WO2020008366A1 - Transdermal dosage form - Google Patents
Transdermal dosage form Download PDFInfo
- Publication number
- WO2020008366A1 WO2020008366A1 PCT/IB2019/055644 IB2019055644W WO2020008366A1 WO 2020008366 A1 WO2020008366 A1 WO 2020008366A1 IB 2019055644 W IB2019055644 W IB 2019055644W WO 2020008366 A1 WO2020008366 A1 WO 2020008366A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fentanyl
- naltrexone
- transdermal dosage
- dosage system
- less
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4468—Non condensed piperidines, e.g. piperocaine having a nitrogen directly attached in position 4, e.g. clebopride, fentanyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
- A61K9/7023—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms
- A61K9/703—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms characterised by shape or structure; Details concerning release liner or backing; Refillable patches; User-activated patches
- A61K9/7092—Transdermal patches having multiple drug layers or reservoirs, e.g. for obtaining a specific release pattern, or for combining different drugs
Definitions
- the present disclosure relates to transdermal dosage forms having reduced potential for abuse.
- the disclosure relates to a system of transdermal administration of an active agent or agonist for example fentanyl to a subject over an extended period of time, wherein when subject to abuse, the system is capable of providing an adverse agent : active agent release ratio sufficient to prevent or discourage the abuse of the active agent.
- the present disclosure further relates to tamper-resistant transdermal dosage forms comprising: an active agent and a barrier which separates an antagonist in the form of a salt from the antagonist in base form.
- Pain is the most frequent reported symptom and is a common clinical problem confronting the clinician.
- Opioids have long been recognized as one of the most effective treatments of pain whether for treating or preventing cancer pain, central pain, myocardial infarction pain, pancreatic pain, colic pain, post-operative pain, headache pain, muscle pain, bone pain, and pain associated with intensive care.
- the US societal costs of prescription opioid abuse were estimated at more than $50 billion in 2007 and have only grown since.
- Transdermal dosage forms offer a favorable route of administration by providing a method of administering sustained release of a drug for an extended period of time, while increasing patient compliance and decreasing extreme peaks and troughs in blood plasma.
- these dosage forms also contain large amounts of active agent and therefore also have a high potential for abuse.
- U.S. Pat. No. 5,236,714 discloses a dosage form comprising an abusable substance formulated with an antagonist for the abusable substance.
- U.S. Pat. No. 5,149,538 discloses a transdermal patch comprising an opioid and an antagonist for the opioid that is releasable upon ingestion or solvent immersion, wherein the two reservoirs are separated by an impermeable barrier.
- U.S. Pat. Nos. 8,747,889, 8,790,689, 7,182,955 each discloses a transdermal patch system comprising an opioid and an antagonist with different methods for the antagonist to leave the patch.
- US Pub. No. 20040126323 discloses a transdermal system with an opioid layer and an antagonist layer comprising antagonist salt and base form, both with or without a barrier separating the opioid and antagonist layer.
- transdermal dosage forms that are effective for preventing abuse yet useful for delivering an active agent, such as an opioid or a pharmaceutically acceptable salt thereof.
- the present disclosure relates to an abuse deterrent transdermal dosage form wherein when contacted with skin, allows for the transdermal administration of an active agent, such as an opioid, but either (a) allows for the transdermal administration of only an amount of an antagonist that is ineffective for inhibiting the effect of the active agent, or (b) does not allow for the transdermal administration of the antagonist.
- an active agent such as an opioid
- the transdermal dosage form of the disclosure is used to deliver an active agent via a route other than transdermal, such as buccal, nasal, oral, parenteral, rectal and/or vaginal, or if the transdermal dosage form is subjected to abuse or misuse, then the antagonist inhibits the effect of the active agent.
- a transdermal dosage form having reduced potential for abuse comprising an active agent and more than one antagonist reservoir or source.
- the transdermal dosage comprises a first antagonist component comprising an active agent and a first antagonist of the active agent, and a second antagonist component comprising a second antagonist of the active agent.
- the present disclosure is directed to an abuse deterrent transdermal dosage forms wherein the first and second antagonist are separated by a barrier.
- the first antagonist is in the form of a pharmaceutically acceptable salt and the second antagonist is in the form of a free base.
- the first antagonist component has a proximal and distal surface; the second antagonist component is disposed distal to the first antagonist component and the barrier is interposed between the first and second antagonist components.
- the present disclosure is directed to transdermal dosage forms wherein the first antagonist component comprises a homogenous mixture of the active agent and the first antagonist or alternatively, wherein the active agent and the first antagonist are separated by one or more spacers.
- the disclosure relates to a transdermal system for administering an active agent through the skin, the system having a reduced potential for abuse, comprising:
- a first antagonist component comprising a first antagonist salt and an active agent in base form, wherein the active agent may be for example fentanyl or an analog thereof and the analog is selected from the group consisting of alfentanil, lofentanil, remifentanil, sufentanil and trefentanil;
- a second antagonist component comprising a second antagonist in base form, optionally in amorphous base form, the antagonist being releasable from system upon being ingested or substantially immersed in a solvent, and further wherein the antagonist is selected from the group consisting of naltrexone, methylnaltrexone, naloxone, nalbuphine, nalorphine, nalorphine dinicotinate, nalmefene, nadide, levallorphan, cyclozocine and pharmaceutically acceptable salts thereof; and
- a barrier layer separating said first and second antagonist components, said barrier layer being substantially impermeable to said active agents and/or excipients, wherein the release of the antagonist from the system when used transdermally is such that levels are sufficiently low that the active agent's effect is maintained for more than about two days or about three days; and the system provides release of the antagonist at a rate sufficient to provide an abuse limiting release rate ratio of the antagonist to the active agent when the dosage form is subject to abuse, e.g., upon transmucosal applications or substantial immersion of the system in the solvent.
- the disclosure is further directed to a kit for treating pain in a patient, comprising: a) the transdermal-delivery device as disclosed above; and b) a printed set of instructions directing the use of the transdermal dosage form to treat pain.
- the disclosure is further directed to methods for treating or preventing pain in an animal comprising contacting the skin of an animal in need thereof with any one or more of the transdermal dosage forms described above.
- a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a C max less than 50% the C max of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when administered buccally.
- the present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a C max less than 50% the C max of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when chewed.
- the present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a AUCo-t less than 50% the AUCo- t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when chewed.
- the present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.7 for the first three hours after buccal administration.
- the present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.7 for the first three hours after being chewed.
- the present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a AUCo-t less than 50% the AUCo-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when administered buccally.
- the transdermal dosage form will inhibit the euphoric effect of an opioid if the device is used other than transdermally whether before or after the device is used by an animal or human for treating or preventing pain.
- the present disclosure relates to a method of treating pain in opioid-tolerant human patients in need of continuous, "around-the-clock," long-term opioid treatment by administering to a patient in need thereof, a transdermal dosage system comprising fentanyl and naltrexone.
- the long-term treatment can be for at least 12 weeks.
- the present disclosure also relates to a transdermal dosage system for administering fentanyl to a human, the system providing for the safe release of fentanyl after exposure to various extrinsic factors such as, for example, heat sources, exercise and/or showering.
- the transdermal dosage system provides for a fentanyl Cio of 110% or more and/or 180% or less, after exposure to an extrinsic factor.
- the transdermal dosage system provides for a naltrexone Ci 0 of 105% or more and/or 180% or less, after exposure to an extrinsic factor.
- the transdermal dosage system provides for a fentanyl AUCio of 110% or more and/or 250% or less, after exposure to an extrinsic factor.
- the transdermal dosage system provides for a naltrexone AUCio of 105% or more and/or 250% or less, after exposure to an extrinsic factor.
- the transdermal dosage system provides for a transformed fentanyl Cio to naltrexone Cio ratio and/or a transformed fentanyl AUCio to naltrexone AUCio ratio of 1:1 or less after exposure to an extrinsic factor.
- the transdermal dosage system provides for a fentanyl Cio that is 80% or less than the fentanyl Cio and/or the fentanyl AUCio of a reference fentanyl transdermal dosage system after exposure to an extrinsic factor.
- the disclosure is further directed to methods for reducing or preventing misuse of any one or more transdermal patches de scribed above.
- FIG. 1 is a schematic cross-section of a transdermal dosage system of the disclosure.
- FIG. 2 is a schematic cross-section of another transdermal dosage system of the disclosure.
- FIG. 3 is a schematic cross-section of a prototype of Example 1.
- FIG. 4 is a schematic cross-section of a prototype of Example 2.
- FIGs. 5-9 are manufacturing process steps for patches B1 and B2.
- FIG. 10 is a line graph showing in vitro cumulative delivery per cm 2 of Patch B2 versus the Duragesic ® patch.
- FIGs. 11 and 12 are line graphs showing the cumulative [pg/patch] and skin flux profile [pg/patch/hr] of Patch B2 versus the Duragesic ® patch.
- FIG. 13 is a line graph of showing in vitro delivery of a Patch B2 24 cm 2 100 mcg/h central strip versus a 42 cm 2 100 mcg/h Duragesic ® patch.
- FIG. 14 is a line graph showing the in vitro fentanyl skin flux [pg/patch/hr] from Patch B2 24 cm 2 100 mcg/h versus a 100 mcg/h Duragesic” patch.
- FIG. 15 is a line graph showing in vitro results for patch B1 (solid line), patch A1 (dotted line;— ⁇ — ), and a Duragesic ® patch (dotted line; ⁇ ⁇ ).
- FIG. 16 is a schematic of an in vitro permeation study set-up.
- FIG. 17 is a line graph showing the transbuccal delivery rate of fentanyl from Patch B2 as compared to the Duragesic ® patch.
- FIG. 18 is a bar graph showing the extraction of fentanyl and naltrexone from cut Patch B2 (14.06 cm 2 ; 25 mcg/h) at room temperature using water, ethanol, phosphate buffer, vinegar, and artificial saliva.
- FIG. 19 is a bar graph showing the extraction of fentanyl and naltrexone from cut Patch B2 (14.06 cm 2 ; 25 mcg/h) at 70°C using water, ethanol, phosphate buffer, vinegar, and artificial saliva.
- transdermal dosage form and “dosage form”, as used herein, refer to any dosage form that, when contacted with a patient's skin for a sufficient period of time, can transdermally deliver an effective amount of any biologically active agent, such as a pharmaceutical agent, e.g., an opioid, through the patient's skin whether the type of transdermal dosage type is reservoir-type, polymer-matrix-type, drug-in-adhesive-type, or other.
- transmucosal refers to buccal, nasally, sublingual, topical, rectal, and/or vaginal.
- uccal administration refers to a topical route of administration by which a composition is held or applied to the buccal area of the inner cheek.
- chew or “chewed” refers to a method of administration of a composition whereby said composition is chewed over a pre-defined period of time in the oral cavity without being swallowed.
- a "patient” or “animal” or “human” is a mammal, and includes, but is not limited to, a cow, monkey, horse, sheep, pig, chicken, turkey, quail, cat, dog, mouse, rat, rabbit, and guinea pig. In some embodiments, the "patient” or “animal” is a human.
- abuse resistant and “abuse deterrent” are synonymous and shall mean any transdermal dosage form that when misused, inhibits or deters the abuser from achieving the non-therapeutic effects sought from misuse of the composition, formulation or dosage form, such as opioid induced euphoria.
- Abuse or misuse shall mean any means including but not limited to being administered buccally, nasally, sublingually, parenterally, rectally, and/or vaginally to an animal.
- active agent refers to a pharmaceutical agent, therapeutic agent, drug, and/or agonist that causes a biological effect when absorbed in sufficient quantity into the blood stream of a patient.
- the active agent of the present disclosure may be any drug substance that is capable of being abused.
- Many drugs have a potential for abuse, and include, for example, narcotics, such as morphine, fentanyl, codeine, sufentanil, and oxycodone;
- psychostimulants such as amphetamine, methamphetamine, and methylphenidate; methoxy substituted amphetamines, such as 3,4-methylenedioxymethamphetamine (MDMA); and
- benzodiazepines such as diazepam, oxazepam, and lorazepam.
- examples include but are not limited to: alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, buprenorphine, butorphanol, clonitazene, codeine, desomorphine, dextromoramide, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dihydromorphone, dihydroisomorphine,
- the active agent is a narcotic such as fentanyl, alfentanil, carfentanil, lofentanil, remifentanil, sufentanil, trefentanil, and the like. In further embodiments, the active is fentanyl.
- fentanyl refers to the chemical compound N-Phenyl-N-(l-(2- phenylethyl)-4-piperidinyl) propanamide as either free base or a pharmaceutically acceptable salt. In some embodiments, the fentanyl is in the form of its free base.
- naltrexone refers to the chemical compound morphinan-6- one, 17 (cyclopropylmethyl) 4,5-epoxy3,14-dihydroxy-(5ct) as free base, co-crystal, or a pharmaceutically acceptable salt.
- two forms of naltrexone are present in the transdermal dosage form.
- the naltrexone is in the form of its free base.
- the naltrexone is in the form of its HCI salt.
- biological effect refers to a physical reaction in a patient.
- the effect is analgesic, euphoria, respiratory, anti- depressive, or combinations thereof.
- the phrase "adverse agent” or “antagonist” refers to a pharmaceutical agent, drug, and/or antagonist that partially or completely prevents, negates, diminishes, delays or reverses at least one biological effect of the active agent present in the dosage form, e.g. euphoric effect, or produces one or more unpleasant physiological reactions, e.g., vomiting, nausea, diarrhea, bad taste, when absorbed in sufficient amount into the blood stream of a patient.
- an opioid agonist is used as the active agent in the dosage form of the present disclosure, an opioid antagonist can be used as the adverse agent.
- opioid or "opioid agonist” refer to an active agent which exhibits opium- or morphine-like properties when absorbed in sufficient amounts into the bloodstream of a patient.
- Opioid agonists bind, optionally stereo-specifically, to any one or more of several subspecies of opioid receptors and produce agonist activity.
- opioid antagonist refers to an adverse agent that either partially or completely prevents, negates, diminishes, delays or reverses at least one biological effect of an opioid agonist, e.g., euphoric effect, when absorbed in sufficient amounts into the blood stream of a patient.
- the phrase "pharmaceutically acceptable salt,” as used herein, is a salt formed from an acid and the basic nitrogen group of an opioid.
- the salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucoronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p- toluenesulfonate, glubionate and pamoate (i.e., l,l'-
- Suitable bases include, but are not limited to, hydroxides of alkali metals such as sodium, potassium, and lithium; hydroxides of alkaline earth metal such as calcium and magnesium; hydroxides of other metals, such as aluminum and zinc; ammonia, and organic amines, such as unsubstituted or hydroxy-substituted mono-, di-, or trialkylamines; dicyclohexylamine; tributyl amine; pyridine; N-methyl, N-ethylamine; diethylamine; triethylamine; mono-, bis-, or tris-(2-hydroxy-lower alkyl amines), such as mono-, bis-, or tris-(2-hydroxyethyl)amine, 2-hydroxy-tert-but
- the phrase "active agent" refers to the drug which action is needed, intended or desired as the result of administration of the transdermal dosage form of the present invention.
- the active agent may be in any form which provides the desired biological effect.
- the active agent may be utilized in any solid state form such as amorphic or polymorphic form of the active agent.
- the active agent is amorphous.
- the active agent is crystalline.
- the active agent includes the fentanyl polymorphs and amorphic forms described in US Patent Publication No. 2010/0076198. The term “active agent” therefore encompasses all amorphic forms or polymorphic forms existing under any possible crystal morphology.
- the active agent such as fentanyl is in the form of a base. In further embodiments, the active agent such as fentanyl base is in amorphous form when formulated with excipients in the final transdermal dosage form.
- the antagonist such as Naltrexone is in salt form such as HCI. In further embodiments, the antagonist such as Naltrexone salt is in micronized form. In further embodiments, the micronized Naltrexone salt is maintained in crystalline form within the transdermal dosage form. In further embodiments, the transdermal dosage form comprises crystalline ionic antagonist such as Naltrexone salt, admixed with povidone and silicone.
- the antagonist such as Naltrexone is in base form.
- the antagonist such as Naltrexone base is in amorphous form when formulated with excipients in the final transdermal dosage form.
- the transdermal dosage form comprises amorphous antagonist in base form such as Naltrexone base in povidone.
- the active agent or salt thereof also may be in the form of a prodrug.
- prodrugs may include, without limitation, esters, carbamates sulfate, oximes, sulfamites, carbonates and other conventional "pro-drug" forms, which, when administered in such form, convert to the active agent in vivo.
- the prodrugs are esters.
- proximal refers to the location of a component, when considered as a whole, at a position which is relatively near to a site for application of the transdermal dosage form.
- proximal surface refers to the surface of a component which, when considered as a whole, is relatively near to a site for application of the transdermal dosage form, as compared to other surfaces of the component.
- the proximal surface of a component can be either continuous or discontinuous.
- distal refers to the location of a component, when considered as a whole, at a position which is relatively distant from a site for application of the transdermal dosage form.
- distal surface refers to the surface of a component which, when considered as a whole, is relatively distant from a site for application of the transdermal dosage form, as compared to other surfaces of the component.
- the proximal surface of a component can be either continuous or discontinuous.
- a "spacer" is meant to include a channel, pore, orifice, opening, void, gap, hole, crack and/or slit which to some degree provides a distance between two components. It may be made up of air, inactive ingredient, a barrier material or other.
- a “strip” is a formulation comprising either an active agent, antagonist or both in any desired geometry and may be either continuous or discontinuous and may be disposed in a pattern.
- an "adhesive strip” is a formulation able to adhere to human skin and comprising one or more adhesives but substantially free of an active agent, antagonist, or both.
- the adhesive strip may have any desired geometry and may be either continuous or discontinuous and may be disposed in a pattern.
- the terms "commercially available fentanyl transdermal dosage system” or “reference fentanyl transdermal dosage system” refers to DuragesicTM (as commercially available in 2015) or a transdermally bioequivalent dosage form thereof, as determined by guidance from the U.S. Food and Drug Administration.
- the terms ""commercially available fentanyl transdermal dosage system” and “reference fentanyl transdermal dosage system” are used interchangeably herein.
- DuragesicTM fentanyl patch is used interchangeably with “DurogesicTM fentanyl patch”, “DuragesicTM” and “DurogesicTM” and refers to a fentanyl patch (Janssen Pharmaceuticals, Inc.).
- treatment of pain includes amelioration of pain or the cessation of pain in an animal.
- prevention of pain includes the avoidance of the onset of pain in an animal.
- dispersed refers to dispersed, mixed, and/or dissolved either homogenously and/or heterogeneously.
- the phrase "component” refers to a layer, a stratum, a coating, a sheet, a film, a deposit, a sediment, a residue, and/or a cover.
- An "antagonist component” is a component comprising an antagonist.
- An antagonist component may or may not additional comprise an active agent.
- An “antagonist salt component” is a component comprising an antagonist salt.
- An “antagonist base component” is a component comprising an antagonist base.
- a “first antagonist component” is a component comprising an active agent and an antagonist.
- the term “opposed” as used with reference to two surfaces of a component refers to two surfaces which are generally facing in opposite directions regardless of whether one or both of the two surfaces are planar and/or parallel to each other.
- porous medium and “porous material” are used interchangeably.
- a layer refers to a layer which contains one or more active or adverse agents.
- a layer is a layer of the transdermal dosage system.
- said layer forms part of a reservoir-type transdermal dosage form, in another embodiment said layer forms part of a polymer-matrix type transdermal dosage form and in yet another embodiment said layer forms part of a drug-in-adhesive type transdermal dosage form.
- Room temperature refers to a typical indoor temperature. In some embodiments, room temperature is about 15 to about 25°C. In further embodiments, room temperature is about 20 °C.
- resistant to transdermal absorption refers to the tendency of a compound to cross the epidermis layer. In some embodiments, resistant to transdermal absorption means that no amount of a compound discussed herein crosses the epidermis layer. In other embodiments, “resistant to transdermal absorption” includes a “biologically insignificant” or negligible amount.
- the phase “biologically significant” refers to an amount of an active agent or antagonist described herein which results in one or more intended effect in a subject after administration.
- the "biologically significant effect relates to a physiological symptom, an interaction between the active agent and/or antagonist and at least one component of the subject, among others.
- the biologically significant effect is the binding or lack thereof of the active agent or agonist to a relevant receptor in a patient. In still further embodiments, the biologically significant effect is physically observed. In other embodiments, the biologically significant effect physically observed is less than the full effect of the active agent.
- the phrase “biologically insignificant” refers to an amount of an active agent or antagonist described herein which results in no or less than an intended effect in a subject after administration.
- the "biologically insignificant" effect relates to a physiological symptom, an interaction between the active agent and/or antagonist and at least one component of the subject, among others.
- the biologically insignificant effect refers to the binding or lack thereof of the active agent or agonist to a relevant receptor in a patient.
- the biologically insignificant effect is not physically observed.
- Similar in effect when used for comparing one or more compound refers to its ability to have a similar biological result, PK profile, PD profile, or combinations thereof.
- transdermal dosage system is similar in effect to the DuragesicTM transdermal patch.
- Conformable describes the ability of the transdermal dosage system to adapt in shape to the skin of the patient.
- the patch is flexible. When applied as instructed, the transdermal dosage system moves as the skin of the patient moves. In some embodiments, the transdermal dosage system does not become displaced from the patient as the skin moves or shifts.
- bioequivalent means that two products, i.e., transdermal patches, are expected to be the same.
- bioequivalent means that, when two products are compared, there is no significant difference in the rate and/or extent to which the active agent becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study.”
- transdermal bioequivalence to fentanyl may be determined as specified by the Food and Drug Administration using (FDA) bioequivalent with pharmacokinetic endpoints, adhesion studies, skin irritation studies, skin sensitization studies, or combinations thereof. See, e.g., the "Draft Guidance on Fentanyl", recommended Oct.
- the transdermal dosage system parallels the FDA PK standard of about 90% Cl compared to the DuragesicTM transdermal patch.
- the transdermal dosage system has a mean cumulative adhesion score of less than or equal to 0 as defined in the "Draft Guidance on Fentanyl” provided by the FDA and discussed above.
- the transdermal dosage system has a mean cumulative irritation score of less than or equal to 0 as defined in the "Draft Guidance on Fentanyl” provided by the FDA and discussed above.
- the transdermal dosage system has (a) one sensitizing response occurring at more than 24 hours after removal of the patch during the challenge phase, (b) the combined "dermal response” and “other effects” numeric score is at least 2 during the challenge phase, (c) the combined "dermal response” and “other effects” numeric scores obtained during the challenge phase are general higher than during the induction phase as defined in the "Draft Guidance on Fentanyl” provided by the FDA and discussed above.
- the term "therapeutically equivalent dose” refers to two products where the AUCo- t of the test product is between 80 - 120% that of the reference product.
- opioid-tolerant human patient shall refer to a human patient receiving, for one week or longer, at least 60 mg oral morphine/day, 25 meg transdermal fentanyl/hour, 30 mg oral oxycodone/day, 8 mg oral hydromorphone/day, 25 mg oral
- oxymorphone/day 60 mg oral hydrocodone per day, or an equianalgesic dose of another opioid.
- C max shall refer to the mean (average) observed maximum plasma concentration of the drug assayed.
- Cio shall refer to the mean (average) observed maximum partial plasma concentration of the drug assayed at 10 hours after transdermal administration.
- the term "transformed Cio" shall refer to the ratio of the Cio of a drug as found with the application of an extrinsic factor such as heat, exercise or showering, relative to the Cio of said drug as found after transdermal administration without the application of an extrinsic factor and at a therapeutically equivalent dose.
- AUC shall refer to the area under the plasma concentration/time curve.
- AUCio shall refer to the area under the drug plasma concentration/time curve from time 0 to 10 hours after transdermal administration
- AUCo- t shall refer to the area under the plasma concentration/time curve from time 0 to the last quantifiable drug concentration
- AUCo- inf shall refer to the area under the plasma concentration/time curve from time 0 until the extrapolated drug concentration at infinity.
- the term "transformed AUCio" shall refer to the ratio of the AUCio of a drug as found with the application of an extrinsic factor such as heat, exercise or showering, relative to the AUCio of said drug as found after transdermal administration without the application of an extrinsic factor and at a therapeutically equivalent dose.
- naltrexone to fentanyl plasma concentration shall mean the relative average naltrexone plasma concentration to the average fentanyl plasma concentration, as measured at any fixed time point.
- the term "when heat is applied” shall refer to the application of an external heat source such as, for example, a heating pad, covering the transdermal dosage system of the invention and set to keep the temperature at the skin surface to approximately 38°C, wherein said external heat source is held in place for at least 8 consecutive hours and wherein application of said external heat source is immediately after the transdermal application of said transdermal dosage system.
- an external heat source such as, for example, a heating pad
- the term "after exercise” shall refer to the time period following the subject's undergoing of moderate physical exercise, such as, for example, treadmill or stationary bicycle exercise, wherein the subject's heart rate reaches between 50 - 80% their maximum heart rate, wherein said exercise occurs between 2-7 hours after administration of the transdermal dosage system of the invention and wherein said exercise lasts for at least 30 minutes.
- the term “prior to exercise” shall refer to the application of said transdermal dosage system of the invention without the subject undergoing additional exercise for the entire time period of said application.
- the term "after showering” shall refer to the time period following the subject's undergoing of a lukewarm or hot shower, such as, for example, between 36 and 42°C, which lasts for between 5 and 30 minutes, wherein the water jet of said shower is held directly over the transdermal dosage system of the invention for at least 5 minutes and wherein said shower occurs between 2-7 hours after administration of said transdermal dosage system of the invention.
- the term “prior to showering” shall refer to the application of said transdermal dosage system of the invention without the subject showering for the entire time period of said application.
- micronized refers to particles wherein the particles are less than 10 pm in diameter.
- D 5 o is less than about 10 pm or Dio is less than about 5 pm or Dg 0 is less than about 30 pm.
- D 5 o is less than about 10 pm and Dio is less than about 5 pm and Dg 0 is less than about 30 pm.
- a number of micronization techniques may be utilized to micronize one or more components of the transdermal dosage system including, without limitation, conventional jet mills.
- the particle size or particle size distribution may be determined using techniques in the art such as light diffraction methods such as devices of Malvern Instruments, mechanical sieve shaking method, or air jet sieve analyses.
- the present disclosure is directed to a transdermal dosage system having reduced potential for abuse, without blocking the therapeutic or beneficial effects of the active agent when the system is applied to the skin.
- the system of the present disclosure provides for the controlled release of the antagonist at a rate sufficient to provide an abuse limiting release rate ratio of the antagonist to the active when the dosage form is subject to abuse whether via buccal administration or solvent extraction for injection, wherein the system provides for a substantially minimized/negligible skin sensitization response from antagonist exposure.
- the present disclosure is directed to a transdermal dosage system having reduced potential for abuse, without blocking the therapeutic or beneficial effects of fentanyl when the system is applied to the skin.
- the system of the present disclosure provides for the controlled release of naltrexone at a rate sufficient to provide an abuse limiting release rate ratio of the naltrexone to the fentanyl when the dosage form is subject to abuse whether via buccal administration or chewed, wherein the system provides for a substantially minimized/negligible skin sensitization response from naltrexone exposure.
- This transdermal dosage form when contacted with an animal's skin, allows for the transdermal administration of an active such as an opioid, but either (a) allows for the transdermal administration of only an amount of an antagonist salt that is ineffective for inhibiting the analgesic effect of the opioid, or (b) does not allow for the transdermal administration of the antagonist.
- an active such as an opioid
- the transdermal dosage form of the disclosure is used to deliver an opioid via a route other than transdermal, such as buccal, nasal, oral, parenteral, rectal and/or vaginal, or if the transdermal dosage form is subjected to abuse or misuse, then the antagonist, present in sufficient amounts to counter the effect of active, inhibits the euphoric effect of the opioid.
- the transdermal dosage form will inhibit the euphoric effect of an opioid if the device is used other than transdermally whether before or after the device is used by an animal or human for treating or preventing pain.
- the active is fentanyl and the antagonist naltrexone and the transdermal dosage form, when contacted with an animal's skin, allows for the transdermal administration of fentanyl, but either (a) allows for the transdermal administration of only an amount of naltrexone that is ineffective for inhibiting the analgesic effect of the fentanyl, or (b) does not allow for the transdermal administration of the naltrexone.
- the transdermal dosage form of the disclosure is used to deliver fentanyl via a route other than transdermal, such as buccal, nasal, oral, parenteral, rectal and/or vaginal, or if the transdermal dosage form is subjected to abuse or misuse, then the naltrexone, present in sufficient amounts to counter the effect of fentanyl and inhibits the euphoric effect of the fentanyl.
- the transdermal dosage form will inhibit the euphoric effect of fentanyl if used other than transdermally whether before or after it is used by an animal or human for treating or preventing pain.
