウム塩などのアルカリ金属塩;マグネシウム塩、カルシウム塩などのアルカリ土類金属塩;アンモニウム塩、アルキルアンモニウム塩、ジアルキルアンモニウム塩、トリアルキルアンモニウム塩、テトラアルキルアンモニウム塩などのアンモニウム塩などが含まれる。これらの塩は、たとえば、当該化合物と、医薬品の製造に使用可能である酸または塩基とを接触させることにより製造される。本発明において、化合物の溶媒和物とは、化合物が溶媒とともに、一つの分子集団を形成したものをさし、溶媒により形成された溶媒和物であれば特に限定されない。溶媒が水であれば水和物と言う。本発明の化合物の溶媒和物としては、水和物が好ましく、そのような水和物として具体的には1~10水和物、好ましくは1~5水和物、さらに好ましくは1~3水和物が挙げられる。本発明の化合物の溶媒和物には、水、アルコール(例えば、メタノール、エタノール、1-プロパノール、2-プロパノールなど)、ジメチルホルムアミドなどの単独の溶媒との溶媒和物だけでなく、複数の溶媒との溶媒和物も含まれる。 The compound of the present invention may be a salt thereof, preferably a chemically acceptable salt thereof. The compound of the present invention or a salt thereof may be a solvate thereof, preferably a chemically acceptable solvate thereof. The salt of the compound of the present invention may be, for example, hydrochloride; hydrobromide; hydroiodide; phosphate; phosphonate; sulfate; sulfonate such as methanesulfonate and p-toluenesulfonate; carboxylate such as acetate, citrate, malate, tartrate, succinate, salicylate; or alkali metal salt such as sodium salt and potassium salt; alkaline earth metal salt such as magnesium salt and calcium salt; ammonium salt such as ammonium salt, alkylammonium salt, dialkylammonium salt, trialkylammonium salt, tetraalkylammonium salt, etc. These salts are produced, for example, by contacting the compound with an acid or base that can be used in the manufacture of pharmaceuticals. In the present invention, a solvate of a compound refers to a compound that forms a molecular group together with a solvent, and is not particularly limited as long as it is a solvate formed by a solvent. If the solvent is water, it is called a hydrate. The solvates of the compounds of the present invention are preferably hydrates, and specific examples of such hydrates include monohydrates to decahydrates, preferably monohydrates to pentahydrates, and more preferably monohydrates to trihydrates. The solvates of the compounds of the present invention include solvates with a single solvent such as water, alcohol (e.g., methanol, ethanol, 1-propanol, 2-propanol, etc.), and dimethylformamide, as well as solvates with multiple solvents.
本発明に係る化合物がフリー体として得られる場合、当該化合物は、その水和物もしくは溶媒和物の状態に、常法に従って変換することができる。また、本発明に係る化合物がフリー体として得られる場合、当該化合物は、当該化合物が形成してもよい塩またはその水和物もしくは溶媒和物の状態に、常法に従って変換することができる。例えば、式(1a)で表される環状ペプチド化合物もしくはその塩の水和物、エタノール和物等が挙げられる。具体的には、式(1a)で表される環状ペプチド化合物の半水和物、1水和物、2水和物、3水和物、4水和物、5水和物、6水和物、7水和物、8水和物、9水和物、10水和物もしくは1エタノール和物、または式(1a)で表される環状ペプチド化合物のナトリウム塩の半水和物、1水和物、2水和物、3水和物、4水和物、5水和物、6水和物、7水和物、8水和物、9水和物、10水和物もしくは1エタノール和物、または式(1a)で表される環状ペプチド化合物の塩酸塩の水和物もしくはエタノール和物等が挙げられるが、これらに限定されるものではない。水和物または溶媒和物は、結晶形または非結晶形で製造されてもよく、結晶形の場合、結晶多形をとりうる。水和物または溶媒和物の製造方法として、例えば、式(1a)で表される環状ペプチド化合物や本明細書に記載のペプチド化合物に、エタノール等の溶媒および/または水を加え、攪拌、冷却、濃縮、および/または乾燥を行うなど、常法によって水和物または溶媒和物を得ることができる。
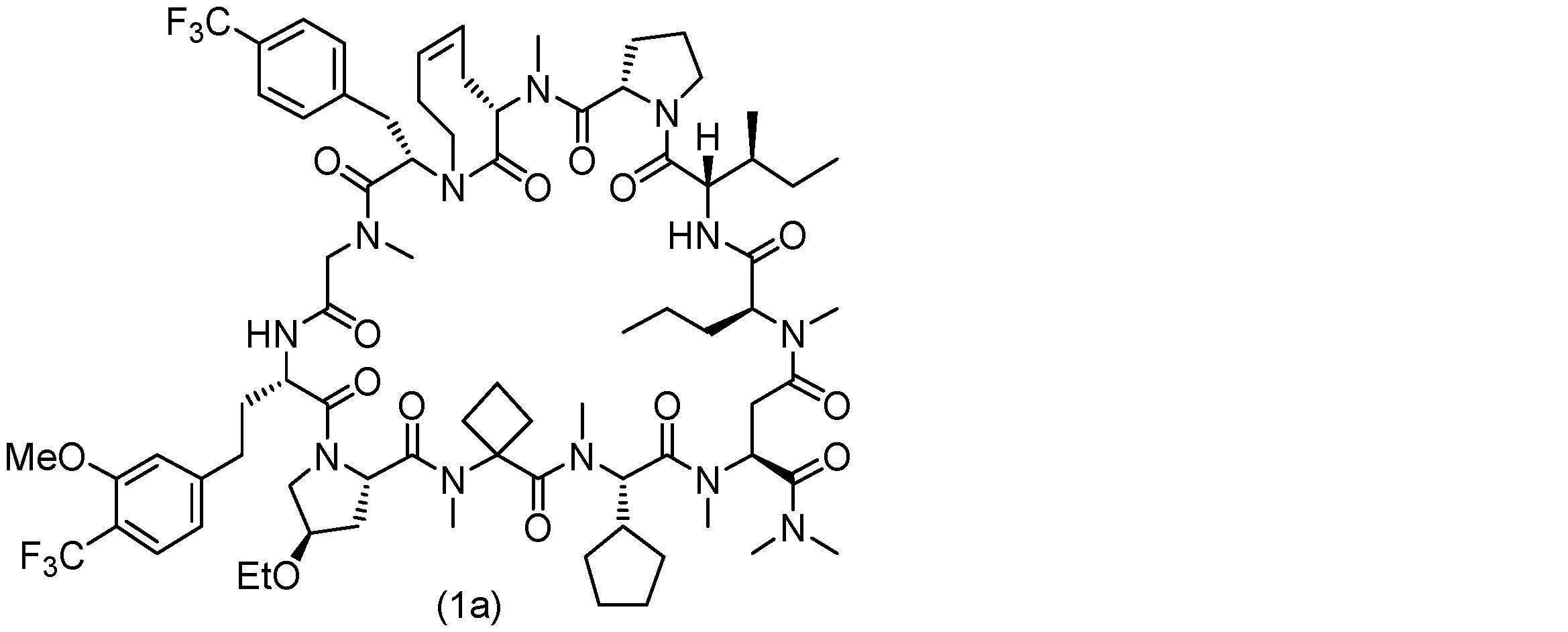
また、本発明に係る化合物が、当該化合物の塩、水和物、または溶媒和物として得られる場合、当該化合物は、そのフリー体に常法に従って変換することができる。 In addition, when the compound according to the present invention is obtained as a salt, hydrate, or solvate of the compound, the compound can be converted to its free form by a conventional method.
本明細書において、「溶媒A/水和物結晶」は、化合物の結晶格子中に溶媒A分子および水分子が含まれる結晶を意味する。本明細書において、「溶媒A/溶媒B/水和物結晶」は、化合物の結晶格子中に溶媒A分子、溶媒B分子および水分子が含まれる結晶を意味する。具体的には、例えば「アセトン/へプタン/水和物結晶は」は、化合物の結晶格子中にアセトン、へプタンおよび水が含まれる結晶を意味する。 In this specification, "solvent A/hydrate crystal" means a crystal in which solvent A molecules and water molecules are contained in the crystal lattice of a compound. In this specification, "solvent A/solvent B/hydrate crystal" means a crystal in which solvent A molecules, solvent B molecules, and water molecules are contained in the crystal lattice of a compound. Specifically, for example, "acetone/heptane/hydrate crystal" means a crystal in which acetone, heptane, and water are contained in the crystal lattice of a compound.
本明細書において、「および/または」との用語の意義は、「および」と「または」が適宜組み合わされたあらゆる組合せを含む。具体的には、例えば、「A、B、および/またはC」には、以下の7通りのバリエーションが含まれる;
(i) A、(ii) B、(iii) C、(iv) AおよびB、(v) AおよびC、(vi) BおよびC、(vii) A、B、およびC。
In this specification, the meaning of the term "and/or" includes any combination of "and" and "or". Specifically, for example, "A, B, and/or C" includes the following seven variations:
(i) A, (ii) B, (iii) C, (iv) A and B, (v) A and C, (vi) B and C, (vii) A, B, and C.
本明細書において「エピマー」とは、環状ペプチド化合物を構成するアミノ酸残基の、α-炭素に結合した側鎖の立体が反転した化合物(エピマー)を意味する。「エピマー」には、直鎖ペプチド化合物を環化して環状ペプチド化合物を製造する際に、直鎖ペプチド化合物のC末端のアミノ酸残基のα-炭素が立体反転した環状ペプチド化合物が含まれる。本発明の方法で製造される環状ペプチド化合物を含む総生成物中におけるエピマーは、たとえば、HPLC分析による210nmもしくは220nmでのUVarea値により決定することができる。 In this specification, "epimer" refers to a compound (epimer) in which the configuration of the side chain bonded to the α-carbon of an amino acid residue constituting a cyclic peptide compound is inverted. "Epimer" includes cyclic peptide compounds in which the α-carbon of the C-terminal amino acid residue of a linear peptide compound is inverted when the linear peptide compound is cyclized to produce the cyclic peptide compound. The epimer in the total product containing the cyclic peptide compound produced by the method of the present invention can be determined, for example, by the UVarea value at 210 nm or 220 nm by HPLC analysis.
本明細書において「環状ダイマー」とは、環状ペプチド化合物の原料であるペプチド化合物同士が直鎖状に結合した後、さらにそれが環化した化合物を意味する。本発明の方法で製造される環状ペプチド化合物を含む総生成物中における環状ダイマーは、たとえば、HPLC分析による210nmもしくは220nmでのUVarea値により決定することができる。 As used herein, the term "cyclic dimer" refers to a compound in which peptide compounds, the raw materials of a cyclic peptide compound, are bonded together in a linear chain and then cyclized. The amount of cyclic dimers in the total product containing the cyclic peptide compound produced by the method of the present invention can be determined, for example, by the UVarea value at 210 nm or 220 nm by HPLC analysis.
環状ペプチド化合物を製造する方法
ある態様において、本発明は、式(1)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法に関する。該方法は、溶媒中、式(2)または(3)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを反応させて環化する工程(環化工程)を含む(以下、「態様1」ともいう)。
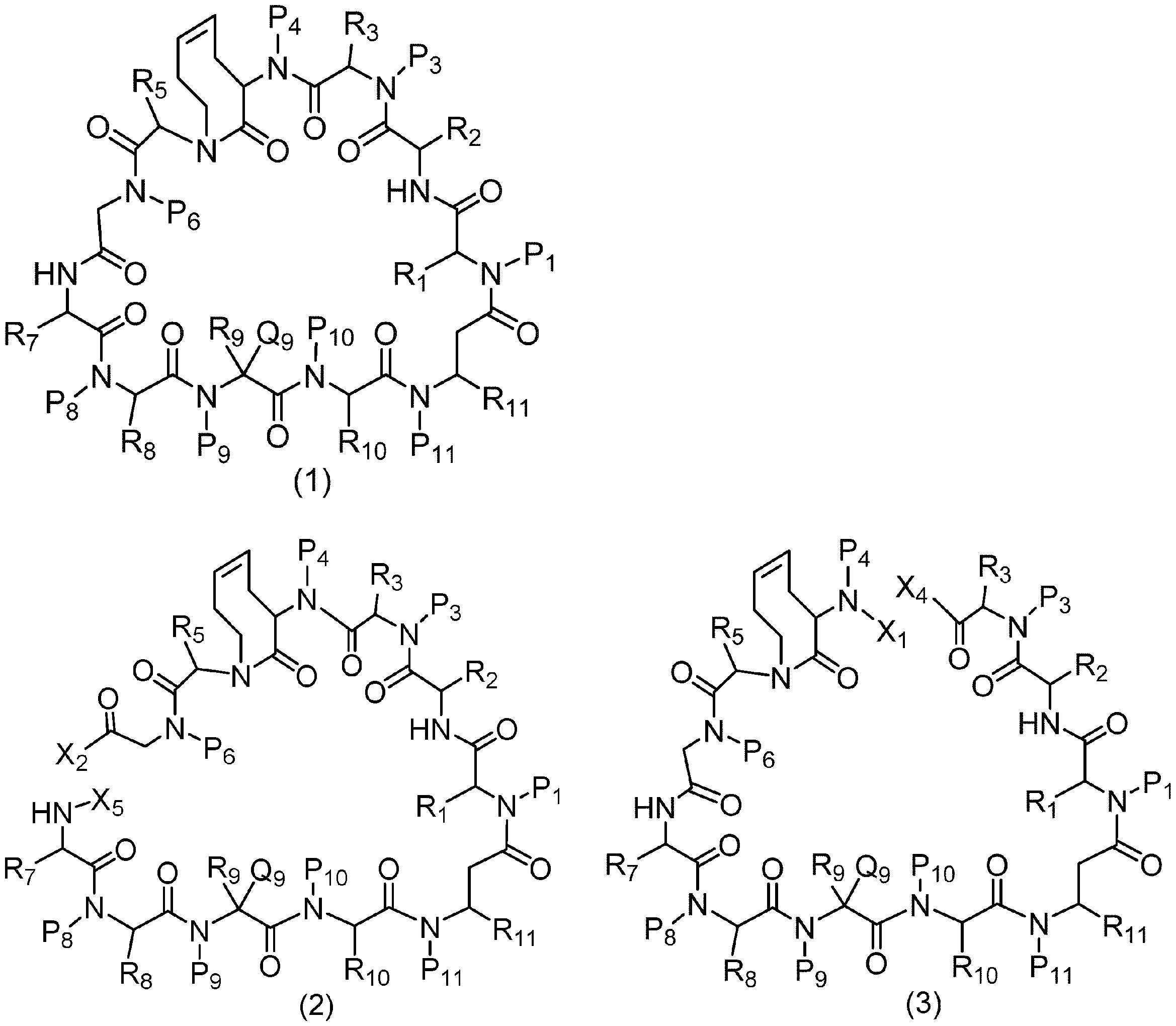
ある態様において、本発明は、式(1)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法に関する。該方法は、
(a) 式(4)~(6)で表されるペプチド化合物、もしくはその塩または前記ペプチド化合物もしくは塩の溶媒和物を用意する工程、
(b) 式(4)~(6)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて連結する工程(連結工程)、および
(c) (b)工程で得られたペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む(以下、「態様2」ともいう)。
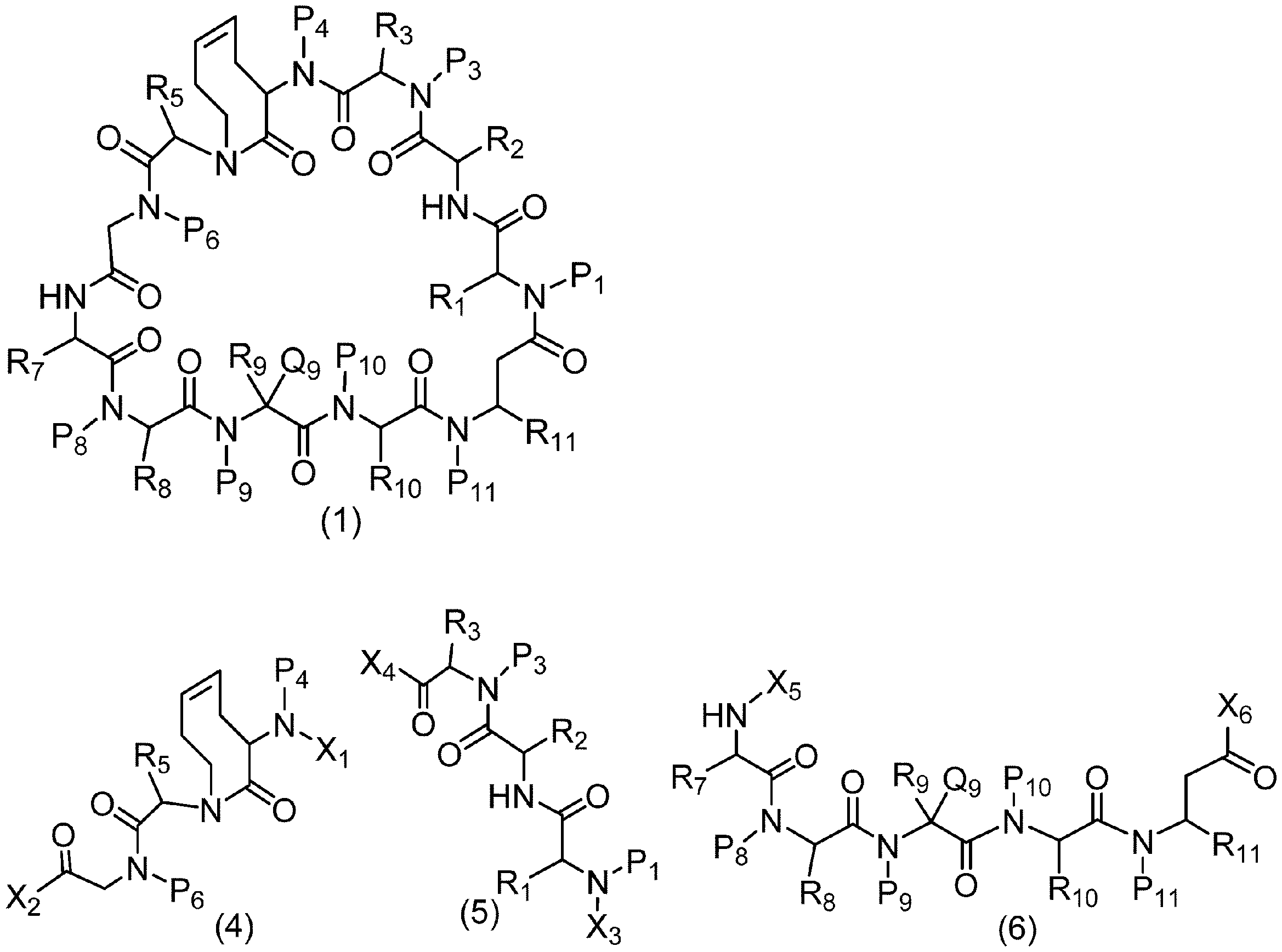
(a) preparing a peptide compound represented by any one of formulas (4) to (6), or a salt thereof, or a solvate of said peptide compound or salt;
(b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4) to (6) in a solvent (linking step); and
(c) A step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in step (b) by reacting them in a solvent (cyclization step) (hereinafter, also referred to as "Mode 2").
態様2において、(b)の工程は、(b-1)式(5)で表されるペプチド化合物のN末端のアミノ酸残基と、式(6)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(7)で表されるペプチド化合物に変換する工程(連結工程)を含んでもよい。
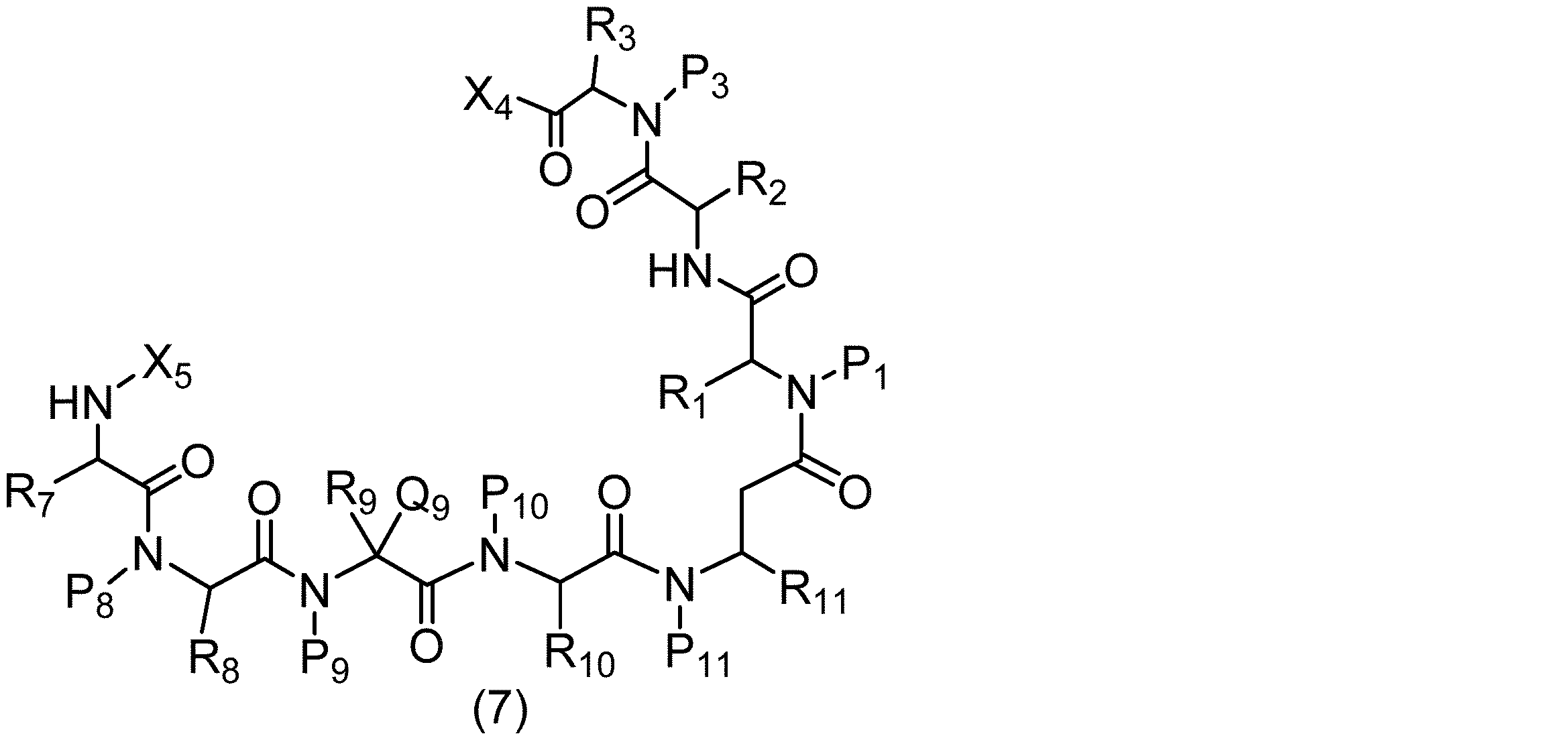
態様2において、(b)の工程は、(b-1)の工程に加えて、
(b-2)式(4)で表されるペプチド化合物のN末端のアミノ酸残基と、式(7)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(2)で表されるペプチド化合物に変換する工程(連結工程)を含んでもよく、かつ(c)の工程は、
(c-1)式(2)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含んでもよい。
In the embodiment 2, the step (b) includes, in addition to the step (b-1),
(b-2) a step (linking step) of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4) and the C-terminal amino acid residue of the peptide compound represented by formula (7) in a solvent to convert them into the peptide compound represented by formula (2), and the step (c) may include
(c-1) The method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2) by reaction in a solvent (cyclization step).
態様2において、(b)の工程は、(b-1)の工程に加えて、
(b-3)式(7)で表されるペプチド化合物のN末端のアミノ酸残基と、式(4)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(3)で表される化合物に変換する工程(連結工程)を含んでもよく、かつ(c)の工程は、
(c-2)式(3)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含んでもよい。
In the embodiment 2, the step (b) includes, in addition to the step (b-1),
(b-3) a step (linking step) of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7) and the C-terminal amino acid residue of the peptide compound represented by formula (4) in a solvent to convert them into the compound represented by formula (3), and the step (c) may include
(c-2) The method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3) by reaction in a solvent (cyclization step).
態様1および2において、好ましくはペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基との連結が、N末端のアミノ酸残基のアミノ基とC末端のアミノ酸残基のカルボキシル基との連結であり、より好ましくはペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基との連結が、N末端のアミノ酸残基のアミノ基とC末端のアミノ酸残基のカルボキシル基とのアミド結合による連結である。 In embodiments 1 and 2, the link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is preferably a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue, and more preferably a link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue via an amide bond.
ある態様において、環化工程の溶媒は、好ましくはニトリル系溶媒、ハロゲン系溶媒、エーテル系溶媒、アミド系溶媒、エステル系溶媒、および炭酸エステル系溶媒からなる群より選択される1つ以上を含む。ニトリル系溶媒として、具体的にはアセトニトリル、プロピオニトリル等が例示される。ハロゲン系溶媒として、具体的にはジクロロメタン、クロロホルム、1,2-ジクロロエタンが例示される。エーテル系溶媒として、具体的にはジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、シクロペンチルメチルエーテル、4-メチルテトラヒドロピラン、1,3-ジオキソラン、1,4-ジオキサン、1,2-ジメトキシエタン、ジイソプロピルエーテル、t-ブチルメチルエーテル、ジグリム、トリグリム、アニソール、テトラグリム等が例示される。アミド系溶媒として、具体的にはDMF、NMP、DMA、NEP、NBP、ホルムアミド等が例示される。エステル系溶媒として、具体的には酢酸メチル、酢酸エチル、プロピオン酸メチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、酢酸イソブチル、酢酸ペンチル、γ-バレロラクトンが例示される。炭酸エステル系溶媒として、具体的には炭酸ジメチル、炭酸ジエチル、炭酸ジブチルが例示される。環化工程の溶媒は、好ましくはアセトニトリル、炭酸ジメチル、2-メチルテトラヒドロフラン、4-メチルテトラヒドロピラン、テトラヒドロフラン、酢酸エチル、酢酸イソプロピル、ジクロロメタン、DMF、およびアニソールからなる群より選択される1種または複数であり、より好ましくはアセトニトリル、2-メチルテトラヒドロフラン、酢酸エチル、およびジクロロメタンからなる群より選択される1種または複数であり、さらに好ましくはアセトニトリル、2-メチルテトラヒドロフラン、または酢酸エチルである。 In one embodiment, the solvent for the cyclization step preferably includes one or more selected from the group consisting of nitrile solvents, halogen solvents, ether solvents, amide solvents, ester solvents, and carbonate solvents. Specific examples of nitrile solvents include acetonitrile and propionitrile. Specific examples of halogen solvents include dichloromethane, chloroform, and 1,2-dichloroethane. Specific examples of ether solvents include diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, 4-methyltetrahydropyran, 1,3-dioxolane, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, t-butyl methyl ether, diglyme, triglyme, anisole, and tetraglyme. Specific examples of amide solvents include DMF, NMP, DMA, NEP, NBP, and formamide. Specific examples of ester solvents include methyl acetate, ethyl acetate, methyl propionate, butyl acetate, propyl acetate, isopropyl acetate, isobutyl acetate, pentyl acetate, and γ-valerolactone. Specific examples of carbonate ester solvents include dimethyl carbonate, diethyl carbonate, and dibutyl carbonate. The solvent for the cyclization step is preferably one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, dichloromethane, DMF, and anisole, more preferably one or more selected from the group consisting of acetonitrile, 2-methyltetrahydrofuran, ethyl acetate, and dichloromethane, and even more preferably acetonitrile, 2-methyltetrahydrofuran, or ethyl acetate.
ある態様において、環化工程は、溶媒中、縮合試薬の存在下または不存在下、塩基の存在下または不存在下、-20℃~溶媒の沸点付近の温度、好ましくは-20℃~100℃、好ましくは-5℃~60℃の温度で、反応混合物を10分~48時間攪拌することで行うことができる。 In one embodiment, the cyclization step can be carried out in a solvent, in the presence or absence of a condensation reagent, in the presence or absence of a base, at a temperature between -20°C and the boiling point of the solvent, preferably between -20°C and 100°C, and preferably between -5°C and 60°C, by stirring the reaction mixture for 10 minutes to 48 hours.
環化工程において使用される縮合試薬、塩基およびそれらの使用量としては特に限定されず、ペプチド合成で一般に使用される縮合試薬、塩基及び使用量が好ましい(例えば、Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.))。一方、環化工程において縮合試薬を用いない場合、あらかじめカルボキシル基を活性エステルに変換したものを利用してもよい。 The condensation reagent, base, and amounts thereof used in the cyclization step are not particularly limited, and condensation reagents, bases, and amounts thereof generally used in peptide synthesis are preferred (e.g., Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.)). On the other hand, when no condensation reagent is used in the cyclization step, a carboxyl group that has been converted into an active ester in advance may be used.
環化工程の縮合試薬としては、具体的には例えば、N,N’-ジシクロヘキシルカルボジイミド(DCC)、N,N’-ジイソプロピルカルボジイミド(DIC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI HCl)、1-ヒドロキシ-1H-ベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、2-シアノ-2-(ヒドロキシイミノ)酢酸エチル(oxyma)、3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(HOOBtまたはHODhbt)、N-ヒドロキシ-5-ノルボルネン-2,3-ジカルボキシミド(HONB)、2,3,4,5,6-ペンタフルオロフェノール(HOPfp)、N-ヒドロキシスクシンイミド(HOSu)、6-クロロ-1-ヒドロキシ-1H-ベンゾトリアゾール(Cl-HOBt)、O-(1H-ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩(HBTU)、O-(7-アザ-1H-ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩(HATU)、N-[1-(シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ(モルホリノ)]ウロニウムヘキサフルオロリン酸塩(COMU)、O-[(エトキシカルボニル)シアノメチレンアミノ]-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩(HOTU)、O-(1H-ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムテトラフルオロホウ酸塩(TBTU)、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムテトラフルオロホウ酸塩(TATU)、[エチルシアノ(ヒドロキシイミノ)アセタト-O2]トリ-1-ピロリジニルホスホニウムヘキサフルオロリン酸塩(PyOxim)、2-ブロモ-1-エチルピリジニウムテトラフルオロホウ酸塩(BEP)、1H-ベンゾトリアゾール-1-イルオキシ-トリ(ピロリジノ)ホスホニウムヘキサフルオロリン酸塩(PyBOP)、1H-ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロリン酸塩(BOP)、ブロモトリ(ピロリジノ)ホスホニウムヘキサフルオロリン酸塩(PyBroP)、クロロトリ(ピロリジノ)ホスホニウムヘキサフルオロリン酸塩(PyCloP)、(7-アザベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウムヘキサフルオロリン酸(PyAOP)、ブロモトリス(ジメチルアミノ)ホスホニウムヘキサフルオロリン酸(Brop)、3-(ジエトキシホスホリルオキシ)-1,2,3-ベンゾトリアジン-4(3H)-オン(DEPBT)、N,N,N’,N’-テトラメチル-O-(N-スクシンイミジル)ウロニウムテトラフルオロホウ酸(TSTU)、N,N,N’,N’-テトラメチル-O-(N-スクシンイミジル)ウロニウムヘキサフルオロリン酸(HSTU)、O-(3,4-ジヒドロ-4-オキソ-1,2,3-ベンゾトリアジン-3-イル)-N,N,N’,N’-テトラメチルウロニウムテトラフルオロホウ酸塩(TDBTU)、テトラメチルチウロニウムS-(1-オキシド-2-ピリジル)-N,N,N’,N’-テトラフルオロホウ酸塩(TOTT)、O-(2-オキソ-1(2H)ピリジル)-N,N,N’,N’-テトラメチルウロニウムテトラフルオロホウ酸(TPTU)、N,N’-カルボニルジイミダゾール(CDI)、1,1’-カルボニル-ジ-(1,2,4-トリアゾール)(CDT)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウム塩化物(DMT-MM)、プロピルホスホン酸無水物(T3P)などが挙げられる。これらの中でも、副生成物を抑制する観点から、環化工程の縮合試薬としては、HATU、COMU、DMT-MM、PyOxim、PyBOP、HCTU、T3P、EDCI、BEP、およびPyClopからなる群より選択される1種または複数が好ましく、HATU、COMU、PyOxim、PyBOP、HCTU、およびT3Pからなる群より選択される1つがより好ましく、HATU、COMUがさらに好ましい。また、溶媒と縮合試薬の組合せは、副生成物をより一層抑制できることから、HATUとアセトニトリルまたは2-メチルテトラヒドロフラン;COMUとアセトニトリルまたは2-メチルテトラヒドロフランが好ましい。 Condensation reagents for the cyclization step include, for example, N,N'-dicyclohexylcarbodiimide (DCC), N,N'-diisopropylcarbodiimide (DIC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI), HCl), 1-hydroxy-1H-benzotriazole (HOBt), 1-hydroxy-7-azabenzotriazole (HOAt), ethyl 2-cyano-2-(hydroxyimino)acetate (oxyma), 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HOOBt or HODhbt), N-hydroxy-5-norbornene-2,3-dicarboximide (HONB), 2,3,4,5,6-pentafluorophenol (HOPfp), N-hydroxysuccinimide (HOSu), 6-chloro-1-hydroxy-1H-benzotriazole (Cl-HOBt), O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HBTU), O-(7-aza-1H -benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU), N-[1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)dimethylamino(morpholino)]uronium hexafluorophosphate (COMU), O-[(ethoxycarbonyl)cyanomethyleneamino]-N,N,N',N'-tetramethyluronium hexafluorophosphate (HOTU), O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (TBTU), O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (TATU), [ethylcyano(hydroxyimino)acetato-O 2 ]tri-1-pyrrolidinylphosphonium hexafluorophosphate (PyOxim), 2-bromo-1-ethylpyridinium tetrafluoroborate (BEP), 1H-benzotriazol-1-yloxy-tri(pyrrolidino)phosphonium hexafluorophosphate (PyBOP), 1H-benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP), bromotri(pyrrolidino)phosphonium hexafluorophosphate phosphate (PyBroP), chlorotri(pyrrolidino)phosphonium hexafluorophosphate (PyCloP), (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP), bromotris(dimethylamino)phosphonium hexafluorophosphate (Brop), 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT), N,N,N',N'-tetramethyl-O- (N-succinimidyl)uronium tetrafluoroborate (TSTU), N,N,N',N'-tetramethyl-O-(N-succinimidyl)uronium hexafluorophosphate (HSTU), O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (TDBTU), tetramethylthiuronium S-(1-oxido-2-pyridyl)-N,N,N',N'-tetramethylthiuronium fluoroborate (TOTT), O-(2-oxo-1(2H)pyridyl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (TPTU), N,N'-carbonyldiimidazole (CDI), 1,1'-carbonyl-di-(1,2,4-triazole) (CDT), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM), propylphosphonic anhydride (T3P), and the like. Among these, from the viewpoint of suppressing by-products, the condensation reagent for the cyclization step is preferably one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop, more preferably one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P, and even more preferably HATU and COMU. Furthermore, as a combination of a solvent and a condensation reagent, HATU and acetonitrile or 2-methyltetrahydrofuran; COMU and acetonitrile or 2-methyltetrahydrofuran are preferred, since by-products can be further suppressed.
環化工程の塩基としては、有機塩基が好適に用いられ、中でも3級アミンを含む有機塩基が好ましい。このような塩基としては、具体的には例えば、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、ピリジン、1,8-ジアザビシクロ〔5.4.0〕-7-ウンデセン(DBU)、2,3,6,7-テトラヒドロ-1H,5H-9-アザベンゾ[ij]キノリジン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,5-ジアザビシクロ[4.3.0]-5-ノネン(DBN)、7-メチル-1,5,7-トリアザビシクロ[4.4.0]デカ-5-エン、1,1,3,3-テトラメチルグアニジン(TMG)、1,8-ビス(テトラメチルグアニジノ)ナフタレン(TMGN)、2-tert-ブチル-1,1,3,3-テトラメチルグアニジン(BTMG)、トリエチルアミン(TEA)、トリメチルアミン、1-メチルピぺリジン、N,N’-ジメチルピペラジン、N-エチルモルホリン、p-ジメチルアミノピリジン(DMAP)などが挙げられる。これらの中でも、副生成物を抑制する観点から、環化工程の塩基としては、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、およびピリジンからなる群より選択される1種または複数が好ましく、N,N-ジイソプロピルエチルアミン(DIPEA)、2,6-ルチジンが好ましい。また、溶媒と縮合試薬と塩基の組合せは、副生成物をより一層抑制できることから、HATUとアセトニトリルとN,N-ジイソプロピルエチルアミン(DIPEA);HATUと2-メチルテトラヒドロフランとN,N-ジイソプロピルエチルアミン(DIPEA);COMUとアセトニトリルと2,6-ルチジン;COMUと2-メチルテトラヒドロフランと2,6-ルチジンが好ましい。 As the base for the cyclization step, an organic base is preferably used, and among them, an organic base containing a tertiary amine is preferable. Specific examples of such bases include 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonane (NDABCO), and 1,5-diazabicyclo[4.3.0]-5-nonane (NDABCO). Examples of bis(tetramethylguanidino)naphthalene (DBN), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene, 1,1,3,3-tetramethylguanidine (TMG), 1,8-bis(tetramethylguanidino)naphthalene (TMGN), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG), triethylamine (TEA), trimethylamine, 1-methylpiperidine, N,N'-dimethylpiperazine, N-ethylmorpholine, and p-dimethylaminopyridine (DMAP). Among these, from the viewpoint of suppressing by-products, the base for the cyclization step is preferably one or more selected from the group consisting of 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, and pyridine, and N,N-diisopropylethylamine (DIPEA) and 2,6-lutidine are preferred. In addition, the combinations of solvent, condensation reagent, and base are more effective in suppressing by-products, and therefore are preferably HATU, acetonitrile, and N,N-diisopropylethylamine (DIPEA); HATU, 2-methyltetrahydrofuran, and N,N-diisopropylethylamine (DIPEA); COMU, acetonitrile, and 2,6-lutidine; and COMU, 2-methyltetrahydrofuran, and 2,6-lutidine.
ある態様において、環化工程は液相法で行われる。 In one embodiment, the cyclization step is carried out in a liquid phase process.
ある態様において、環化工程は溶媒と縮合試薬とを混合して得られた混合液中に、ペプチド化合物および任意で塩基を混合することで行われる。本明細書においては、当該操作を「逆滴下」という場合がある。ペプチド化合物および塩基を長時間、例えば、数時間~数日間、好ましくは1~24時間、より好ましくは1~10時間かけて逆滴下することで、希釈のために多量の溶媒を用いることなく副生成物の副生を抑制することができる。 In one embodiment, the cyclization step is carried out by mixing the peptide compound and, optionally, a base into a mixture obtained by mixing a solvent and a condensation reagent. In this specification, this operation is sometimes referred to as "reverse dripping." By reverse dripping the peptide compound and base over a long period of time, for example, several hours to several days, preferably 1 to 24 hours, and more preferably 1 to 10 hours, it is possible to suppress the production of by-products without using a large amount of solvent for dilution.
本発明の方法により製造される環状ペプチド化合物は、以下に述べるように副生成物(例えば、エピマー、環状ダイマーなど)の含有率が小さく高純度である。 The cyclic peptide compounds produced by the method of the present invention have a low content of by-products (e.g., epimers, cyclic dimers, etc.) and are highly pure, as described below.
ある態様において、環化工程において生成する総副生成物の含有率は、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、20%未満、15%未満、10%未満、5%未満、または3%未満である。 In some embodiments, the content of total by-products produced in the cyclization step is less than 20%, less than 15%, less than 10%, less than 5%, or less than 3% based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
ある態様において、環化工程において生成する副生成物各々の含有率は、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、3%未満、1%未満、または検出不能な量である。 In some embodiments, the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
ある態様において、環化工程において生成する副生成物各々の含有率は、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、3%未満、1%未満、または検出不能な量であり、該副生成物はエピマーおよび/または環状ダイマーを含む。 In some embodiments, the content of each by-product produced in the cyclization step is less than 15%, less than 10%, less than 5%, less than 3%, less than 1%, or an undetectable amount based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis, and the by-products include epimers and/or cyclic dimers.
ある態様において、環化工程において生成する副生成物は、エピマーを含み、該エピマーの含有率は、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、10%未満、7.5%未満、5%未満、2.5%未満、または1%未満である。 In some embodiments, the by-product produced in the cyclization step includes an epimer, and the epimer content is less than 10%, less than 7.5%, less than 5%, less than 2.5%, or less than 1% based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
ある態様において、環化工程において生成する副生成物は、環状ダイマーを含み、該環状ダイマーの含有率は、生成物の総量を基準として、HPLC分析による220nmでのUVarea値により決定される、15%未満、10%未満、5%未満、2.5%未満、または1%未満である。 In some embodiments, the by-products produced in the cyclization step include cyclic dimers, and the content of the cyclic dimers is less than 15%, less than 10%, less than 5%, less than 2.5%, or less than 1% based on the total amount of product, as determined by the UVarea value at 220 nm by HPLC analysis.
ある態様において、連結工程の溶媒は、好ましくはニトリル系溶媒、ハロゲン系溶媒、エーテル系溶媒、アミド系溶媒、エステル系溶媒、および炭酸エステル系溶媒からなる群より選択される1つ以上を含む。ニトリル系溶媒、ハロゲン系溶媒、エーテル系溶媒、アミド系溶媒、エステル系溶媒、および炭酸エステル系溶媒としては、上記環化工程の溶媒として例示されたものが挙げられる。連結工程の溶媒は、好ましくはアセトニトリル、炭酸ジメチル、2-メチルテトラヒドロフラン、4-メチルテトラヒドロピラン、テトラヒドロフラン、酢酸エチル、酢酸イソプロピル、DMF、およびアニソールからなる群より選択される1種または複数であり、より好ましくはアセトニトリル、炭酸ジメチル、2-メチルテトラヒドロフラン、4-メチルテトラヒドロピラン、テトラヒドロフラン、酢酸エチル、酢酸イソプロピル、およびアニソールからなる群より選択される1種または複数と、DMFとの混合溶媒であり、さらに好ましくはアセトニトリル、2-メチルテトラヒドロフラン、およびDMFの混合溶媒であり、特に好ましくは2-メチルテトラヒドロフラン、およびDMFの混合溶媒である。 In one embodiment, the solvent in the linking step preferably includes one or more selected from the group consisting of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents. Examples of nitrile-based solvents, halogen-based solvents, ether-based solvents, amide-based solvents, ester-based solvents, and carbonate-based solvents include those exemplified as the solvent in the cyclization step. The solvent in the linking step is preferably one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, DMF, and anisole, more preferably a mixed solvent of one or more selected from the group consisting of acetonitrile, dimethyl carbonate, 2-methyltetrahydrofuran, 4-methyltetrahydropyran, tetrahydrofuran, ethyl acetate, isopropyl acetate, and anisole and DMF, even more preferably a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF, and particularly preferably a mixed solvent of 2-methyltetrahydrofuran and DMF.
ある態様において、連結工程は、溶媒中、縮合試薬の存在下または不存在下、塩基の存在下または不存在下、-20℃~溶媒の沸点付近の温度、好ましくは-20℃~100℃、好ましくは-5℃~60℃の温度で、反応組成物を10分~48時間攪拌することで行うことができる。 In one embodiment, the linking step can be carried out in a solvent, in the presence or absence of a condensation reagent, in the presence or absence of a base, at a temperature between -20°C and the boiling point of the solvent, preferably between -20°C and 100°C, and preferably between -5°C and 60°C, by stirring the reaction composition for 10 minutes to 48 hours.
連結工程において使用される縮合試薬、塩基およびそれらの使用量としては特に限定されず、ペプチド合成で一般に使用される縮合試薬、塩基及び使用量が好ましい(例えば、Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.))。一方、連結工程において縮合試薬を用いない場合、あらかじめカルボキシル基を活性エステルに変換したものを利用してもよい。 The condensation reagent, base, and amounts thereof used in the coupling step are not particularly limited, and condensation reagents, bases, and amounts thereof generally used in peptide synthesis are preferred (e.g., Peptide Coupling Reagents, More than a Letter Soup (Chem. Rev. 2011, 111, 6557-6602.)). On the other hand, when no condensation reagent is used in the coupling step, a reagent in which the carboxyl group has been converted into an active ester in advance may be used.
連結工程の縮合試薬としては、上記環化工程の縮合試薬として例示されたものが挙げられる。これらの中でも、副生成物を抑制する観点から、連結工程の縮合試薬としては、HATU、COMU、DMT-MM、PyOxim、PyBOP、HCTU、T3P、EDCI、BEP、およびPyClopからなる群より選択される1種または複数が好ましく、HATU、COMU、PyOxim、PyBOP、HCTU、およびT3Pからなる群より選択される1つがより好ましく、HATU、COMUがさらに好ましい。また、溶媒と縮合試薬の組合せは、副生成物をより一層抑制できることから、HATUとアセトニトリルまたは2-メチルテトラヒドロフラン;HATUとアセトニトリル、2-メチルテトラヒドロフランおよびDMFの混合溶媒;COMUとアセトニトリルまたは2-メチルテトラヒドロフラン;COMUとアセトニトリル、2-メチルテトラヒドロフランおよびDMFの混合溶媒が好ましい。 Condensation reagents for the linking step include those exemplified as condensation reagents for the cyclization step. Among these, from the viewpoint of suppressing by-products, the condensation reagent for the linking step is preferably one or more selected from the group consisting of HATU, COMU, DMT-MM, PyOxim, PyBOP, HCTU, T3P, EDCI, BEP, and PyClop, more preferably one selected from the group consisting of HATU, COMU, PyOxim, PyBOP, HCTU, and T3P, and even more preferably HATU and COMU. In addition, the following combinations of solvents and condensation reagents are preferred, since they can further suppress by-products: HATU and acetonitrile or 2-methyltetrahydrofuran; a mixed solvent of HATU and acetonitrile, 2-methyltetrahydrofuran, and DMF; COMU and acetonitrile or 2-methyltetrahydrofuran; and a mixed solvent of COMU and acetonitrile, 2-methyltetrahydrofuran, and DMF.
連結工程の塩基としては、上記環化工程の塩基として例示されたものが挙げられる。これらの中でも、副生成物を抑制する観点から、連結工程の塩基としては、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、ピリジン、1,8-ジアザビシクロ〔5.4.0〕-7-ウンデセン(DBU)、2,3,6,7-テトラヒドロ-1H,5H-9-アザベンゾ[ij]キノリジン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,5-ジアザビシクロ[4.3.0]-5-ノネン(DBN)、7-メチル-1,5,7-トリアザビシクロ[4.4.0]デカ-5-エン、1,1,3,3-テトラメチルグアニジン(TMG)、1,8-ビス(テトラメチルグアニジノ)ナフタレン(TMGN)、2-tert-ブチル-1,1,3,3-テトラメチルグアニジン(BTMG)、トリエチルアミン(TEA)、トリメチルアミン、1-メチルピぺリジン、N,N’-ジメチルピペラジン、N-エチルモルホリン、およびp-ジメチルアミノピリジン(DMAP)からなる群より選択される1種または複数が好ましく、2,2,6,6-テトラメチルピペリジン、N-メチルモルホリン、N,N-ジイソプロピルエチルアミン(DIPEA)、2,4,6-コリジン、2,6-ルチジン、およびピリジンからなる群より選択される1種または複数が好ましい。また、溶媒と縮合試薬と塩基の組合せは、副生成物をより一層抑制できることから、HATUとアセトニトリルまたは2-メチルテトラヒドロフランとN,N-ジイソプロピルエチルアミン(DIPEA);HATUとアセトニトリル、2-メチルテトラヒドロフランおよびDMFの混合溶媒とN,N-ジイソプロピルエチルアミン(DIPEA);COMUとアセトニトリルまたは2-メチルテトラヒドロフランとN-メチルモルホリンまたは2,6-ルチジン;COMUとアセトニトリル、2-メチルテトラヒドロフランおよびDMFの混合溶媒とN-メチルモルホリンまたは2,6-ルチジンが好ましい。 The bases in the linking step include those exemplified as the bases in the cyclization step above. Among these, from the viewpoint of suppressing by-products, the bases in the linking step include 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, pyridine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 2,3,6,7-tetrahydro-1H,5H-9-azabenzo[ij]quinolizine, 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene, 1,1,3,3-tetramethyl One or more selected from the group consisting of guanidine (TMG), 1,8-bis(tetramethylguanidino)naphthalene (TMGN), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG), triethylamine (TEA), trimethylamine, 1-methylpiperidine, N,N'-dimethylpiperazine, N-ethylmorpholine, and p-dimethylaminopyridine (DMAP) are preferred, and one or more selected from the group consisting of 2,2,6,6-tetramethylpiperidine, N-methylmorpholine, N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, 2,6-lutidine, and pyridine are preferred. In addition, the combinations of solvent, condensation reagent, and base can further suppress by-products, so the following are preferred: HATU and acetonitrile or 2-methyltetrahydrofuran and N,N-diisopropylethylamine (DIPEA); HATU and a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF and N,N-diisopropylethylamine (DIPEA); COMU and acetonitrile or 2-methyltetrahydrofuran and N-methylmorpholine or 2,6-lutidine; and COMU and a mixed solvent of acetonitrile, 2-methyltetrahydrofuran, and DMF and N-methylmorpholine or 2,6-lutidine.
ある態様において、連結工程は液相法で行われる。 In one embodiment, the linking step is carried out using a liquid phase method.
ある態様において、本発明の方法は式(4)~(6)で表されるペプチド化合物、もしくはその塩またはそれらの溶媒和物を用意する工程をさらに含む。式(4)~(6)で表されるペプチド化合物は、たとえば、以下の一般製法により製造することができる。 In one embodiment, the method of the present invention further comprises the step of preparing a peptide compound represented by formulas (4) to (6), or a salt thereof, or a solvate thereof. The peptide compound represented by formulas (4) to (6) can be produced, for example, by the following general method.
式(4)で表されるペプチド化合物(ペプチド化合物(4))の一般製法
以下に、ペプチド化合物(4)の一般製法を示す。以下のスキームにおいてPg4およびPg5はアミノ基の保護基、Xg6は酸素原子とそれに結合する保護基、R5はアミノ酸の側鎖、P4およびP6は窒素原子の置換基をそれぞれ表す。
General method for producing a peptide compound represented by formula (4) (peptide compound (4)) is shown below. In the following scheme, Pg4 and Pg5 represent protecting groups for the amino group, Xg6 represents an oxygen atom and a protecting group bonded thereto, R5 represents the side chain of the amino acid, and P4 and P6 represent substituents for the nitrogen atom.
ペプチド化合物(4)は、以下の方法を用いて製造することができる。

ペプチド化合物(4)は、以下の方法を用いて製造することもできる。
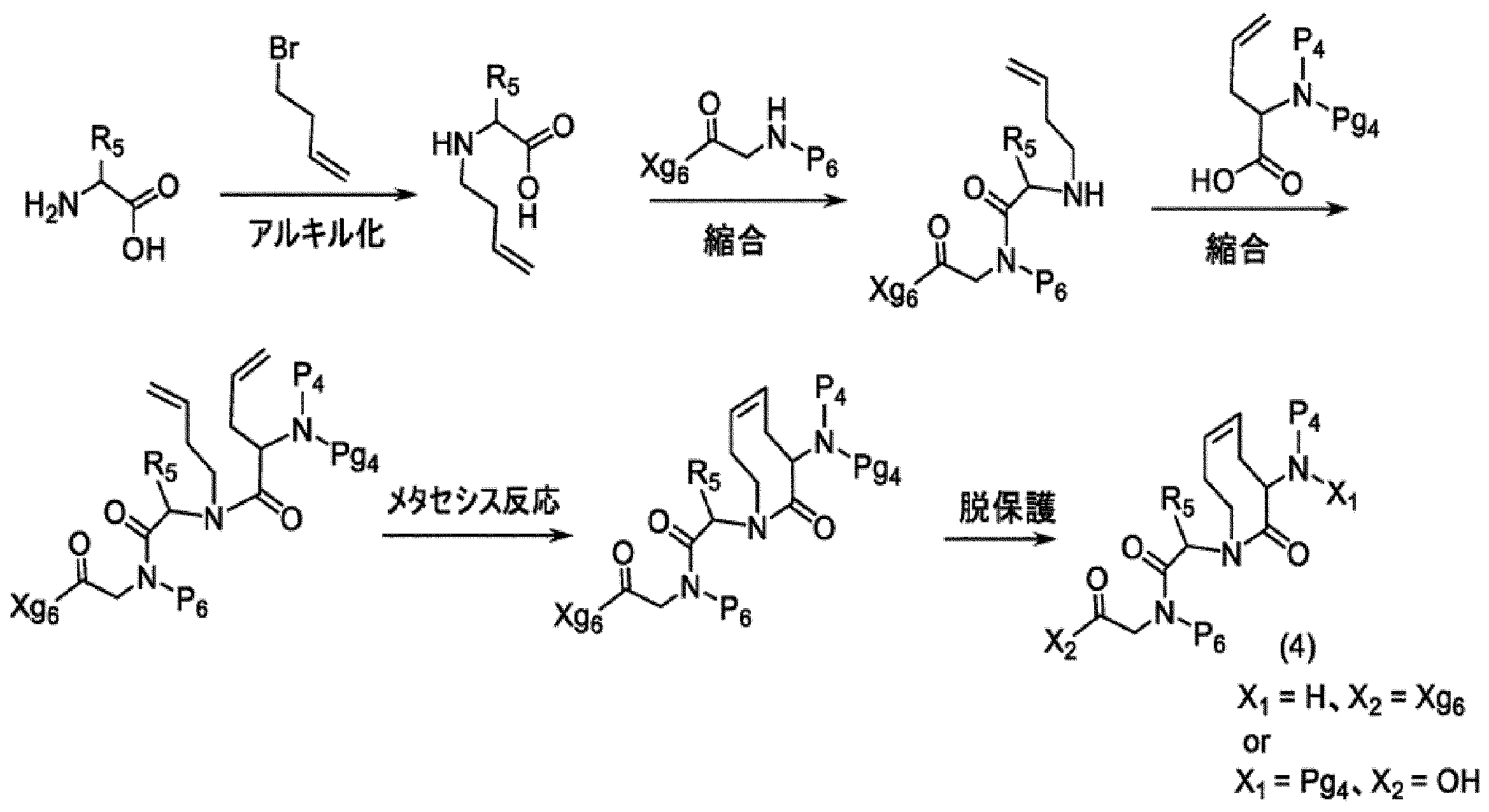
式(5)で表されるペプチド化合物(ペプチド化合物(5))の一般製法
以下に、ペプチド化合物(5)の一般製法を示す。以下のスキームにおいてPg1およびPg2はアミノ基の保護基、Xg3は酸素原子とそれに結合する保護基、R1、R2、およびR3はアミノ酸の側鎖、P1およびP3は窒素原子の置換基をそれぞれ表す。
General method for producing a peptide compound represented by formula (5) (peptide compound (5)) A general method for producing peptide compound (5) is shown below. In the following scheme, Pg1 and Pg2 represent protecting groups for amino groups, Xg3 represents an oxygen atom and a protecting group bonded thereto, R1 , R2 , and R3 represent side chains of an amino acid, and P1 and P3 represent substituents for a nitrogen atom.
ペプチド化合物(5)は、以下の方法を用いて製造することができる。

ペプチド化合物(5)は、固相合成により合成することもできる。その場合、上記式におけるXg3は酸素原子とそれに結合するリンカーを介して固相担体に結合しているアミノ酸であり、これに対して保護アミノ酸を縮合することにより、N末端側にアミノ酸を伸長することができる。縮合反応は、上述の連結工程において使用される縮合試薬、塩基を用いることが可能であり、例えばカルボキシル基の活性化剤として、DICとOxymaの組み合わせ、DICとHOAtの組み合わせ、HATUとDIPEAとの組み合わせ、混合酸無水物や酸ハロゲン化物を経由するなど様々な方法が可能である。続いてアミノ基の保護基の脱保護、保護アミノ酸の縮合を順次行うことにより、3アミノ酸からなるフラグメントを合成することができる。次いで固相からの切り出しを行うことで、C末端無保護のペプチド化合物(5)を製造することができる。 Peptide compound (5) can also be synthesized by solid-phase synthesis. In that case, Xg 3 in the above formula is an amino acid bound to a solid-phase support via an oxygen atom and a linker bound thereto, and an amino acid can be extended to the N-terminus by condensing a protected amino acid with this. The condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a combination via a mixed acid anhydride or acid halide as an activator of the carboxyl group. Subsequently, a fragment consisting of three amino acids can be synthesized by sequentially deprotecting the protecting group of the amino group and condensing the protected amino acid. Then, the peptide compound (5) with an unprotected C-terminus can be produced by cutting out from the solid phase.
式(6)で表されるペプチド化合物(ペプチド化合物(6))の一般製法
以下に、ペプチド化合物(6)の一般製法を示す。以下のスキームにおいて、Pg7、Pg8、Pg9、およびPg10はアミノ基の保護基、Xg11は酸素原子とそれに結合する保護基、R7、R8、R9、Q9、R10、およびR11はアミノ酸の側鎖、P8、P9、P10、およびP11は窒素原子の置換基をそれぞれ表す。
General method for producing a peptide compound represented by formula (6) (peptide compound (6)) The general method for producing peptide compound (6) is shown below. In the following scheme, Pg7 , Pg8 , Pg9 , and Pg10 represent protecting groups for amino groups, Xg11 represents an oxygen atom and a protecting group bonded thereto, R7 , R8 , R9 , Q9 , R10 , and R11 represent side chains of amino acids, and P8 , P9 , P10 , and P11 represent substituents for nitrogen atoms.
ペプチド化合物(6)は、以下の方法を用いて製造することができる。

