WO2020212832A1 - Process of preparation of benzimidazole compounds - Google Patents
Process of preparation of benzimidazole compounds Download PDFInfo
- Publication number
- WO2020212832A1 WO2020212832A1 PCT/IB2020/053489 IB2020053489W WO2020212832A1 WO 2020212832 A1 WO2020212832 A1 WO 2020212832A1 IB 2020053489 W IB2020053489 W IB 2020053489W WO 2020212832 A1 WO2020212832 A1 WO 2020212832A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- binimetinib
- formula
- compound
- acid
- solvate
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 49
- 238000002360 preparation method Methods 0.000 title claims abstract description 41
- 125000003785 benzimidazolyl group Chemical class N1=C(NC2=C1C=CC=C2)* 0.000 title 1
- 229950003054 binimetinib Drugs 0.000 claims abstract description 86
- ACWZRVQXLIRSDF-UHFFFAOYSA-N binimetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1F ACWZRVQXLIRSDF-UHFFFAOYSA-N 0.000 claims abstract description 84
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 claims abstract description 21
- 229950010746 selumetinib Drugs 0.000 claims abstract description 19
- 150000001875 compounds Chemical class 0.000 claims description 70
- 238000006243 chemical reaction Methods 0.000 claims description 38
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 33
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 21
- 239000012453 solvate Substances 0.000 claims description 20
- 150000007524 organic acids Chemical group 0.000 claims description 17
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 16
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 15
- 239000007787 solid Substances 0.000 claims description 15
- -1 Binimetinib organic acid Chemical class 0.000 claims description 14
- 239000002253 acid Substances 0.000 claims description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 12
- 235000011054 acetic acid Nutrition 0.000 claims description 10
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 9
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 9
- 229910052736 halogen Inorganic materials 0.000 claims description 9
- 239000000203 mixture Substances 0.000 claims description 9
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical group COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 8
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 8
- 235000019253 formic acid Nutrition 0.000 claims description 8
- 239000002904 solvent Substances 0.000 claims description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 8
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Substances BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 claims description 7
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 6
- 125000002102 aryl alkyloxo group Chemical group 0.000 claims description 6
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 6
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 6
- 125000005843 halogen group Chemical group 0.000 claims description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 5
- 125000001246 bromo group Chemical group Br* 0.000 claims description 5
- 239000003795 chemical substances by application Substances 0.000 claims description 5
- 125000006239 protecting group Chemical group 0.000 claims description 5
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 claims description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 4
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 claims description 4
- IRJKSAIGIYODAN-ISLYRVAYSA-N benzyl (ne)-n-phenylmethoxycarbonyliminocarbamate Chemical compound C=1C=CC=CC=1COC(=O)/N=N/C(=O)OCC1=CC=CC=C1 IRJKSAIGIYODAN-ISLYRVAYSA-N 0.000 claims description 4
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 claims description 4
- KLKFAASOGCDTDT-UHFFFAOYSA-N ethoxymethoxyethane Chemical group CCOCOCC KLKFAASOGCDTDT-UHFFFAOYSA-N 0.000 claims description 4
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 claims description 4
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 claims description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 4
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 claims description 3
- 239000003153 chemical reaction reagent Substances 0.000 claims description 3
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 claims description 3
- 238000001914 filtration Methods 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 150000007522 mineralic acids Chemical class 0.000 claims description 3
- 239000003960 organic solvent Substances 0.000 claims description 3
- 239000003880 polar aprotic solvent Substances 0.000 claims description 3
- 235000019260 propionic acid Nutrition 0.000 claims description 3
- 238000000746 purification Methods 0.000 claims description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 3
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 claims description 3
- 150000003839 salts Chemical class 0.000 claims description 3
- 238000003756 stirring Methods 0.000 claims description 3
- ZNOVTXRBGFNYRX-UHFFFAOYSA-N 2-[[4-[(2-amino-5-methyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 ZNOVTXRBGFNYRX-UHFFFAOYSA-N 0.000 claims description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 claims description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 claims description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 claims description 2
- 125000005604 azodicarboxylate group Chemical group 0.000 claims description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 claims description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 claims description 2
- 230000002140 halogenating effect Effects 0.000 claims description 2
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 claims description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 claims description 2
- 150000007530 organic bases Chemical class 0.000 claims description 2
- AQSJGOWTSHOLKH-UHFFFAOYSA-N phosphite(3-) Chemical class [O-]P([O-])[O-] AQSJGOWTSHOLKH-UHFFFAOYSA-N 0.000 claims description 2
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 claims description 2
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 claims 3
- VLSDXINSOMDCBK-BQYQJAHWSA-N (E)-1,1'-azobis(N,N-dimethylformamide) Chemical compound CN(C)C(=O)\N=N\C(=O)N(C)C VLSDXINSOMDCBK-BQYQJAHWSA-N 0.000 claims 1
- 229960004365 benzoic acid Drugs 0.000 claims 1
- 235000019445 benzyl alcohol Nutrition 0.000 claims 1
- 230000003197 catalytic effect Effects 0.000 claims 1
- XZTSFVPMMQNIAJ-UHFFFAOYSA-N o-(2-ethenoxyethyl)hydroxylamine Chemical compound NOCCOC=C XZTSFVPMMQNIAJ-UHFFFAOYSA-N 0.000 claims 1
- 229960003424 phenylacetic acid Drugs 0.000 claims 1
- 150000001556 benzimidazoles Chemical class 0.000 abstract description 5
- 239000000543 intermediate Substances 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 39
- 239000011541 reaction mixture Substances 0.000 description 23
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 16
- 239000000243 solution Substances 0.000 description 15
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 239000000463 material Substances 0.000 description 10
- 239000012044 organic layer Substances 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- YBTSZTNYTYAQKE-UHFFFAOYSA-N 6-(4-bromo-2-fluoroanilino)-7-fluoro-3-methylbenzimidazole-5-carboxylic acid Chemical compound OC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1F YBTSZTNYTYAQKE-UHFFFAOYSA-N 0.000 description 4
- KMJNEXZZHQIVEF-UHFFFAOYSA-N 6-(4-bromo-2-fluoroanilino)-n-(2-ethenoxyethoxy)-7-fluoro-3-methylbenzimidazole-5-carboxamide Chemical compound C=COCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1F KMJNEXZZHQIVEF-UHFFFAOYSA-N 0.000 description 4
- IIAGNFGQWMNNMA-UHFFFAOYSA-N NC1=C(C(=C(C(=O)O)C=C1N)NC1=C(C=C(C=C1)Br)F)F Chemical compound NC1=C(C(=C(C(=O)O)C=C1N)NC1=C(C=C(C=C1)Br)F)F IIAGNFGQWMNNMA-UHFFFAOYSA-N 0.000 description 4
- JIOIUVZEVGGYPU-UHFFFAOYSA-N NC1=C(C(=C(C(=O)O)C=C1[N+](=O)[O-])NC1=C(C=C(C=C1)Br)F)F Chemical compound NC1=C(C(=C(C(=O)O)C=C1[N+](=O)[O-])NC1=C(C=C(C=C1)Br)F)F JIOIUVZEVGGYPU-UHFFFAOYSA-N 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 4
- 0 *C(c(c(Nc1c(*)cccc1)c1F)cc(N)c1N)=N Chemical compound *C(c(c(Nc1c(*)cccc1)c1F)cc(N)c1N)=N 0.000 description 3
- WXXHOWQPFHXRDY-UHFFFAOYSA-N 284030-57-5 Chemical compound NC1=C(F)C(F)=C(C(O)=O)C=C1[N+]([O-])=O WXXHOWQPFHXRDY-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 239000002798 polar solvent Substances 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 239000011260 aqueous acid Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- XZALEBBEJBRERO-UHFFFAOYSA-N methyl 4,5-diamino-3-fluoro-2-(2-fluoroanilino)benzoate Chemical compound COC(=O)C1=CC(N)=C(N)C(F)=C1NC1=CC=CC=C1F XZALEBBEJBRERO-UHFFFAOYSA-N 0.000 description 2
- NOGALSZXSQWRDJ-UHFFFAOYSA-N methyl 4-amino-3-fluoro-2-(2-fluoroanilino)-5-nitrobenzoate Chemical compound COC(=O)C1=CC([N+]([O-])=O)=C(N)C(F)=C1NC1=CC=CC=C1F NOGALSZXSQWRDJ-UHFFFAOYSA-N 0.000 description 2
- CLBKHICAWHYQLM-UHFFFAOYSA-N methyl 6-(4-bromo-2-fluoroanilino)-7-fluoro-3-methylbenzimidazole-5-carboxylate Chemical compound COC(=O)C1=CC=2N(C)C=NC=2C(F)=C1NC1=CC=C(Br)C=C1F CLBKHICAWHYQLM-UHFFFAOYSA-N 0.000 description 2
- NYPSEISJJRRIAX-UHFFFAOYSA-N methyl 7-fluoro-6-(2-fluoroanilino)-3-methylbenzimidazole-5-carboxylate Chemical compound COC(=O)C1=CC=2N(C)C=NC=2C(F)=C1NC1=CC=CC=C1F NYPSEISJJRRIAX-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- ISNICOKBNZOJQG-UHFFFAOYSA-N 1,1,2,3,3-pentamethylguanidine Chemical compound CN=C(N(C)C)N(C)C ISNICOKBNZOJQG-UHFFFAOYSA-N 0.000 description 1
