WO2019208043A1 - Method for producing n-unprotected imine compound - Google Patents
Method for producing n-unprotected imine compound Download PDFInfo
- Publication number
- WO2019208043A1 WO2019208043A1 PCT/JP2019/012006 JP2019012006W WO2019208043A1 WO 2019208043 A1 WO2019208043 A1 WO 2019208043A1 JP 2019012006 W JP2019012006 W JP 2019012006W WO 2019208043 A1 WO2019208043 A1 WO 2019208043A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- otf
- reaction
- mmol
- producing
- Prior art date
Links
- AAFFTDXPYADISO-UHFFFAOYSA-N C1CC#CCC1 Chemical compound C1CC#CCC1 AAFFTDXPYADISO-UHFFFAOYSA-N 0.000 description 1
- XRDWXQSOWSFXOB-UHFFFAOYSA-N CC[O]#CCCN Chemical compound CC[O]#CCCN XRDWXQSOWSFXOB-UHFFFAOYSA-N 0.000 description 1
- BUZFUYFNPNXACJ-UHFFFAOYSA-N N=C(c1ccccc1)c(cc1)ccc1[N+]([O-])=O Chemical compound N=C(c1ccccc1)c(cc1)ccc1[N+]([O-])=O BUZFUYFNPNXACJ-UHFFFAOYSA-N 0.000 description 1
- ZYMCBJWUWHHVRX-UHFFFAOYSA-N [O-][N+](c(cc1)ccc1C(c1ccccc1)=O)=O Chemical compound [O-][N+](c(cc1)ccc1C(c1ccccc1)=O)=O ZYMCBJWUWHHVRX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/02—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of compounds containing imino groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/02—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups
- C07C251/20—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups having carbon atoms of imino groups being part of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/02—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups
- C07C251/24—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups having carbon atoms of imino groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/40—Nitrogen atoms, not forming part of a nitro radical, e.g. isatin semicarbazone
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/44—Radicals substituted by doubly-bound oxygen, sulfur, or nitrogen atoms, or by two such atoms singly-bound to the same carbon atom
- C07D213/53—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/78—Ring systems having three or more relevant rings
- C07D311/80—Dibenzopyrans; Hydrogenated dibenzopyrans
- C07D311/82—Xanthenes
- C07D311/84—Xanthenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 9
- C07D311/88—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a method for producing (synthesizing) an unprotected imine compound on nitrogen.
- Patent Document 1 requires a high temperature of reaction temperature of 100 ° C. or higher and an operation of blowing a large amount of ammonia gas that is highly toxic and difficult to handle, and is not easy to implement.
- the reaction substrate is limited to benzophenone imine, and the substrate generality is poor.
- An object of the present invention is to provide a method for easily and inexpensively producing various unprotected imine compounds on nitrogen.
- the present inventors have eliminated various limitations in the conventional method for producing an unprotected imine compound on nitrogen, and can synthesize various unprotected imine compounds on nitrogen easily and inexpensively, As a result of intensive studies, it was found that this can be solved by using a specific catalyst, and the present invention has been completed.
- the present invention is as follows.
- the metal salt having Lewis acidity is a triflate salt, nonaflate salt or trifluoromethanesulfonylimide salt of at least one metal selected from rare earth metals, Fe, In, Sn and Bi, or Sc (NO 3 ) 3 or characterized in that it is a BiBr 3 [1] production method of nitrogen on unprotected imine compounds described. [3] The method for producing an unprotected imine compound on nitrogen according to [2], wherein the rare earth metal is Sc, Y, Sm, Eu, Gd, Er, or Yb.
- the metal salt having Lewis acidity is Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf). ) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3
- the method for producing an unprotected imine compound on nitrogen as described in any one of [1] to [3].
- a nitrogen source compound (2) represented by Following formula (3) (R 1 and R 2 have the same meanings as those in the ketone compound (1) represented by the formula (1).)
- the nitrogen compound is characterized by producing the imine compound (3) or a salt thereof. A method for producing a protected imine compound.
- the method for producing an unprotected imine compound on nitrogen comprises a ketone in the presence of at least one catalyst selected from a catalyst containing a metal salt having Lewis acidity and a catalyst containing a fluorine-containing anion salt of tetraalkylammonium.
- the imine compound (3) or a salt thereof is produced by reacting the compound (1) with the nitrogen source compound (2).
- the production method of the present invention has a larger substrate selection range than conventional methods for producing unprotected imine compounds on nitrogen, and various inexpensive unprotected imine compounds on nitrogen can be obtained from commercially available compounds and catalysts that are readily available. It can be manufactured safely. Moreover, since it can implement under a normal pressure, a special reaction instrument and apparatus are not required and the unprotected imine compound on nitrogen can be obtained with a high yield on reaction conditions with easy implementation. In addition, the conventional method generally requires purification of an unprotected imine compound in order to remove a by-product that can inhibit the following reaction, whereas the present invention provides an unprotected imine compound on nitrogen in a high yield.
- the imine compound can be used as it is in the next reaction without removing it from the reaction system (Example) 6, 7 and 16 (one-pot synthesis)).
- Catalyst (Catalyst containing metal salt having Lewis acidity)
- the metal salt having Lewis acid include a rare earth metal and a metal triflate, nonaflate, or trifluoromethanesulfonylimide salt selected from Fe, In, Sn, and Bi, Sc (NO 3 ) 3 , and BiBr 3. it can.
- At least one metal triflate, nonaflate or trifluoromethanesulfonylimide salt selected from Sc, Y, Sm, Eu, Gd, Er, Yb, Fe, In, Sn and Bi, or Sc (NO 3 ) 3 or BiBr 3 is preferred, and is a triflate, nonaflate or trifluoromethanesulfonylimide salt of at least one metal selected from Sc, Y, Sm, Eu, Gd, Er, Yb, Fe, In, Sn and Bi.
- a triflate salt of at least one metal selected from Sc, Y, Eu, Er, Yb, Fe, Sn, and Bi, or a nonaflate salt is more preferable, and at least one selected from Sc, Er, Sn, and Bi.
- Some metal triflate or nonaflate salts are particularly preferable.
- the metal salt having Lewis acidity Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , Sc (NTf 2 ) 3 , Sc (NO 3 ) 3 , BiBr 3 etc. can be mentioned.
- the amount of the catalyst can be appropriately adjusted so that the reaction proceeds appropriately.
- 0.001 per 1 mol of the ketone compound (1) about 0.3 mol, preferably about 0.002 to 0.2 mol, more preferably about 0.005 to 0.1 mol.
- the alkyl part of the fluorine-containing anion salt of tetraalkylammonium preferably has 1 to 6 carbon atoms.
- Specific examples of the fluorine-containing anion salt of tetraalkylammonium include tetrabutylammonium fluoride (TBAF) and tetrabutylammonium dihydrogen trifluoride (TBAH 2 F 3 ). Tetrabutylammonium fluoride Is preferred. It may be an anhydride or a hydrate.
- the amount of the catalyst can be appropriately adjusted so that the reaction proceeds appropriately, but relative to 1 mol of the ketone compound (1) For example, it is about 0.005 to 0.3 mol, preferably about 0.01 to 0.2 mol, and more preferably about 0.03 to 0.1 mol.
- the ketone compound (1) used in the production method of the present invention is a compound represented by the following formula (1).
- R 1 and R 2 each represents an organic group and may be linked to each other to form a ring.
- examples of R 1 and R 2 include an aromatic group including an aromatic ring, a heterocyclic group including a heterocyclic ring, and an aliphatic group.
- examples of the aromatic ring of the aromatic group include benzene and naphthalene.
- examples of the heterocyclic ring of the heterocyclic group include pyridine, pyrazine, quinoline and the like.
- the aliphatic group may be a linear, branched or cyclic aliphatic group, and may be saturated or unsaturated. In addition, the aliphatic group may be blocked with a heteroatom (a heteroatom may be present between the carbon linkages).
- aromatic groups, heterocyclic groups and aliphatic groups may have a substituent.
- substituents include an alkyl group having 1 to 4 carbon atoms which may be substituted with a halogen atom, an alkoxy group having 1 to 4 carbon atoms which may be substituted with a halogen atom, a hydroxyl group and a halogen atom. it can.
- R 7 to R 11 each represents an aliphatic group, preferably an alkyl group having 1 to 4 carbon atoms.
- examples of the ketone compound (1) include the following.
- ketone compounds (1) a ketone compound having the following structure is extremely difficult to synthesize an imine compound by a conventional method using a Grignard reagent, etc., but according to the production method of the present invention, An imine compound can be obtained with a high yield.
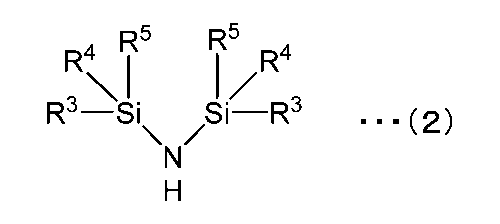
- the nitrogen source compound (2) used in the production method of the present invention is a compound represented by the following formula (2).
- R 3 and R 4 each represent a hydrogen atom or an aliphatic group.
- the aliphatic group may be a linear, branched or cyclic aliphatic group, and may be saturated or unsaturated. Among these, a saturated or unsaturated linear aliphatic group having 1 to 4 carbon atoms is preferable.
- R 5 represents a hydrogen atom, an aliphatic group, or a substituent having the following structure bonded to each other.
- the aliphatic group is the same as R 3 and R 4 .
- R 6 represents a hydrogen atom or an aliphatic group, and the aliphatic group is the same as R 3 and R 4 .
- nitrogen source compound (2) examples include the following.
- the amount of the nitrogen source compound (2) used in the production method of the present invention is, for example, about 1.0 to 3.0 mol, preferably about 1.0 to 1.5 mol, relative to 1 mol of the ketone compound (1). .
- the nitrogen source compound may be used in the same amount or slightly more, and it is not necessary to use it excessively.
- the imine compound (3) to be produced is a compound represented by the following formula (3).
- the imine compound (3) may be in the form of a salt.
- the kind of salt is not particularly limited, and examples thereof include halides (fluorides, chlorides, bromides, iodides), sulfonates, and the like.
- Solvents used in the production method of the present invention include chlorobenzene (preferably monochlorobenzene), toluene, tetrahydrofuran, dioxane (preferably 1,4-dioxane), fluorobenzene, dichloroethane, acetononitrile, alcohol, N, N-dimethyl.
- Formamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropylene urea (DMPU) ), 1,3-dimethyl-2-imidazolidinone (DMI) and the like, and these may be used as a mixture.
- DMF dimethyl sulfoxide
- NMP N-methyl-2-pyrrolidone
- DMA N-dimethylacetamide
- HMPA hexamethylphosphoric triamide
- DMPU N-dimethylpropylene urea
- DI 1,3-dimethyl-2-imidazolidinone
- chlorobenzene, toluene, tetrahydrofuran, fluorobenzene and acetononitrile are preferred.
- chlorobenzene, toluene, tetrahydrofuran, fluorobenzene, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N-dimethylacetamide, hexamethyl Phosphoric triamide, N, N-dimethylpropyleneurea and 1,3-dimethyl-2-imidazolidinone are preferred, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N-dimethyl Polar solvents such as acetamide, hexamethylphosphoric triamide, N, N-dimethylpropyleneurea and 1,3-dimethyl-2-imidazolidinone are more preferred.
- the reaction can be promoted by adding a polar solvent as an additional solvent to the nonpolar solvent.
- reaction accelerator in the present invention, a reaction accelerator can be used.
- the reaction accelerator include water, alcohol, silanol and the like.
- the alcohol include methanol, ethanol, butanol and the like.
- silanols include trimethylsilanol.
- the addition amount of the reaction accelerator is, for example, about 0.01 to 1.0 mol, preferably about 0.02 to 0.8 mol, preferably 0.05 to 0, per 1 mol of the ketone compound (1). More preferably, it is about 5 mol.
- the temperature (reaction temperature) for reacting the ketone compound (1) and the nitrogen source compound (2) in the production method of the present invention is preferably about 0 to 150 ° C.
- a metal salt having Lewis acidity it is more preferably from room temperature (25 ° C.) to about 150 ° C., more preferably from about 50 to 100 ° C.
- a catalyst containing a fluorine-containing anion salt of tetraalkylammonium is used, it is more preferably about 10 to 100 ° C, and further preferably about 15 to 70 ° C.
- the reaction in the production method of the present invention does not require an extremely low temperature or high temperature, and the production cost is low.
- the reaction time depends on other conditions such as the reaction temperature, but is, for example, about 0.25 to 48 hours, preferably about 0.5 to 24 hours, and more preferably about 0.75 to 15 hours. Preferably, about 5 to 15 hours is more preferable.
- the conditions of reaction temperature and reaction time can be determined optimally while confirming the yield.
- the yield is preferably 40 mol% or more, more preferably 60 mol% or more, further preferably 80 mol% or more, particularly preferably 90 mol% or more, and 95 mol% or more. Is most preferred.
- the reaction of the ketone compound (1) and the nitrogen source compound (2) can be carried out substantially without using a solvent.
- performing the reaction substantially without using a solvent means that the reaction is performed without using a general solvent amount of the solvent, for example, a reaction using a small amount of solvent that dissolves the catalyst. Is included in the reaction substantially using no solvent.
- the amount of solvent in the reaction using substantially no solvent in the production method of the present invention is, for example, 0.1 mL or less with respect to 1 mmol of the ketone compound (1).
- the yield exceeded 20% in all cases.
- Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3 were able to achieve a yield of 40% or more.
- the yield is 50% or more, and for Sm (OTf) 3 , the yield is 60% or more.
- Y (OTf), Eu (OTf) 3 the yield is 80% or more, and Sc (OTf) 3 , Er (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 and Sc (ONf)
- the yield was 95% or more.
- Sc (OTf) 3 achieves a yield of 80% or more despite being a low temperature and short time reaction condition of 70 ° C. for 1 hour, and is a particularly excellent catalyst. I understand.
- Example 4-6 Synthesis of di-p-tolylmethanimine
- the reaction was carried out using 0.20 mmol of 4,4′-dimethylbenzophenone under the above basic reaction conditions for 3 hours.
- Example 4-19-2 Synthesis of 2,2,2-trifluoro-1-phenylethaneimine on a large scale (10 mmol)
- a 50 mL flask was charged with scandim (III) trifluoromethanesulfonate (49.2 mg, 0.10 mmol, 1.0 mol%), 2,2,2-trifluoroacetophenone ( 1.4 mL, 10 mmol), fluorobenzene (10 mL, 1.0 M), bis (trimethylsilyl) amine (2.3 mL, 11 mmol, 1.1 equivalents) were added, and the mixture was heated and stirred at 90 ° C. for 24 hours.
- scandim (III) trifluoromethanesulfonate 49.2 mg, 0.10 mmol, 1.0 mol
- 2,2,2-trifluoroacetophenone 1.4 mL, 10 mmol
- fluorobenzene 10 mL, 1.0 M
- bis (trimethylsilyl) amine 2.3
- reaction accelerator [Examination of reaction accelerator] A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (1.0 mL, 1.0 M) as the solvent, nitrogen source compound (2) (0.22 mmol, 1 0.1 equivalent), a reaction accelerator (0.010 mmol, 5.0 mol%) was added. Trimethylsilanol (TMSOH) and methanol (MeOH) were used as reaction accelerators. After the mixture was stirred at 50 ° C., the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 7.
- the imine was obtained as a yellow oil (162 mg, 90% yield).
- unprotected ketimine on nitrogen was produced by using a catalyst containing a fluorine-containing anion salt of tetraalkylammonium.
- TBAF was able to achieve a yield of 80% or more.
- Additional solvents include N, N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropyleneurea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI) were used.
- the mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 10.
- N, N-dimethylformamide (DMF) is improved by the addition of 10 mol% to 200 mol% (0.1 to 2 equivalents), and 100 mol% is particularly excellent.
- Additional solvents include N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropylene Urea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI) were used. After the mixture was stirred at room temperature for 2 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 11.
- N N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N -An improvement in yield was observed by adding dimethylpropyleneurea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI). In particular, it can be seen that DMF and DMPU are excellent.
- the reaction proceeded even at a low temperature of 0 ° C.
- the reaction proceeded particularly well at room temperature (25 ° C.).
- Example 14-1 (Synthesis of cyclohexyl (phenyl) methanimine) Stirring was performed for 6 hours using 1.0 mmol of cyclohexyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclohexyl (phenyl) methanimine was produced in a yield of> 99%.
- Example 14-2 (Synthesis of cyclobutyl (phenyl) methanimine) Stirring was performed for 6 hours using 1.0 mmol of cyclobutyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclobutyl (phenyl) methanimine was produced in a yield of> 99%.
- Example 14-7 (Synthesis of 1-phenylpentane-1-imine) Stirring was performed for 24 hours using 1.0 mmol of valerophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylpentane-1-imine was produced in a yield of 81%, and 10% of unreacted valerophenone remained.
- Example 14-8 (Synthesis of dicyclopropylmethanimine) Stirring was carried out for 24 hours using 1.0 mmol of dicyclopropyl ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target dicyclopropylmethanimine was produced in a yield of 60%, and 35% of unreacted dicyclopropylketone remained.
- reaction proceeded without any problem even when the cyano source was reduced to 1.5 equivalents.
- reaction proceeded similarly when TMSCN was used as the cyano source.
- reaction accelerator (1) A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), reaction Accelerator (1.0 mmol, 1.0 eq) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) as catalyst were added.
- argon cyclohexyl (phenyl) ketone
- bis (trimethylsilyl) amine 2.0 mmol, 2.0 equivalents
- reaction Accelerator 1.0 mmol, 1.0 eq
- TBAF tetrabutylammonium fluoride
- TMSOH trimethylsilanol
- MeOH methanol
- EtOH ethanol
- the reaction is accelerated (the yield is improved) by adding trimethylsilanol, methanol, and ethanol, and it can be seen that ethanol is particularly excellent. It can be seen that the yield is improved when the amount of ethanol is 10 to 50 mol%, and particularly 10 mol% is excellent. Moreover, it turns out that a yield improves by making the quantity of a nitrogen source into 1.5 equivalent. Further, it can be seen that the reaction proceeds almost quantitatively by extending the reaction time to 24 hours. It can be seen that trimethylsilanol gives better results when extended to 24 hours.
- reaction accelerator (2) [Examination of reaction accelerator (2)] A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), solvent As N, N-dimethylformamide (1.0 mmol), tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) as a catalyst, and H 2 O (0 .10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 18.
- the present invention is industrially useful because an unprotected imine compound on nitrogen can be produced.
Abstract
[Problem] To provide a method for simply producing various N-unprotected imine compounds inexpensively and safely. [Solution] This method for producing a N-unprotected imine compound comprises reacting a ketone compound and a nitrogen source compound in the presence of at least one catalyst selected from among: a catalyst containing a Lewis acidic metal salt such as triflate salts of Sc, Y, Sm, Eu, Gd, Er, Yb, Fe, In, Sn, and Bi; and a catalyst containing a fluorine-containing anion salt of tetraalkylammonium.
Description
本発明は、窒素上無保護イミン化合物の製造(合成)方法に関する。
The present invention relates to a method for producing (synthesizing) an unprotected imine compound on nitrogen.
従来より、ケトン化合物からイミン化合物を合成することが行われているが、窒素上無保護イミン化合物を合成することは比較的難しいとされている。このような窒素上無保護イミン化合物を合成する方法としては、鉄塩若しくは鉄錯体の存在下、ベンゾフェノン類とアンモニアを反応させるベンゾフェノンイミンの合成方法が提案されている(例えば、特許文献1参照)。
Conventionally, synthesis of an imine compound from a ketone compound has been performed, but synthesis of an unprotected imine compound on nitrogen is considered to be relatively difficult. As a method for synthesizing such an unprotected imine compound on nitrogen, a method for synthesizing benzophenone imine in which benzophenone and ammonia are reacted in the presence of an iron salt or an iron complex has been proposed (for example, see Patent Document 1). .
この特許文献1に記載されている手法は、反応温度が100℃以上と高温であると共に、毒性が高く取り扱いが困難なアンモニアガスを大量に吹き込む操作が必要であり、その実施が容易ではない。また、反応基質がベンゾフェノンイミンに限定されており、基質一般性に乏しい。
The method described in Patent Document 1 requires a high temperature of reaction temperature of 100 ° C. or higher and an operation of blowing a large amount of ammonia gas that is highly toxic and difficult to handle, and is not easy to implement. Moreover, the reaction substrate is limited to benzophenone imine, and the substrate generality is poor.
また、Grignard試薬などの有機金属試薬を用いる手法や、LiN(SiMe3)2を用いる手法(例えば非特許文献1参照)も提案されているが、強塩基性条件のため、これらの手法も、基質一般性に乏しいという問題がある。さらに、イミノフォスフォラン(Ph3P=NH)を用いる方法(例えば非特許文献2参照)も提案されているが、この方法は、イミノフォスフォランの調製が別途必要であり、大量の除去困難な副生成物(Ph3P=O)を生じることから、大量合成への適用が困難である。
In addition, a method using an organometallic reagent such as a Grignard reagent and a method using LiN (SiMe 3 ) 2 (see, for example, Non-Patent Document 1) have been proposed. There is a problem that the generality of the substrate is poor. Furthermore, a method using iminophosphorane (Ph 3 P = NH) has also been proposed (for example, see Non-Patent Document 2). However, this method requires preparation of iminophosphorane separately, and a large amount of removal is difficult. Since a by-product (Ph 3 P═O) is generated, it is difficult to apply to mass synthesis.
本発明の課題は、安価かつ安全に、種々の窒素上無保護イミン化合物を簡便に製造する方法を提供することにある。
An object of the present invention is to provide a method for easily and inexpensively producing various unprotected imine compounds on nitrogen.
本発明者らは、従来の窒素上無保護イミン化合物の製法における様々な制限を排除して、安価かつ安全に、種々の窒素上無保護イミン化合物を簡便に合成することを可能とすべく、鋭意検討した結果、特定の触媒を用いることにより、これを解決できることを見いだし、本発明を完成するに至った。
The present inventors have eliminated various limitations in the conventional method for producing an unprotected imine compound on nitrogen, and can synthesize various unprotected imine compounds on nitrogen easily and inexpensively, As a result of intensive studies, it was found that this can be solved by using a specific catalyst, and the present invention has been completed.
すなわち、本発明は、以下のとおりのものである。
That is, the present invention is as follows.
[1]ルイス酸性を有する金属塩を含む触媒の存在下、
下記式(1)

(R1及びR2は、それぞれ有機基を表し、互いに連結して環を形成してもよい。)で示されるケトン化合物(1)と、
下記式(2)

[R3及びR4は、それぞれ水素原子又は脂肪族基を表し、R5は、水素原子、脂肪族基、又は互いに結合した下記構造の基

(R6は、水素原子又は脂肪族基を表す。)を表す。]で示される窒素源化合物(2)とを反応させて、
下記式(3)

(R1及びR2は、式(1)で示されるケトン化合物(1)におけるものと同義である。)で示されるイミン化合物(3)又はその塩を製造することを特徴とする窒素上無保護イミン化合物の製造方法。
[1] In the presence of a catalyst containing a metal salt having Lewis acidity,
Following formula (1)
(R 1 and R 2 each represent an organic group, and may be linked to each other to form a ring);
Following formula (2)
[R 3 and R 4 each represent a hydrogen atom or an aliphatic group, and R 5 represents a hydrogen atom, an aliphatic group, or a group having the following structure bonded to each other:
(R 6 represents a hydrogen atom or an aliphatic group). And a nitrogen source compound (2) represented by
Following formula (3)
(R 1 and R 2 have the same meanings as those in the ketone compound (1) represented by the formula (1).) The nitrogen compound is characterized by producing the imine compound (3) or a salt thereof. A method for producing a protected imine compound.
下記式(1)
下記式(2)
下記式(3)
Following formula (1)
Following formula (2)
Following formula (3)
[2]ルイス酸性を有する金属塩が、希土類金属,Fe,In,Sn及びBiから選ばれる少なくとも1種の金属のトリフラート塩、ノナフラート塩若しくはトリフルオロメタンスルホニルイミド塩、又はSc(NO3)3若しくはBiBr3であることを特徴とする[1]記載の窒素上無保護イミン化合物の製造方法。
[3]希土類金属が、Sc,Y,Sm,Eu,Gd,Er又はYbであることを特徴とする[2]記載の窒素上無保護イミン化合物の製造方法。
[4]ルイス酸性を有する金属塩が、Sc(OTf)3,Y(OTf)3,Sm(OTf)3,Eu(OTf)3,Gd(OTf)3,Er(OTf)3,Yb(OTf)3,Fe(OTf)3,In(OTf)3,Sn(OTf)2、Bi(OTf)3,Sc(ONf)3,及びSc(NTf2)3から選ばれる少なくとも1種であることを特徴とする[1]~[3]のいずれか記載の窒素上無保護イミン化合物の製造方法。 [2] The metal salt having Lewis acidity is a triflate salt, nonaflate salt or trifluoromethanesulfonylimide salt of at least one metal selected from rare earth metals, Fe, In, Sn and Bi, or Sc (NO 3 ) 3 or characterized in that it is a BiBr 3 [1] production method of nitrogen on unprotected imine compounds described.
[3] The method for producing an unprotected imine compound on nitrogen according to [2], wherein the rare earth metal is Sc, Y, Sm, Eu, Gd, Er, or Yb.
[4] The metal salt having Lewis acidity is Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf). ) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3 The method for producing an unprotected imine compound on nitrogen as described in any one of [1] to [3].
[3]希土類金属が、Sc,Y,Sm,Eu,Gd,Er又はYbであることを特徴とする[2]記載の窒素上無保護イミン化合物の製造方法。
[4]ルイス酸性を有する金属塩が、Sc(OTf)3,Y(OTf)3,Sm(OTf)3,Eu(OTf)3,Gd(OTf)3,Er(OTf)3,Yb(OTf)3,Fe(OTf)3,In(OTf)3,Sn(OTf)2、Bi(OTf)3,Sc(ONf)3,及びSc(NTf2)3から選ばれる少なくとも1種であることを特徴とする[1]~[3]のいずれか記載の窒素上無保護イミン化合物の製造方法。 [2] The metal salt having Lewis acidity is a triflate salt, nonaflate salt or trifluoromethanesulfonylimide salt of at least one metal selected from rare earth metals, Fe, In, Sn and Bi, or Sc (NO 3 ) 3 or characterized in that it is a BiBr 3 [1] production method of nitrogen on unprotected imine compounds described.
[3] The method for producing an unprotected imine compound on nitrogen according to [2], wherein the rare earth metal is Sc, Y, Sm, Eu, Gd, Er, or Yb.
[4] The metal salt having Lewis acidity is Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf). ) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3 The method for producing an unprotected imine compound on nitrogen as described in any one of [1] to [3].
[5]R1及び/又はR2が、芳香環及び/又は脂肪族基を含むことを特徴とする[1]~[4]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[6]クロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン、フルオロベンゼン、ジクロロエタン及びアセトニトリルから選ばれる少なくとも1種を含む溶媒中で、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[1]~[5]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[7]実質的に溶媒を用いることなく、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[1]~[5]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[8]水、アルコール及びシラノールから選ばれる少なくとも1種の反応促進剤の存在下、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[1]~[7]のいずれか記載の窒素上無保護イミン化合物の製造方法。 [5] The process for producing an unprotected imine compound on nitrogen as described in any one of [1] to [4], wherein R 1 and / or R 2 contains an aromatic ring and / or an aliphatic group.
[6] The reaction of the ketone compound (1) and the nitrogen source compound (2) is performed in a solvent containing at least one selected from chlorobenzene, toluene, tetrahydrofuran, dioxane, fluorobenzene, dichloroethane, and acetonitrile. [1] A process for producing an unprotected imine compound on nitrogen as described in any one of [5].
[7] The unprotected imine on nitrogen according to any one of [1] to [5], wherein the reaction of the ketone compound (1) and the nitrogen source compound (2) is carried out substantially without using a solvent. A method for producing a compound.
[8] The reaction between the ketone compound (1) and the nitrogen source compound (2) in the presence of at least one reaction accelerator selected from water, alcohol and silanol [1] to [7] The manufacturing method of the unprotected imine compound on nitrogen in any one of these.
[6]クロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン、フルオロベンゼン、ジクロロエタン及びアセトニトリルから選ばれる少なくとも1種を含む溶媒中で、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[1]~[5]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[7]実質的に溶媒を用いることなく、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[1]~[5]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[8]水、アルコール及びシラノールから選ばれる少なくとも1種の反応促進剤の存在下、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[1]~[7]のいずれか記載の窒素上無保護イミン化合物の製造方法。 [5] The process for producing an unprotected imine compound on nitrogen as described in any one of [1] to [4], wherein R 1 and / or R 2 contains an aromatic ring and / or an aliphatic group.
[6] The reaction of the ketone compound (1) and the nitrogen source compound (2) is performed in a solvent containing at least one selected from chlorobenzene, toluene, tetrahydrofuran, dioxane, fluorobenzene, dichloroethane, and acetonitrile. [1] A process for producing an unprotected imine compound on nitrogen as described in any one of [5].
[7] The unprotected imine on nitrogen according to any one of [1] to [5], wherein the reaction of the ketone compound (1) and the nitrogen source compound (2) is carried out substantially without using a solvent. A method for producing a compound.
[8] The reaction between the ketone compound (1) and the nitrogen source compound (2) in the presence of at least one reaction accelerator selected from water, alcohol and silanol [1] to [7] The manufacturing method of the unprotected imine compound on nitrogen in any one of these.
[9]テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒の存在下、
下記式(1)

