WO2024029511A1 - 評価方法、複合体及びキット - Google Patents
評価方法、複合体及びキット Download PDFInfo
- Publication number
- WO2024029511A1 WO2024029511A1 PCT/JP2023/028056 JP2023028056W WO2024029511A1 WO 2024029511 A1 WO2024029511 A1 WO 2024029511A1 JP 2023028056 W JP2023028056 W JP 2023028056W WO 2024029511 A1 WO2024029511 A1 WO 2024029511A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- target protein
- nucleic acid
- stranded nucleic
- specific binding
- acid fragment
- Prior art date
Links
- 238000011156 evaluation Methods 0.000 title abstract description 16
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 364
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 363
- 150000007523 nucleic acids Chemical group 0.000 claims abstract description 254
- 239000000126 substance Substances 0.000 claims abstract description 164
- 230000009870 specific binding Effects 0.000 claims abstract description 143
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 95
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 95
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 54
- 238000001514 detection method Methods 0.000 claims abstract description 43
- 238000000034 method Methods 0.000 claims description 106
- 239000003153 chemical reaction reagent Substances 0.000 claims description 86
- 239000007788 liquid Substances 0.000 claims description 51
- 238000007789 sealing Methods 0.000 claims description 30
- 230000014616 translation Effects 0.000 claims description 29
- 238000001243 protein synthesis Methods 0.000 claims description 24
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 claims description 23
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 claims description 23
- 238000003556 assay Methods 0.000 claims description 9
- 238000003776 cleavage reaction Methods 0.000 claims description 8
- 230000007017 scission Effects 0.000 claims description 8
- 238000005406 washing Methods 0.000 claims description 7
- 238000009396 hybridization Methods 0.000 claims description 6
- 230000002194 synthesizing effect Effects 0.000 claims description 6
- 238000006243 chemical reaction Methods 0.000 description 123
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 67
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 description 67
- 239000000758 substrate Substances 0.000 description 35
- 230000000694 effects Effects 0.000 description 26
- 238000000021 kinase assay Methods 0.000 description 26
- 235000018102 proteins Nutrition 0.000 description 24
- 239000000203 mixture Substances 0.000 description 19
- 230000003993 interaction Effects 0.000 description 17
- 239000012530 fluid Substances 0.000 description 16
- 108091000080 Phosphotransferase Proteins 0.000 description 14
- 102000020233 phosphotransferase Human genes 0.000 description 14
- 239000000523 sample Substances 0.000 description 12
- 108010021466 Mutant Proteins Proteins 0.000 description 11
- 102000008300 Mutant Proteins Human genes 0.000 description 11
- 241000283973 Oryctolagus cuniculus Species 0.000 description 11
- 210000004027 cell Anatomy 0.000 description 11
- 239000002245 particle Substances 0.000 description 11
- 239000000243 solution Substances 0.000 description 11
- 102000004150 Flap endonucleases Human genes 0.000 description 10
- 108090000652 Flap endonucleases Proteins 0.000 description 10
- 230000035772 mutation Effects 0.000 description 10
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 9
- 108091034117 Oligonucleotide Proteins 0.000 description 9
- 238000010586 diagram Methods 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- 229920005989 resin Polymers 0.000 description 9
- 239000011347 resin Substances 0.000 description 9
- -1 A-RAF Proteins 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 230000035484 reaction time Effects 0.000 description 7
- 108020004414 DNA Proteins 0.000 description 6
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 6
- 230000027455 binding Effects 0.000 description 6
- 230000000903 blocking effect Effects 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000011259 mixed solution Substances 0.000 description 6
- 239000003656 tris buffered saline Substances 0.000 description 6
- 238000002965 ELISA Methods 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000013519 translation Methods 0.000 description 5
- 102100033072 DNA replication ATP-dependent helicase DNA2 Human genes 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 101000927313 Homo sapiens DNA replication ATP-dependent helicase DNA2 Proteins 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- 230000003321 amplification Effects 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- 238000002372 labelling Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 238000003199 nucleic acid amplification method Methods 0.000 description 4
- 239000013076 target substance Substances 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical group C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 3
- 239000000232 Lipid Bilayer Substances 0.000 description 3
- 238000004364 calculation method Methods 0.000 description 3
- 239000003431 cross linking reagent Substances 0.000 description 3
- 230000002939 deleterious effect Effects 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 238000012252 genetic analysis Methods 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 230000037435 normal mutation Effects 0.000 description 3
- 108091005981 phosphorylated proteins Proteins 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 238000001179 sorption measurement Methods 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 229920005992 thermoplastic resin Polymers 0.000 description 3
- DSNRWDQKZIEDDB-SQYFZQSCSA-N 1,2-dioleoyl-sn-glycero-3-phospho-(1'-sn-glycerol) Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@@H](O)CO)OC(=O)CCCCCCC\C=C/CCCCCCCC DSNRWDQKZIEDDB-SQYFZQSCSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- 229920000089 Cyclic olefin copolymer Polymers 0.000 description 2
- 101001005602 Homo sapiens Mitogen-activated protein kinase kinase kinase 11 Proteins 0.000 description 2
- 101001018196 Homo sapiens Mitogen-activated protein kinase kinase kinase 5 Proteins 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- 102000043136 MAP kinase family Human genes 0.000 description 2
- 108091054455 MAP kinase family Proteins 0.000 description 2
- 102100025207 Mitogen-activated protein kinase kinase kinase 11 Human genes 0.000 description 2
- 102100033127 Mitogen-activated protein kinase kinase kinase 5 Human genes 0.000 description 2
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 101150048834 braF gene Proteins 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000005018 casein Substances 0.000 description 2
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 2
- 235000021240 caseins Nutrition 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 210000004748 cultured cell Anatomy 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 238000005530 etching Methods 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 230000005484 gravity Effects 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000031146 intracellular signal transduction Effects 0.000 description 2
- 238000010030 laminating Methods 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000003753 real-time PCR Methods 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 108091008743 testicular receptors 4 Proteins 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- MWRBNPKJOOWZPW-NYVOMTAGSA-N 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCC\C=C/CCCCCCCC MWRBNPKJOOWZPW-NYVOMTAGSA-N 0.000 description 1
- 102000052866 Amino Acyl-tRNA Synthetases Human genes 0.000 description 1
- 108700028939 Amino Acyl-tRNA Synthetases Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 229940124647 MEK inhibitor Drugs 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102000002508 Peptide Elongation Factors Human genes 0.000 description 1
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 1
- 102000005877 Peptide Initiation Factors Human genes 0.000 description 1
- 108010044843 Peptide Initiation Factors Proteins 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 241000209140 Triticum Species 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 150000007824 aliphatic compounds Chemical class 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000012742 biochemical analysis Methods 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000007847 digital PCR Methods 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 125000004185 ester group Chemical class 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000001746 injection moulding Methods 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000002923 metal particle Substances 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000007918 pathogenicity Effects 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000006916 protein interaction Effects 0.000 description 1
- 238000010384 proximity ligation assay Methods 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 210000001995 reticulocyte Anatomy 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 238000003466 welding Methods 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
Images


















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M1/00—Apparatus for enzymology or microbiology
- C12M1/34—Measuring or testing with condition measuring or sensing means, e.g. colony counters
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
Definitions
- the present invention relates to a method for evaluating the activity of a mutant protein. More specifically, the present invention provides a method for evaluating whether a target protein binds to a target protein, a complex, a kit for evaluating whether a target protein binds to a target protein, and a method for evaluating whether a target protein binds to a target protein.
- the present invention relates to a method for evaluating whether or not a protein is phosphorylated.
- VUS unknown clinical significance
- VUS VUS protein-binding protein
- a method has been established for qualitative evaluation by detecting the cell extract containing the cells by Western blotting or the like.
- evaluation methods require a lot of time, effort, and cost.
- Digital measurement includes digital ELISA, digital PCR, and the like.
- the sample solution is divided into an extremely large number of microsolutions. Then, the signals from each microsolution are binarized, only the presence or absence of the target substance is determined, and the number of molecules of the target substance is measured. Digital measurement can significantly improve detection sensitivity and quantitative performance compared to conventional ELISA, real-time PCR, and the like.
- Non-Patent Document 1 describes a microwell array having microwells and channels for supplying reagents, etc., and describes that digital ELISA was performed using the microwell array.
- Patent Document 1 reports Proximity Ligation Assay (also referred to as PLA), which is a method for detecting proteins using antibodies modified with oligonucleotides. This method utilizes the PCR method or the RCA method for detection.
- PLA Proximity Ligation Assay
- Patent Document 2 describes a method for detecting protein interactions between two molecules using antibodies modified with oligonucleotides. This method also utilizes the PCR method or RCA method for detection.
- Non-Patent Document 3 describes a microwell array having microwells and flow channels for supplying reagents, etc., in which cells are used to express signal transduction and detection of phosphorylated proteins. It says what happened. Signal transduction is monitored by detecting phosphorylated proteins.
- Alpha SureFire https://www.perkinelmer.co.jp/assays/tabid/346/Default.aspx, PerkinElmer is an assay system that can detect kinase activity on a cell basis without washing. This system detects phosphorylated proteins from cell extracts.
- Kan C. W., et al. Isolation and detection of single molecules on paramagnetic beads using sequential fluid flows in microfabricated polymer array assemblies., Lab on a Chip, 12 (5), 977-985, 2012.
- Mohammed H., et al. Approaches for Assessing and Discovering Protein Interactions in Cancer., Mol Cancer Res, 11 (11), 1295-1302, 2013.
- Blazek M., et al. Proximity Ligation Assay for High-content Profiling of Cell Signaling Pathways on a Microfluidic Chip., Molecular & Cellular Proteomics 12: 10.1074/mcp.M113.032821, 3898-3907, 2013 .
- FIG. 1 shows, by way of example, part of mitogen-activated protein kinase (MAPK) signaling.
- MAPK mitogen-activated protein kinase
- An object of the present invention is to provide a technique for easily and quickly evaluating the activity of mutant proteins.
- a method for evaluating whether or not a target protein binds to a target protein which comprises, in a container, labeling the target protein with the target protein, the target protein, and a first single-stranded nucleic acid fragment. a first specific binding substance and a second specific binding substance for the target protein labeled with a second single-stranded nucleic acid fragment, and as a result, the target protein binds to the target protein.
- a complex containing the target protein, the target protein, the first specific binding substance, and the second specific binding substance is formed, and at least a portion of the first single-stranded nucleic acid fragment is and a step (a) of hybridizing at least a portion of the second single-stranded nucleic acid fragment to form a double-stranded nucleic acid, and a step (b) of detecting the formation of the double-stranded nucleic acid. , wherein the detection of the formation of the double-stranded nucleic acid indicates that the protein of interest binds to the target protein.
- the step (a) includes a step (a1) of synthesizing the target protein using a cell-free protein synthesis system in the container, and a step (a1) of synthesizing the target protein, the target protein, and the first protein in the container.
- a specific binding substance and the second specific binding substance are brought into contact, and as a result, the target protein binds to the target protein, the target protein, the target protein, and the first specific binding A complex containing the substance and the second specific binding substance is formed, and at least a portion of the first single-stranded nucleic acid fragment and at least a portion of the second single-stranded nucleic acid fragment hybridize.
- the method according to [1], comprising the step (a2) of forming a double-stranded nucleic acid.
- the target protein, the target protein, the first specific binding substance, and the second specific binding substance are further contacted with adenosine triphosphate (ATP). , the method described in [2].
- ATP adenosine triphosphate
- [4] The method according to any one of [1] to [3], which does not include a washing step.
- [5] The method according to any one of [1] to [4], wherein the container is a well, and the well has a volume of 10 fL to 100 pL.
- [6] The method according to any one of [1] to [5], wherein the first single-stranded nucleic acid fragment and the second single-stranded nucleic acid fragment each have a base length of 10 to 200 bases.
- the step of detecting the formation of double-stranded nucleic acid is performed by Invasive Cleavage Assay.
- a kit for evaluating whether a target protein binds to a target protein comprising a well array having a plurality of wells, a first well array for the target protein labeled with a first single-stranded nucleic acid fragment, and a well array having a plurality of wells. and a second specific binding substance for the target protein labeled with a second single-stranded nucleic acid fragment.
- a reagent for detecting a double-stranded nucleic acid formed by hybridization of at least a portion of the first single-stranded nucleic acid fragment and at least a portion of the second single-stranded nucleic acid fragment is provided.
- a second specific binding substance for the protein, a third specific binding substance for the phosphorylated target protein labeled with a third single-stranded nucleic acid fragment, and ATP are brought into contact with each other, so that the When the target protein is phosphorylated, a complex containing the target protein, the second specific binding substance, and the third specific binding substance is formed, and the second single-stranded nucleic acid fragment is phosphorylated.
- VUS can be easily and efficiently functionally analyzed (that is, evaluated) in a short period of time.
- FIG. 1 is a diagram illustrating normal mutations and deleterious mutations.
- FIG. 2 is a schematic diagram illustrating a method for evaluating whether a target protein binds to a target protein.
- FIG. 3 is a schematic cross-sectional view showing an example of a fluidic device.
- FIG. 4 is a schematic cross-sectional view illustrating a method for evaluating whether a target protein binds to a target protein.
- FIG. 5 is a schematic cross-sectional view illustrating a method for evaluating whether a target protein binds to a target protein.
- FIG. 6 is a schematic cross-sectional view showing an example of a fluidic device.
- FIG. 1 is a diagram illustrating normal mutations and deleterious mutations.
- FIG. 2 is a schematic diagram illustrating a method for evaluating whether a target protein binds to a target protein.
- FIG. 3 is a schematic cross-sectional view showing an example of a fluidic device.
- FIG. 4 is
- FIG. 7 is a schematic cross-sectional view illustrating a method for evaluating whether a target protein binds to a target protein.
- FIG. 8 is a schematic cross-sectional view illustrating a method for evaluating whether a target protein binds to a target protein.
- FIG. 9 is a schematic diagram illustrating an example of the Invasive Cleavage Assay (ICA) method.
- FIG. 10 is a schematic diagram illustrating a method for evaluating the activity of a target protein.
- FIG. 11 is a schematic diagram illustrating a method for evaluating whether a target protein phosphorylates a target protein.
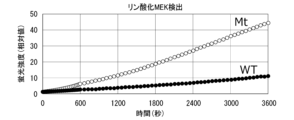
- FIG. 12 is a graph showing the results of Experimental Example 1.
- FIG. 13 is a graph showing the results of Experimental Example 1.
- FIG. 14 is a graph showing the results of Experimental Example 1.
- FIG. 15 is a graph showing the results of Experimental Example 2.
- FIG. 16 is a graph showing the results of Experimental Example 3.
- FIG. 17 is a graph showing the results of Pattern 1 of Experimental Example 4.
- FIG. 18 is a graph showing the results of Pattern 2 of Experimental Example 4.
- FIG. 19 is a histogram of fluorescence intensity measured over time in the immuno-ICA reaction of Experimental Example 5.
- the present invention provides a method for evaluating whether or not a target protein binds to a target protein, wherein the target protein, the target protein, and a first single-stranded nucleic acid fragment are labeled in a container.
- a first specific binding substance for the target protein and a second specific binding substance for the target protein labeled with a second single-stranded nucleic acid fragment are brought into contact with each other, and as a result, the target protein binds to the target protein, a complex containing the target protein, the target protein, the first specific binding substance, and the second specific binding substance is formed, and the first single-stranded a step (a) of hybridizing at least a portion of the nucleic acid fragment and at least a portion of the second single-stranded nucleic acid fragment to form a double-stranded nucleic acid; and a step (a) of detecting the formation of the double-stranded nucleic acid. b), wherein the detection of the formation of the double-stranded nucleic acid indicates that the protein of interest binds to the target protein.
- FIG. 2 is a schematic diagram illustrating the method of this embodiment.
- target protein 110, target protein 120, and first single-stranded nucleic acid fragment 131 are labeled with target protein 110, target protein 120, and first single-stranded nucleic acid fragment 131.
- a first specific binding substance 130 for target protein 110 and a second specific binding substance 140 for target protein 120 labeled with a second single-stranded nucleic acid fragment 141 are contacted.
- the target protein 110 binds to the target protein 120, a complex containing the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140 is formed, and the At least a portion of the first single-stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141 hybridize to form a double-stranded nucleic acid (also referred to as a double-stranded nucleic acid region) 150.
- a double-stranded nucleic acid also referred to as a double-stranded nucleic acid region
- step (b) the formation of double-stranded nucleic acid 150 is detected. If formation of double-stranded nucleic acid 150 is detected, it can be said that target protein 110 binds (that is, interacts) with target protein 120.
- the method of this embodiment may be applied when the target protein 110 does not bind (that is, does not interact) with the target protein 120, and in that case, the formation of the double-stranded nucleic acid 150 is not detected. Detection of the formation of double-stranded nucleic acid 150 will be described later.
- activity evaluation of mutant proteins can be carried out easily and in a short period of time.
- VUS can be easily and efficiently functionally analyzed (ie, evaluated) in a short period of time.
- the first single-stranded nucleic acid fragment 131 is The base length may be 10 to 200 bases.
- the base length of the second single-stranded nucleic acid fragment 141 may also be 10 to 200 bases.
- the length of the double-stranded nucleic acid 150 formed by hybridization of at least a portion of the first single-stranded nucleic acid fragment 121 and at least a portion of the second single-stranded nucleic acid fragment 131 is 7 to 30 mm. It is preferable that the number of bases is about a base, and for example, it may be 9 bases, 12 bases, or 15 bases.
- the target protein 110 may be a protein having a genetic mutation of unknown clinical significance (ie, VUS).
- VUS a genetic mutation of unknown clinical significance
- Target protein 110 may be, for example, a kinase. More specific target proteins include proteins in intracellular signal transduction pathways, such as BRAF, A-RAF, Raf, MAP3K 4/12, MAP3K11, ASK1, and TAK1.
- BRAF protein having a genetic mutation of unknown clinical significance
- the container is preferably a well, and the well preferably constitutes a well array in which a plurality of wells are arranged. Further, the well array is preferably arranged within the flow path of the fluidic device.
- FIG. 3 is a schematic cross-sectional view showing an example of a fluidic device that can suitably implement the method of this embodiment.
- the fluidic device 200 includes a substrate 210 and a lid member 220 disposed opposite to the substrate 210.
- the lid member 220 has a convex portion 221.
- the tip of the protrusion 221 is in contact with the substrate 210.
- the well array 240 is integrally molded with the substrate 210 on one side of the substrate 210, and faces the lid member 220.
- Well array 240 has a plurality of wells 241.
- the lid member 220 may be welded or bonded to the substrate 210.
- the well 241 is open to the surface of the substrate 210. Although the shape, dimensions, and arrangement of the well 241 are not particularly limited, it is preferable that the well 241 be a microwell with a small volume. For example, the volume of one well 241 may be about 10 fL to 100 pL.
- a plurality of wells 241 having the same shape and size constitute a well array 240.
- the same shape and same size may be defined as having the same shape and the same capacity to the extent required for digital measurement, and variations to the extent of manufacturing errors are acceptable.
- the diameter of the well 241 may be, for example, about 1 to 10 ⁇ m.
- the depth of the well 241 may be, for example, about 1 to 10 ⁇ m.
- the arrangement of the wells 241 is not particularly limited; for example, they may be arranged in a triangular lattice shape, a square lattice shape, or randomly arranged.
- a space is formed between the well array 240 and the lid member 220 due to the presence of the convex portion 221.
- the space constitutes a flow path 230.
- the channel 230 functions as a path for transporting a liquid in which the target protein, target protein, first specific binding substance, second specific binding substance, etc. are dispersed, and a sealing liquid to be described later.
- the shape, structure, capacity, etc. of the flow path 230 are not particularly limited, the height of the flow path 230 (distance between the surface of the substrate 210 and the surface of the lid member 220 facing the substrate 210) is, for example, 500 ⁇ m or less. It may be, for example, 300 ⁇ m or less, for example, 200 ⁇ m or less, or, for example, 100 ⁇ m or less.
- the convex portion 221 may be integrally formed with the lid member 220.
- the lid member 220 can be formed into a plate shape having a convex portion 221 by, for example, molding a fluid of thermoplastic resin using a mold. Further, the lid member 220 may be formed with a reagent introduction port 222 and a reagent discharge port 223.
- the lid member 220 When the lid member 220 has the convex portion 221, the lid member 220 and the substrate 210 are stacked so that the convex portion 221 contacts the surface of the substrate 210 where the well 241 opens. As a result, the space between the lid member 220 and the substrate 210 becomes a flow path 230.
- the lid member 220 and the substrate 210 may be welded together by laser welding or the like.
- FIG. 6 is a schematic cross-sectional view showing an example of a fluidic device.
- the fluidic device 500 includes a substrate 210 and a wall member 510.
- the well array 240 is integrally molded with the substrate 210 on one side of the substrate 210.
- Well array 240 has a plurality of wells 241.
- Fluid device 500 differs from fluid device 200 described above primarily in that it does not include lid member 220.
- the lid member 220 and the convex portion 221 are integrally molded.
- the lid member 220 and the convex portion 221 may be molded separately.
- the well array 240 is integrally molded with the substrate 210 on one side of the substrate 210.
- the well array does not have to be integrally molded with the substrate 210.
- a well array 240 molded separately from the fluidic device, may be placed on the substrate 210 of the fluidic device.
- a resin layer may be laminated on the surface of the substrate 210, and a well array may be formed on the resin layer by etching or the like.
- the substrate 210 is formed using resin, for example.
- the type of resin is not particularly limited, it is preferably a resin that is resistant to reagents and sealing liquid.
- a resin with little autofluorescence is preferable.
- resins with low autofluorescence include, but are not limited to, cycloolefin polymers, cycloolefin copolymers, silicones, polypropylene, polycarbonates, polystyrene, polyethylene, polyvinyl acetate, fluororesins, and amorphous fluororesins.
- a plurality of wells 241 may be formed on one surface of the substrate 210 in the thickness direction.
- Methods for forming wells using resin include injection molding, thermal imprinting, optical imprinting, and the like.
- a well array may be formed by laminating a fluororesin on the substrate 210 and processing the fluororesin by etching or the like.
- a fluororesin for example, CYTOP (registered trademark) (Asahi Glass) or the like can be used.
- the material of the lid member 220 is preferably a resin with low autofluorescence, and may be, for example, a thermoplastic resin such as a cycloolefin polymer or a cycloolefin copolymer.
- the lid member 220 may be made of a material that does not transmit light of a wavelength near the wavelength detected when observing signals by fluorescence, or may be made of a material that does not transmit light completely.
- the lid member 220 may be made of thermoplastic resin to which carbon or metal particles are added.
- the method of this embodiment is a method for evaluating whether or not a target protein binds to a target protein, in which a well 241 is labeled with a target protein 110, a target protein 120, and a first single-stranded nucleic acid fragment 131.
- a first specific binding substance 130 for the target protein 110 and a second specific binding substance 140 for the target protein 120 labeled with a second single-stranded nucleic acid fragment 141 are brought into contact with each other, and as a result, the target protein 110 binds to the target protein 120, a complex 100 containing the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140 is formed, and the first one Step (a) in which at least a portion of the stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141 hybridize to form a double-stranded nucleic acid 150; and detecting the formation of the double-stranded nucleic acid 150. (b), and the detection of the formation of double-stranded nucleic acid 150 indicates that the target protein 110 binds to the target protein 120.
- the evaluation method may be the following detection method.
- the detection method of the present embodiment includes, in the well 241, the target protein 110, the target protein 120, the first specific binding substance 130 for the target protein 110 labeled with the first single-stranded nucleic acid fragment 131, and , contacting a second specific binding substance 140 for the target protein 120 labeled with a second single-stranded nucleic acid fragment 141; binding the target protein 110 to the target protein 120;
- a complex 100 is formed that includes a target protein 120, a first specific binding substance 130, and a second specific binding substance 140, and includes at least a portion of the first single-stranded nucleic acid fragment 131 and the second single-stranded nucleic acid fragment 131.
- the method may include hybridizing at least a portion of the nucleic acid fragment 141 to form a double-stranded nucleic acid 150, and detecting the formation of the double-stranded nucleic acid 150.
- mutant proteins include proteins with VUS.
- the reagent liquid L210 is introduced from the introduction port 222 of the fluidic device 200 and sent to the channel 230.
- Reagent liquid L210 is a liquid in which target protein 110, target protein 120, first specific binding substance 130, and second specific binding substance 140 are dispersed, and is used to detect the formation of double-stranded nucleic acid 150. Also includes reagents.
- the reagent solution L210 sent to the channel 230 comes into contact with the well array 240. Then, the reagent solution L210 is accommodated inside the well 241. As a result, the target protein 110, the target protein 120, the first specific binding substance 130, the second specific binding substance 140, and a reagent for detecting the formation of the double-stranded nucleic acid 150 are introduced into the well 241. be done.
- the reagent solution L210 contains the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140. A complex 100 is included.
- the number of complexes 100 introduced into one well 241 is not particularly limited, but preferably one or less, that is, zero or one complex 100, is introduced into one well 241. Thereby, detection of the complex 100 can be performed one by one, that is, digital measurement becomes possible. Furthermore, it is not necessary to introduce the complex 100 into all wells of the well array.
- the means for introducing the complex 100 into the well is not particularly limited, and examples thereof include a method of allowing the complex 100 to settle within the fluid device (specifically, within the channel 230) by its own weight and then distributing it to the well 241. .
- a substance that captures the complex 100 also referred to as a captured substance
- the efficiency of introducing the complex 100 into the well can also be improved by trapping the complex 100 that has been sent.
- the step of binding the capture substance to the complex 100 can be performed at any point in the method of this embodiment.
- this step may be performed by bringing the complex 100 into contact with the capture object in the sample tube before the step of introducing the complex 100 into the well 241.
- the complex 100 may be introduced into the well, and the captured substance and the complex 100 may be brought into contact within the well.
- a capture substance is a substance that can capture the complex 100.
- the capture material may be, for example, a combination of a solid phase and a substance that specifically binds to the complex 100.
- Examples of the solid phase include particles, films, and substrates.
- the number of specific binding substances for the complex 100 may be one or two or more.
- the number of specific binding substances may be three, four, or five or more.
- the particles are not particularly limited, and include polymer particles, magnetic particles, glass particles, and the like.
- the particles are surface-treated to avoid non-specific adsorption.
- particles having a functional group such as a carboxyl group on the surface are preferable. More specifically, a product such as "Magnosphere LC300" manufactured by JSR Corporation can be used.
- Specific binding substances in the first specific binding substance 130, second specific binding substance 140, and capture material include antibodies, antibody fragments, aptamers, and the like.
- antibody fragments include Fab, F(ab') 2 , Fab', single chain antibodies (scFv), disulfide stabilized antibodies (dsFv), dimer V region fragments (diabodies), and peptides containing CDRs. It will be done.
- the antibody may be a monoclonal antibody or a polyclonal antibody. Alternatively, a commercially available antibody may be used.
- a method for labeling a specific binding substance with a single-stranded nucleic acid fragment includes a method using a crosslinking agent.
- a single-stranded nucleic acid fragment may be labeled with a specific binding substance via a linker molecule.
- the linker is not particularly limited, and examples thereof include polyethylene chains, hydrocarbon chains, peptides, and the like.
- a single-stranded nucleic acid fragment may be DNA or RNA. Furthermore, it may contain artificial nucleic acids such as BNA and LNA.
- Methods for immobilizing specific binding substances on particle surfaces are not particularly limited, and include physical adsorption, chemical bonding, avidin-biotin bonding, and protein G or protein A bonding with antibodies.
- a method using examples include methods in which a specific binding substance is immobilized on the particle surface by hydrophobic interaction or electrostatic interaction.
- Examples of the method using chemical bonding include a method using a crosslinking agent. For example, when the surface of the particle has a hydroxyl group, the carboxyl group of the specific binding substance is reacted with a crosslinking agent to form an active ester, and then the hydroxyl group and this ester group are reacted to form a specific binding substance on the particle. It can be immobilized on a surface. Furthermore, it is preferable to provide a spacer between the specific binding substance and the particle surface so as not to inhibit the ability of the specific binding substance to recognize the target molecule.
- the combination of the captured substance and the complex 100 is formed under the condition that 0 or 1 complex 100 is captured in one captured substance. It is preferable to form Furthermore, it is preferable that one well 241 be configured so that zero or one captured substance is introduced. This allows digital measurement.
- a complex 100 containing them is formed. is formed, and at least a portion of the first single-stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141 hybridize to form a double-stranded nucleic acid 150. Formation of the complex 100 may be performed within the sample tube or within the well 241.
- a step of sealing the opening of the well 241 may be performed.
- the method of sealing the opening of the well 241 is not particularly limited as long as it can prevent the liquid contained in one well 241 from mixing with the liquid contained in another well 241.
- the opening of the well 241 may be sealed by covering it with a sealing liquid.
- the opening of the well 241 may be sealed by laminating a plate-like member such as a glass plate.
- the sealing liquid L220 is sent from the introduction port 222 of the lid member 220 to the flow path 230 between the substrate 210 and the lid member 220.
- the sealing liquid L220 sent to the channel 230 contacts the well array 240.
- the sealing liquid L220 sweeps away and replaces the reagent liquid L210 that is not accommodated in the well 241 among the reagent liquid L210 sent to the flow path 230.
- the sealing liquid L220 individually seals the plurality of wells 241 containing the reagent liquid L210 containing the target substance 110, and the wells 241 become independent reaction spaces (also referred to as microcompartments 242).
- FIG. 5 shows a state in which all the wells 241 of the well array 240 are sealed with the sealing liquid L220, and sealed wells (that is, microcompartments) 242 are formed.
- lipid bilayer membrane is formed at the opening of the well 241. It is also possible to form a sealed well 242 by forming a plurality of wells 241 and individually sealing each of the plurality of wells 241 with the lipid bilayer membrane.
- lipids that form a lipid bilayer include 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (also referred to as DOPE) and 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG). ) and mixtures thereof, but are not limited to these.
- the sealing liquid is a liquid that can individually seal the liquids introduced into the plurality of wells 241 so that they do not mix with each other to form droplets (also referred to as microdroplets), and is preferably an oil-based solution. and more preferably oil.
- oil fluorine oil, silicone oil, hydrocarbon oil, or a mixture thereof can be used. More specifically, a product such as "FC-40” manufactured by Sigma Corporation can be used.
- FC-40 (CAS number: 86508-42-1) is a fluorinated aliphatic compound with a specific gravity of 1.85 g/mL at 25°C.
- Detection of the formation of double-stranded nucleic acid 150 is preferably performed using a signal amplification reaction.
- signal amplification reactions include Invasive Cleavage Assay (also referred to as ICA).
- the ICA reaction is related to the principle that signal amplification proceeds through a cycle of two reactions: (1) complementary binding between nucleic acids, and (2) recognition and cleavage of a triplex structure by an enzyme.
- the ICA reaction is less affected by reaction cycle inhibition caused by impurities. Therefore, by using the ICA reaction, the formation of the double-stranded nucleic acid 150 can be detected with high accuracy.
- the reagent solution L210 that is, the liquid containing the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140
- the reagent solution L210 is used for the ICA reaction. Contains the reaction reagents necessary for
- Reaction reagents necessary for the ICA reaction include ICA reaction reagents such as flap probes, flap endonucleases (also referred to as FEN), and fluorescent substrates.
- the flap probe is a nucleic acid fragment designed to hybridize to the first single-stranded nucleic acid fragment 131 or the second single-stranded nucleic acid fragment 141 to form a flap structure with the double-stranded nucleic acid 150.
- FIG. 9 is a schematic diagram illustrating an example of the ICA method.
- a double-stranded nucleic acid 150 is formed by hybridizing at least a portion of the first single-stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141 by the ICA method. Detect.
- a flap probe is hybridized to the first single-stranded nucleic acid fragment 131 or the second single-stranded nucleic acid fragment 141.
- flap probe 810 hybridizes to first single-stranded nucleic acid 131.
- a first flap portion 811 is formed.
- the first flap site 811 when the first flap site 811 is reacted with FEN, the first flap site 811 is cleaved and a nucleic acid fragment 811 is generated. Subsequently, the nucleic acid fragment 811 hybridizes to a fluorescent substrate (ie, the nucleic acid fragment 820) to form a second flap region 821.
- a fluorescent substrate ie, the nucleic acid fragment 820
- a fluorescent substance F is bound to the 5' end of the nucleic acid fragment 820, and a quenching substance Q is bound to the 3' side of the 5' end of the nucleic acid fragment 820.
- the second flap site 821 is reacted with FEN, the second flap site 821 is cleaved and a nucleic acid fragment 821 is generated.
- the fluorescent substance F separates from the quencher Q and generates a fluorescent signal. By detecting this fluorescent signal, the formation of double-stranded nucleic acid 150 can be detected.
- reagent liquid L210 a general liquid used in biochemical analysis performed using a fluidic device can be used, and preferably an aqueous solution. Further, by including a surfactant or the like in the reagent liquid L210, it may be possible to easily seal the liquid in the well.
- fluorescent substance F is liberated from quencher Q by an enzymatic reaction based on an isothermal reaction, and corresponds to excitation light. to emit a predetermined fluorescent signal.
- a known appropriate method can be selected depending on the type of signal to be detected. For example, when observing a fluorescent signal, the sealed well 242 is irradiated with excitation light corresponding to the fluorescent substance, and the fluorescence emitted by the fluorescent substance is observed. For example, as shown in FIG. 5, a predetermined reaction is performed in a sealed well 242, and the generated signal is observed.
- a sealed well 242R is a well in which a signal was detected
- a sealed well 242 is a well in which no signal was detected.
- the reagent liquid L210 is introduced into the fluidic device 500.
- Reagent liquid L210 is a liquid in which target protein 110, target protein 120, first specific binding substance 130, and second specific binding substance 140 are dispersed, and is used to detect the formation of double-stranded nucleic acid 150.
- the reagent solution L210 contains the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140.
- a complex 100 is included.
- the concentration of the complex 100 is preferably adjusted to a concentration such that one molecule or less of the complex 100 enters the well 241 per well.
- the sealing liquid L220 is introduced into the fluidic device 500.
- the specific gravity of the sealing liquid L220 is greater than that of the reagent liquid L210. Therefore, the sealing liquid L220 sinks below the reagent liquid L210 that is not accommodated in the well 241 among the reagent liquids L210 and comes into contact with the well array 240. Then, the sealing liquid L220 individually seals the plurality of wells 241 containing the reagent liquid L210 containing the complex 100, thereby forming independent reaction spaces (also referred to as microcompartments) 242.
- microcompartment 242R is a well in which a signal was detected
- microcompartment 242 is a well in which no signal was detected.
- step (a) the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140 are brought into contact with each other in a container.
- the target protein 110 binds to the target protein 120, a complex 100 containing the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140 is formed.
- Step (a) of hybridizing at least a portion of the first single-stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141 to form the double-stranded nucleic acid 150 is performed in the container, Step (a1) of synthesizing the target protein 110 using a cell-free protein synthesis system, and combining the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140 in a container. contact, and as a result, when the target protein 110 binds to the target protein 120, the complex 100 containing the target protein 110, the target protein 120, the first specific binding substance 130, and the second specific binding substance 140 is formed.
- the target protein 110 may be synthesized using a cell-free protein synthesis system, and the formation of the complex 100 may be performed continuously with the synthesis of the target protein 110.
- the target protein is a kinase or the like containing a genetic mutation, the activity evaluation of the target protein can be carried out easily and in a short period of time.
- a cell-free protein synthesis system does not synthesize proteins within cells, but rather uses ribosomes, transcription and translation factors, etc. derived from living cells or artificially synthesized to synthesize proteins from nucleic acid templates in vitro. Refers to the synthetic system to be synthesized.
- the cell-free protein synthesis system may include a transcription process in addition to the translation process.
- the nucleic acid that encodes a protein is DNA
- the cell-free protein synthesis system may contain factors that enable transcription. Examples of factors that enable transcription include, but are not limited to, RNA polymerase and nucleotides, and factors known to those skilled in the art can be used.
- RNA may be synthesized in advance using DNA encoding a protein as a template, and the RNA may be added to a cell-free protein synthesis system.
- artificially chemically synthesized RNA may be used.
- the nucleic acid fragment that serves as a template for cell-free protein synthesis may be a biologically derived nucleic acid fragment, a cultured cell-derived nucleic acid fragment, or a virus-derived nucleic acid fragment.
- the nucleic acid fragment may be artificially synthesized based on the results of genetic analysis.
- Cell-free protein synthesis systems are not particularly limited, and include, for example, synthetic systems that utilize cell extracts obtained from wheat germ, yeast, insect cells, cultured mammalian cells, rabbit reticulocytes, Escherichia coli, etc.; Examples include synthetic systems in which factors are reconstituted. Among these, human expression cell-free protein synthesis is preferred.
- the cell-free protein synthesis system may contain at least one of factors involved in translation, such as tRNA, aminoacylated tRNA synthetase, translation initiation factor, translation elongation factor, and translation termination factor.
- factors involved in translation such as tRNA, aminoacylated tRNA synthetase, translation initiation factor, translation elongation factor, and translation termination factor.
- the target protein, the target protein, the first specific binding substance, and the second specific binding substance may be further contacted with adenosine triphosphate (ATP). good. This makes it possible to evaluate whether the target protein is phosphorylated. That is, it becomes possible to perform a kinase assay.
- ATP adenosine triphosphate
- FIG. 10 is a schematic diagram showing an example of the method of this embodiment.
- the mutant protein is BRAF
- cell-free protein synthesis kinase assay, antigen-antibody reaction, and ICA reaction are performed sequentially. This makes it possible to evaluate the activity of the target protein more easily and in a shorter period of time. Furthermore, for example, by adding an inhibitor to the reaction system, the effect of the inhibitor can be determined.
- the timing of adding the reagents necessary for each reaction and the timing of introducing the reagent solution into the fluidic device can be appropriately selected. For example, after performing cell-free protein synthesis, reagents necessary for a kinase assay and antigen-antibody reaction are added, and the kinase assay and antigen-antibody reaction are performed.
- An ICA reaction reagent may be added to the mixture after the antigen-antibody reaction and introduced into a fluidic device to perform the ICA reaction.
- reagents necessary for the kinase assay, antigen-antibody reaction, and ICA reaction may be added and introduced into a fluidic device to perform the kinase assay, antigen-antibody reaction, and ICA reaction.
- reagents necessary for the kinase assay, antigen-antibody reaction, and ICA reaction are added, and after performing the kinase assay and antigen-antibody reaction, this is introduced into a fluidic device to perform the ICA reaction. You may go.
- reagents necessary for the kinase assay and antigen-antibody reaction are added, and the kinase assay and antigen-antibody reaction are performed. Thereafter, reagents necessary for the ICA reaction may be added and introduced into the fluidic device to perform the ICA reaction. Further, after performing cell-free protein synthesis, reagents necessary for the kinase assay are added to perform the kinase assay. Thereafter, reagents necessary for antigen-antibody reaction and ICA reaction may be added to perform antigen-antibody reaction, and this may be introduced into a fluidic device to perform ICA reaction. Since the detection sensitivity is good, it is preferable to add the reagents necessary for the ICA reaction after performing the kinase assay and introduce the reagents into the fluidic device.
- the method of this embodiment preferably does not include a washing step.
- a washing step even when the target protein 110 is synthesized using a cell-free protein synthesis system, if it is possible to evaluate whether or not the target protein binds to the target protein without including a washing step, it will be easier to evaluate the activity of the target protein. And it can be implemented in a short period of time.
- the present invention provides a target protein 110, a target protein 120, a first specific binding substance 130 for the target protein 110 labeled with a first single-stranded nucleic acid fragment 131, and a second target protein 130. It includes a second specific binding substance 140 for the target protein 120 labeled with a full-stranded nucleic acid fragment 141, and includes at least a portion of the first single-stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141.
- a complex 100 is provided, a portion of which has hybridized to form a double-stranded nucleic acid 150.
- a flap probe may further hybridize to the first single-stranded nucleic acid fragment 131 or the second single-stranded nucleic acid fragment 141.
- the activity evaluation of a mutant protein can be carried out easily and in a short period of time.
- the present invention is a kit for evaluating whether or not a target protein 110 binds to a target protein 120, which comprises a well array 240 having a plurality of wells 241, a first single-stranded nucleic acid fragment 131
- a kit comprising a first specific binding substance 130 for the target protein 110 labeled with , and a second specific binding substance 140 for the target protein 120 labeled with a second single-stranded nucleic acid fragment 141 I will provide a.
- the kit of this embodiment it is possible to suitably evaluate whether or not the target protein 110 binds to the target protein 120.
- the kit of this embodiment includes, in the well 241, the target protein 110, the target protein 120, the first specific binding substance 130 for the target protein 110 labeled with the first single-stranded nucleic acid fragment 131, and When a second specific binding substance 140 labeled with a second single-stranded nucleic acid fragment 141 for the target protein 120 is introduced, and as a result, the target protein 110 binds to the target protein 120, the target protein 110, A complex 100 is formed that includes a target protein 120, a first specific binding substance 130, and a second specific binding substance 140, and includes at least a portion of the first single-stranded nucleic acid fragment 131 and the second single-stranded nucleic acid fragment 131.
- the method includes a step of hybridizing at least a portion of the nucleic acid fragment 141 to form a double-stranded nucleic acid 150, and a step of detecting the formation of the double-stranded nucleic acid 150, wherein the formation of the double-stranded nucleic acid 150 is detected. It can also be said that this is for use in a method of showing that the target protein 110 binds to the target protein 120.
- the well array may be placed inside the fluidic device described above.
- the target protein, target protein, first single-stranded nucleic acid fragment, first specific binding substance, second single-stranded nucleic acid fragment, and second specific binding substance are This is the same as described above.
- the kit of this embodiment may further contain ATP. This makes it possible to evaluate whether the target protein is phosphorylated. That is, it becomes possible to perform a kinase assay.
- the kit of this embodiment may further include a sealing liquid L220 that seals the opening of the well 241.
- the sealing liquid L220 is the same as that described above.
- the kit of this embodiment detects a double-stranded nucleic acid 150 formed by hybridization of at least a portion of the first single-stranded nucleic acid fragment 131 and at least a portion of the second single-stranded nucleic acid fragment 141. It may further contain a reagent.
- reagents include the above-mentioned reagents for the ICA reaction, and specific examples include flap probes, flap endonucleases, fluorescent substrates, and the like.
- the present invention provides a method for evaluating whether a target protein phosphorylates a target protein, the method comprising: a target protein, a second single-stranded nucleic acid fragment, a target protein, and a second single-stranded nucleic acid fragment in a container; contacting a labeled second specific binding substance for the target protein, a third specific binding substance for the phosphorylated target protein labeled with a third single-stranded nucleic acid fragment, and ATP;
- the target protein is phosphorylated, a complex containing the target protein, the second specific binding substance, and the third specific binding substance is formed, and the second specific binding substance a step of hybridizing at least a portion of the single-stranded nucleic acid fragment and at least a portion of the third single-stranded nucleic acid fragment to form a double-stranded nucleic acid; and a step of
- the evaluation method may be the following detection method.
- the detection method of the present embodiment includes, in a container, a target protein, a target protein, a second specific binding substance for the target protein labeled with a second single-stranded nucleic acid fragment, a third single-stranded contacting a third specific binding substance for the phosphorylated target protein labeled with a nucleic acid fragment with ATP, the target protein, the second specific binding substance, and the third specific binding substance;
- a complex containing a target binding substance is formed, and at least a portion of the second single-stranded nucleic acid fragment and at least a portion of the third single-stranded nucleic acid fragment hybridize to form a double-stranded nucleic acid. and detecting the formation of the double-stranded nucleic acid.
- FIG. 11 is a schematic diagram illustrating the method of this embodiment.
- target protein 110, target protein 120, and second single-stranded nucleic acid fragment 141 are labeled with target protein 110, target protein 120, and second single-stranded nucleic acid fragment 141.
- a third specific binding substance 170 and ATP are contacted.
- a complex 900 containing the target protein 120, the second specific binding substance 140, and the third specific binding substance 170 is formed.
- At least a portion of the second single-stranded nucleic acid fragment 141 and at least a portion of the third single-stranded nucleic acid fragment 171 hybridize to form a double-stranded nucleic acid (also referred to as a double-stranded nucleic acid region) 150. do.
- Step (a') is similar to step (a) in the method for evaluating whether a target protein binds to a target protein described above, but a third specific binding substance 130 is used instead of the first specific binding substance 130.
- the main difference is that a specific binding substance 170 is brought into contact.
- step (b) the formation of double-stranded nucleic acid 150 is detected. If formation of double-stranded nucleic acid 150 is detected, it can be determined that target protein 120 is phosphorylated.
- Step (b) is similar to step (b) in the above-described method for evaluating whether a target protein binds to a target protein. In the method of this embodiment, the amount of double-stranded nucleic acid 150 formed corresponds to the amount of phosphorylated target protein 120 present.
- the method of this embodiment may be applied when the target protein 110 does not phosphorylate the target protein 120, and in that case, the formation of the double-stranded nucleic acid 150 is not detected.
- activity evaluation of mutant proteins can be carried out easily and in a short period of time.
- VUS can be easily and efficiently functionally analyzed (ie, evaluated) in a short period of time.
- the target protein 110 exerts a kinase activity on the target protein 120, first, the target protein 110 and the target protein 120 bind to form a complex 900, and then the target protein 120 is activated by the kinase activity of the target protein 110. is thought to be phosphorylated.
- the function of the target protein 110 can be analyzed (ie, evaluated) by the above-described method of evaluating whether the target protein binds to the target protein or the method of this embodiment.
- the second single-stranded nucleic acid fragment 141 is The base length may be 10 to 200 bases.
- the base length of the third single-stranded nucleic acid fragment 171 may also be 10 to 200 bases.
- the length of the double-stranded nucleic acid 150 formed by hybridization of at least a portion of the second single-stranded nucleic acid fragment 141 and at least a portion of the third single-stranded nucleic acid fragment 171 is 7 to 30 mm. It is preferable that the number of bases is about a base, and for example, it may be 9 bases, 12 bases, or 15 bases.
- the target protein 110 may be a kinase having a genetic mutation of unknown clinical significance. More specific target proteins include proteins in intracellular signal transduction pathways, such as BRAF, A-RAF, Raf, MAP3K 4/12, MAP3K11, ASK1, TAK1, and the like. In this case, whether a protein with VUS is in a constitutively activated state can be evaluated by whether or not it phosphorylates a target protein.
- the present invention provides a kit for evaluating whether a target protein 110 phosphorylates a target protein 120, the kit comprising: a well array 240 having a plurality of wells 241; a second single-stranded nucleic acid; a second specific binding substance 140 for the target protein 120 labeled with a fragment 141; a third specific binding substance 170 for the phosphorylated target protein 120 labeled with a third single-stranded nucleic acid fragment 171; and ATP.
- the kit of this embodiment it is possible to suitably evaluate whether or not the target protein 110 phosphorylates the target protein 120.
- the target protein 110, the target protein 120, the second specific binding substance 140 for the target protein 120 labeled with the second single-stranded nucleic acid fragment 141, the third A third specific binding substance 170 for the phosphorylated target protein 120 labeled with a single-stranded nucleic acid fragment 171 and ATP were introduced, and as a result, the target protein 110 phosphorylated the target protein 120.
- a complex 900 is formed that includes the target protein 120, the second specific binding substance 140, and the third specific binding substance 170, and includes at least a portion of the second single-stranded nucleic acid fragment 141 and the third specific binding substance 140.
- the formation of the double-stranded nucleic acid 150 includes a step of hybridizing at least a portion of the single-stranded nucleic acid fragment 171 to form a double-stranded nucleic acid 150, and a step of detecting the formation of the double-stranded nucleic acid 150. It can also be said that what is detected is for use in a method showing that the target protein 110 phosphorylates the target protein 120.
- the well array may be placed inside the fluidic device described above.
- the target protein, target protein, second single-stranded nucleic acid fragment, second specific binding substance, third single-stranded nucleic acid fragment, and third specific binding substance This is the same as described above.
- the kit of this embodiment may further include a sealing liquid L220 that seals the opening of the well 241.
- the sealing liquid L220 is the same as that described above.
- the kit of this embodiment detects a double-stranded nucleic acid 150 formed by hybridization of at least a portion of the second single-stranded nucleic acid fragment 141 and at least a portion of the third single-stranded nucleic acid fragment 171. It may further contain a reagent.
- reagents include the above-mentioned ICA reaction reagents, and specific examples include flap probes, flap endonucleases, fluorescent substrates, and the like.
- the present invention includes the following aspects.
- a target protein In a container, a target protein, a target protein, a first specific binding substance for the target protein labeled with a first single-stranded nucleic acid fragment, and a second single-stranded nucleic acid fragment labeled contacting a second specific binding substance for the target protein,
- the target protein binds to the target protein to form a complex containing the target protein, the target protein, the first specific binding substance, and the second specific binding substance; At least a portion of the first single-stranded nucleic acid fragment and at least a portion of the second single-stranded nucleic acid fragment hybridize to form a double-stranded nucleic acid; Detecting the formation of the double-stranded nucleic acid.
- Forming the double-stranded nucleic acid comprises further contacting the target protein, the target protein, the first specific binding substance, and the second specific binding substance with adenosine triphosphate.
- [6] The detection method according to any one of [1] to [5], wherein the first single-stranded nucleic acid fragment and the second single-stranded nucleic acid fragment each have a base length of 10 to 200 bases. . [7] Further comprising adding a reagent for detecting the formation of the double-stranded nucleic acid into the container after the formation of the complex and before the formation of the double-stranded nucleic acid, [1] to The detection method according to any one of [6]. [8] The detection method according to any one of [1] to [7], wherein detecting the formation of the double-stranded nucleic acid is performed by Invasive Cleavage Assay.
- a target protein In a container, a target protein, a target protein, labeled with a second single-stranded nucleic acid fragment, a second specific binding substance for the target protein, labeled with a third single-stranded nucleic acid fragment. , a third specific binding substance for the phosphorylated target protein, and ATP;
- the target protein is phosphorylated to form a complex containing the target protein, the second specific binding substance, and the third specific binding substance; At least a portion of the second single-stranded nucleic acid fragment and at least a portion of the third single-stranded nucleic acid fragment hybridize to form a double-stranded nucleic acid; Detecting the formation of the double-stranded nucleic acid.
- oligonucleotides DNA1 (5'-TTTGTCACTGTTCCTCCTTTTGTTTTCCTTTCTGTGAGCAATTTCACCCAA-3', SEQ ID NO: 1) and DNA2 (5'-GCATGGTTCCAATTTGGGTGAT) were added to the above anti-phosphorylated MEK rabbit polyclonal antibody and anti-BRAF rabbit polyclonal antibody. -3' and SEQ ID NO: 2) were linked to each other.
- Anti-phosphorylated MEK rabbit polyclonal antibody (pMEK1/2 (S217/221) rabbit antibody, Cell Signaling Technology Co., Ltd.) was prepared using an antibody-oligonucleotide conjugation tool (product name "oYo-Link antibody labeling reagent", Funakoshi Co., Ltd.).
- anti-MEK rabbit monoclonal antibody (MEK1 Detector Antibody, Abcam) was conjugated with different oligonucleotides (Integrated DNA Technologies).
- oligonucleotides DNA1 (5'-TTTGTCACTGTTCCTCCTTTTGTTTTCCTTTCTGTGAGCAATTTCACCCAA-3', SEQ ID NO: 1) and DNA2 (5'-GCATGGTTCCAATTTGGGTGAT) were added to the above anti-phosphorylated MEK rabbit polyclonal antibody and anti-MEK rabbit polyclonal antibody. -3' and SEQ ID NO: 2) were linked to each other.
- a mixed solution was prepared by mixing the target protein (mutant BRAF and wild type BRAF), the target protein (inactive MEK), the above two types of oligonucleotide-modified antibodies, ATP, and blocking buffer. The volume of each mixture was 10 ⁇ L.
- the target proteins were prepared to have final concentrations of 0 pM, 2.82 pM, 28.2 pM, and 2821 pM, respectively.
- the target protein was prepared to a final concentration of 28210 pM.
- the two types of oligonucleotide-modified antibodies were prepared to have a final concentration of 8.56 nM.
- Tris-buffered saline also referred to as TBS
- BSA bovine serum albumin
- an ICA reaction reagent for detection was prepared.
- the ICA reaction reagents in this experiment included 2 ⁇ M allele probe (5'-CGCGCCGAGGAATTGCTCACAGAAAGGA-3') (Fasmac, SEQ ID NO: 3), 4 ⁇ M FRET cassette 1 (fluorescent substrate, Alexa488-BHQ:5'-X-TTCT- Y-AGCCGGTTTTCCGGCTGAGACCTCGGCGCG-3', , 50mM Tris-HCl (pH 8.5), 20mM MgCl2 , and 0.05% Tween20. Note that the concentrations of each component in these ICA reaction reagents are the final concentrations in the experimental examples.
- FIG. 12 to 14 are graphs showing the results of Experimental Example 1.
- Figure 12 shows the results of immuno-ICA reaction using 2821 pM mutant BRAF (Mt) and wild type BRAF (WT) as target proteins to detect interaction (i.e. binding) between BRAF and phosphorylated MEK. be.
- the vertical axis shows the fluorescence intensity (relative value) of Alexa488, and the horizontal axis shows the time (seconds) after the start of the ICA reaction.
- FIG. 13 shows the results of detecting phosphorylated MEK in an immuno-ICA reaction using 2821 pM of mutant BRAF (Mt) and wild type BRAF (WT) as target proteins.
- the vertical axis shows the fluorescence intensity (relative value) of Alexa488, and the horizontal axis shows the time (seconds) after the start of the ICA reaction.
- the fluorescence intensity when phosphorylated MEK is detected when wild type BRAF is reacted is N (noise)
- the fluorescence intensity when phosphorylated MEK is detected when mutant BRAF is reacted is S (signal).
- the signal-to-noise ratio (S/N ratio) at the end point was calculated to be 3.9.
- Figure 14 shows the results of Figures 12 and 13, where the fluorescence intensity when the antigen concentration is 0 nM is N (noise), the fluorescence intensity at each other concentration is S (signal), and the signal-to-noise ratio (S /N ratio) is a graph showing the results of calculation.
- the vertical axis shows the maximum value of the S/N ratio in each reaction
- the horizontal axis shows the time (seconds) or enzyme concentration after the start of the ICA reaction.
- Example 2 Kermantine activity evaluation of target proteins (mutant BRAF and wild type BRAF) using fluidic device 1) The interaction (that is, binding) between mutant BRAF and phosphorylated MEK and the interaction between wild type BRAF and phosphorylated MEK were conducted in the same manner as in Experimental Example 1 except that fluidic devices as shown in FIGS. 3 to 5 were used. The effect was detected.
- the fluidic device was set in an aluminum block constant temperature bath (model "DTU-Mini”, Taitec Co., Ltd.), heated at 66°C for 7 minutes or 30 minutes, and then placed in a microscope (product name "All-in-one Fluorescence Microscope", model "BZ"). -X810'' (Keyence Corporation). Subsequently, the brightness of each well was calculated based on the microscopic observation image.
- FIG. 15 is a graph showing the calculation results of the brightness of each well.
- the immuno-ICA reaction was performed using mutant BRAF (Mt) and wild type BRAF (WT) as target proteins, and the results of detecting the interaction between BRAF and phosphorylated MEK are shown.
- the vertical axis in FIG. 15 indicates the number of wells, and the horizontal axis indicates the brightness of the wells.
- the left side of FIG. 15 shows the results when the ICA reaction time was 7 minutes, and the right side of FIG. 15 shows the results when the ICA reaction time was 30 minutes.
- the upper row of FIG. 15 shows the results of detecting the interaction between mutant BRAF (Mt) and phosphorylated MEK
- the lower row of FIG. 15 shows the results of detecting the interaction between wild type BRAF (WT) and phosphorylated MEK.
- Table 1 below is a table summarizing the median brightness values of the wells shown in FIG. 15.
- S/N is the fluorescence intensity when detecting the interaction between wild-type BRAF and phosphorylated MEK, which is N (noise), and the interaction between mutant BRAF and phosphorylated MEK was detected.
- S/N ratio signal-to-noise ratio
- the fluidic device was set in an aluminum block constant temperature bath (model "DTU-Mini”, Taitec Co., Ltd.), heated at 66°C for 7 minutes or 30 minutes, and then placed in a microscope (product name "All-in-one Fluorescence Microscope", model "BZ"). -X810'' (Keyence Corporation). Subsequently, the brightness of each well was calculated based on the microscopic observation image.
- FIG. 16 is a graph showing the calculation results of the brightness of each well.