ペプチド化合物(6)は、固相合成により合成することもできる。その場合、上記式におけるXg11は酸素原子とそれに結合するリンカーを介して固相担体に結合しているβアミノ酸骨格を有するアミノ酸であり、これに対して保護アミノ酸を縮合することにより、N末端側にアミノ酸を伸長することができる。縮合反応は、上述の連結工程において使用される縮合試薬、塩基を用いることが可能であり、例えばカルボキシル基の活性化剤として、DICとOxymaの組み合わせ、DICとHOAtの組み合わせ、HATUとDIPEAとの組み合わせ、混合酸無水物や酸ハロゲン化物を経由するなど様々な方法が可能である。続いてアミノ基の保護基の脱保護、保護アミノ酸の縮合を順次行うことにより、5アミノ酸からなるフラグメントを合成することができる。次いで固相からの切り出しを行うことで、C末端無保護のペプチド化合物(6)を製造することができる。 Peptide compound (6) can also be synthesized by solid-phase synthesis. In that case, Xg 11 in the above formula is an amino acid having a β-amino acid skeleton bound to a solid-phase support via an oxygen atom and a linker bound thereto, and an amino acid can be extended to the N-terminus by condensing a protected amino acid with this. The condensation reaction can be performed using the condensation reagent and base used in the above-mentioned linking step, and various methods are possible, such as a combination of DIC and Oxyma, a combination of DIC and HOAt, a combination of HATU and DIPEA, or a combination via a mixed acid anhydride or acid halide as an activator of the carboxyl group. Subsequently, a fragment consisting of five amino acids can be synthesized by sequentially deprotecting the protecting group of the amino group and condensing the protected amino acid. Then, the peptide compound (6) with an unprotected C-terminus can be produced by cutting out from the solid phase.
上記に示した式(4)~(6)で表されるペプチド化合物の製造方法では、目的の官能基以外が化学反応を起こしてしまう場合がある。このような場合は、目的としない官能基に保護基を導入することにより、所望の反応のみを進行させることができる。このような保護基の脱着反応は、例えば、「Greene’s,“Protective Groups in Organic Synthesis”(第5版,John Wiley & Sons 2014)」に記載の方法を挙げることができる。化合物の官能基の変換反応は、LarockのComprehensive Organic Transformations: A Guide to Functional Group Preparations(第5版)やSmithのMarch's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure(第8版)を参照することができる。 In the manufacturing method of peptide compounds represented by formulas (4) to (6) shown above, functional groups other than the intended functional group may undergo chemical reactions. In such cases, a protecting group can be introduced to the unintended functional group to allow only the desired reaction to proceed. Examples of such protecting group removal reactions include the method described in Greene's, "Protective Groups in Organic Synthesis" (5th ed., John Wiley & Sons 2014). For transformation reactions of the functional groups of compounds, refer to Larock's Comprehensive Organic Transformations: A Guide to Functional Group Preparations (5th ed.) and Smith's March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (8th ed.).
ある態様において、本発明の方法により製造される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の単離および/または精製には、カラムクロマトグラフィーを用いることもでき、カラムクロマトグラフィーを用いないこともできる。 In some embodiments, the cyclic peptide compound, or a salt thereof, or a solvate thereof produced by the method of the present invention may or may not be isolated and/or purified using column chromatography.
本発明の方法により製造される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物は、晶析により結晶化することにより、単離および/または精製することができる。具体的には、例えば、縮合反応後の反応溶液を分液操作に供し、必要に応じて有機層を濃縮、および/またはろ過した後、得られた残渣に晶析に適した溶媒を加え、任意で種晶を加えて、必要に応じて攪拌することで、環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の結晶を得ることができる。晶析の際に添加される溶媒は、環状ペプチド化合物が結晶を形成することができる溶媒であれば特に制限はないが、環状ペプチド化合物が溶解した溶液に対し、環状ペプチド化合物の溶解度を低下させる操作を行うことのできる溶媒が好ましい。例えば貧溶媒の添加や溶液の冷却により、環状ペプチド化合物の溶解度を低下させて結晶化が可能な場合は、そのような操作が可能な溶媒が例示される。また、環状ペプチド化合物の粗結晶を懸濁液状態下、任意の時間懸濁状態を保つことで環状ペプチド化合物の結晶を得ることができる場合は、そのような操作が可能な溶媒を、結晶化に用いることができる。晶析の際に添加される溶媒として、具体的には、例えば、アセトン、水、DMSO、アセトニトリル、またはエタノール、およびこれらの混合溶媒などが挙げられる。 The cyclic peptide compound, or its salt, or its solvate produced by the method of the present invention can be isolated and/or purified by crystallization by crystallization. Specifically, for example, the reaction solution after the condensation reaction is subjected to a liquid separation operation, and the organic layer is concentrated and/or filtered as necessary, and then a solvent suitable for crystallization is added to the obtained residue, and seed crystals are optionally added and stirred as necessary to obtain crystals of the cyclic peptide compound, or its salt, or its solvate. The solvent added during crystallization is not particularly limited as long as it is a solvent in which the cyclic peptide compound can form crystals, but a solvent in which the solubility of the cyclic peptide compound can be reduced in the solution in which the cyclic peptide compound is dissolved is preferable. For example, when the solubility of the cyclic peptide compound can be reduced by adding a poor solvent or cooling the solution, examples of solvents that allow such operations are exemplified. In addition, when the crystals of the cyclic peptide compound can be obtained by maintaining the crude crystals of the cyclic peptide compound in a suspension state for any period of time, a solvent that allows such operations can be used for crystallization. Specific examples of solvents that can be added during crystallization include acetone, water, DMSO, acetonitrile, ethanol, and mixtures of these solvents.
式(1)で表される環状ペプチド化合物の構造式、および式(2)~(7)で表されるペプチド化合物の構造式において使用される各記号について説明する。 We will explain each symbol used in the structural formula of the cyclic peptide compound represented by formula (1) and the structural formulas of the peptide compounds represented by formulas (2) to (7).
R1は、C1-C6アルキルである。R1は好ましくはC3-C4アルキルであり、より好ましくはn-プロピル、2-メチルプロピルである。 R 1 is C 1 -C 6 alkyl. R 1 is preferably C 3 -C 4 alkyl, more preferably n-propyl, 2-methylpropyl.
P1は、C1-C6アルキルである。P1は好ましくはC1-C4アルキルであり、より好ましくはメチルである。 P1 is C1 - C6 alkyl. P1 is preferably C1 - C4 alkyl, more preferably methyl.
R2は、C1-C6アルキルである。R2は好ましくはC3-C4アルキルであり、より好ましくは1-メチルプロピルである。 R2 is C1 - C6 alkyl. R2 is preferably C3 - C4 alkyl, more preferably 1-methylpropyl.
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成している。好ましくは、R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している。ある態様において、R3は、好ましくは水素である。ある態様において、R3は、好ましくはP3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している。 R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached forms a 4- to 7-membered saturated heterocyclic ring. Preferably, R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached forms a 5-membered saturated heterocyclic ring. In certain embodiments, R 3 is preferably hydrogen. In certain embodiments, R 3 is preferably together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached forms a 5- membered saturated heterocyclic ring.
P3は、C1-C6アルキル、またはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成している。好ましくは、P3は、C1-C4アルキルであるか、またはP3、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している。ある態様において、P3は、好ましくはメチルである。ある態様において、P3は、好ましくはR3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成している。 P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached forms a 4-7 membered saturated heterocyclic ring. Preferably, P3 is C1 - C4 alkyl, or P3 together with R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached forms a 5 membered saturated heterocyclic ring. In certain embodiments, P3 is preferably methyl. In certain embodiments, P3 is preferably together with R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached forms a 5 membered saturated heterocyclic ring.
P4は、C1-C6アルキルである。P4は、好ましくはC1-C4アルキルであり、より好ましくはメチルである。 P4 is C 1 -C 6 alkyl. P4 is preferably C 1 -C 4 alkyl, more preferably methyl.
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルである。R5は、好ましくはC1-C4ハロアルキルによって置換されていてもよいベンジルであり、より好ましくは4-トリフルオロメチルベンジルである。 R 5 is benzyl optionally substituted by one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl. R 5 is preferably benzyl optionally substituted by C 1 -C 4 haloalkyl, more preferably 4-trifluoromethylbenzyl.
P6は、C1-C6アルキルである。P6は、好ましくはC1-C4アルキルであり、より好ましくはメチルである。 P6 is C 1 -C 6 alkyl. P6 is preferably C 1 -C 4 alkyl, more preferably methyl.
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルである。R7は、好ましくはハロゲン、トリフルオロメチル、およびメトキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり、より好ましくは3-メトキシ-4-トリフルオロメチルフェネチル、または3,5-ジフルオロ-4-トリフルオロメチルフェネチルである。 R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy. R 7 is preferably phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy, and is more preferably 3-methoxy-4-trifluoromethylphenethyl or 3,5-difluoro-4-trifluoromethylphenethyl.
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1-C6アルコキシによって置換されていてもよい。R8は、好ましくはP8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はC1-C4アルキルによって置換されており、より好ましくはP8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はエトキシによって置換されている。 R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocyclic ring, which may be substituted by C 1 -C 6 alkoxy. R 8 preferably together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocyclic ring, which is substituted by C 1 -C 4 alkyl, and more preferably together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocyclic ring, which is substituted by ethoxy.
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよい。R9は、好ましくはQ9、並びにR9およびQ9が結合している炭素原子と一緒になって4~6員脂環式環を形成している。ある態様において、R9は、好ましくはQ9、並びにR9およびQ9が結合している炭素原子と一緒になって4員脂環式環を形成している。ある態様において、R9は、好ましくはQ9、並びにR9およびQ9が結合している炭素原子と一緒になって5員脂環式環を形成している。 R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 3-8 membered alicyclic ring, which may be substituted by one or more C 1 -C 6 alkyls. R 9 preferably together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 4-6 membered alicyclic ring. In some embodiments, R 9 preferably together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 4 membered alicyclic ring. In some embodiments, R 9 preferably together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 5 membered alicyclic ring.
P9は、水素またはC1-C6アルキルである。P9は、好ましくは水素またはC1-C4アルキルである。ある態様において、P9は、好ましくは水素である。ある態様において、P9は、好ましくはメチルである。 P 9 is hydrogen or C 1 -C 6 alkyl. P 9 is preferably hydrogen or C 1 -C 4 alkyl. In some embodiments, P 9 is preferably hydrogen. In some embodiments, P 9 is preferably methyl.
R10は、C1-C6アルキル、またはC3-C8シクロアルキルである。R10は、好ましくはC4-C6シクロアルキルであり、より好ましくはシクロペンチルである。 R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl. R 10 is preferably C 4 -C 6 cycloalkyl, more preferably cyclopentyl.
P10は、C1-C6アルキルである。P10は、好ましくはC1-C4アルキルであり、より好ましくはメチルである。 P 10 is C 1 -C 6 alkyl. P 10 is preferably C 1 -C 4 alkyl, more preferably methyl.
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルである。R11は、好ましくはジC1-C4アルキルアミノカルボニル、または5~6員環状アミノカルボニルであり、より好ましくはジメチルアミノカルボニルである。 R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4- to 8-membered cyclic aminocarbonyl. R 11 is preferably diC 1 -C 4 alkylaminocarbonyl, or 5- to 6-membered cyclic aminocarbonyl, more preferably dimethylaminocarbonyl.
P11は、C1-C6アルキルである。P11は、好ましくはC1-C4アルキルであり、より好ましくはメチルである。 P 11 is C 1 -C 6 alkyl. P 11 is preferably C 1 -C 4 alkyl, more preferably methyl.
X1、X3、およびX5は、それぞれ独立して水素、またはアミノ基の保護基である。X1、X3、およびX5は、好ましくはそれぞれ独立して水素、カルバメート系保護基、アシル系保護基、スルホンアミド系保護基、およびシリル系保護基からなる群より選択される1つである。ある態様において、カルバメート系保護基は、Fmoc基、Cbz基、Troc基、Alloc基、Teoc基、TSoc基、BIBSoc基、IPCSoc基、BBSoc基、CHBSoc基、CDBSoc基、およびBoc基からなる群より選択される1つである。ある態様において、アシル系保護基は、トリフルオロアセチル基、アセチル基、およびベンゾイル基からなる群より選択される1つである。ある態様において、スルホンアミド系保護基は、2-ニトロベンゼンスルホニル基、4-ニトロベンゼンスルホニル基、および2,4-ジニトロベンゼンスルホニル基からなる群より選択される1つである。ある態様において、シリル系保護基は、TMS基、TBDMS基、TES基、TIPS基、およびTBDPS基からなる群より選択される1つである。 X1 , X3 , and X5 are each independently hydrogen or a protecting group of an amino group. X1 , X3 , and X5 are preferably each independently one selected from the group consisting of hydrogen, a carbamate-based protecting group, an acyl-based protecting group, a sulfonamide-based protecting group, and a silyl-based protecting group. In one embodiment, the carbamate-based protecting group is one selected from the group consisting of an Fmoc group, a Cbz group, a Troc group, an Alloc group, a Teoc group, a TSoc group, a BIBSoc group, an IPCSoc group, a BBSoc group, a CHBSoc group, a CDBSoc group, and a Boc group. In one embodiment, the acyl-based protecting group is one selected from the group consisting of a trifluoroacetyl group, an acetyl group, and a benzoyl group. In one embodiment, the sulfonamide protecting group is one selected from the group consisting of 2-nitrobenzenesulfonyl, 4-nitrobenzenesulfonyl, and 2,4-dinitrobenzenesulfonyl groups. In one embodiment, the silyl protecting group is one selected from the group consisting of TMS, TBDMS, TES, TIPS, and TBDPS groups.
ある態様において、X1は、水素、またはカルバメート系保護基である。ある態様において、X1は、好ましくは水素である。ある態様において、X1は、好ましくはFmoc基である。 In some embodiments, X 1 is hydrogen or a carbamate-based protecting group. In some embodiments, X 1 is preferably hydrogen. In some embodiments, X 1 is preferably an Fmoc group.
ある態様において、X3は、水素、またはカルバメート系保護基である。ある態様において、X3は、好ましくは水素である。ある態様において、X3は、好ましくはCbz基である。 In some embodiments, X3 is hydrogen or a carbamate-based protecting group. In some embodiments, X3 is preferably hydrogen. In some embodiments, X3 is preferably a Cbz group.
ある態様において、X5は、水素、またはカルバメート系保護基である。ある態様において、X5は、好ましくは水素である。ある態様において、X5は、好ましくはCbz基である。 In some embodiments, X5 is hydrogen or a carbamate protecting group. In some embodiments, X5 is preferably hydrogen. In some embodiments, X5 is preferably a Cbz group.
X2、X4、およびX6は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。X2、X4、およびX6は、好ましくはそれぞれ独立してハロゲン、水酸基、置換されていてもよいC1-C6アルコキシ、置換されていてもよいC6-C10アリールオキシ、置換されていてもよいC7-C14アラルコキシ、置換されていてもよい4~8員環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、C1-C6アルキル、またはC6-C10アリールである)で表される基である。ある態様において、ハロゲンは塩素、または臭素である。ある態様において、置換されていてもよいアルコキシは、t-ブトキシ、メトキシ、エトキシ、またはイソプロポキシである。ある態様において、置換されていてもよいアリールオキシは、ペンタフルオロフェニルオキシ、またはニトロフェニルオキシである。ある態様において、置換されていてもよいアラルコキシは、置換されていてもよいベンジルオキシである。ある態様において、置換されていてもよい環状アミノオキシは、N-ヒドロキシコハク酸イミノオキシである。ある態様において、-OSiRxRyRzで表される基は、トリメチルシリルオキシ、トリエチルシリルオキシ、トリイソプロピルシリルオキシ、トリフェニルシリルオキシ、トリ-t-ブチルシリルオキシ、ジ-t-ブチルイソブチルシリルオキシ、またはトリス(トリエチルシリル)シリルオキシである。 X 2 , X 4 , and X 6 are each independently a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently an alkyl or an aryl). X 2 , X 4 , and X 6 are preferably each independently a halogen, a hydroxyl group, an optionally substituted C 1 -C 6 alkoxy, an optionally substituted C 6 -C 10 aryloxy, an optionally substituted C 7 -C 14 aralkoxy, an optionally substituted 4- to 8-membered cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently a C 1 -C 6 alkyl, or a C 6 -C 10 aryl). In one embodiment, the halogen is chlorine or bromine. In one embodiment, the optionally substituted alkoxy is t-butoxy, methoxy, ethoxy, or isopropoxy. In one embodiment, the optionally substituted aryloxy is pentafluorophenyloxy, or nitrophenyloxy. In one embodiment, the optionally substituted aralkoxy is an optionally substituted benzyloxy. In one embodiment, the optionally substituted cyclic aminooxy is N- hydroxysuccinimoxy . In one embodiment, the group represented by -OSiRxRyRz is trimethylsilyloxy, triethylsilyloxy, triisopropylsilyloxy, triphenylsilyloxy, tri -t-butylsilyloxy, di-t-butylisobutylsilyloxy, or tris(triethylsilyl)silyloxy.
ある態様において、X2は、水酸基、t-ブトキシ、またはベンジルオキシである。ある態様において、X2は、好ましくは水酸基である。ある態様において、X2は、好ましくはt-ブトキシである。 In some embodiments, X2 is hydroxyl, t-butoxy, or benzyloxy. In some embodiments, X2 is preferably hydroxyl. In some embodiments, X2 is preferably t-butoxy.
ある態様において、X4は、水酸基、t-ブトキシ、またはベンジルオキシである。ある態様において、X4は、好ましくは水酸基である。ある態様において、X4は、好ましくはt-ブトキシである。 In some embodiments, X 4 is hydroxyl, t-butoxy, or benzyloxy. In some embodiments, X 4 is preferably hydroxyl. In some embodiments, X 4 is preferably t-butoxy.
ある態様において、X6は、水酸基、t-ブトキシ、またはベンジルオキシである。ある態様において、X6は、好ましくは水酸基である。ある態様において、X6は、好ましくはt-ブトキシである。 In some embodiments, X 6 is hydroxyl, t-butoxy, or benzyloxy. In some embodiments, X 6 is preferably hydroxyl. In some embodiments, X 6 is preferably t-butoxy.
式(1)で表される環状ペプチド化合物の構造式、および式(2)、式(3)で表されるペプチド化合物の構造式のある態様において、
R1は、C1-C6アルキルであり;
P1は、C1-C6アルキルであり;
R2は、C1-C6アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P3は、C1-C6アルキル、もしくはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P4は、C1-C6アルキルであり;
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルであり;
P6は、C1-C6アルキルであり;
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1-C6アルコキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよく;
P9は、水素またはC1-C6アルキルであり;
R10は、C1-C6アルキル、またはC3-C8シクロアルキルであり;
P10は、C1-C6アルキルであり;
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルであり;
P11は、C1-C6アルキルであり;
X1およびX5は、それぞれ独立して水素、またはアミノ基の保護基であり;
X2およびX4は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。
In one embodiment of the structural formula of the cyclic peptide compound represented by formula (1) and the structural formula of the peptide compound represented by formula (2) and formula (3),
R 1 is C 1 -C 6 alkyl;
P1 is C1 - C6 alkyl;
R2 is C1 - C6 alkyl;
R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring;
P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle ;
P4 is C1 - C6 alkyl;
R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl;
P6 is C1 - C6 alkyl;
R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocycle, which is optionally substituted by C 1 -C 6 alkoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 3- to 8-membered alicyclic ring, which is optionally substituted by one or more C 1 -C 6 alkyl;
P9 is hydrogen or C1 - C6 alkyl;
R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl;
P10 is C1 - C6 alkyl;
R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4-8 membered cyclic aminocarbonyl;
P11 is C1 - C6 alkyl;
X1 and X5 are each independently hydrogen or an amino-protecting group;
X2 and X4 each independently represent a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz each independently represent an alkyl or an aryl ) .
式(1)で表される環状ペプチド化合物の構造式、および式(2)、式(3)で表されるペプチド化合物の構造式のある態様において、好ましくは
R1は、C3-C4アルキルであり;
P1は、C1-C4アルキルであり;
R2は、C3-C4アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P3は、C1-C4アルキルであるであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P4は、C1-C4アルキルであり;
R5は、C1-C4ハロアルキルによって置換されていてもよいベンジルであり;
P6は、C1-C4アルキルであり;
R7は、ハロゲン、トリフルオロメチル、およびメトキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はC1-C4アルキルによって置換されており;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって4~6員脂環式環を形成しており;
P9は、水素またはC1-C4アルキルであり;
R10は、C4-C6シクロアルキルであり;
P10は、C1-C4アルキルであり;
R11は、ジC1-C4アルキルアミノカルボニル、または5~6員環状アミノカルボニル;
P11は、C1-C4アルキルであり;
X1およびX5は、それぞれ独立して水素、カルバメート系保護基、アシル系保護基、スルホンアミド系保護基、およびシリル系保護基からなる群より選択される1つであり;
X2およびX4は、それぞれ独立してハロゲン、水酸基、置換されていてもよいC1-C6アルコキシ、置換されていてもよいC6-C10アリールオキシ、置換されていてもよいC7-C14アラルコキシ、置換されていてもよい4~8員環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、C1-C6アルキル、またはC6-C10アリールである)で表される基である。
In an embodiment of the structural formula of the cyclic peptide compound represented by formula (1) and the structural formula of the peptide compound represented by formula (2) and formula (3), preferably, R 1 is C 3 -C 4 alkyl;
P1 is C1 - C4 alkyl;
R2 is C3 - C4 alkyl;
R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring;
P3 is C 1 -C 4 alkyl or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocycle;
P4 is C1 - C4 alkyl;
R 5 is benzyl optionally substituted by C 1 -C 4 haloalkyl;
P6 is C1 - C4 alkyl;
R7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocycle, which is substituted with C 1 -C 4 alkyl;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 4- to 6-membered alicyclic ring;
P9 is hydrogen or C1 - C4 alkyl;
R 10 is C4 - C6 cycloalkyl;
P10 is C1 - C4 alkyl;
R 11 is diC 1 -C 4 alkylaminocarbonyl, or 5- to 6-membered cyclic aminocarbonyl;
P11 is C1 - C4 alkyl;
X1 and X5 are each independently one selected from the group consisting of hydrogen, a carbamate-based protecting group, an acyl-based protecting group, a sulfonamide-based protecting group, and a silyl-based protecting group;
X2 and X4 are each independently a halogen, a hydroxyl group, an optionally substituted C1 - C6 alkoxy, an optionally substituted C6 - C10 aryloxy, an optionally substituted C7 - C14 aralkoxy, an optionally substituted 4- to 8-membered cyclic aminooxy , or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz are each independently C1 - C6 alkyl, or C6 - C10 aryl ) .
式(1)で表される環状ペプチド化合物の構造式、および式(2)、式(3)で表されるペプチド化合物の構造式のある態様において、より好ましくは、
R1は、n-プロピル、2-メチルプロピルであり;
P1は、メチルであり;
R2は、1-メチルプロピルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P3は、メチルであるか、またはP3、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P4は、メチルであり;
R5は、4-トリフルオロメチルベンジルであり;
P6は、メチルであり;
R7は、3-メトキシ-4-トリフルオロメチルフェネチル、または3,5-ジフルオロ-4-トリフルオロメチルフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はエトキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって4員脂環式環または5員脂環式環を形成しており;
P9は、水素またはメチルであり;
R10は、シクロペンチルであり;
P10は、メチルであり;
R11は、ジメチルアミノカルボニルであり;
P11は、メチルであり;
X1は、水素、またはカルバメート系保護基であり;
X5は、水素、またはカルバメート系保護基であり;
X2は、水酸基、t-ブトキシ、またはベンジルオキシであり;
X4は、水酸基、t-ブトキシ、またはベンジルオキシである。
In an embodiment of the structural formula of the cyclic peptide compound represented by formula (1) and the structural formula of the peptide compound represented by formula (2) and formula (3), more preferably,
R 1 is n-propyl, 2-methylpropyl;
P1 is methyl;
R2 is 1-methylpropyl;
R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring;
P3 is methyl or forms together with P3 , R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached a 5-membered saturated heterocycle;
P4 is methyl;
R5 is 4-trifluoromethylbenzyl;
P6 is methyl;
R 7 is 3-methoxy-4-trifluoromethylphenethyl, or 3,5-difluoro-4-trifluoromethylphenethyl;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocycle, which may be substituted by ethoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 4- or 5-membered alicyclic ring;
P9 is hydrogen or methyl;
R 10 is cyclopentyl;
P10 is methyl;
R 11 is dimethylaminocarbonyl;
P11 is methyl;
X1 is hydrogen or a carbamate protecting group;
X5 is hydrogen or a carbamate protecting group;
X2 is hydroxyl, t-butoxy, or benzyloxy;
X4 is a hydroxyl group, t-butoxy, or benzyloxy.
式(4)~(6)で表されるペプチド化合物の構造式のある態様において、
R1は、C1-C6アルキルであり;
P1は、C1-C6アルキルであり;
R2は、C1-C6アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P3は、C1-C6アルキル、もしくはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P4は、C1-C6アルキルであり;
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルであり;
P6は、C1-C6アルキルであり;
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1-C6アルコキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよく;
P9は、水素またはC1-C6アルキルであり;
R10は、C1-C6アルキル、またはC3-C8シクロアルキルであり;
P10は、C1-C6アルキルであり;
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルであり;
P11は、C1-C6アルキルであり;
X1、X3およびX5は、それぞれ独立して水素、またはアミノ基の保護基であり;
X2、X4およびX6は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。
In one embodiment of the structural formula of the peptide compound represented by formulas (4) to (6),
R 1 is C 1 -C 6 alkyl;
P1 is C1 - C6 alkyl;
R2 is C1 - C6 alkyl;
R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring;
P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle ;
P4 is C1 - C6 alkyl;
R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl;
P6 is C1 - C6 alkyl;
R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocycle, which is optionally substituted by C 1 -C 6 alkoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 3- to 8-membered alicyclic ring, which is optionally substituted by one or more C 1 -C 6 alkyl;
P9 is hydrogen or C1 - C6 alkyl;
R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl;
P10 is C1 - C6 alkyl;
R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4-8 membered cyclic aminocarbonyl;
P11 is C1 - C6 alkyl;
X 1 , X 3 and X 5 are each independently hydrogen or a protecting group for an amino group;
X 2 , X 4 and X 6 are each independently a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y and R z are each independently an alkyl or an aryl).
式(4)~(6)で表されるペプチド化合物の構造式のある態様において、好ましくは、
R1は、C3-C4アルキルであり;
P1は、C1-C4アルキルであり;
R2は、C3-C4アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P3は、C1-C4アルキルであるであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P4は、C1-C4アルキルであり;
R5は、C1-C4ハロアルキルによって置換されていてもよいベンジルであり;
P6は、C1-C4アルキルであり;
R7は、ハロゲン、トリフルオロメチル、およびメトキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はC1-C4アルキルによって置換されており;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって4~6員脂環式環を形成しており;
P9は、水素またはC1-C4アルキルであり;
R10は、C4-C6シクロアルキルであり;
P10は、C1-C4アルキルであり;
R11は、ジC1-C4アルキルアミノカルボニル、または5~6員環状アミノカルボニル;
P11は、C1-C4アルキルであり;
X1、X3およびX5は、それぞれ独立して水素、カルバメート系保護基、アシル系保護基、スルホンアミド系保護基、およびシリル系保護基からなる群より選択される1つであり;
X2、X4およびX6は、それぞれ独立してハロゲン、水酸基、置換されていてもよいC1-C6アルコキシ、置換されていてもよいC6-C10アリールオキシ、置換されていてもよいC7-C14アラルコキシ、置換されていてもよい4~8員環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、C1-C6アルキル、またはC6-C10アリールである)で表される基である。
In one embodiment of the structural formula of the peptide compound represented by formulas (4) to (6), preferably,
R 1 is C 3 -C 4 alkyl;
P1 is C1 - C4 alkyl;
R2 is C3 - C4 alkyl;
R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring;
P3 is C 1 -C 4 alkyl or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocycle;
P4 is C1 - C4 alkyl;
R 5 is benzyl optionally substituted by C 1 -C 4 haloalkyl;
P6 is C1 - C4 alkyl;
R7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, trifluoromethyl, and methoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocycle, which is substituted with C 1 -C 4 alkyl;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 4- to 6-membered alicyclic ring;
P9 is hydrogen or C1 - C4 alkyl;
R 10 is C4 - C6 cycloalkyl;
P10 is C1 - C4 alkyl;
R 11 is diC 1 -C 4 alkylaminocarbonyl, or 5- to 6-membered cyclic aminocarbonyl;
P11 is C1 - C4 alkyl;
X 1 , X 3 and X 5 are each independently one selected from the group consisting of hydrogen, a carbamate-based protecting group, an acyl-based protecting group, a sulfonamide-based protecting group, and a silyl-based protecting group;
X 2 , X 4 and X 6 are each independently a halogen, a hydroxyl group, an optionally substituted C 1 -C 6 alkoxy, an optionally substituted C 6 -C 10 aryloxy, an optionally substituted C 7 -C 14 aralkoxy, an optionally substituted 4- to 8-membered cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y and R z are each independently a C 1 -C 6 alkyl or a C 6 -C 10 aryl).
式(4)~(6)で表されるペプチド化合物の構造式のある態様において、より好ましくは、
R1は、n-プロピル、または2-メチルプロピルであり;
P1は、メチルであり;
R2は、1-メチルプロピルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P3は、メチルであるか、またはP3、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって5員飽和複素環を形成し;
P4は、メチルであり;
R5は、4-トリフルオロメチルベンジルであり;
P6は、メチルであり;
R7は、3-メトキシ-4-トリフルオロメチルフェネチル、または3,5-ジフルオロ-4-トリフルオロメチルフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって5員飽和複素環を形成し、該5員飽和複素環はエトキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって4員脂環式環または5員脂環式環を形成しており;
P9は、水素またはメチルであり;
R10は、シクロペンチルであり;
P10は、メチルであり;
R11は、ジメチルアミノカルボニルであり;
P11は、メチルであり;
X1は、水素、またはカルバメート系保護基であり;
X3は、水素、またはカルバメート系保護基であり;
X5は、水素、またはカルバメート系保護基であり;
X2は、水酸基、t-ブトキシ、またはベンジルオキシであり;
X4は、水酸基、t-ブトキシ、またはベンジルオキシであり;
X6は、水酸基、t-ブトキシ、またはベンジルオキシである。
In one embodiment of the structural formula of the peptide compound represented by formulas (4) to (6), more preferably
R 1 is n-propyl or 2-methylpropyl;
P1 is methyl;
R2 is 1-methylpropyl;
R3 is hydrogen or R3 together with P3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 5-membered saturated heterocyclic ring;
P3 is methyl or forms together with P3 , R3 , the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached a 5-membered saturated heterocycle;
P4 is methyl;
R5 is 4-trifluoromethylbenzyl;
P6 is methyl;
R 7 is 3-methoxy-4-trifluoromethylphenethyl, or 3,5-difluoro-4-trifluoromethylphenethyl;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 5-membered saturated heterocycle, which may be substituted by ethoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached forms a 4- or 5-membered alicyclic ring;
P9 is hydrogen or methyl;
R 10 is cyclopentyl;
P10 is methyl;
R 11 is dimethylaminocarbonyl;
P11 is methyl;
X1 is hydrogen or a carbamate protecting group;
X3 is hydrogen or a carbamate protecting group;
X5 is hydrogen or a carbamate protecting group;
X2 is hydroxyl, t-butoxy, or benzyloxy;
X4 is hydroxyl, t-butoxy, or benzyloxy;
X6 is a hydroxyl group, t-butoxy, or benzyloxy.
ある態様において、本発明の方法により製造される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物は、溶媒和物であることが好ましく、水和物であることがより好ましい。 In one embodiment, the cyclic peptide compound, or a salt thereof, or a solvate thereof produced by the method of the present invention is preferably a solvate, more preferably a hydrate.
ある態様において、本発明の方法により製造される環状ペプチド化合物は、下記式(1a):
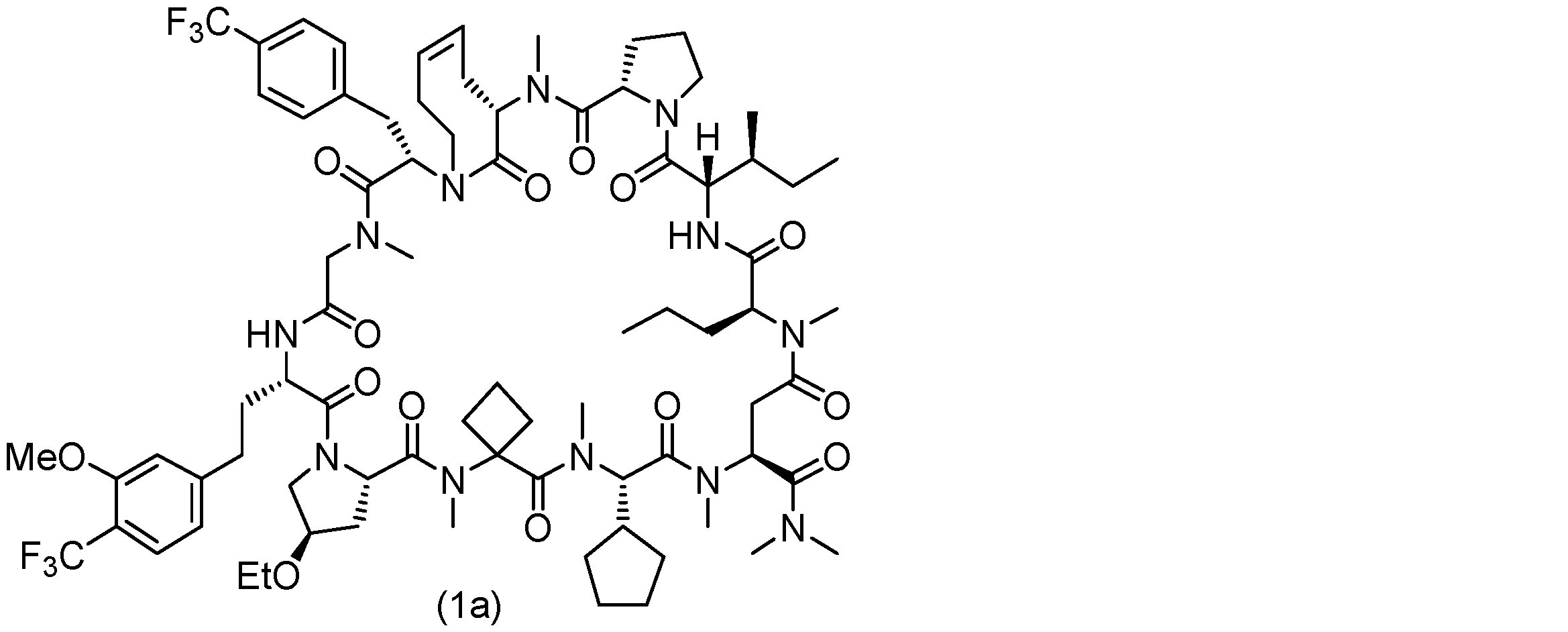
ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法に関する。該方法は、溶媒中、式(2a)または(3a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを反応させて環化する工程(環化工程)を含む(以下、「態様1’」ともいう)。

ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法に関する。該方法は、
(a) 式(4a)~(6a)で表されるペプチド化合物、もしくはその塩または前記ペプチド化合物もしくは塩の溶媒和物を用意する工程、
(b) 式(4a)~(6a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを反応させて連結する工程(連結工程)、および
(c) (b)工程で得られたペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを反応させて環化する工程(環化工程)を含む(以下、「態様2’」ともいう)。

(a) providing a peptide compound represented by any one of formulas (4a) to (6a), or a salt thereof, or a solvate of said peptide compound or salt;
(b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4a) to (6a) (linking step); and
(c) A step of cyclizing the peptide compound obtained in step (b) by reacting the N-terminal amino acid residue with the C-terminal amino acid residue (cyclization step) (hereinafter, also referred to as "Mode 2'").
態様2’において、(b)の工程は、(b-1)式(5a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(6a)で表されるペプチド化合物のC末端のアミノ酸残基とを、連結することにより式(7a)で表されるペプチド化合物に変換する工程を含んでもよい。

態様2’において、(b)の工程は、(b-1)の工程に加えて、
(b-2)式(4a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(7a)で表されるペプチド化合物のC末端のアミノ酸残基とを連結することにより式(2a)で表されるペプチド化合物に変換する工程(連結工程)を含んでもよく、かつ(c)の工程は、
(c-1)式(2a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含んでもよい。
In the embodiment 2', the step (b) includes, in addition to the step (b-1),
(b-2) a step (linking step) of converting a peptide compound represented by formula (4a) to a peptide compound represented by formula (7a) by linking the N-terminal amino acid residue with the C-terminal amino acid residue of the peptide compound represented by formula (7a) to obtain a peptide compound represented by formula (2a), and the step (c) may include
(c-1) The method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2a) by reaction in a solvent (cyclization step).
態様2’において、(b)の工程は、(b-1)の工程に加えて、
(b-3)式(7a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(4a)で表されるペプチド化合物のC末端のアミノ酸残基とを連結することにより式(3a)で表されるペプチド化合物に変換する工程(連結工程)を含んでもよく、かつ(c)の工程は、
(c-2)式(3a)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含んでもよい。
In the embodiment 2', the step (b) includes, in addition to the step (b-1),
(b-3) a step (linking step) of converting a peptide compound represented by formula (7a) to a peptide compound represented by formula (4a) by linking the N-terminal amino acid residue with the C-terminal amino acid residue of the peptide compound represented by formula (4a) to obtain a peptide compound represented by formula (3a), and the step (c) may include
(c-2) The method may include a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3a) by reaction in a solvent (cyclization step).
態様1’および2’において、好ましくはペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基との連結が、N末端のアミノ酸残基のアミノ基とC末端のアミノ酸残基のカルボキシル基との連結であり、より好ましくはペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基との連結が、N末端のアミノ酸残基のアミノ基とC末端のアミノ酸残基のカルボキシル基とのアミド結合による連結である。 In embodiments 1' and 2', the link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is preferably a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue, and more preferably a link between the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound is a link between the amino group of the N-terminal amino acid residue and the carboxyl group of the C-terminal amino acid residue via an amide bond.
態様1’および2’における、環化工程の溶媒、環化工程の縮合試薬、環化工程の塩基、環化工程において生成する副生成物、連結工程の溶媒、連結工程の縮合試薬、連結工程の塩基等は、上記態様1及び2において説明したものと同一である。 In embodiments 1' and 2', the solvent in the cyclization step, the condensation reagent in the cyclization step, the base in the cyclization step, the by-products generated in the cyclization step, the solvent in the linking step, the condensation reagent in the linking step, the base in the linking step, etc. are the same as those described in embodiments 1 and 2 above.
態様1’および2’において、式(1a)で表される環状ペプチド化合物の構造式、および式(2a)~(7a)で表されるペプチド化合物の構造式において使用される各記号は、上記態様1及び2において説明したものと同一である。 In embodiments 1' and 2', the symbols used in the structural formula of the cyclic peptide compound represented by formula (1a) and the structural formula of the peptide compounds represented by formulas (2a) to (7a) are the same as those described in embodiments 1 and 2 above.
ペプチド化合物
ある態様において、本発明は、式(4a)で表される化合物に関する。

式(4a)において、X1は、水素またはアミノ基の保護基である。ここで、X1における保護基は上記態様1及び2において説明したものと同一である。ある態様におい、X1は、好ましくは水素である。ある態様において、X1は、好ましくはFmoc基である。 In formula (4a), X 1 is hydrogen or a protecting group of an amino group. Here, the protecting group in X 1 is the same as that described in the above embodiments 1 and 2. In one embodiment, X 1 is preferably hydrogen. In one embodiment, X 1 is preferably an Fmoc group.
式(4a)において、X2は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。ここで、X2における置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、および-OSiRxRyRzは上記態様1及び2において説明したものと同一である。ある態様において、X2は、好ましくはt-ブトキシである。 In formula (4a), X 2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl). Here, the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 2 are the same as those explained in the above embodiments 1 and 2. In one embodiment, X 2 is preferably t-butoxy.
ある態様において、式(4a)で表される化合物は、好ましくはtert-ブチル2-[メチル-[(2S)-2-[(4Z,7S)-7-(メチルアミノ)-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]アミノ]酢酸(化合物9)である。 In one embodiment, the compound represented by formula (4a) is preferably tert-butyl 2-[methyl-[(2S)-2-[(4Z,7S)-7-(methylamino)-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]amino]acetic acid (compound 9).
ある態様において、式(4a)で表される化合物(ペプチド化合物(4a))は、以下の方法を用いて製造することができる。

ペプチド化合物(4a)は、以下の方法を用いて製造することもできる。

ある態様において、本発明は、式(5a)で表される化合物に関する。

式(5a)において、X3は、水またはアミノ基の保護基である。ここで、X3におけるアミノ基の保護基は上記態様1及び2において説明したものと同一である。ある態様において、X3は、好ましくは水素である。ある態様において、X3は、好ましくはFmoc基またはCbz基である。 In formula (5a), X3 is water or an amino-protecting group. Here, the amino-protecting group in X3 is the same as that described in the above embodiments 1 and 2. In one embodiment, X3 is preferably hydrogen. In one embodiment, X3 is preferably an Fmoc group or a Cbz group.
式(5a)において、X4は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。ここで、X2における置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、および-OSiRxRyRzは上記態様1及び2において説明したものと同一である。ある態様において、X4は、好ましくはt-ブトキシである。 In formula (5a), X 4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl). Here, the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 2 are the same as those explained in the above embodiments 1 and 2. In one embodiment, X 4 is preferably t-butoxy.
ある態様において、式(5a)で表される化合物は、好ましくはtert-ブチル(2S)-1-[(2S,3S)-3-メチル-2-[[(2S)-2-(メチルアミノ)ペンタノイル]アミノ]ペンタノイル]ピロリジン-2-酢酸(化合物13)である。 In one embodiment, the compound represented by formula (5a) is preferably tert-butyl(2S)-1-[(2S,3S)-3-methyl-2-[[(2S)-2-(methylamino)pentanoyl]amino]pentanoyl]pyrrolidine-2-acetic acid (compound 13).
ある態様において、式(5a)で表される化合物(ペプチド化合物(5a))は、以下の方法を用いて製造することができる。

ある態様において、本発明は、式(6a)で表される化合物に関する。
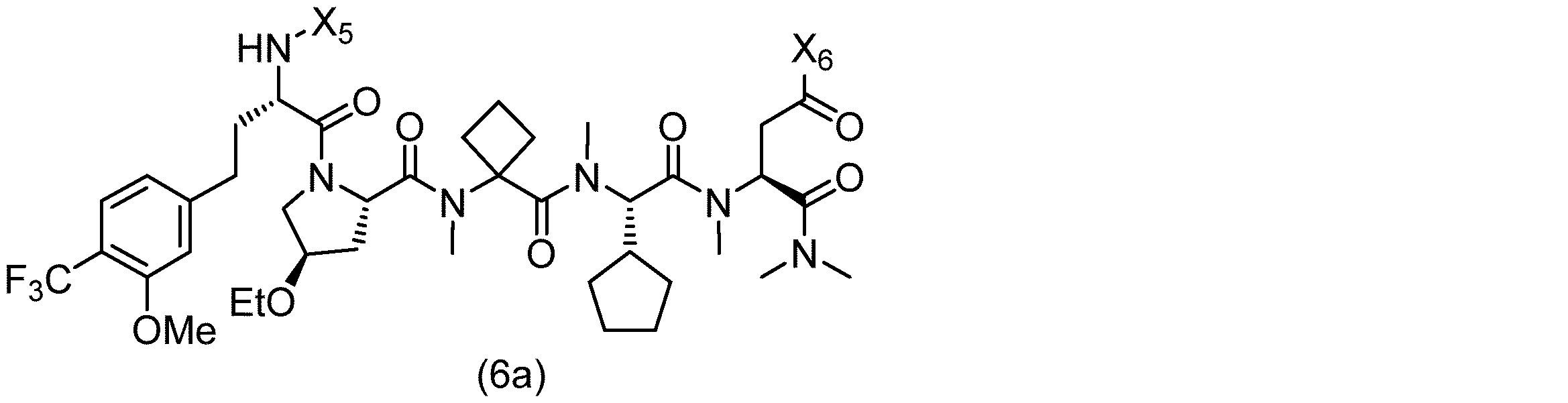
式(6a)において、X5は、水素またはアミノ基の保護基である。ここで、X5におけるアミノ基の保護基は上記態様1及び2において説明したものと同一である。ある態様において、X5は、好ましくは水素である。ある態様において、X5は、好ましくはCbz基である。 In formula (6a), X5 is hydrogen or a protecting group for an amino group. Here, the protecting group for an amino group in X5 is the same as that described in the above embodiments 1 and 2. In one embodiment, X5 is preferably hydrogen. In one embodiment, X5 is preferably a Cbz group.
式(6a)において、X6は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。ここで、X6における置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、および-OSiRxRyRzは上記態様1及び2において説明したものと同一である。ある態様において、X6は、好ましくはt-ブトキシである。 In formula (6a), X 6 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl). Here, the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 6 are the same as those explained in the above embodiments 1 and 2. In one embodiment, X 6 is preferably t-butoxy.
ある態様において、式(6a)で表される化合物は、好ましくは(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸(化合物20)である。 In one embodiment, the compound represented by formula (6a) is preferably (3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid (compound 20).
ある態様において、式(6a)で表される化合物(ペプチド化合物(6a))は、以下の方法を用いて製造することができる。

ある態様において、本発明は、式(7a)で表される化合物に関する。

式(7a)において、X5は、水素またはアミノ基の保護基である。ここで、X5におけるアミノ基の保護基は上記態様1及び2において説明したものと同一である。ある態様において、X5は、好ましくは水素である。ある態様において、X5は、好ましくはCbz基である。 In formula (7a), X5 is hydrogen or a protecting group for an amino group. Here, the protecting group for an amino group in X5 is the same as that described in the above embodiments 1 and 2. In one embodiment, X5 is preferably hydrogen. In one embodiment, X5 is preferably a Cbz group.
式(7a)において、X4は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。ここで、X4における置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、および-OSiRxRyRzは上記態様1及び2において説明したものと同一である。ある態様において、X4は、好ましくは水酸基またはt-ブトキシである。 In formula (7a), X 4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl). Here, the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 4 are the same as those explained in the above embodiments 1 and 2. In one embodiment, X 4 is preferably a hydroxyl group or t-butoxy.
ある態様において、式(7a)で表される化合物は、好ましくは(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチルアセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸(化合物22)である。また、ある態様において、式(7a)で表される化合物は、好ましくはtert-ブチル (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-アミノ-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボキシラート(化合物37)である。 In one embodiment, the compound represented by formula (7a) is preferably (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentylacetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylic acid (compound 22). Also, in one embodiment, the compound represented by formula (7a) is preferably tert-butyl (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate (compound 37).
ある態様において、式(7a)で表される化合物は、式(5a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(6a)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結すること(連結工程)により製造することができる。連結工程の溶媒、連結工程の縮合試薬、連結工程の塩基等は、上記態様1及び2において説明したものと同一である。 In one embodiment, the compound represented by formula (7a) can be produced by reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (5a) and the C-terminal amino acid residue of the peptide compound represented by formula (6a) in a solvent (linking step). The solvent in the linking step, the condensation reagent in the linking step, the base in the linking step, etc. are the same as those described in the above embodiments 1 and 2.
ある態様において、本発明は、式(2a)で表される化合物に関する。
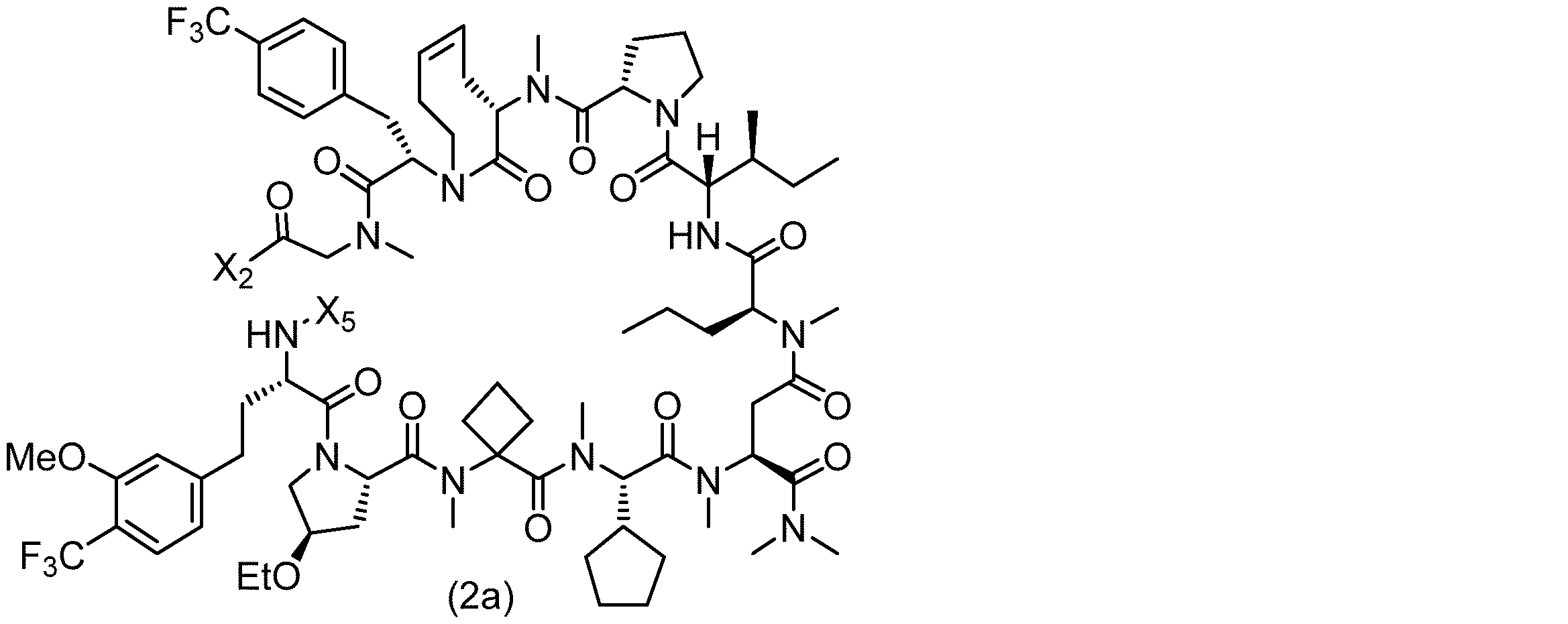
式(2a)において、X5は、水素またはアミノ基の保護基である。ここで、X5におけるアミノ基の保護基は上記態様1及び2において説明したものと同一である。ある態様において、X5は、好ましくは水素である。ある態様において、X5は、好ましくはCbz基である。 In formula (2a), X5 is hydrogen or a protecting group for an amino group. Here, the protecting group for an amino group in X5 is the same as that described in the above embodiments 1 and 2. In one embodiment, X5 is preferably hydrogen. In one embodiment, X5 is preferably a Cbz group.
式(2a)において、X2は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。ここで、X2における置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、および-OSiRxRyRzは上記態様1及び2において説明したものと同一である。ある態様において、X2は、好ましくは水酸基またはt-ブトキシである。 In formula (2a), X 2 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl). Here, the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 2 are the same as those explained in the above embodiments 1 and 2. In one embodiment, X 2 is preferably a hydroxyl group or t-butoxy.
ある態様において、式(2a)で表される化合物は、好ましくは2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-アミノ-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボニル]-メチル-アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸(化合物24)である。 In one embodiment, the compound represented by formula (2a) is preferably 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbo Nyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carbonyl]-methyl-amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetic acid (compound 24).
ある態様において、式(2a)で表される化合物は、式(4a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(7a)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結すること(連結工程)により製造することができる。連結工程の溶媒、連結工程の縮合試薬、連結工程の塩基等は、上記態様1及び2において説明したものと同一である。 In one embodiment, the compound represented by formula (2a) can be produced by reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4a) and the C-terminal amino acid residue of the peptide compound represented by formula (7a) in a solvent (linking step). The solvent for the linking step, the condensation reagent for the linking step, the base for the linking step, etc. are the same as those described in the above embodiments 1 and 2.
ある態様において、本発明は、式(3a)で表される化合物に関する。

式(3a)において、X1は、水素またはアミノ基の保護基である。ここで、X1におけるアミノ基の保護基は上記態様1及び2において説明したものと同一である。ある態様において、X1は、好ましくは水素である。ある態様において、X1は、好ましくはFmoc基である。 In formula (3a), X1 is hydrogen or an amino-protecting group. Here, the amino-protecting group in X1 is the same as that described in the above embodiments 1 and 2. In one embodiment, X1 is preferably hydrogen. In one embodiment, X1 is preferably an Fmoc group.
式(3a)において、X4は、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。ここで、X4における置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、および-OSiRxRyRzは上記態様1及び2において説明したものと同一である。ある態様において、X4は、好ましくは水酸基またはt-ブトキシである。 In formula (3a), X 4 is a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y , and R z are each independently alkyl or aryl). Here, the optionally substituted alkoxy, the optionally substituted aryloxy, the optionally substituted aralkoxy, the optionally substituted cyclic aminooxy, and -OSiR x R y R z in X 4 are the same as those explained in the above embodiments 1 and 2. In one embodiment, X 4 is preferably a hydroxyl group or t-butoxy.
ある態様において、式(3a)で表される化合物は、好ましくは(S)-2-[(S)-3-[(S)-2-シクロペンチル-2-[1-[(2S,4R)-4-エトキシ-1-[(S)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]-2-[(2-[(S)-N-メチル-2-[(R,Z)-3-(メチルアミノ)-2-オキソ-3,4,7,8-テトラヒドロアゾシン-1(2H)-イル]-3-[4-(トリフルオロメチル)フェニル]プロパンアミド]アセトアミド)ブタノイル]-Nメチルピロリジン-2-カルボキシアミド]-N-メチルシクロブタン-1-カルボキシアミド]-N-メチルアセトアミド]-4-(ジメチルアミノ)-N-メチル-4-オキソブタンアミド]ペンタノイル]-L-イソロイシル-L-プロリン(化合物40)である。 In one embodiment, the compound represented by formula (3a) is preferably (S)-2-[(S)-3-[(S)-2-cyclopentyl-2-[1-[(2S,4R)-4-ethoxy-1-[(S)-4-[3-methoxy-4-(trifluoromethyl)phenyl]-2-[(2-[(S)-N-methyl-2-[(R,Z)-3-(methylamino)-2-oxo-3,4,7,8-tetrahydroazocin-1(2H)-yl]-3-[4-(trifluoromethyl)phenyl]propanamido]acetamido)butanoyl]-N-methylpyrrolidine-2-carboxamido]-N-methylcyclobutane-1-carboxamido]-N-methylacetamido]-4-(dimethylamino)-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleucyl-L-proline (compound 40).
ある態様において、式(3a)で表される化合物は、式(7a)で表されるペプチド化合物のN末端のアミノ酸残基と、式(4a)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結すること(連結工程)により製造することができる。連結工程の溶媒、連結工程の縮合試薬、連結工程の塩基等は、上記態様1及び2において説明したものと同一である。 In one embodiment, the compound represented by formula (3a) can be produced by reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7a) and the C-terminal amino acid residue of the peptide compound represented by formula (4a) in a solvent (linking step). The solvent in the linking step, the condensation reagent in the linking step, the base in the linking step, etc. are the same as those described in the above embodiments 1 and 2.
式(2a)~(7a)で表される化合物の製造方法においては、固相合成を用いないことが好ましい。 In the method for producing the compounds represented by formulas (2a) to (7a), it is preferable not to use solid-phase synthesis.
式(1a)で表される環状ペプチド化合物の製造方法においては、固相合成を用いないことが好ましい。 In the method for producing the cyclic peptide compound represented by formula (1a), it is preferable not to use solid-phase synthesis.
環状ペプチド化合物の結晶
ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の結晶に関する。この化合物の結晶として、具体的には、この化合物の非溶媒和物結晶、もしくは溶媒和物結晶、またはこの化合物の塩の非溶媒和物結晶、もしくは溶媒和物結晶が挙げられる。これらの中でも式(1a)で表される環状ペプチド化合物の溶媒和物結晶が好ましい。溶媒和物結晶として好ましくは水和物結晶が挙げられる。
Crystal of Cyclic Peptide Compound In one embodiment, the present invention relates to a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. Specific examples of the crystal of this compound include a nonsolvate crystal or a solvate crystal of this compound, or a nonsolvate crystal or a solvate crystal of a salt of this compound. Among these, a solvate crystal of the cyclic peptide compound represented by formula (1a) is preferred. A preferred example of the solvate crystal is a hydrate crystal.
粉末X線回折における回折角2θは、CuKα、またはCuKα1放射線を用いて測定した回折ピークである。これらの溶媒和物結晶がさらに粉末X線回折における回折角2θで特定された結晶を、例えば以下に示す水和物の「Form A結晶」と呼ぶこともあるが、単に「Form A」と呼ぶこともある。 The diffraction angle 2θ in powder X-ray diffraction is the diffraction peak measured using CuKα or CuKα1 radiation. These solvate crystals further specified by the diffraction angle 2θ in powder X-ray diffraction are sometimes called "Form A crystals" of the hydrate shown below, for example, but are sometimes simply called "Form A".
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm A結晶である。なお、下記回折角(2θ値)は、好ましくは10%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは10%の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form A crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably those of a hydrate crystal stored at a relative humidity of 10% or more for 15 minutes or more, more preferably those of a hydrate crystal stored at a relative humidity of 10% for 15 minutes.
6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm A結晶である。なお、下記回折角(2θ値)は、好ましくは10%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは10%の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form A crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the hydrate crystal stored at a relative humidity of 10% or more for 15 minutes or more, more preferably the diffraction angles (2θ values) of the hydrate crystal stored at a relative humidity of 10% for 15 minutes.
6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm A結晶である。なお、下記回折角(2θ値)は、好ましくは30%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは30%の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form A crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably the diffraction angles (2θ values) of the hydrate crystal stored at a relative humidity of 30% for 15 minutes.
6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm B結晶である。なお、下記回折角(2θ値)は、好ましくは30%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは30%の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form B crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably those of a hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably those of a hydrate crystal stored at a relative humidity of 30% for 15 minutes.
4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm B結晶である。なお、下記回折角(2θ値)は、好ましくは30%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは30%の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form B crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably those of a hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably those of a hydrate crystal stored at a relative humidity of 30% for 15 minutes.
4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm B結晶である。なお、下記回折角(2θ値)は、好ましくは30%以上の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは30%の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form B crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the hydrate crystal stored at a relative humidity of 30% or more for 15 minutes or more, more preferably the diffraction angles (2θ values) of the hydrate crystal stored at a relative humidity of 30% for 15 minutes.
4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm F結晶である。
6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form F crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as a diffraction angle 2θ in powder X-ray diffraction.
6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm F結晶である。
6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form F crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as a diffraction angle 2θ in powder X-ray diffraction.
6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm F結晶である。
6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form F crystal having a powder X-ray diffraction pattern including the following peaks as a diffraction angle 2θ in powder X-ray diffraction.
6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm J結晶である。なお、下記回折角(2θ値)は、好ましくは10%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは10%未満の相対湿度で15分間保存した溶媒和物結晶の回折角(2θ値)である。
6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes.
6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm J結晶である。なお、下記回折角(2θ値)は、好ましくは10%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは10%未満の相対湿度で15分間保存した溶媒和物結晶の回折角(2θ値)である。
6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes.
6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm J結晶である。なお、下記回折角(2θ値)は、好ましくは10%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは10%未満の相対湿度で15分間保存した溶媒和物結晶の回折角(2θ値)である。
6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form J crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 10% for 15 minutes.
6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm J結晶である。なお、下記回折角(2θ値)は、好ましくは10%未満の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは10%未満の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more, more preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes.
6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm J結晶である。なお、下記回折角(2θ値)は、好ましくは10%未満の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは10%未満の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form J crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes.
6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm J結晶である。なお、下記回折角(2θ値)は、好ましくは10%未満の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは10%未満の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form J crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 10% for 15 minutes.
6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、10%未満の相対湿度では、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm J結晶を含み、10%以上の相対湿度では、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm A結晶を含む。 In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has a diffraction angle 2θ of 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°) in powder X-ray diffraction at a relative humidity of less than 10%. It includes Form J crystals having a powder X-ray diffraction pattern containing at least seven peaks, and at a relative humidity of 10% or greater, it includes Form A crystals having a powder X-ray diffraction pattern containing at least seven peaks at the following positions: 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°).
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、10%未満の相対湿度では、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm J結晶を含み、10%以上の相対湿度では、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm A結晶を含む。 In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has a diffraction angle 2θ of 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97° (±0.2°) in powder X-ray diffraction at a relative humidity of less than 10%. It includes Form J crystals having a powder X-ray diffraction pattern containing at least eight peaks, and at a relative humidity of 10% or greater, it includes Form A crystals having a powder X-ray diffraction pattern containing at least eight peaks at the following positions: 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°).
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、10%未満の相対湿度では、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°(±0.2°)のピークを含む粉末X線回折パターンを有するForm J結晶を含み、10%以上の相対湿度では、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°(±0.2°)のピークを含む粉末X線回折パターンを有するForm A結晶を含む。 In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has the following diffraction angles 2θ in powder X-ray diffraction at a relative humidity of less than 10%: 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97°. (±0.2°) at a relative humidity of 10% or greater, and Form A crystals having a powder X-ray diffraction pattern including peaks at 6.93°, 7.56°, 8.26°, 9.00°, 9.58°, 10.35°, 11.35°, 12.26°, 12.85°, 13.51°, 14.12°, 14.69°, 15.46°, 15.92°, 17.43°, and 17.73° (±0.2°) at a relative humidity of 10% or greater.
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm Y結晶である。なお、下記回折角(2θ値)は、好ましくは30%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは30%未満の相対湿度で15分間保存した溶媒和物結晶の回折角(2θ値)である。
5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2θ in powder X-ray diffraction. The following diffraction angles (2θ values) are preferably diffraction angles (2θ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, more preferably diffraction angles (2θ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes.
5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm Y結晶である。なお、下記回折角(2θ値)は、好ましくは30%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは30%未満の相対湿度で15分間保存した溶媒和物結晶の回折角(2θ値)である。
5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, more preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes.
5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、溶媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm Y結晶である。なお、下記回折角(2θ値)は、好ましくは30%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは30%未満の相対湿度で15分間保存した溶媒和物結晶の回折角(2θ値)である。
5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a solvate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of the solvate crystal stored at a relative humidity of less than 30% for 15 minutes.
5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm Y結晶である。なお、下記回折角(2θ値)は、好ましくは30%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは30%未満の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes.
5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm Y結晶である。なお、下記回折角(2θ値)は、好ましくは30%未満の相対湿度で15分間以上保存した溶媒和物結晶の回折角(2θ値)であり、より好ましくは30%未満の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including at least 8 of the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of a solvate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes.
5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm Y結晶である。なお、下記回折角(2θ値)は、好ましくは30%未満の相対湿度で15分間以上保存した水和物結晶の回折角(2θ値)であり、より好ましくは30%未満の相対湿度で15分間保存した水和物結晶の回折角(2θ値)である。
5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal is a Form Y crystal having a powder X-ray diffraction pattern including the following peaks as diffraction angles 2θ in powder X-ray diffraction: The following diffraction angles (2θ values) are preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes or more, and more preferably the diffraction angles (2θ values) of a hydrate crystal stored at a relative humidity of less than 30% for 15 minutes.
5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、30%未満の相対湿度では、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm Y結晶を含み、30%以上の相対湿度では、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm B結晶を含む。 In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal exhibits, in powder X-ray diffraction, a diffraction angle 2θ of at least one of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°) at a relative humidity of less than 30%. At a relative humidity of 30% or greater, it contains Form Y crystals having a powder X-ray diffraction pattern containing at least seven peaks at 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°).
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、30%未満の相対湿度では、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm Y結晶を含み、30%以上の相対湿度では、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm B結晶を含む。 In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal exhibits, in powder X-ray diffraction, a diffraction angle 2θ of at least one of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.2°) at a relative humidity of less than 30%. At a relative humidity of 30% or greater, it contains Form Y crystals having a powder X-ray diffraction pattern containing at least eight peaks at 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°).
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、30%未満の相対湿度では、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°(±0.2°)のピークを含む粉末X線回折パターンを有するForm Y結晶を含み、30%以上の相対湿度では、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°(±0.2°)のピークを含む粉末X線回折パターンを有するForm B結晶を含む。 In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a hydrate crystal, the crystal has diffraction angles 2θ of 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24° (±0.05°) in powder X-ray diffraction at a relative humidity of less than 30%. At a relative humidity of 30% or greater, it contains Form Y crystals having a powder X-ray diffraction pattern including peaks at 4.99°, 8.65°, 9.85°, 10.84°, 11.32°, 12.35°, 13.20°, 14.44°, 15.20°, 16.03°, 16.69°, 17.21°, 18.82°, 19.49°, and 20.03° (±0.2°).
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも7個のピークを含む粉末X線回折パターンを有するForm K結晶である。
7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a water solvate crystal, the crystal is a Form K crystal having a powder X-ray diffraction pattern including at least seven of the following peaks as a diffraction angle 2θ in powder X-ray diffraction.
7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のうちの少なくとも8個のピークを含む粉末X線回折パターンを有するForm K結晶である。
7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a water solvate crystal, the crystal is a Form K crystal having a powder X-ray diffraction pattern including at least eight of the following peaks at a diffraction angle 2θ in powder X-ray diffraction:
7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° (±0.2°)
ある態様において、式(1a)の環状ペプチド化合物の結晶が、水媒和物結晶である場合、該結晶は、粉末X線回折において、回折角2θとして、下記のピークを含む粉末X線回折パターンを有するForm K結晶である。
7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°(±0.2°)
In one embodiment, when the crystal of the cyclic peptide compound of formula (1a) is a water solvate crystal, the crystal is a Form K crystal having a powder X-ray diffraction pattern including the following peaks at a diffraction angle 2θ in powder X-ray diffraction.
7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51° (±0.2°)
ある態様において、本発明は式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の結晶を製造する方法に関する。該方法は、該環状ペプチド化合物を該環状ペプチド化合物が溶解可能な量の極性有機溶媒に溶解させて溶液を得る工程、および該溶液に炭化水素系溶媒または水を加えて、該環状ペプチド化合物の結晶を得る工程を含む(以下、「態様3」ともいう)。態様3において、溶解させる環状ペプチド化合物の性状は特に制限されず、例えば、固体状態、アモルファス状態、結晶状態の環状ペプチド化合物を用いることができる。 In one embodiment, the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The method includes the steps of dissolving the cyclic peptide compound in an amount of a polar organic solvent in which the cyclic peptide compound can be dissolved to obtain a solution, and adding a hydrocarbon solvent or water to the solution to obtain a crystal of the cyclic peptide compound (hereinafter, also referred to as "embodiment 3"). In embodiment 3, the nature of the cyclic peptide compound to be dissolved is not particularly limited, and for example, a cyclic peptide compound in a solid state, an amorphous state, or a crystalline state can be used.
ある態様において、本発明は式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の結晶を製造する方法に関する。該方法は、アモルファス状態または結晶状態の該環状ペプチド化合物に、炭化水素系溶媒と極性有機溶媒の混合液、または水と極性有機溶媒の混合液を加えて、該環状ペプチド化合物の結晶を得る工程を含む(以下、「態様4」ともいう)。 In one embodiment, the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The method includes the step of adding a mixture of a hydrocarbon solvent and a polar organic solvent, or a mixture of water and a polar organic solvent, to the cyclic peptide compound in an amorphous or crystalline state to obtain a crystal of the cyclic peptide compound (hereinafter, also referred to as "embodiment 4").
態様3および4で用いられる極性有機溶媒として、具体的には、DMSO、アセトン、2-ブタノン、メタノール、エタノール、1-プロパノール、2-プロパノール、酢酸エチル、プロピレングリコールおよびそれらの混合溶媒などが好ましく例示され、アセトンがより好ましく例示される。態様3における「環状ペプチド化合物が溶解可能な量」として、式(1a)の環状ペプチド化合物に対し、3~10v/wの範囲、好ましくは3~7v/wの範囲の極性有機溶媒を用いることができる。 Specific examples of the polar organic solvent used in embodiments 3 and 4 include DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, ethyl acetate, propylene glycol, and mixtures thereof, with acetone being a more preferred example. In embodiment 3, the "amount capable of dissolving the cyclic peptide compound" can be in the range of 3 to 10 v/w, preferably 3 to 7 v/w, relative to the cyclic peptide compound of formula (1a).
態様3および4で用いられる炭化水素系溶媒として、具体的には、ヘプタン、ヘキサン、ペンタン、トルエン、キシレン、およびそれらの混合溶媒などが好ましく例示され、ヘプタンがより好ましく例示される。 Specific preferred examples of the hydrocarbon solvent used in embodiments 3 and 4 include heptane, hexane, pentane, toluene, xylene, and mixed solvents thereof, with heptane being a more preferred example.
態様4において、炭化水素系溶媒と極性有機溶媒の混合液における炭化水素系溶媒と極性有機溶媒の混合比としては、極性有機溶媒1重量部に対し、炭化水素系溶媒0.5~10重量部用いることができ、好ましくは、炭化水素系溶媒1~7重量部、さらに好ましくは、炭化水素系溶媒1~5重量部用いることが好ましい。また、態様4において、水と極性有機溶媒の混合液における水と極性有機溶媒の混合比としては、極性有機溶媒1重量部に対し、水0.5~10重量部用いることができ、好ましくは、水1~7重量部、さらに好ましくは、水1~5重量部用いることが好ましい。 In aspect 4, the mixture ratio of the hydrocarbon solvent and the polar organic solvent in the mixture of the hydrocarbon solvent and the polar organic solvent can be 0.5 to 10 parts by weight of the hydrocarbon solvent per 1 part by weight of the polar organic solvent, preferably 1 to 7 parts by weight of the hydrocarbon solvent, and more preferably 1 to 5 parts by weight of the hydrocarbon solvent. Also, in aspect 4, the mixture ratio of the water and the polar organic solvent in the mixture of the water and the polar organic solvent can be 0.5 to 10 parts by weight of water per 1 part by weight of the polar organic solvent, preferably 1 to 7 parts by weight of water, and more preferably 1 to 5 parts by weight of water.
態様4のうち、さらにある態様においては、アモルファス状態または結晶状態の該環状ペプチド化合物に、炭化水素系溶媒と極性有機溶媒の混合液、または水と極性有機溶媒の混合液を加える操作において、結晶にガラスビーズ(例えば、1~5粒)を加えて振とうしてもよい。 In a further embodiment of embodiment 4, in the step of adding a mixture of a hydrocarbon solvent and a polar organic solvent, or a mixture of water and a polar organic solvent, to the cyclic peptide compound in an amorphous or crystalline state, glass beads (e.g., 1 to 5 beads) may be added to the crystals and shaken.
ある態様において、本発明は式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の結晶を製造する方法に関する。該方法は、アモルファス状態の該環状ペプチド化合物をDMSOに溶解させて溶液を得る工程、該溶液を凍結乾燥して該環状ペプチド化合物の凍結乾燥体を得る工程、および、該凍結乾燥体に水と極性有機溶媒の混合液を加えて、該環状ペプチド化合物の結晶を得る工程を含む(以下、「態様5」ともいう)。 In one embodiment, the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The method includes the steps of dissolving the cyclic peptide compound in an amorphous state in DMSO to obtain a solution, freeze-drying the solution to obtain a freeze-dried product of the cyclic peptide compound, and adding a mixture of water and a polar organic solvent to the freeze-dried product to obtain a crystal of the cyclic peptide compound (hereinafter, also referred to as "embodiment 5").
態様5で用いられる極性有機溶媒として、具体的には、DMSO、アセトン、2-ブタノン、メタノール、エタノール、1-プロパノール、2-プロパノール、プロピレングリコール、およびそれらの混合溶媒などが好ましく例示され、アセトンがより好ましく例示される。 Specific preferred examples of the polar organic solvent used in embodiment 5 include DMSO, acetone, 2-butanone, methanol, ethanol, 1-propanol, 2-propanol, propylene glycol, and mixed solvents thereof, with acetone being a more preferred example.
態様5において、水と極性有機溶媒の混合液における水と極性有機溶媒の混合比としては、極性有機溶媒1重量部に対し、水0.5~10重量部用いることができ、好ましくは、水1~7重量部、さらに好ましくは、水1~5重量部用いることが好ましい。 In aspect 5, the mixing ratio of water to polar organic solvent in the mixture of water and polar organic solvent can be 0.5 to 10 parts by weight of water per 1 part by weight of polar organic solvent, preferably 1 to 7 parts by weight of water, and more preferably 1 to 5 parts by weight of water.
ある態様において、本発明は式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の結晶を製造する方法に関する。該方法は、該環状ペプチド化合物の結晶を加熱して、該環状ペプチド化合物の別の結晶多形を得る工程を含む(以下、「態様6」ともいう)。加熱温度は、例えば30~350℃であり、好ましくは30~120℃である。 In one embodiment, the present invention relates to a method for producing a crystal of a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The method includes a step of heating the crystal of the cyclic peptide compound to obtain another crystalline polymorph of the cyclic peptide compound (hereinafter, also referred to as "embodiment 6"). The heating temperature is, for example, 30 to 350°C, and preferably 30 to 120°C.
態様3~6において、環状ペプチド化合物の結晶を得る工程の後に、さらに該結晶をろ過する工程を含んでもよい。 In aspects 3 to 6, after the step of obtaining crystals of the cyclic peptide compound, a step of filtering the crystals may be further included.
態様3~6において、環状ペプチド化合物の結晶を得る工程の後に、さらに該結晶を乾燥する工程を含んでもよい。 In aspects 3 to 6, after the step of obtaining crystals of the cyclic peptide compound, a step of drying the crystals may be further included.
ある態様において、本発明の方法により製造される環状ペプチド化合物の結晶は、溶媒和物結晶であることが好ましく、水和物結晶であることがより好ましい。 In one embodiment, the crystals of the cyclic peptide compound produced by the method of the present invention are preferably solvate crystals, and more preferably hydrate crystals.
ある態様において、本発明の方法により製造される環状ペプチド化合物の結晶は、溶媒中で溶媒和物結晶として形成され、ろ過する工程および/または乾燥する工程の後に、水和物結晶として得られる。また、ある態様において、本発明の方法により製造される環状ペプチド化合物の結晶は、ろ過する工程および/または乾燥する工程の後に大気中の水分を取り込むことで水和物結晶として得られる。 In one embodiment, the crystals of the cyclic peptide compound produced by the method of the present invention are formed as solvate crystals in a solvent, and are obtained as hydrate crystals after the filtering and/or drying steps. In another embodiment, the crystals of the cyclic peptide compound produced by the method of the present invention are obtained as hydrate crystals by absorbing moisture from the atmosphere after the filtering and/or drying steps.
環状ペプチド化合物を含む組成物
ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を含む組成物に関する。該化合物は不純物である式(1a)の環状ダイマーを1.5w/w%以下の割合で含有する。
Compositions containing cyclic peptide compounds In one embodiment, the present invention relates to compositions containing a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof, which contains 1.5 w/w% or less of the cyclic dimer of formula (1a) as an impurity.
ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を含む組成物に関する。該化合物は不純物である式(1a)の環状ダイマーを0.001w/w%以上の割合で含有する。 In one embodiment, the present invention relates to a composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The compound contains the cyclic dimer of formula (1a) as an impurity at a ratio of 0.001 w/w% or more.
ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を含む組成物に関する。該化合物はアセトンを2.0w/w%以下の割合で含有する。 In one embodiment, the present invention relates to a composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The compound contains acetone in an amount of 2.0 w/w% or less.
ある態様において、本発明は、式(1a)で表される環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を含む組成物に関する。該化合物はアセトンを0.001w/w%以上の割合で含有する。 In one embodiment, the present invention relates to a composition comprising a cyclic peptide compound represented by formula (1a), or a salt thereof, or a solvate thereof. The compound contains acetone in an amount of 0.001 w/w% or more.
本発明の内容を以下の実施例でさらに説明するが、本発明はその内容に限定されるものではない。特に記載したものを除き、出発物質、出発原料、溶媒、および試薬は商業的供給業者から入手、もしくは公知の方法を用いて合成した。後述する実施例1-26で用いる化合物25は、国際公開第2022/234853号に記載の方法により合成した。 The present invention is further described in the following examples, but is not limited thereto. Unless otherwise specified, starting materials, starting materials, solvents, and reagents were obtained from commercial suppliers or synthesized using known methods. Compound 25 used in Examples 1-26 below was synthesized by the method described in WO 2022/234853.
LCMSによる分析条件は以下に示した。
LCMS分析条件 method 1
装置:Shimadzu LCMS2020
カラム:CORTECS C18 Column, 3,0 mm IDx50 mm, 2,7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 5%(0 min)→95%(2.0min)→95%(2.8 min)→5%(2.81 min)→5%(3min)
流速:1.5mL/min
カラム温度:40℃
検出波長:190-800nm(PDA)
The analytical conditions by LCMS are shown below.
LCMS analysis conditions method 1
Equipment: Shimadzu LCMS2020
Column: CORTECS C18 Column, 3.0 mm IDx50 mm, 2.7 μm
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Dissolution method: B) 5% (0 min) → 95% (2.0 min) → 95% (2.8 min) → 5% (2.81 min) → 5% (3 min)
Flow rate: 1.5mL/min
Column temperature: 40°C
Detection wavelength: 190-800 nm (PDA)
LCMS分析条件 method 2
装置:Waters UPLC/SQD
カラム:Ascentis Express RP 90A amide, 2.1 mm IDx50 mm, 2.7 μm(PDA)m
移動相:0.1%FA/water (A)、0.1%FA/MeCN (B)
溶出法:B) 5%(0 min)→100%(4.5 min)→100%(5.0 min)→5%(5.01min)→5%(7 min)
流速:0.5mL/min
カラム温度:40℃
検出波長:210-400nm(PDA)
LCMS analysis conditions method 2
Instrument: Waters UPLC/SQD
Column: Ascentis Express RP 90A amide, 2.1 mm ID x 50 mm, 2.7 μm (PDA)
Mobile phase: 0.1% FA/water (A), 0.1% FA/MeCN (B)
Dissolution method: B) 5% (0 min) → 100% (4.5 min) → 100% (5.0 min) → 5% (5.01 min) → 5% (7 min)
Flow rate: 0.5mL/min
Column temperature: 40°C
Detection wavelength: 210-400 nm (PDA)
LCMS分析条件 method 3
装置:Waters UPLC/SQD
カラム:Ascentis Express 90A C18, 2.1 mm IDx50 mm, 2.7 μm
移動相:0.1%FA/water (A)、0.1%FA/MeCN (B)
溶出法:B) 5%(0 min)→100%(5 min)→5%(5.01 min)→5%(7 min)
流速:0.5mL/min
カラム温度:Off
検出波長:210-400nm(PDA)
LCMS analysis conditions method 3
Instrument: Waters UPLC/SQD
Column: Ascentis Express 90A C18, 2.1 mm ID x 50 mm, 2.7 μm
Mobile phase: 0.1% FA/water (A), 0.1% FA/MeCN (B)
Elution method: B) 5% (0 min) → 100% (5 min) → 5% (5.01 min) → 5% (7 min)
Flow rate: 0.5mL/min
Column temperature: Off
Detection wavelength: 210-400 nm (PDA)
LCMS分析条件 method 4
装置:Waters UPLC/SQD
カラム:ACQUITY UPLC BEH C18 Column, 2.1 mm IDx50 mm, 1.7 μm
移動相:0.1% FA/water (A)、0.1% FA/MeCN (B)
溶出法:B) 5%(0 min)→98%(8 min)→98%(10 min)→5%(10.01min)→5%(12 min)
流速:0.5mL/min
カラム温度:60℃
検出波長:210-400nm(PDA)
LCMS analysis conditions method 4
Instrument: Waters UPLC/SQD
Column: ACQUITY UPLC BEH C18 Column, 2.1 mm ID x 50 mm, 1.7 μm
Mobile phase: 0.1% FA/water (A), 0.1% FA/MeCN (B)
Dissolution method: B) 5% (0 min) → 98% (8 min) → 98% (10 min) → 5% (10.01 min) → 5% (12 min)
Flow rate: 0.5mL/min
Column temperature: 60°C
Detection wavelength: 210-400 nm (PDA)
LCMS分析条件 method 5
装置:Waters UPLC/SQD
カラム: Ascentis Express C18 2.1x 50mm, 5μm
移動相:10mM ammonium acetate in water (A)、MeOH (B)
溶出法:B) 50%(0 min)→100%(1 min)→100%(1.99 min)→50%(2.01min)→50%(2.5 min)
流速:1.0mL/min
カラム温度:35℃
検出波長:210-400nm(PDA)
LCMS analysis conditions method 5
Instrument: Waters UPLC/SQD
Column: Ascentis Express C18 2.1 x 50 mm, 5 μm
Mobile phase: 10mM ammonium acetate in water (A), MeOH (B)
Dissolution method: B) 50% (0 min) → 100% (1 min) → 100% (1.99 min) → 50% (2.01 min) → 50% (2.5 min)
Flow rate: 1.0mL/min
Column temperature: 35°C
Detection wavelength: 210-400 nm (PDA)
LCMS分析条件 method M
装置:Waters Acquity UPLC/QDa
カラム:Ascentis Express 90A C18, 2.1 mm IDx50 mm, 2.7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 5%(0 min)→100%(5 min)→5%(5.01 min)→5%(7 min)
流速:0.5mL/min
カラム温度:35℃
検出波長:210nm(PDA)
LCMS analysis conditions: Method M
Instrument: Waters Acquity UPLC/QDa
Column: Ascentis Express 90A C18, 2.1 mm ID x 50 mm, 2.7 μm
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 5% (0 min) → 100% (5 min) → 5% (5.01 min) → 5% (7 min)
Flow rate: 0.5mL/min
Column temperature: 35°C
Detection wavelength: 210 nm (PDA)
LCMS分析条件 method K-1
装置:Waters Acquity UPLC/QDa
カラム:CAPCELL CORE ADME, 2.1 mmx50 mm, 2.7 μm (Osaka Soda)
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 5%(0 min)→100%(10 min)→5%(10.1 min)→5%(12 min)
流速:0.5mL/min
カラム温度:35℃
検出波長:210nm(PDA)
LCMS analysis conditions: Method K-1
Equipment: Waters Acquity UPLC/QDa
Column: CAPCELL CORE ADME, 2.1 mmx50 mm, 2.7 μm (Osaka Soda)
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 5% (0 min) → 100% (10 min) → 5% (10.1 min) → 5% (12 min)
Flow rate: 0.5mL/min
Column temperature: 35°C
Detection wavelength: 210 nm (PDA)
LCMS分析条件 method K-2
装置:Waters Acquity UPLC/QDa
カラム:CAPCELL CORE ADME, 2.1 mm×50 mm, 2.7 μm (Osaka Soda)
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 5%(0 min)→100%(5 min)→5%(5.1 min)→5%(7 min)
流速:0.5mL/min
カラム温度:35℃
検出波長:210nm(PDA)
LCMS analysis conditions: Method K-2
Equipment: Waters Acquity UPLC/QDa
Column: CAPCELL CORE ADME, 2.1 mm x 50 mm, 2.7 μm (Osaka Soda)
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 5% (0 min) → 100% (5 min) → 5% (5.1 min) → 5% (7 min)
Flow rate: 0.5mL/min
Column temperature: 35°C
Detection wavelength: 210 nm (PDA)
LCMS分析条件 method P3-4
装置:Waters Acquity UPLC/QDa
カラム:Ascentis Express 90A C18, 2.1 mm IDx50 mm, 2.7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 5%(0 min)→100%(5 min)→5%(5.01 min)→5%(7 min)
流速:0.5mL/min
カラム温度:35℃
検出波長:210nm(PDA)
LCMS analysis conditions: method P3-4
Instrument: Waters Acquity UPLC/QDa
Column: Ascentis Express 90A C18, 2.1 mm ID x 50 mm, 2.7 μm
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 5% (0 min) → 100% (5 min) → 5% (5.01 min) → 5% (7 min)
Flow rate: 0.5mL/min
Column temperature: 35°C
Detection wavelength: 210 nm (PDA)
LCMS分析条件 method FC 2
装置:Waters UPLC
カラム:ACQUITY UPLC CSH C18, 2.1 mm IDx100 mm, 1.7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 20%(0 min)→100%(10 min)→100%(13.5 min)→20%(13.6 min)→20%(15.5 min)
流速:0.3mL/min
カラム温度:50℃
検出波長:210nm(PDA)
LCMS analysis conditions: method FC 2
Instrument: Waters UPLC
Column: ACQUITY UPLC CSH C18, 2.1 mm ID x 100 mm, 1.7 μm
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 20% (0 min) → 100% (10 min) → 100% (13.5 min) → 20% (13.6 min) → 20% (15.5 min)
Flow rate: 0.3mL/min
Column temperature: 50°C
Detection wavelength: 210 nm (PDA)
LC分析条件 method cyc
装置:Waters UPLC
カラム:ACQUITY UPLC CSH Phenyl-Hexyl, 2.1 mm ID×150 mm, 1.7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 20%(0 min)→100%(24 min)→100%(29 min)→20%(29.1 min)→20%(34 min)
流速:0.3mL/min
カラム温度:50℃
検出波長:220nm(PDA)
LC analysis conditions: method cyc
Instrument: Waters UPLC
Column: ACQUITY UPLC CSH Phenyl-Hexyl, 2.1 mm ID x 150 mm, 1.7 μm
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 20% (0 min) → 100% (24 min) → 100% (29 min) → 20% (29.1 min) → 20% (34 min)
Flow rate: 0.3mL/min
Column temperature: 50°C
Detection wavelength: 220 nm (PDA)
LC分析条件 method H
装置:Waters UPLC
カラム:Ascentis Express RP-Amide, 3.0 mm ID×50mm×,2.7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 5%(0 min)→95%(10.0 min)→95%(12.0 min)→5%(12.1 min)→5%(15.0 min)
流速:0.7mL/min
カラム温度:30℃
検出波長:210nm(PDA)
LC analysis conditions: Method H
Instrument: Waters UPLC
Column: Ascentis Express RP-Amide, 3.0 mm ID x 50 mm x 2.7 μm
Mobile phase: 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
Elution method: B) 5% (0 min) → 95% (10.0 min) → 95% (12.0 min) → 5% (12.1 min) → 5% (15.0 min)
Flow rate: 0.7mL/min
Column temperature: 30°C
Detection wavelength: 210 nm (PDA)
HPLCによる分析条件は以下に示した。
HPLC分析条件 method 1
装置:Waters H-Class
カラム:ACQUITY UPLC BEH C18 Column, 2.1 mm ID×50 mm, 1.7 μm
移動相:0.1% FA/water (A)、0.1% FA/MeCN (B)
溶出法:B) 5%(0 min)→98%(4.0 min)→98%(6.0 min)→5%(6.01 min)→5%(8 min)
流速:0.5 mL/min
カラム温度:60℃
検出波長:210nm(PDA)
The HPLC analysis conditions are shown below.
HPLC analysis conditions: Method 1
Equipment: Waters H-Class
Column: ACQUITY UPLC BEH C18 Column, 2.1 mm ID x 50 mm, 1.7 μm
Mobile phase: 0.1% FA/water (A), 0.1% FA/MeCN (B)
Elution method: B) 5% (0 min) → 98% (4.0 min) → 98% (6.0 min) → 5% (6.01 min) → 5% (8 min)
Flow rate: 0.5 mL/min
Column temperature: 60°C
Detection wavelength: 210 nm (PDA)
qNMRによる測定法は、目的化合物を含む残渣と内部標準物質をCDCl3またはDMSO‐d6に溶解させ、以下の分析条件により行った。収率の算出は、qNMRまたはHPLCにより算出された残渣中の目的物の含量の値により、以下の式により行った。
![]()
![]()
測定装置:Bruker Avance III 400
内部標準物質:3,5-ビス(トリフルオロメチル)安息香酸
測定条件(19F-NMR):CDCl3またはDMSO-d6、24.8℃、パルス角度 90°、デジタル分解能 0.24Hz、緩和時間 15秒、スピンなし、積算回数64回
Measuring device: Bruker Avance III 400
Internal standard substance: 3,5-bis(trifluoromethyl)benzoic acid Measurement conditions ( 19 F-NMR): CDCl 3 or DMSO-d 6 , 24.8° C., pulse angle 90°, digital resolution 0.24 Hz, relaxation Time: 15 seconds, no spin, 64 times
測定装置:JEOL JNM-ECZ500R/S1
測定条件(1H-NMR):methanol-d4、25.3℃、パルス角度 45°、デジタル分解能 0.76Hz、緩和時間 5秒、スピンあり、積算回数8回
Measuring device: JEOL JNM-ECZ500R/S1
Measurement conditions ( 1 H-NMR): methanol-d 4 , 25.3° C., pulse angle 45°, digital resolution 0.76 Hz, relaxation time 5 seconds, spin on, accumulation 8 times
LCMS、LC、およびHPLCによる測定法は、目的化合物を含む混合液を以下のいずれかの方法にてサンプル調製し、上述の分析条件により行った。
サンプル調製法1:目的化合物を含む混合液を、アセトニトリルで希釈した。
サンプル調製法2:目的化合物を含む混合液を、アセトニトリルと水を9対1の比率で混ぜた混合液で希釈した。
サンプル調製法K:目的化合物を含む混合液を、アセトニトリルとノルマルプロピルアミンを100対1の比率で混ぜた混合液で希釈した。
The measurement methods by LCMS, LC, and HPLC were carried out by preparing a sample of a mixture containing the target compound by one of the following methods and under the analytical conditions described above.
Sample preparation method 1: A mixture containing the target compound was diluted with acetonitrile.
Sample preparation method 2: The mixture containing the target compound was diluted with a mixture of acetonitrile and water in a ratio of 9:1.
Sample preparation method K: A mixture containing the target compound was diluted with a mixture of acetonitrile and n-propylamine in a ratio of 100:1.
反応転換率の算出は、HPLC分析により算出された原料の面積値と目的物の面積値、又は原料の面積値と原料の面積値と目的物の面積値、又は反応前の原料の面積値と反応後の原料の面積値を用いて、以下のいずれかの式により行った。
式1:反応転換率(%)=目的物の面積値/(原料の面積値+目的物の面積値)×100
式2:反応転換率(%)=100-(反応後の原料の面積値/反応前の原料の面積値×100)
The reaction conversion rate was calculated by one of the following formulas using the area values of the raw materials and the target product, or the area values of the raw materials, the area values of the raw materials and the target product, or the area values of the raw materials before the reaction and the area values of the raw materials after the reaction, all calculated by HPLC analysis.
Formula 1: Reaction conversion rate (%) = area value of target product / (area value of raw material + area value of target product) x 100
Equation 2: Reaction conversion rate (%) = 100 - (area value of raw material after reaction / area value of raw material before reaction x 100)
環化の選択性(化合物1/環状ダイマー)の算出は、HPLC分析により算出された目的物の面積値と環状ダイマーの面積値を用いて、以下の式により行った。
式1:化合物1比率(%)=化合物1の面積値/(化合物1の面積値+環状ダイマーの面積値)×100
式2:環状ダイマー比率(%)=環状ダイマーの面積値/(化合物1の面積値+環状ダイマーの面積値)×100
The cyclization selectivity (compound 1/cyclic dimer) was calculated according to the following formula using the area value of the target compound and the area value of the cyclic dimer calculated by HPLC analysis.
Formula 1: Compound 1 ratio (%) = area value of compound 1/(area value of compound 1 + area value of cyclic dimer) x 100
Formula 2: Cyclic dimer ratio (%) = area value of cyclic dimer / (area value of compound 1 + area value of cyclic dimer) × 100
実施例1-1Example 1-1
化合物2:9H-フルオレン-9-イルメチル (4S)-5-オキソ-4-[[4-(トリフルオロメチル)フェニル]メチル]オキサゾリジン-3-カルボン酸の合成Compound 2: Synthesis of 9H-fluoren-9-ylmethyl (4S)-5-oxo-4-[[4-(trifluoromethyl)phenyl]methyl]oxazolidine-3-carboxylic acid
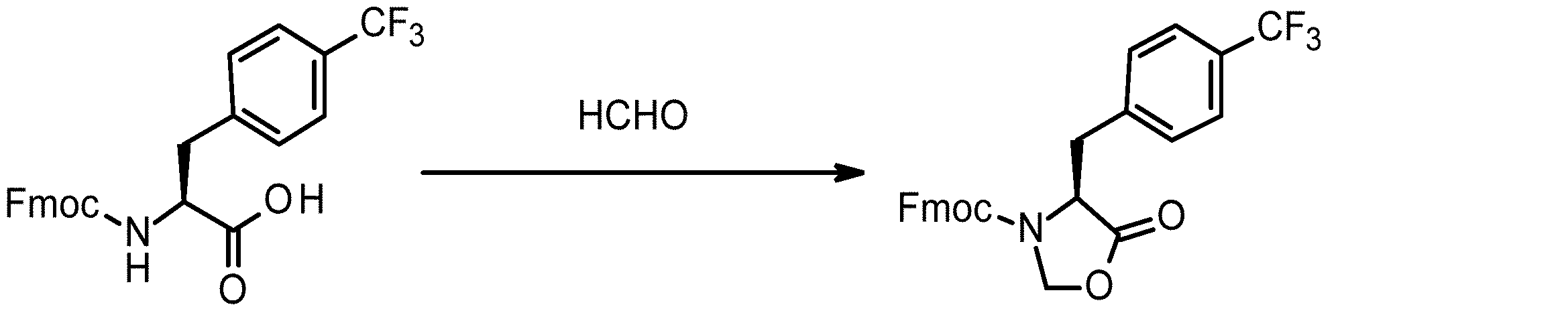
窒素で置換した反応釜に、室温にてDCM(45L)、(2S)-2-{[(9H-フルオレン-9-イルメトキシ)カルボニル]アミノ}-3-[4-(トリフルオロメチル)フェニル]プロピオン酸(3.05kg)を加えて攪拌した。続いてパラホルムアルデヒド(0.90kg)、MgSO4(2.02kg)を加えて20℃で10分間撹拌した。反応釜の外温を15℃に設定し、BF3OEt2(0.95kg)を内温15-20℃でゆっくり滴下した。反応混合物を20-25℃で12時間撹拌した後、シリカゲル(3.05kg)を敷いたろ過器でろ過し、DCM(15.3Lx2)で洗浄した。ろ液を外温30℃で減圧濃縮し、化合物1を含む粗生成物をカラム精製(石油エーテル/酢酸エチル=4/1)し、減圧濃縮することで化合物1(2.76kg)を得た。 In a reaction vessel purged with nitrogen, DCM (45 L) and (2S)-2-{[(9H-fluoren-9-ylmethoxy)carbonyl]amino}-3-[4-(trifluoromethyl)phenyl]propionic acid (3.05 kg) were added at room temperature and stirred. Then, paraformaldehyde (0.90 kg) and MgSO 4 (2.02 kg) were added and stirred at 20°C for 10 minutes. The external temperature of the reaction vessel was set to 15°C, and BF 3 OEt 2 (0.95 kg) was slowly added dropwise at an internal temperature of 15-20°C. The reaction mixture was stirred at 20-25°C for 12 hours, then filtered through a filter covered with silica gel (3.05 kg) and washed with DCM (15.3 L x 2). The filtrate was concentrated under reduced pressure at an external temperature of 30° C., and the crude product containing compound 1 was purified by column chromatography (petroleum ether/ethyl acetate=4/1) and concentrated under reduced pressure to obtain compound 1 (2.76 kg).
実施例1-2Example 1-2
化合物3:(2S)-2-[ブタ-3-エニル(9H-フルオレン-9-イルメトキシカルボニル)アミノ]-3-[4-(トリフルオロメチル)フェニル]プロピオン酸の合成Synthesis of Compound 3: (2S)-2-[but-3-enyl(9H-fluoren-9-ylmethoxycarbonyl)amino]-3-[4-(trifluoromethyl)phenyl]propionic acid

窒素で置換した反応釜に、室温にてトルエン(22.4L)、化合物2(2.80kg)を加えて攪拌した。続いてアリルトリメチルシラン(1.37kg)、ZnBr2(1.35kg)を加えて20℃で10分間撹拌した。反応釜の内温を40-45℃に設定し、反応混合物を10時間撹拌した。反応液を内温10±5℃にて氷水(28.0L)中に加え撹拌した。水層を排出し、有機層を5%食塩水(28.0L)で洗浄した。得られた有機層を外温45-50℃で減圧濃縮し、化合物3(2.44kg)を得た。 Toluene (22.4 L) and compound 2 (2.80 kg) were added to a reaction vessel purged with nitrogen at room temperature and stirred. Allyltrimethylsilane (1.37 kg) and ZnBr 2 (1.35 kg) were then added and stirred at 20°C for 10 minutes. The internal temperature of the reaction vessel was set to 40-45°C, and the reaction mixture was stirred for 10 hours. The reaction liquid was added to ice water (28.0 L) at an internal temperature of 10±5°C and stirred. The aqueous layer was discharged, and the organic layer was washed with 5% saline (28.0 L). The obtained organic layer was concentrated under reduced pressure at an external temperature of 45-50°C to obtain compound 3 (2.44 kg).
実施例1-3Examples 1-3
化合物4:tert-ブチル 2-[[(2S)-2-[ブタ-3-エニル (9H-フルオレン-9-イルメトキシカルボニル)アミノ]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸の合成Compound 4: Synthesis of tert-butyl 2-[[(2S)-2-[but-3-enyl (9H-fluoren-9-ylmethoxycarbonyl)amino]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetate

窒素で置換した反応釜に、室温にてN-メチル-2-ピロリドン(17.0L)、化合物3(2.44kg)を加えて攪拌した。続いてサルコシンtert-ブチルエステル 塩酸塩(0.87kg)、HATU(2.18kg)を20℃で加えて30分間撹拌した。DIPEA(1.85kg)を内温15-20℃で60分間かけて滴下した。反応混合物を20-25℃で3時間撹拌した後、メチルtert-ブチルエーテル(48.8L)で希釈した。有機層を水(48.8Lx2)、5%食塩水(24.4L)で洗浄した後、減圧濃縮することで化合物4を含む濃縮乾固物を得た。得られた濃縮乾固物をカラム精製(石油エーテル/酢酸エチル=4/1)し、35℃で減圧濃縮することで暗黄色のオイルである化合物4(2.79kg)を得た。 N-methyl-2-pyrrolidone (17.0 L) and compound 3 (2.44 kg) were added to a nitrogen-purged reaction vessel at room temperature and stirred. Sarcosine tert-butyl ester hydrochloride (0.87 kg) and HATU (2.18 kg) were then added at 20°C and stirred for 30 minutes. DIPEA (1.85 kg) was added dropwise over 60 minutes at an internal temperature of 15-20°C. The reaction mixture was stirred at 20-25°C for 3 hours and then diluted with methyl tert-butyl ether (48.8 L). The organic layer was washed with water (48.8 L x 2) and 5% saline (24.4 L), and then concentrated under reduced pressure to obtain a concentrated, dry product containing compound 4. The concentrated, dry product was purified by column (petroleum ether/ethyl acetate = 4/1) and concentrated under reduced pressure at 35°C to obtain compound 4 (2.79 kg) as a dark yellow oil.
実施例1-4Examples 1-4
化合物5:tert-ブチル2-[[(2S)-2-(ブタ-3-エニルアミノ)-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸の合成Synthesis of Compound 5: tert-Butyl 2-[[(2S)-2-(but-3-enylamino)-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetate
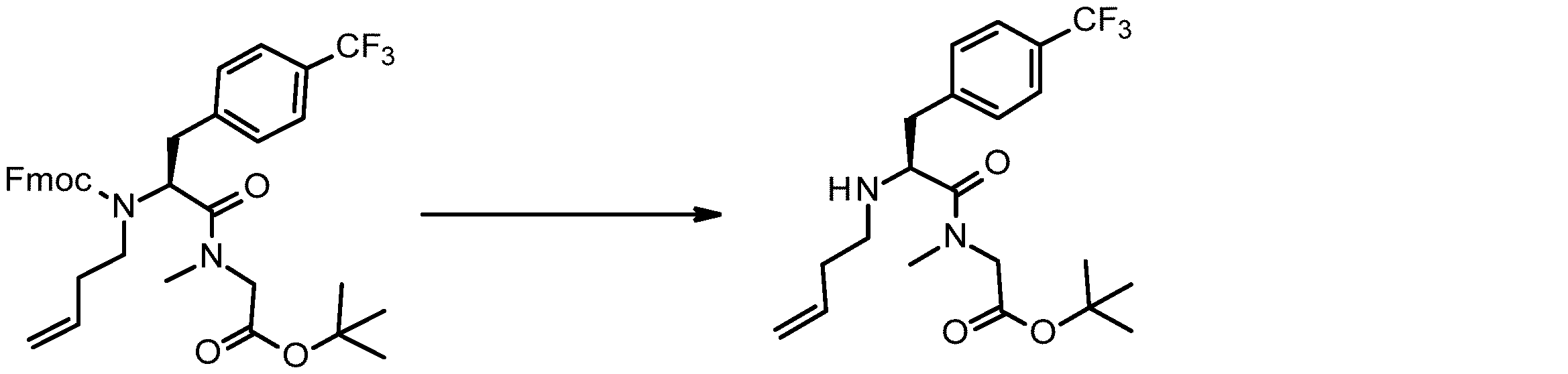
窒素で置換した反応釜に、室温にてトルエン(28.0L)、化合物4(2.79kg)を加えて攪拌した。続いてDBU(0.67kg)を内温20℃で加えて2時間撹拌した。反応混合物を内温20±5℃にて氷水(27.9L)に加え撹拌した。酢酸エチル(14.0L)を加え撹拌した後、水層を排出した。有機層を5%食塩水(28.0L)で洗浄し、有機層を45±5℃で減圧濃縮した。化合物5を含む濃縮物をカラム精製(石油エーテル/酢酸エチル=4/1)した後、減圧濃縮することで淡黄色のオイルである化合物5(1.49kg)を得た。 Toluene (28.0 L) and compound 4 (2.79 kg) were added to a reaction vessel purged with nitrogen at room temperature and stirred. DBU (0.67 kg) was then added at an internal temperature of 20°C and stirred for 2 hours. The reaction mixture was added to ice water (27.9 L) at an internal temperature of 20±5°C and stirred. Ethyl acetate (14.0 L) was added and stirred, after which the aqueous layer was discharged. The organic layer was washed with 5% saline (28.0 L) and concentrated under reduced pressure at 45±5°C. The concentrate containing compound 5 was purified by column (petroleum ether/ethyl acetate = 4/1) and then concentrated under reduced pressure to obtain compound 5 (1.49 kg) as a pale yellow oil.
実施例1-5Examples 1-5
化合物6:9H-フルオレン-9-イルメチルN-[(1S)-1-クロロカルボニルブタ-3-エニル]-N-メチル-カーバメートの合成Synthesis of Compound 6: 9H-fluoren-9-ylmethyl N-[(1S)-1-chlorocarbonylbut-3-enyl]-N-methyl-carbamate

窒素で置換した反応釜に、室温にてDCM(10L)、(2S)-2-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]ペンタ-4-エン酸(1.7kg)、DMF(0.02kg)を内温20℃で加えて撹拌した。内温10℃で塩化オキサリル(1.84kg)を2時間かけて加え、内温10℃のまま2時間撹拌した。混合溶液を30-35℃で減圧濃縮した。濃縮物にDCM(3.4L)添加と減圧濃縮を2回行い、黄色のオイルである化合物6(1.65kg)を得た。 In a reaction vessel purged with nitrogen, DCM (10 L), (2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]pent-4-enoic acid (1.7 kg), and DMF (0.02 kg) were added at room temperature and stirred at an internal temperature of 20°C. Oxalyl chloride (1.84 kg) was added over 2 hours at an internal temperature of 10°C, and the mixture was stirred for 2 hours while maintaining the internal temperature at 10°C. The mixed solution was concentrated under reduced pressure at 30-35°C. DCM (3.4 L) was added to the concentrate and concentrated under reduced pressure twice to obtain compound 6 (1.65 kg) as a yellow oil.
実施例1-6Examples 1-6
化合物7:tert-ブチル2-[[(2S)-2-[ブタ-3-エニル-[(2S)-2-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]ペンタ-4-エノイル]アミノ]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸の合成Synthesis of Compound 7: tert-butyl 2-[[(2S)-2-[but-3-enyl-[(2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]pent-4-enoyl]amino]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetic acid
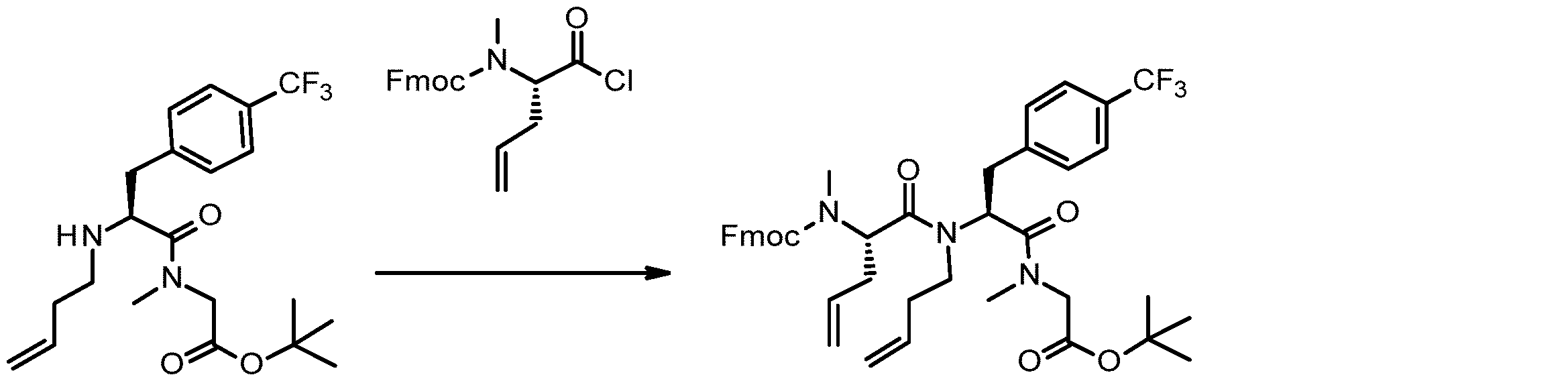
窒素で置換した反応釜に、室温にてDCM(15.0L)、化合物5(1.49kg)を加えて攪拌した。続いてDCM(2.96L)に溶解させた化合物6(1.72kg)を内温0-10℃で1時間かけて滴下した。内温10℃で30分攪拌し、続いてDIPEA(0.926kg)を内温0-10℃で1時間かけて滴下した。20℃で2時間撹拌した後に、反応液を内温20℃で氷水(14.8L)に加え撹拌した。液々分離で有機層1を取り置いた。水層をDCM(7.4L)で抽出し、取り置いた有機層1と合わせた。合わせた有機層を5%食塩水(14.9Lx2)で洗浄し、有機層を30±5℃で減圧濃縮した。化合物7を含む濃縮物をカラム精製(石油エーテル/酢酸エチル=5/1)し、減圧濃縮することで無色の粉末である化合物7(1.97kg)を得た。 DCM (15.0 L) and compound 5 (1.49 kg) were added to a reaction vessel purged with nitrogen at room temperature and stirred. Compound 6 (1.72 kg) dissolved in DCM (2.96 L) was then added dropwise over 1 hour at an internal temperature of 0-10°C. Stirring was continued for 30 minutes at an internal temperature of 10°C, followed by dropwise addition of DIPEA (0.926 kg) over 1 hour at an internal temperature of 0-10°C. After stirring for 2 hours at 20°C, the reaction solution was added to ice water (14.8 L) at an internal temperature of 20°C and stirred. Organic layer 1 was set aside by liquid-liquid separation. The aqueous layer was extracted with DCM (7.4 L) and combined with the set aside organic layer 1. The combined organic layer was washed with 5% saline (14.9 L x 2), and the organic layer was concentrated under reduced pressure at 30±5°C. The concentrate containing compound 7 was purified by column (petroleum ether/ethyl acetate = 5/1) and concentrated under reduced pressure to obtain compound 7 (1.97 kg) as a colorless powder.
実施例1-7Examples 1-7
化合物8:tert-ブチル2-[[(2S)-2-[(4Z,7S)-7-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸の合成Synthesis of Compound 8: tert-Butyl 2-[[(2S)-2-[(4Z,7S)-7-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetic acid

窒素で置換した反応釜に、室温にてトルエン(59.0L)、化合物7(0.655kg)を加えて攪拌した。続いてp‐ベンゾキノン(28.4g)を加え、内温100℃に加温した。内温100℃に加温した混合溶液に対して、トルエン(100mL)に溶解させた第1世代 HOVEYDA-GRUBBS触媒(42.0g)を20分かけて滴下した。上記の反応を3バッチに分けて繰り返した。3バッチ分の反応液を混合し45℃で減圧濃縮し、化合物8を含む濃縮物をカラム精製(石油エーテル/酢酸エチル=4/1)し、減圧濃縮することで黄色のオイルである化合物8(1.05kg)を得た。 Toluene (59.0 L) and compound 7 (0.655 kg) were added to a reaction vessel purged with nitrogen at room temperature and stirred. Next, p-benzoquinone (28.4 g) was added and the internal temperature was heated to 100°C. To the mixed solution heated to an internal temperature of 100°C, first-generation HOVEYDA-GRUBBS catalyst (42.0 g) dissolved in toluene (100 mL) was added dropwise over 20 minutes. The above reaction was repeated in three batches. The reaction liquid from the three batches was mixed and concentrated under reduced pressure at 45°C. The concentrate containing compound 8 was purified by column (petroleum ether/ethyl acetate = 4/1) and concentrated under reduced pressure to obtain compound 8 (1.05 kg) as a yellow oil.
実施例1-8Examples 1-8
化合物9:tert-ブチル2-[メチル-[(2S)-2-[(4Z,7S)-7-(メチルアミノ)-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]アミノ]酢酸の合成Compound 9: Synthesis of tert-butyl 2-[methyl-[(2S)-2-[(4Z,7S)-7-(methylamino)-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]amino]acetic acid

窒素で置換した反応釜に、室温にてトルエン(11.0L)、化合物8(1.05kg)を加えて攪拌した。続いて混合溶液に対して、DBU(223g)を10℃で加え、内温20℃で2時間撹拌した。反応液を10±5℃で氷水(11.0L)に加え撹拌した。混合溶液に酢酸エチル(5.25L)を加え撹拌後、液々分離により水層を排出した。有機層を5%食塩水(5.25L)で洗浄し、有機層を40℃で減圧濃縮し、約1.1Lとなるまで濃縮した。濃縮溶液にトルエン(9.5L)、1Mリン酸二水素カリウム水溶液(6.3L)を加え、20℃で3時間撹拌した。混合溶液をろ過し、トルエン/n-ヘプタン=1/1(11.0L)でろ過物を洗浄した。得られたろ取物にエタノール(31.5L)加え、40℃に加温することで溶解させた。D(-)-酒石酸(215g)を加えて、20℃で3時間撹拌し、析出物を得た。得られた析出物をろ過し、n-ヘプタン(3.15Lx2)で洗浄した。洗浄した析出物を反応釜に移し、1Mリン酸三カリウム水溶液を加え、pH=7-8に調整した。DCM(10.5L)を加え、水層排出後、有機層を回収した。有機層を5%食塩水(5.25L)で洗浄し、有機層を40℃で約0.5Lとなるまで減圧濃縮した。濃縮溶液に対してn-ヘプタン(10.5L)を加えて、析出物を得た。得られた析出物をろ取し、n-ヘプタン(3.15L)で洗浄した。ろ取物を減圧乾燥することで無色の固体である化合物9(449g)を得た。
化合物9のLCMS(ESI):保持時間:1.24分、m/z=498 [M+H]+(LCMS分析条件 method 1)
Toluene (11.0 L) and compound 8 (1.05 kg) were added to a reaction vessel substituted with nitrogen at room temperature and stirred. Next, DBU (223 g) was added to the mixed solution at 10° C., and the mixture was stirred at an internal temperature of 20° C. for 2 hours. The reaction solution was added to ice water (11.0 L) at 10±5° C. and stirred. Ethyl acetate (5.25 L) was added to the mixed solution and stirred, and the aqueous layer was discharged by liquid-liquid separation. The organic layer was washed with 5% saline (5.25 L), and the organic layer was concentrated under reduced pressure at 40° C. until it became about 1.1 L. Toluene (9.5 L) and 1M potassium dihydrogen phosphate aqueous solution (6.3 L) were added to the concentrated solution, and the mixture was stirred at 20° C. for 3 hours. The mixed solution was filtered, and the filtrate was washed with toluene/n-heptane = 1/1 (11.0 L). Ethanol (31.5 L) was added to the obtained filtrate, and the mixture was dissolved by heating to 40° C. D(-)-tartaric acid (215 g) was added and stirred at 20°C for 3 hours to obtain a precipitate. The obtained precipitate was filtered and washed with n-heptane (3.15 L x 2). The washed precipitate was transferred to a reaction vessel, and 1M tripotassium phosphate aqueous solution was added to adjust the pH to 7-8. DCM (10.5 L) was added, and the aqueous layer was discharged and the organic layer was collected. The organic layer was washed with 5% saline (5.25 L), and the organic layer was concentrated under reduced pressure at 40°C to about 0.5 L. n-heptane (10.5 L) was added to the concentrated solution to obtain a precipitate. The obtained precipitate was collected by filtration and washed with n-heptane (3.15 L). The filtered product was dried under reduced pressure to obtain compound 9 (449 g) as a colorless solid.
LCMS (ESI) of compound 9: Retention time: 1.24 minutes, m/z = 498 [M+H] + (LCMS analysis conditions method 1)
実施例1-9Examples 1-9
化合物10:tert-ブチル (2S)-1-[(2S,3S)-2-(ベンジロキシカルボニルアミノ)-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸の合成Compound 10: Synthesis of tert-butyl (2S)-1-[(2S,3S)-2-(benzyloxycarbonylamino)-3-methyl-pentanoyl]pyrrolidine-2-carboxylate
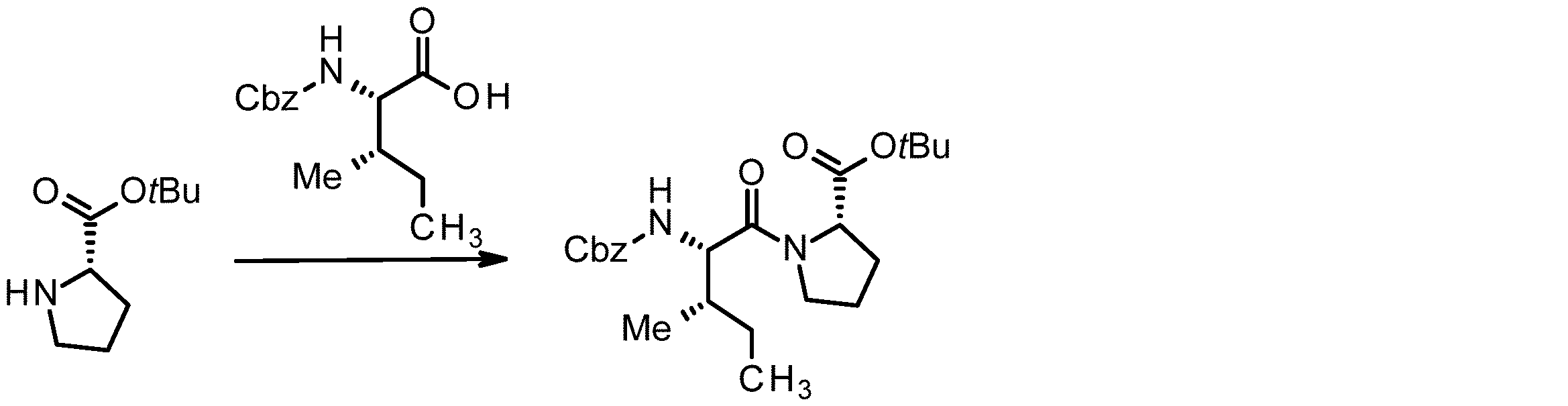
窒素で置換した反応釜に、tert-ブチル(2S)-ピロリジン-2-カルボン酸(18.8g)、(2S,3S)-2-(ベンジロキシカルボニルアミノ)-3-メチル-ペンタン酸(20.0g)、DMF(140mL)を室温で加えて攪拌した。完全溶解を確認後、0℃まで冷却し、DIPEA(52.7mL)を加えた。混合溶液に対して50 wt.%プロピルホスホン酸無水物 酢酸エチル溶液(58.3mL)を0℃で20分かけて加え、0℃で1.5時間撹拌した。混合溶液に対して水(100mL)、酢酸エチル(200mL)をこの順番で加えた。液々分離により水層1と有機層1を分離し、水層1を取り分けた。有機層1を5%硫酸水素カリウム水溶液(100mL)、5%炭酸ナトリウム水溶液(100mL)、10%食塩水(100mL)で洗浄した。取り分けた水層1に対して水(100mL)、酢酸エチル(200mL)を加えて撹拌後、液々分離により有機層2を得た。有機層1と有機層2を合わせて減圧濃縮し、化合物10(33.8g)を得た。
化合物10のLCMS(ESI):保持時間:2.75分、m/z=419 [M+H]+(LCMS分析条件 method 2)
In a reaction vessel purged with nitrogen, tert-butyl (2S)-pyrrolidine-2-carboxylate (18.8 g), (2S,3S)-2-(benzyloxycarbonylamino)-3-methyl-pentanoic acid (20.0 g), and DMF (140 mL) were added at room temperature and stirred. After confirming complete dissolution, the mixture was cooled to 0°C, and DIPEA (52.7 mL) was added. To the mixture, 50 wt.% propylphosphonic anhydride ethyl acetate solution (58.3 mL) was added over 20 minutes at 0°C, and stirred at 0°C for 1.5 hours. To the mixture, water (100 mL) and ethyl acetate (200 mL) were added in this order. Aqueous layer 1 and organic layer 1 were separated by liquid-liquid separation, and aqueous layer 1 was set aside. Organic layer 1 was washed with 5% potassium hydrogen sulfate aqueous solution (100 mL), 5% sodium carbonate aqueous solution (100 mL), and 10% saline solution (100 mL). Water (100 mL) and ethyl acetate (200 mL) were added to the separated aqueous layer 1, and the mixture was stirred, and then separated into liquids to obtain an organic layer 2. The organic layers 1 and 2 were combined and concentrated under reduced pressure to obtain compound 10 (33.8 g).
LCMS (ESI) of compound 10: Retention time: 2.75 minutes, m/z = 419 [M+H] + (LCMS analysis conditions method 2)
実施例1-10Examples 1-10
化合物11:tert-ブチル(2S)-1-[(2S,3S)-2-アミノ-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸の合成Synthesis of Compound 11: tert-butyl (2S)-1-[(2S,3S)-2-amino-3-methyl-pentanoyl]pyrrolidine-2-carboxylate

窒素で置換した反応釜に実施例1-9で得られた化合物10(31.6g)と2-MeTHF(221mL)を加え、10℃に冷却した。混合溶液に対して5% Pd/C(6.32g、50%含水品)を加えた後、トリエチルシラン(60.3mL)を20分かけて加えた。内温15℃で6時間撹拌後、室温でさらに17時間撹拌した。混合溶液をろ過し、ろ液を減圧濃縮し化合物11を得た。化合物11はこれ以上の精製を行うことなく実施例1-11に用いた。
化合物11のLCMS(ESI):保持時間:1.14分、m/z=285 [M+H]+(LCMS分析条件 method 2)
Compound 10 (31.6 g) obtained in Example 1-9 and 2-MeTHF (221 mL) were added to a reaction vessel purged with nitrogen, and cooled to 10°C. 5% Pd/C (6.32 g, 50% water content) was added to the mixed solution, and triethylsilane (60.3 mL) was added over 20 minutes. After stirring at an internal temperature of 15°C for 6 hours, the mixture was stirred at room temperature for an additional 17 hours. The mixed solution was filtered, and the filtrate was concentrated under reduced pressure to obtain compound 11. Compound 11 was used in Example 1-11 without further purification.
LCMS (ESI) of compound 11: Retention time: 1.14 minutes, m/z = 285 [M+H] + (LCMS analysis conditions method 2)
実施例1-11Examples 1-11
化合物12:tert-ブチル(2S)-1-[(2S,3S)-2-[[(2S)-2-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸の合成Synthesis of Compound 12: tert-butyl(2S)-1-[(2S,3S)-2-[[(2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate
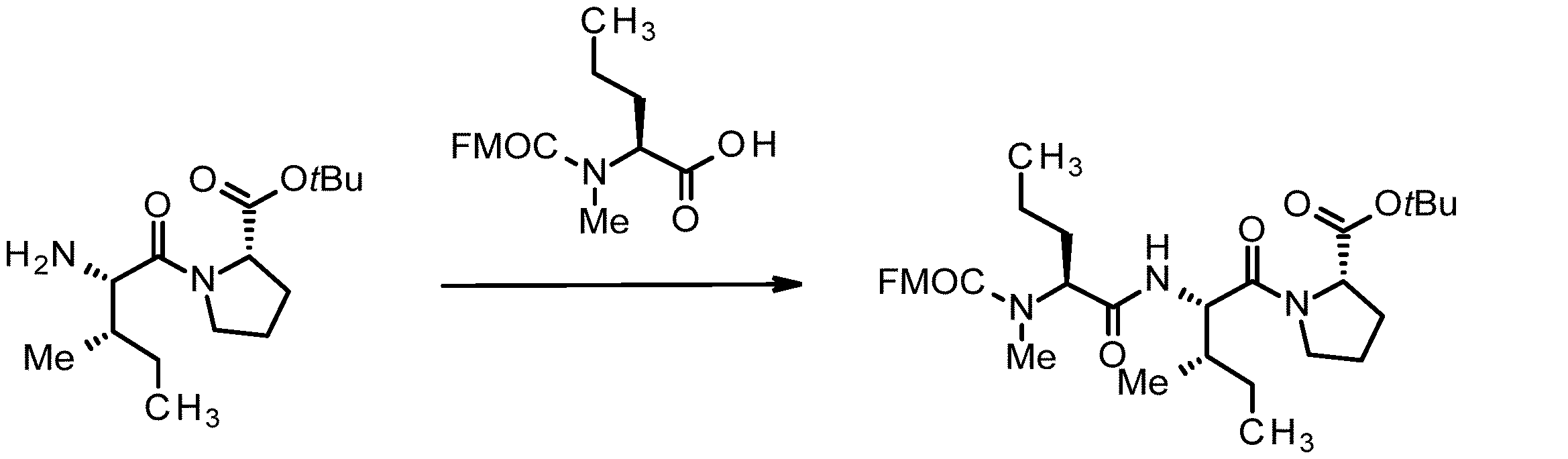
実施例1-10で得られた化合物11に対して、アセトニトリル(221mL)を5℃で加えた後、(2S)-2-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]ペンタン酸(28.0g)、DIPEA(39.6mL)を加えた。混合溶液に対してHATU(34.5g)を2.5℃でゆっくり加えた。2.5℃で1時間撹拌した後、室温で2.5時間撹拌した。混合溶液に対して5%炭酸ナトリウム水溶液(189mL)、水(150mL)をこの順番に加えた。トルエン(80mL)と2-MeTHF(140mL)を加え撹拌後、液々分離した。有機層を5%硫酸水素カリウム水溶液(190mLx2)、10%食塩水(190mLx2)で洗浄した。得られた有機層を減圧濃縮することで化合物12を得た。化合物12はこれ以上の精製を行うことなく実施例1-12に用いた。
化合物12のLCMS(ESI):保持時間:3.43分、m/z=620 [M+H]+(LCMS分析条件 method 2)
Acetonitrile (221 mL) was added to compound 11 obtained in Example 1-10 at 5° C., and then (2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]pentanoic acid (28.0 g) and DIPEA (39.6 mL) were added. HATU (34.5 g) was slowly added to the mixed solution at 2.5° C. After stirring at 2.5° C. for 1 hour, the mixture was stirred at room temperature for 2.5 hours. 5% aqueous sodium carbonate solution (189 mL) and water (150 mL) were added to the mixed solution in this order. Toluene (80 mL) and 2-MeTHF (140 mL) were added and stirred, and then the liquid was separated. The organic layer was washed with 5% aqueous potassium hydrogen sulfate solution (190 mL x 2) and 10% saline (190 mL x 2). The obtained organic layer was concentrated under reduced pressure to obtain compound 12. Compound 12 was used in Example 1-12 without further purification.
LCMS (ESI) of compound 12: Retention time: 3.43 minutes, m/z = 620 [M+H] + (LCMS analysis conditions method 2)
実施例1-12Examples 1-12
化合物13:tert-ブチル(2S)-1-[(2S,3S)-3-メチル-2-[[(2S)-2-(メチルアミノ)ペンタノイル]アミノ]ペンタノイル]ピロリジン-2-酢酸の合成Synthesis of Compound 13: tert-butyl(2S)-1-[(2S,3S)-3-methyl-2-[[(2S)-2-(methylamino)pentanoyl]amino]pentanoyl]pyrrolidine-2-acetic acid

窒素で置換した反応釜に実施例1-11と同様の手法で合成した化合物12(296mg)とトルエン(2.07mL)を加え、撹拌した。混合溶液に対してDBU(0.072mL)を加え30分撹拌した。混合溶液に対してアセトニトリル(1.00mL)を加えた後30分攪拌し、DBU(0.072mL)を加え、さらに30分撹拌した。1N 塩酸(2.00mL)、n-ヘプタン(1.00mL)を加え撹拌後、水層1と有機層1を分離した。有機層1を1N 塩酸(1.00mL)で抽出し、化合物13を含む水層2を得た。水層1と水層2を合わせ、5%炭酸カリウム水溶液(2.00mL)、トルエン(4.00mL)で抽出し、化合物13を含む有機層を分離した。得られた有機層を10%食塩水(2.00mL)で洗浄した後、減圧濃縮することで化合物13(166mg)を得た。
化合物13のLCMS(ESI):保持時間:1.36分、m/z=398 [M+H]+(LCMS分析条件 method 2)
Compound 12 (296 mg) synthesized in the same manner as in Example 1-11 and toluene (2.07 mL) were added to a reaction vessel purged with nitrogen and stirred. DBU (0.072 mL) was added to the mixed solution and stirred for 30 minutes. Acetonitrile (1.00 mL) was added to the mixed solution, which was then stirred for 30 minutes, DBU (0.072 mL) was added, and the solution was further stirred for 30 minutes. 1N hydrochloric acid (2.00 mL) and n-heptane (1.00 mL) were added and stirred, after which aqueous layer 1 and organic layer 1 were separated. Organic layer 1 was extracted with 1N hydrochloric acid (1.00 mL) to obtain aqueous layer 2 containing compound 13. Aqueous layer 1 and aqueous layer 2 were combined and extracted with 5% potassium carbonate aqueous solution (2.00 mL) and toluene (4.00 mL), and the organic layer containing compound 13 was separated. The obtained organic layer was washed with 10% saline (2.00 mL) and then concentrated under reduced pressure to obtain compound 13 (166 mg).
LCMS (ESI) of compound 13: Retention time: 1.36 minutes, m/z = 398 [M+H] + (LCMS analysis conditions method 2)
実施例1-13Examples 1-13
化合物14-レジンの合成Synthesis of Compound 14-Resin
化合物14:(3S)-4-(ジメチルアミノ)-3-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]-4-オキソ-酪酸Compound 14: (3S)-4-(dimethylamino)-3-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]-4-oxo-butyric acid
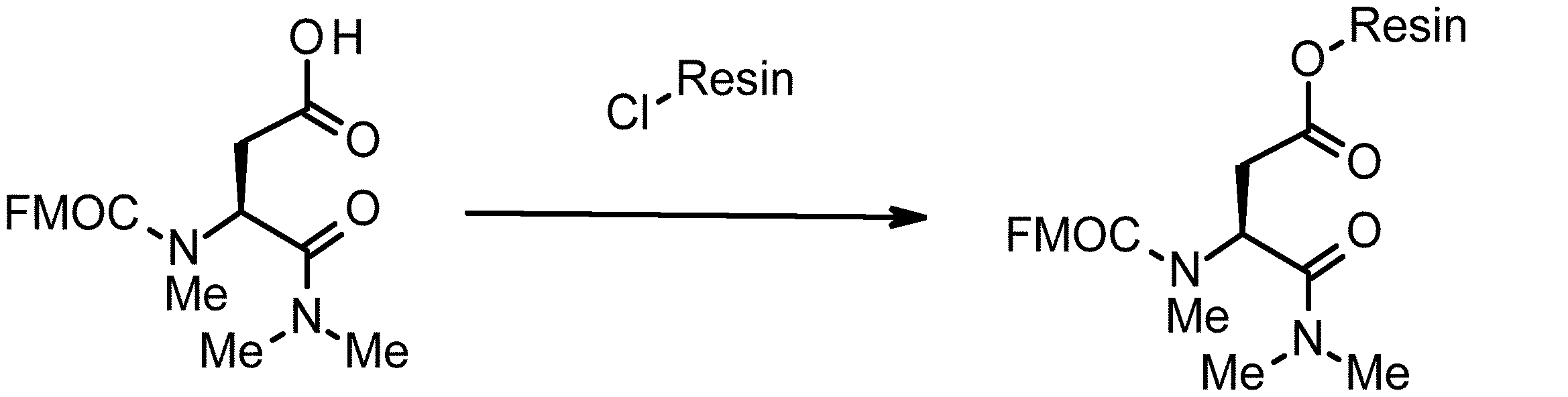
フィルター付きの反応容器(1L)に2-クロロトリチルクロライドレジン(1.12mmol/g、70g, 78.4mmol)とDCM(560mL)を入れ、室温にて30分静置した。DCMを減圧ろ過後、DCM(140mL)に溶解させた(3S)-4-(ジメチルアミノ)-3-[9H-フルオレン-9-イルメトキシカルボニル (メチル)アミノ]-4-オキシ-ブタン酸(23.4g、56.0mmol)を加えDCM(140mL)で洗いこんだ。DIPEA(27.4mL)を反応容器に添加し、45分撹拌後にDCM(140mL)を添加し、室温で105分間撹拌した。反応液を減圧ろ過後、DCM(280mLx2)で洗浄を行った。メタノール(28.0mL)及びDIPEA(14.0mL)のDMF(238mL)溶液を反応容器に添加し、室温で120分間撹拌した。反応液を減圧ろ過後、IPA(280mL)を入れて撹拌した。15分後に反応液を減圧ろ過し、DMF(280mL)を添加し15分撹拌した。反応液を減圧ろ過後、化合物14-レジンを担持量測定分を除いた全量を用いて実施例1-14に進めた。
レジン上のアミノ酸の担持量の算出は以下のとおり行った。得られた化合物14-レジン(4.76mg)を反応容器に入れ、20%Pip/DMF溶液(50mL)を加えて、室温にて1時間振とうした。その溶液の吸光度(301nm)を測定し(Shimadzu、UV-1600PC(セル長1.0cm)を用いて測定)、化合物14-レジンの担持量を0.632mmol/gと算出した。
2-Chlorotrityl chloride resin (1.12 mmol/g, 70 g, 78.4 mmol) and DCM (560 mL) were placed in a filter-equipped reaction vessel (1 L) and left to stand at room temperature for 30 minutes. After the DCM was filtered under reduced pressure, (3S)-4-(dimethylamino)-3-[9H-fluoren-9-ylmethoxycarbonyl (methyl)amino]-4-oxy-butanoic acid (23.4 g, 56.0 mmol) dissolved in DCM (140 mL) was added and washed with DCM (140 mL). DIPEA (27.4 mL) was added to the reaction vessel, and after stirring for 45 minutes, DCM (140 mL) was added and stirred at room temperature for 105 minutes. The reaction solution was filtered under reduced pressure and washed with DCM (280 mL x 2). A solution of methanol (28.0 mL) and DIPEA (14.0 mL) in DMF (238 mL) was added to a reaction vessel and stirred at room temperature for 120 minutes. The reaction solution was filtered under reduced pressure, and then IPA (280 mL) was added and stirred. After 15 minutes, the reaction solution was filtered under reduced pressure, and DMF (280 mL) was added and stirred for 15 minutes. After the reaction solution was filtered under reduced pressure, the entire amount of Compound 14-resin, excluding the amount used for measuring the loading amount, was used to proceed to Example 1-14.
The amount of amino acid supported on the resin was calculated as follows. The obtained compound 14-resin (4.76 mg) was placed in a reaction vessel, and 20% Pip/DMF solution (50 mL) was added and shaken at room temperature for 1 hour. The absorbance (301 nm) of the solution was measured (Shimadzu, UV-1600PC (cell length 1.0 cm)), and the amount of compound 14-resin supported was calculated to be 0.632 mmol/g.
実施例1-14Examples 1-14
化合物15-レジンの合成Synthesis of Compound 15-Resin
化合物15:(3S)-3-[[(2S)-2-シクロペンチル-2-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸Compound 15: (3S)-3-[[(2S)-2-cyclopentyl-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid

フィルター付きの反応容器(1L)に化合物14-レジン(0.632mmol/g)をいれ、20% Pip/DMF溶液(280mL)を加えて室温にて10分間撹拌し、脱Fmoc反応をおこなった。反応液を減圧ろ過し、再度20% Pip/DMF溶液(280mL)を加えて室温にて10分間撹拌し、脱Fmoc反応をおこなった。反応液を減圧ろ過し、レジンをDMF(280mL)で10回洗浄した。得られたレジンに対し、Fmoc-MeGly(cPent)-OH(Cas番号187475-29-2)の伸長反応を実施した。伸長反応はFmoc-MeGly(cPent)-OH(Cas番号187475-29-2、42.5g)、oxyma(7.96g)とDIC(34.9mL)のDMF溶液(280mL)をレジンに加え5分撹拌し、室温にて16時間静置することで実施した。伸長反応の溶液を減圧ろ過し、IPA(280mL)を入れて撹拌した。10分後に反応液を減圧ろ過し、DMF(280mL)を添加し10分撹拌した。反応液を減圧ろ過後、化合物15-レジンを担持量測定分を除いた全量を用いて実施例1-15に進めた。
レジン上のアミノ酸の担持量の算出は以下のとおり行った。得られた化合物15-レジン(4.91mg)を反応容器に入れ、20%Pip/DMF溶液(50mL)を加えて、室温にて1時間振とうした。その溶液の吸光度(301nm)を測定し(Shimadzu、UV-1600PC(セル長1.0cm)を用いて測定)、化合物15-レジンの担持量を0.635mmol/gと算出した。
レジンに担持された化合物15を少量用いてTFE/DCM(1/1)でレジンからの切り出しを行い、LC/MSで構造確認を行った。
化合物15のLCMS(ESI):保持時間:2.53分、m/z=536 [M+H]+(LCMS分析条件 method 2)
Compound 14-resin (0.632 mmol/g) was placed in a reaction vessel (1 L) equipped with a filter, and 20% Pip/DMF solution (280 mL) was added and stirred at room temperature for 10 minutes to carry out the Fmoc-removal reaction. The reaction solution was filtered under reduced pressure, and 20% Pip/DMF solution (280 mL) was added again and stirred at room temperature for 10 minutes to carry out the Fmoc-removal reaction. The reaction solution was filtered under reduced pressure, and the resin was washed 10 times with DMF (280 mL). The obtained resin was subjected to an extension reaction of Fmoc-MeGly(cPent)-OH (Cas number 187475-29-2). The extension reaction was carried out by adding Fmoc-MeGly(cPent)-OH (Cas number 187475-29-2, 42.5 g), oxyma (7.96 g) and DIC (34.9 mL) in a DMF solution (280 mL) to the resin, stirring for 5 minutes, and leaving it at room temperature for 16 hours. The solution of the extension reaction was filtered under reduced pressure, and IPA (280 mL) was added and stirred. After 10 minutes, the reaction solution was filtered under reduced pressure, and DMF (280 mL) was added and stirred for 10 minutes. After the reaction solution was filtered under reduced pressure, the entire amount of compound 15-resin except for the amount loaded for measurement was used to proceed to Example 1-15.
The amount of amino acid supported on the resin was calculated as follows. The obtained compound 15-resin (4.91 mg) was placed in a reaction vessel, and 20% Pip/DMF solution (50 mL) was added and shaken at room temperature for 1 hour. The absorbance (301 nm) of the solution was measured (Shimadzu, UV-1600PC (cell length 1.0 cm)), and the amount of compound 15-resin supported was calculated to be 0.635 mmol/g.
A small amount of compound 15 supported on the resin was used to cleave the compound from the resin with TFE/DCM (1/1), and the structure was confirmed by LC/MS.
LCMS (ESI) of compound 15: Retention time: 2.53 minutes, m/z = 536 [M+H] + (LCMS analysis conditions method 2)
実施例1-15Examples 1-15
化合物16-レジンの合成Synthesis of Compound 16-Resin
化合物16:(3S)-3-[[(2S)-2-シクロペンチル-2-[[1-[9H-フルオレン-9-イルメトキシカルボニル(メチル)アミノ]シクロブタンカルボニル]-メチル-アミノ]アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸Compound 16: (3S)-3-[[(2S)-2-cyclopentyl-2-[[1-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]cyclobutanecarbonyl]-methyl-amino]acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid

フィルター付きの反応容器(1L)に化合物15-レジン(0.635mmol/g)をいれ、20% Pip/DMF溶液(280mL)を加えて室温にて5分間撹拌し、脱Fmoc反応をおこなった。反応液を減圧ろ過後、再度20% Pip/DMF溶液(280mL)を加えて室温にて5分間撹拌し、脱Fmoc反応をおこなった。反応液を減圧ろ過後、レジンをDMF(280mL)で10回洗浄した。得られたレジンに対し、Fmoc-MecVal-OH(Cas番号1700368-07-5)の伸長反応を実施した。伸長反応はFmoc-MecVal-OH(Cas番号1700368-07-5、39.4g)とoxyma(7.96g)のDMF溶液(280 mL)を加えた後、DIC(34.9mL)を加えて5分撹拌後、94時間静置することで実施した。伸長反応の溶液を減圧ろ過後、レジンをDMF(280mL)、IPA(280mL)、DMF(280mL)それぞれで洗浄を実施した。洗浄液を減圧ろ過後、得られた化合物16-レジンを担持量測定分を除いた全量を用いて実施例1-17に進めた。
レジン上のアミノ酸の担持量の算出は以下のとおり行った。得られた化合物16-レジン(5.24mg)を反応容器に入れ、20%Pip/DMF溶液(50mL)を加えて、室温にて1時間振とうした。その溶液の吸光度(301nm)を測定し(Shimadzu、UV-1600PC(セル長1.0cm)を用いて測定)、化合物16-レジンの担持量を0.513mmol/gと算出した。
レジンに担持された化合物16の少量を用いてTFE/DCM(1/1)でレジンからの切り出しを行い、LC/MSで構造確認を行った。
化合物16のLCMS(ESI):保持時間:2.75分、m/z=647 [M+H]+(LCMS分析条件 method 2)
Compound 15-resin (0.635 mmol/g) was placed in a reaction vessel (1 L) equipped with a filter, and 20% Pip/DMF solution (280 mL) was added and stirred at room temperature for 5 minutes to carry out the Fmoc-removal reaction. After the reaction solution was filtered under reduced pressure, 20% Pip/DMF solution (280 mL) was added again and stirred at room temperature for 5 minutes to carry out the Fmoc-removal reaction. After the reaction solution was filtered under reduced pressure, the resin was washed 10 times with DMF (280 mL). The obtained resin was subjected to an extension reaction of Fmoc-MecVal-OH (Cas number 1700368-07-5). The extension reaction was carried out by adding Fmoc-MecVal-OH (Cas No. 1700368-07-5, 39.4 g) and oxyma (7.96 g) in DMF solution (280 mL), adding DIC (34.9 mL), stirring for 5 minutes, and then leaving it to stand for 94 hours. The solution of the extension reaction was filtered under reduced pressure, and the resin was washed with DMF (280 mL), IPA (280 mL), and DMF (280 mL). The washing solution was filtered under reduced pressure, and the obtained compound 16-resin was used in its entirety except for the amount measured for the loading amount, and then proceeded to Example 1-17.
The amount of amino acid supported on the resin was calculated as follows. The obtained compound 16-resin (5.24 mg) was placed in a reaction vessel, and 20% Pip/DMF solution (50 mL) was added and shaken at room temperature for 1 hour. The absorbance (301 nm) of the solution was measured (Shimadzu, UV-1600PC (cell length 1.0 cm)), and the amount of compound 16-resin supported was calculated to be 0.513 mmol/g.
A small amount of compound 16 supported on the resin was cleaved from the resin with TFE/DCM (1/1), and the structure was confirmed by LC/MS.
LCMS (ESI) of compound 16: Retention time: 2.75 minutes, m/z = 647 [M+H] + (LCMS analysis conditions method 2)
実施例1-16Examples 1-16
化合物17:9H-フルオレン-9-イルメチル (2S,4R)-2-クロロカルボニル-4-エトキシ-ピロリジン-1-カルボン酸の合成Compound 17: Synthesis of 9H-fluoren-9-ylmethyl (2S,4R)-2-chlorocarbonyl-4-ethoxy-pyrrolidine-1-carboxylic acid

窒素で置換した反応容器に、(2S,4R)-4-エトキシ-1-(9H-フルオレン-9-イルメトキシカルボニル)ピロリジン-2-カルボン酸(Cas番号1446478-31-4、42.7g)、トルエン(128mL)、塩化チオニル(12.3mL)を室温にて順次加えた。外温を60℃で0.5時間撹拌したのち、外温を55℃で3時間撹拌した。反応混合物を一部サンプリングしてMeOHに希釈し、5分静置し対応する化合物18に変換することでHPLC分析により化合物17への反応転換率が99.9%で進行していることを確認した(反応転換率の算出式1)。反応容器の外温を40℃に設定し、減圧濃縮した。減圧濃縮後、DCM(214mL)を加えて減圧濃縮する操作を2回繰り返した。得られた濃縮液を終夜減圧乾燥し、化合物17を含む濃縮乾固物(45.5g、95%収率)を得た。 (2S,4R)-4-ethoxy-1-(9H-fluoren-9-ylmethoxycarbonyl)pyrrolidine-2-carboxylic acid (Cas number 1446478-31-4, 42.7 g), toluene (128 mL), and thionyl chloride (12.3 mL) were added in sequence at room temperature to a reaction vessel purged with nitrogen. After stirring at an external temperature of 60°C for 0.5 hours, the mixture was stirred at an external temperature of 55°C for 3 hours. A portion of the reaction mixture was sampled and diluted with MeOH, left to stand for 5 minutes, and converted to the corresponding compound 18. By HPLC analysis, it was confirmed that the reaction conversion rate to compound 17 had progressed at 99.9% (reaction conversion rate calculation formula 1). The external temperature of the reaction vessel was set to 40°C, and the mixture was concentrated under reduced pressure. After concentration under reduced pressure, DCM (214 mL) was added and the operation of concentrating under reduced pressure was repeated twice. The resulting concentrate was dried under reduced pressure overnight to obtain a concentrated, dried product containing compound 17 (45.5 g, 95% yield).
化合物18:O1-(9H-フルオレン-9-イルメチル) O2-メチル (2S,4R)-4-エトキシピロリジン-1,2-ジカルボン酸Compound 18: O1-(9H-fluoren-9-ylmethyl) O2-methyl (2S,4R)-4-ethoxypyrrolidine-1,2-dicarboxylate

化合物18のLCMS(ESI):保持時間:2.68分、m/z=396 [M+H]+(LCMS分析条件 method 2) LCMS (ESI) of compound 18: Retention time: 2.68 minutes, m/z = 396 [M+H] + (LCMS analysis conditions method 2)
実施例1-17Examples 1-17
化合物19-レジンの合成Synthesis of Compound 19-Resin
化合物19:(3S)-3-[[(2S)-2-シクロペンチル-2-[[1-[[(2S,4R)-4-エトキシ-1-(9H-フルオレン-9-イルメトキシカルボニル)ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸Compound 19: (3S)-3-[[(2S)-2-cyclopentyl-2-[[1-[[(2S,4R)-4-ethoxy-1-(9H-fluoren-9-ylmethoxycarbonyl)pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid
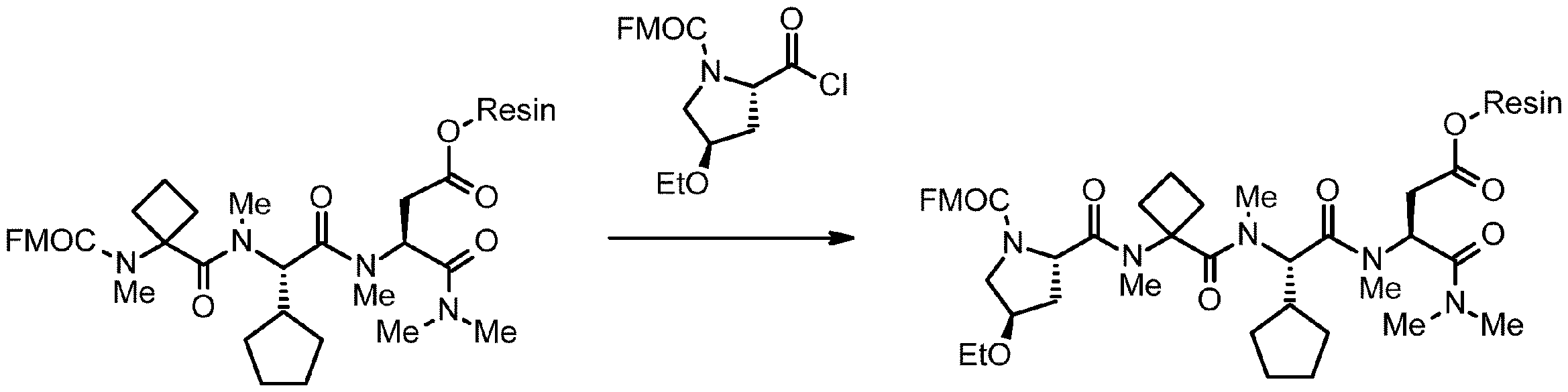
フィルター付きの反応容器(1L)に化合物16-レジン(0.513mmol/g)をいれ、DMF(280mL)での15分間の膨潤と溶液排出を2回行った。膨潤させたレジンに対して、20% Pip/DMF溶液(280mL)を加えて室温にて15分間撹拌し、脱Fmoc反応をおこなった。反応液を減圧ろ過後、再度20% Pip/DMF溶液(280mL)を加えて室温にて15分間撹拌し、脱Fmoc反応をおこなった。反応液を減圧ろ過後、レジンをDCM(280mL)で13回洗浄した。得られたレジンに対し、化合物17の伸長反応を実施した。伸長反応は実施例1-16で得た化合物17(46.0g)のDCM溶液(280mL)とCollidine(74.0mL)を順次加え5分撹拌し、室温にて4時間静置することで実施した。伸長反応の溶液を減圧ろ過後、レジンをDCM(280mL)で洗浄し、得られた化合物19-レジンを担持量測定分を除いた全量を用いて実施例1-18に進めた。
レジン上のアミノ酸の担持量の算出は以下のとおり行った。得られた化合物17-レジン(7.27mg)を反応容器に入れ、20%Pip/DMF溶液(50mL)を加えて、室温にて1時間振とうした。その溶液の吸光度(301nm)を測定し(Shimadzu、UV-1600PC(セル長1.0cm)化合物19-レジンの担持量を0.473mmol/gと算出した。
レジンに担持された化合物19を少量用いてTFE/DCM(1/1)でレジンからの切り出しを行い、LC/MSで構造確認を行った。
化合物17のLCMS(ESI):保持時間:2.68分、m/z=788 [M+H]+(LCMS分析条件 method 2)
Compound 16-resin (0.513 mmol/g) was placed in a reaction vessel (1 L) with a filter, and swelling with DMF (280 mL) for 15 minutes and discharging the solution were performed twice. 20% Pip/DMF solution (280 mL) was added to the swollen resin and stirred at room temperature for 15 minutes to perform the Fmoc-removal reaction. After the reaction solution was filtered under reduced pressure, 20% Pip/DMF solution (280 mL) was added again and stirred at room temperature for 15 minutes to perform the Fmoc-removal reaction. After the reaction solution was filtered under reduced pressure, the resin was washed 13 times with DCM (280 mL). The obtained resin was subjected to an elongation reaction of compound 17. The elongation reaction was performed by sequentially adding a DCM solution (280 mL) of compound 17 (46.0 g) obtained in Example 1-16 and Collidine (74.0 mL) and stirring for 5 minutes, and leaving it at room temperature for 4 hours. After the extension reaction solution was filtered under reduced pressure, the resin was washed with DCM (280 mL), and the entire amount of the obtained Compound 19-resin, excluding the amount used for measuring the loading amount, was used in Example 1-18.
The amount of amino acid supported on the resin was calculated as follows. The obtained compound 17-resin (7.27 mg) was placed in a reaction vessel, and 20% Pip/DMF solution (50 mL) was added and shaken at room temperature for 1 hour. The absorbance (301 nm) of the solution was measured (Shimadzu, UV-1600PC (cell length 1.0 cm) and the amount of compound 19-resin supported was calculated to be 0.473 mmol/g.
A small amount of compound 19 supported on the resin was used to cleave the compound from the resin with TFE/DCM (1/1), and the structure was confirmed by LC/MS.
LCMS (ESI) of compound 17: Retention time: 2.68 minutes, m/z = 788 [M+H] + (LCMS analysis conditions method 2)
実施例1-18Examples 1-18
化合物20-レジンの合成Synthesis of Compound 20-Resin
化合物20:(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸Compound 20: (3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid
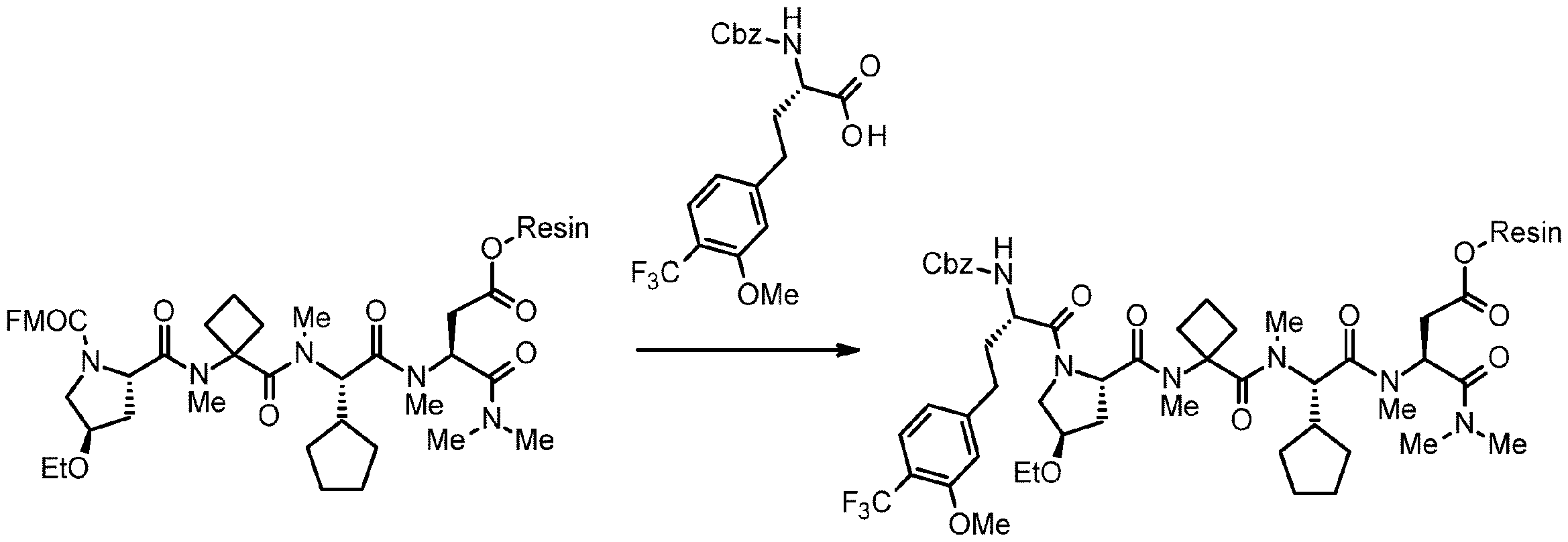
フィルター付きの反応容器(1L)に化合物19-レジン(0.473mmol/g)をいれ、DMF(280mL)での15分間の膨潤と溶液排出を2回行った。膨潤させたレジンに対して、20% Pip/DMF溶液(280mL)を加えて室温にて15分間振とうし、脱Fmoc反応をおこなった。反応液を減圧ろ過後、再度20% Pip/DMF溶液(280mL)を加えて室温にて15分間振とうし、脱Fmoc反応をおこなった。反応液を減圧ろ過後、レジンをDMF(280mL)で13回洗浄した。得られたレジンに対し、Cbz-Hph(4-CF3-3-OMe)-OHの伸長反応を実施した。伸長反応はCbz-Hph(4-CF3-3-OMe)-OH(46.1g)とoxyma(7.96g)のDMF溶液(280mL)とDIC(34.9mL)をレジンに加え、5分撹拌後3.5時間静置することで実施した。伸長反応の溶液を減圧ろ過後、レジンをDMF(280mL)で2回洗浄した。IPA(280mL)を加えた後、15分間撹拌した。溶液を排出したあとDCM(280mL)を加え、15分間撹拌した。IPA(280mL)とDCM(280mL)の洗浄と溶液排出を再度繰り返した。IPA(280mLx3)での洗浄後、レジンを減圧乾燥した。減圧乾燥後、得られた化合物20-レジンは122gだった。
レジンに担持された化合物20を少量用いてTFE/DCM(1/1)でレジンからの切り出しを行い、LC/MSで構造確認を行った。
化合物20のLCMS(ESI):保持時間:2.91分、m/z=960 [M+H]+(LCMS分析条件 method 2)
Compound 19-resin (0.473 mmol/g) was placed in a reaction vessel (1 L) equipped with a filter, and swelling with DMF (280 mL) for 15 minutes and discharging the solution were performed twice. 20% Pip/DMF solution (280 mL) was added to the swollen resin, and the mixture was shaken at room temperature for 15 minutes to perform the Fmoc-removal reaction. After the reaction solution was filtered under reduced pressure, 20% Pip/DMF solution (280 mL) was added again, and the mixture was shaken at room temperature for 15 minutes to perform the Fmoc-removal reaction. After the reaction solution was filtered under reduced pressure, the resin was washed 13 times with DMF (280 mL). The obtained resin was subjected to the extension reaction of Cbz-Hph(4-CF3-3-OMe)-OH. The extension reaction was carried out by adding a DMF solution (280 mL) of Cbz-Hph(4-CF3-3-OMe)-OH (46.1 g) and oxyma (7.96 g) and DIC (34.9 mL) to the resin, stirring for 5 minutes, and then leaving it to stand for 3.5 hours. After the extension reaction solution was filtered under reduced pressure, the resin was washed twice with DMF (280 mL). IPA (280 mL) was added and stirred for 15 minutes. After draining the solution, DCM (280 mL) was added and stirred for 15 minutes. Washing with IPA (280 mL) and DCM (280 mL) and draining the solution were repeated again. After washing with IPA (280 mL x 3), the resin was dried under reduced pressure. After drying under reduced pressure, the compound 20-resin obtained was 122 g.
A small amount of compound 20 supported on the resin was used to cleave the compound from the resin using TFE/DCM (1/1), and the structure was confirmed by LC/MS.
LCMS (ESI) of compound 20: Retention time: 2.91 minutes, m/z = 960 [M+H] + (LCMS analysis conditions method 2)
実施例1-19Examples 1-19
化合物20の合成Synthesis of Compound 20
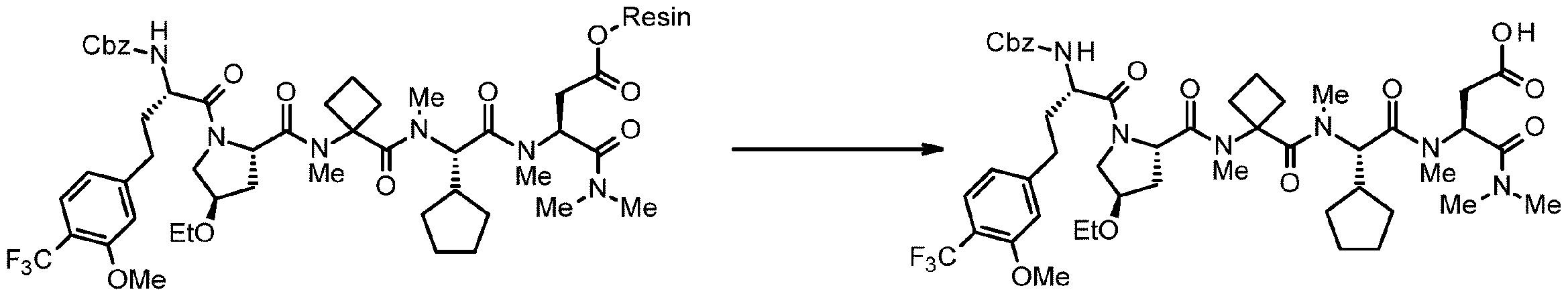
窒素で置換した反応釜に、実施例1-18で得られた化合物20-レジン(122g)と同様の手法で得られた化合物20-レジン(60.0g)、2-MeTHF(1.10L)を室温にて順次加えた。反応釜の外温を0℃に設定し、ヘキサメチルジシラザン(77.3mL)を加えた。20分撹拌後、トリフルオロメタンスルホン酸トリメチルシリル(50.0mL)を内温7℃をこえないように添加した。添加終了20分後に、反応混合物を減圧ろ過し、残渣を2-MeTHF(364mLx3)で洗浄した。ろ液を5%リン酸水素二ナトリウム水溶液(1.10L)、5%硫酸水素カリウム水溶液(1.10L)、5%塩化ナトリウム水溶液(1.10L)で洗浄した。得られた有機層を減圧濃縮し濃縮乾固物を77.6g得た。得られた濃縮乾固物の一部と3,5-ビス(トリフルオロメチル)安息香酸をCDCl3に溶解させ、qNMR分析に付した。含量補正後の化合物20は69.0g含まれていると算出された。
化合物20のLCMS(ESI):保持時間:3.90分、m/z=960 [M+H]+(LCMS分析条件 method 3)
Compound 20-resin (60.0 g) obtained by the same method as compound 20-resin (122 g) obtained in Example 1-18 and 2-MeTHF (1.10 L) were added in sequence at room temperature to a reaction vessel replaced with nitrogen. The external temperature of the reaction vessel was set to 0°C, and hexamethyldisilazane (77.3 mL) was added. After stirring for 20 minutes, trimethylsilyl trifluoromethanesulfonate (50.0 mL) was added so that the internal temperature did not exceed 7°C. 20 minutes after the addition was completed, the reaction mixture was filtered under reduced pressure, and the residue was washed with 2-MeTHF (364 mL x 3). The filtrate was washed with a 5% aqueous solution of disodium hydrogen phosphate (1.10 L), a 5% aqueous solution of potassium hydrogen sulfate (1.10 L), and a 5% aqueous solution of sodium chloride (1.10 L). The obtained organic layer was concentrated under reduced pressure to obtain 77.6 g of a concentrated solid to dryness. A part of the obtained concentrated and dried product and 3,5-bis(trifluoromethyl)benzoic acid were dissolved in CDCl 3 and subjected to qNMR analysis. After correcting for the content, it was calculated that the compound 20 contained was 69.0 g.
LCMS (ESI) of compound 20: Retention time: 3.90 minutes, m/z = 960 [M+H] + (LCMS analysis conditions method 3)
実施例1-20Examples 1-20
化合物21:tert-ブチル (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸の合成Compound 21: Synthesis of tert-butyl (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylic acid

反応釜に化合物20(80.7g)を含む濃縮乾固物と化合物13(45.8g)を加えた。窒素置換した後に、2-MeTHF(484mL)、DIPEA(64.5mL)及びDMF(121mL)を室温で加え撹拌した。完全溶解を確認後、HATU(48.0g)を室温で加えた。1時間後、反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が98%以上であることを確認した(反応転換率の算出式1)。反応釜の外温を5℃に設定し、反応釜に2.5%アンモニア水溶液(484mL)を加えた。分液操作により水層を排出したのち、有機層に対して10%硫酸水素ナトリウム水溶液(484mL)を加えた。分液操作後に液々分離し、水層を排出した。得られた有機層を5%炭酸ナトリウム水溶液(484mL)、5%食塩水(484mL)でそれぞれ洗浄した。得られた有機層を外温40℃に設定し減圧濃縮した。得られた濃縮物に2-MeTHF(161mLx2)を加えて減圧濃縮を繰り返し、濃縮乾固物を134g得た。濃縮乾固物の収率を算出せず、実施例1-21に用いた。
化合物21のLCMS(ESI):保持時間:5.02分、m/z=1340 [M+H]+(LCMS分析条件 method 3)
The concentrated dry matter containing compound 20 (80.7 g) and compound 13 (45.8 g) were added to the reaction vessel. After replacing with nitrogen, 2-MeTHF (484 mL), DIPEA (64.5 mL) and DMF (121 mL) were added at room temperature and stirred. After confirming complete dissolution, HATU (48.0 g) was added at room temperature. After 1 hour, the reaction mixture was sampled and sample preparation (sample preparation method 2) was performed, and it was confirmed that the reaction conversion rate was 98% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). The external temperature of the reaction vessel was set to 5°C, and 2.5% aqueous ammonia solution (484 mL) was added to the reaction vessel. After the aqueous layer was discharged by liquid separation, 10% aqueous sodium hydrogen sulfate solution (484 mL) was added to the organic layer. After liquid separation, the liquid was separated and the aqueous layer was discharged. The obtained organic layer was washed with 5% aqueous sodium carbonate solution (484 mL) and 5% saline solution (484 mL), respectively. The obtained organic layer was concentrated under reduced pressure with the external temperature set to 40° C. 2-MeTHF (161 mL×2) was added to the obtained concentrate, and the concentration under reduced pressure was repeated to obtain 134 g of a concentrated, dried product. The yield of the concentrated, dried product was not calculated, and was used in Example 1-21.
LCMS (ESI) of compound 21: Retention time: 5.02 minutes, m/z = 1340 [M+H] + (LCMS analysis conditions method 3)
実施例1-21Example 1-21
化合物22:(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチルアセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボン酸の合成Synthesis of Compound 22: (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentylacetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylic acid
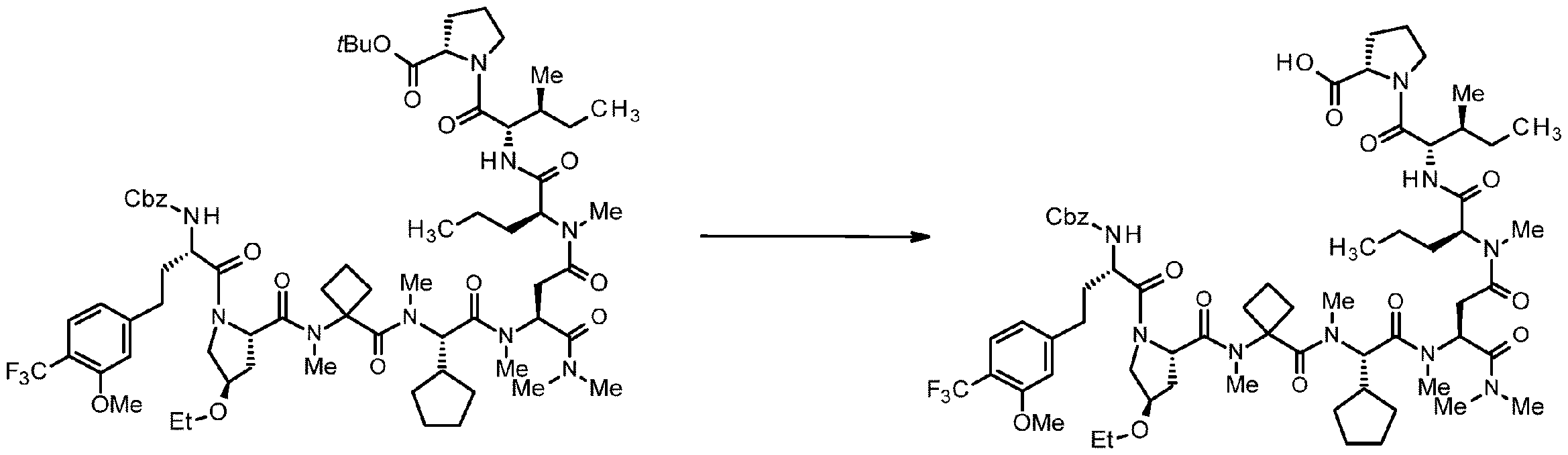
窒素で置換した反応釜に、実施例1-20で得られた化合物19の濃縮乾固物(111g)を含む濃縮乾固物、2-MeTHF(666mL)を順次加え室温で撹拌した。溶解を確認後、外温5℃に冷却しヘキサメチルジシラザン(69.5mL)を加えた後、トリフルオロメタンスルホン酸トリメチルシリル(44.9mL)を内温15℃をこえないように添加した。添加終了後、外温10℃まで昇温し、1時間20分攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により化合物22への反応転換率が99%であることを確認した(反応転換率の算出式1)。外温0℃にて、5%炭酸ナトリウム水溶液(555mL)と5%食塩水(333mL)の混合溶液を内温25℃をこえないように加えた。添加終了後、分液操作にて水層を排出した。得られた有機層を10%硫酸水素ナトリウム水溶液(555mL)、5%炭酸ナトリウム水溶液(555mL)、5%食塩水(555mL)で洗浄した。得られた有機層を外温40℃減圧濃縮した。得られた濃縮物に2-MeTHF(222mLx2)を加えて減圧濃縮を繰り返し、濃縮乾固物を128g得た。得られた濃縮乾固物と3,5-ビス(トリフルオロメチル)安息香酸をCDCl3に溶解させ、qNMR分析に付した結果、96.1g含まれていると算出された。
化合物22のLCMS(ESI):保持時間:4.24分、m/z=1283 [M+H]+(LCMS分析条件 method 3)
In a reaction vessel purged with nitrogen, the concentrated dry product including the concentrated dry product of compound 19 obtained in Example 1-20 (111 g) and 2-MeTHF (666 mL) were added in sequence and stirred at room temperature. After confirming dissolution, the mixture was cooled to an external temperature of 5°C, hexamethyldisilazane (69.5 mL) was added, and trimethylsilyl trifluoromethanesulfonate (44.9 mL) was added so that the internal temperature did not exceed 15°C. After the addition was completed, the external temperature was raised to 10°C and stirred for 1 hour and 20 minutes. The reaction mixture was sampled and sample preparation (sample preparation method 2) was performed, and it was confirmed by HPLC analysis that the reaction conversion rate to compound 22 was 99% (calculation formula 1 for reaction conversion rate). At an external temperature of 0°C, a mixed solution of 5% aqueous sodium carbonate solution (555 mL) and 5% saline (333 mL) was added so that the internal temperature did not exceed 25°C. After the addition was completed, the aqueous layer was discharged by liquid separation. The obtained organic layer was washed with a 10% aqueous sodium hydrogen sulfate solution (555 mL), a 5% aqueous sodium carbonate solution (555 mL), and a 5% saline solution (555 mL). The obtained organic layer was concentrated under reduced pressure at an external temperature of 40°C. 2-MeTHF (222 mL x 2) was added to the obtained concentrate, and the concentration under reduced pressure was repeated to obtain 128 g of a concentrated, dried product. The obtained concentrated, dried product and 3,5-bis(trifluoromethyl)benzoic acid were dissolved in CDCl 3 and subjected to qNMR analysis, and it was calculated that the amount contained was 96.1 g.
LCMS (ESI) of compound 22: Retention time: 4.24 minutes, m/z = 1283 [M+H] + (LCMS analysis conditions method 3)
実施例1-22Example 1-22
化合物23:tert-ブチル2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジロキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボニル]-メチル-アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸Compound 23: tert-butyl 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanoyl 2-(4-(trifluoromethyl)phenyl)propanoyl)-methyl-amino]-2-cyclopentyl-acetyl-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl-methyl-amino]pentanoyl-amino]-3-methyl-pentanoyl]pyrrolidine-2-carbonyl-methyl-amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetic acid

反応釜に実施例1-21で得られた化合物22(94.0g)を含む濃縮乾固物と化合物9(40.1g)を加え窒素置換した後に、2-MeTHF(564mL)、DMF(141mL)を加え室温で撹拌した。DIPEA(56.2mL)を加え、外温43℃で1時間撹拌した。外温33℃に冷却後、反応釜にHATU(41.8g)を加えた。1時間後、室温に冷却し6時間撹拌した。反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が98%以上であることを確認した(反応転換率の算出式1)。反応釜に2.8%アンモニア水溶液(564mL)を室温で加えた。分液操作により水層を排出したのち、有機層に対して10%硫酸水素ナトリウム水溶液(564mL)を加え撹拌した。分液操作後に液々分離し、水層を排出した。得られた有機層を5%炭酸ナトリウム水溶液(564mL)、5%食塩水(564mL)で順次洗浄した。得られた有機層を外温40℃で減圧濃縮し157gの濃縮乾固物を得た。得られた化合物23を含む濃縮乾固物は3,5-ビス(トリフルオロメチル)安息香酸をCDCl3に溶解させ、qNMR分析に付した結果、127.8g(98.9%収率)含まれていると算出された。
化合物23のLCMS(ESI):保持時間:5.34分、m/z=1763 [M+H]+(LCMS分析条件 method 3)
The concentrated dry product containing compound 22 (94.0 g) obtained in Example 1-21 and compound 9 (40.1 g) were added to the reaction vessel, and the mixture was replaced with nitrogen. Then, 2-MeTHF (564 mL) and DMF (141 mL) were added and stirred at room temperature. DIPEA (56.2 mL) was added and stirred at an external temperature of 43 ° C for 1 hour. After cooling to an external temperature of 33 ° C, HATU (41.8 g) was added to the reaction vessel. After 1 hour, the reaction vessel was cooled to room temperature and stirred for 6 hours. The reaction mixture was sampled and sample preparation (sample preparation method 2) was performed, and the reaction conversion rate was confirmed to be 98% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). A 2.8% aqueous ammonia solution (564 mL) was added to the reaction vessel at room temperature. After the aqueous layer was discharged by liquid separation, a 10% aqueous sodium hydrogen sulfate solution (564 mL) was added to the organic layer and stirred. After liquid separation, the liquid was separated and the aqueous layer was discharged. The obtained organic layer was washed successively with a 5% aqueous sodium carbonate solution (564 mL) and a 5% saline solution (564 mL). The obtained organic layer was concentrated under reduced pressure at an external temperature of 40° C. to obtain 157 g of a concentrated, dried product. The concentrated, dried product containing the obtained compound 23 was dissolved in 3,5-bis(trifluoromethyl)benzoic acid in CDCl 3 and subjected to qNMR analysis, and it was calculated that the compound contained 127.8 g (98.9% yield).
LCMS (ESI) of compound 23: Retention time: 5.34 minutes, m/z = 1763 [M+H] + (LCMS analysis conditions method 3)
実施例1-23Example 1-23
化合物24:2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-アミノ-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボニル]-メチル-アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]-メチル-アミノ]酢酸の合成Compound 24: 2-[[(2S)-2-[(4Z,7S)-7-[[(2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl- Synthesis of amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carbonyl]-methyl-amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanoyl]-methyl-amino]acetic acid
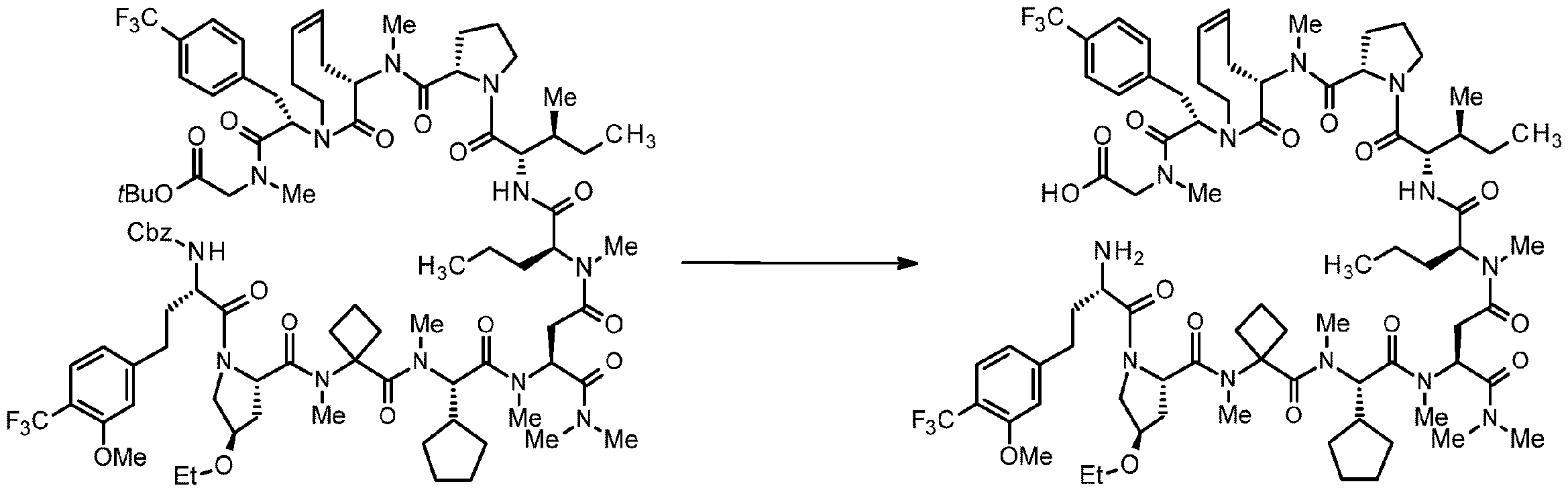
反応釜に化合物23(90.8g)を含む濃縮乾固物とL-システイン(6.3g)、2-MeTHF(363mL)を順次加えた。反応釜を窒素で置換後、外温を20℃に設定した。撹拌しながらヘキサメチルジシラザン(119mL)を加えた。続いてトリフルオロメタンスルホン酸トリメチルシリル(93.5mL)を20分かけて添加した。25分撹拌後、内温50℃になるように昇温した。内温50℃で8時間撹拌した後、反応釜を室温まで冷却した。室温で終夜保管後、再度内温50℃になるように温調を開始し、50℃で4時間撹拌した。外温0℃に冷却後、5%炭酸ナトリウム水溶液(272mL)を内温40℃をこえないようにゆっくり滴下した。その後、分液操作により化合物24を含む水層を取り置いた。取り置いた水層に2-MeTHF(727mL)、MeCN(182mL)及び10%硫酸水素ナトリウム(545mL)を加え、分液操作を行った。水層を排出後、有機層を5%リン酸水素二ナトリウム水溶液(545mLx2)、10%塩化ナトリウム水溶液(273mLx2)で洗浄した。得られた有機層を外温40℃に設定し減圧濃縮した。得られた濃縮物に2-MeTHF(182mLx2)を加えて減圧濃縮を繰り返し、濃縮乾固物を115g得た。濃縮乾固物の化合物24と3,5-ビス(トリフルオロメチル)安息香酸をDMSO‐d6に溶解させ、qNMR分析に付した結果、68.2g(83.9%収率)含まれていると算出された。
化合物24のLCMS(ESI):保持時間:4.37分、m/z=1572 [M+H]+(LCMS分析条件 method 4)
The concentrated dry solid containing compound 23 (90.8 g), L-cysteine (6.3 g), and 2-MeTHF (363 mL) were added in sequence to the reaction vessel. After replacing the reaction vessel with nitrogen, the external temperature was set to 20 ° C. Hexamethyldisilazane (119 mL) was added while stirring. Then, trimethylsilyl trifluoromethanesulfonate (93.5 mL) was added over 20 minutes. After stirring for 25 minutes, the temperature was raised to 50 ° C. After stirring for 8 hours at an internal temperature of 50 ° C, the reaction vessel was cooled to room temperature. After storing at room temperature overnight, temperature control was started again so that the internal temperature was 50 ° C, and stirring was continued at 50 ° C for 4 hours. After cooling to an external temperature of 0 ° C, 5% aqueous sodium carbonate solution (272 mL) was slowly added dropwise so that the internal temperature did not exceed 40 ° C. Then, the aqueous layer containing compound 24 was set aside by a liquid separation operation. 2-MeTHF (727 mL), MeCN (182 mL) and 10% sodium hydrogen sulfate (545 mL) were added to the reserved aqueous layer, and a separation operation was performed. After discharging the aqueous layer, the organic layer was washed with a 5% aqueous disodium hydrogen phosphate solution (545 mL x 2) and a 10% aqueous sodium chloride solution (273 mL x 2). The obtained organic layer was concentrated under reduced pressure with an external temperature of 40°C. 2-MeTHF (182 mL x 2) was added to the obtained concentrate, and the concentration under reduced pressure was repeated to obtain 115 g of a concentrated, dried product. Compound 24 and 3,5-bis(trifluoromethyl)benzoic acid from the concentrated, dried product were dissolved in DMSO-d 6 and subjected to qNMR analysis, and it was calculated that 68.2 g (83.9% yield) was contained.
LCMS (ESI) of compound 24: Retention time: 4.37 minutes, m/z = 1572 [M+H] + (LCMS analysis conditions method 4)
Cbz基の脱保護反応は一般的に、パラジウム/炭素に例示される金属触媒の存在下、接触水素還元条件により実施される。しかしながら、分子内にオレフィンを有する化合物23のCbz基の脱保護反応において、従来法である接触水素還元の条件を適用すると、分子内のオレフィンが還元されるという課題があった。これに対して本発明者は、実施例1-23に記載した条件、すなわちTMSOTf/HMDS条件により、分子内オレフィンを還元することなくCbz基の脱保護が可能である方法を見出した。 The deprotection reaction of the Cbz group is generally carried out under catalytic hydrogen reduction conditions in the presence of a metal catalyst such as palladium/carbon. However, when the conventional catalytic hydrogen reduction conditions are applied to the deprotection reaction of the Cbz group of compound 23, which has an olefin in the molecule, there is a problem that the olefin in the molecule is reduced. In response to this, the present inventors have discovered a method that makes it possible to deprotect the Cbz group without reducing the olefin in the molecule by using the conditions described in Example 1-23, i.e., the TMSOTf/HMDS conditions.
実施例1-24-1Example 1-24-1
化合物1:(1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-シクロペンチル-28-エトキシ-32-[2-[3-メトキシ-4-(トリフルオロメチル)フェニル]エチル]-N,N,2,14,18,21,24,36-オクタメチル-10-[(1S)-1-メチルプロピル]-3,9,12,15,19,22,25,31,34,37,45-ウンデカオキソ-13-プロピル-38-[[4-(トリフルオロメチル)フェニル]メチル]スピロ[2,8,11,14,18,21,24,30,33,36,39-ウンデカザテトラシクロ[37.5.1.0Compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.5.1.0
4,84,8
.0.0
26,3026,30
]ペンタテトラコンタ-42-エン-23,1'-シクロブタン]-17-カルボキサミドの合成(環化位置B)Synthesis of ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide (cyclization position B)
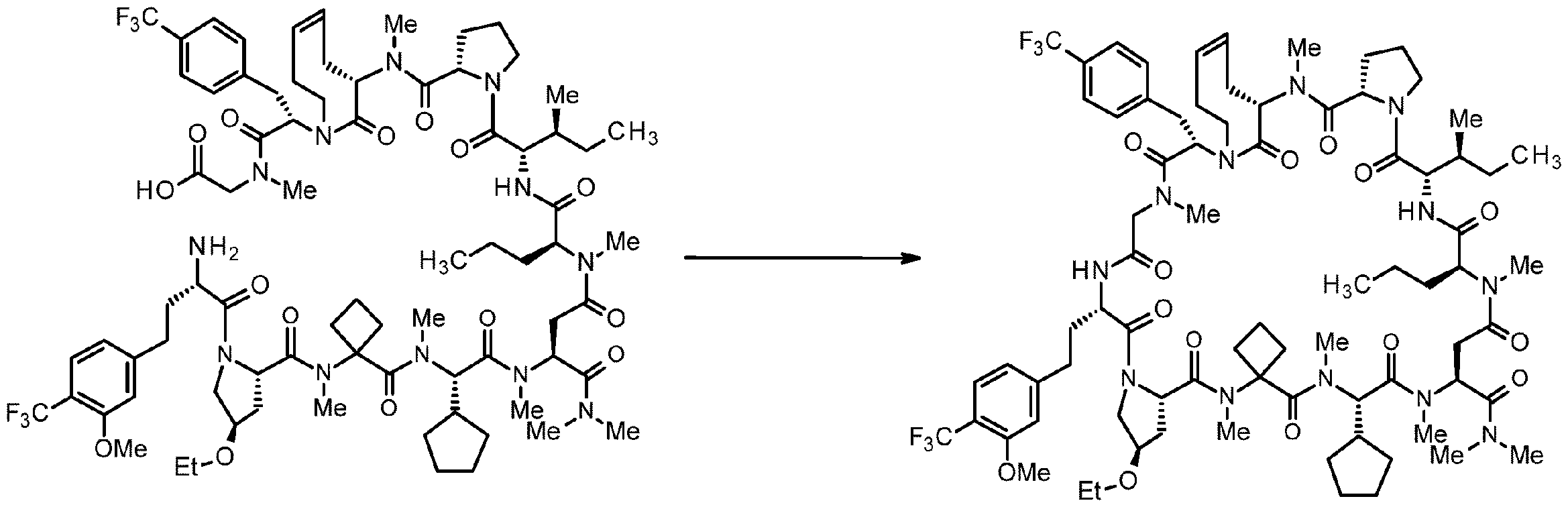
窒素で置換した反応釜に、COMU(65.0g)とアセトニトリル(1.9L)を加えた。COMUの溶解を確認後、反応釜の外温を0℃に設定し、実施例1-23で得られた化合物24(62.7g)を含む濃縮乾固物とLutidine(21.3mL)を含むアセトニトリル(0.941L)溶液をろ過したろ液を4.4mL/分で滴下した。添加完了直後に反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が99%(反応転換率の算出式1)、化合物1/環状ダイマー=98/2(環化選択性の算出式1,2)であることを確認した。反応釜の外温を40℃に設定し、撹拌しながら濃縮乾固した。反応釜の外温を25℃に冷却し、得られた濃縮物に酢酸イソプロピル(627mL)、2.5%アンモニア水溶液(627mL)を加え撹拌した。液々分離にて水層を排出後、得られた有機層に10%硫酸水素ナトリウム水溶液(627mL)を加え撹拌した。液々分離にて水層を排出後、得られた有機層を5%リン酸水素二ナトリウム水溶液(627mLx2)、5%塩化ナトリウム水溶液(627mL)で順次洗浄した。得られた有機層を0.5%塩化ナトリウム水溶液(627mLx2)で洗浄した。外温を40℃に設定し、得られた有機層を減圧濃縮し、化合物1を含む92.23gの濃縮乾固物を得た得られた化合物1を含む濃縮乾固物は実施例1-25に用いた。 COMU (65.0 g) and acetonitrile (1.9 L) were added to a reaction vessel purged with nitrogen. After confirming the dissolution of COMU, the external temperature of the reaction vessel was set to 0°C, and the filtrate obtained by filtering the concentrated and dried product containing compound 24 (62.7 g) obtained in Example 1-23 and a solution of acetonitrile (0.941 L) containing lutidine (21.3 mL) was added dropwise at 4.4 mL/min. Immediately after the addition was completed, the reaction mixture was sampled and prepared as a sample (sample preparation method 2), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% (calculation formula 1 for reaction conversion rate) and compound 1/cyclic dimer = 98/2 (calculation formulas 1 and 2 for cyclization selectivity). The external temperature of the reaction vessel was set to 40°C, and the mixture was concentrated and dried while stirring. The external temperature of the reaction vessel was cooled to 25°C, and isopropyl acetate (627 mL) and 2.5% aqueous ammonia solution (627 mL) were added to the obtained concentrate and stirred. After discharging the aqueous layer by liquid-liquid separation, 10% aqueous sodium hydrogen sulfate solution (627 mL) was added to the obtained organic layer and stirred. After discharging the aqueous layer by liquid-liquid separation, the obtained organic layer was washed successively with 5% aqueous disodium hydrogen phosphate solution (627 mL x 2) and 5% aqueous sodium chloride solution (627 mL). The obtained organic layer was washed with 0.5% aqueous sodium chloride solution (627 mL x 2). The external temperature was set to 40°C, and the obtained organic layer was concentrated under reduced pressure to obtain 92.23 g of a concentrated and dried product containing compound 1. The obtained concentrated and dried product containing compound 1 was used in Example 1-25.
環状ダイマー:

化合物1のLCMS(ESI):保持時間:6.07分、m/z=1555 [M+H]+(LCMS分析条件 method 4)
環状ダイマーのLCMS(ESI):保持時間:7.63分、m/z=1555 [M+H]2+(LCMS分析条件 method 4)
化合物1のHPLC:保持時間:4.09分、(HPLC分析条件 method 1)
環状ダイマーのHPLC:保持時間:4.91分、(HPLC分析条件 method 1)
LCMS (ESI) of compound 1: Retention time: 6.07 minutes, m/z = 1555 [M+H] + (LCMS analysis conditions method 4)
LCMS (ESI) of cyclic dimer: Retention time: 7.63 minutes, m/z = 1555 [M+H] 2+ (LCMS analysis conditions method 4)
HPLC of compound 1: retention time: 4.09 minutes (HPLC analysis condition method 1)
Cyclic dimer HPLC: Retention time: 4.91 minutes (HPLC analysis condition method 1)
実施例1-24-2Example 1-24-2
化合物1:(1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-シクロペンチル-28-エトキシ-32-[2-[3-メトキシ-4-(トリフルオロメチル)フェニル]エチル]-N,N,2,14,18,21,24,36-オクタメチル-10-[(1S)-1-メチルプロピル]-3,9,12,15,19,22,25,31,34,37,45-ウンデカオキソ-13-プロピル-38-[[4-(トリフルオロメチル)フェニル]メチル]スピロ[2,8,11,14,18,21,24,30,33,36,39-ウンデカザテトラシクロ[37.5.1.0Compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.5.1.0
4,84,8
.0.0
26,3026,30
]ペンタテトラコンタ-42-エン-23,1'-シクロブタン]-17-カルボキサミドの合成(環化位置B)Synthesis of ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide (cyclization position B)
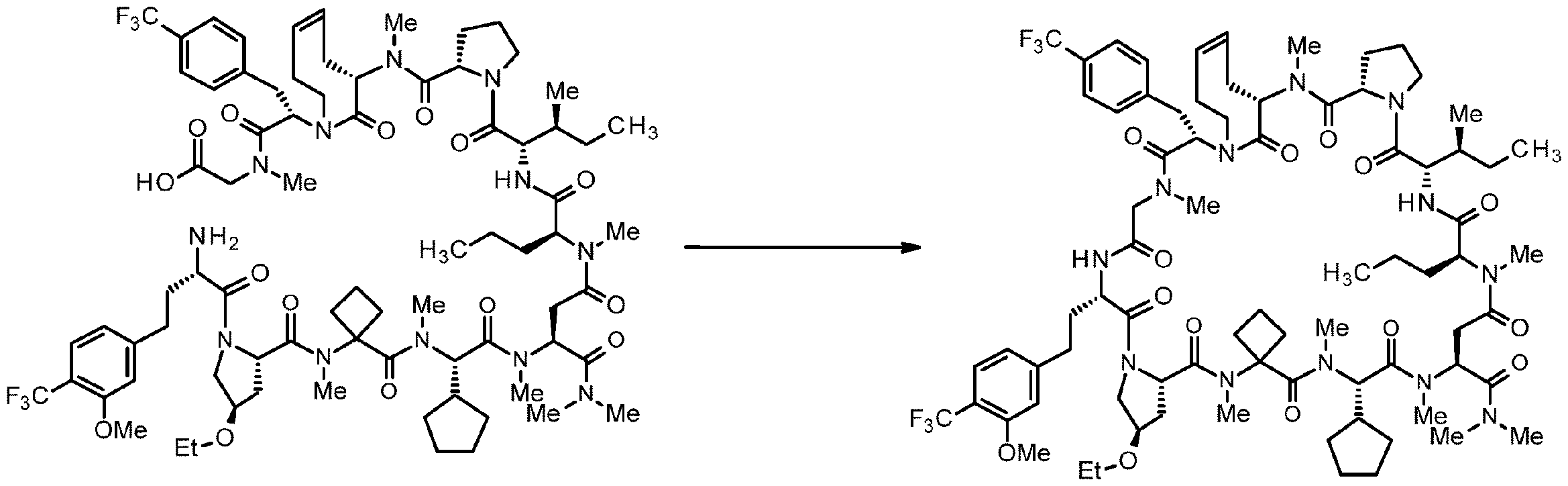
反応容器に、HATU(72.6mg)とアセトニトリル(1.80mL)を加えた。実施例1-23で得られた化合物24(100mg)を含む濃縮乾固物とDIPEA(40μL)を含むアセトニトリル(5.5mL)溶液を、5時間42分で滴下した。添加完了直後に反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が99%(反応転換率の算出式1)、化合物1/環状ダイマー=98/2(環化選択性の算出式1,2)であることを確認した。
化合物1のHPLC:保持時間:3.99分、(HPLC分析条件 method 1)
環状ダイマーのHPLC:保持時間:4.80分、(HPLC分析条件 method 1)
HATU (72.6 mg) and acetonitrile (1.80 mL) were added to the reaction vessel. A solution of the concentrated dry product containing compound 24 (100 mg) obtained in Example 1-23 and acetonitrile (5.5 mL) containing DIPEA (40 μL) was added dropwise over 5 hours and 42 minutes. Immediately after the addition was completed, the reaction mixture was sampled and prepared as a sample (sample preparation method 2). HPLC analysis confirmed that the reaction conversion rate was 99% (calculation formula 1 for reaction conversion rate) and compound 1/cyclic dimer = 98/2 (calculation formulas 1 and 2 for cyclization selectivity).
HPLC of compound 1: retention time: 3.99 minutes (HPLC analysis condition method 1)
Cyclic dimer HPLC: Retention time: 4.80 minutes (HPLC analysis condition method 1)
実施例1-25
化合物1の結晶化:(1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-シクロペンチル-28-エトキシ-32-[2-[3-メトキシ-4-(トリフルオロメチル)フェニル]エチル]-N,N,2,14,18,21,24,36-オクタメチル-10-[(1S)-1-メチルプロピル]-3,9,12,15,19,22,25,31,34,37,45-ウンデカオキソ-13-プロピル-38-[[4-(トリフルオロメチル)フェニル]メチル]スピロ[2,8,11,14,18,21,24,30,33,36,39-ウンデカザテトラシクロ[37.5.1.0 4,8 .0 26,30 ]ペンタテトラコンタ-42-エン-23,1'-シクロブタン]-17-カルボキサミドの水和物結晶(Form A)の合成
実施例1-24-1で得られた化合物1を含む濃縮乾固物24gをBiotage Sfar C18 Duo 100 Å 30μm 240gにローディングし、0.1%ギ酸-水/0.1%ギ酸-アセトニトリル=90/10→18/82)にて展開し、精製した。得られた化合物1(9.70g)にアセトン(29.0mL)とヘプタン(29.0mL)を外温35℃で加えた。溶解を確認後、実施例3-8と同様の操作により得られた、化合物1のアセトン/へプタン/水和物結晶(Form F)(約1.00mg)を反応釜に加え、35℃で23時間撹拌した。25℃に冷却し、さらに6時間撹拌した。ヘプタン(4.90mL)を1時間かけて加え、25℃で14時間撹拌した。さらにヘプタン(4.90mL)を1時間かけて加え、3時間撹拌した。最後にヘプタン(4.90mL)を2時間かけて加え、3時間撹拌した。反応混合物を減圧下ろ取し、得られた結晶をアセトン(7.76mL)とヘプタン(11.6mL)の混合液で洗浄した。得られた結晶を外温を40℃に設定して16時間乾燥した。乾燥末を回収し、白色の粉末(6.8g、Form A)を得た。
Examples 1-25
Crystallization of compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.0, 38.0, 39.5, 40.0 ]. Synthesis of hydrate crystals (Form A) of 26,30 ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide 24 g of the concentrated dry product containing compound 1 obtained in Example 1-24-1 was loaded onto 240 g of Biotage Sfar C18 Duo 100 Å 30 μm, and developed with 0.1% formic acid-water/0.1% formic acid-acetonitrile = 90/10 → 18/82) and purified. Acetone (29.0 mL) and heptane (29.0 mL) were added to the obtained compound 1 (9.70 g) at an external temperature of 35 ° C. After confirming dissolution, acetone/heptane/hydrate crystals (Form F) (about 1.00 mg) of compound 1 obtained by the same operation as in Example 3-8 were added to the reaction vessel and stirred at 35 ° C. for 23 hours. The mixture was cooled to 25°C and stirred for another 6 hours. Heptane (4.90mL) was added over 1 hour and stirred at 25°C for 14 hours. Heptane (4.90mL) was further added over 1 hour and stirred for 3 hours. Finally, heptane (4.90mL) was added over 2 hours and stirred for 3 hours. The reaction mixture was filtered under reduced pressure, and the obtained crystals were washed with a mixture of acetone (7.76mL) and heptane (11.6mL). The obtained crystals were dried for 16 hours with the external temperature set to 40°C. The dried powder was collected, and a white powder (6.8g, Form A) was obtained.
実施例1-26Example 1-26
化合物1:(1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-シクロペンチル-28-エトキシ-32-[2-[3-メトキシ-4-(トリフルオロメチル)フェニル]エチル]-N,N,2,14,18,21,24,36-オクタメチル-10-[(1S)-1-メチルプロピル]-3,9,12,15,19,22,25,31,34,37,45-ウンデカオキソ-13-プロピル-38-[[4-(トリフルオロメチル)フェニル]メチル]スピロ[2,8,11,14,18,21,24,30,33,36,39-ウンデカザテトラシクロ[37.5.1.0Compound 1: (1S,4S,10S,13S,17S,20S,26S,28R,32S,38S,42Z)-20-cyclopentyl-28-ethoxy-32-[2-[3-methoxy-4-(trifluoromethyl)phenyl]ethyl]-N,N,2,14,18,21,24,36-octamethyl-10-[(1S)-1-methylpropyl]-3,9,12,15,19,22,25,31,34,37,45-undecaoxo-13-propyl-38-[[4-(trifluoromethyl)phenyl]methyl]spiro[2,8,11,14,18,21,24,30,33,36,39-undecazatetracyclo[37.5.1.0
4,84,8
.0.0
26,3026,30
]ペンタテトラコンタ-42-エン-23,1'-シクロブタン]-17-カルボキサミドの合成(環化位置A)Synthesis of ]pentatetracont-42-ene-23,1'-cyclobutane]-17-carboxamide (cyclization position A)

窒素で置換した反応釜に、HATU(6.94g)とアセトニトリル(180mL)を加えた。HATUの溶解を確認後、化合物25(9.56g)とDIPEA(3.82mL)を含むアセトニトリル(540mL)溶液を6時間かけて滴下した。添加完了直後に反応混合物を10μLサンプリングして2μLのエタノールアミンを含む0.1mLのアセトニトリルに希釈させた。LCMS分析により、化合物1/環状ダイマー=75/25(環化選択性の算出式1,2)であることを確認した。
化合物1のLCMS(ESI):保持時間:0.78分、m/z=1553 [M-H]-(LCMS分析条件 method 5)
環状ダイマーのLCMS(ESI):保持時間:0.94分、m/z=1553 [M-H]2-(LCMS分析条件 method 5)
HATU (6.94 g) and acetonitrile (180 mL) were added to a reaction vessel purged with nitrogen. After confirming the dissolution of HATU, a solution of compound 25 (9.56 g) and DIPEA (3.82 mL) in acetonitrile (540 mL) was added dropwise over 6 hours. Immediately after the addition was completed, 10 μL of the reaction mixture was sampled and diluted with 0.1 mL of acetonitrile containing 2 μL of ethanolamine. LCMS analysis confirmed that compound 1/cyclic dimer = 75/25 (cyclization selectivity calculation formula 1, 2).
LCMS (ESI) of compound 1: Retention time: 0.78 minutes, m/z = 1553 [MH] - (LCMS analysis conditions method 5)
LCMS (ESI) of cyclic dimer: Retention time: 0.94 minutes, m/z = 1553 [MH] 2- (LCMS analysis conditions method 5)
化合物25の化学名と構造式:
(3S)-3-[[(2S)-2-シクロペンチル-2-[[1-[[(2S,4R)-4-エトキシ-1-[(2S)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]-2-[[2-[メチル-[(2S)-2-[(4Z,7S)-7-[メチル-[(2S)-1-[(2S,3S)-3-メチル-2-[[(2S)-2-(メチルアミノ)ペンタノイル]アミノ]ペンタノイル]ピロリジン-2-カルボニル]アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパノイル]アミノ]アセチル]アミノ]ブタノイル]ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-酪酸

(3S)-3-[[(2S)-2-cyclopentyl-2-[[1-[[(2S,4R)-4-ethoxy-1-[(2S)-4-[3-methoxy-4-(trifluoromethyl)phenyl]-2-[[2-[methyl-[(2S)-2-[(4Z,7S)-7-[methyl-[(2S)-1-[(2S,3S)-3-methyl-2-[[(2S)-2-(methylamino)pentanoyl]amino]pentanoyl 3-[4-(trifluoromethyl)phenyl]propanoyl]amino]acetyl]amino]butanoyl]pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butyric acid
実施例2-1Example 2-1
化合物26:(S)-2-(ブタ-3-エン-1-イルアミノ)-3-(4-(トリフルオロメチル)フェニル)プロピオン酸Compound 26: (S)-2-(but-3-en-1-ylamino)-3-(4-(trifluoromethyl)phenyl)propionic acid

窒素で置換した反応釜に、外温25℃にてアセトニトリル(822mL)、4-ブロモ-1-ブテン(235.07g)、トリエチルアミン(17.66g)を加えて1時間攪拌した。続いて(S)-2-アミノ-3-(4-(トリフルオロメチル)フェニル)プロピオン酸(135.46g)、水(676mL)、トリエチルアミン(158.19g)を加えて、外温70℃に昇温し、3.5時間攪拌した。25℃に冷却し、析出した固体を桐山ろうとで濾過し、アセトニトリルと水の混合物(1:1、676mL)で洗浄した。続いて、アセトニトリル(676mL)でさらに洗浄した。得られた湿性末を外温を40℃に設定して乾燥した。乾燥末を回収し、白色の固体(124.73g)を得た。
化合物26のLCMS(ESI):保持時間:2.34分、m/z=288 [M+H]+(LCMS分析条件 method P3-4)
Acetonitrile (822 mL), 4-bromo-1-butene (235.07 g), and triethylamine (17.66 g) were added to a reaction vessel purged with nitrogen at an external temperature of 25° C. and stirred for 1 hour. Then, (S)-2-amino-3-(4-(trifluoromethyl)phenyl)propionic acid (135.46 g), water (676 mL), and triethylamine (158.19 g) were added, and the external temperature was raised to 70° C. and stirred for 3.5 hours. The mixture was cooled to 25° C., and the precipitated solid was filtered through a Kiriyama funnel and washed with a mixture of acetonitrile and water (1:1, 676 mL). Then, it was further washed with acetonitrile (676 mL). The obtained wet powder was dried by setting the external temperature to 40° C. The dried powder was collected, and a white solid (124.73 g) was obtained.
LCMS (ESI) of compound 26: Retention time: 2.34 minutes, m/z = 288 [M+H] + (LCMS analysis conditions method P3-4)
実施例2-2Example 2-2
化合物27:tert-ブチル (S)-N-(2-(ブタ-3-エン-1-イルアミノ)-3-(4-(トリフルオロメチル)フェニル)プロパノイル)-N-メチルグリシネート ヒドロクロライドCompound 27: tert-Butyl (S)-N-(2-(but-3-en-1-ylamino)-3-(4-(trifluoromethyl)phenyl)propanoyl)-N-methylglycinate hydrochloride

窒素で置換した反応釜に、外温25℃にて(S)-2-(ブタ-3-エン-1-イルアミノ)-3-(4-(トリフルオロメチル)フェニル)プロピオン酸(107.26g)、サルコシンtert-ブチルエステル塩酸塩(102.00g)、アセトニトリル(751mL)、ジアザビシクロウンデセン(233.03g)を加えて10分間攪拌した。均一溶液になることを確認した後に、外温を2℃に設定し、ホスホン酸無水プロパンの2-メチルテトラヒドロフラン 50%溶液(309.03g)を2時間14分かけて滴下した。反応が完結したことを確認した後に、反応混合物にトルエン(751mL)、1N NaOH水溶液(536mL)を加え、30分間攪拌した後、液々分離により水層を排出した。有機層を室温にて終夜保管した。保管後に有機層に5% 炭酸ナトリウム水溶液(536mL)を加え、10分間攪拌した後、液々分離により水層を排出した。続いて、有機層に5% リン酸二水素ナトリウム水溶液(751mL)を加え、10分間攪拌した後、液々分離により水層を排出した。さらにもう一回有機層に5% リン酸二水素ナトリウム水溶液(751mL)を加え、10分間攪拌した後、液々分離により水層を排出した。続いて、有機層に5% 食塩水(751mL)を加え、10分間攪拌した後、液々分離により水層を排出した。有機層を外温5℃にて終夜保管した。保管後に有機層を40℃で減圧濃縮し、約215mLとなるまで濃縮した。濃縮液にトルエン(215mL)を加え、40℃で約215mLとなるまで減圧濃縮し、さらにこの操作を2回繰り返した。析出した無機塩をろ過し、得られたろ液中の目的物とトルエンを定量し、さらにトルエンが296mLになるように添加した。ほかの反応容器にて、ピリジン塩酸塩(43.28g)、アセトニトリル(148mL)を加え、調製された溶液を外温25℃にて目的物のトルエン溶液に45分にかけて滴下した。滴下中に結晶の析出が確認された。さらにアセトニトリル(74mL)でリンスし、1時間攪拌した。その後、トルエン(1.7L)を加え、1時間攪拌した後に、外温を0℃まで下げて、さらに2時間攪拌した。得られた結晶を桐山ろうとで濾過し、0℃まで冷却したトルエン(296mL)で2回洗浄した。得られた湿性末外温5℃にて週末保管した。保管後に湿性末を外温を40℃に設定して乾燥した。乾燥末を回収し、白色の固体(98.66g)を得た。
化合物27のLCMS(ESI):保持時間:3.03分、m/z=415 [M+H]+(LCMS分析条件 method P3-4)
In a reaction vessel purged with nitrogen, (S)-2-(but-3-en-1-ylamino)-3-(4-(trifluoromethyl)phenyl)propionic acid (107.26 g), sarcosine tert-butyl ester hydrochloride (102.00 g), acetonitrile (751 mL), and diazabicycloundecene (233.03 g) were added at an external temperature of 25° C. and stirred for 10 minutes. After confirming that the solution was homogeneous, the external temperature was set to 2° C., and a 50% solution of phosphonic acid anhydride propane in 2-methyltetrahydrofuran (309.03 g) was added dropwise over 2 hours and 14 minutes. After confirming that the reaction was complete, toluene (751 mL) and 1N NaOH aqueous solution (536 mL) were added to the reaction mixture, and the mixture was stirred for 30 minutes, after which the aqueous layer was discharged by liquid-liquid separation. The organic layer was stored overnight at room temperature. After storage, 5% aqueous sodium carbonate solution (536 mL) was added to the organic layer, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation. Next, 5% aqueous sodium dihydrogen phosphate solution (751 mL) was added to the organic layer, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation. 5% aqueous sodium dihydrogen phosphate solution (751 mL) was added to the organic layer once more, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation. Next, 5% saline solution (751 mL) was added to the organic layer, stirred for 10 minutes, and the aqueous layer was discharged by liquid-liquid separation. The organic layer was stored overnight at an external temperature of 5° C. After storage, the organic layer was concentrated under reduced pressure at 40° C. until it became about 215 mL. Toluene (215 mL) was added to the concentrated liquid, and concentrated under reduced pressure at 40° C. until it became about 215 mL, and this operation was repeated twice. The precipitated inorganic salt was filtered, the target substance and toluene in the obtained filtrate were quantified, and toluene was added so that the amount was 296 mL. In another reaction vessel, pyridine hydrochloride (43.28 g) and acetonitrile (148 mL) were added, and the prepared solution was dropped into the toluene solution of the target substance at an external temperature of 25 ° C. over 45 minutes. Crystal precipitation was confirmed during the dropwise addition. Further rinse with acetonitrile (74 mL) and stir for 1 hour. Then, toluene (1.7 L) was added, and after stirring for 1 hour, the external temperature was lowered to 0 ° C. and stirred for another 2 hours. The obtained crystals were filtered with a Kiriyama funnel and washed twice with toluene (296 mL) cooled to 0 ° C. The obtained wet powder was stored over the weekend at an external temperature of 5 ° C. After storage, the wet powder was dried by setting the external temperature to 40 ° C. The dried powder was collected, and a white solid (98.66 g) was obtained.
LCMS (ESI) of compound 27: Retention time: 3.03 minutes, m/z = 415 [M+H] + (LCMS analysis conditions method P3-4)
実施例2-3Example 2-3
化合物28:2-[[(2S)-2-[(4Z,7S)-7-[9H-フルオレン-9-イルメトキシカルボニル (メチル)アミノ]-8-オキソ-2,3,6,7-テトラヒドロアゾシン-1-イル]-3-[4-(トリフルオロメチル)フェニル]プロパニル]-メチル-アミノ]酢酸Compound 28: 2-[[(2S)-2-[(4Z,7S)-7-[9H-Fluoren-9-ylmethoxycarbonyl (methyl)amino]-8-oxo-2,3,6,7-tetrahydroazocin-1-yl]-3-[4-(trifluoromethyl)phenyl]propanyl]-methyl-amino]acetic acid

反応容器に、室温にて2-MeTHF(1.35mL)、化合物8(145mg)、HMDS(98μL)を加えて攪拌した。続いて混合溶液に対して、TMSOTf(51μL)を室温で加え、4時間撹拌した。HPLCを測定し、反応変換率が99%以上であることを確認したのち、外温を5℃に冷却し、内温15℃以下で5%リン酸二カリウム水溶液(1.35mL)をゆっくり加え、室温で撹拌した。液々分離により水層を排出した。有機層を5%リン酸二水素ナトリウム水溶液(1.35mL×2)、5%食塩水(1.35mL)で洗浄し、有機層を外温40℃で減圧濃縮し、化合物28(133mg)を得た。
化合物28のLCMS(ESI):保持時間:4.34分、m/z=664 [M+H]+(LCMS分析条件 method P3-4)
2-MeTHF (1.35 mL), compound 8 (145 mg), and HMDS (98 μL) were added to the reaction vessel at room temperature and stirred. Then, TMSOTf (51 μL) was added to the mixed solution at room temperature and stirred for 4 hours. After measuring HPLC and confirming that the reaction conversion rate was 99% or more, the external temperature was cooled to 5° C., and 5% aqueous potassium phosphate solution (1.35 mL) was slowly added at an internal temperature of 15° C. or less and stirred at room temperature. The aqueous layer was discharged by liquid-liquid separation. The organic layer was washed with 5% aqueous sodium dihydrogen phosphate solution (1.35 mL x 2) and 5% saline (1.35 mL), and the organic layer was concentrated under reduced pressure at an external temperature of 40° C. to obtain compound 28 (133 mg).
LCMS (ESI) of compound 28: Retention time: 4.34 minutes, m/z = 664 [M+H] + (LCMS analysis conditions method P3-4)
反応釜に室温にて、化合物8(3.89kg)の2-MeTHF(20.5kg)溶液を加えたのち、外温15℃に冷却し、HMDS(2.19kg)を加えて撹拌した。続いて混合溶媒にTMSOTf(1.80kg)を内温25℃以下でゆっくり添加し、内温25℃で2時間撹拌した。2-MeTHF(16.6kg)とアセトニトリル(4.60kg)を加え、内温15℃以下に冷却後、5%炭酸水素ナトリウム水溶液(27.3kg)を内温30℃でゆっくり加え、室温で撹拌し、液々分離により水層を排出した。有機層を5%硫酸水素ナトリウム水溶液(27.2kg)、5%食塩水(27.2kg×3)で洗浄し、外温40℃で10Lまで濃縮した。濃縮液にトルエン(56.0kg)を加えて外温40℃で55Lまで濃縮したのち、トルエン(15.6kg)を加えて外温40℃で54Lまで濃縮することを2回繰り返した。結晶の析出を確認後、シクロヘキサン(14.0kg)を添加して一晩撹拌し、ろ過を行い、結晶をトルエン/シクロヘキサン=3:1混液(15.2kg)で洗浄した。外温50℃で得られた湿結晶を減圧乾燥し、化合物28・1トルエン和物(3.21kg)を白色固体として得た。 A solution of compound 8 (3.89 kg) in 2-MeTHF (20.5 kg) was added to the reaction vessel at room temperature, then cooled to an external temperature of 15°C, and HMDS (2.19 kg) was added and stirred. Next, TMSOTf (1.80 kg) was slowly added to the mixed solvent at an internal temperature of 25°C or less, and stirred at an internal temperature of 25°C for 2 hours. 2-MeTHF (16.6 kg) and acetonitrile (4.60 kg) were added, and the mixture was cooled to an internal temperature of 15°C or less, after which 5% aqueous sodium hydrogen carbonate solution (27.3 kg) was slowly added at an internal temperature of 30°C, stirred at room temperature, and the aqueous layer was discharged by liquid-liquid separation. The organic layer was washed with 5% aqueous sodium hydrogen sulfate solution (27.2 kg) and 5% saline solution (27.2 kg x 3), and concentrated to 10 L at an external temperature of 40°C. Toluene (56.0 kg) was added to the concentrated liquid and concentrated to 55 L at an external temperature of 40°C, and then toluene (15.6 kg) was added and concentrated to 54 L at an external temperature of 40°C twice. After confirming that crystals had precipitated, cyclohexane (14.0 kg) was added and stirred overnight, filtered, and the crystals were washed with a toluene/cyclohexane = 3:1 mixture (15.2 kg). The wet crystals obtained at an external temperature of 50°C were dried under reduced pressure to obtain compound 28 monotoluene solvate (3.21 kg) as a white solid.
実施例2-4Example 2-4
化合物10:tert-ブチル N-[(ベンジルオキシ)カルボニル]-L-イソロイシル-L-プロリナートの合成Compound 10: Synthesis of tert-butyl N-[(benzyloxy)carbonyl]-L-isoleucyl-L-prolinate
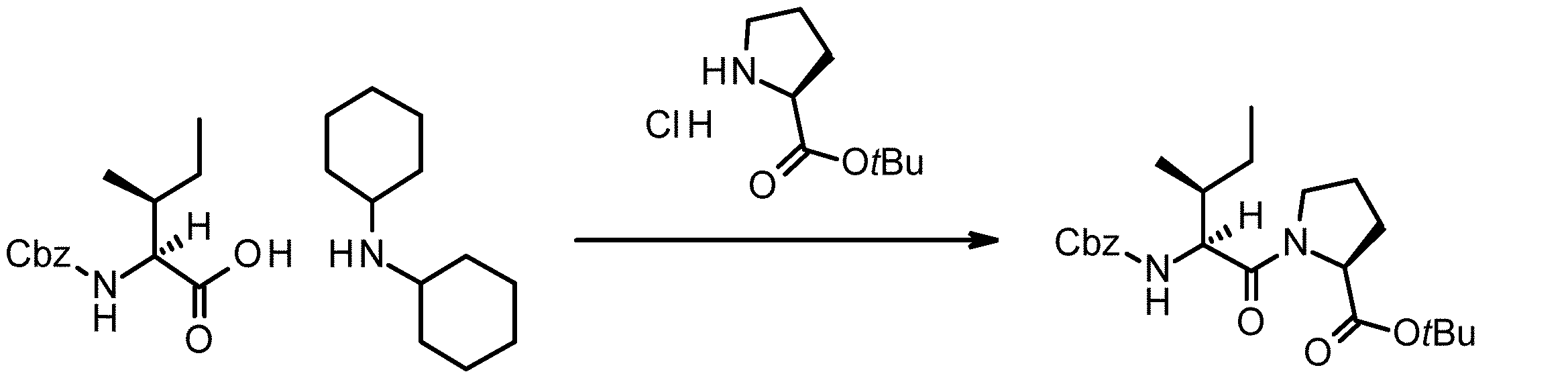
N-シクロヘキシルシクロヘキサナミニウム (2S,3S)-2-{[(ベンジルオキシ)カルボニル]アミノ}-3-メチルペンタノアート (135g)にトルエン(583g)および5%硫酸水素ナトリウム水(2066g)を加え室温で10分間撹拌後、有機層を分離した。得られた有機層を5%硫酸水素ナトリウム水(2066g)、5%食塩水(1397g)の順で洗浄、減圧下溶媒を留去した。得られた残渣にトルエン(55mL)、1,3-ジメチル-2-イミダゾリジノン(270mL)、(2S)-2-(tert-ブトキシカルボニル)ピロリジン-1-イウム クロリド (75.3g)、2-メチルテトラヒドロフラン(540mL)、4-メチルモルホリン(133mL)を加え外温15℃で撹拌した。この混合物に、50%-プロピルホスホン酸無水物 2-メチルテトラヒドロフラン溶液(370mL)を約1時間かけて滴下し、得られた反応混合物を外温20℃で1時間撹拌した。この反応混合物に、5%炭酸水素ナトリウム水(993g)、1-メチルイミダゾール(24mL)を内温20℃以下で加えた後、外温20℃で約30分撹拌後、有機層を分離した。得られた有機層を、10%硫酸水素ナトリウム水(709g)、10%硫酸水素ナトリウム水(639g)、10%炭酸水素ナトリウム水(710g)の順に洗浄、減圧下溶媒を留去して、表題の化合物を含む溶液(137g)を得た。
化合物10のLCMS(ESI):保持時間:4.21分、m/z=419 [M+H]+(LCMS分析条件 method M)
Toluene (583 g) and 5% aqueous sodium hydrogen sulfate (2066 g) were added to N-cyclohexylcyclohexanaminium (2S,3S)-2-{[(benzyloxy)carbonyl]amino}-3-methylpentanoate (135 g), and the mixture was stirred at room temperature for 10 minutes, and the organic layer was separated. The resulting organic layer was washed with 5% aqueous sodium hydrogen sulfate (2066 g) and 5% saline (1397 g) in that order, and the solvent was distilled off under reduced pressure. Toluene (55 mL), 1,3-dimethyl-2-imidazolidinone (270 mL), (2S)-2-(tert-butoxycarbonyl)pyrrolidin-1-ium chloride (75.3 g), 2-methyltetrahydrofuran (540 mL), and 4-methylmorpholine (133 mL) were added to the resulting residue, and the mixture was stirred at an external temperature of 15°C. A 50% solution of propylphosphonic anhydride in 2-methyltetrahydrofuran (370 mL) was added dropwise to this mixture over about 1 hour, and the resulting reaction mixture was stirred at an external temperature of 20° C. for 1 hour. 5% aqueous sodium hydrogen carbonate (993 g) and 1-methylimidazole (24 mL) were added to this reaction mixture at an internal temperature of 20° C. or lower, and the mixture was stirred at an external temperature of 20° C. for about 30 minutes, after which the organic layer was separated. The resulting organic layer was washed with 10% aqueous sodium hydrogen sulfate (709 g), 10% aqueous sodium hydrogen sulfate (639 g), and 10% aqueous sodium hydrogen carbonate (710 g) in that order, and the solvent was distilled off under reduced pressure to obtain a solution (137 g) containing the title compound.
LCMS (ESI) of compound 10: Retention time: 4.21 minutes, m/z = 419 [M+H] + (LCMS analysis conditions method M)
実施例2-5Example 2-5
化合物11:tert-ブチル L-イソロイシル-L-プロリナートの合成Compound 11: Synthesis of tert-butyl L-isoleucyl-L-prolinate
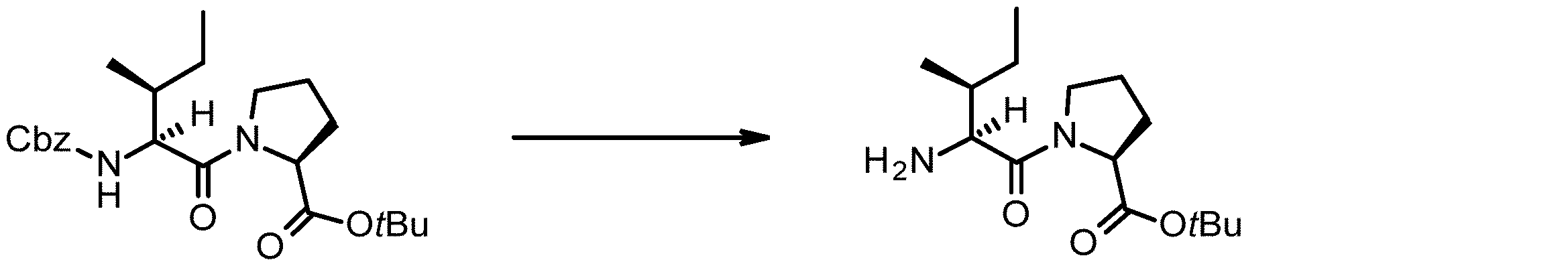
Pd/C(50% wet、36.7g)および2-メチルテトラヒドロフラン(253mL)を外温25℃、水素加圧(0.4MPa)下で2時間撹拌した。得られた混合物に、実施例2-4で得られた化合物10を含む溶液(137g)と2-メチルテトラヒドロフラン(495mL)を加え、外温25℃、水素加圧(0.2MPaG)下2時間撹拌した。反応混合物をろ過、固体を2-メチルテトラヒドロフラン(127mL)で3回洗浄し、ろ過液をすべて合わせて減圧下溶媒を留去し、表題の化合物を含む溶液(160g)を得た。
化合物11のLCMS(ESI):保持時間:2.40分、m/z=285 [M+H]+(LCMS分析条件 method M)
Pd/C (50% wet, 36.7 g) and 2-methyltetrahydrofuran (253 mL) were stirred at an external temperature of 25° C. under hydrogen pressure (0.4 MPa) for 2 hours. To the resulting mixture, a solution containing compound 10 obtained in Example 2-4 (137 g) and 2-methyltetrahydrofuran (495 mL) were added, and the mixture was stirred at an external temperature of 25° C. under hydrogen pressure (0.2 MPaG) for 2 hours. The reaction mixture was filtered, and the solid was washed three times with 2-methyltetrahydrofuran (127 mL). The filtrate was combined and the solvent was distilled off under reduced pressure to obtain a solution containing the title compound (160 g).
LCMS (ESI) of compound 11: Retention time: 2.40 minutes, m/z = 285 [M+H] + (LCMS analysis conditions method M)
実施例2-6Example 2-6
化合物29:tert-ブチル N-[(ベンジルオキシ)カルボニル]-N-メチル-L-ノルバリル-L-イソロイシル-L-プロリナートの合成Synthesis of Compound 29: tert-Butyl N-[(benzyloxy)carbonyl]-N-methyl-L-norvalyl-L-isoleucyl-L-prolinate
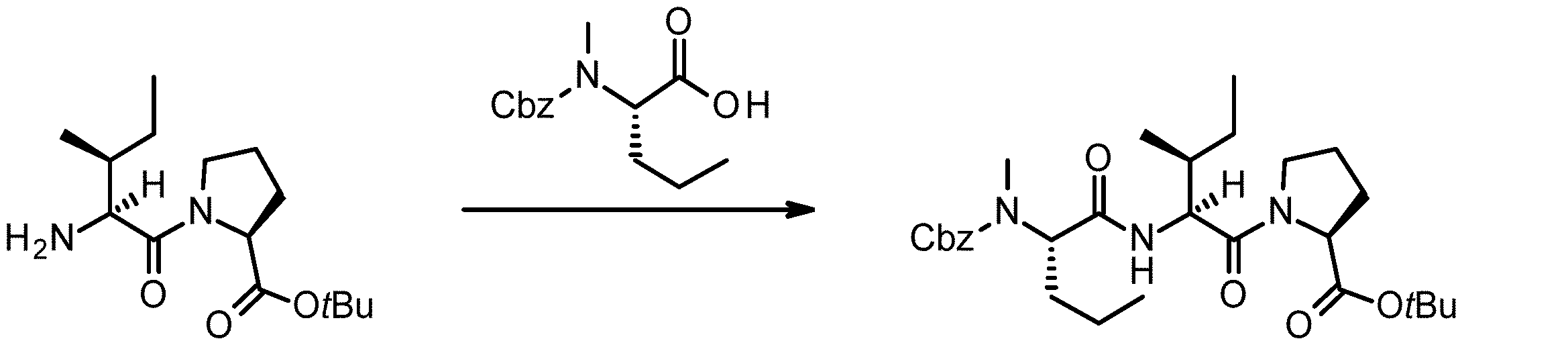
実施例2-5で得られた化合物11を含む溶液(120g)、N-[(ベンジルオキシ)カルボニル]-N-メチル-L-ノルバリン (72.12g)、2-メチルテトラヒドロフラン(257mL)、4-メチルモルホリン(100mL)の混合物を外温15℃で撹拌した。この混合物に、50%-プロピルホスホン酸無水物 2-メチルテトラヒドロフラン溶液(277mL)を約40分かけて滴下し、得られた反応混合物を外温20℃で1時間撹拌した。この反応混合物に、5%炭酸水素ナトリウム水(472g)、1-メチルイミダゾール(18mL)を内温30℃以下で加えた後、外温15℃で約30分撹拌後、有機層を分離した。得られた有機層を、外温20℃にて10%硫酸水素ナトリウム水(338g)、10%硫酸水素ナトリウム水(338g)、10%炭酸水素ナトリウム水(340g)の順に洗浄、減圧下溶媒を留去して、表題の化合物を含む溶液(219g)を得た。
化合物29のLCMS(ESI):保持時間:4.58分、m/z=532 [M+H]+(LCMS分析条件 method M)
A mixture of the solution containing compound 11 obtained in Example 2-5 (120 g), N-[(benzyloxy)carbonyl]-N-methyl-L-norvaline (72.12 g), 2-methyltetrahydrofuran (257 mL), and 4-methylmorpholine (100 mL) was stirred at an external temperature of 15° C. To this mixture, a 50% propylphosphonic anhydride 2-methyltetrahydrofuran solution (277 mL) was added dropwise over about 40 minutes, and the resulting reaction mixture was stirred at an external temperature of 20° C. for 1 hour. To this reaction mixture, 5% sodium bicarbonate water (472 g) and 1-methylimidazole (18 mL) were added at an internal temperature of 30° C. or less, and the mixture was stirred at an external temperature of 15° C. for about 30 minutes, and then the organic layer was separated. The obtained organic layer was washed successively with 10% aqueous sodium hydrogen sulfate (338 g), 10% aqueous sodium hydrogen sulfate (338 g), and 10% aqueous sodium hydrogen carbonate (340 g) at an external temperature of 20° C., and the solvent was distilled off under reduced pressure to obtain a solution containing the title compound (219 g).
LCMS (ESI) of compound 29: Retention time: 4.58 minutes, m/z = 532 [M+H] + (LCMS analysis conditions method M)
実施例2-7Example 2-7
化合物13:tert-ブチル N-メチル-L-ノルバリル-L-イソロイシル-L-プロリナートの合成Synthesis of Compound 13: tert-Butyl N-methyl-L-norvalyl-L-isoleucyl-L-prolinate
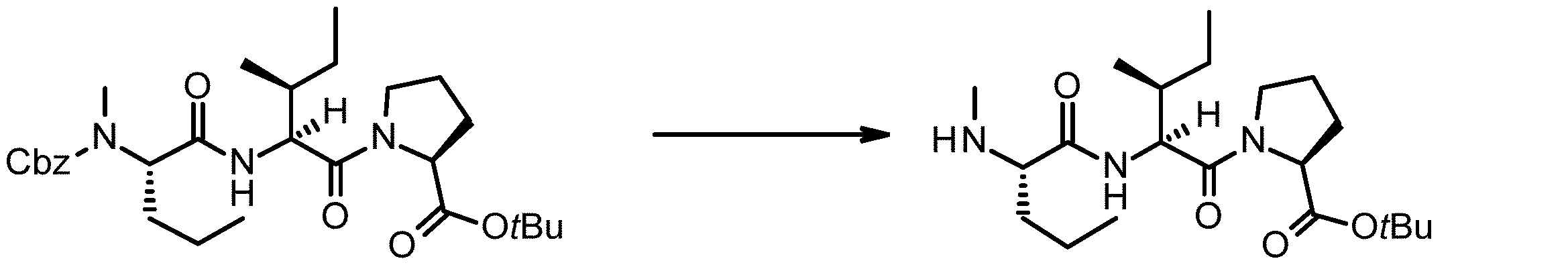
Pd/C(50% wet、22.9g)および2-メチルテトラヒドロフラン(400mL)を外温25℃、水素加圧(0.4MPaG)下で2時間撹拌した。得られた混合物に、実施例2-6で得られた化合物29を含む溶液(182g)と2-メチルテトラヒドロフラン(50mL)を加え、外温25℃、水素加圧(0.4MPaG)下2時間撹拌した。反応混合物をろ過、固体を2-メチルテトラヒドロフラン(100mL)で3回洗浄し、ろ過液をすべて合わせて減圧下濃縮し、表題の化合物を含む溶液(109g)を得た。この溶液のうち9.5346gを減圧下溶媒を留去し、得られた残渣にヘプタン(100mL)を加えた。混合物を外温50℃で溶解し、内温40℃にて実施例2-7-1で得られた種晶(11.0mg)を添加した。混合物を外温42℃で15分、外温43℃で13分、外温44℃で17分撹拌後、外温を1時間あたり12℃の速度で0℃まで冷却、さらに外温0℃で1.5時間撹拌した。得られた固体をろ取し、冷ヘプタン(25mL)で洗浄、外温30℃から40℃、減圧下乾燥し、表題の化合物(4.8474g)を得た。
化合物13のLCMS(ESI):保持時間:2.56分、m/z=398 [M+H]+(LCMS分析条件 method M)
Pd/C (50% wet, 22.9 g) and 2-methyltetrahydrofuran (400 mL) were stirred at an external temperature of 25° C. under hydrogen pressure (0.4 MPaG) for 2 hours. To the resulting mixture, a solution containing compound 29 obtained in Example 2-6 (182 g) and 2-methyltetrahydrofuran (50 mL) were added, and the mixture was stirred at an external temperature of 25° C. under hydrogen pressure (0.4 MPaG) for 2 hours. The reaction mixture was filtered, the solid was washed three times with 2-methyltetrahydrofuran (100 mL), and the filtrate was combined and concentrated under reduced pressure to obtain a solution containing the title compound (109 g). The solvent was distilled off from 9.5346 g of this solution under reduced pressure, and heptane (100 mL) was added to the resulting residue. The mixture was dissolved at an external temperature of 50° C., and the seed crystals (11.0 mg) obtained in Example 2-7-1 were added at an internal temperature of 40° C. The mixture was stirred at an external temperature of 42° C. for 15 minutes, at an external temperature of 43° C. for 13 minutes, and at an external temperature of 44° C. for 17 minutes, then the external temperature was cooled to 0° C. at a rate of 12° C. per hour, and further stirred for 1.5 hours at an external temperature of 0° C. The obtained solid was collected by filtration, washed with cold heptane (25 mL), and dried under reduced pressure at an external temperature of 30° C. to 40° C. to obtain the title compound (4.8474 g).
LCMS (ESI) of compound 13: Retention time: 2.56 minutes, m/z = 398 [M+H] + (LCMS analysis conditions method M)
実施例2-7-1
化合物13:tert-ブチル N-メチル-L-ノルバリル-L-イソロイシル-L-プロリナートの種晶の合成
実施例2-7の反応で得られた表題の化合物を含む溶液の一部を減圧下濃縮し、得られた残渣(0.3811g)にヘプタン(7622μL)を加えた。生成した固体を外温50℃で溶解後、撹拌しながら室温に冷却、得られたスラリーにヘプタン(3811μL)を加えて撹拌を継続した。固体をろ取、ヘプタン(1906μL)で洗浄後、減圧下室温で乾燥し、表題の化合物(0.2187g)を得た。
化合物13の種晶のLCMS(ESI):保持時間:2.60分、m/z=398 [M+H]+(LCMS分析条件 method M)
Example 2-7-1
Compound 13: Synthesis of seed crystals of tert-butyl N-methyl-L-norvalyl-L-isoleucyl-L-prolinate A part of the solution containing the title compound obtained in the reaction of Example 2-7 was concentrated under reduced pressure, and heptane (7622 μL) was added to the resulting residue (0.3811 g). The resulting solid was dissolved at an external temperature of 50° C., cooled to room temperature with stirring, and heptane (3811 μL) was added to the resulting slurry and stirring was continued. The solid was collected by filtration, washed with heptane (1906 μL), and then dried at room temperature under reduced pressure to obtain the title compound (0.2187 g).
LCMS (ESI) of seed crystals of compound 13: Retention time: 2.60 minutes, m/z = 398 [M+H] + (LCMS analysis conditions method M)
実施例2-8-1Example 2-8-1
化合物A11:(tert-ブチル (3S)-3-[ベンジルオキシカルボニル(メチル)アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound A11: (tert-Butyl (3S)-3-[benzyloxycarbonyl(methyl)amino]-4-(dimethylamino)-4-oxo-butanoate)
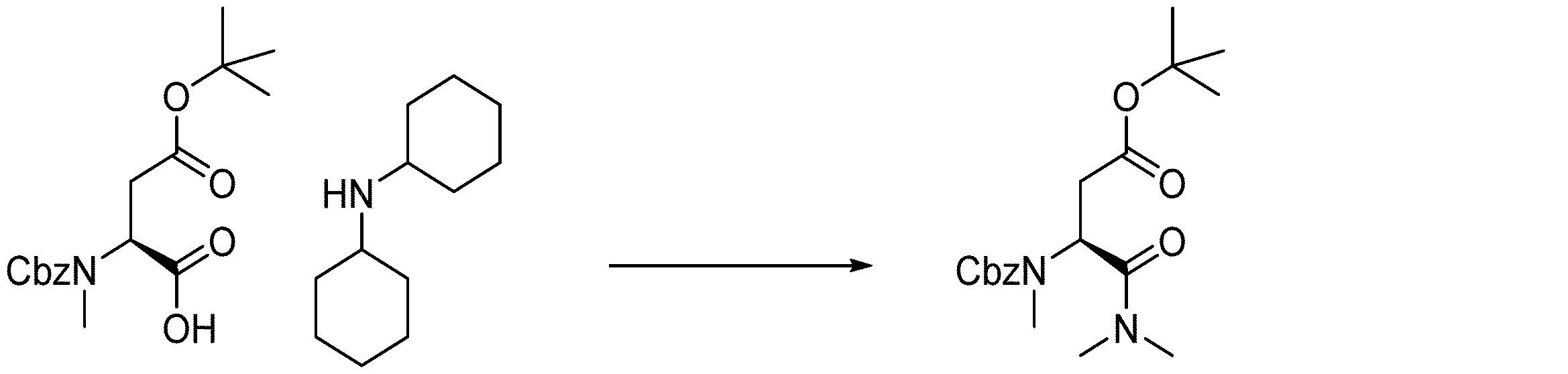
反応容器に(2S)-2-(((ベンジルオキシ)カルボニル)(メチル)アミノ)-4-tert-ブトキシ-4-オキソブタン酸ジシクロヘキシルアミン塩(Cas番号42417-70-9、25.00g、48.2mmol)と2-メチルテトラヒドロフラン(126g)を25℃にて加えた。10%硫酸水素ナトリウム一水和物水溶液(150g)による分液洗浄を2回繰り返した後、5%塩化ナトリウム水溶液(150g)により洗浄した。得られた有機層を減圧条件下で濃縮した。得られた残渣に2-メチルテトラヒドロフラン(95g)を加え、減圧条件下で濃縮する操作を2回繰り返した。得られた残渣(47.91g)に2-メチルテトラヒドロフラン(95g)、アセトニトリル(75g)、DIPEA(35.46g、274mmol)、ジメチルアミン塩酸塩(7.88g、96.6mmol)を25℃にて加えた。プロピルホスホン酸無水物の2-メチルテトラヒドロフラン溶液(50.4wt%、61.33g、97.1mmol)を1時間30分かけて滴下した。滴下終了後1時間攪拌した後にサンプリングし、HPLC分析で反応完結を確認した。2M水酸化ナトリウム水溶液(150g)を加えた。10分撹拌した後に静置し、水層を除去した。得られた有機層を2M水酸化ナトリウム水溶液(150g)、13%硫酸水溶液(150g)、10%硫酸水素ナトリウム一水和物水溶液(150g)、5%炭酸ナトリウム水溶液(150g)により洗浄した後、減圧条件下で濃縮した。2-メチルテトラヒドロフラン(125g)を加えて減圧条件下で濃縮する操作を2回繰り返し、化合物A11を含む溶液(42.39g)を得た。
化合物A11のLCMS(ESI):保持時間:3.37分、m/z=387 [M+Na]+(LCMS分析条件 method K-2)
(2S)-2-(((benzyloxy)carbonyl)(methyl)amino)-4-tert-butoxy-4-oxobutanoic acid dicyclohexylamine salt (Cas number 42417-70-9, 25.00 g, 48.2 mmol) and 2-methyltetrahydrofuran (126 g) were added to a reaction vessel at 25° C. After separation and washing with a 10% aqueous solution of sodium hydrogen sulfate monohydrate (150 g) was repeated twice, washing was performed with a 5% aqueous solution of sodium chloride (150 g). The obtained organic layer was concentrated under reduced pressure. 2-Methyltetrahydrofuran (95 g) was added to the obtained residue, and the operation of concentrating under reduced pressure was repeated twice. 2-Methyltetrahydrofuran (95 g), acetonitrile (75 g), DIPEA (35.46 g, 274 mmol), and dimethylamine hydrochloride (7.88 g, 96.6 mmol) were added to the obtained residue (47.91 g) at 25° C. A solution of propylphosphonic anhydride in 2-methyltetrahydrofuran (50.4 wt %, 61.33 g, 97.1 mmol) was added dropwise over 1 hour and 30 minutes. After the completion of the dropwise addition, the mixture was stirred for 1 hour, sampled, and the completion of the reaction was confirmed by HPLC analysis. 2M aqueous sodium hydroxide solution (150 g) was added. After stirring for 10 minutes, the mixture was allowed to stand, and the aqueous layer was removed. The obtained organic layer was washed with 2M aqueous sodium hydroxide solution (150 g), 13% aqueous sulfuric acid solution (150 g), 10% aqueous sodium hydrogen sulfate monohydrate solution (150 g), and 5% aqueous sodium carbonate solution (150 g), and then concentrated under reduced pressure. The operation of adding 2-methyltetrahydrofuran (125 g) and concentrating under reduced pressure was repeated twice to obtain a solution containing compound A11 (42.39 g).
LCMS (ESI) of compound A11: Retention time: 3.37 minutes, m/z = 387 [M+Na] + (LCMS analysis conditions method K-2)
実施例2-8-2Example 2-8-2
化合物A12:(tert-ブチル (3S)-4-(ジメチルアミノ)-3-(メチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound A12: (tert-Butyl (3S)-4-(dimethylamino)-3-(methylamino)-4-oxo-butanoate)
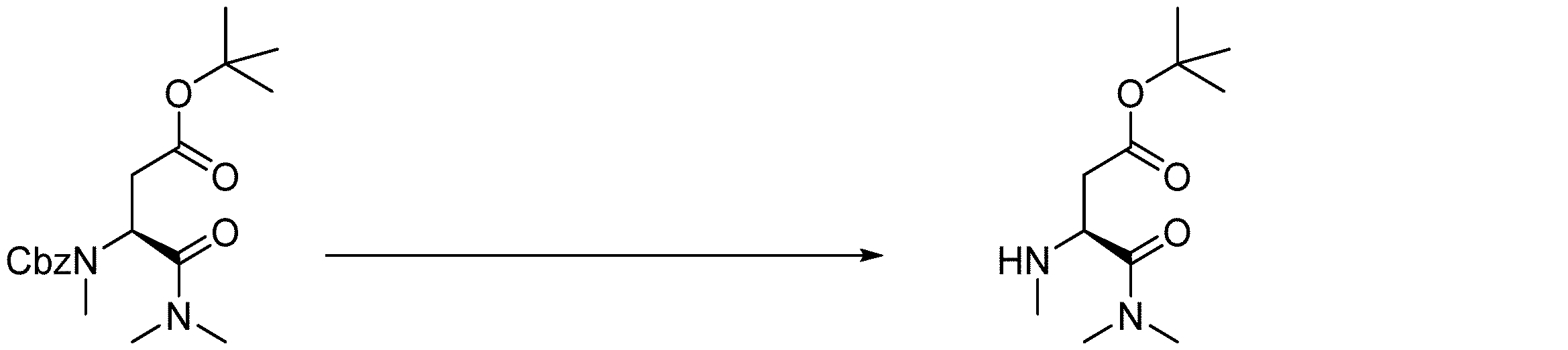
反応容器に10%パラジウム炭素(54.33% wet、3.39g、1.45mmol、3mol% on Pd metal basis)と2-メチルテトラヒドロフラン(75g)を加えた。25℃で窒素置換し、水素置換して水素雰囲気下(0.40MPaG)で2時間攪拌した。実施例2-8-1で得られた化合物A11の溶液(42.39g)と2-メチルテトラヒドロフラン(22g)を加えた。水素雰囲気下(0.20MPaG)で1時間30分攪拌した後にサンプリングし、HPLC分析で反応完結を確認した。反応混合物を濾過した後、ケーキを2-メチルテトラヒドロフラン(75g)により2回洗浄した。濾液と洗浄液の混合溶液を減圧条件下で濃縮し、化合物A12を含む溶液(30.76g)を得た。
化合物A12のLCMS(ESI):保持時間:1.44分、m/z=231 [M+H]+(LCMS分析条件 method K-2)
A reaction vessel was charged with 10% palladium carbon (54.33% wet, 3.39 g, 1.45 mmol, 3 mol% on Pd metal basis) and 2-methyltetrahydrofuran (75 g). The atmosphere was replaced with nitrogen and hydrogen at 25°C, and the mixture was stirred under a hydrogen atmosphere (0.40 MPaG) for 2 hours. A solution of compound A11 (42.39 g) obtained in Example 2-8-1 and 2-methyltetrahydrofuran (22 g) were added. After stirring for 1 hour and 30 minutes under a hydrogen atmosphere (0.20 MPaG), a sample was taken, and the completion of the reaction was confirmed by HPLC analysis. The reaction mixture was filtered, and the cake was washed twice with 2-methyltetrahydrofuran (75 g). The mixture of the filtrate and washings was concentrated under reduced pressure to obtain a solution containing compound A12 (30.76 g).
LCMS (ESI) of compound A12: Retention time: 1.44 minutes, m/z = 231 [M+H] + (LCMS analysis conditions method K-2)
実施例2-8-3Example 2-8-3
化合物A14:(tert-ブチル (3S)-3-[[(2S)-2-[ベンジルオキシカルボニル(メチル)アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound A14: (tert-Butyl (3S)-3-[[(2S)-2-[benzyloxycarbonyl(methyl)amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoate)

反応容器に実施例2-8-2で得られた化合物A12の溶液(30.76g)、(S)-2-(((ベンジルオキシ)カルボニル)(メチル)アミノ)-2-シクロペンチル酢酸(Cas番号2411591-78-9、16.96g、58.2mmol)、2-メチルテトラヒドロフラン(40g)、アセトニトリル(17g)、DIPEA(27.74g、215mmol)を25℃にて加えた。HATU(27.49g、72.3mmol)を10分かけて添加した。添加終了後3時間攪拌した後にサンプリングし、HPLC分析で反応完結を確認した。トルエン(30g)、5%炭酸カリウム水溶液(23g)、1-メチルイミダゾール(3.97g、48.4mmol)を加え、30分攪拌した。2.5%アンモニア水溶液(88g)、2-メチルテトラヒドロフラン(25g)を加えた。10分撹拌した後に静置し、水層を除去した。得られた有機層を2.5%アンモニア水溶液(113g)、10%硫酸水素ナトリウム一水和物水溶液(113g、2回)、5%炭酸カリウム水溶液(113g)により洗浄した後、減圧条件下で濃縮した。2-メチルテトラヒドロフラン(42g)を加えて減圧条件下で濃縮し、化合物A14を含む溶液(52.78g)を得た。
化合物A14のLCMS(ESI):保持時間:4.10分、m/z=526 [M+Na]+(LCMS分析条件 method K-2)
The solution of compound A12 obtained in Example 2-8-2 (30.76 g), (S)-2-(((benzyloxy)carbonyl)(methyl)amino)-2-cyclopentylacetic acid (Cas number 2411591-78-9, 16.96 g, 58.2 mmol), 2-methyltetrahydrofuran (40 g), acetonitrile (17 g), and DIPEA (27.74 g, 215 mmol) were added to a reaction vessel at 25 ° C. HATU (27.49 g, 72.3 mmol) was added over 10 minutes. After stirring for 3 hours after the completion of the addition, the mixture was sampled and the completion of the reaction was confirmed by HPLC analysis. Toluene (30 g), 5% aqueous potassium carbonate solution (23 g), and 1-methylimidazole (3.97 g, 48.4 mmol) were added and stirred for 30 minutes. 2.5% aqueous ammonia solution (88 g) and 2-methyltetrahydrofuran (25 g) were added. After stirring for 10 minutes, the mixture was allowed to stand and the aqueous layer was removed. The resulting organic layer was washed with 2.5% aqueous ammonia solution (113 g), 10% aqueous sodium hydrogen sulfate monohydrate solution (113 g, twice), and 5% aqueous potassium carbonate solution (113 g), and then concentrated under reduced pressure. 2-Methyltetrahydrofuran (42 g) was added and the mixture was concentrated under reduced pressure to obtain a solution containing compound A14 (52.78 g).
LCMS (ESI) of compound A14: Retention time: 4.10 minutes, m/z = 526 [M+Na] + (LCMS analysis conditions method K-2)
実施例2-8-4Example 2-8-4
化合物A15:(tert-ブチル (3S)-3-[[(2S)-2-シクロペンチル-2-(メチルアミノ)アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound A15: (tert-Butyl (3S)-3-[[(2S)-2-cyclopentyl-2-(methylamino)acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoate)