(R1及びR2は、それぞれ有機基を表し、互いに連結して環を形成してもよい。)で示されるケトン化合物(1)と、
下記式(2)

[R3及びR4は、それぞれ水素原子又は脂肪族基を表し、R5は、水素原子、脂肪族基、又は互いに結合した下記構造の基

(R6は、水素原子又は脂肪族基を表す。)を表す。]で示される窒素源化合物(2)とを反応させて、
下記式(3)

(R1及びR2は、式(1)で示されるケトン化合物(1)におけるものと同義である。)で示されるイミン化合物(3)又はその塩を製造することを特徴とする窒素上無保護イミン化合物の製造方法。
[9] In the presence of a catalyst comprising a fluorine-containing anion salt of tetraalkylammonium,
Following formula (1)
(R 1 and R 2 each represent an organic group, and may be linked to each other to form a ring);
Following formula (2)
[R 3 and R 4 each represent a hydrogen atom or an aliphatic group, and R 5 represents a hydrogen atom, an aliphatic group, or a group having the following structure bonded to each other:
(R 6 represents a hydrogen atom or an aliphatic group). And a nitrogen source compound (2) represented by
Following formula (3)
(R 1 and R 2 have the same meanings as those in the ketone compound (1) represented by the formula (1).) The nitrogen compound is characterized by producing the imine compound (3) or a salt thereof. A method for producing a protected imine compound.
下記式(1)
下記式(2)
下記式(3)
Following formula (1)
Following formula (2)
Following formula (3)
[10]テトラアルキルアンモニウムのフッ素含有アニオン塩のアルキル部分の炭素数が、1~6であることを特徴とする[9]記載の窒素上無保護イミン化合物の製造方法。
[11]テトラアルキルアンモニウムのフッ素含有アニオン塩が、テトラブチルアンモニウムフルオリド、又はテトラブチルアンモニウムジヒドロゲントリフルオリドであることを特徴とする[9]又は[10]記載の窒素上無保護イミン化合物の製造方法。
[12]R1及び/又はR2が、芳香環及び/又は脂肪族基を含むことを特徴とする[9]~[11]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[13]クロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン、フルオロベンゼン、ジクロロエタン、N,N-ジメチルホルムアミド、ジメチルスルホキシド、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、N,N-ジメチルプロピレン尿素、及び1,3-ジメチル-2-イミダゾリジノンから選ばれる少なくとも1種を含む溶媒中で、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[9]~[12]記載の窒素上無保護イミン化合物の製造方法。
[14]実質的に溶媒を用いることなく、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[9]~[12]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[15]水、アルコール及びシラノールから選ばれる少なくとも1種の反応促進剤の存在下、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[9]~[14]のいずれか記載の窒素上無保護イミン化合物の製造方法。 [10] The method for producing an unprotected imine compound on nitrogen according to [9], wherein the alkyl moiety of the fluorine-containing anion salt of tetraalkylammonium has 1 to 6 carbon atoms.
[11] The unprotected imine compound on nitrogen according to [9] or [10], wherein the fluorine-containing anion salt of tetraalkylammonium is tetrabutylammonium fluoride or tetrabutylammonium dihydrogentrifluoride. Production method.
[12] The process for producing an unprotected imine compound on nitrogen as described in any one of [9] to [11], wherein R 1 and / or R 2 contains an aromatic ring and / or an aliphatic group.
[13] Chlorobenzene, toluene, tetrahydrofuran, dioxane, fluorobenzene, dichloroethane, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N-dimethylacetamide, hexamethylphosphoric triamide, N, N A reaction of a ketone compound (1) and a nitrogen source compound (2) in a solvent containing at least one selected from dimethylpropyleneurea and 1,3-dimethyl-2-imidazolidinone [9] A process for producing an unprotected imine compound on nitrogen as described in [12].
[14] The unprotected imine on nitrogen as described in any one of [9] to [12], wherein the reaction of the ketone compound (1) and the nitrogen source compound (2) is carried out substantially without using a solvent. A method for producing a compound.
[15] The reaction between the ketone compound (1) and the nitrogen source compound (2) is carried out in the presence of at least one reaction accelerator selected from water, alcohol and silanol [9] to [14] The manufacturing method of the unprotected imine compound on nitrogen in any one of these.
[11]テトラアルキルアンモニウムのフッ素含有アニオン塩が、テトラブチルアンモニウムフルオリド、又はテトラブチルアンモニウムジヒドロゲントリフルオリドであることを特徴とする[9]又は[10]記載の窒素上無保護イミン化合物の製造方法。
[12]R1及び/又はR2が、芳香環及び/又は脂肪族基を含むことを特徴とする[9]~[11]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[13]クロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン、フルオロベンゼン、ジクロロエタン、N,N-ジメチルホルムアミド、ジメチルスルホキシド、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、N,N-ジメチルプロピレン尿素、及び1,3-ジメチル-2-イミダゾリジノンから選ばれる少なくとも1種を含む溶媒中で、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[9]~[12]記載の窒素上無保護イミン化合物の製造方法。
[14]実質的に溶媒を用いることなく、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[9]~[12]のいずれか記載の窒素上無保護イミン化合物の製造方法。
[15]水、アルコール及びシラノールから選ばれる少なくとも1種の反応促進剤の存在下、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする[9]~[14]のいずれか記載の窒素上無保護イミン化合物の製造方法。 [10] The method for producing an unprotected imine compound on nitrogen according to [9], wherein the alkyl moiety of the fluorine-containing anion salt of tetraalkylammonium has 1 to 6 carbon atoms.
[11] The unprotected imine compound on nitrogen according to [9] or [10], wherein the fluorine-containing anion salt of tetraalkylammonium is tetrabutylammonium fluoride or tetrabutylammonium dihydrogentrifluoride. Production method.
[12] The process for producing an unprotected imine compound on nitrogen as described in any one of [9] to [11], wherein R 1 and / or R 2 contains an aromatic ring and / or an aliphatic group.
[13] Chlorobenzene, toluene, tetrahydrofuran, dioxane, fluorobenzene, dichloroethane, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N-dimethylacetamide, hexamethylphosphoric triamide, N, N A reaction of a ketone compound (1) and a nitrogen source compound (2) in a solvent containing at least one selected from dimethylpropyleneurea and 1,3-dimethyl-2-imidazolidinone [9] A process for producing an unprotected imine compound on nitrogen as described in [12].
[14] The unprotected imine on nitrogen as described in any one of [9] to [12], wherein the reaction of the ketone compound (1) and the nitrogen source compound (2) is carried out substantially without using a solvent. A method for producing a compound.
[15] The reaction between the ketone compound (1) and the nitrogen source compound (2) is carried out in the presence of at least one reaction accelerator selected from water, alcohol and silanol [9] to [14] The manufacturing method of the unprotected imine compound on nitrogen in any one of these.
本発明によれば、安価かつ安全に、種々の窒素上無保護イミン化合物を簡便に製造することができる。
According to the present invention, various unprotected imine compounds on nitrogen can be easily and inexpensively produced.
本発明に係る窒素上無保護イミン化合物の製造方法は、ルイス酸性を有する金属塩を含む触媒、及びテトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒から選ばれる少なくとも1種の触媒の存在下、ケトン化合物(1)と窒素源化合物(2)を反応させて、イミン化合物(3)又はその塩を製造することを特徴とする。
The method for producing an unprotected imine compound on nitrogen according to the present invention comprises a ketone in the presence of at least one catalyst selected from a catalyst containing a metal salt having Lewis acidity and a catalyst containing a fluorine-containing anion salt of tetraalkylammonium. The imine compound (3) or a salt thereof is produced by reacting the compound (1) with the nitrogen source compound (2).
本発明の製造方法は、従来の窒素上無保護イミン化合物の製造方法に比べて基質の選択幅が大きく、容易に入手可能な市販の化合物及び触媒から種々の窒素上無保護イミン化合物を安価かつ安全に製造することができる。また、常圧下で実施可能なため、特殊な反応器具や装置を必要とせず、実施容易な反応条件にて高い収率で窒素上無保護イミン化合物を得ることができる。さらに、従来法では次の反応を阻害しうる副生成物を取り除くために一般に無保護イミン化合物の精製が必要であるのに対し、本発明では高い収率で窒素上無保護イミン化合物が得られ、かつ共生成物(ヘキサメチルジシロキサン等)の反応性が低く次の反応を阻害しづらいことから、イミン化合物を反応系から取り出すことなく、そのまま次の反応に利用することもできる(実施例6,7及び16(ワンポット合成)参照)。
The production method of the present invention has a larger substrate selection range than conventional methods for producing unprotected imine compounds on nitrogen, and various inexpensive unprotected imine compounds on nitrogen can be obtained from commercially available compounds and catalysts that are readily available. It can be manufactured safely. Moreover, since it can implement under a normal pressure, a special reaction instrument and apparatus are not required and the unprotected imine compound on nitrogen can be obtained with a high yield on reaction conditions with easy implementation. In addition, the conventional method generally requires purification of an unprotected imine compound in order to remove a by-product that can inhibit the following reaction, whereas the present invention provides an unprotected imine compound on nitrogen in a high yield. In addition, since the reactivity of the co-product (hexamethyldisiloxane, etc.) is low and it is difficult to inhibit the next reaction, the imine compound can be used as it is in the next reaction without removing it from the reaction system (Example) 6, 7 and 16 (one-pot synthesis)).
[触媒]
(ルイス酸性を有する金属塩を含む触媒)
ルイス酸性を有する金属塩としては、希土類金属並びにFe,In,Sn及びBiから選ばれる金属のトリフラート塩、ノナフラート塩若しくはトリフルオロメタンスルホニルイミド塩や、Sc(NO3)3、BiBr3を挙げることができる。 [catalyst]
(Catalyst containing metal salt having Lewis acidity)
Examples of the metal salt having Lewis acid include a rare earth metal and a metal triflate, nonaflate, or trifluoromethanesulfonylimide salt selected from Fe, In, Sn, and Bi, Sc (NO 3 ) 3 , and BiBr 3. it can.
(ルイス酸性を有する金属塩を含む触媒)
ルイス酸性を有する金属塩としては、希土類金属並びにFe,In,Sn及びBiから選ばれる金属のトリフラート塩、ノナフラート塩若しくはトリフルオロメタンスルホニルイミド塩や、Sc(NO3)3、BiBr3を挙げることができる。 [catalyst]
(Catalyst containing metal salt having Lewis acidity)
Examples of the metal salt having Lewis acid include a rare earth metal and a metal triflate, nonaflate, or trifluoromethanesulfonylimide salt selected from Fe, In, Sn, and Bi, Sc (NO 3 ) 3 , and BiBr 3. it can.
これらの中でも、Sc,Y,Sm,Eu,Gd,Er,Yb,Fe,In,Sn及びBiから選ばれる少なくとも1種の金属のトリフラート塩、ノナフラート塩若しくはトリフルオロメタンスルホニルイミド塩、又はSc(NO3)3若しくはBiBr3が好ましく、Sc,Y,Sm,Eu,Gd,Er,Yb,Fe,In,Sn及びBiから選ばれる少なくとも1種の金属のトリフラート塩、ノナフラート塩若しくはトリフルオロメタンスルホニルイミド塩がより好ましく、Sc,Y,Eu,Er,Yb,Fe,Sn及びBiから選ばれる少なくとも1種の金属のトリフラート塩、若しくはノナフラート塩がさらに好ましく、Sc,Er,Sn及びBiから選ばれる少なくとも1種の金属のトリフラート塩若しくはノナフラート塩が特に好ましい。
Among these, at least one metal triflate, nonaflate or trifluoromethanesulfonylimide salt selected from Sc, Y, Sm, Eu, Gd, Er, Yb, Fe, In, Sn and Bi, or Sc (NO 3 ) 3 or BiBr 3 is preferred, and is a triflate, nonaflate or trifluoromethanesulfonylimide salt of at least one metal selected from Sc, Y, Sm, Eu, Gd, Er, Yb, Fe, In, Sn and Bi. Is more preferable, and a triflate salt of at least one metal selected from Sc, Y, Eu, Er, Yb, Fe, Sn, and Bi, or a nonaflate salt is more preferable, and at least one selected from Sc, Er, Sn, and Bi. Some metal triflate or nonaflate salts are particularly preferable.
具体的に、ルイス酸性を有する金属塩としては、Sc(OTf)3,Y(OTf)3,Sm(OTf)3,Eu(OTf)3,Gd(OTf)3,Er(OTf)3,Yb(OTf)3,Fe(OTf)3,In(OTf)3,Sn(OTf)2、Bi(OTf)3,Sc(ONf)3、Sc(NTf2)3,Sc(NO3)3,BiBr3等を挙げることができる。
Specifically, as the metal salt having Lewis acidity, Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , Sc (NTf 2 ) 3 , Sc (NO 3 ) 3 , BiBr 3 etc. can be mentioned.
本発明の製造方法においてルイス酸性を有する金属塩を用いる場合、その触媒量としては、反応が適切に進むよう適宜調整することができるが、ケトン化合物(1)1molに対して、例えば0.001~0.3mol程度であり、0.002~0.2mol程度であることが好ましく、0.005~0.1mol程度であることがより好ましい。
When a metal salt having Lewis acidity is used in the production method of the present invention, the amount of the catalyst can be appropriately adjusted so that the reaction proceeds appropriately. For example, 0.001 per 1 mol of the ketone compound (1). About 0.3 mol, preferably about 0.002 to 0.2 mol, more preferably about 0.005 to 0.1 mol.
(テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒)
テトラアルキルアンモニウムのフッ素含有アニオン塩のアルキル部分は炭素数1~6であることが好ましい。テトラアルキルアンモニウムのフッ素含有アニオン塩としては、具体的には、テトラブチルアンモニウムフルオリド(TBAF)、テトラブチルアンモニウムジヒドロゲントリフルオリド(TBAH2F3)等を挙げることができ、テトラブチルアンモニウムフルオリドが好ましい。なお、無水物であっても、水和物であってもよい。 (Catalyst containing a fluorine-containing anion salt of tetraalkylammonium)
The alkyl part of the fluorine-containing anion salt of tetraalkylammonium preferably has 1 to 6 carbon atoms. Specific examples of the fluorine-containing anion salt of tetraalkylammonium include tetrabutylammonium fluoride (TBAF) and tetrabutylammonium dihydrogen trifluoride (TBAH 2 F 3 ). Tetrabutylammonium fluoride Is preferred. It may be an anhydride or a hydrate.
テトラアルキルアンモニウムのフッ素含有アニオン塩のアルキル部分は炭素数1~6であることが好ましい。テトラアルキルアンモニウムのフッ素含有アニオン塩としては、具体的には、テトラブチルアンモニウムフルオリド(TBAF)、テトラブチルアンモニウムジヒドロゲントリフルオリド(TBAH2F3)等を挙げることができ、テトラブチルアンモニウムフルオリドが好ましい。なお、無水物であっても、水和物であってもよい。 (Catalyst containing a fluorine-containing anion salt of tetraalkylammonium)
The alkyl part of the fluorine-containing anion salt of tetraalkylammonium preferably has 1 to 6 carbon atoms. Specific examples of the fluorine-containing anion salt of tetraalkylammonium include tetrabutylammonium fluoride (TBAF) and tetrabutylammonium dihydrogen trifluoride (TBAH 2 F 3 ). Tetrabutylammonium fluoride Is preferred. It may be an anhydride or a hydrate.
本発明の製造方法においてテトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒を用いる場合、その触媒量としては、反応が適切に進むよう適宜調整することができるが、ケトン化合物(1)1molに対して、例えば0.005~0.3mol程度であり、0.01~0.2mol程度であることが好ましく、0.03~0.1mol程度であることがより好ましい。
When a catalyst containing a fluorine-containing anion salt of tetraalkylammonium is used in the production method of the present invention, the amount of the catalyst can be appropriately adjusted so that the reaction proceeds appropriately, but relative to 1 mol of the ketone compound (1) For example, it is about 0.005 to 0.3 mol, preferably about 0.01 to 0.2 mol, and more preferably about 0.03 to 0.1 mol.
テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒を用いることにより、ルイス酸性を有する金属塩を含む触媒を用いた手法では高収率で合成することが困難だったアルキルケチミンについても高収率で合成することができる。
By using a catalyst containing a fluorine-containing anion salt of tetraalkylammonium, it is also possible to synthesize alkyl ketimine in high yield, which was difficult to synthesize in high yield using a catalyst containing a Lewis acid metal salt. can do.
[ケトン化合物(1)]
本発明の製造方法で用いるケトン化合物(1)は、下記式(1)で示される化合物である。 [Ketone compound (1)]
The ketone compound (1) used in the production method of the present invention is a compound represented by the following formula (1).
本発明の製造方法で用いるケトン化合物(1)は、下記式(1)で示される化合物である。 [Ketone compound (1)]
The ketone compound (1) used in the production method of the present invention is a compound represented by the following formula (1).

R1及びR2は、それぞれ有機基を表し、互いに連結して環を形成してもよい。具体的に、R1及びR2としては、芳香環を含む芳香族基、複素環を含む複素環基、脂肪族基等を挙げることができる。芳香族基の芳香環としては、ベンゼン、ナフタレン等を挙げることができる。複素環基の複素環としては、ピリジン、ピラジン、キノリン等を挙げることができる。脂肪族基としては、直鎖状、分岐状、環状のいずれの脂肪族基でもよく、飽和、不飽和のいずれであってもよい。また、脂肪族基は、ヘテロ原子で遮断されていてもよい(炭素連結間にヘテロ原子が存在していてもよい)。
R 1 and R 2 each represents an organic group and may be linked to each other to form a ring. Specifically, examples of R 1 and R 2 include an aromatic group including an aromatic ring, a heterocyclic group including a heterocyclic ring, and an aliphatic group. Examples of the aromatic ring of the aromatic group include benzene and naphthalene. Examples of the heterocyclic ring of the heterocyclic group include pyridine, pyrazine, quinoline and the like. The aliphatic group may be a linear, branched or cyclic aliphatic group, and may be saturated or unsaturated. In addition, the aliphatic group may be blocked with a heteroatom (a heteroatom may be present between the carbon linkages).
これらの芳香族基、複素環基、脂肪族基は、置換基を有していてもよい。置換基としては、ハロゲン原子で置換されていてもよい炭素数1~4のアルキル基、ハロゲン原子で置換されていてもよい炭素数1~4のアルコキシ基、水酸基、ハロゲン原子等を挙げることができる。また、以下に示す置換基を挙げることができる。なお、R7~R11は、それぞれ脂肪族基を表し、炭素数1~4のアルキル基が好ましい。
These aromatic groups, heterocyclic groups and aliphatic groups may have a substituent. Examples of the substituent include an alkyl group having 1 to 4 carbon atoms which may be substituted with a halogen atom, an alkoxy group having 1 to 4 carbon atoms which may be substituted with a halogen atom, a hydroxyl group and a halogen atom. it can. Moreover, the substituent shown below can be mentioned. R 7 to R 11 each represents an aliphatic group, preferably an alkyl group having 1 to 4 carbon atoms.

具体的に、ケトン化合物(1)としては、次に示すものを挙げることができる。
Specifically, examples of the ketone compound (1) include the following.

上記ケトン化合物(1)の中でも、下記構造のケトン化合物は、Grignard試薬を用いる従来の方法等でも、イミン化合物を合成することが極めて困難なものであるが、本発明の製造方法によれば、高い収率でイミン化合物を得ることができる。
Among the ketone compounds (1), a ketone compound having the following structure is extremely difficult to synthesize an imine compound by a conventional method using a Grignard reagent, etc., but according to the production method of the present invention, An imine compound can be obtained with a high yield.

[窒素源化合物(2)]
本発明の製造方法で用いる窒素源化合物(2)は、下記式(2)で示される化合物である。 [Nitrogen source compound (2)]
The nitrogen source compound (2) used in the production method of the present invention is a compound represented by the following formula (2).
本発明の製造方法で用いる窒素源化合物(2)は、下記式(2)で示される化合物である。 [Nitrogen source compound (2)]
The nitrogen source compound (2) used in the production method of the present invention is a compound represented by the following formula (2).
R3及びR4は、それぞれ水素原子又は脂肪族基を表す。脂肪族基としては、直鎖状、分岐状、環状のいずれの脂肪族基でもよく、飽和、不飽和のいずれであってもよい。これらの中でも、飽和又は不飽和の炭素数1~4の直鎖状脂肪族基が好ましい。
R5は、水素原子、脂肪族基、又は互いに結合した下記構造の置換基を表す。脂肪族基については、R3及びR4と同様である。 R 3 and R 4 each represent a hydrogen atom or an aliphatic group. The aliphatic group may be a linear, branched or cyclic aliphatic group, and may be saturated or unsaturated. Among these, a saturated or unsaturated linear aliphatic group having 1 to 4 carbon atoms is preferable.
R 5 represents a hydrogen atom, an aliphatic group, or a substituent having the following structure bonded to each other. The aliphatic group is the same as R 3 and R 4 .
R5は、水素原子、脂肪族基、又は互いに結合した下記構造の置換基を表す。脂肪族基については、R3及びR4と同様である。 R 3 and R 4 each represent a hydrogen atom or an aliphatic group. The aliphatic group may be a linear, branched or cyclic aliphatic group, and may be saturated or unsaturated. Among these, a saturated or unsaturated linear aliphatic group having 1 to 4 carbon atoms is preferable.
R 5 represents a hydrogen atom, an aliphatic group, or a substituent having the following structure bonded to each other. The aliphatic group is the same as R 3 and R 4 .

R6は、水素原子又は脂肪族基を表し、脂肪族基については、R3及びR4と同様である。
R 6 represents a hydrogen atom or an aliphatic group, and the aliphatic group is the same as R 3 and R 4 .
具体的に、窒素源化合物(2)としては、次に示すものを挙げることができる。
Specific examples of the nitrogen source compound (2) include the following.

本発明の製造方法において用いる窒素源化合物(2)の量としては、ケトン化合物(1)1molに対して、例えば1.0~3.0mol程度であり、1.0~1.5mol程度が好ましい。本発明の製造方法においては、窒素源化合物を同量又は若干多い程度用いればよく、過剰に用いる必要はない。
The amount of the nitrogen source compound (2) used in the production method of the present invention is, for example, about 1.0 to 3.0 mol, preferably about 1.0 to 1.5 mol, relative to 1 mol of the ketone compound (1). . In the production method of the present invention, the nitrogen source compound may be used in the same amount or slightly more, and it is not necessary to use it excessively.
[イミン化合物(3)]
製造されるイミン化合物(3)は、下記式(3)で示される化合物である。 [Imine Compound (3)]
The imine compound (3) to be produced is a compound represented by the following formula (3).
製造されるイミン化合物(3)は、下記式(3)で示される化合物である。 [Imine Compound (3)]
The imine compound (3) to be produced is a compound represented by the following formula (3).

R1及びR2は、ケトン化合物(1)で説明したのと同義である。
また、イミン化合物(3)は、塩の形態であってもよい。塩の種類は、特に限定されず、例えば、ハロゲン化物(フッ化物、塩化物、臭化物、ヨウ化物)、スルホン酸塩などを挙げることができる。 R 1 and R 2 have the same meaning as described for the ketone compound (1).
The imine compound (3) may be in the form of a salt. The kind of salt is not particularly limited, and examples thereof include halides (fluorides, chlorides, bromides, iodides), sulfonates, and the like.
また、イミン化合物(3)は、塩の形態であってもよい。塩の種類は、特に限定されず、例えば、ハロゲン化物(フッ化物、塩化物、臭化物、ヨウ化物)、スルホン酸塩などを挙げることができる。 R 1 and R 2 have the same meaning as described for the ketone compound (1).
The imine compound (3) may be in the form of a salt. The kind of salt is not particularly limited, and examples thereof include halides (fluorides, chlorides, bromides, iodides), sulfonates, and the like.
[溶媒]
本発明の製造方法で用いる溶媒としては、クロロベンゼン(好ましくはモノクロロベンゼン)、トルエン、テトラヒドロフラン、ジオキサン(好ましくは1,4-ジオキサン)、フルオロベンゼン、ジクロロエタン、アセトノニトリル、アルコール、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)等の有機溶媒を挙げることができ、これらは混合して用いてもよい。 [solvent]
Solvents used in the production method of the present invention include chlorobenzene (preferably monochlorobenzene), toluene, tetrahydrofuran, dioxane (preferably 1,4-dioxane), fluorobenzene, dichloroethane, acetononitrile, alcohol, N, N-dimethyl. Formamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropylene urea (DMPU) ), 1,3-dimethyl-2-imidazolidinone (DMI) and the like, and these may be used as a mixture.
本発明の製造方法で用いる溶媒としては、クロロベンゼン(好ましくはモノクロロベンゼン)、トルエン、テトラヒドロフラン、ジオキサン(好ましくは1,4-ジオキサン)、フルオロベンゼン、ジクロロエタン、アセトノニトリル、アルコール、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)等の有機溶媒を挙げることができ、これらは混合して用いてもよい。 [solvent]
Solvents used in the production method of the present invention include chlorobenzene (preferably monochlorobenzene), toluene, tetrahydrofuran, dioxane (preferably 1,4-dioxane), fluorobenzene, dichloroethane, acetononitrile, alcohol, N, N-dimethyl. Formamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropylene urea (DMPU) ), 1,3-dimethyl-2-imidazolidinone (DMI) and the like, and these may be used as a mixture.
ルイス酸性を有する金属塩を含む触媒を用いる場合、クロロベンゼン、トルエン、テトラヒドロフラン、フルオロベンゼン、アセトノニトリルが好ましい。
When using a catalyst containing a metal salt having Lewis acidity, chlorobenzene, toluene, tetrahydrofuran, fluorobenzene and acetononitrile are preferred.
また、テトラアルキルアンモニウムのフッ素含有アニオン塩を用いる場合、クロロベンゼン、トルエン、テトラヒドロフラン、フルオロベンゼン、N,N-ジメチルホルムアミド、ジメチルスルホキシド、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、N,N-ジメチルプロピレン尿素、及び1,3-ジメチル-2-イミダゾリジノンが好ましく、N,N-ジメチルホルムアミド、ジメチルスルホキシド、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、N,N-ジメチルプロピレン尿素、1,3-ジメチル-2-イミダゾリジノンなどの極性溶媒がより好ましい。例えば、無極性溶媒に、追加溶媒として極性溶媒を添加することにより、反応を促進することができる。
When a fluorine-containing anion salt of tetraalkylammonium is used, chlorobenzene, toluene, tetrahydrofuran, fluorobenzene, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N-dimethylacetamide, hexamethyl Phosphoric triamide, N, N-dimethylpropyleneurea and 1,3-dimethyl-2-imidazolidinone are preferred, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N-dimethyl Polar solvents such as acetamide, hexamethylphosphoric triamide, N, N-dimethylpropyleneurea and 1,3-dimethyl-2-imidazolidinone are more preferred. For example, the reaction can be promoted by adding a polar solvent as an additional solvent to the nonpolar solvent.
[反応促進剤]
本発明においては、反応促進剤を用いることができる。反応促進剤としては、水、アルコール、シラノール等を挙げることができる。アルコールとしては、メタノール、エタノール、ブタノール等を例示することができる。シラノールとしては、トリメチルシラノール等を例示することができる。 [Reaction accelerator]
In the present invention, a reaction accelerator can be used. Examples of the reaction accelerator include water, alcohol, silanol and the like. Examples of the alcohol include methanol, ethanol, butanol and the like. Examples of silanols include trimethylsilanol.
本発明においては、反応促進剤を用いることができる。反応促進剤としては、水、アルコール、シラノール等を挙げることができる。アルコールとしては、メタノール、エタノール、ブタノール等を例示することができる。シラノールとしては、トリメチルシラノール等を例示することができる。 [Reaction accelerator]
In the present invention, a reaction accelerator can be used. Examples of the reaction accelerator include water, alcohol, silanol and the like. Examples of the alcohol include methanol, ethanol, butanol and the like. Examples of silanols include trimethylsilanol.
反応促進剤の添加量としては、ケトン化合物(1)1molに対して、例えば0.01~1.0mol程度であり、0.02~0.8mol程度であることが好ましく、0.05~0.5mol程度であることがより好ましい。
The addition amount of the reaction accelerator is, for example, about 0.01 to 1.0 mol, preferably about 0.02 to 0.8 mol, preferably 0.05 to 0, per 1 mol of the ketone compound (1). More preferably, it is about 5 mol.
[その他の条件]
本発明の製造方法におけるケトン化合物(1)と窒素源化合物(2)を反応させる温度(反応温度)としては、0~150℃程度であることが好ましい。ルイス酸性を有する金属塩を用いる場合、常温(25℃)~150℃程度であることがより好ましく、50~100℃程度であることがさらに好ましい。テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒を用いる場合、10~100℃程度であることがより好ましく、15~70℃程度であることがさらに好ましい。本発明の製造方法における反応は、極端な低温や高温を必要とせず、製造コストが安価なものとなる。 [Other conditions]
The temperature (reaction temperature) for reacting the ketone compound (1) and the nitrogen source compound (2) in the production method of the present invention is preferably about 0 to 150 ° C. When a metal salt having Lewis acidity is used, it is more preferably from room temperature (25 ° C.) to about 150 ° C., more preferably from about 50 to 100 ° C. When a catalyst containing a fluorine-containing anion salt of tetraalkylammonium is used, it is more preferably about 10 to 100 ° C, and further preferably about 15 to 70 ° C. The reaction in the production method of the present invention does not require an extremely low temperature or high temperature, and the production cost is low.
本発明の製造方法におけるケトン化合物(1)と窒素源化合物(2)を反応させる温度(反応温度)としては、0~150℃程度であることが好ましい。ルイス酸性を有する金属塩を用いる場合、常温(25℃)~150℃程度であることがより好ましく、50~100℃程度であることがさらに好ましい。テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒を用いる場合、10~100℃程度であることがより好ましく、15~70℃程度であることがさらに好ましい。本発明の製造方法における反応は、極端な低温や高温を必要とせず、製造コストが安価なものとなる。 [Other conditions]
The temperature (reaction temperature) for reacting the ketone compound (1) and the nitrogen source compound (2) in the production method of the present invention is preferably about 0 to 150 ° C. When a metal salt having Lewis acidity is used, it is more preferably from room temperature (25 ° C.) to about 150 ° C., more preferably from about 50 to 100 ° C. When a catalyst containing a fluorine-containing anion salt of tetraalkylammonium is used, it is more preferably about 10 to 100 ° C, and further preferably about 15 to 70 ° C. The reaction in the production method of the present invention does not require an extremely low temperature or high temperature, and the production cost is low.
反応時間としては、反応温度等の他の条件にもよるが、例えば0.25~48時間程度であり、0.5~24時間程度であることが好ましく、0.75~15時間程度がより好ましく、5~15時間程度がさらに好ましい。
The reaction time depends on other conditions such as the reaction temperature, but is, for example, about 0.25 to 48 hours, preferably about 0.5 to 24 hours, and more preferably about 0.75 to 15 hours. Preferably, about 5 to 15 hours is more preferable.
反応温度や反応時間の条件は、収率を確認しつつ最適な条件を決定することができる。収率としては、40mol%以上であることが好ましく、60mol%以上であることがより好ましく、80mol%以上であることがさらに好ましく、90mol%以上であることが特に好ましく、95mol%以上であることが最も好ましい。
The conditions of reaction temperature and reaction time can be determined optimally while confirming the yield. The yield is preferably 40 mol% or more, more preferably 60 mol% or more, further preferably 80 mol% or more, particularly preferably 90 mol% or more, and 95 mol% or more. Is most preferred.
本発明の製造方法においては、実質的に溶媒を用いずにケトン化合物(1)と窒素源化合物(2)の反応を行うことも可能である。ここで、実質的に溶媒を用いずに反応を行うとは、一般的な溶媒量の溶媒を用いずに反応を行うことをいい、例えば、触媒を溶解する程度の少量の溶媒を用いた反応は、実質的に溶媒を用いない反応に含まれる。具体的に、本発明の製造方法における実質的に溶媒を用いない反応での溶媒量は、ケトン化合物(1)1mmolに対して、例えば0.1mL以下である。
In the production method of the present invention, the reaction of the ketone compound (1) and the nitrogen source compound (2) can be carried out substantially without using a solvent. Here, performing the reaction substantially without using a solvent means that the reaction is performed without using a general solvent amount of the solvent, for example, a reaction using a small amount of solvent that dissolves the catalyst. Is included in the reaction substantially using no solvent. Specifically, the amount of solvent in the reaction using substantially no solvent in the production method of the present invention is, for example, 0.1 mL or less with respect to 1 mmol of the ketone compound (1).
[触媒の検討(1)]
4mLのバイアルに撹拌子を入れ、触媒(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒としてのモノクロロベンゼン(0.20mL,1.0M)、窒素源化合物(2)としてのビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を90℃で12時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表1に示す。 [Examination of catalyst (1)]
A stir bar was placed in a 4 mL vial, a catalyst (0.010 mmol, 5.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (0.20 mL, 1.0 M) as the solvent, bis (trimethylsilyl) as the nitrogen source compound (2) Amine (0.22 mmol, 1.1 eq) was added. After the mixture was stirred at 90 ° C. for 12 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 1.
4mLのバイアルに撹拌子を入れ、触媒(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒としてのモノクロロベンゼン(0.20mL,1.0M)、窒素源化合物(2)としてのビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を90℃で12時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表1に示す。 [Examination of catalyst (1)]
A stir bar was placed in a 4 mL vial, a catalyst (0.010 mmol, 5.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (0.20 mL, 1.0 M) as the solvent, bis (trimethylsilyl) as the nitrogen source compound (2) Amine (0.22 mmol, 1.1 eq) was added. After the mixture was stirred at 90 ° C. for 12 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 1.

表1に示すように、本発明の製造方法で用いられる触媒(entry1-15)の場合、いずれも20%を超える収率であった。これらの中でも、Sc(OTf)3,Y(OTf)3,Sm(OTf)3,Eu(OTf)3,Gd(OTf)3,Er(OTf)3,Yb(OTf)3,Fe(OTf)3,In(OTf)3,Sn(OTf)2、Bi(OTf)3、Sc(ONf)3、Sc(NTf2)3は、収率40%以上を達成することができた。Gd(OTf)3及びSc(NTf2)3については50%以上の収率であり、Sm(OTf)3については60%以上の収率であり、Y(OTf)、Eu(OTf)3、Fe(OTf)3及びYb(OTf)3については80%以上の収率であり、Sc(OTf)3、Er(OTf)3、Sn(OTf)2、Bi(OTf)3及びSc(ONf)3については95%以上の収率であった。
なお、以上の結果は、無触媒(entry 16)、及び類似の反応を触媒することが報告(T. Morimoto, M. Sekiya, Chem. Lett. 1985, 1371.)されているTMSOTf(entry 17)よりも優れていた。 As shown in Table 1, in the case of the catalyst (entry 1-15) used in the production method of the present invention, the yield exceeded 20% in all cases. Among these, Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3 were able to achieve a yield of 40% or more. For Gd (OTf) 3 and Sc (NTf 2 ) 3 , the yield is 50% or more, and for Sm (OTf) 3 , the yield is 60% or more. Y (OTf), Eu (OTf) 3 , For Fe (OTf) 3 and Yb (OTf) 3 , the yield is 80% or more, and Sc (OTf) 3 , Er (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 and Sc (ONf) For No. 3 , the yield was 95% or more.
The above results indicate that TMSOTf (entry 17) is reported to be non-catalytic (entry 16) and to catalyze a similar reaction (T. Morimoto, M. Sekiya, Chem. Lett. 1985, 1371.). Was better than.
なお、以上の結果は、無触媒(entry 16)、及び類似の反応を触媒することが報告(T. Morimoto, M. Sekiya, Chem. Lett. 1985, 1371.)されているTMSOTf(entry 17)よりも優れていた。 As shown in Table 1, in the case of the catalyst (entry 1-15) used in the production method of the present invention, the yield exceeded 20% in all cases. Among these, Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3 were able to achieve a yield of 40% or more. For Gd (OTf) 3 and Sc (NTf 2 ) 3 , the yield is 50% or more, and for Sm (OTf) 3 , the yield is 60% or more. Y (OTf), Eu (OTf) 3 , For Fe (OTf) 3 and Yb (OTf) 3 , the yield is 80% or more, and Sc (OTf) 3 , Er (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 and Sc (ONf) For No. 3 , the yield was 95% or more.
The above results indicate that TMSOTf (entry 17) is reported to be non-catalytic (entry 16) and to catalyze a similar reaction (T. Morimoto, M. Sekiya, Chem. Lett. 1985, 1371.). Was better than.
[触媒の検討(2)]
特に結果が良好であったSc(OTf)3、Er(OTf)3、Yb(OTf)3、Sn(OTf)2及びBi(OTf)3について、ケトン化合物(1)及び窒素源化合物(2)等の混合物を70℃で1時間撹拌した以外は、上記「触媒の検討(1)」と同様に実験を行った。結果を表2に示す。 [Examination of catalyst (2)]
Regarding the Sc (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Sn (OTf) 2 and Bi (OTf) 3 with particularly good results, the ketone compound (1) and the nitrogen source compound (2) An experiment was conducted in the same manner as in the above "Examination of catalyst (1)" except that the mixture was stirred at 70 ° C for 1 hour. The results are shown in Table 2.
特に結果が良好であったSc(OTf)3、Er(OTf)3、Yb(OTf)3、Sn(OTf)2及びBi(OTf)3について、ケトン化合物(1)及び窒素源化合物(2)等の混合物を70℃で1時間撹拌した以外は、上記「触媒の検討(1)」と同様に実験を行った。結果を表2に示す。 [Examination of catalyst (2)]
Regarding the Sc (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , Sn (OTf) 2 and Bi (OTf) 3 with particularly good results, the ketone compound (1) and the nitrogen source compound (2) An experiment was conducted in the same manner as in the above "Examination of catalyst (1)" except that the mixture was stirred at 70 ° C for 1 hour. The results are shown in Table 2.

表2に示すように、Sc(OTf)3は、70℃1時間という低温短時間の反応条件であるにもかかわらず、収率80%以上を達成しており、特に優れた触媒であることがわかる。
As shown in Table 2, Sc (OTf) 3 achieves a yield of 80% or more despite being a low temperature and short time reaction condition of 70 ° C. for 1 hour, and is a particularly excellent catalyst. I understand.
[溶媒の検討]
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒(0.20mL,1.0M)、窒素源化合物(2)としてのビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を70℃で1時間撹拌した後、生成物の収率を反応粗生成物の1HNMR測定により求めた。結果を表3に示す。 [Examination of solvent]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as ketone compound (1), solvent (0.20 mL, 1.0 M), bis (trimethylsilyl) amine (0. 22 mmol, 1.1 eq.) Was added. After the mixture was stirred at 70 ° C. for 1 hour, the yield of the product was determined by 1 HNMR measurement of the reaction crude product. The results are shown in Table 3.
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒(0.20mL,1.0M)、窒素源化合物(2)としてのビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を70℃で1時間撹拌した後、生成物の収率を反応粗生成物の1HNMR測定により求めた。結果を表3に示す。 [Examination of solvent]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as ketone compound (1), solvent (0.20 mL, 1.0 M), bis (trimethylsilyl) amine (0. 22 mmol, 1.1 eq.) Was added. After the mixture was stirred at 70 ° C. for 1 hour, the yield of the product was determined by 1 HNMR measurement of the reaction crude product. The results are shown in Table 3.

表3に示すように、溶媒として、モノクロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン(1,4-ジオキサン)、フルオロベンゼン、ジクロロエタン、アセトニトリルが使用可能であり、モノクロロベンゼン、トルエン、テトラヒドロフラン、フルオロベンゼン、アセトニトリルが特に優れていることがわかる。
As shown in Table 3, monochlorobenzene, toluene, tetrahydrofuran, dioxane (1,4-dioxane), fluorobenzene, dichloroethane, and acetonitrile can be used as the solvent, and monochlorobenzene, toluene, tetrahydrofuran, fluorobenzene, and acetonitrile can be used. It turns out that it is especially excellent.
[窒素源の検討]
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒としてのモノクロロベンゼン(1.0mL,1.0M)、窒素源化合物(2)(0.22mmol,1.1当量)を加えた。混合物を90℃で2時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表4に示す。 [Investigation of nitrogen source]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (1.0 mL, 1.0 M) as the solvent, nitrogen source compound (2) (0.22 mmol, 1 0.1 equivalent) was added. After the mixture was stirred at 90 ° C. for 2 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 4.
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒としてのモノクロロベンゼン(1.0mL,1.0M)、窒素源化合物(2)(0.22mmol,1.1当量)を加えた。混合物を90℃で2時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表4に示す。 [Investigation of nitrogen source]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (1.0 mL, 1.0 M) as the solvent, nitrogen source compound (2) (0.22 mmol, 1 0.1 equivalent) was added. After the mixture was stirred at 90 ° C. for 2 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 4.

表4に示すように、窒素源化合物としては、ビス(トリメチルシリル)アミン及び2,2,4,4,6,6-ヘキサメチルシクロトリシラザンが、収率が95%以上であり、特に優れた窒素源化合物であることがわかる。
As shown in Table 4, bis (trimethylsilyl) amine and 2,2,4,4,6,6-hexamethylcyclotrisilazane were particularly excellent as the nitrogen source compound with a yield of 95% or more. It turns out that it is a nitrogen source compound.
[様々な窒素上無保護イミン化合物の合成]
(基本反応条件)
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)(0.20mmol)、溶媒としてのモノクロロベンゼン(0.20mL,1.0M)、窒素源化合物(2)としてのビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を90℃で所定時間撹拌した後、原料の消失を1H NMR測定により確認し、シリカゲルカラムクロマトグラフィーによって窒素上無保護イミン化合物を単離した。以上の反応条件を基本反応条件という。 [Synthesis of various unprotected imine compounds on nitrogen]
(Basic reaction conditions)
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, and the ketone compound (1) (0.20 mmol), monochlorobenzene (0.20 mL, 1.0 M) as a solvent, bis (trimethylsilyl) amine (0) as a nitrogen source compound (2) .22 mmol, 1.1 eq) was added. After the mixture was stirred at 90 ° C. for a predetermined time, disappearance of the raw materials was confirmed by 1 H NMR measurement, and the unprotected imine compound on nitrogen was isolated by silica gel column chromatography. The above reaction conditions are called basic reaction conditions.
(基本反応条件)
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)(0.20mmol)、溶媒としてのモノクロロベンゼン(0.20mL,1.0M)、窒素源化合物(2)としてのビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を90℃で所定時間撹拌した後、原料の消失を1H NMR測定により確認し、シリカゲルカラムクロマトグラフィーによって窒素上無保護イミン化合物を単離した。以上の反応条件を基本反応条件という。 [Synthesis of various unprotected imine compounds on nitrogen]
(Basic reaction conditions)
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, and the ketone compound (1) (0.20 mmol), monochlorobenzene (0.20 mL, 1.0 M) as a solvent, bis (trimethylsilyl) amine (0) as a nitrogen source compound (2) .22 mmol, 1.1 eq) was added. After the mixture was stirred at 90 ° C. for a predetermined time, disappearance of the raw materials was confirmed by 1 H NMR measurement, and the unprotected imine compound on nitrogen was isolated by silica gel column chromatography. The above reaction conditions are called basic reaction conditions.


[実施例4-1-1]
(ベンゾフェノンイミンの合成)
上記基本反応条件で0.20mmolのベンゾフェノンを用いて2時間反応を行った。得られた粗生成物に対し、ヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のベンゾフェノンイミンを黄色のオイルとして得た(31.2mg、86%収率)。 [Example 4-1-1]
(Synthesis of benzophenone imine)
The reaction was carried out for 2 hours using 0.20 mmol of benzophenone under the above basic reaction conditions. The obtained crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone imine as a yellow oil (31.2 mg, 86% yield).
(ベンゾフェノンイミンの合成)
上記基本反応条件で0.20mmolのベンゾフェノンを用いて2時間反応を行った。得られた粗生成物に対し、ヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のベンゾフェノンイミンを黄色のオイルとして得た(31.2mg、86%収率)。 [Example 4-1-1]
(Synthesis of benzophenone imine)
The reaction was carried out for 2 hours using 0.20 mmol of benzophenone under the above basic reaction conditions. The obtained crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone imine as a yellow oil (31.2 mg, 86% yield).

1H NMR (500 MHz, CDCl3) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
13C NMR (125 MHz, CDCl3) δ 178.38, 139.35, 130.33, 128.45, 128.36. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.38, 139.35, 130.33, 128.45, 128.36.
13C NMR (125 MHz, CDCl3) δ 178.38, 139.35, 130.33, 128.45, 128.36. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.38, 139.35, 130.33, 128.45, 128.36.
[実施例4-1-2]
(大スケール(10mmol)でのベンゾフェノンイミンの合成)
上記基本反応条件に従って、50mLのフラスコにトリフルオロメタンスルホン酸スカンジム(III)(12.3mg,0.025mmol,0.25mol%)、ベンゾフェノン(1.82g,10mmol)、モノクロロベンゼン(10mL,1.0M)、ビス(トリメチルシリル)アミン(2.3mL,11mmol,1.1当量)を加えて、90℃で24時間加熱撹拌した。溶媒を留去後、粗生成物をヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを用いて精製し、目的のベンゾフェノンイミンを黄色のオイルとして得た(1.79g、99%収率)。 [Example 4-1-2]
(Synthesis of benzophenone imine on large scale (10 mmol))
In accordance with the above basic reaction conditions, a 50 mL flask was charged with scandium trifluoromethanesulfonate (III) (12.3 mg, 0.025 mmol, 0.25 mol%), benzophenone (1.82 g, 10 mmol), monochlorobenzene (10 mL, 1.0 M). ), Bis (trimethylsilyl) amine (2.3 mL, 11 mmol, 1.1 equivalents) was added, and the mixture was stirred with heating at 90 ° C. for 24 hours. After the solvent was distilled off, the crude product was purified using silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone imine as a yellow oil (1.79 g, 99% yield). rate).
(大スケール(10mmol)でのベンゾフェノンイミンの合成)
上記基本反応条件に従って、50mLのフラスコにトリフルオロメタンスルホン酸スカンジム(III)(12.3mg,0.025mmol,0.25mol%)、ベンゾフェノン(1.82g,10mmol)、モノクロロベンゼン(10mL,1.0M)、ビス(トリメチルシリル)アミン(2.3mL,11mmol,1.1当量)を加えて、90℃で24時間加熱撹拌した。溶媒を留去後、粗生成物をヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを用いて精製し、目的のベンゾフェノンイミンを黄色のオイルとして得た(1.79g、99%収率)。 [Example 4-1-2]
(Synthesis of benzophenone imine on large scale (10 mmol))
In accordance with the above basic reaction conditions, a 50 mL flask was charged with scandium trifluoromethanesulfonate (III) (12.3 mg, 0.025 mmol, 0.25 mol%), benzophenone (1.82 g, 10 mmol), monochlorobenzene (10 mL, 1.0 M). ), Bis (trimethylsilyl) amine (2.3 mL, 11 mmol, 1.1 equivalents) was added, and the mixture was stirred with heating at 90 ° C. for 24 hours. After the solvent was distilled off, the crude product was purified using silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone imine as a yellow oil (1.79 g, 99% yield). rate).

1H NMR (500 MHz, CDCl3) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
[実施例4-1-3]
(大スケール(50mmol)でのベンゾフェノンイミンの合成)
上記基本反応条件に従って、100mLのフラスコにトリフルオロメタンスルホン酸スカンジム(III)(49.2mg,0.10mmol,0.20mol%)、ベンゾフェノン(9.11g,50mmol)、ビス(トリメチルシリル)アミン(11.5mL,55mmol,1.1当量)を加えて、90℃で24時間加熱撹拌した。反応混合物を室温で減圧留去後、粗生成物を減圧蒸留して目的のベンゾフェノンイミンを黄色のオイルとして得た(7.66g、85%収率)。 [Example 4-1-3]
(Synthesis of benzophenone imine on large scale (50 mmol))
In accordance with the above basic reaction conditions, in a 100 mL flask, scandim (III) trifluoromethanesulfonate (49.2 mg, 0.10 mmol, 0.20 mol%), benzophenone (9.11 g, 50 mmol), bis (trimethylsilyl) amine (11. 5 mL, 55 mmol, 1.1 equivalents) was added, and the mixture was stirred with heating at 90 ° C. for 24 hours. The reaction mixture was evaporated under reduced pressure at room temperature, and the crude product was distilled under reduced pressure to obtain the desired benzophenone imine as a yellow oil (7.66 g, 85% yield).
(大スケール(50mmol)でのベンゾフェノンイミンの合成)
上記基本反応条件に従って、100mLのフラスコにトリフルオロメタンスルホン酸スカンジム(III)(49.2mg,0.10mmol,0.20mol%)、ベンゾフェノン(9.11g,50mmol)、ビス(トリメチルシリル)アミン(11.5mL,55mmol,1.1当量)を加えて、90℃で24時間加熱撹拌した。反応混合物を室温で減圧留去後、粗生成物を減圧蒸留して目的のベンゾフェノンイミンを黄色のオイルとして得た(7.66g、85%収率)。 [Example 4-1-3]
(Synthesis of benzophenone imine on large scale (50 mmol))
In accordance with the above basic reaction conditions, in a 100 mL flask, scandim (III) trifluoromethanesulfonate (49.2 mg, 0.10 mmol, 0.20 mol%), benzophenone (9.11 g, 50 mmol), bis (trimethylsilyl) amine (11. 5 mL, 55 mmol, 1.1 equivalents) was added, and the mixture was stirred with heating at 90 ° C. for 24 hours. The reaction mixture was evaporated under reduced pressure at room temperature, and the crude product was distilled under reduced pressure to obtain the desired benzophenone imine as a yellow oil (7.66 g, 85% yield).

1H NMR (500 MHz, CDCl3) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
13C NMR (125 MHz, CDCl3) δ 178.23, 140.25, 138.29, 130.29, 129.16, 128.28, 127.60. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.23, 140.25, 138.29, 130.29, 129.16, 128.28, 127.60.
13C NMR (125 MHz, CDCl3) δ 178.23, 140.25, 138.29, 130.29, 129.16, 128.28, 127.60. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.50-7.46 (m, 2H), 7.44-7.41 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.23, 140.25, 138.29, 130.29, 129.16, 128.28, 127.60.
[実施例4-2]
(9-イミノフルオレンの合成)
上記基本反応条件で0.20mmolの9-フルオレノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の9-イミノフルオレンを黄色の固体として得た(34.7mg、97%収率)。 [Example 4-2]
(Synthesis of 9-iminofluorene)
The reaction was performed for 2 hours using 0.20 mmol of 9-fluorenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired 9-iminofluorene as a yellow solid (34.7 mg, 97% yield).
(9-イミノフルオレンの合成)
上記基本反応条件で0.20mmolの9-フルオレノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の9-イミノフルオレンを黄色の固体として得た(34.7mg、97%収率)。 [Example 4-2]
(Synthesis of 9-iminofluorene)
The reaction was performed for 2 hours using 0.20 mmol of 9-fluorenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired 9-iminofluorene as a yellow solid (34.7 mg, 97% yield).

1H NMR (500 MHz, CDCl3) δ 10.27 (br, 1H), 7.91 (br, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.47 (td, J = 1.0, 7.5 Hz, 2H), 7.34 (td, J = 1.0, 7.5 Hz, 2H).
13C NMR (125 MHz, CDCl3) δ 173.17, 142.21, 134.67, 128.17, 122.25, 120.09. 1 H NMR (500 MHz, CDCl 3 ) δ 10.27 (br, 1H), 7.91 (br, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.47 (td, J = 1.0, 7.5 Hz, 2H) , 7.34 (td, J = 1.0, 7.5 Hz, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 173.17, 142.21, 134.67, 128.17, 122.25, 120.09.
13C NMR (125 MHz, CDCl3) δ 173.17, 142.21, 134.67, 128.17, 122.25, 120.09. 1 H NMR (500 MHz, CDCl 3 ) δ 10.27 (br, 1H), 7.91 (br, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.47 (td, J = 1.0, 7.5 Hz, 2H) , 7.34 (td, J = 1.0, 7.5 Hz, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 173.17, 142.21, 134.67, 128.17, 122.25, 120.09.
[実施例4-3]
(3-イミノインドリン-2-オンの合成)
上記基本反応条件で0.20mmolのイサチンを用いて室温で10時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=3/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の3-イミノインドリン-2-オンを黄色の固体として得た(26.7mg、91%収率)。 [Example 4-3]
(Synthesis of 3-iminoindoline-2-one)
The reaction was carried out at room temperature for 10 hours using 0.20 mmol of isatin under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / dichloromethane / triethylamine = 3/1/1 to obtain the desired 3-iminoindolin-2-one as a yellow solid (26. 7 mg, 91% yield).
(3-イミノインドリン-2-オンの合成)
上記基本反応条件で0.20mmolのイサチンを用いて室温で10時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=3/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の3-イミノインドリン-2-オンを黄色の固体として得た(26.7mg、91%収率)。 [Example 4-3]
(Synthesis of 3-iminoindoline-2-one)
The reaction was carried out at room temperature for 10 hours using 0.20 mmol of isatin under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / dichloromethane / triethylamine = 3/1/1 to obtain the desired 3-iminoindolin-2-one as a yellow solid (26. 7 mg, 91% yield).