- KYVBNYUBXIEUFW-UHFFFAOYSA-N 1,1,3,3-tetramethylguanidine Chemical compound CN(C)C(=N)N(C)C KYVBNYUBXIEUFW-UHFFFAOYSA-N 0.000 description 1
- AVQQQNCBBIEMEU-UHFFFAOYSA-N 1,1,3,3-tetramethylurea Chemical compound CN(C)C(=O)N(C)C AVQQQNCBBIEMEU-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WWWTWPXKLJTKPM-UHFFFAOYSA-N 2-aminooxyethanol Chemical compound NOCCO WWWTWPXKLJTKPM-UHFFFAOYSA-N 0.000 description 1
- FTZQXOJYPFINKJ-UHFFFAOYSA-N 2-fluoroaniline Chemical compound NC1=CC=CC=C1F FTZQXOJYPFINKJ-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- GZRMNMGWNKSANY-UHFFFAOYSA-N 4-bromo-2-fluoroaniline Chemical compound NC1=CC=C(Br)C=C1F GZRMNMGWNKSANY-UHFFFAOYSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- GXMHFZUMJZYEOJ-UHFFFAOYSA-N 6-(4-bromo-2-fluoroanilino)-7-fluoro-3h-benzimidazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=2NC=NC=2C(F)=C1NC1=CC=C(Br)C=C1F GXMHFZUMJZYEOJ-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- OCRPZASHKZCJQT-UHFFFAOYSA-N C(C)OC(=O)C1=CC2=C(N=CN2C)C(=C1NC1=C(C=C(C=C1)Br)F)F Chemical compound C(C)OC(=O)C1=CC2=C(N=CN2C)C(=C1NC1=C(C=C(C=C1)Br)F)F OCRPZASHKZCJQT-UHFFFAOYSA-N 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical class [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 201000005969 Uveal melanoma Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000012351 deprotecting agent Substances 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 229950001969 encorafenib Drugs 0.000 description 1
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethyl cyclohexane Natural products CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 1
- 231100001261 hazardous Toxicity 0.000 description 1
- 201000008298 histiocytosis Diseases 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- CMJCXYNUCSMDBY-ZDUSSCGKSA-N lgx818 Chemical compound COC(=O)N[C@@H](C)CNC1=NC=CC(C=2C(=NN(C=2)C(C)C)C=2C(=C(NS(C)(=O)=O)C=C(Cl)C=2)F)=N1 CMJCXYNUCSMDBY-ZDUSSCGKSA-N 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- HOJFIOHGPQOQBF-UHFFFAOYSA-N methyl 4-amino-2,3-difluoro-5-nitrobenzoate Chemical compound COC(=O)C1=CC([N+]([O-])=O)=C(N)C(F)=C1F HOJFIOHGPQOQBF-UHFFFAOYSA-N 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- BATOBWCJYQFPTK-UHFFFAOYSA-N o-(2-ethoxyethyl)hydroxylamine Chemical compound CCOCCON BATOBWCJYQFPTK-UHFFFAOYSA-N 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical class [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical class [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 238000000634 powder X-ray diffraction Methods 0.000 description 1
- 238000001144 powder X-ray diffraction data Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 102200055464 rs113488022 Human genes 0.000 description 1
- 102220197820 rs121913227 Human genes 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 238000005549 size reduction Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 150000003458 sulfonic acid derivatives Chemical class 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 150000003738 xylenes Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/04—Formation of amino groups in compounds containing carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/04—Formation of amino groups in compounds containing carboxyl groups
- C07C227/10—Formation of amino groups in compounds containing carboxyl groups with simultaneously increasing the number of carbon atoms in the carbon skeleton
Definitions
- present application describes an improved process of preparation of benzimidazole compounds such as Binimetinib and Selumetinib. Further, present application provides intermediates that are useful in the preparation of Binimetinib and Selumetinib. BACKGROUND OF THE INVENTION
- Binimetinib is chemically known as 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N- (2-hydroxyethoxy)-l-methyl-lH-benzimidazole-6-carboxamide. Binimetinib is represented by the following chemical structure according to Formula (I).
- Binimetinib is indicated, in combination with encorafenib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation.
- Selumetinib of Formula (II) is chemically known as 5-[(4-bromo-2- chlorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-l-methyl-lH -benzimidazole-6- carboxamide.
- Selumetinib is under clinical trials for various types of cancer such as biliary cancer, colorectal cancer, gastric cancer, gastrointestinal stromal tumours, glioma, histiocytosis, neurofibromatoses, non-hodgkin's lymphoma, non-small cell lung cancer, Solid tumours, Thyroid cancer, Uveal melanoma, Astrocytoma, Kaposi's sarcoma, Precursor cell lymphoblastic leukaemia-lymphoma.
- cancer such as biliary cancer, colorectal cancer, gastric cancer, gastrointestinal stromal tumours, glioma, histiocytosis, neurofibromatoses, non-hodgkin's lymphoma, non-small cell lung cancer, Solid tumours, Thyroid cancer, Uveal melanoma, Astrocytoma, Kaposi's sarcoma, Precursor cell lymphoblastic leukaemia-lymphoma.
- Patents US 7,425,637 and US 9,156,795 provide processes for the preparation of Selumetinib; and Patents US 7,777,050, US 8,039,637, and US 9,238,627 provide processes for the preparation of Binimetinib.
- the process in US‘050 is as shown in below scheme:
- US 9,562,016 directed to process for the preparation of crystallized Form of Binimetinib which comprises crystallizing Binimetinib from aqueous ether solution by adding water.
- W02016131406A1 patent application discloses polymorph forms designated as Form A and Form B.
- An aspect of the present invention is to provide a process of preparation of Binimetinib comprises treating Binimetinib solvate with water.
- Another aspect of the invention is to provide a process of preparation of Binimetinib or Selumetinib comprising steps of: a. treating a compound of Formula (1) with C1-C6 dialkoxymethane in the presence of an acid to obtain a compound of Formula (2);
- Still another aspect of the invention is to provide the invention provides compound of compound of Formula-2 and/or Formula-4 and use thereof in the preparation of Binimetinib and/or Selumetinib.
- X is halogen and Xi is chloro or bromo.
- Binimetinib with organic acid
- step (a) reaction mass followed by stirring; c. filtering the obtained solid;
- the organic acid used in step (a) is selected from formic acid, acetic acid, propionic acid and the like; preferably acetic acid.
- the amount of organic acid in the Binimetinib isolated may be either in stoichiometric level like 0.5, 1, 1.5. 2 mole equivalent OR in non- stoichiometric level.
- the solvent used in step (b) includes, but not limited to, toluene, anisole, MTBE (methyl tert-butyl ether), diisopropyl ether, methyl THF, THF (tetrhydrofuran) and the like.
- the step (b) solution is stirred at a temperature in the range of 20- 50 deg for about 1-15 hours, preferably stirred at ambient temperatures.
- the compound isolated in step (d) is taken to next stages with or without drying.
- Binimetinib containing organic acid is selected from Binimetinib acetic acid solvate, Binimetinib formic acid solvate, Binimetinib propionic acid solvate, Binimetinib diacetate and the like.
- Another embodiment of the invention relates to a process of preparation of Binimetinib comprises treating Binimetinib containing organic acid in stoichiometric or non- stoichiometric level with water.
- the process of preparation of Binimetinib comprises treating Binimetinib containing acetic acid in stoichiometric or non-stoichiometric level with water. Accordingly the said treatment with water is carried out at a temperature in the range of 30 to 80°C.
- the reaction mass containing Binimetinib acetic acid solvate in water stirred for about 30 min to 3 hour.
- the pure Binimetinib obtained is optionally again slurry washed to get pharmaceutically acceptable level of acid molecule.
- the isolation is carried out by conventional method, preferably by filtration.
- the Binimetinib thus isolated is in crystalline Form; preferably having PXRD that corresponds to Form A.
- the isolated Binimetinib is substantially free of solvate, which means the isolated Bininetinib is anhydrous and may contain the residual solvent of which is pharmaceutically acceptable level.
- the process for the preparation of crystalline form of Binimetinib comprising treating solvated form of Binimetinib with water.
- the solvate form of Binimetinib may be prepared from conventional methods and the solvate includes but not limited to propylene glycol, ethylene glycol, ethanol, benzyl alcohol, IP A and the like.
- Binimetinib may be obtained by treating Binimetinib containing organic acid in stoichiometric or non- stoichiometric level with polar solvent or aqueous polar solvent.
- the polar solvent includes, but not limited to, methanol, ethanol and the like.
- the Binimetinib may be prepared by the process described herein below, in the present application.
- another embodiment of the present invention relates to compound of Formula (2) and its use in preparation of Benzimidazole compounds like Binimetinib and/or Selumetinib.
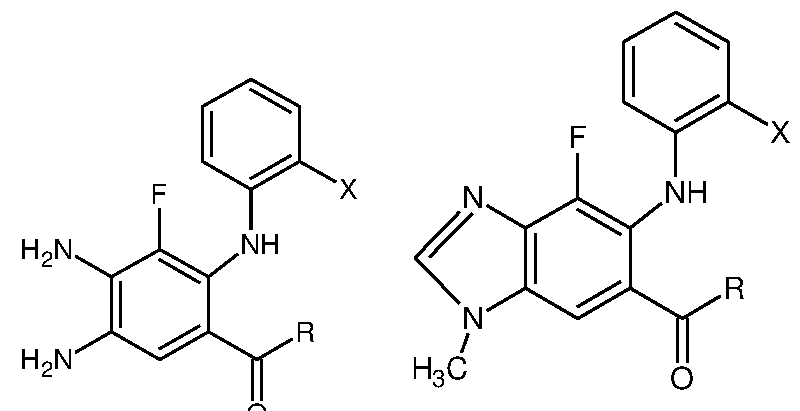
- X is any halogen, and preferably selected from the group consisting of fluoro or chloro and R is selected from group consisting of hydroxy, C ⁇ -C alkoxy, arylalkyloxy such as benzyloxy, 0-(2-vinyloxy-ethyl)-hydroxylamino, or O- (hydroxyethyl)- hydroxylamino.
- C1-C6 alkoxy refers to a linear or a branched chain alkoxy group having C1-C6 carbon atoms which include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, butoxy, sec-butoxy, and tert-butoxy.
- C1-C6 dialkoxymethane is selected from diethoxymethane and dimethoxy methane.
- the acid used in imidazole formation is selected from the group consisting of an inorganic acid or an organic acid.
- the inorganic acid includes but not limited to hydrochloric acid and sulfuric acid.
- the organic acid includes but not limited to sulfonic acid derivatives such as methane sulfonic acid, p-toluene sulfonic acid, and benzene sulfonic acid; or carboxylic acid such as formic acid and acetic acid.
- the compound of Formula (2) is converted in to compound of Formula-3 using a brominating agent such as NBS optionally in the presence of an acid like p-TsOH.
- a brominating agent such as NBS optionally in the presence of an acid like p-TsOH.
- the reaction may be effected in the presence of suitable organic solvent. For that matter any solvent that does not affect the course of the reaction can be used.
- reaction is represented by below scheme:
- the compound of Formula (3) when R is OCH 2 CH 3 is converted to Binimetinib by first converting the ethyl ester compound to acid compound as represented by below scheme, and then converting acid compound to Binimetinib according to the at least one of the suitable known processes in the art as described above.
- the invention relates to Benzimidazole compounds like Binimetinib and/or Selumetinib, prepared by using Formula (4)
- the process of preparation of Binimetinib or Selumetinib comprising steps of:
- the conversion of compound of Formula-4 into Binimetinib or Selumetinib according to step (b) is carried out by reacting the compound of Formula-4 with optionally protected ethylene glycol preferably under Mitsunobu conditions followed by removing the protecting groups if any in polar aprotic solvent.
- the suitable reagent for the said Mitsunobu conditions includes triphenylphosphine, trialkylphosphines or phosphites, diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), dibenzyl azodicarboxylate (DBAD), N,N,N',N'- tetramethylazodicarbonamide (TMAD), and dipiperidyl azodicarboxylate (DPAD), tosyl chloride, mesyl chloride and mixture thereof.
- DEAD diethyl azodicarboxylate
- DIAD diisopropyl azodicarboxylate
- DBAD dibenzyl azodicarboxylate
- TMAD N,N,N',N'- tetramethylazodicarbonamide
- DPAD dipiperidyl azodicarboxylate
- the polar aprotic solvent is selected from the group consisting of tetrahydrofuran, acetonitrile, dioxane, dimethyl sulfoxide, dimethylformamide, dimethylacetamide, N-methylpyrrolidon, dimethoxymethane, methylene chloride, ethyl acetate and mixtures thereof.
- Binimetinib or Selumetinib is prepared by treating compound of Formula-3, wherein R is C1-C6 alkoxy, preferably ethoxy or methoxy or tert-butoxy with O- (2-hydroxyethyl) hydroxylamine with or without formation of acid compound as intermediate.
- R is C1-C6 alkoxy, preferably ethoxy or methoxy or tert-butoxy with O- (2-hydroxyethyl) hydroxylamine
- Binimetinib or Selumetinib is also prepared by treating compound of Formula (3), wherein R is C1-C6 alkoxy; preferably ethoxy or methoxy or tert-butoxy with 0-(2- hydroxyethyl) hydroxyl amine compound protected with PG, without formation of acid compound.
- the above reaction may proceed in solvent such as xylene, toluene, tetrahydrofuran, 2-methyl tetrahydrofuran, NMP, sulfolane, N,N-dimethylimidazolinone, cyclopentylmethyl ether, DMF, DMAc, NMP, tetramethylurea, tetramethyl guanidine, and pentamethyl guanidine.
- solvent such as xylene, toluene, tetrahydrofuran, 2-methyl tetrahydrofuran, NMP, sulfolane, N,N-dimethylimidazolinone, cyclopentylmethyl ether, DMF, DMAc, NMP, tetramethylurea, tetramethyl guanidine, and pentamethyl guanidine.
- the above reaction may proceed with or without use of base.
- the base can be organic or inorganic base.
- the above reaction may proceed preferably under close conditions such as in autoclave under pressure at higher temperature.
- the protecting group PG is selected from alkyl groups, such as tertiary alkyls (e.g., tertiary C4-C7 alkyls such as t-butyl or tertiary amyl); alkenyl groups; tertiary aryl-alkyl groups, such as 1 -methyl- 1-phenylethyl (cumyl) or triphenylmethyl (trityl); groups that result in acetals, such as methoxymethyl, 1-ethoxyethyl, 2-tetrahydropyranyl or 2- tetrahydrofuranyl; and silyl groups, such as trimethylsilyl, triethylsilyl or tert-butyl- dimethyldilyl.
- alkyl groups such as tertiary alkyls (e.g., tertiary C4-C7 alkyls such as t-butyl or tertiary amyl); alkeny
- the protecting group PG may be removed using any suitable deprotecting agent and deprotection conditions and will vary with the choice of protecting group.
- the suitable deprotection agents may include, but are not limited to, an aqueous acid such as phosphoric acid, hydrochloric acid or sulfuric acid, non-aqueous acids such as hydrogen chloride acid.
- the Binimetinib may be prepared by the process described herein above or below, in the present application.
- the invention relates to a process for preparation of Binimetinib comprising steps of: a. converting compound of Formula (A) to compound of Formula (B), wherein the reaction proceeds in the absence of solvent and use of quinolone or pyridine as catalyst poisoning agent;
- Formula C Formula (C) c. converting compound of Formula (C) to compound of Formula (D) using 2- [(aminooxy)ethoxy] ethane, wherein the obtained compound is purified using ethanol and n-heptane;
- Binimetinib used herein is prepared by any conventional methods or by following the processes given as shown by following scheme or by following the examples described herein.
- the Binimetinib may be prepared by the process described herein above or by any conventional process and further purified as below.
- the DIO, D50, and D90 values are useful ways for indicating a particle size distribution.
- D90 refers to the value for the particle size for which at least 90 volume percent of the particles have a size smaller than the value.
- D50 and D10 refer to the values for the particle size for which 50 volume percent, and 10 volume percent, of the particles have a size smaller than the value.
- Methods for determining D10, D50 and D90 include laser diffraction, such as using Malvern Instruments Ltd. (of Malvern) equipment.
- Binimetinib obtained according to the process described herein or after performing size reduction operations such as milling or micronization has a particle size distribution pattern of D10 less than or equal to about 150 pm, D50 less than or equal to about 300 pm, and D90 less than or equal to about 500 pm. There is no specific lower limit for any of the D values.
- Example 9 Preparation of 4-amino-2-[(4-bromo-2-fluorophenyl)amino]-3-fluoro-5- nitrobenzoic acid.
- Binimetinib (100 g) was stirred in acetic acid (400 mL) at room temperature. Toluene (1250 mL) was added to the reaction mass and stirred for 12 hours at room temperature. Filtered the solid and washed with toluene. The material was dried in VTD for 8 hours at 50°C. (acetic acid content: 7 %).
- Binimetinib obtained in above step was added to water at room temperature; temperature was raised to 65+3°C and stirred for 2 hours. The solid was filtered at 65°C and washed with hot water. The material was dried in VTD for 8 hours at 50°C to obtain pure Binimetinib. (Yield: -80 %).
- Binimetinib diacetate (Form B) solvate was added to water at room temperature; temperature was raised to 65+3 °C and stirred for about 1-3 hours. The solid was filtered at ambient temperature and washed with water at ambient temperature. The material was dried in VTD for 8 hours at 50°C to obtain pure Binimetinib. (Yield: -80 %).
- Binimetinib alcohol solvate disclosed herein was added to water at room temperature; temperature was raised to 70°C and stirred for about 3 hours. The solid was filtered at ambient temperature and washed with water at ambient temperature. The material was dried in VTD for 8 hours at 50°C to obtain pure Binimetinib. (Yield: ⁇ 80 %).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The application provides process of preparation of benzimidazole compounds such as Binimetinib and Selumetinib. Also, the present application provides intermediates for process of preparation of Binimetinib and Selumetinib.
Description
TITLE: Process of Preparation of benzimidazole compounds
RELATED APPLICATION:
This application claims the benefit of the earlier filing date of Indian Provisional Patent Application No. 201921015257 filed on Apr. 16, 2019 and Indian Provisional Patent Application No. 202021001198 filed on Jan. 10, 2020.
FIELD OF THE INVENTION
The present application describes an improved process of preparation of benzimidazole compounds such as Binimetinib and Selumetinib. Further, present application provides intermediates that are useful in the preparation of Binimetinib and Selumetinib. BACKGROUND OF THE INVENTION
Binimetinib, is chemically known as 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N- (2-hydroxyethoxy)-l-methyl-lH-benzimidazole-6-carboxamide. Binimetinib is represented by the following chemical structure according to Formula (I).

