WO2023091921A1 - Methods and compositions for treating cognitive impairment - Google Patents
Methods and compositions for treating cognitive impairment Download PDFInfo
- Publication number
- WO2023091921A1 WO2023091921A1 PCT/US2022/079920 US2022079920W WO2023091921A1 WO 2023091921 A1 WO2023091921 A1 WO 2023091921A1 US 2022079920 W US2022079920 W US 2022079920W WO 2023091921 A1 WO2023091921 A1 WO 2023091921A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- cognitive impairment
- impaired
- cognitive
- subject
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 79
- 208000028698 Cognitive impairment Diseases 0.000 title claims abstract description 52
- 208000010877 cognitive disease Diseases 0.000 title claims abstract description 52
- 239000000203 mixture Substances 0.000 title description 48
- 230000003920 cognitive function Effects 0.000 claims abstract description 20
- 150000001875 compounds Chemical class 0.000 claims description 66
- 150000003839 salts Chemical class 0.000 claims description 35
- 230000001771 impaired effect Effects 0.000 claims description 31
- 230000015654 memory Effects 0.000 claims description 10
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 10
- 230000001149 cognitive effect Effects 0.000 claims description 9
- 230000006870 function Effects 0.000 claims description 9
- 230000013016 learning Effects 0.000 claims description 9
- 230000005764 inhibitory process Effects 0.000 claims description 7
- 230000004044 response Effects 0.000 claims description 7
- 230000003936 working memory Effects 0.000 claims description 7
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 claims description 6
- 230000002708 enhancing effect Effects 0.000 claims description 6
- 239000002664 nootropic agent Substances 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 208000024827 Alzheimer disease Diseases 0.000 claims description 3
- 206010003805 Autism Diseases 0.000 claims description 3
- 208000020706 Autistic disease Diseases 0.000 claims description 3
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 claims description 3
- 201000000980 schizophrenia Diseases 0.000 claims description 3
- NJVZDURTFWBXAM-VIFPVBQESA-N [(2S)-1-hydroxy-3-phenylpropan-2-yl]urea Chemical class C(N)(=O)N[C@@H](CC1=CC=CC=C1)CO NJVZDURTFWBXAM-VIFPVBQESA-N 0.000 abstract description 2
- 239000003826 tablet Substances 0.000 description 53
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 49
- 239000002552 dosage form Substances 0.000 description 40
- 238000009472 formulation Methods 0.000 description 25
- 239000000314 lubricant Substances 0.000 description 20
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 18
- UCTRAOBQFUDCSR-SECBINFHSA-N [(2r)-2-amino-3-phenylpropyl] carbamate Chemical compound NC(=O)OC[C@H](N)CC1=CC=CC=C1 UCTRAOBQFUDCSR-SECBINFHSA-N 0.000 description 17
- 239000003814 drug Substances 0.000 description 17
- 239000011230 binding agent Substances 0.000 description 16
- 229940070119 solriamfetol Drugs 0.000 description 16
- 229940068196 placebo Drugs 0.000 description 14
- 239000000902 placebo Substances 0.000 description 14
- 238000011282 treatment Methods 0.000 description 12
- 229940079593 drug Drugs 0.000 description 11
- 239000006186 oral dosage form Substances 0.000 description 10
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- -1 hemisulfate Chemical compound 0.000 description 9
- 235000019359 magnesium stearate Nutrition 0.000 description 9
- 239000000651 prodrug Substances 0.000 description 9
- 229940002612 prodrug Drugs 0.000 description 9
- 206010041349 Somnolence Diseases 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 208000007590 Disorders of Excessive Somnolence Diseases 0.000 description 6
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 239000002775 capsule Substances 0.000 description 5
- 238000007906 compression Methods 0.000 description 5
- 230000006835 compression Effects 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- 238000005550 wet granulation Methods 0.000 description 5
- 239000007894 caplet Substances 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 239000007884 disintegrant Substances 0.000 description 4
- 230000037406 food intake Effects 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 238000007792 addition Methods 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 230000004888 barrier function Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 239000007891 compressed tablet Substances 0.000 description 3
- 230000006735 deficit Effects 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 208000001797 obstructive sleep apnea Diseases 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 208000001573 Cataplexy Diseases 0.000 description 2
- 229920001353 Dextrin Polymers 0.000 description 2
- 239000004375 Dextrin Substances 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 2
- 229920002774 Maltodextrin Polymers 0.000 description 2
- 239000005913 Maltodextrin Substances 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- 208000032140 Sleepiness Diseases 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- 150000001241 acetals Chemical class 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 125000002843 carboxylic acid group Chemical group 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229920001688 coating polymer Polymers 0.000 description 2
- 230000004633 cognitive health Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000002537 cosmetic Substances 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 235000019425 dextrin Nutrition 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229940035034 maltodextrin Drugs 0.000 description 2
- 201000003631 narcolepsy Diseases 0.000 description 2
- 230000003557 neuropsychological effect Effects 0.000 description 2
- 229920000058 polyacrylate Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000193 polymethacrylate Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000037321 sleepiness Effects 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 229920003169 water-soluble polymer Polymers 0.000 description 2
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 1
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- LYONXVJRBWWGQO-UHFFFAOYSA-N 2-(2-oxo-4-phenylpyrrolidin-1-yl)acetamide Chemical compound C1C(=O)N(CC(=O)N)CC1C1=CC=CC=C1 LYONXVJRBWWGQO-UHFFFAOYSA-N 0.000 description 1
- YFGHCGITMMYXAQ-UHFFFAOYSA-N 2-[(diphenylmethyl)sulfinyl]acetamide Chemical compound C=1C=CC=CC=1C(S(=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 244000187129 Bacopa monnieria Species 0.000 description 1
- 235000015418 Bacopa monnieria Nutrition 0.000 description 1
