WO2020240392A1 - Process for preparation of pyroxasulfone - Google Patents
Process for preparation of pyroxasulfone Download PDFInfo
- Publication number
- WO2020240392A1 WO2020240392A1 PCT/IB2020/054914 IB2020054914W WO2020240392A1 WO 2020240392 A1 WO2020240392 A1 WO 2020240392A1 IB 2020054914 W IB2020054914 W IB 2020054914W WO 2020240392 A1 WO2020240392 A1 WO 2020240392A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- methyl
- reacting
- dimethyl
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 66
- 238000002360 preparation method Methods 0.000 title claims abstract description 23
- CASLETQIYIQFTQ-UHFFFAOYSA-N 3-[[5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)pyrazol-4-yl]methylsulfonyl]-5,5-dimethyl-4h-1,2-oxazole Chemical compound CN1N=C(C(F)(F)F)C(CS(=O)(=O)C=2CC(C)(C)ON=2)=C1OC(F)F CASLETQIYIQFTQ-UHFFFAOYSA-N 0.000 title claims abstract description 19
- 150000001875 compounds Chemical class 0.000 claims abstract description 61
- 150000003839 salts Chemical class 0.000 claims abstract description 49
- 239000007800 oxidant agent Substances 0.000 claims abstract description 12
- -1 hydroxycarbonimidic dibromide compound Chemical class 0.000 claims abstract description 7
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims abstract description 5
- 238000006243 chemical reaction Methods 0.000 claims description 37
- 239000002904 solvent Substances 0.000 claims description 34
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 33
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 claims description 28
- PBZUAIHRZUBBAJ-UHFFFAOYSA-N 2-hydroxyiminoacetic acid Chemical compound ON=CC(O)=O PBZUAIHRZUBBAJ-UHFFFAOYSA-N 0.000 claims description 23
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 18
- 239000000203 mixture Substances 0.000 claims description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 17
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 16
- IDPUHROBLIJORN-UHFFFAOYSA-N (5,5-dimethyl-4h-1,2-oxazol-3-yl) carbamimidothioate Chemical compound CC1(C)CC(SC(N)=N)=NO1 IDPUHROBLIJORN-UHFFFAOYSA-N 0.000 claims description 15
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 15
- 238000004128 high performance liquid chromatography Methods 0.000 claims description 15
- ARJPLYZBOHQOCK-UHFFFAOYSA-N 3-[[5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)pyrazol-4-yl]methylsulfanyl]-5,5-dimethyl-4h-1,2-oxazole Chemical compound CN1N=C(C(F)(F)F)C(CSC=2CC(C)(C)ON=2)=C1OC(F)F ARJPLYZBOHQOCK-UHFFFAOYSA-N 0.000 claims description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 12
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 12
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 11
- 229910052794 bromium Inorganic materials 0.000 claims description 11
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 10
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical group ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 10
- YMCWJQIZJIKFHO-UHFFFAOYSA-N 3-chloro-5,5-dimethyl-4h-1,2-oxazole Chemical compound CC1(C)CC(Cl)=NO1 YMCWJQIZJIKFHO-UHFFFAOYSA-N 0.000 claims description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 8
- VAWFDIIAHPIUDB-UHFFFAOYSA-N [5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)pyrazol-4-yl]methanol Chemical compound CN1N=C(C(F)(F)F)C(CO)=C1OC(F)F VAWFDIIAHPIUDB-UHFFFAOYSA-N 0.000 claims description 8
- 239000012320 chlorinating reagent Substances 0.000 claims description 8
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 8
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 claims description 8
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 claims description 8
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 7
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 claims description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 7
- 239000000460 chlorine Substances 0.000 claims description 7
- 229910052801 chlorine Inorganic materials 0.000 claims description 7
- 150000007529 inorganic bases Chemical class 0.000 claims description 7
- WQRHIGNAKDJJKN-UHFFFAOYSA-N 2-methyl-5-(trifluoromethyl)-1h-pyrazol-3-one Chemical compound CN1NC(C(F)(F)F)=CC1=O WQRHIGNAKDJJKN-UHFFFAOYSA-N 0.000 claims description 6
- XZDWVMFSTKIDHG-UHFFFAOYSA-N 4-(chloromethyl)-5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)pyrazole Chemical compound CN1N=C(C(F)(F)F)C(CCl)=C1OC(F)F XZDWVMFSTKIDHG-UHFFFAOYSA-N 0.000 claims description 6
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 6
- 150000007530 organic bases Chemical group 0.000 claims description 6
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 6
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 claims description 5
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 claims description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 5
- 238000004064 recycling Methods 0.000 claims description 5
- 229910021653 sulphate ion Inorganic materials 0.000 claims description 5
- XTEGARKTQYYJKE-UHFFFAOYSA-M Chlorate Chemical compound [O-]Cl(=O)=O XTEGARKTQYYJKE-UHFFFAOYSA-M 0.000 claims description 4
- VOPWNXZWBYDODV-UHFFFAOYSA-N Chlorodifluoromethane Chemical compound FC(F)Cl VOPWNXZWBYDODV-UHFFFAOYSA-N 0.000 claims description 4
- MIRMXMHHBRZJCN-UHFFFAOYSA-N N1(C)N=C(C(F)(F)F)C(CO)=C1O Chemical compound N1(C)N=C(C(F)(F)F)C(CO)=C1O MIRMXMHHBRZJCN-UHFFFAOYSA-N 0.000 claims description 4
- GQPLMRYTRLFLPF-UHFFFAOYSA-N Nitrous Oxide Chemical compound [O-][N+]#N GQPLMRYTRLFLPF-UHFFFAOYSA-N 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 4
- WFPZPJSADLPSON-UHFFFAOYSA-N dinitrogen tetraoxide Chemical compound [O-][N+](=O)[N+]([O-])=O WFPZPJSADLPSON-UHFFFAOYSA-N 0.000 claims description 4
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Chemical compound Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 claims description 4
- 239000002798 polar solvent Substances 0.000 claims description 4
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 claims description 4
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 claims description 4
- XMVONEAAOPAGAO-UHFFFAOYSA-N sodium tungstate Chemical compound [Na+].[Na+].[O-][W]([O-])(=O)=O XMVONEAAOPAGAO-UHFFFAOYSA-N 0.000 claims description 4
- POPVUKGJWNLYGW-UHFFFAOYSA-N (hydroxyamino) hydrogen sulfate Chemical compound ONOS(O)(=O)=O POPVUKGJWNLYGW-UHFFFAOYSA-N 0.000 claims description 3
- PNOQDONZIUXXMM-UHFFFAOYSA-N 3,4-dichloropyrrolidine-2,5-dione Chemical compound ClC1C(Cl)C(=O)NC1=O PNOQDONZIUXXMM-UHFFFAOYSA-N 0.000 claims description 3
- ZKQDCIXGCQPQNV-UHFFFAOYSA-N Calcium hypochlorite Chemical compound [Ca+2].Cl[O-].Cl[O-] ZKQDCIXGCQPQNV-UHFFFAOYSA-N 0.000 claims description 3
- 230000002378 acidificating effect Effects 0.000 claims description 3
- 239000006172 buffering agent Substances 0.000 claims description 3
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 claims description 3
- 239000003880 polar aprotic solvent Substances 0.000 claims description 3
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 claims description 3
- 229960002218 sodium chlorite Drugs 0.000 claims description 3
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 claims description 2
- MGWGWNFMUOTEHG-UHFFFAOYSA-N 4-(3,5-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C)=CC(C=2N=C(N)SC=2)=C1 MGWGWNFMUOTEHG-UHFFFAOYSA-N 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 2
- 239000005708 Sodium hypochlorite Substances 0.000 claims description 2
- 239000003638 chemical reducing agent Substances 0.000 claims description 2
- 229910001919 chlorite Inorganic materials 0.000 claims description 2
- 229910052619 chlorite group Inorganic materials 0.000 claims description 2
- QBWCMBCROVPCKQ-UHFFFAOYSA-N chlorous acid Chemical compound OCl=O QBWCMBCROVPCKQ-UHFFFAOYSA-N 0.000 claims description 2
- 229940117975 chromium trioxide Drugs 0.000 claims description 2
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 claims description 2
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 239000011737 fluorine Substances 0.000 claims description 2
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 claims description 2
- JCXJVPUVTGWSNB-UHFFFAOYSA-N nitrogen dioxide Inorganic materials O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 claims description 2
- 239000001272 nitrous oxide Substances 0.000 claims description 2
- 239000012454 non-polar solvent Substances 0.000 claims description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 claims description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 claims description 2
- JRKICGRDRMAZLK-UHFFFAOYSA-N peroxydisulfuric acid Chemical compound OS(=O)(=O)OOS(O)(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-N 0.000 claims description 2
- FHHJDRFHHWUPDG-UHFFFAOYSA-N peroxysulfuric acid Chemical compound OOS(O)(=O)=O FHHJDRFHHWUPDG-UHFFFAOYSA-N 0.000 claims description 2
- 239000004323 potassium nitrate Substances 0.000 claims description 2
- 235000010333 potassium nitrate Nutrition 0.000 claims description 2
- 239000012286 potassium permanganate Substances 0.000 claims description 2
- PNYYBUOBTVHFDN-UHFFFAOYSA-N sodium bismuthate Chemical compound [Na+].[O-][Bi](=O)=O PNYYBUOBTVHFDN-UHFFFAOYSA-N 0.000 claims description 2
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 claims description 2
- 229960001922 sodium perborate Drugs 0.000 claims description 2
- YKLJGMBLPUQQOI-UHFFFAOYSA-M sodium;oxidooxy(oxo)borane Chemical compound [Na+].[O-]OB=O YKLJGMBLPUQQOI-UHFFFAOYSA-M 0.000 claims description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims 1
- 229910017604 nitric acid Inorganic materials 0.000 claims 1
- 238000005112 continuous flow technique Methods 0.000 abstract description 6
- 239000000243 solution Substances 0.000 description 32
- 239000011541 reaction mixture Substances 0.000 description 20
- 239000002585 base Substances 0.000 description 14
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 14
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 10
- 235000011121 sodium hydroxide Nutrition 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 7
- AWBKQZSYNWLCMW-UHFFFAOYSA-N n-(dibromomethylidene)hydroxylamine Chemical compound ON=C(Br)Br AWBKQZSYNWLCMW-UHFFFAOYSA-N 0.000 description 7
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 238000002955 isolation Methods 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 235000010265 sodium sulphite Nutrition 0.000 description 5
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- NXPHCVPFHOVZBC-UHFFFAOYSA-N hydroxylamine;sulfuric acid Chemical compound ON.OS(O)(=O)=O NXPHCVPFHOVZBC-UHFFFAOYSA-N 0.000 description 4
- 150000002923 oximes Chemical class 0.000 description 4
- 235000011118 potassium hydroxide Nutrition 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- UBOXGVDOUJQMTN-UHFFFAOYSA-N 1,1,2-trichloroethane Chemical compound ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- QPRQEDXDYOZYLA-UHFFFAOYSA-N 2-methylbutan-1-ol Chemical compound CCC(C)CO QPRQEDXDYOZYLA-UHFFFAOYSA-N 0.000 description 3
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- XPFVYQJUAUNWIW-UHFFFAOYSA-N furfuryl alcohol Chemical compound OCC1=CC=CO1 XPFVYQJUAUNWIW-UHFFFAOYSA-N 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 3
- JIRJHEXNDQBKRZ-UHFFFAOYSA-N phosgene oxime Chemical compound ON=C(Cl)Cl JIRJHEXNDQBKRZ-UHFFFAOYSA-N 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 2
- ARXKVVRQIIOZGF-UHFFFAOYSA-N 1,2,4-butanetriol Chemical compound OCCC(O)CO ARXKVVRQIIOZGF-UHFFFAOYSA-N 0.000 description 2
- JYYNAJVZFGKDEQ-UHFFFAOYSA-N 2,4-Dimethylpyridine Chemical compound CC1=CC=NC(C)=C1 JYYNAJVZFGKDEQ-UHFFFAOYSA-N 0.000 description 2
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 2
- YIWUKEYIRIRTPP-UHFFFAOYSA-N 2-ethylhexan-1-ol Chemical compound CCCCC(CC)CO YIWUKEYIRIRTPP-UHFFFAOYSA-N 0.000 description 2
- PFNHSEQQEPMLNI-UHFFFAOYSA-N 2-methyl-1-pentanol Chemical compound CCCC(C)CO PFNHSEQQEPMLNI-UHFFFAOYSA-N 0.000 description 2
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- FZERHIULMFGESH-UHFFFAOYSA-N N-phenylacetamide Chemical compound CC(=O)NC1=CC=CC=C1 FZERHIULMFGESH-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- KUHLVNZNNTXIIC-UHFFFAOYSA-M [K+].Cn1nc(c(CO)c1[O-])C(F)(F)F Chemical compound [K+].Cn1nc(c(CO)c1[O-])C(F)(F)F KUHLVNZNNTXIIC-UHFFFAOYSA-M 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- DIKBFYAXUHHXCS-UHFFFAOYSA-N bromoform Chemical compound BrC(Br)Br DIKBFYAXUHHXCS-UHFFFAOYSA-N 0.000 description 2
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- HRYZWHHZPQKTII-UHFFFAOYSA-N chloroethane Chemical compound CCCl HRYZWHHZPQKTII-UHFFFAOYSA-N 0.000 description 2
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 2
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 2
- 235000019797 dipotassium phosphate Nutrition 0.000 description 2
- 229960003750 ethyl chloride Drugs 0.000 description 2
- 239000004009 herbicide Substances 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 2
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 2
- WGTYBPLFGIVFAS-UHFFFAOYSA-M tetramethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C WGTYBPLFGIVFAS-UHFFFAOYSA-M 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- QVLAWKAXOMEXPM-UHFFFAOYSA-N 1,1,1,2-tetrachloroethane Chemical compound ClCC(Cl)(Cl)Cl QVLAWKAXOMEXPM-UHFFFAOYSA-N 0.000 description 1
- UOCLXMDMGBRAIB-UHFFFAOYSA-N 1,1,1-trichloroethane Chemical compound CC(Cl)(Cl)Cl UOCLXMDMGBRAIB-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- LGXVIGDEPROXKC-UHFFFAOYSA-N 1,1-dichloroethene Chemical compound ClC(Cl)=C LGXVIGDEPROXKC-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- KFUSEUYYWQURPO-UHFFFAOYSA-N 1,2-dichloroethene Chemical compound ClC=CCl KFUSEUYYWQURPO-UHFFFAOYSA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- 229940035437 1,3-propanediol Drugs 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- KIZQNNOULOCVDM-UHFFFAOYSA-M 2-hydroxyethyl(trimethyl)azanium;hydroxide Chemical compound [OH-].C[N+](C)(C)CCO KIZQNNOULOCVDM-UHFFFAOYSA-M 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- JTNCEQNHURODLX-UHFFFAOYSA-N 2-phenylethanimidamide Chemical compound NC(=N)CC1=CC=CC=C1 JTNCEQNHURODLX-UHFFFAOYSA-N 0.000 description 1
- ITQTTZVARXURQS-UHFFFAOYSA-N 3-methylpyridine Chemical compound CC1=CC=CN=C1 ITQTTZVARXURQS-UHFFFAOYSA-N 0.000 description 1
- ZNBNBTIDJSKEAM-UHFFFAOYSA-N 4-[7-hydroxy-2-[5-[5-[6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3-methyloxolan-2-yl]-5-methyloxolan-2-yl]-2,8-dimethyl-1,10-dioxaspiro[4.5]decan-9-yl]-2-methyl-3-propanoyloxypentanoic acid Chemical compound C1C(O)C(C)C(C(C)C(OC(=O)CC)C(C)C(O)=O)OC11OC(C)(C2OC(C)(CC2)C2C(CC(O2)C2C(CC(C)C(O)(CO)O2)C)C)CC1 ZNBNBTIDJSKEAM-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- UGVAQFVQIHNCJV-UHFFFAOYSA-N C[n]1nc(C(F)(F)F)cc1O Chemical compound C[n]1nc(C(F)(F)F)cc1O UGVAQFVQIHNCJV-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- PBZUAIHRZUBBAJ-HNQUOIGGSA-N OC(/C=N/O)=O Chemical compound OC(/C=N/O)=O PBZUAIHRZUBBAJ-HNQUOIGGSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- CYTYCFOTNPOANT-UHFFFAOYSA-N Perchloroethylene Chemical compound ClC(Cl)=C(Cl)Cl CYTYCFOTNPOANT-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- 229960001413 acetanilide Drugs 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- HFACYLZERDEVSX-UHFFFAOYSA-N benzidine Chemical compound C1=CC(N)=CC=C1C1=CC=C(N)C=C1 HFACYLZERDEVSX-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 229950005228 bromoform Drugs 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- NEHMKBQYUWJMIP-NJFSPNSNSA-N chloro(114C)methane Chemical compound [14CH3]Cl NEHMKBQYUWJMIP-NJFSPNSNSA-N 0.000 description 1
- 229960001701 chloroform Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- ZCDOYSPFYFSLEW-UHFFFAOYSA-N chromate(2-) Chemical class [O-][Cr]([O-])(=O)=O ZCDOYSPFYFSLEW-UHFFFAOYSA-N 0.000 description 1
- JOPOVCBBYLSVDA-UHFFFAOYSA-N chromium(6+) Chemical class [Cr+6] JOPOVCBBYLSVDA-UHFFFAOYSA-N 0.000 description 1
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000011928 denatured alcohol Substances 0.000 description 1
- 239000011929 di(propylene glycol) methyl ether Substances 0.000 description 1
- 125000000950 dibromo group Chemical group Br* 0.000 description 1
- CMMUKUYEPRGBFB-UHFFFAOYSA-L dichromic acid Chemical class O[Cr](=O)(=O)O[Cr](O)(=O)=O CMMUKUYEPRGBFB-UHFFFAOYSA-L 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- GGSUCNLOZRCGPQ-UHFFFAOYSA-N diethylaniline Chemical compound CCN(CC)C1=CC=CC=C1 GGSUCNLOZRCGPQ-UHFFFAOYSA-N 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- KVFVBPYVNUCWJX-UHFFFAOYSA-M ethyl(trimethyl)azanium;hydroxide Chemical compound [OH-].CC[N+](C)(C)C KVFVBPYVNUCWJX-UHFFFAOYSA-M 0.000 description 1
- 230000004136 fatty acid synthesis Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 150000002366 halogen compounds Chemical class 0.000 description 1
- 230000002363 herbicidal effect Effects 0.000 description 1
- VHHHONWQHHHLTI-UHFFFAOYSA-N hexachloroethane Chemical compound ClC(Cl)(Cl)C(Cl)(Cl)Cl VHHHONWQHHHLTI-UHFFFAOYSA-N 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- QWARLPGIFZKIQW-UHFFFAOYSA-N hydrogen peroxide;nitric acid Chemical compound OO.O[N+]([O-])=O QWARLPGIFZKIQW-UHFFFAOYSA-N 0.000 description 1
- 229910000378 hydroxylammonium sulfate Inorganic materials 0.000 description 1
- 230000003116 impacting effect Effects 0.000 description 1
- 150000002547 isoxazolines Chemical class 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- NBTOZLQBSIZIKS-UHFFFAOYSA-N methoxide Chemical compound [O-]C NBTOZLQBSIZIKS-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 238000007040 multi-step synthesis reaction Methods 0.000 description 1
- OOHAUGDGCWURIT-UHFFFAOYSA-N n,n-dipentylpentan-1-amine Chemical compound CCCCCN(CCCCC)CCCCC OOHAUGDGCWURIT-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- BNIXVQGCZULYKV-UHFFFAOYSA-N pentachloroethane Chemical compound ClC(Cl)C(Cl)(Cl)Cl BNIXVQGCZULYKV-UHFFFAOYSA-N 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000006479 redox reaction Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- HYHCSLBZRBJJCH-UHFFFAOYSA-M sodium hydrosulfide Chemical compound [Na+].[SH-] HYHCSLBZRBJJCH-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 1
- 229950011008 tetrachloroethylene Drugs 0.000 description 1
- 229940073455 tetraethylammonium hydroxide Drugs 0.000 description 1
- LRGJRHZIDJQFCL-UHFFFAOYSA-M tetraethylazanium;hydroxide Chemical compound [OH-].CC[N+](CC)(CC)CC LRGJRHZIDJQFCL-UHFFFAOYSA-M 0.000 description 1
- LPSKDVINWQNWFE-UHFFFAOYSA-M tetrapropylazanium;hydroxide Chemical compound [OH-].CCC[N+](CCC)(CCC)CCC LPSKDVINWQNWFE-UHFFFAOYSA-M 0.000 description 1
- 150000004992 toluidines Chemical class 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- 229960002415 trichloroethylene Drugs 0.000 description 1
- GRNRCQKEBXQLAA-UHFFFAOYSA-M triethyl(2-hydroxyethyl)azanium;hydroxide Chemical compound [OH-].CC[N+](CC)(CC)CCO GRNRCQKEBXQLAA-UHFFFAOYSA-M 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910000406 trisodium phosphate Inorganic materials 0.000 description 1
- 235000019801 trisodium phosphate Nutrition 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/04—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
Definitions
- the present invention relates to a process for the preparation of Pyroxasulfone of Formula (I) or salt thereof. Particularly, the present invention relates to an improved process for the preparation of hydroxycarbonimidicdibromide, compound of Formula (III) or salt thereof, wherein bromide anion is recycled by using a suitable oxidizing agent. Moreover, the present invention relates to a continuous flow process for preparing of compound of Formula (I) or salt thereof.
- Pyroxasulfone belongs to isoxazoline class of herbicides, which inhibits fatty acid synthesis. Pyroxasulfone herbicide is classified in Group 15 of WSSA. It is chemically known as 3-[5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-ylmethylsulfonyl]-4,5- dihydro-5,5-dimethylisoxazole, compound of Formula (I) and represent as:
- Yet another objective of the present invention is to provide a continuous flow process for preparing the compound of Formula (I) or salt thereof by compound of Formula V (1- methyl-3-(trifluoromethyl)-1H-pyrazol-5-ol)
- a further objective of the present invention is to provide a continuous flow process for preparing the compound of Formula IX (5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate)
- the present invention relates to a new method for the preparation of compound of Formula (I) or salt thereof said method comprises an integrated continuous flow process for reactions wherein a succession of integrated flow reactors are used to perform a series of reaction steps to yield the final product.
- the process is a multistep synthesis of compound of Formula (I) or salt thereof in a continuous flow without isolation of intermediates produced during the flow.
- the purity of Pyroxasulfoneof Formula (I) or salt thereof obtained according to the present invention is more than 99 %, measured by HPLC.
- a condition A“or” B is satisfied by any one of the following: A is true (or present) and B is false (or not present), A is false (or not present) and B is true (or present), and both A and B is true (or present).
- the indefinite articles "a” and “an” preceding an element or component of the present invention are intended to be nonrestrictive regarding the number of instances (i.e. occurrences) of the element or component. Therefore “a” or “an” should be read to include one or at least one, and the singular word form of the element or component also includes the plural unless the number is obviously meant to be singular. All amounts are percent by weight (“% wt”), unless otherwise noted. All ranges are inclusive.
- step (a) and (b) comprising reacting glyoxalic acid with hydroxy amine sulphate to obtain 2-(hydroxyimino)acetic acid compound of Formula (II) or salt thereof,in a suitable solvent at a suitable pH,followed by reacting with bromine in a suitable solvent at a suitable pH, wherein the suitable solvent is a polar solvent and suitable pH is in the range of about 8 to 14, preferably in the range of about 10 to 14.
- the bromination reaction is carried out at temperature of about 5°C to 35°C.
- the amount of bromine used is in the range of about 0.25 to 3 mole equivalents, based on the amount of 2-(hydroxyimino)acetic acid or salt thereof, preferably the amount of bromine is in the range of about 0.25 to 2.0 mole equivalents.
- the step (c) comprises recycling bromine anionby addition of a suitableoxidizing agent, wherein the oxidizing agent is selected from the group comprising of fluorine; chlorine; hydrogen peroxide; nitric acid or nitrate compounds; sulfuric acid; peroxydisulfuric acid; peroxymonosulfuric acid; chlorite, chlorate, perchlorate and other analogus of halogen compounds; hypochlorite and other hypohalite compounds such as sodium hypochlorite; hexavalent chromium compounds such as chromic and dichromic acids, chromium trioxide, pyridinium chlorochromate, chromate/dichromate compounds; permanganate compounds such as potassium permanganate; sodium perborate
- the addition of a suitable oxidizing agentin the reaction mixture is carried out at temperature of about 5°C to 35°C.
- the step (d) optionally comprises the isolation of the compound of Formula (III) or salt thereof, wherein the isolation may be carried out by any conventional method, such as by filtration or solvent extraction. After the completion of the reaction, traces of bromine were decomposed by the addition of a solution of sodium sulphite to the reaction mixture.
- the step (e) comprises the conversion of compound of Formula (III) or salt thereof to Pyroxasulfone or salt thereofby process as described in U.S. Patent No.7,238,689.
- an integrated, continuous flow method for the preparation of compound of Formula (I) or salt thereof there is provided an integrated, continuous flow method for the preparation of compound of Formula (I) or salt thereof
- An aspect of the present invention provides an integrated, continuous flow method for step A. for the preparation of 4-(hydroxymethyl)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-olate preferably potassium 4-(hydroxymethyl)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-olate (Formula VI) by reacting with a solution of formaldehyde, a base preferably potassium hydroxide in a suitable solvent preferably a polar solvent, and 1-methyl-3-(trifluoromethyl)- 1H-pyrazol-5-ol (Formula V).
- the residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 01 minute depending on the temperature.
- the temperature in the reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 80°C and even more preferably between 20°C and 50°C, specifically 40°C.
- step B for the preparation of (5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H- pyrazol-4-yl)methanol (Formula VII) by reacting with potassium salt of 4-(hydroxymethyl)- 1-methyl-3-(trifluoromethyl)-1H- pyrazol-5-olate (Formula VI) with chlorodifluoromethane in a suitable solvent (preferably chlorinated solvent or polar aprotic solvent) in the presence of a base.
- a suitable solvent preferably chlorinated solvent or polar aprotic solvent
- the residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 05 minutes depending on the temperature.
- the temperature in the reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 80°C and even more preferably between 20°C and 50°C, specifically 25°C.
- an integrated continuous flow method is provided for step C. for the preparation of 4-(chloromethyl)-5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazole (Formula VIII) by reacting with a chlorinating agent selected from thionyl chloride, phosphorus oxychloride, phosphorus pentachloride, dichlorosuccinimide, calcium hypochlorite, sodium chlorite, carbon tetrachloride, hydrochloric acid or chlorine preferably thionyl chloride and (5-(difluoromethoxy)-1-methyl- 3-(trifluoromethyl)-1H-pyrazol-4-yl)methanol (Formula VII).
- a chlorinating agent selected from thionyl chloride, phosphorus oxychloride, phosphorus pentachloride, dichlorosuccinimide, calcium hypochlorite, sodium chlorite, carbon tetrachloride, hydrochloric
- the residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 02 minutes depending on the temperature.
- the temperature in the reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 80°C and even more preferably between 20°C and 50°C, specifically 20°C.
- an integrated, continuous flow method for step D. for the preparation of compound of Formula X (3-(((5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl- 4,5-dihydroisoxazole) by reacting with a solution of 5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate (Formula IX) and 4-(chloromethyl)-5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazole (Formula VIII) in presence of a base preferably sodium hydroxide and a suitable solvent preferably polar protic solvent.
- a base preferably sodium hydroxide
- a suitable solvent preferably polar protic solvent.
- the residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 05 minutes depending on the temperature.
- the temperature in the reactor is typically anywhere between 10°C and 100°C, preferably 100°C.
- Suitable solvent refers to polar protic or polar aprotic or non-polar solvent, preferably selected from the group comprising of, acetonitrile, water, alcohol or chlorinated solvents or a mixture thereof, wherein alcohol is selected from tert-amyl alcohol, benzyl alcohol, 1,4- butanediol, 1,2,4-butanetriol, n-butanol, 2-butanol, tert-butyl alcohol, denatured alcohol, di(propylene glycol) methyl ether, diethylene glycol, ethanol, ethylene glycol, 2- ethylhexanol, furfuryl alcohol, methanol,2-(2-methoxyethoxy)ethanol, 2-methyl-1-butanol, 2-methyl-1-pentanol, neopentyl alcohol, 2-pentanol, 1,3 propanediol, n-proponol, and propylene glycol; chlorinated solvent is selected from carbon tet
- the solvent is a polar solvent such as water.
- Other solvent that can also employ in the present invention is selected from the group the comprising of, bromoform, carbon tetrabromide, ethylene dibromide, toluene, xylene, benzotrifluorideor a mixture thereof. pH of the reaction mixture may be maintained by using a suitable base, wherein the base is selected from organic base or inorganic base.
- Organic base is selected from the group comprising of, but not limited to, alkyl ammonium hydroxide such as tetramethyl ammonium hydroxide, tetraethyl ammonium hydroxide, tetrapropyl ammonium hydroxide, tetrabutyl ammonium hydroxide, trimethyl-2-hydroxyethyl ammonium hydroxide (Choline), triethyl-2-hydroxy ethyl ammonium hydroxide, ethyltrimethyl ammonium hydroxide; alkyl amine such as trimethylamine, triethylamine, tributylamine, tripentylamine, monoethanolamine, diethylamine; pyridine, 4- dimethylaminopyridine, 2,4-lutidine, 2,6-lutidine, collidine, alpha-picoline, beta-picoline, gamma.-picoline, quinoline, isoquinoline, aniline, diemthylaniline, N, N-
- Inorganic base is selected from the group comprising of, but not limited to, alkali or alkaline earth metal hydroxides, carbonates, bicarbonates, bisulfates, acetate, methoxide, ethoxide, phosphate, sulfite, sulfate preferably selected from sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium bisulfate, potassium bisulfate, sodium acetate, potassium acetate, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, trisodium phosphate, tripotassium phosphate, sodium dihydrogen phosphate, potassium dihydrogen phosphate, sodium hydrogen sulfide, sodium sulfate, and mixtures thereof.
- alkali or alkaline earth metal hydroxides carbonates, bicarbonates, bisulfates, acetate, methoxide, ethoxide, phosphate, sul
- the base is selected from triethylamine, pyridine, potassium hydroxide, sodium hydroxide, and sodium carbonate.
- an integrated continuous flow method is provided for step E. for the preparation of compound of Formula I or salt thereof by reacting with sodium tungstate, and a oxidizing agent preferably hydrogen peroxide, and 3-(((5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl- 4,5-dihydroisoxazole (Formula IX).
- the residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 10 minutes depending on the temperature.
- the temperature in the reactor is typically anywhere between 10°C and 100°C, preferably 100°C.
- the purity of Pyroxasulfone or salt thereof of Formula (I) obtained according to the present invention process is more than 95%, measured by HPLC.
- All the reactions in steps A. to E. as described above are performed in flow reactors connected to each other in such a way to provide an integrated system. There are many configurations of such connected reactor system, that a person skilled in the art is aware of.
- the oxidizing agent refers to a reactant that removes electrons from other reactants during a redox reaction. Suitable oxidizing agent is selected from but not limited to, hydrogen peroxide or metachloroperbenzoic acid preferablyhydrogen peroxide.
- the compound of Formula I, Formula VI, Formula VII, Formula VIII, or Formula X having HPLC purity of at least 95% is obtained by the process as described above.
- the improved process as disclosed in the present invention is used to prepare Pyroxasulfone of Formula (I) by reacting glyoxalic acid with hydroxy amine sulphate to obtain 2- (hydroxyimino)acetic acid of Formula (II) or salt thereof, in the presence of an organic base or inorganic baseto maintain the pH of the reaction mixture.
- the reaction mixture was further reacted with bromine in a suitable solvent at a suitable pH and recycling of bromine anionis carried out by addition of a suitable oxidizing agent such as chlorine to obtainhydroxycarbonimidic dibromide compound of Formula (III).
- Hydroxycarbonimidicdibromide is reacted with isobutylene gas in the presence of a suitable base at a suitable pH to produce compound of Formula (IV) as shown below, which is further utilized in the preparation of Pyroxasulfone of Formula (I) or salt thereof as described in U.S. Patent No.7,238,689.
- 1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-ol (Formula V) is one of the important starting material useful for the synthesis of compounds of Formula I, Formula VI, Formula VII, Formula VIII, or Formula X or agrochemically acceptable salt thereof.
- the residence time of the fluid within the system is determined by the internal diameter and length of the reactor coil.
- Mixers and unions connect reactor coils together and allow the addition of new reagents to the continuous-flow stream.
- the solution can be flowed through packed bed reactors to ensure efficient mixing, or to provide exposure to immobilized reagents for synthetic transformations.
- a continuous-flow system allows the possibility of in-line purification and reagent introduction at set points in the continuous-flow sequence.
- Another embodiment of the present invention provides a process for the preparation of an intermediate compound of Formula IX (5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate) comprising the steps of:
- the operation temperature in the reactor is typically anywhere between 5°C and 80°C, preferably between 5°C and 40°C more preferably 15°C and 20°C for step 2), preferably between 10°C and 70°C more preferably 40°C for step 3) and preferably between 10°C and 40°C more preferably 25°C and 40°C for step 4).
- Another aspect of the present invention provides a process for the preparation of an intermediate compound of Formula IX (5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate) by a multistep process, including a process of reacting glyoxalic acid with hydroxyamino sulfate in presence of a base preferably sodium hydroxide and/or buffering agent preferably potassium hydrogen phosphate to obtain hydroxyimino acetic acid and hydroxyimino acetic acid which is further reacted with a chlorinating agent preferably chlorine at a suitable temperature of about 5-40°C preferably about 15-20°C to obtain hydroxycarbodimic dichloride.
- a base preferably sodium hydroxide and/or buffering agent preferably potassium hydrogen phosphate
- a chlorinating agent preferably chlorine at a suitable temperature of about 5-40°C preferably about 15-20°C to obtain hydroxycarbodimic dichloride.
- Hydroxycarbodimic dichloride was reacted with isobutylene and a base preferably potassium carbonate at a suitable temperature of about 10-70°C preferably about 40°C to obtain 3-chloro-5,5-dimethyl-4,5-dihydroisoxazole.
- 3-chloro-5,5- dimethyl-4,5-dihydroisoxazole was then reacted with thiourea in acidic medium preferably hydrogen bromide or hydrogen chloride at a suitable temperature of about 10 to 40°C preferably about 25°C to 40°C to obtain compound of Formula IX (5,5-dimethyl-4,5- dihydroisoxazol-3-yl carbamimidothioate).
- Yet another aspect of the present invention provides the subsequent conversion of 5,5- dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate (Formula IX) to Formula X (3-(((5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl- 4,5-dihydroisoxazole).
- the purity of compound of Formula (IX) or salt thereof is more than 95 %, as measured by HPLC.
- provided herein is the compound of Formula IX having HPLC purity of at least 95% or atleast 98% obtained by the process as described above.
- the continuous flow process of the present invention has many advantages over the batch process as follows:- 1. Minimizes handling of intermediates, toxic and corrosive reagents and solvents.
- the temperature used in each module of the reactor can be adapted at the kinetic rate of reaction.
- the continuous flow reactions are performed in flow reactors connected to each other in such a way to provide an integrated system. There are many configurations of such connected reactor system, that a person skilled in the art is aware of.
- the compounds in continuous flow reaction may be prepared according to the following examples. The following examples are presented to illustrate further aspects of the present invention, but are not intended to limit the scope of the invention in any aspect. Examples Example 1:
- Example 3 50% aqueous solution of glyoxylic acid (74.05 g) was slowly added to a solution of 17.38% hydroxylamine sulphate in water at 0-5 °C for 1h. 25% sodium hydroxide (164 g) solution was added to the reaction mixture at 10-15 °C in 1 h. pH was adjusted to 10 to 14 by means of addition of 13% sodium carbonate (100 g).
- the obtained oxime solution was added to a solution of water (50 g) and liquid bromine (96 g) at 10-15 °C in 1 h. Chlorine gas (36 g) was purged into the reaction mixture at 10-15 °C for 4 h. After completion of reaction, a solution of 17% sodium sulphite (50 g) was added to the reaction mixture. n-butyl acetate (150 g) solvent was added. The organic and aqueous layers were separated to obtain hydroxycarbonimidicdibromide.
- the obtained oxime solution was added to a solution of water (50 g) and liquid bromine (96 g) at 10-15 °C in 1 h.50% hydrogen peroxide (30 g) was added to the reaction mixture at 10- 15 °C for 4 h. After completion of reaction, a solution of 17% sodium sulphite (50 g) was added to the reaction mixture. n-butyl acetate (176 g) solvent was added. The organic and aqueous layer were separated to obtain hydroxycarbonimidic dibromide.
- the resulted reaction stream was directed towards a 24ml flow with a static mixture (MoC–PFA) reactor, operating at 15-20°C and converted to hydroxycarbonimidic dichloride (Conversion by GC > 87%).
- Hydroxycarbonimidic dichloride in the aqueous solvent was extracted into n-butyl acetate solvent.
- Hydroxycarbonimidic dichloride in n-butyl acetate was reacted with isobutylene gas in n- butyl acetate with potassium carbonate as base at 40 °C to ensure full conversion to 3-chloro- 5,5-dimethyl-4,5-dihydroisoxazole (Conversion by GC > 87%).
- reaction mass was filtered and the filtrate containing 3-chloro-5,5-dimethyl-4,5- dihydroisoxazole was transferred to the another reactor having a solution of thio-urea and hydrogen bromide solution at 25-40°C to ensure full conversion to 5,5-dimethyl-4,5- dihydroisoxazol-3-yl carbamimidothioate (Conversion by GC > 45%).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
The present invention discloses a process for the preparation of Pyroxasulfone of Formula (I) or salt thereof. Particularly, the present invention discloses an improved process for the preparation of hydroxycarbonimidic dibromide compound of Formula (III) or salt thereof, wherein bromine anion is recycled by using a suitable oxidizing agent. Moreover, the present invention relates to a continuous flow process for preparing of compound of Formula (I) or salt thereof.
Description
PROCESS FOR PREPARATION OF
PYROXASULFONE FIELD OF THE INVENTION: The present invention relates to a process for the preparation of Pyroxasulfone of Formula (I) or salt thereof. Particularly, the present invention relates to an improved process for the preparation of hydroxycarbonimidicdibromide, compound of Formula (III) or salt thereof, wherein bromide anion is recycled by using a suitable oxidizing agent.Moreover, the present invention relates to a continuous flow process for preparing of compound of Formula (I) or salt thereof.


Pyroxasulfone or salt thereof is disclosed in U.S. Patent No. 7,238,689, which is assigned to Ihara Chemical Industry Co. Ltd.
Dibromo formimine of formula (III) is a key building block for the preparation of compound of formula (III), is disclosed in U.S. Patent No.6,207,863. The presently know methods for preparing hydroxycarbonimidicdibromide, compound of Formula (III)or salt thereof generate significant levels of effluent as by-products such as sodium bromide and hydrogen bromide, which impacts on product yield as well as cost. Therefore, there is a need to develop an effective process for the preparation of compound of Formula (I) or its intermediate hydroxycarbonimidicdibromide, compound of Formula (III) or salt thereof, wherein by-products can be recycled to reduce effluent and improvement in the yield, without impacting purity of hydroxycarbonimidicdibromide. SUMMARY OF THE INVENTION: The present invention provides an improved process for the preparation of Pyroxasulfone of Formula (I) or salt thereof;
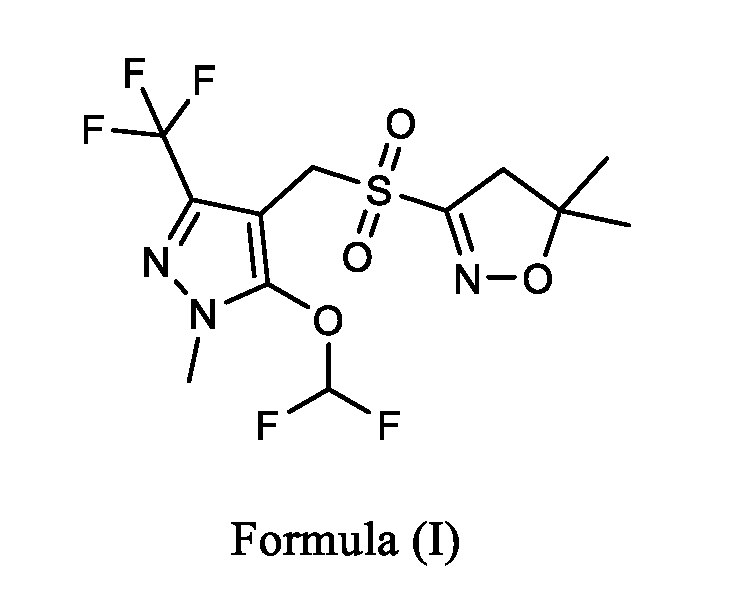
wherein, the process comprising the steps of:
a) reacting glyoxalic acid with hydroxy amine sulphate to obtain 2-(hydroxyimino)acetic acid compound of Formula (II)or salt thereof, in a suitable solvent at a suitable pH;

b) reacting 2-(hydroxyimino)acetic acid with bromine in a suitable solvent at a suitable pH;
c) recycling of bromine anion by addition of a suitable oxidizing agent;
d) optionally, isolating the compound of Formula (III) or salt thereof;

e) convertingthe compound of Formula (III) or salt thereof, to Pyroxasulfone of Formula (I) or salt thereof. Yet another objective of the present invention is to provide a continuous flow process for preparing the compound of Formula (I) or salt thereof by compound of Formula V (1- methyl-3-(trifluoromethyl)-1H-pyrazol-5-ol)

as a starting material and with intermediates thereof of Formula VI, Formula VII, Formula VIII, or Formula X. A further objective of the present invention is to provide a continuous flow process for preparing the compound of Formula IX (5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate)

by a multistep process using glyoxalic acid as a starting material and preparation of intermediate thereof. The present invention relates to a new method for the preparation of compound of Formula (I) or salt thereof said method comprises an integrated continuous flow process for reactions wherein a succession of integrated flow reactors are used to perform a series of reaction steps to yield the final product.
In one aspect the process is a multistep synthesis of compound of Formula (I) or salt thereof in a continuous flow without isolation of intermediates produced during the flow. The purity of Pyroxasulfoneof Formula (I) or salt thereof obtained according to the present invention is more than 99 %, measured by HPLC. DETAILED DESCRIPTION OF THE INVENTION: DEFINITIONS: The foregoing definitions provided herein for the terminologies used in the present disclosure are for illustrative purpose only and in no manner limit the scope of the present invention disclosed in the present disclosure. As used herein, the terms "comprises", "comprising", "includes", "including",“consisting” or any other variation thereof, are intended to cover a non-exclusive inclusion, subject to any limitation explicitly indicated. For example, a process or method that comprises a list of elements is not necessarily limited to only those elements but may include other elements not expressly listed or inherent to such process or method. Further, unless expressly stated to the contrary, "or" refers to an inclusive“or” and not to an exclusive“or”. For example, a condition A“or” B is satisfied by any one of the following: A is true (or present) and B is false (or not present), A is false (or not present) and B is true (or present), and both A and B is true (or present). Also, the indefinite articles "a" and "an" preceding an element or component of the present invention are intended to be nonrestrictive regarding the number of instances (i.e. occurrences) of the element or component. Therefore "a" or "an" should be read to include one or at least one, and the singular word form of the element or component also includes the plural unless the number is obviously meant to be singular. All amounts are percent by weight (“% wt”), unless otherwise noted. All ranges are inclusive. As used throughout the specification, the following abbreviations are applied: g=gram; and °C = Centigrade.
The numerical values mentioned in the description and the foregoing claims though might form a critical part of the present invention of the present disclosure, any deviation from such numerical values shall still fall within the scope of the present disclosure if that deviation follows the same scientific principle as that of the present invention disclosed in the present disclosure. The present invention is directed to an improved process for the preparation of Pyroxasulfone of Formula (I) or salt thereof;

wherein, the process comprising the steps of:
a) reacting glyoxalic acid with hydroxy amine sulphate to obtain 2-(hydroxyimino)acetic acid (Formula II) or salt thereof, in a suitable solvent at a suitable pH;

b) reacting 2-(hydroxyimino)acetic acid with bromine in a suitable solvent at a suitable pH;
c) recycling of bromine anion by addition of a suitable oxidizing agent;
d) optionally, isolating the compound of Formula (III) or salt thereof;

e) converting the compound of Formula (III) or salt thereof, to Pyroxasulfone of Formula (I) or salt thereof.
The step (a) and (b) comprising reacting glyoxalic acid with hydroxy amine sulphate to obtain 2-(hydroxyimino)acetic acid compound of Formula (II) or salt thereof,in a suitable solvent at a suitable pH,followed by reacting with bromine in a suitable solvent at a suitable pH, wherein the suitable solvent is a polar solvent and suitable pH is in the range of about 8 to 14, preferably in the range of about 10 to 14. The bromination reaction is carried out at temperature of about 5°C to 35°C. The amount of bromine used is in the range of about 0.25 to 3 mole equivalents, based on the amount of 2-(hydroxyimino)acetic acid or salt thereof, preferably the amount of bromine is in the range of about 0.25 to 2.0 mole equivalents. The step (c) comprises recycling bromine anionby addition of a suitableoxidizing agent, wherein the oxidizing agent is selected from the group comprising of fluorine; chlorine; hydrogen peroxide; nitric acid or nitrate compounds; sulfuric acid; peroxydisulfuric acid; peroxymonosulfuric acid; chlorite, chlorate, perchlorate and other analogus of halogen compounds; hypochlorite and other hypohalite compounds such as sodium hypochlorite; hexavalent chromium compounds such as chromic and dichromic acids, chromium trioxide, pyridinium chlorochromate, chromate/dichromate compounds; permanganate compounds such as potassium permanganate; sodium perborate; nitrous oxide, nitrogen dioxide, dinitrogen tetroxide; potassium nitrate; sodium bismuthate and mixture thereof. The addition of a suitable oxidizing agentin the reaction mixture is carried out at temperature of about 5°C to 35°C. The step (d) optionally comprises the isolation of the compound of Formula (III) or salt thereof, wherein the isolation may be carried out by any conventional method, such as by filtration or solvent extraction. After the completion of the reaction, traces of bromine were decomposed by the addition of a solution of sodium sulphite to the reaction mixture. The step (e) comprises the conversion of compound of Formula (III) or salt thereof to Pyroxasulfone or salt thereofby process as described in U.S. Patent No.7,238,689. In an embodiment of the present invention, there is provided an integrated, continuous flow method for the preparation of compound of Formula (I) or salt thereof
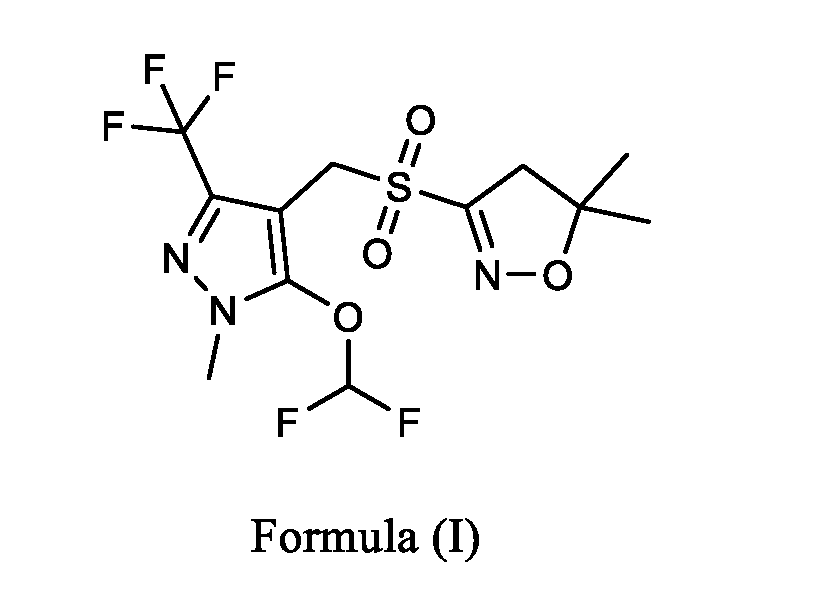
comprising the steps of
A. reacting a compound of Formula V (1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-ol)

with formaldehyde in presence of a base to obtain a compound of Formula VI (4- (hydroxymethyl)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-olate) or salt thereof;

B. reacting the compound of Formula VI or salt thereof with chlorodifluoromethane in a suitable solvent in presence of a base to obtain a compound of Formula VII (5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methanol;

C. reacting the compound of Formula VII (5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-4-yl)methanol) with chlorinating agent to obtain a compound of Formula VIII (3-(((5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H- pyrazol-4-yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole)

D. reacting the compound of Formula VIII (4-(chloromethyl)-5-(difluoromethoxy)-1-methyl- 3-(trifluoromethyl)-1H-pyrazole) with 5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate (Formula IX)

to obtain a compound of Formula X (3-(((5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H- pyrazol-4-yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole)

E. reacting the compound of Formula X (3-(((5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole) with sodium tungstate and a reducing agent to obtain the compound of Formula (I) or salt thereof,
wherein said process is performed using continuous flow reaction conditions. An aspect of the present invention provides an integrated, continuous flow method for step A. for the preparation of 4-(hydroxymethyl)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-olate preferably potassium 4-(hydroxymethyl)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-olate (Formula VI) by reacting with a solution of formaldehyde, a base preferably potassium hydroxide in a suitable solvent preferably a polar solvent, and 1-methyl-3-(trifluoromethyl)- 1H-pyrazol-5-ol (Formula V). The residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 01 minute depending on the temperature. The temperature in the
reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 80°C and even more preferably between 20°C and 50°C, specifically 40°C.


In a further aspect of the present invention, an integrated continuous flow method is provided for step C. for the preparation of 4-(chloromethyl)-5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazole (Formula VIII) by reacting with a chlorinating agent selected from thionyl chloride, phosphorus oxychloride, phosphorus pentachloride, dichlorosuccinimide, calcium hypochlorite, sodium chlorite, carbon tetrachloride,
hydrochloric acid or chlorine preferably thionyl chloride and (5-(difluoromethoxy)-1-methyl- 3-(trifluoromethyl)-1H-pyrazol-4-yl)methanol (Formula VII). The residence time of the mixture in the reactor is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes, and more preferably about 02 minutes depending on the temperature. The temperature in the reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 80°C and even more preferably between 20°C and 50°C, specifically 20°C.





1) reacting glyoxalic acid with hydroxyamino sulfate in presence of a base and/or a buffering agent to obtain hydroxyimino acetic acid;

2) reacting hydroxyimino acetic acid with chlorinating agent to obtain hydroxycarbodimic dichloride;

3) reacting hydroxycarbodimic dichloride with isobutylene and a base to obtain 3- chloro-5,5-dimethyl-4,5-dihydroisoxazole;

4) reacting 3-chloro-5,5-dimethyl-4,5-dihydroisoxazole with thiourea in acidic medium to obtain 5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate.

wherein said process is performed using continuous flow reaction conditions. The operation temperature in the reactor is typically anywhere between 5°C and 80°C, preferably between 5°C and 40°C more preferably 15°C and 20°C for step 2), preferably between 10°C and 70°C more preferably 40°C for step 3) and preferably between 10°C and 40°C more preferably 25°C and 40°C for step 4).
Another aspect of the present invention provides a process for the preparation of an intermediate compound of Formula IX (5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate) by a multistep process, including a process of reacting glyoxalic acid with hydroxyamino sulfate in presence of a base preferably sodium hydroxide and/or buffering agent preferably potassium hydrogen phosphate to obtain hydroxyimino acetic acid and hydroxyimino acetic acid which is further reacted with a chlorinating agent preferably chlorine at a suitable temperature of about 5-40°C preferably about 15-20°C to obtain hydroxycarbodimic dichloride. Hydroxycarbodimic dichloride was reacted with isobutylene and a base preferably potassium carbonate at a suitable temperature of about 10-70°C preferably about 40°C to obtain 3-chloro-5,5-dimethyl-4,5-dihydroisoxazole. 3-chloro-5,5- dimethyl-4,5-dihydroisoxazole was then reacted with thiourea in acidic medium preferably hydrogen bromide or hydrogen chloride at a suitable temperature of about 10 to 40°C preferably about 25°C to 40°C to obtain compound of Formula IX (5,5-dimethyl-4,5- dihydroisoxazol-3-yl carbamimidothioate).

Yet another aspect of the present invention, provides the subsequent conversion of 5,5- dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate (Formula IX) to Formula X (3-(((5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl- 4,5-dihydroisoxazole). The purity of compound of Formula (IX) or salt thereof is more than 95 %, as measured by HPLC. In an embodiment, provided herein is the compound of Formula IX having HPLC purity of at least 95% or atleast 98% obtained by the process as described above.
The continuous flow process of the present invention has many advantages over the batch process as follows:- 1. Minimizes handling of intermediates, toxic and corrosive reagents and solvents.
2. Reduces solvent load, minimizes effluents and waste generation.
3. Dramatically reduced reaction times, less down streaming processing and increases process efficiency. The other advantages of this continuous reactor system are:
• The temperature used in each module of the reactor can be adapted at the kinetic rate of reaction.
• Overall reaction time is reduced from about hours to about minutes.
• Avoids the use and handling of toxic reagents.
• Avoids the isolation of intermediates at each stage and tedious workup procedure. • Improve yield and purity. The continuous flow reactions are performed in flow reactors connected to each other in such a way to provide an integrated system. There are many configurations of such connected reactor system, that a person skilled in the art is aware of. The compounds in continuous flow reaction may be prepared according to the following examples. The following examples are presented to illustrate further aspects of the present invention, but are not intended to limit the scope of the invention in any aspect. Examples Example 1:

50% aqueous solution of glyoxylic acid (74.05 g) was slowly added to a solution of 41.05% hydroxylamine sulphate in water at 0-5 °C for 1h. 25% sodium hydroxide (164 g) solution
was added to the reaction mixture at 10-15 °C in 1 h. pH was adjusted to 10 to 14 by means of addition of 13% sodium carbonate (100 g). The obtained oxime solution was added to a solution of water (50 g) and liquid bromine (96 g) at 10-15 °C in 1 h. Chlorine gas (36 g) was purged into the reaction mixture at 10-15 °Cfor 4 h. After completion of reaction, a solution of 17% sodium sulphite (50 g) was added to the reaction mixture.n-butyl acetate (176 g) solvent was added. The organic and aqueous layers were separated to obtainhydroxycarbonimidicdibromide.
Yield: 95%
HPLC Purity: 90% Example 2:

50% aqueous solution of glyoxylic acid (74.05 g) was slowly added to a solution of 17.38% hydroxylamine sulphate in water at 0-5 °C for 1h. 25% sodium hydroxide (164 g) solution was added to the reaction mixture at 10-15 °C in 1 h. pH was adjusted to 10 to 14 by means of addition of 13% sodium carbonate (100 g). The obtained oxime solution was added to a solution of water (50 g) and liquid bromine (96 g) at 10-15 °C in 1 h. Chlorine gas (36 g) was purged into the reaction mixture at 10-15 °C for 4 h. After completion of reaction, a solution of 17% sodium sulphite (50 g) was added to the reaction mixture. n-butyl acetate (176 g) solvent was added. The organic and aqueous layers were separated to obtainhydroxycarbonimidicdibromide.
Yield: 96%
HPLC Purity: 92% Example 3:
 50% aqueous solution of glyoxylic acid (74.05 g) was slowly added to a solution of 17.38% hydroxylamine sulphate in water at 0-5 °C for 1h. 25% sodium hydroxide (164 g) solution was added to the reaction mixture at 10-15 °C in 1 h. pH was adjusted to 10 to 14 by means of addition of 13% sodium carbonate (100 g).
50% aqueous solution of glyoxylic acid (74.05 g) was slowly added to a solution of 17.38% hydroxylamine sulphate in water at 0-5 °C for 1h. 25% sodium hydroxide (164 g) solution was added to the reaction mixture at 10-15 °C in 1 h. pH was adjusted to 10 to 14 by means of addition of 13% sodium carbonate (100 g).
The obtained oxime solution was added to a solution of water (50 g) and liquid bromine (96 g) at 10-15 °C in 1 h. Chlorine gas (36 g) was purged into the reaction mixture at 10-15 °C for 4 h. After completion of reaction, a solution of 17% sodium sulphite (50 g) was added to the reaction mixture. n-butyl acetate (150 g) solvent was added. The organic and aqueous layers were separated to obtain hydroxycarbonimidicdibromide.
Yield: 94%
HPLC Purity: 89% Example 4:

50% aqueous solution of glyoxylic acid (74.05 g) was slowly added to a solution of 17.38% hydroxylamine sulphate in water at 0-5 °C for 1h. 25% sodium hydroxide (164 g) solution was added to the reaction mixture at 10-15 °C in 1 h. pH was adjusted to 10 to 14 by means of addition of 13% sodium carbonate (100 g).
The obtained oxime solution was added to a solution of water (50 g) and liquid bromine (96 g) at 10-15 °C in 1 h.50% hydrogen peroxide (30 g) was added to the reaction mixture at 10- 15 °C for 4 h. After completion of reaction, a solution of 17% sodium sulphite (50 g) was added to the reaction mixture. n-butyl acetate (176 g) solvent was added. The organic and aqueous layer were separated to obtain hydroxycarbonimidic dibromide.
Yield: 90%
HPLC Purity: 93%
Example 5:

To an aqueous stream of 1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-ol (Formula V) and potassium hydroxide (2.5 M in water) were combined through a T-piece with an aqueous stream of formaldehyde solution (12.5 M in water). The resulting reaction stream was directed toward a 40ml flow reactor, with the residence time of 1 min and an operating temperature of 40°C. The resulting potassium 4-(hydroxymethyl)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-5-olate was combined with difluorochloromethane gas in an acetonitrile solution directed towards another flow reactor, with residence time of 5 min and operating room temperature 25°C to yield (5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-4-yl)methanol (conversion by HPLC 95%). Example 6:

To an organic stream of (5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4- yl)methanol was combined with thionyl chloride (10M in acetonitrile) in flow reactor with residence time of 2 min and an operating temperature of 20°C to yield 4-(chloromethyl)-5- (difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazole (conversion by HPLC > 98%). Example 7:

To an organic stream of 3-(((5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol- 4-yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole (0.45 M) was combined through a T-
piece with an aqueous solution of 5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate (1.0M) with residence time of 5 min and an operating temperature of 100°C in a flow reactor to yield 3-(((5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4- yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole (conversion by HPLC > 85%). Example 8:

To an organic solution of 3-(((5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol- 4-yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole and sodium tungstate was combined through a T-mixture with an aqueous solution of hydrogen peroxide with the residence time of 10 min and operating temperature 100°C in flow reactor to yield compound of Formula I. Example 9:
Synthesis of 5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate

To an aqueous solution of hydroxylamine sulfate (2M in water), glyoxalic acid (5M in water) was added at 25-30°C. The reaction mixture was directed towards a 24ml flow reactor, operating at 25-30°C. Sodium hydroxide (7M solution) and potassium hydrogen phosphate (3M solution) were added to the reaction mixture. After, the completion of the reaction, the reaction mixture was converted to hydroxyimino acetic acid at pH 7 (Conversion by HPLC > 82%).
A stream of chlorine gas was combined through T-piece with an aqueous stream of hydroxyimino acetic acid. The resulted reaction stream was directed towards a 24ml flow with a static mixture (MoC–PFA) reactor, operating at 15-20°C and converted to hydroxycarbonimidic dichloride (Conversion by GC > 87%). Hydroxycarbonimidic dichloride in the aqueous solvent was extracted into n-butyl acetate solvent. Hydroxycarbonimidic dichloride in n-butyl acetate was reacted with isobutylene gas in n- butyl acetate with potassium carbonate as base at 40 °C to ensure full conversion to 3-chloro- 5,5-dimethyl-4,5-dihydroisoxazole (Conversion by GC > 87%). The reaction mass was filtered and the filtrate containing 3-chloro-5,5-dimethyl-4,5- dihydroisoxazole was transferred to the another reactor having a solution of thio-urea and hydrogen bromide solution at 25-40°C to ensure full conversion to 5,5-dimethyl-4,5- dihydroisoxazol-3-yl carbamimidothioate (Conversion by GC > 45%).
Claims
1. An improved process for the preparation of Pyroxasulfone of Formula (I) or salts thereof;


b) reacting 2-(hydroxyimino)acetic acid with bromine in a suitable solvent at a suitable pH;
c) recycling of unreacted bromine anion by addition of a suitable oxidizing agent;
d) optionally, isolating the compound of Formula (III) or salts thereof;

e) converting the compound of Formula (III) or salts thereof, to Pyroxasulfone or salts thereof.
2. The process according to claim 1, wherein the oxidizing agent is selected from the group comprising of fluorine, chlorine, hydrogen peroxide, nitric acid, sulfuric acid, peroxydisulfuric acid, peroxymonosulfuric acid, chlorite, chlorate, perchlorate,
hypochlorite, sodium hypochlorite, chromium trioxide, pyridiniumchlorochromate, potassium permanganate, sodium perborate, nitrous oxide, nitrogen dioxide, dinitrogen tetroxide, potassium nitrate, sodium bismuthate and mixture thereof.
3. The process according to claim 1, wherein the solvent is a polar solvent.
4. The process according to claim 1, wherein the solvent is selected from the group comprising of water, alcohol, chlorinated solvent or a mixture thereof.
5. The process according to claim 4, wherein the solvent is water.
6. The process according to claim 1, wherein the amount of the bromine used is in the range of 0.25 to 3 mole equivalents, based on the amount of 2-(hydroxyimino)acetic acid.
7. The process according to claim 1, wherein the base is selected from organic or inorganic base.
8. The process according to claim 1, wherein the pH of the reaction is maintained in the range of 8 to 14.
9. A process of preparing a compound of Formula (I) or salt thereof:

comprising the steps of:
A. reacting a compound of Formula V (1-methyl-3-(trifluoromethyl)-1H-pyrazol-5- ol)


B. reacting the compound of Formula VI or salt thereof with chlorodifluoromethane in a suitable solvent in presence of a base to obtain a compound of Formula VII (5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methanol;
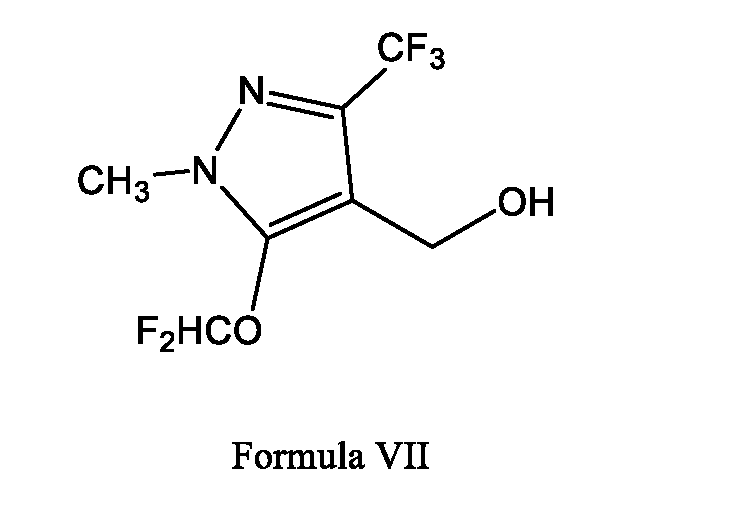
C. reacting the compound of Formula VII (5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-4-yl)methanol) with chlorinating agent to obtain a compound of Formula VIII (3-(((5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl-4,5- dihydroisoxazole)

D. reacting the compound of Formula VIII (4-(chloromethyl)-5-(difluoromethoxy)-1- methyl-3-(trifluoromethyl)-1H-pyrazole) with 5,5-dimethyl-4,5-dihydroisoxazol- 3-yl carbamimidothioate (Formula IX)


E. reacting the compound of Formula X (3-(((5-(difluoromethoxy)-1-methyl-3- (trifluoromethyl)-1H-pyrazol-4-yl)methyl)thio)-5,5-dimethyl-4,5- dihydroisoxazole) with sodium tungstate and an oxidizing agent to obtain the compound of Formula (I) or salt thereof;
wherein said process is performed using continuous flow reaction conditions.
10. The process as claimed in claim 9, wherein the base is an organic or inorganic base, preferably an inorganic base and more preferably selected from potassium hydroxide, sodium hydroxide, sodium carbonate, and/or potassium carbonate.
11. The process as claimed in claim 9, wherein the solvent is selected from polar protic or polar aprotic or non-polar solvent, preferably selected from acetonitrile, water, alcohol or chlorinated solvents or a mixture thereof.
12. The process as claimed in claim 9, wherein the chlorinating agent is selected from thionyl chloride, phosphorus oxychloride, phosphorus pentachloride, dichlorosuccinimide, calcium hypochlorite, sodium chlorite, carbon tetrachloride, hydrochloric acid or chlorine.
13. The process as claimed in claim 9, wherein the reducing agent is selected from hydrogen peroxide or meta chloroperbenzoic acid.
14. The process as claimed in claim 9, wherein the residence time of said mixture in the reactor in step A. or step B. or step C., is typically anywhere between 30 seconds and 20 minutes, preferably about 30 seconds to 05 minutes depending on the temperature.
15. The process as claimed in claim 13, wherein the operation temperature in the reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 80°C and even more preferably between 20°C and 50°C.
16. The process as claimed in claim 9, wherein the residence time of said mixture in the reactor in step D or step E, is typically anywhere between 30 seconds and 20 minutes, preferably about 02 minutes to 10 minutes depending on the temperature.
17. The process as claimed in claim 16, wherein the operation temperature in the reactor is typically anywhere between 10°C and 100°C, preferably between 20°C and 100°C.
18. The process as claimed in claim 9, wherein purity of compound of Formula (I) or salt thereof is more than 95 %, as measured by HPLC.
19. The process for the preparation of an intermediate compound of Formula IX (5,5- dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate) comprising the steps of: 1) reacting glyoxalic acid with hydroxyamino sulfate in presence of a base and/or a buffering agent to obtain hydroxyimino acetic acid;

2) reacting hydroxyimino acetic acid reacted with chlorinating agent to obtain hydroxycarbodimic dichloride;


4) reacting 3-chloro-5,5-dimethyl-4,5-dihydroisoxazole with thiourea in acidic medium to obtain 5,5-dimethyl-4,5-dihydroisoxazol-3-yl carbamimidothioate.

wherein said process is performed using continuous flow reaction conditions.
20. The process as claimed in claim 19, wherein the operation temperature in the reactor is typically anywhere between 5°C and 80°C, preferably between 5°C and 40°C more preferably 15°C and 20°C for step 2), preferably between 10°C and 70°C more preferably 40°C for step 3) and preferably between 10°C and 40°C more preferably 25°C and 40°C for step 4).
21. The process as claimed in claim 19, wherein subsequently 5,5-dimethyl-4,5- dihydroisoxazol-3-yl carbamimidothioate (Formula IX) is converted into Formula X (3-(((5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-4- yl)methyl)thio)-5,5-dimethyl-4,5-dihydroisoxazole).
22. The process as claimed in claim 19, wherein the base is an organic or inorganic base, preferably an inorganic base and more preferably selected from potassium hydroxide, sodium hydroxide, sodium carbonate, and/or potassium carbonate.
23. The process as claimed in claim 19, wherein the chlorinating agent is selected from thionyl chloride, phosphorus oxychloride, phosphorus pentachloride, dichlorosuccinimide, calcium hypochlorite, sodium chlorite, carbon tetrachloride, hydrochloric acid or chlorine.
24. The process as claimed in claim 19, wherein purity of compound of Formula (IX) or salt thereof is more than 95 %, as measured by HPLC.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201911020660 | 2019-05-24 | ||
| IN201911020660 | 2019-05-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020240392A1 true WO2020240392A1 (en) | 2020-12-03 |
Family
ID=71670306
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2020/054914 WO2020240392A1 (en) | 2019-05-24 | 2020-05-23 | Process for preparation of pyroxasulfone |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2020240392A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113372288A (en) * | 2021-06-02 | 2021-09-10 | 安徽久易农业股份有限公司 | Synthetic method of topramezone pesticide intermediate |
| WO2022000603A1 (en) * | 2020-06-28 | 2022-01-06 | 安徽久易农业股份有限公司 | Synthesis method for pyroxasulfone, and application of pyroxasulfone |
| EP4043456A1 (en) | 2021-02-11 | 2022-08-17 | Rotam Agrochem International Company Limited | Crystalline form of pyroxasulfone, methods for its preparation and use of the same |
| CN115073441A (en) * | 2021-03-11 | 2022-09-20 | 山东润博生物科技有限公司 | Synthetic method of topramezone intermediate |
| CN116023295A (en) * | 2022-12-19 | 2023-04-28 | 青岛润农化工有限公司 | A kind of synthetic method of 1,1-dihaloformaldoxime |
| CN116102510A (en) * | 2021-11-10 | 2023-05-12 | 江西仰立新材料有限公司 | Preparation method of 3-bromo-5, 5-dimethyl-4, 5-dihydro-isoxazole |
| EP4053125A4 (en) * | 2019-10-31 | 2023-07-05 | Kumiai Chemical Industry Co., Ltd. | Herbicide and production method for intermediate thereof |
| CN116410138A (en) * | 2021-12-30 | 2023-07-11 | 山东润博生物科技有限公司 | A kind of synthetic method of pyrazoxazole intermediate |
| WO2023194957A1 (en) * | 2022-04-08 | 2023-10-12 | Upl Limited | A process for preparation of 3-[5-(difluoromethoxy)-1- methyl-3-(trifluoromethyl)pyrazol-4- ylmethylsulfonyl]-4,5-dihydro-5,5-dimethyl-1,2-oxazole and its intermediates |
| CN117486821A (en) * | 2023-09-18 | 2024-02-02 | 湖北泰盛化工有限公司 | A continuous flow method for synthesizing sulfonazole intermediates |
| CN118221663A (en) * | 2024-05-24 | 2024-06-21 | 吉林凯莱英医药化学有限公司 | Continuous synthesis method of pyribenzoxim |
| CN118255757A (en) * | 2024-05-28 | 2024-06-28 | 吉林凯莱英医药化学有限公司 | Continuous synthesis method of pyribenzoxim |
| WO2024194794A1 (en) * | 2023-03-20 | 2024-09-26 | Upl Limited | Process for preparation of pyroxasulfone and intermediates thereof |
| WO2024209278A1 (en) * | 2023-04-03 | 2024-10-10 | Gharda Chemicals Limited | A process for the preparation of pyroxasulfone |
| CN119241526A (en) * | 2024-12-03 | 2025-01-03 | 天津凯莱英医药科技发展有限公司 | Continuous synthesis method of sulfonepyraclostrobin |
| CN119258947A (en) * | 2024-12-09 | 2025-01-07 | 天津凯莱英医药科技发展有限公司 | Continuous preparation system and method of sulfonepyraclostrobin |
| CN119318925A (en) * | 2024-12-18 | 2025-01-17 | 天津凯莱英医药科技发展有限公司 | Continuous production system and continuous production method of 5- (difluoromethoxy) -4-chloromethyl-1-methyl-3- (trifluoromethyl) -1H-pyrazole |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6207863B1 (en) | 1998-08-11 | 2001-03-27 | Rohm And Haas Company | Synthesis of haloformimine compounds |
| US20050256004A1 (en) * | 2002-08-07 | 2005-11-17 | Satoru Takahashi | Herbicide compositions |
| US7238689B2 (en) | 2001-02-08 | 2007-07-03 | Ihara Chemical Industry Co., Ltd. | Isoxazoline derivative and herbicide comprising the same as active ingredient |
| US20120264947A1 (en) * | 2009-11-26 | 2012-10-18 | Basf Se | Method for Producing 5,5-Disubstituted 4,5-Dihydroisoxazol-3-Thiocarboxamidine Salts |
-
2020
- 2020-05-23 WO PCT/IB2020/054914 patent/WO2020240392A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6207863B1 (en) | 1998-08-11 | 2001-03-27 | Rohm And Haas Company | Synthesis of haloformimine compounds |
| US7238689B2 (en) | 2001-02-08 | 2007-07-03 | Ihara Chemical Industry Co., Ltd. | Isoxazoline derivative and herbicide comprising the same as active ingredient |
| US20050256004A1 (en) * | 2002-08-07 | 2005-11-17 | Satoru Takahashi | Herbicide compositions |
| US20120264947A1 (en) * | 2009-11-26 | 2012-10-18 | Basf Se | Method for Producing 5,5-Disubstituted 4,5-Dihydroisoxazol-3-Thiocarboxamidine Salts |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4053125A4 (en) * | 2019-10-31 | 2023-07-05 | Kumiai Chemical Industry Co., Ltd. | Herbicide and production method for intermediate thereof |
| WO2022000603A1 (en) * | 2020-06-28 | 2022-01-06 | 安徽久易农业股份有限公司 | Synthesis method for pyroxasulfone, and application of pyroxasulfone |
| EP4043456A1 (en) | 2021-02-11 | 2022-08-17 | Rotam Agrochem International Company Limited | Crystalline form of pyroxasulfone, methods for its preparation and use of the same |
| CN115073441A (en) * | 2021-03-11 | 2022-09-20 | 山东润博生物科技有限公司 | Synthetic method of topramezone intermediate |
| CN115073441B (en) * | 2021-03-11 | 2024-04-02 | 山东润博生物科技有限公司 | Synthesis method of topiramate intermediate |
| CN113372288A (en) * | 2021-06-02 | 2021-09-10 | 安徽久易农业股份有限公司 | Synthetic method of topramezone pesticide intermediate |
| CN116102510A (en) * | 2021-11-10 | 2023-05-12 | 江西仰立新材料有限公司 | Preparation method of 3-bromo-5, 5-dimethyl-4, 5-dihydro-isoxazole |
| CN116102510B (en) * | 2021-11-10 | 2025-03-25 | 江西仰立新材料有限公司 | A preparation method of 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole |
| CN116410138A (en) * | 2021-12-30 | 2023-07-11 | 山东润博生物科技有限公司 | A kind of synthetic method of pyrazoxazole intermediate |
| CN116410138B (en) * | 2021-12-30 | 2025-05-06 | 山东润博生物科技有限公司 | A method for synthesizing sulfonepyraclostrobin intermediate |
| WO2023194957A1 (en) * | 2022-04-08 | 2023-10-12 | Upl Limited | A process for preparation of 3-[5-(difluoromethoxy)-1- methyl-3-(trifluoromethyl)pyrazol-4- ylmethylsulfonyl]-4,5-dihydro-5,5-dimethyl-1,2-oxazole and its intermediates |
| CN116023295B (en) * | 2022-12-19 | 2025-07-08 | 青岛润农化工有限公司 | Synthesis method of 1, 1-dihalogen aldoxime |
| CN116023295A (en) * | 2022-12-19 | 2023-04-28 | 青岛润农化工有限公司 | A kind of synthetic method of 1,1-dihaloformaldoxime |
| WO2024194794A1 (en) * | 2023-03-20 | 2024-09-26 | Upl Limited | Process for preparation of pyroxasulfone and intermediates thereof |
| WO2024209278A1 (en) * | 2023-04-03 | 2024-10-10 | Gharda Chemicals Limited | A process for the preparation of pyroxasulfone |
| CN117486821A (en) * | 2023-09-18 | 2024-02-02 | 湖北泰盛化工有限公司 | A continuous flow method for synthesizing sulfonazole intermediates |
| CN118221663A (en) * | 2024-05-24 | 2024-06-21 | 吉林凯莱英医药化学有限公司 | Continuous synthesis method of pyribenzoxim |
| CN118255757B (en) * | 2024-05-28 | 2024-09-27 | 吉林凯莱英医药化学有限公司 | Continuous synthesis method of pyribenzoxim |
| CN118255757A (en) * | 2024-05-28 | 2024-06-28 | 吉林凯莱英医药化学有限公司 | Continuous synthesis method of pyribenzoxim |
| CN119241526A (en) * | 2024-12-03 | 2025-01-03 | 天津凯莱英医药科技发展有限公司 | Continuous synthesis method of sulfonepyraclostrobin |
| CN119258947A (en) * | 2024-12-09 | 2025-01-07 | 天津凯莱英医药科技发展有限公司 | Continuous preparation system and method of sulfonepyraclostrobin |
| CN119318925A (en) * | 2024-12-18 | 2025-01-17 | 天津凯莱英医药科技发展有限公司 | Continuous production system and continuous production method of 5- (difluoromethoxy) -4-chloromethyl-1-methyl-3- (trifluoromethyl) -1H-pyrazole |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2020240392A1 (en) | Process for preparation of pyroxasulfone | |
| EP0966452B1 (en) | Process for preparing sulphurous 2-chloro-3-(4,5-dihydroisoxazol-3-yl)-benzoic acids | |
| US20130023668A1 (en) | Processes for the preparation of pyrazoles | |
| CN110483438A (en) | A kind of duct type continuous production method of 3- iso thiazoline ketone compound | |
| EP3812369B1 (en) | A bromination method for m-diamide compounds | |
| JP6911143B2 (en) | New method for producing pyrazole or pyrimidinone | |
| Boruah et al. | KF/Al2O3: Solid-Supported Reagent Used in 1, 3-Dipolar Cycloaddition Reaction of Nitrile Oxide | |
| US6548677B1 (en) | Method of producing 2-alkyl-3-(4,5-dihydroisoxazole-3-yl)-halobenzenes | |
| CN116102510B (en) | A preparation method of 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole | |
| AU2023288874A1 (en) | Method for producing sulfone derivative using haloacetic acid | |
| TWI734776B (en) | Method for manufacturing nitrobenzene compound | |
| US20220185810A1 (en) | Novel Process for the Preparation of Filgotinib and Intermediates Thereof | |
| CN117343023B (en) | Preparation method of 3-bromo-5, 5-dimethyl-4, 5-dihydro-isoxazole | |
| WO2014203855A2 (en) | Manufacturing method for 2-amino-2-hydroxyimino-n-alkoxy acetoimidoyl cyanide, and manufacturing intermediate thereof | |
| CN119173499A (en) | Method for chlorinating benzaldehyde oxime | |
| JP3148226B2 (en) | Process for preparing isoxazole-4,5-dicarboxylic acid diesters and diesters of this kind | |
| Sonavane et al. | Improved, Solvent Free, Atom Efficient Commercial Process for the Synthesis of Diphenhydramine Hydrochloride | |
| CN113717120A (en) | Preparation method of 3- (2-methyl-6-nitrophenyl) -4, 5-dihydroisoxazole | |
| CN116375660A (en) | Preparation method of isoxazoline pesticide fluorine Lei Lana intermediate for pets | |
| US5371264A (en) | Process for preparing N,O-disubstituted hydroxylamine compounds | |
| WO2024194794A1 (en) | Process for preparation of pyroxasulfone and intermediates thereof | |
| CN118638011A (en) | A kind of synthetic method of chloramphenicol intermediate | |
| JP4482165B2 (en) | Process for producing cyanobenzaldehyde compounds | |
| JPS5899438A (en) | 2-halogenopropionic acid ester and its preparation | |
| JP4841032B2 (en) | Process for producing 2-methoxy-4-nitrobenzaldehyde |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20742882 Country of ref document: EP Kind code of ref document: A1 |
|
| WA | Withdrawal of international application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20742882 Country of ref document: EP Kind code of ref document: A1 |