- the results of immuno-ICA reaction performed using mutant BRAF (Mt) and wild type BRAF (WT) as target proteins and detection of phosphorylated MEK are shown.
- the vertical axis in FIG. 16 indicates the number of wells, and the horizontal axis indicates the brightness of the wells.
- the left side of FIG. 16 shows the results when the ICA reaction time was 7 minutes, and the right side of FIG. 16 shows the results when the ICA reaction time was 30 minutes.
- the upper row of FIG. 16 shows the results of detecting phosphorylated MEK when reacting with mutant BRAF (Mt)
- the lower row of FIG. 16 shows the results of detecting phosphorylated MEK when reacting with wild-type BRAF (WT). Show the results.
- Table 2 below is a table summarizing the median brightness values of the wells shown in FIG. 16.
- S/N is the fluorescence intensity when reacting with wild type BRAF, N (noise), and the fluorescence intensity when reacting with mutant BRAF, S (signal), and at the end point. The results of calculating the signal-to-noise ratio (S/N ratio) are shown.
- Pattern 1 is a case in which phosphorylated MEK is detected by mixing all reagents for the kinase assay reaction, antigen-antibody reaction, and immuno-ICA reaction before performing the kinase assay. After performing the kinase assay, reagents for antigen-antibody reaction and immuno-ICA reaction are mixed and introduced into a tube, and pattern 2 is when phosphorylated MEK is detected.
- a mixed solution was prepared by mixing target proteins (mutant BRAF and wild type BRAF), target proteins (inactive MEK and active MEK), the above two types of oligonucleotide-modified antibodies, ATP, and blocking buffer.
- the volume of each mixture was 10 ⁇ L.
- the target proteins were each prepared at a final concentration of 2821 pM.
- the target proteins were each prepared at a final concentration of 7 nM.
- the two types of oligonucleotide-modified antibodies were prepared so that the final concentration was 1 nM when DNA1 was bound, and the final concentration was 4 nM when DNA2 was bound.
- Tris-buffered saline also referred to as TBS
- casein Tris-buffered saline
- ATP Tris-buffered saline
- the ICA reaction reagent in Pattern 1 was prepared with the same composition and final concentration as in Experimental Examples 1 to 3, except that it was 0.108 ⁇ M flap endonuclease (FEN)-1. Note that the concentrations of each component in these ICA reaction reagents are the final concentrations in the experimental examples.
- a mixed solution was prepared by mixing the target protein (mutant BRAF and wild type BRAF), target protein (inactive MEK and active MEK), ATP, 20mM MgCl2, and blocking buffer. The volume of each mixture was 7.5 ⁇ L. The target proteins were each prepared at a final concentration of 2821 pM. The target proteins were each prepared at a final concentration of 7 nM. Further, as a blocking buffer, Tris-buffered saline (also referred to as TBS) containing casein and a nonionic surfactant was used. As shown in Table 3, six patterns of combinations of BRAF, MEK, and ATP were used as reaction compositions. In Table 3, ⁇ indicates that the corresponding component is included, and ⁇ indicates that the corresponding component is not included.
- the mixed solution of the antigen-antibody reaction reagent and the ICA reaction reagent in pattern 2 contained the above two types of oligonucleotide-modified antibodies, 2 ⁇ M allele probe (5'-CGCGCCGAGGAATTGCTCACAGAAAGGA-3') (Fasmac, SEQ ID NO: 3), and 4 ⁇ M.
- FRET cassette 1 fluorescent substrate, Alexa488-BHQ:5'-X-TTCT-Y-AGCCGGTTTTCCGGCTGAGACCTCGGCGCG-3', X: Alexa488+AminoC6, Y: black hole quencher (BHQ) 1-dT) (Japan Bioservices, SEQ ID NO: 4), 0.108 ⁇ M flap endonuclease (FEN)-1, 50 mM Tris-HCl (pH 8.5), and 0.05% Tween20. Furthermore, the two types of oligonucleotide-modified antibodies were prepared so that the final concentration was 1 nM when DNA1 was bound, and the final concentration was 4 nM when DNA2 was bound.
- each reaction reagent was 2.5 ⁇ L for the antigen-antibody reaction reagent and 10 ⁇ L for the ICA reaction reagent. Note that the concentrations of each component in these ICA reaction reagents are the final concentrations in the experimental examples.
- the kinase assay solution was introduced into the tube, the kinase assay was allowed to react at 30° C. for 60 minutes, and then the antigen-antibody reaction reagent and the ICA reaction reagent were introduced into the tube.
- Immuno-ICA reaction was performed in the same manner as in Experimental Example 1 by reacting at 37°C for 60 minutes and at 66°C for 60 to 120 minutes. Mutant BRAF and wild type BRAF were used as target proteins.
- FIG. 17 is a graph showing the results of Pattern 1 of Experimental Example 4.
- FIG. 18 is a graph showing the results of Pattern 2 of Experimental Example 4. Labels 1 to 6 in FIGS. 17 and 18 correspond to reaction compositions 1 to 6 in Table 3, respectively.
- FIG. 17 and FIG. 18 show the results of detecting phosphorylation by performing an immuno-ICA reaction for 60 minutes using 2821 pM of mutant BRAF (Mt) or wild type BRAF (WT) as the target protein.
- Mt mutant BRAF
- WT wild type BRAF
- Example 5 Kininase activity evaluation of target protein (mutant BRAF and wild type BRAF) in fluidic device 3) Phosphorylated MEK was detected in the same manner as in Experimental Example 4, except that the immuno-ICA reaction was performed using a fluidic device as shown in FIGS. 3 to 5. Mutant BRAF and wild type BRAF were used as target proteins.
- kinase assays and antigen-antibody reactions were performed in tubes, and ICA reactions were performed in fluidic devices.
- pattern 1 the mixed solution and the ICA reaction reagent were mixed, a kinase assay and an antigen-antibody reaction were performed, and then the mixture was introduced into a fluidic device. After reacting at 30° C. for 60 minutes, phosphorylated MEK was detected in the same manner as in Experimental Examples 2 and 3.
- the reaction time of the immuno-ICA reaction was from 60 minutes to 180 minutes.
- pattern 2 after the kinase assay was reacted at 30° C. for 60 minutes, the antigen-antibody reaction reagent and the ICA reaction reagent were mixed, reacted at 37° C. for 60 minutes, and introduced into the fluidic device. The reaction was carried out at 66° C. for 60 to 120 minutes, and phosphorylated MEK was detected in the same manner as in Experimental Examples 2 and 3 (Pattern 2).
- FIG. 19 is a histogram of the fluorescence intensity measured over time in the immuno-ICA reaction. From the left side of Figure 19, fluorescence intensity 120 minutes after the start of the immuno-ICA reaction in pattern 1, fluorescence intensity 180 minutes after the start of the immuno-ica reaction in pattern 1, fluorescence intensity 90 minutes after the start of the immuno-ICA reaction in pattern 2 and the fluorescence intensity 120 minutes after the start of the immuno-ICA reaction in pattern 2 are shown. 1 to 6 in FIG. 19 correspond to reaction compositions 1 to 6 in Table 3, respectively.
- pattern 2 that is, adding the antigen-antibody reaction reagent and the ICA reaction reagent after the kinase assay, resulted in better reactivity.
- the presence of antibodies during kinase assays may inhibit the phosphorylation reaction.
- Sealing liquid 242R... Well in which signal was detected, 510... Wall member, 810... Flap probe, 811... First flap site (nucleic acid fragment), 821... Second flap site (nucleic acid fragment), 820, 820' ...Nucleic acid fragment, F...fluorescent substance, Q...quencher.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Analytical Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Sustainable Development (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
この評価方法は、対象タンパク質が標的タンパク質と結合する否かを評価する方法であって、容器中で、対象タンパク質、標的タンパク質、第1の一本鎖核酸断片で標識された、対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、標的タンパク質に対する第2の特異的結合物質を接触させ、その結果、対象タンパク質が標的タンパク質と結合した場合に、対象タンパク質、標的タンパク質、第1及び第2の特異的結合物質を含む複合体が形成され、第1の一本鎖核酸断片及び第2の一本鎖核酸断片がハイブリダイズして二本鎖核酸を形成する工程(a)と、二本鎖核酸の形成を検出する工程(b)と、を含み、二本鎖核酸の形成が検出されたことが、対象タンパク質が標的タンパク質と結合することを示す。
Description
本発明は、変異タンパク質の活性評価方法に関する。より詳細には、本発明は、対象タンパク質が標的タンパク質と結合する否かを評価する方法、複合体、対象タンパク質が標的タンパク質と結合する否かを評価するためのキット、及び、対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法に関する。
本願は、2022年8月1日に日本に出願された特願2022-122819号について優先権を主張し、その内容をここに援用する。
本願は、2022年8月1日に日本に出願された特願2022-122819号について優先権を主張し、その内容をここに援用する。
次世代シークエンサーに代表される網羅的な遺伝子解析技術の登場により、多くの遺伝子の変化と疾患発症のメカニズムが明らかになりつつある。これと同時に数多くの臨床的意義不明の遺伝子変異(variants of unknown significance:VUS、以下「VUS」という。)が同定された。
現在のゲノム医療において、遺伝子解析による診断率及び薬剤到達性が低い。この原因の一つとして、VUSが多数報告されていることが挙げられ、VUSの意義付けとその評価が急務である。
VUSの意義付けとその評価には、Combined Annotation Dependent Depletion(https://cadd.gs.washington.edu/)スコアやMutpred2(https://mutpred.mutdb.org/)スコアをはじめとする各種スコアが算出されており、参照することができる。しかしながら、コンピュータによる予測は、実際の表現型と一致しない場合があるため、病原性を評価するための唯一のエビデンスとして使用することはできない。
VUSを実験的に評価する方法の一つとしては、目的遺伝子に変異のあるプラスミドを作製し、細胞に導入してタンパク質を合成して細胞内で合成したタンパク質をリン酸化させ、リン酸化タンパク質を含んだ細胞抽出液をウエスタンブロッティング等で検出することで定性的に評価する方法が確立されている。しかしながら、このような評価方法には多くの時間、労力及びコストを要する。
従来、タンパク質の定量検出は、酵素結合免疫吸着アッセイ(ELISAともいう)等により行われ、核酸の定量は、リアルタイムPCR法等により行われてきた。
標的物質を精度よく検出する手法として、多数の微小区画内で酵素反応を行う技術が検討されている。これらの手法は、デジタル計測と呼ばれている。デジタル計測には、デジタルELISA及びデジタルPCR等が存在する。
デジタル計測では、試料溶液を極めて多数の微小溶液に分割する。そして、各微小溶液からの信号を2値化し、標的物質が存在するか否かのみを判別して、標的物質の分子数を計測する。デジタル計測によれば、従来のELISAやリアルタイムPCR法等と比較して、検出感度及び定量性を格段に向上させることができる。
例えば、非特許文献1には、微小ウェル及び試薬等を供給する流路を有する微小ウェルアレイが記載されており、当該微小ウェルアレイを用いてデジタルELISAを行ったことが記載されている。
特許文献1には、オリゴヌクレオチドを修飾した抗体を用いてタンパク質を検出する方法である、Proximity Ligation Assay(PLAともいう)が報告されている。この方法は、検出にPCR法又はRCA法を利用している。
特許文献2には、オリゴヌクレオチドを修飾した抗体を用いて2分子間のタンパク質相互作用を検出する方法が記載されている。この方法も、検出にPCR法又はRCA法を利用している。
非特許文献3には、微小ウェル及び試薬等を供給する流路を有する微小ウェルアレイが記載されており、当該微小ウェルアレイ内で細胞を用いてシグナル伝達を表現し、リン酸化タンパク質の検出を行ったことが記載されている。シグナル伝達の様子を、リン酸化タンパク質を検出することでモニタリングしている。
Alpha SureFire(https://www.perkinelmer.co.jp/assays/tabid/346/Default.aspx、パーキンエルマー社)は、洗浄操作なしに、細胞ベースでキナーゼ活性を検出可能なアッセイシステムである。本システムでは、細胞抽出液からリン酸化タンパク質を検出している。
Kan C. W., et al., Isolation and detection of single molecules on paramagnetic beads using sequential fluid flows in microfabricated polymer array assemblies., Lab on a Chip, 12 (5), 977-985, 2012.
Mohammed H., et al., Approaches for Assessing and Discovering Protein Interactions in Cancer., Mol Cancer Res, 11 (11), 1295-1302, 2013.
Blazek M., et al., Proximity Ligation Assay for High-content Profiling of Cell Signaling Pathways on a Microfluidic Chip., Molecular & Cellular Proteomics 12: 10.1074/mcp.M113.032821, 3898-3907, 2013.
生体内には外部の刺激を細胞内に伝達する機構が存在する。その際には様々なタンパク質同士が一時的に結合し、情報を伝達する。この情報伝達システムをシグナル伝達システムといい、刺激を媒介する様々なタンパク質分子が担っている。図1は、一例として、マイトジェン活性化プロテインキナーゼ(MAPKともいう)シグナル伝達の一部を示している。
図1では、一例として、変異タンパク質がBRAFである場合に、BRAF遺伝子における臨床的意義不明の遺伝子変異(つまりVUS)が「正常変異」である場合、及び、「有害変異」である場合を説明している。VUSが正常変異である場合、上流からの刺激がない限り、図1左に示すように、BRAFの基質であるMEKはリン酸化されない。これに対し、VUSが有害変異である場合、図1右に示すように、BRAFは恒常的に活性化しており、上流からの刺激がなくても、基質であるMEKをリン酸化してしまう。このようなBRAFは癌の原因となる。
本発明は、変異タンパク質の活性評価を簡便かつ短期間に実施する技術を提供することを目的とする。
本発明は以下の態様を含む。
[1]対象タンパク質が標的タンパク質と結合する否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a)と、前記二本鎖核酸の形成を検出する工程(b)と、を含み、前記二本鎖核酸の形成が検出されたことが、前記対象タンパク質が前記標的タンパク質と結合することを示す、方法。
[2]前記工程(a)が、前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成する工程(a1)と、前記容器中で、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a2)と、を含む、[1]に記載の方法。
[3]前記工程(a2)において、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質に、アデノシン三リン酸(ATP)を更に接触させる、[2]に記載の方法。
[4]洗浄工程を含まない、[1]~[3]のいずれかに記載の方法。
[5]前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、[1]~[4]のいずれかに記載の方法。
[6]前記第1の一本鎖核酸断片及び前記第2の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、[1]~[5]のいずれかに記載の方法。
[7]前記二本鎖核酸の形成を検出する工程が、Invasive Cleavage Assayにより行われる、[1]~[6]のいずれかに記載の方法。
[8]対象タンパク質、標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を含み、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成した、複合体。
[9]対象タンパク質が標的タンパク質と結合する否かを評価するためのキットであって、複数のウェルを有するウェルアレイ、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、を含む、キット。
[10]ATPを更に含む、[9]に記載のキット。
[11]前記ウェルの開口部を封止する封止液を更に含む、[9]又は[10]に記載のキット。
[12]前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして形成された二本鎖核酸を検出する試薬を更に含む、[9]~[11]のいずれかに記載のキット。
[13]対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させ、その結果、前記標的タンパク質がリン酸化されていた場合に、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体が形成され、前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a’)と、前記二本鎖核酸の形成を検出する工程(b)と、を含み、前記二本鎖核酸の形成が検出されたことが、前記標的タンパク質がリン酸化されていることを示す、方法。
[1]対象タンパク質が標的タンパク質と結合する否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a)と、前記二本鎖核酸の形成を検出する工程(b)と、を含み、前記二本鎖核酸の形成が検出されたことが、前記対象タンパク質が前記標的タンパク質と結合することを示す、方法。
[2]前記工程(a)が、前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成する工程(a1)と、前記容器中で、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a2)と、を含む、[1]に記載の方法。
[3]前記工程(a2)において、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質に、アデノシン三リン酸(ATP)を更に接触させる、[2]に記載の方法。
[4]洗浄工程を含まない、[1]~[3]のいずれかに記載の方法。
[5]前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、[1]~[4]のいずれかに記載の方法。
[6]前記第1の一本鎖核酸断片及び前記第2の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、[1]~[5]のいずれかに記載の方法。
[7]前記二本鎖核酸の形成を検出する工程が、Invasive Cleavage Assayにより行われる、[1]~[6]のいずれかに記載の方法。
[8]対象タンパク質、標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を含み、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成した、複合体。
[9]対象タンパク質が標的タンパク質と結合する否かを評価するためのキットであって、複数のウェルを有するウェルアレイ、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、を含む、キット。
[10]ATPを更に含む、[9]に記載のキット。
[11]前記ウェルの開口部を封止する封止液を更に含む、[9]又は[10]に記載のキット。
[12]前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして形成された二本鎖核酸を検出する試薬を更に含む、[9]~[11]のいずれかに記載のキット。
[13]対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させ、その結果、前記標的タンパク質がリン酸化されていた場合に、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体が形成され、前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a’)と、前記二本鎖核酸の形成を検出する工程(b)と、を含み、前記二本鎖核酸の形成が検出されたことが、前記標的タンパク質がリン酸化されていることを示す、方法。
本発明によれば、変異タンパク質の活性評価を簡便かつ短期間に実施する技術を提供することができる。本発明によれば、例えば、VUSを簡便かつ短期間に効率よく機能解析(つまり評価)することができる。
以下、場合により図面を参照しつつ、本発明の実施形態について詳細に説明する。なお、図面中、同一又は相当部分には同一又は対応する符号を付し、重複する説明は省略する。なお、各図における寸法比は、説明のため誇張している部分があり、必ずしも実際の寸法比とは一致しない。
[対象タンパク質が標的タンパク質と結合する否かを評価する方法]
一実施形態において、本発明は、対象タンパク質が標的タンパク質と結合する否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a)と、前記二本鎖核酸の形成を検出する工程(b)と、を含み、前記二本鎖核酸の形成が検出されたことが、前記対象タンパク質が前記標的タンパク質と結合することを示す、方法を提供する。
一実施形態において、本発明は、対象タンパク質が標的タンパク質と結合する否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a)と、前記二本鎖核酸の形成を検出する工程(b)と、を含み、前記二本鎖核酸の形成が検出されたことが、前記対象タンパク質が前記標的タンパク質と結合することを示す、方法を提供する。
図2は、本実施形態の方法を説明する模式図である。図2に示すように、本実施形態の方法では、まず、工程(a)において、容器中で、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を接触させる。
その結果、対象タンパク質110が標的タンパク質120と結合した場合に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸(二本鎖核酸領域ともいう)150を形成する。
続いて、工程(b)において、二本鎖核酸150の形成を検出する。二本鎖核酸150の形成が検出された場合、対象タンパク質110が標的タンパク質120と結合する(つまり相互作用する)ということができる。
本実施形態の方法は、対象タンパク質110が標的タンパク質120と結合しない(つまり相互作用しない)場合に適用してもよく、その場合には、二本鎖核酸150の形成は検出されない。二本鎖核酸150の形成の検出については後述する。
実施例において後述するように、本発明によれば、変異タンパク質の活性評価を簡便かつ短期間に実施することができる。例えば、VUSを簡便かつ短期間に効率よく機能解析(つまり評価)することができる。
第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズすることができる必要がある観点から、第1の一本鎖核酸断片131の塩基長は10~200塩基であってもよい。同様に、第2の一本鎖核酸断片141の塩基長も10~200塩基であってもよい。また、第1の一本鎖核酸断片121の少なくとも一部及び第2の一本鎖核酸断片131の少なくとも一部がハイブリダイズして形成された二本鎖核酸150の長さは、7~30塩基程度であることが好ましく、例えば9塩基であってもよく、12塩基であってもよく、15塩基であってもよい。
本実施形態の方法において、対象タンパク質110は、臨床的意義不明の遺伝子変異(つまりVUS)を有するタンパク質であってもよい。対象タンパク質110は、例えばキナーゼであってもよい。より具体的な対象タンパク質としては、細胞内シグナル伝達経路のタンパク質が挙げられ、例えば、BRAF、A-RAF、Raf、MAP3K 4/12、MAP3K11、ASK1及びTAK1等が挙げられる。この場合、VUSを有するタンパク質が恒常的な活性化状態にあるか否かを、標的タンパク質と結合するか否かにより評価することができる。
(容器)
本実施形態の方法は、デジタル計測により実施することができる。本実施形態の方法をデジタル計測で行う場合、容器はウェルであることが好ましく、ウェルは複数のウェルが配置されたウェルアレイを構成していることが好ましい。また、ウェルアレイは、流体デバイスの流路内に配置されていることが好ましい。
本実施形態の方法は、デジタル計測により実施することができる。本実施形態の方法をデジタル計測で行う場合、容器はウェルであることが好ましく、ウェルは複数のウェルが配置されたウェルアレイを構成していることが好ましい。また、ウェルアレイは、流体デバイスの流路内に配置されていることが好ましい。
(流体デバイス)
図3は、本実施形態の方法を好適に実施することができる流体デバイスの一例を示す模式断面図である。図3に示すように、流体デバイス200は、基板210と、基板210に対向して配置される蓋部材220とを備えている。蓋部材220は、凸部221を有している。凸部221の先端は、基板210に接している。流体デバイス200においては、ウェルアレイ240は、基板210の一方面上に基板210と一体成型されており、蓋部材220と対向している。ウェルアレイ240は、複数のウェル241を有する。蓋部材220は、基板210に溶着又は接着されていてもよい。
図3は、本実施形態の方法を好適に実施することができる流体デバイスの一例を示す模式断面図である。図3に示すように、流体デバイス200は、基板210と、基板210に対向して配置される蓋部材220とを備えている。蓋部材220は、凸部221を有している。凸部221の先端は、基板210に接している。流体デバイス200においては、ウェルアレイ240は、基板210の一方面上に基板210と一体成型されており、蓋部材220と対向している。ウェルアレイ240は、複数のウェル241を有する。蓋部材220は、基板210に溶着又は接着されていてもよい。
ウェル241は、基板210の表面に開口している。ウェル241の形状、寸法、及び配置は特に限定されないが、ウェル241は、容積が小さい微小ウェルであることが好ましい。例えば、1つのウェル241の容積は、10fL~100pL程度であってもよい。流体デバイス200では、同形同大の複数のウェル241がウェルアレイ240を構成している。同形同大とは、デジタル計測を行うために要求される程度に同一の形状で同一の容量であればよく、製造上の誤差程度のばらつきであれば許容される。
ウェル241の直径は、例えば1~10μm程度であってもよい。ウェル241の深さは、例えば1~10μm程度であってもよい。また、ウェル241の配置は特に制限されず、例えば三角格子状に整列していてもよいし、正方格子状も整列していてもよいし、ランダムに配置されていてもよい。
流体デバイス200では、凸部221の存在により、ウェルアレイ240と蓋部材220との間に空間が形成されている。当該空間は、流路230を構成している。流路230は、対象タンパク質、標的タンパク質、第1の特異的結合物質及び第2の特異的結合物質等が分散した液体及び後述する封止液を送液するための経路として機能する。流路230の形状、構造及び容量等は特に限定されないが、流路230の高さ(基板210の表面と、蓋部材220の基板210と対向する面との間の距離)は、例えば500μm以下であってもよいし、例えば300μm以下であってもよいし、例えば200μm以下であってもよいし、例えば100μm以下であってもよい。
凸部221は、蓋部材220と一体に成形されていてもよい。蓋部材220は、例えば、熱可塑性樹脂の流動体を、成形型を用いて成形することで、凸部221を有する板状に成形することができる。また、蓋部材220には試薬の導入ポート222及び排出ポート223が形成されていてもよい。
蓋部材220が凸部221を有する場合、基板210のウェル241が開口する面に凸部221が接するように、蓋部材220と基板210とが重ねられる。この結果、蓋部材220と基板210との間の空間が流路230となる。蓋部材220と基板210とは、レーザー溶着等により溶着されてもよい。
(流体デバイスの変形例1)
本実施形態の方法に用いられる流体デバイスは、上述した流体デバイス200に限られない。図6は、流体デバイスの一例を示す模式断面図である。図6に示すように、流体デバイス500は、基板210と、壁部材510とを備えている。流体デバイス500においては、ウェルアレイ240は、基板210の一方面上に基板210と一体成形されている。ウェルアレイ240は、複数のウェル241を有する。流体デバイス500は、蓋部材220を有していない点で上述した流体デバイス200と主に異なっている。
本実施形態の方法に用いられる流体デバイスは、上述した流体デバイス200に限られない。図6は、流体デバイスの一例を示す模式断面図である。図6に示すように、流体デバイス500は、基板210と、壁部材510とを備えている。流体デバイス500においては、ウェルアレイ240は、基板210の一方面上に基板210と一体成形されている。ウェルアレイ240は、複数のウェル241を有する。流体デバイス500は、蓋部材220を有していない点で上述した流体デバイス200と主に異なっている。
(流体デバイスの変形例2)
上述した流体デバイス200においては、蓋部材220と凸部221は一体成形されている。しかしながら、蓋部材220と凸部221は、別体として成形されていてもよい。
上述した流体デバイス200においては、蓋部材220と凸部221は一体成形されている。しかしながら、蓋部材220と凸部221は、別体として成形されていてもよい。
また、上述した流体デバイス200及び流体デバイス500においては、ウェルアレイ240は、基板210の一方面上に基板210と一体成形されている。しかしながら、ウェルアレイは、基板210と一体成形されていなくてもよい。例えば、流体デバイスとは別体として成形されたウェルアレイ240を、流体デバイスの基板210上に配置してもよい。あるいは、基板210の表面に樹脂層を積層し、エッチング等により、樹脂層にウェルアレイを形成してもよい。
(流体デバイスの材質)
基板210は、例えば樹脂を用いて形成される。樹脂の種類は特に限定されないが、試薬及び封止液に対して耐性のある樹脂であることが好ましい。また、検出するシグナルが蛍光である場合には、自家蛍光が少ない樹脂であることが好ましい。例えば、自家蛍光が少ない樹脂として、シクロオレフィンポリマーや、シクロオレフィンコポリマー、シリコーン、ポリプロピレン、ポリカーボネート、ポリスチレン、ポリエチレン、ポリ酢酸ビニル、フッ素樹脂及びアモルファスフッ素樹脂等が挙げられるがこれらに限定されない。
基板210は、例えば樹脂を用いて形成される。樹脂の種類は特に限定されないが、試薬及び封止液に対して耐性のある樹脂であることが好ましい。また、検出するシグナルが蛍光である場合には、自家蛍光が少ない樹脂であることが好ましい。例えば、自家蛍光が少ない樹脂として、シクロオレフィンポリマーや、シクロオレフィンコポリマー、シリコーン、ポリプロピレン、ポリカーボネート、ポリスチレン、ポリエチレン、ポリ酢酸ビニル、フッ素樹脂及びアモルファスフッ素樹脂等が挙げられるがこれらに限定されない。
基板210の板厚方向の一方面に複数のウェル241が形成されていてもよい。樹脂を用いたウェルの形成方法としては、射出成形、熱インプリント及び光インプリント等が挙げられる。
あるいは、例えば、基板210の上にフッ素樹脂を積層し、当該フッ素樹脂をエッチング等により加工してウェルアレイを形成してもよい。フッ素樹脂としては、例えばCYTOP(登録商標)(旭硝子)等を用いることができる。
また、流体デバイスが蓋部材220を有する場合、蓋部材220の材質は、自家蛍光の少ない樹脂であることが好ましく、例えば、シクロオレフィンポリマー、シクロオレフィンコポリマー等の熱可塑性樹脂であってもよい。
また、蓋部材220は、シグナルを蛍光観察する際に検出される波長の近傍の波長の光を透過しない材料から形成されていてもよく、完全に光を透過しない材料から形成されていてもよい。例えば、蓋部材220は、カーボン又は金属粒子等を添加した熱可塑性樹脂から形成されていてもよい。
(本実施形態の方法)
続いて、場合により図3~図5を参照しながら、流体デバイス200を用いた場合を例に、本実施形態の方法について説明する。本実施形態の方法は、対象タンパク質が標的タンパク質と結合する否かを評価する方法であり、ウェル241に、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を接触させ、その結果、対象タンパク質110が標的タンパク質120と結合した場合に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する工程(a)と、二本鎖核酸150の形成を検出する工程(b)と、を含み、二本鎖核酸150の形成が検出されたことが、対象タンパク質110が標的タンパク質120と結合することを示す方法である。
続いて、場合により図3~図5を参照しながら、流体デバイス200を用いた場合を例に、本実施形態の方法について説明する。本実施形態の方法は、対象タンパク質が標的タンパク質と結合する否かを評価する方法であり、ウェル241に、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を接触させ、その結果、対象タンパク質110が標的タンパク質120と結合した場合に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する工程(a)と、二本鎖核酸150の形成を検出する工程(b)と、を含み、二本鎖核酸150の形成が検出されたことが、対象タンパク質110が標的タンパク質120と結合することを示す方法である。
対象タンパク質110が標的タンパク質120と結合する場合において、前記評価する方法は、以下の検出方法であってもよい。つまり、本実施形態の検出方法は、ウェル241に、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を接触させることと、対象タンパク質110を標的タンパク質120と結合させることと、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成することと、二本鎖核酸150の形成を検出することと、を含んでいてもよい。
本実施形態の方法によれば、変異タンパク質の活性評価を簡便かつ短期間に実施することができる。変異タンパク質としては、VUSを有するタンパク質が挙げられる。
《導入工程》
まず、図3に示すように、流体デバイス200の導入ポート222から試薬液L210を導入し、流路230に送液する。試薬液L210は、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、及び、第2の特異的結合物質140が分散した液体であり、二本鎖核酸150の形成を検出するための試薬も含む。
まず、図3に示すように、流体デバイス200の導入ポート222から試薬液L210を導入し、流路230に送液する。試薬液L210は、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、及び、第2の特異的結合物質140が分散した液体であり、二本鎖核酸150の形成を検出するための試薬も含む。
流路230に送液された試薬液L210は、ウェルアレイ240に接触する。そして、ウェル241の内部に試薬液L210が収容される。この結果、ウェル241に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、第2の特異的結合物質140、及び、二本鎖核酸150の形成を検出するための試薬が導入される。ここで、対象タンパク質110が標的タンパク質120と結合する場合には、試薬液L210内に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が含まれている。
1つのウェル241に導入される複合体100の数は特に制限されないが、好ましくは、1つのウェル241に1つ以下、すなわち、0個又は1個の複合体100が導入される。これにより、複合体100の検出を1個単位で行うことができ、すなわちデジタル計測が可能となる。また、ウェルアレイの全てのウェルに複合体100が導入される必要はない。
複合体100をウェルに導入する手段は、特に制限されず、例えば、複合体100を自重により流体デバイス内(具体的には流路内230)で沈降させ、ウェル241に分配する方法が挙げられる。あるいは、複合体100を捕捉する物質(捕捉物ともいう)を利用し、自重で沈降しにくい複合体100に捕捉物を結合させて送液してもよく、予めウェルに捕捉物を固定化させておき、送液された複合体100を捕捉させることで、複合体100のウェルへの導入効率を向上させることもできる。
捕捉物を複合体100に結合させる工程は、本実施形態の方法の任意の時点で行うことができる。例えば、この工程は、ウェル241に複合体100を導入する工程の前に、サンプルチューブ内で複合体100と捕捉物を接触させることにより行ってもよい。あるいは、捕捉物をウェル241に導入した後に、複合体100をウェルに導入し、ウェル内で捕捉物と複合体100とを接触させてもよい。
捕捉物は、複合体100を捕捉することができる物質である。捕捉物は、例えば、固相と複合体100に対する特異的結合物質との結合体であってもよい。
固相としては、粒子、膜及び基板等が挙げられる。また、複合体100に対する特異的結合物質は、1種類でもよいし、2種類以上であってもよい。例えば特異的結合物質は、3種類であってもよいし、4種類であってもよいし、5種類以上であってもよい。
粒子としては、特に制限されず、ポリマー粒子、磁気粒子及びガラス粒子等が挙げられる。粒子は、非特異的な吸着を避けるための表面処理が施された粒子が好ましい。また、特異的結合物質を固定化するために、表面にカルボキシル基等の官能基を有する粒子が好ましい。より具体的には、JSR社製の商品名「Magnosphere LC300」等を用いることができる。
第1の特異的結合物質130、第2の特異的結合物質140及び捕捉物における特異的結合物質としては、抗体、抗体断片及びアプタマー等が挙げられる。抗体断片としては、Fab、F(ab’)2、Fab’、一本鎖抗体(scFv)、ジスルフィド安定化抗体(dsFv)、2量化体V領域断片(Diabody)及びCDRを含むペプチド等が挙げられる。抗体は、モノクローナル抗体であってもよく、ポリクローナル抗体であってもよい。また、市販の抗体であってもよい。
一本鎖核酸断片で特異的結合物質を標識する方法としては、架橋剤を使用する方法が挙げられる。一本鎖核酸断片は、リンカーである分子を介して特異的結合物質に標識されてもよい。リンカーとしては、特に限定されず、例えば、ポエリエチレン鎖、炭化水素鎖及びペプチド等が挙げられる。一本鎖核酸断片は、DNAであってもよいし、RNAであってもよい。また、BNA及びLNA等の人工核酸を含んでいてもよい。
特異的結合物質を粒子表面に固定化する方法としては、特に制限されず、物理吸着による方法、化学結合による方法、アビジン-ビオチンの結合を利用する方法及びプロテインG又はプロテインAと抗体との結合を利用する方法等が挙げられる。物理吸着による方法としては、疎水的相互作用又は静電的相互作用により、特異的結合物質を粒子表面に固定化する方法が挙げられる。化学結合による方法としては、架橋剤を使用する方法が挙げられる。例えば、粒子の表面が水酸基を有する場合、特異的結合物質が有するカルボキシル基に架橋剤を反応させて活性エステル化した後、水酸基とこのエステル基とを反応させることにより、特異的結合物質を粒子表面に固定化させることができる。また、特異的結合物質による標的分子の認識能を阻害しないよう、特異的結合物質と粒子表面との間にスペーサーを設けることが好ましい。
上述したように、捕捉物を用いて複合体100をウェル241に導入する場合、捕捉物1個に複合体100が0個又は1個捕捉される条件で捕捉物と複合体100との結合体を形成することが好ましい。さらに、1つのウェル241には、捕捉物が0個又は1個導入されるように構成されることが好ましい。これによりデジタル計測が可能となる。
対象タンパク質110が標的タンパク質120と結合する場合には、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を接触させると、これらを含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する。複合体100の形成は、サンプルチューブ内で行われてもよいし、ウェル241内で行われてもよい。
《封入工程》
ウェル241に、試薬液L210を導入した後、ウェル241の開口部を封止する工程を実施してもよい。ウェル241の開口部を封止する方法は、あるウェル241の内部に収容された液体が、別のウェル241の内部に収容された液体と互いに混合しない状態にすることができる限り特に限定されず、例えば、封止液でウェル241の開口部を覆うことにより封止してもよい。あるいは、ウェル241の開口部にガラス板等の板状部材を積層することにより封止してもよい。
ウェル241に、試薬液L210を導入した後、ウェル241の開口部を封止する工程を実施してもよい。ウェル241の開口部を封止する方法は、あるウェル241の内部に収容された液体が、別のウェル241の内部に収容された液体と互いに混合しない状態にすることができる限り特に限定されず、例えば、封止液でウェル241の開口部を覆うことにより封止してもよい。あるいは、ウェル241の開口部にガラス板等の板状部材を積層することにより封止してもよい。
例えば、図4に示すように、蓋部材220の導入ポート222から、基板210と蓋部材220との間の流路230に、封止液L220を送液する。流路230に送液された封止液L220は、ウェルアレイ240に接触する。そして、封止液L220は、流路230に送液された試薬液L210のうち、ウェル241に収容されていない試薬液L210を押し流して置換する。これにより、封止液L220が、標的物質110を含む試薬液L210を収容した複数のウェル241をそれぞれ個別に封止し、ウェル241は独立した反応空間(微小区画242ともいう)となる。流路230が封止液L220で満たされると、余分な封止液L220は排出ポート223から排出される。図5は、ウェルアレイ240のウェル241の全てが封止液L220で封止され、封止されたウェル(つまり微小区画)242が形成された状態を示す。
また、試薬液L210に脂質を溶解させておき、封止液L220を流路230に送液した後、再び脂質を含む液体を送液することにより、ウェル241の開口部に脂質二重膜を形成し、当該脂質二重膜によって複数のウェル241をそれぞれ個別に封止し、封止されたウェル242を形成することもできる。脂質二重膜を形成する脂質としては、例えば、1,2-ジオレオイル-sn-グリセロ-3-ホスホエタノールアミン(DOPEともいう)、1,2-ジオレオイル-sn-グリセロ-3-ホスホグリセロール(DOPGともいう)及びこれらの混合物等が挙げられるがこれらに限定されない。
封止液は、複数のウェル241に導入された液体同士を互いに混合しないように個別に封止して液滴(微小液滴ともいう)を形成することができる液であり、好ましくは油性溶液であり、より好ましくはオイルである。オイルとしては、フッ素系オイル、シリコーン系オイル、炭化水素系オイル又はこれらの混合物等を使用することができる。より具体的には、シグマ社製の商品名「FC-40」等を用いることができる。FC-40(CAS番号:86508-42-1)はフッ素化脂肪族化合物であり、25℃における比重が1.85g/mLである。
《検出工程》
続いて、二本鎖核酸150の形成を検出する。二本鎖核酸150の形成の検出は、シグナル増幅反応を用いて行うことが好ましい。シグナル増幅反応としては、例えば、Invasive Cleavage Assay(ICAともいう)が挙げられる。
続いて、二本鎖核酸150の形成を検出する。二本鎖核酸150の形成の検出は、シグナル増幅反応を用いて行うことが好ましい。シグナル増幅反応としては、例えば、Invasive Cleavage Assay(ICAともいう)が挙げられる。
ICA反応は、(1)核酸同士の相補的結合と、(2)酵素による三重鎖構造の認識及び切断との2つの反応のサイクルによってシグナル増幅が進行するという原理に関連する。
ICA反応は、夾雑物による反応サイクル阻害の影響が小さい。したがって、ICA反応を用いることにより、前記二本鎖核酸150の形成を精度よく検出することができる。シグナル増幅反応にICA反応を用いる場合、試薬液L210(つまり対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、及び、第2の特異的結合物質140を含む液体)は、ICA反応に必要な反応試薬を含む。
ICA反応に必要な反応試薬としては、フラッププローブ、フラップエンドヌクレアーゼ(FENともいう)及び蛍光基質等のICA反応試薬が挙げられる。フラッププローブは、第1の一本鎖核酸断片131又は第2の一本鎖核酸断片141にハイブリダイズして二本鎖核酸150とフラップ構造を形成するように設計された核酸断片である。
図9は、ICA法の一例を説明する模式図である。図9の例では、ICA法により、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして形成された二本鎖核酸150を検出する。
まず、第1の一本鎖核酸断片131又は第2の一本鎖核酸断片141にフラッププローブをハイブリダイズさせる。図9の例では、フラッププローブ810は、第1の一本鎖核酸131にハイブリダイズする。その結果、第1フラップ部位811が形成される。
続いて、第1フラップ部位811にFENを反応させると、第1フラップ部位811が切断され、核酸断片811が生成される。続いて、核酸断片811は、蛍光基質(つまり核酸断片820)にハイブリダイズして第2フラップ部位821を形成する。
図9の例では、核酸断片820の5’末端には蛍光物質Fが結合されており、核酸断片820の5’末端から数塩基3’側に消光物質Qが結合されている。続いて、第2フラップ部位821にFENを反応させると、第2フラップ部位821が切断され、核酸断片821が生成される。その結果、蛍光物質Fが消光物質Qから離れ、蛍光シグナルを発生する。この蛍光シグナルを検出することにより、二本鎖核酸150の形成を検出することができる。
試薬液L210としては、流体デバイスを用いて行われる生化学分析において使用される一般的な液体を用いることができ、好ましくは水性溶液である。また、試薬液L210に界面活性剤等を含ませることにより、ウェル内に液体を封入しやすくしてもよい。
二本鎖核酸150の形成の検出にICA反応を用いる場合、二本鎖核酸150が存在する場合には、等温反応による酵素反応によって、蛍光物質Fが消光物質Qから遊離し、励起光に対応して所定の蛍光シグナルを発する。
二本鎖核酸150の形成の検出は、検出するシグナルの種類に応じて公知の適切な方法を選択することができる。例えば、蛍光シグナルを観察する場合は、蛍光物質に対応する励起光を封止されたウェル242に照射し、蛍光物質が発する蛍光を観察する。例えば、図5に示すように、封止されたウェル242内で所定の反応を行い、発生したシグナルを観察する。図5において、封止されたウェル242Rはシグナルが検出されたウェルであり、封止されたウェル242はシグナルが検出されなかったウェルである。
続いて、図6~図8を参照しながら、流体デバイス500を用いた場合を例に、本実施形態の方法について説明する。
まず、図6に示すように、流体デバイス500の内部に試薬液L210を導入する。試薬液L210は、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、及び、第2の特異的結合物質140が分散した液体であり、二本鎖核酸150の形成を検出するための試薬も含む。ここで、対象タンパク質110が標的タンパク質120と結合する場合には、試薬液L210内に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が含まれている。試薬液L210において、複合体100の濃度は、ウェル241に1ウェルあたり1分子以下の複合体100が入る濃度に調整されていることが好ましい。
続いて、図7に示すように、流体デバイス500の内部に封止液L220を導入する。封止液L220の比重は試薬液L210よりも大きい。このため、封止液L220は、試薬液L210のうち、ウェル241に収容されていない試薬液L210よりも下に沈み、ウェルアレイ240に接する。そして、封止液L220は、複合体100を含む試薬液L210を収容した複数のウェル241をそれぞれ個別に封止し、独立した反応空間(微小区画ともいう)242となる。
続いて、図8に示すように、微小区画242内で所定の反応を行い、発生したシグナルを観察する。図8中、微小区画242Rはシグナルが検出されたウェルであり、微小区画242はシグナルが検出されなかったウェルである。
本実施形態の方法において、工程(a)、すなわち、容器中で、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、及び、第2の特異的結合物質140を接触させ、その結果、対象タンパク質110が標的タンパク質120と結合した場合に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する工程(a)は、前記容器中で、無細胞タンパク質合成系により対象タンパク質110を合成する工程(a1)と、容器中で、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130、及び、第2の特異的結合物質140を接触させ、その結果、対象タンパク質110が標的タンパク質120と結合した場合に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する工程(a2)と、を含んでいてもよい。
すなわち、対象タンパク質110を無細胞タンパク質合成系により合成し、複合体100の形成を、対象タンパク質110の合成と連続的に行ってもよい。これにより、対象タンパク質が遺伝子変異を含むキナーゼ等である場合に、対象タンパク質の活性評価を簡便かつ短期間に実施することができる。
無細胞タンパク質合成系とは、細胞内でタンパク質を合成するのではなく、生細胞由来又は人工的に合成された、リボソームや転写及び翻訳因子等を用いて、鋳型である核酸からタンパク質をインビトロで合成する合成系をいう。
無細胞タンパク質合成系は、翻訳の過程に加えて、転写の過程を含む合成系であってもよい。タンパク質をコードする核酸がDNAである場合、DNAを転写することによって、タンパク質をコードするRNAを合成する必要がある。この場合、無細胞タンパク質合成系は、転写を可能にする因子を含んでいてもよい。転写を可能にする因子としては、例えば、RNAポリメラーゼ及びヌクレオチド等が挙げられるが、これに限定されず、当業者に公知の因子を用いることができる。
あるいは、予め、タンパク質をコードするDNAを鋳型としてRNAを合成し、そのRNAを、無細胞タンパク質合成系に添加してもよい。あるいは、人工的に化学合成されたRNAを用いてもよい。
無細胞タンパク質合成の鋳型となる核酸断片は、生体由来の核酸断片であってもよく、培養細胞由来の核酸断片であってもよく、ウイルス由来の核酸断片であってもよい。あるいは、核酸断片は、遺伝子解析結果に基づいて人工的に合成したものであってもよい。
無細胞タンパク質合成系としては、特に限定されず、例えば、コムギ胚芽、酵母、昆虫細胞、哺乳類培養細胞、ウサギ網状赤血球又は大腸菌等から得られた細胞抽出液を利用した合成系;翻訳に必要な因子を再構成した合成系等が挙げられる。なかでも、ヒト発現系無細胞タンパク質合成が好ましい。
無細胞タンパク質合成系は、翻訳に関わる因子として、例えば、tRNA、アミノアシル化tRNA合成酵素、翻訳開始因子、翻訳伸長因子及び翻訳終結因子等の少なくとも1つを含んでいてもよい。
上記の工程(a2)において、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質に、アデノシン三リン酸(ATP)を更に接触させてもよい。これにより、標的タンパク質がリン酸化されるか否かを評価することが可能になる。すなわち、キナーゼアッセイを行うことが可能になる。
図10は、本実施形態の方法の一例を示す模式図である。ここでは、一例として、変異タンパク質がBRAFである場合に、BRAF遺伝子における臨床的意義不明の遺伝子変異が、恒常的な活性化状態をもたらすものであるか否かを判定している。
図10では、無細胞タンパク質合成、キナーゼアッセイ、抗原抗体反応及びICA反応を連続的に行う。これにより、対象タンパク質の活性評価をより簡便かつ短期間に実施することができる。また、例えば、反応系に阻害剤を加えることで、阻害剤の効果を判定することもできる。
図10において、各反応に必要な試薬を添加するタイミング及び流体デバイスに試薬液を導入するタイミングは、適切に選択することができる。例えば、無細胞タンパク質合成を行った後、キナーゼアッセイ及び抗原抗体反応に必要な試薬を添加し、キナーゼアッセイ及び抗原抗体反応を行う。抗原抗体反応後の混合液にICA反応試薬を添加し、これを流体デバイスに導入してICA反応を行ってもよい。また、無細胞タンパク質合成を行った後、キナーゼアッセイ、抗原抗体反応及びICA反応に必要な試薬を添加し、これを流体デバイスに導入してキナーゼアッセイ、抗原抗体反応及びICA反応を行ってもよい。また、無細胞タンパク質合成を行った後、キナーゼアッセイ、抗原抗体反応及びICA反応に必要な試薬を添加し、キナーゼアッセイ及び抗原抗体反応を行った後、これを流体デバイスに導入してICA反応を行ってもよい。
別の方法として、無細胞タンパク質合成を行った後、キナーゼアッセイ及び抗原抗体反応に必要な試薬を添加しキナーゼアッセイ及び抗原抗体反応を行う。その後、ICA反応に必要な試薬を添加し、これを流体デバイスに導入してICA反応を行ってもよい。また、無細胞タンパク質合成を行った後、キナーゼアッセイに必要な試薬を添加しキナーゼアッセイを行う。その後、抗原抗体反応及びICA反応に必要な試薬を添加して抗原抗体反応を行い、これを流体デバイスに導入してICA反応を行ってもよい。検出感度がよいことから、キナーゼアッセイを行った後にICA反応に必要な試薬を添加し、これを流体デバイスに導入することが好ましい。
本実施形態の方法は、洗浄工程を含まないことが好ましい。特に、対象タンパク質110を無細胞タンパク質合成系により合成する場合においても、洗浄工程を含むことなく、対象タンパク質が標的タンパク質と結合する否かを評価することができれば、対象タンパク質の活性評価を更に簡便かつ短期間に実施することができる。
[複合体]
1実施形態において、本発明は、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を含み、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成した、複合体100を提供する。
1実施形態において、本発明は、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を含み、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成した、複合体100を提供する。
複合体100において、第1の一本鎖核酸断片131又は第2の一本鎖核酸断片141にフラッププローブが更にハイブリダイズしていてもよい。
上述したように、本実施形態の複合体を利用することにより、変異タンパク質の活性評価を簡便かつ短期間に実施することができる。
[キット]
1実施形態において、本発明は、対象タンパク質110が標的タンパク質120と結合する否かを評価するためのキットであって、複数のウェル241を有するウェルアレイ240、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を含む、キットを提供する。
1実施形態において、本発明は、対象タンパク質110が標的タンパク質120と結合する否かを評価するためのキットであって、複数のウェル241を有するウェルアレイ240、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を含む、キットを提供する。
本実施形態のキットにより、対象タンパク質110が標的タンパク質120と結合する否かを好適に評価することができる。
したがって、本実施形態のキットは、ウェル241に、対象タンパク質110、標的タンパク質120、第1の一本鎖核酸断片131で標識された、対象タンパク質110に対する第1の特異的結合物質130、及び、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140を導入し、その結果、対象タンパク質110が標的タンパク質120と結合した場合に、対象タンパク質110、標的タンパク質120、第1の特異的結合物質130及び第2の特異的結合物質140を含む複合体100が形成され、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する工程と、二本鎖核酸150の形成を検出する工程と、を含み、二本鎖核酸150の形成が検出されたことが、対象タンパク質110が標的タンパク質120と結合することを示す方法に用いるためのものであるということもできる。
ウェルアレイは、上述した流体デバイスの内部に配置されていてもよい。また、本実施形態のキットにおいて、対象タンパク質、標的タンパク質、第1の一本鎖核酸断片、第1の特異的結合物質、第2の一本鎖核酸断片及び第2の特異的結合物質については上述したものと同様である。
本実施形態のキットは、ATPを更に含んでいてもよい。これにより、標的タンパク質がリン酸化されるか否かを評価することが可能になる。すなわち、キナーゼアッセイを行うことが可能になる。
本実施形態のキットは、ウェル241の開口部を封止する封止液L220を更に含んでいてもよい。封止液L220については、上述したものと同様である。
本実施形態のキットは、第1の一本鎖核酸断片131の少なくとも一部及び第2の一本鎖核酸断片141の少なくとも一部がハイブリダイズして形成された二本鎖核酸150を検出する試薬を更に含んでいてもよい。
このような試薬としては、上述したICA反応用の試薬が挙げられ、具体的には、フラッププローブ、フラップエンドヌクレアーゼ及び蛍光基質等が挙げられる。
[対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法]
一実施形態において、本発明は、対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させ、その結果、前記標的タンパク質がリン酸化されていた場合に、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体が形成され、前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程と、前記二本鎖核酸の形成を検出する工程と、を含み、前記二本鎖核酸の形成が検出されたことが、前記標的タンパク質がリン酸化されていることを示す、方法を提供する。
一実施形態において、本発明は、対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法であって、容器中で、前記対象タンパク質、前記標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させ、その結果、前記標的タンパク質がリン酸化されていた場合に、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体が形成され、前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程と、前記二本鎖核酸の形成を検出する工程と、を含み、前記二本鎖核酸の形成が検出されたことが、前記標的タンパク質がリン酸化されていることを示す、方法を提供する。
すなわち、標的タンパク質をリン酸化する対象タンパク質であるとき、前記評価する方法は、以下の検出方法であってもよい。つまり、本実施形態の検出方法は、容器中で、対象タンパク質、標的タンパク質、第2の一本鎖核酸断片で標識された、標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させることと、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体が形成され、前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成することと、前記二本鎖核酸の形成を検出することと、を含んでいてもよい。
図11は、本実施形態の方法を説明する模式図である。図11に示すように、本実施形態の方法では、まず、工程(a’)において、容器中で、対象タンパク質110、標的タンパク質120、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140、第3の一本鎖核酸断片171で標識された、リン酸化された(図11中、リン酸基を符号160で示す。)前記標的タンパク質120に対する第3の特異的結合物質170、及び、ATP(図示せず)を接触させる。
その結果、対象タンパク質110のキナーゼ活性により、標的タンパク質120がリン酸化された場合に、標的タンパク質120、第2の特異的結合物質140及び第3の特異的結合物質170を含む複合体900が形成され、第2の一本鎖核酸断片141の少なくとも一部及び第3の一本鎖核酸断片171の少なくとも一部がハイブリダイズして二本鎖核酸(二本鎖核酸領域ともいう)150を形成する。
工程(a’)は、上述した、対象タンパク質が標的タンパク質と結合する否かを評価する方法における工程(a)と類似しているが、第1の特異的結合物質130の代わりに第3の特異的結合物質170を接触させる点が主に異なっている。
続いて、工程(b)において、二本鎖核酸150の形成を検出する。二本鎖核酸150の形成が検出された場合、標的タンパク質120がリン酸化されていると判断することができる。工程(b)は、上述した、対象タンパク質が標的タンパク質と結合する否かを評価する方法における工程(b)と同様である。本実施形態の方法において、二本鎖核酸150の形成量は、リン酸化された標的タンパク質120の存在量に対応する。
本実施形態の方法は、対象タンパク質110が標的タンパク質120をリン酸化しない場合に適用してもよく、その場合には、二本鎖核酸150の形成は検出されない。
実施例において後述するように、本発明によれば、変異タンパク質の活性評価を簡便かつ短期間に実施することができる。例えば、VUSを簡便かつ短期間に効率よく機能解析(つまり評価)することができる。
対象タンパク質110が標的タンパク質120に対してキナーゼ活性を発揮する場合、まず、対象タンパク質110と標的タンパク質120が結合して複合体900を形成し、続いて対象タンパク質110のキナーゼ活性により、標的タンパク質120がリン酸化されることが考えられる。
したがって、上述した、対象タンパク質が標的タンパク質と結合する否かを評価する方法、又は、本実施形態の方法により、対象タンパク質110の機能を解析(評価つまり)することができる。
第2の一本鎖核酸断片141の少なくとも一部及び第3の一本鎖核酸断片171の少なくとも一部がハイブリダイズすることができる必要がある観点から、第2の一本鎖核酸断片141の塩基長は10~200塩基であってもよい。同様に、第3の一本鎖核酸断片171の塩基長も10~200塩基であってもよい。また、第2の一本鎖核酸断片141の少なくとも一部及び第3の一本鎖核酸断片171の少なくとも一部がハイブリダイズして形成された二本鎖核酸150の長さは、7~30塩基程度であることが好ましく、例えば9塩基であってもよく、12塩基であってもよく、15塩基であってもよい。
本実施形態の方法において、対象タンパク質110は、臨床的意義不明の遺伝子変異を有するキナーゼであってもよい。より具体的な対象タンパク質としては、細胞内シグナル伝達経路のタンパク質が挙げられ、例えば、BRAF、A-RAF、Raf、MAP3K 4/12、MAP3K11、ASK1、TAK1等が挙げられる。この場合、VUSを有するタンパク質が恒常的な活性化状態にあるか否かを、標的タンパク質をリン酸化する否かにより評価することができる。
1実施形態において、本発明は、対象タンパク質110が標的タンパク質120をリン酸化するか否かを評価するためのキットであって、複数のウェル241を有するウェルアレイ240、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140、第3の一本鎖核酸断片171で標識された、リン酸化された標的タンパク質120に対する第3の特異的結合物質170、及び、ATPを含む、キットを提供する。
本実施形態のキットにより、対象タンパク質110が標的タンパク質120をリン酸化するか否かを好適に評価することができる。
したがって、本実施形態のキットは、ウェル241に、対象タンパク質110、標的タンパク質120、第2の一本鎖核酸断片141で標識された、標的タンパク質120に対する第2の特異的結合物質140、第3の一本鎖核酸断片171で標識された、リン酸化された標的タンパク質120に対する第3の特異的結合物質170、及び、ATPを導入し、その結果、対象タンパク質110が標的タンパク質120をリン酸化した場合に、標的タンパク質120、第2の特異的結合物質140及び第3の特異的結合物質170を含む複合体900が形成され、第2の一本鎖核酸断片141の少なくとも一部及び第3の一本鎖核酸断片171の少なくとも一部がハイブリダイズして二本鎖核酸150を形成する工程と、二本鎖核酸150の形成を検出する工程と、を含み、二本鎖核酸150の形成が検出されたことが、対象タンパク質110が標的タンパク質120をリン酸化することを示す方法に用いるためのものであるということもできる。
ウェルアレイは、上述した流体デバイスの内部に配置されていてもよい。また、本実施形態のキットにおいて、対象タンパク質、標的タンパク質、第2の一本鎖核酸断片、第2の特異的結合物質、第3の一本鎖核酸断片及び第3の特異的結合物質については上述したものと同様である。
本実施形態のキットは、ウェル241の開口部を封止する封止液L220を更に含んでいてもよい。封止液L220については、上述したものと同様である。
本実施形態のキットは、第2の一本鎖核酸断片141の少なくとも一部及び第3の一本鎖核酸断片171の少なくとも一部がハイブリダイズして形成された二本鎖核酸150を検出する試薬を更に含んでいてもよい。
このような試薬としては、上述したICA反応用の試薬が挙げられ、具体的には、フラッププローブ、フラップエンドヌクレアーゼ、蛍光基質等が挙げられる。
もう一つの態様として、本発明は以下の態様を含む。
[1]容器中で、対象タンパク質、標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させることと、
前記対象タンパク質が前記標的タンパク質と結合し、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体を形成することと、
前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成することと、
前記二本鎖核酸の形成を検出することと、を含む検出方法。
[2]さらに、前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成することを含む、[1]に記載の検出方法。
[3]前記二本鎖核酸を形成することが、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質に、アデノシン三リン酸を更に接触させることを含む、[2]に記載の検出方法。
[4]洗浄工程を含まない、[1]~[3]のいずれかに記載の検出方法。
[5]前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、[1]~[4]のいずれかに記載の検出方法。
[6]前記第1の一本鎖核酸断片及び前記第2の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、[1]~[5]のいずれかに記載の検出方法。
[7]前記複合体の形成後かつ前記二本鎖核酸の形成の前に、前記容器中に前記二本鎖核酸の形成を検出するための試薬を添加することをさらに含む、[1]~[6]のいずれかに記載の検出方法。
[8]前記二本鎖核酸の形成を検出することが、Invasive Cleavage Assayにより行われる、[1]~[7]のいずれかに記載の検出方法。
[9]容器中で、対象タンパク質、標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させることと、
前記標的タンパク質がリン酸化され、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体を形成することと、
前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成することと、
前記二本鎖核酸の形成を検出することと、を含む検出方法。
[10]さらに、前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成することを含む、[9]に記載の検出方法。
[11]洗浄工程を含まない、[9]又は[10]に記載の検出方法。
[12]前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、[9]又は[11]のいずれかに記載の検出方法。
[13]前記第2の一本鎖核酸断片及び前記第3の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、[9]~[12]のいずれかに記載の検出方法。
[14]前記複合体の形成後かつ前記二本鎖核酸の形成の前に、前記容器中に前記二本鎖核酸の形成を検出するための試薬を添加することをさらに含む、[9]~[13]のいずれかに記載の検出方法。
[15]前記二本鎖核酸の形成を検出することが、Invasive Cleavage Assayにより行われる、[9]~[14]のいずれかに記載の検出方法。
[1]容器中で、対象タンパク質、標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させることと、
前記対象タンパク質が前記標的タンパク質と結合し、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体を形成することと、
前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成することと、
前記二本鎖核酸の形成を検出することと、を含む検出方法。
[2]さらに、前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成することを含む、[1]に記載の検出方法。
[3]前記二本鎖核酸を形成することが、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質に、アデノシン三リン酸を更に接触させることを含む、[2]に記載の検出方法。
[4]洗浄工程を含まない、[1]~[3]のいずれかに記載の検出方法。
[5]前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、[1]~[4]のいずれかに記載の検出方法。
[6]前記第1の一本鎖核酸断片及び前記第2の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、[1]~[5]のいずれかに記載の検出方法。
[7]前記複合体の形成後かつ前記二本鎖核酸の形成の前に、前記容器中に前記二本鎖核酸の形成を検出するための試薬を添加することをさらに含む、[1]~[6]のいずれかに記載の検出方法。
[8]前記二本鎖核酸の形成を検出することが、Invasive Cleavage Assayにより行われる、[1]~[7]のいずれかに記載の検出方法。
[9]容器中で、対象タンパク質、標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させることと、
前記標的タンパク質がリン酸化され、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体を形成することと、
前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成することと、
前記二本鎖核酸の形成を検出することと、を含む検出方法。
[10]さらに、前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成することを含む、[9]に記載の検出方法。
[11]洗浄工程を含まない、[9]又は[10]に記載の検出方法。
[12]前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、[9]又は[11]のいずれかに記載の検出方法。
[13]前記第2の一本鎖核酸断片及び前記第3の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、[9]~[12]のいずれかに記載の検出方法。
[14]前記複合体の形成後かつ前記二本鎖核酸の形成の前に、前記容器中に前記二本鎖核酸の形成を検出するための試薬を添加することをさらに含む、[9]~[13]のいずれかに記載の検出方法。
[15]前記二本鎖核酸の形成を検出することが、Invasive Cleavage Assayにより行われる、[9]~[14]のいずれかに記載の検出方法。
[材料及び方法]
(抗体へのオリゴヌクレオチド修飾《BRAF-MEK相互作用検出》)
抗体-オリゴヌクレオチドコンジュゲーションツール(製品名「oYo-Link抗体ラベリング試薬」、フナコシ社)を用いて、抗リン酸化MEKウサギポリクローナル抗体(pMEK1/2(S217/221)ウサギ抗体、セルシグナリングテクノロジー社)、抗BRAFウサギポリクローナル抗体(プロテインテック社)に、それぞれ異なるオリゴヌクレオチド(インテグレーテッドDNAテクノロジーズ社)を結合させた。具体的には、上記の抗リン酸化MEKウサギポリクローナル抗体及び抗BRAFウサギポリクローナル抗体に、オリゴヌクレオチドである、DNA1(5’-TTTGTCACTGTTCCTCCTTTTGTTTTCCTTTCTGTGAGCAATTTCACCCAA-3’、配列番号1)、及び、DNA2(5’-GCATGGTTCCAATTTGGGTGAT-3’、配列番号2)をそれぞれ結合させた。
(抗体へのオリゴヌクレオチド修飾《BRAF-MEK相互作用検出》)
抗体-オリゴヌクレオチドコンジュゲーションツール(製品名「oYo-Link抗体ラベリング試薬」、フナコシ社)を用いて、抗リン酸化MEKウサギポリクローナル抗体(pMEK1/2(S217/221)ウサギ抗体、セルシグナリングテクノロジー社)、抗BRAFウサギポリクローナル抗体(プロテインテック社)に、それぞれ異なるオリゴヌクレオチド(インテグレーテッドDNAテクノロジーズ社)を結合させた。具体的には、上記の抗リン酸化MEKウサギポリクローナル抗体及び抗BRAFウサギポリクローナル抗体に、オリゴヌクレオチドである、DNA1(5’-TTTGTCACTGTTCCTCCTTTTGTTTTCCTTTCTGTGAGCAATTTCACCCAA-3’、配列番号1)、及び、DNA2(5’-GCATGGTTCCAATTTGGGTGAT-3’、配列番号2)をそれぞれ結合させた。
(抗体へのオリゴヌクレオチド修飾《リン酸化MEK検出》)
抗体-オリゴヌクレオチドコンジュゲーションツール(製品名「oYo-Link抗体ラベリング試薬」、フナコシ社)を用いて、抗リン酸化MEKウサギポリクローナル抗体(pMEK1/2(S217/221)ウサギ抗体、セルシグナリングテクノロジー社)、抗MEKウサギモノクローナル抗体(MEK1 Detector Antibody、アブカム社)に、それぞれ異なるオリゴヌクレオチド(インテグレーテッドDNAテクノロジーズ社)を結合させた。具体的には、上記の抗リン酸化MEKウサギポリクローナル抗体及び抗MEKウサギポリクローナル抗体に、オリゴヌクレオチドである、DNA1(5’-TTTGTCACTGTTCCTCCTTTTGTTTTCCTTTCTGTGAGCAATTTCACCCAA-3’、配列番号1)、及び、DNA2(5’-GCATGGTTCCAATTTGGGTGAT-3’、配列番号2)をそれぞれ結合させた。
抗体-オリゴヌクレオチドコンジュゲーションツール(製品名「oYo-Link抗体ラベリング試薬」、フナコシ社)を用いて、抗リン酸化MEKウサギポリクローナル抗体(pMEK1/2(S217/221)ウサギ抗体、セルシグナリングテクノロジー社)、抗MEKウサギモノクローナル抗体(MEK1 Detector Antibody、アブカム社)に、それぞれ異なるオリゴヌクレオチド(インテグレーテッドDNAテクノロジーズ社)を結合させた。具体的には、上記の抗リン酸化MEKウサギポリクローナル抗体及び抗MEKウサギポリクローナル抗体に、オリゴヌクレオチドである、DNA1(5’-TTTGTCACTGTTCCTCCTTTTGTTTTCCTTTCTGTGAGCAATTTCACCCAA-3’、配列番号1)、及び、DNA2(5’-GCATGGTTCCAATTTGGGTGAT-3’、配列番号2)をそれぞれ結合させた。
(無細胞タンパク質合成系による変異型BRAF、野生型BRAFの合成)
野生型又は臨床的意義不明の遺伝子変異を有する変異型BRAF遺伝子を導入したプラスミドDNA(ベクター:pT7CFE1-CHis Expression Vector、サーモフィッシャーサイエンティフィック社)を鋳型とし、ヒト無細胞タンパク質合成系(1-Step Human Coupled IVT Kit-DNA(サーモフィッシャーサイエンティフィック社)又はHuman Cell-Free Protein Expression System(タカラバイオ社))に添加し、30~32℃、90分~6時間タンパク質合成を行い、BRAFタンパク質を得た。
野生型又は臨床的意義不明の遺伝子変異を有する変異型BRAF遺伝子を導入したプラスミドDNA(ベクター:pT7CFE1-CHis Expression Vector、サーモフィッシャーサイエンティフィック社)を鋳型とし、ヒト無細胞タンパク質合成系(1-Step Human Coupled IVT Kit-DNA(サーモフィッシャーサイエンティフィック社)又はHuman Cell-Free Protein Expression System(タカラバイオ社))に添加し、30~32℃、90分~6時間タンパク質合成を行い、BRAFタンパク質を得た。
(キナーゼアッセイ及び抗原抗体反応)
対象タンパク質(変異型BRAF及び野生型BRAF)、標的タンパク質(inactive MEK)、上記の2種類のオリゴヌクレオチド修飾抗体、ATP、及び、ブロッキングバッファーを混合し、混合液を調製した。各混合液の用量はそれぞれ10μLであった。対象タンパク質は、終濃度が、それぞれ、0pM、2.82pM、28.2pM及び2821pMとなるように調製した。標的タンパク質は、終濃度が、28210pMとなるように調製した。また、2種類のオリゴヌクレオチド修飾抗体は、終濃度が、それぞれ8.56nMとなるように調製した。また、ブロッキングバッファーとしては、1%ウシ血清アルブミン(BSAともいう)を含むトリス緩衝生理食塩水(TBSともいう)を使用した。続いて、各混合液を30℃で45分間、次に37℃で60分間反応させた。
対象タンパク質(変異型BRAF及び野生型BRAF)、標的タンパク質(inactive MEK)、上記の2種類のオリゴヌクレオチド修飾抗体、ATP、及び、ブロッキングバッファーを混合し、混合液を調製した。各混合液の用量はそれぞれ10μLであった。対象タンパク質は、終濃度が、それぞれ、0pM、2.82pM、28.2pM及び2821pMとなるように調製した。標的タンパク質は、終濃度が、28210pMとなるように調製した。また、2種類のオリゴヌクレオチド修飾抗体は、終濃度が、それぞれ8.56nMとなるように調製した。また、ブロッキングバッファーとしては、1%ウシ血清アルブミン(BSAともいう)を含むトリス緩衝生理食塩水(TBSともいう)を使用した。続いて、各混合液を30℃で45分間、次に37℃で60分間反応させた。
(Invasive Cleavage Assay反応試薬の調製)
上記の抗体に修飾したオリゴヌクレオチドを用いてICA反応を行うため、検出用ICA反応試薬を調製した。本実験例におけるICA反応試薬は、2μMアレルプローブ(5’-CGCGCCGAGGAATTGCTCACAGAAAGGA-3’)(ファスマック社、配列番号3)、4μM FRETカセット1(蛍光基質、Alexa488-BHQ:5’-X-TTCT-Y-AGCCGGTTTTCCGGCTGAGACCTCGGCGCG-3’,X:Alexa488+AminoC6,Y:ブラックホールクエンチャー(BHQ)1-dT)(日本バイオサービス社、配列番号4)、0.108μM~1.73μMフラップエンドヌクレアーゼ(FEN)-1、50mM Tris-HCl(pH8.5)、20mM MgCl2、及び0.05% Tween20を含んでいた。なお、これらのICA反応試薬における各成分の濃度は、実験例における終濃度である。
上記の抗体に修飾したオリゴヌクレオチドを用いてICA反応を行うため、検出用ICA反応試薬を調製した。本実験例におけるICA反応試薬は、2μMアレルプローブ(5’-CGCGCCGAGGAATTGCTCACAGAAAGGA-3’)(ファスマック社、配列番号3)、4μM FRETカセット1(蛍光基質、Alexa488-BHQ:5’-X-TTCT-Y-AGCCGGTTTTCCGGCTGAGACCTCGGCGCG-3’,X:Alexa488+AminoC6,Y:ブラックホールクエンチャー(BHQ)1-dT)(日本バイオサービス社、配列番号4)、0.108μM~1.73μMフラップエンドヌクレアーゼ(FEN)-1、50mM Tris-HCl(pH8.5)、20mM MgCl2、及び0.05% Tween20を含んでいた。なお、これらのICA反応試薬における各成分の濃度は、実験例における終濃度である。
[実験例1]
(チューブ内での対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価)
上記の抗原抗体反応後の混合液10μLと上記のICA反応試薬10μLを混合し、Rotor-Gene Q(キアゲン社)を用いて66℃、1時間反応させ、Alexa488の蛍光を検出した。以下、この反応を「イムノICA反応」という場合がある。
(チューブ内での対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価)
上記の抗原抗体反応後の混合液10μLと上記のICA反応試薬10μLを混合し、Rotor-Gene Q(キアゲン社)を用いて66℃、1時間反応させ、Alexa488の蛍光を検出した。以下、この反応を「イムノICA反応」という場合がある。
図12~図14は、実験例1の結果を示すグラフである。図12は、対象タンパク質として、2821pMの変異型BRAF(Mt)及び野生型BRAF(WT)を用いて、イムノICA反応を行い、BRAF及びリン酸化MEKの相互作用(つまり結合)を検出した結果である。図12中、縦軸はAlexa488の蛍光強度(相対値)を示し、横軸はICA反応開始後の時間(秒)を示す。
その結果、変異型BRAFとリン酸化MEKの相互作用を検出することができることが明らかとなった。野生型BRAFとリン酸化MEKとの相互作用を検出した時の蛍光強度をN(ノイズ)とし、変異型BRAFとリン酸化MEKとの相互作用を検出した時の蛍光強度をS(シグナル)とし、エンドポイントにおけるシグナル・ノイズ比(S/N比)を算出した結果、2.4であった。
図13は、対象タンパク質として、2821pMの変異型BRAF(Mt)及び野生型BRAF(WT)を用いて、イムノICA反応を行い、リン酸化MEKを検出した結果である。図13中、縦軸はAlexa488の蛍光強度(相対値)を示し、横軸はICA反応開始後の時間(秒)を示す。
その結果、変異型BRAFのキナーゼ活性を検出することができることが明らかとなった。野生型BRAFを反応させた場合にリン酸化MEKを検出した時の蛍光強度をN(ノイズ)とし、変異型BRAFを反応させた場合にリン酸化MEKを検出した時の蛍光強度をS(シグナル)とし、エンドポイントにおけるシグナル・ノイズ比(S/N比)を算出した結果、3.9であった。
図14は、図12及び図13の結果において、抗原濃度が0nMの時の蛍光強度をN(ノイズ)とし、その他の各濃度での蛍光強度をS(シグナル)とし、シグナル・ノイズ比(S/N比)を算出した結果を示すグラフである。図14中、縦軸は各反応でのS/N比最大値を示し、横軸はICA反応開始後の時間(秒)又は酵素濃度を示す。その結果、短い反応時間で、低濃度の酵素条件で反応が可能なのは、酵素濃度0.432μMの条件であった。その際のS/N比最大値は、BRAF-MEK相互作用検出で3.9、リン酸化MEK検出で5.4であった。
[実験例2]
(流体デバイスを用いた対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価1)
図3~図5に示すような流体デバイスを用いた以外は実験例1と同様にして、変異型BRAF及びリン酸化MEKの相互作用、つまり結合)、並びに、野生型BRAF及びリン酸化MEKの相互作用を検出した。
(流体デバイスを用いた対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価1)
図3~図5に示すような流体デバイスを用いた以外は実験例1と同様にして、変異型BRAF及びリン酸化MEKの相互作用、つまり結合)、並びに、野生型BRAF及びリン酸化MEKの相互作用を検出した。
上記の抗原抗体反応後の混合液10μLと上記のICA反応試薬10μLを混合し、ウェルアレイを備えた流体デバイスのウェルに導入した。ウェルアレイは約100万個のウェルを有しており、ウェル1つあたりの容積は約60fLであった。
続いて、流体デバイスに封止液(FC-40、CAS番号:86508-42-1)を導入した。その結果、ウェルアレイの各ウェルがそれぞれ個別に封止され、独立した反応空間となった。
続いて、流体デバイスをアルミブロック恒温槽(型式「DTU-Mini」、タイテック社)にセットし、66℃で7分間又は30分間加熱した後、顕微鏡(製品名「オールインワン蛍光顕微鏡」、型式「BZ-X810」、キーエンス社)を用いて観察した。続いて、顕微鏡観察画像に基づいて、各ウェルの輝度を算出した。
図15は、各ウェルの輝度の算出結果を示すグラフである。対象タンパク質として、変異型BRAF(Mt)及び野生型BRAF(WT)を用いて、イムノICA反応を行い、BRAF及びリン酸化MEKの相互作用を検出した結果を示す。図15の縦軸はウェル数を示し、横軸はウェルの輝度を示す。また、図15左は、ICA反応時間7分間の結果を示し、図15右は、ICA反応時間30分間の結果を示す。また、図15上段は変異型BRAF(Mt)とリン酸化MEKの相互作用を検出した結果を示し、図15下段は野生型BRAF(WT)とリン酸化MEKの相互作用を検出した結果を示す。
下記表1は、図15に示すウェルの輝度の中央値をまとめた表である。表1中、「S/N」は、野生型BRAFとリン酸化MEKとの相互作用を検出した時の蛍光強度をN(ノイズ)とし、変異型BRAFとリン酸化MEKとの相互作用を検出した時の蛍光強度をS(シグナル)とし、エンドポイントにおけるシグナル・ノイズ比(S/N比)を算出した結果を示す。

その結果、変異型BRAF及びリン酸化MEKの相互作用と、野生型BRAF及びリン酸化MEKの相互作用との間に差が認められ、変異型BRAFとリン酸化MEKの相互作用を検出することができることが明らかとなった。
[実験例3]
(流体デバイスを用いた対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価2)
図3~図5に示すような流体デバイスを用いた以外は実験例1と同様にして、リン酸化MEKを検出した。対象タンパク質として、変異型BRAF及び野生型BRAFを用いた。
(流体デバイスを用いた対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価2)
図3~図5に示すような流体デバイスを用いた以外は実験例1と同様にして、リン酸化MEKを検出した。対象タンパク質として、変異型BRAF及び野生型BRAFを用いた。
上記の抗原抗体反応後の混合液10μLと上記のICA反応試薬10μLを混合し、ウェルアレイを備えた流体デバイスのウェルに導入した。ウェルアレイは約100万個のウェルを有しており、ウェル1つあたりの容積は約60fLであった。
続いて、流体デバイスに封止液(FC-40、CAS番号:86508-42-1)を導入した。その結果、ウェルアレイの各ウェルがそれぞれ個別に封止され、独立した反応空間となった。
続いて、流体デバイスをアルミブロック恒温槽(型式「DTU-Mini」、タイテック社)にセットし、66℃で7分間又は30分間加熱した後、顕微鏡(製品名「オールインワン蛍光顕微鏡」、型式「BZ-X810」、キーエンス社)を用いて観察した。続いて、顕微鏡観察画像に基づいて、各ウェルの輝度を算出した。
図16は、各ウェルの輝度の算出結果を示すグラフである。対象タンパク質として、変異型BRAF(Mt)及び野生型BRAF(WT)を用いて、イムノICA反応を行い、リン酸化MEKを検出した結果を示す。図16の縦軸はウェル数を示し、横軸はウェルの輝度を示す。また、図16左は、ICA反応時間7分間の結果を示し、図16右は、ICA反応時間30分間の結果を示す。また、図16上段は変異型BRAF(Mt)を反応させた場合にリン酸化MEKを検出した結果を示し、図16下段は野生型BRAF(WT)を反応させた場合にリン酸化MEKを検出した結果を示す。
下記表2は、図16に示すウェルの輝度の中央値をまとめた表である。表2中、「S/N」は、野生型BRAFを反応させた場合の蛍光強度をN(ノイズ)とし、変異型BRAFを反応させた場合の蛍光強度をS(シグナル)とし、エンドポイントにおけるシグナル・ノイズ比(S/N比)を算出した結果を示す。

その結果、変異型BRAFを反応させた場合のリン酸化MEKの存在量と、野生型BRAFを反応させた場合のリン酸化MEKの存在量との間に差が認められ、BRAFのキナーゼ活性を検出することができることが明らかとなった。
[実験例4]
(チューブ内での対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価3)
試薬添加の順番による活性の違いを評価した。キナーゼアッセイを行う前に、キナーゼアッセイ反応、抗原抗体反応、イムノICA反応のための試薬をすべて混合して、リン酸MEKを検出した場合をパターン1とする。キナーゼアッセイを行ったあとに、抗原抗体反応とイムノICA反応を行うための試薬を混合してチューブに導入し、リン酸化MEKを検出した場合をパターン2とする。
(チューブ内での対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価3)
試薬添加の順番による活性の違いを評価した。キナーゼアッセイを行う前に、キナーゼアッセイ反応、抗原抗体反応、イムノICA反応のための試薬をすべて混合して、リン酸MEKを検出した場合をパターン1とする。キナーゼアッセイを行ったあとに、抗原抗体反応とイムノICA反応を行うための試薬を混合してチューブに導入し、リン酸化MEKを検出した場合をパターン2とする。
パターン1の詳細な手順を説明する。対象タンパク質(変異型BRAF及び野生型BRAF)、標的タンパク質(inactive MEK及びactive MEK)、上記の2種類のオリゴヌクレオチド修飾抗体、ATP、及び、ブロッキングバッファーを混合し、混合液を調製した。各混合液の用量は、それぞれ10μLであった。対象タンパク質は、終濃度がそれぞれ2821pMとなるように調製した。標的タンパク質は、終濃度がそれぞれ7nMとなるように調製した。また、2種類のオリゴヌクレオチド修飾抗体は、DNA1を結合した場合は終濃度が1nM、DNA2を結合した場合は終濃度が4nMとなるように調製した。また、ブロッキングバッファーとしては、カゼイン及びノニオン系界面活性剤を含むトリス緩衝生理食塩水(TBSともいう)を使用した。表3に示す通り、反応組成として6パターンのBRAF、MEK及びATPの組み合わせを使用した。表3中、○は該当する成分が含まれることを示し、×は該当する成分が含まれないことを示す。

(ICA反応試薬の調製)
パターン1におけるICA反応試薬は、0.108μMフラップエンドヌクレアーゼ(FEN)-1であること以外は実験例1~3と同じ組成及び終濃度で調整した。なお、これらのICA反応試薬における各成分の濃度は、実験例における終濃度である。
パターン1におけるICA反応試薬は、0.108μMフラップエンドヌクレアーゼ(FEN)-1であること以外は実験例1~3と同じ組成及び終濃度で調整した。なお、これらのICA反応試薬における各成分の濃度は、実験例における終濃度である。
チューブ内で、キナーゼアッセイ、抗原抗体反応及びICA反応を行った。混合液とICA反応試薬を混合し、チューブに導入した。30℃で60分間反応させた後、実験例1と同様にして、リン酸化MEKを検出した(パターン1とする)。イムノICA反応の反応時間は、60分間から180分間までとした。
パターン2の詳細な手順を説明する。対象タンパク質(変異型BRAF及び野生型BRAF)、標的タンパク質(inactive MEK及びactive MEK)、ATP、20mM MgCl2、及び、ブロッキングバッファーを混合し、混合液を調製した。各混合液の用量は、それぞれ7.5μLであった。対象タンパク質は、終濃度がそれぞれ2821pMとなるように調製した。標的タンパク質は、終濃度がそれぞれ7nMとなるように調製した。また、ブロッキングバッファーとしては、カゼイン及びノニオン系界面活性剤を含むトリス緩衝生理食塩水(TBSともいう)を使用した。表3に示す通り、反応組成として6パターンのBRAF、MEK及びATPの組み合わせを使用した。表3中、○は該当する成分が含まれることを示し、×は該当する成分が含まれないことを示す。
(抗原抗体反応試薬とICA反応試薬の調製)
パターン2における、抗原抗体反応試薬とICA反応試薬の混合液は、上記の2種類のオリゴヌクレオチド修飾抗体、2μMアレルプローブ(5’-CGCGCCGAGGAATTGCTCACAGAAAGGA-3’)(ファスマック社、配列番号3)、4μM FRETカセット1(蛍光基質、Alexa488-BHQ:5’-X-TTCT-Y-AGCCGGTTTTCCGGCTGAGACCTCGGCGCG-3’,X:Alexa488+AminoC6,Y:ブラックホールクエンチャー(BHQ)1-dT)(日本バイオサービス社、配列番号4)、0.108μMフラップエンドヌクレアーゼ(FEN)-1、50mM Tris-HCl(pH8.5)、及び0.05% Tween20を含んでいた。また、2種類のオリゴヌクレオチド修飾抗体は、DNA1を結合した場合は終濃度が1nM、DNA2を結合した場合は終濃度が4nMとなるように調製した。各反応試薬の用量は、抗原抗体反応試薬が2.5μL、ICA反応試薬が10μLであった。なお、これらのICA反応試薬における各成分の濃度は、実験例における終濃度である。
パターン2における、抗原抗体反応試薬とICA反応試薬の混合液は、上記の2種類のオリゴヌクレオチド修飾抗体、2μMアレルプローブ(5’-CGCGCCGAGGAATTGCTCACAGAAAGGA-3’)(ファスマック社、配列番号3)、4μM FRETカセット1(蛍光基質、Alexa488-BHQ:5’-X-TTCT-Y-AGCCGGTTTTCCGGCTGAGACCTCGGCGCG-3’,X:Alexa488+AminoC6,Y:ブラックホールクエンチャー(BHQ)1-dT)(日本バイオサービス社、配列番号4)、0.108μMフラップエンドヌクレアーゼ(FEN)-1、50mM Tris-HCl(pH8.5)、及び0.05% Tween20を含んでいた。また、2種類のオリゴヌクレオチド修飾抗体は、DNA1を結合した場合は終濃度が1nM、DNA2を結合した場合は終濃度が4nMとなるように調製した。各反応試薬の用量は、抗原抗体反応試薬が2.5μL、ICA反応試薬が10μLであった。なお、これらのICA反応試薬における各成分の濃度は、実験例における終濃度である。
パターン2では、キナーゼアッセイ液をチューブに導入し、キナーゼアッセイを30℃で60分間反応させた後、抗原抗体反応試薬とICA反応試薬を、チューブに導入した。37℃で60分間反応させ、66℃で60分間から120分間反応させて、実験例1と同様にしてイムノICA反応を行った。対象タンパク質として、変異型BRAF及び野生型BRAFを用いた。
図17は、実験例4のパターン1の結果を示すグラフである。図18は、実験例4のパターン2の結果を示すグラフである。図17及び図18におけるラベル1~6は、それぞれ表3の反応組成1~6に対応している。図17及び図18は、対象タンパク質として、2821pMの変異型BRAF(Mt)又は野生型BRAF(WT)を用いて、イムノICA反応を60分間行い、リン酸化を検出した結果である。
図17に示される反応組成5のパターン1と図18に示される反応組成5のパターン2とを比較すると、パターン2、つまりキナーゼアッセイ後に抗体反応液とICA反応試薬を添加した方が、反応性が良くなることが明らかとなった。また、ICA反応は、少なくとも反応開始から60分間で検出を行うことが可能であることが明らかになった。さらに反応組成3のパターン2の結果から、バックグラウンド反応の上昇も見られないことが分かった。なお、反応組成5と反応組成3のパターン2の結果から算出したS/N比は、最大で約4となることが分かった。
[実験例5]
(流体デバイス内での対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価3)
図3~図5に示すような流体デバイスを用いて、イムノICA反応を行った以外は実験例4と同様にして、リン酸化MEKを検出した。対象タンパク質として、変異型BRAF及び野生型BRAFを用いた。
(流体デバイス内での対象タンパク質(変異型BRAF及び野生型BRAF)のキナーゼ活性評価3)
図3~図5に示すような流体デバイスを用いて、イムノICA反応を行った以外は実験例4と同様にして、リン酸化MEKを検出した。対象タンパク質として、変異型BRAF及び野生型BRAFを用いた。
チューブ内で、キナーゼアッセイ、抗原抗体反応を、流体デバイス内でICA反応を行った。パターン1では、混合液とICA反応試薬を混合し、キナーゼアッセイ及び抗原抗体反応を行った後、流体デバイスに導入した。30℃で60分間反応させた後、実験例2、3と同様にして、リン酸化MEKを検出した。イムノICA反応の反応時間は、60分間から180分間までとした。また、パターン2では、キナーゼアッセイを30℃で60分間反応させた後、抗原抗体反応試薬とICA反応試薬を混合して、37℃で60分間反応させ、流体デバイスに導入した。66℃で60分間から120分間反応させて、実験例2、3と同様にして、リン酸化MEKを検出した(パターン2とする)。
図19は、イムノICA反応における時間ごとの蛍光強度を測定したヒストグラムである。図19の左側から、パターン1におけるイムノICA反応開始から120分後の蛍光強度、パターン1におけるイムノICA反応開始から180分後の蛍光強度、パターン2におけるイムノICA反応開始から90分後の蛍光強度及びパターン2におけるイムノICA反応開始から120分後の蛍光強度をそれぞれ示す。図19の1~6は、それぞれ表3の反応組成1~6に対応している。
図19の結果からもパターン2、つまりキナーゼアッセイ後に抗原抗体反応試薬とICA反応試薬を添加した方が、反応性が良くなることが明らかとなった。キナーゼアッセイ時に抗体が存在することで、リン酸化反応を阻害することが考えられる。
本発明によれば、変異タンパク質の活性評価を簡便かつ短期間に実施する技術を提供することができる。
100,900…複合体、110…対象タンパク質、120…標的タンパク質、131…第1の一本鎖核酸断片、130…第1の特異的結合物質、141…第2の一本鎖核酸断片、140…第2の特異的結合物質、150…二本鎖核酸、171…第3の一本鎖核酸断片、170…第3の特異的結合物質、200,500…流体デバイス、210…基板、220…蓋部材、221…凸部、222…導入ポート、223…排出ポート、230…流路、240…ウェルアレイ、241…ウェル、242…封止されたウェル(微小区画)、L210…試薬液、L220…封止液、242R…シグナルが検出されたウェル、510…壁部材、810…フラッププローブ、811…第1フラップ部位(核酸断片)、821…第2フラップ部位(核酸断片)、820,820’…核酸断片、F…蛍光物質、Q…消光物質。
Claims (13)
- 対象タンパク質が標的タンパク質と結合する否かを評価する方法であって、
容器中で、前記対象タンパク質、前記標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a)と、
前記二本鎖核酸の形成を検出する工程(b)と、を含み、
前記二本鎖核酸の形成が検出されたことが、前記対象タンパク質が前記標的タンパク質と結合することを示す、方法。 - 前記工程(a)が、
前記容器中で、無細胞タンパク質合成系により前記対象タンパク質を合成する工程(a1)と、
前記容器中で、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質を接触させ、その結果、前記対象タンパク質が前記標的タンパク質と結合した場合に、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質及び前記第2の特異的結合物質を含む複合体が形成され、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a2)と、を含む、請求項1に記載の方法。 - 前記工程(a2)において、前記対象タンパク質、前記標的タンパク質、前記第1の特異的結合物質、及び、前記第2の特異的結合物質に、アデノシン三リン酸(ATP)を更に接触させる、請求項2に記載の方法。
- 洗浄工程を含まない、請求項1~3のいずれか一項に記載の方法。
- 前記容器がウェルであり、前記ウェルの容積が10fL~100pLである、請求項1~3のいずれか一項に記載の方法。
- 前記第1の一本鎖核酸断片及び前記第2の一本鎖核酸断片の塩基長が、それぞれ10~200塩基である、請求項1~3のいずれか一項に記載の方法。
- 前記二本鎖核酸の形成を検出する工程が、Invasive Cleavage Assayにより行われる、請求項1~3のいずれか一項に記載の方法。
- 対象タンパク質、標的タンパク質、第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質を含み、前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成した、複合体。
- 対象タンパク質が標的タンパク質と結合する否かを評価するためのキットであって、
複数のウェルを有するウェルアレイ、
第1の一本鎖核酸断片で標識された、前記対象タンパク質に対する第1の特異的結合物質、及び、
第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、を含む、キット。 - ATPを更に含む、請求項9に記載のキット。
- 前記ウェルの開口部を封止する封止液を更に含む、請求項9又は10に記載のキット。
- 前記第1の一本鎖核酸断片の少なくとも一部及び前記第2の一本鎖核酸断片の少なくとも一部がハイブリダイズして形成された二本鎖核酸を検出する試薬を更に含む、請求項9又は10に記載のキット。
- 対象タンパク質が標的タンパク質をリン酸化するか否かを評価する方法であって、
容器中で、前記対象タンパク質、前記標的タンパク質、第2の一本鎖核酸断片で標識された、前記標的タンパク質に対する第2の特異的結合物質、第3の一本鎖核酸断片で標識された、リン酸化された前記標的タンパク質に対する第3の特異的結合物質、及び、ATPを接触させ、その結果、前記標的タンパク質がリン酸化されていた場合に、前記標的タンパク質、前記第2の特異的結合物質及び前記第3の特異的結合物質を含む複合体が形成され、前記第2の一本鎖核酸断片の少なくとも一部及び前記第3の一本鎖核酸断片の少なくとも一部がハイブリダイズして二本鎖核酸を形成する工程(a’)と、
前記二本鎖核酸の形成を検出する工程(b)と、を含み、
前記二本鎖核酸の形成が検出されたことが、前記標的タンパク質がリン酸化されていることを示す、方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022122819 | 2022-08-01 | ||
| JP2022-122819 | 2022-08-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024029511A1 true WO2024029511A1 (ja) | 2024-02-08 |
Family
ID=89849333
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/028056 WO2024029511A1 (ja) | 2022-08-01 | 2023-08-01 | 評価方法、複合体及びキット |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024029511A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007020526A (ja) * | 2005-07-20 | 2007-02-01 | Japan Science & Technology Agency | 特異的結合を利用した標的分子の高感度検出法、そのキット |
| JP2020122799A (ja) * | 2014-05-15 | 2020-08-13 | メソ スケール テクノロジーズ エルエルシー | 改善されたアッセイ方法 |
| WO2023080137A1 (ja) * | 2021-11-04 | 2023-05-11 | 凸版印刷株式会社 | 標的物質の検出方法 |
-
2023
- 2023-08-01 WO PCT/JP2023/028056 patent/WO2024029511A1/ja unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007020526A (ja) * | 2005-07-20 | 2007-02-01 | Japan Science & Technology Agency | 特異的結合を利用した標的分子の高感度検出法、そのキット |
| JP2020122799A (ja) * | 2014-05-15 | 2020-08-13 | メソ スケール テクノロジーズ エルエルシー | 改善されたアッセイ方法 |
| WO2023080137A1 (ja) * | 2021-11-04 | 2023-05-11 | 凸版印刷株式会社 | 標的物質の検出方法 |
Non-Patent Citations (1)
| Title |
|---|
| MOHAMMED HISHAM, CARROLL JASON S.: "Approaches for Assessing and Discovering Protein Interactions in Cancer", MOLECULAR CANCER RESEARCH, AMERICAN ASSOCIATION FOR CANCER RESEARCH, US, vol. 11, no. 11, 1 November 2013 (2013-11-01), US , pages 1295 - 1302, XP093135026, ISSN: 1541-7786, DOI: 10.1158/1541-7786.MCR-13-0454 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11828753B2 (en) | Biosensor microarray compositions and methods | |
| US11261484B2 (en) | Biomolecule analysis kit and biomolecule analysis method | |
| Rödiger et al. | Nucleic acid detection based on the use of microbeads: a review | |
| US20210382002A1 (en) | Detection methods for epitachophoresis workflow automation | |
| US10718776B2 (en) | Method for detecting biological substance | |
| JP2023106482A (ja) | 標的分子の検出方法 | |
| EP4047368A1 (en) | Method and kit for target molecule detection | |
| US20200363406A1 (en) | Highly-specific assays | |
| EP3974844A1 (en) | Method for detecting target molecule | |
| JP2024012501A (ja) | 試料分析のための装置および方法 | |
| US20240279717A1 (en) | Method of detecting target substance | |
| US10914735B2 (en) | Protein detection using FET | |
| WO2019079334A1 (en) | DISEASE PROTEASE PROTEIN NETWORKS AND USES THEREOF | |
| WO2024029511A1 (ja) | 評価方法、複合体及びキット | |
| US20220340953A1 (en) | Detection method | |
| US20220088598A1 (en) | Method of introducing liquid into wells | |
| WO2016141040A1 (en) | Homogenous entropy-driven biomolecular assay (heba) | |
| WO2009150583A1 (en) | Diagnostic device | |
| US20210285937A1 (en) | Multiplexed colocalization-by-linkage assays for the detection and analysis of analytes | |
| WO2024162347A1 (ja) | 標的rnaの検出方法及びキット | |
| WO2024085132A1 (ja) | 生体物質処理方法及び反応検出方法並びに生体物質処理装置及び反応検出装置 | |
| WO2024084958A1 (ja) | 検出デバイス及び標的分子の検出方法 | |
| JP2024129127A (ja) | ウェルに液体を導入する方法 | |
| WO2024141901A1 (en) | Heat-based transfer of reaction products made in situ to a planar support | |
| US20100233697A1 (en) | Method for standarizing surface binding of a nucleic acid sample for sequencing analysis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23850065 Country of ref document: EP Kind code of ref document: A1 |