WO2019229255A1 - Photocatalyst and photocatalytic methods for producing hydrogen - Google Patents
Photocatalyst and photocatalytic methods for producing hydrogen Download PDFInfo
- Publication number
- WO2019229255A1 WO2019229255A1 PCT/EP2019/064221 EP2019064221W WO2019229255A1 WO 2019229255 A1 WO2019229255 A1 WO 2019229255A1 EP 2019064221 W EP2019064221 W EP 2019064221W WO 2019229255 A1 WO2019229255 A1 WO 2019229255A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- photocatalyst
- ncn
- catalyst
- biomass
- carbon
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen; Reversible storage of hydrogen
- C01B3/02—Production of hydrogen; Production of gaseous mixtures containing hydrogen
- C01B3/04—Production of hydrogen; Production of gaseous mixtures containing hydrogen by decomposition of inorganic compounds
- C01B3/042—Decomposition of water
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/18—Carbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/24—Nitrogen compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/39—Photocatalytic properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/613—10-100 m2/g
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen; Reversible storage of hydrogen
- C01B3/02—Production of hydrogen; Production of gaseous mixtures containing hydrogen
- C01B3/22—Production of hydrogen; Production of gaseous mixtures containing hydrogen by decomposition of gaseous or liquid organic compounds
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/02—Processes for making hydrogen or synthesis gas
- C01B2203/0266—Processes for making hydrogen or synthesis gas containing a decomposition step
- C01B2203/0277—Processes for making hydrogen or synthesis gas containing a decomposition step containing a catalytic decomposition step
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1052—Nickel or cobalt catalysts
- C01B2203/1058—Nickel catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1064—Platinum group metal catalysts
- C01B2203/107—Platinum catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/36—Hydrogen production from non-carbon containing sources, e.g. by water electrolysis
Definitions
- the present invention provides photocatalysts and the use of those photocatalysts in photocatalytic methods for degrading an organic substrate, such as a biomass, or a species, such as pollutant, in untreated water. Also provided is the use of the photocatalysts in photocatalytic methods for generating hydrogen, and methods for preparing those photocatalysts.
- An emerging strategy is to replace the water oxidation reaction by the direct reformation of unprocessed biomass such as lignocellulosic waste, which does not compete with food production (see Kuehnel et al.).
- the biomass is oxidized instead of water and enables sustainable hydrogen production driven by solar light in the presence of a photocatalyst.
- Photocatalytic reforming of biomass is thermodynamically more favourable (thus potentially enabling higher hydrogen generation rates than classical water splitting), and prevents the formation of potentially unwanted oxygen.
- Lignocellulosic biomass is the most abundant form of biomass and its photoreformation has been previously achieved using CdS/CdOx and Pt/TiC>2 photocatalysts.
- these photocatalysts are problematic as they can be toxic (for example, owing to the presence of Cd), expensive (for example, owing to the use of Pt), inefficient and/or require harsh experimental conditions (e.g., strongly alkaline solutions).
- Wakerley et al. describe the use of a CdS quantum dot-based photocatalyst that has activity to generate hydrogen from biomass under incident simulated solar-light (see also PCT/EP2017/080371 ).
- the photocatalyst has an oxygenated surface, and this is generated by treatment of the CdS semiconductor material under highly alkaline conditions, such as pH 10 or more, and preferably pH 14 or more.
- the exemplified photocatalyst also makes use of Cd, and there is a desire to avoid the use of this toxic species, where possible.
- the present inventors have identified a particular class of catalysts that have useful properties when exposed to visible and/or UV light, such as sunlight or simulated visible light, in combination with a co-catalyst. These photocatalysts may be reliably and efficiently used in photocatalytic methods for degrading organic substrates, such as biomass, and photocatalytic methods for producing hydrogen.
- the catalysts are carbonaceous, and therefore of low cost and low toxicity, and are additionally well suited to large scale applications.
- the catalyst is therefore a photocatalyst.
- the photocatalyst of the invention is a carbon- containing catalyst, where the carbon may be present in a graphitic and/or amorphous phase.
- the photocatalyst may be a carbon nitride (CN X ) photocatalyst or a carbon dot (CD) photocatalyst.
- the photocatalyst may be a carbon- and nitrogen-containing catalyst, such as a carbon nitride (CN X ) photocatalyst or a nitrogen-doped carbon dot (CD) photocatalyst.
- the photocatalyst is a NCN CN X photocatalyst, and more preferably such a photocatalyst having a high surface area, such as obtainable from sonication, such as ultrasonication, of a bulk sample of NCN CN x .
- the photocatalyst may have a surface area that is at least 60 m 2 g 1 .
- the photocatalyst is a carbon dot photocatalyst, and more preferably such a photocatalyst is obtained or obtainable from a biomass, for example by calcination of the biomass.
- the biomass may be a readily available and an abundant natural or processed resource.
- the carbon dot photocatalyst is obtained or obtainable from readily available and low cost organic molecular precursors.
- the photocatalysts of the invention have excellent photocatalytic activity in methods for degrading (or reform) biomass, such as those biomasses having a high cellulose content, and this degradation may be accomplished at ambient temperatures and pressures using natural light or simulated natural light, such as solar-light.
- the photocatalyst may be used to degrade tree material, such as branches, as well as processed biomass products such as paper and cardboard, which may be obtained as waste materials.
- the photocatalysts are also capable of catalysing the degradation of lignin, which is also a component of many biomasses, and which is difficult to process on an industrial scale.
- the photocatalyst may also be used to degrade semi-refined biomass, for example the photocatalyst may be used to degrade other biomass components, such as cellulose and xylan, which have been extracted from a biomass.
- the photocatalysts of the invention may be used in a photocatalytic method for the generation of hydrogen, for example from biomass.
- the photocatalysts are therefore capable of producing a useful fuel from abundant raw and waste biomass.
- the photocatalysts of the invention are active under solar-light, including simulated solar- light, and may be used for sustained periods, for example for more than two days, such as more than 12 days, without appreciable loss in activity.
- the photocatalysts are therefore have long term photocatalytic activity and stability.
- the rate of substrate degradation is remarkably high, and the photocatalyst of the invention has a significant advantage in that no substrate chemical processing is required in order to obtain the hydrogen product.
- the photocatalyst of the invention has a significant advantage in that no substrate chemical processing is required in order to obtain the hydrogen product.
- photocatalyst is able to catalyse biomass reformation without the need for pre-treatment under highly basic conditions.
- the photocatalysts of the invention are for use together with a co-catalyst for the generation of hydrogen.
- the co-catalyst is a hydrogen evolution catalyst, such as those well known in the art, including NiP-, Pt- and Mo-based co-catalysts.
- the co-catalyst is a noble metal-free co-catalyst, such as NiP.
- a method for degrading an organic substrate comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of the organic substrate and a co-catalyst that is a hydrogen evolution catalyst.
- the degradation of the organic substrate may produce hydrogen, and the hydrogen is optionally collected.
- a method for producing hydrogen comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of an organic substrate and a co-catalyst that is a hydrogen evolution catalyst.
- the method may also comprise the step of collecting the hydrogen. Carbon monoxide may be optionally produced in this method.
- the photocatalyst and the organic substrate may be provided as an aqueous mixture, together with a co-catalyst that is a hydrogen evolution catalyst.
- the photocatalyst may be used within a broad pH range, and the aqueous mixture may have a pH in the range pH 2 to 15, such as a pH in the range pH 2 to 9.
- the aqueous mixture may be basic, acidic or substantially neutral.
- the aqueous mixture may contain untreated water.
- water for the aqueous mixture may be obtained, without treatment, from a river, lake, canal, or a sea, or from industrial waste water, such as sewage water.
- the photocatalysts of the invention therefore allow the production of clean hydrogen under real-world conditions, without the need for purified drinking water or laboratory quality water.
- the organic substrate may be or comprise a hydroxyl-functionalised compound, such as a hydroxyl-functionalised compound having a molecular weight of 50 or more, such as 500 or more.
- the organic substrate may be or comprise a polyol.
- the organic substrate may be or comprise a monosaccharide, an oligosaccharide or a polysaccharide, such as cellulose, and preferably the organic substrate may be or comprise a polysaccharide.
- the organic substrate may be a biomass, such as wood, paper, cardboard or grass.
- the organic substrate may contain cellulose, xylan and lignin.
- the organic substrate may be selected from the group consisting of general waste, plastics, packing materials, waste food, aliphatic polyols, algae, sugars, starches, biomass, sewage and/or domestic waste.
- the organic substrate may be mechanically processed, such as chopped (including chipped), prior to use in the methods of the invention. However, in a preferred method of the invention, no mechanical processing is required, and the organic substrate may be used directly.
- the organic substrate may be pre-treated, for example with an aqueous base, to at least partially degrade the organic substrate and/or to improve the aqueous solubility of the organic substrate.
- a pre-treatment is associated with an increased hydrogen evolution rate in the subsequent photocatalytic reaction.
- the organic substrate may be an organic polymer, such as a polymer having hydroxyl, ester, amide, ether, carbonate, and/or urethane functionality, either in the backbone chain or a side group.
- organic polymer such as a polymer having hydroxyl, ester, amide, ether, carbonate, and/or urethane functionality, either in the backbone chain or a side group.
- the organic polymer may be selected from polyester, substituted polyethylene, polyether, polycarbonate, and polyurethane.
- the organic substrate may be a polymer selected from the group consisting of poly(ethylene terephthalate) (PET), poly(lactic acid) (PLA), poly(vinyl pyrrolidone) (PVP), and polyethylene glycol) (PEG). Additionally or alternatively, the organic substrate may be selected from the group consisting of polyurethane (PUR), a polypeptide and a polysaccharide.
- the polymer may be further selected from low density polyethylene (LDPE), polyvinyl chloride (PVC), poly(methyl methacrylate), polystyrene (PS) and polycarbonate.
- the polymers may require pre-treatment, such as with aqueous base, prior to the photocatalytic reaction.
- the organic polymer may have a molecular weight of 500 or more, such as 1 ,000 or more, such as 5,000 or more.
- the organic substrate may be a component that is a pollutant in an untreated water source.
- the organic substrate may be selected from chloromethylphenol,
- the component may additionally or alternatively be selected from atrazine, desmetryn, dicamba, chloromehtylphenoxyacetic acid, mecoprop, prometryn, propazine, simazine, terbutryn, and trietazine.
- the component may also be an inorganic component, such as chloride. These components may be found in river water, also including canal and lake water (or“fresh water” in contrast to sea water).
- the incident light in the photocatalytic reaction may be ultraviolet light, such as light having a wavelength in the range from 100 to less than 400 nm, and it may be visible light, such as natural light (such as sunlight), such as light having a wavelength in the range from 400 to 700 nm, or a combination of visible and ultraviolet light, such as natural light (such as sunlight, which has visible and UV components).
- the incident light may be a combination of natural and non-natural light.
- the intensity of the light may be in the range 1 to 150 mW/cm 2 , such as 10 to 150 mW/cm 2 , such as 50 to 150 mW/cm 2 , such as 90 to 150 mW/cm 2 , such as 130 to 145 mW/cm 2 .
- the light intensity may be around 100 mW/cm 2 .
- the photocatalyst as a photocatalyst.
- the photocatalyst may be for use under incident light, including natural light, such as solar light.
- Also provided by the present invention are methods of preparing the photocatalysts.
- a method of preparing NCN CN x comprising the step of forming a bulk sample of NCN CN X and subsequently treating the bulk NCN CN X thereby to increase the surface area of the NCN CN x .
- the bulk NCN CN X may be sonicated, such as ultrasonicated, to increase its surface area.
- the bulk NCN CN X may be obtained or obtainable from H2N CN X .
- a method of preparing a carbon dot comprising the step of calcining an organic substrate.
- the organic substrate may be or comprise an oligosaccharide or a polysaccharide, such as cellulose, and preferably the organic substrate may be or comprise a polysaccharide.
- the method may provide a product that is a plurality of carbon dots.
- the organic substrate may be the source for the photocatalyst as well as a source - an electron donor - in the hydrogen production method.
- the biomass may be sourced locally together with local untreated water, to provide a substantial proportion of the chemical input for the method from the immediate environment, thereby minimising transport costs for the user.
- the invention also provides photocatalysts obtained or obtainable from the methods of preparation described above.
- Figure 1 is a schematic representation of a photocatalytic system according to an
- lignocellulose may be replaced with a component, such as a pollutant, present within untreated water.
- Figure 2 shows the characterisation data for bulk and sonicated NCN CN x , where (a) is the UV-vis absorption spectra for bulk and sonicated NCN CN X at 0.0125 mg ml. 1 ; (b) is the photoluminescence emission spectra for bulk and sonicated NCN CN X at 0.67 mg ml.
- Figure 3 shows the characterising data for CDs synthesised from purified and waste biomass, where (a) are the HR-XPS C 1 s and N 1 s spectra of ocellulose (top) and
- G. /V/Va//s-derived CDs bottom.
- the black traces show the‘as recorded’ spectra and the colored plots (red, blue and green) are the deconvoluted bands for each element;
- the white lines denote the (100) intralayer spacings;
- Figure 4 shows the recorded photocatalytic Pl 2 evolution using CDs from purified and waste biomass, in purified water in the presence of EDTA (3 ml_, 0.1 M, pFH 6) and 50 nmol of NiP, where (a) shows the absolute quantities of Fh produced (in pmol) from ocellulose, xylan and lignin-derived photoabsorbers after 6 h (solid bars) and 24 hrs of irradiation (empty bars); (b) specific activities of ocellulose CDs, xylan and lignin light-harvesters after 24 h of irradiation; and (c) quantities of Pl 2 produced (in pmol) from waste biomass photoabsorbers after 24 h of irradiation.
- Figure 5 shows the photocatalytic performances of CDs from purified and waste biomass in untreated sea, river and sewage water, where (a) shows photo-Fh production with ocellulose CDs in river, sea, sewage and pure water, in the absence (filled symbols) and presence (open symbols) of EDTA (0.1 M, pH 6, 3 ml_); (b) shows solar-H2 generation of ocellulose CDs in KPi (0.1 M, pH 6, 3 ml_), in the absence of EDTA, but instead in the existence of organic pollutants (200 pmol) and chloride ions (3.2 wt %), found in river (open symbols) and sea water (filled symbols).
- Figure 6 shows solar-light driven H 2 evolution using ocellulose CDs at quantities between 0.03 and 2.8 mg. All photocatalytic experiments were carried out at 50 nmol NiP and in aqueous EDTA solutions (0.1 M, pH 6, 3 ml_), under full solar spectrum irradiation
- Figure 7 shows the results of control photocatalytic experiments in the absence of ocellulose CDs, NiP and EDTA, carried out under full solar spectrum irradiation (AM 1.5 G, 100 mW cm 2 ), at 25°C.
- Figure 8 shows solar-induced H 2 production using CDs from cotton.
- cotton from different sources such as cotton wool, pads and cotton T-shirt, were employed as precursors for this purpose.
- the photocatalytic systems were carried out in aqueous EDTA solutions (0.1 M, pH 6, 3 ml_), for 24 h, under full solar spectrum irradiation (AM 1.5 G,
- Figure 9 shows (a) light-driven H 2 evolution (24 h) using ocellulose CDs as photoabsorbers in untreated river, sea and sewage water, in the absence of NiP and in the presence of EDTA (0.1 M, 3 ml_, pH 6); (b) the chemical structures of organic water pollutants, which exist in sea (Gulf of Mexico) and river water (River Cam, UK), based on reports of the US Department of Interior (Torres et al.) and the British environmental agency (Huber et al.), respectively; (c) photo-H 2 production (24 h) using ocellulose CDs, in KPi (0.1 M, pH 6,
- Figure 10 shows the H 2 production using activated (10 min ultra-sonicated) NCN CN X (5 mg) with NiP (50 nmol) and polymer substrates (100 mg) in KPi solution (0.1 M, pH 4.5, 3 ml.) under 1 sun irradiation for 24 hours (100 mW cm -2 , AM1.5G, 25°C, where PET: polyethylene terephthalate; PUR: polyurethane; and PS: polystyrene.
- the present inventors have found that a particular class of photocatalysts may be used to degrade organic substrates under incident light, such as under incident solar light, so as to provide hydrogen and optionally other products, including carbon monoxide.
- a photocatalyst of the invention is a carbon-containing catalyst, and more specifically the photocatalyst contains carbon in a graphitic and/or amorphous phase.
- the photocatalysts typically have a predominant carbon sp 2 character.
- the photocatalyst may be a carbon nitride (CN X ) photocatalyst or a carbon dot (CD)
- Lau et al. (Nat. Commun. 2016) have previously described the design and optimisation of carbon nitride photocatalysts. Lau et at. do not suggest the use of the photocatalyst to degrade biomass.
- CN 107961808 describes the preparation of a carbon nitride photocatalyst, and the use of the photocatalyst to generate hydrogen from water. There is no suggestion anywhere that the catalyst should be used to degrade an organic substrate. There is also no suggestion that the photocatalyst could be used to degrade pollutants within waste water. There is also no reference to the use of the carbon nitride photocatalyst together with a hydrogen evolution catalyst, which is a requirement of the methods of the present case.
- CN 107413378 describes the preparation of a carbon nitride photocatalyst. This catalyst is for use in generating hydrogen from water. There is no mention of the catalyst for use together with a hydrogen evolution catalyst, and there is no mention of any organic substrate, such as a polysaccharide, or a pollutant from waste water.
- CN 107297217 describes the preparation of a platinum-based carbon nitride catalyst.
- This catalyst is for use in the generation of hydrogen from water. This water is not said to be waste water, and the action of the catalyst is on water as a substrate not any pollutant.
- the worked examples show the use of the carbon nitride catalyst together with water and sacrificial triethanolamine to generate hydrogen. There is no mention of organic substrates (other than the triethanolamine), and there is no mention of a co-catalyst for generation of hydrogen.
- CN 107008484 describes a metal sulfide-modified carbon-nitride photocatalyst. This photocatalyst is used to generate hydrogen from water. There is no reference to any biomass materials and there is no reference to the treatment of waste water and its pollutants. There is no disclosure in this Chinese application of the use of two separate catalysts species in the reaction mixture.
- CN 106902803 describes the modification of carbon dots with KNbC>3 to give a composite photocatalyst.
- the photocatalyst has use in the degradation of organic pollutants, whilst also producing hydrogen.
- a biomass materials such as paper, or large organic molecules such as
- CN 107837817 describes a composite material that is a combination of carbon dots, carbon nitride and titanium dioxide. This material is said to be useful for the photocatalytic generation of hydrogen from water.
- the carbon dots are prepared from the soot of a burning candle. There is no suggestion that these dots could be prepared from saccharides.
- CN 106902857 refers to the use of a carbon nitride photocatalyst to degrade organic pollutants, and it also refers to the use of the photocatalyst to generate hydrogen. However, it does not suggest the two together.
- the organic dye Rhodamine B is degraded in the presence of a photocatalyst. There is no description of the degradation products, and the example only looks at the loss of absorbance at a single wavelengths as indicative of a degradation event. There is no suggestion that hydrogen is generated.
- the carbon nitride photocatalyst is used in water together with ascorbic acid and chloroplatinic acid to generate hydrogen.
- ascorbic acid and chloroplatinic acid there is no reference to anything that that is a biomass.
- the carbon nitride photocatalyst itself is called an expanded carbon nitride, which apparently has an increased surface area. There is no reference to ultrasonication as a method to treat bulk carbon nitride.
- CN 107552083 describes a carbon nitride photocatalyst combined with FeP.
- the photocatalytic activity is only briefly described: the photocatalyst is for use in producing hydrogen from water. There is no mention of any other substrate.
- the photocatalyst of the invention is a material, which may be in the form of a particle, which may be at least partially aggregated with other photocatalyst particles.
- the photocatalyst is a carbonaceous photocatalyst such as a carbon nitride or a carbon dot. The use of a carbonaceous photocatalyst to degrade biomass has not previously been reported in the literature.
- the photocatalyst for use in the present invention may be a carbon nitride catalyst, and in particular a NCN CN X catalyst.
- NCN CN X catalysts are known, their use to degrade organic substrates such as a-cellulose and biomass components such as paper, sawdust, and wood, has not previously been described.
- the methods of the invention use a known NCN CN X catalyst to generate hydrogen from a biomass.
- the inventors have also developed new NCN CN X catalysts having greater activity than those NCN CN X catalysts reported to date.
- these catalysts have a greater surface area than those known in the art. This increase in surface area is achieved by treating NCN CN X to reduce the size of agglomerates within a bulk sample, for example by sonicating, such as ultrasonicating, the bulk.
- the surface area of the carbon nitride catalyst is at least 60, at least 70, at least 80, at least 90 or at least 95 m 2 g 1 .
- the surface area of the carbon nitride catalyst may be about 97 m 2 g 1 .
- a bulk NCN CN X catalyst produced by methods know in the art has a surface area of 59.5 m 2 g -1 .
- the carbon nitride catalyst produced in the present case has a surface area of 97.4 m 2 g 1 .
- the surface area may be determined by BET measurement, such as described herein.
- the photocatalyst for use in the invention may be a carbon dot, which is optionally doped, such as nitrogen, sulfur, or phosphorous doped, such as nitrogen doped.
- a carbon dot may also be referred to as a carbon quantum dot (CQD).
- CQD carbon quantum dot
- Carbon dots are described by some of the present inventors in Martindale et al. ( Angew . Chem. Int. Ed. 2017) and Guo et al.
- a carbon dot may be a particle having a largest dimension of at most 10 nm, such as at most 5 nm, such as at most 3 nm.
- the size of a quantum dot may be determined by, for example, analysis of TEM imagery of a collection of carbon dots.
- a carbon dot is obtained or is obtainable by calcination (pyrolysis) of an organic substrate, such as a described herein.
- carbon dot photocatalysts are known, their use to degrade organic substrates such as ocellulose has not previously been described.
- preparation of carbon dots is well known from small organic starting materials, such as amino acids, the use of biomass materials, such as ocellulose, for the preparations of carbon dots has not previously been described.
- the photocatalyst is provided in an aqueous mixture together with an organic substrate and a co-catalyst.
- the photocatalyst may be used within a broad pH range, and the aqueous mixture may have a pH in the range pH 2 to 15, such as pH 2 to 9.
- the aqueous mixture may be basic, acidic or substantially neutral.
- the aqueous mixture may be acidic, such as a pH in the range pH 2-6, such as a pH in the range pH 3-5. In one embodiment, the aqueous mixture has a pH of about pH 4.5.
- the aqueous mixture may have a pH that is pH 9 or less, such as pH 8 or less.
- the aqueous mixture may have a pH that is around pH 7.
- the aqueous mixture may be provided with a buffer to maintain a stable pH during the photocatalytic reaction.
- the buffer may be a phosphate buffer, such as potassium phosphate buffer (KPi buffer), and such are readily available from commercial sources.
- the aqueous mixture may contain additional co-solvents, however such are not typically required in the methods of the present case, which may advantageously avoid the need to use any co-solvent.
- the inventors have found that the photocatalysts of the invention tolerate the presence of many different components within the aqueous mixture, making them suitable for use in a variety of situations, particularly where the water for the aqueous mixture is untreated water.
- the aqueous mixture may contain water that is treated, for example such that the water is suitable for drinking or suitable for use in laboratory.
- the aqueous mixture may alternatively or additionally contain water that is untreated, for example the water may be obtained from a river, lake, canal, or a sea, or from industrial waste water, such as sewage water.
- the photocatalysts of the invention can be used under real-world conditions, without the need for purified drinking water or laboratory quality water for the degradation of an organic substrate and the production of hydrogen.
- the aqueous mixture, where it contains untreated water typically contains one or more organic components, optionally together with inorganic components, which are pollutants within the untreated water. These organic components may be used as sacrificial electron donors in the photocatalytic reaction.
- the photocatalysts of the invention are used together with a co-catalyst, which is a hydrogen evolution catalyst.
- the co-catalyst is water soluble.
- the co-catalyst may be provided as a separe component within the aqueous mixture.
- the photocatalyst and the co-catalyst are not provided together, for example, they are not provided together in a composite.
- the co-catalyst may also not be physically adsorbed onto the photocatalyst.
- the co-catalyst is not a photocatalyst of the invention.
- the co-catalyst is provided as a hydrogen evolution catalyst for the generation of hydrogen (for example, as illustrated in Figure 1 ).
- the photocatalyst is not used directly to generate hydrogen. Rather, irradiation of the photocatalyst results in the formation of a photoexcited state, in which the holes are quenched by the substrate or pollutant, and the photoexcited electrons are effectively transferred from the photocatalyst to the hydrogen evolution catalyst, resulting in hydrogen formation at the hydrogen evolution catalyst (see Kasap et al.).
- co-catalysts together with a photocatalyst for the generation of hydrogen are well known in the art.
- co-catalysts are described for use by Kasap et al., Lau et al. (Angew. Chemie Int. Ed. 2016), and Martindale et al. ( Angew . Chemie Int. Ed. 2017).
- the co-catalyst may be selected from a Ni-, Pt- or Mo-containing hydrogen evolution catalyst.
- the co-catalyst is a Ni catalyst, such as NiP (molecular Ni
- NiP bis(diphosphine), also written as NiP.
- the present inventors have previously disclosed a Ni co-catalyst for use as a hydrogen generation catalyst. See, for example, Kasap et al.
- the co-catalyst is a Pt catalyst, such as HhPtCle.
- the co-catalyst is a Mo catalyst, such as a MoS2-based catalyst, such as H 8 N 2 M0S 4 .
- the present invention provides for the use of the photocatalyst as such, for example in the degradation of an organic substrate.
- the degradation may also be referred to as
- the invention provides a method for degrading an organic substrate, the method comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of the organic substrate.
- the photocatalyst and the organic substrate may be provided in an aqueous mixture, and preferably a substantially neutral aqueous mixture.
- Degradation may refer to a reduction in the molecular weight of the organic substrate due, for example, to the oxidative cleavage of bonds within the organic substrate.
- degradation may be accompanied by the formal loss of hydrogen and/or carbon monoxide from the organic substrate.
- the methods of the invention also provide for the generation of hydrogen and/or a product such as carbon monoxide, the method comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of the organic substrate.
- the photocatalyst and the organic substrate may both be provided in an aqueous mixture.
- the aqueous mixture may have a pH in the range 2 to 15
- the aqueous mixture may be a basic aqueous mixture, such as a mixture having a pH of 10 or more, such as 1 1 or more, such as 12 or more, such as 13 or more, such as 14 or more.
- the aqueous mixture may have a pH in the range 8 to 12, such as 9 to 10.
- the aqueous mixture may be an acidic mixture, such as a mixture having a pH of 6 or less, such as 5 or less.
- the aqueous mixture may have a pH in the range 2 to 6, such as 4 to 5.
- the aqueous mixture may be substantially neutral.
- the source of hydrogen may be the organic substrate, the basic aqueous mixture or both.
- the present inventors have found from deuterium experiments that the predominant source of hydrogen is water.
- the source of carbon is the organic substrate.
- the photocatalyst is active (“excited”) under incident light in the visible and/or ultraviolet range, and preferably the photocatalyst is used under incident light, including incident light having a visible light component, such as solar light.
- incident light refers to light having a wavelength in the range 400 to 700 nm
- ultraviolet light refers to light having a wavelength in the range 100 to 400 nm, such as 315 to 400 nm.
- the incident light may be natural light, such as sunlight. Natural light may include light in both the visible and ultraviolet ranges.
- the incident light may be artificial light, such as provided by a solar simulator, or light from a xenon or mercury lamp, or an LED.
- simulated solar light include AM1 5D, AM1.5G and AMO.
- the incident light may be monochromatic.
- the incident light may be a combination of natural and artificial light.
- the methods of the present invention may be performed under light having an intensity that is similar to that of natural light. Thus, in the methods of the invention it is not necessary to expose the photocatalyst to intense light.
- the intensity of the incident light may be at most 140, at most 150, at most 160 or at most 200 mW/cm 2 .
- the intensity of the incident light may be at least 1 , at least 10, at least 20, at least 50, at least 70, at least 80, at least 90, or at least 100 mW/cm 2 .
- the incident light maybe of an intensity within a range where the upper and lower limits are selected from the values given above.
- the intensity of the incident light may be in the range 1 to 150 mW/cm 2 , such as 10 to 150 mW/cm 2 , such as 50 to 150 mW/cm 2 , such as 90 to 150 mW/cm 2 , such as 130 to 145 mW/cm 2 .
- the light intensity may be around 100 mW/cm 2 .
- artificial light 100 mW/cm 2 is used (AM 1 .5G).
- the methods of the invention may be conducted at ambient temperatures, for example at a temperature in the range 5 to 30°C. Accordingly, it is not necessary to apply heat to the reaction mixture in order to promote the reaction. Photocatalytic reactions previously reported in the art described heating the reaction mixture, and such is not required in the methods of the present invention.
- the methods of the invention may be performed at an elevated temperature, such as a temperature greater than 30°C, and for example at a temperature no more than 60°C, such as no more than 50°C, such as no more than 40°C.
- the organic substrate may be pre-heated prior to its contact with the photocatalyst or prior to irradiation, and preferably prior to its contact with the photocatalyst.
- the mixture may be a heated to a temperature that is no more than 100°C, such as no more than 80°C, such as no more than 60°C, such as no more than 50°C, such as no more than 40°C.
- the methods of the invention do not require the substrate to pre-heated, and the photocatalytic reaction is performed at ambient
- the heating of the mixture may be maintained during and after the addition of the photocatalyst, and throughout the catalytic reaction. Alternatively, the heating of the mixture may be discontinued during or after the addition of the photocatalyst. Indeed, the mixture may be allowed to cool, for example to a temperature in the range 5 to 30°C, prior to the addition of the photocatalyst.
- the methods of the invention may be performed as a batch process, or as a flow process where the organic substrate is permitted to flow across the photocatalyst, which may be immobilised to a support.
- the photocatalyst may be recovered for future further use.
- the photocatalyst may be separated from remaining unreacted organic substrate and any reaction intermediates, such as partially degraded organic substrate. If appropriate, the recovered photocatalyst may be rejuvenated prior to reuse, such as described in further detail below.
- the photocatalyst of the invention may be used to degrade, or reform, an organic substrate, and preferably the degradation reaction yields hydrogen and/or carbon monoxide.
- the methods of the invention may be used to degrade waste material as part of the disposal process of that material.
- the methods of the invention may be intended for the generation of a fuel material, such as hydrogen, for downstream use.
- the methods of the invention may be used to dispose of unwanted biomass and also to generate commercially relevant fuels.
- the organic substrate typically contains both hydrogen and carbon atoms, and most typically contains multiple carbon-carbon and multiple carbon-hydrogen bonds.
- the photocatalyst is most useful in the degradation of higher molecular weight organic substrates, and is also most useful in the degradation of hydroxyl functionalised organic substrates.
- the photocatalyst may also be used to directly degrade biomass without any prior refining of the biomass.
- the photocatalyst is suitable for use with relatively simple organic materials as well as complex biomass mixtures, which contain a wide variety of different chemical structures, often with a wider distribution of molecular weights.
- the photocatalyst has tolerance to a range of substrates, and such tolerance has not previously been reported in the art.
- the ability to use the photocatalyst with unrefined substrates therefore provides flexibility for the process, which may be used at any location where there is a source of biomass materials, such as wood and grass.
- biomass materials such as wood and grass.
- many of the degradation and hydrogen evolution reactions reported to date rely on the use of refined substrates for use in the photocatalytic reaction, and therefore the methods can only be performed where there it the ability to refine biomass materials, or where there is a ready commercial source of the refined material.
- the degradation of the organic substrate may yield hydrogen, which has use as a commercial fuel and a chemical feedstock. Accordingly, the degradation reaction allows for the valorisation of the organic substrate.
- degradation products are also produced, such as carbon monoxide.
- the production of degradation products therefore provides an additional or alternative pathway for creating value from the organic substrate.
- the production of ethanol from cellulose has been described previously as a route to preparing a fuel stock.
- cellulose-derived ethanol requires purification of glucose from lignocellulose, followed by the subsequent fermentation of that glucose to yield ethanol.
- the present invention allows for the production of hydrogen directly from any source of unprocessed lignocellulose.
- the organic substrate may contain one or more, such as two or more, hydroxymethyl groups (-CH2OH).
- cellulose, hemi-cellulose and lignin contain a plurality of hydroxymethyl groups within their core structures.
- the organic substrate may be a polyol, for example a polyol having two or more, such as five or more, such as ten or more hydroxyl groups.
- the photocatalyst of the invention may be used to degrade a high molecular weight component within the organic substrate.
- the photocatalyst is shown to be useful in degrading cellulose, hemi-cellulose, xylan and lignin. Indeed, the photocatalyst may be used to degrade biomass directly, and without any refining.
- the photocatalyst may be used to degrade an organic substrate that is or contains a component having a molecular weight of 70 or more, such as 100 or more, such as 200 or more, such as 500 or more, such as 1 ,000 or more.
- the organic substrate may have a molecular weight of at most 1 ,500.
- the photocatalyst may be used to degrade an organic substrate that is or contains a component having a molecular weight of 2 kDa or more, 5 kDa or more, 10 kDa or more,
- the organic substrate may be a biomass or a refined biomass.
- the organic substrate may be a biomass or a refined biomass.
- refining of the biomass is not required, and a biomass may be used directly (or after mechanical processing).
- refining refers to a process where components of the biomass, such as lignin, cellulose and hemicellulose are separated from each other, of from other components of the biomass.
- Refining may refer to thermal, chemical or biochemical processes for achieving this separation.
- the organic substrate may be or comprise a polysaccharide.
- the organic substrate is cellulose, such as a-cellulose.
- the organic substrate may be or comprise a polypeptide.
- a mixture of different organic substrates, such as those described above, may be present.
- the organic substrate is a complex biomass material, such as paper or wood.
- the organic substrate may contain many different types of biomass in a mixture.
- a mixture of lignin, cellulose and hemicellulose may be present as organic substrates.
- Such materials typically feature within biomasses such as paper and wood or a biomass that is plant matter.
- the organic substrate may be wood, paper, cardboard, bagasse, grass and/or sawdust.
- the organic substrate can be general waste, plastics, packing materials, waste food, aliphatic polyols, algae, sugars, starches, biomass, sewage and/or domestic waste.
- the organic substrate preferably comprises lignocellulose or one or more of its constituent components.
- a biomass for use in a method of the invention may be mechanically treated prior to used, for example to reduce the size of the biomass and optionally to provide the biomass in a uniform size distribution.
- Such mechanical treatments are for optimal processing of the biomass in the methods of the invention, for example to increase the available surface area of the substrate or simply to ensure that the organic substrate can be handled and distributed within a processing facility.
- the photocatalyst of the invention can be used directly to catalyse the reaction of biomass, and each of the major components of lignocellulose is reactive in the presence of the photocatalyst when it is exposed to light. Accordingly, the methods of the invention do not require the biomass to be chemically treated prior to use. Many methods contained in the prior art have described the preparation of monosaccharides from cellulose components as a preliminary step, prior to the use of the photocatalyst.
- the photocatalysts of the invention have broad application in degrading, or reforming, organic substrates that are organic polymers, and preferably the degradation reaction yields hydrogen.
- the photocatalyst is also found to have use in the degradation of other organic polymers, and may be used to generate hydrogen from those organic polymers.
- PET, PUR and PS may be used as feedstocks to generate hydrogen, as shown in the worked examples of the present case.
- organic polymers described here may be used in the same way as the organic substrates described above.
- An organic polymer may refer to a polymer that contains multiple monomer units, and typically each monomer unit contains at least one carbon-carbon bond in the backbone chain of the polymer. Where a monomer does not contain a carbon-carbon bond in the backbone chain of the polymer, it may contain a carbon-oxygen in the backbone, for example as seen with paraformaldehyde polymers. An organic polymer may contain both carbon-carbon and carbon-oxygen bonds within the backbone.
- the monomer units may be covalently linked by carbon-carbon bonds, such as in a substituted polyethylene, but this is not necessary, and monomers may be linked by carbon- oxygen or carbon-nitrogen bonds.
- carbon-oxygen bonds are present in polyethers and polyethylene terephthalates.
- Carbon-oxygen and carbon-nitrogen bonds are present in polyurethanes.
- the organic polymer may be a homopolymer or a copolymer.
- each monomer (repeat) unit within the organic polymer contains one or more heteroatoms, such as one or more heteroatoms selected from oxygen, nitrogen and sulfur.
- a heteroatom may be present within the covalent bond connecting monomers, and/or a heteroatom may be present within a side group of the monomer.
- an organic substrate such as an organic polymer may be at least partially degraded or solubilised prior to its treatment with the photocatalyst. This pre-treatment of the organic substrate may be undertaken to improve the solubility of the substrate in the aqueous mixture.
- the organic polymer may therefore comprise functionality, either in the backbone chain or the side chain, which is hydrolysable, for example under basic or acidic conditions.
- the organic polymer may contain ester, amide, ether, carbonate, and/or urethane (carbamate) functionality.
- the organic polymer may be a polyester, such as polyethylene terephthalate (PET) or polylactic acid (PLA), a substituted polyethylene, such as polyvinyl pyrrolidone (PVP) and optionally also polyethylene, a polyether, such as polyethylene glycol (PEG), a
- PET polyethylene terephthalate
- PLA polylactic acid
- PVP polyvinyl pyrrolidone
- PEG polyethylene glycol
- PC polycarbonate
- PUR polyurethane
- the organic polymer may be a polyamide, such as a protein.
- the organic polymer may be a polyester, a substituted polyethylene, or a polyether.
- the photocatalyst may be used together with an organic polymer that is a polyester.
- the polyester may be an aryl-containing polyester (aromatic polyester) or an alkyl-containing polyester (aliphatic polyester).
- the polyester may contain both aryl and alkyl functionality.
- Examples of aryl-containing polyesters include polyethylene terephthalate, polyethylene naphthalate, polybutylene terephthalate and polytrimethylene terephthalate.
- Examples of alkyl-containing polyesters include the homopolymers polylactic acid, polycaprolactone (PCL), polyglycolic acid, poly-3-hydroxyvalerate, poly-3-hydroxybutyrate, poly-4-hydroxybutyrate.
- Examples of alkyl-containing polyesters include the copolymer polybutylene succinate and poly(3-hydroxybutyrate-co-3-hydroxyvalerate).
- the photocatalyst may be used together with an organic polymer that is a substituted polyethylene.
- the polyethylene has a side chain.
- the substituted polyethylene has a side chain.
- polyethylene is a polymer having one or more heteroatoms, such as selected from oxygen, sulfur or nitrogen, within the side chain.
- the heteroatom functionally may be a component of a functional group that is hydrolysable under the reaction conditions, or it may assist in the dissolution of the polymer in the reaction mixture.
- the polyethylene may be substituted with, for example, a heterocycle, such as a C4-7 heterocycle having one ring heteroatom.
- a heterocycle such as a C4-7 heterocycle having one ring heteroatom.
- the heterocycle may be pyrrolidone.
- the polyethylene may be substituted with, for example, a cyano group.
- the polymer may be a polyacrylonitrile (PAN).
- the polyethylene may be an acrylate polymer, for example a poly(methyl acrylate).
- the photocatalyst may be used together with an organic polymer that is a polyether, including for example polyethylene glycol.
- a polyether may be an alkyl-containing polyether (aliphatic polyether) or an aryl-containing polyether (aromatic polyether).
- the polyether may contain both aryl and alkyl functionality.
- alkyl-containing polyethers examples include polyethylene glycol, polypropylene glycol, paraformaldehyde (PFA) and poly(tetramethylene ether) glycol (also known as
- aryl-containing polyethers examples include polyphenyl ether and poly(p-phenylene ether) (PPE).
- lignin is a complex polyether containing both alkyl and aryl functionality, together with further substituent groups.
- lignin is a polymer crosslinked phenolic units.
- a polyether is not lignin.
- the organic polymer may be a component of a consumer product.
- polyethylene terephthalate is found in fibres for clothing and containers for foods and liquids.
- a polysaccharide may not be considered a polyether.
- polyamide is a polypeptide.
- Example polypeptides include bovine serum albumin and casein.
- the organic polymer may have a molecular weight that 500 or more, such as 1 ,000 or more.
- the photocatalyst may be used to degrade an organic substrate that is or contains a component having a molecular weight of 2 kDa or more, 5 kDa or more, 10 kDa or more, 20 kDa or more, 25 kDa or more, 30 kDa or more, or 50 kDa or more.
- the organic substrate such as the organic polymer
- the organic substrate may be pre-treated prior to its mixing with the photocatalyst.
- the organic substrate may be pre-heated to assist the hydrolysis or dissolution of the organic polymer.
- the organic polymer may be pre-treated with base to assist the hydrolysis or dissolution of the organic polymer.
- the organic polymer may also be mechanically treated, for example by cutting, grinding and milling and other such techniques.
- the mechanical treatment may be particularly
- the organic polymer is a component of a consumer product, and a size reduction in the product will assist in the rate of degrading the product and its organic polymer component.
- the photocatalysts of the invention may be used in untreated water to generate hydrogen.
- the method of generating hydrogen may use the photocatalyst together with a co-catalyst and a sacrificial electron donor, which donor is typically a component, such as an organic or inorganic pollutant, within the untreated water.
- the hydrogen generation method has the advantage of providing useful fuel, in the form of hydrogen, whilst also degrading pollutants within the untreated water, thereby to at least partially purify the untreated water.
- the sacrificial electron donor is a compound, which may be an organic compound or an ionic species, within the water that is capable of quenching holes, by the donation of electrons, that are photochemically generated in the photocatalyst.
- sacrificial electron donor compounds that are found in river water include one or more compounds selected from atrazine, desmetryn, dicamba, chloromehtylphenoxyacetic acid, mecoprop, prometryn, propazine, simazine, terbutryn, and trietazine.
- sacrificial electron donor compounds that are found in sea water include one or more compounds selected from chloromethylphenol, pentachlorophenol, benzaldehyde, trichlorophenol, nitroanaline, dichlorphenol, dimethyl phenol, atrazine, dinitrophenol and naphthylamine.
- a sacrificial electron donor compound may be selected from the group consisting of from atrazine, prometryn, terbutryn and benzaldehyde.
- the untreated water may contain chloride, as an example of an ionic sacrificial electron donor.
- Untreated water may contain one or more components identified in sea water and river water as noted by the US Department of Interior (Torres et al.) and the British environmental agency (Huber et a!.).
- the untreated water may be supplemented by the addition of material that is capable of acting as a sacrificial electron donor.
- material that is capable of acting as a sacrificial electron donor for example, an organic substrate, such as those substrates described above, including biomass, may be added to the untreated water.
- the untreated water and the biomass are taken from the same locality, for example within 5 km from each other, such as 1 km from each other.
- the methods for generating hydrogen and optionally also purifying the untreated water do not require large movement of materials, and hydrogen may be generated close to the source of the water and the biomass. This provides a great efficiency to these methods.
- the photocatalyst for use here is a carbon dot photocatalyst.
- Such a photocatalyst may itself be generated from an organic substrate, such as a biomass.
- This same organic substrate may be used as a sacrificial electron donor in the methods for generating hydrogen from untreated water.
- many of the components for use in the method may be sourced locally, and typically at minimum cost also, and may be prepared from local components. This provides a further efficiency to the system.
- the present case provides methods for preparing a photocatalyst, including methods for preparing a carbon nitride photocatalyst and a carbon dot photocatalyst.
- biomass waste may be used as a starting material for preparing carbon dots having photocatalytic activity.
- carbon dots may be prepared from cotton, including cotton products, G Nivalis, G. Elliptica, T. Baccata, Elaeagnus X ebbingei and olive leaves.
- biomass components isolated, such as at least partially purified, from plants may also be used as staring materials for preparing carbon dots having photocatalytic activity.
- carbon dots may be prepared from ocellulose, xylan and lignin.
- the invention provides a method of preparing a carbon dot, the method comprising the step of calcining an organic substrate.
- the organic substrate may be or comprise a monosaccharide, an oligosaccharide or a polysaccharide, such as cellulose, and preferably the organic substrate may be or comprise an oligosaccharide or a polysaccharide, such as a polysaccharide.
- the organic substrate may be lignin, cellulose or hemicellulose.
- the organic substrate may be wood, leaves, paper, cardboard, bagasse, cotton, grass and/or sawdust.
- the organic substrate can be general waste, packing materials, waste food, aliphatic polyols, algae, sugars, starches, and/or biomass.
- the organic substrate preferably comprises lignocellulose or one or more of its constituent components.
- the calcining step is a heating step and the temperature and duration of the heating step may be selected for optimal production of the carbon dot.
- the minimum heating temperature may be at least 200°C, at least 250°C, at least 300°C or at least 350°C.
- the maximum heating temperature may be at most 400°C, at most 450°C or at most 500°C.
- the heating temperature may be in a range selected from the minimum and maximum temperature given above.
- the heating temperature may be in the range 250 to 450°C. If necessary the temperature can be varied within this range, for example with an initial lower heating temperature followed by a subsequent higher heating temperature.
- the heating may be performed for a total of at least 12 hours, at least 24 hours, at least 2 days, at least 3 days or at least 4 days.
- the heating may be performed for a total at most 5 days, at most 6 days, at most 7 days or at most 1 week.
- the worked examples in the present case demonstrate that the products formed from the calcination reactions have excellent activity as photocatalysts, and these photocatalysts may be used to generate hydrogen from a range of organic substrates provided in purified and untreated water, including ocellulose, EDTA (as an exemplary sacrificial electron donor), and pollutants in sea water and river water, such as atrazine, terbutyrn, benzaldehyde and prometryn.
- Other species, such as chloride, which is ionic and inorganic, may also act as sacrificial electron donors.
- NCN CN X may be used as a photocatalyst in the degradation of organic substrates, as described herein.
- the inventors have also found that the activity of the carbon nitride photocatalyst may be enhanced when the surface area of a catalyst sample is increased.
- NCN CN x from H2N CN X as described to date, provides a bulk sample of material where product particles are provided as aggregates (as shown in the TEM images of Figure 2(f) in the present case).
- the inventors have found that by disrupting the aggregates, such as by sonication, including ultrasonication, to reduce the aggregate size, increases the surface area of a sample, as judged by BET analysis. This increase in surface area is shown to increase the photocatalytic activity of the material.
- the invention therefore provides a method of preparing NCN CN x , the method comprising the step of forming a bulk sample of NCN CN X and subsequently treating the bulk NCN CN X thereby to increase the surface area of the NCN CN x .
- the bulk NCN CN X may be sonicated to increase its surface area.
- the bulk NCN CN X may be obtained or obtainable from H2N CN X .
- the pure biomass components a-cellulose, xylan and lignin, as well as natural sea water (from Gulf of Mexico) and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA), were purchased from Sigma-Aldrich and used without any further purification.
- Cotton wool was purchased from Stores, Department of Chemistry, University of Cambridge, UK, whereas cotton pads from Waitrose, UK and cotton T-shirt from an open market in
- Buffer solutions were prepared using analytical grade reagents and titrated to the desired pH with a pH meter (Mettler Toledo; SevenEasy).
- the amount of H 2 accumulation was quantified via periodic headspace gas injections (20 pl_) to gas chromatography (GC), Agilent 7890A equipped with a 5 A molecular sieve column.
- GC gas chromatography
- Agilent 7890A equipped with a 5 A molecular sieve column.
- the temperature of the GC oven was maintained at 45°C and N 2 was used as the carrier gas, with a flow rate of 3 mL min 1 .
- H 2 N CN X was prepared by heating melamine at 550°C for 4 h under air. The yellow solid obtained was then thoroughly ground using a pestle and mortar prior the surface
- NCN CIM x was prepared from grounded H 2 N CN X upon reaction with KSCN (mass ratio 1 :2) at 400°C for 1 h, and then at 500°C for 30 min under Ar. After cooling to room temperature, the resulting solid was excessively washed with water to remove the residual KSCN and then dried in vacuo to yield NCN CN x .
- the material was suspended in solvent (KPi, water, iPrOH and MeOH; at 1 or 5 mg/ml_), followed by ultrasonication for 1 , 10, or 30 min, using a Sonic Dismembrator (Model 120, Fischer Scientific).
- NiP was synthesised following a reported procedure: see Gross et al.
- NCN CN X was characterised before and after ultrasonication by UV-visible (UV-vis) absorption and photoluminescence (PL) spectroscopy, Brunauer-Emmett-Teller (BET) surface area analysis, X-ray diffraction (XRD), Scanning (SEM) and Transmission Electron Microscopy (TEM), Attenuated Total Reflectance - Fourier Transform Infrared (ATR-FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS), to reveal the effect of the ultrasonication on the structural and optical properties of the bulk material, and thus its photocatalytic performance.
- UV-Vis absorption spectra were collected by a 50 Varian Cary UV-vis spectrophotometer in the wavelength range between 200 and 800 nm (in water) using quartz cuvettes with 1 cm pathlength. Photoluminescence emission spectra of bulk and
- ultrasonicated NCN CNX were recorded on a FS5 spectrofluorometer (Edinburgh Instruments) equipped with an integrating sphere.
- Brunauer-Emmett-Teller (BET) analysis was carried out with a 3Flex Surface Characterisation Analyzer (Micromeritics) at -196°C, using N 2 as the absorbing gas. Prior to measurements, the samples were outgassed under vacuum for 6 h at 150°C. The specific surface areas were calculated by fitting the data to the Brunauer-Emmett-Teller (BET) isotherm.
- XRD X-ray diffraction
- PANalytical BV X’Pert PRO diffractometer
- SEM and TEM, respectively Scanning and Transmission Electron Microscopy (SEM and TEM, respectively) images, were recorded with MIRA3 FEG-SEM (TESCAN) and Tecnai
- ATR-FTIR Attenuated Total Reflectance - Fourier Transform Infrared
- n H2 is the moles of photogenerated H 2
- h the Planck constant
- c the speed of light
- t irr the time of irradiation
- A the cross-sectional area of irradiation (0.28 cm 2 ).
- BET Brunauer-Emmett-Teller
- the increase in surface area is due to the presence of smaller aggregate sizes in the sample after ultrasonication, compared to the bulk NCN CN x , as indicated by TEM images (see Figure 2(f)), which also show negligible changes in particle size and morphology (only a small fraction of rod-like particles was observed), in accordance with SEM images ( Figure 2(g)).
- the external quantum efficiency (EQE) of the NCN CN x -NiP photosystems was determined, and the EQE of the ultrasonicated and bulk systems are respectively 22.2% ⁇ 1.1 % and 13.2% ⁇ 1.2%.
- the former is approximately 68% higher than latter, which is highly consistent with the increase of BET surface area.
- N CN CN X -NIP systems are much higher than the previously reported H2N CN x -NiP and RuP-NiP systems (Caputo et al., Gross et a!.).
- the material preserves its surface functionalities (Figure 2(e)) after ultrasonication. More specifically, both the bulk and ultrasonicated NCN CN X samples show similar features in their FTIR spectra; a broad peak at about 3300 cm -1 assigned to N-H stretching, a pair of sharp peaks at 2175 cm 1 and 2144 cm 1 which signify the preservation of the CoN groups after ultrasonication, the bands at 1639 cm 1 and 1557 cm 1 which are attributed to secondary amine -C-N bending, whereas the signal at 807 cm 1 is due to the heptazine core vibration (Kasap et al., Lau et al. Nat. Commun. 2016).
- Photocatalysis was carried out in a borosilicate glass photoreactor (total volume 7.74 ml.) in a water bath thermostated at 25°C.
- the suspensions were prepared by mixing for 10 min NCN CN X (such as the ultasonicated form) or CDs and NiP (or hhPtCh and H8N2M0S4 as precursors for Pt and M0S2, respectively) in aqueous 0.1 M KPi solution (at pH 4.5, 3 ml.) containing an electron donor (4-MBA or 100 mg lignocellulosic substrates) in a photoreactor equipped with a stirrer bar (total solvent volume 3 ml_).
- the container was then tightly sealed with a rubber septum and purged with N2 containing 2% CH 4 as the internal gas chromatography standard.
- the photoreactor was stirred and kept at a constant temperature (25°C) with continuous water circulation through a water-jacketed reservoir during the light experiments.
- the solar light simulator was equipped with an air mass 1.5 global filter (AM 1 5G) and water filter to remove the infrared irradiation.
- the photocatalytic activity of the photoreforming system to produce H2 was optimised with respect to the activity of NCN CNX using 4-MBA as an electron donor, NiP as H2 evolution catalyst and irradiation by simulated solar light (AM1.5G, 100 mW cm 2 ) at room
- the parameters are duration and temperature of
- the photocatalytic reaction conditions were further optimised systematically for specific activity with respect to carbon nitride, and high TOFNIP and high TONNIP with respect to NiP, as well as the yield of H 2 production and alcohol conversion to oxidation product (determined after 6 h of irradiation).
- NiP catalyst was varied at various NCN CN X loadings, 0.5 mg and 5 mg, with 4-MBA (30 pmol), and the hydrogen evolution was monitored for the first 6 hours.
- Table 6 the total H 2 evolution and percentage of conversion of 4-MBA to the oxidation product 4-MBAd raised significantly as the amount of NiP catalyst increased from 10 nmol to 100 nmol, where >99% selective alcohol conversion was also observed.
- the production of H 2 and 4-MBAd starts to be limited by the availability of 4-MBA electron donors at NiP loadings above 100 nmol, the first-hour rate of H 2 production kept climbing.
- the TOFNIP is higher than many previously reported NiP systems with carbon-based photosensitiser, and it is comparable to the reported N-doped carbon dot system (Hutton et al. Chem. Soc. Rev. 2017). Both TOFNIP and TONNIP are in good agreement with previously reported NCN CN x -NiP and the RuP-NiP systems (Kasap et a! , Gross et a!.).
- Table 6 shows the changes in hydrogen productions with changes in the relative and absolute amounts of the NCN CN X photocatalyst and the NiP co-catalyst.
- NCN CN X can still generate excited electron-hole pairs and drive the 4-MBA oxidation, even in absence of any catalyst.
- the kinetics of electron transfer processes in the NCN CN x -NiP photosystem were extensively studied for the bulk material using transient absorption spectroscopy (TAS), and the electron transfer to NiP was identified as the rate-limiting step (Kasap et ai).
- NCN CN X The bulk NCN CN X was exploited for time-delayed‘dark’ photocatalysis (Kasap et air, Lau et al. Angew. Chemie Int. Ed. 2017).
- the colour of NCN CN X changes from yellow to blue upon irradiation in presence of a suitable electron donor such as 4-MBA. This blue state has a lifetime of up to hours and is representative of trapping of long-lived electrons in NCN CN X .
- H 2 yield increases from 2.0 pmol to 2.5 pmol as the irradiation length increases from 2 hours to 4 hours; however, no further rise in H 2 yield was observed when irradiation increased to 18 hours, suggesting saturation is reached.
- Biomass conversion is both an affordable and sustainable approach to produce hydrogen.
- Described below is the use of carbon nitride photocatalyst for hydrogen evolution from lignocellulosic biomass under mild conditions with no pre-treatment.
- the carbon nitride photocatalyst was used together with a co-catalyst for photocatalytic reforming of biomass over a wide spectrum of pH values.
- the loadings of biomass, carbon nitride and NiP were optimised with respect to hydrogen production.
- Cellulose is the main component of wood-derived biomass (Sun et ai), so it was used in all initial optimisations.
- the ultrasonicated NCN CN x -NiP photosystem (3 ml_, 0.1 M KPi, pH 4.5) was used based on the merit of its high specific activity.
- NiP shows the best activity amongst all for first 24 h, demonstrating the first example of molecular catalyst-based biomass reformation system. After 24 hours of irradiation, the activity of NiP terminated due to UV-induced degradation caused by ligand displacement and ligand oxidation caused by photogenerated holes (Martindale et al. Angew. Chemie 2016). NCN CN X can be easily platinised by in situ photodeposition of H2PtCl6 (Lau et al. Nat. Commun. 2016), which accounts for the relatively slow H2-evolving rate of the first day. Similarly, M0S2 can be generated by in situ
- Lignocellulose is the most abundant constituent of biomass (Azadi et al.), and it is composed of cellulose, hemicellulose and lignin.
- Cellulose is in the form of microfibrils, which are coated with hemicellulose to form an open network whose empty spaces are filled up with lignin (Rubin et al.).
- Cellulose, the major component is a polymeric chain of glucose units mainly linked by b (1 4) glycosidic bonds (van Wyk et al.).
- Cellobiose and glucose are respectively the disaccharide and monosaccharide units of cellulose.
- Hemicellulose is a branched polysaccharide of various pentose and hexose sugars.
- xylan a type of hemicellulose that accounts for up to a third of carbohydrate content in many lignocellulosic materials (Hinman et al.), and consist of xylose units
- Lignocellulose components ocellulose, xylan and lignin
- Lignocellulose components as well as their subunits were used as electron donors for photocatalytic H 2 evolution, and the data are presented in Table 1 1.
- the photocatalytic reforming yields of the subunits are much higher than those of ocellulose, hemicellulose and lignin.
- NiP was chosen instead of NiP because the photocatalytic performance of NiP is largely impaired at extreme pH (Caputo et ai).
- using the benchmark Pt H 2 -evoluting catalyst can provide a direct comparison between this NCN CN X -Pt photosystem and many previously reported systems (Kuehnel et ai).
- ocellulose was suspended in different solvents and stirred for 24 h under 1 sun irradiation (AM 1 5G) at 25°C before the addition of H 2 PtCl 6 and NCN CN X .
- Table 14 illustrates the photocatalytic H 2 evolution performance of the NCN CN X -Pt
- Photoreactors were prepared using 10 min ultra-sonicated NCN CN X (5 mg) with H2PtCl2 (4 wt %) and cellulose (1.65 or 0.81 mg) in 10 M KOH. The samples were pre-treated in 10 M KOH prior to injection of NCN CN X and H2PtCl2. Percentage conversion yields were calculated after irradiating (AM 1.5G, 100 mW cm 2 , 25°C) the samples for 6 days.
- this NCN CN X -Pt photosystem can operate over a wide range of pH with reasonable yield. This can be attributed to the stability of NCN CN X as well as facilitated electron transfer to Pt (Kasap et air, Lau et al. Nat. Commun.
- NCN CN X (5 mg) was used together with NiP (50 nmol) to produce hydrogen from a range of polymer substrates (100 mg) in KPi solution (0.1 M, pH 4.5, 3 ml_).
- the substrates included PET (polyethylene terephthalate), PUR (polyurethane) and PS (polystyrene). The reaction was irradiated under 1 sun for 24 hours (100 mW cm -2 , AM1.5G, 25°C).
- CDs were prepared as previously described by Martindale et at. Angew. Chemie Int. Ed. 2017, or as described below, from biomass organic precursors. Pyrolysis of organic precursors under air to form CDs was optimized for high photocatalytic activity by varying the calcination temperatures and times (data for thermogravimetric analysis, TGA, and photocatalytic screening not shown). All organic precursors were calcinated in muffle Carbolite furnaces for 4 days under air, at optimum temperatures, to obtain CDs with good photocatalytic properties.
- ocellulose and cotton wool were treated thermally at 320°C, xylan at 250°C, lignin at 300°C, cotton pads and T-shirt at 340°C, G. Nivalis, G. Elliptica and T. Baccata at 230°C, Elaeagnus X ebbingei at 275°C and olive leaves at 250°C.
- the samples after carbonization were used in their crude form for all types of characterization, without any further treatment.
- Cotton contains mainly cellulose ( ⁇ 93-97%) with the remaining part consisting mostly of hemicellulose ( ⁇ 7%) (Chen; Zhu et al.). It was therefore an excellent carbon source for this purpose, and cotton-derived CDs were prepared from four-day pyrolysis of commercial cotton wool, pads and recyclable cotton fabric (T-shirt) at 320, 340 and 340°C, respectively.
- CDs from cotton resources have been prepared previously for ion detection, imaging, patterning and sensing applications (Wen et ai. ⁇ Wang et al. and Alves et al.), either by hydrothermal or strong-acid mediated approaches.
- the synthesis of such materials with great potential in photocatalysis, via facile and simple calcination approaches, is unprecedented.
- inedible plant waste was also used as precursors.
- the species selected are in great abundance in various climates, have no special growth requirements, and can be obtained at no cost when the plants complete their life-cycle and/or from tree trimming. More specifically, pruning of the olive trees worldwide, results in 25 kg of waste/tree annually (Herrero et al., Peralbo-Molina et al., Abaza et a!.).
- the optimal conditions for CD synthesis from crude biomass depends on the relative content of the samples in ocellulose, xylan and lignin as well as their morphology (George et al., Zhang et al.).
- the TGA traces of the plants show three main bands: xylan degrades first at 210-300°C, followed by ocellulose (300-350°C), and lignin as a shoulder at higher temperatures (380-430°C) (Carrier et al., Biagini et al., Barneto et al.). Olive leaves show additional bands between 460 and 490°C, possibly due to the presence of other polyphenols in the sample (Abaza et al.).
- CDs were characterized by X-ray photoelectron spectroscopy (XPS) upon drop casting the CD aqueous solutions on clean fluorine-doped indium tin oxide (FTO)-coated glass slides, at RT, under atmospheric pressure. The spectra were collected at NEXUS
- ATR-FTIR Fourier Transform Infrared
- X-ray diffraction (XRD) patterns for all materials were recorded with a PANalytical BV X’Pert PRO, at a scan rate of 0.9 min -1 .
- TEM Transmission electron microscopy
- Thermogravimetric analysis (TGA) measurements were performed with a Mettler Toledo TGA analyzer, under a nitrogen atmosphere, at a scan rate of 10°C/min and in the temperature range from 50 to 800°C.
- Xylan and lignin-derived carbonaceous materials show a significantly higher surface coverage by alkoxy/epoxy groups (532.6-532.7 eV, 63-65%) compared to carboxylates (531.3 eV, 29-37%). (Qin et al., Yang et al., Qu et al.).
- N-containing CDs with a pyrrolic (N-5, 399.3 eV) and graphitic quaternary N (N-Q, 400.2 eV) content Zhang et al., Li et al.) ( Figure 3(a), bottom).
- the carboxylate content is 67, 64, 61 and 46% for G. Elliptica, G. Nivalis, T. Baccata and for Elaeagnus X. ebbingei, respectively.
- TEM images show that CDs synthesized from ocellulose and G. Nivalis are highly graphitic with an average diameter of 9.4 and 7 nm, respectively ( Figure 3(b)).
- the lattice fringes allow for the assignments of the (100) intralayer spacings (3.0 A for ocellulose and 2.7 A for G. Nivalis CDs), which is larger than for graphite (2.4 A) (Martindale et al. Angew. Chem. Int. Ed. 2017; Qu et a! , Qu et al.).
- CDs derived from G. Elliptica and lignin are relatively large colloids with a diameter of 96 and 115 nm, respectively (data not shown).
- Raman spectroscopy supports highly graphitic nanostructures for ocellulose
- G A//Va//s-derived CDs. Specifically, the D (1579.3 cm -1 , defective sp 2 carbon) to G
- Elaeagnus X. ebbingei displays ID/I G ratios of 0.35 and 0.56, respectively, indicating more defective graphitic structures.
- the Raman results are in good agreement with XPS analysis, which shows sp 3 /sp 2 C ratios of 0.3 and 0.5 for ocellulose and G.
- XRD patterns of CDs indicate variations in the (002) interlayer spacing of the graphitic structures.
- oCellulose CDs show an interlayer spacing of 3.8 A (23° 2Q), which deviates from that of pure graphite (3.3 A), and suggest material with turbostratic disorder (data not shown) (Bourlinos et al.] Zhang et al.] Mewada et al.).
- the samples for photocatalytic experiments were prepared in borosilicate glass vials (7.74 ml_), by first dissolving 0.03-2.8 mg of the as calcinated ocellulose CDs and 50 nmol of the hydrogen evolution catalyst (NiP), in aqueous EDTA solutions (0.1 M, 3 ml_, pH 6). Next, the vessels were sealed with rubber septa (Subaseal), purged with N 2 containing 2 % CH 4 (internal gas chromatography (GC) standard) for about 20 mins, and irradiated using a Newport Oriel Solar Light Simulator (100 mW cm -2 ) equipped with an Air Mass (AM) 1.5G filter. The samples were kept at 25°C and stirred constantly during irradiation.
- GC internal gas chromatography
- Carbon nanodots were also prepared from biomass organic precursors, and the use of these CDs is also described below.
- Undoped and amorphous (undoped, a-CDs) and nitrogen-doped and graphitic (N-doped, g-CDs) carbon nanodots in combination with NiP as the H 2 evolution catalyst are active for photoreforming ocellulose into H 2 under benign conditions (0.1 M KPi, pH 6, 3 ml.) (see Table 17).
- the CDs used in this work which include an a-CD (amorphous CD) and a nitrogen-doped CD (g-N-CD; graphitic nitrogen-doped CD), were prepared as described Martindale et al. Angew. Chemie Int. Ed. 2017. The experimental methods for the preparation of these CD photocatalysts are described on pages S1 to S5 of the Supporting Text for this reference.
- CDs were prepared from ocellulose (ocell CDs) and these were also used as photocatalysts to degrade ocellulose and glucose.
- CDs prepared from purified and waste biomass were used as photoabsorbers to drive H 2 evolution.
- Photocatalytic experiments were carried out for 24 hrs, using NiP (50 nmol) as the H 2 evolution catalysts and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA 0.1 M, pH 6) as the electron donor. All experiments were carried out at as described above.
- the quantity of CDs was systematically varied between 0.03 and 2.8 mg ( Figure 6) during photocatalytic experiments and optimized for maximum H 2 yield (in /vmol), from which the specific activities (/vmol H 2 (gcDs) -1 h -1 ) and NiP turnovers (TON NR, mol H 2 (mol NiP) -1 ) were determined.
- a-Cellulose CDs (2.2 mg), after 24 hrs of irradiation, showed the best H 2 yield (15.6 ⁇
- NCN CNx is a reference to a bulk sample that has not been ultrasonically treated.
- CDs produced from ocellulose have been reported for use in bioimaging (Shen et ai), this is the first example of CDs from a cellulosic precursor being employed in
- CDs from xylan and lignin are limited and have been previously used only in drug-delivery and bioimaging applications, and thus this is the first demonstration of their use in energy production (Rai ef a/.; Chen ef a/.; Liang et ai).
- a-Cellulose is a linear and crystalline polymer (100,000 g mol -1 ) made of glucopyranose units
- xylan is an amorphous and branched polysaccharide (30,000 g mol -1 ), made of sugars such as glucose, mannose, xylose and arabinose.
- Lignin is also amorphous and consists of randomly cross-linked phenolic monomers (p-coumaryl, coniferyl, and sinapyl alcohols) (Lee et ai).
- Our photocatalytic experiments suggest that pyrolysis of a crystalline homopolymer with regular microstructure might support the formation of high-performance CD photocatalysts.
- Cotton is predominantly made from ocellulose with no lignin content (Krasig et ai), and thus cotton wool-derived CDs showed indeed almost identical photocatalytic performance as ocellulose CDs: 11.8 ⁇ 0.29 /vmol H 2 are photo-produced after 6 hrs of irradiation
- Untreated sea water (Gulf of Mexico) contains a cocktail of organic compounds (Nowell et al.), among which benzaldehyde and atrazine can potentially act as electron donors ( Figure 9(b)) (Nowell et al.). Chloride anions that are abundantly present in seawater can also serve the same role (Iguchi et al.).
- untreated river water (River Cam) is rich in organic herbicides/pesticides (River Cam), among which prometryn and terbutryn can quench the photogenerated holes (Figure 9(b)).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Combustion & Propulsion (AREA)
- Inorganic Chemistry (AREA)
- Catalysts (AREA)
Abstract
The invention provides methods for producing hydrogen using a carbon nitride (CN x ) photocatalyst or a carbon dot (CD) photocatalyst. The method may include exposing a photocatalyst to visible and/or ultraviolet light in the presence of an organic substrate, such as a biomass or an organic component having a molecular weight of 200 or more, and a co-catalyst that is a hydrogen evolution catalyst. The method may include exposing a photocatalyst within untreated water to visible and/or ultraviolet light in the presence of a co-catalyst that is a hydrogen evolution catalyst.
Description
PHOTOCATALYST AND PHOTOCATALYTIC METHODS FOR
PRODUCING HYDROGEN
Abstract
The present case claims the benefit and priority of GB 1808905.2 filed on 31 May 2018, the contents of which are hereby incorporated by reference in their entirety.
Field of the Invention
The present invention provides photocatalysts and the use of those photocatalysts in photocatalytic methods for degrading an organic substrate, such as a biomass, or a species, such as pollutant, in untreated water. Also provided is the use of the photocatalysts in photocatalytic methods for generating hydrogen, and methods for preparing those photocatalysts.
Background
Solar-driven hydrogen production with semiconductor particles is a sustainable strategy to generate clean and renewable energy carriers. Research in this area typically focuses on coupling hydrogen production to the oxidation of water into oxygen, which is the kinetically demanding half-reaction in the water splitting process. In addition, overall water splitting generates a hydrogen and oxygen gas mixture in a single-pot photoreactor, which is a potentially explosive combination, and requires costly separation of the components.
An emerging strategy is to replace the water oxidation reaction by the direct reformation of unprocessed biomass such as lignocellulosic waste, which does not compete with food production (see Kuehnel et al.). Here, the biomass is oxidized instead of water and enables sustainable hydrogen production driven by solar light in the presence of a photocatalyst.
Such a process has the potential to provide large scale, affordable and clean hydrogen generation. Photocatalytic reforming of biomass is thermodynamically more favourable (thus potentially enabling higher hydrogen generation rates than classical water splitting), and prevents the formation of potentially unwanted oxygen. Lignocellulosic biomass is the most abundant form of biomass and its photoreformation has been previously achieved using CdS/CdOx and Pt/TiC>2 photocatalysts. However, these photocatalysts are problematic as they can be toxic (for example, owing to the presence of Cd), expensive (for example, owing to the use of Pt), inefficient and/or require harsh experimental conditions (e.g., strongly alkaline solutions). Furthermore, the reported photocatalysts suffer from a combination of limited visible-light absorption, low surface area and poor interfacial interactions, as well as photoinstability, which limit their potential for real-world applications.
Most currently studied solar-driven catalytic systems, do not only rely on non-scalable photocatalysts, but also only operate in organic solvents, purified water and the presence of unsustainable sacrificial reagents (see Martindale et al. J. Am. Chem. Soc. 2015; Hutton et al. J. Am. Chem. Soc. 2016; Hutton et al. Chem. Soc. Rev. 2017; Martindale et at. Angew. Chemie tnt. Ed. 2017, 56, 6459). This bottleneck is critical for the future development as it prevents the implementation of an economic process
Photoreformation of lignocellulose with low cost and non-toxic photocatalysts in a benign environment has not yet been reported and is at the focus of this invention disclosure.
Some of the present inventors have previously described the use of photocatalysts in the degradation of organic substrates, including biomass and organic polymers.
For example Wakerley et al. describe the use of a CdS quantum dot-based photocatalyst that has activity to generate hydrogen from biomass under incident simulated solar-light (see also PCT/EP2017/080371 ).
The photocatalyst has an oxygenated surface, and this is generated by treatment of the CdS semiconductor material under highly alkaline conditions, such as pH 10 or more, and preferably pH 14 or more. The exemplified photocatalyst also makes use of Cd, and there is a desire to avoid the use of this toxic species, where possible.
There is a need for alternative photocatalysts that work under environmentally benign conditions, at ambient temperature, and do not require the use of toxic metal species.
Summary of the Invention
The present inventors have identified a particular class of catalysts that have useful properties when exposed to visible and/or UV light, such as sunlight or simulated visible light, in combination with a co-catalyst. These photocatalysts may be reliably and efficiently used in photocatalytic methods for degrading organic substrates, such as biomass, and photocatalytic methods for producing hydrogen. The catalysts are carbonaceous, and therefore of low cost and low toxicity, and are additionally well suited to large scale applications.
The catalyst is therefore a photocatalyst. The photocatalyst of the invention is a carbon- containing catalyst, where the carbon may be present in a graphitic and/or amorphous phase. For example, the photocatalyst may be a carbon nitride (CNX) photocatalyst or a carbon dot (CD) photocatalyst.
The photocatalyst may be a carbon- and nitrogen-containing catalyst, such as a carbon nitride (CNX) photocatalyst or a nitrogen-doped carbon dot (CD) photocatalyst.
In a preferred embodiment, the photocatalyst is a NCNCNX photocatalyst, and more preferably such a photocatalyst having a high surface area, such as obtainable from sonication, such as ultrasonication, of a bulk sample of NCNCNx. The photocatalyst may have a surface area that is at least 60 m2 g 1.
In a further preferred embodiment, the photocatalyst is a carbon dot photocatalyst, and more preferably such a photocatalyst is obtained or obtainable from a biomass, for example by calcination of the biomass. The biomass may be a readily available and an abundant natural or processed resource. Alternatively, the carbon dot photocatalyst is obtained or obtainable from readily available and low cost organic molecular precursors.
The photocatalysts of the invention have excellent photocatalytic activity in methods for degrading (or reform) biomass, such as those biomasses having a high cellulose content, and this degradation may be accomplished at ambient temperatures and pressures using natural light or simulated natural light, such as solar-light. Thus, the photocatalyst may be used to degrade tree material, such as branches, as well as processed biomass products such as paper and cardboard, which may be obtained as waste materials. Advantageously, the photocatalysts are also capable of catalysing the degradation of lignin, which is also a component of many biomasses, and which is difficult to process on an industrial scale.
Further, the photocatalyst may also be used to degrade semi-refined biomass, for example the photocatalyst may be used to degrade other biomass components, such as cellulose and xylan, which have been extracted from a biomass.
The photocatalysts of the invention may be used in a photocatalytic method for the generation of hydrogen, for example from biomass. Thus, the photocatalysts are therefore capable of producing a useful fuel from abundant raw and waste biomass.
The photocatalysts of the invention are active under solar-light, including simulated solar- light, and may be used for sustained periods, for example for more than two days, such as more than 12 days, without appreciable loss in activity. The photocatalysts are therefore have long term photocatalytic activity and stability.
The rate of substrate degradation, such as biomass degradation, is remarkably high, and the photocatalyst of the invention has a significant advantage in that no substrate chemical processing is required in order to obtain the hydrogen product. In particular, the
photocatalyst is able to catalyse biomass reformation without the need for pre-treatment under highly basic conditions.
The photocatalysts of the invention are for use together with a co-catalyst for the generation of hydrogen. The co-catalyst is a hydrogen evolution catalyst, such as those well known in
the art, including NiP-, Pt- and Mo-based co-catalysts. Preferably, the co-catalyst is a noble metal-free co-catalyst, such as NiP.
Accordingly, in a first aspect of the invention there is provided a method for degrading an organic substrate, the method comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of the organic substrate and a co-catalyst that is a hydrogen evolution catalyst. The degradation of the organic substrate may produce hydrogen, and the hydrogen is optionally collected.
In a second aspect of the invention there is provided a method for producing hydrogen, the method comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of an organic substrate and a co-catalyst that is a hydrogen evolution catalyst. The method may also comprise the step of collecting the hydrogen. Carbon monoxide may be optionally produced in this method.
In the methods of the invention, the photocatalyst and the organic substrate may be provided as an aqueous mixture, together with a co-catalyst that is a hydrogen evolution catalyst. Advantageously, the photocatalyst may be used within a broad pH range, and the aqueous mixture may have a pH in the range pH 2 to 15, such as a pH in the range pH 2 to 9. For example, the aqueous mixture may be basic, acidic or substantially neutral.
In a further embodiment, the aqueous mixture may contain untreated water. Thus, water for the aqueous mixture may be obtained, without treatment, from a river, lake, canal, or a sea, or from industrial waste water, such as sewage water. The photocatalysts of the invention therefore allow the production of clean hydrogen under real-world conditions, without the need for purified drinking water or laboratory quality water.
Organic pollutants and chloride anions, which inherently exist in these untreated media, serve as sacrificial electron donors, promote high activity and thereby enable simultaneously substantial fuel generation and water purification. As noted previously, current solar-driven catalysis systems only operate in organic solvents, purified water and in the presence of unsustainable sacrificial reagents. The present photocatalysts may address these limitations in the art.
The organic substrate may be or comprise a hydroxyl-functionalised compound, such as a hydroxyl-functionalised compound having a molecular weight of 50 or more, such as 500 or more. The organic substrate may be or comprise a polyol.
The organic substrate may be or comprise a monosaccharide, an oligosaccharide or a polysaccharide, such as cellulose, and preferably the organic substrate may be or comprise a polysaccharide.
The organic substrate may be a biomass, such as wood, paper, cardboard or grass. The organic substrate may contain cellulose, xylan and lignin.
The organic substrate may be selected from the group consisting of general waste, plastics, packing materials, waste food, aliphatic polyols, algae, sugars, starches, biomass, sewage and/or domestic waste.
The organic substrate may be mechanically processed, such as chopped (including chipped), prior to use in the methods of the invention. However, in a preferred method of the invention, no mechanical processing is required, and the organic substrate may be used directly.
The organic substrate may be pre-treated, for example with an aqueous base, to at least partially degrade the organic substrate and/or to improve the aqueous solubility of the organic substrate. Such a pre-treatment is associated with an increased hydrogen evolution rate in the subsequent photocatalytic reaction.
Additionally or alternatively, the organic substrate may be an organic polymer, such as a polymer having hydroxyl, ester, amide, ether, carbonate, and/or urethane functionality, either in the backbone chain or a side group.
The organic polymer may be selected from polyester, substituted polyethylene, polyether, polycarbonate, and polyurethane.
The organic substrate may be a polymer selected from the group consisting of poly(ethylene terephthalate) (PET), poly(lactic acid) (PLA), poly(vinyl pyrrolidone) (PVP), and polyethylene glycol) (PEG). Additionally or alternatively, the organic substrate may be selected from the group consisting of polyurethane (PUR), a polypeptide and a polysaccharide. Optionally the polymer may be further selected from low density polyethylene (LDPE), polyvinyl chloride (PVC), poly(methyl methacrylate), polystyrene (PS) and polycarbonate. Here, the polymers may require pre-treatment, such as with aqueous base, prior to the photocatalytic reaction.
The organic polymer may have a molecular weight of 500 or more, such as 1 ,000 or more, such as 5,000 or more.
The organic substrate may be a component that is a pollutant in an untreated water source. For example, the organic substrate may be selected from chloromethylphenol,
pentachlorophenol, benzaldehyde, trichlorophenol, nitroanaline, dichlorphenol, dimethyl phenol, atrazine, dinitrophenol and naphthylamine. These components may be found in sea water. The component may additionally or alternatively be selected from atrazine, desmetryn, dicamba, chloromehtylphenoxyacetic acid, mecoprop, prometryn, propazine, simazine, terbutryn, and trietazine. The component may also be an inorganic component, such as chloride. These components may be found in river water, also including canal and lake water (or“fresh water” in contrast to sea water).
The incident light in the photocatalytic reaction may be ultraviolet light, such as light having a wavelength in the range from 100 to less than 400 nm, and it may be visible light, such as natural light (such as sunlight), such as light having a wavelength in the range from 400 to 700 nm, or a combination of visible and ultraviolet light, such as natural light (such as sunlight, which has visible and UV components). The incident light may be a combination of natural and non-natural light.
The intensity of the light may be in the range 1 to 150 mW/cm2, such as 10 to 150 mW/cm2, such as 50 to 150 mW/cm2, such as 90 to 150 mW/cm2, such as 130 to 145 mW/cm2. The light intensity may be around 100 mW/cm2.
In yet another aspect of the invention there is provided a photocatalyst, which may be used in the methods of the invention.
In a further aspect of the invention there is provided the use of the photocatalyst as a photocatalyst. The photocatalyst may be for use under incident light, including natural light, such as solar light.
Also provided by the present invention are methods of preparing the photocatalysts.
In one aspect there is provided a method of preparing NCNCNx, the method comprising the step of forming a bulk sample of NCNCNX and subsequently treating the bulk NCNCNX thereby to increase the surface area of the NCNCNx.
The bulk NCNCNX may be sonicated, such as ultrasonicated, to increase its surface area.
The bulk NCNCNX may be obtained or obtainable from H2NCNX.
In one aspect there is provided a method of preparing a carbon dot, the method comprising the step of calcining an organic substrate. The organic substrate may be or comprise an oligosaccharide or a polysaccharide, such as cellulose, and preferably the organic substrate may be or comprise a polysaccharide. The method may provide a product that is a plurality of carbon dots.
This method of the invention provides a complete and self-contained approach to dealing with organic substrate waste and hydrogen production: the organic substrate may be the source for the photocatalyst as well as a source - an electron donor - in the hydrogen production method. In these systems the biomass may be sourced locally together with local untreated water, to provide a substantial proportion of the chemical input for the method from the immediate environment, thereby minimising transport costs for the user.
The invention also provides photocatalysts obtained or obtainable from the methods of preparation described above.
These and other aspects and embodiments of the invention are described in further detail herein.
Summary of the Figures
Figure 1 is a schematic representation of a photocatalytic system according to an
embodiment of the invention for photoreforming lignocellulose to Fh based on carbon nitride and carbon nanodot photocatalysts. In alternative methods of the invention, the
lignocellulose may be replaced with a component, such as a pollutant, present within untreated water.
Figure 2 shows the characterisation data for bulk and sonicated NCNCNx, where (a) is the UV-vis absorption spectra for bulk and sonicated NCNCNX at 0.0125 mg ml. 1; (b) is the photoluminescence emission spectra for bulk and sonicated NCNCNX at 0.67 mg ml. 1, where lbc = 360 nm, 25°C; (c) is the N2 adsorption-desorption isotherms for ultrasonicated and bulk NCNCNX from which BET surface areas were calculated; (d) is the XRD patterns for bulk and sonicated NCNCNx; (e) is the IR spectra for bulk and sonicated NCNCNx; (f) is are TEM images for a sample of bulk (top) and sonicated (bottom) NCNCNx. The measurement bars in (f) are 20 nm; and (g) are SEM images for a sample of bulk (top) and sonicated (bottom) NCNCNx. The measurement bars in (g) are 250 nm.
Figure 3 shows the characterising data for CDs synthesised from purified and waste biomass, where (a) are the HR-XPS C 1 s and N 1 s spectra of ocellulose (top) and
G. /V/Va//s-derived CDs (bottom). The black traces show the‘as recorded’ spectra and the colored plots (red, blue and green) are the deconvoluted bands for each element; (b) HR-TEM images of ocellulose (top) and G. /V/Va//s-derived CDs (bottom). The white lines denote the (100) intralayer spacings; (c) Raman spectra of the CDs derived from purified and waste biomass. The intensity was normalized with respect to the G band for all samples to allow the easy determination of the ID/IG ratio; and (d) UV/vis spectra of CDs recorded in water (0.1 mg mL_1) at 25°C.
Figure 4 shows the recorded photocatalytic Pl2 evolution using CDs from purified and waste biomass, in purified water in the presence of EDTA (3 ml_, 0.1 M, pFH 6) and 50 nmol of NiP, where (a) shows the absolute quantities of Fh produced (in pmol) from ocellulose, xylan and lignin-derived photoabsorbers after 6 h (solid bars) and 24 hrs of irradiation (empty bars); (b) specific activities of ocellulose CDs, xylan and lignin light-harvesters after 24 h of irradiation; and (c) quantities of Pl2 produced (in pmol) from waste biomass photoabsorbers after 24 h of irradiation. For comparison, the hydrogen yield of ocellulose CDs is shown (red close symbols) under the same conditions. The numbers on the right indicate the RFA of ocellulose CDs for each biomass sample. All experiments were carried out upon simulated
solar light irradiation (100 mW cm-2, AM1.5G) for 24 h, under N2 atmosphere with 2% ChU as internal GC standard, at 25°C.
Figure 5 shows the photocatalytic performances of CDs from purified and waste biomass in untreated sea, river and sewage water, where (a) shows photo-Fh production with ocellulose CDs in river, sea, sewage and pure water, in the absence (filled symbols) and presence (open symbols) of EDTA (0.1 M, pH 6, 3 ml_); (b) shows solar-H2 generation of ocellulose CDs in KPi (0.1 M, pH 6, 3 ml_), in the absence of EDTA, but instead in the existence of organic pollutants (200 pmol) and chloride ions (3.2 wt %), found in river (open symbols) and sea water (filled symbols). The results in the absence of any electron donor (ED) are also provided for comparison; and (c) and (d) show the photo-H2 production in sea and river water, respectively, using CDs form ocellulose and waste biomass, in the presence of EDTA (0.1 M, pH 6, 3 ml_). All experiments were carried out under full solar light irradiation (AM 1.5 G, 100 mW cm-2), for 24 h, in the presence of NiP (50 nmol), under N2 atmosphere containing 2% CH4, at 25°C.
Figure 6 shows solar-light driven H2 evolution using ocellulose CDs at quantities between 0.03 and 2.8 mg. All photocatalytic experiments were carried out at 50 nmol NiP and in aqueous EDTA solutions (0.1 M, pH 6, 3 ml_), under full solar spectrum irradiation
(AM 1.5 G, 100 mW cm 2), at 25°C.
Figure 7 shows the results of control photocatalytic experiments in the absence of ocellulose CDs, NiP and EDTA, carried out under full solar spectrum irradiation (AM 1.5 G, 100 mW cm 2), at 25°C.
Figure 8 shows solar-induced H2 production using CDs from cotton. Namely, cotton from different sources, such as cotton wool, pads and cotton T-shirt, were employed as precursors for this purpose. The photocatalytic systems were carried out in aqueous EDTA solutions (0.1 M, pH 6, 3 ml_), for 24 h, under full solar spectrum irradiation (AM 1.5 G,
100 mW cm-2), in the presence of 50 nmol NiP.
Figure 9 shows (a) light-driven H2 evolution (24 h) using ocellulose CDs as photoabsorbers in untreated river, sea and sewage water, in the absence of NiP and in the presence of EDTA (0.1 M, 3 ml_, pH 6); (b) the chemical structures of organic water pollutants, which exist in sea (Gulf of Mexico) and river water (River Cam, UK), based on reports of the US Department of Interior (Torres et al.) and the British environmental agency (Huber et al.), respectively; (c) photo-H2 production (24 h) using ocellulose CDs, in KPi (0.1 M, pH 6,
3 ml_), in the absence of EDTA, but instead in the presence of the organic pollutants (200 pmol), prometryn (PT), terbutryn (TT), benzaldehyde (BA) and atrazine (AZ), as well as chloride ions (3.2 wt %), which all exist in sea and river water; and (d) photo-H2 production (24 h) using CDs from ocellulose and crude biomass, in purified and untreated river and sea water, in the presence of EDTA (0.1 M, 3 ml_, pH 6). Biomass waste samples; Cotton Wool,
G. Nivalis (G. N.), Olive leaves (Ol. leav.), G. Elliptica (G. E.), T. Baccata (T. B.) and Elaeagnus X. ebbingei (E. X. E). All experiments were carried out upon full solar light irradiation (AM 1.5 G, 100 mW cm-2) and under N2 atmosphere (containing 2% CH4), at 25°C.
Figure 10 shows the H2 production using activated (10 min ultra-sonicated) NCNCNX (5 mg) with NiP (50 nmol) and polymer substrates (100 mg) in KPi solution (0.1 M, pH 4.5, 3 ml.) under 1 sun irradiation for 24 hours (100 mW cm-2, AM1.5G, 25°C, where PET: polyethylene terephthalate; PUR: polyurethane; and PS: polystyrene.
Detailed Description of the Invention
The present inventors have found that a particular class of photocatalysts may be used to degrade organic substrates under incident light, such as under incident solar light, so as to provide hydrogen and optionally other products, including carbon monoxide.
Generally, a photocatalyst of the invention is a carbon-containing catalyst, and more specifically the photocatalyst contains carbon in a graphitic and/or amorphous phase. The photocatalysts typically have a predominant carbon sp2 character. For example, the photocatalyst may be a carbon nitride (CNX) photocatalyst or a carbon dot (CD)
photocatalyst.
Meyer et al. have previously described the use of carbon nitride as photocatalysts, including Melon-type amino terminated carbon nitride (H2NCNX), Urea-CNX and NCNCNx. Here, the photocatalysts were used together with an oxidant to catalyse the sulfonylation reaction between a sulfinate and an alkene. There is no disclosure of suggestion that a carbon nitride photocatalyst could or should be used to degrade a biomass.
Kasap et al. have previously described the use of the carbon nitride photocatalyst NCNCNx. Here, the photocatalyst was used together with NiP to generate hydrogen from 4-methyl benzaldehyde (4-MBA). There is no disclosure of suggestion that NCNCNX could or should be used to degrade a biomass.
Lau et al. ( Angew . Chemie int. Ed. 2016) describes the use of carbon nitride for time- delayed hydrogen generation. Here, the carbon nitride catalyses the reaction of 4-methyl benzaldehyde (4-MBA). There is no disclosure of suggestion that NCNCNX could or should be used to degrade a biomass.
Long et al. have previously described the use of carbon nitride photocatalysts, and in particular mesoporous graphitic carbon nitride (mpg-CN). Here, the photocatalyst acts to selectively oxidise aromatic alcohols in water under visible light. Long et al. do not suggest
the use of carbon nitride photocatalysts for the use in degrading biomass, or for the use in generating hydrogen from biomass.
Lau et al. (Nat. Commun. 2016) have previously described the design and optimisation of carbon nitride photocatalysts. Lau et at. do not suggest the use of the photocatalyst to degrade biomass.
Martindale et at. have previously described the use of carbon dots photocatalysts together with the hydrogen evolution catalyst NiP for the generation of hydrogen using EDTA as a sacrificial donor. There is no suggestion that EDTA could or should be replaced with a biomass, and there is no suggestion that the hydrogen evolution reaction could be performed in untreated water, with pollutants used as sacrificial donors.
Guo et at. have previously described the use of carbon dots in seawater. However, the carbon dots act as a draw solute in a forward osmosis method for seawater desalination. There is no suggestion that the carbon dots could be used to generate hydrogen from components, such as pollutants, within untreated seawater.
CN 107961808 describes the preparation of a carbon nitride photocatalyst, and the use of the photocatalyst to generate hydrogen from water. There is no suggestion anywhere that the catalyst should be used to degrade an organic substrate. There is also no suggestion that the photocatalyst could be used to degrade pollutants within waste water. There is also no reference to the use of the carbon nitride photocatalyst together with a hydrogen evolution catalyst, which is a requirement of the methods of the present case.
CN 107413378 describes the preparation of a carbon nitride photocatalyst. This catalyst is for use in generating hydrogen from water. There is no mention of the catalyst for use together with a hydrogen evolution catalyst, and there is no mention of any organic substrate, such as a polysaccharide, or a pollutant from waste water.
CN 107297217 describes the preparation of a platinum-based carbon nitride catalyst. This catalyst is for use in the generation of hydrogen from water. This water is not said to be waste water, and the action of the catalyst is on water as a substrate not any pollutant. The worked examples show the use of the carbon nitride catalyst together with water and sacrificial triethanolamine to generate hydrogen. There is no mention of organic substrates (other than the triethanolamine), and there is no mention of a co-catalyst for generation of hydrogen.
CN 107008484 describes a metal sulfide-modified carbon-nitride photocatalyst. This photocatalyst is used to generate hydrogen from water. There is no reference to any biomass materials and there is no reference to the treatment of waste water and its
pollutants. There is no disclosure in this Chinese application of the use of two separate catalysts species in the reaction mixture.
CN 106902803 describes the modification of carbon dots with KNbC>3 to give a composite photocatalyst. The photocatalyst has use in the degradation of organic pollutants, whilst also producing hydrogen. However, there is not reference to the use of the photocatalyst to degrade a biomass materials, such as paper, or large organic molecules such as
polysaccharides.
CN 107837817 describes a composite material that is a combination of carbon dots, carbon nitride and titanium dioxide. This material is said to be useful for the photocatalytic generation of hydrogen from water. The carbon dots are prepared from the soot of a burning candle. There is no suggestion that these dots could be prepared from saccharides.
The worked examples refers to the use of water to generate hydrogen, in the presence of the electron donor triethanolamine. There is no suggestion that waste water could be used, and there is no reference to any biomass or any polymeric organic material, such as a polysaccharide.
CN 106902857 refers to the use of a carbon nitride photocatalyst to degrade organic pollutants, and it also refers to the use of the photocatalyst to generate hydrogen. However, it does not suggest the two together.
In a worked example, the organic dye Rhodamine B is degraded in the presence of a photocatalyst. There is no description of the degradation products, and the example only looks at the loss of absorbance at a single wavelengths as indicative of a degradation event. There is no suggestion that hydrogen is generated.
In a second worked example, the carbon nitride photocatalyst is used in water together with ascorbic acid and chloroplatinic acid to generate hydrogen. There is no reference to anything that that is a biomass.
The carbon nitride photocatalyst itself is called an expanded carbon nitride, which apparently has an increased surface area. There is no reference to ultrasonication as a method to treat bulk carbon nitride.
CN 107552083 describes a carbon nitride photocatalyst combined with FeP. The photocatalytic activity is only briefly described: the photocatalyst is for use in producing hydrogen from water. There is no mention of any other substrate.
The use of carbonaceous-based catalysts for the degradation of biomass, and the use of carbonaceous-based catalysts for the generation of hydrogen from untreated water, is believed to be non-obvious in view of the disclosures within the prior art.
Photocatalyst
The photocatalyst of the invention is a material, which may be in the form of a particle, which may be at least partially aggregated with other photocatalyst particles. The photocatalyst is a carbonaceous photocatalyst such as a carbon nitride or a carbon dot. The use of a carbonaceous photocatalyst to degrade biomass has not previously been reported in the literature.
The photocatalyst for use in the present invention may be a carbon nitride catalyst, and in particular a NCNCNX catalyst.
Although NCNCNX catalysts are known, their use to degrade organic substrates such as a-cellulose and biomass components such as paper, sawdust, and wood, has not previously been described.
Thus, in one embodiment, the methods of the invention use a known NCNCNX catalyst to generate hydrogen from a biomass.
The inventors have also developed new NCNCNX catalysts having greater activity than those NCNCNX catalysts reported to date. In particular, these catalysts have a greater surface area than those known in the art. This increase in surface area is achieved by treating NCNCNX to reduce the size of agglomerates within a bulk sample, for example by sonicating, such as ultrasonicating, the bulk.
In one embodiment, the surface area of the carbon nitride catalyst is at least 60, at least 70, at least 80, at least 90 or at least 95 m2 g 1. The surface area of the carbon nitride catalyst may be about 97 m2 g 1.
For reference, a bulk NCNCNX catalyst produced by methods know in the art has a surface area of 59.5 m2 g-1. The carbon nitride catalyst produced in the present case has a surface area of 97.4 m2 g 1.
The surface area may be determined by BET measurement, such as described herein.
The photocatalyst for use in the invention may be a carbon dot, which is optionally doped, such as nitrogen, sulfur, or phosphorous doped, such as nitrogen doped.
A carbon dot may also be referred to as a carbon quantum dot (CQD). Carbon dots are described by some of the present inventors in Martindale et al. ( Angew . Chem. Int. Ed. 2017) and Guo et al.
A carbon dot may be a particle having a largest dimension of at most 10 nm, such as at most 5 nm, such as at most 3 nm. The size of a quantum dot may be determined by, for example, analysis of TEM imagery of a collection of carbon dots.
A carbon dot is obtained or is obtainable by calcination (pyrolysis) of an organic substrate, such as a described herein.
Although carbon dot photocatalysts are known, their use to degrade organic substrates such as ocellulose has not previously been described. Furthermore, although the preparation of carbon dots is well known from small organic starting materials, such as amino acids, the use of biomass materials, such as ocellulose, for the preparations of carbon dots has not previously been described.
Aqueous Mixture
The photocatalyst is provided in an aqueous mixture together with an organic substrate and a co-catalyst.
The photocatalyst may be used within a broad pH range, and the aqueous mixture may have a pH in the range pH 2 to 15, such as pH 2 to 9. For example, the aqueous mixture may be basic, acidic or substantially neutral.
In one embodiment, the aqueous mixture may be acidic, such as a pH in the range pH 2-6, such as a pH in the range pH 3-5. In one embodiment, the aqueous mixture has a pH of about pH 4.5.
In one embodiment, the aqueous mixture may have a pH that is pH 9 or less, such as pH 8 or less.
In one embodiment, the aqueous mixture may have a pH that is around pH 7.
The aqueous mixture may be provided with a buffer to maintain a stable pH during the photocatalytic reaction. The buffer may be a phosphate buffer, such as potassium phosphate buffer (KPi buffer), and such are readily available from commercial sources.
The aqueous mixture may contain additional co-solvents, however such are not typically required in the methods of the present case, which may advantageously avoid the need to use any co-solvent.
The inventors have found that the photocatalysts of the invention tolerate the presence of many different components within the aqueous mixture, making them suitable for use in a variety of situations, particularly where the water for the aqueous mixture is untreated water.
The aqueous mixture may contain water that is treated, for example such that the water is suitable for drinking or suitable for use in laboratory.
However, the aqueous mixture may alternatively or additionally contain water that is untreated, for example the water may be obtained from a river, lake, canal, or a sea, or from industrial waste water, such as sewage water. The photocatalysts of the invention can be used under real-world conditions, without the need for purified drinking water or laboratory quality water for the degradation of an organic substrate and the production of hydrogen.
The aqueous mixture, where it contains untreated water, typically contains one or more organic components, optionally together with inorganic components, which are pollutants within the untreated water. These organic components may be used as sacrificial electron donors in the photocatalytic reaction.
Co-Catalyst
The photocatalysts of the invention are used together with a co-catalyst, which is a hydrogen evolution catalyst.
Preferably, the co-catalyst is water soluble.
The co-catalyst may be provided as a separe component within the aqueous mixture. Thus, the photocatalyst and the co-catalyst are not provided together, for example, they are not provided together in a composite. The co-catalyst may also not be physically adsorbed onto the photocatalyst.
The co-catalyst is not a photocatalyst of the invention.
As is apparent from the description of hydrogen generation herein, together with the worked examples, the co-catalyst is provided as a hydrogen evolution catalyst for the generation of hydrogen (for example, as illustrated in Figure 1 ). Here, the photocatalyst is not used directly to generate hydrogen. Rather, irradiation of the photocatalyst results in the formation of a photoexcited state, in which the holes are quenched by the substrate or pollutant, and the photoexcited electrons are effectively transferred from the photocatalyst to the hydrogen evolution catalyst, resulting in hydrogen formation at the hydrogen evolution catalyst (see Kasap et al.).
The use of co-catalysts together with a photocatalyst for the generation of hydrogen is well known in the art. For example, co-catalysts are described for use by Kasap et al., Lau et al. (Angew. Chemie Int. Ed. 2016), and Martindale et al. ( Angew . Chemie Int. Ed. 2017).
The co-catalyst may be selected from a Ni-, Pt- or Mo-containing hydrogen evolution catalyst.
In one embodiment, the co-catalyst is a Ni catalyst, such as NiP (molecular Ni
bis(diphosphine), also written as NiP). The present inventors have previously disclosed a Ni co-catalyst for use as a hydrogen generation catalyst. See, for example, Kasap et al.
In one embodiment, the co-catalyst is a Pt catalyst, such as HhPtCle.
In one embodiment, the co-catalyst is a Mo catalyst, such as a MoS2-based catalyst, such as H8N2M0S4.
Use of the Photocatalyst
The present invention provides for the use of the photocatalyst as such, for example in the degradation of an organic substrate. The degradation may also be referred to as
reformation or reforming, and more specifically as photoreformation or photoreforming.
Accordingly, the invention provides a method for degrading an organic substrate, the method comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of the organic substrate. The photocatalyst and the organic substrate may be provided in an aqueous mixture, and preferably a substantially neutral aqueous mixture.
Degradation may refer to a reduction in the molecular weight of the organic substrate due, for example, to the oxidative cleavage of bonds within the organic substrate. The
degradation may be accompanied by the formal loss of hydrogen and/or carbon monoxide from the organic substrate.
The methods of the invention also provide for the generation of hydrogen and/or a product such as carbon monoxide, the method comprising the step of exposing a photocatalyst of the invention to visible and/or ultraviolet light in the presence of the organic substrate.
The photocatalyst and the organic substrate may both be provided in an aqueous mixture.
The aqueous mixture may have a pH in the range 2 to 15
The aqueous mixture may be a basic aqueous mixture, such as a mixture having a pH of 10 or more, such as 1 1 or more, such as 12 or more, such as 13 or more, such as 14 or more. The aqueous mixture may have a pH in the range 8 to 12, such as 9 to 10.
The aqueous mixture may be an acidic mixture, such as a mixture having a pH of 6 or less, such as 5 or less.
The aqueous mixture may have a pH in the range 2 to 6, such as 4 to 5.
The aqueous mixture may be substantially neutral.
In the methods for generating hydrogen, the source of hydrogen may be the organic substrate, the basic aqueous mixture or both. The present inventors have found from deuterium experiments that the predominant source of hydrogen is water.
In the methods for generating carbon monoxide the source of carbon is the organic substrate.
The photocatalyst is active (“excited”) under incident light in the visible and/or ultraviolet range, and preferably the photocatalyst is used under incident light, including incident light having a visible light component, such as solar light. Here, visible light refers to light having a wavelength in the range 400 to 700 nm, and ultraviolet light refers to light having a wavelength in the range 100 to 400 nm, such as 315 to 400 nm.
The incident light may be natural light, such as sunlight. Natural light may include light in both the visible and ultraviolet ranges.
The incident light may be artificial light, such as provided by a solar simulator, or light from a xenon or mercury lamp, or an LED. Examples of simulated solar light include AM1 5D, AM1.5G and AMO.
The incident light may be monochromatic.
The incident light may be a combination of natural and artificial light.
The methods of the present invention may be performed under light having an intensity that is similar to that of natural light. Thus, in the methods of the invention it is not necessary to expose the photocatalyst to intense light.
The intensity of the incident light may be at most 140, at most 150, at most 160 or at most 200 mW/cm2.
The intensity of the incident light may be at least 1 , at least 10, at least 20, at least 50, at least 70, at least 80, at least 90, or at least 100 mW/cm2.
The incident light maybe of an intensity within a range where the upper and lower limits are selected from the values given above. For example, the intensity of the incident light may be in the range 1 to 150 mW/cm2, such as 10 to 150 mW/cm2, such as 50 to 150 mW/cm2, such as 90 to 150 mW/cm2, such as 130 to 145 mW/cm2.
The light intensity may be around 100 mW/cm2. In the worked examples of the present case, artificial light of 100 mW/cm2 is used (AM 1 .5G).
The methods of the invention may be conducted at ambient temperatures, for example at a temperature in the range 5 to 30°C. Accordingly, it is not necessary to apply heat to the reaction mixture in order to promote the reaction. Photocatalytic reactions previously reported in the art described heating the reaction mixture, and such is not required in the methods of the present invention.
However, if needed, the methods of the invention may be performed at an elevated temperature, such as a temperature greater than 30°C, and for example at a temperature no more than 60°C, such as no more than 50°C, such as no more than 40°C.
Additionally or alternatively, the organic substrate may be pre-heated prior to its contact with the photocatalyst or prior to irradiation, and preferably prior to its contact with the photocatalyst. The mixture may be a heated to a temperature that is no more than 100°C, such as no more than 80°C, such as no more than 60°C, such as no more than 50°C, such as no more than 40°C. Generally, however, the methods of the invention do not require the substrate to pre-heated, and the photocatalytic reaction is performed at ambient
temperature.
The heating of the mixture may be maintained during and after the addition of the photocatalyst, and throughout the catalytic reaction. Alternatively, the heating of the mixture may be discontinued during or after the addition of the photocatalyst. Indeed, the mixture may be allowed to cool, for example to a temperature in the range 5 to 30°C, prior to the addition of the photocatalyst.
The methods of the invention may be performed as a batch process, or as a flow process where the organic substrate is permitted to flow across the photocatalyst, which may be immobilised to a support.
After a degradation reaction is deemed complete the photocatalyst may be recovered for future further use. Here, the photocatalyst may be separated from remaining unreacted organic substrate and any reaction intermediates, such as partially degraded organic substrate. If appropriate, the recovered photocatalyst may be rejuvenated prior to reuse, such as described in further detail below.
Organic Substrate
The photocatalyst of the invention may be used to degrade, or reform, an organic substrate, and preferably the degradation reaction yields hydrogen and/or carbon monoxide. The methods of the invention may be used to degrade waste material as part of the disposal process of that material. Alternatively, the methods of the invention may be intended for the generation of a fuel material, such as hydrogen, for downstream use. Advantageously the
methods of the invention may be used to dispose of unwanted biomass and also to generate commercially relevant fuels.
The organic substrate typically contains both hydrogen and carbon atoms, and most typically contains multiple carbon-carbon and multiple carbon-hydrogen bonds. The photocatalyst is most useful in the degradation of higher molecular weight organic substrates, and is also most useful in the degradation of hydroxyl functionalised organic substrates. The photocatalyst may also be used to directly degrade biomass without any prior refining of the biomass. Thus, the photocatalyst is suitable for use with relatively simple organic materials as well as complex biomass mixtures, which contain a wide variety of different chemical structures, often with a wider distribution of molecular weights.
The photocatalyst has tolerance to a range of substrates, and such tolerance has not previously been reported in the art. The ability to use the photocatalyst with unrefined substrates therefore provides flexibility for the process, which may be used at any location where there is a source of biomass materials, such as wood and grass. In contrast, many of the degradation and hydrogen evolution reactions reported to date rely on the use of refined substrates for use in the photocatalytic reaction, and therefore the methods can only be performed where there it the ability to refine biomass materials, or where there is a ready commercial source of the refined material.
The degradation of the organic substrate may yield hydrogen, which has use as a commercial fuel and a chemical feedstock. Accordingly, the degradation reaction allows for the valorisation of the organic substrate.
The ability to generate hydrogen from any organic substrate, including wood and cardboard as well as refined cellulose, xylan and lignin, is highly advantageous, as the methods of the invention are not limited to locals where refined materials are available, which is a clear limitation of the prior art methods.
It is possible that other degradation products are also produced, such as carbon monoxide. The production of degradation products therefore provides an additional or alternative pathway for creating value from the organic substrate.
The production of ethanol from cellulose has been described previously as a route to preparing a fuel stock. However, in many cases cellulose-derived ethanol requires purification of glucose from lignocellulose, followed by the subsequent fermentation of that glucose to yield ethanol. As noted above, the present invention allows for the production of hydrogen directly from any source of unprocessed lignocellulose.
The organic substrate may contain one or more, such as two or more, hydroxymethyl groups (-CH2OH). For example, cellulose, hemi-cellulose and lignin contain a plurality of hydroxymethyl groups within their core structures.
The organic substrate may be a polyol, for example a polyol having two or more, such as five or more, such as ten or more hydroxyl groups.
The photocatalyst of the invention may be used to degrade a high molecular weight component within the organic substrate. In the worked examples of the present case, the photocatalyst is shown to be useful in degrading cellulose, hemi-cellulose, xylan and lignin. Indeed, the photocatalyst may be used to degrade biomass directly, and without any refining.
Thus, the photocatalyst may be used to degrade an organic substrate that is or contains a component having a molecular weight of 70 or more, such as 100 or more, such as 200 or more, such as 500 or more, such as 1 ,000 or more. Here, the organic substrate may have a molecular weight of at most 1 ,500.
The photocatalyst may be used to degrade an organic substrate that is or contains a component having a molecular weight of 2 kDa or more, 5 kDa or more, 10 kDa or more,
20 kDa or more, 25 kDa or more, 30 kDa or more, or 50 kDa or more.
It is noted that many of the reported uses of carbon dots and carbon nitride photocatalyst are with small organic substrates, and many documents use the photocatalysts not for degradation but to catalyse organic bond-forming reactions.
The organic substrate may be a biomass or a refined biomass. In the preferred
embodiments of the invention, refining of the biomass is not required, and a biomass may be used directly (or after mechanical processing). Here, refining refers to a process where components of the biomass, such as lignin, cellulose and hemicellulose are separated from each other, of from other components of the biomass. Refining may refer to thermal, chemical or biochemical processes for achieving this separation.
The organic substrate may be or comprise a polysaccharide.
In the preferred methods of the invention, the organic substrate is cellulose, such as a-cellulose.
The organic substrate may be or comprise a polypeptide.
A mixture of different organic substrates, such as those described above, may be present.
In the worked examples of the present case, the organic substrate is a complex biomass material, such as paper or wood. The organic substrate may contain many different types of biomass in a mixture.
In one embodiment, a mixture of lignin, cellulose and hemicellulose may be present as organic substrates. Such materials typically feature within biomasses such as paper and wood or a biomass that is plant matter.
The organic substrate may be wood, paper, cardboard, bagasse, grass and/or sawdust.
The organic substrate can be general waste, plastics, packing materials, waste food, aliphatic polyols, algae, sugars, starches, biomass, sewage and/or domestic waste. The organic substrate preferably comprises lignocellulose or one or more of its constituent components.
A biomass for use in a method of the invention may be mechanically treated prior to used, for example to reduce the size of the biomass and optionally to provide the biomass in a uniform size distribution. Such mechanical treatments are for optimal processing of the biomass in the methods of the invention, for example to increase the available surface area of the substrate or simply to ensure that the organic substrate can be handled and distributed within a processing facility.
The inventors have found that the photocatalyst of the invention can be used directly to catalyse the reaction of biomass, and each of the major components of lignocellulose is reactive in the presence of the photocatalyst when it is exposed to light. Accordingly, the methods of the invention do not require the biomass to be chemically treated prior to use. Many methods contained in the prior art have described the preparation of monosaccharides from cellulose components as a preliminary step, prior to the use of the photocatalyst.
The present inventors have also found that the photocatalysts of the invention have broad application in degrading, or reforming, organic substrates that are organic polymers, and preferably the degradation reaction yields hydrogen.
The inventors found that the photocatalysts could be used to degrade organic polymers such as polysaccharides, including cellulose, hemicellulose, and lignin. The photocatalyst is also found to have use in the degradation of other organic polymers, and may be used to generate hydrogen from those organic polymers. For example, PET, PUR and PS may be used as feedstocks to generate hydrogen, as shown in the worked examples of the present case.
The organic polymers described here may be used in the same way as the organic substrates described above.
An organic polymer may refer to a polymer that contains multiple monomer units, and typically each monomer unit contains at least one carbon-carbon bond in the backbone chain of the polymer. Where a monomer does not contain a carbon-carbon bond in the backbone chain of the polymer, it may contain a carbon-oxygen in the backbone, for
example as seen with paraformaldehyde polymers. An organic polymer may contain both carbon-carbon and carbon-oxygen bonds within the backbone.
The monomer units may be covalently linked by carbon-carbon bonds, such as in a substituted polyethylene, but this is not necessary, and monomers may be linked by carbon- oxygen or carbon-nitrogen bonds. For example, carbon-oxygen bonds are present in polyethers and polyethylene terephthalates. Carbon-oxygen and carbon-nitrogen bonds are present in polyurethanes.
The organic polymer may be a homopolymer or a copolymer.
Typically each monomer (repeat) unit within the organic polymer contains one or more heteroatoms, such as one or more heteroatoms selected from oxygen, nitrogen and sulfur.
A heteroatom may be present within the covalent bond connecting monomers, and/or a heteroatom may be present within a side group of the monomer.
In certain methods of the invention, an organic substrate such as an organic polymer may be at least partially degraded or solubilised prior to its treatment with the photocatalyst. This pre-treatment of the organic substrate may be undertaken to improve the solubility of the substrate in the aqueous mixture.
The organic polymer may therefore comprise functionality, either in the backbone chain or the side chain, which is hydrolysable, for example under basic or acidic conditions. For example, the organic polymer may contain ester, amide, ether, carbonate, and/or urethane (carbamate) functionality.
The organic polymer may be a polyester, such as polyethylene terephthalate (PET) or polylactic acid (PLA), a substituted polyethylene, such as polyvinyl pyrrolidone (PVP) and optionally also polyethylene, a polyether, such as polyethylene glycol (PEG), a
polycarbonate (PC) or a polyurethane (PUR).
Additionally or alternatively, the organic polymer may be a polyamide, such as a protein.
For example, the organic polymer may be a polyester, a substituted polyethylene, or a polyether.
The photocatalyst may be used together with an organic polymer that is a polyester. The polyester may be an aryl-containing polyester (aromatic polyester) or an alkyl-containing polyester (aliphatic polyester). The polyester may contain both aryl and alkyl functionality.
Examples of aryl-containing polyesters include polyethylene terephthalate, polyethylene naphthalate, polybutylene terephthalate and polytrimethylene terephthalate.
Examples of alkyl-containing polyesters include the homopolymers polylactic acid, polycaprolactone (PCL), polyglycolic acid, poly-3-hydroxyvalerate, poly-3-hydroxybutyrate, poly-4-hydroxybutyrate. Examples of alkyl-containing polyesters include the copolymer polybutylene succinate and poly(3-hydroxybutyrate-co-3-hydroxyvalerate).
The photocatalyst may be used together with an organic polymer that is a substituted polyethylene. Thus, the polyethylene has a side chain. Typically, the substituted
polyethylene is a polymer having one or more heteroatoms, such as selected from oxygen, sulfur or nitrogen, within the side chain. The heteroatom functionally may be a component of a functional group that is hydrolysable under the reaction conditions, or it may assist in the dissolution of the polymer in the reaction mixture.
The polyethylene may be substituted with, for example, a heterocycle, such as a C4-7 heterocycle having one ring heteroatom. The heterocycle may be pyrrolidone.
The polyethylene may be substituted with, for example, a cyano group. Thus, the polymer may be a polyacrylonitrile (PAN).
The polyethylene may be an acrylate polymer, for example a poly(methyl acrylate).
The photocatalyst may be used together with an organic polymer that is a polyether, including for example polyethylene glycol.
A polyether may be an alkyl-containing polyether (aliphatic polyether) or an aryl-containing polyether (aromatic polyether). The polyether may contain both aryl and alkyl functionality.
Examples of alkyl-containing polyethers include polyethylene glycol, polypropylene glycol, paraformaldehyde (PFA) and poly(tetramethylene ether) glycol (also known as
polytetrahydrofuran).
Examples of aryl-containing polyethers include polyphenyl ether and poly(p-phenylene ether) (PPE).
An example of a complex polyether containing both alkyl and aryl functionality, together with further substituent groups, is lignin. Here, lignin is a polymer crosslinked phenolic units. In one embodiment, a polyether is not lignin.
The organic polymer may be a component of a consumer product. For example, polyethylene terephthalate is found in fibres for clothing and containers for foods and liquids.
For the purposes of the present case, a polysaccharide may not be considered a polyether.
An example of a polyamide is a polypeptide. Example polypeptides include bovine serum albumin and casein.
As with the organic substrates previously described, the organic polymer may have a molecular weight that 500 or more, such as 1 ,000 or more. The photocatalyst may be used to degrade an organic substrate that is or contains a component having a molecular weight of 2 kDa or more, 5 kDa or more, 10 kDa or more, 20 kDa or more, 25 kDa or more, 30 kDa or more, or 50 kDa or more.
As previously noted, the organic substrate, such as the organic polymer, may be pre-treated prior to its mixing with the photocatalyst. For example, the organic substrate may be pre-heated to assist the hydrolysis or dissolution of the organic polymer. In a further example, the organic polymer may be pre-treated with base to assist the hydrolysis or dissolution of the organic polymer.
The organic polymer may also be mechanically treated, for example by cutting, grinding and milling and other such techniques. The mechanical treatment may be particularly
advantageous where the organic polymer is a component of a consumer product, and a size reduction in the product will assist in the rate of degrading the product and its organic polymer component.
Water Treatment
The photocatalysts of the invention may be used in untreated water to generate hydrogen. Here, the method of generating hydrogen may use the photocatalyst together with a co-catalyst and a sacrificial electron donor, which donor is typically a component, such as an organic or inorganic pollutant, within the untreated water.
The hydrogen generation method has the advantage of providing useful fuel, in the form of hydrogen, whilst also degrading pollutants within the untreated water, thereby to at least partially purify the untreated water.
The sacrificial electron donor is a compound, which may be an organic compound or an ionic species, within the water that is capable of quenching holes, by the donation of electrons, that are photochemically generated in the photocatalyst.
Examples of sacrificial electron donor compounds that are found in river water include one or more compounds selected from atrazine, desmetryn, dicamba, chloromehtylphenoxyacetic acid, mecoprop, prometryn, propazine, simazine, terbutryn, and trietazine. These
components may also be found in untreated water from lakes and canals.
Examples of sacrificial electron donor compounds that are found in sea water include one or more compounds selected from chloromethylphenol, pentachlorophenol, benzaldehyde,
trichlorophenol, nitroanaline, dichlorphenol, dimethyl phenol, atrazine, dinitrophenol and naphthylamine.
A sacrificial electron donor compound may be selected from the group consisting of from atrazine, prometryn, terbutryn and benzaldehyde.
The untreated water may contain chloride, as an example of an ionic sacrificial electron donor.
Untreated water may contain one or more components identified in sea water and river water as noted by the US Department of Interior (Torres et al.) and the British environmental agency (Huber et a!.).
The untreated water may be supplemented by the addition of material that is capable of acting as a sacrificial electron donor. For example, an organic substrate, such as those substrates described above, including biomass, may be added to the untreated water.
Where an untreated water is used together with a biomass, it is preferable that the untreated water and the biomass are taken from the same locality, for example within 5 km from each other, such as 1 km from each other. Here, the methods for generating hydrogen and optionally also purifying the untreated water, do not require large movement of materials, and hydrogen may be generated close to the source of the water and the biomass. This provides a great efficiency to these methods.
In a preferred embodiment, the photocatalyst for use here is a carbon dot photocatalyst.
Such a photocatalyst may itself be generated from an organic substrate, such as a biomass. This same organic substrate may be used as a sacrificial electron donor in the methods for generating hydrogen from untreated water. Thus, many of the components for use in the method may be sourced locally, and typically at minimum cost also, and may be prepared from local components. This provides a further efficiency to the system.
Methods of Preparation
The present case provides methods for preparing a photocatalyst, including methods for preparing a carbon nitride photocatalyst and a carbon dot photocatalyst.
The preparation of carbon dot photocatalysts from simple organic starting material is well known in the art. For example, Martindale et al. (Angew. Chemie Int. Ed. 2017) describe the preparation of amorphous carbon dots (a-CD) by calcination of citric acid at 180°C for 40 h. Martindale et al also describe the preparation of graphitic nitrogen-doped carbon dots (g-N-CD) by calcination of aspartic acid at 320°C. Graphitic undoped carbon dots were
prepared by calcination of citric acid at 180°C for 40 h, then further heating at 320°C for 100 h.
The present inventors have now found that more complex organic substrates may be used to generate carbon dots. In particular, the present inventors have shown that biomass waste may be used as a starting material for preparing carbon dots having photocatalytic activity. By way of example, the inventors have shown that carbon dots may be prepared from cotton, including cotton products, G Nivalis, G. Elliptica, T. Baccata, Elaeagnus X ebbingei and olive leaves.
The present inventors have also found that biomass components isolated, such as at least partially purified, from plants may also be used as staring materials for preparing carbon dots having photocatalytic activity. By way of example, the inventors have shown that carbon dots may be prepared from ocellulose, xylan and lignin.
The invention provides a method of preparing a carbon dot, the method comprising the step of calcining an organic substrate.
The organic substrate may be or comprise a monosaccharide, an oligosaccharide or a polysaccharide, such as cellulose, and preferably the organic substrate may be or comprise an oligosaccharide or a polysaccharide, such as a polysaccharide.
The organic substrate may be lignin, cellulose or hemicellulose.
The organic substrate may be wood, leaves, paper, cardboard, bagasse, cotton, grass and/or sawdust.
The organic substrate can be general waste, packing materials, waste food, aliphatic polyols, algae, sugars, starches, and/or biomass. The organic substrate preferably comprises lignocellulose or one or more of its constituent components.
The calcining step is a heating step and the temperature and duration of the heating step may be selected for optimal production of the carbon dot.
The minimum heating temperature may be at least 200°C, at least 250°C, at least 300°C or at least 350°C.
The maximum heating temperature may be at most 400°C, at most 450°C or at most 500°C. The heating temperature may be in a range selected from the minimum and maximum temperature given above. For example, the heating temperature may be in the range 250 to 450°C. If necessary the temperature can be varied within this range, for example with an initial lower heating temperature followed by a subsequent higher heating temperature.
The heating may be performed for a total of at least 12 hours, at least 24 hours, at least 2 days, at least 3 days or at least 4 days.
The heating may be performed for a total at most 5 days, at most 6 days, at most 7 days or at most 1 week.
The worked examples in the present case demonstrate that the products formed from the calcination reactions have excellent activity as photocatalysts, and these photocatalysts may be used to generate hydrogen from a range of organic substrates provided in purified and untreated water, including ocellulose, EDTA (as an exemplary sacrificial electron donor), and pollutants in sea water and river water, such as atrazine, terbutyrn, benzaldehyde and prometryn. Other species, such as chloride, which is ionic and inorganic, may also act as sacrificial electron donors.
The preparation of carbon nitride catalysts is well known in the art, and the NCNCNX has previously been reported for use as a photocatalyst, for example in sulfonylation reactions (see Meyer et al.).
The inventors have found that NCNCNX may be used as a photocatalyst in the degradation of organic substrates, as described herein. The inventors have also found that the activity of the carbon nitride photocatalyst may be enhanced when the surface area of a catalyst sample is increased.
The preparation of NCNCNx from H2NCNX as described to date, provides a bulk sample of material where product particles are provided as aggregates (as shown in the TEM images of Figure 2(f) in the present case). The inventors have found that by disrupting the aggregates, such as by sonication, including ultrasonication, to reduce the aggregate size, increases the surface area of a sample, as judged by BET analysis. This increase in surface area is shown to increase the photocatalytic activity of the material.
The invention therefore provides a method of preparing NCNCNx, the method comprising the step of forming a bulk sample of NCNCNX and subsequently treating the bulk NCNCNX thereby to increase the surface area of the NCNCNx.
The bulk NCNCNX may be sonicated to increase its surface area.
The bulk NCNCNX may be obtained or obtainable from H2NCNX.
Other Preferences
Each and every compatible combination of the embodiments described above is explicitly disclosed herein, as if each and every combination was individually and explicitly recited.
Various further aspects and embodiments of the present invention will be apparent to those skilled in the art in view of the present disclosure.
“and/or” where used herein is to be taken as specific disclosure of each of the two specified features or components with or without the other. For example“A and/or B” is to be taken as specific disclosure of each of (i) A, (ii) B and (iii) A and B, just as if each is set out individually herein.
Unless context dictates otherwise, the descriptions and definitions of the features set out above are not limited to any particular aspect or embodiment of the invention and apply equally to all aspects and embodiments which are described.
Certain aspects and embodiments of the invention will now be illustrated by way of example and with reference to the figures described above.
Experimental and Results
Materials
The pure biomass components, a-cellulose, xylan and lignin, as well as natural sea water (from Gulf of Mexico) and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA), were purchased from Sigma-Aldrich and used without any further purification. Cotton wool was purchased from Stores, Department of Chemistry, University of Cambridge, UK, whereas cotton pads from Waitrose, UK and cotton T-shirt from an open market in
Trumpington, UK. The crude plant biomass samples, Galanthus Nivalis (G. Nivalis), Garrya Elliptica (G. Elliptica), Taxus Baccata ( T . Baccata) and ElaeagnusX ebbingei, were collected from St John's College garden, University of Cambridge, UK, whereas the olive leaves from D. S. Achilleos house garden, Trachoni, Cyprus. The untreated river water was obtained from the River Cam (Midsummer Common, September 2017), Cambridge, UK and used without any purification.
Buffer solutions were prepared using analytical grade reagents and titrated to the desired pH with a pH meter (Mettler Toledo; SevenEasy).
Quantification of Photocatalyst Products
The amount of H2 accumulation was quantified via periodic headspace gas injections (20 pl_) to gas chromatography (GC), Agilent 7890A equipped with a 5 A molecular sieve column. The temperature of the GC oven was maintained at 45°C and N2 was used as the carrier gas, with a flow rate of 3 mL min 1.
The amounts of alcohol (4-MBA) and aldehyde (4-methylbenzyl aldehyde, 4-MBAd) were quantified by nuclear magnetic resonance (1H-NMR) spectroscopy (Bruker DPX 400 spectrometer). Thus, the reaction suspension was extracted with CH2CI2 (2 x 3 mL), and combined extracts were concentrated in vacuo. The residue was then characterised by 1H
NMR spectroscopy on a Bruker DPX 400 spectrometer at 298 K. Comparison of the methyl peak integrals of 4-MBA and 4-MBAd, as well as the distinctive aldehyde (COH) peak, allowed for the determination of a quantitative ratio of the starting material to product.
Treatment of Analytical Data
All measurements were performed as triplicates and the data is reported as mean value ± standard deviation (o).
NCNCNX
Synthesis of CNX and NiP
H2NCNX was prepared by heating melamine at 550°C for 4 h under air. The yellow solid obtained was then thoroughly ground using a pestle and mortar prior the surface
functionalisation of the material. NCNCIMx was prepared from grounded H2NCNX upon reaction with KSCN (mass ratio 1 :2) at 400°C for 1 h, and then at 500°C for 30 min under Ar. After cooling to room temperature, the resulting solid was excessively washed with water to remove the residual KSCN and then dried in vacuo to yield NCNCNx.
To prepare a higher surface area NCNCN, the material was suspended in solvent (KPi, water, iPrOH and MeOH; at 1 or 5 mg/ml_), followed by ultrasonication for 1 , 10, or 30 min, using a Sonic Dismembrator (Model 120, Fischer Scientific).
NiP was synthesised following a reported procedure: see Gross et al.
Analysis and Characterisation of CNx
NCNCNX was characterised before and after ultrasonication by UV-visible (UV-vis) absorption and photoluminescence (PL) spectroscopy, Brunauer-Emmett-Teller (BET) surface area analysis, X-ray diffraction (XRD), Scanning (SEM) and Transmission Electron Microscopy (TEM), Attenuated Total Reflectance - Fourier Transform Infrared (ATR-FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS), to reveal the effect of the ultrasonication on the structural and optical properties of the bulk material, and thus its photocatalytic performance.
Ultraviolet-visible (UV-Vis) absorption spectra were collected by a 50 Varian Cary UV-vis spectrophotometer in the wavelength range between 200 and 800 nm (in water) using quartz cuvettes with 1 cm pathlength. Photoluminescence emission spectra of bulk and
ultrasonicated NCNCNX were recorded on a FS5 spectrofluorometer (Edinburgh Instruments) equipped with an integrating sphere.
Brunauer-Emmett-Teller (BET) analysis was carried out with a 3Flex Surface Characterisation Analyzer (Micromeritics) at -196°C, using N2 as the absorbing gas. Prior to measurements, the samples were outgassed under vacuum for 6 h at 150°C. The specific surface areas were calculated by fitting the data to the Brunauer-Emmett-Teller (BET) isotherm.
The X-ray diffraction (XRD) patterns were recorded with a BV X’Pert PRO diffractometer (PANalytical). The Scanning and Transmission Electron Microscopy (SEM and TEM, respectively) images, were recorded with MIRA3 FEG-SEM (TESCAN) and Tecnai
G2 80-200kV transmission electron (FEI) microscopes, respectively. Attenuated Total Reflectance - Fourier Transform Infrared (ATR-FTIR) spectra were recorded on a Nicolet iS50 FTIR spectrometer (ThermoScientific) and analysed using the Omnic software.
The external quantum efficiency (EQE) of a NCNCNX-NIP photosystems was measured using a solar light simulator (LOT LSN 254) equipped with a monochromator (LOT MSH 300) to irradiate the samples at a wavelength of A = 360 ± 10 nm, at a light intensity (I) of I =
4.05 mW cm-2. EQE was calculated using the following equation: 100
where nH2 is the moles of photogenerated H2, NA he Avogadro’s constant, h the Planck constant, c the speed of light, tirr the time of irradiation, and A the cross-sectional area of irradiation (0.28 cm 2).
Brunauer-Emmett-Teller (BET) measurements showed a 60% increase in the surface area of NCNCNX after ultrasonication (97.4 ± 0.6 m2 g 1), compared to bulk (59.5 ± 0.5 m2 g 1) (Figure 2(c)), in accordance with the literature (Meyer et al.). The BET values were obtained from the recorded absorption data by fitting to the UPAC Type IV isotherm.
The increase in surface area is due to the presence of smaller aggregate sizes in the sample after ultrasonication, compared to the bulk NCNCNx, as indicated by TEM images (see Figure 2(f)), which also show negligible changes in particle size and morphology (only a small fraction of rod-like particles was observed), in accordance with SEM images (Figure 2(g)).
This also explains the almost identical X-ray diffraction (XRD) patterns of the two materials, and the preservation of the characteristic inter-layer (28° 2Q) and intra-layer (7.9° 2Q) periodicities of bulk NCNCNX after ultrasonication (see Figure 2(d)) (Lau et al.).
Optical characterisation of the bulk and ultrasonicated NCNCNX by UV-vis spectroscopy, at the same concentration (0.0125 mg ml. 1), revealed mainly UV absorption attributed to the TT-TT* transitions of the heptazine units (Zhuo et a!.), which tails into the vis region for both
materials. However, after ultrasonication, although the spectroscopic features of the two materials remain identical (Figure 2(a)), there is a dramatic increase in the UV absorption of the ultrasonicated sample by about 9 times (Figure 2(a)). This is due to the increased surface area of the material and the smaller aggregate sizes (that minimize light scattering), which both maximize light absorption (Wu et al.).
PL studies (/\ex = 360 nm) revealed increased emission intensity and no significant shift in the emission peak position ( l = 438 nm, corresponding to a band gap of approximately 2.8 eV) for the ultrasonicated NCNCNx, which implies no change in the band levels, but instead a higher number of delocalised C=N TT-TT* transitions and photoexcited states (Figure 2(b)). The increased intensity, according to literature, also signifies smaller aggregates size and higher dispersity of the ultrasonicated sample, in agreement with the observed change in BET surface area (Shiravand et al.).
The external quantum efficiency (EQE) of the NCNCNx-NiP photosystems was determined, and the EQE of the ultrasonicated and bulk systems are respectively 22.2% ± 1.1 % and 13.2% ± 1.2%. The former is approximately 68% higher than latter, which is highly consistent with the increase of BET surface area. The EQE of both the bulk and ultrasonicated
NCNCNX-NIP systems are much higher than the previously reported H2NCNx-NiP and RuP-NiP systems (Caputo et al., Gross et a!.).
Moreover, the material preserves its surface functionalities (Figure 2(e)) after ultrasonication. More specifically, both the bulk and ultrasonicated NCNCNX samples show similar features in their FTIR spectra; a broad peak at about 3300 cm-1 assigned to N-H stretching, a pair of sharp peaks at 2175 cm 1 and 2144 cm 1 which signify the preservation of the CºN groups after ultrasonication, the bands at 1639 cm 1 and 1557 cm 1 which are attributed to secondary amine -C-N bending, whereas the signal at 807 cm 1 is due to the heptazine core vibration (Kasap et al., Lau et al. Nat. Commun. 2016).
Finally, XPS measurements (spectra not shown) show no significant changes between the high-resolution C 1 s spectra of bulk and ultrasonicated samples, indicating that
ultrasonication does not change the chemical properties of NCNCNX, which is consistent with FTIR characterization. The two C 1 s environments at 288.01 eV and 287.02 eV are assigned to sp2 C in the heptazine units and terminal N-CºN groups respectively (Miller et al.), and the peak at 284.4 eV is typical of adventitious carbon contamination (Algara-Siller et al.).
Overall, there is no evidence that ultrasonication induced any significant structural or chemical change to the bulk NCNCNx. AS such, the enhancement in photocatalytic activity of the ultrasonicated NCNCNX is mainly attributed to the increased surface area due to aggregate breakage in the bulk NCNCNx, the better light absorption (less light scattering) and the higher number of photoexcited charge generation in the ultrasonicated sample.
Photocatalytic Experiments
Photocatalysis was carried out in a borosilicate glass photoreactor (total volume 7.74 ml.) in a water bath thermostated at 25°C. In a typical experiment, the suspensions were prepared by mixing for 10 min NCNCNX (such as the ultasonicated form) or CDs and NiP (or hhPtCh and H8N2M0S4 as precursors for Pt and M0S2, respectively) in aqueous 0.1 M KPi solution (at pH 4.5, 3 ml.) containing an electron donor (4-MBA or 100 mg lignocellulosic substrates) in a photoreactor equipped with a stirrer bar (total solvent volume 3 ml_). The container was then tightly sealed with a rubber septum and purged with N2 containing 2% CH4 as the internal gas chromatography standard.
The photoreactor was stirred and kept at a constant temperature (25°C) with continuous water circulation through a water-jacketed reservoir during the light experiments.
The vials were then irradiated using a calibrated solar light simulated (Xe lamp,
100 mW cm-2, 1 Sun, Newport Oriel) whilst stirring. The solar light simulator was equipped with an air mass 1.5 global filter (AM 1 5G) and water filter to remove the infrared irradiation.
Use of NCNCNX
Optimising Performance of Activated NCNCNX
The photocatalytic activity of the photoreforming system to produce H2 was optimised with respect to the activity of NCNCNX using 4-MBA as an electron donor, NiP as H2 evolution catalyst and irradiation by simulated solar light (AM1.5G, 100 mW cm 2) at room
temperature. An optimised specific activity of 39 mmol H2 (g NCNCNx) 1 h 1 was obtained, which is twice the highest activity previously reported with respect to any CNX.
Simultaneous proton reduction and 4-MBA oxidation to 4-MBAd was investigated using bulk and activated NCNCNX through sonication (data not shown). The activated (sonicated) NCNCNX samples showed more than three times higher photocatalytic activity than previously reported bulk NCNCNX samples. Thus, sonication was demonstrated as a means to activate NCNCNx. After 2 h of irradiation, the system becomes limited by the availability of the electron donor and complete and selective 4-MBA conversions to 4-MBAd was achieved in less than 6 hours. Here, the bulk was sonicated for 10 min. as described previously.
The conditions of ultrasonication were optimised systematically for achieving high
photocatalytic performance. The parameters are duration and temperature of
ultrasonication, as well as concentration of NCNCNX and solvent used during ultrasonication. As shown in Table 1 , over the first 4 hours, the photocatalytic H2 production rate and the NiP-based turnover number are enhanced by approximately 5 times after ultrasonication of 10 and 30 minutes. The material shows similar photocatalytic activity when ultrasonicated
for 10 and 30 minutes, and thus 10 min ultrasonication was chosen as the optimum sonication time.
Table 1 - Changes in H2 production with change in ultrasonication time
CO
c

ro
co
H
H
C * Solar light driven (AM 1.5G, 100 mW cm-2, 25°C) H2 production with bulk and 1 , 10 and 30 min ultra-sonicated NCNCNX (0.5 mg) and NiP (10 nmol) in
H
m aqueous potassium phosphate buffer, KPi, (0.1 M, pH 4.5, 3 ml_) containing 4-methyl benzyl alcohol, 4-MBA, (30 pmol). The suspension was assembled in a c
i photoreactor with a total volume of 7.74 ml_.
m
m
H
¾
C
I- m
IΌ
05
Ultrasonic acoustic cavitation can lead to a dramatic increase in the temperature of ultrasonicated suspension (see Han et al.) so the effect of sonication temperature on the photocatalytic performance of NCNCNX was investigated. In this case, the temperature of dispersion after 10 min ultrasonication was 40°C. Another dispersion was kept at 25°C using a water circulator throughout 10 min ultrasonication. As the difference between ultrasonication at 40°C and 25°C was insignificant, the water circulator setup was not used in later experiments.
There was no significant enhancement in photocatalytic performance when ultrasonication was carried out at 0°C. This suggests that there is a temperature threshold for cavitation, i.e. ultrasonic acoustic cavitation can only happen within a definite temperature range, which has been observed for water cavitation (Niemczewski). Bath sonication (without the mechanical shearing of a tip) at 40°C was applied for comparison, and this led to no enhancement in the photocatalytic activity of NCNCNX, because it generates cavitation of a relatively low concentration and thus smaller magnitude of mechanical force, due to its relatively low frequency (Jiang et ai). This indicates that the enhancement in photocatalytic activity is not thermally induced. Table 2 contains the data from these experiments.
A range of solvents have been reported as the optimum dispersion media for ultrasonication of NCNCNX (Yang et at. and Hong et ai). Accordingly, the photocatalytic performances of NCNCNX dispersions in KPi, isopropanol (iPrOH), methanol (MeOH) and water were tested (see Figure 6). There is no significant difference in the photocatalytic activity of NCNCNX dispersions in different solvents, thus KPi buffer was chosen, as it provides a stable pH at which the optimal NiP activity is achieved (Kasap et ai).
Table 2 - Changes in H2 production with changes in sonication temperature

CO ‘Standard sonication bath.
c
ro
co * Solar light driven (AM 1.5G, 100 mW cnr2, 25°C) H2 production with 10 min ultra-sonicated NCNCNX (0.5 mg) at 0°C, 25°C, 40°C and NiP (10 nmol)
H
H in KPi (0.1 M, pH 4.5, 3 ml_) containing 4-MBA, (30 pmol). Control experiment for 10 min sonication of NCNCNX using standard sonication bath at 40°C is also
C
H shown.
m
c
i
m Table 3 - Changes in H2 Production with Changes in Solvent
m
H
¾
C
I- m
IΌ
s>

Solar light driven (AM 1.5G, 100 mW cm 2, 25°C) H2 production using 10 min ultra-sonicated NCNCNX (0.5 mg) in KPi, isopropanol, methanol or water with NiP (10 nmol) in KPi (0.1 M, pH 4.5, 3 ml_) containing 4-MBA, (30 pmol). Control experiment with different sonication concentration is also presented.
Further experiments were also conducted to compare EDTA with KPi as reaction solutions for the photosynthesis reaction, using both bulk and sonicated NCNCNX. The results are shown in Table 4. The NCNCNX photocatalyst is useable in different solvent systems, and the sonicated sample is superior to the bulk sample in each of the different solvent systems.
In some studies, a relatively large loading of NCNCNX was used in photocatalyst experiments and ultrasonication of dispersions at a higher concentration was therefore needed. The effect of dispersion concentration on photocatalytic performance was studied. As shown in Table 5, despite a 5-fold increase of dispersion concentration from 1 to 5 mg mL 1, the H2 production was barely affected.
Table 4 - Changes in H2 production with Changes in Solvent

CO
c * Solar light driven (AM 1.5G, 100 mW cm 2, 25°C) H2 production with bulk and 10 min ultra-sonicated NCNCNX (0.5 mg) and NiP (10 nmol) in KPi, (0.1 ro
co M, pH 4.5, 3 ml_) containing 4-MBA, (30 pmol) or in aqueous ethylenediamine tetraacetic acid (EDTA) (0.1 M, pH 4.5, 3 ml_) solution.
H
H
C
H
m
c Table 5 - Changes in H2 production with Changes in Sonication Concentration
i
m
m
H
¾
C
I- m
IΌ
05

* Photocatalytic hydrogen production with NCNCNX (0.5 mg) dispersed at an initial concentration of 5 mg/mL and 1 mg/mL, NiP (10 nmol), 4-MBA (30 pmol) in KPi (0.1 M, pH 4.5, 3 ml_) under 1 sun irradiation (AM 1 .5G) at 25°C.
The photocatalytic reaction conditions were further optimised systematically for specific activity with respect to carbon nitride, and high TOFNIP and high TONNIP with respect to NiP, as well as the yield of H2 production and alcohol conversion to oxidation product (determined after 6 h of irradiation).
The loading of NiP catalyst was varied at various NCNCNX loadings, 0.5 mg and 5 mg, with 4-MBA (30 pmol), and the hydrogen evolution was monitored for the first 6 hours. As shown in Table 6, the total H2 evolution and percentage of conversion of 4-MBA to the oxidation product 4-MBAd raised significantly as the amount of NiP catalyst increased from 10 nmol to 100 nmol, where >99% selective alcohol conversion was also observed. Although the production of H2 and 4-MBAd starts to be limited by the availability of 4-MBA electron donors at NiP loadings above 100 nmol, the first-hour rate of H2 production kept climbing. The maximum TOFNIP, 254.16 ± 18.97 mol H2 (mol NiP)-1 h 1, and maximum TONNIP, 700.91 ± 20.24 mol H2 (mol NiP) 1, are obtained when 30 nmol NiP and 5 mg NCNCNX were added to the system (Table 6). The TOFNIP is higher than many previously reported NiP systems with carbon-based photosensitiser, and it is comparable to the reported N-doped carbon dot system (Hutton et al. Chem. Soc. Rev. 2017). Both TOFNIP and TONNIP are in good agreement with previously reported NCNCNx-NiP and the RuP-NiP systems (Kasap et a! , Gross et a!.).
Similar optimisation was carried out at NCNCNX loading of 0.5 mg. Under these conditions, higher material specific activities were obtained. It is clearly shown that the specific activity increases dramatically as the amount of NiP increases at both NCNCNX loadings. This growth of specific activity does not reach saturation until NiP loading reaches 400 nmol when 0.5 mg NCNCNX is in the system.
Table 6 shows the changes in hydrogen productions with changes in the relative and absolute amounts of the NCNCNX photocatalyst and the NiP co-catalyst.
Table 6 - Changes in H2 production with changes in NCNCNX Loading
O
O ©
C/I
co
c
ro
co
H
H
C
H
m

co
i
m * Solar light driven (AM 1.5G, 100 mW cnr2, 25°C) H2 production with 10 min ultra-sonicated NCNCNX and NiP at various loadings in KPi (0.1 M, pH m
H 4.5, 3 mL) containing 4-MBA, (30 pmol).
¾
C
I- m
IΌ
s>
n
H
o o o
Table 7 - Changes in H2 production with changes in NCNCNX Loading
CO
c
ro
co
H
H
C
H
m
c
i
m
m

H
¾
C * Solar light driven (AM 1.5G, 100 mW cm-2, 25°C) simultaneous H2 and 4-MBA oxidation to 4-methyl benzaldehyde (4-MBAd) with 10 min ultra-
I- m sonicated NCNCNX and NiP at various loadings in KPi (0.1 M, pH 4.5, 3 mL) containing 4-MBA, (30 pmol).
IΌ
05
To date, the highest activity of carbon nitride photocatalytic system, from urea-derived CNX with Pt catalyst and TEOA as sacrificial electron donor, was reported to be approx.
20 mmol H2 g_1 lr1 (Martin et ai). In contrast, a specific activity of 39.310 ±
0.263 mmol H2 g 1 lr1 was achieved in the designed system (with 400 nmol NiP and 0.5 mg NCNCNX) in this work.
At the loading of 300 nmol NiP and 0.5 mg NCNCNX, a 3-fold enhancement of first-hour H2 production rate was observed for ultrasonicated NCNCNx, compared to the bulk material (see Table 7 above).
The control experiment in absence of NiP showed no H2 production, indicating the catalyst is an essential component to the photocatalytic system (see Table 7). Degradation of NiP under UV/Vis solar irradiation (A < 400 nm) has been reported (Matindale et ai), and the inferred degradation product is Ni(ll), which can act as H2-evoluting catalyst.
Therefore, a control experiment without NiP but in presence of NiCI2 was performed, and it yielded small amounts of H2 and 4-MBAd over 6 h (see Table 7).
Another experiment used FeCh in place of NiP to rule out the possibility of Fe deposition from the homogeniser tip during ultrasonication, however there was no measurable H2 evolution (see Table 7). Hence, the improvement in photocatalytic performance must be attributed to improved activity of NCNCNX rather than the catalyst. In all three control experiments, even the one with no metal at all, some 4-MBAd production was observed.
This means that NCNCNX can still generate excited electron-hole pairs and drive the 4-MBA oxidation, even in absence of any catalyst.
The kinetics of electron transfer processes in the NCNCNx-NiP photosystem were extensively studied for the bulk material using transient absorption spectroscopy (TAS), and the electron transfer to NiP was identified as the rate-limiting step (Kasap et ai).
The bulk NCNCNX was exploited for time-delayed‘dark’ photocatalysis (Kasap et air, Lau et al. Angew. Chemie Int. Ed. 2017). The colour of NCNCNX changes from yellow to blue upon irradiation in presence of a suitable electron donor such as 4-MBA. This blue state has a lifetime of up to hours and is representative of trapping of long-lived electrons in NCNCNX. Upon addition of an electron acceptor such as the H2-evoluting catalyst NiP in dark, a decay of this blue colour as well as H2 evolution was observed, suggesting the photogenerated electrons are transferred from NCNCNX to the NiP and reductively produce H2 in absence of light. This means that NCNCNX can efficiently decouple photon reduction and 4-MBA oxidation, allowing these two processes to be studied independently.
This property of NCNCNX was utilised to investigate the mechanistic changes of the photosystem upon ultrasonication of the materials. The data collected in the experiments described below is shown in Table 8. As NCNCNX is irradiated for a longer time, the more electrons are stored in the heptazine framework, the more H2 can be produced until saturation is reached. As shown in Table 8 H2 yield increases from 2.0 pmol to 2.5 pmol as the irradiation length increases from 2 hours to 4 hours; however, no further rise in H2 yield was observed when irradiation increased to 18 hours, suggesting saturation is reached.
When the photosystem was irradiated for only 30 minutes before addition of NiP, 60% more H2 was yielded from the ultrasonicated NCNCNX than from the bulk material, suggesting that there are more photoexcited electrons stored in ultrasonicated NCNCNX and the reductive quenching of the photogenerated holes in the ultrasonicated material by 4-MBA is faster than in bulk. The initial H2 evolution rate of ultrasonicated NCNCNX was approx. 33% faster than bulk although the total amounts of electrons trapped in them are the same. This indicates that the rate-limiting electron transfer from NCNCNX to NiP was accelerated, which led to improved photocatalytic performance.
Table 8 - Dark H2 production
CO
c
ro
co
H
H
C
H
m
c
i
m
m
H
¾
C
I- m
IΌ
05

* Solar light driven (AM 1.5G, 100 mW cm-2, 25°C) dark H2 production with bulk and 10 min ultrasonicated NCNCNX (5 mg) in KPi solution (0.1 M, pH 4.5, 3 ml_) containing 4-MBA (30 pmol) in the absence of NiP. The samples were irradiated under 1 sun (100 mW cm 2, AM1.5G, 25°C) for (a) 30 min, (b) 2 h and (c) 4 h, moved to dark and NiP (in KPi solution) was injected under anaerobic conditions.
Photocatalysis - a-Cellulose
Biomass conversion is both an affordable and sustainable approach to produce hydrogen.
So far, only systems comprising of noble and toxic metals as light harvesters and catalysts have been reported for photocatalytic reforming of biomass, and most of these have involved pre-treatment of the biomass under alkaline conditions (Young Park et a! , Roy et ai).
Described below is the use of carbon nitride photocatalyst for hydrogen evolution from lignocellulosic biomass under mild conditions with no pre-treatment. The carbon nitride photocatalyst was used together with a co-catalyst for photocatalytic reforming of biomass over a wide spectrum of pH values.
The loadings of biomass, carbon nitride and NiP were optimised with respect to hydrogen production. Cellulose is the main component of wood-derived biomass (Sun et ai), so it was used in all initial optimisations. a-Cellulose, the most unreactive form of cellulose also with the highest degree of polymerisation (DP = 1450) (Sweet et ai), was chosen. In all initial optimisations, the ultrasonicated NCNCNx-NiP photosystem (3 ml_, 0.1 M KPi, pH 4.5) was used based on the merit of its high specific activity.
The data in Table 9 shows photocatalytic hydrogen evolution at different loadings of ocellulose. Since NCNCNX is a heterogeneous photosensitiser and ocellulose is a nearly insoluble electron donor, stirring becomes critical for photocatalytic hydrogen evolution in this system. Increasing the ocellulose loading scarifies stirring efficiency while lower ocellulose amount reduces electron donor availability; thus, 100 mg is the optimum loading for a balance between providing effective stirring and sufficient electron donor. Table 9 shows that the H2 evolution at different loadings of NCNCNX and NiP. The system with 0.5 mg NCNCNX and 50 nmol NiP gave a high specific activity of 1694.87 ± 104.89 pmol H2
(g NCNCNX) 1 hr1, whereas the highest H2 production over 4 h was observed with 5 mg NCNCNX and 50 nmol NiP. The latter (5 mg ultrasonicated NCNCNX, 50 nmol NiP and 100 mg lignocellulosic substrate) was used in the following photocatalytic experiments, unless specified otherwise.
Table 9 - Changes in H2 Production with Change in CNX and Substrate Loading
CO
c
ro
co
H
H
C
H
m

c
i
m
m * Solar light driven (AM 1.5G, 100 mW cm-2, 25°C) H2 production with bulk H2NCNX, bulk and 10 min ultra-sonicated NCNCNX and NiP at different
H
concentrations in KPi (0.1 M, pH 4.5, 3 mL) containing ocellulose.
¾
C
I- m
IΌ
s>
Moreover, the photocatalytic reforming of cellulose was studied using previously reported H2NCNx-NiP and bulk NCNCNx-NiP photosystems (Caputo ef a/.; Martindale et al. Angew. Chemie Int. Ed. 2017). As the data in Table 9 shows, the H2NCNX system produced a trace amount of H2 after 4 h of irradiation while bulk and ultrasonicated NCNCNX systems respectively yielded approximately 20 or 30 times more. This enhancement in photocatalytic performance is attributed to improved charge separation as well as efficient substrate oxidation brought by the insertion of the cyanamide groups (Zhang ef a/.; Lau et al. Nat. Commun.). Different H2-evolving catalysts were investigated for photocatalytic reforming of ocellulose. Pt, the benchmark H2-evolving catalyst, as well as M0S2 and the molecular catalyst NiP were used, and the results are presented in Table 10. NiP shows the best activity amongst all for first 24 h, demonstrating the first example of molecular catalyst-based biomass reformation system. After 24 hours of irradiation, the activity of NiP terminated due to UV-induced degradation caused by ligand displacement and ligand oxidation caused by photogenerated holes (Martindale et al. Angew. Chemie 2016). NCNCNX can be easily platinised by in situ photodeposition of H2PtCl6 (Lau et al. Nat. Commun. 2016), which accounts for the relatively slow H2-evolving rate of the first day. Similarly, M0S2 can be generated by in situ
photodeposition of [MoS4]2- precursor (Nguyen et al.). Both the NCNCNX-MOS2 and the NCNCNX-Pt photosystems outperform the NCNCNx-NiP system over long term due to their robustness.
Table 10 - Change in H2 Production with Change in Catalyst

* Solar light driven (AM 1.5G, 100 mW cnr2, 25°C) H2 production with 10 min ultra-sonicated NCNCNx (5 mg) and NiP (50 nmol), HSRKOIS (4 wt.%) or H8N2M0S4 (4 wt.%) in the presence of ocellulose (100 mg) in KPi (0.1 M, pH 4.5, 3 mL) over 6 days.
Photocatalysis - Lignocellulose
Lignocellulose is the most abundant constituent of biomass (Azadi et al.), and it is composed of cellulose, hemicellulose and lignin. Cellulose is in the form of microfibrils, which are coated with hemicellulose to form an open network whose empty spaces are filled up with lignin (Rubin et al.). Cellulose, the major component, is a polymeric chain of glucose units mainly linked by b (1 4) glycosidic bonds (van Wyk et al.). Cellobiose and glucose are respectively the disaccharide and monosaccharide units of cellulose. Hemicellulose is a branched polysaccharide of various pentose and hexose sugars. In this project, xylan, a type of hemicellulose that accounts for up to a third of carbohydrate content in many lignocellulosic materials (Hinman et al.), and consist of xylose units, was used as the representative hemicellulose.
Lignocellulose components (ocellulose, xylan and lignin) as well as their subunits were used as electron donors for photocatalytic H2 evolution, and the data are presented in Table 1 1. As expected, the photocatalytic reforming yields of the subunits are much higher than those of ocellulose, hemicellulose and lignin.
Cellobiose, the only disaccharide substrate, showed lower PR yield than the
monosaccharides, which is consistent with previous reports.35 3.74 ± 0.12 pmol and 4.92 ± 0.17 pmol H2 were produced from ocellulose and xylan respectively over 24 h. Lignin is strongly light-absorbing and its UV-Vis spectrum shows a broad peak at 300 nm and a shoulder at 350 nm (Wakerley et ai), the region over which the photosensitiser NCNCNX absorbs most strongly. Therefore, due to this competition for light absorption in the UV-vis region, no measurable amount of H2 was produced from this NCNCNx-NiP photosystem with lignin as an electron donor at 100 mg loading. However, changing lignin loading to 0.5 mg resulted in H2 production of 0.42 ± 0.03 pmol over 24 h, due to reduced concentration and light absorption.
Some of these experiments were repeated using H2PtCI2 in place of NiP. The results are shown in Table 12 in relation to the use of ocellulose, xylan and lignin as substrates. These results are reported in an aqueous mixture at 10 M KOH, after a basic pre-treatment of the substrate.
Table 1 1 - H2 Production with NCNCNX and Changes in Substrate
CO
c
ro
co
H
H
C
H
m
c
i
m
m

H *0.5 mg lignin is used.
¾
C
I- m
I Solar light driven (AM 1.5G, 100 mW cm-2, 25°C) H2 production using 10 min ultra-sonicated NCNCNX (5 mg) and NiP (50 nmol) in the presence of sΌ> lignocellulosic substrates (100 mg) in KPi (0.1 M, pH 4.5, 3 ml_).
- w— » ro
CO
_Q
3
C
c
CO
CD
O
c
CO
_c
O
"O c
CO
O -— »
CL
CM
X
5
C
g

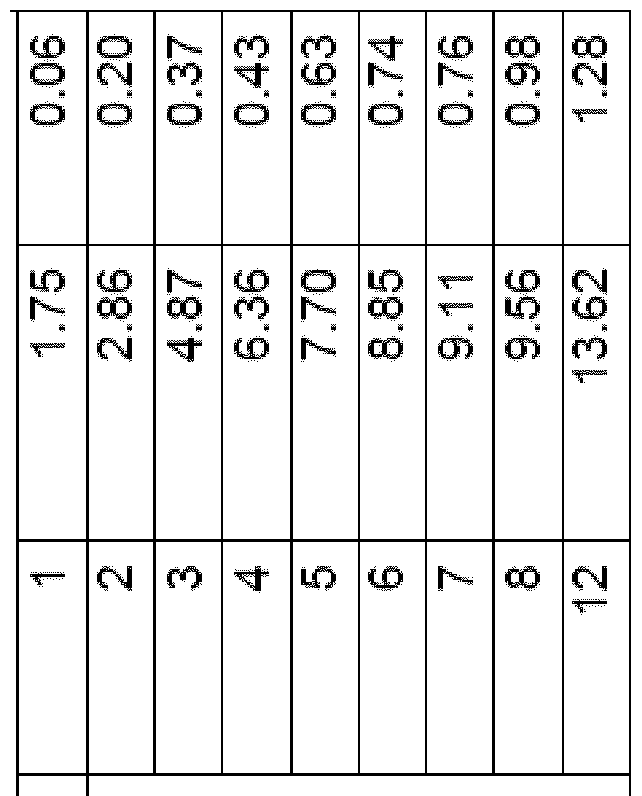


The photocatalytic reforming of raw biomass was also investigated. Three natural lignocellulosic materials (bagasse, wooden branch and sawdust) and two waste materials (waste paper and cardboard) were used as electron donors for photocatalytic H2 evolution and the 24-hour H2 production yields are shown in Table 13. Lignin is the most challenging component of lignocellulose for photoreforming, and paper, wood and bagasse have lignin content of <15%, 18-35% and 20%, respectively (Sun et a! , Lavarack et ai). However, the difference in lignin content does not dictate the trend in H2 production in these
photosystems. Sawdust is the only powder and it stirs the best with the heterogeneous photosensitiser, so it achieved the highest PR yield. Cardboard, turned into small crumbs after grinding, is the second best, while the remaining samples, paper, bagasse and wooden branch, remain as large pieces after grinding. Therefore, the bulkiness of solid biomass has a more significant impact than the lignin content on photocatalytic H2 evolution in the case of this NCNCNx-NiP photosystem.
Table 13 - H2 Production with Change in Substrate
O
C/I
CO
c
ro
co
H

H
C
H
m * Solar light driven (AM 1.5G, 100 mW cnr2, 25°C) H2 production using 10 min ultra-sonicated NCNCNX (5 mg) and NiP (50 nmol) in the presence of
c Ux i raw biomass substrates (100 mg) in KPi (0.1 M, pH 4.5, 3 ml_).
m
m
H
¾
C
I- m
I 0Ό5
n
H
t¾ o o o
Photocatalysis - Pre-Treatment
Since the conversion of lignocellulose materials is often hindered by the crystallinity of cellulose, a variety of physical, physio-chemical, chemical and biological pre-treatment methods, e.g. pyrolysis, gas explosion, ozonolysis and use of cellulase, have been studied to make cellulose accessible for hydrolysis to improve its solubility (Sun et air, Kumar et ai). In this project, a combination of hydrolysis at extreme pH ranges and irradiation was used as the pre-treatment (Chosdu et al.). The cellulose pre-treatment was explored over a broad range of pH. Pt was chosen instead of NiP because the photocatalytic performance of NiP is largely impaired at extreme pH (Caputo et ai). In addition, using the benchmark Pt H2-evoluting catalyst can provide a direct comparison between this NCNCNX-Pt photosystem and many previously reported systems (Kuehnel et ai). In the following experiments, ocellulose was suspended in different solvents and stirred for 24 h under 1 sun irradiation (AM 1 5G) at 25°C before the addition of H2PtCl6 and NCNCNX.
Table 14 illustrates the photocatalytic H2 evolution performance of the NCNCNX-Pt
photosystem with ocellulose pre-treated at different pH values. The system with no pre-treatment is shown by the orange line for comparison. Pre-treatment at pH 15 (with 10 M KOH) gives the highest H2-evoluting activity. The total H2 production yield over 6 days is 39.50 ± 1.11 pmol, which is twice as that of pre-treatment with 5 M KOH and is 4 to 5 times higher than that of systems in more acidic conditions. This is consistent with previous studies that showed cellulosic material is more easily solubilised in more alkaline conditions
(Budtova et ai).
Table 14 - Changes in H2 Production with Changes in Pre-Treatment
O
O
S©
C/I
co
c
ro
co
H
H
C
H m
co
i
m
m
H
¾
C
I- m
I 0Ό
n H t¾ o o o

O
o
'Jl
'Jl
co
c
ro
co
H
H
C
H
m
co
1
m
m
H
¾
C
I- m
IΌ
s>

tPre-treatment v/ith 10 M KOH for 24 h, followed by neutralization to pH 7 O
H
W
o
* Solar light driven (AM 1.5G, 100 mW cnr2, 25°C) H2 production using 10 min ultra-sonicated NCNCNX (5 mg) with hhPtCk (4 wt.%) and a-cellulose
(100 mg) in 1 , 5, 10 M KOH, pH 2 H2SO4 or pH 4.5 KPi solution (0.1 M, pH 4.5, 3 ml_). All the samples were pre-treated prior to injection of NCNCNX and o H2PtCI2.
The conversion yield determined with different ocellulose loadings in the presence of NCNCNx indicates that 22% conversion is reached after 6 days of irradiation in 10 M KOH solution (see Table 15). This is more than twice (9.7%) the yield reported previously with a CdS/CdOx photocatalyst system (Wakerley et a!.).
Table 15 - Changes in H2 Production with Changes in Pre-Treatment

* Photoreactors were prepared using 10 min ultra-sonicated NCNCNX (5 mg) with H2PtCl2 (4 wt %) and cellulose (1.65 or 0.81 mg) in 10 M KOH. The samples were pre-treated in 10 M KOH prior to injection of NCNCNX and H2PtCl2. Percentage conversion yields were calculated after irradiating (AM 1.5G, 100 mW cm 2, 25°C) the samples for 6 days.
[a] Moles of anhydroglucose monomer units are calculated by dividing the total amount of o cellulose used with the molecular mass of an anhydroglucose repeating unit (162.14 g mol 1)
[b] The theoretical maximum H2 is calculated based on the assumption that maximum 12 equivalent of H2 can be produced per anhydroglucose unit in ocellulose structure.
Unlike many reported systems that can only photocatalytically reform cellulose in extremely alkaline conditions (Wakerley ef a/.; Kawai et al.), this NCNCNX-Pt photosystem can operate over a wide range of pH with reasonable yield. This can be attributed to the stability of NCNCNX as well as facilitated electron transfer to Pt (Kasap et air, Lau et al. Nat. Commun.
2016).
Photocatalysis - Polymer Substrates
NCNCNX (5 mg) was used together with NiP (50 nmol) to produce hydrogen from a range of polymer substrates (100 mg) in KPi solution (0.1 M, pH 4.5, 3 ml_). The substrates included PET (polyethylene terephthalate), PUR (polyurethane) and PS (polystyrene). The reaction was irradiated under 1 sun for 24 hours (100 mW cm-2, AM1.5G, 25°C).
The results are shown in Figure 10.
Carbon Dots
Synthesis of CDs
CDs were prepared as previously described by Martindale et at. Angew. Chemie Int. Ed. 2017, or as described below, from biomass organic precursors.
Pyrolysis of organic precursors under air to form CDs was optimized for high photocatalytic activity by varying the calcination temperatures and times (data for thermogravimetric analysis, TGA, and photocatalytic screening not shown). All organic precursors were calcinated in muffle Carbolite furnaces for 4 days under air, at optimum temperatures, to obtain CDs with good photocatalytic properties.
Namely, ocellulose and cotton wool were treated thermally at 320°C, xylan at 250°C, lignin at 300°C, cotton pads and T-shirt at 340°C, G. Nivalis, G. Elliptica and T. Baccata at 230°C, Elaeagnus X ebbingei at 275°C and olive leaves at 250°C. The samples after carbonization, were used in their crude form for all types of characterization, without any further treatment.
Cotton contains mainly cellulose (< 93-97%) with the remaining part consisting mostly of hemicellulose (< 7%) (Chen; Zhu et al.). It was therefore an excellent carbon source for this purpose, and cotton-derived CDs were prepared from four-day pyrolysis of commercial cotton wool, pads and recyclable cotton fabric (T-shirt) at 320, 340 and 340°C, respectively.
CDs from cotton resources have been prepared previously for ion detection, imaging, patterning and sensing applications (Wen et ai.\ Wang et al. and Alves et al.), either by hydrothermal or strong-acid mediated approaches. However, the synthesis of such materials with great potential in photocatalysis, via facile and simple calcination approaches, is unprecedented.
To demonstrate the potential of various biomass sources for CDs synthesis, inedible plant waste was also used as precursors. The species selected are in great abundance in various climates, have no special growth requirements, and can be obtained at no cost when the plants complete their life-cycle and/or from tree trimming. More specifically, pruning of the olive trees worldwide, results in 25 kg of waste/tree annually (Herrero et al., Peralbo-Molina et al., Abaza et a!.).
The optimal conditions for CD synthesis from crude biomass depends on the relative content of the samples in ocellulose, xylan and lignin as well as their morphology (George et al., Zhang et al.). The TGA traces of the plants show three main bands: xylan degrades first at 210-300°C, followed by ocellulose (300-350°C), and lignin as a shoulder at higher temperatures (380-430°C) (Carrier et al., Biagini et al., Barneto et al.). Olive leaves show additional bands between 460 and 490°C, possibly due to the presence of other polyphenols in the sample (Abaza et al.).
Deconvolution of the xylan, cellulose and lignin bands in the TGA plots allowed to determine the relative fractional areas (RFAs) for each component (cellulose, lignin and xylan;
(Table 16). The calcination temperature of the sample was optimized below the
decomposition temperature of the dominating component, and CDs with the best
photocatalytic activity were obtained upon the following heat treatment; 230°C for G. Nivalis, G. Elliptica and T. Baccata, 275°C for Elaeagnus X. ebbingei and 290°C for olive leaves. Calcination resulted in color change to black and shrinkage for all leaves, but the overall macroscopic morphology remained unchanged (images not shown).
Table 16 - Decomposition Temperatures and Relative Fractional Areas (RFAs) of xylan, ocellulose and lignin in crude biomass samples as determined by TGA
Precursor Xylan Cellulose Lignin
T max (°C) RFAx 1 T max (°C) RFAc 1 T max (°C) RFA,1
G. Nivalis 209.6 0.21 317.7 0.55 434.0 0.23 Cyprus Olive Tree 268.1 0.30 332.4 0.43 402.8 0.27 G. Elliptica 240.4 0.34 303.0 0.31 412.4 0.35 T. Baccata 233.7 0.41 308.5 0.14 381.3 0.45
ElaeagnusX. ebbingei 299.3 0.56 344.4 0.14 413.5 0.30
1 Relative Fractional Area; Ratio of the area of a single component divided by the overall areas of all components; RFAX= A x/ Ax+ Ac+ Ai; RFAC= Ac/ Ax+ Ac+ Ai; RFAF AI/ Ax+ Ac+ AI (X: xylan, c: «-cellulose, I: lignin).
Analysis and Characterisation of CDs
All CDs were characterized by X-ray photoelectron spectroscopy (XPS) upon drop casting the CD aqueous solutions on clean fluorine-doped indium tin oxide (FTO)-coated glass slides, at RT, under atmospheric pressure. The spectra were collected at NEXUS
(Newcastle University, UK). Analysis was performed using a K-Alpha (Thermo Scientific, East Grinstead, UK) spectrometer utilizing a monochromatic AI Ka X-ray source (1486.6 eV, 400 pm spot size, 36 W). High resolution (HR) spectra were collected at a pass energy of 40 eV with 10 sweeps. Measurements were taken at 3 points on each sample surface. The data were processed using CasaXPS.
Fourier Transform Infrared (ATR-FTIR) spectra were recorded on a ThermoScientific Nicolet iS50 FTIR spectrometer, in the range of 400 to 4000 cm 1 and analyzed using the Omnic software. Raman measurements were carried out on a Horiba LabRAM HR Evolution microscope using a 473 nm laser.
1H and 13C NMR spectra for all CD samples were measured using a 400 MHz Bruker NMR spectrometer in D2O.
The optical properties of the CDs were investigated with a Varian Cary 50 UV/vis
spectrophotometer, in the wavelength range between 250 and 850 nm (in water), using quartz cuvettes with 1 cm pathlength. The X-ray diffraction (XRD) patterns for all materials were recorded with a PANalytical BV X’Pert PRO, at a scan rate of 0.9 min-1.
The Transmission electron microscopy (TEM) images of the carbonaceous materials were obtained on a JEOL 200-CX instrument operating at 120 kV. For this purpose, a droplet from the aqueous solutions of the samples, was placed on a carbon-coated holey grid and dried under atmospheric pressure.
Thermogravimetric analysis (TGA) measurements, were performed with a Mettler Toledo TGA analyzer, under a nitrogen atmosphere, at a scan rate of 10°C/min and in the temperature range from 50 to 800°C.
XPS confirmed that the carbonaceous photoabsorbers synthesized from pure biomass components contain predominantly C and O species. In particular, the high-resolution (HR)
C 1s XPS spectrum of a-cellulose CDs displays peaks at 284.8 eV, 286.3 and 288.5 eV (Figure 3(a), top), which correspond to C=C, C-0 (alkoxy and epoxy) (Yang et al .; Qin et al.) and C=0 groups, respectively (Martindale et al. Angew. Chem. Int. Ed. 2017; Oza et al., Young Park et al., Roy et al.). This suggests nanoparticles with a graphitic sp2 core structure, decorated with alkoxy, epoxy, and carboxyl functionalities. Carboxylates (531.
8 eV, 89.5 %) are the predominant surface functionalities, which coexist with C-0 groups (534.1 eV, 10.5 %) but to a less extend, as indicated by their relative area ratio in the HR O 1s spectrum (data not shown).
Xylan and lignin-derived carbonaceous materials show a significantly higher surface coverage by alkoxy/epoxy groups (532.6-532.7 eV, 63-65%) compared to carboxylates (531.3 eV, 29-37%). (Qin et al., Yang et al., Qu et al.).
Elemental analysis showed that CDs produced from real biomass samples contained significant amounts of N, with C/N ratios varying between 5.5 to 11.1. XPS analysis of CDs from G. Nivalis, T. Baccata and Elaeagnus X. Ebbingei, showed mainly graphitic
N-containing CDs with a pyrrolic (N-5, 399.3 eV) and graphitic quaternary N (N-Q, 400.2 eV) content (Zhang et al., Li et al.) (Figure 3(a), bottom). G. £///pf/ca-derived CDs displayed pyridinic (N-6, 398.7 eV) functionalities in addition to graphitic N-Q. The C 1s and O 1s environments are similar for CDs from pure and real biomass precursors. All samples show bands assigned to C=C (284.7-284.8 eV), C-0 (Gupta et al.) and C=0 groups (287.9-288.5 eV). The CDs are surface-covered by C=0 groups (531.3-531.7 eV) and C-0 (532.8- 533eV) moieties, which provide good water solubility to the nanoparticles. The carboxylate content is 67, 64, 61 and 46% for G. Elliptica, G. Nivalis, T. Baccata and for Elaeagnus X. ebbingei, respectively.
FTIR spectroscopy (data not shown) of the CDs confirms the coexistence of C=0, C-OH and O-H functionalities at the CD surface (u = 1319, 1380, 1560 and 3000-3500 cm-1) with skeletal vibrations from the graphitic core tentatively assigned at 161 1 cm-1 (Martindale et al. J. Am. Chem. Soc. 2015; Martindale et al. Angew. Chem. Int. Ed. 2017; Roy et al.; Mondal et al.; Yang et al.).
TEM images show that CDs synthesized from ocellulose and G. Nivalis are highly graphitic with an average diameter of 9.4 and 7 nm, respectively (Figure 3(b)). The lattice fringes allow for the assignments of the (100) intralayer spacings (3.0 A for ocellulose and 2.7 A for G. Nivalis CDs), which is larger than for graphite (2.4 A) (Martindale et al. Angew. Chem. Int. Ed. 2017; Qu et a! , Qu et al.). CDs derived from G. Elliptica and lignin are relatively large colloids with a diameter of 96 and 115 nm, respectively (data not shown).
Raman spectroscopy supports highly graphitic nanostructures for ocellulose and
G. A//Va//s-derived CDs. Specifically, the D (1579.3 cm-1, defective sp2 carbon) to G
(1320.7 cm-1, intact sp2 carbon) band intensity ratio (ID/IG) for both ocellulose and G. Nivalis CDs is about 0.18 (Figure 3(c)) (Ferrari; Pimenta et al.). This value is far lower than what reported previously for other graphitic CD systems (> 0.75) (Martindale et al. Angew. Chem. Int. Ed. 2017; Qin et a /.; Roy et al.] Wang et al.). The ID/IG ratios of our biomass-derived CDs suggests low-defect crystalline structures (Yang et al.] Yang et al.). T. Baccata and
Elaeagnus X. ebbingei displays ID/IG ratios of 0.35 and 0.56, respectively, indicating more defective graphitic structures. The Raman results are in good agreement with XPS analysis, which shows sp3/sp2 C ratios of 0.3 and 0.5 for ocellulose and G. A//Va//s-derived CDs, respectively, and sp3/sp2 C > 0.6 for the other CD samples.
NMR studies confirm predominant sp2 (C=C, C=N) core environments for the CDs from ocellulose and G. Nivalis (data not shown) with peaks between 120-140 ppm and 7-8 ppm in 13C and 1H NMR spectra, respectively (Martindale et al. Angew. Chem. Int. Ed. 2017; Gao et al.] Yu et al.). 13C NMR spectra further show peaks in the 175 to 180 ppm range for surface carboxylates, whereas the absence of signals between 8 and 80 ppm, suggests limited sp3 character for these CDs, in agreement with the Raman measurements. The other CDs exhibit significantly higher sp3 character than ocellulose and G. Nivalis CDs.
XRD patterns of CDs indicate variations in the (002) interlayer spacing of the graphitic structures. oCellulose CDs show an interlayer spacing of 3.8 A (23° 2Q), which deviates from that of pure graphite (3.3 A), and suggest material with turbostratic disorder (data not shown) (Bourlinos et al.] Zhang et al.] Mewada et al.).
CDs from G. Nivalis show an interlayer spacing closer to that of graphite (3.5 A; 25.3° 2Q), which is similar to that obtained for the other CDs produce from real biomass (data not shown).
UV-vis absorption spectra (Figure 3(d)) of all CDs show a predominant absorption in the UV region with a featureless tail extending into the visible spectrum (Baker et ai). These properties are assigned to p-p* (C=C, core) and h-p* (C=0, surface) transitions of the carbonaceous structures (Yang et ai). Increased graphitic character results in enhanced absorption, as indicated by the stronger absorption profiles of ocellulose and G. Nivalis CDs, compared to G. Elliptica, T. Baccata, olive leave and Elaeagnus X. ebbingei-demed CDs. Moreover, CDs from waste biomass, presumably due to N-doping/graphitic N-centres (N-Q), (Figure 3(a), bottom), exhibit a stronger absorption in the visible spectrum compared to ocellulose CDs. It had been shown that graphitic N centers are associated with electron doping that causes a decrease in the magnitude of the electronic gap (Sarkar et ai).
Photocatalytic Experiments
The samples for photocatalytic experiments were prepared in borosilicate glass vials (7.74 ml_), by first dissolving 0.03-2.8 mg of the as calcinated ocellulose CDs and 50 nmol of the hydrogen evolution catalyst (NiP), in aqueous EDTA solutions (0.1 M, 3 ml_, pH 6). Next, the vessels were sealed with rubber septa (Subaseal), purged with N2 containing 2 % CH4 (internal gas chromatography (GC) standard) for about 20 mins, and irradiated using a Newport Oriel Solar Light Simulator (100 mW cm-2) equipped with an Air Mass (AM) 1.5G filter. The samples were kept at 25°C and stirred constantly during irradiation.
Samples of the headspace gas (20 pL) were taken from the photoreactor and analyzed by gas chromatography (GC) as described above.
The photocatalytic performances of the rest CDs synthesized in this study, were evaluated as described above, upon dissolving 2.2 mg of the as calcinated CDs along with NiP (50 nmol) in EDTA solutions (0.1 M). For the investigation of the photocatalytic activities of the CDs in sea, river and sewage water, the aqueous phases were first passed through a 0.20 pm filter, to remove any solids and/or microorganisms prior the addition of EDTA and photocatalytic experiments.
Photocatalytic experiments in purified water in the presence of organic pollutants (200 pmol) and chloride ions (3.2 wt %), were carried out by dissolving 2.2. mg of ocellulose CDs in phosphate solution (KPi, 0.1 M, pH 6, 3 mL), in the absence of EDTA and in the presence of 50 nmol NiP.
Use of CDs
Photocatalytic biomass reformation into H2 was also studied using a carbon-based alternative to carbon nitrides, namely carbon nanodots. Carbon nanodots can be cheaply prepared from abundant organic precursors on a large scale (see the review Hutton et al. Chem. Soc. Rev. 2017.). The use of these known CDs is described below.
Carbon nanodots were also prepared from biomass organic precursors, and the use of these CDs is also described below.
Photocatalysis - Known CDs
Undoped and amorphous (undoped, a-CDs) and nitrogen-doped and graphitic (N-doped, g-CDs) carbon nanodots in combination with NiP as the H2 evolution catalyst are active for photoreforming ocellulose into H2 under benign conditions (0.1 M KPi, pH 6, 3 ml.) (see Table 17).
The CDs used in this work, which include an a-CD (amorphous CD) and a nitrogen-doped CD (g-N-CD; graphitic nitrogen-doped CD), were prepared as described Martindale et al. Angew. Chemie Int. Ed. 2017. The experimental methods for the preparation of these CD photocatalysts are described on pages S1 to S5 of the Supporting Text for this reference.
Additionally, CDs were prepared from ocellulose (ocell CDs) and these were also used as photocatalysts to degrade ocellulose and glucose.
Table 17 - H2 Production with CDs

* Solar light driven (AM 1.5G, 100 mW cnr2, 25°C) H2 production using a variety of CDs and NiP (50 nmol) in the presence of lignocellulosic substrates (100 mg) in KPi (0.1 M, pH 6, 3 ml_).
Photocatalysis - CDs in Pure Water
CDs prepared from purified and waste biomass were used as photoabsorbers to drive H2 evolution. Photocatalytic experiments were carried out for 24 hrs, using NiP (50 nmol) as
the H2 evolution catalysts and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA 0.1 M, pH 6) as the electron donor. All experiments were carried out at as described above. The quantity of CDs was systematically varied between 0.03 and 2.8 mg (Figure 6) during photocatalytic experiments and optimized for maximum H2 yield (in /vmol), from which the specific activities (/vmol H2 (gcDs)-1 h-1) and NiP turnovers (TONNR, mol H2 (mol NiP)-1) were determined. a-Cellulose CDs (2.2 mg), after 24 hrs of irradiation, showed the best H2 yield (15.6 ±
0.73 pmol) and TONNIR (312 ± 14.6) among all photoabsorbers synthesized from purified biomass components (Figure 4(a) and Figure 6). The highest specific activity of
13,450 pmol H2 (gcDs)-1 h-1, was achieved at 0.03 mg a-cellulose CDs (Figure 4(b)), which outperformed previously reported carbonaceous photocatalysts (CDs and carbon nitrides) when combined with noble-metal free catalysts (Table 18) (Matindale et al. J. Am. Chem. Soc. 2015; Hutton et al., Martindale et al. Angew. Chem. Int. Ed. 2017; Caputo et al.).
These systems reach activities only up to 7,950 /vmol H2 (gcDs)-1 h-1 when N-doped CDs are combined with NiP and EDTA.
Table 18 - Photocatalytic performances of noble-metal free carbonaceous photoabsorbers in water using sacrificial electron donors and molecular as well as noble-metal catalysts.
Entry Photoabsorbers Catalyst ED ft tjumol) Activity/ Mmol g-1 h-1 TON™t GE/% Ref.
Noble-metal free carbonaceous
1 a-CD NiP EDTA 0.64 397 64 1 .4 1
2 a-CD NiP TCEP/AA3 10/9 53 1094 n.d. 2
3 g-co NiP EDTA 0.5 1 ,548 45 n.d. 3
4 g-W-CD NiP EDTA 2.8 7,950 277 5.2 3
5 CD (yeast) Pt TEOAb n.d 31 n.d. n.d. 4
6 r CNx NiP EDTA 3.32 437 166 0.4 5
7 N NCNX NiP 4-MBAc 213 311 425 15.2 6
8 sr-CsN4 Pt TEOAb n.d. 20,000 641 26.5 7 aTCEP = tris(carboxyethyl jphosphine; AA = ascorbic acid; bTEOA = triethanolamine; C4-MBA= 4-methylbenzyl alcohol
* NCNCNx is a reference to a bulk sample that has not been ultrasonically treated.
Control experiments in the absence of CDs, NiP and EDTA, resulted in no H2 generation, suggesting that ocellulose CDs themselves are not catalytic for H+ reduction and do not serve as sacrificial electron donors via autooxidation (Figure 7).
Although CDs produced from ocellulose have been reported for use in bioimaging (Shen et ai), this is the first example of CDs from a cellulosic precursor being employed in
photocatalytic applications.
Photocatalytic H2 generation with CDs synthesized from xylan and lignin in sacrificial EDTA solution, after 24 h of irradiation, showed lower activities than with ocellulose CDs. Xylan- and lignin-derived photoabsorbers yielded 4.7 ± 0.34 and 0.5 ± 0.07 pmol H2 after 24 hrs of irradiation (Figure 4(a)) and specific activities of 625 and 87 pmol H2 (gcDs)-1 h-1, respectively (Figure 4(b)). CDs from xylan and lignin are limited and have been previously used only in drug-delivery and bioimaging applications, and thus this is the first demonstration of their use in energy production (Rai ef a/.; Chen ef a/.; Liang et ai).
The distinct photocatalytic activity of CDs produced from different biomass components is presumably associated with the distinct chemical composition, polymeric architecture and microstructure of the biopolymer. a-Cellulose is a linear and crystalline polymer (100,000 g mol-1) made of glucopyranose units, whereas xylan is an amorphous and branched polysaccharide (30,000 g mol-1), made of sugars such as glucose, mannose, xylose and arabinose. Lignin is also amorphous and consists of randomly cross-linked phenolic monomers (p-coumaryl, coniferyl, and sinapyl alcohols) (Lee et ai). Our photocatalytic experiments suggest that pyrolysis of a crystalline homopolymer with regular microstructure might support the formation of high-performance CD photocatalysts.
Cotton is predominantly made from ocellulose with no lignin content (Krasig et ai), and thus cotton wool-derived CDs showed indeed almost identical photocatalytic performance as ocellulose CDs: 11.8 ± 0.29 /vmol H2 are photo-produced after 6 hrs of irradiation
(Figure 4(c)). The photocatalytic performances of CDs derived from cotton pads and a T-shirt, after 6 hrs of irradiation, do not exceed 4.86 ± 0.16 and 1.74 ± 0.17 /vmol H2, respectively (Figure 8). This discrepancy between photocatalytic properties of CDs prepared from ocellulose precursors may be due to additives in the commercial materials (Rivero et ai, Barker et ai).
The photocatalytic performances of CDs obtained from real biomass samples, leaves of G. Nivalis, G. Elliptica, T. Baccata, ElaeagnusX ebbingei and“Cyprus olive tree", were subsequently studied.
Photocatalytic experiments using the leave-derived CDs (Figure 4(c)), after 6 hrs of irradiation, reveal lower H2 evolution activity than with ocellulose CDs (1 1 ± 0.49 /vmol H2, Figure 4(a)), which can be explained by their composite nature (see below).
The best performances were observed for CDs from G. Nivalis and olive leaves, which over 6 hrs of irradiation, were driven the production of 6.15 ± 0.11 and 5.29 ± 0.27 pmol H2 (Figure 5 and Table 19), respectively, which is consistent with their highest RFAs for ocellulose, and lowest for xylan and lignin (data not shown). The H2 evolution activity of CDs prepared from the other leaves, declined as the RFA of ocellulose decreases. In particular, after 6 hrs of irradiation, CDs from G. Elliptica photo-generate 4.47 ± 0.03 pmo\
H2, since the RFA of ocellulose drops to 0.31 , whereas that of xylan and lignin increase to 0.34 and 0.35, respectively (Table 19). The H2 evolution capacities of CDs from T. Baccata and Elaeagnus X. ebbingei are even lower, and do not exceed the 3.50 ± 0.07 and
3.19 ± 0.15 pmol H2, after 6 h of irradiation, respectively. For both samples, the RFA of ocellulose decrease to its minimum (0.14) whereas that for lignin and xylan increase significantly (data not shown). The correlation between the RFAs of the components and the photocatalytic performances of the derived CDs gives therefore a qualitative predictor for photocatalytic efficiency (Table 19).
Table 19 - Experimentally and theoretically-determined H2 evolution capacities of the photocatalytic systems, which use CDs from crude biomass as light-harvesters.
Sample Photocatalytic Activity (pmol Hz}
Experimental1 Theoretical2
G. Nivalis 8.74 ± 0.13 971
Cyprus Olive Tree 7.93 ± 0.97 8.17
G. Elliptica 6.49 ± 0.03 6.53
T. Baccata 4.70 ± 0.13 4.30
BaeagnusX. ebbingei 3.94 ± 0.10 4.90
1 Experimentally-determined H2 evolution capacities (in pmol), as obtained from photocatalytic experiments using CDs from crude biomass
2Theoretically-determined H 2 evolution capacities, calculated by multiplying the RFA for each component (obtained by TGA) with the capacity of the respective pure constituent to produce H2 after 24 hrs of irradiation (ocellulose; 15.6 pmol, xylan; 4.7 pmol and lignin; 0.5 pmol, Figure 4(a)). The derived values for all components were then summed up, to result the overall activity for each plant species.
The results discussed above, suggest that the best photocatalytic performances are associated with CDs of smaller sizes and low-defect graphitic cores, which are bestowed mainly with carboxylates, whereas they contain C-0 functionalities in less extend. N- doping
of the CDs from biomass waste, does not seem to have a significant effect in the
photocatalytic activities.
Photocatalysis - CDs in Untreated Waste Water
A major limitation in photocatalysis is the requirement of (/') pure water to avoid catalyst poisoning at high salinities and/or impurities (Li et al.), and (/'/) an external, costly sacrificial reagent (Pellegrin et al.). Given the global shortage of clean water supplies (Service et al.), and challenges of water purification in many developing countries, we sought to establish the photocatalytic activity of our biomass-derived CDs in sea, river and sewage water, first in the presence and later absence of EDTA (Figure 5).
Simulated solar irradiation of the ocellulose CDs in untreated water for 6 hrs, in the presence of both EDTA and NiP, resulted in the production of 12.57 ± 0.17 and
12.61 ± 0.58 pmol Fh in sea and river water (Figure 5(a)), respectively, which indicates a slight improvement in the photocatalytic performance of the systems in untreated water (14.4 - 14.7%), compared to pure water (11 ± 0.491 /vmol Fh, Figure 4(c)). Flowever, under the same conditions, when sewage water was used as the reaction medium, after 6 hrs of irradiation, only 1.12 ± 0.263 /vmol Fhwere photogenerated, suggesting the intolerance of the NiP catalyst to sewage impurities.
On the other hand, the photocatalytic properties of ocellulose CDs in untreated water, in the absence of EDTA but existence of NiP, deviate significantly than those in purified water under the same conditions; 2.85 ± 0.46 and 3.38 ± 0.23 /vmol Fh were photo-evolved over a period of 6 hrs in sea and river water (Figure 5(a)), respectively, compared to no Ph evolution in purified water (Figure 7). This indicated the presence of additives in the untreated aqueous media which could serve as sacrificial electron donors to extract the photogenerated holes (Martindale et al. Angew. Chem. Int. Ed. 2017). On the other hand, elimination of NiP in the presence of EDTA (Figure 9(a)), resulted in no PI2 production neither in pure or untreated water, suggesting that no species (i.e. metal traces) exist in sea, river and sewage water that could drive fuel formation.
Untreated sea water (Gulf of Mexico) contains a cocktail of organic compounds (Nowell et al.), among which benzaldehyde and atrazine can potentially act as electron donors (Figure 9(b)) (Nowell et al.). Chloride anions that are abundantly present in seawater can also serve the same role (Iguchi et al.). Similarly, untreated river water (River Cam), is rich in organic herbicides/pesticides (River Cam), among which prometryn and terbutryn can quench the photogenerated holes (Figure 9(b)).
To confirm this, an aqueous ocellulose CD phosphate solution (KPi, 0.1 M, pFH 6, 3 mL), in the absence of EDTA, was bestowed with these species (200 pmol), in the presence of NiP. After 6 hrs of full solar light irradiation (AM 1.5 G, 100 mW cm-2), in the presence of the
organic pollutants and Cl· anions, the H2 yields were in the range between 2.05 ± 0.20 and 4.99 ± 0.15 /vmol (Figure 5(b)), which are close to those recorded in the absence of any electron donor in untreated water media (Figure 5(a) and Figure 9(c)). As such, the unwanted toxic substances and/or the abundant Cl·, eliminate the need for additional chemicals to serve as electron donors, allowing simultaneously for fuel synthesis and water treatment. To the best of our knowledge, this is the first time that photogenerated CD holes drive the oxidation of water pollutants, a process that traditionally is catalyzed by reactive oxygen species (Xiao et a!.).
Next, the photocatalytic performances of the photoabsorbers synthesized from biomass waste, were evaluated in sea (Figure 5(c)) and river water (Figure 5(d)), in the presence of EDTA and NiP. In sea water, after 24 hrs of irradiation, most of the CDs show higher photocatalytic performances (Figure 5(c)) than those measured in purified water under the same conditions (Figure 4(c) and 9(d)).
The only exception are the CDs from ElaeagnusX ebbingei, which within error, show the same performances in both pure and sea water. On the other hand, all systems show slightly lower performances in river compared to sea water, except for the CDs from T. Baccata and Elaeagnus X Ebbingei, which show slightly higher H2 evolution yields in river water
(Figure 5(d) and 9(d)).
These results suggest the robust character of our photocatalytic systems, which maintain their good photocatalytic properties, independently of the purity and origin of the aqueous phase. As such, the toxic water contaminants do not restrict the performances of the CDs by hindering photoabsorption and/or electron transfer, and more importantly do not poison the NiP molecular catalyst. The performances of the photocatalytic systems described herein, even in untreated water media, are believed to be the highest among all photocatalytic systems based on carbonaceous systems reported up-to-date (see Table 17).
References
All documents mentioned in this specification are incorporated herein by reference in their entirety.
Abaza et al. Antioxidants 2015, 4, 682
Algara-Siller et al. Angew. Chemie Int. Ed. 2014, 53, 7450
Alves et al. Appl. Surf. Sci. 2016, 370, 486
Azadi et al. Appl. Catal. B Environ. 2012, 117-118, 330
Barneto et al. Energy Fuels 2009, 23, 951
Biagini et al. Ind. Eng. Chem. Res. 2006, 45, 4486
Bourlinos et al. Chem. Mater. 2008, 20, 4539
Budtova et al. Cellulose 2016, 23, 5
Caputo et al. Angew. Chemie Int. Ed. 2014, 53, 1 1538
Caputo et al. Angewandte Chemie International Edition 2014, 53, 1 1538
Carrier et al. Biomass Bioenergy 2011 , 35, 298
Chen et al. Materials 2016, 9, 184
Chen, H. In Biotechnology of Lignocellulose: Theory and Practice, Springer Netherlands: Dordrecht, 2014, p 25.
Chosdu et al. Radiat. Phys. Chem. 1993, 42, 695
CN 106902803
CN 106902857
CN 107008484
CN 107297217
CN 107413378
CN 107552083
CN 107837817
CN 107961808
Ferrari Solid State Commun. 2007, 143, 47
Gao et al. Nature Chemistry 2009, 1, 403
George et al. Energy Fuels 2014, 28, 6918
Gross et al. J. Am. Chem. Soc. 2014, 136, 356
Gupta et al. Scientific Reports 2017, 7, 45030
Han et al. Sci. Rep. 2014, 4, 5133
Herrero et al. J. Chromatogr. A 2011 , 1218, 751 1
Hinman et al. Appl. Biochem. Biotechnol. 1992, 34-35, 639
Hong et al. Nanoscale 2014, 6, 14984
Huber et al. Chemical Reviews 2006, 106, 4044
Hutton et al. Chem. Soc. Rev. 2017, 46, 61 1 1
Hutton et al. J. Am. Chem. Soc. 2016, 138, 16722
Iguchi et al. Phys. Chem. Chem. Phys. 2015, 17, 17995
Jiang et al. J. Nanoparticle Res. 2009, 11, 77
Kasap et al. J. Am. Chem. Soc. 2016, 138, 9183
Kawai et al. Chem. Lett. 1981 , 10, 81
Krassig et al. In Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA: 2000
Kuehnel et al. Angew. Chemie Int. Ed. 2018, 57, 3290
Kumar et al. Ind. Eng. Chem. Res. 2009, 48, 3713
Lau et al. Adv. Energy Mater. 2017, 7, 1602251
Lau et al. Angew. Chemie Int. Ed. 2016, 55, 1
Lau et al. Angew. Chemie Int. Ed. 2017, 56, 510
Lau et al. Nat. Commun. 2016, 7, 12165
Lavarack et al. Biomass and Bioenergy 2002, 23, 367
Lee et al. The Scientific World Journal 2014, 2014, 20
Li et al. Journal of Materials Chemistry A 2015, 3, 23307
Martin et al. Angew. Chemie Int. Ed. 2014, 53, 9240
Martindale et al. Angew. Chemie Int. Ed. 2016, 55, 9402
Martindale et al. Angew. Chemie Int. Ed. 2017, 56, 6459
Martindale et al. J. Am. Chem. Soc. 2015, 137, 6018
Mewada et al. Materials Science and Engineering: C 2013, 33, 2914
Meyer et al. European J. Org. Chem. 2017, 2017, 2179
Miller et al. Phys. Chem. Chem. Phys. 2017, 19, 15613
Mondal et al. Sensors and Actuators B: Chemical 2018, 257, 586
Nguyen et al. Nanoscale 2013, 5, 1479
Niemczewski Ultrason. Sonochem. 2014, 21, 354
Nowell et al. Report 2012-5228, 96 p, plus appendixes
Nowell et al. USGS Open-File Report 2011-1271, 201 1
Oza et al. Scientific Reports 2016, 6, 21286
PCT/EP2017/080371
Pellegrin et al. R. Chim. 2017, 20, 283
Peralbo-Molina et al. Trends Food Sci. Technol. 2013, 32, 16
Pimenta et al. Phys. Chem. Chem. Phys. 2007, 9, 1276
Qin et al. J. Phys. Chem. C 2017
Qu et al. Advanced Optical Materials 2015, 3, 360
Qu et al. Nanoscale 2013, 5, 12272
Rai et al. J. Lumin. 2017, 190, 492
River Cam water quality data, British environmental agency, November 2017
Rivero et al. Nanoscale Research Letters 2015, 10, 501
Roy et al. Plant leaf-derived graphene quantum dots and applications for white LEDs, 2014; Vol. 38
Rubin Nature 2008, 454, 841
Sarkar et al. J. Phys. Chem. C 2016, 120, 1303
Service Science 2006, 313, 1088
Shen et al. ChemistrySelect 2016, 1, 1314
Shiravand et al. Sensors Actuators B Chem. 2017, 242, 244
Sun, Y.; Cheng, J. Bioresour. Technol. 2002, 83, 1
Sweet et al. Holzforschung 1999, 53, 31 1
Torres et al. Catalysis Reviews 2007, 49, 407
van Wyk, J. P. Trends Biotechnol. 2001, 19, 172
Wakerley et al. Nat. Energy 2017, 2, 17021
Wang et al. J. Lumin. 2017, 188, 230
Wang et al. RSC Advances 2017, 7, 21969
Wen et al. Sensors and Actuators B: Chemical 2015, 221, 769
Wu et al. J. Mater. Chem. A 2015, 3, 15710
Xiao et al. Environ. Sci. Technol. 2017, 51, 13380
Yang et al. Adv. Mater. 2013, 25, 2452
Yang et al. Journal of Materials Chemistry 2012, 22, 4391
Young Park et al. Scientific Reports 2015, 5, 12420 Yu et al. RSC Advances 2016, 6, 18217
Zhang et al. >4CS Sustainable Chemistry & Engineering 2015, 3, 293 Zhang et al. J. Am. Chem. Soc. 2013, 135, 18
Zhang et al. New J. Chem. 2010, 34, 591
Zhang et al. Science Advances 2015, 1
Zhou et al. Carbon N. Y. 2016, 99, 1 1 1
Zhu et al. Chemical Reviews 2016, 776, 9305
Claims
1 . A method for producing hydrogen, the method comprising the step of exposing a photocatalyst to visible and/or ultraviolet light in the presence of an organic substrate and a co-catalyst that is a hydrogen evolution catalyst, wherein the photocatalyst is a
carbon-containing catalyst selected from a carbon nitride (CNX) photocatalyst and a carbon dot (CD) photocatalyst.
2. A method for producing hydrogen, the method comprising the step of exposing a photocatalyst within untreated water to visible and/or ultraviolet light in the presence of a co- catalyst that is a hydrogen evolution catalyst, wherein the photocatalyst is a carbon- containing catalyst selected from a carbon nitride (CNX) photocatalyst and a carbon dot (CD) photocatalyst.
3. The method of 1 or 2, wherein the catalyst is a carbon nitride photocatalyst.
4. The method of claim 3, wherein the catalyst is a NCNCNX photocatalyst.
5. The method of claim 5, wherein the catalyst is a NCNCNX photocatalyst having a surface area that is at least 60 m2 g 1, such as at least 90 m2 g 1.
6. The method of claim 1 or 2, wherein the catalyst is a carbon dot, which is optionally doped.
7. The method of claim 6, wherein the catalyst is a carbon dot, which is optionally doped, such as nitrogen doped, which catalyst is obtained or obtainable by calcination of an organic substrate, such as a biomass.
8. The method of claim 1 , wherein the organic substrate is or contains a component having a molecular weight of 200 or more, such as 500 or more, such as 2 kDa or more.
9. The method of claim 1 , wherein the organic substrate is selected from ocellulose, xylan and lignin, such as ocellulose.
10. The method of claim 1 , wherein the organic substrate is a biomass.
1 1 . The method of claim 10, wherein the biomass is selected from wood, paper, cardboard, bagasse, grass and/or sawdust.
12. The method of claim 1 , wherein the organic substrate is selected from general waste, plastics, packing materials, waste food, aliphatic polyols, algae, sugars, starches, biomass, sewage and/or domestic waste.
13. The method of claim 2, wherein the untreated water is sea water.
14. The method of claim 2, wherein the untreated water is sewage, or river, lake, or canal water.
15. The method of claim 2, wherein the untreated water contains a sacrificial electron donor that is an organic compound, such as selected from atrazine, prometryn, terbutryn and benzaldehyde, or an inorganic species, such as chloride.
16. The method of claim 1 or claim 2, wherein the hydrogen evolution catalyst is selected from a Ni-, Pt- or Mo-containing hydrogen evolution catalyst.
17. A method of preparing NCNCNx, the method comprising the step of forming a bulk sample of NCNCNX and subsequently treating the bulk NCNCNX thereby to increase the surface area of the NCNCNx.
18. The method of claim 17, wherein the bulk NCNCNX is sonicated, such as
ultrasonicated, to increase its surface area.
19. A method of preparing a carbon dot, the method comprising the step of calcining an organic substrate comprising an oligosaccharide or a polysaccharide, such as calcining at a temperature in the range 250 to 450°C.
20. The method of claim 19, wherein the organic substrate is or comprises a
polysaccharide.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1808905.2A GB201808905D0 (en) | 2018-05-31 | 2018-05-31 | Photocatalyst and photocatalytic methods |
| GB1808905.2 | 2018-05-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019229255A1 true WO2019229255A1 (en) | 2019-12-05 |
Family
ID=62872686
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2019/064221 Ceased WO2019229255A1 (en) | 2018-05-31 | 2019-05-31 | Photocatalyst and photocatalytic methods for producing hydrogen |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB201808905D0 (en) |
| WO (1) | WO2019229255A1 (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111363543A (en) * | 2020-03-03 | 2020-07-03 | 温州医科大学 | Method for preparing carbon dots from catkins, carbon dots and application thereof |
| CN111871445A (en) * | 2020-08-13 | 2020-11-03 | 郑州大学 | CN/BOS van der Waals heterostructure photocatalyst and preparation method thereof |
| CN112371160A (en) * | 2020-12-03 | 2021-02-19 | 安徽中医药大学 | Preparation method and use method of high-crystallization wide-absorption carbon nitride photocatalytic material |
| CN112371161A (en) * | 2020-12-25 | 2021-02-19 | 郑州大学 | Carbon-point-modified graphite-phase carbon nitride hollow sphere photocatalyst and preparation method and application thereof |
| CN112517043A (en) * | 2020-12-17 | 2021-03-19 | 中南大学 | Nitrogen vacancy and hydroxyl synergistically modified graphite-phase carbon nitride photocatalyst, preparation method thereof and application thereof in photocatalytic hydrogen production |
| CN112717974A (en) * | 2020-11-23 | 2021-04-30 | 北京工业大学 | Phosphorus-doped ultrathin hollow carbon nitride nanosphere catalyst for efficient photocatalytic water splitting hydrogen production |
| CN112808313A (en) * | 2020-12-30 | 2021-05-18 | 陕西科技大学 | Nitrogen-doped carbon quantum dot/metal organic framework material MOF-5 photocatalyst and preparation method and application thereof |
| CN112973750A (en) * | 2021-01-29 | 2021-06-18 | 南京工业大学 | Carbon quantum dot coated metal monoatomic-carbon nitride composite material and preparation method thereof |
| CN113413913A (en) * | 2021-08-02 | 2021-09-21 | 哈尔滨工业大学 | Preparation method, product and application of graphene photocatalyst |
| CN113413870A (en) * | 2021-05-17 | 2021-09-21 | 中国科学院青海盐湖研究所 | Magnesium oxide-metal sulfide-biomass charcoal composite material and preparation method and application thereof |
| CN113680366A (en) * | 2021-08-26 | 2021-11-23 | 上海电力大学 | A kind of graphitic carbon nitride based composite photocatalyst and preparation method and application thereof |
| CN113707894A (en) * | 2021-08-20 | 2021-11-26 | 中国第一汽车股份有限公司 | Fuel cell catalyst and preparation method and application thereof |
| CN113816501A (en) * | 2021-08-25 | 2021-12-21 | 福建农林大学 | Bio-photoelectrochemical reagent for synchronously realizing plastic degradation and heavy metal reduction and preparation method thereof |
| CN113842939A (en) * | 2021-09-24 | 2021-12-28 | 郑州大学 | Photocatalyst and preparation method thereof |
| CN114160178A (en) * | 2021-11-12 | 2022-03-11 | 华南理工大学 | Carbon nitride nanosheet-gold nanoparticle composite material and preparation method and application thereof |
| CN114160202A (en) * | 2021-11-25 | 2022-03-11 | 华南理工大学 | In-situ modified MIL-68(Fe) photocatalyst and preparation method and application thereof |
| CN114229808A (en) * | 2021-11-22 | 2022-03-25 | 苏州科技大学 | Synthesis of g-C3N4/CQDs materials |
| CN114917942A (en) * | 2022-04-14 | 2022-08-19 | 华南理工大学 | Preparation method of one-dimensional nanorod carbon nitride photocatalyst and application of photocatalyst in synthesis of lactic acid through photocatalytic oxidation of monosaccharide |
| CN115337954A (en) * | 2022-10-17 | 2022-11-15 | 山东环投环境工程有限公司 | Composite photocatalyst based on carbon nitride and preparation method and application thereof |
| CN115466761A (en) * | 2022-08-25 | 2022-12-13 | 哈尔滨工业大学(深圳) | Method and equipment for preparing hydrogen by catalytically converting biomass through membrane-assisted bio-enzyme catalysis coupling |
| CN115634703A (en) * | 2022-09-15 | 2023-01-24 | 贵州黔大生态环境与健康研究院有限公司 | A kind of catalyst and its application |
| CN115786971A (en) * | 2022-10-14 | 2023-03-14 | 聊城大学 | A kind of preparation method of ruthenium phosphide porous nitrogen and phosphorus doped carbon |
| CN115779943A (en) * | 2022-10-27 | 2023-03-14 | 广东省科学院测试分析研究所(中国广州分析测试中心) | KSCN (KSCN) -based modified carbon nitride, preparation method thereof and photocatalytic H production method 2 O 2 In (1) |
| CN115957792A (en) * | 2023-01-03 | 2023-04-14 | 浙江芯科半导体有限公司 | Method for preparing photocatalyst by using waste cigarette ends, photocatalyst and application |
| CN116239103A (en) * | 2023-02-20 | 2023-06-09 | 中国石油大学(华东) | Carbon dots and preparation method thereof, photocatalytic hydroboration method |
| EP4212477A1 (en) | 2022-01-12 | 2023-07-19 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Novel carbon nitride compositions and their use as photoelectrodes |
| RU2800948C1 (en) * | 2022-12-22 | 2023-08-01 | Федеральное государственное бюджетное учреждение науки "Федеральный исследовательский центр " Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук" (ИК СО РАН, Институт катализа СО РАН) | Catalyst, method for its preparation, and method for photocatalytic production of hydrogen |
| CN116689006A (en) * | 2023-03-28 | 2023-09-05 | 上海大学 | Carbon nitride nano-sheet composite material and preparation method and application thereof |
| CN117088771A (en) * | 2022-05-13 | 2023-11-21 | 北京大学 | A method for recycling polystyrene |
| CN117720067A (en) * | 2024-01-29 | 2024-03-19 | 四川大学 | A method for photochemically preparing H2 from benzyl compounds |
| CN118237064A (en) * | 2024-05-28 | 2024-06-25 | 江西省科学院能源研究所 | Composite photocatalyst and preparation method and application thereof |
| WO2024160923A1 (en) * | 2023-02-01 | 2024-08-08 | Cambridge Enterprise Limited | Method for upcycling plastic waste by enzymatic degradation and photoreforming |
| CN118994599A (en) * | 2024-08-07 | 2024-11-22 | 华侨大学 | Sulfate lignin-heptazine conjugated polymer, and preparation method and application thereof |
| WO2025189296A1 (en) * | 2024-03-13 | 2025-09-18 | Hydrofuel Canada Inc. | Process of photochemical reforming of plastics using a carbon enabler |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112295586B (en) * | 2020-10-28 | 2023-07-25 | 温州医科大学 | Phosphorus-sulfur co-doped carbon nitride nanomaterial, preparation method and application thereof |
| CN113651312B (en) * | 2021-08-30 | 2023-01-17 | 新疆大学 | A kind of carbon quantum dot gas sensitive sensing material and its preparation method and application |
| CN115181265B (en) * | 2022-07-29 | 2023-10-31 | 华侨大学 | A methylene-modified covalent triazine framework material and its preparation method and application |
| CN115920943A (en) * | 2022-12-12 | 2023-04-07 | 湖南易净环保科技有限公司 | Photocatalyst for degradation of tetracycline in wastewater, preparation method and application thereof |
| CN117960224B (en) * | 2024-01-17 | 2026-04-03 | 河南大学 | A method for preparing porous carbon nitride/carbon composite photocatalyst and its application. |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106902857A (en) | 2017-02-27 | 2017-06-30 | 兰州大学 | One kind expansion g C3N4Photochemical catalyst and preparation method thereof |
| CN106902803A (en) | 2017-03-13 | 2017-06-30 | 辽宁大学 | Compound photocatalytic system CQDS‑KNbO3And its preparation method and application |
| CN107008484A (en) | 2017-04-17 | 2017-08-04 | 武汉理工大学 | A kind of binary metal sulfide/carbonitride composite photocatalyst material and preparation method thereof |
| CN107297217A (en) | 2017-06-01 | 2017-10-27 | 西安交通大学 | A kind of thin porous layer graphite phase carbon nitride loaded platinum photo catalyst and its preparation method and application |
| CN107413378A (en) | 2016-05-23 | 2017-12-01 | 中国科学院上海硅酸盐研究所 | A kind of preparation method for the graphite phase carbon nitride visible light catalyst that combined polymerization is modified |
| CN107552083A (en) | 2017-10-11 | 2018-01-09 | 肇庆市华师大光电产业研究院 | A kind of FeP/C3N4Composite photo-catalyst and its preparation method and application |
| CN107597166A (en) * | 2017-09-29 | 2018-01-19 | 南昌航空大学 | A kind of carbon point/cadmiumsulfide quantum dot/carbon nitride catalyst and preparation method thereof |
| CN107837817A (en) | 2017-11-21 | 2018-03-27 | 华中农业大学 | A kind of carbon point/carbonitride/composite titania material and its preparation method and application |
| CN107961808A (en) | 2017-12-04 | 2018-04-27 | 蚌埠医学院 | A kind of preparation method of visible light catalyst and application |
-
2018
- 2018-05-31 GB GBGB1808905.2A patent/GB201808905D0/en not_active Ceased
-
2019
- 2019-05-31 WO PCT/EP2019/064221 patent/WO2019229255A1/en not_active Ceased
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107413378A (en) | 2016-05-23 | 2017-12-01 | 中国科学院上海硅酸盐研究所 | A kind of preparation method for the graphite phase carbon nitride visible light catalyst that combined polymerization is modified |
| CN106902857A (en) | 2017-02-27 | 2017-06-30 | 兰州大学 | One kind expansion g C3N4Photochemical catalyst and preparation method thereof |
| CN106902803A (en) | 2017-03-13 | 2017-06-30 | 辽宁大学 | Compound photocatalytic system CQDS‑KNbO3And its preparation method and application |
| CN107008484A (en) | 2017-04-17 | 2017-08-04 | 武汉理工大学 | A kind of binary metal sulfide/carbonitride composite photocatalyst material and preparation method thereof |
| CN107297217A (en) | 2017-06-01 | 2017-10-27 | 西安交通大学 | A kind of thin porous layer graphite phase carbon nitride loaded platinum photo catalyst and its preparation method and application |
| CN107597166A (en) * | 2017-09-29 | 2018-01-19 | 南昌航空大学 | A kind of carbon point/cadmiumsulfide quantum dot/carbon nitride catalyst and preparation method thereof |
| CN107552083A (en) | 2017-10-11 | 2018-01-09 | 肇庆市华师大光电产业研究院 | A kind of FeP/C3N4Composite photo-catalyst and its preparation method and application |
| CN107837817A (en) | 2017-11-21 | 2018-03-27 | 华中农业大学 | A kind of carbon point/carbonitride/composite titania material and its preparation method and application |
| CN107961808A (en) | 2017-12-04 | 2018-04-27 | 蚌埠医学院 | A kind of preparation method of visible light catalyst and application |
Non-Patent Citations (90)
| Title |
|---|
| ABAZA ET AL., ANTIOXIDANTS, vol. 4, 2015, pages 682 |
| ALGARA-SILLER ET AL., ANGEW. CHEMIE INT. ED., vol. 53, 2014, pages 11538 |
| ALVES ET AL., APPL. SURF. SCI., vol. 370, 2016, pages 486 |
| AZADI ET AL., APPL. CATAL. B ENVIRON., vol. 330, 2012, pages 117 - 118 |
| BARNETO ET AL., ENERGY FUELS, vol. 23, 2009, pages 951 |
| BENJAMIN C. M. MARTINDALE ET AL: "Clean Donor Oxidation Enhances the H 2 Evolution Activity of a Carbon Quantum Dot-Molecular Catalyst Photosystem", ANGEWANDTE CHEMIE, INTERNATIONAL EDITION, vol. 55, no. 32, 29 June 2016 (2016-06-29), DE, pages 9402 - 9406, XP055622885, ISSN: 1433-7851, DOI: 10.1002/anie.201604355 * |
| BENJAMIN C. M. MARTINDALE ET AL: "Solar Hydrogen Production Using Carbon Quantum Dots and a Molecular Nickel Catalyst", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 137, no. 18, 28 April 2015 (2015-04-28), US, pages 6018 - 6025, XP055623012, ISSN: 0002-7863, DOI: 10.1021/jacs.5b01650 * |
| BIAGINI ET AL., IND. ENG. CHEM. RES., vol. 45, 2006, pages 4486 |
| BOURLINOS ET AL., CHEM. MATER., vol. 20, 2008, pages 4539 |
| BUDTOVA ET AL., CELLULOSE, vol. 23, 2016, pages 5 |
| CAPUTO ET AL., ANGEWANDTE CHEMIE INTERNATIONAL EDITION, vol. 53, 2014, pages 11538 |
| CARRIER ET AL., BIOMASS BIOENERGY, vol. 35, 2011, pages 298 |
| CHEN ET AL., MATERIALS, vol. 9, 2016, pages 184 |
| CHOSDU ET AL., RADIAT. PHYS. CHEM., vol. 42, 1993, pages 695 |
| FERRARI, SOLID STATE COMMUN, vol. 143, 2007, pages 47 |
| GAO ET AL., NATURE CHEMISTRY, vol. 1, 2009, pages 403 |
| GEORGE ET AL., ENERGY FUELS, vol. 28, 2014, pages 6918 |
| GROSS ET AL., J. AM. CHEM. SOC., vol. 136, 2014, pages 356 |
| GUPTA ET AL., SCIENTIFIC REPORTS, vol. 7, 2017, pages 45030 |
| HAN ET AL., SCI. REP., vol. 4, 2014, pages 5133 |
| HATICE KASAP ET AL: "Solar-Driven Reduction of Aqueous Protons Coupled to Selective Alcohol Oxidation with a Carbon Nitride-Molecular Ni Catalyst System", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 138, no. 29, 14 July 2016 (2016-07-14), US, pages 9183 - 9192, XP055622827, ISSN: 0002-7863, DOI: 10.1021/jacs.6b04325 * |
| HERRERO ET AL., J. CHROMATOGR. A, vol. 1218, 2011, pages 7511 |
| HINMAN ET AL., APPL. BIOCHEM. BIOTECHNOL., vol. 34-35, 1992, pages 639 |
| HONG ET AL., NANOSCALE, vol. 6, 2014, pages 14984 |
| HUBER ET AL., CHEMICAL REVIEWS, vol. 106, 2006, pages 4044 |
| HUI ZHU ET AL: "Microwave synthesis of fluorescent carbon nanoparticles with electrochemiluminescence properties", CHEMICAL COMMUNICATIONS (CAMBRIDGE, ENGLAND), 14 September 2009 (2009-09-14), England, pages 5118 - 5120, XP055622662, Retrieved from the Internet <URL:https://pubs.rsc.org/en/content/articlepdf/2009/cc/b907612c> [retrieved on 20190917], DOI: 10.1039/b907612c * |
| HUIJUN YU ET AL: "Carbon quantum dots/TiO2 composites for efficient photocatalytic hydrogen evolution", JOURNAL OF MATERIALS CHEMISTRY A, vol. 2, no. 10, 14 March 2014 (2014-03-14), GB, pages 3344, XP055622587, ISSN: 2050-7488, DOI: 10.1039/c3ta14108j * |
| HUTTON ET AL., CHEM. SOC. REV., vol. 46, 2017, pages 6111 |
| HUTTON ET AL., J. AM. CHEM. SOC., vol. 138, 2016, pages 16722 |
| IGUCHI ET AL., PHYS. CHEM. CHEM. PHYS., vol. 17, 2015, pages 17995 |
| JIANG ET AL., J. NANOPARTICLE RES., vol. 11, 2009, pages 77 |
| KAWAI ET AL., CHEM. LETT., vol. 10, 1981, pages 81 |
| KRASSIG ET AL.: "In Ullmann's Encyclopedia of Industrial Chemistry", 2000, WILEY-VCH VERLAG GMBH & CO. KGAA |
| KUEHNEL ET AL., ANGEW. CHEMIE INT. ED., vol. 57, 2018, pages 3290 |
| KUMAR ET AL., IND. ENG. CHEM. RES., vol. 48, 2009, pages 3713 |
| LAU ET AL., ADV. ENERGY MATER., vol. 7, 2017, pages 1602251 |
| LAU ET AL., NAT. COMMUN |
| LAU ET AL., NAT. COMMUN., vol. 7, 2016, pages 12165 |
| LAVARACK ET AL., BIOMASS AND BIOENERGY, vol. 23, 2002, pages 367 |
| LEE ET AL., THE SCIENTIFIC WORLD JOURNAL, vol. 2014, 2014, pages 20 |
| LI ET AL., JOURNAL OF MATERIALS CHEMISTRY A, vol. 3, 2015, pages 23307 |
| MARTINDALE ET AL., ANGEW. CHEM. INT. ED., 2017 |
| MARTINDALE ET AL., ANGEW. CHEMIE INT. ED., vol. 55, 2016, pages 9402 |
| MARTINDALE ET AL., ANGEW. CHEMIE INT. ED., vol. 56, 2017, pages 6459 |
| MARTINDALE ET AL., ANGEW. CHEMIE, 2016 |
| MARTINDALE ET AL., J. AM. CHEM. SOC., vol. 137, 2015, pages 6018 |
| MEWADA ET AL., MATERIALS SCIENCE AND ENGINEERING: C, vol. 33, 2013, pages 2914 |
| MEYER ET AL., EUROPEAN J. ORG. CHEM., vol. 2017, 2017, pages 2179 |
| MILLER ET AL., PHYS. CHEM. CHEM. PHYS., vol. 19, 2017, pages 15613 |
| MONDAL ET AL., SENSORS AND ACTUATORS B: CHEMICAL, vol. 257, 2018, pages 586 |
| NGUYEN ET AL., NANOSCALE, vol. 5, 2013, pages 12272 |
| NIEMCZEWSKI ULTRASON, SONOCHEM, vol. 21, 2014, pages 354 |
| NOWELL ET AL., REPORT, vol. 5228, 2012, pages 96 |
| NOWELL ET AL., USGS OPEN-FILE REPORT, vol. 1271, 2011, pages 2011 |
| OZA ET AL., SCIENTIFIC REPORTS, vol. 6, 2016, pages 21286 |
| PELLEGRIN ET AL., R. CHIM., vol. 20, 2017, pages 283 |
| PERALBO-MOLINA ET AL., TRENDS FOOD SCI. TECHNOL., vol. 32, 2013, pages 16 |
| PIMENTA ET AL., PHYS. CHEM. CHEM. PHYS., vol. 9, 2007, pages 1276 |
| PRATHIK ROY ET AL: "Photoluminescent carbon nanodots: synthesis, physicochemical properties and analytical applications", MATERIALS TODAY, vol. 18, no. 8, October 2015 (2015-10-01), AMSTERDAM, NL, pages 447 - 458, XP055622666, ISSN: 1369-7021, DOI: 10.1016/j.mattod.2015.04.005 * |
| QIN ET AL., J. PHYS. CHEM. C, 2017 |
| QU ET AL., ADVANCED OPTICAL MATERIALS, vol. 3, 2015, pages 360 |
| RIVER, CAM WATER QUALITY DATA, BRITISH ENVIRONMENTAL AGENCY, November 2017 (2017-11-01) |
| RIVERO ET AL., NANOSCALE RESEARCH LETTERS, vol. 10, 2015, pages 501 |
| ROY ET AL., PLANT LEAF-DERIVED GRAPHENE QUANTUM DOTS AND APPLICATIONS FOR WHITE LEDS, vol. 38, 2014 |
| RUBIN, NATURE, vol. 454, 2008, pages 841 |
| SARKAR ET AL., J. PHYS. CHEM. C, vol. 120, 2016, pages 1303 |
| SERVICE, SCIENCE, vol. 313, 2006, pages 1088 |
| SHEN ET AL., CHEMISTRYSELECT, vol. 1, 2016, pages 1314 |
| SHIRAVAND ET AL., SENSORS ACTUATORS B CHEM, vol. 242, 2017, pages 244 |
| SUN, Y.CHENG, J., BIORESOUR. TECHNOL., vol. 83, 2002, pages 1 |
| SWEET ET AL., HOLZFORSCHUNG, vol. 53, 1999, pages 311 |
| TORRES ET AL., CATALYSIS REVIEWS, vol. 49, 2007, pages 407 |
| VAN WYK, J. P, TRENDS BIOTECHNOL., vol. 19, 2001, pages 172 |
| WAKERLEY ET AL., NAT. ENERGY, vol. 2, 2017, pages 17021 |
| WANG ET AL., J. LUMIN., vol. 188, 2017, pages 230 |
| WANG ET AL., RSC ADVANCES, vol. 7, 2017, pages 21969 |
| WEN ET AL., SENSORS AND ACTUATORS B: CHEMICAL, vol. 221, 2015, pages 769 |
| WU ET AL., J. MATER. CHEM. A, vol. 3, 2015, pages 15710 |
| XIAO ET AL., ENVIRON. SCI. TECHNOL., vol. 51, 2017, pages 13380 |
| XIAOWEI SHI ET AL: "Faster Electron Injection and More Active Sites for Efficient Photocatalytic H 2 Evolution in g- C3N4 /MoS2 Hybrid", SMALL, vol. 14, no. 11, 26 January 2018 (2018-01-26), DE, pages 1703277, XP055622642, ISSN: 1613-6810, DOI: 10.1002/smll.201703277 * |
| YANG ET AL., ADV. MATER., vol. 25, 2013, pages 2452 |
| YANG ET AL., JOURNAL OF MATERIALS CHEMISTRY, vol. 22, 2012, pages 4391 |
| YOUNG PARK ET AL., SCIENTIFIC REPORTS, vol. 5, 2015, pages 12420 |
| YU ET AL., RSC ADVANCES, vol. 6, 2016, pages 18217 |
| ZHANG ET AL., ACS SUSTAINABLE CHEMISTRY & ENGINEERING, vol. 3, 2015, pages 293 |
| ZHANG ET AL., J. AM. CHEM. SOC., vol. 135, 2013, pages 18 |
| ZHANG ET AL., NEW J. CHEM., vol. 34, 2010, pages 591 |
| ZHANG ET AL., SCIENCE ADVANCES, vol. 1, 2015 |
| ZHOU ET AL., CARBON N. Y., vol. 99, 2016, pages 111 |
| ZHU ET AL., CHEMICAL REVIEWS, vol. 116, 2016, pages 9305 |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111363543A (en) * | 2020-03-03 | 2020-07-03 | 温州医科大学 | Method for preparing carbon dots from catkins, carbon dots and application thereof |
| CN111871445A (en) * | 2020-08-13 | 2020-11-03 | 郑州大学 | CN/BOS van der Waals heterostructure photocatalyst and preparation method thereof |
| CN112717974A (en) * | 2020-11-23 | 2021-04-30 | 北京工业大学 | Phosphorus-doped ultrathin hollow carbon nitride nanosphere catalyst for efficient photocatalytic water splitting hydrogen production |
| CN112717974B (en) * | 2020-11-23 | 2023-04-07 | 北京工业大学 | Phosphorus-doped ultrathin hollow carbon nitride nanosphere catalyst for efficient photocatalytic water splitting hydrogen production |
| CN112371160A (en) * | 2020-12-03 | 2021-02-19 | 安徽中医药大学 | Preparation method and use method of high-crystallization wide-absorption carbon nitride photocatalytic material |
| CN112517043B (en) * | 2020-12-17 | 2021-11-26 | 中南大学 | Nitrogen vacancy and hydroxyl synergistically modified graphite-phase carbon nitride photocatalyst, preparation method thereof and application thereof in photocatalytic hydrogen production |
| CN112517043A (en) * | 2020-12-17 | 2021-03-19 | 中南大学 | Nitrogen vacancy and hydroxyl synergistically modified graphite-phase carbon nitride photocatalyst, preparation method thereof and application thereof in photocatalytic hydrogen production |
| CN112371161A (en) * | 2020-12-25 | 2021-02-19 | 郑州大学 | Carbon-point-modified graphite-phase carbon nitride hollow sphere photocatalyst and preparation method and application thereof |
| CN112808313A (en) * | 2020-12-30 | 2021-05-18 | 陕西科技大学 | Nitrogen-doped carbon quantum dot/metal organic framework material MOF-5 photocatalyst and preparation method and application thereof |
| CN112973750A (en) * | 2021-01-29 | 2021-06-18 | 南京工业大学 | Carbon quantum dot coated metal monoatomic-carbon nitride composite material and preparation method thereof |
| CN112973750B (en) * | 2021-01-29 | 2023-09-26 | 南京工业大学 | Carbon quantum dot coated metal monoatomic-carbon nitride composite material and preparation method thereof |
| CN113413870B (en) * | 2021-05-17 | 2022-11-25 | 中国科学院青海盐湖研究所 | Magnesium oxide-metal sulfide-biomass charcoal composite material and preparation method and application thereof |
| CN113413870A (en) * | 2021-05-17 | 2021-09-21 | 中国科学院青海盐湖研究所 | Magnesium oxide-metal sulfide-biomass charcoal composite material and preparation method and application thereof |
| CN113413913A (en) * | 2021-08-02 | 2021-09-21 | 哈尔滨工业大学 | Preparation method, product and application of graphene photocatalyst |
| CN113707894A (en) * | 2021-08-20 | 2021-11-26 | 中国第一汽车股份有限公司 | Fuel cell catalyst and preparation method and application thereof |
| CN113816501A (en) * | 2021-08-25 | 2021-12-21 | 福建农林大学 | Bio-photoelectrochemical reagent for synchronously realizing plastic degradation and heavy metal reduction and preparation method thereof |
| CN113816501B (en) * | 2021-08-25 | 2022-08-12 | 福建农林大学 | A kind of biophotoelectrochemical reagent for realizing plastic degradation and heavy metal reduction simultaneously and preparation method thereof |
| CN113680366B (en) * | 2021-08-26 | 2023-12-01 | 上海电力大学 | A graphite-phase carbon nitride-based composite photocatalyst and its preparation method and application |
| CN113680366A (en) * | 2021-08-26 | 2021-11-23 | 上海电力大学 | A kind of graphitic carbon nitride based composite photocatalyst and preparation method and application thereof |
| CN113842939A (en) * | 2021-09-24 | 2021-12-28 | 郑州大学 | Photocatalyst and preparation method thereof |
| CN113842939B (en) * | 2021-09-24 | 2023-09-01 | 郑州大学 | Photocatalyst and preparation method thereof |
| CN114160178A (en) * | 2021-11-12 | 2022-03-11 | 华南理工大学 | Carbon nitride nanosheet-gold nanoparticle composite material and preparation method and application thereof |
| CN114229808A (en) * | 2021-11-22 | 2022-03-25 | 苏州科技大学 | Synthesis of g-C3N4/CQDs materials |
| CN114160202A (en) * | 2021-11-25 | 2022-03-11 | 华南理工大学 | In-situ modified MIL-68(Fe) photocatalyst and preparation method and application thereof |
| EP4212477A1 (en) | 2022-01-12 | 2023-07-19 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Novel carbon nitride compositions and their use as photoelectrodes |
| CN114917942B (en) * | 2022-04-14 | 2023-09-26 | 华南理工大学 | Preparation method of one-dimensional nanorod-shaped carbon nitride photocatalyst and its application in photocatalytic oxidation of monosaccharides to synthesize lactic acid |
| CN114917942A (en) * | 2022-04-14 | 2022-08-19 | 华南理工大学 | Preparation method of one-dimensional nanorod carbon nitride photocatalyst and application of photocatalyst in synthesis of lactic acid through photocatalytic oxidation of monosaccharide |
| CN117088771A (en) * | 2022-05-13 | 2023-11-21 | 北京大学 | A method for recycling polystyrene |
| CN115466761A (en) * | 2022-08-25 | 2022-12-13 | 哈尔滨工业大学(深圳) | Method and equipment for preparing hydrogen by catalytically converting biomass through membrane-assisted bio-enzyme catalysis coupling |
| CN115466761B (en) * | 2022-08-25 | 2023-11-07 | 哈尔滨工业大学(深圳) | Method and equipment for preparing hydrogen by coupling membrane-assisted biological enzyme catalysis and photocatalysis to convert biomass |
| CN115634703A (en) * | 2022-09-15 | 2023-01-24 | 贵州黔大生态环境与健康研究院有限公司 | A kind of catalyst and its application |
| CN115786971A (en) * | 2022-10-14 | 2023-03-14 | 聊城大学 | A kind of preparation method of ruthenium phosphide porous nitrogen and phosphorus doped carbon |
| CN115337954A (en) * | 2022-10-17 | 2022-11-15 | 山东环投环境工程有限公司 | Composite photocatalyst based on carbon nitride and preparation method and application thereof |
| CN115337954B (en) * | 2022-10-17 | 2023-01-03 | 山东环投环境工程有限公司 | Composite photocatalyst based on carbon nitride and preparation method and application thereof |
| CN115779943A (en) * | 2022-10-27 | 2023-03-14 | 广东省科学院测试分析研究所(中国广州分析测试中心) | KSCN (KSCN) -based modified carbon nitride, preparation method thereof and photocatalytic H production method 2 O 2 In (1) |
| RU2800948C1 (en) * | 2022-12-22 | 2023-08-01 | Федеральное государственное бюджетное учреждение науки "Федеральный исследовательский центр " Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук" (ИК СО РАН, Институт катализа СО РАН) | Catalyst, method for its preparation, and method for photocatalytic production of hydrogen |
| CN115957792A (en) * | 2023-01-03 | 2023-04-14 | 浙江芯科半导体有限公司 | Method for preparing photocatalyst by using waste cigarette ends, photocatalyst and application |
| WO2024160923A1 (en) * | 2023-02-01 | 2024-08-08 | Cambridge Enterprise Limited | Method for upcycling plastic waste by enzymatic degradation and photoreforming |
| CN116239103A (en) * | 2023-02-20 | 2023-06-09 | 中国石油大学(华东) | Carbon dots and preparation method thereof, photocatalytic hydroboration method |
| CN116689006A (en) * | 2023-03-28 | 2023-09-05 | 上海大学 | Carbon nitride nano-sheet composite material and preparation method and application thereof |
| CN117720067A (en) * | 2024-01-29 | 2024-03-19 | 四川大学 | A method for photochemically preparing H2 from benzyl compounds |
| WO2025189296A1 (en) * | 2024-03-13 | 2025-09-18 | Hydrofuel Canada Inc. | Process of photochemical reforming of plastics using a carbon enabler |
| CN118237064A (en) * | 2024-05-28 | 2024-06-25 | 江西省科学院能源研究所 | Composite photocatalyst and preparation method and application thereof |
| CN118994599A (en) * | 2024-08-07 | 2024-11-22 | 华侨大学 | Sulfate lignin-heptazine conjugated polymer, and preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| GB201808905D0 (en) | 2018-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019229255A1 (en) | Photocatalyst and photocatalytic methods for producing hydrogen | |
| Ashraf et al. | Photoreforming of waste polymers for sustainable hydrogen fuel and chemicals feedstock: waste to energy | |
| He et al. | Hydrothermal and pyrolytic conversion of biomasses into catalysts for advanced oxidation treatments | |
| US11738330B2 (en) | Doped carbonaceous materials for photocatalytic removal of pollutants under visible light, making methods and applications of same | |
| Varma | Biomass-derived renewable carbonaceous materials for sustainable chemical and environmental applications | |
| Achilleos et al. | Photocatalytic hydrogen generation coupled to pollutant utilisation using carbon dots produced from biomass | |
| Jawad et al. | Adsorptive removal of methylene blue by chemically treated cellulosic waste banana (Musa sapientum) peels | |
| Anusuyadevi et al. | Floating photocatalysts for effluent refinement based on stable pickering cellulose foams and graphitic carbon nitride (g-C3N4) | |
| Rafatullah et al. | Oil palm biomass as a precursor of activated carbons: a review | |
| Pi et al. | g-C3N4 Modified biochar as an adsorptive and photocatalytic material for decontamination of aqueous organic pollutants | |
| WO2018096103A1 (en) | Photocatalyst and photocatalytic methods | |
| Shaik et al. | Titanium dioxide/graphene‐based nanocomposites as photocatalyst for environmental applications: A review | |
| Zhou et al. | Enhance photocatalytic hydrogen evolution by using alkaline pretreated corn stover as a sacrificial agent | |
| Shen et al. | Review on catalytic cracking of lignin for the production of fuels and high-value-added chemicals: advances, challenges, opportunities, and outlook | |
| Zerga et al. | Biobased kapok fiber nano-structure for energy and environment application: A critical review | |
| Xu et al. | Efficient photocatalytic cleavage of C–C bonds in β-1 lignin models and aromatic vicinal diols by combining alkali metal-treated graphitic carbon nitride and persulfate | |
| Katre et al. | Photocatalytic nanomaterials and their implications towards biomass conversion for renewable chemical and fuel production | |
| Saki et al. | Photoswitchable catalytic aerobic oxidation of biomass-based furfural: a selective route for the synthesis of 5-hydroxy-2 (5H)-furanone and maleic acid by using the CdS/MOF photocatalyst | |
| Iuliano et al. | A new nanocomposite from vesuvian slope pinecones for azo-dyes removal | |
| Yin et al. | Fe immobilized within accordion-like tubular carbon nitride as a catalyst for the Fenton-like degradation of ranitidine: synergetic roles of• OH and 1O2 | |
| He et al. | Recycling strategy for bioaqueous phase via catalytic wet air oxidation to biobased acetic acid solution | |
| Liu et al. | Photocatalytic technology in lignin: from traditional depolymerization to actively light-driven conversion | |
| Bui et al. | Delignified sugarcane bagasse decorated with hybrid ZIF-67/Ag/AgCl as efficient photocatalysts for water remediation under visible light irradiation | |
| Xu et al. | Distillery‐Waste‐Derived C− SiO2 Catalyst Support Reinforces Phenol Adsorption and Selective Hydrogenation | |
| Mokhtar et al. | Biomass and Plastic Photoreforming for Hydrogen and Valuable Chemicals Production |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19728043 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19728043 Country of ref document: EP Kind code of ref document: A1 |