- the transdermal dosage form of the disclosure is also tamper-resistant in that if an abuser attempts to extract or separate an opioid from the transdermal dosage form, and self-administer the opioid via another route, such as, but not limited to, oral, parenteral, nasal, or buccal, rectal or vaginal, i.e., a route of administration that can result in a quick euphoric rush, the abuser would self- administer an amount of an antagonist along with the opioid, the amount of antagonist being effective to inhibit the euphoric effect of the opioid.
- another route such as, but not limited to, oral, parenteral, nasal, or buccal, rectal or vaginal, i.e., a route of administration that can result in a quick euphoric rush
- transdermal dosage form having reduced potential for abuse comprising an antagonist to agonist weight ratio of more than 3:1.
- transdermal dosage form having reduced potential for abuse comprising an antagonist to agonist weight ratio of about 4:1.
- the transdermal dosage form of the present disclosure would also have stability, adhesive properties as required by pharmaceutical regulatory approval.
- transdermal dosage form having reduced potential for abuse comprising an active agent and more than one antagonist reservoir or source.
- transdermal dosage form having reduced potential for abuse comprising fentanyl and more than one naltrexone layers.
- a transdermal dosage form comprises an active agent, a first antagonist component, and a second antagonist component.
- the first antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable salt
- the second antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable base.
- the first antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable salt and the active agent and the second antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable base.
- the present disclosure is directed to an abuse deterrent transdermal dosage forms wherein the first and second antagonist are separated by a barrier.
- a transdermal dosage form comprises fentanyl, a first naltrexone component, and a second naltrexone component.
- the first layer comprises naltrexone in the form of a pharmaceutically acceptable salt
- the second layer comprises naltrexone in the form of a pharmaceutically acceptable base.
- the first layer comprises naltrexone in the form of a pharmaceutically acceptable salt and fentanyl
- the second layer comprises naltrexone in the form of a pharmaceutically acceptable base.
- the present disclosure is directed to an abuse deterrent transdermal dosage forms wherein the first and second layers are separated by a barrier.
- the transdermal dosage form the first antagonist component has a proximal and distal surface; the second antagonist component is disposed distal to the first antagonist component and the barrier is interposed between the first and second antagonist components.
- the dosage form may also include a backing layer located distal to the second antagonist component. In another embodiment, the backing layer is permeable to the second antagonist.
- the proximal surface of the first antagonist component has an area of about 5 to about 150 cm 2 .
- the surface area of the dosage form is about 5 to about 60 cm 2 .
- the surface area of the dosage form is about 25 to about 35 cm 2 or about 100 to about 125 cm 2 .
- the transdermal dosage form releases about 10 to about 100 meg active agent per hour to skin.
- the transdermal dosage form releases about 12.5, 25, 50, 75 or 100 meg active agent per hour to skin. Any amount of active is possible, for example the transdermal dosage form may contain anywhere from 0.1 to 500 mg of active agent.
- the transdermal dosage form comprises fentanyl base or alkaloid in an amount of about 1 to about lOmg.
- the antagonist salt and active agent are present in a single layer between the barrier and the skin-contacting surface.
- the layer may comprise the antagonist salt and the active agent dispersed, mixed and/or dissolved to some degree homogenously throughout a polymeric material.
- the layer may comprise the antagonist salt and active agent where they are individually dispersed, mixed and/or dissolved prior to being set in alternating strips of active agent and antagonist salt.
- the active agent and the first antagonist are separated by one or more spacers.
- the active agent and the first antagonist are in a ratio of about 1:2 or 1:1.81.
- the active agent and the first antagonist are in a ratio of about 1:2 or 1:1.81
- the active agent is fentanyl and the first antagonist, naltrexone.
- the fentanyl is in the form of its free base
- the Naltrexone is in the form of a salt
- the fentanyl base and the Naltrexone salt are in a ratio of about 1:2 or 1:1.81.
- the single layer comprises about 8% ionic naltrexone such as naltrexone HCI.
- the single layer comprises ionic naltrexone such as naltrexone HCI in crystalline form. Additional excipients such as solubilizers like povidone or adhesives such as silicone adhesives may also be present.
- the disclosure relates to a transdermal system for administering an active agent through the skin, the system having a reduced potential for abuse, comprising:
- a first antagonist component comprising a first antagonist salt and an active agent in base form
- the active agent may be for example amorphous or crystalline fentanyl or an analog thereof and the analog is selected from the group consisting of alfentanil, lofentanil, remifentanil, sufentanil and trefentanil;
- a second antagonist component comprising a second antagonist in base form, optionally in amorphous base form, the antagonist being releasable from system upon being ingested or substantially immersed in a solvent, and further wherein the antagonist is selected from the group consisting of naltrexone, methylnaltrexone, naloxone, nalbuphine, nalorphine, nalorphine dinicotinate, nalmefene, nadide, levallorphan, cyclozocine and pharmaceutically acceptable salts thereof; and
- a barrier layer said barrier layer separating said first and second antagonist components, said barrier layer optionally being substantially impermeable to said active agents and/or excipients, wherein: the release of the antagonist from the system when used transdermally such that levels are sufficiently low that the active agent's biological effect is maintained; and the system provides release of the antagonist at a rate sufficient to provide an abuse limiting release rate ratio of the antagonist to the active agent when the dosage form is subject to abuse, e.g., upon transmucosal applications or substantial immersion of the system in the solvent.
- the disclosure relates to a transdermal system for administering an active agent through the skin, wherein the active agent, such as fentanyl, is formulated in amorphous form while the first antagonist salt, such as Naltrexone HCI, is in crystalline form and the second antagonist, such as naltrexone base, is formulated in amorphous form.
- the active agent such as fentanyl
- the first antagonist salt such as Naltrexone HCI
- the second antagonist such as naltrexone base
- the disclosure relates to a transdermal system for administering an active agent through the skin, wherein the active agent, such as fentanyl, is formulated in crystalline form while the first antagonist salt, such as Naltrexone HCI, is in amorphous form and the second antagonist, such as naltrexone base, is formulated in crystalline form.
- the active agent such as fentanyl
- the first antagonist salt such as Naltrexone HCI
- the second antagonist such as naltrexone base
- the transdermal dosage form will inhibit the euphoric effect of an opioid if the device is used other than transdermally whether before or after the device is used by an animal or human for treating or preventing pain.
- release of the antagonists from the system, when used transdermally, is controlled so that antagonist levels are sufficiently low while maintaining the active agent's effect.
- the active agent's effects are maintained for more than about two days. In other embodiments, the active agent's effects are maintained for more than about three days.
- an abuser tries to extract an opioid, for example, fentanyl, from the transdermal dosage form by placing it in a solvent, including saliva
- an amount of antagonist for example, naltrexone
- Antagonist free bases such as naltrexone free base
- antagonist salts for example, naltrexone HCI
- antagonist salts for example, naltrexone HCI
- naltrexone if an abuser attempts to extract fentanyl from the transdermal dosage form, whether aqueous or non-aqueous solvent, at least one form of naltrexone will be released along with the opioid. If a mixture of fentanyl and naltrexone is administered via a route other than the intended transdermal route, such a transmucosally, at least one form of the naltrexone would exert its antagonistic effect to inhibit the euphoric effect of the opioid.
- the present disclosure comprises a transdermal dosage form 100 comprising: a first antagonist component 120, optionally comprising a polymeric material and optionally in the form of a continuous, planar component in the form of a slab; an second antagonist component 140 comprising an antagonist in free base form; and optionally a barrier 130.
- the first antagonist component has a proximal surface 115, which may be a skin contacting surface and is optionally covered with a release liner 110, and a distal surface 125 which is opposed to the proximal surface 115.
- the barrier 130 is disposed between the distal surface 125 of the first antagonist component 120 and the second antagonist component 140.
- a backing 150 is disposed adjacent to the second antagonist component 140 at a location which provides an outer surface 155 of the dosage form 100.
- a permeable backing layer 150 is adjacent to the second antagonist component 140.
- a structured release liner 110 is located proximal to the first antagonist component 120 and functions to protect the surface of the skin-contacting first antagonist component 120 prior to use of the dosage form.
- transdermal dosage form 200 comprising:
- a first antagonist component 120 comprising alternating strips of an active agent 121 and an antagonist in base form 122, optionally wherein the active agent and first antagonist is discontinuous having an alternating spacers 123 made up of air or voided spaces or inactive material between the active agent and antagonist strip; a second antagonist component 140 comprising an adverse agent; and a barrier 130.
- the first antagonist component 120 has a proximal surface 115, which may be a skin contacting surface and is optionally covered with a release liner 110, and a distal surface 125 which is opposed to the proximal surface 115.
- the barrier 130 is disposed between the distal surface 125 of the first antagonist component 120 and the second antagonist component 140.
- a structured release liner 110 is located proximal to the first antagonist component 120 and functions to protect the structured surface of the skin-contacting first antagonist component 120 prior to use of the dosage form.
- the width of the strips of active agent component 121 is greater than about 0.1 cm. In another embodiment, the width of said 121 is greater than about 0.2 cm. In another embodiment, the width of said 121 is greater than about 0.4 cm.
- the width of said 121 is less than about 2.0 cm. In another embodiment, the width of the strips is less than 1.0 cm. In another embodiment, the width of said 121 is less than 0.6 cm. Although a specific configuration is described above, it should be understood that the alternating strips may be of any shape such as but not limited to squares, diamonds, ovals, triangles, pentagons, or hexagons.
- the active agent is an opioid and the antagonist is an opioid antagonist.
- Opioid antagonists useful in the present disclosure include, but are not limited to, naloxone, naltrexone, nalmefene, nalbuphine, nalorphine, cyclazacine, cyclazocine, levallorphan, pharmaceutically acceptable salts thereof, and mixtures thereof.
- the opioid antagonist is nalmefene, naloxone, naltrexone, or a pharmaceutically acceptable salt thereof.
- the opioid antagonist is a naltrexone.
- the antagonist free base is naltrexone in amorphous form.
- Useful opioid antagonist salts include salts formed from an acid and the basic nitrogen group of an opioid antagonist.
- opioid antagonist salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucoronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p- toluenesulfonate, and pamoate (i.e., l,l'-methylene-bis-(2-hydroxy-3-naphtho
- opioid antagonist salts include salts prepared from an antagonist having an acidic functional group, such as a carboxylic acid or sulfonic acid functional group, and a pharmaceutically acceptable inorganic or organic base.
- the antagonist salt is naltrexone hydrochloride.
- the amount of active agent present in the transdermal drug delivery composition of the disclosure is greater than about 0.01 wt-%, based on the total weight of the composition of the active agent component. In another embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is greater than about 1.0 wt-%, based on the total weight of the composition of the active agent component. In another embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is less than about 40 wt-%, based on the total weight of the composition of the active agent component. In another embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is less than about 20.0 wt-%, based on the total weight of the composition of the active agent component.
- the analgesically effective amount of an opioid present in the transdermal dosage form typically ranges from about 0.01 to about 50 mg/cm 2 in one embodiment, from about 0.05 to about 15mg/cm 2 in another embodiment, and from about 0.05 to about 5.0mg/ cm 2 in another embodiment. It is well within the purview of one skilled in the art to readily determine the analgesically effective amount of an opioid needed for a particular indication.
- the antagonist free base and an antagonist salt are present in an amount sufficient to inhibit at least one biological effect of an active agent.
- the antagonist free base and an antagonist salt are provided in a total amount sufficient to inhibit the euphoric effect of an opioid when the transdermal dosage form is subjected to abuse or misuse.
- the naltrexone free base and naltrexone salt are present in an amount sufficient to inhibit at least one biological effect of fentanyl.
- the naltrexone free base and naltrexone salt are provided in a total amount sufficient to inhibit the euphoric effect of fentanyl when the transdermal dosage form is subjected to abuse or misuse.
- the adverse agent present in transdermal patch 100 or 200 the adverse agent is capable of containing a sufficient amount of adverse agent to blunt or block at least one biological effect of the active agent or to cause at least one unpleasant side effect in a patient or animal when the patch is subjected to abuse or misuse. This amount can vary according to the amount and type of active agent in the dosage form. The amount may be included in each adverse agent component individually or combined in component 140 and component 120 depending on desired effect and form of the formulation.
- the transdermal patch is capable of containing a sufficient amount of naltrexone to blunt or block at least one biological effect of the fentanyl or to cause at least one unpleasant side effect in a patient or animal when the patch is subjected to abuse or misuse.
- This amount can vary according to the amount and type of fentanyl in the dosage form. The amount may be included in each layer individually or combined in second antagonist component 140 and first antagonist component 120 depending on desired effect and form of the formulation.
- the antagonist components comprise an adverse agent in any form or composition or reservoir which allows the antagonist to be at least partially extracted in the presence of a solvent, including but not limited to, water, ethanol or ether, or mixtures thereof.
- a solvent including but not limited to, water, ethanol or ether, or mixtures thereof.
- the antagonist can be dispersed, mixed and/or dissolved in a polymeric material, including but not limited to, the polymeric materials which are suitable for incorporation into the active agent component.
- the dosage form is provided such that the antagonist, such as naltrexone, is not absorbed to any biologically significant degree into a blood stream when administered transdermally.
- the dosage form is provided such that the ratio of adverse agent to active agent in the dosage form is from about 1:10 to about 10:1.
- the dosage form is provided such that the ratio of naltrexone to fentanyl in the dosage form is from about 1:10 to about 10:1.
- the dosage form is provided such that the ratio of adverse agent to active agent in the dosage form is more than 3:1.
- the dosage form is provided such that the ratio of naltrexone to fentanyl in the dosage form is more than 3:1.
- a transdermal dosage form having reduced potential for abuse comprising an antagonist to agonist weight ratio of about 4:1.
- transdermal dosage form having reduced potential for abuse comprising naltrexone to fentanyl weight ratio of about 4:1.
- the transdermal dosage form of the present disclosure would also have stability, adhesive properties as required by pharmaceutical regulatory approval.
- the ratio of adverse agent to active agent released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated is at least 1:5, 1:4, 1:3, 1:2, or 1:1.
- the ratio of adverse agent to active agent released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated is at least 1:1 at numerous time points between 5 min. to 4 hours.
- the ratio of naltrexone to fentanyl released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated is at least 1:5, 1:4, 1:3, 1:2, or 1:1.
- the ratio of naltrexone to fentanyl released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated is at least 1:1 at time points between 5 min. to 4 hours.
- the C max of the dosage form when administered buccally, is less than 50% the buccally administered C max of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
- the buccally administered C max of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the buccally administered C max of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
- the C max of the dosage form when chewed, is less than 50% the chewed C max of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses. In another embodiment, the chewed C max of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the chewed C max of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
- the AUCo- t of the dosage form when administered buccally, is less than 50% the buccally administered AUC 0-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
- the buccally administered AUCo- t of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the buccally administered
- the AUCo- t of the dosage form when chewed, is less than 50% the chewed AUCo- t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
- the chewed AUC 0-t of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the chewed AUC 0-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent trans
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first fifteen minutes after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first thirty minutes after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first hour after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75,
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first two hours after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first three hours after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first fifteen minutes after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first thirty minutes after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first hour after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first two hours after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than
- the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first three hours after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than
- the fentanyl Cio of the dosage form when heat is applied, is 110% or more than the fentanyl Cio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl Cio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the fentanyl Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form when heat is applied, is 180% or less than the fentanyl Cio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl Cio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form when heat is applied, is 105% or more than the naltrexone Cio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form when heat is applied, is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the naltrexone Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form when heat is applied, is 180% or less than the naltrexone Cio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl of the dosage form when heat is applied, is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, 115% or less, or 110% or less than the naltrexone Cio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form when heat is applied, is 110% or more than the fentanyl AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form when heat is applied, is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the fentanyl Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form when heat is applied, is 250% or less than the fentanyl AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl of the dosage form when heat is applied, is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form when heat is applied, is 105% or more than the naltrexone AUCio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the naltrexone AUCio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more,
- the naltrexone AUCio of the dosage form when heat is applied, is 250% or less than the naltrexone AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl of the dosage form when heat is applied, is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, 115% or less, or 110% or less than the naltrexone AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form is 110% or more than the fentanyl Cio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the fentanyl Cio of the dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form is 180% or less than the fentanyl Cio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the fentanyl of the dosage form is less than 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl Cio of said dosage form prior to exercise at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form is 105% or more than the naltrexone Cio of said dosage form prior to exercise at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the naltrexone Cio of the dosage form prior to exercise at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form is 180% or less than the naltrexone Cio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, 115% or less, or 110% or less than the naltrexone Cio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form is 110% or more than the fentanyl AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the fentanyl Cio of the dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form is 250% or less than the fentanyl AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, or 115% or less than the fentanyl AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form is 105% or more than the naltrexone AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the naltrexone Cio of the dosage form prior to exercise at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form is 250% or less than the naltrexone AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, 115% or less, or 110% or less than the naltrexone AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form is 110% or more than the fentanyl Cio of said dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the fentanyl Cio of the dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl Cio of the dosage form is 180% or less than the fentanyl Cio of said dosage form prior to showering at therapeutically equivalent doses. In another embodiment, after showering, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl Cio of said dosage form prior to showering at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form is 105% or more than the naltrexone Cio of said dosage form prior to showering at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the naltrexone Cio of the dosage form prior to showering at therapeutically equivalent doses.
- the naltrexone Cio of the dosage form is 180% or less than naltrexone Cio of said dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, 115% or less, or 110% or less than the naltrexone Cio of said dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form is 110% or more than the fentanyl AUCio of said dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the fentanyl Cio of the dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl AUCio of the dosage form is 250% or less than the fentanyl AUCio of said dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, or 115% or less than the fentanyl AUCio of said dosage form prior to showering at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form is 105% or more than the naltrexone AUCio of said dosage form prior to showering at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the naltrexone Cio of the dosage form prior to showering at therapeutically equivalent doses.
- the naltrexone AUCio of the dosage form is 250% or less than the naltrexone AUCio of said dosage form prior to showering at therapeutically equivalent doses.
- the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, 115% or less, or 110% or less than the naltrexone AUCio of said dosage form prior to showering at therapeutically equivalent doses.
- the transformed fentanyl Cio when heat is applied, the transformed fentanyl Cio, i.e., the ratio of the Cio of fentanyl as found with the application of heat relative to the fentanyl Cio as found when heat is not applied at a therapeutically equivalent dose, is 1.1. In another embodiment, when heat is applied, the transformed fentanyl Cio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or
- the transformed naltrexone Cio when heat is applied, the transformed naltrexone Cio, i.e., the ratio of the Cio of naltrexone as found with the application of heat relative to the naltrexone Cio as found when heat is not applied at a therapeutically equivalent dose, is 1.05.
- the transformed naltrexone Cio when heat is applied, is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
- the transformed fentanyl Cio to naltrexone Cio ratio when heat is applied, is 1:1 or less. In another embodiment, when heat is applied, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1:1.25 or less, 1:1.3 or less, 1:1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1:1.55 or less, or 1:1.6 or less.
- the transformed fentanyl AUCio when heat is applied, is 1.1. In another embodiment, when heat is applied, the transformed fentanyl AUCio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55,
- the transformed naltrexone AUCio when heat is applied, is 1.05.
- the transformed naltrexone AUCio when heat is applied, is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
- the transformed fentanyl AUCio to naltrexone AUCio ratio when heat is applied, is less than 1: 1. In another embodiment, when heat is applied, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1:1.05 or less, 1: 1.1 or less, 1:1.15 or less, 1: 1.2 or less, 1:1.25 or less, 1: 1.3 or less, 1:1.35 or less, 1: 1.4 or less, 1: 1.45 or less, 1: 1.5 or less, 1:1.55 or less, 1:1.6 or less, 1:1.65 or less, 1: 1.7 or less, 1:1.75 or less, 1:1.8 or less, 1:1.85 or less, 1:1.9 or less, 1:1.95 or less, 1:2 or less, 1:2.05 or less, 1:2.1 or less, 1:2.15 or less, 1:2.2 or less, or 1:2.25 or less.
- the transformed fentanyl Cio i.e., the ratio of the Cio of fentanyl as found after exercise relative to the fentanyl Cio as found prior to exercise at a therapeutically equivalent dose, is 1.1.
- the transformed fentanyl Cio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
- the transformed naltrexone Cio i.e., the ratio of the Cio of naltrexone as found after exercise relative to the naltrexone Cio as found prior to exercise at a therapeutically equivalent dose, is 1.05.
- the transformed naltrexone Cio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
- the transformed fentanyl Cio to naltrexone Cio ratio is 1:1 or less.
- the transformed fentanyl Cio to naltrexone Cio ratio is 1:1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1:1.25 or less, 1:1.3 or less, 1:1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1:1.55 or less, or 1:1.6 or less.
- the transformed fentanyl AUCio i.e., the ratio of the AUCio of fentanyl as found after exercise relative to the fentanyl AUCio as found prior to exercise at a therapeutically equivalent dose
- the transformed fentanyl AUCio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7, 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
- the transformed naltrexone AUCio i.e., the ratio of the AUCio of naltrexone as found after exercise relative to the naltrexone AUCio as found prior to exercise at a therapeutically equivalent dose, is 1.05.
- the transformed naltrexone AUCio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 1.75,
- the transformed fentanyl AUCio to naltrexone AUCio ratio is 1:1 or less. In another embodiment, after exercise, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1: 1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1: 1.25 or less, 1:1.3 or less, 1: 1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1: 1.55 or less, 1:1.6 or less, 1: 1.65 or less, 1:1.7 or less, 1: 1.75 or less, 1:1.8 or less, 1:1.85 or less, 1:1.9 or less, 1: 1.95 or less, 1:2 or less, 1:2.05 or less, 1:2.1 or less, 1:2.15 or less, 1:2.2 or less, or 1:2.25 or less.
- the transformed fentanyl Cio i.e., the ratio of the Cio of fentanyl as found after showering relative to the fentanyl Cio as found prior to showering at a therapeutically equivalent dose, is 1.1.
- the transformed fentanyl Cio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
- the transformed naltrexone Cio i.e., the ratio of the Cio of naltrexone as found after showering relative to the naltrexone Cio as found prior to showering at a therapeutically equivalent dose, is 1.05.
- the transformed naltrexone Cio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
- the transformed fentanyl Cio to naltrexone Cio ratio is 1:1 or less. In another embodiment, after showering, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1:1.25 or less, 1:1.3 or less, 1:1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1:1.55 or less, or 1:1.6 or less.
- the transformed fentanyl AUCio i.e., the ratio of the AUCio of fentanyl as found after showering relative to the fentanyl AUCio as found prior to showering at a therapeutically equivalent dose, is 1.1.
- the transformed fentanyl AUCio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7, 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
- the transformed naltrexone AUCio i.e., the ratio of the AUCio of naltrexone as found after showering relative to the naltrexone AUCio as found prior to showering at a therapeutically equivalent dose, is 1.05.
- the transformed naltrexone AUCio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 1.75,
- the transformed fentanyl AUCio to naltrexone AUCio ratio is 1:1 or less. In another embodiment, after showering, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1: 1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1: 1.25 or less, 1:1.3 or less, 1: 1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1: 1.55 or less, 1:1.6 or less, 1: 1.65 or less, 1:1.7 or less, 1: 1.75 or less, 1:1.8 or less, 1:1.85 or less, 1:1.9 or less, 1: 1.95 or less, 1:2 or less, 1:2.05 or less, 1:2.1 or less, 1:2.15 or less, 1:2.2 or less, or 1:2.25 or less.
- the fentanyl Cio of the dosage form when heat is applied, is less than 80% the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, when heat is applied, the fentanyl Cio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at
- the fentanyl Cio of the dosage form is less than 80% the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl Cio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl Cio of the dosage form is less than 80% the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl Cio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at
- the fentanyl AUCio of the dosage form when heat is applied, is less than 80% the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, when heat is applied, the fentanyl AUCio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl AUCio of the dosage form is less than 80% the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl AUCio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl AUCio of the dosage form is less than 80% the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the fentanyl AUCio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
- the proximal surface 115 has a release liner 110 which is removed to reveal a skin-contacting surface which should be sufficiently conformable when placed on a skin surface so as to make intimate contact with at least a portion of the skin surface.
- substantially all of the polymeric material of the proximal surface of the first antagonist component 120 will make intimate contact with the skin surface of a patient.
- Suitable release liners include conventional release liners comprising a known sheet material such as a polyester web, a polyethylene web, a polypropylene web, or a polyethylene-coated paper coated with a suitable fluoropolymer or silicone based coating.
- the release liner that has been coated with the first antagonist component 120 can be dried and laminated onto a barrier component 120 using conventional methods.
- the first antagonist component 120 comprises a polymeric material, an active agent and an antagonist salt.
- Suitable polymeric materials or matrices for use in the first antagonist or adverse agent component include, but are not limited to, acrylates, natural rubbers, polyisobutylenes, polyisoprenes, styrenic block copolymers, polyvinylethers, silicone polymers, polyurethanes, and polyurethane-ureas.
- the active agent or adverse agent is preferably dispersed substantially homogeneously throughout a polymeric material.
- the active agent or adverse agent is dissolved in the polymeric material.
- the active agent is substantially in amorphous form.
- the adverse agent component includes a crystalline form substantially dispersed throughout the polymeric material.
- the first antagonist component 120 comprises a polymeric material, fentanyl as free base and naltrexone HCI.
- Suitable polymeric materials or matrices for use in the first antagonist or adverse agent component include, but are not limited to, acrylates, natural rubbers, polyisobutylenes, polyisoprenes, styrenic block copolymers, polyvinylethers, silicone polymers, polyurethanes, and polyurethane-ureas.
- the fentanyl or naltrexone is preferably dispersed substantially homogeneously throughout a polymeric material.
- the fentanyl or naltrexone is dissolved in the polymeric material.
- fentanyl is substantially in amorphous form.
- the naltrexone includes a crystalline form substantially dispersed throughout the polymeric material.
- the polymeric matrix is a pressure sensitive adhesive.
- Suitable pressure-sensitive adhesives include those suitable for use as the polymeric material of the active agent component. Additionally, pressure-sensitive adhesives that are not suitable for direct skin contact can be suitable for use as the polymeric material of the active agent or adverse agent.
- pressure-sensitive adhesives for use in the dosage forms of the disclosure include acrylates, polyisobutylenes, silicone polymers, and mixtures thereof. Examples of useful polyisobutylene pressure-sensitive adhesives are described in U.S. Pat. No. 5,985,317, the disclosure of which is incorporated herein by reference in its entirety for all purposes. Examples of useful acrylate and silicone polymer pressure-sensitive adhesives, and mixtures thereof, are described in U.S. Pat. No. 5,474,783, the disclosure of which is incorporated herein by reference in its entirety for all purposes.
- Acrylate polymers and copolymers may include pressure-sensitive adhesives.
- Suitable monomers for use in acrylate copolymers include alkyl acrylates, such as isooctyl, 2-ethylhexyl, n-butyl, ethyl, methyl, and dimethylhexyl, and alkyl methacrylates, such as lauryl, isodecyl, and tridecyl.
- alkyl acrylates such as isooctyl, 2-ethylhexyl, n-butyl, ethyl, methyl, and dimethylhexyl
- alkyl methacrylates such as lauryl, isodecyl, and tridecyl.
- Monomers containing functional groups, such as carboxylic acid, hydroxy, amide, and amino may also be incorporated into an acrylate copolymer.
- Suitable monomers containing functional groups include acrylic acid, hydroxyalkyl acrylates containing 2 to 4 carbon atoms in the hydroxyalkyl group, acrylamide, N-vinyl-2-pyrrolidone, vinyl acetate, and alkoxyethyl acrylates.
- Acrylate copolymers may optionally further comprise a substantially linear macromonomer copolymerizable with the other monomers.
- Suitable macromonomers include polymethylmethacrylate, styrene/acrylonitrile copolymer, polyether, and polystyrene macromonomers. Examples of useful macromonomers and their preparation are described in U.S. Pat. No. 4,693,776 (Krampe et al.), the disclosure of which is incorporated herein by reference in its entirety for all purposes.
- polymer materials of the first antagonist component may include but are not limited to polyethylene; polypropylene; ethylene/propylene copolymers; ethylene/ethylacrylate copolymers; ethylene/vinyl acetate copolymers; silicone elastomers, especially the medical-grade polydimethylsiloxanes; neoprene rubber; polyisobutylene; chlorinated polyethylene; polyvinyl chloride; vinyl chloride-vinyl acetate copolymer; polymethacrylate polymer (hydrogel); polyvinylidene chloride; poly(ethylene terephthalate); butyl rubber; epichlorohydrin rubber; ethylene-vinyl alcohol copolymer; ethylene-vinyloxyethanol copolymer; silicone copolymers, for example, polysiloxane-polycarbonate copolymers, polysiloxane-polyethyleneoxide copolymers, polysiloxane-polymethacryl
- the polymer matrix has a glass- transition temperature below room temperature.
- the polymer can, but need not necessarily, have a degree of crystallinity at room temperature.
- Cross-linking monomeric units or sites can be incorporated into the polymers.
- cross-linking monomers can be incorporated into polyacrylate polymers.
- the cross-linking monomers provide sites for cross-linking the polymer matrix after microdispersing the active agent into the polymer.
- Known cross-linking monomers for polyacrylate polymers include, but are not limited to, polymethacrylic esters of polyols such as butylene diacrylate and dimethacrylate, trimethylol propane trimethacrylate, and the like.
- polymer matrix does not allow any, or any detectable amount, of an active agent or adverse agent to diffuse out of it, particularly in those instances in which the active agent or adverse agent can penetrate a patient's skin.
- the first antagonist component can also comprise a porous medium, such as a woven fabric, porous or microporous film, or other open, mesh-like material, wherein at least a portion of the pores contain active agent or adverse agent.
- the active agent or adverse agent can be present within the pores in any form, including but not limited to a liquid, a gel or a solid, such as a solid crystalline or powdered material.
- the active agent or adverse agent can be mixed with a carrier, such as a viscous liquid, semi-solid or gel material.
- suitable materials for incorporation into the active agent or adverse agent component include, but are not limited to, microporous films formed by extruding polyethylene or polypropylene with mineral oil as described in U.S. Pat. No. 4,539,256, the disclosure of which is incorporated herein by reference in its entirety.
- Each of the layers comprising active agent or antagonist may comprise a number of additional components. Additional components of the active agent or first antagonist component can include skin penetration enhancers, drug solubilizers, plasticizers, anti-oxidants, colorants, bittering agent and the like.
- the first antagonist component will typically comprise a skin penetration enhancer.
- excipients useful as skin penetration enhancers or solubilizers in transdermal drug delivery systems include C 8 -C 24 fatty acids such as isostearic acid, octanoic acid, and oleic acid; C 8 -C 24 fatty alcohols such as oleyl alcohol and lauryl alcohol; lower alkyl esters of C 8 -C 24 fatty acids such as ethyl oleate, isopropyl myristate, butyl stearate, and methyl laurate; monoglycerides of C 8 -C 24 fatty acids such as ethyl oleate, isopropyl myristate, butyl stearate, and methyl laurate; monoglycerides of C 8 -C 24 fatty acids such as glyceryl monolaurate; tetraglycol (tetrahydrofurfuryl alcohol polyethylene glycol
- the skin penetration enhancers, drug solubilizers, plasticizers, and other additives can be dispersed or mixed, optionally substantially uniformly, or optionally dissolved in the composition.
- the additive is a penetration enhancer, it is present in an amount that enhances active agent permeation through the skin compared to a like composition not containing the penetration enhancer(s) when this phenomenon is measured using a standard skin penetration model, such as set forth in U.S. Pat. No. 5,585,111, the disclosure of which is herein incorporated by reference in its entirety.
- the total amount of penetration enhancer and solubilizer is less than about 40% by weight based on the total weight of the composition. In another embodiment, the total amount of penetration enhancer and solubilizer is less than about 30% based on the total weight of the composition.
- a solubility enhancer may also be included.
- the solubility enhancer is in an amount more than 2%. In another embodiment, it is in an amount of between 2.5 to 3.5%.
- polyvinylpyrrolidone (PVP) is used.
- the active agent and antagonist are optionally dispersed homogeneously throughout the polymeric material, or optionally dissolved within the polymeric material.
- the proximal or skin contacting surface 115 should be sufficiently conformable when placed on a skin surface so as to make intimate contact with at least a portion of the skin surface. In one embodiment, substantially all of the polymeric material at the proximal surface 115 will make intimate contact with the skin surface.
- the first antagonist component and the antagonist each have a thickness of no less than about 10pm.
- the first antagonist agent component has a thickness of no less than about 20pm.
- the first antagonist component has a thickness of no less than about 50pm.
- the first antagonist component has a thickness of no greater than about 250pm.
- the first antagonist component has a thickness of no greater than about 200pm.
- the first antagonist component has a thickness of no greater than about 150pm.
- the barrier 130 is a substantially continuous component adjacent to the distal surface of the first antagonist component 120 on one side and the second antagonist component 140 on the other side.
- the barrier layer is impermeable to the antagonist and the active agent; and comprises a material which is insoluble in water, alcohol and organic solvents.
- the barrier layer comprises a polymer such as polyolefin laminates (Dow Chemical, Midland, Ml), acrylonitrile copolymer films (BAREX, BP Chemicals, Koln, Germany), polyethylnapthalene (PEN), polyethylene terephthalate (PET), polyimide, polyurethane, polyethylene, metallized films and glass coated films where these films can include ethylene copolymers such as ethylene-vinyl acetate copolymer (EVA), and combinations thereof.
- the barrier layer comprises polyester such as PET laminated to a polymer such as polyurethane, polyethylene, and ethylene copolymers.
- the barrier layer comprises polyester such as PET laminated to ethylene copolymers such as ethylene-vinyl acetate copolymer (EVA).
- the barrier layer as a multilaminate layer has a thickness of about 0.075 mm (0.3 mil) to about 0.125 mm (5 mil); optionally 0.025 mm (1 mil) to about 0.1 mm (4 mil); optionally 0. 0625 mm (1.5 mil) to about 0.0875 mm (3.5 mil); and optionally 0.025 mm (1 mil) to about 0.05 mm (2 mil).
- the polyethylene or EVA laminated layer of the PET-PE laminates improves the adhesion of the antagonist component to the backing, and serves to prevent the facile removal of the antagonist base layer from the system by the potential abuser.
- the backing is laminated to the surface of the antagonist base component, optionally using heat, pressure and/or an additional tie component to ensure adequate contact between the reservoir component and backing.
- the backing is non sticking and hydrophobic.
- the transdermal dosage form can be a reservoir-type transdermal dosage form, a polymer-matrix type transdermal dosage form, or a drug-in-adhesive type transdermal dosage form.
- the transdermal dosage form is designed so that when contacted with the animal's skin, an analgesically effective amount of the active therapeutic agent, such as an opioid, is transdermally administered to the animal while the antagonist either remains in the transdermal dosage form and is not administered to the animal or is administered to the animal in an amount insufficient to inhibit the analgesic effect of the active agent.
- transdermal dosage forms of the disclosure can be made in the form of an article such as a tape, a patch, a sheet, a dressing or any other form known to those skilled in the art.
- the dosage form will be in the form of a patch of a size suitable to deliver a preselected amount of active agent through the skin.
- the dosage form will have a surface area greater than 5cm 2 . In another embodiment, the dosage form will have a surface area of greater than 10cm 2 . In another embodiment, the dosage form will have a surface area of less than 100 cm 2 . In another embodiment, the dosage form will have a surface area of less than 40 cm 2 .
- Dosage forms of the present disclosure are typically packaged individually in a foil- lined pouch for storage. Dosage forms of the present disclosure may alternatively be provided in a rolled or stacked form suitable for use with a dispensing apparatus. An optional tie component, heat, and/or pressure may be used to connect the skin-contacting component with the barrier component. In addition, the skin-contacting component compositions may be directly coated onto the barrier component and subsequently dried and laminated to a release liner.
- the transdermal dosage form is contacted with the skin of the patient and an opioid is released by the transdermal dosage form and becomes absorbed through the skin. Once absorbed into the patient, the opioid is provided in an analgesically effective amount.
- the transdermal dosage form can provide sustained and continuous delivery of an analgesically effective amount of the opioid.
- the transdermal dosage form on administration over the skin, the transdermal dosage form exhibits a steady state drug flux of about 1 to about 10pg/cm 2 /hr. In one embodiment, the transdermal dosage form exhibits a steady state drug flux of about 1 to about 8 pg/cm 2 /hr.
- the transdermal dosage form exhibits a steady state drug flux of about 1 to about 5 pg/cm 2 /hr. In one embodiment, the transdermal dosage form exhibits a steady state drug flux of about 2 to about 3 pg/cm 2 /hr. In one embodiment the transdermal dosage form is contacted with the skin of the patient and fentanyl is released by the transdermal dosage form and becomes absorbed through the skin. Once absorbed into the patient, the fentanyl is provided in an analgesically effective amount. The transdermal dosage form can provide sustained and continuous delivery of an analgesically effective amount of fentanyl.
- the transdermal dosage form exhibits a nominal flux (i.e., the average amount of drug delivered to the systemic circulation per hour across average skin) of about 12.5 mcg/hr. In one embodiment, the transdermal dosage form exhibits a nominal flux of about 50 mcg/hr.
- the transdermal dosage form exhibits a nominal flux of about 75 mcg/hr. In one embodiment, the transdermal dosage form exhibits a nominal flux of about 100 mcg/hr.
- the method of treating pain with any one of the dosage forms described herein, wherein said dosage form can provide a ratio of adverse agent to active agent released, or alternatively absorbed into a blood stream, from about 1:10 to about 10:1 when the dosage form is used in an inappropriate manner.
- a solvent such as a quid or gas.
- the dosage form when tampered with in such a manner, will release both adverse and active agent.
- the ratio of adverse agent to active agent released when tampered with is about is 1:5, 1:4, 1:3, 1:2, or 1:1.
- the method of treating pain comprises applying a dosage form as described herein, wherein said dosage from comprises a ratio of adverse agent to active agent from about 1:10 to about 10:1. In other embodiments, the ratio of adverse agent to active agent is about is 1:5, 1:4, 1:3, 1:2, or 1:1.
- an abuser may attempt to extract the fentanyl from the transdermal dosage form with a solvent, such as a liquid or gas.
- the dosage form when tampered with in such a manner, will release both naltrexone and fentanyl.
- the ratio of naltrexone to fentanyl released from a transdermal dosage form when tampered with is about is 1:5, 1:4, 1:3, 1:2, or 1:1.
- the method of treating pain comprises applying to the skin of a human subject a transdermal dosage form as described herein, wherein said dosage from comprises a ratio of naltrexone to fentanyl from about 1:10 to about 10:1.
- the ratio of naltrexone to fentanyl released into the plasma is about is 1:5, 1:4, 1:3, 1:2, or 1:1.
- the present disclosure is also directed to a kit comprising at least one dosage form of the disclosure.
- the dosage form is present in a container, e.g., a box.
- the kit further comprises a set of instructions directing the use of the dosage form to treat a patient, e.g., for pain.
- the instructions may be a printed label affixed to or printed on the container.
- the instructions may comprise a printed sheet inserted into the container or into the packaging which contains the container. The instructions may also state that the dosage form and/or its usage are designed to reduce abuse, misuse or diversion of the dosage form.
- Example 1 The transdermal dosage form Patch A1 as depicted in Figures 3 and 4 were prepared according to the manufacturing process steps described below. The quantitative composition of component of the Patch A1 is provided below in Tables.
- Patch A1 was manufactured according to the amounts below in Table 1 and according to the following process steps:
- transdermal dosage form Patch B2 as depicted in Figures 1 and 2 and Patch B1 as depicted in Figures 3 and 4 were prepared according to the manufacturing process steps described in Figures 5-9.
- Figure 5 is a schematic of the manufacturing process steps for transdermal patch B1 and B2.
- Figure 6 is a manufacturing flow diagram of the skin contact layer (central) active strip containing fentanyl base and naltrexone HCI (layer B).
- Figure 7 is a schematic of the manufacturing process steps of the skin contact layer adhesive strips (Layer B) for patch B2.
- Figure 8 is a manufacturing flow diagram for the non-skin contact layer (layer D) containing naltrexone (step 3).
- Figure 9 is a manufacturing flow diagram for the integration of the non-skin contact layer, skin contact adhesive side strips and skin contact central active strip into the finished transdermal patch (step 4).
- the quantitative composition of component of the Patch B1 and B2 is provided in Table 2.
- Other doses for Patch B1 can be calculated.
- 12.5 mcg/hr is 12.5% of each mg/patch of the 100 mcg/hr fentanyl amounts.
- the drug product manufacturing process consists of a four step process as described in Figure 5. For each of the first three steps, a wet blend is coated onto an in-process release liner, dried and laminated to another release liner or a barrier film, resulting in one of the three separate intermediate adhesive laminates. During the last two steps of the manufacturing process, each of the intermediate laminates is die-cut to the appropriate size for each patch strength and assembled into the final dosage form.
- the manufacturing process for Step 1 is shown in Figure 6 and according to the following:
- the test samples were transdermal patches: Patch A1 (6.25 cm 2 ) and Patch B2 (6 cm 2 ).
- the extraction solution was chosen from one of the following solutions at room temperature and 70°C: water; ethanol (USP, absolute); pH6.8 Phosphate Buffer; cooking vinegar; and artificial saliva.
- Example 6 In vitro skin flux study of Patch B2: This study was designed to obtain in vitro transdermal flux data.
- FIGs 10-14 show that the patches prepared by Example 5 deliver more fentanyl per cm 2 active patch than DuragesicTM fentanyl transdermal system based on in vitro skin flux study experiments using modified Franz cell and human cadaver epidermis. Transdermal delivery is proportional to patch size. As shown in Figure 10, 72 hours of in vitro cumulative delivery per cm 2 of Patch B2 17.3 cm 2 100 mcg/h central strip is greater than the 42 cm 2 100 mcg/h DuragesicTM patch.
- Figures 11 and 12 show that the cumulative [pg/patch] and skin flux profile [pg/patch/hr] of the Patch B2 17.3 cm2 100 mcg/h central strip matches that of the 42 cm 2 100 mcg/h DuragesicTM patch.
- Figure 13 shows that Patch B2 24 cm 2 100 mcg/h central strip delivers more fentanyl [in pg/patch] than the 42 cm 2 100 mcg/h DuragesicTM patch in vitro.
- Figure 14 shows the in vitro fentanyl skin flux [pg/patch/hr] from Patch B2 24 cm 2 100 mcg/h is higher than the 100 mcg/h DuragesicTM patch.
- transbuccal delivery was simulated by permeation of naltrexone and fentanyl from a section of patch through human cadaver buccal tissue of 500 pm in thickness into the receptor solution, which is pH 6.8 commercially available artificial saliva stirred at 300 rpm at 37°C.
- the receptor fluid was stirred by means of a magnetic stirrer throughout the experiment to assure a uniform sample and a reduced diffusion barrier on the dermal side of the skin.
- the entire volume of receptor fluid was withdrawn at specified time intervals (0.25, 0.5, 1.0, 1.5, 2, 2.5, 3, 4, 7, and 24 hours) and immediately replaced with fresh fluid.
- the withdrawn fluid was filtered through a 0.45pm filter.
- the last 1-2 mL was then analyzed for the active agent, e.g., fentanyl, using conventional high performance liquid chromatography methods.
- the cumulative amount of fentanyl penetrating through the cadaver buccal was calculated and reported as mcg/cm 2 .
- the results are mean of 6 donors, 1 replicate per donor for Patch B2 while reference product DuragesicTM calculations are mean of 18 donors, 1 or 2 replicates per donor.
- the transbuccal delivery rate of fentanyl from Patch B2 was comparable to the reference product DuragesicTM.
- the transbuccal delivery rate of naltrexone in the non-skin contact layer and that of naltrexone in the skin contact layer are listed in the Table 5.
- the Patch B2 non-skin contact layer provides additional naltrexone transbuccal delivery.
- the total combined transbuccal delivery rate of naltrexone from Patch B2 from the 2 layers is thus even higher than the fentanyl delivery rate from the Patch B2 patch.
- the ratio of naltrexone delivery to fentanyl delivery is 4.1 and 2.6 in the early time points (0.25 h, 0.5 h) and 3.5 to 6.3 in later time points (1 to 24 h).
- Patch B2, 25 mcg/h strength is cut into halves. The liner from each half is peeled off. Both halves are transferred into a 20-mL glass vial. Ten mL of appropriate extraction medium is pipetted into the glass vial with screw cap. The vial was shaken for 2 hours at 180 osc/minute at room temperature or at 125 strike/minute in 70-C water bath. Samples (20 pL) are pipetted into separated HPLC vials at time points of 10 min, 30 min and 120 min, and diluted with 980 pL of Diluent for analysis by HPLC.
- Extraction media were Dl Water, Ethanol, Cooking Vinegar, pH 6.8 phosphate buffer and artificial saliva (Dissolve 16 g of sodium chloride, 0.38 g of sodium phosphate monobasic and 4.76 g of sodium phosphate dibasic into 2000 mL of water. Adjust pH to 6.8 with sodium hydroxide. Mix well and filter the solution through a 0.45-pm membrane filter. Degas the solution for 15 minutes.
- Figure 18 shows the simulated small volume solvent extraction from cut Patch B2 (14.06 cm 2 ; 25 mcg/h) at room temperature.
- Figure 19 shows the simulated small volume solvent extraction from cut Patch B2 (14.06 cm 2 ; 25 mcg/h Patch) at 70°C.
- naloxone HCI was administered intravenously (IV) into the cephalic vein followed by an approximately 1 mL flush with normal saline.
- the tested patch was cut in half and the release liner on each half patch was removed by hand and a sticky adhesive side applied to a separate cheek (between the gum and cheek), with sticky adhesive in direct contact with the cheek.
- All animals were anesthetized with isoflurane 3-5 minutes prior to patch application. The anesthesia was maintained for approximately 30 minutes. The dog's mouth was held closed, and pressure was applied to hold the cheek against the teeth/gum for 30 minutes. At 5, 10, and 20 minutes post patch application, the dog's mouth was briefly opened to observed the location of the patches and ensure that the patch was still flat against the buccal region. After each check, the patch was manipulated to return it to its original orientation if necessary, the mouth was carefully closed, and pressure reapplied. All observations were recorded. At 30 minutes post application, the patches were removed and the left and the right buccal regions were checked.
- Example 10 In a single-center, randomized, open-label, single-dose, crossover study, the relative bioavailability following a single dose dermal application of Patch B2 as compared to DuragesicTM was assessed under standard exposure conditions.
- naltrexone concentration averaged 4.42 ng/mL (ranging from 3.60 ng/mL to 5.98 ng/mL) throughout the duration of the study (0 hour to 192 hours).
- Subjects were administered with a single 12.5mcg fentanyl/hr Patch B2 applied dermally with overlay to the upper arm for 72 hours. Subsequently, the subjects were administered with a single 25mcg fentanyl/hr Patch B2 applied dermally with overlay to the upper arm for 72 hours.
- both naltrexone and 6-b- naltrexol concentrations were identified in amounts ranging from 5.1 - 71.9pg/ml amongst most of the subjects assayed. More subjects with identifiable levels of either naltrexone or e-b-naltrexol were found after the 25mcg/hr than in the 12.5mcg/hr patch.
- a 2-period sequential treatment phase in which the dosage form of Patch B2 is to be administered via unintended routes (buccal administration of an intact 12.5 mcg/hr patch or chewed administration of 1 ⁇ 4 of a 25 mcg/hr patch) in unblocked non-dependent, opioid-experienced, recreational drug users is assessed.
- Rental drug users who meet inclusion/exclusion criteria based on the results of screening assessments receive the first application of study drug within 28 days of their first screening assessments.
- Subjects are randomly assigned to sample groups to test either intact buccal administration over 30 minutes of a 12.5pg/h releasing patch or a 1 ⁇ 4 of a 25pg/h releasing patch chewed over 10 minutes and then removed. After a minimum 7 day wash-out period, the procedure is repeated with the second patch.
- the study consists of a 4-week screening period, followed by either an 8 single application treatment periods for group A (each lasting 9 days separated by at least a 2-week washout period) or 2 single-application treatment periods for group B (each lasting 6 days separated by at least a 10-day washout period), and a follow-up visit 7 to 10 days after completing the final treatment period.
- Groups A and B may run in parallel.
- 50mg naltrexone is administered every 12 hours until 117 hours after patch removal.
- a naloxone challenge test is performed at least 12 hours prior to patch application, to confirm that he/she was not physically dependent on opioids.
- Subjects in group A are randomly assigned to 1 of 4 treatment groups (A, B, C or D) and rotated through the 4 treatment sequences, with each treatment sequence receiving a 25mcg/hr transdermal dosage system of Patch B2 followed by a second series of the same treatment groups with the subjects receiving a 25mcg/hr Duragesic patch;
- Subjects in group B are randomly assigned to 1 of 2 treatment groups (E or F) and rotated through the 2 treatment sequences receiving a 25mcg/hr transdermal dosage system of Patch B2;
- Study drug ie, a 25mcg/hr transdermal dosage system of Patch B2 or a naltrexone-only patch
- Study drug are applied dermally to the upper-outer arm according to the procedures specified below and remain affixed to the skin for 72 hours.
- Safety monitoring and serial blood sampling for quantification of plasma fentanyl and/or naltrexone and e-b-naltrexol (depending on the group) is performed. All study patches are removed 72 hours ( ⁇ 10 minutes) after application, with safety monitoring and pharmacokinetic sampling continuing through 192 hours after application of a study patch for groups A and B.
- the subjects being assessed for applied daily heat undergo application of heat to a study patch using a standardized, conventional heating pad, covering the transdermal system and kept in place using a gauze bandage, set to increase the temperature at the skin surface to 38 ⁇ 3°C (setting on low, with a temperature probe placed under the pad for the entire 10 hours of heat application, the skin temperature being monitored and recorded at 0, 2, 4, 6, 8, and 10 hours post application).
- the heating pad is applied over the study patch for 10 hours on day 1 (0 to 10 hours), day 2 (24 to 34 hours), and day 3 (48 to 58 hours).
- the subjects being assessed for exercise undergo moderate physical exercise (ie, treadmill, weights, rowing machine, stationary bicycles, elliptical machine) for 1 hour on day 1 (over 4-5 hours post application), day 2 (over 28-29 hours post application), and day 3 (over 52-53 hours post application); rest breaks will be allowed.
- Heart rate is monitored continuously by subjects and monitored and recorded by study personnel every 10 minutes to reach >60% to ⁇ 80% of maximum heart rate, as calculated below:
- a constant heart rate is maintained as much as possible during the entire hour. If the heart rate reaches >80% of maximum heart rate, the intensity of the exercise is lowered and if the heart rate lowers to ⁇ 60% of maximum heart rate, the intensity of the exercise is increased.
- Example 15 A controlled, randomized, double blind study evaluating the clinical efficacy of the transdermal dosage system of the invention is performed in subjects with moderate to severe pain severe enough to require around-the-clock long term opioid treatment. 140 patients enter into a 15- day dose-titration period, followed by a 9-day double-blind period wherein patients are randomized to receive either the 25mcg/hr or 50mcg/hr fentanyl transdermal dosage system of the invention or of a reference fentanyl transdermal dosage system every 72 hours with a study duration of 12 weeks. Patient's responses are monitored for changes in their daily and weekly worst pain intensity scores at weeks 1 through 12 using a numerical 11-point rating scale and for changes from baseline to the final on-treatment visit in the Roland Morris Disability Questionnaire (RMDQ) score
- RMDQ Roland Morris Disability Questionnaire
Abstract
The disclosure generally relates to tamper-resistant transdermal dosage forms. The dosage forms can comprise an active agent and more than one antagonist layer.
Description
TRANSDERMAL DOSAGE FORM
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of International application No.
PCT/US2018/040536, filed July 2, 2018, U.S. Provisional Application No. 62/692,904, filed July 2, 2018, and U.S. Provisional Application No. 62/692,959, filed July 2, 2018, the entireties of which are incorporated by reference herein.
TECHNICAL FI ELD
[0002] The present disclosure relates to transdermal dosage forms having reduced potential for abuse. In particular, the disclosure relates to a system of transdermal administration of an active agent or agonist for example fentanyl to a subject over an extended period of time, wherein when subject to abuse, the system is capable of providing an adverse agent : active agent release ratio sufficient to prevent or discourage the abuse of the active agent. The present disclosure further relates to tamper-resistant transdermal dosage forms comprising: an active agent and a barrier which separates an antagonist in the form of a salt from the antagonist in base form.
BACKGROU ND
[0003] Pain is the most frequent reported symptom and is a common clinical problem confronting the clinician. Opioids have long been recognized as one of the most effective treatments of pain whether for treating or preventing cancer pain, central pain, myocardial infarction pain, pancreatic pain, colic pain, post-operative pain, headache pain, muscle pain, bone pain, and pain associated with intensive care. However, the US societal costs of prescription opioid abuse were estimated at more than $50 billion in 2007 and have only grown since.
[0004] Transdermal dosage forms offer a favorable route of administration by providing a method of administering sustained release of a drug for an extended period of time, while increasing patient compliance and decreasing extreme peaks and troughs in blood plasma. However, these dosage forms also contain large amounts of active agent and therefore also have a high potential for abuse.
[0005] There are many different approaches proposed to prevent abuse of transdermal patches. In particular, U.S. Pat. No. 5,236,714 discloses a dosage form comprising an abusable substance formulated with an antagonist for the abusable substance. U.S. Pat. No. 5,149,538 discloses a
transdermal patch comprising an opioid and an antagonist for the opioid that is releasable upon ingestion or solvent immersion, wherein the two reservoirs are separated by an impermeable barrier. U.S. Pat. Nos. 8,747,889, 8,790,689, 7,182,955 each discloses a transdermal patch system comprising an opioid and an antagonist with different methods for the antagonist to leave the patch.
[0006] US Pub. No. 20040126323 discloses a transdermal system with an opioid layer and an antagonist layer comprising antagonist salt and base form, both with or without a barrier separating the opioid and antagonist layer.
[0007] Although there seems to have been much research in the area, no one has been successful in bringing an abuse deterrent transdermal patch to the market even while the inadvertent misuse or abuse of transdermal dosage forms remains a significant health problem. Thus, there remains a need in the art for improved transdermal dosage forms that are effective for preventing abuse yet useful for delivering an active agent, such as an opioid or a pharmaceutically acceptable salt thereof.
SUMMARY
[0008] The present disclosure relates to an abuse deterrent transdermal dosage form wherein when contacted with skin, allows for the transdermal administration of an active agent, such as an opioid, but either (a) allows for the transdermal administration of only an amount of an antagonist that is ineffective for inhibiting the effect of the active agent, or (b) does not allow for the transdermal administration of the antagonist. However, if the transdermal dosage form of the disclosure is used to deliver an active agent via a route other than transdermal, such as buccal, nasal, oral, parenteral, rectal and/or vaginal, or if the transdermal dosage form is subjected to abuse or misuse, then the antagonist inhibits the effect of the active agent.
[0009] In one embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising an active agent and more than one antagonist reservoir or source. In some embodiments, the transdermal dosage comprises a first antagonist component comprising an active agent and a first antagonist of the active agent, and a second antagonist component comprising a second antagonist of the active agent.
[0010] In one embodiment, the present disclosure is directed to an abuse deterrent transdermal dosage forms wherein the first and second antagonist are separated by a barrier. In a further embodiment, the first antagonist is in the form of a pharmaceutically acceptable salt and the second antagonist is in the form of a free base.
[0011] In another embodiment of the present disclosure, the first antagonist component has a proximal and distal surface; the second antagonist component is disposed distal to the first antagonist component and the barrier is interposed between the first and second antagonist components.
[0012] In another embodiment, the present disclosure is directed to transdermal dosage forms wherein the first antagonist component comprises a homogenous mixture of the active agent and the first antagonist or alternatively, wherein the active agent and the first antagonist are separated by one or more spacers.
[0013] In a more specific embodiment, the disclosure relates to a transdermal system for administering an active agent through the skin, the system having a reduced potential for abuse, comprising:
(a) a first antagonist component comprising a first antagonist salt and an active agent in base form, wherein the active agent may be for example fentanyl or an analog thereof and the analog is selected from the group consisting of alfentanil, lofentanil, remifentanil, sufentanil and trefentanil;
(b) a second antagonist component comprising a second antagonist in base form, optionally in amorphous base form, the antagonist being releasable from system upon being ingested or substantially immersed in a solvent, and further wherein the antagonist is selected from the group consisting of naltrexone, methylnaltrexone, naloxone, nalbuphine, nalorphine, nalorphine dinicotinate, nalmefene, nadide, levallorphan, cyclozocine and pharmaceutically acceptable salts thereof; and
(c) a barrier layer, said barrier layer separating said first and second antagonist components, said barrier layer being substantially impermeable to said active agents and/or excipients, wherein the release of the antagonist from the system when used transdermally is such that levels are sufficiently low that the active agent's effect is maintained for more than about two days or about three days; and the system provides release of the antagonist at a rate sufficient to provide an abuse limiting release rate ratio of the antagonist to the active agent when the dosage form is subject to abuse, e.g., upon transmucosal applications or substantial immersion of the system in the solvent.
[0014] The disclosure is further directed to a kit for treating pain in a patient, comprising: a) the transdermal-delivery device as disclosed above; and b) a printed set of instructions directing the use of the transdermal dosage form to treat pain.
[0015] The disclosure is further directed to methods for treating or preventing pain in an animal comprising contacting the skin of an animal in need thereof with any one or more of the transdermal dosage forms described above. In one embodiment of the present disclosure, there is provided a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a Cmax less than 50% the Cmax of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when administered buccally.
[0016] The present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a Cmax less than 50% the Cmax of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when chewed.
[0017] The present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a AUCo-t less than 50% the AUCo-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when chewed.
[0018] The present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each
other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.7 for the first three hours after buccal administration.
[0019] The present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.7 for the first three hours after being chewed.
[0020] The present disclosure also relates to a transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits a AUCo-t less than 50% the AUCo-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses, when administered buccally.
[0021] When an opioid is used as the active agent, release of the antagonist from one or both of the antagonist sources can inhibit the euphoric effect of the opioid. In some embodiments, the transdermal dosage form will inhibit the euphoric effect of an opioid if the device is used other than transdermally whether before or after the device is used by an animal or human for treating or preventing pain.
[0022] The present disclosure relates to a method of treating pain in opioid-tolerant human patients in need of continuous, "around-the-clock," long-term opioid treatment by administering to a patient in need thereof, a transdermal dosage system comprising fentanyl and naltrexone. For example, the long-term treatment can be for at least 12 weeks.
[0023] The present disclosure also relates to a transdermal dosage system for administering fentanyl to a human, the system providing for the safe release of fentanyl after exposure to various extrinsic factors such as, for example, heat sources, exercise and/or showering.
[0024] In one embodiment of the invention, the transdermal dosage system provides for a fentanyl Cio of 110% or more and/or 180% or less, after exposure to an extrinsic factor.
[0025] In another embodiment of the invention, the transdermal dosage system provides for a naltrexone Ci0of 105% or more and/or 180% or less, after exposure to an extrinsic factor.
[0026] In another embodiment of the invention, the transdermal dosage system provides for a fentanyl AUCio of 110% or more and/or 250% or less, after exposure to an extrinsic factor.
[0027] In another embodiment of the invention, the transdermal dosage system provides for a naltrexone AUCio of 105% or more and/or 250% or less, after exposure to an extrinsic factor.
[0028] In yet another embodiment of the invention, the transdermal dosage system provides for a transformed fentanyl Cio to naltrexone Cio ratio and/or a transformed fentanyl AUCio to naltrexone AUCio ratio of 1:1 or less after exposure to an extrinsic factor.
[0029] In yet another embodiment of the invention, the transdermal dosage system provides for a fentanyl Cio that is 80% or less than the fentanyl Cio and/or the fentanyl AUCio of a reference fentanyl transdermal dosage system after exposure to an extrinsic factor.
[0030] The disclosure is further directed to methods for reducing or preventing misuse of any one or more transdermal patches de scribed above.
[0031] The present disclosure may be understood more fully by reference to the following figures, detailed description and examples, which are intended to exemplify non-limiting embodiments of the disclosure.
BRIEF DESCRIPTION OF THE DRAWINGS
[0032] FIG. 1 is a schematic cross-section of a transdermal dosage system of the disclosure.
[0033] FIG. 2 is a schematic cross-section of another transdermal dosage system of the disclosure.
[0034] FIG. 3 is a schematic cross-section of a prototype of Example 1.
[0035] FIG. 4 is a schematic cross-section of a prototype of Example 2.
[0036] FIGs. 5-9 are manufacturing process steps for patches B1 and B2.
[0037] FIG. 10 is a line graph showing in vitro cumulative delivery per cm2 of Patch B2 versus the Duragesic® patch.
[0038] FIGs. 11 and 12 are line graphs showing the cumulative [pg/patch] and skin flux profile [pg/patch/hr] of Patch B2 versus the Duragesic® patch.
[0039] FIG. 13 is a line graph of showing in vitro delivery of a Patch B2 24 cm2 100 mcg/h central strip versus a 42 cm2 100 mcg/h Duragesic® patch.
[0040] FIG. 14 is a line graph showing the in vitro fentanyl skin flux [pg/patch/hr] from Patch B2 24 cm2 100 mcg/h versus a 100 mcg/h Duragesic” patch.
[0041] FIG. 15 is a line graph showing in vitro results for patch B1 (solid line), patch A1 (dotted line;— ¨— ), and a Duragesic® patch (dotted line; ···¨···).
[0042] FIG. 16 is a schematic of an in vitro permeation study set-up.
[0043] FIG. 17 is a line graph showing the transbuccal delivery rate of fentanyl from Patch B2 as compared to the Duragesic® patch.
[0044] FIG. 18 is a bar graph showing the extraction of fentanyl and naltrexone from cut Patch B2 (14.06 cm2; 25 mcg/h) at room temperature using water, ethanol, phosphate buffer, vinegar, and artificial saliva.
[0045] FIG. 19 is a bar graph showing the extraction of fentanyl and naltrexone from cut Patch B2 (14.06 cm2; 25 mcg/h) at 70°C using water, ethanol, phosphate buffer, vinegar, and artificial saliva.
DETAILED DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
[0046] The phrases "transdermal dosage form" and "dosage form", as used herein, refer to any dosage form that, when contacted with a patient's skin for a sufficient period of time, can transdermally deliver an effective amount of any biologically active agent, such as a pharmaceutical agent, e.g., an opioid, through the patient's skin whether the type of transdermal dosage type is reservoir-type, polymer-matrix-type, drug-in-adhesive-type, or other.
[0047] As used herein, the term "transmucosal" refers to buccal, nasally, sublingual, topical, rectal, and/or vaginal.
[0048] As used herein, the term "buccal administration" refers to a topical route of administration by which a composition is held or applied to the buccal area of the inner cheek.
[0049] As used herein, the term "chew" or "chewed" refers to a method of administration of a composition whereby said composition is chewed over a pre-defined period of time in the oral cavity without being swallowed.
[0050] A "patient" or "animal" or "human" is a mammal, and includes, but is not limited to, a cow, monkey, horse, sheep, pig, chicken, turkey, quail, cat, dog, mouse, rat, rabbit, and guinea pig. In some embodiments, the "patient" or "animal" is a human.
[0051] As used herein the terms "dosage form" and "delivery device" are synonymous and interchangeable.
[0052] As used herein, the terms "abuse resistant" and "abuse deterrent" are synonymous and shall mean any transdermal dosage form that when misused, inhibits or deters the abuser from achieving the non-therapeutic effects sought from misuse of the composition, formulation or dosage form, such as opioid induced euphoria. Abuse or misuse shall mean any means including but not limited to being administered buccally, nasally, sublingually, parenterally, rectally, and/or vaginally to an animal.
[0053] As used herein, the phrase "active agent" refers to a pharmaceutical agent, therapeutic agent, drug, and/or agonist that causes a biological effect when absorbed in sufficient quantity into the blood stream of a patient. The active agent of the present disclosure unless otherwise indicated may be any drug substance that is capable of being abused. Many drugs have a potential for abuse, and include, for example, narcotics, such as morphine, fentanyl, codeine, sufentanil, and oxycodone;
psychostimulants, such as amphetamine, methamphetamine, and methylphenidate; methoxy substituted amphetamines, such as 3,4-methylenedioxymethamphetamine (MDMA); and
benzodiazepines, such as diazepam, oxazepam, and lorazepam. Examples include but are not limited to: alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, buprenorphine, butorphanol, clonitazene, codeine, desomorphine, dextromoramide, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dihydromorphone, dihydroisomorphine,
dimenoxadol, dimepheptanol, dimethylthiambutene, dioxaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmorphine, etonitazene, etorphine, dihydroetorphine,
fentanyl, heroin, hydrocodone, hydromorphone, hydromorphodone, hydroxypethidine, isomethadone, ketobemidone, levorphanol, levophenacylmorphan, lofentanil, meperidine, meptazinol, metazocine, methadone, metopon, morphine, myrophine, narceine, nicomorphine, norlevorphanol, normethadone, nalorphine, nalbuphene, normorphine, norpipanone, opium, oxycodone, oxymorphone, pantopon, papaveretum, paregoric, pentazocine, phenadoxone, phendimetrazine, phendimetrazone,
phenomorphan, phenazocine, phenoperidine, piminodine, piritramide, propheptazine, promedol, properidine, propoxyphene, propylhexedrine, sufentanil, tilidine, tramadol, pharmaceutically acceptable salts or prodrugs thereof and mixtures of any two or more thereof. In some embodiments, the active agent is a narcotic such as fentanyl, alfentanil, carfentanil, lofentanil, remifentanil, sufentanil, trefentanil, and the like. In further embodiments, the active is fentanyl.
[0054] As used herein, the term "fentanyl" refers to the chemical compound N-Phenyl-N-(l-(2- phenylethyl)-4-piperidinyl) propanamide as either free base or a pharmaceutically acceptable salt. In some embodiments, the fentanyl is in the form of its free base.
[0055] As used herein, the term "naltrexone" refers to the chemical compound morphinan-6- one, 17 (cyclopropylmethyl) 4,5-epoxy3,14-dihydroxy-(5ct) as free base, co-crystal, or a pharmaceutically acceptable salt. In some embodiments, two forms of naltrexone are present in the transdermal dosage form. In one preferred embodiment, the naltrexone is in the form of its free base. In another preferred embodiment, the naltrexone is in the form of its HCI salt.
[0056] The term "biological effect" or "biological result" as used herein refers to a physical reaction in a patient. In some embodiments, the effect is analgesic, euphoria, respiratory, anti- depressive, or combinations thereof.
[0057] As used herein, the phrase "adverse agent" or "antagonist" refers to a pharmaceutical agent, drug, and/or antagonist that partially or completely prevents, negates, diminishes, delays or reverses at least one biological effect of the active agent present in the dosage form, e.g. euphoric effect, or produces one or more unpleasant physiological reactions, e.g., vomiting, nausea, diarrhea, bad taste, when absorbed in sufficient amount into the blood stream of a patient. When an opioid agonist is used as the active agent in the dosage form of the present disclosure, an opioid antagonist can be used as the adverse agent.
[0058] As used herein, the terms "opioid" or "opioid agonist" refer to an active agent which exhibits opium- or morphine-like properties when absorbed in sufficient amounts into the bloodstream
of a patient. Opioid agonists bind, optionally stereo-specifically, to any one or more of several subspecies of opioid receptors and produce agonist activity.
[0059] As used herein, the phrase "opioid antagonist" refers to an adverse agent that either partially or completely prevents, negates, diminishes, delays or reverses at least one biological effect of an opioid agonist, e.g., euphoric effect, when absorbed in sufficient amounts into the blood stream of a patient.
[0060] The phrase "pharmaceutically acceptable salt," as used herein, is a salt formed from an acid and the basic nitrogen group of an opioid. In some embodiments, the salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucoronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p- toluenesulfonate, glubionate and pamoate (i.e., l,l'-methylene-bis-(2-hydroxy-3-naphthoate)) salts. The term "pharmaceutically acceptable salt" also refers to a salt prepared from an opioid having an acidic functional group, such as a carboxylic acid or sulfonic acid functional group, and a pharmaceutically acceptable inorganic or organic base. Suitable bases include, but are not limited to, hydroxides of alkali metals such as sodium, potassium, and lithium; hydroxides of alkaline earth metal such as calcium and magnesium; hydroxides of other metals, such as aluminum and zinc; ammonia, and organic amines, such as unsubstituted or hydroxy-substituted mono-, di-, or trialkylamines; dicyclohexylamine; tributyl amine; pyridine; N-methyl, N-ethylamine; diethylamine; triethylamine; mono-, bis-, or tris-(2-hydroxy-lower alkyl amines), such as mono-, bis-, or tris-(2-hydroxyethyl)amine, 2-hydroxy-tert-butylamine, or tris- (hydroxymethyl)methylamine, N, N, -di-lower alkyl-N-(hydroxy lower alkyl)-amines, such as N, N,- dimethyl-N-(2-hydroxyethyl)amine, or tri-(2-hydroxyethyl)amine; N-methyl-D-glucamine; and amino acids such as arginine, lysine, and the like. The list is not meant to be exhaustive but merely illustrative as a person of ordinary skill in the art would appreciate that other salts of opioids may be prepared.
[0061] As used herein, the phrase "active agent" refers to the drug which action is needed, intended or desired as the result of administration of the transdermal dosage form of the present invention. The active agent may be in any form which provides the desired biological effect. In addition to pharmaceutically acceptable salts, the active agent may be utilized in any solid state form such as amorphic or polymorphic form of the active agent. In some embodiments, the active agent is amorphous. In further embodiments, the active agent is crystalline. In further embodiments, the active agent includes the fentanyl polymorphs and amorphic forms described in US Patent Publication No.
2010/0076198.The term "active agent" therefore encompasses all amorphic forms or polymorphic forms existing under any possible crystal morphology. In further embodiments, the active agent such as fentanyl is in the form of a base. In further embodiments, the active agent such as fentanyl base is in amorphous form when formulated with excipients in the final transdermal dosage form. In further embodiments, the antagonist such as Naltrexone is in salt form such as HCI. In further embodiments, the antagonist such as Naltrexone salt is in micronized form. In further embodiments, the micronized Naltrexone salt is maintained in crystalline form within the transdermal dosage form. In further embodiments, the transdermal dosage form comprises crystalline ionic antagonist such as Naltrexone salt, admixed with povidone and silicone. In further embodiments, the antagonist such as Naltrexone is in base form. In further embodiments, the antagonist such as Naltrexone base is in amorphous form when formulated with excipients in the final transdermal dosage form. In further embodiments, the transdermal dosage form comprises amorphous antagonist in base form such as Naltrexone base in povidone.
[0062] The active agent or salt thereof also may be in the form of a prodrug. Such prodrugs may include, without limitation, esters, carbamates sulfate, oximes, sulfamites, carbonates and other conventional "pro-drug" forms, which, when administered in such form, convert to the active agent in vivo. In some embodiment, the prodrugs are esters.
[0063] As used herein, the term "proximal" refers to the location of a component, when considered as a whole, at a position which is relatively near to a site for application of the transdermal dosage form. The term "proximal surface" refers to the surface of a component which, when considered as a whole, is relatively near to a site for application of the transdermal dosage form, as compared to other surfaces of the component. In certain embodiments, the proximal surface of a component can be either continuous or discontinuous.
[0064] As used herein, the term "distal" refers to the location of a component, when considered as a whole, at a position which is relatively distant from a site for application of the transdermal dosage form. The term "distal surface" refers to the surface of a component which, when considered as a whole, is relatively distant from a site for application of the transdermal dosage form, as compared to other surfaces of the component. In certain embodiments, the proximal surface of a component can be either continuous or discontinuous.
[0065] As used herein, a "spacer" is meant to include a channel, pore, orifice, opening, void, gap, hole, crack and/or slit which to some degree provides a distance between two components. It may be made up of air, inactive ingredient, a barrier material or other.
[0066] As used herein, a "strip" is a formulation comprising either an active agent, antagonist or both in any desired geometry and may be either continuous or discontinuous and may be disposed in a pattern. As used herein, an "adhesive strip" is a formulation able to adhere to human skin and comprising one or more adhesives but substantially free of an active agent, antagonist, or both. The adhesive strip may have any desired geometry and may be either continuous or discontinuous and may be disposed in a pattern.
[0067] As used herein, the terms "commercially available fentanyl transdermal dosage system" or "reference fentanyl transdermal dosage system" refers to Duragesic™ (as commercially available in 2015) or a transdermally bioequivalent dosage form thereof, as determined by guidance from the U.S. Food and Drug Administration. The terms ""commercially available fentanyl transdermal dosage system" and "reference fentanyl transdermal dosage system" are used interchangeably herein.
[0068] As used herein, the "Duragesic™ fentanyl patch" is used interchangeably with "Durogesic™ fentanyl patch", "Duragesic™" and "Durogesic™" and refers to a fentanyl patch (Janssen Pharmaceuticals, Inc.).
[0069] The phrase "treatment of pain" or "treating pain," as used herein, includes amelioration of pain or the cessation of pain in an animal.
[0070] The phrase "prevention of pain" or "preventing pain," as used herein, includes the avoidance of the onset of pain in an animal.
[0071] As used herein, the phrase "dispersed" unless otherwise specified refers to dispersed, mixed, and/or dissolved either homogenously and/or heterogeneously.
[0072] As used herein, the phrase "component" refers to a layer, a stratum, a coating, a sheet, a film, a deposit, a sediment, a residue, and/or a cover. An "antagonist component" is a component comprising an antagonist. An antagonist component may or may not additional comprise an active agent. An "antagonist salt component" is a component comprising an antagonist salt. An "antagonist base component" is a component comprising an antagonist base. A "first antagonist component" is a component comprising an active agent and an antagonist.
[0073] As used herein, the term "opposed" as used with reference to two surfaces of a component refers to two surfaces which are generally facing in opposite directions regardless of whether one or both of the two surfaces are planar and/or parallel to each other.
[0074] As used herein, the terms "porous medium" and "porous material" are used interchangeably.
[0075] The term "layer" refers to a layer which contains one or more active or adverse agents. In some embodiments, a layer is a layer of the transdermal dosage system. In one embodiment, said layer forms part of a reservoir-type transdermal dosage form, in another embodiment said layer forms part of a polymer-matrix type transdermal dosage form and in yet another embodiment said layer forms part of a drug-in-adhesive type transdermal dosage form.
[0076] "Room temperature" refers to a typical indoor temperature. In some embodiments, room temperature is about 15 to about 25°C. In further embodiments, room temperature is about 20 °C.
[0077] The modifier "about" or "substantially" should be considered as disclosing the range defined by the absolute values of the two endpoints. For example, the expression "from about 2 to about 4" also discloses the range "from 2 to 4." When used to modify a single number, the term "about" may refer to plus or minus 10% of the indicated number and includes the indicated number.
For example, "about 10%" may indicate a range of 9% to 11%, and "about 1" means from 0.9-1.1.
[0078] The term "resistant to transdermal absorption" as used herein refers to the tendency of a compound to cross the epidermis layer. In some embodiments, resistant to transdermal absorption means that no amount of a compound discussed herein crosses the epidermis layer. In other embodiments, "resistant to transdermal absorption" includes a "biologically insignificant" or negligible amount.
[0079] The phase "biologically significant" refers to an amount of an active agent or antagonist described herein which results in one or more intended effect in a subject after administration. In some embodiments, the "biologically significant effect relates to a physiological symptom, an interaction between the active agent and/or antagonist and at least one component of the subject, among others.
In other embodiments, the biologically significant effect is the binding or lack thereof of the active agent or agonist to a relevant receptor in a patient. In still further embodiments, the biologically significant
effect is physically observed. In other embodiments, the biologically significant effect physically observed is less than the full effect of the active agent.
[0080] Alternatively, the phrase "biologically insignificant" refers to an amount of an active agent or antagonist described herein which results in no or less than an intended effect in a subject after administration. In some embodiments, the "biologically insignificant" effect relates to a physiological symptom, an interaction between the active agent and/or antagonist and at least one component of the subject, among others. In other embodiments, the biologically insignificant effect refers to the binding or lack thereof of the active agent or agonist to a relevant receptor in a patient. In still further embodiments, the biologically insignificant effect is not physically observed.
[0081] "Similar in effect" when used for comparing one or more compound refers to its ability to have a similar biological result, PK profile, PD profile, or combinations thereof. In some
embodiments, "similar in effect" means bioequivalent as defined below. In other embodiments, the transdermal dosage system is similar in effect to the Duragesic™ transdermal patch.
[0082] "Conformable" describes the ability of the transdermal dosage system to adapt in shape to the skin of the patient. In some embodiments, the patch is flexible. When applied as instructed, the transdermal dosage system moves as the skin of the patient moves. In some embodiments, the transdermal dosage system does not become displaced from the patient as the skin moves or shifts.
[0083] The term "bioequivalent" means that two products, i.e., transdermal patches, are expected to be the same. In some embodiments, "bioequivalent" means that, when two products are compared, there is no significant difference in the rate and/or extent to which the active agent becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study." In further embodiments, transdermal bioequivalence to fentanyl may be determined as specified by the Food and Drug Administration using (FDA) bioequivalent with pharmacokinetic endpoints, adhesion studies, skin irritation studies, skin sensitization studies, or combinations thereof. See, e.g., the "Draft Guidance on Fentanyl", recommended Oct. 2016 provided by the FDA which is incorporated by reference. In other embodiments, the transdermal dosage system parallels the FDA PK standard of about 90% Cl compared to the Duragesic™ transdermal patch. In further embodiments, the transdermal dosage system has a mean cumulative adhesion score of less than or equal to 0 as defined in the "Draft Guidance on Fentanyl" provided by the FDA and discussed above. In still other embodiments, the transdermal dosage system has a mean cumulative irritation
score of less than or equal to 0 as defined in the "Draft Guidance on Fentanyl" provided by the FDA and discussed above. In yet further embodiments, the transdermal dosage system has (a) one sensitizing response occurring at more than 24 hours after removal of the patch during the challenge phase, (b) the combined "dermal response" and "other effects" numeric score is at least 2 during the challenge phase, (c) the combined "dermal response" and "other effects" numeric scores obtained during the challenge phase are general higher than during the induction phase as defined in the "Draft Guidance on Fentanyl" provided by the FDA and discussed above.
[0084] As used herein, the term "therapeutically equivalent dose" refers to two products where the AUCo-t of the test product is between 80 - 120% that of the reference product.
[0001] As used herein, the term "opioid-tolerant human patient" shall refer to a human patient receiving, for one week or longer, at least 60 mg oral morphine/day, 25 meg transdermal fentanyl/hour, 30 mg oral oxycodone/day, 8 mg oral hydromorphone/day, 25 mg oral
oxymorphone/day, 60 mg oral hydrocodone per day, or an equianalgesic dose of another opioid.
[0085] As used herein and unless stated so explicitly otherwise, the term "Cmax" shall refer to the mean (average) observed maximum plasma concentration of the drug assayed. As used herein, the term "Cio" shall refer to the mean (average) observed maximum partial plasma concentration of the drug assayed at 10 hours after transdermal administration.
[0002] As used herein, the term "transformed Cio" shall refer to the ratio of the Cio of a drug as found with the application of an extrinsic factor such as heat, exercise or showering, relative to the Cio of said drug as found after transdermal administration without the application of an extrinsic factor and at a therapeutically equivalent dose.
[0086] As used herein and unless stated so explicitly otherwise, the term "AUC" shall refer to the area under the plasma concentration/time curve. The term "AUCio" shall refer to the area under the drug plasma concentration/time curve from time 0 to 10 hours after transdermal administration, the term "AUCo-t" shall refer to the area under the plasma concentration/time curve from time 0 to the last quantifiable drug concentration and the term "AUCo-inf" shall refer to the area under the plasma concentration/time curve from time 0 until the extrapolated drug concentration at infinity.
[0087] As used herein, the term "transformed AUCio" shall refer to the ratio of the AUCio of a drug as found with the application of an extrinsic factor such as heat, exercise or showering, relative to
the AUCio of said drug as found after transdermal administration without the application of an extrinsic factor and at a therapeutically equivalent dose.
[0088] As used herein, the term "naltrexone to fentanyl plasma concentration" shall mean the relative average naltrexone plasma concentration to the average fentanyl plasma concentration, as measured at any fixed time point.
[0089] As used herein, the term "when heat is applied" shall refer to the application of an external heat source such as, for example, a heating pad, covering the transdermal dosage system of the invention and set to keep the temperature at the skin surface to approximately 38°C, wherein said external heat source is held in place for at least 8 consecutive hours and wherein application of said external heat source is immediately after the transdermal application of said transdermal dosage system. As used herein, the term "when heat is not applied" shall refer to the application of the transdermal dosage system of the invention without the additional application of an external heat source.
[0090] As used herein, the term "after exercise" shall refer to the time period following the subject's undergoing of moderate physical exercise, such as, for example, treadmill or stationary bicycle exercise, wherein the subject's heart rate reaches between 50 - 80% their maximum heart rate, wherein said exercise occurs between 2-7 hours after administration of the transdermal dosage system of the invention and wherein said exercise lasts for at least 30 minutes. As used herein, the term "prior to exercise" shall refer to the application of said transdermal dosage system of the invention without the subject undergoing additional exercise for the entire time period of said application.
[0091] As used herein, the term "after showering" shall refer to the time period following the subject's undergoing of a lukewarm or hot shower, such as, for example, between 36 and 42°C, which lasts for between 5 and 30 minutes, wherein the water jet of said shower is held directly over the transdermal dosage system of the invention for at least 5 minutes and wherein said shower occurs between 2-7 hours after administration of said transdermal dosage system of the invention. As used herein, the term "prior to showering" shall refer to the application of said transdermal dosage system of the invention without the subject showering for the entire time period of said application.
[0092] The term "micronized" refers to particles wherein the particles are less than 10 pm in diameter. In other embodiments, D5o is less than about 10 pm or Dio is less than about 5 pm or Dg0 is less than about 30 pm. In further embodiments D5o is less than about 10 pm and Dio is less than about 5 pm and Dg0 is less than about 30 pm. A number of micronization techniques may be utilized to micronize
one or more components of the transdermal dosage system including, without limitation, conventional jet mills. The particle size or particle size distribution may be determined using techniques in the art such as light diffraction methods such as devices of Malvern Instruments, mechanical sieve shaking method, or air jet sieve analyses.
[0093] The present disclosure is directed to a transdermal dosage system having reduced potential for abuse, without blocking the therapeutic or beneficial effects of the active agent when the system is applied to the skin. In particular, the system of the present disclosure provides for the controlled release of the antagonist at a rate sufficient to provide an abuse limiting release rate ratio of the antagonist to the active when the dosage form is subject to abuse whether via buccal administration or solvent extraction for injection, wherein the system provides for a substantially minimized/negligible skin sensitization response from antagonist exposure.
[0094] The present disclosure is directed to a transdermal dosage system having reduced potential for abuse, without blocking the therapeutic or beneficial effects of fentanyl when the system is applied to the skin. In particular, the system of the present disclosure provides for the controlled release of naltrexone at a rate sufficient to provide an abuse limiting release rate ratio of the naltrexone to the fentanyl when the dosage form is subject to abuse whether via buccal administration or chewed, wherein the system provides for a substantially minimized/negligible skin sensitization response from naltrexone exposure.
[0095] This transdermal dosage form, when contacted with an animal's skin, allows for the transdermal administration of an active such as an opioid, but either (a) allows for the transdermal administration of only an amount of an antagonist salt that is ineffective for inhibiting the analgesic effect of the opioid, or (b) does not allow for the transdermal administration of the antagonist.
However, if the transdermal dosage form of the disclosure is used to deliver an opioid via a route other than transdermal, such as buccal, nasal, oral, parenteral, rectal and/or vaginal, or if the transdermal dosage form is subjected to abuse or misuse, then the antagonist, present in sufficient amounts to counter the effect of active, inhibits the euphoric effect of the opioid. In some embodiments, the transdermal dosage form will inhibit the euphoric effect of an opioid if the device is used other than transdermally whether before or after the device is used by an animal or human for treating or preventing pain.
[0096] In some embodiments, the active is fentanyl and the antagonist naltrexone and the transdermal dosage form, when contacted with an animal's skin, allows for the transdermal
administration of fentanyl, but either (a) allows for the transdermal administration of only an amount of naltrexone that is ineffective for inhibiting the analgesic effect of the fentanyl, or (b) does not allow for the transdermal administration of the naltrexone. However, if the transdermal dosage form of the disclosure is used to deliver fentanyl via a route other than transdermal, such as buccal, nasal, oral, parenteral, rectal and/or vaginal, or if the transdermal dosage form is subjected to abuse or misuse, then the naltrexone, present in sufficient amounts to counter the effect of fentanyl and inhibits the euphoric effect of the fentanyl. In some embodiments, the transdermal dosage form will inhibit the euphoric effect of fentanyl if used other than transdermally whether before or after it is used by an animal or human for treating or preventing pain.
[0097] The transdermal dosage form of the disclosure is also tamper-resistant in that if an abuser attempts to extract or separate an opioid from the transdermal dosage form, and self-administer the opioid via another route, such as, but not limited to, oral, parenteral, nasal, or buccal, rectal or vaginal, i.e., a route of administration that can result in a quick euphoric rush, the abuser would self- administer an amount of an antagonist along with the opioid, the amount of antagonist being effective to inhibit the euphoric effect of the opioid.
[0098] In another embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising an antagonist to agonist weight ratio of more than 3:1. In another embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising an antagonist to agonist weight ratio of about 4:1. The transdermal dosage form of the present disclosure would also have stability, adhesive properties as required by pharmaceutical regulatory approval.
[0099] In one embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising an active agent and more than one antagonist reservoir or source.
[00100] In one embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising fentanyl and more than one naltrexone layers.
[00101] In one embodiment, a transdermal dosage form comprises an active agent, a first antagonist component, and a second antagonist component. In one embodiment, the first antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable salt, and the second antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable base. In one embodiment, the first antagonist component comprises an
antagonist of the active agent in the form of a pharmaceutically acceptable salt and the active agent and the second antagonist component comprises an antagonist of the active agent in the form of a pharmaceutically acceptable base. In one embodiment the present disclosure is directed to an abuse deterrent transdermal dosage forms wherein the first and second antagonist are separated by a barrier.
[00102] In one embodiment, a transdermal dosage form comprises fentanyl, a first naltrexone component, and a second naltrexone component. In one embodiment, the first layer comprises naltrexone in the form of a pharmaceutically acceptable salt, and the second layer comprises naltrexone in the form of a pharmaceutically acceptable base. In one embodiment, the first layer comprises naltrexone in the form of a pharmaceutically acceptable salt and fentanyl and the second layer comprises naltrexone in the form of a pharmaceutically acceptable base. In one embodiment the present disclosure is directed to an abuse deterrent transdermal dosage forms wherein the first and second layers are separated by a barrier.
[00103] In another embodiment of the present disclosure, the transdermal dosage form the first antagonist component has a proximal and distal surface; the second antagonist component is disposed distal to the first antagonist component and the barrier is interposed between the first and second antagonist components. The dosage form may also include a backing layer located distal to the second antagonist component. In another embodiment, the backing layer is permeable to the second antagonist.
[00104] In another embodiment, the proximal surface of the first antagonist component has an area of about 5 to about 150 cm2. In another embodiment, the surface area of the dosage form is about 5 to about 60 cm2. In another embodiment, the surface area of the dosage form is about 25 to about 35 cm2 or about 100 to about 125 cm2. In another embodiment, the transdermal dosage form releases about 10 to about 100 meg active agent per hour to skin. In another embodiment, the transdermal dosage form releases about 12.5, 25, 50, 75 or 100 meg active agent per hour to skin. Any amount of active is possible, for example the transdermal dosage form may contain anywhere from 0.1 to 500 mg of active agent. In one embodiment, the transdermal dosage form comprises fentanyl base or alkaloid in an amount of about 1 to about lOmg.
[00105] In one embodiment of this disclosure, the antagonist salt and active agent are present in a single layer between the barrier and the skin-contacting surface. The layer may comprise the antagonist salt and the active agent dispersed, mixed and/or dissolved to some degree homogenously throughout a polymeric material. In another embodiment, the layer may comprise the antagonist salt
and active agent where they are individually dispersed, mixed and/or dissolved prior to being set in alternating strips of active agent and antagonist salt. In another embodiment, the active agent and the first antagonist are separated by one or more spacers. In another embodiment, the active agent and the first antagonist are in a ratio of about 1:2 or 1:1.81.
[00106] In one embodiment, the active agent and the first antagonist are in a ratio of about 1:2 or 1:1.81, the active agent is fentanyl and the first antagonist, naltrexone. In another embodiment, the fentanyl is in the form of its free base, the Naltrexone is in the form of a salt and the fentanyl base and the Naltrexone salt are in a ratio of about 1:2 or 1:1.81. In another embodiment, the single layer comprises about 8% ionic naltrexone such as naltrexone HCI. In another embodiment, the single layer comprises ionic naltrexone such as naltrexone HCI in crystalline form. Additional excipients such as solubilizers like povidone or adhesives such as silicone adhesives may also be present.
[00107] In a more specific embodiment, the disclosure relates to a transdermal system for administering an active agent through the skin, the system having a reduced potential for abuse, comprising:
(a) a first antagonist component comprising a first antagonist salt and an active agent in base form, wherein the active agent may be for example amorphous or crystalline fentanyl or an analog thereof and the analog is selected from the group consisting of alfentanil, lofentanil, remifentanil, sufentanil and trefentanil;
(b) a second antagonist component comprising a second antagonist in base form, optionally in amorphous base form, the antagonist being releasable from system upon being ingested or substantially immersed in a solvent, and further wherein the antagonist is selected from the group consisting of naltrexone, methylnaltrexone, naloxone, nalbuphine, nalorphine, nalorphine dinicotinate, nalmefene, nadide, levallorphan, cyclozocine and pharmaceutically acceptable salts thereof; and
(c) a barrier layer, said barrier layer separating said first and second antagonist components, said barrier layer optionally being substantially impermeable to said active agents and/or excipients, wherein: the release of the antagonist from the system when used transdermally such that levels are sufficiently low that the active agent's biological effect is maintained; and
the system provides release of the antagonist at a rate sufficient to provide an abuse limiting release rate ratio of the antagonist to the active agent when the dosage form is subject to abuse, e.g., upon transmucosal applications or substantial immersion of the system in the solvent.
[00108] In another embodiment, the disclosure relates to a transdermal system for administering an active agent through the skin, wherein the active agent, such as fentanyl, is formulated in amorphous form while the first antagonist salt, such as Naltrexone HCI, is in crystalline form and the second antagonist, such as naltrexone base, is formulated in amorphous form.
[00109] In another embodiment, the disclosure relates to a transdermal system for administering an active agent through the skin, wherein the active agent, such as fentanyl, is formulated in crystalline form while the first antagonist salt, such as Naltrexone HCI, is in amorphous form and the second antagonist, such as naltrexone base, is formulated in crystalline form.
[00110] When an opioid is used as the active agent, release of the antagonist from one or both of the antagonist sources can inhibit the euphoric effect of the opioid. In some embodiments, the transdermal dosage form will inhibit the euphoric effect of an opioid if the device is used other than transdermally whether before or after the device is used by an animal or human for treating or preventing pain.
[00111] Release of the antagonists from the system, when used transdermally, is controlled so that antagonist levels are sufficiently low while maintaining the active agent's effect. In some embodiments, the active agent's effects are maintained for more than about two days. In other embodiments, the active agent's effects are maintained for more than about three days.
[00112] For example, if an abuser tries to extract an opioid, for example, fentanyl, from the transdermal dosage form by placing it in a solvent, including saliva, then an amount of antagonist, for example, naltrexone, would also be extracted, providing a mixture of the opioid and the antagonist. Antagonist free bases, such as naltrexone free base, generally exhibit greater solubility, and in one embodiment bioavailability, in non-aqueous solvents than in aqueous solvents, while antagonist salts, for example, naltrexone HCI, generally exhibits greater solubility and, in one embodiment, bioavailability in aqueous solvents than in non-aqueous solvents. Thus, for example, depending on the solvent of choice, if an abuser attempts to extract fentanyl from the transdermal dosage form, whether aqueous or non-aqueous solvent, at least one form of naltrexone will be released along with the opioid. If a mixture of fentanyl and naltrexone is administered via a route other than the intended transdermal route, such a
transmucosally, at least one form of the naltrexone would exert its antagonistic effect to inhibit the euphoric effect of the opioid.
[00113] Referring now to Figure 1, in one embodiment, the present disclosure comprises a transdermal dosage form 100 comprising: a first antagonist component 120, optionally comprising a polymeric material and optionally in the form of a continuous, planar component in the form of a slab; an second antagonist component 140 comprising an antagonist in free base form; and optionally a barrier 130.
[00114] The first antagonist component has a proximal surface 115, which may be a skin contacting surface and is optionally covered with a release liner 110, and a distal surface 125 which is opposed to the proximal surface 115. The barrier 130 is disposed between the distal surface 125 of the first antagonist component 120 and the second antagonist component 140. A backing 150 is disposed adjacent to the second antagonist component 140 at a location which provides an outer surface 155 of the dosage form 100. In some embodiments, a permeable backing layer 150 is adjacent to the second antagonist component 140. In other embodiments, a structured release liner 110 is located proximal to the first antagonist component 120 and functions to protect the surface of the skin-contacting first antagonist component 120 prior to use of the dosage form.
[00115] In another embodiment, shown in FIG. 4, the present disclosure comprises a transdermal dosage form 200 comprising:
[00116] a first antagonist component 120, comprising alternating strips of an active agent 121 and an antagonist in base form 122, optionally wherein the active agent and first antagonist is discontinuous having an alternating spacers 123 made up of air or voided spaces or inactive material between the active agent and antagonist strip; a second antagonist component 140 comprising an adverse agent; and a barrier 130.
[00117] The first antagonist component 120 has a proximal surface 115, which may be a skin contacting surface and is optionally covered with a release liner 110, and a distal surface 125 which is opposed to the proximal surface 115. The barrier 130 is disposed between the distal surface 125 of the
first antagonist component 120 and the second antagonist component 140. In some embodiments, a structured release liner 110 is located proximal to the first antagonist component 120 and functions to protect the structured surface of the skin-contacting first antagonist component 120 prior to use of the dosage form.
[00118] In one embodiment of transdermal dosage form 200, the width of the strips of active agent component 121 is greater than about 0.1 cm. In another embodiment, the width of said 121 is greater than about 0.2 cm. In another embodiment, the width of said 121 is greater than about 0.4 cm.
In another embodiment, the width of said 121 is less than about 2.0 cm. In another embodiment, the width of the strips is less than 1.0 cm. In another embodiment, the width of said 121 is less than 0.6 cm. Although a specific configuration is described above, it should be understood that the alternating strips may be of any shape such as but not limited to squares, diamonds, ovals, triangles, pentagons, or hexagons.
[00119] The following relates to both figures 1 and 4 above and the disclosure in general.
[00120] In one embodiment, the active agent is an opioid and the antagonist is an opioid antagonist. Opioid antagonists useful in the present disclosure include, but are not limited to, naloxone, naltrexone, nalmefene, nalbuphine, nalorphine, cyclazacine, cyclazocine, levallorphan, pharmaceutically acceptable salts thereof, and mixtures thereof. In certain embodiments, the opioid antagonist is nalmefene, naloxone, naltrexone, or a pharmaceutically acceptable salt thereof. In another embodiment, the opioid antagonist is a naltrexone. In some embodiments of this disclosure, the antagonist free base is naltrexone in amorphous form.
[00121] Useful opioid antagonist salts include salts formed from an acid and the basic nitrogen group of an opioid antagonist. Examples of opioid antagonist salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucoronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p- toluenesulfonate, and pamoate (i.e., l,l'-methylene-bis-(2-hydroxy-3-naphthoate)) salts. Other opioid antagonist salts include salts prepared from an antagonist having an acidic functional group, such as a carboxylic acid or sulfonic acid functional group, and a pharmaceutically acceptable inorganic or organic base. In some embodiments, the antagonist salt is naltrexone hydrochloride.
[00122] The active agent or agonist will be present in an amount such that the composition delivers a therapeutically effective amount for the condition being treated. This amount will vary according to the type of active agent used, the condition to be treated, the amount of time the composition is allowed to remain in contact with the skin of the subject, and other factors known to those of skill in the art. For example, information on dosing and the amount of opioid agonist active agent present in a transdermal dosage form is set forth in U.S. Published Patent Application No.
2002/0119187 and U.S. Published Patent Application No. 2003/0026829 each of which are incorporated by reference herein in their entirety for all purposes. In one embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is greater than about 0.01 wt-%, based on the total weight of the composition of the active agent component. In another embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is greater than about 1.0 wt-%, based on the total weight of the composition of the active agent component. In another embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is less than about 40 wt-%, based on the total weight of the composition of the active agent component. In another embodiment, the amount of active agent present in the transdermal drug delivery composition of the disclosure is less than about 20.0 wt-%, based on the total weight of the composition of the active agent component.
[00123] The analgesically effective amount of an opioid present in the transdermal dosage form typically ranges from about 0.01 to about 50 mg/cm2 in one embodiment, from about 0.05 to about 15mg/cm2 in another embodiment, and from about 0.05 to about 5.0mg/ cm2 in another embodiment. It is well within the purview of one skilled in the art to readily determine the analgesically effective amount of an opioid needed for a particular indication.
[00124] In one embodiment, the antagonist free base and an antagonist salt are present in an amount sufficient to inhibit at least one biological effect of an active agent. In a further embodiment, the antagonist free base and an antagonist salt are provided in a total amount sufficient to inhibit the euphoric effect of an opioid when the transdermal dosage form is subjected to abuse or misuse.
[00125] In one embodiment, the naltrexone free base and naltrexone salt are present in an amount sufficient to inhibit at least one biological effect of fentanyl. In a further embodiment, the naltrexone free base and naltrexone salt are provided in a total amount sufficient to inhibit the euphoric effect of fentanyl when the transdermal dosage form is subjected to abuse or misuse.
[00126] Depending on the type of abuse, the adverse agent present in transdermal patch 100 or 200, the adverse agent is capable of containing a sufficient amount of adverse agent to blunt or block at least one biological effect of the active agent or to cause at least one unpleasant side effect in a patient or animal when the patch is subjected to abuse or misuse. This amount can vary according to the amount and type of active agent in the dosage form. The amount may be included in each adverse agent component individually or combined in component 140 and component 120 depending on desired effect and form of the formulation.
[00127] In one embodiment of the present disclosure, the transdermal patch is capable of containing a sufficient amount of naltrexone to blunt or block at least one biological effect of the fentanyl or to cause at least one unpleasant side effect in a patient or animal when the patch is subjected to abuse or misuse. This amount can vary according to the amount and type of fentanyl in the dosage form. The amount may be included in each layer individually or combined in second antagonist component 140 and first antagonist component 120 depending on desired effect and form of the formulation.
[00128] The antagonist components comprise an adverse agent in any form or composition or reservoir which allows the antagonist to be at least partially extracted in the presence of a solvent, including but not limited to, water, ethanol or ether, or mixtures thereof. In certain embodiments, the antagonist can be dispersed, mixed and/or dissolved in a polymeric material, including but not limited to, the polymeric materials which are suitable for incorporation into the active agent component.
[00129] In one embodiment of the present disclosure, the dosage form is provided such that the antagonist, such as naltrexone, is not absorbed to any biologically significant degree into a blood stream when administered transdermally. In other embodiments, the dosage form is provided such that the ratio of adverse agent to active agent in the dosage form is from about 1:10 to about 10:1. In other embodiments, the dosage form is provided such that the ratio of naltrexone to fentanyl in the dosage form is from about 1:10 to about 10:1. In other embodiments, the dosage form is provided such that the ratio of adverse agent to active agent in the dosage form is more than 3:1. In other embodiments, the dosage form is provided such that the ratio of naltrexone to fentanyl in the dosage form is more than 3:1. In another embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising an antagonist to agonist weight ratio of about 4:1.
In another embodiment of the present disclosure, there is provided a transdermal dosage form having reduced potential for abuse comprising naltrexone to fentanyl weight ratio of about 4:1. The
transdermal dosage form of the present disclosure would also have stability, adhesive properties as required by pharmaceutical regulatory approval.
[00130] In another embodiment, the ratio of adverse agent to active agent released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated, is at least 1:5, 1:4, 1:3, 1:2, or 1:1. In another embodiment, the ratio of adverse agent to active agent released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated, is at least 1:1 at numerous time points between 5 min. to 4 hours. In another embodiment, the ratio of naltrexone to fentanyl released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated, is at least 1:5, 1:4, 1:3, 1:2, or 1:1. In another embodiment, the ratio of naltrexone to fentanyl released from the dosage form when the dosage form is tampered with, e.g., chewed, extracted, mechanically violated, is at least 1:1 at time points between 5 min. to 4 hours.
[00131] In one embodiment, when administered buccally, the Cmax of the dosage form is less than 50% the buccally administered Cmax of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses. In another embodiment, the buccally administered Cmax of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the buccally administered Cmax of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
[00132] In one embodiment, when chewed, the Cmax of the dosage form is less than 50% the chewed Cmax of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses. In another embodiment, the chewed Cmax of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the chewed Cmax of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
[00133] In one embodiment, when administered buccally, the AUCo-t of the dosage form is less than 50% the buccally administered AUC0-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses. In another embodiment, the buccally administered AUCo-t of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the buccally administered
AUCo-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
[00134] In one embodiment, when chewed, the AUCo-t of the dosage form is less than 50% the chewed AUCo-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses. In another embodiment, the chewed AUC0-t of the dosage form is less than 49%, less than 48%, less than 47%, less than 46%, less than 45%, less than 44%, less than 43%, less than 42%, less than 41%, less than 40%, less than 39%, less than 38%, less than 37%, less than 36%, less than 35%, less than 34%, less than 33%, less than 32%, less than 31%, less than 30%, less than 29%, less than 28%, less than 27%, less than 26%, less than 25%, less than 24%, less than 23%, less than 22%, less than 21% or less than 20% the chewed AUC0-t of the commercially available fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
[00135] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first fifteen minutes after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater
than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first fifteen minutes after buccal administration.
[00136] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first thirty minutes after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first thirty minutes after buccal administration.
[00137] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first hour after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first hour after buccal administration.
[00138] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first two hours after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first two hours after buccal administration.
[00139] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first three hours after buccal administration. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first three hours after buccal administration.
[00140] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first fifteen minutes after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater
than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first fifteen minutes after being chewed.
[00141] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first thirty minutes after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first thirty minutes after being chewed.
[00142] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first hour after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75,
greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first hour after being chewed.
[00143] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first two hours after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than
1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than 2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first two hours after being chewed.
[00144] In one embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.7 for the first three hours after being chewed. In another embodiment, the naltrexone to fentanyl plasma concentration ratio is greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, greater than 0.95, greater than 1, greater than 1.05, greater than 1.1, greater than 1.15, greater than 1.2, greater than 1.25, greater than 1.3, greater than 1.35, greater than 1.4, greater than 1.45, greater than 1.5, greater than 1.55, greater than 1.6, greater than 1.65, greater than 1.7, greater than 1.75, greater than 1.8, greater than 1.85, greater than 1.9, greater than 1.95, greater than 2, greater than 2.05, greater than 2.1, greater than 2.15, greater than 2.2, greater than 2.25, greater than
2.3, greater than 2.35, greater than 2.4, greater than 2.45, greater than 2.5, greater than 2.55, greater
than 2.6, greater than 2.65, greater than 2.7, greater than 2.75, greater than 2.8, greater than 2.85, greater than 2.9, greater than 2.95, greater than 3, greater than 3.05, greater than 3.1, greater than 3.15, greater than 3.2, greater than 3.25, greater than 3.3, greater than 3.35, greater than 3.4, greater than 3.45, greater than 3.5, greater than 3.55, greater than 3.6, greater than 3.65, greater than 3.7, greater than 3.75, greater than 3.8, greater than 3.85, greater than 3.9, greater than 3.95 or greater than 4 for the first three hours after being chewed.
[00145] In one embodiment, when heat is applied, the fentanyl Cio of the dosage form is 110% or more than the fentanyl Cio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl Cio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the fentanyl Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
[00146] In one embodiment, when heat is applied, the fentanyl Cio of the dosage form is 180% or less than the fentanyl Cio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl Cio of said dosage form when heat is not applied at therapeutically equivalent doses.
[00147] In one embodiment, when heat is applied, the naltrexone Cio of the dosage form is 105% or more than the naltrexone Cio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the naltrexone Cio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the naltrexone Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
[00148] In one embodiment, when heat is applied, the naltrexone Cio of the dosage form is 180% or less than the naltrexone Cio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, 115% or less, or 110% or less than the naltrexone Cio of said dosage form when heat is not applied at therapeutically equivalent doses.
[00149] In one embodiment, when heat is applied, the fentanyl AUCio of the dosage form is 110% or more than the fentanyl AUCio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl AUCio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the fentanyl Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
[00150] In one embodiment, when heat is applied, the fentanyl AUCio of the dosage form is 250% or less than the fentanyl AUCio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
[00151] In one embodiment, when heat is applied, the naltrexone AUCio of the dosage form is 105% or more than the naltrexone AUCio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the naltrexone AUCio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more,
140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the naltrexone Cio of the dosage form when heat is not applied at therapeutically equivalent doses.
[00152] In one embodiment, when heat is applied, the naltrexone AUCio of the dosage form is 250% or less than the naltrexone AUCio of said dosage form when heat is not applied at therapeutically equivalent doses. In another embodiment, when heat is applied, the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or
less, 130% or less, 125% or less 120% or less, 115% or less, or 110% or less than the naltrexone AUCio of said dosage form when heat is not applied at therapeutically equivalent doses.
[00153] In one embodiment, after exercise, the fentanyl Cio of the dosage form is 110% or more than the fentanyl Cio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the fentanyl Cio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the fentanyl Cio of the dosage form prior to exercise at therapeutically equivalent doses.
[00154] In one embodiment, after exercise, the fentanyl Cio of the dosage form is 180% or less than the fentanyl Cio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the fentanyl of the dosage form is less than 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl Cio of said dosage form prior to exercise at therapeutically equivalent doses.
[00155] In one embodiment, after exercise, the naltrexone Cio of the dosage form is 105% or more than the naltrexone Cio of said dosage form prior to exercise at therapeutically equivalent doses.
In another embodiment, after exercise, the naltrexone Cio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the naltrexone Cio of the dosage form prior to exercise at therapeutically equivalent doses.
[00156] In one embodiment, after exercise, the naltrexone Cio of the dosage form is 180% or less than the naltrexone Cio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, 115% or less, or 110% or less than the naltrexone Cio of said dosage form prior to exercise at therapeutically equivalent doses.
[00157] In one embodiment, after exercise, the fentanyl AUCio of the dosage form is 110% or more than the fentanyl AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
In another embodiment, after exercise, the fentanyl AUCio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more,
190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the fentanyl Cio of the dosage form prior to exercise at therapeutically equivalent doses.
[00158] In one embodiment, after exercise, the fentanyl AUCio of the dosage form is 250% or less than the fentanyl AUCio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, or 115% or less than the fentanyl AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
[00159] In one embodiment, after exercise, the naltrexone AUCio of the dosage form is 105% or more than the naltrexone AUCio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the naltrexone AUCio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the naltrexone Cio of the dosage form prior to exercise at therapeutically equivalent doses.
[00160] In one embodiment, after exercise, the naltrexone AUCio of the dosage form is 250% or less than the naltrexone AUCio of said dosage form prior to exercise at therapeutically equivalent doses. In another embodiment, after exercise, the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, 115% or less, or 110% or less than the naltrexone AUCio of said dosage form prior to exercise at therapeutically equivalent doses.
[00161] In one embodiment, after showering, the fentanyl Cio of the dosage form is 110% or more than the fentanyl Cio of said dosage form prior to showering at therapeutically equivalent doses.
In another embodiment, after showering, the fentanyl Cio of the dosage form is 115% or more, 120% or
more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the fentanyl Cio of the dosage form prior to showering at therapeutically equivalent doses.
[00162] In one embodiment, after showering, the fentanyl Cio of the dosage form is 180% or less than the fentanyl Cio of said dosage form prior to showering at therapeutically equivalent doses. In another embodiment, after showering, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, or 115% or less than the fentanyl Cio of said dosage form prior to showering at therapeutically equivalent doses.
[00163] In one embodiment, after showering, the naltrexone Cio of the dosage form is 105% or more than the naltrexone Cio of said dosage form prior to showering at therapeutically equivalent doses. In another embodiment, after showering, the naltrexone Cio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, or 175% or more than the naltrexone Cio of the dosage form prior to showering at therapeutically equivalent doses.
[00164] In one embodiment, after showering, the naltrexone Cio of the dosage form is 180% or less than naltrexone Cio of said dosage form prior to showering at therapeutically equivalent doses.
In another embodiment, after showering, the fentanyl of the dosage form is 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less, 120% or less, 115% or less, or 110% or less than the naltrexone Cio of said dosage form prior to showering at therapeutically equivalent doses.
[00165] In one embodiment, after showering, the fentanyl AUCio of the dosage form is 110% or more than the fentanyl AUCio of said dosage form prior to showering at therapeutically equivalent doses. In another embodiment, after showering, the fentanyl AUCio of the dosage form is 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the fentanyl Cio of the dosage form prior to showering at therapeutically equivalent doses.
[00166] In one embodiment, after showering, the fentanyl AUCio of the dosage form is 250% or less than the fentanyl AUCio of said dosage form prior to showering at therapeutically equivalent
doses. In another embodiment, after showering, the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, or 115% or less than the fentanyl AUCio of said dosage form prior to showering at therapeutically equivalent doses.
[00167] In one embodiment, after showering, the naltrexone AUCio of the dosage form is 105% or more than the naltrexone AUCio of said dosage form prior to showering at therapeutically equivalent doses. In another embodiment, after showering, the naltrexone AUCio of the dosage form is 110% or more, 115% or more, 120% or more, 125% or more, 130% or more, 135% or more, 140% or more, 145% or more, 150% or more, 155% or more, 160% or more, 165% or more, 170% or more, 175% or more, 180% or more, 185% or more, 190% or more, 195% or more, 200% or more, 205% or more, 210% or more, 215% or more, 220% or more, 225% or more, 230% or more, 235% or more, 240% or more, or 245% or more than the naltrexone Cio of the dosage form prior to showering at therapeutically equivalent doses.
[00168] In one embodiment, after showering, the naltrexone AUCio of the dosage form is 250% or less than the naltrexone AUCio of said dosage form prior to showering at therapeutically equivalent doses. In another embodiment, after showering, the fentanyl of the dosage form is 245% or less, 240% or less, 235% or less, 230% or less, 225% or less, 220% or less, 215% or less, 210% or less, 205% or less, 200% or less, 195% or less, 190% or less, 185% or less, 180% or less, 175% or less, 170% or less, 165% or less, 160% or less, 155% or less, 150% or less, 145% or less, 140% or less, 135% or less, 130% or less, 125% or less 120% or less, 115% or less, or 110% or less than the naltrexone AUCio of said dosage form prior to showering at therapeutically equivalent doses.
[00169] In one embodiment, when heat is applied, the transformed fentanyl Cio, i.e., the ratio of the Cio of fentanyl as found with the application of heat relative to the fentanyl Cio as found when heat is not applied at a therapeutically equivalent dose, is 1.1. In another embodiment, when heat is applied, the transformed fentanyl Cio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or
1.75.
[00170] In one embodiment, when heat is applied, the transformed naltrexone Cio, i.e., the ratio of the Cio of naltrexone as found with the application of heat relative to the naltrexone Cio as found when heat is not applied at a therapeutically equivalent dose, is 1.05. In another embodiment,
when heat is applied, the transformed naltrexone Cio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
[00171] In one embodiment, when heat is applied, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1 or less. In another embodiment, when heat is applied, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1:1.25 or less, 1:1.3 or less, 1:1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1:1.55 or less, or 1:1.6 or less.
[00172] In one embodiment, when heat is applied, the transformed fentanyl AUCio, i.e., the ratio of the AUCio of fentanyl as found with the application of heat relative to the fentanyl AUCio as found when heat is not applied at a therapeutically equivalent dose, is 1.1. In another embodiment, when heat is applied, the transformed fentanyl AUCio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55,
1.6, 1.65, 1.7, 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
[00173] In one embodiment, when heat is applied, the transformed naltrexone AUCio, i.e., the ratio of the AUCio of naltrexone as found with the application of heat relative to the naltrexone AUCio as found when heat is not applied at a therapeutically equivalent dose, is 1.05. In another embodiment, when heat is applied, the transformed naltrexone AUCio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
[00174] In one embodiment, when heat is applied, the transformed fentanyl AUCio to naltrexone AUCio ratio is less than 1: 1. In another embodiment, when heat is applied, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1:1.05 or less, 1: 1.1 or less, 1:1.15 or less, 1: 1.2 or less, 1:1.25 or less, 1: 1.3 or less, 1:1.35 or less, 1: 1.4 or less, 1: 1.45 or less, 1: 1.5 or less, 1:1.55 or less, 1:1.6 or less, 1:1.65 or less, 1: 1.7 or less, 1:1.75 or less, 1:1.8 or less, 1:1.85 or less, 1:1.9 or less, 1:1.95 or less, 1:2 or less, 1:2.05 or less, 1:2.1 or less, 1:2.15 or less, 1:2.2 or less, or 1:2.25 or less.
[00175] In one embodiment, after exercise, the transformed fentanyl Cio, i.e., the ratio of the Cio of fentanyl as found after exercise relative to the fentanyl Cio as found prior to exercise at a therapeutically equivalent dose, is 1.1. In another embodiment, after exercise, the transformed fentanyl Cio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
[00176] In one embodiment, after exercise, the transformed naltrexone Cio, i.e., the ratio of the Cio of naltrexone as found after exercise relative to the naltrexone Cio as found prior to exercise at a therapeutically equivalent dose, is 1.05. In another embodiment, after exercise, the transformed naltrexone Cio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
[00177] In one embodiment, after exercise, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1 or less. In another embodiment, after exercise, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1:1.25 or less, 1:1.3 or less, 1:1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1:1.55 or less, or 1:1.6 or less.
[00178] In one embodiment, after exercise, the transformed fentanyl AUCio, i.e., the ratio of the AUCio of fentanyl as found after exercise relative to the fentanyl AUCio as found prior to exercise at a therapeutically equivalent dose, is 1.1. In another embodiment, after exercise, the transformed fentanyl AUCio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7, 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
[00179] In one embodiment, after exercise, the transformed naltrexone AUCio, i.e., the ratio of the AUCio of naltrexone as found after exercise relative to the naltrexone AUCio as found prior to exercise at a therapeutically equivalent dose, is 1.05. In another embodiment, after exercise, the transformed naltrexone AUCio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 1.75,
1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
[00180] In one embodiment, after exercise, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1:1 or less. In another embodiment, after exercise, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1: 1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1: 1.25 or less, 1:1.3 or less, 1: 1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1: 1.55 or less, 1:1.6 or less, 1: 1.65 or less, 1:1.7 or less, 1: 1.75 or less, 1:1.8 or less, 1:1.85 or less, 1:1.9 or less, 1: 1.95 or less, 1:2 or less, 1:2.05 or less, 1:2.1 or less, 1:2.15 or less, 1:2.2 or less, or 1:2.25 or less.
[00181] In one embodiment, after showering, the transformed fentanyl Cio, i.e., the ratio of the Cio of fentanyl as found after showering relative to the fentanyl Cio as found prior to showering at a therapeutically equivalent dose, is 1.1. In another embodiment, after showering, the transformed fentanyl Cio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
[00182] In one embodiment, after showering, the transformed naltrexone Cio, i.e., the ratio of the Cio of naltrexone as found after showering relative to the naltrexone Cio as found prior to showering at a therapeutically equivalent dose, is 1.05. In another embodiment, after showering, the transformed naltrexone Cio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 or 1.75.
[00183] In one embodiment, after showering, the transformed fentanyl Cio to naltrexone Cio ratio is 1:1 or less. In another embodiment, after showering, the transformed fentanyl Cio to naltrexone
Cio ratio is 1:1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1:1.25 or less, 1:1.3 or less, 1:1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1:1.55 or less, or 1:1.6 or less.
[00184] In one embodiment, after showering, the transformed fentanyl AUCio, i.e., the ratio of the AUCio of fentanyl as found after showering relative to the fentanyl AUCio as found prior to showering at a therapeutically equivalent dose, is 1.1. In another embodiment, after showering, the transformed fentanyl AUCio is 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7, 1.75, 1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
[00185] In one embodiment, after showering, the transformed naltrexone AUCio, i.e., the ratio of the AUCio of naltrexone as found after showering relative to the naltrexone AUCio as found prior to showering at a therapeutically equivalent dose, is 1.05. In another embodiment, after showering, the transformed naltrexone AUCio is 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5, 1.55, 1.6, 1.65, 1.7 1.75,
1.8, 1.85, 1.9, 1.95, 2, 2.05, 2.1, 2.15, 2.2, 2.25, 2.3, 2.35, 2.4 or 2.45.
[00186] In one embodiment, after showering, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1:1 or less. In another embodiment, after showering, the transformed fentanyl AUCio to naltrexone AUCio ratio is 1: 1.05 or less, 1:1.1 or less, 1:1.15 or less, 1:1.2 or less, 1: 1.25 or less, 1:1.3 or less, 1: 1.35 or less, 1:1.4 or less, 1:1.45 or less, 1:1.5 or less, 1: 1.55 or less, 1:1.6 or less, 1: 1.65 or less, 1:1.7 or less, 1: 1.75 or less, 1:1.8 or less, 1:1.85 or less, 1:1.9 or less, 1: 1.95 or less, 1:2 or less, 1:2.05 or less, 1:2.1 or less, 1:2.15 or less, 1:2.2 or less, or 1:2.25 or less.
[00187] In one embodiment, when heat is applied, the fentanyl Cio of the dosage form is less than 80% the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, when heat is applied, the fentanyl Cio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at
therapeutically equivalent transdermal doses.
[00188] In one embodiment, after exercise, the fentanyl Cio of the dosage form is less than 80% the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, when heat is applied, the fentanyl Cio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less the fentanyl Cio of a
reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
[00189] In one embodiment, after showering, the fentanyl Cio of the dosage form is less than 80% the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, when heat is applied, the fentanyl Cio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at
therapeutically equivalent transdermal doses.
[00190] In one embodiment, when heat is applied, the fentanyl AUCio of the dosage form is less than 80% the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, when heat is applied, the fentanyl AUCio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
[00191] In one embodiment, after exercise, the fentanyl AUCio of the dosage form is less than 80% the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, after exercise is applied, the fentanyl AUCio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
[00192] In one embodiment, after showering, the fentanyl AUCio of the dosage form is less than 80% the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses. In another embodiment, after showering is applied, the fentanyl AUCio of the dosage form is 75% or less, 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, or 25% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
[00193] In some embodiments, the proximal surface 115 has a release liner 110 which is removed to reveal a skin-contacting surface which should be sufficiently conformable when placed on a skin surface so as to make intimate contact with at least a portion of the skin surface. In one embodiment, substantially all of the polymeric material of the proximal surface of the first antagonist component 120 will make intimate contact with the skin surface of a patient. Suitable release liners include conventional release liners comprising a known sheet material such as a polyester web, a polyethylene web, a polypropylene web, or a polyethylene-coated paper coated with a suitable fluoropolymer or silicone based coating. The release liner that has been coated with the first antagonist component 120 can be dried and laminated onto a barrier component 120 using conventional methods.
[00194] In one embodiment, the first antagonist component 120 comprises a polymeric material, an active agent and an antagonist salt. Suitable polymeric materials or matrices for use in the first antagonist or adverse agent component include, but are not limited to, acrylates, natural rubbers, polyisobutylenes, polyisoprenes, styrenic block copolymers, polyvinylethers, silicone polymers, polyurethanes, and polyurethane-ureas. In one embodiment, the active agent or adverse agent is preferably dispersed substantially homogeneously throughout a polymeric material. In one embodiment, the active agent or adverse agent is dissolved in the polymeric material. In another embodiment, the active agent is substantially in amorphous form. In another embodiment, the adverse agent component includes a crystalline form substantially dispersed throughout the polymeric material.
[00195] In one embodiment, the first antagonist component 120 comprises a polymeric material, fentanyl as free base and naltrexone HCI. Suitable polymeric materials or matrices for use in the first antagonist or adverse agent component include, but are not limited to, acrylates, natural rubbers, polyisobutylenes, polyisoprenes, styrenic block copolymers, polyvinylethers, silicone polymers, polyurethanes, and polyurethane-ureas. In one embodiment, the fentanyl or naltrexone is preferably dispersed substantially homogeneously throughout a polymeric material. In one embodiment, the fentanyl or naltrexone is dissolved in the polymeric material. In another embodiment, fentanyl is substantially in amorphous form. In another embodiment, the naltrexone includes a crystalline form substantially dispersed throughout the polymeric material.
[00196] In certain embodiments, the polymeric matrix is a pressure sensitive adhesive.
Suitable pressure-sensitive adhesives include those suitable for use as the polymeric material of the active agent component. Additionally, pressure-sensitive adhesives that are not suitable for direct skin contact can be suitable for use as the polymeric material of the active agent or adverse agent. In some embodiments, pressure-sensitive adhesives for use in the dosage forms of the disclosure include
acrylates, polyisobutylenes, silicone polymers, and mixtures thereof. Examples of useful polyisobutylene pressure-sensitive adhesives are described in U.S. Pat. No. 5,985,317, the disclosure of which is incorporated herein by reference in its entirety for all purposes. Examples of useful acrylate and silicone polymer pressure-sensitive adhesives, and mixtures thereof, are described in U.S. Pat. No. 5,474,783, the disclosure of which is incorporated herein by reference in its entirety for all purposes.
[00197] Acrylate polymers and copolymers may include pressure-sensitive adhesives.
Examples of suitable monomers for use in acrylate copolymers include alkyl acrylates, such as isooctyl, 2-ethylhexyl, n-butyl, ethyl, methyl, and dimethylhexyl, and alkyl methacrylates, such as lauryl, isodecyl, and tridecyl. Monomers containing functional groups, such as carboxylic acid, hydroxy, amide, and amino may also be incorporated into an acrylate copolymer. Examples of suitable monomers containing functional groups include acrylic acid, hydroxyalkyl acrylates containing 2 to 4 carbon atoms in the hydroxyalkyl group, acrylamide, N-vinyl-2-pyrrolidone, vinyl acetate, and alkoxyethyl acrylates.
[00198] Acrylate copolymers may optionally further comprise a substantially linear macromonomer copolymerizable with the other monomers. Suitable macromonomers include polymethylmethacrylate, styrene/acrylonitrile copolymer, polyether, and polystyrene macromonomers. Examples of useful macromonomers and their preparation are described in U.S. Pat. No. 4,693,776 (Krampe et al.), the disclosure of which is incorporated herein by reference in its entirety for all purposes.
[00199] Other polymer materials of the first antagonist component may include but are not limited to polyethylene; polypropylene; ethylene/propylene copolymers; ethylene/ethylacrylate copolymers; ethylene/vinyl acetate copolymers; silicone elastomers, especially the medical-grade polydimethylsiloxanes; neoprene rubber; polyisobutylene; chlorinated polyethylene; polyvinyl chloride; vinyl chloride-vinyl acetate copolymer; polymethacrylate polymer (hydrogel); polyvinylidene chloride; poly(ethylene terephthalate); butyl rubber; epichlorohydrin rubber; ethylene-vinyl alcohol copolymer; ethylene-vinyloxyethanol copolymer; silicone copolymers, for example, polysiloxane-polycarbonate copolymers, polysiloxane-polyethyleneoxide copolymers, polysiloxane-polymethacrylate copolymers, polysiloxane-alkylene copolymers (e.g., polysiloxane-ethylene copolymers), polysiloxane-alkylenesilane copolymers (e.g., polysiloxane-ethylenesilane copolymers), and the like; cellulose polymers, for example methyl or ethyl cellulose, hydroxypropyl methylcellulose, and cellulose esters; polycarbonates;
polytetrafluoroethylene; and combinations thereof. In one embodiment, the polymer matrix has a glass- transition temperature below room temperature. The polymer can, but need not necessarily, have a degree of crystallinity at room temperature. Cross-linking monomeric units or sites can be incorporated
into the polymers. For example, cross-linking monomers can be incorporated into polyacrylate polymers. The cross-linking monomers provide sites for cross-linking the polymer matrix after microdispersing the active agent into the polymer. Known cross-linking monomers for polyacrylate polymers include, but are not limited to, polymethacrylic esters of polyols such as butylene diacrylate and dimethacrylate, trimethylol propane trimethacrylate, and the like. Other monomers that provide cross-linking sites include allyl acrylate, allyl methacrylate, diallyl maleate, and the like. In one embodiment the polymer matrix does not allow any, or any detectable amount, of an active agent or adverse agent to diffuse out of it, particularly in those instances in which the active agent or adverse agent can penetrate a patient's skin.
[00200] The first antagonist component can also comprise a porous medium, such as a woven fabric, porous or microporous film, or other open, mesh-like material, wherein at least a portion of the pores contain active agent or adverse agent. The active agent or adverse agent can be present within the pores in any form, including but not limited to a liquid, a gel or a solid, such as a solid crystalline or powdered material. For example, the active agent or adverse agent can be mixed with a carrier, such as a viscous liquid, semi-solid or gel material. Examples of suitable materials for incorporation into the active agent or adverse agent component include, but are not limited to, microporous films formed by extruding polyethylene or polypropylene with mineral oil as described in U.S. Pat. No. 4,539,256, the disclosure of which is incorporated herein by reference in its entirety.
[00201] Each of the layers comprising active agent or antagonist may comprise a number of additional components. Additional components of the active agent or first antagonist component can include skin penetration enhancers, drug solubilizers, plasticizers, anti-oxidants, colorants, bittering agent and the like.
[00202] The first antagonist component will typically comprise a skin penetration enhancer. Examples of excipients useful as skin penetration enhancers or solubilizers in transdermal drug delivery systems include C8-C24 fatty acids such as isostearic acid, octanoic acid, and oleic acid; C8-C24 fatty alcohols such as oleyl alcohol and lauryl alcohol; lower alkyl esters of C8-C24 fatty acids such as ethyl oleate, isopropyl myristate, butyl stearate, and methyl laurate; monoglycerides of C8-C24 fatty acids such as ethyl oleate, isopropyl myristate, butyl stearate, and methyl laurate; monoglycerides of C8-C24 fatty acids such as glyceryl monolaurate; tetraglycol (tetrahydrofurfuryl alcohol polyethylene glycol ether); tetraethylene glycol (ethanol, 2, 2'-(oxybis(ethylenoxy))diglycol); polyethylene glycol; propylene glycol; N,N-dimethyldodecylamine-N-oxide; terpenes, such as d-limonene, menthol, and terpineol.
[00203] The skin penetration enhancers, drug solubilizers, plasticizers, and other additives can be dispersed or mixed, optionally substantially uniformly, or optionally dissolved in the composition. Where the additive is a penetration enhancer, it is present in an amount that enhances active agent permeation through the skin compared to a like composition not containing the penetration enhancer(s) when this phenomenon is measured using a standard skin penetration model, such as set forth in U.S. Pat. No. 5,585,111, the disclosure of which is herein incorporated by reference in its entirety. In one embodiment, the total amount of penetration enhancer and solubilizer is less than about 40% by weight based on the total weight of the composition. In another embodiment, the total amount of penetration enhancer and solubilizer is less than about 30% based on the total weight of the composition.
[00204] A solubility enhancer may also be included. In one embodiment, the solubility enhancer is in an amount more than 2%. In another embodiment, it is in an amount of between 2.5 to 3.5%. In one embodiment, polyvinylpyrrolidone (PVP) is used.
[00205] In the first antagonist component, the active agent and antagonist, whether formulated together or separately in strips are optionally dispersed homogeneously throughout the polymeric material, or optionally dissolved within the polymeric material. The proximal or skin contacting surface 115 should be sufficiently conformable when placed on a skin surface so as to make intimate contact with at least a portion of the skin surface. In one embodiment, substantially all of the polymeric material at the proximal surface 115 will make intimate contact with the skin surface.
[00206] In one embodiment, the first antagonist component and the antagonist each have a thickness of no less than about 10pm. In another embodiment, the first antagonist agent component has a thickness of no less than about 20pm. In another embodiment, the first antagonist component has a thickness of no less than about 50pm. In another embodiment, the first antagonist component has a thickness of no greater than about 250pm. In another embodiment, the first antagonist component has a thickness of no greater than about 200pm. In another embodiment, the first antagonist component has a thickness of no greater than about 150pm.
[00207] The barrier 130, as shown in FIGS. 1 and 4, is a substantially continuous component adjacent to the distal surface of the first antagonist component 120 on one side and the second antagonist component 140 on the other side. The barrier layer is impermeable to the antagonist and the active agent; and comprises a material which is insoluble in water, alcohol and organic solvents. The barrier layer comprises a polymer such as polyolefin laminates (Dow Chemical, Midland, Ml), acrylonitrile copolymer films (BAREX, BP Chemicals, Koln, Germany), polyethylnapthalene (PEN),
polyethylene terephthalate (PET), polyimide, polyurethane, polyethylene, metallized films and glass coated films where these films can include ethylene copolymers such as ethylene-vinyl acetate copolymer (EVA), and combinations thereof. In some embodiments, the barrier layer comprises polyester such as PET laminated to a polymer such as polyurethane, polyethylene, and ethylene copolymers. In other embodiments, the barrier layer comprises polyester such as PET laminated to ethylene copolymers such as ethylene-vinyl acetate copolymer (EVA). The barrier layer as a multilaminate layer has a thickness of about 0.075 mm (0.3 mil) to about 0.125 mm (5 mil); optionally 0.025 mm (1 mil) to about 0.1 mm (4 mil); optionally 0. 0625 mm (1.5 mil) to about 0.0875 mm (3.5 mil); and optionally 0.025 mm (1 mil) to about 0.05 mm (2 mil). The polyethylene or EVA laminated layer of the PET-PE laminates improves the adhesion of the antagonist component to the backing, and serves to prevent the facile removal of the antagonist base layer from the system by the potential abuser.
[00208] In some embodiments, the backing is laminated to the surface of the antagonist base component, optionally using heat, pressure and/or an additional tie component to ensure adequate contact between the reservoir component and backing. In some embodiments, the backing is non sticking and hydrophobic.
[00209] Any device known to those skilled in the art for transdermally delivering a therapeutic agent, particularly an opioid, to an animal can be used for the transdermal dosage form of the disclosure. For example, the transdermal dosage form can be a reservoir-type transdermal dosage form, a polymer-matrix type transdermal dosage form, or a drug-in-adhesive type transdermal dosage form. The transdermal dosage form is designed so that when contacted with the animal's skin, an analgesically effective amount of the active therapeutic agent, such as an opioid, is transdermally administered to the animal while the antagonist either remains in the transdermal dosage form and is not administered to the animal or is administered to the animal in an amount insufficient to inhibit the analgesic effect of the active agent.
[00210] The transdermal dosage forms of the disclosure can be made in the form of an article such as a tape, a patch, a sheet, a dressing or any other form known to those skilled in the art.
Generally, the dosage form will be in the form of a patch of a size suitable to deliver a preselected amount of active agent through the skin.
[00211] In one embodiment, the dosage form will have a surface area greater than 5cm2. In another embodiment, the dosage form will have a surface area of greater than 10cm2. In another
embodiment, the dosage form will have a surface area of less than 100 cm2. In another embodiment, the dosage form will have a surface area of less than 40 cm2.
[00212] Dosage forms of the present disclosure are typically packaged individually in a foil- lined pouch for storage. Dosage forms of the present disclosure may alternatively be provided in a rolled or stacked form suitable for use with a dispensing apparatus. An optional tie component, heat, and/or pressure may be used to connect the skin-contacting component with the barrier component. In addition, the skin-contacting component compositions may be directly coated onto the barrier component and subsequently dried and laminated to a release liner.
[00213] One skilled in the art will appreciate that it may be preferred to vary the order of lamination steps depending on the types and thickness of the components comprising the dosage form.
[00214] According to the methods of the disclosure, in one embodiment the transdermal dosage form is contacted with the skin of the patient and an opioid is released by the transdermal dosage form and becomes absorbed through the skin. Once absorbed into the patient, the opioid is provided in an analgesically effective amount. The transdermal dosage form can provide sustained and continuous delivery of an analgesically effective amount of the opioid. In another embodiment, on administration over the skin, the transdermal dosage form exhibits a steady state drug flux of about 1 to about 10pg/cm2/hr. In one embodiment, the transdermal dosage form exhibits a steady state drug flux of about 1 to about 8 pg/cm2/hr. In one embodiment, the transdermal dosage form exhibits a steady state drug flux of about 1 to about 5 pg/cm2/hr. In one embodiment, the transdermal dosage form exhibits a steady state drug flux of about 2 to about 3 pg/cm2/hr. In one embodiment the transdermal dosage form is contacted with the skin of the patient and fentanyl is released by the transdermal dosage form and becomes absorbed through the skin. Once absorbed into the patient, the fentanyl is provided in an analgesically effective amount. The transdermal dosage form can provide sustained and continuous delivery of an analgesically effective amount of fentanyl.
[00215] In one embodiment, the transdermal dosage form exhibits a nominal flux (i.e., the average amount of drug delivered to the systemic circulation per hour across average skin) of about 12.5 mcg/hr. In one embodiment, the transdermal dosage form exhibits a nominal flux of about 50 mcg/hr.
In one embodiment, the transdermal dosage form exhibits a nominal flux of about 75 mcg/hr. In one embodiment, the transdermal dosage form exhibits a nominal flux of about 100 mcg/hr.
[00216] In one embodiment of the present disclosure, the method of treating pain with any one of the dosage forms described herein, wherein said dosage form can provide a ratio of adverse
agent to active agent released, or alternatively absorbed into a blood stream, from about 1:10 to about 10:1 when the dosage form is used in an inappropriate manner. For example, it may be attempted to extract the active agent from the dosage form with a solvent, such as a quid or gas. In certain embodiments, the dosage form, when tampered with in such a manner, will release both adverse and active agent. In certain embodiments, the ratio of adverse agent to active agent released when tampered with is about is 1:5, 1:4, 1:3, 1:2, or 1:1. In other embodiments, the method of treating pain comprises applying a dosage form as described herein, wherein said dosage from comprises a ratio of adverse agent to active agent from about 1:10 to about 10:1. In other embodiments, the ratio of adverse agent to active agent is about is 1:5, 1:4, 1:3, 1:2, or 1:1.
[00217] In one embodiment of the present disclosure, the method of treating pain with any one of the transdermal dosage forms described herein, wherein said transdermal dosage form can provide a ratio of naltrexone to fentanyl released, or alternatively absorbed into a blood stream, from about 1:10 to about 10:1 when the transdermal dosage form is used in an inappropriate manner. For example, an abuser may attempt to extract the fentanyl from the transdermal dosage form with a solvent, such as a liquid or gas. In certain embodiments, the dosage form, when tampered with in such a manner, will release both naltrexone and fentanyl. In certain embodiments, the ratio of naltrexone to fentanyl released from a transdermal dosage form when tampered with is about is 1:5, 1:4, 1:3, 1:2, or 1:1. In other embodiments, the method of treating pain comprises applying to the skin of a human subject a transdermal dosage form as described herein, wherein said dosage from comprises a ratio of naltrexone to fentanyl from about 1:10 to about 10:1. In other embodiments, the ratio of naltrexone to fentanyl released into the plasma is about is 1:5, 1:4, 1:3, 1:2, or 1:1.
[00218] The present disclosure is also directed to a kit comprising at least one dosage form of the disclosure. In one embodiment, the dosage form is present in a container, e.g., a box. In another embodiment, the kit further comprises a set of instructions directing the use of the dosage form to treat a patient, e.g., for pain. In one embodiment, the instructions may be a printed label affixed to or printed on the container. In another embodiment, the instructions may comprise a printed sheet inserted into the container or into the packaging which contains the container. The instructions may also state that the dosage form and/or its usage are designed to reduce abuse, misuse or diversion of the dosage form.
EXAMPLES
Example 1
[00219] The transdermal dosage form Patch A1 as depicted in Figures 3 and 4 were prepared according to the manufacturing process steps described below. The quantitative composition of component of the Patch A1 is provided below in Tables.
[00220] Patch A1 was manufactured according to the amounts below in Table 1 and according to the following process steps:
1. Manufacturing skin contact layer fentanyl wet blend and then coat to make laminate containing fentanyl base, die-cut to 10 mm strip.
2. Manufacturing skin contact layer naltrexone hydrochloride wet blend and then coat to make laminate containing naltrexone hydrochloride, die cut to 10 mm strip.
3. Manufacturing non-skin contact layer wet blend and then laminate containing naltrexone base.
4. Die-cut non skin contact layer to appropriate sized patches.
5. Apply one or more fentanyl adhesive strips to the barrier film of a non-skin contact layer patch. Apply one of more naltrexone hydrochloride adhesive strips to the barrier film of the non-skin contact layer patch with about 1mm gap between a fentanyl strip and a naltrexone HCI strip.
6 Pouch to give finished product.
Table 1


Example 2
[00221] The transdermal dosage form Patch B2 as depicted in Figures 1 and 2 and Patch B1 as depicted in Figures 3 and 4 were prepared according to the manufacturing process steps described in Figures 5-9. Figure 5 is a schematic of the manufacturing process steps for transdermal patch B1 and B2. Figure 6 is a manufacturing flow diagram of the skin contact layer (central) active strip containing fentanyl base and naltrexone HCI (layer B). Figure 7 is a schematic of the manufacturing process steps of the skin contact layer adhesive strips (Layer B) for patch B2. Figure 8 is a manufacturing flow diagram for the non-skin contact layer (layer D) containing naltrexone (step 3). Figure 9 is a manufacturing flow diagram for the integration of the non-skin contact layer, skin contact adhesive side strips and skin contact central active strip into the finished transdermal patch (step 4).
[00222] The quantitative composition of component of the Patch B1 and B2 is provided in Table 2. Other doses for Patch B1 can be calculated. For example, 12.5 mcg/hr is 12.5% of each mg/patch of the 100 mcg/hr fentanyl amounts.
[00223] The drug product manufacturing process consists of a four step process as described in Figure 5. For each of the first three steps, a wet blend is coated onto an in-process release liner, dried and laminated to another release liner or a barrier film, resulting in one of the three separate intermediate adhesive laminates. During the last two steps of the manufacturing process, each of the intermediate laminates is die-cut to the appropriate size for each patch strength and assembled into the final dosage form.
[00224] The manufacturing process for Step 1 is shown in Figure 6 and according to the following:
(i) Prepare naltrexone hydrochloride suspension in three silicone adhesives: Charge Bio-PSA"7- 4301 and Bio-PSA" 7-4202 into a Ross mixer bowl. Cool to approximately 20°C. Charge naltrexone hydrochloride and Bio-PSA" 7-4302. Homogenize to form a uniform suspension.
(ii) Prepare fentanyl and povidone solution in a mixed solvent of ethanol and ethyl acetate: Charge fentanyl base, Povidone" K90, ethyl acetate and ethanol into a Ross mixer bowl. Heat to approximately 50°C and mix to form a clear, viscous solution.
(iii) Prepare final wet blend: Add the naltrexone hydrochloride suspension to the Ross mixer bowl containing fentanyl and povidone K90 solution. Mix at low speed for a minimum of 4 hours. This is the final wet blend.
(iv) Transfer the final wet blend to four glass containers. Place the containers in jar rollers. Rotate the jar at slow speed to remove air bubbles and to keep the naltrexone hydrochloride crystals uniformly suspended.
(v) Form Skin Contact Layer Active Laminate Strip of coat weight: Coat the final wet blend to a fluoropolymer coated polyester in-process release liner (Scotchpak™ 9755) in a continuous coater with slot-die coating knife and three-zone drying ovens at 0.3 meter/min web speed and zone temperatures (zone 1: ~65°C, zone 2: ~85°C and zone 3: ~109°C). Laminate to a fluoropolymer coated polyester in-process release liner (Scotchpak™ 9744) to form the skin contact active intermediate laminate.
(vi) Using a rotary die press, die-cut into individual [central] active strips.
[00225] The manufacturing process for Step 2 is shown in Figure 7 and in the following:
(i) Charge Duro-Tak” 87-4287 and ethyl acetate to a Ross mixer bowl. Mix for approximately 30 min.
(ii) Charge titanium dioxide to the Ross mixer bowl. Homogenize to form a uniform suspension.
(iii) Form Skin Contact Layer Adhesive Laminate of 105 GSM coat weight: Coat the wet blend to an in-process polyester release liner with a fluoropolymer coating (Scotchpak™1022) in the continuous coater with slot-die coating knife and three-zone drying ovens at 0.3 meter/min web
speed and zone temperature (zone 1: ~65 °C, zone 2: ~85 °C, and zone 3: ~109 °C). Laminate to an in-process silicone coated polyester release liner (SG4130).
(iv) Using a rotary die press, die-cut into individual adhesive strips.
[00226] The manufacturing process for Step 3 is shown in Figure 8 and in the following:
(i) Prepare 3 kg wet blend: Charge naltrexone base, Povidone K90, ethyl acetate and ethyl alcohol to a 3 kg glass container. Heat and mix at 55°C for about 1 hour until a clear solution is formed. Add Duro-Tak" 387-2510, Brij" CS20-SO-MH and Bio-PSA" 7-4302. Mix to form a 3 kg wet blend.
(ii) Repeat to prepare six 3 kg wet blends.
(iii) Transfer the six 3 kg wet blends to a 10 gallon Ross mixer bowl. Mix until uniform.
(iv) Transfer the wet blend from the 10 gallon mixer to six 3 kg containers. Place the 3 kg containers to roller and roll to remove air bubbles.
(v) Perform in-process testing: Description, Blend CU and Viscosity
(vi) Form Non-skin Contact Layer Adhesive Laminate of 105 GSM coat weight: coat the wet blend to release liner Scotchpak " 9755 in the continuous coater with slot-die coating knife and three zone drying ovens. Laminate to Barrier film Scotchpak " 9735 to form the Non-skin Contact Intermediate Laminate.
(vii) In a Slitter machine, slit the wide laminate from the coater to narrower laminate.
(viii) In a Converter machine, remove the release liner (Scotchpak™ 9755) and laminate the drug- adhesive layer to silicone coated elastic polyester fabric KOB051 backing.
(ix) Using a rotary die press, die-cut into individual non-skin contact layer patches.
[00227] The manufacturing process for Step 4 is shown in Figure 9 and in the following:
(i) Place a non-skin contact layer patch on a vacuum board with the barrier film (Scotchpak " 9735) facing up.
(ii) Remove one release liner (SG4130) from a skin contact adhesive strip. Adhere the adhesive layer to the right side of the non-skin contact patch.
(iii) Remove one release liner (Scotchpak™ 9755) from a skin contact central active strip. Adhere the adhesive layer to the center of the non-skin contact layer.
(iv) Remove one release liner (SG4130) from a second skin contact adhesive strip. Adhere the adhesive layer to the left side of the non-skin contact layer.
(v) Remove all the 3 release liners (Scotchpak™ 1022, Scotchpak™ 9744 and Scotchpak 1022) from the three strips already adhered on the barrier film (Scotchpak™ 9735). Laminate one oversized release liner (Scotchpak™ 9755) to the left side. Laminate a second piece of release liner (Scotchpak™ 9755) to the right side of the patch overlapping the first piece of the same release liner. This forms the finished product (transdermal patch).
(vi) Insert one patch into an open pouch. Heat-seal the open side of the pouch.

Layer D: Non Skin Contact Layer Formulation



Layer B: Skin Contact Adhesive Strip Formulation


Example 3
[00228] Solvent extraction in buffered saline was determined using the test method described for Patch A1 and B2.
Extraction Method
[00229] The test samples were transdermal patches: Patch A1 (6.25 cm2) and Patch B2 (6 cm2). The extraction solution was chosen from one of the following solutions at room temperature and 70°C: water; ethanol (USP, absolute); pH6.8 Phosphate Buffer; cooking vinegar; and artificial saliva.
[00230] The patch and a 15 mL extraction solution were added into a 40 mL vial. The sealed vial was vigorously shaken with a wrist-action shaker (Burrel, Model 75, speed setting: 10). At fixed time intervals of 5, 15, and 30 minutes aliquots were removed. Each aliquot was placed into an analysis vial. If the extraction solvent was ethyl acetate or ether, then it was evaporated to dryness and methanol (HPLC grade) was added to the sample and mixed. Samples were assayed for active drug substance by reverse-phase HPLC.

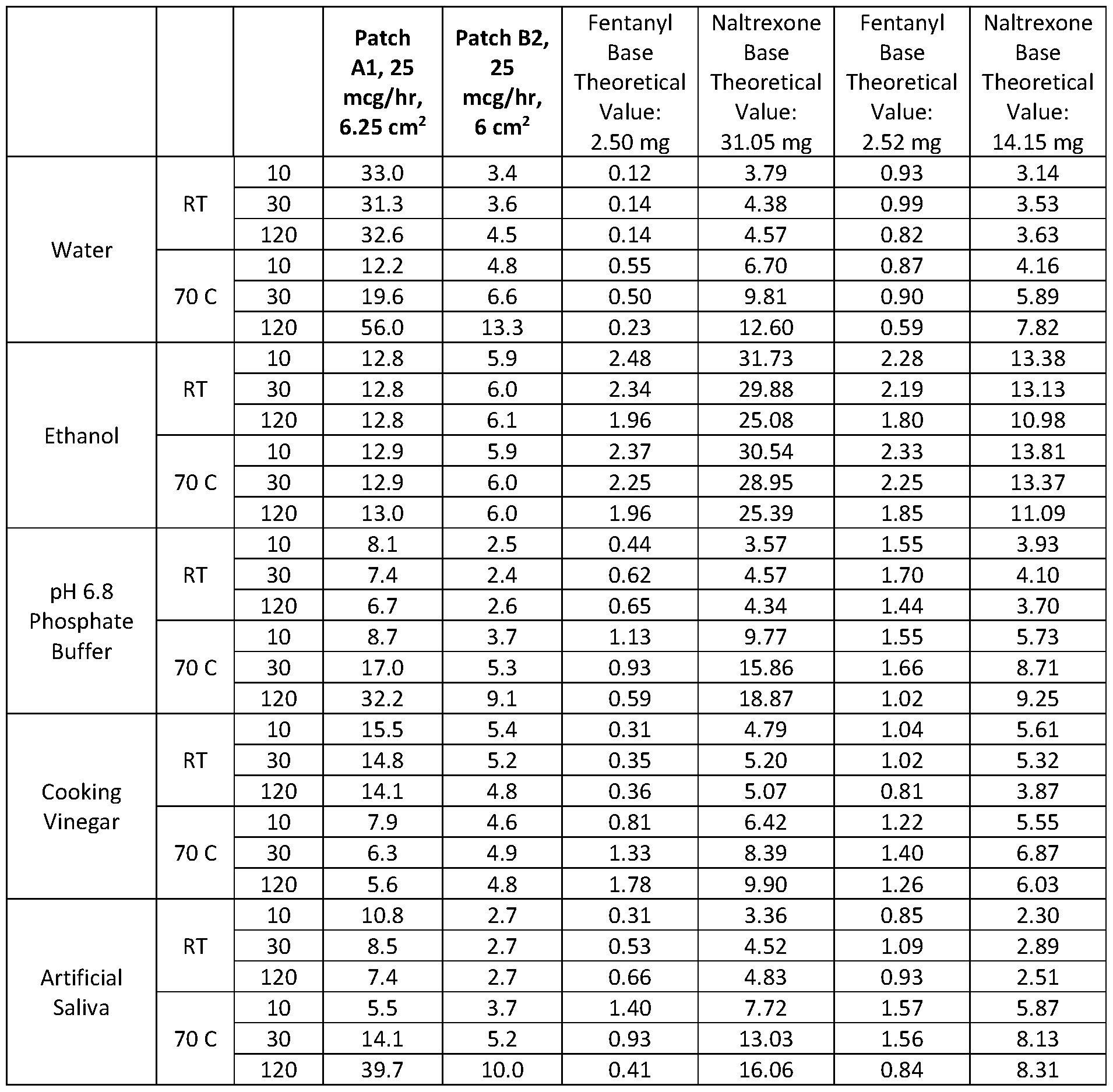
Example 4
[00231] Both Patch A1 and Patch B1 were shown to be similar to the Duragesic™ product as tested in a standard in vitro skin flux tests. Figure 15 shows the in vitro results where the solid line is for patch Bl, dotted line (— ¨— ) is for patch Al, and dotted line (· ·· ¨·· ·) is for Duragesic™. Figure 10 shows in vitro cumulative delivery per cm2 of Patch B2 versus the Duragesic™ patch.
Example 5

Example 6
[00232] In vitro skin flux study of Patch B2: This study was designed to obtain in vitro transdermal flux data.
Results
[00233] Figures 10-14 show that the patches prepared by Example 5 deliver more fentanyl per cm2 active patch than Duragesic™ fentanyl transdermal system based on in vitro skin flux study experiments using modified Franz cell and human cadaver epidermis. Transdermal delivery is proportional to patch size. As shown in Figure 10, 72 hours of in vitro cumulative delivery per cm2 of Patch B2 17.3 cm2 100 mcg/h central strip is greater than the 42 cm2 100 mcg/h Duragesic™ patch. Figures 11 and 12 show that the cumulative [pg/patch] and skin flux profile [pg/patch/hr] of the Patch B2 17.3 cm2 100 mcg/h central strip matches that of the 42 cm2 100 mcg/h Duragesic™ patch. Figure 13 shows that Patch B2 24 cm2 100 mcg/h central strip delivers more fentanyl [in pg/patch] than the 42 cm2 100 mcg/h Duragesic™ patch in vitro. Figure 14 shows the in vitro fentanyl skin flux [pg/patch/hr] from Patch B2 24 cm2 100 mcg/h is higher than the 100 mcg/h Duragesic™ patch.
Example 7
[00234] With reference to Figure 16, transbuccal delivery was simulated by permeation of naltrexone and fentanyl from a section of patch through human cadaver buccal tissue of 500 pm in thickness into the receptor solution, which is pH 6.8 commercially available artificial saliva stirred at 300 rpm at 37°C. The receptor fluid was stirred by means of a magnetic stirrer throughout the experiment to assure a uniform sample and a reduced diffusion barrier on the dermal side of the skin. The entire volume of receptor fluid was withdrawn at specified time intervals (0.25, 0.5, 1.0, 1.5, 2, 2.5, 3, 4, 7, and 24 hours) and immediately replaced with fresh fluid. The withdrawn fluid was filtered through a 0.45pm filter. The last 1-2 mL was then analyzed for the active agent, e.g., fentanyl, using conventional high performance liquid chromatography methods. The cumulative amount of fentanyl penetrating through the cadaver buccal was calculated and reported as mcg/cm2. Unless noted, the results are mean of 6 donors, 1 replicate per donor for Patch B2 while reference product Duragesic™ calculations are mean of 18 donors, 1 or 2 replicates per donor.
[00235] Because the impermeable polyester barrier between skin contact layer and the non skin contact layer of the patch prevents permeation through the buccal tissue in this in vitro test system, each side of the patch (skin contact layer and non-skin contact layer) was tested separately in this model
in order to evaluate the conditions of human oral use, where both layers would be in contact with oral tissue.
[00236] Permeation through skin contact layer - To assess buccal permeation of fentanyl and naltrexone from Duragesic™ and Patch B2 skin contact layer, a 7 cm2 die cut of the fentanyl and naltrexone HCI skin contact strip was applied to a 500 pm thick buccal cell. The patch-buccal tissue was then assembled to a modified Franz donor cell with the buccal tissue in contact with the receptor solution.
[00237] Permeation through non-skin contact layer - To assess buccal permeation of naltrexone from the non-skin contact layer, the semipermeable backing film side of the non-skin contact layer was applied to buccal tissue and then assembled to a modified Franz cell with the buccal tissue in contact with the receptor solution.
[00238] As illustrated in Figure 17, the transbuccal delivery rate of fentanyl from Patch B2 was comparable to the reference product Duragesic™. The transbuccal delivery rate of naltrexone in the non-skin contact layer and that of naltrexone in the skin contact layer are listed in the Table 5. The Patch B2 non-skin contact layer provides additional naltrexone transbuccal delivery. The total combined transbuccal delivery rate of naltrexone from Patch B2 from the 2 layers is thus even higher than the fentanyl delivery rate from the Patch B2 patch. The ratio of naltrexone delivery to fentanyl delivery is 4.1 and 2.6 in the early time points (0.25 h, 0.5 h) and 3.5 to 6.3 in later time points (1 to 24 h).


Example 8
Solvent Extraction Method
[00239] Patch B2, 25 mcg/h strength is cut into halves. The liner from each half is peeled off. Both halves are transferred into a 20-mL glass vial. Ten mL of appropriate extraction medium is pipetted into the glass vial with screw cap. The vial was shaken for 2 hours at 180 osc/minute at room temperature or at 125 strike/minute in 70-C water bath. Samples (20 pL) are pipetted into separated HPLC vials at time points of 10 min, 30 min and 120 min, and diluted with 980 pL of Diluent for analysis by HPLC. Extraction media were Dl Water, Ethanol, Cooking Vinegar, pH 6.8 phosphate buffer and artificial saliva (Dissolve 16 g of sodium chloride, 0.38 g of sodium phosphate monobasic and 4.76 g of sodium phosphate dibasic into 2000 mL of water. Adjust pH to 6.8 with sodium hydroxide. Mix well and filter the solution through a 0.45-pm membrane filter. Degas the solution for 15 minutes.
[00240] Figure 18 shows the simulated small volume solvent extraction from cut Patch B2 (14.06 cm2; 25 mcg/h) at room temperature. Figure 19 shows the simulated small volume solvent extraction from cut Patch B2 (14.06 cm2; 25 mcg/h Patch) at 70°C.



Abbreviations: %LC = percent of label claim (percent of the amount in the patch claimed by the prescribing information); HCI = hydrochloride;
 = Weight of Naltrexone extracted from the Patch, = Weight of Fentanyl extracted from the Patch; min = minute
= Weight of Naltrexone extracted from the Patch, = Weight of Fentanyl extracted from the Patch; min = minute
Example 9
[00241] The pharmacokinetics of 30 minutes of buccal administration of a 25pg/h patch of either Patch B2 or Duragesic™ was evaluated in fasted naive male beagle dogs. The study was conducted with a crossover design with the same dogs receiving each of the scheduled treatments with at least a seven-day washout period between each dose.
[00242] 30 minutes prior to dosing with the test articles, naloxone HCI was administered intravenously (IV) into the cephalic vein followed by an approximately 1 mL flush with normal saline.
Just prior to its buccal application, the tested patch was cut in half and the release liner on each half patch was removed by hand and a sticky adhesive side applied to a separate cheek (between the gum and cheek), with sticky adhesive in direct contact with the cheek. All animals were anesthetized with isoflurane 3-5 minutes prior to patch application. The anesthesia was maintained for approximately 30 minutes. The dog's mouth was held closed, and pressure was applied to hold the cheek against the teeth/gum for 30 minutes. At 5, 10, and 20 minutes post patch application, the dog's mouth was briefly opened to observed the location of the patches and ensure that the patch was still flat against the buccal region. After each check, the patch was manipulated to return it to its original orientation if necessary, the mouth was carefully closed, and pressure reapplied. All observations were recorded. At 30 minutes post application, the patches were removed and the left and the right buccal regions were checked.
[00243] Blood samples were collected for pharmacokinetic evaluation during and up to 24 hours post patch application. Plasma concentrations of fentanyl and naltrexone, as listed in Table 7, were determined by LC-MS/MS using a qualified method and pharmacokinetic parameters were determined using WinNonlin software.
Table 7
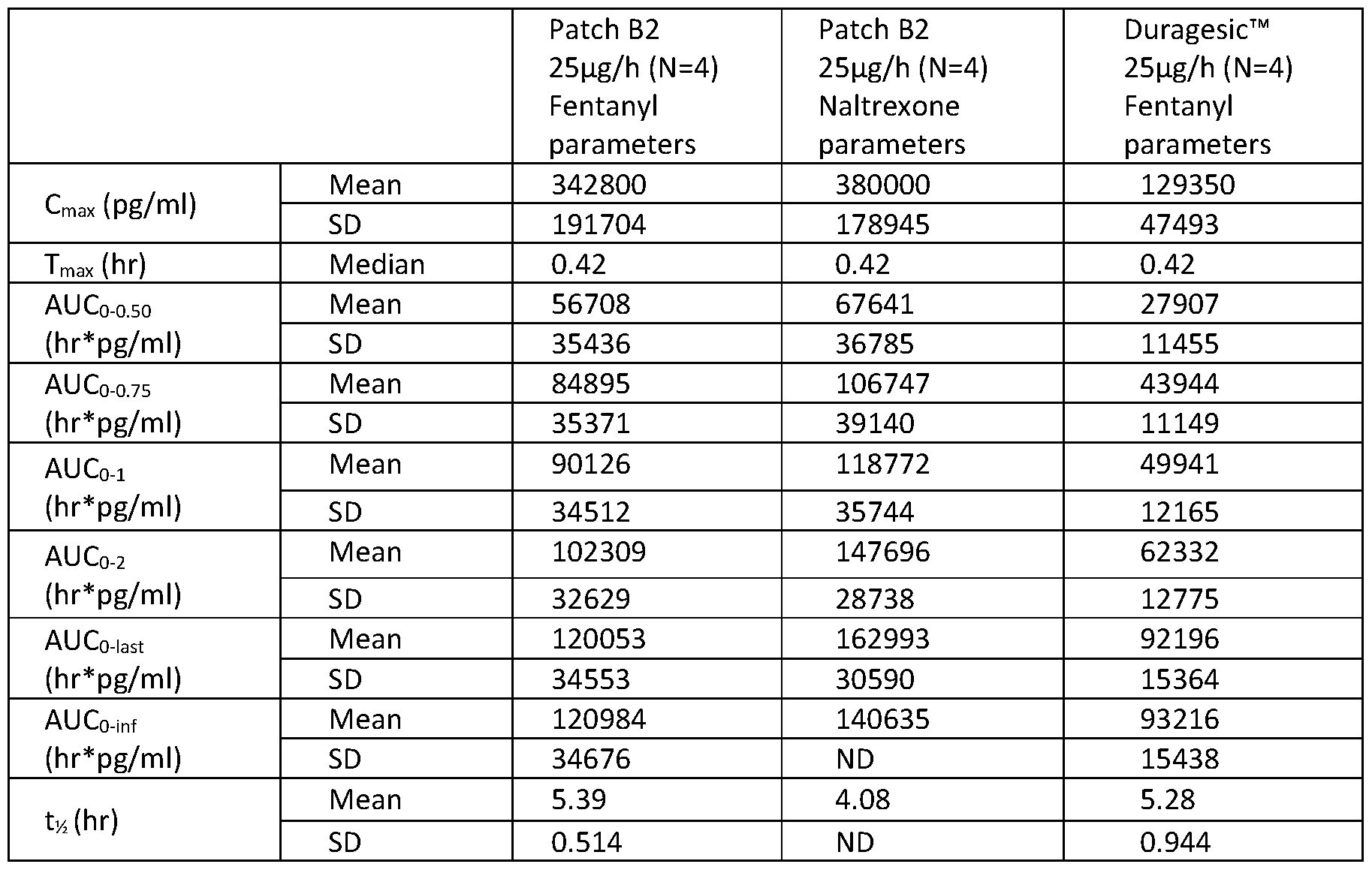
[00244] The average naltrexone to fentanyl plasma concentration ratios from the study for
Patch B2 are shown in Table 8.
Table 8

Example 10
[00245] In a single-center, randomized, open-label, single-dose, crossover study, the relative bioavailability following a single dose dermal application of Patch B2 as compared to Duragesic™ was assessed under standard exposure conditions.
[00246] In healthy subjects who met inclusion/exclusion criteria based on the results of screening assessments, the first application of study drug for each subject was administered within 28 days of their first screening assessments. 50mg naltrexone was administered orally every 12 hours beginning 15 hours prior to application of the patch in an identical manner for all treatment groups. Subjects were randomly assigned to sample groups to begin a round of treatment. Each subsequent patch application was separated by a minimum of 10 days. Patches were removed approximately 72 hours after application.
[00247] Blood samples for measurement of the concentrations of fentanyl and naltrexone (and its metabolite e-b-naltrexol, if needed) were obtained prior to and over 8 days following each patch application (including the 72 hours while the patch was affixed to the skin plus 5 days after its removal). The results of the study are listed in Table 9.
Table 9


[00248] When measured 3 hours post dose on 17 occasions, across all dose groups, naltrexone concentration averaged 4.42 ng/mL (ranging from 3.60 ng/mL to 5.98 ng/mL) throughout the duration of the study (0 hour to 192 hours).
[00249] Over a 192-hour period, the relative bioavailability of fentanyl from Patch B2 (test) relative to that from Duragesic™ patch (reference) was similar for both patch strengths and is presented in Table 10.
Table 10

Example 11
[00250] In a single-center, fixed sequence open label, single-dose, crossover study, the relative bioavailability following a single dose dermal application of Patch B2 was assessed under standard exposure conditions in non-dependent recreational opioid users.
[00251] Subjects who met inclusion/exclusion criteria based on the results of screening assessments with the first application of study drug for each subject administered within 28 days of their first screening assessments. After confirming the subject was otherwise eligible for study participation, a naloxone challenge test was performed at least 12 hours prior to patch application, which occurred on the following day, to confirm that he/she was not physically dependent on opioids.
[00252] Subjects were administered with a single 12.5mcg fentanyl/hr Patch B2 applied dermally with overlay to the upper arm for 72 hours. Subsequently, the subjects were administered with a single 25mcg fentanyl/hr Patch B2 applied dermally with overlay to the upper arm for 72 hours.
[00253] Each patch application was separated by a minimum of 10 days. Patches were removed approximately 72 hours after application. Blood samples for measurement of the
concentrations of fentanyl and naltrexone (and its metabolite e-b-naltrexol, if needed) were obtained prior to and over 8 days following each patch application (including the 72 hours while the patch was affixed to the skin plus 5 days after its removal). The results of the study are listed in Table 11.
Table 11

[00254] When measured post dose across both dose groups, both naltrexone and 6-b- naltrexol concentrations were identified in amounts ranging from 5.1 - 71.9pg/ml amongst most of the
subjects assayed. More subjects with identifiable levels of either naltrexone or e-b-naltrexol were found after the 25mcg/hr than in the 12.5mcg/hr patch.
Example 12
[00255] In a single-center, fixed-sequence, open-label, single-dose, parallel group, 2 study period sequential study, the relative bioavailability following a single dose administration via unintended uses (buccal, chewing) of Patch B2 as compared to Duragesic™ was assessed in naltrexone-blocked healthy subjects.
[00256] In healthy subjects who met inclusion/exclusion criteria based on the results of screening assessments, the first application of study drug for each subject was administered within 28 days of their first screening assessments. Subjects were randomly assigned to sample groups to test either intact buccal administration over 30 minutes of a 12.5pg/h releasing patch or a ¼ of a 25pg/h releasing patch chewed over 10 minutes and then removed. All subjects in received one 50 mg tablet of naltrexone HCI orally with 240 mL of water every 12 hours, starting approximately 15 hours before each patch administration through approximately 69 hours after each patch administration.
[00257] Blood samples for measurement of the concentrations of fentanyl and naltrexone (and its metabolite e-b-naltrexol, if needed) were obtained prior to and over 48 hours following each dose with the results of the study listed in Table 12. There was a minimum 7-day washout between the 2 doses administered.
Table 12


[00258] As presented in Table 13, the relative bioavailability of fentanyl in Patch B2 (test) was found to be considerably lower relative to that from Duragesic™ patch (reference) when applied either buccally or chewed.
Table 13


Example 13
[00259] A 2-period sequential treatment phase in which the dosage form of Patch B2 is to be administered via unintended routes (buccal administration of an intact 12.5 mcg/hr patch or chewed administration of ¼ of a 25 mcg/hr patch) in unblocked non-dependent, opioid-experienced, recreational drug users is assessed. Recreational drug users who meet inclusion/exclusion criteria based on the results of screening assessments receive the first application of study drug within 28 days of their first screening assessments. Subjects are randomly assigned to sample groups to test either intact buccal administration over 30 minutes of a 12.5pg/h releasing patch or a ¼ of a 25pg/h releasing patch chewed over 10 minutes and then removed. After a minimum 7 day wash-out period, the procedure is repeated with the second patch.
[00260] Blood samples for measurement of the concentrations of fentanyl and naltrexone (and its metabolite e-b-naltrexol, if needed) are obtained prior to and over 48 hours following each dose. There is a minimum 7-day washout between the 2 doses administered.
Example 14
[00261] In a randomized, single-dose, crossover, open-label, two-group study, the effect of various extrinsic factors on a 25mcg/hr transdermal dosage system of Patch B2 are assessed in comparison to a naltrexone only patch in both healthy volunteers and non-dependent recreational opioid users.
[00262] The study is divided into 2 groups;
group A) Naltrexone-Blocked Healthy Adult Subjects
group B) Non-Dependent Opioid-Experienced, Recreational Drug Users.
[00263] The study consists of a 4-week screening period, followed by either an 8 single application treatment periods for group A (each lasting 9 days separated by at least a 2-week washout period) or 2 single-application treatment periods for group B (each lasting 6 days separated by at least a 10-day washout period), and a follow-up visit 7 to 10 days after completing the final treatment period. Groups A and B may run in parallel.
[00264] Subjects who met inclusion/exclusion criteria based on the results of screening assessments with the first application of study drug for each subject administered within 28 days of their first screening assessments. For the subjects of group A, 50mg naltrexone is administered every 12 hours until 117 hours after patch removal. After confirming that the subjects of group B are otherwise eligible for study participation, a naloxone challenge test is performed at least 12 hours prior to patch application, to confirm that he/she was not physically dependent on opioids.
[00265] Subjects in group A are randomly assigned to 1 of 4 treatment groups (A, B, C or D) and rotated through the 4 treatment sequences, with each treatment sequence receiving a 25mcg/hr transdermal dosage system of Patch B2 followed by a second series of the same treatment groups with the subjects receiving a 25mcg/hr Duragesic patch;
Normal conditions
Applied daily heat
Daily exercise
Daily showering.
[00266] Subjects in group B are randomly assigned to 1 of 2 treatment groups (E or F) and rotated through the 2 treatment sequences receiving a 25mcg/hr transdermal dosage system of Patch B2;
Normal conditions
Applied daily heat
[00267] On day 1 of period 1 for each group, following an overnight fast of at least 10 hours, subjects ingest a standardized light breakfast 1 to 2 hours before topical application of study patch.
Study drug (ie, a 25mcg/hr transdermal dosage system of Patch B2 or a naltrexone-only patch) are
applied dermally to the upper-outer arm according to the procedures specified below and remain affixed to the skin for 72 hours. Safety monitoring and serial blood sampling for quantification of plasma fentanyl and/or naltrexone and e-b-naltrexol (depending on the group) is performed. All study patches are removed 72 hours (±10 minutes) after application, with safety monitoring and pharmacokinetic sampling continuing through 192 hours after application of a study patch for groups A and B.
[00268] The subjects being assessed for applied daily heat undergo application of heat to a study patch using a standardized, conventional heating pad, covering the transdermal system and kept in place using a gauze bandage, set to increase the temperature at the skin surface to 38 ±3°C (setting on low, with a temperature probe placed under the pad for the entire 10 hours of heat application, the skin temperature being monitored and recorded at 0, 2, 4, 6, 8, and 10 hours post application). The heating pad is applied over the study patch for 10 hours on day 1 (0 to 10 hours), day 2 (24 to 34 hours), and day 3 (48 to 58 hours).
[00269] The subjects being assessed for exercise undergo moderate physical exercise (ie, treadmill, weights, rowing machine, stationary bicycles, elliptical machine) for 1 hour on day 1 (over 4-5 hours post application), day 2 (over 28-29 hours post application), and day 3 (over 52-53 hours post application); rest breaks will be allowed. Heart rate is monitored continuously by subjects and monitored and recorded by study personnel every 10 minutes to reach >60% to <80% of maximum heart rate, as calculated below:
(220 - [subject age in years]) * 0.6 = XX bpm;
example for 35-year old subject: (220-35) * 0.6 = 111 bpm
[00270] A constant heart rate is maintained as much as possible during the entire hour. If the heart rate reaches >80% of maximum heart rate, the intensity of the exercise is lowered and if the heart rate lowers to <60% of maximum heart rate, the intensity of the exercise is increased.
[00271] The subjects being assessed for daily showers undergo a lukewarm (between 98°F [36.7°C] and 105°F [40.6°C]), 15-minute shower with the water jet positioned over the patch at maximum output for 10 minutes (between 4 and 5 hours post application) on day 1 and at the same approximate time on days 2 and 3. Water temperature is recorded prior to and after the subject use of the shower; however, flow rate is not measured or controlled.
Example 15
[00272] A controlled, randomized, double blind study evaluating the clinical efficacy of the transdermal dosage system of the invention is performed in subjects with moderate to severe pain severe enough to require around-the-clock long term opioid treatment. 140 patients enter into a 15- day dose-titration period, followed by a 9-day double-blind period wherein patients are randomized to receive either the 25mcg/hr or 50mcg/hr fentanyl transdermal dosage system of the invention or of a reference fentanyl transdermal dosage system every 72 hours with a study duration of 12 weeks. Patient's responses are monitored for changes in their daily and weekly worst pain intensity scores at weeks 1 through 12 using a numerical 11-point rating scale and for changes from baseline to the final on-treatment visit in the Roland Morris Disability Questionnaire (RMDQ) score
[00273] Those skilled in the art will appreciate that numerous changes and modifications can be made to the preferred embodiments of the disclosure and that such changes and modifications can be made without departing from the spirit of the disclosure. It is, therefore, intended that the appended claims cover all such equivalent variations as fall within the true spirit and scope of the disclosure.
Claims
1. A transdermal dosage system for administering an active agent to a human, the system having reduced potential for abuse and comprising:
a first antagonist layer comprising an active agent and a first antagonist of the active agent; and a second antagonist layer comprising a second antagonist of the active agent; wherein:
the first and second antagonist layers are separated from each other by a barrier that is impermeable to the active agent and the antagonists.
2. The transdermal dosage system of claim 1, wherein the first antagonist is in salt form.
3. The transdermal dosage system of claim 1, wherein the second antagonist is in free base form.
4. The transdermal dosage system of claim 3, wherein the second antagonist free base is in
amorphous form.
5. The transdermal dosage system of claim 1, further comprising a permeable backing layer distal to the second antagonist layer.
6. The transdermal dosage system of claim 2, wherein the first antagonist and the second antagonist are the same antagonist and the second antagonist is in free base form.
7. The transdermal dosage system of any one of the preceding claims, wherein the active agent is an opioid agonist.
8. The transdermal dosage system of any one of the preceding claims, wherein the proximal surface of the first antagonist layer comprises a release liner.
9. The transdermal dosage system of any one of the preceding claims, wherein the active agent and the first antagonist are separated by one or more spacers.
10. The transdermal dosage system of claim 9, wherein the first antagonist layer comprises alternating strips of the active agent and the first antagonist.
11. The transdermal dosage system of any one of the preceding claims, wherein the first antagonist layer comprises one or more skin penetration enhancer, silicone or acrylic adhesive.
12. The transdermal dosage system of claim 11, wherein the skin penetration enhancer is
polyvinylpyrrolidone.
13. The transdermal dosage system of claim 12, wherein the polyvinylpyrrolidone is in an amount of about 2.5 to 3.5%, by weight based on the total weight of the transdermal dosage system.
14. The transdermal dosage system of any one of the preceding claims, wherein the second antagonist layer allows for greater release of the second antagonist in the presence of an organic solvent than in the presence of water.
15. The transdermal dosage system of any one of the preceding claims, wherein the first antagonist of the active agent and the active agent are in a homogenous mixture.
16. The transdermal dosage system of any one of the preceding claims, wherein the first antagonist and the second antagonist are independently naltrexone, methylnaltrexone, naloxone, nalbuphine, nalorphine, nalorphine dinicotinate, nalmefene, nadide, levallorphan, or cyclozocine.
17. The transdermal dosage system of any one of the preceding claims, wherein the active agent is selected from the group consisting of alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, buprenorphine, butorphanol, clonitazene, codeine, desomorphine, dextromoramide, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dihydromorphone, dihydroisomorphine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmorphine, etonitazene, etorphine, dihydroetorphine, fentanyl, heroin, hydrocodone, hydromorphone, hydromorphodone, hydroxypethidine, isomethadone, ketobemidone, levorphanol, levophenacylmorphan, lofentanil, meperidine, meptazinol, metazocine, methadone, metopon, morphine, myrophine, narceine, nicomorphine, norlevorphanol, normethadone, nalorphine, nalbuphene, normorphine, norpipanone, opium, oxycodone, oxymorphone, pantopon, papaveretum, paregoric, pentazocine, phenadoxone, phendimetrazine, phendimetrazone, phenomorphan, phenazocine, phenoperidine, piminodine, piritramide, propheptazine, promedol, properidine, propoxyphene, propylhexedrine, sufentanil, tilidine, tramadol, a pharmaceutically acceptable salt thereof , a prodrug thereof, a derivative thereof and a mixture of any two or more thereof.
18. The transdermal dosage system of claim 17, wherein the active agent is fentanyl, alfentanil,
carfentanil, lofentanil, remifentanil, sufentanil, trefentnanil, or a pharmaceutically acceptable salt thereof, or a prodrug thereof, or a derivative thereof, or a mixture of any two or more thereof.
19. The transdermal dosage system of claim 18, wherein the active agent is fentanyl or a
pharmaceutically acceptable salt thereof.
20. The transdermal dosage system of claim 19, which releases about 5 to about 30 meg of the fentanyl per hour to the skin of the human.
21. The transdermal dosage form of system 20, wherein the fentanyl is in free base form.
22. The transdermal dosage system of claim 21, wherein the fentanyl free base is in a crystalline form.
23. The transdermal dosage system of claim 19 wherein the weight per area of fentanyl is about 0.05 to about 15 mg/cm2.
24. The transdermal dosage system of claim 19, herein the weight per area of fentanyl is about 0.05 to about 5.0 mg/cm2.
25. The transdermal dosage system of claim 19, which releases the fentanyl at a rate of about 75 to about 125 meg per hour to the skin of the human.
26. The transdermal dosage system of claim 19, wherein the first antagonist is naltrexone or a
pharmaceutically acceptable salt thereof; and wherein the second antagonist is naltrexone or a pharmaceutically acceptable salt thereof.
27. The transdermal dosage system of claim 26, wherein the ratio of the weight of the active agent to the weight of the first antagonist and the second antagonist is about 15:1 to about 1:5.
28. The transdermal dosage system of claim 26, wherein the first antagonist is naltrexone free base, the second antagonist is naltrexone hydrochloride and the active agent is fentanyl in free base form.
29. The transdermal dosage system of claim 26, wherein the active agent is fentanyl in amorphous form, the first antagonist is naltrexone HCI in crystalline form, and the second antagonist is naltrexone base in amorphous form.
30. The transdermal dosage system of claim 29, wherein the ratio of fentanyl to naltrexone released when the dosage from is exposed to a liquid is from about 10:1 to about 2:1.
31. The transdermal dosage system of claim 29, which releases about 5 to about 30 meg of the fentanyl per hour to the skin of the human.
32. The transdermal dosage system of claim 29, which releases about 75 to about 125 meg of the fentanyl per hour to the skin of the human.
33. The transdermal dosage system of claim 28, wherein the active agent is fentanyl in crystalline form, the first antagonist is naltrexone HCI in amorphous form, and the second antagonist is naltrexone base in crystalline form.
34. The transdermal dosage system of claim 28, wherein the weight ratio of total naltrexone to fentanyl is more than 3:1.
35. The transdermal dosage system of claim 34, wherein the weight ratio of total naltrexone to fentanyl is about 4:1.
36. A transdermal dosage system for administering fentanyl to a human, the system having reduced potential for abuse and comprising:
a first antagonist layer comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone, or a pharmaceutically acceptable salt thereof; and
a second antagonist layer comprising naltrexone, or a pharmaceutically acceptable salt thereof; wherein the first and second antagonist layers are separated from each other by a barrier that is impermeable to the fentanyl, or the pharmaceutically acceptable salt thereof, and the naltrexone, or the pharmaceutically acceptable salt thereof; and
wherein the ratio of naltrexone, or a pharmaceutically acceptable salt thereof, to fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the system when the system is tampered with is at least 1:1.
37. The transdermal dosage system of Claim 36, wherein the tampering comprises immersion or substantial immersion of the system in a solvent.
38. The transdermal dosage system of Claim 37, wherein the solvent is an aqueous solvent or an
organic solvent.
39. The transdermal dosage system of Claim 38, wherein the solvent is water.
40. The transdermal system of Claim 39, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature is at least 3.4:1 after 10 minutes of tampering.
41. The transdermal system of Claim 39, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°Cis at least 4.8:1 after 10 minutes of tampering.
42. The transdermal system of Claim 39, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C is at least 6.6:1 after 30 minutes of tampering.
43. The transdermal system of Claim 39, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature is at least 3.6:1 after 30 minutes of tampering.
44. The transdermal dosage system of Claim 38, wherein the aqueous solvent is cooking vinegar.
45. The transdermal system of Claim 44, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature is at least 5.4:1 after 10 minutes of tampering.
46. The transdermal system of Claim 44, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C is at least 4.6:1 after 10 minutes of tampering.
47. The transdermal system of Claim 44, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C is at least 4.9:1 after 30 minutes of tampering.
48. The transdermal system of Claim 44, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature is at least 5.2:1 after 30 minutes of tampering.
49. The transdermal dosage system of Claim 38, wherein the organic solvent is ethanol.
50. The transdermal system of Claim 49, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature is at least 5.9:1 after 10 minutes of tampering.
51. The transdermal system of Claim 49, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C is at least 5.9:1 after 10 minutes of tampering.
52. The transdermal system of Claim 49, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C is at least 6.0:1 after 30 minutes of tampering.
53. The transdermal system of Claim 49, wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature is at least 6.0:1 after 30 minutes of tampering.
54. The transdermal dosage system of claim 38, wherein the aqueous solvent is pH 6.8 phosphate buffer.
55. The transdermal dosage system of claim 37, wherein the solvent is artificial saliva.
56. The transdermal dosage system of Claim 37, wherein the solvent is water, ethanol, pH 6.8
phosphate buffer, cooking vinegar, or artificial saliva and wherein the ratio of the naltrexone, or a pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage system by each of water, ethanol, pH 6.8 phosphate buffer, cooking vinegar, or artificial saliva, is at least 1:1 after between 5 minutes and 4 hours of tampering.
57. The transdermal dosage system of any one of claims 36 to 56, wherein the tampering occurs at room temperature.
58. The transdermal dosage system of any one of claims 36 to 56, wherein the tampering occurs at 70°C.
59. The transdermal dosage system of Claim 57, wherein the ratio of the naltrexone, or a
pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at room temperature after 10 minutes of tampering is at least 3.4:1 when the tampering comprises water and at least 5.9:1 when the tampering comprises ethanol.
60. The transdermal dosage system of Claim 58, wherein the ratio of the naltrexone, or a
pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C after 10 minutes of tampering is at least 4.8:1 when the tampering comprises water and at least 5.9:1 when the tapering comprises ethanol.
61. The transdermal dosage system of Claim 57, wherein the ratio of the naltrexone, or a
pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt
thereof, extracted from the dosage form at room temperature after 30 minutes of tampering is at least 3.6:lwhen the tampering comprises water and at least 6.0:1 when the tampering comprises ethanol.
62. The transdermal dosage system of Claim 58, wherein the ratio of the naltrexone, or a
pharmaceutically acceptable salt thereof, to the fentanyl, or a pharmaceutically acceptable salt thereof, extracted from the dosage form at 70°C after 30 minutes of tampering is at least 6.6:1 when the tampering comprises water and at least 6.0:1 when the tampering comprises ethanol.
63. A transdermal dosage system for administering fentanyl to a human comprising a first layer
comprising fentanyl, or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. when administered buccally, a fentanyl Cmax that is 50% or less than the fentanyl Cmax of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, b. when administered buccally, a fentanyl AUCo-tthat is 50% or less than the fentanyl AUCo- t of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, c. when chewed, a fentanyl Cmax that is 50% or less than the fentanyl Cmax of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, d. when chewed, a fentanyl AUCo-t that is 50% or less than the fentanyl AUCo-t of a
reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, e. a naltrexone to fentanyl plasma concentration ratio of 0.7 or greater for the first three hours after buccal administration, and f. a naltrexone to fentanyl plasma concentration ratio of 0.7 or greater for the first three hours after being chewed by a human.
64. The transdermal dosage system of Claim 63, wherein the naltrexone of the first layer is in a pharmaceutically acceptable salt form.
65. The transdermal dosage system of Claim 63, wherein the naltrexone of the second layer is in a free base form.
66. The transdermal dosage system of Claim 64, wherein the naltrexone of the first layer is naltrexone HCI.
67. The transdermal dosage system of Claim 66, wherein the naltrexone HCI is in a crystalline form.
68. The transdermal dosage system of Claim 65, wherein the naltrexone free base is in an amorphous form.
69. The transdermal dosage system of Claim 63, wherein the fentanyl is in a free base form.
70. The transdermal dosage system of Claim 69, wherein the fentanyl is in an amorphous form.
71. The transdermal dosage system of Claim 63, wherein the fentanyl is in a free base form, the
naltrexone in the first layer is naltrexone HCI and the naltrexone in the second layer is in a free base form.
72. The transdermal dosage system of Claim 71, wherein the fentanyl is in an amorphous form, the naltrexone HCI is in a crystalline form, and the naltrexone base is in an amorphous form.
73. The transdermal dosage system of any one of claims 63 to 72, wherein the ratio of the weight of the fentanyl to the weight of the total naltrexone in the first and second layers is about 15:1 to about 1:5.
74. The transdermal dosage system of any one of claims 63 to 72 further comprising a permeable backing layer distal to the second layer.
75. The transdermal dosage system of any one of claims 63 to 74, wherein the proximal surface of the first layer comprises a release liner.
76. The transdermal dosage system of any one of claims 63 to 75, wherein the first layer further
comprises one or more skin penetration enhancers, silicone, or acrylic adhesives.
77. The transdermal dosage system of Claim 76, wherein the skin penetration enhancer is
polyvinylpyrrolidone.
78. The transdermal dosage system of Claim 77, wherein the polyvinylpyrrolidone is in an amount of about 2.5 to 3.5%, by weight based on the total weight of the transdermal dosage system.
79. The transdermal dosage system of any one of claims 63 to 78, wherein the fentanyl and naltrexone of the first layer are in a homogenous mixture.
80. The transdermal dosage system of any one of claims 63 to 78, wherein when administered buccally, exhibits a fentanyl Cmax that is less than 45% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
81. The transdermal dosage system of any one of claims 63 to 78, wherein when administered buccally, exhibits a fentanyl Cmax less than 40% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
82. The transdermal dosage system of any one of claims 63 to 78, wherein when administered buccally, exhibits a fentanyl Cmax less than 35% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
83. The transdermal dosage system of any one of claims 63 to 78, wherein when administered buccally, exhibits a fentanyl Cmax less than 30% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
84. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl Cmax less than 45% the Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
85. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl Cmax less than 40% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
86. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl Cmax less than 35% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
87. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl Cmax less than 30% the fentanyl Cmax of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
88. The transdermal dosage system of any one of claims 63 to 83, wherein when administered buccally, exhibits a fentanyl AUCo-t less than 45% the fentanyl AUCo-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
89. The transdermal dosage system of any one of claims 63 to 83, wherein when administered buccally, exhibits a fentanyl AUCo-t less than 40% the fentanyl AUC0-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
90. The transdermal dosage system of any one of claims 63 to 83, wherein when administered buccally, exhibits a fentanyl AUC0-t less than 35% the fentanyl AUC0-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
91. The transdermal dosage system of any one of claims 63 to 83, wherein when administered buccally, exhibits a fentanyl AUC0-t less than 30% the fentanyl AU M of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
92. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl AUCo-t less than 45% the fentanyl AUCo-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
93. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl AUC0-t less than 40% the fentanyl AUC0-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
94. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl AUC0-t less than 35% the fentanyl AUC0-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
95. The transdermal dosage system of any one of claims 63 to 83, wherein when chewed, exhibits a fentanyl AUC0-t less than 30% the fentanyl AUC0-t of the reference fentanyl transdermal dosage system at therapeutically equivalent transdermal doses.
96. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to
fentanyl plasma concentration ratio of greater than 0.75 for the first three hours after buccal administration.
97. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.80 for the first three hours after buccal administration.
98. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.85 for the first three hours after buccal administration.
99. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.90 for the first three hours after buccal administration.
100. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.95 for the first three hours after buccal administration.
101. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.0 for the first three hours after buccal administration.
102. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.1 for the first three hours after buccal administration.
103. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.2 for the first three hours after buccal administration.
104. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.3 for the first three hours after buccal administration.
105. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.4 for the first three hours after buccal administration.
106. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.5 for the first three hours after buccal administration.
107. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.7 for the first three hours after buccal administration.
108. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 2 for the first three hours after buccal administration.
109. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 2.5 for the first three hours after buccal administration.
110. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 3 for the first three hours after buccal administration.
111. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.75 for the first three hours after being chewed.
112. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.80 for the first three hours after being chewed.
113. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.85 for the first three hours after being chewed.
114. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.90 for the first three hours after being chewed.
115. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 0.95 for the first three hours after being chewed.
116. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.0 for the first three hours after being chewed.
117. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.1 for the first three hours after being chewed.
118. The transdermal dosage system of Claim 63, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.2 for the first three hours after being chewed.
119. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.3 for the first three hours after being chewed.
120. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.4 for the first three hours after being chewed.
121. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.5 for the first three hours after being chewed.
122. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 1.7 for the first three hours after being chewed.
123. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 2 for the first three hours after being chewed.
124. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 2.5 for the first three hours after being chewed.
125. The transdermal dosage system of any one of claims 63 to 83, which exhibits a naltrexone to fentanyl plasma concentration ratio of greater than 3 for the first three hours after being chewed.
126. A method of treating pain in an opioid-tolerant human patient in need thereof comprising
administering to the patient a transdermal dosage system comprising a therapeutically effective amount of fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a
pharmaceutically acceptable salt thereof.
127. The method of Claim 126, wherein the transdermal dosage system comprises a
pharmaceutically acceptable salt of fentanyl.
128. The transdermal dosage system of Claim 127 wherein the pharmaceutically acceptable salt of fentanyl is fentanyl hydrochloride.
129. The method of any one of the preceding claims, wherein the transdermal dosage system
comprises a pharmaceutically acceptable salt of naltrexone.
130. The transdermal dosage system of Claim 129 wherein the pharmaceutically acceptable salt of naltrexone is naltrexone hydrochloride.
131. The method of any one of the preceding claims, wherein the transdermal dosage system
comprises naltrexone base.
132. The method of any one of the preceding claims, wherein the administration of the transdermal dosage system results in a fentanyl Cmax of > 250pg/ml in said patient.
133. The method of Claim 132, wherein the administration of the transdermal dosage system results in a circulating naltrexone plasma concentration of > 6pg/ml in said patient.
134. The method of Claim 132, wherein the administration of the transdermal dosage system results in a circulating e-b-naltrexol plasma concentration of > 6pg/ml in said patient.
135. The method of Claim 132, wherein the administration of the transdermal dosage system results in a circulating naltrexone plasma concentration of > 6pg/ml and a circulating e-b-naltrexol plasma concentration of > 5pg/ml in said patient.
138. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. when heat is applied, a fentanyl Cio that is 110% or more than the Cio of said
transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, b. when heat is applied, a naltrexone Cio that is 105% or more than the Cio of said
transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, c. when heat is applied, a fentanyl Cio that is 180% or less than the Cio of said transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, d. when heat is applied, a naltrexone Cio that is 180% or less than the Cio of said
transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, e. when heat is applied, a fentanyl Cio that is 110% or more and 180% or less than the Cio of said transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses,
f. when heat is applied, a naltrexone Cio that is 105% or more and 180% or less than the Cio of said transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, g. when heat is applied, a transformed fentanyl Cio to naltrexone Cio ratio of 1: 1 or less.
137. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. when heat is applied, a fentanyl AUCio that is 110% or more than the Cio of said
transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, b. when heat is applied, a naltrexone AUCio that is 105% or more than the Cio of said transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, c. when heat is applied, a fentanyl AUCio that is 250% or less than the Cio of said
transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, d. when heat is applied, a naltrexone AUCio that is 250% or less than Cio of said
transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, e. when heat is applied, a fentanyl AUCio that isll0% or more and 250% or less than the AUCio of said transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses, f. when heat is applied, a naltrexone AUCio that is 105% or more and 250% or less than the AUCio of said transdermal dosage system when heat is not applied and when the systems are administered at therapeutically equivalent doses,
g. when heat is applied, a transformed fentanyl AUCio to naltrexone AUCio ratio ofl:l or less.
138. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. after exercise, a fentanyl Cio that is 110% or more than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, b. after exercise, a naltrexone Cio that is 105% or more than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, c. after exercise, a fentanyl Cio that is 180% or less than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, d. after exercise, a naltrexone Cio that is 180% or less than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, e. after exercise, a fentanyl Cio that is 110% or more and 180% or less than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, f. after exercise, a naltrexone Cio that is 105% or more and 180% or less than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, g. after exercise, a transformed fentanyl Cio to naltrexone Cio ratio of 1:1 or less.
139. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. after exercise, a fentanyl AUCio that is 110% or more than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, b. after exercise, a naltrexone AUCio that is 105% or more than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, c. after exercise, a fentanyl AUCio that is 250% or less than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, d. after exercise, a naltrexone AUCio that is250% or less than the Cio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, e. after exercise, a fentanyl AUCio that is 110% or more and 250% or less than the AUCio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, f. after exercise, a naltrexone AUCio that is 105% or more and 250% or less than the AUCio of said transdermal dosage system prior to exercise and when the systems are administered at therapeutically equivalent doses, g. after exercise, a transformed fentanyl AUCio to naltrexone AUCio ratio of 1:1 or less.
140. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from
each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. after showering, a fentanyl Cio that is 110% or more than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, b. after showering, a naltrexone Cio that is 105% or more than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, c. after showering, a fentanyl Cio that is 180% or less than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, d. after showering, a naltrexone Cio that is 180% or less than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, e. after showering, a fentanyl Cio that is 110% or more and 180% or less than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, f. after showering, a naltrexone Cio that is 105% or more and 180% or less than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, g. after showering, a transformed fentanyl Cio to naltrexone Cio ratio of 1:1 or less.
141. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following :
a. after showering, a fentanyl AUCio that is 110% or more than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, b. after showering, a naltrexone AUCio that is 105% or more than the Cio of said
transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, c. after showering, a fentanyl AUCio that is 250% or less than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, d. after showering, a naltrexone AUCio that is 250% or less than the Cio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, e. after showering, a fentanyl AUCio that is 110% or more and 250% or less than the AUCio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, f. after showering, a naltrexone AUCio that is 105% or more and 250% or less than the AUCio of said transdermal dosage system prior to showering and when the systems are administered at therapeutically equivalent doses, g. after showering, a transformed fentanyl AUCio to naltrexone AUCio ratio of 1: 1 or less.
142. A transdermal dosage system for administering fentanyl to a human comprising a first layer comprising fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and a second layer comprising naltrexone or a pharmaceutically acceptable salt thereof, wherein the first and second layers are separated from each other by a barrier that is impermeable to fentanyl or a pharmaceutically acceptable salt thereof, and naltrexone or a pharmaceutically acceptable salt thereof, and wherein said transdermal dosage system exhibits at least one of the following : a. when heat is applied, a fentanyl Cio that is 80% or less than the fentanyl Cio of a
reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses,
b. after exercise, a fentanyl Cio that is 80% or less than the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, c. after showering, a fentanyl Cio that is 80% or less than the fentanyl Cio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, d. when heat is applied, a fentanyl AUCio that is 80% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, e. after exercise, a fentanyl AUCio that is 80% or less than the fentanyl AUCio of a reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses, f. after showering, a fentanyl AUCio that is 80% or less than the fentanyl AUCio of a
reference fentanyl transdermal dosage system when the systems are administered at therapeutically equivalent transdermal doses.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862692904P | 2018-07-02 | 2018-07-02 | |
| US201862692959P | 2018-07-02 | 2018-07-02 | |
| US62/692,904 | 2018-07-02 | ||
| US62/692,959 | 2018-07-02 | ||
| PCT/US2018/040536 WO2020009685A1 (en) | 2018-07-02 | 2018-07-02 | Transdermal dosage form |
| USPCT/US2018/040536 | 2018-07-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020008366A1 true WO2020008366A1 (en) | 2020-01-09 |
Family
ID=67402984
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2019/055644 WO2020008366A1 (en) | 2018-07-02 | 2019-07-02 | Transdermal dosage form |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2020008366A1 (en) |
Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4539256A (en) | 1982-09-09 | 1985-09-03 | Minnesota Mining And Manufacturing Co. | Microporous sheet material, method of making and articles made therewith |
| US4693776A (en) | 1985-05-16 | 1987-09-15 | Minnesota Mining And Manufacturing Company | Macromer reinforced pressure sensitive skin adhesive |
| US5149538A (en) | 1991-06-14 | 1992-09-22 | Warner-Lambert Company | Misuse-resistive transdermal opioid dosage form |
| US5236714A (en) | 1988-11-01 | 1993-08-17 | Alza Corporation | Abusable substance dosage form having reduced abuse potential |
| US5474783A (en) | 1988-03-04 | 1995-12-12 | Noven Pharmaceuticals, Inc. | Solubility parameter based drug delivery system and method for altering drug saturation concentration |
| US5585111A (en) | 1993-12-08 | 1996-12-17 | Minnesota Mining And Manufacturing Company | Transdermal delivery device |
| US5985317A (en) | 1996-09-06 | 1999-11-16 | Theratech, Inc. | Pressure sensitive adhesive matrix patches for transdermal delivery of salts of pharmaceutical agents |
| US20020119187A1 (en) | 2000-09-29 | 2002-08-29 | Cantor Adam S. | Composition for the transdermal delivery of fentanyl |
| WO2002087482A1 (en) * | 2001-05-01 | 2002-11-07 | Euro-Celtique | Abuse resistant opioid containing transdermal systems |
| US20030026829A1 (en) | 2001-03-16 | 2003-02-06 | Venkatraman Subramanian S. | Transdermal administration of fentanyl and analogs thereof |
| US20040126323A1 (en) | 2002-08-20 | 2004-07-01 | Ihor Shevchuk | Transdermal dosage form comprising an active agent and a salt and free-base form of an adverse agent |
| US20040219196A1 (en) * | 2003-04-30 | 2004-11-04 | 3M Innovative Properties Company | Abuse-resistant transdermal dosage form |
| US20050095279A1 (en) * | 2003-10-30 | 2005-05-05 | Gale Robert M. | Transdermal analgesic systems having reduced abuse potential |
| US20080020028A1 (en) * | 2003-08-20 | 2008-01-24 | Euro-Celtique S.A. | Transdermal dosage form comprising an active agent and a salt and a free-base form of an adverse agent |
| US20100076198A1 (en) | 2008-09-19 | 2010-03-25 | Mallinckrodt Inc. | Crystalline forms of Fentanyl Alkaloid |
| US8747889B2 (en) | 2002-04-23 | 2014-06-10 | Durect Corporation | Transdermal analgesic systems with reduced abuse potential |
| US8778382B2 (en) * | 2003-04-30 | 2014-07-15 | Purdue Pharma L.P. | Tamper resistant transdermal dosage form |
| US8790689B2 (en) | 2003-04-30 | 2014-07-29 | Purdue Pharma L.P. | Tamper resistant transdermal dosage form |
| WO2017125455A1 (en) * | 2016-01-18 | 2017-07-27 | Buzzz Pharmaceuticals Limited | Transdermal patch |
-
2019
- 2019-07-02 WO PCT/IB2019/055644 patent/WO2020008366A1/en active Application Filing
Patent Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4539256A (en) | 1982-09-09 | 1985-09-03 | Minnesota Mining And Manufacturing Co. | Microporous sheet material, method of making and articles made therewith |
| US4693776A (en) | 1985-05-16 | 1987-09-15 | Minnesota Mining And Manufacturing Company | Macromer reinforced pressure sensitive skin adhesive |
| US5474783A (en) | 1988-03-04 | 1995-12-12 | Noven Pharmaceuticals, Inc. | Solubility parameter based drug delivery system and method for altering drug saturation concentration |
| US5236714A (en) | 1988-11-01 | 1993-08-17 | Alza Corporation | Abusable substance dosage form having reduced abuse potential |
| US5149538A (en) | 1991-06-14 | 1992-09-22 | Warner-Lambert Company | Misuse-resistive transdermal opioid dosage form |
| US5585111A (en) | 1993-12-08 | 1996-12-17 | Minnesota Mining And Manufacturing Company | Transdermal delivery device |
| US5985317A (en) | 1996-09-06 | 1999-11-16 | Theratech, Inc. | Pressure sensitive adhesive matrix patches for transdermal delivery of salts of pharmaceutical agents |
| US20020119187A1 (en) | 2000-09-29 | 2002-08-29 | Cantor Adam S. | Composition for the transdermal delivery of fentanyl |
| US20030026829A1 (en) | 2001-03-16 | 2003-02-06 | Venkatraman Subramanian S. | Transdermal administration of fentanyl and analogs thereof |
| WO2002087482A1 (en) * | 2001-05-01 | 2002-11-07 | Euro-Celtique | Abuse resistant opioid containing transdermal systems |
| US8747889B2 (en) | 2002-04-23 | 2014-06-10 | Durect Corporation | Transdermal analgesic systems with reduced abuse potential |
| US20040126323A1 (en) | 2002-08-20 | 2004-07-01 | Ihor Shevchuk | Transdermal dosage form comprising an active agent and a salt and free-base form of an adverse agent |
| US7182955B2 (en) | 2003-04-30 | 2007-02-27 | 3M Innovative Properties Company | Abuse-resistant transdermal dosage form |
| US20040219196A1 (en) * | 2003-04-30 | 2004-11-04 | 3M Innovative Properties Company | Abuse-resistant transdermal dosage form |
| US8778382B2 (en) * | 2003-04-30 | 2014-07-15 | Purdue Pharma L.P. | Tamper resistant transdermal dosage form |
| US8790689B2 (en) | 2003-04-30 | 2014-07-29 | Purdue Pharma L.P. | Tamper resistant transdermal dosage form |
| US20080020028A1 (en) * | 2003-08-20 | 2008-01-24 | Euro-Celtique S.A. | Transdermal dosage form comprising an active agent and a salt and a free-base form of an adverse agent |
| US20050095279A1 (en) * | 2003-10-30 | 2005-05-05 | Gale Robert M. | Transdermal analgesic systems having reduced abuse potential |
| US20100076198A1 (en) | 2008-09-19 | 2010-03-25 | Mallinckrodt Inc. | Crystalline forms of Fentanyl Alkaloid |
| WO2017125455A1 (en) * | 2016-01-18 | 2017-07-27 | Buzzz Pharmaceuticals Limited | Transdermal patch |
Non-Patent Citations (1)
| Title |
|---|
| DRAFT GUIDANCE ON FENTANYL, October 2016 (2016-10-01) |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2522529C (en) | Tamper resistant transdermal dosage form | |
| US8790689B2 (en) | Tamper resistant transdermal dosage form | |
| JP4642467B2 (en) | Transdermal dosage form containing active agent and salt and free base form of side effect substance | |
| ES2592277T3 (en) | Transdermal deterrent formulations of abuse of opioid agonists and agonists | |
| US20080020028A1 (en) | Transdermal dosage form comprising an active agent and a salt and a free-base form of an adverse agent | |
| US20190328681A1 (en) | Transdermal patch | |
| JP2018080197A (en) | Systems and methods for treating opioid-induced adverse pharmacodynamic response | |
| CN111447927A (en) | Naloxone transdermal drug delivery device and method of use thereof | |
| US10406154B2 (en) | Transdermal dosage form | |
| WO2020008366A1 (en) | Transdermal dosage form | |
| CA2961009A1 (en) | Systems and methods for attenuating opioid-induced euphoria | |
| ZA200507862B (en) | Tamper resistant transdermal dosage form | |
| WO2020009685A1 (en) | Transdermal dosage form | |
| WO2020008370A1 (en) | Transdermal patch | |
| KR20180074797A (en) | Transdermal transdermal delivery devices and compositions comprising non-transdermal delivery of N-oxide derivatives of opioid agonists and opioid antagonists for the treatment of pain |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19742952 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19742952 Country of ref document: EP Kind code of ref document: A1 |