1H NMR (500 MHz, DMSO-d6) δ 12.35* (s, 1H), 11.27 (s, 1H), 10.92 (br, 1H), 10.83* (br, 1H), 7.73* (d. J = 7.5 Hz, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.46 (td, J = 1.0, 7.5 Hz, 1H), 7.40* (td, J = 1.0, 7.5 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 7.03* (t, J = 7.5 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.86* (d, J = 8.0 Hz, 1H).
13C NMR (125 MHz, DMSO-d6) δ 165.01*, 163.82*, 163.54, 159.35, 146.47, 144.92*, 143.39, 133.93*, 123.59*, 123.14, 122.59, 122.01*, 120.45, 117.96*, 111.33, 110.90*.
[* denotes the minor diastereomer] 1 H NMR (500 MHz, DMSO-d 6 ) δ 12.35 * (s, 1H), 11.27 (s, 1H), 10.92 (br, 1H), 10.83 * (br, 1H), 7.73 * (d.J = 7.5 Hz, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.46 (td, J = 1.0, 7.5 Hz, 1H), 7.40 * (td, J = 1.0, 7.5 Hz, 1H), 7.08 (t , J = 7.5 Hz, 1H), 7.03 * (t, J = 7.5 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.86 * (d, J = 8.0 Hz, 1H).
13 C NMR (125 MHz, DMSO-d 6 ) δ 165.01 *, 163.82 *, 163.54, 159.35, 146.47, 144.92 *, 143.39, 133.93 *, 123.59 *, 123.14, 122.59, 122.01 *, 120.45, 117.96 *, 111.33, 110.90 *.
[* designate the minor diastereomer]
13C NMR (125 MHz, DMSO-d6) δ 165.01*, 163.82*, 163.54, 159.35, 146.47, 144.92*, 143.39, 133.93*, 123.59*, 123.14, 122.59, 122.01*, 120.45, 117.96*, 111.33, 110.90*.
[* denotes the minor diastereomer] 1 H NMR (500 MHz, DMSO-d 6 ) δ 12.35 * (s, 1H), 11.27 (s, 1H), 10.92 (br, 1H), 10.83 * (br, 1H), 7.73 * (d.J = 7.5 Hz, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.46 (td, J = 1.0, 7.5 Hz, 1H), 7.40 * (td, J = 1.0, 7.5 Hz, 1H), 7.08 (t , J = 7.5 Hz, 1H), 7.03 * (t, J = 7.5 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.86 * (d, J = 8.0 Hz, 1H).
13 C NMR (125 MHz, DMSO-d 6 ) δ 165.01 *, 163.82 *, 163.54, 159.35, 146.47, 144.92 *, 143.39, 133.93 *, 123.59 *, 123.14, 122.59, 122.01 *, 120.45, 117.96 *, 111.33, 110.90 *.
[* designate the minor diastereomer]
[実施例4-4]
(2-(イミノ(フェニル)メチル)フェノールの合成)
上記基本反応条件で0.20mmolの2-ヒドロキシベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=4/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2-(イミノ(フェニル)メチル)フェノールを黄色の固体として得た(27.8mg、70%収率)。 [Example 4-4]
(Synthesis of 2- (imino (phenyl) methyl) phenol)
The reaction was performed for 2 hours using 0.20 mmol of 2-hydroxybenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 4/1 to obtain the desired 2- (imino (phenyl) methyl) phenol as a yellow solid (27.8 mg, 70% yield).
(2-(イミノ(フェニル)メチル)フェノールの合成)
上記基本反応条件で0.20mmolの2-ヒドロキシベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=4/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2-(イミノ(フェニル)メチル)フェノールを黄色の固体として得た(27.8mg、70%収率)。 [Example 4-4]
(Synthesis of 2- (imino (phenyl) methyl) phenol)
The reaction was performed for 2 hours using 0.20 mmol of 2-hydroxybenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 4/1 to obtain the desired 2- (imino (phenyl) methyl) phenol as a yellow solid (27.8 mg, 70% yield).

1H NMR (500 MHz, CDCl3) δ 8.33 (br, 1H), 7.53-7.47 (m, 3H), 7.42-7.40 (m, 2H), 7.36 (ddd, J = 1.5, 7.5, 8.5 Hz, 1H), 7.20 (dd, J = 2.0, 8.0 Hz, 1H), 7.05 (dd, J = 1.0, 8.5 Hz, 1H), 6.74 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H).
13C NMR (125 MHz, CDCl3) δ 181.32, 163.57, 139.11, 133.46, 132.14, 129.91, 128.69, 127.21, 118.41, 118.31, 117.62. 1 H NMR (500 MHz, CDCl 3 ) δ 8.33 (br, 1H), 7.53-7.47 (m, 3H), 7.42-7.40 (m, 2H), 7.36 (ddd, J = 1.5, 7.5, 8.5 Hz, 1H ), 7.20 (dd, J = 2.0, 8.0 Hz, 1H), 7.05 (dd, J = 1.0, 8.5 Hz, 1H), 6.74 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H).
13 C NMR (125 MHz, CDCl 3 ) δ 181.32, 163.57, 139.11, 133.46, 132.14, 129.91, 128.69, 127.21, 118.41, 118.31, 117.62.
13C NMR (125 MHz, CDCl3) δ 181.32, 163.57, 139.11, 133.46, 132.14, 129.91, 128.69, 127.21, 118.41, 118.31, 117.62. 1 H NMR (500 MHz, CDCl 3 ) δ 8.33 (br, 1H), 7.53-7.47 (m, 3H), 7.42-7.40 (m, 2H), 7.36 (ddd, J = 1.5, 7.5, 8.5 Hz, 1H ), 7.20 (dd, J = 2.0, 8.0 Hz, 1H), 7.05 (dd, J = 1.0, 8.5 Hz, 1H), 6.74 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H).
13 C NMR (125 MHz, CDCl 3 ) δ 181.32, 163.57, 139.11, 133.46, 132.14, 129.91, 128.69, 127.21, 118.41, 118.31, 117.62.
[実施例4-5]
(ビス(4-クロロフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジクロロベンゾフェノン用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-クロロフェニル)メタンイミンを白色の固体として得た(47.6mg、95%収率)。 [Example 4-5]
(Synthesis of bis (4-chlorophenyl) methanimine)
The reaction was performed for 2 hours using 0.20 mmol of 4,4′-dichlorobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (4-chlorophenyl) methanimine as a white solid (47.6 mg, 95% yield). rate).
(ビス(4-クロロフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジクロロベンゾフェノン用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-クロロフェニル)メタンイミンを白色の固体として得た(47.6mg、95%収率)。 [Example 4-5]
(Synthesis of bis (4-chlorophenyl) methanimine)
The reaction was performed for 2 hours using 0.20 mmol of 4,4′-dichlorobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (4-chlorophenyl) methanimine as a white solid (47.6 mg, 95% yield). rate).

1H NMR (500 MHz, CDCl3) δ 9.76 (br, 1H), 7.58 (br, 4H), 7.41 (d, J = 8.0 Hz, 4H).
13C NMR (125 MHz, CDCl3) δ 176.07, 137.23, 136.71, 129.67, 128.72. 1 H NMR (500 MHz, CDCl3) δ 9.76 (br, 1H), 7.58 (br, 4H), 7.41 (d, J = 8.0 Hz, 4H).
13 C NMR (125 MHz, CDCl3) δ 176.07, 137.23, 136.71, 129.67, 128.72.
13C NMR (125 MHz, CDCl3) δ 176.07, 137.23, 136.71, 129.67, 128.72. 1 H NMR (500 MHz, CDCl3) δ 9.76 (br, 1H), 7.58 (br, 4H), 7.41 (d, J = 8.0 Hz, 4H).
13 C NMR (125 MHz, CDCl3) δ 176.07, 137.23, 136.71, 129.67, 128.72.
[実施例4-6]
(ジ-p-トリルメタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジメチルベンゾフェノン用いて3時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のジ-p-トリルメタンイミンを無色のオイルとして得た(39.7mg、95%収率)。 [Example 4-6]
(Synthesis of di-p-tolylmethanimine)
The reaction was carried out using 0.20 mmol of 4,4′-dimethylbenzophenone under the above basic reaction conditions for 3 hours. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired di-p-tolylmethanimine as a colorless oil (39.7 mg, 95% yield). rate).
(ジ-p-トリルメタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジメチルベンゾフェノン用いて3時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のジ-p-トリルメタンイミンを無色のオイルとして得た(39.7mg、95%収率)。 [Example 4-6]
(Synthesis of di-p-tolylmethanimine)
The reaction was carried out using 0.20 mmol of 4,4′-dimethylbenzophenone under the above basic reaction conditions for 3 hours. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired di-p-tolylmethanimine as a colorless oil (39.7 mg, 95% yield). rate).

1H NMR (500 MHz, CDCl3) δ 9.56 (br, 1H), 7.47 (br, 4H), 7.22 (d, J = 8.0 Hz, 4H), 2.41 (s, 6H).
13C NMR (125 MHz, CDCl3) δ 178.12, 140.40, 136.71, 128.93, 128.43, 21.39. 1 H NMR (500 MHz, CDCl 3 ) δ 9.56 (br, 1H), 7.47 (br, 4H), 7.22 (d, J = 8.0 Hz, 4H), 2.41 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.12, 140.40, 136.71, 128.93, 128.43, 21.39.
13C NMR (125 MHz, CDCl3) δ 178.12, 140.40, 136.71, 128.93, 128.43, 21.39. 1 H NMR (500 MHz, CDCl 3 ) δ 9.56 (br, 1H), 7.47 (br, 4H), 7.22 (d, J = 8.0 Hz, 4H), 2.41 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.12, 140.40, 136.71, 128.93, 128.43, 21.39.
[実施例4-7]
(ビス(4-メトキシフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジクロロベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-メトキシフェニル)メタンイミンを白色の固体として得た(46.8mg、97%収率)。 [Example 4-7]
(Synthesis of bis (4-methoxyphenyl) methanimine)
The reaction was performed for 24 hours using 0.20 mmol of 4,4′-dichlorobenzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / dichloromethane / triethylamine = 8/1/1 to obtain the desired bis (4-methoxyphenyl) methanimine as a white solid (46. 8 mg, 97% yield).
(ビス(4-メトキシフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジクロロベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-メトキシフェニル)メタンイミンを白色の固体として得た(46.8mg、97%収率)。 [Example 4-7]
(Synthesis of bis (4-methoxyphenyl) methanimine)
The reaction was performed for 24 hours using 0.20 mmol of 4,4′-dichlorobenzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / dichloromethane / triethylamine = 8/1/1 to obtain the desired bis (4-methoxyphenyl) methanimine as a white solid (46. 8 mg, 97% yield).
1H NMR (500 MHz, CDCl3) δ 9.31 (br, 1H), 7.53 (d, J = 6.5 Hz, 4H), 6.94-6.91 (m, 4H), 3.86 (s, 6H).
13C NMR (125 MHz, CDCl3) δ 177.05, 161.18. 132.14, 130.11, 113.52, 55.37. 1 H NMR (500 MHz, CDCl 3 ) δ 9.31 (br, 1H), 7.53 (d, J = 6.5 Hz, 4H), 6.94-6.91 (m, 4H), 3.86 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.05, 161.18. 132.14, 130.11, 113.52, 55.37.
13C NMR (125 MHz, CDCl3) δ 177.05, 161.18. 132.14, 130.11, 113.52, 55.37. 1 H NMR (500 MHz, CDCl 3 ) δ 9.31 (br, 1H), 7.53 (d, J = 6.5 Hz, 4H), 6.94-6.91 (m, 4H), 3.86 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.05, 161.18. 132.14, 130.11, 113.52, 55.37.
[実施例4-8]
((4-ブロモフェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-ブロモベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-ブロモフェニル)(フェニル)メタンイミンを黄色のオイルとして得た(50.2mg、96%収率)。 [Example 4-8]
(Synthesis of (4-Bromophenyl) (phenyl) methanimine)
The reaction was performed for 2 hours using 0.20 mmol of 4-bromobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired (4-bromophenyl) (phenyl) methanimine as a yellow oil (50.2 mg, 96% yield).
((4-ブロモフェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-ブロモベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-ブロモフェニル)(フェニル)メタンイミンを黄色のオイルとして得た(50.2mg、96%収率)。 [Example 4-8]
(Synthesis of (4-Bromophenyl) (phenyl) methanimine)
The reaction was performed for 2 hours using 0.20 mmol of 4-bromobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired (4-bromophenyl) (phenyl) methanimine as a yellow oil (50.2 mg, 96% yield).

1H NMR (500 MHz, CDCl3) δ 9.74 (br, 1H), 7.56 (dd, J = 2.0, 6.5 Hz, 2H), 7.55-7.47 (m, 4H), 7.45-7.41 (m, 4H).
13C NMR (125 MHz, CDCl3) δ 177.29, 139.01, 137.92, 131.52, 130.44, 130.18, 128.48, 128.12, 124.95. 1 H NMR (500 MHz, CDCl 3 ) δ 9.74 (br, 1H), 7.56 (dd, J = 2.0, 6.5 Hz, 2H), 7.55-7.47 (m, 4H), 7.45-7.41 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.29, 139.01, 137.92, 131.52, 130.44, 130.18, 128.48, 128.12, 124.95.
13C NMR (125 MHz, CDCl3) δ 177.29, 139.01, 137.92, 131.52, 130.44, 130.18, 128.48, 128.12, 124.95. 1 H NMR (500 MHz, CDCl 3 ) δ 9.74 (br, 1H), 7.56 (dd, J = 2.0, 6.5 Hz, 2H), 7.55-7.47 (m, 4H), 7.45-7.41 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.29, 139.01, 137.92, 131.52, 130.44, 130.18, 128.48, 128.12, 124.95.
[実施例4-9]
(ビス(3-(トリフルオロメチル)フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの3,3’-ビス(トリフルオロメチル)ベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて48時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(3-(トリフルオロメチル)フェニル)メタンイミンを無色のオイルとして得た(56.0mg、88%収率)。 [Example 4-9]
(Synthesis of bis (3- (trifluoromethyl) phenyl) methanimine)
The reaction was carried out for 48 hours using 0.20 mmol of 3,3′-bis (trifluoromethyl) benzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (3- (trifluoromethyl) phenyl) methanimine as a colorless oil (56. 0 mg, 88% yield).
(ビス(3-(トリフルオロメチル)フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの3,3’-ビス(トリフルオロメチル)ベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて48時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(3-(トリフルオロメチル)フェニル)メタンイミンを無色のオイルとして得た(56.0mg、88%収率)。 [Example 4-9]
(Synthesis of bis (3- (trifluoromethyl) phenyl) methanimine)
The reaction was carried out for 48 hours using 0.20 mmol of 3,3′-bis (trifluoromethyl) benzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (3- (trifluoromethyl) phenyl) methanimine as a colorless oil (56. 0 mg, 88% yield).

1H NMR (500 MHz, DMSO-d6) δ 11.26 (br, 1H), 8.00 (s, 1H), 7.93-7.90 (m, 2H), 7.83-7.82 (m, 2H), 7.76-7.68 (m, 3H).
13C NMR (125 MHz, DMSO-d6) δ 172.61, 139.27, 139.02, 132.76, 131.72, 129.76, 129.73, 129.43 (q, JC-F = 32 Hz), 129.27 (q, JC-F = 32 Hz), 127.18 (q, JC-F = 3.5 Hz), 126.68 (q, JC-F = 3.5 Hz), 124.63 (q, JC-F = 3.8 Hz), 124.43 (q, JC-F = 3.8 Hz), 124.05 (q, JC-F = 271 Hz), 123.97 (q, JC-F = 271 Hz).
19F NMR (470 MHz, DMSO-d6) δ-61.14 (s, 3F), -61.27 (s, 3F). 1 H NMR (500 MHz, DMSO-d 6 ) δ 11.26 (br, 1H), 8.00 (s, 1H), 7.93-7.90 (m, 2H), 7.83-7.82 (m, 2H), 7.76-7.68 (m , 3H).
13 C NMR (125 MHz, DMSO-d 6 ) δ 172.61, 139.27, 139.02, 132.76, 131.72, 129.76, 129.73, 129.43 (q, J CF = 32 Hz), 129.27 (q, J CF = 32 Hz), 127.18 (q, J CF = 3.5 Hz), 126.68 (q, J CF = 3.5 Hz), 124.63 (q, J CF = 3.8 Hz), 124.43 (q, J CF = 3.8 Hz), 124.05 (q, J CF = 271 Hz), 123.97 (q, J CF = 271 Hz).
19 F NMR (470 MHz, DMSO-d 6 ) δ-61.14 (s, 3F), -61.27 (s, 3F).
13C NMR (125 MHz, DMSO-d6) δ 172.61, 139.27, 139.02, 132.76, 131.72, 129.76, 129.73, 129.43 (q, JC-F = 32 Hz), 129.27 (q, JC-F = 32 Hz), 127.18 (q, JC-F = 3.5 Hz), 126.68 (q, JC-F = 3.5 Hz), 124.63 (q, JC-F = 3.8 Hz), 124.43 (q, JC-F = 3.8 Hz), 124.05 (q, JC-F = 271 Hz), 123.97 (q, JC-F = 271 Hz).
19F NMR (470 MHz, DMSO-d6) δ-61.14 (s, 3F), -61.27 (s, 3F). 1 H NMR (500 MHz, DMSO-d 6 ) δ 11.26 (br, 1H), 8.00 (s, 1H), 7.93-7.90 (m, 2H), 7.83-7.82 (m, 2H), 7.76-7.68 (m , 3H).
13 C NMR (125 MHz, DMSO-d 6 ) δ 172.61, 139.27, 139.02, 132.76, 131.72, 129.76, 129.73, 129.43 (q, J CF = 32 Hz), 129.27 (q, J CF = 32 Hz), 127.18 (q, J CF = 3.5 Hz), 126.68 (q, J CF = 3.5 Hz), 124.63 (q, J CF = 3.8 Hz), 124.43 (q, J CF = 3.8 Hz), 124.05 (q, J CF = 271 Hz), 123.97 (q, J CF = 271 Hz).
19 F NMR (470 MHz, DMSO-d 6 ) δ-61.14 (s, 3F), -61.27 (s, 3F).
[実施例4-10]
(フェニル(ピリジン-2-イル)メタンイミンの合成)
上記基本反応条件で0.20mmolの2-ベンゾイルピリジンと1.5当量のビス(トリメチルシリル)アミンを用いて12時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のフェニル(ピリジン-2-イル)メタンイミンとケトンの混合物を無色のオイルとして得た(31.6mg、イミン/ケトン=92/8、イミンの収率81%)。 [Example 4-10]
(Synthesis of phenyl (pyridin-2-yl) methanimine)
The reaction was carried out for 12 hours using 0.20 mmol of 2-benzoylpyridine and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain a desired mixture of phenyl (pyridin-2-yl) methanimine and ketone as a colorless oil (31 .6 mg, imine / ketone = 92/8, imine yield 81%).
(フェニル(ピリジン-2-イル)メタンイミンの合成)
上記基本反応条件で0.20mmolの2-ベンゾイルピリジンと1.5当量のビス(トリメチルシリル)アミンを用いて12時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のフェニル(ピリジン-2-イル)メタンイミンとケトンの混合物を無色のオイルとして得た(31.6mg、イミン/ケトン=92/8、イミンの収率81%)。 [Example 4-10]
(Synthesis of phenyl (pyridin-2-yl) methanimine)
The reaction was carried out for 12 hours using 0.20 mmol of 2-benzoylpyridine and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain a desired mixture of phenyl (pyridin-2-yl) methanimine and ketone as a colorless oil (31 .6 mg, imine / ketone = 92/8, imine yield 81%).

1H NMR (500 MHz, CDCl3) δ 11.05 (br, 1H), 8.75 (d, J = 4.0 Hz, 1H), 7.77 (dd, J = 7.5, 7.5 Hz, 1H), 7.66 (br, 2H), 7.51-7.44 (m, 3H), 7.41-7.38 (m, 1H).
13C NMR (125 MHz, CDCl3) δ 175.35, 155.35, 149.75, 137.88, 136.77, 130.24, 128.70, 128.34, 124.80. 1 H NMR (500 MHz, CDCl 3 ) δ 11.05 (br, 1H), 8.75 (d, J = 4.0 Hz, 1H), 7.77 (dd, J = 7.5, 7.5 Hz, 1H), 7.66 (br, 2H) , 7.51-7.44 (m, 3H), 7.41-7.38 (m, 1H).
13 C NMR (125 MHz, CDCl 3 ) δ 175.35, 155.35, 149.75, 137.88, 136.77, 130.24, 128.70, 128.34, 124.80.
13C NMR (125 MHz, CDCl3) δ 175.35, 155.35, 149.75, 137.88, 136.77, 130.24, 128.70, 128.34, 124.80. 1 H NMR (500 MHz, CDCl 3 ) δ 11.05 (br, 1H), 8.75 (d, J = 4.0 Hz, 1H), 7.77 (dd, J = 7.5, 7.5 Hz, 1H), 7.66 (br, 2H) , 7.51-7.44 (m, 3H), 7.41-7.38 (m, 1H).
13 C NMR (125 MHz, CDCl 3 ) δ 175.35, 155.35, 149.75, 137.88, 136.77, 130.24, 128.70, 128.34, 124.80.
[実施例4-11]
(5H-ジベンゾ[a,d][7]アヌレン-5-イミンの合成)
上記基本反応条件で0.20mmolのジベンゾスベレノンと1.5当量のビス(トリメチルシリル)アミンを用いて12時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の5H-ジベンゾ[a,d][7]アヌレン-5-イミンを無色のオイルとして得た(40.1mg、98%収率)。 [Example 4-11]
(Synthesis of 5H-dibenzo [a, d] [7] annulene-5-imine)
The reaction was performed for 12 hours using 0.20 mmol of dibenzosuberenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product is subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired 5H-dibenzo [a, d] [7] annulene-5-imine as a colorless oil. (40.1 mg, 98% yield).
(5H-ジベンゾ[a,d][7]アヌレン-5-イミンの合成)
上記基本反応条件で0.20mmolのジベンゾスベレノンと1.5当量のビス(トリメチルシリル)アミンを用いて12時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の5H-ジベンゾ[a,d][7]アヌレン-5-イミンを無色のオイルとして得た(40.1mg、98%収率)。 [Example 4-11]
(Synthesis of 5H-dibenzo [a, d] [7] annulene-5-imine)
The reaction was performed for 12 hours using 0.20 mmol of dibenzosuberenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product is subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired 5H-dibenzo [a, d] [7] annulene-5-imine as a colorless oil. (40.1 mg, 98% yield).

1H NMR (500 MHz, CDCl3) δ 9.72 (br, 1H), 7.70 (br, 2H), 7.48-7.44 (m, 4H), 7.40-7.38 (m, 2H), 6.93 (s, 2H).
13C NMR (125 MHz, CDCl3) δ 178.49, 139.60, 133.16, 130.83, 129.42, 129.11, 129.03, 127.35. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.70 (br, 2H), 7.48-7.44 (m, 4H), 7.40-7.38 (m, 2H), 6.93 (s, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.49, 139.60, 133.16, 130.83, 129.42, 129.11, 129.03, 127.35.
13C NMR (125 MHz, CDCl3) δ 178.49, 139.60, 133.16, 130.83, 129.42, 129.11, 129.03, 127.35. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.70 (br, 2H), 7.48-7.44 (m, 4H), 7.40-7.38 (m, 2H), 6.93 (s, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.49, 139.60, 133.16, 130.83, 129.42, 129.11, 129.03, 127.35.
[実施例4-12]
(9H-キサンテン-9-イミンの合成)
上記基本反応条件で0.20mmolのキサントンと1.5当量のビス(トリメチルシリル)アミンを用いて48時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の9H-キサンテン-9-イミンを白色の固体として得た(38.5mg、99%収率)。 [Example 4-12]
(Synthesis of 9H-xanthene-9-imine)
The reaction was carried out for 48 hours using 0.20 mmol of xanthone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / dichloromethane / triethylamine = 8/1/1 to obtain the desired 9H-xanthene-9-imine as a white solid (38.5 mg). 99% yield).
(9H-キサンテン-9-イミンの合成)
上記基本反応条件で0.20mmolのキサントンと1.5当量のビス(トリメチルシリル)アミンを用いて48時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の9H-キサンテン-9-イミンを白色の固体として得た(38.5mg、99%収率)。 [Example 4-12]
(Synthesis of 9H-xanthene-9-imine)
The reaction was carried out for 48 hours using 0.20 mmol of xanthone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / dichloromethane / triethylamine = 8/1/1 to obtain the desired 9H-xanthene-9-imine as a white solid (38.5 mg). 99% yield).

1H NMR (500 MHz, CDCl3) δ 9.49 (br, 1H), 8.17 (br, 2H), 7.58 (ddd, J = 1.5, 7.0, 8.5 Hz, 2H), 7.35 (dd, J = 1.0, 8.5 Hz, 2H), 7.31 (ddd, J = 1.0, 7.0, 8.0 Hz, 2H).
13C NMR (125 MHz, CDCl3) δ 159.06, 152.84, 132.58, 124.56, 123.68, 119.73, 117.93. 1 H NMR (500 MHz, CDCl 3 ) δ 9.49 (br, 1H), 8.17 (br, 2H), 7.58 (ddd, J = 1.5, 7.0, 8.5 Hz, 2H), 7.35 (dd, J = 1.0, 8.5 Hz, 2H), 7.31 (ddd, J = 1.0, 7.0, 8.0 Hz, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 159.06, 152.84, 132.58, 124.56, 123.68, 119.73, 117.93.
13C NMR (125 MHz, CDCl3) δ 159.06, 152.84, 132.58, 124.56, 123.68, 119.73, 117.93. 1 H NMR (500 MHz, CDCl 3 ) δ 9.49 (br, 1H), 8.17 (br, 2H), 7.58 (ddd, J = 1.5, 7.0, 8.5 Hz, 2H), 7.35 (dd, J = 1.0, 8.5 Hz, 2H), 7.31 (ddd, J = 1.0, 7.0, 8.0 Hz, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 159.06, 152.84, 132.58, 124.56, 123.68, 119.73, 117.93.
[実施例4-13]
(2-(4-((4-クロロフェニル)(イミノ)メチル)フェノキシ)-2-メチルプロピオン酸イソプロピルの合成)
上記基本反応条件で0.20mmolのフェノフィブレートと1.5当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2-(4-((4-クロロフェニル)(イミノ)メチル)フェノキシ)-2-メチルプロピオン酸イソプロピルを黄色のオイルとして得た(67.2 mg、93%収率)。 [Example 4-13]
(Synthesis of 2- (4-((4-chlorophenyl) (imino) methyl) phenoxy) -2-methylpropionate)
The reaction was carried out for 24 hours using 0.20 mmol of fenofibrate and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained crude reaction product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired 2- (4-((4-chlorophenyl) (imino) methyl) phenoxy) -2-methyl. Isopropyl propionate was obtained as a yellow oil (67.2 mg, 93% yield).
(2-(4-((4-クロロフェニル)(イミノ)メチル)フェノキシ)-2-メチルプロピオン酸イソプロピルの合成)
上記基本反応条件で0.20mmolのフェノフィブレートと1.5当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2-(4-((4-クロロフェニル)(イミノ)メチル)フェノキシ)-2-メチルプロピオン酸イソプロピルを黄色のオイルとして得た(67.2 mg、93%収率)。 [Example 4-13]
(Synthesis of 2- (4-((4-chlorophenyl) (imino) methyl) phenoxy) -2-methylpropionate)
The reaction was carried out for 24 hours using 0.20 mmol of fenofibrate and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained crude reaction product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired 2- (4-((4-chlorophenyl) (imino) methyl) phenoxy) -2-methyl. Isopropyl propionate was obtained as a yellow oil (67.2 mg, 93% yield).

1H NMR (500 MHz, CDCl3) δ 7.46 (br, 4H), 7.39 (dd, J = 1.5, 6.5 Hz, 2H), 6.85-6.82 (m, 2H), 5.09 (septet, J = 6.0 Hz, 1H), 1.64 (s, 6H), 1.22 (d, J = 6.0 Hz, 6H).
13C NMR (125 MHz, CDCl3) δ 276.46, 173.33, 157.68, 137.94, 136.23, 129.58, 128.53, 117.77, 79.20, 69.18, 59.34, 25.34, 21.54, 8.49. 1 H NMR (500 MHz, CDCl 3 ) δ 7.46 (br, 4H), 7.39 (dd, J = 1.5, 6.5 Hz, 2H), 6.85-6.82 (m, 2H), 5.09 (septet, J = 6.0 Hz, 1H), 1.64 (s, 6H), 1.22 (d, J = 6.0 Hz, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 276.46, 173.33, 157.68, 137.94, 136.23, 129.58, 128.53, 117.77, 79.20, 69.18, 59.34, 25.34, 21.54, 8.49.
13C NMR (125 MHz, CDCl3) δ 276.46, 173.33, 157.68, 137.94, 136.23, 129.58, 128.53, 117.77, 79.20, 69.18, 59.34, 25.34, 21.54, 8.49. 1 H NMR (500 MHz, CDCl 3 ) δ 7.46 (br, 4H), 7.39 (dd, J = 1.5, 6.5 Hz, 2H), 6.85-6.82 (m, 2H), 5.09 (septet, J = 6.0 Hz, 1H), 1.64 (s, 6H), 1.22 (d, J = 6.0 Hz, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 276.46, 173.33, 157.68, 137.94, 136.23, 129.58, 128.53, 117.77, 79.20, 69.18, 59.34, 25.34, 21.54, 8.49.
[実施例4-14]
(2-(3-(イミノ(フェニル)メチル)フェニル)プロピオン酸の合成)
上記基本反応条件で0.20mmolのケトプロフェンと2.0当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてジクロロメタン/メタノール=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2-(3-(イミノ(フェニル)メチル)フェニル)プロピオン酸を黄色のオイルとして得た(44.4mg、88%収率)。 [Example 4-14]
(Synthesis of 2- (3- (imino (phenyl) methyl) phenyl) propionic acid)
The reaction was carried out for 24 hours using 0.20 mmol of ketoprofen and 2.0 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product is subjected to silica gel column chromatography with a developing solvent of dichloromethane / methanol = 9/1 to obtain the desired 2- (3- (imino (phenyl) methyl) phenyl) propionic acid as a yellow oil. (44.4 mg, 88% yield).
(2-(3-(イミノ(フェニル)メチル)フェニル)プロピオン酸の合成)
上記基本反応条件で0.20mmolのケトプロフェンと2.0当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてジクロロメタン/メタノール=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2-(3-(イミノ(フェニル)メチル)フェニル)プロピオン酸を黄色のオイルとして得た(44.4mg、88%収率)。 [Example 4-14]
(Synthesis of 2- (3- (imino (phenyl) methyl) phenyl) propionic acid)
The reaction was carried out for 24 hours using 0.20 mmol of ketoprofen and 2.0 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product is subjected to silica gel column chromatography with a developing solvent of dichloromethane / methanol = 9/1 to obtain the desired 2- (3- (imino (phenyl) methyl) phenyl) propionic acid as a yellow oil. (44.4 mg, 88% yield).