反応容器に10%パラジウム炭素(54.33% wet、3.39g、1.45mmol、3mol% on Pd metal basis)と2-メチルテトラヒドロフラン(75g)を加えた。25℃で窒素置換し、水素置換して水素雰囲気下(0.40MPaG)で2時間攪拌した。実施例2-8-3で得られた化合物A14の溶液(52.78g)と2-メチルテトラヒドロフラン(15g)を加えた後、30℃に昇温した。水素雰囲気下(0.20MPaG)で2時間攪拌した後にサンプリングし、HPLC分析で反応完結を確認した。反応混合物を濾過した後、ケーキを2-メチルテトラヒドロフラン(75g)により2回洗浄した。濾液と洗浄液の混合溶液を減圧条件下で濃縮した後、アセトニトリル(75g)を加えて減圧条件下で濃縮する操作を2回繰り返し、化合物A15を含む溶液(42.5mL)を得た。
化合物A15のLCMS(ESI):保持時間:2.96分、m/z=370 [M+H]+(LCMS分析条件 method K-1)
A reaction vessel was charged with 10% palladium carbon (54.33% wet, 3.39 g, 1.45 mmol, 3 mol% on Pd metal basis) and 2-methyltetrahydrofuran (75 g). The atmosphere was replaced with nitrogen at 25°C, and the mixture was stirred under a hydrogen atmosphere (0.40 MPaG) for 2 hours. The solution of compound A14 obtained in Example 2-8-3 (52.78 g) and 2-methyltetrahydrofuran (15 g) were added, and the temperature was raised to 30°C. After stirring for 2 hours under a hydrogen atmosphere (0.20 MPaG), a sample was taken, and the completion of the reaction was confirmed by HPLC analysis. The reaction mixture was filtered, and the cake was washed twice with 2-methyltetrahydrofuran (75 g). The mixed solution of the filtrate and washings was concentrated under reduced pressure, and then acetonitrile (75 g) was added and concentrated under reduced pressure. This operation was repeated twice to obtain a solution containing compound A15 (42.5 mL).
LCMS (ESI) of compound A15: Retention time: 2.96 minutes, m/z = 370 [M+H] + (LCMS analysis conditions method K-1)
実施例2-8-5Example 2-8-5
化合物A16:(tert-ブチル (3S)-3-[[(2S)-2-シクロペンチル-2-(メチルアミノ)アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノアート)塩酸塩の合成Synthesis of Compound A16: (tert-Butyl (3S)-3-[[(2S)-2-cyclopentyl-2-(methylamino)acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoate) hydrochloride
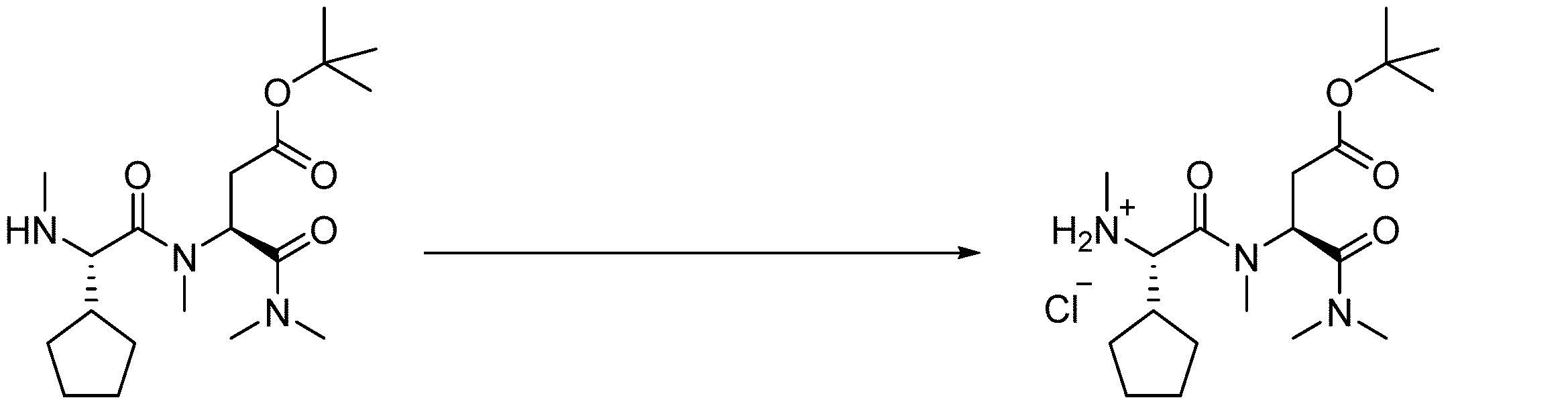
反応容器に実施例2-8-4で得られた化合物A15の溶液(42.5mL)とアセトニトリル(8.0g)を加えた。40℃にてMTBE(65g)を加えた後、ピリジン塩酸塩のアセトニトリル溶液(16.94w/w%、4.50g)を30分かけて滴下した。1時間攪拌した後、ピリジン塩酸塩のアセトニトリル溶液(16.94w/w%、31.34g)を3時間30分かけて滴下し、アセトニトリル(14g)を加えた。1時間攪拌した後、6時間かけて10℃まで冷却した。10℃にてさらに11時間攪拌した後、スラリーを濾過した。得られた固体をMTBE(38g)により2回洗浄した後、減圧条件下にて乾燥し、化合物 A16(15.68g)を得た。
化合物A16のLCMS(ESI):保持時間:2.92分、m/z=370 [M+H]+(LCMS分析条件 method K-1)
The solution of compound A15 obtained in Example 2-8-4 (42.5 mL) and acetonitrile (8.0 g) were added to the reaction vessel. After adding MTBE (65 g) at 40°C, a solution of pyridine hydrochloride in acetonitrile (16.94 w/w%, 4.50 g) was added dropwise over 30 minutes. After stirring for 1 hour, a solution of pyridine hydrochloride in acetonitrile (16.94 w/w%, 31.34 g) was added dropwise over 3 hours and 30 minutes, and acetonitrile (14 g) was added. After stirring for 1 hour, the mixture was cooled to 10°C over 6 hours. After stirring for an additional 11 hours at 10°C, the slurry was filtered. The obtained solid was washed twice with MTBE (38 g) and then dried under reduced pressure to obtain compound A16 (15.68 g).
LCMS (ESI) of compound A16: Retention time: 2.92 minutes, m/z = 370 [M+H] + (LCMS analysis conditions method K-1)
実施例2-8-6Example 2-8-6
化合物30:(tert-ブチル (S)-3-((S)-2-(1-(((ベンジルオキシ)カルボニル)(メチル)アミノ)-N-メチルシクロブタン-1-カルボキサミド)-2-シクロペンチル-N-メチルアセトアミド)-4-(ジメチルアミノ)-3-オキソ-ブタノアート)の合成Synthesis of Compound 30: (tert-Butyl (S)-3-((S)-2-(1-(((benzyloxy)carbonyl)(methyl)amino)-N-methylcyclobutane-1-carboxamido)-2-cyclopentyl-N-methylacetamido)-4-(dimethylamino)-3-oxo-butanoate)
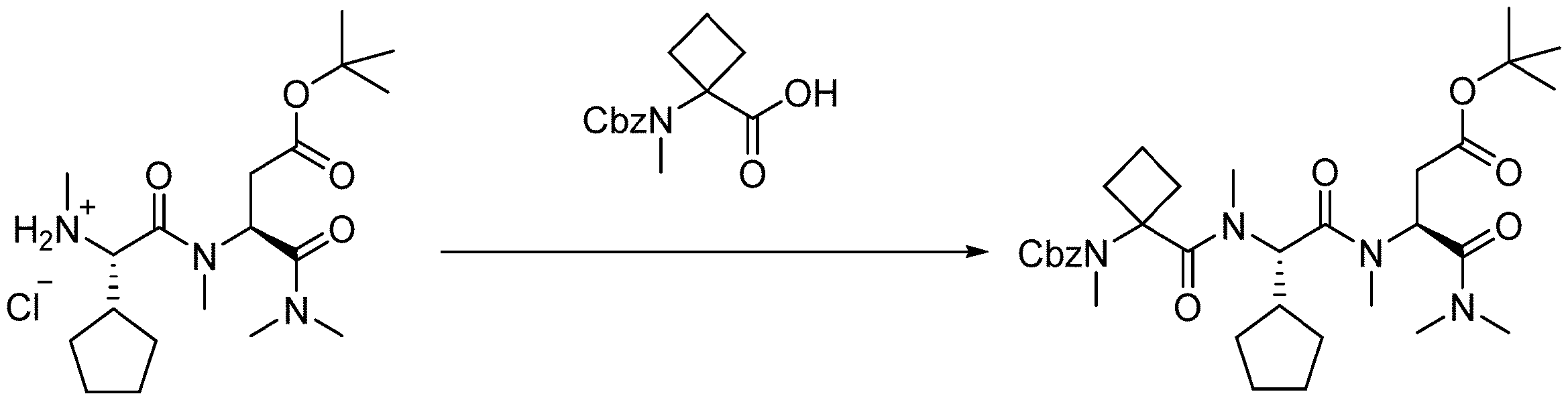
窒素で置換した反応釜(2L)にアセトニトリル(224mL)、DIPEA(150g)、実施例2-8-5で得られた化合物A16(74.56g)、及び1-(ベンジルオキシカルボニル(メチル)アミノ)シクロブタン酸(Cas番号1408729-60-1、131g)をいれ、室温にて10分間撹拌した。完全溶解を確認後、混合溶液に対して50 wt.%プロピルホスホン酸無水物 2-メチルテトラヒドロフラン溶液(339g)を15分間で添加した。添加終了後、内温60℃まで昇温し、2時間攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調整法K)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。5%炭酸カリウム水溶液(447mL)、N,N-ジメチル-4-アミノピリジン(90g)を加え、さらに1時間30分攪拌した。内温を25℃に冷却し、トルエン(373mL)を加え、分液操作により水層を排出した。有機層を4%硫酸水溶液(447mLx2)、5%炭酸ナトリウム水溶液(447mLx2)で洗浄した。得られた有機層を外温60℃で226mLまで減圧濃縮した。減圧濃縮後、THF(447mL)を加えて、226mLまで減圧濃縮を2回繰り返し、化合物30のTHF溶液(167.76g)を得た。得られた溶液は収率算出せず、実施例2-9に用いた
化合物30のLCMS(ESI):保持時間:7.02分、m/z=637.43 [M+Na]+(LCMS分析条件 method K-1)
Acetonitrile (224 mL), DIPEA (150 g), compound A16 (74.56 g) obtained in Example 2-8-5, and 1-(benzyloxycarbonyl(methyl)amino)cyclobutanoic acid (Cas number 1408729-60-1, 131 g) were placed in a reaction vessel (2 L) purged with nitrogen, and stirred at room temperature for 10 minutes. After confirming complete dissolution, 50 wt.% propylphosphonic anhydride 2-methyltetrahydrofuran solution (339 g) was added to the mixed solution over 15 minutes. After the addition was completed, the internal temperature was raised to 60°C and stirred for 2 hours. The reaction mixture was sampled and sample preparation (sample preparation method K) was performed, and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (reaction conversion rate calculation formula 1). 5% potassium carbonate aqueous solution (447 mL) and N,N-dimethyl-4-aminopyridine (90 g) were added, and the mixture was further stirred for 1 hour and 30 minutes. The internal temperature was cooled to 25°C, toluene (373mL) was added, and the aqueous layer was discharged by liquid separation. The organic layer was washed with 4% aqueous sulfuric acid solution (447mL x 2) and 5% aqueous sodium carbonate solution (447mL x 2). The obtained organic layer was concentrated under reduced pressure to 226mL at an external temperature of 60°C. After the concentration under reduced pressure, THF (447mL) was added, and the concentration under reduced pressure to 226mL was repeated twice to obtain a THF solution of compound 30 (167.76g). The yield of the obtained solution was not calculated, and the LCMS (ESI) of compound 30 used in Example 2-9: retention time: 7.02 minutes, m/z = 637.43 [M+Na] + (LCMS analysis conditions, method K-1)
実施例2-9Example 2-9
化合物31:(tert-ブチル (S)-3-((S)-2-シクロペンチル-N-メチル-2-(N-メチル-1-(メチルアミノ)シクロブタン-1-カルボキサミド)アセトアミド)-4-(ジメチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound 31: (tert-Butyl (S)-3-((S)-2-cyclopentyl-N-methyl-2-(N-methyl-1-(methylamino)cyclobutane-1-carboxamido)acetamido)-4-(dimethylamino)-4-oxo-butanoate)

耐圧反応釜(5L)に実施例2-8で得られた化合物26のTHF溶液(163.97g)、THF(665mL)、4.57wt.%Pd/C(14.71g)をいれ、外温を25℃に設定し、撹拌した。0.20MPaの窒素で3回置換し、さらに0.20MPaの水素で3回置換し、0.20MPaの水素雰囲気下、1時間攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応容器内を0.20MPaの窒素で3回置換し、濾過によってPd/Cを取り除いた。取り除いたPd/CをTHF(444mL)で3回洗浄した。ろ液と洗浄液を混合し、外温50℃で174mLまで減圧濃縮した。アセトニトリル(444mL)を加え、174mLまで減圧濃縮を2回繰り返し化合物31のアセトニトリル溶液(100.91g)を得た。得られた溶液は収率算出せず、実施例2-10に用いた
化合物31のLCMS(ESI):保持時間:2.63分、m/z=481.43 [M+H]+(LCMS分析条件 method K-2)
A THF solution (163.97 g) of compound 26 obtained in Example 2-8, THF (665 mL), and 4.57 wt. % Pd/C (14.71 g) were placed in a pressure-resistant reaction vessel (5 L), and the external temperature was set to 25 ° C. and stirred. The atmosphere was replaced with 0.20 MPa nitrogen three times, and further replaced with 0.20 MPa hydrogen three times, and stirred for 1 hour under a 0.20 MPa hydrogen atmosphere. The reaction mixture was sampled and prepared as a sample (sample preparation method 1), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). The inside of the reaction vessel was replaced with 0.20 MPa nitrogen three times, and Pd/C was removed by filtration. The removed Pd/C was washed three times with THF (444 mL). The filtrate and washings were mixed and concentrated under reduced pressure to 174 mL at an external temperature of 50 ° C. Acetonitrile (444 mL) was added, and the mixture was concentrated under reduced pressure to 174 mL twice to obtain an acetonitrile solution (100.91 g) of compound 31. The yield of the obtained solution was not calculated, and the LCMS (ESI) of compound 31 used in Example 2-10: retention time: 2.63 minutes, m/z = 481.43 [M+H] + (LCMS analysis condition: method K-2)
実施例2-10Example 2-10
化合物32:(ベンジル(2S,4R)-2-((1-(((S)-2-(((S)-4-(tert-ブトキシ)-1-(ジメチルアミノ)-1,4-ジオキサブタン-2-イル)(メチル)アミノ)-1-シクロペンチル-2-オキソエチル)(メチル)カルバモイル)シクロブチル)(メチル)カルバモイル)-4-エトキシピロリジン-1-カルボキシラート)の合成Synthesis of Compound 32: (benzyl (2S,4R)-2-((1-(((S)-2-(((S)-4-(tert-butoxy)-1-(dimethylamino)-1,4-dioxabutan-2-yl)(methyl)amino)-1-cyclopentyl-2-oxoethyl)(methyl)carbamoyl)cyclobutyl)(methyl)carbamoyl)-4-ethoxypyrrolidine-1-carboxylate)
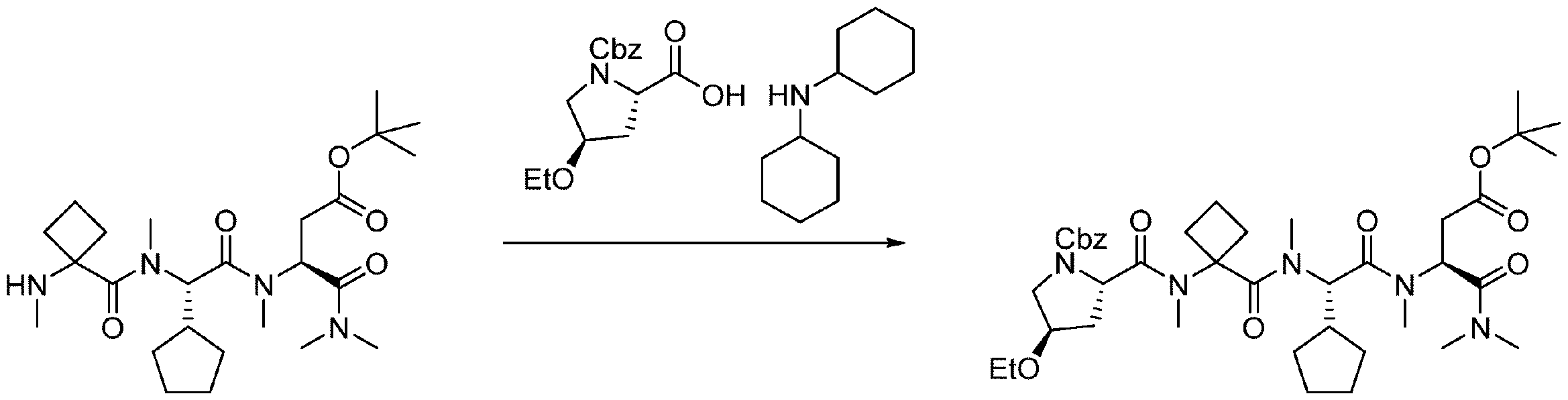
反応釜(2L)に(2S,4R)-1-((ベンジルオキシ)カルボニル)-4-エトキシピロリジン-2-カルボン酸ジシクロヘキシルアミン塩(227g)、2-メチルテトラヒドロフラン(767mL)を室温で加え攪拌した。4%硫酸水溶液(1151mL)を加え、分液操作により水層を排出し、有機層を4%硫酸水溶液(1151mL)、5%塩化ナトリウム水溶液(1151mL)でそれぞれ洗浄した。得られた有機層を外温50℃に設定し153mLまで減圧濃縮した。得られた濃縮液にアセトニトリル(384mL)を加え、153mLまで減圧濃縮した。反応釜に実施例2-9で得られた化合物31(89.31g)、アセトニトリル(384mL)、DIPEA(103g)を加え、室温にて撹拌した。TCFH(121g)を加え、反応釜の外温を50℃に昇温し5時間攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調整法K)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。水(307mL)、NMI(52.4g)を加え30分攪拌し、攪拌を停止し室温にて終夜保管した。トルエン(767mL)を室温で加え、分液操作により水層を排出した。有機層を10%アンモニア水(767mLx2)、4%硫酸水溶液(767mLx2)、5%炭酸ナトリウム水溶液(767mL)で洗浄した。得られた有機層を外温50℃で242mLまで減圧濃縮した。減圧濃縮後、THF(537mL)を加えて、242mLまで減圧濃縮を2回繰り返し、化合物32のTHF溶液(195.54g)を得た。得られた溶液は収率算出せず、実施例2-11に用いた
化合物32のLCMS(ESI):保持時間:4.30分、m/z=778.59 [M+Na]+(LCMS分析条件 method K-2)
(2S,4R)-1-((benzyloxy)carbonyl)-4-ethoxypyrrolidine-2-carboxylic acid dicyclohexylamine salt (227 g) and 2-methyltetrahydrofuran (767 mL) were added to a reaction vessel (2 L) at room temperature and stirred. 4% aqueous sulfuric acid solution (1151 mL) was added, the aqueous layer was discharged by a separation operation, and the organic layer was washed with 4% aqueous sulfuric acid solution (1151 mL) and 5% aqueous sodium chloride solution (1151 mL), respectively. The obtained organic layer was set to an external temperature of 50° C. and concentrated under reduced pressure to 153 mL. Acetonitrile (384 mL) was added to the obtained concentrated liquid, and the mixture was concentrated under reduced pressure to 153 mL. Compound 31 (89.31 g), acetonitrile (384 mL), and DIPEA (103 g) obtained in Example 2-9 were added to the reaction vessel, and the mixture was stirred at room temperature. TCFH (121 g) was added, the external temperature of the reaction vessel was raised to 50 ° C., and the mixture was stirred for 5 hours. The reaction mixture was sampled to prepare a sample (sample preparation method K), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). Water (307 mL) and NMI (52.4 g) were added and stirred for 30 minutes, and the stirring was stopped and the mixture was stored at room temperature overnight. Toluene (767 mL) was added at room temperature, and the aqueous layer was discharged by liquid separation. The organic layer was washed with 10% aqueous ammonia (767 mL x 2), 4% aqueous sulfuric acid (767 mL x 2), and 5% aqueous sodium carbonate (767 mL). The obtained organic layer was concentrated under reduced pressure to 242 mL at an external temperature of 50 ° C. After the concentration under reduced pressure, THF (537 mL) was added, and the concentration under reduced pressure to 242 mL was repeated twice to obtain a THF solution of compound 32 (195.54 g). The yield of the obtained solution was not calculated. Compound 32 used in Example 2-11: LCMS (ESI): retention time: 4.30 minutes, m/z = 778.59 [M+Na] + (LCMS analysis condition: method K-2)
実施例2-11Example 2-11
化合物33:(tert-ブチル (S)-3-((S)-2-シクロペンチル-2-(1-((2S,4R)-4-エトキシ-N-メチルピロリジン-2-カルボキサミド)-N-メチルシクロブタン-1-カルボキサミド)-N-メチルアセトアミド)-4-(ジメチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound 33: (tert-Butyl (S)-3-((S)-2-cyclopentyl-2-(1-((2S,4R)-4-ethoxy-N-methylpyrrolidine-2-carboxamido)-N-methylcyclobutane-1-carboxamido)-N-methylacetamido)-4-(dimethylamino)-4-oxo-butanoate)

耐圧反応釜(5L)に4.57wt.%Pd/C(8.53g)、THF(388.5mL)をいれ、外温を25℃に設定し撹拌した。0.20MPaの窒素で3回置換し、さらに0.20MPaの水素で3回置換し、0.40MPaの水素雰囲気下、3時間攪拌した。実施例2-10で得られた化合物32のTHF溶液(179.08g)、THF(531mL)を加えた。0.20MPaの窒素で3回置換し、さらに0.20MPaの水素で3回置換し、0.20MPaの水素雰囲気下、6時間攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応容器内を0.20MPaの窒素で3回置換し、濾過によってPd/Cを取り除いた。取り除いたPd/Cを2-メチルテトラヒドロフラン(232mL)で3回洗浄した。ろ液と洗浄液を混合し、外温50℃で182mLまで減圧濃縮した。2-メチルテトラヒドロフラン(774mL)を加え、182mLまで減圧濃縮した。得られた濃縮液に2-メチルテトラヒドロフラン(55.2mL)を加え外温54℃に昇温し攪拌した。ヘプタン158mLを加え、実施例2-11と同様の操作により得られた、化合物33の結晶(0.396g)をヘプタン(15.85mL)に懸濁させ反応釜に加えた。外温54℃で3時間攪拌し、外温を6.5時間かけて22℃に冷却し、さらに10時間攪拌した。ヘプタン(143mL)を1時間かけて加え、1時間攪拌した。さらにヘプタン(634mL)を4時間かけて加え、1時間攪拌した。外温を2.5時間かけて10℃に冷却し、14時間攪拌した。反応混合物を桐山ろうとで濾過し、得られた結晶をヘプタン(396mL)で2回洗浄した。得られた結晶を外温を40℃に設定して減圧条件にて2.5時間乾燥した。乾燥末を回収し、白色の粉末(72.9g)を得た。
化合物33のLCMS(ESI):保持時間:4.50分、m/z=622.53 [M+H]+(LCMS分析条件 method K-1)
In a pressure-resistant reactor (5 L), 4.57 wt. % Pd/C (8.53 g) and THF (388.5 mL) were placed, and the external temperature was set to 25 ° C. and stirred. The mixture was purged with 0.20 MPa nitrogen three times, further purged with 0.20 MPa hydrogen three times, and stirred for 3 hours under a 0.40 MPa hydrogen atmosphere. A THF solution (179.08 g) of compound 32 obtained in Example 2-10 and THF (531 mL) were added. The mixture was purged with 0.20 MPa nitrogen three times, further purged with 0.20 MPa hydrogen three times, and stirred for 6 hours under a 0.20 MPa hydrogen atmosphere. The reaction mixture was sampled and prepared as a sample (sample preparation method 1), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (calculation formula 1 for reaction conversion rate). The inside of the reaction vessel was replaced with nitrogen at 0.20 MPa three times, and Pd/C was removed by filtration. The removed Pd/C was washed three times with 2-methyltetrahydrofuran (232 mL). The filtrate and the washings were mixed and concentrated under reduced pressure to 182 mL at an external temperature of 50° C. 2-Methyltetrahydrofuran (774 mL) was added, and the mixture was concentrated under reduced pressure to 182 mL. 2-Methyltetrahydrofuran (55.2 mL) was added to the resulting concentrated solution, and the external temperature was raised to 54° C. and stirred. 158 mL of heptane was added, and the crystals of compound 33 (0.396 g), obtained by the same operation as in Example 2-11, were suspended in heptane (15.85 mL) and added to the reaction vessel. The mixture was stirred at an external temperature of 54° C. for 3 hours, cooled to 22° C. over 6.5 hours, and further stirred for 10 hours. Heptane (143 mL) was added over 1 hour, and stirred for 1 hour. Further, heptane (634 mL) was added over 4 hours, and the mixture was stirred for 1 hour. The external temperature was cooled to 10°C over 2.5 hours, and the mixture was stirred for 14 hours. The reaction mixture was filtered through a Kiriyama funnel, and the obtained crystals were washed twice with heptane (396 mL). The obtained crystals were dried for 2.5 hours under reduced pressure with the external temperature set to 40°C. The dried powder was collected, and a white powder (72.9 g) was obtained.
LCMS (ESI) of compound 33: Retention time: 4.50 minutes, m/z = 622.53 [M+H] + (LCMS analysis conditions method K-1)
耐圧反応釜(100mL)に4.57wt.%Pd/C(324mg)、THF(9.00mL)、をいれ、外温を25℃に設定し撹拌した。0.20MPaの窒素で3回置換し、さらに0.20MPaの水素で3回置換し、0.40MPaの水素雰囲気下、2時間攪拌した。実施例2-10で得られた化合物32のTHF溶液(6.28g)、THF(24.0mL)を加えた。0.20MPaの窒素で3回置換し、さらに0.20MPaの水素で3回置換し、0.20MPaの水素雰囲気下、反応釜の内温を40℃に昇温し2時間攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応容器内を0.20MPaの窒素で3回置換し、濾過によってPd/Cを取り除いた。取り除いたPd/Cをテトラヒドロフラン(6.30mL)で3回洗浄した。ろ液と洗浄液を混合し、外温50℃で6.3mLまで減圧濃縮した。2-メチルテトラヒドロフラン(21.0mL)を加え、6.3mLまで減圧濃縮を3回繰り返した。得られた濃縮液の一部(5.5g)を反応釜(50mL)にいれ、2-メチルテトラヒドロフラン(1.34mL)を加えた。得られた溶液を内温67.5℃に昇温し攪拌した。ヘプタン(35.6mL)を加え、実施例2-11と同様の操作により得られた、化合物33の結晶(322mg)をヘプタン(0.40mL)に懸濁させ反応釜に加えた。内温67.5℃で1時間以上攪拌し、内温を2時間以上かけて57.5℃に冷却し、さらに1時間以上攪拌した。さらに内温を1時間以上かけて47.5℃に冷却し、さらに1時間以上攪拌した。さらに内温を2時間以上かけて10℃以下に冷却し、さらに1時間以上攪拌した。反応混合物を桐山ろうとで濾過し、得られた結晶をヘプタン(10.0mL)で2回、10℃に冷却したヘプタン/2-メチルテトラヒドロフラン(6/1)溶液(10.0mL)で洗浄した。得られた結晶を外温を40℃に設定して減圧条件にて2時間乾燥した。乾燥末を回収し、白色の粉末(1.39g)を得た。 4.57 wt. % Pd/C (324 mg) and THF (9.00 mL) were placed in a pressure-resistant reaction vessel (100 mL), and the external temperature was set to 25°C and stirred. The mixture was purged with 0.20 MPa nitrogen three times, then purged with 0.20 MPa hydrogen three times, and stirred for 2 hours under a hydrogen atmosphere of 0.40 MPa. A THF solution (6.28 g) of compound 32 obtained in Example 2-10 and THF (24.0 mL) were added. The mixture was purged with 0.20 MPa nitrogen three times, then purged with 0.20 MPa hydrogen three times, and stirred for 2 hours under a hydrogen atmosphere of 0.20 MPa, with the internal temperature of the reaction vessel raised to 40°C. The reaction mixture was sampled and sample preparation (sample preparation method 1) was performed, and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). The inside of the reaction vessel was replaced with nitrogen at 0.20 MPa three times, and Pd/C was removed by filtration. The removed Pd/C was washed three times with tetrahydrofuran (6.30 mL). The filtrate and the washings were mixed and concentrated under reduced pressure to 6.3 mL at an external temperature of 50°C. 2-Methyltetrahydrofuran (21.0 mL) was added, and the concentration under reduced pressure to 6.3 mL was repeated three times. A part of the obtained concentrated liquid (5.5 g) was placed in a reaction vessel (50 mL), and 2-methyltetrahydrofuran (1.34 mL) was added. The obtained solution was heated to an internal temperature of 67.5°C and stirred. Heptane (35.6 mL) was added, and the crystals of compound 33 (322 mg) obtained by the same operation as in Example 2-11 were suspended in heptane (0.40 mL) and added to the reaction vessel. The mixture was stirred at an internal temperature of 67.5°C for 1 hour or more, cooled to 57.5°C over 2 hours or more, and further stirred for 1 hour or more. The internal temperature was further cooled to 47.5°C over 1 hour or more, and further stirred for 1 hour or more. The internal temperature was further cooled to below 10°C over 2 hours or more, and further stirred for 1 hour or more. The reaction mixture was filtered through a Kiriyama funnel, and the obtained crystals were washed twice with heptane (10.0 mL) and with a heptane/2-methyltetrahydrofuran (6/1) solution (10.0 mL) cooled to 10°C. The obtained crystals were dried for 2 hours under reduced pressure with an external temperature set to 40°C. The dried powder was collected, and a white powder (1.39 g) was obtained.
実施例2-12Example 2-12
化合物34:((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸ジシクロヘキシルアミン塩)の合成Synthesis of Compound 34: ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt)

国際公開第2020/189540号に記載の方法に従い、4-ブロモ-2-メトキシ-1-(トリフルオロメチル)ベンゼン(190g)から、化合物34(206.8g)を得た。
化合物34のNMR:1H NMR (500 MHz, methanol-d4) 7.38-7.30 (m, 5H), 7.27-7.26 (m, 1H), 6.97 (s, 1H), 6.84-6.82 (m, 1H), 5.07 (d, J = 13.0 Hz, 1H), 5.04 (d, J = 12.5 Hz, 1H), 4.06-4.03 (m, 1H), 3.83 (s, 3H), 3.14-3.11 (m, 2H), 2.69 (t, J = 8.0 Hz, 2H), 2.15-2.05 (m, 1H), 2.04-2.01 (m, 4H), 2.00-1.90 (m, 1H), 1.84-1.82 (m, 4H), 1.70-1.64 (m, 2H), 1.36-1.23 (m, 8H), 1.20-1.12 (m, 2H).
According to the method described in WO 2020/189540, compound 34 (206.8 g) was obtained from 4-bromo-2-methoxy-1-(trifluoromethyl)benzene (190 g).
NMR of compound 34: 1 H NMR (500 MHz, methanol-d 4 ) 7.38-7.30 (m, 5H), 7.27-7.26 (m, 1H), 6.97 (s, 1H), 6.84-6.82 (m, 1H), 5.07 (d, J = 13.0 Hz, 1H), (d, J = 12.5 Hz, 1H), 4.06-4.03 (m, 1H), 3.83 (s, 3H), 3.14-3.11 (m, 2H), 2.69 (t, J = 8.0 Hz, 2H), 2.15-2.05 (m, 1H), 2.04-2.01 (m, 4H) , 2.00-1.90 (m, 1H), 1.84-1.82 (m, 4H), 1.70-1.64 (m, 2H), 1.36-1.23 (m, 8H), 1.20-1.12 (m, 2H).
実施例2-12-1
化合物34-α1:ベンジル(S)-2-((tert-ブトキシカルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノアートの合成
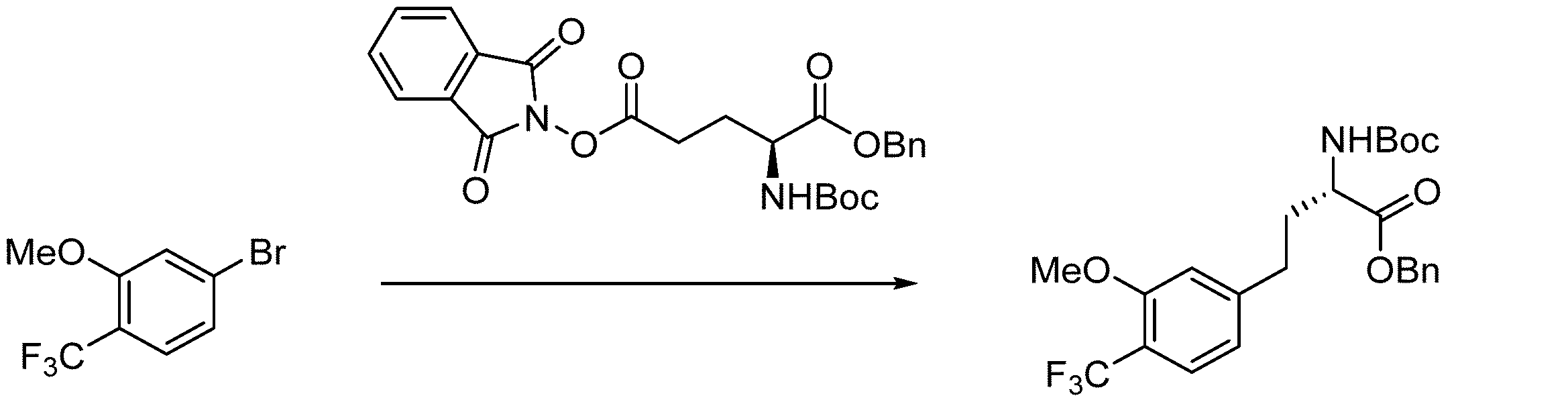
Synthesis of compound 34-α1: benzyl (S)-2-((tert-butoxycarbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoate
反応釜(1000L)に臭化ニッケル三水和物(1.23kg)、1,3-ジメチル-2-イミダゾリジノン(98.8kg)を混ぜスラリーとして仕込み、その後4,4’-ジ-tert-ブチル-2,2-ジピリジル(1.21kg)を仕込み、20.8~22.7℃で28分間撹拌した。国際公開第2020/189540号に従い合成した1-ベンジル 5-(1,3-ジオキソイソインドリン-2-イル) (tert-ブトキシカルボニル)-L-グルタメート(31.0kg)と4-ブロモ-2-メトキシ-1-(トリフルオロメチル)ベンゼン(24.57kg)、1,3-ジメチル-2-イミダゾリジノン(230kg)、N-メチルモルホリン(3.25kg)を仕込んだ後に内温8.7℃まで冷却し、亜鉛(12.6kg)を仕込んだ。撹拌し、温度が安定したのちにクロロトリメチルシラン(21.0kg)を内温8.7~11.9℃の範囲において3時間かけて滴下した。反応混合物をサンプリングしてサンプル調整(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。内温を2.6℃まで冷却したのちに、15%塩化アンモニウム水溶液(332kg)を滴下した。得られた混合液にトルエン(135kg)を、加え30分以上撹拌したのちに、セライトを用いてろ過した。トルエン(135kg)にてセライトを洗浄したのち、水層を廃棄した。有機層をエチレンジアミン四酢酸二ナトリウム(24.3kg)-2.5%アンモニア水混合溶液(465kg)、続く10%塩化ナトリウム水溶液(332kg)にて洗浄した。得られた有機層を外温40℃設定にて93Lまで濃縮したのち、表題の化合物を含む溶液87.3kgを得た。
化合物34-α1のLCMS(ESI):保持時間:7.23分、m/z=368.38 [M-Boc+H]+(LCMS分析条件 method H)
Nickel bromide trihydrate (1.23 kg) and 1,3-dimethyl-2-imidazolidinone (98.8 kg) were mixed as a slurry and charged into a reaction vessel (1000 L), followed by charging 4,4'-di-tert-butyl-2,2-dipyridyl (1.21 kg) and stirring at 20.8-22.7°C for 28 minutes. 1-benzyl 5-(1,3-dioxoisoindolin-2-yl) (tert-butoxycarbonyl)-L-glutamate (31.0 kg) synthesized according to WO 2020/189540, 4-bromo-2-methoxy-1-(trifluoromethyl)benzene (24.57 kg), 1,3-dimethyl-2-imidazolidinone (230 kg), and N-methylmorpholine (3.25 kg) were charged, and then cooled to an internal temperature of 8.7 ° C., and zinc (12.6 kg) was charged. After stirring and stabilizing the temperature, chlorotrimethylsilane (21.0 kg) was added dropwise over 3 hours at an internal temperature of 8.7 to 11.9 ° C. The reaction mixture was sampled and sample preparation (sample preparation method 1), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). After cooling the inside temperature to 2.6°C, 15% aqueous ammonium chloride solution (332 kg) was added dropwise. Toluene (135 kg) was added to the resulting mixture, which was stirred for 30 minutes or more, and then filtered using Celite. After washing the Celite with toluene (135 kg), the aqueous layer was discarded. The organic layer was washed with a mixed solution of disodium ethylenediaminetetraacetate (24.3 kg)-2.5% aqueous ammonia (465 kg), followed by a 10% aqueous sodium chloride solution (332 kg). The resulting organic layer was concentrated to 93 L with an external temperature set at 40°C, and 87.3 kg of a solution containing the title compound was obtained.
LCMS (ESI) of compound 34-α1: Retention time: 7.23 minutes, m/z = 368.38 [M-Boc+H] + (LCMS analysis conditions method H)
実施例2-12-2
化合物34-α2:(S)-2-((tert-ブトキシカルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸の合成
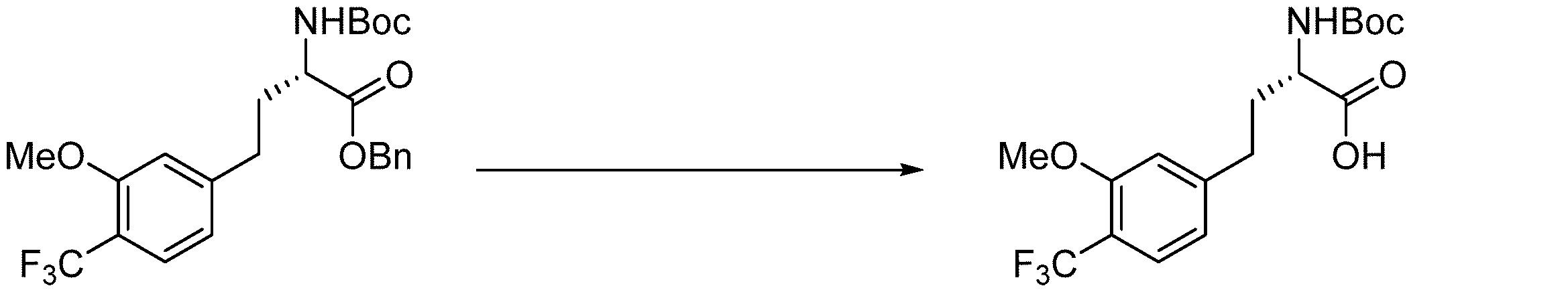
Synthesis of compound 34-α2: (S)-2-((tert-butoxycarbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid
耐圧反応釜(150L)に、ベンジル(S)-2-((tert-ブトキシカルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノアートを含む溶液(43.7kg)をトルエン(40.2kg)にて洗い込みながら仕込み、10%Pd/C(8.37kg)を加え撹拌を開始した。水素にて0.15MPaGまで加圧し、気相を水素へと置換したのちに内温22.6~29.2℃にて2時間2分撹拌した。反応混合物をサンプリングしてサンプル調整(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。混合液をろ過し、Pd/C残渣をトルエン(26.7kg)を用いて洗浄した。同様の操作にて再度ろ液を取得したのちに合一し、外温40℃設定にて155Lまで濃縮することで、表題の化合物を含む溶液155kgを得た。
化合物34-α2のLCMS(ESI):保持時間:5.72分、m/z=278.37 [M-Boc+H]+(LCMS分析条件 method H)
A solution (43.7 kg) containing benzyl (S)-2-((tert-butoxycarbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoate was charged into a pressure-resistant reactor (150 L) while washing with toluene (40.2 kg), 10% Pd/C (8.37 kg) was added, and stirring was started. The mixture was pressurized to 0.15 MPaG with hydrogen, the gas phase was replaced with hydrogen, and then stirring was performed for 2 hours and 2 minutes at an internal temperature of 22.6 to 29.2 ° C. The reaction mixture was sampled and sample preparation (sample preparation method 1), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (reaction conversion rate calculation formula 1). The mixture was filtered, and the Pd/C residue was washed with toluene (26.7 kg). The filtrate was obtained again by the same operation, then combined, and concentrated to 155 L at an external temperature setting of 40 ° C., to obtain 155 kg of a solution containing the title compound.
LCMS (ESI) of compound 34-α2: Retention time: 5.72 minutes, m/z = 278.37 [M-Boc+H] + (LCMS analysis conditions method H)
実施例2-12-3
化合物34-α2:(S)-2-((tert-ブトキシカルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸の合成

Synthesis of compound 34-α2: (S)-2-((tert-butoxycarbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid
反応容器(100mL)に臭化ニッケル三水和物(0.198g)、1,3-ジメチル-2-イミダゾリジノン(15mL)を混ぜスラリーとして仕込み、その後4,4’-ジ-tert-ブチル-2,2-ジピリジル(0.195g)を仕込み、25℃で30分以上間撹拌した。1-ベンジル 5-(1,3-ジオキソイソインドリン-2-イル) (tert-ブトキシカルボニル)-L-グルタメート(5.00g)、4-ブロモ-2-メトキシ-1-(トリフルオロメチル)ベンゼン(5.29g)、1,3-ジメチル-2-イミダゾリジノン(35mL)、N-メチルモルホリン(0.524g)を仕込んだ後に内温5~25℃の範囲で、亜鉛(2.03g)を仕込んだ。撹拌し、温度が安定したのちにクロロトリメチルシラン(3.38g)を内温5~25℃の範囲において2時間以上かけて滴下した。反応混合物をサンプリングしてサンプル調整(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応率適合を確認後、10%水酸化カリウム水溶液(35mL)を内温0~25℃の範囲で滴下した。反応混合物をサンプリングしてサンプル調整(サンプル調整法H)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式H)。得られた混合液にトルエン(15mL)を加えて撹拌したのちに、セライトを用いてろ過し、その後セライト残渣を水(5mL)-1,3-ジメチル-2-イミダゾリジノン(5mL)混合溶液にて洗浄することで、ろ液を得た。得られたろ液にトルエン(15mL)及び35%クエン酸水溶液(25g)を加え、pH4.5以下となっていることを確認したのちに水層を廃棄した。得られた有機層をエチレンジアミン四酢酸二ナトリウム(3.93g)-水混合溶液(74.2g)にて洗浄することで、表題の化合物を含む溶液25.6gを得た。
化合物34-α2のLCMS(ESI):保持時間:5.72分、m/z=278.37 [M-Boc+H]+(LCMS分析条件 method H)
Nickel bromide trihydrate (0.198 g) and 1,3-dimethyl-2-imidazolidinone (15 mL) were mixed and charged as a slurry in a reaction vessel (100 mL), then 4,4'-di-tert-butyl-2,2-dipyridyl (0.195 g) was charged and stirred at 25°C for 30 minutes or more. 1-Benzyl 5-(1,3-dioxoisoindolin-2-yl) (tert-butoxycarbonyl)-L-glutamate (5.00 g), 4-bromo-2-methoxy-1-(trifluoromethyl)benzene (5.29 g), 1,3-dimethyl-2-imidazolidinone (35 mL), and N-methylmorpholine (0.524 g) were charged, and then zinc (2.03 g) was charged at an internal temperature of 5 to 25°C. After stirring and stabilizing the temperature, chlorotrimethylsilane (3.38 g) was added dropwise over 2 hours or more at an internal temperature of 5 to 25°C. The reaction mixture was sampled and sample preparation (sample preparation method 1) was performed, and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). After confirming the reaction rate conformity, 10% potassium hydroxide aqueous solution (35 mL) was added dropwise at an internal temperature of 0 to 25°C. The reaction mixture was sampled and sample preparation (sample preparation method H) was performed, and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula H for reaction conversion rate). Toluene (15 mL) was added to the obtained mixture and stirred, followed by filtration using Celite, and the Celite residue was washed with a mixed solution of water (5 mL)-1,3-dimethyl-2-imidazolidinone (5 mL) to obtain a filtrate. Toluene (15 mL) and 35% citric acid aqueous solution (25 g) were added to the obtained filtrate, and after confirming that the pH was 4.5 or less, the aqueous layer was discarded. The obtained organic layer was washed with a mixed solution of disodium ethylenediaminetetraacetate (3.93 g) and water (74.2 g) to obtain 25.6 g of a solution containing the title compound.
LCMS (ESI) of compound 34-α2: Retention time: 5.72 minutes, m/z = 278.37 [M-Boc+H] + (LCMS analysis conditions method H)
実施例2-12-4
化合物35:((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノアート)の合成
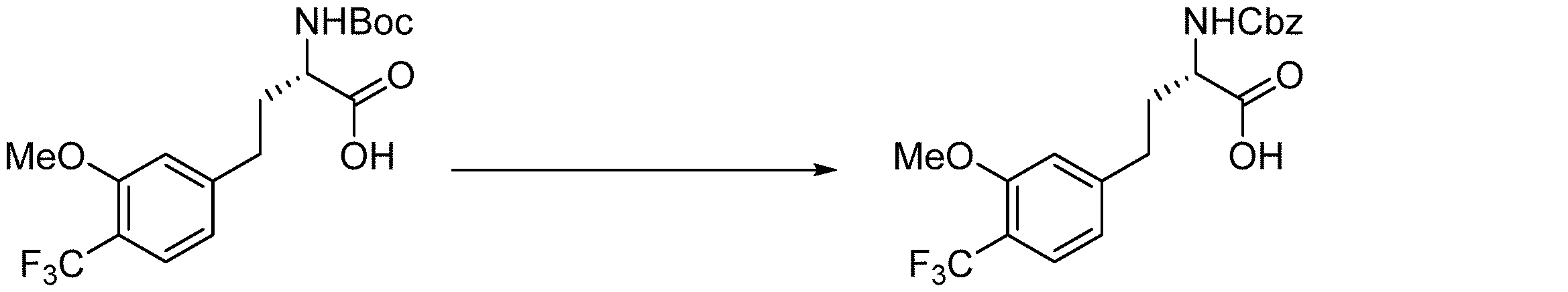
Synthesis of Compound 35: (S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoate
反応釜(1000L)に、(S)-2-((tert-ブトキシカルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸を含む溶液(155kg)を加え、メタンスルホン酸(24.7kg)を内温20.4℃~27.6℃の範囲で32分間かけて滴下した。その後昇温し、内温が59.5℃となった時点で反応開始とした。反応混合物をサンプリングしてサンプル調整(サンプル調整法H)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出1)。反応混合物を冷却し、水(186kg)を内温25.9℃~32.0℃の範囲で80分かけて滴下した。有機層を除去したのち、トルエン(124kg)で3回洗浄したのち、40%リン酸三カリウム水溶液(95.7kg)にてpHを6.52に調整した。その後アセトニトリル(61.2kg)を加え、再度40%リン酸三カリウム水溶液(25.4kg)を用いてpHを7.83に調整した。pH調整後の反応混合液に対して、N-(ベンジルオキシカルボニルオキシ)コハク酸イミド(16.0kg)を加え、反応開始とした。反応混合物をサンプリングしてサンプル調整(サンプル調整法1)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式H)。得られた反応混合物に対して再度40%リン酸三カリウム水溶液(44.7kg)を用いてpHを7.73に調整したのちに、メチル-tert-ブチルエーテル(46.3kg)及びヘプタン(42.4kg)を加え、三層に分離したことを確認した。上層のみを廃棄し、下層二層に対して再度メチル-tert-ブチルエーテル(115kg)を加えたのちに、24%水酸化ナトリウム水溶液(2.1kg)-塩化ナトリウム(12.4kg)-水(110.0kg)混合溶液を加え、水層を廃棄した。得られた有機層を24%水酸化ナトリウム水溶液(2.1kg)-塩化ナトリウム(12.4kg)-水(110.0kg)混合溶液で4回洗浄したのちに、有機層をサンプリングしてサンプル調整(サンプル調整法1)し、NMR分析により残留アセトニトリル量が0.44v/w以下であることを確認した(残留アセトニトリルの算出式H)。得られた有機層を0.2M水酸化ナトリウム水溶液(62.1kg)で洗浄したのちに、24%水酸化ナトリウム水溶液(2.1kg)-塩化ナトリウム(3.1kg)-水(72.3kg)混合溶液で洗浄した。洗浄後の有機層をサンプリングしてサンプル調整(サンプル調整法1)し、HPLC分析により残留不純物が0.10%以下であることを確認した。得られた有機層を1M塩酸(310kg)にて洗浄したのちに、浄後の有機層をサンプリングしてサンプル調整(サンプル調整法H)し、HPLC分析により残留不純物が0.10%以下であることを確認した。得られた有機層を10%食塩水(166kg)にて洗浄し、62Lまで濃縮した後に、トルエン(80.9kg)を加え再度62Lまで濃縮することでトルエンへと溶媒置換を実施した。得られた濃縮残渣にトルエン(204kg)を加えることで、表題の化合物を含む溶液(260kg)を得た。
化合物35のLCMS(ESI):保持時間:5.913分、m/z=368.39 [M-C02+H]+(LCMS分析条件 method H)
A solution (155 kg) containing (S)-2-((tert-butoxycarbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid was added to a reaction vessel (1000 L), and methanesulfonic acid (24.7 kg) was added dropwise over 32 minutes at an internal temperature of 20.4°C to 27.6°C. The temperature was then raised, and the reaction was started when the internal temperature reached 59.5°C. The reaction mixture was sampled and subjected to sample preparation (sample preparation method H), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation of reaction conversion rate 1). The reaction mixture was cooled, and water (186 kg) was added dropwise over 80 minutes at an internal temperature of 25.9°C to 32.0°C. After removing the organic layer, the mixture was washed three times with toluene (124 kg), and the pH was adjusted to 6.52 with 40% aqueous potassium phosphate tripotassium solution (95.7 kg). Acetonitrile (61.2 kg) was then added, and the pH was adjusted to 7.83 again using 40% tripotassium phosphate aqueous solution (25.4 kg). N-(benzyloxycarbonyloxy)succinimide (16.0 kg) was added to the reaction mixture after pH adjustment to start the reaction. The reaction mixture was sampled and sample preparation (sample preparation method 1), and it was confirmed by HPLC analysis that the reaction conversion rate was 99% or more (calculation formula H for reaction conversion rate). The pH of the obtained reaction mixture was adjusted to 7.73 again using 40% tripotassium phosphate aqueous solution (44.7 kg), and then methyl-tert-butyl ether (46.3 kg) and heptane (42.4 kg) were added, and it was confirmed that the mixture was separated into three layers. Only the upper layer was discarded, and methyl-tert-butyl ether (115 kg) was added again to the lower two layers, after which a mixed solution of 24% sodium hydroxide aqueous solution (2.1 kg)-sodium chloride (12.4 kg)-water (110.0 kg) was added, and the aqueous layer was discarded. The obtained organic layer was washed four times with a mixed solution of 24% sodium hydroxide aqueous solution (2.1 kg)-sodium chloride (12.4 kg)-water (110.0 kg), and the organic layer was sampled and sampled (sample preparation method 1), and it was confirmed by NMR analysis that the amount of residual acetonitrile was 0.44 v/w or less (calculation formula H for residual acetonitrile). The obtained organic layer was washed with a 0.2 M sodium hydroxide aqueous solution (62.1 kg), and then washed with a mixed solution of 24% sodium hydroxide aqueous solution (2.1 kg)-sodium chloride (3.1 kg)-water (72.3 kg). The organic layer after washing was sampled and subjected to sample preparation (sample preparation method 1), and it was confirmed by HPLC analysis that the residual impurities were 0.10% or less. The obtained organic layer was washed with 1M hydrochloric acid (310 kg), and then the organic layer after washing was sampled and subjected to sample preparation (sample preparation method H), and it was confirmed by HPLC analysis that the residual impurities were 0.10% or less. The obtained organic layer was washed with 10% saline (166 kg), concentrated to 62 L, and then toluene (80.9 kg) was added and concentrated again to 62 L to carry out solvent replacement with toluene. Toluene (204 kg) was added to the obtained concentrated residue to obtain a solution (260 kg) containing the title compound.
LCMS (ESI) of compound 35: Retention time: 5.913 minutes, m/z = 368.39 [M-C02+H] + (LCMS analysis conditions method H)
実施例2-12-5Example 2-12-5
化合物34:((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸ジシクロヘキシルアミン塩)の合成Synthesis of Compound 34: ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt)

反応釜(1000L)に((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノアート)を含む溶液(260kg)を加え内温を59.5℃まで昇温したのちに、ジシクロヘキシルアミン(16.1kg)を添加した。反応混合物を66.3℃まで昇温したのちに、ヘプタン(49.9kg)を1時間7分かけて滴下した。種晶として((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸ジシクロヘキシルアミン塩)(91.0g)をトルエン(2.51kg)-ヘプタン(511g)混合溶液に懸濁して仕込んだ。内温を67.0℃から52.1℃まで1時間38分かけて冷却したのちに、1時間以上撹拌を継続し、その後内温を50.1℃から23.0℃まで1時間55分かけて冷却した。冷却後14時間保温撹し、上澄み液をサンプリングしてサンプル調整(サンプル調整法H)し、HPLC分析により上澄み濃度が2mg/mL以下であることを確認した。得られたスラリーをろ過し、トルエン(47.4kg)-ヘプタン(37.2kg)混合溶液で洗浄することで、湿性末を得た。得られた湿性末を外温38℃にて50時間乾燥することで、((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸ジシクロヘキシルアミン塩)(25.6kg)を得た。
化合物34のLCMS(ESI):保持時間:5.89分、m/z=368.41 [M-CO2-DCHA+H]+(LCMS分析条件 method H)
A solution (260 kg) containing ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoate) was added to a reaction vessel (1000 L), and the internal temperature was raised to 59.5°C, after which dicyclohexylamine (16.1 kg) was added. The reaction mixture was heated to 66.3°C, and heptane (49.9 kg) was added dropwise over 1 hour and 7 minutes. As seed crystals, ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt) (91.0 g) was suspended in a toluene (2.51 kg)-heptane (511 g) mixed solution and charged. After cooling the internal temperature from 67.0 ° C. to 52.1 ° C. over 1 hour 38 minutes, stirring was continued for more than 1 hour, and then the internal temperature was cooled from 50.1 ° C. to 23.0 ° C. over 1 hour 55 minutes. After cooling, the mixture was stirred for 14 hours, and the supernatant was sampled and sample preparation (sample preparation method H) was performed. The supernatant concentration was confirmed to be 2 mg / mL or less by HPLC analysis. The obtained slurry was filtered and washed with a toluene (47.4 kg) - heptane (37.2 kg) mixed solution to obtain wet powder. The obtained wet powder was dried at an external temperature of 38 ° C. for 50 hours to obtain ((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt) (25.6 kg).
LCMS (ESI) of compound 34: Retention time: 5.89 minutes, m/z = 368.41 [M-CO2-DCHA+H] + (LCMS analysis conditions method H)
実施例2-13Example 2-13
化合物35:((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノアート)の合成Synthesis of Compound 35: (S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoate

反応釜(2L)に(S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタン酸ジシクロヘキシルアミン塩(57.8g)、2-メチルテトラヒドロフラン(253mL)を室温で加え攪拌した。4%硫酸水溶液(253mL)を加え、分液操作により水層を排出し、有機層を4%硫酸水溶液(253mL)、5%塩化ナトリウム水溶液(253mL)でそれぞれ洗浄した。得られた有機層を外温40℃に設定し100.8mLまで減圧濃縮した。得られた濃縮液に2-メチルテトラヒドロフラン(253mL)を加え、100.8mLまで減圧濃縮を2回繰り返し、表題の化合物を含む溶液(94.29g)を得た。
化合物35のLCMS(ESI):保持時間:5.92分、m/z=368.18 [M-CO2+H]+(LCMS分析条件LCMS分析条件 method K-1)
(S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoic acid dicyclohexylamine salt (57.8 g) and 2-methyltetrahydrofuran (253 mL) were added to a reaction vessel (2 L) at room temperature and stirred. 4% aqueous sulfuric acid solution (253 mL) was added, the aqueous layer was discharged by a separation operation, and the organic layer was washed with 4% aqueous sulfuric acid solution (253 mL) and 5% aqueous sodium chloride solution (253 mL). The obtained organic layer was set to an external temperature of 40° C. and concentrated under reduced pressure to 100.8 mL. 2-Methyltetrahydrofuran (253 mL) was added to the obtained concentrated solution, and reduced pressure concentration to 100.8 mL was repeated twice to obtain a solution containing the title compound (94.29 g).
LCMS (ESI) of compound 35: Retention time: 5.92 minutes, m/z = 368.18 [M-CO 2 +H] + (LCMS analysis conditions LCMS analysis conditions method K-1)
実施例2-14Example 2-14
化合物36:(tert-ブチル-(S)-3-((S)-2-(1-((2S,4R)-1-((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノイル)-4-エトキシ-N-メチルピロリジン-2-カルボキサミド)-N-メチルシクロブタン-1-カルボキサミド)-2-シクロペンチル-N-メチルアセトアミド)-4-(ジメチルアミノ)-4-オキソ-ブタノアート)の合成Synthesis of Compound 36: (tert-butyl-(S)-3-((S)-2-(1-((2S,4R)-1-((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoyl)-4-ethoxy-N-methylpyrrolidine-2-carboxamide)-N-methylcyclobutane-1-carboxamide)-2-cyclopentyl-N-methylacetamide)-4-(dimethylamino)-4-oxo-butanoate)

反応釜(2L)に実施例2-11で得られた化合物33(94.29g)、2-メチルテトラヒドロフラン(253mL)、DIPEA(46.2g)、実施例2-13で得られた化合物35(50.5g)を加え、外温を10℃に冷却して撹拌した。混合溶液に対して50 wt.%プロピルホスホン酸無水物 2-メチルテトラヒドロフラン溶液(124g)を15分間で添加した。反応釜の外温を25℃に昇温し1時間攪拌した。反応混合物をサンプリングしてサンプル調製(サンプル調整法K)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。5%炭酸カリウム水溶液(303mL)、NMI(6.67g)を加え30分攪拌し、攪拌を停止し室温にて終夜保管した。分液操作により水層を排出し、有機層を4%硫酸水溶液(303mLx2)洗浄した。有機層にヘプタン(182mL)、MTBE(121mL)、アセトニトリル(116mL)、2.5%炭酸カリウム水溶液(288mL)を加え分液操作により水層を排出した。得られた有機層にアセトニトリル(172mL)、2-メチルテトラヒドロフラン(101mL)、2.5%炭酸カリウム水溶液(434mL)を加え分液操作により水層を排出した。さらに得られた有機層にアセトニトリル(172mL)、2-メチルテトラヒドロフラン(101mL)、2.5%炭酸カリウム水溶液(434mL)を加え分液操作により水層を排出した。有機層を外温50℃で167mLまで減圧濃縮した。減圧濃縮後、IPAc(354mL)を加えて、167mLまで減圧濃縮を2回繰り返し、表題の化合物を含む溶液(170.11g)を得た。
化合物36のLCMS(ESI):保持時間:8.10分、m/z=1037.62 [M+H]+(LCMS分析条件 method K-1)
Compound 33 (94.29 g) obtained in Example 2-11, 2-methyltetrahydrofuran (253 mL), DIPEA (46.2 g), and compound 35 (50.5 g) obtained in Example 2-13 were added to a reaction vessel (2 L), and the external temperature was cooled to 10° C. and stirred. 50 wt.% propylphosphonic anhydride 2-methyltetrahydrofuran solution (124 g) was added to the mixed solution over 15 minutes. The external temperature of the reaction vessel was raised to 25° C. and stirred for 1 hour. The reaction mixture was sampled and prepared as a sample (sample preparation method K), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). 5% potassium carbonate aqueous solution (303 mL) and NMI (6.67 g) were added and stirred for 30 minutes, and the stirring was stopped and the mixture was stored at room temperature overnight. The aqueous layer was discharged by liquid separation, and the organic layer was washed with 4% sulfuric acid aqueous solution (303 mL x 2). To the organic layer, heptane (182 mL), MTBE (121 mL), acetonitrile (116 mL), and 2.5% potassium carbonate aqueous solution (288 mL) were added, and the aqueous layer was discharged by a liquid separation operation. To the obtained organic layer, acetonitrile (172 mL), 2-methyltetrahydrofuran (101 mL), and 2.5% potassium carbonate aqueous solution (434 mL) were added, and the aqueous layer was discharged by a liquid separation operation. Further, to the obtained organic layer, acetonitrile (172 mL), 2-methyltetrahydrofuran (101 mL), and 2.5% potassium carbonate aqueous solution (434 mL) were added, and the aqueous layer was discharged by a liquid separation operation. The organic layer was concentrated under reduced pressure to 167 mL at an external temperature of 50°C. After the concentration under reduced pressure, IPAc (354 mL) was added, and the concentration under reduced pressure to 167 mL was repeated twice, to obtain a solution containing the title compound (170.11 g).
LCMS (ESI) of compound 36: Retention time: 8.10 minutes, m/z = 1037.62 [M+H] + (LCMS analysis conditions method K-1)
実施例2-15Example 2-15
化合物20:(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジルオキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタン酸の合成Synthesis of compound 20: (3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoic acid

窒素で置換した反応釜に、室温にて(tert-ブチル-(S)-3-((S)-2-(1-((2S,4R)-1-((S)-2-(((ベンジルオキシ)カルボニル)アミノ)-4-(3-メトキシ-4-(トリフルオロメチル)フェニル)ブタノイル)-4-エトキシ-N-メチルピロリジン-2-カルボキサミド)-N-メチルシクロブタン-1-カルボキサミド)-2-シクロペンチル-N-メチルアセトアミド)-4-(ジメチルアミノ)-4-オキソ-ブタノアート)(47.8g)の酢酸イソプロピル(53.1g)溶液、酢酸イソプロピル(186mL)を加えて攪拌した。続いてHMDS(19.0g)を加え、反応釜の外温を0℃に設定し、15分撹拌した。TMSOTf (15.4g)を内温が20℃を超えないようにゆっくり滴下した。反応釜の外温を20℃に設定し、反応混合物を1時間撹拌した後、2-メチルテトラヒドロフラン(244mL)で希釈し、反応釜の外温を10℃に設定した。5%リン酸水素二カリウム水溶液(478mL)をゆっくり滴下したのち、撹拌を止め水層を排出した。得られた有機層を5%リン酸二水素ナトリウム(478mL)で洗浄した後、ジイソプロピルエチルアミン(9.0mL)を添加し、減圧濃縮することで化合物20の2-メチルテトラヒドロフラン溶液(120.1g)を得た。
化合物20のLCMS(ESI):保持時間:4.30 分、m/z=981.66 [M+Na]+(LCMS分析条件 method K-2)
In a reaction vessel replaced with nitrogen, a solution of (tert-butyl-(S)-3-((S)-2-(1-((2S,4R)-1-((S)-2-(((benzyloxy)carbonyl)amino)-4-(3-methoxy-4-(trifluoromethyl)phenyl)butanoyl)-4-ethoxy-N-methylpyrrolidine-2-carboxamide)-N-methylcyclobutane-1-carboxamide)-2-cyclopentyl-N-methylacetamide)-4-(dimethylamino)-4-oxo-butanoate) (47.8 g) in isopropyl acetate (53.1 g) and isopropyl acetate (186 mL) were added at room temperature and stirred. Then, HMDS (19.0 g) was added, the external temperature of the reaction vessel was set to 0 ° C, and the mixture was stirred for 15 minutes. TMSOTf (15.4 g) was slowly added dropwise so that the internal temperature did not exceed 20 ° C. The external temperature of the reaction vessel was set to 20°C, and the reaction mixture was stirred for 1 hour, then diluted with 2-methyltetrahydrofuran (244mL), and the external temperature of the reaction vessel was set to 10°C. 5% aqueous solution of dipotassium hydrogen phosphate (478mL) was slowly added dropwise, and stirring was stopped and the aqueous layer was discharged. The resulting organic layer was washed with 5% sodium dihydrogen phosphate (478mL), and then diisopropylethylamine (9.0mL) was added and concentrated under reduced pressure to obtain a 2-methyltetrahydrofuran solution of compound 20 (120.1g).
LCMS (ESI) of compound 20: Retention time: 4.30 minutes, m/z = 981.66 [M+Na] + (LCMS analysis conditions method K-2)
実施例2-16Example 2-16
化合物21:tert-ブチル (2S)-1-[(2s,3s)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジルオキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボキラートの合成Compound 21: Synthesis of tert-butyl (2S)-1-[(2s,3s)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate

窒素で置換した反応釜に、室温にて、2-メチルテトラヒドロフラン(70mL)と(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(ベンジルオキシカルボニルアミノ)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタン酸(40.6g)の2-メチルテトラヒドロフラン溶液(113.85g)を加えて攪拌した。続いてジイソプロピルエチルアミン(25.3mL)と2-メチルテトラヒドロフラン(180mL)に溶かした化合物13(21.9g)を加えたのち、アセトニトリル(40mL)を加えて撹拌した。HATU(24.2g)をアセトニトリル(53mL)に懸濁し、反応釜へ加え1時間撹拌した。酢酸(1.2mL)を加え、20分撹拌したのち終夜静置した。撹拌を再開した後、MeTHF(122mL)で希釈し、10%アンモニア水(282mL)を加えた。撹拌を止め水層を排出した後、得られた有機層を10%アンモニア水(280mL)、4%希硫酸(280mL)、4%希硫酸(280mL)、5%炭酸ナトリウム水溶液(280mL)の順で洗浄した。得られた有機層を減圧濃縮し、化合物21の2-メチルテトラヒドロフラン溶液(132.34g)を得た。 In a reaction vessel purged with nitrogen, 2-methyltetrahydrofuran (70 mL) and a solution (113.85 g) of (3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-(benzyloxycarbonylamino)-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoic acid (40.6 g) in 2-methyltetrahydrofuran were added at room temperature and stirred. Next, compound 13 (21.9 g) dissolved in diisopropylethylamine (25.3 mL) and 2-methyltetrahydrofuran (180 mL) was added, and then acetonitrile (40 mL) was added and stirred. HATU (24.2 g) was suspended in acetonitrile (53 mL) and added to the reaction vessel and stirred for 1 hour. Acetic acid (1.2 mL) was added, and the mixture was stirred for 20 minutes and then left to stand overnight. After resuming stirring, the mixture was diluted with MeTHF (122 mL) and 10% aqueous ammonia (282 mL) was added. After stopping stirring and discharging the aqueous layer, the obtained organic layer was washed in the order of 10% aqueous ammonia (280 mL), 4% dilute sulfuric acid (280 mL), 4% dilute sulfuric acid (280 mL), and 5% aqueous sodium carbonate (280 mL). The obtained organic layer was concentrated under reduced pressure to obtain a 2-methyltetrahydrofuran solution of compound 21 (132.34 g).
実施例2-17Example 2-17
化合物37:tert-ブチル (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-アミノ-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-ピロリジン-2-カルボニル]-メチル-アミノ]シクロブタンカルボニル]-メチル-アミノ]-2-シクロペンチル-アセチル]-メチル-アミノ]-4-(ジメチルアミノ)-4-オキソ-ブタノイル]-メチル-アミノ]ペンタノイル]アミノ]-3-メチル-ペンタノイル]ピロリジン-2-カルボキシラートの合成Compound 37: Synthesis of tert-butyl (2S)-1-[(2S,3S)-2-[[(2S)-2-[[(3S)-3-[[(2S)-2-[[1-[[(2S,4R)-1-[(2S)-2-amino-4-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-pyrrolidine-2-carbonyl]-methyl-amino]cyclobutanecarbonyl]-methyl-amino]-2-cyclopentyl-acetyl]-methyl-amino]-4-(dimethylamino)-4-oxo-butanoyl]-methyl-amino]pentanoyl]amino]-3-methyl-pentanoyl]pyrrolidine-2-carboxylate
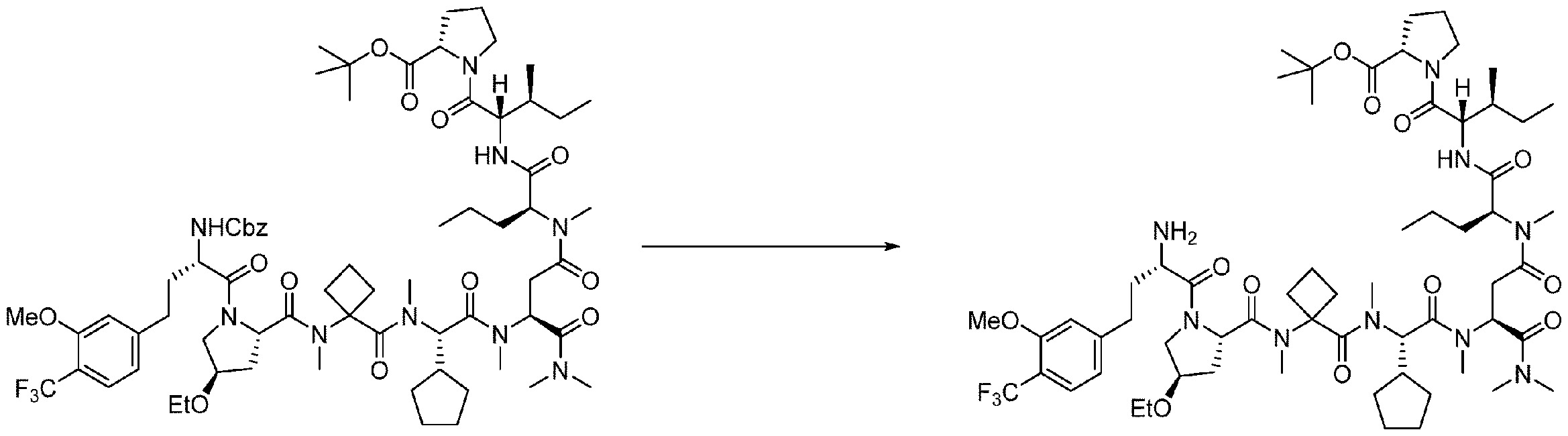
窒素で置換した反応釜に、室温にて2-メチルテトラヒドロフラン(1.0mL)と10%活性炭担持型パラジウム(90.3mg)を加えた。水素で反応釜を置換し、水素加圧(0.40MPa)下1時間撹拌した。水素を排出し窒素で置換した後、2-メチルテトラヒドロフラン(1.5mL)に溶かした化合物21(514mg)を加え撹拌した。窒素を排出後に水素で置換し、水素加圧(0.20MPa)下で合計8時間30分撹拌した。水素を排出し窒素置換した後、ろ過により活性炭担持型パラジウムを除去した。2-メチルテトラヒドロフラン(500μL)で3回ケークを洗浄し、ろ液と合わせて減圧下濃縮乾固することで化合物37(503mg)を得た。
化合物37のLCMS(ESI):保持時間:8.04 分、m/z=1205.0 [M+H]+(LCMS分析条件 method FC2)
2-Methyltetrahydrofuran (1.0 mL) and 10% palladium on activated carbon (90.3 mg) were added to the reaction vessel replaced with nitrogen at room temperature. The reaction vessel was replaced with hydrogen and stirred for 1 hour under hydrogen pressure (0.40 MPa). After discharging hydrogen and replacing with nitrogen, compound 21 (514 mg) dissolved in 2-methyltetrahydrofuran (1.5 mL) was added and stirred. After discharging nitrogen, it was replaced with hydrogen and stirred for a total of 8 hours and 30 minutes under hydrogen pressure (0.20 MPa). After discharging hydrogen and replacing with nitrogen, palladium on activated carbon was removed by filtration. The cake was washed three times with 2-methyltetrahydrofuran (500 μL), combined with the filtrate, and concentrated to dryness under reduced pressure to obtain compound 37 (503 mg).
LCMS (ESI) of compound 37: Retention time: 8.04 minutes, m/z = 1205.0 [M+H] + (LCMS analysis conditions method FC2)
実施例2-18Example 2-18
化合物38:tert-ブチル[(S)-2-[(S)-3-[(S)-2-(1-[(2S,4R)-1-[(S)-2-アミノ-[3-メトキシ-4-(トリフルオロメチル)フェニル]ブタノイル]-4-エトキシ-N-メチルピロリジン-2-カルボキシアミド]-N-メチルシクロブタン-1-カルボキシアミド)-2-シクロペンチル-N-メチルアセトアミド]-4-[ジメチルアミノ]-N-メチル-4-オキソブタナミド]ペンタノイル]-L-イソロイシル-L-プロリン酸tert-ブチルの合成Compound 38: Synthesis of tert-butyl [(S)-2-[(S)-3-[(S)-2-(1-[(2S,4R)-1-[(S)-2-amino-[3-methoxy-4-(trifluoromethyl)phenyl]butanoyl]-4-ethoxy-N-methylpyrrolidine-2-carboxamido]-N-methylcyclobutane-1-carboxamido)-2-cyclopentyl-N-methylacetamido]-4-[dimethylamino]-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleucyl-L-proline tert-butyl
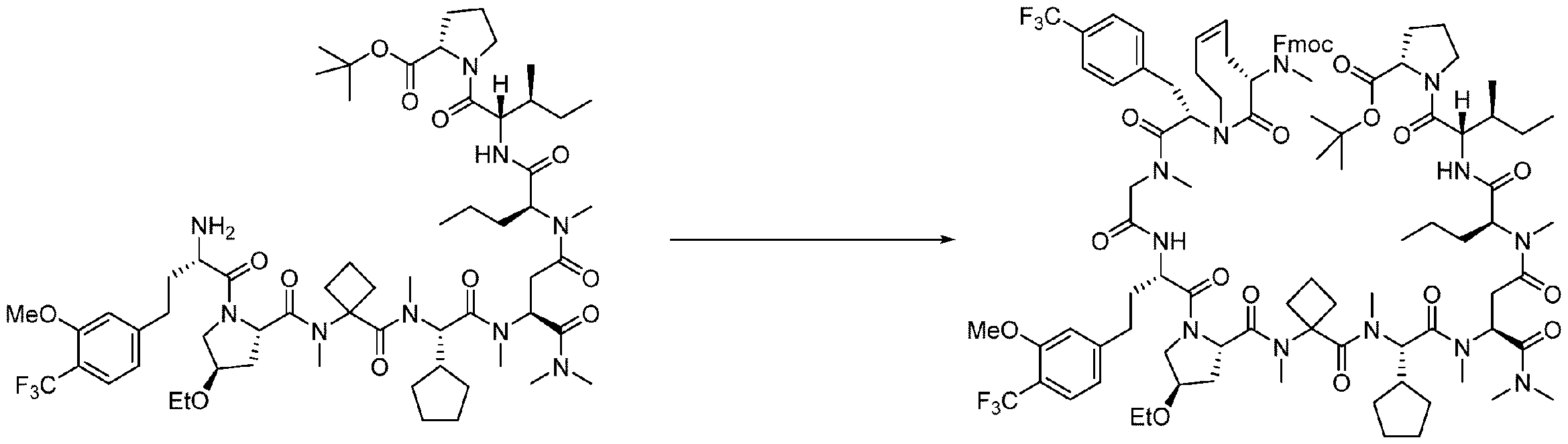
反応容器に実施例2-3で得られた化合物28(122mg)と2-MeTHF(1.82mL)、アセトニトリル(460μL)、化合物37(200mg)を加え、室温で撹拌した。NMM(80μL)、HATU(95.4mg)を加え、外温25℃で2時間撹拌した。2時間後、反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。5%炭酸ナトリウム水溶液(1.40mL)、1-メチルイミダゾール(13.2μL)を加えて30分間撹拌した。30分後、10%アンモニア水(1.4mL)を室温で加えて撹拌した。分液操作により水層を排出したのち、有機層に対して5%硫酸水素ナトリウム水溶液(1.4mL)を加え撹拌した。分液操作後に液々分離し、水層を排出した。得られた有機層を5%炭酸ナトリウム水溶液(1.4mL)で洗浄した。得られた有機層を外温を40℃で減圧濃縮し、粗体の化合物38を290mg(見かけ収率94.0%)で得た。
化合物38のLCMS(ESI):保持時間:12.0分、(LCMS分析条件 method FC2)
Compound 28 (122 mg) obtained in Example 2-3, 2-MeTHF (1.82 mL), acetonitrile (460 μL), and compound 37 (200 mg) were added to a reaction vessel and stirred at room temperature. NMM (80 μL) and HATU (95.4 mg) were added and stirred at an external temperature of 25 ° C. for 2 hours. After 2 hours, the reaction mixture was sampled to prepare a sample (sample preparation method 2), and it was confirmed that the reaction conversion rate was 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). 5% aqueous sodium carbonate solution (1.40 mL) and 1-methylimidazole (13.2 μL) were added and stirred for 30 minutes. After 30 minutes, 10% aqueous ammonia (1.4 mL) was added at room temperature and stirred. After the aqueous layer was discharged by liquid separation, 5% aqueous sodium hydrogen sulfate solution (1.4 mL) was added to the organic layer and stirred. After liquid separation, the liquid was separated and the aqueous layer was discharged. The obtained organic layer was washed with a 5% aqueous sodium carbonate solution (1.4 mL) and concentrated under reduced pressure at an external temperature of 40° C. to obtain 290 mg of crude compound 38 (apparent yield 94.0%).
LCMS (ESI) of compound 38: Retention time: 12.0 minutes, (LCMS analysis conditions method FC2)
反応釜に室温下で化合物37(3.41kg)の2-MeTHF溶液(17.6kg)を加え、2-MeTHF(12.3kg)、化合物28・1トルエン和物(2.38kg)、アセトニトリル(6.20kg)を順次添加した。外温を5℃に設定し、NMM(1.26kg)、HATU(1.65kg)を内温30℃以下で添加し、内温25℃で1時間撹拌した。反応転換率が99%以上であることを確認後、5%炭酸カリウム水溶液(23.9kg)と1-メチルイミダゾール(234g)を加えて室温下で30分撹拌し、液々分離により水層を排出した。得られた有機層を10%アンモニア水(23.0kg)、5%硫酸水素ナトリウム水溶液(23.9kg)、5%炭酸ナトリウム水溶液(23.9kg)で洗浄したのち、外温40℃で濃縮し、10Lまで濃縮した。濃縮液にアセトニトリル(16.2kg)を加えて10Lまで濃縮を2回繰り返し、化合物38アセトニトリル溶液(19.4kg)を得た。 A 2-MeTHF solution (17.6 kg) of compound 37 (3.41 kg) was added to the reaction vessel at room temperature, followed by the addition of 2-MeTHF (12.3 kg), compound 28 monotoluene solvate (2.38 kg), and acetonitrile (6.20 kg) in that order. The external temperature was set to 5°C, and NMM (1.26 kg) and HATU (1.65 kg) were added at an internal temperature of 30°C or less, followed by stirring for 1 hour at an internal temperature of 25°C. After confirming that the reaction conversion rate was 99% or more, a 5% aqueous potassium carbonate solution (23.9 kg) and 1-methylimidazole (234 g) were added and stirred at room temperature for 30 minutes, and the aqueous layer was discharged by liquid-liquid separation. The resulting organic layer was washed with 10% aqueous ammonia (23.0 kg), 5% aqueous sodium hydrogen sulfate (23.9 kg), and 5% aqueous sodium carbonate (23.9 kg), and then concentrated at an external temperature of 40°C until the volume reached 10 L. Acetonitrile (16.2 kg) was added to the concentrated solution, and the concentration was repeated twice until the volume reached 10 L, yielding a solution of Compound 38 in acetonitrile (19.4 kg).
実施例2-19Example 2-19
化合物39:(S)-2-[(S)-3-[(S)-2-シクロペンチル-2-[1-[(2S,4R)-4-エトキシ-1-[(S)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]-2-[(2-[(S)-N-メチル-2-[(R,Z)-3-(メチルアミノ)-2-オキソ-3,4,7,8-テトラヒドロアゾシン-1(2H)-イル]-3-[4-(トリフルオロメチル)フェニル]プロパンアミド]アセトアミド)ブタノイル]-Nメチルピロリジン-2-カルボキシアミド]-N-メチルシクロブタン-1-カルボキシアミド]-N-メチルアセトアミド]-4-(ジメチルアミノ)-N-メチル-4-オキソブタンアミド]ペンタノイル]-L-イソロイシル-L-プロリン酸tert-ブチルの合成Compound 39: Synthesis of (S)-2-[(S)-3-[(S)-2-cyclopentyl-2-[1-[(2S,4R)-4-ethoxy-1-[(S)-4-[3-methoxy-4-(trifluoromethyl)phenyl]-2-[(2-[(S)-N-methyl-2-[(R,Z)-3-(methylamino)-2-oxo-3,4,7,8-tetrahydroazocin-1(2H)-yl]-3-[4-(trifluoromethyl)phenyl]propanamido]acetamido)butanoyl]-N-methylpyrrolidine-2-carboxamido]-N-methylcyclobutane-1-carboxamido]-N-methylacetamido]-4-(dimethylamino)-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleucyl-L-proline tert-butyl
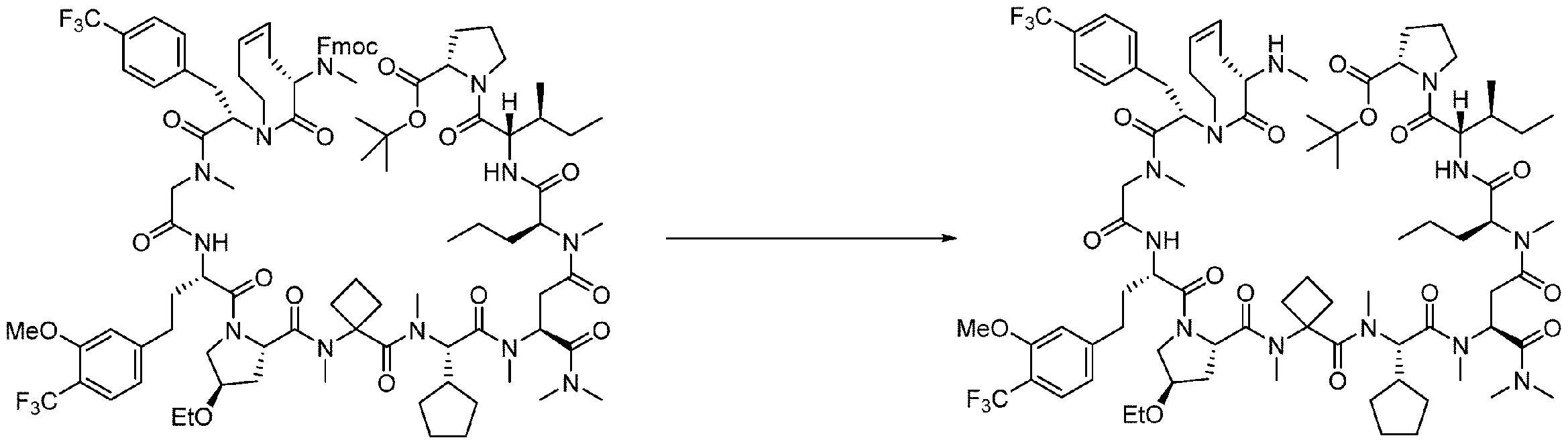
反応釜に化合物38(113mg)とアセトニトリル(453μL)、DBU(27.5μL)を順次加え、外温25℃で1時間撹拌した。1時間後、反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応容器に亜硫酸水素ナトリウム(23.1mg)、トリエチルアミン(34.1μL)、水(27.6μL)を加えて2時間撹拌し、HPLC分析により、ジベンゾフルベンの反応転換率が99%以上であることを確認した。反応容器にトルエン(565μL)、10%アンモニア水(1.13mL)を加えて撹拌し、分液操作により水層を排出した。有機層を10%アンモニア水(1.13mL)でさらに2回洗浄した。得られた有機層を外温を40℃に設定し減圧濃縮し、粗体の化合物39を94.1mg(見かけ収率94.0%)で得た。化合物39のLCMS:保持時間:8.14分(LCMS分析条件 method FC2) Compound 38 (113 mg), acetonitrile (453 μL), and DBU (27.5 μL) were added to the reaction vessel in order, and the mixture was stirred at an external temperature of 25°C for 1 hour. After 1 hour, the reaction mixture was sampled to prepare a sample (sample preparation method 2), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula for reaction conversion rate 1). Sodium hydrogen sulfite (23.1 mg), triethylamine (34.1 μL), and water (27.6 μL) were added to the reaction vessel and stirred for 2 hours, and the reaction conversion rate of dibenzofulvene was confirmed to be 99% or more by HPLC analysis. Toluene (565 μL) and 10% aqueous ammonia (1.13 mL) were added to the reaction vessel and stirred, and the aqueous layer was discharged by a separation operation. The organic layer was washed twice more with 10% aqueous ammonia (1.13 mL). The obtained organic layer was concentrated under reduced pressure with the external temperature set to 40°C, and crude compound 39 was obtained in an amount of 94.1 mg (apparent yield 94.0%). LCMS of compound 39: Retention time: 8.14 minutes (LCMS analysis conditions method FC2)
反応釜に化合物38(4.67kg)のアセトニトリル(14.9kg)溶液を加え、アセトニトリル(4.53kg)を追加して内温15℃以下に冷却し、内温30℃以下でDBU(1.65kg)を添加した。25℃で1時間撹拌し、反応変換率が99%以上であることを確認後、再度内温15℃以下に冷却し、トリエチルアミン(1.02kg)、水(1.14kg)、亜硫酸水素ナトリウム(925g)を内温30℃を超えない範囲で順次添加し、1時間撹拌した。撹拌後、反応液にトルエン(20.4kg)、10%アンモニア水(46.7kg)を加えて撹拌し、液々分離により水層を排出した。有機層にアセトニトリル(18.4kg)、10%アンモニア水(46.7kg)を加えて再度撹拌し、液々分離で水層を排出するのを2回繰り返した。有機層に5%食塩水(23.4kg)を加えて洗浄し、液々分離したのち、有機層を外温60℃で9.3Lまで濃縮した。濃縮液に2-MeTHF(16.0kg)を加え、外温60℃で9.3Lまで濃縮を2回繰り返し、化合物39の2-MeTHF溶液(7.72kg)を得た。 A solution of compound 38 (4.67 kg) in acetonitrile (14.9 kg) was added to the reaction vessel, acetonitrile (4.53 kg) was added, the internal temperature was cooled to below 15°C, and DBU (1.65 kg) was added when the internal temperature was below 30°C. After stirring at 25°C for 1 hour and confirming that the reaction conversion rate was 99% or more, the internal temperature was cooled again to below 15°C, triethylamine (1.02 kg), water (1.14 kg), and sodium hydrogen sulfite (925 g) were added in sequence so that the internal temperature did not exceed 30°C, and the mixture was stirred for 1 hour. After stirring, toluene (20.4 kg) and 10% ammonia water (46.7 kg) were added to the reaction liquid and stirred, and the aqueous layer was discharged by liquid-liquid separation. Acetonitrile (18.4 kg) and 10% ammonia water (46.7 kg) were added to the organic layer, stirred again, and the aqueous layer was discharged by liquid-liquid separation, which was repeated twice. The organic layer was washed with 5% saline (23.4 kg) and separated into liquids, after which the organic layer was concentrated to 9.3 L at an external temperature of 60°C. 2-MeTHF (16.0 kg) was added to the concentrated solution, and the solution was concentrated twice to 9.3 L at an external temperature of 60°C to obtain a 2-MeTHF solution (7.72 kg) of compound 39.
実施例2-20Example 2-20
化合物40:(S)-2-[(S)-3-[(S)-2-シクロペンチル-2-[1-[(2S,4R)-4-エトキシ-1-[(S)-4-[3-メトキシ-4-(トリフルオロメチル)フェニル]-2-[(2-[(S)-N-メチル-2-[(R,Z)-3-(メチルアミノ)-2-オキソ-3,4,7,8-テトラヒドロアゾシン-1(2H)-イル]-3-[4-(トリフルオロメチル)フェニル]プロパンアミド]アセトアミド)ブタノイル]-Nメチルピロリジン-2-カルボキシアミド]-N-メチルシクロブタン-1-カルボキシアミド]-N-メチルアセトアミド]-4-(ジメチルアミノ)-N-メチル-4-オキソブタンアミド]ペンタノイル]-L-イソロイシル-L-プロリンの合成Compound 40: Synthesis of (S)-2-[(S)-3-[(S)-2-cyclopentyl-2-[1-[(2S,4R)-4-ethoxy-1-[(S)-4-[3-methoxy-4-(trifluoromethyl)phenyl]-2-[(2-[(S)-N-methyl-2-[(R,Z)-3-(methylamino)-2-oxo-3,4,7,8-tetrahydroazocin-1(2H)-yl]-3-[4-(trifluoromethyl)phenyl]propanamido]acetamido)butanoyl]-N-methylpyrrolidine-2-carboxamide]-N-methylcyclobutane-1-carboxamide]-N-methylacetamide]-4-(dimethylamino)-N-methyl-4-oxobutanamido]pentanoyl]-L-isoleucyl-L-proline
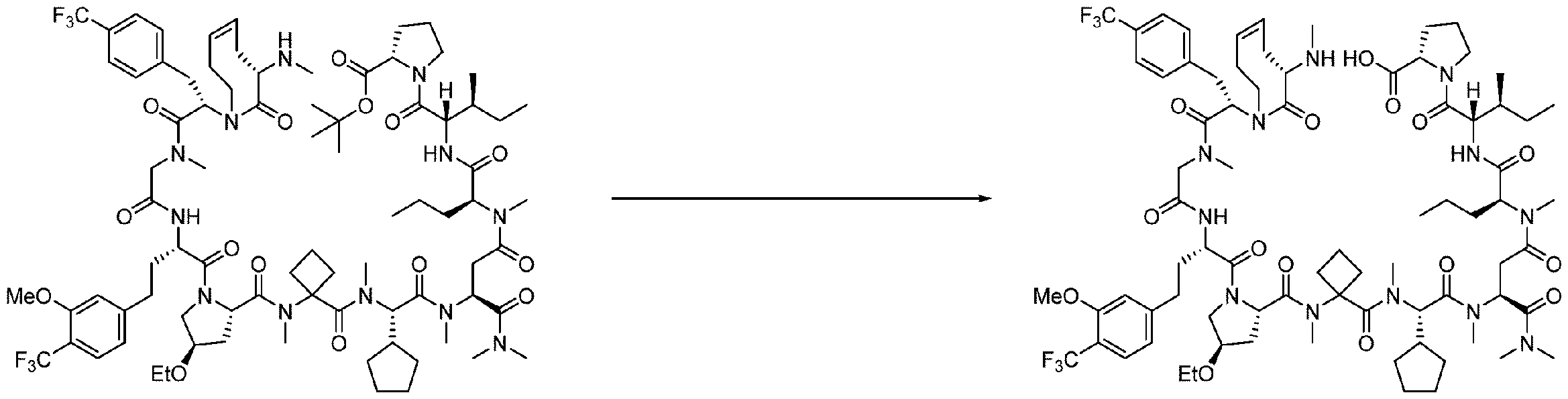
反応釜に化合物39(94.0mg)と2-MeTHF(470μL)を加え、外温を5℃に冷却した。HMDS(37.0μL)とTMSOTf(21.0μL)を順次加え、外温25℃に昇温し、1時間撹拌した。1時間後、反応混合物をサンプリングしてサンプル調製(サンプル調製法2)し、HPLC分析により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応容器に反応容器に2-MeTHF(752μL)を加えた後、再度外温を5℃に冷却し、5%リン酸水素二カリウム水溶液(658μL)を内温10℃で加えて撹拌し、分液操作により水層を排出した。有機層にアセトニトリル(188μL)、5%硫酸水素ナトリウム水溶液(658μL)を加えて撹拌し、分液操作により水層を排出した。有機層に5%炭酸ナトリウム水溶液(658μL)を添加して撹拌し分液操作により水層を排出した。得られた有機層を5%食塩水(658μL)で洗浄し、外温を40℃に設定して減圧濃縮した。標品を用いたHPLC測定の結果、得られた化合物40は72.4mg(3工程収率62.8%)であった。化合物40のLCMS:保持時間:6.86分(LCMS分析条件 method FC2) Compound 39 (94.0 mg) and 2-MeTHF (470 μL) were added to the reaction vessel, and the external temperature was cooled to 5°C. HMDS (37.0 μL) and TMSOTf (21.0 μL) were added in sequence, and the external temperature was raised to 25°C and stirred for 1 hour. After 1 hour, the reaction mixture was sampled and prepared (sample preparation method 2), and the reaction conversion rate was confirmed to be 99% or more by HPLC analysis (calculation formula 1 for reaction conversion rate). 2-MeTHF (752 μL) was added to the reaction vessel, and the external temperature was cooled again to 5°C. 5% dipotassium hydrogen phosphate aqueous solution (658 μL) was added at an internal temperature of 10°C and stirred, and the aqueous layer was discharged by liquid separation. Acetonitrile (188 μL) and 5% sodium hydrogen sulfate aqueous solution (658 μL) were added to the organic layer, and the mixture was stirred, and the aqueous layer was discharged by liquid separation. A 5% aqueous solution of sodium carbonate (658 μL) was added to the organic layer, which was then stirred and the aqueous layer was removed by a separation operation. The resulting organic layer was washed with 5% saline (658 μL) and concentrated under reduced pressure with the external temperature set to 40°C. As a result of HPLC measurement using a standard sample, the amount of compound 40 obtained was 72.4 mg (3-step yield 62.8%). LCMS of compound 40: Retention time: 6.86 minutes (LCMS analysis conditions method FC2)
反応釜に化合物39(3.74kg)の2-MeTHF(11.9kg)溶液を加え、2-MeTHF(7.78kg)を追加し、HMDS(1.12kg)。内温5℃以下に冷却した後、内温30℃を超えないようにTMSOTf(1.03kg)を添加し、内温25℃に昇温して2時間撹拌した。反応転換率99%以上であることを確認した後、2-MeTHF(25.6kg)、アセトニトリル(5.88kg)を加えた後、内温を5℃以下に冷却して5%リン酸水素二カリウム水溶液(26.2kg)を内温30℃を超えない様にゆっくり添加した。撹拌し、液々分離により水層を排出後、有機層を5%硫酸水素ナトリウム水溶液(26.2kg)、5%炭酸ナトリウム水溶液(26.2kg)、5%食塩水(26.2kg)で順次洗浄した。外温50℃で得られた有機層を9.4Lまで濃縮し、アセトニトリル(7.36kg)を加えて再度9.4Lまで濃縮することを2回繰り返した。濃縮液にアセトニトリル(8.82kg)、ヘプタン(25.6kg)を加えて撹拌し、液々分離して下層を回収し、化合物40のアセトニトリル溶液(17.7kg)を得た。qNMRによる定量より、化合物40の収率は3工程93.4%であった。 A solution of compound 39 (3.74 kg) in 2-MeTHF (11.9 kg) was added to the reaction vessel, 2-MeTHF (7.78 kg) was added, and HMDS (1.12 kg) was added. After cooling to an internal temperature of 5°C or less, TMSOTf (1.03 kg) was added so that the internal temperature did not exceed 30°C, and the internal temperature was raised to 25°C and stirred for 2 hours. After confirming that the reaction conversion rate was 99% or more, 2-MeTHF (25.6 kg) and acetonitrile (5.88 kg) were added, and the internal temperature was cooled to 5°C or less, and 5% dipotassium hydrogen phosphate aqueous solution (26.2 kg) was slowly added so that the internal temperature did not exceed 30°C. After stirring and liquid-liquid separation, the aqueous layer was discharged, and the organic layer was washed successively with 5% sodium hydrogen sulfate aqueous solution (26.2 kg), 5% sodium carbonate aqueous solution (26.2 kg), and 5% saline solution (26.2 kg). The organic layer obtained at an external temperature of 50°C was concentrated to 9.4 L, acetonitrile (7.36 kg) was added, and the mixture was concentrated again to 9.4 L, and this process was repeated twice. Acetonitrile (8.82 kg) and heptane (25.6 kg) were added to the concentrated liquid, and the mixture was stirred. The liquid was separated, and the lower layer was collected, yielding an acetonitrile solution of compound 40 (17.7 kg). Quantitative analysis by qNMR revealed that the yield of compound 40 was 93.4% over the three steps.
実施例2-21
化合物1の合成(環化位置C)

Synthesis of Compound 1 (Cyclization Position C)
反応容器に、化合物40(72.4mg)とアセトニトリル(17.8mL)、DIPEA(59μL)を加えた。外温25℃でHATU(65.1mg)を加え、2時間撹拌した。HPLC分析(LCMS分析条件method FC2)により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応混合物をサンプリングしてサンプル調製(サンプル調製法1)し、HPLC分析により化合物1/環状ダイマー=97.6/2.4(環化選択性の算出式1,2)であることを確認した。反応釜の外温を40℃に設定し、2.14mLまで濃縮した。得られた濃縮物に室温でMTBE(1.25mL)、ヘプタン(89μL)、2.5%アンモニア水溶液(890μL)を加え撹拌した。液々分離にて水層を排出後、得られた有機層を4%硫酸(1.25mL)、5%リン酸水素二カリウム水溶液(890μL)で順次洗浄した。得られた有機層を0.5%塩化ナトリウム水溶液(890μLLx2)で洗浄した。得られた有機層について、標品を用いたHPLC分析(分析条件:method cyc)の結果、得られた化合物1は61.1mg(85.4%収率)であった。
化合物1のLCMS:保持時間:6.88分、(LCMS分析条件method FC2)、化合物1のHPLC:保持時間:18.40分、環状dimerのHPLC:保持時間23.96分(method cyc)
Compound 40 (72.4 mg), acetonitrile (17.8 mL), and DIPEA (59 μL) were added to the reaction vessel. HATU (65.1 mg) was added at an external temperature of 25° C., and the mixture was stirred for 2 hours. The reaction conversion rate was confirmed to be 99% or more by HPLC analysis (LCMS analysis condition method FC2) (calculation formula 1 for reaction conversion rate). The reaction mixture was sampled and prepared as a sample (sample preparation method 1), and the HPLC analysis confirmed that compound 1/cyclic dimer = 97.6/2.4 (calculation formulas 1 and 2 for cyclization selectivity). The external temperature of the reaction vessel was set to 40° C., and the mixture was concentrated to 2.14 mL. MTBE (1.25 mL), heptane (89 μL), and 2.5% aqueous ammonia solution (890 μL) were added to the obtained concentrate at room temperature and stirred. After discharging the aqueous layer by liquid-liquid separation, the obtained organic layer was washed with 4% sulfuric acid (1.25 mL) and 5% dipotassium hydrogen phosphate aqueous solution (890 μL) in that order. The obtained organic layer was washed with 0.5% sodium chloride aqueous solution (890 μL x 2). The obtained organic layer was analyzed by HPLC using a standard sample (analysis conditions: method cyc), and the obtained compound 1 was 61.1 mg (85.4% yield).
LCMS of compound 1: retention time: 6.88 minutes (LCMS analysis condition: method FC2), HPLC of compound 1: retention time: 18.40 minutes, HPLC of cyclic dimer: retention time 23.96 minutes (method cyc)
反応釜に、HATU(949g)とアセトニトリル(25.6kg)を加え溶解させ、HATUのアセトニトリル溶液を調製した。ほかの容器に化合物40(1.31kg)のアセトニトリル(5.52kg)溶液、アセトニトリル(19.1kg)、N-メチルモルホリン(504g)を加え、化合物40のアセトニトリル溶液を調製した。外温25℃でHATUのアセトニトリル溶液に調製された化合物40のアセトニトリル溶液を4時間22分かけて滴下し、アセトニトリル(1.04kg)を用いて洗いこみを実施した。滴下後にさらに1時間撹拌した。HPLC分析(LC分析条件method cyc)により反応転換率が99%以上であることを確認した(反応転換率の算出式1)。反応混合物をサンプリングしてサンプル調製(サンプル調製法1)し、HPLC分析により化合物1/環状ダイマー=98.5/1.5(環化選択性の算出式1,2)であることを確認した。外温を40℃に設定し、反応混合物をおよそ12.5 Lまで濃縮した。得られた濃縮物に室温でMTBE(14.5kg)、2.5%アンモニア水溶液(13.3kg)を加え34分間撹拌した。液々分離にて水層を排出後、得られた有機層を5%硫酸 (13.1kg)、5%リン酸水素二カリウム水溶液(13.1kg)、0.5%塩化ナトリウム水溶液(13.07kg)で順次洗浄した。得られた有機層にアセトニトリル(2.00kg)を加え、さらに0.5%塩化ナトリウム水溶液(13.1kg)で洗浄した。得られた有機層をエタノールで溶媒置換した後に、標品を用いたHPLC分析(分析条件:method cyc)の結果、得られた化合物1は1.14kg(88.3%収率)であった。
化合物1のHPLC:保持時間:18.67分、環状dimerのHPLC:保持時間24.07分(method cyc)
HATU (949 g) and acetonitrile (25.6 kg) were added to the reaction vessel and dissolved to prepare an acetonitrile solution of HATU. Compound 40 (1.31 kg) in acetonitrile (5.52 kg), acetonitrile (19.1 kg), and N-methylmorpholine (504 g) were added to another vessel to prepare an acetonitrile solution of compound 40. The acetonitrile solution of compound 40 prepared in the acetonitrile solution of HATU at an external temperature of 25° C. was dropped over 4 hours and 22 minutes, and acetonitrile (1.04 kg) was used to wash it in. After dropping, the mixture was stirred for another hour. The reaction conversion rate was confirmed to be 99% or more by HPLC analysis (LC analysis condition method cyc) (calculation formula 1 for reaction conversion rate). The reaction mixture was sampled to prepare a sample (sample preparation method 1), and HPLC analysis confirmed that compound 1/cyclic dimer = 98.5/1.5 (calculation formulas 1 and 2 for cyclization selectivity). The external temperature was set to 40°C, and the reaction mixture was concentrated to approximately 12.5 L. MTBE (14.5 kg) and 2.5% aqueous ammonia (13.3 kg) were added to the resulting concentrate at room temperature and stirred for 34 minutes. After discharging the aqueous layer by liquid-liquid separation, the resulting organic layer was washed successively with 5% sulfuric acid (13.1 kg), 5% aqueous dipotassium hydrogen phosphate (13.1 kg), and 0.5% aqueous sodium chloride (13.07 kg). Acetonitrile (2.00 kg) was added to the resulting organic layer, which was then washed with 0.5% aqueous sodium chloride (13.1 kg). After the organic layer was solvent-substituted with ethanol, the compound 1 was analyzed by HPLC (analysis conditions: method cyc) using a standard sample, and the compound 1 obtained was 1.14 kg (88.3% yield).
HPLC of compound 1: retention time: 18.67 min, HPLC of cyclic dimer: retention time 24.07 min (method cyc)
環化位置の違いによる環状ダイマーの抑制について考察する。 We will consider how differences in the cyclization position affect the inhibition of cyclic dimers.
化合物1の合成法として国際公開第2022/234853号による合成法が知られているが、環化反応における副生成物として分子間反応に由来する環状ダイマーを生じる。国際公開第2022/234853号に記載されている環化位置Aの環化反応において、化合物23を6時間かけて逆滴下することによって疑似希釈条件での環化を実施しているものの、環状ダイマーが25%生成するものであった(実施例1-26[化合物1/環状ダイマー = 75/25])。本発明者らは、環化位置を変更して検討したところ、環化位置Bによる環化は、滴下時間が短いにもかかわらず、環状ダイマー量が低減できることを見出した(実施例1-24-2[化合物1/環状ダイマー=98/2])。さらに環化位置Cによる環化においては、逆滴下を使用しない高濃度条件であるものの、環状ダイマー量が低減された結果が得られた(実施例2-21[化合物1/環状ダイマー=97.6/2.4])。以上の事実は本発明での環化位置Bおよび環化位置Cが、既存の環化位置Aと比較して、不純物である環状ダイマー量が少なく、化合物1の製造において効率的な環化位置であることを示している。 The synthesis method of compound 1 described in WO 2022/234853 is known, but in the cyclization reaction, a cyclic dimer resulting from an intermolecular reaction is generated as a by-product. In the cyclization reaction at cyclization position A described in WO 2022/234853, cyclization was performed under pseudo-dilution conditions by back-dropping compound 23 over a period of 6 hours, but only 25% of the cyclic dimer was produced (Example 1-26 [Compound 1/cyclic dimer = 75/25]). The inventors changed the cyclization position and found that cyclization at cyclization position B could reduce the amount of cyclic dimer despite the short drop time (Example 1-24-2 [Compound 1/cyclic dimer = 98/2]). Furthermore, in the cyclization at cyclization position C, the amount of cyclic dimer was reduced despite the high concentration conditions without back-dropping (Example 2-21 [Compound 1/cyclic dimer = 97.6/2.4]). The above facts indicate that cyclization positions B and C in the present invention have a smaller amount of cyclic dimer impurities than the existing cyclization position A, and are therefore efficient cyclization positions for the production of compound 1.
実施例3 結晶化
実施例3-1
アモルファス状態の化合物1(77.4mg)をジメチルスルホキシド(0.387mL)に溶解させ、この溶解液(0.015mL)を-20℃で3日間凍結乾燥した。得られた凍結乾燥物に2-ブタノン-ヘプタン混合液(1:4(v/v)、0.015mL)を加え、室温にて7日間振とう攪拌することで化合物1の結晶を得た。得られた結晶は単結晶X線構造解析(実施例4-7)によりジメチルスルホキシド/ヘプタン/水和物結晶(Form G)であることを確認した。図1に構造を示す。
Example 3 Crystallization
Example 3-1
Amorphous compound 1 (77.4 mg) was dissolved in dimethyl sulfoxide (0.387 mL), and the resulting solution (0.015 mL) was freeze-dried at -20°C for 3 days. A 2-butanone-heptane mixture (1:4 (v/v), 0.015 mL) was added to the resulting freeze-dried product, and the mixture was shaken and stirred at room temperature for 7 days to obtain crystals of compound 1. The resulting crystals were confirmed to be dimethyl sulfoxide/heptane/hydrate crystals (Form G) by single crystal X-ray structural analysis (Examples 4-7). The structure is shown in Figure 1.
実施例3-2
アモルファス状態の化合物1(11.4mg)をアセトン(34.2μL)に溶解させ、実施例3-1の結晶を約0.1mg添加した。その後ヘプタン(22.8μL)を添加し、室温で3時間振とう攪拌した。さらにヘプタン(22.8μL)を添加し、室温で2時間振とう攪拌した。さらにヘプタン(22.8μL)を添加し、室温で18時間振とう攪拌した後、ろ過して減圧乾燥を行った。得られた結晶は、化合物1の水和物結晶(Form A)であった。得られた結晶の粉末X線回折測定(実施例4-3)を図2に示す。
Example 3-2
Amorphous Compound 1 (11.4 mg) was dissolved in acetone (34.2 μL), and about 0.1 mg of the crystals of Example 3-1 was added. Heptane (22.8 μL) was then added, and the mixture was shaken and stirred at room temperature for 3 hours. Heptane (22.8 μL) was further added, and the mixture was shaken and stirred at room temperature for 2 hours. Heptane (22.8 μL) was further added, and the mixture was shaken and stirred at room temperature for 18 hours, after which it was filtered and dried under reduced pressure. The obtained crystals were hydrate crystals of Compound 1 (Form A). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in FIG. 2.
実施例3-3
アモルファス状態の化合物1(10.7mg)を2-プロパノール(32.1μL)に溶解させ、実施例3-1で得られた結晶を約0.1mg添加した。その後ヘプタン(21.4μL)を添加し、室温で3時間振とう攪拌した。さらにヘプタン(21.4μL)を添加し、室温で2時間振とう後にヘプタン(21.4μL)を添加し、室温で3時間振とう攪拌した。さらにヘプタン(21.4μL)を添加し、室温で15時間振とう攪拌することで化合物1の結晶(Form E)を得た。得られた結晶は単結晶X線構造解析(実施例4-8)を実施し、2-プロパノール/へプタン/水和物であることを確認した。図3に結晶構造を示す。得られたForm Eを減圧乾燥し、粉末X線回折測定(実施例4-3)を実施した。図4に結果を示す。回折パターンはForm Aを示しており、減圧乾燥により2-プロパノール/へプタン/水和物(Form E)が水和物結晶(Form A)に転移することを確認した。
Example 3-3
Amorphous compound 1 (10.7 mg) was dissolved in 2-propanol (32.1 μL), and about 0.1 mg of the crystals obtained in Example 3-1 was added. Heptane (21.4 μL) was then added, and the mixture was shaken and stirred at room temperature for 3 hours. Heptane (21.4 μL) was further added, and the mixture was shaken at room temperature for 2 hours, after which heptane (21.4 μL) was added, and the mixture was shaken and stirred at room temperature for 3 hours. Heptane (21.4 μL) was further added, and the mixture was shaken and stirred at room temperature for 15 hours to obtain a crystal of compound 1 (Form E). The obtained crystal was subjected to single crystal X-ray structural analysis (Example 4-8), and it was confirmed that the crystal was 2-propanol/heptane/hydrate. The crystal structure is shown in FIG. 3. The obtained Form E was dried under reduced pressure, and powder X-ray diffraction measurement (Example 4-3) was performed. The results are shown in FIG. 4. The diffraction pattern showed Form A, and it was confirmed that 2-propanol/heptane/hydrate (Form E) was transformed into a hydrate crystal (Form A) by drying under reduced pressure.
実施例3-4
化合物1のForm A結晶(5.9mg)にエタノール-ヘプタン混合液(1:1(v/v)、0.017mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、37℃、2000rpmで7日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図5に示す。
Examples 3-4
To the Form A crystals of Compound 1 (5.9 mg), an ethanol-heptane mixture (1:1 (v/v), 0.017 mL) and one glass bead were added. The mixture was stirred at 37° C. and 2000 rpm for 7 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). The powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in FIG. 5.
実施例3-5
化合物1のForm A結晶(5.8mg)に酢酸エチル-ヘプタン混合液(1:1(v/v)、0.017mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、37℃、2000rpmで7日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図6に示す。
Examples 3-5
To the Form A crystals of Compound 1 (5.8 mg), an ethyl acetate-heptane mixture (1:1 (v/v), 0.017 mL) and one glass bead were added. The mixture was stirred at 37° C. and 2000 rpm for 7 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). The powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in FIG. 6.
実施例3-6
アモルファス状態の化合物1(9.0mg)をエタノール(27.0μL)に溶解させ、ヘプタン(27.0μL)を添加し、室温で4分間振とう攪拌した。実施例3-4で得られた結晶を約0.1mg添加し、室温で1分間振とう攪拌したところ、化合物1の結晶を得た。単結晶X線構造解析(実施例4-9)を実施し、エタノール/水和物(Form H)であることを確認した。図7に結晶構造を示す。得られたForm Hを減圧乾燥し、粉末X線回折測定(実施例4-3)を実施した。図8に結果を示す。回折パターンはForm Bを示しており、減圧乾燥によりエタノール/水和物(Form H)が水和物結晶(Form B)に転移することを確認した。
Examples 3-6
Amorphous compound 1 (9.0 mg) was dissolved in ethanol (27.0 μL), heptane (27.0 μL) was added, and the mixture was shaken and stirred at room temperature for 4 minutes. When about 0.1 mg of the crystals obtained in Example 3-4 was added and the mixture was shaken and stirred at room temperature for 1 minute, a crystal of compound 1 was obtained. Single crystal X-ray structure analysis (Example 4-9) was performed, and it was confirmed that the crystal was an ethanol/hydrate (Form H). The crystal structure is shown in FIG. 7. The obtained Form H was dried under reduced pressure, and powder X-ray diffraction measurement (Example 4-3) was performed. The results are shown in FIG. 8. The diffraction pattern shows Form B, and it was confirmed that the ethanol/hydrate (Form H) was transformed into a hydrate crystal (Form B) by drying under reduced pressure.
実施例3-7
アモルファス状態の化合物1(10.9mg)を酢酸エチル(32.7μL)に溶解させ、ヘプタン(32.7μL)を添加し、室温で4分間振とう攪拌した。実施例3-5の結晶を約0.1mg添加し、室温で1分間振とう攪拌することで、化合物1の結晶を得た。単結晶X線構造解析(実施例4-9)を実施し、酢酸エチル/水和物(Form C)であることを確認した。図9に結晶構造を示す。得られたForm Cを減圧乾燥し、粉末X線回折測定(実施例4-3)を実施した。図10に結果を示す。回折パターンはForm Bを示しており、減圧乾燥により酢酸エチル/水和物(Form C)が水和物結晶(Form B)に転移することを確認した。
Examples 3-7
Amorphous compound 1 (10.9 mg) was dissolved in ethyl acetate (32.7 μL), heptane (32.7 μL) was added, and the mixture was shaken and stirred at room temperature for 4 minutes. About 0.1 mg of the crystals of Example 3-5 was added, and the mixture was shaken and stirred at room temperature for 1 minute to obtain crystals of compound 1. Single crystal X-ray structure analysis (Example 4-9) was performed, and it was confirmed that the crystals were ethyl acetate/hydrate (Form C). The crystal structure is shown in FIG. 9. The obtained Form C was dried under reduced pressure, and powder X-ray diffraction measurement (Example 4-3) was performed. The results are shown in FIG. 10. The diffraction pattern shows Form B, and it was confirmed that the ethyl acetate/hydrate (Form C) was transformed into a hydrate crystal (Form B) by drying under reduced pressure.
実施例3-8
アモルファス状態の化合物1(112.3mg)をアセトン(337μL)に溶解させ、Form A結晶を種結晶として約0.1mg添加後、ヘプタン(112μL)を添加し、スターラーを用いて室温で2分間攪拌した。再びヘプタン(112μL)を添加し、室温で10分間攪拌した。種結晶の溶解を確認したため、再びForm A結晶約0.1mgを添加し、室温で1時間攪拌した。その後、ヘプタン(225μL)を添加し、室温で20分間攪拌した。さらにForm A結晶を約0.1mg添加し、室温で40分間攪拌した。ヘプタン(337μL)を添加し、室温で16時間振とう攪拌した。反応物を減圧濾過し、1日真空乾燥することでForm A結晶を86.3mg得た。Form Aについて、粉末X線回折測定(実施例4-1)を実施した。図11に測定結果を示す。また、Form Aの熱重量・示差熱同時測定の結果を図12に、1H-NMR測定の結果を図13に示す。図12において、3wt%の重量減少が見られた一方、図13において、アセトンが1wt%のみ観測されたことから、Form Aは水和物であり、残留溶媒としてアセトンが存在していると判断した。
Examples 3-8
Amorphous compound 1 (112.3 mg) was dissolved in acetone (337 μL), and about 0.1 mg of Form A crystals was added as seed crystals. Heptane (112 μL) was added and stirred at room temperature for 2 minutes using a stirrer. Heptane (112 μL) was added again and stirred at room temperature for 10 minutes. After confirming the dissolution of the seed crystals, about 0.1 mg of Form A crystals was added again and stirred at room temperature for 1 hour. Heptane (225 μL) was then added and stirred at room temperature for 20 minutes. About 0.1 mg of Form A crystals were further added and stirred at room temperature for 40 minutes. Heptane (337 μL) was added and shaken and stirred at room temperature for 16 hours. The reactant was filtered under reduced pressure and dried in vacuum for 1 day to obtain 86.3 mg of Form A crystals. Powder X-ray diffraction measurement (Example 4-1) was performed on Form A. The measurement results are shown in FIG. 11. The results of simultaneous thermogravimetry and differential thermal analysis of Form A are shown in Figure 12, and the results of 1 H-NMR analysis are shown in Figure 13. A weight loss of 3 wt % was observed in Figure 12, while only 1 wt % of acetone was observed in Figure 13. Therefore, it was determined that Form A was a hydrate and that acetone was present as a residual solvent.
実施例3-9
アモルファス状態の化合物1(61.7mg)を2-プロパノール(185μL)に溶解させ、ヘプタン(123μL)を添加し、スターラーを用いて室温で3分間攪拌した。Form A結晶を種結晶として約0.1mg添加後、室温で30分間攪拌した。その後、ヘプタン(123μL)を添加し、室温で2時間攪拌した。さらに、ヘプタン(185μL)を添加し、室温で18時間振とう攪拌した。反応物を減圧濾過し、1日真空乾燥することでForm A結晶を23.3mg得た。Form Aについて、粉末X線回折測定(実施例4-3)を実施した。図14に測定結果を示す。また、Form Aの熱重量・示差熱同時測定の結果を図15に、1H-NMR測定の結果を図16に示す。図14の粉末X線回折パターンが実施例3-8のパターンと一致したこと、図15において、2wt%の重量減少が見られたこと、図16において、2-プロパノールが1wt%のみ観測されたことから、Form Aは水和物であり、残留溶媒として2-プロパノールが存在していると判断した。
Examples 3-9
Amorphous Compound 1 (61.7 mg) was dissolved in 2-propanol (185 μL), heptane (123 μL) was added, and the mixture was stirred at room temperature for 3 minutes using a stirrer. After adding about 0.1 mg of Form A crystals as seed crystals, the mixture was stirred at room temperature for 30 minutes. Thereafter, heptane (123 μL) was added, and the mixture was stirred at room temperature for 2 hours. Furthermore, heptane (185 μL) was added, and the mixture was shaken and stirred at room temperature for 18 hours. The reaction product was filtered under reduced pressure and dried in vacuum for 1 day to obtain 23.3 mg of Form A crystals. Powder X-ray diffraction measurement (Example 4-3) was performed on Form A. The measurement results are shown in FIG. 14. The results of simultaneous thermogravimetry and differential thermal analysis of Form A are shown in FIG. 15, and the results of 1 H-NMR measurement are shown in FIG. 16. Since the powder X-ray diffraction pattern in FIG. 14 coincided with the pattern of Example 3-8, a weight loss of 2 wt % was observed in FIG. 15, and only 1 wt % of 2-propanol was observed in FIG. 16, it was determined that Form A was a hydrate and that 2-propanol was present as a residual solvent.
実施例3-10
アモルファス状態の化合物1(60.1mg)をアセトン(180μL)に溶解させ、スターラーを用いて攪拌した。ヘプタン(120μL)を添加し、スターラーを用いて室温で1分間攪拌した。Form A結晶約0.1mgを添加し、30分間攪拌した。再びヘプタン(120μL)を添加し、室温で1分間攪拌した。Form A結晶約0.1mgを再び添加し、室温で2.5時間攪拌した。さらにヘプタン(180μL)を添加し、室温で18時間攪拌した。反応混合物にジルコニアビーズ(1mm)を約10粒加え、室温で一晩攪拌し、化合物1のアセトン/へプタン/水和物結晶(Form F)を湿性粉末として得た。得られたForm Fをガラスキャピラリーに封入し、粉末X線回折測定(実施例4-2)を実施したところ、主要なピークとして、6.99°、8.49°、9.49°、9.88°、10.21°、11.81°、12.32°、12.75°、13.17°、13.94°、14.92°、15.20°、15.64°、16.78°、17.01°、および17.47°が確認された。測定結果を図17に示す。
Example 3-10
Amorphous compound 1 (60.1 mg) was dissolved in acetone (180 μL) and stirred using a stirrer. Heptane (120 μL) was added and stirred at room temperature for 1 minute. About 0.1 mg of Form A crystals was added and stirred for 30 minutes. Heptane (120 μL) was added again and stirred at room temperature for 1 minute. About 0.1 mg of Form A crystals was added again and stirred at room temperature for 2.5 hours. Heptane (180 μL) was further added and stirred at room temperature for 18 hours. About 10 grains of zirconia beads (1 mm) were added to the reaction mixture and stirred at room temperature overnight, and acetone/heptane/hydrate crystals of compound 1 (Form F) were obtained as wet powder. The obtained Form F was enclosed in a glass capillary, and powder X-ray diffraction measurement (Example 4-2) was performed, confirming the following main peaks: 6.99°, 8.49°, 9.49°, 9.88°, 10.21°, 11.81°, 12.32°, 12.75°, 13.17°, 13.94°, 14.92°, 15.20°, 15.64°, 16.78°, 17.01°, and 17.47°. The measurement results are shown in FIG.
実施例3-11
アモルファス状態の化合物1(10.0mg)をアセトン(30μL)に溶解させ、スターラーを用いて攪拌した。ヘプタン(20μL)を添加し、スターラーを用いて室温で1分間攪拌した。Form A結晶約0.1mgを添加し、5分間攪拌した。再びヘプタン(20μL)を添加し、室温で2時間攪拌したところ、良質な単結晶を得た。得られた結晶は単結晶構造解析(実施例4-8)によりアセトン/へプタン/水和物(Form F)であることを確認した。結晶構造を図18に示す。
Example 3-11
Amorphous Compound 1 (10.0 mg) was dissolved in acetone (30 μL) and stirred using a stirrer. Heptane (20 μL) was added and stirred at room temperature for 1 minute using a stirrer. Approximately 0.1 mg of Form A crystal was added and stirred for 5 minutes. Heptane (20 μL) was added again and stirred at room temperature for 2 hours, resulting in a high-quality single crystal. The obtained crystal was confirmed to be acetone/heptane/hydrate (Form F) by single crystal structure analysis (Example 4-8). The crystal structure is shown in FIG. 18.
実施例3-12
実施例1-25で得られた結晶(Form A)の粉末X線回折測定(実施例4-1)を実施したところ、主要なピークとして、6.93°、7.56°、8.26°、9.00°、9.58°、10.35°、11.35°、12.26°、12.85°、13.51°、14.12°、14.69°、15.46°、15.92°、17.43°、および17.73°が確認された。測定結果を図19に示す。また、熱重量・示差熱同時測定の結果を図20に示す。Form Aの湿度における結晶形への影響を確認するため、各湿度における粉末X線回折測定(実施例4-4)を実施した。結果を図21に示す。相対湿度10%から90%ではForm Aが維持しているが、相対湿度0%ではForm Jに転移することを確認した。Form Jは主要なピークとして、6.95°、7.33°、7.93°、8.84°、9.45°、9.97°、10.44°、11.19°、12.43°、12.93°、13.46°、14.36°、14.74°、15.21°、15.87°、16.76°、20.87°、および22.97°を有している。
Example 3-12
When the powder X-ray diffraction measurement (Example 4-1) of the crystal (Form A) obtained in Example 1-25 was carried out, the main peaks were confirmed as 6.93 °, 7.56 °, 8.26 °, 9.00 °, 9.58 °, 10.35 °, 11.35 °, 12.26 °, 12.85 °, 13.51 °, 14.12 °, 14.69 °, 15.46 °, 15.92 °, 17.43 °, and 17.73 °. The measurement results are shown in FIG. 19. The results of simultaneous thermogravimetry and differential thermal analysis are shown in FIG. 20. In order to confirm the effect of humidity on the crystal form of Form A, powder X-ray diffraction measurement (Example 4-4) was carried out at each humidity. The results are shown in FIG. 21. It was confirmed that Form A was maintained at a relative humidity of 10% to 90%, but transition to Form J occurred at a relative humidity of 0%. Form J has major peaks at 6.95°, 7.33°, 7.93°, 8.84°, 9.45°, 9.97°, 10.44°, 11.19°, 12.43°, 12.93°, 13.46°, 14.36°, 14.74°, 15.21°, 15.87°, 16.76°, 20.87°, and 22.97°.
実施例3-13
実施例1-24-1で得られた化合物1(29.4g)を含む濃縮乾固物をアセトン(90mL)に溶解させた。溶解した化合物1をBiotage Sfar C18 Duo 100 Å 30μm 240gにローディングし、0.1%ギ酸-水/0.1%ギ酸-アセトニトリル=9/1→0/10)にて展開した。溶出してきた混合溶液を採取し、2日間静置したところ、良質な単結晶を得た。得られた結晶を40℃で16時間、減圧乾燥することで結晶(Form B)の乾燥末を15mg取得した。得られた結晶(Form B)の粉末X線回折測定(実施例4-1)を実施したところ、主要なピークとして、4.99°、8.65°、9.85°、10.84°、11.32°、12.35°、13.20°、14.44°、15.20°、16.03°、16.69°、17.21°、18.82°、19.49°、および20.03°が確認された。測定結果を図22に示す。また、熱重量・示差熱同時測定の結果を図23に示す。Form Bの湿度における結晶形への影響を確認するため、各湿度における粉末X線回折測定(実施例4-5)を実施した。結果を図24に示す。相対湿度30%から90%ではForm Bが維持しているが、相対湿度0%ではForm Yに転移することを確認した。Form Yは主要なピークとして、5.13°、8.33°、8.82°、9.80°、10.32°、11.39°、12.58°、13.28°、14.80°、15.40°、15.88°、17.12°、17.67°、19.18°、19.54°、および21.24°を有する。
Example 3-13
The concentrated, dried product containing compound 1 (29.4 g) obtained in Example 1-24-1 was dissolved in acetone (90 mL). The dissolved compound 1 was loaded onto 240 g of Biotage Sfar C18 Duo 100 Å 30 μm and developed with 0.1% formic acid-water/0.1% formic acid-acetonitrile = 9/1 → 0/10). The eluted mixed solution was collected and left to stand for 2 days, resulting in high-quality single crystals. The obtained crystals were dried under reduced pressure at 40 ° C. for 16 hours to obtain 15 mg of dried powder of crystals (Form B). When the obtained crystal (Form B) was subjected to powder X-ray diffraction measurement (Example 4-1), the main peaks were confirmed to be 4.99 °, 8.65 °, 9.85 °, 10.84 °, 11.32 °, 12.35 °, 13.20 °, 14.44 °, 15.20 °, 16.03 °, 16.69 °, 17.21 °, 18.82 °, 19.49 °, and 20.03 °. The measurement results are shown in FIG. 22. The results of simultaneous thermogravimetry and differential thermal analysis are shown in FIG. 23. In order to confirm the effect of humidity on the crystal form of Form B, powder X-ray diffraction measurement (Example 4-5) was performed at each humidity. The results are shown in FIG. 24. It was confirmed that Form B was maintained at a relative humidity of 30% to 90%, but was transformed to Form Y at a relative humidity of 0%. Form Y has major peaks at 5.13°, 8.33°, 8.82°, 9.80°, 10.32°, 11.39°, 12.58°, 13.28°, 14.80°, 15.40°, 15.88°, 17.12°, 17.67°, 19.18°, 19.54°, and 21.24°.
実施例3-14
実施例3-13で得られた結晶は単結晶X線構造解析(実施例4-9)により水和物結晶(Form B)であることを確認した。結晶構造を図25に示す。
Example 3-14
The crystal obtained in Example 3-13 was confirmed to be a hydrate crystal (Form B) by single crystal X-ray structure analysis (Example 4-9). The crystal structure is shown in FIG.
実施例3-15
アモルファス状態の化合物1(77.4mg)をジメチルスルホキシド(0.387mL)に溶解させ、この溶解液(0.015mL)を-20℃で3日間凍結乾燥した。得られた凍結乾燥物に1,4-ジオキサン-ヘプタン混合液(1:4(v/v)、0.015mL)を加え、室温にて7日間振とう攪拌することで化合物1の結晶を得た。得られた結晶は単結晶X線構造解析(実施例4-8)により1,4-ジオキサン/水和物結晶(Form D)であることを確認した。結晶構造を図26に示す。
Example 3-15
Amorphous compound 1 (77.4 mg) was dissolved in dimethyl sulfoxide (0.387 mL), and the solution (0.015 mL) was freeze-dried at -20°C for 3 days. A 1,4-dioxane-heptane mixture (1:4 (v/v), 0.015 mL) was added to the freeze-dried product and stirred with shaking at room temperature for 7 days to obtain crystals of compound 1. The obtained crystals were confirmed to be 1,4-dioxane/hydrate crystals (Form D) by single crystal X-ray structural analysis (Example 4-8). The crystal structure is shown in Figure 26.
実施例3-16
実施例3-8で得られた化合物1の結晶(Form A、6.8mg)をジメチルスルホキシド(0.12mL)に溶解させ、室温にて13日振とう攪拌した。さらに、水(1μL)を加えて室温にて34日間攪拌することで化合物1の結晶を得た。得られた結晶は単結晶X線構造解析(実施例4-8)によりジメチルスルホキシド/水和物結晶(Form L)であることを確認した。結晶構造を図27に示す。同様の操作を行って得られた結晶の粉末X線回折測定(実施例4-3)により、Form Lは減圧乾燥でアモルファス化する溶媒和物結晶であることを確認した。粉末X線回折パターンを図28に示す。
Example 3-16
The crystals of Compound 1 obtained in Example 3-8 (Form A, 6.8 mg) were dissolved in dimethyl sulfoxide (0.12 mL) and stirred at room temperature for 13 days by shaking. Water (1 μL) was then added and stirred at room temperature for 34 days to obtain crystals of Compound 1. The obtained crystals were confirmed to be dimethyl sulfoxide/hydrate crystals (Form L) by single crystal X-ray structure analysis (Example 4-8). The crystal structure is shown in FIG. 27. Powder X-ray diffraction measurement of the crystals obtained by the same procedure (Example 4-3) confirmed that Form L was a solvate crystal that became amorphous by drying under reduced pressure. The powder X-ray diffraction pattern is shown in FIG. 28.
実施例3-17
化合物1の結晶(Form B、45.8mg)にプロピレングリコール(0.12mL)を添加し、室温で5分間攪拌した。さらに、プロピレングリコール(0.020mL)を添加し、40℃で5分間攪拌した後、プロピレングリコール(0.070mL)を添加して40℃で5分間攪拌し、さらに5時間静置した。さらにプロピレングリコール(0.070mL)を添加して35℃で一晩時間攪拌した後、プロピレングリコール(0.070mL)を添加して約1分間攪拌することで化合物1の湿性末を得た。得られた湿性末は、粉末X線回折測定(実施例4-3)によりForm M結晶であることを確認した。Form M結晶を室温で一晩減圧乾燥すると、Form N結晶に転移することを粉末X線回折測定により確認した。Form MおよびForm Nの粉末X線回折パターンを図29に示す。熱重量・示差熱同時測定の結果を図30に示す。157℃で約11%の重量減少が見られ、これは化合物1に対して2.5分子のプロピレングリコールに相当する。以上より、Form MおよびForm Nがプロピレングリコール和物であることを確認した。
Example 3-17
Propylene glycol (0.12 mL) was added to the crystals of Compound 1 (Form B, 45.8 mg) and stirred at room temperature for 5 minutes. Propylene glycol (0.020 mL) was further added and stirred at 40° C. for 5 minutes, then propylene glycol (0.070 mL) was added and stirred at 40° C. for 5 minutes, and the mixture was allowed to stand for another 5 hours. Propylene glycol (0.070 mL) was further added and stirred at 35° C. overnight, then propylene glycol (0.070 mL) was added and stirred for about 1 minute to obtain wet powder of Compound 1. The obtained wet powder was confirmed to be Form M crystals by powder X-ray diffraction measurement (Example 4-3). It was confirmed by powder X-ray diffraction measurement that Form M crystals were transformed to Form N crystals when dried under reduced pressure at room temperature overnight. The powder X-ray diffraction patterns of Form M and Form N are shown in FIG. 29. The results of simultaneous thermogravimetry and differential thermal analysis are shown in FIG. 30. A weight loss of about 11% was observed at 157° C., which corresponds to 2.5 molecules of propylene glycol per compound 1. From the above, it was confirmed that Form M and Form N were propylene glycol solvates.
実施例3-18
化合物1の結晶(Form B、12.66mg)をプロピレングリコール(0.10mL)に溶解させ、室温で20日間攪拌した。さらに、実施例3-17で得られた湿性末を約0.1mg添加して室温で2.5時間攪拌した。プロピレングリコール(0.10mL)を添加し、ホットプレートで60℃、30分加熱攪拌した後、室温で4.5時間振とう攪拌することで、化合物1の結晶を得た。得られた結晶は単結晶X線構造解析(実施例4-8)により、プロピレングリコール/水和物結晶(Form M)であることを確認した。結晶構造を図31に示す。
Example 3-18
The crystals of Compound 1 (Form B, 12.66 mg) were dissolved in propylene glycol (0.10 mL) and stirred at room temperature for 20 days. Furthermore, about 0.1 mg of the wet powder obtained in Example 3-17 was added and stirred at room temperature for 2.5 hours. Propylene glycol (0.10 mL) was added, and the mixture was heated and stirred at 60° C. for 30 minutes on a hot plate, and then shaken and stirred at room temperature for 4.5 hours to obtain crystals of Compound 1. The obtained crystals were confirmed to be propylene glycol/hydrate crystals (Form M) by single crystal X-ray structure analysis (Example 4-8). The crystal structure is shown in FIG. 31.
実施例3-19
実施例1-25で得られた結晶(Form A)を、オープンアルミパンに7.773mg、7.258mg、8.665mg、13.047mg(計36.7mg)それぞれ秤量し、熱重量・示差熱同時測定装置を用いて加熱を行った(実施例4-11)。得られた結晶は、粉末X線回折測定(実施例4-2)を実施したところ、主要なピークとして、7.49°、7.91°、8.14°、9.11°、9.33°、11.04°、11.71°、12.52°、13.21°、13.70°、14.82°、15.13°、15.52°、15.68°、17.22°、および17.51°が確認された。Form Kの粉末X線回折パターンを図32に示す。また、得られたForm K結晶の熱重量・示差熱同時測定の結果を図33に、1H-NMR測定の結果を図34に示す。図33において約1.8wt%の重量減少が見られた一方、1H-NMR測定では測定溶媒のDMSO(テトラメチルシランを含む)、化合物1、および水に相当するピーク以外が見られなかった。以上の結果より、Form Kは水和物と判断した。
Example 3-19
The crystals (Form A) obtained in Example 1-25 were weighed in amounts of 7.773 mg, 7.258 mg, 8.665 mg, and 13.047 mg (total 36.7 mg) in open aluminum pans, and heated using a simultaneous thermogravimetric and differential thermal analyzer (Example 4-11). The obtained crystals were subjected to powder X-ray diffraction measurement (Example 4-2), and the main peaks were confirmed to be 7.49°, 7.91°, 8.14°, 9.11°, 9.33°, 11.04°, 11.71°, 12.52°, 13.21°, 13.70°, 14.82°, 15.13°, 15.52°, 15.68°, 17.22°, and 17.51°. The powder X-ray diffraction pattern of Form K is shown in FIG. 32. The results of simultaneous thermogravimetry and differential thermal analysis of the obtained Form K crystals are shown in Figure 33, and the results of 1 H-NMR measurement are shown in Figure 34. A weight loss of about 1.8 wt % was observed in Figure 33, while the 1 H-NMR measurement showed no peaks other than those corresponding to the measurement solvent DMSO (containing tetramethylsilane), Compound 1, and water. From these results, it was determined that Form K was a hydrate.
実施例3-20
純度の低い粗体溶液を濃縮乾固、40℃で減圧乾燥し粗体のアモルファス固体を調製した。粗体アモルファス固体(20mg)を2-プロパノール(60μL)に溶解させ、スターラーを用いて撹拌した。ヘプタン(60μL)を添加した。Form A種結晶を約0.1mg添加し、室温で1時間撹拌した。結晶の析出が認められなかったため、さらに2-プロパノール(20μL)を添加したところ、オイルアウトし結晶は得られなかった。
Example 3-20
The crude solution with low purity was concentrated to dryness and dried under reduced pressure at 40°C to prepare a crude amorphous solid. The crude amorphous solid (20 mg) was dissolved in 2-propanol (60 μL) and stirred using a stirrer. Heptane (60 μL) was added. Approximately 0.1 mg of Form A seed crystals were added and stirred at room temperature for 1 hour. As no crystal precipitation was observed, further 2-propanol (20 μL) was added, which resulted in oiling out and no crystals being obtained.
実施例3-21
粗体溶液を濃縮乾固、40℃で減圧乾燥し粗体のアモルファス固体を調製した。粗体アモルファス固体(20mg)をエタノール(60μL)に溶解させ、スターラーを用いて撹拌した。ヘプタン(60μL)を添加した。同じ溶液を2種調製し、一方にはForm A種結晶、もう一方にはForm B種結晶をそれぞれ約0.1mg添加し、室温で1時間撹拌した。いずれも結晶が析出し、ろ過し湿性粉末を得た。Form A種結晶を添加した条件にも関わらずForm Bが得られた。一方、Form B種結晶を添加した条件ではFormBが得られた。
Example 3-21
The crude solution was concentrated to dryness and dried under reduced pressure at 40°C to prepare a crude amorphous solid. The crude amorphous solid (20 mg) was dissolved in ethanol (60 μL) and stirred using a stirrer. Heptane (60 μL) was added. Two identical solutions were prepared, one of which was added with about 0.1 mg of Form A seed crystals, and the other was added with about 0.1 mg of Form B seed crystals, and the mixture was stirred at room temperature for 1 hour. In both cases, crystals precipitated, and the mixture was filtered to obtain a wet powder. Form B was obtained despite the conditions in which Form A seed crystals were added. On the other hand, Form B was obtained under the conditions in which Form B seed crystals were added.
実施例3-22
粗体溶液(17.7kg)を外温50℃で7Lまで濃縮した。濃縮液の一部(4.31kg)を反応缶に投入し、エタノール(1.16kg)を加えた。その後外温42℃とし撹拌した。精製水(1.01kg)と種結晶(Form B、5.17g)を添加し1時間撹拌した。続いて精製水(2.08kg)を80分かけて滴下した。さらに精製水(1.04kg)を40分かけて滴下した。48分かけて内温を25℃以下まで降温した。一晩撹拌し、ろ過を行い、結晶をエタノール/精製水(2.50L/1.66L混合)で2回に分けて洗浄した。外温50℃で得られた湿結晶を減圧乾燥し、化合物1(467g)を白色固体として得た。得られた化合物1の結晶(Form B)の粉末X線回折測定(実施例4-3)を実施した。結果を図36に示す。Form B結晶であることを確認した。
Example 3-22
The crude solution (17.7 kg) was concentrated to 7 L at an external temperature of 50° C. A part of the concentrated solution (4.31 kg) was charged into a reactor, and ethanol (1.16 kg) was added. The external temperature was then set to 42° C. and the mixture was stirred. Purified water (1.01 kg) and seed crystals (Form B, 5.17 g) were added and stirred for 1 hour. Purified water (2.08 kg) was then added dropwise over 80 minutes. Purified water (1.04 kg) was further added dropwise over 40 minutes. The internal temperature was lowered to 25° C. or less over 48 minutes. The mixture was stirred overnight, filtered, and the crystals were washed twice with ethanol/purified water (2.50 L/1.66 L mixture). The wet crystals obtained at an external temperature of 50° C. were dried under reduced pressure to obtain compound 1 (467 g) as a white solid. Powder X-ray diffraction measurement (Example 4-3) of the obtained crystals of compound 1 (Form B) was carried out. The results are shown in Figure 36. It was confirmed that the crystal was Form B.
実施例4 結晶の評価
実施例4-1
実施例3-8、実施例3-12、実施例3-13、実施例3-19の粉末X線測定は下記の条件で実施した。
測定装置:SmartLab System、D/Tex Ultra detector(リガク社製)
線源:CuKα1
管電圧:45kV
管電流:200mA
走査範囲:4~50°
サンプリング幅:0.005°
Example 4. Evaluation of crystals
Example 4-1
Powder X-ray measurements of Examples 3-8, 3-12, 3-13, and 3-19 were carried out under the following conditions.
Measurement device: SmartLab System, D/Tex Ultra detector (manufactured by Rigaku Corporation)
Radiation source: CuKα1
Tube voltage: 45 kV
Tube current: 200mA
Scanning range: 4 to 50 degrees
Sampling width: 0.005°
実施例4-2
実施例3-10の粉末X線測定は下記の条件で実施し、懸濁液中の結晶形を評価した。
測定装置:SmartLab System、D/Tex Ultra detector(リガク社製)
線源:CuKα1
管電圧:45kV
管電流:200mA
走査範囲:4~50°
サンプリング幅:0.005°
測定:サンプリングした懸濁液をX線結晶解析用キャピラリーに詰め、測定した。
Example 4-2
Powder X-ray measurements of Examples 3 to 10 were carried out under the following conditions to evaluate the crystal forms in the suspensions.
Measurement device: SmartLab System, D/Tex Ultra detector (manufactured by Rigaku Corporation)
Radiation source: CuKα1
Tube voltage: 45 kV
Tube current: 200mA
Scanning range: 4 to 50 degrees
Sampling width: 0.005°
Measurement: The sampled suspension was packed into a capillary for X-ray crystallography and measured.
実施例4-3
実施例3-2、実施例3-3、実施例3-4、実施例3-5、実施例3-6、実施例3-7、実施例3-9、実施例3-16、実施例3-17、実施例3-22、実施例5-1、実施例5-2、実施例5-3、実施例5-4、実施例5-5、実施例5-6、実施例5-7、実施例5-8、実施例5-9の粉末X線測定は下記の条件で実施した。
測定装置:D8 Discover、2D VANTEC-500 solid state detector(Bruker社製)
線源:CuKα
管電圧、管電流:50kV、1000μA
測定範囲:5~31°
露光時間:40秒
Example 4-3
Powder X-ray measurement of Example 3-2, Example 3-3, Example 3-4, Example 3-5, Example 3-6, Example 3-7, Example 3-9, Example 3-16, Example 3-17, Example 3-22, Example 5-1, Example 5-2, Example 5-3, Example 5-4, Example 5-5, Example 5-6, Example 5-7, Example 5-8, and Example 5-9 was carried out under the following conditions.
Measuring device: D8 Discover, 2D VANTEC-500 solid state detector (manufactured by Bruker)
Radiation source: CuKα
Tube voltage, tube current: 50 kV, 1000 μA
Measurement range: 5 to 31 degrees
Exposure time: 40 seconds
実施例4-4
実施例3-12の粉末X線回折測定は下記の条件で実施し、各相対湿度における結晶形を評価した。相対湿度0%に落としてから、90%に上げるまでの間の時点で、0%(図21(A))、10%(図21(B))、20%(図21(C))、50%(図21(D))、90%(図21(E))を測定した。
測定装置:SmartLab System、D/Tex Ultra detector、水蒸気発生装置HUM-SL(リガク社製)
対陰極:Cu
管電圧:45kV
管電流:200mA
走査範囲:4~35°
走査速度:5°/分
サンプリング幅:0.02°
湿度変化条件:
Example 4-4
Powder X-ray diffraction measurements of Example 3-12 were carried out under the following conditions, and the crystal form at each relative humidity was evaluated. Measurements were taken at 0% (FIG. 21(A)), 10% (FIG. 21(B)), 20% (FIG. 21(C)), 50% (FIG. 21(D)), and 90% (FIG. 21(E)) at points between when the relative humidity was lowered to 0% and when it was raised to 90%.
Measurement equipment: SmartLab System, D/Tex Ultra detector, water vapor generator HUM-SL (manufactured by Rigaku Corporation)
Anticathode: Cu
Tube voltage: 45 kV
Tube current: 200mA
Scanning range: 4 to 35°
Scanning speed: 5°/min Sampling width: 0.02°
Humidity change conditions:

実施例4-5
実施例3-13の粉末X線回折測定は下記の条件で実施し、各相対湿度における結晶形を評価した。相対湿度0%に落としてから、90%に上げるまでの間の時点で、0%(図24(A))、30%(図24(B))、50%(図24(C))、90%(図24(D))を測定した。
測定装置:SmartLab System、D/Tex Ultra detector、水蒸気発生装置HUM-SL(リガク社製)
対陰極:Cu
管電圧:45kV
管電流:200mA
走査範囲:4~35°
走査速度:5°/分
サンプリング幅:0.02°
湿度変化条件:
Examples 4-5
Powder X-ray diffraction measurements of Example 3-13 were carried out under the following conditions, and the crystal form at each relative humidity was evaluated. Measurements were taken at 0% (FIG. 24(A)), 30% (FIG. 24(B)), 50% (FIG. 24(C)), and 90% (FIG. 24(D)) at points between when the relative humidity was lowered to 0% and when it was raised to 90%.
Measurement equipment: SmartLab System, D/Tex Ultra detector, water vapor generator HUM-SL (manufactured by Rigaku Corporation)
Anticathode: Cu
Tube voltage: 45 kV
Tube current: 200mA
Scanning range: 4 to 35°
Scanning speed: 5°/min Sampling width: 0.02°
Humidity change conditions:
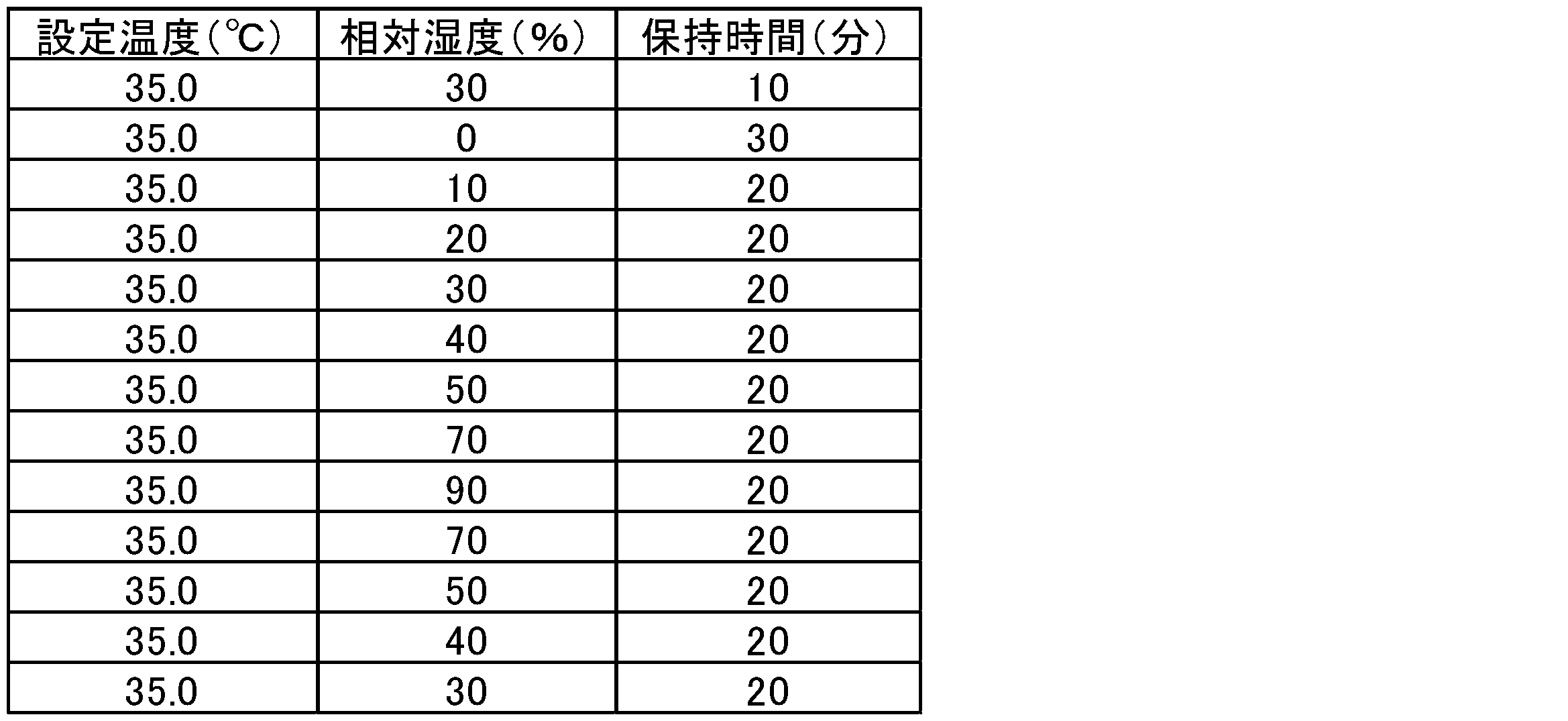
実施例4-6
実施例3-8、実施例3-9、実施例3-12、実施例3-13、実施例3-17、実施例3-19の熱重量・示差熱同時測定(TG-DTA)は下記の条件で実施した。
測定装置:STA7200RV+AS-3T(日立ハイテクサイエンス製)
測定範囲:30~350℃
昇温速度:10℃/分
雰囲気:窒素
測定:サンプルをオープンアルミパンに秤量し、メッシュを被せて測定を行った。
Examples 4-6
Thermogravimetry and differential thermal analysis (TG-DTA) of Examples 3-8, 3-9, 3-12, 3-13, 3-17, and 3-19 were carried out under the following conditions.
Measurement device: STA7200RV+AS-3T (Hitachi High-Tech Science)
Measurement range: 30 to 350°C
Heating rate: 10° C./min. Atmosphere: nitrogen Measurement: A sample was weighed in an open aluminum pan, covered with a mesh and then the measurement was performed.
実施例4-7
実施例3-1の単結晶X線構造解析は下記の条件で実施した。
測定装置:Rigaku XtaLAB Synergy Custom with a VariMax Cu Diffractometer(リガク社製)
対陰極:Cu
管電圧:40kV
管電流:30mA
温度:-180℃
測定:構造解析に十分な回折斑点が得られると考えられるストラテジー、露光時間で測定を行った。
構造解析:Olex2プログラムを用い、初期構造決定はSIR2008、構造精密化はFull-matrix least-squares method(SHELXL-2018/3)で行った。
Examples 4-7
The single crystal X-ray structure analysis of Example 3-1 was carried out under the following conditions.
Measurement device: Rigaku XtaLAB Synergy Custom with a VariMax Cu Diffractometer (manufactured by Rigaku Corporation)
Anticathode: Cu
Tube voltage: 40 kV
Tube current: 30mA
Temperature: -180°C
Measurement: Measurement was performed using a strategy and exposure time that was believed to provide sufficient diffraction spots for structural analysis.
Structural analysis: The Olex2 program was used, the initial structure was determined using SIR2008, and the structure was refined using the Full-matrix least-squares method (SHELXL-2018/3).
実施例4-8
実施例3-3、実施例3-11実施例3-15、実施例3-16、実施例3-18の単結晶X線構造解析は下記の条件で実施した。
測定装置:Rigaku XtaLAB Synergy Custom with a VariMax Cu Diffractometer(リガク社製)
対陰極:Cu
管電圧:40kV
管電流:30mA
温度:-180℃
測定:構造解析に十分な回折斑点が得られると考えられるストラテジー、露光時間で測定を行った。
構造解析:Olex2プログラムを用い、初期構造決定はDual Space method(SHELXD-2008)、構造精密化はFull-matrix least-squares method(SHELXL-2018/3)で行った。
Examples 4-8
The single crystal X-ray structure analyses of Examples 3-3, 3-11, 3-15, 3-16, and 3-18 were carried out under the following conditions.
Measurement device: Rigaku XtaLAB Synergy Custom with a VariMax Cu Diffractometer (manufactured by Rigaku Corporation)
Anticathode: Cu
Tube voltage: 40 kV
Tube current: 30mA
Temperature: -180°C
Measurement: Measurement was performed using a strategy and exposure time that was believed to provide sufficient diffraction spots for structural analysis.
Structural analysis: Using the Olex2 program, initial structure determination was performed using the Dual Space method (SHELXD-2008), and structure refinement was performed using the Full-matrix least-squares method (SHELXL-2018/3).
実施例4-9
実施例3-6、実施例3-7、実施例3-14の単結晶X線構造解析は下記の条件で実施した。
測定装置:高エネルギー加速器研究機構 フォトンファクトリー BL-5A
線源:放射光(λ=0.92Å)
温度:-178℃
測定:構造解析に十分な回折斑点が得られると考えられるストラテジー、露光時間で測定を行った。
構造解析:Olex2プログラムを用い、初期構造決定はDual Space method(SHELXD-2008)、構造精密化はFull-matrix least-squares method(SHELXL-2018/3)で行った。
Examples 4-9
The single crystal X-ray structure analyses of Examples 3-6, 3-7, and 3-14 were carried out under the following conditions.
Measurement equipment: High Energy Accelerator Research Organization Photon Factory BL-5A
Line source: synchrotron radiation (λ=0.92Å)
Temperature: -178°C
Measurement: Measurement was performed using a strategy and exposure time that was believed to provide sufficient diffraction spots for structural analysis.
Structural analysis: Using the Olex2 program, initial structure determination was performed using the Dual Space method (SHELXD-2008), and structure refinement was performed using the Full-matrix least-squares method (SHELXL-2018/3).
実施例4-10
実施例3-8、実施例3-9、実施例3-19の1H-NMR測定は下記の条件で実施した。
測定装置:JNM-ECX500II(JEOL社製)
測定溶媒:DMSO-d6,contains 0.03%(v/v)TMS
測定温度:295K
サンプル調製:市販の重水素化溶媒を、測定対象化合物と混合して調製した。
積算回数、緩和待ち時間:十分なs/n比が得られると考えられる回数(8、16または512回)、緩和時間より十分長い時間(60秒)で測定を実施した。
積分値:各シグナルのシグナル面積強度比をもとに、残留溶媒比率(重量%)を算出した。
Examples 4-10
The 1 H-NMR measurements of Examples 3-8, 3-9, and 3-19 were carried out under the following conditions.
Measuring device: JNM-ECX500II (manufactured by JEOL)
Measurement solvent: DMSO-d6, contains 0.03% (v/v) TMS
Measurement temperature: 295K
Sample preparation: A commercially available deuterated solvent was mixed with the compound to be measured to prepare a sample.
Accumulation number and relaxation waiting time: Measurement was performed the number of times (8, 16 or 512) that was considered to provide a sufficient s/n ratio, and for a time (60 seconds) sufficiently longer than the relaxation time.
The residual solvent ratio (wt %) was calculated based on the integral value: signal area intensity ratio of each signal.
実施例4-11
実施例3-19のサンプル調製は下記の条件で実施した。
使用装置:STA7200RV+AS-3T(日立ハイテクサイエンス製)
温度範囲:30~120℃
昇温速度:20℃/分(30~120℃)、120℃で60分維持
雰囲気:窒素
調製:サンプルをオープンアルミパンに秤量し、加熱を行った。
Example 4-11
The sample preparation for Example 3-19 was carried out under the following conditions.
Equipment used: STA7200RV+AS-3T (Hitachi High-Tech Science)
Temperature range: 30 to 120°C
Heating rate: 20° C./min (30-120° C.), maintained at 120° C. for 60 minutes. Atmosphere: nitrogen. Preparation: A sample was weighed in an open aluminum pan and heated.
実施例5 スラリーコンバージョン実験
実施例5-1
化合物1のForm A結晶(3.0mg)、Form B結晶(3.0mg)、Form K結晶(3.0mg)を混合し、アセトン-ヘプタン混合液(1:1(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(A)に示す。アセトン-ヘプタン混合液(1:1(v/v))中ではForm B結晶が安定形であることが確認された。
Example 5 Slurry conversion experiment
Example 5-1
Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.0 mg) of Compound 1 were mixed, and an acetone-heptane mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (A). It was confirmed that Form B crystals were the stable form in the acetone-heptane mixture (1:1 (v/v)).
実施例5-2
化合物1のForm A結晶(3.1mg)、Form B結晶(3.1mg)、Form K結晶(3.1mg)を混合し、2-プロパノール-ヘプタン混合液(1:1(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(B)に示す。2-プロパノール-ヘプタン混合液(1:1(v/v))中ではForm B結晶が安定形であることが確認された。
Example 5-2
Form A crystals (3.1 mg), Form B crystals (3.1 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and a 2-propanol-heptane mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (B). It was confirmed that Form B crystals were the stable form in the 2-propanol-heptane mixture (1:1 (v/v)).
実施例5-3
化合物1のForm A結晶(3.0mg)、Form B結晶(3.0mg)、Form K結晶(3.1mg)を混合し、エタノール-ヘプタン混合液(1:1(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(C)に示す。エタノール-ヘプタン混合液(1:1(v/v))中ではForm B結晶が安定形であることが確認された。
Example 5-3
Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and an ethanol-heptane mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred and shaken at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (C). It was confirmed that Form B crystals were the stable form in the ethanol-heptane mixture (1:1 (v/v)).
実施例5-4
化合物1のForm A結晶(3.0mg)、Form B結晶(3.0mg)、Form K結晶(3.0mg)を混合し、アセトン-水混合液(1:1(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(D)に示す。アセトン-水混合液(1:1(v/v))中ではForm B結晶が安定形であることが確認された。
Example 5-4
Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.0 mg) of Compound 1 were mixed, and an acetone-water mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (D). It was confirmed that Form B crystals were the stable form in the acetone-water mixture (1:1 (v/v)).
実施例5-5
化合物1のForm A結晶(3.0mg)、Form B結晶(3.1mg)、Form K結晶(3.1mg)を混合し、2-プロパノール-水混合液(1:1(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(E)に示す。2-プロパノール-水混合液(1:1(v/v))中ではForm B結晶が安定形であることが確認された。
Example 5-5
Form A crystals (3.0 mg), Form B crystals (3.1 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and a 2-propanol-water mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (E). It was confirmed that Form B crystals were the stable form in the 2-propanol-water mixture (1:1 (v/v)).
実施例5-6
化合物1のForm A結晶(3.1mg)、Form B結晶(3.0mg)、Form K結晶(3.1mg)を混合し、エタノール-水混合液(1:1(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(F)に示す。エタノール-水混合液(1:1(v/v))中ではForm B結晶が安定形であることが確認された。
Examples 5-6
Form A crystals (3.1 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and an ethanol-water mixture (1:1 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (F). It was confirmed that Form B crystals were the stable form in the ethanol-water mixture (1:1 (v/v)).
実施例5-7
化合物1のForm A結晶(3.0mg)、Form B結晶(3.0mg)、Form K結晶(3.1mg)を混合し、アセトン-水混合液(1:4(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(G)に示す。アセトン-水混合液(1:4(v/v))中ではForm B結晶が安定形であることが確認された。
Examples 5-7
Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and an acetone-water mixture (1:4 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (G). It was confirmed that Form B crystals were the stable form in the acetone-water mixture (1:4 (v/v)).
実施例5-8
化合物1のForm A結晶(3.0mg)、Form B結晶(3.0mg)、Form K結晶(3.1mg)を混合し、2-プロパノール-水混合液(1:4(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで9日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(H)に示す。2-プロパノール-水混合液(1:4(v/v))中ではForm B結晶が安定形であることが確認された。
Examples 5-8
Form A crystals (3.0 mg), Form B crystals (3.0 mg), and Form K crystals (3.1 mg) of Compound 1 were mixed, and a 2-propanol-water mixture (1:4 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 9 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (H). It was confirmed that Form B crystals were the stable form in the 2-propanol-water mixture (1:4 (v/v)).
実施例5-9
化合物1のForm A結晶(3.0mg)、Form B結晶(3.1mg)、Form K結晶(3.0mg)を混合し、エタノール-水混合液(1:4(v/v)、0.030mL)とガラスビーズ1粒を加えた。振とう機(TAITEC製BioShaker M・BR-022UP)を用い、25℃、2000rpmで7日間振とう攪拌し、ろ過して減圧乾燥することで化合物1の結晶(Form B)を得た。得られた結晶の粉末X線回折測定(実施例4-3)を図35(I)に示す。エタノール-水混合液(1:4(v/v))中ではForm B結晶が安定形であることが確認された。
Examples 5-9
Form A crystals (3.0 mg), Form B crystals (3.1 mg), and Form K crystals (3.0 mg) of Compound 1 were mixed, and an ethanol-water mixture (1:4 (v/v), 0.030 mL) and one glass bead were added. The mixture was stirred at 25°C and 2000 rpm for 7 days using a shaker (TAITEC BioShaker M.BR-022UP), filtered, and dried under reduced pressure to obtain crystals of Compound 1 (Form B). Powder X-ray diffraction measurement of the obtained crystals (Example 4-3) is shown in Figure 35 (I). It was confirmed that Form B crystals were the stable form in the ethanol-water mixture (1:4 (v/v)).
本発明により、医薬品として有用な環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物を製造する方法、および該環状ペプチド化合物、もしくはその塩またはそれらの溶媒和物の製造に用いられるペプチド化合物を製造する方法が提供される。
The present invention provides a method for producing a cyclic peptide compound, or a salt thereof, or a solvate thereof that is useful as a pharmaceutical, and a method for producing a peptide compound used in the production of the cyclic peptide compound, or a salt thereof, or a solvate thereof.
Claims (16)
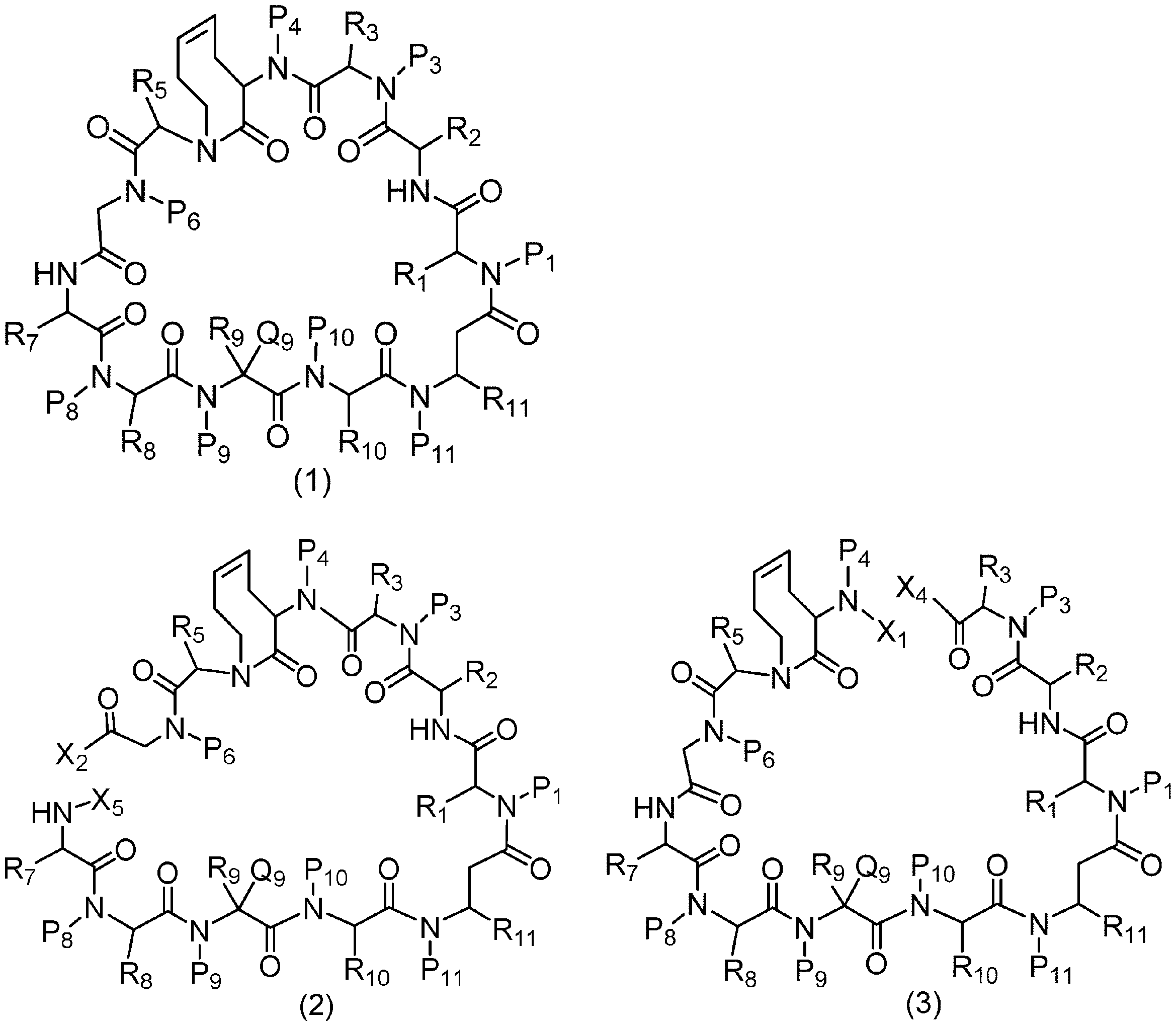
P1は、C1-C6アルキルであり;
R2は、C1-C6アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P3は、C1-C6アルキル、またはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P4は、C1-C6アルキルであり;
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルであり;
P6は、C1-C6アルキルであり;
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1~C6アルコキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよく;
P9は、水素またはC1-C6アルキルであり;
R10は、C1-C6アルキル、またはC3-C8シクロアルキルであり;
P10は、C1-C6アルキルであり;
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルであり;
P11は、C1-C6アルキルであり;
X1およびX5は、それぞれ独立して水素、またはアミノ基の保護基であり;
X2およびX4は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。] A method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof, comprising a step of cyclization (cyclization step) by reacting an N-terminal amino acid residue with a C-terminal amino acid residue of a peptide compound represented by formula (2) or (3) in a solvent.
P1 is C1 - C6 alkyl;
R2 is C1 - C6 alkyl;
R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring;
P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle ;
P4 is C1 - C6 alkyl;
R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl;
P6 is C1 - C6 alkyl;
R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4-7 membered saturated heterocycle, which is optionally substituted by C 1 -C 6 alkoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 3- to 8-membered alicyclic ring, which is optionally substituted by one or more C 1 -C 6 alkyl;
P9 is hydrogen or C1 - C6 alkyl;
R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl;
P10 is C1 - C6 alkyl;
R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4-8 membered cyclic aminocarbonyl;
P11 is C1 - C6 alkyl;
X1 and X5 are each independently hydrogen or an amino-protecting group;
X2 and X4 each independently represent a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiRxRyRz (wherein Rx , Ry , and Rz each independently represent an alkyl or an aryl ) .
(a) 式(4)~(6)で表されるペプチド化合物、もしくはその塩または前記ペプチド化合物もしくは塩の溶媒和物を用意する工程、
(b) 式(4)~(6)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて連結する工程(連結工程)、および
(c) (b)工程で得られたペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、前記方法。
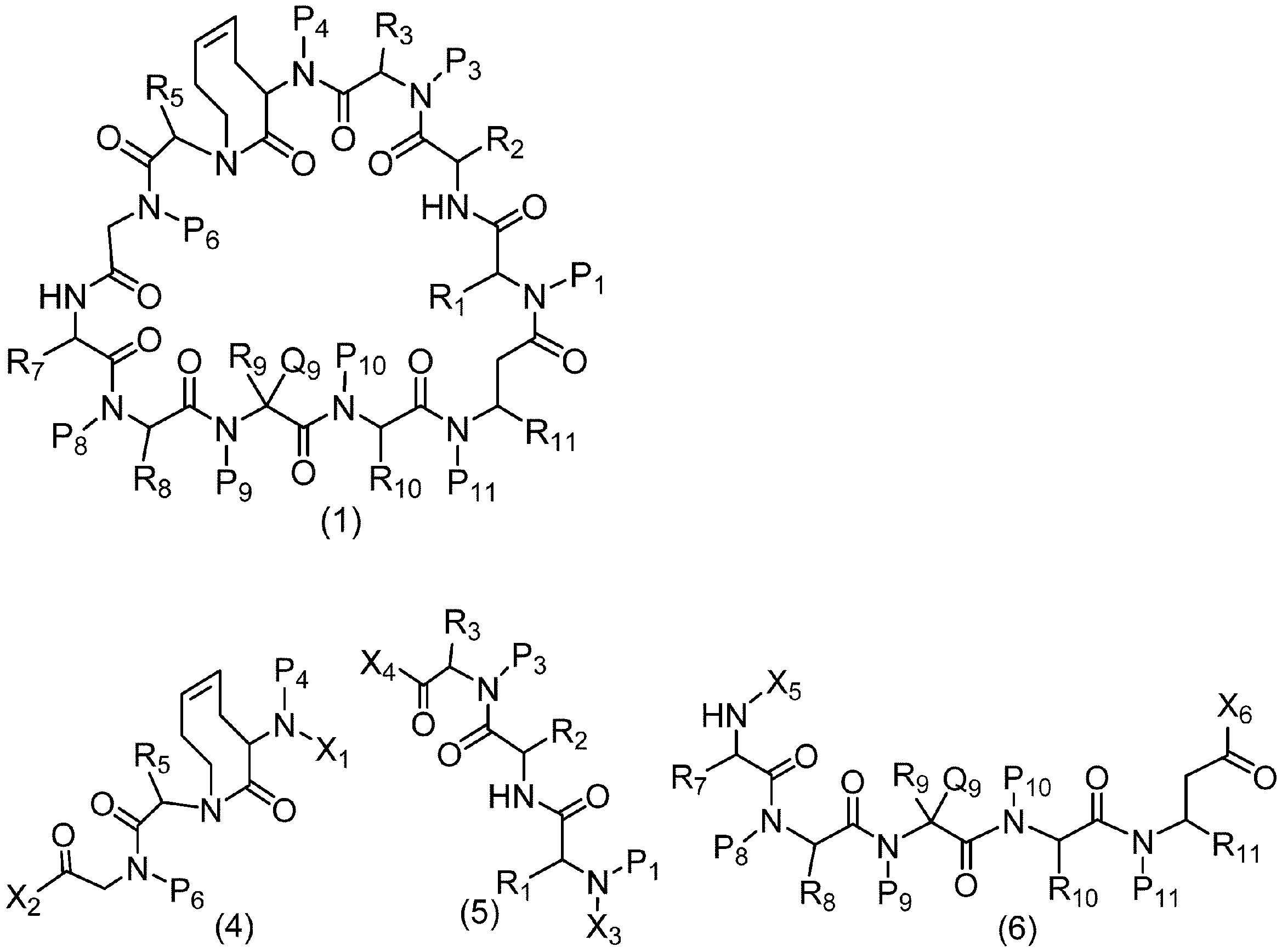
P1は、C1-C6アルキルであり;
R2は、C1-C6アルキルであり;
R3は、水素であるか、またはR3は、P3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P3は、C1-C6アルキル、またはC3-C8シクロアルキルであるか、またはP3は、R3、R3が結合している炭素原子、およびP3が結合している窒素原子と一緒になって4~7員飽和複素環を形成し;
P4は、C1-C6アルキルであり;
R5は、C1-C6アルキル、C1-C6ハロアルキル、およびC3-C8シクロアルキルからなる群より選択される1つまたは複数の基によって置換されていてもよいベンジルであり;
P6は、C1-C6アルキルであり;
R7は、ハロゲン、C1-C6ハロアルキル、およびC1-C6アルコキシからなる群より選択される1つまたは複数の基によって置換されていてもよいフェネチルであり;
R8は、P8、R8が結合している炭素原子、およびP8が結合している窒素原子と一緒になって4~7員飽和複素環を形成し、該4~7員飽和複素環はC1-C6アルコキシによって置換されていてもよく;
R9は、Q9、並びにR9およびQ9が結合している炭素原子と一緒になって3~8員脂環式環を形成し、該3~8員脂環式環は、1つまたは複数のC1-C6アルキルによって置換されていてもよく;
P9は、水素またはC1-C6アルキルであり;
R10は、C1-C6アルキル、またはC3-C8シクロアルキルであり;
P10は、C1-C6アルキルであり;
R11は、ジC1-C6アルキルアミノカルボニル、または4~8員環状アミノカルボニルであり;
P11は、C1-C6アルキルであり;
X1、X3およびX5は、それぞれ独立して水素、またはアミノ基の保護基であり;
X2、X4およびX6は、それぞれ独立してハロゲン、水酸基、置換されていてもよいアルコキシ、置換されていてもよいアリールオキシ、置換されていてもよいアラルコキシ、置換されていてもよい環状アミノオキシ、または-OSiRxRyRz(式中、Rx、Ry、およびRzは、それぞれ独立して、アルキル、またはアリールである)で表される基である。] A method for producing a cyclic peptide compound represented by formula (1), or a salt thereof, or a solvate thereof, comprising the steps of:
(a) preparing a peptide compound represented by any one of formulas (4) to (6), or a salt thereof, or a solvate of said peptide compound or salt;
(b) a step of reacting and linking the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formulae (4) to (6) in a solvent (linking step); and
(c) the step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound obtained in the step (b) by reacting them in a solvent (cyclization step).
P1 is C1 - C6 alkyl;
R2 is C1 - C6 alkyl;
R 3 is hydrogen or R 3 together with P 3 , the carbon atom to which R 3 is attached, and the nitrogen atom to which P 3 is attached form a 4- to 7-membered saturated heterocyclic ring;
P3 is C1 - C6 alkyl, or C3 - C8 cycloalkyl, or P3 together with R3, the carbon atom to which R3 is attached, and the nitrogen atom to which P3 is attached form a 4- to 7-membered saturated heterocycle ;
P4 is C1 - C6 alkyl;
R 5 is benzyl optionally substituted with one or more groups selected from the group consisting of C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 3 -C 8 cycloalkyl;
P6 is C1 - C6 alkyl;
R 7 is phenethyl optionally substituted with one or more groups selected from the group consisting of halogen, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
R 8 together with P 8 , the carbon atom to which R 8 is attached, and the nitrogen atom to which P 8 is attached form a 4- to 7-membered saturated heterocycle, which is optionally substituted by C 1 -C 6 alkoxy;
R 9 together with Q 9 and the carbon atom to which R 9 and Q 9 are attached form a 3- to 8-membered alicyclic ring, which is optionally substituted by one or more C 1 -C 6 alkyl;
P9 is hydrogen or C1 - C6 alkyl;
R 10 is C 1 -C 6 alkyl, or C 3 -C 8 cycloalkyl;
P10 is C1 - C6 alkyl;
R 11 is diC 1 -C 6 alkylaminocarbonyl, or 4-8 membered cyclic aminocarbonyl;
P11 is C1 - C6 alkyl;
X 1 , X 3 and X 5 are each independently hydrogen or a protecting group for an amino group;
X 2 , X 4 and X 6 each independently represent a halogen, a hydroxyl group, an optionally substituted alkoxy, an optionally substituted aryloxy, an optionally substituted aralkoxy, an optionally substituted cyclic aminooxy, or a group represented by -OSiR x R y R z (wherein R x , R y and R z each independently represent an alkyl or an aryl).
(b-1)式(5)で表されるペプチド化合物のN末端のアミノ酸残基と、式(6)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(7)で表されるペプチド化合物に変換する工程(連結工程)を含む、請求項2に記載の方法。
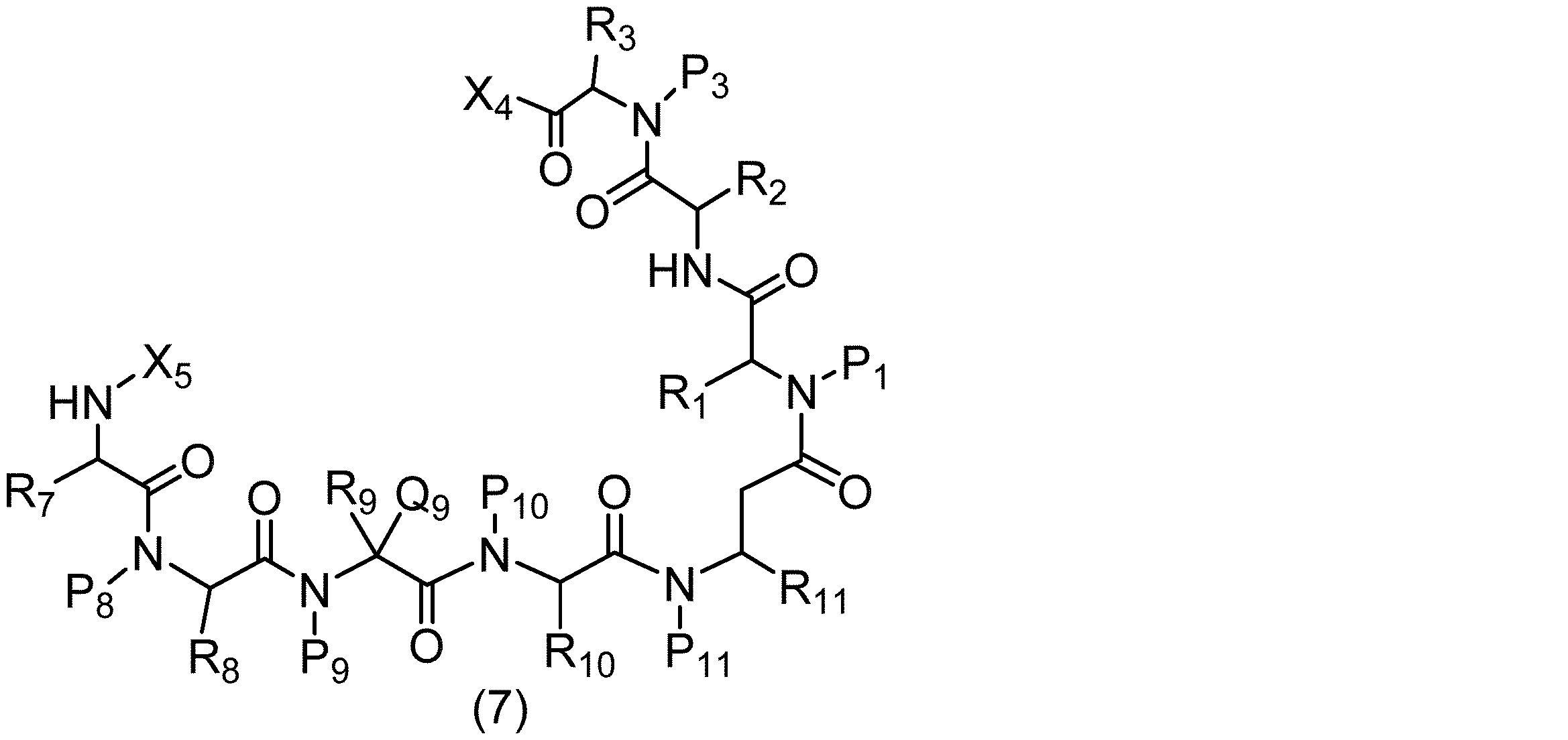
The method according to claim 2, comprising: (b-1) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (5) and the C-terminal amino acid residue of the peptide compound represented by formula (6) in a solvent to convert them into the peptide compound represented by formula (7) (linking step).
(b-2)式(4)で表されるペプチド化合物のN末端のアミノ酸残基と、式(7)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(2)で表されるペプチド化合物に変換する工程(連結工程)、および、
前記(c)の工程において、
(c-1)式(2)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、請求項3に記載の方法。 In the step (b),
(b-2) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (4) and the C-terminal amino acid residue of the peptide compound represented by formula (7) in a solvent to convert them into the peptide compound represented by formula (2) (linking step); and
In the step (c),
The method according to claim 3, comprising (c-1) a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (2) in a solvent (cyclization step).
(b-3)式(7)で表されるペプチド化合物のN末端のアミノ酸残基と、式(4)で表されるペプチド化合物のC末端のアミノ酸残基とを、溶媒中、反応させて連結することにより式(3)で表される化合物に変換する工程(連結工程)、および、
前記(c)の工程において、
(c-2)式(3)で表されるペプチド化合物のN末端のアミノ酸残基とC末端のアミノ酸残基とを、溶媒中、反応させて環化する工程(環化工程)を含む、請求項3に記載の方法。 In the step (b),
(b-3) a step of reacting and linking the N-terminal amino acid residue of the peptide compound represented by formula (7) and the C-terminal amino acid residue of the peptide compound represented by formula (4) in a solvent to convert them into a compound represented by formula (3) (linking step); and
In the step (c),
The method according to claim 3, further comprising: (c-2) a step of cyclizing the N-terminal amino acid residue and the C-terminal amino acid residue of the peptide compound represented by formula (3) in a solvent (cyclization step).

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023069189 | 2023-04-20 | ||
| JP2023-069189 | 2023-04-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024219480A1 true WO2024219480A1 (en) | 2024-10-24 |
Family
ID=93152939
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/015529 WO2024219480A1 (en) | 2023-04-20 | 2024-04-19 | Method for producing cyclic peptide compound |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202506706A (en) |
| WO (1) | WO2024219480A1 (en) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11505208A (en) * | 1995-03-29 | 1999-05-18 | 藤沢薬品工業株式会社 | Cyclic peptide compounds and their derivatives |
| WO2022138891A1 (en) * | 2020-12-25 | 2022-06-30 | 中外製薬株式会社 | Method for producing peptide compound containing n-substituted-amino acid residue |
| WO2022234853A1 (en) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Cyclic compound having inhibitory effect selective for kras but not for hras and nras |
| WO2022234850A1 (en) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Preparation containing cyclic peptide compound and method for producing same |
| WO2022234864A1 (en) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Method for producing cyclic compound containing n-substituted amino acid residue |
-
2024
- 2024-04-19 TW TW113114671A patent/TW202506706A/en unknown
- 2024-04-19 WO PCT/JP2024/015529 patent/WO2024219480A1/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11505208A (en) * | 1995-03-29 | 1999-05-18 | 藤沢薬品工業株式会社 | Cyclic peptide compounds and their derivatives |
| WO2022138891A1 (en) * | 2020-12-25 | 2022-06-30 | 中外製薬株式会社 | Method for producing peptide compound containing n-substituted-amino acid residue |
| WO2022234853A1 (en) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Cyclic compound having inhibitory effect selective for kras but not for hras and nras |
| WO2022234850A1 (en) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Preparation containing cyclic peptide compound and method for producing same |
| WO2022234864A1 (en) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Method for producing cyclic compound containing n-substituted amino acid residue |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202506706A (en) | 2025-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2022234864A1 (en) | Method for producing cyclic compound containing n-substituted amino acid residue | |
| CN114630835B (en) | Method for preparing peptide compounds containing sterically hindered amino acids | |
| TWI873263B (en) | Synthesis of peptide compounds | |
| WO2005118620A2 (en) | Methods for preparing internally constraied peptides and peptidomimetics | |
| EP1805187A1 (en) | Inhibitors of hepatitis c virus protease, and compositions and treatments using the same | |
| TW201245149A (en) | Processes and intermediates | |
| EP4269422A1 (en) | Method for producing peptide compound containing n-substituted-amino acid residue | |
| JP7430297B2 (en) | Method for producing N-alkylamino acids and peptides containing N-alkylamino acids | |
| JP2012525392A (en) | Methods and intermediates | |
| TW202229306A (en) | Peptide synthetic method to suppress deletions caused by diketopiperazine formation | |
| KR20230019120A (en) | Efficient peptide condensation of difficult sequences | |
| CN106456613A (en) | Process for the preparation of cyclic depsipeptides | |
| WO2024219480A1 (en) | Method for producing cyclic peptide compound | |
| CN120051480A (en) | Method for producing cyclic peptide compounds containing N-substituted amino acid residues | |
| JP7165289B1 (en) | Method for Producing Cyclic Compounds Containing N-Substituted Amino Acid Residues | |
| CN117279928A (en) | Process for the preparation of cyclic compounds comprising N-substituted amino acid residues | |
| EA049862B1 (en) | METHOD FOR PRODUCING A CYCLIC COMPOUND CONTAINING AN N-SUBSTITUTED AMINO ACID RESIDURE | |
| TW202523345A (en) | Method for producing peptide compounds using mixed acid anhydride method | |
| JP2024539131A (en) | Process for preparing bicyclic glycine-proline compounds and their monocyclic glycine-proline intermediates | |
| EA049473B1 (en) | COMPOSITIONS OF TROPHINETIDE | |
| JPH04261198A (en) | Endothelin antagonistic cyclic pentapeptide | |
| EP1476427A4 (en) | 1,2-Dihydro-3 (6H) -pyricin-based peptide-base reactants |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24792752 Country of ref document: EP Kind code of ref document: A1 |