WO2024145659A1 - Heterocycle fused gamma-carbolines acting on the serotonine 5-ht2a receptor - Google Patents
Heterocycle fused gamma-carbolines acting on the serotonine 5-ht2a receptor Download PDFInfo
- Publication number
- WO2024145659A1 WO2024145659A1 PCT/US2023/086562 US2023086562W WO2024145659A1 WO 2024145659 A1 WO2024145659 A1 WO 2024145659A1 US 2023086562 W US2023086562 W US 2023086562W WO 2024145659 A1 WO2024145659 A1 WO 2024145659A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- cycloalkyl
- methyl
- receptor
- Prior art date
Links
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 title abstract description 48
- 125000000623 heterocyclic group Chemical group 0.000 title abstract description 10
- 102000049773 5-HT2A Serotonin Receptor Human genes 0.000 title description 4
- 108010072564 5-HT2A Serotonin Receptor Proteins 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 456
- 238000000034 method Methods 0.000 claims abstract description 195
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 76
- 102000000072 beta-Arrestins Human genes 0.000 claims abstract description 58
- 108010080367 beta-Arrestins Proteins 0.000 claims abstract description 58
- 150000003839 salts Chemical class 0.000 claims abstract description 58
- 239000000556 agonist Substances 0.000 claims abstract description 51
- 238000011282 treatment Methods 0.000 claims abstract description 45
- 239000005557 antagonist Substances 0.000 claims abstract description 37
- 230000008484 agonism Effects 0.000 claims abstract description 32
- -1 methoxy, ethoxy Chemical group 0.000 claims description 208
- 230000011664 signaling Effects 0.000 claims description 52
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 49
- 239000003814 drug Substances 0.000 claims description 47
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 31
- 208000015114 central nervous system disease Diseases 0.000 claims description 30
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 29
- 208000012902 Nervous system disease Diseases 0.000 claims description 24
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 23
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 23
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 20
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 claims description 18
- 239000003085 diluting agent Substances 0.000 claims description 18
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims description 17
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 claims description 16
- 230000008485 antagonism Effects 0.000 claims description 16
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 15
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 claims description 15
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 15
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 claims description 13
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 12
- 125000003118 aryl group Chemical group 0.000 claims description 10
- 125000000217 alkyl group Chemical group 0.000 claims description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 8
- 125000001072 heteroaryl group Chemical group 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 7
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 6
- 125000001475 halogen functional group Chemical group 0.000 claims description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 6
- 238000011321 prophylaxis Methods 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 4
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 4
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 claims description 4
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 claims description 4
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 claims description 4
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 4
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 claims description 4
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 4
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 claims description 4
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 claims description 4
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 4
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 claims description 4
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 claims description 4
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 claims description 3
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 3
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000004536 indazol-1-yl group Chemical group N1(N=CC2=CC=CC=C12)* 0.000 claims description 3
- 125000004245 indazol-3-yl group Chemical group [H]N1N=C(*)C2=C([H])C([H])=C([H])C([H])=C12 0.000 claims description 3
- 125000000593 indol-1-yl group Chemical group [H]C1=C([H])C([H])=C2N([*])C([H])=C([H])C2=C1[H] 0.000 claims description 3
- 125000000814 indol-3-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C([*])C2=C1[H] 0.000 claims description 3
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 3
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 2
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 claims description 2
- 125000001607 1,2,3-triazol-1-yl group Chemical group [*]N1N=NC([H])=C1[H] 0.000 claims description 2
- CSNIZNHTOVFARY-UHFFFAOYSA-N 1,2-benzothiazole Chemical compound C1=CC=C2C=NSC2=C1 CSNIZNHTOVFARY-UHFFFAOYSA-N 0.000 claims description 2
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 claims description 2
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 claims description 2
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 claims description 2
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 claims description 2
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 2
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 2
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 2
- 125000004414 alkyl thio group Chemical group 0.000 claims description 2
- 125000004104 aryloxy group Chemical group 0.000 claims description 2
- 125000005605 benzo group Chemical group 0.000 claims description 2
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 claims description 2
- 239000012964 benzotriazole Substances 0.000 claims description 2
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 2
- 125000001188 haloalkyl group Chemical group 0.000 claims description 2
- 125000004531 indol-5-yl group Chemical group [H]N1C([H])=C([H])C2=C([H])C(*)=C([H])C([H])=C12 0.000 claims description 2
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 claims description 2
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 claims description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 claims description 2
- 125000001624 naphthyl group Chemical group 0.000 claims description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 claims description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 claims description 2
- 125000004553 quinoxalin-5-yl group Chemical group N1=CC=NC2=C(C=CC=C12)* 0.000 claims description 2
- 229930192474 thiophene Natural products 0.000 claims description 2
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 38
- 230000003400 hallucinatory effect Effects 0.000 abstract description 29
- 230000019491 signal transduction Effects 0.000 abstract description 23
- 229940076279 serotonin Drugs 0.000 abstract description 20
- 208000019901 Anxiety disease Diseases 0.000 abstract description 18
- 230000036506 anxiety Effects 0.000 abstract description 18
- 239000007787 solid Substances 0.000 abstract description 15
- 201000010099 disease Diseases 0.000 abstract description 10
- 208000019022 Mood disease Diseases 0.000 abstract description 6
- 208000007415 Anhedonia Diseases 0.000 abstract description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 238
- 102000005962 receptors Human genes 0.000 description 85
- 108020003175 receptors Proteins 0.000 description 85
- 230000000694 effects Effects 0.000 description 74
- 239000000203 mixture Substances 0.000 description 74
- 238000001308 synthesis method Methods 0.000 description 73
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 68
- 238000005160 1H NMR spectroscopy Methods 0.000 description 67
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 59
- 239000010410 layer Substances 0.000 description 52
- LQDHVNHVHAYANB-UHFFFAOYSA-N 1-(2-bromoethyl)-3-methoxybenzene Chemical compound COC1=CC=CC(CCBr)=C1 LQDHVNHVHAYANB-UHFFFAOYSA-N 0.000 description 51
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 51
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 46
- 229920000642 polymer Polymers 0.000 description 32
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- 238000012360 testing method Methods 0.000 description 30
- 208000035475 disorder Diseases 0.000 description 28
- 230000027455 binding Effects 0.000 description 26
- 229940079593 drug Drugs 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 238000003556 assay Methods 0.000 description 24
- 239000011159 matrix material Substances 0.000 description 24
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 24
- 235000019439 ethyl acetate Nutrition 0.000 description 23
- 230000001404 mediated effect Effects 0.000 description 22
- 210000004027 cell Anatomy 0.000 description 21
- 230000005764 inhibitory process Effects 0.000 description 21
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 20
- 230000015572 biosynthetic process Effects 0.000 description 20
- 230000037361 pathway Effects 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 239000011859 microparticle Substances 0.000 description 18
- 229940124597 therapeutic agent Drugs 0.000 description 18
- 239000000935 antidepressant agent Substances 0.000 description 17
- 239000000243 solution Substances 0.000 description 17
- BYQQRDWZLNYPPA-UHFFFAOYSA-N α-methylserotonin Chemical compound C1=CC(O)=C[C]2C(CC(N)C)=CN=C21 BYQQRDWZLNYPPA-UHFFFAOYSA-N 0.000 description 17
- JXSPKRUNMHMICQ-UHFFFAOYSA-N 1-(2-bromoethoxy)-4-fluorobenzene Chemical compound FC1=CC=C(OCCBr)C=C1 JXSPKRUNMHMICQ-UHFFFAOYSA-N 0.000 description 16
- 208000028017 Psychotic disease Diseases 0.000 description 16
- 238000009472 formulation Methods 0.000 description 16
- 230000001337 psychedelic effect Effects 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- 108091006027 G proteins Proteins 0.000 description 15
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 15
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 15
- 102000030782 GTP binding Human genes 0.000 description 15
- 108091000058 GTP-Binding Proteins 0.000 description 15
- 102000051367 mu Opioid Receptors Human genes 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- 108020001612 μ-opioid receptors Proteins 0.000 description 15
- PAEWLYMIIHHNKB-UHFFFAOYSA-N 1,9-diazatricyclo[6.3.1.04,12]dodeca-2,4(12),5,7,9-pentaene Chemical compound C1C=NC2=CC=CC3=C2N1C=C3 PAEWLYMIIHHNKB-UHFFFAOYSA-N 0.000 description 14
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 14
- 239000007903 gelatin capsule Substances 0.000 description 14
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Substances [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 14
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 13
- HOIIHACBCFLJET-SFTDATJTSA-N 4-((6br,10as)-3-methyl-2,3,6b,9,10,10a-hexahydro-1h-pyrido-[3',4':4,5]-pyrrolo[1,2,3-de]quinoxalin-8-(7h)-yl)-1-(4-fluorophenyl)-1-butanone Chemical compound C([C@@H]1N2CCN(C=3C=CC=C(C2=3)[C@@H]1C1)C)CN1CCCC(=O)C1=CC=C(F)C=C1 HOIIHACBCFLJET-SFTDATJTSA-N 0.000 description 12
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 230000004913 activation Effects 0.000 description 12
- 239000004031 partial agonist Substances 0.000 description 12
- 229910000027 potassium carbonate Inorganic materials 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- 241000700159 Rattus Species 0.000 description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 11
- 239000007924 injection Substances 0.000 description 11
- 238000002347 injection Methods 0.000 description 11
- 238000007920 subcutaneous administration Methods 0.000 description 11
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 238000013270 controlled release Methods 0.000 description 10
- 230000036461 convulsion Effects 0.000 description 10
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 10
- 201000000980 schizophrenia Diseases 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 9
- 230000001430 anti-depressive effect Effects 0.000 description 9
- 230000004888 barrier function Effects 0.000 description 9
- 229950003467 lumateperone Drugs 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 235000018102 proteins Nutrition 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- AAHQPLJUSLMHHR-UHFFFAOYSA-N 3-chloro-1-(4-fluorophenyl)propan-1-one Chemical compound FC1=CC=C(C(=O)CCCl)C=C1 AAHQPLJUSLMHHR-UHFFFAOYSA-N 0.000 description 8
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 8
- 208000004547 Hallucinations Diseases 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 8
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 8
- 229940005513 antidepressants Drugs 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 229920000747 poly(lactic acid) Polymers 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- 241000282414 Homo sapiens Species 0.000 description 7
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- 210000004556 brain Anatomy 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 230000015556 catabolic process Effects 0.000 description 7
- 229920001577 copolymer Polymers 0.000 description 7
- 239000013078 crystal Substances 0.000 description 7
- 239000003480 eluent Substances 0.000 description 7
- 229960003299 ketamine Drugs 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 208000024714 major depressive disease Diseases 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 235000019198 oils Nutrition 0.000 description 7
- 210000002442 prefrontal cortex Anatomy 0.000 description 7
- 230000007115 recruitment Effects 0.000 description 7
- 230000000697 serotonin reuptake Effects 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- 239000007929 subcutaneous injection Substances 0.000 description 7
- 238000013268 sustained release Methods 0.000 description 7
- 239000012730 sustained-release form Substances 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 6
- 102000040125 5-hydroxytryptamine receptor family Human genes 0.000 description 6
- 108091032151 5-hydroxytryptamine receptor family Proteins 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 208000020925 Bipolar disease Diseases 0.000 description 6
- 108010078791 Carrier Proteins Proteins 0.000 description 6
- 101150049660 DRD2 gene Proteins 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- VAYOSLLFUXYJDT-RDTXWAMCSA-N Lysergic acid diethylamide Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N(CC)CC)C2)=C3C2=CNC3=C1 VAYOSLLFUXYJDT-RDTXWAMCSA-N 0.000 description 6
- SPCIYGNTAMCTRO-UHFFFAOYSA-N Psilocine Natural products C1=CC(O)=C2C(CCN(C)C)=CNC2=C1 SPCIYGNTAMCTRO-UHFFFAOYSA-N 0.000 description 6
- 102000019208 Serotonin Plasma Membrane Transport Proteins Human genes 0.000 description 6
- 108010012996 Serotonin Plasma Membrane Transport Proteins Proteins 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 238000006731 degradation reaction Methods 0.000 description 6
- 230000003111 delayed effect Effects 0.000 description 6
- 238000010494 dissociation reaction Methods 0.000 description 6
- 230000005593 dissociations Effects 0.000 description 6
- 239000012458 free base Substances 0.000 description 6
- 239000007927 intramuscular injection Substances 0.000 description 6
- 229950002454 lysergide Drugs 0.000 description 6
- 239000002207 metabolite Substances 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- ZBWSBXGHYDWMAK-UHFFFAOYSA-N psilocin Chemical compound C1=CC=C(O)[C]2C(CCN(C)C)=CN=C21 ZBWSBXGHYDWMAK-UHFFFAOYSA-N 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 102000004076 Dopamine D1 Receptors Human genes 0.000 description 5
- 108090000511 Dopamine D1 Receptors Proteins 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- BKRGVLQUQGGVSM-KBXCAEBGSA-N Revanil Chemical compound C1=CC(C=2[C@H](N(C)C[C@H](C=2)NC(=O)N(CC)CC)C2)=C3C2=CNC3=C1 BKRGVLQUQGGVSM-KBXCAEBGSA-N 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 239000013543 active substance Substances 0.000 description 5
- 230000001800 adrenalinergic effect Effects 0.000 description 5
- 239000000164 antipsychotic agent Substances 0.000 description 5
- 239000002249 anxiolytic agent Substances 0.000 description 5
- 208000028683 bipolar I disease Diseases 0.000 description 5
- 208000025307 bipolar depression Diseases 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 229960003587 lisuride Drugs 0.000 description 5
- 230000003204 osmotic effect Effects 0.000 description 5
- 229920000728 polyester Polymers 0.000 description 5
- 150000003077 polyols Chemical group 0.000 description 5
- 239000013641 positive control Substances 0.000 description 5
- 239000003196 psychodysleptic agent Substances 0.000 description 5
- 239000002287 radioligand Substances 0.000 description 5
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 5
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 5
- 230000000862 serotonergic effect Effects 0.000 description 5
- 208000019116 sleep disease Diseases 0.000 description 5
- 230000002459 sustained effect Effects 0.000 description 5
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 description 4
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 description 4
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 4
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 description 4
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 4
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 4
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 4
- 102000015554 Dopamine receptor Human genes 0.000 description 4
- 108050004812 Dopamine receptor Proteins 0.000 description 4
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 4
- 102000004310 Ion Channels Human genes 0.000 description 4
- 108090000862 Ion Channels Proteins 0.000 description 4
- JVTAAEKCZFNVCJ-REOHCLBHSA-N L-lactic acid Chemical compound C[C@H](O)C(O)=O JVTAAEKCZFNVCJ-REOHCLBHSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 4
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 description 4
- 229920000954 Polyglycolide Polymers 0.000 description 4
- 229920001710 Polyorthoester Polymers 0.000 description 4
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 4
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 description 4
- 206010041250 Social phobia Diseases 0.000 description 4
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 4
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 4
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 4
- 230000003213 activating effect Effects 0.000 description 4
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 4
- 229960004538 alprazolam Drugs 0.000 description 4
- 230000000561 anti-psychotic effect Effects 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 4
- 230000006399 behavior Effects 0.000 description 4
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 4
- 229960002495 buspirone Drugs 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 230000000747 cardiac effect Effects 0.000 description 4
- 229960001653 citalopram Drugs 0.000 description 4
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 4
- 229960003529 diazepam Drugs 0.000 description 4
- 229960003638 dopamine Drugs 0.000 description 4
- 229960002866 duloxetine Drugs 0.000 description 4
- 229960004341 escitalopram Drugs 0.000 description 4
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 229960002464 fluoxetine Drugs 0.000 description 4
- 229960004038 fluvoxamine Drugs 0.000 description 4
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 description 4
- 208000011331 hallucinogen-persisting perception disease Diseases 0.000 description 4
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 4
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 238000007918 intramuscular administration Methods 0.000 description 4
- 238000007912 intraperitoneal administration Methods 0.000 description 4
- 239000007928 intraperitoneal injection Substances 0.000 description 4
- 238000007913 intrathecal administration Methods 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000004310 lactic acid Substances 0.000 description 4
- 235000014655 lactic acid Nutrition 0.000 description 4
- 229960004391 lorazepam Drugs 0.000 description 4
- 238000004020 luminiscence type Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 4
- 230000003551 muscarinic effect Effects 0.000 description 4
- 229960005017 olanzapine Drugs 0.000 description 4
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 229960002296 paroxetine Drugs 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000002035 prolonged effect Effects 0.000 description 4
- 229960004431 quetiapine Drugs 0.000 description 4
- URKOMYMAXPYINW-UHFFFAOYSA-N quetiapine Chemical compound C1CN(CCOCCO)CCN1C1=NC2=CC=CC=C2SC2=CC=CC=C12 URKOMYMAXPYINW-UHFFFAOYSA-N 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 229960001534 risperidone Drugs 0.000 description 4
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 description 4
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 description 4
- 229960002073 sertraline Drugs 0.000 description 4
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 4
- 230000003997 social interaction Effects 0.000 description 4
- 230000009870 specific binding Effects 0.000 description 4
- 238000010254 subcutaneous injection Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 4
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 description 4
- 229960004688 venlafaxine Drugs 0.000 description 4
- VSWBSWWIRNCQIJ-GJZGRUSLSA-N (R,R)-asenapine Chemical compound O1C2=CC=CC=C2[C@@H]2CN(C)C[C@H]2C2=CC(Cl)=CC=C21 VSWBSWWIRNCQIJ-GJZGRUSLSA-N 0.000 description 3
- UIIJKBSUGZNVQN-UHFFFAOYSA-N 1-(2-bromoethyl)-2-methoxybenzene Chemical compound COC1=CC=CC=C1CCBr UIIJKBSUGZNVQN-UHFFFAOYSA-N 0.000 description 3
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 3
- SMYALUSCZJXWHG-UHFFFAOYSA-N Altanserin Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCN2C(C3=CC=CC=C3NC2=S)=O)CC1 SMYALUSCZJXWHG-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 description 3
- DPRWLTPMPVJESF-KBPBESRZSA-N C1CCCN2[C@H]3CCNC[C@H]3C3=CC=CC1=C32 Chemical compound C1CCCN2[C@H]3CCNC[C@H]3C3=CC=CC1=C32 DPRWLTPMPVJESF-KBPBESRZSA-N 0.000 description 3
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 description 3
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 3
- 206010012289 Dementia Diseases 0.000 description 3
- 208000020401 Depressive disease Diseases 0.000 description 3
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- QGWNDRXFNXRZMB-UUOKFMHZSA-N GDP Chemical group C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O QGWNDRXFNXRZMB-UUOKFMHZSA-N 0.000 description 3
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 3
- 208000018737 Parkinson disease Diseases 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- 239000004372 Polyvinyl alcohol Substances 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 208000028552 Treatment-Resistant Depressive disease Diseases 0.000 description 3
- MMWCIQZXVOZEGG-HOZKJCLWSA-N [(1S,2R,3S,4S,5R,6S)-2,3,5-trihydroxy-4,6-diphosphonooxycyclohexyl] dihydrogen phosphate Chemical compound O[C@H]1[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H](O)[C@H]1OP(O)(O)=O MMWCIQZXVOZEGG-HOZKJCLWSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 229950009005 altanserin Drugs 0.000 description 3
- 230000000202 analgesic effect Effects 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000000949 anxiolytic effect Effects 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000012300 argon atmosphere Substances 0.000 description 3
- 229960004372 aripiprazole Drugs 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 229960005245 asenapine Drugs 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 230000003542 behavioural effect Effects 0.000 description 3
- 229940049706 benzodiazepine Drugs 0.000 description 3
- 229960001210 brexpiprazole Drugs 0.000 description 3
- ZKIAIYBUSXZPLP-UHFFFAOYSA-N brexpiprazole Chemical compound C1=C2NC(=O)C=CC2=CC=C1OCCCCN(CC1)CCN1C1=CC=CC2=C1C=CS2 ZKIAIYBUSXZPLP-UHFFFAOYSA-N 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- PCDHSSHKDZYLLI-UHFFFAOYSA-N butan-1-one Chemical compound CCC[C]=O PCDHSSHKDZYLLI-UHFFFAOYSA-N 0.000 description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 description 3
- 229960005123 cariprazine Drugs 0.000 description 3
- KPWSJANDNDDRMB-QAQDUYKDSA-N cariprazine Chemical compound C1C[C@@H](NC(=O)N(C)C)CC[C@@H]1CCN1CCN(C=2C(=C(Cl)C=CC=2)Cl)CC1 KPWSJANDNDDRMB-QAQDUYKDSA-N 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 229960004606 clomipramine Drugs 0.000 description 3
- 229960004170 clozapine Drugs 0.000 description 3
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 229910052805 deuterium Inorganic materials 0.000 description 3
- 150000001982 diacylglycerols Chemical class 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- FAGHUWIRBRMPCV-STQMWFEESA-N ethyl (6br, 10as)-3-methyl-2-oxo-2,3,6b,9,10,10a-hexahydro-1h-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline-8(7h)-carboxylate Chemical compound CN1C(=O)CN2[C@H]3CCN(C(=O)OCC)C[C@H]3C3=CC=CC1=C32 FAGHUWIRBRMPCV-STQMWFEESA-N 0.000 description 3
- 230000000848 glutamatergic effect Effects 0.000 description 3
- QGWNDRXFNXRZMB-UHFFFAOYSA-N guanidine diphosphate Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(O)=O)C(O)C1O QGWNDRXFNXRZMB-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 239000007972 injectable composition Substances 0.000 description 3
- 238000010255 intramuscular injection Methods 0.000 description 3
- 229960001432 lurasidone Drugs 0.000 description 3
- PQXKDMSYBGKCJA-CVTJIBDQSA-N lurasidone Chemical compound C1=CC=C2C(N3CCN(CC3)C[C@@H]3CCCC[C@H]3CN3C(=O)[C@@H]4[C@H]5CC[C@H](C5)[C@@H]4C3=O)=NSC2=C1 PQXKDMSYBGKCJA-CVTJIBDQSA-N 0.000 description 3
- 239000003595 mist Substances 0.000 description 3
- 229960001800 nefazodone Drugs 0.000 description 3
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 description 3
- 229940127240 opiate Drugs 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 208000019906 panic disease Diseases 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 229920005862 polyol Polymers 0.000 description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000000952 serotonin receptor agonist Substances 0.000 description 3
- 238000009097 single-agent therapy Methods 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000006190 sub-lingual tablet Substances 0.000 description 3
- 229940098466 sublingual tablet Drugs 0.000 description 3
- 229960003991 trazodone Drugs 0.000 description 3
- PHLBKPHSAVXXEF-UHFFFAOYSA-N trazodone Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1 PHLBKPHSAVXXEF-UHFFFAOYSA-N 0.000 description 3
- 230000000007 visual effect Effects 0.000 description 3
- 229960000607 ziprasidone Drugs 0.000 description 3
- MVWVFYHBGMAFLY-UHFFFAOYSA-N ziprasidone Chemical compound C1=CC=C2C(N3CCN(CC3)CCC3=CC=4CC(=O)NC=4C=C3Cl)=NSC2=C1 MVWVFYHBGMAFLY-UHFFFAOYSA-N 0.000 description 3
- IGLYMJRIWWIQQE-QUOODJBBSA-N (1S,2R)-2-phenylcyclopropan-1-amine (1R,2S)-2-phenylcyclopropan-1-amine Chemical compound N[C@H]1C[C@@H]1C1=CC=CC=C1.N[C@@H]1C[C@H]1C1=CC=CC=C1 IGLYMJRIWWIQQE-QUOODJBBSA-N 0.000 description 2
- QLGUCSLWLPCOTR-RYUDHWBXSA-N (6br,10as)-3-methyl-2,3,6b,7,8,9,10,10a-octahydro-1h-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline Chemical compound C1([C@@H]2CNCC[C@@H]22)=CC=CC3=C1N2CCN3C QLGUCSLWLPCOTR-RYUDHWBXSA-N 0.000 description 2
- QXNQHNWWRRMQPS-AAEUAGOBSA-N (6br,10as)-ethyl 2,3,6b,9,10,10a-hexahydro-2-oxo-1h-pyrido[3',4':4,5]-pyrrolo[1,2,3-de]quinoxaline-8-carboxylate Chemical compound N1C(=O)CN2[C@H]3CCN(C(=O)OCC)C[C@H]3C3=CC=CC1=C32 QXNQHNWWRRMQPS-AAEUAGOBSA-N 0.000 description 2
- YQEZLKZALYSWHR-ZDUSSCGKSA-N (S)-ketamine Chemical compound C=1C=CC=C(Cl)C=1[C@@]1(NC)CCCCC1=O YQEZLKZALYSWHR-ZDUSSCGKSA-N 0.000 description 2
- RKDVKSZUMVYZHH-UHFFFAOYSA-N 1,4-dioxane-2,5-dione Chemical compound O=C1COC(=O)CO1 RKDVKSZUMVYZHH-UHFFFAOYSA-N 0.000 description 2
- AECBVDLERUETKG-UHFFFAOYSA-N 1-(2-bromoethyl)-2-chlorobenzene Chemical compound ClC1=CC=CC=C1CCBr AECBVDLERUETKG-UHFFFAOYSA-N 0.000 description 2
- AKMIGKNTCCSJNQ-UHFFFAOYSA-N 1-(3-bromopropyl)-2-chlorobenzene Chemical compound ClC1=CC=CC=C1CCCBr AKMIGKNTCCSJNQ-UHFFFAOYSA-N 0.000 description 2
- UQWRGHGMXHIGIV-UHFFFAOYSA-N 1-(3-bromopropyl)-2-methylbenzene Chemical compound CC1=CC=CC=C1CCCBr UQWRGHGMXHIGIV-UHFFFAOYSA-N 0.000 description 2
- IQLGEKQZDZVBKY-UHFFFAOYSA-N 1-(3-bromopropyl)-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC(CCCBr)=C1 IQLGEKQZDZVBKY-UHFFFAOYSA-N 0.000 description 2
- OHERYFKQOIVITI-UHFFFAOYSA-N 1-(3-bromopropyl)-3-chlorobenzene Chemical compound ClC1=CC=CC(CCCBr)=C1 OHERYFKQOIVITI-UHFFFAOYSA-N 0.000 description 2
- LISAIUHOJQKVHK-UHFFFAOYSA-N 1-(3-bromopropyl)-3-methoxybenzene Chemical compound COC1=CC=CC(CCCBr)=C1 LISAIUHOJQKVHK-UHFFFAOYSA-N 0.000 description 2
- DFFWYMMOMUTKOI-UHFFFAOYSA-N 1-(3-chloropropoxy)-4-fluorobenzene Chemical compound FC1=CC=C(OCCCCl)C=C1 DFFWYMMOMUTKOI-UHFFFAOYSA-N 0.000 description 2
- IPJLXAZYIJLXHL-UHFFFAOYSA-N 1-(3-chloropropyl)-2-methoxybenzene Chemical compound COC1=CC=CC=C1CCCCl IPJLXAZYIJLXHL-UHFFFAOYSA-N 0.000 description 2
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 2
- HOZLOOPIXHWKCI-UHFFFAOYSA-N 2-chloro-n-methylacetamide Chemical compound CNC(=O)CCl HOZLOOPIXHWKCI-UHFFFAOYSA-N 0.000 description 2
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 2
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 description 2
- PMXMIIMHBWHSKN-UHFFFAOYSA-N 3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-9-hydroxy-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a]pyrimidin-4-one Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCC(O)C4=NC=3C)=NOC2=C1 PMXMIIMHBWHSKN-UHFFFAOYSA-N 0.000 description 2
- DMAYBPBPEUFIHJ-UHFFFAOYSA-N 4-bromobut-1-ene Chemical compound BrCCC=C DMAYBPBPEUFIHJ-UHFFFAOYSA-N 0.000 description 2
- HXAOUYGZEOZTJO-UHFFFAOYSA-N 4-chloro-1-(4-fluorophenyl)butan-1-one Chemical compound FC1=CC=C(C(=O)CCCCl)C=C1 HXAOUYGZEOZTJO-UHFFFAOYSA-N 0.000 description 2
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 2
- XKFPYPQQHFEXRZ-UHFFFAOYSA-N 5-methyl-N'-(phenylmethyl)-3-isoxazolecarbohydrazide Chemical compound O1C(C)=CC(C(=O)NNCC=2C=CC=CC=2)=N1 XKFPYPQQHFEXRZ-UHFFFAOYSA-N 0.000 description 2
- 102000003678 AMPA Receptors Human genes 0.000 description 2
- 108090000078 AMPA Receptors Proteins 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 2
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 229940126062 Compound A Drugs 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 206010012218 Delirium Diseases 0.000 description 2
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 2
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 2
- PLDUPXSUYLZYBN-UHFFFAOYSA-N Fluphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 PLDUPXSUYLZYBN-UHFFFAOYSA-N 0.000 description 2
- 102000034353 G alpha subunit Human genes 0.000 description 2
- 108091006099 G alpha subunit Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102100022630 Glutamate receptor ionotropic, NMDA 2B Human genes 0.000 description 2
- 206010019070 Hallucination, auditory Diseases 0.000 description 2
- 206010019075 Hallucination, visual Diseases 0.000 description 2
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 102000043136 MAP kinase family Human genes 0.000 description 2
- 108091054455 MAP kinase family Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 2
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 2
- KLPWJLBORRMFGK-UHFFFAOYSA-N Molindone Chemical compound O=C1C=2C(CC)=C(C)NC=2CCC1CN1CCOCC1 KLPWJLBORRMFGK-UHFFFAOYSA-N 0.000 description 2
- 206010028347 Muscle twitching Diseases 0.000 description 2
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 description 2
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 description 2
- KYYIDSXMWOZKMP-UHFFFAOYSA-N O-desmethylvenlafaxine Chemical compound C1CCCCC1(O)C(CN(C)C)C1=CC=C(O)C=C1 KYYIDSXMWOZKMP-UHFFFAOYSA-N 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- RGCVKNLCSQQDEP-UHFFFAOYSA-N Perphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 RGCVKNLCSQQDEP-UHFFFAOYSA-N 0.000 description 2
- RXBKMJIPNDOHFR-UHFFFAOYSA-N Phenelzine sulfate Chemical compound OS(O)(=O)=O.NNCCC1=CC=CC=C1 RXBKMJIPNDOHFR-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 229920002732 Polyanhydride Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ZGUGWUXLJSTTMA-UHFFFAOYSA-N Promazinum Chemical compound C1=CC=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZGUGWUXLJSTTMA-UHFFFAOYSA-N 0.000 description 2
- 102000003923 Protein Kinase C Human genes 0.000 description 2
- 108090000315 Protein Kinase C Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- KLBQZWRITKRQQV-UHFFFAOYSA-N Thioridazine Chemical compound C12=CC(SC)=CC=C2SC2=CC=CC=C2N1CCC1CCCCN1C KLBQZWRITKRQQV-UHFFFAOYSA-N 0.000 description 2
- GFBKORZTTCHDGY-UWVJOHFNSA-N Thiothixene Chemical compound C12=CC(S(=O)(=O)N(C)C)=CC=C2SC2=CC=CC=C2\C1=C\CCN1CCN(C)CC1 GFBKORZTTCHDGY-UWVJOHFNSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 102000030621 adenylate cyclase Human genes 0.000 description 2
- 108060000200 adenylate cyclase Proteins 0.000 description 2
- 230000001270 agonistic effect Effects 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229960000836 amitriptyline Drugs 0.000 description 2
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical group C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 2
- 229960002519 amoxapine Drugs 0.000 description 2
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 description 2
- 239000003098 androgen Substances 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- 239000003420 antiserotonin agent Substances 0.000 description 2
- 230000007529 anxiety like behavior Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229960002430 atomoxetine Drugs 0.000 description 2
- VHGCDTVCOLNTBX-QGZVFWFLSA-N atomoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=CC=C1C VHGCDTVCOLNTBX-QGZVFWFLSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 238000006065 biodegradation reaction Methods 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 2
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 2
- 229960001058 bupropion Drugs 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229930003827 cannabinoid Natural products 0.000 description 2
- 239000003557 cannabinoid Substances 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 239000005018 casein Substances 0.000 description 2
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 2
- 235000021240 caseins Nutrition 0.000 description 2
- 229960001076 chlorpromazine Drugs 0.000 description 2
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 2
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 150000004292 cyclic ethers Chemical class 0.000 description 2
- 229960003914 desipramine Drugs 0.000 description 2
- 229960001623 desvenlafaxine Drugs 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 2
- 229960005426 doxepin Drugs 0.000 description 2
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 2
- RMEDXOLNCUSCGS-UHFFFAOYSA-N droperidol Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC=C(N2C(NC3=CC=CC=C32)=O)CC1 RMEDXOLNCUSCGS-UHFFFAOYSA-N 0.000 description 2
- 229960000394 droperidol Drugs 0.000 description 2
- 125000006575 electron-withdrawing group Chemical group 0.000 description 2
- 229960000450 esketamine Drugs 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- YKRFDXKYULKKPS-JQWIXIFHSA-N ethyl (4as,9br)-6-bromo-1,3,4,4a,5,9b-hexahydropyrido[4,3-b]indole-2-carboxylate Chemical compound N([C@H]1CCN(C[C@H]11)C(=O)OCC)C2=C1C=CC=C2Br YKRFDXKYULKKPS-JQWIXIFHSA-N 0.000 description 2
- WHCUAJCNKSIETM-KBPBESRZSA-N ethyl (6br, 10as)-3-methyl-2,3,6b,9,10,10a-hexahydro-1h-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline-8(7h)-carboxylate Chemical compound CN1CCN2[C@H]3CCN(C(=O)OCC)C[C@H]3C3=CC=CC1=C32 WHCUAJCNKSIETM-KBPBESRZSA-N 0.000 description 2
- 239000010408 film Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229960002690 fluphenazine Drugs 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 238000002825 functional assay Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000000380 hallucinogen Substances 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 229960003878 haloperidol Drugs 0.000 description 2
- 208000018578 heart valve disease Diseases 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229960001340 histamine Drugs 0.000 description 2
- 229920002674 hyaluronan Polymers 0.000 description 2
- 229960003160 hyaluronic acid Drugs 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 229960004801 imipramine Drugs 0.000 description 2
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000000415 inactivating effect Effects 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 229960002672 isocarboxazid Drugs 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- LHAPOGAFBLSJJQ-GUTACTQSSA-N iti007 Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1.C([C@@H]1N2CCN(C=3C=CC=C(C2=3)[C@@H]1C1)C)CN1CCCC(=O)C1=CC=C(F)C=C1 LHAPOGAFBLSJJQ-GUTACTQSSA-N 0.000 description 2
- JJTUDXZGHPGLLC-UHFFFAOYSA-N lactide Chemical compound CC1OC(=O)C(C)OC1=O JJTUDXZGHPGLLC-UHFFFAOYSA-N 0.000 description 2
- 229960000685 levomilnacipran Drugs 0.000 description 2
- 230000006742 locomotor activity Effects 0.000 description 2
- 229960000423 loxapine Drugs 0.000 description 2
- XJGVXQDUIWGIRW-UHFFFAOYSA-N loxapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2OC2=CC=C(Cl)C=C12 XJGVXQDUIWGIRW-UHFFFAOYSA-N 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- FRIJBUGBVQZNTB-UHFFFAOYSA-M magnesium;ethane;bromide Chemical compound [Mg+2].[Br-].[CH2-]C FRIJBUGBVQZNTB-UHFFFAOYSA-M 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 229960004090 maprotiline Drugs 0.000 description 2
- QSLMDECMDJKHMQ-GSXCWMCISA-N maprotiline Chemical compound C12=CC=CC=C2[C@@]2(CCCNC)C3=CC=CC=C3[C@@H]1CC2 QSLMDECMDJKHMQ-GSXCWMCISA-N 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229960000300 mesoridazine Drugs 0.000 description 2
- SLVMESMUVMCQIY-UHFFFAOYSA-N mesoridazine Chemical compound CN1CCCCC1CCN1C2=CC(S(C)=O)=CC=C2SC2=CC=CC=C21 SLVMESMUVMCQIY-UHFFFAOYSA-N 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 229960000600 milnacipran Drugs 0.000 description 2
- 229960001785 mirtazapine Drugs 0.000 description 2
- RONZAEMNMFQXRA-UHFFFAOYSA-N mirtazapine Chemical compound C1C2=CC=CN=C2N2CCN(C)CC2C2=CC=CC=C21 RONZAEMNMFQXRA-UHFFFAOYSA-N 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 229960004938 molindone Drugs 0.000 description 2
- 230000036651 mood Effects 0.000 description 2
- 229960005181 morphine Drugs 0.000 description 2
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000002105 nanoparticle Substances 0.000 description 2
- 229920005615 natural polymer Polymers 0.000 description 2
- 229930014626 natural product Natural products 0.000 description 2
- 229960001158 nortriptyline Drugs 0.000 description 2
- 238000012346 open field test Methods 0.000 description 2
- 229940096978 oral tablet Drugs 0.000 description 2
- 239000007935 oral tablet Substances 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000002357 osmotic agent Substances 0.000 description 2
- 229960001057 paliperidone Drugs 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 229960000762 perphenazine Drugs 0.000 description 2
- 229960004790 phenelzine sulfate Drugs 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 229960003634 pimozide Drugs 0.000 description 2
- YVUQSNJEYSNKRX-UHFFFAOYSA-N pimozide Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)CCCN1CCC(N2C(NC3=CC=CC=C32)=O)CC1 YVUQSNJEYSNKRX-UHFFFAOYSA-N 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 229920001432 poly(L-lactide) Polymers 0.000 description 2
- 239000002745 poly(ortho ester) Substances 0.000 description 2
- 229920002463 poly(p-dioxanone) polymer Polymers 0.000 description 2
- 229920001281 polyalkylene Polymers 0.000 description 2
- 229920001610 polycaprolactone Polymers 0.000 description 2
- 239000004632 polycaprolactone Substances 0.000 description 2
- 229920000515 polycarbonate Polymers 0.000 description 2
- 239000004417 polycarbonate Substances 0.000 description 2
- 239000000622 polydioxanone Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000004626 polylactic acid Substances 0.000 description 2
- 208000028173 post-traumatic stress disease Diseases 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- 229960003111 prochlorperazine Drugs 0.000 description 2
- WIKYUJGCLQQFNW-UHFFFAOYSA-N prochlorperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 WIKYUJGCLQQFNW-UHFFFAOYSA-N 0.000 description 2
- 229960003598 promazine Drugs 0.000 description 2
- UFUASNAHBMBJIX-UHFFFAOYSA-N propan-1-one Chemical compound CC[C]=O UFUASNAHBMBJIX-UHFFFAOYSA-N 0.000 description 2
- KRIOVPPHQSLHCZ-UHFFFAOYSA-N propiophenone Chemical compound CCC(=O)C1=CC=CC=C1 KRIOVPPHQSLHCZ-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229960002601 protriptyline Drugs 0.000 description 2
- BWPIARFWQZKAIA-UHFFFAOYSA-N protriptyline Chemical compound C1=CC2=CC=CC=C2C(CCCNC)C2=CC=CC=C21 BWPIARFWQZKAIA-UHFFFAOYSA-N 0.000 description 2
- 208000020016 psychiatric disease Diseases 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 238000001525 receptor binding assay Methods 0.000 description 2
- 238000004064 recycling Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000013643 reference control Substances 0.000 description 2
- 230000029058 respiratory gaseous exchange Effects 0.000 description 2
- 239000002399 serotonin 2A agonist Substances 0.000 description 2
- 239000002400 serotonin 2A antagonist Substances 0.000 description 2
- 239000003244 serotonin 2B agonist Substances 0.000 description 2
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 2
- 229960004425 sibutramine Drugs 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 238000000935 solvent evaporation Methods 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 238000012453 sprague-dawley rat model Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 229960002784 thioridazine Drugs 0.000 description 2
- 229960005013 tiotixene Drugs 0.000 description 2
- 229960003741 tranylcypromine Drugs 0.000 description 2
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 2
- ZEWQUBUPAILYHI-UHFFFAOYSA-N trifluoperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 ZEWQUBUPAILYHI-UHFFFAOYSA-N 0.000 description 2
- 229960002324 trifluoperazine Drugs 0.000 description 2
- 229960002431 trimipramine Drugs 0.000 description 2
- ZSCDBOWYZJWBIY-UHFFFAOYSA-N trimipramine Chemical compound C1CC2=CC=CC=C2N(CC(CN(C)C)C)C2=CC=CC=C21 ZSCDBOWYZJWBIY-UHFFFAOYSA-N 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- PGVSRLLNTZQDLH-QWRGUYRKSA-N (10R,15S)-4-thia-1,12-diazatetracyclo[7.6.1.05,16.010,15]hexadeca-5,7,9(16)-triene Chemical compound S1CCN2[C@H]3CCNC[C@H]3C3=CC=CC1=C32 PGVSRLLNTZQDLH-QWRGUYRKSA-N 0.000 description 1
- WYDUSKDSKCASEF-LJQANCHMSA-N (1s)-1-cyclohexyl-1-phenyl-3-pyrrolidin-1-ylpropan-1-ol Chemical compound C([C@](O)(C1CCCCC1)C=1C=CC=CC=1)CN1CCCC1 WYDUSKDSKCASEF-LJQANCHMSA-N 0.000 description 1
- KHEUWLQKCXGVEL-LLVKDONJSA-N (2R)-1-(5-methoxyindol-1-yl)-N,N-dimethylpropan-2-amine Chemical compound COC=1C=C2C=CN(C2=CC=1)C[C@@H](C)N(C)C KHEUWLQKCXGVEL-LLVKDONJSA-N 0.000 description 1
- IVTMXOXVAHXCHI-YXLMWLKOSA-N (2s)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid;(2s)-3-(3,4-dihydroxyphenyl)-2-hydrazinyl-2-methylpropanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1.NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 IVTMXOXVAHXCHI-YXLMWLKOSA-N 0.000 description 1
- GEZMEIHVFSWOCA-UHFFFAOYSA-N (4-fluorophenyl)methanol Chemical compound OCC1=CC=C(F)C=C1 GEZMEIHVFSWOCA-UHFFFAOYSA-N 0.000 description 1
- CNCZLLXGSLADGW-STQMWFEESA-N (7as,11ar)-5,6,7a, 8,9,10,11,11a-octahydro-4h-pyrido[3',4':4,5]pyrrolo[3,2,1-ij]quinoline Chemical compound C1([C@@H]2CNCC[C@@H]22)=CC=CC3=C1N2CCC3 CNCZLLXGSLADGW-STQMWFEESA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 229920003178 (lactide-co-glycolide) polymer Polymers 0.000 description 1
- YNGDWRXWKFWCJY-UHFFFAOYSA-N 1,4-Dihydropyridine Chemical compound C1C=CNC=C1 YNGDWRXWKFWCJY-UHFFFAOYSA-N 0.000 description 1
- PBRPKYRJVDJZTF-UHFFFAOYSA-N 1-(2-bromoethoxy)-2-methoxybenzene Chemical compound COC1=CC=CC=C1OCCBr PBRPKYRJVDJZTF-UHFFFAOYSA-N 0.000 description 1
- SHZCYCKEZBSEAV-UHFFFAOYSA-N 1-(2-bromoethyl)-2-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=CC=C1CCBr SHZCYCKEZBSEAV-UHFFFAOYSA-N 0.000 description 1
- AMQPULMQCZXNPM-UHFFFAOYSA-N 1-(2-bromoethyl)-2-ethylbenzene Chemical compound CCC1=CC=CC=C1CCBr AMQPULMQCZXNPM-UHFFFAOYSA-N 0.000 description 1
- XEBOYHYBVZJAGJ-UHFFFAOYSA-N 1-(2-bromoethyl)-3-ethylbenzene Chemical compound CCC1=CC=CC(CCBr)=C1 XEBOYHYBVZJAGJ-UHFFFAOYSA-N 0.000 description 1
- QONMBCKMZUHDQJ-UHFFFAOYSA-N 1-(2-bromoethyl)-4-fluoro-2-methoxybenzene Chemical compound COC1=CC(F)=CC=C1CCBr QONMBCKMZUHDQJ-UHFFFAOYSA-N 0.000 description 1
- OXHPTABOQVHKLN-UHFFFAOYSA-N 1-(2-bromoethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCBr)C=C1 OXHPTABOQVHKLN-UHFFFAOYSA-N 0.000 description 1
- KPDMESZOWPPZMA-UHFFFAOYSA-N 1-(2-bromoethyl)benzimidazole Chemical compound C1=CC=C2N(CCBr)C=NC2=C1 KPDMESZOWPPZMA-UHFFFAOYSA-N 0.000 description 1
- PTYVNIAAGXTKBQ-UHFFFAOYSA-N 1-(2-bromoethyl)benzotriazole Chemical compound C1=CC=C2N(CCBr)N=NC2=C1 PTYVNIAAGXTKBQ-UHFFFAOYSA-N 0.000 description 1
- JGBYWOLITOFHFC-UHFFFAOYSA-N 1-(2-bromoethyl)indazole Chemical compound C1=CC=C2N(CCBr)N=CC2=C1 JGBYWOLITOFHFC-UHFFFAOYSA-N 0.000 description 1
- NATIFIFIDLQXIT-UHFFFAOYSA-N 1-(2-bromoethyl)pyridin-2-one Chemical compound BrCCN1C=CC=CC1=O NATIFIFIDLQXIT-UHFFFAOYSA-N 0.000 description 1
- VULUPRKDRIRLLH-UHFFFAOYSA-N 1-(2-chloroethoxy)-2-methylbenzene Chemical compound CC1=CC=CC=C1OCCCl VULUPRKDRIRLLH-UHFFFAOYSA-N 0.000 description 1
- OODHDKJVMWONSL-UHFFFAOYSA-N 1-(2-chloroethyl)-2-ethoxybenzene Chemical compound CCOC1=CC=CC=C1CCCl OODHDKJVMWONSL-UHFFFAOYSA-N 0.000 description 1
- VBFWRQKEAXBSMU-PMACEKPBSA-N 1-(4-fluorophenyl)-3-[(10R,15S)-4-methyl-1,4,12-triazatetracyclo[7.6.1.05,16.010,15]hexadeca-5,7,9(16)-trien-12-yl]propan-1-one Chemical compound [H][C@]12CCN(CCC(=O)C3=CC=C(F)C=C3)C[C@@]1([H])C1=CC=CC3=C1N2CCN3C VBFWRQKEAXBSMU-PMACEKPBSA-N 0.000 description 1
- HIINSSPLKGECTJ-PMACEKPBSA-N 1-(4-fluorophenyl)-3-[(11S,16R)-6-thia-10,14-diazatetracyclo[8.6.1.05,17.011,16]heptadeca-1(17),2,4-trien-14-yl]propan-1-one Chemical compound C1=CC(F)=CC=C1C(=O)CCN1C[C@@H](C=2C=3N4CCCSC=3C=CC=2)[C@@H]4CC1 HIINSSPLKGECTJ-PMACEKPBSA-N 0.000 description 1
- MLOYGHJCLXLZTL-UHFFFAOYSA-N 1-(bromomethyl)-2-ethylbenzene Chemical compound CCC1=CC=CC=C1CBr MLOYGHJCLXLZTL-UHFFFAOYSA-N 0.000 description 1
- WAUNFWSVXRPCDQ-UHFFFAOYSA-N 1-(bromomethyl)-2-methoxybenzene Chemical compound COC1=CC=CC=C1CBr WAUNFWSVXRPCDQ-UHFFFAOYSA-N 0.000 description 1
- HNUZAFPGSNEQDM-UHFFFAOYSA-N 1-(bromomethyl)-3-ethylbenzene Chemical compound CCC1=CC=CC(CBr)=C1 HNUZAFPGSNEQDM-UHFFFAOYSA-N 0.000 description 1
- ZKSOJQDNSNJIQW-UHFFFAOYSA-N 1-(bromomethyl)-3-methoxybenzene Chemical compound COC1=CC=CC(CBr)=C1 ZKSOJQDNSNJIQW-UHFFFAOYSA-N 0.000 description 1
- YYPPFEKHXPADAV-UHFFFAOYSA-N 1-(bromomethyl)-4-ethylbenzene Chemical compound CCC1=CC=C(CBr)C=C1 YYPPFEKHXPADAV-UHFFFAOYSA-N 0.000 description 1
- GIGRWGTZFONRKA-UHFFFAOYSA-N 1-(bromomethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CBr)C=C1 GIGRWGTZFONRKA-UHFFFAOYSA-N 0.000 description 1
- YPWUBYFSOOJNTB-UHFFFAOYSA-N 1-[2-(2-chloroethoxy)ethoxy]-4-fluorobenzene Chemical compound FC1=CC=C(OCCOCCCl)C=C1 YPWUBYFSOOJNTB-UHFFFAOYSA-N 0.000 description 1
- BXFBZVYWFSEXCS-UHFFFAOYSA-N 1-ethyl-3-(3-methylbutylamino)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile Chemical compound C1CCCC2=C1C(C#N)=C(NCCC(C)C)N=C2CC BXFBZVYWFSEXCS-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 1
- MGTUVUVRFJVHAL-UHFFFAOYSA-N 2,8-dibenzyl-6-(4-hydroxyphenyl)imidazo[1,2-a]pyrazin-3-ol Chemical compound Oc1c(Cc2ccccc2)nc2c(Cc3ccccc3)nc(cn12)-c1ccc(O)cc1 MGTUVUVRFJVHAL-UHFFFAOYSA-N 0.000 description 1
- OMALKMWOCZAJEA-UHFFFAOYSA-N 2-(2-bromoethyl)-1,4-dimethoxybenzene Chemical compound COC1=CC=C(OC)C(CCBr)=C1 OMALKMWOCZAJEA-UHFFFAOYSA-N 0.000 description 1
- IYZRKPDDKSEWCW-UHFFFAOYSA-N 2-(2-bromoethyl)benzonitrile Chemical compound BrCCC1=CC=CC=C1C#N IYZRKPDDKSEWCW-UHFFFAOYSA-N 0.000 description 1
- GZPRFUSTANBATG-UHFFFAOYSA-N 2-(2-bromoethyl)phenol Chemical compound OC1=CC=CC=C1CCBr GZPRFUSTANBATG-UHFFFAOYSA-N 0.000 description 1
- DRRNLZXCGPJUMC-UHFFFAOYSA-N 2-(4-chlorophenyl)-n,n-bis(2-methylpropyl)quinazolin-4-amine Chemical compound N=1C2=CC=CC=C2C(N(CC(C)C)CC(C)C)=NC=1C1=CC=C(Cl)C=C1 DRRNLZXCGPJUMC-UHFFFAOYSA-N 0.000 description 1
- YFGHCGITMMYXAQ-UHFFFAOYSA-N 2-[(diphenylmethyl)sulfinyl]acetamide Chemical compound C=1C=CC=CC=1C(S(=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 1
- IJVRPNIWWODHHA-UHFFFAOYSA-N 2-cyanoprop-2-enoic acid Chemical class OC(=O)C(=C)C#N IJVRPNIWWODHHA-UHFFFAOYSA-N 0.000 description 1
- XBBVURRQGJPTHH-UHFFFAOYSA-N 2-hydroxyacetic acid;2-hydroxypropanoic acid Chemical compound OCC(O)=O.CC(O)C(O)=O XBBVURRQGJPTHH-UHFFFAOYSA-N 0.000 description 1
- GHCZTIFQWKKGSB-UHFFFAOYSA-N 2-hydroxypropane-1,2,3-tricarboxylic acid;phosphoric acid Chemical compound OP(O)(O)=O.OC(=O)CC(O)(C(O)=O)CC(O)=O GHCZTIFQWKKGSB-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- NTLAICDKHHQUGC-UHFFFAOYSA-N 3-(2-bromoethyl)-1h-indole Chemical compound C1=CC=C2C(CCBr)=CNC2=C1 NTLAICDKHHQUGC-UHFFFAOYSA-N 0.000 description 1
- ZLWFSTBZPKLELA-UHFFFAOYSA-N 3-(2-bromoethyl)-6-fluoro-1h-indole Chemical compound FC1=CC=C2C(CCBr)=CNC2=C1 ZLWFSTBZPKLELA-UHFFFAOYSA-N 0.000 description 1
- ZGHDXEGECZAFAT-UHFFFAOYSA-N 3-(2-chloroethyl)-2H-indazole Chemical compound N1N=C(C2=CC=CC=C12)CCCl ZGHDXEGECZAFAT-UHFFFAOYSA-N 0.000 description 1
- TVHBMGLNQYHPBK-UHFFFAOYSA-N 3-chloro-1-(2-methylphenyl)propan-1-one Chemical compound CC1=CC=CC=C1C(=O)CCCl TVHBMGLNQYHPBK-UHFFFAOYSA-N 0.000 description 1
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 1
- NNNRGWOWXNCGCV-UHFFFAOYSA-N 4-(2-bromoethyl)benzonitrile Chemical compound BrCCC1=CC=C(C#N)C=C1 NNNRGWOWXNCGCV-UHFFFAOYSA-N 0.000 description 1
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 1
- 125000004801 4-cyanophenyl group Chemical group [H]C1=C([H])C(C#N)=C([H])C([H])=C1* 0.000 description 1
- 125000004860 4-ethylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- UGAKAFQJJSSKJM-UHFFFAOYSA-N 5-chloro-1-(4-fluorophenyl)pentan-1-one Chemical compound FC1=CC=C(C(=O)CCCCCl)C=C1 UGAKAFQJJSSKJM-UHFFFAOYSA-N 0.000 description 1
- ZSTKHSQDNIGFLM-UHFFFAOYSA-N 5-methoxy-N,N-dimethyltryptamine Chemical compound COC1=CC=C2NC=C(CCN(C)C)C2=C1 ZSTKHSQDNIGFLM-UHFFFAOYSA-N 0.000 description 1
- RDMFHRSPDKWERA-UHFFFAOYSA-N 5H-Pyrido[4,3-b]indole Chemical compound C1=NC=C2C3=CC=CC=C3NC2=C1 RDMFHRSPDKWERA-UHFFFAOYSA-N 0.000 description 1
- NMKHCGOYOKIXRW-UHFFFAOYSA-N 6-chloro-1-(4-fluorophenyl)hexan-1-one Chemical compound FC1=CC=C(C(=O)CCCCCCl)C=C1 NMKHCGOYOKIXRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- 108060003345 Adrenergic Receptor Proteins 0.000 description 1
- 102000017910 Adrenergic receptor Human genes 0.000 description 1
- 102100024349 Alpha-1A adrenergic receptor Human genes 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 102000003916 Arrestin Human genes 0.000 description 1
- 108090000328 Arrestin Proteins 0.000 description 1
- 206010003805 Autism Diseases 0.000 description 1
- 208000020706 Autistic disease Diseases 0.000 description 1
- 102100023006 Basic leucine zipper transcriptional factor ATF-like 2 Human genes 0.000 description 1
- 102100021942 C-C motif chemokine 28 Human genes 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- 102100025841 Cholecystokinin Human genes 0.000 description 1
- 101800001982 Cholecystokinin Proteins 0.000 description 1
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 1
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 description 1
- 208000026331 Disruptive, Impulse Control, and Conduct disease Diseases 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 102000006441 Dopamine Plasma Membrane Transport Proteins Human genes 0.000 description 1
- 108010044266 Dopamine Plasma Membrane Transport Proteins Proteins 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 102000027484 GABAA receptors Human genes 0.000 description 1
- 108091008681 GABAA receptors Proteins 0.000 description 1
- 230000009508 GABAergic inhibition Effects 0.000 description 1
- 102000018899 Glutamate Receptors Human genes 0.000 description 1
- 108010027915 Glutamate Receptors Proteins 0.000 description 1
- 101710195187 Glutamate receptor ionotropic, NMDA 2B Proteins 0.000 description 1
- 108091006068 Gq proteins Proteins 0.000 description 1
- 102000052606 Gq-G11 GTP-Binding Protein alpha Subunits Human genes 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 1
- 102000000543 Histamine Receptors Human genes 0.000 description 1
- 108010002059 Histamine Receptors Proteins 0.000 description 1
- 101000903615 Homo sapiens Basic leucine zipper transcriptional factor ATF-like 2 Proteins 0.000 description 1
- 101000897477 Homo sapiens C-C motif chemokine 28 Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000030990 Impulse-control disease Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 102000004086 Ligand-Gated Ion Channels Human genes 0.000 description 1
- 108090000543 Ligand-Gated Ion Channels Proteins 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 102000010909 Monoamine Oxidase Human genes 0.000 description 1
- 108010062431 Monoamine oxidase Proteins 0.000 description 1
- 108010085220 Multiprotein Complexes Proteins 0.000 description 1
- 102000007474 Multiprotein Complexes Human genes 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- 229940099433 NMDA receptor antagonist Drugs 0.000 description 1
- 208000009668 Neurobehavioral Manifestations Diseases 0.000 description 1
- 102000008092 Norepinephrine Plasma Membrane Transport Proteins Human genes 0.000 description 1
- 108010049586 Norepinephrine Plasma Membrane Transport Proteins Proteins 0.000 description 1
- 240000001090 Papaver somniferum Species 0.000 description 1
- 108010089430 Phosphoproteins Proteins 0.000 description 1
- 102000007982 Phosphoproteins Human genes 0.000 description 1
- 229920005372 Plexiglas® Polymers 0.000 description 1
- 229920002730 Poly(butyl cyanoacrylate) Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 108091008611 Protein Kinase B Proteins 0.000 description 1
- QVDSEJDULKLHCG-UHFFFAOYSA-N Psilocybine Natural products C1=CC(OP(O)(O)=O)=C2C(CCN(C)C)=CNC2=C1 QVDSEJDULKLHCG-UHFFFAOYSA-N 0.000 description 1
- 108091005682 Receptor kinases Proteins 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 206010038678 Respiratory depression Diseases 0.000 description 1
- 108010038912 Retinoid X Receptors Proteins 0.000 description 1
- 101150001535 SRC gene Proteins 0.000 description 1
- 108010052164 Sodium Channels Proteins 0.000 description 1
- 102000018674 Sodium Channels Human genes 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 102000014384 Type C Phospholipases Human genes 0.000 description 1
- 108010079194 Type C Phospholipases Proteins 0.000 description 1
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 description 1
- 102000002852 Vasopressins Human genes 0.000 description 1
- 108010004977 Vasopressins Proteins 0.000 description 1
- VOXIUXZAOFEFBL-UHFFFAOYSA-N Voacangin Natural products CCC1CC2CN3CC1C(C2)(OC(=O)C)c4[nH]c5ccc(OC)cc5c4C3 VOXIUXZAOFEFBL-UHFFFAOYSA-N 0.000 description 1
- 102000003734 Voltage-Gated Potassium Channels Human genes 0.000 description 1
- 108090000013 Voltage-Gated Potassium Channels Proteins 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- CGNMLOKEMNBUAI-UHFFFAOYSA-N adrafinil Chemical compound C=1C=CC=CC=1C(S(=O)CC(=O)NO)C1=CC=CC=C1 CGNMLOKEMNBUAI-UHFFFAOYSA-N 0.000 description 1
- 229960002820 adrafinil Drugs 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 230000002009 allergenic effect Effects 0.000 description 1
- 108020004102 alpha-1 Adrenergic Receptor Proteins 0.000 description 1
- WOLHOYHSEKDWQH-UHFFFAOYSA-N amantadine hydrochloride Chemical compound [Cl-].C1C(C2)CC3CC2CC1([NH3+])C3 WOLHOYHSEKDWQH-UHFFFAOYSA-N 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940125688 antiparkinson agent Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- YFGHCGITMMYXAQ-LJQANCHMSA-N armodafinil Chemical compound C=1C=CC=CC=1C([S@](=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-LJQANCHMSA-N 0.000 description 1
- 229960004823 armodafinil Drugs 0.000 description 1
- 239000003693 atypical antipsychotic agent Substances 0.000 description 1
- 229960001081 benzatropine Drugs 0.000 description 1
- GIJXKZJWITVLHI-PMOLBWCYSA-N benzatropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(C=1C=CC=CC=1)C1=CC=CC=C1 GIJXKZJWITVLHI-PMOLBWCYSA-N 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- YSXKPIUOCJLQIE-UHFFFAOYSA-N biperiden Chemical compound C1C(C=C2)CC2C1C(C=1C=CC=CC=1)(O)CCN1CCCCC1 YSXKPIUOCJLQIE-UHFFFAOYSA-N 0.000 description 1
- 229960003003 biperiden Drugs 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 description 1
- 229960002802 bromocriptine Drugs 0.000 description 1
- 229940046011 buccal tablet Drugs 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- BFTSQLRGEOKCJS-RYUDHWBXSA-N chembl2337103 Chemical compound S1CCCN2[C@H]3CCNC[C@H]3C3=CC=CC1=C32 BFTSQLRGEOKCJS-RYUDHWBXSA-N 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 229960004782 chlordiazepoxide Drugs 0.000 description 1
- ANTSCNMPPGJYLG-UHFFFAOYSA-N chlordiazepoxide Chemical compound O=N=1CC(NC)=NC2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 ANTSCNMPPGJYLG-UHFFFAOYSA-N 0.000 description 1
- 229940107137 cholecystokinin Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000009194 climbing Effects 0.000 description 1
- 229940121657 clinical drug Drugs 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003001 depressive effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960002069 diamorphine Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 description 1
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 description 1
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 230000009429 distress Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003291 dopaminomimetic effect Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- JRURYQJSLYLRLN-BJMVGYQFSA-N entacapone Chemical compound CCN(CC)C(=O)C(\C#N)=C\C1=CC(O)=C(O)C([N+]([O-])=O)=C1 JRURYQJSLYLRLN-BJMVGYQFSA-N 0.000 description 1
- 229960003337 entacapone Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 238000012048 forced swim test Methods 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 230000005176 gastrointestinal motility Effects 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 229920000578 graft copolymer Polymers 0.000 description 1
- 230000003370 grooming effect Effects 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108091006093 heterotrimeric G proteins Proteins 0.000 description 1
- 102000034345 heterotrimeric G proteins Human genes 0.000 description 1
- MOMDHKVDJYHNRF-UHFFFAOYSA-N hexan-1-one Chemical compound CCCCC[C]=O MOMDHKVDJYHNRF-UHFFFAOYSA-N 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000003326 hypnotic agent Substances 0.000 description 1
- 239000005554 hypnotics and sedatives Substances 0.000 description 1
- HSIBGVUMFOSJPD-CFDPKNGZSA-N ibogaine Chemical compound N1([C@@H]2[C@H]3C[C@H](C1)C[C@@H]2CC)CCC1=C3NC2=CC=C(OC)C=C12 HSIBGVUMFOSJPD-CFDPKNGZSA-N 0.000 description 1
- OLOCMRXSJQJJPL-UHFFFAOYSA-N ibogaine Natural products CCC1CC2CC3C1N(C2)C=Cc4c3[nH]c5ccc(OC)cc45 OLOCMRXSJQJJPL-UHFFFAOYSA-N 0.000 description 1
- AREITJMUSRHSBK-UHFFFAOYSA-N ibogamine Natural products CCC1CC2C3CC1CN2CCc4c3[nH]c5ccccc45 AREITJMUSRHSBK-UHFFFAOYSA-N 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 208000015046 intermittent explosive disease Diseases 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- CXHHAWACKSPTFF-UHFFFAOYSA-N iodomethylcyclohexane Chemical compound ICC1CCCCC1 CXHHAWACKSPTFF-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 229950007462 lorpiprazole Drugs 0.000 description 1
- BNRMWKUVWLKDQJ-YJBOKZPZSA-N lorpiprazole Chemical compound FC(F)(F)C1=CC=CC(N2CCN(CCC=3N4C[C@@H]5CCC[C@@H]5C4=NN=3)CC2)=C1 BNRMWKUVWLKDQJ-YJBOKZPZSA-N 0.000 description 1
- 230000031852 maintenance of location in cell Effects 0.000 description 1
- DOTIMEKVTCOGED-UHFFFAOYSA-N mepiprazole Chemical compound N1C(C)=CC(CCN2CCN(CC2)C=2C=C(Cl)C=CC=2)=N1 DOTIMEKVTCOGED-UHFFFAOYSA-N 0.000 description 1
- 229950004808 mepiprazole Drugs 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- FJQXCDYVZAHXNS-UHFFFAOYSA-N methadone hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 FJQXCDYVZAHXNS-UHFFFAOYSA-N 0.000 description 1
- VUZPPFZMUPKLLV-UHFFFAOYSA-N methane;hydrate Chemical compound C.O VUZPPFZMUPKLLV-UHFFFAOYSA-N 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960001165 modafinil Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000002969 morbid Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 description 1
- DAZXVJBJRMWXJP-UHFFFAOYSA-N n,n-dimethylethylamine Chemical compound CCN(C)C DAZXVJBJRMWXJP-UHFFFAOYSA-N 0.000 description 1
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 1
- UZHSEJADLWPNLE-GRGSLBFTSA-N naloxone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4CC=C UZHSEJADLWPNLE-GRGSLBFTSA-N 0.000 description 1
- 229960004127 naloxone Drugs 0.000 description 1
- 239000003887 narcotic antagonist Substances 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003401 opiate antagonist Substances 0.000 description 1
- 229940124636 opioid drug Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000002746 orthostatic effect Effects 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 235000006502 papoula Nutrition 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N pentanal Chemical compound CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 229960004851 pergolide Drugs 0.000 description 1
- YEHCICAEULNIGD-MZMPZRCHSA-N pergolide Chemical compound C1=CC([C@H]2C[C@@H](CSC)CN([C@@H]2C2)CCC)=C3C2=CNC3=C1 YEHCICAEULNIGD-MZMPZRCHSA-N 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000003905 phosphatidylinositols Chemical class 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- FASDKYOPVNHBLU-ZETCQYMHSA-N pramipexole Chemical compound C1[C@@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-ZETCQYMHSA-N 0.000 description 1
- 229960003089 pramipexole Drugs 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 230000003518 presynaptic effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229960005253 procyclidine Drugs 0.000 description 1
- 210000001176 projection neuron Anatomy 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- QKTAAWLCLHMUTJ-UHFFFAOYSA-N psilocybin Chemical compound C1C=CC(OP(O)(O)=O)=C2C(CCN(C)C)=CN=C21 QKTAAWLCLHMUTJ-UHFFFAOYSA-N 0.000 description 1
- 230000001107 psychogenic effect Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 230000001698 pyrogenic effect Effects 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 238000003653 radioligand binding assay Methods 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000011369 resultant mixture Substances 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000007151 ring opening polymerisation reaction Methods 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- UHSKFQJFRQCDBE-UHFFFAOYSA-N ropinirole Chemical compound CCCN(CCC)CCC1=CC=CC2=C1CC(=O)N2 UHSKFQJFRQCDBE-UHFFFAOYSA-N 0.000 description 1
- 229960001879 ropinirole Drugs 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 208000022610 schizoaffective disease Diseases 0.000 description 1
- 230000009291 secondary effect Effects 0.000 description 1
- MEZLKOACVSPNER-GFCCVEGCSA-N selegiline Chemical compound C#CCN(C)[C@H](C)CC1=CC=CC=C1 MEZLKOACVSPNER-GFCCVEGCSA-N 0.000 description 1
- 229960003946 selegiline Drugs 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000002891 serotonin 2B antagonist Substances 0.000 description 1
- 239000002485 serotonin 2C agonist Substances 0.000 description 1
- 239000002484 serotonin 2C antagonist Substances 0.000 description 1
- 239000003478 serotonin 5-HT2 receptor agonist Substances 0.000 description 1
- 239000003762 serotonin receptor affecting agent Substances 0.000 description 1
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 230000007727 signaling mechanism Effects 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 1
- 230000007958 sleep Effects 0.000 description 1
- 208000020685 sleep-wake disease Diseases 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 229940103422 stalevo Drugs 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- MIQPIUSUKVNLNT-UHFFFAOYSA-N tolcapone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC(O)=C(O)C([N+]([O-])=O)=C1 MIQPIUSUKVNLNT-UHFFFAOYSA-N 0.000 description 1
- 229960004603 tolcapone Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 1
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960003726 vasopressin Drugs 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/16—Peri-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
Definitions
- the invention relates to particular substituted heterocycle fused gamma-carbolines, in free, solid, pharmaceutically acceptable salt and/or substantially pure form as described herein, pharmaceutical compositions thereof, and methods of use as non-hallucinogenic biased agonists or antagonists at the serotonin (5-HT 2A ) receptor, in particularly having agonism biased towards the beta-arrestin signaling pathway, or as serotonin (5-HT 2A ) receptor antagonists.
- Such compounds may be useful for the treatment of mood disorders, general anxiety, social anxiety, depression, schizophrenia, anhedonia, and other neuropsychiatric diseases.
- the hallucinogenic psychedelics can cause overwhelming distress (i.e., “bad trips”) or prolonged or intermittent psychoses, can promote self-harm or harm to others, and are prone to abuse and dependence.
- serotonergic agents having 5-HT 2B agonist activity may cause valvular heart disease and/or pulmonary arterial hypertension. Therefore, there has been an effort to develop novel compounds having a pharmacological profile for treatment of mood and other CNS disorders, similar to the hallucinogenic psychedelics, but without the hallucinations, abuse liability, or valvular heart disease risk.
- GPCRs G- protein coupled receptors
- the G protein consists of alpha-, beta-, and gamma-subunits in a complex.
- the G-protein heterotrimer is normally in an inactive state, bound to a guanosine diphosphate ligand.
- GPCR G-protein alpha subunit
- G-alpha proteins including G i , G o , and G s , regulate levels of the cytosolic second messenger cAMP (cyclic adenosine monophosphate), by activating or inactivating adenylyl cyclase enzymes.
- the G-alpha-q variant (Gq) operates by activating phospholipase C enzyme, which cleaves phosphatidylinositol diphosphate (PIP 2 ) to form the two second messengers inositol triphosphate (IP 3 ) and diacylglycerol (DAG).
- IP3 stimulates calcium release from intracellular storage locations, resulting in the activation of various calcium-dependent kinases, and DAG stimulates protein kinase C (PKC) activation.
- PKC protein kinase C
- G-proteins are abundant soluble cytosolic proteins, an agonist-activated GPCR which has activated and released its G-protein may bind to another GDP-bound heterotrimer, thus activating and releasing a second GTP-bound G-alpha subunit, and so on, until the receptor is deactivated.
- GPCRs are deactivated in a two-step process. First, they are phosphorylated by a family of enzymes called G receptor kinases (GRKs). This greatly reduces the affinity of the GPCR’s active site for G-protein. In the second step, beta-arrestin proteins can competitively bind to the active site of the phosphorylated GPCR, resulting in complete inhibition of G-protein binding. The arrestin-GPCR complex then targets the inactivated receptor for
- beta-arrestin proteins were originally thought to only play a role in inactivating and targeting the inactivated GPCR for recycling or degradation. However, in the last few years, it has become increasingly apparent that beta-arrestin proteins also play a novel role in G-protein independent signaling mechanisms. Hence, the GPCR-arrestin complex created following agonist-induced receptor activation and deactivation may serve as a scaffold which can attract many other proteins to form large multi-protein complexes. The complexes can then cause the activation of a variety of kinases involved in cell signaling, including Src, ERKs, and MAPKs.
- beta-arrestin signaling provides an alternative mechanism for GPCR signal transduction that does not rely on G-protein-induced signaling cascades and second messengers.
- Signaling bias is the concept that some ligands will bind to a GPCR in such a manner as to bias the receptor towards or away from one of the two signaling pathway: G-protein mediated signaling and beta-arrestin mediated signaling.
- G-protein mediated signaling and beta-arrestin mediated signaling.
- MOR mu-opioid receptor
- beta-arrestin recruitment to the receptor that is the cause of the major opioid side effects, respiratory depression and inhibition of gastrointestinal motility.
- MOR ligands have been discovered which show bias towards the G-protein signaling and away from the beta-arrestin signaling, resulting in enhanced analgesic efficacy with a reduced side effect profile. This can either reflect a higher efficacy for agonism via one pathway or the other, or even agonism of one pathway and antagonism of the other pathway.
- serotonin agonists must agonize both the G-protein signaling pathway and the beta-arrestin signaling pathway of the 5-HT 2A receptor in order to cause hallucinations, while agonists which selectively agonize the beta-arrestin pathway without agonizing the G-protein coupled pathway may provide relief of mood disorders, anxiety, and other CNS disorders without hallucinogenic side effects.
- agonists which selectively agonize the beta-arrestin pathway without agonizing the G-protein coupled pathway may provide relief of mood disorders, anxiety, and other CNS disorders without hallucinogenic side effects.
- Cao et al. recently studied the binding of numerous compounds, including serotonin, psilocin, LSD, the non-hallucinogenic psychedelic analog lisuride, and lumateperone, to the 5-HT 2A receptor using high-resolution X-ray crystallography. Science, 375:403-11 (2022).
- Substituted heterocycle fused gamma-carbolines are known to be agonists or antagonists of 5-HT 2 receptors, particularly 5-HT 2A receptors, and are useful in treating central nervous system disorders. These compounds have been generally disclosed in U.S. Pat. No. 6,548,493; 7,238,690; 6,552,017; 6,713,471; 7,183,282; U.S. RE 39.680, and U.S. RE 39,679.
- Patent 7,081,455 disclose methods of making such substituted heterocycle fused gamma-carbolines and uses of these gamma-carbolines as serotonin agonists and antagonists useful for the control and prevention of central nervous system disorders such as addictive behavior and sleep disorders.
- U.S. Patent 8,598,119 discloses use of particular substituted heterocycle fused gamma-carbolines for the treatment of a combination of psychosis and depressive disorders as well as sleep, depressive and/or mood disorders in patients with psychosis or Parkinson's disease.
- this patent application discloses and claims use of these compounds at a low dose to selectively antagonize 5-HT 2A receptors without affecting or minimally affecting dopamine D 2 receptors, thereby useful for the treatment of sleep disorders without the side effects associated with high occupancy of the dopamine D2 pathways or side effects of other pathways (e.g., GABAA receptors) associated with conventional sedative-hypnotic agents (e.g., benzodiazepines).
- U.S. Patent 8,648,077 discloses methods of preparing toluenesulfonic acid addition salt crystals of these substituted heterocycle fused gamma-carbolines.
- Ketamine directly antagonizes extrasynaptic glutamatergic NMDA receptors, which also decreases GABAergic inhibition resulting in indirect activation of AMPA-type glutamate receptors.
- the downstream effects of AMPA receptor activation involve increase levels of brain-derived neurotrophic factor (BDNF) and activation of mTORC1 kinase pathways.
- BDNF brain-derived neurotrophic factor
- mTORC1 kinase pathways activation of mTORC1 kinase pathways.
- U.S.10,245,260 discloses novel oxo-metabolites of the substituted heterocycle fused gamma-carbolines disclosed in the above-mentioned publications. These new oxo-metabolites retain much of the unique pharmacologic activity of the parent compounds, including serotonin receptor inhibition, SERT inhibition, and dopamine receptor modulation. However, these oxo- metabolites were found to unexpectedly also show significant activity at mu-opioid receptors. Analogs of these novel compounds have also been disclosed, for example, in publications U.S. 10, 906,906 and U.S.10,961,245.
- This compound is principally known to be useful for the treatment of positive and negative symptoms of schizophrenia, depression (especially acute depression and bipolar depression), anxiety and traumatic disorders (including acute anxiety and post-traumatic stress disorder), and dementias (including Alzheimer's disease and the symptoms associated therewith).
- Lumateperone's effect as an anti-depressant is tied to its antagonism at the 5-HT 2A receptor and its inhibition of the serotonin transporter (SERT), pharmacological features which it shares with the SARI class of antidepressants (Serotonin Antagonist and Reuptake Inhibitor), which includes trazodone, nefazodone, lorpiprazole, and mepiprazole.
- lumateperone At dopamine D2 receptors, lumateperone has dual properties and acts as both a post- synaptic antagonist and a pre-synaptic partial agonist of the D2 receptor. It also stimulates phosphorylation of glutamatergic NMDA NR2B, or GluN2B, receptors in a mesolimbic specific manner. It is believed that this regional selectivity in the brain areas is thought to mediate the efficacy of antipsychotic drugs, together with the serotonergic, glutamatergic, and dopaminergic interactions, may result in antipsychotic efficacy for positive, negative, affective and cognitive symptoms associated with schizophrenia.
- the compound also exhibits serotonin reuptake inhibition, providing antidepressant activity for the treatment of schizoaffective disorder, co- morbid depression, and/or as a stand-alone treatment for major depressive disorder.
- Lumateperone is also useful for the treatment of bipolar disorder and other psychiatric and neurodegenerative disorders, particularly behavioral disturbances associated with dementia, autism and other CNS diseases. These features may be able to improve the quality of life of patients with schizophrenia and enhance social function to allow them to more fully integrate into their families and their workplace.
- Lumateperone tosylate Caplyta®
- Caplyta® is currently approved in the United States for the treatment of schizophrenia and bipolar depression. It is currently in clinical trials and development for additional indications, including major depressive disorder (MDD).
- spiro-joined is meant to clarify that the stated C 3- 6 cycloalkyl group or 3-6-membered heterocycloalkyl is present in a spiro-junction, meaning that one atom of said cyclic group is an atom of the ring to which the group is attached.
- the follow are examples of compounds of Formula I having spiro-joined cyclic groups within the scope of the present disclosure:
- the cyclopropane, cyclobutane, aziridine, azetidine, or oxetane may be replaced by any other C 3-6 cycloalkyl or 3-6-membered heterocycloalkyl, including, but not limited to, cyclopentane, cyclohexane, tetrahydrofuran, tetrahydropyran, pyrrolidine, piperidine, piperazine, or morpholine.
- Method 1.22 wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-1000mg, for example 2.5mg-50mg, or for a long-acting formulation, 25mg-1500mg, for example, 50mg to 500mg, or 250mg to 1000mg, or 250mg to 750mg, or 75mg to 300mg; 1.25 Method 1.22, where therapeutically in the effective amount of the Compound of the Invention is 1 mg-100mg per day, for example 2.5mg-50mg per day; 1.26 Method 1.22, wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-5mg, preferably 2.5-5mg, per day; 1.27 Method 1.22, wherein the therapeutically effective amount of the Compound of the Invention is 2.5mg or 5mg, per day; 1.28 Method 1 or any of 1.1-1.27, wherein the pharmaceutical composition is a sustained release or delayed release formulation, e.g., according to Pharmaceutical Composition 1-A as described herein; 1.
- the invention provides a method (Method 2) for the treatment or prophylaxis of a central nervous system disorder, or more than one central nervous system disorder, the method comprising administering to a patient in need thereof a therapeutically effective amount of a Compound of the Invention (e.g., a Compound of Formula I), wherein the Compound of the Invention is an antagonist of the 5-HT 2A receptor.
- a Compound of the Invention e.g., a Compound of Formula I
- the Compound of the Invention is an antagonist of the 5-HT 2A receptor.
- the Pharmaceutical Composition comprising a Compound of the Invention may be administered for controlled- and/or sustained-release of the Compounds of the Invention over a period of from about 14 days, about 30 to about 180 days, preferably over the period of about 30, about 60 or about 90 days.
- Controlled- and/or sustained-release is particularly useful for circumventing premature discontinuation of therapy, particularly for antipsychotic drug therapy where non-compliance or non-adherence to medication regimes is a common occurrence.
- hallucinogen refers to a compound which causes hallucinogenic symptoms, which are any one or more symptoms selected from visual hallucinations, auditory hallucinations, visual distortions (such as drifting, morphing, breathing or melting of objects and surfaces in the field of view), detachment from reality, dissociation, delirium, and undesired altered states of consciousness.
- a compound of the present disclosure is considered “non- hallucinogenic” if at doses which are therapeutically effective for the treatment of neuropsychiatric disorders described herein (e.g., depression, anxiety, etc.) the compound does not cause hallucinogenic symptoms.
- alkyl contains 1 to 21 carbon atoms, preferably straight chain and optionally saturated or unsaturated, for example in some embodiments wherein R 1 is an alkyl chain containing 1 to 21 carbon atoms, preferably 6-15 carbon atoms, 16-21 carbon atoms, e.g., so that together with the -C(O)- to which it attaches, e.g., when cleaved from the compound of Formula I, forms the residue of a natural or unnatural, saturated or unsaturated fatty acid.
- Suitable pharmaceutically acceptable diluents and carriers can be found in any of several well-known treatises on pharmaceutical formulations, for example Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001; Remington's Pharmaceutical Sciences, 20 th Ed., Lippincott Williams & Wilkins., 2000; and Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); all of which are incorporated by reference herein in their entirety.
- the compounds thus exist in individual isomeric, e.g., enantiomeric or diastereomeric form or as mixtures of individual forms, e.g., racemic/diastereomeric mixtures.
- Any isomer may be present in which the asymmetric center is in the (R)-, (S)-, or (R,S)- configuration.
- the invention is to be understood as embracing both individual optically active isomers as well as mixtures (e.g., racemic/diastereomeric mixtures) thereof.
- the Compounds of the Invention may be a racemic mixture or it may be predominantly, e.g., in pure, or substantially pure, isomeric form, e.g., greater than 70% enantiomeric/diastereomeric excess (“ee”), preferably greater than 80% ee, more preferably greater than 90% ee, most preferably greater than 95% ee.
- ee enantiomeric/diastereomeric excess
- the purification of said isomers and the separation of said isomeric mixtures may be accomplished by standard techniques known in the art (e.g., column chromatography, preparative TLC, preparative HPLC, simulated moving bed and the like).
- Geometric isomers by nature of substituents about a double bond or a ring may be present in cis (Z) or trans (E) form, and both isomeric forms are encompassed within the scope of this invention.
- the compounds of the present disclosure encompass their stable and unstable isotopes.
- Stable isotopes are nonradioactive isotopes which contain one additional neutron compared to the abundant nuclides of the same species (i.e., element). It is expected that the activity of compounds comprising such isotopes would be retained, and such compound would also have utility for measuring pharmacokinetics of the non-isotopic analogs.
- the hydrogen atom at a certain position on the compounds of the disclosure may be replaced with deuterium (a stable isotope which is non-radioactive).
- stable isotopes include, but not limited to, deuterium ( 2 H or D), 13 C, 15 N, 18 O.
- unstable isotopes which are radioactive isotopes which contain additional neutrons compared to the abundant nuclides of the same species (i.e., element), e.g., 123 I, 131 I, 125 I, 14 C, 18 F, may replace the corresponding abundant species of I, C and F.
- Another example of useful isotope of the compound of the invention is the 14 C isotope.
- radio isotopes are useful for radio-imaging and/or pharmacokinetic studies of the compounds of the invention.
- substitution of atoms of having the natural isotopic distributing with heavier isotopes can result in desirable change in pharmacokinetic rates when these substitutions are made at metabolically liable sites.
- the incorporation of deuterium ( 2 H) in place of hydrogen can slow metabolic degradation when the position of the hydrogen is a site of enzymatic or metabolic activity.
- the pharmaceutical depot delivers the Compounds of the Invention to the subject at concentrations effective for treatment of the particular disease or medical condition over a sustained period of time, e.g., 14-180 days, preferably about 30, about 60 or about 90 days.
- Polymers useful for the polymeric matrix in the Composition of the Invention may include a polyester of a hydroxyfatty acid and derivatives thereof or other agents such as polylactic acid, polyglycolic acid, polycitric acid, polymalic acid, poly-beta.-hydroxybutyric acid, epsilon.-capro-lactone ring opening polymer, lactic acid-glycolic acid copolymer, 2-hydroxybutyric acid-glycolic acid copolymer, polylactic acid-polyethylene glycol copolymer or polyglycolic acid-polyethylene glycol copolymer), a polymer of an alkyl alpha-cyanoacrylate (for example poly(butyl 2-
- the polymers are copolymers, they may be any of random, block and/or graft copolymers.
- any one of D-isomers, L- isomers and/or DL-isomers may be used.
- alpha-hydroxycarboxylic acid polymer preferably lactic acid-glycolic acid polymer
- its ester preferably lactic acid-glycolic acid polymer
- poly-alpha-cyanoacrylic acid esters etc.
- the polymer useful for the polymeric matrix is PLGA.
- the term PLGA includes polymers of lactic acid (also referred to as polylactide, poly(lactic acid), or PLA).
- the polymer is the biodegradable poly(d,l-lactide-co-glycolide) polymer.
- the polymeric matrix of the invention is a biocompatible and biodegradable polymeric material.
- biocompatible is defined as a polymeric material that is not toxic, is not carcinogenic, and does not significantly induce inflammation in body tissues.
- the matrix material should be biodegradable wherein the polymeric material should degrade by bodily processes to products readily disposable by the body and should not accumulate in the body.
- the products of the biodegradation should also be biocompatible with the body in that the polymeric matrix is biocompatible with the body.
- lactic acid and glycolic acid arc water-soluble, non-toxic products of normal metabolism, which may further biodegrade to form carbon dioxide and water.
- PLGA is believed to degrade by means of hydrolysis of its ester groups in the presence of water, for example in the body of a warm-blooded animal such as man, to produce lactic acid and glycolic acid and create the acidic microclimate.
- Lactic and glycolic acid are by-products of various metabolic pathways in the body of a warm-blooded animal such as man under normal physiological conditions and therefore are well tolerated and produce minimal systemic toxicity.
- the polymeric matrix useful for the invention may comprise a star polymer wherein the structure of the polyester is star-shaped.
- polyesters have a single polyol residue as a central moiety surrounded by acid residue chains.
- the polyol moiety may be, e. g., glucose or, e. g., mannitol.
- esters are known and described in GB 2,145,422 and in U. S. Patent No. 5,538,739, the contents of which are incorporated by reference.
- the depot compositions of the invention may comprise the polymer in the form of microparticles or nanoparticles, or in a liquid form, with the Compounds of the Invention dispersed or encapsulated therein.
- “Microparticles” is meant solid particles that contain the Compounds of the Invention either in solution or in solid form wherein such compound is dispersed or dissolved within the polymer that serves as the matrix of the particle.
- the microparticles may be prepared using any appropriate method, such as by a solvent evaporation or solvent extraction method.
- a solvent evaporation method the Compounds of the Invention and the polymer may be dissolved in a volatile organic solvent (for example a ketone such as acetone, a halogenated hydrocarbon such as chloroform or methylene chloride, a halogenated aromatic hydrocarbon, a cyclic ether such as dioxane, an ester such as ethyl acetate, a nitrile such as acetonitrile, or an alcohol such as ethanol) and dispersed in an aqueous phase containing a suitable emulsion stabilizer (for example polyvinyl alcohol, PVA).
- a suitable emulsion stabilizer for example polyvinyl alcohol, PVA
- the organic solvent is then evaporated to provide microparticles with the Compounds of the Invention encapsulated therein.
- the Compounds of the Invention and polymer may be dissolved in a polar solvent (such as acetonitrile, dichloromethane, methanol, ethyl acetate or methyl formate) and then dispersed in an aqueous phase (such as a water/PVA solution).
- a polar solvent such as acetonitrile, dichloromethane, methanol, ethyl acetate or methyl formate
- an aqueous phase such as a water/PVA solution
- Spray drying is an alternative manufacturing technique for preparing the microparticles.
- Solvents for making such compositions comprising the Compounds of the Invention and the polymeric matrix material that can be employed in the practice of the present invention include organic solvents, such as acetone; halogenated hydrocarbons, such as chloroform, methylene chloride, and the like; aromatic hydrocarbon compounds; halogenated aromatic hydrocarbon compounds; cyclic ethers; alcohols, such as, benzyl alcohol; ethyl acetate; and the like.
- the solvent for use in the practice of the present invention may be a mixture of benzyl alcohol and ethyl acetate.
- the amount of the Compounds of the present disclosure incorporated in the microparticles usually ranges from about 1 wt. % to about 90 wt. %, preferably 30 to 50 wt. %, more preferably 35 to 40 wt. %.
- weight % is meant parts of the Compounds of the present disclosure per total weight of microparticle.
- the pharmaceutical depot compositions may comprise a pharmaceutically-acceptable diluent or carrier, such as a water miscible diluent or carrier.
- Details of Osmotic-controlled Release Oral Delivery System composition may be found in EP 1539115 (U.S. Pub.
- a “therapeutically effective amount” is any amount of the Compounds of the invention (for example as contained in the pharmaceutical depot) which, when administered to a subject suffering from a disease or disorder, is effective to cause a reduction, remission, or regression of the disease or disorder over the period of time as intended for the treatment.
- Dosages employed in practicing the present invention will of course vary depending, e.g., on the particular disease or condition to be treated, the particular Compound of the Invention used, the mode of administration, and the therapy desired.
- an amount of the Compound of the Invention for administration refers to or is based on the amount of the Compound of the Invention in free base form (i.e., the calculation of the amount is based on the free base amount).
- Compounds of the Invention may be administered by any satisfactory route, including orally, parenterally (intravenously, intramuscular, intranasal, pulmonary or subcutaneous) or transdermally.
- the Compounds of the Invention e.g., in depot formulation, is preferably administered parenterally, e.g., by injection, for example, intramuscular or subcutaneous injection.
- the dosages will be higher relative to the shorter action composition, e.g., higher than 1-100mg, e.g., 25mg, 50mg, 100mg, 500mg, 1,000mg, or greater than 1000mg.
- Duration of action of the Compounds of the present disclosure may be controlled by manipulation of the polymer composition, i.e., the polymer:drug ratio and microparticle size. Wherein the composition of the invention is a depot composition, administration by injection is preferred.
- the pharmaceutically acceptable salts of the Compounds of the present disclosure can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free base forms of these compounds with a stoichiometric amount of the appropriate acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Further details for the preparation of these salts, e.g., toluenesulfonic salt in amorphous or crystal form, may be found in U.S. 8,309,722, 8.648,077, 9,199,995, and 9,586,960.
- compositions comprising Compounds of the present disclosure may be prepared using conventional diluents or excipients (an example include, but is not limited to sesame oil) and techniques known in the galenic art.
- oral dosage forms may include tablets, capsules, solutions, suspensions and the like.
- Concurrently when referring to a therapeutic use means administration of two or more active ingredients to a patient as part of a regimen for the treatment of a disease or disorder, whether the two or more active agents are given at the same or different times or whether given by the same or different routes of administrations. Concurrent administration of the two or more active ingredients may be at different times on the same day, or on different dates or at different frequencies.
- Isolation or purification of the diastereomers of the Compounds of the Invention may be achieved by conventional methods known in the art, e.g., column purification, preparative thin layer chromatography, preparative HPLC, crystallization, trituration, simulated moving beds and the like.
- Salts of the Compounds of the present disclosure may be prepared as similarly described in U.S. Pat. No. 6,548,493; 7,238,690; 6,552,017; 6,713,471; 7,183,282, 8,648,077; 9,199,995; 9,586,860; U.S. RE39680; and U.S. RE39679, the contents of each of which are incorporated by reference in their entirety.
- Typical reagents and conditions (a) RX, DIPEA, KI, 18-crown-6, dioxane, 95 °C, or RX, K 2 CO 3 , dioxane, 60 °C.
- the tetracyclic starting material can be made according to known methods, for example, according to the following Scheme 3-A: Typical reagents and conditions: (a) N-methyl chloroacetamide, DIPEA, KI, dioxane, reflux, 48 h; (b) CuI, K 2 CO 3 , DMEDA, dioxane, reflux, 24 h; (c) BH 3 -THF, THF, 60 °C, 20 h; (d) KOH, n- BuOH, 120 °C, 3 h.
- the resulting mixture is heated to 95 °C and stirred for 6.5 hours. After cooling to room temperature, the solvent is removed, and the residue is suspended in ethyl acetate (50 mL) and water (50 mL). The aqueous phase is separated and extracted twice with ethyl acetate (30 mL). The combined organic phase is dried over MgSO 4 and concentrated. The residue is purified by silica gel column chromatography using a gradient of 0 – 20% mixed solvents [ethyl acetate/methanol/7N NH 3 in methanol (10 : 1 : 0.1 v/v)] in ethyl acetate to obtain the title product as a brown solid (0.8 g, yield 16%).
- the synthesis method is analogous to the synthesis of the compound of Example 8 according to Scheme 2 wherein 4-chloro-1-(4-fluorophenyl)butan-1-one is added in Step E instead of 1-(3-chloropropoxy)-4-fluorobenzene.
- Step B ethyl (6bR,10aS)-2-oxo-2,3,6b,9,10,10a-hexahydro-1H- pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline-8(7H)-carboxylate.
- Step C (7a'S,11a'R)-ethyl 5',6',8',9',11',11a'-hexahydrospiro[cyclopropane-1,4'- pyrido[3',4':4,5]pyrrolo[3,2,1-ij]quinoline]-10'(7a'H)-carboxylate.
- CH 2 I 2 (600 mg, 2.23 mmol) is added dropwise to a stirred solution of ZnEt 2 (1.5 M in toluene, 0.7 mL, 1.1 mmol) in dichloromethane (0.5 mL) at 0 °C under an argon atmosphere, and the mixture is stirred at 0 °C for 50 minutes.
- Step D (7a'S,11a'R)-5',6',7a',8',9',10',11',11a'-octahydrospiro[cyclopropane-1,4'- pyrido[3',4':4,5]pyrrolo[3,2,1-ij]quinoline].
- the eluent is concentrated to a solid residue (320 g), and then redissolved in hot ethanol (80 ml). This mixture is allowed to cool to ambient temperature and stirred overnight, and then cooled to 0-5°C, aged for 1h and filtered. The filtered cake is washed with cold ethanol (15 ml) and allowed to air dry to afford the title compound (17.0 g, 70% yield) as a white solid.
- Example 81 (6bR,10aS)-8-(2-ethylphenethyl)-3-methyl-2,3,6b,7,8,9,10,10a- octahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline
- the synthesis method is analogous to Example 71, with 1-(2-bromoethyl)-2- ethylbenzene added in Step D instead of 1-(2-bromoethyl)-3-methoxybenzene.64% isolated yield.
- Example 180 13 C NMR (126 MHz, CDCl 3 ) ⁇ 138.2, 135.1, 130.3, 120.4, 112.9, 109.0, 64.8, 61.1, 56.6, 50.8, 49.2, 44.5, 41.9, 37.7, 25.2, 20.3, 12.2.
- the remaining compounds through Example 180 may be prepared according to analogous procedures.
- receptor binding assays can be performed via competitive assay against the agonist radioligand 125 I-(+/-)- DOI, to determine Ki by displacement, using human recombinant HEK-293 cells expressing human 5-HT 2A , 5-HT 2B , and/or 5-HT 2C receptors.
- the results are expressed as a percent of control specific binding: and as a percent inhibition of control specific binding: obtained in the presence of the test compounds.
- IC 50 values concentration causing a half-maximal inhibition of control specific binding
- Ki inhibition constants
- L concentration of radioligand in the assay
- K D affinity of the radioligand for the receptor.
- a Scatchard plot is used to determine the K D .
- the compound of Formula A (ITI-007, lumateperone) is used as a comparison in the assays.
- the following receptor affinity results are obtained (with the Compound of Formula A for comparison): [000274] As shown, many of the compounds of the present disclosure show significantly more receptor selectivity than the reference compound of Formula A. For example, the compounds of Examples 14, 15, and 16 show little D1, D2, or Mu receptor affinity, but retain strong affinity at the serotonin 2A receptor.
- Some compounds are additionally tested in a receptor profiling panel consisting agonist and/or antagonist radioligand binding assays. Assays are conducted using a 100 nM concentration of the test compound. Compound binding is calculated as a percent inhibition of the binding of the specific radioligand for the receptor (or ion channel) tested, which can be an agonist or antagonist. The following receptors and ion channels are included in the panel:
- the compounds of the present disclosure have high selectivity with few off- target interactions.
- the compounds interact significantly (> 45% inhibition) only with the following receptors: alpha-1A (e.g., 40-80% inhibition), serotonin-2A (e.g., 50- 100% inhibition), and serotonin-2B (50-100% inhibition), with insignificant activity ( ⁇ 40% inhibition) at other receptors which are commonly associated with side effects, such as the serotonin-1A, serotonin-1B, serotonin-3, muscarinic, other adrenergic, and histamine receptors.
- alpha-1A e.g., 40-80% inhibition
- serotonin-2A e.g., 50- 100% inhibition
- serotonin-2B 50-100% inhibition
- insignificant activity ⁇ 40% inhibition
- Agonist Assay CHO-K1 cells expressing human 5-HT 2A (ES-313-AF) are obtained from PerkinElmer and used according to the supplier's recommendations. Frozen cells are thawed in a water bath at 37 °C, and then are resuspended in 10 mL Ham's F-12 medium containing 10% FBS.
- Cells are recovered by centrifugation at 150g for 5 minutes and are then resuspended in pre-warmed assay buffer (DMEM/HAM's F-12 with HEPES) at 3x10 5 cells/L in a Falcon tube. Under sterile conditions, Coelenterazine H is added to a final concentration of 5 ⁇ M to the cell suspension. The Falcon tube is wrapped in aluminum foil and placed on a rotating wheel (about 45° angle and 7 rpm/min speed) for 4 hours at room temperature. Cells are then diluted to 1x10 5 cells/mL in assay buffer and transferred to a beaker wrapped in aluminum foil on a magnetic stirrer.
- pre-warmed assay buffer DMEM/HAM's F-12 with HEPES
- Coelenterazine H is added to a final concentration of 5 ⁇ M to the cell suspension.
- the Falcon tube is wrapped in aluminum foil and placed on a rotating wheel (about 45° angle and 7 rpm/min speed) for 4 hours at room temperature. Cells are
- A is the concentration of the reference agonist alpha-methylserotonin
- EC 50A is the EC 50 value of the reference agonist alpha-methylserotonin.
- Beta-arrestin Signaling Agonism/Antagonism at the 5-HT 2A Receptor Selected compounds are submitted to 5-HT 2A agonist and antagonist assays for beta-arrestin recruitment. Alpha-methylserotonin is used as the reference control for agonist assays, and altanserin is used as the control for antagonist assays.
- Bias score (or bias ratio) is calculated as the ratio of the intrinsic relative activity (RA i ) for agonism of beta-arrestin signaling over the intrinsic relative activity (RA i ) for agonism of G-q signaling.
- RA i Intrinsic Relative Activity, compared to the positive controls methylserotonin or altanserin):
- the compounds of Examples 2, 4, and 5 are each strong (full) antagonists of the G-q signaling pathway, with the compound of Example 4 having comparable antagonist activity to the non-biased reference compound of Formula A.
- compounds should have either antagonistic activity at the G-q signaling pathway, or partial agonist activity with low intrinsic efficacy.
- Full agonist activity at the G-q signaling pathway i.e., high intrinsic activity
- the compounds of the present disclosure are preferably agonists of beta-arrestin mediated signaling, either as partial agonists or as full agonists.
- they should be strongly biased towards beta-arrestin signaling.
- the compounds of Examples 2, 4, and 5, however, are antagonists of beta-arrestin signaling.
- the compound of Example 4 for example, much like the compound of Formula A, has equivalent strong antagonistic activity at both G-q and beta-arrestin signaling pathways.
- it could be a potent antidepressant, antipsychotic, like lumateperone (ITI-007), it would not display the unique hallmarks of the psychedelic antidepressant family.
- the compounds of Examples 14, 15, and 16 in particular show partial agonist activity in the beta-arrestin assay and significant bias towards beta-arrestin mediated agonism, especially the compound of Example 15, which has zero G-q mediated agonist activity.
- the compounds of Examples 24, 25, and 40 also show zero G-q mediated agonist activity, but while the compound of Example 24 is a beta-arrestin antagonist, the compounds of Examples 25 and 40 are beta-arrestin partial agonists.
- the results together show a wide variety of functional activity profiles for the compounds according to the present disclosure, which will provide them each with the potential for varying uses and secondary effect or side effect profiles.
- the data further shows a trend that compound having a 2 or 3-atom side chain linker preferentially active the beta-arrestin signaling pathway, with varying levels of intrinsic activity.
- n 2 or 3
- Z is a bond
- compounds wherein n is 1 or 2 and Z is a group one-atom across e.g., a -C(O)-, -O-, or a carbonyl equivalent group
- the side chain linker is preferably 3 atoms in length, thus wherein n is 3 and Z is a bond, or n is 2 and Z is a group one-atom across.
- the data also suggests that the substituent pattern around the A ring may impact binding modes of the compounds to the 5-HT 2A receptor, as can the connecting group Z. For some series of compounds agonist versus antagonist binding may depend on the choice of group Z or on the presence or absence of electron-donating or electron-withdrawing groups on the ring A.
- Example 183 In Vivo Characterization
- Selected compounds are submitted to rodent functional model assays to determine in vivo efficacy.
- Head Twitch Assay The stereotyped head twitch response induced by 5-HT 2A agonists is used as a behavioral proxy for hallucinations. See Halberstadt, et al., Neuropharmacology, 167, 107933 (2020).
- Test compound (0.3, 1.0, 3 and 3.0 mg/kg, or alternatively, 1, 3, and 10 mg/kg) or vehicle (0.5% aqueous CMC) is injected SC 30 minutes before behavioral testing.
- a pair of rats Sprague-Dawley males
- Social interactions include sniffing the other rat, grooming the other rat, climbing over, under or around the other rat, following the other rat, and exploring the ano- genital area of the other rat.
- the time the rats spend interacting with each other during the 6- minute test is recorded by a trained observer.
- Chlordiazepoxide (IP, 5 mg/kg) is used as a positive control.
- Male adult mice are injected SC with either test compound (1 mg/kg) or vehicle.
- samples from the PFC region of the brain are collected, and a synaptoneurosome-enriched fraction is collected and prepared for Western blotting.
- Quantitative analysis of phospho (p) protein immunoblots are determined relative to total levels of each protein. Change in the amount of phosphorylated ERK, Akt, mTOR, and P70S6K proteins, in the PFC are determined relative to vehicle-treated mice, as previously described (Dutheil, et al., J.
- test compounds unlike the serotonergic psychedelic DOI, even high doses of a test compound up to 10 mg/kg does not elicit hallucinogenic behavior, as shown by a rate of head twitch comparable to control (e.g., ⁇ 10 head twitches per 5 minutes, or less than 5 head twitches or less than 1 head twitch (mean results)) and substantially less than that induced by DOI (p ⁇ 0.0001). Test compounds also show a dose dependent increase in social interaction between rats, and the data show that even the lowest test dose of 0.3 mg/kg is effective.
- Example 184 Pharmacokinetic evaluation
- Compounds according to the present disclosure are submitted to a standard testing protocol for oral pharmacokinetics in Sprague-Dawley rats (males, 200-400 g). Test compounds are administered to rats either IV at 1 mg/kg or PO at 10 mg/kg, using a 0.05M citrate phosphate buffer as vehicle. Other potential vehicles include PEG-400 and aqueous 10% Trapposol/1% Tween 80, depending on compound solubility.
- a third arm may utilize subcutaneous dosing (e.g., SC at 1 mg/kg). Plasma samples are collected at 2, 5, 15, and 30 minutes, and 1 , 2, 4, 8, and 24 hours, post-dosing.
- Plasma samples are analyzed for the presence of the test compound, and for some cases, for the presence of major expected metabolites (e.g., N-des-methyl metabolite).
- Time to maximum concentration (Tmax), maximal plasma concentration (Cmax), and area-under-the-curve (AUC) are calculated from the data. Comparison between the AUC values for oral versus IV dosing provides the oral bioavailability of the test compound.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Neurology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Neurosurgery (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention relates to particular substituted heterocycle fused gamma-carbolines, in free, solid, pharmaceutically acceptable salt and/or substantially pure form as described herein, pharmaceutical compositions thereof, and methods of use as non-hallucinogenic biased agonists or antagonists at the serotonin (5-HT2A) receptor, in particularly having agonism biased towards the beta-arrestin signaling pathway. Such compounds are useful for the treatment of mood disorders, general anxiety, social anxiety, depression, anhedonia, and other neuropsychiatric diseases.
Description
HETEROCYCLE FUSED GAMMA-CARBOLINES ACTING ON THE SEROTONINE 5-HT2A RECEPTOR
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is an international application which claims priority to, and the benefit of, U.S. Provisional Application Ser. No. 63/478,010, filed on Dec. 30, 2022, and U.S. Provisional Application Ser. No. 63/603,617, filed on Nov. 28, 2023, the contents of each of which are hereby incorporated by reference in their entireties.
TECHNICAL FIELD
[0001] The invention relates to particular substituted heterocycle fused gamma-carbolines, in free, solid, pharmaceutically acceptable salt and/or substantially pure form as described herein, pharmaceutical compositions thereof, and methods of use as non-hallucinogenic biased agonists or antagonists at the serotonin (5-HT2A) receptor, in particularly having agonism biased towards the beta-arrestin signaling pathway, or as serotonin (5-HT2A) receptor antagonists. Such compounds may be useful for the treatment of mood disorders, general anxiety, social anxiety, depression, schizophrenia, anhedonia, and other neuropsychiatric diseases.
BACKGROUND
[0002] Serotonin, also known as 5 -hydroxy tryptamine (5-HT), is a neurotransmitter widely distributed in the brain. The dysregulation of serotonergic signaling in the brain has been implicated in the pathogenesis of several neuropsychiatric diseases. Drugs that directly or indirectly target 5-HT, such as 5-HT2 receptor agonists and selective serotonin reuptake inhibitors, are widely used in psychiatry, finding use in the treatment of numerous mood disorders as well as in the treatment of psychosis. However, strong 5-HT2 agonists tend to cause hallucinations, which is a dangerous side effect. Serotonergic psychedelic hallucinogens are powerful psychoactive substances that alter both perception and mood, and they may be useful in restoring function to dysregulated serotonergic networks in the brain. There have been studies suggesting that the classic hallucinogenic psychedelic serotonin agonists, such as LSD (D- lysergic acid diethylamide) and psilocybin (via its active metabolite psilocin) could be very effective in the treatment of numerous neuropsychiatric disorders, especially depression, but these drugs are not feasible in practice because of their hallucinogenic side effects. The hallucinogenic psychedelics can cause overwhelming distress (i.e., “bad trips”) or prolonged or
intermittent psychoses, can promote self-harm or harm to others, and are prone to abuse and dependence. In addition, it has been reported that serotonergic agents having 5-HT2B agonist activity may cause valvular heart disease and/or pulmonary arterial hypertension. Therefore, there has been an effort to develop novel compounds having a pharmacological profile for treatment of mood and other CNS disorders, similar to the hallucinogenic psychedelics, but without the hallucinations, abuse liability, or valvular heart disease risk.

[0003] One well-known non-hallucinogenic psychedelic analog is lisuride, developed in the
1970’s, which has been used for the treatment of Parkinson’s disease and migraine attacks. Two non-hallucinogenic psychedelic analogs have been recently described as having anti-depressant like behavior. Cameron et al. generated the novel compound tabemanthalog by modifying the skeletal core of the natural hallucinogenic psychedelic ibogaine. Nature, 589:474-79 (2021).
Cell, 184:2779-92.e18 (2021). Similarly, based on the well-known hallucinogenic psychedelic 5- methoxy-dimethyltryptamine, Dong et al. arrived at the novel analog AAZ-A-154. However, neither of these groups developed any structure-activity guidance for these compounds, and it thus remained unclear how to rationally design non-hallucinogenic psychedelic analogs.


[0006] The beta-arrestin proteins were originally thought to only play a role in inactivating and targeting the inactivated GPCR for recycling or degradation. However, in the last few years, it has become increasingly apparent that beta-arrestin proteins also play a novel role in G-protein independent signaling mechanisms. Apparently, the GPCR-arrestin complex created following agonist-induced receptor activation and deactivation may serve as a scaffold which can attract many other proteins to form large multi-protein complexes. The complexes can then cause the activation of a variety of kinases involved in cell signaling, including Src, ERKs, and MAPKs. For example, some signaling cascades, such as the MAPK cascade, require two kinase proteins to approach each other closely so that one may phosphorylate the other. Arrestin complexes may facilitate such phosphorylation by binding to both kinase proteins at the same time. Therefore, beta-arrestin signaling provides an alternative mechanism for GPCR signal transduction that does not rely on G-protein-induced signaling cascades and second messengers.
[0007] Signaling bias is the concept that some ligands will bind to a GPCR in such a manner as to bias the receptor towards or away from one of the two signaling pathway: G-protein mediated signaling and beta-arrestin mediated signaling. Several examples of such biased GPCR ligands have been discovered, and it creates the potential for functionally selective receptor agonism. For example, the mu-opioid receptor (MOR) is a GPCR coupled to the Gi-type alpha subunit, so that agonist binding results in inhibition of adenylate cyclase activity and a reduction in cytosolic cAMP levels. It is found that this G-protein mediated pathway is responsible for morphine and other opioid drugs’ analgesic activity. However, it is beta-arrestin recruitment to the receptor that is the cause of the major opioid side effects, respiratory depression and inhibition of gastrointestinal motility. Various MOR ligands have been discovered which show bias towards the G-protein signaling and away from the beta-arrestin signaling, resulting in enhanced analgesic efficacy with a reduced side effect profile. This can either reflect a higher
efficacy for agonism via one pathway or the other, or even agonism of one pathway and antagonism of the other pathway. [0008] While the 5-HT1, 5-HT4, 5-HT5, 5-HT6, and 5-HT7 receptors are coupled to the cAMP-regulating G proteins, the 5-HT2 family of receptors are GPCRs coupled to Gq proteins, thus resulting in a classic signaling cascade mediated by the phosphatidylinositol pathway. It has also been found that the 5-HT2 receptors are also capable of activating a beta-arrestin signaling cascade as well. It is believed that serotonin agonists must agonize both the G-protein signaling pathway and the beta-arrestin signaling pathway of the 5-HT2A receptor in order to cause hallucinations, while agonists which selectively agonize the beta-arrestin pathway without agonizing the G-protein coupled pathway may provide relief of mood disorders, anxiety, and other CNS disorders without hallucinogenic side effects. [0009] For example, Cao et al. recently studied the binding of numerous compounds, including serotonin, psilocin, LSD, the non-hallucinogenic psychedelic analog lisuride, and lumateperone, to the 5-HT2A receptor using high-resolution X-ray crystallography. Science, 375:403-11 (2022). They identified a new binding mode for serotonin and psilocin. Traditionally, LSD, lisuride, serotonin and psilocin were known to bind in the so-called “orthostatic binding pocket (OBP).” In this binding mode, the polycyclic core of LSD and lisuride binds low in the OBP, while the side chains of LSD and lisuride protrude into the so- called “extended-binding pocket (EBP).” In contrast, the indole core of serotonin and psilocin sit higher in the OBP, closer to the EBP, but without significant protrusion into the EBP. [00010] Surprisingly, Cao et al. discovered that serotonin and psilocin have a second binding mode which is flipped so that the indole core sits in the EBP with only minimal presence in the upper part of the OBP. Cao further generated data suggesting that this second binding mode is responsible for increased beta-arrestin recruitment by the receptor, thus explaining the existence of biased serotonin receptor agonism. They also presented evidence that the Gq-mediated signaling is responsible for the hallucinogenic effects of the traditional psychedelics, and that ligands which are biased towards beta-arrestin recruitment may provide the therapeutic benefits of the psychedelics, such as antidepressant action, without hallucinogenic side effects. [00011] Substituted heterocycle fused gamma-carbolines are known to be agonists or antagonists of 5-HT2 receptors, particularly 5-HT2A receptors, and are useful in treating central nervous system disorders. These compounds have been generally disclosed in U.S. Pat. No.
6,548,493; 7,238,690; 6,552,017; 6,713,471; 7,183,282; U.S. RE 39.680, and U.S. RE 39,679. U.S. Patent 8,309,722, and U.S. Patent 7,081,455, disclose methods of making such substituted heterocycle fused gamma-carbolines and uses of these gamma-carbolines as serotonin agonists and antagonists useful for the control and prevention of central nervous system disorders such as addictive behavior and sleep disorders. [00012] In addition, U.S. Patent 8,598,119 discloses use of particular substituted heterocycle fused gamma-carbolines for the treatment of a combination of psychosis and depressive disorders as well as sleep, depressive and/or mood disorders in patients with psychosis or Parkinson's disease. In addition to disorders associated with psychosis and/or depression, this patent application discloses and claims use of these compounds at a low dose to selectively antagonize 5-HT2A receptors without affecting or minimally affecting dopamine D2 receptors, thereby useful for the treatment of sleep disorders without the side effects associated with high occupancy of the dopamine D2 pathways or side effects of other pathways (e.g., GABAA receptors) associated with conventional sedative-hypnotic agents (e.g., benzodiazepines). U.S. Patent 8,648,077 discloses methods of preparing toluenesulfonic acid addition salt crystals of these substituted heterocycle fused gamma-carbolines. [00013] US 2021/006009 discloses evidence showing that the aforementioned substituted fused heterocycle gamma carbolines may operate, in part, through glutamate (NMDA and AMPA receptor) receptor activation leading to enhanced mTOR1 signaling, in a manner similar to that of ketamine. Ketamine is a selective NMDA receptor antagonist. Ketamine acts through a system that is unrelated to the common psychogenic monoamines (serotonin, norepinephrine and dopamine), and this may be a major reason for its much more rapid effects. Ketamine directly antagonizes extrasynaptic glutamatergic NMDA receptors, which also decreases GABAergic inhibition resulting in indirect activation of AMPA-type glutamate receptors. The downstream effects of AMPA receptor activation involve increase levels of brain-derived neurotrophic factor (BDNF) and activation of mTORC1 kinase pathways. Like ketamine, recent evidence suggests that compounds related to those of the present disclosure enhance both NMDA and AMPA- induced currents in rat medial prefrontal cortex pyramidal neurons via activation of D1 receptors, and that this is associated with increased mTORC1 signaling. [00014] U.S.10,245,260 discloses novel oxo-metabolites of the substituted heterocycle fused gamma-carbolines disclosed in the above-mentioned publications. These new oxo-metabolites
retain much of the unique pharmacologic activity of the parent compounds, including serotonin receptor inhibition, SERT inhibition, and dopamine receptor modulation. However, these oxo- metabolites were found to unexpectedly also show significant activity at mu-opioid receptors. Analogs of these novel compounds have also been disclosed, for example, in publications U.S. 10, 906,906 and U.S.10,961,245. [00015] One such substituted heterocycle fused gamma-carboline of the aforementioned art is lumateperone shown below,
 with the chemical name (4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4': 4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-l-(4-fluorophenyl)-1-butanone). It is known to be an extremely potent serotonin receptor (5-HT2A) antagonist, as well as a modulator of dopamine receptor (D1 and/or D2) signaling, and a serotonin transporter (SERT) antagonist, and it is useful in treating a variety of central nervous system disorders. It has also been known as ITI-007. [00016] Lumateperone antagonizes the serotonin-2A (5-HT2A) receptor, and/or modulates dopamine receptor signaling at the level of key intracellular phosphoproteins. This compound is principally known to be useful for the treatment of positive and negative symptoms of schizophrenia, depression (especially acute depression and bipolar depression), anxiety and traumatic disorders (including acute anxiety and post-traumatic stress disorder), and dementias (including Alzheimer's disease and the symptoms associated therewith). Lumateperone's effect as an anti-depressant is tied to its antagonism at the 5-HT2A receptor and its inhibition of the serotonin transporter (SERT), pharmacological features which it shares with the SARI class of antidepressants (Serotonin Antagonist and Reuptake Inhibitor), which includes trazodone, nefazodone, lorpiprazole, and mepiprazole. [00017] At dopamine D2 receptors, lumateperone has dual properties and acts as both a post- synaptic antagonist and a pre-synaptic partial agonist of the D2 receptor. It also stimulates phosphorylation of glutamatergic NMDA NR2B, or GluN2B, receptors in a mesolimbic specific manner. It is believed that this regional selectivity in the brain areas is thought to mediate the
efficacy of antipsychotic drugs, together with the serotonergic, glutamatergic, and dopaminergic interactions, may result in antipsychotic efficacy for positive, negative, affective and cognitive symptoms associated with schizophrenia. The compound also exhibits serotonin reuptake inhibition, providing antidepressant activity for the treatment of schizoaffective disorder, co- morbid depression, and/or as a stand-alone treatment for major depressive disorder. Lumateperone is also useful for the treatment of bipolar disorder and other psychiatric and neurodegenerative disorders, particularly behavioral disturbances associated with dementia, autism and other CNS diseases. These features may be able to improve the quality of life of patients with schizophrenia and enhance social function to allow them to more fully integrate into their families and their workplace. [00018] Lumateperone tosylate (Caplyta®) is currently approved in the United States for the treatment of schizophrenia and bipolar depression. It is currently in clinical trials and development for additional indications, including major depressive disorder (MDD). [00019] The functional effects of lumateperone on the beta-arrestin recruitment and Gq- mediated signaling pathways induced by binding to the 5-HT2A receptor have not been previously disclosed. The present inventors have found that lumateperone is a potent antagonist of 5-HT2A receptors in both the beta-arrestin and Gq functional assays. [00020] There remains a need for new compounds with efficacy in treating neuropsychiatric disorders, especially depression, anxiety, and schizophrenia. It would be particularly beneficial to have compounds with strong biased agonism (e.g., full agonism or partial agonism) towards beta-arrestin recruitment at the 5-HT2A receptor in order to provide a non-hallucinogenic anti- depressant or anxiolytic effect. BRIEF SUMMARY [00021] In a first aspect, the present disclosure relates to a compound (Compound I) of Formula I:
with the chemical name (4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4': 4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-l-(4-fluorophenyl)-1-butanone). It is known to be an extremely potent serotonin receptor (5-HT2A) antagonist, as well as a modulator of dopamine receptor (D1 and/or D2) signaling, and a serotonin transporter (SERT) antagonist, and it is useful in treating a variety of central nervous system disorders. It has also been known as ITI-007. [00016] Lumateperone antagonizes the serotonin-2A (5-HT2A) receptor, and/or modulates dopamine receptor signaling at the level of key intracellular phosphoproteins. This compound is principally known to be useful for the treatment of positive and negative symptoms of schizophrenia, depression (especially acute depression and bipolar depression), anxiety and traumatic disorders (including acute anxiety and post-traumatic stress disorder), and dementias (including Alzheimer's disease and the symptoms associated therewith). Lumateperone's effect as an anti-depressant is tied to its antagonism at the 5-HT2A receptor and its inhibition of the serotonin transporter (SERT), pharmacological features which it shares with the SARI class of antidepressants (Serotonin Antagonist and Reuptake Inhibitor), which includes trazodone, nefazodone, lorpiprazole, and mepiprazole. [00017] At dopamine D2 receptors, lumateperone has dual properties and acts as both a post- synaptic antagonist and a pre-synaptic partial agonist of the D2 receptor. It also stimulates phosphorylation of glutamatergic NMDA NR2B, or GluN2B, receptors in a mesolimbic specific manner. It is believed that this regional selectivity in the brain areas is thought to mediate the
efficacy of antipsychotic drugs, together with the serotonergic, glutamatergic, and dopaminergic interactions, may result in antipsychotic efficacy for positive, negative, affective and cognitive symptoms associated with schizophrenia. The compound also exhibits serotonin reuptake inhibition, providing antidepressant activity for the treatment of schizoaffective disorder, co- morbid depression, and/or as a stand-alone treatment for major depressive disorder. Lumateperone is also useful for the treatment of bipolar disorder and other psychiatric and neurodegenerative disorders, particularly behavioral disturbances associated with dementia, autism and other CNS diseases. These features may be able to improve the quality of life of patients with schizophrenia and enhance social function to allow them to more fully integrate into their families and their workplace. [00018] Lumateperone tosylate (Caplyta®) is currently approved in the United States for the treatment of schizophrenia and bipolar depression. It is currently in clinical trials and development for additional indications, including major depressive disorder (MDD). [00019] The functional effects of lumateperone on the beta-arrestin recruitment and Gq- mediated signaling pathways induced by binding to the 5-HT2A receptor have not been previously disclosed. The present inventors have found that lumateperone is a potent antagonist of 5-HT2A receptors in both the beta-arrestin and Gq functional assays. [00020] There remains a need for new compounds with efficacy in treating neuropsychiatric disorders, especially depression, anxiety, and schizophrenia. It would be particularly beneficial to have compounds with strong biased agonism (e.g., full agonism or partial agonism) towards beta-arrestin recruitment at the 5-HT2A receptor in order to provide a non-hallucinogenic anti- depressant or anxiolytic effect. BRIEF SUMMARY [00021] In a first aspect, the present disclosure relates to a compound (Compound I) of Formula I:
 wherein:
X is S, S(O), S(O)2, O, CH2, CHRb, C(Rb)2, NH, N(Ra) (e.g., N(CH3)), N-C(O)-Ra, N- C(O)-O-Ra, N-C(O)-O-CH2-O-Ra, N-CH2-O-C(O)-Ra, N+(=O–), a spiro-joined C3- 6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3- 6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Y is CH2, CHRc, -C(O)-, C(Rc)2, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3- 6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Z is a bond, -S-, S(O), S(O)2, -O-, -NH, N(Rd), -C(O)-, -C(OH)-, -C(OC1-6alkyl), -C(=N- OH)-, -C(=N-OC1-6alkyl)-, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), a spiro- joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), or -O(CH2)pO- wherein p is 2, 3, or 4 (e.g., p is 2), wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; A is H, C3-6cycloalkyl (e.g., cyclopropyl or cyclohexyl), aryl (e.g., phenyl), or heteroaryl, wherein said cycloalkyl, aryl, or heteroaryl is substituted by 0-5 groups R; each R is independently selected from aryl (e.g., phenyl), aryloxy (e.g., phenoxy), heteroaryl (e.g., pyridyl), C1-6alkyl (e.g., methyl, ethyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy, ethoxy), C1-6alkylthio (e.g., methylthio), halo (e.g., F), cyano, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), or hydroxy, wherein each of said aryl, heteroaryl, alkyl, haloalkyl, alkylsulfonyl, alkoxy, alkylthio, cycloalkyl, or cycloalkoxy is optionally further substituted by one or more groups selected from aryl (optionally substituted with halo), halo, C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy), C1-
6alkylthio (e.g., methylthio), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), amino, C1-6alkylamino (e.g., methylamino), di(C1-6alkyl)amino (e.g., dimethylamino), (C1-6alkyl)(C1-6alkyl)amino (e.g., methylethylamino), and hydroxy; Ra and Rd, are each independently selected from C1-20alkyl (e.g., methyl or tert-butyl), and C1-2alkylaryl (e.g., benzyl or phenethyl); Rb and Rc are each independently selected from C1-6alkyl (e.g., methyl, ethyl, tert-butyl), C1-6alkoxy, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and C1-2alkylaryl (e.g., benzyl or phenethyl); m is 1 or 2; n is 1, 2, 3, 4, or 5; in free or salt form (e.g., pharmaceutically acceptable salt form); provided that n is not 3 when Z is -C(O)-, X is CH2 or O, and m is 2; and provided that n is not 3 when Z is -C(O)-, X is CH2, and m is 1; and provided that n is not 3 when Z is -C(O)- or -O-, X is NH or N(Ra), and m is 1; and provided that n is not 3 when Z is O, X is NCH3, Y is -C(O)-, and m is 1. [00022] The present disclosure further provides additional embodiments of first aspect, including: 1.1 Compound I, wherein X is S, S(O), or S(O)2; 1.2 Compound I, wherein X is O; 1.3 Compound I, wherein X is CH2, CHRb, or C(Rb)2; 1.4 Compound 1.3, wherein Rb is independently C1-6alkyl (e.g., methyl); 1.5 Compound I, wherein X is CH2; 1.6 Compound I, wherein X is NH; 1.7 Compound I, wherein X is N(Ra); 1.8 Compound I, wherein X is N-C(O)-Ra; 1.9 Compound I, wherein X is N-C(O)-O-Ra; 1.10 Compound I, wherein X is N-C(O)-O-CH2-O-Ra; 1.11 Compound I, wherein X is N-CH2-O-C(O)-Ra; 1.12 Compound I, or any of 1.7-1.11, wherein Ra is C1-2alkylaryl (e.g., benzyl or phenethyl);
1.13 Compound I, or any of 1.7-1.11, wherein Ra is C1-20alkyl (e.g., methyl or tert- butyl); 1.14 Compound I, or any of 1.7-1.11, wherein Ra is C10-20alkyl (e.g., decyl or dodecyl); 1.15 Compound I, or any of 1.7-1.11, wherein Ra is C1-15alkyl (e.g., hexyl or octyl); 1.16 Compound I, or any of 1.7-1.11, wherein Ra is C7-15alkyl (e.g., heptyl or nonyl); 1.17 Compound I, or any of 1.7-1.11, wherein Ra is C1-6alkyl (e.g., butyl or hexyl); 1.18 Compound I, or any of 1.7-1.11, wherein Ra is C1-4alkyl (e.g., n-butyl or tert- butyl); 1.19 Compound I, or any of 1.7-1.11, wherein Ra is C1-3alkyl (e.g., propyl or isopropyl); 1.20 Compound I, or any of 1.7-1.11, wherein Ra is C1-2alkyl (e.g., methyl or ethyl); 1.21 Compound I, or any of 1.7-1.11, wherein X is N(CH3); 1.22 Compound I, wherein X is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane); 1.23 Compound 1.22, wherein the spiro-joined C3-6cycloalkyl is selected from cyclopropane, cyclobutane, cyclopentane, and cyclohexane; 1.24 Compound 1.22, wherein the spiro-joined C3-6cycloalkyl is cyclopropane; 1.25 Compound I, wherein X is spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane); 1.26 Compound 1.25, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine, azetidine, oxetane, pyrrolidine, tetrahydrofuran, piperidine, tetrahydropyran, piperazine, and morpholine; 1.27 Compound 1.25, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine; 1.28 Compound I, or any of 1.22-1.27, wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is unsubstituted; 1.29 Compound I, or any of 1.22-1.27, wherein said spiro-joined C3-6cycloalkyl or 3- 6-membered heterocycloalkyl is substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; 1.30 Compound I, or any of 1.1-1.29, wherein Y is CH2;
1.31 Compound I, or any of 1.1-1.29, wherein Y is -C(O)-; 1.32 Compound I, or any of 1.1-1.29, wherein Y is CHRc or C(Rc)2; 1.33 Compound 1.32, wherein each Rc is independently C1-6alkyl; 1.34 Compound 1.32, wherein each Rc is independently selected from methyl, ethyl and propyl; 1.35 Compound I, or any of 1.1-1.29, wherein Y is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane); 1.36 Compound 1.35, wherein the spiro-joined C3-6cycloalkyl is selected from cyclopropane, cyclobutane, cyclopentane, and cyclohexane; 1.37 Compound 1.35, wherein the spiro-joined C3-6cycloalkyl is cyclopropane; 1.38 Compound I, or any of 1.1-1.29, wherein Y is spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane); 1.39 Compound 1.38, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine, azetidine, oxetane, pyrrolidine, tetrahydrofuran, piperidine, tetrahydropyran, piperazine, and morpholine; 1.40 Compound 1.39, wherein the spiro-joined 3-6-membered heterocycloalkyl is aziridine; 1.41 Compound I, or any of 1.35-1.40, wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is unsubstituted; 1.42 Compound I, or any of 1.35-1.40, wherein said spiro-joined C3-6cycloalkyl or 3- 6-membered heterocycloalkyl is substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; 1.43 Compound I, or any of 1.1-1.42, wherein Z is a bond; 1.44 Compound I, or any of 1.1-1.42, wherein Z is S, S(O), or S(O)2; 1.45 Compound I, or any of 1.1-1.42, wherein Z is O; 1.46 Compound I, or any of 1.1-1.42, wherein Z is NH; 1.47 Compound I, or any of 1.1-1.42, wherein Z is N(Ra), e.g., N(CH3); 1.48 Compound I, or any of 1.1-1.42, wherein Z is -C(O)-;
1.49 Compound I, or any of 1.1-1.42, wherein Z is -C(OH)-, -C(OC1-6alkyl), -C(=N- OH)-, -C(=N-OC1-6alkyl)-, optionally wherein said C1-6alkyl is methyl; 1.50 Compound I, or any of 1.1-1.42, wherein Z is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane); 1.51 Compound 1.50, wherein the spiro-joined C3-6cycloalkyl is selected from cyclopropane, cyclobutane, cyclopentane, and cyclohexane; 1.52 Compound 1.50, wherein the spiro-joined C3-6cycloalkyl is cyclopropane 1.53 Compound I, or any of 1.1-1.42, wherein Z is spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane); 1.54 Compound 1.53, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine, azetidine, oxetane, pyrrolidine, tetrahydrofuran, piperidine, tetrahydropyran, piperazine, and morpholine; 1.55 Compound 1.53, wherein the spiro-joined 3-6-membered heterocycloalkyl is aziridine; 1.56 Compound I, or any of 1.49-1.55, wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is unsubstituted; 1.57 Compound I, or any of 1.49-1.55, wherein said spiro-joined C3-6cycloalkyl or 3- 6-membered heterocycloalkyl is substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; 1.58 Compound I, or any of 1.1-1.57, wherein A is a 6-10 membered aryl ring, e.g., selected from phenyl and naphthyl, substituted by 0-5 groups R; 1.59 Compound I, or any of 1.1-1.57, wherein A is a 5-10 membered heteroaryl ring, substituted by 0-5 groups R; 1.60 Compound 1.59, wherein A is selected from furan, thiophene (e.g., thiophen-2- yl), pyrrole, oxazole, thiazole, imidazole, isoxazole, isothiazole, pyrazole, pyridine (e.g., pyrid-4-yl), 2-oxopyridine (e.g., 2-oxopyridin-1(2H)-yl), pyrimidine, pyridazine, pyrazine, benzofuran (e.g., benzofuran-4-yl, or benzofuran-7-yl, or 2-methylbenzofuran-4-yl), dihydrobenzofuran (e.g., 2,3- dihydrobenzofuran-7-yl), benzothiophene, indole (e.g., indol-1-yl, indol-3-yl, or
indol-5-yl), benzoxazole, benzothiazole, benzimidazole (e.g., benzo[d]imidazol-1- yl), benzisoxazole (e.g., benzo[d]isoxazol-3-yl, or benzo[d]isoxazol-4-yl), benzisothiazole (e.g., benzo[d]isothiazol-3-yl), benzotriazole (e.g., benzo[d][1,2,3-triazol-1-yl), indazole (e.g., indazol-1-yl, indazol-3-yl, or indazol- 7-yl), quinoline (e.g., quinolin-8-yl), isoquinoline (e.g., isoquinolin-7-yl), quinazoline (e.g., quinazolin-7-yl), and quinoxaline (e.g., quinoxalin-5-yl); 1.61 Compound 1.60, wherein A is substituted by 0 groups R; 1.62 Compound 1.60, wherein A is substituted by 1 group R; 1.63 Compound 1.60, wherein A is substituted by 2 groups R; 1.64 Compound 1.58, wherein A is a phenyl ring, substituted by 0-5 groups R; 1.65 Compound 1.64, wherein there is one group R; 1.66 Compound 1.65, wherein the group R is positioned at the para position of the phenyl ring; 1.67 Compound 1.65, wherein the group R is positioned at the meta position of the phenyl ring; 1.68 Compound 1.65, wherein the group R is positioned at the ortho position of the phenyl ring; 1.69 Compound 1.64, wherein there are two groups R; 1.70 Compound 1.69, wherein the groups R are positioned at the ortho and para positions of the phenyl ring; 1.71 Compound 1.69, wherein the groups R are positioned at the meta and para positions of the phenyl ring; 1.72 Compound 1.69, wherein the groups R are positioned at the ortho and meta positions on the same side of the phenyl ring; 1.73 Compound 1.69, wherein the groups R are positioned at the ortho and meta positions on opposite sides of the phenyl ring; 1.74 Compound 1.69, wherein the groups R are positioned at the two ortho positions of the phenyl ring; 1.75 Compound 1.69, wherein the groups R are positioned at the two meta positions of the phenyl ring; 1.76 Compound 1.64, wherein there are three groups R;
1.77 Compound 1.76, wherein the groups R are positioned at the two ortho positions and the para position of the phenyl ring; 1.78 Compound 1.64, wherein there are four groups R; 1.79 Compound 1.64, wherein there are five groups R; 1.80 Compound I, or any of 1.1-1.79, wherein each group R is independently selected from methyl, ethyl, trifluoromethyl, methoxy, ethoxy, F, Cl, cyano, hydroxy, 2- methoxyethoxy, methylsulfonyl, methylthio, cyclopropoxy, cyclopropylmethoxy, methylamino, 4-fluorophenoxy, and (4-fluorobenzyl)oxy; 1.81 Compound I, or any of 1.1-1.80, wherein A is selected from the group consisting of: phenyl, 2-cyanophenyl, 3-cyanophenyl, 4-cyanophenyl, 2-methylphenyl, 2- ethylphenyl, 3-ethylphenyl, 4-ethylphenyl, 2-chlorophenyl, 3-chlorophenyl, 4- fluorophenyl, 3-chloro-4-fluorophenyl, 2-cyano-4-fluorophenyl, 3-cyano-4- fluorophenyl, 2-methyl-4-fluorophenyl, 3-methyl-4-fluorophenyl, 2-methoxy-4- fluorophenyl, 2-methoxy-5-fluorophenyl, 2-fluoro-4-methylphenyl, 2- methoxyphenyl, 3-methoxyphenyl, 4-methoxyphenyl, 2-ethoxyphenyl, 3- ethoxyphenyl, 2-hydroxyphenyl, 2,5-dimethoxyphenyl, 2- trifluoromethyoxyphenyl, 3-trifluoromethylphenyl, 2-(methylsulfonyl)phenyl, 3- (methylthio)phenyl, 4-(methoxyethoxy)phenyl, 4-(4-fluorobenzyloxy)phenyl, 4- (4-fluorophenoxy)phenyl, 3-cyclopropoxyphenyl, 3-(cyclopropylmethoxy)phenyl, and 2-(methylamino)phenyl; 1.82 Compound I, or any of 1.1-1.80, wherein A is selected from the group consisting of: pyrid-4-yl, thiophen-2-yl, indol-1-yl, indol-3-yl, 5-fluoroindol-3-yl, indazol-1- yl, indazol-3-yl, indazol-7-yl, benzofuran-4-yl, benzofuran-7-yl, 2,3- dihydrobenzofuran-7-yl, 2-methylbenzofuran-7-yl, benzo[d]isoxazol-3-yl, benzo[d]isoxazol-4-yl, benzo[d]isoxazol-7-yl, 6-fluorobenzo[d]isoxazol-3-yl, benzo[d]isothiazol-3-yl, benzo[d]imidazole-1-yl, benzo[d][1,2,3]triazol-1-yl, isoquinolin-7-yl, quinolin-8-yl, quinoxaline-5-yl, quinazolin-7-yl, and 2- oxopyridin-1(2H)-yl; 1.83 Compound I, or any of 1.1-1.80, wherein A is selected from the group consisting of: phenyl, 2-ethylphenyl, 4-fluorophenyl, 2-methoxyphenyl, 3-methoxyphenyl, benzofuran-7-yl, benzo[d]isoxazol-3-yl, and benzo[d]isothiazol-3-yl;
1.84 Compound I, or any of 1.1-1.83, wherein m is 1; 1.85 Compound I, or any of 1.1-1.83, wherein m is 2; 1.86 Compound I, or any of 1.1-1.85, wherein n is 2; 1.87 Compound I, or any of 1.1-1.85, wherein n is 3; 1.88 Compound I, or any of 1.1-1.85, wherein n is 4; 1.89 Compound I, or any of 1.1-1.85, wherein n is 5; 1.90 Compound I, or any of 1.1-1.89, wherein X is S, O, CH2, NH, N(CH3), or spiro- joined cyclopropyl; Y is CH2, C(O), or spiro-joined cyclopropyl, and Z is a bond, -O-, -C(O)-, -O(CH2)2O-, or -C(=NOCH3)-; 1.91 Compound I, or any of 1.1-1.89, wherein X is N(CH3) or spiro-joined cyclopropyl; Y is CH2, C(O), or spiro-joined cyclopropyl, and Z is a bond, -O-, or -C(O)-; 1.92 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ia:
wherein:
X is S, S(O), S(O)2, O, CH2, CHRb, C(Rb)2, NH, N(Ra) (e.g., N(CH3)), N-C(O)-Ra, N- C(O)-O-Ra, N-C(O)-O-CH2-O-Ra, N-CH2-O-C(O)-Ra, N+(=O–), a spiro-joined C3- 6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3- 6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Y is CH2, CHRc, -C(O)-, C(Rc)2, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3- 6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Z is a bond, -S-, S(O), S(O)2, -O-, -NH, N(Rd), -C(O)-, -C(OH)-, -C(OC1-6alkyl), -C(=N- OH)-, -C(=N-OC1-6alkyl)-, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), a spiro- joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), or -O(CH2)pO- wherein p is 2, 3, or 4 (e.g., p is 2), wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; A is H, C3-6cycloalkyl (e.g., cyclopropyl or cyclohexyl), aryl (e.g., phenyl), or heteroaryl, wherein said cycloalkyl, aryl, or heteroaryl is substituted by 0-5 groups R; each R is independently selected from aryl (e.g., phenyl), aryloxy (e.g., phenoxy), heteroaryl (e.g., pyridyl), C1-6alkyl (e.g., methyl, ethyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy, ethoxy), C1-6alkylthio (e.g., methylthio), halo (e.g., F), cyano, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), or hydroxy, wherein each of said aryl, heteroaryl, alkyl, haloalkyl, alkylsulfonyl, alkoxy, alkylthio, cycloalkyl, or cycloalkoxy is optionally further substituted by one or more groups selected from aryl (optionally substituted with halo), halo, C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy), C1-
6alkylthio (e.g., methylthio), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), amino, C1-6alkylamino (e.g., methylamino), di(C1-6alkyl)amino (e.g., dimethylamino), (C1-6alkyl)(C1-6alkyl)amino (e.g., methylethylamino), and hydroxy; Ra and Rd, are each independently selected from C1-20alkyl (e.g., methyl or tert-butyl), and C1-2alkylaryl (e.g., benzyl or phenethyl); Rb and Rc are each independently selected from C1-6alkyl (e.g., methyl, ethyl, tert-butyl), C1-6alkoxy, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and C1-2alkylaryl (e.g., benzyl or phenethyl); m is 1 or 2; n is 1, 2, 3, 4, or 5; in free or salt form (e.g., pharmaceutically acceptable salt form); provided that n is not 3 when Z is -C(O)-, X is CH2 or O, and m is 2; and provided that n is not 3 when Z is -C(O)-, X is CH2, and m is 1; and provided that n is not 3 when Z is -C(O)- or -O-, X is NH or N(Ra), and m is 1; and provided that n is not 3 when Z is O, X is NCH3, Y is -C(O)-, and m is 1. [00022] The present disclosure further provides additional embodiments of first aspect, including: 1.1 Compound I, wherein X is S, S(O), or S(O)2; 1.2 Compound I, wherein X is O; 1.3 Compound I, wherein X is CH2, CHRb, or C(Rb)2; 1.4 Compound 1.3, wherein Rb is independently C1-6alkyl (e.g., methyl); 1.5 Compound I, wherein X is CH2; 1.6 Compound I, wherein X is NH; 1.7 Compound I, wherein X is N(Ra); 1.8 Compound I, wherein X is N-C(O)-Ra; 1.9 Compound I, wherein X is N-C(O)-O-Ra; 1.10 Compound I, wherein X is N-C(O)-O-CH2-O-Ra; 1.11 Compound I, wherein X is N-CH2-O-C(O)-Ra; 1.12 Compound I, or any of 1.7-1.11, wherein Ra is C1-2alkylaryl (e.g., benzyl or phenethyl);
1.13 Compound I, or any of 1.7-1.11, wherein Ra is C1-20alkyl (e.g., methyl or tert- butyl); 1.14 Compound I, or any of 1.7-1.11, wherein Ra is C10-20alkyl (e.g., decyl or dodecyl); 1.15 Compound I, or any of 1.7-1.11, wherein Ra is C1-15alkyl (e.g., hexyl or octyl); 1.16 Compound I, or any of 1.7-1.11, wherein Ra is C7-15alkyl (e.g., heptyl or nonyl); 1.17 Compound I, or any of 1.7-1.11, wherein Ra is C1-6alkyl (e.g., butyl or hexyl); 1.18 Compound I, or any of 1.7-1.11, wherein Ra is C1-4alkyl (e.g., n-butyl or tert- butyl); 1.19 Compound I, or any of 1.7-1.11, wherein Ra is C1-3alkyl (e.g., propyl or isopropyl); 1.20 Compound I, or any of 1.7-1.11, wherein Ra is C1-2alkyl (e.g., methyl or ethyl); 1.21 Compound I, or any of 1.7-1.11, wherein X is N(CH3); 1.22 Compound I, wherein X is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane); 1.23 Compound 1.22, wherein the spiro-joined C3-6cycloalkyl is selected from cyclopropane, cyclobutane, cyclopentane, and cyclohexane; 1.24 Compound 1.22, wherein the spiro-joined C3-6cycloalkyl is cyclopropane; 1.25 Compound I, wherein X is spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane); 1.26 Compound 1.25, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine, azetidine, oxetane, pyrrolidine, tetrahydrofuran, piperidine, tetrahydropyran, piperazine, and morpholine; 1.27 Compound 1.25, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine; 1.28 Compound I, or any of 1.22-1.27, wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is unsubstituted; 1.29 Compound I, or any of 1.22-1.27, wherein said spiro-joined C3-6cycloalkyl or 3- 6-membered heterocycloalkyl is substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; 1.30 Compound I, or any of 1.1-1.29, wherein Y is CH2;
1.31 Compound I, or any of 1.1-1.29, wherein Y is -C(O)-; 1.32 Compound I, or any of 1.1-1.29, wherein Y is CHRc or C(Rc)2; 1.33 Compound 1.32, wherein each Rc is independently C1-6alkyl; 1.34 Compound 1.32, wherein each Rc is independently selected from methyl, ethyl and propyl; 1.35 Compound I, or any of 1.1-1.29, wherein Y is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane); 1.36 Compound 1.35, wherein the spiro-joined C3-6cycloalkyl is selected from cyclopropane, cyclobutane, cyclopentane, and cyclohexane; 1.37 Compound 1.35, wherein the spiro-joined C3-6cycloalkyl is cyclopropane; 1.38 Compound I, or any of 1.1-1.29, wherein Y is spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane); 1.39 Compound 1.38, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine, azetidine, oxetane, pyrrolidine, tetrahydrofuran, piperidine, tetrahydropyran, piperazine, and morpholine; 1.40 Compound 1.39, wherein the spiro-joined 3-6-membered heterocycloalkyl is aziridine; 1.41 Compound I, or any of 1.35-1.40, wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is unsubstituted; 1.42 Compound I, or any of 1.35-1.40, wherein said spiro-joined C3-6cycloalkyl or 3- 6-membered heterocycloalkyl is substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; 1.43 Compound I, or any of 1.1-1.42, wherein Z is a bond; 1.44 Compound I, or any of 1.1-1.42, wherein Z is S, S(O), or S(O)2; 1.45 Compound I, or any of 1.1-1.42, wherein Z is O; 1.46 Compound I, or any of 1.1-1.42, wherein Z is NH; 1.47 Compound I, or any of 1.1-1.42, wherein Z is N(Ra), e.g., N(CH3); 1.48 Compound I, or any of 1.1-1.42, wherein Z is -C(O)-;
1.49 Compound I, or any of 1.1-1.42, wherein Z is -C(OH)-, -C(OC1-6alkyl), -C(=N- OH)-, -C(=N-OC1-6alkyl)-, optionally wherein said C1-6alkyl is methyl; 1.50 Compound I, or any of 1.1-1.42, wherein Z is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane); 1.51 Compound 1.50, wherein the spiro-joined C3-6cycloalkyl is selected from cyclopropane, cyclobutane, cyclopentane, and cyclohexane; 1.52 Compound 1.50, wherein the spiro-joined C3-6cycloalkyl is cyclopropane 1.53 Compound I, or any of 1.1-1.42, wherein Z is spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane); 1.54 Compound 1.53, wherein the spiro-joined 3-6-membered heterocycloalkyl is selected from aziridine, azetidine, oxetane, pyrrolidine, tetrahydrofuran, piperidine, tetrahydropyran, piperazine, and morpholine; 1.55 Compound 1.53, wherein the spiro-joined 3-6-membered heterocycloalkyl is aziridine; 1.56 Compound I, or any of 1.49-1.55, wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is unsubstituted; 1.57 Compound I, or any of 1.49-1.55, wherein said spiro-joined C3-6cycloalkyl or 3- 6-membered heterocycloalkyl is substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; 1.58 Compound I, or any of 1.1-1.57, wherein A is a 6-10 membered aryl ring, e.g., selected from phenyl and naphthyl, substituted by 0-5 groups R; 1.59 Compound I, or any of 1.1-1.57, wherein A is a 5-10 membered heteroaryl ring, substituted by 0-5 groups R; 1.60 Compound 1.59, wherein A is selected from furan, thiophene (e.g., thiophen-2- yl), pyrrole, oxazole, thiazole, imidazole, isoxazole, isothiazole, pyrazole, pyridine (e.g., pyrid-4-yl), 2-oxopyridine (e.g., 2-oxopyridin-1(2H)-yl), pyrimidine, pyridazine, pyrazine, benzofuran (e.g., benzofuran-4-yl, or benzofuran-7-yl, or 2-methylbenzofuran-4-yl), dihydrobenzofuran (e.g., 2,3- dihydrobenzofuran-7-yl), benzothiophene, indole (e.g., indol-1-yl, indol-3-yl, or
indol-5-yl), benzoxazole, benzothiazole, benzimidazole (e.g., benzo[d]imidazol-1- yl), benzisoxazole (e.g., benzo[d]isoxazol-3-yl, or benzo[d]isoxazol-4-yl), benzisothiazole (e.g., benzo[d]isothiazol-3-yl), benzotriazole (e.g., benzo[d][1,2,3-triazol-1-yl), indazole (e.g., indazol-1-yl, indazol-3-yl, or indazol- 7-yl), quinoline (e.g., quinolin-8-yl), isoquinoline (e.g., isoquinolin-7-yl), quinazoline (e.g., quinazolin-7-yl), and quinoxaline (e.g., quinoxalin-5-yl); 1.61 Compound 1.60, wherein A is substituted by 0 groups R; 1.62 Compound 1.60, wherein A is substituted by 1 group R; 1.63 Compound 1.60, wherein A is substituted by 2 groups R; 1.64 Compound 1.58, wherein A is a phenyl ring, substituted by 0-5 groups R; 1.65 Compound 1.64, wherein there is one group R; 1.66 Compound 1.65, wherein the group R is positioned at the para position of the phenyl ring; 1.67 Compound 1.65, wherein the group R is positioned at the meta position of the phenyl ring; 1.68 Compound 1.65, wherein the group R is positioned at the ortho position of the phenyl ring; 1.69 Compound 1.64, wherein there are two groups R; 1.70 Compound 1.69, wherein the groups R are positioned at the ortho and para positions of the phenyl ring; 1.71 Compound 1.69, wherein the groups R are positioned at the meta and para positions of the phenyl ring; 1.72 Compound 1.69, wherein the groups R are positioned at the ortho and meta positions on the same side of the phenyl ring; 1.73 Compound 1.69, wherein the groups R are positioned at the ortho and meta positions on opposite sides of the phenyl ring; 1.74 Compound 1.69, wherein the groups R are positioned at the two ortho positions of the phenyl ring; 1.75 Compound 1.69, wherein the groups R are positioned at the two meta positions of the phenyl ring; 1.76 Compound 1.64, wherein there are three groups R;
1.77 Compound 1.76, wherein the groups R are positioned at the two ortho positions and the para position of the phenyl ring; 1.78 Compound 1.64, wherein there are four groups R; 1.79 Compound 1.64, wherein there are five groups R; 1.80 Compound I, or any of 1.1-1.79, wherein each group R is independently selected from methyl, ethyl, trifluoromethyl, methoxy, ethoxy, F, Cl, cyano, hydroxy, 2- methoxyethoxy, methylsulfonyl, methylthio, cyclopropoxy, cyclopropylmethoxy, methylamino, 4-fluorophenoxy, and (4-fluorobenzyl)oxy; 1.81 Compound I, or any of 1.1-1.80, wherein A is selected from the group consisting of: phenyl, 2-cyanophenyl, 3-cyanophenyl, 4-cyanophenyl, 2-methylphenyl, 2- ethylphenyl, 3-ethylphenyl, 4-ethylphenyl, 2-chlorophenyl, 3-chlorophenyl, 4- fluorophenyl, 3-chloro-4-fluorophenyl, 2-cyano-4-fluorophenyl, 3-cyano-4- fluorophenyl, 2-methyl-4-fluorophenyl, 3-methyl-4-fluorophenyl, 2-methoxy-4- fluorophenyl, 2-methoxy-5-fluorophenyl, 2-fluoro-4-methylphenyl, 2- methoxyphenyl, 3-methoxyphenyl, 4-methoxyphenyl, 2-ethoxyphenyl, 3- ethoxyphenyl, 2-hydroxyphenyl, 2,5-dimethoxyphenyl, 2- trifluoromethyoxyphenyl, 3-trifluoromethylphenyl, 2-(methylsulfonyl)phenyl, 3- (methylthio)phenyl, 4-(methoxyethoxy)phenyl, 4-(4-fluorobenzyloxy)phenyl, 4- (4-fluorophenoxy)phenyl, 3-cyclopropoxyphenyl, 3-(cyclopropylmethoxy)phenyl, and 2-(methylamino)phenyl; 1.82 Compound I, or any of 1.1-1.80, wherein A is selected from the group consisting of: pyrid-4-yl, thiophen-2-yl, indol-1-yl, indol-3-yl, 5-fluoroindol-3-yl, indazol-1- yl, indazol-3-yl, indazol-7-yl, benzofuran-4-yl, benzofuran-7-yl, 2,3- dihydrobenzofuran-7-yl, 2-methylbenzofuran-7-yl, benzo[d]isoxazol-3-yl, benzo[d]isoxazol-4-yl, benzo[d]isoxazol-7-yl, 6-fluorobenzo[d]isoxazol-3-yl, benzo[d]isothiazol-3-yl, benzo[d]imidazole-1-yl, benzo[d][1,2,3]triazol-1-yl, isoquinolin-7-yl, quinolin-8-yl, quinoxaline-5-yl, quinazolin-7-yl, and 2- oxopyridin-1(2H)-yl; 1.83 Compound I, or any of 1.1-1.80, wherein A is selected from the group consisting of: phenyl, 2-ethylphenyl, 4-fluorophenyl, 2-methoxyphenyl, 3-methoxyphenyl, benzofuran-7-yl, benzo[d]isoxazol-3-yl, and benzo[d]isothiazol-3-yl;
1.84 Compound I, or any of 1.1-1.83, wherein m is 1; 1.85 Compound I, or any of 1.1-1.83, wherein m is 2; 1.86 Compound I, or any of 1.1-1.85, wherein n is 2; 1.87 Compound I, or any of 1.1-1.85, wherein n is 3; 1.88 Compound I, or any of 1.1-1.85, wherein n is 4; 1.89 Compound I, or any of 1.1-1.85, wherein n is 5; 1.90 Compound I, or any of 1.1-1.89, wherein X is S, O, CH2, NH, N(CH3), or spiro- joined cyclopropyl; Y is CH2, C(O), or spiro-joined cyclopropyl, and Z is a bond, -O-, -C(O)-, -O(CH2)2O-, or -C(=NOCH3)-; 1.91 Compound I, or any of 1.1-1.89, wherein X is N(CH3) or spiro-joined cyclopropyl; Y is CH2, C(O), or spiro-joined cyclopropyl, and Z is a bond, -O-, or -C(O)-; 1.92 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ia:
 wherein n, Z, and A are defined as in any preceding embodiment; 1.93 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ib:
wherein n, Z, and A are defined as in any preceding embodiment; 1.93 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ib:
 wherein n, Z, and A are defined as in any preceding embodiment; 1.94 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ic:
wherein n, Z, and A are defined as in any preceding embodiment; 1.94 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ic:
 wherein n, Z, and A are defined as in any preceding embodiment;
1.95 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Id:
wherein n, Z, and A are defined as in any preceding embodiment;
1.95 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Id:
 wherein n, Z, and A are defined as in any preceding embodiment; 1.96 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ie:
wherein n, Z, and A are defined as in any preceding embodiment; 1.96 Compound I, or any of 1.1-1.91, wherein the compound of Formula I is a compound of Formula Ie:
 wherein n, Z, and A are defined as in any preceding embodiment; 1.97 Compound I, or any of 1.1-1.96, wherein n is 4 and Z is a bond; 1.98 Compound I, or any of 1.1-1.96, wherein n is 3 and Z is -O- or -C(O)-; 1.99 Compound I, or any of 1.1-1.96, wherein n is 3 and Z is a bond; 1.100 Compound I, or any of 1.1-1.96, wherein n is 2 and Z is -O- or -C(O)-; 1.101 Compound I, or any of 1.1-1.96, wherein n is 2 and Z is a bond; 1.102 Compound I, or any of 1.1-1.96, wherein n is 1 and Z is -O- or -C(O)-; 1.103 Compound I, or any of 1.1-1.96, wherein n is 1 and Z is a bond; 1.104 Compound I, or any of 1.1-1.103, wherein A is H or C3-6cycloalkyl (e.g., cyclopropyl or cyclohexyl), Z is a bond or -C(O)-, and n is 1, 2, or 3; 1.105 Compound I, or any of 1.1-1.103, wherein the compound is selected from the group consisting of:
wherein n, Z, and A are defined as in any preceding embodiment; 1.97 Compound I, or any of 1.1-1.96, wherein n is 4 and Z is a bond; 1.98 Compound I, or any of 1.1-1.96, wherein n is 3 and Z is -O- or -C(O)-; 1.99 Compound I, or any of 1.1-1.96, wherein n is 3 and Z is a bond; 1.100 Compound I, or any of 1.1-1.96, wherein n is 2 and Z is -O- or -C(O)-; 1.101 Compound I, or any of 1.1-1.96, wherein n is 2 and Z is a bond; 1.102 Compound I, or any of 1.1-1.96, wherein n is 1 and Z is -O- or -C(O)-; 1.103 Compound I, or any of 1.1-1.96, wherein n is 1 and Z is a bond; 1.104 Compound I, or any of 1.1-1.103, wherein A is H or C3-6cycloalkyl (e.g., cyclopropyl or cyclohexyl), Z is a bond or -C(O)-, and n is 1, 2, or 3; 1.105 Compound I, or any of 1.1-1.103, wherein the compound is selected from the group consisting of:


1.106 Compound I, or any of 1.1-1.105, wherein the compound is the compound of any one of Examples 1 to 180, or is selected from the group consisting of:
 wherein the variables are defined as provided any of the following embodiments:
wherein the variables are defined as provided any of the following embodiments:






 each independently in free or pharmaceutically acceptable salt or form, wherein Cyp refers to a spiro-joined cyclopropyl ring; 1.107 Compound I, or any of 1.1-1.105, wherein the compound is selected from the group consisting of:
each independently in free or pharmaceutically acceptable salt or form, wherein Cyp refers to a spiro-joined cyclopropyl ring; 1.107 Compound I, or any of 1.1-1.105, wherein the compound is selected from the group consisting of:
 wherein the variables are defined as provided any of the following embodiments:
wherein the variables are defined as provided any of the following embodiments:

 ; 1.108 Compound I, or any of 1.1-1.107, in free form; 1.109 Compound I, or any of 1.1-1.107, in salt form, e.g., pharmaceutically acceptable salt form; 1.110 Compound I, or any of 1.1-1.107, wherein the compound is in acid addition salt form, for example, hydrochloric or toluenesulfonic acid salt form; 1.111 Compound I, or any of 1.1-1.10, in substantially pure diastereomeric form (i.e., substantially free from other diastereomers); 1.112 Compound I or any of 1.1-1.110, having a diastereomeric excess of greater than 70%, preferably greater than 80%, more preferably greater than 90% and most preferably greater than 95%; 1.113 Compound I or any of 1.1-1.112, in solid form, e.g., in crystal form;
1.114 Compound I or any of 1.1-1.113, in isolated or purified form (e.g., in at least 90% pure form, or at least 95% or at least 98% or at least 99%). 1.115 Compound I or any of 1.1-1.114, wherein the compound has 5-HT2A receptor binding affinity of at least 60% at 100 nM concentration, e.g., at least 70%, or at least 75%, or at least 80%, or at least 85%, or at least 90%, or at least 95%, or at least 98%, at 100 nM concentration; 1.116 Compound I or any of 1.1-1.115, wherein the compound has a 5-HT2A receptor dissociation constant (Kd) of less than 250 nM, or less than 100 nM, or less than 70 nM, or less than 60 nM, or less than 50 nM, or less than 40 nM, or less than 30 nM, or less than 20 nM, or less than 10 nM; 1.117 Compound I or any of 1.1-1.116, wherein the compound is an agonist of beta- arrestin signaling via the 5-HT2A receptor, e.g., a partial agonist or a full agonist; 1.118 Compound 1.116, wherein the compound is a partial agonist of beta-arrestin signaling having an Emax of less than 90%, or less than 80%, or less than 70%, or less than 60%, or less than 50%, or less than 40%, or less than 30%, or less than 20%, or less than 10%, relative to a full agonist (e.g., alpha-methylserotonin); 1.119 Compound 1.117 or 1.118, wherein the compound has an EC50 for 5-HT2A receptor beta-arrestin agonism of less than 500 nM, or less than 200 nM, or less than 150 nM, or less than 100 nM, or less than 70 nM, or less than 60 nM, or less than 50 nM, or less than 40 nM, or less than 30 nM, or less than 20 nM, or less than 10 nM; 1.120 Compound 1.117, 1.118 or 1.119, wherein the compound has a beta-arrestin signaling relative intrinsic activity (RAi) of less than 1.0 compared to the reference compound alpha-methylserotonin, e.g., a relative intrinsic activity of less than 0.8, or less than 0.6, or less than 0.5, or less than 0.4, or less than 0.3, or less than 0.2, or less than 0.1, or 0.1 to 0.8, or 0.2 to 0.8, or 0.4 to 0.8, or 0.5 to 0.8, or 0.2 to 0.6, or 0.2 to 0.5, or 0.2 to 0.4, or 0.5 to 1.0, or 0.5 to 0.9, or 0.5 to 0.8, or 0.6 to 0.9, or 0.6 to 0.8; 1.121 Compound 1.117, 1.118, or 1.119, wherein the compound has a beta-arrestin signaling relative intrinsic activity (RAi) of greater than 1.0 compared to the
reference compound alpha-methylserotonin, e.g., a relative intrinsic activity of 1.0 to 1.2, or 1.0 to 1.4, or 1.0 to 1.6; 1.122 Compound I or any of 1.1-1.116, wherein the compound is an antagonist of beta- arrestin signaling via the 5-HT2A receptor; 1.123 Compound 1.122, wherein the compound has an IC50 for 5-HT2A receptor beta- arrestin antagonism of less than 300 nM, or less than 200 nM, or less than 100 nM, or less than 70 nM, or less than 60 nM, or less than 50 nM, or less than 40 nM, or less than 30 nM, or less than 20 nM, or less than 10 nM; 1.124 Compound I or any of 1.1-1.116, wherein the compound is not an antagonist of beta-arrestin signaling via the 5-HT2A receptor; 1.125 Compound 1.124, wherein the compound has an IC50 for 5-HT2A receptor beta- arrestin antagonism of greater than 10 nM, or greater than 50 nM, or greater than 100 nM, or greater than 250 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 5000 nM, or greater than 10,000 nM; 1.126 Compound I or any of 1.1-1.125, wherein the compound is not an agonist of G-q signaling via the 5-HT2A receptor, or is a weak agonist thereof; 1.127 Compound 1.126, wherein the compound is a partial agonist of G-q signaling having an Emax of less than 90%, or less than 80%, or less than 70%, or less than 60%, or less than 50%, or less than 40%, or less than 30%, or less than 20%, or less than 10%, relative to a full agonist (e.g., alpha-methylserotonin), preferably an Emax of less than 50% or less than 30% or less than 10%; 1.128 Compound 1.125 or 1.126, wherein the compound has an EC50 for 5-HT2A receptor G-q agonism of greater than 10 nM, or greater than 25 nM, or greater than 50 nM, or greater than 100 nM, or greater than 150 nM, or greater than 200 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 2000 nM, or greater than 5000 nM, or greater than 10,000 nM; 1.129 Compound 1.125, 1.126 or 1.127, wherein the compound has a G-q signaling relative intrinsic activity (RAi) of less than 1.0 compared to the reference compound alpha-methylserotonin, e.g., a relative intrinsic activity of less than 0.8, or less than 0.6, or less than 0.5, or less than 0.4, or less than 0.3, or less than 0.2, or less than 0.1, or 0.1 to 0.8, or 0.2 to 0.8, or 0.4 to 0.8, or 0.5 to 0.8, or 0.2 to
0.6, or 0.2 to 0.5, or 0.2 to 0.4, or 0.5 to 1.0, or 0.5 to 0.9, or 0.5 to 0.8, or 0.6 to 0.9, or 0.6 to 0.8; 1.130 Compound I or any of 1.1-1.129, wherein the compound is an antagonist of G-q signaling via the 5-HT2A receptor; 1.131 Compound 1.130, wherein the compound has an IC50 for 5-HT2A receptor G-q antagonism of less than 10 nM, or less than 25 nM, or less than 50 nM, or less than 100 nM, or less than 150 nM, or less than 200 nM, or less than 500 nM; 1.132 Compound I or any of 1.1-1.131, wherein the compound has a bias ratio (beta- arrestin/G-q) for agonism 5-HT2A receptor of at least 2, or at least 5, or at least 10, or at least 25, or at least 50, or at least 100, or at least 150, or at least 200 or at least 500, or at least 1000, or at least 10,000, or undefined (i.e., where the compound has any degree of beta-arrestin agonism and zero G-q agonism); 1.133 Compound I or any of 1.1-1.132, wherein the compound is an antagonist or agonist of the D1 and/or D2 dopamine receptor (e.g., having at least 70% receptor affinity at 100 nM concentration or an IC50 of less than 100 nM); 1.134 Compound I or any of 1.1-1.132, wherein the compound is not active at the D1 and/or D2 dopamine receptor (e.g., having less than 50% receptor affinity at 100 nM concentration and/or an EC50 or IC50 of more than 500 nM); 1.135 Compound I or any of 1.1-1.134, wherein the compound is an antagonist of the serotonin transporter (e.g., having at least 70% receptor binding affinity at 100 nM concentration or an IC50 of less than 100 nM); 1.136 Compound I or any of 1.1-1.134, wherein the compound is not active at the serotonin transporter (e.g., having less than 50% receptor binding affinity at 100 nM concentration and/or an EC50 or IC50 of more than 500 nM); 1.137 Compound I or any of 1.1-1.136, wherein the compound is an agonist, antagonist, or partial agonist of the mu-opioid receptor (e.g., having at least 70% receptor binding affinity at 100 nM concentration or an EC50 or IC50 of less than 100 nM); 1.138 Compound I or any of 1.1-1.136, wherein the compound is not active at the mu- opioid receptor (e.g., having at less than 50% receptor binding affinity at 100 nM concentration and/or an EC50 or IC50 of more than 500 nM);
1.139 Compound I, or any of 1.1-1.138, wherein the compound is non-hallucinogenic, e.g., at therapeutic doses for the treatment of a neuropsychiatric disorder described herein (e.g., depression, anxiety, etc.) the compound does not cause visual or auditory hallucinations, visual distortions (such as drifting, morphing, breathing or melting of objects and surfaces in the field of view), detachment from reality, dissociation, delirium, undesired altered states of consciousness; 1.140 Compound I, or any of 1.1-1.139, wherein the compound does not stimulate head twitch response in an animal test model, or is antagonist of DOI-induced head twitch response; 1.141 Compound I, or any of 1.1-1.140, wherein the compound is effective in a murine model of depression (tail suspension or forced swim test); 1.142 Compound I, or any of 1.1-1.141, wherein the compound is effective in an animal model of social anxiety disorder or anhedonia; 1.143 Compound I, or any of 1.1-1.142, wherein the compound does not have 5-HT2B agonist activity (e.g., an EC50 of greater than 100 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 10,000 nM); 1.144 Compound I, or any of 1.1-1.143, wherein the compound has 5-HT2B antagonist activity (e.g., an IC50 of less than 1000 nM, or less than 500 nM, or less than 250 nM, or less than 100 nM, or less than 50 nM, or less than 25 nM, or less than 15 nM); 1.145 Compound I, or any of 1.1-1.144, wherein the compound has 5-HT2c agonist activity (e.g., an EC50 of less than 1000 nM, or less than 500 nM, or less than 250 nM, or less than 100 nM, or less than 50 nM, or less than 25 nM, or less than 15 nM); 1.146 Compound I, or any of 1.1-1.144, wherein the compound does not have 5-HT2C antagonist activity (e.g., an IC50 of greater than 100 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 10,000 nM); 1.147 Compound I, or any of 1.1-1.146, wherein the compound binds to the alpha-1A adrenergic receptor (e.g., with a binding affinity Ki of less than 1000 nM, or less than 500 nM, or less than 250 nM, or less than 200 nM, or less than 150 nM, or less than 100 nM, or less than 50 nM, or less than 25 nM);
1.148 Compound I, or any of 1.1-1.147, wherein the compound does not cause psychoses (e.g., prolonged or intermittent psychoses); 1.149 Compound I, or any of 1.1-1.148, wherein the compound does not promote self- harm or harm to others in the patient; 1.150 Compound I, or any of 1.1-1.149, wherein the compound does not cause heart valvulopathy or pulmonary arterial hypertension, e.g., wherein the compound is safe to administer to a patient having cardiac comorbidities; 1.151 Compound I, or any of 1.1-1.150, wherein the compound does not cause abuse or dependence (e.g., physical or psychological dependence); 1.152 Compound I, or any of 1.1-1.151, wherein the compound is functionally inactive at one or more of the following receptors and ion channels: adenosine A2A, alpha-1A adrenergic, alpha-2A adrenergic, beta-1 adrenergic, beta-2 adrenergic, GABA-A benzodiazepine site (BZD, central), CB1 cannabinoid, CB2 cannabinoid, cholecystokinin CCK1, endothelin-A (ETA), NMDA, histamine H1, histamine H2, MAO-A, muscarinic M1, muscarinic M2, muscarinic M3, nicotinic acetylcholine (neuronal alpha-4-beta-2), delta opioid, kappa opioid, mu opioid, serotonin-1A, serotonin-1B, serotonin-3, glucocorticoid (GR), androgen (AR), vasopressin V1A, cardiac calcium channel (dihydropyridine site), hERG potassium channel, voltage-gated potassium channel KV, sodium channel (site 2), norepinephrine transporter, dopamine transporter, and/or serotonin transporter; 1.153 Compound 1.152, wherein the compound has an in vitro receptor activity (for agonism or antagonism) of less than 60% inhibition of radioligand binding (e.g., at 100 nM test concentration) for any one or more of said receptor or ion channels, e.g., less than 50%, less than 40%, less than 30%, less than 20%, or less than 10%; 1.154 Compound I, or any of 1.1-1.153, wherein the compound is orally bioavailable (e.g., oral bioavailability of at least 10%, or at least 15%, or at least 20%, or at least 30%, or at least 40%). [00023] As used herein, the term “spiro-joined” is meant to clarify that the stated C3- 6cycloalkyl group or 3-6-membered heterocycloalkyl is present in a spiro-junction, meaning that one atom of said cyclic group is an atom of the ring to which the group is attached. For example,
the follow are examples of compounds of Formula I having spiro-joined cyclic groups within the scope of the present disclosure:
; 1.108 Compound I, or any of 1.1-1.107, in free form; 1.109 Compound I, or any of 1.1-1.107, in salt form, e.g., pharmaceutically acceptable salt form; 1.110 Compound I, or any of 1.1-1.107, wherein the compound is in acid addition salt form, for example, hydrochloric or toluenesulfonic acid salt form; 1.111 Compound I, or any of 1.1-1.10, in substantially pure diastereomeric form (i.e., substantially free from other diastereomers); 1.112 Compound I or any of 1.1-1.110, having a diastereomeric excess of greater than 70%, preferably greater than 80%, more preferably greater than 90% and most preferably greater than 95%; 1.113 Compound I or any of 1.1-1.112, in solid form, e.g., in crystal form;
1.114 Compound I or any of 1.1-1.113, in isolated or purified form (e.g., in at least 90% pure form, or at least 95% or at least 98% or at least 99%). 1.115 Compound I or any of 1.1-1.114, wherein the compound has 5-HT2A receptor binding affinity of at least 60% at 100 nM concentration, e.g., at least 70%, or at least 75%, or at least 80%, or at least 85%, or at least 90%, or at least 95%, or at least 98%, at 100 nM concentration; 1.116 Compound I or any of 1.1-1.115, wherein the compound has a 5-HT2A receptor dissociation constant (Kd) of less than 250 nM, or less than 100 nM, or less than 70 nM, or less than 60 nM, or less than 50 nM, or less than 40 nM, or less than 30 nM, or less than 20 nM, or less than 10 nM; 1.117 Compound I or any of 1.1-1.116, wherein the compound is an agonist of beta- arrestin signaling via the 5-HT2A receptor, e.g., a partial agonist or a full agonist; 1.118 Compound 1.116, wherein the compound is a partial agonist of beta-arrestin signaling having an Emax of less than 90%, or less than 80%, or less than 70%, or less than 60%, or less than 50%, or less than 40%, or less than 30%, or less than 20%, or less than 10%, relative to a full agonist (e.g., alpha-methylserotonin); 1.119 Compound 1.117 or 1.118, wherein the compound has an EC50 for 5-HT2A receptor beta-arrestin agonism of less than 500 nM, or less than 200 nM, or less than 150 nM, or less than 100 nM, or less than 70 nM, or less than 60 nM, or less than 50 nM, or less than 40 nM, or less than 30 nM, or less than 20 nM, or less than 10 nM; 1.120 Compound 1.117, 1.118 or 1.119, wherein the compound has a beta-arrestin signaling relative intrinsic activity (RAi) of less than 1.0 compared to the reference compound alpha-methylserotonin, e.g., a relative intrinsic activity of less than 0.8, or less than 0.6, or less than 0.5, or less than 0.4, or less than 0.3, or less than 0.2, or less than 0.1, or 0.1 to 0.8, or 0.2 to 0.8, or 0.4 to 0.8, or 0.5 to 0.8, or 0.2 to 0.6, or 0.2 to 0.5, or 0.2 to 0.4, or 0.5 to 1.0, or 0.5 to 0.9, or 0.5 to 0.8, or 0.6 to 0.9, or 0.6 to 0.8; 1.121 Compound 1.117, 1.118, or 1.119, wherein the compound has a beta-arrestin signaling relative intrinsic activity (RAi) of greater than 1.0 compared to the
reference compound alpha-methylserotonin, e.g., a relative intrinsic activity of 1.0 to 1.2, or 1.0 to 1.4, or 1.0 to 1.6; 1.122 Compound I or any of 1.1-1.116, wherein the compound is an antagonist of beta- arrestin signaling via the 5-HT2A receptor; 1.123 Compound 1.122, wherein the compound has an IC50 for 5-HT2A receptor beta- arrestin antagonism of less than 300 nM, or less than 200 nM, or less than 100 nM, or less than 70 nM, or less than 60 nM, or less than 50 nM, or less than 40 nM, or less than 30 nM, or less than 20 nM, or less than 10 nM; 1.124 Compound I or any of 1.1-1.116, wherein the compound is not an antagonist of beta-arrestin signaling via the 5-HT2A receptor; 1.125 Compound 1.124, wherein the compound has an IC50 for 5-HT2A receptor beta- arrestin antagonism of greater than 10 nM, or greater than 50 nM, or greater than 100 nM, or greater than 250 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 5000 nM, or greater than 10,000 nM; 1.126 Compound I or any of 1.1-1.125, wherein the compound is not an agonist of G-q signaling via the 5-HT2A receptor, or is a weak agonist thereof; 1.127 Compound 1.126, wherein the compound is a partial agonist of G-q signaling having an Emax of less than 90%, or less than 80%, or less than 70%, or less than 60%, or less than 50%, or less than 40%, or less than 30%, or less than 20%, or less than 10%, relative to a full agonist (e.g., alpha-methylserotonin), preferably an Emax of less than 50% or less than 30% or less than 10%; 1.128 Compound 1.125 or 1.126, wherein the compound has an EC50 for 5-HT2A receptor G-q agonism of greater than 10 nM, or greater than 25 nM, or greater than 50 nM, or greater than 100 nM, or greater than 150 nM, or greater than 200 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 2000 nM, or greater than 5000 nM, or greater than 10,000 nM; 1.129 Compound 1.125, 1.126 or 1.127, wherein the compound has a G-q signaling relative intrinsic activity (RAi) of less than 1.0 compared to the reference compound alpha-methylserotonin, e.g., a relative intrinsic activity of less than 0.8, or less than 0.6, or less than 0.5, or less than 0.4, or less than 0.3, or less than 0.2, or less than 0.1, or 0.1 to 0.8, or 0.2 to 0.8, or 0.4 to 0.8, or 0.5 to 0.8, or 0.2 to
0.6, or 0.2 to 0.5, or 0.2 to 0.4, or 0.5 to 1.0, or 0.5 to 0.9, or 0.5 to 0.8, or 0.6 to 0.9, or 0.6 to 0.8; 1.130 Compound I or any of 1.1-1.129, wherein the compound is an antagonist of G-q signaling via the 5-HT2A receptor; 1.131 Compound 1.130, wherein the compound has an IC50 for 5-HT2A receptor G-q antagonism of less than 10 nM, or less than 25 nM, or less than 50 nM, or less than 100 nM, or less than 150 nM, or less than 200 nM, or less than 500 nM; 1.132 Compound I or any of 1.1-1.131, wherein the compound has a bias ratio (beta- arrestin/G-q) for agonism 5-HT2A receptor of at least 2, or at least 5, or at least 10, or at least 25, or at least 50, or at least 100, or at least 150, or at least 200 or at least 500, or at least 1000, or at least 10,000, or undefined (i.e., where the compound has any degree of beta-arrestin agonism and zero G-q agonism); 1.133 Compound I or any of 1.1-1.132, wherein the compound is an antagonist or agonist of the D1 and/or D2 dopamine receptor (e.g., having at least 70% receptor affinity at 100 nM concentration or an IC50 of less than 100 nM); 1.134 Compound I or any of 1.1-1.132, wherein the compound is not active at the D1 and/or D2 dopamine receptor (e.g., having less than 50% receptor affinity at 100 nM concentration and/or an EC50 or IC50 of more than 500 nM); 1.135 Compound I or any of 1.1-1.134, wherein the compound is an antagonist of the serotonin transporter (e.g., having at least 70% receptor binding affinity at 100 nM concentration or an IC50 of less than 100 nM); 1.136 Compound I or any of 1.1-1.134, wherein the compound is not active at the serotonin transporter (e.g., having less than 50% receptor binding affinity at 100 nM concentration and/or an EC50 or IC50 of more than 500 nM); 1.137 Compound I or any of 1.1-1.136, wherein the compound is an agonist, antagonist, or partial agonist of the mu-opioid receptor (e.g., having at least 70% receptor binding affinity at 100 nM concentration or an EC50 or IC50 of less than 100 nM); 1.138 Compound I or any of 1.1-1.136, wherein the compound is not active at the mu- opioid receptor (e.g., having at less than 50% receptor binding affinity at 100 nM concentration and/or an EC50 or IC50 of more than 500 nM);
1.139 Compound I, or any of 1.1-1.138, wherein the compound is non-hallucinogenic, e.g., at therapeutic doses for the treatment of a neuropsychiatric disorder described herein (e.g., depression, anxiety, etc.) the compound does not cause visual or auditory hallucinations, visual distortions (such as drifting, morphing, breathing or melting of objects and surfaces in the field of view), detachment from reality, dissociation, delirium, undesired altered states of consciousness; 1.140 Compound I, or any of 1.1-1.139, wherein the compound does not stimulate head twitch response in an animal test model, or is antagonist of DOI-induced head twitch response; 1.141 Compound I, or any of 1.1-1.140, wherein the compound is effective in a murine model of depression (tail suspension or forced swim test); 1.142 Compound I, or any of 1.1-1.141, wherein the compound is effective in an animal model of social anxiety disorder or anhedonia; 1.143 Compound I, or any of 1.1-1.142, wherein the compound does not have 5-HT2B agonist activity (e.g., an EC50 of greater than 100 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 10,000 nM); 1.144 Compound I, or any of 1.1-1.143, wherein the compound has 5-HT2B antagonist activity (e.g., an IC50 of less than 1000 nM, or less than 500 nM, or less than 250 nM, or less than 100 nM, or less than 50 nM, or less than 25 nM, or less than 15 nM); 1.145 Compound I, or any of 1.1-1.144, wherein the compound has 5-HT2c agonist activity (e.g., an EC50 of less than 1000 nM, or less than 500 nM, or less than 250 nM, or less than 100 nM, or less than 50 nM, or less than 25 nM, or less than 15 nM); 1.146 Compound I, or any of 1.1-1.144, wherein the compound does not have 5-HT2C antagonist activity (e.g., an IC50 of greater than 100 nM, or greater than 500 nM, or greater than 1000 nM, or greater than 10,000 nM); 1.147 Compound I, or any of 1.1-1.146, wherein the compound binds to the alpha-1A adrenergic receptor (e.g., with a binding affinity Ki of less than 1000 nM, or less than 500 nM, or less than 250 nM, or less than 200 nM, or less than 150 nM, or less than 100 nM, or less than 50 nM, or less than 25 nM);
1.148 Compound I, or any of 1.1-1.147, wherein the compound does not cause psychoses (e.g., prolonged or intermittent psychoses); 1.149 Compound I, or any of 1.1-1.148, wherein the compound does not promote self- harm or harm to others in the patient; 1.150 Compound I, or any of 1.1-1.149, wherein the compound does not cause heart valvulopathy or pulmonary arterial hypertension, e.g., wherein the compound is safe to administer to a patient having cardiac comorbidities; 1.151 Compound I, or any of 1.1-1.150, wherein the compound does not cause abuse or dependence (e.g., physical or psychological dependence); 1.152 Compound I, or any of 1.1-1.151, wherein the compound is functionally inactive at one or more of the following receptors and ion channels: adenosine A2A, alpha-1A adrenergic, alpha-2A adrenergic, beta-1 adrenergic, beta-2 adrenergic, GABA-A benzodiazepine site (BZD, central), CB1 cannabinoid, CB2 cannabinoid, cholecystokinin CCK1, endothelin-A (ETA), NMDA, histamine H1, histamine H2, MAO-A, muscarinic M1, muscarinic M2, muscarinic M3, nicotinic acetylcholine (neuronal alpha-4-beta-2), delta opioid, kappa opioid, mu opioid, serotonin-1A, serotonin-1B, serotonin-3, glucocorticoid (GR), androgen (AR), vasopressin V1A, cardiac calcium channel (dihydropyridine site), hERG potassium channel, voltage-gated potassium channel KV, sodium channel (site 2), norepinephrine transporter, dopamine transporter, and/or serotonin transporter; 1.153 Compound 1.152, wherein the compound has an in vitro receptor activity (for agonism or antagonism) of less than 60% inhibition of radioligand binding (e.g., at 100 nM test concentration) for any one or more of said receptor or ion channels, e.g., less than 50%, less than 40%, less than 30%, less than 20%, or less than 10%; 1.154 Compound I, or any of 1.1-1.153, wherein the compound is orally bioavailable (e.g., oral bioavailability of at least 10%, or at least 15%, or at least 20%, or at least 30%, or at least 40%). [00023] As used herein, the term “spiro-joined” is meant to clarify that the stated C3- 6cycloalkyl group or 3-6-membered heterocycloalkyl is present in a spiro-junction, meaning that one atom of said cyclic group is an atom of the ring to which the group is attached. For example,
the follow are examples of compounds of Formula I having spiro-joined cyclic groups within the scope of the present disclosure:
 In each of the above examples, the cyclopropane, cyclobutane, aziridine, azetidine, or oxetane, may be replaced by any other C3-6cycloalkyl or 3-6-membered heterocycloalkyl, including, but not limited to, cyclopentane, cyclohexane, tetrahydrofuran, tetrahydropyran, pyrrolidine, piperidine, piperazine, or morpholine. [00024] As used hereinbelow, the “Compound of the Invention” refers to a Compound of Formula I or any of 1.1-1.154. [00025] In a second aspect, the present disclosure provides a pharmaceutical composition (Pharmaceutical Composition 1) comprising Compound of the Invention, e.g., in admixture with a pharmaceutically acceptable diluent or carrier. In a particular embodiment, the Compound of the Invention is in pharmaceutically acceptable salt form. In some embodiments, the pharmaceutical composition is in the form of a tablet or capsule, e.g., for gastroenteric absorption (i.e., absorption through the stomach and/or large and small intestines). In some embodiments, the pharmaceutical composition is an oral transmucosal composition, e.g., an orally dissolving tablet, wafer, film, gel or spray. For example, the composition may be a rapidly-dissolving sublingual or buccal tablet, wafer, film, or gel. In some embodiments, the pharmaceutical composition is formulated for intranasal or intrapulmonary administration (e.g., as an aerosol, mist, or powder for inhalation). In some embodiments, the pharmaceutical composition is formulated for intravenous, intrathecal, intramuscular, subcutaneous or intraperitoneal injection. In particular, pharmaceutical compositions for intramuscular or subcutaneous injection may be in the form of long-acting injectable compositions or depot compositions, e.g., providing for sustained or delayed release of the Compound of the Invention into the blood stream and body tissues. Alternatively, particularly as formulated for intravenous, intrathecal, intraperitoneal, or subcutaneous injection, the composition may be an immediate-acting composition, e.g., providing immediate release into the body fluids of the majority or entirety of the dose. [00026] In a further embodiment, the Pharmaceutical Compositions of the present disclosure, are for a sustained or delayed release formulation (Pharmaceutical Composition 1-A), e.g., a depot formulation. In some embodiments, the Compound of the Invention is provided, preferably in free or pharmaceutically acceptable salt form, in admixture with a pharmaceutically acceptable diluent or carrier, in the form of an injectable depot, which provides sustained or delayed release of the compound.
[00027] In a particular embodiment, the Pharmaceutical Composition 1-A comprises a Compound of the Invention, in free base or pharmaceutically acceptable salt form, optionally in crystal form, wherein the compound has been milled to, or the compound crystallized to, microparticle or nanoparticle size, e.g., particles or crystals having a volume-based particle size (e.g., diameter or Dv50) of 0.5 to 100 microns, for example, for example, 5-30 microns, 10-20 microns, 20-100 microns, 20-50 microns or 30-50 microns. Such particles or crystals may be combined with a suitable pharmaceutically acceptable diluent or carrier, for example water, to form a depot formulation for injection. For example, the depot formulation may be formulated for intramuscular or subcutaneous injection with a dosage of drug suitable for 4 to 6 weeks of treatment. In some embodiments, the particles or crystals have a surface area of 0.1 to 5 m2/g, for example, 0.5 to 3.3 m2/g or from 0.8 to 1.2 m2/g. [00028] In another embodiment, the present disclosure provides a Pharmaceutical Composition 1-B, which is Pharmaceutical Composition 1, wherein Compound of the Invention, is in a polymeric matrix. In one embodiment, the Compound of the present disclosure is dispersed or dissolved within the polymeric matrix. In a further embodiment, the polymeric matrix comprises standard polymers used in depot formulations such as polymers selected from a polyester of a hydroxy fatty acid and derivatives thereof, or a polymer of an alkyl alpha- cyanoacrylate, a polyalkylene oxalate, a polyortho ester, a polycarbonate, a polyortho-carbonate, a polyamino acid, a hyaluronic acid ester, and mixtures thereof. In a further embodiment, the polymer is selected from a group consisting of polylactide, poly d,l-lactide, poly glycolide, PLGA 50:50, PLGA 85:15 and PLGA 90:10 polymer. In another embodiment, the polymer is selected form poly(glycolic acid), poly-D,L-lactic acid, poly-L-lactic acid, copolymers of the foregoing, poly(aliphatic carboxylic acids), copolyoxalates, polycaprolactone, polydioxanone, poly(ortho carbonates), poly(acetals), poly(lactic acid-caprolactone), polyorthoesters, poly(glycolic acid-caprolactone), polyanhydrides, and natural polymers including albumin, casein, and waxes, such as, glycerol mono- and distearate, and the like. In a preferred embodiment, the polymeric matrix comprises poly(d,l-lactide-co-glycolide). [00029] The Pharmaceutical Composition 1-B is particularly useful for sustained or delayed release, wherein the Compound of the present disclosure is released upon degradation of the polymeric matrix. These Compositions may be formulated for controlled- and/or sustained- release of the Compounds of the present disclosure (e.g., as a depot composition) over a period
of up to 180 days, e.g., from about 14 to about 30 to about 180 days. For example, the polymeric matrix may degrade and release the Compounds of the present disclosure over a period of about 30, about 60 or about 90 days. In another example, the polymeric matrix may degrade and release the Compounds of the present disclosure over a period of about 120, or about 180 days. [00030] In still another embodiment, the Pharmaceutical Composition 1 or 1-A or 1-B may be formulated for administration by injection, for example, as a sterile solution. [00031] In another embodiment, the present disclosure provides a Pharmaceutical Composition (Pharmaceutical Composition 1-C) comprising a Compound of the Invention as hereinbefore described, in an osmotic controlled release oral delivery system (OROS), which is described in US 2001/0036472 and US 2009/0202631, the contents of each of which applications are incorporated by reference in their entirety. Therefore in one embodiment, the present disclosure provides a pharmaceutical composition or device comprising (a) a gelatin capsule containing a Compound of any of Formulae I et seq. in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier; (b) a multilayer wall superposed on the gelatin capsule comprising, in outward order from the capsule: (i) a barrier layer, (ii) an expandable layer, and (iii) a semipermeable layer; and (c) and orifice formed or formable through the wall (Pharmaceutical Composition P.1). [00032] In another embodiment, the invention provides a pharmaceutical composition comprising a gelatin capsule containing a liquid, the Compound of the Invention in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier, the gelatin capsule being surrounded by a composite wall comprising a barrier layer contacting the external surface of the gelatin capsule, an expandable layer contacting the barrier layer, a semi-permeable layer encompassing the expandable layer, and an exit orifice formed or formable in the wall (Pharmaceutical Composition P.2). [00033] In still another embodiment, the invention provides a composition comprising a gelatin capsule containing a liquid, the Compound of the Invention in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier , the gelatin capsule being surrounded by a composite wall comprising a barrier layer contacting the external surface of the gelatin capsule, an expandable layer contacting the barrier layer, a semipermeable layer encompassing the expandable layer, and an exit orifice formed or
formable in the wall, wherein the barrier layer forms a seal between the expandable layer and the environment at the exit orifice (Pharmaceutical Composition P.3).
In each of the above examples, the cyclopropane, cyclobutane, aziridine, azetidine, or oxetane, may be replaced by any other C3-6cycloalkyl or 3-6-membered heterocycloalkyl, including, but not limited to, cyclopentane, cyclohexane, tetrahydrofuran, tetrahydropyran, pyrrolidine, piperidine, piperazine, or morpholine. [00024] As used hereinbelow, the “Compound of the Invention” refers to a Compound of Formula I or any of 1.1-1.154. [00025] In a second aspect, the present disclosure provides a pharmaceutical composition (Pharmaceutical Composition 1) comprising Compound of the Invention, e.g., in admixture with a pharmaceutically acceptable diluent or carrier. In a particular embodiment, the Compound of the Invention is in pharmaceutically acceptable salt form. In some embodiments, the pharmaceutical composition is in the form of a tablet or capsule, e.g., for gastroenteric absorption (i.e., absorption through the stomach and/or large and small intestines). In some embodiments, the pharmaceutical composition is an oral transmucosal composition, e.g., an orally dissolving tablet, wafer, film, gel or spray. For example, the composition may be a rapidly-dissolving sublingual or buccal tablet, wafer, film, or gel. In some embodiments, the pharmaceutical composition is formulated for intranasal or intrapulmonary administration (e.g., as an aerosol, mist, or powder for inhalation). In some embodiments, the pharmaceutical composition is formulated for intravenous, intrathecal, intramuscular, subcutaneous or intraperitoneal injection. In particular, pharmaceutical compositions for intramuscular or subcutaneous injection may be in the form of long-acting injectable compositions or depot compositions, e.g., providing for sustained or delayed release of the Compound of the Invention into the blood stream and body tissues. Alternatively, particularly as formulated for intravenous, intrathecal, intraperitoneal, or subcutaneous injection, the composition may be an immediate-acting composition, e.g., providing immediate release into the body fluids of the majority or entirety of the dose. [00026] In a further embodiment, the Pharmaceutical Compositions of the present disclosure, are for a sustained or delayed release formulation (Pharmaceutical Composition 1-A), e.g., a depot formulation. In some embodiments, the Compound of the Invention is provided, preferably in free or pharmaceutically acceptable salt form, in admixture with a pharmaceutically acceptable diluent or carrier, in the form of an injectable depot, which provides sustained or delayed release of the compound.
[00027] In a particular embodiment, the Pharmaceutical Composition 1-A comprises a Compound of the Invention, in free base or pharmaceutically acceptable salt form, optionally in crystal form, wherein the compound has been milled to, or the compound crystallized to, microparticle or nanoparticle size, e.g., particles or crystals having a volume-based particle size (e.g., diameter or Dv50) of 0.5 to 100 microns, for example, for example, 5-30 microns, 10-20 microns, 20-100 microns, 20-50 microns or 30-50 microns. Such particles or crystals may be combined with a suitable pharmaceutically acceptable diluent or carrier, for example water, to form a depot formulation for injection. For example, the depot formulation may be formulated for intramuscular or subcutaneous injection with a dosage of drug suitable for 4 to 6 weeks of treatment. In some embodiments, the particles or crystals have a surface area of 0.1 to 5 m2/g, for example, 0.5 to 3.3 m2/g or from 0.8 to 1.2 m2/g. [00028] In another embodiment, the present disclosure provides a Pharmaceutical Composition 1-B, which is Pharmaceutical Composition 1, wherein Compound of the Invention, is in a polymeric matrix. In one embodiment, the Compound of the present disclosure is dispersed or dissolved within the polymeric matrix. In a further embodiment, the polymeric matrix comprises standard polymers used in depot formulations such as polymers selected from a polyester of a hydroxy fatty acid and derivatives thereof, or a polymer of an alkyl alpha- cyanoacrylate, a polyalkylene oxalate, a polyortho ester, a polycarbonate, a polyortho-carbonate, a polyamino acid, a hyaluronic acid ester, and mixtures thereof. In a further embodiment, the polymer is selected from a group consisting of polylactide, poly d,l-lactide, poly glycolide, PLGA 50:50, PLGA 85:15 and PLGA 90:10 polymer. In another embodiment, the polymer is selected form poly(glycolic acid), poly-D,L-lactic acid, poly-L-lactic acid, copolymers of the foregoing, poly(aliphatic carboxylic acids), copolyoxalates, polycaprolactone, polydioxanone, poly(ortho carbonates), poly(acetals), poly(lactic acid-caprolactone), polyorthoesters, poly(glycolic acid-caprolactone), polyanhydrides, and natural polymers including albumin, casein, and waxes, such as, glycerol mono- and distearate, and the like. In a preferred embodiment, the polymeric matrix comprises poly(d,l-lactide-co-glycolide). [00029] The Pharmaceutical Composition 1-B is particularly useful for sustained or delayed release, wherein the Compound of the present disclosure is released upon degradation of the polymeric matrix. These Compositions may be formulated for controlled- and/or sustained- release of the Compounds of the present disclosure (e.g., as a depot composition) over a period
of up to 180 days, e.g., from about 14 to about 30 to about 180 days. For example, the polymeric matrix may degrade and release the Compounds of the present disclosure over a period of about 30, about 60 or about 90 days. In another example, the polymeric matrix may degrade and release the Compounds of the present disclosure over a period of about 120, or about 180 days. [00030] In still another embodiment, the Pharmaceutical Composition 1 or 1-A or 1-B may be formulated for administration by injection, for example, as a sterile solution. [00031] In another embodiment, the present disclosure provides a Pharmaceutical Composition (Pharmaceutical Composition 1-C) comprising a Compound of the Invention as hereinbefore described, in an osmotic controlled release oral delivery system (OROS), which is described in US 2001/0036472 and US 2009/0202631, the contents of each of which applications are incorporated by reference in their entirety. Therefore in one embodiment, the present disclosure provides a pharmaceutical composition or device comprising (a) a gelatin capsule containing a Compound of any of Formulae I et seq. in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier; (b) a multilayer wall superposed on the gelatin capsule comprising, in outward order from the capsule: (i) a barrier layer, (ii) an expandable layer, and (iii) a semipermeable layer; and (c) and orifice formed or formable through the wall (Pharmaceutical Composition P.1). [00032] In another embodiment, the invention provides a pharmaceutical composition comprising a gelatin capsule containing a liquid, the Compound of the Invention in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier, the gelatin capsule being surrounded by a composite wall comprising a barrier layer contacting the external surface of the gelatin capsule, an expandable layer contacting the barrier layer, a semi-permeable layer encompassing the expandable layer, and an exit orifice formed or formable in the wall (Pharmaceutical Composition P.2). [00033] In still another embodiment, the invention provides a composition comprising a gelatin capsule containing a liquid, the Compound of the Invention in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier , the gelatin capsule being surrounded by a composite wall comprising a barrier layer contacting the external surface of the gelatin capsule, an expandable layer contacting the barrier layer, a semipermeable layer encompassing the expandable layer, and an exit orifice formed or
formable in the wall, wherein the barrier layer forms a seal between the expandable layer and the environment at the exit orifice (Pharmaceutical Composition P.3).
[00034] In still another embodiment, the invention provides a composition comprising a gelatin capsule containing a liquid, the Compound of the Invention in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier, the gelatin capsule being surrounded by a barrier layer contacting the external surface of the gelatin capsule, an expandable layer contacting a portion of the barrier layer, a semi- permeable layer encompassing at least the expandable layer, and an exit orifice formed or formable in the dosage form extending from the external surface of the gelatin capsule to the environment of use (Pharmaceutical Composition P.4). The expandable layer may be formed in one or more discrete sections, such as for example, two sections located on opposing sides or ends of the gelatin capsule.
[00035] In a particular embodiment, the Compound of the Invention in the Osmotic-controlled Release Oral Delivery System (i.e., in Composition P.1-P.4) is in a liquid formulation, which formulation may be neat, liquid active agent, liquid active agent in a solution, suspension, emulsion or self-emulsifying composition or the like.
[00036] Further information on Osmotic-controlled Release Oral Delivery System composition including characteristics of the gelatin capsule, barrier layer, an expandable layer, a semi-permeable layer; and orifice may be found in US 2001/0036472, the contents of which are incorporated by reference in their entirety.
[00037] Other Osmotic-controlled Release Oral Delivery System for the Compound of Formulas I et seq. or the Pharmaceutical Composition of the present disclosure may be found in US 2009/0202631, the contents of which are incorporated by reference in their entirety. Therefore, in another embodiment, the invention provides a composition or device comprising (a) two or more layers, said two or more layers comprising a first layer and a second layer, said first layer comprises the Compound of the Invention in free or pharmaceutically acceptable salt form, optionally in admixture with a pharmaceutically acceptable diluent or carrier, said second layer comprises a polymer; (b) an outer wall surrounding said two or more layers; and (c) an orifice in said outer wall (Pharmaceutical Composition P.5).
[00038] Pharmaceutical Composition P.5 preferably utilizes a semi-permeable membrane surrounding a three-layer-core: in these embodiments, the first layer is referred to as a first drug
layer and contains low amounts of drug (e.g., the Compound of the Invention) and an osmotic agent such as salt, the middle layer referred to as the second drug layer contains higher amounts of drug, excipients and no salt; and the third layer referred to as the push layer contains osmotic agents and no drug (Pharmaceutical Composition P.6). At least one orifice is drilled through the membrane on the first drug layer end of the capsule-shaped tablet. [00039] Pharmaceutical Composition P.5 or P.6 may comprise a membrane defining a compartment, the membrane surrounding an inner protective subcoat, at least one exit orifice formed or formable therein and at least a portion of the membrane being semi-permeable; an expandable layer located within the compartment remote from the exit orifice and in fluid communication with the semi-permeable portion of the membrane; a first drug layer located adjacent the exit orifice; and a second drug layer located within the compartment between the first drug layer and the expandable layer, the drug layers comprising the Compound of the Invention in free or pharmaceutically acceptable salt thereof (Pharmaceutical Composition P.7). Depending upon the relative viscosity of the first drug layer and second drug layer, different release profiles are obtained. It is imperative to identify the optimum viscosity for each layer. In the present invention, viscosity is modulated by addition of salt, sodium chloride. The delivery Profile from the core is dependent on the weight, formulation and thickness of each of the drug layers. [00040] In a particular embodiment, the invention provides Pharmaceutical Composition P.7 wherein the first drug layer comprises salt and the second drug layer contains no salt. Pharmaceutical Composition P.5-P.7 may optionally comprise a flow-promoting layer between the membrane and the drug layers. [00041] Pharmaceutical Compositions P.1-P.7 will generally be referred to as Osmotic- controlled Release Oral Delivery System Composition. [00042] In a third aspect, the invention provides a method (Method 1) for the treatment or prophylaxis of a central nervous system disorder, or more than one central nervous system disorder, the method comprising administering to a patient in need thereof a therapeutically effective amount of a Compound of the Invention (e.g., a Compound of Formula I), wherein the Compound of the Invention is a biased agonist of the 5-HT2A receptor. In further embodiments of Method 1, the present disclosure provides:
1.1 Method 1, wherein the Compound of the invention is a Compound of Formula I, in free form; 1.2 Method 1, wherein the Compound of the invention is a Compound of Formula I, in pharmaceutically acceptable salt form; 1.3 Method 1.2, wherein the pharmaceutically acceptable salt form is a toluenesulfonic acid addition salt. 1.4 Any of Methods 1 or 1.1-1.3, wherein the Compound of the Invention is administered in the form of a pharmaceutical composition comprising Compound of the Invention in admixture with a pharmaceutically acceptable diluent or carrier (e.g., Pharmaceutical Composition I or any of 1-A, 1-B, 1-C, or P.1 to P.7); 1.5 Method 1.4, wherein the pharmaceutical composition is a Pharmaceutical Composition 1-A, 1-B or 1-C; 1.6 Method 1.4, wherein the pharmaceutical composition is any of the Pharmaceutical Compositions P.1 to P.7; 1.7 Method 1 or any of Methods 1.1-1.6, wherein the central nervous system disorder is a disorder which is susceptible to treatment by agonism of the beta-arrestin signaling pathway and/or agonism or antagonism of the G-q signaling pathway, mediated by the 5-HT2A receptor; 1.8 Method 1 or any of Methods 1.1-1.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the serotonin 5-HT2A receptor, the dopamine D1 receptor, and/or D2 receptor systems, and/or the serotonin reuptake transporter (SERT) pathways, and/or the mu-opioid receptor pathway; 1.9 Method 1, or any of 1.1-1.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the serotonin 5-HT2A receptor, the serotonin reuptake transporter (SERT), but not the dopamine D1 receptor or D2 receptor systems, and not the mu-opioid receptor pathway; 1.10 Method 1, or any of 1.1-1.7, wherein the central nervous system disorder is a disorder not involving, mediated by, or affected (directly or indirectly) by, one or
more of the dopamine D1 receptor, dopamine D2 receptor, serotonin reuptake transporter (SERT), or mu-opioid receptor pathway; 1.11 Method 1, or any of 1.1-1.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the serotonin 5-HT2A receptor; 1.12 Method 1, or any of 1.1-1.11, wherein central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, signaling via the beta-arrestin signaling pathway of the serotonin 5-HT2A receptor; 1.13 Method 1 or any of Methods 1.1-1.12, wherein the central nervous system disorder is a disorder selected from a group consisting of anxiety (including general anxiety, social anxiety, and panic disorders), and depression (for example refractory depression and major depressive disorder, bipolar depression); 1.14 Method 1 or any of Methods 1.1-1.13, wherein the central nervous system disorder is anxiety, such as general anxiety, social anxiety, and panic disorders; 1.15 Method 1 or any of Methods 1.1-1.13, wherein the central nervous system disorder is depression, such as refractory depression, major depressive disorder, and bipolar depression, and/or anhedonia; 1.16 Method 1, or any of 1.1-1.15, wherein the patient suffers from any combination of the disorders recited in Methods 1.7-1.15; 1.17 Method 1, or any of 1.1-1.15, wherein the method is a method for the treatment or prophylaxis of any combination of the disorders recited in Methods 1.7-1.15; 1.18 Method 1 or any of Methods 1.1-1.17, wherein said patient is not responsive to or is unable to tolerate the side effects of conventional anti-depressant agents; 1.19 Method 1 or any of Methods 1.1-1.18, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with selective serotonin reuptake inhibitors (SSRIs), such as citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline; 1.20 Method 1 or any of Methods 1.1-1.19, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with serotonin-norepinephrine reuptake inhibitors (SNRIs), such as venlafaxine, sibutramine, duloxetine, atomoxetine, desvenlafaxine, milnacipran, and levomilnacipran;
1.21 Method 1 or any of Methods 1.1-1.20, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with conventional anxiolytic agents, such as lorazepam, diazepam, alprazolam, and buspirone; 1.22 Any of the foregoing methods, wherein the therapeutically effective amount of the Compound of the Invention (e.g., the Compound of Formula I) is 1 mg-1000mg, preferably 2.5mg-50mg, or for a long-acting formulation, 25mg-1500mg, for example, 50mg to 500mg, or 250mg to 1000mg, or 250mg to 750mg, or 75mg to 300mg; 1.23 Method 1.22, wherein the therapeutically effective amount is 1 mg-100mg per day, preferably 2.5mg-50mg per day. 1.24 Method 1.22, wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-1000mg, for example 2.5mg-50mg, or for a long-acting formulation, 25mg-1500mg, for example, 50mg to 500mg, or 250mg to 1000mg, or 250mg to 750mg, or 75mg to 300mg; 1.25 Method 1.22, where therapeutically in the effective amount of the Compound of the Invention is 1 mg-100mg per day, for example 2.5mg-50mg per day; 1.26 Method 1.22, wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-5mg, preferably 2.5-5mg, per day; 1.27 Method 1.22, wherein the therapeutically effective amount of the Compound of the Invention is 2.5mg or 5mg, per day; 1.28 Method 1 or any of 1.1-1.27, wherein the pharmaceutical composition is a sustained release or delayed release formulation, e.g., according to Pharmaceutical Composition 1-A as described herein; 1.29 Method 1 or any of 1.1-1.28, wherein the pharmaceutical composition comprises the Compound of the Invention in a polymeric matrix, e.g., according to Pharmaceutical Composition 1-B as described herein; 1.30 Method 1 or any of 1.1-1.29, wherein the pharmaceutical composition is in the form of a tablet or capsule; 1.31 Method 1 or any of 1.1-1.30, wherein the pharmaceutical composition is formulated for oral, sublingual, or buccal administration;
1.32 Method 1 or any of 1.1-1.31, wherein the pharmaceutical composition is a rapidly-dissolving oral tablet (e.g., a rapidly dissolving sublingual tablet); 1.33 Method 1 or any of 1.1-1.32, wherein the pharmaceutical composition is formulated for intranasal or intrapulmonary administration (e.g., as an aerosol, mist, or powder for inhalation); 1.34 Method 1 or any of 1.1-1.33, wherein the pharmaceutical composition is formulated for administration by injection, for example, as a sterile aqueous solution; 1.35 Method 1.34, wherein the pharmaceutical composition is formulated for intravenous, intrathecal, intramuscular, subcutaneous or intraperitoneal injection 1.36 Any of Method 1 or 1.1-1.35, wherein the method further comprises the concurrent administration of one or more other therapeutic agents, e.g., administered simultaneously, separately or sequentially; 1.37 Method 1.36, wherein the additional therapeutic agent is an anti-depressant agent, optionally wherein the anti-depressant agent is selected from amitriptyline, amoxapine, bupropion, citalopram, clomipramine, desipramine, doxepin, duloxetine, escitalopram, fluoxetine, fluvoxamine, imipramine, isocarboxazid, maprotiline, mirtazapine, nefazodone, nortriptyline, paroxetine, phenelzine sulfate, protriptyline, sertraline, tranylcypromine, trazodone, trimipramine, venlafaxine, ketamine, and esketamine; 1.38 Method 1.36, wherein the additional therapeutic agent is an anxiolytic agent, optionally selected from lorazepam, diazepam, alprazolam, and buspirone; 1.39 Method 1, or any of 1.1-1.38, wherein the method does not cause psychoses (e.g., prolonged or intermittent psychoses); 1.40 Method 1, or any of 1.1-1.39, wherein the method does not promote self-harm or harm to others in the patient; 1.41 Method 1, or any of 1.1-1.40, wherein the method does not cause heart valvulopathy or pulmonary arterial hypertension, e.g., wherein the method is safe for treatment of a patient having cardiac comorbidities; 1.42 Method 1, or any of 1.1-1.41, wherein the method does not cause abuse or dependence (e.g., physical or psychological dependence);
1.43 Method 1, or any of 1.1-1.42, wherein the patient suffers from or has previously diagnosed with hallucinogen persisting perception disorder (HPPD); 1.44 Method 1, or any of 1.1-1.43, wherein the patient suffers from or has previously diagnosed with psychosis (e.g., schizophrenia). [00043] In a fourth aspect, the invention provides a method (Method 2) for the treatment or prophylaxis of a central nervous system disorder, or more than one central nervous system disorder, the method comprising administering to a patient in need thereof a therapeutically effective amount of a Compound of the Invention (e.g., a Compound of Formula I), wherein the Compound of the Invention is an antagonist of the 5-HT2A receptor. In particular embodiments, Method 2 comprises administering: 2.1 Method 2, wherein the Compound of the invention is a Compound of Formula I, in free form; 2.2 Method 2, wherein the Compound of the invention is a Compound of Formula I, in pharmaceutically acceptable salt form; 2.3 Method 2.2, wherein the pharmaceutically acceptable salt form is a toluenesulfonic acid addition salt. 2.4 Any of Methods 2 or 2.1-2.3, wherein the Compound of the Invention is administered in the form of a pharmaceutical composition comprising Compound of the Invention in admixture with a pharmaceutically acceptable diluent or carrier (e.g., Pharmaceutical Composition I or any of 1-A, 1-B, 1-C, or P.1 to P.7); 2.5 Method 2.4, wherein the pharmaceutical composition is a Pharmaceutical Composition 1-A, 1-B or 1-C; 2.6 Method 2.4, wherein the pharmaceutical composition is any of the Pharmaceutical Compositions P.1 to P.7; 2.7 Method 2 or any of Methods 2.1-2.6, wherein the central nervous system disorder is a disorder which is susceptible to treatment by antagonism of the 5-HT2A receptor, e.g., antagonism of the beta-arrestin signaling pathway and antagonism of the G-q signaling pathway, mediated by the 5-HT2A receptor; 2.8 Method 2 or any of Methods 2.1-2.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the
serotonin 5-HT2A receptor, dopamine D1 receptor, and/or D2 receptor systems, and/or the serotonin reuptake transporter (SERT) pathways, and/or the mu-opioid receptor pathway; 2.9 Method 2, or any of 2.1-2.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the serotonin 5-HT2A receptor or serotonin reuptake transporter (SERT), but not the dopamine D1 receptor or D2 receptor systems, and not the mu-opioid receptor pathway; 2.10 Method 2, or any of 2.1-2.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the serotonin 5-HT2A receptor or serotonin reuptake transporter (SERT), but not the mu-opioid receptor pathway; 2.11 Method 2, or any of 2.1-2.7, wherein the central nervous system disorder is a disorder involving, mediated by, or affected (directly or indirectly) by, the serotonin 5-HT2A receptor; 2.12 Method 2 or any of Methods 2.1-2.12, wherein the central nervous system disorder is a disorder selected from a group consisting of anxiety, depression, psychosis, schizophrenia, sleep disorders, impulse control disorder, post-traumatic stress disorder, intermittent explosive disorder, and dementia; 2.13 Method 2 or any of Methods 2.1-2.13, wherein the central nervous system disorder is anxiety, such as general anxiety, social anxiety, and panic disorders; 2.14 Method 2 or any of Methods 2.1-2.13, wherein the central nervous system disorder is depression, such as refractory depression, major depressive disorder, and bipolar depression; 2.15 Method 2 or any of Methods 2.1-2.13, wherein the central nervous system disorder is psychosis, e.g., schizophrenia; 2.16 Method 2.16, wherein the method is effective to treat the positive and/or negative symptoms of schizophrenia; 2.17 Method 2, or any of 2.1-2.16, wherein the patient suffers from any combination of the disorders recited in Methods 2.7-2.16;
2.18 Method 2, or any of 2.1-2.46, wherein the method is a method for the treatment or prophylaxis of any combination of the disorders recited in Methods 2.7-2.16; 2.19 Method 2 or any of Methods 2.1-2.18, wherein said patient is not responsive to or is unable to tolerate the side effects of conventional antipsychotic drugs, e.g., chlorpromazine, haloperidol, droperidol, fluphenazine, loxapine, mesoridazine molindone, perphenazine, pimozide, prochlorperazine promazine, thioridazine, thiothixene, trifluoperazine, brexpiprazole, cariprazine, asenapine, lurasidone, clozapine, aripiprazole, olanzapine, quetiapine, risperidone and ziprasidone; 2.20 Method 2 or any of Methods 2.1-2.19, wherein said patient is not responsive to or is unable to tolerate the side effects of conventional anti-depressant agents; 2.21 Method 2 or any of Methods 2.1-2.20, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with selective serotonin reuptake inhibitors (SSRIs), such as citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline; 2.22 Method 2 or any of Methods 2.1-2.21, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with serotonin-norepinephrine reuptake inhibitors (SNRIs), such as venlafaxine, sibutramine, duloxetine, atomoxetine, desvenlafaxine, milnacipran, and levomilnacipran; 2.23 Method 2 or any of Methods 2.1-2.22, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with conventional anxiolytic agents, such as lorazepam, diazepam, alprazolam, and buspirone; 2.24 Method 2 or any of Methods 2.1-2.23, wherein said patient is not responsive to or cannot tolerate the side effects from, treatment with antipsychotic agents, such as clomipramine, risperidone, quetiapine and olanzapine; 2.25 Any of the foregoing methods, wherein the therapeutically effective amount of the Compound of the Invention (e.g., the Compound of Formula I) is 1 mg-1000mg, preferably 2.5mg-50mg, or for a long-acting formulation, 25mg-1500mg, for example, 50mg to 500mg, or 250mg to 1000mg, or 250mg to 750mg, or 75mg to 300mg; 2.26 Method 2.26, wherein the therapeutically effective amount is 1 mg-100mg per day, preferably 2.5mg-50mg per day.
2.27 Method 2.26, wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-1000mg, for example 2.5mg-50mg, or for a long-acting formulation, 25mg-1500mg, for example, 50mg to 500mg, or 250mg to 1000mg, or 250mg to 750mg, or 75mg to 300mg; 2.28 Method 2.26, wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-100mg per day, for example 2.5mg-50mg per day; 2.29 Method 2.26, wherein the therapeutically effective amount of the Compound of the Invention is 1 mg-5mg, preferably 2.5-5mg, per day; 2.30 Method 2.26, wherein the therapeutically effective amount of the Compound of the Invention is 2.5mg or 5mg, per day; 2.31 Method 2 or any of Methods 2.1-2.30, wherein the pharmaceutical composition is a sustained release or delayed release formulation, e.g., according to Pharmaceutical Composition 1-A as described herein; 2.32 Method 2 or any of Methods 2.1-2.30, wherein the pharmaceutical composition comprises the Compound of the Invention in a polymeric matrix, e.g., according to Pharmaceutical Composition 1-B as described herein; 2.33 Method 2 or any of Methods 2.1-2.32, wherein the pharmaceutical composition is in the form of a tablet or capsule; 2.34 Method 2 or any of Methods 2.1-2.33, wherein the pharmaceutical composition is formulated for oral, sublingual, or buccal administration; 2.35 Method 2 or any of Methods 2.1-2.34, wherein the pharmaceutical composition is a rapidly-dissolving oral tablet (e.g., a rapidly dissolving sublingual tablet); 2.36 Method 2 or any of Methods 2.1-2.35, wherein the pharmaceutical composition is formulated for intranasal or intrapulmonary administration (e.g., as an aerosol, mist, or powder for inhalation); 2.37 Method 2 or any of Methods 2.1-2.36, wherein the pharmaceutical composition is formulated for administration by injection, for example, as a sterile aqueous solution; 2.38 Method 2.37, wherein the pharmaceutical composition is formulated for intravenous, intrathecal, intramuscular, subcutaneous or intraperitoneal injection
2.39 Method 2 or any of Methods 2.1-2.38, wherein the method further comprises the concurrent administration of one or more other therapeutic agents, e.g., administered simultaneously, separately or sequentially; 2.40 Method 2.39, wherein the additional therapeutic agent is an antipsychotic agent, optionally wherein the antipsychotic agent is selected from a group consisting of chlorpromazine, haloperidol, droperidol, fluphenazine, loxapine, mesoridazine, molindone, perphenazine, pimozide, prochlorperazine promazine, thioridazine, thiothixene, trifluoperazine, brexpiprazole, cariprazine, asenapine, lurasidone, clozapine, aripiprazole, olanzapine, quetiapine, risperidone, ziprasidone and paliperidone; 2.41 Method 2.39, wherein the additional therapeutic agent is an anti-depressant agent, optionally wherein the anti-depressant agent is selected from amitriptyline, amoxapine, bupropion, citalopram, clomipramine, desipramine, doxepin, duloxetine, escitalopram, fluoxetine, fluvoxamine, imipramine, isocarboxazid, maprotiline, mirtazapine, nefazodone, nortriptyline, paroxetine, phenelzine sulfate, protriptyline, sertraline, tranylcypromine, trazodone, trimipramine, venlafaxine, ketamine, and esketamine; 2.42 Method 2.39, wherein the additional therapeutic agent is an atypical antipsychotic agent, optionally wherein the agent is selected from a group consisting of brexpiprazole, cariprazine, asenapine, lurasidone, clozapine, aripiprazole, olanzapine, quetiapine, risperidone, ziprasidone, and paliperidone; 2.43 Method 2.39, wherein the additional therapeutic agent is an atypical stimulant, optionally selected from modafinil, adrafinil, and armodafinil; 2.44 Method 2.39, wherein the additional therapeutic agent is an anti-Parkinson's agent, optionally selected from L- dopa, co-careldopa, duodopa, stalevo, Symmetrel, benztropine, biperiden, bromocriptine, entacapone, pergolide, pramipexole, procyclidine, ropinirole, selegiline and tolcapone; 2.45 Method 2.39 wherein the additional therapeutic agent is an anxiolytic agent, optionally selected from lorazepam, diazepam, alprazolam, and buspirone; 2.46 Method 2, or any of 2.1-2.45, wherein the method does not cause psychoses (e.g., prolonged or intermittent psychoses);
2.47 Method 2, or any of 2.1-2.46, wherein the method does not promote self-harm or harm to others in the patient; 2.48 Method 2, or any of 2.1-2.47, wherein the method does not cause heart valvulopathy or pulmonary arterial hypertension, e.g., wherein the method is safe for treatment of a patient having cardiac comorbidities; 2.49 Method 2, or any of 2.1-2.48, wherein the method does not cause abuse or dependence (e.g., physical or psychological dependence. [00044] In some embodiments of the methods described hereinbelow, the Pharmaceutical Composition comprising a Compound of the Invention may be administered for controlled- and/or sustained-release of the Compounds of the Invention over a period of from about 14 days, about 30 to about 180 days, preferably over the period of about 30, about 60 or about 90 days. Controlled- and/or sustained-release is particularly useful for circumventing premature discontinuation of therapy, particularly for antipsychotic drug therapy where non-compliance or non-adherence to medication regimes is a common occurrence. [00045] In some embodiments of the methods described hereinbelow, the Pharmaceutical Composition comprising a Compound of the Invention may be a Depot Composition of the present disclosure which is administered for controlled- and/or sustained-release of the Compounds of the Invention over a period of time. [00046] The Compounds of the present disclosure (i.e., Compounds of the Invention) and the Pharmaceutical Compositions of the present disclosure may be used in combination with a second therapeutic agent, particularly at lower dosages than when the individual agents are used as a monotherapy so as to enhance the therapeutic activities of the combined agents without causing the undesirable side effects commonly occur in conventional monotherapy. Therefore, the Compounds of the present disclosure may be simultaneously, sequentially, or contemporaneously administered with other therapeutic agents as described hereinabove, such as opiate, opioid, analgesic, anti-depressant, anti-psychotic, other hypnotic agents, and/or agents use to treat Parkinson's disease or mood disorders. [00047] In any of the embodiments of Method 1 et seq. or Method 2 et seq. wherein the Compound of the present disclosure is administered along with one or more second therapeutic agents, the one or more second therapeutic agents may be administered as a part of the pharmaceutical composition comprising the Compound of the present disclosure. Alternatively,
the one or more second therapeutic agents may be administered in separate pharmaceutical compositions (such as pills, tablets, capsules and injections) administered simultaneously, sequentially or separately from the administration of the Compound of the present disclosure. [00048] In some further embodiments of the present disclosure, the Pharmaceutical Compositions of the present disclosure may be used in combination with a second therapeutic agent, particularly at lower dosages than when the individual agents are used as a monotherapy so as to enhance the therapeutic activities of the combined agents without causing the undesirable side effects, wherein the second therapeutic agent is an opioid antagonist or inverse agonist (e.g., naloxone). The Compounds of the present disclosure may be simultaneously, sequentially, or contemporaneously administered with such opioid antagonists or opioid inverse agonists. [00049] In a fifth aspect, the present disclosure provides use of a Compound of the Invention, in the manufacture of a medicament for use according to Method 1 or any of Methods 1.1-1.42 or Method 2 or any of Methods 2.1-2.49. In another embodiment, the present disclosure provides a Compound of the Invention, for use in the treatment of a disease or disorder according to Method 1 or any of Methods 1.1-1.42 or Method 2 or any of Methods 2.1-2.49. DETAILED DESCRIPTION [00050] The term “biased agonist” as used herein, is used in reference to a compound having activity at the serotonin 5-HT2A receptor with either partial or full agonism for beta-arrestin signaling via the receptor, but with either antagonism or weak partial agonism for G-q mediated signaling. A useful measure of bias is the “bias ratio”, which is calculated as the ratio of the intrinsic relative activity (RAi) for beta-arrestin signaling over the RAi for G-q signaling. A non- biased agonist has a bias ratio of 1.0. A biased agonist has a non-zero bias ratio. In some embodiments, compounds of the present disclosure are preferably biased towards beta-arrestin signaling, and thus have a bias ratio greater than 1.0. More preferably, the bias ratio towards beta-arrestin signaling is greater than 10, or greater than 100, or greater than 1000, or 10,000 or more. [00051] As used herein, the term “partial agonist” is understood to refer to a compound having agonism to any extent that is lesser than that of a reference standard full agonist. For example, the reference compound for 5-HT2A receptor agonism is alpha-methylserotonin. A
compound which has a maximum efficacy (Emax) that is less than 100% of the maximum efficacy for alpha-methylserotonin is a partial agonist. [00052] The term “hallucinogen” refers to a compound which causes hallucinogenic symptoms, which are any one or more symptoms selected from visual hallucinations, auditory hallucinations, visual distortions (such as drifting, morphing, breathing or melting of objects and surfaces in the field of view), detachment from reality, dissociation, delirium, and undesired altered states of consciousness. A compound of the present disclosure is considered “non- hallucinogenic” if at doses which are therapeutically effective for the treatment of neuropsychiatric disorders described herein (e.g., depression, anxiety, etc.) the compound does not cause hallucinogenic symptoms. [00053] It is understood that the terms “opiate” and “opioid” are distinct, in that “opiate” refers to natural products derived from the opium poppy, such as morphine, codeine and heroin, but “opioid” refers to these natural compounds as well as semi-synthetic and synthetic derivatives thereof, such as fentanyl and its analogs. [00054] “Alkyl” as used herein is a saturated or unsaturated hydrocarbon moiety, e.g., one to twenty-one carbon atoms in length, unless indicated otherwise; any such alkyl may be linear or branched (e.g., n-butyl or tert-butyl), preferably linear, unless otherwise specified. For example, “C1-21 alkyl” denotes alkyl having 1 to 21 carbon atoms. In one embodiment, alkyl is optionally substituted with one or more hydroxy or C1-22alkoxy (e.g., ethoxy) groups. In another embodiment, alkyl contains 1 to 21 carbon atoms, preferably straight chain and optionally saturated or unsaturated, for example in some embodiments wherein R1 is an alkyl chain containing 1 to 21 carbon atoms, preferably 6-15 carbon atoms, 16-21 carbon atoms, e.g., so that together with the -C(O)- to which it attaches, e.g., when cleaved from the compound of Formula I, forms the residue of a natural or unnatural, saturated or unsaturated fatty acid. [00055] The term “pharmaceutically acceptable diluent or carrier” is intended to mean diluents and carriers that are useful in pharmaceutical preparations, and that are free of substances that are allergenic, pyrogenic or pathogenic, and that are known to potentially cause or promote illness. Pharmaceutically acceptable diluents or carriers thus exclude bodily fluids such as example blood, urine, spinal fluid, saliva, and the like, as well as their constituent components such as blood cells and circulating proteins. Suitable pharmaceutically acceptable diluents and carriers can be found in any of several well-known treatises on pharmaceutical
formulations, for example Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001; Remington's Pharmaceutical Sciences, 20th Ed., Lippincott Williams & Wilkins., 2000; and Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); all of which are incorporated by reference herein in their entirety. [00056] The terms “purified,” “in purified form” or “in isolated and purified form” for a compound refers to the physical state of said compound after being isolated from a synthetic process (e.g., from a reaction mixture), or natural source or combination thereof. Thus, the term “purified,” “in purified form” or “in isolated and purified form” for a compound refers to the physical state of said compound after being obtained from a purification process or processes described herein or well known to the skilled artisan (e.g., chromatography, recrystallization, LC-MS and LC-MS/MS techniques and the like), in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan. [00057] Unless otherwise indicated, the Compounds of the Invention may exist in free base form or in salt form, such as a pharmaceutically acceptable salt form, e.g., as acid addition salts. An acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric acid or toluenesulfonic acid. In addition, a salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, or a salt with an organic base which affords a physiologically-acceptable cation. In a particular embodiment, the salt of a Compound of the Invention is a toluenesulfonic acid addition salt or the hydrochloric acid addition salt. [00058] The Compounds of the Invention are intended for use as pharmaceuticals, therefore pharmaceutically acceptable salts are preferred. Salts which are unsuitable for pharmaceutical uses may be useful, for example, for the isolation or purification of free Compounds of the Invention, and are therefore also included within the scope of the compounds of the present disclosure.
[00059] The Compounds of the Invention may comprise one or more chiral carbon atoms. The compounds thus exist in individual isomeric, e.g., enantiomeric or diastereomeric form or as mixtures of individual forms, e.g., racemic/diastereomeric mixtures. Any isomer may be present in which the asymmetric center is in the (R)-, (S)-, or (R,S)- configuration. The invention is to be understood as embracing both individual optically active isomers as well as mixtures (e.g., racemic/diastereomeric mixtures) thereof. Accordingly, the Compounds of the Invention may be a racemic mixture or it may be predominantly, e.g., in pure, or substantially pure, isomeric form, e.g., greater than 70% enantiomeric/diastereomeric excess (“ee”), preferably greater than 80% ee, more preferably greater than 90% ee, most preferably greater than 95% ee. The purification of said isomers and the separation of said isomeric mixtures may be accomplished by standard techniques known in the art (e.g., column chromatography, preparative TLC, preparative HPLC, simulated moving bed and the like). [00060] Geometric isomers by nature of substituents about a double bond or a ring may be present in cis (Z) or trans (E) form, and both isomeric forms are encompassed within the scope of this invention. [00061] It is also intended that the compounds of the present disclosure encompass their stable and unstable isotopes. Stable isotopes are nonradioactive isotopes which contain one additional neutron compared to the abundant nuclides of the same species (i.e., element). It is expected that the activity of compounds comprising such isotopes would be retained, and such compound would also have utility for measuring pharmacokinetics of the non-isotopic analogs. For example, the hydrogen atom at a certain position on the compounds of the disclosure may be replaced with deuterium (a stable isotope which is non-radioactive). Examples of known stable isotopes include, but not limited to, deuterium (2H or D), 13C, 15N, 18O. Alternatively, unstable isotopes, which are radioactive isotopes which contain additional neutrons compared to the abundant nuclides of the same species (i.e., element), e.g., 123I, 131I, 125I, 14C, 18F, may replace the corresponding abundant species of I, C and F. Another example of useful isotope of the compound of the invention is the 14C isotope. These radio isotopes are useful for radio-imaging and/or pharmacokinetic studies of the compounds of the invention. In addition, the substitution of atoms of having the natural isotopic distributing with heavier isotopes can result in desirable change in pharmacokinetic rates when these substitutions are made at metabolically liable sites.
For example, the incorporation of deuterium (2H) in place of hydrogen can slow metabolic degradation when the position of the hydrogen is a site of enzymatic or metabolic activity. [00062] Compounds of the Invention may be included as a depot formulation, e.g., by dispersing, dissolving or encapsulating the Compounds of the Invention in a polymeric matrix as described hereinbefore, such that the Compound is continually released as the polymer degrades over time. The release of the Compounds of the Invention from the polymeric matrix provides for the controlled- and/or delayed- and/or sustained-release of the Compounds, e.g., from the pharmaceutical depot composition, into a subject, for example a warm-blooded animal such as man, to which the pharmaceutical depot is administered. Thus, the pharmaceutical depot delivers the Compounds of the Invention to the subject at concentrations effective for treatment of the particular disease or medical condition over a sustained period of time, e.g., 14-180 days, preferably about 30, about 60 or about 90 days. [00063] Polymers useful for the polymeric matrix in the Composition of the Invention (e.g., Depot composition of the Invention) may include a polyester of a hydroxyfatty acid and derivatives thereof or other agents such as polylactic acid, polyglycolic acid, polycitric acid, polymalic acid, poly-beta.-hydroxybutyric acid, epsilon.-capro-lactone ring opening polymer, lactic acid-glycolic acid copolymer, 2-hydroxybutyric acid-glycolic acid copolymer, polylactic acid-polyethylene glycol copolymer or polyglycolic acid-polyethylene glycol copolymer), a polymer of an alkyl alpha-cyanoacrylate (for example poly(butyl 2-cyanoacrylate)), a polyalkylene oxalate (for example polytrimethylene oxalate or polytetramethylene oxalate), a polyortho ester, a polycarbonate (for example polyethylene carbonate or polyethylene-propylene carbonate), a polyortho-carbonate, a polyamino acid (for example poly-gamma.-L-alanine, poly- .gamma.-benzyl-L-glutamic acid or poly-y-methyl-L-glutamic acid), a hyaluronic acid ester, and the like, and one or more of these polymers can be used. [00064] If the polymers are copolymers, they may be any of random, block and/or graft copolymers. When the above alpha-hydroxycarboxylic acids, hydroxydicarboxylic acids and hydroxytricarboxylic acids have optical activity in their molecules, any one of D-isomers, L- isomers and/or DL-isomers may be used. Among others, alpha-hydroxycarboxylic acid polymer (preferably lactic acid-glycolic acid polymer), its ester, poly-alpha-cyanoacrylic acid esters, etc. may be used, and lactic acid-glycolic acid copolymer (also referred to as poly(lactide-alpha- glycolide) or poly(lactic-co-glycolic acid), and hereinafter referred to as PLGA) are preferred.
Thus, in one aspect the polymer useful for the polymeric matrix is PLGA. As used herein, the term PLGA includes polymers of lactic acid (also referred to as polylactide, poly(lactic acid), or PLA). Most preferably, the polymer is the biodegradable poly(d,l-lactide-co-glycolide) polymer. [00065] In a preferred embodiment, the polymeric matrix of the invention is a biocompatible and biodegradable polymeric material. The term “biocompatible” is defined as a polymeric material that is not toxic, is not carcinogenic, and does not significantly induce inflammation in body tissues. The matrix material should be biodegradable wherein the polymeric material should degrade by bodily processes to products readily disposable by the body and should not accumulate in the body. The products of the biodegradation should also be biocompatible with the body in that the polymeric matrix is biocompatible with the body. Particular useful examples of polymeric matrix materials include poly(glycolic acid), poly-D,L-lactic acid, poly-L-lactic acid, copolymers of the foregoing, poly(aliphatic carboxylic acids), copolyoxalates, polycaprolactone, polydioxanone, poly(ortho carbonates), poly(acetals), poly(lactic acid- caprolactone), polyorthoesters, poly(glycolic acid-caprolactone), polyanhydrides, and natural polymers including albumin, casein, and waxes, such as, glycerol mono- and distearate, and the like. The preferred polymer for use in the practice of this invention is dl(polylactide-co- glycolide). It is preferred that the molar ratio of lactide to glycolide in such a copolymer be in the range of from about 75:25 to 50:50. [00066] Useful PLGA polymers may have a weight-average molecular weight of from about 5,000 to 500,000 Daltons, preferably about 150,000 Daltons. Dependent on the rate of degradation to be achieved, different molecular weight of polymers may be used. For a diffusional mechanism of drug release, the polymer should remain intact until all of the drug is released from the polymeric matrix and then degrade. The drug can also be released from the polymeric matrix as the polymeric excipient bioerodes. The PLGA may be prepared by any conventional method, or may be commercially available. For example, PLGA can be produced by ring-opening polymerization with a suitable catalyst from cyclic lactide, glycolide, etc. (see EP-0058481B2; Effects of polymerization variables on PLGA properties: molecular weight, composition and chain structure). [00067] It is believed that PLGA is biodegradable by means of the degradation of the entire solid polymer composition, due to the break-down of hydrolysable and enzymatically cleavable ester linkages under biological conditions (for example in the presence of water and biological
enzymes found in tissues of warm-blooded animals such as humans) to form lactic acid and glycolic acid. Both lactic acid and glycolic acid arc water-soluble, non-toxic products of normal metabolism, which may further biodegrade to form carbon dioxide and water. In other words, PLGA is believed to degrade by means of hydrolysis of its ester groups in the presence of water, for example in the body of a warm-blooded animal such as man, to produce lactic acid and glycolic acid and create the acidic microclimate. Lactic and glycolic acid are by-products of various metabolic pathways in the body of a warm-blooded animal such as man under normal physiological conditions and therefore are well tolerated and produce minimal systemic toxicity. [00068] In another embodiment, the polymeric matrix useful for the invention may comprise a star polymer wherein the structure of the polyester is star-shaped. These polyesters have a single polyol residue as a central moiety surrounded by acid residue chains. The polyol moiety may be, e. g., glucose or, e. g., mannitol. These esters are known and described in GB 2,145,422 and in U. S. Patent No. 5,538,739, the contents of which are incorporated by reference.
[00069] The star polymers may be prepared using polyhydroxy compounds, e. g., polyol, e.g., glucose or mannitol as the initiator. The polyol contains at least 3 hydroxy groups and has a molecular weight of up to about 20,000 Daltons, with at least 1, preferably at least 2, e.g., as a mean 3 of the hydroxy groups of the polyol being in the form of ester groups, which contain polylactide or co-polylactide chains. The branched polyesters, e.g., poly (d, 1-lactide-co- glycolide) have a central glucose moiety having rays of linear polylactide chains.
[00070] The depot compositions of the invention (long-acting injectable compositions having a Compound of the Invention in a polymeric matrix) as hereinbefore described may comprise the polymer in the form of microparticles or nanoparticles, or in a liquid form, with the Compounds of the Invention dispersed or encapsulated therein. “Microparticles” is meant solid particles that contain the Compounds of the Invention either in solution or in solid form wherein such compound is dispersed or dissolved within the polymer that serves as the matrix of the particle. By an appropriate selection of polymeric materials, a microparticle formulation can be made in which the resulting microparticles exhibit both diffusional release and biodegradation release properties.
[00071] When the polymer is in the form of microparticles, the microparticles may be prepared using any appropriate method, such as by a solvent evaporation or solvent extraction method. For example, in the solvent evaporation method, the Compounds of the Invention and
the polymer may be dissolved in a volatile organic solvent (for example a ketone such as acetone, a halogenated hydrocarbon such as chloroform or methylene chloride, a halogenated aromatic hydrocarbon, a cyclic ether such as dioxane, an ester such as ethyl acetate, a nitrile such as acetonitrile, or an alcohol such as ethanol) and dispersed in an aqueous phase containing a suitable emulsion stabilizer (for example polyvinyl alcohol, PVA). The organic solvent is then evaporated to provide microparticles with the Compounds of the Invention encapsulated therein. In the solvent extraction method, the Compounds of the Invention and polymer may be dissolved in a polar solvent (such as acetonitrile, dichloromethane, methanol, ethyl acetate or methyl formate) and then dispersed in an aqueous phase (such as a water/PVA solution). An emulsion is produced to provide microparticles with the Compounds of the Invention encapsulated therein. Spray drying is an alternative manufacturing technique for preparing the microparticles.
[00072] Another method for preparing the microparticles of the invention is also described in both U.S. Pat. No. 4,389,330 and U.S. Pat. No. 4,530,840.
[00073] The microparticle of the present invention can be prepared by any method capable of producing microparticles in a size range acceptable for use in an injectable composition. One preferred method of preparation is that described in U.S. Pat. No. 4,389,330. In this method the active agent is dissolved or dispersed in an appropriate solvent. To the agent-containing medium is added the polymeric matrix material in an amount relative to the active ingredient that provides a product having the desired loading of active agent. Optionally, all of the ingredients of the microparticle product can be blended in the solvent medium together.
[00074] Solvents for making such compositions comprising the Compounds of the Invention and the polymeric matrix material that can be employed in the practice of the present invention include organic solvents, such as acetone; halogenated hydrocarbons, such as chloroform, methylene chloride, and the like; aromatic hydrocarbon compounds; halogenated aromatic hydrocarbon compounds; cyclic ethers; alcohols, such as, benzyl alcohol; ethyl acetate; and the like. In one embodiment, the solvent for use in the practice of the present invention may be a mixture of benzyl alcohol and ethyl acetate. Further information for the preparation of microparticles useful for the invention can be found in U.S. Patent Publication Number 2008/0069885, the contents of which are incorporated herein by reference in their entirety.
[00075] The amount of the Compounds of the present disclosure incorporated in the microparticles usually ranges from about 1 wt. % to about 90 wt. %, preferably 30 to 50 wt. %,
more preferably 35 to 40 wt. %. By weight % is meant parts of the Compounds of the present disclosure per total weight of microparticle. [00076] The pharmaceutical depot compositions may comprise a pharmaceutically-acceptable diluent or carrier, such as a water miscible diluent or carrier. [00077] Details of Osmotic-controlled Release Oral Delivery System composition may be found in EP 1539115 (U.S. Pub. No.2009/0202631) and WO 2000/35419 (US 2001/0036472), the contents of each of which are incorporated by reference in their entirety. [00078] A “therapeutically effective amount” is any amount of the Compounds of the invention (for example as contained in the pharmaceutical depot) which, when administered to a subject suffering from a disease or disorder, is effective to cause a reduction, remission, or regression of the disease or disorder over the period of time as intended for the treatment. [00079] Dosages employed in practicing the present invention will of course vary depending, e.g., on the particular disease or condition to be treated, the particular Compound of the Invention used, the mode of administration, and the therapy desired. Unless otherwise indicated, an amount of the Compound of the Invention for administration (whether administered as a free base or as a salt form) refers to or is based on the amount of the Compound of the Invention in free base form (i.e., the calculation of the amount is based on the free base amount). [00080] Compounds of the Invention may be administered by any satisfactory route, including orally, parenterally (intravenously, intramuscular, intranasal, pulmonary or subcutaneous) or transdermally. In certain embodiments, the Compounds of the Invention, e.g., in depot formulation, is preferably administered parenterally, e.g., by injection, for example, intramuscular or subcutaneous injection. [00081] In general, satisfactory results for the methods of treatment disclosed herein, or use of the Compounds of the Invention as hereinbefore described, as set forth above are indicated to be obtained on oral administration at dosages of the order from about 1 mg to 100 mg once daily, preferably 2.5 mg-50 mg, e.g., 2.5 mg, 5 mg, 10 mg, 20 mg, 30 mg, 40 mg or 50 mg, once daily, preferably via oral administration. [00082] For some disease treatments, lower doses are satisfactory, particularly for sleep disorder treatment, such as from about 2.5mg-5mg, e.g., 2.5mg, 3mg, 4mg or 5mg, of a Compound of the Invention, in free or pharmaceutically acceptable salt form, once daily, preferably via oral administration.
[00083] Satisfactory results for methods of treatment involving co-administration of a second therapeutic agent may be obtained at doses of less than 100mg, preferably less than 50mg, e.g., less than 40mg, less than 30mg, less than 20mg, less than 10mg, less than 5mg, less than 2.5mg, once daily. [00084] For treatment of the disorders disclosed herein wherein the depot composition is used to achieve longer duration of action, the dosages will be higher relative to the shorter action composition, e.g., higher than 1-100mg, e.g., 25mg, 50mg, 100mg, 500mg, 1,000mg, or greater than 1000mg. Duration of action of the Compounds of the present disclosure may be controlled by manipulation of the polymer composition, i.e., the polymer:drug ratio and microparticle size. Wherein the composition of the invention is a depot composition, administration by injection is preferred. [00085] The pharmaceutically acceptable salts of the Compounds of the present disclosure can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free base forms of these compounds with a stoichiometric amount of the appropriate acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Further details for the preparation of these salts, e.g., toluenesulfonic salt in amorphous or crystal form, may be found in U.S. 8,309,722, 8.648,077, 9,199,995, and 9,586,960. [00086] Pharmaceutical compositions comprising Compounds of the present disclosure may be prepared using conventional diluents or excipients (an example include, but is not limited to sesame oil) and techniques known in the galenic art. Thus, oral dosage forms may include tablets, capsules, solutions, suspensions and the like. [00087] The term “concurrently” when referring to a therapeutic use means administration of two or more active ingredients to a patient as part of a regimen for the treatment of a disease or disorder, whether the two or more active agents are given at the same or different times or whether given by the same or different routes of administrations. Concurrent administration of the two or more active ingredients may be at different times on the same day, or on different dates or at different frequencies. [00088] The term “simultaneously” when referring to a therapeutic use means administration of two or more active ingredients at or about the same time by the same route of administration.
[00089] The term “separately” when referring to a therapeutic use means administration of two or more active ingredients at or about the same time by different route of administration Methods of Making the Compounds of the Invention: [00090] The Compound of Formula A, and methods for its synthesis, including the synthesis of intermediates used in the synthetic schemes described below, have been disclosed in, for example, U.S.10,245,260, and US 2022/0041600, and US 2022/0064166, the contents of which are hereby incorporated by reference in their entireties. [00091] The synthesis of similar fused gamma-carbolines has been disclosed in, for example, U.S.8,309,722, U.S. 8,993,572, US 2017/0183350, WO 2018/126140 and WO 2018/126143, the contents of each of which are incorporated by reference in their entireties. Compounds of the present disclosure can be prepared using similar procedures. [00092] Other Compounds of the present disclosure came be made by analogous procedures known to those skilled in the art. [00093] Isolation or purification of the diastereomers of the Compounds of the Invention may be achieved by conventional methods known in the art, e.g., column purification, preparative thin layer chromatography, preparative HPLC, crystallization, trituration, simulated moving beds and the like. [00094] Salts of the Compounds of the present disclosure may be prepared as similarly described in U.S. Pat. No. 6,548,493; 7,238,690; 6,552,017; 6,713,471; 7,183,282, 8,648,077; 9,199,995; 9,586,860; U.S. RE39680; and U.S. RE39679, the contents of each of which are incorporated by reference in their entirety. [00095] Diastereomers of prepared compounds can be separated by, for example, HPLC using CHIRALPAK® AY-H, 5μ, 30x250mm at room temperature and eluted with 10% ethanol / 90% hexane / 0.1% dimethylethylamine. Peaks can be detected at 230 nm to produce 98-99.9%ee of the diastereomer. EXAMPLES [00096] Method for the synthesis of the compounds of the present disclosure are known in the art. In particular, methods for synthesis of the tetracyclic core structure, and for various modifications and variations of the pendant side chain on the piperidine ring have been
published. For example, see Li, et al., Journal of Medicinal Chemistry 57:2670-2682 (2014), U.S.6,713,471, U.S. 6,552,017, U.S. 7,071,186, U.S.8,309,722, U.S., 9,708,322, U.S. 10,245,260, U.S. 10,688,097, U.S.10,961,245, U.S. 10,906,906, U.S.11,427,587, U.S. 11,453,670, and US 2022/0048910, the contents of each of which are hereby incorporated by reference in their entireties. [00097] Compounds of the present disclosure may be prepared according to the following general schemes: [00098] Scheme 1
 Typical reagents and conditions: (a) ethylmagnesium bromide, titanium isopropoxide, THF, 25 °C; (b) sat. KOH in 90% EtOH solution, 100 °C; (c) RX, KI, DIPEA, DMF, 75 °C. [00099] Scheme 2
Typical reagents and conditions: (a) ethylmagnesium bromide, titanium isopropoxide, THF, 25 °C; (b) sat. KOH in 90% EtOH solution, 100 °C; (c) RX, KI, DIPEA, DMF, 75 °C. [00099] Scheme 2
 Typical reagents and conditions: (a) 4-bromo-1-butene, DIPEA, KI, dioxane, 110 °C; (b) DBU, Pd(OAc)2, tricyclohexylphosphine, DMF, 140 °C; (c) ZnEt2, CH2l2, CH2Cl2, 0 °C; (d) sat. KOH in 90% EtOH solution, 100 °C; (e) RX, KI, DIPEA, DMF, 75 °C. [000100] Scheme 3
Typical reagents and conditions: (a) 4-bromo-1-butene, DIPEA, KI, dioxane, 110 °C; (b) DBU, Pd(OAc)2, tricyclohexylphosphine, DMF, 140 °C; (c) ZnEt2, CH2l2, CH2Cl2, 0 °C; (d) sat. KOH in 90% EtOH solution, 100 °C; (e) RX, KI, DIPEA, DMF, 75 °C. [000100] Scheme 3


















































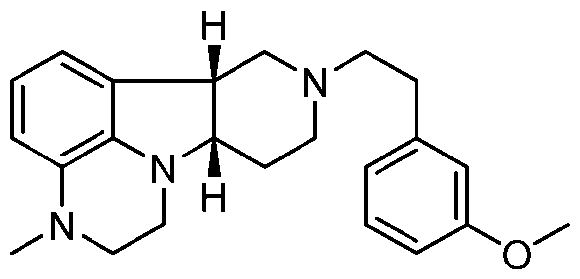















































[000277] Some compounds are additionally tested in a receptor profiling panel consisting agonist and/or antagonist radioligand binding assays. Assays are conducted using a 100 nM concentration of the test compound. Compound binding is calculated as a percent inhibition of the binding of the specific radioligand for the receptor (or ion channel) tested, which can be an agonist or antagonist. The following receptors and ion channels are included in the panel:


It is unexpectedly found that the compounds of the present disclosure have high selectivity with few off- target interactions. For example, the compounds interact significantly (> 45% inhibition) only with the following receptors: alpha-1A (e.g., 40-80% inhibition), serotonin-2A (e.g., 50-
100% inhibition), and serotonin-2B (50-100% inhibition), with insignificant activity (< 40% inhibition) at other receptors which are commonly associated with side effects, such as the serotonin-1A, serotonin-1B, serotonin-3, muscarinic, other adrenergic, and histamine receptors. [000278] These results are particularly surprising because it is found that compounds having a tetracyclic core corresponding to, or related to, that of the instantly disclosed compounds, but without the side chain (i.e., compounds of Formula I wherein n is 0 and Z-A is H) have significant activity in the receptor assays for serotonin-1A (> 70% inhibition), serotonin- 1B (> 50% inhibition), serotonin-2A (> 70% inhibition), and serotonin-2B (> 80% inhibition). [000279] Example 182: Biased Agonism/Antagonism [000280] G-q Signaling Agonism/Antagonism at the 5-HT2A Receptor. Selected compounds are submitted to 5-HT2A agonist and antagonist assays for G-q recruitment. Alpha- methylserotonin is used as the reference control for agonist assays, and altanserin is used as the control for antagonist assays. [000281] Agonist Assay: CHO-K1 cells expressing human 5-HT2A (ES-313-AF) are obtained from PerkinElmer and used according to the supplier's recommendations. Frozen cells are thawed in a water bath at 37 °C, and then are resuspended in 10 mL Ham's F-12 medium containing 10% FBS. Cells are recovered by centrifugation at 150g for 5 minutes and are then resuspended in pre-warmed assay buffer (DMEM/HAM's F-12 with HEPES) at 3x105 cells/L in a Falcon tube. Under sterile conditions, Coelenterazine H is added to a final concentration of 5 µM to the cell suspension. The Falcon tube is wrapped in aluminum foil and placed on a rotating wheel (about 45° angle and 7 rpm/min speed) for 4 hours at room temperature. Cells are then diluted to 1x105 cells/mL in assay buffer and transferred to a beaker wrapped in aluminum foil on a magnetic stirrer. After 1 hour of incubation, 50 µL of cells (5,000 cells/well) are injected into 50 µL of the test compound at increasing concentrations in the wells of a 96-well white plate. Light emission is recorded for 20 seconds immediately with FLUOstar Omega luminescence detector (BMG LABTECH). Digitonin at a final concentration of 50 µM in assay buffer is used as a positive control to measure the receptor independent cellular calcium response. Curve fitting was performed using GraphPad Prism and EC50 values are determined using a 4-parameter logistic fit, and maximum response (Emax) values are calculated by subtracting the bottom value from the top value of the dose-response curve.
[000282] Antagonist Assay: 50 µL of cells (5,000 cells/well) are mixed with 50 µL of the test compound at increasing concentrations in the wells of a 96-well white plate, and then the plates are incubated 15 minutes at room temperature. Thereafter, 50 µL of alpha- methylserotonin (final assay concentration corresponding to its measured EC80) is injected and the light emission is recorded for 20 seconds immediately with FLUOstar Omega luminescence detector (BMG LABTECH). Curve fitting is performed using Prism and IC50 values are determined using a 4-parameter logistic fit. For the antagonists, the apparent dissociation constants (KB) are calculated using the modified Cheng Prusoff equation—KB = IC50 / (1+(A/EC50A)) – where A is the concentration of the reference agonist alpha-methylserotonin, and EC50A is the EC50 value of the reference agonist alpha-methylserotonin. [000283] Beta-arrestin Signaling Agonism/Antagonism at the 5-HT2A Receptor. Selected compounds are submitted to 5-HT2A agonist and antagonist assays for beta-arrestin recruitment. Alpha-methylserotonin is used as the reference control for agonist assays, and altanserin is used as the control for antagonist assays. [000284] Agonist Assay: U2OS cells expressing human 5-HT2A receptor (93- 0401E3CP19L) are obtained from Eurofins DiscoverX and used according to the supplier's recommendations. Frozen cells are thawed in a water bath at 37 °C, and then mixed with 0.5 mL of pre-warmed Cell Plating Reagent. Cells are pipetted up and down gently several times to ensure even distribution before transfer to 11.5 mL of pre-warmed Cell Plating Reagent and poured into a disposable reagent reservoir.100 µL of cells are plated into each well of the 96- well tissue culture plate, and the plates are incubated for 24 hours at 37 °C (5% CO2). 10 µL of the test compound at increasing concentrations is added into the cells in the 96-well plate, and then the plates are incubated for 3 hours at room temperature. After addition of 55 µL of prepared detection reagent and a further 1-hour incubation, the samples are read on an Envision luminescence plate reader. All assay points are determined in duplicate and the data is presented as average values. Curve fitting is performed using Prism software (Graphpad), EC50 values are determined using a 4-parameter logistic fit, and maximum response (Emax) values are calculated by subtracting the bottom value from the top value of the dose-response curve. [000285] Antagonist Assay: 5 µL of the test compound at increasing concentrations is added into the cells in the 96-well plate, and the plates are incubated for 30 minutes at 37 °C (5% CO2). Thereafter 5 µL of α-methylserotonin (final assay concentration Thereafter, 50 µL of
alpha-methylserotonin (final assay concentration corresponding to its measured EC80) is added and the plates are incubated for 3 hours at room temperature. After addition of 55 µL of prepared detection reagent and a further 1-hour incubation, the samples are read on an Envision luminescence plate reader. All assay points are determined in duplicate and the data is presented as average values. Curve fitting is performed using Prism software (Graphpad), EC50 values are determined using a 4-parameter logistic fit. For the antagonists, the apparent dissociation constants (KB) are calculated using the modified Cheng Prusoff equation—KB = IC50 / (1+(A/EC50A)) – where A is the concentration of the reference agonist alpha-methylserotonin, and EC50A is the EC50 value of the reference agonist alpha-methylserotonin. [000286] For both the G-q signaling agonism assay and the beta-arrestin signaling agonism assay, the EC50 (effective concentration for 50% activation), and the Emax (maximum activation) are determined. [000287] The maximum efficacy (Emax) for the compound is calculated as a percentage of the maximal signaling activity induced by the compound compared to that induced by the full agonist alpha-methylserotonin. This is an indication of the compound's intrinsic activity. Any degree of maximal activity less than that of the full agonist reference indicates that the test compound is a partial agonist. [000288] Like intrinsic activity (Emax), intrinsic relative activity (RAi) is another way to quantify the partial versus full agonism of receptor activity, but it also takes into account the efficacy of the compound. It is calculated using the following formula:
 [000289] Bias Score. Bias score (or bias ratio) is calculated as the ratio of the intrinsic relative activity (RAi) for agonism of beta-arrestin signaling over the intrinsic relative activity (RAi) for agonism of G-q signaling. [000290] The results are shown in the table below (RAi = Intrinsic Relative Activity, compared to the positive controls methylserotonin or altanserin):
[000289] Bias Score. Bias score (or bias ratio) is calculated as the ratio of the intrinsic relative activity (RAi) for agonism of beta-arrestin signaling over the intrinsic relative activity (RAi) for agonism of G-q signaling. [000290] The results are shown in the table below (RAi = Intrinsic Relative Activity, compared to the positive controls methylserotonin or altanserin):




 [000291] The compounds of Examples 2, 4, and 5 are each strong (full) antagonists of the G-q signaling pathway, with the compound of Example 4 having comparable antagonist activity to the non-biased reference compound of Formula A. Preferably, for non-hallucinogenic activity, compounds should have either antagonistic activity at the G-q signaling pathway, or partial agonist activity with low intrinsic efficacy. Full agonist activity at the G-q signaling pathway (i.e., high intrinsic activity) is believed to cause hallucinogenic side effects. [000292] For maximum effectiveness, the compounds of the present disclosure are preferably agonists of beta-arrestin mediated signaling, either as partial agonists or as full agonists. Thus, in combination with the lack of agonist activity at the G-q pathway, they should be strongly biased towards beta-arrestin signaling. The compounds of Examples 2, 4, and 5, however, are antagonists of beta-arrestin signaling. The compound of Example 4, for example, much like the compound of Formula A, has equivalent strong antagonistic activity at both G-q and beta-arrestin signaling pathways. Thus, while it could be a potent antidepressant, antipsychotic, like lumateperone (ITI-007), it would not display the unique hallmarks of the psychedelic antidepressant family. [000293] In contrast, the compounds of Examples 14, 15, and 16 in particular show partial agonist activity in the beta-arrestin assay and significant bias towards beta-arrestin mediated agonism, especially the compound of Example 15, which has zero G-q mediated agonist activity. The compounds of Examples 24, 25, and 40, also show zero G-q mediated agonist activity, but while the compound of Example 24 is a beta-arrestin antagonist, the compounds of Examples 25 and 40 are beta-arrestin partial agonists. [000294] The results together show a wide variety of functional activity profiles for the compounds according to the present disclosure, which will provide them each with the potential for varying uses and secondary effect or side effect profiles. [000295] The data further shows a trend that compound having a 2 or 3-atom side chain linker preferentially active the beta-arrestin signaling pathway, with varying levels of intrinsic activity. For example, compounds wherein n is 2 or 3, and Z is a bond, or compounds wherein n is 1 or 2 and Z is a group one-atom across (e.g., a -C(O)-, -O-, or a carbonyl equivalent group). For some embodiments, the side chain linker is preferably 3 atoms in length, thus wherein n is 3 and Z is a bond, or n is 2 and Z is a group one-atom across.
[000296] For example, comparison of the results for the homologous compounds of Examples 2, 3, 4, and 129, and the reference Compound A, each having Z= -C(O)- with n of 1-5, suggests that having a linker that is shorter or longer may reduce or eliminate beta-arrestin agonist activity (n=1, 3, 4, 5), compared to a linker of optimal length (n =2). Indeed, in this series, even though all compounds bind strongly to the 5-HT2A receptor as antagonists (Ki = 0.5- 53 nM), only the compound of Example 3 shows beta-arrestin agonist activity but Gq antagonist activity. In contrast, the Compound A, wherein n = 3, is a potent beta-arrestin signaling antagonist, rather than an agonist, as well as a Gq signaling antagonist. [000297] The data also suggests that the substituent pattern around the A ring may impact binding modes of the compounds to the 5-HT2A receptor, as can the connecting group Z. For some series of compounds agonist versus antagonist binding may depend on the choice of group Z or on the presence or absence of electron-donating or electron-withdrawing groups on the ring A. This permits one to tune the desired activity of the molecule by optimizing these various groups, achieving either strong agonism and/or strong antagonism in the signaling pathways, including mixed agonist/antagonist activities for beta-arrestin signaling (e.g., the compound of Example 34. The data suggests that small electron-donating groups on ring A may promote agonistic beta-arrestin activity, while larger groups or electron-withdrawing groups on ring A may abolish agonistic beta arrestin activity. [000298] While the compounds of the present disclosure provide a range of related receptor binding activities, it should be noted that the desirable antidepressant, anxiolytic, and other CNS therapeutic properties of the hallucinogenic psychedelics is presently believed to be associated with beta-arrestin signaling at the 5-HT2A receptor, it cannot yet be ruled that some degree of Gq signaling is also desirable. While strong Gq signaling results in hallucinogenic effects, it remains possible that there is a degree of Gq signaling that may be beneficial to the therapeutic use of the present compounds without causing an undue risk of hallucinatory behavior. Indeed, in patients without a history of psychosis or hallucinogen persisting perception disorder (HPPD), higher levels of Gq signaling may be permissible, whereas in patients with such history, a compound that is more completely devoid of Gq signaling may be optimal. [000299] Example 183: In Vivo Characterization
[000300] Selected compounds (test compounds) are submitted to rodent functional model assays to determine in vivo efficacy. [000301] Head Twitch Assay. The stereotyped head twitch response induced by 5-HT2A agonists is used as a behavioral proxy for hallucinations. See Halberstadt, et al., Neuropharmacology, 167, 107933 (2020). The head-twitch response is an indicator of hallucinogenic potency of a compound in humans and occurs nearly immediately after the administration of a classical psychedelic in rodents. By definition, a head-twitch is a rapid movement of the head from one side to the other. Head twitch is used to evaluate the potential hallucinogenic liability of compounds (at up to 10 mg/kg) relative to the positive control DOI (2.5 mg/kg). Male C57bl/6 mice (9 weeks of age) are administered test compounds and vehicle via subcutaneous (SC) injection. Mice receiving the positive control DOI receive an intraperitoneal (IP) injection. At 30 minutes post-treatment, the number of head twitch responses is recorded by a blinded observer for 5 minutes. [000302] Open Field Test of Anxiety-Like Behaviors. Adult mice are habituated to the test room for one hour, and then administered a SC injection of test compound (1, 3, or 10 mg/kg) or methylcellulose vehicle, in the hind flank. For testing, animals are placed in one of four arenas in a square apparatus measuring 500 cm x 500 cm for 15 minutes. Sessions are filmed using Anymaze software (Stoelting Co, IL), using a camera mounted to the ceiling. Locomotor activity is measured by the software within the pre-defined arena. An additional center arena is pre- defined for each zone, all with an area of 100 cm x 100 cm. [000303] Rat Social Interaction Test. Test compound (0.3, 1.0, 3 and 3.0 mg/kg, or alternatively, 1, 3, and 10 mg/kg) or vehicle (0.5% aqueous CMC) is injected SC 30 minutes before behavioral testing. During the testing phase, a pair of rats (Sprague-Dawley males) receiving the same treatment are placed in a white Plexiglass open field arena and are allowed to interact for 6 minutes. Social interactions include sniffing the other rat, grooming the other rat, climbing over, under or around the other rat, following the other rat, and exploring the ano- genital area of the other rat. The time the rats spend interacting with each other during the 6- minute test is recorded by a trained observer. Chlordiazepoxide (IP, 5 mg/kg) is used as a positive control. [000304] mTOR signaling in the pre-frontal cortex (PFC). Male adult mice are injected SC with either test compound (1 mg/kg) or vehicle. At 24 hours post-injection, samples from the
PFC region of the brain are collected, and a synaptoneurosome-enriched fraction is collected and prepared for Western blotting., Quantitative analysis of phospho (p) protein immunoblots are determined relative to total levels of each protein. Change in the amount of phosphorylated ERK, Akt, mTOR, and P70S6K proteins, in the PFC are determined relative to vehicle-treated mice, as previously described (Dutheil, et al., J. Neuroscience, 43(5):863-77, 2023). [000305] It is found that compounds according to the disclosure (e.g., Examples 3, 14, 15, 25, 40, 69, 70, 71, 72, 92, 98, 99, 107, 112, 117, 118) elicit non-hallucinogenic activity, increase social interaction, and/or reduce measures of anxiety in test animals. For example, unlike the serotonergic psychedelic DOI, even high doses of a test compound up to 10 mg/kg does not elicit hallucinogenic behavior, as shown by a rate of head twitch comparable to control (e.g., < 10 head twitches per 5 minutes, or less than 5 head twitches or less than 1 head twitch (mean results)) and substantially less than that induced by DOI (p < 0.0001). Test compounds also show a dose dependent increase in social interaction between rats, and the data show that even the lowest test dose of 0.3 mg/kg is effective. In the open-field test, the test compounds are found to dose dependently reduce anxiety-like behaviors, including time in the center arena and number of entries into the center arena, without altering levels of locomotor activity or immobility. The lowest dose tested, 1 mg/kg, is found to be effective. These results demonstrate functional anxiolytic activity. [000306] The test compounds are also found to stimulate mTOR signaling in the mouse medial PFC, as demonstrated by increases in p-ERK, p-mTOR, and p-P70s6k in the tested brain regions. The mTOR signaling pathway has been shown to contribute to neuroplasticity and enhanced cognitive function and it is altered in brain regions associated with major depressive disorder. Rapid-acting antidepressants have been reported to stimulate this pathway in the prefrontal cortex. [000307] Example 184: Pharmacokinetic evaluation [000308] Compounds according to the present disclosure are submitted to a standard testing protocol for oral pharmacokinetics in Sprague-Dawley rats (males, 200-400 g). Test compounds are administered to rats either IV at 1 mg/kg or PO at 10 mg/kg, using a 0.05M citrate phosphate buffer as vehicle. Other potential vehicles include PEG-400 and aqueous 10% Trapposol/1% Tween 80, depending on compound solubility. In some studies, a third arm may utilize subcutaneous dosing (e.g., SC at 1 mg/kg). Plasma samples are collected at 2, 5, 15, and 30
minutes, and 1 , 2, 4, 8, and 24 hours, post-dosing. After processing, plasma samples are analyzed for the presence of the test compound, and for some cases, for the presence of major expected metabolites (e.g., N-des-methyl metabolite). Time to maximum concentration (Tmax), maximal plasma concentration (Cmax), and area-under-the-curve (AUC) are calculated from the data. Comparison between the AUC values for oral versus IV dosing provides the oral bioavailability of the test compound.
[000291] The compounds of Examples 2, 4, and 5 are each strong (full) antagonists of the G-q signaling pathway, with the compound of Example 4 having comparable antagonist activity to the non-biased reference compound of Formula A. Preferably, for non-hallucinogenic activity, compounds should have either antagonistic activity at the G-q signaling pathway, or partial agonist activity with low intrinsic efficacy. Full agonist activity at the G-q signaling pathway (i.e., high intrinsic activity) is believed to cause hallucinogenic side effects. [000292] For maximum effectiveness, the compounds of the present disclosure are preferably agonists of beta-arrestin mediated signaling, either as partial agonists or as full agonists. Thus, in combination with the lack of agonist activity at the G-q pathway, they should be strongly biased towards beta-arrestin signaling. The compounds of Examples 2, 4, and 5, however, are antagonists of beta-arrestin signaling. The compound of Example 4, for example, much like the compound of Formula A, has equivalent strong antagonistic activity at both G-q and beta-arrestin signaling pathways. Thus, while it could be a potent antidepressant, antipsychotic, like lumateperone (ITI-007), it would not display the unique hallmarks of the psychedelic antidepressant family. [000293] In contrast, the compounds of Examples 14, 15, and 16 in particular show partial agonist activity in the beta-arrestin assay and significant bias towards beta-arrestin mediated agonism, especially the compound of Example 15, which has zero G-q mediated agonist activity. The compounds of Examples 24, 25, and 40, also show zero G-q mediated agonist activity, but while the compound of Example 24 is a beta-arrestin antagonist, the compounds of Examples 25 and 40 are beta-arrestin partial agonists. [000294] The results together show a wide variety of functional activity profiles for the compounds according to the present disclosure, which will provide them each with the potential for varying uses and secondary effect or side effect profiles. [000295] The data further shows a trend that compound having a 2 or 3-atom side chain linker preferentially active the beta-arrestin signaling pathway, with varying levels of intrinsic activity. For example, compounds wherein n is 2 or 3, and Z is a bond, or compounds wherein n is 1 or 2 and Z is a group one-atom across (e.g., a -C(O)-, -O-, or a carbonyl equivalent group). For some embodiments, the side chain linker is preferably 3 atoms in length, thus wherein n is 3 and Z is a bond, or n is 2 and Z is a group one-atom across.
[000296] For example, comparison of the results for the homologous compounds of Examples 2, 3, 4, and 129, and the reference Compound A, each having Z= -C(O)- with n of 1-5, suggests that having a linker that is shorter or longer may reduce or eliminate beta-arrestin agonist activity (n=1, 3, 4, 5), compared to a linker of optimal length (n =2). Indeed, in this series, even though all compounds bind strongly to the 5-HT2A receptor as antagonists (Ki = 0.5- 53 nM), only the compound of Example 3 shows beta-arrestin agonist activity but Gq antagonist activity. In contrast, the Compound A, wherein n = 3, is a potent beta-arrestin signaling antagonist, rather than an agonist, as well as a Gq signaling antagonist. [000297] The data also suggests that the substituent pattern around the A ring may impact binding modes of the compounds to the 5-HT2A receptor, as can the connecting group Z. For some series of compounds agonist versus antagonist binding may depend on the choice of group Z or on the presence or absence of electron-donating or electron-withdrawing groups on the ring A. This permits one to tune the desired activity of the molecule by optimizing these various groups, achieving either strong agonism and/or strong antagonism in the signaling pathways, including mixed agonist/antagonist activities for beta-arrestin signaling (e.g., the compound of Example 34. The data suggests that small electron-donating groups on ring A may promote agonistic beta-arrestin activity, while larger groups or electron-withdrawing groups on ring A may abolish agonistic beta arrestin activity. [000298] While the compounds of the present disclosure provide a range of related receptor binding activities, it should be noted that the desirable antidepressant, anxiolytic, and other CNS therapeutic properties of the hallucinogenic psychedelics is presently believed to be associated with beta-arrestin signaling at the 5-HT2A receptor, it cannot yet be ruled that some degree of Gq signaling is also desirable. While strong Gq signaling results in hallucinogenic effects, it remains possible that there is a degree of Gq signaling that may be beneficial to the therapeutic use of the present compounds without causing an undue risk of hallucinatory behavior. Indeed, in patients without a history of psychosis or hallucinogen persisting perception disorder (HPPD), higher levels of Gq signaling may be permissible, whereas in patients with such history, a compound that is more completely devoid of Gq signaling may be optimal. [000299] Example 183: In Vivo Characterization
[000300] Selected compounds (test compounds) are submitted to rodent functional model assays to determine in vivo efficacy. [000301] Head Twitch Assay. The stereotyped head twitch response induced by 5-HT2A agonists is used as a behavioral proxy for hallucinations. See Halberstadt, et al., Neuropharmacology, 167, 107933 (2020). The head-twitch response is an indicator of hallucinogenic potency of a compound in humans and occurs nearly immediately after the administration of a classical psychedelic in rodents. By definition, a head-twitch is a rapid movement of the head from one side to the other. Head twitch is used to evaluate the potential hallucinogenic liability of compounds (at up to 10 mg/kg) relative to the positive control DOI (2.5 mg/kg). Male C57bl/6 mice (9 weeks of age) are administered test compounds and vehicle via subcutaneous (SC) injection. Mice receiving the positive control DOI receive an intraperitoneal (IP) injection. At 30 minutes post-treatment, the number of head twitch responses is recorded by a blinded observer for 5 minutes. [000302] Open Field Test of Anxiety-Like Behaviors. Adult mice are habituated to the test room for one hour, and then administered a SC injection of test compound (1, 3, or 10 mg/kg) or methylcellulose vehicle, in the hind flank. For testing, animals are placed in one of four arenas in a square apparatus measuring 500 cm x 500 cm for 15 minutes. Sessions are filmed using Anymaze software (Stoelting Co, IL), using a camera mounted to the ceiling. Locomotor activity is measured by the software within the pre-defined arena. An additional center arena is pre- defined for each zone, all with an area of 100 cm x 100 cm. [000303] Rat Social Interaction Test. Test compound (0.3, 1.0, 3 and 3.0 mg/kg, or alternatively, 1, 3, and 10 mg/kg) or vehicle (0.5% aqueous CMC) is injected SC 30 minutes before behavioral testing. During the testing phase, a pair of rats (Sprague-Dawley males) receiving the same treatment are placed in a white Plexiglass open field arena and are allowed to interact for 6 minutes. Social interactions include sniffing the other rat, grooming the other rat, climbing over, under or around the other rat, following the other rat, and exploring the ano- genital area of the other rat. The time the rats spend interacting with each other during the 6- minute test is recorded by a trained observer. Chlordiazepoxide (IP, 5 mg/kg) is used as a positive control. [000304] mTOR signaling in the pre-frontal cortex (PFC). Male adult mice are injected SC with either test compound (1 mg/kg) or vehicle. At 24 hours post-injection, samples from the
PFC region of the brain are collected, and a synaptoneurosome-enriched fraction is collected and prepared for Western blotting., Quantitative analysis of phospho (p) protein immunoblots are determined relative to total levels of each protein. Change in the amount of phosphorylated ERK, Akt, mTOR, and P70S6K proteins, in the PFC are determined relative to vehicle-treated mice, as previously described (Dutheil, et al., J. Neuroscience, 43(5):863-77, 2023). [000305] It is found that compounds according to the disclosure (e.g., Examples 3, 14, 15, 25, 40, 69, 70, 71, 72, 92, 98, 99, 107, 112, 117, 118) elicit non-hallucinogenic activity, increase social interaction, and/or reduce measures of anxiety in test animals. For example, unlike the serotonergic psychedelic DOI, even high doses of a test compound up to 10 mg/kg does not elicit hallucinogenic behavior, as shown by a rate of head twitch comparable to control (e.g., < 10 head twitches per 5 minutes, or less than 5 head twitches or less than 1 head twitch (mean results)) and substantially less than that induced by DOI (p < 0.0001). Test compounds also show a dose dependent increase in social interaction between rats, and the data show that even the lowest test dose of 0.3 mg/kg is effective. In the open-field test, the test compounds are found to dose dependently reduce anxiety-like behaviors, including time in the center arena and number of entries into the center arena, without altering levels of locomotor activity or immobility. The lowest dose tested, 1 mg/kg, is found to be effective. These results demonstrate functional anxiolytic activity. [000306] The test compounds are also found to stimulate mTOR signaling in the mouse medial PFC, as demonstrated by increases in p-ERK, p-mTOR, and p-P70s6k in the tested brain regions. The mTOR signaling pathway has been shown to contribute to neuroplasticity and enhanced cognitive function and it is altered in brain regions associated with major depressive disorder. Rapid-acting antidepressants have been reported to stimulate this pathway in the prefrontal cortex. [000307] Example 184: Pharmacokinetic evaluation [000308] Compounds according to the present disclosure are submitted to a standard testing protocol for oral pharmacokinetics in Sprague-Dawley rats (males, 200-400 g). Test compounds are administered to rats either IV at 1 mg/kg or PO at 10 mg/kg, using a 0.05M citrate phosphate buffer as vehicle. Other potential vehicles include PEG-400 and aqueous 10% Trapposol/1% Tween 80, depending on compound solubility. In some studies, a third arm may utilize subcutaneous dosing (e.g., SC at 1 mg/kg). Plasma samples are collected at 2, 5, 15, and 30
minutes, and 1 , 2, 4, 8, and 24 hours, post-dosing. After processing, plasma samples are analyzed for the presence of the test compound, and for some cases, for the presence of major expected metabolites (e.g., N-des-methyl metabolite). Time to maximum concentration (Tmax), maximal plasma concentration (Cmax), and area-under-the-curve (AUC) are calculated from the data. Comparison between the AUC values for oral versus IV dosing provides the oral bioavailability of the test compound.
[000309] Compounds tested include Examples 40, 99, 107, 112, 117, and 118. The compounds are found to have acceptable oral bioavailability.
[000310] The forgoing examples are merely exemplary and are not meant to limit the scope of the present disclosure in any way.
Claims
CLAIMS What is claimed: 1. A compound of a Formula I
 wherein: X is S, S(O), S(O)2, O, CH2, CHRb, C(Rb)2, NH, N(Ra) (e.g., N(CH3)), N-C(O)-Ra, N- C(O)-O-Ra, N-C(O)-O-CH2-O-Ra, N-CH2-O-C(O)-Ra, N+(=O–), a spiro-joined C3- 6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3- 6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Y is CH2, CHRc, -C(O)-, C(Rc)2, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3- 6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Z is a bond, -S-, S(O), S(O)2, -O-, -NH, N(Rd), -C(O)-, -C(OH)-, -C(OC1-6alkyl), -C(=N- OH)-, -C(=N-OC1-6alkyl)-, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), a spiro- joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), or -O(CH2)pO- wherein p is 2, 3, or 4 (e.g., p is 2), wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; A is H, a C3-6cycloalkyl (e.g., cyclopropyl or cyclohexyl), aryl (e.g., phenyl), or heteroaryl, wherein said cycloalkyl, aryl, or heteroaryl is substituted by 0-5 groups R;
each R is independently selected from aryl (e.g., phenyl), aryloxy (e.g., phenoxy), heteroaryl (e.g., pyridyl), C1-6alkyl (e.g., methyl, ethyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy, ethoxy), C1-6alkylthio (e.g., methylthio), halo (e.g., F), cyano, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), or hydroxy, wherein each of said aryl, heteroaryl, alkyl, haloalkyl, alkylsulfonyl, alkoxy, alkylthio, cycloalkyl, or cycloalkoxy is optionally further substituted by one or more groups selected from aryl (optionally substituted with halo), halo, C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy), C1- 6alkylthio (e.g., methylthio), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), amino, C1-6alkylamino (e.g., methylamino), di(C1-6alkyl)amino (e.g., dimethylamino), (C1-6alkyl)(C1-6alkyl)amino (e.g., methylethylamino), and hydroxy; Ra and Rd, are each independently selected from C1-20alkyl (e.g., methyl or tert-butyl), and C1-2alkylaryl (e.g., benzyl or phenethyl); Rb and Rc are each independently selected from C1-6alkyl (e.g., methyl, ethyl, tert-butyl), C1-6alkoxy, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and C1-2alkylaryl (e.g., benzyl or phenethyl); m is 1 or 2; n is 1, 2, 3, 4, or 5; in free or salt form (e.g., pharmaceutically acceptable salt form); provided that n is not 3 when Z is -C(O)-, X is CH2 or O, and m is 2; and provided that n is not 3 when Z is -C(O)-, X is CH2, and m is 1; and provided that n is not 3 when Z is -C(O)- or -O-, X is NH or N(Ra), and m is 1; and provided that n is not 3 when Z is O, X is NCH3, Y is -C(O)-, and m is 1. 2. The compound according to claim 1, wherein X is NH or N(Ra) (e.g., N(CH3)). 3. The compound according to claim 1, wherein X is S, S(O), S(O)2, or O. 4. The compound according to claim 1, wherein X is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane). 5. The compound according to any of claims 1-4, wherein Y is CH2 or -C(O)-.
6. The compound according to any of claims 1-4, wherein Y is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane). 7. The compound according to any of claims 1-6, wherein Z is a bond, CH2, -C(O)-, or -O-. 8. The compound according to any of claims 1-7, wherein A is a 6-10 membered aryl ring, e.g., selected from phenyl and naphthyl, substituted by 0-5 groups R. 9. The compound according to any of claims 1-7, wherein A is a 5-10 membered heteroaryl ring, substituted by 0-5 groups R. 10. The compound according to any one of claims 1-7, wherein A is selected from furan, thiophene (e.g., thiophen-2-yl), pyrrole, oxazole, thiazole, imidazole, isoxazole, isothiazole, pyrazole, pyridine (e.g., pyrid-4-yl), 2-oxopyridine (e.g., 2-oxopyridin-1(2H)- yl), pyrimidine, pyridazine, pyrazine, benzofuran (e.g., benzofuran-4-yl, or benzofuran-7- yl, or 2-methylbenzofuran-4-yl), dihydrobenzofuran (e.g., 2,3-dihydrobenzofuran-7-yl), benzothiophene, indole (e.g., indol-1-yl, indol-3-yl, or indol-5-yl), benzoxazole, benzothiazole, benzimidazole (e.g., benzo[d]imidazol-1-yl), benzisoxazole (e.g., benzo[d]isoxazol-3-yl, or benzo[d]isoxazol-4-yl), benzisothiazole (e.g., benzo[d]isothiazol-3-yl), benzotriazole (e.g., benzo[d][1,2,3-triazol-1-yl), indazole (e.g., indazol-1-yl, indazol-3-yl, or indazol-7-yl), quinoline (e.g., quinolin-8-yl), isoquinoline (e.g., isoquinolin-7-yl), quinazoline (e.g., quinazolin-7-yl), and quinoxaline (e.g., quinoxalin-5-yl), each substituted by 0-5 groups R. 11. The compound according to any of claims 1-7, wherein A is phenyl substituted by 0-5 group R wherein each group R is independently selected from methyl, trifluoromethyl, methoxy, F, Cl, cyano, hydroxy, 2-methoxyethoxy, and (4-fluorobenzyl)oxy. 12. The compound according to any of claims 1-11, wherein the compound is selected from any one of the compounds of Examples 1 to 180. 13. The compound according to any of claims 1-12, in pharmaceutically acceptable salt form. 14. The compound according to any one of claims 1-13, wherein the compound is an agonist of beta-arrestin signaling via the 5-HT2A receptor. 15. The compound according to any one of claims 1-13, wherein the compound is an antagonist of beta-arrestin signaling via the 5-HT2A receptor.
16. The compound according to any one of claims 1-15, wherein the compound is not an agonist or antagonist of G-q signaling via the 5-HT2A receptor, or is a weak agonist or antagonist thereof. 17. The compound according to any one of claims 1-16, wherein the compound has a bias ratio (beta-arrestin/G-q) for agonism or antagonism 5-HT2A receptor of the of at least 2, or at least 5, or at least 10, or at least 25, or at least 50, or at least 100, or at least 150, or at least 200. 18. A pharmaceutical composition comprising a compound according to any one of claims 1- 17, in free or pharmaceutically acceptable salt form (e.g., pharmaceutically acceptable salt form), in admixture with a pharmaceutically acceptable diluent or carrier. 19. A method for the treatment or prophylaxis of a central nervous system disorder, comprising administering to a patient in need thereof a compound according to any one of claims 1-17, in free or pharmaceutically acceptable salt form, or a pharmaceutical composition according to claim 18. 20. Use of a compound according to any of claims 1-17, in free or pharmaceutically acceptable salt form, or a pharmaceutical composition according to claim 18, in free or pharmaceutically acceptable salt form, in the manufacture of a medicament for the treatment or prophylaxis of a central nervous system disorder.
wherein: X is S, S(O), S(O)2, O, CH2, CHRb, C(Rb)2, NH, N(Ra) (e.g., N(CH3)), N-C(O)-Ra, N- C(O)-O-Ra, N-C(O)-O-CH2-O-Ra, N-CH2-O-C(O)-Ra, N+(=O–), a spiro-joined C3- 6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3- 6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Y is CH2, CHRc, -C(O)-, C(Rc)2, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), or a spiro-joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), wherein said spiro-joined C3-6cycloalkyl or 3-6-membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3- 6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; Z is a bond, -S-, S(O), S(O)2, -O-, -NH, N(Rd), -C(O)-, -C(OH)-, -C(OC1-6alkyl), -C(=N- OH)-, -C(=N-OC1-6alkyl)-, a spiro-joined C3-6cycloalkyl (e.g., cyclopropane), a spiro- joined 3-6-membered heterocycloalkyl (e.g., aziridine or oxetane), or -O(CH2)pO- wherein p is 2, 3, or 4 (e.g., p is 2), wherein said spiro-joined C3-6cycloalkyl or 3-6- membered heterocycloalkyl is optionally substituted by one or more groups selected from C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkoxy (e.g., methoxy), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and hydroxy; A is H, a C3-6cycloalkyl (e.g., cyclopropyl or cyclohexyl), aryl (e.g., phenyl), or heteroaryl, wherein said cycloalkyl, aryl, or heteroaryl is substituted by 0-5 groups R;
each R is independently selected from aryl (e.g., phenyl), aryloxy (e.g., phenoxy), heteroaryl (e.g., pyridyl), C1-6alkyl (e.g., methyl, ethyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy, ethoxy), C1-6alkylthio (e.g., methylthio), halo (e.g., F), cyano, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), or hydroxy, wherein each of said aryl, heteroaryl, alkyl, haloalkyl, alkylsulfonyl, alkoxy, alkylthio, cycloalkyl, or cycloalkoxy is optionally further substituted by one or more groups selected from aryl (optionally substituted with halo), halo, C1-6alkyl (e.g., methyl), haloC1-6alkyl (e.g., trifluoromethyl), C1-6alkylsulfonyl (e.g., methylsulfonyl), C1-6alkoxy (e.g., methoxy), C1- 6alkylthio (e.g., methylthio), C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), amino, C1-6alkylamino (e.g., methylamino), di(C1-6alkyl)amino (e.g., dimethylamino), (C1-6alkyl)(C1-6alkyl)amino (e.g., methylethylamino), and hydroxy; Ra and Rd, are each independently selected from C1-20alkyl (e.g., methyl or tert-butyl), and C1-2alkylaryl (e.g., benzyl or phenethyl); Rb and Rc are each independently selected from C1-6alkyl (e.g., methyl, ethyl, tert-butyl), C1-6alkoxy, C3-6cycloalkyl (e.g., cyclopropyl), C3-6cycloalkoxy (e.g., cyclopropoxy), and C1-2alkylaryl (e.g., benzyl or phenethyl); m is 1 or 2; n is 1, 2, 3, 4, or 5; in free or salt form (e.g., pharmaceutically acceptable salt form); provided that n is not 3 when Z is -C(O)-, X is CH2 or O, and m is 2; and provided that n is not 3 when Z is -C(O)-, X is CH2, and m is 1; and provided that n is not 3 when Z is -C(O)- or -O-, X is NH or N(Ra), and m is 1; and provided that n is not 3 when Z is O, X is NCH3, Y is -C(O)-, and m is 1. 2. The compound according to claim 1, wherein X is NH or N(Ra) (e.g., N(CH3)). 3. The compound according to claim 1, wherein X is S, S(O), S(O)2, or O. 4. The compound according to claim 1, wherein X is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane). 5. The compound according to any of claims 1-4, wherein Y is CH2 or -C(O)-.
6. The compound according to any of claims 1-4, wherein Y is a spiro-joined C3-6cycloalkyl (e.g., cyclopropane). 7. The compound according to any of claims 1-6, wherein Z is a bond, CH2, -C(O)-, or -O-. 8. The compound according to any of claims 1-7, wherein A is a 6-10 membered aryl ring, e.g., selected from phenyl and naphthyl, substituted by 0-5 groups R. 9. The compound according to any of claims 1-7, wherein A is a 5-10 membered heteroaryl ring, substituted by 0-5 groups R. 10. The compound according to any one of claims 1-7, wherein A is selected from furan, thiophene (e.g., thiophen-2-yl), pyrrole, oxazole, thiazole, imidazole, isoxazole, isothiazole, pyrazole, pyridine (e.g., pyrid-4-yl), 2-oxopyridine (e.g., 2-oxopyridin-1(2H)- yl), pyrimidine, pyridazine, pyrazine, benzofuran (e.g., benzofuran-4-yl, or benzofuran-7- yl, or 2-methylbenzofuran-4-yl), dihydrobenzofuran (e.g., 2,3-dihydrobenzofuran-7-yl), benzothiophene, indole (e.g., indol-1-yl, indol-3-yl, or indol-5-yl), benzoxazole, benzothiazole, benzimidazole (e.g., benzo[d]imidazol-1-yl), benzisoxazole (e.g., benzo[d]isoxazol-3-yl, or benzo[d]isoxazol-4-yl), benzisothiazole (e.g., benzo[d]isothiazol-3-yl), benzotriazole (e.g., benzo[d][1,2,3-triazol-1-yl), indazole (e.g., indazol-1-yl, indazol-3-yl, or indazol-7-yl), quinoline (e.g., quinolin-8-yl), isoquinoline (e.g., isoquinolin-7-yl), quinazoline (e.g., quinazolin-7-yl), and quinoxaline (e.g., quinoxalin-5-yl), each substituted by 0-5 groups R. 11. The compound according to any of claims 1-7, wherein A is phenyl substituted by 0-5 group R wherein each group R is independently selected from methyl, trifluoromethyl, methoxy, F, Cl, cyano, hydroxy, 2-methoxyethoxy, and (4-fluorobenzyl)oxy. 12. The compound according to any of claims 1-11, wherein the compound is selected from any one of the compounds of Examples 1 to 180. 13. The compound according to any of claims 1-12, in pharmaceutically acceptable salt form. 14. The compound according to any one of claims 1-13, wherein the compound is an agonist of beta-arrestin signaling via the 5-HT2A receptor. 15. The compound according to any one of claims 1-13, wherein the compound is an antagonist of beta-arrestin signaling via the 5-HT2A receptor.
16. The compound according to any one of claims 1-15, wherein the compound is not an agonist or antagonist of G-q signaling via the 5-HT2A receptor, or is a weak agonist or antagonist thereof. 17. The compound according to any one of claims 1-16, wherein the compound has a bias ratio (beta-arrestin/G-q) for agonism or antagonism 5-HT2A receptor of the of at least 2, or at least 5, or at least 10, or at least 25, or at least 50, or at least 100, or at least 150, or at least 200. 18. A pharmaceutical composition comprising a compound according to any one of claims 1- 17, in free or pharmaceutically acceptable salt form (e.g., pharmaceutically acceptable salt form), in admixture with a pharmaceutically acceptable diluent or carrier. 19. A method for the treatment or prophylaxis of a central nervous system disorder, comprising administering to a patient in need thereof a compound according to any one of claims 1-17, in free or pharmaceutically acceptable salt form, or a pharmaceutical composition according to claim 18. 20. Use of a compound according to any of claims 1-17, in free or pharmaceutically acceptable salt form, or a pharmaceutical composition according to claim 18, in free or pharmaceutically acceptable salt form, in the manufacture of a medicament for the treatment or prophylaxis of a central nervous system disorder.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263478010P | 2022-12-30 | 2022-12-30 | |
| US63/478,010 | 2022-12-30 | ||
| US202363603617P | 2023-11-28 | 2023-11-28 | |
| US63/603,617 | 2023-11-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024145659A1 true WO2024145659A1 (en) | 2024-07-04 |
Family
ID=89898023
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2023/086562 WO2024145659A1 (en) | 2022-12-30 | 2023-12-29 | Heterocycle fused gamma-carbolines acting on the serotonine 5-ht2a receptor |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024145659A1 (en) |
Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4389330A (en) | 1980-10-06 | 1983-06-21 | Stolle Research And Development Corporation | Microencapsulation process |
| GB2145422A (en) | 1983-08-26 | 1985-03-27 | Sandoz Ltd | Novel poly-esters, their preparation and pharmacological use thereof |
| US4530840A (en) | 1982-07-29 | 1985-07-23 | The Stolle Research And Development Corporation | Injectable, long-acting microparticle formulation for the delivery of anti-inflammatory agents |
| US5538739A (en) | 1989-07-07 | 1996-07-23 | Sandoz Ltd. | Sustained release formulations of water soluble peptides |
| WO2000035419A3 (en) | 1998-12-17 | 2000-11-09 | Alza Corp | Conversion of liquid filled gelatin capsules into controlled release systems by multiple coatings |
| US6548493B1 (en) | 1999-06-15 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| EP0058481B2 (en) | 1981-02-16 | 2003-05-21 | AstraZeneca AB | Continuous release pharmaceutical compositions |
| US6713471B1 (en) | 1999-06-15 | 2004-03-30 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| EP1539115A1 (en) | 2002-07-29 | 2005-06-15 | ALZA Corporation | Methods and dosage forms for controlled delivery of paliperidone |
| US7071186B2 (en) | 1999-06-15 | 2006-07-04 | Bristol-Myers Squibb Pharma Co. | Substituted heterocycle fused gamma-carbolines |
| US20080069885A1 (en) | 1993-11-19 | 2008-03-20 | Alkermes, Inc. | Microencapsulated 3-piperidinyl-substituted 1,2-benzisoxazoles and 1,2-benzisothiazoles |
| US20090202631A1 (en) | 2002-07-29 | 2009-08-13 | Yam Nyomi V | Methods and dosage forms for controlled delivery of paliperidone and risperidone |
| US8309722B2 (en) | 2007-03-12 | 2012-11-13 | Intra-Cellular Therapies, Inc. | Substituted heterocycle gamma-carbolines synthesis |
| US20130202692A1 (en) * | 2010-04-22 | 2013-08-08 | Sharon Mates | Organic compounds |
| US8598119B2 (en) | 2008-05-27 | 2013-12-03 | Intra-Cellular Therapies, Inc. | Methods and compositions for sleep disorders and other disorders |
| US8648077B2 (en) | 2008-03-12 | 2014-02-11 | Intra-Cellular Therapies, Inc. | 4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-1-(4-fluorophenyl)-1-butanone toluenesulfonic acid addition salt and salt crystals |
| US9586860B2 (en) | 2012-11-20 | 2017-03-07 | Corning Incorporated | Method of making three dimensional glass ceramic article |
| US20170183350A1 (en) | 2014-04-04 | 2017-06-29 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9708322B2 (en) | 2013-03-15 | 2017-07-18 | Intra-Cellular Therapies, Inc. | Substituted pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalines for inhibiting serotonin reuptake transporter activity |
| WO2017165755A1 (en) * | 2016-03-25 | 2017-09-28 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2018126143A1 (en) | 2016-12-29 | 2018-07-05 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2018126140A1 (en) | 2016-12-29 | 2018-07-05 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10245260B2 (en) | 2016-01-26 | 2019-04-02 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10688097B2 (en) | 2016-03-25 | 2020-06-23 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2020154519A2 (en) * | 2019-01-23 | 2020-07-30 | Intra-Cellular Therapies, Inc. | Methods of treating addiction |
| US20210006009A1 (en) | 2019-07-03 | 2021-01-07 | Eaton Intelligent Power Limited | Locking connector cable secureness attachment assemblies and methods for protecting electrical connections in a hazardous environment |
| US20220041600A1 (en) | 2018-12-17 | 2022-02-10 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines synthesis |
| US20220048910A1 (en) | 2018-12-21 | 2022-02-17 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20220064166A1 (en) | 2018-12-17 | 2022-03-03 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines synthesis |
| US11427587B2 (en) | 2017-07-26 | 2022-08-30 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20220296591A1 (en) * | 2017-07-26 | 2022-09-22 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US11453670B2 (en) | 2018-06-11 | 2022-09-27 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines synthesis |
| CN116444520A (en) * | 2022-01-14 | 2023-07-18 | 上海科技大学 | And cyclic piperidine compound, and preparation method and application thereof |
| CN117069720A (en) * | 2022-05-17 | 2023-11-17 | 上海翊石医药科技有限公司 | 5-HT 2A Receptor agonist, preparation method and application thereof |
-
2023
- 2023-12-29 WO PCT/US2023/086562 patent/WO2024145659A1/en unknown
Patent Citations (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4389330A (en) | 1980-10-06 | 1983-06-21 | Stolle Research And Development Corporation | Microencapsulation process |
| EP0058481B2 (en) | 1981-02-16 | 2003-05-21 | AstraZeneca AB | Continuous release pharmaceutical compositions |
| US4530840A (en) | 1982-07-29 | 1985-07-23 | The Stolle Research And Development Corporation | Injectable, long-acting microparticle formulation for the delivery of anti-inflammatory agents |
| GB2145422A (en) | 1983-08-26 | 1985-03-27 | Sandoz Ltd | Novel poly-esters, their preparation and pharmacological use thereof |
| US5538739A (en) | 1989-07-07 | 1996-07-23 | Sandoz Ltd. | Sustained release formulations of water soluble peptides |
| US20080069885A1 (en) | 1993-11-19 | 2008-03-20 | Alkermes, Inc. | Microencapsulated 3-piperidinyl-substituted 1,2-benzisoxazoles and 1,2-benzisothiazoles |
| WO2000035419A3 (en) | 1998-12-17 | 2000-11-09 | Alza Corp | Conversion of liquid filled gelatin capsules into controlled release systems by multiple coatings |
| US20010036472A1 (en) | 1998-12-17 | 2001-11-01 | Wong Patrick S.-L. | Conversion of liquid filled gelatin capsules into controlled release systems by multiple coatings |
| USRE39679E1 (en) | 1999-06-15 | 2007-06-05 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| US6713471B1 (en) | 1999-06-15 | 2004-03-30 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| US7071186B2 (en) | 1999-06-15 | 2006-07-04 | Bristol-Myers Squibb Pharma Co. | Substituted heterocycle fused gamma-carbolines |
| US7081455B2 (en) | 1999-06-15 | 2006-07-25 | Bristol-Myers Squibb Company | Substituted heterocycle fused gamma-carbolines |
| US7183282B2 (en) | 1999-06-15 | 2007-02-27 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused γ-carbolines |
| USRE39680E1 (en) | 1999-06-15 | 2007-06-05 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| US6552017B1 (en) | 1999-06-15 | 2003-04-22 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| US7238690B2 (en) | 1999-06-15 | 2007-07-03 | Bristol-Myers Squibb Company | Substituted heterocycle fused gamma-carbolines |
| US6548493B1 (en) | 1999-06-15 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| EP1539115A1 (en) | 2002-07-29 | 2005-06-15 | ALZA Corporation | Methods and dosage forms for controlled delivery of paliperidone |
| US20090202631A1 (en) | 2002-07-29 | 2009-08-13 | Yam Nyomi V | Methods and dosage forms for controlled delivery of paliperidone and risperidone |
| US8309722B2 (en) | 2007-03-12 | 2012-11-13 | Intra-Cellular Therapies, Inc. | Substituted heterocycle gamma-carbolines synthesis |
| US8648077B2 (en) | 2008-03-12 | 2014-02-11 | Intra-Cellular Therapies, Inc. | 4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-1-(4-fluorophenyl)-1-butanone toluenesulfonic acid addition salt and salt crystals |
| US9199995B2 (en) | 2008-03-12 | 2015-12-01 | Intra-Cellular Therapies, Inc. | 4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-1-(4-fluorophenyl)-1-butanone toluenesulfonic acid addition salt and salt crystals |
| US9586960B2 (en) | 2008-03-12 | 2017-03-07 | Intra-Cellular Therapies, Inc. | 4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de] quinoxalin-8(7H)-yl)-1-(4-fluorophenyl)-1-butanone toluenesulfonic acid salt crystal forms |
| US8598119B2 (en) | 2008-05-27 | 2013-12-03 | Intra-Cellular Therapies, Inc. | Methods and compositions for sleep disorders and other disorders |
| US20130202692A1 (en) * | 2010-04-22 | 2013-08-08 | Sharon Mates | Organic compounds |
| US8993572B2 (en) | 2010-04-22 | 2015-03-31 | Intra-Cellular Therapies, Inc. | Pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalines derivatives and [1,4]oxazino[2,3,4-hi]pyrido[4,3-b]indole derivatives |
| US9586860B2 (en) | 2012-11-20 | 2017-03-07 | Corning Incorporated | Method of making three dimensional glass ceramic article |
| US9708322B2 (en) | 2013-03-15 | 2017-07-18 | Intra-Cellular Therapies, Inc. | Substituted pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalines for inhibiting serotonin reuptake transporter activity |
| US20170183350A1 (en) | 2014-04-04 | 2017-06-29 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10245260B2 (en) | 2016-01-26 | 2019-04-02 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10688097B2 (en) | 2016-03-25 | 2020-06-23 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2017165755A1 (en) * | 2016-03-25 | 2017-09-28 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2018126143A1 (en) | 2016-12-29 | 2018-07-05 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10906906B2 (en) | 2016-12-29 | 2021-02-02 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10961245B2 (en) | 2016-12-29 | 2021-03-30 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines for treatment of central nervous system disorders |
| WO2018126140A1 (en) | 2016-12-29 | 2018-07-05 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20220296591A1 (en) * | 2017-07-26 | 2022-09-22 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20220372035A1 (en) * | 2017-07-26 | 2022-11-24 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US11427587B2 (en) | 2017-07-26 | 2022-08-30 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US11453670B2 (en) | 2018-06-11 | 2022-09-27 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines synthesis |
| US20220041600A1 (en) | 2018-12-17 | 2022-02-10 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines synthesis |
| US20220064166A1 (en) | 2018-12-17 | 2022-03-03 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines synthesis |
| US20220048910A1 (en) | 2018-12-21 | 2022-02-17 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2020154519A2 (en) * | 2019-01-23 | 2020-07-30 | Intra-Cellular Therapies, Inc. | Methods of treating addiction |
| US20210006009A1 (en) | 2019-07-03 | 2021-01-07 | Eaton Intelligent Power Limited | Locking connector cable secureness attachment assemblies and methods for protecting electrical connections in a hazardous environment |
| CN116444520A (en) * | 2022-01-14 | 2023-07-18 | 上海科技大学 | And cyclic piperidine compound, and preparation method and application thereof |
| CN117069720A (en) * | 2022-05-17 | 2023-11-17 | 上海翊石医药科技有限公司 | 5-HT 2A Receptor agonist, preparation method and application thereof |
Non-Patent Citations (18)
| Title |
|---|
| "Handbook of Clinical Drug Data", 2002, MCGRAW-HILL |
| "Remington's Pharmaceutical Sciences", 2000, LIPPINCOTT WILLIAMS & WILKINS |
| "The Pharmacological Basis of Therapeutics", 2001, MCGRAW HILL |
| BRYANT, H.U. ET AL., LIFE SCI., vol. 15, 1996, pages 1259 - 1268 |
| CELL, vol. 184, 2021, pages 2779 - 92 |
| DATABASE CAPLUS [online] Chemical Abstract Service, Columbus, Ohio, US; 16 May 2023 (2023-05-16), SONG YUNLONG ET AL: "Preparation of quinoxaline derivative and application as 5-HT2A receptoragonist", XP093136355, retrieved from STN Database accession no. 2023:2377458 * |
| DATABASE CAPLUS [online] Chemical Abstract Service, Columbus, Ohio, US; 18 July 2023 (2023-07-18), CHENG JIANJUN ET AL: "Preparation of cyclopiperidine compounds as 5-HT2A receptor for depressiontreatment", XP093136359, retrieved from STN Database accession no. 2023:1474543 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICES, COLUMBUS OHIO; 8 August 2013 (2013-08-08), MATES SHARON ET AL: "Organic compounds", XP093136380, Database accession no. 2016:2120038 * |
| DUTHEIL ET AL., J. NEUROSCIENCE, vol. 43, no. 5, 2023, pages 863 - 77 |
| HALBERSTADT ET AL., NEUROPHARMACOLOGY, vol. 167, 2020, pages 107933 |
| HALL, D.A.STRANGE, P.G., BRIT. J. PHARMACOL., vol. 121, 1997, pages 731 - 736 |
| LI ET AL., JOURNAL OF MEDICINAL CHEMISTRY, vol. 57, 2014, pages 2670 - 2682 |
| NATURE, vol. 589, 2021, pages 474 - 79 |
| PARK, Y.M. ET AL., ANAL. BIOCHEM., vol. 269, 1999, pages 94 - 104 |
| PENG LI ET AL: "Discovery of a Tetracyclic Quinoxaline Derivative as a Potent and Orally Active Multifunctional Drug Candidate for the Treatment of Neuropsychiatric and Neurological Disorders", JOURNAL OF MEDICINAL CHEMISTRY, vol. 57, no. 6, 5 March 2014 (2014-03-05), US, pages 2670 - 2682, XP055545362, ISSN: 0022-2623, DOI: 10.1021/jm401958n * |
| SCIENCE, vol. 375, 2022, pages 403 - 11 |
| WANG, J.B. ET AL., FEBS LETT., vol. 338, 1994, pages 217 - 222 |
| ZHOU, Q.Y., NATURE, vol. 347, 1990, pages 76 - 80 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2017211791B2 (en) | Organic compounds | |
| CA3016353C (en) | Deuterated fused heterocycle gamma-carbolines and compositions thereof useful in the treatment of nervous system disorders | |
| EP2560676B1 (en) | Organic compounds | |
| EP3125893B1 (en) | Deuterated heterocycle fused gamma-carbolines as antagonists of 5-ht2a receptors | |
| EP3562484B1 (en) | Pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline derivatives useful in the treatment of cns disorders | |
| IL303857B1 (en) | Organic compounds | |
| EP3897621A1 (en) | Organic compounds | |
| JP7377871B2 (en) | organic compound | |
| WO2024145659A1 (en) | Heterocycle fused gamma-carbolines acting on the serotonine 5-ht2a receptor | |
| AU2019236860B2 (en) | Organic compounds | |
| AU2011243202B2 (en) | Organic compounds | |
| AU2011243202A1 (en) | Organic compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23851086 Country of ref document: EP Kind code of ref document: A1 |