WO2023069727A1 - Compositions and methods for treatment of hyperproliferative, inflammatory, and immunological diseases, and infections - Google Patents
Compositions and methods for treatment of hyperproliferative, inflammatory, and immunological diseases, and infections Download PDFInfo
- Publication number
- WO2023069727A1 WO2023069727A1 PCT/US2022/047447 US2022047447W WO2023069727A1 WO 2023069727 A1 WO2023069727 A1 WO 2023069727A1 US 2022047447 W US2022047447 W US 2022047447W WO 2023069727 A1 WO2023069727 A1 WO 2023069727A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- elesclomol
- group
- derivative
- analog
- optionally substituted
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- This invention is directed to compositions and methods employing elesclomol or derivatives or analogs thereof or related redox agents for treatment of benign and neoplastic hyperproliferative diseases, infections, inflammatory, and immunological diseases.
- biotherapeutics designed to stimulate the human immune system with adoptive immune cell transfers e.g., CAR-T
- vaccines e.g., therapeutic antibodies, drug-antibody conjugates, cytokines, lymphokines, cytokines, active peptides, inhibitors of tumor blood vessel development (angiogenesis) or gene and antisense therapies to alter the genetic makeup of cancer cells or alter the immune system, as well as agents with other mechanisms.
- new methods employing combinatorial chemistry and high- throughput screening have expanded the options for discovering and employing new medications.
- cancer is a collection of diseases with a multitude of etiologies, biological phenotypes or genotype with high rise for drug resistance and susceptible genomic mutations and that a patient’s response and survival from therapeutic intervention is complex with many factors playing a role in the success or failure of treatment including disease indication, pathology stage related to invasion and metastatic spread, patient gender, age, health conditions, previous therapies or other illnesses, genetic background, other diseases or conditions affecting the patient, especially diseases or conditions that tend to induce immunosuppression, the opportunity for significant cures rates without treatment morbidity in the near term remains elusive.
- the present invention meets the needs described above by providing new methods and compositions employing elesclomol or derivatives or analogs thereof for treatment of a number of diseases and conditions, particularly including, but not limited to, malignancies.
- the malignancies that can be treatable by methods or compositions employing elesclomol or derivatives or analogs thereof include, but are not limited to, ovarian epithelial cancer (OEC).
- OEC ovarian epithelial cancer
- One aspect of the invention is a method to improve the efficacy and/or reduce the side effects of the administration of elesclomol or a derivative, analog, salt, or solvate of elesclomol for treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases or conditions comprising the steps of:
- the factor or parameter is selected from the group consisting of: (i) dose modification; (ii) route of administration;
- the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
- the elesclomol is in the form of a coordinate- covalent complex with a transition metal cation selected from the group consisting of Ni 2+ , Cu + , Cu 2+ , Co 2+ , Co 3+ , Fe 2+ , Fe 3+ , Zn 2+ , Pt 2+ , Pd 2+ , V 4+ , V 5+ , Cr 2+ , Cr 3+ , Cr 4+ , Mn 2+ , Mn 3+ , Mn 4+ , and Mn 5+ .
- a transition metal cation selected from the group consisting of Ni 2+ , Cu + , Cu 2+ , Co 2+ , Co 3+ , Fe 2+ , Fe 3+ , Zn 2+ , Pt 2+ , Pd 2+ , V 4+ , V 5+ , Cr 2+ , Cr 3+
- the divalent metal cation is a divalent transition metal cation selected from the group consisting of Ni 2+ , Cu 2+ , Co 2+ , Fe 2+ , Zn 2+ , Pt 2+ , and Pd 2+ . More preferably, the divalent metal cation is selected from the group consisting of Cu 2+ and Ni +2 . Most preferably, the divalent metal cation is Cu 2+ .
- the treatment is treatment of a malignancy.
- the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
- OEC ovarian epithelial cancer
- OCCC ovarian clear-cell carcinoma
- uterine corpus endothelial carcinoma stomach adenocarcinoma
- bladder urothelial carcinoma adenoid cystic carcinoma
- cholangiocarcinoma cholangiocarcinoma
- pancreatic cancer metastatic esophagogastric cancer
- the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
- OEC ovarian epithelial cancer
- OCCC ovarian clear-cell carcinoma
- the malignancy is selected from the group consisting of human sarcomas and carcinomas.
- the treatment is treatment of a disease or condition selected from the group consisting of: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; and scleroderma.
- a disease or condition selected from the group consisting of: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or
- the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol and the treatment comprises administration of a therapeutically effective quantity of elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 mg/mm 2 /day to about 10 g/mm 2 /day. Typically, the therapeutically effective quantity of elesclomol is from about 2 mg/mm 2 /day to about 10 g/mm 2 /day.
- the therapeutically effective quantity of elesclomol is from about 1 pg/kg to about 500 mg/kg, typically from about 500 pg/kg to about 250 mg/kg, preferably from about 1 mg/kg to about 100 mg/kg, more preferably from about 10 mg/kg to about 50 mg/kg.
- the therapeutically effective quantity of elesclomol or the derivative, analog, salt, or solvate of elesclomol is administered in a pharmaceutical composition.
- Another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Still another aspect of the invention is a method for treatment of a malignancy treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising: (1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
- Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
- compositions to improve the efficacy or reduce the side effects of treatment with elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol wherein the composition comprises:
- the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
- the elesclomol is in the form of a coordinate-covalent complex with a transition metal cation selected from the group consisting of Ni 2+ , Cu + , Cu 2+ , Co 2+ , Co 3+ , Fe 2+ , Fe 3+ , Zn 2+ , Pt 2+ , Pd 2+ , V 4+ , V 5+ , Cr 2+ , Cr 3+ , Cr 4+ , Mn 2+ , Mn 3+ , Mn 4+ , and Mn 5+ .
- a transition metal cation selected from the group consisting of Ni 2+ , Cu + , Cu 2+ , Co 2+ , Co 3+ , Fe 2+ , Fe 3+ , Zn 2+ , Pt 2+ , Pd 2+ , V 4+ , V 5+ , Cr 2+ , Cr 3+ , Cr 4+ , Mn 2+ , Mn 3+ , Mn 4+ , and Mn 5+ .
- the composition is formulated for treatment of a malignancy. Malignancies that can be treated by methods or compositions according to the present invention are described above.
- composition is formulated for treatment of a disease or condition other than a malignancy as described above.
- compositions according to the present invention can be formulated for administration of therapeutically effective dosages such as from about 1 mg/mm 2 /day to about 10 g/mm 2 /day or, alternatively, from about 1 pg/kg to about 500 mg/kg.
- composition comprises an additional therapeutic agent
- additional therapeutic agent is selected from the group consisting of:
- biological therapies selected from the group consisting of Avastin, Rituxan, Herceptin, Erbitux, PD-1 inhibitors, and PD-L1 inhibitors;
- the additional therapeutic agent can be selected from the group consisting of a microtubulin stabilizer, a microtubulin inhibitor, a PARP inhibitor, an LDH inhibitor, 2-deoxyglucose or an analog or derivative thereof, a glutamine metabolism inhibitor, a DNA-damaging agent, an agent that inhibits the SWI/SNF complex, an agent that causes tumor cells to rely on oxidative phosphorylation, an agent that is an inhibitor of the base excision repair (BER) pathway, an agent that acts as an inhibitor of the homologous repair pathway, an agent that acts as an activator of the homologous repair pathway, an agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation, and an agent that inhibits cysteine uptake.
- BER base excision repair
- composition can comprise a pharmaceutically acceptable diluent, a pharmaceutically acceptable solvent, or a pharmaceutically acceptable excipient.
- Figure 1 is a graph showing the results for progression-free survival (PFS) and overall survival (OS) for serous adenocarcinoma, endometrioid adenocarcinoma, mixed epithelial carcinoma, clear-cell carcinoma, and other types of ovarian cancer.
- PFS progression-free survival
- OS overall survival
- Figure 2 is a diagram showing the involvement of ARID1A in pathways of tumor growth, tumor migration, tumor invasion, and angiogenesis.
- Figure 3 is a graph showing that progression free survival in patients exhibiting an ARIDIA-deficient phenotype is significantly reduced as compared with ARID1 A wild-type in patients treated with PARP inhibitors: (A) showing the impact of ARID1 A loss, determined via mutation, homozygous deletion, or loss of expression, and survival in TCGA serous ovarian cancers; (B) showing the progression-free survival of relapsed, platinum-sensitive, high-grade ovarian carcinomas in a clinical trial of rucaparib (a PARP inhibitor), stratified based on ARID1A mutation status.
- Figure 5 is a graph showing that loss of ARID1 A results in downregulation of cystine transporter SLC7A11 and impairment of GSH pathway leading to ROS accumulation and cell death:
- A ARID1A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway.
- B Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
- Figure 6 is a graph that shows the difference between cells with ARID1A mutations and cells with active ARID1 A in terms of the effect of accumulation of ROS:
- A ARID1A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway.
- B Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
- FIG. 7 is a graph that shows a therapeutic window for cancer cells in the presence of an OXPHOS inhibitor.
- Top Panel Normal body cells have moderate ATP demand and adequate levels of oxygen and glucose and survive in presence of OXPHOS inhibitors by upregulating glycolysis to meet their ATP demands.
- Middle Panel Highly proliferating cancer cells have extraordinarily high ATP demand and adequate levels of oxygen and glucose. Despite glycolytic pathway upregulation, OXPHOS inhibition results in failure to meet ATP demand and cell death.
- Quiescent cancer cells have low ATP demand but live in a highly compromised microenvironment (low glucose and hypoxia). Inhibition of OXPHOS is lethal as insufficient glucose is present to compensate for the loss of ATP production by oxidative phosphorylation.
- Figure 8 is a diagram showing the conformational change undergone by elesclomol upon chelation with copper.
- Figure 9 is a graph showing the synergy of elesclomol with PARP inhibitors.
- drug sensitivity was assessed following treatment of mutant BRCA1 cells (SUM149) with elesclomol including paclitaxel (A) and PARP inhibitors talazoparib (B) and rucaparib (C).
- A paclitaxel
- B PARP inhibitors talazoparib
- C rucaparib
- Figure 10 is a diagram showing gene screens indicating genes whose activity is affected by elesclomol:
- A Gene scores in elesclomol-1 (100 nM) and elesclomol-2 (1 ⁇ M) treated K562 cells. The gene score is the median Iog2 fold change in abundance of all sgRNAs targeting that gene during the culture period.
- B Corrected p-values (-logio) of KS tests of the sgRNA distribution for each gene versus the distribution of all sgRNAs in the screen of elesclomol-1 (100 nM) and elesclomol-2 (1 ⁇ M) screens. Values are ordered on the x-axis by chromosome and location; the dotted line indicates a corrected p-value of 0.05.
- Figure 11 is a graph showing results that indicate elesclomol inhibits the natural function of FDX1 in FE-S cluster biosynthesis,
- the [2Fe-2S] cluster in FDX1 is indicated by spheres,
- Reduced FDX1 was used as a reducing agent in the presence or absence of either 5x (green) or 10x (blue) elesclomol. Data are representative of two independent experiments, (d) Electron transfer from reduced FDX1 to the cysteine desulfurase complex was measured on the addition of cysteine in the presence or absence of elesclomol. Data are representative of two independent experiments, (e) The molecular structure of elesclomol analogs and their corresponding calculated EC50 (from T47D cells grown in the Hi-Mito state). The reactive sulfur substitution with an oxygen is indicated with an asterisk, (f) The UV/visible spectra of reduced FDX1 before and after incubation with elesclomol-Cu(ll) or Cu(ll) alone.
- Figure 12 is a graph showing that sensitivity to elesclomol as a single agent is highly specific to ARID1A mutation in cancer cell lines.
- A Cell growth of endometrial and ovarian cancer cell lines treated with elesclomol for 72 h as measured using the WST-1 assay.
- B IC 5 0 values of elesclomol in these cell lines.
- Figure 13 is a diagram showing that ARIDIA-mutant tumors are highly dependent on OXPHOS offering a potential strategy for synthetic lethality employing targeted agents interrupting mitochondrial metabolism.
- Figure 14 is a diagram showing that the protein target of elesclomol is ferredoxin-1 , a key component of the OXPHOS pathway establishing a synthetically lethal therapeutic strategy in ARIDIA-mutant tumors.
- Figure 15 is a diagram showing the occurrence of ARID1 A mutations in a number of types of malignancies, including mutations, deletions, amplification, and multiple alterations.
- Figure 16 is a series of graphs showing the effects of a 72-hour treatment with elesclomol on the viability of either the OVCA429 NTC (non-targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line.
- Figure 17 is a series of graphs showing the effects of a 72-hour treatment with elesclomol on the viability of either the RMG1 NTC (non-targeted control) clear cell ovarian cancer cell line or the RMG1 ARID1 A mutant cell line.
- Figure 18 is a series of graphs showing the results over time of treatment of either the OVCA429 NTC (non-targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line with 1 ng, 10 ng, 100 ng, or 1000 ng of elesclomol for a time interval of up to 72 hours.
- Figure 18A shows the results for OVCA429 NTC;
- Figure 18B shows the results for OVCA429 ARID1 A mutant;
- Figure 18C overlays the results from Figures 18A and Figure 18B. In all cases, the mutant cells were more sensitive to elesclomol as a function of time than were the OVCA429 ARID1A NTC cells.
- Figures 16 and 17 do show a clear difference in results between the ARID1 A wild-type and the ARID1A mutant phenotype, the difference in results is not clear in Figure 18. This is simply an artifact of the assays.
- the Incucyte analyses shown in Figure 18 are based on bright-field images which provide an analysis of confluence so does not differentiate between viable and dying cells, whereas Figures 16 and 17 measure only viable cells.
- Figure 19 shows photomicrographs of either the OVCA429 NTC (non- targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line that were either not treated with elesclomol or treated with 10 nM of elesclomol.
- A OVCA429 NTC not treated with elesclomol
- B OVCA429 ARID1 A mutant cell line not treated with elesclomol
- C OVCA429 NTC treated with 10 nM of elesclomol
- D OVCA429 ARID1 A mutant cell line treated with 10 nM of elesclomol.
- treatment with elesclomol resulted in cell death visible in the photomicrographs.
- Figure 20 is a schematic diagram showing the mechanisms of action of elesclomol in cells with wild-type ARID1 A and in mutant ARID1 A, in particular the occurrence of reactive oxygen species (ROS) in cells with mutant ARID1 A induced by elesclomol, leading to cytotoxicity.
- ROS reactive oxygen species
- Figure 21 is a schematic diagram showing that loss-of-function mutations in ARID1 A promotes oncogenesis and reliance on OXPHOS.
- Figure 22 is a schematic diagram showing that elesclomol induces synthetic lethality in tumor cells with loss-of-function mutations in ARID1A.
- the terms “comprise,” “include,” and linguistic variations thereof denote the presence of recited features, elements, method steps, or other components of the invention without the exclusion of the presence of additional /recited features, elements, method steps, or other components.
- the terms “consisting of’ and linguistic variations thereof denote the presence of recited features, elements, method steps, or other components of the invention and exclude any unrecited recited features, elements, method steps, or other components of the invention except for ordinarily-associated impurities.
- a methylamine substituent is while an aminomethyl substituent is
- the term “subject” broadly refers to any animal, including, but not limited to, humans and non-human mammals.
- the reference to non-human mammals includes, but is not limited to, socially or economically important animals or animals used for research including cattle, sheep, goats, horses, pigs, llamas, alpacas, dogs, cats, rabbits, guinea pigs, rats, and mice.
- methods and compositions according to the present invention are not limited to treatment of humans. In general, when treatment of humans is intended, the term “patient” can used in place of “subject.”
- the terms “effective amount,” “therapeutically effective amount,” or other equivalent terminology refer to the amount of a compound or compounds or to the amount of a composition sufficient to effect beneficial or desired results.
- the beneficial or desired results are typically a reduction in severity, symptoms, or duration of a disease or condition being treated and can generally be characterized as an amount of a therapeutic agent or composition effective to treat, ameliorate, or prevent a desired disease or condition, or to exhibit a detectable therapeutic or preventative effect.
- the use of such terminology cannot, unless specifically indicated, be interpreted as implying a complete cure for any disease or condition as recited herein.
- An effective amount can be administered in one or more administrations, applications, or dosages, and is not intended to be limited to a particular formulation or administration route unless a particular formulation or administration route is specified.
- the effect induced by the administration of a therapeutically effective amount can be detected by, for example, chemical markers, antigen levels, or changes in pathological indicators such as tumor burden.
- Therapeutic effects also can include subjective improvements in well-being, reduction of fatigue, or increased energy noted by the subjects or their caregivers.
- the precise therapeutically effective amount for a subject will depend upon the subject’s size, weight, and health, the nature and extent of the condition affecting the subject, the administration of other therapeutics administered to treat the particular disease or condition being treated or other diseases or conditions affecting the subject, as well as variables such as liver and kidney function that affect the pharmacokinetics of administered therapeutics. Thus, it is not useful to specify an exact effective amount in advance. However, the therapeutically effective amount for a given situation can be determined by routine experimentation and is within the judgment of the clinician.
- administering refers to the act of giving a drug, prodrug, pharmaceutical composition, or other agent intended to provide therapeutic treatment to a subject or in vivo, in vitro, or ex vivo to cells, tissues, or organs.
- Exemplary routes of administration to the human body can be through space under the arachnoid membrane of the brain or spinal cord (intrathecal), the eyes (ophthalmic), mouth (oral), skin (topical or transdermal), nose (nasal), lungs or other portions of the respiratory tract (inhalant), oral mucosa (buccal), ear, rectal, vaginal, by injection (such as, but not limited to, intravenously, subcutaneously, intraperitoneally, or by other injection routes as known in the art).
- injection such as, but not limited to, intravenously, subcutaneously, intraperitoneally, or by other injection routes as known in the art).
- co-administration refers to the administration of at least two agents, such as, for example, elesclomol or a derivative or analog thereof and a PARP inhibitor, or therapies to a subject.
- the co-administration of two or more agents or therapies is concurrent.
- a first agent/therapy is administered prior to a second agent/therapy.
- the appropriate dosage for co-administration can be readily determined by one skilled in the art.
- when agents or therapies are co-administered the respective agents or therapies are administered at lower dosages than appropriate for their administration alone.
- co-administration is especially desirable in embodiments where the co-administration of the agents or therapies lowers the requisite dosage of a potentially harmful agent or agent, and/or when co-administration of two or more agents results in sensitization of a subject to beneficial effects of one of the agents via co- administration of the other agent.
- concurrent administration refers to the administration of two or more active agents sufficiently close in time to achieve a combined therapeutic effect that is preferably greater than that which would be achieved by the administration of either agent alone.
- Such concurrent administration can be carried out simultaneously, e.g., by administering the active agents together in a common pharmaceutically acceptable carrier, thereby forming a pharmaceutical composition with two or more active agents, in one or more doses of the pharmaceutical composition.
- the term “pharmaceutical composition” refers to the combination of one or more therapeutically active agents with at least one carrier, inert or active, making the composition especially suitable for diagnostic or therapeutic use in vitro, in vivo or ex vivo.
- the term “pharmaceutically acceptable carrier” refers to any of the standard pharmaceutical carriers including, but not limited to, phosphate buffered saline solution, water, emulsions, such as oil/water or water/oil emulsions), and various types of wetting agents, any and all solvents, dispersion media, coatings, sodium lauryl sulfate, isotonic and absorption delaying agents, disintegrants such as potato starch or sodium starch glycolate), and the like.
- the carriers also can include stabilizers and preservatives.
- the term “pharmaceutically acceptable salt” refers to any pharmaceutically acceptable salt (e.g., acid or base) of a compound that is used in a method of the present invention or is a component of a composition of the present invention, which, upon administration to a subject, is capable of providing a compound of the present invention or an active metabolite or residue thereof.
- salts of the compounds of the present invention may be derived from inorganic or organic acids and bases.
- acids include, but are not limited to, hydrochloric, hydrobromic, sulfuric, nitric, perchloric, fumaric, maleic, phosphoric, glycolic, lactic, salicylic, succinic, toluene-p-sulfonic, tartaric, acetic, citric, methanesulfonic, ethanesulfonic, formic, benzoic, malonic, naphthalene-2-sulfonic, benzenesulfonic acid, and other acids known in the art as suitable for formation of pharmaceutically acceptable salts.
- acids such as oxalic
- bases include, but are not limited to, alkali metals (such as sodium or potassium) hydroxides, alkaline earth metals (such as calcium or magnesium), hydroxides, ammonia, and compounds of formula NW4 + , wherein W is C 1 -C 4 alkyl, and the like.
- salts include, but are not limited to: acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, flucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2- hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, palmoate, pectinate, persulfate, phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate
- salts include anions of the compounds of the present invention compounded with a suitable cation such as Na + , NH4 + , and NW4 + , wherein W is a C 1 -C 4 alkyl group), and the like.
- a suitable cation such as Na + , NH4 + , and NW4 + , wherein W is a C 1 -C 4 alkyl group
- salts of the compounds herein are contemplated as being pharmaceutically acceptable.
- salts of acids and bases that are non-pharmaceutically acceptable may also find use, for example, in the preparation or purification of a pharmaceutically acceptable compound.
- the term “instructions for administering a compound to a subject,” and grammatical equivalents thereof, includes instructions for using the compositions contained in a kit for the treatment of conditions. Such instructions, for example, provide dosing, routes of administration, or decision trees for treating physicians for correlating patient-specific characteristics with therapeutic courses of action. Such instructions may be part of a kit according to the present invention.
- analogs and derivatives of the compounds described in further detail below including elesclomol and other therapeutically active agents described herein.
- analogue refers to a chemical compound that is structurally similar to a parent compound, but differs slightly in composition (e.g., one atom or functional group is different, added, or removed).
- the analogue may or may not have different chemical or physical properties than the original compound and may or may not have improved biological and/or chemical activity.
- the analogue may be more hydrophilic or hydrophobic or it may have altered reactivity as compared to the parent compound.
- the analogue may mimic the chemical and/or biologically activity of the parent compound (i.e.
- analogue may be a naturally or non-naturally occurring variant of the original compound.
- Other types of analogues include isomers (enantiomers, diastereomers, and the like) and other types of chiral variants of a compound, as well as structural isomers.
- “derivative” refers to a chemically or biologically modified version of a chemical compound that is structurally similar to a parent compound and (actually or theoretically) derivable from that parent compound.
- a “derivative” differs from an “analog” in that a parent compound may be the starting material to generate a “derivative,” whereas the parent compound may not necessarily be used as the starting material to generate an “analog.”
- a derivative may or may not have different chemical or physical properties than the parent compound. For example, the derivative may be more hydrophilic or hydrophobic or it may have altered reactivity as compared to the parent compound. Derivatization (i.e. , modification) may involve substitution of one or more moieties within the molecule (e.g., a change in functional group).
- the term “derivative” also includes conjugates and prodrugs of a parent compound (i.e., chemically modified derivatives which can be converted into the original compound under physiological conditions).
- alkyl refers to an unbranched, branched, or cyclic saturated hydrocarbyl residue, or a combination thereof, of from 1 to 12 carbon atoms, or in some cases up to 50 or more carbon atoms, that can be optionally substituted; the alkyl residues contain only C and H when unsubstituted.
- the unbranched or branched saturated hydrocarbyl residue is from 1 to 6 carbon atoms, preferably 1 to 4 carbon atoms, more preferably 1 to 3 carbon atoms, which is referred to herein as “lower alkyl.”
- the alkyl residue is cyclic and includes a ring, it is understood that the hydrocarbyl residue includes at least three carbon atoms, which is the minimum number to form a ring.
- An alkyl group can be linear, branched, cyclic, or a combination thereof, and may contain from 1 to 50 or more carbon atoms, such as a straight chain or branched C 1 -C 2 0 alkane.
- alkyl groups include but are not limited to methyl, ethyl, propyl, isopropyl, cyclopropyl, butyl isomers (e.g. n-butyl, isobutyl, and te/Y-butyl), cyclobutyl isomers (e.g. cyclobutyl, methylcyclopropyl, or other isomers), pentyl isomers, cyclopentane isomers, hexyl isomers, cyclohexane isomers, and the like.
- butyl isomers e.g. n-butyl, isobutyl, and te/Y-butyl
- cyclobutyl isomers e.g. cyclobutyl, methylcyclopropyl, or other isomers
- pentyl isomers cyclopentane isomers
- hexyl isomers cyclohexan
- an alkyl group contains carbon and hydrogen atoms only.
- linear alkyl refers to a chain of carbon and hydrogen atoms (e.g., ethane, propane, butane, pentane, hexane, or other examples).
- a linear alkyl group may be referred to by the designation - -(CH 2 ) q CH 3 , where q is 0-49.
- C 1 -C 12 alkyl refers to alkyl having from 1 to 12 carbon atoms such as methyl, ethyl, propyl isomers (e.g. n-propyl or isopropyl), butyl isomers, cyclobutyl isomers (e.g.
- C x -C y when used in conjunction with a chemical moiety, such as alkyl, alkenyl, alkynyl, or carbocycle is meant to include groups that contain from x to y carbons in the chain or ring.
- C x -C y alkyl refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups that contain from x to y carbons in the chain, including haloalkyl groups such as trifluoromethyl and 2,2,2-trifluoroethyl, or other alternatives.
- C x -C y alkenyl and “C x -C y alkynyl” refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- C x -C y carbocycle refers to a substituted or unsubstituted carbocycle, that contain from x to y ring carbons.
- branched alkyl refers to a chain of carbon and hydrogen atoms, without double or triple bonds, that contains a fork, branch, and/or split in the chain (e.g., 3,5-dimethyl-2-ethylhexane, 2-methyl-pentane, 1-methyl-cyclobutane, ortho -diethyl-cyclohexane, or other alternatives).
- Branching refers to the divergence of a carbon chain
- substitution refers to the presence of non-carbon/non-hydrogen atoms in a moiety.
- a branched alkyl group contains carbon and hydrogen atoms only.
- the term “carbocycle,” “carbocyclyl,” or “carbocyclic” refers to a cyclic ring containing only carbon atoms in the ring, whereas the term “heterocycle” or “heterocyclic” refers to a ring comprising a heteroatom.
- the carbocycle can be fully saturated or partially saturated, but non-aromatic.
- carbocyclyl encompasses cycloalkyl.
- the carbocyclic and heterocyclic structures encompass compounds having monocyclic, bicyclic or multiple (polycyclic) ring systems; and such systems may mix aromatic, heterocyclic, and carbocyclic rings. Mixed ring systems are described according to the ring that is attached to the rest of the compound being described.
- Bicyclic or polycyclic rings may include fused or spiro rings.
- Carbocycles may include 3- to 10-membered monocyclic rings, 6- to 12- membered bicyclic rings, and 6- to 12-membered bridged rings. Each ring of a bicyclic or polycyclic carbocycle may be selected from saturated, unsaturated, and aromatic rings.
- an aromatic carbocycle e.g., phenyl
- a saturated or unsaturated ring e.g., cyclohexane, cyclopentane, or cyclohexene.
- the carbocycle is an aromatic carbocycle .
- the carbocycle is a cycloalkyl.
- the carbocycle is a cycloalkenyl.
- Exemplary carbocycles include cyclopentyl, cyclohexyl, cyclohexenyl, adamantyl, phenyl, indanyl, and naphthyl.
- an alkenyl group can be optionally substituted by one or more substituents such as those substituents described herein.
- a “non-aromatic carbocycle” includes rings and ring systems that are saturated, unsaturated, substituted or unsubstituted, but not aromatic or aryl rings or ring systems.
- cycloalkyl refers to a completely saturated mono- or multi-cyclic hydrocarbon ring system. When composed of two or more rings, the rings may be joined together in a fused, bridged or spiro-connected fashion. Cycloalkyl groups of the present application may range from three to ten carbons (C 3 to C 10 ). A cycloalkyl group may be unsubstituted, substituted, branched, and/or unbranched. Typical cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
- the substituent(s) may be an alkyl or can be selected from those indicated above with regard to substitution of an alkyl group unless otherwise indicated. While “alkyl” as used herein includes cycloalkyl and cycloalkylalkyl groups, the term “cycloalkyl” may be used herein to describe a carbocyclic non-aromatic group that is connected via a ring carbon atom, and “cycloalkylalkyl” may be used to describe a carbocyclic non-aromatic group that is connected to the molecule through an alkyl linker.
- heteroalkyl refers to an alkyl group, as defined herein, wherein one or more carbon atoms are independently replaced by one or more heteroatoms (e.g., oxygen, sulfur, nitrogen, phosphorus, selenium, silicon, or combinations thereof).
- the alkyl group containing the non-carbon substitution(s) may be a linear alkyl, branched alkyl, cycloalkyl (e.g., cycloheteroalkyl), or combinations thereof.
- Non-carbons may be at terminal locations (e.g., 2-hexanol) or integral to an alkyl group (e.g., diethyl ether).
- hetero terms refer to groups that typically contain 1-3 O, S or N heteroatoms or combinations thereof within the backbone residue; thus at least one carbon atom of a corresponding alkyl, alkenyl, or alkynyl group is replaced by one of the specified heteroatoms to form, respectively, a heteroalkyl, heteroalkenyl, or heteroalkynyl group. In some cases, more than three heteroatoms may be present. Unless stated otherwise specifically in the specification, the heteroalkyl group may be optionally substituted as described herein. Representative heteroalkyl groups include, but are not limited to -OCH 2 OMe, - OCH 2 CH 2 OMe, or --OCH 2 CH 2 OCH 2 CH 2 NH 2 . For reasons of chemical stability, it is also understood that, unless otherwise specified, such groups do not include more than two contiguous heteroatoms except where an oxo group is present on N or S as in a nitro or sulfonyl group.
- heteroalkylene refers to an alkyl radical as described above where one or more carbon atoms of the alkyl is replaced with a heteroatom, e.g., O, N or S, or another heteroatom as described above.
- heteroatom e.g., O, N or S
- heteroalkylene or heteroalkylene chain refers to a straight or branched divalent heteroalkyl chain linking the rest of the molecule to a radical group. Unless stated otherwise specifically in the specification, the heteroalkylene group may be optionally substituted as described herein.
- heteroalkylene groups include, but are not limited to -OCH 2 CH 2 O-, -OCH 2 CH 2 OCH 2 CH 2 O-, or - OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 O-.
- optionally substituted indicates that the particular group or groups referred to as optionally substituted may have no non- hydrogen substituents, or the group or groups may have one or more non-hydrogen substituents consistent with the chemistry and pharmacological activity of the resulting molecule and such that a stable compound is formed thereby, i.e. , a compound that does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, hydrolysis, lactone or lactam formation, or other reaction.
- the total number of such substituents that may be present is equal to the total number of hydrogen atoms present on the unsubstituted form of the group being described; fewer than the maximum number of such substituents may be present.
- the group takes up two available valences on the carbon atom to which the optional substituent is attached, so the total number of substituents that may be included is reduced according to the number of available valences.
- substituted when used to modify a specific group, moiety, or radical, means that one or more hydrogen atoms are, each, independently of each other, replaced with the same or different substituent or substituents. Substitution of a structure depicted herein may result in removal or moving of a double bond or other bond, as will be understood by one in the field. In certain embodiments, substituted refers to moieties having substituents replacing two hydrogen atoms on the same carbon atom, such as substituting the two hydrogen atoms on a single carbon with an oxo, imino or thioxo group.
- the term “substituted” is contemplated to include all permissible substituents of organic compounds that do not significantly alter the pharmacological activity of the compound in the context of the present invention.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
- haloalkyl or “haloalkane” refers to an alkyl radical, as defined above, that is substituted by one or more halogen radicals, for example, trifluoromethyl, dichloromethyl, bromomethyl, 2,2,2-trifluoroethyl, 1- fluoromethyl-2-fluoroethyl, and the like.
- the alkyl part of the fluoroalkyl radical is optionally further substituted.
- halogen substituted alkanes include halomethane (e.g., chloromethane, bromomethane, fluoromethane, iodomethane), di-and trihalomethane (e.g., trichloromethane, tribromomethane, trifluoromethane, triiodomethane), 1-haloethane, 2-haloethane, 1 ,2- dihaloethane, 1-halopropane, 2-halopropane, 3-halopropane, 1 ,2-dihalopropane, 1 ,3- dihalopropane, 2,3-dihalopropane, 1 ,2,3-trihalopropane, and any other suitable combinations of alkanes (or substituted alkanes) and halogens (e.g., Cl, Br, F, or I).
- halogen substituted alkanes e.g., Cl, Br, F, or I.
- aryl refers to a monocyclic or fused bicyclic moiety having the well-known characteristics of aromaticity; examples include phenyl and naphthyl, which can be optionally substituted. Additional examples of aromatic rings include furan, benzofuran, isobenzofuran, pyrrole, indole, isoindole, thiophene, benzothiophene, benzo(c)thiophene, imidazole, benzimidazole, purine, pyrazole, indazole, oxazole, benzooxazole, isoxazole, benzisoxazole, thiazole, benzothiazole, benzene, naphthalene, pyridine, quinolone, isoquinoline, pyrazine, quinoxaline, pyrimidine, quinazoline, pyridazine, cinnoline, phthalazine, tri
- aromatic carbocycle refers to an aromatic ring without heteroatoms present within the ring structure, such as, but not limited to benzene or naphthalene.
- aromatic ring refers to an aromatic ring without heteroatoms present within the ring structure, such as, but not limited to benzene or naphthalene.
- Other terms that can be used include “aromatic ring,” “aryl group,” or “aryl ring.”
- heterocycle As used herein, the term “heterocycle,” “heterocyclyl,” “heterocyclic ring” or “heterocyclic group” is intended to mean a stable 4-, 5-, 6-, or 7-membered monocyclic or 7-, 8-, 9-, 10-, 11 -, 12-, 13-, or 14-membered bicyclic heterocyclic ring which is saturated, partially unsaturated, or fully unsaturated or aromatic, and which consists of carbon atoms and 1 , 2, 3 or 4 heteroatoms independently selected from N, O, and S; and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- heteroatoms such as P, Se, B, or Si
- the nitrogen and sulfur heteroatoms may optionally be oxidized.
- the nitrogen atom may be substituted or unsubstituted (i.e. , N or NR wherein R is H or another substituent, if defined).
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable.
- a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and 0 atoms in the heterocycle exceeds 1 , then these heteroatoms are not adjacent to one another.
- heterocycle When the term “heterocycle,” “heterocyclyl,” “heterocyclic ring” or “heterocyclic group” is used, it is intended to include heteroaryl unless heteroaryl is excluded.
- heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H-
- other heteroatoms including P, Se, B, or Si can be included.
- Non-limiting examples of non-aromatic heterocycles include morpholino, pyrrol idiny I , pyrrolidinyl-2-one, piperazinyl, piperid iny I, piperidinylone, 1 ,4-dioxa-8-aza- spiro(4.5)dec-8-yl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, 1 ,3-dioxolanyl, 2-imidazolinyl, imidazolidinyl, 2-pyrazolinyl, pyrazolidinyl, 1 ,4-dioxanyl, 1 ,4-dithianyl, thiomorpholinyl, azepanyl, hexahydro-1 ,4-diazepinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl
- a non-aromatic heterocyclic ring is aziridine, thiirane, oxirane, oxaziridine, dioxirane, azetidine, oxetan, thietane, diazetidine, dioxetane, dithietane, pyrrolidine, tetrahydrofuran, thiolane, imidazolidine, pyrazolidine, oxazolidine, isoxazolidine, thiazolidine, isothiazolidine, dioxolane, dithiolane, piperdine, oxane, thiane, piperazine, morpholine, thiomorpholine, dioxane, dithiane, trioxane, thithiane, azepane, oxepane, thiepane, homopiperazine, or azocane.
- heteroaryl or “heteroaromatic” refer to monocyclic, bicyclic, or polycyclic ring systems, wherein at least one ring in the system is aromatic and contains at least one heteroatom, for example, nitrogen, oxygen and sulfur.
- Each ring of the heteroaromatic ring systems may contain 3 to 7 ring atoms.
- Exemplary heteroaromatic monocyclic ring systems include 5- to 7-membered rings whose ring structures include one to four heteroatoms, for example, one or two heteroatoms. The inclusion of a heteroatom permits aromaticity in 5-membered rings as well as in 6-membered rings.
- Typical heteroaromatic systems include monocyclic C 5 -C 6 heteroaromatic groups such as pyridyl, pyrim idyl, pyrazinyl, thienyl, furanyl, pyrrolyl, pyrazolyl, thiazolyl, oxazolyl, triazolyl, triazinyl, tetrazolyl, tetrazinyl, and imidazolyl, as well as the fused bicyclic moieties formed by fusing one of these monocyclic heteroaromatic groups with a phenyl ring or with any of the heteroaromatic monocyclic groups to form a C 8 -C- 10 bicyclic group such as indolyl, benzimidazolyl, indazolyl, benzotriazolyl, isoquinolyl, quinolyl, benzothiazolyl, benzofuranyl, pyrazolylpyridyl, quinazolinyl, qui
- any monocyclic or fused ring bicyclic system that has the characteristics of aromaticity in terms of delocalized electron distribution throughout the ring system is included in this definition.
- This definition also includes bicyclic groups where at least the ring that is directly attached to the remainder of the molecule has the characteristics of aromaticity, including the delocalized electron distribution that is characteristic of aromaticity.
- the ring systems contain 5 to 12 ring member atoms and up to four heteroatoms, wherein the heteroatoms are selected from the group consisting of N, 0, and S.
- the monocyclic heteroaryls contain 5 to 6 ring members and up to three heteroatoms selected from the group consisting of N, 0, and S; frequently, the bicyclic heteroaryls contain 8 to 10 ring members and up to four heteroatoms selected from the group consisting of N, 0, and S.
- the number and placement of heteroatoms in heteroaryl ring structures is in accordance with the well-known limitations of aromaticity and stability, where stability requires the heteroaromatic group to be stable enough to be exposed to water at physiological temperatures without rapid degradation.
- the term “hydroxyheteroaryl” refers to a heteroaryl group including one or more hydroxyl groups as substituents; as further detailed below, further substituents can be optionally included.
- haloaryl and haloheteroaryl refer to aryl and heteroaryl groups, respectively, substituted with at least one halo group, where “halo” refers to a halogen selected from the group consisting of fluorine, chlorine, bromine, and iodine, typically, the halogen is selected from the group consisting of chlorine, bromine, and iodine; as detailed below, further substituents can be optionally included.
- haloalkyl refers to alkyl, alkenyl, and alkynyl groups, respectively, substituted with at least one halo group
- halo refers to a halogen selected from the group consisting of fluorine, chlorine, bromine, and iodine, typically, the halogen is selected from the group consisting of chlorine, bromine, and iodine; as detailed below, further substituents can be optionally included.
- halo refers to a halogen selected from the group consisting of fluorine, chlorine, bromine, and iodine
- the halogen is selected from the group consisting of chlorine, bromine, and iodine
- further substituents can be optionally included.
- C 1 - C 6 alkyl includes alkyl groups with 1 , 2, 3, 4, 5, or 6 carbon atoms and all possible subranges.
- hydroxyaryl refers to an aryl group including one or more hydroxyl groups as substituents; as further detailed below, further substituents can be optionally included.
- solvate means a compound formed by solvation (the combination of solvent molecules with molecules or ions of the solute), or an aggregate that consists of a solute ion or molecule, i.e., a compound of the invention, with one or more solvent molecules.
- solvate typically means a physical association of a compound involving varying degrees of ionic and/or covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent atoms are incorporated into the crystal lattice of the crystalline solid.
- solvate encompasses both solution-phase and isolatable solvates.
- Suitable solvates in which the solvent is other than water include, but are not limited to, ethanolates or methanolates.
- water is the solvent
- the corresponding solvate is a “hydrate.”
- examples of hydrates include, but are not limited to, hemihydrate, monohydrate, dihydrate, trihydrate, hexahydrate, and other hydrated forms.
- the pharmaceutically acceptable salt and/or prodrug of compounds described herein for use in methods or compositions according to the present invention may also exist in a solvate form.
- the solvate is a hydrate
- the hydrate is typically formed via hydration which is either part of the preparation of the present compound or through natural absorption of moisture by the anhydrous compound of the present invention.
- esters means any ester of a present compound in which any of the --COOH functions of the molecule is replaced by a --COOR function, in which the R moiety of the ester is any carbon-containing group which forms a stable ester moiety, including but not limited to alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl and substituted derivatives thereof.
- hydrolyzable esters of the present compounds are the compounds whose carboxyls are present in the form of hydrolyzable ester groups. That is, these esters are pharmaceutically acceptable and can be hydrolyzed to the corresponding carboxyl acid in vivo.
- alkenyl refers to an unbranched, branched or cyclic hydrocarbyl residue having one or more carbon-carbon double bonds. Typically, the hydrocarbyl residue has from 2 to 12 carbon atoms (C 2 -C 12 alkenyl). In certain embodiments, an alkenyl comprises two to eight carbon atoms (C 2 -C 8 alkenyl). In certain embodiments, an alkenyl comprises two to six carbon atoms (i.e. , C 2 -C 6 alkenyl). In other embodiments, an alkenyl comprises two to four carbon atoms (i.e., C 2 -C 4 alkenyl).
- alkenyl is attached to the rest of the molecule by a single bond, for example, ethenyl (i.e., vinyl), prop-1 -enyl (i.e., allyl), but-1 -enyl, pent-1 -enyl, penta-1 ,4- dienyl, and the like.
- An alkenyl group can be optionally substituted by one or more substituents such as those substituents described herein. With respect to the use of “alkenyl,” the presence of multiple double bonds cannot product an aromatic ring structure.
- alkynyl refers to an unbranched, branched, or cyclic hydrocarbyl residue having one or more carbon-carbon triple bonds; the residue can also include one or more double bonds.
- the hydrocarbyl residue has from 2 to 12 carbon atoms (C 2 -C 12 alkynyl).
- an alkenyl comprises two to eight carbon atoms (C 2 -C 8 alkynyl).
- an alkenyl comprises two to six carbon atoms (i.e., C 2 -C 6 alkynyl).
- an alkenyl comprises two to four carbon atoms (i.e., C 2 -C 4 alkynyl).
- the alkynyl is attached to the rest of the molecule by a single bond, for example, ethynyl, propynyl, butynyl, pentynyl, hexynyl, and the like.
- alkynyl the presence of multiple double bonds in addition to the one or more triple bonds cannot produce an aromatic ring structure.
- alkylene or “alkylene chain” refers to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing no unsaturation, and preferably having from one to twelve carbon atoms, for example, methylene, ethylene, propylene, n-butylene, and the like.
- the alkylene chain is attached to the rest of the molecule through a single bond and to the radical group through a single bond.
- the points of attachment of the alkylene chain to the rest of the molecule and to the radical group may be through any two carbons within the chain.
- an alkylene comprises one to ten carbon atoms (i.e.
- an alkylene comprises one to eight carbon atoms (i.e., C 1 -C 8 alkylene). In other embodiments, an alkylene comprises one to five carbon atoms (i.e., C 1 -C 5 alkylene). In other embodiments, an alkylene comprises one to four carbon atoms (i.e., C 1 -C 4 alkylene). In other embodiments, an alkylene comprises one to three carbon atoms (i.e., C 1 -C 3 alkylene). In other embodiments, an alkylene comprises one to two carbon atoms (i.e., C 1 -C 2 alkylene).
- an alkylene comprises only one carbon atom (i.e., C 1 alkylene or a -CH 2 — group).
- An alkylene group can be optionally substituted by one or more substituents such as those substituents described herein.
- alkenylene or “alkenylene chain” refers to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing at least one carbon- carbon double bond, and preferably having from two to twelve carbon atoms.
- the alkenylene chain is attached to the rest of the molecule through a single bond and to the radical group through a single bond.
- the points of attachment of the alkenylene chain to the rest of the molecule and to the radical group may be through any two carbons within the chain.
- an alkenylene comprises two to ten carbon atoms (i.e., C 2 -C 10 alkenylene).
- an alkenylene comprises two to eight carbon atoms (i.e., C 2 -C 8 alkenylene). In other embodiments, an alkenylene comprises two to five carbon atoms (i.e., C 2 -C 5 alkenylene). In other embodiments, an alkenylene comprises two to four carbon atoms (i.e. , C 2 -C 4 alkenylene). In other embodiments, an alkenylene comprises two to three carbon atoms (i.e., C 2 -C 3 alkenylene). In other embodiments, an alkenylene comprises two carbon atom (i.e., C 2 alkenylene). An alkenylene group can be optionally substituted by one or more substituents such as those substituents described herein.
- alkynylene or “alkynylene chain” refers to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing at least one carbon-carbon triple bond, and preferably having from two to twelve carbon atoms.
- the alkynylene chain is attached to the rest of the molecule through a single bond and to the radical group through a single bond.
- the points of attachment of the alkynylene chain to the rest of the molecule and to the radical group may be through any two carbons within the chain.
- an alkynylene comprises two to ten carbon atoms (i.e., C 2 -C 10 alkynylene).
- an alkynylene comprises two to eight carbon atoms (i.e., C 2 -C 8 alkynylene). In other embodiments, an alkynylene comprises two to five carbon atoms (i.e., C 2 -C 5 alkynylene). In other embodiments, an alkynylene comprises two to four carbon atoms (i.e., C 2 -C 4 alkynylene). In other embodiments, an alkynylene comprises two to three carbon atoms (i.e., C 2 -C 3 alkynylene). In other embodiments, an alkynylene comprises two carbon atom (i.e., C 2 alkynylene).
- An alkenylene group can be optionally substituted by one or more substituents such as those substituents described herein.
- amine or “amino” includes primary, secondary, and tertiary amines wherein each non-hydrogen group on nitrogen may be selected from alkyl, aryl, and the like.
- Amines include but are not limited to --NH 2 , --NH-phenyl, -- NH--CH 3 , --NH--CH 2 CH 3 , and --N(CH 3 )benzyl. The amino group can be optionally substituted.
- the term can include NR'R" wherein each R' and R" is independently H, or is an alkyl, alkenyl, alkynyl, acyl, aryl, or arylalkyl group, and each of the alkyl, alkenyl, alkynyl, acyl, aryl, or arylalkyl groups is optionally substituted with the substituents described herein as suitable for the corresponding group; the R' and R" groups and the nitrogen atom to which they are attached can optionally form a 3- to 8- membered ring which may be saturated, unsaturated or aromatic and which contains 1- 3 heteroatoms independently selected from N, 0 and S as ring members, and which is optionally substituted with the substituents described as suitable for alkyl groups or, if NR'R" is an aromatic group, it is optionally substituted with the substituents described as typical for heteroaryl groups.
- amide or “amido” includes C- and N-amide groups, e.g., --C(O)NR 2 , and --NRC(O)R groups, respectively, where R can be H, alkyl, aryl, or other groups, which can be optionally substituted.
- Amide groups therefore include but are not limited to -C(O)NH 2 , -NHC(O)H, -C(O)NHCH 2 CH 3 , - NHC(O)CH 3 ,or -C(O)N(CH 2 CH 3 )phenyl.
- acyl encompasses groups comprising an alkyl, alkenyl, alkynyl, aryl or arylalkyl radical attached at one of the two available valence positions of a carbonyl carbon atom
- heteroacyl refers to the corresponding groups wherein at least one carbon other than the carbonyl carbon has been replaced by a heteroatom chosen from N, O and S.
- arylalkyl and “heteroarylalkyl” refer to aromatic and heteroaromatic ring systems which are bonded to their attachment point through a linking group such as an alkylene, including substituted or unsubstituted, saturated or unsaturated, cyclic or acyclic linkers.
- the linker is C 1 -C 8 alkyl.
- These linkers may also include a carbonyl group, thus making them able to provide substituents as an acyl or heteroacyl moiety.
- An aryl or heteroaryl ring in an arylalkyl or heteroarylalkyl group may be substituted with the same substituents described above for aryl groups.
- an arylalkyl group includes a phenyl ring optionally substituted with the groups defined above for aryl groups and a C 1 -C 4 alkylene that is unsubstituted or is substituted with one or two C 1 -C 4 alkyl groups or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
- a heteroarylalkyl group preferably includes a C 5 -C 6 monocyclic heteroaryl group that is optionally substituted with the groups described above as substituents typical on aryl groups and a C 1 -C 4 alkylene that is unsubstituted or is substituted with one or two C 1 -C 4 alkyl groups or heteroalkyl groups, or it includes an optionally substituted phenyl ring or C 5 -C 6 monocyclic heteroaryl and a C 1 -C 4 heteroalkylene that is unsubstituted or is substituted with one or two C 1 -C 4 alkyl or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
- heteroatom refers to any atom that is not carbon or hydrogen, such as nitrogen, oxygen or sulfur. When it is part of the backbone or skeleton of a chain or ring, a heteroatom must be at least divalent, and will typically be selected from N, O, P, and S, more typically from N, O, and P.
- heteroatom can include, in some contexts, other atoms, including selenium, silicon, or boron.
- lower alkanoyl refers to an alkanoyl group in which the alkyl portion of the alkanoyl group is C 1 -C 6 .
- the alkyl portion of the alkanoyl group can be optionally substituted as described above.
- alkylcarbonyl can alternatively be used.
- alkenylcarbonyl and alkynylcarbonyl refer to an alkenyl or alkynyl group, respectively, linked to a carbonyl group.
- alkoxy refers to an alkyl group covalently linked to an oxygen atom; the alkyl group can be considered as replacing the hydrogen atom of a hydroxyl group.
- lower alkoxy refers to an alkoxy group in which the alkyl portion of the alkoxy group is C 1 -C 6 .
- the alkyl portion of the alkoxy group can be optionally substituted as described above.
- haloalkoxy refers to an alkoxy group in which the alkyl portion is substituted with one or more halo groups.
- sulfo refers to a sulfonic acid ( — SO3H) substituent.
- sulfamoyl refers to a substituent with the structure — S(O2)NH 2 , wherein the nitrogen of the NH 2 portion of the group can be optionally substituted as described above.
- carboxyl refers to a group of the structure — C(O 2 )H.
- carboxyl refers to a group of the structure — C(O 2 )NH 2 , wherein the nitrogen of the NH 2 portion of the group can be optionally substituted as described above.
- the terms “monoalkylaminoalkyl” and “dialkylaminoalkyl” refer to groups of the structure — Alk 1 -NH-Alk 2 and — Alk 1 -N(Alk 2 )( Alk 3 ), wherein Alk 1 , Alk 2 , and Alk 3 refer to alkyl groups as described above.
- alkylsulfonyl refers to a group of the structure — S(O) 2 -Alk wherein Aik refers to an alkyl group as described above.
- alkenylsulfonyl and alkynylsulfonyl refer analogously to sulfonyl groups covalently bound to alkenyl and alkynyl groups, respectively.
- arylsulfonyl refers to a group of the structure — S(O) 2 -Ar wherein Ar refers to an aryl group as described above.
- aryloxyalkylsulfonyl refers to a group of the structure — S(O) 2 -Alk-O-Ar, where Aik is an alkyl group as described above and Ar is an aryl group as described above.
- arylalkylsulfonyl refers to a group of the structure — S(O) 2 -AlkAr, where Aik is an alkyl group as described above and Ar is an aryl group as described above.
- alkyloxycarbonyl refers to an ester substituent including an alkyl group wherein the carbonyl carbon is the point of attachment to the molecule.
- An example is ethoxycarbonyl, which is CH 3 CH 2 OC(O) — .
- alkenyloxycarbonyl,” “alkynyloxycarbonyl,” and “cycloalkylcarbonyl” refer to similar ester substituents including an alkenyl group, alkenyl group, or cycloalkyl group respectively.
- aryloxycarbonyl refers to an ester substituent including an aryl group wherein the carbonyl carbon is the point of attachment to the molecule.
- aryloxyalkylcarbonyl refers to an ester substituent including an alkyl group wherein the alkyl group is itself substituted by an aryloxy group.
- the term “absent” when used in reference to a functional group or substituent, particularly in reference to the chemical structure of a compound, means that the particular functional group or substituent is not present in the compound being described.
- the absence of the substituent typically means that the bond to the substituent is absent and that absence of the bond is compensated for with a H atom.
- the absence of the position typically means that the two positions otherwise connected by the absent position are instead directly connected by a covalent bond.
- alkylidene and similar terminology refer to an alkyl group, alkenyl group, alkynyl group, or cycloalkyl group, as specified, that has two hydrogen atoms removed from a single carbon atom so that the group is double-bonded to the remainder of the structure.
- Certain compounds described herein for use in methods and compositions according to the present invention possess asymmetric carbon atoms (optical or chiral centers) or double bonds; the enantiomers, racemates, diastereomers, tautomers, geometric isomers, stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)-or (S)- or, as (D)- or (L)- for amino acids, and individual isomers are encompassed within the scope of the present invention unless specific isomers are excluded.
- the present disclosure is meant to include compounds in racemic and optically pure forms.
- Optically active (R)- and (S)-, or (D)- and (L)-isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques.
- the compounds described herein contain olefinic bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers.
- the term “tautomer,” as used herein, refers to one of two or more structural isomers that exist in equilibrium and that are readily converted from one form to another. Examples of tautomerism include, but are not limited to, keto-enol tautomerism, enamine-imine tautomerism, and lactam- lactim tautomerism. Unless one of the tautomeric alternatives is expressly excluded, all such tautomeric forms are intended to be within the scope of the invention.
- Elesclomol -dimethyl- bis(phenylcarbonothioyl)propanedihydrazide) is a novel, injectable, drug candidate that kills cancer cells by elevating oxidative stress levels beyond a breaking point, triggering programmed cell death.
- elesclomol showed potent killing of a broad range of cancer cell types at high doses, and an ability to strongly enhance the efficacy of certain chemotherapy agents, with minimal additional toxicity, at moderate doses.
- Elesclomol induces oxidative stress by provoking a buildup of reactive oxygen species within cancer cells (J.R. Kirshner et al., “Elesclomol Induces Cancer Cell Apoptosis Through Oxidative Stress,” Mol. Cancer Ther. 7: 2319-2327 (2008)). Elesclomol requires a redox-active metal ion to function.
- the Cu(ll) complex is 34 times more potent than the Ni(ll) complex and 1040-fold more potent than the Pt(ll) complex (A. A. Yadav et al., “Molecular mechanisms of the biological activity of the anticancer drug elesclomol and its complexes with Cu(ll), Ni(ll) and Pt(ll),” J.
- Elesclomol can also directly or indirectly induce apoptosis. Elesclomol can also act as an oxidative phosphorylation inhibitor (A.P. Nayak et al., “Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies against Ovarian Cancer,” Cancers (Basel) 10: 337 (2016)).
- Elesclomol is an anticancer drug that targets mitochondrial metabolism. In the past, elesclomol was recognized as an inducer of oxidative stress, but now it has also been found to suppress cancer by inducing cuproptosis. Elesclomol’s anticancer activity is determined by the dependence of cancer on mitochondrial metabolism.
- Elesclomol exhibited tremendous toxicity to all three kinds of cells. Elesclomol’s toxicity to cells is highly dependent on its transport of extracellular copper ions, a process involved in cuproptosis (P. Zheng et al., “Elesclomol, a Copper Ionophore Targeting Mitochondrial Metabolism for Cancer Therapy,” J. Exp. Clin. Cancer Res. 41 : 271 (2022)).
- elesclomol includes reference to elesclomol itself as well as derivatives and analogs of elesclomol and also salts, solvates, or prodrugs of elesclomol unless derivatives, analogs, salts, solvates, or prodrugs are clearly excluded by the context of the reference.
- Elesclomol and derivatives or analogs thereof described further below can be used to treat a number of diseases and conditions, particularly including, but not limited to, malignancies.
- OEC ovarian epithelial cancer
- Type I OEC low-grade serous, endometrial, mucinous and ovarian clear-cell carcinoma (OCCC)
- OCCC clear-cell carcinoma
- OCCC and other Type I OEC as well as endometrial and uterine tumors are characterized by high prevalence of ARID1A mutations, which are mutually exclusive to BRCA and TP53 mutations commonly seen in Type II OEC subtypes such as HGSOC (high grade serous ovarian cancer).
- ARID1A mutations result in cells that are highly susceptible to synthetic lethality by agents that interrupt OXPHOS or the mitochondrial electron transport chain (mETC).
- Elesclomol is an agent that may: (a) have high specificity for cells expressing ARID1 A mutations; and (b) act against a specific protein target within the mETC, thereby providing a potential novel therapeutic approach for OCCC and other tumors expressing ARID1A mutations.
- OECs can be generally divided into two types: Type I and Type II.
- Type I tumors are low-grade serous, endometrioid, mucinous, or clear-cell and typically, have the mass confined to the ovary. They are slower growing, not driven by p53 or BRCA mutations, and are often associated with endometriosis.
- Type II tumors are high-grade serous ovarian carcinomas that are more aggressive and frequently present at a later stage. These tumors have often spread to other tissues in the peritoneal cavity by the time they are diagnosed. Typically, these tumors express TP53 and/or BRCA mutations.
- Elesclomol may also provide benefit to Type II OEC patients who have become resistant to PARP inhibitors.
- HGSOC accounts for 68% of ovarian cancer diagnoses. Platinum-based therapy is frequently administered to treat these malignancies; disease control rates as high as 81 .7% have been observed, with 27% complete responses and 45% partial responses seen in one study. Mutations in the tumor suppressor genes BRCA1 and BRCA2 have been associated with an increased risk for the development of breast and ovarian cancer.
- PARP poly(ADP-ribose) polymerase
- OCCC is the second most common OEC subtype and the second leading cause of death from ovarian cancer. OCCC represents up to 10% of all OEC cases in North America with an increased prevalence in Asian populations, as high as 25% in Japan and 50% in Thailand.
- OCCC Compared to HGSOC, treatment for OCCC has not benefited from recent advances in treatment strategies. [0128] OCCC is often detected at an early stage, presenting with a large unilateral pelvic mass confined to the ovary and causing symptoms of abdominal pain and distortion. Despite the earlier diagnosis, OCCC exhibits poorer survival compared to HGSOC across all FIGO stages. When diagnosed at an advanced stage, OCCC exhibits the worst prognosis and lowest survival rates among all OEC subtypes.
- PFS progression-free survival
- OS overall survival
- OCCC is highly resistant to chemotherapy. In one study, only 11.1 % responded to platinum- based chemotherapy with median overall survival less than 18 months, compared to a response rate of 72.5% for advanced HGSOC patients in the same study. For the minority of OCCC patients who initially responded, less than 9% responded to retreatment with platinum-based chemotherapy following disease recurrence.
- OCCC patients generally do not have BRCA mutations and thus are not considered to be within the treatment scope of recent FDA-approved PARP inhibitors. Therefore, at present, the extremely poor prognosis and lack of an effective systemic therapy for OCCC remains a significant unmet medical need, especially when surgical cytoreduction is insufficient.
- ARID1A is the most highly mutated gene with PIK3CA mutations the second most prevalent.
- Loss of ARID1 A frequently co-exists with PIK3CA leading to hypothesis that they cooperate to promote tumor growth, possibly via induction of IL- 6 and downstream JAK/STAT3 signaling, which has a prominent role in tumor cell growth and differentiation (Chandler 2015).
- the coexistence of these two mutations has been reported in gastric cancer and other cancers; however, the rate of co- occurrence reported to date is highest in OCCC.
- PI3KCA is an important component of AKT/mTOR signaling pathways commonly altered in OCCC
- mTOR gained interest as a potentially promising target for OCCC therapy.
- a 90-patient clinical trial of the mTOR inhibitor temsirolimus failed to demonstrate an improvement in outcomes compared to historical controls in patients with Stage lll-IV OCCC.
- the gene ARID1A is a haplo-insufficient tumor suppressor gene associated with regulation of DNA damage repair (DDR) processes, as well as the SWI/SNF (Switch-Sucrose Non-fermenting) pathway, which is involved in sugar (sucrose) metabolism.
- DDR DNA damage repair
- SWI/SNF Switch-Sucrose Non-fermenting pathway
- the loss of ARID1 A function is thought to enhance cell cycle progression and the formation of cancer; however, therapeutic strategies targeting ARID1A mutant tumors are limited at this time.
- ARID1A is frequently mutated in liver, breast and gastric cancer, but the highest mutation frequency is in gynecologic cancers (Kwan, 2016), particularly in OCCC where mutations are observed in up to 56% of cases (Jones, 2010).
- ARID1A mutations are less common in HGSOC where they are largely mutually exclusive to highly prominent mutations in BRCA and TP53 (Hu, 2018). The mutual exclusivity between ARID1A and TP53 mutations has also been observed in several other major cancer types such as prostate, stomach, colon and uterine cancers (Wu, 2017).
- ARID1A mutations are associated with treatment resistance and reduced sensitivity to paclitaxel, cisplatin and PARP inhibitors (Hu, Clovis, 2018). Mutations resulting in loss of ARID1 A function leads to activation of PI3K-AKT pathway. AKT activates the anti-apoptotic pathway by phosphorylating downstream targets to inhibit IKB and release NF- K B which activates genes promoting cellular survival and conferring resistance to platinum-based regimens (Lyu, 2016).
- ARID1 A loss also correlates with poor clinical outcomes in HGSOC cases receiving Pt-based chemotherapy regimens or PARP inhibitor treatment. Tumors accumulate ARID1A mutations in response to treatment as a mechanism of acquired resistance (Berns, 2016). Progression free survival in patients exhibiting an ARID1A- deficient phenotype is significantly reduced as compared with ARID1 A wild-type in patients treated with PARP inhibitors. These results are shown in Figure 3.
- Figure 3(A) shows the impact of ARID1A loss, determined via mutation, homozygous deletion, or loss of expression, and survival in TCGA serous ovarian cancers.
- Figure 3(B) shows the progression-free survival of relapsed, platinum-sensitive, high-grade ovarian carcinomas in a clinical trial of rucaparib (a PARP inhibitor), stratified based on ARID1A mutation status.
- ARID1 A The activity of ARID1 A is essential for establishing an open chromatin state upon DNA damage and facilitating binding of TOPO-2 to DNA (Park, 2019).
- Cells with mutated or downregulated ARID1 A exhibit compromised homologous (HR) and non-homologous end-joining (NHEJ) DNA repair mechanisms and are resistant to treatment with single-agent PARP inhibitors.
- ARID1 A /_ cells are deficient in NHEJ repair resulting in a partial cytotoxic response to radiation. Synthetic lethality is activated to synergistically potentiate cytotoxicity in ARID1A /_ cells when PARP inhibitors are combined with DNA damaging agents such as XRT (Yakovlev, 2019).
- ARID1A encodes SLC7A11 , a SWI/SNF chromatin-remodeling factor that is frequently mutated in various cancers. Cancers with SWI/SNF mutations tend to be aggressive in nature and have a poor prognosis (Kwan, 2015). ARID1A loss leads to downregulation of multiple genes involved in oxidative reduction . ARID1 A maintains GSH homeostasis by enhancing SLC7A11 transcription. ARID1 A-deficient cells exhibit impaired expression of the cysteine transporter SLC7A11 , which is a critical component in the antioxidant glutathione (GSH) pathway and the glutamate-cysteine ligase synthetase catalytic subunit (GCLC). Loss of ARID1A prevents cystine uptake leading to increased reactive oxygen species (ROS) in the cell. Accumulation of ROS contributes to oncogenesis through increased mitotic signaling and cell growth (Kwan, 2016).
- ROS reactive oxygen species
- Figure 5 is a graph showing that loss of ARID1A results in downregulation of cystine transporter SLC7A11 and impairment of GSH pathway leading to ROS accumulation and cell death:
- A ARID1A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway.
- B Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
- ROS can have pro-survival or pro-death effects depending on amount of ROS and effectiveness of the antioxidant system resident in the cell.
- Cancer cells show persistently high levels of reactive oxygen species (ROS) due to oncogenic transformation including alteration in genetic, metabolic, and tumor microenvironment. Higher baseline levels of ROS in tumor versus normal cells are known to contribute to the development and/or maintenance of the malignant phenotype and render cancer cells more vulnerable to irreversible oxidative damage and consequent cell death.
- ROS reactive oxygen species
- ROS leads to activation of the nuclear factor NRF2, which binds within the Xc promoter region and activates transcription increasing the uptake of cystine and expression of GSH contributing to the stability of cancer cells in their naturally high-ROS environment.
- GSH is an antioxidant involved in scavenging ROS and detoxifying the cell to promote cancer progression by avoiding activation of cell-death signaling pathways.
- GSH depletion has been largely unsuccessful as a therapeutic strategy.
- cells with GSH abnormalities could be susceptible to synthetic lethality.
- Conditions of oxidative stress e.g. high ROS
- GSSG accumulation can be potentially toxic and can result in activation of redox mediated cell death (Couto, 2016) and therefore is rapidly converted back to the reduced form (GSH) by NADPH highlighting the dependence on oxidative phosphorylation.
- Cells with deficiency in ARID1 A have reduced GSH due to downregulation of SLC7A11 and related reduction in the GSH precursor cystine thereby rendering cancer cells highly dependent on oxidative phosphorylation and susceptible to agents that may tip the balance toward redox- induced apoptosis while sparing normal cells.
- ARID1A mutated cells have been demonstrated to be significantly more sensitive to inhibition of OXPHOS versus wild- type cells (Emmings, 2019).
- Figure 6 shows the difference between cells with ARID1A mutations and cells with active ARID1 A in terms of the effect of accumulation of ROS:
- A ARID1 A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway.
- B Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
- Figure 7 shows a therapeutic window for cancer cells in the presence of an oxidative phosphorylation (OXPHOS) inhibitor.
- OXPHOS oxidative phosphorylation
- Top Panel Normal body cells have moderate ATP demand and adequate levels of oxygen and glucose and survive in presence of OXPHOS inhibitors by upregulating glycolysis to meet their ATP demands.
- Middle Panel Highly proliferating cancer cells have extraordinarily high ATP demand and adequate levels of oxygen and glucose. Despite glycolytic pathway upregulation, OXPHOS inhibition results in failure to meet ATP demand and cell death.
- Quiescent cancer cells have low ATP demand but live in a highly compromised microenvironment (low glucose and hypoxia). Inhibition of OXPHOS is lethal as insufficient glucose is present to compensate for the loss of ATP production by oxidative phosphorylation.
- elesclomol shown above as Formula (I) is a small molecule drug candidate previously studied extensively in clinical trials as an anti- cancer agent as a single agent and in combination with the tubulin inhibitor paclitaxel. Its safety and pharmacokinetics have been established.
- Elesclomol was initially believed not to be active as a single agent based on mouse xenograft data; however, it was later observed that the drug strongly binds copper and has no activity in the absence of bound copper (Blackman, 2012). Upon chelation with copper in the blood stream, elesclomol undergoes a conformational change allowing the drug to permeate cellular and sub-cellular membranes. As stated above, elesclomol can also bind other divalent metal ions. This conformational change is shown in Figure 8.
- elesclomol induces apoptosis in cancer cells through a non-specific induction of oxidative stress, possibly through the generation of reactive oxygen species (ROS) due to cycling of Cu( 11 ) ⁇ Cu( I ).
- ROS reactive oxygen species
- comparison of elesclomol to over 3000 drug profiles demonstrated that the elesclomol profile was unique suggesting that elesclomol acts by a novel mechanism not shared by other tested compounds. Inhibition of activity of genes associated with antioxidant activity and protection against ROS and upregulation of TP53-regulated proapoptotic signaling was observed.
- LDH lactate dehydrogenase
- Elesclomol-Cu directly binds and inhibits FDX-1 function to block iron- sulfur cluster formation in complex I, a critical component of the mitochondrial electron transport chain. Further, loss of the FDX1 protein confers resistance to elesclomol consistent with the direct inhibition of FDX1 function by elesclomol.
- FIG. 10 The results of these gene screens are shown in Figure 10.
- Iron-sulfur clusters play a critical role in the oxidation-reduction reactions of electron transport in mitochondria relied on by cancer cells that have made an adaptive shift from glycolysis to high mitochondrial dependence (oxidative phosphorylation).
- Complex I plays a role in redox control and the biosynthesis of macromolecules and nucleic acids necessary for cell proliferation. It is suggested that these complex l-dependent events contribute to tumor formation, resistance to cell death, and metastasis of cancer cells in part by causing an increase in ROS levels.
- FIG. 11 shows that elesclomol inhibits the natural function of FDX1 in FE-S cluster biosynthesis
- the [2Fe-2S] cluster in FDX1 is indicated by spheres
- the mitochondrial ISC core complex catalyzes the conversion of cysteine to alanine and generates S° for iron-sulfur cluster assembly. S° is reduced by FDX1 .
- a [2Fe-2S] cluster is subsequently formed on the scaffold protein ISCII.
- Inhibitors of mitochondrial ETC complex I have been shown to increase mitochondrial ROS production and, consequently, to induce cell death via apoptosis. Recently, Fe-S clusters have emerged as an area of interest as potential therapeutic targets. Because Fe-S clusters are particularly sensitive to ROS, the represent key targets of oxidative stress.
- Tumors such as OCCC with high prevalence of inactivating mutations in the ARID1A subunit of SWI/SNF complex may represent an ideal biomarker-derived population for targeted therapy with elesclomol. These tumors would have a profound dependency on OXPHOS compared to wild type cells revealing a valuable therapeutic target for elesclomol-driven synthetic lethality in an area of significant unmet medical need.
- elesclomol exhibits the highest differentiation in activity in an in vitro screen of drugs observed to have a significantly lower IC 5 0 in ARID1 A-mutated cells versus wild-type as shown in Table 1 .
- Figure 12 shows that sensitivity to elesclomol as a single agent is highly specific to ARID1A mutation in cancer cell lines.
- A Cell growth of endometrial and ovarian cancer cell lines treated with elesclomol for 72 h as measured using the WST-1 assay.
- B IC 5 0 values of elesclomol in these cell lines.
- FIG. 13 is a diagram showing that ARIDIA-mutant tumors are highly dependent on OXPHOS offering a potential strategy for synthetic lethality employing targeted agents interrupting mitochondrial metabolism.
- Figure 14 is a diagram showing that the protein target of elesclomol is ferredoxin-1 , a key component of the OXPHOS pathway establishing a synthetically lethal therapeutic strategy in ARIDIA-mutant tumors.
- a previous single agent study was a dose escalation design evaluating a starting dose of 200 mg/m 2 elesclomol via a 60-m inute infusion once weekly in a four- week cycle.
- Six patients were accrued and a dose level of 400 mg/m 2 was achieved without observation of dose limiting toxicity (DLT). No adverse events higher than Grade 1 related to the study drug were observed. at this dose.
- DLT dose limiting toxicity
- Pharmacokinetic studies were not conducted in this single-agent Phase I trial. It was suggested that dosages higher than 400 mg/m 2 will be well tolerated.
- Figure 15 shows the occurrence of ARID1 A mutations in a number of types of malignancies, including mutations, deletions, amplification, and multiple alterations.
- ARID1A loss is also associated with treatment resistance and poor patient outcomes in a range of tumors including HER2 + breast cancer. Recently published findings suggest that tumors accumulate ARID1A mutations in response to treatment as a mechanism of acquired resistance. Such tumors may be susceptible to synthetic lethality treatment strategies with elesclomol.
- elesclomol can be employed as either the free acid or as a salt, typically a sodium salt. However, also, as detailed further below, elesclomol is typically employed as a coordinate covalent complex with a divalent transition state metal ion.
- Elesclomol is one of a number of therapeutic agents that is considered an agent that has previously been considered one of suboptimal importance.
- the present invention describes novel improvements, pharmaceutical ingredients, dosage forms, excipients, solvents, diluents, drug delivery systems, preservatives, more accurate drug administrations, improved dose determination and schedules, toxicity monitoring and amelioration, techniques or agents to circumvent or reduce toxicity, and techniques and tools to identify/predict those patients who might have a better outcome with a therapeutic agent by the use of phenotype or genotype determination through the use of diagnostic kits or pharmacokinetic or metabolism monitoring approaches.
- the present invention also relates to the use of drug delivery systems, novel prodrugs, polymer conjugates, novel routes of administration, other agents to potentiate the activity of the compounds or inhibit the repair of suboptimal cellular effects or sublethal damage or to “push” the cell into more destructive cellular phases such as immune stimulation and apoptosis.
- these suboptimal therapeutics in conjunction with radiation or other conventional chemotherapeutic agents or biotherapeutic agents such as antibodies, vaccines, cytokines, lymphokines, gene and antisense RNA therapies, as detailed further below, would provide novel approaches and potential significant treatment improvement.
- the term suboptimal therapy includes agents where Phase I toxicity precluded further human clinical evaluation.
- suboptimal therapy includes those agents, the subject of Phase III clinical trials the outcome of which was either medically or statistically not significant to warrant regulatory submission or approval by government agencies for commercialization for commercialized agents whose clinical performance (i.e. response rates) as a monotherapy are less than 25%, or whose side effects are severe enough to limit wide utility.
- Agents with suboptimal clinical activity include but are not limited to the following: small chemical therapeutics, natural products, biologies such as peptide, protein antibody drug conjugates, or vaccines, including cell based therapies. More specifically, the inventive methods and compositions also focus on improvements for redox modulating agents including elesclomol and derivatives or analogs of elesclomol, as well as salts, solvates, or prodrugs of elesclomol.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the time that the compound is administered, the use of dose-modifying agents that control the rate of metabolism of the compound, normal tissue protective agents, or other relevant factors affecting dosages.
- General examples include: variations of infusion schedules (e.g., bolus i.v.
- lymphokines e.g., G-CSF, GM-CSF, EPO
- rescue agents such as leucovorin for 5-Fll or thiosulfate for cisplatin treatment.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: i.v.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the route that the compound is administered.
- General examples include: changing the route of administration from oral to intravenous administration and vice versa, or the use of specialized routes such as subcutaneous, intramuscular, intraarterial, intraperitoneal, intralesional, intralymphatic, intratumoral, intrathecal, intravesicular, or intracranial administration.
- redox modulating agent compounds such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: topical administration; intravesicular administration for bladder cancer; oral administration; slow release oral delivery; intrathecal administration; intraarterial administration; continuous infusion; intermittent infusion, administration via large volume oral solution; buccal administration; or rectal administration.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the time that the compound is administered.
- General examples include: changing from a monthly administration to a weekly or daily dosing or variations of the schedule.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: daily; weekly for three weeks; weekly for two weeks; biweekly; biweekly for three weeks with a 1-2 week rest period; intermittent boost dose administration; daily for one week then once per week for multiple weeks; daily on days 1-5, 8-12 every three weeks; or daily on days 1-3, 8-11 per cycle.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the types of disease, clinical stage of disease that the compound is administered.
- General examples include: the use of solid tumor agents for leukemias and vice versa, the use of antitumor agents for the treatment of benign hyperproliferative disease such as psoriasis or benign prostate hypertrophy.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- leukemias acute and chronic, AML, ALL, CLL, CML
- MDS myelodysplastic syndrome
- angiogenic diseases or conditions benign prostate hypertrophy
- psoriasis gout
- autoimmune conditions prevention of transplantation rejection
- mycosis fungoides use in bone marrow transplantation; as an antiinfective; treatment for AIDS; treatment for lymphoma; mantle cell lymphoma; meningeal leukemia; malignant meningitis; cutaneous T cell lymphoma; Barrett’s esophagus; anaplastic gliomas; triple negative breast cancer; Braf- mutated melanoma; BTK resistant CLL; chordoma; Kras-mutated colon cancer; pediatric tumors including brain and s
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the stage of disease at diagnosis/progression that the compound is administered.
- General examples include: the use of chemotherapy for non-resectable local disease, prophylactic use to prevent metastatic spread or inhibit disease progression or conversion to more malignant stages.
- redox modulating agent compounds such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use for the treatment of localized polyp stage colon cancer; leukoplakia in the oral cavity; angiogenesis inhibition to prevent or limit metastatic spread; use against HIV with AZT, DDI, or reverse transcriptase inhibitors.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by using the compound for non-malignant diseases and conditions.
- General examples include: premalignant conditions, benign hyperproliferative conditions, treatment of infections, parasites, usage to relieve pain, control of pleural effusions.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the type of patient that would best tolerate or benefit from the use of the compound.
- General examples include: use of pediatric doses for elderly patients, altered doses for obese patients; exploitation of co-morbid disease conditions such as diabetes, cirrhosis, or other conditions that may uniquely exploit a feature of the compound.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- patients with disease conditions with high levels of metabolic enzymes, reactive oxygen species, histone deacetylase, protein kinases, or ornithine decarboxylase include: patients with disease conditions with high levels of metabolic enzymes, reactive oxygen species, histone deacetylase, protein kinases, or ornithine decarboxylase; patients with disease conditions with low levels of metabolic enzymes, histone deacetylase, protein kinases, or ornithine decarboxylase; patients with low or high susceptibility to thrombocytopenia or neutropenia; patients intolerant of Gl toxicities; patients with deficiencies in DNA repair capacity including BRCA, ARID1 A or other deficiencies in the SWI/SWF pathway or mitochondrial electron transport, over- or under- expression of jun, GPCR’s and signal transduction proteins, VEGF, prostate specific genes, protein
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by more precise identification of a patient’s ability to tolerate, metabolize and exploit the use of the compound.
- General examples include: use of diagnostic tools and kits to better characterize a patients ability to process/metabolize a chemotherapeutic agent or their susceptibility to toxicity caused by potential specialized cellular, metabolic, organ system phenotypes.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- the gene ARID1A is a member of the SWI/SNF family, whose members have helicase and ATPase activities and are thought to regulate transcription of certain genes by altering the chromatin structure around those genes.
- the encoded protein is part of the large ATP-dependent chromatin remodeling complex SWI/SNF, which is required for transcriptional activation of genes normally repressed by chromatin. It possesses at least two conserved domains that could be important for its function. First, it has an ARID domain, which is a DNA-binding domain that can specifically bind an AT-rich DNA sequence known to be recognized by a SWI/SNF complex at the beta- globin locus.
- the C-terminus of the protein can stimulate glucocorticoid receptor-dependent transcriptional activation. It is thought that the protein encoded by this gene confers specificity to the SWI/SNF complex and may recruit the complex to its targets through either protein-DNA or protein-protein interactions. Two transcript variants encoding different isoforms have been found for this gene. This gene has been commonly found mutated in gastric cancers, ovarian clear cell carcinoma, and pancreatic cancer. In breast cancer distant metastases acquire inactivation mutations in ARID1A not seen in the primary tumor, and reduced ARID1 A expression confers resistance to different drugs such as trastuzumab and mTOR inhibitors.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by testing and analyzing a patient’s genotype for unique features that may be of value to predict efficacy, toxicity, metabolism, or other factors involving efficacy.
- General examples include: biopsy samples of tumors or normal tissues (e.g., white blood cells) may also be taken and analyzed to specifically tailor or monitor the use of a particular drug against a gene target, unique tumor gene expression pattern, SNP’s (single nucleotide polymorphisms), to enhance efficacy or to avoid particular drug-sensitive normal tissue toxicities.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- diagnostic tools, techniques, kits and assays to confirm a patient’s particular genotype gene/protein expression chips and analysis; single nucleotide polymorphism (SNP) assessment; SNP’s for histone deacetylase, ornithine decarboxylase, genes affecting S-adenosyl methionine metabolism, GPCR’s, protein kinases, telomerase, jun; identification and measurement of metabolism enzymes and metabolites, mutations in wild type and mutated genes, epigenetics via methylation and acetylation, ARID1A mutation, ARID1 A deficiency, mTor signaling, PI3K-AKT activating mutation, PARPi resistance, HR deficiency, DDR deficiency, SWI/SNF pathway alteration, P53 status/mutation, BRCA
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by specialized preparation of a patient prior to or after the use of a therapeutic agent.
- General examples include: induction or inhibition of metabolizing enzymes, specific protection of sensitive normal tissues or organ systems.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: use of colchicine or analogs; use of diuretics such as probenecid; use of uricase; non-oral use of nicotinamide; use of sustained release forms of nicotinamide; use of inhibitors of polyADP ribose polymerase; use of caffeine; leucovorin rescue; infection control; use of antihypertensives; alteration of stem cell populations; pretreatment to limit or prevent graft vs. host (GVH) cytokine storm reactions; use of anti-inflammatories; anaphylactic reaction suppression.
- GVH graft vs. host
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by use of additional drugs or procedures to prevent or reduce potential side-effects or toxicities.
- General examples include: the use of anti-emetics, anti-nausea, hematological support agents to limit or prevent neutropenia, anemia, thrombocytopenia, vitamins, antidepressants, treatments for sexual dysfunction, or other agents or regimens known in the art.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of monitoring drug levels after dosing in an effort to maximize a patient’s drug plasma level, to monitor the generation of toxic metabolites, monitoring of ancillary medicines that could be beneficial or harmful in terms of drug- drug interactions.
- General examples include: the monitoring of drug plasma protein binding.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: multiple determinations of drug plasma levels; multiple determinations of metabolites in the blood or urine, measurement of polyamines, LAT-1 surface receptors, gene sequencing, immune effectors.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting unique drug combinations that may provide a more than additive or synergistic improvement in efficacy or side-effect management.
- General examples include: alkylating agents with anti-metabolites, topoisomerase inhibitors with antitubulin agents.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting them as chemopotentiators where minimal therapeutic activity is observed alone but in combination with other therapeutics a more than additive or synergistic improvement in efficacy is observed.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- General examples include: amonafide with cisplatin or 5-Fll.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by drugs, treatments and diagnostics to allow for the maximum benefit to patients treated with a compound.
- General examples include: pain management, nutritional support, anti-emetics, anti-nausea therapies, anti-anemia therapy, anti-inflammatories.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: use with therapies associated with pain management; nutritional support; anti- emetics; anti-nausea therapies; anti-anemia therapy; anti-inflammatories: antipyretics; immune stimulants; anti diarrhea medicines, famotidine, antihistamines, suppository lubricants, soothing agents, lidocaine, hydrocortisone.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of therapeutics or methods to enhance effectiveness or reduce side effects.
- General examples include: herbal medications and extracts.
- NF- K B inhibitors such as parthenolide, curcumin, rosmarinic acid
- natural antiinflammatories including rhein, parthenolide
- immunostimulants such as those found in Echinacea
- antimicrobials such as berberine
- flavonoids and flavones such as apigenenin, genistein
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the pharmaceutical bulk substance.
- General examples include: salt formation, homogeneous crystalline structure, pure isomers.
- Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: salt formation; homogeneous crystalline structure; pure isomers; increased purity; lower residual solvents and heavy metals.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the diluents used to solubilize and deliver/present the compound for administration.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- General examples include: Cremophor-EL, cyclodextrins for poorly water soluble compounds.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: use of emulsions; DMSO; NMF; DMF; DMA; ethanol; benzyl alcohol; dextrose-containing water for injection; Cremophor; cyclodextrins; PEG; a sweetening agent such as saccharin; agents to thicken an oral dosage form such as glycerol; taste masking effectors such as menthol, rum flavor, fruit flavorings.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the solvents used or required to solubilize a compound for administration or for further dilution.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- General examples include: ethanol, dimethylacetamide (DMA).
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: the use of emulsions; dimethyl sulfoxide (DMSO); N-methylformamide (NMF); dimethylformamide (DMF); dimethylacetamide (DMA); ethanol; benzyl alcohol; dextrose-containing water for injection; Cremophor; PEG; glycerol, cocoa butter for suppositories.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the materials/excipients, buffering agents, preservatives required to stabilize and present a chemical compound for proper administration.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- General examples include: mannitol, albumin, EDTA, sodium bisulfite, benzyl alcohol.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: the use of mannitol; albumin; EDTA; sodium bisulfite; benzyl alcohol; carbonate buffers; phosphate buffers; glycerol; sweeteners; taste masking agents such as rum flavor, menthol; substituted celluloses; sodium azide as a preservative.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the potential dosage forms of the compound dependent on the route of administration, duration of effect, plasma levels required, exposure to side effects for normal tissues and metabolizing enzymes.
- General examples include: tablets, capsules, topical gels, creams, patches, suppositories, oral solutions, suspensions, syrups.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the dosage forms, container/closure systems, accuracy of mixing and dosage preparation and presentation.
- General examples include: amber vials to protect from light, stoppers with specialized coatings.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: the use of amber vials to protect from light; stoppers with specialized coatings to improve shelf-life stability; special dropper measuring devices; single-use or multiple-use container closure systems; testing for allergies; suppository delivery devices, epinephrine pens for side effect management; physician and nurse assistance gloves; dosage measuring devices.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of delivery systems to improve the potential attributes of a pharmaceutical product such as convenience, duration of effect, reduction of toxicities.
- General examples include: nanocrystals, bioerodible polymers, liposomes, slow release injectable gels, microspheres.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: the use of nanocrystals; bioerodible polymers; liposomes; slow release injectable gels; microspheres; suspensions with glycerol, meltable drug release suppositories with polymers such as cocoa butter alone or in combination with PEG, lecithin; polylactide/polyglycolide; rectal plugs for drug delivery.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the parent molecule with covalent, ionic, or hydrogen bonded moieties to alter the efficacy, toxicity, pharmacokinetics, metabolism, or route of administration.
- General examples include: polymer systems such as polyethylene glycols, polylactides, polyglycolides, amino acids, peptides, multivalent linkers.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- polymer systems such as polyethylene glycols; polylactides; polyglycolides; amino acids; peptides; multivalent linkers.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the parent structure of a molecule with additional chemical functionalities that may alter efficacy or reduce toxicity, pharmacological performance, route of administration, or other factors that alter efficacy or reduce toxicity.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- additional chemical functionalities that may alter efficacy or reduce toxicity, pharmacological performance, route of administration, or other factors that alter efficacy or reduce toxicity.
- General examples include: alteration of side chains to increase or decrease lipophilicity, additional chemical functionalities to alter reactivity, electron affinity, binding capacity, salt forms.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: alteration of side chains to increase or decrease lipophilicity; additional chemical functionalities to alter reactivity; alteration of electron affinity; alteration of binding capacity; salt forms.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the molecule such that improved pharmaceutical performance is gained with a variant of the active molecule in that after introduction into the body a portion of the molecule is cleaved to reveal the preferred active molecule.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- General examples include: enzyme sensitive esters, dimers, Schiff bases.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of additional compounds, biological agents that when administered in the proper fashion, a unique and beneficial effect can be realized.
- General examples include: inhibitors of multi-drug resistance, specific drug resistance inhibitors, specific inhibitors of selective enzymes, signal transduction inhibitors, repair inhibition.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of inhibitors of multi-drug resistance; specific drug resistance inhibitors; specific inhibitors of selective enzymes; signal transduction inhibitors; repair inhibition; topoisomerase inhibitors with non-overlapping side effects.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by its use in combination as sensitizers/potentiators with biological response modifiers.
- General examples include: use in combination as sensitizers/potentiators with biological response modifiers, cytokines, lymphokines, therapeutic antibodies, antisense therapies, gene therapies.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting their selective use to overcome developing or complete resistance to the efficient use of biotherapeutics.
- General examples include: tumors resistant to the effects of biological response modifiers, cytokines, lymphokines, therapeutic antibodies, antisense therapies, gene therapies.
- redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol
- redox modulating agents include: the use against tumors resistant to the effects of biological response modifiers; cytokines; lymphokines; therapeutic antibodies; antisense therapies such as bevacizumab, trastuzumab, rituximab, and cetuximab; gene therapies; ribozymes; RNA interference.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting their use in combination with ionizing radiation, phototherapies, heat therapies, radio-frequency generated therapies.
- General examples include: hypoxic cell sensitizers, radiation sensitizers/protectors, photosensitizers, radiation repair inhibitors.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by optimizing their utility by determining the various mechanisms of actions, biological targets of a compound for greater understanding and precision to better exploit the utility of the molecule.
- General examples include: Gleevec for chronic myelocytic leukemia (CML), arsenic trioxide for acute promyelocytic leukemia (APL), retinoic acid for APL.
- Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by more precise identification and exposure of the compound to those select cell populations where the compounds effect can be maximally exploited.
- General examples include: tirapazamine and mitomycin c for hypoxic cells, vinca alkaloids for cells entering mitosis.
- Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use against radiation sensitive cells; radiation resistant cells; energy depleted cells; endothelial cells.
- Another aspect of the present invention is use of elesclomol or a derivative or analog of elesclomol to reverse resistance to platinum-containing anti- neoplastic agents or PARP inhibitor anti-neoplastic agents, particularly resistance modulated by mutations in ARID1A. Suitable platinum-containing and PARP inhibitor anti-neoplastic agents are described below. [0246] Use to Induce Synthetic Lethality
- Yet another aspect of the present invention is induction of synthetic lethality by administration of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1 A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol renders the malignant cells susceptible to a targeted agent that otherwise would have no effect.
- an onco-driving mutation that allows for tumor formation such as a mutation causing a defect in DNA damage repair, promotion of an open chromosome, or enhanced transcription, can also create an “Achilles heel” for the same tumor cells by its very existence.
- the mutation in ARID1 A leads to tumor formation, but also leads to a dependence on OXPHOS — the Achilles heel — as such tumor cells are uniquely susceptible to mitochondrial targeting agents, such as, but not necessarily limited to, elesclomol or a derivative or analog of elesclomol, which targets ferredoxin-1 , because of the defect that allows the tumor to form in the first place. If the tumor-causing mutation had not formed, such mitochondrial-targeting agents would have had minimal or no activity.
- mitochondrial targeting agents such as, but not necessarily limited to, elesclomol or a derivative or analog of elesclomol, which targets ferredoxin-1 , because of the defect that allows the tumor to form in the first place. If the tumor-causing mutation had not formed, such mitochondrial-targeting agents would have had minimal or no activity.
- Yet another aspect of the present invention is use of elesclomol or a derivative or analog of elesclomol to target ferredoxin-l (FDX1 ) in order to inhibit oxidative phosphorylation (OXPHOS) in circumstances in which inhibition of oxidative phosphorylation would be desirable.
- FDX1 target ferredoxin-l
- OXPHOS oxidative phosphorylation
- transition metal cation refers to a cation of a metal in Groups 3-12 of the Periodic Table.
- the transition metal cation is preferably divalent, such as, but not limited to, Ni 2+ , Cu 2+ , Co 2+ , Fe 2+ , Zn 2+ , Pt 2+ , Pd 2+ , V 4+ , V 5+ , Cr 2+ , Cr 3+ , Cr 4+ , Mn 2+ , Mn 3+ , Mn 4+ , or Mn 5+ .
- the transition metal cation is preferably divalent, such as, but not limited to, Ni 2+ , Cu 2+ , Co 2+ , Fe 2+ , Zn 2+ , Pt 2+ , and Pd 2+ . More preferably, the divalent transition metal cation is Cu 2+ or Ni +2 . Still more preferably, the divalent transition metal cation is Cu 2+ .
- the molar ratio of elesclomol to the transition metal cation is equal to or greater than 0.5 and equal to or less than 2.0; typically, the molar ratio of elesclomol to the transition metal cation is 1.0.
- Analogs and derivatives of elesclomol include, but are not limited to, compounds of Formula (IV): (IV), wherein:
- R 1 is a substituted or unsubstituted aliphatic group or a substituted or unsubstituted non-aromatic heterocyclic group
- R 2 , R 3 , and R 4 are independently hydrogen, a substituted or unsubstituted aliphatic group, a substituted or unsubstituted non-aromatic heterocyclic group, a substituted or unsubstituted aryl group, or R 1 and R 3 and/or R 2 and R 4 taken together with the carbon and nitrogen atoms to which they are bonded, form a non-aromatic heterocyclic ring optionally fused to an aromatic ring;
- R 5 and R 6 are independently hydrogen, a substituted or unsubstituted aliphatic group, or a substituted or unsubstituted non-aromatic heterocyclic group;
- R 7 and R 8 are independently hydrogen or a substituted or unsubstituted aliphatic group, or R 7 is hydrogen and R 8 is a substituted or unsubstituted aryl group, or R 7 and R 8 taken together are C 2 -C 6 substituted or unsubstituted alkylene group;
- Y is a covalent bond or - C(R 7 R 8 ) ⁇ .
- a particular derivative or analog of elesclomol is a compound of Formula (V):
- each Z is independently S, 0, or Se, provided that both Z moieties cannot be 0;
- R 1 and R 2 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted alkynyl, an optionally substituted cycloalkyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group wherein the heterocyclic group is bonded to the thiocarbonyl via a carbon-carbon linkage, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to seven-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl wherein the heteroaryl is bonded to the thiocarbonyl via a carbon- carbon linkage, --NR 12 R 13 , --OR 14 , SR 14 , and S(O)pR 15 ; (3) R 3 and R 4 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally
- R 6 and R 7 are both hydrogen or an optionally substituted lower alkyl
- R 8 is selected from the group consisting of hydroxyl, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxylalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic aryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, — N10R 11 , and -COR 9 ;
- R 9 is an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted alkyl, an optionally substituted cycloalkyl, or an optionally substituted heterocyclic group;
- R 10 and R 11 are each independently selected from the group consisting of hydrogen, hydroxyl, alkylamino, dialkylamino, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxyalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, and - COR 9 ; or R 10 and R 11 , taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group; (9) R 12 , R 11
- R 15 is an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl
- elesclomol includes all coordinate covalent complexes of elesclomol.
- one aspect of the present invention is a method for treating a malignancy comprising the step of administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug thereof to a subject with a malignancy to treat the malignancy.
- the therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug thereof is a therapeutically effective quantity of elesclomol.
- the therapeutically effective quantity of elesclomol is administered in a coordinate-covalent complex with a transition metal selected from the group consisting of Ni 2+ , Cu + , Cu 2+ , Co 2+ , Co 3+ , Fe 2+ , Fe 3+ , Zn 2+ , Pt 2+ , Pd 2+ , V 4+ , V 5+ , Cr 2+ , Cr 3+ , Cr 4+ , Mn 2+ , Mn 3+ , Mn 4+ , and Mn 5+ .
- the transition metal is selected from the group consisting of Ni 2+ , Cu 2+ , Co 2+ , Fe 2+ , Zn 2+ , Pt 2+ , and Pd 2+ . More preferably, the transition metal is selected from the group consisting of Ni 2+ and Cu 2+ . Most preferably, the transition metal is Cu +2 .
- the molar ratio of elesclomol to the transition metal cation is equal to or greater than 0.5 and equal to or less than 2.0; preferably, the molar ratio of elesclomol to the transition metal cation is 1 .0.
- the term “therapeutically effective quantity” or equivalent terminology refers to the quantity of a therapeutic agent in which a beneficial clinical outcome is achieved when the quantity of the therapeutic agent is administered to a subject in need thereof.
- beneficial clinical outcome refers to a detectable or observable improvement in a clinical parameter as a result of the administration of the therapeutic agent; the detectable or observable improvement can be objective or subjective.
- a “beneficial clinical outcome” can include, but is not necessarily limited to: a reduction in tumor mass or tumor burden; a reduction in tumor spread or metastasis; a reduction in pain; a reduction of symptoms associated with the malignancy such as seizures for central nervous system malignancies; a reduction of fatigue; a reduction of malaise; an increase in longevity; or an improved Karnofsky performance score.
- Other determinants of a beneficial clinical outcome are known in the art, including determinants for diseases or conditions other than malignancies.
- the therapeutically effective quantity will depend on the type and severity of the malignancy and on the characteristics of the subject, such as the general health, the age, the sex, the body weight, the tolerance to drugs, the relevant pharmacokinetic factors such as liver and kidney function.
- the therapeutically effective quantity will also depend on other therapeutic agents being concurrently administered to the subject to treat the malignancy or to treat other co- morbid conditions affecting the subject.
- One of ordinary skill in the art will be able to determine the therapeutically effective quantity based on this and other factors.
- the therapeutically effective quantity typically ranges between about 1 mg/mm 2 /day to about 10 g/mm 2 /day; more typically, the therapeutically effective quantity ranges between about 2 mg/mm 2 /day to about 5 g/mm 2 /day. In some alternatives, the therapeutically effective quantity is from about 1 pg/kg to about 500 mg/kg, from about 500 pg/kg to about 250 mg/kg, from about 1 mg/kg to about 100 mg/kg, or from about 10 mg/kg to about 50 mg/kg.
- co-administration refers to the administration of at least two agents, such as elesclomol or a derivative, analog, or prodrug of elesclomol and an inhibitor, or therapies to a subject.
- the co-administration of two or more agents or therapies is concurrent.
- a first agent/therapy is administered prior to a second agent/therapy.
- the appropriate dosage for co-administration can be readily determined by one skilled in the art.
- when agents or therapies are co-administered the respective agents or therapies are administered at lower dosages than appropriate for their administration alone.
- co-administration is especially desirable in embodiments where the co-administration of the agents or therapies lowers the requisite dosage of a potentially harmful agent or agent, and/or when co-administration of two or more agents results in sensitization of a subject to beneficial effects of one of the agents via co-administration of the other agent.
- concurrent administration refers to the administration of two or more active agents sufficiently close in time to achieve a combined therapeutic effect that is preferably greater than that which would be achieved by the administration of either agent alone.
- Such concurrent administration can be carried out simultaneously, e.g., by administering the active agents together in a common pharmaceutically acceptable carrier, thereby forming a pharmaceutical composition with two or more active agents, in one or more doses of the pharmaceutical composition.
- the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol can be administered in a pharmaceutically acceptable composition.
- suitable pharmaceutical compositions that comprise elesclomol or a derivative, analog, salt, solvate, or prodrug are as described above.
- the malignancy to be treated can be, but is not limited to, ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, or lymphoid diffuse large B-cell lymphoma.
- OEC ovarian epithelial cancer
- OCCC ovarian clear-cell carcinoma
- uterine corpus endothelial carcinoma stomach adenocarcinoma
- bladder urothelial carcinoma adenoid cystic carcinoma
- adenoid cystic carcinoma uterine carcinosarcoma
- cholangiocarcinoma pancreatic cancer
- metastatic esophagogastric cancer recurrent or meta
- Methods according to the present invention can alternatively be used to treat other malignancies, including, but not limited to, human sarcomas and carcinomas.
- malignancies include, but are not limited to: fibrosarcoma; myxosarcoma; liposarcoma, chondrosarcoma; osteogenic sarcoma; chordoma; angiosarcoma; endotheliosarcoma; lymphangiosarcoma; lymphangioendotheliosarcoma; synovioma; mesothelioma; Ewing’s tumor; leiomyosarcoma; rhabdomyosarcoma; colon carcinoma, including Kras-mutated colon carcinoma; colorectal cancer; anal carcinoma; esophageal cancer; gastric cancer; hepatocellular cancer; bladder cancer; endometrial cancer; pancreatic cancer; breast cancer, including triple-negative breast cancer; ovarian cancer; prostate cancer; stomach cancer; atrial myx
- methods according to the present invention can be used to treat other diseases and conditions, including, but not limited to: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; or scleroderma.
- diseases and conditions including, but not limited to: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses,
- Methods as described above can further comprise administering a therapeutically effective quantity of at least one additional therapeutic agent.
- additional therapeutic agents are described below.
- microtubulin stabilizers refers to an agent, typically an anti-cancer agent, that acts by arresting cells in the G2/M phase of cell division by stabilization of microtubules. Elesclomol or derivatives or analogs of elesclomol as described herein can contribute to the G2/M arrest associated with microtubulin stabilizers or agents with similar mechanisms of action. Examples of microtubulin stabilizers include paclitaxel and paclitaxel analogs.
- microtubulin stabilizers include, but are not limited to: discodermolide; epothilone A; epothilone B; epothilone C; epothilone D; epothilone E; epothilone F; epothilone B N-oxide; epothilone A N-oxide; 16-aza-epothilone B; 21- aminoepothilone B; 21-hydroxyepothilone D; 26-fluoroepothilone; sagopilone; ixabepilone; uditelone; (1 R,2S,4S,7S,8S,9S, 10R, 11 S, 12S, 13R, 17R, 18S)-8, 10- dihydroxy-1 ,5,9, 18-tetramethyl-16,20- dioxahexacyclo[15.3.2.02, 13.04, 12.07, 11.014,19]docosa-5, 14(19)-dien
- Paclitaxel is an antineoplastic drug that functions by enhancing and stabilizing microtubule formation, thus inhibiting mitosis.
- the structure of paclitaxel is shown in Formula P-l, below:
- paclitaxel Many analogs and derivatives of paclitaxel are known. These compounds have the basic taxane skeleton as a common structural feature and have the ability to arrest cells in the G2/M phase of the cell cycle due to microtubule stabilization. A wide variety of substituents can be made on the taxane skeleton while retaining the microtubule-stabilizing activity. It is also the case that zero, one, or two of the cyclohexane rings of a paclitaxel analog can have a double bond (paclitaxel itself has one double bond in such a six-membered ring). Other substitutions can be made at various positions, including moieties including an oxygen atom such as hydroxyl, acyl, or alkoxy.
- One particularly useful compound with the basic taxane skeleton is docetaxel.
- paclitaxel analogs useful in methods or compositions according to the present invention are represented by Formulas (P-l I) or (P-lll):
- R 10 is substituted or unsubstituted lower alkyl, substituted or unsubstituted phenyl; --SR 19 , --NHR 19 , or -OR 19 ;
- R 11 is substituted or unsubstituted lower alkyl or substituted or unsubstituted aryl
- R 12 is hydrogen, hydroxyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkoxy, --O-C(O)-(lower alkyl)--, --O-C(O)-(substituted lower alkyl)--, --O-CH 2 -O-(lower alkyl), or --S-CH 2 -O-(lower alkyl);
- R 13 is hydrogen, methyl, or, taken together with R 14 , --CH 2 --;
- R 14 is hydrogen, hydroxyl, substituted or unsubstituted lower alkoxy, -0- C(O)-(substituted lower alkyl), -O-CH 2 -O-P(O)(OH) 2 , -O-CH 2 -O-( lower alkyl), -O-CH 2 - S-(lower alkyl), or taken together with R 20 , a double bond;
- R 15 is hydrogen, lower acyl, substituted or unsubstituted lower alkyl, alkoxymethyl, alkylthiomethyl, --C(O)-O-(lower alkyl), --C(O)-O-(substituted lower alkyl), --C(O)-NH-(lower alkyl), or --C(O)-NH-(substituted lower alkyl);
- R 16 is substituted or unsubstituted phenyl
- R 17 is hydrogen, substituted or unsubstituted lower acyl, substituted or unsubstituted lower alkyl, (lower alkoxy)methyl, or (lower alkoxy)thiomethyl;
- R 18 is hydrogen, methyl, or taken together with R 17 and the carbon atoms to which R 17 and R 18 are bonded, a five-membered or six-membered non-aromatic heterocyclic ring;
- R 19 is substituted or unsubstituted lower alkyl or substituted or unsubstituted phenyl
- R 20 is hydrogen or a halogen
- R 21 is substituted or unsubstituted lower alkyl or substituted or unsubstituted acyl.
- Paclitaxel analogs or derivatives can also be covalently linked to a pharmaceutically acceptable polymer such as a polyacrylamide.
- microtubulin inhibitor refers to an anti-cancer agent that acts by inhibiting tubulin polymerization or microtubule assembly.
- microtubulin inhibitors include, but are not limited to: erbulozole; dolastatin 10; mivobulin isethionate; vincristine; vinblastine; vinorelbine; vinflunine; vindesine; N-[[4-(5- bromopyrimidin-2-yl)oxy-3-methylphenyl]carbamoyl]-2-(dimethylamino)benzamide (NSC-639829); N-[2-(4-hydroxyanilino)pyridin-3-yl]-4-methoxybenzenesulfonamide (ABT-751 ); liabilityhytin A; perpetuhytin C; spongistatin 1 , spongistatin 2, spongistatin 3, spongistatin 4, spongistatin 5,
- SPIKET-P (a synthetic spiroketal pyran); 3-(iodoacetamido)-benzoylurea (MF-569); narcosine; nascapine; 1 -[5-(4-amino-3-methylphenyl)-2-(3,4,5-trimethoxyphenyl)-2H- 1 ,3,4-oxadiazol-3-yl]ethenone (A-105972); hemiasterlin; 3-bromoacetylamino benzoylurea (MF-191 ); benzyl 2,2,4,4-tetramethylpentanoate; vanadocene acetylacetonate; T-138026; (1 S,3S)-1 -ethenyl-3-[(3-hydroxy-4-methoxyphenyl)methyl]- 7-methyl-2,3-dihydro-1 H-inden-4-ol (RPR-115781 ); desmethyleleutherobin; dese
- microtubule-disrupting antineoplastics include, but are not necessarily limited to, cabazitaxel, larotaxel, vedotin, belantamab mafodotin, ortataxel, and tesetaxel.
- a second category of suitable additional therapeutic agents is PARP inhibitors.
- Inhibitors of the enzyme poly-ADP ribose polymerase (PARP) have been developed for multiple indications, especially for treatment of malignancies. Several forms of cancer are more dependent on the activity of PARP than are non-malignant cells.
- PARP poly-ADP ribose polymerase
- the enzyme PARP catalyzes the polymerization of poly-ADP ribose chains, typically attached to a single-strand break in cellular DNA.
- the coenzyme NAD + is required as a substrate for generating ADP-ribose monomers to be polymerized; nicotinamide is the leaving group during polymerization, in contrast to pyrophosphate which is the leaving group during normal DNA or RNA synthesis, which leaves a pyrophosphate as the linking group between adjacent ribose sugars in the chain rather than phosphate as occurs in normal DNA or RNA.
- the PARP enzyme comprises four domains: a DNA-binding domain, a caspase-cleaved domain, an auto-modification domain, and a catalytic domain.
- the DNA-binding domain comprises two zinc finger motifs. In the presence of damaged DNA, the DNA-binding domain will bind the DNA and induce a conformational shift.
- PARP can be inactivated by caspase-3 cleavage, which is a step that occurs in programmed cell death (apoptosis).
- PARP1 is responsible for most cellular PARP activity.
- PARP2 has been shown to oligomerize with PARP1 , and the oligomerization stimulates catalytic activity. PARP2 is also therefore implicated in BER.
- PARP1 inhibitors inhibit the activity of PARP1 and thus inhibit the repair of single-strand breaks in DNA. When such breaks are unrepaired, subsequent DNA replication can induce double-strand breaks.
- the proteins BRCA1 , BRCA2, and PALB2 can repair double-strand breaks in DNA by the error-free homologous recombinational repair (HRR) pathway. In tumors with mutations in the genes BRCA1, BRCA2, or PALB1, these double-strand breaks cannot be efficiently repaired, leading to cell death.
- HRR homologous recombinational repair
- Normal cells do not replicate their DNA as frequently as tumor cells, and normal cells that lack mutated BRCA1 or BRCA2 proteins can still repair these double-strand breaks through homologous repair. Therefore, normal cells are less sensitive to the activity of PARP inhibitors than tumor cells.
- Some tumor cells that lack the tumor suppressor PTEN may be sensitive to PARP inhibitors because of downregulation of Rad51 , a critical homologous recombination component. Tumor cells that are low in oxygen are also sensitive to PARP inhibitors.
- PARP inhibitors are also considered potential treatments for other life- threatening diseases, including stroke and myocardial infarction, as well as for long- term neurodegenerative diseases (G. Graziani & C. Szabo, “Clinical Perspectives of PARP Inhibitors,” Pharmacol. Res. 52: 109-118 (2005)).
- PARP inhibitors include, but are not limited to, iniparib, talazoparib, olaparib, rucaparib, veliparib, CEP- 9722 (a prodrug of CEP-8983 (11 -methoxy-4,5,6,7-tetrahydro-1 H- cyclopenta[a]pyrrolo[3,4-c]carbazole-1 ,3(2H)-dione), MK 4827 ((S)-2-(4-(piperidin-3- yl)phenyl)-2H-indazole-7-carboxamide), and BGB-290.
- Other PARP inhibitors are described below.
- R 1 represents a halogen atom, a lower alkyl group, a hydroxy group, a lower alkoxy group, an amino group, a nitro group or a cyano group;
- R 2 and R 3 may be the same or different and each represent a hydrogen atom, a halogen atom or a lower alkyl group;
- R 4 and R 5 may be the same or different and each represent a hydrogen atom, a deuterium atom or a lower alkyl group, or R 4 and R 5 may form an oxo group;
- R a and R b may be the same or different and each represent a hydrogen atom, a lower alkyl group optionally having a substituent or an aryl group optionally having a substituent;
- R a and R b may bind to each other to form a nitrogen-containing heterocyclic ring which may be substituted by one or plural R c ;
- R c represents a lower alkyl group optionally having a substituent, a lower cycloalkyl group optionally having a substituent, an aryl group optionally having a substituent, a heterocyclic group optionally having a substituent, a hydroxy group, a lower alkoxy group optionally having a substituent, a lower alkylcarbonyl group optionally having a substituent, a lower cycloalkylcarbonyl group optionally having a substituent, a lower alkylaminocarbonyl group optionally having a substituent, a lower cycloalkylaminocarbonyl group optionally having a substituent, a lower alkoxycarbonyl group optionally having a substituent, an amino group, a lower alkylamino group or a carboxyl group;
- ring A represents a benzene ring or an unsaturated heteromonocyclic ring
- m 0, 1 or 2.
- United States Patent No. 8,993,594 to Papeo et al. discloses substituted isoquinolin-1 (2H)-one derivatives as inhibitors of PARP.
- United States Patent No. 8,980,902 to Brown et al. discloses substituted benzamide PARP inhibitors.
- United States Patent No. 8,946,221 to Mevellec et al. discloses phthalazine derivatives as PARP inhibitors.
- United States Patent No. 8,889,866 to Angibaud et al. discloses tetrahydrophenanthridinones and tetrahydrocyclopentaquinolinones as PARP inhibitors.
- United States Patent No. 8,877,944 to Papeo et al. discloses substituted 3-oxo-2,3-dihydro-1 H-isoindole-4-carboxamide derivatives as PARP inhibitors.
- United States Patent No. 8,546,368 to Penning et al. discloses pyrazoquinolones as PARP inhibitors, including 7,9-dimethyl-1 ,2,3,4,6,7-hexahydro-5H- pyrazolo[3,4-h]-1 ,6-naphthyridin-5-one.
- United States Patent No. 8,541 ,417 to Brown et al. discloses PARP inhibitors, including: 3-(hydroxymethyl)pyrido[2,3-e]pyrrolo[1 ,2-c]pyrimidin-6(5H)-one; N- ethyl-4-(4-((6-oxo-5,6-dihydropyrido[2,3-e]pyrrolo[1 ,2-c]pyrimidin-3-yl)methyl)piperazin- 1-yl)benzamide; and N-methyl-4-(4-((6-oxo-5,6-dihydropyrido[2,3-e]pyrrolo[1 ,2- c]pyrimidin-3-yl)methyl)piperazin-1-yl)benzamide, as well as additional compounds.
- United States Patent No. 8,541 ,403 to Chu et al. discloses dihydropyridophthalazinone derivatives as PARP inhibitors.
- United States Patent No. 8,513,433 to Panicker et al. discloses inhibitors of PARP, including benzyl 2-(4-carbamoyl-1 H-benzo[d]imidazol-2-yl)indoline-1- carboxylate; 2-(indolin-2-yl)-1 H-benzo[d]imidazole-4-carboxamide; tert-butyl 2-(4- carbamoyl-1 H-benzo[d]imidazol-2-yl)-3,4-dihydroquinoline-1 (2H)-carboxylate; and 2- (1 ,2,3,4-tetrahydroquinolin-2-yl)-1 H-benzo[d]imidazole-4-carboxamide, as well as additional compounds.
- United States Patent No. 8,362,030 to Ingenito et al. discloses tricyclic PARP inhibitors, including: N-methyl[4-(6-oxo-3,4,5,6-tetrahydro-2H-azepino[5,4,3- cd]indazol-2-yl)phenyl]methanaminium trifluoroacetate; N, N-dimethyl[4-(6-oxo-3, 4,5,6- tetrahydro-2H-azepino[5,4,3-cd]indazol-2-yl)phenyl]methanaminium trifluoroacetate; and N 2 ,N 2 -dimethyl-N-[4-(1 -oxo-1 ,2,3,4-tetrahydroazepino[3,4,5-hi]indolizin-5- yl)phenyl]glycinamide, as well as additional compounds.
- United States Patent No. 8,354,413 to Jones et al. discloses quinoline- one and 4-oxodihydrocinnoline derivatives as PARP inhibitors, including: 1-[3-(8-aza-1- azoniaspiro[4.5]dec-8-ylcarbonyl)-4-fluorobenzyl]-4-oxo-1 ,4-dihydroquinolinium bis(trifluoroacetate); 1 -[4-fluoro-3-( ⁇ 4-[2-(4-fluorobenzyl)prolyl]piperazin-1 - yl ⁇ carbonyl)benzyl]quinolin-4(1 H)-one; and 1-[3-(8-aza-1-azoniaspiro[4.5]dec-8- ylcarbonyl)-4-fluorobenzyl]-4-oxo-1 ,4-dihydrocinnolin-1-ium bis(trifluoroacetate), as well as additional compounds.
- United States Patent No. 8,268,827 to Branca et al. discloses pyridazinone derivatives as PARP inhibitors, including: 6- ⁇ 4-fluoro-3-[(3-oxo-4- phenylpiperazin-1-yl)carbonyl]benzyl ⁇ -4,5-dimethyl-3-oxo-2,3-dihydropyridazin-1-ium trifluoroacetate; 6- ⁇ 3-[(4-cyclohexyl-3-oxopiperazin-1-yl)carbonyl]-4-fluorobenzyl ⁇ -4,5- dimethyl-3-oxo-2,3-dihydropyridazin-1 -ium trifluoroacetate; 6- ⁇ 3-[(4-cyclopentyl-3- oxopiperazin-1 -yl)carbonyl]-4-fluorobenzyl ⁇ -4,5-dimethylpyridazin-3(2H)-one; and 6- ⁇ 4- fluoro
- United States Patent No. 8,217,070 to Zhu et al. discloses 2-substituted- 1 H-benzimidazole-4-carboxamides as PARP inhibitors, including: 2-(1- aminocyclopropyl)-1 H-benzimidazole-4-carboxamide; 2-[1 -(isopropylamino)cyclopropyl]- 1 H-benzimidazole-4-carboxamide; 2-[1 -(cyclobutylamino)cyclopropyl]-1 H- benzimidazole-4-carboxamide; and 2- ⁇ 1 -[(3,5-dimethylbenzyl)amino]cyclopropyl ⁇ -1 H- benzimidazole-4-carboxamide, as well as additional compounds.
- United States Patent No. 8,173,682 to Weintraub et al. discloses 2,3,5- substituted pyridone derivatives as PARP inhibitors, including: 5-(5-ethyl-2-methyl-6- oxo-1 ,6-dihydro-pyridin-3-yl)-thiophene-2-sulfonic acid [3-(3-hydroxy-pyrrolidin-1-yl)- propyl]-amide hydrochloride; and 5-(5-ethyl-2-methyl-6-oxo-1 ,6-dihydropyridin-3- yl)thiophene-2-sulfonic acid [2-(1 -methylpyrrolidin-2-yl)ethyl]amide hydrochloride, as well as additional compounds.
- R 1 is H, halogen, alkoxy, or lower alkyl
- R 2 is H, halogen, alkoxy, or lower alkyl
- R3 is independently H, amino, hydroxy, --N--N, halogen-substituted amino, -- O-alkyl, --O-aryl, or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, --COR8, where R8 is H, --OH an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or --OR6 or -- NR6R7 where R6 and R7 are each independently hydrogen or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl;
- R4 is independently H, amino, hydroxy, --N--N, --CO--N--N, halogen- substituted amino, - O-alkyl, --O-aryl, or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, --COR8, where R8 is H, --OH an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or -- OR6 or --NR6R7 where R6 and R7 are each independently hydrogen or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl; and
- R5 is independently H, amino, hydroxy, --N--N, --CO--N--N, halogen- substituted amino, --O-alkyl, --O-aryl, or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, --COR8, where R8 is H, --OH an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or -- OR6 or --NR6R7 where R6 and R7 are each independently hydrogen or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl.
- United States Patent No. 8,088,760 to Chu et al. discloses benzoxazole carboxamide inhibitors of PARP, including: 2-(4- ((methylamino)methyl)phenyl)benzo[d]oxazole-4-carboxamide; 2-(2-methylpyrrolidin-2- yl)benzo[d]oxazole-4-carboxamide; 2-(4-((methylamino)methyl)phenyl)benzo[d]oxazole- 7-carboxamide; 2-(2-methylpyrrolidin-2-yl)benzo[d]oxazole-7-carboxamide; and 2- (pyrrolidin-2-yl)benzo[d]oxazole-4-carboxamide, as well as additional compounds.
- United States Patent No. 8,071 ,623 to Jones et al. discloses amide- substituted indazoles as PARP inhibitors, including: 2-(4-piperidin-3-ylphenyl)-2H- indazole-7-carboxamide; 2- ⁇ 4-[(3R)-piperidin-3-yl]phenyl ⁇ -2H-indazole-7-carboxamide; 2- ⁇ 4-[(3S)-piperidin-3-yl]phenyl ⁇ -2H-indazole-7-carboxamide; 5-fluoro-2-(4-piperidin-3- ylphenyl)-2H-indazole-7-carboxamide; and 5-fluoro-2- ⁇ 4-[(3S)-piperidin-3-yl]phenyl ⁇ -2H- indazole-7-carboxamide, as well as additional compounds.
- United States Patent No. 8,058,275 to Xu et al. discloses diazabenzo[de]anthracen-3-one compounds as PARP inhibitors.
- United States Patent No. 8,012,976 to Wang et al. discloses dihydropyridophthalazinone compounds as PARP inhibitors, including 5-fluoro-8-(4- fluorophenyl)-9-(1 -methyl-1 H-1 ,2,4-triazol-5-yl)-8,9-dihydro-2H-pyrido[4,3,2- de]phthalazin-3(7H)-one.
- United States Patent No. 8,008,491 to Jiang et al. discloses substituted aza-indole derivatives as PARP inhibitors, including: 1 -phenyl-2-(piperazin-1 -yl)-1 ,3- dihydropyrrolo[2,3-b]pyridine-3-carboxaldehyde, 1 -phenyl-2-(piperazin-1 -y l)-1 H- pyrrolo[2,3-c]pyridine-3-carboxaldehyde, 2-[1 ,4]diazepan-1 -y 1-1 -phenyl-1 H-pyrrolo[2,3- b]pyridine-3-carbaldehyde trifluoroacetic acid salt, and 2-piperazin-1 -yl-1 -pyridin-3-yl- 1 H-pyrrolo[2,3-b]pyridine-3-carbaldehyde bis-trifluoroacetic acid salt, as well as additional compounds.
- United States Patent No. 7,999,117 to Giranda et al. discloses 1 H- benzimidazole-4-carboxamides as PARP inhibitors, including: 6-fluoro-2-[4-((S)-2- hydroxymethylpyrrolidin-1-ylmethyl)phenyl]-1 H-benzimidazole-4-carboxamide; 6-fluoro- 2-[4-(2-trifluoromethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-benzimidazole-4-carboxamide; 6- fluoro-2-[4-((R)-2-hydroxymethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-benzimidazole-4- carboxamide; 2-[4-((S)-2-hydroxymethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-benzimidazole- 4-carboxamide; and 2-[4-(2-trifluoromethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-
- R 1 is hydrogen or a moiety of Subformula (PA-IV(a)):
- (2) k is 1 , 2, 3, or 4;
- n 0 or 1 ;
- Q is an oxyl group or hydrogen
- R a and R b are independently hydrogen or C 1 -C 6 alkyl
- R b and R d are independently C 1 -C 6 alkyl
- the broken line in the six-membered ring is an optional valence bond (the bond is either a single or a double bond);
- R 2 is either:
- R 1 is other than hydrogen, hydrogen or C 1 -C 6 alkyl
- R 1 is hydrogen, a group of Subformula (PA-IV(b)), Subformula (PA-IV(c)), or Subformula (PA-IV(d)):
- k is 1 , 2, or 3, and R 3 and R 4 are independently C 1 -C 6 alkyl;
- Y is selected from sulfur, nitrogen, and oxygen
- R 1 , R 2 , R 3 , R 4 , R 5 and R 6 are the same or different, and each represent hydrogen, hydroxy, OR 7 , COOR 7 , carboxy, amino, NHR 7 or halogen, or R5 and R 6 taken together form a fused non-aromatic 5- or 6-membered carbocylic ring; and
- R 7 is C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 3 -C7 cycloalkyl optionally substituted with one or more group selected from hydroxyl, C 1 -C 4 alkoxy, carboxy, C 1 -C 6 alkoxycarbonyl, amino, C 1 -C 6 mono-alkylamino, C 1 -C 6 di-alkylamino and halogen.
- 1 H-benzimidazole-4-carboxamides as PARP inhibitors including 2-(1 -amino-1 - methylethyl)-1 H-benzimidazole-4-carboxamide; 2-[1-methyl-1 -(propylamino)ethyl]-1 H- benzim idazole-4-carboxam ide; 2-[1 -(butylam ino)-1 -methylethyl]-1 H-benzim idazole-4- carboxamide; and 2- ⁇ 1 -methyl-1-[(2-phenylethyl)amino]ethyl ⁇ -1 H-benzim idazole-4- carboxamide, as well as additional compounds.
- substituted 1 H-benzimidazole-4-carboxamides as PARP inhibitors, including 2- ⁇ 4-[1 - (cyclohexylmethylamino)ethyl]phenyl ⁇ -1 H-benzimidazole-4-carboxamide; 2-[4-(1 - cyclobutylam inoethyl)phenyl]-1 H-benzim idazole-4-carboxam ide; 2-[3-(2- cyclopropylaminoethyl)phenyl]-1 H-benzim idazole-4-carboxam ide; and 2-(4- cyclopropylaminomethylphenyl)-1 H-benzim idazole-4-carboxam ide, as well as additional compounds.
- United States Patent No. 7,550,603 to Zhu et al. discloses 1 H- benzimidazole-4-carboxamides substituted with a quaternary carbon at the 2-position as PARP inhibitors, including 2-(2-methylpyrrolidin-2-yl)-1 H-benzim idazole-4-carboxam ide; 2-[(2R)-2-methylpyrrolidin-2-yl]-1 H-benzim idazole-4-carboxam ide; 2-[(2S)-2- methylpyrrolidin-2-yl]-1 H-benzim idazole-4-carboxam ide; 2-(1 ,2-dimethylpyrrolidin-2-yl)- 1 H-benzim idazole-4-carboxam ide; 2-(1 -ethyl-2-methylpyrrolidin-2-yl)-1 H-benzim idazole- 4-carboxamide; and 2-(2-methyl-1 -propylpyrrolidin-2
- United States Patent No. 7,405,300 to Jiang et al. discloses substituted indoles as PARP inhibitors, including 2-(piperazin-1 -yl)-1 -(3-nitrophenyl)-1 H-indole-3- carboxaldehyde; 2-(piperazin-1 -yl)-1 -(4-methoxyphenyl)-1 H-indole-3-carboxaldehyde; 2-(piperazin-1 -yl)-1 -(4-tert-butylphenyl)-1 H-indole-3-carboxaldehyde; 2-(piperazin-1 -yl)- 1 -(4-bromophenyl)-1 H-indole-3-carboxaldehyde; and 2-(piperazin-1 -yl)-1 -(4- chlorophenyl)-1 H-indole-3-carboxaldehyde, as well as additional compounds.
- United States Patent No. 7,087,637 to Grandel et al. discloses indole derivatives as PARP inhibitors, including: 2-(4(4-/?-propyl-piperazin-1 -yl)-phenyl)-1 H- indol-4-carboxamide; 2-(4-piperazin-1-yl-phenyl)-1 H-indol-4-carboxamide; 2-(4(4- isopropyl-piperazin-1 -yl)-phenyl)-1 H-indol-4-carboxamide; 2-(4(4-benzyl-piperazin-1 -yl)- phenyl)-1 H-indol-4-carboxamide; 2-(4(4-/?-butyl-piperazin-1 -yl)-phenyl)-1 H-indol-4- carboxamide; and 2-(4(4-ethyl-piperazin-1 -yl)-phenyl)-1 H-in
- United States Patent No. 7,041 ,675 to Lubisch et al. discloses substituted pyridine carboxamides as PARP inhibitors, including 2-phenylimidazo[1 ,2-a]pyridine-8- carboxamide; 2-(4-nitrophenyl)imidazo[1 ,2-a]pyridine-8-carboxamide; 2-(4- aminophenyl)imidazo[1 ,2-a]pyridine-8-carboxamide; 2-(2-benzothienyl)imidazo[1 ,2- a]pyridine-8-carboxamide; 2-(4-bromophenyl)-imidazo[1 ,2-a]pyridine-8-carboxamide; and 2-(4-imidazol-1-ylphenyl)imidazo[1 ,2-a]pyridine-8-carboxamide, as well as additional compounds.
- United States Patent No. 6,635,642 to Jackson et al. discloses phthalazinone derivatives as PARP inhibitors, including 4-(3-nitro-4-(piperidin-1 - yl)phenyl-phthalazin-1 (2H)-one; 4-(4-(dimethylamino)-3-nitrophenyl)-phthalazin-1 (2H)- one; 4-(3-amino-4-(dimethylamino)phenyl)-phthalazin-1 (2H)-one; 4-(4-phenylpiperazin- 1 -yl)-phthalazin-1 (2H)-one; and 4-(4-(4-chlorophenyl)-piperazin-1 -yl)-phthalazin-1 (2H)- one, as well as additional compounds.
- United States Patent No. 6,448,271 to Lubisch et al. discloses substituted benzimidazoles as PARP inhibitors, including 2-(piperidin-4-yl)benzimidazole-4- carboxamide dihydrochloride; 2-(N-acetylpiperidin-4-yl)benzimidazole-4-carboxamide; 2-(N-propylpiperidin-4-yl)benzimidazole-4-carboxamide; 2-piperidin-3-ylbenzimidazole-
- United States Patent No. 6,426,415 to Jackson et al. discloses alkoxy- substituted PARP inhibitors, including 1 -(benzyloxy)-5-methylphthalazine; l -(methoxy)-
- R 1 -R 9 and Z are independently hydrogen, hydroxy, halo, haloalkyl, thiocarbonyl, cyano, nitro, amino, imino, alkylamino, aminoalkyl, sulfhydryl, thioalkyl, alkylthio, sulfonyl, alkylsulfonyl, C 1 -C 9 straight or branched chain alkyl, C 2 -C 9 straight or branched chain alkenyl, C 2 -C 9 straight or branched chain alkynyl, C 1 -C 6 straight or branched chain alkoxy, C 2 -C 6 straight or branched chain alkenoxy, C 2 -C 6 straight or branched chain alkynoxy, aryl, carbocycle, heterocycle, aralkyl, alkylaryl, alkylaryloxy, aryloxy, aralkyloxy, aralkylsulfonyl, aralky
- PA-VI(a) wherein U is C or N; R 7 and R 8 are as defined in (1 ); and X and Y are independently aryl, carbocycle, or heterocycle.
- United States Patent No. 6,380,211 to Jackson et al. discloses alkoxy- substituted PARP inhibitors, including 1 -(methoxy)-5-methylisoquinoline, 1 -(ethoxy)-5- methyl-isoquinoline, 1 -(propoxy)-5-methylisoquinoline, 1 -(butoxy)-5-methylisoquinoline, 1 -(ethoxy)-5-hydroxy-isoquinoline, 1 -(propoxy)-5-hydroxyisoquinoline, 1 -(butoxy)-5- hydroxyisoquinoline, 1-(benzyloxy)-5-methylphthalazine and 1 -(benzyloxy)-5- methylisoquinoline, as well as additional compounds.
- United States Patent No. 6,358,975 to Eliasson et al. discloses PARP inhibitors, including 6(5H)-phenanthridinone, 2-nitro-6(5H)-phenanthridinone, 4- hydroxyquinazoline, 2-methyl-4(3H)-quinazoline, 2-mercapto-4(3H)-quinazoline, benzoyleneurea, 6-amino-1 ,2-benzopyrone, trp-P-1 (3-amino-1 ,4-dimethyl-5H- pyrido[4,3-b]indole), juglone, luminol, 1 (2H)-phthalazinone, phthalhydrazide, and chlorothenoxazin.
- 6(5H)-phenanthridinone 2-nitro-6(5H)-phenanthridinone
- 4- hydroxyquinazoline 2-methyl-4(3H)-quinazoline
- 2-mercapto-4(3H)-quinazoline benzo
- United States Patent No. 6,235,748 to Li et al. discloses oxo-substituted compounds containing at least one ring nitrogen as PARP inhibitors.
- A is O or S
- R is C 1 -C 10 straight or branched chain alkyl, C 2 -C 10 straight or branched chain alkenyl, C 2 -C 10 straight or branched chain alkynyl, aryl, heteroaryl, carbocycle, or heterocycle;
- D is a bond, or a C 1 -C 3 straight or branched chain alkyl, C 2 -C 3 straight or branched chain alkenyl, C 2 -C 3 straight or branched chain alkynyl, wherein any of the carbon atoms of said alkyl, alkenyl, or alkynyl of D are optionally replaced with oxygen, nitrogen, or sulfur; and (4) X is aryl, heteroaryl, carbocycle, or heterocycle.
- United States Patent No. 5,756,510 to Griffin et al. discloses benzamide analogs that are PARP inhibitors, including: 3-benzyloxybenzamide; 3-(4- methoxybenzyloxy)benzamide; 3-(4-nitrobenzyloxy)benzamide; 3-(4- azidobenzyloxy)benzamide; 3-(4-bromobenzyloxy)benzamide; 3-(4- fluorobenzyloxy)benzamide; 3-(4-aminobenzyloxy)benzamide; 3-(3- nitrobenzyloxy)benzamide; 3-(3,4-methylenedioxyphenylmethyloxy)benzamide; 3- (piperonyloxy)benzamide; 3-(N-acetyl-4-aminobenzyloxy)benzamide; and 3-(4- trifluoromethylbenzyloxy)benzamide; and 3-(4-cyanobenzyloxy)benzamide, as well as additional compounds.
- United States Patent Application Publication No. 2015/0175617 by Zhou et al. discloses fused tetra or penta-cyclic dihydrodiazepinocarbazolones as PARP inhibitors, including: 2,3,5, 10-tetrahydro-[1 ,2]diazepino[3,4:5,6-def]carbazol-6(1 H)-one; 5,6,7,8-tetrahydro-4H-4,9, 10-triazaindeno[2, 1 ,7-kla]heptalen-11 (10H)-one; 2-methyl- 2,3,5, 10-tetrahydro-[1 ,2]diazepino[3,4:5,6-def]carbazol-6(1 H)-one; and 3,3-dimethyl- 2,3,5, 10-tetrahydro-[1 ,2]diazepino[3, 4:5, 6-def]carbazol-6(1 H)-one, as well as additional compounds.
- United States Patent Application Publication No. 2015/0152118 by Jana et al. discloses tetrahydroquinazolinone derivatives as PARP inhibitors, including: 2'-(3- (4-(4-fluorophenyl)piperazin-1-yl)propyl)-6',7'-dihydro-3'H-spiro[cyclopropane-1 ,8'- quinazolin]-4'(5'H)-one; 2'-(3-(4-(4-chlorophenyl)piperazin-1-yl)propyl)-6',7'-dihydro-3'H- spiro[cyclopropane-1 ,8'-quinazolin]-4'(5'H)-one; 2'-(3-(4-phenyl-5,6-dihydropyridin- 1 (2H)-yl)propyl)-6',7'-dihydro-3'H-spiro[
- United States Patent Application Publication No. 2015/0025071 by Buchstaller et al. discloses tetrahydroquinazolinone derivatives as PARP inhibitors, including: 2-[4-(4-methoxy-phenyl)-piperazin-1-yl]-5,6,7,8-tetrahydro-3H-quinazolin-4- one; 2-[4-(3-fluorophenyl)piperazin-1 -yl]-5,6,7,8-tetrahydro-3H-quinazolin-4-one; 2-[4-(4- fluorophenyl)piperazin-1 -yl]-5,6,7,8-tetrahydro-3H-quinazolin-4-one; and 2-[4-(3- methoxyphenyl)piperazin-1-yl]-5,6,7,8-tetrahydro-3H-quinazolin-4-one, as well as additional compounds.
- United States Patent Application Publication No. 2015/0018356 by Zhou et al. discloses fused tetra- or pentacyclic pyridophthalazinones as PARP inhibitors.
- United States Patent Application Publication No. 2014/0023642 by Cai et al. discloses 1-(arylmethyl)quinazoline-2,4(1 H,3H)-diones as PARP inhibitors, including: 1 -(3-methoxycarbonylbenzyl)quinazoline-2,4(1 H,3H)-dione; 1 -(3- carboxybenzyl)quinazoline-2,4(1 H,3H)-dione; 1 -(3-(4-(pyridin-2-yl)piperazine-1 - carbonyl)benzyl)quinazoline-2,4(1 H,3H)-dione; 1 -(3-(4-(pyrimidin-2-yl)piperazine-1 - carbonyl)benzyl)quinazoline-2,4(1 H,3H)-dione; and 1 -(3-(4-cyclohexylpiperazine-1 - carbonyl)
- R 1 , R 2 , and R 3 are independently selected from the group consisting of hydrogen, alkenyl, alkoxy, alkoxycarbonyl, alkyl, alkynyl, cyano, haloalkoxy, haloalkyl, halogen, hydroxy, hydroxyalkyl, nitro, NRARB, and (NRARB)carbonyl;
- A is a nonaromatic 4, 5, 6, 7, or 8-membered ring that contains 1 or 2 nitrogen atoms and, optionally, one sulfur or oxygen atom, wherein the nonaromatic ring is optionally substituted with 1 , 2, or 3 substituents selected from the group consisting of alkenyl, alkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkynyl, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, cyano, haloalkoxy, haloalkyl, halogen, heterocycle, heterocyclylalkyl, heteroaryl, heteroarylalkyl, hydroxy, hydroxyalkyl, nitro, NRCRD, (NR C R D )alkyl, (NRcRD)carbonyl, (NRcRD)carbonylalkyl, and (NR C R D )sulfonyl; and;
- RA, RB, RC, and RD are independently selected from the group consisting of hydrogen, alkyl, and alkycarbonyl.
- PARP inhibitors are known in the art and can be employed in methods and compositions according to the present invention. These additional PARP inhibitors include, but are not limited to: (1 ) derivatives of tetracycline as described in United States Patent No. 8,338,477 to Duncan et al.; (2) 3,4-dihydro-5-methyl-1(2/-/)- isoquinoline, 3-aminobenzamide, 6-aminonicotinamide, and 8-hydroxy-2-methyl-4(3/-/)- quinazolinone, as described in United States Patent No.
- a third category of suitable additional therapeutic agents is lactate dehydrogenase (LDH) inhibitors.
- Lactate dehydrogenase (LDH) catalyzes the interconversion of pyruvate and lactate with the concomitant interconversion of NADH and NAD + .
- LDH lactate dehydrogenase
- pyruvate is converted to lactate, pyruvate is reduced by reduction of a keto moiety in pyruvate to the hydroxyl moiety in lactate.
- the enzyme converts pyruvate, the final product of glycolysis, to lactate when oxygen is absent or in short supply, and also regenerates NAD + , the oxidized form of NAD, maintaining its concentration so that further glycolysis can occur.
- Cancer cells may rely on increased glycolysis in certain metabolic states, resulting in increased lactate production, in addition to aerobic respiration in the mitochondria, a process known as oxidative phosphorylation (OXPHOS), even under oxygen-sufficient conditions, a phenomenon known as the Warburg effect.
- OXPHOS oxidative phosphorylation
- This state of fermentative glycolysis is catalyzed by the A form of LDH. This mechanism allows tumor cells to convert the majority of their glucose stores into lactate regardless of oxygen availability, shifting use of glucose metabolites from simple energy production to the promotion of accelerated cell growth and replication.
- Oxamate is a cytosolic inhibitor of the A form of LDH that significantly decreases adenosine triphosphate (ATP) production in tumor cells as well as increasing production of reactive oxygen species (ROS).
- ROS include peroxides, superoxide, hydroxyl radical, singlet oxygen, and alpha-oxygen. These reactive oxygen species can drive cancer cell proliferation by activating kinases that drive cell cycle progression growth factors at low concentrations, but can damage DNA through oxidative stress at higher concentrations.
- elesclomol can potentiate the activity of PARP inhibitors, inhibitors of histone deacetylase, and hypomethylating agents; inhibitors of histone deacetylase and hypomethylating agents are epigenetic targets.
- Histone deacetylase inhibitors include, but are not limited to, parthenolide, abexinostat, allyl mercaptan, apicidin, belinostat, chidamide, 3,3'- diindolylmethane, entinostat, givinostat, martinostat, mocetinostat, panobinostat, pracinostat, resminostat, romidepsin, trichostatin A, and vorinostat.
- Hypomethylating agents include, but are not limited to, azacitidine and decitabine.
- Inhibitors of the A form of lactate dehydrogenase disclosed in this reference include: 1-(phenylseleno)-4-bromobenzene, 1- (phenylseleno)-4-methylbenzene, 1 -(phenylseleno)-4-methoxybenzene, 1 - (phenylseleno)-4-phenylbenzene, 3-(phenylseleno)tetrahydrothiophene, and 1- (phenylmethylseleno-4-methoxybenzene.
- Still other inhibitors of the A form of lactate dehydrogenase include 3-[[3- (cyclopropylsulfamoyl)-7-(2,4-dimethoxypyrimidin-5-yl)quinolin-4-yl]amino]-5-(3,5- difluorophenoxy)benzoic acid (GSK2837808A), 7-benzyl-2,3-dihydroxy-6-methyl-4- propylnaphthalene-1 -carboxylic acid (FX-11 ), (2R)-5-(2-chlorophenyl)sulfanyl-4-hydroxy- 2-(4-morpholin-4-ylphenyl)-2-thiophen-3-yl-1 ,3-dihydropyridin-6-one ((R)-GNE-140), (LDH-IN-1 ); 3-dehydrotrametanolic acid, nifurtimox, bromoalanine, and glomeratose A.
- R a is hydrogen, halogen, or alkoxy optionally substituted with one or more halogens
- R to is hydrogen, halogen, alkoxy optionally substituted with one or more halogens, or nitro, or is a group which together with R c , R d , or R g forms a ring structure optionally having substituents;
- R c is a hydrogen atom or carboxyl, or is a group which together with R b , R e , R f or R g forms a ring structure optionally having substituents;
- R d is a hydrogen atom or a moiety -X 11 -R 11 , or is a group which together with R to or R g forms a ring structure optionally having substituents;
- X 11 is alkylene, -NHCO-, -CH 2 — NR 12 — CO-, or -S-;
- R 11 is optionally substituted aryl, alkyl optionally substituted with one or more halogens, or hydroxyalkyl;
- R 12 is hydroxyalkyl
- R 21 is hydroxy or is a group which together with R c or R g forms a ring structure optionally having substituents;
- R 22 is nitro or aminocarbonyl in which a hydrogen atom bound to a nitrogen atom may be substituted with alkyl;
- X 31 is a single bond, alkenylene, --CH 2 — O-CH 2 — CO — NH--, --CH 2 — O- CH 2 — CO— , or -CO—;
- R 31 is optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, optionally substituted heterocyclic amino, hydroxy, alkoxy optionally substituted with one or more halogens, or hydroxyalkyl, wherein, when X 31 is a single bond and R 31 is heterocyclic amino optionally having substituents, the single bond is bound to an atom other than the nitrogen atom of the heterocyclic amino;
- X 32 represents a single bond or -CH 2 -CO-NH-;
- R 32 is optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, alkyl optionally substituted with one or more halogen atoms;
- X 33 and X 34 each are a single bond, alkylene, -CH 2 — CO — NH, or -SO2-;
- R 33 and R 34 each independently represent a hydrogen atom, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, hydroxyalkyl, carboxyl, alkyl optionally substituted with one or more halogens, or alkynyl group, or represent a group which together with R b , R c , and R d or R e and Reforms a ring structure optionally having substituents, or R 33 and R 34 , together with the nitrogen atom to which they are bound through X 33 and X 34 which are single bonds, represent optionally substituted heterocyclic amino;
- R 35 , R 36 , and R 37 are each a hydrogen atom or X 31 — R 31 , or represent a group which together with R b , R c , and R d or R e and Reforms an optionally substituted ring structure;
- R h is hydrogen or halogen; and (19) wherein the number of ring structures formed by binding between R b and R c , the ring structure formed by binding between R b and R d , the ring structure formed by binding between R b and R g , the ring structure formed by binding between R c and R e or R f , the ring structure formed by binding between R c and R g , the ring structure formed by binding between R d and R g , or the ring structure formed by binding between R e or R f and Rg formed in Formula (LI) is no more than one.
- a fourth category of suitable additional therapeutic agents is 2- deoxyglucose and analogs or derivatives thereof.
- the additional therapeutic agent is 2-deoxyglucose (H. Pelicano et al., “Glycolysis Inhibition for Anticancer Treatment,” Oncogene 25: 4633-4646 (2006)).
- a fifth category of suitable additional agents is glutamine metabolism inhibitors.
- Glutamine metabolism inhibitors include compound 968 (5-(3-bromo-4- (dimethylamino)phenyl)-2,2-dimethyl-2,3,5,6-tetrahydrobenzo[a]phenanthridin-4(1 H)- one), BPTES (bis-2-(5-phenylacetamido-1 ,2,4-thiadiazoyl-2-yl)ethyl sulfide), L- asparaginase, and phenylbutyrate.
- glutamine metabolism inhibitors are disclosed in United States Patent No. 10,947,598 by Ricci et al., including: (i) inhibitors of glutamine synthase (GS) such as methionine sulfoximine, methionine sulfone, phosphinothricin, tabtoxinin- ⁇ - lactam, methionine sulfoximine phosphate, a-methyl methionine sulfoximine, a-ethyl methionine sulfoximine, ethionine sulfoximine, a-methyl ethionine sulfoximine, prothionine sulfoximine, a-methyl prothionine sulfoximine, y-hydroxy phosphinothricin, gamma-methyl phosphinothricin, y-acetoxy phosphinothricin, a-methyl phosphinothricin, a-e
- GS
- X is selected from the group consisting of a bond, -0--, and -(CH 2 ) n --, wherein n is an integer selected from the group consisting of 1 , 2, 3, 4, 5, 6, 7, and 8;
- R 1 is selected from the group consisting of hydrogen and a first prodrug- forming moiety capable of forming a salt or an ester;
- R 2 is selected from the group consisting of hydrogen and a second prodrug- forming moiety capable of forming an amide linkage, a carbamate linkage, a phosphoram idate linkage or a phosphorodiamidate linkage with the nitrogen adjacent to R 2 ;
- R 1 is phenyl optionally substituted with at least one R 3 , benzyl optionally substituted with at least one R 3 , or pyridinyl optionally substituted with at least one R 3 ;
- R2 is phenyl optionally substituted with at least one R 3 , benzyl optionally substituted with at least one R 3 , or pyridinyl optionally substituted with at least one R 3 ;
- R 3 is independently hydrogen, methyl, alkyl, methoxy, alkoxy, halogen, or trifluoromethyl
- n 0-6.
- These compounds include (S)-2-amino-4-(bis(2-((3- methylbenzyl)oxy)benzyl)amino)butanoic acid.
- a sixth category of suitable additional agents is DNA-damaging agents, in particular, DNA-damaging anti-neoplastic agents.
- DNA-damaging anti-neoplastic agents are disclosed in K. Cheung-Ong et al., “DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology,” Chem. Biol. 20: 648-659 (2013). These agents fall into several categories. One category is agents that damage DNA directly; these agents employ one of several mechanisms, including direct modification of DNA bases, intercalation between DNA bases, and formation of crosslinks in DNA. Another category is agents that interfere with DNA synthesis. Yet another category is agents that inhibit topoisomerases; some of these agents also have additional activities that can damage DNA or interfere with DNA replication in various other ways.
- nitrogen mustards act by directly alkylating DNA on purine bases, leading to stalled replication fork progression and subsequent cell death via apoptosis.
- Derivatives of the nitrogen mustards were subsequently developed, including the DNA alkylators cyclophosphamide, chlorambucil, and melphalan, all of which are currently used in clinical therapeutics.
- DNA-alkylating agents used in cancer treatment include nitrosoureas (e.g., carmustine, lomustine, and semustine) and triazenes (e.g., dacarbazine and temozolomide).
- Natural products which alkylate DNA bases were also discovered, such as mitomycin C and streptozotocin.
- interstrand cross-links crosslink DNA on opposite strands of the double helix (interstrand cross-links), resulting in a more potent effect against cancer cells compared to monofunctional alkylation.
- carmustine binds to the N1 atom of guanine on one DNA strand and the N3 atom of cytosine of the opposite strand to form interstrand crosslinks, which block DNA replication and can cause cell death if not repaired.
- Additional agents that damage DNA include the alkylating-like platinum agents that act by forming adducts on DNA. When two platinum adducts form on adjacent bases on the same DNA strand, they form intrastrand crosslinks. These agents include cisplatin, carboplatin, oxaliplatin, and picoplatin, as well as other platinum-containing analogs.
- Antimetabolites typically do not damage DNA molecules directly, but interfere with DNA replication. Examples include the pyrimidine analogs 5-fluorouracil, capecitabine, floxuridine, and gemcitabine, and the purine analogs 6-mercaptopurine, 8- azaguanine, fludarabine, and cladribine. Another class of antimetabolites inhibit enzymes important for DNA synthesis; this class of antimetabolites include antifolates such as methotrexate, aminopterin, pemetrexed, and ralitrexed. [0376] Topoisomerase inhibitors inhibit replication fork progression in replicating DNA and cause double-strand breaks. These agents include etoposide, camptothecin, and anthracycline antibiotics.
- the anthracyclines such as, but not limited to, doxorubicin and daunorubicin, are also able to intercalate into DNA, generate free radicals, bind and alkylate DNA, crosslink DNA, interfere with helicase activity, and induce apoptosis.
- DNA-damaging agents are also described in the following patents or published patent applications: United States Patent No. 9,097,722 to Yu; United States Patent No. 9,096,602 to Everitt et al.; United States Patent No. 8,840,898 to Goldmakher; United States Patent No. 8,735,590 to Adejare et al.; United States Patent No. 8,415,357 to Kawabe et al.; United States Patent No. 8,476,025 to Clifford; United States Patent No. 7,902,165 to Kim; United States Patent No. 7,875,586 to Kovbasnjuk et al.; United States Patent No. 7,652,042 to Kawabe et al.; United States Patent No.
- DNA-damaging anti-neoplastic agents can act by a variety of mechanisms, including modification of DNA bases such as by alkylation, intercalation into the DNA structure, formation of crosslinks in DNA, prevention of unwinding or replication of DNA to induce double-strand breaks, incorporation into DNA in place of normal nucleosides, and other mechanisms.
- DNA-damaging anti-neoplastic agents include, but are not limited to: cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, tetraplatin, doxorubicin, daunorubicin, methotrexate, 5-fluorouracil, gemcitabine, podophyllotoxin, etoposide, teniposide, cyclophosphamide, chlorambucil, melphalan, carmustine, lomustine, estramustine, semustine, bendamustine, prednamustine, uramustine, chlornaphazine, dacarbazine, altretamine, temozolomide, mitomycin C, streptozotocin, chlorozotocin, capecitabine, floxuridine, 6-mercaptopurine, 8-azaguanine, azathiopurine, 5-ethynyluracil, thiogu
- a seventh category of suitable additional agents is agents that inhibit the SWI/SNF complex.
- SWI/SNF (BAF) complexes are a diverse family of ATP- dependent chromatin remodelers produced by combinatorial assembly that are mutated in and thought to contribute to 20% of human cancers and a large number of neurologic diseases.
- SWI/SNF inhibitors are compounds that inhibit one or more members of a subfamily of ATP-dependent chromatin remodeling complexes; the activity of these proteins affects nucleosome rearrangement, which, in turn, affects access to the chromatin, allowing genes to be activated or repressed.
- SWI/SNF inhibitors include BD98 (1-isopropyl-3-((4R,5S,8S)-4-methoxy- 2 , 5, 8-trim ethy 1-1 -oxo-7-(4-pyridyl-2-yl)benzyl)-1 ,2, 3, 4, 5, 6, 7, 8, 9, 10- decahydrobenzo[h][1 ,6]diazacyclododecin-13-yl)urea) (E.J. Chory et al., “Chemical Inhibitors of a Selective SWI/SNF Function Synergize with ATR Inhibition in Cancer Cell Killing,” ACS Chem. Biol. 15: 1685-1696 (2020)).
- the ATR inhibitor described in this reference is 3-amino-6-(4-(methylsulfonyl)phenyl)-N-phenylpyrazine-2-carboxamide.
- SWI/SNF inhibitors are disclosed in United States Patent Application Publication No. 2020/0147120 by Iba et al. and United States Patent No. 9,850,543 to Zainab et al.
- An eighth category of additional agents is agents that cause the tumor cells to rely heavily on oxidative phosphorylation (OXPHOS). It had been previously shown that cancer cells exhibit an alteration in their metabolism when compared with non-malignant cells. Normal cells in the presence of oxygen use primarily the mitochondrial tricarboxylic acid (TCA) cycle and oxidative phosphorylation for the production of energy and rely on glycolysis only when their oxygen supply is limited. In contrast, cancer cells frequently utilize glycolysis even in the presence of sufficient amounts of oxygen. The fact that cancer cells reduce their dependence on oxidative phosphorylation on mitochondrial oxidative phosphorylation and thus are substantially more reliant on glycolysis can potentially provide a range of therapeutic targets.
- TCA mitochondrial tricarboxylic acid
- the glycolytic pathway includes a total of 10 reactions.
- the enzymes that catalyze reactions in the glycolytic pathway include: hexokinase; glucose-6- phosphate isomerase; phosphofructokinase 1 ; fructose-bisphosphate aldolase; triosephosphate isomerase; glyceraldehyde 3-phosphate dehydrogenase; phosphoglycerate kinase; phosphoglycerate mutase; enolase; and pyruvate kinase.
- extracellular glucose is transported into the cell by the glucose transporter GLUT1 , and the activity of this glucose transporter may also be a potential therapeutic target.
- Inhibitors of GLUT 1 include the flavonoids phloretin and quercetin.
- Another inhibitor of GLUT1 is STF31 (4-[[[[4-(1 ,1- dimethylethyl)phenyl]sulfonyl]amino]methyl]-A/-3-pyridinylbenzamide).
- Still another inhibitor of GLUT 1 is WZB117 ((2-fluoro-6-(m -hydroxybenzoyloxy) phenyl m- hydroxybenzoate).
- Inhibitors of hexokinase include 3-bromopyruvate.
- Inhibitors of phosphofructokinase 1 include 3-(3-pyridinyl)-1 -(4-pyridinyl)- 2-propen-1-one and PFK158 (1 -(4-pyridinyl)-3-[7-(trifluoromethyl)-2E-quino!inyl]-2- propen-1 -one.
- dichloroacetate is an inhibitor of pyruvate decarboxylase kinase, an enzyme that inhibits the mitochondrial pyruvate dehydrogenase. In this way, it suppresses glycolysis and stimulates oxidative phosphorylation.
- Oxamic acid is a pyruvate analog and is a competitive lactate dehydrogenase inhibitor that inhibits glycolysis.
- each of R 1 , R 2 , R 3 , and R 4 is independently H or COR 5 ;
- each of R 5 is independently selected from the group consisting of C 1 -C 10 straight-chain or branched chain alkyl, C 4 -C 10 alkylcycloalkyl, and C 3 -C 7 cycloalkyl.
- (1 ) A is selected from the group consisting of Subformulas (G-lla) and (G-ll(b): (G-ll(a));
- Ring B is a five- or six-membered ring containing 1 or 2 heteroatoms selected from the group consisting of N, 0 and S;
- Ring C is a five- or six-membered aryl or heteroaryl ring containing from 0 to 2 heteroatoms selected from the group consisting of N, 0 and S;
- each R 1 is independently selected from the group consisting of halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 1 -C 6 alkoxy, C 3 -C 6 cycloalkyl, oxo, hydroxy, amino, cyano and C 1 - C 3 perfluoroalkyl;
- n is selected from 0, 1 , or 2;
- each R 2 is independently selected from the group consisting of halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 1 -C 6 alkoxy, C 3 -C 6 cycloalkyl, oxo, hydroxy, amino, cyano and C 1 - C 3 perfluoroalkyl;
- n is selected from 0, 1 , or 2;
- R 3 is selected from the group consisting of halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 1 -C 6 alkoxy, C 3 -C 6 cycloalkyl, oxo, hydroxy, amino, cyano and C 1 -C 3 perfluoroalkyl;
- R 4 is selected from the group consisting of halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 1 -C 6 alkoxy, C 3 -C 6 cycloalkyl, oxo, hydroxy, amino, cyano and C 1 -C 3 perfluoroalkyl;
- (11 ) y is selected from 1 , 2, or 3;
- R 5 and R 6 are independently selected from the group consisting of H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, --( C 1 -C 6 alkyl)-O--( C 1 -C 6 alkyl), aryl, aralkyl, heteroaryl, and C 3 -C 6 cycloalkyl, or R 5 and R 6 may be taken together with the nitrogen to which they are attached to form a 5- to 6-membered heterocyclic ring having up to 3 heteroatoms selected from N, O, and S, and which is optionally substituted by from 1 to 3 substituents independently selected from halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 1 -C 6 alkoxy, C 3 -C 6 cycloalkyl, oxo, hydroxy, amino, cyano and C 1 -C 3 perfluoroalkyl; and
- R 7 is selected from the group consisting of aryl, heteroaryl, and a heterocyclic group.
- X is selected from the group consisting of nitro, an imidazole, a halide, sulfonate, a carboxylate, an alkoxide, and amine oxide;
- R is selected from the group consisting of OR', N(R") 2 , C(O)R"', C 1 -C 6 alkyl, C 6 -C 12 aryl, C 1 -C 6 heteroalkyl, C 6 -C 12 heteroaryl, hydrogen, and an alkali metal;
- R' is hydrogen, an alkali metal, C 1 -C 6 alkyl, C 6 -C 12 aryl, or C(O)R"';
- R" is hydrogen, C 1 -C 6 alkyl, or C 6 -C 12 aryl
- R'" is hydrogen, C 1 -C 20 alkyl, or C 6 -C 12 aryl.
- United States Patent No. 8,927,506 to Priebe et al. discloses acetates of 2-deoxymonosaccharides as inhibitors of glycolysis.
- United States Patent No. 8,329,753 to Newell et al. discloses bifunctional compounds that covalently link a glycolysis inhibitor and a fatty acid metabolism inhibitor.
- the anti-neoplastic agent lonidamine can be used together with elesclomol or a derivative or analog of elesclomol; in this context, lonidamine acts as an additional agent that causes the tumor cells to rely heavily on oxidative phosphorylation.
- Lonidamine is an agent that suppresses glycolysis in tumor cells through the inhibition of the mitochondrially bound hexokinase.
- Another agent that acts as an additional agent that causes the tumor cells to rely heavily on oxidative phosphorylation is the alkylating agent dianhydrogalactitol. Dianhydrogalactitol is described in PCT Patent Application Publication No. WO 2012/024367 by Brown.
- a ninth category of additional agents is agents that act as inhibitors of the base excision repair (BER) pathway.
- Inhibitors of the base excision repair (BER) pathway as anti-neoplastic agents are disclosed in A.M. Reed et al., “Small-Molecule Inhibitors of Proteins Involved in Base Excision Repair Potentiate the Anti-Tumorigenic Effect of Existing Chemotherapeutics and Irradiation,” Future Oncol. 5: 713-726 (2009).
- inhibitors include: (i) PARP inhibitors, specifically INO-1001 (3-aminobenzamide); AG14361 (2-[4- [(dimethylamino)methyl]phenyl]-1 ,3, 10-triazatricyclo[6.4.1 .04, 13]trideca-2 , 4, 6, 8( 13)- tetraen-9-one); AG014699 (rucaparib phosphate); ABT-888 (veliparib); and AZD2281 (olaparib); (ii) inhibitors of the Apel enzyme, including methoxyamine; lucanthone; 7- nitroindole-2-carboxylic acid; an arylstilbonic acid derivative (A.
- United States Patent No. 10,220,053 to Bellacosa et al. discloses inhibitors of thymine DNA glycosylase as inhibitors of the BER pathway.
- BER inhibitors including AP endonuclease inhibitor, a DNA glycosylase inhibitor, a DNA polymerase inhibitor, a PARP inhibitor, a DNA alkyltransferase inhibitor, and a DNA ligase inhibitor.
- Preferred BER inhibitors include methoxyamine and a compound of Formula (B-l):
- the AP endonuclease inhibitor can be methoxyamine, the compound of Formula (B-l), or N-ethylmaleimide.
- the PARP inhibitor can be 5-methyl-3,4-dihydro- 2H-isoquinolin-1 -one (PD128763), 3-aminobenzamide, 6-aminonicotinamide, 8- hydroxy-2-methyl-3H-quinazolin-4-one (NU 1025), or 4-amino-1 ,8-naphthalimide.
- the DNA polymerase inhibitor can be prunasin, aphidicolin, ddCTP, ddTTP, ddATP, ddGTP, or arabinocytidine.
- a tenth category of additional agents is agents that act as inhibitors of the homologous repair (HR) pathway (S.B. Chernikova, “Inhibiting Homologous Recombination for Cancer Therapy,” Cancer Biol. Ther. 13: 61-68 (2012)).
- HR homologous repair
- agents include imatinib mesylate, erlotinib, valproic acid, abexinostat, tanespimycin, ⁇ - lactacystin, benzyl N-[(2S)-4-methyl-1 -[[(2S)-4-methyl-1 -[[(2S)-4-methyl-1 -oxopentan-2- yl]amino]-1 -oxopentan-2 -yl]amino]-1 -oxopentan-2 -yl]carbamate (MG-132), bortezomib, nelfinavir, mirin, 6-(cyclohexylmethoxy)-5-nitrosopyrimidine-2,4-diamine (NU6027), 3- (carbamoylamino)-5-(3-fluorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide (AZD7762), (5Z)-5
- United States Patent No. 10,927,075 to Mills discloses inhibitors of homologous repair including 4,4'-diisothiocyanatostilbene-2,2'-disulfonate.
- United States Patent No. 10,590,122 to Castro et al. discloses substituted thiazole derivatives as RAD51 inhibitors that function as inhibitors of homologous repair; the substituted thiazole derivatives can include a cycloalkyl, heterocyclyl, or heteroaromatic moiety.
- An eleventh category of additional agents is antineoplastic agents that can activate homologous repair as part of their mechanism of antineoplastic activity or as a consequence of inducing DNA damage.
- additional agents include carboplatin, cisplatin, dianhydrogalactitol, and dibromodulcitol.
- a twelfth category of additional agents is antineoplastic agents that are activated by bioreductases under acute conditions of hypoxia or that function to sensitize hypoxic cells to antineoplastic agents or radiation.
- agents include hypoxic cell sensitizers and cytotoxic agents.
- Hypoxic cell sensitizers include, but are not necessarily limited to, misonidazole, metronidazole, nimorazole, benznidazole, desmethylmisonidazole, etanidazole, pimonidazole, and 1 -(aziridin-1 -yl)-3-(2- nitroimidazol-1-yl)propan-2-ol (RSU-1069) (S.
- Cytotoxic agents that can be activated by bioreductases include, but are not limited to, tirapazamine and mitomycin C.
- Tirapazamine produces hydroxyl and/or benzotriazinyl radicals as DNA-damaging reactive species.
- Mitomycin C is an alkylating agent that crosslinks DNA; it is reductively activated to form a mitosene, which reacts successively via /V-alky lation of two DNA bases. Both alkylations are sequence-specific for a guanine nucleoside in the sequence 5'-CpG-3'.
- a thirteenth category of additional agents is agents that inhibit cysteine uptake.
- One class of agents that inhibits cysteine uptake is peptides derived from digestion of human p-casein, bovine p-casein, and gliadin. These peptides include human p-casomorphin-7 (hBCM7) (YPFVEPL) (SEQ ID NO: 1 ); bovine p-casomorphin- 7 (bBCM7) (YPFPGPL) (SEQ ID NO: 2); and gliadinomorphin-7 (GM7) (YPQPQPF) (SEQ ID NO: 3) (M.S. Trivedi, “Food-Derived Opioid Peptides Inhibit Cysteine Uptake with Redox and Epigenetic Consequences,” J. Nutr.
- Another class of agents that inhibits cysteine uptake is inhibitors of the excitatory amino acid transporters (EAATs) EAAT2 and EAAT3. These inhibitors include L-glutamate, L-aspartate, and the synthetic inhibitors threo-p-hydroxyaspartate, which is a non-selective EAAT inhibitor, and dihydrokainate, which is a selective EAAT2 inhibitor.
- Another cysteine uptake inhibitor is threo-p-benzyloxyaspartate (Y. Chen & R.A. Swanson, “The Glutamate Transporters EAAT2 and EAAT3 Mediate Cysteine Uptake in Cortical Neuron Cultures,” J.
- EAAT2 inhibitor is WAY-855 (3-amino-tricyclo[2.2.1 ,0 26 ]heptane-1 ,3- dicarboxylic acid) (J. Dunlop et al., “WAY-855 (3-Amino-tricyclo[2.2.1 ,0 26 ]heptane-1 ,3- Dicarboxylic Acid), a Novel, EAAT2-Preferring, Nonsubstrate Inhibitor of High-Affinity Glutamate Uptake,” Br. J. Pharmacol. 140: 839-846 (2003)).
- Erastin analogs are known in the art and are disclosed in United States Patent No. 7,615,554 to Selliah et al. These analogs include erastin A, erastin B, and compounds of Formula (E-l):
- R 1 is selected from the group consisting of hydrogen, -Z-Q-Z-, --(C 1 - C 8 )alkyl-N(R 2 )(R 4 ), --(C 1 -C 8 )alkyl-OR 3 , 3- to 8-membered carbocyclyl, 3- to 8-membered heterocyclyl, aryl, heteroaryl, and (C 1 -C 4 ) aralkyl;
- R 2 and R 4 are each independently selected from the group consisting of hydrogen, (C 1 -C 4 ) alkyl, (C 1 -C 4 ) aralkyl, aryl, heteroaryl, acyl, alkylsulfonyl, and arylsulfonyl, provided that when both R 2 and R 4 are on the same nitrogen atom and not both hydrogen they are different and that when both R 2 and R 4 are on the same nitrogen atom and either R 2 or R 4 is acyl, alkylsulfonyl
- R 3 is selected from the group consisting of hydrogen, (C 1 -C 4 ) alkyl, (C 1 -C 4 ) aralkyl, aryl, and heteroaryl;
- W is selected from a moiety of Subformula (E-I(a)), (E-I(b)), or (E-I(c)):
- Q is selected from 0 and NR 2 ;
- (6) Z is independently selected for each occurrence from the group consisting of (C 1 -C 6 ) alkyl, (C 2 -C 6 ) alkenyl, and (C 2 -C 6 ) alkynyl.
- erastin analogs include piperazine erastin (2-[[4-[2-(4- chlorophenoxy)acetyl]piperazin-1-yl]methy!]-3-[5-(piperazin-1-ylmethyl)-2-propan-2- yloxyphenyl]quinazolin-4-one) (United States Patent No. 9,695,133 to Stockwell et al.).
- R 1 is selected from the group consisting of hydrogen, (C 1 -C 8 ) alkyl, (C 1 -C 8 ) alkoxy, 3- to 8-membered carbocyclyl, 3- to 8-membered heterocyclyl, aryl, heteroaryl, residues of glycolic acid, residues of ethylene glycol/propylene glycol copolymers, carboxylate, ester, amide, carbohydrate, amino acid, alditol, OC(R 7 ) 2 COOH, SC(R 7 ) 2 COOH, NHCHR 7 COOH, COR 8 , CO2R 8 , sulfate, sulfonamide, sulfoxide, sulfonate, sulfone, thioalkyl, thioester, and thioether;
- R 2 , R 3 , R 4 , R 5 , and R 6 are each independently selected from the group consisting of hydrogen, halogen, (C 1 -C 4 ) alkyl, (C 1 -C 4 ) alkylamino, acyl, and alkylsulfonyl;
- R 7 is selected from the group consisting of hydrogen, (C 1 -C 8 ) alkyl, optionally substituted carbocyclyl, aryl, heteroaryl, heterocyclyl, alkylaryl, alkylheteroaryl, and alkylheterocyclyl; and
- R 8 is selected from the group consisting of optionally substituted (C 1 -C 8 ) alkyl, (C 2 -C 8 ) alkenyl, (C 2 -C 8 ) alkynyl, aryl, carbocyclyl, heteroaryl, heterocyclyl, alkylaryl, alkylheteroaryl, and alkylheterocyclyl; with the proviso that R 1 is not methyl when R 4 is chloro.
- Yet another class of inhibitors of cysteine uptake includes inhibitors of other transporters of cysteine, or, in some cases, its oxidized form cystine.
- additional transporters include LAT1 , ASCT2, and the X c ’ system (B. Daher et al., “Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptic Cell Death,” Front. Oncol. 10:723 (2020)).
- LAT1 is responsible for transport of essential amino acids, general amino acid homeostasis, and, consequently tumor growth.
- ASCT2 is a transporter that exchanges small neutral amino acids and plays a crucial role in glutamine uptake and the promotion of tumor growth, independently of LAT1 activity; however, there is evidence that this transporter may not actually be involved in the transport of cysteine in vivo and that cysteine may actually be a competitive inhibitor of ASCT2.
- the third transporter of relevance is the X c ’ system, which is an exchanger that imports cystine (the oxidized form of cysteine) and exports glutamate.
- This sodium- independent antiporter comprises two subunits: xCT (encoded by the gene SLC7A11), a subunit responsible for the amino acid exchange, and a chaperone CD98 (encoded by the gene SLC3A2).
- Inhibitors of the X c ’ system include erastin, imidazole ketone erastin (IKE), sorafenib, and sulfasalazine (SSZ).
- GCN2 protein kinase General Control Nonderepressible
- GCN2 represses general protein synthesis and activates the transcription of genes involved in synthesis and transport of amino acids through activation of the ATF4 transcription factor.
- Yet another transcription factor that regulates the X c ’ system is the nuclear factor erythroid 2-related factor (NRF2) that acts on the antioxidant response element (ARE) present in the promoter region of the xCT gene.
- NRF2 nuclear factor erythroid 2-related factor
- ARE antioxidant response element
- One aspect of the present invention is a method to improve the efficacy and/or reduce the side effects of the administration of elesclomol or a derivative, analog, salt, or solvate of elesclomol for treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases or conditions comprising the steps of:
- the factor or parameter is selected from the group consisting of:
- the dose modification can be, but is not limited to, at least one dose modification selected from the group consisting of:
- the route of administration can be, but is not limited to, at least one route of administration selected from the group consisting of:
- the schedule of administration can be, but is not limited to, a schedule of administration selected from the group consisting of:
- the indication for use can be, but is not limited to, an indication for use selected from the group consisting of:
- leukemias including acute and chronic, leukemias, including AML, ALL, CLL, CML
- MDS myelodysplastic syndrome
- the disease stage can be, but is not limited to, a disease stage selected from the group consisting of:
- the other indication can be, but is not limited to, another indication selected from the group consisting of:
- the patient selection can be, but is not limited to, a method of patient selection selected from the group consisting of:
- the analysis of patient or disease phenotype can be, but is not limited to, an analysis of patient or disease phenotype selected from the group consisting of:
- the cellular proto-oncogene c-Jun encodes a protein that, in combination with c-Fos, forms the AP-1 early response transcription factor.
- This proto-oncogene plays a key role in transcription and interacts with a large number of proteins affecting transcription and gene expression. It is also involved in proliferation and apoptosis of cells that form part of a number of tissues, including cells of the endometrium and glandular epithelial cells.
- G-protein coupled receptors GPCRs
- the superfamily of G protein coupled receptors includes a large number of receptors.
- receptors are integral membrane proteins characterized by amino acid sequences that contain seven hydrophobic domains, predicted to represent the transmembrane spanning regions of the proteins. They are found in a wide range of organisms and are involved in the transmission of signals to the interior of cells as a result of their interaction with heterotrimeric G proteins. They respond to a diverse range of agents including lipid analogues, amino acid derivatives, small molecules such as epinephrine and dopamine, and various sensory stimuli. The properties of many known GPCR are summarized in S. Watson & S. Arkinstall, “The G-Protein Linked Receptor Facts Book” (Academic Press, London, 1994), incorporated herein by this reference.
- GPCR receptors include, but are not limited to, acetylcholine receptors, ⁇ - adrenergic receptors, 03-adrenergic receptors, serotonin (5-hydroxytryptamine) receptors, dopamine receptors, adenosine receptors, angiotensin Type II receptors, bradykinin receptors, calcitonin receptors, calcitonin gene-related receptors, cannabinoid receptors, cholecystokinin receptors, chemokine receptors, cytokine receptors, gastrin receptors, endothelin receptors, y-aminobutyric acid (GABA) receptors, galanin receptors, glucagon receptors, glutamate receptors, luteinizing hormone receptors, choriogonadotrophin receptors, follicle-stimulating hormone receptors, thyroid-stimulating hormone receptors, gonadotrophin-releasing hormone receptors, leukotriene receptors, Neuro
- the analysis of patient or disease genotype can be, but is not limited to, an analysis of patient or disease genotype selected from the group consisting of:
- the SNP analysis can be carried out on a gene selected from the group consisting of histone deacetylase, ornithine decarboxylase, VEGF, a prostate specific gene, c-Jun, and a protein kinase.
- SNP analysis is described in S. Levy and Y.-H. Rogers, “DNA Sequencing for the Detection of Human Genome Variation” in Essentials of Genomic and Personalized Medicine (G.S. Ginsburg & H.F. Willard, eds., Academic Press, Amsterdam, 2010), ch. 3, pp. 27-37.
- the pre/post treatment preparation can be, but is not limited to, a method selected from the group consisting of:
- Uricosurics include, but are not limited to, probenecid, benzbromarone, and sulfinpyrazone. A particularly preferred uricosuric is probenecid. Uricosurics, including probenecid, may also have diuretic activity. Other diuretics are well known in the art, and include, but are not limited to, hydrochlorothiazide, carbonic anhydrase inhibitors, furosemide, ethacrynic acid, amiloride, and spironolactone.
- Poly-ADP ribose polymerase inhibitors are described in G.J. Southan & C. Szabo, “Poly(ADP-Ribose) Inhibitors,” Curr. Med. Chem. 10: 321-240 (2003), and include nicotinamide, 3-aminobenzamide, substituted 3,4-dihydroisoquinolin-1 (2H)-ones and isoquinolin-1 (2H)-ones, benzimidazoles, indoles, phthalazin-1 (2H)-ones, quinazolinones, isoindolinones, phenanthridinones, and other compounds.
- Leucovorin rescue comprises administration of folinic acid (leucovorin) to patients in which methotrexate has been administered.
- Leucovorin is a reduced form of folic acid that bypasses dihydrofolate reductase and restores hematopoietic function.
- Leucovorin can be administered either intravenously or orally.
- the uricosuric is probenecid or an analog thereof.
- the toxicity management can be, but is not limited to, a method selected from the group consisting of: (a) use of colchicine or analogs;
Abstract
The present invention is directed to methods and compositions employing elesclomol or derivatives or analogs thereof for treatment of malignancies as well as benign hyperproliferative diseases or conditions, infections, inflammatory diseases or conditions, or immunological diseases or conditions.
Description
COMPOSITIONS AND METHODS TO IMPROVE THE THERAPEUTIC BENEFIT OF REDOX MODULATING AGENTS SUCH AS ELESCLOMOL AND ANALOGS AND DERIVATIVES THEREOF FOR TREATMENT OF BENIGN AND NEOPLASTIC HYPERPROLIFERATIVE DISEASES, INFECTIONS, INFLAMMATORY, AND IMMUNOLOGICAL DISEASES by
Jeffrey A. Bacha & Dennis Brown
CROSS-REFERENCES TO RELATED APPLICATIONS
[0001] This application claims the benefit of United States Provisional Patent Application Serial No. 63/270,382 for Compositions and Methods to Improve the Therapeutic Benefit of Redox Modulating Agents Such as Elesclomol and Analogs and Derivatives Thereof for Treatment of Benign and Neoplastic Hyperproliferative Diseases, Infections, Inflammatory, and Immunological Diseases by Jeffrey A. Bacha and Dennis Brown, filed on October 21 , 2021 , the contents of which are incorporated herein in their entirety by this reference.
FIELD OF THE INVENTION
[0002] This invention is directed to compositions and methods employing elesclomol or derivatives or analogs thereof or related redox agents for treatment of benign and neoplastic hyperproliferative diseases, infections, inflammatory, and immunological diseases.
BACKGROUND OF THE INVENTION
[0003] The search for and identification of cures for many life-threatening diseases that plague humans still remains an empirical and sometimes serendipitous process. While many advances have been made from basic scientific research to improvements in practical patient management, there still remains tremendous frustration in the rational and successful discovery and application of useful therapies,
particularly for life-threatening diseases such as cancer, immune-mediated diseases, inflammatory conditions, and infections, as well as other diseases and conditions such as neurodegenerative conditions.
[0004] Since the “War on Cancer” began in the early 1970’s by the United States National Cancer Institute (NCI) of the National Institutes of Health (NIH), a wide variety of strategies and programs have been created and implemented to prevent, diagnose, treat and cure cancer and other life threatening disease conditions. One of the oldest and arguably most successful programs has been the synthesis and screening of small chemical entities, mostly with molecular weights of less than about 1500 daltons) for biological activity against cancer. These programs were organized to improve and streamline the progression of discovery and development events from chemical synthesis and molecular biology and biological screening to preclinical studies for the logical progression into human clinical trials with the hope of finding cures for the many types of life-threatening diseases including cancer. The synthesis and screening of hundreds of thousands chemical compounds from academic and industrial sources, in addition to the screening of natural products and extracts from prokaryotes, invertebrate animals, plant collections, or other sources, from all over the world as well as novel products exploited by molecular and synthetic biology methodologies has been and continues to be a major approach for the identification of novel lead structures as potential new and useful medicines. This is in addition to other programs including biotherapeutics designed to stimulate the human immune system with adoptive immune cell transfers (e.g., CAR-T), vaccines, therapeutic antibodies, drug-antibody conjugates, cytokines, lymphokines, cytokines, active peptides, inhibitors of tumor blood vessel development (angiogenesis) or gene and antisense therapies to alter the genetic makeup of cancer cells or alter the immune system, as well as agents with other mechanisms. Additionally, new methods employing combinatorial chemistry and high- throughput screening have expanded the options for discovering and employing new medications.
[0005] The work supported by the NCI, other governmental agencies both domestic and foreign in academic or industrial research and development laboratories has resulted in an extraordinary body of biological, genomic, pharmacologic,
biochemical, chemical and clinical information. In addition, large chemical and biological libraries have been created, as well as highly characterized in silico, in vitro and in vivo biological screening systems that have been successfully used. However, from the tens of billions of dollars spent over the past fifty years supporting these programs both preclinically and clinically, only a limited number of therapeutics have been identified or discovered that have resulted in the successful development of useful pharmaceutical products. Nevertheless, the biological systems, including work in silico, in vitro, and in vivo and the “decision trees” used to warrant further preclinical studies leading to Phase l-lll clinical trials have been validated. These drug screening programs, biological models, clinical trial protocols, and other analytical and testing methods, remain critical for the discovery and development of any new therapeutic agent.
[0006] Unfortunately, many of the compounds that have successfully met the preclinical testing and federal regulatory requirements for clinical evaluation were either unsuccessful or disappointing in human clinical trials. Many compounds were found to have untoward or idiosyncratic side effects that were discovered during human clinical Phase I dose-escalation studies used to determine the maximum tolerated dose (MTD) and side-effect profile. In some cases, these toxicities or the magnitude of their toxicity were not identified or predicted in preclinical toxicology studies. In other cases, therapeutic agents where in vitro and in vivo studies suggested a potentially unique activity against a particular tumor type, molecular target or biological pathway were not successful in human Phase II clinical trials where specific examination of particular disease indications/types were evaluated in government sanctioned (e.g., U.S. FDA), IRB approved clinical trials. In addition, there are those cases where potential new agents were evaluated in randomized Phase III clinical trials where a significant clinical benefit could not be demonstrated; these cases have also been the cause of great frustration and disappointment. Finally, a number of compounds have reached regulatory approved commercialization but their ultimate clinical utility has been limited by poor efficacy as monotherapy (e.g., <25% response rates) and for untoward Grade III or IV dose-limiting side-effects (e.g., myelosuppression, cardiotoxicity,
gastrointestinal toxicities, cytokine storm effects) not clearly identified in the course of regulatory clinical trials.
[0007] In many cases, after the great time and expense of developing and moving an investigational compound into human clinical trials and where clinical failure has occurred, the tendency has been to return to the laboratory to create a better analog, look for agents with different structures but potentially related mechanisms of action, or attempt to research agents with substantially different mechanisms of action. In some cases, efforts have been made to try additional Phase I or II clinical trials in an attempt to make some improvement with the side-effect profile or therapeutic effect in selected patients or other disease indications. In many of those cases, the results did not realize a significant enough improvement to warrant further clinical development toward product registration. Even for commercialized products, their ultimate use can still be limited by suboptimal performance in particular clinical contexts.
[0008] For example in oncology, with so few therapeutics approved for cancer patients and the realization that cancer is a collection of diseases with a multitude of etiologies, biological phenotypes or genotype with high rise for drug resistance and susceptible genomic mutations and that a patient’s response and survival from therapeutic intervention is complex with many factors playing a role in the success or failure of treatment including disease indication, pathology stage related to invasion and metastatic spread, patient gender, age, health conditions, previous therapies or other illnesses, genetic background, other diseases or conditions affecting the patient, especially diseases or conditions that tend to induce immunosuppression, the opportunity for significant cures rates without treatment morbidity in the near term remains elusive. Moreover, the incidence of cancer continues to rise such that over 1.9 million new cancer cases are estimated for 2021 in the United States by the American Cancer Society. In addition, with advances in diagnosis such as BRCA genetic testing and mammography for breast cancer and PSA tests for prostate cancer, more patients are being diagnosed at a younger age or at an earlier stage of disease. For difficult to treat cancers, a patient’s treatment options are often exhausted quickly resulting in a desperate need for additional treatment regimens. Even for the most limited of patient populations, any additional treatment opportunities would be of considerable value.
This invention focuses on inventive compositions and methods for improving the therapeutic benefit of suboptimally administered therapeutic agents including redox modulating agents, particularly compounds such as elesclomol or derivatives or analogs thereof.
[0009] Relevant literature includes Foye, W.O., “Cancer Chemotherapeutic Agents,” American Chemical Society, 1995 and Von Hoff, D.D., “Cancer Chemotherapy Handbook,” Appleton and Lange, 1994.
SUMMARY OF THE INVENTION
[0010] The present invention meets the needs described above by providing new methods and compositions employing elesclomol or derivatives or analogs thereof for treatment of a number of diseases and conditions, particularly including, but not limited to, malignancies. The malignancies that can be treatable by methods or compositions employing elesclomol or derivatives or analogs thereof include, but are not limited to, ovarian epithelial cancer (OEC).
[0011] One aspect of the invention is a method to improve the efficacy and/or reduce the side effects of the administration of elesclomol or a derivative, analog, salt, or solvate of elesclomol for treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases or conditions comprising the steps of:
(1 ) identifying at least one factor or parameter associated with the efficacy and/or occurrence of side effects of the administration of the elesclomol or a derivative, analog, salt, or solvate of elesclomol for the treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases; and
(2) modifying the factor or parameter to improve the efficacy and/or reduce the side effects of the administration of the elesclomol or a derivative, analog, salt, or solvate of elesclomol for the treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases.
[0012] Typically, the factor or parameter is selected from the group consisting of: (i) dose modification;
(ii) route of administration;
(iii) schedule of administration;
(iv) indications for use;
(v) disease stages;
(vi) other indications;
(vii) patient selection;
(viii) patient/disease phenotype;
(ix) patient/disease genotype;
(x) pre/post-treatment preparation;
(xi) toxicity management;
(xii) pharmacokinetic/pharmacodynamic monitoring;
(xiii) drug combinations;
(xiv) chemosensitization;
(xv) chemopotentiation;
(xvi) post-treatment patient management;
(xvii) bulk drug product improvements;
(xviii) diluent systems;
(xix) solvent systems;
(xx) excipients;
(xxi) dosage forms;
(xxii) dosage kits and packaging;
(xxiii) drug delivery systems;
(xxiv) drug conjugate forms;
(xxv) compound analogs;
(xxvi) prodrug systems;
(xxvii) multiple drug systems;
(xxviii) biotherapeutic enhancement;
(xxix) biotherapeutic resistance modulation;
(xxx) radiation therapy enhancement;
(xxxi) novel mechanisms of action;
(xxxii) selective target cell population therapeutics;
(xxxiii) reversal of resistance to an agent selected from the group consisting of a platinum-containing anti-neoplastic agent and a PARP inhibitor anti- neoplastic agent;
(xxxiv) induction of synthetic lethality by administration of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1 A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol renders the malignant cells susceptible to a targeted agent that otherwise would have no effect;
(xxxv) modulation of activity of FDX1 to inhibit OXPHOS.
[0013] Typically, the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol. Typically, the elesclomol is in the form of a coordinate- covalent complex with a transition metal cation selected from the group consisting of Ni2+, Cu+, Cu2+, Co2+, Co3+, Fe2+, Fe3+, Zn2+, Pt2+, Pd2+, V4+, V5+, Cr2+, Cr3+, Cr4+, Mn2+, Mn3+, Mn4+, and Mn5+. Preferably, the divalent metal cation is a divalent transition metal cation selected from the group consisting of Ni2+, Cu2+, Co2+, Fe2+, Zn2+, Pt2+, and Pd2+. More preferably, the divalent metal cation is selected from the group consisting of Cu2+ and Ni+2. Most preferably, the divalent metal cation is Cu2+.
[0014] Typically, the treatment is treatment of a malignancy. In one alternative, the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma. Typically, the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC). In another alternative, the malignancy is selected from the group consisting of human sarcomas and carcinomas.
[0015] In another alternative, the treatment is treatment of a disease or condition selected from the group consisting of: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in
cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; and scleroderma.
[0016] In one alternative, the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol and the treatment comprises administration of a therapeutically effective quantity of elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 mg/mm2/day to about 10 g/mm2/day. Typically, the therapeutically effective quantity of elesclomol is from about 2 mg/mm2/day to about 10 g/mm2/day. Alternatively, the therapeutically effective quantity of elesclomol is from about 1 pg/kg to about 500 mg/kg, typically from about 500 pg/kg to about 250 mg/kg, preferably from about 1 mg/kg to about 100 mg/kg, more preferably from about 10 mg/kg to about 50 mg/kg.
[0017] In one alternative, the therapeutically effective quantity of elesclomol or the derivative, analog, salt, or solvate of elesclomol is administered in a pharmaceutical composition.
[0018] Another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of a PARP inhibitor.
[0019] Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an LDH inhibitor.
[0020] Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of 2-deoxyglucose or an analog or derivative thereof.
[0021] Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of a glutamine metabolism inhibitor.
[0022] Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of a DNA- damaging agent.
[0023] Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that inhibits the SWI/SNF complex.
[0024] Still another aspect of the invention is a method for treatment of a malignancy treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that causes tumor cells to rely on oxidative phosphorylation.
[0025] Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that is an inhibitor of the base excision repair (BER) pathway.
[0026] Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that acts as an inhibitor of the homologous repair pathway.
[0027] Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that activates the homologous repair pathway.
[0028] Still another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation.
[0029] Yet another aspect of the invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(1 ) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(2) administering a therapeutically effective quantity of an agent that inhibits cysteine uptake.
[0030] Yet another aspect of the invention is a composition to improve the efficacy or reduce the side effects of treatment with elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol, wherein the composition comprises:
(1 ) an alternative selected from the group consisting of:
(a) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol;
(b) two or more therapeutically active ingredients comprising:
(i) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol; and
(ii) at least one additional therapeutic agent, therapeutic agent subject to chemosensitization, therapeutic agent subject to chemopotentiation, or component of a multiple drug system;
(c) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a dosage form;
(d) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol that is incorporated into a dosage kit and packaging;
(e) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol that is subjected to a bulk drug product improvement;
(f) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug delivery system;
(g) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug conjugate form; and
(h) a therapeutically effective quantity of a prodrug of elesclomol or a derivative or analog of elesclomol; and
(2) at least one pharmaceutically acceptable diluent, solvent or excipient.
[0031] Typically, in the composition, the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
[0032] Typically, as described above, the elesclomol is in the form of a coordinate-covalent complex with a transition metal cation selected from the group consisting of Ni2+, Cu+, Cu2+, Co2+, Co3+, Fe2+, Fe3+, Zn2+, Pt2+, Pd2+, V4+, V5+, Cr2+, Cr3+, Cr4+, Mn2+, Mn3+, Mn4+, and Mn5+.
[0033] Typically, the composition is formulated for treatment of a malignancy. Malignancies that can be treated by methods or compositions according to the present invention are described above.
[0034] Alternatively, the composition is formulated for treatment of a disease or condition other than a malignancy as described above.
[0035] Compositions according to the present invention can be formulated for administration of therapeutically effective dosages such as from about 1 mg/mm2/day to about 10 g/mm2/day or, alternatively, from about 1 pg/kg to about 500 mg/kg.
[0036] When the composition comprises an additional therapeutic agent, in one alternative the additional therapeutic agent is selected from the group consisting of:
(i) topoisomerase inhibitors;
(ii) fraudulent nucleosides;
(iii) fraudulent nucleotides;
(iv) thymidylate synthetase inhibitors;
(v) signal transduction inhibitors;
(vi) cisplatin or gallium analogs;
(vii) nitrosourea alkylating agents (BCNU , Gliadel wafers, CCNU);
(viii) bendamustine (Treanda);
(ix) anti-tubulin agents;
(x) antimetabolites;
(xi) berberine;
(xii) apigenin;
(xiii) amonafide;
(xiv) colchicine or an analog thereof;
(xv) genistein;
(xvi) etoposide;
(xvii) cytarabine;
(xviii) a camptothecin;
(xix) vinca alkaloids;
(xx) 5-fluorouracil;
(xxi) curcumin;
(xxii) NF-KB inhibitors;
(xxiii) rosmarinic acid;
(xxiv) biological therapies selected from the group consisting of Avastin, Rituxan, Herceptin, Erbitux, PD-1 inhibitors, and PD-L1 inhibitors;
(xxv) prednimustine;
(xxvi) DNA or RNA therapeutics;
(xxvii) Braf inhibitors;
(xxviii) BTK inhibitors;
(xxix) 5-azacytidine;
(xxx) decitabine;
(xxxi) PARP inhibitors;
(xxxii) agents inducing hypomethylation; and
(xxxiii) histone deacetylase inhibitors.
[0037] In another alternative, the additional therapeutic agent can be selected from the group consisting of a microtubulin stabilizer, a microtubulin inhibitor, a PARP inhibitor, an LDH inhibitor, 2-deoxyglucose or an analog or derivative thereof, a glutamine metabolism inhibitor, a DNA-damaging agent, an agent that inhibits the SWI/SNF complex, an agent that causes tumor cells to rely on oxidative phosphorylation, an agent that is an inhibitor of the base excision repair (BER) pathway, an agent that acts as an inhibitor of the homologous repair pathway, an agent that acts as an activator of the homologous repair pathway, an agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation, and an agent that inhibits cysteine uptake.
[0038] The composition can comprise a pharmaceutically acceptable diluent, a pharmaceutically acceptable solvent, or a pharmaceutically acceptable excipient.
BRIEF DESCRIPTION OF THE DRAWINGS
[0039] The following invention will become better understood with reference to the specification, appended claims, and accompanying drawings, where:
[0040] Figure 1 is a graph showing the results for progression-free survival (PFS) and overall survival (OS) for serous adenocarcinoma, endometrioid adenocarcinoma, mixed epithelial carcinoma, clear-cell carcinoma, and other types of ovarian cancer.
[0041] Figure 2 is a diagram showing the involvement of ARID1A in pathways of tumor growth, tumor migration, tumor invasion, and angiogenesis.
[0042] Figure 3 is a graph showing that progression free survival in patients exhibiting an ARIDIA-deficient phenotype is significantly reduced as compared with ARID1 A wild-type in patients treated with PARP inhibitors: (A) showing the impact of ARID1 A loss, determined via mutation, homozygous deletion, or loss of expression, and survival in TCGA serous ovarian cancers; (B) showing the progression-free survival of relapsed, platinum-sensitive, high-grade ovarian carcinomas in a clinical trial of rucaparib (a PARP inhibitor), stratified based on ARID1A mutation status.
[0043] Figure 4 is a graph showing that loss of ARID1 A expression correlates with shorter progression-free survival in ovarian clear cell carcinoma patients who
received primary cytoreductive surgery followed by standard platinum-based chemotherapy: (a) Kaplan-Meier survival analysis showing that negative ARID1 A expression (solid line, n=9) is associated with a shorter progression-free survival than positive ARID1 A expression (dashed line, n=51 ) ( P<0.01 , Log-rank test), (b) Negative ARID1A expression (solid line, n= 9) had a statistically nonsignificant effect on overall survival in ovarian clear cell carcinoma patients who received primary cytoreductive surgery followed by standard platinum-based chemotherapy ( P =0.15, Log-rank test).
[0044] Figure 5 is a graph showing that loss of ARID1 A results in downregulation of cystine transporter SLC7A11 and impairment of GSH pathway leading to ROS accumulation and cell death: (A) ARID1A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway. (B) Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
[0045] Figure 6 is a graph that shows the difference between cells with ARID1A mutations and cells with active ARID1 A in terms of the effect of accumulation of ROS: (A) ARID1A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway. (B) Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
[0046] Figure 7 is a graph that shows a therapeutic window for cancer cells in the presence of an OXPHOS inhibitor. (Top Panel) Normal body cells have moderate ATP demand and adequate levels of oxygen and glucose and survive in presence of OXPHOS inhibitors by upregulating glycolysis to meet their ATP demands. (Middle Panel). Highly proliferating cancer cells have extraordinarily high ATP demand and adequate levels of oxygen and glucose. Despite glycolytic pathway upregulation, OXPHOS inhibition results in failure to meet ATP demand and cell death. (Bottom Panel) Quiescent cancer cells have low ATP demand but live in a highly compromised microenvironment (low glucose and hypoxia). Inhibition of OXPHOS is lethal as
insufficient glucose is present to compensate for the loss of ATP production by oxidative phosphorylation.
[0047] Figure 8 is a diagram showing the conformational change undergone by elesclomol upon chelation with copper.
[0048] Figure 9 is a graph showing the synergy of elesclomol with PARP inhibitors. In Figure 9, drug sensitivity was assessed following treatment of mutant BRCA1 cells (SUM149) with elesclomol including paclitaxel (A) and PARP inhibitors talazoparib (B) and rucaparib (C). After generating a line of additivity ( — ) using the IC50 values for each drug alone (•), the IC50 values for each combined treatment («) are plotted as data points on a Cartesian plot. Data points below the line of additivity represent synergism, whereas those above or on the line indicate antagonism or an additive effect, respectively.
[0049] Figure 10 is a diagram showing gene screens indicating genes whose activity is affected by elesclomol: (A) Gene scores in elesclomol-1 (100 nM) and elesclomol-2 (1 μM) treated K562 cells. The gene score is the median Iog2 fold change in abundance of all sgRNAs targeting that gene during the culture period. (B) Corrected p-values (-logio) of KS tests of the sgRNA distribution for each gene versus the distribution of all sgRNAs in the screen of elesclomol-1 (100 nM) and elesclomol-2 (1 μM) screens. Values are ordered on the x-axis by chromosome and location; the dotted line indicates a corrected p-value of 0.05.
[0050] Figure 11 is a graph showing results that indicate elesclomol inhibits the natural function of FDX1 in FE-S cluster biosynthesis, (a) Chemical shift perturbation results mapped onto a diagram of the structure of FDX1 . Color code: gray, not significantly affected (A6NH < 0.01 ppm); blue, significant chemical shift changes (A6NH > 0.01 ppm); red, severe line broadening; gray, no assignments. The [2Fe-2S] cluster in FDX1 is indicated by spheres, (b) Schematic describing mitochondrial Fe-S cluster biosynthesis. The mitochondrial ISC core complex catalyzes the conversion of cysteine to alanine and generates S° for iron-sulfur cluster assembly. S° is reduced by FDX1 . A [2Fe-2S] cluster is subsequently formed on the scaffold protein ISCII. (c) In vitro Fe-S cluster assembly was monitored by following the increase of absorbance at 456 nm. Reduced FDX1 was used as a reducing agent in the presence or absence of either 5x
(green) or 10x (blue) elesclomol. Data are representative of two independent experiments, (d) Electron transfer from reduced FDX1 to the cysteine desulfurase complex was measured on the addition of cysteine in the presence or absence of elesclomol. Data are representative of two independent experiments, (e) The molecular structure of elesclomol analogs and their corresponding calculated EC50 (from T47D cells grown in the Hi-Mito state). The reactive sulfur substitution with an oxygen is indicated with an asterisk, (f) The UV/visible spectra of reduced FDX1 before and after incubation with elesclomol-Cu(ll) or Cu(ll) alone, that elesclomol inhibits the natural function of FDX1 in FE-S cluster biosynthesis, (a) Chemical shift perturbation results mapped onto a diagram of the structure of FDX1 . Color code: gray, not significantly affected (A6NH < 0.01 ppm); blue, significant chemical shift changes (A6NH > 0.01 ppm); red, severe line broadening; gray, no assignments. The [2Fe-2S] cluster in FDX1 is indicated by spheres, (b) Schematic describing mitochondrial Fe-S cluster biosynthesis. The mitochondrial ISC core complex catalyzes the conversion of cysteine to alanine and generates S° for iron-sulfur cluster assembly. S° is reduced by FDX1 . A [2Fe-2S] cluster is subsequently formed on the scaffold protein ISCII. (c) In vitro Fe-S cluster assembly was monitored by following the increase of absorbance at 456 nm. Reduced FDX1 was used as a reducing agent in the presence or absence of either 5x (green) or 10x (blue) elesclomol. Data are representative of two independent experiments, (d) Electron transfer from reduced FDX1 to the cysteine desulfurase complex was measured on the addition of cysteine in the presence or absence of elesclomol. Data are representative of two independent experiments, (e) The molecular structure of elesclomol analogs and their corresponding calculated EC50 (from T47D cells grown in the Hi-Mito state). The reactive sulfur substitution with an oxygen is indicated with an asterisk, (f) The UV/visible spectra of reduced FDX1 before and after incubation with elesclomol-Cu(ll) or Cu(ll) alone.
[0051] Figure 12 is a graph showing that sensitivity to elesclomol as a single agent is highly specific to ARID1A mutation in cancer cell lines. (A) Cell growth of endometrial and ovarian cancer cell lines treated with elesclomol for 72 h as measured using the WST-1 assay. (B) IC50 values of elesclomol in these cell lines.
[0052] Figure 13 is a diagram showing that ARIDIA-mutant tumors are highly dependent on OXPHOS offering a potential strategy for synthetic lethality employing targeted agents interrupting mitochondrial metabolism.
[0053] Figure 14 is a diagram showing that the protein target of elesclomol is ferredoxin-1 , a key component of the OXPHOS pathway establishing a synthetically lethal therapeutic strategy in ARIDIA-mutant tumors.
[0054] Figure 15 is a diagram showing the occurrence of ARID1 A mutations in a number of types of malignancies, including mutations, deletions, amplification, and multiple alterations.
[0055] Figure 16 is a series of graphs showing the effects of a 72-hour treatment with elesclomol on the viability of either the OVCA429 NTC (non-targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line. Figure 16A shows the viability of these cell lines after 72 hours of treatment with 10 nM or 100 nM of elesclomol; viability is determined by the MTT assay (• = OVCA429 NTC; ■ = OVCA429 ARID1 A mutant). Figure 16B shows the viability of these cell lines after 72 hours of treatment with 1 nM, 2 nM, 3 nM, 4 nM, 5 nM, 6 nM, 7 nM, 8 nM, 9 nM, 10 nM, or 100 nm of elesclomol; viability is determined by the MTT assay (• = OVCA429 NTC; ■ = OVCA429 ARID1 A mutant). Figure 16C shows the viability of these cell lines after 72 hours of treatment with 10 nM, 20 nM, 30 nM, 40 nM, 50 nM, 60 nM, 70 nM, 80 nM, 90 nM, or 100 nM of elesclomol; viability is determined by the MTT assay (• = OVCA429 NTC; ■ = OVCA429 ARID1 A mutant). In all cases, OVCA429 ARID1 A mutant cells are more sensitive to elesclomol than the OVCA429 NTC cells.
[0056] Figure 17 is a series of graphs showing the effects of a 72-hour treatment with elesclomol on the viability of either the RMG1 NTC (non-targeted control) clear cell ovarian cancer cell line or the RMG1 ARID1 A mutant cell line. Figure 17A shows the viability of these cell lines after 72 hours of treatment with 10 nM, 100 nM, or 1000 nM of elesclomol; viability is determined by the MTT assay (• = RMG1 NTC; ■ = RMG1 ARID1 A mutant). Figure 17B shows the viability of these cell lines after 72 hours of treatment with 10 nM, 20 nM, 30 nM, 40 nM, 50 nM, 60 nM, 70 nM, 80 nM, 90 nM, or 100 nM of elesclomol; viability is determined by the MTT assay (• = RMG1 NTC; ■ =
RMG1 ARID1 A mutant). In all cases, the RMG1 ARID1 A mutant cells were more sensitive to elesclomol than the RMG1 NTC cells.
[0057] Figure 18 is a series of graphs showing the results over time of treatment of either the OVCA429 NTC (non-targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line with 1 ng, 10 ng, 100 ng, or 1000 ng of elesclomol for a time interval of up to 72 hours. Figure 18A shows the results for OVCA429 NTC; Figure 18B shows the results for OVCA429 ARID1 A mutant; Figure 18C overlays the results from Figures 18A and Figure 18B. In all cases, the mutant cells were more sensitive to elesclomol as a function of time than were the OVCA429 ARID1A NTC cells. Although Figures 16 and 17 do show a clear difference in results between the ARID1 A wild-type and the ARID1A mutant phenotype, the difference in results is not clear in Figure 18. This is simply an artifact of the assays. The Incucyte analyses shown in Figure 18 are based on bright-field images which provide an analysis of confluence so does not differentiate between viable and dying cells, whereas Figures 16 and 17 measure only viable cells.
[0058] Figure 19 shows photomicrographs of either the OVCA429 NTC (non- targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line that were either not treated with elesclomol or treated with 10 nM of elesclomol. (A) OVCA429 NTC not treated with elesclomol; (B) OVCA429 ARID1 A mutant cell line not treated with elesclomol; (C) OVCA429 NTC treated with 10 nM of elesclomol; and (D) OVCA429 ARID1 A mutant cell line treated with 10 nM of elesclomol. In all cases, treatment with elesclomol resulted in cell death visible in the photomicrographs.
[0059] Figure 20 is a schematic diagram showing the mechanisms of action of elesclomol in cells with wild-type ARID1 A and in mutant ARID1 A, in particular the occurrence of reactive oxygen species (ROS) in cells with mutant ARID1 A induced by elesclomol, leading to cytotoxicity.
[0060] Figure 21 is a schematic diagram showing that loss-of-function mutations in ARID1 A promotes oncogenesis and reliance on OXPHOS.
[0061] Figure 22 is a schematic diagram showing that elesclomol induces synthetic lethality in tumor cells with loss-of-function mutations in ARID1A.
DEFINITIONS
[0062] Although any methods and materials similar to or equivalent to those described herein can be used in the practice or testing of embodiments described herein or other embodiments within the scope of the invention, some preferred methods, compositions, materials, and devices are described herein. However, in this context, it must be understood that this invention is not limited to the particular molecules, compositions, methodologies, or protocols described herein, as these aspects of the invention may vary in accordance with routine experimentation and optimization as is generally known in the art. It is also to be understood that the terminology used in the description and the claims is for the purpose of describing the particular versions or embodiments only, and is not intended to limit the scope of the embodiments as described herein as understood by one of skill in the art.
[0063] Unless otherwise defined, all technical and scientific terms used herein have the same meanings as commonly understood by one of ordinary skill in the art to which this invention belongs. However, in case of any conflict of meanings, the present specification and claims, including definitions therein, shall control. Accordingly, in the context of the embodiments described herein, the following definitions apply.
[0064] As used herein and in the appended claims, the singular forms “a,” “an,” and “the” include references to the plural unless the context clearly dictates otherwise. Thus, for example, a reference to “a PARP inhibitor” is a reference to one or more PARP inhibitors or equivalents thereof known to those skilled in the art.
[0065] As used herein, the terms “comprise,” “include,” and linguistic variations thereof denote the presence of recited features, elements, method steps, or other components of the invention without the exclusion of the presence of additional /recited features, elements, method steps, or other components. Conversely, the terms “consisting of’ and linguistic variations thereof denote the presence of recited features, elements, method steps, or other components of the invention and exclude any unrecited recited features, elements, method steps, or other components of the invention except for ordinarily-associated impurities. The phrase “consisting essentially of” and linguistic variations thereof denote the presence of recited features, elements, method steps, or other components of the invention and any additional features,
elements, method steps, or other components of the invention that do not materially affect the basic nature of the composition, system, or method. Many embodiments herein are described using open “comprising” language; such embodiments also encompass embodiments described in terms of “consisting essentially of” or “consisting of” language, which may be alternatively claimed or described using such language, unless the context clearly excludes “consisting essentially of’ or “consisting of” language.
[0066] All chemical names used herein, including names of substituents, should be interpreted in light of the chemical nomenclature conventions of IUPAC and/or a modified format in which functional groups within a substituent are read in the order in which they branch from the scaffold or main structure. For example, in the modified nomenclature, methylsulfonylpropanol refers to CH2SO2CH2CH2CH2OH or

As another example, according to the modified nomenclature, a methylamine substituent is
 while an aminomethyl substituent is
while an aminomethyl substituent is
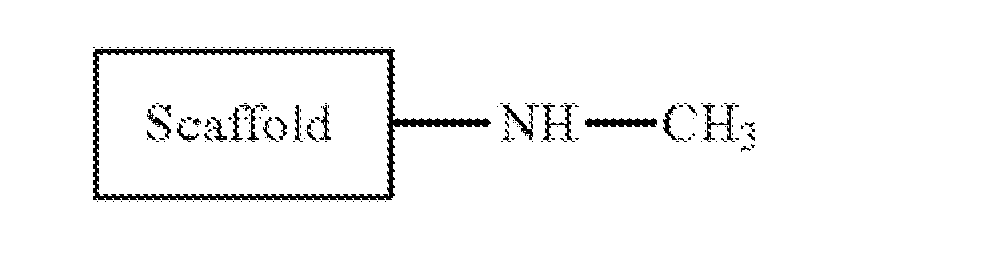
[0067] As used herein, the term “subject” broadly refers to any animal, including, but not limited to, humans and non-human mammals. The reference to non-human mammals includes, but is not limited to, socially or economically important animals or animals used for research including cattle, sheep, goats, horses, pigs, llamas, alpacas, dogs, cats, rabbits, guinea pigs, rats, and mice. Unless specified, methods and
compositions according to the present invention are not limited to treatment of humans. In general, when treatment of humans is intended, the term “patient” can used in place of “subject.”
[0068] As used herein, the terms “effective amount,” “therapeutically effective amount,” or other equivalent terminology refer to the amount of a compound or compounds or to the amount of a composition sufficient to effect beneficial or desired results. The beneficial or desired results are typically a reduction in severity, symptoms, or duration of a disease or condition being treated and can generally be characterized as an amount of a therapeutic agent or composition effective to treat, ameliorate, or prevent a desired disease or condition, or to exhibit a detectable therapeutic or preventative effect. The use of such terminology cannot, unless specifically indicated, be interpreted as implying a complete cure for any disease or condition as recited herein. An effective amount can be administered in one or more administrations, applications, or dosages, and is not intended to be limited to a particular formulation or administration route unless a particular formulation or administration route is specified. The effect induced by the administration of a therapeutically effective amount can be detected by, for example, chemical markers, antigen levels, or changes in pathological indicators such as tumor burden. Therapeutic effects also can include subjective improvements in well-being, reduction of fatigue, or increased energy noted by the subjects or their caregivers. The precise therapeutically effective amount for a subject will depend upon the subject’s size, weight, and health, the nature and extent of the condition affecting the subject, the administration of other therapeutics administered to treat the particular disease or condition being treated or other diseases or conditions affecting the subject, as well as variables such as liver and kidney function that affect the pharmacokinetics of administered therapeutics. Thus, it is not useful to specify an exact effective amount in advance. However, the therapeutically effective amount for a given situation can be determined by routine experimentation and is within the judgment of the clinician.
[0069] As used herein, the terms “administration,” “administering,” or other equivalent terminology, refer to the act of giving a drug, prodrug, pharmaceutical composition, or other agent intended to provide therapeutic treatment to a subject or in
vivo, in vitro, or ex vivo to cells, tissues, or organs. Exemplary routes of administration to the human body can be through space under the arachnoid membrane of the brain or spinal cord (intrathecal), the eyes (ophthalmic), mouth (oral), skin (topical or transdermal), nose (nasal), lungs or other portions of the respiratory tract (inhalant), oral mucosa (buccal), ear, rectal, vaginal, by injection (such as, but not limited to, intravenously, subcutaneously, intraperitoneally, or by other injection routes as known in the art).
[0070] As used herein, the terms “co-administration” and “co-administering” refer to the administration of at least two agents, such as, for example, elesclomol or a derivative or analog thereof and a PARP inhibitor, or therapies to a subject. In some embodiments, the co-administration of two or more agents or therapies is concurrent. In other embodiments, a first agent/therapy is administered prior to a second agent/therapy. Those of skill in the art understand that the formulations and/or routes of administration of the various agents or therapies used may vary. The appropriate dosage for co-administration can be readily determined by one skilled in the art. In some embodiments, when agents or therapies are co-administered, the respective agents or therapies are administered at lower dosages than appropriate for their administration alone. Thus, co-administration is especially desirable in embodiments where the co-administration of the agents or therapies lowers the requisite dosage of a potentially harmful agent or agent, and/or when co-administration of two or more agents results in sensitization of a subject to beneficial effects of one of the agents via co- administration of the other agent. As used herein, the term “concurrent administration” refers to the administration of two or more active agents sufficiently close in time to achieve a combined therapeutic effect that is preferably greater than that which would be achieved by the administration of either agent alone. Such concurrent administration can be carried out simultaneously, e.g., by administering the active agents together in a common pharmaceutically acceptable carrier, thereby forming a pharmaceutical composition with two or more active agents, in one or more doses of the pharmaceutical composition.
[0071] As used herein, the term “pharmaceutical composition” refers to the combination of one or more therapeutically active agents with at least one carrier, inert
or active, making the composition especially suitable for diagnostic or therapeutic use in vitro, in vivo or ex vivo.
[0072] As used herein, the terms “pharmaceutically acceptable” or “pharmacologically acceptable,” as used herein, refer to compositions, or components within compositions, that do not substantially produce adverse reactions, such as, but not limited to, toxic, allergic, or unwanted immunological reactions, when administered to a subject.
[0073] As used herein, the term “pharmaceutically acceptable carrier” refers to any of the standard pharmaceutical carriers including, but not limited to, phosphate buffered saline solution, water, emulsions, such as oil/water or water/oil emulsions), and various types of wetting agents, any and all solvents, dispersion media, coatings, sodium lauryl sulfate, isotonic and absorption delaying agents, disintegrants such as potato starch or sodium starch glycolate), and the like. The carriers also can include stabilizers and preservatives.
[0074] As used herein, the term “pharmaceutically acceptable salt” refers to any pharmaceutically acceptable salt (e.g., acid or base) of a compound that is used in a method of the present invention or is a component of a composition of the present invention, which, upon administration to a subject, is capable of providing a compound of the present invention or an active metabolite or residue thereof. As is known to those of skill in the art, salts of the compounds of the present invention may be derived from inorganic or organic acids and bases. Examples of acids include, but are not limited to, hydrochloric, hydrobromic, sulfuric, nitric, perchloric, fumaric, maleic, phosphoric, glycolic, lactic, salicylic, succinic, toluene-p-sulfonic, tartaric, acetic, citric, methanesulfonic, ethanesulfonic, formic, benzoic, malonic, naphthalene-2-sulfonic, benzenesulfonic acid, and other acids known in the art as suitable for formation of pharmaceutically acceptable salts. Other acids, such as oxalic, while not in themselves pharmaceutically acceptable, may be employed in the preparation of salts useful as intermediates in obtaining the compounds of the invention and their pharmaceutically acceptable acid addition salts. Examples of bases include, but are not limited to, alkali metals (such as sodium or potassium) hydroxides, alkaline earth metals (such as calcium or magnesium), hydroxides, ammonia, and compounds of formula NW4+,
wherein W is C1-C4 alkyl, and the like. Examples of salts include, but are not limited to: acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, flucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2- hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, palmoate, pectinate, persulfate, phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, tosylate, undecanoate, and the like. Other examples of salts include anions of the compounds of the present invention compounded with a suitable cation such as Na+, NH4+, and NW4+, wherein W is a C1-C4 alkyl group), and the like. For therapeutic use, salts of the compounds herein are contemplated as being pharmaceutically acceptable. However, salts of acids and bases that are non-pharmaceutically acceptable may also find use, for example, in the preparation or purification of a pharmaceutically acceptable compound.
[0075] As used herein, the term “instructions for administering a compound to a subject,” and grammatical equivalents thereof, includes instructions for using the compositions contained in a kit for the treatment of conditions. Such instructions, for example, provide dosing, routes of administration, or decision trees for treating physicians for correlating patient-specific characteristics with therapeutic courses of action. Such instructions may be part of a kit according to the present invention.
[0076] The following applies to analogs and derivatives of the compounds described in further detail below, including elesclomol and other therapeutically active agents described herein. As used herein, “analog” refers to a chemical compound that is structurally similar to a parent compound, but differs slightly in composition (e.g., one atom or functional group is different, added, or removed). The analogue may or may not have different chemical or physical properties than the original compound and may or may not have improved biological and/or chemical activity. For example, the analogue may be more hydrophilic or hydrophobic or it may have altered reactivity as compared to the parent compound. The analogue may mimic the chemical and/or biologically activity of the parent compound (i.e. , it may have similar or identical activity), or, in some cases, may have increased or decreased activity. The analogue may be a
naturally or non-naturally occurring variant of the original compound. Other types of analogues include isomers (enantiomers, diastereomers, and the like) and other types of chiral variants of a compound, as well as structural isomers. As used herein, “derivative” refers to a chemically or biologically modified version of a chemical compound that is structurally similar to a parent compound and (actually or theoretically) derivable from that parent compound. A “derivative” differs from an “analog” in that a parent compound may be the starting material to generate a “derivative,” whereas the parent compound may not necessarily be used as the starting material to generate an “analog.” A derivative may or may not have different chemical or physical properties than the parent compound. For example, the derivative may be more hydrophilic or hydrophobic or it may have altered reactivity as compared to the parent compound. Derivatization (i.e. , modification) may involve substitution of one or more moieties within the molecule (e.g., a change in functional group). The term “derivative” also includes conjugates and prodrugs of a parent compound (i.e., chemically modified derivatives which can be converted into the original compound under physiological conditions).
[0077] As used herein, the term “alkyl” refers to an unbranched, branched, or cyclic saturated hydrocarbyl residue, or a combination thereof, of from 1 to 12 carbon atoms, or in some cases up to 50 or more carbon atoms, that can be optionally substituted; the alkyl residues contain only C and H when unsubstituted. Typically, the unbranched or branched saturated hydrocarbyl residue is from 1 to 6 carbon atoms, preferably 1 to 4 carbon atoms, more preferably 1 to 3 carbon atoms, which is referred to herein as “lower alkyl.” When the alkyl residue is cyclic and includes a ring, it is understood that the hydrocarbyl residue includes at least three carbon atoms, which is the minimum number to form a ring. An alkyl group can be linear, branched, cyclic, or a combination thereof, and may contain from 1 to 50 or more carbon atoms, such as a straight chain or branched C1-C20 alkane. Examples of alkyl groups include but are not limited to methyl, ethyl, propyl, isopropyl, cyclopropyl, butyl isomers (e.g. n-butyl, isobutyl, and te/Y-butyl), cyclobutyl isomers (e.g. cyclobutyl, methylcyclopropyl, or other isomers), pentyl isomers, cyclopentane isomers, hexyl isomers, cyclohexane isomers, and the like. Unless specified otherwise (e.g., substituted alkyl group, heteroalkyl, alkoxy group, haloalkyl, alkylamine, thioalkyl, or other possible groups), an alkyl group
contains carbon and hydrogen atoms only. As used herein, the term “linear alkyl” refers to a chain of carbon and hydrogen atoms (e.g., ethane, propane, butane, pentane, hexane, or other examples). A linear alkyl group may be referred to by the designation - -(CH2)qCH3, where q is 0-49. The designation “C1-C12 alkyl” or a similar designation refers to alkyl having from 1 to 12 carbon atoms such as methyl, ethyl, propyl isomers (e.g. n-propyl or isopropyl), butyl isomers, cyclobutyl isomers (e.g. cyclobutyl or methylcyclopropyl), pentyl isomers, cyclopentyl isomers, hexyl isomers, cyclohexyl isomers, heptyl isomers, cycloheptyl isomers, octyl isomers, cyclooctyl isomers, nonyl isomers, cyclononyl isomers, decyl isomers, cyclodecyl isomers, or other alternatives known in the art. Similar designations refer to alkyl with a number of carbon atoms in a different range. As used herein, the term “Cx-Cy” when used in conjunction with a chemical moiety, such as alkyl, alkenyl, alkynyl, or carbocycle is meant to include groups that contain from x to y carbons in the chain or ring. For example, the term “Cx-Cy alkyl” refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups that contain from x to y carbons in the chain, including haloalkyl groups such as trifluoromethyl and 2,2,2-trifluoroethyl, or other alternatives. The terms “Cx-Cy alkenyl” and “Cx-Cy alkynyl” refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively. The term “Cx-Cy carbocycle” refers to a substituted or unsubstituted carbocycle, that contain from x to y ring carbons. As used herein, the term "branched alkyl" refers to a chain of carbon and hydrogen atoms, without double or triple bonds, that contains a fork, branch, and/or split in the chain (e.g., 3,5-dimethyl-2-ethylhexane, 2-methyl-pentane, 1-methyl-cyclobutane, ortho -diethyl-cyclohexane, or other alternatives). “Branching” refers to the divergence of a carbon chain, whereas “substitution” refers to the presence of non-carbon/non-hydrogen atoms in a moiety. Unless specified otherwise (e.g., substituted branched alkyl group, branched heteroalkyl, branched alkoxy group, branched haloalkyl, branched alkylamine, branched thioalkyl, or other alternatives), a branched alkyl group contains carbon and hydrogen atoms only.
[0078] As used herein, the term “carbocycle,” “carbocyclyl,” or “carbocyclic” refers to a cyclic ring containing only carbon atoms in the ring, whereas the term “heterocycle” or “heterocyclic” refers to a ring comprising a heteroatom. The carbocycle can be fully saturated or partially saturated, but non-aromatic. For example, the general term “carbocyclyl” encompasses cycloalkyl. The carbocyclic and heterocyclic structures encompass compounds having monocyclic, bicyclic or multiple (polycyclic) ring systems; and such systems may mix aromatic, heterocyclic, and carbocyclic rings. Mixed ring systems are described according to the ring that is attached to the rest of the compound being described. Bicyclic or polycyclic rings may include fused or spiro rings. Carbocycles may include 3- to 10-membered monocyclic rings, 6- to 12- membered bicyclic rings, and 6- to 12-membered bridged rings. Each ring of a bicyclic or polycyclic carbocycle may be selected from saturated, unsaturated, and aromatic rings. In an exemplary embodiment, an aromatic carbocycle, e.g., phenyl, may be fused to a saturated or unsaturated ring, e.g., cyclohexane, cyclopentane, or cyclohexene. In some embodiments, the carbocycle is an aromatic carbocycle . In some embodiments, the carbocycle is a cycloalkyl. In some embodiments, the carbocycle is a cycloalkenyl. Exemplary carbocycles include cyclopentyl, cyclohexyl, cyclohexenyl, adamantyl, phenyl, indanyl, and naphthyl. An alkenyl group can be optionally substituted by one or more substituents such as those substituents described herein. A “non-aromatic carbocycle” includes rings and ring systems that are saturated, unsaturated, substituted or unsubstituted, but not aromatic or aryl rings or ring systems.
[0079] As used herein, the term “cycloalkyl” refers to a completely saturated mono- or multi-cyclic hydrocarbon ring system. When composed of two or more rings, the rings may be joined together in a fused, bridged or spiro-connected fashion. Cycloalkyl groups of the present application may range from three to ten carbons (C3 to C10). A cycloalkyl group may be unsubstituted, substituted, branched, and/or unbranched. Typical cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like. If substituted, the substituent(s) may be an alkyl or can be selected from those indicated above with regard to substitution of an alkyl group unless otherwise indicated. While “alkyl” as used herein includes cycloalkyl and cycloalkylalkyl groups, the term “cycloalkyl” may be used herein to
describe a carbocyclic non-aromatic group that is connected via a ring carbon atom, and “cycloalkylalkyl” may be used to describe a carbocyclic non-aromatic group that is connected to the molecule through an alkyl linker.
[0080] As used herein, the term “heteroalkyl” refers to an alkyl group, as defined herein, wherein one or more carbon atoms are independently replaced by one or more heteroatoms (e.g., oxygen, sulfur, nitrogen, phosphorus, selenium, silicon, or combinations thereof). The alkyl group containing the non-carbon substitution(s) may be a linear alkyl, branched alkyl, cycloalkyl (e.g., cycloheteroalkyl), or combinations thereof. Non-carbons may be at terminal locations (e.g., 2-hexanol) or integral to an alkyl group (e.g., diethyl ether). In general, the “hetero” terms refer to groups that typically contain 1-3 O, S or N heteroatoms or combinations thereof within the backbone residue; thus at least one carbon atom of a corresponding alkyl, alkenyl, or alkynyl group is replaced by one of the specified heteroatoms to form, respectively, a heteroalkyl, heteroalkenyl, or heteroalkynyl group. In some cases, more than three heteroatoms may be present. Unless stated otherwise specifically in the specification, the heteroalkyl group may be optionally substituted as described herein. Representative heteroalkyl groups include, but are not limited to -OCH2OMe, - OCH2CH2OMe, or --OCH2CH2OCH2CH2NH2. For reasons of chemical stability, it is also understood that, unless otherwise specified, such groups do not include more than two contiguous heteroatoms except where an oxo group is present on N or S as in a nitro or sulfonyl group.
[0081] As used herein, the term “heteroalkylene” refers to an alkyl radical as described above where one or more carbon atoms of the alkyl is replaced with a heteroatom, e.g., O, N or S, or another heteroatom as described above. “Heteroalkylene” or “heteroalkylene chain” refers to a straight or branched divalent heteroalkyl chain linking the rest of the molecule to a radical group. Unless stated otherwise specifically in the specification, the heteroalkylene group may be optionally substituted as described herein. Representative heteroalkylene groups include, but are not limited to -OCH2CH2O-, -OCH2CH2OCH2CH2O-, or - OCH2CH2OCH2CH2OCH2CH2O-.
[0082] As used herein, the term “optionally substituted” indicates that the particular group or groups referred to as optionally substituted may have no non- hydrogen substituents, or the group or groups may have one or more non-hydrogen substituents consistent with the chemistry and pharmacological activity of the resulting molecule and such that a stable compound is formed thereby, i.e. , a compound that does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, hydrolysis, lactone or lactam formation, or other reaction. If not otherwise specified, the total number of such substituents that may be present is equal to the total number of hydrogen atoms present on the unsubstituted form of the group being described; fewer than the maximum number of such substituents may be present. Where an optional substituent is attached via a double bond, such as a carbonyl oxygen (C=O), the group takes up two available valences on the carbon atom to which the optional substituent is attached, so the total number of substituents that may be included is reduced according to the number of available valences. As used herein, the term “substituted,” whether used as part of “optionally substituted” or otherwise, when used to modify a specific group, moiety, or radical, means that one or more hydrogen atoms are, each, independently of each other, replaced with the same or different substituent or substituents. Substitution of a structure depicted herein may result in removal or moving of a double bond or other bond, as will be understood by one in the field. In certain embodiments, substituted refers to moieties having substituents replacing two hydrogen atoms on the same carbon atom, such as substituting the two hydrogen atoms on a single carbon with an oxo, imino or thioxo group. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds that do not significantly alter the pharmacological activity of the compound in the context of the present invention. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds. The permissible substituents can be one or more and the same or different for appropriate organic compounds. The heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
[0083] As used herein, the term “haloalkyl” or “haloalkane” refers to an alkyl radical, as defined above, that is substituted by one or more halogen radicals, for example, trifluoromethyl, dichloromethyl, bromomethyl, 2,2,2-trifluoroethyl, 1- fluoromethyl-2-fluoroethyl, and the like. In some embodiments, the alkyl part of the fluoroalkyl radical is optionally further substituted. Examples of halogen substituted alkanes (“haloalkanes”) include halomethane (e.g., chloromethane, bromomethane, fluoromethane, iodomethane), di-and trihalomethane (e.g., trichloromethane, tribromomethane, trifluoromethane, triiodomethane), 1-haloethane, 2-haloethane, 1 ,2- dihaloethane, 1-halopropane, 2-halopropane, 3-halopropane, 1 ,2-dihalopropane, 1 ,3- dihalopropane, 2,3-dihalopropane, 1 ,2,3-trihalopropane, and any other suitable combinations of alkanes (or substituted alkanes) and halogens (e.g., Cl, Br, F, or I). When an alkyl group is substituted with more than one halogen radical, each halogen may be independently selected e.g., 1 -chloro, 2-fluoroethane.
[0084] As used herein, the term “aryl” refers to a monocyclic or fused bicyclic moiety having the well-known characteristics of aromaticity; examples include phenyl and naphthyl, which can be optionally substituted. Additional examples of aromatic rings include furan, benzofuran, isobenzofuran, pyrrole, indole, isoindole, thiophene, benzothiophene, benzo(c)thiophene, imidazole, benzimidazole, purine, pyrazole, indazole, oxazole, benzooxazole, isoxazole, benzisoxazole, thiazole, benzothiazole, benzene, naphthalene, pyridine, quinolone, isoquinoline, pyrazine, quinoxaline, pyrimidine, quinazoline, pyridazine, cinnoline, phthalazine, triazine (e.g., 1 ,2,3-triazine; 1 ,2,4-triazine; 1 ,3,5 triazine), and thiadiazole. The term “aromatic carbocycle” refers to an aromatic ring without heteroatoms present within the ring structure, such as, but not limited to benzene or naphthalene. Other terms that can be used include “aromatic ring,” “aryl group,” or “aryl ring.”
[0085] As used herein, the term “heterocycle,” “heterocyclyl,” “heterocyclic ring” or “heterocyclic group” is intended to mean a stable 4-, 5-, 6-, or 7-membered monocyclic or 7-, 8-, 9-, 10-, 11 -, 12-, 13-, or 14-membered bicyclic heterocyclic ring which is saturated, partially unsaturated, or fully unsaturated or aromatic, and which consists of carbon atoms and 1 , 2, 3 or 4 heteroatoms independently selected from N, O, and S; and including any bicyclic group in which any of the above-defined
heterocyclic rings is fused to a benzene ring. Other heteroatoms, such as P, Se, B, or Si, can be included in some alternatives. The nitrogen and sulfur heteroatoms may optionally be oxidized. The nitrogen atom may be substituted or unsubstituted (i.e. , N or NR wherein R is H or another substituent, if defined). The heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure. The heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. A nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and 0 atoms in the heterocycle exceeds 1 , then these heteroatoms are not adjacent to one another. When the term “heterocycle,” “heterocyclyl,” “heterocyclic ring" or “heterocyclic group” is used, it is intended to include heteroaryl unless heteroaryl is excluded. Examples of heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H-
1 .5.2-dithiazinyl, dihydrofuro[2,3-b]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1 H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H-indolyl, isatinoyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isothiazolopyridinyl, isoxazolyl, isoxazolopyridinyl, methylenedioxyphenyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl,
1 .2.3-oxadiazolyl, 1 ,2,4-oxadiazolyl, 1 ,2,5-oxadiazolyl, 1 ,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxindolyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, piperidonyl, 4-piperidonyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, 2-pyrrolidonyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H-1 ,2,5-thiadiazinyl, 1 ,2,3- thiadiazolyl, 1 ,2,4-thiadiazolyl, 1 ,2,5-thiadiazolyl, 1 ,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl,
1 ,2,3-triazolyl, 1 ,2,4-triazolyl, 1 ,2,5-triazolyl, 1 ,3,4-triazolyl, and xanthenyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
[0086] As used herein, the term “non-aromatic heterocycle” refers to a cycloalkyl or cycloalkenyl, as defined herein, wherein one or more of the ring carbons are replaced by a moiety selected from -0--, --N=, --NR--, --C(O)--, --S--, --S(O)-- or --S(O)2--, wherein R is hydrogen, C1-C8 alkyl or a nitrogen protecting group, with the proviso that the ring of such a group does not contain two adjacent O or S atoms. In some alternatives, other heteroatoms including P, Se, B, or Si can be included. Non-limiting examples of non-aromatic heterocycles, as used herein, include morpholino, pyrrol idiny I , pyrrolidinyl-2-one, piperazinyl, piperid iny I, piperidinylone, 1 ,4-dioxa-8-aza- spiro(4.5)dec-8-yl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, 1 ,3-dioxolanyl, 2-imidazolinyl, imidazolidinyl, 2-pyrazolinyl, pyrazolidinyl, 1 ,4-dioxanyl, 1 ,4-dithianyl, thiomorpholinyl, azepanyl, hexahydro-1 ,4-diazepinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, thioxanyl, azetidinyl, oxetanyl, thietanyl, oxepanyl, thiepanyl, 1 ,2,3,6-tetrahydropyridinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1 ,3-dioxolanyl, dithianyl, dithiolanyl, dihydropyranyl, dihydrothienyl, dihydrofuranyl, imidazolinyl, imidazolidinyl, 3-azabicyclo(3.1.0)hexanyl, and 3- azabicyclo(4.1.0)heptanyl, 3,8-diazabicyclo(3.2.1 )octanyl, and 2,5- diazabicyclo(2.2.1 )heptanyl. In certain embodiments, a non-aromatic heterocyclic ring is aziridine, thiirane, oxirane, oxaziridine, dioxirane, azetidine, oxetan, thietane, diazetidine, dioxetane, dithietane, pyrrolidine, tetrahydrofuran, thiolane, imidazolidine, pyrazolidine, oxazolidine, isoxazolidine, thiazolidine, isothiazolidine, dioxolane, dithiolane, piperdine, oxane, thiane, piperazine, morpholine, thiomorpholine, dioxane, dithiane, trioxane, thithiane, azepane, oxepane, thiepane, homopiperazine, or azocane.
[0087] As used herein, the terms “heteroaryl” or “heteroaromatic” refer to monocyclic, bicyclic, or polycyclic ring systems, wherein at least one ring in the system is aromatic and contains at least one heteroatom, for example, nitrogen, oxygen and sulfur. Each ring of the heteroaromatic ring systems may contain 3 to 7 ring atoms. Exemplary heteroaromatic monocyclic ring systems include 5- to 7-membered rings whose ring structures include one to four heteroatoms, for example, one or two heteroatoms. The inclusion of a heteroatom permits aromaticity in 5-membered rings as
well as in 6-membered rings. Typical heteroaromatic systems include monocyclic C5-C6 heteroaromatic groups such as pyridyl, pyrim idyl, pyrazinyl, thienyl, furanyl, pyrrolyl, pyrazolyl, thiazolyl, oxazolyl, triazolyl, triazinyl, tetrazolyl, tetrazinyl, and imidazolyl, as well as the fused bicyclic moieties formed by fusing one of these monocyclic heteroaromatic groups with a phenyl ring or with any of the heteroaromatic monocyclic groups to form a C8-C-10 bicyclic group such as indolyl, benzimidazolyl, indazolyl, benzotriazolyl, isoquinolyl, quinolyl, benzothiazolyl, benzofuranyl, pyrazolylpyridyl, quinazolinyl, quinoxalinyl, cinnolinyl, and other ring systems known in the art. Any monocyclic or fused ring bicyclic system that has the characteristics of aromaticity in terms of delocalized electron distribution throughout the ring system is included in this definition. This definition also includes bicyclic groups where at least the ring that is directly attached to the remainder of the molecule has the characteristics of aromaticity, including the delocalized electron distribution that is characteristic of aromaticity. Typically the ring systems contain 5 to 12 ring member atoms and up to four heteroatoms, wherein the heteroatoms are selected from the group consisting of N, 0, and S. Frequently, the monocyclic heteroaryls contain 5 to 6 ring members and up to three heteroatoms selected from the group consisting of N, 0, and S; frequently, the bicyclic heteroaryls contain 8 to 10 ring members and up to four heteroatoms selected from the group consisting of N, 0, and S. The number and placement of heteroatoms in heteroaryl ring structures is in accordance with the well-known limitations of aromaticity and stability, where stability requires the heteroaromatic group to be stable enough to be exposed to water at physiological temperatures without rapid degradation. As used herein, the term “hydroxyheteroaryl” refers to a heteroaryl group including one or more hydroxyl groups as substituents; as further detailed below, further substituents can be optionally included. As used herein, the terms “haloaryl” and “haloheteroaryl” refer to aryl and heteroaryl groups, respectively, substituted with at least one halo group, where “halo” refers to a halogen selected from the group consisting of fluorine, chlorine, bromine, and iodine, typically, the halogen is selected from the group consisting of chlorine, bromine, and iodine; as detailed below, further substituents can be optionally included. As used herein, the terms “haloalkyl,” “haloalkenyl,” and “haloalkynyl” refer to alkyl, alkenyl, and alkynyl groups, respectively, substituted with at least one halo group,
where “halo” refers to a halogen selected from the group consisting of fluorine, chlorine, bromine, and iodine, typically, the halogen is selected from the group consisting of chlorine, bromine, and iodine; as detailed below, further substituents can be optionally included. When a range of values is listed, such as for the number of carbon atoms in an alkyl group, it is intended to encompass each value and subrange within the range. For example, “C1- C6 alkyl” includes alkyl groups with 1 , 2, 3, 4, 5, or 6 carbon atoms and all possible subranges.
[0088] As used herein, the term “hydroxyaryl” refers to an aryl group including one or more hydroxyl groups as substituents; as further detailed below, further substituents can be optionally included.
[0089] As used herein, the term “solvate” means a compound formed by solvation (the combination of solvent molecules with molecules or ions of the solute), or an aggregate that consists of a solute ion or molecule, i.e., a compound of the invention, with one or more solvent molecules. The term “solvate” typically means a physical association of a compound involving varying degrees of ionic and/or covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent atoms are incorporated into the crystal lattice of the crystalline solid. The term “solvate” encompasses both solution-phase and isolatable solvates. Suitable solvates in which the solvent is other than water include, but are not limited to, ethanolates or methanolates. When water is the solvent, the corresponding solvate is a “hydrate.” Examples of hydrates include, but are not limited to, hemihydrate, monohydrate, dihydrate, trihydrate, hexahydrate, and other hydrated forms. It should be understood by one of ordinary skill in the art that the pharmaceutically acceptable salt and/or prodrug of compounds described herein for use in methods or compositions according to the present invention may also exist in a solvate form. When the solvate is a hydrate, the hydrate is typically formed via hydration which is either part of the preparation of the present compound or through natural absorption of moisture by the anhydrous compound of the present invention. Additionally, compounds may exist as clathrates or other complexes, which are therapeutic agent-host inclusion complexes wherein the therapeutic agent and the host are present in stoichiometric or non-stoichiometric amounts.
[0090] As used herein, the term “ester” means any ester of a present compound in which any of the --COOH functions of the molecule is replaced by a --COOR function, in which the R moiety of the ester is any carbon-containing group which forms a stable ester moiety, including but not limited to alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl and substituted derivatives thereof. The hydrolyzable esters of the present compounds are the compounds whose carboxyls are present in the form of hydrolyzable ester groups. That is, these esters are pharmaceutically acceptable and can be hydrolyzed to the corresponding carboxyl acid in vivo.
[0091] As used herein, the term “alkenyl” refers to an unbranched, branched or cyclic hydrocarbyl residue having one or more carbon-carbon double bonds. Typically, the hydrocarbyl residue has from 2 to 12 carbon atoms (C2-C12 alkenyl). In certain embodiments, an alkenyl comprises two to eight carbon atoms (C2-C8 alkenyl). In certain embodiments, an alkenyl comprises two to six carbon atoms (i.e. , C2-C6 alkenyl). In other embodiments, an alkenyl comprises two to four carbon atoms (i.e., C2-C4 alkenyl). The alkenyl is attached to the rest of the molecule by a single bond, for example, ethenyl (i.e., vinyl), prop-1 -enyl (i.e., allyl), but-1 -enyl, pent-1 -enyl, penta-1 ,4- dienyl, and the like. An alkenyl group can be optionally substituted by one or more substituents such as those substituents described herein. With respect to the use of “alkenyl,” the presence of multiple double bonds cannot product an aromatic ring structure.
[0092] As used herein, the term “alkynyl” refers to an unbranched, branched, or cyclic hydrocarbyl residue having one or more carbon-carbon triple bonds; the residue can also include one or more double bonds. Typically, the hydrocarbyl residue has from 2 to 12 carbon atoms (C2-C12 alkynyl). In certain embodiments, an alkenyl comprises two to eight carbon atoms (C2-C8 alkynyl). In certain embodiments, an alkenyl comprises two to six carbon atoms (i.e., C2-C6 alkynyl). In other embodiments, an alkenyl comprises two to four carbon atoms (i.e., C2-C4 alkynyl). The alkynyl is attached to the rest of the molecule by a single bond, for example, ethynyl, propynyl, butynyl, pentynyl, hexynyl, and the like. With respect to the use of “alkynyl,” the presence of
multiple double bonds in addition to the one or more triple bonds cannot produce an aromatic ring structure.
[0093] As used herein, the term “alkylene” or “alkylene chain” refers to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing no unsaturation, and preferably having from one to twelve carbon atoms, for example, methylene, ethylene, propylene, n-butylene, and the like. The alkylene chain is attached to the rest of the molecule through a single bond and to the radical group through a single bond. The points of attachment of the alkylene chain to the rest of the molecule and to the radical group may be through any two carbons within the chain. In certain embodiments, an alkylene comprises one to ten carbon atoms (i.e. , C1-C10 alkylene). In certain embodiments, an alkylene comprises one to eight carbon atoms (i.e., C1-C8 alkylene). In other embodiments, an alkylene comprises one to five carbon atoms (i.e., C1-C5 alkylene). In other embodiments, an alkylene comprises one to four carbon atoms (i.e., C1-C4 alkylene). In other embodiments, an alkylene comprises one to three carbon atoms (i.e., C1-C3 alkylene). In other embodiments, an alkylene comprises one to two carbon atoms (i.e., C1-C2 alkylene). In other embodiments, an alkylene comprises only one carbon atom (i.e., C1 alkylene or a -CH2 — group). An alkylene group can be optionally substituted by one or more substituents such as those substituents described herein.
[0094] As used herein, the term “alkenylene” or “alkenylene chain” refers to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing at least one carbon- carbon double bond, and preferably having from two to twelve carbon atoms. The alkenylene chain is attached to the rest of the molecule through a single bond and to the radical group through a single bond. The points of attachment of the alkenylene chain to the rest of the molecule and to the radical group may be through any two carbons within the chain. In certain embodiments, an alkenylene comprises two to ten carbon atoms (i.e., C2-C10 alkenylene). In certain embodiments, an alkenylene comprises two to eight carbon atoms (i.e., C2-C8 alkenylene). In other embodiments, an alkenylene comprises two to five carbon atoms (i.e., C2-C5 alkenylene). In other embodiments, an
alkenylene comprises two to four carbon atoms (i.e. , C2-C4 alkenylene). In other embodiments, an alkenylene comprises two to three carbon atoms (i.e., C2-C3 alkenylene). In other embodiments, an alkenylene comprises two carbon atom (i.e., C2 alkenylene). An alkenylene group can be optionally substituted by one or more substituents such as those substituents described herein.
[0095] As used herein, “alkynylene” or “alkynylene chain” refers to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing at least one carbon-carbon triple bond, and preferably having from two to twelve carbon atoms. The alkynylene chain is attached to the rest of the molecule through a single bond and to the radical group through a single bond. The points of attachment of the alkynylene chain to the rest of the molecule and to the radical group may be through any two carbons within the chain. In certain embodiments, an alkynylene comprises two to ten carbon atoms (i.e., C2-C10 alkynylene). In certain embodiments, an alkynylene comprises two to eight carbon atoms (i.e., C2-C8 alkynylene). In other embodiments, an alkynylene comprises two to five carbon atoms (i.e., C2-C5 alkynylene). In other embodiments, an alkynylene comprises two to four carbon atoms (i.e., C2-C4 alkynylene). In other embodiments, an alkynylene comprises two to three carbon atoms (i.e., C2-C3 alkynylene). In other embodiments, an alkynylene comprises two carbon atom (i.e., C2 alkynylene). An alkenylene group can be optionally substituted by one or more substituents such as those substituents described herein.
[0096] As used herein, the term “amine” or “amino” includes primary, secondary, and tertiary amines wherein each non-hydrogen group on nitrogen may be selected from alkyl, aryl, and the like. Amines include but are not limited to --NH2, --NH-phenyl, -- NH--CH3, --NH--CH2CH3, and --N(CH3)benzyl. The amino group can be optionally substituted. For example, the term can include NR'R" wherein each R' and R" is independently H, or is an alkyl, alkenyl, alkynyl, acyl, aryl, or arylalkyl group, and each of the alkyl, alkenyl, alkynyl, acyl, aryl, or arylalkyl groups is optionally substituted with the substituents described herein as suitable for the corresponding group; the R' and R" groups and the nitrogen atom to which they are attached can optionally form a 3- to 8- membered ring which may be saturated, unsaturated or aromatic and which contains 1-
3 heteroatoms independently selected from N, 0 and S as ring members, and which is optionally substituted with the substituents described as suitable for alkyl groups or, if NR'R" is an aromatic group, it is optionally substituted with the substituents described as typical for heteroaryl groups.
[0097] As used herein, the term “amide” or “amido” includes C- and N-amide groups, e.g., --C(O)NR2, and --NRC(O)R groups, respectively, where R can be H, alkyl, aryl, or other groups, which can be optionally substituted. Amide groups therefore include but are not limited to -C(O)NH2, -NHC(O)H, -C(O)NHCH2CH3, - NHC(O)CH3,or -C(O)N(CH2CH3)phenyl.
[0098] As used herein, “acyl” encompasses groups comprising an alkyl, alkenyl, alkynyl, aryl or arylalkyl radical attached at one of the two available valence positions of a carbonyl carbon atom, and heteroacyl refers to the corresponding groups wherein at least one carbon other than the carbonyl carbon has been replaced by a heteroatom chosen from N, O and S.
[0099] As used herein, similarly, “arylalkyl” and “heteroarylalkyl” refer to aromatic and heteroaromatic ring systems which are bonded to their attachment point through a linking group such as an alkylene, including substituted or unsubstituted, saturated or unsaturated, cyclic or acyclic linkers. Typically the linker is C1-C8 alkyl. These linkers may also include a carbonyl group, thus making them able to provide substituents as an acyl or heteroacyl moiety. An aryl or heteroaryl ring in an arylalkyl or heteroarylalkyl group may be substituted with the same substituents described above for aryl groups. Preferably, an arylalkyl group includes a phenyl ring optionally substituted with the groups defined above for aryl groups and a C1-C4 alkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl groups or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane. Similarly, a heteroarylalkyl group preferably includes a C5-C6 monocyclic heteroaryl group that is optionally substituted with the groups described above as substituents typical on aryl groups and a C1-C4 alkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl groups or heteroalkyl groups, or it includes an optionally substituted phenyl ring or C5-C6 monocyclic heteroaryl and a C1-C4 heteroalkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl
or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
[0100] As used herein, the term “heteroatom” refers to any atom that is not carbon or hydrogen, such as nitrogen, oxygen or sulfur. When it is part of the backbone or skeleton of a chain or ring, a heteroatom must be at least divalent, and will typically be selected from N, O, P, and S, more typically from N, O, and P. The term “heteroatom” can include, in some contexts, other atoms, including selenium, silicon, or boron.
[0101] As used herein, the term “alkanoyl” refers to an alkyl group covalently linked to a carbonyl (C=O) group. The term “lower alkanoyl” refers to an alkanoyl group in which the alkyl portion of the alkanoyl group is C1-C6. The alkyl portion of the alkanoyl group can be optionally substituted as described above. The term “alkylcarbonyl” can alternatively be used. Similarly, the terms “alkenylcarbonyl” and “alkynylcarbonyl” refer to an alkenyl or alkynyl group, respectively, linked to a carbonyl group.
[0102] As used herein, the term “alkoxy” refers to an alkyl group covalently linked to an oxygen atom; the alkyl group can be considered as replacing the hydrogen atom of a hydroxyl group. The term “lower alkoxy” refers to an alkoxy group in which the alkyl portion of the alkoxy group is C1-C6. The alkyl portion of the alkoxy group can be optionally substituted as described above. As used herein, the term “haloalkoxy” refers to an alkoxy group in which the alkyl portion is substituted with one or more halo groups.
[0103] As used herein, the term “sulfo” refers to a sulfonic acid ( — SO3H) substituent.
[0104] As used herein, the term “sulfamoyl” refers to a substituent with the structure — S(O2)NH2, wherein the nitrogen of the NH2 portion of the group can be optionally substituted as described above.
[0105] As used herein, the term “carboxyl” refers to a group of the structure — C(O2)H.
[0106] As used herein, the term “carbamyl” refers to a group of the structure — C(O2)NH2, wherein the nitrogen of the NH2 portion of the group can be optionally substituted as described above.
[0107] As used herein, the terms “monoalkylaminoalkyl” and “dialkylaminoalkyl” refer to groups of the structure — Alk1 -NH-Alk2 and — Alk1 -N(Alk2)( Alk3 ), wherein Alk1 , Alk2, and Alk3 refer to alkyl groups as described above.
[0108] As used herein, the term “alkylsulfonyl” refers to a group of the structure — S(O)2-Alk wherein Aik refers to an alkyl group as described above. The terms “alkenylsulfonyl” and “alkynylsulfonyl” refer analogously to sulfonyl groups covalently bound to alkenyl and alkynyl groups, respectively. The term “arylsulfonyl” refers to a group of the structure — S(O)2-Ar wherein Ar refers to an aryl group as described above. The term “aryloxyalkylsulfonyl” refers to a group of the structure — S(O)2-Alk-O-Ar, where Aik is an alkyl group as described above and Ar is an aryl group as described above. The term “arylalkylsulfonyl” refers to a group of the structure — S(O)2-AlkAr, where Aik is an alkyl group as described above and Ar is an aryl group as described above.
[0109] As used herein, the term “alkyloxycarbonyl” refers to an ester substituent including an alkyl group wherein the carbonyl carbon is the point of attachment to the molecule. An example is ethoxycarbonyl, which is CH3CH2OC(O) — . Similarly, the terms “alkenyloxycarbonyl,” “alkynyloxycarbonyl,” and “cycloalkylcarbonyl” refer to similar ester substituents including an alkenyl group, alkenyl group, or cycloalkyl group respectively. Similarly, the term “aryloxycarbonyl” refers to an ester substituent including an aryl group wherein the carbonyl carbon is the point of attachment to the molecule. Similarly, the term “aryloxyalkylcarbonyl” refers to an ester substituent including an alkyl group wherein the alkyl group is itself substituted by an aryloxy group.
[0110] As used herein, the term “absent” when used in reference to a functional group or substituent, particularly in reference to the chemical structure of a compound, means that the particular functional group or substituent is not present in the compound being described. When used in reference to a substituent, the absence of the substituent typically means that the bond to the substituent is absent and that absence of the bond is compensated for with a H atom. When used in reference to a position
within a chain or ring, the absence of the position typically means that the two positions otherwise connected by the absent position are instead directly connected by a covalent bond.
[0111] Other combinations of substituents are known in the art and, are described, for example, in United States Patent No. 8,344,162 to Jung et al. or in PCT Patent Application Publication No. WO 2019/204768 by Kobilka et al. For example, the term “thiocarbonyl” and combinations of substituents including “thiocarbonyl” include a carbonyl group in which a double-bonded sulfur replaces the normal double-bonded oxygen in the group. The term “alkylidene” and similar terminology refer to an alkyl group, alkenyl group, alkynyl group, or cycloalkyl group, as specified, that has two hydrogen atoms removed from a single carbon atom so that the group is double-bonded to the remainder of the structure.
[0112] Certain compounds described herein for use in methods and compositions according to the present invention possess asymmetric carbon atoms (optical or chiral centers) or double bonds; the enantiomers, racemates, diastereomers, tautomers, geometric isomers, stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)-or (S)- or, as (D)- or (L)- for amino acids, and individual isomers are encompassed within the scope of the present invention unless specific isomers are excluded. The present disclosure is meant to include compounds in racemic and optically pure forms. Optically active (R)- and (S)-, or (D)- and (L)-isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. When the compounds described herein contain olefinic bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. The term “tautomer,” as used herein, refers to one of two or more structural isomers that exist in equilibrium and that are readily converted from one form to another. Examples of tautomerism include, but are not limited to, keto-enol tautomerism, enamine-imine tautomerism, and lactam- lactim tautomerism. Unless one of the tautomeric alternatives is expressly excluded, all such tautomeric forms are intended to be within the scope of the invention.
[0113] For a number of compounds and classes of compounds disclosed in methods and compositions according to the present invention, alternative structures are
presented in terms of Markush groups; an example is a ring moiety in a structure that includes one or more substituents which are described in the alternative as, for example, ethyl, methyl, or propyl. It is specifically contemplated that each member of the Markush group should be considered separately, thereby comprising another embodiment, and the Markush group is not to be read as a single unit.
DETAILED DESCRIPTION OF THE INVENTION
[0114] The structure of elesclomol is shown below as Formula (I):

[0115] Elesclomol
 -dimethyl- bis(phenylcarbonothioyl)propanedihydrazide) is a novel, injectable, drug candidate that kills cancer cells by elevating oxidative stress levels beyond a breaking point, triggering programmed cell death. In preclinical models elesclomol showed potent killing of a broad range of cancer cell types at high doses, and an ability to strongly enhance the efficacy of certain chemotherapy agents, with minimal additional toxicity, at moderate doses.
-dimethyl- bis(phenylcarbonothioyl)propanedihydrazide) is a novel, injectable, drug candidate that kills cancer cells by elevating oxidative stress levels beyond a breaking point, triggering programmed cell death. In preclinical models elesclomol showed potent killing of a broad range of cancer cell types at high doses, and an ability to strongly enhance the efficacy of certain chemotherapy agents, with minimal additional toxicity, at moderate doses.
[0116] Elesclomol induces oxidative stress by provoking a buildup of reactive oxygen species within cancer cells (J.R. Kirshner et al., “Elesclomol Induces Cancer Cell Apoptosis Through Oxidative Stress,” Mol. Cancer Ther. 7: 2319-2327 (2008)). Elesclomol requires a redox-active metal ion to function. The Cu(ll) complex is 34 times more potent than the Ni(ll) complex and 1040-fold more potent than the Pt(ll) complex (A. A. Yadav et al., “Molecular mechanisms of the biological activity of the anticancer drug elesclomol and its complexes with Cu(ll), Ni(ll) and Pt(ll),” J. Inorq. Biochem. 126: 1-6 (2013)). Elesclomol can also directly or indirectly induce apoptosis. Elesclomol can also act as an oxidative phosphorylation inhibitor (A.P. Nayak et al., “Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer,” Cancers (Basel) 10: 337 (2018)).
[0117] Elesclomol is an anticancer drug that targets mitochondrial metabolism. In the past, elesclomol was recognized as an inducer of oxidative stress, but now it has also been found to suppress cancer by inducing cuproptosis. Elesclomol’s anticancer activity is determined by the dependence of cancer on mitochondrial metabolism. The mitochondrial metabolism of cancer stem cells, cancer cells resistant to platinum drugs, proteasome inhibitors, molecularly targeted drugs, and cancer cells with inhibited glycolysis was significantly enhanced. Elesclomol exhibited tremendous toxicity to all three kinds of cells. Elesclomol’s toxicity to cells is highly dependent on its transport of extracellular copper ions, a process involved in cuproptosis (P. Zheng et al., “Elesclomol, a Copper Ionophore Targeting Mitochondrial Metabolism for Cancer Therapy,” J. Exp. Clin. Cancer Res. 41 : 271 (2022)). As used herein, reference to “elesclomol” includes reference to elesclomol itself as well as derivatives and analogs of elesclomol and also salts, solvates, or prodrugs of elesclomol unless derivatives, analogs, salts, solvates, or prodrugs are clearly excluded by the context of the reference.
[0118] Elesclomol and derivatives or analogs thereof described further below can be used to treat a number of diseases and conditions, particularly including, but not limited to, malignancies.
[0119] A particularly significant malignancy that can be treated with elesclomol is ovarian epithelial cancer (OEC). OEC is the deadliest of gynecological cancers. Type I OEC (low-grade serous, endometrial, mucinous and ovarian clear-cell carcinoma (OCCC)) lack an effective systemic therapy and remain a significant unmet medical need especially when surgical resection is insufficient. When diagnosed at an advanced stage, OCCCs have the worst prognosis and lowest survival rates among all OEC subtypes. OCCC and other Type I OEC as well as endometrial and uterine tumors are characterized by high prevalence of ARID1A mutations, which are mutually exclusive to BRCA and TP53 mutations commonly seen in Type II OEC subtypes such as HGSOC (high grade serous ovarian cancer).
[0120] ARID1A mutations result in cells that are highly susceptible to synthetic lethality by agents that interrupt OXPHOS or the mitochondrial electron transport chain (mETC).
[0121] Elesclomol is an agent that may: (a) have high specificity for cells expressing ARID1 A mutations; and (b) act against a specific protein target within the mETC, thereby providing a potential novel therapeutic approach for OCCC and other tumors expressing ARID1A mutations.
[0122] OECs can be generally divided into two types: Type I and Type II. Type I tumors are low-grade serous, endometrioid, mucinous, or clear-cell and typically, have the mass confined to the ovary. They are slower growing, not driven by p53 or BRCA mutations, and are often associated with endometriosis. Type II tumors are high-grade serous ovarian carcinomas that are more aggressive and frequently present at a later stage. These tumors have often spread to other tissues in the peritoneal cavity by the time they are diagnosed. Typically, these tumors express TP53 and/or BRCA mutations.
[0123] Elesclomol may also provide benefit to Type II OEC patients who have become resistant to PARP inhibitors.
[0124] HGSOC accounts for 68% of ovarian cancer diagnoses. Platinum-based therapy is frequently administered to treat these malignancies; disease control rates as high as 81 .7% have been observed, with 27% complete responses and 45% partial responses seen in one study. Mutations in the tumor suppressor genes BRCA1 and BRCA2 have been associated with an increased risk for the development of breast and ovarian cancer.
[0125] Inhibitors of poly(ADP-ribose) polymerase (PARP) have recently established a new treatment paradigm for OEC tumors with mutated BRCA. In one study, patients treated with PARP inhibitors exhibited a 70% reduced risk of disease progression or death, and 60% of patients remained progression-free at 36 months compared to 27% in the placebo arm of the trial.
[0126] OCCC is the second most common OEC subtype and the second leading cause of death from ovarian cancer. OCCC represents up to 10% of all OEC cases in North America with an increased prevalence in Asian populations, as high as 25% in Japan and 50% in Thailand.
[0127] Compared to HGSOC, treatment for OCCC has not benefited from recent advances in treatment strategies.
[0128] OCCC is often detected at an early stage, presenting with a large unilateral pelvic mass confined to the ovary and causing symptoms of abdominal pain and distortion. Despite the earlier diagnosis, OCCC exhibits poorer survival compared to HGSOC across all FIGO stages. When diagnosed at an advanced stage, OCCC exhibits the worst prognosis and lowest survival rates among all OEC subtypes.
[0129] The results for progression-free survival (PFS) and overall survival (OS) for serous adenocarcinoma, endometrioid adenocarcinoma, mixed epithelial carcinoma, clear-cell carcinoma, and other types of ovarian cancer are shown in Figure 1 based on a study of 845 ovarian cancer patients. Patients with clear cell histology were associated with decreased PFS (HR = 2.66; 95% Cl, 1.47-4.82; P = 0.001) and OS (HR = 3.88; 95% Cl, 2.11-7.12; P = <0.001). PFS is shown in the left panel and OS in the right panel.
[0130] Based on both clinical observations and laboratory research, OCCC is highly resistant to chemotherapy. In one study, only 11.1 % responded to platinum- based chemotherapy with median overall survival less than 18 months, compared to a response rate of 72.5% for advanced HGSOC patients in the same study. For the minority of OCCC patients who initially responded, less than 9% responded to retreatment with platinum-based chemotherapy following disease recurrence.
[0131] OCCC patients generally do not have BRCA mutations and thus are not considered to be within the treatment scope of recent FDA-approved PARP inhibitors. Therefore, at present, the extremely poor prognosis and lack of an effective systemic therapy for OCCC remains a significant unmet medical need, especially when surgical cytoreduction is insufficient.
[0132] In contrast with HGSOC, BRCA mutations occur at a very low frequency in OCCC (Arts-de-Jong, 2016) and mutations in TP53 occur much less frequently compared to other OECs (Ho, 2001 ) highlighting important differences in these OEC histological subtypes.
[0133] The most frequent mutations observed in OCCC include ARID1A and PIK3CA (Kwan 2015). ARID1A is the most highly mutated gene with PIK3CA mutations the second most prevalent. Loss of ARID1 A frequently co-exists with PIK3CA leading to hypothesis that they cooperate to promote tumor growth, possibly via induction of IL-
6 and downstream JAK/STAT3 signaling, which has a prominent role in tumor cell growth and differentiation (Chandler 2015). The coexistence of these two mutations has been reported in gastric cancer and other cancers; however, the rate of co- occurrence reported to date is highest in OCCC.
[0134] Because PI3KCA is an important component of AKT/mTOR signaling pathways commonly altered in OCCC, mTOR gained interest as a potentially promising target for OCCC therapy. Unfortunately, a 90-patient clinical trial of the mTOR inhibitor temsirolimus failed to demonstrate an improvement in outcomes compared to historical controls in patients with Stage lll-IV OCCC.
[0135] The gene ARID1A is a haplo-insufficient tumor suppressor gene associated with regulation of DNA damage repair (DDR) processes, as well as the SWI/SNF (Switch-Sucrose Non-fermenting) pathway, which is involved in sugar (sucrose) metabolism. The loss of ARID1 A function is thought to enhance cell cycle progression and the formation of cancer; however, therapeutic strategies targeting ARID1A mutant tumors are limited at this time. ARID1A is frequently mutated in liver, breast and gastric cancer, but the highest mutation frequency is in gynecologic cancers (Kwan, 2016), particularly in OCCC where mutations are observed in up to 56% of cases (Jones, 2010). ARID1A mutations are less common in HGSOC where they are largely mutually exclusive to highly prominent mutations in BRCA and TP53 (Hu, 2018). The mutual exclusivity between ARID1A and TP53 mutations has also been observed in several other major cancer types such as prostate, stomach, colon and uterine cancers (Wu, 2017).
[0136] ARID1A mutations are associated with treatment resistance and reduced sensitivity to paclitaxel, cisplatin and PARP inhibitors (Hu, Clovis, 2018). Mutations resulting in loss of ARID1 A function leads to activation of PI3K-AKT pathway. AKT activates the anti-apoptotic pathway by phosphorylating downstream targets to inhibit IKB and release NF-KB which activates genes promoting cellular survival and conferring resistance to platinum-based regimens (Lyu, 2016).
[0137] The involvement of ARID1A in pathways of tumor growth, tumor migration, tumor invasion, and angiogenesis is shown in Figure 2.
[0138] ARID1 A loss also correlates with poor clinical outcomes in HGSOC cases receiving Pt-based chemotherapy regimens or PARP inhibitor treatment. Tumors accumulate ARID1A mutations in response to treatment as a mechanism of acquired resistance (Berns, 2016). Progression free survival in patients exhibiting an ARID1A- deficient phenotype is significantly reduced as compared with ARID1 A wild-type in patients treated with PARP inhibitors. These results are shown in Figure 3. Figure 3(A) shows the impact of ARID1A loss, determined via mutation, homozygous deletion, or loss of expression, and survival in TCGA serous ovarian cancers. Figure 3(B) shows the progression-free survival of relapsed, platinum-sensitive, high-grade ovarian carcinomas in a clinical trial of rucaparib (a PARP inhibitor), stratified based on ARID1A mutation status.
[0139] Loss of ARID1A correlated with shorter progression-free survival (PFS, p<0.01 ) and tended to correlate with shorter overall survival in patients with OCCC treated with platinum-based chemotherapy. Figure 4 shows that loss of ARID1 A expression correlates with shorter progression-free survival in ovarian clear cell carcinoma patients who received primary cytoreductive surgery followed by standard platinum-based chemotherapy: (a) Kaplan-Meier survival analysis showing that negative ARID1A expression (solid line, n=9) is associated with a shorter progression- free survival than positive ARID1A expression (dashed line, n=51) (P<0.01 , Log-rank test), (b) Negative ARID1A expression (solid line, n= 9) had a statistically nonsignificant effect on overall survival in ovarian clear cell carcinoma patients who received primary cytoreductive surgery followed by standard platinum-based chemotherapy (P =0.15, Log-rank test).
[0140] The activity of ARID1 A is essential for establishing an open chromatin state upon DNA damage and facilitating binding of TOPO-2 to DNA (Park, 2019). Cells with mutated or downregulated ARID1 A exhibit compromised homologous (HR) and non-homologous end-joining (NHEJ) DNA repair mechanisms and are resistant to treatment with single-agent PARP inhibitors. ARID1 A/_ cells are deficient in NHEJ repair resulting in a partial cytotoxic response to radiation. Synthetic lethality is activated to synergistically potentiate cytotoxicity in ARID1A/_ cells when PARP inhibitors are combined with DNA damaging agents such as XRT (Yakovlev, 2019).
[0141] ARID1A encodes SLC7A11 , a SWI/SNF chromatin-remodeling factor that is frequently mutated in various cancers. Cancers with SWI/SNF mutations tend to be aggressive in nature and have a poor prognosis (Kwan, 2015). ARID1A loss leads to downregulation of multiple genes involved in oxidative reduction . ARID1 A maintains GSH homeostasis by enhancing SLC7A11 transcription. ARID1 A-deficient cells exhibit impaired expression of the cysteine transporter SLC7A11 , which is a critical component in the antioxidant glutathione (GSH) pathway and the glutamate-cysteine ligase synthetase catalytic subunit (GCLC). Loss of ARID1A prevents cystine uptake leading to increased reactive oxygen species (ROS) in the cell. Accumulation of ROS contributes to oncogenesis through increased mitotic signaling and cell growth (Kwan, 2016).
[0142] Figure 5 is a graph showing that loss of ARID1A results in downregulation of cystine transporter SLC7A11 and impairment of GSH pathway leading to ROS accumulation and cell death: (A) ARID1A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway. (B) Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
[0143] ROS can have pro-survival or pro-death effects depending on amount of ROS and effectiveness of the antioxidant system resident in the cell. Cancer cells show persistently high levels of reactive oxygen species (ROS) due to oncogenic transformation including alteration in genetic, metabolic, and tumor microenvironment. Higher baseline levels of ROS in tumor versus normal cells are known to contribute to the development and/or maintenance of the malignant phenotype and render cancer cells more vulnerable to irreversible oxidative damage and consequent cell death. However, by their very nature, tumor cells have evolved mechanisms to adapt to elevated levels of ROS by activating antioxidant pathways. ROS leads to activation of the nuclear factor NRF2, which binds within the Xc promoter region and activates transcription increasing the uptake of cystine and expression of GSH contributing to the stability of cancer cells in their naturally high-ROS environment. GSH is an antioxidant involved in scavenging ROS and detoxifying the cell to promote cancer progression by
avoiding activation of cell-death signaling pathways. GSH depletion has been largely unsuccessful as a therapeutic strategy. However, cells with GSH abnormalities could be susceptible to synthetic lethality. Conditions of oxidative stress (e.g. high ROS) cause an increase in GSSG, the oxidative form of GSH. GSSG accumulation can be potentially toxic and can result in activation of redox mediated cell death (Couto, 2016) and therefore is rapidly converted back to the reduced form (GSH) by NADPH highlighting the dependence on oxidative phosphorylation. Cells with deficiency in ARID1 A have reduced GSH due to downregulation of SLC7A11 and related reduction in the GSH precursor cystine thereby rendering cancer cells highly dependent on oxidative phosphorylation and susceptible to agents that may tip the balance toward redox- induced apoptosis while sparing normal cells. ARID1A mutated cells have been demonstrated to be significantly more sensitive to inhibition of OXPHOS versus wild- type cells (Emmings, 2019).
[0144] Figure 6 shows the difference between cells with ARID1A mutations and cells with active ARID1 A in terms of the effect of accumulation of ROS: (A) ARID1 A complex associated with NRF2 in high ROS environment promotes transcription of SLC7A11 and increased cystine uptake and activation of GHS antioxidant pathway. (B) Loss of ARID1A inhibits cystine uptake resulting in reduced antioxidant capacity within the cell and increased susceptibility to ROS-driven cell death.
[0145] High ROS accumulation has different effects on cell fate depending on p53 status; with more apoptosis in cells with functional WTp53 (Lyu, 2016). In cells with intact TP53, this phenomenon tips the balance toward apoptotic pathways. Under conditions of high ROS, mutant p53 cancer cells decrease expression of detoxifying enzymes to potentiate aberrant signaling and uncontrolled tumor growth whereas intact p53 tips the balance toward cell death. Exploiting this vulnerability in reliance on OXPHOS, by interrupting or uncoupling members of the mitochondrial electron transport chain, for example, would be a viable strategy of synthetic lethality. Selecting patients whose tumors express vulnerabilities rendering them susceptible to synthetic lethality is of critical importance. Such a strategy would be expected to be particularly effective in tumors such as OCCC which express ARID1A mutations and are generally devoid of
mutations in TP53, thereby providing a novel therapeutic strategy against these underserved tumors.
[0146] Figure 7 shows a therapeutic window for cancer cells in the presence of an oxidative phosphorylation (OXPHOS) inhibitor. (Top Panel) Normal body cells have moderate ATP demand and adequate levels of oxygen and glucose and survive in presence of OXPHOS inhibitors by upregulating glycolysis to meet their ATP demands. (Middle Panel). Highly proliferating cancer cells have extraordinarily high ATP demand and adequate levels of oxygen and glucose. Despite glycolytic pathway upregulation, OXPHOS inhibition results in failure to meet ATP demand and cell death. (Bottom Panel) Quiescent cancer cells have low ATP demand but live in a highly compromised microenvironment (low glucose and hypoxia). Inhibition of OXPHOS is lethal as insufficient glucose is present to compensate for the loss of ATP production by oxidative phosphorylation.
[0147] As stated above, elesclomol (shown above as Formula (I)) is a small molecule drug candidate previously studied extensively in clinical trials as an anti- cancer agent as a single agent and in combination with the tubulin inhibitor paclitaxel. Its safety and pharmacokinetics have been established.
[0148] Elesclomol was initially believed not to be active as a single agent based on mouse xenograft data; however, it was later observed that the drug strongly binds copper and has no activity in the absence of bound copper (Blackman, 2012). Upon chelation with copper in the blood stream, elesclomol undergoes a conformational change allowing the drug to permeate cellular and sub-cellular membranes. As stated above, elesclomol can also bind other divalent metal ions. This conformational change is shown in Figure 8.
[0149] Previously, it was believed that elesclomol induces apoptosis in cancer cells through a non-specific induction of oxidative stress, possibly through the generation of reactive oxygen species (ROS) due to cycling of Cu( 11 )^Cu( I ). However, comparison of elesclomol to over 3000 drug profiles demonstrated that the elesclomol profile was unique suggesting that elesclomol acts by a novel mechanism not shared by other tested compounds. Inhibition of activity of genes associated with antioxidant
activity and protection against ROS and upregulation of TP53-regulated proapoptotic signaling was observed.
[0150] The primary cytotoxic effect of elesclomol appeared to be confined to mitochondria and does not involve a cytoplasmic component. Deletion of SOD2 (mitochondrial superoxide dismutase) is highly sensitizing to elesclomol, whereas deletion of the cytosol-resident SOD1 has no impact on sensitivity to the drug.
[0151] Development had previously been undertaken of the use of elesclomol with paclitaxel; however, efficacy endpoints were not achieved, and subsequent studies had demonstrated that that the combination of elesclomol and paclitaxel was not synergistic and potentially antagonistic. However, subsequently, single-agent activity for elesclomol has been suggested in tumors exhibiting DNA-repair deficiency, including BRCA1 or BRCA2-deficient tumors in vitro. Strong synergy with PARP inhibitors was also observed, including talazoparib and rucaparib, as shown in Figure 9. In Figure 9, drug sensitivity was assessed following treatment of mutant BRCA1 cells (SUM 149) with elesclomol including paclitaxel (A) and PARP inhibitors talazoparib (B) and rucaparib (C). After generating a line of additivity ( — ) using the IC50 values for each drug alone (•), the IC50 values for each combined treatment («) are plotted as data points on a Cartesian plot. Data points below the line of additivity represent synergism, whereas those above or on the line indicate antagonism or an additive effect, respectively.
[0152] In prior clinical trials, it was observed that patients with low lactate dehydrogenase (LDH) achieved superior outcomes as compared to those with high LDH; high LDH indicates a tumor burden with increased reliance on glycolysis, while low LDH indicates a tumor burden with increased reliance on oxidative phosphorylation.
[0153] In subsequent research, ROS production and cytotoxicity induced by elesclomol was suggested to be associated with the uncoupling of mitochondrial oxidative phosphorylation or inhibition of electron transport activity, or both. More recently, it has been demonstrated that elesclomol inhibits electron transport activity in isolated, intact mammalian mitochondria, and induces a dose-dependent inhibition of mitochondrial NADH-ubiquinone oxidoreductase activity (complex I).
[0154] CRISPR/CAS9-based screening identified ferredoxin 1 (FDX1), a mitochondrial enzyme critical to the iron-sulfur (Fe-S) biosynthesis pathway, as the single protein target of the elesclomol. In both screens, this gene scored higher than any other gene. Elesclomol-Cu directly binds and inhibits FDX-1 function to block iron- sulfur cluster formation in complex I, a critical component of the mitochondrial electron transport chain. Further, loss of the FDX1 protein confers resistance to elesclomol consistent with the direct inhibition of FDX1 function by elesclomol.
[0155] The results of these gene screens are shown in Figure 10. In Figure 10: (A) Gene scores in elesclomol-1 (100 nM) and elesclomol-2 (1 μM) treated K562 cells. The gene score is the median Iog2 fold change in abundance of all sgRNAs targeting that gene during the culture period. (B) Corrected p-values (-log10) of KS tests of the sgRNA distribution for each gene versus the distribution of all sgRNAs in the screen of elesclomol-1 (100 nM) and elesclomol-2 (1 μM) screens. Values are ordered on the x- axis by chromosome and location; the dotted line indicates a corrected p-value of 0.05.
[0156] Iron-sulfur clusters play a critical role in the oxidation-reduction reactions of electron transport in mitochondria relied on by cancer cells that have made an adaptive shift from glycolysis to high mitochondrial dependence (oxidative phosphorylation). Complex I plays a role in redox control and the biosynthesis of macromolecules and nucleic acids necessary for cell proliferation. It is suggested that these complex l-dependent events contribute to tumor formation, resistance to cell death, and metastasis of cancer cells in part by causing an increase in ROS levels.
[0157] Figure 11 shows that elesclomol inhibits the natural function of FDX1 in FE-S cluster biosynthesis, (a) Chemical shift perturbation results mapped onto a diagram of the structure of FDX1 . Color code: gray, not significantly affected (A6NH < 0.01 ppm); blue, significant chemical shift changes (A6NH > 0.01 ppm); red, severe line broadening; gray, no assignments. The [2Fe-2S] cluster in FDX1 is indicated by spheres, (b) Schematic describing mitochondrial Fe-S cluster biosynthesis. The mitochondrial ISC core complex catalyzes the conversion of cysteine to alanine and generates S° for iron-sulfur cluster assembly. S° is reduced by FDX1 . A [2Fe-2S] cluster is subsequently formed on the scaffold protein ISCII. (c) In vitro Fe-S cluster assembly was monitored by following the increase of absorbance at 456 nm. Reduced
FDX1 was used as a reducing agent in the presence or absence of either 5x (green) or 10x (blue) elesclomol. Data are representative of two independent experiments, (d) Electron transfer from reduced FDX1 to the cysteine desulfurase complex was measured on the addition of cysteine in the presence or absence of elesclomol. Data are representative of two independent experiments, (e) The molecular structure of elesclomol analogs and their corresponding calculated EC50 (from T47D cells grown in the Hi-Mito state). The reactive sulfur substitution with an oxygen is indicated with an asterisk, (f) The UV/visible spectra of reduced FDX1 before and after incubation with elesclomol-Cu(ll) or Cu(ll) alone.
[0158] Inhibitors of mitochondrial ETC complex I have been shown to increase mitochondrial ROS production and, consequently, to induce cell death via apoptosis. Recently, Fe-S clusters have emerged as an area of interest as potential therapeutic targets. Because Fe-S clusters are particularly sensitive to ROS, the represent key targets of oxidative stress.
[0159] Because many cancer drugs exhibit ROS-increasing effects, selecting for patients with deficient Fe-S machinery may have synergistic effects. Likewise, inhibiting Fe-S related ECT function in cells already highly dependent on OXPHOS, for example, due to a deficiency in ARID1A, would be a viable strategy to induce synthetic lethality.
[0160] Interestingly, these results support the clinical observation of increased anti-tumor activity by elesclomol in patients with low serum LDH because low LDH is an indicator of reliance on OXPHOS. This also suggests the possibility that differences between the mitochondria of normal versus cancer cells (e.g., in membrane permeability, response to uncoupling, and complex I structure and function) contribute to the selective cytotoxicity exhibited by elesclomol for certain cancer cell types.
[0161] Tumors such as OCCC with high prevalence of inactivating mutations in the ARID1A subunit of SWI/SNF complex may represent an ideal biomarker-derived population for targeted therapy with elesclomol. These tumors would have a profound dependency on OXPHOS compared to wild type cells revealing a valuable therapeutic target for elesclomol-driven synthetic lethality in an area of significant unmet medical need.
[0162] Importantly, elesclomol exhibits the highest differentiation in activity in an in vitro screen of drugs observed to have a significantly lower IC50 in ARID1 A-mutated cells versus wild-type as shown in Table 1 .
Table 1

[0163] As expected, based on these data, elesclomol sensitivity has been demonstrated to be higher in ARID1 A mutants in a panel of 11 ovarian cancer cell lines, including OCCC cell lines. Markers for apoptosis were upregulated in ARIDIA-mutant tumors but not in ARID1A-wild type cells.
[0164] Figure 12 shows that sensitivity to elesclomol as a single agent is highly specific to ARID1A mutation in cancer cell lines. (A) Cell growth of endometrial and ovarian cancer cell lines treated with elesclomol for 72 h as measured using the WST-1 assay. (B) IC50 values of elesclomol in these cell lines.
[0165] Recently published work identified elesclomol as one of only three hits in a screen of 4300 small molecules for their ability to kill proteasome inhibitor-resistant cells. This work revealed that cancer cell adaptation to proteotoxic stress renders the cells more sensitive to elesclomol in a manner that depends on Cu.
[0166] These observations suggest ARID1 A mutant OCCC as an important underserved cancer population for targeting with elesclomol therapy, which can be rapidly investigated in clinical trials based on the significant clinical history with the drug.
[0167] Figure 13 is a diagram showing that ARIDIA-mutant tumors are highly dependent on OXPHOS offering a potential strategy for synthetic lethality employing targeted agents interrupting mitochondrial metabolism.
[0168] Figure 14 is a diagram showing that the protein target of elesclomol is ferredoxin-1 , a key component of the OXPHOS pathway establishing a synthetically lethal therapeutic strategy in ARIDIA-mutant tumors.
[0169] The preclinical pharmacokinetics, distribution, metabolism and toxicity of elesclomol has been well established based on a series of nonclinical, Good Laboratory Practices (GLP)-compliant safety assessment studies. In general, elesclomol (in combination with paclitaxel and/or paclitaxel + carboplatin) was well-tolerated and the pharmacokinetics of elesclomol were consistent across trials. When summarized for all completed combination clinical trials, the most frequently observed >Grade 3 adverse events in elesclomol-treated subjects were neutropenia (17%), fatigue, thrombocytopenia, anemia, and leukopenia (4% each), none of which resulted in death.
[0170] Early clinical studies investigated doses up to 525 mg/m2 elesclomol weekly in combination with paclitaxel and identified a maximum tolerated dose of 438 mg/m2. In advanced studies, a dose of 213-260 mg/m2 of elesclomol was most commonly used in combination with paclitaxel. Pharmacokinetic analyses of elesclomol when administered in combination with paclitaxel via infusion for solid tumors reported a half-life of about 1 hour for elesclomol at doses in the range of 44-525 mg/m2. Observed Cmax was about 11 pmol/L and the AUC was about 21 pmol h/L at a dose of 264 mg/m2.
[0171] A previous single agent study was a dose escalation design evaluating a starting dose of 200 mg/m2 elesclomol via a 60-m inute infusion once weekly in a four- week cycle. Six patients were accrued and a dose level of 400 mg/m2 was achieved without observation of dose limiting toxicity (DLT). No adverse events higher than Grade 1 related to the study drug were observed. at this dose. Although higher dose levels were planned, the trial was stopped due to the difficulty in recruiting patients, which primarily related to the stringent inclusion criteria requiring LDH level < 0.8 normal levels. Pharmacokinetic studies were not conducted in this single-agent Phase I trial. It was suggested that dosages higher than 400 mg/m2 will be well tolerated.
[0172] Figure 15 shows the occurrence of ARID1 A mutations in a number of types of malignancies, including mutations, deletions, amplification, and multiple alterations.
[0173] ARID1A loss is also associated with treatment resistance and poor patient outcomes in a range of tumors including HER2+ breast cancer. Recently published findings suggest that tumors accumulate ARID1A mutations in response to treatment as a mechanism of acquired resistance. Such tumors may be susceptible to synthetic lethality treatment strategies with elesclomol.
[0174] As detailed further below, elesclomol can be employed as either the free acid or as a salt, typically a sodium salt. However, also, as detailed further below, elesclomol is typically employed as a coordinate covalent complex with a divalent transition state metal ion.
[0175] Elesclomol is one of a number of therapeutic agents that is considered an agent that has previously been considered one of suboptimal importance. The present invention describes novel improvements, pharmaceutical ingredients, dosage forms, excipients, solvents, diluents, drug delivery systems, preservatives, more accurate drug administrations, improved dose determination and schedules, toxicity monitoring and amelioration, techniques or agents to circumvent or reduce toxicity, and techniques and tools to identify/predict those patients who might have a better outcome with a therapeutic agent by the use of phenotype or genotype determination through the use of diagnostic kits or pharmacokinetic or metabolism monitoring approaches. The present invention also relates to the use of drug delivery systems, novel prodrugs, polymer conjugates, novel routes of administration, other agents to potentiate the activity of the compounds or inhibit the repair of suboptimal cellular effects or sublethal damage or to “push” the cell into more destructive cellular phases such as immune stimulation and apoptosis. In some case, the use of these suboptimal therapeutics in conjunction with radiation or other conventional chemotherapeutic agents or biotherapeutic agents such as antibodies, vaccines, cytokines, lymphokines, gene and antisense RNA therapies, as detailed further below, would provide novel approaches and potential significant treatment improvement.
[0176] In the inventive compositions and methods, the term suboptimal therapy includes agents where Phase I toxicity precluded further human clinical evaluation. It also includes those agents from Phase II trials where limited (e.g., <25% response rates) or no significant treatment responses were identified. Also, suboptimal therapy includes those agents, the subject of Phase III clinical trials the outcome of which was either medically or statistically not significant to warrant regulatory submission or approval by government agencies for commercialization for commercialized agents whose clinical performance (i.e. response rates) as a monotherapy are less than 25%, or whose side effects are severe enough to limit wide utility. Agents with suboptimal clinical activity include but are not limited to the following: small chemical therapeutics, natural products, biologies such as peptide, protein antibody drug conjugates, or vaccines, including cell based therapies. More specifically, the inventive methods and compositions also focus on improvements for redox modulating agents including elesclomol and derivatives or analogs of elesclomol, as well as salts, solvates, or prodrugs of elesclomol.
[0177] Dose Modification
[0178] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the time that the compound is administered, the use of dose-modifying agents that control the rate of metabolism of the compound, normal tissue protective agents, or other relevant factors affecting dosages. General examples include: variations of infusion schedules (e.g., bolus i.v. versus continuous infusion), intermittent infusions for 1-3, 3-6, 6-12, or 12-24 hours, the modification of suitable dosages in conjunction with the use of lymphokines (e.g., G-CSF, GM-CSF, EPO) to increase leukocyte count for or prevent anemia caused by potentially myelosuppressive agents, or the modification of suitable dosages in conjunction with the use of rescue agents such as leucovorin for 5-Fll or thiosulfate for cisplatin treatment. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: i.v. infusion for hours to days; biweekly, triweekly, monthly administration; doses greater than 100 mg/m2/day; progressive escalation of dosing from 100 mg/m2/day based on patient
tolerance; doses less than 2 mg/m2 for greater than 14 days; modification of dosage in conjunction with use of polyamine to modulate metabolism; modification of dosage in conjunction with use of eflornithine to modulate metabolism; selected and intermittent boost dose administration; bolus single and multiple doses escalating from 100 mg/m2; oral doses below 30 or above 130 mg/m2.
[0179] Route of Administration
[0180] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the route that the compound is administered. General examples include: changing the route of administration from oral to intravenous administration and vice versa, or the use of specialized routes such as subcutaneous, intramuscular, intraarterial, intraperitoneal, intralesional, intralymphatic, intratumoral, intrathecal, intravesicular, or intracranial administration. Specific inventive examples for redox modulating agent compounds such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: topical administration; intravesicular administration for bladder cancer; oral administration; slow release oral delivery; intrathecal administration; intraarterial administration; continuous infusion; intermittent infusion, administration via large volume oral solution; buccal administration; or rectal administration.
[0181] Schedule of Administration
[0182] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the time that the compound is administered. General examples include: changing from a monthly administration to a weekly or daily dosing or variations of the schedule. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: daily; weekly for three weeks; weekly for two weeks; biweekly; biweekly for three weeks with a 1-2 week rest period; intermittent boost dose administration; daily for one week then once per week for multiple weeks; daily on days 1-5, 8-12 every three weeks; or daily on days 1-3, 8-11 per cycle.
[0183] Indications for Use
[0184] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the types of disease, clinical stage of disease that the compound is administered. General examples include: the use of solid tumor agents for leukemias and vice versa, the use of antitumor agents for the treatment of benign hyperproliferative disease such as psoriasis or benign prostate hypertrophy. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use for the treatment of leukemias (acute and chronic, AML, ALL, CLL, CML); myelodysplastic syndrome (MDS); angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; mycosis fungoides; use in bone marrow transplantation; as an antiinfective; treatment for AIDS; treatment for lymphoma; mantle cell lymphoma; meningeal leukemia; malignant meningitis; cutaneous T cell lymphoma; Barrett’s esophagus; anaplastic gliomas; triple negative breast cancer; Braf- mutated melanoma; BTK resistant CLL; chordoma; Kras-mutated colon cancer; pediatric tumors including brain and sarcoma; neuroblastoma; rhabdomyosarcoma; Ewing’s sarcoma; medulloblastoma; thyroid cancer; melanoma, lymphoma; multiple myeloma; ovarian osteogenic sarcoma; bladder cancer; prostate cancer; bone metastases; bone pain; ovarian clear cell carcinoma; high-grade serous ovarian carcinoma; small-cell carcinoma of the ovary, particularly the hypercalcemic type.
[0185] Disease Stages
[0186] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the stage of disease at diagnosis/progression that the compound is administered. General examples include: the use of chemotherapy for non-resectable local disease, prophylactic use to prevent metastatic spread or inhibit disease progression or conversion to more malignant stages. Specific inventive examples for redox modulating agent compounds such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use for the treatment of localized polyp stage colon cancer; leukoplakia in the oral cavity; angiogenesis inhibition
to prevent or limit metastatic spread; use against HIV with AZT, DDI, or reverse transcriptase inhibitors.
[0187] Other Indications
[0188] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by using the compound for non-malignant diseases and conditions. General examples include: premalignant conditions, benign hyperproliferative conditions, treatment of infections, parasites, usage to relieve pain, control of pleural effusions. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use as antiinfectives; use as antivirals; use as antibacterials; use for pleural effusions; use as antifungals; use as antiparasitics; use for eczema; use for shingles; use for condylomata; use as anti HPV; use as anti-HSV; use for early and late stage MDS (myelodysplastic syndrome); use for polycythemia vera; use for Paget’s disease.
[0189] Patient Selection
[0190] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the type of patient that would best tolerate or benefit from the use of the compound. General examples include: use of pediatric doses for elderly patients, altered doses for obese patients; exploitation of co-morbid disease conditions such as diabetes, cirrhosis, or other conditions that may uniquely exploit a feature of the compound. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: patients with disease conditions with high levels of metabolic enzymes, reactive oxygen species, histone deacetylase, protein kinases, or ornithine decarboxylase; patients with disease conditions with low levels of metabolic enzymes, histone deacetylase, protein kinases, or ornithine decarboxylase; patients with low or high susceptibility to thrombocytopenia or neutropenia; patients intolerant of Gl toxicities; patients with deficiencies in DNA repair capacity including BRCA, ARID1 A or other deficiencies in the SWI/SWF pathway or mitochondrial electron transport, over- or under- expression of jun, GPCR’s and signal transduction proteins, VEGF, prostate
specific genes, protein kinases, telomerase, abnormalities in gallium scans, abnormalities in bone scans.
[0191] Patient/Disease Phenotype
[0192] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by more precise identification of a patient’s ability to tolerate, metabolize and exploit the use of the compound. General examples include: use of diagnostic tools and kits to better characterize a patients ability to process/metabolize a chemotherapeutic agent or their susceptibility to toxicity caused by potential specialized cellular, metabolic, organ system phenotypes. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: diagnostic tools, techniques, kits and assays to confirm a patient’s particular phenotype and for the measurement of metabolism enzymes and metabolites, histone deacetylase, protein kinases, ornithine decarboxylase, VEGF, products of prostate specific genes, telomerase, jun GPCR’s; ARID1 A mutant phenotype; surrogate compound dosing; or low dose drug pre-testing for enzymatic status.
[0193] The gene ARID1A is a member of the SWI/SNF family, whose members have helicase and ATPase activities and are thought to regulate transcription of certain genes by altering the chromatin structure around those genes. The encoded protein is part of the large ATP-dependent chromatin remodeling complex SWI/SNF, which is required for transcriptional activation of genes normally repressed by chromatin. It possesses at least two conserved domains that could be important for its function. First, it has an ARID domain, which is a DNA-binding domain that can specifically bind an AT-rich DNA sequence known to be recognized by a SWI/SNF complex at the beta- globin locus. Second, the C-terminus of the protein can stimulate glucocorticoid receptor-dependent transcriptional activation. It is thought that the protein encoded by this gene confers specificity to the SWI/SNF complex and may recruit the complex to its targets through either protein-DNA or protein-protein interactions. Two transcript variants encoding different isoforms have been found for this gene. This gene has been commonly found mutated in gastric cancers, ovarian clear cell carcinoma, and
pancreatic cancer. In breast cancer distant metastases acquire inactivation mutations in ARID1A not seen in the primary tumor, and reduced ARID1 A expression confers resistance to different drugs such as trastuzumab and mTOR inhibitors.
[0194] Patient/Disease Genotype
[0195] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by testing and analyzing a patient’s genotype for unique features that may be of value to predict efficacy, toxicity, metabolism, or other factors involving efficacy. General examples include: biopsy samples of tumors or normal tissues (e.g., white blood cells) may also be taken and analyzed to specifically tailor or monitor the use of a particular drug against a gene target, unique tumor gene expression pattern, SNP’s (single nucleotide polymorphisms), to enhance efficacy or to avoid particular drug-sensitive normal tissue toxicities. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: diagnostic tools, techniques, kits and assays to confirm a patient’s particular genotype; gene/protein expression chips and analysis; single nucleotide polymorphism (SNP) assessment; SNP’s for histone deacetylase, ornithine decarboxylase, genes affecting S-adenosyl methionine metabolism, GPCR’s, protein kinases, telomerase, jun; identification and measurement of metabolism enzymes and metabolites, mutations in wild type and mutated genes, epigenetics via methylation and acetylation, ARID1A mutation, ARID1 A deficiency, mTor signaling, PI3K-AKT activating mutation, PARPi resistance, HR deficiency, DDR deficiency, SWI/SNF pathway alteration, P53 status/mutation, BRCA-1 independence/mutation, NAC1 mutation, mitochondrial targeting.
[0196] Pre/Post-Treatment Preparation
[0197] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by specialized preparation of a patient prior to or after the use of a therapeutic agent. General examples include: induction or inhibition of metabolizing enzymes, specific protection of sensitive normal tissues or organ systems. Specific inventive examples for redox modulating agents such as elesclomol or derivatives,
analogs, salts, solvates, or prodrugs of elesclomol include: use of colchicine or analogs; use of diuretics such as probenecid; use of uricase; non-oral use of nicotinamide; use of sustained release forms of nicotinamide; use of inhibitors of polyADP ribose polymerase; use of caffeine; leucovorin rescue; infection control; use of antihypertensives; alteration of stem cell populations; pretreatment to limit or prevent graft vs. host (GVH) cytokine storm reactions; use of anti-inflammatories; anaphylactic reaction suppression.
[0198] Toxicity Management
[0199] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by use of additional drugs or procedures to prevent or reduce potential side-effects or toxicities. General examples include: the use of anti-emetics, anti-nausea, hematological support agents to limit or prevent neutropenia, anemia, thrombocytopenia, vitamins, antidepressants, treatments for sexual dysfunction, or other agents or regimens known in the art. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of colchicine or analogs; use of diuretics such as probenecid; use of uricase; non-oral use of nicotinamide; sustained release forms of nicotinamide; use of inhibitors of polyADP-ribose polymerase; use of caffeine; leucovorin rescue; use of sustained release allopurinol; non-oral use of allopurinol; use of bone marrow transplant stimulants, blood, platelet infusions, Neupogen, G-CSF, GM- CSF; pain management; use of anti-inflammatories; fluids; corticosteroids; insulin control medications; antipyretics; anti-nausea treatments; anti-diarrhea treatment; N- acetylcysteine, antihistamines, use of agents to limit or prevent mucositis, limit or prevent GVH reactions, cytokine storm reactions; use of anti-fungal agents, sodium thiosulfate, glutathione; use of platelet transfusions; use of anti-diarrheal therapeutics; use of epinephrine for allergic and anaphylactic reactions, lidocaine, vasoconstrictors, vasodilators.
[0200] Pharmacokinetic/Pharmacodynamic Monitoring
[0201] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of
elesclomol are made by the use of monitoring drug levels after dosing in an effort to maximize a patient’s drug plasma level, to monitor the generation of toxic metabolites, monitoring of ancillary medicines that could be beneficial or harmful in terms of drug- drug interactions. General examples include: the monitoring of drug plasma protein binding. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: multiple determinations of drug plasma levels; multiple determinations of metabolites in the blood or urine, measurement of polyamines, LAT-1 surface receptors, gene sequencing, immune effectors.
[0202] Drug Combinations
[0203] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting unique drug combinations that may provide a more than additive or synergistic improvement in efficacy or side-effect management. General examples include: alkylating agents with anti-metabolites, topoisomerase inhibitors with antitubulin agents. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use with topoisomerase inhibitors; use with fraudulent nucleosides; use with fraudulent nucleotides; use with thymidylate synthetase inhibitors; use with signal transduction inhibitors; use with cisplatin or gallium analogs; use with alkylating agents such as the nitrosoureas (BCNU , Gliadel wafers, CCNU ); use with bendamustine (Treanda); use with anti-tubulin agents; use with antimetabolites; use with berberine; apigenin; amonafide; colchicine and analogs; genistein; etoposide; cytarabine; camptothecins; vinca alkaloids; topoisomerase inhibitors; 5-fluorouracil; curcumin; NF-KB inhibitors; rosmarinic acid; in combination with biological therapies such as antibodies such as Avastin, Rituxan, Herceptin, Erbitux, PD-1 and PD-L1 inhibitors, prednimustine, DNA and RNA therapeutics, Braf inhibitors; BTK inhibitors, 5- azacytidine, decitabine, PARP inhibitors, agents inducing hypomethylation, histone deacetylase inhibitors.
[0204] Chemosensitization
[0205] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting them as chemosensitizers where no measurable activity is observed when used alone but in combination with other therapeutics a more than additive or synergistic improvement in efficacy is observed. General examples include: misonidazole with alkylating agents, tirapazamine with cisplatin. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: as a chemosensitizer in combination with topoisomerase inhibitors; use with fraudulent nucleosides; use with fraudulent nucleotides; use with thymidylate synthetase inhibitors; use with signal transduction inhibitors; use with cisplatin or gallium analogs; use with alkylating agents such as BCNU Gliadel wafers, CCNU , bendamustine (Treanda), temozolomide (Temodar); use with anti-tubulin agents; use with antimetabolites; use with berberine; apigenin; amonafide; colchicine and analogs; genistein; etoposide; cytarabine; camptothecins; vinca alkaloids; topoisomerase inhibitors; 5-fluorouracil; curcumin; NF- KB inhibitors; rosmarinic acid.
[0206] Chemopotentiation
[0207] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting them as chemopotentiators where minimal therapeutic activity is observed alone but in combination with other therapeutics a more than additive or synergistic improvement in efficacy is observed. General examples include: amonafide with cisplatin or 5-Fll. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: as a chemopotentiator in combination with topoisomerase inhibitors; use with fraudulent nucleosides; use with fraudulent nucleotides; use with thymidylate synthetase inhibitors; use with signal transduction inhibitors; use with cisplatin or gallium analogs; use with alkylating agents such as BCNU , BCNU wafers, Gliadel, bendamustine (Treanda); use with anti-tubulin agents; use with antimetabolites; use with berberine; apigenin; amonafide; colchicine and
analogs; genistein; etoposide; cytarabine; camptothecins; vinca alkaloids; topoisomerase inhibitors; 5-fluorouracil; curcumin; NF-KB inhibitors; rosmarinic acid.
[0208] Post-Treatment Patient Management
[0209] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by drugs, treatments and diagnostics to allow for the maximum benefit to patients treated with a compound. General examples include: pain management, nutritional support, anti-emetics, anti-nausea therapies, anti-anemia therapy, anti-inflammatories. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use with therapies associated with pain management; nutritional support; anti- emetics; anti-nausea therapies; anti-anemia therapy; anti-inflammatories: antipyretics; immune stimulants; anti diarrhea medicines, famotidine, antihistamines, suppository lubricants, soothing agents, lidocaine, hydrocortisone.
[0210] Alternative Medicine/Therapeutic Support
[0211] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of therapeutics or methods to enhance effectiveness or reduce side effects. General examples include: herbal medications and extracts. Specific inventive examples for redox modulating agent compounds such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: herbal medications created either synthetically or through extraction including NF- KB inhibitors (such as parthenolide, curcumin, rosmarinic acid); natural antiinflammatories (including rhein, parthenolide); immunostimulants (such as those found in Echinacea); antimicrobials (such as berberine); flavonoids and flavones (such as apigenenin, genistein).
[0212] Bulk Drug Product Improvements
[0213] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the pharmaceutical bulk substance. General examples include: salt formation, homogeneous crystalline structure, pure isomers.
Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: salt formation; homogeneous crystalline structure; pure isomers; increased purity; lower residual solvents and heavy metals.
[0214] Diluent Systems
[0215] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the diluents used to solubilize and deliver/present the compound for administration. General examples include: Cremophor-EL, cyclodextrins for poorly water soluble compounds. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use of emulsions; DMSO; NMF; DMF; DMA; ethanol; benzyl alcohol; dextrose-containing water for injection; Cremophor; cyclodextrins; PEG; a sweetening agent such as saccharin; agents to thicken an oral dosage form such as glycerol; taste masking effectors such as menthol, rum flavor, fruit flavorings.
[0216] Solvent Systems
[0217] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the solvents used or required to solubilize a compound for administration or for further dilution. General examples include: ethanol, dimethylacetamide (DMA). Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of emulsions; dimethyl sulfoxide (DMSO); N-methylformamide (NMF); dimethylformamide (DMF); dimethylacetamide (DMA); ethanol; benzyl alcohol; dextrose-containing water for injection; Cremophor; PEG; glycerol, cocoa butter for suppositories.
[0218] Excipients
[0219] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the materials/excipients, buffering agents, preservatives required to stabilize and present a chemical compound for proper
administration. General examples include: mannitol, albumin, EDTA, sodium bisulfite, benzyl alcohol. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of mannitol; albumin; EDTA; sodium bisulfite; benzyl alcohol; carbonate buffers; phosphate buffers; glycerol; sweeteners; taste masking agents such as rum flavor, menthol; substituted celluloses; sodium azide as a preservative.
[0220] Dosage Forms
[0221] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the potential dosage forms of the compound dependent on the route of administration, duration of effect, plasma levels required, exposure to side effects for normal tissues and metabolizing enzymes. General examples include: tablets, capsules, topical gels, creams, patches, suppositories, oral solutions, suspensions, syrups. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of tablets; capsules; topical gels; topical creams; patches; suppositories; lyophilized dosage fills, suppositories with quick release <15 min or long melt times >15 min release time; temperature adjusted suppositories.
[0222] Dosage Kits and Packaging
[0223] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations in the dosage forms, container/closure systems, accuracy of mixing and dosage preparation and presentation. General examples include: amber vials to protect from light, stoppers with specialized coatings. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of amber vials to protect from light; stoppers with specialized coatings to improve shelf-life stability; special dropper measuring devices; single-use or multiple-use container closure systems; testing for allergies; suppository delivery devices, epinephrine pens for side effect management; physician and nurse assistance gloves; dosage measuring devices.
[0224] Drug Delivery Systems
[0225] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of delivery systems to improve the potential attributes of a pharmaceutical product such as convenience, duration of effect, reduction of toxicities. General examples include: nanocrystals, bioerodible polymers, liposomes, slow release injectable gels, microspheres. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of nanocrystals; bioerodible polymers; liposomes; slow release injectable gels; microspheres; suspensions with glycerol, meltable drug release suppositories with polymers such as cocoa butter alone or in combination with PEG, lecithin; polylactide/polyglycolide; rectal plugs for drug delivery.
[0226] Drug Conjugate Forms
[0227] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the parent molecule with covalent, ionic, or hydrogen bonded moieties to alter the efficacy, toxicity, pharmacokinetics, metabolism, or route of administration. General examples include: polymer systems such as polyethylene glycols, polylactides, polyglycolides, amino acids, peptides, multivalent linkers. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of polymer systems such as polyethylene glycols; polylactides; polyglycolides; amino acids; peptides; multivalent linkers.
[0228] Compound Analogs
[0229] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the parent structure of a molecule with additional chemical functionalities that may alter efficacy or reduce toxicity, pharmacological performance, route of administration, or other factors that alter efficacy or reduce toxicity. General examples include: alteration of side chains to increase or decrease lipophilicity, additional chemical functionalities to alter reactivity, electron affinity, binding capacity, salt forms. Specific inventive examples for redox modulating agents such as
elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: alteration of side chains to increase or decrease lipophilicity; additional chemical functionalities to alter reactivity; alteration of electron affinity; alteration of binding capacity; salt forms.
[0230] Prodrug Systems
[0231] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by alterations to the molecule such that improved pharmaceutical performance is gained with a variant of the active molecule in that after introduction into the body a portion of the molecule is cleaved to reveal the preferred active molecule. General examples include: enzyme sensitive esters, dimers, Schiff bases. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of enzyme sensitive esters; dimers; Schiff bases; pyridoxal complexes; caffeine complexes; bioreductive analogs including nitroso-substituted analogs.
[0232] Multiple Drug Systems
[0233] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by the use of additional compounds, biological agents that when administered in the proper fashion, a unique and beneficial effect can be realized. General examples include: inhibitors of multi-drug resistance, specific drug resistance inhibitors, specific inhibitors of selective enzymes, signal transduction inhibitors, repair inhibition. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use of inhibitors of multi-drug resistance; specific drug resistance inhibitors; specific inhibitors of selective enzymes; signal transduction inhibitors; repair inhibition; topoisomerase inhibitors with non-overlapping side effects.
[0234] Biotherapeutic Enhancement
[0235] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by its use in combination as sensitizers/potentiators with biological
response modifiers. General examples include: use in combination as sensitizers/potentiators with biological response modifiers, cytokines, lymphokines, therapeutic antibodies, antisense therapies, gene therapies. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use in combination as sensitizers/potentiators with biological response modifiers; cytokines; lymphokines; therapeutic antibodies such as bevacizumab, trastuzumab, rituximab, and cetuximab; gene therapies; ribozymes; RNA interference, cell based therapeutics.
[0236] Biotherapeutic Resistance Modulation
[0237] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting their selective use to overcome developing or complete resistance to the efficient use of biotherapeutics. General examples include: tumors resistant to the effects of biological response modifiers, cytokines, lymphokines, therapeutic antibodies, antisense therapies, gene therapies. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use against tumors resistant to the effects of biological response modifiers; cytokines; lymphokines; therapeutic antibodies; antisense therapies such as bevacizumab, trastuzumab, rituximab, and cetuximab; gene therapies; ribozymes; RNA interference.
[0238] Radiation Therapy Enhancement
[0239] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by exploiting their use in combination with ionizing radiation, phototherapies, heat therapies, radio-frequency generated therapies. General examples include: hypoxic cell sensitizers, radiation sensitizers/protectors, photosensitizers, radiation repair inhibitors. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use with hypoxic cell sensitizers; radiation sensitizers/protectors; photosensitizers; radiation repair inhibitors; thiol depletion; vaso-targeted agents; use with radioactive seeds, radionuclides, radiolabeled antibodies, brachytherapy.
[0240] Novel Mechanisms of Action
[0241] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by optimizing their utility by determining the various mechanisms of actions, biological targets of a compound for greater understanding and precision to better exploit the utility of the molecule. General examples include: Gleevec for chronic myelocytic leukemia (CML), arsenic trioxide for acute promyelocytic leukemia (APL), retinoic acid for APL. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: the use with inhibitors of poly-ADP ribose polymerase; agents that affect vasculature; vasodilation; oncogenic targeted agents; signal transduction inhibitors; EGFR inhibition; Protein Kinase C inhibition; Phospholipase C downregulation; jun downregulation; histone genes, VEGF, ornithine decarboxylase, jun D, v-jun, GPCR’s, protein kinase A, telomerase, prostate specific genes, protein kinases, histone deacetylase.
[0242] Selective Target Cell Population Therapeutics
[0243] Improvements for suboptimal therapeutics including redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol are made by more precise identification and exposure of the compound to those select cell populations where the compounds effect can be maximally exploited. General examples include: tirapazamine and mitomycin c for hypoxic cells, vinca alkaloids for cells entering mitosis. Specific inventive examples for redox modulating agents such as elesclomol or derivatives, analogs, salts, solvates, or prodrugs of elesclomol include: use against radiation sensitive cells; radiation resistant cells; energy depleted cells; endothelial cells.
[0244] Use to Reverse Resistance to Platinum-Containing Anti-Neoplastic Agents and PARP Inhibitor Anti-Neoplastic Agents
[0245] Another aspect of the present invention is use of elesclomol or a derivative or analog of elesclomol to reverse resistance to platinum-containing anti- neoplastic agents or PARP inhibitor anti-neoplastic agents, particularly resistance modulated by mutations in ARID1A. Suitable platinum-containing and PARP inhibitor anti-neoplastic agents are described below.
[0246] Use to Induce Synthetic Lethality
[0247] Yet another aspect of the present invention is induction of synthetic lethality by administration of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1 A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol renders the malignant cells susceptible to a targeted agent that otherwise would have no effect. In particular, an onco-driving mutation that allows for tumor formation, such as a mutation causing a defect in DNA damage repair, promotion of an open chromosome, or enhanced transcription, can also create an “Achilles heel” for the same tumor cells by its very existence. In the case of elesclomol, the mutation in ARID1 A leads to tumor formation, but also leads to a dependence on OXPHOS — the Achilles heel — as such tumor cells are uniquely susceptible to mitochondrial targeting agents, such as, but not necessarily limited to, elesclomol or a derivative or analog of elesclomol, which targets ferredoxin-1 , because of the defect that allows the tumor to form in the first place. If the tumor-causing mutation had not formed, such mitochondrial-targeting agents would have had minimal or no activity.
[0248] Use to Target Ferredoxin-I to Inhibit OXPHOS
[0249] Yet another aspect of the present invention is use of elesclomol or a derivative or analog of elesclomol to target ferredoxin-l (FDX1 ) in order to inhibit oxidative phosphorylation (OXPHOS) in circumstances in which inhibition of oxidative phosphorylation would be desirable.
[0250] The structure of elesclomol as the free acid is shown above in Formula (I).
[0251] However, the activity of elesclomol is, as stated above, dependent on the formation of a coordinate-covalent complex with a transition metal cation, preferably a divalent transition metal cation. As used herein, the term “transition metal cation” refers to a cation of a metal in Groups 3-12 of the Periodic Table. Examples include, but are not necessarily limited to, Ni2+, Cu+, Cu2+, Co2+, Co3+, Fe2+, Fe3+, Zn2+, Pt2+, Pd2+, V4+, V5+, Cr2+, Cr3+, Cr4+, Mn2+, Mn3+, Mn4+, or Mn5+. As stated above, the transition metal cation is preferably divalent, such as, but not limited to, Ni2+, Cu2+, Co2+, Fe2+, Zn2+, Pt2+,
and Pd2+. More preferably, the divalent transition metal cation is Cu2+ or Ni+2. Still more preferably, the divalent transition metal cation is Cu2+.
[0252] The molar ratio of elesclomol to the transition metal cation is equal to or greater than 0.5 and equal to or less than 2.0; typically, the molar ratio of elesclomol to the transition metal cation is 1.0.
[0253] A coordinate-covalent complex of elesclomol and divalent copper is shown in Formula (II):

(II).
[0254] A coordinate-covalent complex of elesclomol and divalent nickel is shown in Formula (III):

(HI).
[0255] Analogs and derivatives of elesclomol include, but are not limited to, compounds of Formula (IV):
 (IV), wherein:
(IV), wherein:
(1 ) Y is a covalent bond, a phenylene group or a substituted or unsubstituted straight-chain hydrocarbyl group, or Y taken together with both >C=Z groups with which it is bonded is a substituted or unsubstituted aromatic group;
(2) R1 is a substituted or unsubstituted aliphatic group or a substituted or unsubstituted non-aromatic heterocyclic group;
(3) R2, R3, and R4 are independently hydrogen, a substituted or unsubstituted aliphatic group, a substituted or unsubstituted non-aromatic heterocyclic group, a substituted or unsubstituted aryl group, or R1 and R3 and/or R2 and R4 taken together with the carbon and nitrogen atoms to which they are bonded, form a non-aromatic heterocyclic ring optionally fused to an aromatic ring;
(4) R5 and R6 are independently hydrogen, a substituted or unsubstituted aliphatic group, or a substituted or unsubstituted non-aromatic heterocyclic group;
(5) R7 and R8 are independently hydrogen or a substituted or unsubstituted aliphatic group, or R7 is hydrogen and R8 is a substituted or unsubstituted aryl group, or R7 and R8 taken together are C2-C6 substituted or unsubstituted alkylene group; and
(6) Z is =0 or =S.
[0256] In compounds of Formula (IV), preferably Y is a covalent bond or - C(R7R8)~.
[0257] A particular derivative or analog of elesclomol is a compound of Formula (V):

(V).
[0258] Additional derivatives or analogs of elesclomol are compounds of Formulas (VI) and (VII):

(VII).
[0259] Additional derivatives and analogs of elesclomol are compounds of
Formula (VIII):

(VIII), wherein:
(1 ) each Z is independently S, 0, or Se, provided that both Z moieties cannot be 0;
(2) R1 and R2 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted alkynyl, an optionally substituted cycloalkyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group wherein the heterocyclic group is bonded to the thiocarbonyl via a carbon-carbon linkage, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to seven-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl wherein the heteroaryl is bonded to the thiocarbonyl via a carbon- carbon linkage, --NR12R13, --OR14, SR14, and S(O)pR15;
(3) R3 and R4 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group, and an optionally substituted five-membered or six-membered aryl or heteroaryl group; or, alternatively, R1 and R3, and/or R2 and R4, taken together with the atoms to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group; in this alternative, R3 and R4 can also be hydrogen;
(4) R5 is -CR6R7, -C(=CHR8), or -C(=NR8);
(5) R6 and R7 are both hydrogen or an optionally substituted lower alkyl;
(6) R8 is selected from the group consisting of hydroxyl, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxylalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic aryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, — N10R11 , and -COR9;
(7) R9 is an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted alkyl, an optionally substituted cycloalkyl, or an optionally substituted heterocyclic group;
(8) R10 and R11 are each independently selected from the group consisting of hydrogen, hydroxyl, alkylamino, dialkylamino, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxyalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, and - COR9; or R10 and R11, taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group;
(9) R12, R13, and R14 are each independently hydrogen, an optionally substituted alkyl, an optionally substituted phenyl, or an optionally substituted benzyl, or R12 and R13, taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group;
(10) R15 is an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl; and
(11 ) p is 1 or 2; provided that when both Z are S and R3 and R4 are both methyl, then R1 and R2 are not both unsubstituted phenyl; alternatively, for compounds of Formula (VIII), R10 and R11 are not both hydrogen.
[0260] Additional derivatives and analogs of elesclomol are described in United States Patent No. 6,762,204 to Koya et al. and in PCT Patent Application Publication No. WO 2010/066512 by Jiang et al.
[0261] As used herein, the term “elesclomol,” unless further defined or limited, includes all coordinate covalent complexes of elesclomol.
[0262] The synthesis of the coordinate covalent complex of Cu+2 with elesclomol is described in N.H. Vo et al., “Synthesis, Crystallographic Characterization and Electrochemical Property of a Copper(ll) Complex of the Anticancer Agent Elesclomol,” J. Inorg. Biochem. 130: 69-73 (2014).
[0263] Accordingly, one aspect of the present invention is a method for treating a malignancy comprising the step of administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug thereof to a subject with a malignancy to treat the malignancy.
[0264] Typically, the therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug thereof is a therapeutically effective quantity of elesclomol. Typically, the therapeutically effective quantity of elesclomol is administered in a coordinate-covalent complex with a transition metal selected from the group consisting of Ni2+, Cu+, Cu2+, Co2+, Co3+, Fe2+, Fe3+, Zn2+, Pt2+, Pd2+, V4+, V5+, Cr2+, Cr3+, Cr4+, Mn2+, Mn3+, Mn4+, and Mn5+. Preferably, the transition metal is selected from the group consisting of Ni2+, Cu2+, Co2+, Fe2+, Zn2+, Pt2+, and Pd2+. More preferably, the transition metal is selected from the group consisting of Ni2+ and Cu2+.
Most preferably, the transition metal is Cu+2. Typically, the molar ratio of elesclomol to the transition metal cation is equal to or greater than 0.5 and equal to or less than 2.0; preferably, the molar ratio of elesclomol to the transition metal cation is 1 .0.
[0265] As used herein, the term “therapeutically effective quantity” or equivalent terminology refers to the quantity of a therapeutic agent in which a beneficial clinical outcome is achieved when the quantity of the therapeutic agent is administered to a subject in need thereof. The term “beneficial clinical outcome” refers to a detectable or observable improvement in a clinical parameter as a result of the administration of the therapeutic agent; the detectable or observable improvement can be objective or subjective. For example, when the therapeutic agent is administered to treat a malignancy, a “beneficial clinical outcome” can include, but is not necessarily limited to: a reduction in tumor mass or tumor burden; a reduction in tumor spread or metastasis; a reduction in pain; a reduction of symptoms associated with the malignancy such as seizures for central nervous system malignancies; a reduction of fatigue; a reduction of malaise; an increase in longevity; or an improved Karnofsky performance score. Other determinants of a beneficial clinical outcome are known in the art, including determinants for diseases or conditions other than malignancies.
[0266] For administration of the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol to the subject, the therapeutically effective quantity will depend on the type and severity of the malignancy and on the characteristics of the subject, such as the general health, the age, the sex, the body weight, the tolerance to drugs, the relevant pharmacokinetic factors such as liver and kidney function. The therapeutically effective quantity will also depend on other therapeutic agents being concurrently administered to the subject to treat the malignancy or to treat other co- morbid conditions affecting the subject. One of ordinary skill in the art will be able to determine the therapeutically effective quantity based on this and other factors. In general, and subject to the factors recited above, the therapeutically effective quantity typically ranges between about 1 mg/mm2/day to about 10 g/mm2/day; more typically, the therapeutically effective quantity ranges between about 2 mg/mm2/day to about 5 g/mm2/day. In some alternatives, the therapeutically effective quantity is from about 1 pg/kg to about 500 mg/kg, from about 500 pg/kg to about 250 mg/kg, from about 1
mg/kg to about 100 mg/kg, or from about 10 mg/kg to about 50 mg/kg. As used herein, the terms “co-administration” and “co-administering” refer to the administration of at least two agents, such as elesclomol or a derivative, analog, or prodrug of elesclomol and an inhibitor, or therapies to a subject. In some embodiments, the co-administration of two or more agents or therapies is concurrent. In other embodiments, a first agent/therapy is administered prior to a second agent/therapy. Those of skill in the art understand that the formulations and/or routes of administration of the various agents or therapies used may vary. The appropriate dosage for co-administration can be readily determined by one skilled in the art. In some embodiments, when agents or therapies are co-administered, the respective agents or therapies are administered at lower dosages than appropriate for their administration alone. Thus, co-administration is especially desirable in embodiments where the co-administration of the agents or therapies lowers the requisite dosage of a potentially harmful agent or agent, and/or when co-administration of two or more agents results in sensitization of a subject to beneficial effects of one of the agents via co-administration of the other agent. As used herein, the term “concurrent administration” refers to the administration of two or more active agents sufficiently close in time to achieve a combined therapeutic effect that is preferably greater than that which would be achieved by the administration of either agent alone. Such concurrent administration can be carried out simultaneously, e.g., by administering the active agents together in a common pharmaceutically acceptable carrier, thereby forming a pharmaceutical composition with two or more active agents, in one or more doses of the pharmaceutical composition.
[0267] In some alternatives, the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol can be administered in a pharmaceutically acceptable composition. Suitable pharmaceutical compositions that comprise elesclomol or a derivative, analog, salt, solvate, or prodrug are as described above.
[0268] The malignancy to be treated can be, but is not limited to, ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer,
metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, or lymphoid diffuse large B-cell lymphoma.
[0269] Methods according to the present invention can alternatively be used to treat other malignancies, including, but not limited to, human sarcomas and carcinomas. These malignancies include, but are not limited to: fibrosarcoma; myxosarcoma; liposarcoma, chondrosarcoma; osteogenic sarcoma; chordoma; angiosarcoma; endotheliosarcoma; lymphangiosarcoma; lymphangioendotheliosarcoma; synovioma; mesothelioma; Ewing’s tumor; leiomyosarcoma; rhabdomyosarcoma; colon carcinoma, including Kras-mutated colon carcinoma; colorectal cancer; anal carcinoma; esophageal cancer; gastric cancer; hepatocellular cancer; bladder cancer; endometrial cancer; pancreatic cancer; breast cancer, including triple-negative breast cancer; ovarian cancer; prostate cancer; stomach cancer; atrial myxomas; squamous cell carcinoma; basal cell carcinoma; adenocarcinoma; sweat gland carcinoma; sebaceous gland carcinoma; thyroid and parathyroid neoplasms; papillary carcinoma; papillary adenocarcinoma; cystadenocarcinoma; medullary carcinoma; bronchogenic carcinoma; renal cell carcinoma; hepatoma; bile duct carcinoma; choriocarcinoma; seminoma; embryonal carcinoma; Wilms’ tumor; cervical cancer; testicular tumor; lung carcinoma, including small-cell lung carcinoma and non-small-cell lung carcinoma; bladder carcinoma; epithelial carcinoma; glioma; pituitary neoplasms; astrocytoma; medulloblastoma; craniopharyngioma; ependymoma; pinealoma; hemangioblastoma; acoustic neuroma; schwannoma; oligodendroglioma; meningioma; spinal cord tumors; melanoma, including Braf-mutated melanoma; neuroblastoma; pheochromocytoma; endocrine neoplasia, Types 1-3; retinoblastoma; leukemias, including acute lymphocytic leukemia and acute myelocytic leukemia (including myeloblastic, promyelocytic, myelomonocytic, monocytic, and erythroleukemia), chronic leukemia (including chronic myelocytic (granulocytic) leukemia and chronic lymphocytic leukemia, including BTK- resistant chronic lymphocytic leukemia), and meningeal leukemia; polycythemia vera; lymphoma, including Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, mantle cell lymphoma, and cutaneous T-cell lymphoma; multiple myeloma; Waldenstrom’s macroglobulinemia; mycosis fungoides; leptomeningeal cancer; pediatric brain tumors;
pediatric sarcoma; ovarian osteogenic sarcoma; small-cell carcinoma of the ovary, including the hypercalcemic type, and heavy chain disease.
[0270] In addition, methods according to the present invention can be used to treat other diseases and conditions, including, but not limited to: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; or scleroderma.
[0271] Methods as described above can further comprise administering a therapeutically effective quantity of at least one additional therapeutic agent. Suitable additional therapeutic agents are described below.
[0272] One category of suitable additional therapeutic agents is microtubulin stabilizers and/or microtubulin inhibitors. As used herein, the term “microtubulin stabilizer” refers to an agent, typically an anti-cancer agent, that acts by arresting cells in the G2/M phase of cell division by stabilization of microtubules. Elesclomol or derivatives or analogs of elesclomol as described herein can contribute to the G2/M arrest associated with microtubulin stabilizers or agents with similar mechanisms of action. Examples of microtubulin stabilizers include paclitaxel and paclitaxel analogs. Additional examples of microtubulin stabilizers include, but are not limited to: discodermolide; epothilone A; epothilone B; epothilone C; epothilone D; epothilone E; epothilone F; epothilone B N-oxide; epothilone A N-oxide; 16-aza-epothilone B; 21- aminoepothilone B; 21-hydroxyepothilone D; 26-fluoroepothilone; sagopilone; ixabepilone; uditelone; (1 R,2S,4S,7S,8S,9S, 10R, 11 S, 12S, 13R, 17R, 18S)-8, 10- dihydroxy-1 ,5,9, 18-tetramethyl-16,20- dioxahexacyclo[15.3.2.02, 13.04, 12.07, 11.014,19]docosa-5, 14(19)-dien-15-one (FR- 182877); tasidotin hydrochloride; 2-methoxy-5-[(Z)-2-(3,4,5- trimethoxyphenyl)ethenyl]aniline; hydrochloride (AC-7739); (2S)-2-amino-3-hydroxy-N- [2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenyl]propanamide;hydrochloride (AC-7700); fijianolide B; laulimalide; caribaeoside; caribaeolin; taccalonolide;
eleutherobin; sarcodictyin; dictyostatin-1 ; jatrophane esters; abraxane; and analogs and derivatives of these compounds.
[0273] Paclitaxel is an antineoplastic drug that functions by enhancing and stabilizing microtubule formation, thus inhibiting mitosis. The structure of paclitaxel is shown in Formula P-l, below:

(P-l).
[0274] Many analogs and derivatives of paclitaxel are known. These compounds have the basic taxane skeleton as a common structural feature and have the ability to arrest cells in the G2/M phase of the cell cycle due to microtubule stabilization. A wide variety of substituents can be made on the taxane skeleton while retaining the microtubule-stabilizing activity. It is also the case that zero, one, or two of the cyclohexane rings of a paclitaxel analog can have a double bond (paclitaxel itself has one double bond in such a six-membered ring). Other substitutions can be made at various positions, including moieties including an oxygen atom such as hydroxyl, acyl, or alkoxy.
[0275] One particularly useful compound with the basic taxane skeleton is docetaxel.
[0276] Typically, paclitaxel analogs useful in methods or compositions according to the present invention are represented by Formulas (P-l I) or (P-lll):

(P-lll).
[0277] In the compounds of Formulas (P-ll) and (P-lll):
(1 ) R10 is substituted or unsubstituted lower alkyl, substituted or unsubstituted phenyl; --SR19, --NHR19, or -OR19;
(2) R11 is substituted or unsubstituted lower alkyl or substituted or unsubstituted aryl;
(3) R12 is hydrogen, hydroxyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkoxy, --O-C(O)-(lower alkyl)--, --O-C(O)-(substituted lower alkyl)--, --O-CH2-O-(lower alkyl), or --S-CH2-O-(lower alkyl);
(4) R13 is hydrogen, methyl, or, taken together with R14, --CH2--;
(5) R14 is hydrogen, hydroxyl, substituted or unsubstituted lower alkoxy, -0- C(O)-(substituted lower alkyl), -O-CH2-O-P(O)(OH)2, -O-CH2-O-( lower alkyl), -O-CH2- S-(lower alkyl), or taken together with R20, a double bond;
(6) R15 is hydrogen, lower acyl, substituted or unsubstituted lower alkyl, alkoxymethyl, alkylthiomethyl, --C(O)-O-(lower alkyl), --C(O)-O-(substituted lower alkyl), --C(O)-NH-(lower alkyl), or --C(O)-NH-(substituted lower alkyl);
(7) R16 is substituted or unsubstituted phenyl;
(8) R17 is hydrogen, substituted or unsubstituted lower acyl, substituted or unsubstituted lower alkyl, (lower alkoxy)methyl, or (lower alkoxy)thiomethyl;
(9) R18 is hydrogen, methyl, or taken together with R17 and the carbon atoms to which R17 and R18 are bonded, a five-membered or six-membered non-aromatic heterocyclic ring;
(10) R19 is substituted or unsubstituted lower alkyl or substituted or unsubstituted phenyl;
(11 ) R20 is hydrogen or a halogen; and
(12) R21 is substituted or unsubstituted lower alkyl or substituted or unsubstituted acyl.
[0278] Paclitaxel analogs or derivatives can also be covalently linked to a pharmaceutically acceptable polymer such as a polyacrylamide.
[0279] Paclitaxel analogs and derivatives are disclosed in PCT Patent Application Publication No. WO 2010/065512 by Jiang et al.
[0280] As used herein, the term “microtubulin inhibitor” refers to an anti-cancer agent that acts by inhibiting tubulin polymerization or microtubule assembly. Examples of microtubulin inhibitors include, but are not limited to: erbulozole; dolastatin 10; mivobulin isethionate; vincristine; vinblastine; vinorelbine; vinflunine; vindesine; N-[[4-(5- bromopyrimidin-2-yl)oxy-3-methylphenyl]carbamoyl]-2-(dimethylamino)benzamide (NSC-639829); N-[2-(4-hydroxyanilino)pyridin-3-yl]-4-methoxybenzenesulfonamide (ABT-751 ); altorhytin A; altorhytin C; spongistatin 1 , spongistatin 2, spongistatin 3, spongistatin 4, spongistatin 5, spongistatin 6, spongistatin 7, spongistatin 8, spongistatin 9; cemadotin hydrochloride; solbidotin; LS-4559-P; LS-4578; LS-4477 (an N-acyl- aminoalkyl phenyl ether and an analog of estramustine); LS-4559 (RPRan N-acyl- aminoalkyl phenyl ether and an analog of estramustine); RPR-112378; RPR-115781 ; 1- [5-methyl-1 -(2-pyrim id iny I) -4-pyrazolyl]-3-[4-(3-chlorophenyl)-1 -piperaziny l]-1 -trans- propene hydrochloride (DZ-3358); estramustine phosphate; [3,5-bis(4-fluorophenyl)- 1 ,3,5,7-tetrahydro-[1 ,3]oxazolo[3,4-c][1 ,3]oxazol-7a-yl]methanol (GS-164); GS-198; KAR-2; SAH-49960; SDZ-268970; (E)-2-methyl-3-(6-methyl-1 H-indol-3-yl)-1 -(3,4,5- trimethoxyphenyl)prop-2-en-1-one (AM-97); (E)-3-[5-(2,3-dihydroxypropylsulfanyl)-1 H- indol-3-y l]-1 -(3,4,5-trimethoxyphenyl)prop-2-en-1 -one (AM-132); (E)-3-[5-(2,3-
dihydroxypropylsulfanyl)-6-methyl-1 H-indol-3-y l]-1 -(3,4,5-trimethoxyphenyl)prop-2-en-1 - one (AM-138); 2,2,2-trifluoro-N-[(7S)-1 , 2, 3-trim ethoxy-10-methylsulfanyl-9-oxo-6,7- dihydro-5H-benzo[a]heptalen-7-yl]acetamide (IDN-5005); cryptophycin 52; vitilevuamide; tubulysin A; canadensol; centaureidin; batabulin; (1 R)-1 -[(2R,5R)-5- (hydroxymethyl)oxolan-2-yl]tridecan-1 -ol (WH 1-261 ); 6-bromo-3-(5-chloro-2- ethylsulfonylanilino)-7-[(4-pentan-3-ylpiperazin-1 -yl)methyl]quinazolin-4-one (H10); 7- [[(3R)-3-aminopyrrolidin-1-yl]methyl]-8-bromo-3-[(5-chloro-2- ethylsulfonylphenyl)methyl]-6-(trifluoromethoxy)-1 H-quinazoline-2, 4-dione (H-16); oncocidin A1 ; DDE-313 (an epoxy tetrahydrofuran-containing compound); SPA-2;
SPIKET-P (a synthetic spiroketal pyran); 3-(iodoacetamido)-benzoylurea (MF-569); narcosine; nascapine; 1 -[5-(4-amino-3-methylphenyl)-2-(3,4,5-trimethoxyphenyl)-2H- 1 ,3,4-oxadiazol-3-yl]ethenone (A-105972); hemiasterlin; 3-bromoacetylamino benzoylurea (MF-191 ); benzyl 2,2,4,4-tetramethylpentanoate; vanadocene acetylacetonate; T-138026; (1 S,3S)-1 -ethenyl-3-[(3-hydroxy-4-methoxyphenyl)methyl]- 7-methyl-2,3-dihydro-1 H-inden-4-ol (RPR-115781 ); desmethyleleutherobin; desethyleleutherobin, isoeleutherobin A; Z-eleutherobin; halichondrin B; (5-methoxy-1 H- indol-2-yl)-phenylmethanone (D-64131 ); D-68144 (a 2-aroylindole); diazonamide A; NPI-2350; NPI-2358 (a synthetic diketopiperazine); A-259754; diazostatin; (-)- phenylahistin; polyandrocarpidine C; D-68836; myoseverin B; D-43411 ; (5S)-2-(1 - methylindol-5-yl)-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1 ,3-oxazole (A-289099); A- 318315; taltobulin; D-82318; 2-phenyl-1 H-indol-6-amine; resverastatin phosphate sodium; BPR-OY-007 (a bis-benzylidenecyclopentanone derivative); SSR-250411 ; and combretastatin A4.
[0281] Other microtubule-disrupting antineoplastics include, but are not necessarily limited to, cabazitaxel, larotaxel, vedotin, belantamab mafodotin, ortataxel, and tesetaxel.
[0282] A second category of suitable additional therapeutic agents is PARP inhibitors. Inhibitors of the enzyme poly-ADP ribose polymerase (PARP) have been developed for multiple indications, especially for treatment of malignancies. Several forms of cancer are more dependent on the activity of PARP than are non-malignant cells.
[0283] The enzyme PARP catalyzes the polymerization of poly-ADP ribose chains, typically attached to a single-strand break in cellular DNA. The coenzyme NAD+ is required as a substrate for generating ADP-ribose monomers to be polymerized; nicotinamide is the leaving group during polymerization, in contrast to pyrophosphate which is the leaving group during normal DNA or RNA synthesis, which leaves a pyrophosphate as the linking group between adjacent ribose sugars in the chain rather than phosphate as occurs in normal DNA or RNA. The PARP enzyme comprises four domains: a DNA-binding domain, a caspase-cleaved domain, an auto-modification domain, and a catalytic domain. The DNA-binding domain comprises two zinc finger motifs. In the presence of damaged DNA, the DNA-binding domain will bind the DNA and induce a conformational shift. PARP can be inactivated by caspase-3 cleavage, which is a step that occurs in programmed cell death (apoptosis).
[0284] Several PARP enzymes are known, including PARP1 and PARP2. Of these two enzymes, PARP1 is responsible for most cellular PARP activity. The binding of PARP1 to single-strand breaks in DNA through the amino-terminal zinc finger motifs recruits XRCC1 , DNA ligase III, DNA polymerase β, and a kinase to the nick. This is known as base excision repair (BER). PARP2 has been shown to oligomerize with PARP1 , and the oligomerization stimulates catalytic activity. PARP2 is also therefore implicated in BER.
[0285] PARP1 inhibitors inhibit the activity of PARP1 and thus inhibit the repair of single-strand breaks in DNA. When such breaks are unrepaired, subsequent DNA replication can induce double-strand breaks. The proteins BRCA1 , BRCA2, and PALB2 can repair double-strand breaks in DNA by the error-free homologous recombinational repair (HRR) pathway. In tumors with mutations in the genes BRCA1, BRCA2, or PALB1, these double-strand breaks cannot be efficiently repaired, leading to cell death. Normal cells do not replicate their DNA as frequently as tumor cells, and normal cells that lack mutated BRCA1 or BRCA2 proteins can still repair these double-strand breaks through homologous repair. Therefore, normal cells are less sensitive to the activity of PARP inhibitors than tumor cells.
[0286] Some tumor cells that lack the tumor suppressor PTEN may be sensitive to PARP inhibitors because of downregulation of Rad51 , a critical homologous
recombination component. Tumor cells that are low in oxygen are also sensitive to PARP inhibitors.
[0287] PARP inhibitors are also considered potential treatments for other life- threatening diseases, including stroke and myocardial infarction, as well as for long- term neurodegenerative diseases (G. Graziani & C. Szabo, “Clinical Perspectives of PARP Inhibitors,” Pharmacol. Res. 52: 109-118 (2005)).
[0288] A number of PARP inhibitors are known in the art. PARP inhibitors include, but are not limited to, iniparib, talazoparib, olaparib, rucaparib, veliparib, CEP- 9722 (a prodrug of CEP-8983 (11 -methoxy-4,5,6,7-tetrahydro-1 H- cyclopenta[a]pyrrolo[3,4-c]carbazole-1 ,3(2H)-dione), MK 4827 ((S)-2-(4-(piperidin-3- yl)phenyl)-2H-indazole-7-carboxamide), and BGB-290. Other PARP inhibitors are described below.
[0289] United States Patent No. 9,073,893 to Papeo et al. discloses 3-oxo-2,3- dihydro-1 H-indazole-4-carboxamide derivatives as PARP inhibitors.
[0290] United States Patent No. 9,062,061 by Honda et al. discloses a PARP inhibitor of Formula (PA-I):

(PA-I), wherein:
(1 ) R1 represents a halogen atom, a lower alkyl group, a hydroxy group, a lower alkoxy group, an amino group, a nitro group or a cyano group;
(2) R2 and R3 may be the same or different and each represent a hydrogen atom, a halogen atom or a lower alkyl group;
(3) R4 and R5 may be the same or different and each represent a hydrogen atom, a deuterium atom or a lower alkyl group, or R4 and R5 may form an oxo group; Ra and
Rb may be the same or different and each represent a hydrogen atom, a lower alkyl group optionally having a substituent or an aryl group optionally having a substituent; ; Ra and Rb may bind to each other to form a nitrogen-containing heterocyclic ring which may be substituted by one or plural Rc;
(4) Rc represents a lower alkyl group optionally having a substituent, a lower cycloalkyl group optionally having a substituent, an aryl group optionally having a substituent, a heterocyclic group optionally having a substituent, a hydroxy group, a lower alkoxy group optionally having a substituent, a lower alkylcarbonyl group optionally having a substituent, a lower cycloalkylcarbonyl group optionally having a substituent, a lower alkylaminocarbonyl group optionally having a substituent, a lower cycloalkylaminocarbonyl group optionally having a substituent, a lower alkoxycarbonyl group optionally having a substituent, an amino group, a lower alkylamino group or a carboxyl group;
(5) ring A represents a benzene ring or an unsaturated heteromonocyclic ring; and
(6) m represents 0, 1 or 2.
[0291] United States Patent No. 9,062,043 to Chua et al. discloses fused tricyclic PARP inhibitors, including a compound of Formula (PA-II):

(P-ll).
[0292] United States Patent No. 9,018,201 to Chu et al discloses dihydropyridophthalazinone inhibitors of PARP.
[0293] United States Patent No. 8,993,594 to Papeo et al. discloses substituted isoquinolin-1 (2H)-one derivatives as inhibitors of PARP.
[0294] United States Patent No. 8,980,902 to Brown et al. discloses substituted benzamide PARP inhibitors.
[0295] United States Patent No. 8,946,221 to Mevellec et al. discloses phthalazine derivatives as PARP inhibitors.
[0296] United States Patent No. 8,889,866 to Angibaud et al. discloses tetrahydrophenanthridinones and tetrahydrocyclopentaquinolinones as PARP inhibitors.
[0297] United States Patent No. 8,883,787 to Xu et al. discloses diazabenzo[de]anthracen-3-one derivatives as PARP inhibitors.
[0298] United States Patent No. 8,877,944 to Papeo et al. discloses substituted 3-oxo-2,3-dihydro-1 H-isoindole-4-carboxamide derivatives as PARP inhibitors.
[0299] United States Patent No. 8,778,966 to Vialard et al. discloses substituted quinolinone derivatives as PARP inhibitors.
[0300] United States Patent No. 8,697,736 to Penning et al. discloses 1 H- benzimidazole-4-carboxamides as PARP inhibitors.
[0301] United States Patent No. 8,669,249 to Brown et al. discloses PARP inhibitors including: 2-methyl-6-((4-phenylpiperidin-1 -yl)methyl)-2H-benzo[b][1 ,4]oxazin- 3(4H)-one; 2-methyl-6-((4-phenylpiperazin-1 -yl)methyl)-2H-benzo[b][1 ,4]oxazin-3(4H)- one; and 6-((4-(4-fluorophenyl)-5,6-dihydropyridin-1 (2H)-yl)methyl)-2-methyl-2H- benzo[b][1 ,4]oxazin-3(4H)-one, as well as additional compounds.
[0302] United States Patent No. 8,663,884 to Kennis et al. discloses quinazolinedione derivatives as PARP inhibitors.
[0303] United States Patent No. 8,623,872 to Guillemont et al. discloses quinazolinone derivatives as PARP inhibitors.
[0304] United States Patent No. 8,546,368 to Penning et al. discloses pyrazoquinolones as PARP inhibitors, including 7,9-dimethyl-1 ,2,3,4,6,7-hexahydro-5H- pyrazolo[3,4-h]-1 ,6-naphthyridin-5-one.
[0305] United States Patent No. 8,541 ,417 to Brown et al. discloses PARP inhibitors, including: 3-(hydroxymethyl)pyrido[2,3-e]pyrrolo[1 ,2-c]pyrimidin-6(5H)-one; N- ethyl-4-(4-((6-oxo-5,6-dihydropyrido[2,3-e]pyrrolo[1 ,2-c]pyrimidin-3-yl)methyl)piperazin- 1-yl)benzamide; and N-methyl-4-(4-((6-oxo-5,6-dihydropyrido[2,3-e]pyrrolo[1 ,2- c]pyrimidin-3-yl)methyl)piperazin-1-yl)benzamide, as well as additional compounds.
[0306] United States Patent No. 8,541 ,403 to Chu et al. discloses dihydropyridophthalazinone derivatives as PARP inhibitors.
[0307] United States Patent No. 8,513,433 to Panicker et al. discloses inhibitors of PARP, including benzyl 2-(4-carbamoyl-1 H-benzo[d]imidazol-2-yl)indoline-1- carboxylate; 2-(indolin-2-yl)-1 H-benzo[d]imidazole-4-carboxamide; tert-butyl 2-(4- carbamoyl-1 H-benzo[d]imidazol-2-yl)-3,4-dihydroquinoline-1 (2H)-carboxylate; and 2- (1 ,2,3,4-tetrahydroquinolin-2-yl)-1 H-benzo[d]imidazole-4-carboxamide, as well as additional compounds.
[0308] United States Patent No. 8,470,825 to Xu et al. discloses substituted diazabenzo[de]anthracen-3-one compounds as PARP inhibitors.
[0309] United States Patent No. 8,420,650 to Wang et al. discloses dihydropyridophthalazinone inhibitors of PARP.
[0310] United States Patent No. 8,362,030 to Ingenito et al. discloses tricyclic PARP inhibitors, including: N-methyl[4-(6-oxo-3,4,5,6-tetrahydro-2H-azepino[5,4,3- cd]indazol-2-yl)phenyl]methanaminium trifluoroacetate; N, N-dimethyl[4-(6-oxo-3, 4,5,6- tetrahydro-2H-azepino[5,4,3-cd]indazol-2-yl)phenyl]methanaminium trifluoroacetate; and N2,N2-dimethyl-N-[4-(1 -oxo-1 ,2,3,4-tetrahydroazepino[3,4,5-hi]indolizin-5- yl)phenyl]glycinamide, as well as additional compounds.
[0311] United States Patent No. 8,354,413 to Jones et al. discloses quinoline- one and 4-oxodihydrocinnoline derivatives as PARP inhibitors, including: 1-[3-(8-aza-1- azoniaspiro[4.5]dec-8-ylcarbonyl)-4-fluorobenzyl]-4-oxo-1 ,4-dihydroquinolinium bis(trifluoroacetate); 1 -[4-fluoro-3-({4-[2-(4-fluorobenzyl)prolyl]piperazin-1 - yl}carbonyl)benzyl]quinolin-4(1 H)-one; and 1-[3-(8-aza-1-azoniaspiro[4.5]dec-8- ylcarbonyl)-4-fluorobenzyl]-4-oxo-1 ,4-dihydrocinnolin-1-ium bis(trifluoroacetate), as well as additional compounds.
[0312] United States Patent No. 8,268,827 to Branca et al. discloses pyridazinone derivatives as PARP inhibitors, including: 6-{4-fluoro-3-[(3-oxo-4- phenylpiperazin-1-yl)carbonyl]benzyl}-4,5-dimethyl-3-oxo-2,3-dihydropyridazin-1-ium trifluoroacetate; 6-{3-[(4-cyclohexyl-3-oxopiperazin-1-yl)carbonyl]-4-fluorobenzyl}-4,5- dimethyl-3-oxo-2,3-dihydropyridazin-1 -ium trifluoroacetate; 6-{3-[(4-cyclopentyl-3- oxopiperazin-1 -yl)carbonyl]-4-fluorobenzyl}-4,5-dimethylpyridazin-3(2H)-one; and 6-{4-
fluoro-3-[(3-oxo-4-phenylpiperazin-1 -yl)carbonyl]benzyl}-4,5-dimethylpyridazin-3(2H)- one hydrochloride, as well as additional compounds.
[0313] United States Patent No. 8,217,070 to Zhu et al. discloses 2-substituted- 1 H-benzimidazole-4-carboxamides as PARP inhibitors, including: 2-(1- aminocyclopropyl)-1 H-benzimidazole-4-carboxamide; 2-[1 -(isopropylamino)cyclopropyl]- 1 H-benzimidazole-4-carboxamide; 2-[1 -(cyclobutylamino)cyclopropyl]-1 H- benzimidazole-4-carboxamide; and 2-{1 -[(3,5-dimethylbenzyl)amino]cyclopropyl}-1 H- benzimidazole-4-carboxamide, as well as additional compounds.
[0314] United States Patent No. 8,188,103 to Van der Aa et al. discloses substituted 2-alkyl quinazolinone derivatives as PARP inhibitors.
[0315] United States Patent No. 8,173,682 to Weintraub et al. discloses 2,3,5- substituted pyridone derivatives as PARP inhibitors, including: 5-(5-ethyl-2-methyl-6- oxo-1 ,6-dihydro-pyridin-3-yl)-thiophene-2-sulfonic acid [3-(3-hydroxy-pyrrolidin-1-yl)- propyl]-amide hydrochloride; and 5-(5-ethyl-2-methyl-6-oxo-1 ,6-dihydropyridin-3- yl)thiophene-2-sulfonic acid [2-(1 -methylpyrrolidin-2-yl)ethyl]amide hydrochloride, as well as additional compounds.
[0316] United States Patent No. 8,129,382 to Kalish et al. discloses PARP inhibitors of Formula (PA-III)

(PA-III), wherein:
(1 ) R1 is H, halogen, alkoxy, or lower alkyl;
(2) R2 is H, halogen, alkoxy, or lower alkyl;
(3) R3 is independently H, amino, hydroxy, --N--N, halogen-substituted amino, -- O-alkyl, --O-aryl, or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl,
heterocycloalkyl, aryl, heteroaryl, --COR8, where R8 is H, --OH an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or --OR6 or -- NR6R7 where R6 and R7 are each independently hydrogen or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl;
(4) R4 is independently H, amino, hydroxy, --N--N, --CO--N--N, halogen- substituted amino, - O-alkyl, --O-aryl, or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, --COR8, where R8 is H, --OH an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or -- OR6 or --NR6R7 where R6 and R7 are each independently hydrogen or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl; and
(5) R5 is independently H, amino, hydroxy, --N--N, --CO--N--N, halogen- substituted amino, --O-alkyl, --O-aryl, or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, --COR8, where R8 is H, --OH an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or -- OR6 or --NR6R7 where R6 and R7 are each independently hydrogen or an optionally substituted alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl.
[0317] United States Patent No. 8,088,760 to Chu et al. discloses benzoxazole carboxamide inhibitors of PARP, including: 2-(4- ((methylamino)methyl)phenyl)benzo[d]oxazole-4-carboxamide; 2-(2-methylpyrrolidin-2- yl)benzo[d]oxazole-4-carboxamide; 2-(4-((methylamino)methyl)phenyl)benzo[d]oxazole- 7-carboxamide; 2-(2-methylpyrrolidin-2-yl)benzo[d]oxazole-7-carboxamide; and 2- (pyrrolidin-2-yl)benzo[d]oxazole-4-carboxamide, as well as additional compounds.
[0318] United States Patent No. 8,071 ,623 to Jones et al. discloses amide- substituted indazoles as PARP inhibitors, including: 2-(4-piperidin-3-ylphenyl)-2H- indazole-7-carboxamide; 2-{4-[(3R)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide; 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide; 5-fluoro-2-(4-piperidin-3- ylphenyl)-2H-indazole-7-carboxamide; and 5-fluoro-2-{4-[(3S)-piperidin-3-yl]phenyl}-2H- indazole-7-carboxamide, as well as additional compounds.
[0319] United States Patent No. 8,058,275 to Xu et al. discloses diazabenzo[de]anthracen-3-one compounds as PARP inhibitors.
[0320] United States Patent No. 8,012,976 to Wang et al. discloses dihydropyridophthalazinone compounds as PARP inhibitors, including 5-fluoro-8-(4- fluorophenyl)-9-(1 -methyl-1 H-1 ,2,4-triazol-5-yl)-8,9-dihydro-2H-pyrido[4,3,2- de]phthalazin-3(7H)-one.
[0321] United States Patent No. 8,008,491 to Jiang et al. discloses substituted aza-indole derivatives as PARP inhibitors, including: 1 -phenyl-2-(piperazin-1 -yl)-1 ,3- dihydropyrrolo[2,3-b]pyridine-3-carboxaldehyde, 1 -phenyl-2-(piperazin-1 -y l)-1 H- pyrrolo[2,3-c]pyridine-3-carboxaldehyde, 2-[1 ,4]diazepan-1 -y 1-1 -phenyl-1 H-pyrrolo[2,3- b]pyridine-3-carbaldehyde trifluoroacetic acid salt, and 2-piperazin-1 -yl-1 -pyridin-3-yl- 1 H-pyrrolo[2,3-b]pyridine-3-carbaldehyde bis-trifluoroacetic acid salt, as well as additional compounds.
[0322] United States Patent No. 7,999,117 to Giranda et al. discloses 1 H- benzimidazole-4-carboxamides as PARP inhibitors, including: 6-fluoro-2-[4-((S)-2- hydroxymethylpyrrolidin-1-ylmethyl)phenyl]-1 H-benzimidazole-4-carboxamide; 6-fluoro- 2-[4-(2-trifluoromethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-benzimidazole-4-carboxamide; 6- fluoro-2-[4-((R)-2-hydroxymethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-benzimidazole-4- carboxamide; 2-[4-((S)-2-hydroxymethylpyrrolidin-1 -ylmethyl)phenyl]-1 H-benzimidazole- 4-carboxamide; and 2-[4-(2-trifluoromethylpyrrolidin-1 -ylmethyl)phenyl]-1 H- benzimidazole-4-carboxamide, as well as additional compounds.
[0323] United States Patent No. 7,994,182 to Sumegi et al. discloses quinazolinone derivatives as PARP inhibitors of Formula (PA-IV):

(PA-IV), wherein:
(1 ) R1 is hydrogen or a moiety of Subformula (PA-IV(a)):

(2) k is 1 , 2, 3, or 4;
(3) n is 0 or 1 ;
(4) Q is an oxyl group or hydrogen;
(5) Ra and Rb are independently hydrogen or C1-C6 alkyl;
(6) Rb and Rd are independently C1-C6 alkyl;
(7) the broken line in the six-membered ring is an optional valence bond (the bond is either a single or a double bond);
(8) R2 is either:
(8a) if R1 is other than hydrogen, hydrogen or C1-C6 alkyl;
(8b) if R1 is hydrogen, a group of Subformula (PA-IV(b)), Subformula (PA-IV(c)), or Subformula (PA-IV(d)):


(9) in Subformula (PA-IV(b)), k, n, Ra, Rb, Rc, Rd, and the broken line are as defined above in (2), (3), (5), (6), and (7);
(10) in Subformula (PA-IV(c)), k is 1 , 2, or 3, and R3 and R4 are independently C1-C6 alkyl;
(11 ) or together with the attached nitrogen form a group of Subformula (P-IV(e)), wherein p is 0 or 1 and Rab , Rd', Rc', and Rd' are independently hydrogen or C1-C6 alkyl;

(PA-IV(e)); and
(12) in Subformula (P-IV(d), n, Ra, Rb, Rc, Rd, and the broken line are as defined above in (3), (5), (6), and (7).
[0324] United States Patent No. 7,834,015 to Jones et al. discloses pyrrolo[1 ,2- a] pyrazin-1 (2H)-one and pyrrolo[1 ,2-d][1 ,2,4]triazin-1 (2H)-one derivatives as PARP inhibitors.
[0325] United States Patent No. 7,825,129 to Pellicciari et al. discloses thieno[2,3-c]quinolones as PARP inhibitors, including compounds of Formula (PA-V):
 wherein:
wherein:
(1 ) Y is selected from sulfur, nitrogen, and oxygen;
(2) R1, R2, R3, R4, R5 and R6 are the same or different, and each represent hydrogen, hydroxy, OR7, COOR7, carboxy, amino, NHR7 or halogen, or R5 and R6 taken together form a fused non-aromatic 5- or 6-membered carbocylic ring; and
(3) R7 is C1-C6 alkyl, C2-C6 alkenyl or C3-C7 cycloalkyl optionally substituted with one or more group selected from hydroxyl, C1-C4 alkoxy, carboxy, C1-C6 alkoxycarbonyl, amino, C1-C6 mono-alkylamino, C1-C6 di-alkylamino and halogen.
[0326] United States Patent No. 7,820,668 to Xu et al. discloses diazabenzo[de]anthracen-3-one compounds as PARP inhibitors.
[0327] United States Patent No. 7,732,491 to Sherman et al. discloses 4-iodo-3- nitrobenzamide as a PARP inhibitor.
[0328] United States Patent No. 7,728,026 to Zhu et al. discloses 2-substituted
1 H-benzimidazole-4-carboxamides as PARP inhibitors, including 2-(1 -amino-1 - methylethyl)-1 H-benzimidazole-4-carboxamide; 2-[1-methyl-1 -(propylamino)ethyl]-1 H- benzim idazole-4-carboxam ide; 2-[1 -(butylam ino)-1 -methylethyl]-1 H-benzim idazole-4- carboxamide; and 2-{1 -methyl-1-[(2-phenylethyl)amino]ethyl}-1 H-benzim idazole-4- carboxamide, as well as additional compounds.
[0329] United States Patent No. 7,595,406 to Penning et al. discloses substituted 1 H-benzimidazole-4-carboxamides as PARP inhibitors, including 2-{4-[1 - (cyclohexylmethylamino)ethyl]phenyl}-1 H-benzimidazole-4-carboxamide; 2-[4-(1 - cyclobutylam inoethyl)phenyl]-1 H-benzim idazole-4-carboxam ide; 2-[3-(2- cyclopropylaminoethyl)phenyl]-1 H-benzim idazole-4-carboxam ide; and 2-(4- cyclopropylaminomethylphenyl)-1 H-benzim idazole-4-carboxam ide, as well as additional compounds.
[0330] United States Patent No. 7,550,603 to Zhu et al. discloses 1 H- benzimidazole-4-carboxamides substituted with a quaternary carbon at the 2-position as PARP inhibitors, including 2-(2-methylpyrrolidin-2-yl)-1 H-benzim idazole-4-carboxam ide; 2-[(2R)-2-methylpyrrolidin-2-yl]-1 H-benzim idazole-4-carboxam ide; 2-[(2S)-2- methylpyrrolidin-2-yl]-1 H-benzim idazole-4-carboxam ide; 2-(1 ,2-dimethylpyrrolidin-2-yl)- 1 H-benzim idazole-4-carboxam ide; 2-(1 -ethyl-2-methylpyrrolidin-2-yl)-1 H-benzim idazole- 4-carboxamide; and 2-(2-methyl-1 -propylpyrrolidin-2-yl)-1 H-benzim idazole-4- carboxamide, as well as additional compounds.
[0331] United States Patent No. 7,405,300 to Jiang et al. discloses substituted indoles as PARP inhibitors, including 2-(piperazin-1 -yl)-1 -(3-nitrophenyl)-1 H-indole-3- carboxaldehyde; 2-(piperazin-1 -yl)-1 -(4-methoxyphenyl)-1 H-indole-3-carboxaldehyde; 2-(piperazin-1 -yl)-1 -(4-tert-butylphenyl)-1 H-indole-3-carboxaldehyde; 2-(piperazin-1 -yl)- 1 -(4-bromophenyl)-1 H-indole-3-carboxaldehyde; and 2-(piperazin-1 -yl)-1 -(4- chlorophenyl)-1 H-indole-3-carboxaldehyde, as well as additional compounds.
[0332] United States Patent No. 7,087,637 to Grandel et al. discloses indole derivatives as PARP inhibitors, including: 2-(4(4-/?-propyl-piperazin-1 -yl)-phenyl)-1 H- indol-4-carboxamide; 2-(4-piperazin-1-yl-phenyl)-1 H-indol-4-carboxamide; 2-(4(4- isopropyl-piperazin-1 -yl)-phenyl)-1 H-indol-4-carboxamide; 2-(4(4-benzyl-piperazin-1 -yl)- phenyl)-1 H-indol-4-carboxamide; 2-(4(4-/?-butyl-piperazin-1 -yl)-phenyl)-1 H-indol-4- carboxamide; and 2-(4(4-ethyl-piperazin-1 -yl)-phenyl)-1 H-indol-4-carboxamide, as well as additional compounds.
[0333] United States Patent No. 7,041 ,675 to Lubisch et al. discloses substituted pyridine carboxamides as PARP inhibitors, including 2-phenylimidazo[1 ,2-a]pyridine-8- carboxamide; 2-(4-nitrophenyl)imidazo[1 ,2-a]pyridine-8-carboxamide; 2-(4-
aminophenyl)imidazo[1 ,2-a]pyridine-8-carboxamide; 2-(2-benzothienyl)imidazo[1 ,2- a]pyridine-8-carboxamide; 2-(4-bromophenyl)-imidazo[1 ,2-a]pyridine-8-carboxamide; and 2-(4-imidazol-1-ylphenyl)imidazo[1 ,2-a]pyridine-8-carboxamide, as well as additional compounds.
[0334] United States Patent No. 6,924,284 to Beaton et al. discloses substituted bicyclic aryl PARP inhibitors, including: N-[3-(4-oxo-3,4-dihydro-phthalazin-1 -ylamino)- propyl]-3-[3-(1 H-pyrrol-2-yl)-[1 ,2,4]oxadiaol-5-yl]propionamide; N-[3-(4-oxo-3,4-dihydro- phthalazin-1 -ylamino)-propyl]-3-(3-thiophen-3-yl-[1 ,2,4]oxadiazol-5-yl)propionamide; 3- (3-furan-2-yl-[1 ,2,4]oxadiazol-5-yl)-N-[3-(4-oxo-3,4-dihydro-phthalazin-1-ylamino)- propyl]-propionamide; and N-[3-(4-oxo-3,4-dihydro-phthalazin-1 -ylamino)-propyl]-3-(3- thiophen-2-yl-[1 ,2,4]oxadiazol-5-yl)-propionamide, as well as additional compounds.
[0335] United States Patent No. 6,635,642 to Jackson et al. discloses phthalazinone derivatives as PARP inhibitors, including 4-(3-nitro-4-(piperidin-1 - yl)phenyl-phthalazin-1 (2H)-one; 4-(4-(dimethylamino)-3-nitrophenyl)-phthalazin-1 (2H)- one; 4-(3-amino-4-(dimethylamino)phenyl)-phthalazin-1 (2H)-one; 4-(4-phenylpiperazin- 1 -yl)-phthalazin-1 (2H)-one; and 4-(4-(4-chlorophenyl)-piperazin-1 -yl)-phthalazin-1 (2H)- one, as well as additional compounds.
[0336] United States Patent No. 6,448,271 to Lubisch et al. discloses substituted benzimidazoles as PARP inhibitors, including 2-(piperidin-4-yl)benzimidazole-4- carboxamide dihydrochloride; 2-(N-acetylpiperidin-4-yl)benzimidazole-4-carboxamide; 2-(N-propylpiperidin-4-yl)benzimidazole-4-carboxamide; 2-piperidin-3-ylbenzimidazole-
4-carboxamide dihydrochloride; and 2-(N-(O-f-butoxycarbonyl)piperidin-3- yl)benzimidazole-4-carboxamide; 2-(N-benzylpiperidin-3-yl)benzimidazole-4- carboxamide, as well as additional compounds.
[0337] United States Patent No. 6,426,415 to Jackson et al. discloses alkoxy- substituted PARP inhibitors, including 1 -(benzyloxy)-5-methylphthalazine; l -(methoxy)-
5-methyl-phthalazine; 1 -(ethoxy)-5-methylphthalazine; 1 -(propoxy)-5-methylphthalazine; 1 -(butoxy)-5-methyl-phthalazine; 1 -(methoxy)-5-hydroxyphthalazine; 1 -(ethoxy)-5- hydroxyphthalazine; 1 -(propoxyoxy)-5-hydroxy-phthalazine; and 1 -(butoxy)-5- hydroxyphthalazine, as well as additional compounds.
[0338] United States Patent No. 6,395,749 to Li et al. discloses substituted carboxamides as PARP inhibitors, including 5-carbamoylquinoline-4-carboxylic acid.
[0339] United States Patent No. 6,387,902 to Zhang et al. discloses substituted phenazines as PARP inhibitors, including compounds of Formula (PA-VI):

(PA-VI) wherein:
(1 ) R1-R9 and Z are independently hydrogen, hydroxy, halo, haloalkyl, thiocarbonyl, cyano, nitro, amino, imino, alkylamino, aminoalkyl, sulfhydryl, thioalkyl, alkylthio, sulfonyl, alkylsulfonyl, C1-C9 straight or branched chain alkyl, C2-C9 straight or branched chain alkenyl, C2-C9 straight or branched chain alkynyl, C1-C6 straight or branched chain alkoxy, C2-C6 straight or branched chain alkenoxy, C2-C6 straight or branched chain alkynoxy, aryl, carbocycle, heterocycle, aralkyl, alkylaryl, alkylaryloxy, aryloxy, aralkyloxy, aralkylsulfonyl, aralkylamino, arylamino, arylazo, arylthio, or aralkylthio; or
(2) Z is a moiety of Subformula (PA-VI(a))
(PA-VI(a), wherein U is C or N; R7 and R8 are as defined in (1 ); and X and Y are independently aryl, carbocycle, or heterocycle.
[0340] United States Patent No. 6,380,211 to Jackson et al. discloses alkoxy- substituted PARP inhibitors, including 1 -(methoxy)-5-methylisoquinoline, 1 -(ethoxy)-5-
methyl-isoquinoline, 1 -(propoxy)-5-methylisoquinoline, 1 -(butoxy)-5-methylisoquinoline, 1 -(ethoxy)-5-hydroxy-isoquinoline, 1 -(propoxy)-5-hydroxyisoquinoline, 1 -(butoxy)-5- hydroxyisoquinoline, 1-(benzyloxy)-5-methylphthalazine and 1 -(benzyloxy)-5- methylisoquinoline, as well as additional compounds.
[0341] United States Patent No. 6,358,975 to Eliasson et al. discloses PARP inhibitors, including 6(5H)-phenanthridinone, 2-nitro-6(5H)-phenanthridinone, 4- hydroxyquinazoline, 2-methyl-4(3H)-quinazoline, 2-mercapto-4(3H)-quinazoline, benzoyleneurea, 6-amino-1 ,2-benzopyrone, trp-P-1 (3-amino-1 ,4-dimethyl-5H- pyrido[4,3-b]indole), juglone, luminol, 1 (2H)-phthalazinone, phthalhydrazide, and chlorothenoxazin.
[0342] United States Patent No. 6,235,748 to Li et al. discloses oxo-substituted compounds containing at least one ring nitrogen as PARP inhibitors.
[0343] United States Patent No. 6,201 ,020 to Zhang et al. discloses ortho- diphenol compounds as PARP inhibitors, including compounds of Formula (PA-VII):

(PA-VII), wherein:
(1 ) A is O or S;
(2) R is C1-C10 straight or branched chain alkyl, C2-C10 straight or branched chain alkenyl, C2-C10 straight or branched chain alkynyl, aryl, heteroaryl, carbocycle, or heterocycle;
(3) D is a bond, or a C1-C3 straight or branched chain alkyl, C2-C3 straight or branched chain alkenyl, C2-C3 straight or branched chain alkynyl, wherein any of the carbon atoms of said alkyl, alkenyl, or alkynyl of D are optionally replaced with oxygen, nitrogen, or sulfur; and
(4) X is aryl, heteroaryl, carbocycle, or heterocycle.
[0344] United States Patent No. 5,756,510 to Griffin et al. discloses benzamide analogs that are PARP inhibitors, including: 3-benzyloxybenzamide; 3-(4- methoxybenzyloxy)benzamide; 3-(4-nitrobenzyloxy)benzamide; 3-(4- azidobenzyloxy)benzamide; 3-(4-bromobenzyloxy)benzamide; 3-(4- fluorobenzyloxy)benzamide; 3-(4-aminobenzyloxy)benzamide; 3-(3- nitrobenzyloxy)benzamide; 3-(3,4-methylenedioxyphenylmethyloxy)benzamide; 3- (piperonyloxy)benzamide; 3-(N-acetyl-4-aminobenzyloxy)benzamide; and 3-(4- trifluoromethylbenzyloxy)benzamide; and 3-(4-cyanobenzyloxy)benzamide, as well as additional compounds.
[0345] United States Patent Application Publication No. 2015/0175617 by Zhou et al. discloses fused tetra or penta-cyclic dihydrodiazepinocarbazolones as PARP inhibitors, including: 2,3,5, 10-tetrahydro-[1 ,2]diazepino[3,4:5,6-def]carbazol-6(1 H)-one; 5,6,7,8-tetrahydro-4H-4,9, 10-triazaindeno[2, 1 ,7-kla]heptalen-11 (10H)-one; 2-methyl- 2,3,5, 10-tetrahydro-[1 ,2]diazepino[3,4:5,6-def]carbazol-6(1 H)-one; and 3,3-dimethyl- 2,3,5, 10-tetrahydro-[1 ,2]diazepino[3, 4:5, 6-def]carbazol-6(1 H)-one, as well as additional compounds.
[0346] United States Patent Application Publication No. 2015/0152118 by Jana et al. discloses tetrahydroquinazolinone derivatives as PARP inhibitors, including: 2'-(3- (4-(4-fluorophenyl)piperazin-1-yl)propyl)-6',7'-dihydro-3'H-spiro[cyclopropane-1 ,8'- quinazolin]-4'(5'H)-one; 2'-(3-(4-(4-chlorophenyl)piperazin-1-yl)propyl)-6',7'-dihydro-3'H- spiro[cyclopropane-1 ,8'-quinazolin]-4'(5'H)-one; 2'-(3-(4-phenyl-5,6-dihydropyridin- 1 (2H)-yl)propyl)-6',7'-dihydro-3'H-spiro[cyclopropane-1 ,8'-quinazolin]-4'(5'H)-one; and 2'-(3-(3-(4-fluorophenyl)-3,8-diazabicyclo[3.2.1]octan-8-yl)propyl)-4a',5',6',7'-tetrahydro- 3'H-spiro[cyclopropane-1 ,8'-quinazolin]-4'(8a'H)-one, as well as additional compounds.
[0347] United States Patent Application Publication No. 2015/0031652 by Gangloff et al. discloses substituted 1 ,2,3,4-tetrahydropyrido[2,3-b]pyrazines as PARP inhibitors, including (S)-3-((4-(4-chlorophenyl)piperazin-1 -yl)methyl)-6a, 7,8,9- tetrahydropyrido[3,2-e]pyrrolo[1 ,2-a]pyrazin-6(5H)-one; (S)-3-((4-(4-chlorophenyl)-5,6- dihydropyridin-1(2H)-yl)methyl)-6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1 ,2-a]pyrazin-
6(5H)-one; (S)-3-((4-(4-chlorophenyl)piperidin-1-yl)methyl)-6a, 7,8,9- tetrahydropyrido[3,2-e]pyrrolo[1 ,2-a]pyrazin-6(5H)-one; and (S)-4-(4-((6-oxo- 5,6,6a,7,8,9-hexahydropyrido[3,2-e]pyrrolo[1 ,2-a]pyrazin-3-yl)methyl)piperazin-1- yl)benzonitrile, as well as additional compounds.
[0348] United States Patent Application Publication No. 2015/0025071 by Buchstaller et al. discloses tetrahydroquinazolinone derivatives as PARP inhibitors, including: 2-[4-(4-methoxy-phenyl)-piperazin-1-yl]-5,6,7,8-tetrahydro-3H-quinazolin-4- one; 2-[4-(3-fluorophenyl)piperazin-1 -yl]-5,6,7,8-tetrahydro-3H-quinazolin-4-one; 2-[4-(4- fluorophenyl)piperazin-1 -yl]-5,6,7,8-tetrahydro-3H-quinazolin-4-one; and 2-[4-(3- methoxyphenyl)piperazin-1-yl]-5,6,7,8-tetrahydro-3H-quinazolin-4-one, as well as additional compounds.
[0349] United States Patent Application Publication No. 2015/0018356 by Zhou et al. discloses fused tetra- or pentacyclic pyridophthalazinones as PARP inhibitors.
[0350] United States Patent Application Publication No. 2014/0336192 to Papeo et al. discloses substituted 3-phenyl-isoquinolin-1 (2H)-one derivatives as PARP inhibitors, including: 4-(2-amino-ethoxy)-3-(4-bromo-phenyl)-7-fluoro-2H-isoquinolin-1 - one; 4-(2-amino-ethoxy)-7-fluoro-3-(3-trifluoromethyl-phenyl)-2H-isoquinolin-1-one; 4-(2- amino-ethoxy)-7-fluoro-3-(4-morpholin-4-yl-phenyl)-2H-isoquinolin-1-one; 4-(2-amino- ethoxy)-3-(3-bromo-4-morpholin-4-yl-phenyl)-7-fluoro-2H-isoquinolin-1-one; 4-(2-amino- ethoxy)-3-(3-bromo-phenyl)-7-fluoro-2H-isoquinolin-1 -one; and 4-[4-(2-amino-ethoxy)-7- fluoro-1 -oxo-1 ,2-dihydro-isoquinolin-3-yl]-benzonitrile, as well as additional compounds.
[0351] United States Patent Application Publication No. 2014/0235675 by Papeo et al. discloses 3-oxo-2,3-dihydro-1 H-indazole-4-carboxamide derivatives as PARP inhibitors, including: 3-oxo-2-(piperidin-4-yl)-2,3-dihydro-1 H-indazole-4-carboxamide; 2- (1 -cyclopentylpiperidin-4-yl)-3-oxo-2,3-dihydro-1 H-indazole-4-carboxamide; 2-(1 - cyclohexylpiperidin-4-yl)-3-oxo-2,3-dihydro-1 H-indazole-4-carboxamide; and 2-[1 -(4,4- difluorocyclohexyl)piperidin-4-yl]-3-oxo-2,3-dihydro-1 H-indazole-4-carboxamide, as well as additional compounds.
[0352] United States Patent Application Publication No. 2014/0023642 by Cai et al. discloses 1-(arylmethyl)quinazoline-2,4(1 H,3H)-diones as PARP inhibitors, including: 1 -(3-methoxycarbonylbenzyl)quinazoline-2,4(1 H,3H)-dione; 1 -(3-
carboxybenzyl)quinazoline-2,4(1 H,3H)-dione; 1 -(3-(4-(pyridin-2-yl)piperazine-1 - carbonyl)benzyl)quinazoline-2,4(1 H,3H)-dione; 1 -(3-(4-(pyrimidin-2-yl)piperazine-1 - carbonyl)benzyl)quinazoline-2,4(1 H,3H)-dione; and 1 -(3-(4-cyclohexylpiperazine-1 - carbonyl)benzyl)quinazoline-2,4(1 H,3H)-dione, as well as additional compounds.
[0353] United States Patent Application Publication No. 2013/0225647 by Donawho et al. discloses PARP inhibitors of Formula (PA-VIII):

(PA-VIII), wherein:
(1 ) R1, R2, and R3 are independently selected from the group consisting of hydrogen, alkenyl, alkoxy, alkoxycarbonyl, alkyl, alkynyl, cyano, haloalkoxy, haloalkyl, halogen, hydroxy, hydroxyalkyl, nitro, NRARB, and (NRARB)carbonyl;
(2) A is a nonaromatic 4, 5, 6, 7, or 8-membered ring that contains 1 or 2 nitrogen atoms and, optionally, one sulfur or oxygen atom, wherein the nonaromatic ring is optionally substituted with 1 , 2, or 3 substituents selected from the group consisting of alkenyl, alkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkynyl, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, cyano, haloalkoxy, haloalkyl, halogen, heterocycle, heterocyclylalkyl, heteroaryl, heteroarylalkyl, hydroxy, hydroxyalkyl, nitro, NRCRD, (NRCRD)alkyl, (NRcRD)carbonyl, (NRcRD)carbonylalkyl, and (NRCRD)sulfonyl; and;
(3) RA, RB, RC, and RD are independently selected from the group consisting of hydrogen, alkyl, and alkycarbonyl.
[0354] United States Patent Application Publication No. 2013/0129841 by Ciavolella et al. discloses PARP inhibitors including 2-[1 -(4,4- difluorocyclohexyl)piperidin-4-yl]-3-oxo-2,3-dihydro-1 H-isoindole-4-carboxamide; 2-[1 - (4,4-difluorocyclohexy)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-
carboxamide; 6-fluoro-3-oxo-2-[1 -(4-oxocyclohexy)piperidin-4-yl]-2,3-dihydro-1 H- isoindole-4-carboxamide, and 2-[1 -(4,4-dichlorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo- 2,3-dihydro-1-H-isoindole-4 carboxamide, as well as additional compounds.
[0355] Other PARP inhibitors are known in the art and can be employed in methods and compositions according to the present invention. These additional PARP inhibitors include, but are not limited to: (1 ) derivatives of tetracycline as described in United States Patent No. 8,338,477 to Duncan et al.; (2) 3,4-dihydro-5-methyl-1(2/-/)- isoquinoline, 3-aminobenzamide, 6-aminonicotinamide, and 8-hydroxy-2-methyl-4(3/-/)- quinazolinone, as described in United States Patent No. 8,324,282 by Gerson et al.; (3) 6-(5H)- phenanthridinone and 1 ,5-isoquinolinediol, as described in United States Patent No. 8,324,262 by Yuan et al.; (4) (R)-3-[2-(2-hydroxymethylpyrrolidin-1-yl)ethyl]-5- methyl-2H-isoquinolin-1-one, as described in United States Patent No. 8,309,573 to Fujio et al.; (5) 6-alkenyl-substituted 2-quinolinones, 6-phenylalkyl-substituted quinolinones, 6-alkenyl-substituted 2-quinoxalinones, 6-phenylalkyl-substituted 2- quinoxalinones, substituted 6-cyclohexylalkyl substituted 2-quinolinones, 6- cyclohexylalkyl substituted 2-quinoxalinones, substituted pyridones, quinazolinone derivatives, phthalazine derivatives, quinazolinedione derivatives, and substituted 2- alkyl quinazolinone derivatives, as described in United States Patent No. 8,299,256 to Vialard et al.; (6) 5-bromoisoquinoline, as described in United States Patent No. 8,299,088 to Mateucci et al.; (7) 5-bis-(2-chloroethyl)amino]-1-methyl-2- benzimidazolebutyric acid, 4-iodo-3-nitrobenzamide, 8-fluoro-5-(4- ((methylamino)methyl)phenyl)-3,4-dihydro-2H-azepino[5,4,3-cd]indol-1 (6H)-one phosphoric acid, and N-[3-(3,4-dihydro-4-oxo-1-phthalazinyl)phenyl]-4- morpholinebutanamide methanesulfonate, as described in United States Patent No. 8,227,807 to Gallagher et al.; (8) pyridazinone derivatives, as described in United States Patent No. 8,268,827 to Branca et al.; (9) 4-[3-(4-cyclopropanecarbonyl-piperazine-1- carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one, as described in United States Patent No. 8,247,416 to Menear et al.; (10) tetraaza phenalen-3-one compounds, as described in United States Patent No. 8,236,802 to Xu et al.; (11 ) 2-substituted-1/-/-benzimidazole-4- carboxamides, as described in United States Patent No. 8,217,070 to Zhu et al.; (12) substituted 2-alkyl quinazolinones, as described in United States Patent No. 8,188,103
to Van der Aa et al.; (13) 1/-/-benzimidazole-4-carboxamides, as described in United States Patent No. 8,183,250 to Penning et al.; (14) indenoisoquinolinone analogs, as described in United States Patent No. 8,119,654 to Jagtap et al.; (15) benzoxazole carboxamides, described in United States Patent No. 8,088,760 to Chu et al; (16) diazabenzo[de] anthracen-3-one compounds, described in United States Patent No. 8,058,075 to Xu et al.; (17) dihydropyridophthalazinones, described in United States Patent No. 8,012,976 to Wang et al., (18) substituted azaindoles, described in United States Patent No. 8,008,491 to Jiang et al.; (19) fused tricyclic compounds, described in United States Patent No. 7,956,064 to Chua et al.; (20) substituted 6a, 7,8,9- tetrahydropyrido[3,2-e]pyrrolo[1 ,2-a]pyrazin-6(5/-/)-ones, described in United States Patent No. 7,928,105 to Gangloff et al.; and (21 ) thieno[2,3-c] isoquinolines, described in United States Patent No. 7,825,129, all of which patents are incorporated herein by this reference. Still other PARP inhibitors are known in the art. Additionally, derivatives and analogs of PARP inhibitors described above that have PARP-inhibiting activity that is sufficiently great that they could replace the PARP inhibitors described above in methods or compositions according to the present invention can also be employed in methods and compositions according to the present invention.
[0356] A third category of suitable additional therapeutic agents is lactate dehydrogenase (LDH) inhibitors. Lactate dehydrogenase (LDH) catalyzes the interconversion of pyruvate and lactate with the concomitant interconversion of NADH and NAD+. When pyruvate is converted to lactate, pyruvate is reduced by reduction of a keto moiety in pyruvate to the hydroxyl moiety in lactate. The enzyme converts pyruvate, the final product of glycolysis, to lactate when oxygen is absent or in short supply, and also regenerates NAD+, the oxidized form of NAD, maintaining its concentration so that further glycolysis can occur.
[0357] Cancer cells may rely on increased glycolysis in certain metabolic states, resulting in increased lactate production, in addition to aerobic respiration in the mitochondria, a process known as oxidative phosphorylation (OXPHOS), even under oxygen-sufficient conditions, a phenomenon known as the Warburg effect. This state of fermentative glycolysis is catalyzed by the A form of LDH. This mechanism allows tumor cells to convert the majority of their glucose stores into lactate regardless of
oxygen availability, shifting use of glucose metabolites from simple energy production to the promotion of accelerated cell growth and replication.
[0358] Therefore, interest has arisen in inhibiting the activity of the A form of LDH as a promising target in cancer treatment, in particular, preventing the proliferation of cancer cells by interfering with their energy metabolism. One inhibitor of the activity of the A form of LDH is oxamate. Oxamate is a cytosolic inhibitor of the A form of LDH that significantly decreases adenosine triphosphate (ATP) production in tumor cells as well as increasing production of reactive oxygen species (ROS). ROS include peroxides, superoxide, hydroxyl radical, singlet oxygen, and alpha-oxygen. These reactive oxygen species can drive cancer cell proliferation by activating kinases that drive cell cycle progression growth factors at low concentrations, but can damage DNA through oxidative stress at higher concentrations. In addition, generation of reactive oxygen species induces lipid peroxidation. Generation of reactive oxygen species can cause DNA damage through reaction of the free radicals generated with DNA molecules. This can further impact DNA repair systems and chromatin structure and also interfere with cell cycling and thus block cell replication. Therefore, elesclomol can potentiate the activity of PARP inhibitors, inhibitors of histone deacetylase, and hypomethylating agents; inhibitors of histone deacetylase and hypomethylating agents are epigenetic targets. Histone deacetylase inhibitors include, but are not limited to, parthenolide, abexinostat, allyl mercaptan, apicidin, belinostat, chidamide, 3,3'- diindolylmethane, entinostat, givinostat, martinostat, mocetinostat, panobinostat, pracinostat, resminostat, romidepsin, trichostatin A, and vorinostat. Hypomethylating agents include, but are not limited to, azacitidine and decitabine.
[0359] Other inhibitors of the A form of lactate dehydrogenase are described in E.-Y. Kim, “A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4- (Trifluoromethyl) Benzene, Suppresses Tumor Cell Growth Through Apoptotic Cell Death,” Sci. Rep. 9, 3069 (2019). Inhibitors of the A form of lactate dehydrogenase disclosed in this reference include: 1-(phenylseleno)-4-bromobenzene, 1- (phenylseleno)-4-methylbenzene, 1 -(phenylseleno)-4-methoxybenzene, 1 - (phenylseleno)-4-phenylbenzene, 3-(phenylseleno)tetrahydrothiophene, and 1- (phenylmethylseleno-4-methoxybenzene.
[0360] Still other inhibitors of the A form of lactate dehydrogenase include 3-[[3- (cyclopropylsulfamoyl)-7-(2,4-dimethoxypyrimidin-5-yl)quinolin-4-yl]amino]-5-(3,5- difluorophenoxy)benzoic acid (GSK2837808A), 7-benzyl-2,3-dihydroxy-6-methyl-4- propylnaphthalene-1 -carboxylic acid (FX-11 ), (2R)-5-(2-chlorophenyl)sulfanyl-4-hydroxy- 2-(4-morpholin-4-ylphenyl)-2-thiophen-3-yl-1 ,3-dihydropyridin-6-one ((R)-GNE-140), (LDH-IN-1 ); 3-dehydrotrametanolic acid, nifurtimox, bromoalanine, and glomeratose A.
[0361] Still other inhibitors of the A form of lactate dehydrogenase are disclosed in United States Patent Application Publication No. 2018/1105068 by Inoue et al. These inhibitors include isosafrole and a compound of Formula (L-l):

(L-l), wherein:
(1 ) Ra is hydrogen, halogen, or alkoxy optionally substituted with one or more halogens;
(2) Rto is hydrogen, halogen, alkoxy optionally substituted with one or more halogens, or nitro, or is a group which together with Rc, Rd, or Rg forms a ring structure optionally having substituents;
(3) Rc is a hydrogen atom or carboxyl, or is a group which together with Rb, Re, Rf or Rg forms a ring structure optionally having substituents;
(4) Rd is a hydrogen atom or a moiety -X11-R11, or is a group which together with Rto or Rg forms a ring structure optionally having substituents;
(5) X11 is alkylene, -NHCO-, -CH2— NR12— CO-, or -S-;
(6) R11 is optionally substituted aryl, alkyl optionally substituted with one or more halogens, or hydroxyalkyl;
(7) R12 is hydroxyalkyl;
(8) for Re, R7, and Rg:
(i) all are hydrogen; or
(ii) Re and R7 together are =0, =NR21, or =CR21R22, and Rg is hydrogen, -- X31— R31, 0— X32— R32, — N(— X33— R33)(— X34— R34), or -CR35R36R37;
(9) R21 is hydroxy or is a group which together with Rc or Rg forms a ring structure optionally having substituents;
(10) R22 is nitro or aminocarbonyl in which a hydrogen atom bound to a nitrogen atom may be substituted with alkyl;
(11 ) X31 is a single bond, alkenylene, --CH2 — O-CH2 — CO — NH--, --CH2 — O- CH2— CO— , or -CO—;
(12) R31 is optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, optionally substituted heterocyclic amino, hydroxy, alkoxy optionally substituted with one or more halogens, or hydroxyalkyl, wherein, when X31 is a single bond and R31 is heterocyclic amino optionally having substituents, the single bond is bound to an atom other than the nitrogen atom of the heterocyclic amino;
(13) X32 represents a single bond or -CH2-CO-NH-;
(14) R32 is optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, alkyl optionally substituted with one or more halogen atoms;
(15) X33 and X34 each are a single bond, alkylene, -CH2 — CO — NH, or -SO2-;
(16) R33 and R34 each independently represent a hydrogen atom, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, hydroxyalkyl, carboxyl, alkyl optionally substituted with one or more halogens, or alkynyl group, or represent a group which together with Rb, Rc, and Rd or Re and Reforms a ring structure optionally having substituents, or R33 and R34, together with the nitrogen atom to which they are bound through X33 and X34 which are single bonds, represent optionally substituted heterocyclic amino;
(17) R35, R36, and R37 are each a hydrogen atom or X31 — R31, or represent a group which together with Rb, Rc, and Rd or Re and Reforms an optionally substituted ring structure;
(18) Rh is hydrogen or halogen; and
(19) wherein the number of ring structures formed by binding between Rb and Rc, the ring structure formed by binding between Rb and Rd, the ring structure formed by binding between Rb and Rg, the ring structure formed by binding between Rc and Re or Rf, the ring structure formed by binding between Rc and Rg, the ring structure formed by binding between Rd and Rg, or the ring structure formed by binding between Re or Rf and Rg formed in Formula (LI) is no more than one.
[0362] Other inhibitors of Form A of LDH are known in the art.
[0363] A fourth category of suitable additional therapeutic agents is 2- deoxyglucose and analogs or derivatives thereof. Preferably, the additional therapeutic agent is 2-deoxyglucose (H. Pelicano et al., “Glycolysis Inhibition for Anticancer Treatment,” Oncogene 25: 4633-4646 (2006)).
[0364] A fifth category of suitable additional agents is glutamine metabolism inhibitors.
[0365] Glutamine metabolism inhibitors include compound 968 (5-(3-bromo-4- (dimethylamino)phenyl)-2,2-dimethyl-2,3,5,6-tetrahydrobenzo[a]phenanthridin-4(1 H)- one), BPTES (bis-2-(5-phenylacetamido-1 ,2,4-thiadiazoyl-2-yl)ethyl sulfide), L- asparaginase, and phenylbutyrate.
[0366] Other glutamine metabolism inhibitors are disclosed in United States Patent No. 10,947,598 by Ricci et al., including: (i) inhibitors of glutamine synthase (GS) such as methionine sulfoximine, methionine sulfone, phosphinothricin, tabtoxinin-β- lactam, methionine sulfoximine phosphate, a-methyl methionine sulfoximine, a-ethyl methionine sulfoximine, ethionine sulfoximine, a-methyl ethionine sulfoximine, prothionine sulfoximine, a-methyl prothionine sulfoximine, y-hydroxy phosphinothricin, gamma-methyl phosphinothricin, y-acetoxy phosphinothricin, a-methyl phosphinothricin, a-ethyl phosphinothricin, cyclohexane phosphinothricin, cyclopentane phosphinothricin, tetrahydrofuran phosphinothricin, s-phosphonomethylhomocysteine, s- phosphonomethyl homocysteine sulfoxide, s-phosphonomethyl homocysteine sulfone, 4-(phosphonoacetyl)-L-a-aminobutyrate, threo-4-hydroxy-D-glutamic acid, threo-4- fluoro-D,L-glutamic acid, erythro-4-fluoro-D,L-glutamic acid, 2-amino-4- [(phosphonomethyl)hydroxyphosphinyl)]butanoic acid, alanosine, 2-amino-4-
phosphonobutanoic acid, 2-amino-2-methyl-4-phosphonobutanoic acid, 4-amino-4- phosphonobutanoic acid, 4-amino-4-(hydroxymethylphosphinyl)butanoic acid, 4-amino- 4-methyl-4-phosphonobutanoic acid, 4-amino-4-(hydroxymethylphosphinyl)-4-methyl butanoic acid, 4-amino-4 phosphonobutanamide, 2-amido-4-phosphonobutanoic acid, 2-methoxycarbonyl-4-phosphonobutanoic acid, methyl 4-amino-4-phosphonobutanoate oxetin, and bis-2-(5-phenylacetamido-1 ,3,4-thiadiazol-2-yl)ethyl sulfide; and (ii) inhibitors of glutamine transporters ASCT2 and LAT1 such as L-asparaginase and phenylbutyrate.
[0367] United States Patent No. 10,954,257 to Slusher et al. discloses glutamine antagonists including 6-diazo-5-oxo-L-norleucine and azaserine, as well as prodrugs of glutamine analogs of Formula (G-l):
 wherein:
wherein:
(1 ) X is selected from the group consisting of a bond, -0--, and -(CH2 )n--, wherein n is an integer selected from the group consisting of 1 , 2, 3, 4, 5, 6, 7, and 8;
(2) R1 is selected from the group consisting of hydrogen and a first prodrug- forming moiety capable of forming a salt or an ester;
(3) R2 is selected from the group consisting of hydrogen and a second prodrug- forming moiety capable of forming an amide linkage, a carbamate linkage, a phosphoram idate linkage or a phosphorodiamidate linkage with the nitrogen adjacent to R2;
(4) R2' is selected from the group consisting of hydrogen, optionally substituted C1-C6 alkyl; or R2 and R2' together form a ring structure comprising -C(=O) — G — C(=O)- -, wherein G is selected from the group consisting of C1-C8 alkylene, C1-C8 heteroalkylene, C5-C8 cycloalkylene, C6-C12 arylene, C5-C14 heteroarylene, and bivalent C4-C10 heterocyclyl, each of which can be optionally substituted; or R1 and R2' together
form a 4- to 6-membered heterocylic ring comprising the oxygen atom adjacent to R1 and the nitrogen atom adjacent to R2 ; provided that the compound has at least one prodrug-forming moiety selected from the group consisting of the first and the second prodrug-forming moieties.
[0368] United States Patent No. 10,793,514 to Manning et al. discloses glutamine transport inhibitors that are 2-amino-4-bis(aryloxybenzyl)aminobutanoic acids of Formula (G-ll):

(G-ll), wherein:
(1 ) R1 is phenyl optionally substituted with at least one R3, benzyl optionally substituted with at least one R3, or pyridinyl optionally substituted with at least one R3;
(2) R2 is phenyl optionally substituted with at least one R3, benzyl optionally substituted with at least one R3, or pyridinyl optionally substituted with at least one R3;
(3) R3 is independently hydrogen, methyl, alkyl, methoxy, alkoxy, halogen, or trifluoromethyl; and
(4) n is 0-6.
[0369] These compounds include (S)-2-amino-4-(bis(2-((3- methylbenzyl)oxy)benzyl)amino)butanoic acid.
[0370] Other inhibitors of glutamine metabolism are known in the art.
[0371] A sixth category of suitable additional agents is DNA-damaging agents, in particular, DNA-damaging anti-neoplastic agents.
[0372] DNA-damaging anti-neoplastic agents are disclosed in K. Cheung-Ong et al., “DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology,” Chem. Biol. 20: 648-659 (2013). These agents fall into several categories.
One category is agents that damage DNA directly; these agents employ one of several mechanisms, including direct modification of DNA bases, intercalation between DNA bases, and formation of crosslinks in DNA. Another category is agents that interfere with DNA synthesis. Yet another category is agents that inhibit topoisomerases; some of these agents also have additional activities that can damage DNA or interfere with DNA replication in various other ways.
[0373] For example, nitrogen mustards act by directly alkylating DNA on purine bases, leading to stalled replication fork progression and subsequent cell death via apoptosis. Derivatives of the nitrogen mustards were subsequently developed, including the DNA alkylators cyclophosphamide, chlorambucil, and melphalan, all of which are currently used in clinical therapeutics. Other examples of DNA-alkylating agents used in cancer treatment include nitrosoureas (e.g., carmustine, lomustine, and semustine) and triazenes (e.g., dacarbazine and temozolomide). Natural products which alkylate DNA bases were also discovered, such as mitomycin C and streptozotocin. These compounds and several of the alkylators mentioned above crosslink DNA on opposite strands of the double helix (interstrand cross-links), resulting in a more potent effect against cancer cells compared to monofunctional alkylation. For example, carmustine binds to the N1 atom of guanine on one DNA strand and the N3 atom of cytosine of the opposite strand to form interstrand crosslinks, which block DNA replication and can cause cell death if not repaired.
[0374] Additional agents that damage DNA include the alkylating-like platinum agents that act by forming adducts on DNA. When two platinum adducts form on adjacent bases on the same DNA strand, they form intrastrand crosslinks. These agents include cisplatin, carboplatin, oxaliplatin, and picoplatin, as well as other platinum-containing analogs.
[0375] Antimetabolites typically do not damage DNA molecules directly, but interfere with DNA replication. Examples include the pyrimidine analogs 5-fluorouracil, capecitabine, floxuridine, and gemcitabine, and the purine analogs 6-mercaptopurine, 8- azaguanine, fludarabine, and cladribine. Another class of antimetabolites inhibit enzymes important for DNA synthesis; this class of antimetabolites include antifolates such as methotrexate, aminopterin, pemetrexed, and ralitrexed.
[0376] Topoisomerase inhibitors inhibit replication fork progression in replicating DNA and cause double-strand breaks. These agents include etoposide, camptothecin, and anthracycline antibiotics. The anthracyclines, such as, but not limited to, doxorubicin and daunorubicin, are also able to intercalate into DNA, generate free radicals, bind and alkylate DNA, crosslink DNA, interfere with helicase activity, and induce apoptosis.
[0377] DNA-damaging agents are also described in the following patents or published patent applications: United States Patent No. 9,097,722 to Yu; United States Patent No. 9,096,602 to Everitt et al.; United States Patent No. 8,840,898 to Goldmakher; United States Patent No. 8,735,590 to Adejare et al.; United States Patent No. 8,415,357 to Kawabe et al.; United States Patent No. 8,476,025 to Clifford; United States Patent No. 7,902,165 to Kim; United States Patent No. 7,875,586 to Kovbasnjuk et al.; United States Patent No. 7,652,042 to Kawabe et al.; United States Patent No. 7,465,542 to Chu et al.; United States Patent No. 7,070,968 to Kufe et al.; United States Patent Application Publication No. 2011/0028422 by Aloyz et al.; and United States Patent Application Publication No. 2007/0032502 by Mallams et al.
[0378] Therefore, DNA-damaging anti-neoplastic agents can act by a variety of mechanisms, including modification of DNA bases such as by alkylation, intercalation into the DNA structure, formation of crosslinks in DNA, prevention of unwinding or replication of DNA to induce double-strand breaks, incorporation into DNA in place of normal nucleosides, and other mechanisms. DNA-damaging anti-neoplastic agents include, but are not limited to: cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, tetraplatin, doxorubicin, daunorubicin, methotrexate, 5-fluorouracil, gemcitabine, podophyllotoxin, etoposide, teniposide, cyclophosphamide, chlorambucil, melphalan, carmustine, lomustine, estramustine, semustine, bendamustine, prednamustine, uramustine, chlornaphazine, dacarbazine, altretamine, temozolomide, mitomycin C, streptozotocin, chlorozotocin, capecitabine, floxuridine, 6-mercaptopurine, 8-azaguanine, azathiopurine, 5-ethynyluracil, thioguanine, fludarabine, cytarabine, cladribine, 2-fluoro-arabinosyl-adenine, aminopterin, pemetrexed, ralitrexed, camptothecin, epirubicin, idarubicin, methylnitronitrosoguanidine, topotecan, irinotecan, mechlorethamine, ifosfamide, trofosfamide, busulfan, procarbazine, mitoxantrone,
actinomycin, calicheamicin, Tegafur (R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil), 2', 2'- difluoro-2'-deoxycytidine, bischloroethylsulfide, thiotepa, aziridinylbenzoquinone, BCNU , CCNU , 4-methyl-CCNU, ACNU , rebeccamycin, bleomycin, pepleomycin, ethylmethanesulfonate, methylmethanesulfonate, dimethylnitrosamine, dimethyl sulfate, and N'-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1 ,3- propanediamine dihydrochloride.
[0379] A seventh category of suitable additional agents is agents that inhibit the SWI/SNF complex. The SWI/SNF (BAF) complexes are a diverse family of ATP- dependent chromatin remodelers produced by combinatorial assembly that are mutated in and thought to contribute to 20% of human cancers and a large number of neurologic diseases.
[0380] SWI/SNF inhibitors are compounds that inhibit one or more members of a subfamily of ATP-dependent chromatin remodeling complexes; the activity of these proteins affects nucleosome rearrangement, which, in turn, affects access to the chromatin, allowing genes to be activated or repressed.
[0381] SWI/SNF inhibitors include BD98 (1-isopropyl-3-((4R,5S,8S)-4-methoxy- 2 , 5, 8-trim ethy 1-1 -oxo-7-(4-pyridyl-2-yl)benzyl)-1 ,2, 3, 4, 5, 6, 7, 8, 9, 10- decahydrobenzo[h][1 ,6]diazacyclododecin-13-yl)urea) (E.J. Chory et al., “Chemical Inhibitors of a Selective SWI/SNF Function Synergize with ATR Inhibition in Cancer Cell Killing,” ACS Chem. Biol. 15: 1685-1696 (2020)). The ATR inhibitor described in this reference is 3-amino-6-(4-(methylsulfonyl)phenyl)-N-phenylpyrazine-2-carboxamide.
[0382] Further SWI/SNF inhibitors are disclosed in United States Patent Application Publication No. 2020/0147120 by Iba et al. and United States Patent No. 9,850,543 to Zainab et al.
[0383] An eighth category of additional agents is agents that cause the tumor cells to rely heavily on oxidative phosphorylation (OXPHOS). It had been previously shown that cancer cells exhibit an alteration in their metabolism when compared with non-malignant cells. Normal cells in the presence of oxygen use primarily the mitochondrial tricarboxylic acid (TCA) cycle and oxidative phosphorylation for the production of energy and rely on glycolysis only when their oxygen supply is limited. In contrast, cancer cells frequently utilize glycolysis even in the presence of sufficient
amounts of oxygen. The fact that cancer cells reduce their dependence on oxidative phosphorylation on mitochondrial oxidative phosphorylation and thus are substantially more reliant on glycolysis can potentially provide a range of therapeutic targets.
[0384] The glycolytic pathway includes a total of 10 reactions. The enzymes that catalyze reactions in the glycolytic pathway include: hexokinase; glucose-6- phosphate isomerase; phosphofructokinase 1 ; fructose-bisphosphate aldolase; triosephosphate isomerase; glyceraldehyde 3-phosphate dehydrogenase; phosphoglycerate kinase; phosphoglycerate mutase; enolase; and pyruvate kinase. In addition, extracellular glucose is transported into the cell by the glucose transporter GLUT1 , and the activity of this glucose transporter may also be a potential therapeutic target. (C. Xintaropoulou et al., “A Comparative Analysis of Inhibitors of the Glycolysis Pathway in Breast and Ovarian Cancer Models,” Oncotarget 6: 25677-25695 (2015)).
[0385] Inhibitors of GLUT 1 include the flavonoids phloretin and quercetin. Another inhibitor of GLUT1 is STF31 (4-[[[[4-(1 ,1- dimethylethyl)phenyl]sulfonyl]amino]methyl]-A/-3-pyridinylbenzamide). Still another inhibitor of GLUT 1 is WZB117 ((2-fluoro-6-(m -hydroxybenzoyloxy) phenyl m- hydroxybenzoate).
[0386] Inhibitors of hexokinase include 3-bromopyruvate.
[0387] Inhibitors of phosphofructokinase 1 include 3-(3-pyridinyl)-1 -(4-pyridinyl)- 2-propen-1-one and PFK158 (1 -(4-pyridinyl)-3-[7-(trifluoromethyl)-2E-quino!inyl]-2- propen-1 -one.
[0388] Other inhibitors of glycolysis do not directly inhibit the enzymes that are part of the glycolysis pathway itself but can interfere with glycolysis by other routes. For example, dichloroacetate is an inhibitor of pyruvate decarboxylase kinase, an enzyme that inhibits the mitochondrial pyruvate dehydrogenase. In this way, it suppresses glycolysis and stimulates oxidative phosphorylation. Oxamic acid is a pyruvate analog and is a competitive lactate dehydrogenase inhibitor that inhibits glycolysis. NH 1-1 (1- hydroxy-6-phenyl-4-trifluoromethyl-1 H-indole-2-carboxylic acid) and NHI-2 (methyl 1- hydroxy-6-phenyl-4-trifluoromethyl-1 H-indole-2-carboxylate) are lactate dehydrogenase inhibitors; by diminishing lactate production, these compounds stimulate oxidative phosphorylation and inhibit glycolysis by feedback inhibition.
[0389] United States Patent No. 11 ,026,960 to Priebe et al. discloses esters of 2-deoxymonosaccharides of Formula (G-l) or of a salt, ester, or prodrug of a compound of Formula (G-l):
(G-l), wherein:
(1 ) each of R1, R2, R3, and R4 is independently H or COR5; and
(2) each of R5, where present, is independently selected from the group consisting of C1-C10 straight-chain or branched chain alkyl, C4-C10 alkylcycloalkyl, and C3-C7 cycloalkyl.
[0390] United States Patent No. 10,988,484 to Olszewski et al. discloses glucose uptake inhibitors of Formula (G-ll):

(G-ll), wherein:
(1 ) A is selected from the group consisting of Subformulas (G-lla) and (G-ll(b):
 (G-ll(a));
(G-ll(a));

(G-ll(b));
(2) Ring B is a five- or six-membered ring containing 1 or 2 heteroatoms selected from the group consisting of N, 0 and S;
(3) Ring C is a five- or six-membered aryl or heteroaryl ring containing from 0 to 2 heteroatoms selected from the group consisting of N, 0 and S;
(4) each R1 is independently selected from the group consisting of halo, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, C3-C6 cycloalkyl, oxo, hydroxy, amino, cyano and C1- C3 perfluoroalkyl;
(5) n is selected from 0, 1 , or 2;
(6) each R2 is independently selected from the group consisting of halo, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, C3-C6 cycloalkyl, oxo, hydroxy, amino, cyano and C1- C3 perfluoroalkyl;
(7) m is selected from 0, 1 , or 2;
(8) R3 is selected from the group consisting of halo, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, C3-C6 cycloalkyl, oxo, hydroxy, amino, cyano and C1-C3 perfluoroalkyl;
(9) R4 is selected from the group consisting of halo, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, C3-C6 cycloalkyl, oxo, hydroxy, amino, cyano and C1-C3 perfluoroalkyl;
(10) D is selected from the group consisting of --O--(CH2)y--C(=O)NR5R6, -O-- C(=O)— (CH2)y— NR5R6, -O-(CH2)y-NR5R6, -NH-C(=O)-(CH2)y-NR5R6, -NH-C(=O)- (CH2)y-R7, and -NH-(CH2)y-NR5R6;
(11 ) y is selected from 1 , 2, or 3;
(12) R5 and R6 are independently selected from the group consisting of H, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, --( C1-C6 alkyl)-O--( C1-C6 alkyl), aryl, aralkyl, heteroaryl, and C3-C6 cycloalkyl, or R5 and R6 may be taken together with the nitrogen to which they are attached to form a 5- to 6-membered heterocyclic ring having up to 3 heteroatoms selected from N, O, and S, and which is optionally substituted by from 1 to
3 substituents independently selected from halo, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, C3-C6 cycloalkyl, oxo, hydroxy, amino, cyano and C1-C3 perfluoroalkyl; and
(13) R7 is selected from the group consisting of aryl, heteroaryl, and a heterocyclic group.
[0391] United States Patent No. 10,500,175 to Ko discloses a glycolysis inhibitor of Formula (G-lll):

(G-lll), wherein:
(1 ) X is selected from the group consisting of nitro, an imidazole, a halide, sulfonate, a carboxylate, an alkoxide, and amine oxide;
(2) R is selected from the group consisting of OR', N(R")2, C(O)R"', C1-C6 alkyl, C6-C12 aryl, C1-C6 heteroalkyl, C6-C12 heteroaryl, hydrogen, and an alkali metal;
(3) R' is hydrogen, an alkali metal, C1-C6 alkyl, C6-C12 aryl, or C(O)R"';
(4) R" is hydrogen, C1-C6 alkyl, or C6-C12 aryl; and
(5) R'" is hydrogen, C1-C20 alkyl, or C6-C12 aryl.
[0392] United States Patent No. 8,927,506 to Priebe et al. discloses acetates of 2-deoxymonosaccharides as inhibitors of glycolysis.
[0393] United States Patent No. 8,329,753 to Newell et al. discloses bifunctional compounds that covalently link a glycolysis inhibitor and a fatty acid metabolism inhibitor.
[0394] The anti-neoplastic agent lonidamine can be used together with elesclomol or a derivative or analog of elesclomol; in this context, lonidamine acts as an additional agent that causes the tumor cells to rely heavily on oxidative phosphorylation. Lonidamine is an agent that suppresses glycolysis in tumor cells through the inhibition of the mitochondrially bound hexokinase.
[0395] Another agent that acts as an additional agent that causes the tumor cells to rely heavily on oxidative phosphorylation is the alkylating agent dianhydrogalactitol. Dianhydrogalactitol is described in PCT Patent Application Publication No. WO 2012/024367 by Brown.
[0396] A ninth category of additional agents is agents that act as inhibitors of the base excision repair (BER) pathway.
[0397] Inhibitors of the base excision repair (BER) pathway as anti-neoplastic agents are disclosed in A.M. Reed et al., “Small-Molecule Inhibitors of Proteins Involved in Base Excision Repair Potentiate the Anti-Tumorigenic Effect of Existing Chemotherapeutics and Irradiation,” Future Oncol. 5: 713-726 (2009). These inhibitors include: (i) PARP inhibitors, specifically INO-1001 (3-aminobenzamide); AG14361 (2-[4- [(dimethylamino)methyl]phenyl]-1 ,3, 10-triazatricyclo[6.4.1 .04, 13]trideca-2 , 4, 6, 8( 13)- tetraen-9-one); AG014699 (rucaparib phosphate); ABT-888 (veliparib); and AZD2281 (olaparib); (ii) inhibitors of the Apel enzyme, including methoxyamine; lucanthone; 7- nitroindole-2-carboxylic acid; an arylstilbonic acid derivative (A. Bapat et al., “Novel Small-Molecule Inhibitor of Apurinic/Apyrimidinic Endonuclease 1 Blocks Proliferation and Reduces Viability of Glioblastoma Cells,” J. Pharmacol. Exp. Ther 334: 988-998 (2010)); E3330 ((2E)-2-[(4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1 ,4-dien-1- yl)methylidene]undecanoic acid); and (iii) inhibitors of Pol β, including oleanolic acid, edgeworin, betulinic acid, stigmasterol, and kohamaic acid.
[0398] Other inhibitors of Ape1 are disclosed in A. Bapat et al., “Novel Small- Molecule Inhibitor of Apurinic/Apyrimidinic Endonuclease 1 Blocks Proliferation and Reduces Viability of Glioblastoma Cells,” J. Pharmacol. Exp. Ther. 334: 988-998 (2010), including 2-(4-(2,5-dimethyl-1 H-pyrrol-1 -yl)phenoxy)acetic acid; 2,4,9- trimethylbenzo[b][1 ,8]-naphthyridin-5-amine; and N-(3-chlorophenyl)-5,6-dihydro-4H- cyclopenta[d]isoxazole-3-carboxamide.
[0399] United States Patent No. 10,220,053 to Bellacosa et al. discloses inhibitors of thymine DNA glycosylase as inhibitors of the BER pathway.
[0400] United States Patent No. 8,716,346 to Gerson et al. discloses BER inhibitors including AP endonuclease inhibitor, a DNA glycosylase inhibitor, a DNA polymerase inhibitor, a PARP inhibitor, a DNA alkyltransferase inhibitor, and a DNA
ligase inhibitor. Preferred BER inhibitors include methoxyamine and a compound of Formula (B-l):

(B-l).
[0401] The AP endonuclease inhibitor can be methoxyamine, the compound of Formula (B-l), or N-ethylmaleimide. The PARP inhibitor can be 5-methyl-3,4-dihydro- 2H-isoquinolin-1 -one (PD128763), 3-aminobenzamide, 6-aminonicotinamide, 8- hydroxy-2-methyl-3H-quinazolin-4-one (NU 1025), or 4-amino-1 ,8-naphthalimide. The DNA polymerase inhibitor can be prunasin, aphidicolin, ddCTP, ddTTP, ddATP, ddGTP, or arabinocytidine.
[0402] A tenth category of additional agents is agents that act as inhibitors of the homologous repair (HR) pathway (S.B. Chernikova, “Inhibiting Homologous Recombination for Cancer Therapy,” Cancer Biol. Ther. 13: 61-68 (2012)). These agents include imatinib mesylate, erlotinib, valproic acid, abexinostat, tanespimycin, β- lactacystin, benzyl N-[(2S)-4-methyl-1 -[[(2S)-4-methyl-1 -[[(2S)-4-methyl-1 -oxopentan-2- yl]amino]-1 -oxopentan-2 -yl]amino]-1 -oxopentan-2 -yl]carbamate (MG-132), bortezomib, nelfinavir, mirin, 6-(cyclohexylmethoxy)-5-nitrosopyrimidine-2,4-diamine (NU6027), 3- (carbamoylamino)-5-(3-fluorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide (AZD7762), (5Z)-5-(quinolin-6-ylmethylidene)-2-(thiophen-2-ylmethylimino)-1 ,3- thiazolidin-4-one (RO-3306), N-[[5-[3-(4,6-difluoro-1 H-benzimidazol-2-yl)-1 H-indazol-5- yl]-4-methylpyridin-3-yl]methyl]ethanamine (AG-024322), wortmannin, N-(2- aminoethyl)isoquinoline-5-sulfonamide (H-9), alsterpaulone, and curcumin.
[0403] United States Patent No. 10,927,075 to Mills discloses inhibitors of homologous repair including 4,4'-diisothiocyanatostilbene-2,2'-disulfonate.
[0404] United States Patent No. 10,590,122 to Castro et al. discloses substituted thiazole derivatives as RAD51 inhibitors that function as inhibitors of homologous repair; the substituted thiazole derivatives can include a cycloalkyl, heterocyclyl, or heteroaromatic moiety.
[0405] An eleventh category of additional agents is antineoplastic agents that can activate homologous repair as part of their mechanism of antineoplastic activity or as a consequence of inducing DNA damage. These additional agents include carboplatin, cisplatin, dianhydrogalactitol, and dibromodulcitol.
[0406] A twelfth category of additional agents is antineoplastic agents that are activated by bioreductases under acute conditions of hypoxia or that function to sensitize hypoxic cells to antineoplastic agents or radiation. These agents include hypoxic cell sensitizers and cytotoxic agents. Hypoxic cell sensitizers include, but are not necessarily limited to, misonidazole, metronidazole, nimorazole, benznidazole, desmethylmisonidazole, etanidazole, pimonidazole, and 1 -(aziridin-1 -yl)-3-(2- nitroimidazol-1-yl)propan-2-ol (RSU-1069) (S. Dische, “Chemical Sensitizers for Hypoxic Cells: A Decade of Experience in Clinical Radiotherapy,” Radiother. Oncol. 3: 97-115 (1985)). Cytotoxic agents that can be activated by bioreductases include, but are not limited to, tirapazamine and mitomycin C. Tirapazamine produces hydroxyl and/or benzotriazinyl radicals as DNA-damaging reactive species. Mitomycin C is an alkylating agent that crosslinks DNA; it is reductively activated to form a mitosene, which reacts successively via /V-alky lation of two DNA bases. Both alkylations are sequence-specific for a guanine nucleoside in the sequence 5'-CpG-3'.
[0407] A thirteenth category of additional agents is agents that inhibit cysteine uptake.
[0408] One class of agents that inhibits cysteine uptake is peptides derived from digestion of human p-casein, bovine p-casein, and gliadin. These peptides include human p-casomorphin-7 (hBCM7) (YPFVEPL) (SEQ ID NO: 1 ); bovine p-casomorphin- 7 (bBCM7) (YPFPGPL) (SEQ ID NO: 2); and gliadinomorphin-7 (GM7) (YPQPQPF) (SEQ ID NO: 3) (M.S. Trivedi, “Food-Derived Opioid Peptides Inhibit Cysteine Uptake with Redox and Epigenetic Consequences,” J. Nutr. Biochem. 25: 1011-1018 (2014)).
[0409] Another class of agents that inhibits cysteine uptake is inhibitors of the excitatory amino acid transporters (EAATs) EAAT2 and EAAT3. These inhibitors include L-glutamate, L-aspartate, and the synthetic inhibitors threo-p-hydroxyaspartate, which is a non-selective EAAT inhibitor, and dihydrokainate, which is a selective EAAT2 inhibitor. Another cysteine uptake inhibitor is threo-p-benzyloxyaspartate (Y. Chen & R.A. Swanson, “The Glutamate Transporters EAAT2 and EAAT3 Mediate Cysteine Uptake in Cortical Neuron Cultures,” J. Neurochem. 84: 1332-1339 (2003)). Yet another EAAT2 inhibitor is WAY-855 (3-amino-tricyclo[2.2.1 ,026]heptane-1 ,3- dicarboxylic acid) (J. Dunlop et al., “WAY-855 (3-Amino-tricyclo[2.2.1 ,026]heptane-1 ,3- Dicarboxylic Acid), a Novel, EAAT2-Preferring, Nonsubstrate Inhibitor of High-Affinity Glutamate Uptake,” Br. J. Pharmacol. 140: 839-846 (2003)).
[0410] Yet another inhibitor of cysteine transport is erastin or analogs of erastin (S.J. Dixon et al., “Pharmacological Inhibition of Cystine-Glutamine Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis,” eLife 2014:3: e2523).
[0411] Erastin analogs are known in the art and are disclosed in United States Patent No. 7,615,554 to Selliah et al. These analogs include erastin A, erastin B, and compounds of Formula (E-l):

(E-l), wherein:
(1 ) R1 is selected from the group consisting of hydrogen, -Z-Q-Z-, --(C1- C8)alkyl-N(R2)(R4), --(C1-C8)alkyl-OR3, 3- to 8-membered carbocyclyl, 3- to 8-membered heterocyclyl, aryl, heteroaryl, and (C1-C4) aralkyl;
(2) R2 and R4 are each independently selected from the group consisting of hydrogen, (C1-C4) alkyl, (C1-C4) aralkyl, aryl, heteroaryl, acyl, alkylsulfonyl, and arylsulfonyl, provided that when both R2 and R4 are on the same nitrogen atom and not both hydrogen they are different and that when both R2 and R4 are on the same nitrogen atom and either R2 or R4 is acyl, alkylsulfonyl, or arylsulfonyl, then the other is selected from the group consisting of hydrogen, (C1-C8) alkyl, aryl, (C1-C4) aralkyl, and heteroaryl;
(3) R3 is selected from the group consisting of hydrogen, (C1-C4) alkyl, (C1-C4) aralkyl, aryl, and heteroaryl;
(4) W is selected from a moiety of Subformula (E-I(a)), (E-I(b)), or (E-I(c)):

((E-I(a)), (E-I(b)), or (E-I(c));
(5) Q is selected from 0 and NR2; and
(6) Z is independently selected for each occurrence from the group consisting of (C1-C6) alkyl, (C2-C6) alkenyl, and (C2-C6) alkynyl.
[0412] Other erastin analogs include piperazine erastin (2-[[4-[2-(4- chlorophenoxy)acetyl]piperazin-1-yl]methy!]-3-[5-(piperazin-1-ylmethyl)-2-propan-2- yloxyphenyl]quinazolin-4-one) (United States Patent No. 9,695,133 to Stockwell et al.).
[0413] United States Patent Application Publication No. 2007/0161644 by Stockwell discloses erastin analogs. These erastin analogs include compounds of Formula (E-II)

(1 ) R1 is selected from the group consisting of hydrogen, (C1-C8) alkyl, (C1-C8) alkoxy, 3- to 8-membered carbocyclyl, 3- to 8-membered heterocyclyl, aryl, heteroaryl, residues of glycolic acid, residues of ethylene glycol/propylene glycol copolymers, carboxylate, ester, amide, carbohydrate, amino acid, alditol, OC(R7)2COOH, SC(R7)2COOH, NHCHR7COOH, COR8, CO2R8, sulfate, sulfonamide, sulfoxide, sulfonate, sulfone, thioalkyl, thioester, and thioether;
(2) R2, R3, R4, R5, and R6 are each independently selected from the group consisting of hydrogen, halogen, (C1-C4) alkyl, (C1-C4) alkylamino, acyl, and alkylsulfonyl;
(3) R7 is selected from the group consisting of hydrogen, (C1-C8) alkyl, optionally substituted carbocyclyl, aryl, heteroaryl, heterocyclyl, alkylaryl, alkylheteroaryl, and alkylheterocyclyl; and
(4) R8 is selected from the group consisting of optionally substituted (C1-C8) alkyl, (C2-C8) alkenyl, (C2-C8) alkynyl, aryl, carbocyclyl, heteroaryl, heterocyclyl, alkylaryl, alkylheteroaryl, and alkylheterocyclyl; with the proviso that R1 is not methyl when R4 is chloro.
[0414] Yet another class of inhibitors of cysteine uptake includes inhibitors of other transporters of cysteine, or, in some cases, its oxidized form cystine. These additional transporters include LAT1 , ASCT2, and the Xc’ system (B. Daher et al.,
“Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptic Cell Death,” Front. Oncol. 10:723 (2020)). LAT1 is responsible for transport of essential amino acids, general amino acid homeostasis, and, consequently tumor growth. ASCT2 is a transporter that exchanges small neutral amino acids and plays a crucial role in glutamine uptake and the promotion of tumor growth, independently of LAT1 activity; however, there is evidence that this transporter may not actually be involved in the transport of cysteine in vivo and that cysteine may actually be a competitive inhibitor of ASCT2. The third transporter of relevance is the Xc’ system, which is an exchanger that imports cystine (the oxidized form of cysteine) and exports glutamate. This sodium- independent antiporter comprises two subunits: xCT (encoded by the gene SLC7A11), a subunit responsible for the amino acid exchange, and a chaperone CD98 (encoded by the gene SLC3A2). Inhibitors of the Xc’ system include erastin, imidazole ketone erastin (IKE), sorafenib, and sulfasalazine (SSZ). Expression of the genes involved in the Xc’ system is controlled by the protein kinase General Control Nonderepressible (GCN2), which is a sensor of amino acids that is activated by the intracellular accumulation of uncharged tRNA. GCN2 represses general protein synthesis and activates the transcription of genes involved in synthesis and transport of amino acids through activation of the ATF4 transcription factor. Yet another transcription factor that regulates the Xc’ system is the nuclear factor erythroid 2-related factor (NRF2) that acts on the antioxidant response element (ARE) present in the promoter region of the xCT gene.
[0415] One aspect of the present invention is a method to improve the efficacy and/or reduce the side effects of the administration of elesclomol or a derivative, analog, salt, or solvate of elesclomol for treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases or conditions comprising the steps of:
(1 ) identifying at least one factor or parameter associated with the efficacy and/or occurrence of side effects of the administration of the elesclomol or a derivative, analog, salt, or solvate of elesclomol for the treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases; and
(2) modifying the factor or parameter to improve the efficacy and/or reduce the side effects of the administration of the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol for the treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases.
[0416] Typically, the factor or parameter is selected from the group consisting of:
(1 ) dose modification;
(2) route of administration;
(3) schedule of administration;
(4) indications for use;
(5) disease stages;
(6) other indications;
(7) patient selection;
(8) patient/disease phenotype;
(9) patient/disease genotype;
(10) pre/post-treatment preparation;
(11 ) toxicity management;
(12) pharmacokinetic/pharmacodynamic monitoring;
(13) drug combinations;
(14) chemosensitization;
(15) chemopotentiation;
(16) post-treatment patient management;
(17) bulk drug product improvements;
(18) diluent systems;
(19) solvent systems;
(20) excipients;
(21 ) dosage forms;
(22) dosage kits and packaging;
(23) drug delivery systems;
(24) drug conjugate forms;
(25) compound analogs;
(26) prodrug systems;
(27) multiple drug systems;
(28) biotherapeutic enhancement;
(29) biotherapeutic resistance modulation;
(30) radiation therapy enhancement;
(31 ) novel mechanisms of action;
(32) selective target cell population therapeutics;
(33) reversal of resistance to an agent selected from the group consisting of a platinum-containing anti-neoplastic agent and a PARP inhibitor anti- neoplastic agent;
(34) induction of synthetic lethality by administration of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1 A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol renders the malignant cells susceptible to a targeted agent that otherwise would have no effect; and
(35) modulation of activity of FDX1 to inhibit OXPHOS.
[0417] When the improvement made is by dose modification, the dose modification can be, but is not limited to, at least one dose modification selected from the group consisting of:
(a) i.v. infusion for hours to days;
(b) biweekly administration;
(c) triweekly administration;
(d) monthly administration;
(e) doses greater than 100 mg/m2/day;
(f) progressive escalation of dosing from 100 mg/m2/day based on patient tolerance;
(g) doses less than 2 mg/m2 for greater than 14 days;
(h) modification of dosage in conjunction with use of polyamine to modulate metabolism;
(i) modification of dosage in conjunction with use of eflornithine to modulate metabolism;
(j) selected and intermittent boost dose administration;
(k) bolus single and multiple doses escalating from 100 mg/m2; and
(l) oral doses below 30 or above 130 mg/m2.
[0418] When the improvement made is by route of administration, the route of administration can be, but is not limited to, at least one route of administration selected from the group consisting of:
(a) topical administration;
(b) intravesicular administration for bladder cancer;
(c) oral administration;
(d) slow release oral delivery;
(e) intrathecal administration;
(f) intraarterial administration;
(g) continuous infusion;
(h) intermittent infusion
(i) administration via large volume oral solution;
(j) buccal administration; and
(k) rectal administration.
[0419] When the improvement is made by schedule of administration, the schedule of administration can be, but is not limited to, a schedule of administration selected from the group consisting of:
(a) daily administration;
(b) weekly administration for three weeks;
(c) weekly administration for two weeks;
(d) biweekly administration;
(e) biweekly administration for three weeks with a 1 -2 week rest period;
(f) intermittent boost dose administration;
(g) daily administration for one week then administration once per week for multiple weeks;
(h) daily administration on days 1-5, 8-12 every three weeks; and
(i) daily administration on days 1-3, 8-11 per cycle.
[0420] When the improvement is made by indication for use, the indication for use can be, but is not limited to, an indication for use selected from the group consisting of:
(a) use for the treatment of leukemias including acute and chronic, leukemias, including AML, ALL, CLL, CML);
(b) use for the treatment of myelodysplastic syndrome (MDS);
(c) use for the treatment of angiogenic diseases or conditions;
(d) use for the treatment of benign prostate hypertrophy;
(e) use for the treatment of psoriasis;
(f) use for the treatment of gout;
(g) use for the treatment of autoimmune conditions;
(h) use for the prevention of transplantation rejection;
(i) use for restenosis prevention in cardiovascular disease;
(j) use for the treatment of mycosis fungoides;
(k) use in bone marrow transplantation;
(l) use as an anti-infective;
(m) use for the treatment of AIDS;
(n) use for the treatment of lymphoma;
(o) use for the treatment of mantle cell lymphoma;
(p) use for the treatment of meningeal leukemia;
(q) use for the treatment of malignant meningitis;
(r) use for the treatment of cutaneous T cell lymphoma;
(s) use for the treatment of Barrett’s esophagus;
(t) use for the treatment of anaplastic gliomas;
(u) use for the treatment of triple negative breast cancer;
(v) use for the treatment of Braf-mutated melanoma;
(w) use for the treatment of BTK resistant CLL;
(x) use for the treatment of chordoma;
(y) use for the treatment of Kras-mutated colon cancer;
(z) use for the treatment of pediatric tumors including brain and sarcoma;
(aa) use for the treatment of neuroblastoma;
(ab) use for the treatment of rhabdomyosarcoma;
(ac) use for the treatment of Ewing’s sarcoma;
(ad) use for the treatment of medulloblastoma;
(ae) use for the treatment of thyroid cancer;
(af) use for the treatment of melanoma,
(ag) use for the treatment of lymphoma;
(ah) use for the treatment of multiple myeloma;
(ai) use for the treatment of ovarian osteogenic sarcoma;
(aj) use for the treatment of bladder cancer;
(ak) use for the treatment of prostate cancer;
(al) use for the treatment of bone metastases;
(am) use for the treatment of bone pain;
(an) use for the treatment of ovarian clear cell carcinoma;
(ao) use for the treatment of high-grade serous ovarian carcinoma; and
(ap) use for the treatment of small-cell carcinoma of the ovary, particularly the hypercalcemic type.
[0421] When the improvement is made by selection of a disease stage, the disease stage can be, but is not limited to, a disease stage selected from the group consisting of:
(a) use for the treatment of localized polyp stage colon cancer;
(b) use for the treatment of leukoplakia in the oral cavity;
(c) use for angiogenesis inhibition to prevent or limit metastatic spread; and
(d) use against HIV with AZT, DDI, or reverse transcriptase inhibitors.
[0422] When the improvement is made by use for other indications, the other indication can be, but is not limited to, another indication selected from the group consisting of:
(a) use as an antiinfective;
(b) use as an antiviral;
(c) use as an antibacterial;
(d) use for pleural effusions;
(e) use as an antifungal;
(f) use as an antiparasitic;
(g) use for eczema;
(h) use for shingles;
(i) use for condylomata;
(j) use as an anti HPV agent;
(k) use as an anti-HSV agent;
(l) use for early or late stage MDS (myelodysplastic syndrome);
(m) use for polycythemia vera; and
(n) use for Paget’s disease.
[0423] When the improvement is made by use with patient selection, the patient selection can be, but is not limited to, a method of patient selection selected from the group consisting of:
(a) selecting patients with disease conditions with high levels of metabolic enzymes;
(b) selecting patients with disease conditions with high levels of reactive oxygen species;
(c) selecting patients with disease conditions with high levels of histone deacetylase;
(d) selecting patients with disease conditions with high levels of protein kinases;
(e) selecting patients with disease conditions with high levels of ornithine decarboxylase;
(f) selecting patients with disease conditions with low levels of metabolic enzymes;
(g) selecting patients with disease conditions with low levels of reactive oxygen species;
(h) selecting patients with disease conditions with low levels of histone deacetylase;
(i) selecting patients with disease conditions with low levels of protein kinases;
(j) selecting patients with disease conditions with low levels of ornithine decarboxylase;
(k) selecting patients with low or high susceptibility to thrombocytopenia or neutropenia;
(l) selecting patients intolerant of Gl toxicities;
(m) selecting patients with deficiencies in DNA repair capacity including BRCA or ARID1 A or other deficiencies in the SWI/SWF pathway or mitochondrial electron transport;
(n) selecting patients with over- or under-expression of jun;
(o) selecting patients with over- or under-expression of GPCR’s or signal transduction proteins;
(p) selecting patients with over- or under-expression of VEGF;
(q) selecting patients with over- or under-expression of prostate specific genes;
(r) selecting patients with over- or under-expression of protein kinases;
(s) selecting patients with over- or under-expression of telomerase;
(t) selecting patients with abnormalities in gallium scans; and
(u) selecting patients with abnormalities in bone scans.
[0424] When the improvement is made by use determined by analysis of patient or disease phenotype, the analysis of patient or disease phenotype can be, but is not limited to, an analysis of patient or disease phenotype selected from the group consisting of:
(a) use of diagnostic tools, techniques, kits and assays to confirm a patient’s particular phenotype;
(b) measurement of metabolism enzymes and metabolites;
(c) measurement of histone deacetylase;
(d) measurement of protein kinases;
(e) measurement of ornithine decarboxylase;
(f) measurement of VEGF;
(g) measurement of products of prostate specific genes;
(h) measurement of protein kinases;
(i) measurement of telomerase;
(j) measurement of jun;
(k) measurement of GPCR’s;
(l) use of surrogate compound dosing;
(m) low dose drug pre-testing for enzymatic status; and
(n) determination of ARID1 A phenotypic status.
[0425] The cellular proto-oncogene c-Jun encodes a protein that, in combination with c-Fos, forms the AP-1 early response transcription factor. This proto-oncogene plays a key role in transcription and interacts with a large number of proteins affecting transcription and gene expression. It is also involved in proliferation and apoptosis of cells that form part of a number of tissues, including cells of the endometrium and glandular epithelial cells. G-protein coupled receptors (GPCRs) are important signal transducing receptors. The superfamily of G protein coupled receptors includes a large number of receptors. These receptors are integral membrane proteins characterized by amino acid sequences that contain seven hydrophobic domains, predicted to represent the transmembrane spanning regions of the proteins. They are found in a wide range of organisms and are involved in the transmission of signals to the interior of cells as a result of their interaction with heterotrimeric G proteins. They respond to a diverse range of agents including lipid analogues, amino acid derivatives, small molecules such as epinephrine and dopamine, and various sensory stimuli. The properties of many known GPCR are summarized in S. Watson & S. Arkinstall, “The G-Protein Linked Receptor Facts Book” (Academic Press, London, 1994), incorporated herein by this reference. GPCR receptors include, but are not limited to, acetylcholine receptors, β- adrenergic receptors, 03-adrenergic receptors, serotonin (5-hydroxytryptamine) receptors, dopamine receptors, adenosine receptors, angiotensin Type II receptors, bradykinin receptors, calcitonin receptors, calcitonin gene-related receptors, cannabinoid receptors, cholecystokinin receptors, chemokine receptors, cytokine receptors, gastrin receptors, endothelin receptors, y-aminobutyric acid (GABA) receptors, galanin receptors, glucagon receptors, glutamate receptors, luteinizing
hormone receptors, choriogonadotrophin receptors, follicle-stimulating hormone receptors, thyroid-stimulating hormone receptors, gonadotrophin-releasing hormone receptors, leukotriene receptors, Neuropeptide Y receptors, opioid receptors, parathyroid hormone receptors, platelet activating factor receptors, prostanoid (prostaglandin) receptors, somatostatin receptors, thyrotropin-releasing hormone receptors, vasopressin and oxytocin receptors.
[0426] When the improvement is made by use determined by analysis of patient or disease genotype, the analysis of patient or disease genotype can be, but is not limited to, an analysis of patient or disease genotype selected from the group consisting of:
(a) use of diagnostic tools, techniques, kits and assays to confirm a patient’s particular genotype;
(b) use of gene/protein expression chips and analysis;
(c) single nucleotide polymorphism (SNP) assessment;
(d) determination of SNP’s for histone deacetylase;
(e) determination of SNP’s for ornithine decarboxylase;
(f) determination of SNP’s for genes affecting S-adenosyl methionine metabolism;
(g) determination of SNP’s for GPCR’s;
(h) determination of SNP’s for protein kinases;
(i) determination of SNP’s for telomerase;
(j) determination of SNP’s for jun;
(k) identification and measurement of genes for metabolism enzymes and metabolites;
(l) identification of mutations in wild type and mutated genes;
(m) analysis of epigenetics via analysis of methylation and acetylation,
(n) analysis of ARID1 A mutation or ARID1 A deficiency;
(o) analysis of mTor signaling;
(p) analysis of mutations activating PI3K-AKT;
(q) analysis of mutations affecting PARPi resistance;
(r) analysis of mutations affecting HR deficiency;
(s) analysis of mutations affecting DDR deficiency;
(t) analysis of mutations causing SWI/SNF pathway alteration;
(u) analysis of P53 status/mutation;
(v) analysis of BRCA-1 independence/mutation;
(w) analysis of NAC1 mutation; and
(x) analysis of mutations affecting mitochondrial targeting.
[0427] The use of gene chips is described in A. J. Lee & S. Ramaswamy, “DNA Microarrays in Biological Discovery and Patient Care” in Essentials of Genomic and Personalized Medicine (G.S. Ginsburg & H.F. Willard, eds., Academic Press, Amsterdam, 2010), ch. 7, pp. 73-88.
[0428] When the method is the use of single nucleotide polymorphism (SNP) analysis, the SNP analysis can be carried out on a gene selected from the group consisting of histone deacetylase, ornithine decarboxylase, VEGF, a prostate specific gene, c-Jun, and a protein kinase. The use of SNP analysis is described in S. Levy and Y.-H. Rogers, “DNA Sequencing for the Detection of Human Genome Variation” in Essentials of Genomic and Personalized Medicine (G.S. Ginsburg & H.F. Willard, eds., Academic Press, Amsterdam, 2010), ch. 3, pp. 27-37.
[0429] Still other genomic techniques such as copy number variation analysis and analysis of DNA methylation can be employed. Copy number variation analysis is described in C. Lee et al., “Copy Number Variation and Human Health” in Essentials of Genomic and Personalized Medicine (G.S. Ginsburg & H.F. Willard, eds., Academic Press, Amsterdam, 2010), ch. 5, pp. 46-59. DNA methylation analysis is described in S. Cottrell et al., “DNA Methylation Analysis: Providing New Insight into Human Disease” in Essentials of Genomic and Personalized Medicine (G.S. Ginsburg & H.F. Willard, eds., Academic Press, Amsterdam, 2010), ch. 6, pp. 60-72.
[0430] When the improvement is made by pre/post-treatment preparation, the pre/post treatment preparation can be, but is not limited to, a method selected from the group consisting of:
(a) use of colchicine or analogs;
(b) use of diuretics such as probenecid;
(c) use of uricase;
(d) non-oral use of nicotinamide;
(e) use of sustained release forms of nicotinamide;
(f) use of inhibitors of poly-ADP ribose polymerase;
(g) use of caffeine;
(h) use of leucovorin rescue;
(i) use of infection control;
(j) use of antihypertensives;
(k) alteration of stem cell populations;
(l) pretreatment to limit or prevent graft versus host (GVH) cytokine storm reactions;
(m) use of anti-inflammatories; and
(n) anaphylactic reaction suppression.
[0431] Uricosurics include, but are not limited to, probenecid, benzbromarone, and sulfinpyrazone. A particularly preferred uricosuric is probenecid. Uricosurics, including probenecid, may also have diuretic activity. Other diuretics are well known in the art, and include, but are not limited to, hydrochlorothiazide, carbonic anhydrase inhibitors, furosemide, ethacrynic acid, amiloride, and spironolactone.
[0432] Poly-ADP ribose polymerase inhibitors are described in G.J. Southan & C. Szabo, “Poly(ADP-Ribose) Inhibitors,” Curr. Med. Chem. 10: 321-240 (2003), and include nicotinamide, 3-aminobenzamide, substituted 3,4-dihydroisoquinolin-1 (2H)-ones and isoquinolin-1 (2H)-ones, benzimidazoles, indoles, phthalazin-1 (2H)-ones, quinazolinones, isoindolinones, phenanthridinones, and other compounds.
[0433] Leucovorin rescue comprises administration of folinic acid (leucovorin) to patients in which methotrexate has been administered. Leucovorin is a reduced form of folic acid that bypasses dihydrofolate reductase and restores hematopoietic function. Leucovorin can be administered either intravenously or orally.
[0434] In one alternative, wherein the pre/post treatment is the use of a uricosuric, the uricosuric is probenecid or an analog thereof.
[0435] When the improvement is made by toxicity management, the toxicity management can be, but is not limited to, a method selected from the group consisting of:
(a) use of colchicine or analogs;
(b) use of diuretics such as probenecid;
(c) use of uricase;
(d) non-oral use of nicotinamide;
(e) use of sustained release forms of nicotinamide;
(f) use of inhibitors of poly-ADP-ribose polymerase;
(g) use of caffeine;
(h) use of leucovorin rescue;
(i) use of sustained release allopurinol;
(j) non-oral use of allopurinol;
(k) use of bone marrow transplant stimulants;
(l) administration of blood;
(m) administration of platelet infusions;
(n) administration of Neupogen;
(o) administration of G-CSF or GM-CSF;
(p) administration of agents for pain management;
(q) use of anti-inflammatories;
(r) administration of fluids;
(s) use of corticosteroids;
(t) use of insulin control medications;
(u) use of antipyretics;
(v) use of anti-nausea treatments;
(w) use of anti-diarrhea treatments;
(x) use of N-acetylcysteine;
(y) use of antihistamines;
(z) use of agents to limit or prevent mucositis;
(aa) use of agents to limit or prevent GVH reactions or cytokine storm reactions;
(ab) use of anti-fungal agents;
(ac) use of sodium thiosulfate;
(ad) use of glutathione;
(ae) use of platelet transfusions;
(af) use of anti-diarrheal therapeutics;
(ag) use of epinephrine for allergic and anaphylactic reactions;
(ah) use of lidocaine;
(ai) use of vasoconstrictors; and
(aj) use of vasodilators.
[0436] Filgrastim is a granulocytic colony-stimulating factor (G-CSF) analog produced by recombinant DNA technology that is used to stimulate the proliferation and differentiation of granulocytes and is used to treat neutropenia; G-CSF can be used in a similar manner. GM-CSF is granulocyte macrophage colony-stimulating factor and stimulates stem cells to produce granulocytes (eosinophils, neutrophils, and basophils) and monocytes; its administration is useful to prevent or treat infection.
[0437] Anti-inflammatory agents are well known in the art and include corticosteroids and non-steroidal anti-inflammatory agents (NSAIDs). Corticosteroids with anti-inflammatory activity include, but are not limited to, hydrocortisone, cortisone, beclomethasone dipropionate, betamethasone, dexamethasone, prednisone, methylprednisolone, triamcinolone, fluocinolone acetonide, and fludrocortisone. Non- steroidal anti-inflammatory agents include, but are not limited to, acetylsalicylic acid (aspirin), sodium salicylate, choline magnesium trisalicylate, salsalate, diflunisal, sulfasalazine, olsalazine, acetaminophen, indomethacin, sulindac, tolmetin, diclofenac, ketorolac, ibuprofen, naproxen, flurbiprofen, ketoprofen, fenoprofin, oxaprozin, mefenamic acid, meclofenamic acid, piroxicam, meloxicam, nabumetone, rofecoxib, celecoxib, etodolac, nimesulide, aceclofenac, alclofenac, alminoprofen, amfenac, ampiroxicam, apazone, araprofen, azapropazone, bendazac, benoxaprofen, benzydamine, bermoprofen, benzpiperylon, bromfenac, bucloxic acid, bumadizone, butibufen, carprofen, cimicoxib, cinmetacin, cinnoxicam, clidanac, clofezone, clonixin, clopirac, darbufelone, deracoxib, droxicam, eltenac, enfenamic acid, epirizole, esflurbiprofen, ethenzamide, etofenamate, etoricoxib, felbinac, fenbufen, fenclofenac, fenclozic acid, fenclozine, fendosal, fentiazac, feprazone, filenadol, flobufen, florifenine, flosulide, flubichin methanesulfonate, flufenamic acid, flufenisal, flunixin, flunoxaprofen, fluprofen, fluproquazone, furofenac, ibufenac, imrecoxib, indoprofen, isofezolac,
isoxepac, isoxicam, licofelone, lobuprofen, lomoxicam, lonazolac, loxaprofen, lumaricoxib, mabuprofen, miroprofen, mofebutazone, mofezolac, morazone, nepafanac, niflumic acid, nitrofenac, nitroflurbiprofen, nitronaproxen, orpanoxin, oxaceprol, oxindanac, oxpinac, oxyphenbutazone, pamicogrel, parcetasal, parecoxib, parsalmide, pelubiprofen, pemedolac, phenylbutazone, pirazolac, pirprofen, pranoprofen, salicin, salicylamide, salicylsalicylic acid, satigrel, sudoxicam, suprofen, talmetacin, talniflumate, tazofelone, tebufelone, tenidap, tenoxicam, tepoxalin, tiaprofenic acid, tiaramide, tilmacoxib, tinoridine, tiopinac, tioxaprofen, tolfenamic acid, triflusal, tropesin, ursolic acid, valdecoxib, ximoprofen, zaltoprofen, zidometacin, and zomepirac, and the salts, solvates, analogues, congeners, bioisosteres, hydrolysis products, metabolites, precursors, and prodrugs thereof.
[0438] The clinical use of corticosteroids is described in B.P. Schimmer & K.L. Parker, “Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones” in Goodman & Gilman’s The Pharmacological Basis of Therapeutics (L.L. Brunton, ed., 11th ed., McGraw-Hill, New York, 2006), ch. 59, pp. 1587-1612.
[0439] Anti-nausea treatments include, but are not limited to, ondansetron, metoclopramide, promethazine, cyclizine, hyoscine, dronabinol, dimenhydrinate, diphenhydramine, hydroxyzine, medizine, dolasetron, granisetron, palonosetron, ramosetron, domperidone, haloperidol, chlorpromazine, fluphenazine, perphenazine, prochlorperazine, betamethasone, dexamethasone, lorazepam, and thiethylperazine.
[0440] Anti-diarrheal treatments include, but are not limited to, diphenoxylate, difenoxin, loperamide, codeine, racecadotril, octreoside, and berberine.
[0441] N-acetylcysteine is an antioxidant and mucolytic that also provides biologically accessible sulfur.
[0442] When the improvement is made by pharmacokinetic/pharmacodynamic monitoring, the pharmacokinetic/pharmacodynamic monitoring can be, but is not limited to, a method selected from the group consisting of:
(a) multiple determinations of drug plasma levels;
(b) multiple determinations of metabolites in the blood or urine;
(c) measurement of polyamines;
(d) measurement of LAT-1 surface receptors;
(e) use of gene sequencing; and
(f) measurement of immune effectors.
[0443] Typically, determination of blood plasma levels or determination of at least one metabolite in blood or urine is carried out by immunoassays. Methods for performing immunoassays are well known in the art, and include radioimmunoassay, ELISA (enzyme-linked immunosorbent assay), competitive immunoassay, immunoassay employing lateral flow test strips, and other assay methods.
[0444] When the improvement is made by drug combination, the drug combination can be, but is not limited to, a drug combination selected from the group consisting of:
(a) use with topoisomerase inhibitors;
(b) use with fraudulent nucleosides;
(c) use with fraudulent nucleotides;
(d) use with thymidylate synthetase inhibitors;
(e) use with signal transduction inhibitors;
(f) use with cisplatin or gallium analogs;
(g) use with nitrosourea alkylating agents (BCNU , Gliadel wafers, CCNU);
(h) use with bendamustine (Treanda);
(i) use with anti-tubulin agents;
(j) use with antimetabolites;
(k) use with berberine;
(l) use with apigenin;
(m) use with amonafide;
(n) colchicine or an analog thereof;
(o) use with genistein;
(p) use with etoposide;
(q) use with cytarabine;
(r) use with a camptothecin;
(s) use with vinca alkaloids;
(t) use with 5-fluorouracil;
(u) use with curcumin;
(v) use with NF-KB inhibitors;
(w) use with rosmarinic acid;
(x) use in combination with biological therapies such as antibodies such as Avastin, Rituxan, Herceptin, Erbitux, PD-1 , or PD-L1 inhibitors;
(y) use with prednimustine;
(z) use with DNA and RNA therapeutics;
(aa) use with Braf inhibitors;
(ab) use with BTK inhibitors;
(ac) use with 5-azacytidine;
(ad) use with decitabine;
(ae) use with PARP inhibitors;
(af) use with agents inducing hypomethylation; and
(ag) use with histone deacetylase inhibitors.
[0445] Topoisomerase inhibitors include, but are not limited to, irinotecan, topotecan, camptothecin, lamellarin D, amsacrine, etoposide, etoposide phosphate, teniposide, doxorubicin, and 4-[2-(3,5-dioxo-1-piperazinyl)-1-methylpropyl]piperazine- 2, 6-dione (ICRF-193).
[0446] Fraudulent nucleosides include, but are not limited to, cytosine arabinoside, gemcitabine, and fludarabine; other fraudulent nucleosides are known in the art.
[0447] Fraudulent nucleotides include, but are not limited to, tenofovir disoproxil fumarate and adefovir dipivoxil; other fraudulent nucleotides are known in the art.
[0448] Thymidylate synthetase inhibitors include, but are not limited to, raltitrexed, pemetrexed, nolatrexed, ZD9331 , GS7094L, fluorouracil, and BGC 945.
[0449] Signal transduction inhibitors are described in A.V. Lee et al., “New Mechanisms of Signal Transduction Inhibitor Action: Receptor Tyrosine Kinase Down- Regulation and Blockade of Signal Transactivation,” Clin. Cancer Res. 9: 516s (2003).
[0450] Alkylating agents include, but are not limited to, Shionogi 254-S, aldo- phosphamide analogues, altretamine, anaxirone, Boehringer Mannheim BBR-2207,
bendamustine, bestrabucil, budotitane, Wakunaga CA-102, carboplatin, carmustine, Chinoin-139, Chinoin-153, chlorambucil, cisplatin, cyclophosphamide, American Cyanamid CL-286558, Sanofi CY-233, cyplatate, Degussa D-19-384, Sumimoto DACHP(Myr)2, diphenylspiromustine, diplatinum cytostatic, Erba distamycin derivatives, Chugai DWA-2114R, ITI E09, elmustine, Erbamont FCE-24517, estramustine phosphate sodium, fotemustine, Unimed G-6-M, Chinoin GYKI-17230, hepsul-fam, ifosfamide, iproplatin, lomustine, mafosfamide, melphalan, mitolactol, Nippon Kayaku NK-121 , NCI NSC-264395, NCI NSC-342215, oxaliplatin, Upjohn PCNU, prednimustine, Proter PTT-119, ranimustine, semustine, SmithKline SK&F-101772, Yakult Honsha SN-22, spiromustine, Tanabe Seiyaku TA-077, tauromustine, temozolomide, teroxirone, tetraplatin and trimelamol, as described in United States Patent No. 7,446,122 by Chao et al. Bendamustine is a chemotherapeutic alkylating agent for the treatment of chronic lymphocytic leukemia, multiple myeloma, and non- Hodgkin’s lymphoma and can be administered in combination with etoposide, fludarabine, mitoxantrone, methotrexate, prednisone, rituximab, vincristine, and 90Y- ibritumomab tiuxetan. Alkylating agents can include nitroso-containing alkylating agents.
[0451] Gallium analogs of cisplatin are described in U. Ndagi et al., “Metal Complexes in Cancer Therapy — An Update from Drug Design Perspective,” Drug Des. Devel. Ther. 11 : 599-616 (2017).
[0452] Berberine has antibiotic activity and prevents and suppresses the expression of pro-inflammatory cytokines and E-selectin, as well as increasing adiponectin expression.
[0453] Apigenin is a flavone that can reverse the adverse effects of cyclosporine and has chemoprotective activity, either alone or derivatized with a sugar.
[0454] Colchicine is a tricyclic alkaloid that exerts its activity by binding to the protein tubulin. Analogs of colchicine include, but are not limited to, colchiceinamide, A/- desacetylthiocolchicine, demecolcine, A/-acetyliodocolchinol, trimethylcolchicinic acid (TMCA) methyl ether, A/-acetylcolchinol, TMCA ethyl ether, isocolchicine, isocolchiceinamide, iso-TMCA methyl ether, colchiceine, TMCA, A/-benzoyl TMCA, colchicosamide, colchicoside, colchinol and colchinoic acid (M.H. Zweig & C.F. Chignell,
“Interaction of Some Colchicine Analogs, Vinblastine and Podophyllotoxin with Rat Brain Microtubule Protein,” Biochem. Pharmacol. 22: 2141-2150 (1973) and B. Yang et al., “Syntheses and Biological Evaluation of Ring C-Modified Colchicine Analogs,” Bioorg. Med. Chem. Lett. 20: 3831-3833 (2010)).
[0455] Genistein is an isoflavone with the systemic name 5,7-dihydroxy-3-(4- hydroxyphenyl)chromen-4-one. Genistein has a number of biological activities, including activation of PPARs, inhibition of several tyrosine kinases, inhibition of topoisomerase, antioxidative activity, activation of Nrf2 antioxidative response, activation of estrogen receptor beta, and inhibition of the mammalian hexose transporter GLUT2.
[0456] Etoposide is an anticancer agent that acts primarily as a topoisomerase II inhibitor. Etoposide forms a ternary complex with DNA and the topoisomerase II enzyme, prevents re-ligation of the DNA strands and thus induces DNA strand breakage and promotes apoptosis of the cancer cells.
[0457] Cytarabine is a nucleoside analog replacing the ribose with arabinose. It can be incorporated into DNA and also inhibits both DNA and RNA polymerases and nucleotide reductase. It is particularly useful in the treatment of acute myeloid leukemia and acute lymphocytic leukemia, but can be used for other malignancies and in various drug combinations.
[0458] Camptothecins include camptothecin, homocamptothecin, topotecan, irinotecan, DB 67, BNP 1350, exatecan, lurtotecan, ST 1481 , and CKD 602. These compounds act as topoisomerase I inhibitors and block DNA synthesis in cancer cells.
[0459] Vinca alkaloids include vinblastine, vincristine, vindesine, and vinorelbine.
[0460] Topoisomerase inhibitors include topoisomerase I inhibitors and topoisomerase II inhibitors. Topoisomerase I inhibitors include the camptothecins and lamellarin D. Topoisomerase II inhibitors include, in addition to amonafide and derivatives and analogs thereof, etoposide, teniposide, doxorubicin, daunorubicin, mitoxantrone, amsacrine, ellipticines, and aurintricarboxylic acid. A number of plant- derived naturally-occurring phenolic compounds, such as genistein, quercetin, and resveratrol, exhibit inhibitory activity toward both topoisomerase I and topoisomerase II.
[0461] The compound 5-fluorouracil is a base analog that acts as a thymidylate synthase inhibitor and thereby inhibits DNA synthesis. When deprived of a sufficient supply of thymidine, rapidly dividing cancer cells die by a process known as thymineless death.
[0462] NF-KB is a protein complex that controls transcription of DNA, cytokine production, and cell survival. NF-KB is involved in cellular responses to stimuli such as stress, cytokines, free radicals, heavy metals, ultraviolet radiation, oxidized low-density lipoprotein, and antigens of bacterial or viral antigens. NF-KB inhibitors include, but are not limited, to, bortezomib, denosumab, disulfiram, olmesartan, dithiocarbamates, (-)- DHMEQ, PBS-1086, IT-603, IT-901 , BAY-11-7082, palmitoylethanolamide, and iguratimod.
[0463] Curcumin is believed to have anti-neoplastic, anti-inflammatory, antioxidant, anti-ischemic, anti-arthritic, and anti-amyloid properties and also has hepatoprotective activity.
[0464] Rosmarinic acid is a naturally-occurring phenolic antioxidant that also has anti-inflammatory activity.
[0465] Avastin (bevacizumab) is a recombinant humanized monoclonal antibody that blocks angiogenesis by inhibiting vascular endothelial growth factor A (VEGF) and that is used to treat a number of malignancies, including colorectal cancer, lung cancer, breast cancer, renal cancers, ovarian cancer, and cervical cancer, as well as a number of non-malignant conditions such as age-related macular degeneration and diabetic retinopathy. Rituxan (rituximab) is a chimeric monoclonal antibody that binds to the B cell surface antigen CD20 and that is used to treat non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and a number of non-malignant conditions including rheumatoid arthritis, vasculitis, and pemphigus vulgaris. Herceptin (trastuzumab) is a monoclonal antibody targeting HER2 that induces an immune-mediated response that causes internalization and recycling of HER2 and may upregulate cell cycle inhibitors; it is used to treat breast cancer. Erbitux (cetuximab) is a chimeric monoclonal antibody that inhibits epidermal growth factor receptor (EGFR) and is used to treat squamous cell carcinoma of the head and neck.
[0466] PD-1 inhibitors include pembrolizumab, nivolumab, cemiplimab, JTX- 4014, spartalizumab, camrelizumab, sintilimab, tislelizumab, toripalimab, dostarlimab, MGA012, AMP-224, and AMP-514. PD-L1 inhibitors include atezolizumab, avelumab, durvalumab, KN035, AUNP12, CA-170, and BMS-986189. PL-1 and PDL-1 inhibitors are checkpoint inhibitors and can be used to treat malignancies by preventing the malignancy from evading the immune system.
[0467] Prednimustine is an alkylating agent that is an ester formed from prednisolone and chlorambucil and is used in the treatment of leukemias and lymphomas.
[0468] Braf inhibitors include vemurafenib, GDC-0879, PLX-4720, sorafenib, dabrafenib, and LGX818 and are used to treat metastatic melanoma.
[0469] BTK inhibitors include ibrutinib, acalabrutinib, zanubrutinib, tirabrutinib, tolebrutinib, evobrutinib, ABBV-105, fenebrutinib, pirtobrutinib, GS-4059, spebrutinib, and HM71224.
[0470] 5-azacytidine and decitabine are antimetabolites that are analogs of cytidine or 2'-deoxycitidine and are used in the treatment of myelodysplastic syndrome.
[0471] PARP inhibitors are described above; further details regarding PARP inhibitors are provided below.
[0472] Agents inducing hypomethylation include 5-azacytidine and decitabine, as well as pseudoisocytidine and 5-fluoro-2'-deoxycytidine. Histone deacetylase inhibitors include vorinostat and romidepsin. The use of histone deacetylase inhibitors is also described in United States Patent Application Publication No. 2011/0105474 by Thaler et al. These histone deacetylase inhibitors include, but are not limited to, (E)-N- hydroxy-3-{4-[(E)-3-(4-methyl-piperazin-1-yl)-3-oxo-propenyl]-phenyl}-acrylamide; (E)-N- hydroxy-3-{3-[(E)-3-(4-methyl-piperazin-1-yl)-3-oxo-propenyl]-phenyl}-acrylamide; (E)-N- hydroxy-3-{3-[(E)-3-oxo-3-(4-phenyl-piperazin-1-yl)-propenyl]-phenyl}-acrylamide; and (E)-3-[3-((E)-3-[1 ,4']bipiperidinyl-1 '-yl-3-oxo-propenyl)-phenyl]-N-hydroxy-acrylamide. Additional histone deacetylase inhibitors, including spirocyclic derivatives, are described in United States Patent Application Publication No. 2011/039840 by Varasi et al. Prodrugs of histone deacetylase inhibitors are described in United States Patent No. 8,227,636 to Miller et al. Histone deacetylase inhibitors are described in United States
Patent No. 8,222,451 to Kozikowski et al. Histone deacetylase inhibitors, including disubstituted aniline compounds, are also described in United States Patent No. 8,119,685 to Heidebrecht et al. Histone deacetylase inhibitors, including aryl-fused spirocyclic compounds, are also described in United States Patent No. 8,119,852 to Hamblett et al.
[0473] When the improvement is made by chemosensitization, the chemosensitization can be, but is not limited to, use as chemosensitizer with an additional agent selected from the group consisting of:
(a) fraudulent nucleosides;
(b) fraudulent nucleotides;
(c) thymidylate synthetase inhibitors;
(d) signal transduction inhibitors;
(e) cisplatin or gallium analogs;
(f) an alkylating agent such as BCNU, Gliadel wafers, CCNU, bendamustine (Treanda), or temozolomide (Temodar);
(g) anti-tubulin agents;
(h) antimetabolites;
(i) berberine;
(j) apigenin;
(k) amonafide;
(l) colchicine and analogs;
(m) genistein;
(n) etoposide;
(o) cytarabine;
(p) camptothecins;
(q) vinca alkaloids;
(r) topoisomerase inhibitors;
(s) 5-fluorouracil;
(t) curcumin;
(u) NF-KB inhibitors; and
(v) rosmarinic acid.
[0474] When the improvement is made by chemopotentiation, the chemopotentiation can be, but is not limited to, use as chemopotentiator with an additional agent selected from the group consisting of:
(a) fraudulent nucleosides;
(b) fraudulent nucleotides;
(c) thymidylate synthetase inhibitors;
(d) signal transduction inhibitors;
(e) cisplatin or gallium analogs;
(f) an alkylating agent such as BCNU , Gliadel wafers, CCNU , bendamustine (Treanda), or temozolomide (Temodar);
(g) anti-tubulin agents;
(h) antimetabolites;
(i) berberine;
(j) apigenin;
(k) amonafide;
(l) colchicine and analogs;
(m) genistein;
(n) etoposide;
(o) cytarabine;
(p) camptothecins;
(q) vinca alkaloids;
(r) topoisomerase inhibitors;
(s) 5-fluorouracil;
(t) curcumin;
(u) NF-KB inhibitors; and
(v) rosmarinic acid.
[0475] When the improvement is by post-treatment management, the post- treatment management can be, but is not limited to, a method selected from the group consisting of:
(a) use with therapies associated with pain management;
(b) nutritional support;
(c) administration of anti-emetics;
(d) anti-nausea therapies;
(e) anti-anemia therapy;
(f) administration of anti-inflammatories;
(g) administration of antipyretics;
(h) administration of immune stimulants;
(i) administration of anti-diarrhea medicines;
(j) administration of famotidine;
(k) administration of antihistamines;
(l) administration of suppository lubricants;
(m) administration of soothing agents;
(n) administration of lidocaine; and
(o) administration of hydrocortisone.
[0476] When the improvement is by alternative medicine/therapeutic support, the alternative medicine/therapeutic support can be, but is not limited to, an agent selected from the group consisting of:
(a) an herbal medication created either synthetically or through extraction that is a NF-KB inhibitor such as parthenolide, curcumin, or rosmarinic acid;
(b) an herbal medication created either synthetically or through extraction that is a natural anti-inflammatory such as rhein or parthenolide;
(c) an herbal medication created either synthetically or through extraction that is an immunostimulant such as those found in Echinacea;
(d) an herbal medication created either synthetically or through extraction that is an antimicrobial such as berberine; and
(e) an herbal medication created either synthetically or through extraction that is a flavonoid or flavone such as apigenin, genistein, apigenenin, genistin, 6"-O-malonylgenistin, 6"-O-acetylgenistin, daidzein, daidzin, 6"-O- malonyldaidzin, 6"-O-acetylgenistin, glycitein, glycitin, 6"-O-malonylglycitin, or 6-0- acetylglycitin.
[0477] When the improvement is made by a bulk drug product improvement, the bulk drug product improvement can be, but is not limited to, a bulk drug product improvement selected from the group consisting of:
(a) salt formation;
(b) homogeneous crystalline structure;
(c) pure isomers;
(d) increased purity;
(e) lower residual solvents; and
(f) lower residual heavy metals.
[0478] When the improvement is made by use of a diluent system, the diluent system can be, but is not limited to, a diluent system selected from the group consisting of:
(a) an emulsion;
(b) DMSO;
(c) NMF;
(d) DMF;
(e) DMA;
(f) ethanol;
(g) benzyl alcohol;
(h) dextrose-containing water for injection;
(i) Cremophor;
(j) cyclodextrins;
(k) PEG;
(l) a sweetening agent such as saccharin;
(m) glycerol; and
(n) a taste masking effector such as menthol, rum flavor, or fruit flavorings.
[0479] When the improvement is made by use of a solvent system, the diluent system can be, but is not limited to, a solvent system selected from the group consisting of:
(a) emulsions;
(b) dimethyl sulfoxide (DMSO);
(c) N-methylformamide (NMF);
(d) dimethylformamide (DMF);
(e) DMA;
(f) ethanol;
(g) benzyl alcohol;
(h) dextrose-containing water for injection;
(i) Cremophor;
(j) PEG;
(k) glycerol; and
(l) cocoa butter for suppositories.
[0480] When the improvement is made by the use of an excipient, the excipient can be, but is not limited to, an excipient selected from the group consisting of:
(a) mannitol;
(b) albumin;
(c) EDTA;
(d) sodium bisulfite;
(e) benzyl alcohol;
(f) carbonate buffers;
(g) phosphate buffers;
(h) glycerin;
(i) sweeteners;
(j) a taste masking agent;
(k) substituted celluloses; and
(l) sodium azide as a preservative.
[0481] When the improvement is made by the use of a dosage form, the excipient can be, but is not limited to, a dosage form selected from the group consisting of:
(a) tablets;
(b) capsules;
(c) topical gels;
(d) topical creams;
(e) patches;
(f) suppositories;
(g) lyophilized dosage fills,
(h) suppositories with quick release of <15 min or long melt times of >15 min release time; and
(i) temperature adjusted suppositories.
[0482] Formulation of pharmaceutical compositions in tablets, capsules, and topical gels, topical creams or suppositories is well known in the art and is described, for example, in United States Patent Application Publication No. 2004/0023290 by Griffin et al.
[0483] Formulation of pharmaceutical compositions as patches such as transdermal patches is well known in the art and is described, for example, in United States Patent No. 7,728,042 to Eros et al.
[0484] Lyophilized dosage fills are also well known in the art. One general method for the preparation of such lyophilized dosage fills, applicable to many therapeutic agents, comprises the following steps:
(1 ) Dissolve the drug in water for injection precooled to below 10° C. Dilute to final volume with cold water for injection to yield a 40 mg/mL solution.
(2) Filter the bulk solution through an 0.2-pm filter into a receiving container under aseptic conditions. The formulation and filtration should be completed in 1 hour.
(3) Fill nominal 1 .0 mL filtered solution into sterilized glass vials in a controlled target range under aseptic conditions.
(4) After the filling, all vials are placed with rubber stoppers inserted in the “lyophilization position” and loaded in the prechilled lyophilizer. For the lyophilizer, shelf temperature is set at +5° C and held for 1 hour; shelf temperature is then adjusted to -5° C and held for one hour, and the condenser, set to -60° C, turned on.
(5) The vials are then frozen to 30° C or below and held for no less than 3 hours, typically 4 hours.
(6) Vacuum is then turned on, the shelf temperature is adjusted to -5° C, and primary drying is performed for 8 hours; the shelf temperature is again adjusted to - 5° C and drying is carried out for at least 5 hours.
(7) Secondary drying is started after the condenser (set at -60° C) and vacuum are turned on. In secondary drying, the shelf temperature is controlled at +5° C for 1 to 3 hours, typically 1.5 hours, then at 25°C for 1 to 3 hours, typically 1.5 hours, and finally at 35-40° C for at least 5 hours, typically for 9 hours, or until the product is completely dried.
(8) Break the vacuum with filtered inert gas (e.g., nitrogen). Stopper the vials in the lyophilizer.
(9) Vials are removed from the lyophilizer chamber and sealed with aluminum flip-off seals. All vials are visually inspected and labeled with approved labels.
[0485] When the improvement is made by dosage kits and packaging, the dosage kits and packaging can be, but are not limited to, a dosage kit or packaging selected from the group consisting of:
(a) amber vials to protect from light;
(b) stoppers with specialized coatings to improve shelf-life stability;
(c) special dropper measuring devices;
(d) single-use or multiple-use container closure systems;
(e) suppository delivery devices; and
(f) dosage measuring devices.
[0486] When the improvement is made by drug delivery systems, the drug delivery system can be, but is not limited to, a drug delivery system selected from the group consisting of:
(a) nanocrystals;
(b) bioerodible polymers;
(c) liposomes;
(d) slow release injectable gels;
(e) microspheres;
(f) suspensions with glycerol;
(g) meltable drug release suppositories with a cocoa butter polymer alone or in combination with PEG;
(h) lecithin;
(i) polylactide/polyglycolide; and
(j) rectal plugs for drug delivery.
[0487] Nanocrystals are described in United States Patent No. 7,101 ,576 to Hovey et al.
[0488] Bioerodible polymers, also known as biodegradable polymers, are disclosed in N. Kamaly et al., “Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release,” Chem. Rev. 116: 2602-2663 (2016). Bioerodible polymers are also described in United States Patent No. 7,318,931 to Okumu et al. A bioerodible polymer decomposes when placed inside an organism, as measured by a decline in the molecular weight of the polymer over time. Polymer molecular weights can be determined by a variety of methods including size exclusion chromatography (SEC), and are generally expressed as weight averages or number averages. A polymer is bioerodible if, when in phosphate buffered saline (PBS) of pH 7.4 and a temperature of 37° C, its weight-average molecular weight is reduced by at least 25% over a period of 6 months as measured by SEC. Useful bioerodible polymers include polyesters, such as poly(caprolactone), poly(glycolic acid), poly(lactic acid), and poly(hydroxybutryate); polyanhydrides, such as poly(adipic anhydride) and poly(maleic anhydride); polydioxanone; polyamines; polyamides; polyurethanes; polyesteramides; polyorthoesters; polyacetals; polyketals; polycarbonates; polyorthocarbonates; polyphosphazenes; poly(malic acid); poly(amino acids); polyvinylpyrrolidone; poly(methyl vinyl ether); poly(alkylene oxalate); poly(alkylene succinate); polyhydroxycellulose; chitin; chitosan; and copolymers and mixtures thereof.
[0489] Liposomes are well known as drug delivery vehicles. Liposome preparation is described in European Patent Application Publication No. EP 1332755 by Weng et al. Liposomes can incorporate short oligopeptide sequences capable of targeting the EGFR receptor, as described in United States Patent Application Publication 2012/0213844 by Huang et al. Alternatively, liposomes can include nuclear
localization signal/fusogenic peptide conjugates and form targeted liposome complexes, as described in United States Patent Application Publication 2012/0183596 to Boulikas.
[0490] The use of microspheres for drug delivery is known in the art and is 82described, for example, in H. Okada & H. Taguchi, “Biodegradable Microspheres in Drug Delivery,” Crit. Rev. Ther. Drug Carrier Sys. 12: 1-99 (1995).
[0491] When the improvement is made by drug conjugate forms, the drug conjugate form can be, but is not limited to, a drug conjugate form selected from the group consisting of:
(a) polyethylene glycols;
(b) polylactides;
(c) polyglycolides;
(d) amino acids;
(e) peptides; and
(f) multivalent linkers.
[0492] Polylactide conjugates are well known in the art and are described, for example, in R. Tong & C. Cheng, “Controlled Synthesis of Camptothecin-Polylactide Conjugates and Nanoconjugates,” Bioconjugate Chem. 21 : 111-121 (2010).
[0493] Polyglycolide conjugates are also well known in the art and are described, for example, in PCT Patent Application Publication No. WO 2003/070823 by Elmaleh et al.
[0494] Multivalent linkers are known in the art and are described, for example, in United States Patent Application Publication No. 2007/0207952 by Silva et al. For example, multivalent linkers can contain a thiophilic group for reaction with a reactive cysteine, and multiple nucleophilic groups (such as NH2 or OH) or electrophilic groups (such as activated esters) that permit attachment of a plurality of biologically active moieties to the linker.
[0495] Suitable reagents for cross-linking many combinations of functional groups are known in the art. For example, electrophilic groups can react with many functional groups, including those present in proteins or polypeptides. Various combinations of reactive amino acids and electrophiles are known in the art and can be used. For example, N-terminal cysteines, containing thiol groups, can be reacted with
halogens or maleimides. Thiol groups are known to have reactivity with a large number of coupling agents, such as alkyl halides, haloacetyl derivatives, maleimides, aziridines, acryloyl derivatives, arylating agents such as aryl halides, and others. These are described in G. T. Hermanson, “Bioconjugate Techniques” (Academic Press, San Diego, 1996), pp. 146-150. The reactivity of the cysteine residues can be optimized by appropriate selection of the neighboring amino acid residues. For example, a histidine residue adjacent to the cysteine residue will increase the reactivity of the cysteine residue. Other combinations of reactive amino acids and electrophilic reagents are known in the art. For example, maleimides can react with amino groups, such as the ε- amino group of the side chain of lysine, particularly at higher pH ranges. Aryl halides can also react with such amino groups. Haloacetyl derivatives can react with the imidazolyl side chain nitrogens of histidine, the thioether group of the side chain of methionine, and the ε-amino group of the side chain of lysine. Many other electrophilic reagents are known that will react with the ε-amino group of the side chain of lysine, including, but not limited to, isothiocyanates, isocyanates, acyl azides, N- hydroxysuccinimide esters, sulfonyl chlorides, epoxides, oxiranes, carbonates, imidoesters, carbodiimides, and anhydrides. These are described in G.T. Hermanson, “Bioconjugate Techniques” (Academic Press, San Diego, 1996), pp. 137-146.
Additionally, electrophilic reagents are known that will react with carboxylate side chains such as those of aspartate and glutamate, such as diazoalkanes and diazoacetyl compounds, carbonydilmidazole, and carbodiimides. These are described in G. T. Hermanson, "Bioconjugate Techniques" (Academic Press, San Diego, 1996), pp. 152- 154. Furthermore, electrophilic reagents are known that will react with hydroxyl groups such as those in the side chains of serine and threonine, including reactive haloalkane derivatives. These are described in G. T. Hermanson, “Bioconjugate Techniques” (Academic Press, San Diego, 1996), pp. 154-158. In another alternative embodiment, the relative positions of electrophile and nucleophile (i.e. , a molecule reactive with an electrophile) are reversed so that the protein has an amino acid residue with an electrophilic group that is reactive with a nucleophile and the targeting molecule includes therein a nucleophilic group. This includes the reaction of aldehydes (the electrophile) with hydroxylamine (the nucleophile), described above, but is more general
than that reaction; other groups can be used as electrophile and nucleophile. Suitable groups are well known in organic chemistry and need not be described further in detail.
[0496] Additional combinations of reactive groups for cross-linking are known in the art. For example, amino groups can be reacted with isothiocyanates, isocyanates, acyl azides, N-hydroxysuccinimide (NHS) esters, sulfonyl chlorides, aldehydes, glyoxals, epoxides, oxiranes, carbonates, alkylating agents, imidoesters, carbodiimides, and anhydrides. Thiol groups can be reacted with haloacetyl or alkyl halide derivatives, maleimides, aziridines, acryloyl derivatives, acylating agents, or other thiol groups by way of oxidation and the formation of mixed disulfides. Carboxy groups can be reacted with diazoalkanes, diazoacetyl compounds, carbonyldiimidazole, carbodiimides. Hydroxyl groups can be reacted with epoxides, oxiranes, carbonyldiimidazole, N,N'- disuccinimidyl carbonate, N-hydroxysuccinimidyl chloroformate, periodate (for oxidation), alkyl halogens, or isocyanates. Aldehyde and ketone groups can react with hydrazines, reagents forming Schiff bases, and other groups in reductive amination reactions or Mannich condensation reactions. Still other reactions suitable for cross- linking reactions are known in the art. Such cross-linking reagents and reactions are described in G.T. Hermanson, “Bioconjugate Techniques” (Academic Press, San Diego, 1996).
[0497] When the improvement is made by use of a compound analog, the compound analog can be, but is not limited to, a compound analog selected from the group consisting of:
(a) alteration of side chains to increase or decrease lipophilicity;
(b) additional chemical functionalities to alter reactivity;
(c) alteration of electron affinity;
(d) alteration of binding capacity; and
(e) salt forms.
[0498] When the improvement is made by use of a prodrug system, the prodrug system can be, but is not limited to, a prodrug system selected from the group consisting of:
(a) enzyme sensitive esters;
(b) dimers;
(c) Schiff bases;
(d) pyridoxal complexes;
(e) caffeine complexes; and
(f) bioreductive analogs as prodrugs including nitroso-substituted analogs.
[0499] As used herein, the term “prodrug” refers to compounds that are transformed in vivo to yield a disclosed compound or a pharmaceutically acceptable form of the compound. In some embodiments, a prodrug is a compound that may be converted under physiological conditions or by solvolysis to a biologically active compound as described herein. Thus, the term “prodrug” refers to a precursor of a biologically active compound that is pharmaceutically acceptable. A prodrug can be inactive when administered to a subject, but is then converted in vivo to an active compound, for example, by hydrolysis (e.g., hydrolysis in blood or a tissue). In certain cases, a prodrug has improved physical and/or delivery properties over a parent compound from which the prodrug has been derived. The prodrug often offers advantages of solubility, tissue compatibility, or delayed release in a mammalian organism (H. Bundgard, Design of Prodrugs (Elsevier, Amsterdam, 1988), pp. 7-9, 21- 24), incorporated herein by this reference. A discussion of prodrugs is provided in T. Higuchi et al., “Pro-Drugs as Novel Delivery Systems,” ACS Symposium Series, Vol. 14 and in E.B. Roche, ed., Bioreversible Carriers in Drug Design (American Pharmaceutical Association & Pergamon Press, 1987). Exemplary advantages of a prodrug can include, but are not limited to, its physical properties, such as enhanced water solubility for parenteral administration at physiological pH compared to the parent compound, enhanced absorption from the digestive tract, or enhanced drug stability for long-term storage.
[0500] The term “prodrug” is also meant to include any covalently bonded carriers which release the active compound in vivo when the prodrug is administered to a subject. Prodrugs of a therapeutically active compound, as described herein, can be prepared by modifying one or more functional groups present in the therapeutically active compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to yield the parent therapeutically active compound. Prodrugs
include compounds wherein a hydroxy, amino, or mercapto group is covalently bonded to any group that, when the prodrug of the active compound is administered to a subject, cleaves to form a free hydroxy, free amino, or free mercapto group, respectively. Examples of prodrugs include, but are not limited to, formate or benzoate derivatives of an alcohol or acetamide, formamide or benzamide derivatives of a therapeutically active agent possessing an amine functional group available for reaction, and the like.
[0501] For example, if a therapeutically active agent or a pharmaceutically acceptable form of a therapeutically active agent contains a carboxylic acid functional group, a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the carboxylic acid group with a group such as C1-8 alkyl, C2-12 alkanoyloxymethyl, 1 -(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1 -methyl-1 - (alkanoyloxy)ethyl having from 5 to 10 carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1 -(alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1 -methyl-1 -(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N- (alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1 -(N- (alkoxycarbonyl)amino)ethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4- crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N(C1-C2)alkylamino(C2-C3)alkyl (such as (3-dimethylaminoethyl), carbamoyl-(C1-C2)alkyl, N, N-di (C1-C2)alkylcarbamoyl-(C1- C2)alkyl and piperidino-, pyrrolidino-, or morpholino(C2-C3)alkyl.
[0502] Similarly, if a disclosed compound or a pharmaceutically acceptable form of the compound contains an alcohol functional group, a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as (C1- C6)alkanoyloxymethyl, 1 -((C1-C6))alkanoyloxy)ethyl, 1-methyl-1 -((C1- C6)alkanoyloxy)ethyl (C1- C6)alkoxycarbonyloxymethyl, N(C1- C6)alkoxycarbonylaminomethyl, succinoyl, (C1-C6)alkanoyl, α-amino(C1-C4)alkanoyl, arylacyl and a-aminoacyl, or a-aminoacyl-a-aminoacyl, where each a-aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)(OH)2, P(O)(O(C1-C6)alkyl)2 or glycosyl (the radical resulting from the removal of a hydroxyl group of the hemiacetal form of a carbohydrate).
[0503] If a disclosed compound or a pharmaceutically acceptable form of the compound incorporates an amine functional group, a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as R-carbonyl, RO-carbonyl, NRR'-carbonyl where R and R' are each independently (C1-C10)alkyl, (C3- C7)cycloalkyl, benzyl, or R-carbonyl is a natural a-aminoacyl or natural a-aminoacyl- natural a-aminoacyl, C(OH)C(O)OY1 wherein Y1 is H, (C1-C6)alkyl or benzyl, C(OY2)Y3 wherein Y2 is (C1-C4) alkyl and Y3 is (C1-C6)alkyl, carboxy(C1-C6)alkyl, amino(C1-C4)alkyl or mono-N or di-N,N(C1-C6)alkylaminoalkyl, C(Y4)Y5 wherein Y4 is H or methyl and Y5 is mono-N or di-N,N(C1-C6)alkylamino, morpholino, piperidin-1 -yl or pyrrolidin-1 -yl.
[0504] The use of prodrug systems is described in T. Jarvinen et al., “Design and Pharmaceutical Applications of Prodrugs” in Drug Discovery Handbook (S.C. Gad, ed., Wiley-lnterscience, Hoboken, NJ, 2005), ch. 17, pp. 733-796. This publication describes the use of enzyme sensitive esters as prodrugs. The use of dimers as prodrugs is described in United States Patent No. 7,879,896 to Allegretti et al. The use of peptides in prodrugs is described in S. Prasad et al., “Delivering Multiple Anticancer Peptides as a Single Prodrug Using Lysyl-Lysine as a Facile Linker,” J. Peptide Sci. 13: 458-467 (2007). The use of Schiff bases as prodrugs is described in United States Patent No. 7,619,005 to Epstein et al. The use of caffeine complexes as prodrugs is described in United States Patent No. 6,443,898 to Unger et al. The use of nitric oxide- releasing prodrugs is described in N. Nath et al., “JS-K, a Nitric Oxide-Releasing Prodrug, Modulates [3-Catenin/TCF Signaling in Leukemic Jurkat Cells: Evidence of an S-Nitrosylated Mechanism,” Biochem. Pharmacol. 80: 1641 -1649 (2010). The use of prodrugs that are subject to redox activation is described in S.H. van Rijt & P.J. Sadler, “Current Applications and Future Potential for Bioinorganic Chemistry in the Development of Anticancer Drugs,” Drug Discov. Today 14: 1089-1097 (2009).
[0505] When the improvement is made by use of a multiple drug system, the multiple drug system can be, but is not limited to, a multiple drug system selected from the group consisting of:
(a) inhibitors of multi-drug resistance;
(b) specific drug resistance inhibitors;
(c) specific inhibitors of selective enzymes;
(d) signal transduction inhibitors;
(e) repair inhibition; and
(f) topoisomerase inhibitors with non-overlapping side effects.
[0506] Multi-drug resistance inhibitors are described in United States Patent No. 6,011 ,069 to Inomata et al.
[0507] Specific drug resistance inhibitors are described in T. Hideshima et al., “The Proteasome Inhibitor PS-341 Inhibits Growth, Induces Apoptosis, and Overcomes Drug Resistance in Human Multiple Myeloma Cells,” Cancer Res. 61 : 3071-3076 (2001 ).
[0508] Selective inhibitors of specific enzymes are described in D. Leung et al., “Discovering Potent and Selective Reversible Inhibitors of Enzymes in Complex Proteomes,” Nature Biotechnol. 21 : 687-691 (2003).
[0509] Repair inhibition is described in N.M. Martin, “DNA Repair Inhibition and Cancer Therapy,” J. Photochem. Photobiol. B 63: 162-170 (2001 ).
[0510] When the improvement is made by biotherapeutic enhancement, the biotherapeutic enhancement can be, but is not limited to, use as a sensitizer or potentiator with a biological response modifier selected from the group consisting of:
(a) cytokines;
(b) lymphokines;
(c) therapeutic antibodies such as bevacizumab, trastuzumab, rituximab, and cetuximab;
(d) gene therapies;
(e) ribozymes;
(f) RNA interference, and
(g) cell based therapeutics.
[0511] When the improvement is made by biotherapeutic resistance modulation, the biotherapeutic resistance modulation can be used to modulate biotherapeutic resistance that can develop to a biotherapeutic agent that can be, but is not limited to, a biotherapeutic agent selected from the group consisting of:
(a) cytokines;
(b) lymphokines;
(c) therapeutic antibodies such as bevacizumab, trastuzumab, rituximab, and cetuximab;
(d) gene therapies;
(e) ribozymes; and
(f) RNA interference.
[0512] Antisense therapies are described, for example, in B. Weiss et al., “Antisense RNA Gene Therapy for Studying and Modulating Biological Processes,” Cell. Mol. Life Sci. 55: 334-358 (1999).
[0513] Ribozymes are described, for example, in S. Pascolo, “RNA-Based Therapies” in Drug Discovery Handbook (S.C. Gad, ed., Wiley-lnterscience, Hoboken, NJ, 2005), ch.27, pp. 1273-1278.
[0514] RNA interference is described, for example, in S. Pascolo, “RNA-Based Therapies” in Drug Discovery Handbook (S.C. Gad, ed., Wiley-lnterscience, Hoboken, NJ, 2005), ch.27, pp. 1278-1283.
[0515] When the improvement is made by radiation therapy enhancement, the radiation therapy enhancement can be used in combination with a method or agent for radiation therapy that can be, but is not limited to, a method or agent for radiation therapy selected from the group consisting of:
(a) hypoxic cell sensitizers;
(b) radiation sensitizers/protectors;
(c) photosensitizers;
(d) radiation repair inhibitors;
(e) thiol depletion;
(f) vaso-targeted agents;
(g) use with radioactive seeds;
(h) use with radionuclides;
(i) use with radiolabeled antibodies; and
(j) use with brachytherapy.
[0516] Hypoxic cell sensitizers are described in C.C. Ling et al., “The Effect of Hypoxic Cell Sensitizers at Different Irradiation Dose Rates,” Radiation Res. 109: 396- 406 (1987). Radiation sensitizers are described in T.S. Lawrence, “Radiation
Sensitizers and Targeted Therapies,” Oncology 17 (Suppl. 13) 23-28 (2003). Radiation protectors are described in S.B. Vuyyuri et al., “Evaluation of D-Methionine as a Novel Oral Radiation Protector for Prevention of Mucositis,” Clin. Cancer Res. 14: 2161-2170 (2008). Photosensitizers are described in R.R. Allison & C.H. Sibata, “Oncologic Photodynamic Therapy Photosensitizers: A Clinical Review,” Photodiagnosis Photodynamic Ther. 7: 61-75 (2010). Radiation repair inhibitors and DNA repair inhibitors are described in M. Hingorani et al., “Evaluation of Repair of Radiation- Induced DNA Damage Enhances Expression from Replication-Defective Adenoviral Vectors,” Cancer Res. 68: 9771-9778 (2008). Thiol depleters are described in K.D. Held et al., “Postirradiation Sensitization of Mammalian Cells by the Thiol-Depleting Agent Dimethyl Fumarate,” Radiation Res. 127: 75-80 (1991 ). Vaso-targeted agents are described in A.L. Seynhaeve et al., “Tumor Necrosis Factor a Mediates Homogeneous Distribution of Liposomes in Murine Melanoma that Contributes to a Better Tumor Response,” Cancer Res. 67: 9455-9462 (2007).
[0517] When the improvement is made by use in connection with a novel mechanism of action, the novel mechanism of action can be, but is not limited to, a novel mechanism of action selected from the group consisting of:
(a) inhibitors of poly-ADP ribose polymerase;
(b) agents that effect vasculature;
(c) vasodilation;
(d) oncogenic targeted agents;
(e) signal transduction inhibitors;
(f) EGFR inhibition;
(g) Protein Kinase C inhibition;
(h) Phospholipase C downregulation;
(i) jun downregulation;
(j) histone genes;
(k) VEGF:
(l) ornithine decarboxylase,
(m) jun D;
(n) v-jun;
(o) GPCR’s
(p) protein kinase A;
(q) telomerase;
(r) prostate specific genes;
(s) protein kinases; and
(t) histone deacetylase.
[0518] EGFR inhibition is described in G. Giaccone & J. A. Rodriguez, “EGFR Inhibitors: What Have We Learned from the Treatment of Lung Cancer,” Nat. Clin. Pract. Oncol. 11 : 554-561 (2005). Protein kinase C inhibition is described in H.C. Swannie & S.B. Kaye, “Protein Kinase C Inhibitors,” Curr. Oncol. Rep. 4: 37-46 (2002). Phospholipase C downregulation is described in A.M. Martelli et al., “Phosphoinositide Signaling in Nuclei of Friend Cells: Phospholipase C β Downregulation Is Related to Cell Differentiation,” Cancer Res. 54: 2536-2540 (1994). Downregulation of Jun (specifically, c-Jun) is described in A. A. P. Zada et al., “Downregulation of c-Jun Expression and Cell Cycle Regulatory Molecules in Acute Myeloid Leukemia Cells Upon CD44 Ligation,” Oncogene 22: 2296-2308 (2003). The role of histone genes as a target for therapeutic intervention is described in B. Calabretta et al., “Altered Expression of G1 -Specific Genes in Human Malignant Myeloid Cells,” Proc. Natl. Acad. Sci. USA 83: 1495-1498 (1986). The role of VEGF as a target for therapeutic intervention is described in A. Zielke et al., “VEGF-Mediated Angiogenesis of Human Pheochromocytomas Is Associated to Malignancy and Inhibited by anti-VEGF Antibodies in Experimental Tumors,” Surgery 132: 1056-1063 (2002). The role of ornithine decarboxylase as a target for therapeutic intervention is described in J. A. Nilsson et al., “Targeting Ornithine Decarboxylase in Myc-lnduced Lymphomagenesis Prevents Tumor Formation,” Cancer Cell 7: 433-444 (2005). The role of ubiquitin C as a target for therapeutic intervention is described in C. Aghajanian et al., “A Phase I Trial of the Novel Proteasome Inhibitor PS341 in Advanced Solid Tumor Malignancies,” Clin. Cancer Res. 8: 2505-2511 (2002). The role of Jun D as a target for therapeutic intervention is described in M.M. Caffarel et al., “JunD Is Involved in the Antiproliferative Effect of A9-Tetrahydrocannibinol on Human Breast Cancer Cells,” Oncogene 27: 5033- 5044 (2008). The role of v-Jun as a target for therapeutic intervention is described in M.
Gao et al., “Differential and Antagonistic Effects of v-Jun and c-Jun,” Cancer Res. 56: 4229-4235 (1996). The role of protein kinase A as a target for therapeutic intervention is described in P.C. Gordge et al., “Elevation of Protein Kinase A and Protein Kinase C in Malignant as Compared With Normal Breast Tissue,” Eur. J. Cancer 12: 2120-2126 (1996). The role of telomerase as a target for therapeutic intervention is described in E.K. Parkinson et al., “Telomerase as a Novel and Potentially Selective Target for Cancer Chemotherapy,” Ann. Med. 35: 466-475 (2003). The role of histone deacetylase as a target for therapeutic intervention is described in A. Melnick & J.D. Licht, “Histone Deacetylases as Therapeutic Targets in Hematologic Malignancies,” Curr. Opin. Hematol. 9: 322-332 (2002).
[0519] When the improvement is made by selective target cell therapeutics, the improvement can be, but is not limited to, use against a specific type or class of cells selected from the group consisting of:
(a) radiation sensitive cells;
(b) radiation resistant cells;
(c) energy depleted cells; and
(d) endothelial cells.
[0520] When the improvement is made by use to reverse resistance to platinum- containing anti-neoplastic agents or PARP inhibitor anti-neoplastic agents, the improvement can be, but is not limited to, administration of a therapeutically effective quantity of elesclomol or a derivative or analog of elesclomol to reverse resistance to either platinum-containing anti-neoplastic agents or PARP inhibitor anti-neoplastic agents in malignant cells. Typically, the platinum-containing anti-neoplastic agent is selected from the group consisting of cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, and tetraplatin. Typically, the PARP inhibitor is selected from the group consisting of iniparib, talazoparib, olaparib, rucaparib, veliparib, CEP- 9722 (a prodrug of CEP-8983 (11-methoxy-4,5,6,7-tetrahydro-1 H-cyclopenta[a]pyrrolo[3,4- c]carbazole-1 ,3(2H)-dione), MK 4827 ((S)-2-(4-(piperidin-3-yl)phenyl)-2H-indazole-7- carboxamide), and BGB-290. Other PARP inhibitors are described herein.
[0521] When the improvement is made by use to induce synthetic lethality, the improvement can be, but is not limited to, administration of a therapeutically effective
quantity of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1 A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol renders the malignant cells susceptible to a targeted agent that otherwise would have no effect. In particular, an onco-driving mutation that allows for tumor formation, such as a mutation causing a defect in DNA damage repair, promotion of an open chromosome, or enhanced transcription, can also create an “Achilles heel” for the same tumor cells by its very existence. In the case of elesclomol, the mutation in ARID1 A leads to tumor formation, but also leads to a dependence on OXPHOS — the Achilles heel — as such tumor cells are uniquely susceptible to mitochondrial targeting agents, such as, but not necessarily limited to, elesclomol or a derivative or analog of elesclomol, which targets ferredoxin-1 , because of the defect that allows the tumor to form in the first place. If the tumor- causing mutation had not formed, such mitochondrial-targeting agents would have had minimal or no activity. A similar mechanism applies when the tumor cells have a mutation in a component of the SWI/SNF complex that increases the dependence of the tumor cells on OXPHOS.
[0522] The general mechanism of action for the use of elesclomol to induce synthetic lethality is shown in Figures 21 and 22. Figure 21 shows that loss-of-function mutations in ARID1A both promote oncogenesis and increase reliance on OXPHOS for tumor cells. The loss-of-function mutation significantly enhances DNA replication, diminishes the capacity for DNA repair, and causes increased mitotic activity and cell division, leading to malignant transformation. However, the loss-of function mutation also causes greater cellular energy dependence on OXPHOS as compared with glycolysis. Figure 22 shows the binding of elesclomol (“EO3001”) to ferredoxin, inhibiting Fe-S cluster biogenesis and interrupting OXPHOS, which leads to the generation of reactive oxygen species (ROS), which, in turn, mediated by wild-type p53, leads to apoptosis.
[0523] Synthetic lethality was originally described as a situation in which mutations may occur as a result of mutations in two genes in which the mutation of only one of the genes is not lethal for the cell, whereas the mutations of both of the genes is lethal to the cell. The concept has since been extended to situations in which the
combination of a mutation and the action of chemical compound, such as a small molecule drug, causes lethality, while the occurrence of only the mutation or the action of the chemical compound is not lethal to the cell. This latter alternative mechanism of synthetic lethality is the mechanism involved here. Synthetic lethality is described in S.M.B. Nijman, “Synthetic Lethality: General Principles, Utility and Detection Using Genetic Screens in Human Cells,” FEBS Lett. 585: 1-6 (2011 ).
[0524] When the improvement is made by use to target ferredoxin-1 to inhibit OXPHOS, the improvement can be, but is not limited to, administration of a therapeutically effective quantity of elesclomol or a derivative or analog of elesclomol to target ferredoxin-1 (FDX1) in order to inhibit oxidative phosphorylation (OXPHOS) in circumstances in which inhibition of oxidative phosphorylation would be desirable. Circumstances in which inhibition of oxidative phosphorylation would be desirable include, but are not limited to, treatment of malignancies such as melanomas (M. Pollak, “Targeting Oxidative Phosphorylation: Why, When, and How,” Cancer Cell 23: 263-264 (2013)), leukemias, lymphomas, pancreatic ductal carcinoma, and endometrial carcinoma (T.M. Ashton et al., “Oxidative Phosphorylation as an Emerging Target in Cancer Therapy,” J. Clin. Cancer Res. 24: 2482-2490 (2018)).
[0525] Another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising the steps of:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of a microtubulin stabilizer or a microtubulin inhibitor.
[0526] In this method, the microtubulin stabilizer is typically paclitaxel or an analog of paclitaxel. Alternatively, the microtubulin stabilizer can be docetaxel.
[0527] In this method, the microtubulin inhibitor is typically a vinca alkaloid selected from the group consisting of vincristine, vinblastine, vinorelbine, vinflunine, and vindesine.
[0528] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of a PARP inhibitor.
[0529] Suitable PARP inhibitors are described above. Typically, the PARP inhibitor is in iparib, talazoparib, olaparib, rucaparib, or vel iparib.
[0530] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of a LDH inhibitor.
[0531] Suitable LDH inhibitors are described above. Typically, the LDH inhibitor is oxamate.
[0532] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of 2- deoxyglucose or an analog or derivative thereof.
[0533] Typically, the 2-deoxyglucose or the analog or derivative thereof is 2- deoxyglucose itself.
[0534] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of a glutamine metabolism inhibitor.
[0535] Suitable glutamine metabolism inhibitors are described above. Typically, the glutamine metabolism inhibitor is phenylbutyrate.
[0536] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of a DNA- damaging anti-neoplastic agent.
[0537] Suitable DNA-damaging anti-neoplastic agents are as described above. Typically, the DNA-damaging anti-neoplastic agent is selected from the group consisting of: (1 ) an agent that damages DNA directly; (2) an agent that interferes with DNA synthesis; and (3) an agent that inhibits a topoisomerase. An agent that damages DNA can act by a mechanism selected from the group consisting of: (a) direct modification of DNA bases, (b) intercalation between DNA bases, and (c) formation of crosslinks in DNA. An agent that interferes with DNA synthesis can be an antimetabolite or inhibit an enzyme required for DNA synthesis.
[0538] Typically, the DNA-damaging anti-neoplastic agent is selected from the group consisting of cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, tetraplatin, doxorubicin, daunorubicin, methotrexate, 5-fluorouracil, gemcitabine, podophyllotoxin, etoposide, teniposide, cyclophosphamide, chlorambucil, melphalan, carmustine, lomustine, estramustine, semustine, bendamustine, prednamustine,
uramustine, chlornaphazine, dacarbazine, altretamine, temozolomide, mitomycin C, streptozotocin, chlorozotocin, capecitabine, floxuridine, 6-mercaptopurine, 8- azaguanine, azathiopurine, 5-ethynyluracil, thioguanine, fludarabine, cytarabine, cladribine, 2-fluoro-arabinosyl-adenine, aminopterin, pemetrexed, ralitrexed, camptothecin, epirubicin, idarubicin, methylnitronitrosoguanidine, topotecan, irinotecan, mechlorethamine, ifosfamide, trofosfamide, busulfan, procarbazine, mitoxantrone, actinomycin, calicheamicin, Tegafur (R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil), 2', 2'- difluoro-2'-deoxycytidine, bischloroethylsulfide, thiotepa, aziridinylbenzoquinone, BCNU , CCNU , 4-methyl CCNU , ACNU , rebeccamycin, bleomycin, pepleomycin, ethylmethanesulfonate, methylmethanesulfonate, dimethylnitrosamine, dimethyl sulfate, and N'-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1 ,3- propanediamine dihydrochloride.
[0539] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of an agent that inhibits the SWI/SNF complex.
[0540] Typically, the agent that inhibits the SWI/SNF complex is BD98.
[0541] Still another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of an agent that causes tumor cells to rely on oxidative phosphorylation.
[0542] The agent that causes tumor cells to rely on oxidative phosphorylation can be, but is not limited to: (i) an inhibitor of GLUT 1 ; (ii) an inhibitor of hexokinase; (iii)
an inhibitor of phosphofructokinase 1 ; (iv) an inhibitor of pyruvate decarboxylase kinase; and (v) an inhibitor of lactate dehydrogenase.
[0543] Still another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of an agent that inhibits the base excision repair pathway.
[0544] The inhibitor of the base excision repair pathway can be, but is not limited to: (i) a PARP inhibitor; (ii) an inhibitor of the Ape1 enzyme; (iii) an inhibitor of Pol ; (iv) an AP endonuclease inhibitor; (v) a DNA glycosylase inhibitor; (vi) a DNA alkyltransferase inhibitor; or (vii) a DNA ligase inhibitor.
[0545] Still another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of an agent that inhibits the homologous repair pathway.
[0546] Typically, the agent that inhibits the homologous repair pathway is imatinib mesylate or erlotinib.
[0547] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of an agent that activates homologous repair as part of its mechanism of antineoplastic activity or as a consequence of inducing DNA damage.
[0548] Typically, the agent that activates homologous repair as part of its mechanism of antineoplastic activity or as a consequence of inducing DNA damage is selected from the group consisting of carboplatin, cisplatin, dianhydrogalactitol, and dibromodulcitol.
[0549] Still another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administration of a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administration of a therapeutically effective quantity of an agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation.
[0550] Typically, the agent that is activated by bioreductases under acute conditions of hypoxia is selected from the group consisting of tirapazamine and mitomycin C.
[0551] Typically, the agent that functions to sensitize hypoxic cells to antineoplastic agents or radiation is selected from the group consisting of misonidazole, metronidazole, nimorazole, benznidazole, desmethylmisonidazole, etanidazole, pimonidazole, and 1-(aziridin-1-yl)-3-(2-nitroimidazol-1-yl)propan-2-ol (RSU-1069).
[0552] Yet another aspect of the claimed invention is a method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol as described above comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that inhibits cysteine uptake.
[0553] Agents that inhibit cysteine uptake are described above and include L- glutamate, L-aspartate, threo-β-hydroxyaspartate, dihydrokainate, threo-p- benzyloxyaspartate, peptides derived from digestion of human p-casein, bovine p- casein, and gliadin, erastin, analogs of erastin, sorafenib, and sulfasalazine.
[0554] The methods described above in which an additional agent is administered can be used to treat a malignancy. Typically, the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma. The methods can alternatively be used to treat other malignancies as described above.
[0555] In other alternatives, particularly in alternatives in which the additional agent has therapeutic activities other than anti-neoplastic activity when administered as a single agent, the methods described above can be used to treat diseases and conditions other than malignancies as described above.
[0556] Still another aspect of the present invention is a composition to improve the efficacy or reduce the side effects of treatment with elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol wherein the composition comprises:
(a) an alternative selected from the group consisting of:
(i) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol;
(ii) two or more therapeutically active ingredients comprising:
(A) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol; and
(B) at least one additional therapeutic agent, therapeutic agent subject to chemosensitization, therapeutic agent subject to chemopotentiation, or component of a multiple drug system;
(iii) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a dosage form;
(iv) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a dosage kit and packaging;
(v) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is subjected to a bulk drug product improvement;
(vi) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug delivery system; and
(vii) a therapeutically effective quantity of a prodrug of elesclomol or a derivative or analog of elesclomol; and
(b) at least one pharmaceutically acceptable diluent, solvent or excipient.
[0557] Typically, the elesclomol, the derivative or analog of elesclomol, or the salt, solvate, or prodrug of elesclomol is elesclomol itself as described above. Suitable derivatives and analogs of elesclomol are described above.
[0558] Typically, the composition is formulated for treatment of a malignancy. Malignancies treatable by administration of compositions are described above. In another alternative, the composition is formulated for a treatment of a disease or condition selected from the group consisting of: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; and scleroderma as described above.
[0559] When the composition is formulated for treatment of a malignancy, and wherein the elesclomol or the derivative, analog, salt, solvate or prodrug of elesclomol is elesclomol itself, typically, the composition is formulated for administration of a therapeutically effective quantity of elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 mg/mm2/day to about 10 g/mm2/day. More
typically, the therapeutically effective quantity of elesclomol is from about 2 mg/mm2/day to about 10 g/mm2/day. In another alternative, the therapeutically effective quantity of elesclomol is from about 1 pg/kg to about 500 mg/kg. In this alternative, typically, the therapeutically effective quantity of elesclomol is from about 500 pg/kg to about 250 mg/kg. In this alternative, preferably, the therapeutically effective quantity of elesclomol is from about 1 mg/kg to about 100 mg/kg. In this alternative, more preferably, the therapeutically effective quantity of elesclomol is from about 10 mg/kg to about 50 mg/kg.
[0560] Suitable additional therapeutic agents, therapeutic agents subject to chemosensitization, or therapeutic agents subject to chemopotentiation are also described above. These additional therapeutic agents, therapeutic agents subject to chemosensitization, and therapeutic agents subject to chemopotentiation, in order to be incorporated in a single pharmaceutical composition with the elesclomol or the derivative or analog of elesclomol, do not interact negatively with the elesclomol or the derivative or analog of elesclomol in such a manner that the therapeutic activity or the bioavailability of either agent is significantly reduced.
[0561] Suitable dosage forms, dosage kits and packaging, bulk drug product improvements, and drug delivery systems are as stated above.
[0562] In compositions according to the present invention, diluents, solvents, or excipients can include, in addition to components described above, components generally described as pharmaceutically acceptable carriers. Such pharmaceutically acceptable carriers can include, but are not limited to, phosphate buffered saline solution, water, emulsions, such as oil/water or water/oil emulsions), and various types of wetting agents, any and all solvents, dispersion media, coatings, sodium lauryl sulfate, isotonic and absorption delaying agents, disintegrants such as potato starch or sodium starch glycolate), and the like, depending on the physical form of the pharmaceutical composition. The carriers also can include stabilizers and preservatives. Still other pharmaceutical excipients and carriers are known in the art, and include, but are not limited to: preservatives; sweetening agents for oral administration; thickening agents; buffers; liquid carriers; wetting, solubilizing, or emulsifying agents; acidifying agents; antioxidants; alkalinizing agents; carrying agents;
chelating agents; colorants; complexing agents; suspending or viscosity-increasing agents; flavors or perfumes; oils; penetration enhancers; polymers; stiffening agents; proteins; carbohydrates; bulking agents; and lubricating agents. The use of such agents for pharmaceutically active substances is well known in the art, and suitable agents for inclusion into dosage forms can be chosen according to factors such as the quantity of elesclomol or derivative, analog, salt, solvate, or prodrug of elesclomol, and, if present, other active agent or agents to be included per unit dose, the intended route of administration, the physical form of the dosage form, and optimization of patient compliance with administration. Except insofar as any conventional medium, carrier, or agent is incompatible with the active ingredient or ingredients, its use in a composition according to the present invention is contemplated.
[0563] Pharmaceutical compositions according to the present invention can be formulated for oral, sustained-release oral, buccal, sublingual, inhalation, insufflation, or parenteral administration. Suitable routes for administration of pharmaceutical compositions according to the present invention can be chosen based on factors known to one of skill in the art including the unit dose of the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol, and, if present, the other active agent or agents, the particular carriers or excipients included in the composition, the intended route of administration, the disease or condition to be treated, its severity, other diseases or conditions affecting the and other factors known in the art.
[0564] If a pharmaceutical composition according to the present invention is intended for oral administration, it is typically administered in a conventional unit dosage form such as a tablet, a capsule, a pill, a troche, a wafer, a powder, or a liquid such as a solution, a suspension, a tincture, or a syrup. Oral formulations typically include such normally employed excipients as, for example, pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharin, cellulose, magnesium carbonate, and other conventional pharmaceutical excipients. In certain defined embodiments, oral pharmaceutical compositions will comprise an inert diluent and/or assimilable edible carrier, and/or they may be enclosed in hard or soft shell gelatin capsules. Alternatively, they may be compressed into tablets. As another alternative, particularly for veterinary practice, they can be incorporated directly into food. For oral
therapeutic administration, they can be incorporated with excipients or used in the form of ingestible tablets, buccal tablets, dragees, pills, troches, capsules, wafers, or other conventional dosage forms. The tablets, pills, troches, capsules, wafers, or other conventional dosage forms can also contain the following: a binder, such as gum tragacanth, acacia, cornstarch, sorbitol, mucilage of starch, polyvinylpyrrolidone, or gelatin; excipients or fillers such as dicalcium phosphate, lactose, microcrystalline cellulose, or sugar; a disintegrating agent such as potato starch, croscarmellose sodium, or sodium starch glycolate, or alginic acid; a lubricant such as magnesium stearate, stearic acid, talc, polyethylene glycol, or silica; a sweetening agent, such as sucrose, lactose, or saccharin; a wetting agent such as sodium lauryl sulfate; or a flavoring agent, such as peppermint, oil of Wintergreen, orange flavoring, or cherry flavoring. When the dosage unit form is a capsule, it can contain, in addition to materials of the above types, a liquid carrier. Various other materials can be present as coatings or to otherwise modify the physical form and properties of the dosage unit. For instance, tablets, pills, or capsules can be coated with shellac, sugar, or both. The pharmaceutical compositions of the present invention may be manufactured in a manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levitating, emulsifying, encapsulating, entrapping or lyophilizing processes.
[0565] In one alternative, a sustained-release formulation is used. Sustained- release formulations are well-known in the art. For example, they can include the use of polysaccharides such as xanthan gum and locust bean gum in conjunction with carriers such as dimethylsiloxane, silicic acid, a mixture of mannans and galactans, xanthans, and micronized seaweed, as disclosed in U.S. Patent No. 6,039,980 to Baichwal. Other sustained-release formulations incorporate a biodegradable polymer, such as the lactic acid-glycolic acid polymer disclosed in U.S. Patent No. 6,740,634 to Saikawa et al. Still other sustained-release formulations incorporate an expandable lattice that includes a polymer based on polyvinyl alcohol and polyethylene glycol, as disclosed in U.S. Patent No. 4,428,926 to Keith. Still other sustained-release formulations are based on the Eudragit™ polymers of Rohm & Haas that include copolymers of acrylate and
methacrylates with quaternary ammonium groups as functional groups as well as ethylacrylate methylmethacrylate copolymers with a neutral ester group.
[0566] Oral liquid preparations can be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups, tinctures, or elixirs, or can be presented as a dry product for reconstitution with water or other suitable vehicles before use. Such liquid preparations can contain conventional additives such as suspending agents, for example, sorbitol syrup, methylcellulose, glucose/sugar syrup, gelatin, hydroxymethylcellulose, carboxymethylcellulose, aluminum stearate gel, or hydrogenated edible fats; emulsifying agents, such as lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example, almond oil, fractionated coconut oil, oily esters, propylene glycol, or ethyl alcohol; or preservatives, for example, methylparaben, propylparaben, or sorbic acid. The preparations can also contain buffer salts, flavoring, coloring, or sweetening agents (e.g., mannitol) as appropriate.
[0567] When compositions according to the present invention are formulated for parenteral administration, e.g., formulated for injection via the intravenous, intramuscular, subcutaneous, intralesional, or intraperitoneal routes or other routes known in the art, many options are possible. The preparation of an aqueous composition as described above will be known to those of skill in the art. Typically, such compositions can be prepared as injectables, either as liquid solutions and/or suspensions. Solid forms suitable for use to prepare solutions and/or suspensions upon the addition of a liquid prior to injection can also be prepared. The preparations can also be emulsified. The pharmaceutical forms suitable for injectable use include sterile aqueous solutions and/or dispersions; formulations including sesame oil, peanut oil, synthetic fatty acid esters such as ethyl oleate, triglycerides, and/or aqueous propylene glycol; and/or sterile powders for the extemporaneous preparation of sterile injectable solutions and/or dispersions. Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions. In all cases the form must be sterile
and/or must be fluid to the extent that the solution will pass readily through a syringe and needle of suitable diameter for administration. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms, such as bacteria or fungi.
[0568] For administration of the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol or of a pharmaceutical composition containing the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol, various factors must be taken into account in setting suitable dosages. These factors include other medications being administered to the subject, which, in some cases, may alter the pharmacokinetics of the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol, either causing it to be degraded more rapidly or more slowly. These medications can, for example, affect either liver or kidney function or may induce the synthesis of one or more cytochrome P450 enzymes that can metabolize the elesclomol or the derivative, analog, salt, solvate, or prodrug of elesclomol.
[0569] The exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition. (See, e.g., Fingl et al., in The Pharmacological Basis of Therapeutics, 1975, ch. 1 p. 1). It should be noted that the attending physician would know how to and when to terminate, interrupt, or adjust administration due to toxicity, or to organ dysfunctions. Conversely, the attending physician would also know to adjust treatment to higher levels if the clinical response were not adequate (precluding toxicity). The magnitude of an administered dose in the management of the disorder of interest will vary with the severity of the condition to be treated, such as, but not limited to, a malignancy, and to the route of administration. The severity of the condition may, for example, be evaluated, in part, by standard prognostic evaluation methods. Further, the dose and perhaps the dose frequency, will also vary according to the age, body weight, and response of the individual patient. A program comparable to that discussed above may be used in veterinary medicine.
[0570] The invention is illustrated by the following Examples. These Examples are included for illustrative purposes only, and are not intended to limit the invention.
EXAMPLES
Example 1
Results of Treatment of Clear Cell Ovarian Cancer Cell Lines with Elesclomol [0571] The clear cell ovarian cancer cell lines OVCA429 and RMG1 were treated with elesclomol in order to determine the cytotoxicity of elesclomol for these cell lines. For both cell lines, variants with wild-type ARID1 A and mutated ARID1 A were tested.
[0572] Figure 16 is a series of graphs showing the effects of a 72-hour treatment with elesclomol on the viability of either the OVCA429 NTC (non-targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line. Figure 16A shows the viability of these cell lines after 72 hours of treatment with 10 nM or 100 nM of elesclomol; viability is determined by the MTT assay (• = OVCA429 NTC; ■ = OVCA429 ARID1 A mutant). Figure 16B shows the viability of these cell lines after 72 hours of treatment with 1 nM, 2 nM, 3 nM, 4 nM, 5 nM, 6 nM, 7 nM, 8 nM, 9 nM, 10 nM, or 100 nm of elesclomol; viability is determined by the MTT assay (• = OVCA429 NTC; ■ = OVCA429 ARID1 A mutant). Figure 16C shows the viability of these cell lines after 72 hours of treatment with 10 nM, 20 nM, 30 nM, 40 nM, 50 nM, 60 nM, 70 nM, 80 nM, 90 nM, or 100 nM of elesclomol; viability is determined by the MTT assay (• = OVCA429 NTC; ■ = OVCA429 ARID1 A mutant). In all cases, OVCA429 ARID1 A mutant cells are more sensitive to elesclomol than the OVCA429 NTC cells.
[0573] Figure 17 is a series of graphs showing the effects of a 72-hour treatment with elesclomol on the viability of either the RMG1 NTC (non-targeted control) clear cell ovarian cancer cell line or the RMG1 ARID1 A mutant cell line. Figure 17A shows the viability of these cell lines after 72 hours of treatment with 10 nM, 100 nM, or 1000 nM of elesclomol; viability is determined by the MTT assay (• = RMG1 NTC; ■ = RMG1 ARID1 A mutant). Figure 17B shows the viability of these cell lines after 72 hours of treatment with 10 nM, 20 nM, 30 nM, 40 nM, 50 nM, 60 nM, 70 nM, 80 nM, 90 nM, or 100 nM of elesclomol; viability is determined by the MTT assay (• = RMG1 NTC; ■ = RMG1 ARID1 A mutant). In all cases, the RMG1 ARID1 A mutant cells were more sensitive to elesclomol than the RMG1 NTC cells.
[0574] Figure 18 is a series of graphs showing the results over time of treatment of either the OVCA429 NTC (non-targeted control) clear cell ovarian cancer cell line or
the OVCA429 ARID1 A mutant cell line with 1 ng, 10 ng, 100 ng, or 1000 ng of elesclomol for a time interval of up to 72 hours. Figure 18A shows the results for OVCA429 NTC; Figure 18B shows the results for OVCA429 ARID1 A mutant; Figure 18C overlays the results from Figures 18A and Figure 18B. In all cases, the mutant cells were more sensitive to elesclomol as a function of time than were the OVCA429 ARID1A NTC cells. Although Figures 16 and 17 do show a clear difference in results between the ARID1 A wild-type and the ARID1A mutant phenotype, the difference in results is not clear in Figure 18. This is simply an artifact of the assays. The Incucyte analyses shown in Figure 18 are based on bright-field images which provide an analysis of confluence so does not differentiate between viable and dying cells, whereas Figures 16 and 17 measure only viable cells.
[0575] Figure 19 shows photomicrographs of either the OVCA429 NTC (non- targeted control) clear cell ovarian cancer cell line or the OVCA429 ARID1 A mutant cell line that were either not treated with elesclomol or treated with 10 nM of elesclomol. (A) OVCA429 NTC not treated with elesclomol; (B) OVCA429 ARID1 A mutant cell line not treated with elesclomol; (C) OVCA429 NTC treated with 10 nM of elesclomol; and (D) OVCA429 ARID1 A mutant cell line treated with 10 nM of elesclomol. In all cases, treatment with elesclomol resulted in cell death visible in the photomicrographs.
[0576] Figure 20 is a schematic diagram showing the mechanisms of action of elesclomol in cells with wild-type ARID1 A and in mutant ARID1 A, in particular the occurrence of reactive oxygen species (ROS) in cells with mutant ARID1 A induced by elesclomol, leading to cytotoxicity in such cells.
[0577] The results of this example show the cytotoxicity of elesclomol against two cell lines of clear cell ovarian cancer cells. The cytotoxicity is enhanced in mutants of these cell lines with mutant ARID1 A. The confluence assay demonstrates activity against both OVCA429 with wild-type AR1 D1 A and OVCA429 with mutant AR1 D1 A, but does not show a difference between the isogenic pair; however, the cell viability assay (as determined by MTT) does demonstrate increased sensitivity for OVCA429 with mutant AR1 D1 A. Review of the mutational profile for OVCA429 in Harmonizome shows a mutation in FDX1 , which has been demonstrated to be the protein binding content. There are also several mutations related to DNA repair and SWI/SNF, which could also
be implicated in the lack of differentiation in the ARID1 a knockout phenotype for the cell confluence assay. The results of this example demonstrates the utility of the use of elesclomol, particularly in the treatment of clear cell ovarian cancer.
Example 2
Therapeutic Potential of Elesclomol in SWI/SNF-Mutant Cancer (Prophetic Example)
[0578] The SWI/SNF complex is a key regulator of multiple cellular pathways frequently mutated in cancer. The switch/sucrose non-fermenting (SWI/SNF) chromatin remodeling complexes are evolutionarily conserved multi-subunit protein complexes that were first discovered in yeast as necessary for growth in the presence of different carbon sources (1 -3). Mammalian SWI/SNF complexes are heterogenous due to incorporation of mutually exclusive paralogue subunits, resulting in numerous combinations of subcomplexes in individual cells (4). All complexes include a catalytic ATPase subunit (mutually exclusive SMARCA4/BRG1 or SMARCA2/BRM) and core subunits including SMARCC, SMARCD and SMARCE, SMARCB1 (5). All SWI/SNF complexes utilize the energy of ATP hydrolysis to mobilize nucleosomes and remodel chromatin (6-10) Given its pivotal role in regulating a broad range of normal functions (11 ), it is not surprising that loss of function mutations impacting a number of SWI/SNF subunits occur in about 20% of cancers, ranking it among the most highly mutated protein complexes in cancer (4, 12, 13).
[0579] Directly relevant to this application, SMARCA4 is recurrently mutated in lung cancer (14, 15), medulloblastoma (16), pancreatic cancer (17, 18) small cell carcinoma of the ovary hypercalcemic type (SCCOHT) (19-21 ), and dedifferentiated carcinoma (DDC) of various organs (22). Regardless of the reported synthetic lethal interaction between two SWI/SNF ATPases (23-25), SMARCA2 is epigenetically silenced in SCCOHT and a subset of lung and ovarian/endometrial DDC (26-27). Such dual loss of SMARCA4 and SMARCA2 (referred to as “SMARCA4/A2-loss” hereafter) results in no compensatory SWI/SNF ATPase function and is accompanied by extremely poor clinical outcomes in affected patients. Furthermore, in addition to SMARCA4/2- loss, dual loss of ARIDIA and ARID1 B, two mutually exclusive subunits of the canonical
SWI/SNF BAF complex, or loss of SMARCB1 also frequently occurs in DDC, further supporting that SWI/SNF abnormality is one key event driving cancer progression.
[0580] Therefore, effective targeted treatment strategies are needed for cancers bearing mutations in SWI/SNF subunits. Previous research has demonstrated the role of SWI/SNF in enhancer regulation and its functional antagonism with the polycomb repressive complex 2 (PRC2 ) at these sites (4, 28-32). These studies have identified transcriptional programs reliant on SWI/SNF and have determined the susceptibility of SWI/SNF-mutant cancers to epigenetic drugs targeting PRC2 and histone deacetylases (HDACs) (4, 33-36). Several other targeted vulnerabilities, such as receptor tyrosine kinases (37), CDK4/6 (38-39), and MDM2 (40), have been discovered in SWI/SNF- mutant cancers. However, their clinical potency is either lacking or remains to be demonstrated. Therefore, further studies are required to identify additional therapeutic vulnerabilities in order to develop potential single agent and combination therapy approaches.
[0581] In particular, SMARCA4/2 cells are hypersensitive to elesclomol. Notably, compared to high mutation burden tumors, such as lung cancers, where many background mutations may obscure the effect of a single genetic alteration, SCCOHT (median age of onset in twenties) is solely driven by SMARCA4/A2-loss and thus represents a powerful tool to understanding the role of chromatin remodelers that can be translated to other SWI/SNF-mutant cancers. Through an unbiased functional genomics screen, we identified unique metabolic vulnerabilities in SMARCA4/2-deficient cancer cells. Notably, in line with a report showing that SMARCA4 loss induces reactive oxygen species (ROS) (41 ), SCCOHT cells are more sensitive to ROS inducing agents, including hydrogen peroxide and elesclomol, than SWI/SNF-intact ovarian cancer cells.
[0582] This example is intended to evaluate the therapeutic potential of elesclomol in SWI/SNF-mutant cancers and to understand the mechanism of action underlying the selective effect of elesclomol in SWI/SNF-mutant cancers.
[0583] Based on a preliminary study of the effect of elesclomol on SCCOHT cells, the effect of elesclomol in a panel of SWI/SNF-mutant cancer cell lines will be evaluated. This will include: (1 ) SMARCA4-deficient ovarian and lung cancer cell lines with or without SMARCA2 deficiency; (2) SMARCB1 -deficient malignant rhabdoid
tumor, epithelioid sarcoma and poorly differentiated chordoma cell lines; (3) ARID1A/B- dual deficient ovarian and uterine cancer cell lines. SWI/SNF-intact ovarian, uterine, lung and brain cancer cell lines and non-transformed cell lines will be used as controls.
[0584] In vitro testing will be performed. In particular, short-term growth and long-term clonogeneic assays will be performed to evaluate the potency of elesclomol on SWI/SNF-deficient cancer cell lines. To measure the effect of elesclomol on ROS generation, fluorescent microscopy and FACS analyses using H2 DCFDA and CellROX reagents will be performed. Cell death will be measured by live cell imaging using fluorescent probes detecting activated caspase-3 and western blotting analysis. To address the dependency on relevant SWI/SNF deficiency, a full collection of well- established isogenic cell line models will be utilized for all these cancer types to address whether correction of SWI/SNF defects will decrease the reliance on elesclomol.
[0585] To evaluate the effect of elesclomol in vivo, it will be evaluated in a patient-derived xenograft (PDX) model. 2 SCCOHT PDX models, 2 ARID1A/B-dual deficient DDC PDX models have been established; furthermore, access exists to PDX models of SMARCA4/2-deficent lung cancers and SMARCB1 -deficient cancers. Elesclomol will then be tested in at least 6 PDX models with at least 2 doses per model. The potential synergism between elesclomol and conventional chemotherapy both in vitro and in vivo will thus be evaluated.
[0586] Another aim is to investigate the mechanism of action of elesclomol in SWI/SNF mutant cancer. The focus will be on SMARCA4/2-deficient SCCOHT, DDC and lung cancer cells. Transcriptom ic, proteomic and global metabolomic profiles of isogenic SCCOHT, DDC and lung cancer cells, -/+ SMARCA4 re-expression, will be obtained by RNA sequencing and mass spectrometry to identify how elesclomol impacts the molecular profiles of SMARCA4/2-mutant cancer cells. Integrated analysis of these dataset will identify key molecular pathways that explains the SWI/SNF-dependent hypersensitivity to elesclomol.
[0587] Additionally, direct targets of elesclomol will be identified by thermal proteome profiling (TPP). TPP entails subjecting intact cells or protein lysates, in the presence and absence of a small molecule, to temperature gradient to precipitate insoluble heat-denatured proteins. It is expected that proteins interacting with the
compound of interest will be stabilized or destabilized, leading to a change in the temperature at which the target protein denatures and precipitates out of solution. Through profiling the abundance of soluble proteins by performing tandem mass spectrometry, TPP exploits the thermodynamic changes to protein stability that occur upon binding to a small molecule and can be used to identify even rare proteins that are thermally affected by such interactions (42). Here, the advantage of hypersensitivity of SCCOHT cells to elesclomol and the low mutation burden of these cells and perform TPP analysis on two SCCOHT cells -/+SMARCA4, in the absence or presence of elesclomol will be exploited to determine the proteomic effect of elesclomol. Shared hits will be prioritized for validation using biochemical assays.
[0588] Additionally, resistance mechanisms to elesclomol will be identified using genome-wide CRISPR/cas9 screens. To complement the effort with TPP, a genome- wide CRISPR/cas9 loss of function screen will be performed to identify potential mediators of elesclomol whose disruption confers resistance to the treatment. A genome-wide CRISPR library (Toronto Knockout library v3, TKOv3) targets over 18k protein-coding genes (43) will be used. A CRISPR genome-wide screen in one SCCOHT line treated with elesclomol will be performed. Briefly, 100 million cells will be infected with TKOv3 at a MOI of 0.3-0.4 to ensure the individual cell being infected with 1 or less virus with a coverage of at least 200 cells per guide RNA (43, 44). Cells will be selected for stable integration with puromycin for 48 hours and then exposed to 10 nM elesclomol for about 3 weeks to allow selection of emerged resistance clones. DNA will be extracted from cells right after puromycin selection (baseline) and 3 weeks post selection. Guide RNA (gRNA) sequences will be determined by targeted sequencing with Illumina pair-end sequencing platform through BC Genome Science Centre. Genes with multiple gRNAs (>=2) that are enriched in endpoint samples compared to baseline samples in 3 independent experiments will be considered as candidates that promote drug resistance to elesclomol. Top hits will be validated using conventional genetic and pharmacologic approaches across various SWI/SNF-mutant cell lines. Such study will not only provide candidates for developing combinational therapy but suggest additional biomarker for predicting the responses to elesclomol that warrant future studies.
[0589] The references for Example 2 are as follows:
Neigeborn, L. & Carlson, M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 108, 845-858 (1984).
Stern, M., Jensen, R. & Herskowitz, I. Five SWI genes are required for expression of the HO gene in yeast. J. Mol. Biol. 178, 853-868, doi:10.1016/0022-2836(84)90315-2 (1984).
Cairns, B. R., Kim, Y. J., Sayre, M. H., Laurent, B. C. & Kornberg, R. D. A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proc. Natl. Acad. Sci. U. S. A. 91, 1950-1954, doi:10.1073/pnas.91.5.1950 (1994).
Mittal, P. & Roberts, C. W. M. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat. Rev. Clin. Oncol., doi:10.1038/s41571-020-0357-3 (2020).
Reisman, D. N., Sciarrotta, J., Bouldin, T. W., Weissman, B. E. & Funkhouser, W. K. The expression of the SWI/SNF ATPase subunits BRG1 and BRM in normal human tissues. Appl Immunohistochem Mol Morphol 13, 66-74 (2005).
Kassabov, S. R., Zhang, B., Persinger, J. & Bartholomew, B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol Cell 11, 391-403 (2003).
Phelan, M. L., Sif, S., Narlikar, G. J. & Kingston, R. E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 3, 247-253 (1999).
Hu, G. et al. Regulation of nucleosome landscape and transcription factor targeting at tissue- specific enhancers by BRG1. Genome Res 21, 1650-1658, doi:10.1101/gr.l21145.111 (2011). Tolstorukov, M. Y. et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. U. S. A. 110, 10165-10170, doi:10.1073/pnas.1302209110 (2013).
You, J. S. et al. SNF5 is an essential executor of epigenetic regulation during differentiation. PLoS Genet 9, el003459, doi:10.1371/journal.pgen.1003459 (2013).
Euskirchen, G., Auerbach, R. K. & Snyder, M. SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J Biol Chem 287, 30897-30905, doi:10.1074/jbc.Rlll.309302 (2012).
Kadoch, C. & Crabtree, G. R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1, el500447, doi:10.1126/sciadv,1500447 (2015).
Biegel, J. A., Busse, T. M. & Weissman, B. E. SWI/SNF chromatin remodeling complexes and cancer. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 350-366, doi:10.1002/ajmg.c.31410 (2014).
Medina, P. P. et al. Frequent BRGl/SMARCA4-inactivating mutations in human lung cancer cell lines. Human Mutation 29, 617-622, doi:10.1002/humu.20730 (2008).
Reisman, D. N., Sciarrotta, J., Wang, W., Funkhouser, W. K. & Weissman, B. E. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis.
Cancer research 63, 560-566 (2003).
Parsons, D. W. et al. The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435-439, doi:10.1126/science.H98056 (2011).
Shain, A. H. et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proceedings of the National Academy of Sciences 109, E252-259, doi:10.1073/pnas.1114817109 (2012).
Wong, A. K. et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res 60, 6171-6177. (2000).
Jelinic, P. et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nature genetics 46, 424-426 (2014).
Ramos, P. et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat. Genet. 46, 427-429, doi:10.1038/ng.2928 (2014).
Witkowski, L. et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nature genetics 46, 438-443 (2014).
Karnezis, A. N. et al. Loss of switch/sucrose non-fermenting complex protein expression is associated with dedifferentiation in endometrial carcinomas. Mod. Pathol. 29, 302-314, doi:10.1038/modpathol.2015.155 (2016).
Oike, T. et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 73, 5508-5518, doi:10.1158/0008- 5472.CAN-12-4593 (2013).
Wilson, B. G. et al. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol. Cell. Biol. 34, 1136-1144, doi:10.1128/MCB.01372-13 (2014).
Hoffman, G. R. et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRGl-deficient cancers. Proc. Natl. Acad. Sci. U. S. A. Ill, 3128-3133, doi:10.1073/pnas.1316793111 (2014).
Karnezis, A. N. et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 238, 389-400, doi:10.1002/path.4633 (2016).
Jelinic, P. et al. Concomitant loss of SMARCA2 and SMARCA4 expression in small cell carcinoma of the ovary, hypercalcemic type. Mod. Pathol. 29, 60-66, doi:10.1038/modpathol.2015.129 (2016).
Pan, J. et al. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat. Genet., doi:10.1038/s41588-019-0363-5 (2019).
Stanton, B. Z. et al. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin.
Nat. Genet. 49, 282-288, doi:10.1038/ng.3735 (2017).
Wang, X. et al. SMARCBl-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289-295, doi:10.1038/ng.3746 (2017).
Mathur, R. et al. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 49, 296-302, doi:10.1038/ng.3744 (2017).
Wilson, B. G. et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell l8, 316-328, doi:10.1016/j.ccr.2010.09.006 (2010).
Wang, Y. et al. Histone Deacetylase Inhibitors Synergize with Catalytic Inhibitors of EZH2 to Exhibit Antitumor Activity in Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Mol.
Cancer Ther. 17, 2767-2779, doi:10.1158/1535-7163.MCT-18-0348 (2018).
Watanabe, M. et al. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer 124, 55-67, doi:10.1002/ijc.23897 (2009).
Wang, Y. et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 242, 371-383, doi:10.1002/path.4912 (2017).
36 Chan-Penebre, E. et al. Selective Killing of SMARCA2- and SMARCA4-deficient Small Cell Carcinoma of the Ovary, Hypercalcemic Type Cells by Inhibition of EZH2: In Vitro and In Vivo Preclinical Models. Mol. Cancer Ther. 16, 850-860, doi:10.1158/1535-7163.MCT-16-0678 (2017).
37 Lang, J. D. et al. Ponatinib Shows Potent Antitumor Activity in Small Cell Carcinoma of the Ovary Hypercalcemic Type (SCCOHT) through Multikinase Inhibition. Clin. Cancer Res. 24, 1932-1943, doi:10.1158/1078-0432. CCR-17-1928 (2018).
38 Xue, Y. et al. CDK4/6 inhibitors target SMARCA4-determined cyclin DI deficiency in hypercalcemic small cell carcinoma of the ovary. Nat Common 10, 558, doi:10.1038/s41467-018- 06958-9 (2019).
39 Xue, Y. et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat Common 10, 557, doi:10.1038/s41467-019-08380-l (2019).
40 Howard, T. P. et al. MDM2 and MDM4 Are Therapeutic Vulnerabilities in Malignant Rhabdoid Tumors. Cancer Res. 79, 2404-2414, doi:10.1158/0008-5472.CAN-18-3066 (2019).
41 Lissanu Deribe, Y. et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 24, 1047-1057, doi:10.1038/s41591-018- 0019-5 (2018).
42 Johnson, F. D. et al. Characterization of a small molecule inhibitor of disulfide reductases that induces oxidative stress and lethality in lung cancer cells. Cell Rep 38, 110343, doi:10.1016/j.celrep.2022.110343 (2022).
43 Hart, T. et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 7, 2719-2727, doi:10.1534/g3.117.041277 (2017).
44 Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84- 87, doi:10.1126/science.1247005 (2014).
Example 3 Investigation of the Therapeutic Potential of Elesclomol in Clear Cell Ovarian Cancer (Prophetic Example)
[0590] The impact of elesclomol on proliferation, migration/invasion, and tumorigenic potential of clear cell ovarian cancer (CCOC) cell lines will be investigated. The initial focus will be on CCOC cell lines harboring an ARID1A mutation; specifically, OVISE, OVMAMA, and JHOC-7 cell lines. The IC50 for elesclomol in these cells will be determined. A determination of whether elesclomol impacts the aggressive phenotype of CCOC cells will be made; it is postulated that elesclomol impairs cell adaptation to stress and thus inhibits a migratory/invasiveness phenotype. The effects of elesclomol on cell viability will be determined using crystal violet, cell proliferation using Ki67, cell death using caspase-3 activation assay and western blotting, generation of reactive oxygen species (ROS) using H2 DCFDA, CellROX and FACS analysis, independent cell growth using soft agar colony formation assay, migration using scratch assay and in vitro migration assay, as well as invasiveness capacity using an in vitro invasion assay.
These assays will be performed under various culture conditions including ambient, oxidative stress (cells treated with Piperiongum ine, a potent inducer of oxidative stressdependent cell killing), and hypoxia (1 % O2 short-term and longer incubation periods).
[0591] An additional aim will be to address the impact of elesclomol on the molecular signature of the cells. Transcriptional profiling of -/+ treated cells, -/+ stress using RNA sequencing will be conducted to investigate the potential shift in cells’ molecular signature. Expression profiles will be interrogated using pathway analysis tools such as Ingenuity or Pathway Studio to identify potential pathways of interest. Potential targets with known clinically relevant inhibitors will also be preferentially evaluated in future studies, as our major long-term goal is to provide novel clinical options for patients with metastatic disease.
[0592] Furthermore, the in vivo activity of elesclomol in relevant murine models will be assessed.
Example 4 Pulmonary Metastatic Assay (PUMA) (Prophetic Example)
[0593] The efficacy of elesclomol against malignancies is to be tested using the pulmonary metastatic assay (PUMA). Tumor cells (expressing EGFP or RFP) are injected into the tail vein of mice. The cells are allowed to grow and form lung metastases, typically over a period of 2.5 months. The mice are then euthanized, the lungs are insufflated with agarose, and then allowed to solidify. The lungs are then cut into sections and grown for 2-3 weeks in TC plates. The cells are treated in vitro with elesclomol with controls not treated with elesclomol. The cells are monitored by fluorescence microscopy to determine how treatment with elesclomol impacts the metastatic lesions.
ADVANTAGES OF THE INVENTION
[0594] The present invention provides improved methods and compositions for treatment of malignancies and other diseases and conditions, including, but not limited
to, benign hyperproliferative diseases and conditions, infections, inflammatory diseases and conditions, and immunological diseases and conditions. Elesclomol functions by elevating oxidative stress levels, particularly in cancer cells. Methods and compositions according to the present invention are well-tolerated and can be used together with other methods and therapeutic agents for treating malignancy, as well as other diseases.
[0595] As used herein in the specification and claims, the transitional phrase “comprising” and equivalent language also encompasses the transitional phrases “consisting essentially of’ and “consisting of” with respect to the scope of any claims presented herein, unless the narrower transitional phrases are explicitly excluded.
[0596] Methods according to the present invention possess industrial applicability for the preparation of a medicament for the treatment of diseases or conditions described herein, including, but not limited to, malignancy. Methods according to the present invention also possess industrial applicability for use in treating such diseases and conditions, including, but not limited to, malignancy. Compositions according to the present invention possess industrial applicability as pharmaceutical compositions, particularly for the treatment of malignancy, as well as for other diseases and conditions described above.
[0597] The method claims of the present invention provide specific method steps that are more than general applications of laws of nature and require that those practicing the method steps employ steps other than those conventionally known in the art, in addition to the specific applications of laws of nature recited or implied in the claims, and thus confine the scope of the claims to the specific applications recited therein. In some contexts, these claims are directed to new ways of using an existing drug.
[0598] The inventions illustratively described herein can suitably be practiced in the absence of any element or elements, limitation or limitations, not specifically disclosed herein. Thus, for example, the terms “comprising,” “including,” “containing,” or similar expressions shall be read expansively and without limitation. Additionally, the terms and expressions employed herein have been used as terms of description and not of limitation, and there is no intention in the use of such terms and expressions of
excluding any equivalents of the future shown and described or any portion thereof, and it is recognized that various modifications are possible within the scope of the invention claimed. Thus, it should be understood that although the present invention has been specifically disclosed by preferred embodiments and optional features, modification and variation of the inventions herein disclosed can be resorted to by those skilled in the art, and that such modifications and variations are considered to be within the scope of the inventions disclosed herein. The inventions have been described broadly and generically herein. Each of the narrower species and subgeneric groupings falling within the scope of the generic disclosure also form part of these inventions. This includes the generic description of each invention with a proviso or negative limitation removing any subject matter from the genus, regardless of whether or not the excised materials specifically resided therein.
[0589] In addition, where features or aspects of an invention are described in terms of the Markush group, those schooled in the art will recognize that the invention is also thereby described in terms of any individual member or subgroup of members of the Markush group. It is also to be understood that the above description is intended to be illustrative and not restrictive. Many embodiments will be apparent to those of in the art upon reviewing the above description. The scope of the invention should therefore, be determined not with reference to the above description, but should instead be determined with reference to the appended claims, along with the full scope of equivalents to which such claims are entitled. The disclosures of all articles and references, including patent publications, are incorporated herein by reference.
Claims
1 . A method to improve the efficacy and/or reduce the side effects of the administration of elesclomol or a derivative, analog, salt, or solvate of elesclomol for treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases or conditions comprising the steps of:
(a) identifying at least one factor or parameter associated with the efficacy and/or occurrence of side effects of the administration of the elesclomol or a derivative, analog, salt, or solvate of elesclomol for the treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases; and
(b) modifying the factor or parameter to improve the efficacy and/or reduce the side effects of the administration of the elesclomol or a derivative, analog, salt, or solvate of elesclomol for the treatment of benign or neoplastic hyperproliferative diseases, infections, inflammatory disease or conditions, or immunological diseases.
2. The method of claim 1 wherein the factor or parameter is selected from the group consisting of:
(a) dose modification;
(b) route of administration;
(c) schedule of administration;
(d) indications for use;
(e) disease stages;
(f) other indications;
(g) patient selection;
(h) patient/disease phenotype;
(i) patient/disease genotype;
(j) pre/post-treatment preparation;
(k) toxicity management;
(l) pharmacokinetic/pharmacodynamic monitoring;
(m) drug combinations;
(n) chemosensitization;
(o) chemopotentiation;
(p) post-treatment patient management;
(q) bulk drug product improvements;
(r) diluent systems;
(s) solvent systems;
(t) excipients;
(u) dosage forms;
(v) dosage kits and packaging;
(w) drug delivery systems;
(x) drug conjugate forms;
(y) compound analogs;
(z) prodrug systems;
(aa) multiple drug systems;
(ab) biotherapeutic enhancement;
(ac) biotherapeutic resistance modulation;
(ad) radiation therapy enhancement;
(ae) novel mechanisms of action;
(af) selective target cell population therapeutics;
(ag) reversal of resistance to an agent selected from the group consisting of a platinum-containing anti-neoplastic agent and a PARP inhibitor anti- neoplastic agent;
(ah) induction of synthetic lethality by administration of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1 A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol renders the malignant cells susceptible to a targeted agent that otherwise would have no effect; and
(ai) modulation of activity of FDX1 to inhibit OXPHOS.
3. The method of claim 1 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
4. The method of claim 3 wherein the elesclomol is in the form of a coordinate-covalent complex with a transition metal cation selected from the group
consisting of Ni2+, Cu+, Cu2+, Co2+, Co3+, Fe2+, Fe3+, Zn2+, Pt2+, Pd2+, V4+, V5+, Cr2+, Cr3+, Cr4+, Mn2+, Mn3+, Mn4+, and Mn5+.
5. The method of claim 4 wherein the transition metal cation is a divalent transition metal cation selected from the group consisting of Ni2+, Cu2+, Co2+, Fe2+, Zn2+, Pt2+, and Pd2+.
6. The method of claim 5 wherein the divalent transition metal cation is selected from the group consisting of Cu2+ and Ni+2.
7. The method of claim 6 wherein the divalent transition metal cation is Cu2+.
8. The method of claim 1 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is a derivative of elesclomol.
9. The method of claim 8 wherein the derivative of elesclomol is a compound of Formula (IV):

(IV), wherein:
(1 ) Y is a covalent bond, a phenylene group or a substituted or unsubstituted straight-chain hydrocarbyl group, or Y taken together with both >C=Z groups with which it is bonded is a substituted or unsubstituted aromatic group;
(2) R1 is a substituted or unsubstituted aliphatic group or a substituted or unsubstituted non-aromatic heterocyclic group;
(3) R2, R3, and R4 are independently hydrogen, a substituted or unsubstituted aliphatic group, a substituted or unsubstituted non-aromatic heterocyclic group, a substituted or unsubstituted aryl group, or R1 and R3 and/or R2 and R4 taken together with the carbon and nitrogen atoms to which they are bonded, form a non-aromatic heterocyclic ring optionally fused to an aromatic ring;
(4) R5 and R6 are independently hydrogen, a substituted or unsubstituted aliphatic group, or a substituted or unsubstituted non-aromatic heterocyclic group;
(5) R7 and R8 are independently hydrogen or a substituted or unsubstituted aliphatic group, or R7 is hydrogen and R8 is a substituted or unsubstituted aryl group, or R7 and R8 taken together are C2-C6 substituted or unsubstituted alkylene group; and
(6) Z is =0 or =S.
10. The method of claim 8 wherein the derivative of elesclomol is a compound of Formula (V):

(V).
11 . The method of claim 8 wherein the derivative of elesclomol is a compound of Formula (VI) or Formula (VII):

(VII).
12. The method of claim 8 wherein the derivative of elesclomol is a compound of Formula (VIII):

(VIII), wherein:
(1 ) each Z is independently S, 0, or Se, provided that both Z moieties cannot be 0;
(2) R1 and R2 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted alkynyl, an optionally substituted cycloalkyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group wherein the heterocyclic group is bonded to the thiocarbonyl via a carbon-carbon linkage, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to seven-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl wherein the heteroaryl is bonded to the thiocarbonyl via a carbon- carbon linkage, --NR12R13, --OR14, SR14, and S(O)pR15;
(3) R3 and R4 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group, and an optionally substituted five-membered or six-membered aryl or heteroaryl group; or, alternatively, R1 and R3, and/or R2 and R4, taken together with the atoms to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group; in this alternative, R3 and R4 can also be hydrogen;
(4) R5 is -CR6R7, -C(=CHR8), or -C(=NR8);
(5) R6 and R7 are both hydrogen or an optionally substituted lower alkyl;
(6) R8 is selected from the group consisting of hydroxyl, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxylalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic aryl,
an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, - N10R11 , and -COR9;
(7) R9 is an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted alkyl, an optionally substituted cycloalkyl, or an optionally substituted heterocyclic group;
(8) R10 and R11 are each independently selected from the group consisting of hydrogen, hydroxyl, alkylamino, dialkylamino, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxyalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, and - COR9; or R10 and R11, taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group;
(9) R12, R13, and R14 are each independently hydrogen, an optionally substituted alkyl, an optionally substituted phenyl, or an optionally substituted benzyl, or R12 and R13, taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group;
(10) R15 is an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl; and
(11 ) p is 1 or 2; provided that when both Z are S and R3 and R4 are both methyl, then R1 and R2 are not both unsubstituted phenyl; alternatively, for compounds of Formula (VIII), R10 and R11 are not both hydrogen.
13. The method of claim 1 wherein the treatment is treatment of a malignancy.
14. The method of claim 13 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
15. The method of claim 14 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
16. The method of claim 13 wherein the malignancy is selected from the group consisting of human sarcomas and carcinomas.
17. The method of claim 16 wherein the malignancy is selected from the group consisting of fibrosarcoma; myxosarcoma; liposarcoma, chondrosarcoma; osteogenic sarcoma; chordoma; angiosarcoma; endotheliosarcoma; lymphangiosarcoma; lymphangioendotheliosarcoma; synovioma; mesothelioma; Ewing’s tumor; leiomyosarcoma; rhabdomyosarcoma; Kras-mutated colon carcinoma; colorectal cancer; anal carcinoma; esophageal cancer; gastric cancer; hepatocellular cancer; bladder cancer; endometrial cancer; pancreatic cancer; triple-negative breast cancer; ovarian cancer; prostate cancer; stomach cancer; atrial myxomas; squamous cell carcinoma; basal cell carcinoma; adenocarcinoma; sweat gland carcinoma; sebaceous gland carcinoma; thyroid and parathyroid neoplasms; papillary carcinoma; papillary adenocarcinoma; cystadenocarcinoma; medullary carcinoma; bronchogenic carcinoma; renal cell carcinoma; hepatoma; bile duct carcinoma; choriocarcinoma; seminoma; embryonal carcinoma; Wilms’ tumor; cervical cancer; testicular tumor; small- cell lung carcinoma; non-small-cell lung carcinoma; bladder carcinoma; epithelial carcinoma; glioma; pituitary neoplasms; astrocytoma; medulloblastoma; craniopharyngioma; ependymoma; pinealoma; hemangioblastoma; acoustic neuroma; schwannoma; oligodendroglioma; meningioma; spinal cord tumors; Braf-mutated melanoma; neuroblastoma; pheochromocytoma; endocrine neoplasia, Types 1-3; retinoblastoma; acute lymphocytic leukemia and acute myelocytic leukemia; chronic myelocytic (granulocytic) leukemia; chronic lymphocytic leukemia; meningeal leukemia;
polycythemia vera; Hodgkin’s lymphoma; non-Hodgkin’s lymphoma; mantle cell lymphoma; cutaneous T-cell lymphoma; multiple myeloma; Waldenstrom’s macroglobulinemia; mycosis fungoides; leptomeningeal cancer; pediatric brain tumors; pediatric sarcoma; ovarian osteogenic sarcoma; hypercalcemic small-cell carcinoma of the ovary; and heavy chain disease.
18. The method of claim 1 wherein the treatment is treatment of a disease or condition selected from the group consisting of: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; and scleroderma.
19. The method of claim 13 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol and wherein the treatment comprises administration of a therapeutically effective quantity of elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 mg/mm2/day to about 10 g/mm2/day.
20. The method of claim 19 wherein the therapeutically effective quantity of elesclomol is from about 2 mg/mm2/day to about 10 g/mm2/day.
21 . The method of claim 13 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol and wherein the treatment comprises administration of a therapeutically effective quantity of elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 pg/kg to about 500 mg/kg.
22. The method of claim 21 wherein the therapeutically effective quantity of elesclomol is from about 500 pg/kg to about 250 mg/kg.
23. The method of claim 22 wherein the therapeutically effective quantity of elesclomol is from about 1 mg/kg to about 100 mg/kg.
24. The method of claim 23 wherein the therapeutically effective quantity of elesclomol is from about 10 mg/kg to about 50 mg/kg.
25. The method of claim 1 wherein the treatment comprises administration of the therapeutically effective quantity of elesclomol or the derivative, analog, salt, or solvate of elesclomol in a pharmaceutical composition.
26. The method of claim 25 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
27. The method of claim 13 wherein the treatment comprises administration of the therapeutically effective quantity of elesclomol or the derivative, analog, salt, or solvate of elesclomol in a pharmaceutical composition.
28. The method of claim 27 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
29. The method of claim 2 wherein the factor or parameter is dose modification, and wherein the dose modification is a dose modification selected from the group consisting of:
(a) i.v. infusion for hours to days;
(b) biweekly administration;
(c) triweekly administration;
(d) monthly administration;
(e) doses greater than 100 mg/m2/day;
(f) progressive escalation of dosing from 100 mg/m2/day based on patient tolerance;
(g) doses less than 2 mg/m2 for greater than 14 days;
(h) modification of dosage in conjunction with use of polyamine to modulate metabolism;
(i) modification of dosage in conjunction with use of eflornithine to modulate metabolism;
(j) selected and intermittent boost dose administration;
(k) bolus single and multiple doses escalating from 100 mg/m2; and
(l) oral doses below 30 or above 130 mg/m2.
30. The method of claim 2 wherein the factor or parameter is route of administration, and wherein the route of administration is a route of administration selected from the group consisting of:
(a) topical administration;
(b) intravesicular administration for bladder cancer;
(c) oral administration;
(d) slow release oral delivery;
(e) intrathecal administration;
(f) intraarterial administration;
(g) continuous infusion;
(h) intermittent infusion
(i) administration via large volume oral solution;
(j) buccal administration; and
(k) rectal administration.
31 . The method of claim 2 wherein the factor or parameter is schedule of administration, and wherein the schedule of administration is selected from the group consisting of:
(a) daily administration;
(b) weekly administration for three weeks;
(c) weekly administration for two weeks;
(d) biweekly administration;
(e) biweekly administration for three weeks with a 1 -2 week rest period;
(f) intermittent boost dose administration;
(g) daily administration for one week then administration once per week for multiple weeks;
(h) daily administration on days 1-5, 8-12 every three weeks; and
(i) daily administration on days 1-3, 8-11 per cycle.
32. The method of claim 2 wherein the factor or parameter is indication for use, and wherein the indication for use is selected from the group consisting of:
(a) use for the treatment of leukemias, wherein the leukemia is a leukemia selected from the group consisting of AML, ALL, CLL, and CML;
(b) use for the treatment of myelodysplastic syndrome (MDS);
(c) use for the treatment of angiogenic diseases or conditions;
(d) use for the treatment of benign prostate hypertrophy;
(e) use for the treatment of psoriasis;
(f) use for the treatment of gout;
(g) use for the treatment of autoimmune conditions;
(h) use for the prevention of transplantation rejection;
(i) use for restenosis prevention in cardiovascular disease;
(j) use for the treatment of mycosis fungoides;
(k) use in bone marrow transplantation;
(l) use as an antiinfective;
(m) use for the treatment of AIDS;
(n) use for the treatment of lymphoma;
(o) use for the treatment of mantle cell lymphoma;
(p) use for the treatment of meningeal leukemia;
(q) use for the treatment of malignant meningitis;
(r) use for the treatment of cutaneous T cell lymphoma;
(s) use for the treatment of Barrett’s esophagus;
(t) use for the treatment of anaplastic gliomas;
(u) use for the treatment of triple negative breast cancer;
(v) use for the treatment of Braf-mutated melanoma;
(w) use for the treatment of BTK resistant CLL;
(x) use for the treatment of chordoma;
(y) use for the treatment of Kras-mutated colon cancer;
(z) use for the treatment of pediatric tumors including brain and sarcoma;
(aa) use for the treatment of neuroblastoma;
(ab) use for the treatment of rhabdomyosarcoma;
(ac) use for the treatment of Ewing’s sarcoma;
(ad) use for the treatment of medulloblastoma;
(ae) use for the treatment of thyroid cancer;
(af) use for the treatment of melanoma,
(ag) use for the treatment of lymphoma;
(ah) use for the treatment of multiple myeloma;
(ai) use for the treatment of ovarian osteogenic sarcoma;
(aj) use for the treatment of bladder cancer;
(ak) use for the treatment of prostate cancer;
(al) use for the treatment of bone metastases;
(am) use for the treatment of bone pain;
(an) use for the treatment of ovarian clear cell carcinoma;
(ao) use for the treatment of high-grade serous ovarian carcinoma; and
(ap) use for the treatment of small-cell carcinoma of the ovary.
33. The method of claim 2 wherein the factor or parameter is selection of a disease stage and wherein the disease stage is a disease stage selected from the group consisting of:
(a) use for the treatment of localized polyp stage colon cancer;
(b) use for the treatment of leukoplakia in the oral cavity;
(c) use for angiogenesis inhibition to prevent or limit metastatic spread; and
(d) use against HIV with AZT, DDI, or reverse transcriptase inhibitors.
34. The method of claim 2 wherein the factor or parameter is use for other indications and wherein the other indication is an indication selected from the group consisting of:
(a) use as an antiinfective;
(b) use as an antiviral;
(c) use as an antibacterial;
(d) use for pleural effusions;
(e) use as an antifungal;
(f) use as an antiparasitic;
(g) use for eczema;
(h) use for shingles;
(i) use for condylomata;
(j) use as an anti HPV agent;
(k) use as an anti-HSV agent;
(l) use for early or late stage MDS (myelodysplastic syndrome);
(m) use for polycythemia vera; and
(n) use for Paget’s disease.
35. The method of claim 2 wherein the factor or parameter is patient selection, and wherein the patient selection is a method of patient selection selected from the group consisting of:
(a) selecting patients with disease conditions with high levels of metabolic enzymes;
(b) selecting patients with disease conditions with high levels of reactive oxygen species;
(c) selecting patients with disease conditions with high levels of histone deacetylase;
(d) selecting patients with disease conditions with high levels of protein kinases;
(e) selecting patients with disease conditions with high levels of ornithine decarboxylase;
(f) selecting patients with disease conditions with low levels of metabolic enzymes;
(g) selecting patients with disease conditions with low levels of reactive oxygen species;
(h) selecting patients with disease conditions with low levels of histone deacetylase;
(i) selecting patients with disease conditions with low levels of protein kinases;
(j) selecting patients with disease conditions with low levels of ornithine decarboxylase;
(k) selecting patients with low or high susceptibility to thrombocytopenia or neutropenia;
(l) selecting patients intolerant of Gl toxicities;
(m) selecting patients with deficiencies in DNA repair capacity including BRCA or ARID1 A or other deficiencies in the SWI/SWF pathway or mitochondrial electron transport;
(n) selecting patients with over- or under-expression of jun;
(o) selecting patients with over- or under-expression of GPCR’s or signal transduction proteins;
(p) selecting patients with over- or under-expression of VEGF;
(q) selecting patients with over- or under-expression of prostate specific genes;
(r) selecting patients with over- or under-expression of protein kinases;
(s) selecting patients with over- or under-expression of telomerase;
(t) selecting patients with abnormalities in gallium scans; and
(u) selecting patients with abnormalities in bone scans.
36. The method of claim 2 wherein the factor or parameter is use determined by analysis of patient or disease phenotype, and wherein the analysis of patient or disease phenotype is selected from the group consisting of:
(a) use of diagnostic tools, techniques, kits and assays to confirm a patient’s particular phenotype;
(b) measurement of metabolism enzymes and metabolites;
(c) measurement of histone deacetylase;
(d) measurement of protein kinases;
(e) measurement of ornithine decarboxylase;
(f) measurement of VEGF;
(g) measurement of products of prostate specific genes;
(h) measurement of protein kinases;
(i) measurement of telomerase;
(j) measurement of jun;
(k) measurement of GPCR’s;
(l) use of surrogate compound dosing;
(m) low dose drug pre-testing for enzymatic status; and
(n) determination of ARIDIa mutant phenotype.
37. The method of claim 2 wherein the factor or parameter is use determined by analysis of patient or disease genotype, and wherein the analysis of patient or disease genotype is selected from the group consisting of:
(a) use of diagnostic tools, techniques, kits and assays to confirm a patient’s particular genotype;
(b) use of gene/protein expression chips and analysis;
(c) single nucleotide polymorphism (SNP) assessment;
(d) determination of SNP’s for histone deacetylase;
(e) determination of SNP’s for ornithine decarboxylase;
(f) determination of SNP’s for genes affecting S-adenosyl methionine metabolism;
(g) determination of SNP’s for GPCR’s;
(h) determination of SNP’s for protein kinases;
(i) determination of SNP’s for telomerase;
(j) determination of SNP’s for jun;
(k) identification and measurement of genes for metabolism enzymes and metabolites;
(l) identification of mutations in wild type and mutated genes;
(m) analysis of epigenetics via analysis of methylation and acetylation,
(n) analysis of ARID1A mutation or ARID1 a deficiency;
(o) analysis of mTor signaling;
(p) analysis of mutations activating PI3K-AKT;
(q) analysis of mutations affecting PARPi resistance;
(r) analysis of mutations affecting HR deficiency;
(s) analysis of mutations affecting DDR deficiency;
(t) analysis of mutations causing SWI/SNF pathway alteration;
(u) analysis of P53 status/mutation;
(v) analysis of BRCA-1 independence/mutation;
(w) analysis of NAC1 mutation; and
(x) analysis of mutations affecting mitochondrial targeting.
38. The method of claim 2 wherein the factor or parameter is pre/post- treatment preparation, and wherein the pre/post-treatment preparation is a method selected from the group consisting of:
(a) use of colchicine or analogs;
(b) use of diuretics such as probenecid;
(c) use of uricase;
(d) non-oral use of nicotinamide;
(e) use of sustained release forms of nicotinamide;
(f) use of inhibitors of polyADP ribose polymerase;
(g) use of caffeine;
(h) use of leucovorin rescue;
(i) use of infection control;
(j) use of antihypertensives;
(k) alteration of stem cell populations;
(l) pretreatment to limit or prevent graft versus host (GVH) cytokine storm reactions;
(m) use of anti-inflammatories; and
(n) anaphylactic reaction suppression.
39. The method of claim 2 wherein the factor or parameter is toxicity management, and wherein the toxicity management is a method selected from the group consisting of:
(a) use of colchicine or analogs;
(b) use of diuretics;
(c) use of uricase;
(d) non-oral use of nicotinamide;
(e) use of sustained release forms of nicotinamide;
(f) use of inhibitors of polyADP-ribose polymerase;
(g) use of caffeine;
(h) use of leucovorin rescue;
(i) use of sustained release allopurinol;
(j) non-oral use of allopurinol;
(k) use of bone marrow transplant stimulants;
(l) administration of blood;
(m) administration of platelet infusions;
(n) administration of Neupogen;
(o) administration of G-CSF or GM-CSF;
(p) administration of agents for pain management;
(q) use of anti-inflammatories;
(r) administration of fluids;
(s) use of corticosteroids;
(t) use of insulin control medications;
(u) use of antipyretics;
(v) use of anti-nausea treatments;
(w) use of anti-diarrhea treatments;
(x) use of N-acetylcysteine;
(y) use of antihistamines;
(z) use of agents to limit or prevent mucositis;
(aa) use of agents to limit or prevent GVH reactions or cytokine storm reactions;
(ab) use of anti-fungal agents;
(ac) use of sodium thiosulfate;
(ad) use of glutathione;
(ae) use of platelet transfusions;
(af) use of anti-diarrheal therapeutics;
(ag) use of epinephrine for allergic and anaphylactic reactions;
(ah) use of lidocaine;
(ai) use of vasoconstrictors; and
(aj) use of vasodilators.
40. The method of claim 2 wherein the factor or parameter is pharmacokinetic/pharmacodynamic monitoring, and wherein the pharmacokinetic/pharmacodynamic monitoring is a method selected from the group consisting of:
(a) multiple determinations of drug plasma levels;
(b) multiple determinations of metabolites in the blood or urine;
(c) measurement of polyamines;
(d) measurement of LAT-1 surface receptors;
(e) use of gene sequencing; and
(f) measurement of immune effectors.
41 . The method of claim 2 wherein the factor or parameter is drug combination, and wherein the drug combination is selected from the group consisting of:
(a) use with topoisomerase inhibitors;
(b) use with fraudulent nucleosides;
(c) use with fraudulent nucleotides;
(d) use with thymidylate synthetase inhibitors;
(e) use with signal transduction inhibitors;
(f) use with cisplatin or gallium analogs;
(g) use with nitrosourea alkylating agents (BCNU , Gliadel wafers, CCNU);
(h) use with bendamustine (Treanda);
(i) use with anti-tubulin agents;
(j) use with antimetabolites;
(k) use with berberine;
(l) use with apigenin;
(m) use with amonafide;
(n) use with colchicine or an analog thereof;
(o) use with genistein;
(p) use with etoposide;
(q) use with cytarabine;
(r) use with a camptothecin;
(s) use with vinca alkaloids;
(t) use with topoisomerase inhibitors;
(u) use with 5-fluorouracil;
(v) use with curcumin;
(w) use with NF-KB inhibitors;
(x) use with rosmarinic acid;
(y) use with an agent selected from the group consisting of Avastin, Rituxan, Herceptin, Erbitux, PD-1 inhibitors, and PD-L1 inhibitors;
(z) use with prednimustine;
(aa) use with DNA and RNA therapeutics;
(ab) use with Braf inhibitors;
(ac) use with BTK inhibitors;
(ad) use with 5-azacytidine;
(ae) use with decitabine;
(af) use with PARP inhibitors;
(ag) use with agents inducing hypomethylation; and
(ah) use with histone deacetylase inhibitors.
42. The method of claim 2 wherein the factor or parameter is chemosensitization, and wherein the chemosensitization is use as chemosensitizer with an additional agent selected from the group consisting of:
(a) fraudulent nucleosides;
(b) fraudulent nucleotides;
(c) thymidylate synthetase inhibitors;
(d) signal transduction inhibitors;
(e) cisplatin or gallium analogs;
(f) an alkylating agent selected from the group consisting of BCNU , Gliadel wafers, CCNU , bendamustine (Treanda), or temozolomide (Temodar);
(g) anti-tubulin agents;
(h) antimetabolites;
(i) berberine;
(j) apigenin;
(k) amonafide;
(l) colchicine and analogs;
(m) genistein;
(n) etoposide;
(o) cytarabine;
(p) camptothecins;
(q) vinca alkaloids;
(r) topoisomerase inhibitors;
(s) 5-fluorouracil;
(t) curcumin;
(u) NF-KB inhibitors;and
(v) rosmarinic acid.
43. The method of claim 2 wherein the factor or parameter is chemopotentiation, and wherein the chemopotentiation is use as chemopotentiator with an additional agent selected from the group consisting of:
(a) fraudulent nucleosides;
(b) fraudulent nucleotides;
(c) thymidylate synthetase inhibitors;
(d) signal transduction inhibitors;
(e) cisplatin or gallium analogs;
(f) an alkylating agent selected from the group consisting of BCNU , Gliadel wafers, CCNU , bendamustine (Treanda), or temozolomide (Temodar);
(g) anti-tubulin agents;
(h) antimetabolites;
(i) berberine;
(j) apigenin;
(k) amonafide;
(l) colchicine and analogs;
(m) genistein;
(n) etoposide;
(o) cytarabine;
(p) camptothecins;
(q) vinca alkaloids;
(r) topoisomerase inhibitors;
(s) 5-fluorouracil;
(t) curcumin;
(u) NF-KB inhibitors;and
(v) rosmarinic acid.
44. The method of claim 2 wherein the factor or parameter is post- treatment management, and wherein the post-treatment management is a method selected from the group consisting of:
(a) use with therapies associated with pain management;
(b) nutritional support;
(c) administration of anti-emetics;
(d) anti-nausea therapies;
(e) anti-anemia therapy;
(f) administration of anti-inflammatories;
(g) administration of antipyretics;
(h) administration of immune stimulants;
(i) administration of anti-diarrhea medicines;
(j) administration of famotidine;
(k) administration of antihistamines;
(l) administration of suppository lubricants;
(m) administration of soothing agents;
(n) administration of lidocaine; and
(o) administration of hydrocortisone.
45. The method of claim 2 wherein the factor or parameter is alternative medicine/therapeutic support, and wherein the alternative medicine/therapeutic support is an agent selected from the group consisting of: selected from the group consisting of:
(a) an herbal medication created either synthetically or through extraction that is a NF-KB inhibitor selected from the group consisting of parthenolide, curcumin, and rosmarinic acid;
(b) an herbal medication created either synthetically or through extraction that is a natural anti-inflammatory selected from the group consisting of rhein and parthenolide;
(c) an herbal medication created either synthetically or through extraction that is an immunostimulant from Echinacea;
(d) berberine; and
(e) an herbal medication created either synthetically or through extraction that is a flavonoid or flavone selected from the group consisting of apigenin or genistein.
46. The method of claim 2 wherein the factor or parameter is a bulk drug product improvement, and wherein the bulk drug product improvement is selected from the group consisting of:
(a) salt formation;
(b) homogeneous crystalline structure;
(c) pure isomers;
(d) increased purity;
(e) lower residual solvents; and
(f) lower residual heavy metals.
47. The method of claim 2 wherein the factor or parameter is a diluent system, and wherein the diluent system is selected from the group consisting of:
(a) an emulsion;
(b) DMSO;
(c) NMF;
(d) DMF;
(e) DMA;
(f) ethanol;
(g) benzyl alcohol;
(h) dextrose-containing water for injection;
(i) Cremophor;
(j) cyclodextrins;
(k) PEG;
(l) saccharin;
(m) glycerin; and
(n) a taste masking effector selected from the group consisting of menthol, rum flavor, and fruit flavorings.
48. The method of claim 2 wherein the factor or parameter is a solvent system, and wherein the solvent system is selected from the group consisting of:
(a) emulsions;
(b) dimethyl sulfoxide (DMSO);
(c) N-methylformamide (NMF);
(d) dimethylformamide (DMF);
(e) DMA;
(f) ethanol;
(g) benzyl alcohol;
(h) dextrose-containing water for injection;
(i) Cremophor;
(j) PEG;
(k) glycerol; and
(l) cocoa butter for suppositories.
49. The method of claim 2 wherein the factor or parameter is an excipient, and wherein the excipient is selected from the group consisting of: can be, but is not limited to, an excipient selected from the group consisting of:
(a) mannitol;
(b) albumin;
(c) EDTA;
(d) sodium bisulfite;
(e) benzyl alcohol;
(f) carbonate buffers;
(g) phosphate buffers;
(h) glycerin;
(i) sweeteners;
(j) a taste masking agent;
(k) substituted celluloses; and
(l) sodium azide as a preservative.
50. The method of claim 2 wherein the factor or parameter is a dosage form, and wherein the dosage form is selected from the group consisting of:
(a) tablets;
(b) capsules;
(c) topical gels;
(d) topical creams;
(e) patches;
(f) suppositories;
(g) lyophilized dosage fills,
(h) suppositories with quick release of <15 min or long melt times of >15 min release time; and
(i) temperature adjusted suppositories.
51 . The method of claim 2 wherein the factor or parameter is dosage kits and packaging, and wherein the dosage kit or packaging is selected from the group consisting of:
(a) amber vials to protect from light;
(b) stoppers with specialized coatings to improve shelf-life stability;
(c) special dropper measuring devices;
(d) single-use or multiple-use container closure systems;
(e) suppository delivery devices; and
(f) dosage measuring devices.
52. The method of claim 2 wherein the factor or parameter is drug delivery systems, and wherein the drug delivery system is selected from the group consisting of:
(a) nanocrystals;
(b) bioerodible polymers;
(c) liposomes;
(d) slow release injectable gels;
(e) microspheres;
(f) suspensions with glycerol;
(g) meltable drug release suppositories with a cocoa butter polymer alone or in combination with PEG;
(h) lecithin;
(i) polylactide/polyglycolide; and
(j) rectal plugs for drug delivery.
53. The method of claim 2 wherein the factor or parameter is drug conjugate forms, and wherein the drug conjugate form is selected from the group consisting of:
(a) polyethylene glycols;
(b) polylactides;
(c) polyglycolides;
(d) amino acids;
(e) peptides; and
(f) multivalent linkers.
54. The method of claim 2 wherein the factor or parameter is use of a compound analog, and wherein the compound analog is selected from the group consisting of:
(a) alteration of side chains to increase or decrease lipophilicity;
(b) additional chemical functionalities to alter reactivity;
(c) alteration of electron affinity;
(d) alteration of binding capacity; and
(e) salt forms.
55. The method of claim 2 wherein the factor or parameter is use of a prodrug system, and wherein the prodrug system is selected from the group consisting of:
(a) enzyme sensitive esters;
(b) dimers;
(c) Schiff bases;
(d) pyridoxal complexes;
(e) caffeine complexes; and
(f) nitroso-substituted analogs as prodrugs.
56. The method of claim 2 wherein the factor or parameter is use of a multiple drug system, and wherein the multiple drug system is selected from the group consisting of:
(a) inhibitors of multi-drug resistance;
(b) specific drug resistance inhibitors;
(c) specific inhibitors of selective enzymes;
(d) signal transduction inhibitors;
(e) repair inhibition; and
(f) topoisomerase inhibitors with non-overlapping side effects.
57. The method of claim 2 wherein the factor or parameter is biotherapeutic enhancement, and wherein the biotherapeutic enhancement is use as a sensitizer or potentiator with a biological response modifier selected from the group consisting of:
(a) cytokines;
(b) lymphokines;
(c) a therapeutic antibody selected from the group consisting of bevacizumab, trastuzumab, rituximab, and cetuximab;
(d) gene therapies;
(e) ribozymes;
(f) RNA interference, and
(g) cell based therapeutics.
58. The method of claim 2 wherein the factor or parameter is biotherapeutic resistance modulation, and wherein the biotherapeutic resistance modulation is use to modulate biotherapeutic resistance that can develop to a biotherapeutic agent that is a biotherapeutic agent selected from the group consisting of:
(a) cytokines;
(b) lymphokines;
(c) therapeutic antibodies selected from the group consisting of consisting bevacizumab, trastuzumab, rituximab, and cetuximab;
(d) gene therapies;
(e) ribozymes; and
(f) RNA interference.
59. The method of claim 2 wherein the factor or parameter is radiation therapy enhancement, and wherein the radiation therapy enhancement is use in
combination with a method or agent for radiation therapy selected from the group consisting of:
(a) hypoxic cell sensitizers;
(b) radiation sensitizers/protectors;
(c) photosensitizers;
(d) radiation repair inhibitors;
(e) thiol depletion;
(f) vaso-targeted agents;
(g) use with radioactive seeds;
(h) use with radionuclides;
(i) use with radiolabeled antibodies; and
(j) use with brachytherapy.
60. The method of claim 2 wherein the factor or parameter is use in connection with a novel mechanism of action, wherein the novel mechanism of action is a novel mechanism of action selected from the group consisting of:
(a) inhibitors of poly-ADP ribose polymerase;
(b) agents that effect vasculature;
(c) vasodilation;
(d) oncogenic targeted agents;
(e) signal transduction inhibitors;
(f) EGFR inhibition;
(g) Protein Kinase C inhibition;
(h) Phospholipase C downregulation;
(i) jun downregulation;
(j) histone genes;
(k) VEGF:
(l) ornithine decarboxylase,
(m) jun D;
(n) v-jun;
(o) GPCR’s
(p) protein kinase A;
(q) telomerase;
(r) prostate specific genes;
(s) protein kinases; and
(t) histone deacetylase.
61 . The method of claim 2 wherein the factor or parameter is selective target cell therapeutics, and wherein the target cell therapeutics is use against a specific type or class of cells selected from the group consisting of:
(a) radiation sensitive cells;
(b) radiation resistant cells;
(c) energy depleted cells; and
(d) endothelial cells.
62. The method of claim 2 wherein the factor or parameter is use to reverse resistance to platinum-containing anti-neoplastic agents or PARP inhibitor anti- neoplastic agents.
63. The method of claim 62 wherein the improvement is use to reverse resistance to a platinum-containing anti-neoplastic agent.
64. The method of claim 63 wherein the platinum-containing anti- neoplastic agent is selected from the group consisting of cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, and tetraplatin.
65. The method of claim 62 wherein the improvement is use to reverse resistance to a PARP inhibitor anti-neoplastic agent.
66. The method of claim 65 wherein the PARP inhibitor anti-neoplastic agent is selected from the group consisting of iniparib, talazoparib, olaparib, rucaparib, vel iparib, CEP- 9722 (a prodrug of CEP-8983 (11 -methoxy-4,5,6,7-tetrahydro-1 H- cyclopenta[a]pyrrolo[3,4-c]carbazole-1 ,3(2H)-dione), MK 4827 ((S)-2-(4-(piperidin-3- yl)phenyl)-2H-indazole-7-carboxamide), and BGB-290.
67. The method of claim 2 wherein the factor or parameter is induction of synthetic lethality by administration of elesclomol or a derivative or analog of elesclomol to a malignancy characterized by a mutation in ARID1A or in a component of the SWI/SNF complex wherein the elesclomol or the derivative or analog of elesclomol
renders the malignant cells susceptible to a targeted agent that otherwise would have no effect.
68. The method of claim 67 wherein the loss-of-function mutation is a mutation in ARID1A.
69. The method of claim 67 wherein the loss-of-function mutation is a mutation in a component of the SWI/SNF complex.
70. The method of claim 2 wherein the factor or parameter is modulation of activity of FDX1 to inhibit OXPHOS.
71 . The method of claim 70 wherein the modulation of activity of FDX1 to inhibit OXPHOS is to treat a malignancy.
72. The method of claim 71 wherein the malignancy is selected from the group consisting of melanoma, leukemia, lymphoma, pancreatic ductal carcinoma, and endometrial carcinoma.
73. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of a microtubulin stabilizer or a microtubulin inhibitor.
74. The method of claim 73 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
75. The method of claim 73 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
76. The method of claim 75 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
77. The method of claim 76 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
78. The method of claim 77 wherein the microtubulin stabilizer or the microtubulin inhibitor is a microtubulin stabilizer.
79. The method of claim 78 wherein the microtubulin stabilizer is selected from the group consisting of paclitaxel, an analog of paclitaxel, and docetaxel.
80. The method of claim 78 wherein the microtubulin stabilizer or the microtubulin inhibitor is a microtubulin inhibitor.
81 . The method of claim 80 wherein the microtubulin inhibitor is selected from the group consisting of vincristine, vinblastine, vinorelbine, vinflunine, and vindesine.
82. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of a PARP inhibitor.
83. The method of claim 82 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
84. The method of claim 82 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
85. The method of claim 84 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
86. The method of claim 85 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
87. The method of claim 82 wherein the PARP inhibitor is selected from the group consisting of iniparib, talazoparib, olaparib, rucaparib, and veliparib.
88. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an LDH inhibitor.
89. The method of claim 88 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
90. The method of claim 88 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
91 . The method of claim 90 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
92. The method of claim 91 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
93. The method of claim 88 wherein the LDH inhibitor is oxamate.
94. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of 2-deoxyglucose or an analog or derivative thereof.
95. The method of claim 94 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
96. The method of claim 94 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
97. The method of claim 96 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
98. The method of claim 97 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
99. The method of claim 94 wherein the 2-deoxyglucose or the analog or derivative thereof is 2-deoxyglucose.
100. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of a glutamine metabolism inhibitor.
101 . The method of claim 100 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
102. The method of claim 100 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
103. The method of claim 102 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
104. The method of claim 103 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
105. The method of claim 100 wherein the glutamine metabolism inhibitor is phenylbutyrate.
106. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of a DNA- damaging agent.
107. The method of claim 106 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
108. The method of claim 106 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
101 . The method of claim 108 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma,
cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
110. The method of claim 109 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
111. The method of claim 106 wherein the DNA-damaging agent is selected from the group consisting of:
(a) an agent that damages DNA directly;
(b) an agent that interferes with DNA synthesis; and
(c) an agent that inhibits a topoisomerase.
112. The method of claim 106 wherein the DNA-damaging agent is selected from the group consisting of cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, tetraplatin, doxorubicin, daunorubicin, methotrexate, 5- fluorouracil, gemcitabine, podophyllotoxin, etoposide, teniposide, cyclophosphamide, chlorambucil, melphalan, carmustine, lomustine, estramustine, semustine, bendamustine, prednamustine, uramustine, chlornaphazine, dacarbazine, altretamine, temozolomide, mitomycin C, streptozotocin, chlorozotocin, capecitabine, floxuridine, 6- mercaptopurine, 8-azaguanine, azathiopurine, 5-ethynyluracil, thioguanine, fludarabine, cytarabine, cladribine, 2-fluoro-arabinosyl-adenine, aminopterin, pemetrexed, ralitrexed, camptothecin, epirubicin, idarubicin, methylnitronitrosoguanidine, topotecan, irinotecan, mechlorethamine, ifosfamide, trofosfamide, busulfan, procarbazine, mitoxantrone, actinomycin, calicheamicin, Tegafur (R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil), 2', 2'- difluoro-2'-deoxycytidine, bischloroethylsulfide, thiotepa, aziridinylbenzoquinone, BCNU , CCNU , 4-methyl CCNU , ACNU , rebeccamycin, bleomycin, pepleomycin, ethylmethanesulfonate, methylmethanesulfonate, dimethylnitrosamine, dimethyl sulfate, and N'-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1 ,3- propanediamine dihydrochloride.
113. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that inhibits the SWI/SNF complex.
114. The method of claim 113 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
115. The method of claim 113 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
116. The method of claim 115 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
117. The method of claim 116 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
118. The method of claim 113 wherein the agent that inhibits the SWI/SNF complex is BD98 (1-isopropyl-3-((4R,5S,8S)-4-methoxy-2,5,8-trimethyl-1-oxo- 7-(4-pyridyl-2-yl)benzyl)-1 ,2, 3, 4, 5, 6, 7, 8, 9, 10-decahydrobenzo[h][1 ,6]diazacyclododecin- 13-yl)urea).
119. A method for treatment of a malignancy treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that causes tumor cells to rely on oxidative phosphorylation.
120. The method of claim 119 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
121. The method of claim 119 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
122. The method of claim 121 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
123. The method of claim 119 wherein the agent that causes tumor cells to rely on oxidative phosphorylation is selected from the group consisting of:
(a) an inhibitor of GLUT 1 ;
(b) an inhibitor of hexokinase;
(c) an inhibitor of pyruvate decarboxylase kinase; and
(d) an inhibitor of lactate dehydrogenase.
124. The method of claim 123 wherein the agent that causes tumor cells to rely on oxidative phosphorylation is selected from the group consisting of ph loretin , quercetin, STF31 (4-[[[[4-(1 ,1-dimethylethyl)phenyl]sulfonyl]amino]methyl]-A/-3- pyridinylbenzamide), WZB117 ((2-fluoro-6-(m -hydroxybenzoyloxy) phenyl m- hydroxybenzoate), 3-bromopyruvate, 3-(3-pyridinyl)-1 -(4-pyridinyl)-2-propen-1 -one, PFK158 (1 -(4-pyridinyl)-3-[7-(trifluoromethyl)-2E-quinolinyl]-2-propen-1 -one, dichloroacetate, oxamic acid, NHI-1 (1-hydroxy-6-phenyl-4-trifluoromethyl-1 H-indole-2- carboxylic acid), and NHI-2 (methyl 1-hydroxy-6-phenyl-4-trifluoromethyl-1 H-indole-2- carboxylate).
125. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that is an inhibitor of the base excision repair (BER) pathway.
126. The method of claim 125 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
127. The method of claim 125 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
128. The method of claim 127 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
129. The method of claim 128 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
130. The method of claim 129 wherein the BER inhibitor is selected from the group consisting of: 3-aminobenzamide; AG14361 (2-[4- [(dimethylamino)methyl]phenyl]-1 ,3, 10-triazatricyclo[6.4.1 .04, 13]trideca-2 , 4, 6, 8( 13)- tetraen-9-one); rucaparib phosphate; veliparib; olaparib; methoxyamine; lucanthone; 7- nitroindole-2-carboxylic acid; arylstilbonic acid derivatives; E3330 ((2E)-2-[(4,5- dimethoxy-2-methyl-3,6-dioxocyclohexa-1 ,4-dien-1 -yl)methylidene]undecanoic acid); oleanolic acid; edgeworin; betulinic acid; stigmasterol; kohamaic acid; 2-(4-(2,5- dimethyl-1 H-pyrrol-1-yl)phenoxy)acetic acid; 2,4,9-trimethylbenzo[b][1 ,8]-naphthyridin-5- amine; A/-(3-chlorophenyl)-5,6-dihydro-4H-cyclopenta[d]isoxazole-3-carboxamide; methoxyamine; N-ethylmaleimide; 5-methyl-3,4-dihydro-2H-isoquinolin-1 -one
(PD128763); 3-aminobenzamide; 6-aminonicotinamide; 8-hydroxy-2-methyl-3H- quinazolin-4-one (NU 1025); and 4-amino-1 ,8-naphthalimide.
131. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that acts as an inhibitor of the homologous repair pathway.
132. The method of claim 131 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
133. The method of claim 131 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
134. The method of claim 133 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
135. The method of claim 134 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
136. The method of claim 131 wherein the inhibitor of the homologous repair pathway is selected from the group consisting of: imatinib mesylate; erlotinib; valproic acid; abexinostat; tanespimycin; β-lactacystin; benzyl N-[(2S)-4-methyl-1-[[(2S)- 4-methyl-1-[[(2S)-4-methyl-1 -oxopentan-2 -yl]amino]-1 -oxopentan-2 -yl]amino]-1- oxopentan-2-yl]carbamate (MG-132); bortezomib; nelfinavir; mirin; 6- (cyclohexylmethoxy)-5-nitrosopyrimidine-2,4-diamine (NU6027); 3-(carbamoylamino)-5- (3-fluorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide (AZD7762); (5Z)-5- (quinolin-6-ylmethylidene)-2-(thiophen-2-ylmethylimino)-1 ,3-thiazolidin-4-one (RO- 3306); N-[[5-[3-(4,6-difluoro-1 H-benzimidazol-2-yl)-1 H-indazol-5-yl]-4-methylpyridin-3- yl]methyl]ethanamine (AG-024322); wortmannin; N-(2-aminoethyl)isoquinoline-5- sulfonamide (H-9); alsterpaulone; curcumin; and 4,4'-diisothiocyanatostilbene-2,2'- disulfonate.
137. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that activates homologous repair as part of its mechanism of antineoplastic activity or as a consequence of inducing DNA damage.
138. The method of claim 137 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
139. The method of claim 137 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
140. The method of claim 139 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
141 . The method of claim 140 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
142. The method of claim 137 wherein the agent that activates homologous repair as part of its mechanism of antineoplastic activity or as a consequence of inducing DNA damage is selected from the group consisting of carboplatin, cisplatin, dianhydrogalactitol, and dibromodulcitol.
143. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation.
144. The method of claim 143 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
145. The method of claim 144 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
146. The method of claim 145 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
147. The method of claim 146 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
148. The method of claim 143 wherein the agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation is an agent that is activated by bioreductases under acute conditions of hypoxia, and wherein the agent that is activated by bioreductases under acute conditions of hypoxia is selected from the group consisting of tirapazamine and mitomycin C.
149. The method of claim 143 wherein the agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation is an agent that functions to sensitize hypoxic cells to antineoplastic agents or radiation, and wherein the agent that functions to sensitize hypoxic cells to antineoplastic agents or radiation is selected from the group consisting of misonidazole, metronidazole, nimorazole, benznidazole, desmethylmisonidazole, etanidazole, pimonidazole, and 1 -(aziridin-1 -yl)-3-(2- nitroimidazol-1-yl)propan-2-ol (RSU-1069).
150. A method for treatment of a malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol comprising:
(a) administering a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol; and
(b) administering a therapeutically effective quantity of an agent that inhibits cysteine uptake.
151 . The method of claim 150 wherein the elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is elesclomol.
152. The method of claim 151 wherein the malignancy or another disease or condition treatable by administration of elesclomol or a derivative, analog, salt, solvate, or prodrug of elesclomol is a malignancy.
153. The method of claim 152 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
154. The method of claim 153 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
155. The method of claim 150 wherein the agent that inhibits cysteine uptake is selected from the group consisting of:
(i) a peptide derived from digestion of human β-casein, bovine P-casein, or gliadin;
(ii) an inhibitor of the excitatory amino acid transporter EAAT2 or EAAT3;
(iii) an inhibitor of a transporter of cysteine selected from the group consisting of LAT1 , ASCT2, and the Xc’ system.
156. The method of claim 155 wherein the agent that inhibits cysteine uptake is selected from the group consisting of erastin, imidazole ketone erastin (IKE), sorafenib, and sulfasalazine (SSZ).
157. The method of claim 150 wherein the agent that inhibits cysteine uptake is selected from the group consisting of erastin, erastin A, erastin B, piperazine erastin, and imidazole ketone erastin.
158. A composition to improve the efficacy or reduce the side effects of treatment with elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol, wherein the composition comprises:
(a) an alternative selected from the group consisting of:
(i) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol;
(ii) two or more therapeutically active ingredients comprising:
(A) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol; and
(B) at least one additional therapeutic agent, therapeutic agent subject to chemosensitization, therapeutic agent subject to chemopotentiation, or component of a multiple drug system;
(iii) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a dosage form;
(iv) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol that is incorporated into a dosage kit and packaging;
(v) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, solvate or prodrug of elesclomol that is subjected to a bulk drug product improvement;
(vi) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug delivery system;
(vii) a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug conjugate form; and
(viii) a therapeutically effective quantity of a prodrug of elesclomol or a derivative or analog of elesclomol; and
(b) at least one pharmaceutically acceptable diluent, solvent or excipient.
159. The composition of claim 158 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
160. The composition of claim 159 wherein the elesclomol is in the form of a coordinate-covalent complex with a transition metal cation selected from the group consisting of Ni2+, Cu+, Cu2+, Co2+, Co3+, Fe2+, Fe3+, Zn2+, Pt2+, Pd2+, V4+, V5+, Cr2+, Cr3+, Cr4+, Mn2+, Mn3+, Mn4+, and Mn5+.
161 . The composition of claim 160 wherein the transition metal cation is a divalent transition metal cation selected from the group consisting of Ni2+, Cu2+, Co2+, Fe2+, Zn2+, Pt2+, and Pd2+.
162. The composition of claim 161 wherein the divalent transition metal cation is selected from the group consisting of Cu2+ and Ni+2.
163. The composition of claim 162 wherein the divalent transition metal cation is Cu2+.
164. The composition of claim 158 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is a derivative of elesclomol.
165. The composition of claim 164 wherein the derivative of elesclomol is a compound of Formula (IV):

(IV), wherein:
(1 ) Y is a covalent bond, a phenylene group or a substituted or unsubstituted straight-chain hydrocarbyl group, or Y taken together with both >C=Z groups with which it is bonded is a substituted or unsubstituted aromatic group;
(2) R1 is a substituted or unsubstituted aliphatic group or a substituted or unsubstituted non-aromatic heterocyclic group;
(3) R2, R3, and R4 are independently hydrogen, a substituted or unsubstituted aliphatic group, a substituted or unsubstituted non-aromatic heterocyclic group, a substituted or unsubstituted aryl group, or R1 and R3 and/or R2 and R4 taken together with the carbon and nitrogen atoms to which they are bonded, form a non-aromatic heterocyclic ring optionally fused to an aromatic ring;
(4) R5 and R6 are independently hydrogen, a substituted or unsubstituted aliphatic group, or a substituted or unsubstituted non-aromatic heterocyclic group;
(5) R7 and R8 are independently hydrogen or a substituted or unsubstituted aliphatic group, or R7 is hydrogen and R8 is a substituted or unsubstituted aryl group, or R7 and R8 taken together are C2-C6 substituted or unsubstituted alkylene group; and
(6) Z is =0 or =S.
166. The composition of claim 164 wherein the derivative of elesclomol is a compound of Formula (V):

(V).
167. The composition of claim 164 wherein the derivative of elesclomol is a compound of Formula (VI) or Formula (VII):
(VI); and


(VII).
168. The composition of claim 164 wherein the derivative of elesclomol is a compound of Formula (VIII):

(VIII), wherein:
(1 ) each Z is independently S, 0, or Se, provided that both Z moieties cannot be 0;
(2) R1 and R2 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted alkynyl, an optionally substituted cycloalkyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group wherein the heterocyclic group is bonded to the thiocarbonyl via a carbon-carbon linkage, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to seven-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl wherein the heteroaryl is bonded to the thiocarbonyl via a carbon- carbon linkage, --NR12R13, --OR14, SR14, and S(O)pR15;
(3) R3 and R4 are each independently selected from the group consisting of an optionally substituted alkyl, an optionally substituted alkenyl, an optionally substituted cycloalkenyl, an optionally substituted heterocyclic group, and an optionally substituted five-membered or six-membered aryl or heteroaryl group; or, alternatively, R1 and R3, and/or R2 and R4, taken together with the atoms to which they are attached, form an
optionally substituted heterocyclic group or an optionally substituted heteroaryl group; in this alternative, R3 and R4 can also be hydrogen;
(4) R5 is -CR6R7, -C(=CHR8), or -C(=NR8);
(5) R6 and R7 are both hydrogen or an optionally substituted lower alkyl;
(6) R8 is selected from the group consisting of hydroxyl, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxylalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic aryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, — N10R11 , and -COR9;
(7) R9 is an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted alkyl, an optionally substituted cycloalkyl, or an optionally substituted heterocyclic group;
(8) R10 and R11 are each independently selected from the group consisting of hydrogen, hydroxyl, alkylamino, dialkylamino, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, hydroxyalkyl, hydroxyalkenyl, hydroxyalkynyl, haloalkyl, haloalkenyl, haloalkynyl, an optionally substituted phenyl, an optionally substituted bicyclic aryl, an optionally substituted five-membered to six-membered monocyclic heteroaryl, an optionally substituted nine-membered to fourteen-membered bicyclic heteroaryl, an optionally substituted cycloalkyl, an optionally substituted heterocyclic group, and - COR9; or R10 and R11, taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group;
(9) R12, R13, and R14 are each independently hydrogen, an optionally substituted alkyl, an optionally substituted phenyl, or an optionally substituted benzyl, or R12 and R13, taken together with the nitrogen atom to which they are attached, form an optionally substituted heterocyclic group or an optionally substituted heteroaryl group;
(10) R15 is an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl; and
(11 ) p is 1 or 2; provided that when both Z are S and R3 and R4 are both methyl, then R1 and R2 are not both unsubstituted phenyl; alternatively, for compounds of Formula (VIII), R10 and R11 are not both hydrogen.
169. The composition of claim 158 wherein the composition is formulated for treatment of a malignancy.
170. The composition of claim 169 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC), ovarian clear-cell carcinoma (OCCC), uterine corpus endothelial carcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cholangiocarcinoma, pancreatic cancer, metastatic esophagogastric cancer, recurrent or metastatic head and neck cancer, and lymphoid diffuse large B-cell lymphoma.
171. The composition of claim 170 wherein the malignancy is selected from the group consisting of ovarian epithelial cancer (OEC) and ovarian clear-cell carcinoma (OCCC).
172. The composition of claim 169 wherein the malignancy is selected from the group consisting of human sarcomas and carcinoma.
173. The composition of claim 169 wherein the malignancy is selected from the group consisting of fibrosarcoma; myxosarcoma; liposarcoma, chondrosarcoma; osteogenic sarcoma; chordoma; angiosarcoma; endotheliosarcoma; lymphangiosarcoma; lymphangioendotheliosarcoma; synovioma; mesothelioma; Ewing’s tumor; leiomyosarcoma; rhabdomyosarcoma; Kras-mutated colon carcinoma; colorectal cancer; anal carcinoma; esophageal cancer; gastric cancer; hepatocellular cancer; bladder cancer; endometrial cancer; pancreatic cancer; triple-negative breast cancer; ovarian cancer; prostate cancer; stomach cancer; atrial myxomas; squamous cell carcinoma; basal cell carcinoma; adenocarcinoma; sweat gland carcinoma; sebaceous gland carcinoma; thyroid and parathyroid neoplasms; papillary carcinoma; papillary adenocarcinoma; cystadenocarcinoma; medullary carcinoma; bronchogenic carcinoma; renal cell carcinoma; hepatoma; bile duct carcinoma; choriocarcinoma;
seminoma; embryonal carcinoma; Wilms’ tumor; cervical cancer; testicular tumor; small- cell lung carcinoma; non-small-cell lung carcinoma; bladder carcinoma; epithelial carcinoma; glioma; pituitary neoplasms; astrocytoma; medulloblastoma; craniopharyngioma; ependymoma; pinealoma; hemangioblastoma; acoustic neuroma; schwannoma; oligodendroglioma; meningioma; spinal cord tumors; Braf-mutated melanoma; neuroblastoma; pheochromocytoma; endocrine neoplasia, Types 1-3; retinoblastoma; acute lymphocytic leukemia and acute myelocytic leukemia; chronic myelocytic (granulocytic) leukemia; chronic lymphocytic leukemia; meningeal leukemia; polycythemia vera; Hodgkin’s lymphoma; non-Hodgkin’s lymphoma; mantle cell lymphoma; cutaneous T-cell lymphoma; multiple myeloma; Waldenstrom’s macroglobulinemia; mycosis fungoides; leptomeningeal cancer; pediatric brain tumors; pediatric sarcoma; ovarian osteogenic sarcoma; hypercalcemic small-cell carcinoma of the ovary; and heavy chain disease.
174. The composition of claim 158 wherein the composition is formulated for treatment of a disease or condition selected from the group consisting of: angiogenic diseases or conditions; benign prostate hypertrophy; psoriasis; gout; autoimmune conditions; use in bone marrow transplantation; prevention of transplantation rejection; restenosis prevention in cardiovascular disease; infections caused by bacteria, viruses, or fungi, including viral infections caused by HPV or HSV; AIDS; Barrett’s esophagus; eczema; shingles; condylomata; Reiter’s syndrome; pityriasis rubra pilaris; actinic keratosis; senile keratosis; and scleroderma.
175. The composition of claim 158 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol and wherein the composition is formulated for administration of a therapeutically effective quantity of elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 mg/mm2/day to about 10 g/mm2/day.
176. The composition of claim 175 wherein the therapeutically effective quantity of elesclomol is from about 1 mg/mm2/day to about 10 g/mm2/day.
177. The composition of claim 158 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol and wherein the composition is formulated for administration of a therapeutically effective quantity of
elesclomol, wherein the therapeutically effective quantity of elesclomol is from about 1 pg/kg to about 500 mg/kg.
178. The composition of claim 177 wherein the therapeutically effective quantity of elesclomol is from about 500 pg/kg to about 250 mg/kg.
179. The composition of claim 178 wherein the therapeutically effective quantity of elesclomol is from about 1 mg/kg to about 100 mg/kg.
180. The composition of claim 179 wherein the therapeutically effective quantity of elesclomol is from about 10 mg/kg to about 50 mg/kg.
181 . The composition of claim 158 wherein the composition comprises a therapeutically effective quantity of an additional therapeutic agent.
182. The composition of claim 181 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
183. The composition of claim 182 wherein the additional therapeutic agent is selected from the group consisting of:
(i) topoisomerase inhibitors;
(ii) fraudulent nucleosides;
(iii) fraudulent nucleotides;
(iv) thymidylate synthetase inhibitors;
(v) signal transduction inhibitors;
(vi) cisplatin or gallium analogs;
(vii) nitrosourea alkylating agents (BCNU , Gliadel wafers, CCNU);
(viii) bendamustine (Treanda);
(ix) anti-tubulin agents;
(x) antimetabolites;
(xi) berberine;
(xii) apigenin;
(xiii) amonafide;
(xiv) colchicine or an analog thereof;
(xv) genistein;
(xvi) etoposide;
(xvii) cytarabine;
(xviii) a camptothecin;
(xix) vinca alkaloids;
(xx) topoisomerase inhibitors;
(xxi) 5-fluorouracil;
(xxii) curcumin;
(xxiii) NF-KB inhibitors;
(xxiv) rosmarinic acid;
(xxv) biological therapies selected from the group consisting of Avastin, Rituxan, Herceptin, Erbitux, PD-1 inhibitors, and PD-L1 inhibitors;
(xxvi) prednimustine;
(xxvii) DNA or RNA therapeutics;
(xxviii) Braf inhibitors;
(xxix) BTK inhibitors;
(xxx) 5-azacytidine;
(xxxi) decitabine;
(xxxii) PARP inhibitors;
(xxxiii) agents inducing hypomethylation; and
(xxxiv) histone deacetylase inhibitors.
184. The composition of claim 181 wherein the additional therapeutic agent is a microtubulin stabilizer.
185. The composition of claim 184 wherein the microtubulin stabilizer is selected from the group consisting of paclitaxel, an analog of paclitaxel, and docetaxel.
186. The composition of claim 181 wherein the additional therapeutic agent is a microtubulin inhibitor.
187. The composition of claim 186 wherein the microtubulin inhibitor is selected from the group consisting of vincristine, vinblastine, vinorelbine, vinflunine, and vindesine.
188. The composition of claim 181 wherein the additional therapeutic agent is a PARP inhibitor.
189. The composition of claim 188 wherein the PARP inhibitor is selected from the group consisting of iniparib, talazoparib, olaparib, rucaparib, and veliparib.
190. The composition of claim 181 wherein the additional therapeutic agent is an LDH inhibitor.
191 . The composition of claim 190 wherein the LDH inhibitor is oxamate.
192. The composition of claim 181 wherein the additional therapeutic agent is 2-deoxyglucose or an analog or derivative thereof.
193. The composition of claim 192 wherein the 2-deoxyglucose or an analog or derivative thereof is 2-deoxyglucose.
194. The composition of claim 181 wherein the additional therapeutic agent is a glutamine metabolism inhibitor.
195. The composition of claim 194 wherein the glutamine metabolism inhibitor is phenylbutyrate.
196. The composition of claim 181 wherein the additional therapeutic agent is a DNA-damaging agent.
197. The composition of claim 196 wherein the DNA-damaging agent is selected from the group consisting of:
(a) an agent that damages DNA directly;
(b) an agent that interferes with DNA synthesis; and
(c) an agent that inhibits a topoisomerase.
198. The composition of claim 196 wherein the DNA-damaging agent is selected from the group consisting of cisplatin, carboplatin, oxaliplatin, picoplatin, nedaplatin, satraplatin, tetraplatin, doxorubicin, daunorubicin, methotrexate, 5- fluorouracil, gemcitabine, podophyllotoxin, etoposide, teniposide, cyclophosphamide, chlorambucil, melphalan, carmustine, lomustine, estramustine, semustine, bendamustine, prednamustine, uramustine, chlornaphazine, dacarbazine, altretamine, temozolomide, mitomycin C, streptozotocin, chlorozotocin, capecitabine, floxuridine, 6- mercaptopurine, 8-azaguanine, azathiopurine, 5-ethynyluracil, thioguanine, fludarabine, cytarabine, cladribine, 2-fluoro-arabinosyl-adenine, aminopterin, pemetrexed, ralitrexed, camptothecin, epirubicin, idarubicin, methylnitronitrosoguanidine, topotecan, irinotecan,
mechlorethamine, ifosfamide, trofosfamide, busulfan, procarbazine, mitoxantrone, actinomycin, calicheamicin, Tegafur (R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil), 2', 2'- difluoro-2'-deoxycytidine, bischloroethylsulfide, thiotepa, aziridinylbenzoquinone, BCNU , CCNU , 4-methyl CCNU , ACNU , rebeccamycin, bleomycin, pepleomycin, ethylmethanesulfonate, methylmethanesulfonate, dimethylnitrosamine, dimethyl sulfate, and N'-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1 ,3- propanediamine dihydrochloride.
199. The composition of claim 181 wherein the additional agent is an agent that inhibits the SWI/SNF complex.
200. The composition of claim 199 wherein the agent that inhibits the SWI/SNF complex is BD98 (1-isopropyl-3-((4R,5S,8S)-4-methoxy-2,5,8-trimethyl-1-oxo- 7-(4-pyridyl-2-yl)benzyl)-1 ,2, 3, 4, 5, 6, 7, 8, 9, 10-decahydrobenzo[h][1 ,6]diazacyclododecin- 13-yl)urea).
201 . The composition of claim 181 wherein the additional agent is an agent that causes tumor cells to rely on oxidative phosphorylation.
202. The composition of claim 201 wherein the agent that causes tumor cells to rely on oxidative phosphorylation is selected from the group consisting of:
(a) an inhibitor of GLUT 1 ;
(b) an inhibitor of hexokinase;
(c) an inhibitor of pyruvate decarboxylase kinase; and
(d) an inhibitor of lactate dehydrogenase.
203. The composition of claim 202 wherein the agent that causes tumor cells to rely on oxidative phosphorylation is selected from the group consisting of phloretin, quercetin, STF31 (4-[[[[4-(1 ,1-dimethylethyl)phenyl]sulfonyl]amino]methyl]-A/- 3-pyridinylbenzamide), WZB117 ((2-fluoro-6-(m -hydroxybenzoyloxy) phenyl m- hydroxybenzoate), 3-bromopyruvate, 3-(3-pyridinyl)-1 -(4-pyridinyl)-2-propen-1 -one, PFK158 (1 -(4-pyridinyl)-3-[7-(trifluoromethyl)-2E-quinolinyl]-2-propen-1 -one, dichloroacetate, oxamic acid, NHI-1 (1-hydroxy-6-phenyl-4-trifluoromethyl-1 H-indole-2- carboxylic acid), and NHI-2 (methyl 1-hydroxy-6-phenyl-4-trifluoromethyl-1 H-indole-2- carboxylate).
204. The composition of claim 181 wherein the additional agent is an agent that is an inhibitor of the base excision repair (BER) pathway.
205. The composition of claim 204 wherein the agent that is an inhibitor of the base excision repair (BER) pathway inhibitor is selected from the group consisting of: 3-aminobenzamide; AG14361 (2-[4-[(dimethylamino)methyl]phenyl]-1 ,3,10- triazatricyclo[6.4.1 .04, 13]trideca-2,4,6,8(13)-tetraen-9-one); rucaparib phosphate; veliparib; olaparib; methoxyamine; lucanthone; 7-nitroindole-2-carboxylic acid; arylstilbonic acid derivatives; E3330 ((2E)-2-[(4,5-dimethoxy-2-methyl-3,6- dioxocyclohexa-1 ,4-dien-1-yl)methylidene]undecanoic acid); oleanolic acid; edgeworin; betulinic acid; stigmasterol; kohamaic acid; 2-(4-(2,5-dimethyl-1 H-pyrrol-1 - yl)phenoxy)acetic acid; 2,4,9-trimethylbenzo[b][1 ,8]-naphthyridin-5-amine; N-(3- chlorophenyl)-5,6-dihydro-4H-cyclopenta[c/]isoxazole-3-carboxamide; methoxyamine; N- ethylmaleimide; 5-methyl-3,4-dihydro-2H-isoquinolin-1 -one (PD128763); 3- aminobenzamide; 6-aminonicotinamide; 8-hydroxy-2-methyl-3H-quinazolin-4-one (NU 1025); and 4-amino-1 ,8-naphthalimide.
206. The composition of claim 181 wherein the additional therapeutic agent is an agent that acts as an inhibitor of the homologous repair pathway.
207. The composition of claim 206 wherein the agent that acts as an inhibitor of the homologous repair pathway is selected from the group consisting of: imatinib mesylate; erlotinib; valproic acid; abexinostat; tanespimycin; β-lactacystin; benzyl N-[(2S)-4-methyl-1 -[[(2S)-4-methyl-1 -[[(2S)-4-methyl-1 -oxopentan-2-yl]amino]-1 - oxopentan-2-yl]amino]-1 -oxopentan-2-yl]carbamate (MG-132); bortezomib; nelfinavir; mirin; 6-(cyclohexylmethoxy)-5-nitrosopyrimidine-2,4-diamine (NU6027); 3- (carbamoylamino)-5-(3-fluorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide (AZD7762); (5Z)-5-(quinolin-6-ylmethylidene)-2-(thiophen-2-ylmethylimino)-1 ,3- thiazolidin-4-one (RO-3306); N-[[5-[3-(4,6-difluoro-1 H-benzimidazol-2-yl)-1 H-indazol-5- yl]-4-methylpyridin-3-yl]methyl]ethanamine (AG-024322); wortmannin; N-(2- aminoethyl)isoquinoline-5-sulfonamide (H-9); alsterpaulone; curcumin; and 4,4'- diisothiocyanatostilbene-2,2'-disulfonate.
208. The composition of claim 181 wherein the additional therapeutic agent is an agent that activates homologous repair as part of its mechanism of antineoplastic activity or as a consequence of inducing DNA damage.
209. The composition of claim 208 wherein the agent that activates homologous repair as part of its mechanism of antineoplastic activity or as a consequence of inducing DNA damage is selected from the group consisting of carboplatin, cisplatin, dianhydrogalactitol, and dibromodulcitol.
210. The composition of claim 181 wherein the additional therapeutic agent is an agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation.
211 . The composition of claim 200 wherein the agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation is an agent that is activated by bioreductases under acute conditions of hypoxia, and wherein the agent that is activated by bioreductases under acute conditions of hypoxia is selected from the group consisting of tirapazamine and mitomycin C.
212. The composition of claim 210 wherein the agent that is activated by bioreductases under acute conditions of hypoxia or that functions to sensitize hypoxic cells to antineoplastic agents or radiation is an agent that functions to sensitize hypoxic cells to antineoplastic agents or radiation, and wherein the agent that functions to sensitize hypoxic cells to antineoplastic agents or radiation is selected from the group consisting of misonidazole, metronidazole, nimorazole, benznidazole, desmethylmisonidazole, etanidazole, pimonidazole, and 1 -(aziridin-1 -yl)-3-(2- nitroimidazol-1-yl)propan-2-ol (RSU-1069).
213. The composition of claim 181 wherein the additional therapeutic agent is an agent that inhibits cysteine uptake.
214. The composition of claim 213 wherein the agent that inhibits cysteine uptake is selected from the group consisting of:
(i) a peptide derived from digestion of human β-casein, bovine β-casein, or gliadin;
(ii) an inhibitor of the excitatory amino acid transporter EAAT2 or EAAT3;
(iii) an inhibitor of a transporter of cysteine selected from the group consisting of LAT1 , ASCT2, and the Xc’ system.
215. The composition of claim 214 wherein the agent that inhibits cysteine uptake is selected from the group consisting of erastin, imidazole ketone erastin (IKE), sorafenib, and sulfasalazine (SSZ).
216. The composition of claim 214 wherein the agent that inhibits cysteine uptake is selected from the group consisting of erastin, erastin A, erastin B, piperazine erastin, and imidazole ketone erastin.
217. The composition of claim 158 wherein the composition comprises a therapeutic agent subject to chemosensitization.
218. The composition of claim 217 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
219. The composition of claim 217 wherein the agent subject to chemosensitization is selected from the group consisting of:
(i) fraudulent nucleosides;
(ii) fraudulent nucleotides;
(iii) thymidylate synthetase inhibitors;
(iv) signal transduction inhibitors;
(v) cisplatin or gallium analogs;
(vi) an alkylating agent selected from the group consisting of BCNU , Gliadel wafers, CCNU , bendamustine (Treanda), or temozolomide (Temodar);
(vii) anti-tubulin agents;
(viii) antimetabolites;
(ix) berberine;
(x) apigenin;
(xi) amonafide;
(xii) colchicine and analogs;
(xiii) genistein;
(xiv) etoposide;
(xv) cytarabine;
(xvi) camptothecins;
(xvii) vinca alkaloids;
(xviii) topoisomerase inhibitors;
(xix) 5-fluorouracil;
(xx) curcumin;
(xxi) NF-KB inhibitors;and
(xxii) rosmarinic acid.
220. The composition of claim 158 wherein the composition comprises a therapeutic agent subject to chemopotentiation.
221 . The composition of claim 220 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
222. The composition of claim 220 wherein the agent subject to chemopotentiation is selected from the group consisting of:
(i) fraudulent nucleosides;
(ii) fraudulent nucleotides;
(iii) thymidylate synthetase inhibitors;
(iv) signal transduction inhibitors;
(v) cisplatin or gallium analogs;
(vi) an alkylating agent selected from the group consisting of BCNU , Gliadel wafers, CCNU , bendamustine (Treanda), or temozolomide (Temodar);
(vii) anti-tubulin agents;
(viii) antimetabolites;
(ix) berberine;
(x) apigenin;
(xi) amonafide;
(xii) colchicine and analogs;
(xiii) genistein;
(xiv) etoposide;
(xv) cytarabine;
(xvi) camptothecins;
(xvii) vinca alkaloids;
(xviii) topoisomerase inhibitors;
(xix) 5-fluorouracil;
(xx) curcumin;
(xxi) NF-KB inhibitors;and
(xxii) rosmarinic acid.
223. The composition of claim 158 wherein the therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol is incorporated into a dosage form.
224. The composition of claim 223 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
225. The composition of claim 223 wherein the dosage form is selected from the group consisting of:
(i) amber vials to protect from light;
(ii) stoppers with specialized coatings to improve shelf-life stability;
(iii) special dropper measuring devices;
(iv) single-use or multiple-use container closure systems;
(v) suppository delivery devices; and
(vi) dosage measuring devices.
226. The composition of claim 158 wherein the composition comprises a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is subjected to a bulk drug product improvement.
227. The composition of claim 226 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
228. The composition of claim 226 wherein the bulk drug product improvement is selected from the group consisting of:
(i) salt formation;
(ii) homogeneous crystalline structure;
(iii) pure isomers;
(iv) increased purity;
(v) lower residual solvents; and
(vi) lower residual heavy metals.
229. The composition of claim 158 wherein the composition comprises a therapeutically effective quantity of a component of a multiple drug system.
230. The composition of claim 229 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
231 . The composition of claim 229 wherein the component of the multiple drug system is selected from the group consisting of:
(i) inhibitors of multi-drug resistance;
(ii) specific drug resistance inhibitors;
(iii) specific inhibitors of selective enzymes;
(iv) signal transduction inhibitors;
(v) repair inhibition; and
(vi) topoisomerase inhibitors with non-overlapping side effects.
232. The composition of claim 158 wherein the composition comprises a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug delivery system.
233. The composition of claim 232 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
234. The composition of claim 232 wherein the drug delivery system is selected from the group consisting of:
(i) nanocrystals;
(ii) bioerodible polymers;
(iii) liposomes;
(iv) slow release injectable gels;
(v) microspheres;
(vi) suspensions with glycerol;
(vii) meltable drug release suppositories with a cocoa butter polymer alone or in combination with PEG;
(viii) lecithin; and
(ix) polylactide/polyglycolide.
235. The composition of claim 158 wherein the composition comprises a therapeutically effective quantity of elesclomol or a derivative, analog, salt, or solvate of elesclomol that is incorporated into a drug conjugate form.
236. The composition of claim 235 wherein the elesclomol or the derivative, analog, salt, or solvate of elesclomol is elesclomol.
237. The composition of claim 235 wherein the drug conjugate form is selected from the group consisting of:
(i) polyethylene glycols;
(ii) polylactides;
(iii) polyglycolides;
(iv) amino acids;
(v) peptides; and
(vi) multivalent linkers.
238. The composition of claim 158 wherein the composition comprises a therapeutically effective quantity of a prodrug of elesclomol or a derivative or analog of elesclomol.
239. The composition of claim 238 wherein the prodrug is selected from the group consisting of:
(i) enzyme sensitive esters;
(ii) dimers;
(iii) Schiff bases;
(iv) pyridoxal complexes;
(v) caffeine complexes; and
(vi) nitroso-substituted analogs as prodrugs.
240. The composition of claim 158 wherein the composition comprises a pharmaceutically acceptable diluent.
241 . The composition of claim 240 wherein the pharmaceutically acceptable diluent is selected from the group consisting of:
(i) an emulsion;
(ii) DMSO;
(iii) NMF;
(iv) DMF;
(v) DMA;
(vi) ethanol;
(vii) benzyl alcohol;
(viii) dextrose-containing water for injection;
(ix) Cremophor;
(x) cyclodextrins;
(xi) PEG;
(xii) saccharin;
(xiii) glycerin; and
(xiv) a taste masking effector selected from the group consisting of menthol, rum flavor, and fruit flavorings.
242. The composition of claim 158 wherein the composition comprises a pharmaceutically acceptable solvent.
243. The composition of claim 242 wherein the pharmaceutically acceptable solvent is selected from the group consisting of:
(i) emulsions;
(ii) dimethyl sulfoxide (DMSO);
(iii) N-methylformamide (NMF);
(iv) dimethylformamide (DMF);
(v) DMA;
(vi) ethanol;
(vii) benzyl alcohol;
(viii) dextrose-containing water for injection;
(ix) Cremophor;
(x) PEG;
(xi) glycerol; and
(xii) cocoa butter for suppositories.
244. The pharmaceutical composition of claim 158 wherein the composition comprises a pharmaceutically acceptable excipient.
245. The composition of claim 244 wherein the pharmaceutically acceptable excipient is selected from the group consisting of:
(a) mannitol;
(b) albumin;
(c) EDTA;
(d) sodium bisulfite;
(e) benzyl alcohol;
(f) carbonate buffers;
(g) phosphate buffers;
(h) glycerin;
(i) sweeteners;
(j) a taste masking agent;
(k) substituted celluloses; and
(l) sodium azide as a preservative.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163270382P | 2021-10-21 | 2021-10-21 | |
| US63/270,382 | 2021-10-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023069727A1 true WO2023069727A1 (en) | 2023-04-27 |
Family
ID=86059594
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2022/047447 WO2023069727A1 (en) | 2021-10-21 | 2022-10-21 | Compositions and methods for treatment of hyperproliferative, inflammatory, and immunological diseases, and infections |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023069727A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120128580A1 (en) * | 2010-11-18 | 2012-05-24 | Synta Pharmaceuticals Corp. | Preselection of subjects for therapeutic treatment with elesclomol based on hypoxic status |
| US20130150440A1 (en) * | 2010-04-20 | 2013-06-13 | Synta Pharmaceuticals Corp. | Use of bis [thiohydrazide amide] compounds such as elesclomol for treating cancers |
| US20200289618A1 (en) * | 2014-10-21 | 2020-09-17 | Sciclone Pharmaceuticals International Ltd. | Treatment of cancer with immune stimulators |
-
2022
- 2022-10-21 WO PCT/US2022/047447 patent/WO2023069727A1/en active Search and Examination
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130150440A1 (en) * | 2010-04-20 | 2013-06-13 | Synta Pharmaceuticals Corp. | Use of bis [thiohydrazide amide] compounds such as elesclomol for treating cancers |
| US20120128580A1 (en) * | 2010-11-18 | 2012-05-24 | Synta Pharmaceuticals Corp. | Preselection of subjects for therapeutic treatment with elesclomol based on hypoxic status |
| US20200289618A1 (en) * | 2014-10-21 | 2020-09-17 | Sciclone Pharmaceuticals International Ltd. | Treatment of cancer with immune stimulators |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20200206188A1 (en) | Compositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigo | |
| AU2019283893B2 (en) | Use of dianhydrogalactitol and analogs and derivatives thereof to treat recurrent malignant glioma or progressive secondary brain tumor | |
| AU2021257978A1 (en) | Therapeutic benefit of suboptimally administered chemical compounds | |
| AU2019272044A1 (en) | Combinatorial methods to improve the therapeutic benefit of bisantrene | |
| JP2021004244A (en) | Use of substituted hexitols including dianhydrogalactitol and analogs to treat neoplastic disease and cancer stem cells including glioblastoma multiforme and medulloblastoma | |
| WO2014179528A2 (en) | Compositions and methods to improve the therapeutic benefit of suboptimally administered chemical compounds including substituted naphthalimides such as amonafide for the treatment of immunological, metabolic, infectious, and benign or neoplastic hyperproliferative disease conditions | |
| WO2017042634A2 (en) | Use of dianhydrogalactitol or derivatives and analogs thereof for treatment of non-small-cell lung carcinoma, glioblastoma, and ovarian carcinoma by induction of dna damage and stalling of cell cycle | |
| IL305418A (en) | Angiopoietin-like 3 (angptl3) irna compositions and methods of use thereof | |
| WO2023069727A1 (en) | Compositions and methods for treatment of hyperproliferative, inflammatory, and immunological diseases, and infections | |
| CA3209512A1 (en) | Compositions and methods to improve the therapeutic benefit of suboptimally administered chemical compounds and biological therapies including substituted camptothecins such as irinotecan and topotecan for the treatment of benign and neoplastic hyperproliferative disease conditions, infections, inflammatory and immunological diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22884534 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) |