WO2022008025A1 - 2-hydroxyiminopyrimidine nucleosides and derivitives and antiviral uses thereto - Google Patents
2-hydroxyiminopyrimidine nucleosides and derivitives and antiviral uses thereto Download PDFInfo
- Publication number
- WO2022008025A1 WO2022008025A1 PCT/EG2021/000021 EG2021000021W WO2022008025A1 WO 2022008025 A1 WO2022008025 A1 WO 2022008025A1 EG 2021000021 W EG2021000021 W EG 2021000021W WO 2022008025 A1 WO2022008025 A1 WO 2022008025A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- formula
- optionally substituted
- halogen
- Prior art date
Links
- 0 C*(C)C(*1)*O[C@@]1(*)N** Chemical compound C*(C)C(*1)*O[C@@]1(*)N** 0.000 description 13
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
- C07H19/11—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids containing cyclic phosphate
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/12—Triazine radicals
Definitions
- This disclosure relates to 2-hydroxyiminopyrimidine nucleoside derivatives, as well as compositions and methods related thereto.
- the disclosure relates to the treatment or prophylaxis of viral infections, in particular respiratory viral infections including coronaviruses, influenza viruses, and respiratory syncytial virus.
- MERS-CoV Middle East respiratory syndrome coronavirus
- MERS- CoV quickly spread around the globe with a case fatality rate of 38% according to the World Health Organization (WHO) (http://www. who.int/mediacentre/factsheets/mers-cov/en/).
- WHO World Health Organization
- HRSV human respiratory virus
- the present invention provides agents, compositions, and methods for treating or preventing respiratory viral infections.
- Nucleoside analogs as a class of small molecules have a well-established regulatory history, with several of them currently approved by the US Food and Drug Administration (US FDA) for treating hepatitis C (HCV), hepatitis B virus (HBV), human immunodeficiency virus (HIV), and herpes simplex C virus (HSV).
- HCV hepatitis C
- HBV hepatitis B virus
- HSV human immunodeficiency virus
- HSV herpes simplex C virus
- Examples of compounds useful for treating viral infections are described in AU 2020/202600 A1 (Cho et al), AU 2020/200499 A1 (Beigelman et al.), US 2018/009861652 B2 (Schinazi et al.), US 2017/009809616 B2 (Amblard et al.), WO 2010/091386 A3 (Cho et.
- the present invention provides compounds, methods, and compositions for treating or preventing respiratory viral infections in a host.
- a compound of Formula I or a pharmaceutically acceptable salt thereof: Wherein:
- R 1 is selected from hydrogen, COR 1* , COOR 1* , CONHR 1* .
- R 2 is, independently, selected from NH, CH, C-halogen, N(CO)NH 2 , the dashed and the solid lines combined represent an optional double bond.
- R 4 is independently, selected from H, F, Cl, Br, I, CH 3 , CF 3 , CH 2 F, CHF 2 , CH 2 OH, CH 2 Cl, CH 2 Br, CH 2 I, CN, an optionally substituted alkyl C1-C 10 , an optionally substituted C 1 - C 10 cycloalkyl, an optionally substituted C 2 -C 10 alkenyl, an optionally substituted C 2 -C 10 alkynyl.
- X is independently, selected from CH, C-(halogen; F; Cl, Br, I), C-CN, C-CH 3 , C-CF 3 , C-
- Sugar for formula I is ribose or a modified ribose of the general formula (I-
- R 6 is independently selected from, H, OH, halogen (F, Cl, Br, I), Nitrile (CN), Nitro (NO 2 ), methyl (CH 3 ), C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is independently selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 8 is selected from absent, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester, Wherein:
- Z is independently selected from O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’, CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid
- Sugar for formula I is ribose or a modified ribose of the general Formula
- R 6 is independently selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is independently selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 8 is independently selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- Z is independently selected from O, S, NOH, NOR 9 ;
- R 9 is independently selected from C(O)alkyl (C1-C17), C(O)cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), C(O)aryl, C(O)heteroaryl;
- R 15 is independently selected from O-aryl, O-heteroaryl,
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, pyridinyl CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- R 6 is independently selected from, H, halogen (F, Cl, Br, I), CH 3 , NO 2 , CN, C 1 -C 22 an optionally substituted methyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is independently selected from, H, halogen (F, Cl, Br, I), N 3 , CN; NO 2 , C 1 -C 22 an optionally substituted methyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 8 is independently selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,
- Z is independently selected from O, S, NOH, NOR 9 ;
- n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid
- R 6 is independently selected from, H, halogen (F, Cl, Br, I), CH 3 , NO 2 , CN, C 1 -C 22 an optionally substituted methyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is independently selected from, H, halogen (F, Cl, Br, I), N 3 , CN; NO 2 , C 1 -C 22 an optionally substituted methyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- Z is independently selected from O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid
- R 6 is independently selected from, H, halogen (F, Cl, Br, I), CH 3 , NO 2 , CN, C 1 -C 22 an optionally substituted methyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is independently selected from, H, halogen (F, Cl, Br, I), N 3 , CN; NO 2 , C 1 -C 22 an optionally substituted methyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 8 is independently selected from, H, halogen (F, Cl, Br, I), N 3 , NO 2 , methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- Z is independently selected from O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
- sugar for formula I is formula I-F:
- R 8 is independently selected from, H, methyl, an optionally substituted methyl, CN; alkyl (C1- C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester, Wherein:
- Z is independently selected from O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid
- R 8 is independently selected from, H, methyl, an optionally substituted methyl, CN; alkyl (C1- C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- Z is independently selected from O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
- sugar for formula I is the formula I-H
- X is independently selected from absent, CH 2 , CHR 16 ;
- Z is independently selected from O, S, NOH, NOR 9 .
- X is independently selected from absent, CH 2 , CHR 16 ;
- Z is independently selected from O, S, NOH, NOR 9 .
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,
- Z is independently selected from O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14 independently is H, CH 3 , CH 2 SR 9 , CH 2 SC(O)R 9 , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR’ , CH 2 OC(O)R 9 , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CH 2 CO 2 R’ , CH 2 -imidazolyl;
- Lipid is a C 6 -C 22 alkyl, alkoxy, polyethylene glycol, or sphingolipid
- the disclosure relates to a compound of formula I having formula I-L,
- R 1 is as defined in Formula I
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate,
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to a compound of formula I having formula I-M,
- E is selected from absent, O, CH 2 , NH;
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate,
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to a compound of formula I having formula I-N,
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I; monophosphate, diphosphate, triphosphate,
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to a compound of formula I having formula I-O,
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 9 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I;
- the disclosure relates to a compound of formula I having formula I-P,
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 6 and R 7 all together, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl;
- R 8 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN;
- R 9 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate, Wherein: X is O, S, NOH, NOR 9 ; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to a compound of formula I having formula I-Q,
- the dashed and the solid lines combined represent an optional double bond;
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C 3 -C 6 ), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 9 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate,
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to a compound of formula I having formula I-R,
- R 1 isselected as described in Formula I.
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 8 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN;
- R 9 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate,
- X is O, S, NOH, NOR 9 ;
- n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to a compound of formula I having formula I-S,
- E is selected from absent, O, CH 2 , NH;
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate, wherein:
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- V is independently N, CH, C(F, Cl, Br, I), CCN,CCO 2 H, CO 2 R 9 , CCH 3 , CCF 3
- R 3 is selected from H, OH, NH 2 , NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
- R 4 is selected from H, F, Cl, I, CF 3 , an optionally substituted alkyl C1-C3, cyclopropyl;
- R 5 is selected from, H, halogen (F, Cl, Br, I), N 3 , methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
- R 6 is selected from, H, OH, halogen (F, Cl, Br, I), CH 3 , C 1 -C 22 an optionally substituted methyl; cyclopropyl, C 2 -C 22 alkenyl, C 2 -C 22 an optionally substituted alkenyl, C 2 -C 22 an optionally substituted alkynyl;
- R 7 is selected from, H, OH, halogen (F, Cl, Br, I), N 3 , CN;
- R 9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- W 2 is H, COR 1* , COOR 1* , CONHR 1* , where R 1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate,
- X is O, S, NOH, NOR 9 ;
- n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
- R 9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
- R 10 is H, alkyl (C1-C6) and substituted alkyl
- R 11 is H, OCH 3 , halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
- R 12 is alkyl, aryl, heteroaryl;
- R 13 and R 14’ independently is H, CH 3 , CH 2 SR 9’ , CH 2 SC(O)R 9’ , CH 2 CH 3 , isobutyl, CH 2 CH 2 SMe, benzyl, pyrolinyl, CH 2 OR 9’ , CH 2 OC(O)R 9’ , CH 2 -indolyl, 4-hydroxylbenzyl, isopropyl, CH 2 CO 2 R 9’ , CH 2 CH 2 CO 2 R 9’ , CH 2 -imidazolyl;
- Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
- the disclosure relates to methods of treating or preventing a viral infection by administering in effective amount of a compound disclosed herein to a subject in need thereof.
- the disclosure relates to methods to a method of treating and/or preventing an infection caused by a Coronaviridae virus that can include administering to a subject an effective amount of one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), or a pharmaceutical composition that includes a compound described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof).
- a method of treating and/or preventing an infection caused by a Coronaviridae virus that can include administering to a subject an effective amount of one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), or a pharmaceutical composition that includes a compound described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof).
- the disclosure relates to using one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), in the manufacture of a medicine for preventing and/or treating an infection caused by a Coronaviridae virus that can include administering to a subject an effective amount of one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof).
- Still other embodiments described herein relate to one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), that can be used for ameliorating and/or treating an infection caused by a Coronaviridae virus by contacting a cell infected with the virus with an effective amount of said compound(s).
- a Coronaviridae virus include, but not limited to, 2019-novel coronavirus (2019 nCoV), Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS coronavirus (SARS-CoV), and human coronavirus.
- 2019-novel coronavirus 2019 nCoV
- MERS-CoV Middle East respiratory syndrome coronavirus
- SARS-CoV SARS coronavirus
- a compound described herein can prevent and/or treat a 2019-nCoV infection.
- a compound described herein can prevent and/or treat a 2019-nCoV infection.
- a compound described herein can inhibit replication of 2019-nCo V.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against 2019-nCoV.
- a compound described herein can prevent and/or treat a MERS-CoV infection.
- a compound described herein can prevent and/or treat a MERS-CoV infection.
- a compound described herein can inhibit replication of MERS-CoV.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against MERS-CoV.
- a compound described herein can prevent and/or treat a SARS-CoV (SARS-CoV- 1, SARSCoV-2) infection.
- SARS-CoV- 1, SARSCoV-2 SARS-CoV- 1
- a compound described herein can inhibit replication of MERS-Co V.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against SARS-CoV.
- a compound described herein can prevent and/or treat a human coronavirus infection.
- a compound described herein can prevent and/or treat a human coronavirus infection.
- a compound described herein can inhibit replication of a human coronavirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against a human coronavirus.
- the viral infection is, or is caused by, Respiratory syncytial virus (RSV).
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- RSV Respiratory Syncytial Virus
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein can inhibit replication of RSV.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against RSV.
- the viral infection is, or is caused by, Hepatitis B virus (HBV).
- HBV Hepatitis B virus
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against HBV.
- the viral infection is, or is caused by, influenza A virus including subtype H1N1, H1N2, H2N2, H2N3, H3N1, H3N2,H3N8, H5N1 (low bath), H5N1 (high bath), H5N2, H5N3, H5N6, H5N6, H5N8, H5N9, H6N1, H6N2, H7N1, H7N2, H7N3, H7N4, H7N7, H7N9, H9N2, H10N7, influenza B virus, and influenza C virus.
- influenza A virus including subtype H1N1, H1N2, H2N2, H2N3, H3N1, H3N2,H3N8, H5N1 (low bath), H5N1 (high bath), H5N2, H5N3, H5N6, H5N6, H5N8, H5N9, H6N1, H6N2, H7N1, H7N2, H7N3, H7N4, H7N7, H7N9, H9N2, H10
- a compound described herein can prevent and/or treat an influenza A virus , influenza B virus, and influenza C virus infection.
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against an influenza A virus , influenza B virus, and influenza C virus.
- the viral infection is, or is caused by, a flavivirus such as a Dengue virus, West Nile virus, Yellow fever virus, Japanese encephalitis virus, Powassen virus, Zika (ZIKA) virus, Usutu virus, hepatitis C virus (HCV).
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein can inhibit replication of a flavivirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against a flavivirus.
- the viral infection is, or is caused by, an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus.
- a compound described herein can prevent and/or treat an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus infection.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can prevent and/or treat an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus infection.
- administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof to a subject infected with an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus and/or by contacting a cell infected with an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus.
- a compound described herein can inhibit replication of an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus.
- the viral infection is, or is caused by, a Picornaviridae viruses such as polio virus, Enterovirus-71, Enterovius-68, Coxsackie virus B3.
- a compound described herein can prevent and/or treat a Picornaviruse infection.
- a compound described herein can prevent and/or treat a Picornaviruse infection.
- a compound described herein can inhibit replication of a Picornaviruse.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against Picornaviruses.
- the viral infection is, or is caused by, a Papovaviridae viruses such as BK virus, JC virus, and human Papillomavirus.
- a compound described herein can prevent and/or treat a Papovavirus infection.
- a compound described herein can inhibit replication of a Papovavirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against Papovaviruses.
- the viral infection is, or is caused by, a Togaviridae viruses such as Venezuelan equine encephalitis virus, Eastern equine encephalitis virus, and Chikungyunya virus.
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein can inhibit replication of a Togavivirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against Togaviviruses.
- the viral infection is, or is caused by, an Arenaviridae viruses such as Tacaribe virus, Pichinde virus, Junin virus, Lassa fever virus, and Lymphocytic choriomeningtis virus.
- an Arenaviridae viruses such as Tacaribe virus, Pichinde virus, Junin virus, Lassa fever virus, and Lymphocytic choriomeningtis virus.
- a compound described herein can prevent and/or treat an Arenavirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with an Arenavirus and/or by contacting a cell infected with an Arenavirus.
- a compound described herein can inhibit replication of an Arenavirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against Arenaviruses.
- the viral infection is, or is caused by, a Herpesviridae viruses such as Herpes simples viruse-1, Herpes simples viruse-2, Herpes simples viruse-6b, Herpes simples viruse-8, human cytomegalovirus, Murine cytomegalovirus, Varicella Zoster virus, Guinae pig cytomegalovirus, and Epstein-Barr virus.
- a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a Herpesvirus infection.
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein can inhibit replication of a Herpesvirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against Herpesviruses.
- the viral infection is, or is caused by, a Bunyaviridae viruses such as Rift fever virus, Punta Toto virus, La Crosse virus, Maporal virus, Heartland virus, Sever Fever Thrombocytopenia syndrome virus.
- a compound described herein for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof
- a compound described herein can inhibit replication of a Bunyavirus.
- a compound of Formula (I), or a pharmaceutical acceptable salt thereof can be effective against Bunyaviruses.
- the subject is diagnosed with coronavirus, respiratory syncytial virus, hepatitis B, human immunodeficiency virus, eastern equine encephalitis virus, western equine encephalitis virus, California encephalitis virus, Japanese encephalitis virus, Rift Valley fever virus, hantavirus, Dengue virus serotypes 1, 2, 3 and 4, Zika virus, Junin, rabies virus, influenza B virus, influenza C virus, rotavirus A, rotavirus B, rotavirus C, rotavirus D, rotavirus E, human papillomavirus, parvovirus B1 9, molluscum contagiosum virus, JC virus, Merkel cell polyomavirus, Rubella virus, lymphocytic choriomeningitis virus, mumps virus, respiratory syncytial virus, parainfluenza viruses I and 3, rinderpest virus, chikungunya, ebola virus, marburg virus, herpes
- the compounds disclosed herein such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition that includes a compound described herein, or a pharmaceutically acceptable salt thereof, can be used in combination with one or more additional agent(s) for treating, preventing and/or inhibiting a Coronaviridae, a Togaviridae, a Hepeviridae and/or a Bunyaviridae viral infection.
- compounds disclosed herein can be administered alone or in combination with other the antiviral agent(s) such as but not limited to, acyclovir, adefovir, amprenavir, arbidol, atripla, abacavir, boceprevir, cidofovir, ampligen, combivir, amantadine, daclatasvir, danmavir, dasabuvir, delavirdine, didanosine, efavirenz, emtricitabine, enfuvirtide, entecavir, famciclovir, fomivirsen, fosamprenavir, pyramidine, foscamet, fosfonet, ganciclovir, ibacitabine, imunovir, idoxuridine, imiquimod, indinavir, inosine, interferon type III, interferon type II, interferon type I, lamivudine, tenofovir disoproxil
- a compound of formula (I) or a pharmaceutically accepted formulation is administered by inhalation through the lungs.
- a pharmaceutical composition for treating or preventing a Coronavirus, influenza A viruses, an Influenza B viruses and RSV infection comprises an antiviral agent and an effective amount of a compound selected from the group consisting of but not limited to:
- a pharmaceutical composition for treating or preventing an influenza A virus, an Influenza a B virus and RSV infection comprises an antiviral agent and an effective amount of a compound selected from the group consisting of but not limited to:
- a pharmaceutical composition for treating or preventing a Coronaviridae virus, a Caliciviridae virus, an Orthomyxoviridae virus, a Herpesviridae virus, a Flaviviridae virus, a Filoviridae virus, and a Pneumoviridae virus infection comprises an antiviral agent with an effective amount of a compound selected from the group consisting of:
- 2-Hydroxylimino nucleosides derivatives, modified monophosphate and phosphonates prodrugs analogs disclosed herein can be prepared as described in detail below, or by other methods known to those skilled in the art.
- Nucleoside 4 can be prepared by coupling of a pie-modified sugar 1 with a protected or silylated pyrimidine base 2 in the presence of a Lewis acid such as but not limited to SnCl 4 or TMSOTf. Replacement of the TP group with hydroxylamine followed by deprotection of the sugar hydroxyl groups gives nucleosides 4 (Scheme 1).
- nucleoside 4 An alternate synthesis of the nucleoside 4 can be achieved by the coupling of silylated 2- thiopyrimidine derivatives 2 with sugar 1 in the presence of a Lewis acid such as, but not limited to, SnCl 4 or TMSOTf to give compound 3. Activation of the 2-position of compound 3 to the corresponding alkylthio derivative 3’, followed by treating with hydroxylamine or hydroxyl protected hydroxylamine and deprotection of the hydroxyl groups give compound 4 (Scheme 2).
- a Lewis acid such as, but not limited to, SnCl 4 or TMSOTf
- 2-Hydroxylimino nucleosides 4 can also be prepared from pre-modified nucleosides as described in Scheme 3. Protection of nucleosides 5 with a silyl reagent such as but not limited to tert- butyldimethylsily chloride (TBDMSCl) followed by selective deprotection of the 5 ’-hydroxyl group gives nucleoside 7. Applying Mistunobu reaction on nucleoside 7 gives 2,5’-anhydro nucleoside derivatives 8. Nucleoside 8 can also be prepared in many ways including, but not limited to tosylation of the 5 ‘-hydroxyl group followed by reacting the 5’tosyl derivative of compound 6 with a base such as, but not limited to, AgCO 3 .
- a base such as, but not limited to, AgCO 3 .
- nucleoside monophosphate prodrug derivatives 14 (Scheme 6).
- reacting the nucleoside derivative 9 with the phosphorchloridite derivative 13 give the nucleotide derivative 14.
- Reagents and conditions a) NaOH, Mel, H 2 O ; b) BSA, DCE, 30 min, rt then SnCl 4 , rt; c) NH 2 OH- HCl, Pyridine, rt; d) NaOMe, MeOH;
- T4 A mixture of T3 (2 g, 3.4 mmol) and hydroxylamine hydrochloride (4.78 g, 68.18 mmol) were dissolved in dry pyridine (25 mL) and the reaction mixture was stirred for 15 hours at 30- 35 °C. The solvent was evaporated and co-evaporated with toluene. The residue was partitioned between EtOAc and H 2 O, the organic phase was dried over Na 2 SO 4 , and evaporated. The residue was purified with column chromatography (eluate: 2% methanol in dichloromethane) to give T4 (1.55 g, 79.5 %) as a white solid: MP.
- AH-01 Sodium methoxide (1M, 8.26 mL) was added to a suspension of T4 (1 g, 1.75 mmol) in anhydrous methanol (20 mL) at 0 °C. The reaction mixture stirred for 2 hours at room temperature, then neutralized with glacial acetic acid. The solvents were evaporated under reduced pressure and the residue was purified with column chromatography (eluate: methanol: DCM, 15%) to give AH-01 (400 mg, 88 %), as a pale yellow solid, MP.
- T5 A solution of T4 (1.2 g, 2.1 mmol), bn. (0.43 g, 6.3 mmol), and TBSCl (0.47 g, 3.15 mmol) in dry DMF (2 mL) was stirred for 2 h at rt under argon atmosphere. Ice-H 2 0 (5 g) was added and the whole was partitioned between EtOAc (30 mL) and H 2 O (10 mL), washed with H 2 O (10 mL x 3 times), brine (10 mL), dried over MgSO 4 , filtered, and evaporated under reduced pressure.
- T6 Triethylamine (0.46 mL, 3.2 mmol) was added to a solution of T5 (0.9 g, 1.3 mmol), TIPSCl (0.97 g, 3.2 mmol), and DMAP (0.93 g, 3.2 mmol) in dry CH 3 CN (10 mL) at 0 °C under argon atmosphere. The mixture was stirred at rt overnight. Ammonia gas was bubbled into the reaction mixture for 15 min. at 0 °C. The mixture was stirred at rt for 30 min. and the solvent was evaporated to dryness. The residue was partitioned between EtOAc and H 2 O.
- AH-02. Sodium methoxide (1M, 2.6 mL) was added to a suspension of T6 (0.6 g, 0.88 mmol) in anhydrous methanol (20 mL) at 0 °C. The reaction mixture stirred for 2 h at rt, then neutralized with glacial acetic acid. The solvent was evaporated under reduced pressure and the residue was purified with column chromatography (eluate: gradient of 5-15% methanol in DCM) to give AH- 02 (0.16 g, 71 %) as a pale yellow solid: ESI-MS m/z 259.1 [M+1] + .
- AH-03. A solution of AH-01 (150 mg, 0.58 mmol) in anhydrous THE (3 mL) at 20 0°C was treated with a 1.7 M THF solution of tert-butylmagnesium chloride (0.68 mL, 0.31 mmol). After 1 h at 0°C, the mixture was treated dropwise with a solution of T7 (0.26 mg, 1.16 mmol) in anhydrous THE (3 mL) over a 5 min period. The mixture was allowed to warm to rt and was stirred for 48h at room temperature. The mixture was quenched with sat. aq. NH 4 CI (5 mL) and then extracted with ethyl acetate (60 mL).
- Reagents and conditions a) NaOEt, EtOH, rt; b) cone. H 2 SO 4 , acetone; c) Isobutyryl chloride, pyridine; d) TBAF, THE; e) 80% aq. HCO 2 H.
- T9 A suspension of T8 (600 mg, 1.6 mmol) in dry acetone (20 mL) was treated with 1M co cone. H 2 SO 4 in dry acetone (0.16 mL) and the mixture was stirred for 24 h at room temperature. The reaction mixture was neutralized with IN NH 4 OH and the solvents were evaporated under reduced pressure. The residue was purified with a silica column chromatography (eluate: gradient of 1-7% methanol in DCM) to give T9 (0.522 g, 79%) as a white solid: ESI-MS m/z 415.5 [M+1] + .
- T10 A solution of T9 (0.5 g, 1.2 mmol) in dry pyridine (10 mL) was treated with isobutyryl chloride (0.18 mL, 1.8 mmol) at 0 C. The reaction mixture was stirred for 3h at rt and the solvent was removed under reduced pressure. The residue was partitioned between EtOAc and H 2 O. The organic phase was washed with H 2 O (10 mL), brine (10 mL) and dried over Na 2 SO 4 , filtered and evaporated.
- T11 A solution of T10 (0.4 g, 0.83 mmol) in THF (3 mL) was treated with 1M solution of TBAF (1 mL) and the mixture was stirred at room temperature for 1h . The solvent was removed under reduced pressure to give crude T11 which was used entirely in the next step without further purification.
- AH-04 The crude T11 was dissolved in 80% aq. Formic acid (10 mL) and stirred at room temperature for 6 hours. The solvent was evaporated under reduced pressure and the residue was purified by a silica gel column chromatography (eluate: gradient of 1-7% MeOH in DCM) to give AH (0.0.17 g, 61%) as a white solid: ESI-MS m/z 330.1 [M+1] + .
- T13 To a solution of T1 (1.1 g, 7.58 mmol) in DCE (15 mL) was added BSA (2.25 mL, 9.1 mmol) and the reaction was stirred for 30 min. at room temperature under argon atmosphere. A solution of T12 (4.0 g, 6.9 mmol) in DCE (15 mL) via a cannula. The reaction was cooled to 0 °C and SnCl 4 (1.6 mL, 13. 8 mmol) was added dropwise. The reaction mixture was stirred for 12 hours at 40 °C then cooled to room temperature and diluted with DCM (50 mL).
- T13 A mixture of T13 (3.5 g, 5.8 mmol) and hydroxylamine hydrochloride (4.03 g, 58 mmol) was dissolved in dry pyridine (20 mL) and stirred for 48 hours at room temperature under argon atmosphere. The solvent was removed in vacuo and the residue was partitioned between EtOAc and H 2 O. The organic phase was washed with H 2 O, brine, dried over anhydrous MgSO 4 , filtered off, and evaporated under reduced pressure. The residue was purified by a silica gel column (eluate; gradient 0-7% MeOH in DCM) to give T14 (3.0 g, 89% yield) as a white solid.
- AH-05 Sodium methoxide (1M, 15 mL) was added to a suspension of T14 (2.5 g, 4.27 mmol) in anhydrous methanol (15 mL) at 0 °C. The reaction mixture stirred for 12 h at rt, then neutralized with glacial acetic acid. The solvent was evaporated under reduced pressure and the residue was purified by a silica gel column chromatography (eluate: gradient of 5-12% methanol in DCM) to give AH-05 (1 g, 85%), as a white solid.
- Reagents and conditions a) EtOH, reflux, 2 h; b) NaOAc. AcOH, reflux; c) i) BSA, DCE, rt, 30 min; ii) SnCl 4 , - 10 °C, 3 h, then NaCHO 3 , 0 °C, 0.5 h; d)DBU, Mel, DMF; e) NH 2 OH-HCl, dry pyridine, rt; f) NaOMe, MeOH, rt, 3 h.
- Example 7 Synthesis of 6-aza-2-hydroxyiminouridine AH-06 T15. To a solution of thiosemicarbazide (10 g, 0.11 mole) in absolute ethanol (30 mL) was added ethyl glycosylate (40% in toluene, 28.1 mL). Then the solution was stirred for 2 hours at reflux temperature.
- T16 A solution of T15 (17 g, 0.097 mol) and anhydrous sodium acetate (63.67 g, 0.776 mol) in glacial acetic acid (80 mL) was heated for 3 h at 110 °C. The reaction mixture was cooled to room temperature and the solvent was evaporated under reduced pressure. The residue was taken in EtOAc (200 mL) and neutralized with cold saturated NaHCO 3 solution. The organic phase was separated, dried over anhydrous Na 2 SO 4 , filtered, and evaporated under vacuum.
- T17 To a suspension of T21 (3 g, 0.023 mol) and 1-O-acetyl-2,3,5-tri-O-benzoyl- ⁇ -D- ribofuranose (11.05 g 0.022 mol) in dry DCE (300 mL) was added BSA (6.75 mL, 0.0276 mol) drop wise at room temperature. The solution was stirred for 30 minutes at room temperature until complete dissolution of the solid material. Then, SnCl 4 (3.86 mL, 0.033 mol) was added drop wise at -10 °C. The reaction mixture was stirred for 3 hours at 0 °C, then wormed up to room temperature and stirred for further three hours.
- reaction mixture was diluted with DCM 100 mL) and neutralized with saturated sodium bicarbonate solution.
- the produced white cake was filtered over a Celite pad, thoroughly washed with DCM.
- the organic phase was separated, dried over anhydrous Na 2 SO 4 , and evaporated under vacuum.
- T18 To a solution of T17 (3.8 g, 6.63 mmol) in dry DMF (35 mL) was added Mel (1.03 mL, 16.58 mmol) and the mixture was cooled down to 0 °C. The reaction mixture was treated with DBU (1.48 mL, 9.95 mmol) and stirred for further 1h at 0 °C under argon atmosphere. The reaction mixture was diluted with EtOAc (100 mL) and washed with H20 (30 mL x 3 times). The organic phase was separated, was separated, dried over anhydrous Na 2 SO 4 , and evaporated under vacuum.
- T19 A mixture of T18 (6.15 g, 10.73 mmol) and hydroxylamine hydrochloride (14.92 g, 0.214 mol) in dry pyridine (80 mL) was stirred over night at 45 °C. The solvent was evaporated under reduced pressure, co-evaporated with toluene, and the residue was partitioned between EtOAc and H 2 O. The organic phase was separated, dried over anhydrous Na 2 SO 4 , and evaporated under vacuum.
- AH-07 A solution of AH-06 (0.13 g, 0.498 mmol) in anhydrous THF (1.5 mL) was treated with a 2 M THF solution of tert-butylmagnesium chloride (0.62 mL, 1.05 mmol) at 0°C and the mixture was stirred for 1h at 0°C. Next, the mixture was treated dropwise with a solution of T7 (0.405 g, 1.0 mmol) in anhydrous THF (1.5 mL) over a 5 min period. The mixture was allowed to warm to rt and was stirred for 4 days at room temperature. The mixture was quenched with sat. aq.
- Reagents and conditions (a) KOH, CH 3 I, H 2 0; (b) BSA, DCE, 1h , rt, then SnCl 4 , 12 h, rt; pyridine, rt; (d) NaOMe, MeOH, rt.
- T30 A mixture of T2 (9.23 g, 18.29 mmol) and T29 (3 g, 19.2 mmol) were dissolved in dry DCE (100 mL). BSA (4.7 mL, 19.2 mmol) was added and the reaction mixture was stirred for 1 hour at room temperature. The reaction mixture was cooled to -10 °C, and then SnCl 4 (3.2 mL, 27.44 mmol) was added and the reaction mixture for 12 hours at room temperature. The reaction mixture was diluted with DCM (100 mL) and poured into saturated solution of aqueous sodium bicarbonate. The mixture was filtered over Celite pad, and the filtrate was extracted with dichloromethane.
- T31 A mixture of T30 (1.5 g, 2.50 mmol) and hydroxylamine hydrochloride (3.5 g, 68.18 mmol) were dissolved in dry pyridine (25 mL) and the reaction mixture was stirred for 15 hours at 45 °C. The solvent was evaporated and co-evaporated with toluene. The residue was partitioned between EtOAc and H 2 O, the organic phase was dried over Na 2 SO 4 , and evaporated.
- Reagents and conditions (a) BSA, DCE, then SnCl 4 ; (b) NH 3 /MeOH; c) cone. H 2 SO 4 , acetone; d) i) TsCl, DCE, DMAP, rt; ii) DBU, CH 3 CN; e) NH 2 OH+HCl, DIEA, CH 3 CN, rt; f) aq. HCOOH.
- Reagents and conditions (a) BSA, DCE, 1h , rt, then SnCl 4 , 12h, rt; (b) NH 3 /MeOH, rt; c) cone. H 2 SO 4 , acetone, rt; d) i) TsCl, DCE, DMAP, rt, ii) AgOAc, DMF, rt; e) NH 2 OH+HCI, DIEA, CH 3 CN, rt; f) aq. HCOOH, rt.
- Reagents and conditions a) NaOH, Mel, H 2 O, rt; b) BSA, DCE, 1h , rt, then SnCl 4 , rt; c) NH 2 OH+HCI, pyridine, rt; d) NaOMe, MeOH, rt.
- Reagents and conditions a) TBSCl, Im, DMF, rt; b) aq. TFA, 0 °C, c) i) TsCl, pyridine, ii) AgOAc, DMF; NH 2 OH+HCl, DIEA, rt; d) TBAF, THF, rt.
- Reagents and conditions a) TBSCl, Im, DMF, rt; b) aq. TFA, 0 °C, c) i) TsCl, pyridine, ii) AgOAc, DMF; NH 2 OH+HCl, DIEA, rt; d) TBAF, THF, rt.
- Reagents and conditions a) TBSCl, Im, DMF, rt; b) aq. TFA, 0 °C, c) i) TsCl, pyridine, ii)
- Reagents and conditions a) BSA, DCE, 1h , rt, then SnCl 4 , rt; b) NaOMe, MeOH, rt; c) i)TBSCl, Im., DMF, rt; ii)50% aq. TFA, 0 °C; d) i) TsCl, pyridine, ii)AgOAc, DMF; e) NH 2 OH.HCI, DIEA, rt; f) TBAF, THF, rt. ydroxyiminouridines.
- Reagents and conditions a) i) Ac 2 O, CH 3 CN, reflux; ii) AcBr, CH 3 CN; iii) H 2 P(O)OH, Et 3 N, CaCO 3 , H 2 O, CH 3 CN; b) TBSCl, Im, DMF, rt; c) aq. TFA, 0 °C, d) i) TsCl, DMAP, DCM, ii) AgOAc, DMF; e) NH 2 OH+HCl, DIE A, rt; f) TBAF, THF, rt.
- T74 To a solution of T73 (0.52 g, 2.98 mmol) in DCE (10 mL) was added BSA (0.59 mL, 2.38 mmol) and the reaction was stirred for 30 min. at room temperature under argon atmosphere. A solution of T12 (1.73 g, 2.97 mmol) in DCE (10 mL) via a cannula. The reaction was cooled to 0 °C and SnCl 4 (0.6 mL, 8.91 mmol) was added dropwise. The reaction mixture was stirred for 8 hours at 40 °C then cooled to room temperature and diluted with DCM (50 mL).
- T75 A mixture of T74 (1.5 g, 2.37 mmol) and hydroxylamine hydrochloride (3.3 g, 47.4 mmol) was dissolved in dry pyridine (25 mL). The reaction mixture was stirred for 48 hours at 40 °C. Then pyridine was evaporated and co-evaporated with toluene.
- Reagents and conditions (a) cone. H 2 SO 4 , acetone, rt; (b) I 2 , TTP, THF; (c) NaOMe, MeOH, reflux; d) AgF, I 2 , DCM; (e) AgOAc, DMF, rt; (f) NH 2 OH+HCl, DIEA, CH 3 CN, rt; f) aq. HCOOH, rt.
- Reagents and conditions (a) i) I 2 , Im., TPP, THF; ii) NaOMe, MeOH, reflux; (b) i) BTEAN 3 /MeCN, I 2 , THF, NaS 2 O 3 ; ii) BzCl, NMM, DMAP, MeCN; c) AgOAc, DMF; (e) NaOMe, MeOH.
- Reagents and conditions (a) cone. H 2 SO 4 , acetone, rt; (b) i) TsCl, DMAP, DCM, ii) AgOAc, DMF; c) NH 2 OH+HCI, DIEA, rt; d) aq. HCOOH, rt.
- Reagents and conditions (a) i) TMM, p-MeC 6 H 4 SO 3 H, DMF; ii) DMSO, TFA, Pyridine, EDC, DMF; iii) Et 3 N, DMF, rt; iv) NaBH 4 , MeOH; v), MeNH 2 , EtOH; (b) NaNO 2 , AcOH; (c) i) TBSCl, Im., DMF; ii) aq.
- TFA THF
- d) ) i) TsCl, DMAP, DCM, ii) AgOAc, DMF; e) i) NH 2 OH+HCl, DIEA, rt; ii) TBAF, THF, rt; f) t-BuMgCl, THF, rt.
- g) i) NaSH, Et 3 N- HCl, DMF; ii) TBAF, THF.
- Reagents and conditions a) TBSCl, Im., DMF, rt; b) i) TPP, DIAD, Toluene; ii) NaBH 4 , (PhSe) 2 , EtOH, rt; c) m-CPBA, DCM; d) TBAF, THF; e) T7, t-BuMgCl, THF, rt; f, TIPSCl, DMAP, Et 3 N, CH 3 CN, then NH 3 .
- T111 A mixture of T110 (0.14 g, 0.51 mmol) and NH 2 OH-HCl (0.71 g, 10.2 mmol) was dissolved in dry pyridine (5 mL). The reaction mixture was stirred for 48 hours at 40 °C. The solvent was evaporated and co-evaporated with toluene.
- T112 A solution of T111 (40 mg, 0.16 mmol) in dry methanol (5 mL) was treated with NaOCH 3 (1M, 1.1 mL) at 0 °C, then the mixture was stirred for 2 hours at room temperature. The reaction mixture was neutralized with acetic acid, and then the solvents were evaporated.
- T114 A solution of T108 (0.15 g, 0.95 mmol) in dry THF (7 mL) was treated with LiHMDS (1M, 1.1 mL) at rt and the mixture was further stirred for 30 minute at the same temperature. 4- Bromobutyl acetate (0.74 g, 3.82 mmol) was added dropwise at rt then the mixture was stirred overnight at 80 °C under argon atmosphere. The mixture was cooled to rt and then aq. NH 4 CI (1 mL) was added. The whole was partitioned between EtOAc water. The organic phase work washed with water, brine, and separated. The combined organic phases were dried over anhydrous (Na 2 SO 4 ), and filtered off.
- 4- Bromobutyl acetate (0.74 g, 3.82 mmol) was added dropwise at rt then the mixture was stirred overnight at 80 °C under argon atmosphere. The mixture was cooled to rt and then aq.
- T115 A mixture of compound T114 (90 mg, 0.33 mmol) and NH 2 OH-HCl (0.47g, 6.63 mmol) was dissolved in dry pyridine (3 mL). The reaction mixture was stirred for 48 hours at 40 °C. Then pyridine was evaporated and co-evaporated with toluene.
- T116 A solution of compound T115 (75 mg, 0.29 mmol) in dry methanol (3 mL) was treated with NaOCH 3 (1M, 2.06 mL) at 0 °C, then the mixture was stirred for 3 hours at room temperature. The reaction mixture was neutralized with acetic acid, and the solvents were evaporated.
- Reagents and conditions a) BSA, DCE, O.Sh , rt, then SnCl 4 , rt; b) NH 2 OH+HCI, pyridine, rt; c) NaOMe, MeOH, rt.
- T117 A mixture of T108 (110 mg, 0.7 mmol) and T12 (435 mg, 0.84 mmol) was suspended in DCE (5 mL), then treated with BSA (0.21 mL, 0.84 mmol) and the mixture was stirred for 30 min, until complete dissolution. SnCl 4 (0.246 mL, 2.1 mmol) added drop wise and the mixture was stirred for 24 h at room temperature. The reaction mixture was quenched with sat. NaHCO 3 and the white cake was filtrated on a Celite pad and was washed with DCM (50 mL). The separated organic layers were combined, dried over Na 2 SO 4 and filtered.
- T119 A solution of compound T118 (150 mg, 0.25 mmol) in dry methanol (5mL) was treated with sodium methoxide (1M, 2.0 mL) at 0 °C, then the mixture was stirred for 24 hours at room temperature. The reaction mixture was neutralized with acetic acid and the volatiles were evaporated under reduced pressure.
- T120 A solution of AH-09 (115 mg, 0.42 mmol) in anhydrous THF (1.5 mL) was treated with a 1.7M THF solution of tert-butylmagnesium chloride (0.52 mL, 0.88 mmol) at 0°C and the mixture was stirred for 1h at 0°C. Next, the mixture was treated dropwise with a solution of T7 (342 mg, 0.84 mmol) in anhydrous THF (1.5 mL) over a 5 min period. The mixture was allowed to warm to rt and was stirred for 2 days at room temperature. The mixture was quenched with sat. aq.
- CPE Primary Cytopathic Effect
- Four-concentration CPE inhibition assays are performed. Confluent or near-confluent cell culture monolayers in 96-well disposable microplates are prepared. Cells are maintained in MEM or DMEM supplemented with FBS as required for each cell line. For antiviral assays the same medium is used but with FBS reduced to 2% or less and supplemented with 50 ⁇ g/ml gentamicin. The test compound is prepared at four log10 final concentrations, usually 0.1, 1.0, 10, and 100 ⁇ g/ml or ⁇ (depending upon sponsor’s preference). Lower concentrations are used when insufficient compound is supplied for the usual concentrations. Four compounds can be tested per microplate.
- Controls for the experiment consist of six microwells that are infected (virus controls) and six that are untreated (cell controls).
- the virus control and cell control wells are on every microplate.
- a known active drug is tested as a positive control drug using the same method as is applied for test compounds.
- the positive control is tested with each test run.
- the assay is initiated by first removing growth media from the 96-well plates of cells. Then the test compound is applied in 0.1 ml volume to wells at 2X concentration. Virus, normally at ⁇ 100 50% cell culture infectious doses (CCID50) in 0.1 ml volume, is placed in those wells designated for virus infection.
- CCID50 cell culture infectious doses
- Virus control wells are treated similarly with virus. Plates are incubated at 37 °C with 5% CO 2 until maximum CPE is observed in virus control wells. The plates are then stained with 0.011% neutral red for approximately two hours at 37oC in a 5% CO 2 incubator. The neutral red medium is removed by complete aspiration, and the cells rinsed 1X with phosphate buffered solution (PBS) to remove residual dye. The PBS is completely removed and the incorporated neutral red is eluted with 50% Sorensen’s citrate buffer/50% ethanol for at least 30 minutes. Neutral red dye penetrates into living cells, thus, the more intense the red color, the larger the number of viable cells present in the wells.
- PBS phosphate buffered solution
- the dye content in each well is quantified using a 96- well spectrophotometer at 540 nm wavelength.
- the dye content in each set of wells is converted to a percentage of dye present in untreated control wells using a Microsoft Excel computer-based spreadsheet.
- the 50% effective (EC50, virus-inhibitory) concentrations and 50% cytotoxic (CC50, cell-inhibitory) concentrations are then calculated by linear regression analysis.
- the quotient of CC50 divided by EC50 gives the selectivity index (SI) value.
- SI selectivity index
- VYR Secondary CPE/ Viral Yield Reduction Assay. This assay involves similar methodology to what is described in in CPE assay using 96-well microplates of cells. The differences are noted in this section. Eight half-log10 concentrations of inhibitor are tested for antiviral activity and cytotoxicity. Only two compounds can be evaluated per 96-well microplate. One positive control drug is tested per batch of compounds evaluated. After sufficient virus replication occurs, a sample of supernatant is taken from each infected well (three replicate wells are pooled) and frozen for the VYR portion of this test, if needed. Alternately, a separate plate may be prepared and the plate may be frozen for the VYR assay.
- VYR assay is a direct determination of the concentration of the test compound that inhibits virus replication. Virus that was replicated in the presence of test compound is titrated and compared to virus from untreated, infected controls. Titration of pooled viral samples (collected as described in the paragraph above) is performed by endpoint dilution.
- the virus control and cell control will be run in parallel with each tested compound. Further, a known active drug is tested as a positive control drug using the same experimental set-up as described for the virus and cell control. The positive control is tested with each test run. Test compounds and positive controls are typically tested in biological triplicates. The assay is initiated by first removing growth media from the 12-well plates of cells, and infecting cells with 0.01 MOI of virus or about 50 to 100 plaque forming units (pfu). Cells will be incubated for 60 min: ⁇ inoculum/ well, at 37°C, 5% CO 2 with constant gentle rocking.
- Virus inoculum will be removed, cells washed and overlaid with either 1% agarose or 1% methylcellulose diluted 1:1 with 2X MEM and supplemented with 2% FBS and 1 %penicillin/streptomycin and supplemented with the corresponding drug concentration. Cells will be incubated at 370C with 5% CO 2 for 5 (Lassa fever), 10 (Ebola), or 3 (Nipah) days. The overlay is then removed and plates stained with 0.05% crystal violet in 10% buffered formalin for approximately twenty minutes at room temperature. The plates are then washed, dried and the number of plaques counted. The number of plaques is in each set of compound dilution is converted to a percentage relative to the untreated virus control.
- the 50%effective (EC50, virus- inhibitory) concentrations are then calculated by linear regression analysis. Cytotoxicity is evaluated in parallel to the actual primary PR assay.
- the cytotoxicity assay (In vitro Toxicology Assay Kit, Neutral red based; Sigma) is being performed in 96-well plates following the manufacturer’s instructions. Briefly, growth medium will be removed from confluent cell monolayer’s and replaced with fresh medium (total of 100 ⁇ ) containing the test compound with the concentrations as indicated for the primary assay. Control wells will contain medium with the positive control or medium devoid of compound. Wells without cells and growth medium only will serve as blank. A total of up to five replicates will be performed for each condition. Plates are then incubated for 3, 5, or 10 days at 37°C with 5% CO2.
- the plates are then stained with 0.033% neutral red for approximately two hours at 37oC in a 5% CO2 incubator.
- the neutral red medium is removed by complete aspiration, and the cells rinsed 1X with phosphate buffered solution (PBS) to remove residual dye.
- PBS phosphate buffered solution
- the PBS is completely removed and the incorporated neutral red is eluted with 1% acetic acid/50% ethanol for at least 30 minutes.
- Neutral red dye penetrates into living cells, thus, the more intense the red color, the larger the number of viable cells present in the wells.
- the dye content in each well is quantified using a 96-well spectrophotometer at 540 nm wavelength and 690 nm wavelength (background reading).
- the 50% cytotoxic (CC50, cell-inhibitory) concentrations are then calculated by linear regression analysis.
- the quotient of CC50 divided by EC50 gives the selectivity index (SI) value.
- the secondary assay involves similar methodology to what is described in the previous paragraphs using 12-well plates of cells. The differences are noted in this section. Cells are being infected as described above. The test compound is prepared at eight half-log10 final concentrations, usually 0.032, 0.1, 0.32, 1.0, 3.2, 10, 32 and 100 ⁇ g/ml or ⁇ . Test compound is applied in 1 ml of total volume of media. Tissue culture supernatant (TCS) aliquots will be collected at appropriate time points and then be used to determine the compounds inhibitory effect on virus replication. Virus that was replicated in the presence of test compound is titrated and compared to virus from untreated, infected controls.
Abstract
Disclosed herein are nucleosides and nucleotides analogs, methods for preparing the same, and methods for treating and/or ameliorating infection caused by a Coronaviridae virus, a Caliciviridae virus, an Orthomyxoviridae virus, a Herpesviridae virus, a Flaviviridae virus, a Filoviridae virus,and a Pneumoviridae virus with one or more nucleoside and nucleotide analogs of formula I. In certain embodiments, compounds and compositions of nucleoside or nucleotide derivatives are disclosed, which can be administered either alone or in combination with other anti-viral agents. In certain embodiments, the compounds are according to Formula (I): or a pharmaceutically acceptable salt, solvate, stereoisomeric form, a tautomeric form or polymorphic form thereof, wherein R1, R2, R3, R4, X, and sugar are as described herein.
Description
2-HYDROXYIMINOPYRIMIDINE NUCLEOSIDES AND DERIVITIVES AND
ANTIVIRAL USES THERETO
Field of the Invention :
This disclosure relates to 2-hydroxyiminopyrimidine nucleoside derivatives, as well as compositions and methods related thereto. In certain embodiments, the disclosure relates to the treatment or prophylaxis of viral infections, in particular respiratory viral infections including coronaviruses, influenza viruses, and respiratory syncytial virus.
Background of the Invention:
Emerged outbreaks of respiratory viral infections caused by viruses belong to Coronaviridae, Orthomyxoviridae, and Paramyxoviridae are causing global health concerns and economic impact worldwide. Middle East respiratory syndrome coronavirus (MERS-CoV) is a highly pathogenic human coronavirus causes severe acute pneumonia and renal failure. MERS- CoV has recently emerged in Saudi Arabia (Assiri et al., N Engl. J. Med. 2013, 369, 407-416) and outbreaks in South Korea (Zaki, et.al., N. Engl. J. Med. 2012, 367, 1814-1820.). MERS- CoV quickly spread around the globe with a case fatality rate of 38% according to the World Health Organization (WHO) (http://www. who.int/mediacentre/factsheets/mers-cov/en/). No clinically effective vaccines or specific antiviral drugs are currently available for the prevention and treatment of coronavirus infections. Therefore, there is a need for anti -coronavirus therapeutics.
Pneumoviridae viruses, including human respiratory virus (HRSV) are responsible for many prevalent human and animal diseases. HRSV is the major cause of lower respiratory tract infections in infa cy and childhood. Adults and elders with chronic heart, lung disease or those that are immunosuppressed also have a high risk for developing severe HRSV disease (http://www.cdc.gov/rsv/index.html). No vaccine infection is currently available for HRSV and the only antiviral agent that has been approved to treat HRSV infections, Ribavirin, a nucleoside analogue has limited efficacy. Therefore, there is a need for anti-pneumovirinae therapeutics. The present invention provides agents, compositions, and methods for treating or preventing respiratory viral infections.
In late December 2019, a cluster of pneumonia cases caused by a novel coronavirus (nCoV) was reported in Wuhan, China (Zhu et. al., N. Engl. J. Med. 2020, 382, 727-733). This novel coronavirus was named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and was designated as COVID-
19 by the World Health Organization (WHO). COVID-19 has claimed hundreds of thousands of lives worldwide and have caused great economic impact on the global economy. No clinically effective vaccines or specific antiviral drugs are currently available for the prevention and treatment of coronavirus infections.
Nucleoside analogs as a class of small molecules have a well-established regulatory history, with several of them currently approved by the US Food and Drug Administration (US FDA) for treating hepatitis C (HCV), hepatitis B virus (HBV), human immunodeficiency virus (HIV), and herpes simplex C virus (HSV). Examples of compounds useful for treating viral infections are described in AU 2020/202600 A1 (Cho et al), AU 2020/200499 A1 (Beigelman et al.), US 2018/009861652 B2 (Schinazi et al.), US 2017/009809616 B2 (Amblard et al.), WO 2010/091386 A3 (Cho et. al.), US 2016/009382286 B2 (Schinazi et al.), US2013/OO8415321 B2 (Schinazi et al.), WO 2001/0196353 A3 (Bryant et al.), WO 2001/091737 A2 (Sommadossi et al.), WO 2012/037038 (Clarke et al.), WO 2017/162169 (Guocheng et al.), U.S. 2012/0009147 Al (Cho et al.), WO 2008/141079 A1 (Babu et al.), WO 2010/002877 A2 (Ffancom), WO 2012/142075 A1 (Girijavallabhan et al.), WO 2012/087596 A1 (Delaney et al.), WO 2011/035231 A1 (Cho et al.), US 2014/0057863 A1 (Stuyver et. al.), and US 2018/OORE46762 E (Butler et. al).
Disclosure Of The Invention :
Summary of the Invention
The present invention provides compounds, methods, and compositions for treating or preventing respiratory viral infections in a host. Provided is a compound of Formula I, or a pharmaceutically acceptable salt thereof:
 Wherein:
Wherein:
R1 is selected from hydrogen, COR1*, COOR1*, CONHR1*.
R1* is, independently, C1-C22 alkyl, the carbon chain is derived from fatty alcohol or amino acid, C3-C7 cycloalkyl, aryl, heterocyclyl, CH2OCO-C1-C22 alkyl, CH2OCO-C3-C7 cycloalkyl, CH2OCO-C3-C7 heterocyclyl, C1-C22 alkoxy, C3-C7 cycloalkoxy, aryloxy, heterocyclyloxy, C2-C18 alkenyl, C2-C18 alkynyl, substituted C1-C22 alkyl with halogen or C1- C6 carbon chain, (C=O)N(alkyl)2,NH(aryl), NH(Heroaryl), NH(C=O)-alkyl (C1-C22), NHO(C=O)alkyloxy (C1-C22).
R2 is, independently, selected from NH, CH, C-halogen, N(CO)NH2, the dashed and the solid lines combined represent an optional double bond.

R3 is independently, selected from H, OH, NHOH, NHO(C=O) C1-C22 alkyl, NHO(C=O)-C1-C22 alkyloxy, NH2, SH, CN, S-C1-C10 alkyl, S-C2-C10 alkenyl, S-C2-C10 alkynyl, S-aryl, NH-C1-C22 alkyl, NH- C3-C10 cycloalkyl, N(alkyl)2,NH(aryl), NH(Heroaryl), NH(C=O)- C1-C22 alkyl, NHO(C=O)-C1-C22 alkyloxy.
R4 is independently, selected from H, F, Cl, Br, I, CH3, CF3, CH2F, CHF2, CH2OH, CH2Cl, CH2Br, CH2I, CN, an optionally substituted alkyl C1-C10, an optionally substituted C1- C10 cycloalkyl, an optionally substituted C2-C10 alkenyl, an optionally substituted C2-C10 alkynyl.
X is independently, selected from CH, C-(halogen; F; Cl, Br, I), C-CN, C-CH3, C-CF3, C-
CHF2, C-CH2F, C-CH2OH, C-CCH, C-CHCH2, N, C(C=R1*, C-NO2.
In one embodiment, Sugar for formula I is ribose or a modified ribose of the general formula (I-
A):

Wherein:
R5 is, independently, selected from H, halogen (F, Cl, Br, I), CN, N3, CH3, substituted methyl, CCH, C=CH2;
R6 is independently selected from, H, OH, halogen (F, Cl, Br, I), Nitrile (CN), Nitro (NO2), methyl (CH3), C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is independently selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from absent, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3, CFCH3, cyclopropyl;;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3
R20 is independently selected from lower alkyl (C1-C-6), cycloalkyl, higher alkyl (C7-C16), alkenyl (C=CH2), alkynyl (CCH), aryl, heteroaryl, heterocyclyl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,
 Wherein:
Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’, CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In another embodiment, Sugar for formula I is ribose or a modified ribose of the general Formula
(I-B):

Formula l-B
Wherein:
R5 is independently selected from H, CN, N3, CH3, substituted methyl, CCH, C=CH2;
R6 is independently selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is independently selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is independently selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3,
CFCH3,cyclopropyl;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
Z is independently selected from O, S, NOH, NOR9;
R9 is independently selected from C(O)alkyl (C1-C17), C(O)cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), C(O)aryl, C(O)heteroaryl;
R15 is independently selected from O-aryl, O-heteroaryl,

Wherein:
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, pyridinyl CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
In still another embodiments sugar for the general formula is formula I-C:

Wherein:
R5 is independently selected from H, CN, N3, CH3, substituted methyl, CCH, C=CH2;
R6 is independently selected from, H, halogen (F, Cl, Br, I), CH3, NO2, CN, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is independently selected from, H, halogen (F, Cl, Br, I), N3, CN; NO2, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R8 is independently selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3, CFCH3,cyclopropyl;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3;
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,

Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In still another embodiments sugar for formula I is ribose or a modified ribose of Formula I-D

Formula I-D
Wherein:
R5 is independently selected from H, CN, N3, CH3, substituted methyl, CCH, C=CH2;
R6 is independently selected from, H, halogen (F, Cl, Br, I), CH3, NO2, CN, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is independently selected from, H, halogen (F, Cl, Br, I), N3, CN; NO2, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R8 is independently selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3, CFCH3,cyclopropyl;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3;
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH; W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,

Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In still another embodiments sugar for formula I is formula I-E:

Formula l-E
Wherein:
R5 is independently selected from H, halogen (F, Cl, Br, I), CN, N3, CH3, substituted methyl, CCH, C=CH2;
R6 is independently selected from, H, halogen (F, Cl, Br, I), CH3, NO2, CN, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is independently selected from, H, halogen (F, Cl, Br, I), N3, CN; NO2, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is independently selected from, H, halogen (F, Cl, Br, I), N3, NO2, methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3,
CFCH3,cyclopropyl;
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,

Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In still another embodiments sugar for formula I is formula I-F:

Formula l-F
Wherein:
R5 is independently selected from H, CN, CH3, substituted methyl, CCH, C=CH2;
R8 is independently selected from, H, methyl, an optionally substituted methyl, CN; alkyl (C1- C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3,
CFCH3,cyclopropyl;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3;
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,
 Wherein:
Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In still another embodiments sugar for formula I is the formula I-G:

Formula l-G
Wherein:
R5 is independently selected from H, CN, CH3, substituted methyl, CCH, C=CH2;
R8 is independently selected from, H, methyl, an optionally substituted methyl, CN; alkyl (C1- C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3,
CFCH3,cyclopropyl;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3;
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,

Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In still another embodiments sugar for formula I is the formula I-H

Formula l-H
Wherein:
X is independently selected from absent, CH2, CHR16; R16 (is independently selected from CH3, CH2OH, CH2NH2, CH2CN, CH2NO2, CH2F, CH2CI, CH2I, CH2Br, CF3, CCH, CH=CH2, CH2, CH2CO2H, CH2O-protected monophosphate;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3, CFCH3;
Z is independently selected from O, S, NOH, NOR9.
R15 is independently selected from (C=O)-alkyl (C1-C22), optionally substituted (C=O)-alkyl, (C=O)-aryl, (C=O)-CH2 aryl, (C=O)-Heteroaryl,

In still another embodiments sugar for formula I is the formula I-J

Formula l-J
Wherein:
X is independently selected from absent, CH2, CHR16; R16 (is independently selected from CH3, CH2OH, CH2NH2, CH2CN, CH2NO2, CH2F, CH2CI, CH2I, CH2Br, CF3, CCH, CH=CH2, CH2, CH2CO2H, CH2O-protected monophosphate;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3, CFCH3;
Z is independently selected from O, S, NOH, NOR9.
R15 is independently selected from O-C(=O)alkyl (C1-C22), optionally substituted O- C(=O)
 alkyl, O-C(=O)aryl, O- C(=O)CH2aryl, O- C(=O)Heteroaryl, R16 is independently selected
alkyl, O-C(=O)aryl, O- C(=O)CH2aryl, O- C(=O)Heteroaryl, R16 is independently selected

In still another embodiments sugar for formula I is the formula I-K

Formula l-K
Wherein:
Y is independently selected from O, S, NH, NR9, CH2, CF2, C(=O), C=CH2, COHCH3, CFCH3; R17 is independently selected from Hydrogen, CH3, CF3, CH2OH, CH2F, CH2CI, CH2Br, CH2I, CH2N3, CH2NH2, CH=CH2, CCH;
R18 is independently selected from Hydrogen, CH3, CF3, CH2OH, CH2F, CH2CI, CH2Br, CH2I, CH2N3, CH2NH2, CH=CH2, CCH;
Y with absent R18 is selected from C=CH, C=CF, C=CCH3;
W1 is independently selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate ester, diphosphate ester, triphosphate ester,

Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl. R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl;
Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid;
In certain embodiments, the disclosure relates to a compound of formula I having formula I-L,

Formula l-L or a salt thereof,
Wherein:
Y is O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3;
R1 is as defined in Formula I
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent R8 is selected from C=CH, C=CF, C=CCH3 ;
R9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I,
 monophosphate, diphosphate, triphosphate,
monophosphate, diphosphate, triphosphate,
Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl; R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In certain embodiments, the disclosure relates to a compound of formula I having formula I-M,

Formula l-M or a salt thereof, Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3;
E is selected from absent, O, CH2, NH;
The dashed and the solid lines combined represent an optional double bond;

R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent R8 is selected from C=CH, C=CF, C=CCH3 ;
R9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I, monophosphate, diphosphate, triphosphate,

Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In certain embodiments, the disclosure relates to a compound of formula I having formula I-N,

Formula l-N or a salt thereof,
Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3;
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent R8 is selected from C=CH, C=CF, C=CCH3 ;
R9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I;
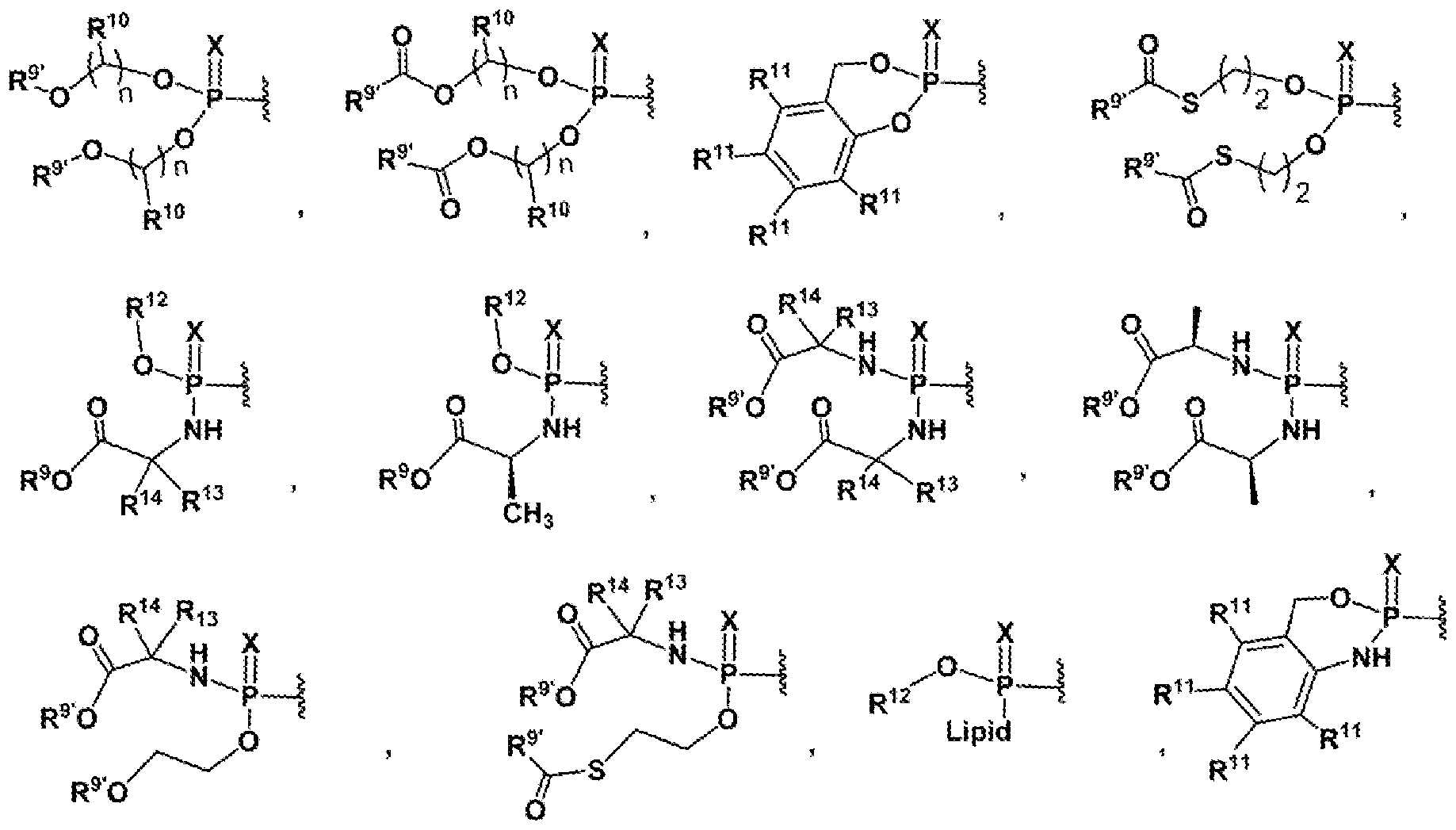 monophosphate, diphosphate, triphosphate,
monophosphate, diphosphate, triphosphate,
Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl;
R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In certain embodiments, the disclosure relates to a compound of formula I having formula I-O,

Formula I-O or a salt thereof,
Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3;
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent R8 is selected from C=CH, C=CF, C=CCH3 ;
R9 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I;
R17* is selected from hydrogen, methyl, isopropyl, benzyl, p-hydroxybenzyl, CH(CH3)CH2CH3, butyl, tert-butyl, isobutyl, pentyl, hydroxymethyl, mercaptomethyl, -CH2-OR9, -CH2-SR9, -CH2-, -CH2-SMe; -CH2-indolyl, -CH2-imidazolyl, -CH2-(CH2)3-NHC(=O)R9, -CH2- CH2CH2CH2NHC(=NH)NH2, -CH(OH)CH3;
R18* is selected from -C(=O)R9, -SO2-R9;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CHCCH, CHCH2OH;
In certain embodiments, the disclosure relates to a compound of formula I having formula I-P,

Formula l-P or a salt thereof, Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3;
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl;
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3
R9 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I;
R17* is selected from hydrogen, methyl, isopropyl, benzyl, p-hydroxybenzyl, CH(CH3)CH2CH3, butyl, tert-butyl, isobutyl, pentyl, hydroxymethyl, mercaptomethyl, -CH2-OR9, -CH2-SR9, -CH2-, -CH2-SMe; -CH2-indolyl, -CH2-imidazolyl, -CH2-(CH2)3-NHC(=O)R9, -CH2- CH2CH2CH2NHC(=NH)NH2, -CH(OH)CH3;
R18* is selected from -C(=O)R9, -SO2-R9;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I,
 monophosphate, diphosphate, triphosphate, Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
monophosphate, diphosphate, triphosphate, Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl; R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In certain embodiments, the disclosure relates to a compound of formula I having formula I-Q,

Formula l-Q or a salt thereof, Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3; all together with C- The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent R8 is selected from C=CH, C=CF, C=CCH3;
R9 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I;
R17* is selected from hydrogen, methyl, isopropyl, benzyl, p-hydroxybenzyl, CH(CH3)CH2CH3, butyl, tert-butyl, isobutyl, pentyl, hydroxymethyl, mercaptomethyl, -CH2-OR9, -CH2-SR9, -CH2-, -CH2-SMe; -CH2-indolyl, -CH2-imidazolyl, -CH2-(CH2)3-NHC(=O)R9, -CH2- CH2CH2CH2NHC(=NH)NH2, -CH(OH)CH3;
R18* is selected from -C(=O)R9, -SO2-R9;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I,
 monophosphate, diphosphate, triphosphate,
monophosphate, diphosphate, triphosphate,
Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl; R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In certain embodiments, the disclosure relates to a compound of formula I having formula I-R,

Formula l-R or a salt thereof, Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3; all together with C- R8 is CH=CH, CF=CH; CH= CF;
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R1isselected as described in Formula I.
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3
R9 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I,
 monophosphate, diphosphate, triphosphate,
monophosphate, diphosphate, triphosphate,
Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl; R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In certain embodiments, the disclosure relates to a compound of formula I having formula I-S,

Formula I-S or a salt thereof,
Wherein:
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3; all together with C- R8 is CH=CH, CF=CH; CH= CF;
E is selected from absent, O, CH2, NH;
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN,
alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent R8 is selected from C=CH, C=CF, C=CCH3
R9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I,
 monophosphate, diphosphate, triphosphate,
Wherein:
monophosphate, diphosphate, triphosphate,
Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl; R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
In another embodiment this disclosure relates to a compound of formula II

Formula II or salts thereof Wherein;
Q is independently O, S
V is independently N, CH, C(F, Cl, Br, I), CCN,CCO2H, CO2R9, CCH3, CCF3
Y is selected from O, S, NH, CH2, CHF, CF2, C=CH2, C(OH)CH3, CFCH3;
The dashed and the solid lines combined
 represent an optional double bond;
represent an optional double bond;
R3 is selected from H, OH, NH2, NH-alkyl (C1-C22), NH-C(O)-alkyl(C1-C22);
R4 is selected from H, F, Cl, I, CF3, an optionally substituted alkyl C1-C3, cyclopropyl;
R5 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN,
alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
R6 is selected from, H, OH, halogen (F, Cl, Br, I), CH3, C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (= CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; Y with absent H8 is selected from C=CH, C=CF, C=CCH3
R9 is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I,
 monophosphate, diphosphate, triphosphate,
monophosphate, diphosphate, triphosphate,
Wherein:
X is O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl; R12 is alkyl, aryl, heteroaryl;
R13 and R14’ independently is H, CH3, CH2SR9’, CH2SC(O)R9’, CH2CH3, isobutyl, CH2CH2SMe, benzyl, pyrolinyl, CH2OR9’ , CH2OC(O)R9’ , CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CO2R9’, CH2CH2CO2R9’, CH2-imidazolyl;
Lipid is a C6-22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
Methods of Use
In certain embodiments, the disclosure relates to methods of treating or preventing a viral infection by administering in effective amount of a compound disclosed herein to a subject in need thereof.
In certain embodiments, the disclosure relates to methods to a method of treating and/or preventing an infection caused by a Coronaviridae virus that can include administering to a subject an effective amount of one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), or a pharmaceutical composition that includes a compound described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof).
In certain embodiments, the disclosure relates to using one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), in the manufacture of a medicine for preventing and/or treating an infection caused by a Coronaviridae virus that can include administering to a subject an effective amount of one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof).
Still other embodiments described herein relate to one or more compounds described herein (such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof), that can be used for ameliorating and/or treating an infection caused by a Coronaviridae virus by contacting a cell infected with the virus with an effective amount of said compound(s).
In certain embodiments, a Coronaviridae virus include, but not limited to, 2019-novel coronavirus (2019 nCoV), Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS coronavirus (SARS-CoV), and human coronavirus.
In certain embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a 2019-nCoV infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with 2019-nCoV and/or by contacting a cell infected with 2019-nCo V. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of 2019-nCo V. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against 2019-nCoV.
In certain embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a MERS-CoV infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with MERS-CoV and/or by contacting a cell infected with MERS-Co V. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of MERS-CoV. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against MERS-CoV.
In certain embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a SARS-CoV (SARS-CoV- 1, SARSCoV-2) infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with SARS-CoV
and/or by contacting a cell infected with SARS-CoV. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of MERS-Co V. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against SARS-CoV.
In certain embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a human coronavirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a human coronavirus and/or by contacting a cell infected with a human coronavirus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a human coronavirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against a human coronavirus.
In certain embodiments, the viral infection is, or is caused by, Respiratory syncytial virus (RSV). In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat Respiratory Syncytial Virus (RSV) infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with RSV and/or by contacting a cell infected with a RSV. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of RSV. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against RSV.
In certain embodiments, the viral infection is, or is caused by, Hepatitis B virus (HBV). In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat HBV infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with HBV and/or by contacting a cell infected with a HBV. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of HBV. In some embodiments, a
compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against HBV.
In certain embodiments, the viral infection is, or is caused by, influenza A virus including subtype H1N1, H1N2, H2N2, H2N3, H3N1, H3N2,H3N8, H5N1 (low bath), H5N1 (high bath), H5N2, H5N3, H5N6, H5N6, H5N8, H5N9, H6N1, H6N2, H7N1, H7N2, H7N3, H7N4, H7N7, H7N9, H9N2, H10N7, influenza B virus, and influenza C virus. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat an influenza A virus , influenza B virus, and influenza C virus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with an influenza A virus , influenza B virus, and influenza C virus and/or by contacting a cell infected with an influenza A virus , influenza B virus, and influenza C virus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of influenza A virus , influenza B virus, and influenza C virus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against an influenza A virus , influenza B virus, and influenza C virus.
In certain embodiments, the viral infection is, or is caused by, a flavivirus such as a Dengue virus, West Nile virus, Yellow fever virus, Japanese encephalitis virus, Powassen virus, Zika (ZIKA) virus, Usutu virus, hepatitis C virus (HCV). In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a flavivirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a flavivirus and/or by contacting a cell infected with a flavivirus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a flavivirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against a flavivirus.
In In certain embodiments, the viral infection is, or is caused by, an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus and/or by contacting a cell infected with an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against an Adenovirus, Measles virus, Ebola virus, Human Norovirus, Murine Norovirus, and Nipah virus. In certain embodiments, the viral infection is, or is caused by, a Picornaviridae viruses such as polio virus, Enterovirus-71, Enterovius-68, Coxsackie virus B3. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a Picornaviruse infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a Picornaviruse and/or by contacting a cell infected with a Picornaviruse. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a Picornaviruse. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against Picornaviruses.
In certain embodiments, the viral infection is, or is caused by, a Papovaviridae viruses such as BK virus, JC virus, and human Papillomavirus. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a Papovavirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a Papovavirus and/or by contacting a cell infected with a Papovavirus. In
some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a Papovavirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against Papovaviruses.
In certain embodiments, the viral infection is, or is caused by, a Togaviridae viruses such as Venezuelan equine encephalitis virus, Eastern equine encephalitis virus, and Chikungyunya virus. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a Togavivirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a Togavivirus and/or by contacting a cell infected with a Togavivirus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a Togavivirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against Togaviviruses.
In certain embodiments, the viral infection is, or is caused by, an Arenaviridae viruses such as Tacaribe virus, Pichinde virus, Junin virus, Lassa fever virus, and Lymphocytic choriomeningtis virus. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat an Arenavirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with an Arenavirus and/or by contacting a cell infected with an Arenavirus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of an Arenavirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against Arenaviruses.
In certain embodiments, the viral infection is, or is caused by, a Herpesviridae viruses such as Herpes simples viruse-1, Herpes simples viruse-2, Herpes simples viruse-6b, Herpes simples viruse-8, human cytomegalovirus, Murine cytomegalovirus, Varicella Zoster virus, Guinae pig cytomegalovirus, and Epstein-Barr virus. In still other embodiments, a compound described
herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a Herpesvirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a Herpesvirus and/or by contacting a cell infected with a Herpesvirus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a Herpesvirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against Herpesviruses.
In certain embodiments, the viral infection is, or is caused by, a Bunyaviridae viruses such as Rift fever virus, Punta Toto virus, La Crosse virus, Maporal virus, Heartland virus, Sever Fever Thrombocytopenia syndrome virus. In still other embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can prevent and/or treat a Bunyavirus infection. For example, by administering an effective amount of a compound of Formula (I), or a pharmaceutical acceptable salt thereof, to a subject infected with a Bunyavirus and/or by contacting a cell infected with a Bunyavirus. In some embodiments, a compound described herein (for example, a compound of Formula (I), or a pharmaceutical acceptable salt thereof) can inhibit replication of a Bunyavirus. In some embodiments, a compound of Formula (I), or a pharmaceutical acceptable salt thereof, can be effective against Bunyaviruses.
In certain embodiments, the subject is diagnosed with coronavirus, respiratory syncytial virus, hepatitis B, human immunodeficiency virus, eastern equine encephalitis virus, western equine encephalitis virus, California encephalitis virus, Japanese encephalitis virus, Rift Valley fever virus, hantavirus, Dengue virus serotypes 1, 2, 3 and 4, Zika virus, Junin, rabies virus, influenza B virus, influenza C virus, rotavirus A, rotavirus B, rotavirus C, rotavirus D, rotavirus E, human papillomavirus, parvovirus B1 9, molluscum contagiosum virus, JC virus, Merkel cell polyomavirus, Rubella virus, lymphocytic choriomeningitis virus, mumps virus, respiratory syncytial virus, parainfluenza viruses I and 3, rinderpest virus, chikungunya, ebola virus, marburg virus, herpes simplex virus-I, herpes simplex virus-2, varicella zoster virus, herpes
lymphotropic virus, roseolovirus, or Kaposi's sarcoma- associated herpesvirus, hepatitis A, hepatitis D, hepatitis E, human adenovirus types (HAdV-1 to 55).
In some embodiments, the compounds disclosed herein, such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition that includes a compound described herein, or a pharmaceutically acceptable salt thereof, can be used in combination with one or more additional agent(s) for treating, preventing and/or inhibiting a Coronaviridae, a Togaviridae, a Hepeviridae and/or a Bunyaviridae viral infection.
In certain embodiments, compounds disclosed herein can be administered alone or in combination with other the antiviral agent(s) such as but not limited to, acyclovir, adefovir, amprenavir, arbidol, atripla, abacavir, boceprevir, cidofovir, ampligen, combivir, amantadine, daclatasvir, danmavir, dasabuvir, delavirdine, didanosine, efavirenz, emtricitabine, enfuvirtide, entecavir, famciclovir, fomivirsen, fosamprenavir, pyramidine, foscamet, fosfonet, ganciclovir, ibacitabine, imunovir, idoxuridine, imiquimod, indinavir, inosine, interferon type III, interferon type II, interferon type I, lamivudine, tenofovir disoproxil, ledipasvir, docosanol.lopinavir, loviride, maraviroc, moroxydine, methisazone, nelfinavir, atazanavir, nevirapine, nexavir, ombitasvir, edoxudine, oseltamivir, paritaprevir, peginterferon alfa-2a, penciclovir, peramivir, pleconaril, podophyllotoxin , raltegravir, ribavirin, rimantadine, ritonavir, saquinavir, simeprevir, sofosbuvir, stavudine, telbivudine, tenofovir, tipranavir, trifluridine, trizivir, tromantadine, truvada, valaciclovir, valganciclovir, vicriviroc, telaprevir, vidarabine, viramidine zalcitabine, zanamivir, or zidovudine and combinations thereof.
In certain embodiments, a compound of formula (I) or a pharmaceutically accepted formulation is administered by inhalation through the lungs.
Specific active compounds
In certain exemplary embodiments, a pharmaceutical composition for treating or preventing a Coronavirus, influenza A viruses, an Influenza B viruses and RSV infection comprises an antiviral agent and an effective amount of a compound selected from the group consisting of but not limited to:

In certain exemplary embodiments, a pharmaceutical composition for treating or preventing an influenza A virus, an Influenza a B virus and RSV infection comprises an antiviral agent and an effective amount of a compound selected from the group consisting of but not limited to:
 In some exemplary embodiments, a pharmaceutical composition for treating or preventing a Coronaviridae virus, a Caliciviridae virus, an Orthomyxoviridae virus, a Herpesviridae virus, a Flaviviridae virus, a Filoviridae virus, and a Pneumoviridae virus infection comprises an antiviral agent with an effective amount of a compound selected from the group consisting of:
In some exemplary embodiments, a pharmaceutical composition for treating or preventing a Coronaviridae virus, a Caliciviridae virus, an Orthomyxoviridae virus, a Herpesviridae virus, a Flaviviridae virus, a Filoviridae virus, and a Pneumoviridae virus infection comprises an antiviral agent with an effective amount of a compound selected from the group consisting of:




















General Schemes for preparing active compounds.
Compounds of Formula (I) and those disclosed herein may be prepared in various ways. General synthetic routes to the compound of Formula (I) and some examples of starting materials used to synthesize the compounds of Formula (I) are shown in Scheme 1, 2, 3 and 4, and described herein. The routes shown and described herein are illustrative only and are not intended, nor are they to be interpreted, to limit the scope of the claims in any manner whatsoever. It will be understood by one of ordinary skill in the art that these schemes are in no way limiting and that variations of detail can be made without leaving from the spirit and scope of the present invention.
Methods for the facile preparation of 2-hydroxylimino nucleosides derivatives, modified monophosphate and phosphonates prodrugs analogs are provided. 2-Hydroxylimino nucleosides derivatives, modified monophosphate and phosphonates prodrugs analogs disclosed herein can be prepared as described in detail below, or by other methods known to those skilled in the art. Nucleoside 4 can be prepared by coupling of a pie-modified sugar 1 with a protected or silylated pyrimidine base 2 in the presence of a Lewis acid such as but not limited to SnCl4 or TMSOTf. Replacement of the TP group with hydroxylamine followed by deprotection of the sugar
 hydroxyl groups gives nucleosides 4 (Scheme 1).
hydroxyl groups gives nucleosides 4 (Scheme 1).
Scheme 1. Synthetic approach for the synthesis of nucleosides 4. Y, X, R4, R5, R6, R7, R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
An alternate synthesis of the nucleoside 4 can be achieved by the coupling of silylated 2- thiopyrimidine derivatives 2 with sugar 1 in the presence of a Lewis acid such as, but not limited
 to, SnCl4 or TMSOTf to give compound 3. Activation of the 2-position of compound 3 to the corresponding alkylthio derivative 3’, followed by treating with hydroxylamine or hydroxyl protected hydroxylamine and deprotection of the hydroxyl groups give compound 4 (Scheme 2).
to, SnCl4 or TMSOTf to give compound 3. Activation of the 2-position of compound 3 to the corresponding alkylthio derivative 3’, followed by treating with hydroxylamine or hydroxyl protected hydroxylamine and deprotection of the hydroxyl groups give compound 4 (Scheme 2).
Scheme 2. Alternate synthetic approach for the synthesis of nucleosides 4. Y, X, R4, R5, R6, R7, R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
2-Hydroxylimino nucleosides 4 can also be prepared from pre-modified nucleosides as described in Scheme 3. Protection of nucleosides 5 with a silyl reagent such as but not limited to tert- butyldimethylsily chloride (TBDMSCl) followed by selective deprotection of the 5 ’-hydroxyl group gives nucleoside 7. Applying Mistunobu reaction on nucleoside 7 gives 2,5’-anhydro nucleoside derivatives 8. Nucleoside 8 can also be prepared in many ways including, but not limited to tosylation of the 5 ‘-hydroxyl group followed by reacting the 5’tosyl derivative of compound 6 with a base such as, but not limited to, AgCO3. Reacting nucleosides 7 with NH2OH followed by deprotecting the hydroxyl groups give compound 4 (Scheme 3). An
alternate approach for the synthesis of nucleoside 4 is illustrated in scheme 4. Reacting compound 5 with NaSH· xH2O /Et3Ν· HCl give compound 3. Applying the same procedures as illustrated in scheme 1 give compound 4.

Scheme 3. Alternate synthetic approach for the synthesis of nucleosides 4. Y, X, R4, R5, R6, R7, R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
 Scheme 4. Alternate synthetic approach for the synthesis of nucleosides 4. Y, X, R4 , R5 , R6 , R7 , R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
Scheme 4. Alternate synthetic approach for the synthesis of nucleosides 4. Y, X, R4 , R5 , R6 , R7 , R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
Reacting nucleosides 9 with acyl chlorides provides nucleosides 10. Reacting nucleosides 9 with R9-N=C=O provide nucleoside derivatives 11 (Scheme 5).

Scheme 5. Synthetic approach for the synthesis of nucleosides 10 an11. Y, X, R4, R5, R6, R7, R8, R9, and R20 are as defined in Formula I, PG could be H or suitable protecting group.
The synthesis of the monophosphate prodrugs for nucleosides defined as in formula I is described in Scheme 6. Reacting nucleosides 9 with the phosphoramidite reagent 12 give nucleoside monophosphate prodrug derivatives 14 (Scheme 6). Alternatively, reacting the nucleoside derivative 9 with the phosphorchloridite derivative 13 give the nucleotide derivative
 14.
14.
Scheme 6. Synthetic approach for the synthesis of compounds 13. Y, X, R3, R4, R5, R6, R7, R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
Reacting nucleosides 9 with the phosphoramidite reagent 12 give nucleoside monophosphate prodrug derivatives 14 (Scheme 6). Alternatively, reacting the nucleoside derivative 9 with the phosphorchloridite derivative 13 give the nucleotide derivative 14.
The synthesis of the 4-substituted nucleosides defined as in formula I is described in Scheme 7.

Scheme 7. Synthetic approach for the synthesis of compounds 17. Y, X, R3, R4, R5, R6, R7, R8, and R20 are as defined in Formula I. PG could be H or suitable protecting group.
Example 1
General Methods: All chemical reactions were performed in oven-dried glassware under a Argon atmosphere, except where noted. Chemicals and solvents were reagent-grade and purchased from commercial suppliers (typically Aldrich, Fisher, Alfa-Aesar, Acres organics, Carbosynth Limited, Combi-blocks, Pharma-blocks and Oakwood Chemical) and used as received, excepting where noted. All reactions were monitored by thin layer chromatography (TLC), unless stated otherwise. TLC analysis was performed on silica gel, using visualization with a UV lamp (254 nm) or staining with 5% H2SO4 in ethanol and heating. 1H-NMR spectra were measured on a Broker 400 MHz spectrometer. Chemical shifts were measured relative to the appropriate solvent peak: CDCI3 (δ 7.27), DMSO-d6 (δ 2.50), CD3OD (δ 3.31), D2O (δ 4.79). The following abbreviations were used to describe signal splitting: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. 13C-NMR spectra were measured on a Broker
400 MHz spectrometer at 100 MHz with chemical shifts relative to the appropriate solvent peak: CDCI3 (δ 77.0), DMSO-d6 (δ 39.5), CD3OD (δ 49.0). 19F-NMR and 31P-NMR spectra were measured on Broker 400 MHz spectrometer at 376 MHz and at 162 MHz, respectively.
Example 2
Synthesis of 2-hydroxyimino nucleosides AH-01.

Reagents and conditions, a) NaOH, Mel, H2O ; b) BSA, DCE, 30 min, rt then SnCl4, rt; c) NH2OH- HCl, Pyridine, rt; d) NaOMe, MeOH;
Tl. 2-Thiouracil (10.0 g, 0.078 mol) was dissolved in an aqueous solution of sodium hydroxide (6.25 g 0.156 mol) in water (55 mL) and the reaction mixture was cooled to 0°C. Methyl iodide (5.56 mL, 0.087 mol) was slowly added to the reaction mixture and was then stirred for 20 hours at room temperature. The pale yellow solution was cooled to 0°C and acidified with glacial acetic acid. The precipitate was collected by vacuum filtration, washed with cold water (3 x 50 mL), and dried under vacuum to afford 2-methylthio-3H-pyrimidin-4-one as a white powder (11.1 g, 96 %). 1H-NMR (DMSO-d6) δ 12.71 (1H, s, NH), 7.84 (1H, d, H-6, J = 6.4 Hz), 6.05 (1H, d, H5, J= 6.4 Hz), 2.45 (3H, s, CH3); 13C-NMR (DMSO-d6) δ 163.83, 153.68, 109.39, 12.93.
T3. A mixture of l-O-acetyl-2,3,5-tri-O-benzoyl-D-ribose (5 g, 9.9 mmol) and 2-methylthio-3H- pyrimidin-4-one (1.41 g, 9.9 mmol) were dissolved in dry DCE (80 mL). BSA (1.96 mL, 7.92 mmol) was added and the reaction mixture was stirred for 1 hour at room temperature. The reaction mixture was cooled to -10 °C, and then SnCl4 (2.3 mL, 19.8 mmol) was added and the reaction mixture for 12 hours at room temperature. The reaction mixture was diluted with dichloromethane and poured into saturated solution of aqueous sodium bicarbonate. The mixture was filtered over Celite pad, and the filtrate was extracted with dichloromethane. The organic layer was separated dried over anhydrous sodium sulfate, filtered, and evaporated under vacuum. The residue was purified by silica gel column chromatography (eluate: 20% EtOAc in hexanes) to give T3 (5.69 g, 96% yield) as a colorless foam: IR: 1720 cm-1 (ν C=O), 1645 cm-1 (ν C=N) and 1088 cm-1 (ν C-N); 1H-NMR (CDCl3) δ 8.1 (2H, m, Ar), 7.99-7.93 (4H, m, Ar ), 7.66-7.64 (2H, m, Ar), 7.63-7.58 (2H, m, Ar), 7.58-7.52 (2H, m, Ar), 7.43-7.38 (4H, m, Ar,H-6) , 6.38 (1H, d, J = 5.6 Hz, H-1’), 5.98 (1H, d, J = 7.6 Hz, H-5), 5.86 (1H, dd, J = 6 ,3.6 Hz, H-2’), 5.71 (1H, t, J = 6 Hz, H-3’), 4.87 (1H, dd, J = 12.4, 2.6 Hz, H-5’), 4.77-4.76 (1H, m, H-4’), 4.71 (1H, dd, J = 12.4, 2.8 Hz, H-5’), 2.59 (3H, s, CH3), 13C-NMR (CDCl3) δ 167.64, 166.01, 165.27, 164.86, 162.58, 136.97, 134.11, 134.03), 134.00, 130.03, 129.86, 129.65, 129.03, 128.73, 128.49, 128.17, 110.55, 89.18, 81.27, 74.33,70.83, 63.40, 15.03.
T4. A mixture of T3 (2 g, 3.4 mmol) and hydroxylamine hydrochloride (4.78 g, 68.18 mmol) were dissolved in dry pyridine (25 mL) and the reaction mixture was stirred for 15 hours at 30- 35 °C. The solvent was evaporated and co-evaporated with toluene. The residue was partitioned between EtOAc and H2O, the organic phase was dried over Na2SO4, and evaporated. The residue was purified with column chromatography (eluate: 2% methanol in dichloromethane) to give T4 (1.55 g, 79.5 %) as a white solid: MP. 160-162 °C; UV-vis λmax (MeOH) 230 and 275 nm; IR 1717 cm-1 (νC=O), 1650 cm-1 (ν C=N, oxime), 1250 cm-1 (ν C-O), and 1093 cm-1 (ν C- N); 1HNMR (DMSO-d6) δ 9.63 (1H, s, NH exchangeable with D2O), 9.61 (1H, s, OH exchangeable with D2O), 7.99 (2H, d, J = 7.2 Hz, Ar), 7.87-7.62 (8H, m, Ar), 7.52-7.41 (6H, m, Ar - H-6), 6.06 (1H, d, H-1’, J = 3.6 Hz), 5.94 (1H, dd, H-2’, J = 6.4, 3.6 Hz,), 5.83 (1H, t, H-3', J = 6.4 Hz), 5.12 (1H, d, H-5, J = 8 Hz), 4.73- 4.65 (3H, m, H-5’ a,b and H-4’);13C-NMR
(DMSO-d 6) δ 166.23, 165.28, 161.67, 144.02, 142.72,134.66, 134.55, 134.31, 129.91, 129.82, 129.77, 129.53, 129.40, 129.32 , 128.84, 128.78, 96.82, 90.41 , 78.94, 73.28,70.99, 64.10.
AH-01. Sodium methoxide (1M, 8.26 mL) was added to a suspension of T4 (1 g, 1.75 mmol) in anhydrous methanol (20 mL) at 0 °C. The reaction mixture stirred for 2 hours at room temperature, then neutralized with glacial acetic acid. The solvents were evaporated under reduced pressure and the residue was purified with column chromatography (eluate: methanol: DCM, 15%) to give AH-01 (400 mg, 88 %), as a pale yellow solid, MP. 236-238 °C, IR: 2917 cm-1 (νOH), 1648 cm-1 (VC=N), 1262 cm-1 (νC-O) and 1091 cm-1 (νC-N); 1Η-NMR (DMSO-d6) δ 9.52 (1H, s, NH exchangeable with D2O), 9.31 (1H, s, OH exchangeable with D2O), 7.85 (1H, d, H-6, J = 8 Hz), 5.54 ( 1H, brs, H-1’), 5.26 (1H, d, 2’ -OH, exchangeable with D2O , J = 4 Hz), 5.08 (2H, brd, 3’-OH, exchangeable with D2O , and H5), 3.96 (2H, brs, H-2’ and H3’), 3.81 (1H, brs, 5’-OH, exchangeable with D2O), 3.69 (1H, d, H-4’), 3.56 (2H, d, Jgem = 10.8 Hz, H-5’a,b); 13C-NMR (DMSO-d6) δ 161.77, 145.13, 141.53, 95.44, 89.04, 84.11, 73.67, 69.33, 60.49.
Example 3

Reagents and conditions. a)TBDMSCl, Im., DMF; b) TIPSCl, Et3N, DMAP, CH3CN, then NH3; C) NaOMe, MeOH.
Synthesis of 2-hydroxyinimiocytidine AH-02
T5. A solution of T4 (1.2 g, 2.1 mmol), bn. (0.43 g, 6.3 mmol), and TBSCl (0.47 g, 3.15 mmol) in dry DMF (2 mL) was stirred for 2 h at rt under argon atmosphere. Ice-H20 (5 g) was added and the whole was partitioned between EtOAc (30 mL) and H2O (10 mL), washed with H2O (10
mL x 3 times), brine (10 mL), dried over MgSO4, filtered, and evaporated under reduced pressure. The residue was purified by a silica gel column chromatography (eluate: gradient of 0- 15% EtOAc in hexanes) to give T5 (1.3 g, 92% yield) as a colorless foam: ESI-MS m/z 686 [M+1]+.
T6. Triethylamine (0.46 mL, 3.2 mmol) was added to a solution of T5 (0.9 g, 1.3 mmol), TIPSCl (0.97 g, 3.2 mmol), and DMAP (0.93 g, 3.2 mmol) in dry CH3CN (10 mL) at 0 °C under argon atmosphere. The mixture was stirred at rt overnight. Ammonia gas was bubbled into the reaction mixture for 15 min. at 0 °C. The mixture was stirred at rt for 30 min. and the solvent was evaporated to dryness. The residue was partitioned between EtOAc and H2O. The organic phase was washed with H2O, brine, dried over MgSO4, filtered, and evaporated under reduced pressure. The residue was purified by a flash silica gel column (eluate: gradient of 0-7% MeOH in DCM) to give T6 (0.7 g, 79% yield) as a pale yellow foam: ESI-MS m/z 685.8 [M+1]+.
AH-02. Sodium methoxide (1M, 2.6 mL) was added to a suspension of T6 (0.6 g, 0.88 mmol) in anhydrous methanol (20 mL) at 0 °C. The reaction mixture stirred for 2 h at rt, then neutralized with glacial acetic acid. The solvent was evaporated under reduced pressure and the residue was purified with column chromatography (eluate: gradient of 5-15% methanol in DCM) to give AH- 02 (0.16 g, 71 %) as a pale yellow solid: ESI-MS m/z 259.1 [M+1]+.
Example 4.
Synthesis of 5 ’ -momophosphate conjugate AH-03

Reagents and conditions, a) t-BuMgCl, THF, rt.
AH-03. A solution of AH-01 (150 mg, 0.58 mmol) in anhydrous THE (3 mL) at 20 0°C was treated with a 1.7 M THF solution of tert-butylmagnesium chloride (0.68 mL, 0.31 mmol). After
1 h at 0°C, the mixture was treated dropwise with a solution of T7 (0.26 mg, 1.16 mmol) in anhydrous THE (3 mL) over a 5 min period. The mixture was allowed to warm to rt and was stirred for 48h at room temperature. The mixture was quenched with sat. aq. NH4CI (5 mL) and then extracted with ethyl acetate (60 mL). The organic phase was washed with sat. aq. NaHCO3 (2 x 15 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting crude yellow oil was purified by flash chromatography (eluate: 0-7%% gradient of MeOH in DCM) to give a AH-03 (140 mg, 46%) as an off-white solid as a mixture of two diastereomers: 31P-NMR (CDCI3) δ 3.36, 3.32; 13C-NMR (CDCl3) δ 173.75, 173.70, 161.30, 152.11, 152.08, 141.19, 141.09, 130.51125.59, 125.57, 121.17, 121.15, 96.69, 91.01, 90.92, 83.26, 83.2074.89, 74.81, 70.74, 70.67, 69.33, 69.29, 66.50, 66.45, 51.24, 51.15, 21.94, 21.85, 20.90, 20.85.
Example 5

Reagents and conditions, a) NaOEt, EtOH, rt; b) cone. H2SO4, acetone; c) Isobutyryl chloride, pyridine; d) TBAF, THE; e) 80% aq. HCO2H.
Synthesis of 5 ’-O-isobutyryl-2-hydroxyiminouridine AH-04.
T8. Sodium ethoxide (1M, 4.76 mL) was added to a suspension of T5 (3.0 g, 4.74 mmol) in anhydrous ethanol (50 mL) at 0 °C. The reaction mixture stirred for 12 h at rt, then neutralized with glacial acetic acid. The solvent was evaporated under reduced pressure and the residue was purified by a silica column chromatography (eluate: gradient of 5-10% methanol in DCM) to give T8 (1.34 g, 82 %) as a white solid: ESI-MS m/z 374.5 [M+1]+.
T9. A suspension of T8 (600 mg, 1.6 mmol) in dry acetone (20 mL) was treated with 1M co cone. H2SO4 in dry acetone (0.16 mL) and the mixture was stirred for 24 h at room temperature. The reaction mixture was neutralized with IN NH4OH and the solvents were evaporated under reduced pressure. The residue was purified with a silica column chromatography (eluate: gradient of 1-7% methanol in DCM) to give T9 (0.522 g, 79%) as a white solid: ESI-MS m/z 415.5 [M+1]+.
T10. A solution of T9 (0.5 g, 1.2 mmol) in dry pyridine (10 mL) was treated with isobutyryl chloride (0.18 mL, 1.8 mmol) at 0 C. The reaction mixture was stirred for 3h at rt and the solvent was removed under reduced pressure. The residue was partitioned between EtOAc and H2O. The organic phase was washed with H2O (10 mL), brine (10 mL) and dried over Na2SO4, filtered and evaporated. The residue was purified by a silica column chromatography (eluate: gradient of 10-20% EtOAc in hexanes) to give T10 (0.49 g, 85%) as a colorless foam: ESI-MS m/z 484.6 [M+1]+.
T11. A solution of T10 (0.4 g, 0.83 mmol) in THF (3 mL) was treated with 1M solution of TBAF (1 mL) and the mixture was stirred at room temperature for 1h . The solvent was removed under reduced pressure to give crude T11 which was used entirely in the next step without further purification.
AH-04. The crude T11 was dissolved in 80% aq. Formic acid (10 mL) and stirred at room temperature for 6 hours. The solvent was evaporated under reduced pressure and the residue was purified by a silica gel column chromatography (eluate: gradient of 1-7% MeOH in DCM) to give AH (0.0.17 g, 61%) as a white solid: ESI-MS m/z 330.1 [M+1]+.
Example 6

Reagents and conditions, a) BSA, DCE, then SnCl4, 40 °C; b) NH2OH.HCl, pyridine, rt; c) Synthesis of 2’ -C-methyl-2-hydroxyiminouridine AH-05.
T13. To a solution of T1 (1.1 g, 7.58 mmol) in DCE (15 mL) was added BSA (2.25 mL, 9.1 mmol) and the reaction was stirred for 30 min. at room temperature under argon atmosphere. A solution of T12 (4.0 g, 6.9 mmol) in DCE (15 mL) via a cannula. The reaction was cooled to 0 °C and SnCl4 (1.6 mL, 13. 8 mmol) was added dropwise. The reaction mixture was stirred for 12 hours at 40 °C then cooled to room temperature and diluted with DCM (50 mL). Cold saturated aqueous NaHCO3 (20 mL) was added dropwise to the reaction mixture and the whole was filtered over a Celite pad. The organic phase was separated and dried over anhydrous MgSO4 and filtered off. The filtrate was evaporated under reduced pressure to give T13 (3.9 g, 86%) as a colorless foam. 1Η-NMR (CDCI3) δ 8.07-7.29 (16, h, H-6 and Bz), 6.65 (1H, s, H1’), 5.97 (1H, d, H-5, J = 8.0 Hz), 6.66 (1H, H, H3’, J = 6.0 Hz), 4.90 (1H, dd, H5’a, J = 2.8, J = 12.4 Hz), 4.81 (iH, m, H4’), 4.74 (1H, dd, H5’b, J = 4.8, J = 12.4 Hz), 2.65 (3H, s, SMe), 1.70 (3H, s, 2’-CH3); 13C- NMR (CDCI3) δ 167.71, 166.22, 165.45, 165.31, 162.84, 138.46, 133.97, 130.08, 129.90, 129.74, 129.40, 129.35, 128.90, 128.72, 128.60, 128.36, 109.62, 91.17, 85.54, 80.52, 74.76, 62.60, 19.57, 15.37.
T13. A mixture of T13 (3.5 g, 5.8 mmol) and hydroxylamine hydrochloride (4.03 g, 58 mmol) was dissolved in dry pyridine (20 mL) and stirred for 48 hours at room temperature under argon atmosphere. The solvent was removed in vacuo and the residue was partitioned between EtOAc and H2O. The organic phase was washed with H2O, brine, dried over anhydrous MgSO4, filtered off, and evaporated under
reduced pressure. The residue was purified by a silica gel column (eluate; gradient 0-7% MeOH in DCM) to give T14 (3.0 g, 89% yield) as a white solid. 1Η-NMR (CDCI3) δ 9.74 (1H, s, NH), 9.62 (1H, s, NOH), 8.00-7.31 (16 h, m, H6, Bz), 6.41 (1H, s, HI’), 5.68 (1H, s, H3’), 5.16 (1H, d, H-5, J = 8.0 Hz), 4.73 (3H, m, H4’, H5’a, and H5’b), 1.71 (3H, s, 2’-CH3); 13C-NMR (CDCl3) δ 165.68, 164.86, 164.67, 160.72, 144.36, 134.00, 133.90, 133.76, 129.71, 129.48, 129.35, 128.9 4, 128.71, 128.52, 96.07, 85.12, 78.88, 75.40, 64.04, 48.73, 18.56.
AH-05. Sodium methoxide (1M, 15 mL) was added to a suspension of T14 (2.5 g, 4.27 mmol) in anhydrous methanol (15 mL) at 0 °C. The reaction mixture stirred for 12 h at rt, then neutralized with glacial acetic acid. The solvent was evaporated under reduced pressure and the residue was purified by a silica gel column chromatography (eluate: gradient of 5-12% methanol in DCM) to give AH-05 (1 g, 85%), as a white solid. 1H-ΝΜR (CD3OD) d 7.99 (1H, d, H-6, J = 8.4 Hz), 5.79 (1H, s, HI’), 5.19 (1H, d, H-5, J =8.4 Hz), 3.99-3.96 (1H, dd, H5’a, J = 2.0 , J = 12.4 Hz), 3.90-3.87 (1H, m, H-4’), 3.83 (1H, hr d, H-3’), 3.80-3.76 (1H, hr dd, H-5’b), 1.24 (3H, s, CH3).

Reagents and conditions, a) EtOH, reflux, 2 h; b) NaOAc. AcOH, reflux; c) i) BSA, DCE, rt, 30 min; ii) SnCl4, - 10 °C, 3 h, then NaCHO3, 0 °C, 0.5 h; d)DBU, Mel, DMF; e) NH2OH-HCl, dry pyridine, rt; f) NaOMe, MeOH, rt, 3 h.
Example 7.Synthesis of 6-aza-2-hydroxyiminouridine AH-06
T15. To a solution of thiosemicarbazide (10 g, 0.11 mole) in absolute ethanol (30 mL) was added ethyl glycosylate (40% in toluene, 28.1 mL). Then the solution was stirred for 2 hours at reflux temperature. The reaction mixture was cooled to room temperature and the precipitate was filtered off, dried under vacuum to give T15 (17.73 g, 92% yield) as a pale yellow solid: 1H- NMR (DMSO-d6 δ 11.89 (1H, s, NH), 8.64 (1H, s, NH), 7.83 (1H, S, NH), 7.42 (1H, s, N=CH), 4.20 (2H, q, ,CH2CH3 J = 7.2 Hz), 1.25 (3H, t , CH2CH3, J = 7.2 Hz); 13C-NMR (DMSO-d6) δ 179.76 (C=S), 163.13 (C=O), 132.39 (C=N), 61.24 (CH2CH3) 14.51 (CH3CH2).
T16 A solution of T15 (17 g, 0.097 mol) and anhydrous sodium acetate (63.67 g, 0.776 mol) in glacial acetic acid (80 mL) was heated for 3 h at 110 °C. The reaction mixture was cooled to room temperature and the solvent was evaporated under reduced pressure. The residue was taken in EtOAc (200 mL) and neutralized with cold saturated NaHCO3 solution. The organic phase was separated, dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The residue was purified by silica gel column chromatography (eluate: ethyl acetate) to give T16 (10.9 g, 87%) as a yellow powder: 1H-NMR (DMSO-d6) δ 13.52 (1H, s, NH), 13.16 (1H, s, NH), 7.69 (1H, s, H-5).
T17. To a suspension of T21 (3 g, 0.023 mol) and 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D- ribofuranose (11.05 g 0.022 mol) in dry DCE (300 mL) was added BSA (6.75 mL, 0.0276 mol) drop wise at room temperature. The solution was stirred for 30 minutes at room temperature until complete dissolution of the solid material. Then, SnCl4 (3.86 mL, 0.033 mol) was added drop wise at -10 °C. The reaction mixture was stirred for 3 hours at 0 °C, then wormed up to room temperature and stirred for further three hours. The reaction mixture was diluted with DCM 100 mL) and neutralized with saturated sodium bicarbonate solution. The produced white cake was filtered over a Celite pad, thoroughly washed with DCM. The organic phase was separated, dried over anhydrous Na2SO4, and evaporated under vacuum. The residue was purified with silica gel column chromatography (eluate: 20% ethyl acetate / DCM) to give T17 (10.73 g, 85%) as an off-white solid: 1Η-NMR (DMSO - d6) δ 13.49 (1H, s, NH), 7.95-7.39 (17H, m, Bz, H-5, and H-1’), 6.00 (1H, dd, J = 2.4 Hz, H-2’), 5.90 (1H, dd, H-3’, J = 6.8 Hz),
4.84 (1H, m, H-4’), 4.72-4.68 (1H , dd, H-5'a, J = 3.6, 12.0 Hz), 4.53 (1H, dd, H-5’b ,J = 4.8, 12.0
Hz).
T18. To a solution of T17 (3.8 g, 6.63 mmol) in dry DMF (35 mL) was added Mel (1.03 mL, 16.58 mmol) and the mixture was cooled down to 0 °C. The reaction mixture was treated with DBU (1.48 mL, 9.95 mmol) and stirred for further 1h at 0 °C under argon atmosphere. The reaction mixture was diluted with EtOAc (100 mL) and washed with H20 (30 mL x 3 times). The organic phase was separated, was separated, dried over anhydrous Na2SO4, and evaporated under vacuum. The residue was purified with silica gel column chromatography (eluate: 15% ethyl acetate / DCM) to give T18 (3.56 g, 91.5%) as a colorless foam:1H-NMR (CDCI3) δ 8.06- 7.38 (16H, m, Bz and H5), 6.29 (1H, d, H-1’, J = 2.4 Hz), 6.09 (1H, dd, H-2’, J = 2.4 Hz), 5.99 (1H, dd, H-3’, J= 6.0 Hz), 4.88-4.80 (2H, m, H4’ and H5’a), 4.56-4.52 (1H, dd, H5’b, J = 4.0, J = 12.4 Hz), 2.60 (3h, s, S-Me).
T19 A mixture of T18 (6.15 g, 10.73 mmol) and hydroxylamine hydrochloride (14.92 g, 0.214 mol) in dry pyridine (80 mL) was stirred over night at 45 °C. The solvent was evaporated under reduced pressure, co-evaporated with toluene, and the residue was partitioned between EtOAc and H2O. The organic phase was separated, dried over anhydrous Na2SO4, and evaporated under vacuum. The residue was purified with silica gel column chromatography (eluate: 25% ethyl acetate / DCM) to give T19 (4.85 g, 79%) as pale yellow solid: 1Η-NMR (DMSO-d6) δ 10.72 (1H, s ,OH, exchangeable with D2O), 9.71 (1H, s ,NH ,exchangeable with D2O), 7.99 -7.43 (15H, m, Bz), 6.91 (1H, s, H-5), 6.23 (1H, hr d, H-1’ J = 3.2 Hz), 6.01 (1H, dd, H-2’, J = 2.8, 5.6 Hz), 5.85 (1H, hr dd, H-3’, J = 5.6Hz), 4.75-4.73 (1H, m, H-4’), 4.68 (1H, dd, H-5’a, J = 3.6, 12.0 Hz), 4.54 (1H, dd, H-5’b, J = 4.4, J = 12.0 Hz).
AH-06. A solution of T19 (1.39 g, 2.42 mmol) in dry methanol (15 mL) was treated with sodium methoxide (1M, 7.3 mL) at 0 °C, then the mixture was stirred for 1 hours at room temperature. The reaction mixture was neutralized with acetic acid, and then the solvents were evaporated. The residue was purified by silica gel column chromatography (eluate: 8: 1:1:1 ethyl acetate/acetone/ethanol/H2O) to give AH-06 (0.56 g, 90.3% yield) as a pale yellow solid:
1H-NMR (DMSO-d6) δ 10.87(1H, s, NH, exchangeable with D2O), 9.63 (1H, s, N=OH, exchangeable with D2O), 6.97 (1H, s, H-5), 5.69 (1H, hr d, H-1’, J = 2.8 Hz), 5.20 (1H, d, OH-2’, exchangeable with D2O, J = 5.2 Hz), 4.97 (1H, d, 3 ’-OH, exchangeable with D2O, J = 5.6 Hz), 4.67 (1H, hr t, 5’-OH, exchangeable with D2O), 4.22 (1H, m, H-2’), 4.09 (1H, m, H-3’), 3.94 (1H, m, H-4'), 3.50-3.37 (2H, m, H-5'a,b);
13C-NMR (DMSO-d6) δ 153.48, 143.18, 129.83, 90.34, 84.26, 71.80, 70.22, 62.05, 48.58
Example 8
Synthesis of 5’-momophosphate conjugate AH-07

Reagents and conditions, a) t-BuMgCl, THF, rt.
AH-07. A solution of AH-06 (0.13 g, 0.498 mmol) in anhydrous THF (1.5 mL) was treated with a 2 M THF solution of tert-butylmagnesium chloride (0.62 mL, 1.05 mmol) at 0°C and the mixture was stirred for 1h at 0°C. Next, the mixture was treated dropwise with a solution of T7 (0.405 g, 1.0 mmol) in anhydrous THF (1.5 mL) over a 5 min period. The mixture was allowed to warm to rt and was stirred for 4 days at room temperature. The mixture was quenched with sat. aq. NH4CI (10 mL) and then extracted with ethyl acetate (50 mL). The organic phase was washed with sat. aq. NaHCO3 (2 x 15 mL), dried over Na2SO4, filtered and concentrated to dryness. The resulting crude yellow oil was purified by flash silica gel column chromatography (eluate; 0-3% gradient of MeOH in DCM) to give AH-09 (65 mg, 25%) as pale yellow syrup: 31P-NMR (CDCI3) δ 3.07, 2.96.
Example 9

Reagents and conditions, (a) KOH, CH3I, H20; (b) BSA, DCE, 1h , rt, then SnCl4, 12 h, rt;
 pyridine, rt; (d) NaOMe, MeOH, rt.
pyridine, rt; (d) NaOMe, MeOH, rt.
Synthesis of 2-hydroxyimino-5 -methyluridines AH-12 and AH-13.
T29. S-Methyl-2-thiouracil (5.0 g, 35 mmol) was dissolved in an aqueous solution of sodium hydroxide (2.1 g, 52.5 mmol) in water (25 mL) and the reaction mixture was cooled to 0°C. Methyl iodide (2.4 mL, 38.5 mol) was slowly added to the reaction mixture and was then stirred for 20 h at room temperature. The pale yellow solution was cooled to 0°C and acidified with glacial acetic acid. The precipitate was collected by vacuum filtration, washed with cold water (3 x 20 mL), and dried under vacuum to afford T29 (4.97 g, 91 %). as a white powder: 1Η-NMR (DMSO-d6) δ 7.73(1H, s, H-6), 6.05 (1H, d, H5, J = 6.4 Hz), 2.42(3H, s, CH3), 1.85 (3H, S, 5- CH3); 13C-NMR (DMSO-d6) δ 163.65, 160.08, 150.60, 119.22, 13,21, 12.98.
T30. A mixture of T2 (9.23 g, 18.29 mmol) and T29 (3 g, 19.2 mmol) were dissolved in dry DCE (100 mL). BSA (4.7 mL, 19.2 mmol) was added and the reaction mixture was stirred for 1 hour at room temperature. The reaction mixture was cooled to -10 °C, and then SnCl4 (3.2 mL, 27.44 mmol) was added and the reaction mixture for 12 hours at room temperature. The reaction mixture was diluted with DCM (100 mL) and poured into saturated solution of aqueous sodium bicarbonate. The mixture was filtered over Celite pad, and the filtrate was extracted with dichloromethane. The organic layer was separated dried over anhydrous sodium sulfate, filtered, and evaporated under vacuum. The residue was purified by silica gel column chromatography (eluate: 20% EtOAc in hexanes) to give T30 (10 g, 82% yield) as a colorless foam: 1H-NMR
(CDCl3) δ 8.1 (2H, m, Ar), 7.98-7.94 (4H, m, Ar ), 7.66-7.38 (10H, m, Ar and H6), 6.41(1H, d, H-1’, J = 6.8 Hz), 5.89 (1H, dd, H-2’, J = 3.2, J = 6.0 Hz,), 5.74 (1H, dr dd, H-3’), 4.91 (1H, dd, , H-5’a, J = 2.4, J = 12.4 Hz), 4.77-4.75 (1H, m, H-4’), 4.72 - 4.68 (1H, dd, H5’b, J = 2.6, J = 12.4 Hz), 2.60 (3H, s, SCH3), 1.16 (3H, s, 5-CH3); 13C-NMR (CDCl3) δ 168.71, 185.85, 165.22, 164.82, 161.70, 158.02, 133.95, 133.92, 133.89, 133.29, 129.90, 129.80129.76, 129.73, 129.54, 128.98, 128.89128.83, 128.61, 128.58, 128.43, 128.40, 128.32, 119.72, 88.76, 81.45, 74.08, 71.19, 63.60, 15.07, 13.45.
T31. A mixture of T30 (1.5 g, 2.50 mmol) and hydroxylamine hydrochloride (3.5 g, 68.18 mmol) were dissolved in dry pyridine (25 mL) and the reaction mixture was stirred for 15 hours at 45 °C. The solvent was evaporated and co-evaporated with toluene. The residue was partitioned between EtOAc and H2O, the organic phase was dried over Na2SO4, and evaporated. The residue was purified with column chromatography (eluate: 20% EtOAc in DCM) to give T31 (1.1 g, 74 %) as an off-white: 1HNMR (CDCl3) δ 8.12-7.92 (6H, m, Ar), 7.63-7.32 (9H, m, Ar), 7.11 (1H, d, H-6, J = 1.2 Hz), 6.16 (1H, d, H-1’, J = 4.4 Hz), 5.88-5.83 (2H, m, H-2’, and H- 3’), 4.87 (1H, dd, H5’a, J = 2.8, J = 12.4 Hz), 4.69 (1H, m, H-4’); 4.61 (1H, dd, H5’b, J = 2.8, J = 12.4 Hz); 13C-NMR (CDCI3) δ 166.03, 165.39, 165.24, 161.00, 146.79, 134.86, 133.79, 133.76, 133.73, 133.57, 129.23, 88.07, 80.05, 73.25, 70.63, 63.37, 12.17.
AH-12. Sodium methoxide (1M, 6.2 mL) was added to a suspension of T31 (1.2 g, 2.05 mmol) in anhydrous methanol (20 mL) at 0 °C. The reaction mixture stirred for 2 hours at room temperature, then neutralized with glacial acetic acid. The solvents were evaporated under reduced pressure and the residue was purified with column chromatography (eluate: EtOAc: acetone: EtOH: H2O 8: 1:1:1) to give AH-12 (448 mg, 80 %) as a pale yellow solid: 1Η-NMR (DMSO-d6+ D2O) δ 7.58 (1H, s, H-6), 5.46 ( 1H, hr d, H-1’, J = 4.4 Hz), 4.04 (1H, m, H2’), 3.96 (1H, m, H3’), 3.84 (1H, m, H-4’), 3.70-3.67 (1H, hr dd, H-5’a), 3.58-3.54 (1H, hr dd, H5’-b), 1.69 (3H, s, 5-CH3); 13C-NMR (DMSO-d6) δ 162.09, 145.80, 137.33, 102.63, 88.88, 84.13, 73.67, 69.43, 60.62, 12.77.
Example 10

Reagents and conditions, (a) BSA, DCE, then SnCl4; (b) NH3/MeOH; c) cone. H2SO4, acetone; d) i) TsCl, DCE, DMAP, rt; ii) DBU, CH3CN; e) NH2OH+HCl, DIEA, CH3CN, rt; f) aq. HCOOH.
General synthetic route for the synthesis of substituted-2-hydroxyiminouridine derivatives.
Example 11

Reagents and conditions, (a) BSA, DCE, 1h , rt, then SnCl4, 12h, rt; (b) NH3/MeOH, rt; c) cone. H2SO4, acetone, rt; d) i) TsCl, DCE, DMAP, rt, ii) AgOAc, DMF, rt; e) NH2OH+HCI, DIEA, CH3CN, rt; f) aq. HCOOH, rt.
Synthesis of 2-hydroxyimino-5-fluorouridine, AH-13.
Example 12

Reagents and conditions, a) NaOH, Mel, H2O, rt; b) BSA, DCE, 1h , rt, then SnCl4, rt; c) NH2OH+HCI, pyridine, rt; d) NaOMe, MeOH, rt.
Synthesis of 2-hydroxyimino-6-methyluridine nucleoside analog T47.

Reagents and conditions, a) TBSCl, Im, DMF, rt; b) aq. TFA, 0 °C, c) i) TsCl, pyridine, ii) AgOAc, DMF; NH2OH+HCl, DIEA, rt; d) TBAF, THF, rt.
Example 13
Synthesis of 2’ -deoxy-2’ -fluoro-2-hydroxyiminouridine
Reagents and conditions, a) TBSCl, Im, DMF, rt; b) aq. TFA, 0 °C, c) i) TsCl, pyridine, ii) AgOAc, DMF; NH2OH+HCl, DIEA, rt; d) TBAF, THF, rt.
Example 14
Synthesis of 2’ -deoxy-2’ -fluoro2’ -C-methyl-2-hydroxyiminouridine
Example 15

Reagents and conditions, a) TBSCl, Im, DMF, rt; b) aq. TFA, 0 °C, c) i) TsCl, pyridine, ii)

Synthesis of 2’ -deoxy-2’ -gem-difluoro-2-hydroxyiminouridine
Example 16 General synthetic route for the synthesis of 5-substituted-3 ’ -deoxy-2-

X = F, Cl, Br, I, CN, CH3, CF3, CCH, CH=CH2, CH=CHBr
Reagents and conditions, a) BSA, DCE, 1h , rt, then SnCl4, rt; b) NaOMe, MeOH, rt; c) i)TBSCl, Im., DMF, rt; ii)50% aq. TFA, 0 °C; d) i) TsCl, pyridine, ii)AgOAc, DMF; e) NH2OH.HCI, DIEA, rt; f) TBAF, THF, rt. ydroxyiminouridines.
Example 17

Reagents and conditions, a) i) Ac2O, CH3CN, reflux; ii) AcBr, CH3CN; iii) H2P(O)OH, Et3N, CaCO3, H2O, CH3CN; b) TBSCl, Im, DMF, rt; c) aq. TFA, 0 °C, d) i) TsCl, DMAP, DCM, ii) AgOAc, DMF; e) NH2OH+HCl, DIE A, rt; f) TBAF, THF, rt.
Alternative synthetic route for 3 ’ -deoxy-2-hydroxyiminouridine nucleoside analogs
Example 18
Synthesis of 6-aza-2’ -C-methyl-2-hydoxyiminouridine nucleoside analog, AH-14.
Tie

Reagents and conditions, a) NaOH, Mel, H2O, rt; b) BSA, DCE, lh , rt, then SnCl4, rt; c) NH2OH+HCl, pyridine, rt; d) NaOMe, MeOH, rt.
T73 A solution of 6-aza-2-thiouracil (5.6 g, 0.043 mol) in 1 N NaOH (48 mL) was treated with CH3I (4.08 mL, 0.066 mol) at 0 °C. The reaction mixture was stirred for 14 h at rt and the formed precipitate was filtered off and washed with cold H2O. The precipitate was dried under reduced pressure to give T73 (5.35 g, 87%) as pale yellow solid: 1Η-NMR (DMSO-d6) d 13.97 (1H, br s, NH), 7.58 (1H, s, H5), 2.47 (3H, s, SCH3).
T74 To a solution of T73 (0.52 g, 2.98 mmol) in DCE (10 mL) was added BSA (0.59 mL, 2.38 mmol) and the reaction was stirred for 30 min. at room temperature under argon atmosphere. A solution of T12 (1.73 g, 2.97 mmol) in DCE (10 mL) via a cannula. The reaction was cooled to 0 °C and SnCl4 (0.6 mL, 8.91 mmol) was added dropwise. The reaction mixture was stirred for 8 hours at 40 °C then cooled to room temperature and diluted with DCM (50 mL). Cold saturated aqueous NaHCO3 (20 mL) was added dropwise to the reaction mixture and the whole was filtered over a Celite pad. The organic phase was separated and dried over anhydrous MgSO4 and filtered off. The filtrate was evaporated under reduced pressure. The residue was purified by a silica gel column (eluate: gradient from 10-20% EtOAc in hexanes to give T74 (1.55 g, 82%) as a colorless foam: 1Η-ΝΜR (CDCl3) δ 8.16-7.33 (16, h, H-5 and Bz), 6.89 (1H, s, H1’), 6.13 (1H, d, H- 3’, J = 8.8 Hz), 4.84-4.79 (2H, m, H4’ and H5’a), 4.59-4.54 (1H, m, H5’b), 2.632 (3H, s, SMe), 1.74 (3H, s, 2’-CH3); 13C-NMR (CDCI3) δ 167.08, 166.17, 165.50, 165.30, 158.87, 151.54, 134.00, 130.03, 129.93, 129.78, 129.57, 128.87, 128.49, 91.86, 87.66, 79.00, 76.08, 63.82, 17.20, 14.83.
T75 A mixture of T74 (1.5 g, 2.37 mmol) and hydroxylamine hydrochloride (3.3 g, 47.4 mmol) was dissolved in dry pyridine (25 mL). The reaction mixture was stirred for 48 hours at 40 °C. Then pyridine was evaporated and co-evaporated with toluene. The residue was purified by silica gel column chromatography (eluate: 10% EtOAc/DCM) to give T75 (1.17 g, 84% yield) as a pale yellow foam: 1Η-NMR (CDCl3) δ 8.69 (1H, s ,NH), 8.13 -7.31 (15H, m, Bz), 6.94 (1H, s, H-5), 6.87 (1H, s, H-1’), 6.29 (1H, br s, OH), 6.04 (1H, d, H-3’, J = 8.8 Hz), 4.78-4.48 (3H, m, H-4', H5’a, and H5’b), 1.81 (3H, s, CH3); 13C-NMR (CDCl3) δ 166.32, 165.68, 165.18, 152.01, 144.56, 133.89, 133.75, 133.32, 131.11, 130.42, 130.07, 129.95, 129.86, 129.76, 129.13, 128.76, 128.73, 128.46, 88.74, 87.79, 78.20, 76..66, 64.30, 17.34
AH-14. Sodium methoxide (1M, 5.1 mL) was added to a suspension of T75 (1.0 g, 1.7 mmol) in anhydrous methanol (15 mL) at 0 °C. The reaction mixture stirred for 2 hours at room temperature, then neutralized with glacial acetic acid. The solvents were evaporated under reduced pressure and the residue was purified with column chromatography (eluate: EtOAc: acetone: EtOH: H2O 8: 1:1:1) to give AH-14 (0.39 g, 85 %) as a pale yellow solid: 13C-NMR (CD3COCD3) δ 156.77, 149.02, 137.05, 92.66, 84.99, 79.93, 75.22, 63.43, 20.15.
Example 19

Reagents and conditions, (a) cone. H2SO4, acetone, rt; (b) I2, TTP, THF; (c) NaOMe, MeOH, reflux; d) AgF, I2, DCM; (e) AgOAc, DMF, rt; (f) NH2OH+HCl, DIEA, CH3CN, rt; f) aq. HCOOH, rt.
Synthetic route for 4’ -fluoro-2-hydroxyiminouridine, AH 15.
Example 20

Reagents and conditions, (a) i) I2, Im., TPP, THF; ii) NaOMe, MeOH, reflux; (b) i) BTEAN3/MeCN, I2, THF, NaS2O3; ii) BzCl, NMM, DMAP, MeCN; c) AgOAc, DMF; (e) NaOMe, MeOH.
Synthetic route for 4’-azido-2-hydroxyiminouridine, T85.
Example 21

Reagents and conditions, (a) cone. H2SO4, acetone, rt; (b) i) TsCl, DMAP, DCM, ii) AgOAc, DMF; c) NH2OH+HCI, DIEA, rt; d) aq. HCOOH, rt.
Synthetic route for 1 ’-cyano2-hydroxyiminouridine, T90.
Compound T86 was prepared according to reported procedure (Tetrahedron 1993, pp. 8579- 8588).
Example 22

Reagents and conditions, (a) i) TMM, p-MeC6H4SO3H, DMF; ii) DMSO, TFA, Pyridine, EDC, DMF; iii) Et3N, DMF, rt; iv) NaBH4, MeOH; v), MeNH2, EtOH; (b) NaNO2, AcOH; (c) i) TBSCl, Im., DMF; ii) aq. TFA, THF; d) ) i) TsCl, DMAP, DCM, ii) AgOAc, DMF; e) i) NH2OH+HCl, DIEA, rt; ii) TBAF, THF, rt; f) t-BuMgCl, THF, rt. g) i) NaSH, Et3N- HCl, DMF; ii) TBAF, THF.
Synthesis of 3 ’,4’ -didehydro-2-hydroxyimino-nucleosides
Example 23

Reagents and conditions, a) TBSCl, Im., DMF, rt; b) i) TPP, DIAD, Toluene; ii) NaBH4, (PhSe)2, EtOH, rt; c) m-CPBA, DCM; d) TBAF, THF; e) T7, t-BuMgCl, THF, rt; f, TIPSCl, DMAP, Et3N, CH3CN, then NH3.
Synthesis of 3’, 4’ -didehydro-nucleosides; T103, T104, T106, and T107.
Example 24

Reagents and conditions, (a) NaOH, CH3I, rt; (b) CsCO3, DMF, rt; (c) NH2OH+HCl, Pyridine, 48 h, 40 °C; (d) NaOMe, MeOH, rt.
Synthesis of 2-hydoxyimino-acyclic nucleosides T112
T108. Methyl iodide (3.96 mL) was added to a solution of compound T21 (1.g, 6.36 mmol) in dry DMF (20 mL) and the mixture was cooled to 0 °C. Then, DBU (1.25 mL, 8.45 mmol) was
added drop wise to the reaction mixture at 0 °C. The reaction mixture was stirred for 3 hours at 0 °C. The solvent was evaporated under reduced pressure and the residue was dissolved in EtOAc and with H2O and brine. The combined organic phases were dried over anhydrous (Na2SO4), and filtered off. The filtrate was evaporated under vacuum and the residue was purified by silica el column chromatography (eluate: 3% methanol / DCM) to give compound T108 (0.87 g, 79.3% yield) as a pale white solid; MP = 224-226 °C; UV-vis (MeOH) λmax 235 nm; IR v 2635.12 (, 1727.49, 1602.07, and 1583.08 cm1; 1Η-NMR (DMSO-d6) δ 13.68 (1H, s, NH), 2.47 (3H, s, S- CH3), 2.10 (3H, s, 5-CH3); 13C-NMR (DMSO-d6) δ 164.26, 160.93, 149.68, 16.66, 12.06.
T110. 2-(Bromomethoxy)ethyl acetate (1.128 g, 5.71 mmol) was added dropwise to a solution of T108 (0.3 g, 1.1 mmol) and CsCO3 (1.24 g, 3.82 mmol) in DMF (13 mL) at room temperature under argon atmosphere. The reaction mixture was stirred for 3 days at the same temperature. The solvent was evaporated under vacuum and the residue was partitioned between EtOAc and H2O. The combined organic phases were dried over anhydrous (Na2SO4), and filtered off. The filtrate was evaporated under vacuum and the residue was purified by silica gel column chromatography (eluate: 2.5% EtOAc/DCM) to give T110 (0.15 mg, 28.9% yield) as a white solid: UV-vis (MeOH) λmax 225, 240 nm (shoulder); 1Η-NMR (CDCl3) δ 5.39 (2H, s, 1’-CH2), 4.22 (2H, m, 4’-CH2), 3.82 (2H, m, 3’-CH2), 2.59 (3H, s, 2-SCH3), 2.10 (3H,s, 5-CH3), 2.06 (3H, s, Ac-CH3); 13CNMR (CDCl3) δ 170.95, 166.74 , 160.70, 150.23, 84.07, 67.83, 62.95, 21, 17.7, 14.18.
T111. A mixture of T110 (0.14 g, 0.51 mmol) and NH2OH-HCl (0.71 g, 10.2 mmol) was dissolved in dry pyridine (5 mL). The reaction mixture was stirred for 48 hours at 40 °C. The solvent was evaporated and co-evaporated with toluene. The residue was purified by silica gel column chromatography (eluate: 1.5% MeOH / DCM).to give compound T111 (97 mg, 73.7% yield) as a pale yellow solid: MP =138.5-140.5 °C; UV-vis (MeOH) λmax 330, 230 nm (shoulder); IR: 3336.96, 3140.49, 1692.80, 1659.09 cm-1; 1Η-NMR (DMSO-d6) δ 10.83 (1H, s, NH), 9.45 (1H, s, OH), 5.05 (2H, s, CH2-1’), 4.09 (2H, dd, CH2-4’ J = 4.4, J = 9.2 Hz), 3.73 (2H, dd, 3’- CH2 J = 4.4, 9.2 Hz), 1.99 (3H, s, Ac-CH3), 1.97 (3H, s, 5-CH3); 13C-NMR (DMSO-d6) δ 170.29, 154.14, 143.71, 136.71, 79.93, 66.77, 63.04, 20.63, 15.76.
T112. A solution of T111 (40 mg, 0.16 mmol) in dry methanol (5 mL) was treated with NaOCH3 (1M, 1.1 mL) at 0 °C, then the mixture was stirred for 2 hours at room temperature. The reaction mixture was neutralized with acetic acid, and then the solvents were evaporated. The residue was purified by silica gel column chromatography (eluate: 5% MEOH / DCM) to give T112 (24 mg, 72% yield) as a pale yellow semisolid; UV-vis (MeOH) λmax 330, 230 nm (shoulder); 1Η-NMR (DMSO-d6) δ 10.82 (1H, s, NH), 9.42 (1H, s, OH), 5.04 (2H, s, 1’-CH2), 4.63 (1H, s, OH), 3.54 (2H, m, 3’-CH2), 3.47 (2H , m, 4’-CH2), 1.97 (3H, s, 5-CH3); 13C-NMR (DMSO-d6): 154.33, 144.03, 136.69, 80.11, 78.79, 70.75, 15.77.
Example 25
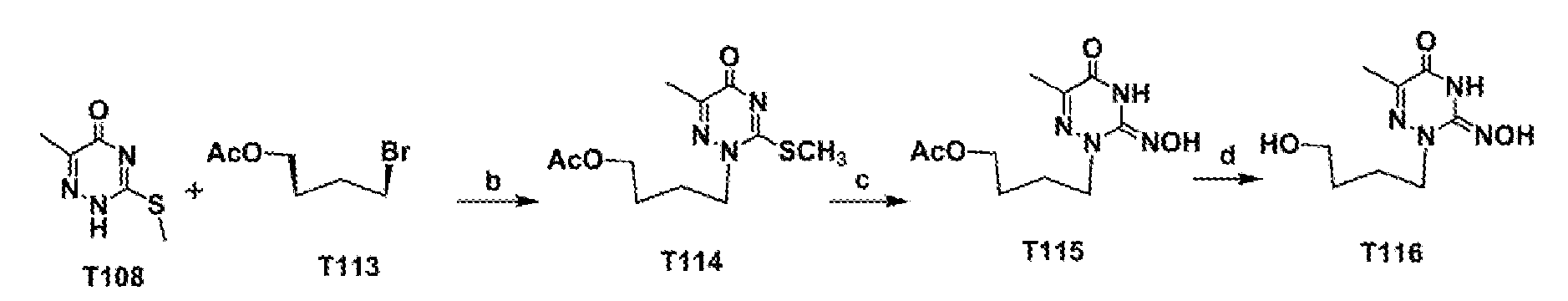
Reagents and conditions, (a) LiHMSD, THF, reflux; (c) NH2OH+HCl, Pyridine, 48 h, 40 °C; (d) NaOMe, MeOH, rt.
Synthesis of 2-hydoxyimino-acyclic nucleosides T116.
T114. A solution of T108 (0.15 g, 0.95 mmol) in dry THF (7 mL) was treated with LiHMDS (1M, 1.1 mL) at rt and the mixture was further stirred for 30 minute at the same temperature. 4- Bromobutyl acetate (0.74 g, 3.82 mmol) was added dropwise at rt then the mixture was stirred overnight at 80 °C under argon atmosphere. The mixture was cooled to rt and then aq. NH4CI (1 mL) was added. The whole was partitioned between EtOAc water. The organic phase work washed with water, brine, and separated. The combined organic phases were dried over anhydrous (Na2SO4), and filtered off. The filtrate was evaporated under vacuum and the residue was purified by silica gel column chromatography (eluate: 12% EtoAc / hexanes) to give compound T114 (112 mg, 44.6 % yield) as a white solid: UV-vis (MeOH) 235 nm; 1H- NMR (CDCI3) δ 4.11 (2H, t, 4’-CH2, J = 6.4 Hz), 4.01 (2H, t, 1’-CH2, J = 7.2 Hz), 2.58 (3H, s,
S-CH3), 2.28 (3H, s, 5-CH3), 2.06 (3H, s, Ac), 1.91 (2H, m, H-2’), 1.70 (2H, m, 3’-CH2); 13C- NMR (CDCl3) δ 171.15, 164.89, 161.03, 150.78, 63.66, 54.63, 26.01, 25.01, 21.09, 17.14; 14.27.
T115. A mixture of compound T114 (90 mg, 0.33 mmol) and NH2OH-HCl (0.47g, 6.63 mmol) was dissolved in dry pyridine (3 mL). The reaction mixture was stirred for 48 hours at 40 °C. Then pyridine was evaporated and co-evaporated with toluene. The residue was purified by silica gel column chromatography (eluate: 2% MeOH / DCM).to give compound T115 (85 mg, 97.1% yield) as a canary yellow solid: MP =127-130 °C; UV-vis (MeOH) λmax340, 235 nm (shoulder); 1H-NMR (DMSO-d6) δ 10.66 (1H, s, NH, exchangeable with D2O), 9.33 (1H, s, OH, exchangeable with D2O), 3.99 (2H, t, 4’-CH2, J = 6.4, HZ), 3.66 (2H, t, H-1’, J = 7.2 Hz), 1.98 (3H, s, CH3-AC), 1.95 (3H, s, 5-CH3), 1.69-1.55 (4H, m, 2’-CH2 and 3’-CH2);.13C-NMR (DMSO-d6) δ 171.01, 161.01,154.14, 143.71, 36.86, 50.81, 24.01, 23.01, 21.01, 16.03.
T116. A solution of compound T115 (75 mg, 0.29 mmol) in dry methanol (3 mL) was treated with NaOCH3 (1M, 2.06 mL) at 0 °C, then the mixture was stirred for 3 hours at room temperature. The reaction mixture was neutralized with acetic acid, and the solvents were evaporated. The residue was purified by silica gel column chromatography (eluate: 4% MeOH / DCM) to give compound T116 (33 mg, 52% yield) as yellow solid: UV-vis (MeOH) λmax 235 (shoulder) and 340 nm; IR 3393.08, 3173.06, 2940.13, 2911.25, 1695.55, 1659.18, and 1494.56 cm-1; 1H-NMR (DMSO-d6) δ 10.65 (1H, s, NH, exchangeable with D2O), 9.30 (1H, s, OH, exchangeable with D2O), 4.40 (1H, s, OH , exchangeable with D2O), 3.65 (2H, t, 4’-CH2, J = 7.2 Hz), 3.37 (2H, t, 1’-CH2, J = 6.4 Hz), 1.96 (3H, s, 5-CH3), 1.65 (2H, m, 2’-CH2), 1.41 (2H, m, 3’-CH2);13C-NMR (DMSO-d6) δ 158.48, 149.92, 144.41, 61.40, 50.48, 29.73, 25.24, 16.82.
Example 26

Reagents and conditions, a) BSA, DCE, O.Sh , rt, then SnCl4, rt; b) NH2OH+HCI, pyridine, rt; c) NaOMe, MeOH, rt.
Synthesis of 6-aza-2’-C-methyl-2-hydoxyimino-5-methyluridine nucleoside analog, T119.
T117. A mixture of T108 (110 mg, 0.7 mmol) and T12 (435 mg, 0.84 mmol) was suspended in DCE (5 mL), then treated with BSA (0.21 mL, 0.84 mmol) and the mixture was stirred for 30 min, until complete dissolution. SnCl4 (0.246 mL, 2.1 mmol) added drop wise and the mixture was stirred for 24 h at room temperature. The reaction mixture was quenched with sat. NaHCO3 and the white cake was filtrated on a Celite pad and was washed with DCM (50 mL). The separated organic layers were combined, dried over Na2SO4 and filtered. The filtrate was evaporated under vacuum to give compound T117 (0.4 g, 92.8% yield) as a colorless foam; UV- vis (MeOH) 234.99 nm IR 2698.40 cm-1 (νN-H), 1721.85 cm-1 (νC=O) and 1664.55 cm-1 (νC5=N) 1604.67 cm-1 (νC=S) ; 1H-NMR (CDCl3) δ 8.15-7.45 (15H, m, Ar), 6.88 (1H, s, Η-1’), 6.15 (1H, d, H-3', J = 8.4 Hz), 4.85-4.79 (2H, m, H-5a’, H-5b’), 4.76 (1H, d, H-3’, J = 4.4 Hz), 4.64 (1H, q, H-4’, J = 6.8,11.6 Hz), 2.64 (3H, s, CH3-S), 2.36 (3H, s, 5-CH3), 1.73 (3H, s, 2’- CH3); 13C-NMR (CDCl3) δ 166.57, 166.27, 165.61, 165.34, 159.93, 151.06, 133.99, 133.33, 130.05, 129.98, 129.84, 129.56, 128.96, 128.81, 128.46, 92.08, 87.79, 78.96, 76.74, 64.87, 17.46, 17.20, 14.93.
T118. A mixture of compound T117 (0.2 g, 0.32 mmol) and hydroxylamine hydrochloride (0.45 g, 6.48 mmol) was dissolved in dry pyridine (5 mL). The reaction mixture was stirred for 48
hours at 40 °C. The volatiles were evaporated and co-evaporated with toluene. The residue was partitioned between EtOAc and H2O. The separated organic phases were dried over Na2SO4 and evaporated. The residue was purified by silica gel column chromatography (eluate:
1.5%methanol / DCM) to give compound T118 (175 mg, 90.1% yield) as a white greenish solid: MP = 226-228 C; UV-vis (MeOH) λmax 230, 270 nm; IR 3356.79, 2915.81, 1703.09, 1660.79, and 1600.60 cm-1; 1H-NMR (CDCl3) δ 8.67 (1H, s ,OH, exchangeable with D2O), 8.14-7.94 (15H, m, Bz), 6.06 (1H, d, H-1’), 5.90 (1H, s, H-3’), 4.74-4.69 (2H, m, H-4’ and H-5’a), 4.61 (1H, m, , H-5'b), 2.22 (3H, s,5-CH3), 1.54 (3H, s, 2’-CH3); 13C-NMR (CDCl3) δ 166, 165.75, 164,
139.59-128.41, 88.87, 87.9, 79, 65.13, 17.33, 16.50.
T119. A solution of compound T118 (150 mg, 0.25 mmol) in dry methanol (5mL) was treated with sodium methoxide (1M, 2.0 mL) at 0 °C, then the mixture was stirred for 24 hours at room temperature. The reaction mixture was neutralized with acetic acid and the volatiles were evaporated under reduced pressure. The residue was purified by silica gel column chromatography (eluate: 6% MeOH / EtOAc) to give compound T119 (50 mg, 69.4% yield) as a white solid: MP = 225-227 °C; UV-vis (MeOH) λmax 205, 270 nm); IR 3464, 3167.98, 3032.54, 2931.25, 2829.94,1691.11, 1661.61, 1455.31cm-1; 1Η-NMR(DMSO-d6) δ 12.12 (1H, s, NH, exchangeable with D2O), 5.92 (1H, s, Η-1’), 4.99 (1H, s, OH-2' exchangeable with D2O), 4.96 (1H, m, OH-3' exchangeable with D2O) 4.50 (1H, t, 5’-OH, exchangeable with D2O, J= 5.6 Hz, J = 11.6 Hz), 3.77 (2H, m, 3’and 5’a), 3.64 (1H, dd, , Η-5’b), 3.52 (1H, m, H-4’), 2.07 (3H, s, 5- CH3); 1.01 (3H, s, 2’-CH3); 13C-NMR(DMSO-d6) δ 156.38, 148.83, 143.71, 83.75,78, 74.33, 62. 80, 87, 19.49, 16.17.
Example 27

Reagents and conditions, a) t-BuMgCl, THF, rt. Synthesis of 5’-momophosphate conjugate T120
T120. A solution of AH-09 (115 mg, 0.42 mmol) in anhydrous THF (1.5 mL) was treated with a 1.7M THF solution of tert-butylmagnesium chloride (0.52 mL, 0.88 mmol) at 0°C and the mixture was stirred for 1h at 0°C. Next, the mixture was treated dropwise with a solution of T7 (342 mg, 0.84 mmol) in anhydrous THF (1.5 mL) over a 5 min period. The mixture was allowed to warm to rt and was stirred for 2 days at room temperature. The mixture was quenched with sat. aq. NH4CI (10 mL) and then extracted with ethyl acetate (50 mL). The organic phase was washed with sat. aq. NaHCO3 (2 x 15 mL), dried over Na2SO4, filtered and concentrated to dryness. The resulting crude yellow oil was purified by flash silica gel column chromatography (eluate; 0-5% gradient of EtOH in DCM) to give T120 (123 mg, 56%) as pale yellow syrup. UV- vis (MeOH) 335 nm; 1Η-NMR (CDCl3) δ 8.76 (1H, s, NH), 7.23-7.07 (5H, m, Ar) , 5.90 (1H, d, H-1’, J= 5.6 Hz), 4.92 (1H, m, CH-(CH3)2), 4.48 (1H, s, H-2’), 4.33 (2H, m, H-3'and H- 4’), 4.22 (2H, m, 2’-OH and 3’-OH), 3.88-3.85 (1H, m, CHCH3) 2.02 (3H, s, 5-CH3), 1.29-1.16 (9H, m, CHCH3 and CH(CH3)3).
Example 28
Antiviral Assay Protocols
(1) Screening assays against Respiratory viruses, Dengue viruses, Yellow Fever virus, West Nile virus, Zika virus, Japanese encephalitis virus, Usutu virus, Chikungunya virus, Enteroviruses.
(a). Primary Cytopathic Effect (CPE) Assay. Four-concentration CPE inhibition assays are performed. Confluent or near-confluent cell culture monolayers in 96-well disposable microplates are prepared. Cells are maintained in MEM or DMEM supplemented with FBS as required for each cell line. For antiviral assays the same medium is used but with FBS reduced to 2% or less and supplemented with 50 μg/ml gentamicin. The test compound is prepared at four log10 final concentrations, usually 0.1, 1.0, 10, and 100 μg/ml or μΜ (depending upon sponsor’s preference). Lower concentrations are used when insufficient compound is supplied for the usual concentrations. Four compounds can be tested per microplate. Five microwells are used per dilution: three for infected cultures and two for uninfected toxicity cultures. Controls for the experiment consist of six microwells that are infected (virus controls) and six that are untreated (cell controls). The virus control and cell control wells are on every microplate. In parallel, a known active drug is tested as a positive control drug using the same method as is applied for test compounds. The positive control is tested with each test run. The assay is initiated by first removing growth media from the 96-well plates of cells. Then the test compound is applied in 0.1 ml volume to wells at 2X concentration. Virus, normally at <100 50% cell culture infectious doses (CCID50) in 0.1 ml volume, is placed in those wells designated for virus infection. Medium devoid of virus is placed in toxicity control wells and cell control wells. Virus control wells are treated similarly with virus. Plates are incubated at 37 °C with 5% CO2 until maximum CPE is observed in virus control wells. The plates are then stained with 0.011% neutral red for approximately two hours at 37oC in a 5% CO2 incubator. The neutral red medium is removed by complete aspiration, and the cells rinsed 1X with phosphate buffered solution (PBS) to remove residual dye. The PBS is completely removed and the incorporated neutral red is eluted with 50% Sorensen’s citrate buffer/50% ethanol for at least 30 minutes. Neutral red dye penetrates into living cells, thus, the more intense the red color, the larger the number of viable cells present in the wells. The dye content in each well is quantified using a 96- well spectrophotometer at 540 nm wavelength. The dye content in each set of wells is converted
to a percentage of dye present in untreated control wells using a Microsoft Excel computer-based spreadsheet. The 50% effective (EC50, virus-inhibitory) concentrations and 50% cytotoxic (CC50, cell-inhibitory) concentrations are then calculated by linear regression analysis. The quotient of CC50 divided by EC50 gives the selectivity index (SI) value. Compounds showing SI values >10 are considered active and are tested in an 8-concentration confirmatory assay. For certain viruses that are difficult to inhibit, such as West Nile virus, we may test compounds with SI values >5 in the confirmatory assay.
(b). Secondary CPE/ Viral Yield Reduction (VYR) Assay. This assay involves similar methodology to what is described in in CPE assay using 96-well microplates of cells. The differences are noted in this section. Eight half-log10 concentrations of inhibitor are tested for antiviral activity and cytotoxicity. Only two compounds can be evaluated per 96-well microplate. One positive control drug is tested per batch of compounds evaluated. After sufficient virus replication occurs, a sample of supernatant is taken from each infected well (three replicate wells are pooled) and frozen for the VYR portion of this test, if needed. Alternately, a separate plate may be prepared and the plate may be frozen for the VYR assay. After maximum CPE is observed, the viable plates are stained with neutral red dye. The incorporated dye content is quantified as described above. The data generated from this portion of the test are neutral red EC50, CC50, and SI values. Uninfected wells are tested in parallel for compound toxicity, as is explained in more detail in the above paragraph. Compounds observed to be active above are further evaluated by VYR assay. The VYR test is a direct determination of the concentration of the test compound that inhibits virus replication. Virus that was replicated in the presence of test compound is titrated and compared to virus from untreated, infected controls. Titration of pooled viral samples (collected as described in the paragraph above) is performed by endpoint dilution. This is accomplished by titrating log10 dilutions of virus using 3 or 4 microwells per dilution on fresh monolayers of cells in 96-well plates. Wells are scored for presence or absence of virus after distinct CPE is observed. Plotting the inhibitor concentration versus log10 of virus produced at each concentration allows calculation of the 90% (one log10) effective concentration by linear regression. Dividing EC90 by the CC50 obtained in part 1 of the assay gives the SI value for this test.
(2). Screening assays against Lassa fever, Ebola, and Nipah viruses.
(a) Primary PR assay. Confluent or near-confluent cell culture monolayers in 12-well disposable cell culture plates are prepared. Cells are maintained in MEM or DMEM supplemented with 10% FBS. For antiviral assays the same medium is used but with FBS reduced to 2% or less and supplemented with 1% penicillin/streptomycin. The test compound is prepared at four log10 final concentrations, usually 0.1, 1.0, 10, and 100 μg/ml or μΜ (depending upon the sponsor’s preference) in 2X MEM or 2X DMEM. Lower concentrations are used when insufficient compound is supplied for the usual concentrations after discussion with the PI and COR. The virus control and cell control will be run in parallel with each tested compound. Further, a known active drug is tested as a positive control drug using the same experimental set-up as described for the virus and cell control. The positive control is tested with each test run. Test compounds and positive controls are typically tested in biological triplicates. The assay is initiated by first removing growth media from the 12-well plates of cells, and infecting cells with 0.01 MOI of virus or about 50 to 100 plaque forming units (pfu). Cells will be incubated for 60 min: ΙΟΟμΙ inoculum/ well, at 37°C, 5% CO2 with constant gentle rocking. Virus inoculum will be removed, cells washed and overlaid with either 1% agarose or 1% methylcellulose diluted 1:1 with 2X MEM and supplemented with 2% FBS and 1 %penicillin/streptomycin and supplemented with the corresponding drug concentration. Cells will be incubated at 370C with 5% CO2 for 5 (Lassa fever), 10 (Ebola), or 3 (Nipah) days. The overlay is then removed and plates stained with 0.05% crystal violet in 10% buffered formalin for approximately twenty minutes at room temperature. The plates are then washed, dried and the number of plaques counted. The number of plaques is in each set of compound dilution is converted to a percentage relative to the untreated virus control. The 50%effective (EC50, virus- inhibitory) concentrations are then calculated by linear regression analysis. Cytotoxicity is evaluated in parallel to the actual primary PR assay. The cytotoxicity assay (In vitro Toxicology Assay Kit, Neutral red based; Sigma) is being performed in 96-well plates following the manufacturer’s instructions. Briefly, growth medium will be removed from confluent cell monolayer’s and replaced with fresh medium (total of 100μΙ) containing the test compound with the concentrations as indicated for the primary assay. Control wells will contain medium with the positive control or medium devoid of compound. Wells without cells and growth medium only will serve as blank. A total of up to five replicates will be performed for each condition. Plates are then incubated for 3, 5, or 10 days at 37°C with 5% CO2. The plates are then stained with
0.033% neutral red for approximately two hours at 37oC in a 5% CO2 incubator. The neutral red medium is removed by complete aspiration, and the cells rinsed 1X with phosphate buffered solution (PBS) to remove residual dye. The PBS is completely removed and the incorporated neutral red is eluted with 1% acetic acid/50% ethanol for at least 30 minutes. Neutral red dye penetrates into living cells, thus, the more intense the red color, the larger the number of viable cells present in the wells. The dye content in each well is quantified using a 96-well spectrophotometer at 540 nm wavelength and 690 nm wavelength (background reading). The 50% cytotoxic (CC50, cell-inhibitory) concentrations are then calculated by linear regression analysis. The quotient of CC50 divided by EC50 gives the selectivity index (SI) value.
We now have the capability to test Ebola virus in human HEK 293T cells, using T-705 as the positive control compound. It typically gives selectivity index values >10. This test is based on a VYR assay instead of PR because the virus does not produce distinct plaques in this cell line.
(b). Secondary VYR assay. The secondary assay involves similar methodology to what is described in the previous paragraphs using 12-well plates of cells. The differences are noted in this section. Cells are being infected as described above. The test compound is prepared at eight half-log10 final concentrations, usually 0.032, 0.1, 0.32, 1.0, 3.2, 10, 32 and 100 μg/ml or μΜ. Test compound is applied in 1 ml of total volume of media. Tissue culture supernatant (TCS) aliquots will be collected at appropriate time points and then be used to determine the compounds inhibitory effect on virus replication. Virus that was replicated in the presence of test compound is titrated and compared to virus from untreated, infected controls. For titration of TCS, serial ten-fold dilutions will be prepared and used to infect fresh monolayers of cells. Cells will be overlaid with 1% agarose mixed 1:1 with 2XMEM supplemented with 2% FBS and 1% penicillin, and the number of plaques determined. Test compounds and positive controls are typically tested in biological triplicates. Plotting the log10 of the inhibitor concentration versus log10 of virus produced at each concentration allows calculation of the 90% (one log10) effective concentration by linear regression.
Example 29

Antiviral activity of AH-01
Example 30
Antiviral activity of AH-06

Claims
1. A compound of formula I
 or pharmaceutically acceptable salt or prodrug thereof, wherein:
or pharmaceutically acceptable salt or prodrug thereof, wherein:
R1 is selected from hydrogen, COR1*, COOR1*, CONHR1*.
R1* is, independently, C1-C22 alkyl, the carbon chain id derived from fatty alcohol or amino acid C3-C7 cycloalkyl, aryl, heterocyclyl, CH2OCO-C1-C22 alkyl, CH2OCO-C3-C7 cycloalkyl, CH2OCO-C3-C7 heterocyclyl, C1-C22 alkoxy, C3-C7 cycloalkoxy, aryloxy, heterocyclyloxy, C2-C18 alkenyl, C2-C18 alkynyl, substituted C1-C22 alkyl with halogen or C1-C6 carbon chain, (C=O)N(alkyl)2,NH(aryl), NH(Heroaryl), NH(C=O)-alkyl (C1-C22), NHO(C=O)alkyloxy (C1-C22).
R2 is, independently, selected from NH, CH, C-halogen, N(CO)NH2, the dashed and the solid lines combined
 represent an optional double bond.
represent an optional double bond.
R3 is independently, selected from H, OH, NHOH, NHO(C=O) C1-C22 alkyl,
NHO(C=O)- C1-C22 alkyloxy, NH2, SH, CN, S-C1-C10 alkyl, S-C2-C10 alkenyl, S-C2-C10 alkynyl, S-aryl, NH-C1-C22 alkyl, NH- C3-C10 cycloalkyl, N(alkyl)2,NH(aryl), NH(Heroaryl), NH(C=O)- C1-C22 alkyl, NHO(C=O)-C1-C22 alkyloxy.
R4 is independently, selected from H, F, Cl, Br, I, CH3, CF3, CH2F, CHF2, CH2OH, CH2CI, CH2Br, CH2I, CN, an optionally substituted alkyl C1-C10, an optionally substituted C1- C10 cycloalkyl, an optionally substituted C2-C10 alkenyl, an optionally substituted C2-C10 alkynyl.
X is independently, selected from CH, C-(halogen; F; Cl, Br, I), C-CN, C-CH3, C-CF3, C-
CHF2, C-CH2F, C-CH2OH, C-CCH, C-CHCH2, N, C(C=O)R1*, C-NO2.
2. The compound in claim 1 , wherein Sugar is ribose or modified ribose of the general formula (I- A)

R5 is, independently, selected from hydrogen, halogen (F, Cl, Br, I), CN, N3, CH3, substituted methyl, CCH, C=CH2;
R6 is independently selected from, H, OH, halogen (F, Cl, Br, I), Nitrile (CN), Nitro (NO2), Methyl (CH3), C1-C22 an optionally substituted methyl; cyclopropyl, C2-C22 alkenyl, C2- C22 an optionally substituted alkenyl, C2-C22 an optionally substituted alkynyl;
R7 is independently selected from, H, OH, halogen (F, Cl, Br, I), N3, CN;
R6 and R7 all together, hydrogen, halogen (F, Cl, Br, I), mixed halogen; cyclopropyl, cyclobutyl, aziridine, epoxide, methylidine (=CH2), halomethylidine (=CHF, =CHCl, =CHBr, =CHI);
R8 is selected from absent, H, halogen (F, Cl, Br, I), N3, methyl, an optionally substituted methyl, CN; alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, alkenyl, alkynyl, heteroaryl;
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3,
CFCH3,cyclopropyl;
Y with absent R8 is selected from C=CH, C=CF, C=CCH3
R20 is independently selected from Hydrogen, lower alkyl (C1-C-6), cycloalkyl, higher alkyl (C7-C16), alkenyl (C=CH2), alkynyl (CCH), aryl, heteroaryl, heterocyclyl;
W1 is selected from CH2, CHCH3, CHCF3, CHC=CH2, CH-CCH;
W2 is H, COR1*, COOR1*, CONHR1*, where R1* is as defined above in Formula I;
 monophosphate, diphosphate, triphosphate,, Wherein:
monophosphate, diphosphate, triphosphate,, Wherein:
Z is independently selected from O, S, NOH, NOR9; n is (1-7) represent the number of carbon atoms, optionally saturated and unsaturated;
R9’ is H, alkyl (C1-C17), cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), aryl, heteroaryl;
R10 is H, alkyl (C1-C6) and substituted alkyl;
R11 is H, OCH3, halogen (F, Cl, Br, I), nitro, cyano, O-alkyl (C2-C4), carboxyl, alkyl carboxyl.
R12 is alkyl, aryl, heteroaryl;
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl,
CH2CH2SMe, benzyl, pyrolinyl, CH2OR’ , CH2OC(O)R9, CH2-indolyl, 4-hydroxylbenzyl, isopropyl, CH2CH2CO2R' , CH2-imidazolyl; Lipid is a C6-C22 alkyl, alkoxy, polyethylene glycol, or sphingolipid.
3. The compound in claim 1 , wherein Sugar is ribose or modified ribose of the general formula (I-B)

Formula l-B or pharmaceutically acceptable salt or prodrug thereof, wherein:
R5, R6, R7, R6 and R7all together, R8, Y, W1 are as defined above in connection with claims 1-2.
Z is independently selected from O, S, NOH, NOR9;
R9 is independently selected from C(O)alkyl (C1-C17), C(O)cycloalkyl (C3-C6), optionally substituted cycloalkyl (C3-C6), C(O)aryl, C(O)heteroaryl;
R15 is independently selected from O-aryl, O-heteroaryl,

Wherein:
R13 and R14 independently is H, CH3, CH2SR9, CH2SC(O)R9, CH2CH3, isobutyl,
CH2CH2SMe, benzyl, pyrolinyl, pyridinyl CH2OR' , CH2OC(O)R9, CH2-indolyl, 4- hydroxylbenzyl, isopropyl, CH2CH2CO2R’ , CH2-imidazolyl.
4. The compound in claim 1 , wherein Sugar is ribose or modified ribose of the general formula (I-C or ID)

Formula I-C Formula l-D or pharmaceutically acceptable salt or prodrug thereof, wherein:
R5, R8, Y, W1, and W2 are as defined above in connections with claims 1-3.
R6 is independently selected from, H, halogen (F, Cl, Br, I), CH3, ΝO2, CN, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2- C22 an optionally substituted alkynyl;
R7 is independently selected from, H, halogen (F, Cl, Br, I), N3, CN; NO2, C1-C22 an optionally substituted methyl, C2-C22 alkenyl, C2-C22 an optionally substituted alkenyl, C2- C22 an optionally substituted alkynyl;
5. The compound in claim 1 , wherein Sugar is ribose or modified ribose of the general formula (I-E)

Formula I-E or pharmaceutically acceptable salt or prodrug thereof, wherein:
R5, R6, R7, R6 and R7all together, R8, Y, W1, and W2 are as defined above in connection with claims 1-3.
6. The compound in claim 1, wherein Sugar is modified L-deoxyribose of the general formula
(I-F or I-G)

Formula l-F Formula 1-G or pharmaceutically acceptable salt or prodrug thereof, wherein:
R5, R8, Y, W1 and W2 are as defined above in connection with claims 1-5.
Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3, CFCH3,cyclopropyl;
Y with absent H8 is selected from C=CH, C=CF, C=CCH3
7. The compound in claim 1, wherein Sugar has one acyclic sugar of the formula I-H or I-J or I-K)
 or pharmaceutically acceptable salt or prodrug thereof, wherein: Wland W2 are as defined above in claims 1-6
or pharmaceutically acceptable salt or prodrug thereof, wherein: Wland W2 are as defined above in claims 1-6
X is independently selected from absent, CH2, CHR16;
R16 (is independently selected from CH3, CH2OH, CH2NH2, CH2CN, CH2NO2, CH2F, CH2Cl, CH2I, CH2Br, CF3, CCH, CH=CH2, CH2, CH2CO2H, CH2O-protected monophosphate; Y is independently selected from O, S, NH, NR9, CH2, C(=O), C=CH2, COHCH3,
CFCH3;
Z is independently selected from O, S, NOH, NOR9.
R15 is independently selected from (C=O)-alkyl (C1-C22), optionally substituted (C=O)-alkyl, (C=O)-aryl, (C=O)-CH2 aryl, (C=O)-Heteroaryl,

8. The compound in claim 1, wherein Sugar is a sugar of the formula I-L

Formula l-L or pharmaceutically acceptable salt or prodrug thereof, wherein:
R5, R6, R7, R6 and R7 all together, W1, and W2 are as defined above in claims 1-3 R5* is independently selected from hydrogen, halogen (F, Cl, Br, I), CN, N3, CH3, substituted methyl, CCH, C=CH2;
R is, independently, hydrogen, halogen (F, Cl, Br, I), azide, amino, NHOH, CN, NO2, C1-C6 lower alkyl, C3-C6 cycloalkyl, C2-C-8 alkenyl, C2-C-8 alkynyl.
R21 is, independently, hydrogen, hydroxyl, halogen (F, Cl, Br, I), azide, amino, NHOH, CN, NO2, C1-C6 lower alkyl, C3-C6 cycloalkyl, C2-C-8 alkenyl, C2-C-8 alkynyl.
9. The compound in claim 1, wherein the compound has the formula I-M

Formula l-M or pharmaceutically acceptable salt or prodrug thereof, wherein:
R1 is selected from hydrogen, COR1*, COOR1*, CONHR1*.
R1* is, independently, C1-C22 alkyl, the carbon chain id derived from fatty alcohol or amino acid C3-C7 cycloalkyl, aryl, heterocyclyl, CH2OCO-C1-C22 alkyl, CH2OCO-C3-C7 cycloalkyl, CH2OCO-C3-C7 heterocyclyl, C1-C22 alkoxy, C3-C7 cycloalkoxy, aryloxy,
heteiocyclyloxy, C2-C18 alkenyl, C2-C18 alkynyl, substituted C1-C22 alkyl with halogen or C1-C6 carbon chain, (C=O)N(alkyl)2,NH(aryl), NH(Heroaryl), NH(C=O)-alkyl (C1-C22), NHO(C=O)alkyloxy (C1-C22).
R3, R4, R5, R6, R6 and R7 all together, R8, R20, R21, W1, W2 are as defined above in connection with claims 1-8.
R4’ is independently, selected from H, F, Cl, Br, I, CH3, CF3, CH2F, CHF2, CH2OH, CH2Cl, CH2Br, CH2I, CN, an optionally substituted alkyl C1-C10, an optionally substituted C1- C10 cycloalkyl, an optionally substituted C2-C10 alkenyl, an optionally substituted C2-C10 alkynyl.
10. The compound in claim 1, wherein the compound has the formula I-N

Formula I-N or pharmaceutically acceptable salt or prodrug thereof, wherein:
R1 is selected from hydrogen, COR1*, COOR1*, CONHR1*.
R1* is, independently, C1-C22 alkyl, the carbon chain is derived from fatty alcohol or amino acid, C3-C7 cycloalkyl, aryl, heterocyclyl, CH2OCO-C1-C22 alkyl, CH2OCO-C3-C7 cycloalkyl, CH2OCO-C3-C7 heterocyclyl, C1-C22 alkoxy, C3-C7 cycloalkoxy, aryloxy, heterocyclyloxy, C2-C18 alkenyl, C2-C18 alkynyl, substituted C1-C22 alkyl with halogen or C1-C6 carbon chain, (C=O)N(alkyl)2,NH(aryl), NH(Heroaryl), NH(C=O)-alkyl (C1-C22), NHO(C=O)alkyloxy (C1-C22).
R3, R4, R5, R6, R7, R6 and R7 all together, R8, R20, R21, W1, W2 are as defined above in connection with claims 1-9.
11. A compound of Formula II

Formula II
Wherein;
X is independently O, S
V is independently N, CH, C(F, Cl, Br, I), CCN,CCO2H, CO2R9, CCH3, CCF3 R3, R4, R5, R6, R7, R6 and R7 all together, R8, W1, W2 are as defined above in Formula I.
12. The compound of any of claims 1-10, wherein the compounds are described herein can be in the form of the β-L- or β-D-configuration, or a mixture thereof, including a racemic mixture thereof.
13. The compound of any of claims 1-11, wherein when the phosphorous portion of the compound described herein contains a chiral center, such chiral center can be in the form of the RP- or Sp-configuration or a mixture thereof, including a racemic mixture thereof.
14. A method for treating or preventing Respiratory Viruses (Coronaviruses, Respiratory Syncytial virus, Influenza viruses) infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 , or in combination with another anti-Respiratory virus agent to a subject in need thereof.
15. A method for treating or preventing Influenza viruses infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12, or in combination with another anti- Influenza virus agent to a subject in need thereof.
16. A method for treating or preventing Coronaviruses (MERS-CoV, SARS-CoV, SARS-2 - CoV, human coronavirus) infections, comprising administering an effective amount of a
compound of formula I or any compound of claims 1 to 12, or in combination with anti- Coronavirus agent to a subject in need thereof.
17. A method for treating or preventing Respiratory Syncytial Virus infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12, or in combination with anti-Respiratory Syncytial Virus agent to a subject in need thereof.
18. A method for treating or preventing Zika Virus infections, comprising administering an effective amount of a compound of formula I or any compound of claims lto 12 or in combination with anti-Zika Virus agent to a subject in need thereof.
19. A method for treating or preventing Ebola Virus infection, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 or in combination with anti-Ebola Virus agent to a subject in need thereof.
20. A method for treating or preventing Hepatitis C infection, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 or in combination with anti- Hepatitis C agent to a subject in need thereof.
21. A method for treating or preventing Hepatitis B infection, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 or in combination with anti- Hepatitis B agent to a subject in need thereof.
22. A method for treating or preventing HIV-1 or HIV-2 infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 to a subject in need thereof.
23. A method for treating or preventing Noroviruses infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12, or in combination with another anti-Norovirus agent to a subject in need thereof.
24. A method for treating or preventing Dengue viruses infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 or in combination with another anti-Dengue virus agent to a subject in need thereof.
25. A method for treating or preventing Epstein Barr virus, Adenoviruses, and Herpes viruses infections, comprising administering an effective amount of a compound of formula I or any compound of claims 1 to 12 or in combination with another anti- Epstein Barr virus agent to a subject in need thereof.\
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EG2020070960 | 2020-07-05 | ||
| EG2020070960 | 2020-07-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022008025A1 true WO2022008025A1 (en) | 2022-01-13 |
Family
ID=79553720
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EG2021/000021 WO2022008025A1 (en) | 2020-07-05 | 2021-07-05 | 2-hydroxyiminopyrimidine nucleosides and derivitives and antiviral uses thereto |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022008025A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022123501A1 (en) * | 2020-12-10 | 2022-06-16 | Victoria Link Limited | Protected deoxydidehydro-nucleosides |
| WO2023085242A1 (en) * | 2021-11-12 | 2023-05-19 | 国立大学法人北海道大学 | Antiviral agent |
Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1193531A (en) * | 1967-01-30 | 1970-06-03 | Upjohn Co | Novel Reduced Pyrimidine Nucleosides and Nucleotides, and Processes for the Production Thereof |
| US4211773A (en) * | 1978-10-02 | 1980-07-08 | Sloan Kettering Institute For Cancer Research | 5-Substituted 1-(2'-Deoxy-2'-substituted-β-D-arabinofuranosyl)pyrimidine nucleosides |
| EP0196185A2 (en) * | 1985-03-16 | 1986-10-01 | The Wellcome Foundation Limited | Antiviral nucleosides |
| EP0375164A1 (en) * | 1988-11-23 | 1990-06-27 | The Wellcome Foundation Limited | Antiviral compounds |
| WO1991001326A1 (en) * | 1989-07-17 | 1991-02-07 | University Of Birmingham | Antiviral pyrimidine nucleosides |
| WO1991004982A1 (en) * | 1989-10-04 | 1991-04-18 | University Of Birmingham | Further antiviral pyrimidine nucleosides |
| EP0453247A2 (en) * | 1990-04-18 | 1991-10-23 | The Wellcome Foundation Limited | Anti-viral compounds |
| EP0461815A1 (en) * | 1990-06-09 | 1991-12-18 | The Wellcome Foundation Limited | Anti-HBV pyrimidine nucleoside |
| WO1994005687A1 (en) * | 1992-09-04 | 1994-03-17 | University Of Birmingham | Antiviral pyrimidine nucleosides |
| WO2001060315A2 (en) * | 2000-02-18 | 2001-08-23 | Shire Biochem Inc. | Method for the treatment or prevention of flavivirus infections using nucleoside analogues |
| WO2001090121A2 (en) * | 2000-05-23 | 2001-11-29 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus |
| WO2002079213A1 (en) * | 2001-03-30 | 2002-10-10 | Triangle Pharmaceuticals, Inc. | Process for the preparation of 2'-halo-$g(b)-l-arabinofuranosyl nucleosides |
| WO2004046159A1 (en) * | 2002-11-19 | 2004-06-03 | F. Hoffmann-La-Roche Ag | Antiviral nucleoside derivatives |
| US20040242599A1 (en) * | 2003-02-27 | 2004-12-02 | Biocryst Pharmaceuticals, Inc. | Cyclopentene nucleosides, preparation thereof and use as inhibitors of RNA viral polymerases |
| US20060040890A1 (en) * | 2004-08-23 | 2006-02-23 | Roche Palo Alto Llc | Anti-viral nucleosides |
| WO2008104408A2 (en) * | 2007-02-27 | 2008-09-04 | K. U. Leuven Research & Development | Phosphate modified nucleosides useful as substrates for polymerases and as antiviral agents |
| EP2332952A1 (en) * | 2002-06-28 | 2011-06-15 | IDENIX Pharmaceuticals, Inc. | Modified 2' and 3'-nucleoside prodrugs for treating flaviridae infections |
| US20130303747A1 (en) * | 2010-09-07 | 2013-11-14 | Junbiao Chang | Pyrimidine nucleoside derivatives, synthesis methods and uses thereof for preparing anti-tumor and anti-virus medicaments |
| WO2014070771A1 (en) * | 2012-10-29 | 2014-05-08 | Rfs Pharma, Llc | Pyrimidine nucleotides and their monophosphate prodrugs for treatment of viral infections and cancer |
| WO2015017713A1 (en) * | 2013-08-01 | 2015-02-05 | Idenix Pharmaceuticals, Inc. | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
-
2021
- 2021-07-05 WO PCT/EG2021/000021 patent/WO2022008025A1/en active Application Filing
Patent Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1193531A (en) * | 1967-01-30 | 1970-06-03 | Upjohn Co | Novel Reduced Pyrimidine Nucleosides and Nucleotides, and Processes for the Production Thereof |
| US4211773A (en) * | 1978-10-02 | 1980-07-08 | Sloan Kettering Institute For Cancer Research | 5-Substituted 1-(2'-Deoxy-2'-substituted-β-D-arabinofuranosyl)pyrimidine nucleosides |
| EP0196185A2 (en) * | 1985-03-16 | 1986-10-01 | The Wellcome Foundation Limited | Antiviral nucleosides |
| EP0375164A1 (en) * | 1988-11-23 | 1990-06-27 | The Wellcome Foundation Limited | Antiviral compounds |
| WO1991001326A1 (en) * | 1989-07-17 | 1991-02-07 | University Of Birmingham | Antiviral pyrimidine nucleosides |
| WO1991004982A1 (en) * | 1989-10-04 | 1991-04-18 | University Of Birmingham | Further antiviral pyrimidine nucleosides |
| EP0453247A2 (en) * | 1990-04-18 | 1991-10-23 | The Wellcome Foundation Limited | Anti-viral compounds |
| EP0461815A1 (en) * | 1990-06-09 | 1991-12-18 | The Wellcome Foundation Limited | Anti-HBV pyrimidine nucleoside |
| WO1994005687A1 (en) * | 1992-09-04 | 1994-03-17 | University Of Birmingham | Antiviral pyrimidine nucleosides |
| WO2001060315A2 (en) * | 2000-02-18 | 2001-08-23 | Shire Biochem Inc. | Method for the treatment or prevention of flavivirus infections using nucleoside analogues |
| WO2001090121A2 (en) * | 2000-05-23 | 2001-11-29 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus |
| WO2002079213A1 (en) * | 2001-03-30 | 2002-10-10 | Triangle Pharmaceuticals, Inc. | Process for the preparation of 2'-halo-$g(b)-l-arabinofuranosyl nucleosides |
| EP2332952A1 (en) * | 2002-06-28 | 2011-06-15 | IDENIX Pharmaceuticals, Inc. | Modified 2' and 3'-nucleoside prodrugs for treating flaviridae infections |
| WO2004046159A1 (en) * | 2002-11-19 | 2004-06-03 | F. Hoffmann-La-Roche Ag | Antiviral nucleoside derivatives |
| US20040242599A1 (en) * | 2003-02-27 | 2004-12-02 | Biocryst Pharmaceuticals, Inc. | Cyclopentene nucleosides, preparation thereof and use as inhibitors of RNA viral polymerases |
| US20060040890A1 (en) * | 2004-08-23 | 2006-02-23 | Roche Palo Alto Llc | Anti-viral nucleosides |
| WO2008104408A2 (en) * | 2007-02-27 | 2008-09-04 | K. U. Leuven Research & Development | Phosphate modified nucleosides useful as substrates for polymerases and as antiviral agents |
| US20130303747A1 (en) * | 2010-09-07 | 2013-11-14 | Junbiao Chang | Pyrimidine nucleoside derivatives, synthesis methods and uses thereof for preparing anti-tumor and anti-virus medicaments |
| WO2014070771A1 (en) * | 2012-10-29 | 2014-05-08 | Rfs Pharma, Llc | Pyrimidine nucleotides and their monophosphate prodrugs for treatment of viral infections and cancer |
| WO2015017713A1 (en) * | 2013-08-01 | 2015-02-05 | Idenix Pharmaceuticals, Inc. | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022123501A1 (en) * | 2020-12-10 | 2022-06-16 | Victoria Link Limited | Protected deoxydidehydro-nucleosides |
| WO2023085242A1 (en) * | 2021-11-12 | 2023-05-19 | 国立大学法人北海道大学 | Antiviral agent |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11166973B2 (en) | Substituted nucleotides and nucleosides for treating viral infections | |
| EP3762372A1 (en) | 4'-halogen containing nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| JP6430364B2 (en) | Substituted nucleosides, nucleotides and analogs thereof | |
| KR102072041B1 (en) | Substituted nucleosides, nucleotides and analogs thereof | |
| GB2590198A (en) | N4-hydroxycytidine and derivatives and anti-viral uses related thereto | |
| WO2016145142A1 (en) | Nucleotide and nucleoside therapeutics compositions and uses related thereto | |
| AU2020220216B2 (en) | 2'-substituted-N6-substituted purine nucleotides for RNA virus treatment | |
| KR102363946B1 (en) | β-D-2′-deoxy-2′-α-fluoro-2′-β-C-substituted-2-modified-N6-substituted purine nucleotides for the treatment of HCV | |
| WO2014124430A1 (en) | Nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| MX2013013570A (en) | Purine monophosphate prodrugs for treatment of viral infections. | |
| WO2022008025A1 (en) | 2-hydroxyiminopyrimidine nucleosides and derivitives and antiviral uses thereto | |
| WO2017155923A1 (en) | Nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| US20230062181A1 (en) | Alkyne containing nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| EP4333859A1 (en) | Nucleotide and nucleoside therapeutic compositions, combinations and uses related thereto | |
| WO2017106710A1 (en) | Nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| AU2020418425A1 (en) | 4'-halogen containing nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| WO2022174179A1 (en) | 4'-halogen containing nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| WO2024086592A2 (en) | 4'-halogen containing nucleotide and nucleoside therapeutic compositions and uses related thereto | |
| JP2024517807A (en) | Nucleotide and nucleoside therapeutic compositions, combinations, and related uses | |
| EA040007B1 (en) | ALKINE-CONTAINING NUCLEOTIDE AND NUCLEOSIDE THERAPEUTIC COMPOSITIONS AND RELATED USES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21838480 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 21838480 Country of ref document: EP Kind code of ref document: A1 |