Selumetinib of Formula (II), is chemically known as 5-[(4-bromo-2- chlorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-l-methyl-lH -benzimidazole-6- carboxamide.

Patents US 7,425,637 and US 9,156,795 provide processes for the preparation of Selumetinib; and Patents US 7,777,050, US 8,039,637, and US 9,238,627 provide processes for the preparation of Binimetinib. The process in US‘050 is as shown in below scheme:

US 9,562,016 directed to process for the preparation of crystallized Form of Binimetinib which comprises crystallizing Binimetinib from aqueous ether solution by adding water.
W02016131406A1 patent application discloses polymorph forms designated as Form A and Form B.
The known processes have one or more of the disadvantages such as low yield, increased number of chemical reaction steps, difficulties in isolation or separation of the products, flash chromatography, costly reagents and use of hazardous.
Since the compounds are potentially useful as therapeutics, there exists a need in the art for the development of an easy, cost-effective, and industrially advantageous process for the preparation of Binimitinib and Selumetinib which overcomes the difficulties of the prior art process. SUMMARY OF THE INVETION
An aspect of the present invention is to provide a process of preparation of Binimetinib comprises treating Binimetinib solvate with water.
Another aspect of the invention is to provide a process of preparation of Binimetinib or Selumetinib comprising steps of: a. treating a compound of Formula (1) with C1-C6 dialkoxymethane in the presence of an acid to obtain a compound of Formula (2);
o
Formula (1) Formula (2) wherein X is halogen.
b. converting the compound Formula (2) to a compound Formula (3), using an halogenating agent selected from NBS or NCS;

Formula (3) wherein R is hydroxy, C1-C6 alkoxy, arylalkyloxy such as benzyloxy; Xi is bromo or chloro ;and X is a halogen;
c. treating the compound of Formula (3) with hydroxylamine or its salt to obtain compound of Formula-4; and


Formula (2) wherein X is any halogen, and R is selected from group consisting of hydroxy, C\-C alkoxy, arylalkyloxy such as benzyloxy, 0-(2-vinyloxy-ethyl)-hydroxylamino, or O-
(hydroxyethyl)-hydroxylamino .

Wherein X is halogen and Xi is chloro or bromo.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig 1. depicts the powder X-ray diffraction (PXRD) pattern of Form AL-1 of Binimetinib obtained in Example- 15.
DETAILED DESCRIPTION OF THE INVENTION
In an embodiment of the invention relates to a process of preparation of Binimetinib comprising
a. treating Binimetinib with organic acid;
b. adding organic solvent to step (a) reaction mass followed by stirring; c. filtering the obtained solid; and
d. isolating the Binimetinib containing organic acid in stoichiometric or non- stoichiometric level.
In an embodiment, the organic acid used in step (a) is selected from formic acid, acetic acid, propionic acid and the like; preferably acetic acid.
In an embodiment, the amount of organic acid in the Binimetinib isolated may be either in stoichiometric level like 0.5, 1, 1.5. 2 mole equivalent OR in non- stoichiometric level.
In an embodiment, the solvent used in step (b) includes, but not limited to, toluene, anisole, MTBE (methyl tert-butyl ether), diisopropyl ether, methyl THF, THF (tetrhydrofuran) and the like.
In an embodiment, the step (b) solution is stirred at a temperature in the range of 20- 50 deg for about 1-15 hours, preferably stirred at ambient temperatures.
In an embodiment, the compound isolated in step (d) is taken to next stages with or without drying.
Binimetinib containing organic acid is selected from Binimetinib acetic acid solvate, Binimetinib formic acid solvate, Binimetinib propionic acid solvate, Binimetinib diacetate and the like.
Another embodiment of the invention relates to a process of preparation of Binimetinib comprises treating Binimetinib containing organic acid in stoichiometric or non- stoichiometric level with water.
In an embodiment, the process of preparation of Binimetinib comprises treating Binimetinib containing acetic acid in stoichiometric or non-stoichiometric level with water.
Accordingly the said treatment with water is carried out at a temperature in the range of 30 to 80°C. The reaction mass containing Binimetinib acetic acid solvate in water stirred for about 30 min to 3 hour. The pure Binimetinib obtained is optionally again slurry washed to get pharmaceutically acceptable level of acid molecule. The isolation is carried out by conventional method, preferably by filtration.
The Binimetinib thus isolated is in crystalline Form; preferably having PXRD that corresponds to Form A. the isolated Binimetinib is substantially free of solvate, which means the isolated Bininetinib is anhydrous and may contain the residual solvent of which is pharmaceutically acceptable level.
In embodiment, the process for the preparation of crystalline form of Binimetinib comprising treating solvated form of Binimetinib with water.
The solvate form of Binimetinib may be prepared from conventional methods and the solvate includes but not limited to propylene glycol, ethylene glycol, ethanol, benzyl alcohol, IP A and the like.
Alternatively the Binimetinib may be obtained by treating Binimetinib containing organic acid in stoichiometric or non- stoichiometric level with polar solvent or aqueous polar solvent. The polar solvent includes, but not limited to, methanol, ethanol and the like.
In certain embodiments, the Binimetinib may be prepared by the process described herein below, in the present application. In another embodiment of the present invention relates to compound of Formula (2) and its use in preparation of Benzimidazole compounds like Binimetinib and/or Selumetinib.

Further embodiment of the invention relates to the compound of Formula (2) is prepared by treating compound of Formula- 1 with C1-C6 dialkoxymethane in the presence of an acid.

C1-C6 dialkoxymethane is selected from diethoxymethane and dimethoxy methane.
In embodiment, the acid used in imidazole formation is selected from the group consisting of an inorganic acid or an organic acid. The inorganic acid includes but not limited to hydrochloric acid and sulfuric acid. The organic acid includes but not limited to sulfonic acid derivatives such as methane sulfonic acid, p-toluene sulfonic acid, and benzene sulfonic acid; or carboxylic acid such as formic acid and acetic acid.
In an embodiment, the compound of Formula (2) is converted in to compound of Formula-3 using a brominating agent such as NBS optionally in the presence of an acid like p-TsOH. The reaction may be effected in the presence of suitable organic solvent. For that matter any solvent that does not affect the course of the reaction can be used.

Formula (3) wherein Xi is bromo or chloro.
In a non-limiting embodiment, the reaction is represented by below scheme:

In an embodiment, the compound of Formula (3), when R is OCH2CH3 is converted to Binimetinib by first converting the ethyl ester compound to acid compound as represented by below scheme, and then converting acid compound to Binimetinib according to the at least one of the suitable known processes in the art as described above.

In another embodiment, the invention relates to Benzimidazole compounds like Binimetinib and/or Selumetinib, prepared by using Formula (4)

In an embodiment, the process of preparation of Binimetinib or Selumetinib comprising steps of:
a) treating the compound of Formula (3), wherein R is selected from group consisting of hydroxy, C1-C6 alkoxy, arylalkyloxy such as benzyloxy; with hydroxylamine or its salt such as hydroxylamine hydrochloride to obtain compound of Formula (4); and
b) converting the compound of Formula (4) into binimitinib.
In an embodiment, the conversion of compound of Formula-4 into Binimetinib or Selumetinib according to step (b) is carried out by reacting the compound of Formula-4 with
optionally protected ethylene glycol preferably under Mitsunobu conditions followed by removing the protecting groups if any in polar aprotic solvent.
In an embodiment, the suitable reagent for the said Mitsunobu conditions includes triphenylphosphine, trialkylphosphines or phosphites, diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), dibenzyl azodicarboxylate (DBAD), N,N,N',N'- tetramethylazodicarbonamide (TMAD), and dipiperidyl azodicarboxylate (DPAD), tosyl chloride, mesyl chloride and mixture thereof.
In an embodiment, the polar aprotic solvent is selected from the group consisting of tetrahydrofuran, acetonitrile, dioxane, dimethyl sulfoxide, dimethylformamide, dimethylacetamide, N-methylpyrrolidon, dimethoxymethane, methylene chloride, ethyl acetate and mixtures thereof.
In a non-limiting illustrative embodiment, the said reaction is represented by below scheme for Binimitinib:

Alternatively, Binimetinib or Selumetinib is prepared by treating compound of Formula-3, wherein R is C1-C6 alkoxy, preferably ethoxy or methoxy or tert-butoxy with O- (2-hydroxyethyl) hydroxylamine with or without formation of acid compound as intermediate. The said reaction is shown below:

SCHEME (A)
Binimetinib or Selumetinib is also prepared by treating compound of Formula (3), wherein R is C1-C6 alkoxy; preferably ethoxy or methoxy or tert-butoxy with 0-(2- hydroxyethyl) hydroxyl amine compound protected with PG, without formation of acid compound.
The above reaction may proceed in solvent such as xylene, toluene, tetrahydrofuran, 2-methyl tetrahydrofuran, NMP, sulfolane, N,N-dimethylimidazolinone, cyclopentylmethyl ether, DMF, DMAc, NMP, tetramethylurea, tetramethyl guanidine, and pentamethyl guanidine.
The above reaction may proceed with or without use of base. The base can be organic or inorganic base.
The above reaction may proceed preferably under close conditions such as in autoclave under pressure at higher temperature.
In a non-limiting illustrative embodiment, the said reaction is shown as below:

The protecting group PG is selected from alkyl groups, such as tertiary alkyls (e.g., tertiary C4-C7 alkyls such as t-butyl or tertiary amyl); alkenyl groups; tertiary aryl-alkyl
groups, such as 1 -methyl- 1-phenylethyl (cumyl) or triphenylmethyl (trityl); groups that result in acetals, such as methoxymethyl, 1-ethoxyethyl, 2-tetrahydropyranyl or 2- tetrahydrofuranyl; and silyl groups, such as trimethylsilyl, triethylsilyl or tert-butyl- dimethyldilyl.
The protecting group PG may be removed using any suitable deprotecting agent and deprotection conditions and will vary with the choice of protecting group. The suitable deprotection agents may include, but are not limited to, an aqueous acid such as phosphoric acid, hydrochloric acid or sulfuric acid, non-aqueous acids such as hydrogen chloride acid.
In certain embodiments, the Binimetinib may be prepared by the process described herein above or below, in the present application.
In another embodiment, the invention relates to a process for preparation of Binimetinib comprising steps of: a. converting compound of Formula (A) to compound of Formula (B), wherein the reaction proceeds in the absence of solvent and use of quinolone or pyridine as catalyst poisoning agent;

F ormula A Formula B Formula (A) Formula (B) b. converting compound of Formula (B) to compound of Formula (C), wherein the obtained compound of Formula (C) is purified using formic acid and acetone;

Formula C Formula (C)
c. converting compound of Formula (C) to compound of Formula (D) using 2- [(aminooxy)ethoxy] ethane, wherein the obtained compound is purified using ethanol and n-heptane;

F ormula D Formula (D)
d. converting compound of Formula (D) to Binimetinib.
In certain embodiments, Binimetinib used herein is prepared by any conventional methods or by following the processes given as shown by following scheme or by following the examples described herein.

SCHEME - B
In certain embodiments, the Binimetinib may be prepared by the process described herein above or by any conventional process and further purified as below.
The DIO, D50, and D90 values are useful ways for indicating a particle size distribution. D90 refers to the value for the particle size for which at least 90 volume percent of the particles have a size smaller than the value. Likewise D50 and D10 refer to the values for the particle size for which 50 volume percent, and 10 volume percent, of the particles have a size smaller than the value. Methods for determining D10, D50 and D90 include laser diffraction, such as using Malvern Instruments Ltd. (of Malvern) equipment.
Binimetinib obtained according to the process described herein or after performing size reduction operations such as milling or micronization, has a particle size distribution pattern of D10 less than or equal to about 150 pm, D50 less than or equal to about 300 pm, and D90 less than or equal to about 500 pm. There is no specific lower limit for any of the D values.
The invention is further exemplified by the following non-limiting examples, which are illustrative representing the preferred modes of carrying out the invention. The invention's scope is not limited to these specific embodiments only but should be read in conjunction with what is disclosed anywhere else in the specification together with those information and knowledge which are within the general understanding of the person skilled in the art.
Examples:
Example 1: Preparation of 4-amino-3-fluoro-2-(2-fluoro-phenylamino)-5-nitro-benzoic acid methyl ester
Mixture of 4-amino-2,3-difluoro-5-nitro-benzoic acid methyl ester (50 g), 2-fluoro- phenylamine (239.31 g) in xylenes (500 mL), was heated at 150-155°C till completion of reaction. After completion of reaction, the reaction mixture was cooled to room temperature and the precipitated product was filtered and then dried under vacuum at 50-55°C to obtain the title compound (54.5 g).
Example 2: Preparation of 4, 5-diamino-3-fluoro-2-(2-fluoro-phenylamino)-benzoic acid methyl ester (Formula-1, X=F, and R=OCH3)
4-Amino-3-fluoro-2-(2-fluoro-phenylamino)-5-nitro-benzoic acid methyl ester (10 g) in methanol (150 mL): THF (150 mL) mixture was hydrogenated using Pd/C (0.5 g) at 50-55°C till completion of reaction. After completion of reaction, hydrogen pressure was released and the reaction mixture was cooled to room temperature, filtered through hyflo to remove the
catalyst. The filtered mass was distilled under vacuum at 45-50°C to obtain the title compound (8.3 g).
Example 3: Preparation of methyl 4-fluoro-5-[(2-fluorophenyl)amino]-l-methyl-lH- benzimidazole-6-carboxylate (Formula-2, X=F, and R=OCH )
The solution of 4,5-diamino-3-fluoro-2-(2-fluoro-phenylamino)-benzoic acid methyl ester (1.8 g), diethoxymethane (1.83 g), p-toluene sulfonic acid monohydrate (1.28 g), water (0.01 mL) and acetonitrile (18 mL) were heated at 60-65°C till completion of reaction. After completion of reaction, pyridine was added into the reaction mixture and cooled to room temperature. The reaction mixture was stirred for 40-5 hours; the precipitated solid was filtered, and dried under vacuum at 45-50°c to obtain the title compound (0.8 g).
Example 4: Preparation of 6-(4-bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole-5-carboxylic acid methyl ester (Formula-3, X=F, Xi=Br and R=OCH )
Mixture of methyl 4-fluoro-5-[(2-fluorophenyl)amino]-l-methyl-lH-benzimidazole-6- carboxylate (0.6), tetrahydrofuran (20 mL) and methanol (20 mL) cooled to -78° C under N2 atmosphere p-toluene sulfonic acid monohydrate (0.54 g) was added followed by N- bromosuccinimide (0.37 g). After 10 minutes, the reaction mixture was warmed to room temperature. The reaction mixture was stirred till completion of reaction, followed by quenching with the addition of aqueous saturated sodium bisulfite solution and diluted with ethyl acetate and water. Layers were separated and the aqueous layer re-extracted using ethyl acetate. The combined organic layers were washed with brine solution and separated out both layers. Organic layer distilled out under vacuum at 45-50°C to obtain solid residue. The resulting solid residue was triturated with methylene chloride, filtered, and dried to obtain the title compound (0.5 g).
Example 6: Preparation of 6-(4-Bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole-5 -carboxylic acid
To the solution of 6-(4-Bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole-5-carboxylic acid methyl ester (0.3 g) in THF (10 mL)/water (7.8 mL), 1M aqueous solution of NaOH (0.12 g) was added and stirred at room temperature. After completion of reaction, the solvent was distilled under vacuum at 50-55°C and cooled to room temperature. The reaction mixture, pH was adjusted to 1-2 by the addition of 1.0 M
aqueous HC1 and the product was extracted with 1:1 tetrahydrofuran: ethyl acetate mixture. Organic layer distilled under vacuum at 45-50°C to obtain the title compound (0.2 g).
Example 6A: Preparation of 6-(4-Bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole-5-carboxylic acid
To the solution of 6-(4-Bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole-5-carboxylic acid ethyl ester (10 g) (said compound is prepared by following the example given in example 1-5 or by any other conventional methods) in THF (100mL)/water (100 mL), 1M aqueous solution of NaOH ( 20 g) was added and stirred at room temperature. After completion of reaction, the solvent was distilled under vacuum at 50-55°C and cooled to room temperature. The reaction mixture, pH was adjusted to 1-2 by the addition of 2.0 M aqueous HC1 and the product was extracted with 1:1 tetrahydrofuran: ethyl acetate mixture. Organic layer distilled under vacuum at 45-50°C to obtain the title compound.
Example 7: Preparation of 6-(4-bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole -5-carboxylic acid (2-vinyloxy-ethoxy)-amide
6-(4-Bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (0.6), 0-(2-vinyloxy-ethyl)-hydroxylamine (0.243 g), HOBt (0.27 g), triethylamine (0.07 g) and EDCI (0.39 g) were dissolved in dimethylformamide (20 mL) and stirred at room temperature till completion of reaction. After the completion of reaction, the reaction mixture was added with ethyl acetate, washed with water, and followed by saturated potassium carbonate aqueous solution. The organic layer washed with aqueous ammonium chloride solution and finally with aqueous sodium chloride solution. The organic layer was distilled under vacuum at 45-50°C to obtain the title compound (0.6 g).
Example 8: Preparation of Binimetinib
To the solution of 6-(4-bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole -5-carboxylic acid (2-vinyloxy-ethoxy)-amide (0.6 g) in ethanol (13.76 mL), hydrochloric acid (6.42 mL, 1.0 M aqueous solution) was added and stirred for 24 hours. After the completion of reaction, the solvent was concentrated till dryness under vacuum at 45-50°C. Ethyl acetate: tetrahydrofuran was added to the residue and washed with aqueous saturated potassium carbonate solution. Aqueous layer re-extracted out using ethyl acetate:
tetrahydrofuran mixture. The organic layer was distilled under vacuum at 45-50°C and isolated using ethyl acetate to obtain the title compound (0.2 g).
Example 9: Preparation of 4-amino-2-[(4-bromo-2-fluorophenyl)amino]-3-fluoro-5- nitrobenzoic acid.
4-amino-2, 3-difluoro-5-nitrobenzoic acid (100 g) was added to the N,N-diisopropyl ethylamine (100 g) and 4-bromo-2-fluoroaniline (262 g) at room temperature, and the temperature of the reaction mixture was raised to 107+5 °C and stirred till the completion of the reaction. The reaction mixture temperature was cooled to 25±5°C, process water was added, then the reaction mixture pH was adjusted 4.0 to 4.5 using acetic acid, stirred, dried and filtered. The obtained wet material was added to acetonitrile at 35±5°C, stirred, filtered and dried under vacuum to obtain title compound. (Yield 87 %).
Purification of 4-amino-2,3-difluoro-5-nitrobenzoic acid (100 g): 4-amino-2.3-difluoro-5- nitrobenzoic acid was added to water at 25±3°C and stirred. pH of the reaction mixture was adjusted to 1-2 using cone hydrochloric acid at 20-30°C, stirred, filtered and dried. To the dried material, 1 -methyl pyrrolidone was added and heated to 60+3 °C, stirred and cooled to 25+3 °C. To this water was added, stirred and filtered the solid. Obtained wet solid was stirred again with water for 3 hours, then filtered and dried to yield title compound. (~ 80 %)
Example 10: Preparation of 4,5-diamino-2-[(4-bromo-2-fluorophenyl)amino]-3-fluoro- benzoic acid
The solution of methanol and tetrahydrofuran in hydrogenator, 4-amino-2-[(4-bromo-2- fluorophenyl)amino]-3-fluoro-5-nitrobenzoic acid (100 g) was added at room temperature quinoline (33.26 g), and 5% Pt/C (50% wet) in methanol was added to the reaction mixture in hydrogenator. 1-1.5 Kg/cm2 hydrogen gas pressure was applied and stirred till the completion of reaction. The reaction mixture was filtered and concentrated. Methanol was added to the obtained residue and stirred at 45+5 °C, cooled, filtered and dried to obtain title compound. (Yield -86%).
Example 11: Preparation of 4,5-diamino-2-[(4-bromo-2-fluorophenyl)amino]-3-fluoro- benzoic acid
4-amino-2-[(4-bromo-2-fluorophenyl)amino]-3-fluoro-5-nitrobenzoic acid (100 g), DMF (300 g) and methanol (150 g) were mixed in to reaction vessel and cooled to 17+5°C. Zinc
dust (58.95 g) was added and after that cone hydrochloride acid (93.53 mL) was added drop wise below 30°C to the reaction mixture. Stir the reaction mixture for till completion. After completion of the reaction water (500 mL) was added and stirred for one hour. Filtered the solid and washed with water. Wet cake was again stirred with water and pH was adjusted to
6.5-7.5 using aq. Sodium bicarbonate solution (500 ml). Stirred for half an hour and filtered the solid and dried. (Yield -87.0%)
Example 12: Preparation of 6-(4-bromo-2-fluoro-phenylamino)-7-fluoro-3H- benzoimidazole-5-carboxylic acid
To acetonitrile, 4,5-diamino-2-[(4-bromo-2-fluorophenyl)amino]-3-fluoro-benzoic acid (lOOg), para toluenesulphonic acid (69.04 g) and water were added and stirred at room temperature. To this reaction mixture diethoxymethane (63.98 g) was added, temperature was raised to 60+3 °C and stirred till the completion of reaction. The reaction mixture was cooled to RT, stirred and filtered. Water was added to the obtained wet material; pH was adjusted to
7.5-8.5 using aq. ammonia solution, stirred at room temperature and filtered. Obtained wet material was added to Formic acid and heated to 65+3 °C, acetone was added to the reaction material, stirred at 65+3 °C, cooled to room temperature, stirred, filtered and dried to obtain title compound. (Yield: -56 %).
Example 13: Preparation of 6-(4-bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H- benzoimidazole -5-carboxylic acid (2-vinyloxy-ethoxy)-amide
6-(4-bromo-2-fluoro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (100 g), 0-(2-vinyloxy-ethyl)-hydroxylamine (40.47 g), HOBt (45.99 g), triethylamine (12.18 g) and EDC.HC1 (65.04 g) were dissolved in dimethylformamide (20 mL) and stirred at room temperature till completion of reaction. The reaction mixture was cooled to 17+3°C, water was added, pH was adjusted between pH 7-7.5 using 7% sodium bicarbonate solution. Ethyl acetate was added into the reaction mixture and layers were separated. The organic layer was concentrated. Ethyl acetate and cyclohexane was added to obtained solid and stirred at 52+3 °C, cooled to room temperature, filtered and dried. To the ethanol (denatured with 0.5% cyclohexane), the obtained wet cake was charged at room temperature, the temperature was raised to 65+3°C. n-heptane was added to the reaction mixture, cooled to room temperature, stirred, filtered and dried under vacuum to obtain title compound. (Yield: -61%).
Example 14: Preparation of Binimetinib
6-(4-bromo-2-fluoro-phenylamino)-7 -fluoro-3-methyl-3H-benzoimidazole -5-carboxylic acid (2-vinyloxy-ethoxy)-amide (100 g) was charged to in ethanol (100 mL) at room temperature, cooled to 0+3 °C. Cone. HC1 (30% aq. solution) was added to the reaction mixture at 0+3 °C and the temperature was raised to 15+3 °C and stirred for completion of reaction. To the obtained reaction material, water was added at below 30 °C and pH was adjusted to 7-7.5 using sodium bicarbonate solution. Ethyl acetate was added to the reaction mass, stirred at room temperature, layers were separated, organic layer was concentrated. Ethyl acetate was added to the obtained solid and stirred at 52+3 °C for 30 min. Filtered and dried to obtain Binimetinib. (Yield: -90 %).
Example 15: Preparation of Binimetinib.
Binimetinib (100 g) was stirred in acetic acid (400 mL) at room temperature. Toluene (1250 mL) was added to the reaction mass and stirred for 12 hours at room temperature. Filtered the solid and washed with toluene. The material was dried in VTD for 8 hours at 50°C. (acetic acid content: 7 %).
Example 16: Preparation of Binimetinib.
Binimetinib obtained in above step (Example 15) was added to water at room temperature; temperature was raised to 65+3°C and stirred for 2 hours. The solid was filtered at 65°C and washed with hot water. The material was dried in VTD for 8 hours at 50°C to obtain pure Binimetinib. (Yield: -80 %).
Example 17: Preparation of Binimetinib.
Binimetinib diacetate (Form B) solvate was added to water at room temperature; temperature was raised to 65+3 °C and stirred for about 1-3 hours. The solid was filtered at ambient temperature and washed with water at ambient temperature. The material was dried in VTD for 8 hours at 50°C to obtain pure Binimetinib. (Yield: -80 %).
Example 17: Preparation of Binimetinib.
Binimetinib alcohol solvate disclosed herein was added to water at room temperature; temperature was raised to 70°C and stirred for about 3 hours. The solid was filtered at
ambient temperature and washed with water at ambient temperature. The material was dried in VTD for 8 hours at 50°C to obtain pure Binimetinib. (Yield: ~80 %).
Claims
1. A process of preparation of Binimetinib comprises treating Binimetinib solvate with water.
2. The process as claimed in claim 1, wherein the Binimetinib solvate is selected from organic acid solvate of Binimetinib, propylene glycol solvate of Binimetinib, IPA solvate of Binimetinib, or benzyl alcohol solvate of Binimetinib.
3. The process as claimed in claim 2, wherein the organic acid in Binimetinib organic acid solvate is selected from formic acid, acetic acid and propionic acid.
4. The process as claimed in claim 1 wherein the said treatment of Binimetinib organic acid solvate with water is carried out at a temperature in the range of 20 to 80°C.
5. The process as claimed in claim 1, further comprising isolating Binimetinib substantially free of solvate.
6. The process as claimed in claim 2, wherein Binimetinib containing organic acid is prepared by steps of:
a. treating Binimetinib with organic acid;
b. adding organic solvent to step (a) reaction mass followed by stirring; and c. isolating the obtained solid .
7. The process as claimed in claim 6, wherein the organic acid in Binimetinib organic acid solvate is either stoichiometric level or non-stochiometric level.
8. The process as claimed in claim 6, wherein the solvent used in step (b) is selected from toluene, anisole, MTBE, diisopropyl ether, methyl THF and the like.
9. The process according to claim 6, wherein the process further comprises step of isolating Binimetinib containing organic acid in stoichiometric or non- stoichiometric level and wherein the amount of organic acid in the isolated Binimetinib is in stoichiometric level 0.5, 1, 1.5, 2 mole equivalent or in non-stoichiometric level by filtration.
10. A process of preparation of Binimetinib or Selumetinib comprising steps of:
a. treating a compound of Formula (1) with C1-C6 dialkoxymethane in the presence of an acid to obtain a compound of Formula (2);
 wherein X is halogen.
wherein X is halogen.
b. converting the compound Formula (2) to a compound Formula-3, using an halogenating agent selected from NBS or NCS;

c. treating the compound of Formula (3) with hydroxylamine or its salt to obtain compound of Formula (4); and

11. The process as claimed in claim 10, wherein the C1-C6 dialkoxymethane is selected from diethoxymethane or dimethoxymethane.
12. The process as claimed in claim 10, wherein the acid is inorganic acid selected from hydrochloric acid or sulfuric acid; or organic acid selected from methane sulfonic acid, p-toluene sulfonic acid, benzene sulfonic acid, formic acid or acetic acid.
13. The process as claimed in claim 10, wherein step (d) is carried out by reacting the compound of Formula-4 with optionally protected ethylene glycol preferably under mitsunobu conditions followed by removing the protecting groups if any in polar aprotic solvent.
14. The process as claimed in claim 13, wherein the mitsunobu conditions is created by using mitsunobu reagents selected from triphenylphosphine, trialkylphosphines or phosphites, diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), dibenzyl azodicarboxylate (DBAD), N,N,N',N'-tetramethylazodicarbonamide (TMAD), and dipiperidyl azodicarboxylate (DPAD), tosyl chloride, mesyl chloride and mixture thereof.
15. A compound of Formula (2),

16. A compound of Formula (4)

Wherein X is halogen and Xi is chloro or bromo.
17. A process for preparation of Binimetinib comprising steps of:
rb converting compound of Formula (A) to compound of Formula (B);



Formula (D) d. converting compound of Formula (D) to Binimetinib.
18. The process as claimed in claim 17, further comprising carrying out step (a) in the presence of catalytic amount of organic base selected from quinolone or pyridine.
19. The process as claimed in claim 17, further comprising carrying out step (b) in the presence of an acid selcted from PTSA.
20. The process as claimed in claim 17, further comprising purification of compound of
Formula (C ) using formic acid and acetone;
21. The process as claimed in claim 17, further comprising purification of compound of Formula (D) using ethanol and n-heptane.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201921015257 | 2019-04-16 | ||
| IN201921015257A IN201921015257A (en) | 2019-04-16 | 2019-04-16 | |
| IN202021001198 | 2020-01-10 | ||
| IN202021001198A IN202021001198A (en) | 2020-01-10 | 2020-04-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020212832A1 true WO2020212832A1 (en) | 2020-10-22 |
Family
ID=72837775
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2020/053489 WO2020212832A1 (en) | 2019-04-16 | 2020-04-14 | Process of preparation of benzimidazole compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2020212832A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112679438A (en) * | 2020-12-31 | 2021-04-20 | 武汉九州钰民医药科技有限公司 | Process for the preparation of semetinib |
| CN112759552A (en) * | 2020-12-31 | 2021-05-07 | 武汉九州钰民医药科技有限公司 | Synthesis method of semetinib |
| WO2023131976A1 (en) * | 2022-01-06 | 2023-07-13 | Msn Laboratories Private Limited, R&D Center | Improved process for the preparation of 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-n-(2hydroxyethoxy)-1-methyl-1h-benzimidazole-6-carboxamide |
| WO2024003942A1 (en) * | 2022-07-01 | 2024-01-04 | Natco Pharma Limited | An improved process for the preparation of selumetinib sulfate |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7777050B2 (en) * | 2002-03-13 | 2010-08-17 | Array Biopharma Inc. | N3 alkylated benzimidazole derivatives as MEK inhibitors |
| WO2014063024A1 (en) * | 2012-10-19 | 2014-04-24 | Novartis Ag | Preparation of and formulation comprising a mek inhibitor |
-
2020
- 2020-04-14 WO PCT/IB2020/053489 patent/WO2020212832A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7777050B2 (en) * | 2002-03-13 | 2010-08-17 | Array Biopharma Inc. | N3 alkylated benzimidazole derivatives as MEK inhibitors |
| WO2014063024A1 (en) * | 2012-10-19 | 2014-04-24 | Novartis Ag | Preparation of and formulation comprising a mek inhibitor |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112679438A (en) * | 2020-12-31 | 2021-04-20 | 武汉九州钰民医药科技有限公司 | Process for the preparation of semetinib |
| CN112759552A (en) * | 2020-12-31 | 2021-05-07 | 武汉九州钰民医药科技有限公司 | Synthesis method of semetinib |
| WO2023131976A1 (en) * | 2022-01-06 | 2023-07-13 | Msn Laboratories Private Limited, R&D Center | Improved process for the preparation of 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-n-(2hydroxyethoxy)-1-methyl-1h-benzimidazole-6-carboxamide |
| EP4460303A4 (en) * | 2022-01-06 | 2025-07-23 | Msn Laboratories Private Ltd R&D Center | Improved process for the preparation of 5-[(4-BROMO-2-FLUORPHENYL)AMINO]-4-FLUORO-N-(2HYDROXYETHOXY)-1-METHYL-1H-BENZIMIDAZOLE-6-CARBOXAMIDE |
| WO2024003942A1 (en) * | 2022-07-01 | 2024-01-04 | Natco Pharma Limited | An improved process for the preparation of selumetinib sulfate |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2020212832A1 (en) | Process of preparation of benzimidazole compounds | |
| TWI752307B (en) | Novel compound and method of preparing compound | |
| JP5269798B2 (en) | Trityl olmesartan medoxomil and method for producing olmesartan medoxomil | |
| EP2190804B1 (en) | Process and intermediates for preparing integrase inhibitors | |
| US8471039B2 (en) | Process for the preparation of indoline derivatives and their intermediates thereof | |
| EP0868423B1 (en) | Method for the synthesis of a benzimidazole compound | |
| EP1926705A2 (en) | Process for preparing valsartan | |
| EP2268634A2 (en) | Processes for the preparation of bosentan and related compounds using novel intermediates | |
| WO2011007368A2 (en) | An improved process for preparation of olmesartan | |
| WO2023137007A1 (en) | Processes for the preparation of 4,6,7-trifluoro-1h-indole-2-carboxylic acid | |
| US20040014983A1 (en) | Method for preparing benzisoxazole methane sulfonyl chloride and its amidation to form zonisamide | |
| WO2011033468A1 (en) | Process for the preparation of a crystalline form of lenalidomid | |
| JP2019507156A (en) | Process for producing 4-alkoxy-3-hydroxypicolinic acid | |
| JP2018090551A (en) | L-carnosine derivative or salt thereof, and method for producing L-carnosine or salt thereof | |
| US20240336603A1 (en) | Process for the preparation of a cyp11a1 inhibitor and intermediates thereof | |
| WO2012056468A1 (en) | A process for the preparation of bosentan | |
| CN109422767B (en) | Methoxycephalosporin intermediate, preparation method thereof and synthesis method of cefminox sodium | |
| HU200459B (en) | New process for producing 7-bromo-beta-carboline compounds | |
| US8093391B2 (en) | Process for the preparation of substantially pure palonosetron and its acid salts | |
| WO2017209035A1 (en) | Method for producing biphenyl benzimidazole derivative | |
| US20250263414A1 (en) | An improved process for the preparation of remimazolam | |
| JP2010526126A (en) | Method for producing valsartan | |
| WO2024084491A1 (en) | Process for synthesis of res metirom and its intermediates thereof | |
| EP4175645A1 (en) | Synthesis of molnupiravir by green chemistry | |
| JP2004536101A (en) | Improved synthesis of branched acyclic nucleosides. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20790845 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20790845 Country of ref document: EP Kind code of ref document: A1 |