- 208000020925 Bipolar disease Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- GJSURZIOUXUGAL-UHFFFAOYSA-N Clonidine Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCN1 GJSURZIOUXUGAL-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 208000001640 Fibromyalgia Diseases 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 235000008100 Ginkgo biloba Nutrition 0.000 description 1
- 244000194101 Ginkgo biloba Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- INJOMKTZOLKMBF-UHFFFAOYSA-N Guanfacine Chemical compound NC(=N)NC(=O)CC1=C(Cl)C=CC=C1Cl INJOMKTZOLKMBF-UHFFFAOYSA-N 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 description 1
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- DUGOZIWVEXMGBE-UHFFFAOYSA-N Methylphenidate Chemical compound C=1C=CC=CC=1C(C(=O)OC)C1CCCCN1 DUGOZIWVEXMGBE-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- DATAGRPVKZEWHA-YFKPBYRVSA-N N(5)-ethyl-L-glutamine Chemical compound CCNC(=O)CC[C@H]([NH3+])C([O-])=O DATAGRPVKZEWHA-YFKPBYRVSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000009668 Neurobehavioral Manifestations Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 description 1
- 240000004371 Panax ginseng Species 0.000 description 1
- 235000002789 Panax ginseng Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- GMZVRMREEHBGGF-UHFFFAOYSA-N Piracetam Chemical compound NC(=O)CN1CCCC1=O GMZVRMREEHBGGF-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 206010062519 Poor quality sleep Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 240000007164 Salvia officinalis Species 0.000 description 1
- 235000002912 Salvia officinalis Nutrition 0.000 description 1
- 239000002262 Schiff base Substances 0.000 description 1
- 150000004753 Schiff bases Chemical class 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 229930003779 Vitamin B12 Natural products 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 229920002494 Zein Polymers 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940025084 amphetamine Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229960000793 aniracetam Drugs 0.000 description 1
- ZXNRTKGTQJPIJK-UHFFFAOYSA-N aniracetam Chemical compound C1=CC(OC)=CC=C1C(=O)N1C(=O)CCC1 ZXNRTKGTQJPIJK-UHFFFAOYSA-N 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 208000008784 apnea Diseases 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 229960004823 armodafinil Drugs 0.000 description 1
- YFGHCGITMMYXAQ-LJQANCHMSA-N armodafinil Chemical compound C=1C=CC=CC=1C([S@](=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-LJQANCHMSA-N 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 229960002430 atomoxetine Drugs 0.000 description 1
- VHGCDTVCOLNTBX-QGZVFWFLSA-N atomoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=CC=C1C VHGCDTVCOLNTBX-QGZVFWFLSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 229960002896 clonidine Drugs 0.000 description 1
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 239000002475 cognitive enhancer Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 229920001531 copovidone Polymers 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 125000004431 deuterium atom Chemical group 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000007922 dissolution test Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 229960003592 fexofenadine Drugs 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 230000009969 flowable effect Effects 0.000 description 1
- 238000009477 fluid bed granulation Methods 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229940014259 gelatin Drugs 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 229960002048 guanfacine Drugs 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 235000008216 herbs Nutrition 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000002346 layers by function Substances 0.000 description 1
- 229960004502 levodopa Drugs 0.000 description 1
- 229940059904 light mineral oil Drugs 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- YAHRDLICUYEDAU-UHFFFAOYSA-N methylhexaneamine Chemical compound CCC(C)CC(C)N YAHRDLICUYEDAU-UHFFFAOYSA-N 0.000 description 1
- 229960001344 methylphenidate Drugs 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229960001165 modafinil Drugs 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 235000020660 omega-3 fatty acid Nutrition 0.000 description 1
- 229940012843 omega-3 fatty acid Drugs 0.000 description 1
- 239000006014 omega-3 oil Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000008184 oral solid dosage form Substances 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 229960001227 oxiracetam Drugs 0.000 description 1
- IHLAQQPQKRMGSS-UHFFFAOYSA-N oxiracetam Chemical compound NC(=O)CN1CC(O)CC1=O IHLAQQPQKRMGSS-UHFFFAOYSA-N 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 150000002993 phenylalanine derivatives Chemical class 0.000 description 1
- 229960004526 piracetam Drugs 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 238000009725 powder blending Methods 0.000 description 1
- 229920003124 powdered cellulose Polymers 0.000 description 1
- 235000019814 powdered cellulose Nutrition 0.000 description 1
- 229960003089 pramipexole Drugs 0.000 description 1
- FASDKYOPVNHBLU-ZETCQYMHSA-N pramipexole Chemical compound C1[C@@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-ZETCQYMHSA-N 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000009490 roller compaction Methods 0.000 description 1
- 235000002020 sage Nutrition 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- XYGBKMMCQDZQOZ-UHFFFAOYSA-M sodium;4-hydroxybutanoate Chemical compound [Na+].OCCCC([O-])=O XYGBKMMCQDZQOZ-UHFFFAOYSA-M 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229960004603 tolcapone Drugs 0.000 description 1
- MIQPIUSUKVNLNT-UHFFFAOYSA-N tolcapone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC(O)=C(O)C([N+]([O-])=O)=C1 MIQPIUSUKVNLNT-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 235000019163 vitamin B12 Nutrition 0.000 description 1
- 239000011715 vitamin B12 Substances 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 229940051225 xyrem Drugs 0.000 description 1
- 239000005019 zein Substances 0.000 description 1
- 229940093612 zein Drugs 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
Definitions
- the present invention relates to carbamoyl phenylalaninol compounds and methods of using the same to enhance one or more cognitive function and to treat one or more cognitive impairment.
- the present invention overcomes shortcomings in the art by providing methods and compositions for enhancing one or more cognitive function and treating one or more cognitive impairment.
- the present invention relates to the development of methods for enhancing one or more cognitive function and treating one or more cognitive impairment.