1H NMR (500 MHz, CDCl3) δ 7.79-7.76 (m, 3H), 7.64 (d, J = 7.0 Hz, 1H), 7.59 (dd, J = 7.0, 7.0 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.47 (dd, J = 7.5, 7.5 Hz, 2H), 3.79 (q, J = 6.0 Hz, 1H), 1.53 (d, J = 6.0 Hz, 3H).
13C NMR (125 MHz, CDCl3) δ 198.86, 180.08, 141.36, 137.69, 137.29, 132.63, 131.84, 130.19, 129.18, 128.97, 128.42, 128.32, 45.99, 18.37. 1 H NMR (500 MHz, CDCl 3 ) δ 7.79-7.76 (m, 3H), 7.64 (d, J = 7.0 Hz, 1H), 7.59 (dd, J = 7.0, 7.0 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.47 (dd, J = 7.5, 7.5 Hz, 2H), 3.79 (q, J = 6.0 Hz, 1H), 1.53 (d, J = 6.0 Hz, 3H).
13 C NMR (125 MHz, CDCl 3 ) δ 198.86, 180.08, 141.36, 137.69, 137.29, 132.63, 131.84, 130.19, 129.18, 128.97, 128.42, 128.32, 45.99, 18.37.
13C NMR (125 MHz, CDCl3) δ 198.86, 180.08, 141.36, 137.69, 137.29, 132.63, 131.84, 130.19, 129.18, 128.97, 128.42, 128.32, 45.99, 18.37. 1 H NMR (500 MHz, CDCl 3 ) δ 7.79-7.76 (m, 3H), 7.64 (d, J = 7.0 Hz, 1H), 7.59 (dd, J = 7.0, 7.0 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.47 (dd, J = 7.5, 7.5 Hz, 2H), 3.79 (q, J = 6.0 Hz, 1H), 1.53 (d, J = 6.0 Hz, 3H).
13 C NMR (125 MHz, CDCl 3 ) δ 198.86, 180.08, 141.36, 137.69, 137.29, 132.63, 131.84, 130.19, 129.18, 128.97, 128.42, 128.32, 45.99, 18.37.
[実施例4-15]
(2,2-ジメチル-3,4-ジヒドロナフタレン-1(2H)-イミンの合成)
上記基本反応条件で0.20mmolの2,2-ジメチル-3,4-ジヒドロナフタレン-1(2H)-オン用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/ジエチルエーテル/トリエチルアミン=20/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2,2-ジメチル-3,4-ジヒドロナフタレン-1(2H)-イミンを黄色のオイルとして得た(33.7mg、97%収率)。 [Example 4-15]
(Synthesis of 2,2-dimethyl-3,4-dihydronaphthalene-1 (2H) -imine)
The reaction was performed for 24 hours using 0.20 mmol of 2,2-dimethyl-3,4-dihydronaphthalen-1 (2H) -one under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / diethyl ether / triethylamine = 20/1/1 to obtain the desired 2,2-dimethyl-3,4-dihydronaphthalene-1 (2H). -The imine was obtained as a yellow oil (33.7 mg, 97% yield).
(2,2-ジメチル-3,4-ジヒドロナフタレン-1(2H)-イミンの合成)
上記基本反応条件で0.20mmolの2,2-ジメチル-3,4-ジヒドロナフタレン-1(2H)-オン用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/ジエチルエーテル/トリエチルアミン=20/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の2,2-ジメチル-3,4-ジヒドロナフタレン-1(2H)-イミンを黄色のオイルとして得た(33.7mg、97%収率)。 [Example 4-15]
(Synthesis of 2,2-dimethyl-3,4-dihydronaphthalene-1 (2H) -imine)
The reaction was performed for 24 hours using 0.20 mmol of 2,2-dimethyl-3,4-dihydronaphthalen-1 (2H) -one under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / diethyl ether / triethylamine = 20/1/1 to obtain the desired 2,2-dimethyl-3,4-dihydronaphthalene-1 (2H). -The imine was obtained as a yellow oil (33.7 mg, 97% yield).

1H NMR (500 MHz, CDCl3) δ 9.42 (br, 1H), 8.08 (br, 1H), 7.35 (td, J = 1.5, 7.5 Hz, 1H), 7.27 (tt, J = 0.5, 7.5 Hz, 1H), 7.17 (dd, J = 0.5, 7.5 Hz, 1H), 2.93 (t, J = 6.5 Hz, 2H), 1.85 (t, J = 6.5 Hz, 2H), 1.22 (s, 6H).
13C NMR (125 MHz, CDCl3) δ 181.16, 139.71, 132.50, 130.40, 128.76, 126.36, 126.11, 38.36, 36.42, 25.93, 25.73. 1 H NMR (500 MHz, CDCl 3 ) δ 9.42 (br, 1H), 8.08 (br, 1H), 7.35 (td, J = 1.5, 7.5 Hz, 1H), 7.27 (tt, J = 0.5, 7.5 Hz, 1H), 7.17 (dd, J = 0.5, 7.5 Hz, 1H), 2.93 (t, J = 6.5 Hz, 2H), 1.85 (t, J = 6.5 Hz, 2H), 1.22 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 181.16, 139.71, 132.50, 130.40, 128.76, 126.36, 126.11, 38.36, 36.42, 25.93, 25.73.
13C NMR (125 MHz, CDCl3) δ 181.16, 139.71, 132.50, 130.40, 128.76, 126.36, 126.11, 38.36, 36.42, 25.93, 25.73. 1 H NMR (500 MHz, CDCl 3 ) δ 9.42 (br, 1H), 8.08 (br, 1H), 7.35 (td, J = 1.5, 7.5 Hz, 1H), 7.27 (tt, J = 0.5, 7.5 Hz, 1H), 7.17 (dd, J = 0.5, 7.5 Hz, 1H), 2.93 (t, J = 6.5 Hz, 2H), 1.85 (t, J = 6.5 Hz, 2H), 1.22 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 181.16, 139.71, 132.50, 130.40, 128.76, 126.36, 126.11, 38.36, 36.42, 25.93, 25.73.
[実施例4-16]
(1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミンの合成)
上記基本反応条件で0.20mmolのDL-カンファーと1.5当量のビス(トリメチルシリル)アミン、10mol%のトリフルオロメタンスルホン酸スカンジム(III)を用いて24時間反応を行った。原料の消失を1HNMR測定で確認後、反応混合物に-50℃に冷却したヘキサンを加え、ヘキサン/トリエチルアミン=100/1で事前に平衡化したショートパッドシリカゲルカラムに添加した。そのパッドを冷却したヘキサンで洗浄した後、得られたヘキサン層を分離し、パッドを-50℃に冷却したジクロロメタンで溶出した。得られたジクロロメタン抽出液から減圧下溶媒を留去し、目的の1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミンを淡黄色の固体として得た(15.3mg、約90%純度、約45%収率)。
なお、本実施例の1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミンの合成は、既知の方法では市販の入手容易な化合物から少なくとも2-3段階の工程が必要であるが、本手法では市販の入手容易な化合物から1段階で合成できる。 [Example 4-16]
(Synthesis of 1,7,7-trimethylbicyclo [2.2.1] heptane-2-imine)
The reaction was carried out for 24 hours using 0.20 mmol of DL-camphor and 1.5 equivalents of bis (trimethylsilyl) amine, 10 mol% of scandium (III) trifluoromethanesulfonate under the above basic reaction conditions. After confirming disappearance of the raw materials by 1 HNMR measurement, hexane cooled to −50 ° C. was added to the reaction mixture, and added to a short pad silica gel column pre-equilibrated with hexane / triethylamine = 100/1. After washing the pad with chilled hexane, the resulting hexane layer was separated and the pad was eluted with dichloromethane cooled to -50 ° C. The solvent was distilled off from the obtained dichloromethane extract under reduced pressure to obtain the desired 1,7,7-trimethylbicyclo [2.2.1] heptane-2-imine as a pale yellow solid (15.3 mg, About 90% purity, about 45% yield).
The synthesis of 1,7,7-trimethylbicyclo [2.2.1] heptan-2-imine of this example requires at least 2-3 steps from a commercially available compound by a known method. However, this method can be synthesized in one step from commercially available compounds.
(1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミンの合成)
上記基本反応条件で0.20mmolのDL-カンファーと1.5当量のビス(トリメチルシリル)アミン、10mol%のトリフルオロメタンスルホン酸スカンジム(III)を用いて24時間反応を行った。原料の消失を1HNMR測定で確認後、反応混合物に-50℃に冷却したヘキサンを加え、ヘキサン/トリエチルアミン=100/1で事前に平衡化したショートパッドシリカゲルカラムに添加した。そのパッドを冷却したヘキサンで洗浄した後、得られたヘキサン層を分離し、パッドを-50℃に冷却したジクロロメタンで溶出した。得られたジクロロメタン抽出液から減圧下溶媒を留去し、目的の1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミンを淡黄色の固体として得た(15.3mg、約90%純度、約45%収率)。
なお、本実施例の1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミンの合成は、既知の方法では市販の入手容易な化合物から少なくとも2-3段階の工程が必要であるが、本手法では市販の入手容易な化合物から1段階で合成できる。 [Example 4-16]
(Synthesis of 1,7,7-trimethylbicyclo [2.2.1] heptane-2-imine)
The reaction was carried out for 24 hours using 0.20 mmol of DL-camphor and 1.5 equivalents of bis (trimethylsilyl) amine, 10 mol% of scandium (III) trifluoromethanesulfonate under the above basic reaction conditions. After confirming disappearance of the raw materials by 1 HNMR measurement, hexane cooled to −50 ° C. was added to the reaction mixture, and added to a short pad silica gel column pre-equilibrated with hexane / triethylamine = 100/1. After washing the pad with chilled hexane, the resulting hexane layer was separated and the pad was eluted with dichloromethane cooled to -50 ° C. The solvent was distilled off from the obtained dichloromethane extract under reduced pressure to obtain the desired 1,7,7-trimethylbicyclo [2.2.1] heptane-2-imine as a pale yellow solid (15.3 mg, About 90% purity, about 45% yield).
The synthesis of 1,7,7-trimethylbicyclo [2.2.1] heptan-2-imine of this example requires at least 2-3 steps from a commercially available compound by a known method. However, this method can be synthesized in one step from commercially available compounds.

1H NMR (500 MHz, CDCl3) δ 2.47-2.42 (m, 1H), 1.97 (d, J = 17.5 Hz, 1H), 1.92 (t, J = 4.5 Hz, 1H), 1.91-1.83 (m, 1H), 1.69-1.63 (m, 1H), 1.38-1.25 (m, 2H), 0.94 (s, 3H), 0.93 (s, 3H), 0.80 (s, 3H).
13C NMR (125 MHz, CDCl3) δ. 194.41, 54.86, 47.37, 43.74, 40.35, 32.06, 27.41, 19.64, 19.04, 10.44. 1 H NMR (500 MHz, CDCl 3 ) δ 2.47-2.42 (m, 1H), 1.97 (d, J = 17.5 Hz, 1H), 1.92 (t, J = 4.5 Hz, 1H), 1.91-1.83 (m, 1H), 1.69-1.63 (m, 1H), 1.38-1.25 (m, 2H), 0.94 (s, 3H), 0.93 (s, 3H), 0.80 (s, 3H).
13 C NMR (125 MHz, CDCl 3 ) δ. 194.41, 54.86, 47.37, 43.74, 40.35, 32.06, 27.41, 19.64, 19.04, 10.44.
13C NMR (125 MHz, CDCl3) δ. 194.41, 54.86, 47.37, 43.74, 40.35, 32.06, 27.41, 19.64, 19.04, 10.44. 1 H NMR (500 MHz, CDCl 3 ) δ 2.47-2.42 (m, 1H), 1.97 (d, J = 17.5 Hz, 1H), 1.92 (t, J = 4.5 Hz, 1H), 1.91-1.83 (m, 1H), 1.69-1.63 (m, 1H), 1.38-1.25 (m, 2H), 0.94 (s, 3H), 0.93 (s, 3H), 0.80 (s, 3H).
13 C NMR (125 MHz, CDCl 3 ) δ. 194.41, 54.86, 47.37, 43.74, 40.35, 32.06, 27.41, 19.64, 19.04, 10.44.
[実施例4-17]
(アダマンタン-2-イミンの合成)
上記基本反応条件で1.0mmolのアダマンタン-2-オンと3.0当量のビス(トリメチルシリル)アミン、10mol%のトリフルオロメタンスルホン酸スカンジム(III)を用いて48時間反応を行った。粗生成物の1HNMR測定を行ったところ、原料と単一の生成物が確認され、その生成物の収率は59%であった。 [Example 4-17]
(Synthesis of adamantane-2-imine)
The reaction was carried out for 48 hours using 1.0 mmol of adamantan-2-one and 3.0 equivalents of bis (trimethylsilyl) amine and 10 mol% of scandium (III) trifluoromethanesulfonate under the above basic reaction conditions. As a result of 1 HNMR measurement of the crude product, the raw material and a single product were confirmed, and the yield of the product was 59%.
(アダマンタン-2-イミンの合成)
上記基本反応条件で1.0mmolのアダマンタン-2-オンと3.0当量のビス(トリメチルシリル)アミン、10mol%のトリフルオロメタンスルホン酸スカンジム(III)を用いて48時間反応を行った。粗生成物の1HNMR測定を行ったところ、原料と単一の生成物が確認され、その生成物の収率は59%であった。 [Example 4-17]
(Synthesis of adamantane-2-imine)
The reaction was carried out for 48 hours using 1.0 mmol of adamantan-2-one and 3.0 equivalents of bis (trimethylsilyl) amine and 10 mol% of scandium (III) trifluoromethanesulfonate under the above basic reaction conditions. As a result of 1 HNMR measurement of the crude product, the raw material and a single product were confirmed, and the yield of the product was 59%.

[実施例4-18]
(2,2-ジメチル-1-フェニルプロパン-1-イミニウムクロライドの合成)
上記基本反応条件で1.0mmolのtert-ブチルフェニルケトンと1.5当量のビス(トリメチルシリル)アミンを用いて6時間反応を行った。原料の消失を1HNMR測定で確認後、ヘキサンで希釈した反応混合物をセライトろ過した。ろ液を減圧下で濃縮後、残渣を1.0mLのジエチルエーテルに溶かし、1.0当量の塩酸(1Mジエチルエーテル溶液、1.0mL)を加え、懸濁液を1時間撹拌した。懸濁液をろ過することで目的の2,2-ジメチル-1-フェニルプロパン-1-イミニウムクロライドを白色の固体として得た(143mg、72%収率、>95%純度)。 [Example 4-18]
(Synthesis of 2,2-dimethyl-1-phenylpropane-1-iminium chloride)
The reaction was performed for 6 hours using 1.0 mmol of tert-butyl phenyl ketone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. After confirming disappearance of the raw material by 1 HNMR measurement, the reaction mixture diluted with hexane was filtered through Celite. After the filtrate was concentrated under reduced pressure, the residue was dissolved in 1.0 mL of diethyl ether, 1.0 equivalent of hydrochloric acid (1M diethyl ether solution, 1.0 mL) was added, and the suspension was stirred for 1 hour. The suspension was filtered to give the desired 2,2-dimethyl-1-phenylpropane-1-iminium chloride as a white solid (143 mg, 72% yield,> 95% purity).
(2,2-ジメチル-1-フェニルプロパン-1-イミニウムクロライドの合成)
上記基本反応条件で1.0mmolのtert-ブチルフェニルケトンと1.5当量のビス(トリメチルシリル)アミンを用いて6時間反応を行った。原料の消失を1HNMR測定で確認後、ヘキサンで希釈した反応混合物をセライトろ過した。ろ液を減圧下で濃縮後、残渣を1.0mLのジエチルエーテルに溶かし、1.0当量の塩酸(1Mジエチルエーテル溶液、1.0mL)を加え、懸濁液を1時間撹拌した。懸濁液をろ過することで目的の2,2-ジメチル-1-フェニルプロパン-1-イミニウムクロライドを白色の固体として得た(143mg、72%収率、>95%純度)。 [Example 4-18]
(Synthesis of 2,2-dimethyl-1-phenylpropane-1-iminium chloride)
The reaction was performed for 6 hours using 1.0 mmol of tert-butyl phenyl ketone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. After confirming disappearance of the raw material by 1 HNMR measurement, the reaction mixture diluted with hexane was filtered through Celite. After the filtrate was concentrated under reduced pressure, the residue was dissolved in 1.0 mL of diethyl ether, 1.0 equivalent of hydrochloric acid (1M diethyl ether solution, 1.0 mL) was added, and the suspension was stirred for 1 hour. The suspension was filtered to give the desired 2,2-dimethyl-1-phenylpropane-1-iminium chloride as a white solid (143 mg, 72% yield,> 95% purity).

1H NMR (500 MHz, DMSO-d6) δ 12.75 (br, 2H), 7.67-7.63 (m, 1H), 7.59-7.58 (m, 4H), 1.35 (s, 9H).
13C NMR (125 MHz, DMSO-d6) δ. 199.86, 132.74, 132.11, 128.93, 127.59, 40.99, 27.93. 1 H NMR (500 MHz, DMSO-d 6 ) δ 12.75 (br, 2H), 7.67-7.63 (m, 1H), 7.59-7.58 (m, 4H), 1.35 (s, 9H).
13 C NMR (125 MHz, DMSO-d 6 ) δ. 199.86, 132.74, 132.11, 128.93, 127.59, 40.99, 27.93.
13C NMR (125 MHz, DMSO-d6) δ. 199.86, 132.74, 132.11, 128.93, 127.59, 40.99, 27.93. 1 H NMR (500 MHz, DMSO-d 6 ) δ 12.75 (br, 2H), 7.67-7.63 (m, 1H), 7.59-7.58 (m, 4H), 1.35 (s, 9H).
13 C NMR (125 MHz, DMSO-d 6 ) δ. 199.86, 132.74, 132.11, 128.93, 127.59, 40.99, 27.93.
[実施例4-19-1]
(2,2,2-トリフルオロ-1-フェニルエタンイミンの合成)
4mLのバイアルに撹拌子を入れ、トリフルオロメタンスルホン酸スカンジウム(III)(0.020mmol,2.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、2,2,2-トリフルオロアセトフェノン(1.0 mmol)、モノクロロベンゼン(1.0mL,1.0M)、ビス(トリメチルシリル)アミン(1.1mmol,1.1当量)を加えた。混合物を90℃で24時間撹拌した後、収率を19FNMR測定により確認したところ、95%以上の2,2,2-トリフルオロ-1-フェニルエタンイミンへの変換が確認された。 [Example 4-19-1]
(Synthesis of 2,2,2-trifluoro-1-phenylethanimine)
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.020 mmol, 2.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon and 2,2,2-trifluoroacetophenone (1.0 mmol), monochlorobenzene (1.0 mL, 1.0 M), bis (trimethylsilyl) amine (1.1 mmol, 1.1 mmol). Equivalent) was added. After the mixture was stirred at 90 ° C. for 24 hours, the yield was confirmed by 19 FNMR measurement, and conversion to 95% or more of 2,2,2-trifluoro-1-phenylethaneimine was confirmed.
(2,2,2-トリフルオロ-1-フェニルエタンイミンの合成)
4mLのバイアルに撹拌子を入れ、トリフルオロメタンスルホン酸スカンジウム(III)(0.020mmol,2.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、2,2,2-トリフルオロアセトフェノン(1.0 mmol)、モノクロロベンゼン(1.0mL,1.0M)、ビス(トリメチルシリル)アミン(1.1mmol,1.1当量)を加えた。混合物を90℃で24時間撹拌した後、収率を19FNMR測定により確認したところ、95%以上の2,2,2-トリフルオロ-1-フェニルエタンイミンへの変換が確認された。 [Example 4-19-1]
(Synthesis of 2,2,2-trifluoro-1-phenylethanimine)
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.020 mmol, 2.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon and 2,2,2-trifluoroacetophenone (1.0 mmol), monochlorobenzene (1.0 mL, 1.0 M), bis (trimethylsilyl) amine (1.1 mmol, 1.1 mmol). Equivalent) was added. After the mixture was stirred at 90 ° C. for 24 hours, the yield was confirmed by 19 FNMR measurement, and conversion to 95% or more of 2,2,2-trifluoro-1-phenylethaneimine was confirmed.

[実施例4-19-2]
(大スケール(10mmol)での2,2,2-トリフルオロ-1-フェニルエタンイミンの合成)
上記実施例4-14-1の反応条件に従って、50mLのフラスコにトリフルオロメタンスルホン酸スカンジム(III)(49.2mg,0.10mmol,1.0mol%)、2,2,2-トリフルオロアセトフェノン(1.4mL,10mmol)、フルオロベンゼン(10mL,1.0M)、ビス(トリメチルシリル)アミン(2.3mL,11mmol,1.1当量)を加えて、90℃で24時間加熱撹拌した。原料の消失を19FNMR測定で確認後、反応混合物に-50℃に冷却したヘキサンを加え、ヘキサン/トリエチルアミン=100/1で事前に平衡化したショートパッドシリカゲルカラムに添加した。そのパッドを冷却したヘキサンで洗浄した後、得られたヘキサン層を分離し、パッドを-50℃に冷却したジクロロメタンで溶出した。得られたジクロロメタン抽出液から減圧下溶媒を留去し、目的の2,2,2-トリフルオロ-1-フェニルエタンイミンを無色のオイルとして得た(1.45g、84%収率)。NHイミンのプロトンのE/Z異性体によるジアステレオ比は19FNMRにより2.6:1と決定した。 [Example 4-19-2]
(Synthesis of 2,2,2-trifluoro-1-phenylethaneimine on a large scale (10 mmol))
According to the reaction conditions of Example 4-14-1 above, a 50 mL flask was charged with scandim (III) trifluoromethanesulfonate (49.2 mg, 0.10 mmol, 1.0 mol%), 2,2,2-trifluoroacetophenone ( 1.4 mL, 10 mmol), fluorobenzene (10 mL, 1.0 M), bis (trimethylsilyl) amine (2.3 mL, 11 mmol, 1.1 equivalents) were added, and the mixture was heated and stirred at 90 ° C. for 24 hours. After confirming the disappearance of the raw materials by 19 FNMR measurement, hexane cooled to −50 ° C. was added to the reaction mixture, and added to a short pad silica gel column pre-equilibrated with hexane / triethylamine = 100/1. After washing the pad with chilled hexane, the resulting hexane layer was separated and the pad was eluted with dichloromethane cooled to -50 ° C. The solvent was distilled off from the obtained dichloromethane extract under reduced pressure to obtain the desired 2,2,2-trifluoro-1-phenylethaneimine as a colorless oil (1.45 g, 84% yield). The diastereo ratio of the NH imine proton due to the E / Z isomer was determined to be 2.6: 1 by 19 FNMR.
(大スケール(10mmol)での2,2,2-トリフルオロ-1-フェニルエタンイミンの合成)
上記実施例4-14-1の反応条件に従って、50mLのフラスコにトリフルオロメタンスルホン酸スカンジム(III)(49.2mg,0.10mmol,1.0mol%)、2,2,2-トリフルオロアセトフェノン(1.4mL,10mmol)、フルオロベンゼン(10mL,1.0M)、ビス(トリメチルシリル)アミン(2.3mL,11mmol,1.1当量)を加えて、90℃で24時間加熱撹拌した。原料の消失を19FNMR測定で確認後、反応混合物に-50℃に冷却したヘキサンを加え、ヘキサン/トリエチルアミン=100/1で事前に平衡化したショートパッドシリカゲルカラムに添加した。そのパッドを冷却したヘキサンで洗浄した後、得られたヘキサン層を分離し、パッドを-50℃に冷却したジクロロメタンで溶出した。得られたジクロロメタン抽出液から減圧下溶媒を留去し、目的の2,2,2-トリフルオロ-1-フェニルエタンイミンを無色のオイルとして得た(1.45g、84%収率)。NHイミンのプロトンのE/Z異性体によるジアステレオ比は19FNMRにより2.6:1と決定した。 [Example 4-19-2]
(Synthesis of 2,2,2-trifluoro-1-phenylethaneimine on a large scale (10 mmol))
According to the reaction conditions of Example 4-14-1 above, a 50 mL flask was charged with scandim (III) trifluoromethanesulfonate (49.2 mg, 0.10 mmol, 1.0 mol%), 2,2,2-trifluoroacetophenone ( 1.4 mL, 10 mmol), fluorobenzene (10 mL, 1.0 M), bis (trimethylsilyl) amine (2.3 mL, 11 mmol, 1.1 equivalents) were added, and the mixture was heated and stirred at 90 ° C. for 24 hours. After confirming the disappearance of the raw materials by 19 FNMR measurement, hexane cooled to −50 ° C. was added to the reaction mixture, and added to a short pad silica gel column pre-equilibrated with hexane / triethylamine = 100/1. After washing the pad with chilled hexane, the resulting hexane layer was separated and the pad was eluted with dichloromethane cooled to -50 ° C. The solvent was distilled off from the obtained dichloromethane extract under reduced pressure to obtain the desired 2,2,2-trifluoro-1-phenylethaneimine as a colorless oil (1.45 g, 84% yield). The diastereo ratio of the NH imine proton due to the E / Z isomer was determined to be 2.6: 1 by 19 FNMR.

