WO2023012263A1 - Solid oral peptide formulations - Google Patents
Solid oral peptide formulations Download PDFInfo
- Publication number
- WO2023012263A1 WO2023012263A1 PCT/EP2022/071913 EP2022071913W WO2023012263A1 WO 2023012263 A1 WO2023012263 A1 WO 2023012263A1 EP 2022071913 W EP2022071913 W EP 2022071913W WO 2023012263 A1 WO2023012263 A1 WO 2023012263A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ethoxy
- glp
- salt
- peptide
- nac
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims description 300
- 108090000765 processed proteins & peptides Proteins 0.000 title claims description 230
- 239000007787 solid Substances 0.000 title claims description 36
- 238000009472 formulation Methods 0.000 title description 6
- 150000003839 salts Chemical class 0.000 claims abstract description 194
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 claims abstract description 132
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 91
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 claims abstract description 66
- 229960002446 octanoic acid Drugs 0.000 claims abstract description 66
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 182
- 230000001225 therapeutic effect Effects 0.000 claims description 113
- 239000003877 glucagon like peptide 1 receptor agonist Substances 0.000 claims description 102
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 claims description 102
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 claims description 94
- 235000005152 nicotinamide Nutrition 0.000 claims description 90
- 239000011570 nicotinamide Substances 0.000 claims description 90
- 229960003966 nicotinamide Drugs 0.000 claims description 90
- FIWQZURFGYXCEO-UHFFFAOYSA-M sodium;decanoate Chemical compound [Na+].CCCCCCCCCC([O-])=O FIWQZURFGYXCEO-UHFFFAOYSA-M 0.000 claims description 90
- 229940122392 PCSK9 inhibitor Drugs 0.000 claims description 85
- 239000000314 lubricant Substances 0.000 claims description 61
- 235000019359 magnesium stearate Nutrition 0.000 claims description 51
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 48
- 239000000194 fatty acid Substances 0.000 claims description 48
- 229930195729 fatty acid Natural products 0.000 claims description 48
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical class N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 48
- 150000004665 fatty acids Chemical class 0.000 claims description 47
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 claims description 46
- 239000008194 pharmaceutical composition Substances 0.000 claims description 42
- 125000004432 carbon atom Chemical group C* 0.000 claims description 35
- 239000000556 agonist Substances 0.000 claims description 33
- 159000000000 sodium salts Chemical class 0.000 claims description 33
- 239000004026 insulin derivative Substances 0.000 claims description 23
- DLSWIYLPEUIQAV-UHFFFAOYSA-N Semaglutide Chemical group CCC(C)C(NC(=O)C(Cc1ccccc1)NC(=O)C(CCC(O)=O)NC(=O)C(CCCCNC(=O)COCCOCCNC(=O)COCCOCCNC(=O)CCC(NC(=O)CCCCCCCCCCCCCCCCC(O)=O)C(O)=O)NC(=O)C(C)NC(=O)C(C)NC(=O)C(CCC(N)=O)NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(CC(C)C)NC(=O)C(Cc1ccc(O)cc1)NC(=O)C(CO)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(Cc1ccccc1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(C)(C)NC(=O)C(N)Cc1cnc[nH]1)C(C)O)C(C)O)C(C)C)C(=O)NC(C)C(=O)NC(Cc1c[nH]c2ccccc12)C(=O)NC(CC(C)C)C(=O)NC(C(C)C)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CCCNC(N)=N)C(=O)NCC(O)=O DLSWIYLPEUIQAV-UHFFFAOYSA-N 0.000 claims description 21
- 229950011186 semaglutide Drugs 0.000 claims description 21
- 108010060325 semaglutide Proteins 0.000 claims description 20
- 229940002612 prodrug Drugs 0.000 claims description 15
- 239000000651 prodrug Substances 0.000 claims description 15
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 13
- 239000011734 sodium Substances 0.000 claims description 13
- 229910052708 sodium Inorganic materials 0.000 claims description 13
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 claims description 8
- 150000003863 ammonium salts Chemical class 0.000 claims description 5
- 150000004671 saturated fatty acids Chemical class 0.000 claims description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 3
- 239000011591 potassium Substances 0.000 claims description 3
- 229910052700 potassium Inorganic materials 0.000 claims description 3
- 238000000034 method Methods 0.000 abstract description 91
- 239000003623 enhancer Substances 0.000 abstract description 46
- 238000010521 absorption reaction Methods 0.000 abstract description 32
- 238000002360 preparation method Methods 0.000 abstract description 20
- 239000003814 drug Substances 0.000 abstract description 14
- 239000008247 solid mixture Substances 0.000 abstract description 7
- 101800004266 Glucagon-like peptide 1(7-37) Proteins 0.000 description 82
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 74
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 72
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 71
- 239000003752 hydrotrope Substances 0.000 description 63
- QUCDWLYKDRVKMI-UHFFFAOYSA-M sodium;3,4-dimethylbenzenesulfonate Chemical compound [Na+].CC1=CC=C(S([O-])(=O)=O)C=C1C QUCDWLYKDRVKMI-UHFFFAOYSA-M 0.000 description 63
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 description 60
- 125000001424 substituent group Chemical class 0.000 description 59
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 56
- 239000008187 granular material Substances 0.000 description 51
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 49
- 238000002156 mixing Methods 0.000 description 41
- 238000011282 treatment Methods 0.000 description 38
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 36
- 150000001875 compounds Chemical class 0.000 description 35
- 239000007916 tablet composition Substances 0.000 description 35
- PBGKTOXHQIOBKM-FHFVDXKLSA-N insulin (human) Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 PBGKTOXHQIOBKM-FHFVDXKLSA-N 0.000 description 30
- 239000000546 pharmaceutical excipient Substances 0.000 description 30
- 101000976075 Homo sapiens Insulin Proteins 0.000 description 29
- 150000001408 amides Chemical class 0.000 description 28
- 102000009027 Albumins Human genes 0.000 description 27
- 108010088751 Albumins Proteins 0.000 description 27
- 241000282472 Canis lupus familiaris Species 0.000 description 23
- 102400000324 Glucagon-like peptide 1(7-37) Human genes 0.000 description 23
- 125000000539 amino acid group Chemical group 0.000 description 20
- 125000005647 linker group Chemical group 0.000 description 20
- 238000005469 granulation Methods 0.000 description 18
- 230000036515 potency Effects 0.000 description 18
- 230000002265 prevention Effects 0.000 description 18
- 230000003179 granulation Effects 0.000 description 17
- 125000003275 alpha amino acid group Chemical group 0.000 description 16
- 150000004667 medium chain fatty acids Chemical class 0.000 description 16
- 102000008100 Human Serum Albumin Human genes 0.000 description 15
- 108091006905 Human Serum Albumin Proteins 0.000 description 15
- 238000003556 assay Methods 0.000 description 15
- 230000008569 process Effects 0.000 description 15
- 238000006467 substitution reaction Methods 0.000 description 15
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 14
- 239000008186 active pharmaceutical agent Substances 0.000 description 14
- 235000001014 amino acid Nutrition 0.000 description 14
- 238000007908 dry granulation Methods 0.000 description 14
- 230000036470 plasma concentration Effects 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 12
- 238000012217 deletion Methods 0.000 description 12
- 230000037430 deletion Effects 0.000 description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 12
- 239000003112 inhibitor Substances 0.000 description 12
- 101001098868 Homo sapiens Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 description 11
- 102000004877 Insulin Human genes 0.000 description 11
- 108090001061 Insulin Proteins 0.000 description 11
- 102100038955 Proprotein convertase subtilisin/kexin type 9 Human genes 0.000 description 11
- 238000007792 addition Methods 0.000 description 11
- 239000004615 ingredient Substances 0.000 description 11
- 102200127556 rs34159654 Human genes 0.000 description 11
- 238000005550 wet granulation Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 description 10
- 208000024172 Cardiovascular disease Diseases 0.000 description 10
- 208000008589 Obesity Diseases 0.000 description 10
- 101710176384 Peptide 1 Proteins 0.000 description 10
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 10
- 238000007906 compression Methods 0.000 description 10
- 230000006835 compression Effects 0.000 description 10
- 206010012601 diabetes mellitus Diseases 0.000 description 10
- 238000003780 insertion Methods 0.000 description 10
- 230000037431 insertion Effects 0.000 description 10
- 229940125396 insulin Drugs 0.000 description 10
- 235000020824 obesity Nutrition 0.000 description 10
- 102000004196 processed proteins & peptides Human genes 0.000 description 10
- 229940089838 Glucagon-like peptide 1 receptor agonist Drugs 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 9
- -1 C20 fatty acid Chemical class 0.000 description 8
- 108010004460 Gastric Inhibitory Polypeptide Proteins 0.000 description 8
- 102100039994 Gastric inhibitory polypeptide Human genes 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 239000011230 binding agent Substances 0.000 description 8
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 8
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 239000004094 surface-active agent Substances 0.000 description 8
- 241000282412 Homo Species 0.000 description 7
- 102000000853 LDL receptors Human genes 0.000 description 7
- 108010001831 LDL receptors Proteins 0.000 description 7
- 239000005557 antagonist Substances 0.000 description 7
- 239000011324 bead Substances 0.000 description 7
- 208000035475 disorder Diseases 0.000 description 7
- 150000002632 lipids Chemical class 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 7
- 238000009490 roller compaction Methods 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 6
- 150000001413 amino acids Chemical class 0.000 description 6
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 6
- DMBUODUULYCPAK-UHFFFAOYSA-N 1,3-bis(docosanoyloxy)propan-2-yl docosanoate Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCCCCCC DMBUODUULYCPAK-UHFFFAOYSA-N 0.000 description 5
- 108010086246 Glucagon-Like Peptide-1 Receptor Proteins 0.000 description 5
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 5
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 5
- 239000013543 active substance Substances 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 239000012943 hotmelt Substances 0.000 description 5
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 5
- 239000008108 microcrystalline cellulose Substances 0.000 description 5
- 229940016286 microcrystalline cellulose Drugs 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 229940069328 povidone Drugs 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 4
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 4
- 102100040214 Apolipoprotein(a) Human genes 0.000 description 4
- 101710115418 Apolipoprotein(a) Proteins 0.000 description 4
- 208000028399 Critical Illness Diseases 0.000 description 4
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 description 4
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 4
- 108010019598 Liraglutide Proteins 0.000 description 4
- YSDQQAXHVYUZIW-QCIJIYAXSA-N Liraglutide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC)C(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 YSDQQAXHVYUZIW-QCIJIYAXSA-N 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 206010003119 arrhythmia Diseases 0.000 description 4
- 238000010241 blood sampling Methods 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 230000000747 cardiac effect Effects 0.000 description 4
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 235000012000 cholesterol Nutrition 0.000 description 4
- 208000029078 coronary artery disease Diseases 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 108010063245 glucagon-like peptide 1 (7-36)amide Proteins 0.000 description 4
- 239000008103 glucose Substances 0.000 description 4
- 238000009474 hot melt extrusion Methods 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 229960002701 liraglutide Drugs 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 244000309715 mini pig Species 0.000 description 4
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 4
- 229940044601 receptor agonist Drugs 0.000 description 4
- 239000000018 receptor agonist Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- 206010007558 Cardiac failure chronic Diseases 0.000 description 3
- 229920002261 Corn starch Polymers 0.000 description 3
- 102100021752 Corticoliberin Human genes 0.000 description 3
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 3
- 108010022152 Corticotropin-Releasing Hormone Proteins 0.000 description 3
- 208000032928 Dyslipidaemia Diseases 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 208000002705 Glucose Intolerance Diseases 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 239000012901 Milli-Q water Substances 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 102000034527 Retinoid X Receptors Human genes 0.000 description 3
- 108010038912 Retinoid X Receptors Proteins 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 230000036528 appetite Effects 0.000 description 3
- 235000019789 appetite Nutrition 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 235000010980 cellulose Nutrition 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000008120 corn starch Substances 0.000 description 3
- 235000019700 dicalcium phosphate Nutrition 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 108010036598 gastric inhibitory polypeptide receptor Proteins 0.000 description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000002473 insulinotropic effect Effects 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 239000006186 oral dosage form Substances 0.000 description 3
- 201000009104 prediabetes syndrome Diseases 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229940076279 serotonin Drugs 0.000 description 3
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 3
- 239000004299 sodium benzoate Substances 0.000 description 3
- 235000010234 sodium benzoate Nutrition 0.000 description 3
- 238000001694 spray drying Methods 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 2
- LDHMAVIPBRSVRG-UHFFFAOYSA-O 1-methylnicotinamide Chemical compound C[N+]1=CC=CC(C(N)=O)=C1 LDHMAVIPBRSVRG-UHFFFAOYSA-O 0.000 description 2
- DVSLBDBGAXXLKZ-UHFFFAOYSA-N 2,3-diethylbenzamide Chemical compound CCC1=CC=CC(C(N)=O)=C1CC DVSLBDBGAXXLKZ-UHFFFAOYSA-N 0.000 description 2
- RUVRGYVESPRHSZ-UHFFFAOYSA-N 2-[2-(2-azaniumylethoxy)ethoxy]acetate Chemical compound NCCOCCOCC(O)=O RUVRGYVESPRHSZ-UHFFFAOYSA-N 0.000 description 2
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 2
- 238000012815 AlphaLISA Methods 0.000 description 2
- 206010002383 Angina Pectoris Diseases 0.000 description 2
- 102000007592 Apolipoproteins Human genes 0.000 description 2
- 108010071619 Apolipoproteins Proteins 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 229920003084 Avicel® PH-102 Polymers 0.000 description 2
- 201000006474 Brain Ischemia Diseases 0.000 description 2
- 208000031229 Cardiomyopathies Diseases 0.000 description 2
- 206010008120 Cerebral ischaemia Diseases 0.000 description 2
- 101800001982 Cholecystokinin Proteins 0.000 description 2
- 102100025841 Cholecystokinin Human genes 0.000 description 2
- 102100032165 Corticotropin-releasing factor-binding protein Human genes 0.000 description 2
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 2
- 206010052337 Diastolic dysfunction Diseases 0.000 description 2
- 208000007530 Essential hypertension Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102000051325 Glucagon Human genes 0.000 description 2
- 108060003199 Glucagon Proteins 0.000 description 2
- 102100032882 Glucagon-like peptide 1 receptor Human genes 0.000 description 2
- 102000018997 Growth Hormone Human genes 0.000 description 2
- 108010051696 Growth Hormone Proteins 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 101001051093 Homo sapiens Low-density lipoprotein receptor Proteins 0.000 description 2
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 2
- 208000035150 Hypercholesterolemia Diseases 0.000 description 2
- 206010058179 Hypertensive emergency Diseases 0.000 description 2
- 206010022562 Intermittent claudication Diseases 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- 108010028554 LDL Cholesterol Proteins 0.000 description 2
- 108010007622 LDL Lipoproteins Proteins 0.000 description 2
- 102000007330 LDL Lipoproteins Human genes 0.000 description 2
- 208000007177 Left Ventricular Hypertrophy Diseases 0.000 description 2
- 108010033266 Lipoprotein(a) Proteins 0.000 description 2
- 102000057248 Lipoprotein(a) Human genes 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 208000035180 MODY Diseases 0.000 description 2
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 2
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 2
- 101710151321 Melanostatin Proteins 0.000 description 2
- 102100040200 Mitochondrial uncoupling protein 2 Human genes 0.000 description 2
- 102400000064 Neuropeptide Y Human genes 0.000 description 2
- 102100038991 Neuropeptide Y receptor type 2 Human genes 0.000 description 2
- 101710197945 Neuropeptide Y receptor type 2 Proteins 0.000 description 2
- 102000028435 Neuropeptide Y4 receptor Human genes 0.000 description 2
- 108010002245 Neuropeptide Y4 receptor Proteins 0.000 description 2
- 229940127355 PCSK9 Inhibitors Drugs 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 2
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 206010038563 Reocclusion Diseases 0.000 description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 2
- 102000007562 Serum Albumin Human genes 0.000 description 2
- 108010071390 Serum Albumin Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 2
- 206010071436 Systolic dysfunction Diseases 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 102000018594 Tumour necrosis factor Human genes 0.000 description 2
- 108050007852 Tumour necrosis factor Proteins 0.000 description 2
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 2
- ASPYKPQOVYJYCA-UHFFFAOYSA-N [Mg].C(C1=CN=CC=C1)(=O)N Chemical compound [Mg].C(C1=CN=CC=C1)(=O)N ASPYKPQOVYJYCA-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000012615 aggregate Substances 0.000 description 2
- 239000012491 analyte Substances 0.000 description 2
- 239000000883 anti-obesity agent Substances 0.000 description 2
- 229940125708 antidiabetic agent Drugs 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 229940125710 antiobesity agent Drugs 0.000 description 2
- 230000006793 arrhythmia Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 206010008118 cerebral infarction Diseases 0.000 description 2
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical group C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 2
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 2
- 229940107137 cholecystokinin Drugs 0.000 description 2
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 2
- 108010083720 corticotropin releasing factor-binding protein Proteins 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 2
- 229960003834 dapagliflozin Drugs 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 229940095079 dicalcium phosphate anhydrous Drugs 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000002706 dry binder Substances 0.000 description 2
- 229960003345 empagliflozin Drugs 0.000 description 2
- OBWASQILIWPZMG-QZMOQZSNSA-N empagliflozin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(O[C@@H]3COCC3)=CC=2)=C1 OBWASQILIWPZMG-QZMOQZSNSA-N 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 2
- 238000001125 extrusion Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 235000012631 food intake Nutrition 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 229960005219 gentisic acid Drugs 0.000 description 2
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 2
- 229960004666 glucagon Drugs 0.000 description 2
- 229940049654 glyceryl behenate Drugs 0.000 description 2
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 2
- 239000000122 growth hormone Substances 0.000 description 2
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 102000043555 human LDLR Human genes 0.000 description 2
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 2
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 2
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 208000021156 intermittent vascular claudication Diseases 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 201000005857 malignant hypertension Diseases 0.000 description 2
- 201000006950 maturity-onset diabetes of the young Diseases 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- IMNDHOCGZLYMRO-UHFFFAOYSA-N n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC=C1 IMNDHOCGZLYMRO-UHFFFAOYSA-N 0.000 description 2
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 208000033808 peripheral neuropathy Diseases 0.000 description 2
- 229920001983 poloxamer Polymers 0.000 description 2
- 201000010065 polycystic ovary syndrome Diseases 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 229940079877 pyrogallol Drugs 0.000 description 2
- 238000003908 quality control method Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000000697 serotonin reuptake Effects 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 229920003109 sodium starch glycolate Polymers 0.000 description 2
- 239000008109 sodium starch glycolate Substances 0.000 description 2
- 229940079832 sodium starch glycolate Drugs 0.000 description 2
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 230000009747 swallowing Effects 0.000 description 2
- 239000008399 tap water Substances 0.000 description 2
- 235000020679 tap water Nutrition 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- 229940071104 xylenesulfonate Drugs 0.000 description 2
- HMJIYCCIJYRONP-UHFFFAOYSA-N (+-)-Isradipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)C1C1=CC=CC2=NON=C12 HMJIYCCIJYRONP-UHFFFAOYSA-N 0.000 description 1
- QKDRXGFQVGOQKS-CRSSMBPESA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methylsulfanyloxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](SC)O2)O)=CC=C1Cl QKDRXGFQVGOQKS-CRSSMBPESA-N 0.000 description 1
- BTCRKOKVYTVOLU-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-(2-cyclopropyloxyethoxy)phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCCOC3CC3)=CC=2)=C1 BTCRKOKVYTVOLU-SJSRKZJXSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- UIAGMCDKSXEBJQ-IBGZPJMESA-N 3-o-(2-methoxyethyl) 5-o-propan-2-yl (4s)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COCCOC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)[C@H]1C1=CC=CC([N+]([O-])=O)=C1 UIAGMCDKSXEBJQ-IBGZPJMESA-N 0.000 description 1
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical class COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 1
- RZTAMFZIAATZDJ-HNNXBMFYSA-N 5-o-ethyl 3-o-methyl (4s)-4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC(Cl)=C1Cl RZTAMFZIAATZDJ-HNNXBMFYSA-N 0.000 description 1
- 101150026173 ARG2 gene Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 229940127438 Amylin Agonists Drugs 0.000 description 1
- 208000031729 Bacteremia Diseases 0.000 description 1
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 108010051479 Bombesin Proteins 0.000 description 1
- 102000013585 Bombesin Human genes 0.000 description 1
- 206010006550 Bulimia nervosa Diseases 0.000 description 1
- 101100335894 Caenorhabditis elegans gly-8 gene Proteins 0.000 description 1
- 101100455752 Caenorhabditis elegans lys-3 gene Proteins 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 206010007556 Cardiac failure acute Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000002249 Diabetes Complications Diseases 0.000 description 1
- 206010012655 Diabetic complications Diseases 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 231100000491 EC50 Toxicity 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 239000004150 EU approved colour Substances 0.000 description 1
- 208000030814 Eating disease Diseases 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- MCIACXAZCBVDEE-CUUWFGFTSA-N Ertugliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@]23O[C@@](CO)(CO2)[C@@H](O)[C@H](O)[C@H]3O)=CC=C1Cl MCIACXAZCBVDEE-CUUWFGFTSA-N 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- 108010011459 Exenatide Proteins 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- 102400001370 Galanin Human genes 0.000 description 1
- 101800002068 Galanin Proteins 0.000 description 1
- 229940099498 Gastric inhibitory polypeptide receptor agonist Drugs 0.000 description 1
- 102400000921 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- 241000237858 Gastropoda Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 102000004366 Glucosidases Human genes 0.000 description 1
- 108010056771 Glucosidases Proteins 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 108091016366 Histone-lysine N-methyltransferase EHMT1 Proteins 0.000 description 1
- 101500028774 Homo sapiens Glucagon-like peptide 1 Proteins 0.000 description 1
- 101001015516 Homo sapiens Glucagon-like peptide 1 receptor Proteins 0.000 description 1
- 101500028773 Homo sapiens Glucagon-like peptide 1(7-37) Proteins 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102000016267 Leptin Human genes 0.000 description 1
- 108010092277 Leptin Proteins 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 208000017170 Lipid metabolism disease Diseases 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHSOLWOTCHFFBK-ZQGJOIPISA-N Luseogliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)S2)O)=C(OC)C=C1C WHSOLWOTCHFFBK-ZQGJOIPISA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108010008364 Melanocortins Proteins 0.000 description 1
- 229920003091 Methocel™ Polymers 0.000 description 1
- 208000004302 Microvascular Angina Diseases 0.000 description 1
- 208000026018 Microvascular coronary artery disease Diseases 0.000 description 1
- 101710112393 Mitochondrial uncoupling protein 2 Proteins 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 241000238367 Mya arenaria Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- 241000275031 Nica Species 0.000 description 1
- ZBBHBTPTTSWHBA-UHFFFAOYSA-N Nicardipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZBBHBTPTTSWHBA-UHFFFAOYSA-N 0.000 description 1
- 102000002512 Orexin Human genes 0.000 description 1
- 102400000319 Oxyntomodulin Human genes 0.000 description 1
- 101800001388 Oxyntomodulin Proteins 0.000 description 1
- 229940122985 Peptide agonist Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 102000004257 Potassium Channel Human genes 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 102100040918 Pro-glucagon Human genes 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000007107 Stomach Ulcer Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 102000011923 Thyrotropin Human genes 0.000 description 1
- 108010061174 Thyrotropin Proteins 0.000 description 1
- ZXOCGDDVNPDRIW-NHFZGCSJSA-N Tofogliflozin Chemical compound O.C1=CC(CC)=CC=C1CC1=CC=C(CO[C@@]23[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)C2=C1 ZXOCGDDVNPDRIW-NHFZGCSJSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 108010021111 Uncoupling Protein 2 Proteins 0.000 description 1
- 102000008200 Uncoupling Protein 3 Human genes 0.000 description 1
- 108010021098 Uncoupling Protein 3 Proteins 0.000 description 1
- ICMGLRUYEQNHPF-UHFFFAOYSA-N Uraprene Chemical compound COC1=CC=CC=C1N1CCN(CCCNC=2N(C(=O)N(C)C(=O)C=2)C)CC1 ICMGLRUYEQNHPF-UHFFFAOYSA-N 0.000 description 1
- 108010059705 Urocortins Proteins 0.000 description 1
- 102000005630 Urocortins Human genes 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 description 1
- 125000002252 acyl group Chemical class 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229960004539 alirocumab Drugs 0.000 description 1
- 239000002160 alpha blocker Substances 0.000 description 1
- 229940124308 alpha-adrenoreceptor antagonist Drugs 0.000 description 1
- 229960002213 alprenolol Drugs 0.000 description 1
- PAZJSJFMUHDSTF-UHFFFAOYSA-N alprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1CC=C PAZJSJFMUHDSTF-UHFFFAOYSA-N 0.000 description 1
- 229940025084 amphetamine Drugs 0.000 description 1
- 239000003392 amylase inhibitor Substances 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000561 anti-psychotic effect Effects 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000003910 behenoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960004530 benazepril Drugs 0.000 description 1
- KKBIUAUSZKGNOA-HNAYVOBHSA-N benzyl (2s)-2-[[(2s)-2-(acetylsulfanylmethyl)-3-(1,3-benzodioxol-5-yl)propanoyl]amino]propanoate Chemical compound O=C([C@@H](NC(=O)[C@@H](CSC(C)=O)CC=1C=C2OCOC2=CC=1)C)OCC1=CC=CC=C1 KKBIUAUSZKGNOA-HNAYVOBHSA-N 0.000 description 1
- 229950003611 bexagliflozin Drugs 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 208000014679 binge eating disease Diseases 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- DNDCVAGJPBKION-DOPDSADYSA-N bombesin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC2=CC=CC=C2C=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DNDCVAGJPBKION-DOPDSADYSA-N 0.000 description 1
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 description 1
- 229960002802 bromocriptine Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 229960001713 canagliflozin Drugs 0.000 description 1
- VHOFTEAWFCUTOS-TUGBYPPCSA-N canagliflozin hydrate Chemical compound O.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1 VHOFTEAWFCUTOS-TUGBYPPCSA-N 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000005465 channeling Effects 0.000 description 1
- 230000001055 chewing effect Effects 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229960001214 clofibrate Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 229960003920 cocaine Drugs 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229920001531 copovidone Polymers 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 229960005168 croscarmellose Drugs 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- NKLCHDQGUHMCGL-UHFFFAOYSA-N cyclohexylidenemethanone Chemical group O=C=C1CCCCC1 NKLCHDQGUHMCGL-UHFFFAOYSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 229960001767 dextrothyroxine Drugs 0.000 description 1
- 229940061607 dibasic sodium phosphate Drugs 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 235000014632 disordered eating Nutrition 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229960001389 doxazosin Drugs 0.000 description 1
- RUZYUOTYCVRMRZ-UHFFFAOYSA-N doxazosin Chemical compound C1OC2=CC=CC=C2OC1C(=O)N(CC1)CCN1C1=NC(N)=C(C=C(C(OC)=C2)OC)C2=N1 RUZYUOTYCVRMRZ-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 201000006549 dyspepsia Diseases 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229950006535 ertugliflozin Drugs 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229960002027 evolocumab Drugs 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 229960001519 exenatide Drugs 0.000 description 1
- 229960003580 felodipine Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 230000030136 gastric emptying Effects 0.000 description 1
- 230000030135 gastric motility Effects 0.000 description 1
- 210000005095 gastrointestinal system Anatomy 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960003627 gemfibrozil Drugs 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 208000004104 gestational diabetes Diseases 0.000 description 1
- 238000007446 glucose tolerance test Methods 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 230000004116 glycogenolysis Effects 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000010005 growth-factor like effect Effects 0.000 description 1
- 229960004198 guanidine Drugs 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 239000003395 histamine H3 receptor antagonist Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229950000991 ipragliflozin Drugs 0.000 description 1
- AHFWIQIYAXSLBA-RQXATKFSSA-N ipragliflozin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(F)C(CC=2SC3=CC=CC=C3C=2)=C1 AHFWIQIYAXSLBA-RQXATKFSSA-N 0.000 description 1
- 229960004427 isradipine Drugs 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229940039781 leptin Drugs 0.000 description 1
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 1
- 102000005861 leptin receptors Human genes 0.000 description 1
- 108010019813 leptin receptors Proteins 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 229950004397 luseogliflozin Drugs 0.000 description 1
- 229940037627 magnesium lauryl sulfate Drugs 0.000 description 1
- HBNDBUATLJAUQM-UHFFFAOYSA-L magnesium;dodecyl sulfate Chemical compound [Mg+2].CCCCCCCCCCCCOS([O-])(=O)=O.CCCCCCCCCCCCOS([O-])(=O)=O HBNDBUATLJAUQM-UHFFFAOYSA-L 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229950004994 meglitinide Drugs 0.000 description 1
- 239000002865 melanocortin Substances 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- SLZIZIJTGAYEKK-CIJSCKBQSA-N molport-023-220-247 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)CN)[C@@H](C)O)C1=CNC=N1 SLZIZIJTGAYEKK-CIJSCKBQSA-N 0.000 description 1
- PXZWGQLGAKCNKD-DPNMSELWSA-N molport-023-276-326 Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 PXZWGQLGAKCNKD-DPNMSELWSA-N 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229960001783 nicardipine Drugs 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960000715 nimodipine Drugs 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 230000002474 noradrenergic effect Effects 0.000 description 1
- 230000000966 norepinephrine reuptake Effects 0.000 description 1
- 229940006093 opthalmologic coloring agent diagnostic Drugs 0.000 description 1
- 108060005714 orexin Proteins 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229960002508 pindolol Drugs 0.000 description 1
- PHUTUTUABXHXLW-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=NC=C[C]12 PHUTUTUABXHXLW-UHFFFAOYSA-N 0.000 description 1
- BXRNXXXXHLBUKK-UHFFFAOYSA-N piperazine-2,5-dione Chemical group O=C1CNC(=O)CN1 BXRNXXXXHLBUKK-UHFFFAOYSA-N 0.000 description 1
- 229960000540 polacrilin potassium Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000018656 positive regulation of gluconeogenesis Effects 0.000 description 1
- 108020001213 potassium channel Proteins 0.000 description 1
- WVWZXTJUCNEUAE-UHFFFAOYSA-M potassium;1,2-bis(ethenyl)benzene;2-methylprop-2-enoate Chemical compound [K+].CC(=C)C([O-])=O.C=CC1=CC=CC=C1C=C WVWZXTJUCNEUAE-UHFFFAOYSA-M 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229920003124 powdered cellulose Polymers 0.000 description 1
- 235000019814 powdered cellulose Nutrition 0.000 description 1
- 229940028952 praluent Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 229960001289 prazosin Drugs 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 230000006920 protein precipitation Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 229950011516 remogliflozin etabonate Drugs 0.000 description 1
- UAOCLDQAQNNEAX-ABMICEGHSA-N remogliflozin etabonate Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](COC(=O)OCC)O[C@H]1OC1=NN(C(C)C)C(C)=C1CC1=CC=C(OC(C)C)C=C1 UAOCLDQAQNNEAX-ABMICEGHSA-N 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 229940017164 repatha Drugs 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000036186 satiety Effects 0.000 description 1
- 235000019627 satiety Nutrition 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 230000002295 serotoninergic effect Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- UOENJXXSKABLJL-UHFFFAOYSA-M sodium;8-[(2-hydroxybenzoyl)amino]octanoate Chemical compound [Na+].OC1=CC=CC=C1C(=O)NCCCCCCCC([O-])=O UOENJXXSKABLJL-UHFFFAOYSA-M 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 229950005268 sotagliflozin Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000012421 spiking Methods 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 229960004274 stearic acid Drugs 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 230000035488 systolic blood pressure Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960001693 terazosin Drugs 0.000 description 1
- VCKUSRYTPJJLNI-UHFFFAOYSA-N terazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1CCCO1 VCKUSRYTPJJLNI-UHFFFAOYSA-N 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 108091004331 tirzepatide Proteins 0.000 description 1
- BTSOGEDATSQOAF-SMAAHMJQSA-N tirzepatide Chemical group CC[C@H](C)[C@@H](C(N[C@@H](C)C(N[C@@H](CCC(N)=O)C(N[C@@H](CCCCNC(COCCOCCNC(COCCOCCNC(CC[C@H](C(O)=O)NC(CCCCCCCCCCCCCCCCCCC(O)=O)=O)=O)=O)=O)C(N[C@@H](C)C(N[C@@H](CC1=CC=CC=C1)C(N[C@@H](C(C)C)C(N[C@@H](CCC(N)=O)C(N[C@@H](CC1=CNC2=C1C=CC=C2)C(N[C@@H](CC(C)C)C(N[C@@H]([C@@H](C)CC)C(N[C@@H](C)C(NCC(NCC(N(CCC1)[C@@H]1C(N[C@@H](CO)C(N[C@@H](CO)C(NCC(N[C@@H](C)C(N(CCC1)[C@@H]1C(N(CCC1)[C@@H]1C(N(CCC1)[C@@H]1C(N[C@@H](CO)C(N)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)NC([C@H](CCCCN)NC([C@H](CC(O)=O)NC([C@H](CC(C)C)NC(C(C)(C)NC([C@H]([C@@H](C)CC)NC([C@H](CO)NC([C@H](CC(C=C1)=CC=C1O)NC([C@H](CC(O)=O)NC([C@H](CO)NC([C@H]([C@@H](C)O)NC([C@H](CC1=CC=CC=C1)NC([C@H]([C@@H](C)O)NC(CNC([C@H](CCC(O)=O)NC(C(C)(C)NC([C@H](CC(C=C1)=CC=C1O)N)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O BTSOGEDATSQOAF-SMAAHMJQSA-N 0.000 description 1
- 229940121512 tirzepatide Drugs 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 229950006667 tofogliflozin Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- 210000001364 upper extremity Anatomy 0.000 description 1
- 229960001130 urapidil Drugs 0.000 description 1
- 239000000777 urocortin Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
- 239000002888 zwitterionic surfactant Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/20—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/455—Nicotinic acids, e.g. niacin; Derivatives thereof, e.g. esters, amides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/26—Glucagons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to solid compositions comprising a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid and a further absorption enhancer, their method of preparation and their use in medicine.
- SEQUENCE LISTING The present application is filed with a Sequence Listing in electronic form. The entire contents of the sequence listing are hereby incorporated by reference. BACKGROUND Oral administration of peptide therapeutics has been an area of intense research and with the recent approval of Rybelsus® a first tablet product has been launched allowing patients to received GLP-1 RA treatment without the need for injection.
- EGF(A) Epidermal Growth Factor-like domain A sequence (40 amino acids) of the LDL-R (LDL-R-(293-332)) is well recognized as the site for PCSK9 binding.
- the isolated wild- type EGF(A) peptide has been shown to inhibit the binding of PCSK9 to the LDL-R with an IC 50 in the low ⁇ M range (Biochemical and Biophysical Research Communications 375 (2008) 69–73). This poor potency has prevented a practical pharmaceutical use of the EGF(A) peptide. Furthermore, the half-life of such peptides would be expected to be too short to be of therapeutic use.
- WO2012177741 and J. Mol. Biol. (2012) 422, 685-696 disclose analogues of the EGF(A) and Fc-Fusion thereof.
- Alternative EGF(A) peptide based PCSK9 inhibitors with an extended half-life have been disclosed in WO2017/121850.
- the percentage of the peptide therapeutic that reaches circulation after oral administration is still in the lower single digits compared to subcutaneous administration and thus further alternative compositions providing increased bioavailability of peptides are highly desirable.
- a more cost-effective product can be obtained by combing absorption enhancers. Considering the low bioavailability of a peptide therapeutic after oral administration, the amounts of API and excipients (particular absorption enhancers) are substantial, and thus the ability to reduce the amount of API and/or excipient(s) needed to reach a given level of exposure of a peptide therapeutic is advantageous.
- the present invention describes solid pharmaceutical compositions which includes a mix of absorption enhancers.
- the solid pharmaceutical composition comprises a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid and a fatty acid such as a salt of capric acid.
- the solid pharmaceutical composition comprises a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid, a fatty acid such as a salt of capric acid and a hydrotrope such as nicotinamide.
- the composition comprises a peptide therapeutic.
- the solid pharmaceutical composition comprises i) a peptide therapeutic ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
- the peptide therapeutic is an acylated peptide, such as a GLP-1 receptor agonist, a PCSK9 inhibitor peptide or an insulin analogue.
- the peptide therapeutics may comprise one or more albumin binding moieties covalently attached to the peptides, such as in the form of a substituent comprising an albumin binding moiety.
- the solid pharmaceutical composition comprises i) a GLP-1 agonist, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
- the solid pharmaceutical composition comprises i) semaglutide or a pro-drug of semaglutide ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
- the solid pharmaceutical composition comprises i) a GLP-1-GIP co-agonist, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
- the solid pharmaceutical composition comprises i) a PCSK9 inhibitor peptide, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
- the solid pharmaceutical composition comprises i) an insulin analogue, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- a fatty acid consisting of 6-14 carbon atoms or a salt hereof iv) nicotinamide
- the invention relates to medical use of a composition as described herein, such as the use in a method of treatment of diabetes, obesity and/or cardiovascular diseases.
- DESCRIPTION Peptide therapeutics refers to a compound which comprises a series of amino acids interconnected by amide (or peptide) bonds.
- a peptide therapeutic may also herein be referred to as a therapeutic peptide and is encompassed by the term active pharmaceutical ingredient as relevant.
- a peptide therapeutic is a peptide-based compound suited for medical treatment, comprising at least 20 amino acid residues. In further embodiments the peptide therapeutic comprises at least 30, such as at least 40, such as at least 50 amino acid residues. In one embodiment the peptide therapeutic comprises 20-500 amino acids residues. In one embodiment the peptide therapeutic may comprise one or more modified amino acid residues and/or non- proteogenic amino acid residues.
- An example of a peptide therapeutics is semaglutide, and other GLP-1 agonists described below, and thus a peptide therapeutic is frequently an analogue of a naturally existing peptide.
- the peptide therapeutic has an extended half-life compared to the naturally existing peptide.
- the peptide therapeutic has a plasma half-life in humans of at least 24 hours, such as at least 48 hours, such as at least 72 hours.
- the peptide therapeutic has a plasma half-life in humans of at least 96 hours, such as 120-200 hours.
- the plasma half-life of a peptide may be extended by attaching an albumin binding fatty acid to a peptide, which has been demonstrated for several peptide and proteins.
- the peptide therapeutic is a fatty acid substituted peptide.
- the peptide therapeutic has an albumin binding substituent.
- the peptide therapeutic comprises an albumin binding moiety. In one embodiment the peptide therapeutic comprises two albumin binding moieties. In one embodiment the peptide therapeutic is a pro-drug. In one embodiment the peptide therapeutic is a pro-drug comprising a pharmaceutical active peptide and a dipeptide moiety which is converted into a diketopiperazine moiety upon liberation of the pharmaceutical active peptide, such as those described in WO2010/071807, WO2010/080605, WO2011/163012, WO2014/152460 and WO2016/049174. In general, the term peptide therapeutic is meant to encompass compound as such and any pharmaceutically acceptable salt, amide, or ester thereof.
- the composition comprises the peptide therapeutic or a pharmaceutically acceptable salt, amide, or ester thereof. In some embodiments the composition comprises the peptide therapeutic and one or more pharmaceutically acceptable counter ions.
- Substituent A substituent is a moiety comprised by the peptide therapeutic. According to the invention it is preferred that the moiety e.g., the substituent has no or minimal effect on the biological functionality of the peptide therapeutic while adding other beneficial properties, such as longer half-life and/or improved exposure after oral dosing.
- the peptide therapeutic comprises a substituent comprising an albumin binding moiety. In one embodiment the peptide therapeutic comprises two substituents comprising an albumin binding moiety.
- the substituent comprising an albumin binding moiety is covalently attached to the peptide. In one embodiment the substituent comprising an albumin binding moiety is attached via a lysine residue. In one embodiment the substituent comprising an albumin binding moiety is attached to the peptide back-bone via an epsilon nitrogen of a lysine residue. In some embodiments the substituent comprises a fatty acid or a fatty diacid. In some embodiments the substituent comprises a C16, C18 or C20 fatty acid. In some embodiments the substituent comprises a C16, C18 or C20 fatty diacid.
- the albumin binding moiety is selected from the group consisting of: Chem.1: HOOC-(CH 2 ) n -CO-* wherein n is an integer in the range of 8-20, Chem.2: 5-tetrazolyl-(CH 2 ) n -CO-* wherein n is an integer in the range of 8-20, Chem.3: HOOC-(C 6 H 4 )-O-(CH 2 ) n -CO-* wherein n is an integer in the range of 6-20, Chem.4: HO-S(O) 2 -(CH 2 ) n -CO-* wherein n is an integer in the range of 8-20, Chem.5: MeS(O) 2 NH(CO)NH-(CH 2 ) n -CO-* wherein n is an integer in the range of 8-20 and Chem.6: 3-HO-Isoxazole-(CH 2 ) n -CO-* wherein n is
- the substituent comprises Chem.1: HOOC-(CH 2 ) n -CO-* wherein n is at least 13, such as n is 13, 14, 15, 16, 17, 18 or 19. In some embodiments n is in the range of 13 to 19, such as in the range of 13 to 17. In some embodiments n is 13, 15 or 17. In some embodiments n is 13. In some embodiments n is 15. In some embodiments n is 17. The diacid part may also be referred to using a systematic name as follows. In some embodiments the substituent comprises Chem.3: HOOC-(C 6 H 4 )-O-(CH 2 ) n -CO-* wherein n is an integer in the range of 6-14.
- the substituent comprises Chem 3b wherein the carboxy group is in position 2, 3 or 4 of the (C 6 H 4 ) group and wherein m is an integer in the range of 8-11. In some embodiments the substituent comprises Chem 3 or Chem 3b wherein n/m is in the range of 6 to 14, such as in the range of 8 to 11. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein n/m is 8, 10 or 12. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein n/m is 9. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein/ m is 11.
- linkers Additional elements may be referred to as linkers, and it follows that the substituent may then comprise or consist of an albumin binding moiety and one or more linker elements which are referred to as linker A, linker B and linker C elements herein.
- the substituent comprises a linker A element selected from Chem.7: *-NH-SO 2 -(CH 2 ) 3 -CO-* or and Chem.8: -NH-CH 2 -(C 6 H 10 )-CO- or . comprises one or more linker B elements.
- linker B element(s) is/are selected from the group consisting of: Glu, ⁇ Glu, Gly, Ser, Ala, Thr, Ado (or OEG), Aeep, Aeeep and TtdSuc. Glu, Gly, Ser, Ala, Thr are amino acid residues well known in the art.
- ⁇ Glu (or gGlu) is of formula Chem.9: *-NH-CH(COOH)-(CH 2 ) 2 -CO-* which is the same as Chem. 9b: TtdSuc is of formula Chem.10: *-NH-(CH 2 ) 3 -O-(CH 2 ) 2 -O-(CH 2 ) 2 O-(CH 2 ) 3 -NHCO* or *-NH-CH 2 CH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 CH 2 NHCO* which is the same as Chem.10b
- Ado (or OEG) is of formula Chem.11: *-NH-(CH 2 ) 2 -O-(CH 2 ) 2 -O-CH 2 -CO-* may also be referred to as 8-amino-3,6-dioxaoctanoic acid and which is the same as Chem.11b Aeep is of formula Chem.12: *NH-CH 2 CH 2
- the substituent comprises one or more ⁇ -Lys, such as two ⁇ -Lys elements.
- the substituent may further comprise a linker C element of Chem. 15: *-NH-CH 2 -(C 6 H 4 )-CH 2 -*, which may also be referred to as ne or two substituent(s) is/are selected from the group of substituents consisting of: HOOC-(CH 2 ) 18 -CO-gGlu-2xAdo HOOC-(CH 2 ) 18 -CO-NH-CH 2 -(C 6 H 10 )-CO-gGlu-2xAdo HOOC-(CH 2 ) 16 -CO-gGlu-2xAdo HOOC-(CH 2 ) 16 -CO-gGlu-2xAdo HOOC-(CH 2 ) 16 -CO-gGlu-2xAdo-NH-CH 2 -(C 6 H 4 )-CH 2 HOOC-(CH 2 ) 16 -CO-gGlu
- the substituent is [2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-(17- carboxyheptadecanoylamino) butyrylamino]ethoxy ⁇ ethoxy)acetylamino] ethoxy ⁇ ethoxy)acetyl]. In some embodiments the substituent is [2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-( ⁇ trans-4-[(19- carboxynonadecanoylamino)methyl]cyclohexanecarbonyl ⁇ amino)butyrylamino]ethoxy ⁇ ethoxy)acetylamino]ethoxy ⁇ ethoxy)acetyl].
- GLP-1 receptor agonists/GLP-1 agonist In one embodiment the peptide therapeutic is a GLP-1 analogue. In one embodiment the peptide therapeutic is a GLP-1 receptor agonist, also referred to as a GLP-1 agonist herein.
- GLP-1 agonist refers to a compound, which fully or partially activates the human GLP-1 receptor. The term is thus equal to the term “GLP-1 receptor agonist” used in other documents.
- GLP-1 agonist as well as the specific GLP-1 agonists described herein also encompass salt forms thereof. It follows that the GLP-1 agonist should display “GLP-1 activity” which refers to the ability of the compound, i.e.
- the “GLP-1 agonist” binds to a GLP-1 receptor, e.g., with an affinity constant (K D ) or activate the receptor with a potency (EC 50 ) of below 1 ⁇ M, e.g. below 100 nM as measured by methods known in the art (see e.g. WO 98/08871) and exhibits insulinotropic activity, where insulinotropic activity may be measured in vivo or in vitro assays known to those of ordinary skill in the art.
- K D affinity constant
- EC 50 potency
- the GLP-1 agonist may be administered to an animal with increased blood glucose (e.g. obtained using an Intravenous Glucose Tolerance Test (IVGTT).
- IVGTT Intravenous Glucose Tolerance Test
- a person skilled in the art will be able to determine a suitable glucose dosage and a suitable blood sampling regime, e.g. depending on the species of the animal, for the IVGTT) and measure the plasma insulin concentration over time. Suitable assays have been described in such as WO2015/155151.
- the term half maximal effective concentration (EC 50 ) generally refers to the concentration which induces a response halfway between the baseline and maximum, by reference to the dose response curve. EC 50 is used as a measure of the potency of a compound and represents the concentration where 50% of its maximal effect is observed.
- the in vitro potency of the GLP-1 agonist may be determined as described in WO2015/155151, example 29 without Human Serum Albumin (HSA), and the EC 50 determined. The lower the EC 50 value, the better the potency.
- the potency (EC 50 ) as determined (without HSA) is 5-1000 pM, such as 10-750 pM, 10-500 pM or 10-200 pM.
- the EC 50 (without HSA) is at most 500 pM, such as at most 300 pM, such as at most 200 pM.
- the EC50 (without HSA) is comparable to human GLP-1(7-37). In one embodiment the EC 50 (without HSA) is at most 50 pM. In a further such embodiment the EC 50 is at most 40 pM, such as at most 30 pM such as at most 20 pM, such as at most 10 pM. In one embodiment the EC 50 is around 10 pM. Also, or alternatively, the binding of the GLP-1 agonist to albumin may be measured using the in vitro potency assay of Example 29 in WO2015/155151 including HSA. An increase of the in vitro potency, EC 50 value, in the presence of serum albumin reflects the affinity to serum albumin.
- the potency (EC 50 ) as determined (with 1 % HSA) is 5-1000 pM, such as 100-750 pM, 200-500 pM or 100-400 pM. In one embodiment the EC 50 (with 1 % HSA) is at most 750 pM, such as at most 500 pM, such as at most 400 pM, such as at most 300 or such as at most 250 pM.
- the fold variation in relation to a known GLP-1 receptor agonist may be calculated as EC 50 (test analogue)/EC 50 (known analogue), and if this ratio is such as 0.5-1.5, or 0.8-1.2 the potencies are considered to be equivalent.
- the potency, EC 50 (without HSA), is equivalent to the potency of liraglutide. In one embodiment the potency, EC 50 (without HSA), is equivalent to the potency of semaglutide. In one embodiment the potency, EC 50 (with 1 % HSA), is equivalent to the potency of liraglutide. In one embodiment the potency, EC 50 (with 1 % HSA), is equivalent to the potency of semaglutide. In some embodiments the GLP-1 agonist is a GLP-1 analogue, optionally comprising one substituent.
- analogue as used herein referring to a GLP-1 peptide (hereafter “peptide”) means a peptide wherein at least one amino acid residue of the peptide has been substituted with another amino acid residue and/or wherein at least one amino acid residue has been deleted from the peptide and/or wherein at least one amino acid residue has been added to the peptide and/or wherein at least one amino acid residue of the peptide has been modified. Such addition or deletion of amino acid residues may take place at the N-terminal of the peptide and/or at the C-terminal of the peptide.
- GLP-1 agonist designates an analogue of GLP-1(7-37) wherein the naturally occurring Ala in position 8 has been substituted with Aib.
- GLP-1 agonist comprises a maximum of twelve, such as a maximum of 10, 8 or 6, amino acids which have been altered, e.g., by substitution, deletion, insertion and/or modification, compared to e.g. GLP-1(7-37).
- the analogue comprises up to 10 substitutions, deletions, additions and/or insertions, such as up to 9 substitutions, deletions, additions and/or insertions, up to 8 substitutions, deletions, additions and/or insertions, up to 7 substitutions, deletions, additions and/or insertions, up to 6 substitutions, deletions, additions and/or insertions, up to 5 substitutions, deletions, additions and/or insertions, up to 4 substitutions, deletions, additions and/or insertions or up to 3 substitutions, deletions, additions and/or insertions, compared to e.g. GLP-1(7-37). Unless otherwise stated the GLP-1 agonist comprises only L-amino acids.
- GLP-1 analogue or “analogue of GLP-1” as used herein refers to a peptide, or a compound, which is a variant of the human Glucagon-Like Peptide-1 (GLP-1(7-37)).
- GLP-1(7-37) has the sequence HAEGTFTSDV SSYLEGQAAKEFIAWLVKGRG (SEQ ID No.: 1).
- variant refers to a compound which comprises one or more amino acid substitutions, deletions, additions and/or insertions.
- the GLP-1 agonist exhibits at least 60%, 65%, 70%, 80% or 90% sequence identity to GLP-1(7-37) over the entire length of GLP-1(7-37).
- sequence identity As an example of a method for determination of sequence identity between two analogues the two peptides [Aib8]GLP-1(7-37) and GLP-1(7-37) are aligned.
- the sequence identity of [Aib8]GLP-1(7-37) relative to GLP-1(7-37) is given by the number of aligned identical residues minus the number of different residues divided by the total number of residues in GLP-1(7-37). Accordingly, in said example the sequence identity is (31-1)/31.
- the C-terminal of the GLP-1 agonist is an amide.
- the GLP-1 agonist is GLP-1(7-37) or GLP-1(7-36)amide.
- the GLP-1 agonist is exendin-4, the sequence of which is HGEGTFITSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS (SEQ ID No.: 2).
- the GLP-1 agonist is an exendin-4 analogue or an engineered peptide thereof, as disclosed in WO2013/009545 and references therein. In order to prolong the effect of the GLP-1 agonist it is preferred that the GLP-1 agonist have an extended half-life.
- the half-life can be determined by method known in the art an in an appropriate model, such as in Male Sprague Dawley rats or minipigs as described in WO2012/140117. Half-life in rats may be determined as in Example 39 and the half-life in minipigs may be determined as in Example 37 therein.
- the GLP-1 agonist according to the invention has a half-life above 2 hours in rat. In one embodiment the GLP-1 agonist according to the invention has a half-life above 4 hours, such as above 6 hours, such as above 8 hours, such as above 10 hours, such as above 12 hours or such as above 15 hours in rat. In one embodiment the GLP-1 agonist according to the invention has a half-life above 24 hours in minipig.
- the GLP-1 agonist according to the invention has a half- life above 30 hours, such as above 36 hours, such as above 42 hours, such as above 48 hours, such as above 54 hours or such as above 60 hours in minipig.
- the GLP-1 agonist has a molecular weight of at most 50000 Da, such as at most 40000 Da, such as at most 30000 Da.
- the GLP-1 agonist has a molecular weight of at most 20000, such as at most 10000 Da, such as at most 7500 Da, such as at most 5000 Da.
- the GLP-1 agonist has a molar mass of at most 50000 g/mol, such as at most 40000 g/mol, such as at most 30000 g/mol.
- the GLP-1 agonist has a molar mass of at most 10000 g/mol, such as at most 8000 g/mol, such as at most 6000 g/mol.
- the GLP-1 agonist comprises one substituent which is covalently attached to the peptide as described herein above.
- the substituent is [2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-(17- carboxyheptadecanoylamino) butyrylamino]ethoxy ⁇ ethoxy)acetylamino] ethoxy ⁇ ethoxy)acetyl].
- the substituent is [2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-( ⁇ trans-4-[(19- carboxynonadecanoylamino)methyl]cyclohexanecarbonyl ⁇ amino)butyrylamino]ethoxy ⁇ ethoxy)acetylamino]ethoxy ⁇ ethoxy)acetyl].
- the GLP-1 agonist is liraglutide.
- the GLP-1 agonist is semaglutide, also known as N-epsilon26-[2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-(17-carboxyheptadecanoylamino) butyrylamino]ethoxy ⁇ ethoxy)acetylamino]ethoxy ⁇ ethoxy)acetyl][Aib8,Arg34]GLP-1(7-37), (SEQ ID No.: 3) which may be prepared as described in WO2006/097537, Example 4 with the following structure:
- the GLP-1 agonist is GLP-1 peptide 1 which is diacylated [Aib8,Arg34,Lys37]GLP-1(7-37) (SEQ ID No.: 4) as shown in Example 2 of WO2011/080103 and named N ⁇ ⁇ ⁇ ⁇ 2-[2-(2- ⁇ (S)-4-Carboxy-4-[10-(4-
- the GLP-1 agonist is selected from one or more of the GLP-1 agonists mentioned in WO93/19175, WO96/29342, WO98/08871, WO99/43707, WO99/43706, WO99/43341, WO99/43708, WO2005/027978, WO2005/058954, WO2005/058958, WO2006/005667, WO2006/037810, WO2006/037811, WO2006/097537, WO2006/097538, WO2008/023050, WO2009/030738, WO2009/030771 and WO2009/030774.
- the GLP-1 agonist is selected from the group consisting of N- epsilon37 ⁇ 2-[2-(2- ⁇ 2-[2-((R)-3-carboxy-3- ⁇ [1-(19-carboxynonadecanoyl) piperidine-4- carbonyl]amino ⁇ propionylamino)ethoxy]ethoxy ⁇ acetylamino)ethoxy]ethoxy ⁇ acetyl [desaminoHis7,Glu22,Arg26,Arg34,Lys37]GLP-1(7-37)amide; N-epsilon26 ⁇ 2-[2-(2- ⁇ 2-[2-((R)-3- carboxy-3- ⁇ [1-(19-carboxynonadecanoyl) piperidine-4-carbonyl]amino ⁇ propionylamino)ethoxy]ethoxy ⁇ acetylamino)ethoxy] ethoxy ⁇ acetyl [desaminoHis7, Arg34]
- GLP-1 agonist as used herein includes pro-drugs of GLP-1 agonists.
- the GLP-1 agonist is a pro-drug of semaglutide.
- the GLP-1 agonist is a pro-drug of semaglutide as described in WO2022/096636, such as in Example 1 therein providing: Gly-N ⁇ -2-[[(4S)-4-carboxy-4-(15-carboxypentadecanoylamino)butanoyl]amino]ethyl-Gly- semaglutide (SEQ ID No.: 26) with the structure
- the GLP-1 agonist is also a Gastric inhibitory polypeptide receptor agonist (GIP agonist).
- the GLP-1 agonist is a GLP-1/GIP receptor co-agonists as described in WO2022/018186. In one embodiment the GLP-1 agonist is selected from the group of GLP-1/GIP receptor co-agonists compounds described in WO2022/018186 In one embodiment the GLP-1 agonist is the GLP-1/GIP receptor co-agonists identical to compound 31 described in WO2022/018186 and having the amino acid sequence included as SEQ ID No.: 25 herein. In one embodiment the GLP-1 agonist is Tirzepatide. PCSK9 inhibitor peptides In one embodiment the peptide therapeutics is a PCSK9 inhibitor peptide.
- PCSK9 inhibitor peptide is a peptide molecule, which fully or partially prevents PCSK9 from binding to the human Low Density Lipoprotein Receptor (LDL-R).
- LDL-R Low Density Lipoprotein Receptor
- the EGF(A) LDL-R(293- 332) peptide binds PCSK9, but is not considered a PCSK9 inhibitor due to a relatively week binding to PCSK9.
- the EGF(A) LDL-R(293-332) peptide may also be referred to as an the EGF(A) domain of LDL-R or in short just as EGF(A) wherein the peptide is identified by the amino acid sequence: Gly-Thr-Asn-Glu-Cys-Leu-Asp-Asn-Asn-Gly-Gly-Cys-Ser-His-Val-Cys- Asn-Asp-Leu-Lys-Ile-Gly-Tyr-Glu-Cys-Leu-Cys-Pro-Asp-Gly-Phe-Gln-Leu-Val-Ala-Gln-Arg-Arg- Cys-Glu (SEQ ID No.: 6).
- the PCSK9i peptide is an EGF(A) analogue.
- EGF(A) analogues are potent PCSK9 inhibitor peptides, which may be measured in an ELISA assay (such as method D.1.1 page 175 of WO2017/121850) providing the apparent binding affinity of the EGF(A) analogue or a compound comprising an EGF(A) analogue reported as a K i .
- a low K i is thus characteristic for compounds with a strong inhibitory function as described in WO2017/121850.
- a suitable PCSK9 inhibitor has a K i below 8 nM, such as below 5 nM.
- the PCSK9 inhibitor has a K i around 0.5-8 nM, or such as 0.5-5 nM or such as 1.0-4 nM when determined as described in WO2017/121850 (D.1.1).
- the term PCSK9 inhibitor peptide herein encompasses EGF(A) peptide analogues and derivatives hereof comprising a substituent as described herein above and may further be referred to as PCSK9 inhibitor peptide therapeutics.
- the PCSK9 inhibitor peptide is selected from one or more of the EGF(A) peptide analogues and derivatives hereof mentioned in WO2017/121850.
- the PCSK9 inhibitor peptide comprises an amino acid sequence identified by the group of sequences defined by SEQ ID No.: 8-20. In one embodiment the PCSK9 inhibitor peptide comprises an amino acid sequence identified by the group of sequences defined by SEQ ID No.: 13-20. In one embodiment the PCSK9 inhibitor peptide comprises an amino acid sequence identified by the group of sequences defined by SEQ ID No.: 16-19. In one embodiment the PCSK9 inhibitor peptide comprises an amino acid sequence identified by the group of sequences defined by SEQ ID No.: 16.
- the PCSK9 inhibitor peptide is selected from: Compounds A to J (disclosed as Examples compounds 31, 95, 128, 133, 143, 144, 150, 151, 152 and 153 in WO2017/121850) having the structures shown below and including the SEQ ID reference for the amino acid sequence in parenthesis.
- the PCSK9 inhibitor peptide is Compound H having the following structure (SEQ ID No.: 16): Insulins
- the peptide therapeutic is an insulin.
- the peptide therapeutic is an insulin analogue.
- the insulin analogue is an acylated insulin, such as further described below.
- the acylated insulin is an insulin analogue comprising one or more substituents as described herein above.
- the insulin analogue comprises by covalent attachment, such as to a lysine residue an albumin binding substituent.
- a lysine residue an albumin binding substituent multiple examples of insulin analogues are known in the art and can be prepared as described such as in WO09115469 and WO 2016/119854. It has also previously been recognized that protease stability is beneficial in order to increase exposure after oral administration of a solid pharmaceutical composition.
- the acylated insulin is a protease stabilised insulin. Insulin analogues are herein defined relative to the wild type sequence of the A and B chains.
- a chain is: Gly-Ile-VaI-GIu-GIn-Cys-Cys-Tre-Ser-Ile-Cys-Ser-Lys Tyr-Gln- Leu- Glu-Asn-Tyr-Cys-Asn or GIVEQCCTSICSLYQLENYCN (SEQ ID No.: 21) and the B chain is: Phe-Val-Asn-Gln-His -Leu-Cys-Gly-Ser-His-Leu-Val-GIu-Ala-Leu-Tyr- Leu-VaI-Cys-Gly-GIu-Arg- Gly-Phe-Phe-Tyr-Tre-Pro-Lys-Thr or FVNQHLCGSHLVEALYLVCGERGFFYTPKT (SEQ ID No.: 22).
- Insulin analogues comprising amino acid changes compared to the native sequences are described by indicating the chain; A or B, followed by a number specifying the position in the respective chain and finally the amino acid residue in that position is specified (by one letter code). Deletion of an amino acid residue may be described by “des”.
- A10C, B1C, desB30 human insulin is an analogue of human insulin where the amino acid in position 10 in the A chain is substituted with cysteine, the amino acid in position 1 in the B chain is substituted with cysteine, and the amino acid in position 30 (threonine, Thr) in the B chain is deleted.
- the insulin analogue comprises a peptide sequence selected from the group consisting of: A14E, B25H, desB30 human insulin, A14E, B16H, B25H, desB30 human insulin, A14E, B25H, desB27, desB3010 human insulin and A14E, desB27, desB30 human insulin.
- the insulin analogue comprises a side chain, generally referred to as a substituent, in the form of an acyl group on the ⁇ -amino group of a Lys residue of the insulin amino acid sequence.
- the substituent is attached to the wild type lysine residue in position 29 of the B chain, referred to B29K.
- a substituent is frequently attached to the peptide and may be described in parenthesis after the amino acid residue of attachment, such as by B29K(N ⁇ Eicosanedioyl- ⁇ Glu), which indicates that the substituent Eicosanedioyl- ⁇ Glu- is attached via the epsylon nitrogen of the lysine residue in position 29 of the B chain.
- the insulin analogue comprises 2-7, such as 1-6, 2-5 or 2-4 amino acid substitutions relative to human insulin.
- the peptide therapeutic is an insulin analogue comprising one or more substituents and 2-8 amino acid substitutions in the A and/or B chain.
- the insulin analogue comprises one or two substituents and 2-7, such as 1-6, 2-5 or 2-4 amino acid substitutions in the A and/or B chain relative to human insulin.
- the peptide therapeutic is an insulin analogue selected from the group consisting of: A14E,B25H,B29K(N ⁇ Octadecanedioyl- ⁇ Glu-OEG-OEG),desB30 human insulin, A14E,B16H,B25H,B29K(N ⁇ Octadecanedioyl- ⁇ Glu-OEG-OEG),desB30 human insulin, A14E,B16H,B25H,B29K(N(eps)Eicosanedioyl- ⁇ Glu-OEG-OEG),desB30 human insulin, A14E,B25H,desB27,B29K(N ⁇ Octadecanedioyl- ⁇ Glu-OEG-OEG),desB30 human insulin, A14E,B16H,B25
- the insulin analogue is insulin analogue comprises and A chain of the sequence GIVEQCCTSICSLEQLENYCN (SEQ ID NO.: 23) and/or a B-Chain of the sequence FVNQHLCGSHLVEALYLVCGERGFHYTP ( SEQ ID NO.:24 ) and the structure:
- the composition of the invention comprises two absorption enhancers, wherein the two enhancers are a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid and a fatty acid consisting of 6-14 carbon atoms or a salt hereof.
- Salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid The structural formula of N-(8-(2-hydroxybenzoyl)amino)caprylate is shown in formula (I).
- the absorption enhancers used in the present invention is a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid (NAC).
- the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid comprises one monovalent cation, two monovalent cations or one divalent cation. In one embodiment the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid is selected from the group consisting of the sodium salt, potassium salt and/or the ammonium salt. In one embodiment the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid is the sodium salt or the potassium salt. In one embodiment the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid is selected from the group consisting of the sodium salt and the ammonium salt.
- Salts of N-(8-(2- hydroxybenzoyl)amino)caprylic acid may be prepared using the method described in e.g. WO96/030036, WO00/046182, WO01/092206, WO2008/028859.
- the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid may be crystalline and/or amorphous.
- the absorption enhancer comprises the anhydrate, monohydrate, dihydrate, trihydrate, a solvate or one third of a hydrate of the salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid as well as combinations thereof.
- one absorption enhancer is the sodium salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid sodium N-(8-(2-hydroxybenzoyl)amino)caprylate (referred to as “SNAC” herein), also known as sodium 8-(salicyloylamino)octanoate.
- SNAC N-(8-(2-hydroxybenzoyl)amino)caprylate
- the compositions according to the invention comprises a second absorption enhancer which is a medium chain fatty acid consisting of 6-14 carbon atoms or a salt hereof.
- the fatty acid consists of 8-12 carbon atoms, such as 8, 10 or 12 carbon atoms.
- the fatty acid is a saturated fatty acid.
- the absorption enhancer is capric acid or a salt hereof.
- Capric acid may also be referred to as decanoic acid (CH 3 (CH 2 ) 8 COOH).
- the salt of capric acid is sodium caprate (i.e. CH 3 (CH 2 ) 8 COONa).
- Composition The composition or pharmaceutical composition of the present invention is a solid or dry composition suited for administration by the oral route as described further herein below.
- the composition comprises at least one pharmaceutically acceptable excipient.
- excipient as used herein broadly refers to any component other than the active therapeutic ingredient(s) or active pharmaceutical ingredient(s) (API(s)) which in the present application is referred to as a peptide therapeutic.
- the excipient may be a pharmaceutically inert substance, an inactive substance, and/or a therapeutically or medicinally nonactive substance.
- the excipient may serve various purposes, e.g. as a carrier, vehicle, filler, binder, lubricant, glidant, disintegrant, flow control agent, crystallization inhibitors solubilizer, stabilizer, colouring agent, flavouring agent, surfactant, emulsifier or combinations of thereof and/or to improve administration, and/or absorption of the therapeutically active substance(s) or active pharmaceutical ingredient(s).
- the amount of each excipient used may vary within ranges conventional in the art.
- Excipients are generally selected from binders, such as polyvinyl pyrrolidone (povidone), etc.; fillers such as cellulose powder, microcrystalline cellulose, cellulose derivatives like hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose and hydroxy- propylmethylcellulose, dibasic calcium phosphate, corn starch, pregelatinized starch, etc.; lubricants and/or glidants such as stearic acid, magnesium stearate, sodium stearyl fumarate, glycerol tribehenate, etc.; flow control agents such as colloidal silica, talc, etc.; crystallization inhibitors such as Povidone, etc.; solubilizers such as Pluronic, Povidone, etc.; colouring agents, including dyes and pigments such as iron oxide red or yellow, titanium dioxide, talc, etc.; pH control agents such as citric acid, tartaric acid, fumaric acid, sodium citrate, dibasic calcium
- the composition comprises a binder, such as povidone; starches; celluloses and derivatives thereof, such as microcrystalline cellulose, e.g., Avicel PH from FMC (Philadelphia, PA), hydroxypropyl cellulose hydroxylethyl cellulose and hydroxylpropylmethyl cellulose METHOCEL from Dow Chemical Corp. (Midland, MI); sucrose; dextrose; corn syrup; polysaccharides; and gelatin.
- the binder may be selected from the group consisting of dry binders and/or wet granulation binders. Suitable dry binders are, e.g., cellulose powder and microcrystalline cellulose, such as Avicel PH 102 and Avicel PH 200.
- the composition comprises Avicel, such as Aavicel PH 102.
- Suitable binders for wet granulation or dry granulation are corn starch, polyvinyl pyrrolidone (povidon), vinylpyrrolidone-vinylacetate copolymer (copovidone) and cellulose derivatives like hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose and hydroxyl-propylmethylcellulose.
- the composition comprises povidone.
- the composition comprises a filler which may be selected from lactose, mannitol, erythritol, sucrose, sorbitol, calcium phosphate, such as calciumhydrogen phosphate, microcrystalline cellulose, powdered cellulose, confectioner's sugar, compressible sugar, dextrates, dextrin and dextrose.
- the composition comprises microcrystalline cellulose, such as Avicel PH 102 or Avicel PH 200.
- the composition comprises a lubricant and/or a glidant.
- the composition comprises a lubricant and/or a glidant, such as talc, magnesium stearate, calcium stearate, zinc stearate, glyceryl behenate, glyceryl debehenate, behenoyl polyoxyl-8 glycerides, polyethylene oxide polymers, sodium lauryl sulfate, magnesium lauryl sulfate, sodium oleate, sodium stearyl fumarate, stearic acid, hydrogenated vegetable oils, silicon dioxide and/or polyethylene glycol etc.
- the composition comprises magnesium stearate or glyceryl debehenate (such as the product Compritol® 888 ATO).
- the composition comprises magnesium stearate.
- the composition comprises a disintegrant, such as sodium starch glycolate, polacrilin potassium, sodium starch glycolate, crospovidon, croscarmellose, sodium carboxymethylcellulose or dried corn starch.
- the composition may comprise one or more surfactants, for example a surfactant, at least one surfactant, or two different surfactants.
- surfactant refers to any molecules or ions that are comprised of a water-soluble (hydrophilic) part, and a fat-soluble (lipophilic) part.
- the surfactant may e.g.
- the composition comprises a hydrotrope, such as Resorcinol, Pyrocatechol, Pyrogallol, Gentisic acid, Xylenesulfonate, p-toluenesulfonate, Nicotinamide, Dimethylbenzamide, Diethylbenzamide, 1-methylnicotinamide, Salicyclic acid and P- Hydroxybenzoic acid.
- a hydrotrope such as Resorcinol, Pyrocatechol, Pyrogallol, Gentisic acid, Xylenesulfonate, p-toluenesulfonate, Nicotinamide, Dimethylbenzamide, Diethylbenzamide, 1-methylnicotinamide, Salicyclic acid and P- Hydroxybenzoic acid.
- the hydrotrope is nicotinamide and/or Resorcinol.
- the hydrotrope is nicotinamide.
- the hydrotrope is not sodium benzoate.
- Dosage form The composition may be administered in several dosage forms, for example as a tablet, a coated tablet, a sachet or a capsule, such as hard or soft shell gelatine capsules, and all such compositions are considered solid oral dosage forms.
- a dose unit refers to a single entity to be administered such as a tablet, and the amounts of each ingredient comprised by a dose unit thus refers to the content of a single entity, such as one tablet.
- the composition may be in the form of a dose unit, such as a tablet.
- the weight of the unit dose is in the range of 50 to 2000 mg, such as 50 mg to 1200 mg, such as in the range of 50-1000 mg, or such as in the range of 100-800 mg. In some embodiments the weight of the unit dose is in the range of 50 mg to 1000 mg, such as in the range of 50-750 mg, or such as in the range of 100-600 mg. In some embodiments the weight of the dose unit is in the range of 75 mg to 400 mg. In an embodiment the weight of the unit dose is in the range of 100-400 mg, such as in the range of 100-300 mg or such as in the range of 150-350 mg.
- the weight of the dose unit is in the range of 300 mg to 800 mg, such as in the range of 400-700 mg or such as in the range of 500-600 mg. In some embodiment, the weight of the unit dose is 300 to 600 mg, such as approximately 350 mg, 450 mg or 550 mg.
- the pharmaceutical composition according to the invention is preferably produced in a dosage form suitable for oral administration as described herein below. In the following the absolute amounts of the ingredients of the composition of the invention are provided with reference to the content in a dosage unit i.e. per tablet, capsule or sachet.
- the pharmaceutical compositions of the invention in an embodiment comprises 0.1-100 mg of the peptide therapeutic per dose unit.
- a dose unit of the composition comprises an amount of the peptide therapeutic is in the range of 1 – 100 mg, 20 to 100 mg, 40 to 100 mg or 50 to 85 mg. In one embodiment a dose unit of the composition comprises an amount of the peptide therapeutic is in the range of 0.1 – 50 mg, 0.2 to 50 mg, 0.5 to 50 mg or 1 to 40 mg. In one embodiment a dose unit of the composition comprises an amount of the peptide therapeutic is in the range of 0.1 – 50 mg, 0.1 – 40 mg, 0.1 – 30 mg or 0.1 – 20 mg.
- the pharmaceutical compositions of the invention in an embodiment comprise comprises 0.1-100 mg of the GLP-1 agonist per dose unit.
- a dose unit of the composition comprises an amount of GLP-1 agonist is in the range of 0.1 – 50 mg, 0.2 to 50 mg, 0.5 to 50 mg or 1 to 40 mg. In one embodiment a dose unit of the composition comprises an amount of GLP-1 agonist is in the range of 0.1 – 50 mg, 0.1 – 40 mg, 0.1 – 30 mg or 0.1 – 20 mg. In one embodiment a dose unit comprises 0.5-5 mg of the GLP-1 agonist, such as 0.75- 4 1 ⁇ 2 mg, such as 1, 1 1 ⁇ 2, 2, 2 1 ⁇ 2 or 3 mg or 3 1 ⁇ 2, 4, 4 1 ⁇ 2 mg, such as 1-3 or 3-5 mg of the GLP-1 agonist per dose unit.
- a dose unit comprises 2 to 20 mg of the GLP-1 agonist, such as 2- 15 mg, such as 2, 3, 4 or 5 mg, or such as 8, 10, 12 or 14 mg, such as 15 mg or such as 20 mg of the GLP-1 agonist per dose unit.
- a dose unit comprises 25 to 100 mg of the GLP-1 agonist, such as 10-90 mg, or such as 20-60 such as 20, 30 or 40 mg, or such as 40-80 mg of the GLP-1 agonist, such as 60, 70, or 80 mg, per dose unit.
- a dose unit comprises 10 to 75 mg of the GLP-1 agonist, such as 10-70 mg, such as 20, 30 or 40 mg, or such as 50, 60, or 65 mg, or such as 20-60 mg or such as 30-50 mg of the GLP-1 agonist per dose unit. In one embodiment a dose unit comprises 5 to 50 mg of the GLP-1 agonist, such as 10- 45 mg, such as 20, 30 or 40 mg, or such as 25, 35, or 45 mg, or such as 30-50 mg or such as 20-40 mg of the GLP-1 agonist per dose unit. In an embodiment, a dose unit of the pharmaceutical compositions of the invention comprises 0.1-200 mg, 0.1-150 mg or 0.2-100 mg of the PCSK9 inhibitor.
- a dose unit of the composition comprises an amount of PCSK9 inhibitor is in the range of 0.5-150 mg, 0.5-120 mg, 0.5-100 mg, 1-80 mg, 1-70 mg, 1-60, 1-50 mg or 1-40 mg. In some embodiments, a dose unit of the composition comprises an amount of PCSK9 inhibitor is in the range of 0.1-50 mg, 0.2-50 mg, 1-50 mg or 10-50 mg. In some embodiments, a dose unit of the composition comprises an amount of PCSK9 inhibitor is in the range of 0.1-50 mg, 0.1-40 mg, 0.1-30 mg or 0.1-20 mg.
- a dose unit comprises 1-80 mg of the PCSK9 inhibitor, such as 2-70 mg, such as 10, 15, 20, 25 or 30 mg or 35, 40, 45 mg, such as 10-30 or 30-50 mg of the PCSK9 inhibitor per dose unit.
- a dose unit comprises 20-200 mg of the PCSK9 inhibitor, such as 20-150, such as 20-120 mg, such as 20-100 mg, such as 20-80 mg, or such as 20, 30, 40, 50, 60, 70 or 80 mg, or such as 90, 95, 100, 105 or 110 mg, or such as 30-100 mg or such as 40-80 mg of the PCSK9 inhibitor per dose unit.
- a dose unit comprises 10-80 mg of the PCSK9 inhibitor, such as 10-70 mg, such as 20, 30 or 40 mg, or such as 50, 60, 70 or 80 mg, or such as 30-60 mg or such as 20-50 mg of the PCSK9 inhibitor per dose unit.
- the amount of PCSK9 inhibitor may be varied depending on identity of the PCSK9 inhibitor and the effect desired.
- the pharmaceutical compositions of the invention in an embodiment comprise at most 1000 mg of said salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as sodium N- (8-(2-hydroxybenzoyl)amino)caprylate (SNAC) per dose unit.
- the composition comprises at most 800, such as at most 600 mg of said salt.
- the amount of salt of NAC is at least 20 mg or 25 mg, such as at least 50 mg, at least 75 mg, at least 100 mg, at least 125 mg, at least 150 mg, at least 175 mg, at least 200 mg, at least 225 mg, at least 250 mg, at least 275 mg and at least 300 mg per dose unit.
- the amount of salt of NAC is up to 800 mg, such as up to 750 mg, up to 700, up to 650 mg, up to 600 mg, up to 550 mg, up to 500 mg, up to 450 mg, up to 400 mg, up to 350 mg, or such as up to 300 mg per dose unit.
- the amount of salt of NAC is in the range of 30-500 mg, such as from 40-400 mg, such as from around 50 to around 300 mg per dose unit.
- the amount of salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC) per dose unit of the composition is up to 3.3 mmol.
- the composition comprises up to 2.66 mmol, such as up to 1.99 mmol of said salt.
- the salt of NAC is at least 0.07 or 0.08 mmol, such as at least 0.17 mmol, at least 0.25 mmol, at least 0.33 mmol, at least 0.41 mmol, at least 0.50 mmol, at least 0.58 mmol, at least 0.66 mmol, at least 0.75 mmol, at least 0.83 mmol, at least 0.91 mmol, at least 1.0 mmol per unit dose.
- the amount of salt of NAC is up to 2.66 mmol, such as up to 2.49 mmol, up to 2.32 mmol, up to 2.16 mmol, up to 1.99 mmol, up to 1.83 mmol, up to 1.66 mmol, such as up to 1.49 mmol, up to 1.33 mmol, up to 1.16 mmol or such as up to 1.00 mmol per dose unit.
- the amount of salt of NAC is in the range of 0.10-1.66 mmol, such as 0.13-1.33 mmol, such as from around 0.17 mmol to around 1.00 mmol per dose unit.
- the salt of NAC is SNAC.
- the amount of SNAC in the composition is at least 20 mg, such as at least 25 mg, such as at least 50 mg, such as at least 75 mg, at least 100 mg, at least 125 mg, at least 150 mg, at least 175 mg, at least 200 mg, at least 225 mg, at least 250 mg, at least 275 mg and at least 300 mg per dose unit.
- the amount of SNAC in the composition is up to 1000 mg, such as up to 800 mg, such as up to 600 mg, such as up to 575 mg, such as up to 550 mg, up to 525 mg, up to 500 mg, up to 475 mg, up to 450 mg, up to 425 mg, up to 400 mg, up to 375 mg, up to 350 mg, up to 325 mg per dose unit, or up to 300 mg per dose unit.
- the amount of SNAC in the composition is in the range of 20-800 mg, such as 25-600 mg, such as 50-500 mg, such as 50-400 mg, such as 75-400 mg, such as from 80-350 mg, such as from around 100 to around 300 mg per dose unit.
- the amount of SNAC is in the range of 20-200 mg, such as 25-175 mg, such as 75-150 mg, such as 80-120 mg such as around 100 mg per dose unit. In one embodiment, where the salt of NAC is SNAC, the amount of SNAC is in the range of 50-400 mg, such as 75-300 mg, such as 100-300 mg, such as 150-250 mg, such as around 200 mg per dose unit. In one embodiment, where the salt of NAC is SNAC, the amount of SNAC is in the range of 200-800 mg, such as 250-400 mg, such as 250-350 mg, such as 275-325 mg, such as around 300 mg per dose unit.
- the amount of the second absorption enhance is to be balanced, with the amount of the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid but in general the formulation according to the invention comprise at most 1000 mg of the second absorption enhancer, such as the fatty acid or a salt thereof, such as sodium caprate.
- the composition comprises at most 800 mg, such as at most 600 mg of said second enhancer.
- the composition comprises at most 1000 mg, such as at most 800 mg, such as at most 600 mg of the fatty acid or a salt hereof, such as sodium caprate.
- the amount of the second enhancer, the fatty acid or a salt thereof is at least 20 or at least 25 mg, such as at least 50 mg, at least 75 mg, at least 100 mg, at least 125 mg, at least 150 mg, at least 175 mg, at least 200 mg, at least 225 mg, at least 250 mg, at least 275 mg and at least 300 mg per dose unit.
- the amount of the second enhancer, the fatty acid or a salt thereof is up to 800 mg, such as up to 750 mg, up to 700 mg, up to 650 mg, up to 600 mg, up to 550 mg, up to 500 mg, up to 450 mg, up to 400 mg, up to 350 mg, or up to 300 mg per dose unit.
- the amount of the second enhancer, the fatty acid or a salt thereof is in the range of 30-500 mg, such as from 40-400 mg, such as from around 80 to around 300 mg per dose unit.
- the pharmaceutical compositions of the invention in an embodiment comprise at most 5.1 mmol of the second absorption enhancer, such as the medium chain fatty acid or a salt thereof, such as sodium caprate.
- the composition comprises at most 4.12 mmol, such as at most 3.09 mmol of said second enhancer.
- the composition comprises at most 5.1 mmol, such as at most 4.12 mmol, such as at most 3.09 mmol of the medium chain fatty acid or a salt hereof, such as sodium caprate.
- the amount of the second enhancer, the medium chain fatty acid or a salt thereof, such as sodium caprate is at least 0.10 or 0.13 mmol, such as at least 0.26 mmol, at least 0.39 mmol, at least 0.51 mmol, at least 0.64 mmol, at least 0.77 mmol, at least 0.90 mmol, at least 1.03 mmol, at least 1.16 mmol, at least 1.29 mmol, at least 1.42 mmol and at least 1.54 mmol per dose unit.
- the amount of the second enhancer, the medium chain fatty acid or a salt thereof, such as sodium caprate is up to 4.12 mmol, such as up to 3.86 mmol, up to 3.60 mmol, up to 3.35 mmol, up to 3.09 mmol, up to 2.83 mmol, up to 2.57 mmol, up to 2.32 mmol, up to 2.06 mmol, up to 1.80 mmol, or up to 1.54 mmol per dose unit.
- the amount of the second enhancer, the medium chain fatty acid or a salt thereof, such as sodium caprate is in the range of 0.15-2.57 mmol, such as from 0.21-2.06 mmol, such as from around 0.41 to around 1.54 mmol per dose unit.
- the second enhancer is a salt of capric acid, such as sodium caprate.
- the amount of sodium caprate in the composition is at least 20 mg or 25 mg, such as at least 50 mg, such as at least 75 mg, at least 100 mg, at least 125 mg, at least 150 mg, at least 175 mg, at least 200 mg, at least 225 mg, at least 250 mg, at least 275 mg and at least 300 mg per dose unit.
- the amount of sodium caprate in the composition is up to 1000 mg, such as up to 800 mg, such as up to 600 mg, such as up to 575 mg, such as up to 550 mg, up to 525 mg, up to 500 mg, up to 475 mg, up to 450 mg, up to 425 mg, up to 400 mg, up to 375 mg, up to 350 mg, up to 325 mg per dose unit, or up to 300 mg per dose unit.
- the amount of sodium caprate in the composition is in the range of 20-800 mg, such as 25-600 mg, such as 50-500 mg, such as 50-400 mg, such as 75-400 mg, such as from 80-350 mg, such as from around 100 to around 300 mg per dose unit.
- the amount of sodium caprate is in the range of 20-200 mg, such as 30-175 mg, such as 75-150 mg, such as 80-120 mg such as around 100 mg per dose unit. In one embodiment, where the salt of capric acid is sodium caprate, the amount of sodium caprate is in the range of 50-400 mg, such as 75-300 mg, such as 100-300 mg, such as 150-250 mg, such as around 200 mg per dose unit. In one embodiment, where the salt of capric acid is sodium caprate, the amount of sodium caprate is in the range of 200-800 mg, such as 250-400 mg, such as 250-350 mg, such as 275-325 mg, such as around 300 mg per dose unit.
- the amounts (w) of the two absorption enhancers differs with at most a factor 10, such as a factor 5, such as a factor 2.
- the ratio of SNAC/sodium caprate (w/w) is at least 0.1.
- the weight ratio (w/w), i.e. the ratio of the amount (w) of a salt of NAC to the amount (w) of a salt of the fatty acid is 0.1-10, such as 0.2-8, such as 0.3-5, such as 0.3-3, such as 0.4-2.5 or such as 0.5-2.
- the weight ratio (w/w) of SNAC to sodium caprate is 0.1-10, such as 0.2-8, such as 0.3-5.
- the weight ratio (w/w) of SNAC to sodium caprate is 0.3-3, such as 0.4-2.5 or such as 0.5-2. In one embodiment the weight ratio (w/w) of SNAC to sodium caprate is 0.6-1.9, or such as 0.5-1.8, or such as 0.8-1.7. In one embodiment the weight ratio (w/w) of SNAC to sodium caprate is 0.8-1.6, or such as 0.8-1.5, or such as 0.8-1.4. In one embodiment the weight ratio (w/w) of SNAC to sodium caprate is 0.9-1.3, or such as 0.9-1.2, or such as 0.8-1.3, or such as 0.8-1.2.
- the amount of the second absorption enhancer is to be approximately 1:1 with the amount (w) of the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid.
- the amount of the hydrotrope is to be balanced with the amount of the absorption enhancers, such as one or more of the salt of NAC and/or the salt of capric acid, but in general a dose unit of the compositions of the invention comprises 10-600 mg of the hydrotrope. In on embodiment the composition comprises 10-400 mg of the hydrotrope. In one embodiment, a dose unit comprises 20-400 mg, such as 40-300, such as 50-200 mg, such as 50-175 mg of the hydrotrope.
- a unit dose comprises 10-600 mg, such as 15-500 mg or such as 20-400 mg of the hydrotrope. In some embodiment, a unit dose comprises 70-155 mg, such as 70-105 mg or 70-100 mg of the hydrotrope. In some embodiment, a unit dose comprises 70-105 mg or 100-155 mg of the hydrotrope. In one embodiment, a dose unit comprises 100-600 mg, such as 100-500, such as 150-400 mg, such as 150-300 mg of the hydrotrope. In one embodiment a dose unit comprises 10-200 mg, such as 15-175, such as 20-150 mg, such as 20-125 mg of the hydrotrope.
- a unit dose comprises 20-200 mg, 30-200 mg, 40-200 mg, 50- 200 or such as 75-200 mg of the hydrotrope. In some embodiment, a unit dose comprises 20-175 mg or 30-150 mg of the hydrotrope. In further such embodiments, a unit dose of the composition according to the invention comprises 50-600 mg nicotinamide and/or resorcinol. In one embodiment, a dose unit comprises 10-200 mg, such as 15-175, such as 20-150 mg, such as 25-150 mg nicotinamide and/or resorcinol.
- a dose unit comprises 50-400 mg, such as 50-300, such as 50-200 mg, such as 50-175 mg nicotinamide and/or resorcinol.
- a unit dose of the composition comprises 50-600 mg nicotinamide.
- a dose unit comprises 50-400 mg, such as 50-300, such as 50-200 mg, such as 50-175 mg nicotinamide.
- a unit dose comprises 70-155 mg, such as 70-105 mg or 70-100 mg nicotinamide.
- a unit dose comprises 70-155 mg, such as 100-155 mg nicotinamide
- a unit dose of the composition according to the invention comprises 10-400 mg nicotinamide and/or resorcinol.
- a dose unit comprises 10-200 mg, such as 20-175, such as 20-150 mg, such as 20-125 mg nicotinamide and/or resorcinol.
- a unit dose comprises 20-200 mg, 30-200 mg, 40-200 mg, 50- 200 or such as 75-200 mg nicotinamide and/or resorcinol.
- a unit dose comprises 20-175 mg or 30-150 mg nicotinamide and/or resorcinol.
- a unit dose of the composition comprises 10-400 mg nicotinamide.
- a dose unit comprises 10-200 mg, such as 20-175, such as 20- 150 mg, such as 20-125 mg nicotinamide.
- a unit dose comprises 20-200 mg, 30-200 mg, 40-200 mg, 50- 200 or such as 75-200 mg nicotinamide.
- a unit dose comprises 20-175 mg or 30-150 mg nicotinamide.
- the weight ratio of the amount (w) of salt of NAC to the amount (w) of the hydrotrope is at least 0.5.
- the weight ratio of the amount (w) of salt of NAC to the amount (w) of the hydrotrope is 0.5-10, such as 0.5-5 or such as 1-5. In one embodiment the weight ratio of the amount (w) of salt of NAC to nicotinamide (w) is 0.5-10, such as 0.5-5 or such as 1-5. In one embodiment, the weight ratio of the amount (w) of salt of NAC to nicotinamide (w) is 0.5-10, such as 0.5-5 or such as 1-5. In one embodiment the ratio of the amount (w) of salt of NAC to the amount (w) of nicotinamide is 1-2, such as 1.2-1.8 or such as 1.3-1.7 or such as 1.4-1.6.
- a unit dose of the composition further comprises 0.5-50 mg of a lubricant, such as 0.10-25 mg, such as 0.25-10 mg, such as 0.5-8 mg lubricant.
- a unit dose of the composition comprises 0.5-50 mg of magnesium stearate, such as 0.10-25 mg, such as 0.25-10 mg, such as 0.5-8 mg or such as 0.5-5 mg magnesium stearate.
- a unit dose of the composition comprises 1-10 mg magnesium stearate, such as 2-8 mg or 3-7 mg.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 0.5-100 mg GLP-1 agonist, 10-600 mg hydrotrope and 0.10-50 mg lubricant.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 1.0-90 mg GLP-1 agonist, 10-500 mg hydrotrope and 0.10-40 mg lubricant.
- a unit dose of the composition comprises 40-600 mg SNAC, 40-600 mg sodium caprate, 5-80 mg GLP-1 agonist, 30-400 mg hydrotrope and 0.10-40 mg lubricant.
- a unit dose of the composition comprises 50-300 mg SNAC, 50-300 mg sodium caprate, 10-80 mg GLP-1 agonist, 40-300 mg hydrotrope and 2-8 mg lubricant.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 10-80 mg GLP-1 agonist, 10-500 mg nicotinamide and 0.10-50 mg magnesium stearate.
- a unit dose of the composition comprises 30-500 mg SNAC, 30-500 mg sodium caprate, 10-50 mg GLP-1 agonist, 20-400 mg nicotinamide and 0.10-25 mg magnesium stearate.
- a unit dose of the composition comprises 50-300 mg SNAC, 50-300 mg sodium caprate, 10-80 mg GLP-1 agonist, 40-250 mg nicotinamide and 2-8 mg magnesium stearate.
- a unit dose of the composition comprises 90-250 mg SNAC, 90-250 mg sodium caprate, 10-80 mg GLP-1 agonist, 50-170 mg nicotinamide and 3-7 mg magnesium stearate.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 0.50-50 mg GLP-1 agonist, 15-500 mg hydrotrope and 0.10-40 mg lubricant.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 0.50-25 mg GLP-1 agonist, 15-500 mg hydrotrope and 0.10-40 mg lubricant.
- a unit dose of the composition comprises 25-600 mg SNAC, 25-600 mg sodium caprate, 1-75 mg GLP-1 agonist, 20-500 mg hydrotrope and 0.10-40 mg lubricant.
- the composition comprises: i) 0.5-100 mg GLP-1 agonist, ii) 20-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 20-1000 mg a fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate iv) 10-750 mg, such as 50-200 mg, nicotinamide or resorcinol and v) 0-25 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: vi) 0.5-100 mg GLP-1 agonist, vii) 25-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), viii) 25-1000 mg a fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate ix) 15-750 mg, such as 50-200 mg, nicotinamide or resorcinol and x) 0-25 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- a fatty acid consisting of 6-14 carbon atoms such capric acid, or a salt hereof such as sodium caprate ix
- 15-750 mg such as 50-200 mg, nicotinamide or resorcinol and x) 0-25 mg lubricant.
- the composition comprises: i) 1-75 mg GLP-1 agonist, ii) 25-300 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 25-300 mg a fatty acid consisting of 6-14 carbon atoms,, such capric acid, or a salt hereof such as sodium caprate iv) 15-200 mg nicotinamide and v) 0-10 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- capric acid or a salt hereof such as sodium caprate iv) 15-200 mg nicotinamide and v) 0-10 mg lubricant.
- the composition comprises: i) 1-75 mg GLP-1 agonist, ii) 25-200 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 25-200 mg sodium caprate iv) 15-150 mg nicotinamide and v) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 1-50 mg GLP-1 agonist, ii) 25-100 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 25-100 mg sodium caprate, iv) 15-75 mg nicotinamide and v) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 1-10 mg GLP-1 agonist, ii) 20-40 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 20-40 mg sodium caprate iv) 15-30 mg nicotinamide and v) 0-5 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 1-20 mg GLP-1 agonist, ii) 50-75 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 50-75 mg sodium caprate iv) 30-50 mg nicotinamide and v) 0-3 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 10-50 mg GLP-1 agonist, ii) 100-150 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 100-150 mg sodium caprate, iv) 70-100 mg nicotinamide and v) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 10-80 mg GLP-1 agonist, ii) 150-250 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 150-250 mg sodium caprate, iv) 100-180 mg nicotinamide and v) 0-10 mg magnesium stearate.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 0.5-100 mg PCSK9 inhibitor, 10-600 mg hydrotrope and 0.10-50 mg lubricant.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 1.0-90 mg PCSK9 inhibitor, 20-500 mg hydrotrope and 0.10-40 mg lubricant.
- a unit dose of the composition comprises 40-600 mg SNAC, 40-600 mg sodium caprate, 5-80 mg PCSK9 inhibitor, 30-400 mg hydrotrope and 0.10-40 mg lubricant.
- a unit dose of the composition comprises 50-300 mg SNAC, 5-300 mg sodium caprate, 10-80 mg PCSK9 inhibitor, 50-300 mg hydrotrope and 2-8 mg lubricant.
- a unit dose of the composition comprises 20-600 mg SNAC, 20-600 mg sodium caprate, 10-80 mg PCSK9 inhibitor, 10-600 mg nicotinamide and 0.10-50 mg magnesium stearate.
- a unit dose of the composition comprises 30-500 mg SNAC, 30-500 mg sodium caprate, 10-50 mg PCSK9 inhibitor,10-600 mg nicotinamide and 0.10-25 mg magnesium stearate.
- a unit dose of the composition comprises 50-300 mg SNAC, 5-300 mg sodium caprate, 10-80 mg PCSK9 inhibitor, 50-300 mg nicotinamide and 2-8 mg magnesium stearate.
- a unit dose of the composition comprises 90-250 mg SNAC, 90-250 mg sodium caprate, 10-80 mg PCSK9 inhibitor, 50-170 mg nicotinamide and 3-7 mg magnesium stearate.
- the composition comprises: i) 0.1-200 mg PCSK9 inhibitor, ii) 25-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 15-750 mg, such as 50-200 mg, nicotinamide or resorcinol, iv) 25-1000 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate, and v) 0-50 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- 15-750 mg such as 50-200 mg,
- the composition comprises: i) 0.1-150 mg PCSK9 inhibitor, ii) 25-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 15-750 mg, such as 50-200 mg, nicotinamide or resorcinol, iv) 25-1000 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate, and v) 0-25 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- 15-750 mg such as 50-200 mg
- nicotinamide or resorcinol iv) 25-1000 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium cap
- the composition comprises: i) 1-100 mg PCSK9 inhibitor, ii) 50-800 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 40-600 mg nicotinamide, iv) 50-800 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate, and v) 0-20 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 1-100 mg PCSK9 inhibitor, ii) 50-600 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 40-400 mg nicotinamide, iv) 50-600 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate, and v) 0-15 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- nicotinamide iv) 50-600 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate
- v) 0-15 mg lubricant 0-15 mg lubricant.
- the composition comprises: i) 5-100 mg PCSK9 inhibitor, ii) 55-400 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 45-300 mg nicotinamide, iv) 55-400 mg sodium caprate, and v) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 10-80 mg PCSK9 inhibitor, ii) 150-230 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 100-160 mg nicotinamide, iv) 150-230 mg sodium caprate, and v) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 10-50 mg PCSK9 inhibitor, ii) 100-160 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 65-110 mg nicotinamide, iv) 100-160 mg sodium caprate, and v) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the composition comprises: i) 30-50 mg PCSK9 inhibitor, ii) 125-175 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 80-120 mg nicotinamide, iv) 125-175 mg sodium caprate, and v) 1-8 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- SNAC sodium salt of NAC
- 80-120 mg nicotinamide iv) 125-175 mg sodium caprate
- v) 1-8 mg magnesium stearate v
- the composition may be granulated prior to being compacted and i.e. compressed into tablets.
- the composition may comprise a granular part (or parts) and/or an extragranular part, wherein the granular part(s) has/have been
- the granular part(s) comprise(s) the two absorption enhancers and the hydrotrope and optionally the therapeutic peptide.
- the granular part(s) comprise(s) a further excipient, such as a lubricant and/or glidant.
- the extra-granular part comprises the therapeutic peptide.
- the extra-granular part comprises a lubricant and/or a glidant, such as magnesium stearate.
- the extra-granular part comprises the therapeutic peptide and a lubricant and/or a glidant, such as magnesium stearate.
- Preparation of solid pharmaceutical compositions for oral administration Preparation of a composition according to the invention may be performed according to methods known in the art. To prepare a dry blend of tabletting material, the various components are optionally delumped or sieved, weighed, and then combined. The mixing of the components may be carried out until a homogeneous blend is obtained.
- the terms “granulate” and “granules” are used interchangeably herein to refer to particles of composition material which may be prepared as described above. The term refers broadly to pharmaceutical ingredients in the form of particles, granules and aggregates which are used in the preparation of solid dose formulations.
- granules are obtained by processing a powder or a blend to obtain a solid which is subsequently broken down to obtain granules of the desired size. If granules are to be used in the tabletting material, granules may be produced in a manner known to a person skilled in the art, for example using wet granulation methods known for the production of "built-up" granules or "broken-down" granules.
- Methods for the formation of built-up granules may operate continuously and comprise, for example simultaneously spraying the granulation mass with granulation solution and drying, for example in a drum granulator, in pan granulators, on disc granulators, in a fluidized bed, by spray-drying, spray-granulation or spray-solidifying, or operate discontinuously, for example in a fluidized bed, in a rotary fluid bed, in a batch mixer, such as a high shear mixer or a low shear mixer, or in a spray-drying drum.
- Methods for the production of broken-down granules which may be carried out discontinuously and in which the granulation mass first forms a wet aggregate with the granulation solution, which is subsequently comminuted or by other means formed into granules of the desired size and the granules may then be dried.
- Suitable equipment for the wet granulation step is, but not limited to, planetary mixers, low shear mixers, high shear mixers, extruders and spheronizers, such as an apparatus from the companies Loedige, Glatt, Diosna, Fielder, Collette, Aeschbach, Alexanderwerk, Ytron, Wyss & Probst, Werner & Pfleiderer, HKD, Loser, Fuji, Nica, Caleva and Gabler.
- planetary mixers such as an apparatus from the companies Loedige, Glatt, Diosna, Fielder, Collette, Aeschbach, Alexanderwerk, Ytron, Wyss & Probst, Werner & Pfleiderer, HKD, Loser, Fuji, Nica, Caleva and Gabler.
- Granules may also be formed by dry granulation techniques in which one or more of the excipient(s) and/or an active pharmaceutical ingredient, such as a peptide therapeutic as described herein, is compressed to form relatively large moldings, for example slugs or ribbons, which are comminuted by grinding, and the ground material serves as the tabletting material to be later compacted.
- Suitable equipment for dry granulation is, but not limited to, roller compaction equipment from Gerteis such as Gerteis MICRO-PACTOR, MINI-PACTOR and MACRO- PACTOR.
- Granules may alternatively be prepared by hot melt extruding techniques in which one or more of the excipient(s) is feed into an extruder, heated and extruded through a die.
- Suitable equipment is such as Thermo Scientific Process 11 twin screw.
- a tablet press may be used to compact the tabletting material into a solid oral dosage form, for example a tablet.
- the tabletting material is filled (e.g. force feeding or gravity feeding) into a die cavity.
- the tabletting material is then compacted by a set of punches applying pressure.
- the resulting compact, or tablet is ejected from the tablet press.
- the above-mentioned tabletting process is subsequently referred to herein as the "compaction process”.
- Suitable tablet presses include, but are not limited to, rotary tablet presses and eccentric tablet presses.
- granulates may be prepared by extrusion, wet or dry granulation. Granules comprising one or more of the two absorption enhancers and nicotinamide may thus be obtained by dry granulation, such as by roller compaction. In an alternative embodiment wet granulation may be used to obtain the granules.
- hot melt extrusion may be used to obtain the granules.
- the ingredients of a pharmaceutical composition according to the invention may thus be mix or blended prior or after granulation.
- the granulates can then be used directly or further refined to obtain the final granules.
- the composition comprises at least one granulate.
- the composition comprises one type of granulate.
- the composition may alternatively comprise two types of granulates. In embodiments where the granular part comprises both the absorption enhancers and the hydrotrope these excipients may be co-processed prior to or in the preparation of the granules.
- the granulation maybe be obtained by various methods as described above, wherein the excipients are initially mixed either as powders or by the preparation of a solution comprising the ingredients.
- granules of the excipients are obtained by feeding all ingredients separately into the process stream, such as into an extruder or a compactor.
- Granules may then be obtained by dry granulation of the blend, such as by roller compaction.
- the excipients may be hot melt extruded to obtain an extrudate which is optionally subsequently milled to obtain the granules. This material can then be used directly or in dry granulation/roller compaction process to obtain the final granules.
- a solution of the excipient(s) is prepared and subject to spray granulation whereby granules are directly obtained.
- the solution can be used in a fluid bed spray granulation process.
- spray drying can be used followed by dry granulation/roller compaction to obtain granules.
- a solution of the excipient(s) is prepared, and granules prepared by wet granulation.
- different processes may be used for different excipient(s).
- the peptide therapeutic may be included at any step in the process except in the hot melt extrusion. In one embodiment the peptide therapeutic is included in the blend prior to dry granulation or wet granulation.
- the peptide therapeutic agonist is comprised by a granule produced by dry granulation, such as roller compaction. In one embodiment the peptide therapeutic is comprised by a granule produced by wet granulation. In one embodiment the peptide therapeutic is added after granulation. In one embodiment the invention relates to a method of preparation a solid pharmaceutical composition according to the invention. In one embodiment the method of preparing a tablet comprises the steps of: a) granulating the absorption enhancers and a hydrotrope b) blending the granulates of a) with a peptide therapeutic, and optionally a lubricant c) compressing the blend of b) into tablets.
- the method of preparing a tablet comprises the steps of: a) granulating a peptide therapeutic, the absorption enhancers and a hydrotrope b) optionally blending of the granulate of a) with a lubricant c) compressing the granules of a) or the blend of b) into tablets.
- the granulation may be a wet, hot or dry granulation.
- a lubricant such as magnesium stearate or glyceryl behenate may be included in any of the steps.
- the invention relates to a method for producing a solid pharmaceutical composition
- a method for producing a solid pharmaceutical composition comprising the steps of: a) obtaining a salt of NAC and a hydrotrope, b) co-processing said salt of NAC and hydrotrope of a), c) obtaining a salt of capric acid d) blending the product of b) with the salt of capric acid of c) and e) optionally include a lubricant in the blending process d) f) preparing said solid pharmaceutical composition using the blend of d) or e) wherein a peptide therapeutic is included in any of the steps.
- a GLP-1 agonist is included in blending step d) or e) or in a separate blending step.
- the method is for producing a solid pharmaceutical composition comprising a peptide therapeutic, wherein the method comprises the steps of: a) obtaining a salt of NAC and a hydrotrope, b) hot melt extruding said salt of NAC and hydrotrope of a) and c) obtaining a salt of capric acid d) granulating the salt of capric acid e) blending the product of b) with the granulate of d) and f) optionally include a lubricant in the blending process e) g) preparing said solid pharmaceutical composition using the blend of e) or f) wherein the GLP-1 agonist is included in any of the steps.
- the peptide therapeutic is included in the granules prepared in d), the blending step e) or f) or in a separate blending step.
- the method may as described herein include further steps, such as a step of admixing the extrudate of b) with an active pharmaceutical ingredient and optionally any further excipients and preparing said solid pharmaceutical composition using the mixture.
- Pharmaceutical Indications The present invention relates to a composition comprising a peptide therapeutic, such as a GLP-1 peptide agonist for use as a medicament.
- the composition may be used for the following medical treatments, all preferably relating one way or the other to diabetes: (i) prevention and/or treatment of all forms of diabetes, such as hyperglycemia, type 2 diabetes, impaired glucose tolerance, type 1 diabetes, non-insulin dependent diabetes, MODY (maturity onset diabetes of the young), gestational diabetes, and/or for reduction of HbA1C; (ii) delaying or preventing diabetic disease progression, such as progression in type 2 diabetes, delaying the progression of impaired glucose tolerance (IGT) to insulin requiring type 2 diabetes, and/or delaying the progression of non-insulin requiring type 2 diabetes to insulin requiring type 2 diabetes; (iii) improving ⁇ -cell function, such as decreasing ⁇ -cell apoptosis, increasing ⁇ -cell function and/or ⁇ -cell mass, and/or for restoring glucose sensitivity to ⁇ -cells; (iv) prevention and/or treatment of cognitive disorders; (v) prevention and/or treatment of eating disorders, such as obesity, e.
- diabetes
- the indication is selected from the group consisting of (i)-(iii) and (v)-(iix), such as indications (i), (ii), and/or (iii); or indication (v), indication (vi), indication (vii), and/or indication (iix).
- the indication is (i).
- the indication is (v).
- the indication is (iix).
- the indications are type 2 diabetes and/or obesity.
- the invention further relates to a method of treatment of an individual in need thereof, comprising administering a therapeutically active amount of a composition comprising a peptide therapeutics, such as GLP-1 agonist according to the present invention to said individual.
- one or more dose units may be administered to said individual in need.
- the invention relates to the use of a solid composition comprising a PCSK9 inhibitor, such as an EGF(A) peptide analogue or an EGF(A) derivative for use in the manufacture of a pharmaceutical composition as described herein.
- the invention relates to a solid composition comprising a PCSK9 inhibitor, such as an EGF(A) peptide analogue or an EGF(A) derivative, for use as a medicament and/or in a method of treatment.
- the composition is for use in a method of treatment, such as for (i) improving lipid parameters, such as prevention and/or treatment of dyslipidaemia, lowering total serum lipids; lowering LDL-C, increasing HDL; lowering small, dense LDL; lowering VLDL; lowering triglycerides; lowering cholesterol; lowering plasma levels of lipoprotein a (Lp(a)); inhibiting generation of apolipoprotein A (apo(A)) ; (ii) the prevention and/or the treatment of cardiovascular diseases, such as cardiac syndrome X, atherosclerosis, myocardial infarction, coronary heart disease, reperfusion injury, stroke, cerebral ischemia, an early cardiac or early cardiovascular disease, left ventricular hypertrophy, coronary artery disease, hypertension, essential hypertension, acute hypertensive emergency, cardiomyopathy, heart insufficiency, exercise intolerance, acute and/or chronic heart failure, arrhythmia, cardiac dysrhythmia, syncopy, angina pectoris, cardiac bypass
- Dyslipidaemia may be such as a high plasm concentration of cholesterols also called hypercholesterolaemia referring to a situation where the plasma cholesterol concentrations is above the normal range of a total cholesterol ⁇ 5.0 mmol/l.
- the compound or composition of the invention may be used for treatment of hypercholesterolaemia.
- Method of treatment further relates to a method of treating a subject in need thereof, comprising administering a therapeutically effective amount of a composition according to the present invention to said subject.
- the peptide therapeutic is a GLP-1 agonist
- the method is for treatment of a disease or disorder, such as diabetes and/or obesity and/or the further indications specified above.
- a method for treating a disease or disorder comprises administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a therapeutic peptide, such a GLP-1 agonist, a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), a hydrotrope, a second absorption enhancer and optionally, a lubricant.
- a therapeutic peptide such as GLP-1 agonist, a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), a hydrotrope, a second absorption enhancer and optionally, a lubricant.
- the method for treating a disease or disorder comprises administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition
- a pharmaceutical composition comprising i) 0.1-100 mg therapeutic peptide ii) 20-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC) iii) 20-1000 mg of a fatty acid consisting of 6-14 carbon atoms, such as capric acid, or a salt hereof, such as sodium caprate iv) 10-500 mg nicotinamide and v) 0-25 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- the therapeutic peptide is a GLP-1 agonist such as semaglutide having a formula of N-epsilon26-[2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-(17-carboxy- heptadecanoylamino)butyrylamino]ethoxy ⁇ ethoxy)acetylamino]ethoxy ⁇ ethoxy)acetyl] [Aib8,Arg34]GLP-1(7-37) and the salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC) is sodium N-(8-(2-hydroxybenzoyl)amino)caprylic acid (SNAC).
- GLP-1 agonist such as semaglutide having a formula of N-epsilon26-[2-(2- ⁇ 2-[2-(2- ⁇ 2-[(S)-4-carboxy-4-(17-carboxy- heptadecanoylamino)butyryl
- the therapeutic peptide is a GLP-1 agonist such as diacylated [Aib8,Arg34,Lys37]GLP-1(7-37) (SEQ ID NO.4) and named N ⁇ 26 ⁇ 2-[2-(2- ⁇ 2-[2-(2- ⁇ (S)-4-Carboxy- 4-[10-(4-carboxyphenoxy)decanoylamino]butyrylamino ⁇ - ethoxy)ethoxy]acetylamino ⁇ ethoxy)ethoxy]acetyl ⁇ , N ⁇ 37 - ⁇ 2-[2-(2- ⁇ 2-[2-(2- ⁇ (S)-4-carboxy-4-[10-(4- carboxyphenoxy)decanoylamino]butyrylamino ⁇ ethoxy)ethoxy]acetylamino ⁇ ethoxy)ethoxy]-acetyl ⁇ - [Aib 8 ,Arg 34 ,Lys 37 ]GLP-1(7-37)–peptide (G)
- the GLP-1 agonist is N ⁇ 27 -[2-[2-[2-[[2-[2-[2-[[(4S)-4-carboxy-4- [10-(4-carboxyphenoxy)decanoylamino]butanoyl]amino] ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy]-acetyl], N ⁇ 36 -[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[(4S)-4-carboxy-4-[10- (4-carboxyphenoxy)decanoylamino]- butanoyl]amino]ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy]acetyl]-[Aib8,Glu22,Arg26,Lys27, Glu30,Arg34,Lys36]-GLP-1-(7-37)-peptidy
- the therapeutic peptide is a PCSK9 inhibitor peptide.
- the invention further relates to a method of treating a subject in need thereof, comprising administering a therapeutically effective amount of a composition according to the present invention to said subject.
- the method of treatment is for treatment of a disease or disorder, such as (i) improving lipid parameters and/or (ii) preventing and/or treating cardiovascular diseases and/or the further indications specified above.
- a method for treating a disease or disorder comprising administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a PCSK9 inhibitor, a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid (NAC), a hydrotrope, a second absorption enhancer and optionally, a lubricant.
- a pharmaceutical composition comprising a PCSK9 inhibitor, a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid (NAC), a hydrotrope, a second absorption enhancer and optionally, a lubricant.
- a method for i) improving lipid parameters and/or ii) treating or preventing cardiovascular diseases comprising administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising i) 0.1-150 mg of a PCSK9 inhibitor, ii) 25-1000 mg of salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt (SNAC), iii) 15-750 mg nicotinamide, iv) 25-1000 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such as capric acid, or a salt hereof, such as sodium caprate, and v) 0-25 mg lubricant.
- a pharmaceutical composition comprising i) 0.1-150 mg of a PCSK9 inhibitor, ii) 25-1000 mg of salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt (SNAC), i
- the PCSK9 inhibitor is selected from compounds A to J described herein above. In some embodiments, the PCSK9 inhibitor is selected from compounds A, L, C, D, E, F, G H, I and J described herein above. In some embodiments, the PCSK9 inhibitor is compound H disclosed above having the following structure: Various examples of a lubricant are described, including magnesium stearate.
- the composition is administered orally and is in a form of a table, capsule or a sachet. In a further such embodiments one or more dose units may be administered to said subject in need. In one embodiment a unit dose is administered orally.
- the treatment with a GLP-1 peptide composition according to the present invention may also be combined with one or more additional active pharmaceutical ingredient(s), e.g. selected from antidiabetic agents, anti-obesity agents, appetite regulating agents, antihypertensive agents, agents for the treatment and/or prevention of complications resulting from or associated with diabetes and agents for the treatment and/or prevention of complications and disorders resulting from or associated with obesity.
- additional active pharmaceutical ingredient(s) e.g. selected from antidiabetic agents, anti-obesity agents, appetite regulating agents, antihypertensive agents, agents for the treatment and/or prevention of complications resulting from or associated with diabetes and agents for the treatment and/or prevention of complications and disorders resulting from or associated with obesity.
- Examples of these pharmacologically active substances are: Insulin, sulphonylureas, biguanides, meglitinides, glucosidase inhibitors, glucagon antagonists, DPP-IV (dipeptidyl peptidase-IV) inhibitors, sodium glucose linked transporter 2 (SGLT2) inhibitors; canagliflozin, dapagliflozin, empagliflozin, ertugliflozin, ipragliflozin, tofogliflozin, luseogliflozin, bexagliflozin, remogliflozin etabonate and sotagliflozin, particulally dapagliflozin and empagliflozin, inhibitors of hepatic enzymes involved in stimulation of gluconeogenesis and/or glycogenolysis, glucose uptake modulators, compounds modifying the lipid metabolism such as antihyperlipidemic agents as HMG CoA inhibitor
- Treatment with a PCSK9 inhibitor in a composition according to the present invention may be combined with treatment with one or more additional pharmacologically active substances, e.g. selected from anti-diabetic agents, anti-obesity agents, appetite regulating agents, antihypertensive agents, agents for the treatment and/or prevention of complications resulting from or associated with diabetes and agents for the treatment and/or prevention of complications and disorders resulting from or associated with obesity.
- additional pharmacologically active substances are: GLP-1 receptor agonists, insulin, DPP-IV (dipeptidyl peptidase-IV) inhibitors, amylin agonists, anti-inflammatory, triglyceride-lowering agents and leptin receptor agonists.
- a solid pharmaceutical composition comprising a) a therapeutic peptide of at least 20 amino acid residues b) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), c) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and d) hydrotrope.
- the therapeutic peptide is at least 30, such as at least 40, such as at least 50 amino acid residues.
- the peptide therapeutic comprises 20-500 amino acids residues. 4.
- composition according to any of the previous embodiments, wherein the peptide therapeutic comprises one or more modified amino acid residue and/or non-proteogenic amino acid residues. 5. The composition according to any of the previous embodiments, wherein the peptide therapeutic has an extended half-life. 6. The composition according to any of the previous embodiments, wherein the peptide therapeutic has a plasma half-life in humans of at least 24 hours, such as at least 48 hours, such as at least 72 hours. 7. The composition according to any of the previous embodiments, wherein the peptide therapeutic has a plasma half-life in humans of at least 96 hours, such as 120-200 hours, 8. The composition according to any of the previous embodiments, wherein the therapeutic peptide is a fatty acid substituted peptide. 9.
- composition according to any of the previous embodiments, wherein the therapeutic peptide has an albumin binding substituent.
- the composition according to any of the previous embodiments, wherein the peptide therapeutic comprises an albumin binding moiety.
- the composition according to any of the previous embodiments, wherein the peptide therapeutic comprises two albumin binding moieties.
- the peptide therapeutic comprises a substituent comprising an albumin binding moiety.
- the composition according to any of the previous embodiments, wherein the peptide therapeutic comprises two substituents comprising an albumin binding moiety.
- the substituent comprising an albumin binding moiety is attached via a lysine residue. 15.
- composition according to any of the previous embodiments wherein the substituent comprising an albumin binding moiety is attached via an epsilon nitrogen of a lysine residue. 16.
- the albumin binding moiety is selected from the group consisting of: Chem.1: HOOC-(CH2)n-CO-* wherein n is an integer in the range of 8-20, Chem.2: 5-tetrazolyl-(CH2)n-CO-* wherein n is an integer in the range of 8-20, Chem.3: HOOC-(C6H4)-O-(CH2)m-CO-* wherein n is an integer in the range of 8-20, Chem.4: HO-S(O)2-(CH2)n-CO-* wherein n is an integer in the range of 8-20, Chem.5: MeS(O)2NH(CO)NH-(CH2)n-CO-* wherein n is an integer in the range of 8-20 and Chem.
- the substituent comprises a linker.
- the linker comprises one or more elements selected from the group consisting of: Glu, ⁇ Glu, Gly, Ser, Ala, Thr, Ado, Aeep, Aeeep and TtdSuc. 19.
- the composition according to embodiment 1-4 wherein the therapeutic peptide is a GLP- 1 agonist.
- 20. The composition according to embodiment 1-5, wherein the therapeutic peptide is a GLP- 1 receptor agonist.
- composition according to embodiment 1, wherein the therapeutic peptide is GLP-1 peptide 1 or GLP-1 peptide 3. 23. The composition according to embodiment 1, wherein the therapeutic peptide is a pro- drug of semaglutide. 24. The composition according to embodiment 1, wherein the therapeutic peptide is an insulin analogue. 25. The composition according to embodiment 1, wherein the therapeutic peptide is a PCSK9 inhibitor. 26. The composition according to embodiment 1, wherein the therapeutic peptide is a PCSK9 inhibitor peptide selected from the group consisting of:
- composition according to any of the previous embodiments, wherein the peptide therapeutic is a PCSK9 inhibitor peptide selected from the group consisting of compounds G, H, I and J. 28.
- the composition according to any of the previous embodiments, wherein the peptide therapeutic is compound H 29.
- the hydrotrope is selected from the group consisting of : Resorcinol, Pyrocatechol, Pyrogallol, Gentisic acid, Xylenesulfonate, p-toluenesulfonate, Nicotinamide, Dimethylbenzamide, Diethylbenzamide, 1-methylnicotinamide, Salicyclic acid and P-Hydroxybenzoic acid. 30.
- composition according to any of the previous embodiments, wherein the hydrotrope is nicotinamide and/or Resorcinol. 31. The composition according to any of the previous embodiments, wherein the hydrotrope is nicotinamide. 32. The composition according to any of the previous embodiments, wherein the hydrotrope is not sodium benzoate. 33. The composition according to any of the previous embodiments, wherein the composition further comprises a lubricant. 34. The composition according to embodiment 13, wherein the lubricant is magnesium stearate or glyceryl dibehenate. 35. The composition according to embodiment 13, wherein the lubricant is magnesium stearate 36. The composition according to any of the previous embodiments, wherein the fatty acid is a saturated fatty acid.
- composition according to any of the previous embodiments, wherein the fatty acid is consisting of 8-12 carbon atoms or 10-12 carbon atoms. 38. The composition according to any of the previous embodiments, wherein the fatty acid is capric acid or a salt thereof. 39. The composition according to any of the previous embodiments, wherein the fatty acid or a salt hereof is sodium caprate. 40. The composition according to any of the previous embodiments, wherein the salt of NAC is selected form the sodium, the potassium and the ammonium salt of NAC. 41. The composition according to any of the previous embodiments, wherein the salt of NAC is sodium N-(8-(2-hydroxybenzoyl)amino)caprylate (SNAC). 42.
- composition according to any one of the preceding embodiments wherein the weight ratio (w/w) of the amount (w) of salt of NAC and the amount (w) of the hydrotrope is at least 0.5. 43. The composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of salt of NAC and the amount (w) of the hydrotrope is 0.5-10, such as 0.5-8 or such as 0.5-5. 44. The composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of the salt of NAC to the amount (w) of nicotinamide is 0.5-10, such as 0.5-8 or such as 0.5-5. 45.
- composition according to any of the previous embodiments wherein the weight ratio (w/w) of the amount (w) of SNAC to the amount (w) of nicotinamide is 0.5-5, such as 0.8- 3 or such as 0.5-5. 46.
- the composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of salt of NAC to the amount (w) of hydrotrope is 0.5-2, such as 1-2, or such as 1.2-1.8.
- the weight ratio (w/w) of the amount (w) of salt of NAC to the amount (w) of nicotinamide is 0.5-2, such as 1-2, or such as 1.2-1.8. 48.
- composition according to any of the previous embodiments wherein the weight ratio (w/w) of the amount (w) of SNAC to the amount (w) of nicotinamide is 0.5-2, such as 1-2, or such as 1.2-1.8 49.
- the composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of salt of NAC to the amount (w) of hydrotrope is 1-2, such as 1.2-1.8 or such as 1.3-1.7, or such as 1.4-1.6. 50.
- composition according to any of the previous embodiments wherein the weight ratio (w/w) of the amount (w) of salt of NAC to the amount (w) of nicotinamide is 1-2, such as 1.2-1.8 or such as 1.3-1.7, or such as 1.4-1.6. 51.
- the composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of SNAC to the amount (w) of nicotinamide is 1-2, such as 1.2- 1.8 or such as 1.3-1.7, or such as 1.4-1.6. 52.
- composition according to any of the previous embodiments wherein the weight ratio (w/w) of the amount (w) of the salt of NAC to the of the amount (w) of the fatty acid or salt hereof is 0.5-10, such as 0.5-5 or such as 0.5-2. 53.
- the composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of the salt of NAC and the amount (w) of capric acid or a salt hereof is 0.5-10, such as 0.5-5 or such as 0.5-2. 54.
- composition according to any of the previous embodiments wherein the weight ratio (w/w) of the amount (w) of the salt of NAC and the amount (w) of sodium caprate is 0.5- 10, such as 0.5-5 or such as 0.5-2. 55.
- the composition according to any of the previous embodiments, wherein the weight ratio (w/w) of the amount (w) of SNAC to the amount (w) of sodium caprate is 0,5-10, such as 0.5-5 or such as 0.5-2.
- the ratio of salt of NAC/medium chain fatty acid or salt thereof is 0.5-2, such as 0.8-1.7 or such as 0.9-1.2. 57.
- composition according to any of the previous embodiments consisting of: a) a therapeutic peptide of at least 10, such as at least 20 amino acid residues, b) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), c) a fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof, such as sodium caprate, d) nicotinamide or resorcinol and e) a lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- a fatty acid consisting of 6-14 carbon atoms such capric acid, or a salt hereof, such as sodium caprate
- nicotinamide or resorcinol and e) a lubricant.
- composition according to any of the previous embodiments wherein the composition comprises 1-50, 1-40 mg, 1-30 mg, 1-25, or 1-20 mg of the peptide therapeutic.
- composition comprises 10-100, 20-100 mg, 30-100 mg, 40-100 or 50-100 mg of the peptide therapeutic.
- amount of the peptide therapeutic is less than 50 % w/w, such as less than 40, 30, 20, 15, 10 or 5 % w/w of the composition.
- a unit dosage comprises 0.1-100 mg of a GLP-1 agonist.
- composition according to any of the previous embodiments, wherein a unit dosage comprises 1-100 mg semaglutide. 67. The composition according to any one of the previous embodiments, wherein a unit dose comprises 0.1-200 mg of a PCSK9 inhibitor. 68. The composition according to any one of the preceding embodiments, wherein a unit dose comprises 1-100 mg of the PCSK9 inhibitor Compound H having the structure: 69. The composition according to any of the previous embodiments, wherein a unit dosage comprises at most 1000 mg of said salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC). 70.
- composition according to any of the previous embodiments wherein a unit dosage comprises at least 20 mg of said salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC).
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- a unit dosage comprises 20-1000 mg of said salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC).
- a unit dosage comprises at most 1000 mg of said fatty acid or salt thereof, such as said salt of capric acid.
- a unit dosage comprises at least 20 mg of said fatty acid or salt thereof, such as said salt of capric acid.
- composition according to any of the previous embodiments, wherein a unit dosage comprises 20-1000 mg of said fatty acid or salt thereof, such as said salt of capric acid. 75. The composition according to any of the previous embodiments, wherein a unit dosage comprises 10-600 mg of the hydrotrope, such as nicotinamide. 76. The composition according to any of the previous embodiments, wherein a unit dosage comprises 0.1-25 mg lubricant, such as magnesium stearate. 77.
- composition according to any of the previous embodiments wherein a unit dosage comprises i) 0.1-100 mg therapeutic peptide of at least 20 amino acid residues, ii) 200-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 20-1000 mg of a fatty acid consisting of 6-14 carbon atoms, such as capric acid, or a salt hereof, such as sodium caprate, iv) 10-500 mg nicotinamide and v) 0-25 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- 20-1000 mg of a fatty acid consisting of 6-14 carbon atoms such as capric acid, or a salt hereof, such as sodium caprate
- 10-500 mg nicotinamide and v) 0-25 mg lubricant 78.
- composition according to any of the previous embodiments, wherein a unit dosage comprises vi) 0.5-100 mg GLP-1 agonist, vii) 20-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), viii) 20-1000 mg a fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate ix) 10-750 mg, such as 50-200 mg nicotinamide or resorcinol and x) 0-25 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- 20-1000 mg a fatty acid consisting of 6-14 carbon atoms such capric acid, or a salt hereof such as sodium caprate ix
- 10-750 mg such as 50-200 mg nicotinamide or resorcinol and x
- composition according to any of the previous embodiments wherein a unit dosage comprises i) 1-75 mg GLP-1 agonist, ii) 25-300 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 25-300 mg a fatty acid consisting of 6-14 carbon atoms, such capric acid, or a salt hereof such as sodium caprate, iv) 15-200 mg nicotinamide and v) 0-10 mg lubricant.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- composition according to any of the previous embodiments, wherein a unit dosage comprises: a) 1-75 mg GLP-1 agonist, b) 20-200 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), c) 20-200 mg sodium caprate, d) 10-150 mg nicotinamide and e) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- composition according to any of the previous embodiments wherein a unit dosage comprises a) 1-50 mg GLP-1 agonist, b) 20-100 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), c) 20-100 mg sodium caprate, d) 10-75 mg nicotinamide and e) 0-10 mg magnesium stearate.
- NAC N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- SNAC sodium salt of NAC
- composition according to any of the previous embodiments, wherein the composition comprises i) 0.1-200 mg of a PCSK9 inhibitor peptide; ii) 20-600 mg, such as 25-400 mg, such as 50-300 mg of a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid; iii) 20-600 mg, such as 25-400 mg, such as 50-300 mg of a medium chain fatty acid consisting of 6-14 carbon atoms, such as capric acid or a salt thereof, such as sodium caprate; iv) 10-400 mg Nicotinamide; and v) 0-50 mg lubricant.
- a PCSK9 inhibitor peptide ii) 20-600 mg, such as 25-400 mg, such as 50-300 mg of a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid
- 20-600 mg such as 25-400 mg, such as 50-300 mg of a medium chain fatty acid
- composition according to any one of the preceding embodiments, wherein the composition comprises: i) 1-100 mg, such as 5-80 mg of a PCSK9 inhibitor peptide; ii) 50-500 mg of a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid; iii) 50-500 mg of a saturated, medium-chain fatty acid consisting of 6-14 carbon atoms or a salt thereof; iv) 10-400 mg Nicotinamide; and v) 0-25 mg lubricant. 84.
- composition according to any one of the preceding embodiments, wherein the composition comprises: i) 10-80 mg of a PCSK9 inhibitor peptide; ii) 50-400 mg of a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid; iii) 50-400 mg of a saturated, medium-chain fatty acid consisting of 6-14 carbon atoms or a salt thereof; iv) 10-300 mg Nicotinamide; and v) 0-20 mg lubricant.
- a PCSK9 inhibitor peptide ii) 50-400 mg of a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid
- iii) 50-400 mg of a saturated, medium-chain fatty acid consisting of 6-14 carbon atoms or a salt thereof iv) 10-300 mg Nicotinamide; and v) 0-20 mg lubricant.
- composition according to any one of the preceding embodiments, wherein the composition comprises: i) 10-50 mg of a PCSK9 inhibitor peptide; ii) 50-300 mg of a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid; iii) 50-300 mg of a saturated, medium-chain fatty acid consisting of 6-14 carbon atoms or a salt thereof; iv) 10-200 mg Nicotinamide; and v) 0-15 mg lubricant.
- the composition is a pharmaceutical composition for oral administration.
- the composition is for use in a method of treatment. 88.
- composition according to any of the previous embodiments wherein the composition is a pharmaceutical composition for use in a method of treating diabetes and/or obesity.
- a method for treatment of diabetes and/or obesity comprising administering to a subject in need a therapeutically effective amount of a composition according to embodiments 1-81.
- the method according to embodiment 89, wherein said composition is administered, once daily or less frequent.
- a method for preparation of a solid pharmaceutical composition comprising the steps of: a) granulating two absorption enhancers and a hydrotrope b) blending the granulates of a) with a therapeutic peptide, such as a GLP-1 peptide or a PCSK9 inhibitor, and optionally a lubricant c) compressing the blend of b) into tablets. 92.
- a method for preparation of a solid pharmaceutical composition comprising the steps of: a) blending a therapeutic peptide, such as a GLP-1 peptide or a PCSK9 inhibitor, two absorption enhancers, a hydrotrope and optionally a lubricant b) granulating the blend of a) c) optionally blending the granules of a) with a lubricant d) compressing the granules of b) or the blend of c) into tablets.
- a method for producing a solid pharmaceutical composition comprising the steps of: a) obtaining a salt of NAC and a hydrotrope, b) co-processing said salt of NAC and hydrotrope, c) obtaining a salt of capric acid d) blending the product of b) with the salt of capric acid of c) and e) optionally include a lubricant in the blending process d) f) preparing said solid pharmaceutical composition using the blend of d) or e) wherein a peptide therapeutic, such as a GLP-1 peptide or a PCSK9 inhibitor, is included in any of the steps a)-f). 94.
- a method for producing a solid pharmaceutical composition comprising a GLP-1 agonist, wherein the method comprises the steps of: a) obtaining a salt of NAC and a hydrotrope, b) hot melt extruding said salt of NAC and hydrotrope of a) and c) obtaining a salt of capric acid d) granulating the salt of capric acid e) blending the product of b) with the granulate of d) and f) optionally include a lubricant in the blending process e) g) preparing said solid pharmaceutical composition using the blend of e) or f) wherein the GLP-1 agonist is included in any of the steps.
- a method for producing a solid pharmaceutical composition comprising a PCSK9 inhibitor, wherein the method comprises the steps of: a) obtaining a salt of NAC and a hydrotrope, b) hot melt extruding said salt of NAC and the hydrotrope of a); c) obtaining a salt of capric acid; d) granulating the salt of capric acid; e) blending the product of b) with the granulate of d); and f) optionally include a lubricant in the blending process e); g) preparing said solid pharmaceutical composition using the blend of e) or f) wherein the PCSK9 inhibitor is included in any of the steps.
- the lubricant is magnesium stearate or glyceryl dibehenate.
- the lubricant is magnesium stearate 105.
- one of the two enhancers are a fatty acid, such as a saturated fatty acid or a salt hereof.
- 106 The method according to any of the previous embodiments 91-105, wherein the fatty acid is consisting of 8-12 carbon atoms or 10-12 carbon atoms.
- the fatty acid is capric acid. 108.
- the tablets are placed in the back of the mouth of the dog to prevent chewing.
- the mouth is then closed, and 10 mL of tap water is given by a syringe to facilitate swallowing of the tablet.
- 40 mL of water is administered by gavage just prior to tablet dosing, where after the tablet is dosed and 10 mL of tap water is given by a syringe to facilitate swallowing of the tablet.
- Blood sampling Blood is sampled at predefined time points for up till 10 hr post dosing to adequately cover the full plasma concentration-time absorption profile of the therapeutic peptide
- For each blood sampling time point approximately 0.8 mL of whole blood is collected in a 1.5 mL EDTA coated tube, and the tube is gently turned to allowing mixing of the sample with the EDTA.
- Blood samples (for example 0.8 mL) are collected in EDTA buffer (8mM) and then centrifuged at 4°C and 2000G for 10 minutes. Plasma is pipetted into Micronic tubes on dry ice and kept at - 20°C until analysis.
- Blood samples are taken as appropriate, for example from a venflon in the cephalic vein in the front leg for the first 2 hours and then with syringe from the jugular vein for the rest of the time points (the first few drops are allowed to drain from the venflon to avoid heparin saline from the venflon in the sample).
- General methods for tablet preparation Method 1 Dry Granulation Prior to dry granulation the enhancer or enhancers is/are blended with magnesium stearate by manual geometric mixing followed by blending on V-shell blender (50 min, 25 rpm). Dry granulation is carried out by roller compaction on a Gerteis MINI-PACTOR.
- Hot melt extrusion Hot melt extrusion is carried out on a Leistritz ZSE Micro 27. The enhancer or enhancers and nicotinamide fed separately into the extruder using gravimetric feeders. The equipment is operated at process temperatures varying between 105 ⁇ C to 200 ⁇ C along the barrel to facilitate the melt extrusion. The screw speed is approximately 300 rpm. The resulting extrudates are milled using a final screen of 0.4 mm.
- Method 3 – Wet granulation Twin screw wet granulation is carried out using a Thermo Scientific Process 11 twin screw extruder at a screw speed of 100 rpm.
- the enhancer is fed into the barrel using a gravimetric feeder at 100 g/h and the granulation medium, water, is added using a peristaltic pump at a rate of 0.25 ml/h.
- the barrel is water-cooled to 30 ⁇ C.
- Milling of the dried extrudates is carried out using a Frewitt FreDrive-Lab oscillating mill at a speed of 300 mm/s through a 0.355x0.14 mm flat screen.
- Method 4 Blending for tablet compression Blending is carried out by manual geometric mixing the intermediate granulate with any further ingredients followed by blending on a turbula mixer (7 min, 25 rpm). In compositions including additional magnesium stearate it was sieved through a 125 ⁇ m or 355 ⁇ m mesh and added in a secondary blending step prior to compression by manual geometric mixing followed by blending on a turbula mixer (2 min, 25 rpm).
- Method 5 Tablet compression Tablets are produced on a Kilian STYL’One or a Fette 102i mounted with a single set of punches, resulting in either 7 mm round or 6.8 mm ⁇ 12 mm or 8 mm x 14 mm oval compound cup tablets having no score.
- Punch size is chosen according to the total tablet weight.
- the press speed is set to 10% and for Fette 102i the press speed is set at 20 rpm.
- the fill volume is adjusted to obtain tablets having target weights based on composition. Compression forces around 1 to 25 kN are applied to obtain tablets with a crushing strength of around 20-120 N respective to the tablet size.
- compositions described herein the content in weight (mg) of the peptide therapeutic is provided.
- the peptide therapeutic preparations may include impurities, and thus the absolute weight of peptide therapeutic preparation in a tablet is higher than the listed amount of the peptide therapeutic, and therefore the total weight of the tablet is usually slightly higher than the sum of the ingredients listed.
- GLP-1 analogue a matched antibody pair (2F6 and 3F15) is involved in the assay, one biotinylated (3F15) and bound to streptavidin-coated Alpha donor beads, and the other conjugated to AlphaLISA acceptor beads (2F6).
- LOCI Luminescence Oxygen Channeling Immunoassay
- This emission signal is measured in the EnVision plate reader for API concentration analysis.
- the amount of light was proportional to the concentration of active peptide ingredient and the lower limit of quantification (LLOQ) in plasma was 100 pM.
- the matched antibody pair is 3F15 and 7F1.
- the antibody pair for use is 1F31 and S1C3D3E3.
- the latter (S1C3D3E3) is biotinylated and bound to streptavidin-coated Alpha donor beads, while the former (1F31) is conjugated to AlphaLISA acceptor beads.
- LC-MS liquid chromatography-mass spectrometry
- the supernatant was diluted with 2 volumes of Milli-Q water containing 1% formic acid before injection on the LC-MS system.
- the system used was a Transcend II Interface Module SRD3200 system from Thermo Scientific (Waltham, MA, USA) coupled to Orbitrap QExactive Plus mass spectrometer from Thermo Scientific.
- the LC was equipped with a Cyclone column (CH-953288, Thermo Scientific) as the first dimensional trapping column and Aeris 3.6 ⁇ m PEPTIDE XB-C18 as the analytical column (2.1 x 50 mm from Phenomenex).
- the mobile phase composition of the loading pump is as below: mobile phase A consists of 95% milli-Q water, 2.5% acetonitrile, 2.5 % methanol and 1% formic acid; mobile phase B consists of 47.5% acetonitrile, 47.5% methanol, 5% milli-Q water, and 1% formic acid.
- the analyte of interest was loaded from the Turbo flow column at 15% B to the second dimensional analytical column.
- the Orbitrap QExactive Plus were operating in positive ionization mode with the parallel reaction monitoring (PRM) scan mode. Linear calibration curves (weighting 1/x 2 ) were used for calculating the concentration in the plasma samples. Quality control samples for analytes were included.
- PCSK9-LDL-R binding - Competitive This assay measures the apparent binding affinity to PCSK9 in competition with LDL-R.
- the assay is used to evaluate the apparent binding affinity of an PCSK9 inhibitor such as an EGF(A) analogue and compounds comprising an EGF(A) analogue
- PCSK9 inhibitor such as an EGF(A) analogue and compounds comprising an EGF(A) analogue
- recombinant human Low Density Lipoprotein Receptor (rhLDL-R; NSO-derived; R & D systems # 2148-LD) is dissolved at 1 ⁇ g/ml in 50 mM sodium carbonate, pH 9.6, and then 100 ⁇ l of the solution is added to each well of the assay plates (Maxisorp 96, NUNC # 439454) and coated overnight at 4 °C.
- 8 point concentration curves of the EGF(A) compounds containing Biotinylated PCSK9 (0.5 ug/ml, BioSite/BPSBioscience cat#71304) are made in duplicate.
- Test compound and biotinylated PCSK9 mixtures are prepared and incubated for 1 hour at room temperature in assay buffer containing 25 mM Hepes, pH 7.2 (15630-056, 100 ml, 1M), 150 mM NaCl (Emsure 1.06404.1000) 1 % HSA (Sigma A1887-25G) 0.05 % Tween 20 (Calbiochem 655205) 2 mM CaCl 2 (Sigma 223506-500G).
- the coated assay plates are then washed 4x in 200 ⁇ l assay buffer, and then 100 ⁇ l of the mixture of test compounds and biotinylated PCSK9 is added to the plates and incubated 2 h at room temperature.
- the plates are washed 4x in 200 ⁇ l assay buffer and then incubated with Streptevadin-HRP (25ng/ml; VWR # 14-30-00) for 1 h at room temperature.
- the reaction is detected by adding 50 ⁇ l TMB-on (KEM-EN-TEC) and incubated 10 min in the dark. Then the reaction is stopped by adding 50 ⁇ l 4 M H 3 PO 4 to the mixture, added by electronic multi pipetting.
- the plates are then read in a Spectramax at 450 and 620 nm within 1 h. The 620 nm read is used for background subtraction.
- IC50 values are calculated using Graphpad Prism, by nonlinear regression log(inhibitor) vs.
- Kd of the biotin-PCSK9 is 1.096727714 ⁇ g/ml
- [Biotin-PCSK9] 0.5 ⁇ g/ml.
- Higher Ki values reflects lower apparent binding affinities to PCSK9 and vice versa. A value above 500 nM, will indicate that the observed binding is not specific.
- Ki values for examples of EGF(A) peptide analogues and derivatives thereof are included below, showing that the high affinity of compounds having an EGF(A) peptide including 301L and optionally one or more of 309R, 312E and 321E is very similar also including compounds with one or two substituents attached to the N-terminal or a Lysine residue.
- EGF(A) peptide analogues Ki EGF(A) peptide derivatives, PCSK9i peptide therapeutics
- EGF(A) peptide SEQ ID NO Substituent Attachment Ki compound # site(s) (nM) 9
- Example 1 - Preparation of compositions In order to evaluate the disintegration time for different solid composition comprising one or more absorption enhancer and optional a hydrotrope a series of tablets comprising SNAC, sodium caprate (NaC10), nicotinamide and magnesium stearate (MgSt) was prepared including the excipients as specified in the table 1.1 below.
- compositions T4-C1 and T4-D1 where prepared by separately granulating SNAC + nicotinamide by method 2 and NaC10 by method 3 followed by blending with magnesium stearate prior to compression, while compositions T4-C2 and T4-D2 were prepared by blending all ingredients prior to granulation by method 1. Tablets not including nicotinamide were punched in size 6,8x12 mm while the tablets including nicotinamide were punched in size 8x14 mm.
- Table 1.1 Tablet compositions expressed as mg per tablet.
- Example 2 Disintegration of enhancer compositions
- Table 2.1 provides % disintegration after 25 and 30 minutes. It was surprisingly found that the disintegration of a tablet comprising SNAC, NaC10 and nicotinamide (T4 tablets) is accelerated compared to tablets including SNAC, SNAC and NaC10 or SNAC and nicotinamide (Ref -T1, -T2 and -T3 tablets).
- Table 2.1 Percent disintegration for the different compositions after 25 and 30 minutes.
- Example 3 Preparation of compositions comprising GLP-1 peptide compound and exposure of GLP-1 peptide after oral administration to beagle dogs Tablets with different amounts of different GLP-1 peptides, SNAC and further excipients were prepared. The content of the prepared compositions is provided in Tables 3.1 and 3.2.
- GLP-1 peptide 1 is Diacylated [Aib8,Arg34,Lys37]GLP-1(7-37) (Example 2 of WO2011/080103)
- GLP-1 peptide 2 is semaglutide which can be prepared as described in WO2011/080103. Semaglutide can be prepared according to the method described in WO2006/097537, Example 4 and SNAC can be prepared as described in WO2008/028859.
- a first series of tablet compositions comprising GLP-1 peptide 1 different ratios of SNAC/NA/NaC10 were prepared.
- the tablet compositions were prepared by a combination of the methods 1-5 described above, whereby granules of SNAC + NA and NaC10 was obtained and subsequently mixed with magnesium stearate prior to compressions.
- a pharmacokinetic study was carried out to evaluate pharmacokinetic parameters after p.o. administration to Beagle dogs as described in assay II.
- Plasma concentration of GLP-1 peptide measured over the first 30 minutes after dosing was used to evaluate the exposure observed for the different tablet compositions.
- the table further includes the observed variability in plasma concentration of the GLP-1 peptide determined as %CV.
- the relative bioavailability is indicated by ++, +++, ++++, +++++, or ++++++, where ++++++ is used for the compositions providing the overall best results.
- Data for all compositions are obtained from dosing of 16 dogs, except Ref-T3 which was dose to 40 dogs.
- Table 3.2 Pharmacokinetic parameters obtained for the GLP-1 peptide 1 compositions of table 3.1. It was surprisingly found that the exposure of the GLP-1 peptide 1 was increased after administration of the compositions T4-F, T4-G, T4-H and, T4-I all comprising sodium caprate, whereof T4-G showed the highest exposure as illustrated in table 3.2.
- T4-G has almost equal amounts of SNAC and NaC10 and a bit less nicotinamide and thus the preferred ratio of SNAC : NaC10 : NA is around 100 : 67 : 100 (w:w:w). It is further observed that increasing the amount of NaC10 beyond 2.5 relative to SNAC is disadvantageous, and thus the preferred SNAC:NaC10 ratio is above 0.4.
- a second series of tablet compositions comprising GLP-1 peptide 2 was prepared to evaluate the exposure of GLP-1 peptide 2. The composition was prepared and tested as the GLP-1 peptide 1 tablet compositions. Table 3.3 GLP-1 peptide 2 tablet compositions. Tablet compositions expressed as mg per tablet. The disintegration of the different tablet compositions of table 3.3 was evaluated using a small volume disintegration test (Assay I). Table 3.4 provides % disintegration after 10 and 15 minutes.
- Example 5 Preparation of compositions comprising PCSK9 inhibitor and exposure of PCSK9 inhibitor after oral administration to beagle dogs Tablets with different amounts of PCSK9 inhibitor, SNAC and further excipients were prepared. The content of the prepared compositions is provided in Table 5.1 and 5.2.
- the PCSK9 inhibitor used is a peptide analogue of LDL-R293-332 comprising two substituents in the form of fatty diacids attached via a hydrophilic linker molecule.
- the EGF(A) derivative is prepared as described in WO2017/121850 (Example 151/page 161) and has the following structure and is described as compound H herein.
- the amino acid sequence is defined by SEQ ID No:16.:
- PCSK9i peptide tablets were prepared similar to as described in WO2021/023855 and WO2021/089761.
- the tablet compositions included in table 5.1 were prepared by a combination of the methods 1-5 described above, whereby granules of SNAC + NA and NaC10 were separately obtained and subsequently mixed with API and magnesium stearate prior to compressions.
- the tablets compositions included in table 5.2 were prepared by physically blending SNAC and NA and separately granulating NaC10 by method 3 prior to mixing with API and magnesium stearate.
- the tablet compositions included in table 5.1 were prepared with the same SNAC/NA/NaC10 ratio but differing in weight.
- the SNAC/NaC10 ratio was varied from about 12 to 0.1 and the SNAC/NA ration from about 20 to 0.1.
- Table 5.1 PCSK9 inhibitor tablet compositions Tablet compositions expressed as mg per tablet.
- PCSK9 inhibitor tablet compositions with different SNAC/NA/NaC10 ratios. Tablet compositions expressed as mg per tablet.
- a pharmacokinetic study was carried out to evaluate pharmacokinetic parameters after p.o. administration to Beagle dogs as described in Assay II above. Blood samples were drawn at predefined time points after dosing, and samples were analysed for concentration of the PCSK9 inhibitor. Based on these measurements plasma concentration versus time profile were plotted and a non-compartmental pharmacokinetic analysis of the data was performed. The plasma concentration of PCSK9 inhibitor measured over the first 30 minutes after dosing was used to evaluate the exposure observed for the different tablet compositions.
- the relative bioavailability is indicated by ++, +++, ++++, +++++, or ++++++, where ++++++ is used for the compositions providing the overall best results. Data for all compositions are obtained from dosing of 16 dogs, except T4-F, where 32 dogs were dosed.
- the content of the prepared compositions is provided in Table 6.1.
- the GLP-1 prodrug is Gly-N ⁇ -2-[[(4S)-4-carboxy-4-(15- carboxypentadecanoylamino)butanoyl]amino]ethyl-Gly-semaglutide (SEQ ID No.: 26) with the structure
- the analogue and preparation thereof have previously been described in WO/2022/096636.
- the tablet compositions were prepared by a combination of the methods 1-5 described above, whereby granules of SNAC + NA and NaC10 were obtained and subsequently mixed with API and magnesium stearate prior to compressions.
- GLP-1 pro-drug tablet compositions Tablet compositions expressed as mg per tablet.
- a pharmacokinetic study was carried out with T4 tablets in comparison with Ref – T2 and Ref – T3 tablets, to evaluate pharmacokinetic parameters after p.o. administration to Beagle dogs as described above and the results are included in table 7.2 below.
- Composition Cp/D 05hr AUC/D 0-05hr Cmax/D Relative Table 7.2 Pharmacokinetic parameters obtained for the GLP-1 prodrug compositions.
- the T4 tablet composition provided higher exposure compared to the reference tablet compositions T2 and T3 demonstrating an improvement by combining SNAC, sodium caprate and nicotinamide.
- SNAC sodium caprate
- nicotinamide good exposure can be obtained with a smaller tablet which comprises less of SNAC, NaC10 and/or nicotinamide in total.
- Example 8 Preparation of compositions comprising a GLP-1 GIP co-agonist and an insulin analogue and exposure after oral administration to beagle dogs Tablets comprising both a GLP-1 GIP co-agonist and an insulin analogue together with different amounts of SNAC and further excipients were prepared.
- the GLP-1 GIP co-agonist used is Y-Aib-EGTFTSDYSILLE-K[(2S)-2-amino-6-[[(2S)-2- amino-6-[[(4S)-4-carboxy-4-(17- carboxyheptadecanoylamino)butanoyl]amino]hexanoyl]amino]hexanoyl]- QAAREFIEWLLAGGPSSGAPPPS-OH (SEQ ID No.:25 with a substituent (HOOC-(CH 2 ) 16 -CO- gGlu-2x ⁇ Lys-) attached via epsilon nitrogen of the lysine (K) in position 16).
- the insulin analogue used is A14E,B25H,B29K(N ⁇ Octadecanedioyl- ⁇ Glu-OEG- OEG),desB30 human insulin (SEQ ID No.: 23 and 24 with a substituent ((HOOC-(CH 2 ) 16 -CO- ⁇ Glu-2xOEG-) attached via epsilon nitrogen of the Lysine (K) in position 29 of the B-chain (B29K)
- the tablet compositions were prepared by a combination of the methods 1-5 described herein above, whereby granules of SNAC + NA and NaC10 were obtained and subsequently mixed with API and magnesium stearate prior to compressions.
- the content and methods used to prepare the compositions are provided in Table 8.1.
- GLP-1-GIP tablet compositions expressed as mg per tablet.
- a pharmacokinetic study was carried out with T4 tablet in comparison with Ref – T2 and Ref – T3, to evaluate pharmacokinetic parameters after p.o. administration to Beagle dogs as described above and the results are included in table 8.3 below.
Abstract
The present invention provides solid compositions comprising a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid and a further absorption enhancer, their method of preparation and use in medicine.
Description
SOLID ORAL PEPTIDE FORMULATIONS TECHNICAL FIELD OF THE INVENTION The present invention relates to solid compositions comprising a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid and a further absorption enhancer, their method of preparation and their use in medicine. SEQUENCE LISTING The present application is filed with a Sequence Listing in electronic form. The entire contents of the sequence listing are hereby incorporated by reference. BACKGROUND Oral administration of peptide therapeutics has been an area of intense research and with the recent approval of Rybelsus® a first tablet product has been launched allowing patients to received GLP-1 RA treatment without the need for injection. Solid oral formulations of semaglutide have been described in various patent documents (WO2011/094531, WO2012/080471, WO2013/139694, WO2016/120378, WO2016/120380) describing the use of the absorption enhancers sodium N-(8-(2-hydroxybenzoyl)amino)caprylate or sodium caprate, whereof sodium N-(8-(2-hydroxybenzoyl)amino)caprylate is used in Rybelsus®. Most recently further improvement in tablet compositions were described in WO2019/149880 and WO/2019/21506, showing that a high ratio of SNAC optionally in combination with a hydrotrope is able to accelerate the bioavailability of semaglutide after oral dosing. Oral administration of peptides and proteins is of general interest such as in the treatment of cardiovascular disease. Two anti-PCSK9 antibodies, alirocumab/Praluent® and evolocumab/Repatha®, have been approved for the treatment of high LDL-C levels. These are administered by 1 ml subcutaneous injections every two weeks. The EGF(A) (Epidermal Growth Factor-like domain A) sequence (40 amino acids) of the LDL-R (LDL-R-(293-332)) is well recognized as the site for PCSK9 binding. The isolated wild- type EGF(A) peptide has been shown to inhibit the binding of PCSK9 to the LDL-R with an IC50 in the low µM range (Biochemical and Biophysical Research Communications 375 (2008) 69–73). This poor potency has prevented a practical pharmaceutical use of the EGF(A) peptide. Furthermore, the half-life of such peptides would be expected to be too short to be of therapeutic use.
WO2012177741 and J. Mol. Biol. (2012) 422, 685-696 disclose analogues of the EGF(A) and Fc-Fusion thereof. Alternative EGF(A) peptide based PCSK9 inhibitors with an extended half-life have been disclosed in WO2017/121850. In order to increase the usability of such drugs it is of interest to develop a suitable oral formulation. Oral administration of therapeutic peptides is challenging due to the rapid degradation of such peptides in the gastrointestinal system. Solid oral formulations of PCSK9 inhibitors have recently been described in WO2021/023855 and WO2021/089761 showing that a high ratio of sodium N-(8-(2- hydroxybenzoyl)amino)caprylate (SNAC) optionally in combination with a hydrotrope is able to increase the plasma concentration of a PCSK9 inhibitor after oral dosing. Although an efficacious GLP-1 treatment has been reached, the bioavailability of the peptide therapeutic i.e. the percentage of the peptide therapeutic that reaches circulation after oral administration is still in the lower single digits compared to subcutaneous administration and thus further alternative compositions providing increased bioavailability of peptides are highly desirable. SUMMARY The present application describes that a more cost-effective product can be obtained by combing absorption enhancers. Considering the low bioavailability of a peptide therapeutic after oral administration, the amounts of API and excipients (particular absorption enhancers) are substantial, and thus the ability to reduce the amount of API and/or excipient(s) needed to reach a given level of exposure of a peptide therapeutic is advantageous. The present invention describes solid pharmaceutical compositions which includes a mix of absorption enhancers. In one embodiment the solid pharmaceutical composition comprises a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid and a fatty acid such as a salt of capric acid. In one embodiment the solid pharmaceutical composition comprises a salt of N-(8-(2- hydroxybenzoyl)amino)caprylic acid, a fatty acid such as a salt of capric acid and a hydrotrope such as nicotinamide. In one embodiment the composition comprises a peptide therapeutic. In one embodiment the solid pharmaceutical composition comprises i) a peptide therapeutic ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
In one embodiment the peptide therapeutic is an acylated peptide, such as a GLP-1 receptor agonist, a PCSK9 inhibitor peptide or an insulin analogue. The peptide therapeutics may comprise one or more albumin binding moieties covalently attached to the peptides, such as in the form of a substituent comprising an albumin binding moiety. In one embodiment the solid pharmaceutical composition comprises i) a GLP-1 agonist, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide. In one embodiment the solid pharmaceutical composition comprises i) semaglutide or a pro-drug of semaglutide ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide. In one embodiment the solid pharmaceutical composition comprises i) a GLP-1-GIP co-agonist, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide. In one embodiment the solid pharmaceutical composition comprises i) a PCSK9 inhibitor peptide, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide. In one embodiment the solid pharmaceutical composition comprises i) an insulin analogue, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide
In further aspects the invention relates to medical use of a composition as described herein, such as the use in a method of treatment of diabetes, obesity and/or cardiovascular diseases. DESCRIPTION Peptide therapeutics The term “peptide” as used in the context of the invention, refers to a compound which comprises a series of amino acids interconnected by amide (or peptide) bonds. A peptide therapeutic may also herein be referred to as a therapeutic peptide and is encompassed by the term active pharmaceutical ingredient as relevant. A peptide therapeutic is a peptide-based compound suited for medical treatment, comprising at least 20 amino acid residues. In further embodiments the peptide therapeutic comprises at least 30, such as at least 40, such as at least 50 amino acid residues. In one embodiment the peptide therapeutic comprises 20-500 amino acids residues. In one embodiment the peptide therapeutic may comprise one or more modified amino acid residues and/or non- proteogenic amino acid residues. An example of a peptide therapeutics is semaglutide, and other GLP-1 agonists described below, and thus a peptide therapeutic is frequently an analogue of a naturally existing peptide. Other examples are insulin analogues and PCSK9 inhibitor peptides. In one embodiment the peptide therapeutic has an extended half-life compared to the naturally existing peptide. In one embodiment the peptide therapeutic has a plasma half-life in humans of at least 24 hours, such as at least 48 hours, such as at least 72 hours. In one embodiment the peptide therapeutic has a plasma half-life in humans of at least 96 hours, such as 120-200 hours. The plasma half-life of a peptide may be extended by attaching an albumin binding fatty acid to a peptide, which has been demonstrated for several peptide and proteins. In one embodiment the peptide therapeutic is a fatty acid substituted peptide. In one embodiment the peptide therapeutic has an albumin binding substituent. In one embodiment the peptide therapeutic comprises an albumin binding moiety. In one embodiment the peptide therapeutic comprises two albumin binding moieties. In one embodiment the peptide therapeutic is a pro-drug. In one embodiment the peptide therapeutic is a pro-drug comprising a pharmaceutical active peptide and a dipeptide moiety which is converted into a diketopiperazine moiety upon liberation of the pharmaceutical active peptide, such as those described in WO2010/071807, WO2010/080605, WO2011/163012, WO2014/152460 and WO2016/049174.
In general, the term peptide therapeutic is meant to encompass compound as such and any pharmaceutically acceptable salt, amide, or ester thereof. In some embodiments the composition comprises the peptide therapeutic or a pharmaceutically acceptable salt, amide, or ester thereof. In some embodiments the composition comprises the peptide therapeutic and one or more pharmaceutically acceptable counter ions. Substituent A substituent is a moiety comprised by the peptide therapeutic. According to the invention it is preferred that the moiety e.g., the substituent has no or minimal effect on the biological functionality of the peptide therapeutic while adding other beneficial properties, such as longer half-life and/or improved exposure after oral dosing. In one embodiment the peptide therapeutic comprises a substituent comprising an albumin binding moiety. In one embodiment the peptide therapeutic comprises two substituents comprising an albumin binding moiety. In one embodiment the substituent comprising an albumin binding moiety is covalently attached to the peptide. In one embodiment the substituent comprising an albumin binding moiety is attached via a lysine residue. In one embodiment the substituent comprising an albumin binding moiety is attached to the peptide back-bone via an epsilon nitrogen of a lysine residue. In some embodiments the substituent comprises a fatty acid or a fatty diacid. In some embodiments the substituent comprises a C16, C18 or C20 fatty acid. In some embodiments the substituent comprises a C16, C18 or C20 fatty diacid. In further embodiments the albumin binding moiety is selected from the group consisting of: Chem.1: HOOC-(CH2)n-CO-* wherein n is an integer in the range of 8-20, Chem.2: 5-tetrazolyl-(CH2)n-CO-* wherein n is an integer in the range of 8-20, Chem.3: HOOC-(C6H4)-O-(CH2)n-CO-* wherein n is an integer in the range of 6-20, Chem.4: HO-S(O)2-(CH2)n-CO-* wherein n is an integer in the range of 8-20, Chem.5: MeS(O)2NH(CO)NH-(CH2)n-CO-* wherein n is an integer in the range of 8-20 and Chem.6: 3-HO-Isoxazole-(CH2)n-CO-* wherein n is an integer in the range of 8-20 wherein the symbol * indicates the attachment point to a linker (see further below) or the peptide. In a further embodiment the substituent comprises Chem.1: HOOC-(CH2)n-CO-* wherein n is at least 13, such as n is 13, 14, 15, 16, 17, 18 or 19. In some embodiments n is in the range of 13 to 19, such as in the range of 13 to 17. In some embodiments n is 13, 15 or 17. In some embodiments n is 13. In some embodiments n is 15. In some embodiments n is 17.
The diacid part may also be referred to using a systematic name as follows.
 In some embodiments the substituent comprises Chem.3: HOOC-(C6H4)-O-(CH2)n-CO-* wherein n is an integer in the range of 6-14. In some embodiments the substituent comprises Chem 3b
In some embodiments the substituent comprises Chem.3: HOOC-(C6H4)-O-(CH2)n-CO-* wherein n is an integer in the range of 6-14. In some embodiments the substituent comprises Chem 3b
 wherein the carboxy group is in position 2, 3 or 4 of the (C6H
wherein the carboxy group is in position 2, 3 or 4 of the (C6H
 4) group and wherein m is an integer in the range of 8-11. In some embodiments the substituent comprises Chem 3 or Chem 3b wherein n/m is in the range of 6 to 14, such as in the range of 8 to 11. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein n/m is 8, 10 or 12. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein n/m is 9. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein/ m is 11. Additional elements may be referred to as linkers, and it follows that the substituent may then comprise or consist of an albumin binding moiety and one or more linker elements which are referred to as linker A, linker B and linker C elements herein. The overall structure of the substituent may thus be described by the formular (I) Formular (I): Albumin binding moiety - linker A - linker B(n=0-5) - linker C -*
wherein linker A and linker C are optional elements, and wherein the linker B element(s) is/are individually selected and n=0-5 indicates that the substituent comprises 0, 1, 2, 3, 4 or 5 linker B elements. In one embodiment the substituent comprises a linker A element selected from Chem.7: *-NH-SO2-(CH2)3-CO-* or and
4) group and wherein m is an integer in the range of 8-11. In some embodiments the substituent comprises Chem 3 or Chem 3b wherein n/m is in the range of 6 to 14, such as in the range of 8 to 11. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein n/m is 8, 10 or 12. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein n/m is 9. In some embodiments the substituent comprises Chem 3 or Chem 3b, wherein/ m is 11. Additional elements may be referred to as linkers, and it follows that the substituent may then comprise or consist of an albumin binding moiety and one or more linker elements which are referred to as linker A, linker B and linker C elements herein. The overall structure of the substituent may thus be described by the formular (I) Formular (I): Albumin binding moiety - linker A - linker B(n=0-5) - linker C -*
wherein linker A and linker C are optional elements, and wherein the linker B element(s) is/are individually selected and n=0-5 indicates that the substituent comprises 0, 1, 2, 3, 4 or 5 linker B elements. In one embodiment the substituent comprises a linker A element selected from Chem.7: *-NH-SO2-(CH2)3-CO-* or and
 Chem.8: -NH-CH2-(C6H10)-CO- or . comprises one or more linker B elements.
Chem.8: -NH-CH2-(C6H10)-CO- or . comprises one or more linker B elements.
 In one embodiment linker B element(s) is/are selected from the group consisting of: Glu, γGlu, Gly, Ser, Ala, Thr, Ado (or OEG), Aeep, Aeeep and TtdSuc. Glu, Gly, Ser, Ala, Thr are amino acid residues well known in the art. γGlu (or gGlu) is of formula Chem.9: *-NH-CH(COOH)-(CH2)2-CO-* which is the same as Chem. 9b:
In one embodiment linker B element(s) is/are selected from the group consisting of: Glu, γGlu, Gly, Ser, Ala, Thr, Ado (or OEG), Aeep, Aeeep and TtdSuc. Glu, Gly, Ser, Ala, Thr are amino acid residues well known in the art. γGlu (or gGlu) is of formula Chem.9: *-NH-CH(COOH)-(CH2)2-CO-* which is the same as Chem. 9b:
 TtdSuc is of formula Chem.10: *-NH-(CH2)3-O-(CH2)2-O-(CH2)2O-(CH2)3-NHCO* or *-NH-CH2CH2CH2OCH2CH2OCH2CH2OCH2CH2CH2NHCO* which is the same as Chem.10b
TtdSuc is of formula Chem.10: *-NH-(CH2)3-O-(CH2)2-O-(CH2)2O-(CH2)3-NHCO* or *-NH-CH2CH2CH2OCH2CH2OCH2CH2OCH2CH2CH2NHCO* which is the same as Chem.10b
 Ado (or OEG) is of formula Chem.11: *-NH-(CH2)2-O-(CH2)2-O-CH2-CO-* may also be referred to as 8-amino-3,6-dioxaoctanoic acid and which is the same as Chem.11b
Ado (or OEG) is of formula Chem.11: *-NH-(CH2)2-O-(CH2)2-O-CH2-CO-* may also be referred to as 8-amino-3,6-dioxaoctanoic acid and which is the same as Chem.11b
 Aeep is of formula Chem.12: *NH-CH2CH2OCH2CH2OCH2CH2CO*, which may also be referred to as Chem.12
Aeep is of formula Chem.12: *NH-CH2CH2OCH2CH2OCH2CH2CO*, which may also be referred to as Chem.12
 Aeeep is of formula Chem.13: *NH-CH2CH2OCH2CH2OCH2CH2OCH2CH2CO*, which may also be referred to as Chem.13b
Aeeep is of formula Chem.13: *NH-CH2CH2OCH2CH2OCH2CH2OCH2CH2CO*, which may also be referred to as Chem.13b
 ε-Lys is defined by Chem.14: *-NH-(CH2)4-CH(NH2)-CO-* which may also be described by Chem.14b:
ε-Lys is defined by Chem.14: *-NH-(CH2)4-CH(NH2)-CO-* which may also be described by Chem.14b:
 In some embodiments the substituent comprises one or more 8-amino-3,6- dioxaoctanoic acid (Ado), such as two Ado elements. In some embodiments the substituent comprises one or more ε-Lys, such as two ε-Lys elements. In one embodiment the substituent may further comprise a linker C element of Chem. 15: *-NH-CH2-(C6H4)-CH2-*, which may also be referred to as
In some embodiments the substituent comprises one or more 8-amino-3,6- dioxaoctanoic acid (Ado), such as two Ado elements. In some embodiments the substituent comprises one or more ε-Lys, such as two ε-Lys elements. In one embodiment the substituent may further comprise a linker C element of Chem. 15: *-NH-CH2-(C6H4)-CH2-*, which may also be referred to as
 ne or two substituent(s) is/are selected from the group of substituents consisting of: HOOC-(CH2)18-CO-gGlu-2xAdo HOOC-(CH2)18-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo HOOC-(CH2)16-CO-gGlu-2xAdo HOOC-(CH2)16-CO-gGlu-2xAdo-NH-CH2-(C6H4)-CH2 HOOC-(CH2)16-CO-gGlu HOOC-(CH2)16-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo HOOC-(CH2)14-CO-gGlu-2xAdo HOOC-(CH2)14-CO-gGlu HOOC-(CH2)14-CO-gGlu-2xAdo- HOOC-(CH2)12-CO-gGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-3xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu 4-HOOC-(C6H4)-O-(CH2)10-CO-2xgGlu 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-3xGly 4-HOOC-(C6H4)-O-(CH2)10-CO-2xgGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-TtdSuc 4-HOOC-(C6H4)-O-(CH2)9-CO 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-4xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)9-CO-gGlu-2xAdo 3-HOOC-(C6H4)-O-(CH2)9-CO-gGlu-2xAdo 3-HO-Isoxazole-(CH2)12-CO-gGlu-2xAdo HOS(O)2-(CH2)15-CO-gGlu-2xAdo-NH-CH2-(C6H4)-CH2 HOS(O)2-(CH2)13-CO-gGlu-2xAdo Tetrazolyl-(CH2)15-CO-NH-SO2-(CH2)3-CO-Ado-Ado-NH-CH2-(C6H4)-CH2 Tetrazolyl-(CH2)12-CO-gGlu-2xAdo Tetrazolyl-(CH2)15-CO-gGlu-2xAdo and MeS(O)2NH(CO)NH-(CH2)12-CO-gGlu-2xAdo.
HOOC-(CH2)14-CO-gGlu-2xεLys- HOOC-(CH2)16-CO-gGlu-2xεLys- HOOC-(CH2)18-CO-gGlu-2xεLys- As an example, 4-HOOC-(C6H4)-O-(CH2)10-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo is a substituent of Formular (I) consisting of: Albumin binder moiety - linker A - linker B(n=3)-* wherein the albumin binder moiety is Chem 3(n=10), linker A is Chem.8 and the linker B elements are gGlu, and 2xAdo. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-(17- carboxyheptadecanoylamino) butyrylamino]ethoxy}ethoxy)acetylamino] ethoxy}ethoxy)acetyl]. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-({trans-4-[(19- carboxynonadecanoylamino)methyl]cyclohexanecarbonyl} amino)butyrylamino]ethoxy}ethoxy)acetylamino]ethoxy}ethoxy)acetyl]. GLP-1 receptor agonists/GLP-1 agonist In one embodiment the peptide therapeutic is a GLP-1 analogue. In one embodiment the peptide therapeutic is a GLP-1 receptor agonist, also referred to as a GLP-1 agonist herein. The term “GLP-1 agonist” as used herein refers to a compound, which fully or partially activates the human GLP-1 receptor. The term is thus equal to the term “GLP-1 receptor agonist” used in other documents. The term GLP-1 agonist as well as the specific GLP-1 agonists described herein also encompass salt forms thereof. It follows that the GLP-1 agonist should display “GLP-1 activity” which refers to the ability of the compound, i.e. a GLP-1 analogue or a compound comprising a GLP-1 analogue, to bind to the GLP-1 receptor and initiate a signal transduction pathway resulting in insulinotropic action or other physiological effects as is known in the art. In some embodiments the “GLP-1 agonist” binds to a GLP-1 receptor, e.g., with an affinity constant (KD) or activate the receptor with a potency (EC50) of below 1 μM, e.g. below 100 nM as measured by methods known in the art (see e.g. WO 98/08871) and exhibits insulinotropic activity, where insulinotropic activity may be measured in vivo or in vitro assays known to those of ordinary skill in the art. For example, the GLP-1 agonist may be administered to an animal with increased blood glucose (e.g. obtained using an Intravenous Glucose Tolerance Test (IVGTT). A person skilled in the art will be able to determine a suitable glucose dosage and a suitable blood sampling regime, e.g. depending on the species of the animal, for the IVGTT) and measure the plasma insulin concentration over time. Suitable assays have been described in such as WO2015/155151.
The term half maximal effective concentration (EC50) generally refers to the concentration which induces a response halfway between the baseline and maximum, by reference to the dose response curve. EC50 is used as a measure of the potency of a compound and represents the concentration where 50% of its maximal effect is observed. Due to the albumin binding effects of GLP-1 agonists comprising a substituent as described herein, it is important to pay attention to if the assay includes human serum albumin or not. The in vitro potency of the GLP-1 agonist may be determined as described in WO2015/155151, example 29 without Human Serum Albumin (HSA), and the EC50 determined. The lower the EC50 value, the better the potency. In one embodiment the potency (EC50) as determined (without HSA) is 5-1000 pM, such as 10-750 pM, 10-500 pM or 10-200 pM. In one embodiment the EC50 (without HSA) is at most 500 pM, such as at most 300 pM, such as at most 200 pM. In one embodiment the EC50 (without HSA) is comparable to human GLP-1(7-37). In one embodiment the EC50 (without HSA) is at most 50 pM. In a further such embodiment the EC50 is at most 40 pM, such as at most 30 pM such as at most 20 pM, such as at most 10 pM. In one embodiment the EC50 is around 10 pM. Also, or alternatively, the binding of the GLP-1 agonist to albumin may be measured using the in vitro potency assay of Example 29 in WO2015/155151 including HSA. An increase of the in vitro potency, EC50 value, in the presence of serum albumin reflects the affinity to serum albumin. In one embodiment the potency (EC50) as determined (with 1 % HSA) is 5-1000 pM, such as 100-750 pM, 200-500 pM or 100-400 pM. In one embodiment the EC50 (with 1 % HSA) is at most 750 pM, such as at most 500 pM, such as at most 400 pM, such as at most 300 or such as at most 250 pM. If desired, the fold variation in relation to a known GLP-1 receptor agonist may be calculated as EC50(test analogue)/EC50(known analogue), and if this ratio is such as 0.5-1.5, or 0.8-1.2 the potencies are considered to be equivalent. In one embodiment the potency, EC50 (without HSA), is equivalent to the potency of liraglutide. In one embodiment the potency, EC50 (without HSA), is equivalent to the potency of semaglutide. In one embodiment the potency, EC50 (with 1 % HSA), is equivalent to the potency of liraglutide. In one embodiment the potency, EC50 (with 1 % HSA), is equivalent to the potency of semaglutide.
In some embodiments the GLP-1 agonist is a GLP-1 analogue, optionally comprising one substituent. The term "analogue" as used herein referring to a GLP-1 peptide (hereafter “peptide”) means a peptide wherein at least one amino acid residue of the peptide has been substituted with another amino acid residue and/or wherein at least one amino acid residue has been deleted from the peptide and/or wherein at least one amino acid residue has been added to the peptide and/or wherein at least one amino acid residue of the peptide has been modified. Such addition or deletion of amino acid residues may take place at the N-terminal of the peptide and/or at the C-terminal of the peptide. In some embodiments a simple nomenclature is used to describe the GLP-1 agonist, e.g., [Aib8] GLP-1(7-37) designates an analogue of GLP-1(7-37) wherein the naturally occurring Ala in position 8 has been substituted with Aib. In some embodiments the GLP-1 agonist comprises a maximum of twelve, such as a maximum of 10, 8 or 6, amino acids which have been altered, e.g., by substitution, deletion, insertion and/or modification, compared to e.g. GLP-1(7-37). In some embodiments the analogue comprises up to 10 substitutions, deletions, additions and/or insertions, such as up to 9 substitutions, deletions, additions and/or insertions, up to 8 substitutions, deletions, additions and/or insertions, up to 7 substitutions, deletions, additions and/or insertions, up to 6 substitutions, deletions, additions and/or insertions, up to 5 substitutions, deletions, additions and/or insertions, up to 4 substitutions, deletions, additions and/or insertions or up to 3 substitutions, deletions, additions and/or insertions, compared to e.g. GLP-1(7-37). Unless otherwise stated the GLP-1 agonist comprises only L-amino acids. In some embodiments the term “GLP-1 analogue” or “analogue of GLP-1” as used herein refers to a peptide, or a compound, which is a variant of the human Glucagon-Like Peptide-1 (GLP-1(7-37)). GLP-1(7-37) has the sequence HAEGTFTSDV SSYLEGQAAKEFIAWLVKGRG (SEQ ID No.: 1). In some embodiments the term “variant” refers to a compound which comprises one or more amino acid substitutions, deletions, additions and/or insertions. In one embodiment the GLP-1 agonist exhibits at least 60%, 65%, 70%, 80% or 90% sequence identity to GLP-1(7-37) over the entire length of GLP-1(7-37). As an example of a method for determination of sequence identity between two analogues the two peptides [Aib8]GLP-1(7-37) and GLP-1(7-37) are aligned. The sequence identity of [Aib8]GLP-1(7-37) relative to GLP-1(7-37) is given by the number of aligned identical residues minus the number of different residues divided by the total number of residues in GLP-1(7-37). Accordingly, in said example the sequence identity is (31-1)/31. In one embodiment the C-terminal of the GLP-1 agonist is an amide.
In some embodiments the GLP-1 agonist is GLP-1(7-37) or GLP-1(7-36)amide. In some embodiments the GLP-1 agonist is exendin-4, the sequence of which is HGEGTFITSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS (SEQ ID No.: 2). In one embodiment the GLP-1 agonist is an exendin-4 analogue or an engineered peptide thereof, as disclosed in WO2013/009545 and references therein. In order to prolong the effect of the GLP-1 agonist it is preferred that the GLP-1 agonist have an extended half-life. The half-life can be determined by method known in the art an in an appropriate model, such as in Male Sprague Dawley rats or minipigs as described in WO2012/140117. Half-life in rats may be determined as in Example 39 and the half-life in minipigs may be determined as in Example 37 therein. In one embodiment the GLP-1 agonist according to the invention has a half-life above 2 hours in rat. In one embodiment the GLP-1 agonist according to the invention has a half-life above 4 hours, such as above 6 hours, such as above 8 hours, such as above 10 hours, such as above 12 hours or such as above 15 hours in rat. In one embodiment the GLP-1 agonist according to the invention has a half-life above 24 hours in minipig. In one embodiment the GLP-1 agonist according to the invention has a half- life above 30 hours, such as above 36 hours, such as above 42 hours, such as above 48 hours, such as above 54 hours or such as above 60 hours in minipig. In one embodiment the GLP-1 agonist has a molecular weight of at most 50000 Da, such as at most 40000 Da, such as at most 30000 Da. In one embodiment the GLP-1 agonist has a molecular weight of at most 20000, such as at most 10000 Da, such as at most 7500 Da, such as at most 5000 Da. In one embodiment the GLP-1 agonist has a molar mass of at most 50000 g/mol, such as at most 40000 g/mol, such as at most 30000 g/mol. In one embodiment the GLP-1 agonist has a molar mass of at most 10000 g/mol, such as at most 8000 g/mol, such as at most 6000 g/mol. In some embodiments the GLP-1 agonist comprises one substituent which is covalently attached to the peptide as described herein above. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-(17- carboxyheptadecanoylamino) butyrylamino]ethoxy}ethoxy)acetylamino] ethoxy}ethoxy)acetyl]. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-({trans-4-[(19- carboxynonadecanoylamino)methyl]cyclohexanecarbonyl} amino)butyrylamino]ethoxy}ethoxy)acetylamino]ethoxy}ethoxy)acetyl]. In one embodiment the GLP-1 agonist is liraglutide.
In one embodiment the GLP-1 agonist is semaglutide, also known as N-epsilon26-[2-(2- {2-[2-(2-{2-[(S)-4-carboxy-4-(17-carboxyheptadecanoylamino) butyrylamino]ethoxy}ethoxy)acetylamino]ethoxy}ethoxy)acetyl][Aib8,Arg34]GLP-1(7-37), (SEQ ID No.: 3) which may be prepared as described in WO2006/097537, Example 4 with the following structure:
ne or two substituent(s) is/are selected from the group of substituents consisting of: HOOC-(CH2)18-CO-gGlu-2xAdo HOOC-(CH2)18-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo HOOC-(CH2)16-CO-gGlu-2xAdo HOOC-(CH2)16-CO-gGlu-2xAdo-NH-CH2-(C6H4)-CH2 HOOC-(CH2)16-CO-gGlu HOOC-(CH2)16-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo HOOC-(CH2)14-CO-gGlu-2xAdo HOOC-(CH2)14-CO-gGlu HOOC-(CH2)14-CO-gGlu-2xAdo- HOOC-(CH2)12-CO-gGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-3xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu 4-HOOC-(C6H4)-O-(CH2)10-CO-2xgGlu 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-3xGly 4-HOOC-(C6H4)-O-(CH2)10-CO-2xgGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-TtdSuc 4-HOOC-(C6H4)-O-(CH2)9-CO 4-HOOC-(C6H4)-O-(CH2)10-CO-gGlu-4xAdo 4-HOOC-(C6H4)-O-(CH2)10-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo 4-HOOC-(C6H4)-O-(CH2)9-CO-gGlu-2xAdo 3-HOOC-(C6H4)-O-(CH2)9-CO-gGlu-2xAdo 3-HO-Isoxazole-(CH2)12-CO-gGlu-2xAdo HOS(O)2-(CH2)15-CO-gGlu-2xAdo-NH-CH2-(C6H4)-CH2 HOS(O)2-(CH2)13-CO-gGlu-2xAdo Tetrazolyl-(CH2)15-CO-NH-SO2-(CH2)3-CO-Ado-Ado-NH-CH2-(C6H4)-CH2 Tetrazolyl-(CH2)12-CO-gGlu-2xAdo Tetrazolyl-(CH2)15-CO-gGlu-2xAdo and MeS(O)2NH(CO)NH-(CH2)12-CO-gGlu-2xAdo.
HOOC-(CH2)14-CO-gGlu-2xεLys- HOOC-(CH2)16-CO-gGlu-2xεLys- HOOC-(CH2)18-CO-gGlu-2xεLys- As an example, 4-HOOC-(C6H4)-O-(CH2)10-CO-NH-CH2-(C6H10)-CO-gGlu-2xAdo is a substituent of Formular (I) consisting of: Albumin binder moiety - linker A - linker B(n=3)-* wherein the albumin binder moiety is Chem 3(n=10), linker A is Chem.8 and the linker B elements are gGlu, and 2xAdo. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-(17- carboxyheptadecanoylamino) butyrylamino]ethoxy}ethoxy)acetylamino] ethoxy}ethoxy)acetyl]. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-({trans-4-[(19- carboxynonadecanoylamino)methyl]cyclohexanecarbonyl} amino)butyrylamino]ethoxy}ethoxy)acetylamino]ethoxy}ethoxy)acetyl]. GLP-1 receptor agonists/GLP-1 agonist In one embodiment the peptide therapeutic is a GLP-1 analogue. In one embodiment the peptide therapeutic is a GLP-1 receptor agonist, also referred to as a GLP-1 agonist herein. The term “GLP-1 agonist” as used herein refers to a compound, which fully or partially activates the human GLP-1 receptor. The term is thus equal to the term “GLP-1 receptor agonist” used in other documents. The term GLP-1 agonist as well as the specific GLP-1 agonists described herein also encompass salt forms thereof. It follows that the GLP-1 agonist should display “GLP-1 activity” which refers to the ability of the compound, i.e. a GLP-1 analogue or a compound comprising a GLP-1 analogue, to bind to the GLP-1 receptor and initiate a signal transduction pathway resulting in insulinotropic action or other physiological effects as is known in the art. In some embodiments the “GLP-1 agonist” binds to a GLP-1 receptor, e.g., with an affinity constant (KD) or activate the receptor with a potency (EC50) of below 1 μM, e.g. below 100 nM as measured by methods known in the art (see e.g. WO 98/08871) and exhibits insulinotropic activity, where insulinotropic activity may be measured in vivo or in vitro assays known to those of ordinary skill in the art. For example, the GLP-1 agonist may be administered to an animal with increased blood glucose (e.g. obtained using an Intravenous Glucose Tolerance Test (IVGTT). A person skilled in the art will be able to determine a suitable glucose dosage and a suitable blood sampling regime, e.g. depending on the species of the animal, for the IVGTT) and measure the plasma insulin concentration over time. Suitable assays have been described in such as WO2015/155151.
The term half maximal effective concentration (EC50) generally refers to the concentration which induces a response halfway between the baseline and maximum, by reference to the dose response curve. EC50 is used as a measure of the potency of a compound and represents the concentration where 50% of its maximal effect is observed. Due to the albumin binding effects of GLP-1 agonists comprising a substituent as described herein, it is important to pay attention to if the assay includes human serum albumin or not. The in vitro potency of the GLP-1 agonist may be determined as described in WO2015/155151, example 29 without Human Serum Albumin (HSA), and the EC50 determined. The lower the EC50 value, the better the potency. In one embodiment the potency (EC50) as determined (without HSA) is 5-1000 pM, such as 10-750 pM, 10-500 pM or 10-200 pM. In one embodiment the EC50 (without HSA) is at most 500 pM, such as at most 300 pM, such as at most 200 pM. In one embodiment the EC50 (without HSA) is comparable to human GLP-1(7-37). In one embodiment the EC50 (without HSA) is at most 50 pM. In a further such embodiment the EC50 is at most 40 pM, such as at most 30 pM such as at most 20 pM, such as at most 10 pM. In one embodiment the EC50 is around 10 pM. Also, or alternatively, the binding of the GLP-1 agonist to albumin may be measured using the in vitro potency assay of Example 29 in WO2015/155151 including HSA. An increase of the in vitro potency, EC50 value, in the presence of serum albumin reflects the affinity to serum albumin. In one embodiment the potency (EC50) as determined (with 1 % HSA) is 5-1000 pM, such as 100-750 pM, 200-500 pM or 100-400 pM. In one embodiment the EC50 (with 1 % HSA) is at most 750 pM, such as at most 500 pM, such as at most 400 pM, such as at most 300 or such as at most 250 pM. If desired, the fold variation in relation to a known GLP-1 receptor agonist may be calculated as EC50(test analogue)/EC50(known analogue), and if this ratio is such as 0.5-1.5, or 0.8-1.2 the potencies are considered to be equivalent. In one embodiment the potency, EC50 (without HSA), is equivalent to the potency of liraglutide. In one embodiment the potency, EC50 (without HSA), is equivalent to the potency of semaglutide. In one embodiment the potency, EC50 (with 1 % HSA), is equivalent to the potency of liraglutide. In one embodiment the potency, EC50 (with 1 % HSA), is equivalent to the potency of semaglutide.
In some embodiments the GLP-1 agonist is a GLP-1 analogue, optionally comprising one substituent. The term "analogue" as used herein referring to a GLP-1 peptide (hereafter “peptide”) means a peptide wherein at least one amino acid residue of the peptide has been substituted with another amino acid residue and/or wherein at least one amino acid residue has been deleted from the peptide and/or wherein at least one amino acid residue has been added to the peptide and/or wherein at least one amino acid residue of the peptide has been modified. Such addition or deletion of amino acid residues may take place at the N-terminal of the peptide and/or at the C-terminal of the peptide. In some embodiments a simple nomenclature is used to describe the GLP-1 agonist, e.g., [Aib8] GLP-1(7-37) designates an analogue of GLP-1(7-37) wherein the naturally occurring Ala in position 8 has been substituted with Aib. In some embodiments the GLP-1 agonist comprises a maximum of twelve, such as a maximum of 10, 8 or 6, amino acids which have been altered, e.g., by substitution, deletion, insertion and/or modification, compared to e.g. GLP-1(7-37). In some embodiments the analogue comprises up to 10 substitutions, deletions, additions and/or insertions, such as up to 9 substitutions, deletions, additions and/or insertions, up to 8 substitutions, deletions, additions and/or insertions, up to 7 substitutions, deletions, additions and/or insertions, up to 6 substitutions, deletions, additions and/or insertions, up to 5 substitutions, deletions, additions and/or insertions, up to 4 substitutions, deletions, additions and/or insertions or up to 3 substitutions, deletions, additions and/or insertions, compared to e.g. GLP-1(7-37). Unless otherwise stated the GLP-1 agonist comprises only L-amino acids. In some embodiments the term “GLP-1 analogue” or “analogue of GLP-1” as used herein refers to a peptide, or a compound, which is a variant of the human Glucagon-Like Peptide-1 (GLP-1(7-37)). GLP-1(7-37) has the sequence HAEGTFTSDV SSYLEGQAAKEFIAWLVKGRG (SEQ ID No.: 1). In some embodiments the term “variant” refers to a compound which comprises one or more amino acid substitutions, deletions, additions and/or insertions. In one embodiment the GLP-1 agonist exhibits at least 60%, 65%, 70%, 80% or 90% sequence identity to GLP-1(7-37) over the entire length of GLP-1(7-37). As an example of a method for determination of sequence identity between two analogues the two peptides [Aib8]GLP-1(7-37) and GLP-1(7-37) are aligned. The sequence identity of [Aib8]GLP-1(7-37) relative to GLP-1(7-37) is given by the number of aligned identical residues minus the number of different residues divided by the total number of residues in GLP-1(7-37). Accordingly, in said example the sequence identity is (31-1)/31. In one embodiment the C-terminal of the GLP-1 agonist is an amide.
In some embodiments the GLP-1 agonist is GLP-1(7-37) or GLP-1(7-36)amide. In some embodiments the GLP-1 agonist is exendin-4, the sequence of which is HGEGTFITSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS (SEQ ID No.: 2). In one embodiment the GLP-1 agonist is an exendin-4 analogue or an engineered peptide thereof, as disclosed in WO2013/009545 and references therein. In order to prolong the effect of the GLP-1 agonist it is preferred that the GLP-1 agonist have an extended half-life. The half-life can be determined by method known in the art an in an appropriate model, such as in Male Sprague Dawley rats or minipigs as described in WO2012/140117. Half-life in rats may be determined as in Example 39 and the half-life in minipigs may be determined as in Example 37 therein. In one embodiment the GLP-1 agonist according to the invention has a half-life above 2 hours in rat. In one embodiment the GLP-1 agonist according to the invention has a half-life above 4 hours, such as above 6 hours, such as above 8 hours, such as above 10 hours, such as above 12 hours or such as above 15 hours in rat. In one embodiment the GLP-1 agonist according to the invention has a half-life above 24 hours in minipig. In one embodiment the GLP-1 agonist according to the invention has a half- life above 30 hours, such as above 36 hours, such as above 42 hours, such as above 48 hours, such as above 54 hours or such as above 60 hours in minipig. In one embodiment the GLP-1 agonist has a molecular weight of at most 50000 Da, such as at most 40000 Da, such as at most 30000 Da. In one embodiment the GLP-1 agonist has a molecular weight of at most 20000, such as at most 10000 Da, such as at most 7500 Da, such as at most 5000 Da. In one embodiment the GLP-1 agonist has a molar mass of at most 50000 g/mol, such as at most 40000 g/mol, such as at most 30000 g/mol. In one embodiment the GLP-1 agonist has a molar mass of at most 10000 g/mol, such as at most 8000 g/mol, such as at most 6000 g/mol. In some embodiments the GLP-1 agonist comprises one substituent which is covalently attached to the peptide as described herein above. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-(17- carboxyheptadecanoylamino) butyrylamino]ethoxy}ethoxy)acetylamino] ethoxy}ethoxy)acetyl]. In some embodiments the substituent is [2-(2-{2-[2-(2-{2-[(S)-4-carboxy-4-({trans-4-[(19- carboxynonadecanoylamino)methyl]cyclohexanecarbonyl} amino)butyrylamino]ethoxy}ethoxy)acetylamino]ethoxy}ethoxy)acetyl]. In one embodiment the GLP-1 agonist is liraglutide.
In one embodiment the GLP-1 agonist is semaglutide, also known as N-epsilon26-[2-(2- {2-[2-(2-{2-[(S)-4-carboxy-4-(17-carboxyheptadecanoylamino) butyrylamino]ethoxy}ethoxy)acetylamino]ethoxy}ethoxy)acetyl][Aib8,Arg34]GLP-1(7-37), (SEQ ID No.: 3) which may be prepared as described in WO2006/097537, Example 4 with the following structure:
 In one embodiment the GLP-1 agonist is GLP-1 peptide 1 which is diacylated [Aib8,Arg34,Lys37]GLP-1(7-37) (SEQ ID No.: 4) as shown in Example 2 of WO2011/080103 and named N ^ ^ ^{2-[2-(2-{2-[2-(2-{(S)-4-Carboxy-4-[10-(4- carboxyphenoxy)decanoylamino]butyrylamino}-ethoxy)ethoxy]acetylamino}ethoxy)ethoxy]acetyl}, N ^ ^ ^-{2-[2-(2-{2-[2-(2-{(S)-4-carboxy-4-[10-(4- carboxyphenoxy)decanoylamino]butyrylamino}ethoxy)ethoxy]acetylamino}ethoxy)ethoxy]-acetyl}- [Aib8,Arg34,Lys37]GLP-1(7-37)–peptide with the following structure.
In one embodiment the GLP-1 agonist is GLP-1 peptide 1 which is diacylated [Aib8,Arg34,Lys37]GLP-1(7-37) (SEQ ID No.: 4) as shown in Example 2 of WO2011/080103 and named N ^ ^ ^{2-[2-(2-{2-[2-(2-{(S)-4-Carboxy-4-[10-(4- carboxyphenoxy)decanoylamino]butyrylamino}-ethoxy)ethoxy]acetylamino}ethoxy)ethoxy]acetyl}, N ^ ^ ^-{2-[2-(2-{2-[2-(2-{(S)-4-carboxy-4-[10-(4- carboxyphenoxy)decanoylamino]butyrylamino}ethoxy)ethoxy]acetylamino}ethoxy)ethoxy]-acetyl}- [Aib8,Arg34,Lys37]GLP-1(7-37)–peptide with the following structure.
 [Aib8,Glu22,Arg26,Lys27,Glu30,Arg34,Lys36]-GLP-1-(7-37)-peptidyl-Glu-Gly (SEQ ID No.: 5) as shown in Example 31 of WO2012/140117 and named Nε27-[2-[2-[2-[[2-[2-[2-[[(4S)-4-carboxy-4- [10-(4-carboxyphenoxy)decanoylamino]butanoyl]amino] ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy]-acetyl], Nε36-[2-[2-[2-[[2-[2-[2-[[(4S)-4-carboxy-4-[10- (4-carboxyphenoxy)decanoylamino]- butanoyl]amino]ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy]acetyl]-[Aib8,Glu22,Arg26,Lys27, Glu30,Arg34,Lys36]-GLP-1-(7-37)-peptidyl-Glu-Gly with the following structure:
[Aib8,Glu22,Arg26,Lys27,Glu30,Arg34,Lys36]-GLP-1-(7-37)-peptidyl-Glu-Gly (SEQ ID No.: 5) as shown in Example 31 of WO2012/140117 and named Nε27-[2-[2-[2-[[2-[2-[2-[[(4S)-4-carboxy-4- [10-(4-carboxyphenoxy)decanoylamino]butanoyl]amino] ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy]-acetyl], Nε36-[2-[2-[2-[[2-[2-[2-[[(4S)-4-carboxy-4-[10- (4-carboxyphenoxy)decanoylamino]- butanoyl]amino]ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy]acetyl]-[Aib8,Glu22,Arg26,Lys27, Glu30,Arg34,Lys36]-GLP-1-(7-37)-peptidyl-Glu-Gly with the following structure:










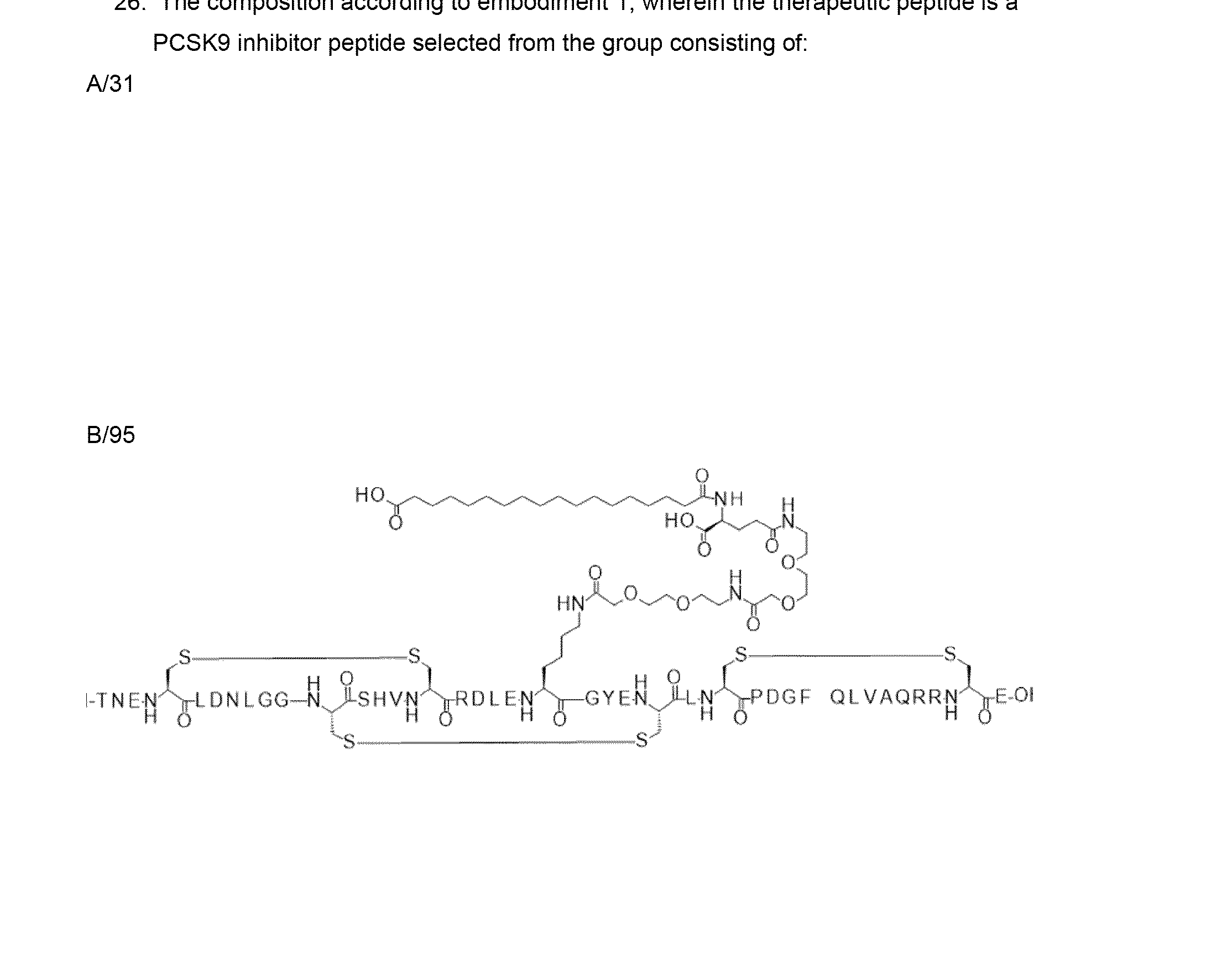

































Claims
CLAIMS 1. A solid pharmaceutical composition comprising i) a peptide therapeutic ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), iii) a fatty acid consisting of 6-14 carbon atoms or a salt hereof and iv) nicotinamide.
2. The composition according to claim 1, wherein the composition further comprises a lubricant, such as magnesium stearate.
3. The composition according to any of the previous claims, wherein the fatty acid is capric acid or a salt hereof, such as sodium caprate.
4. The composition according to any of the previous claims, wherein the salt of NAC is selected from the group of the sodium, the potassium and the ammonium salt of NAC.
5. The composition according to any of the previous claims, wherein the salt of NAC is the sodium salt, sodium N-(8-(2-hydroxybenzoyl)amino)caprylate (SNAC).
6. The composition according to any of the previous claims, wherein the weight ratio (w/w) of the amount (w) of the salt of NAC to the amount (w) of nicotinamide is 0.5-10, such as 0.5-8 or such as 0.5-5.
7. The composition according to any of the previous claims 3-7, wherein the weight ratio (w/w) of the amount (w) of the salt of NAC to the of the amount (w) of capric acid or a salt hereof is 0.5-8, such as 0.5-5 or such as 0.5-2.
8. The composition according to any of the previous claims consisting of: i) a peptide therapeutic, ii) a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) a fatty acid, such as a saturated fatty acid consisting of 6-14 carbon atoms, or a salt hereof, such as capric acid or a salt hereof such as sodium caprate, iv) nicotinamide and
v) a lubricant, such as magnesium stearate.
9. The composition according to any of the previous claims, wherein a unit dosage comprises i) 0.1-100 mg peptide therapeutic, ii) 20-1000 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC) iii) 20-1000 mg fatty acid, such as a saturated fatty acid consisting of 6-14 carbon atoms or a salt hereof, such capric acid, or a salt hereof such as sodium caprate, iv) 10-750 mg nicotinamide and v) 0-25 mg lubricant, such as magnesium stearate.
10. The composition according to any of the previous claims, wherein a unit dosage comprises i) 1-50 mg peptide therapeutic, ii) 20-400 mg salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid (NAC), such as the sodium salt of NAC (SNAC), iii) 20-400 mg capric acid or a salt hereof, such as sodium caprate, iv) 10-250 mg nicotinamide and v) 0-15 mg magnesium stearate.
11. The composition according to any of the previous claims, wherein the peptide therapeutic is a GLP-1 agonist.
12. The composition according to claim 11, wherein the GLP-1 agonist is semaglutide or a pro- drug of semaglutide.
13. The composition according to claim 11, wherein the GLP-1 agonist is a GLP-1-GIP co- agonist.
14. The composition according to any of the claims 1-10, wherein the peptide therapeutic is a PCSK9 inhibitor peptide.
15. The composition according to any of the claims 1-10, wherein the peptide therapeutic is an insulin analogue.
16. The composition according to any of the previous claims wherein the composition is for oral administration.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21189585.9 | 2021-08-04 | ||
| EP21189585 | 2021-08-04 | ||
| EP21189640 | 2021-08-04 | ||
| EP21189640.2 | 2021-08-04 | ||
| EP22161506 | 2022-03-11 | ||
| EP22161506.5 | 2022-03-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023012263A1 true WO2023012263A1 (en) | 2023-02-09 |
Family
ID=83149507
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2022/071913 WO2023012263A1 (en) | 2021-08-04 | 2022-08-04 | Solid oral peptide formulations |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023012263A1 (en) |
Citations (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993019175A1 (en) | 1992-03-25 | 1993-09-30 | Bernard Thorens | Receptor for the glucagon-like-peptide-1 (glp-1) |
| WO1996029342A1 (en) | 1995-03-17 | 1996-09-26 | Novo Nordisk A/S | Lipophilic peptide hormone derivatives |
| WO1996030036A1 (en) | 1995-03-31 | 1996-10-03 | Emisphere Technologies, Inc. | Compounds and compositions for delivering active agents |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1999043708A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | Glp-1 derivatives of glp-1 and exendin with protracted profile of action |
| WO1999043341A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | Glp-1 derivatives with helix-content exceeding 25 %, forming partially structured micellar-like aggregates |
| WO1999043707A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | N-terminally modified glp-1 derivatives |
| WO1999043706A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | Derivatives of glp-1 analogs |
| WO2000046182A1 (en) | 1999-02-05 | 2000-08-10 | Emisphere Technologies, Inc. | Method of preparing alkylated salicylamides |
| WO2001092206A1 (en) | 2000-06-02 | 2001-12-06 | Emisphere Technologies, Inc. | Method of preparing salicylamides |
| WO2005027978A2 (en) | 2003-09-19 | 2005-03-31 | Novo Nordisk A/S | Albumin-binding derivatives of therapeutic peptides |
| WO2005058958A2 (en) | 2003-12-18 | 2005-06-30 | Novo Nordisk A/S | Novel glp-1 analogues linked to albumin-like agents |
| WO2005058954A1 (en) | 2003-12-18 | 2005-06-30 | Novo Nordisk A/S | Novel glp-1 compounds |
| WO2006005667A2 (en) | 2004-07-08 | 2006-01-19 | Novo Nordisk A/S | Polypeptide protracting tags comprising a tetrazole moiety |
| WO2006037811A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted exendin-4 compounds |
| WO2006037810A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted glp-1 compounds |
| WO2006097537A2 (en) | 2005-03-18 | 2006-09-21 | Novo Nordisk A/S | Acylated glp-1 compounds |
| WO2006097538A1 (en) | 2005-03-18 | 2006-09-21 | Novo Nordisk A/S | Extended glp-1 compounds |
| WO2008023050A1 (en) | 2006-08-25 | 2008-02-28 | Novo Nordisk A/S | Acylated exendin-4 compounds |
| WO2008028859A1 (en) | 2006-09-07 | 2008-03-13 | F. Hoffmann-La Roche Ag | A process for the manufacture of snac (salcaprozate sodium) |
| WO2009030774A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Truncated glp-1 derivatives and their therapeutical use |
| WO2009030771A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Peptides derivatized with a-b-c-d- and their therapeutical use |
| WO2009030738A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Glucagon-like peptide-1 derivatives and their pharmaceutical use |
| WO2009115469A1 (en) | 2008-03-18 | 2009-09-24 | Novo Nordisk A/S | Protease stabilized, acylated insulin analogues |
| WO2010071807A1 (en) | 2008-12-19 | 2010-06-24 | Indiana University Research And Technology Corporation | Amide based glucagon superfamily peptide prodrugs |
| WO2010080605A1 (en) | 2008-12-19 | 2010-07-15 | Indiana University Research And Technology Corporation | Dipeptide linked medicinal agents |
| WO2011080103A1 (en) | 2009-12-16 | 2011-07-07 | Novo Nordisk A/S | Double-acylated glp-1 derivatives |
| WO2011094531A1 (en) | 2010-01-28 | 2011-08-04 | Merrion Research Iii Limited | Solid pharmaceutical composition with enhancers and methods of preparing thereof |
| WO2011163012A2 (en) | 2010-06-24 | 2011-12-29 | Indiana University Research And Technology Corporation | Amide based glucagon superfamily peptide prodrugs |
| WO2012080471A1 (en) | 2010-12-16 | 2012-06-21 | Novo Nordisk A/S | Solid compositions comprising a glp-1 agonist and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2012140117A1 (en) | 2011-04-12 | 2012-10-18 | Novo Nordisk A/S | Double-acylated glp-1 derivatives |
| WO2012177741A1 (en) | 2011-06-20 | 2012-12-27 | Genentech, Inc. | Pcsk9-binding polypeptides and methods of use |
| WO2013009545A1 (en) | 2011-07-08 | 2013-01-17 | Amylin Pharmaceuticals, Inc. | Engineered polypeptides having enhanced duration of action with reduced immunogenicity |
| WO2013139694A1 (en) | 2012-03-22 | 2013-09-26 | Novo Nordisk A/S | Compositions of glp-1 peptides and preparation thereof |
| WO2014152460A2 (en) | 2013-03-15 | 2014-09-25 | Indiana University Research And Technology Corporation | Prodrugs with prolonged action |
| WO2015155151A1 (en) | 2014-04-07 | 2015-10-15 | Novo Nordisk A/S | Double-acylated glp-1 compounds |
| WO2016049174A1 (en) | 2014-09-24 | 2016-03-31 | Indiana University Research And Technology Corporation | Lipidated amide-based insulin prodrugs |
| WO2016120378A1 (en) | 2015-01-29 | 2016-08-04 | Novo Nordisk A/S | Tablets comprising glp-1 agonist and enteric coating |
| WO2016120380A1 (en) | 2015-01-29 | 2016-08-04 | Novo Nordisk A/S | Pharmaceutical composition for oral glp-1 administration comprising a tablet core and immediate release coating |
| WO2016119854A1 (en) | 2015-01-29 | 2016-08-04 | Novo Nordisk A/S | Pharmaceutical composition for oral insulin administration comprising a tablet core and a polyvinyl alcohol coating |
| WO2017121850A1 (en) | 2016-01-13 | 2017-07-20 | Novo Nordisk A/S | Egf(a) analogues with fatty acid substituents |
| WO2019021506A1 (en) | 2017-07-26 | 2019-01-31 | シャープ株式会社 | Door opening and closing mechanism |
| WO2019149880A1 (en) | 2018-02-02 | 2019-08-08 | Novo Nordisk A/S | Solid compositions comprising a glp-1 agonist, a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid and a lubricant |
| WO2021023855A1 (en) | 2019-08-07 | 2021-02-11 | Novo Nordisk A/S | Solid compositions comprising an egf(a) derivative and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2021089752A1 (en) * | 2019-11-07 | 2021-05-14 | Novo Nordisk A/S | Solid compositions comprising a glp-1 agonist, an sglt2 inhibitor and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2021089761A1 (en) | 2019-11-07 | 2021-05-14 | Novo Nordisk A/S | Solid compositions comprising a pcsk9 inhibitor and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2022018186A1 (en) | 2020-07-22 | 2022-01-27 | Novo Nordisk A/S | Co-agonists at glp-1 and gip receptors suitable for oral delivery |
| WO2022096636A1 (en) | 2020-11-06 | 2022-05-12 | Novo Nordisk A/S | Glp-1 prodrugs and uses hereof |
-
2022
- 2022-08-04 WO PCT/EP2022/071913 patent/WO2023012263A1/en unknown
Patent Citations (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993019175A1 (en) | 1992-03-25 | 1993-09-30 | Bernard Thorens | Receptor for the glucagon-like-peptide-1 (glp-1) |
| WO1996029342A1 (en) | 1995-03-17 | 1996-09-26 | Novo Nordisk A/S | Lipophilic peptide hormone derivatives |
| WO1996030036A1 (en) | 1995-03-31 | 1996-10-03 | Emisphere Technologies, Inc. | Compounds and compositions for delivering active agents |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1999043708A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | Glp-1 derivatives of glp-1 and exendin with protracted profile of action |
| WO1999043341A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | Glp-1 derivatives with helix-content exceeding 25 %, forming partially structured micellar-like aggregates |
| WO1999043707A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | N-terminally modified glp-1 derivatives |
| WO1999043706A1 (en) | 1998-02-27 | 1999-09-02 | Novo Nordisk A/S | Derivatives of glp-1 analogs |
| WO2000046182A1 (en) | 1999-02-05 | 2000-08-10 | Emisphere Technologies, Inc. | Method of preparing alkylated salicylamides |
| WO2001092206A1 (en) | 2000-06-02 | 2001-12-06 | Emisphere Technologies, Inc. | Method of preparing salicylamides |
| WO2005027978A2 (en) | 2003-09-19 | 2005-03-31 | Novo Nordisk A/S | Albumin-binding derivatives of therapeutic peptides |
| WO2005058958A2 (en) | 2003-12-18 | 2005-06-30 | Novo Nordisk A/S | Novel glp-1 analogues linked to albumin-like agents |
| WO2005058954A1 (en) | 2003-12-18 | 2005-06-30 | Novo Nordisk A/S | Novel glp-1 compounds |
| WO2006005667A2 (en) | 2004-07-08 | 2006-01-19 | Novo Nordisk A/S | Polypeptide protracting tags comprising a tetrazole moiety |
| WO2006037811A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted exendin-4 compounds |
| WO2006037810A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted glp-1 compounds |
| WO2006097537A2 (en) | 2005-03-18 | 2006-09-21 | Novo Nordisk A/S | Acylated glp-1 compounds |
| WO2006097538A1 (en) | 2005-03-18 | 2006-09-21 | Novo Nordisk A/S | Extended glp-1 compounds |
| WO2008023050A1 (en) | 2006-08-25 | 2008-02-28 | Novo Nordisk A/S | Acylated exendin-4 compounds |
| WO2008028859A1 (en) | 2006-09-07 | 2008-03-13 | F. Hoffmann-La Roche Ag | A process for the manufacture of snac (salcaprozate sodium) |
| WO2009030774A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Truncated glp-1 derivatives and their therapeutical use |
| WO2009030771A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Peptides derivatized with a-b-c-d- and their therapeutical use |
| WO2009030738A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Glucagon-like peptide-1 derivatives and their pharmaceutical use |
| WO2009115469A1 (en) | 2008-03-18 | 2009-09-24 | Novo Nordisk A/S | Protease stabilized, acylated insulin analogues |
| WO2010071807A1 (en) | 2008-12-19 | 2010-06-24 | Indiana University Research And Technology Corporation | Amide based glucagon superfamily peptide prodrugs |
| WO2010080605A1 (en) | 2008-12-19 | 2010-07-15 | Indiana University Research And Technology Corporation | Dipeptide linked medicinal agents |
| WO2011080103A1 (en) | 2009-12-16 | 2011-07-07 | Novo Nordisk A/S | Double-acylated glp-1 derivatives |
| WO2011094531A1 (en) | 2010-01-28 | 2011-08-04 | Merrion Research Iii Limited | Solid pharmaceutical composition with enhancers and methods of preparing thereof |
| WO2011163012A2 (en) | 2010-06-24 | 2011-12-29 | Indiana University Research And Technology Corporation | Amide based glucagon superfamily peptide prodrugs |
| WO2012080471A1 (en) | 2010-12-16 | 2012-06-21 | Novo Nordisk A/S | Solid compositions comprising a glp-1 agonist and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2012140117A1 (en) | 2011-04-12 | 2012-10-18 | Novo Nordisk A/S | Double-acylated glp-1 derivatives |
| WO2012177741A1 (en) | 2011-06-20 | 2012-12-27 | Genentech, Inc. | Pcsk9-binding polypeptides and methods of use |
| WO2013009545A1 (en) | 2011-07-08 | 2013-01-17 | Amylin Pharmaceuticals, Inc. | Engineered polypeptides having enhanced duration of action with reduced immunogenicity |
| WO2013139694A1 (en) | 2012-03-22 | 2013-09-26 | Novo Nordisk A/S | Compositions of glp-1 peptides and preparation thereof |
| WO2014152460A2 (en) | 2013-03-15 | 2014-09-25 | Indiana University Research And Technology Corporation | Prodrugs with prolonged action |
| WO2015155151A1 (en) | 2014-04-07 | 2015-10-15 | Novo Nordisk A/S | Double-acylated glp-1 compounds |
| WO2016049174A1 (en) | 2014-09-24 | 2016-03-31 | Indiana University Research And Technology Corporation | Lipidated amide-based insulin prodrugs |
| WO2016120378A1 (en) | 2015-01-29 | 2016-08-04 | Novo Nordisk A/S | Tablets comprising glp-1 agonist and enteric coating |
| WO2016120380A1 (en) | 2015-01-29 | 2016-08-04 | Novo Nordisk A/S | Pharmaceutical composition for oral glp-1 administration comprising a tablet core and immediate release coating |
| WO2016119854A1 (en) | 2015-01-29 | 2016-08-04 | Novo Nordisk A/S | Pharmaceutical composition for oral insulin administration comprising a tablet core and a polyvinyl alcohol coating |
| WO2017121850A1 (en) | 2016-01-13 | 2017-07-20 | Novo Nordisk A/S | Egf(a) analogues with fatty acid substituents |
| WO2019021506A1 (en) | 2017-07-26 | 2019-01-31 | シャープ株式会社 | Door opening and closing mechanism |
| WO2019149880A1 (en) | 2018-02-02 | 2019-08-08 | Novo Nordisk A/S | Solid compositions comprising a glp-1 agonist, a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid and a lubricant |
| WO2021023855A1 (en) | 2019-08-07 | 2021-02-11 | Novo Nordisk A/S | Solid compositions comprising an egf(a) derivative and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2021089752A1 (en) * | 2019-11-07 | 2021-05-14 | Novo Nordisk A/S | Solid compositions comprising a glp-1 agonist, an sglt2 inhibitor and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2021089761A1 (en) | 2019-11-07 | 2021-05-14 | Novo Nordisk A/S | Solid compositions comprising a pcsk9 inhibitor and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| WO2022018186A1 (en) | 2020-07-22 | 2022-01-27 | Novo Nordisk A/S | Co-agonists at glp-1 and gip receptors suitable for oral delivery |
| WO2022096636A1 (en) | 2020-11-06 | 2022-05-12 | Novo Nordisk A/S | Glp-1 prodrugs and uses hereof |
Non-Patent Citations (1)
| Title |
|---|
| J. MOL. BIOL., vol. 422, 2012, pages 685 - 696 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11382957B2 (en) | Solid compositions comprising a GLP-1 agonist and a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid | |
| US11833248B2 (en) | Solid compositions comprising a GLP-1 agonist and a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid | |
| US11622996B2 (en) | Solid compositions comprising a GLP-1 agonist and a salt of N-(8-(2-hydroxybenzoyl)amino)caprylic acid | |
| US20220265777A1 (en) | Solid compositions comprising a glp-1 agonist, an sglt2 inhibitor and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid | |
| US20230165939A1 (en) | Solid compositions comprising a glp-1 agonist and histidine | |
| WO2023012263A1 (en) | Solid oral peptide formulations | |
| RU2807183C2 (en) | Solid compositions containing glp-1 agonist and n-(8-(2-hydroxybenzoyl)amino)caprylic acid salt and lubricant |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22761497 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |