WO2021055863A1 - Pyruvate kinase r (pkr) activating compositions - Google Patents
Pyruvate kinase r (pkr) activating compositions Download PDFInfo
- Publication number
- WO2021055863A1 WO2021055863A1 PCT/US2020/051645 US2020051645W WO2021055863A1 WO 2021055863 A1 WO2021055863 A1 WO 2021055863A1 US 2020051645 W US2020051645 W US 2020051645W WO 2021055863 A1 WO2021055863 A1 WO 2021055863A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- type
- theta
- crystalline
- xrpd
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims description 68
- 102000013009 Pyruvate Kinase Human genes 0.000 title description 6
- 108020005115 Pyruvate Kinase Proteins 0.000 title description 6
- 230000003213 activating effect Effects 0.000 title description 4
- 239000007787 solid Substances 0.000 claims abstract description 373
- 238000010922 spray-dried dispersion Methods 0.000 claims abstract description 132
- 239000006186 oral dosage form Substances 0.000 claims abstract description 41
- KZFFYEPYCVDOGE-LJQANCHMSA-N (2S)-1-[5-(2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-ylsulfonyl)-1,3,4,6-tetrahydropyrrolo[3,4-c]pyrrol-2-yl]-3-hydroxy-2-phenylpropan-1-one Chemical compound O1CCOC2=NC=C(C=C21)S(=O)(=O)N1CC2=C(C1)CN(C2)C([C@H](CO)C1=CC=CC=C1)=O KZFFYEPYCVDOGE-LJQANCHMSA-N 0.000 claims abstract description 31
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 577
- 238000000034 method Methods 0.000 claims description 189
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 174
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 170
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 170
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical group OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 170
- 229920000642 polymer Polymers 0.000 claims description 146
- 238000003860 storage Methods 0.000 claims description 84
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 64
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 64
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 64
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 62
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 62
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 62
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 claims description 56
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 claims description 36
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 claims description 36
- 239000008186 active pharmaceutical agent Substances 0.000 claims description 29
- 150000001875 compounds Chemical class 0.000 claims description 29
- 230000009477 glass transition Effects 0.000 claims description 20
- 239000007921 spray Substances 0.000 claims description 20
- 229920000623 Cellulose acetate phthalate Polymers 0.000 claims description 18
- 239000001856 Ethyl cellulose Substances 0.000 claims description 18
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 claims description 18
- 229940081734 cellulose acetate phthalate Drugs 0.000 claims description 18
- 229920001249 ethyl cellulose Polymers 0.000 claims description 18
- 235000019325 ethyl cellulose Nutrition 0.000 claims description 18
- 238000004128 high performance liquid chromatography Methods 0.000 claims description 17
- KZFFYEPYCVDOGE-IBGZPJMESA-N (2R)-1-[5-(2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-ylsulfonyl)-1,3,4,6-tetrahydropyrrolo[3,4-c]pyrrol-2-yl]-3-hydroxy-2-phenylpropan-1-one Chemical compound O1CCOC2=NC=C(C=C21)S(=O)(=O)N1CC2=C(C1)CN(C2)C([C@@H](CO)C1=CC=CC=C1)=O KZFFYEPYCVDOGE-IBGZPJMESA-N 0.000 claims description 14
- 239000002775 capsule Substances 0.000 claims description 4
- 229940125904 compound 1 Drugs 0.000 abstract description 798
- 239000008194 pharmaceutical composition Substances 0.000 abstract description 235
- 238000002360 preparation method Methods 0.000 abstract description 35
- 239000007962 solid dispersion Substances 0.000 description 171
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 128
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 88
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 84
- 238000002411 thermogravimetry Methods 0.000 description 76
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 74
- 238000004458 analytical method Methods 0.000 description 73
- 238000000113 differential scanning calorimetry Methods 0.000 description 67
- 239000002552 dosage form Substances 0.000 description 67
- 239000000546 pharmaceutical excipient Substances 0.000 description 57
- -1 Solutol® HS15) Chemical compound 0.000 description 43
- 239000000243 solution Substances 0.000 description 40
- 239000002202 Polyethylene glycol Substances 0.000 description 39
- 229920001223 polyethylene glycol Polymers 0.000 description 39
- 239000002904 solvent Substances 0.000 description 37
- 239000000945 filler Substances 0.000 description 33
- 239000000314 lubricant Substances 0.000 description 33
- 238000001757 thermogravimetry curve Methods 0.000 description 33
- 230000004580 weight loss Effects 0.000 description 30
- 238000012512 characterization method Methods 0.000 description 29
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 26
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 26
- 238000002474 experimental method Methods 0.000 description 26
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 26
- 229920002554 vinyl polymer Polymers 0.000 description 26
- 229940125782 compound 2 Drugs 0.000 description 24
- 239000007884 disintegrant Substances 0.000 description 22
- 239000002706 dry binder Substances 0.000 description 22
- 238000001179 sorption measurement Methods 0.000 description 22
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 19
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 17
- 238000009472 formulation Methods 0.000 description 16
- 239000000047 product Substances 0.000 description 16
- 238000002156 mixing Methods 0.000 description 15
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 14
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 13
- 229920003135 Eudragit® L 100-55 Polymers 0.000 description 13
- 101000656751 Haloarcula marismortui (strain ATCC 43049 / DSM 3752 / JCM 8966 / VKM B-1809) 30S ribosomal protein S24e Proteins 0.000 description 13
- SHBUUTHKGIVMJT-UHFFFAOYSA-N Hydroxystearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OO SHBUUTHKGIVMJT-UHFFFAOYSA-N 0.000 description 13
- 229920000578 graft copolymer Polymers 0.000 description 13
- 229940072106 hydroxystearate Drugs 0.000 description 13
- 229960003511 macrogol Drugs 0.000 description 13
- 125000005395 methacrylic acid group Chemical group 0.000 description 13
- 239000000523 sample Substances 0.000 description 13
- 238000001694 spray drying Methods 0.000 description 13
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 239000002002 slurry Substances 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 10
- 238000009792 diffusion process Methods 0.000 description 10
- 238000004090 dissolution Methods 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000012296 anti-solvent Substances 0.000 description 9
- 230000001351 cycling effect Effects 0.000 description 9
- 238000010438 heat treatment Methods 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 8
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 235000019439 ethyl acetate Nutrition 0.000 description 8
- 239000011521 glass Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 229910016860 FaSSIF Inorganic materials 0.000 description 7
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 7
- 235000019359 magnesium stearate Nutrition 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 238000010583 slow cooling Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- JACRWUWPXAESPB-UHFFFAOYSA-N tropic acid Chemical compound OCC(C(O)=O)C1=CC=CC=C1 JACRWUWPXAESPB-UHFFFAOYSA-N 0.000 description 6
- ZENUIXUSKAXIHH-UHFFFAOYSA-N 7-(2,3,4,6-tetrahydro-1H-pyrrolo[3,4-c]pyrrol-5-ylsulfonyl)-2,3-dihydro-[1,4]dioxino[2,3-b]pyridine Chemical compound O1CCOC2=NC=C(C=C21)S(=O)(=O)N1CC=2CNCC=2C1 ZENUIXUSKAXIHH-UHFFFAOYSA-N 0.000 description 5
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 5
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 5
- 229960000913 crospovidone Drugs 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- 239000007888 film coating Substances 0.000 description 5
- 238000009501 film coating Methods 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 229940016286 microcrystalline cellulose Drugs 0.000 description 5
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 5
- 239000008108 microcrystalline cellulose Substances 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 5
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 4
- 241000282693 Cercopithecidae Species 0.000 description 4
- 239000007821 HATU Substances 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 239000012736 aqueous medium Substances 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000011067 equilibration Methods 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 238000005469 granulation Methods 0.000 description 4
- 230000003179 granulation Effects 0.000 description 4
- 229960001021 lactose monohydrate Drugs 0.000 description 4
- 238000011068 loading method Methods 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 4
- 239000004810 polytetrafluoroethylene Substances 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- CFRRNZLVFIRZRJ-UHFFFAOYSA-N tert-butyl 5-(2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-ylsulfonyl)-1,3,4,6-tetrahydropyrrolo[3,4-c]pyrrole-2-carboxylate Chemical compound O1CCOC2=NC=C(C=C21)S(=O)(=O)N1CC2=C(C1)CN(C2)C(=O)OC(C)(C)C CFRRNZLVFIRZRJ-UHFFFAOYSA-N 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- 229920002785 Croscarmellose sodium Polymers 0.000 description 3
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 229960001681 croscarmellose sodium Drugs 0.000 description 3
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 3
- 238000009506 drug dissolution testing Methods 0.000 description 3
- 229940088679 drug related substance Drugs 0.000 description 3
- 230000000968 intestinal effect Effects 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000003801 milling Methods 0.000 description 3
- 239000004570 mortar (masonry) Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 3
- 238000009490 roller compaction Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 239000011877 solvent mixture Substances 0.000 description 3
- 238000013097 stability assessment Methods 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 230000036962 time dependent Effects 0.000 description 3
- FFPRXERCJDFLIL-UHFFFAOYSA-N ClS(=O)(=O)C1=CN=C2OCCOC2=C1 Chemical compound ClS(=O)(=O)C1=CN=C2OCCOC2=C1 FFPRXERCJDFLIL-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 229910005429 FeSSIF Inorganic materials 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 230000000975 bioactive effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 229910021485 fumed silica Inorganic materials 0.000 description 2
- 230000001788 irregular Effects 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 231100000062 no-observed-adverse-effect level Toxicity 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 239000013557 residual solvent Substances 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 239000012086 standard solution Substances 0.000 description 2
- 239000007916 tablet composition Substances 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 231100000041 toxicology testing Toxicity 0.000 description 2
- JACRWUWPXAESPB-QMMMGPOBSA-N (R)-tropic acid Chemical compound OC[C@H](C(O)=O)C1=CC=CC=C1 JACRWUWPXAESPB-QMMMGPOBSA-N 0.000 description 1
- JACRWUWPXAESPB-MRVPVSSYSA-N (S)-tropic acid Chemical compound OC[C@@H](C(O)=O)C1=CC=CC=C1 JACRWUWPXAESPB-MRVPVSSYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- VQNDBXJTIJKJPV-UHFFFAOYSA-N 2h-triazolo[4,5-b]pyridine Chemical compound C1=CC=NC2=NNN=C21 VQNDBXJTIJKJPV-UHFFFAOYSA-N 0.000 description 1
- PMNCDNRJLYPTDB-UHFFFAOYSA-N 7-bromo-2,3-dihydro-[1,4]dioxino[2,3-b]pyridine Chemical compound O1CCOC2=CC(Br)=CN=C21 PMNCDNRJLYPTDB-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000920340 Pion Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- IYKJEILNJZQJPU-UHFFFAOYSA-N acetic acid;butanedioic acid Chemical compound CC(O)=O.OC(=O)CCC(O)=O IYKJEILNJZQJPU-UHFFFAOYSA-N 0.000 description 1
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 239000003517 fume Substances 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 239000006070 nanosuspension Substances 0.000 description 1
- VWBWQOUWDOULQN-UHFFFAOYSA-N nmp n-methylpyrrolidone Chemical compound CN1CCCC1=O.CN1CCCC1=O VWBWQOUWDOULQN-UHFFFAOYSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 238000012429 release testing Methods 0.000 description 1
- 238000004626 scanning electron microscopy Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000011863 silicon-based powder Substances 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000007921 solubility assay Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- OXUPNVQFTWUSGY-UHFFFAOYSA-N tert-butyl 2,3,4,6-tetrahydro-1h-pyrrolo[3,4-c]pyrrole-5-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC2=C1CNC2 OXUPNVQFTWUSGY-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 238000002076 thermal analysis method Methods 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
Definitions
- PYRUVATE KINASE R (PKR) ACTIVATING COMPOSITIONS CROSS-REFERENCE TO RELATED APPLICATIONS [0001]
- PYRUVATE KINASE R (PKR) ACTIVATING COMPOSITIONS CROSS-REFERENCE TO RELATED APPLICATIONS [0001]
- TECHNICAL FIELD [0002] The present disclosure is directed to solid forms, dispersions and pharmaceutical compositions of a pyruvate kinase R (PKR) activating compound.
- PLR pyruvate kinase R
- the present disclosure is directed to crystalline solid forms, spray-dried dispersions and pharmaceutical compositions of (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one, and preparation methods thereof.
- Chemical compounds can form one or more different pharmaceutically acceptable solid forms, including amorphous and crystalline forms.
- Amorphous solid forms include dispersions, such as spray-dried dispersions, of amorphous and crystalline chemical compounds.
- Compound 1 is described in International Publication No. WO 2018/175474 as one of many compounds suitable as small molecule modulators of pyruvate kinase activity. There remains a need for identifying solid forms of Compound 1 useful for various therapeutic applications.
- SUMMARY [0005] One aspect of the disclosure relates to solid oral dosage forms comprising a stabilized amorphous pharmaceutical composition of the compound (S)-1-(5-[2H,3H-[1,4]dioxino[2,3- b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2- phenylpropan-1-one (also referred to as “stabilized amorphous Compound 1”).
- stabilized amorphous Compound 1 refers to an amorphous solid form of Compound 1 that is stabilized (e.g., by combination with certain stabilizing polymers and/or other manufacturing processes) to prevent the formation of crystalline forms of Compound 1 or solid phase separation of Compound 1 under certain storage conditions described herein (e.g., stabilized amorphous pharmaceutical compositions comprising Compound 1 and one or more additional components that do not show crystalline diffraction peaks by XRPD analysis (Method D) after 2 weeks of storage at 60 °C/75% RH (exposed), and/or show a single glass transition temperature (T G ) with no melt endotherm by DSC analysis (Method B) after 2 weeks of storage at 60 °C/75% RH (exposed)).
- the stabilized amorphous Compound 1 is obtained by spray drying a solution of Compound 1 with a stabilizing polymer.
- the inventors discovered that amorphous Compound 1 has higher oral bioavailability than certain crystalline forms of Compound 1, including crystalline form Type A.
- solid oral dosage forms comprising stabilized amorphous Compound 1 advantageously provide superior oral bioavailability of Compound 1 in comparison to solid oral dosage forms comprising certain crystalline forms of Compound 1.
- SDD amorphous spray-dried dispersion
- the present disclosure provides various solid forms of Compound 1, including one or more pharmaceutically acceptable crystalline and amorphous forms for Compound 1, useful for the therapeutic oral administration of Compound 1.
- the various solid forms of Compound 1 can be identified by certain characteristic properties. For example, certain crystalline forms of Compound 1 have distinct characteristic XRPD peaks.
- Another aspect of the disclosure relates to solid forms of Compound 1.
- Solid forms of Compound 1 disclosed herein include various crystalline forms (including Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, and Type M) of Compound 1, preparation methods thereof, and pharmaceutical compositions containing the same.
- a novel Compound 1 crystalline form Type A can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.61, 15.66, 23.19, and 24.76.
- a novel Compound 1 crystalline form Type A can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.7, 23.2, and 24.8.
- a novel Compound 1 crystalline form Type A can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 7.2, 15.7, 21.3, 23.2, and 24.8.
- a novel Compound 1 crystalline form Type B can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.52, 15.57, 22.89, 23.34, and 25.13.
- a novel Compound 1 crystalline form Type B can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.9, 23.3, and 25.1.
- a novel Compound 1 crystalline form Type B can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.2, 22.9, 23.3, and 25.1.
- a novel Compound 1 crystalline form Type C can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.55, 18.85, 23.02, and 24.65.
- a novel Compound 1 crystalline form Type C can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 18.9, 23.0, and 24.7.
- a novel Compound 1 crystalline form Type C can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 7.3, 11.2, 18.9, 23.0, and 24.7.
- a novel Compound 1 crystalline form Type D can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 9.72, 13.08, 15.74, 21.90, and 23.59.
- a novel Compound 1 crystalline form Type D can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 9.7, 13.1, 15.7, 21.9, and 23.6.
- a novel Compound 1 crystalline form Type D can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 6.2, 9.7, 13.1, 15.7, 21.9, and 23.6 and not having a diffraction at an angle (2 theta ⁇ 0.2) of 23.3.
- a novel Compound 1 crystalline form Type E can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.12, 15.75, 17.48, 20.05, 21.93, and 26.72.
- a novel Compound 1 crystalline form Type E can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7.
- a novel Compound 1 crystalline form Type E can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7.
- a novel Compound 1 crystalline form Type F can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 20.01, 21.36, and 22.45.
- a novel Compound 1 crystalline form Type F can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 20.0, 21.4, and 22.5.
- a novel Compound 1 crystalline form Type F can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, and 21.4.
- a novel Compound 1 crystalline form Type G can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 14.34, 16.58, and 21.35.
- a novel Compound 1 crystalline form Type G can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, and 21.4.
- a novel Compound 1 crystalline form Type G can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, 21.3, and 22.3.
- a novel Compound 1 crystalline form Type H can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.8, 14.7, 16.6, 20.0, 21.3, and 25.4.
- a novel Compound 1 crystalline form Type I can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.2, 14.6, 15.5, 20.2, and 21.1.
- a novel Compound 1 crystalline form Type J can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.7, 22.8, 23.1, and 24.5.
- a novel Compound 1 crystalline form Type K can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.4, 15.6, 16.1, 23.2, and 27.4.
- a novel Compound 1 crystalline form Type L can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.9, 11.9, 17.8, 21.6, 23.9, and 36.1.
- a novel Compound 1 crystalline form Type M can be identified by X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.8, 9.7, 15.6, 21.9, and 26.7.
- XRPD X-ray Powder Diffraction
- Another aspect of the present disclosure relates to a pharmaceutical composition comprising a therapeutically effective amount of any of the crystalline solid forms of Compound 1 described above, and one or more pharmaceutically acceptable excipients.
- Yet another aspect of the present disclosure relates to a novel amorphous solid dispersion of Compound 1.
- the novel amorphous solid form of Compound 1 can be prepared by spray-drying a mixture comprising Compound 1 and a polymer.
- Still another aspect of the present disclosure relates to a pharmaceutical composition comprising the novel amorphous solid form of Compound 1 described above.
- the pharmaceutical composition may be in an oral dosage form, such as tablets.
- Another aspect of the present disclosure relates to tablet dosage forms comprising Compound 1.
- BRIEF DESCRIPTION OF THE DRAWINGS [0027] Fig.1 depicts a reaction scheme to prepare Compound 1.
- Fig.2 depicts an alternative reaction scheme to prepare Compound 1.
- Fig.3 depicts an XRPD pattern of Compound 1 crystalline form Type A. [0030] Fig.
- thermogravimetric analysis (TGA) curve upper curve
- DSC differential scanning calorimetry
- Fig.5 depicts a DSC cycling thermogram for Compound 1 crystalline form Type A.
- Fig. 6 depicts a dynamic vapor sorption (DVS) isotherm for Compound 1 crystalline form Type A.
- Fig.7 depicts an XRPD pattern of Compound 1 crystalline form Type B.
- Fig. 8 depicts a thermogravimetric analysis (TGA) curve (upper curve) and a differential scanning calorimetry (DSC) thermogram (lower curve) for Compound 1 crystalline form Type B.
- Fig.9 depicts a DSC cycling thermogram for Compound 1 crystalline form Type B.
- Fig. 10 depicts two thermogravimetric analysis (TGA) curves for Compound 1 crystalline form Type B.
- Fig.11 depicts a dynamic vapor sorption (DVS) isotherm for Compound 1 crystalline form Type B.
- Fig.12 depicts an XRPD pattern of Compound 1 crystalline form Type C.
- Fig. 13 depicts a thermogravimetric analysis (TGA) curve (upper curve) and a differential scanning calorimetry (DSC) thermogram (lower curve) for Compound 1 crystalline form Type C.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- Fig.14 depicts a DSC cycling thermogram for Compound 1 crystalline form Type C.
- Fig.15 depicts thermogravimetric analysis (TGA) curves for Compound 1 crystalline form Type C.
- Fig.16 depicts a dynamic vapor sorption (DVS) isotherm for Compound 1 crystalline form Type C.
- Fig.17 depicts an XRPD pattern of Compound 1 crystalline form Type D.
- Fig. 18 depicts a thermogravimetric analysis (TGA) curve (upper curve) and a differential scanning calorimetry (DSC) thermogram (lower curve) for Compound 1 crystalline form Type D.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- Fig.19 depicts a 1 H NMR spectrum of Type A (upper curve) and Type D (lower curve) crystalline forms of Compound 1.
- Fig.20 depicts an XRPD pattern of Compound 1 crystalline form Type E.
- Fig.21 depicts an XRPD pattern of Compound 1 crystalline form Type F.
- Fig. 22 is a thermogravimetric analysis (TGA) curve (upper curve) and a differential scanning calorimetry (DSC) thermogram (lower curve) for Compound 1 crystalline form Type F.
- Fig.23 depicts an XRPD pattern of Compound 1 crystalline form Type G.
- Fig.24 depicts an XRPD pattern of Compound 1 crystalline form Type H.
- Fig.25 depicts an XRPD pattern of Compound 1 crystalline form Type I.
- Fig.26 depicts an XRPD pattern of Compound 1 crystalline form Type J.
- Fig.27 depicts an XRPD pattern of Compound 1 crystalline form Type K.
- Fig.28 depicts an XRPD pattern of Compound 1 crystalline form Type L.
- Fig.29 depicts an XRPD pattern of Compound 1 crystalline form Type M.
- Fig.30 depicts an XRPD pattern of a spray-dried dispersion (SDD) of Compound 1.
- SDD spray-dried dispersion
- Fig.31 depicts a differential scanning calorimetry (DSC) thermogram for a spray-dried dispersion (SDD) of Compound 1.
- Fig.32 depicts a graph of the plasma concentration over time following administration of three formulations of Compound 1 in rats.
- Fig.33 depicts a graph of the plasma concentration over time following administration of four formulations of Compound 1 in monkeys.
- Fig. 34 depicts a graph of time-dependent solubility of Type A of Compound 1 in biorelevant media.
- Fig.35 depicts a graph of time-dependent solubility of a spray-dried dispersion (SDD) of Compound 1 in biorelevant media.
- Fig.31 depicts a differential scanning calorimetry (DSC) thermogram for a spray-dried dispersion (SDD) of Compound 1.
- Fig.32 depicts a graph of the plasma concentration over time following administration of three formulations of Compound 1 in rats.
- Fig.33 depicts a graph of the plasma
- FIG. 36 depicts overlayed XRPD patterns of five spray-dried dispersions (SDDs) of Compound 1, overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.37 depicts overlayed differential scanning calorimetry (DSC) thermograms of five spray-dried dispersions (SDDs) of Compound 1.
- Fig.38 depicts a graph of the kinetic solubility profiles of five SDDs of Compound 1 at different drug loadings.
- Fig.39 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 0) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, and (c) in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity, overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.40 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 1) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, and (c) in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity, overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.41 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 2) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, and (c) in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity, overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.42 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 3) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, and (c) in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity, overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.43 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 4) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, and (c) in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity, overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.44 depicts overlayed XRPD patterns of two spray dried dispersions of Compound 1 (SDDs 5 and 6), overlayed with the XRPD pattern of crystalline Compound 1 (Type A).
- Fig.43 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 4) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity
- Fig.46 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 5) after storage (a) in a sealed vial for 1 week at 60 °C, (b) in an unsealed vial for 1 week at 25 °C and 60% relative humidity, and (c) in an unsealed vial for 1 week at 40 °C and 75% relative humidity.
- Fig.47 depicts overlayed DSC thermograms of a spray dried dispersion of Compound 1 (SDD 5) after storage (a) in a sealed vial for 1 week at 60 °C, (b) in an unsealed vial for 1 week at 25 °C and 60% relative humidity, and (c) in an unsealed vial for 1 week at 40 °C and 75% relative humidity.
- Fig.48 depicts overlayed DSC thermograms of a spray dried dispersion of Compound 1 (SDD 5) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 25 °C and 60% relative humidity, and (c) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity.
- Fig.49 depicts overlayed XRPD patterns of a spray dried dispersion of Compound 1 (SDD 6) after storage (a) in a sealed vial for 1 week at 60 °C, (b) in an unsealed vial for 1 week at 25 °C and 60% relative humidity, and (c) in an unsealed vial for 1 week at 40 °C and 75% relative humidity.
- Fig.50 depicts overlayed DSC thermograms of a spray dried dispersion of Compound 1 (SDD 6) after storage (a) in a sealed vial for 1 week at 60 °C, (b) in an unsealed vial for 1 week at 25 °C and 60% relative humidity, and (c) in an unsealed vial for 1 week at 40 °C and 75% relative humidity.
- Fig.51 depicts overlayed DSC thermograms of a spray dried dispersion of Compound 1 (SDD 6) after storage (a) in a sealed vial for 2 weeks at 60 °C, (b) in an unsealed vial for 2 weeks at 25 °C and 60% relative humidity, and (c) in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity.
- Fig.52 depicts a graph of the dissolution profile of a tablet formulation of Compound 1.
- Compound 1 is a small molecule modulator of pyruvate kinase.
- the present disclosure provides various solid forms of Compound 1, pharmaceutical compositions thereof, and methods of preparing those novel solid forms of Compound 1.
- the solid forms described herein are associated with favorable characteristics such as favorable or improved solubility, dissolution, bioavailability, stability, and ease of formulation relative to other forms of Compound 1.
- certain amorphous solid dispersions described herein advantageously have high drug loads (e.g., 3 25%, 3 40%, 3 50%, etc.), are free or substantially free of crystalline Compound 1, are physically stable (i.e., remain free or substantially free of crystalline Compound 1 over time in accelerated stability studies), are highly soluble, and/or do not require extensive drying to remove residual solvents.
- certain tablet dosage forms described herein advantageously have high drug loads (e.g., 3 10 weight % of the tablet core, 3 15 weight % of the tablet core, 3 30 weight % of the tablet core), small tablet sizes (e.g., tablet core weight £ 1200 mg, £ 1000 mg, £ 800 mg, £ 700 mg, etc. per tablet), are free or substantially free of crystalline Compound 1, and/or are physically stable (i.e., remain free or substantially free of crystalline Compound 1 over time in accelerated stability studies).
- Compound 1 is in a crystalline solid form (e.g., Type A, Type B, Type C, Type D, Type E, Type F, or Type G).
- Compound 1 is in a crystalline solid form (e.g., Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, or Type M).
- the crystalline solid form is Type A.
- the crystalline solid form is Type B.
- the crystalline solid form is Type C.
- the crystalline solid form is Type D.
- the crystalline solid form is Type E.
- the crystalline solid form is Type F.
- the crystalline solid form is Type G.
- the crystalline solid form is Type H.
- the crystalline solid form is Type I.
- the crystalline solid form is Type J. In some embodiments, the crystalline solid form is Type K. In some embodiments, the crystalline solid form is Type L. In some embodiments, the crystalline solid form is Type M. [0081] In some embodiments, Compound 1 is in amorphous form (e.g., an amorphous solid dispersion). In some embodiments, the amorphous solid dispersion comprises Compound 1 and a polymer.
- a novel Compound 1 crystalline form Type A can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.61, 15.66, 23.19, and 24.76.
- a novel Compound 1 crystalline form Type A can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.7, 23.2, and 24.8.
- Compound 1 crystalline form Type A can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.61, 15.66, 23.19, and 24.76, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.19, 5.66, 3.84, and 3.60, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type A can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.7, 23.2, and 24.8, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.2, 5.7, 3.8, and 3.6, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type A can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 7.2, 15.7, 21.3, 23.2, and 24.8.
- Compound 1 crystalline form Type A can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 7.2, 15.7, 21.3, 23.2, and 24.8, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.2, 12.3, 5.7, 4.2, 3.8, and 3.6, respectively.
- Compound 1 crystalline form Type A can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.61, 7.22, 15.66, 20.48, 21.35, 21.66, 22.47, 23.19, 24.76, and 26.73.
- Compound 1 crystalline form Type A can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 7.2, 15.7, 20.5, 21.4, 21.7, 22.5, 23.2, 24.8, and 26.7.
- Compound 1 crystalline form Type A can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.61, 7.22, 15.66, 20.48, 21.35, 21.66, 22.47, 23.19, 24.76, and 26.73, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.19, 12.25, 5.66, 4.34, 4.16, 4.10, 3.96, 3.84, 3.60, and 3.34, respectively.
- Compound 1 crystalline form Type A can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 7.2, 15.7, 20.5, 21.4, 21.7, 22.5, 23.2, 24.8, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.2, 12.2, 5.7, 4.3, 4.2, 4.1, 4.0, 3.8, 3.6, and 3.3, respectively.
- Compound 1 crystalline form Type A is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.61 5.80 7.22 7.68 11.21 12.31 14.44 15.66 16.95 18.02 19.20 20.48 21.35 21.66 22.47 23.19 24.76 26.73 28.01 28.49 29.35 30.25 32.14 34.12 36.46 [0086] In some embodiments, Compound 1 crystalline form Type A is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.6 5.8 7.2 7.7 11.2 12.3 14.4 15.7 16.9 18.0 19.2 20.5 21.3 21.7 22.5 23.2 24.8 26.7 28.0 28.5 29.4 30.3 32.1 34.1 36.5 [0087] In some embodiments, Compound 1 crystalline form Type A is characterized by an X- ray Power Diffraction pattern
- Compound 1 crystalline form Type A is characterized by a differential scanning calorimetry (DSC) endotherm having a peak temperature of about 85.9 oC and an onset temperature of about 146.0 oC.
- Compound 1 crystalline form Type A is characterized by a dynamic vapor sorption (DVS) of about 3.4% water uptake by weight up to 40% relative humidity.
- Compound 1 crystalline form Type A is characterized by a dynamic vapor sorption (DVS) of about 1.0% water uptake by weight from 40% to 80% relative humidity.
- a novel Compound 1 crystalline form Type B can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.52, 15.57, 22.89, 23.34, and 25.13.
- a novel Compound 1 crystalline form Type B can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.9, 23.3, and 25.1.
- Compound 1 crystalline form Type B can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.52, 15.57, 22.89, 23.34, and 25.13, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.53, 5.69, 3.89, 3.81, and 3.54, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type B can be identified by X- ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.9, 23.3, and 25.1, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 5.7, 3.9, 3.8, and 3.5, respectively.
- XRPD X- ray Powder Diffraction
- Compound 1 crystalline form Type B can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.2, 22.9, 23.3, and 25.1.
- Compound 1 crystalline form Type B can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.2, 22.9, 23.3, and 25.1, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 5.7, 4.0, 3.9, 3.8, and 3.5, respectively.
- Compound 1 crystalline form Type B can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.52, 9.86, 15.57, 19.93, 22.19, 22.89, 23.34, 25.13, and 28.30.
- Compound 1 crystalline form Type B can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 9.9, 15.6, 19.9, 22.2, 22.9, 23.3, 25.1, and 28.3.
- Compound 1 crystalline form Type B can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.52, 9.86, 15.57, 19.93, 22.19, 22.89, 23.34, 25.13, and 28.30, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.53, 8.97, 5.69, 4.45, 4.00, 3.89, 3.81, 3.54, and 3.15, respectively.
- Compound 1 crystalline form Type B can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 9.9, 15.6, 19.9, 22.2, 22.9, 23.3, 25.1, and 28.3, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 9.0, 5.7, 4.5, 4.0, 3.9, 3.8, 3.5, and 3.2, respectively.
- Compound 1 crystalline form Type B is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.52 8.98 9.86 12.37 13.18 15.57 16.86 18.21 19.11 19.93 20.92 22.19 22.89 23.34 25.13 25.80 26.71 28.30 29.39 [0094] In some embodiments, Compound 1 crystalline form Type B is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.5 9.0 9.9 12.4 13.2 15.6 16.9 18.2 19.1 19.9 20.9 22.2 22.9 23.3 25.1 25.8 26.7 28.3 29.4 [0095] In some embodiments, Compound 1 crystalline form Type B is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-
- Compound 1 crystalline form Type B is characterized by a differential scanning calorimetry (DSC) endotherm having an onset temperature of about 138.2- 139.2 oC.
- Compound 1 crystalline form Type B is characterized by a dynamic vapor sorption (DVS) of about 2.9% water uptake by weight up to 60% relative humidity, and a dynamic vapor sorption (DVS) of about 0.4% water uptake by weight from 60% to 80% relative humidity.
- DSC differential scanning calorimetry
- a novel Compound 1 crystalline form Type C can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.55, 18.85, 23.02, and 24.65.
- a novel Compound 1 crystalline form Type C can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 18.9, 23.0, and 24.7.
- Compound 1 crystalline form Type C can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.55, 18.85, 23.02, and 24.65, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.43, 4.71, 3.86, and 3.61, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type C can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 18.9, 23.0, and 24.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.4, 4.7, 3.9, and 3.6, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type C can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 7.3, 11.2, 18.9, 23.0, and 24.7.
- Compound 1 crystalline form Type C can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 7.3, 11.2, 18.9, 23.0, and 24.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.4, 12.0, 7.9, 4.7, 3.9, and 3.6, respectively.
- Compound 1 crystalline form Type C can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.55, 7.34, 9.07, 11.17, 18.34, 18.85, 19.57, 21.66, 23.02, and 24.65.
- Compound 1 crystalline form Type C can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 7.3, 9.1, 11.2, 18.34, 18.9, 19.6, 21.7, 23.0, and 24.7.
- Compound 1 crystalline form Type C can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.55, 7.34, 9.07, 11.17, 18.34, 18.85, 19.57, 21.66, 23.02, and 24.65, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.43, 12.05, 9.75, 7.92, 4.84, 4.71, 4.54, 4.10, 3.86, and 3.61, respectively.
- Compound 1 crystalline form Type C can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 7.3, 9.1, 11.2, 18.3, 18.9, 19.6, 21.7, 23.0, and 24.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.4, 12.0, 9.8, 7.9, 4.8, 4.7, 4.5, 4.1, 3.9, and 3.6, respectively.
- Compound 1 crystalline form Type C is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.55 7.34 9.07 11.17 12.29 14.51 15.66 18.34 18.85 19.57 20.38 21.66 23.02 24.65 26.39 28.28 30.09 32.31 33.91 37.19 [0102] In some embodiments, Compound 1 crystalline form Type C is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.5 7.3 9.1 11.2 12.3 14.5 15.7 18.3 18.9 19.6 20.4 21.7 23.0 24.7 26.4 28.3 30.1 32.3 33.9 37.2 [0103] In some embodiments, Compound 1 crystalline form Type C is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and
- Compound 1 crystalline form Type C is characterized by a differential scanning calorimetry (DSC) endotherm having an onset temperature of about 152.2- 154.2 oC. In some embodiments, Compound 1 crystalline form Type C is characterized by a dynamic vapor sorption (DVS) of about 1.8% water uptake by weight up to 60% relative humidity, and a dynamic vapor sorption (DVS) of about 0.5% water uptake by weight from 60% to 80% relative humidity.
- DSC differential scanning calorimetry
- a novel Compound 1 crystalline form Type D can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 9.72, 13.08, 15.74, 21.90, and 23.59.
- a novel Compound 1 crystalline form Type D can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 9.7, 13.1, 15.7, 21.9, and 23.6.
- Compound 1 crystalline form Type D can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 9.72, 13.08, 15.74, 21.90, and 23.59, corresponding to d-spacing (angstroms ⁇ 0.2) of 9.10, 6.77, 5.63, 4.06 and 3.77, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type D can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 9.7, 13.1, 15.7, 21.9, and 23.6, corresponding to d-spacing (angstroms ⁇ 0.2) of 9.1, 6.8, 5.6, 4.1 and 3.8, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type D can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 6.2, 9.7, 13.1, 15.7, 21.9, and 23.6 and not having a diffraction at an angle (2 theta ⁇ 0.2) of 23.3.
- Compound 1 crystalline form Type D can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 6.2, 9.7, 13.1, 15.7, 21.9, and 23.6, corresponding to d-spacing (angstroms ⁇ 0.2) of 14.4, 9.1, 6.8, 5.6, 4.1 and 3.8, respectively, and not having a diffraction at an angle (2 theta ⁇ 0.2) of 23.3.
- Compound 1 crystalline form Type D can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.27, 6.15, 8.71, 9.72, 12.31, 13.08, 13.76, 15.74, 18.02, 21.90, 23.59, and 26.71.
- Compound 1 crystalline form Type D can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.3, 6.2, 8.7, 9.7, 12.3, 13.1, 13.8, 15.7, 18.0, 21.9, 23.6, and 26.7.
- Compound 1 crystalline form Type D can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.27, 6.15, 8.71, 9.72, 12.31, 13.08, 13.76, 15.74, 18.02, 21.90, 23.59, and 26.71, corresponding to d- spacing (angstroms ⁇ 0.2) of 20.68, 14.36, 10.16, 9.10, 7.19, 6.77, 6.44, 5.63, 4.92, 4.06, 3.77, and 3.34, respectively.
- Compound 1 crystalline form Type D can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.3, 6.2, 8.7, 9.7, 12.3, 13.1, 13.8, 15.7, 18.0, 21.9, 23.6, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 20.7, 14.4, 10.2, 9.1, 7.2, 6.8, 6.4, 5.6, 4.9, 4.1, 3.8, and 3.3, respectively.
- Compound 1 crystalline form Type D is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.27 6.15 8.71 9.72 12.31 13.08 13.76 15.74 18.02 19.55 21.90 23.59 24.79 26.71 29.50 30.82 31.74 35.40 37.84 38.61 [0110] In some embodiments, Compound 1 crystalline form Type D is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.3 6.2 8.7 9.7 12.3 13.1 13.8 15.7 18.0 19.5 21.9 23.6 24.8 26.7 29.5 30.8 31.7 35.4 37.8 38.6 [0111] In some embodiments, Compound 1 crystalline form Type D is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and
- Compound 1 crystalline form Type D is characterized by a differential scanning calorimetry (DSC) endotherm having an onset temperature of about 91.9 oC.
- Compound 1 Crystalline Form Type E [0114]
- a novel Compound 1 crystalline form Type E can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.12, 15.75, 17.48, 20.05, 21.93, and 26.72.
- XRPD X-ray Powder Diffraction
- a novel Compound 1 crystalline form Type E can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7.
- Compound 1 crystalline form Type E can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.12, 15.75, 17.48, 20.05, 21.93, and 26.72, corresponding to d-spacing (angstroms ⁇ 0.2) of 5.86, 5.63, 5.07, 4.43, 4.05, and 3.34, respectively.
- Compound 1 crystalline form Type E can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7, corresponding to d- spacing (angstroms ⁇ 0.2) of 5.9, 5.6, 5.1, 4.4, 4.1, and 3.3, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type E can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7.
- Compound 1 crystalline form Type E can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 19.0, 20.1, 21.9, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 5.9, 5.6, 5.1, 4.7, 4.4, 4.1, and 3.3, respectively.
- Compound 1 crystalline form Type E can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.59, 15.12, 15.75, 17.48, 20.05, 21.93, 23.18, 23.70, and 26.72.
- Compound 1 crystalline form Type E can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.1, 15.8, 17.5, 20.1, 21.9, 23.2, 23.7, and 26.7.
- Compound 1 crystalline form Type E can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.59, 15.12, 15.75, 17.48, 20.05, 21.93, 23.18, 23.70, and 26.72, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.27, 5.86, 5.63, 5.07, 4.43, 4.05, 3.84, 3.75, and 3.34, respectively.
- Compound 1 crystalline form Type E can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.1, 15.8, 17.5, 20.1, 21.9, 23.2, 23.7, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.3, 5.9, 5.6, 5.1, 4.4, 4.1, 3.8, 3.8, and 3.3, respectively.
- Compound 1 crystalline form Type E can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.59, 9.76, 12.36, 13.12, 15.12, 15.75, 16.84, 17.48, 18.06, 19.02, 20.05, 21.93, 23.18, 23.70, 26.72, and 27.81.
- Compound 1 crystalline form Type E can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 9.8, 12.4, 13.1, 15.1, 15.8, 16.8, 17.5, 18.1, 19.0, 20.1, 21.9, 23.2, 23.7, 26.7, and 27.8.
- Compound 1 crystalline form Type E can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.59, 9.76, 12.36, 13.12, 15.12, 15.75, 16.84, 17.48, 18.06, 19.02, 20.05, 21.93, 23.18, 23.70, 26.72, and 27.81, corresponding to d- spacing (angstroms ⁇ 0.2) of 19.27, 9.06, 7.16, 6.75, 5.86, 5.63, 5.27, 5.07, 4.91, 4.67, 4.43, 4.05, 3.84, 3.75, 3.34, and 3.21, respectively.
- Compound 1 crystalline form Type E can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 9.8, 12.4, 13.1, 15.1, 15.8, 16.8, 17.5, 18.1, 19.0, 20.1, 21.9, 23.2, 23.7, 26.7, and 27.8, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.3, 9.1, 7.2, 6.7, 5.9, 5.6, 5.3, 5.1, 4.9, 4.7, 4.4, 4.1, 3.8, 3.8, 3.3, and 3.2, respectively.
- Compound 1 crystalline form Type E is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.59 8.76 9.76 12.36 13.12 13.83 15.12 15.75 16.84 17.48 18.06 19.02 20.05 21.93 23.18 23.70 24.82 26.72 27.81 29.51 30.76 31.74 33.03 34.52 35.39 36.72 37.77 38.66 [0119] In some embodiments, Compound 1 crystalline form Type E is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.6 8.8 9.8 12.4 13.1 13.8 15.1 15.8 16.8 17.5 18.1 19.0 20.1 21.9 23.2 23.7 24.8 26.7 27.8 29.5 30.8 31.7 33.0 34.5 35.4 36.7 37.8 38.7 [0120] In some embodiments, Compound 1 crystalline form Type
- a novel Compound 1 crystalline form Type F can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 20.0, 21.4, and 22.5.
- Compound 1 crystalline form Type F can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 20.01, 21.36, and 22.45, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.23, 6.04, 5.54, 5.28, 4.44, 4.16, and 3.96, respectively.
- Compound 1 crystalline form Type F can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 20.0, 21.4, and 22.5, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.0, 5.5, 5.3, 4.4, 4.2, and 4.0, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type F can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, and 21.4.
- Compound 1 crystalline form Type F can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, and 21.4, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.0, 5.5, 5.3, and 4.2, respectively.
- Compound 1 crystalline form Type F can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 18.99, 20.01, 21.36, 22.45, 23.25, and 25.32.
- Compound 1 crystalline form Type F can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 19.0, 20.0, 21.4, 22.5, 23.2, and 25.3.
- Compound 1 crystalline form Type F can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 18.99, 20.01, 21.36, 22.45, 23.25, and 25.32, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.23, 6.04, 5.54, 5.28, 4.67, 4.44, 4.16, 3.96, 3.83, and 3.52, respectively.
- Compound 1 crystalline form Type F can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 19.0, 20.0, 21.4, 22.5, 23.2, and 25.3, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.0, 5.5, 5.3, 4.7, 4.4, 4.2, 4.0, 3.8, and 3.5, respectively.
- Compound 1 crystalline form Type F can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.45, 12.87, 14.66, 16.00, 16.79, 17.36, 18.99, 20.01, 20.57, 21.36, 22.45, 23.25, 25.32, 26.57, 27.25, 27.97, and 30.02.
- Compound 1 crystalline form Type F can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.9, 14.7, 16.0, 16.8, 17.4, 19.0, 20.0, 20.6, 21.4, 22.5, 23.2, 25.3, 26.6, 27.2, 28.0, and 30.0.
- Compound 1 crystalline form Type F can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.45, 12.87, 14.66, 16.00, 16.79, 17.36, 18.99, 20.01, 20.57, 21.36, 22.45, 23.25, 25.32, 26.57, 27.25, 27.97, and 30.02, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.23, 6.88, 6.04, 5.54, 5.28, 5.11, 4.67, 4.44, 4.32, 4.16, 3.96, 3.83, 3.52, 3.35, 3.27, 3.19, and 2.98, respectively.
- Compound 1 crystalline form Type F can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.9, 14.7, 16.0, 16.8, 17.4, 19.0, 20.0, 20.6, 21.4, 22.5, 23.2, 25.3, 26.6, 27.2, 28.0, and 30.0, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.9, 6.0, 5.5, 5.3, 5.1, 4.7, 4.4, 4.3, 4.2, 4.0, 3.8, 3.5, 3.4, 3.3, 3.2, and 3.0, respectively.
- Compound 1 crystalline form Type F is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.45 10.92 12.87 14.66 16.00 16.79 17.36 18.99 20.01 20.57 21.36 22.45 23.25 25.32 26.57 27.25 27.97 30.02 31.98 32.89 38.29 39.09 [0127] In some embodiments, Compound 1 crystalline form Type F is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.4 10.9 12.9 14.7 16.0 16.8 17.4 19.0 20.0 20.6 21.4 22.5 23.2 25.3 26.6 27.2 28.0 30.0 32.0 32.9 38.3 39.1 [0128] In some embodiments, Compound 1 crystalline form Type F is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇
- Compound 1 crystalline form Type F is characterized by a differential scanning calorimetry (DSC) endotherm having a peak temperature of about 100.4 oC and an onset temperature of 125.9 oC.
- Compound 1 Crystalline Form Type G [0131] A novel Compound 1 crystalline form Type G can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 14.34, 16.58, and 21.35.
- XRPD X-ray Powder Diffraction
- a novel Compound 1 crystalline form Type G can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, and 21.4.
- Compound 1 crystalline form Type G can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 14.34, 16.58, and 21.35, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.48, 6.18, 5.35, and 4.16, respectively.
- Compound 1 crystalline form Type G can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, and 21.4, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.5, 6.2, 5.3, and 4.2, respectively. [0132] In some embodiments, Compound 1 crystalline form Type G can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, 21.3, and 22.3.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type G can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, 21.3, and 22.3, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.5, 6.2, 5.3, 4.2, and 4.0, respectively. [0133] In some embodiments, Compound 1 crystalline form Type G can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 16.58, 19.78, 21.35, 22.35, 25.33, and 26.43.
- Compound 1 crystalline form Type G can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 16.6, 19.8, 21.3, 22.3, 25.3, and 26.4.
- Compound 1 crystalline form Type G can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 16.58, 19.78, 21.35, 22.35, 25.33, and 26.43, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.48, 6.90, 6.18, 5.91, 5.35, 4.49, 4.16, 3.98, 3.52, and 3.37, respectively.
- Compound 1 crystalline form Type G can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 16.6, 19.8, 21.3, 22.3, 25.3, and 26.4, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.5, 6.9, 6.2, 5.9, 5.3, 4.5, 4.2, 4.0, 3.5, and 3.4, respectively.
- Compound 1 crystalline form Type G can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 15.79, 16.58, 19.78, 21.35, 22.35, 25.33, 26.43, 27.35, and 30.21.
- Compound 1 crystalline form Type G can be identified by an XRPD pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.3412.8, 14.3, 15.0, 15.8, 16.6, 19.8, 21.3, 22.3, 25.3, 26.4, 27.4, and 30.2.
- Compound 1 crystalline form Type G can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 15.79, 16.58, 19.78, 21.35, 22.35, 25.33, 26.43, 27.35, and 30.21, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.48, 6.90, 6.18, 5.91, 5.61, 5.35, 4.49, 4.16, 3.98, 3.52, 3.37, 3.26, and 2.96, respectively.
- Compound 1 crystalline form Type G can be identified by XRPD, having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 15.8, 16.6, 19.8, 21.3, 22.3, 25.3, 26.4, 27.4, and 30.2, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.5, 6.9, 6.2, 5.9, 5.6, 5.3, 4.5, 4.2, 4.0, 3.5, 3.4, 3.3, and 3.0, respectively.
- Compound 1 crystalline form Type G is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.36 8.73 12.83 14.34 15.00 15.79 16.58 18.54 19.78 21.35 22.35 23.38 25.33 26.43 27.35 30.21 32.32 38.04 [0136] In some embodiments, Compound 1 crystalline form Type G is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.4 8.7 12.8 14.3 15.0 15.8 16.6 18.5 19.8 21.3 22.3 23.4 25.3 26.4 27.4 30.2 32.3 38.0 [0137] In some embodiments, Compound 1 crystalline form Type G is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstrom).
- Compound 1 crystalline form Type H can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.8, 14.7, 16.6, 20.0, 21.3, and 25.4, corresponding to d- spacing (angstroms ⁇ 0.2) of 15.3, 6.0, 5.4, 4.4, 4.2, and 3.5, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type H is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.8 8.4 11.5 12.4 13.1 13.7 14.7 14.9 16.0 16.2 16.6 16.9 17.3 17.7 18.3 19.5 20.0 21.3 21.9 23.1 23.6 23.9 24.4 24.9 25.1 25.4 26.2 27.4 28.1 28.4 29.3 29.7 30.4 31.0 32.7 33.4 34.1 34.8 35.5 35.8 36.4 37.1 38.5 [0141] In some embodiments, Compound 1 crystalline form Type H is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstroms ⁇ 0.2) of: Pos.
- Compound 1 crystalline form Type I can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.2, 14.6, 15.5, 20.2, and 21.1, corresponding to d-spacing (angstroms ⁇ 0.2) of 17.1, 6.1, 5.7, 4.4, and 4.2, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type I is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.2 8.8 10.3 12.6 14.6 15.5 16.1 16.3 16.6 17.1 17.6 18.7 18.9 20.2 20.5 20.7 21.1 21.5 22.0 22.3 23.7 24.8 25.2 26.0 26.3 26.5 26.8 27.0 27.5 27.7 28.1 29.6 30.0 30.4 31.3 32.0 32.5 33.2 34.0 34.6 36.9 38.2 38.9 39.5 [0144] In some embodiments, Compound 1 crystalline form Type I is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstroms ⁇ 0.2) of: Pos.
- a novel Compound 1 crystalline form Type J can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.7, 22.8, 23.1, and 24.5.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type J can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.7, 22.8, 23.1, and 24.5, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 15.4, 3.9, 3.8, and 3.6, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type J is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.5 5.7 7.1 7.7 9.1 10.5 11.2 11.7 12.3 12.9 14.3 14.5 15.4 15.7 16.3 17.3 18.3 18.7 19.3 19.6 20.5 21.2 21.5 22.8 23.1 23.6 24.1 24.5 25.2 25.9 26.4 27.8 29.3 36.2 37.0 [0147] In some embodiments, Compound 1 crystalline form Type J is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstroms ⁇ 0.2) of: Pos.
- a novel Compound 1 crystalline form Type K can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.4, 15.6, 16.1, 23.2, and 27.4.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type K can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.4, 15.6, 16.1, 23.2, and 27.4, corresponding to d- spacing (angstroms ⁇ 0.2) of 19.2, 5.7, 5.7, 5.5, 3.8, and 3.3, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type K is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.6 9.3 10.1 12.9 13.9 14.7 15.4 15.6 16.1 17.8 18.3 18.6 19.3 20.0 20.7 21.6 21.9 22.9 23.2 24.4 25.0 25.5 26.0 27.4 28.8 29.2 30.7 31.1 32.7 36.3 [0150] In some embodiments, Compound 1 crystalline form Type K is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstroms ⁇ 0.2) of: Pos.
- a novel Compound 1 crystalline form Type L can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.9, 11.9, 17.8, 21.6, 23.9, and 36.1.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type L can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 5.9, 11.9, 17.8, 21.6, 23.9, and 36.1, corresponding to d- spacing (angstroms ⁇ 0.2) of 14.9, 7.5, 5.0, 4.1, 3.7, and 2.5, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type L is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 5.9 8.4 11.9 13.3 14.7 15.0 16.2 16.7 16.9 17.8 18.9 20.4 21.2 21.6 22.2 23.9 24.6 25.5 25.7 26.1 26.8 28.1 28.8 29.9 30.6 31.9 32.4 33.6 34.2 35.6 36.1 38.2 [0153] In some embodiments, Compound 1 crystalline form Type L is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstroms ⁇ 0.2) of: Pos.
- a novel Compound 1 crystalline form Type M can be identified by an X-ray Powder Diffraction (XRPD) pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.8, 9.7, 15.6, 21.9, and 26.7.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type M can be identified by X-ray Powder Diffraction (XRPD), having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.8, 9.7, 15.6, 21.9, and 26.7, corresponding to d- spacing (angstroms ⁇ 0.2) of 19.5, 15.3, 9.1, 5.7, 4.1, and 3.3, respectively.
- XRPD X-ray Powder Diffraction
- Compound 1 crystalline form Type M is characterized by an X- ray Power Diffraction having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) of: 4.5 5.8 6.1 8.7 9.0 9.7 12.3 13.1 13.7 14.5 15.1 15.6 16.8 17.4 18.0 18.5 19.5 20.0 21.4 21.9 22.3 22.9 23.3 23.5 24.1 25.0 25.8 26.3 26.7 27.8 28.1 29.4 30.8 31.7 33.0 35.3 37.8 38.6 [0156] In some embodiments, Compound 1 crystalline form Type M is characterized by an X- ray Power Diffraction pattern having one or more characteristic diffractions at angles (2 theta ⁇ 0.2) and corresponding d-spacing (angstroms ⁇ 0.2) of: Pos.
- compositions Comprising Compound 1 Crystalline Form
- the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, or Type G
- the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, or Type M) of Compound 1 as discussed above, and one or more pharmaceutically acceptable excipients.
- the present disclosure provides a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, or Type G) of Compound 1 as discussed above, and one or more pharmaceutically acceptable excipients.
- the present disclosure provides a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, or Type M) of Compound 1 as discussed above, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition is for oral administration.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, or Type G) of Compound 1 as discussed above, and having a water content of about 0.5-5.0 weight%, preferably about 1.0-4.5 weight%, more preferably about 1.5-4.0 weight%, even more preferably about 2.0-3.5 weight%, still more preferably about 2.5-3.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, or Type M) of Compound 1 as discussed above, and having a water content of about 0.5-5.0 weight%, preferably about 1.0- 4.5 weight%, more preferably about 1.5-4.0 weight%, even more preferably about 2.0-3.5 weight%, still more preferably about 2.5-3.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, or Type G) of Compound 1 as discussed above, and having a water content in an amount selected from the following ranges: about 0.5-1.0 weight%, about 1.0-1.5 weight%, about 1.5-2.0 weight%, about 2.5-3.0 weight%, about 3.0-3.5 weight%, about 3.5-4.0 weight%, about 4.0-4.5 weight%, and about 4.5-5.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, or Type M) of Compound 1 as discussed above, and having a water content in an amount selected from the following ranges: about 0.5-1.0 weight%, about 1.0-1.5 weight%, about 1.5-2.0 weight%, about 2.5-3.0 weight%, about 3.0-3.5 weight%, about 3.5-4.0 weight%, about 4.0-4.5 weight%, and about 4.5-5.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, or Type G) of Compound 1 as discussed above, and having a water content in an amount selected from the weight percentage: about 0.5 weight%, about 1.0 weight%, about 1.5 weight%, about 2.0 weight%, about 2.5 weight%, about 3.0 weight%, about 3.5 weight%, about 4.0 weight%, about 4.5 weight%, and about 5.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising any crystalline solid form (Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, or Type M) of Compound 1 as discussed above, and having a water content in an amount selected from the weight percentage: about 0.5 weight%, about 1.0 weight%, about 1.5 weight%, about 2.0 weight%, about 2.5 weight%, about 3.0 weight%, about 3.5 weight%, about 4.0 weight%, about 4.5 weight%, and about 5.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type A, and one or more pharmaceutically acceptable excipients. In some embodiments, the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type A, and one or more pharmaceutically acceptable excipients. In some embodiments, the pharmaceutical composition is for oral administration. In some embodiments, the pharmaceutical composition is substantially free of other crystalline forms of Compound 1. In some embodiments, the pharmaceutical composition is substantially free of amorphous Compound 1. [0160] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type B, and one or more pharmaceutically acceptable excipients.
- the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type B, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition is for oral administration.
- the pharmaceutical composition is substantially free of other crystalline forms of Compound 1.
- the pharmaceutical composition is substantially free of amorphous Compound 1.
- the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type C, and one or more pharmaceutically acceptable excipients.
- the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type C, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition is for oral administration.
- the pharmaceutical composition is substantially free of other crystalline forms of Compound 1. In some embodiments, the pharmaceutical composition is substantially free of amorphous Compound 1. [0162] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type D, and one or more pharmaceutically acceptable excipients. In some embodiments, the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type D, and one or more pharmaceutically acceptable excipients. In some embodiments, the pharmaceutical composition is for oral administration. In some embodiments, the pharmaceutical composition is substantially free of other crystalline forms of Compound 1. In some embodiments, the pharmaceutical composition is substantially free of amorphous Compound 1.
- the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type E, and one or more pharmaceutically acceptable excipients. In some embodiments, the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type E, and one or more pharmaceutically acceptable excipients. In some embodiments, the pharmaceutical composition is for oral administration. In some embodiments, the pharmaceutical composition is substantially free of other crystalline forms of Compound 1. In some embodiments, the pharmaceutical composition is substantially free of amorphous Compound 1. [0164] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type F, and one or more pharmaceutically acceptable excipients.
- the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type F, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition is for oral administration.
- the pharmaceutical composition is substantially free of other crystalline forms of Compound 1.
- the pharmaceutical composition is substantially free of amorphous Compound 1.
- the present disclosure provides a pharmaceutical composition comprising a therapeutically effective amount of Compound 1 crystalline form Type G, and one or more pharmaceutically acceptable excipients.
- the present disclosure provides a pharmaceutical composition comprising Compound 1 crystalline form Type G, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition is for oral administration.
- the pharmaceutical composition is substantially free of other crystalline forms of Compound 1. In some embodiments, the pharmaceutical composition is substantially free of amorphous Compound 1. [0166] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a crystalline form of Compound 1.
- a pharmaceutical composition comprises a crystalline form of Compound 1 and an amorphous form of Compound 1, wherein the amorphous form of Compound 1 is present in an amount selected from the following ranges: about 90 to about 99%, about 80 to about 89%, about 70 to about 79%, about 60 to about 69%, about 50 to about 59%, about 40 to about 49%, about 30 to about 39%, about 20 to about 29%, about 10 to about 19%, about 1 to about 9% and about 0 to about 0.99%.
- a pharmaceutical composition comprising a crystalline form of Compound 1 is substantially free of amorphous Compound 1.
- a pharmaceutical composition comprising Compound 1 and its enantiomer (“Compound 2”).
- a pharmaceutical composition comprises Compound 1 and its enantiomer Compound 2, wherein Compound 1 has an enantiomeric excess selected from the following ranges: at least about 99%, at least about 95%, at least about 90%, at least about 80%, about 90 to about 99%, about 80 to about 89%, about 70 to about 79%, about 60 to about 69%, about 50 to about 59%, about 40 to about 49%, about 30 to about 39%, about 20 to about 29%, about 10 to about 19%, about 1 to about 9% and about 0 to about 0.99%.
- a pharmaceutical composition comprises Compound 1 and its enantiomer Compound 2, wherein the weight percentage of Compound 1 relative to the total weight of Compound 1 and Compound 2 is in a percentage selected from the following ranges: about 90 to about 99%, about 80 to about 89%, about 70 to about 79%, about 60 to about 69%, about 50 to about 59%, about 40 to about 49%, about 30 to about 39%, about 20 to about 29%, about 10 to about 19%, about 1 to about 9% and about 0 to about 0.99%.
- Pharmaceutical compositions described herein can comprise a pharmaceutically acceptable carrier or one or more excipients.
- compositions described herein can be provided in a unit dosage form container (e.g., in a vial or bag, or the like). In some embodiments, pharmaceutical compositions described herein can be provided in an oral dosage form. In some embodiments, an oral dosage form is a tablet.
- Amorphous Solid Dispersion Comprising Compound 1 [0169] The present disclosure also provides an amorphous solid dispersion comprising Compound 1: , and a polymer.
- the polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam-polyvinyl a
- the polymer is hydroxypropylmethyl cellulose (HPMC) or hydroxypropylmethyl cellulose acetate succinate (HPMC AS). In some embodiments, the polymer is hydroxypropylmethyl cellulose acetate succinate (HPMC AS), including any grade thereof (e.g., HPMC AS MG). [0170] Various amounts of Compound 1 and the polymer can be used in the amorphous solid dispersion.
- the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion can be selected from the following ranges: about 10:1, about 9:1, about 8:1, about 7:1, about 6:1, about 5:1, about 4:1, about 3:1, about 2:1, about 1:1, about 1:2, about 1:3, about 1:4, about 1:5, about 1:6, about 1:7, about 1:8, about 1:9, and about 1:10.
- the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion is in a range of about 3:1 to about 1:3.
- the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion is in a range of about 2:1 to about 1:3.
- the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion is about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion is about 1:1. In some embodiments, the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, about 2:1, or about 3:1. In some embodiments, the weight ratio of Compound 1 to the polymer in the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, or about 2:1. [0171] In some embodiments, the amorphous solid dispersion is free or substantially free of crystalline Compound 1.
- crystalline diffraction peaks are not observable by XRPD analysis (Method D) of the amorphous solid dispersion. In some embodiments, crystalline diffraction peaks are not observable by XRPD analysis (Method D) of the amorphous solid dispersion. In some embodiments, a single glass transition temperature (TG) and no melt endotherm is observable by DSC analysis (Method B) of the amorphous solid dispersion. [0172] In some embodiments, the amorphous solid dispersion is physically stable in that it remains free or substantially free of crystalline Compound 1 over time in accelerated stability studies.
- crystalline diffraction peaks are not observable by XRPD analysis (Method D) of the amorphous solid dispersion after storage in a container as described in Example 20 for 5 months at 2-8 °C and ambient relative humidity, 5 months at 25 °C and 60 % relative humidity, 1 month at 2-8 °C and ambient relative humidity, 1 month at 25 °C and 60 % relative humidity, or 1 month at 40 °C and 75 % relative humidity.
- a single glass transition temperature (TG) and no melt endotherm is observable by DSC analysis (Method B) of the amorphous solid dispersion after storage in a container as described in Example 20 for 5 months at 2-8 °C and ambient relative humidity, 5 months at 25 °C and 60 % relative humidity, 1 month at 2-8 °C and ambient relative humidity, 1 month at 25 °C and 60 % relative humidity, or 1 month at 40 °C and 75 % relative humidity.
- crystalline diffraction peaks are not observable by XRPD analysis (Method D) of the amorphous solid dispersion after storage in a sealed vial for 1 week at 60 °C, storage in a sealed vial for 2 weeks at 60 °C, storage in an unsealed vial for 1 week at 25 °C and 60% relative humidity, storage in an unsealed vial for 2 weeks at 25 °C and 60% relative humidity, storage in an unsealed vial for 1 week at 40 °C and 75% relative humidity, storage in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, storage in an unsealed vial for 1 week at 60 °C and 75% relative humidity, or storage in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity.
- a single glass transition temperature (TG) and no melt endotherm is observable by DSC analysis (Method B) of the amorphous solid dispersion after storage in a sealed vial for 1 week at 60 °C, storage in a sealed vial for 2 weeks at 60 °C, storage in an unsealed vial for 1 week at 25 °C and 60% relative humidity, storage in an unsealed vial for 2 weeks at 25 °C and 60% relative humidity, storage in an unsealed vial for 1 week at 40 °C and 75% relative humidity, storage in an unsealed vial for 2 weeks at 40 °C and 75% relative humidity, storage in an unsealed vial for 1 week at 60 °C and 75% relative humidity, or storage in an unsealed vial for 2 weeks at 60 °C and 75% relative humidity.
- TG glass transition temperature
- Method B DSC analysis
- the amorphous solid dispersion is highly soluble, e.g., Compound 1 dissolves quickly and readily in biorelevant media.
- Compound 1 has a concentration of at least 150 ⁇ g/mL, at least 200 ⁇ g/mL, at least 250 ⁇ g/mL, at least 300 ⁇ g/mL, or at least 350 ⁇ g/mL after 30 minutes in the kinetic solubility experiment described in Example 23.
- Compound 1 has a Cmax of at least 300 ⁇ g/mL, at least 350 ⁇ g/mL, at least 400 ⁇ g/mL, at least 450 ⁇ g/mL, at least 500 ⁇ g/mL, at least 550 ⁇ g/mL, at least 600 ⁇ g/mL, at least 650 ⁇ g/mL, or at least 700 ⁇ g/mL in the kinetic solubility experiment described in Example 23.
- Compound 1 has a concentration of at least 200 ⁇ g/mL, at least 250 ⁇ g/mL, at least 300 ⁇ g/mL, at least 350 ⁇ g/mL, at least 400 ⁇ g/mL, at least 450 ⁇ g/mL, at least 500 ⁇ g/mL, at least 550 ⁇ g/mL, or at least 600 ⁇ g/mL after 4 hours in the kinetic solubility experiment described in Example 23.
- Compound 1 has a concentration of at least 150 ⁇ g/mL, at least 200 ⁇ g/mL, at least 250 ⁇ g/mL, or at least 300 ⁇ g/mL after 16 hours in the kinetic solubility experiment described in Example 23.
- compositions Comprising Compound 1 Amorphous Solid Dispersion
- the present disclosure further provides a pharmaceutical composition comprising a therapeutically effective amount of an amorphous solid dispersion comprising Compound 1, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition is for oral administration.
- the present disclosure provides a pharmaceutical composition comprising an amorphous solid dispersion comprising Compound 1, and having a water content of about 0.5-5.0 weight%, preferably about 1.0-4.5 weight%, more preferably about 1.5-4.0 weight%, even more preferably about 2.0-3.5 weight%, still more preferably about 2.5-3.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising an amorphous solid dispersion comprising Compound 1, and having a water content in an amount selected from the following ranges: about 0.5-1.0 weight%, about 1.0-1.5 weight%, about 1.5-2.0 weight%, about 2.5-3.0 weight%, about 3.0-3.5 weight%, about 3.5-4.0 weight%, about 4.0-4.5 weight%, and about 4.5-5.0 weight% relative to the weight of the pharmaceutical composition.
- the present disclosure provides a pharmaceutical composition comprising an amorphous solid dispersion comprising Compound 1, and having a water content in an amount selected from the weight percentage: about 0.5 weight%, about 1.0 weight%, about 1.5 weight%, about 2.0 weight%, about 2.5 weight%, about 3.0 weight%, about 3.5 weight%, about 4.0 weight%, about 4.5 weight%, and about 5.0 weight% relative to the weight of the pharmaceutical composition.
- the pharmaceutical composition comprises about 10 mg, about 25 mg, about 50 mg, about 100 mg, about 200 mg, or about 300 mg of Compound 1.
- the pharmaceutical composition comprises about 25 mg of Compound 1.
- the pharmaceutical composition comprises about 100 mg of Compound 1.
- the pharmaceutical composition comprises about 200 mg of Compound 1.
- Pharmaceutical compositions described herein can comprise a pharmaceutically acceptable carrier or one or more excipients.
- pharmaceutical compositions described herein can be provided in a unit dosage form container (e.g., in a vial or bag, or the like).
- pharmaceutical compositions described herein can be provided in an oral dosage form.
- an oral dosage form is a tablet.
- the pharmaceutical composition comprises one or more pharmaceutically acceptable excipients which comprise one or more of a filler, a dry binder, a glidant, a lubricant, a disintegrant, and a film coating agent.
- the one or more pharmaceutically acceptable excipients comprise a filler, and the filler comprises microcrystalline cellulose. In some embodiments, the one or more pharmaceutically acceptable excipients comprise a filler, and the filler comprises lactose monohydrate. In some embodiments, the one or more pharmaceutically acceptable excipients comprise a dry binder, and the dry binder comprises crospovidone. In some embodiments, the one or more pharmaceutically acceptable excipients comprise a glidant, and the glidant comprises colloidal silicon dioxide. In some embodiments, the one or more pharmaceutically acceptable excipients comprise a lubricant, and the lubricant comprises magnesium stearate.
- the one or more pharmaceutically acceptable excipients comprise a disintegrant, and the disintegrant comprises croscarmellose sodium. In some embodiments, the one or more pharmaceutically acceptable excipients comprise a lubricant, and the lubricant comprises magnesium stearate.
- a pharmaceutical composition comprises a tablet core. In some embodiments, the tablet core comprises an intra granular portion comprising the amorphous solid dispersion, and an extra granular portion blended with the intra granular portion. In some embodiments, a pharmaceutical composition further comprises a coating disposed on the tablet core.
- amorphous solid dispersion comprising Compound 1 can be about 10 weight%, about 20 weight%, about 30 weight%, about 40 weight%, about 50 weight%, about 60 weight%, about 70 weight%, about 80 weight%, or about 90 weight% of the tablet core.
- the amorphous solid dispersion comprising Compound 1 is at least about 30 weight% of the tablet core. In some embodiments, the amorphous solid dispersion comprising Compound 1 is at least about 50 weight% of the tablet core.
- the amorphous solid dispersion comprising Compound 1 is at least about 60 weight% of the tablet core. In some embodiments, the amorphous solid dispersion comprising Compound 1 is about 50 weight% of the tablet core. In some embodiments, the amorphous solid dispersion comprising Compound 1 is about 50 to about 70 weight% of the tablet core. In some embodiments, the amorphous solid dispersion comprising Compound 1 is about 60 to about 65 weight% of the tablet core. [0181] In some embodiments, the intra granular portion further comprises one or more of a filler, a dry binder, a glidant, and a lubricant.
- the extra granular portion further comprises one or more of a filler, a disintegrant, and a lubricant.
- the tablet core has the following components: [0183] In some embodiments, the tablet core has the following components: [0184] In some embodiments, the tablet core has the following components: [0185] In some embodiments, the tablet core has the following components: [0186] In some embodiments, the Compound 1 oral unit dosage form can be a tablet comprising a total of about 10-35% by weight of Compound 1, with a total dose of about 100 mg or 200 mg, and a total weight of less than about 800 mg.
- a tablet having a composition described in a table above comprises about 50% of an API formed as an amorphous solid dispersion of Compound 1 obtained from a 1:3 SDD process described in the examples below (e.g., about 12.5% of Compound 1 in the tablet). In one embodiment, a tablet having a composition described in a table above comprises about 30% of an API formed as an amorphous solid dispersion of Compound 1 obtained from a 1:1 SDD process described in the examples below (e.g., about 15% of Compound 1 in the tablet, with a total of about 100 mg Compound 1 in the tablet).
- a tablet having a composition described in a table above comprises about 62% of an API formed as an amorphous solid dispersion of Compound 1 obtained from a 1:1 SDD process described in the examples below (e.g., about 31% of Compound 1 in the tablet, with a total of about 200 mg of Compound 1 in the tablet)
- Methods for Preparing Amorphous Solid Dispersions of Compound 1 [0187]
- the present disclosure also provides a method for preparing an amorphous solid dispersion comprising Compound 1: .
- the method comprises mixing Compound 1, a polymer, and a solvent to afford a mixture, and spray-drying the mixture to afford an amorphous solid dispersion comprising Compound 1.
- the polymer used in the method is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam
- HPMC hydroxypropy
- the polymer is hydroxypropylmethyl cellulose (HPMC) or hydroxypropylmethyl cellulose acetate succinate (HPMC AS). In some embodiments, the polymer is hydroxypropylmethyl cellulose acetate succinate (HPMC AS), including any grade thereof (e.g., HPMC AS MG). [0190] Various amounts of Compound 1 and the polymer can be used in the method to prepare the amorphous solid dispersion.
- the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion can be selected from the following ranges: about 10:1, about 9:1, about 8:1, about 7:1, about 6:1, about 5:1, about 4:1, about 3:1, about 2:1, about 1:1, about 1:2, about 1:3, about 1:4, about 1:5, about 1:6, about 1:7, about 1:8, about 1:9, and about 1:10.
- the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is in a range of about 3:1 to about 1:3.
- the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is in a range of about 2:1 to about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:1. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, about 2:1, or about 3:1.
- the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, or about 2:1.
- Various solvents can be used in the method to prepare the amorphous solid dispersion.
- the solvent is dichloromethane and methanol.
- the present disclosure further provides a product prepared by a process comprising mixing Compound 1, a polymer, and a solvent to afford a mixture, and spray-drying the mixture to afford an amorphous solid dispersion comprising Compound 1 .
- the polymer used in the process is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam
- HPMC hydroxypropy
- the polymer is hydroxypropylmethyl cellulose (HPMC) or hydroxypropylmethyl cellulose acetate succinate (HPMC AS). In some embodiments, the polymer is hydroxypropylmethyl cellulose acetate succinate (HPMC AS), including any grade thereof (e.g., HPMC AS MG). [0194] Various amounts of Compound 1 and the polymer can be used in the process to prepare the amorphous solid dispersion.
- the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion can be selected from the following ranges: about 10:1, about 9:1, about 8:1, about 7:1, about 6:1, about 5:1, about 4:1, about 3:1, about 2:1, about 1:1, about 1:2, about 1:3, about 1:4, about 1:5, about 1:6, about 1:7, about 1:8, about 1:9, and about 1:10.
- the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion is in a range of about 3:1 to about 1:3.
- the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion is in a range of about 2:1 to about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion is about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:1. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, about 2:1, or about 3:1.
- the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, or about 2:1.
- Various solvents can be used in the process to prepare the amorphous solid dispersion.
- the solvent is dichloromethane and methanol.
- Pharmaceutical Compositions Comprising Compound 1 [0196] The present disclosure provides a pharmaceutical composition comprising Compound 1: , obtained by a process comprising mixing Compound 1 in a solid form, a polymer, and a solvent to afford a mixture, and spray-drying the mixture to afford an amorphous solid dispersion comprising Compound 1.
- the solid form is Type A of Compound 1. In some embodiments, the solid form is Type B of Compound 1. In some embodiments, the solid form is Type C of Compound 1. In some embodiments, the solid form is Type D of Compound 1. In some embodiments, the solid form is Type E of Compound 1. In some embodiments, the solid form is Type F of Compound 1. In some embodiments, the solid form is Type G of Compound 1. In some embodiments, the solid form is Type H of Compound 1. In some embodiments, the solid form is Type I of Compound 1. In some embodiments, the solid form is Type J of Compound 1. In some embodiments, the solid form is Type K of Compound 1. In some embodiments, the solid form is Type L of Compound 1.
- the solid form is Type M of Compound 1.
- the solid form is selected from the group consisting of Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, and Type M of Compound 1.
- the solid form is amorphous form of Compound 1.
- the pharmaceutical composition obtained by the process has a water content of about 0.5-5.0 weight%, preferably about 1.0-4.5 weight%, more preferably about 1.5-4.0 weight%, even more preferably about 2.0-3.5 weight%, still more preferably about 2.5-3.0 weight% relative to the weight of the pharmaceutical composition.
- the pharmaceutical composition obtained by the process has a water content in an amount selected from the following ranges: about 0.5-1.0 weight%, about 1.0-1.5 weight%, about 1.5-2.0 weight%, about 2.5-3.0 weight%, about 3.0-3.5 weight%, about 3.5-4.0 weight%, about 4.0-4.5 weight%, and about 4.5-5.0 weight% relative to the weight of the pharmaceutical composition.
- the pharmaceutical composition obtained by the process has a water content in an amount selected from the weight percentage: about 0.5 weight%, about 1.0 weight%, about 1.5 weight%, about 2.0 weight%, about 2.5 weight%, about 3.0 weight%, about 3.5 weight%, about 4.0 weight%, about 4.5 weight%, and about 5.0 weight% relative to the weight of the pharmaceutical composition.
- the polymer used in the process is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam
- HPMC hydroxypropy
- the polymer is hydroxypropylmethyl cellulose (HPMC) or hydroxypropylmethyl cellulose acetate succinate (HPMC AS). In some embodiments, the polymer is hydroxypropylmethyl cellulose acetate succinate (HPMC AS), including any grade thereof (e.g., HPMC AS MG). [0200] Various amounts of Compound 1 and the polymer can be used in the process to prepare the amorphous solid dispersion.
- the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion can be selected from the following ranges: about 10:1, about 9:1, about 8:1, about 7:1, about 6:1, about 5:1, about 4:1, about 3:1, about 2:1, about 1:1, about 1:2, about 1:3, about 1:4, about 1:5, about 1:6, about 1:7, about 1:8, about 1:9, and about 1:10.
- the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion is in a range of about 3:1 to about 1:3.
- the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion is in a range of about 2:1 to about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer used in the process to prepare the amorphous solid dispersion is about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:1. In some embodiments, the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, about 2:1, or about 3:1.
- the weight ratio of Compound 1 to the polymer used in the method to prepare the amorphous solid dispersion is about 1:3, about 2:3, about 1:1, about 1.5:1, or about 2:1.
- Various solvents can be used in the process to prepare the amorphous solid dispersion.
- the solvent is dichloromethane and methanol.
- Solid Oral Dosage Forms of Compound 1 [0202] The disclosure also provides solid oral dosage forms of Compound 1, such as tablets and capsules.
- the solid oral dosage form comprises a stabilized amorphous compound (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]- 1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one, wherein the stabilized amorphous compound does not show crystallinity by PXRD (Method D) after 2 weeks of storage at 60 °C/75% RH (exposed).
- the stabilized amorphous compound shows a single glass transition temperature (T G ) and no melt endotherm by DSC (Method B) after 2 weeks of storage at 60 °C/75% RH (exposed).
- the solid oral dosage form contains a total of about 100 mg or about 200 mg of (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one.
- the solid dosage form has a total weight of not more than 700 mg, 800 mg, 900 mg, 1000 mg or 1200 mg.
- the solid oral dosage form is a tablet or capsule.
- the stabilized amorphous compound in the solid oral dosage form is in a spray dried dispersion with a polymer.
- the polymer is selected from the group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co-methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam-polyvinyl acetate-
- the polymer is HPMC AS.
- the (S)-1-(5-[2H,3H- [1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3- hydroxy-2-phenylpropan-1-one is spray dried with HPMC AS in a weight ratio of 1:3 to 2:1.
- the (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]- 1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one is spray dried with HPMC AS in a weight ratio of 1:1.
- the disclosure also relates to a tablet comprising about 100 mg or about 200 mg of stabilized amorphous compound (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]- 1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one as the active pharmaceutical ingredient (API), wherein the stabilized amorphous compound does not show crystallinity by PXRD (Method D) after 2 weeks of storage of the tablet at 60 °C/75% RH (exposed).
- PXRD Metal XRD
- the API comprises less than 5.0% by HPLC of (R)-1-(5- [2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2- yl)-3-hydroxy-2-phenylpropan-1-one.
- the API comprises less than 0.05% by HPLC of (R)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one.
- the tablet has a total weight of less than 700 mg, 800 mg, 900 mg, 1000 mg or 1200 mg. Tablet Dosage Forms of Compound 1 [0207] The disclosure also provides tablet dosage forms of Compound 1.
- the tablet dosage form comprises a tablet core, the tablet core comprising at least 10 weight % of Compound 1 in amorphous form: wherein crystalline Compound 1 (Type A) is not observable by XRPD analysis (Method D) of the tablet core.
- the tablet core comprises at least 15 weight %, at least 20 weight %, at least 25 weight % or at least 30 weight % of Compound 1 in amorphous form.
- the tablet core comprises about 200 mg of Compound 1 per tablet and has a total weight of no more than about 1200 mg, about 1100 mg, about 1000 mg, about 900 mg, about 800 mg, or about 700 mg per tablet per tablet.
- the tablet dosage form comprises a tablet core, the tablet core having a total weight of no more than about 1000 mg and comprising about 200 mg of Compound 1 in amorphous form per tablet, wherein crystalline Compound 1 (Type A) is not observable by XRPD analysis (Method D) of the tablet core.
- the tablet core has a total weight of no more than about 800 mg per tablet.
- the tablet core comprises Compound 1 in highly enantiopure form.
- the tablet core comprises 0.05-5.0 % of Compound 2: based on the total amount of Compound 1 and Compound 2.
- the tablet core comprises 0.05-3.0 % of Compound 2, based on the total amount of Compound 1 and Compound 2. In some embodiments, the tablet core comprises 0.05-2.0 % of Compound 2, based on the total amount of Compound 1 and Compound 2. In some embodiments, the tablet core comprises 0.05-1.0 % of Compound 2, based on the total amount of Compound 1 and Compound 2. [0210] In some embodiments, the tablet dosage form is physically stable in that it remains free or substantially free of crystalline Compound 1 over time in accelerated stability studies.
- crystalline Compound 1 (Type A) is not observable by XRPD analysis (Method D) of the tablet core after storage in a sealed container as described in Example 29 for 1 month at 25 °C and 60 % relative humidity, storage in a sealed container as described in Example 29 for 2 months at 25 °C and 60 % relative humidity, storage in a sealed container as described in Example 29 for 3 months at 25 °C and 60 % relative humidity, storage in a sealed container as described in Example 29 for 1 month at 40 °C and 75 % relative humidity, storage in a sealed container as described in Example 29 for 2 months at 40 °C and 75 % relative humidity, storage in a sealed container as described in Example 29 for 3 months at 40 °C and 75 % relative humidity.
- Compound 1 is present in an amorphous solid dispersion comprising Compound 1 and a polymer.
- the polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co-methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxy
- the polymer is hydroxypropylmethyl cellulose (HPMC) or hydroxypropylmethyl cellulose acetate succinate (HPMC AS). In some embodiments, the polymer is hydroxypropylmethyl cellulose acetate succinate (HPMC AS), including any grade thereof (e.g., HPMC AS MG).
- the weight ratio of Compound 1 to the polymer is in a range of about 3:1 to about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer is in a range of about 2:1 to about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer is about 1:3. In some embodiments, the weight ratio of Compound 1 to the polymer is about 1:1.
- the weight ratio of Compound 1 to the polymer is about 1:3, about 2:3, about 1:1, about 1.5:1, about 2:1, or about 3:1. In some embodiments, the weight ratio of Compound 1 to the polymer is about 1:3, about 2:3, about 1:1, about 1.5:1, or about 2:1.
- the tablet core of the tablet dosage form further comprises one or more pharmaceutically acceptable excipients. In some embodiments, the one or more pharmaceutically acceptable excipients comprise one or more of a filler, a dry binder, a glidant, a lubricant, a disintegrant, and a film coating agent.
- the tablet core comprises an intra granular portion comprising Compound 1; and an extra granular portion blended with the intra granular portion.
- the intragranular portion comprises an amorphous solid dispersion comprising Compound 1 and a polymer and one or more of a filler, a dry binder, a glidant, and a lubricant
- the extragranular portion comprises one or more of a filler, a disintegrant, and a lubricant.
- the intragranular portion comprises: an amorphous solid dispersion of Compound 1 in an amount of 30-70 weight % of the tablet core; one or more fillers in an amount of 15-50 weight % of the tablet core; one or more dry binders in an amount of 2.50-10 weight % of the tablet core; one or more glidants in an amount of 0.50-1.50 weight % of the tablet core; and one or more lubricants in an amount of 0.25-1 weight % of the tablet core; and the extragranular portion comprises: one or more fillers in an amount of 5-15 weight % of the tablet core; one or more disintegrants in an amount of 1.25-5 weight % of the tablet core; and one or more lubricants in an amount of 0.25-1 weight % of the tablet core.
- the tablet dosage form comprises: an amorphous solid dispersion of Compound 1 in an amount of 50-75 weight % of the tablet core; one or more fillers in an amount of 15-50 weight % of the tablet core; one or more dry binders in an amount of 2-10 weight % of the tablet core; one or more glidants in an amount of ⁇ 2 weight % of the tablet core; one or more disintegrants in an amount of 2-10 weight % of the tablet core; and one or more lubricants in an amount of ⁇ 2 weight % of the tablet core.
- the amorphous solid dispersion comprises Compound 1 and a polymer (as described in any of the embodiments set forth herein).
- the one or more fillers comprise microcrystalline cellulose or lactose monohydrate.
- the one or more dry binders comprise crospovidone or crosslinked polyvinylpyrrolidone.
- the one or more glidants comprise colloidal silicon dioxide or fumed silica.
- the one or more lubricants comprise magnesium stearate.
- the one or more disintegrants comprise crocarmellose sodium.
- the disclosure relates to a method of treating a disease associated with decreased activity of PKR in a subject in need thereof which comprises administering to the subject an effective amount of a compound of Formula I in any of the forms described herein, including any embodiment thereof.
- Embodiments [0218] In some embodiments, the disclosure relates to one or more of the following enumerated embodiments: 1. A crystalline solid form of Compound 1: 2.
- Compound 1 3.
- Type A of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.61, 7.22, 15.66, 20.48, 21.35, 21.66, 22.47, 23.19, 24.76, and 26.73. 6.
- Type A of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.61, 7.22, 15.66, 20.48, 21.35, 21.66, 22.47, 23.19, 24.76, and 26.73, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.19, 12.25, 5.66, 4.34, 4.16, 4.10, 3.96, 3.84, 3.60, and 3.34, respectively. 7.
- Type A of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.61 5.80 7.22 7.68 11.21 12.31 14.44 15.66 16.95 18.02 19.20 20.48 21.35 21.66 22.47 23.19 24.76 26.73 28.01 28.49 29.35 30.25 32.14 34.12 36.46 8.
- Type A of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.61 19.19 5.80 15.24 7.22 12.25 7.68 11.50 11.21 7.89 12.31 7.19 14.44 6.13 15.66 5.66 16.95 5.23 18.02 4.92 19.20 4.62 20.48 4.34 21.35 4.16 21.66 4.10 22.47 3.96 23.19 3.84 24.76 3.60 26.73 3.34 28.01 3.19 28.49 3.13 29.35 3.04 30.25 2.95 32.14 2.79 34.12 2.63 36.46 2.46 9.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- XRPD X-ray powder diffraction
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.52, 15.57, 22.89, 23.34, and 25.13, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.53, 5.69, 3.89, 3.81, and 3.54, respectively. 16.
- Type B of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.52, 9.86, 15.57, 19.93, 22.19, 22.89, 23.34, 25.13, and 28.30. 17.
- XRPD X-ray powder diffraction
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.52, 9.86, 15.57, 19.93, 22.19, 22.89, 23.34, 25.13, and 28.30, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.53, 8.97, 5.69, 4.45, 4.00, 3.89, 3.81, 3.54, and 3.15, respectively. 18.
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.52 8.98 9.86 12.37 13.18 15.57 16.86 18.21 19.11 19.93 20.92 22.19 22.89 23.34 25.13 25.80 26.71 28.30 29.39 19.
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.52 19.53 8.98 9.85 9.86 8.97 12.37 7.15 13.18 6.72 15.57 5.69 16.86 5.26 18.21 4.87 19.11 4.64 19.93 4.45 20.92 4.25 22.19 4.00 22.89 3.89 2 theta d-spacing 23.34 3.81 25.13 3.54 25.80 3.45 26.71 3.34 28.30 3.15 29.39 3.04 20.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- DVDS dynamic vapor sorption
- Type C of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.55, 18.85, 23.02, and 24.65.
- XRPD X-ray powder diffraction
- Type C of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.55, 7.34, 9.07, 11.17, 18.34, 18.85, 19.57, 21.66, 23.02, and 24.65. 29.
- XRPD X-ray powder diffraction
- Type C of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.55, 7.34, 9.07, 11.17, 18.34, 18.85, 19.57, 21.66, 23.02, and 24.65, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.43, 12.05, 9.75, 7.92, 4.84, 4.71, 4.54, 4.10, 3.86, and 3.61, respectively. 30.
- Type C of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.55 7.34 9.07 11.17 12.29 14.51 15.66 18.34 18.85 19.57 20.38 21.66 23.02 24.65 26.39 28.28 30.09 32.31 33.91 37.19 31.
- Type C of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.55 19.43 7.34 12.05 9.07 9.75 11.17 7.92 12.29 7.20 14.51 6.11 15.66 5.66 18.34 4.84 18.85 4.71 19.57 4.54 20.38 4.36 21.66 4.10 23.02 3.86 24.65 3.61 26.39 3.38 28.28 3.16 30.09 2.97 32.31 2.77 33.91 2.64 37.19 2.42 32.
- DSC differential scanning calorimetry
- DFS dynamic vapor sorption
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type D of Compound 1. 38.
- Type D of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.27, 6.15, 8.71, 9.72, 12.31, 13.08, 13.76, 15.74, 18.02, 21.90, 23.59, and 26.71. 41.
- XRPD X-ray powder diffraction
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.27, 6.15, 8.71, 9.72, 12.31, 13.08, 13.76, 15.74, 18.02, 21.90, 23.59, and 26.71, corresponding to d-spacing (angstroms ⁇ 0.2) of 20.68, 14.36, 10.16, 9.10, 7.19, 6.77, 6.44, 5.63, 4.92, 4.06, 3.77, and 3.34, respectively. 42.
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.27 6.15 8.71 9.72 12.31 13.08 13.76 15.74 18.02 19.55 21.90 23.59 24.79 26.71 29.50 30.82 31.74 35.40 37.84 38.61 43.
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.27 20.68 6.15 14.36 8.71 10.16 9.72 9.10 12.31 7.19 13.08 6.77 13.76 6.44 15.74 5.63 18.02 4.92 19.55 4.54 21.90 4.06 23.59 3.77 24.79 3.59 26.71 3.34 29.50 3.03 30.82 2.90 31.74 2.82 35.40 2.54 37.84 2.38 38.61 2.33 44.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type E of Compound 1. 47.
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 15.12, 15.75, 17.48, 20.05, 21.93, and 26.72. 48.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 15.12, 15.75, 17.48, 20.05, 21.93, and 26.72, corresponding to d-spacing (angstroms ⁇ 0.2) of 5.86, 5.63, 5.07, 4.43, 4.05, and 3.34, respectively. 49.
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.59, 15.12, 15.75, 17.48, 20.05, 21.93, 23.18, 23.70, and 26.72. 50.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.59, 15.12, 15.75, 17.48, 20.05, 21.93, 23.18, 23.70, and 26.72, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.27, 5.86, 5.63, 5.07, 4.43, 4.05, 3.84, 3.75, and 3.34, respectively. 51.
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.59, 9.76, 12.36, 13.12, 15.12, 15.75, 16.84, 17.48, 18.06, 19.02, 20.05, 21.93, 23.18, 23.70, 26.72, and 27.81. 52.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.59, 9.76, 12.36, 13.12, 15.12, 15.75, 16.84, 17.48, 18.06, 19.02, 20.05, 21.93, 23.18, 23.70, 26.72, and 27.81, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.27, 9.06, 7.16, 6.75, 5.86, 5.63, 5.27, 5.07, 4.91, 4.67, 4.43, 4.05, 3.84, 3.75, 3.34, and 3.21, respectively. 53.
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.59 8.76 9.76 12.36 13.12 13.83 15.12 15.75 16.84 17.48 18.06 19.02 20.05 21.93 23.18 23.70 24.82 26.72 27.81 29.51 30.76 31.74 33.03 34.52 35.39 36.72 37.77 38.66 54.
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.59 19.27 8.76 10.09 9.76 9.06 12.36 7.16 13.12 6.75 13.83 6.40 15.12 5.86 15.75 5.63 16.84 5.27 17.48 5.07 18.06 4.91 2 theta d-spacing 19.02 4.67 20.05 4.43 21.93 4.05 23.18 3.84 23.70 3.75 24.82 3.59 26.72 3.34 27.81 3.21 29.51 3.03 30.76 2.91 31.74 2.82 33.03 2.71 34.52 2.60 35.39 2.54 36.72 2.45 37.77 2.38 38.66 2.33 55.
- XRPD X-ray powder diffraction
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 20.01, 21.36, and 22.45, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.23, 6.04, 5.54, 5.28, 4.44, 4.16, and 3.96, respectively. 58.
- Type F of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 18.99, 20.01, 21.36, 22.45, 23.25, and 25.32. 59.
- XRPD X-ray powder diffraction
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.45, 14.66, 16.00, 16.79, 18.99, 20.01, 21.36, 22.45, 23.25, and 25.32, corresponding to d- spacing (angstroms ⁇ 0.2) of 16.23, 6.04, 5.54, 5.28, 4.67, 4.44, 4.16, 3.96, 3.83, and 3.52, respectively. 60.
- Type F of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.45, 12.87, 14.66, 16.00, 16.79, 17.36, 18.99, 20.01, 20.57, 21.36, 22.45, 23.25, 25.32, 26.57, 27.25, 27.97, and 30.02. 61.
- XRPD X-ray powder diffraction
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.45, 12.87, 14.66, 16.00, 16.79, 17.36, 18.99, 20.01, 20.57, 21.36, 22.45, 23.25, 25.32, 26.57, 27.25, 27.97, and 30.02, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.23, 6.88, 6.04, 5.54, 5.28, 5.11, 4.67, 4.44, 4.32, 4.16, 3.96, 3.83, 3.52, 3.35, 3.27, 3.19, and 2.98, respectively. 62.
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.45 10.92 12.87 14.66 16.00 16.79 17.36 18.99 20.01 20.57 21.36 22.45 23.25 25.32 26.57 27.25 27.97 30.02 31.98 32.89 38.29 39.09 63.
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 5.45 16.23 10.92 8.10 12.87 6.88 14.66 6.04 16.00 5.54 16.79 5.28 17.36 5.11 18.99 4.67 20.01 4.44 20.57 4.32 21.36 4.16 22.45 3.96 23.25 3.83 25.32 3.52 26.57 3.35 27.25 3.27 27.97 3.19 30.02 2.98 31.98 2.80 32.89 2.72 38.29 2.35 39.09 2.30 64.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type G of Compound 1. 67.
- Type G of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 16.58, 19.78, 21.35, 22.35, 25.33, and 26.43. 70.
- XRPD X-ray powder diffraction
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 16.58, 19.78, 21.35, 22.35, 25.33, and 26.43, corresponding to d- spacing (angstroms ⁇ 0.2) of 16.48, 6.90, 6.18, 5.91, 5.35, 4.49, 4.16, 3.98, 3.52, and 3.37, respectively. 71.
- Type G of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 15.79, 16.58, 19.78, 21.35, 22.35, 25.33, 26.43, 27.35, and 30.21. 72.
- XRPD X-ray powder diffraction
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.36, 12.83, 14.34, 15.00, 15.79, 16.58, 19.78, 21.35, 22.35, 25.33, 26.43, 27.35, and 30.21, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.48, 6.90, 6.18, 5.91, 5.61, 5.35, 4.49, 4.16, 3.98, 3.52, 3.37, 3.26, and 2.96, respectively.
- 73 d-spacing
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.36 8.73 12.83 14.34 15.00 15.79 16.58 18.54 19.78 21.35 22.35 23.38 25.33 26.43 27.35 30.21 32.32 38.04 74.
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 5.36 16.48 8.73 10.13 2 theta d-spacing 12.83 6.90 14.34 6.18 15.00 5.91 15.79 5.61 16.58 5.35 18.54 4.79 19.78 4.49 21.35 4.16 22.35 3.98 23.38 3.80 25.33 3.52 26.43 3.37 27.35 3.26 30.21 2.96 32.32 2.77 38.04 2.37 75.
- a pharmaceutical composition comprising a therapeutically effective amount of the crystalline solid form of any one of embodiments 1-74, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition of embodiment 75 or 76, wherein the pharmaceutical composition has a water content of about 0.5-5.0 weight%.
- the pharmaceutical composition of any one of embodiments 75-77, wherein the pharmaceutical composition has a water content of about 1.5-4.0 weight%.
- the pharmaceutical composition of any one of embodiments 75-78, wherein the pharmaceutical composition has a water content of about 2.5-3.0 weight%.
- An amorphous solid dispersion comprising Compound 1: and a polymer. 81.
- the amorphous solid dispersion of embodiment 80 wherein the polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof.
- HPMC hydroxypropylmethyl cellulose
- HPMC AS hydroxypropylmethyl cellulose acetate succinate
- HPMC AS hydroxypropyl methyl cellulose phthalate
- HPPC hydroxypropyl cellulose
- PVP polyvinylpyrrolidone
- a pharmaceutical composition comprising a therapeutically effective amount of the amorphous solid dispersion of any one of embodiments 80-84, and one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition of embodiment 85, wherein the pharmaceutical composition is for oral administration.
- the pharmaceutical composition of embodiment 85 or 86, wherein the pharmaceutical composition is in a tablet dosage form. 88.
- the pharmaceutical composition of any one of embodiments 85-90, wherein the pharmaceutical composition comprises about 10 mg, about 25 mg, about 50 mg, about 100 mg, about 200 mg, or about 300 mg of Compound 1. 92.
- the pharmaceutical composition of any one of embodiments 85-91, wherein the pharmaceutical composition comprises about 25 mg of Compound 1. 93.
- the pharmaceutical composition of embodiment 101 further comprising a coating disposed on the tablet core.
- polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof.
- HPMC hydroxypropylmethyl cellulose
- HPMC AS hydroxypropylmethyl cellulose acetate succinate
- HPMC AS hydroxypropylmethyl cellulose phthalate
- HPPC hydroxypropyl cellulose
- PVP polyvinylpyrrolidone
- polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof.
- HPMC hydroxypropylmethyl cellulose
- HPMC AS hydroxypropylmethyl cellulose acetate succinate
- HPMC AS hydroxypropylmethyl cellulose phthalate
- HPPC hydroxypropyl cellulose
- PVP polyvinylpyrrolidone
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type A of Compound 1.
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type B of Compound 1. 121.
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type C of Compound 1. 122.
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type D of Compound 1. 123.
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type E of Compound 1. 124.
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type F of Compound 1. 125.
- the pharmaceutical composition of embodiment 118, wherein the solid form is Type G of Compound 1. 126.
- the pharmaceutical composition of embodiment 118, wherein the solid form is amorphous form of Compound 1. 127.
- HPMC hydroxypropylmethyl cellulose
- HPMC AS hydroxypropylmethyl cellulose acetate succinate
- HPMC AS hydroxypropylmethyl cellulose phthalate
- HPPC hydroxypropyl cellulose
- PVP polyvinylpyrrolidone
- Compound 1 3.
- XRPD X-ray powder diffraction
- Type A of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 7.2, 15.7, 20.5, 21.3, 21.7, 22.5, 23.2, 24.8, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.2, 12.2, 5.7, 4.3, 4.2, 4.1, 4.0, 3.8, 3.6, and 3.3, respectively.
- Type A of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.6 19.2 5.8 15.2 7.2 12.2 7.7 11.5 11.2 7.9 12.3 7.2 14.4 6.1 15.7 5.7 16.9 5.2 18.0 4.9 19.2 4.6 20.5 4.3 21.3 4.2 21.7 4.1 22.5 4.0 23.2 3.8 24.8 3.6 26.7 3.3 28.0 3.2 28.5 3.1 29.4 3.0 30.3 3.0 32.1 2.8 34.1 2.6 36.5 2.5. 11.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- DSC differential scanning calorimetry
- XRPD X-ray powder diffraction
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 15.6, 22.2, 22.9, 23.3, and 25.1, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 5.7, 4.0, 3.9, 3.8, and 3.5, respectively. 20.
- Type B of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 9.9, 15.6, 19.9, 22.2, 22.9, 23.3, 25.1, and 28.3. 21.
- XRPD X-ray powder diffraction
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 9.9, 15.6, 19.9, 22.2, 22.9, 23.3, 25.1, and 28.3, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 9.0, 5.7, 4.5, 4.0, 3.9, 3.8, 3.5, and 3.2, respectively. 22.
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.5 9.0 9.9 12.4 13.2 15.6 16.9 18.2 19.1 19.9 20.9 22.2 22.9 23.3 25.1 25.8 26.7 28.3 29.4. 23.
- Type B of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.5 19.5 2 theta d-spacing 9.0 9.9 9.9 9.0 12.4 7.2 13.2 6.7 15.6 5.7 16.9 5.3 18.2 4.9 19.1 4.6 19.9 4.5 20.9 4.2 22.2 4.0 22.9 3.9 23.3 3.8 25.1 3.5 25.8 3.5 26.7 3.3 28.3 3.2 29.4 3.0. 24.
- DSC differential scanning calorimetry
- DVDS dynamic vapor sorption
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type C of Compound 1.
- Type C of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 18.9, 23.0, and 24.7.
- XRPD X-ray powder diffraction
- Type C of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 7.3, 9.1, 11.2, 18.3, 18.9, 19.6, 21.7, 23.0, and 24.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.4, 12.0, 9.8, 7.9, 4.8, 4.7, 4.5, 4.1, 3.9, and 3.6, respectively. 36.
- Type C of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.5 19.4 7.3 12.0 9.1 9.8 11.2 7.9 12.3 7.2 14.5 6.1 15.7 5.7 18.3 4.8 2 theta d-spacing 18.9 4.7 19.6 4.5 20.4 4.4 21.7 4.1 23.0 3.9 24.7 3.6 26.4 3.4 28.3 3.2 30.1 3.0 32.3 2.8 33.9 2.6 37.2 2.4. 38.
- DSC differential scanning calorimetry
- DVD dynamic vapor sorption
- Type D of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 9.7, 13.1, 15.7, 21.9, and 23.6.
- XRPD X-ray powder diffraction
- Type D of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 6.2, 9.7, 13.1, 15.7, 21.9, and 23.6 and not having a diffraction at an angle (2 theta ⁇ 0.2) of 23.3. 47.
- XRPD X-ray powder diffraction
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 6.2, 9.7, 13.1, 15.7, 21.9, and 23.6, corresponding to d-spacing (angstroms ⁇ 0.2) of 14.4, 9.1, 6.8, 5.6, 4.1 and 3.8, respectively, and not having a diffraction at an angle (2 theta ⁇ 0.2) of 23.3. 48.
- Type D of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.3, 6.2, 8.7, 9.7, 12.3, 13.1, 13.8, 15.7, 18.0, 21.9, 23.6, and 26.7. 49.
- XRPD X-ray powder diffraction
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.3, 6.2, 8.7, 9.7, 12.3, 13.1, 13.8, 15.7, 18.0, 21.9, 23.6, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 20.7, 14.4, 10.2, 9.1, 7.2, 6.8, 6.4, 5.6, 4.9, 4.1, 3.8, and 3.3, respectively. 50.
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.3 6.2 8.7 9.7 12.3 13.1 13.8 15.7 18.0 19.5 21.9 23.6 24.8 26.7 29.5 30.8 31.7 35.4 37.8 38.6. 51.
- Type D of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.3 20.7 6.2 14.4 8.7 10.2 9.7 9.1 12.3 7.2 13.1 6.8 13.8 6.4 15.7 5.6 18.0 4.9 19.5 4.5 21.9 4.1 23.6 3.8 2 theta d-spacing 24.8 3.6 26.7 3.3 29.5 3.0 30.8 2.9 31.7 2.8 35.4 2.5 37.8 2.4 38.6 2.3. 52.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type E of Compound 1. 55.
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 20.1, 21.9, and 26.7. 58.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 15.1, 15.8, 17.5, 19.0, 20.1, 21.9, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 5.9, 5.6, 5.1, 4.7, 4.4, 4.1, and 3.3, respectively. 59.
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.1, 15.8, 17.5, 20.1, 21.9, 23.2, 23.7, and 26.7. 60.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.1, 15.8, 17.5, 20.1, 21.9, 23.2, 23.7, and 26.7, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.3, 5.9, 5.6, 5.1, 4.4, 4.1, 3.8, 3.8, and 3.3, respectively. 61.
- Type E of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 9.8, 12.4, 13.1, 15.1, 15.8, 16.8, 17.5, 18.1, 19.0, 20.1, 21.9, 23.2, 23.7, 26.7, and 27.8. 62.
- XRPD X-ray powder diffraction
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 9.8, 12.4, 13.1, 15.1, 15.8, 16.8, 17.5, 18.1, 19.0, 20.1, 21.9, 23.2, 23.7, 26.7, and 27.8, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.3, 9.1, 7.2, 6.7, 5.9, 5.6, 5.3, 5.1, 4.9, 4.7, 4.4, 4.1, 3.8, 3.8, 3.3, and 3.2, respectively.
- XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 9.8, 12.4, 13.1, 15.1, 15.8, 16.8, 17.5, 18.1, 19.0, 20.1, 21.9, 23.2, 23.7, 26.7, and 27.8, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.3, 9.1, 7.2, 6.7, 5.9, 5.6, 5.3, 5.1, 4.9,
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.6 8.8 9.8 12.4 13.1 13.8 15.1 15.8 16.8 17.5 18.1 19.0 20.1 21.9 23.2 23.7 24.8 26.7 27.8 29.5 30.8 31.7 33.0 34.5 35.4 36.7 37.8 38.7. 64.
- Type E of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 4.6 19.3 8.8 10.1 9.8 9.1 12.4 7.2 13.1 6.7 13.8 6.4 15.1 5.9 15.8 5.6 16.8 5.3 17.5 5.1 18.1 4.9 2 theta d-spacing 19.0 4.7 20.1 4.4 21.9 4.1 23.2 3.8 23.7 3.8 24.8 3.6 26.7 3.3 27.8 3.2 29.5 3.0 30.8 2.9 31.7 2.8 33.0 2.7 34.5 2.6 35.4 2.5 36.7 2.4 37.8 2.4 38.7 2.3.
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 20.0, 21.4, and 22.5, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.0, 5.5, 5.3, 4.4, 4.2, and 4.0, respectively. 70.
- Type F of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 19.0, 20.0, 21.4, 22.5, 23.2, and 25.3. 71.
- XRPD X-ray powder diffraction
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.7, 16.0, 16.8, 19.0, 20.0, 21.4, 22.5, 23.2, and 25.3, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.0, 5.5, 5.3, 4.7, 4.4, 4.2, 4.0, 3.8, and 3.5, respectively. 72.
- Type F of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.9, 14.7, 16.0, 16.8, 17.4, 19.0, 20.0, 20.6, 21.4, 22.5, 23.2, 25.3, 26.6, 27.2, 28.0, and 30.0. 73.
- XRPD X-ray powder diffraction
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.9, 14.7, 16.0, 16.8, 17.4, 19.0, 20.0, 20.6, 21.4, 22.5, 23.2, 25.3, 26.6, 27.2, 28.0, and 30.0, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.2, 6.9, 6.0, 5.5, 5.3, 5.1, 4.7, 4.4, 4.3, 4.2, 4.0, 3.8, 3.5, 3.4, 3.3, 3.2, and 3.0, respectively. 74.
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.4 10.9 12.9 14.7 16.0 16.8 17.4 19.0 20.0 20.6 21.4 22.5 23.2 25.3 26.6 27.2 28.0 30.0 32.0 32.9 38.3 39.1 75.
- Type F of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: 2 theta d-spacing 5.4 16.23 10.9 8.1 12.9 6.9 14.7 6.0 16.0 5.5 16.8 5.3 17.4 5.1 19.0 4.7 20.0 4.4 20.6 4.3 21.4 4.2 22.5 4.0 23.2 3.8 25.3 3.5 26.6 3.4 27.2 3.3 28.0 3.2 30.0 3.0 2 theta d-spacing 32.0 2.8 32.9 2.7 38.3 2.4 39.1 2.3 76.
- TGA thermogravimetric analysis
- DSC differential scanning calorimetry
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type G of Compound 1. 79.
- the crystalline solid form of embodiment 78 wherein Type G of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 14.3, 16.6, and 21.3.
- XRPD X-ray powder diffraction
- XRPD X-ray powder diffraction
- XRPD X-ray powder diffraction
- Type G of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 16.6, 19.8, 21.3, 22.3, 25.3, and 26.4. 84.
- XRPD X-ray powder diffraction
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 16.6, 19.8, 21.3, 22.3, 25.3, and 26.4, corresponding to d-spacing (angstroms ⁇ 0.2) of 16.5, 6.9, 6.2, 5.9, 5.3, 4.5, 4.2, 4.0, 3.5, and 3.4, respectively. 85.
- Type G of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 15.8, 16.6, 19.8, 21.3, 22.3, 25.3, 26.4, 27.4, and 30.2. 86.
- XRPD X-ray powder diffraction
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.4, 12.8, 14.3, 15.0, 15.8, 16.6, 19.8, 21.3, 22.3, 25.3, 26.4, 27.4, and 30.2, corresponding to d- spacing (angstroms ⁇ 0.2) of 16.5, 6.9, 6.2, 5.9, 5.6, 5.3, 4.5, 4.2, 4.0, 3.5, 3.4, 3.3, and 3.0, respectively. 87.
- Type G of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.4 8.7 12.8 14.3 15.0 15.8 16.6 18.5 19.8 21.3 22.3 23.4 25.3 26.4 27.4 30.2 32.3 38.0 88.
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type H of Compound 1. 90.
- Type H of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.8, 14.7, 16.6, 20.0, 21.3, and 25.4.
- XRPD X-ray powder diffraction
- XRPD X-ray powder diffraction
- Type H of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.8 8.4 11.5 12.4 13.1 13.7 14.7 14.9 16.0 16.2 16.6 16.9 17.3 17.7 18.3 19.5 20.0 21.3 21.9 23.1 23.6 23.9 24.4 24.9 25.1 25.4 26.2 27.4 28.1 28.4 29.3 29.7 30.4 31.0 32.7 33.4 34.1 34.8 35.5 35.8 36.4 37.1 38.5 93.
- Type H of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: Pos. [°2Th.] d-spacing [ ⁇ ] 5.8 15.3 8.4 10.5 11.5 7.7 12.4 7.2 13.1 6.8 13.7 6.5 14.7 6.0 14.9 5.9 16.0 5.6 16.2 5.5 16.6 5.4 16.9 5.3 17.3 5.1 17.7 5.0 18.3 4.8 19.5 4.6 20.0 4.4 21.3 4.2 21.9 4.1 23.1 3.9 Pos.
- the crystalline solid form of embodiment 94 wherein Type I of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 5.2, 14.6, 15.5, 20.2, and 21.1.
- XRPD X-ray powder diffraction
- XRPD X-ray powder diffraction
- Type I of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.2 8.8 10.3 12.6 14.6 15.5 16.1 16.3 16.6 17.1 17.6 18.7 18.9 20.2 20.5 20.7 21.1 21.5 22.0 22.3 23.7 24.8 25.2 26.0 26.3 26.5 26.8 27.0 27.5 27.7 28.1 29.6 30.0 30.4 31.3 32.0 32.5 33.2 34.0 34.6 36.9 38.2 38.9 39.5 98.
- Type I of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: Pos.
- the crystalline solid form of embodiment 1, wherein the crystalline solid form is Type J of Compound 1.
- the crystalline solid form of embodiment 99, wherein Type J of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.7, 22.8, 23.1, and 24.5. 101.
- XRPD X-ray powder diffraction
- Type J of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.7, 22.8, 23.1, and 24.5, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.5, 15.4, 3.9, 3.8, and 3.6, respectively. 102.
- Type J of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.5 5.7 7.1 7.7 9.1 10.5 11.2 11.7 12.3 12.9 14.3 14.5 15.4 15.7 16.3 17.3 18.3 18.7 19.3 19.6 20.5 21.2 21.5 22.8 23.1 23.6 24.1 24.5 25.2 25.9 26.4 27.8 29.3 36.2 37.0 103.
- Type J of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: Pos. [°2Th.] d-spacing [ ⁇ ] 4.5 19.5 5.7 15.4 7.1 12.7 7.7 11.5 9.1 9.7 10.5 8.4 11.2 7.9 11.7 7.5 Pos.
- the crystalline solid form of embodiment 104 wherein Type K of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.4, 15.6, 16.1, 23.2, and 27.4.
- XRPD X-ray powder diffraction
- the crystalline solid form of embodiment 104 or 105 wherein Type K of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.6, 15.4, 15.6, 16.1, 23.2, and 27.4, corresponding to d-spacing (angstroms ⁇ 0.2) of 19.2, 5.7, 5.7, 5.5, 3.8, and 3.3, respectively.
- Type L of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 5.9 8.4 11.9 13.3 14.7 15.0 16.2 16.7 16.9 17.8 18.9 20.4 21.2 21.6 22.2 23.9 24.6 25.5 25.7 26.1 26.8 28.1 28.8 29.9 30.6 31.9 32.4 33.6 34.2 35.6 36.1 38.2 113.
- Type L of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: Pos.
- the crystalline solid form of embodiment 114 wherein Type M of Compound 1 is characterized by an X-ray powder diffraction (XRPD) pattern having diffractions at angles (2 theta ⁇ 0.2) of 4.5, 5.8, 9.7, 15.6, 21.9, and 26.7.
- XRPD X-ray powder diffraction
- Type M of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) of: 4.5 5.8 6.1 8.7 9.0 9.7 12.3 13.1 13.7 14.5 15.1 15.6 16.8 17.4 18.0 18.5 19.5 20.0 21.4 21.9 22.3 22.9 23.3 23.5 24.1 25.0 25.8 26.3 26.7 27.8 28.1 29.4 30.8 31.7 33.0 35.3 37.8 38.6 118.
- Type M of Compound 1 is characterized by an XRPD pattern having diffractions at angles (2 theta ⁇ 0.2) corresponding to d-spacing (angstroms ⁇ 0.2) of: Pos. [°2Th.] d-spacing [ ⁇ ] 4.5 19.5 5.8 15.3 6.1 14.4 8.7 10.2 9.0 9.9 9.7 9.1 12.3 7.2 13.1 6.8 13.7 6.4 14.5 6.1 15.1 5.9 15.6 5.7 16.8 5.3 17.4 5.1 18.0 4.9 18.5 4.8 19.5 4.5 20.0 4.4 21.4 4.1 21.9 4.1 22.3 4.0 22.9 3.9 23.3 3.8 Pos.
- a pharmaceutical composition comprising a therapeutically effective amount of the crystalline solid form of any one of embodiments 1-119, and one or more pharmaceutically acceptable excipients. 121.
- the pharmaceutical composition of embodiment 120, wherein the pharmaceutical composition is for oral administration.
- the pharmaceutical composition of embodiment 120 or 121, wherein the pharmaceutical composition has a water content of about 0.5-5.0 weight%.
- the pharmaceutical composition of any one of embodiments 120-122, wherein the pharmaceutical composition has a water content of about 1.5-4.0 weight%.
- An amorphous solid dispersion comprising Compound 1: and a polymer. 126.
- amorphous solid dispersion of embodiment 125 wherein the polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co-methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprol
- Method D XRPD analysis
- Method D XRPD analysis
- Method D crystalline diffraction peaks are not observable by XRPD analysis
- TG single glass transition temperature
- B DSC analysis
- 140. The amorphous solid dispersion of any one of embodiments 125-139, wherein a single glass transition temperature (T G ) and no melt endotherm is observable by DSC analysis (Method B) of the amorphous solid dispersion after storage in a container as described in Example 20 for 5 months at 2-8 °C and ambient relative humidity.
- T G single glass transition temperature
- T B DSC analysis
- TG single glass transition temperature
- B DSC analysis
- TG single glass transition temperature
- Method B DSC analysis
- Method D XRPD analysis
- Method D XRPD analysis
- Method D crystalline diffraction peaks are not observable by XRPD analysis
- Method D XRPD analysis
- Method D crystalline diffraction peaks are not observable by XRPD analysis
- Method D XRPD analysis
- T G single glass transition temperature
- DSC analysis Methodhod B
- TG single glass transition temperature
- Method B DSC analysis
- a pharmaceutical composition comprising a therapeutically effective amount of the amorphous solid dispersion of any one of embodiments 125-162, and one or more pharmaceutically acceptable excipients.
- 165. The pharmaceutical composition of embodiment 163 or 164, wherein the pharmaceutical composition is in a tablet dosage form.
- the pharmaceutical composition of embodiment 180 further comprising a coating disposed on the tablet core.
- a method for preparing an amorphous solid dispersion comprising Compound 1 comprising: mixing Compound 1, a polymer, and a solvent to afford a mixture; and spray-drying the mixture to afford an amorphous solid dispersion comprising Compound 1. 191.
- polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam-polyvin
- any one of embodiments 190-194, wherein the weight ratio of Compound 1 to the polymer is about 1:1. 197. The method of any one of embodiments 190-194, wherein the weight ratio of Compound 1 to the polymer is about 1:3, about 2:3, about 1:1, about 1.5:1, about 2:1, or about 3:1. 198. The method of any one of embodiments 190-197, wherein the solvent is dichloromethane and methanol. 199. A product prepared by a process comprising: mixing Compound 1, a polymer, and a solvent to afford a mixture; and spray-drying the mixture to afford an amorphous solid dispersion comprising Compound 1: 200.
- polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g., Eudragit® L100-55), macrogol 15 hydroxystearate (e.g., Solutol® HS15), polyvinyl caprolactam-polyvin
- 201 The product of embodiment 199 or 200, wherein the polymer is hydroxypropylmethyl cellulose (HPMC) or hydroxypropylmethyl cellulose acetate succinate (HPMC AS).
- 202 The product of any one of embodiments 199-201, wherein the polymer is hydroxypropylmethyl cellulose acetate succinate (HPMC AS).
- 203 The product of any one of embodiments 199-202, wherein the weight ratio of Compound 1 to the polymer is in a range of about 3:1 to about 1:3 or about 2:1 to about 1:3.
- 204 The product of any one of embodiments 199-203, wherein the weight ratio of Compound 1 to the polymer is about 1:3. 205.
- a pharmaceutical composition comprising Compound 1 obtained by a process comprising: mixing Compound 1 in a solid form, a polymer, and a solvent to afford a mixture; and spray-drying the mixture to afford an amorphous solid dispersion comprising Compound 1. 209.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type A of Compound 1. 210.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type B of Compound 1. 211.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type C of Compound 1. 212.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type D of Compound 1. 213.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type E of Compound 1. 214.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type F of Compound 1. 215.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type G of Compound 1. 216.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type H of Compound 1. 217.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type I of Compound 1. 218.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type J of Compound 1. 219.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type K of Compound 1. 220.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type L of Compound 1. 221.
- the pharmaceutical composition of embodiment 208, wherein the solid form is Type M of Compound 1. 222.
- the pharmaceutical composition of embodiment 208, wherein the solid form is selected from the group consisting of Type A, Type B, Type C, Type D, Type E, Type F, Type G, Type H, Type I, Type J, Type K, Type L, and Type M of Compound 1. 223.
- HPMC
- a tablet dosage form comprising a tablet core, the tablet core comprising at least 10 weight % of Compound 1 in amorphous form: wherein crystalline Compound 1 (Type A) is not observable by XRPD analysis (Method D) of the tablet core. 236.
- the tablet dosage form of embodiment 235 wherein the tablet core comprises at least 15 weight % of Compound 1 in amorphous form. 237.
- a tablet dosage form comprising a tablet core, the tablet core having a total weight of no more than about 1000 mg and comprising about 200 mg of Compound 1 in amorphous form per tablet: wherein crystalline Compound 1 (Type A) is not observable by XRPD analysis (Method D) of the tablet core. 241.
- the tablet dosage form of embodiment 242 wherein the tablet core comprises 0.05-3.0 % of Compound 2, based on the total amount of Compound 1 and Compound 2. 244.
- the polymer is selected from a group consisting of hydroxypropylmethyl cellulose (HPMC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl cellulose (HPC), ethylcellulose, cellulose acetate phthalate, polyvinylpyrrolidone (PVP), and a combination thereof, or is selected from a group consisting of polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), hydroxypropylmethyl cellulose acetate succinate (HPMC AS), hydroxyethylcellulose (HEC), poly(methacrylic acid-co- methyl methacrylates) (e.g.
- the tablet dosage form of embodiment 260, wherein the one or more pharmaceutically acceptable excipients comprise one or more of a filler, a dry binder, a glidant, a lubricant, a disintegrant, and a film coating agent. 262.
- the tablet dosage form of embodiment 262 wherein the intragranular portion comprises an amorphous solid dispersion comprising Compound 1 and a polymer and one or more of a filler, a dry binder, a glidant, and a lubricant, and the extragranular portion comprises one or more of a filler, a disintegrant, and a lubricant.
- the intragranular portion comprises an amorphous solid dispersion comprising Compound 1 and a polymer and one or more of a filler, a dry binder, a glidant, and a lubricant
- the extragranular portion comprises one or more of a filler, a disintegrant, and a lubricant.
- a solid oral dosage form comprising a stabilized amorphous compound (S)-1-(5-[2H,3H- [1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3- hydroxy-2-phenylpropan-1-one, wherein the stabilized amorphous compound does not show crystallinity by PXRD (Method D) after 2 weeks of storage at 60 °C/75% RH (exposed). 278.
- the solid oral dosage form of embodiment 277 wherein the stabilized amorphous compound shows a single glass transition temperature (T G ) and no melt endotherm by DSC (Method B) after 2 weeks of storage at 60 °C/75% RH (exposed). 279.
- the solid oral dosage form of embodiment 284, wherein the (S)-1-(5-[2H,3H- [1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3- hydroxy-2-phenylpropan-1-one is spray dried with HPMC AS in a weight ratio of 1:3 to 2:1. 286.
- a (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one active pharmaceutical ingredient (API) composition comprising 0.05-5.0% by HPLC of (R)-1-(5-[2H,3H-[1,4]dioxino[2,3- b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2- phenylpropan-1-one.
- a tablet comprising 200 mg of stabilized amorphous compound (S)-1-(5-[2H,3H- [1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3- hydroxy-2-phenylpropan-1-one as the active pharmaceutical ingredient (API), wherein the stabilized amorphous compound does not show crystallinity by PXRD (Method D) after 2 weeks of storage of the tablet at 60 °C/75% RH (exposed). 289.
- the present teachings include descriptions provided in the Examples that are not intended to limit the scope of any claim. The following non-limiting examples are provided to further illustrate the present teachings.
- X-ray Powder Diffraction (XRPD or PXRD) [0222]
- Method A XRPD analysis was performed with a Panalytical X’Pert3 Powder XRPD on a Si zero-background holder. The 2q position was calibrated against Panalytical 640 Si powder standard. Details of the XRPD method used in the experiments are listed in Table 1.
- Table 1 [0223]
- Method B XRPD analysis was performed with a Rigaku X-Ray Powder Diffractomer MiniFlex 600 with the parameters listed in Table 2.
- Table 2 [0224]
- Method C XRPD analysis was performed with a Panalytical X’Pert3 powder diffractometer in reflection mode.
- Dissolution of the tablets is performed with USP Apparatus 2 (paddles) by USP ⁇ 711>.
- the determination of assay is achieved by quantitation against an external reference standard using a reversed phase gradient UPLC method.
- the UPLC method utilizes an Acquity UPLC BEH Shield column with two mobile phases, both consisting of acetonitrile, water, and phosphate buffer..
- Example 1 Synthesis of (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]- 1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one (1)
- the PKR Activating Compound 1 can be obtained by the method described herein and the reaction schemes shown in Figures 1 and 2.
- Compound 1 has a molecular weight of 457.50 Da.
- n-BuLi in hexane
- n-Bu2Mg in heptanes
- the resulting solution was stirred overnight at 20 °C.
- the reaction mixture was diluted with 20 mL of water and was then extracted with 3x20 mL of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum.
- the residue was purified by prep-TLC eluted with dichloromethane/methanol (20:1) and further purified by prep-HPLC (Column: XBridge C18 OBD Prep Column, 100 ⁇ , 5 ⁇ m, 19 mm x 250 mm; Mobile Phase A: water (10 mmol/L NH 4 HCO 3 ), Mobile Phase B: MeCN; Gradient: 15% B to 45% B over 8 min; Flow rate: 20 mL/min; UV Detector: 254 nm).
- the two enantiomers were separated by prep-Chiral HPLC (Column, Daicel CHIRALPAK ⁇ IF, 2.0 cm x 25 cm, 5 mm; mobile phase A: DCM, phase B: MeOH (hold 60% MeOH over 15 min); Flow rate: 16 mL/min; Detector, UV 254 & 220 nm).
- Compound 1 can be synthesized using the procedure described here as Step 5.
- the solution was diluted with EtOAc (20 mL), washed sequentially with water (20 mL) and brine (2x20 mL), dried (MgSCL), filtered, treated with silica gel, and evaporated under reduced pressure.
- the material was chromatographed by Biotage MPLC (10 g silica gel column, 0 to 5% MeOH in DCM) to provide a white, slightly sticky solid.
- Example 2- Preparation and Characterization of Type A of Compound 1
- a 1L round-bottom flask with overhead stirring, a temperature probe, and an N 2 inlet was charged with 7-((3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)sulfonyl)-2,3-dihydro- [1,4]dioxino[2,3-b]pyridine (33.24 g, 96 mmol), (S)-3-hydroxy-2-phenylpropanoic acid (19.08 g, 115 mmol), and DMF (361 ml).
- Type A was characterized by XRPD (Method A), TGA (Method A), DSC (Method A), and DVS analysis. [0249] The XRPD pattern for Type A is depicted in Figure 3, and the corresponding data are summarized in the following table: [0250] The foregoing XRPD data for Type A can also be rounded to a single decimal place, as summarized in the following table: [0251] The TGA and DSC curves for Type A are shown in Figure 4.
- Type A showed 1.9% weight loss up to 100 °C by TGA and two endotherms at 85.9 oC (peak temperature) and 146.0 oC (onset temperature) by DSC.
- Type A was heated to 120oC and cooled to 25oC, then heated up to 300oC. No endotherm below 100oC was observed in the second heating cycle.
- XRPD analysis after DSC cycling showed no form change compared to Type A ( Figure 3).
- DVS results showed a 3.4% water uptake up to 40% RH (ambient condition), and 1.0% water uptake from 40% RH to 80%RH at RT, indicating that Type A is hygroscopic (Figure 6).
- Type A is believed to be a channel hydrate.
- Example 3- Polymorph Screening of Compound 1 Polymorph screening experiments were performed using a series of crystallization and solid transition methods. Solid Vapor Diffusion [0256] Solid vapor diffusion experiments were conducted using 13 different solvents. Approximately 15 mg of Compound 1 (Type A) was weighed into a 4-mL vial, which was placed into a 20-mL vial with 3 mL of volatile solvent. The 20-mL vial was sealed with a cap and kept at RT for 7 days, allowing solvent vapor to interact with the sample.
- Table 9 Solvent Solid Form H 2 O Type A EtOH Type A Toluene Type A Acetone Type A ACN Type A DCM Type A THF Type D + Type A CHCl 3 Type A MeOH Type A IPA Type A 1,4-Dioxane Type A DMSO Type A EtOAc Type A Slurry Conversion at 4°C, RT or 50 °C [0257] Slurry experiments were conducted at RT in different solvent systems. About 15 mg of Compound 1 (Type A) was suspended in 0.3 mL of solvent in a 2-mL glass vial.
- Type B was prepared on a 100mg scale from a slurry of Type A in methanol at 50 °C, via a method analogous to the method for slurry conversion described in Example 3.
- Type B was characterized by XRPD (Method A), TGA (Method A), DSC (Method A), and DVS analysis. [0267] The XRPD pattern for Type B is depicted in Figure 7, and the corresponding data are summarized in the following table: [0268] The foregoing XRPD data for Type B can also be rounded to a single decimal place, as summarized in the following table: [0269] The TGA and DSC curves for Type B are shown in Figure 8. As shown in Figure 8, Type B showed 1.8% weight loss up to 100 oC and an endotherm at 138.2 oC (onset temperature) possibly due to melting.
- Type B By DVS analysis (Figure 11), Type B showed a 2.9% water uptake up to 60% RH (ambient condition), and 0.4% water uptake from 60% RH to 80%RH at RT, indicating that Type B is hygroscopic. No form change was observed for Type B before and after DVS test at RT, as determined by XRPD analysis. [0274] Based on the foregoing analytical data, Type B is believed to be a channel hydrate.
- Type C was prepared on a 100 mg scale from a slurry of Type A in 1,4-Dioxane at RT, via a method analogous to the method for slurry conversion described in Example 3. Characterization of Type C of Compound 1 [0276] Type C was characterized by XRPD (Method A), TGA (Method A), DSC (Method A), and DVS analysis.
- the XRPD pattern for Type C is depicted in Figure 12, and the corresponding data are summarized in the following table: [0278]
- the foregoing XRPD data for Type C can also be rounded to a single decimal place, as summarized in the following table: [0279]
- the TGA and DSC curves for Type C are shown in Figure 13. As shown in Figure 13, Type C showed 1.0% weight loss up to 100 oC and an endotherm at 152.2 oC (onset temperature) possibly due to melting.
- DSC cycling RT-120 oC- RT- 250 oC
- Type C showed a 0.7% weight loss up to 130 oC by instant TGA test after heating to 120 o C and exposing to ambient conditions for one minute. The normal TGA curve of the Type C showed 2.3% weight loss up 130 o C without pre-heating treatment. [0282] After heating to 120 oC and cooling to RT by DSC cycling, no form change was observed by XRPD analysis. [0283] By DVS analysis ( Figure 16), Type C showed a 1.8% water uptake up to 60% RH (ambient condition), and 0.5% water uptake from 60% RH to 80%RH at RT, indicating that Type C is hygroscopic.
- Type D was prepared from a slurry of Type A in Tetrahydrofuran (THF) at 4 °C, via a method analogous to the method for slurry conversion described in Example 3. Characterization of Type D of Compound 1 [0286] Type D was characterized by XRPD (Method A), TGA (Method A), DSC (Method A), and 1 H NMR analysis.
- the XRPD pattern for Type D is depicted in Figure 17, and the corresponding data are summarized in the following table: [0288]
- the foregoing XRPD data for Type D can also be rounded to a single decimal place, as summarized in the following table: [0289]
- the TGA and DSC curves for Type D are shown in Figure 18. As shown in Figure 18, Type D showed 9.6% weight loss up to 130 oC by TGA and an endotherm at 91.9oC (onset temperature) by DSC.
- the 1 H NMR spectra of Type A and Type D are shown in Figure 19.
- Type D appears to be a THF solvate, as indicated by the 1 H NMR spectrum (600 MHz, DMSO-d6), which detected the presence of THF protons at ⁇ 1.76 and ⁇ 3.60ppm.
- Example 7- Preparation and Characterization of Type E of Compound 1 Characterization of Type E of Compound 1 [0291] Type E was characterized by XRPD (Method A) analysis.
- Type F of Compound 1 was produced via liquid vapor diffusion in 1,4-Dioxane/heptane at RT. Characterization of Type F of Compound 1 [0295] Type F was characterized by XRPD (Method A), TGA, and DSC analysis (Method A).
- Type F The XRPD pattern for Type F is shown in Figure 21, and the corresponding data are summarized in the following table: [0297]
- the foregoing XRPD data for Type F can also be rounded to a single decimal place, as summarized in the following table: [0298]
- the TGA and DSC curves for Type F are shown in Figure 22. As shown in Figure 22, Type F showed 6.2% weight loss up to 120 oC by TGA and two endotherms at 100.4oC and 125.9 oC (onset temperature) by DSC.
- Type G was prepared from a slurry of Type A in methyl ethyl ketone at room temperature. Characterization of Type G of Compound 1 [0300] Type G was characterized by XRPD (Method A) analysis.
- Type H was prepared by liquid vapor diffusion, as described in Example 3. Characterization of Type H of Compound 1 [0304] Type H was characterized by XRPD (Method C) analysis.
- Type I was prepared by liquid vapor diffusion, as described in Example 3. Characterization of Type I of Compound 1 [0307] Type I was characterized by XRPD (Method C) analysis. [0308] The XRPD pattern for Type I is depicted in Figure 25, and the corresponding data are summarized in the following table: Example 12 - Preparation and Characterization of Type J of Compound 1 Preparation of Type J of Compound 1 [0309] Type J was prepared by liquid vapor diffusion, as described in Example 3.
- Type J was characterized by XRPD (Method C) analysis. [0311] The XRPD pattern for Type J is depicted in Figure 26, and the corresponding data are summarized in the following table: Example 13 - Preparation and Characterization of Type K of Compound 1 Preparation of Type K of Compound 1 [0312] Type K was prepared by slow cooling, as described in Example 3. Characterization of Type K of Compound 1 [0313] Type K was characterized by XRPD (Method C) analysis.
- Type L was prepared by slow cooling, as described in Example 3. Characterization of Type L of Compound 1 [0316] Type L was characterized by XRPD (Method C) analysis. [0317] The XRPD pattern for Type L is depicted in Figure 28, and the corresponding data are summarized in the following table: [0318] Single crystal X-ray analysis revealed that Type L is a THF/water co-solvate of Compound 1, with Compound 1, THF, and water present in a 1:1:1 ratio.
- Example 15 Preparation and Characterization of Type M of Compound 1
- Type M was prepared by liquid vapor diffusion, as described in Example 3. Characterization of Type M of Compound 1 [0320]
- Type M was characterized by XRPD (Method C) analysis. [0321] The XRPD pattern for Type M is depicted in Figure 29, and the corresponding data are summarized in the following table:
- Example 16 Preparation and Characterization of the Spray-Dried Dispersion of Compound 1 [0322] A Spray Dried Dispersion (SDD) of Compound 1 was prepared.
- SDD Spray Dried Dispersion
- the SDD was made up of Compound 1 and a polymer (Hydroxypropylmethyl Cellulose AS-MG) at a 1:3 weight ratio. Compound 1 and the polymer were dissolved in organic solvents (Dichloromethane and Methanol) and spray dried to obtain amorphous an amorphous drug substance.
- the SDD comprising Compound 1 and HPMC AS (1:3) is referred to herein as SDD 0.
- a spray solution was prepared at 7.8% solids content (1:3 Compound 1:HPMC AS- MG) in 80:20 DCM:Methanol per Table 18. An API correction factor of 0.966 was used to prepare the spray solution. The spray solution was prepped by adding DCM and Methanol to a 36L stainless steel mixing vessel.
- HPMC AS-MG was added to the solvent system while mixing with a top down mixer at a medium vortex. Compound 1 was then added to the solution. The solution had a yellow/brown clear appearance, however white fiber particulates were seen in the solution.
- the spray-dried dispersion was dried overnight ( ⁇ 20 hours) in a Shel Vacuum Oven at 50°C and -25 in Hg vacuum under a nitrogen purge at 15 scfh.
- the resulting spray-dried dispersion was confirmed to be dry by GC analysis. This run generated approximately 2.1 kg of spray-dried dispersion.
- Table 19 [0325]
- the SDD was characterized by XRPD (Method B) and DSC analysis (ambient to 200°C, 2°C/minute ramp), as shown in Figures 30 and 31, respectively.
- the SDD was determined to be homogenous and amorphous, as shown by the amorphous diffractogram, lack of a crystalline melt, and a single Tg at 100°C.
- Example 17- Bioavailability of the Spray-Dried Dispersion (SDD) of Compound 1 in Rats and Mice The systemic exposure of Compound 1 in rats and mice was evaluated by dosing a SDD made up of Compound 1 and HPMC AS-MG (1:3) (SDD 0, which can be prepared as described in Example 16) dispersed in an aqueous vehicle (0.5% Hydroxypropylmethyl Cellulose in water).
- the SDD formulation (“500 mpk SDD”) dosed at 500 mg/kg to rats showed an AUClast that was 40X greater than the maximum exposure obtained with the standard formulation (“300 mpk Suspension” made up of Compound 1 (Type A) in 10% Propylene Glycol, 10% Cremophore, 80% Water), as shown in Table 20 and Figure 32. Additionally, the exposure of a 500 mpk Nano- Suspension made up of nanoparticles of Compound 1 (Type A) was evaluated, as shown in Figure 32. Robust exposure was observed with SDD formulation in mouse as well.
- Example 20 Stability Assessment of the Spray-Dried Dispersion of Compound 1
- Stability studies were conducted on two distinct lots of the 1:3 Compound 1: HPMC- AS-MG spray-dried dispersion (SDD 0, which can be prepared as described in Example 16) under the conditions outlined in Table 24.
- SDD spray-dried dispersion
- Table 24 The results of the stability study for each lot and storage condition are reported in the Tables identified in Table 24.
- Spray solutions having varying ratios of Compound 1 to polymer were prepared at 8% solids content in 80:20 DCM:MeOH (Table 30).
- the spray solutions were spray dried using a Procept 4M8-Trix unit with the settings detailed in Table 31.
- the resulting spray dried dispersions (SDDs) were dried at 50 °C at -25 in. Hg in a nitrogen purged vacuum oven for 19 hours.
- the SDDs were evaluated by XRPD analysis (Method D; Figure 36) and DSC analysis (Method B; Figure 37).
- the SDDs appeared amorphous by PXRD analysis, with no crystalline diffraction peaks observed.
- a single well defined T G was seen by DSC for all dispersions. No melt endotherm was observed, further verifying the amorphous nature of all spray dried dispersions.
- Residual solvent analysis of the spray dried dispersions dried for 19 hours showed varying levels for dichloromethane. An observed trend is that levels of dichloromethane increase with increased ratios of Compound 1 to Polymer.
- Spray solutions having varying ratios of Compound 1 to polymer were prepared at 12% solids content in 80:20 DCM:MeOH (Table 32). The spray solutions were sprayed on a GEA Mobile Minor spray dryer, and the SDDs were collected and dried at 50 °C and -25 in Hg under a N 2 purge.
- Standard calibration curves were generated in the respective assay media using a serial addition protocol.
- a stock solution of Compound 1 was prepared in DMSO at ⁇ 20 mg/mL. Calculated aliquots of the stock were added to the respective buffers in order to prepare several standard solutions spanning specific concentration ranges. Concentrations of the standard solutions ranged from ⁇ 50 to ⁇ 300 ⁇ g/mL for channels in both SGF and FaSSIF media respectively. The area under the 2 nd derivative curves was used to calculate the concentrations.
- the wavelength range was selected for the compounds in such a way that sensitivity issues were avoided. Linearity of the standard curves in the selected wavelength regions were characterized by r 2 30.999. [0338] Area under the 2nd derivative curve in 285-300 nm (SGF) and 305-320 nm (FaSSIF) range were used to calculate the standard curves in respective media. The corresponding standard curves were used to determine concentrations of Compound 1 in solubility assays. [0339] Required amount of SDD materials, equivalent to 20 mg of Compound 1 were weighed into 20mL glass vials. The vials were then transferred to the instrument for analysis. A clean stir bar was added to the vial with sample.
- SDD 0 formulation with 25% drug loading reached higher solubility of ⁇ 700 ⁇ g/mL but did not remain in the supersaturated state for longer time.
- solubility of SDD 0 at 4H was slightly lower than the 40% API loading SDD 1
- the equilibrium solubility after ⁇ 16 hours was higher than all other SDD systems.
- SDD 3 and SDD 4 did not reach as high of a concentration (spring effect) as SDD 0.
- SDD 3 and SDD 4 both stayed at supersaturated state for longer time when compared to all other SDD systems tested.
- the equilibrium solubility for SDD 3 and SDD 4 after 16 hours was lower than SDD 0.
- SDD 2 and SDD 3 show marked enhancement in the solubility and prolonged supersaturation.
- SDD 4 also showed two glass transition temperatures by DSC (Method B) after 2 weeks of storage at 60 °C/75% RH (exposed), suggesting phase separation.
- Table 34 Example 25 - Stability of Spray Dried Dispersions of Compound 1 [0345] Spray dried dispersions of Compound 1 (SDDs 5 and 6 (Example 22)) were stored in two different storage configurations at various storage conditions. Samples were set up as “Sealed” and “Exposed”. Sealed samples were stored in an amber crimp sealed vial at the following conditions: 2-8°C, 25°C/60%RH, 40°C/75%RH, and 60°C.
- Table 36 Example 26 - Composition and Preparation of a Tablet Dosage Form of Compound 1 Composition of the Tablet Dosage Form
- a tablet dosage form of Compound 1 comprising an SDD made up of Compound 1 and HPMC AS-MG (1:3) compressed into tablets and film coated with compendial excipients was prepared. The tablets were presented as 25 mg (white coated round shaped tablets) and 100 mg (white coated oval shaped tablets) dose strengths.
- Table 37 1 Quantity based upon a 25.00% (w/w) potency of Compound 1 drug substance in the spray dried intermediate.
- Table 37 1 Quantity based upon a 25.00% (w/w) potency of Compound 1 drug substance in the spray dried intermediate.
- Preparation of the Tablet Dosage Form [0349]
- the Compound 1 tablet formulation manufacturing process consists of four steps: 1) spray dry dispersion, 2) intragranular granulation, roller compaction/milling/blending, 3) extragranular granulation/blending, and 4) tableting and coating.
- the initial step of spray dry dispersion is performed by creating an organic solution containing Compound 1 drug substance, and Hypromellose Acetate Succinate (Hydroxypropyl Methylcellulose Acetate Succinate MG) (HPMCAS-MG).
- Example 27 Dissolution Assessment of the Tablet Dosage Form [0002] The 100 mg tablets described in Example 26 were tested for dissolution. Dissolution testing parameters are provided in Table 38, and results are summarized in Table 39.
- Example 28 - Release Testing of the Tablet Dosage Form Dissolution testing of 25 mg and 100 mg tablets having the composition specified in Example 26 was performed following the dissolution parameters listed in Table 40. Dissolution was determined by UPLC analysis. The results of the dissolution testing are reported in Table 41 and Figure 52. Table 40 Table 41 Example 29- Stability Assessment of the Tablet Dosage Form [0351] Stability studies were conducted under the conditions outlined in Table 42 on two distinct lots of 25 mg and 100 mg tablets having the composition specified in Example 26. The results of the stability study for each lot and storage condition are reported in the Tables identified in Table 42.
- the tablets were prepared for XRPD analysis (Method D) by crushing a tablet with a mortar and pestle and transferring 5-10 mg of material to a sample pan, slightly overfilling and ensuring that powder is spread evenly to cover the bottom of the plate. Weigh paper was placed atop the powder and pressed down gently to even the powder surface.
- the XRPD pattern of the tablet was overlaid with the XRPD pattern of a reference standard (Compound 1, Type A).
- a tablet was deemed to be free of the diffraction peaks that are present in the reference standard only if the peak at ⁇ 15 degrees 2-theta is absent. A small, irregular peak at ⁇ 3 degrees 2-theta is acceptable.
- Example 30 Composition and Preparation of a Tablet Dosage Form of Compound 1
- Tablets comprising a spray dried dispersion (SDD) of Compound 1 and compendial excipients are prepared at 100 mg and 200 mg dosage strengths. The compositions of the tablets are set forth in Tables 51 and 52.
- the tablets are prepared by first manufacturing the SDD (spray drying an organic solution of Compound 1 and HPMC-AS (1:1 w/w) (Table 51) or an organic solution of Compound 1 and HPMC-AS (1.5:1 w/w) (Table 52)), followed by roller compaction/milling with intragranular excipients and blending with extragranular excipients. The final blend is pressed into tablets and then film coated.
- SDD spray drying an organic solution of Compound 1 and HPMC-AS (1:1 w/w)
- Table 52 organic solution of Compound 1 and HPMC-AS
- Tablets having the compositions set forth in Tables 51 and 52 were prepared for XRPD analysis (Method D) by crushing a tablet with a mortar and pestle and transferring 5-10 mg of material to a sample pan, slightly overfilling and ensuring that powder is spread evenly to cover the bottom of the plate. Weigh paper was placed atop the powder and pressed down gently to even the powder surface. The XRPD pattern of the tablet was overlaid with the XRPD pattern of a reference standard (Compound 1, Type A).
- the XRPD pattern of a tablet was deemed to be free of the diffraction peaks that are present in the reference standard only if the peak at ⁇ 15 degrees 2-theta is absent. A small, irregular peak at ⁇ 3 degrees 2-theta is acceptable.
- the tablets were determined to be free of crystalline Type A because the XRPD patterns were free of the diffraction peaks that are present in the reference standard.
- Example 32 Determination of Maximum Dose of Compound 2 [0357] Good Laboratory Practice (GLP) toxicology testing of a Compound 1 test article comprising a pre-determined amount of Compound 2 was performed in rat and cynomolgus monkeys.
- GLP Good Laboratory Practice
- a no-observed-adverse-effect level was determined for each species using standard toxicology techniques, and a resulting dose level for humans was calculated using the FDA Human Equivalent Dose approach based upon dose per body surface area. Based on these experiments and calculations, a maximum recommended starting dose (MRSD) for first-in-human clinical trials was determined based on results from GLP toxicology testing. API compositions of Compound 1 containing less than 5.0% (as determined by percentage area HPLC) of Compound 2 are well within the safe human equivalent dose determined for Compound 2.
Abstract
Description





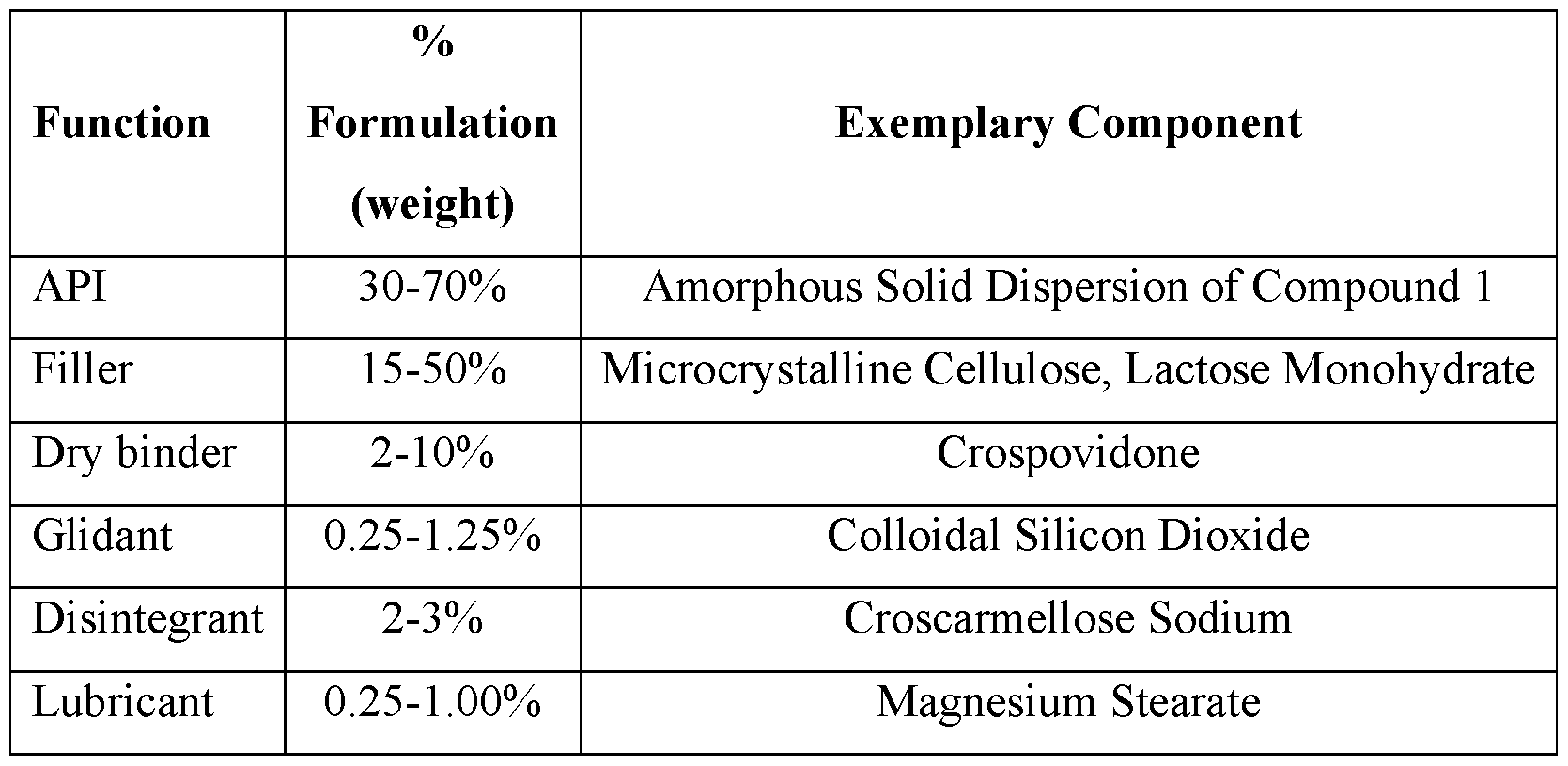



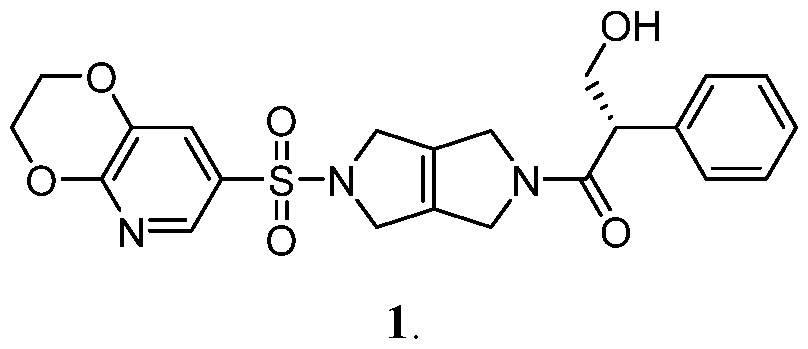










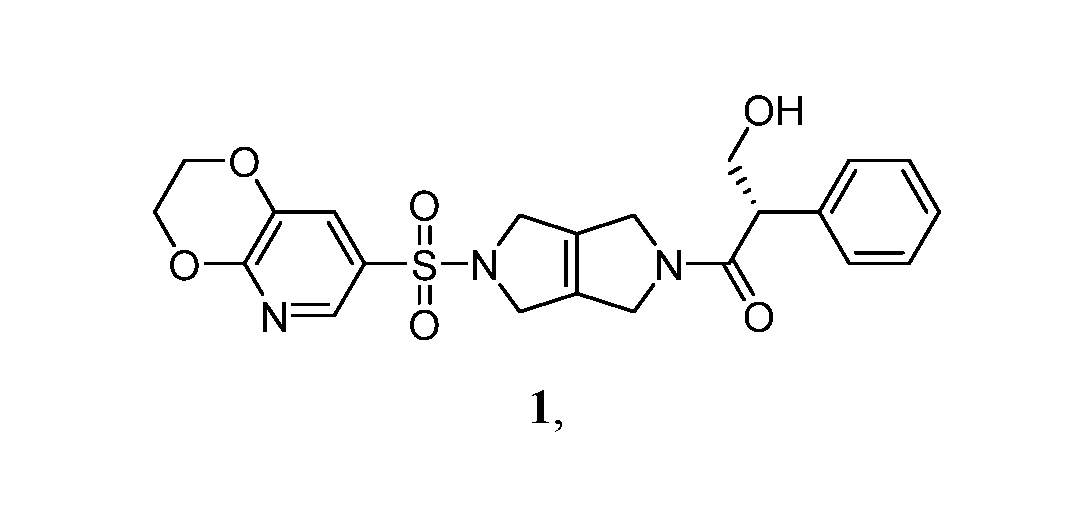

















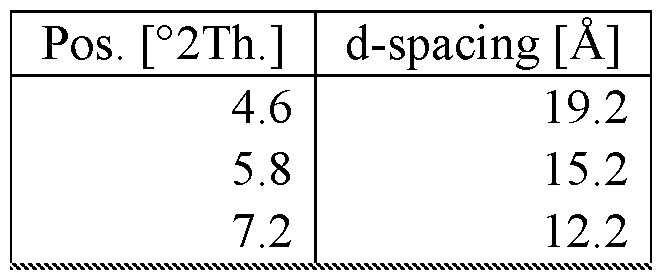













































































Claims
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112022004715A BR112022004715A2 (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase r (pkr) activating compositions |
| AU2020350763A AU2020350763A1 (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase R (PKR) activating compositions |
| CA3151612A CA3151612A1 (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase r (pkr) activating compositions |
| CN202080066237.3A CN114615977A (en) | 2019-09-19 | 2020-09-18 | Pyruvate Kinase R (PKR) activating compositions |
| KR1020227008754A KR20220066058A (en) | 2019-09-19 | 2020-09-18 | Pyruvate Kinase R (PKR) Activating Composition |
| US17/761,788 US20220378755A1 (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase r (pkr) activating compositions |
| MX2022003254A MX2022003254A (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase r (pkr) activating compositions. |
| EP20865925.0A EP4031133A4 (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase r (pkr) activating compositions |
Applications Claiming Priority (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962902887P | 2019-09-19 | 2019-09-19 | |
| US16/576,720 | 2019-09-19 | ||
| PCT/US2019/052024 WO2020061378A1 (en) | 2018-09-19 | 2019-09-19 | Treating sickle cell disease with a pyruvate kinase r activating compound |
| US16/576,720 US20200129485A1 (en) | 2018-09-19 | 2019-09-19 | Treating sickle cell disease with a pyruvate kinase r activating compound |
| USUS2019/052024 | 2019-09-19 | ||
| US16/576,360 US10675274B2 (en) | 2018-09-19 | 2019-09-19 | Activating pyruvate kinase R |
| US16/576,360 | 2019-09-19 | ||
| US62/902,887 | 2019-09-19 | ||
| US201962906437P | 2019-09-26 | 2019-09-26 | |
| US62/906,437 | 2019-09-26 | ||
| US202063024441P | 2020-05-13 | 2020-05-13 | |
| US202063024432P | 2020-05-13 | 2020-05-13 | |
| US63/024,432 | 2020-05-13 | ||
| US63/024,441 | 2020-05-13 | ||
| US202062704785P | 2020-05-28 | 2020-05-28 | |
| US62/704,785 | 2020-05-28 | ||
| US202062705106P | 2020-06-11 | 2020-06-11 | |
| US62/705,106 | 2020-06-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021055863A1 true WO2021055863A1 (en) | 2021-03-25 |
Family
ID=75494917
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2020/051645 WO2021055863A1 (en) | 2019-09-19 | 2020-09-18 | Pyruvate kinase r (pkr) activating compositions |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2021055863A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016046837A1 (en) * | 2014-09-22 | 2016-03-31 | Cadila Healthcare Limited | An improved process for preparation of pyrrolo[3,4- c] pyrrole compounds and intermediates thereof |
| US20160106728A1 (en) * | 2013-05-01 | 2016-04-21 | Academia Sinica | Methods and compositions for treating beta-thalassemia and sickle cell disease |
| WO2018175474A1 (en) * | 2017-03-20 | 2018-09-27 | Forma Therapeutics, Inc. | Pyrrolopyrrole compositions as pyruvate kinase (pkr) activators |
-
2020
- 2020-09-18 WO PCT/US2020/051645 patent/WO2021055863A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160106728A1 (en) * | 2013-05-01 | 2016-04-21 | Academia Sinica | Methods and compositions for treating beta-thalassemia and sickle cell disease |
| WO2016046837A1 (en) * | 2014-09-22 | 2016-03-31 | Cadila Healthcare Limited | An improved process for preparation of pyrrolo[3,4- c] pyrrole compounds and intermediates thereof |
| WO2018175474A1 (en) * | 2017-03-20 | 2018-09-27 | Forma Therapeutics, Inc. | Pyrrolopyrrole compositions as pyruvate kinase (pkr) activators |
| US20190218221A1 (en) * | 2017-03-20 | 2019-07-18 | Forma Therapeutics, Inc. | Compositions for activating pyruvate kinase |
Non-Patent Citations (1)
| Title |
|---|
| DATABASE PUBCHEM COMPOUND [online] 15 December 2018 (2018-12-15), ANONYMOUS: "Compound Summary for CID 135338361", XP055804602, retrieved from NCBI Database accession no. CID 135338361 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI558702B (en) | Solid forms of a pharmaceutically active substance | |
| CA2835332C (en) | Polymorph of linagliptin benzoate | |
| TW200815439A (en) | Hydrochloride salts of 8-[{1-(3,5-bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1,7-diaza-spiro[4.5]decan-2-one and preparation process therefor | |
| HUE028620T2 (en) | Salt and solvates of a tetrahydroisoquinoline derivative | |
| JP5134234B2 (en) | Pyrimidine derivatives for the treatment of abnormal cell proliferation | |
| WO2021055863A1 (en) | Pyruvate kinase r (pkr) activating compositions | |
| AU2020350763A1 (en) | Pyruvate kinase R (PKR) activating compositions | |
| US20240131011A1 (en) | Pyruvate kinase r (pkr) activating compositions | |
| EP1880992A1 (en) | Stable modifications of tegaserod hydrogen maleate | |
| EP3274333B1 (en) | Cabozantinib salts and their use as anti-cancer agents | |
| CN115916751A (en) | Organic acid addition salts of S-pindolol | |
| KR20230003036A (en) | Crystalline form XI of lenvatinib mesylate and method for preparing the same | |
| US20160136148A1 (en) | Solid dispersions of amorphous paroxetine mesylate | |
| CN113372332B (en) | Novel crystal form of octreotide | |
| US20230338338A1 (en) | Pharmaceutical compositions comprising ionic liquids | |
| WO2022199707A1 (en) | Pharmaceutically acceptable salt of pimavanserin, and preparation method therefor, pharmaceutical composition containing same, and use thereof | |
| WO2019105082A1 (en) | Crystal form of galunisertib and preparation method therefor and use thereof | |
| WO2019137027A1 (en) | Crystal form of galunisertib and preparation method and use thereof | |
| WO2023079093A1 (en) | Polymorphs of the mesylate salt of linaprazan glurate | |
| WO2023079094A1 (en) | Polymorphs of the hydrochloride salt of linaprazan glurate | |
| CN116601151A (en) | Crystalline forms of a KRAS G12C inhibitor | |
| ZA200208454B (en) | Zolpidem hemitartrate. | |
| NZ630486B2 (en) | Process for the humanization of animal skim milk and products obtained thereby | |
| KR20070022037A (en) | Pharmaceutical composition comprising a salt of mirtazapine | |
| AU2007254641A1 (en) | Stable modifications of tegaserod hydrogen maleate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20865925 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 3151612 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112022004715 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2020350763 Country of ref document: AU Date of ref document: 20200918 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020865925 Country of ref document: EP Effective date: 20220419 |
|
| ENP | Entry into the national phase |
Ref document number: 112022004715 Country of ref document: BR Kind code of ref document: A2 Effective date: 20220314 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 522431996 Country of ref document: SA |