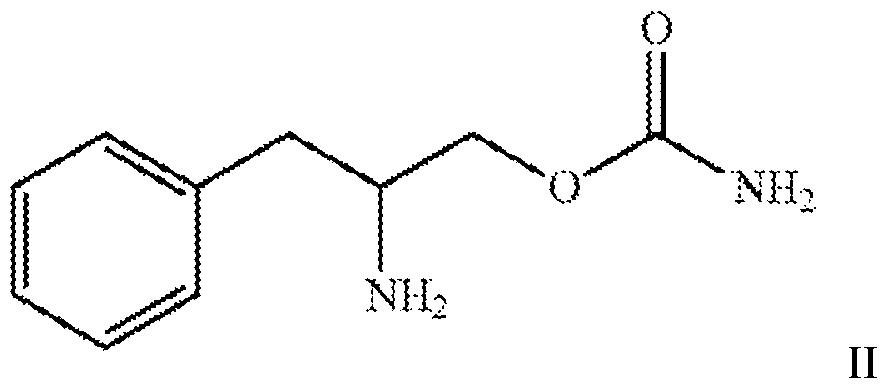
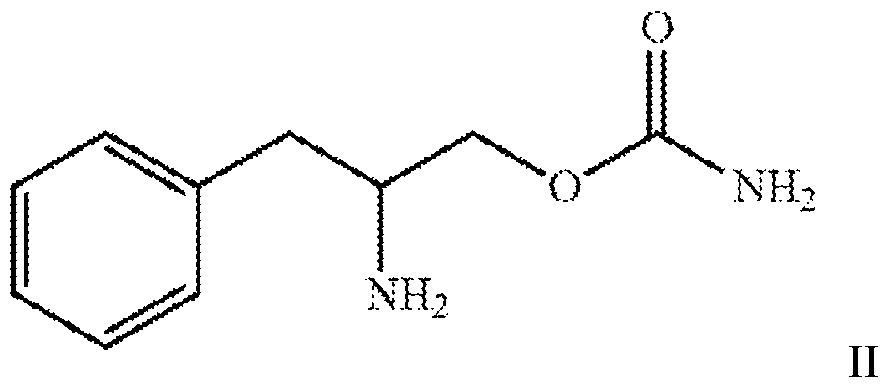
- one aspect of the present invention relates to a method of enhancing one or more cognitive function, said method comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula II or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention relates to a method of treating one or more cognitive impairment, said method comprising administering to a subject having said one or more cognitive impairment a therapeutically effective amount of a compound of formula II or a pharmaceutically acceptable salt thereof.
- a further aspect of the inventio relates to a method of delaying or slowing an increase in one or more cognitive impairment, said method comprising administering to a subject having or at risk of having said one or more cognitive impairment a therapeutically effective amount of a compound of formula II or a pharmaceutically acceptable salt thereof, thereby treating one or more cognitive impairment in the subject.
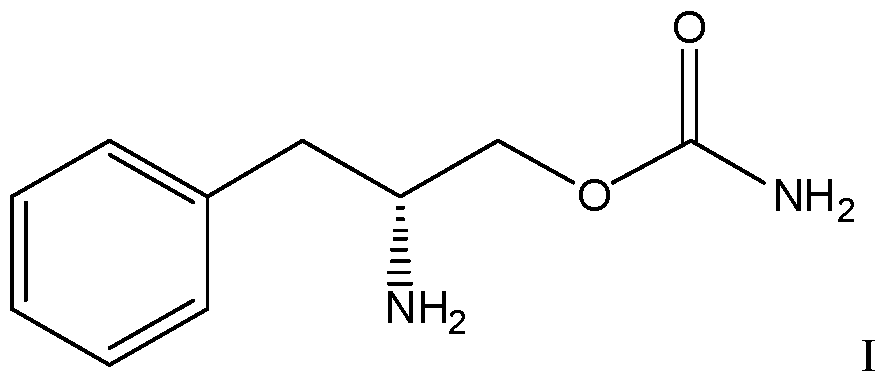
- the compound is the compound of formula I or a pharmaceutically acceptable salt thereof.
- the compound is the hydrochloride salt of the compound of formula II or I.
- a,” “an,” or “the” can mean one or more than one.
- a cell can mean a single cell or a multiplicity of cells.
- the term “about,” as used herein when referring to a measurable value such as an amount of a compound or agent of this invention, dose, time, temperature, and the like, is meant to encompass variations of ⁇ 10%, ⁇ 5%, ⁇ 1%, ⁇ 0.5%, or even ⁇ 0.1% of the specified amount.
- compositions of this invention means the composition can contain additional components as long as the additional components do not materially alter the composition.
- materially altered refers to an increase or decrease in the therapeutic effectiveness of the composition of at least about 20% or more as compared to the effectiveness of a composition consisting of the recited components.
- terapéuticaally effective amount refers to that amount of a composition, compound, or agent of this invention that imparts a modulating effect, which, for example, can be a beneficial effect, to a subject afflicted with a disorder, disease or illness, including improvement in the condition of the subject (e.g., in one or more symptoms), delay or reduction in the progression of the condition, prevention or delay of the onset of the disorder, and/or change in clinical parameters, disease or illness, etc., as would be well known in the art.
- a therapeutically effective amount or effective amount can refer to the amount of a composition, compound, or agent that improves a condition in a subject by at least 5%, e.g., at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- Treat” or “treating” or “treatment” refers to any type of action that imparts a modulating effect, which, for example, can be a beneficial effect, to a subject afflicted with a disorder, disease or illness, including improvement in the condition of the subject (e.g., in one or more symptoms), delay or reduction in the progression of the condition, and/or change in clinical parameters, disease or illness, etc., as would be well known in the art.
- “Pharmaceutically acceptable,” as used herein, means a material that is not biologically or otherwise undesirable, i.e., the material can be administered to an individual along with the compositions of this invention, without causing substantial deleterious biological effects or interacting in a deleterious manner with any of the other components of the composition in which it is contained.
- the material would naturally be selected to minimize any degradation of the active ingredient and to minimize any adverse side effects in the subject, as would be well known to one of skill in the art (see, e.g., Remington's Pharmaceutical Science,' 21 st ed. 2005).
- Concurrently means sufficiently close in time to produce a combined effect (that is, concurrently can be simultaneously, or it can be two or more events occurring within a short time period before or after each other).
- the administration of two or more compounds “concurrently” means that the two compounds are administered closely enough in time that the presence of one alters the biological effects of the other.
- the two compounds can be administered in the same or different formulations or sequentially. Concurrent administration can be carried out by mixing the compounds prior to administration, or by administering the compounds in two different formulations, for example, at the same point in time but at different anatomic sites or using different routes of administration.
- the present invention relates to a method of enhancing one or more cognitive function, said method comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula II
- the one or more cognitive function is selected from executive function, memory, and learning. In some embodiments, the one or more cognitive function is selected from working memory, cognitive flexibility, and response inhibition.
- Another aspect of the invention relates to a method of treating one or more cognitive impairment, said method comprising administering to a subject having said one or more cognitive impairment a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt thereof.
- a further aspect of the inventio relates to a method of delaying or slowing an increase in one or more cognitive impairment, said method comprising administering to a subject having or at risk of having said one or more cognitive impairment a therapeutically effective amount of a compound of formula II or a pharmaceutically acceptable salt thereof, thereby treating one or more cognitive impairment in the subject.
- the subject may be one that has been diagnosed with one or more cognitive impairment and worsening of the impairments is delayed or slowed relative to a subject not treated with the method of the invention.
- the subject may be one that is at risk of developing one or more cognitive impairment and onset of the impairments is delayed relative to a subject not treated with the method of the invention.
- the subject is identified as having a neurological disorder associated with cognitive impairment, e.g., identified as having Alzheimer’s Disease, schizophrenia, autism, or obsessive-compulsive disorder.
- a neurological disorder associated with cognitive impairment e.g., identified as having Alzheimer’s Disease, schizophrenia, autism, or obsessive-compulsive disorder.
- the cognitive impairment is selected from impaired executive function, impaired learning, and impaired memory. In some embodiments, the cognitive impairment is selected from impaired cognitive flexibility, impaired working memory, and impaired response inhibition.
- the compound is the compound of formula I or a pharmaceutically acceptable salt thereof.
- the compound is the hydrochloride salt of the compound of formula II or I.
- the methods of the present invention may be carried out using compounds, formulations and unit dosage forms provided herein.
- the formulations and dosage forms can be utilized to achieve immediate release of APC, as well as pharmaceutically acceptable salts, hydrates, isomers, including tautomers, solvates and complexes of APC.
- Suitable salts of APC include, without limitation, acetate, adipate, alginate, aspartate, benzoate, butyrate, citrate, fumarate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, nicotinate, nitrate, oxalate, palmoate, pectinate, persulfate, hydroxynapthoate, pivalate, propionate, salicylate, succinate, sulfate, tartrate, thiocyanate, tosylate and undecanoate.
- APC compounds include those having quatemization of any basic nitrogen-containing group therein.
- APC can contain one or more asymmetric centers and thus occur as racemates and racemic mixtures and single optical isomers. All such isomeric and deuterated forms of these compounds are expressly included in the present invention.
- the compounds of the invention include prodrugs of the compounds that are converted to the active compound in vivo.
- the compound can be modified to enhance cellular permeability (e.g., by esterification of polar groups) and then converted by cellular enzymes to produce the active agent.
- Methods of masking charged or reactive moieties as a pro-drug are known by those skilled in the art (see, e.g., P. Korgsgaard-Larsen and H. Bundgaard, A Textbook of Drug Design and Development, Reading U.K., Harwood Academic Publishers, 1991).
- prodrug refers to compounds that are rapidly transformed in vivo to yield the parent compound of the above formula, for example, by hydrolysis in blood, see, e.g., T. Higuchi and V. Stella, Prodrugs as Novel delivery Systems, Vol. 14 of the A.C.S. Symposium Series and in Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, both of which are incorporated by reference herein. See also U.S. Patent No. 6,680,299.
- Exemplary prodrugs include a prodrug that is metabolized in vivo by a subject to an active drug having an activity of the compounds as described herein, wherein the prodrug is an ester of an alcohol or carboxylic acid group, if such a group is present in the compound; an amide of an amine group or carboxylic acid group, if such groups are present in the compound; a urethane of an amine group, if such a group is present in the compound; an acetal or ketal of an alcohol group, if such a group is present in the compound; a N-Mannich base or an imine of an amine group, if such a group is present in the compound; or a Schiff base, oxime, acetal, enol ester, oxazolidine, or thiazolidine of a carbonyl group, if such a group is present in the compound, such as described, for example, in U.S. Patent No. 6,680,324 and U.S
- prodrug refers to those prodrugs of APC which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and/or other animals without undue toxicity, irritation, allergic response and the like, commensurate with a reasonable risk/benefit ratio, and effective for their intended use, as well as the zwitterionic forms, where possible, of the compound of the invention.
- APC or a pharmaceutically acceptable salt thereof may be obtained or synthesized by methods known in the art and as described herein. Details of reaction schemes for synthesizing APC have been described in U.S. Patent Nos. 5,705,640; 5,756,817; 5,955,499; 6,140,532; and 10,829,443, all incorporated herein by reference in their entirety.
- compositions e.g., a dosage form, comprising APC that is suitable for used in the methods of the invention.
- the composition is a pharmaceutical composition comprising APC and a pharmaceutically acceptable carrier.
- the dosage form is an oral dosage form, e.g, a tablet or a capsule, e.g., an immediate release dosage form.
- the dosage form is an immediate release tablet that releases at least 85%, e.g. , at least 85%, 90%, 95%, 96%, 97%, 98%, or 99%, of the APC contained therein within a period of less than 15 minutes after administration of the tablet to a subject, e.g, as described in U.S. Patent Nos. 10,195,151; 10,512,609; and 11,439,597.
- Formulations of APC may be processed into unit dosage forms suitable for oral administration, such as for example, filled capsules, compressed tablets or caplets, or other dosage form suitable for oral administration using conventional techniques.
- Immediate release dosage forms prepared as described may be adapted for oral administration, so as to attain and maintain a therapeutic level of the compound over a preselected interval.
- an immediate release dosage form as described herein may comprise a solid oral dosage form of any desired shape and size including round, oval, oblong cylindrical, or polygonal.
- the surfaces of the immediate release dosage form may be flat, round, concave, or convex.
- the immediate release tablets when the immediate release formulations are prepared as a tablet, the immediate release tablets contain a relatively large percentage and absolute amount of the compound and so are expected to improve patient compliance and convenience, by replacing the need to ingest large amounts of liquids or liquid/solid suspensions.
- One or more immediate release tablets as described herein can be administered, by oral ingestion, e.g., closely spaced, in order to provide a therapeutically effective dose of the compound to the subj ect in a relatively short period of time.
- the outer surface of an immediate release dosage form may be coated, e.g., with a color coat or with a moisture barrier layer using materials and methods known in the art.
- the composition is an immediate release compressed tablet, the tablet comprising:
- APC or a pharmaceutically acceptable salt thereof in an amount of about 90-98% by weight of the tablet; at least one binder in an amount of about 1-5% by weight of the tablet; and at least one lubricant in an amount of about 0.1-2% by weight of the tablet; wherein the tablet releases at least 85% of the APC or a pharmaceutically acceptable salt thereof contained therein within a period of less than 15 minutes after administration of the tablet to a subject.
- the tablet comprises:
- APC or a pharmaceutically acceptable salt thereof in an amount of about 91-95% by weight of the tablet at least one binder in an amount of about 2-3% by weight of the tablet; at least one lubricant in an amount of about 0.1-1% by weight of the tablet; and optionally, a cosmetic film coat in an amount of about 3-4% by weight of the tablet; wherein the tablet releases at least 85% of the APC or a pharmaceutically acceptable salt thereof contained therein within a period of less than 15 minutes after administration of the tablet to a subject.
- the tablet comprises:
- the composition is an immediate release oral dosage form of APC, the oral dosage form comprising:
- APC or a pharmaceutically acceptable salt thereof in an amount of about 90-98% by weight of the oral dosage form at least one binder in an amount of about 1-5% by weight of the oral dosage form; and at least one lubricant in an amount of about 0.1-2% by weight of the oral dosage form; wherein the oral dosage form releases at least 85% of the APC or a pharmaceutically acceptable salt thereof contained therein within a period of less than 15 minutes after administration of the oral dosage form to a subject.
- the tablet does not comprise a disintegrant.
- disintegrant refers to an agent added to a tablet to promote the breakup of the tablet in an aqueous environment.
- the tablets of the present invention are advantageous in that they dissolve rather than disintegrate. In the present invention the presence of disintegrant in the formulation may actually slow down release of APC.
- APC or a pharmaceutically acceptable salt thereof is present in an amount of about 90%, 90.5%, 91%, 91.5%, 92%, 92.5%, 93%, 93.5%, 94%, 94.5%, 95%, 95.5%, 96%, 96.5%, 97%, 97.5%, or 98% by weight of the tablet or any value or range therein.
- APC or a pharmaceutically acceptable salt thereof is present in an amount of about 90% to about 98%, about 92% to about 98%, about 94% to about 98%, about 96% to about 98%, about 90% to about 92%, about 90% to about 94%, about 90% to about 96%, about 92% to about 94%, about 92% to about 96%, or about 94% to about 96%.
- the at least one binder is present in an amount of about 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, or 5% by weight of the tablet or any value or range therein. In certain embodiments, the at least one binder is present in an amount of about 1% to about 5%, about 2% to about 5%, about 3% to about 5%, about 4% to about 5%, about 1% to about 2%, about 1% to about 3%, about 1% to about 4%, about 2% to about 3%, about 2% to about 4%, or about 3% to about 4%.
- the tablet may comprise at least one binder, e.g, 1, 2, 3, 4, 5, or more binders.
- the at least one binder is selected from at least one of hydroxypropyl cellulose, ethylcellulose, hydroxypropyl methylcellulose, polyvinyl alcohol, hydroxyethyl cellulose, povidone, copovidone, pregelatinized starch, dextrin, gelatin, maltodextrin, starch, zein, acacia, alginic acid, carbomers (cross-linked polyacrylates), polymethacrylates, sodium carboxymethylcellulose, guar gum, hydrogenated vegetable oil (type 1), methylcellulose, magnesium aluminum silicate, and sodium alginate or any combination thereof.
- the at least one binder is hydroxypropyl cellulose.
- the at least one lubricant is present in an amount of about 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, or 2.0% by weight of the tablet or any value or range therein.
- the at least one lubricant is present in an amount of about 0.1% to about 2.0%, about 0.5% to about 2.0%, about 1.0% to about 2.0%, about 1.5% to about 2.0%, about 0.1% to about 0.5%, about 0.1% to about 1.0%, about 0.1% to about 1.5%, about 0.5% to about 1.0%, about 0.5% to about 1.5%, or about 1.0% to about 1.5%.
- the tablet may comprise at least one lubricant, e.g, 1, 2, 3, 4, 5, or more lubricants. Where the immediate release formulation is provided as a tableted dosage form, still lower lubricant levels may be achieved with use of a “puffer” system during tableting. Such systems are known in the art, commercially available and apply lubricant directly to the punch and die surfaces rather than throughout the formulation.
- the at least one lubricant is selected from at least one of magnesium stearate, stearic acid, calcium stearate, hydrogenated castor oil, hydrogenated vegetable oil, light mineral oil, magnesium stearate, mineral oil, polyethylene glycol, sodium benzoate, sodium stearyl fumarate, and zinc stearate or any combination thereof.
- the at least one lubricant is magnesium stearate.
- magnesium stearate may be used in combination with one or more other lubricants or a surfactant, such as sodium lauryl sulfate.
- sodium lauryl sulfate may also be included when using magnesium stearate (Remington: the Science and Practice of Pharmacy, 20 th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2000)).
- the at least one binder is hydroxypropyl cellulose. In some embodiments, the at least one lubricant is magnesium stearate. In some embodiments, the at least one binder is hydroxypropyl cellulose and the at least one lubricant is magnesium stearate. [0059] In certain embodiments, the tablet is coated. The coating may be, without limitation, a color overcoat.
- the APC or a pharmaceutically acceptable salt thereof is APC hydrochloride.
- the tablet may be any shape that is suitable for immediate release and allows the release of at least 85% of the APC or a pharmaceutically acceptable salt thereof contained therein within a period of less than 15 minutes after administration of the tablet to a subject.
- the tablet maximizes surface area to volume ratio to promote rapid dissolution.
- the tablet is oblong in shape.
- the tablet may contain any amount of APC or a pharmaceutically acceptable salt thereof suitable for administration as a unit dosage form.
- the tablet contains about 1 mg to about 1000 mg of the drug or any range or value therein, e.g., about 100 mg to about 500 mg, e.g., about 37.5 mg, about 75 mg, about 150 mg, or about 300 mg.
- “Immediate release” refers to a composition that releases APC or a pharmaceutically acceptable salt, hydrate, isomer, tautomer, solvate or complex thereof substantially completely into the gastrointestinal tract of the user within a period of less than about 15 minutes, usually between about 1 minute and about 15 minutes from ingestion. Such a delivery rate allows the drug to be absorbed by the gastrointestinal tract in a manner that is bioequivalent to an oral solution. Such rapid absorption will typically occur for an immediate release unit dosage form, such as a tablet, caplet or capsule, if the drug included in such dosage form dissolves in the upper portion the gastrointestinal tract.
- Release rates can be measured using standard dissolution test methods.
- the standard conditions may be those described in FDA guidance (e.g, 50 rpm, 37°C, USP 2 paddles, pH 1.2 and pH 6.8 media, 900 ml, 1 test article per vessel).
- Immediate release formulations suitable for oral administration may comprise unit dosage forms, such as tablets, caplets or filled capsules, which can deliver a therapeutically effective dose of APC upon ingestion thereof by the patient of one or more of said dosage forms, each of which can provide a dosage of, for example, about 1 to about 1000 mg of APC. Additionally, the immediate release dosage forms can be shaped or scored to facilitate dose adjustment through tablet splitting.
- an immediate release dosage form as disclosed herein can be adjusted to provide immediate release performance that suits a particular dosing need.
- the formulation and structure of the dosage forms as described herein can be adjusted to provide any combination of the immediate release performance characteristics described herein.
- an immediate release dosage form as disclosed herein provides rapid onset of action, releasing more than about 85%, such as, for example, more than about 90% or 95%, of the drug contained therein within a period of time selected from less than 15 minutes, less than 12 minutes, less than 10 minutes, and less than 5 minutes after administration.
- the rate of drug release from an immediate release dosage form as disclosed herein may be adjusted as needed to facilitate a desired dosing regimen or achieve targeted dosing.
- the immediate release dosage form may be formulated to deliver as much as 1,000 mg of APC.
- the total amount of drug contained within an immediate release dosage form according to the present description may be between about 50 mg and about 500 mg.
- the total amount of drug may be selected from about 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 725, 750, 775, 800, 825, 850, 875, 900, 925, 950, 975, or 1000 mg or any range or value therein.
- the total amount of drug may be about 10 mg to about 1000 mg, about 10 mg to about 500 mg, about 10 mg to about 300 mg, about 30 mg to about 1000 mg, about 30 mg to about 500 mg, about 30 mg to about 300 mg, about 100 mg to about 1000 mg, about 10 mg to about 500 mg, about 100 mg to about 300 mg, about 150 mg to about 1000 mg, about 150 mg to about 500 mg, or about 150 mg to about 300 mg.
- the immediate release formulations provided herein generally include APC and some level of lubricant to facilitate processing of the formulations into a unit dosage form.
- the formulations described herein include a combination of APC and lubricant, as described herein, and in certain such embodiments, the immediate release formulations are substantially free of other excipients or adjuvants.
- the immediate release formulations described herein include a combination of APC, lubricant, and binder, as described herein, and in certain such embodiments, the immediate release formulations are substantially free of other excipients or adjuvants.
- the immediate release formulations described herein may be formulated using a combination of drug and one or more of a lubricant and binder
- the compositions described herein may include one or more additional excipients selected from, for example, fillers, compression aids, diluents, disintegrants, colorants, flavorants, buffering agents, coatings, glidants, or other suitable excipients.
- the immediate release formulations described herein may be manufactured using standard techniques, such as wet granulation, roller compaction, fluid bed granulation, and dry powder blending. Suitable methods for the manufacture of the immediate release formulations and unit dosage forms described herein are provided, for example, in Remington, 20 th edition, Chapter 45 (Oral Solid Dosage Forms). It has been found that, even without the aid of binders or non-lubricating excipients, such as compression aids, wet granulation techniques can afford flowable granules with compression characteristics suitable for forming unit dosage forms as described herein.
- wet granulation techniques may be used to prepare immediate release formulations as described herein.
- conventional organic or aqueous solvents may be used in the wet granulation process.
- Suitable wet granulation processes can be performed as fluidized bed, high shear, or low shear (wet massing) granulation techniques, as are known in the art.
- the immediate release formulations described herein may also include fillers or compression aids selected from at least one of lactose, calcium carbonate, calcium sulfate, compressible sugars, dextrates, dextrin, dextrose, kaolin, magnesium carbonate, magnesium oxide, maltodextrin, mannitol, microcrystalline cellulose, powdered cellulose, and sucrose.
- fillers or compression aids selected from at least one of lactose, calcium carbonate, calcium sulfate, compressible sugars, dextrates, dextrin, dextrose, kaolin, magnesium carbonate, magnesium oxide, maltodextrin, mannitol, microcrystalline cellulose, powdered cellulose, and sucrose.
- a filler or compression aid may be included in the immediate release formulation in an amount ranging from about 1%-15% by weight.
- Immediate release formulations as described herein may be processed into unit dosage forms suitable for oral administration, such as for example, filled capsules, compressed tablets or caplets, or other dosage form suitable for oral administration using conventional techniques. Immediate release dosage forms prepared as described may be adapted for oral administration, so as to attain and maintain a therapeutic level of APC over a preselected interval.
- an immediate release dosage form as described herein may comprise a solid oral dosage form of any desired shape and size including round, oval, oblong, cylindrical, or polygonal.
- the surfaces of the immediate release dosage form may be flat, round, concave, or convex.
- the shape may be selected to maximize surface area, e.g., to increase the rate of dissolution of the dosage form.
- the immediate release tablets when the immediate release formulations are prepared as a tablet, the immediate release tablets contain a relatively large percentage and absolute amount of APC and so are expected to improve patient compliance and convenience, by replacing the need to ingest large amounts of liquids or liquid/solid suspensions.
- One or more immediate release tablets as described herein can be administered, by oral ingestion, e.g, closely spaced, in order to provide a therapeutically effective dose of APC to the subject in a relatively short period of time.
- dissolution of a 10 mg-1000 mg tablet prepared according to the present description can provide about 80-100% of the APC to the subject in about 10-15 minutes.
- an immediate release dosage form as disclosed herein may be coated with a moisture barrier layer using materials and methods known in the art.
- the APC delivered by the unit dosage form is highly hygroscopic, providing a moisture barrier layer over the immediate release dosage form as disclosed herein may be desirable.
- protection of an immediate release dosage form as disclosed herein from water during storage may be provided or enhanced by coating the tablet with a coating of a substantially water soluble or insoluble polymer.
- Useful waterinsoluble or water-resistant coating polymers include ethyl cellulose and polyvinyl acetates.
- Further water-insoluble or water-resistant coating polymers include polyacrylates, polymethacrylates or the like.
- Suitable water-soluble polymers include polyvinyl alcohol and HPMC.
- Further suitable water-soluble polymers include PVP, HPC, HPEC, PEG, HEC and the like.
- an immediate release dosage form as disclosed herein may be coated with a color overcoat or other aesthetic or functional layer using materials and methods known in the art.
- the dosage forms disclosed herein can also be provided as a kit comprising, separately packaged, a container comprising a plurality of immediate release tablets, which tablets can be individually packaged, as in foil envelopes or in a blister pack.
- the tablets can be packaged in many conformations with or without desiccants or other materials to prevent ingress of water.
- Instruction materials or means, such as printed labeling can also be included for their administration, e.g, sequentially over a preselected time period and/or at preselected intervals, to yield the desired levels of APC in vivo for preselected periods of time, to treat a preselected condition.
- a daily dose of about 1 to about 2000 mg of APC or a pharmaceutically acceptable salt thereof may be administered to accomplish the therapeutic results disclosed herein.
- a daily dosage of about 10-1000 mg, e.g., about 20-500 mg, e.g., about 37.5, 75, 150, or 300 mg, in single or divided doses is administered.
- the daily dose may be about 0.01 to about 150 mg/kg body weight, e.g., about 0.2 to about 18 mg/kg body weight.
- APC is administered to the subject as needed to treat a disorder.
- the compound can be administered continuously or intermittently.
- the compound is administered to the subject more than once a day, e.g, 2, 3, or 4 times per day, or once every 1, 2, 3, 4, 5, 6, or 7 days.
- the compound is administered to the subject no more than once a week, e.g., no more than once every two weeks, once a month, once every two months, once every three months, once every four months, once every five months, once every six months, or longer.
- the compound is administered using two or more different schedules, e.g., more frequently initially (for example to build up to a certain level, e.g, once a day or more) and then less frequently (e.g., once a week or less).
- the compound can be administered by any discontinuous administration regimen.
- the compound can be administered not more than once every three days, every four days, every five days, every six days, every seven days, every eight days, every nine days, or every ten days, or longer.
- the administration can continue for one, two, three, or four weeks or one, two, or three months, or longer.
- the compound can be administered under the same or a different schedule.
- the period of rest can be one, two, three, or four weeks, or longer, according to the pharmacodynamic effects of the compound on the subject.
- the compound can be administered to build up to a certain level, then maintained at a constant level and then a tailing dosage.
- APC is delivered to a subject concurrently with an additional therapeutic agent.
- the additional therapeutic agent can be delivered in the same composition as the compound or in a separate composition.
- the additional therapeutic agent can be delivered to the subject on a different schedule or by a different route as compared to the compound.
- the additional therapeutic agent can be any agent that provides a benefit to the subject.
- nootropic agents e.g., cognitive enhancers
- stimulants e.g, caffeine, dimethylamylamine, methylphenidate, amphetamine, nicotine, modafinil, armodafinil
- racetams e.g, piracetam, oxiracetam, phenylpiracetam, aniracetam
- tolcapone levodopa, atomoxetine, pramipexole, guanfacine, clonidine, and fexofenadine.
- Nootropic agents may also include herbs, such as Bacopa monnieri, Panax ginseng, Ginkgo biloba, and Salvia officinalis, and dietary supplements, such as omega-3 fatty acids, folate, vitamin Be, vitamin B12, vitamin E, and L- theanine.
- herbs such as Bacopa monnieri, Panax ginseng, Ginkgo biloba, and Salvia officinalis
- dietary supplements such as omega-3 fatty acids, folate, vitamin Be, vitamin B12, vitamin E, and L- theanine.
- Xyrem® sold commercially by Jazz Pharmaceuticals, which is used to treat narcolepsy and cataplexy. See U.S. Patent Nos. 8,952,062 and 9,050,302.
- Suitable subjects are generally mammalian subjects.
- mammalian subjects includes, but is not limited to, humans, non-human primates, cattle, sheep, goats, pigs, horses, cats, dog, rabbits, rodents (e.g, rats or mice), etc.
- Human subjects include neonates, infants, juveniles, adults and geriatric subjects.
- the subject can be a subject “in need of’ the methods of the present invention, e.g, in need of the therapeutic effects of the inventive methods.
- the subject can be a subject that is experiencing cognitive impairment, is suspected of having cognitive impairment, and/or is anticipated to experience cognitive impairment, and the methods and compositions of the invention are used for therapeutic and/or prophylactic treatment.
- SHARP Solriamfetol’s Effect on Cognitive Health in Apnea Participants During a Randomized Placebo-controlled Study
- EDS daytime sleepiness
- OSA obstructive sleep apnea
- Solriamfetol was administered orally once daily, starting at 75 mg per day for the first three days and 150 mg per day for the remainder of the 2-week treatment period.
- the primary outcome measure was the Digit Symbol Substitution Test subtest of the Repeatable Battery for the Assessment of Neuropsychological Status (DSST RBANS).
- the Digit Symbol Substitution subtest is also referred to as “Coding.”
- the prespecified primary endpoint was the change from baseline in cognitive function as measured by the DSST RBANS after 2 weeks of treatment (average of the 2-, 4-, 6-, and 8- hour post-dose DSST RBANS scores).
- Secondary endpoints included patient reported measures of cognition including the British Columbia Cognitive Complaints Inventory (BC- CCI) and the Patient Global Impression of Severity (PGI-S) for cognitive symptoms; and the Epworth Sleepiness Scale (ESS) to measure wakefulness.
- BC- CCI British Columbia Cognitive Complaints Inventory
- PKI-S Patient Global Impression of Severity
- ESS Epworth Sleepiness Scale
- the BC-CCI is a patient-reported test that assesses domains of memory, concentration, trouble expressing thoughts, word finding, and problem solving.
- Solriamfetol significantly improved EDS symptoms compared to placebo, as measured by the Epworth Sleepiness Scale (ESS).
- ESS Epworth Sleepiness Scale
Landscapes
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical & Material Sciences (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Description






Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020247018863A KR20240101826A (en) | 2021-11-16 | 2022-11-16 | Methods and compositions for treating cognitive impairment |
| AU2022390879A AU2022390879A1 (en) | 2021-11-16 | 2022-11-16 | Methods and compositions for treating cognitive impairment |
| EP22896667.7A EP4433044A1 (en) | 2021-11-16 | 2022-11-16 | Methods and compositions for treating cognitive impairment |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163279805P | 2021-11-16 | 2021-11-16 | |
| US63/279,805 | 2021-11-16 | ||
| US202263348684P | 2022-06-03 | 2022-06-03 | |
| US63/348,684 | 2022-06-03 | ||
| US202263378162P | 2022-10-03 | 2022-10-03 | |
| US63/378,162 | 2022-10-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023091921A1 true WO2023091921A1 (en) | 2023-05-25 |
Family
ID=86397937
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2022/079920 WO2023091921A1 (en) | 2021-11-16 | 2022-11-16 | Methods and compositions for treating cognitive impairment |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4433044A1 (en) |
| KR (1) | KR20240101826A (en) |
| AU (1) | AU2022390879A1 (en) |
| WO (1) | WO2023091921A1 (en) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996024577A1 (en) * | 1995-02-11 | 1996-08-15 | Yukong Limited | O-carbamoyl-(d)-phenylalanilol compounds and process for preparing the same |
| WO2011005473A2 (en) * | 2009-06-22 | 2011-01-13 | Shionogi Pharma, Inc. | Methods for treating or preventing fatigue |
| WO2016061488A1 (en) * | 2014-10-17 | 2016-04-21 | Concert Pharmaceuticals, Inc. | Amine reuptake inhibitors |
| US20200046674A1 (en) * | 2014-02-28 | 2020-02-13 | Sk Biopharmaceuticals Co., Ltd. | Aminocarbonylcarbamate compounds |
-
2022
- 2022-11-16 EP EP22896667.7A patent/EP4433044A1/en active Pending
- 2022-11-16 KR KR1020247018863A patent/KR20240101826A/en unknown
- 2022-11-16 AU AU2022390879A patent/AU2022390879A1/en active Pending
- 2022-11-16 WO PCT/US2022/079920 patent/WO2023091921A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996024577A1 (en) * | 1995-02-11 | 1996-08-15 | Yukong Limited | O-carbamoyl-(d)-phenylalanilol compounds and process for preparing the same |
| WO2011005473A2 (en) * | 2009-06-22 | 2011-01-13 | Shionogi Pharma, Inc. | Methods for treating or preventing fatigue |
| US20200046674A1 (en) * | 2014-02-28 | 2020-02-13 | Sk Biopharmaceuticals Co., Ltd. | Aminocarbonylcarbamate compounds |
| WO2016061488A1 (en) * | 2014-10-17 | 2016-04-21 | Concert Pharmaceuticals, Inc. | Amine reuptake inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| COREY-BLOOM, J.: "The ABC of Alzheimer`s disease: cognitive changes and their management in Alzheimer`s disease and related dementias", INTERNATIONAL PSYCHOGERIATRICS, SPRINGER PUBLISHING INTERNATIONAL, US, vol. 14, no. Suppl. 1, 1 January 2002 (2002-01-01), US , pages 51 - 75, XP009545470, ISSN: 1041-6102, DOI: 10.1017/S1041610203008664 * |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2022390879A1 (en) | 2024-06-13 |
| EP4433044A1 (en) | 2024-09-25 |
| KR20240101826A (en) | 2024-07-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11998639B2 (en) | Formulations of (R)-2-amino-3-phenylpropyl carbamate | |
| US11865098B1 (en) | Methods and compositions for treating excessive sleepiness | |
| US11149000B2 (en) | Carbamoyl phenylalaninol compounds and uses thereof | |
| WO2023091921A1 (en) | Methods and compositions for treating cognitive impairment |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22896667 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022390879 Country of ref document: AU Ref document number: AU2022390879 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020247018863 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2022390879 Country of ref document: AU Date of ref document: 20221116 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022896667 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022896667 Country of ref document: EP Effective date: 20240617 |