1H NMR (500 MHz, CDCl3) δ 10.78* (br, 1H), 10.69 (br, 1H), 7.99-7.97 (m, 2H + 2H*), 7.59-7.47 (3H + 3H*).
13C NMR (125 MHz, CDCl3) δ 166.76* (q, JC-F = 33 Hz), 163.16 (q, JC-F = 31 Hz), 132.29*, 123.32, 131.84*, 130.93, 129.12*, 128.70, 128.10, 126.56*, 120.27* (q, JC-F = 278 Hz), 118.42 (q, JC-F = 280 Hz).
19F NMR (470 MHz, CDCl3) δ-68.65*, -69.56.
[* denotes the minor diastereomer] 1 H NMR (500 MHz, CDCl 3 ) δ 10.78 * (br, 1H), 10.69 (br, 1H), 7.99-7.97 (m, 2H + 2H *), 7.59-7.47 (3H + 3H *).
13 C NMR (125 MHz, CDCl 3 ) δ 166.76 * (q, J CF = 33 Hz), 163.16 (q, J CF = 31 Hz), 132.29 *, 123.32, 131.84 *, 130.93, 129.12 *, 128.70, 128.10 , 126.56 *, 120.27 * (q, J CF = 278 Hz), 118.42 (q, J CF = 280 Hz).
19 F NMR (470 MHz, CDCl 3 ) δ-68.65 *, -69.56.
[* designate the minor diastereomer]
13C NMR (125 MHz, CDCl3) δ 166.76* (q, JC-F = 33 Hz), 163.16 (q, JC-F = 31 Hz), 132.29*, 123.32, 131.84*, 130.93, 129.12*, 128.70, 128.10, 126.56*, 120.27* (q, JC-F = 278 Hz), 118.42 (q, JC-F = 280 Hz).
19F NMR (470 MHz, CDCl3) δ-68.65*, -69.56.
[* denotes the minor diastereomer] 1 H NMR (500 MHz, CDCl 3 ) δ 10.78 * (br, 1H), 10.69 (br, 1H), 7.99-7.97 (m, 2H + 2H *), 7.59-7.47 (3H + 3H *).
13 C NMR (125 MHz, CDCl 3 ) δ 166.76 * (q, J CF = 33 Hz), 163.16 (q, J CF = 31 Hz), 132.29 *, 123.32, 131.84 *, 130.93, 129.12 *, 128.70, 128.10 , 126.56 *, 120.27 * (q, J CF = 278 Hz), 118.42 (q, J CF = 280 Hz).
19 F NMR (470 MHz, CDCl 3 ) δ-68.65 *, -69.56.
[* designate the minor diastereomer]
[実施例4-20]
(ビス(4-フルオロフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジフルオロベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-フルオロフェニル)メタンイミンを黄色のオイルとして得た(42.8mg、99%収率)。 [Example 4-20]
(Synthesis of bis (4-fluorophenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4,4′-difluorobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (4-fluorophenyl) methanimine as a yellow oil (42.8 mg, 99% yield).
(ビス(4-フルオロフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジフルオロベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-フルオロフェニル)メタンイミンを黄色のオイルとして得た(42.8mg、99%収率)。 [Example 4-20]
(Synthesis of bis (4-fluorophenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4,4′-difluorobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (4-fluorophenyl) methanimine as a yellow oil (42.8 mg, 99% yield).

1H NMR (500 MHz, CDCl3) δ 9.64 (br, 1H), 7.56 (br, 4H), 7.11 (t, J = 8.5 Hz, 4H).
13C NMR (125 MHz, CDCl3) δ 176.07, 164.10 (d, JC-F = 250 Hz), 135.35, 130.43, 115.48 (d, JC-F = 22 Hz).
19F NMR (360 MHz, CDCl3) δ -109.86 (br, 2F) 1 H NMR (500 MHz, CDCl 3 ) δ 9.64 (br, 1H), 7.56 (br, 4H), 7.11 (t, J = 8.5 Hz, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 176.07, 164.10 (d, J CF = 250 Hz), 135.35, 130.43, 115.48 (d, J CF = 22 Hz).
19 F NMR (360 MHz, CDCl 3 ) δ -109.86 (br, 2F)
13C NMR (125 MHz, CDCl3) δ 176.07, 164.10 (d, JC-F = 250 Hz), 135.35, 130.43, 115.48 (d, JC-F = 22 Hz).
19F NMR (360 MHz, CDCl3) δ -109.86 (br, 2F) 1 H NMR (500 MHz, CDCl 3 ) δ 9.64 (br, 1H), 7.56 (br, 4H), 7.11 (t, J = 8.5 Hz, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 176.07, 164.10 (d, J CF = 250 Hz), 135.35, 130.43, 115.48 (d, J CF = 22 Hz).
19 F NMR (360 MHz, CDCl 3 ) δ -109.86 (br, 2F)
[実施例4-21]
(ビス(4-ブロモフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジブロモベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-ブロモフェニル)メタンイミンを白色の固体として得た(65.5mg、97%収率)。 [Example 4-21]
(Synthesis of bis (4-bromophenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4,4′-dibromobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (4-bromophenyl) methanimine as a white solid (65.5 mg, 97% yield).
(ビス(4-ブロモフェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4,4’-ジブロモベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のビス(4-ブロモフェニル)メタンイミンを白色の固体として得た(65.5mg、97%収率)。 [Example 4-21]
(Synthesis of bis (4-bromophenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4,4′-dibromobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography using a developing solvent of hexane / triethylamine = 9/1 to obtain the desired bis (4-bromophenyl) methanimine as a white solid (65.5 mg, 97% yield).

1H NMR (500 MHz, CDCl3) δ 9.77 (br, 1H), 7.56 (d, J = 8.0 Hz, 4H), 7.43 (br, 4H).
13C NMR (125 MHz, CDCl3) δ 176.21, 137.79, 131.70, 129.88, 125.12. 1 H NMR (500 MHz, CDCl 3 ) δ 9.77 (br, 1H), 7.56 (d, J = 8.0 Hz, 4H), 7.43 (br, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 176.21, 137.79, 131.70, 129.88, 125.12.
13C NMR (125 MHz, CDCl3) δ 176.21, 137.79, 131.70, 129.88, 125.12. 1 H NMR (500 MHz, CDCl 3 ) δ 9.77 (br, 1H), 7.56 (d, J = 8.0 Hz, 4H), 7.43 (br, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 176.21, 137.79, 131.70, 129.88, 125.12.
[実施例4-22]
((4-ニトロフェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-ニトロベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/酢酸エチル/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-ニトロフェニル)(フェニル)メタンイミンを黄色のオイルとして得た(44.5mg、98%収率)。NHイミンのプロトンのE/Z異性体によるジアステレオ比は1HNMRにより1.4:1と決定した。 [Example 4-22]
(Synthesis of (4-nitrophenyl) (phenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4-nitrobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / ethyl acetate / triethylamine = 8/1/1 to obtain the desired (4-nitrophenyl) (phenyl) methanimine as a yellow oil. (44.5 mg, 98% yield). The diastereo ratio of the NH imine proton by the E / Z isomer was determined to be 1.4: 1 by 1 HNMR.
((4-ニトロフェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-ニトロベンゾフェノンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/酢酸エチル/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-ニトロフェニル)(フェニル)メタンイミンを黄色のオイルとして得た(44.5mg、98%収率)。NHイミンのプロトンのE/Z異性体によるジアステレオ比は1HNMRにより1.4:1と決定した。 [Example 4-22]
(Synthesis of (4-nitrophenyl) (phenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4-nitrobenzophenone under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / ethyl acetate / triethylamine = 8/1/1 to obtain the desired (4-nitrophenyl) (phenyl) methanimine as a yellow oil. (44.5 mg, 98% yield). The diastereo ratio of the NH imine proton by the E / Z isomer was determined to be 1.4: 1 by 1 HNMR.

1H NMR (500 MHz, DMSO-d6) δ 11.12 (br, 1H), 10.93 (br, 1H*), 8.32-8.23 (m, 2H + 2H*), 7.86 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H*), 7.63 (dd, J = 8.5, 1.5 Hz, 2H*), 7.56-7.44 (m, 5H + 3H*).
13C NMR (125 MHz, DMSO-d6) δ 174.46, 174.18*, 148.93, 148.37*, 145.86*, 145.06, 138.64, 138.02*, 131.30*, 130.71, 130.45, 129.49*, 129.12, 128.94*, 128.90*, 128.06, 124.12*, 123.94.
[* denotes the minor diastereomer] 1 H NMR (500 MHz, DMSO-d 6 ) δ 11.12 (br, 1H), 10.93 (br, 1H *), 8.32-8.23 (m, 2H + 2H *), 7.86 (d, J = 8.5 Hz, 2H ), 7.67 (d, J = 8.5 Hz, 2H *), 7.63 (dd, J = 8.5, 1.5 Hz, 2H *), 7.56-7.44 (m, 5H + 3H *).
13 C NMR (125 MHz, DMSO-d 6 ) δ 174.46, 174.18 *, 148.93, 148.37 *, 145.86 *, 145.06, 138.64, 138.02 *, 131.30 *, 130.71, 130.45, 129.49 *, 129.12, 128.94 *, 128.90 * , 128.06, 124.12 *, 123.94.
[* designate the minor diastereomer]
13C NMR (125 MHz, DMSO-d6) δ 174.46, 174.18*, 148.93, 148.37*, 145.86*, 145.06, 138.64, 138.02*, 131.30*, 130.71, 130.45, 129.49*, 129.12, 128.94*, 128.90*, 128.06, 124.12*, 123.94.
[* denotes the minor diastereomer] 1 H NMR (500 MHz, DMSO-d 6 ) δ 11.12 (br, 1H), 10.93 (br, 1H *), 8.32-8.23 (m, 2H + 2H *), 7.86 (d, J = 8.5 Hz, 2H ), 7.67 (d, J = 8.5 Hz, 2H *), 7.63 (dd, J = 8.5, 1.5 Hz, 2H *), 7.56-7.44 (m, 5H + 3H *).
13 C NMR (125 MHz, DMSO-d 6 ) δ 174.46, 174.18 *, 148.93, 148.37 *, 145.86 *, 145.06, 138.64, 138.02 *, 131.30 *, 130.71, 130.45, 129.49 *, 129.12, 128.94 *, 128.90 * , 128.06, 124.12 *, 123.94.
[* designate the minor diastereomer]
[実施例4-23]
((4-((tert-ブチルシメチルシリル)オキシ)フェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-((tert-ブチルシメチルシリル)オキシ)ベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-((tert-ブチルシメチルシリル)オキシ)フェニル)(フェニル)メタンイミンを黄色のオイルとして得た(49.6mg、80%収率)。 [Example 4-23]
(Synthesis of (4-((tert-butylcymethylsilyl) oxy) phenyl) (phenyl) methanimine)
The reaction was performed for 24 hours using 0.20 mmol of 4-((tert-butylcymethylsilyl) oxy) benzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1, and the target (4-((tert-butylcymethylsilyl) oxy) phenyl) (phenyl) methanimine was converted to yellow. As an oil (49.6 mg, 80% yield).
((4-((tert-ブチルシメチルシリル)オキシ)フェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-((tert-ブチルシメチルシリル)オキシ)ベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて24時間反応を行った。得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-((tert-ブチルシメチルシリル)オキシ)フェニル)(フェニル)メタンイミンを黄色のオイルとして得た(49.6mg、80%収率)。 [Example 4-23]
(Synthesis of (4-((tert-butylcymethylsilyl) oxy) phenyl) (phenyl) methanimine)
The reaction was performed for 24 hours using 0.20 mmol of 4-((tert-butylcymethylsilyl) oxy) benzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1, and the target (4-((tert-butylcymethylsilyl) oxy) phenyl) (phenyl) methanimine was converted to yellow. As an oil (49.6 mg, 80% yield).

1H NMR (500 MHz, CDCl3) δ 9.51 (br, 1H), 7.52 (br, 4H), 7.48-7.44 (m. 1H), 7.43-7.40 (m, 2H), 6.85 (dt, J = 9.5, 2.5 Hz, 2H), 1.00 (s, 9H), 0.23 (s, 6H).
13C NMR (125 MHz, CDCl3) δ 177.92, 157.98, 139.92, 132.03, 130.23, 130.06, 128.30, 119.80, 115.44, 25.64, 18.24. 1 H NMR (500 MHz, CDCl 3 ) δ 9.51 (br, 1H), 7.52 (br, 4H), 7.48-7.44 (m. 1H), 7.43-7.40 (m, 2H), 6.85 (dt, J = 9.5 , 2.5 Hz, 2H), 1.00 (s, 9H), 0.23 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.92, 157.98, 139.92, 132.03, 130.23, 130.06, 128.30, 119.80, 115.44, 25.64, 18.24.
13C NMR (125 MHz, CDCl3) δ 177.92, 157.98, 139.92, 132.03, 130.23, 130.06, 128.30, 119.80, 115.44, 25.64, 18.24. 1 H NMR (500 MHz, CDCl 3 ) δ 9.51 (br, 1H), 7.52 (br, 4H), 7.48-7.44 (m. 1H), 7.43-7.40 (m, 2H), 6.85 (dt, J = 9.5 , 2.5 Hz, 2H), 1.00 (s, 9H), 0.23 (s, 6H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.92, 157.98, 139.92, 132.03, 130.23, 130.06, 128.30, 119.80, 115.44, 25.64, 18.24.
[実施例4-24]
((4-(アジドメチル)フェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-(アジドメチル)ベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-(アジドメチル)フェニル)(フェニル)メタンイミンを黄色のオイルとして得た(39.8mg、84%収率)。 [Example 4-24]
(Synthesis of (4- (azidomethyl) phenyl) (phenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4- (azidomethyl) benzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product is subjected to silica gel column chromatography with a developing solvent of hexane / dichloromethane / triethylamine = 8/1/1 to obtain the desired (4- (azidomethyl) phenyl) (phenyl) methanimine as a yellow oil. (39.8 mg, 84% yield).
((4-(アジドメチル)フェニル)(フェニル)メタンイミンの合成)
上記基本反応条件で0.20mmolの4-(アジドメチル)ベンゾフェノンと1.5当量のビス(トリメチルシリル)アミンを用いて2時間反応を行った。得られた反応粗生成物についてヘキサン/ジクロロメタン/トリエチルアミン=8/1/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的の(4-(アジドメチル)フェニル)(フェニル)メタンイミンを黄色のオイルとして得た(39.8mg、84%収率)。 [Example 4-24]
(Synthesis of (4- (azidomethyl) phenyl) (phenyl) methanimine)
The reaction was carried out for 2 hours using 0.20 mmol of 4- (azidomethyl) benzophenone and 1.5 equivalents of bis (trimethylsilyl) amine under the above basic reaction conditions. The obtained reaction crude product is subjected to silica gel column chromatography with a developing solvent of hexane / dichloromethane / triethylamine = 8/1/1 to obtain the desired (4- (azidomethyl) phenyl) (phenyl) methanimine as a yellow oil. (39.8 mg, 84% yield).

1H NMR (500 MHz, CDCl3) δ 9.76 (br, 1H), 7.62 (br, 4H), 7.50-7.47 (m, 1H), 7.43 (dd, J = 7.5, 7.5 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 4.41 (s, 2H).
13C NMR (125 MHz, CDCl3) δ 177.77, 139.22, 137.67, 130.38, 129.03, 128.42, 128.28, 128.04, 54.41. 1 H NMR (500 MHz, CDCl 3 ) δ 9.76 (br, 1H), 7.62 (br, 4H), 7.50-7.47 (m, 1H), 7.43 (dd, J = 7.5, 7.5 Hz, 2H), 7.38 ( d, J = 8.0 Hz, 2H), 4.41 (s, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.77, 139.22, 137.67, 130.38, 129.03, 128.42, 128.28, 128.04, 54.41.
13C NMR (125 MHz, CDCl3) δ 177.77, 139.22, 137.67, 130.38, 129.03, 128.42, 128.28, 128.04, 54.41. 1 H NMR (500 MHz, CDCl 3 ) δ 9.76 (br, 1H), 7.62 (br, 4H), 7.50-7.47 (m, 1H), 7.43 (dd, J = 7.5, 7.5 Hz, 2H), 7.38 ( d, J = 8.0 Hz, 2H), 4.41 (s, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 177.77, 139.22, 137.67, 130.38, 129.03, 128.42, 128.28, 128.04, 54.41.
[実施例4-25]
(1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミニウムクロライドの合成)
上記基本反応条件で0.20mmolのDL-カンファーと2.0当量のビス(トリメチルシリル)アミン、10mol%のトリフルオロメタンスルホン酸スカンジム(III)を用いて24時間反応を行った。原料の消失を1HNMR測定で確認後、反応混合物を25mLフラスコに移し、4.0当量のメタノールを加えて室温で1時間撹拌した。混合物を減圧下で濃縮後、残渣を2.0mLのジエチルエーテルに溶かし、5.0当量の塩酸(1Mジエチルエーテル溶液、1.0mL)を加え、懸濁液を1時間撹拌した。懸濁液をろ過することで目的の1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミニウムクロライドを白色の固体として得た(25.4mg、68%収率)。 [Example 4-25]
(Synthesis of 1,7,7-trimethylbicyclo [2.2.1] heptane-2-iminium chloride)
The reaction was carried out for 24 hours using 0.20 mmol of DL-camphor and 2.0 equivalents of bis (trimethylsilyl) amine, 10 mol% of scandium (III) trifluoromethanesulfonate under the above basic reaction conditions. After confirming disappearance of the raw materials by 1 HNMR measurement, the reaction mixture was transferred to a 25 mL flask, 4.0 equivalents of methanol was added, and the mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, the residue was dissolved in 2.0 mL of diethyl ether, 5.0 equivalents of hydrochloric acid (1M diethyl ether solution, 1.0 mL) was added, and the suspension was stirred for 1 hour. The suspension was filtered to give the desired 1,7,7-trimethylbicyclo [2.2.1] heptane-2-iminium chloride as a white solid (25.4 mg, 68% yield).
(1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミニウムクロライドの合成)
上記基本反応条件で0.20mmolのDL-カンファーと2.0当量のビス(トリメチルシリル)アミン、10mol%のトリフルオロメタンスルホン酸スカンジム(III)を用いて24時間反応を行った。原料の消失を1HNMR測定で確認後、反応混合物を25mLフラスコに移し、4.0当量のメタノールを加えて室温で1時間撹拌した。混合物を減圧下で濃縮後、残渣を2.0mLのジエチルエーテルに溶かし、5.0当量の塩酸(1Mジエチルエーテル溶液、1.0mL)を加え、懸濁液を1時間撹拌した。懸濁液をろ過することで目的の1,7,7-トリメチルビシクロ[2.2.1]ヘプタン-2-イミニウムクロライドを白色の固体として得た(25.4mg、68%収率)。 [Example 4-25]
(Synthesis of 1,7,7-trimethylbicyclo [2.2.1] heptane-2-iminium chloride)
The reaction was carried out for 24 hours using 0.20 mmol of DL-camphor and 2.0 equivalents of bis (trimethylsilyl) amine, 10 mol% of scandium (III) trifluoromethanesulfonate under the above basic reaction conditions. After confirming disappearance of the raw materials by 1 HNMR measurement, the reaction mixture was transferred to a 25 mL flask, 4.0 equivalents of methanol was added, and the mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, the residue was dissolved in 2.0 mL of diethyl ether, 5.0 equivalents of hydrochloric acid (1M diethyl ether solution, 1.0 mL) was added, and the suspension was stirred for 1 hour. The suspension was filtered to give the desired 1,7,7-trimethylbicyclo [2.2.1] heptane-2-iminium chloride as a white solid (25.4 mg, 68% yield).

1H NMR (500 MHz, DMSO-d6) δ 12.51 (br, 1H), 2.96-2.91 (m, 1H), 2.47 (d, J = 20 Hz, 1H), 2.12 (t, J = 4.5 Hz, 1H), 1.93-1.85 (m, 2H), 1.44-1.34 (m, 2H), 1.11 (s, 3H), 0.95 (s, 3H), 0.79 (s, 3H).
13C NMR (125 MHz, DMSO-d6) δ. 207.11, 57.51, 49.85, 43.32, 38.43, 31.84, 25.96, 19.63, 18.62, 10.19. 1 H NMR (500 MHz, DMSO-d 6 ) δ 12.51 (br, 1H), 2.96-2.91 (m, 1H), 2.47 (d, J = 20 Hz, 1H), 2.12 (t, J = 4.5 Hz, 1H), 1.93-1.85 (m, 2H), 1.44-1.34 (m, 2H), 1.11 (s, 3H), 0.95 (s, 3H), 0.79 (s, 3H).
13 C NMR (125 MHz, DMSO-d 6 ) δ. 207.11, 57.51, 49.85, 43.32, 38.43, 31.84, 25.96, 19.63, 18.62, 10.19.
13C NMR (125 MHz, DMSO-d6) δ. 207.11, 57.51, 49.85, 43.32, 38.43, 31.84, 25.96, 19.63, 18.62, 10.19. 1 H NMR (500 MHz, DMSO-d 6 ) δ 12.51 (br, 1H), 2.96-2.91 (m, 1H), 2.47 (d, J = 20 Hz, 1H), 2.12 (t, J = 4.5 Hz, 1H), 1.93-1.85 (m, 2H), 1.44-1.34 (m, 2H), 1.11 (s, 3H), 0.95 (s, 3H), 0.79 (s, 3H).
13 C NMR (125 MHz, DMSO-d 6 ) δ. 207.11, 57.51, 49.85, 43.32, 38.43, 31.84, 25.96, 19.63, 18.62, 10.19.
[反応促進剤の検討]
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒としてのモノクロロベンゼン(1.0mL,1.0M)、窒素源化合物(2)(0.22mmol,1.1当量)、反応促進剤(0.010mmol、5.0mol%)を加えた。反応促進剤として、トリメチルシラノール(TMSOH)、メタノール(MeOH)を用いた。混合物を50℃で撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表7に示す。 [Examination of reaction accelerator]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (1.0 mL, 1.0 M) as the solvent, nitrogen source compound (2) (0.22 mmol, 1 0.1 equivalent), a reaction accelerator (0.010 mmol, 5.0 mol%) was added. Trimethylsilanol (TMSOH) and methanol (MeOH) were used as reaction accelerators. After the mixture was stirred at 50 ° C., the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 7.
4mLのバイアルに撹拌子を入れ、触媒としてのトリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてのベンゾフェノン(0.20mmol)、溶媒としてのモノクロロベンゼン(1.0mL,1.0M)、窒素源化合物(2)(0.22mmol,1.1当量)、反応促進剤(0.010mmol、5.0mol%)を加えた。反応促進剤として、トリメチルシラノール(TMSOH)、メタノール(MeOH)を用いた。混合物を50℃で撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表7に示す。 [Examination of reaction accelerator]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) as a catalyst was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone (0.20 mmol) as the ketone compound (1), monochlorobenzene (1.0 mL, 1.0 M) as the solvent, nitrogen source compound (2) (0.22 mmol, 1 0.1 equivalent), a reaction accelerator (0.010 mmol, 5.0 mol%) was added. Trimethylsilanol (TMSOH) and methanol (MeOH) were used as reaction accelerators. After the mixture was stirred at 50 ° C., the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 7.

表7に示すように、トリメチルシラノールやメタノールを添加することにより、反応が促進する(収率が向上する)ことがわかる。
As shown in Table 7, it can be seen that addition of trimethylsilanol or methanol promotes the reaction (improves yield).
[窒素上無保護イミン化合物を経由したグリシンシッフ塩基のワンポット合成]
4mLのバイアルに撹拌子を入れ、トリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ベンゾフェノン(36.4mg,0.20mmol)、モノクロロベンゼン(0.20mL,1.0M)、ビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を70℃で5時間撹拌し、原料の消失を1HNMR測定により確認した。 [One-pot synthesis of glycine Schiff base via unprotected imine compound on nitrogen]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon and benzophenone (36.4 mg, 0.20 mmol), monochlorobenzene (0.20 mL, 1.0 M), bis (trimethylsilyl) amine (0.22 mmol, 1.1 eq) were added. . The mixture was stirred at 70 ° C. for 5 hours, and disappearance of raw materials was confirmed by 1 HNMR measurement.
4mLのバイアルに撹拌子を入れ、トリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ベンゾフェノン(36.4mg,0.20mmol)、モノクロロベンゼン(0.20mL,1.0M)、ビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を70℃で5時間撹拌し、原料の消失を1HNMR測定により確認した。 [One-pot synthesis of glycine Schiff base via unprotected imine compound on nitrogen]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon and benzophenone (36.4 mg, 0.20 mmol), monochlorobenzene (0.20 mL, 1.0 M), bis (trimethylsilyl) amine (0.22 mmol, 1.1 eq) were added. . The mixture was stirred at 70 ° C. for 5 hours, and disappearance of raw materials was confirmed by 1 HNMR measurement.
その後、ジクロロメタン(0.80mL,0.25M)およびグリシンt-ブチルエステル塩酸塩(33.5mg,0.20mmol,1.0当量)を加えた。混合物を室温で24時間撹拌し、得られた反応粗生成物についてヘキサン/酢酸エチル=10/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のグリシンシッフ塩基を白色の固体として得た(48.3mg、82%収率(2段階))。
Thereafter, dichloromethane (0.80 mL, 0.25 M) and glycine t-butyl ester hydrochloride (33.5 mg, 0.20 mmol, 1.0 equivalent) were added. The mixture was stirred at room temperature for 24 hours, and the resulting reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / ethyl acetate = 10/1 to obtain the desired glycine Schiff base as a white solid (48 .3 mg, 82% yield (2 steps)).

1H NMR (500 MHz, CDCl3) δ 7.67-7.65 (m, 2H), 7.50-7.42 (m, 3H), 7.41-7.37 (m, 1H), 7.35-7.31 (m, 2H), 7.19-7.18 (m, 2H), 4.12 (s, 2H), 1.46 (s, 9H).
13C NMR (125 MHz, CDCl3) δ 117.53, 169.90, 139.38, 136.18, 130.38, 128.76, 128.76, 128.63, 128.04, 127.74, 81.09, 56.31, 28.12. 1 H NMR (500 MHz, CDCl 3 ) δ 7.67-7.65 (m, 2H), 7.50-7.42 (m, 3H), 7.41-7.37 (m, 1H), 7.35-7.31 (m, 2H), 7.19-7.18 (m, 2H), 4.12 (s, 2H), 1.46 (s, 9H).
13 C NMR (125 MHz, CDCl 3 ) δ 117.53, 169.90, 139.38, 136.18, 130.38, 128.76, 128.76, 128.63, 128.04, 127.74, 81.09, 56.31, 28.12.
13C NMR (125 MHz, CDCl3) δ 117.53, 169.90, 139.38, 136.18, 130.38, 128.76, 128.76, 128.63, 128.04, 127.74, 81.09, 56.31, 28.12. 1 H NMR (500 MHz, CDCl 3 ) δ 7.67-7.65 (m, 2H), 7.50-7.42 (m, 3H), 7.41-7.37 (m, 1H), 7.35-7.31 (m, 2H), 7.19-7.18 (m, 2H), 4.12 (s, 2H), 1.46 (s, 9H).
13 C NMR (125 MHz, CDCl 3 ) δ 117.53, 169.90, 139.38, 136.18, 130.38, 128.76, 128.76, 128.63, 128.04, 127.74, 81.09, 56.31, 28.12.
[窒素上無保護イミン化合物を経由したイサチン付加物のワンポット合成]
4mLのバイアルに撹拌子を入れ、トリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、イサチン(29.4mg,0.20mmol)、モノクロロベンゼン(0.20mL,1.0M)、ビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を室温で12時間撹拌し、原料の消失を1HNMR測定により確認した後、クロロベンゼン(0.60mL,0.25M)およびβ-ケト酸(49.2mg,0.30mmol,1.5当量)を加えた。混合物を室温で9.5時間撹拌し、得られた反応粗生成物についてジクロロメタン/メタノール/トリエチルアミン=100/5/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のイサチン付加物を白色の固体として得た(46.6mg、87%収率(2段階))。 [One-pot synthesis of isatin adducts via unprotected imine compounds on nitrogen]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon and isatin (29.4 mg, 0.20 mmol), monochlorobenzene (0.20 mL, 1.0 M), bis (trimethylsilyl) amine (0.22 mmol, 1.1 eq) were added. . The mixture was stirred at room temperature for 12 hours, and disappearance of the raw materials was confirmed by 1 HNMR measurement, and then chlorobenzene (0.60 mL, 0.25 M) and β-keto acid (49.2 mg, 0.30 mmol, 1.5 eq) Was added. The mixture was stirred at room temperature for 9.5 hours, and the resulting reaction crude product was subjected to silica gel column chromatography with a developing solvent of dichloromethane / methanol / triethylamine = 100/5/1 to obtain the desired isatin adduct as a white solid. (46.6 mg, 87% yield (2 steps)).
4mLのバイアルに撹拌子を入れ、トリフルオロメタンスルホン酸スカンジウム(III)(0.010mmol,5.0mol%)を加えて減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、イサチン(29.4mg,0.20mmol)、モノクロロベンゼン(0.20mL,1.0M)、ビス(トリメチルシリル)アミン(0.22mmol,1.1当量)を加えた。混合物を室温で12時間撹拌し、原料の消失を1HNMR測定により確認した後、クロロベンゼン(0.60mL,0.25M)およびβ-ケト酸(49.2mg,0.30mmol,1.5当量)を加えた。混合物を室温で9.5時間撹拌し、得られた反応粗生成物についてジクロロメタン/メタノール/トリエチルアミン=100/5/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のイサチン付加物を白色の固体として得た(46.6mg、87%収率(2段階))。 [One-pot synthesis of isatin adducts via unprotected imine compounds on nitrogen]
A stir bar was placed in a 4 mL vial, scandium (III) trifluoromethanesulfonate (0.010 mmol, 5.0 mol%) was added, and the mixture was heated and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon and isatin (29.4 mg, 0.20 mmol), monochlorobenzene (0.20 mL, 1.0 M), bis (trimethylsilyl) amine (0.22 mmol, 1.1 eq) were added. . The mixture was stirred at room temperature for 12 hours, and disappearance of the raw materials was confirmed by 1 HNMR measurement, and then chlorobenzene (0.60 mL, 0.25 M) and β-keto acid (49.2 mg, 0.30 mmol, 1.5 eq) Was added. The mixture was stirred at room temperature for 9.5 hours, and the resulting reaction crude product was subjected to silica gel column chromatography with a developing solvent of dichloromethane / methanol / triethylamine = 100/5/1 to obtain the desired isatin adduct as a white solid. (46.6 mg, 87% yield (2 steps)).

1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 1.0 Hz, 2H), 7.56-7.53 (m, 2H), 7.42 (dd, J = 7.5, 8.0 Hz, 2H), 7.34 (d, J = 7.0 Hz, 1H), 7.24 (dd, J = 1.0, 8.0 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 3.89 (d, J = 18 Hz, 1H), 3.62 (d, J = 18 Hz, 1H), 1.90 (br, 2H).
13C NMR (125 MHz, CDCl3) δ 196.30, 181.15, 140.92, 136.18, 133.48, 131.66, 129.24, 128.62, 128.02, 123.81, 122.73, 58.70, 46.79. 1 H NMR (500 MHz, CDCl 3 ) δ 7.88 (d, J = 1.0 Hz, 2H), 7.56-7.53 (m, 2H), 7.42 (dd, J = 7.5, 8.0 Hz, 2H), 7.34 (d, J = 7.0 Hz, 1H), 7.24 (dd, J = 1.0, 8.0 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 3.89 (d, J = 18 Hz, 1H), 3.62 (d, J = 18 Hz, 1H), 1.90 (br, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 196.30, 181.15, 140.92, 136.18, 133.48, 131.66, 129.24, 128.62, 128.02, 123.81, 122.73, 58.70, 46.79.
13C NMR (125 MHz, CDCl3) δ 196.30, 181.15, 140.92, 136.18, 133.48, 131.66, 129.24, 128.62, 128.02, 123.81, 122.73, 58.70, 46.79. 1 H NMR (500 MHz, CDCl 3 ) δ 7.88 (d, J = 1.0 Hz, 2H), 7.56-7.53 (m, 2H), 7.42 (dd, J = 7.5, 8.0 Hz, 2H), 7.34 (d, J = 7.0 Hz, 1H), 7.24 (dd, J = 1.0, 8.0 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 3.89 (d, J = 18 Hz, 1H), 3.62 (d, J = 18 Hz, 1H), 1.90 (br, 2H).
13 C NMR (125 MHz, CDCl 3 ) δ 196.30, 181.15, 140.92, 136.18, 133.48, 131.66, 129.24, 128.62, 128.02, 123.81, 122.73, 58.70, 46.79.
[実施例8-1]
[テトラブチルアンモニウムフルオリド(TBAF)を触媒とする2,2,2-トリフルオロ-1-フェニルエタンイミンの合成(溶媒有)]
4mLのバイアルに撹拌子を入れ、2,2,2-トリフルオロアセトフェノン(1.0mmol)、クロロベンゼン(1.0mL,1.0M)、ビス(トリメチルシリル)アミン(1.1mmol,1.1当量)及びフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.050mL、0.050mmol、5.0mol%)を加えた。混合物を90℃で24時間撹拌し、粗生成物の19FNMR測定を行ったところ、95%以上の反応の進行が確認された。 [Example 8-1]
[Synthesis of 2,2,2-trifluoro-1-phenylethaneimine using tetrabutylammonium fluoride (TBAF) as a catalyst (with solvent)]
A stir bar was placed in a 4 mL vial and 2,2,2-trifluoroacetophenone (1.0 mmol), chlorobenzene (1.0 mL, 1.0 M), bis (trimethylsilyl) amine (1.1 mmol, 1.1 eq) And tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.050 mL, 0.050 mmol, 5.0 mol%) were added. The mixture was stirred at 90 ° C. for 24 hours, and 19 FNMR measurement of the crude product was performed. As a result, the progress of the reaction of 95% or more was confirmed.
[テトラブチルアンモニウムフルオリド(TBAF)を触媒とする2,2,2-トリフルオロ-1-フェニルエタンイミンの合成(溶媒有)]
4mLのバイアルに撹拌子を入れ、2,2,2-トリフルオロアセトフェノン(1.0mmol)、クロロベンゼン(1.0mL,1.0M)、ビス(トリメチルシリル)アミン(1.1mmol,1.1当量)及びフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.050mL、0.050mmol、5.0mol%)を加えた。混合物を90℃で24時間撹拌し、粗生成物の19FNMR測定を行ったところ、95%以上の反応の進行が確認された。 [Example 8-1]
[Synthesis of 2,2,2-trifluoro-1-phenylethaneimine using tetrabutylammonium fluoride (TBAF) as a catalyst (with solvent)]
A stir bar was placed in a 4 mL vial and 2,2,2-trifluoroacetophenone (1.0 mmol), chlorobenzene (1.0 mL, 1.0 M), bis (trimethylsilyl) amine (1.1 mmol, 1.1 eq) And tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.050 mL, 0.050 mmol, 5.0 mol%) were added. The mixture was stirred at 90 ° C. for 24 hours, and 19 FNMR measurement of the crude product was performed. As a result, the progress of the reaction of 95% or more was confirmed.

[実施例8-2]
[TBAFを触媒とする2,2,2-トリフルオロ-1-フェニルエタンイミンの合成(実質的に溶媒無)]
4mLのバイアルに撹拌子を入れ、2,2,2-トリフルオロアセトフェノン(1.0mmol)、ビス(トリメチルシリル)アミン(1.1mmol,1.1当量)およびフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。混合物を70度で12時間撹拌した後、粗生成物の19FNMR測定を行ったところ、約88%の反応の進行が確認された。 [Example 8-2]
[Synthesis of 2,2,2-trifluoro-1-phenylethanimine catalyzed by TBAF (substantially solvent-free)]
A stir bar was placed in a 4 mL vial and 2,2,2-trifluoroacetophenone (1.0 mmol), bis (trimethylsilyl) amine (1.1 mmol, 1.1 eq) and tetrabutylammonium fluoride (TBAF, 1. 0M in THF, 0.10 mL, 0.10 mmol, 10 mol%) was added. After stirring the mixture at 70 ° C. for 12 hours, 19 FNMR measurement of the crude product was performed, and it was confirmed that the reaction progressed by about 88%.
[TBAFを触媒とする2,2,2-トリフルオロ-1-フェニルエタンイミンの合成(実質的に溶媒無)]
4mLのバイアルに撹拌子を入れ、2,2,2-トリフルオロアセトフェノン(1.0mmol)、ビス(トリメチルシリル)アミン(1.1mmol,1.1当量)およびフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。混合物を70度で12時間撹拌した後、粗生成物の19FNMR測定を行ったところ、約88%の反応の進行が確認された。 [Example 8-2]
[Synthesis of 2,2,2-trifluoro-1-phenylethanimine catalyzed by TBAF (substantially solvent-free)]
A stir bar was placed in a 4 mL vial and 2,2,2-trifluoroacetophenone (1.0 mmol), bis (trimethylsilyl) amine (1.1 mmol, 1.1 eq) and tetrabutylammonium fluoride (TBAF, 1. 0M in THF, 0.10 mL, 0.10 mmol, 10 mol%) was added. After stirring the mixture at 70 ° C. for 12 hours, 19 FNMR measurement of the crude product was performed, and it was confirmed that the reaction progressed by about 88%.

[TBAFを触媒とするベンゾフェノンイミンの合成(実質的に溶媒無)]
4mLのバイアルに撹拌子を入れ、ベンゾフェノン(1.0mmol)、ビス(トリメチルシリル)アミン(2.0mmol,2.0当量)、フッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。混合物を室温で2時間撹拌し、原料の消失を1HNMR測定で確認した後、得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のベンゾフェノンイミンを黄色のオイルとして得た(162mg、90%収率)。 [Synthesis of benzophenone imine using TBAF as a catalyst (substantially solvent-free)]
A stir bar was placed in a 4 mL vial, benzophenone (1.0 mmol), bis (trimethylsilyl) amine (2.0 mmol, 2.0 eq), tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 2 hours, and disappearance of the raw materials was confirmed by 1 HNMR measurement. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone. The imine was obtained as a yellow oil (162 mg, 90% yield).
4mLのバイアルに撹拌子を入れ、ベンゾフェノン(1.0mmol)、ビス(トリメチルシリル)アミン(2.0mmol,2.0当量)、フッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。混合物を室温で2時間撹拌し、原料の消失を1HNMR測定で確認した後、得られた反応粗生成物についてヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のベンゾフェノンイミンを黄色のオイルとして得た(162mg、90%収率)。 [Synthesis of benzophenone imine using TBAF as a catalyst (substantially solvent-free)]
A stir bar was placed in a 4 mL vial, benzophenone (1.0 mmol), bis (trimethylsilyl) amine (2.0 mmol, 2.0 eq), tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 2 hours, and disappearance of the raw materials was confirmed by 1 HNMR measurement. The obtained reaction crude product was subjected to silica gel column chromatography with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone. The imine was obtained as a yellow oil (162 mg, 90% yield).

[触媒の検討]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、溶媒としてテトラヒドロフラン(0.1mL)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒(0.10mmol、10mol%)を加えた。触媒としては、テトラブチルアンモニウムフルオリド(TBAF)、テトラブチルアンモニウムジヒドロゲントリフルオリド(TBAH2F3)、TBAFの水和物を用いた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表8に示す。 [Examination of catalyst]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), tetrahydrofuran (0.1 mL) as the solvent, and bis (trimethylsilyl) amine (2. 0 mmol, 2.0 eq) and catalyst (0.10 mmol, 10 mol%) were added. As the catalyst, tetrabutylammonium fluoride (TBAF), tetrabutylammonium dihydrogen trifluoride (TBAH 2 F 3 ), and TBAF hydrate were used. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 8.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、溶媒としてテトラヒドロフラン(0.1mL)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒(0.10mmol、10mol%)を加えた。触媒としては、テトラブチルアンモニウムフルオリド(TBAF)、テトラブチルアンモニウムジヒドロゲントリフルオリド(TBAH2F3)、TBAFの水和物を用いた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表8に示す。 [Examination of catalyst]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), tetrahydrofuran (0.1 mL) as the solvent, and bis (trimethylsilyl) amine (2. 0 mmol, 2.0 eq) and catalyst (0.10 mmol, 10 mol%) were added. As the catalyst, tetrabutylammonium fluoride (TBAF), tetrabutylammonium dihydrogen trifluoride (TBAH 2 F 3 ), and TBAF hydrate were used. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 8.

表8に示すように、テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒を用いることにより、窒素上無保護ケチミンが生成した。これらの中でも、TBAFは収率80%以上を達成することができた。
As shown in Table 8, unprotected ketimine on nitrogen was produced by using a catalyst containing a fluorine-containing anion salt of tetraalkylammonium. Among these, TBAF was able to achieve a yield of 80% or more.
[溶媒の検討(1)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、溶媒(0.1mL)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒としてフッ化テトラブチルアンモニウム水和物(0.10mmol、10mol%)を加えた。溶媒として、テトラヒドロフラン(THF)、N,N-ジメチルホルムアミド(DMF)、モノクロロベンゼン(PhCl)を用いた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表9に示す。 [Study of solvent (1)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), solvent (0.1 mL), bis (trimethylsilyl) amine (2.0 mmol, nitrogen source compound (2)) 2.0 equivalents) and tetrabutylammonium fluoride hydrate (0.10 mmol, 10 mol%) was added as a catalyst. Tetrahydrofuran (THF), N, N-dimethylformamide (DMF), and monochlorobenzene (PhCl) were used as the solvent. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 9.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、溶媒(0.1mL)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒としてフッ化テトラブチルアンモニウム水和物(0.10mmol、10mol%)を加えた。溶媒として、テトラヒドロフラン(THF)、N,N-ジメチルホルムアミド(DMF)、モノクロロベンゼン(PhCl)を用いた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表9に示す。 [Study of solvent (1)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), solvent (0.1 mL), bis (trimethylsilyl) amine (2.0 mmol, nitrogen source compound (2)) 2.0 equivalents) and tetrabutylammonium fluoride hydrate (0.10 mmol, 10 mol%) was added as a catalyst. Tetrahydrofuran (THF), N, N-dimethylformamide (DMF), and monochlorobenzene (PhCl) were used as the solvent. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 9.

表9に示すように、溶媒として、テトラヒドロフラン、N,N-ジメチルホルムアミド、モノクロロベンゼンを用いた場合、反応が良好に進行した。N,N-ジメチルホルムアミドを用いた場合が特に優れていることがわかる。
As shown in Table 9, when tetrahydrofuran, N, N-dimethylformamide, or monochlorobenzene was used as the solvent, the reaction proceeded well. It can be seen that N, N-dimethylformamide is particularly excellent.
[溶媒の検討(2)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒としてフッ化テトラブチルアンモニウム(0.10mmol、10mol%)のTHF溶液、追加溶媒(0.1~2当量)を加えた。追加溶媒として、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)を用いた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表10に示す。 [Study of solvent (2)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), catalyst As a solution, tetrabutylammonium fluoride (0.10 mmol, 10 mol%) in THF and an additional solvent (0.1 to 2 equivalents) were added. Additional solvents include N, N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropyleneurea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI) were used. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 10.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒としてフッ化テトラブチルアンモニウム(0.10mmol、10mol%)のTHF溶液、追加溶媒(0.1~2当量)を加えた。追加溶媒として、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)を用いた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表10に示す。 [Study of solvent (2)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), catalyst As a solution, tetrabutylammonium fluoride (0.10 mmol, 10 mol%) in THF and an additional solvent (0.1 to 2 equivalents) were added. Additional solvents include N, N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropyleneurea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI) were used. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 10.

表10に示すように、N,N-ジメチルホルムアミド(DMF)は、10mol%~200mol%(0.1~2当量)の添加で収率の向上が見られ、100mol%が特に優れていることがわかる。また、N,N-ジメチルホルムアミド(DMF)のほかに、ジメチルスルホキシド(DMSO)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)の添加でも収率の向上が見られた。HMPU、DMPU、DMIが優れており、DMIが特に優れていることがわかる。
As shown in Table 10, the yield of N, N-dimethylformamide (DMF) is improved by the addition of 10 mol% to 200 mol% (0.1 to 2 equivalents), and 100 mol% is particularly excellent. I understand. In addition to N, N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA) , N, N-dimethylpropyleneurea (DMPU), 1,3-dimethyl-2-imidazolidinone (DMI) was also added to improve the yield. It can be seen that HMPU, DMPU, and DMI are excellent, and DMI is particularly excellent.
[溶媒の検討(3)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてベンゾフェノンイミン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒としてフッ化テトラブチルアンモニウム(0.050mmol、5.0mol%)のTHF溶液、追加溶媒(1.0当量)を加えた。追加溶媒として、N,N-ジメチルホルムアミド(DMF)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)を用いた。混合物を室温で2時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表11に示す。 [Study of solvent (3)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone imine (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), and fluoride as the catalyst. Tetrabutylammonium (0.050 mmol, 5.0 mol%) in THF and additional solvent (1.0 eq) were added. Additional solvents include N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropylene Urea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI) were used. After the mixture was stirred at room temperature for 2 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 11.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてベンゾフェノンイミン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol、2.0当量)、触媒としてフッ化テトラブチルアンモニウム(0.050mmol、5.0mol%)のTHF溶液、追加溶媒(1.0当量)を加えた。追加溶媒として、N,N-ジメチルホルムアミド(DMF)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)を用いた。混合物を室温で2時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表11に示す。 [Study of solvent (3)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone imine (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), and fluoride as the catalyst. Tetrabutylammonium (0.050 mmol, 5.0 mol%) in THF and additional solvent (1.0 eq) were added. Additional solvents include N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N-dimethylpropylene Urea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI) were used. After the mixture was stirred at room temperature for 2 hours, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 11.

表11に示すように、N,N-ジメチルホルムアミド(DMF)、N-メチル-2-ピロリドン(NMP)、N,N-ジメチルアセトアミド(DMA)、ヘキサメチルリン酸トリアミド(HMPA)、N,N-ジメチルプロピレン尿素(DMPU)、1,3-ジメチル-2-イミダゾリジノン(DMI)の添加で収率の向上が見られた。特に、DMF、DMPUが優れていることがわかる。
As shown in Table 11, N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), N, N-dimethylacetamide (DMA), hexamethylphosphoric triamide (HMPA), N, N -An improvement in yield was observed by adding dimethylpropyleneurea (DMPU) and 1,3-dimethyl-2-imidazolidinone (DMI). In particular, it can be seen that DMF and DMPU are excellent.
[溶媒の検討(4)]
溶媒の検討(3)の方法において、触媒としてフッ化テトラブチルアンモニウム(0.025mmol、2.5mol%)のTHF溶液を用いると共に、良好な結果が得られたDMFを追加溶媒として用いて、その添加量を変化させた。結果を表12に示す。 [Study of solvent (4)]
In the method of the examination of the solvent (3), while using a THF solution of tetrabutylammonium fluoride (0.025 mmol, 2.5 mol%) as a catalyst and using DMF with good results as an additional solvent, The amount added was varied. The results are shown in Table 12.
溶媒の検討(3)の方法において、触媒としてフッ化テトラブチルアンモニウム(0.025mmol、2.5mol%)のTHF溶液を用いると共に、良好な結果が得られたDMFを追加溶媒として用いて、その添加量を変化させた。結果を表12に示す。 [Study of solvent (4)]
In the method of the examination of the solvent (3), while using a THF solution of tetrabutylammonium fluoride (0.025 mmol, 2.5 mol%) as a catalyst and using DMF with good results as an additional solvent, The amount added was varied. The results are shown in Table 12.

表12に示すように、触媒量を減らしても、比較的多量のDMFを用いることにより十分に反応が進行することがわかる。
なお、DMFを13当量(1.0M)用いると共に、窒素源化合物(2)としてビス(トリメチルシリル)アミン(1.5mmol、1.5当量)用いた系では、TBAFを1.0mol%まで低減させても反応が進行した(79~94%(1HNMR))。 As shown in Table 12, it can be seen that the reaction proceeds sufficiently by using a relatively large amount of DMF even if the amount of the catalyst is reduced.
In the system using 13 equivalents (1.0 M) of DMF and bis (trimethylsilyl) amine (1.5 mmol, 1.5 equivalents) as the nitrogen source compound (2), TBAF was reduced to 1.0 mol%. However, the reaction proceeded (79-94% ( 1 HNMR)).
なお、DMFを13当量(1.0M)用いると共に、窒素源化合物(2)としてビス(トリメチルシリル)アミン(1.5mmol、1.5当量)用いた系では、TBAFを1.0mol%まで低減させても反応が進行した(79~94%(1HNMR))。 As shown in Table 12, it can be seen that the reaction proceeds sufficiently by using a relatively large amount of DMF even if the amount of the catalyst is reduced.
In the system using 13 equivalents (1.0 M) of DMF and bis (trimethylsilyl) amine (1.5 mmol, 1.5 equivalents) as the nitrogen source compound (2), TBAF was reduced to 1.0 mol%. However, the reaction proceeded (79-94% ( 1 HNMR)).
[窒素源化合物量の検討(1)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン、触媒としてフッ化テトラブチルアンモニウム水和物(0.10mmol、10mol%)を加えた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表13に示す。 [Examination of amount of nitrogen source compound (1)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine as the nitrogen source compound (2), and tetrabutylammonium fluoride hydrate ( 0.10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 13.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン、触媒としてフッ化テトラブチルアンモニウム水和物(0.10mmol、10mol%)を加えた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表13に示す。 [Examination of amount of nitrogen source compound (1)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine as the nitrogen source compound (2), and tetrabutylammonium fluoride hydrate ( 0.10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 13.

表13に示すように、窒素源化合物は、3.0~1.1当量で、高収率で反応が進行し、1.5当量用いた場合が特に優れていることがわかる。
As shown in Table 13, it can be seen that the nitrogen source compound is 3.0 to 1.1 equivalents, the reaction proceeds in a high yield, and the use of 1.5 equivalents is particularly excellent.
[窒素源化合物量の検討(2)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてベンゾフェノンイミン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン、触媒としてフッ化テトラブチルアンモニウム(0.025mmol、2.5mol%)を加えた。混合物を室温で1時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表14に示す。 [Examination of amount of nitrogen source compound (2)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone imine (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine as the nitrogen source compound (2), tetrabutylammonium fluoride (0.025 mmol, 2. 5 mol%) was added. After the mixture was stirred at room temperature for 1 hour, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 14.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてベンゾフェノンイミン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン、触媒としてフッ化テトラブチルアンモニウム(0.025mmol、2.5mol%)を加えた。混合物を室温で1時間撹拌した後、生成物の収率を反応粗生成物の1H NMR測定により求めた。結果を表14に示す。 [Examination of amount of nitrogen source compound (2)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, benzophenone imine (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine as the nitrogen source compound (2), tetrabutylammonium fluoride (0.025 mmol, 2. 5 mol%) was added. After the mixture was stirred at room temperature for 1 hour, the yield of the product was determined by 1 H NMR measurement of the reaction crude product. The results are shown in Table 14.

表14に示すように、窒素源化合物は、2.0~1.1当量で、高収率で反応が進行し、1.5当量用いた場合が特に優れていることがわかる。
As shown in Table 14, it can be seen that the nitrogen source compound is 2.0 to 1.1 equivalents, the reaction proceeds in a high yield, and the use of 1.5 equivalents is particularly excellent.
[反応温度の検討]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol)、触媒としてフッ化テトラブチルアンモニウム(0.010mmol、10mol%)を加えた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表15に示す。 [Examination of reaction temperature]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol) as the nitrogen source compound (2), and tetrabutyl fluoride as the catalyst. Ammonium (0.010 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 15.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol)、触媒としてフッ化テトラブチルアンモニウム(0.010mmol、10mol%)を加えた。混合物を室温で24時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表15に示す。 [Examination of reaction temperature]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol) as the nitrogen source compound (2), and tetrabutyl fluoride as the catalyst. Ammonium (0.010 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 24 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 15.

表15に示すように、0℃の低温でも反応が進行した。また、室温(25℃)の場合、反応が特に良好に進行した。
As shown in Table 15, the reaction proceeded even at a low temperature of 0 ° C. In addition, the reaction proceeded particularly well at room temperature (25 ° C.).
[TBAFを触媒とする様々なアルキルケチミンの合成]
(基本反応条件)
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(1.5mmol,1.5当量)、溶媒として1,3-ジメチル-2-イミダゾリジノン(1.0mmol、1.0当量)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。混合物を室温で6時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。以上の反応条件を基本反応条件という。 [Synthesis of various alkyl ketimines catalyzed by TBAF]
(Basic reaction conditions)
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, the ketone compound (1) (1.0 mmol), bis (trimethylsilyl) amine (1.5 mmol, 1.5 equivalents) as the nitrogen source compound (2), and 1,3-dimethyl as the solvent -2-Imidazolidinone (1.0 mmol, 1.0 equivalent) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) were added as a catalyst. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The above reaction conditions are called basic reaction conditions.
(基本反応条件)
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(1.5mmol,1.5当量)、溶媒として1,3-ジメチル-2-イミダゾリジノン(1.0mmol、1.0当量)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。混合物を室温で6時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。以上の反応条件を基本反応条件という。 [Synthesis of various alkyl ketimines catalyzed by TBAF]
(Basic reaction conditions)
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, the ketone compound (1) (1.0 mmol), bis (trimethylsilyl) amine (1.5 mmol, 1.5 equivalents) as the nitrogen source compound (2), and 1,3-dimethyl as the solvent -2-Imidazolidinone (1.0 mmol, 1.0 equivalent) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) were added as a catalyst. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The above reaction conditions are called basic reaction conditions.

[実施例14-1]
(シクロヘキシル(フェニル)メタンイミンの合成)
上記基本反応条件で1.0mmolのシクロヘキシル(フェニル)ケトンを用いて6時間撹拌を行った。1HNMR測定より目的のシクロヘキシル(フェニル)メタンイミンが>99%の収率で生成していることを確認した。 [Example 14-1]
(Synthesis of cyclohexyl (phenyl) methanimine)
Stirring was performed for 6 hours using 1.0 mmol of cyclohexyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclohexyl (phenyl) methanimine was produced in a yield of> 99%.
(シクロヘキシル(フェニル)メタンイミンの合成)
上記基本反応条件で1.0mmolのシクロヘキシル(フェニル)ケトンを用いて6時間撹拌を行った。1HNMR測定より目的のシクロヘキシル(フェニル)メタンイミンが>99%の収率で生成していることを確認した。 [Example 14-1]
(Synthesis of cyclohexyl (phenyl) methanimine)
Stirring was performed for 6 hours using 1.0 mmol of cyclohexyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclohexyl (phenyl) methanimine was produced in a yield of> 99%.

[実施例14-2]
(シクロブチル(フェニル)メタンイミンの合成)
上記基本反応条件で1.0mmolのシクロブチル(フェニル)ケトンを用いて6時間撹拌を行った。1HNMR測定より目的のシクロブチル(フェニル)メタンイミンが>99%の収率で生成していることを確認した。 [Example 14-2]
(Synthesis of cyclobutyl (phenyl) methanimine)
Stirring was performed for 6 hours using 1.0 mmol of cyclobutyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclobutyl (phenyl) methanimine was produced in a yield of> 99%.
(シクロブチル(フェニル)メタンイミンの合成)
上記基本反応条件で1.0mmolのシクロブチル(フェニル)ケトンを用いて6時間撹拌を行った。1HNMR測定より目的のシクロブチル(フェニル)メタンイミンが>99%の収率で生成していることを確認した。 [Example 14-2]
(Synthesis of cyclobutyl (phenyl) methanimine)
Stirring was performed for 6 hours using 1.0 mmol of cyclobutyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclobutyl (phenyl) methanimine was produced in a yield of> 99%.

[実施例14-3]
(シクロプロピル(フェニル)メタンイミンの合成)
上記基本反応条件で1.0mmolのシクロプロピル(フェニル)ケトンを用いて6時間撹拌を行った。1HNMR測定より目的のシクロヘキシル(フェニル)メタンイミンが>99%の収率で生成していることを確認した。 [Example 14-3]
(Synthesis of cyclopropyl (phenyl) methanimine)
Stirring was carried out for 6 hours using 1.0 mmol of cyclopropyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclohexyl (phenyl) methanimine was produced in a yield of> 99%.
(シクロプロピル(フェニル)メタンイミンの合成)
上記基本反応条件で1.0mmolのシクロプロピル(フェニル)ケトンを用いて6時間撹拌を行った。1HNMR測定より目的のシクロヘキシル(フェニル)メタンイミンが>99%の収率で生成していることを確認した。 [Example 14-3]
(Synthesis of cyclopropyl (phenyl) methanimine)
Stirring was carried out for 6 hours using 1.0 mmol of cyclopropyl (phenyl) ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired cyclohexyl (phenyl) methanimine was produced in a yield of> 99%.

[実施例14-4]
(2-メチル-1-フェニルプロパン-1-イミンの合成)
上記基本反応条件で1.0mmolのイソブチロフェノンを用いて6時間撹拌を行った。1HNMR測定より目的の2-メチル-1-フェニルプロパン-1-イミンが89%の収率で生成していることを確認した。 [Example 14-4]
(Synthesis of 2-methyl-1-phenylpropane-1-imine)
Stirring was carried out for 6 hours using 1.0 mmol of isobutyrophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired 2-methyl-1-phenylpropane-1-imine was produced in a yield of 89%.
(2-メチル-1-フェニルプロパン-1-イミンの合成)
上記基本反応条件で1.0mmolのイソブチロフェノンを用いて6時間撹拌を行った。1HNMR測定より目的の2-メチル-1-フェニルプロパン-1-イミンが89%の収率で生成していることを確認した。 [Example 14-4]
(Synthesis of 2-methyl-1-phenylpropane-1-imine)
Stirring was carried out for 6 hours using 1.0 mmol of isobutyrophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the desired 2-methyl-1-phenylpropane-1-imine was produced in a yield of 89%.

[実施例14-5]
(1-フェニルプロパン-1-イミンの合成)
上記基本反応条件で1.0mmolのプロピオフェノンを用いて24時間撹拌を行った。1HNMR測定より目的の1-フェニルプロパン-1-イミンが87%の収率で生成しており、未反応のプロピオフェノンが1%残っていることを確認した。 [Example 14-5]
(Synthesis of 1-phenylpropane-1-imine)
Stirring was performed for 24 hours using 1.0 mmol of propiophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylpropane-1-imine was produced in a yield of 87%, and 1% of unreacted propiophenone remained.
(1-フェニルプロパン-1-イミンの合成)
上記基本反応条件で1.0mmolのプロピオフェノンを用いて24時間撹拌を行った。1HNMR測定より目的の1-フェニルプロパン-1-イミンが87%の収率で生成しており、未反応のプロピオフェノンが1%残っていることを確認した。 [Example 14-5]
(Synthesis of 1-phenylpropane-1-imine)
Stirring was performed for 24 hours using 1.0 mmol of propiophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylpropane-1-imine was produced in a yield of 87%, and 1% of unreacted propiophenone remained.

[実施例14-6]
(1-フェニルブタン-1-イミンの合成)
上記基本反応条件で1.0mmolのブチロフェノンを用いて24時間撹拌を行った。1HNMR測定より目的の1-フェニルブタン-1-イミンが90%の収率で生成しており、未反応のブチロフェノンが6%残っていることを確認した。 [Example 14-6]
(Synthesis of 1-phenylbutane-1-imine)
Stirring was carried out for 24 hours using 1.0 mmol of butyrophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylbutane-1-imine was produced in a yield of 90%, and 6% of unreacted butyrophenone remained.
(1-フェニルブタン-1-イミンの合成)
上記基本反応条件で1.0mmolのブチロフェノンを用いて24時間撹拌を行った。1HNMR測定より目的の1-フェニルブタン-1-イミンが90%の収率で生成しており、未反応のブチロフェノンが6%残っていることを確認した。 [Example 14-6]
(Synthesis of 1-phenylbutane-1-imine)
Stirring was carried out for 24 hours using 1.0 mmol of butyrophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylbutane-1-imine was produced in a yield of 90%, and 6% of unreacted butyrophenone remained.

[実施例14-7]
(1-フェニルペンタン-1-イミンの合成)
上記基本反応条件で1.0mmolのバレロフェノンを用いて24時間撹拌を行った。1HNMR測定より目的の1-フェニルペンタン-1-イミンが81%の収率で生成しており、未反応のバレロフェノンが10%残っていることを確認した。 [Example 14-7]
(Synthesis of 1-phenylpentane-1-imine)
Stirring was performed for 24 hours using 1.0 mmol of valerophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylpentane-1-imine was produced in a yield of 81%, and 10% of unreacted valerophenone remained.
(1-フェニルペンタン-1-イミンの合成)
上記基本反応条件で1.0mmolのバレロフェノンを用いて24時間撹拌を行った。1HNMR測定より目的の1-フェニルペンタン-1-イミンが81%の収率で生成しており、未反応のバレロフェノンが10%残っていることを確認した。 [Example 14-7]
(Synthesis of 1-phenylpentane-1-imine)
Stirring was performed for 24 hours using 1.0 mmol of valerophenone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target 1-phenylpentane-1-imine was produced in a yield of 81%, and 10% of unreacted valerophenone remained.

[実施例14-8]
(ジシクロプロピルメタンイミンの合成)
上記基本反応条件で1.0mmolのジシクロプロピルケトンを用いて24時間撹拌を行った。1HNMR測定より目的のジシクロプロピルメタンイミンが60%の収率で生成しており、未反応のジシクロプロピルケトンが35%残っていることを確認した。 [Example 14-8]
(Synthesis of dicyclopropylmethanimine)
Stirring was carried out for 24 hours using 1.0 mmol of dicyclopropyl ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target dicyclopropylmethanimine was produced in a yield of 60%, and 35% of unreacted dicyclopropylketone remained.
(ジシクロプロピルメタンイミンの合成)
上記基本反応条件で1.0mmolのジシクロプロピルケトンを用いて24時間撹拌を行った。1HNMR測定より目的のジシクロプロピルメタンイミンが60%の収率で生成しており、未反応のジシクロプロピルケトンが35%残っていることを確認した。 [Example 14-8]
(Synthesis of dicyclopropylmethanimine)
Stirring was carried out for 24 hours using 1.0 mmol of dicyclopropyl ketone under the above basic reaction conditions. From 1 HNMR measurement, it was confirmed that the target dicyclopropylmethanimine was produced in a yield of 60%, and 35% of unreacted dicyclopropylketone remained.

[TBAFを触媒とするベンゾフェノンイミンの合成(溶媒有)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(1.5mmol,1.5当量)、追加溶媒としてN,N-ジメチルホルムアミド(1.0mL、1.0M)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.025mL、0.025mmol、2.5mol%)を加えた。混合物を室温で1時間撹拌し、原料の消失を1HNMR測定で確認した。反応溶液を分液ロートに移し、H2O(10mL)を加えた。ジエチルエーテルで抽出後、有機層を減圧下で濃縮し、硫酸ナトリウムで乾燥した。溶媒留去後、ヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のベンゾフェノンイミンを黄色のオイルとして得た(173mg、96%収率)。 [Synthesis of benzophenone imine using TBAF as catalyst (with solvent)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, ketone compound (1) (1.0 mmol), bis (trimethylsilyl) amine (1.5 mmol, 1.5 equivalents) as nitrogen source compound (2), N, N— Dimethylformamide (1.0 mL, 1.0 M) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.025 mL, 0.025 mmol, 2.5 mol%) were added as a catalyst. The mixture was stirred at room temperature for 1 hour, and disappearance of raw materials was confirmed by 1 HNMR measurement. The reaction solution was transferred to a separatory funnel and H 2 O (10 mL) was added. After extraction with diethyl ether, the organic layer was concentrated under reduced pressure and dried over sodium sulfate. After the solvent was distilled off, silica gel column chromatography was performed with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone imine as a yellow oil (173 mg, 96% yield).
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(1.5mmol,1.5当量)、追加溶媒としてN,N-ジメチルホルムアミド(1.0mL、1.0M)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.025mL、0.025mmol、2.5mol%)を加えた。混合物を室温で1時間撹拌し、原料の消失を1HNMR測定で確認した。反応溶液を分液ロートに移し、H2O(10mL)を加えた。ジエチルエーテルで抽出後、有機層を減圧下で濃縮し、硫酸ナトリウムで乾燥した。溶媒留去後、ヘキサン/トリエチルアミン=9/1の展開溶媒でシリカゲルカラムクロマトグラフィーを行い、目的のベンゾフェノンイミンを黄色のオイルとして得た(173mg、96%収率)。 [Synthesis of benzophenone imine using TBAF as catalyst (with solvent)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, ketone compound (1) (1.0 mmol), bis (trimethylsilyl) amine (1.5 mmol, 1.5 equivalents) as nitrogen source compound (2), N, N— Dimethylformamide (1.0 mL, 1.0 M) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.025 mL, 0.025 mmol, 2.5 mol%) were added as a catalyst. The mixture was stirred at room temperature for 1 hour, and disappearance of raw materials was confirmed by 1 HNMR measurement. The reaction solution was transferred to a separatory funnel and H 2 O (10 mL) was added. After extraction with diethyl ether, the organic layer was concentrated under reduced pressure and dried over sodium sulfate. After the solvent was distilled off, silica gel column chromatography was performed with a developing solvent of hexane / triethylamine = 9/1 to obtain the desired benzophenone imine as a yellow oil (173 mg, 96% yield).

1H NMR (500 MHz, CDCl3) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.49-7.46 (m, 2H), 7.44-7.40 (m, 4H).
13C NMR (125 MHz, CDCl3) δ 178.36, 139.35, 130.30, 128.34, 128.34. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.49-7.46 (m, 2H), 7.44-7.40 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.36, 139.35, 130.30, 128.34, 128.34.
13C NMR (125 MHz, CDCl3) δ 178.36, 139.35, 130.30, 128.34, 128.34. 1 H NMR (500 MHz, CDCl 3 ) δ 9.72 (br, 1H), 7.58 (br, 4H), 7.49-7.46 (m, 2H), 7.44-7.40 (m, 4H).
13 C NMR (125 MHz, CDCl 3 ) δ 178.36, 139.35, 130.30, 128.34, 128.34.
[窒素上無保護イミン化合物を経由したアミノニトリルのワンポット合成]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、シクロヘキシル(フェニル)ケトン(1.0mmol)、ビス(トリメチルシリル)アミン(1.5mmol,1.5当量)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加え、混合物を室温で24時間撹拌した。 [One-pot synthesis of aminonitrile via unprotected imine compound on nitrogen]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol), bis (trimethylsilyl) amine (1.5 mmol, 1.5 eq), and tetrabutylammonium fluoride (TBAF, 1.0 M in) as the catalyst. THF, 0.10 mL, 0.10 mmol, 10 mol%) was added and the mixture was stirred at room temperature for 24 hours.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、シクロヘキシル(フェニル)ケトン(1.0mmol)、ビス(トリメチルシリル)アミン(1.5mmol,1.5当量)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加え、混合物を室温で24時間撹拌した。 [One-pot synthesis of aminonitrile via unprotected imine compound on nitrogen]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol), bis (trimethylsilyl) amine (1.5 mmol, 1.5 eq), and tetrabutylammonium fluoride (TBAF, 1.0 M in) as the catalyst. THF, 0.10 mL, 0.10 mmol, 10 mol%) was added and the mixture was stirred at room temperature for 24 hours.
その後、アセトンシアノヒドリン(3.0,mmol、3.0当量)を加えて室温で24時間撹拌した。ケチミン中間体の消失を1HNMR測定で確認したのち、反応粗生成物についてヘキサン/酢酸エチル=6/1の展開溶媒でシリカカラムクロマトグラフィーを行い、目的のアミノニトリルを白色固体として得た(190mg、89%収率(2段階))。
Thereafter, acetone cyanohydrin (3.0, mmol, 3.0 equivalents) was added and stirred at room temperature for 24 hours. After confirming the disappearance of the ketimine intermediate by 1 HNMR measurement, the reaction crude product was subjected to silica column chromatography with a developing solvent of hexane / ethyl acetate = 6/1 to obtain the desired aminonitrile as a white solid (190 mg). , 89% yield (2 steps)).

1H NMR (500 MHz, CDCl3) δ 7.61-7.59 (m, 2H), 7.41-7.33 (m, 3H), 2.08 (br, 2H), 1.93-1.83 (m, 2H), 1.73-1.69 (m, 2H), 1.66-1.64 (m, 1H), 1.49-1.47 (m, 1H), 1.29-1.25 (m, 2H), 1.15-1.05 (m, 3H).
13C NMR (125 MHz, CDCl3) δ 139.40, 128.51, 128.45 (2C), 126.25 (2C), 122.83, 62.33, 48.82, 27.82, 27.37, 25.97 (2C), 25.80. 1 H NMR (500 MHz, CDCl 3 ) δ 7.61-7.59 (m, 2H), 7.41-7.33 (m, 3H), 2.08 (br, 2H), 1.93-1.83 (m, 2H), 1.73-1.69 (m , 2H), 1.66-1.64 (m, 1H), 1.49-1.47 (m, 1H), 1.29-1.25 (m, 2H), 1.15-1.05 (m, 3H).
13 C NMR (125 MHz, CDCl 3 ) δ 139.40, 128.51, 128.45 (2C), 126.25 (2C), 122.83, 62.33, 48.82, 27.82, 27.37, 25.97 (2C), 25.80.
13C NMR (125 MHz, CDCl3) δ 139.40, 128.51, 128.45 (2C), 126.25 (2C), 122.83, 62.33, 48.82, 27.82, 27.37, 25.97 (2C), 25.80. 1 H NMR (500 MHz, CDCl 3 ) δ 7.61-7.59 (m, 2H), 7.41-7.33 (m, 3H), 2.08 (br, 2H), 1.93-1.83 (m, 2H), 1.73-1.69 (m , 2H), 1.66-1.64 (m, 1H), 1.49-1.47 (m, 1H), 1.29-1.25 (m, 2H), 1.15-1.05 (m, 3H).
13 C NMR (125 MHz, CDCl 3 ) δ 139.40, 128.51, 128.45 (2C), 126.25 (2C), 122.83, 62.33, 48.82, 27.82, 27.37, 25.97 (2C), 25.80.
なお、シアノ源は、1.5当量まで低減しても問題なく反応が進行した。また、シアノ源としてTMSCNを用いた場合も同様に反応が進行した。
The reaction proceeded without any problem even when the cyano source was reduced to 1.5 equivalents. The reaction proceeded similarly when TMSCN was used as the cyano source.
[反応促進剤の検討(1)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol,2.0当量)、反応促進剤(1.0mmol、1.0当量)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。反応促進剤として、トリメチルシラノール(TMSOH)、メタノール(MeOH)、エタノール(EtOH)を用いた。混合物を室温で6時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表17に示す。 [Examination of reaction accelerator (1)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), reaction Accelerator (1.0 mmol, 1.0 eq) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) as catalyst were added. As a reaction accelerator, trimethylsilanol (TMSOH), methanol (MeOH), and ethanol (EtOH) were used. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 17.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol,2.0当量)、反応促進剤(1.0mmol、1.0当量)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)を加えた。反応促進剤として、トリメチルシラノール(TMSOH)、メタノール(MeOH)、エタノール(EtOH)を用いた。混合物を室温で6時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表17に示す。 [Examination of reaction accelerator (1)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), reaction Accelerator (1.0 mmol, 1.0 eq) and tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) as catalyst were added. As a reaction accelerator, trimethylsilanol (TMSOH), methanol (MeOH), and ethanol (EtOH) were used. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 17.

表17に示すように、トリメチルシラノール、メタノール、エタノールを添加することにより、反応が促進(収率が向上)しており、特にエタノールが優れていることがわかる。エタノールの量は、10~50mol%で収率が向上し、特に10mol%が優れていることがわかる。また、窒素源の量を1.5当量とすることで収率が改善することがわかる。さらに、反応時間を24時間まで延長することでほぼ定量的に反応が進行することがわかる。24時間まで延長するとトリメチルシラノールがより良い結果を示すことがわかる。
As shown in Table 17, the reaction is accelerated (the yield is improved) by adding trimethylsilanol, methanol, and ethanol, and it can be seen that ethanol is particularly excellent. It can be seen that the yield is improved when the amount of ethanol is 10 to 50 mol%, and particularly 10 mol% is excellent. Moreover, it turns out that a yield improves by making the quantity of a nitrogen source into 1.5 equivalent. Further, it can be seen that the reaction proceeds almost quantitatively by extending the reaction time to 24 hours. It can be seen that trimethylsilanol gives better results when extended to 24 hours.
[反応促進剤の検討(2)]
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol,2.0当量)、溶媒としてN,N-ジメチルホルムアミド(1.0mmol)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)、反応促進剤としてH2O(0.10mmol、10mol%)を加えた。混合物を室温で6時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表18に示す。 [Examination of reaction accelerator (2)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), solvent As N, N-dimethylformamide (1.0 mmol), tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) as a catalyst, and H 2 O (0 .10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 18.
4mLのバイアルに撹拌子を入れ、減圧下ヒートガンで加熱乾燥した。冷却後、バイアルをアルゴンで満たし、ケトン化合物(1)としてシクロヘキシル(フェニル)ケトン(1.0mmol)、窒素源化合物(2)としてビス(トリメチルシリル)アミン(2.0mmol,2.0当量)、溶媒としてN,N-ジメチルホルムアミド(1.0mmol)、触媒としてフッ化テトラブチルアンモニウム(TBAF、1.0M in THF、0.10mL、0.10mmol、10mol%)、反応促進剤としてH2O(0.10mmol、10mol%)を加えた。混合物を室温で6時間撹拌し、内部標準物質として1,2,4,5-テトラメチルベンゼンを用いた粗生成物の1HNMR測定にて収率を決定した。結果を表18に示す。 [Examination of reaction accelerator (2)]
A stir bar was placed in a 4 mL vial and dried with a heat gun under reduced pressure. After cooling, the vial was filled with argon, cyclohexyl (phenyl) ketone (1.0 mmol) as the ketone compound (1), bis (trimethylsilyl) amine (2.0 mmol, 2.0 equivalents) as the nitrogen source compound (2), solvent As N, N-dimethylformamide (1.0 mmol), tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.10 mL, 0.10 mmol, 10 mol%) as a catalyst, and H 2 O (0 .10 mmol, 10 mol%) was added. The mixture was stirred at room temperature for 6 hours, and the yield was determined by 1 HNMR measurement of a crude product using 1,2,4,5-tetramethylbenzene as an internal standard substance. The results are shown in Table 18.

表18に示すように、10mol%の水を加えることにより、反応が促進する(収率が向上する)ことがわかる。
As shown in Table 18, it can be seen that the reaction is promoted (yield is improved) by adding 10 mol% of water.
本発明は、窒素上無保護イミン化合物を製造できることから、産業上有用である。
The present invention is industrially useful because an unprotected imine compound on nitrogen can be produced.
Claims (11)
- ルイス酸性を有する金属塩を含む触媒、及びテトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒から選ばれる少なくとも1種の触媒の存在下、
下記式(1)

下記式(2)


下記式(3)

Following formula (1)
Following formula (2)
Following formula (3)
- ルイス酸性を有する金属塩が、希土類金属,Fe,In,Sn及びBiから選ばれる少なくとも1種の金属のトリフラート塩、ノナフラート塩若しくはトリフルオロメタンスルホニルイミド塩、又はSc(NO3)3若しくはBiBr3であることを特徴とする請求項1記載の窒素上無保護イミン化合物の製造方法。 The metal salt having Lewis acidity is a triflate salt, nonaflate salt or trifluoromethanesulfonylimide salt of at least one metal selected from rare earth metals, Fe, In, Sn and Bi, or Sc (NO 3 ) 3 or BiBr 3 . The method for producing an unprotected imine compound on nitrogen according to claim 1.
- 希土類金属が、Sc,Y,Sm,Eu,Gd,Er又はYbであることを特徴とする請求項2記載の窒素上無保護イミン化合物の製造方法。 The method for producing an unprotected imine compound on nitrogen according to claim 2, wherein the rare earth metal is Sc, Y, Sm, Eu, Gd, Er, or Yb.
- ルイス酸性を有する金属塩が、Sc(OTf)3,Y(OTf)3,Sm(OTf)3,Eu(OTf)3,Gd(OTf)3,Er(OTf)3,Yb(OTf)3,Fe(OTf)3,In(OTf)3,Sn(OTf)2、Bi(OTf)3,Sc(ONf)3,及びSc(NTf2)3から選ばれる少なくとも1種であることを特徴とする請求項1~3のいずれか記載の窒素上無保護イミン化合物の製造方法。 Metal salts having Lewis acidity are Sc (OTf) 3 , Y (OTf) 3 , Sm (OTf) 3 , Eu (OTf) 3 , Gd (OTf) 3 , Er (OTf) 3 , Yb (OTf) 3 , It is at least one selected from Fe (OTf) 3 , In (OTf) 3 , Sn (OTf) 2 , Bi (OTf) 3 , Sc (ONf) 3 , and Sc (NTf 2 ) 3 The method for producing an unprotected imine compound on nitrogen according to any one of claims 1 to 3.
- テトラアルキルアンモニウムのフッ素含有アニオン塩のアルキル部分の炭素数が、1~6であることを特徴とする請求項1記載の窒素上無保護イミン化合物の製造方法。 The method for producing an unprotected imine compound on nitrogen according to claim 1, wherein the alkyl part of the fluorine-containing anion salt of tetraalkylammonium has 1 to 6 carbon atoms.
- テトラアルキルアンモニウムのフッ素含有アニオン塩が、テトラブチルアンモニウムフルオリド、又はテトラブチルアンモニウムジヒドロゲントリフルオリドであることを特徴とする請求項1又は5記載の窒素上無保護イミン化合物の製造方法。 The method for producing an unprotected imine compound on nitrogen according to claim 1 or 5, wherein the fluorine-containing anion salt of tetraalkylammonium is tetrabutylammonium fluoride or tetrabutylammonium dihydrogen trifluoride.
- R1及び/又はR2が、芳香環及び/又は脂肪族基を含むことを特徴とする請求項1~6のいずれか記載の窒素上無保護イミン化合物の製造方法。 The method for producing an unprotected imine compound on nitrogen according to any one of claims 1 to 6, wherein R 1 and / or R 2 contains an aromatic ring and / or an aliphatic group.
- ルイス酸性を有する金属塩を含む触媒の存在下、クロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン、フルオロベンゼン、ジクロロエタン及びアセトニトリルから選ばれる少なくとも1種を含む溶媒中で、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする請求項1~7のいずれか記載の窒素上無保護イミン化合物の製造方法。 In the presence of a catalyst containing a metal salt having Lewis acidity, a ketone compound (1) and a nitrogen source compound (2) in a solvent containing at least one selected from chlorobenzene, toluene, tetrahydrofuran, dioxane, fluorobenzene, dichloroethane and acetonitrile The method for producing an unprotected imine compound on nitrogen according to any one of claims 1 to 7, wherein
- テトラアルキルアンモニウムのフッ素含有アニオン塩を含む触媒の存在下、クロロベンゼン、トルエン、テトラヒドロフラン、ジオキサン、フルオロベンゼン、ジクロロエタン、N,N-ジメチルホルムアミド、ジメチルスルホキシド、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、N,N-ジメチルプロピレン尿素、及び1,3-ジメチル-2-イミダゾリジノンから選ばれる少なくとも1種を含む溶媒中で、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする請求項1~7のいずれか記載の窒素上無保護イミン化合物の製造方法。 In the presence of a catalyst containing a fluorine-containing anion salt of tetraalkylammonium, chlorobenzene, toluene, tetrahydrofuran, dioxane, fluorobenzene, dichloroethane, N, N-dimethylformamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, N, N- A ketone compound (1) and a nitrogen source compound in a solvent containing at least one selected from dimethylacetamide, hexamethylphosphoric triamide, N, N-dimethylpropyleneurea, and 1,3-dimethyl-2-imidazolidinone The process for producing an unprotected imine compound on nitrogen according to any one of claims 1 to 7, wherein the reaction (2) is carried out.
- 実質的に溶媒を用いることなく、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする請求項1~7のいずれか記載の窒素上無保護イミン化合物の製造方法。 The method for producing an unprotected imine compound on nitrogen according to any one of claims 1 to 7, wherein the reaction of the ketone compound (1) and the nitrogen source compound (2) is carried out substantially without using a solvent.
- 水、アルコール及びシラノールから選ばれる少なくとも1種の反応促進剤の存在下、ケトン化合物(1)と窒素源化合物(2)の反応を行うことを特徴とする請求項1~10のいずれか記載の窒素上無保護イミン化合物の製造方法。 11. The reaction of the ketone compound (1) and the nitrogen source compound (2) in the presence of at least one reaction accelerator selected from water, alcohol and silanol. A method for producing an unprotected imine compound on nitrogen.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018082703A JP2021155333A (en) | 2018-04-24 | 2018-04-24 | Method for synthesizing n-unprotected imine compound |
| JP2018-082703 | 2018-04-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019208043A1 true WO2019208043A1 (en) | 2019-10-31 |
Family
ID=68294538
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/012006 WO2019208043A1 (en) | 2018-04-24 | 2019-03-22 | Method for producing n-unprotected imine compound |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2021155333A (en) |
| WO (1) | WO2019208043A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113372238A (en) * | 2021-06-30 | 2021-09-10 | 上海科技大学 | Imine compound and its synthesis method and use |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060052642A1 (en) * | 2004-09-09 | 2006-03-09 | O'shea Paul | Synthesis of alpha fluoroalkyl amines |
-
2018
- 2018-04-24 JP JP2018082703A patent/JP2021155333A/en active Pending
-
2019
- 2019-03-22 WO PCT/JP2019/012006 patent/WO2019208043A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060052642A1 (en) * | 2004-09-09 | 2006-03-09 | O'shea Paul | Synthesis of alpha fluoroalkyl amines |
Non-Patent Citations (2)
| Title |
|---|
| MORI, KEIJI ET AL.: "Synthesis of 3-Aryl-1- trifluoromethyltetrahydroisoquinolines by bronsted acid-catalyzed C(sp3)-H bond functionalization", ADVANCED SYNTHESIS & CATALYSIS, vol. 357, no. 5, 29 December 2014 (2014-12-29) - 2015, pages 901 - 906, XP055647458, ISSN: 1615-4150 * |
| SAKACH, V. ET AL.: "Access to unprotected beta- fluoroalkyl beta-amino acids and their alpha-hydroxy derivatives", ORGANIC LETTERS, vol. 21, no. 7, 15 March 2019 (2019-03-15), pages 2340 - 2345, XP055647491, ISSN: 1523-7052 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113372238A (en) * | 2021-06-30 | 2021-09-10 | 上海科技大学 | Imine compound and its synthesis method and use |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2021155333A (en) | 2021-10-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3781464B2 (en) | Process for producing arylpyrrole insecticides via oxazole intermediates | |
| CN113072436A (en) | Preparation method of benzyl aryl ether | |
| WO2019208043A1 (en) | Method for producing n-unprotected imine compound | |
| CA3058686C (en) | Method for preparing 2-aryl malonamide and applications thereof | |
| JP2005526049A (en) | Preparation of benzisoxazole methanesulfonyl chloride and its method of amidation to form zonisamide | |
| WO2021209269A1 (en) | A process for preparation of substituted enamine compounds | |
| JP2017149686A (en) | Production method of ketoxime compound | |
| RU2224740C2 (en) | Method for preparing intermediate compounds | |
| JP2771994B2 (en) | Method for producing propenoic acid derivative | |
| TWI651306B (en) | PROCESS FOR PREPARING 3,5-BIS(HALOALKYL)PYRAZOLE DERIVATIVES FROM α,α-DIHALOAMINES AND KETIMINES | |
| JPH02289563A (en) | Improved process for producing ortho-carboxypyridyl- and ortho-carboxyquinolylimidazolinones | |
| JP3850471B2 (en) | Preparation of 4-aryl-2-perfluoroalkyl-3-oxazolin-5-one from aryl glycine | |
| JP4538993B2 (en) | Process for producing β-ketonitrile derivatives | |
| JPH09104667A (en) | Production of o-nitrobenzonitrile | |
| JP2527961B2 (en) | Benzoic acid ester derivative and method for producing the same | |
| JP4663105B2 (en) | Method for producing 2-sulfonyl-4-oxypyridine derivative | |
| JP3646223B2 (en) | Method for producing aromatic compound by electrophilic reaction and aromatic compound | |
| EP0881207A1 (en) | 2,3-dihalogeno-6-trifluoromethylbenzene derivatives and processes for the preparation thereof | |
| JP4234967B2 (en) | Novel difluorobenzene derivatives and methods for producing them | |
| JP2003171359A (en) | Method for producing 2-nitrophenylacetonitrile derivative, and its synthetic intermediate | |
| JP4194984B2 (en) | Phenylnaphthylimidazole compound | |
| JP4013772B2 (en) | 2-Hydroxyimino-3-oxopropionitrile and process for producing the same | |
| JP4828740B2 (en) | Process for producing 1,2,5-thiadiazoylmethanone derivative and dioxime derivative | |
| JP4116103B2 (en) | Method for producing pentamethine compound and quaternary salt compound | |
| BR112020019277B1 (en) | PROCESS FOR PRODUCING 2,6-DIALKYLPHENYL ACETIC ACIDS, AND THEIR INTERMEDIATES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19793869 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19793869 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |