WO2019089884A2 - Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy - Google Patents
Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy Download PDFInfo
- Publication number
- WO2019089884A2 WO2019089884A2 PCT/US2018/058635 US2018058635W WO2019089884A2 WO 2019089884 A2 WO2019089884 A2 WO 2019089884A2 US 2018058635 W US2018058635 W US 2018058635W WO 2019089884 A2 WO2019089884 A2 WO 2019089884A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- tgfbr2
- seq
- engineered
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 128
- 238000000034 method Methods 0.000 title claims abstract description 116
- 108010082684 Transforming Growth Factor-beta Type II Receptor Proteins 0.000 title claims abstract description 114
- 210000001744 T-lymphocyte Anatomy 0.000 title claims description 205
- 102000004060 Transforming Growth Factor-beta Type II Receptor Human genes 0.000 title abstract 2
- 238000009169 immunotherapy Methods 0.000 title description 5
- 108091033409 CRISPR Proteins 0.000 claims abstract description 126
- 238000010362 genome editing Methods 0.000 claims abstract description 121
- 230000008685 targeting Effects 0.000 claims abstract description 103
- 210000004027 cell Anatomy 0.000 claims description 447
- 108020005004 Guide RNA Proteins 0.000 claims description 206
- 101710163270 Nuclease Proteins 0.000 claims description 172
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 146
- 108091008874 T cell receptors Proteins 0.000 claims description 144
- 125000003729 nucleotide group Chemical group 0.000 claims description 127
- 150000007523 nucleic acids Chemical class 0.000 claims description 123
- 239000002773 nucleotide Substances 0.000 claims description 123
- 101150093886 TGFBR2 gene Proteins 0.000 claims description 114
- 102000039446 nucleic acids Human genes 0.000 claims description 114
- 108020004707 nucleic acids Proteins 0.000 claims description 114
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims description 112
- 102100033455 TGF-beta receptor type-2 Human genes 0.000 claims description 112
- 239000000427 antigen Substances 0.000 claims description 101
- 108091007433 antigens Proteins 0.000 claims description 101
- 102000036639 antigens Human genes 0.000 claims description 101
- 239000013598 vector Substances 0.000 claims description 92
- 230000014509 gene expression Effects 0.000 claims description 90
- 108090000623 proteins and genes Proteins 0.000 claims description 81
- 206010028980 Neoplasm Diseases 0.000 claims description 68
- 102000004389 Ribonucleoproteins Human genes 0.000 claims description 54
- 108010081734 Ribonucleoproteins Proteins 0.000 claims description 54
- 230000005782 double-strand break Effects 0.000 claims description 52
- 230000000694 effects Effects 0.000 claims description 49
- 230000000295 complement effect Effects 0.000 claims description 48
- 210000002865 immune cell Anatomy 0.000 claims description 42
- 230000011664 signaling Effects 0.000 claims description 35
- 201000011510 cancer Diseases 0.000 claims description 33
- 230000008439 repair process Effects 0.000 claims description 32
- 239000013603 viral vector Substances 0.000 claims description 31
- 238000012217 deletion Methods 0.000 claims description 25
- 230000037430 deletion Effects 0.000 claims description 25
- 230000001965 increasing effect Effects 0.000 claims description 25
- 241000193996 Streptococcus pyogenes Species 0.000 claims description 24
- 230000001404 mediated effect Effects 0.000 claims description 24
- 238000003780 insertion Methods 0.000 claims description 22
- 230000037431 insertion Effects 0.000 claims description 22
- 230000004075 alteration Effects 0.000 claims description 19
- 241000713666 Lentivirus Species 0.000 claims description 18
- 230000002829 reductive effect Effects 0.000 claims description 16
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 claims description 14
- 108091026890 Coding region Proteins 0.000 claims description 13
- 230000002688 persistence Effects 0.000 claims description 13
- 230000005783 single-strand break Effects 0.000 claims description 13
- 108091092724 Noncoding DNA Proteins 0.000 claims description 12
- 102000040430 polynucleotide Human genes 0.000 claims description 12
- 108091033319 polynucleotide Proteins 0.000 claims description 12
- 239000002157 polynucleotide Substances 0.000 claims description 12
- 230000035755 proliferation Effects 0.000 claims description 12
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 claims description 11
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 claims description 11
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 claims description 11
- 241000702421 Dependoparvovirus Species 0.000 claims description 10
- 108700039691 Genetic Promoter Regions Proteins 0.000 claims description 10
- 102000001398 Granzyme Human genes 0.000 claims description 10
- 108060005986 Granzyme Proteins 0.000 claims description 10
- 108010074328 Interferon-gamma Proteins 0.000 claims description 10
- 210000000822 natural killer cell Anatomy 0.000 claims description 10
- 230000000638 stimulation Effects 0.000 claims description 10
- 101710089372 Programmed cell death protein 1 Proteins 0.000 claims description 9
- 102000004887 Transforming Growth Factor beta Human genes 0.000 claims description 9
- 108090001012 Transforming Growth Factor beta Proteins 0.000 claims description 9
- 239000000872 buffer Substances 0.000 claims description 9
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 claims description 9
- 230000003247 decreasing effect Effects 0.000 claims description 8
- 230000008488 polyadenylation Effects 0.000 claims description 8
- 102000008070 Interferon-gamma Human genes 0.000 claims description 7
- 230000009089 cytolysis Effects 0.000 claims description 7
- 239000003623 enhancer Substances 0.000 claims description 7
- 229960003130 interferon gamma Drugs 0.000 claims description 7
- 201000001441 melanoma Diseases 0.000 claims description 7
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 5
- 206010025323 Lymphomas Diseases 0.000 claims description 5
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 5
- 108091036066 Three prime untranslated region Proteins 0.000 claims description 5
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 5
- 210000000988 bone and bone Anatomy 0.000 claims description 5
- 230000002950 deficient Effects 0.000 claims description 5
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 4
- 108091026898 Leader sequence (mRNA) Proteins 0.000 claims description 4
- 210000000481 breast Anatomy 0.000 claims description 4
- 210000001072 colon Anatomy 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 210000004185 liver Anatomy 0.000 claims description 4
- 210000004072 lung Anatomy 0.000 claims description 4
- 230000036210 malignancy Effects 0.000 claims description 4
- 210000002307 prostate Anatomy 0.000 claims description 4
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 3
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 claims description 3
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 3
- 206010005949 Bone cancer Diseases 0.000 claims description 3
- 208000018084 Bone neoplasm Diseases 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 3
- 201000009030 Carcinoma Diseases 0.000 claims description 3
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 3
- 208000017604 Hodgkin disease Diseases 0.000 claims description 3
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 3
- 101000712669 Homo sapiens TGF-beta receptor type-2 Proteins 0.000 claims description 3
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 claims description 3
- 208000000172 Medulloblastoma Diseases 0.000 claims description 3
- 206010027406 Mesothelioma Diseases 0.000 claims description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 3
- 206010029260 Neuroblastoma Diseases 0.000 claims description 3
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 3
- 206010033128 Ovarian cancer Diseases 0.000 claims description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 3
- 208000019065 cervical carcinoma Diseases 0.000 claims description 3
- 201000003444 follicular lymphoma Diseases 0.000 claims description 3
- 230000037433 frameshift Effects 0.000 claims description 3
- 208000005017 glioblastoma Diseases 0.000 claims description 3
- 230000001976 improved effect Effects 0.000 claims description 3
- 201000008968 osteosarcoma Diseases 0.000 claims description 3
- 230000002611 ovarian Effects 0.000 claims description 3
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 claims description 3
- 206010042863 synovial sarcoma Diseases 0.000 claims description 3
- 238000000684 flow cytometry Methods 0.000 claims description 2
- 238000010212 intracellular staining Methods 0.000 claims description 2
- 238000005259 measurement Methods 0.000 claims description 2
- 238000001262 western blot Methods 0.000 claims description 2
- 102100023990 60S ribosomal protein L17 Human genes 0.000 claims 3
- 230000011559 double-strand break repair via nonhomologous end joining Effects 0.000 claims 2
- 238000010354 CRISPR gene editing Methods 0.000 abstract description 16
- -1 with sulfur (S) Chemical compound 0.000 description 63
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 61
- 201000010099 disease Diseases 0.000 description 54
- 102000005962 receptors Human genes 0.000 description 53
- 108020003175 receptors Proteins 0.000 description 53
- 230000027455 binding Effects 0.000 description 50
- 108090000765 processed proteins & peptides Proteins 0.000 description 43
- 229940024606 amino acid Drugs 0.000 description 41
- 235000001014 amino acid Nutrition 0.000 description 40
- 150000001413 amino acids Chemical class 0.000 description 39
- 238000012384 transportation and delivery Methods 0.000 description 39
- 230000004048 modification Effects 0.000 description 37
- 238000012986 modification Methods 0.000 description 37
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 35
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 35
- 102000004169 proteins and genes Human genes 0.000 description 33
- 108020004414 DNA Proteins 0.000 description 31
- 235000018102 proteins Nutrition 0.000 description 30
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 29
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 29
- 125000005647 linker group Chemical group 0.000 description 29
- 241000282414 Homo sapiens Species 0.000 description 26
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 25
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 25
- 239000003550 marker Substances 0.000 description 25
- 102000004196 processed proteins & peptides Human genes 0.000 description 25
- 125000006850 spacer group Chemical group 0.000 description 25
- 239000003795 chemical substances by application Substances 0.000 description 24
- 230000035772 mutation Effects 0.000 description 24
- 230000004068 intracellular signaling Effects 0.000 description 22
- 210000001519 tissue Anatomy 0.000 description 22
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 21
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 21
- 230000015572 biosynthetic process Effects 0.000 description 21
- 229920001184 polypeptide Polymers 0.000 description 21
- 230000034431 double-strand break repair via homologous recombination Effects 0.000 description 20
- 230000006870 function Effects 0.000 description 20
- 230000003612 virological effect Effects 0.000 description 20
- 230000000139 costimulatory effect Effects 0.000 description 19
- 125000002091 cationic group Chemical group 0.000 description 18
- 238000000926 separation method Methods 0.000 description 18
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 17
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 17
- 238000001727 in vivo Methods 0.000 description 17
- 239000008194 pharmaceutical composition Substances 0.000 description 17
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 16
- 108091028043 Nucleic acid sequence Proteins 0.000 description 16
- 238000002659 cell therapy Methods 0.000 description 16
- 230000006780 non-homologous end joining Effects 0.000 description 16
- 230000004936 stimulating effect Effects 0.000 description 16
- 102000006306 Antigen Receptors Human genes 0.000 description 15
- 108010083359 Antigen Receptors Proteins 0.000 description 15
- 230000001086 cytosolic effect Effects 0.000 description 15
- 238000009472 formulation Methods 0.000 description 15
- 230000001177 retroviral effect Effects 0.000 description 15
- 230000004913 activation Effects 0.000 description 14
- 230000003834 intracellular effect Effects 0.000 description 14
- 210000004369 blood Anatomy 0.000 description 13
- 239000008280 blood Substances 0.000 description 13
- 238000003776 cleavage reaction Methods 0.000 description 13
- 238000009826 distribution Methods 0.000 description 13
- 241000894007 species Species 0.000 description 13
- 210000000130 stem cell Anatomy 0.000 description 13
- 238000011282 treatment Methods 0.000 description 13
- 239000003446 ligand Substances 0.000 description 12
- 230000000670 limiting effect Effects 0.000 description 12
- 230000007017 scission Effects 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- 238000012546 transfer Methods 0.000 description 12
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 description 11
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 description 11
- 102000004127 Cytokines Human genes 0.000 description 11
- 108090000695 Cytokines Proteins 0.000 description 11
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 11
- 101001018097 Homo sapiens L-selectin Proteins 0.000 description 11
- 102100033467 L-selectin Human genes 0.000 description 11
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 11
- 101710126859 Single-stranded DNA-binding protein Proteins 0.000 description 11
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 11
- 239000012636 effector Substances 0.000 description 11
- 239000002105 nanoparticle Substances 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 239000000523 sample Substances 0.000 description 11
- 235000019527 sweetened beverage Nutrition 0.000 description 11
- 238000013518 transcription Methods 0.000 description 11
- 108700012439 CA9 Proteins 0.000 description 10
- 101150029707 ERBB2 gene Proteins 0.000 description 10
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 10
- 102100026497 Zinc finger protein 654 Human genes 0.000 description 10
- 230000003213 activating effect Effects 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 238000004520 electroporation Methods 0.000 description 10
- 239000012634 fragment Substances 0.000 description 10
- 238000000338 in vitro Methods 0.000 description 10
- 238000011534 incubation Methods 0.000 description 10
- 210000003071 memory t lymphocyte Anatomy 0.000 description 10
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 10
- 230000003285 pharmacodynamic effect Effects 0.000 description 10
- 229920000642 polymer Polymers 0.000 description 10
- 230000035897 transcription Effects 0.000 description 10
- 230000026683 transduction Effects 0.000 description 10
- 238000010361 transduction Methods 0.000 description 10
- 108091079001 CRISPR RNA Proteins 0.000 description 9
- 102000003735 Mesothelin Human genes 0.000 description 9
- 108090000015 Mesothelin Proteins 0.000 description 9
- 125000003275 alpha amino acid group Chemical group 0.000 description 9
- 230000001413 cellular effect Effects 0.000 description 9
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 9
- 239000013604 expression vector Substances 0.000 description 9
- 238000003197 gene knockdown Methods 0.000 description 9
- 210000000056 organ Anatomy 0.000 description 9
- 230000002085 persistent effect Effects 0.000 description 9
- 235000000346 sugar Nutrition 0.000 description 9
- 230000004083 survival effect Effects 0.000 description 9
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 8
- 241000701022 Cytomegalovirus Species 0.000 description 8
- 102100031940 Epithelial cell adhesion molecule Human genes 0.000 description 8
- 102100031507 Fc receptor-like protein 5 Human genes 0.000 description 8
- 101000920667 Homo sapiens Epithelial cell adhesion molecule Proteins 0.000 description 8
- 108010008707 Mucin-1 Proteins 0.000 description 8
- 102100034256 Mucin-1 Human genes 0.000 description 8
- 101710120463 Prostate stem cell antigen Proteins 0.000 description 8
- 102100036735 Prostate stem cell antigen Human genes 0.000 description 8
- 101001039269 Rattus norvegicus Glycine N-methyltransferase Proteins 0.000 description 8
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- 208000008383 Wilms tumor Diseases 0.000 description 8
- 208000026448 Wilms tumor 1 Diseases 0.000 description 8
- 102100022748 Wilms tumor protein Human genes 0.000 description 8
- 101710127857 Wilms tumor protein Proteins 0.000 description 8
- 238000011467 adoptive cell therapy Methods 0.000 description 8
- 210000000265 leukocyte Anatomy 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 230000037361 pathway Effects 0.000 description 8
- 239000003755 preservative agent Substances 0.000 description 8
- 238000012545 processing Methods 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 7
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 7
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 7
- 108020004705 Codon Proteins 0.000 description 7
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 7
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 7
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 7
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 description 7
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 7
- 108091028113 Trans-activating crRNA Proteins 0.000 description 7
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 235000018417 cysteine Nutrition 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 208000035475 disorder Diseases 0.000 description 7
- 230000001605 fetal effect Effects 0.000 description 7
- 238000001415 gene therapy Methods 0.000 description 7
- 238000010353 genetic engineering Methods 0.000 description 7
- 238000002955 isolation Methods 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 7
- 244000052769 pathogen Species 0.000 description 7
- 210000004986 primary T-cell Anatomy 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 6
- 102100032912 CD44 antigen Human genes 0.000 description 6
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 6
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 6
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 6
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 6
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 6
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 6
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 6
- 101001095088 Homo sapiens Melanoma antigen preferentially expressed in tumors Proteins 0.000 description 6
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 6
- 102000007482 Interleukin-13 Receptor alpha2 Subunit Human genes 0.000 description 6
- 108010085418 Interleukin-13 Receptor alpha2 Subunit Proteins 0.000 description 6
- 102100037020 Melanoma antigen preferentially expressed in tumors Human genes 0.000 description 6
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 6
- 241001529936 Murinae Species 0.000 description 6
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 6
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 6
- 230000006052 T cell proliferation Effects 0.000 description 6
- 238000007792 addition Methods 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 108091092356 cellular DNA Proteins 0.000 description 6
- 108700010039 chimeric receptor Proteins 0.000 description 6
- 238000012937 correction Methods 0.000 description 6
- 108091008034 costimulatory receptors Proteins 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 239000003937 drug carrier Substances 0.000 description 6
- 230000002068 genetic effect Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 230000030648 nucleus localization Effects 0.000 description 6
- 230000001717 pathogenic effect Effects 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 5
- 108010058905 CD44v6 antigen Proteins 0.000 description 5
- 102000016736 Cyclin Human genes 0.000 description 5
- 108050006400 Cyclin Proteins 0.000 description 5
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical class C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 5
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 5
- 101000846908 Homo sapiens Fc receptor-like protein 5 Proteins 0.000 description 5
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 5
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 5
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 5
- 101000884271 Homo sapiens Signal transducer CD24 Proteins 0.000 description 5
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 5
- 108060003951 Immunoglobulin Proteins 0.000 description 5
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 5
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 5
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 5
- 241000124008 Mammalia Species 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 102100023123 Mucin-16 Human genes 0.000 description 5
- 102100038081 Signal transducer CD24 Human genes 0.000 description 5
- 102100033579 Trophoblast glycoprotein Human genes 0.000 description 5
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 5
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 5
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 5
- 210000001185 bone marrow Anatomy 0.000 description 5
- 230000003915 cell function Effects 0.000 description 5
- 238000006471 dimerization reaction Methods 0.000 description 5
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 5
- 210000003743 erythrocyte Anatomy 0.000 description 5
- 102000015694 estrogen receptors Human genes 0.000 description 5
- 108010038795 estrogen receptors Proteins 0.000 description 5
- 208000002672 hepatitis B Diseases 0.000 description 5
- 102000018358 immunoglobulin Human genes 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 150000002632 lipids Chemical class 0.000 description 5
- 239000002502 liposome Substances 0.000 description 5
- 108020004999 messenger RNA Proteins 0.000 description 5
- 239000002953 phosphate buffered saline Substances 0.000 description 5
- 238000003752 polymerase chain reaction Methods 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 102000003998 progesterone receptors Human genes 0.000 description 5
- 108090000468 progesterone receptors Proteins 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 230000000717 retained effect Effects 0.000 description 5
- 238000012163 sequencing technique Methods 0.000 description 5
- 238000001890 transfection Methods 0.000 description 5
- 230000001052 transient effect Effects 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 4
- 102100036841 C-C motif chemokine 1 Human genes 0.000 description 4
- 108010009685 Cholinergic Receptors Proteins 0.000 description 4
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 4
- 101000713104 Homo sapiens C-C motif chemokine 1 Proteins 0.000 description 4
- 101001051490 Homo sapiens Neural cell adhesion molecule L1 Proteins 0.000 description 4
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 4
- 101000801433 Homo sapiens Trophoblast glycoprotein Proteins 0.000 description 4
- 102100020793 Interleukin-13 receptor subunit alpha-2 Human genes 0.000 description 4
- 101710112634 Interleukin-13 receptor subunit alpha-2 Proteins 0.000 description 4
- 108010002350 Interleukin-2 Proteins 0.000 description 4
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 4
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 4
- 108010069196 Neural Cell Adhesion Molecules Proteins 0.000 description 4
- 102100024964 Neural cell adhesion molecule L1 Human genes 0.000 description 4
- 102100023616 Neural cell adhesion molecule L1-like protein Human genes 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Natural products OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- 108091023040 Transcription factor Proteins 0.000 description 4
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 4
- 108091008605 VEGF receptors Proteins 0.000 description 4
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 4
- 102000034337 acetylcholine receptors Human genes 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 230000000735 allogeneic effect Effects 0.000 description 4
- 230000003432 anti-folate effect Effects 0.000 description 4
- 229940127074 antifolate Drugs 0.000 description 4
- 239000011324 bead Substances 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000006172 buffering agent Substances 0.000 description 4
- 230000015556 catabolic process Effects 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 210000005220 cytoplasmic tail Anatomy 0.000 description 4
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 4
- 238000006731 degradation reaction Methods 0.000 description 4
- 238000002716 delivery method Methods 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 239000004052 folic acid antagonist Substances 0.000 description 4
- 210000002443 helper t lymphocyte Anatomy 0.000 description 4
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 4
- 238000011134 hematopoietic stem cell transplantation Methods 0.000 description 4
- 210000000987 immune system Anatomy 0.000 description 4
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 4
- 102000006495 integrins Human genes 0.000 description 4
- 108010044426 integrins Proteins 0.000 description 4
- 238000007912 intraperitoneal administration Methods 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 4
- 210000001616 monocyte Anatomy 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 4
- 102000005912 ran GTP Binding Protein Human genes 0.000 description 4
- 108010005597 ran GTP Binding Protein Proteins 0.000 description 4
- 235000004400 serine Nutrition 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- SSOORFWOBGFTHL-OTEJMHTDSA-N (4S)-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[(2S)-2-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-5-carbamimidamido-1-[[(2S)-5-carbamimidamido-1-[[(1S)-4-carbamimidamido-1-carboxybutyl]amino]-1-oxopentan-2-yl]amino]-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]carbamoyl]pyrrolidin-1-yl]-2-oxoethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-4-[[(2S)-2-[[(2S)-2-[[(2S)-2,6-diaminohexanoyl]amino]-3-methylbutanoyl]amino]propanoyl]amino]-5-oxopentanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H]1CCCN1C(=O)CNC(=O)[C@H](Cc1c[nH]c2ccccc12)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@@H](N)CCCCN)C(C)C)C(C)C)C(C)C)C(C)C)C(C)C)C(C)C)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O SSOORFWOBGFTHL-OTEJMHTDSA-N 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 102100027207 CD27 antigen Human genes 0.000 description 3
- 108010077544 Chromatin Proteins 0.000 description 3
- 108010060273 Cyclin A2 Proteins 0.000 description 3
- 102100025191 Cyclin-A2 Human genes 0.000 description 3
- 101150032879 Fcrl5 gene Proteins 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 102100037931 Harmonin Human genes 0.000 description 3
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 3
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 3
- 101100166600 Homo sapiens CD28 gene Proteins 0.000 description 3
- 101000737793 Homo sapiens Cerebellar degeneration-related antigen 1 Proteins 0.000 description 3
- 101000805947 Homo sapiens Harmonin Proteins 0.000 description 3
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 3
- 102000008100 Human Serum Albumin Human genes 0.000 description 3
- 108091006905 Human Serum Albumin Proteins 0.000 description 3
- 102100037850 Interferon gamma Human genes 0.000 description 3
- 108090000172 Interleukin-15 Proteins 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 101100508818 Mus musculus Inpp5k gene Proteins 0.000 description 3
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 description 3
- 108091034117 Oligonucleotide Proteins 0.000 description 3
- 108700026244 Open Reading Frames Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 3
- 102100038358 Prostate-specific antigen Human genes 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 101100366438 Rattus norvegicus Sphkap gene Proteins 0.000 description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 3
- 108010002687 Survivin Proteins 0.000 description 3
- 230000006044 T cell activation Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 150000003838 adenosines Chemical class 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 125000001769 aryl amino group Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000001363 autoimmune Effects 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000012472 biological sample Substances 0.000 description 3
- 210000001772 blood platelet Anatomy 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 210000003483 chromatin Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 125000004663 dialkyl amino group Chemical group 0.000 description 3
- 125000004986 diarylamino group Chemical group 0.000 description 3
- 125000005240 diheteroarylamino group Chemical group 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 3
- 229940014144 folate Drugs 0.000 description 3
- 239000011724 folic acid Substances 0.000 description 3
- 230000008014 freezing Effects 0.000 description 3
- 238000007710 freezing Methods 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 108020001507 fusion proteins Proteins 0.000 description 3
- 102000037865 fusion proteins Human genes 0.000 description 3
- 210000003714 granulocyte Anatomy 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 125000005241 heteroarylamino group Chemical group 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- 230000004807 localization Effects 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 230000009401 metastasis Effects 0.000 description 3
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 3
- 230000003278 mimic effect Effects 0.000 description 3
- 238000009126 molecular therapy Methods 0.000 description 3
- 230000009437 off-target effect Effects 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 210000003289 regulatory T cell Anatomy 0.000 description 3
- 239000011669 selenium Substances 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- 108020003589 5' Untranslated Regions Proteins 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 102100029457 Adenine phosphoribosyltransferase Human genes 0.000 description 2
- 108010024223 Adenine phosphoribosyltransferase Proteins 0.000 description 2
- 108091023043 Alu Element Proteins 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000271566 Aves Species 0.000 description 2
- 102000052609 BRCA2 Human genes 0.000 description 2
- 108700020462 BRCA2 Proteins 0.000 description 2
- 101150008921 Brca2 gene Proteins 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 101100112922 Candida albicans CDR3 gene Proteins 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102100031437 Cell cycle checkpoint protein RAD1 Human genes 0.000 description 2
- 102100035361 Cerebellar degeneration-related protein 2 Human genes 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 108091060290 Chromatid Proteins 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 230000005778 DNA damage Effects 0.000 description 2
- 231100000277 DNA damage Toxicity 0.000 description 2
- 230000033616 DNA repair Effects 0.000 description 2
- 102100033934 DNA repair protein RAD51 homolog 2 Human genes 0.000 description 2
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 2
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 102000001301 EGF receptor Human genes 0.000 description 2
- 108060006698 EGF receptor Proteins 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 101800003838 Epidermal growth factor Proteins 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 108700024394 Exon Proteins 0.000 description 2
- 108010087819 Fc receptors Proteins 0.000 description 2
- 102000009109 Fc receptors Human genes 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- BCCRXDTUTZHDEU-VKHMYHEASA-N Gly-Ser Chemical compound NCC(=O)N[C@@H](CO)C(O)=O BCCRXDTUTZHDEU-VKHMYHEASA-N 0.000 description 2
- 108091093094 Glycol nucleic acid Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 101001130384 Homo sapiens Cell cycle checkpoint protein RAD1 Proteins 0.000 description 2
- 101000737796 Homo sapiens Cerebellar degeneration-related protein 2 Proteins 0.000 description 2
- 101001132307 Homo sapiens DNA repair protein RAD51 homolog 2 Proteins 0.000 description 2
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 2
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 2
- 101001057156 Homo sapiens Melanoma-associated antigen C2 Proteins 0.000 description 2
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 2
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 2
- 108010002586 Interleukin-7 Proteins 0.000 description 2
- 108091092195 Intron Proteins 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 2
- 108010010995 MART-1 Antigen Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 102100027252 Melanoma-associated antigen C2 Human genes 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 241000713869 Moloney murine leukemia virus Species 0.000 description 2
- 102100025751 Mothers against decapentaplegic homolog 2 Human genes 0.000 description 2
- 101710143123 Mothers against decapentaplegic homolog 2 Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 241000713883 Myeloproliferative sarcoma virus Species 0.000 description 2
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 description 2
- 108091061960 Naked DNA Proteins 0.000 description 2
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 2
- 101710153660 Nuclear receptor corepressor 2 Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- 108020005067 RNA Splice Sites Proteins 0.000 description 2
- 108010068097 Rad51 Recombinase Proteins 0.000 description 2
- 102000002490 Rad51 Recombinase Human genes 0.000 description 2
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 2
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 241000713880 Spleen focus-forming virus Species 0.000 description 2
- 102100036234 Synaptonemal complex protein 1 Human genes 0.000 description 2
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 2
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 2
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 101710171981 Volume-regulated anion channel subunit LRRC8A Proteins 0.000 description 2
- 102100040985 Volume-regulated anion channel subunit LRRC8A Human genes 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 2
- 229960004373 acetylcholine Drugs 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000003044 adaptive effect Effects 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 238000002617 apheresis Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- 229960000686 benzalkonium chloride Drugs 0.000 description 2
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 210000003995 blood forming stem cell Anatomy 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 150000001720 carbohydrates Chemical group 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- 238000007385 chemical modification Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 210000004756 chromatid Anatomy 0.000 description 2
- 230000004186 co-expression Effects 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 238000009295 crossflow filtration Methods 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 210000003162 effector t lymphocyte Anatomy 0.000 description 2
- 210000001671 embryonic stem cell Anatomy 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 210000004700 fetal blood Anatomy 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 235000019152 folic acid Nutrition 0.000 description 2
- 231100000221 frame shift mutation induction Toxicity 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 210000004408 hybridoma Anatomy 0.000 description 2
- 230000033444 hydroxylation Effects 0.000 description 2
- 238000005805 hydroxylation reaction Methods 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 230000002779 inactivation Effects 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000036512 infertility Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000015788 innate immune response Effects 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 210000003738 lymphoid progenitor cell Anatomy 0.000 description 2
- 210000003563 lymphoid tissue Anatomy 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 230000005291 magnetic effect Effects 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 2
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 2
- 229960002216 methylparaben Drugs 0.000 description 2
- 239000011859 microparticle Substances 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 210000004877 mucosa Anatomy 0.000 description 2
- 210000000066 myeloid cell Anatomy 0.000 description 2
- PUPNJSIFIXXJCH-UHFFFAOYSA-N n-(4-hydroxyphenyl)-2-(1,1,3-trioxo-1,2-benzothiazol-2-yl)acetamide Chemical compound C1=CC(O)=CC=C1NC(=O)CN1S(=O)(=O)C2=CC=CC=C2C1=O PUPNJSIFIXXJCH-UHFFFAOYSA-N 0.000 description 2
- 210000000581 natural killer T-cell Anatomy 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- 239000013600 plasmid vector Substances 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 239000001294 propane Substances 0.000 description 2
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 2
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 2
- 229960003415 propylparaben Drugs 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000002271 resection Methods 0.000 description 2
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 210000003705 ribosome Anatomy 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 230000002123 temporal effect Effects 0.000 description 2
- 210000001550 testis Anatomy 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 125000002264 triphosphate group Chemical group [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 229940124676 vascular endothelial growth factor receptor Drugs 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 229920002818 (Hydroxyethyl)methacrylate Polymers 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- OGLIVAIBVXQQHO-UHFFFAOYSA-N 1-(dimethylamino)-4,5-dioctadecoxypentan-2-ol Chemical compound CCCCCCCCCCCCCCCCCCOCC(CC(O)CN(C)C)OCCCCCCCCCCCCCCCCCC OGLIVAIBVXQQHO-UHFFFAOYSA-N 0.000 description 1
- GZEFTKHSACGIBG-UGKPPGOTSA-N 1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)-2-propyloxolan-2-yl]pyrimidine-2,4-dione Chemical compound C1=CC(=O)NC(=O)N1[C@]1(CCC)O[C@H](CO)[C@@H](O)[C@H]1O GZEFTKHSACGIBG-UGKPPGOTSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- FZIIBDOXPQOKBP-UHFFFAOYSA-N 2-methyloxetane Chemical compound CC1CCO1 FZIIBDOXPQOKBP-UHFFFAOYSA-N 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- HYRQVGHJHUTCJL-UHFFFAOYSA-M 3,3-di(tetradecoxy)propyl-(2-hydroxyethyl)-dimethylazanium;bromide Chemical compound [Br-].CCCCCCCCCCCCCCOC(CC[N+](C)(C)CCO)OCCCCCCCCCCCCCC HYRQVGHJHUTCJL-UHFFFAOYSA-M 0.000 description 1
- HJRLNCQDOQJIIB-UHFFFAOYSA-N 3-[4-(3-aminopropylamino)butylamino]propylurea Chemical compound NCCCNCCCCNCCCNC(N)=O HJRLNCQDOQJIIB-UHFFFAOYSA-N 0.000 description 1
- ZAYHVCMSTBRABG-UHFFFAOYSA-N 5-Methylcytidine Natural products O=C1N=C(N)C(C)=CN1C1C(O)C(O)C(CO)O1 ZAYHVCMSTBRABG-UHFFFAOYSA-N 0.000 description 1
- AGFIRQJZCNVMCW-UAKXSSHOSA-N 5-bromouridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 AGFIRQJZCNVMCW-UAKXSSHOSA-N 0.000 description 1
- ZAYHVCMSTBRABG-JXOAFFINSA-N 5-methylcytidine Chemical group O=C1N=C(N)C(C)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 ZAYHVCMSTBRABG-JXOAFFINSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ASUCSHXLTWZYBA-UMMCILCDSA-N 8-Bromoguanosine Chemical compound C1=2NC(N)=NC(=O)C=2N=C(Br)N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O ASUCSHXLTWZYBA-UMMCILCDSA-N 0.000 description 1
- HDZZVAMISRMYHH-UHFFFAOYSA-N 9beta-Ribofuranosyl-7-deazaadenin Natural products C1=CC=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O HDZZVAMISRMYHH-UHFFFAOYSA-N 0.000 description 1
- 241000093740 Acidaminococcus sp. Species 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 102100023003 Ankyrin repeat domain-containing protein 30A Human genes 0.000 description 1
- 101100420868 Anuroctonus phaiodactylus phtx gene Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 102100032306 Aurora kinase B Human genes 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 108700003860 Bacterial Genes Proteins 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 102100033680 Bombesin receptor-activated protein C6orf89 Human genes 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 102000049320 CD36 Human genes 0.000 description 1
- 108010045374 CD36 Antigens Proteins 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 102100037904 CD9 antigen Human genes 0.000 description 1
- 108010083123 CDX2 Transcription Factor Proteins 0.000 description 1
- 102000006277 CDX2 Transcription Factor Human genes 0.000 description 1
- 238000010453 CRISPR/Cas method Methods 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 102100039510 Cancer/testis antigen 2 Human genes 0.000 description 1
- 101100495352 Candida albicans CDR4 gene Proteins 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 102100027668 Carboxy-terminal domain RNA polymerase II polypeptide A small phosphatase 1 Human genes 0.000 description 1
- 101710134395 Carboxy-terminal domain RNA polymerase II polypeptide A small phosphatase 1 Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 1
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 1
- 108010051109 Cell-Penetrating Peptides Proteins 0.000 description 1
- 102000020313 Cell-Penetrating Peptides Human genes 0.000 description 1
- 102100035673 Centrosomal protein of 290 kDa Human genes 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102100032952 Condensin complex subunit 3 Human genes 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241000938605 Crocodylia Species 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 1
- 102100025571 Cutaneous T-cell lymphoma-associated antigen 1 Human genes 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 1
- 102100026846 Cytidine deaminase Human genes 0.000 description 1
- 108010031325 Cytidine deaminase Proteins 0.000 description 1
- 102000000311 Cytosine Deaminase Human genes 0.000 description 1
- 108010080611 Cytosine Deaminase Proteins 0.000 description 1
- 102220605874 Cytosolic arginine sensor for mTORC1 subunit 2_D10A_mutation Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KAZBKCHUSA-N D-altritol Chemical compound OC[C@@H](O)[C@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KAZBKCHUSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 238000010442 DNA editing Methods 0.000 description 1
- 230000007067 DNA methylation Effects 0.000 description 1
- 230000008265 DNA repair mechanism Effects 0.000 description 1
- 230000007018 DNA scission Effects 0.000 description 1
- 102100033587 DNA topoisomerase 2-alpha Human genes 0.000 description 1
- 102100033589 DNA topoisomerase 2-beta Human genes 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 102100031695 DnaJ homolog subfamily C member 2 Human genes 0.000 description 1
- 102100034185 E3 ubiquitin-protein ligase RLIM Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102100031780 Endonuclease Human genes 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 101000809594 Escherichia coli (strain K12) Shikimate kinase 1 Proteins 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102100031812 Fibulin-1 Human genes 0.000 description 1
- 101710170731 Fibulin-1 Proteins 0.000 description 1
- 229920001917 Ficoll Polymers 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 102000010451 Folate receptor alpha Human genes 0.000 description 1
- 108050001931 Folate receptor alpha Proteins 0.000 description 1
- 102100020714 Fragile X mental retardation 1 neighbor protein Human genes 0.000 description 1
- 241000589599 Francisella tularensis subsp. novicida Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 102100035233 Furin Human genes 0.000 description 1
- 108090001126 Furin Proteins 0.000 description 1
- 102100031351 Galectin-9 Human genes 0.000 description 1
- 101710121810 Galectin-9 Proteins 0.000 description 1
- 208000003807 Graves Disease Diseases 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 102100028971 HLA class I histocompatibility antigen, C alpha chain Human genes 0.000 description 1
- 102000006354 HLA-DR Antigens Human genes 0.000 description 1
- 108010058597 HLA-DR Antigens Proteins 0.000 description 1
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 1
- 102100022650 Homeobox protein Hox-A7 Human genes 0.000 description 1
- 102100025056 Homeobox protein Hox-B6 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000757191 Homo sapiens Ankyrin repeat domain-containing protein 30A Proteins 0.000 description 1
- 101000984746 Homo sapiens BRCA1-associated protein Proteins 0.000 description 1
- 101000944524 Homo sapiens Bombesin receptor-activated protein C6orf89 Proteins 0.000 description 1
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 description 1
- 101100005863 Homo sapiens CEP290 gene Proteins 0.000 description 1
- 101000889345 Homo sapiens Cancer/testis antigen 2 Proteins 0.000 description 1
- 101000715664 Homo sapiens Centrosomal protein of 290 kDa Proteins 0.000 description 1
- 101000942622 Homo sapiens Condensin complex subunit 3 Proteins 0.000 description 1
- 101000856239 Homo sapiens Cutaneous T-cell lymphoma-associated antigen 1 Proteins 0.000 description 1
- 101000911952 Homo sapiens Cyclin-dependent kinase 7 Proteins 0.000 description 1
- 101000845887 Homo sapiens DnaJ homolog subfamily C member 2 Proteins 0.000 description 1
- 101000711924 Homo sapiens E3 ubiquitin-protein ligase RLIM Proteins 0.000 description 1
- 101000932499 Homo sapiens Fragile X mental retardation 1 neighbor protein Proteins 0.000 description 1
- 101000986084 Homo sapiens HLA class I histocompatibility antigen, C alpha chain Proteins 0.000 description 1
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 1
- 101001045116 Homo sapiens Homeobox protein Hox-A7 Proteins 0.000 description 1
- 101001077542 Homo sapiens Homeobox protein Hox-B6 Proteins 0.000 description 1
- 101000911772 Homo sapiens Hsc70-interacting protein Proteins 0.000 description 1
- 101100452137 Homo sapiens IGF2BP3 gene Proteins 0.000 description 1
- 101001055145 Homo sapiens Interleukin-2 receptor subunit beta Proteins 0.000 description 1
- 101001008951 Homo sapiens Kinesin-like protein KIF15 Proteins 0.000 description 1
- 101001054842 Homo sapiens Leucine zipper protein 4 Proteins 0.000 description 1
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 1
- 101001065832 Homo sapiens Low-density lipoprotein receptor class A domain-containing protein 4 Proteins 0.000 description 1
- 101000576802 Homo sapiens Mesothelin Proteins 0.000 description 1
- 101000874141 Homo sapiens Probable ATP-dependent RNA helicase DDX43 Proteins 0.000 description 1
- 101000880771 Homo sapiens Protein SSX3 Proteins 0.000 description 1
- 101000880774 Homo sapiens Protein SSX4 Proteins 0.000 description 1
- 101000880775 Homo sapiens Protein SSX5 Proteins 0.000 description 1
- 101000620365 Homo sapiens Protein TMEPAI Proteins 0.000 description 1
- 101000779418 Homo sapiens RAC-alpha serine/threonine-protein kinase Proteins 0.000 description 1
- 101000743264 Homo sapiens RNA-binding protein 6 Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 101000756066 Homo sapiens Serine/threonine-protein kinase RIO1 Proteins 0.000 description 1
- 101000632529 Homo sapiens Shugoshin 1 Proteins 0.000 description 1
- 101000863692 Homo sapiens Ski oncogene Proteins 0.000 description 1
- 101000688996 Homo sapiens Ski-like protein Proteins 0.000 description 1
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 1
- 101000662909 Homo sapiens T cell receptor beta constant 1 Proteins 0.000 description 1
- 101000662902 Homo sapiens T cell receptor beta constant 2 Proteins 0.000 description 1
- 101000946860 Homo sapiens T-cell surface glycoprotein CD3 epsilon chain Proteins 0.000 description 1
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 1
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 1
- 101000784541 Homo sapiens Zinc finger and SCAN domain-containing protein 21 Proteins 0.000 description 1
- 101000785712 Homo sapiens Zinc finger protein 282 Proteins 0.000 description 1
- 101000856240 Homo sapiens cTAGE family member 2 Proteins 0.000 description 1
- 102100027037 Hsc70-interacting protein Human genes 0.000 description 1
- 241000829111 Human polyomavirus 1 Species 0.000 description 1
- 102100027735 Hyaluronan mediated motility receptor Human genes 0.000 description 1
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical class C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 1
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 102100039615 Inactive tyrosine-protein kinase transmembrane receptor ROR1 Human genes 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100037920 Insulin-like growth factor 2 mRNA-binding protein 3 Human genes 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 108010061833 Integrases Proteins 0.000 description 1
- 102100026879 Interleukin-2 receptor subunit beta Human genes 0.000 description 1
- 102100024319 Intestinal-type alkaline phosphatase Human genes 0.000 description 1
- 101710184243 Intestinal-type alkaline phosphatase Proteins 0.000 description 1
- 102100027630 Kinesin-like protein KIF15 Human genes 0.000 description 1
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 102100026910 Leucine zipper protein 4 Human genes 0.000 description 1
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 1
- 102100032094 Low-density lipoprotein receptor class A domain-containing protein 4 Human genes 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102100033247 Lysine-specific demethylase 5B Human genes 0.000 description 1
- 101710105712 Lysine-specific demethylase 5B Proteins 0.000 description 1
- 102000016200 MART-1 Antigen Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 102100025096 Mesothelin Human genes 0.000 description 1
- 101000686985 Mouse mammary tumor virus (strain C3H) Protein PR73 Proteins 0.000 description 1
- 108010085220 Multiprotein Complexes Proteins 0.000 description 1
- 102000007474 Multiprotein Complexes Human genes 0.000 description 1
- 241000701029 Murid betaherpesvirus 1 Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101000753280 Mus musculus Angiopoietin-1 receptor Proteins 0.000 description 1
- 241001508687 Mustela erminea Species 0.000 description 1
- VQAYFKKCNSOZKM-IOSLPCCCSA-N N(6)-methyladenosine Chemical compound C1=NC=2C(NC)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O VQAYFKKCNSOZKM-IOSLPCCCSA-N 0.000 description 1
- OVRNDRQMDRJTHS-KEWYIRBNSA-N N-acetyl-D-galactosamine Chemical compound CC(=O)N[C@H]1C(O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-KEWYIRBNSA-N 0.000 description 1
- VQAYFKKCNSOZKM-UHFFFAOYSA-N NSC 29409 Natural products C1=NC=2C(NC)=NC=NC=2N1C1OC(CO)C(O)C1O VQAYFKKCNSOZKM-UHFFFAOYSA-N 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- 108700019961 Neoplasm Genes Proteins 0.000 description 1
- 102000048850 Neoplasm Genes Human genes 0.000 description 1
- 108010032605 Nerve Growth Factor Receptors Proteins 0.000 description 1
- 208000009869 Neu-Laxova syndrome Diseases 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 108020004485 Nonsense Codon Proteins 0.000 description 1
- 102000007999 Nuclear Proteins Human genes 0.000 description 1
- 108010089610 Nuclear Proteins Proteins 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 101500011382 Oncorhynchus mykiss Corticotropin-like intermediary peptide 1 Proteins 0.000 description 1
- 102100026949 Opioid growth factor receptor Human genes 0.000 description 1
- 101710130302 Opioid growth factor receptor Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 101710124239 Poly(A) polymerase Proteins 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 108010076039 Polyproteins Proteins 0.000 description 1
- 241000605861 Prevotella Species 0.000 description 1
- 102100035724 Probable ATP-dependent RNA helicase DDX43 Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 102100037726 Protein SSX3 Human genes 0.000 description 1
- 102100037727 Protein SSX4 Human genes 0.000 description 1
- 102100037723 Protein SSX5 Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 206010037075 Protozoal infections Diseases 0.000 description 1
- 229930185560 Pseudouridine Chemical group 0.000 description 1
- PTJWIQPHWPFNBW-UHFFFAOYSA-N Pseudouridine C Chemical group OC1C(O)C(CO)OC1C1=CNC(=O)NC1=O PTJWIQPHWPFNBW-UHFFFAOYSA-N 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 1
- 102100038150 RNA-binding protein 6 Human genes 0.000 description 1
- 101500027983 Rattus norvegicus Octadecaneuropeptide Proteins 0.000 description 1
- 108010006700 Receptor Tyrosine Kinase-like Orphan Receptors Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 206010038997 Retroviral infections Diseases 0.000 description 1
- 102100029627 Ribosomal RNA processing protein 1 homolog A Human genes 0.000 description 1
- 101710166144 Ribosomal RNA processing protein 1 homolog A Proteins 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 102100022261 Serine/threonine-protein kinase RIO1 Human genes 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102100028402 Shugoshin 1 Human genes 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 102100029969 Ski oncogene Human genes 0.000 description 1
- 102100024451 Ski-like protein Human genes 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 101800001271 Surface protein Proteins 0.000 description 1
- 101710143177 Synaptonemal complex protein 1 Proteins 0.000 description 1
- 102100035721 Syndecan-1 Human genes 0.000 description 1
- 230000033540 T cell apoptotic process Effects 0.000 description 1
- 102100029452 T cell receptor alpha chain constant Human genes 0.000 description 1
- 102100037272 T cell receptor beta constant 1 Human genes 0.000 description 1
- 102100035794 T-cell surface glycoprotein CD3 epsilon chain Human genes 0.000 description 1
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 1
- 101150002618 TCRP gene Proteins 0.000 description 1
- 108020005038 Terminator Codon Proteins 0.000 description 1
- 108091046915 Threose nucleic acid Proteins 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 101800005109 Triakontatetraneuropeptide Proteins 0.000 description 1
- 101710190034 Trophoblast glycoprotein Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 102100033725 Tumor necrosis factor receptor superfamily member 16 Human genes 0.000 description 1
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 1
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102100039094 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 108020005202 Viral DNA Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 102100020917 Zinc finger and SCAN domain-containing protein 21 Human genes 0.000 description 1
- 102100026417 Zinc finger protein 282 Human genes 0.000 description 1
- FHHZHGZBHYYWTG-INFSMZHSSA-N [(2r,3s,4r,5r)-5-(2-amino-7-methyl-6-oxo-3h-purin-9-ium-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [[[(2r,3s,4r,5r)-5-(2-amino-6-oxo-3h-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] phosphate Chemical compound N1C(N)=NC(=O)C2=C1[N+]([C@H]1[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=C(C(N=C(N)N4)=O)N=C3)O)O1)O)=CN2C FHHZHGZBHYYWTG-INFSMZHSSA-N 0.000 description 1
- FJWGYAHXMCUOOM-QHOUIDNNSA-N [(2s,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6s)-4,5-dinitrooxy-2-(nitrooxymethyl)-6-[(2r,3r,4s,5r,6s)-4,5,6-trinitrooxy-2-(nitrooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-3,5-dinitrooxy-6-(nitrooxymethyl)oxan-4-yl] nitrate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](O[N+]([O-])=O)[C@H]1O[N+]([O-])=O)O[C@H]1[C@@H]([C@@H](O[N+]([O-])=O)[C@H](O[N+]([O-])=O)[C@@H](CO[N+]([O-])=O)O1)O[N+]([O-])=O)CO[N+](=O)[O-])[C@@H]1[C@@H](CO[N+]([O-])=O)O[C@@H](O[N+]([O-])=O)[C@H](O[N+]([O-])=O)[C@H]1O[N+]([O-])=O FJWGYAHXMCUOOM-QHOUIDNNSA-N 0.000 description 1
- KCNVBTZSLOHIKK-YEUCEMRASA-N [2-[(z)-octadec-9-enoyl]oxy-3-(2,4,6-trimethylpyridin-1-ium-1-yl)propyl] (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCCCC)C[N+]1=C(C)C=C(C)C=C1C KCNVBTZSLOHIKK-YEUCEMRASA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- 230000004721 adaptive immunity Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- MXKCYTKUIDTFLY-ZNNSSXPHSA-N alpha-L-Fucp-(1->2)-beta-D-Galp-(1->4)-[alpha-L-Fucp-(1->3)]-beta-D-GlcpNAc-(1->3)-D-Galp Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](NC(C)=O)[C@H](O[C@H]3[C@H]([C@@H](CO)OC(O)[C@@H]3O)O)O[C@@H]2CO)O[C@H]2[C@H]([C@H](O)[C@H](O)[C@H](C)O2)O)O[C@H](CO)[C@H](O)[C@@H]1O MXKCYTKUIDTFLY-ZNNSSXPHSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000002431 aminoalkoxy group Chemical group 0.000 description 1
- 239000002269 analeptic agent Substances 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 208000010928 autoimmune thyroid disease Diseases 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 239000013602 bacteriophage vector Substances 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- CXQCLLQQYTUUKJ-ALWAHNIESA-N beta-D-GalpNAc-(1->4)-[alpha-Neup5Ac-(2->8)-alpha-Neup5Ac-(2->3)]-beta-D-Galp-(1->4)-beta-D-Glcp-(1<->1')-Cer(d18:1/18:0) Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@H](NC(=O)CCCCCCCCCCCCCCCCC)[C@H](O)\C=C\CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@@H](CO)O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)C(O)=O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](CO)O1 CXQCLLQQYTUUKJ-ALWAHNIESA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- WGDUUQDYDIIBKT-UHFFFAOYSA-N beta-Pseudouridine Chemical group OC1OC(CN2C=CC(=O)NC2=O)C(O)C1O WGDUUQDYDIIBKT-UHFFFAOYSA-N 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- HUTDDBSSHVOYJR-UHFFFAOYSA-H bis[(2-oxo-1,3,2$l^{5},4$l^{2}-dioxaphosphaplumbetan-2-yl)oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O HUTDDBSSHVOYJR-UHFFFAOYSA-H 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N butyl alcohol Substances CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 229920006317 cationic polymer Polymers 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 238000003320 cell separation method Methods 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 238000003381 deacetylation reaction Methods 0.000 description 1
- 230000009615 deamination Effects 0.000 description 1
- 238000006481 deamination reaction Methods 0.000 description 1
- 229920006237 degradable polymer Polymers 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000002022 differential scanning fluorescence spectroscopy Methods 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000012361 double-strand break repair Effects 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000008393 encapsulating agent Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 230000000925 erythroid effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000001036 exonucleolytic effect Effects 0.000 description 1
- 210000001808 exosome Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000012595 freezing medium Substances 0.000 description 1
- 238000010230 functional analysis Methods 0.000 description 1
- 230000000799 fusogenic effect Effects 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 238000003500 gene array Methods 0.000 description 1
- 230000030414 genetic transfer Effects 0.000 description 1
- 230000037442 genomic alteration Effects 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 150000002333 glycines Chemical class 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 229940029575 guanosine Drugs 0.000 description 1
- 210000004837 gut-associated lymphoid tissue Anatomy 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 238000012165 high-throughput sequencing Methods 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 230000006195 histone acetylation Effects 0.000 description 1
- 230000006197 histone deacetylation Effects 0.000 description 1
- 108010003425 hyaluronan-mediated motility receptor Proteins 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- MVYQJCPZZBFMLF-UHFFFAOYSA-N hydron;propan-1-amine;bromide Chemical compound [Br-].CCC[NH3+] MVYQJCPZZBFMLF-UHFFFAOYSA-N 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 210000002861 immature t-cell Anatomy 0.000 description 1
- 229940126546 immune checkpoint molecule Drugs 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000002998 immunogenetic effect Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000012744 immunostaining Methods 0.000 description 1
- 230000003308 immunostimulating effect Effects 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000008011 inorganic excipient Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 108010027445 interleukin-22 receptor Proteins 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000000366 juvenile effect Effects 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229960004502 levodopa Drugs 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 210000005210 lymphoid organ Anatomy 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Chemical class 0.000 description 1
- 125000005395 methacrylic acid group Chemical class 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 230000025608 mitochondrion localization Effects 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000002894 multi-fate stem cell Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 230000000869 mutational effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 230000037434 nonsense mutation Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000008012 organic excipient Substances 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 230000005298 paramagnetic effect Effects 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical group NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- 125000005642 phosphothioate group Chemical group 0.000 description 1
- 210000000608 photoreceptor cell Anatomy 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 229920001308 poly(aminoacid) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 210000003240 portal vein Anatomy 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- QSQHZXBTJCXIAV-UHFFFAOYSA-N propan-1-amine;2,2,2-trifluoroacetic acid Chemical compound CCCN.OC(=O)C(F)(F)F QSQHZXBTJCXIAV-UHFFFAOYSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 238000002818 protein evolution Methods 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- PTJWIQPHWPFNBW-GBNDHIKLSA-N pseudouridine Chemical group O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1C1=CNC(=O)NC1=O PTJWIQPHWPFNBW-GBNDHIKLSA-N 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 238000007157 ring contraction reaction Methods 0.000 description 1
- 238000006049 ring expansion reaction Methods 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 238000007480 sanger sequencing Methods 0.000 description 1
- 238000010187 selection method Methods 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 150000003355 serines Chemical class 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000009450 sialylation Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 231100000617 superantigen Toxicity 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 210000004243 sweat Anatomy 0.000 description 1
- 210000001179 synovial fluid Anatomy 0.000 description 1
- 238000012385 systemic delivery Methods 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 230000005100 tissue tropism Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000010474 transient expression Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- AISMNBXOJRHCIA-UHFFFAOYSA-N trimethylazanium;bromide Chemical compound Br.CN(C)C AISMNBXOJRHCIA-UHFFFAOYSA-N 0.000 description 1
- JOPDZQBPOWAEHC-UHFFFAOYSA-H tristrontium;diphosphate Chemical compound [Sr+2].[Sr+2].[Sr+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O JOPDZQBPOWAEHC-UHFFFAOYSA-H 0.000 description 1
- NMEHNETUFHBYEG-IHKSMFQHSA-N tttn Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 NMEHNETUFHBYEG-IHKSMFQHSA-N 0.000 description 1
- HDZZVAMISRMYHH-KCGFPETGSA-N tubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O HDZZVAMISRMYHH-KCGFPETGSA-N 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 230000000381 tumorigenic effect Effects 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
- C12N15/1138—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing against receptors or cell surface proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464416—Receptors for cytokines
- A61K39/464417—Receptors for tumor necrosis factors [TNF], e.g. lymphotoxin receptor [LTR], CD30
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2121/00—Preparations for use in therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
Definitions
- the present disclosure relates to CRISPR Cas9-related methods and components for editing a target nucleic acid sequence, e.g., a Transforming Growth Factor ⁇ Receptor II (TGFBR2) gene, or modulating expression of a target nucleic acid sequence, e.g., a TGFBR2 gene.
- a target nucleic acid sequence e.g., a Transforming Growth Factor ⁇ Receptor II (TGFBR2) gene
- TGFBR2 Transforming Growth Factor ⁇ Receptor II
- Adoptive T cell transfer utilizing genetically modified T cells has entered clinical testing as a therapeutic for solid and hematologic malignancies.
- phase I and II trials involving hematologic malignancies e.g., lymphoma, Chronic Lymphocytic Leukemia (CLL) and Acute Lymphocytic Leukemia (ALL)
- CLL Chronic Lymphocytic Leukemia
- ALL Acute Lymphocytic Leukemia
- the efficacy of adoptive T cell therapies in solid tumor patients may be influenced by a number of factors, such as: (1) T cell proliferation, e.g., limited proliferation of T cells following adoptive transfer; (2) T cell survival, e.g., induction of T cell apoptosis by factors in the target cell, e.g., cancer cell, environment; and (3) T cell function, e.g., inhibition of cytotoxic T cell function by inhibitory factors secreted by host immune cells and target cells, e.g., cancer cells.
- T cell proliferation e.g., limited proliferation of T cells following adoptive transfer
- T cell survival e.g., induction of T cell apoptosis by factors in the target cell, e.g., cancer cell, environment
- T cell function e.g., inhibition of cytotoxic T cell function by inhibitory factors secreted by host immune cells and target cells, e.g., cancer cells.
- TGF- ⁇ transforming growth factor ⁇
- Regs regulatory T cells
- TGFBR2 is a receptor for TGF- ⁇ that is expressed on a myriad of cell types including immune cells. Binding of TGF- ⁇ by TGFBR2 has been demonstrated to down-regulate T cell activation, proliferation and differentiation. Development of TGFBR2 inhibitors that may ameliorate TGFBR2 activity on tumor reactive T cells has been complicated by the lack of pre -clinical mouse models due to the severe autoimmune phenotype observed in mice containing T cells engineered to conditionally lack TGFBR2. Consequently, a need exists for effective strategies to reduce or eliminate the T cell inhibitory impact of TGF- ⁇ , including in the context of T cell mediated immunotherapy.
- CRISPRs Clustered Regularly Interspaced Short Palindromic Repeats
- RNA is transcribed from a portion of the CRISPR locus that includes the viral sequence. That RNA, which contains sequence complementary to the viral genome, mediates targeting of an RNA-guided nuclease to a target sequence in the viral genome. The RNA-guided nuclease, in turn, cleaves and thereby silences the viral target.
- CRISPR/Cas9 system has been adapted for genome editing in eukaryotic cells.
- the introduction of site-specific double strand breaks (DSBs) allows for target sequence alteration through endogenous DNA repair mechanisms, for example non-homologous end -joining (NHEJ) or homology-directed repair (HDR).
- NHEJ non-homologous end -joining
- HDR homology-directed repair
- CRISPR/Cas9 represents a promising avenue for addressing TGF- ⁇ -mediated inhibition of T-cells in the context of tumor therapy, but to date no viable approaches for addressing this issue in T-cells for use in tumor therapy have been identified.
- RNA-guided nuclease protein complexed with a gRNA targeting the TGFBR2 gene a ribonucleoprotein
- the alteration in TGFBR2 expression occurs as a result of a double-stranded break induced by the RNP and subsequent imperfect repair that leads to indels at and/or adjacent to the targeted TGFBR2 sequence.
- the instant disclosure relates to genome editing systems that include a guide RNA with a targeting domain that is complementary to target sequence of a TGFBR2 gene and where the RNA-guided nuclease is a Cas9 nuclease.
- the targeting domain may be 70%, 80%, 85%, 90%, 95%, or 100% complementary.
- the targeting domain has a length of 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, or 26 nucleotides. [0011] In certain embodiments, the targeting domain has at least 18 contiguous nucleotides that are complementary to the TGFBR2 gene.
- the targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096.
- the targeting domain is configured to form a double strand break or a single strand break within about 500bp, about 450bp, about 400bp, about 350bp, about 300bp, about 250bp, about 200bp, about 150bp, about lOObp, about 50bp, about 25bp, or about lObp of the TGFBR2 target position.
- the genome editing system is capable of altering TGFBR2 gene by knocking out the expression of the TGFBR2 gene or knocking down the expression of the TGFBR2 gene.
- the genome editing systems disclosed herein incorporate a gRNA comprising a targeting domain configured to target a coding region or a non-coding region of the TGFBR2 gene, wherein said non-coding region comprises a promoter region, an enhancer region, an intron, the 3' UTR, the 5' UTR, or a polyadenylation signal region of said TGFBR2 gene; and the coding region comprises, e.g., an early coding region of said TGFBR2 gene.
- the genome editing systems disclosed herein have a target sequence of the TGFBR2 gene comprising the sequence selected from the group consisting of SEQ ID NOs: 1, 2, and 3.
- the genome editing systems disclosed herein have a target sequence of the TGFBR2 gene comprising the sequence selected from the group consisting of SEQ ID NOs: 4 to 10.
- the genome editing systems disclosed herein incorporate a targeting domain comprising a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096.
- the targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from the group consisting of: (a) SEQ ID NO: 5041; (b) SEQ ID NO: 5042; (c) SEQ ID NO: 5047; (d) SEQ ID NO: 5050; (e) SEQ ID NO: 5052; (f) SEQ ID NO: 5092; and (g) SEQ ID NO: 5093.
- the genome editing system incorporates pairs of gRNA molecules, including, e.g., gRNA pairs having target sequences SEQ ID NOS: 5042 and 5041, or 5042 and 5092, or SEQ ID NOS: 5042 and 5093, or SEQ ID NOS: 5093 and 5041.
- the present disclosure relates to a composition
- a composition comprising a gRNA molecule comprising a targeting domain that is complementary with a target sequence of a TGFBR2 gene.
- the composition comprises one, two, three, or four gRNA molecules.
- the composition further comprises an RNA- guided nuclease, e.g., a Cas9 molecule.
- the targeting domain incorporated into such compositions comprises a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096.
- the targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from the group consisting of: (a) SEQ ID NO: 5041 ; (b) SEQ ID NO: 5042; (c) SEQ ID NO: 5047; (d) SEQ ID NO: 5050; (e) SEQ ID NO: 5052; (f) SEQ ID NO: 5092; and (g) SEQ ID NO: 5093.
- the composition incorporates pairs of gRNA molecules, including, e.g., gRNA pairs having target sequences SEQ ID NOS: 5042 and 5041, or 5042 and 5092, or SEQ ID NOS: 5042 and 5093, or SEQ ID NOS: 5093 and 5041.
- the present disclosure relates to a vector encoding a gRNA molecule comprising a targeting domain that is complementary with a target sequence of a TGFBR2 gene.
- the vector further encodes for an RNA-guided nuclease, e.g., a Cas9 molecule.
- the targeting domain of the gRNA encoded by the vector comprises a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096.
- the targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than 3 nucleotides from, a nucleotide sequence selected from the group consisting of: (a) SEQ ID NO: 5041 ; (b) SEQ ID NO: 5042; (c) SEQ ID NO: 5047; (d) SEQ ID NO: 5050; (e) SEQ ID NO: 5052; (f) SEQ ID NO: 5092; and (g) SEQ ID NO: 5093.
- the vector is a viral vector.
- the vector is an adeno-associated virus (AAV) vector or a lentivirus (LV) vector.
- the present disclosure is directed to a method of altering a TGFBR2 gene in a cell, comprising administering to said cell one of: (i) a genome editing system comprising a gRNA molecule comprising a targeting domain that is complementary with a target sequence of said TGFBR2 gene, and a Cas9 molecule; (ii) a vector comprising a polynucleotide encoding a gRNA molecule comprising a targeting domain that is complementary with a target sequence of said TGFBR2 gene, and a polynucleotide encoding a Cas9 molecule; or (iii) a composition comprising a gRNA molecule comprising a targeting domain that that is complementary with a target sequence of said TGFBR2 gene, and a Cas9 molecule.
- the present disclosure is directed to a cell comprising a genome editing system as described herein, a gRNA composition of as described herein, or a vector as described herein.
- the cell expresses TGFBR2.
- the cell is a T cell.
- the present disclosure is directed to a gRNA and a RNA-guided nuclease comprising a ribonucleoprotein (RNP) complex.
- RNP ribonucleoprotein
- the present disclosure is directed to administering to a cell two or more RNP complexes comprising gRNAs with different targeting domains.
- the present disclosure is directed to RNP complexes comprising enzymatically active Cas9 (eaCas9) nucleases.
- the present disclosure is directed to RNP complexes comprising eaCas9 nucleases that form double strand breaks in a target nucleic acid or single strand breaks in a target nucleic acid.
- the present disclosure is directed to two RNP complexes comprising distinct gRNAs are used to form offset single strand breaks in the TGFBR2 gene in a cell.
- the present disclosure is directed to a cell that is a T cell or a Natural Killer (NK) cell.
- the cell further comprises an engineered T cell receptor (eTCR) or a chimeric antigen receptor (CAR).
- eTCR engineered T cell receptor
- CAR chimeric antigen receptor
- the present disclosure is directed to an RNA-guided nuclease- mediated method of altering TGFBR2 gene expression in a cell comprising: a) contacting the cell with a sufficient amount of a gRNA that targets TGFBR2 and an RNA-guided nuclease; and b) forming a first DNA double strand break near a TGFBR2 target position in a TGFBR2 gene of the cell, wherein the first DNA double strand break is repaired by NHEJ, wherein said repair alters the expression of the TGFBR2 gene.
- the present disclosure is directed to forming a second DNA double strand break near the TGFBR2 target position.
- a first double strand break is formed within about 500bp, about 450bp, about 400bp, about 350bp, about 300bp, about 250bp, about 200bp, about 150bp, about lOObp, about 50bp, about 25bp, or about lObp of the TGFBR2 target position.
- the first and second double strand breaks are formed within about 500bp, about 450bp, about 400bp, about 350bp, about 300bp, about 250bp, about 200bp, about 150bp, about lOObp, about 50bp, about 25bp, or about lObp of the TGFBR2 target position.
- the first double strand break is formed in a coding region or a non-coding region of said TGFBR2 gene, wherein said non-coding region comprises a promoter region, an enhancer region, an intron, a 3 ' UTR, a 5 ' UTR, or a polyadenylation signal region of said TGFBR2 gene.
- the first and second double strand breaks are formed in a coding region or a non- coding region of said TGFBR2 gene, wherein said non-coding region comprises a promoter region, an enhancer region, an intron, a 3 ' UTR, a 5' UTR, or a polyadenylation signal region of said TGFBR2 gene.
- the coding region is selected from exon 3, exon 4, and exon 5.
- the targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than about 3 nucleotides from, a nucleotide sequence selected from the group consisting of SEQ ID NOS: 5036 to 5096.
- the RNA-guided nuclease is an S. pyogenes Cas9 nuclease
- said targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than about 3 nucleotides from, a nucleotide sequence selected from the group consisting of: (a) SEQ ID NO: 5041 ; (b) SEQ ID NO: 5042; (c) SEQ ID NO: 5047; (d) SEQ ID NO: 5050; (e) SEQ ID NO: 5052; (f) SEQ ID NO: 5092; and (g) SEQ ID NO: 5093.
- the RNA-guided nuclease is an S. aureus Cas9 nuclease.
- the RNA-guided nuclease is a mutant Cas9 nuclease.
- the NHEJ repair produces an insertion or deletion with a frequency of greater than or equal to 20%.
- the insertion or deletion frequency is greater than or equal to 30%, 40%, or 50%.
- the present disclosure is directed to a genome engineered cell comprising an insertion or deletion near or at a target position of a TGFBR2 gene, wherein said target position comprises a nucleotide sequence that is complementary to, or differs by no more than about 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096.
- the insertion or deletion is within about 500bp, about 450bp, about 400bp, about 350bp, about 300bp, about 250bp, about 200bp, about 150bp, about lOObp, about 50bp, about 25bp, or about lObp of the TGFBR2 target position.
- the cell is a T cell or NK cell. In certain embodiments, the cell further comprises a eTCR or CAR.
- the present disclosure is directed to a composition
- a composition comprising: a) a population of genome engineered cells comprising an insertion or deletion near or at a target position of a TGFBR2 gene, wherein said target position comprises a nucleotide sequence that is complementary to, or differs by no more than about 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096; and b) a pharmaceutically acceptable buffer.
- the population of cells comprises T cells or NK cells.
- the cell further comprises a eTCR or CAR.
- the present disclosure is directed to a method of treating cancer in subject, comprising administering to the subject engineered immune cells, wherein the engineered immune cells have reduced expression of TGFBR2, and optionally expresses an engineered T Cell Receptor (eTCR) or a Chimeric Antigen Receptor (CAR), wherein the engineered immune cells have an insertion or a deletion near or at a target position of the TGFBR2 gene.
- eTCR engineered T Cell Receptor
- CAR Chimeric Antigen Receptor
- the engineered immune cells comprise T cells or NK cells.
- the cell further comprises a eTCR or CAR.
- the cancer is selected from the group consisting of: leukemia, lymphoma, e.g., chronic lymphocytic leukemia (CLL), acute-lymphoblastic leukemia (ALL), non- Hodgkin's lymphoma, acute myeloid leukemia, multiple myeloma, refractory follicular lymphoma, mantle cell lymphoma, indolent B cell lymphoma, B cell malignancies, cancers of the colon, lung, liver, breast, prostate, ovarian, skin, melanoma, bone, and brain cancer, ovarian cancer, epithelial cancers, renal cell carcinoma, pancreatic adenocarcinoma, Hodgkin's lymphoma, cervical carcinoma, colorectal cancer, glioblastoma, neuroblastoma, Ewing's sarcoma, medulloblastoma, osteosarcoma, synovial sarcom
- CLL chronic lymphoc
- the T cells are CD4+ and/or CD8+ T cells.
- the engineered immune cells maintain or have enhanced lysis activity against a target cancer cell relative to a non-engineered immune cell.
- the engineered immune cells maintain or have increased expression of granzyme B and/or interferon gamma in the presence of TGFp relative to non-engineered immune cells.
- the engineered immune cells maintain or have improved persistence against repeated antigen stimulation relative to non-engineered immune cells.
- the engineered immune cells maintain or have increased expression of CD25 relative to non-engineered immune cells.
- the engineered immune cells maintain or have decreased expression of PD-1 relative to non-engineered immune cells.
- the engineered immune cells maintain or have increased proliferation relative to non-engineered immune cells.
- the present disclosure is directed to a composition comprising a plurality of engineered T cells, wherein said engineered T cells exhibit reduced TGFBR2 gene expression relative to non-engineered T cells.
- the engineered T cells exhibit a TGFBR2 gene expression level that is about 50%, about 40%, about 30%, about 20%, about 10% or about 5% the level of TGFBR2 expression in non-engineered T cells.
- the engineered T cells further comprise expression of an eTCR or a CAR.
- the T cells are CD4+ T cells and/or CD8+ T cells.
- the engineered T cells are further characterized by possessing: a) enhanced lysis activity against a target cancer cell relative to non-engineered T cells; b) maintained or increased expression of granzyme B and/or interferon gamma in the presence of TGFp relative to non- engineered T cells; c) maintained or increased persistence against repeated antigen stimulation relative to non-engineered T cells; d) maintained or increased expression of CD25 relative to non- engineered T cells; e) maintained or decreased expression of PD-1 relative to non- engineered T cells; and/or f) maintained or increased proliferation relative to non-engineered T cells.
- the present disclosure is directed to a composition
- a composition comprising a plurality of engineered T cells, wherein said engineered T cells exhibit reduced TGFBR2 gene expression relative to non-engineered T cells, said engineered T cells produced by contacting non- engineered T cells with a genome editing system comprising: a gRNA comprising a targeting domain that is complementary with a target sequence of a TGFBR2 gene; and an RNA-guided nuclease.
- the engineered T cells are further transduced with a vector that expresses an eTCR or a CAR.
- the vector is a viral vector.
- the viral vector is an adeno-associated virus (AAV) vector or a lentivirus (LV) vector.
- the RNA-guided nuclease is an S. pyogenes Cas9 nuclease
- said targeting domain comprises a nucleotide sequence that is identical to, or differs by no more than about 3 nucleotides from, a nucleotide sequence selected from the group consisting of: (a) SEQ ID NO: 5041 ; (b) SEQ ID NO: 5042; (c) SEQ ID NO: 5047; (d) SEQ ID NO: 5050; (e) SEQ ID NO: 5052; (f) SEQ ID NO: 5092; and (g) SEQ ID NO: 5093.
- the present disclosure is directed to a composition comprising a plurality of engineered T cells, wherein said engineered T cells are deficient in TGFBR2 signaling.
- the deficient TGFBR2 signaling is mediated by expressing a Dominant Negative (DN) form of the TGFBR2 in said engineered T cells.
- DN Dominant Negative
- the engineered T cells are further transduced with a vector that expresses an eTCR or a CAR.
- the vector is a viral vector.
- the viral vector is an adeno-associated virus (AAV) vector or a lentivirus (LV) vector.
- the T cells are CD4+ T cells and/or CD8+ T cells.
- the engineered T cells are further characterized by possessing: a) enhanced lysis activity against a target cancer cell relative to non-engineered T cells; b) maintained or increased expression of granzyme B and/or interferon gamma in the presence of TGFp relative to non- engineered T cells; c) maintained or increased persistence against repeated antigen stimulation relative to non-engineered T cells; d) maintained or increased expression of CD25 relative to non- engineered T cells; e) maintained or decreased expression of PD-1 relative to non- engineered T cells; and/or f) maintained or increased proliferation relative to non-engineered T cells.
- the engineered immune cells further comprise reduced expression of wild-type TGFBR2.
- the wild-type TGFBR2 expression is reduced by contacting the engineered immune cells with a genome editing system comprising: a gRNA comprising a targeting domain that is complementary with a target sequence of a TGFBR2 gene; and an RNA-guided nuclease.
- the present disclosure is directed to a ribonucleoprotein (RNP) complex comprising a gRNA that comprises a targeting domain that is complementary with a target sequence of a TGFBR2 gene and an RNA-guided nuclease.
- RNP ribonucleoprotein
- the RNP of claim 117, wherein the RNA-guided nuclease is a Cas9 nuclease.
- the RNP of claim 117, wherein the RNP is electroporated into cells are electroporated into cells.
- Headings including numeric and alphabetical headings and subheadings, are for organization and presentation and are not intended to be limiting.
- Fig. 1 depicts a list of exemplary gRNAs targeting Exons 1A-5 of Transforming Growth Factor ⁇ Receptor II (TGFBR2) and their associated % Non-Homologous End Joining (NHEJ) activity.
- TGFBR2 Transforming Growth Factor ⁇ Receptor II
- NHEJ Non-Homologous End Joining
- Fig. 2 depicts the genome editing efficiency of certain exemplary gRNA pairs.
- Fig. 3 depicts % indel formation as determined by miSeq in the TGFBR2 gene. Seven gRNAs were tested at various concentrations of RNPs to produce a dose response curve.
- Fig. 4A-Fig. 4B depicts % edited cells (Fig. 4A) and % indel frequency (Fig. 4B) with several tested gRNAs targeting TGFBR2 and a control gRNA targeting the AAVSl locus.
- Fig. 5 depicts a comparison of two TGFBR2 targeting gRNAs in their ability to produce out- of-frame indel mutations.
- Fig. 6 depicts relative IFN-gamma production in primary T cells transduced to express anti- BCMA CAR in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp. Single or paired gRNAs were compared.
- Fig. 7 depicts relative cell proliferation in primary T cells transduced to express anti-BCMA CAR in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp. Single or paired gRNAs were compared.
- Fig. 8A-Fig. 8B depicts CD25 (Fig. 8A) and PD-1 (Fig. 8B) expression in primary T cells transduced to express anti- BCMA CAR in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp. Single or paired gRNAs were compared. The cells were stimulated with RPMI 8226 cells for 48 hours as well.
- Fig. 9 depicts Smad 2/3 phosphorylation in T cells transduced to express different CARs in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp.
- Fig. 10 depicts relative Granzyme B expression in T cells transduced to express different CARs in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp.
- Fig. 11 depicts IFN-gamma production in T cells transduced to express different CARs in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp.
- Fig. 12 depicts relative T cell proliferation in T cells transduced to express different CARs in a TGFBR2 gene edited background, with (lOng/ml) or without TGFp.
- Fig. 13A-Fig. 13B depicts Granzyme B (Fig. 13 A) and Interferon gamma (Fig. 13B) production in primary T cells transduced to express anti- BCMA CAR in a TGFBR2 gene edited or TGFBR2 DN background, with (lOng/ml) or without TGFp.
- Fig. 14 depicts relative % lysis of RPMI 8226 cells by anti-BCMA CAR-expressing T cells in a TGFBR2 gene edited or TGFBR2 DN background, with (lOng/ml) or without TGFp.
- Fig. 15A-Fig. 15F depicts anti-BCMA CAR-expressing T cells in a TGFBR2 unedited, TGFBR2 gene edited, or TGFBR2 DN background with repeated TGFp antigen stimulation.
- Fig. 15A-Fig. 15C show the projected cell number after stimulation with three different antigens.
- Fig. 15D-Fig. 15F show the % anti-BCMA CAR-expressing T cells over time with repeated TGFp antigen stimulation.
- Fig. 16A-Fig. 16B depicts INF-gamma production in primary T cells transduced to express anti-BCMA CAR in a TGFBR2 gene edited or TGFBR2 DN background, with (lOng/ml) or without TGFp.
- the T cells were co-incubated with either RPMI 8226 cells (Fig. 16A) or OPM2 cells (Fig. 16B).
- Fig. 17A-Fig. 17B depicts CD25 expression in primary T cells transduced to express anti- BCMA CAR in a TGFBR2 gene edited or TGFBR2 DN background, with (lOng/ml) or without TGFp.
- the T cells were co-incubated with either RPMI 8226 cells (Fig. 17A) or OPM2 cells (Fig. 17B).
- Fig. 18A-Fig. 18B depicts PD-1 expression in primary T cells transduced to express anti- BCMA CAR in a TGFBR2 gene edited or TGFBR2 DN background, with (lOng/ml) or without TGFp.
- the T cells were co-incubated with either RPMI 8226 cells (Fig. 18A) or OPM2 cells (Fig. 18B).
- Fig. 19A-Fig. 19B depicts relative cell proliferation (Fig. 19A) and relative IFN-gamma production (Fig. 19B) of anti-BCMA CAR-expressing T cells in a TGFBR2 gene edited or TGFBR2 DN background, with (lOng/ml) or without TGFp.
- the cells were stimulated with RPMI 8226 cells as well.
- Fig. 20A-Fig. 20B depicts % of PD-1 + cells in several CD4+ (Fig. 20A) and CD8+ (Fig. 20B) anti-BCMA CAR-expressing T cells in a TGFBR2 gene edited or TGFBR2 DN background, with increasing concentrations of TGFp.
- the cells were stimulated with RPMI 8226 cells for 48 hours as well.
- Fig. 21 depicts the expression level of TGFp signaling pathway genes PMEPA1, SKIL, SKI, and LDLRAD4 in anti-BCMA CAR-expressing T cells in a TGFBR2 gene edited or TGFBR2 DN background. T cells were isolated from the mouse spleen or from the RPMI 8226 cell-derived tumor.
- Fig. 22 depicts Smad 2/3 phosphorylation levels, proliferation, and Granzyme B expression in anti-CD 19 CAR-expressing T cells in a TGFBR2 DN background, with and without TGFp.
- Fig. 23 depicts a schematic for determining if TGFBR2 gene-editing in T cells confers a selective advantage over wild-type cells.
- Fig. 24 depicts different ratios of anti-BCMA CAR, TGFBR2-KO cells with WT cells (1 , 0.75, 0.5, 0.25).
- the cells were co-cultured with RPMI8226 cells in a 1 : 1 Effector to Target ratio ⁇ lOng/ml TGFp.
- Cells were collected every 7 days for analyzing the % of indel by high throughput sequencing. Cells were re-stimulated with fresh RPMI8226 and re-adjusted to 1 : 1 Effector to Target ratio weekly.
- a module means at least one module, or one or more modules.
- compositions that consisting essentially of means that the species recited are the predominant species, but that other species may be present in trace amounts or amounts that do not affect structure, function or behavior of the subject composition.
- a composition that consists essentially of a particular species will generally comprise 90%, 95%, 96%, or more of that species.
- Domain is used to describe a segment of a protein or nucleic acid. Unless otherwise indicated, a domain is not required to have any specific functional property.
- An "indel” is an insertion and/or deletion in a nucleic acid sequence.
- An indel may be the product of the repair of a DNA double strand break, such as a double strand break formed by a genome editing system of the present disclosure.
- An indel is most commonly formed when a break is repaired by an "error prone” repair pathway such as the NHEJ pathway described below.
- An indel may produce insertions or deletions creating in-frame or out-of-frame mutations in the target sequence.
- "Gene conversion” refers to the alteration of a DNA sequence by incorporation of an endogenous homologous sequence (e.g., a homologous sequence within a gene array).
- Gene correction refers to the alteration of a DNA sequence by incorporation of an exogenous homologous sequence, such as an exogenous single-or double stranded donor template DNA. Gene conversion and gene correction are products of the repair of DNA double-strand breaks by HDR pathways such as those described below.
- DNA samples for sequencing may be prepared by a variety of methods known in the art, and may involve the amplification of sites of interest by polymerase chain reaction (PCR), the capture of DNA ends generated by double strand breaks, as in the GUIDEseq process described in Tsai et al. (Nat. Biotechnol.
- Genome editing outcomes may also be assessed by in situ hybridization methods such as the FiberCombTM system commercialized by Genomic Vision (Bagneux, France), and by any other suitable methods known in the art.
- Alt-HDR Alternative homology-directed repair
- alternative HDR alternative HDR
- canonical HDR canonical HDR in that the process utilizes different pathways from canonical HDR, and can be inhibited by the canonical HDR mediators, RAD51 and BRCA2.
- Alt-HDR is also distinguished by the involvement of a single-stranded or nicked homologous nucleic acid template, whereas canonical HDR generally involves a double- stranded homologous template.
- Canonical HDR refers to the process of repairing DNA damage using a homologous nucleic acid (e.g., an endogenous homologous sequence, e.g., a sister chromatid, or an exogenous nucleic acid, e.g., a template nucleic acid).
- Canonical HDR typically acts when there has been significant resection at the double strand break, forming at least one single stranded portion of DNA.
- HDR In a normal cell, cHDR typically involves a series of steps such as recognition of the break, stabilization of the break, resection, stabilization of single stranded DNA, formation of a DNA crossover intermediate, resolution of the crossover intermediate, and ligation. The process requires RAD51 and BRCA2, and the homologous nucleic acid is typically double- stranded.
- HDR as used herein encompasses both canonical HDR and alt-HDR.
- Non-homologous end joining refers to ligation mediated repair and/or non- template mediated repair including canonical NHEJ (cNHEJ) and alternative NHEJ (altNHEJ), which in turn includes microhomology-mediated end joining (MMEJ), single-strand annealing (SSA), and synthesis-dependent microhomology-mediated end joining (SD-MMEJ).
- cNHEJ canonical NHEJ
- altNHEJ alternative NHEJ
- MMEJ microhomology-mediated end joining
- SSA single-strand annealing
- SD-MMEJ synthesis-dependent microhomology-mediated end joining
- Replacement when used with reference to a modification of a molecule (e.g., a nucleic acid or protein), does not require a process limitation but merely indicates that the replacement entity is present.
- Subject means a human or non-human animal.
- a human subject can be any age (e.g., an infant, child, young adult, or adult), and may suffer from a disease, or may be in need of alteration of a gene.
- the subject may be an animal, which term includes, but is not limited to, mammals, birds, fish, reptiles, amphibians, and more particularly non-human primates, rodents (such as mice, rats, hamsters, etc.), rabbits, guinea pigs, dogs, cats, and so on.
- the subject is livestock, e.g., a cow, a horse, a sheep, or a goat.
- the subject is poultry.
- Treatment mean the treatment of a disease in a subject (e.g., a human subject), including one or more of inhibiting the disease, i.e., arresting or preventing its development or progression; relieving the disease, i.e., causing regression of the disease state; relieving one or more symptoms of the disease; and curing the disease.
- a subject e.g., a human subject
- Prevent refers to the prevention of a disease in a mammal, e.g., in a human, including (a) avoiding or precluding the disease; (b) affecting the predisposition toward the disease; or (c) preventing or delaying the onset of at least one symptom of the disease.
- kits refers to any collection of two or more components that together constitute a functional unit that can be employed for a specific purpose.
- one kit according to this disclosure can include a guide RNA complexed or able to complex with an RNA-guided nuclease, and accompanied by (e.g., suspended in, or suspendable in) a pharmaceutically acceptable carrier.
- the kit can be used to introduce the complex into, for example, a cell or a subject, for the purpose of causing a desired genomic alteration in such cell or subject.
- the components of a kit can be packaged together, or they may be separately packaged.
- Kits according to this disclosure also optionally include directions for use (DFU) that describe the use of the kit e.g., according to a method of this disclosure.
- the DFU can be physically packaged with the kit, or it can be made available to a user of the kit, for instance by electronic means.
- the terms "polynucleotide,” “nucleotide sequence,” “nucleic acid,” “nucleic acid molecule,” “nucleic acid sequence,” and “oligonucleotide” refer to a series of nucleotide bases (also called “nucleotides”) in DNA and RNA, and mean any chain of two or more nucleotides.
- polynucleotides, nucleotide sequences, nucleic acids etc. can be chimeric mixtures or derivatives or modified versions thereof, single-stranded or double-stranded. They can be modified at the base moiety, sugar moiety, or phosphate backbone, for example, to improve stability of the molecule, its hybridization parameters, etc.
- a nucleotide sequence typically carries genetic information, including, but not limited to, the information used by cellular machinery to make proteins and enzymes. These terms include double- or single-stranded genomic DNA, RNA, any synthetic and genetically manipulated polynucleotide, and both sense and antisense polynucleotides. These terms also include nucleic acids containing modified bases.
- protein protein
- peptide and “polypeptide” are used interchangeably to refer to a sequential chain of amino acids linked together via peptide bonds.
- the terms include individual proteins, groups or complexes of proteins that associate together, as well as fragments or portions, variants, derivatives and analogs of such proteins.
- Peptide sequences are presented herein using conventional notation, beginning with the amino or N-terminus on the left, and proceeding to the carboxyl or C-terminus on the right. Standard one-letter or three-letter abbreviations can be used.
- variant refers to an entity such as a polypeptide, polynucleotide or small molecule that shows significant structural identity with a reference entity but differs structurally from the reference entity in the presence or level of one or more chemical moieties as compared with the reference entity. In many embodiments, a variant also differs functionally from its reference entity. In general, whether a particular entity is properly considered to be a "variant" of a reference entity is based on its degree of structural identity with the reference entity.
- the genome editing systems described herein generally include one or more gRNAs comprising targeting domains that are complimentary to one or more TGFBR2 target sequences, which target sequences, in turn, include or are adjacent to protospacer adjacent motif (PAM) sequences recognized by one or more RNA-guided nucleases to which the one or more gRNAs can bind (e.g., complexed).
- PAM protospacer adjacent motif
- the genome editing systems of this disclosure are directed, in a site-specific manner, to the one or more TGFBR2 target sequences, and operate to introduce an alteration within or proximate to those TGFBR2 target sequences.
- Alterations introduced into, or proximate to, the TGFBR2 target sites by the genome editing systems of this disclosure will, most commonly, include DNA single-strand breaks (SSBs or "nicks") and/or double strand breaks (DSBs).
- SSBs or nicks DNA single-strand breaks
- DSBs double strand breaks
- the genome editing systems introduce one or more of a point mutation (e.g., via cysteine deamination), a change in DNA marking (e.g., DNA methylation, histone acetylation or deacetylation, or other chromatin modifications), and/or recruitment of trans-acting factors such as transcription factors.
- a point mutation e.g., via cysteine deamination
- a change in DNA marking e.g., DNA methylation, histone acetylation or deacetylation, or other chromatin modifications
- trans-acting factors such as transcription factors.
- genome editing systems of this disclosure may associate, in a durable (e.g., over an interval of weeks, months or longer) or transient (over an interval of seconds, minutes, hours, or days) manner, with one or more TGFBR2 target sequences, thereby preventing association of other factors (particularly RNA polymerases, but also DNA polymerases, transcription factors, and/or other cis- or trans-acting factors that influence gene expression) with the TGFBR2 target sequences.
- the TGFBR2 target sequences and corresponding gRNA targeting domain sequences are generally, but not necessarily, located in exons, where the introduction of a small indel, or a larger insertion or deletion may result in one or more mutations (e.g., a frameshift mutation, a nonsense mutation, introduction of a codon for an amino acid that disrupts the structure of the surrounding protein, and/or removal of a codon for an amino acid that is necessary for protein activity) that reduce or eliminate function of the TGFBR2 protein.
- Fig. 1 shows a mapping of cutting activity of various S. pyogenes guide RNAs to the locations they target within the exon structure of the TGFBR2 gene.
- TGFBR2 target sequences may be considered "hot spot" target sites for gRNA targeting domain sequences.
- a "hot spot” refers to a site that is preferentially targeted because it produces high % indel frequencies or effective knock down or knock out of the TGFBR2 gene.
- gRNAs targeting one or more of these preferred sites may produce % indel frequencies of 30% or higher.
- a preferred target site in the TGFBR2 gene may have complementary gRNA targeting domains that produce % indel frequencies of 30%, 40%, 50%, 60%, 70%, 80%, 85%, 90%, 95%), or higher. Hot spot target sites within the TGFBR2 gene are described herein.
- Preferred hot spot TGFBR2 regions are shown in table x: Table 2: Preferred TGFBR2 target sites
- a TGFBR2 target sequence can be, for example, located in exon 3, 4, or 5 of the TGFBR2 gene.
- a gRNA targeting domain sequence corresponding to a TGFBR2 target sequence present in exon 3, 4, or 5 of the TGFBR2 gene can comprise a nucleotide sequence that is identical to, or differs by no more than 1, 2, or 3 nucleotides from, a nucleotide sequence selected from SEQ ID NOS: 5036 to 5096.
- an exemplary targeting domain can comprise a nucleotide sequence that is identical to, or differs by no more than 1, 2, or 3 nucleotides from a nucleotide sequence selected from the group consisting of:
- a transcriptional regulatory region e.g., a promoter region (e.g., a promoter region that controls the transcription of the TGFBR2 gene) can be targeted to alter (e.g., knock down) the expression of the gene.
- a targeted knockdown approach can be mediated by a CRISPR/Cas system comprising an enzymatically inactive Cas9 (eiCas9) molecule or an eiCas9 fusion protein (e.g., an eiCas9 fused to a transcription repressor domain or chromatin modifying protein), as described herein.
- one or more gRNA molecules comprising a targeting domain can be configured to target an eiCas9 molecule or an eiCas9 fusion protein, sufficiently close to a transcriptional regulatory region, e.g., a promoter region (e.g., a promoter region that controls the transcription of TGFBR2 gene), such that transcription of the TGFBR2 gene is reduced and/or eliminated.
- a promoter region e.g., a promoter region that controls the transcription of TGFBR2 gene
- an eiCas9 or an eiCas9 fusion protein can be used to knock down TGFBR2 expression in a T cell, e.g., a human T cell.
- TGFBR2 knock-out and/or knock down can be assessed in any suitable way, including without limitation, the examination of the sequence of the TGFBR2 gene, assessment of TGFBR2 protein expression on the surface of cells (e.g., by immunostaining and cell sorting, particularly by fluorescence activated cell sorting or FACS including indirect intracellular staining flow cytometry), detection of cellular or molecular changes mediated by TGFBR2, assessment of the effect of TGF- ⁇ on cell proliferation or survival, or by western blot to detect TGFBR2 protein levels.
- TGFBR2 knockout may be confirmed by (a) sequencing of the TGFBR2 locus or T7E1 primer extension assay, and/or (b) intracellular FACS assessment of SMAD2/3 phosphorylation. Sequencing and T7E1 are described in greater detail below, while intracellular SMAD2/3 phosphorylation assays are described in the literature, e.g., by Chen, et al., J. Experimental Med. Volume 198, Number 12, December 15, 2003 1875-1886 (which reference is incorporated by reference in its entirety and for all purposes herein), particularly the Materials & Methods section at page 1877 and Supplemental Figure S2.
- Knock out and/or knock down of TGFBR2 may correspond to a 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 85%, 90%, 95%, or 100% reduction in TGFBR2 expression relative to a baseline measurement or a wild-type cell.
- compositions and methods include those in which: at least or greater than about 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90% or 95% of cells in a composition of cells into which an agent (e.g. gRNA/Cas9) for knockout or genetic disruption of a TGFBR2 gene was introduced contain the genetic disruption; do not express the endogenous TGFBR2 polypeptide; do not contain a contiguous TGFBR2 gene, a TGFBR2 gene, and/or a functional TGFBR2 gene.
- an agent e.g. gRNA/Cas9
- the methods, compositions and cells according to the present disclosure include those in which at least or greater than about 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90% or 95% of cells in a composition of cells into which an agent (e.g. gRNA/Cas9) for knockout or genetic disruption of a TGFBR2 gene was introduced do not express a TGFBR2 polypeptide, such as on the surface of the cells.
- at least or greater than about 50%, 60%, 65%, 70%, 75%, 80%), 85%), 90%) or 95% of cells in a composition of cells into which an agent (e.g. gRNA/Cas9) for knockout or genetic disruption of a TGFBR2 gene was introduced are knocked out in both alleles, i.e. comprise a biallelic deletion, in such percentage of cells.
- Genome editing systems targeting TGFBR2 may be implemented in a variety of ways, and their implementation may be tailored to the setting in which cells will be edited. Certain embodiments of this disclosure involve the delivery of RNA-guided nucleases and guide RNAs targeting TGFBR2 to cells ex vivo in the form of ribonucleoprotein (RNP) complexes by means of electroporation, e.g., using electroporators and cuvettes available from commercial suppliers such as MaxCyte (Gaithersburg, MD) or Lonza (Basel, Switzerland). Other embodiments, however, may implement in vivo nucleic acids vectors, such as viral vectors or lipid nanoparticles, for either in vivo or ex vivo editing. Details of these implementations are described in greater detail below, under the heading "Implementation of genome editing systems.”
- TGFBR2 is knocked out in an immune cell, such as a T-cell, that will be used in therapy.
- the T-cell may express an engineered receptor such as a chimeric antigen receptor (CAR) or a heterologous T-cell receptor (TCR), which receptor may be configured to recognize an antigen on a cell or tissue that is implicated in a pathology such as a tumor.
- CAR chimeric antigen receptor
- TCR heterologous T-cell receptor
- TGFBR2 knockout T-cells may be employed in the targeting of a tissue or organ in which TGF- ⁇ is present in amounts sufficient to reduce the proliferation or activity of T cells expressing TGFBR2.
- TGFBR2 knock-out and/or knock down cells may be employed in "autologous" cell therapies, in which cells are harvested from a subject, altered to knock-out or knock-down TGFBR2 expression, and then returned to the same subject; alternatively, these cells may be administered to a different subject in an "allogeneic" cell therapy.
- autologous cell therapies in which cells are harvested from a subject, altered to knock-out or knock-down TGFBR2 expression, and then returned to the same subject; alternatively, these cells may be administered to a different subject in an "allogeneic" cell therapy.
- TGFBR2 cells of this disclosure may be manipulated in a variety of ways, such as expanded, stimulated, purified or sorted, transduced with a transgene, frozen and/or thawed.
- Knocking out or knocking down the presence of the TGFBR2 gene as described herein can: (1) improve T cell proliferation; (2) improve T cell survival; and/or (3) improve T cell function. Knocking down the expression of the TGFBR2 gene as described herein can similarly: (1) improve T cell proliferation; (2) improve T cell survival; and/or (3) improve T cell function.
- Genome editing system refers to any system having RNA-guided DNA editing activity.
- Genome editing systems of the present disclosure include at least two components adapted from naturally occurring CRISPR systems: a guide RNA (gRNA) and an RNA-guided nuclease. These two components form a complex that is capable of associating with a specific nucleic acid sequence and editing the DNA in or around that nucleic acid sequence, for instance by making one or more of a single-strand break (an SSB or nick), a double-strand break (a DSB) and/or a point mutation.
- gRNA guide RNA
- a RNA-guided nuclease RNA-guided nuclease
- the double strand or single strand break is within about 500bp, about 450bp, about 400bp, about 350bp, about 300bp, about 250bp, about 200bp, about 150bp, about lOObp, about 50bp, about 25bp, or about lObp of a TGFBR2 target position, thereby inducing an alteration in the expression of the TGFBR2 gene.
- Naturally occurring CRISPR systems are organized evolutionarily into two classes and five types (Makarova et al. Nat Rev Microbiol. 2011 Jun; 9(6): 467-477 (Makarova), incorporated by reference herein), and while genome editing systems of the present disclosure may adapt components of any type or class of naturally occurring CRISPR system, the embodiments presented herein are generally adapted from Class 2, and type II or V CRISPR systems.
- Class 2 systems which encompass types II and V, are characterized by relatively large, multidomain RNA-guided nuclease proteins (e.g., Cas9 or Cpfl) and one or more guide RNAs (e.g., a crRNA and, optionally, a tracrRNA) that form ribonucleoprotein (RNP) complexes that associate with (i.e., target) and cleave specific loci complementary to a targeting (or spacer) sequence of the crRNA.
- RNP ribonucleoprotein
- Genome editing systems similarly target and edit cellular DNA sequences, but differ significantly from CRISPR systems occurring in nature.
- the unimolecular guide RNAs described herein do not occur in nature, and both guide RNAs and RNA-guided nucleases according to this disclosure may incorporate any number of non-naturally occurring modifications.
- Genome editing systems can be implemented (e.g., administered or delivered to a cell or a subject) in a variety of ways, and different implementations may be suitable for distinct applications.
- a genome editing system is implemented, in certain embodiments, as a protein/RNA complex (a ribonucleoprotein, or RNP), which can be included in a pharmaceutical composition that optionally includes a pharmaceutically acceptable carrier and/or an encapsulating agent, such as a lipid or polymer micro- or nano-particle, micelle, liposome, etc.
- a genome editing system is implemented as one or more nucleic acids encoding the RNA-guided nuclease and guide RNA components described above (optionally with one or more additional components); in certain embodiments, the genome editing system is implemented as one or more vectors comprising such nucleic acids, for instance a viral vector such as an adeno-associated virus; and in certain embodiments, the genome editing system is implemented as a combination of any of the foregoing. Additional or modified implementations that operate according to the principles set forth herein will be apparent to the skilled artisan and are within the scope of this disclosure.
- the genome editing systems of the present disclosure can be targeted to a single specific nucleotide sequence, or may be targeted to— and capable of editing in parallel— two or more specific nucleotide sequences through the use of two or more guide RNAs.
- the use of multiple gRNAs is referred to as "multiplexing" throughout this disclosure, and can be employed to target multiple, unrelated target sequences of interest, or to form multiple SSBs or DSBs within a single target domain and, in some cases, to generate specific edits within such target domain.
- multiplexing can be employed to target multiple, unrelated target sequences of interest, or to form multiple SSBs or DSBs within a single target domain and, in some cases, to generate specific edits within such target domain.
- Maeder which is incorporated by reference herein, describes a genome editing system for correcting a point mutation (C.2991+1655A to G) in the human CEP290 gene that results in the creation of a cryptic splice site, which in turn reduces or eliminates the function of the gene.
- the genome editing system of Maeder utilizes two guide RNAs targeted to sequences on either side of (i.e., flanking) the point mutation, and forms DSBs that flank the mutation. This, in turn, promotes deletion of the intervening sequence, including the mutation, thereby eliminating the cryptic splice site and restoring normal gene function.
- Cotta-Ramusino et al.
- Cotta-Ramusino a genome editing system that utilizes two gRNAs in combination with a Cas9 nickase (a Cas9 that makes a single strand nick such as S.
- the dual-nickase system of Cotta-Ramusino is configured to make two nicks on opposite strands of a sequence of interest that are offset by one or more nucleotides, which nicks combine to create a double strand break having an overhang (5' in the case of Cotta-Ramusino, though 3 ' overhangs are also possible).
- the overhang in turn, can facilitate homology directed repair events in some circumstances.
- a gRNA targeted to a nucleotide sequence encoding Cas9 (referred to as a "governing RNA"), which can be included in a genome editing system comprising one or more additional gRNAs to permit transient expression of a Cas9 that might otherwise be constitutively expressed, for example in some virally transduced cells.
- governing RNA nucleotide sequence encoding Cas9
- Genome editing systems can, in some instances, form double strand breaks that are repaired by cellular DNA double-strand break mechanisms such as NHEJ or HDR.
- DNA double-strand break mechanisms such as NHEJ or HDR.
- genome editing systems operate by forming DSBs
- such systems optionally include one or more components that promote or facilitate a particular mode of double-strand break repair or a particular repair outcome.
- Cotta-Ramusino also describes genome editing systems in which a single stranded oligonucleotide "donor template" is added; the donor template is incorporated into a target region of cellular DNA that is cleaved by the genome editing system, and can result in a change in the target sequence.
- genome editing systems modify a target sequence, or modify expression of a gene in or near the target sequence, without causing single- or double-strand breaks.
- a genome editing system may include an RNA-guided nuclease fused to a functional domain that acts on DNA, thereby modifying the target sequence or its expression.
- an RNA-guided nuclease can be connected to (e.g., fused to) a cytidine deaminase functional domain, and may operate by generating targeted C-to-A substitutions. Exemplary nuclease/deaminase fusions are described in Komor et al.
- a genome editing system may utilize a cleavage-inactivated (i.e., a "dead”) nuclease, such as a dead Cas9 (dCas9), and may operate by forming stable complexes on one or more targeted regions of cellular DNA, thereby interfering with functions involving the targeted region(s) including, without limitation, mRNA transcription, chromatin remodeling, etc.
- a cleavage-inactivated nuclease such as a dead Cas9 (dCas9)
- dCas9 dead Cas9
- gRNA Guide RNA
- RNA-guide RNA and “gRNA” refer to any nucleic acid that promotes the specific association (or “targeting") of an RNA-guided nuclease such as a Cas9 or a Cpf 1 to a target sequence such as a genomic or episomal sequence in a cell.
- gRNAs can be unimolecular (comprising a single RNA molecule, and referred to alternatively as chimeric), or modular (comprising more than one, and typically two, separate RNA molecules, such as a crRNA and a tracrRNA, which are usually associated with one another, for instance by duplexing).
- gRNAs and their component parts are described throughout the literature, for instance in Briner et al. (Molecular Cell 56(2), 333-339, October 23, 2014 (Briner), which is incorporated by reference), and in Cotta-Ramusino.
- type II CRISPR systems generally comprise an RNA-guided nuclease protein such as Cas9, a CRISPR RNA (crRNA) that includes a 5' region that is complementary to a foreign sequence, and a trans-activating crRNA (tracrRNA) that includes a 5 ' region that is complementary to, and forms a duplex with, a 3 ' region of the crRNA. While not intending to be bound by any theory, it is thought that this duplex facilitates the formation of— and is necessary for the activity of— the Cas9/gRNA complex.
- Cas9 CRISPR RNA
- tracrRNA trans-activating crRNA
- Guide RNAs include a "targeting domain” that is fully or partially complementary to a target domain within a target sequence, such as a DNA sequence in the genome of a cell where editing is desired.
- Targeting domains are referred to by various names in the literature, including without limitation "guide sequences” (Hsu et al., Nat Biotechnol. 2013 Sep; 31 (9): 827-832, ("Hsu”), incorporated by reference herein), “complementarity regions” (Cotta- Ramusino), “spacers” (Briner) and generically as “crRNAs” (Jiang).
- targeting domains are typically 10-30 nucleotides in length, and in certain embodiments are 16-24 nucleotides in length (for instance, 16, 17, 18, 19, 20, 21, 22, 23 or 24 nucleotides in length), and are at or near the 5' terminus of in the case of a Cas9 gRNA, and at or near the 3' terminus in the case of a Cpf 1 gRNA.
- gRNAs typically (but not necessarily, as discussed below) include a plurality of domains that may influence the formation or activity of gRNA/Cas9 complexes.
- the duplexed structure formed by first and secondary complementarity domains of a gRNA also referred to as a repeat: anti-repeat duplex
- REC recognition
- Cas9/gRNA complexes the duplexed structure formed by first and secondary complementarity domains of a gRNA (also referred to as a repeat: anti-repeat duplex) interacts with the recognition (REC) lobe of Cas9 and can mediate the formation of Cas9/gRNA complexes.
- first and/or second complementarity domains may contain one or more poly- A tracts, which can be recognized by RNA polymerases as a termination signal.
- the sequence of the first and second complementarity domains are, therefore, optionally modified to eliminate these tracts and promote the complete in vitro transcription of gRNAs, for instance through the use of A-G swaps as described in Briner, or A-U swaps.
- Cas9 gRNAs typically include two or more additional duplexed regions that are involved in nuclease activity in vivo but not necessarily in vitro.
- a first stem-loop one near the 3' portion of the second complementarity domain is referred to variously as the "proximal domain,” (Cotta-Ramusino) "stem loop 1" (Nishimasu 2014 and 2015) and the “nexus” (Briner).
- One or more additional stem loop structures are generally present near the 3 ' end of the gRNA, with the number varying by species: s.
- pyogenes gRNAs typically include two 3' stem loops (for a total of four stem loop structures including the repeat: anti-repeat duplex), while s. aureus and other species have only one (for a total of three stem loop structures).
- a description of conserved stem loop structures (and gRNA structures more generally) organized by species is provided in Briner.
- RNA-guided nucleases have been (or may in the future be) discovered or invented which utilize gRNAs that differ in some ways from those described to this point.
- Cpfl CRISPR from Prevotella and Franciscella 1
- a gRNA for use in a Cpfl genome editing system generally includes a targeting domain and a complementarity domain (alternately referred to as a "handle"). It should also be noted that, in gRNAs for use with Cpfl, the targeting domain is usually present at or near the 3 ' end, rather than the 5 ' end as described above in connection with Cas9 gRNAs (the handle is at or near the 5 ' end of a Cpfl gRNA).
- gRNAs can be defined, in broad terms, by their targeting domain sequences, and skilled artisans will appreciate that a given targeting domain sequence can be incorporated in any suitable gRNA, including a unimolecular or chimeric gRNA, or a gRNA that includes one or more chemical modifications and/or sequential modifications (substitutions, additional nucleotides, truncations, etc.). Thus, for economy of presentation in this disclosure, gRNAs may be described solely in terms of their targeting domain sequences.
- gRNA should be understood to encompass any suitable gRNA that can be used with any RNA-guided nuclease, and not only those gRNAs that are compatible with a particular species of Cas9 or Cpf 1.
- the term gRNA can, in certain embodiments, include a gRNA for use with any RNA-guided nuclease occurring in a Class 2 CRISPR system, such as a type II or type V or CRISPR system, or an RNA- guided nuclease derived or adapted therefrom.
- Table 2 below, provides exemplary gRNAs for targeting TGFBR2 with an S. pyogenes Cas9.
- gRNA design may involve the use of a software tool to optimize the choice of potential target sequences corresponding to a user's target sequence, e.g., to minimize total off-target activity across the genome. While off-target activity is not limited to cleavage, the cleavage efficiency at each off -target sequence can be predicted, e.g., using an experimentally-derived weighting scheme. These and other guide selection methods are described in detail in Maeder and Cotta-Ramusino. gRNA modifications
- gRNAs can be altered through the incorporation of certain modifications.
- transiently expressed or delivered nucleic acids can be prone to degradation by, e.g., cellular nucleases.
- the gRNAs described herein can contain one or more modified nucleosides or nucleotides which introduce stability toward nucleases. While not wishing to be bound by theory it is also believed that certain modified gRNAs described herein can exhibit a reduced innate immune response when introduced into cells.
- Those of skill in the art will be aware of certain cellular responses commonly observed in cells, e.g., mammalian cells, in response to exogenous nucleic acids, particularly those of viral or bacterial origin. Such responses, which can include induction of cytokine expression and release and cell death, may be reduced or eliminated altogether by the modifications presented herein.
- Certain exemplary modifications discussed in this section can be included at any position within a gRNA sequence including, without limitation at or near the 5' end (e.g., within 1- 10, 1-5, or 1-2 nucleotides of the 5' end) and/or at or near the 3 ' end (e.g., within 1 -10, 1-5, or 1-2 nucleotides of the 3 ' end).
- modifications are positioned within functional motifs, such as the repeat- anti-repeat duplex of a Cas9 gRNA, a stem loop structure of a Cas9 or Cpf 1 gRNA, and/or a targeting domain of a gRNA.
- the 5' end of a gRNA can include a eukaryotic mRNA cap structure or cap analog (e.g., a G(5 ')ppp(5 ')G cap analog, a m7G(5 ')ppp(5 ')G cap analog, or a 3 '-0-Me- m 7G(5 ')ppp(5 ')G anti reverse cap analog (ARCA)), as shown below:
- a eukaryotic mRNA cap structure or cap analog e.g., a G(5 ')ppp(5 ')G cap analog, a m7G(5 ')ppp(5 ')G cap analog, or a 3 '-0-Me- m 7G(5 ')ppp(5 ')G anti reverse cap analog (ARCA)
- the cap or cap analog can be included during either chemical synthesis or in vitro transcription of the gRNA.
- the 5' end of the gRNA can lack a 5' triphosphate group.
- in vitro transcribed gRNAs can be phosphatase-treated (e.g., using calf intestinal alkaline phosphatase) to remove a 5' triphosphate group.
- polyA tract can be added to a gRNA during chemical synthesis, following in vitro transcription using a polyadenosine polymerase (e.g., E. coli Poly(A)Polymerase), or in vivo by means of a polyadenylation sequence, as described in Maeder.
- a polyadenosine polymerase e.g., E. coli Poly(A)Polymerase
- a gRNA whether transcribed in vivo from a DNA vector, or in vitro transcribed gRNA, can include either or both of a 5' cap structure or cap analog and a 3 ' polyA tract.
- Guide RNAs can be modified at a 3 ' terminal U ribose.
- the two terminal hydroxyl groups of the U ribose can be oxidized to aldehyde groups and a concomitant opening of the ribose ring to afford a modified nucleoside as shown below:
- the 3 ' terminal U ribose can be modified with a 2'3 ' cyclic phosphate as shown below:
- Guide RNAs can contain 3 ' nucleotides which can be stabilized against degradation, e.g., by incorporating one or more of the modified nucleotides described herein.
- uridines can be replaced with modified uridines, e.g., 5-(2-amino)propyl uridine, and 5-bromo uridine, or with any of the modified uridines described herein;
- adenosines and guanosines can be replaced with modified adenosines and guanosines, e.g., with modifications at the 8-position, e.g., 8-bromo guanosine, or with any of the modified adenosines or guanosines described herein.
- sugar-modified ribonucleotides can be incorporated into the gRNA, e.g., wherein the 2' OH-group is replaced by a group selected from H, -OR, -R (wherein R can be, e.g., alkyl, cycloalkyl, aryl, aralkyl, heteroaryl or sugar), halo, -SH, -SR (wherein R can be, e.g., alkyl, cycloalkyl, aryl, aralkyl, heteroaryl or sugar), amino (wherein amino can be, e.g., Nth; alkylamino, dialkylamino, heterocyclyl, arylamino, diarylamino, heteroarylamino, diheteroarylamino, or amino acid); or cyano (-CN).
- R can be, e.g., alkyl, cycloalkyl, aryl, aralkyl, heteroaryl or
- the phosphate backbone can be modified as described herein, e.g., with a phosphothioate (PhTx) group.
- one or more of the nucleotides of the gRNA can each independently be a modified or unmodified nucleotide including, but not limited to 2 '-sugar modified, such as, 2'-0-methyl, 2'-0-methoxyethyl, or 2'-Fluoro modified including, e.g., 2'-F or 2'-0-methyl, adenosine (A), 2'-F or 2'-0-methyl, cytidine (C), 2'-F or 2'-0- methyl, uridine (U), 2'-F or 2'-0-methyl, thymidine (T), 2'-F or 2'-0-methyl, guanosine (G), 2'-0- methoxyethyl-5-methyluridine (Teo), 2'-0-methoxye
- Guide RNAs can also include "locked" nucleic acids (LNA) in which the 2' OH-group can be connected, e.g., by a Cl-6 alkylene or Cl-6 heteroalkylene bridge, to the 4' carbon of the same ribose sugar.
- LNA locked nucleic acids
- any suitable moiety can be used to provide such bridges, include without limitation methylene, propylene, ether, or amino bridges; O-amino (wherein amino can be, e.g., NFh; alkylamino, dialkylamino, heterocyclyl, arylamino, diarylamino, heteroarylamino, or diheteroarylamino, ethylenediamine, or polyamino) and aminoalkoxy or 0(CH2) n -amino (wherein amino can be, e.g., NFb; alkylamino, dialkylamino, heterocyclyl, arylamino, diarylamino, heteroarylamino, or diheteroarylamino, ethylenediamine, or polyamino).
- O-amino wherein amino can be, e.g., NFh; alkylamino, dialkylamino, heterocyclyl, arylamino, diarylamino, heteroary
- a gRNA can include a modified nucleotide which is multicyclic (e.g., tricyclo; and "unlocked" forms, such as glycol nucleic acid (GNA) (e.g., R-GNA or S-GNA, where ribose is replaced by glycol units attached to phosphodiester bonds), or threose nucleic acid (TNA, where ribose is replaced with a-L-threofuranosyl-(3 ' ⁇ 2')).
- GAA glycol nucleic acid
- R-GNA or S-GNA where ribose is replaced by glycol units attached to phosphodiester bonds
- TAA threose nucleic acid
- gRNAs include the sugar group ribose, which is a 5-membered ring having an oxygen.
- exemplary modified gRNAs can include, without limitation, replacement of the oxygen in ribose (e.g., with sulfur (S), selenium (Se), or alkylene, such as, e.g., methylene or ethylene); addition of a double bond (e.g., to replace ribose with cyclopentenyl or cyclohexenyl); ring contraction of ribose (e.g., to form a 4-membered ring of cyclobutane or oxetane); ring expansion of ribose (e.g., to form a 6- or 7-membered ring having an additional carbon or heteroatom, such as for example, anhydrohexitol, altritol, mannitol, cyclohexanyl, cyclohexenyl, and morph
- a gRNA comprises a 4'-S, 4'-Se or a 4'-C-aminomethyl-2'-0-Me modification.
- deaza nucleotides e.g., 7-deaza-adenosine
- O- and N-alkylated nucleotides e.g., N6-methyl adenosine
- one or more or all of the nucleotides in a gRNA are deoxynucleotides.
- RNA-guided nucleases include, but are not limited to, naturally-occurring Class 2 CRISPR nucleases such as Cas9, and Cpfl, as well as other nucleases derived or obtained therefrom.
- RNA-guided nucleases are defined as those nucleases that: (a) interact with (e.g., complex with) a gRNA; and (b) together with the gRNA, associate with, and optionally cleave or modify, a target region of a DNA that includes (i) a sequence complementary to the targeting domain of the gRNA and, optionally, (ii) an additional sequence referred to as a "protospacer adjacent motif,” or "PAM,” which is described in greater detail below.
- PAM protospacer adjacent motif
- RNA-guided nucleases can be defined, in broad terms, by their PAM specificity and cleavage activity, even though variations may exist between individual RNA-guided nucleases that share the same PAM specificity or cleavage activity.
- Skilled artisans will appreciate that some aspects of the present disclosure relate to systems, methods and compositions that can be implemented using any suitable RNA-guided nuclease having a certain PAM specificity and/or cleavage activity.
- the term RNA-guided nuclease should be understood as a generic term, and not limited to any particular type (e.g., Cas9 vs. Cpfl), species (e.g., S.
- RNA-guided nuclease pyogenes vs. S. aureus
- variation e.g., full-length vs. truncated or split; naturally- occurring PAM specificity vs. engineered PAM specificity, etc.
- the PAM sequence takes its name from its sequential relationship to the "protospacer" sequence that is complementary to gRNA targeting domains (or “spacers"). Together with protospacer sequences, PAM sequences define target regions or sequences for specific RNA-guided nuclease / gRNA combinations.
- RNA-guided nucleases may require different sequential relationships between PAMs and protospacers.
- RNA- guided nucleases can also recognize specific PAM sequences.
- S. aureus Cas9 for instance, recognizes a PAM sequence of NNGRRT or NNGRRV, wherein the N residues are immediately 3 ' of the region recognized by the gRNA targeting domain.
- S. pyogenes Cas9 recognizes NGG PAM sequences.
- F. novicida Cpfl recognizes a TTN PAM sequence.
- engineered RNA-guided nucleases can have PAM specificities that differ from the PAM specificities of reference molecules (for instance, in the case of an engineered RNA- guided nuclease, the reference molecule may be the naturally occurring variant from which the RNA- guided nuclease is derived, or the naturally occurring variant having the greatest amino acid sequence homology to the engineered RNA-guided nuclease).
- RNA-guided nucleases can be characterized by their DNA cleavage activity: naturally-occurring RNA-guided nucleases typically form DSBs in target nucleic acids, but engineered variants have been produced that generate only SSBs (discussed above) Ran & Hsu, et al., Cell 154(6), 1380-1389, September 12, 2013 (Ran), incorporated by reference herein), or that that do not cut at all.
- a naturally occurring Cas9 protein comprises two lobes: a recognition (REC) lobe and a nuclease (NUC) lobe; each of which comprise particular structural and/or functional domains.
- the REC lobe comprises an arginine-rich bridge helix (BH) domain, and at least one REC domain (e.g., a REC1 domain and, optionally, a REC2 domain).
- the REC lobe does not share structural similarity with other known proteins, indicating that it is a unique functional domain.
- the BH domain appears to play a role in gRNA:DNA recognition, while the REC domain is thought to interact with the repeat: anti-repeat duplex of the gRNA and to mediate the formation of the Cas9/gRNA complex.
- the NUC lobe comprises a RuvC domain, an HNH domain, and a PAM-interacting (PI) domain.
- the RuvC domain shares structural similarity to retroviral integrase superfamily members and cleaves the non-complementary (i.e., bottom) strand of the target nucleic acid. It may be formed from two or more split RuvC motifs (such as RuvC I, RuvCII, and RuvCIII in s. pyogenes and s. aureus).
- the HNH domain meanwhile, is structurally similar to HNN endonuclease motifs, and cleaves the complementary (i.e., top) strand of the target nucleic acid.
- the PI domain as its name suggests, contributes to PAM specificity.
- Cas9 While certain functions of Cas9 are linked to (but not necessarily fully determined by) the specific domains set forth above, these and other functions may be mediated or influenced by other Cas9 domains, or by multiple domains on either lobe.
- the repeat: antirepeat duplex of the gRNA falls into a groove between the REC and NUC lobes, and nucleotides in the duplex interact with amino acids in the BH, PI, and REC domains.
- Some nucleotides in the first stem loop structure also interact with amino acids in multiple domains (PI, BH and REC1), as do some nucleotides in the second and third stem loops (RuvC and PI domains).
- Cpfl like Cas9, has two lobes: a REC (recognition) lobe, and a NUC (nuclease) lobe.
- the REC lobe includes RECl and REC2 domains, which lack similarity to any known protein structures.
- the NUC lobe includes three RuvC domains (RuvC-I, -II and -III) and a BH domain.
- the Cpfl REC lobe lacks an HNH domain, and includes other domains that also lack similarity to known protein structures: a structurally unique PI domain, three Wedge (WED) domains (WED-I, -II and -III), and a nuclease (Nuc) domain.
- Cpfl While Cas9 and Cpfl share similarities in structure and function, it should be appreciated that certain Cpfl activities are mediated by structural domains that are not analogous to any Cas9 domains. For instance, cleavage of the complementary strand of the target DNA appears to be mediated by the Nuc domain, which differs sequentially and spatially from the HNH domain of Cas9. Additionally, the non-targeting portion of Cpfl gRNA (the handle) adopts a pseudoknot structure, rather than a stem loop structure formed by the repeat: antirepeat duplex in Cas9 gRNAs. Modifications of RNA-guided nucleases
- RNA-guided nucleases described above have activities and properties that can be useful in a variety of applications, but the skilled artisan will appreciate that RNA-guided nucleases can also be modified in certain instances, to alter cleavage activity, PAM specificity, or other structural or functional features.
- inactivation of a RuvC domain of a Cas9 will result in a nickase that cleaves the complementary or top strand.
- inactivation of a Cas9 HNH domain results in a nickase that cleaves the bottom or non-complementary strand.
- RNA-guided nucleases have been split into two or more parts, as described by Zetsche et al. (Nat Biotechnol. 2015 Feb ;33 (2): 139-42 (Zetsche II), incorporated by reference), and by Fine et al. (Sci Rep. 2015 Jul 1 ;5: 10777 (Fine), incorporated by reference).
- RNA-guided nucleases can be, in certain embodiments, size-optimized or truncated, for instance via one or more deletions that reduce the size of the nuclease while still retaining gRNA association, target and PAM recognition, and cleavage activities.
- RNA guided nucleases are bound, covalently or non-covalently, to another polypeptide, nucleotide, or other structure, optionally by means of a linker. Exemplary bound nucleases and linkers are described by Guilinger et al., Nature Biotechnology 32, 577-582 (2014), which is incorporated by reference for all purposes herein.
- RNA-guided nucleases also optionally include a tag, such as, but not limited to, a nuclear localization signal to facilitate movement of RNA-guided nuclease protein into the nucleus.
- a tag such as, but not limited to, a nuclear localization signal to facilitate movement of RNA-guided nuclease protein into the nucleus.
- the RNA-guided nuclease can incorporate C- and/or N-terminal nuclear localization signals. Nuclear localization sequences are known in the art and are described in Maeder and elsewhere.
- Nucleic acids encoding RNA-guided nucleases e.g., Cas9, Cpfl or functional fragments thereof, are provided herein. Exemplary nucleic acids encoding RNA-guided nucleases have been described previously (see, e.g., Cong 2013; Wang 2013; Mali 2013; Jinek 2012).
- a nucleic acid encoding an RNA-guided nuclease can be a synthetic nucleic acid sequence.
- the synthetic nucleic acid molecule can be chemically modified.
- an mRNA encoding an RNA-guided nuclease will have one or more (e.g., all) of the following properties: it can be capped; polyadenylated; and substituted with 5-methylcytidine and/or pseudouridine.
- Synthetic nucleic acid sequences can also be codon optimized, e.g., at least one non-common codon or less-common codon has been replaced by a common codon.
- the synthetic nucleic acid can direct the synthesis of an optimized messenger mRNA, e.g., optimized for expression in a mammalian expression system, e.g., described herein. Examples of codon optimized Cas9 coding sequences are presented in Cotta-Ramusino.
- a nucleic acid encoding an RNA-guided nuclease may comprise a nuclear localization sequence (NLS).
- NLS nuclear localization sequences are known in the art.
- RNA-guided nucleases can be evaluated by standard methods known in the art. See, e.g., Cotta-Ramusino. The stability of RNP complexes may be evaluated by differential scanning fluorimetry, as described below.
- thermostability of ribonucleoprotein (RNP) complexes comprising gRNAs and RNA- guided nucleases can be measured via DSF.
- the DSF technique measures the thermostability of a protein, which can increase under favorable conditions such as the addition of a binding RNA molecule, e.g., a gRNA.
- a DSF assay can be performed according to any suitable protocol, and can be employed in any suitable setting, including without limitation (a) testing different conditions (e.g., different stoichiometric ratios of gRNA: RNA-guided nuclease protein, different buffer solutions, etc.) to identify optimal conditions for RNP formation; and (b) testing modifications (e.g., chemical modifications, alterations of sequence, etc.) of an RNA-guided nuclease and/or a gRNA to identify those modifications that improve RNP formation or stability.
- different conditions e.g., different stoichiometric ratios of gRNA: RNA-guided nuclease protein, different buffer solutions, etc.
- modifications e.g., chemical modifications, alterations of sequence, etc.
- One readout of a DSF assay is a shift in melting temperature of the RNP complex; a relatively high shift suggests that the RNP complex is more stable (and may thus have greater activity or more favorable kinetics of formation, kinetics of degradation, or another functional characteristic) relative to a reference RNP complex characterized by a lower shift.
- a threshold melting temperature shift may be specified, so that the output is one or more RNPs having a melting temperature shift at or above the threshold.
- the threshold can be 5-10°C (e.g., 5°, 6°, 7°, 8°, 9°, 10°) or more, and the output may be one or more RNPs characterized by a melting temperature shift greater than or equal to the threshold.
- a fixed concentration e.g., 2 ⁇
- Cas9 in water+10x SYPRO Orange® (Life Technologies cat#S-6650) is dispensed into a 384 well plate.
- An equimolar amount of gRNA diluted in solutions with varied pH and salt is then added.
- a Bio- Rad CFX384TM Real-Time System CI 000 TouchTM Thermal Cycler with the Bio-Rad CFX Manager software is used to run a gradient from 20°C to 90°C with a 1 °C increase in temperature every 10 seconds.
- the second assay consists of mixing various concentrations of gRNA with fixed concentration (e.g., 2 ⁇ ) Cas9 in optimal buffer from assay 1 above and incubating (e.g., at RT for 10') in a 384 well plate.
- An equal volume of optimal buffer + lOx SYPRO Orange® (Life Technologies cat#S-6650) is added and the plate sealed with Microseal® B adhesive (MSB- 1001).
- MSB- 1001 Microseal® B adhesive
- a Bio-Rad CFX384TM Real-Time System CI 000 TouchTM Thermal Cycler with the Bio-Rad CFX Manager software is used to run a gradient from 20°C to 90°C with a 1°C increase in temperature every 10 seconds.
- the genome editing systems described above are used, in various embodiments of the present disclosure, to generate edits in (i.e., to alter) targeted regions of DNA (e.g., TGFBR2 DNA) within or obtained from a cell.
- targeted regions of DNA e.g., TGFBR2 DNA
- Various strategies are described herein to generate particular edits, and these strategies are generally described in terms of the desired repair outcome, the number and positioning of individual edits (e.g., SSBs or DSBs), and the target sites of such edits.
- Genome editing strategies that involve the formation of SSBs or DSBs are characterized by repair outcomes including: (a) deletion of all or part of a targeted region; (b) insertion into or replacement of all or part of a targeted region; or (c) interruption of all or part of a targeted region.
- This grouping is not intended to be limiting, or to be binding to any particular theory or model, and is offered solely for economy of presentation. Skilled artisans will appreciate that the listed outcomes are not mutually exclusive and that some repairs may result in other outcomes. The description of a particular editing strategy or method should not be understood to require a particular repair outcome unless otherwise specified.
- Replacement of a targeted region generally involves the replacement of all or part of the existing sequence within the targeted region with a homologous sequence, for instance through gene correction or gene conversion, two repair outcomes that are mediated by HDR pathways.
- HDR is promoted by the use of a donor template, which can be single -stranded or double stranded, as described in greater detail below.
- Single or double stranded templates can be exogenous, in which case they will promote gene correction, or they can be endogenous (e.g., a homologous sequence within the cellular genome), to promote gene conversion.
- Exogenous templates can have asymmetric overhangs (i.e., the portion of the template that is complementary to the site of the DSB may be offset in a 3 ' or 5' direction, rather than being centered within the donor template), for instance as described by Richardson et al. (Nature Biotechnology 34, 339-344 (2016), (Richardson), incorporated by reference).
- the template can correspond to either the complementary (top) or non-complementary (bottom) strand of the targeted region.
- Gene conversion and gene correction are facilitated, in some cases, by the formation of one or more nicks in or around the targeted region, as described in Ran and Cotta-Ramusino.
- a dual-nickase strategy is used to form two offset SSBs that, in turn, form a single DSB having an overhang (e.g., a 5' overhang).
- Interruption and/or deletion of all or part of a targeted sequence can be achieved by a variety of repair outcomes.
- a sequence can be deleted by simultaneously generating two or more DSBs that flank a targeted region, which is then excised when the DSBs are repaired, as is described in Maeder for the LCA10 mutation.
- a sequence can be interrupted by a deletion generated by formation of a double strand break with single-stranded overhangs, followed by exonucleolytic processing of the overhangs prior to repair.
- One specific subset of target sequence interruptions is mediated by the formation of an indel within the targeted sequence, where the repair outcome is typically mediated by NHEJ pathways (including Alt-NHEJ).
- NHEJ is referred to as an "error prone" repair pathway because of its association with indel mutations.
- a DSB is repaired by NHEJ without alteration of the sequence around it (a so-called “perfect” or “scarless” repair); this generally requires the two ends of the DSB to be perfectly ligated.
- Indels meanwhile, are thought to arise from enzymatic processing of free DNA ends before they are ligated that adds and/or removes nucleotides from either or both strands of either or both free ends.
- indel mutations tend to be variable, occurring along a distribution, and can be influenced by a variety of factors, including the specific target site, the cell type used, the genome editing strategy used, etc. Even so, it is possible to draw limited generalizations about indel formation: deletions formed by repair of a single DSB are most commonly in the 1-50 bp range, but can reach greater than 100-200 bp. Insertions formed by repair of a single DSB tend to be shorter and often include short duplications of the sequence immediately surrounding the break site. However, it is possible to obtain large insertions, and in these cases, the inserted sequence has often been traced to other regions of the genome or to plasmid DNA present in the cells.
- Indel mutations - and genome editing systems configured to produce indels - are useful for interrupting target sequences, for example, when the generation of a specific final sequence is not required and/or where a frameshift mutation would be tolerated. They can also be useful in settings where particular sequences are preferred, insofar as the certain sequences desired tend to occur preferentially from the repair of an SSB or DSB at a given site. Indel mutations are also a useful tool for evaluating or screening the activity of particular genome editing systems and their components.
- indels can be characterized by (a) their relative and absolute frequencies in the genomes of cells contacted with genome editing systems and (b) the distribution of numerical differences relative to the unedited sequence, e.g., ⁇ 1, ⁇ 2, ⁇ 3, etc.
- multiple gRNAs can be screened to identify those gRNAs that most efficiently drive cutting at a target site based on an indel readout under controlled conditions.
- Guides that produce indels at or above a threshold frequency, or that produce a particular distribution of indels, can be selected for further study and development.
- Indel frequency and distribution can also be useful as a readout for evaluating different genome editing system implementations or formulations and delivery methods, for instance by keeping the gRNA constant and varying certain other reaction conditions or delivery methods.
- genome editing systems may also be employed to generate two or more DSBs, either in the same locus or in different loci.
- Strategies for editing that involve the formation of multiple DSBs, or SSBs, are described in, for instance, Cotta-Ramusino.
- Donor template design is described in detail in the literature, for instance in Cotta-Ramusino.
- DNA oligomer donor templates oligodeoxynucleotides or ODNs
- ssODNs single stranded
- dsODNs double- stranded
- donor templates generally include regions that are homologous to regions of DNA within or near (e.g., flanking or adjoining) a target sequence to be cleaved. These homologous regions are referred to here as "homology arms,” and are illustrated schematically below:
- the homology arms can have any suitable length (including 0 nucleotides if only one homology arm is used), and 3 ' and 5' homology arms can have the same length, or can differ in length.
- the selection of appropriate homology arm lengths can be influenced by a variety of factors, such as the desire to avoid homologies or micro-homologies with certain sequences such as Alu repeats or other very common elements.
- a 5' homology arm can be shortened to avoid a sequence repeat element.
- a 3 ' homology arm can be shortened to avoid a sequence repeat element.
- both the 5' and the 3 ' homology arms can be shortened to avoid including certain sequence repeat elements.
- homology arm designs can improve the efficiency of editing or increase the frequency of a desired repair outcome.
- Richardson et al. Nature Biotechnology 34, 339-344 (2016) (Richardson), which is incorporated by reference, found that the relative asymmetry of 3 ' and 5' homology arms of single stranded donor templates influenced repair rates and/or outcomes.
- a replacement sequence in donor templates have been described elsewhere, including in Cotta-Ramusino et al.
- a replacement sequence can be any suitable length (including zero nucleotides, where the desired repair outcome is a deletion), and typically includes one, two, three or more sequence modifications relative to the naturally-occurring sequence within a cell in which editing is desired.
- One common sequence modification involves the alteration of the naturally- occurring sequence to repair a mutation that is related to a disease or condition of which treatment is desired.
- Another common sequence modification involves the alteration of one or more sequences that are complementary to, or code for, the PAM sequence of the KNA-guided nuclease or the targeting domain of the gKNA(s) being used to generate an SSB or DSB, to reduce or eliminate repeated cleavage of the target site after the replacement sequence has been incorporated into the target site.
- a linear ssODN can be configured to (i) anneal to the nicked strand of the target nucleic acid, (ii) anneal to the intact strand of the target nucleic acid, (iii) anneal to the plus strand of the target nucleic acid, and/or (iv) anneal to the minus strand of the target nucleic acid.
- An ssODN may have any suitable length, e.g., about, at least, or no more than 150-200 nucleotides (e.g., 150, 160, 170, 180, 190, or 200 nucleotides).
- a template nucleic acid can also be a nucleic acid vector, such as a viral genome or circular double stranded DNA, e.g., a plasmid.
- Nucleic acid vectors comprising donor templates can include other coding or non-coding elements.
- a template nucleic acid can be delivered as part of a viral genome (e.g., in an AAV or lentiviral genome) that includes certain genomic backbone elements (e.g., inverted terminal repeats, in the case of an AAV genome) and optionally includes additional sequences coding for a gKNA and/or an RNA-guided nuclease.
- the donor template can be adjacent to, or flanked by, target sites recognized by one or more gRNAs, to facilitate the formation of free DSBs on one or both ends of the donor template that can participate in repair of corresponding SSBs or DSBs formed in cellular DNA using the same gRNAs.
- exemplary nucleic acid vectors suitable for use as donor templates are described in Cotta-Ramusino.
- a template nucleic acid can be designed to avoid undesirable sequences.
- one or both homology arms can be shortened to avoid overlap with certain sequence repeat elements, e.g., Alu repeats, LINE elements, etc.
- Genome editing systems can be used to manipulate or alter a cell, e.g., to edit or alter a target nucleic acid.
- the manipulating can occur, in various embodiments, in vivo or ex vivo.
- a variety of cell types can be manipulated or altered according to the embodiments of this disclosure, and in some cases, such as in vivo applications, a plurality of cell types are altered or manipulated, for example by delivering genome editing systems according to this disclosure to a plurality of cell types. In other cases, however, it may be desirable to limit manipulation or alteration to a particular cell type or types. For instance, it can be desirable in some instances to edit a cell with limited differentiation potential or a terminally differentiated cell, such as a photoreceptor cell in the case of Maeder, in which modification of a genotype is expected to result in a change in cell phenotype.
- the cell may be an embryonic stem cell, induced pluripotent stem cell (iPSC), hematopoietic stem/progenitor cell (HSPC), or other stem or progenitor cell type that differentiates into a cell type of relevance to a given application or indication.
- the cell is a T cell.
- the cell being altered or manipulated is, variously, a dividing cell or a non- dividing cell, depending on the cell type(s) being targeted and/or the desired editing outcome.
- the cells When cells are manipulated or altered ex vivo, the cells can be used (e.g., administered to a subject) immediately, or they can be maintained or stored for later use. Those of skill in the art will appreciate that cells can be maintained in culture or stored (e.g., frozen in liquid nitrogen) using any suitable method known in the art.
- the genome editing systems of this disclosure can be implemented in any suitable manner, meaning that the components of such systems, including without limitation the RNA-guided nuclease, gRNA, and optional donor template nucleic acid, can be delivered, formulated, or administered in any suitable form or combination of forms that results in the transduction, expression or introduction of a genome editing system and/or causes a desired repair outcome in a cell, tissue or subject.
- Tables 3 and 4 set forth several, non-limiting examples of genome editing system implementations. Those of skill in the art will appreciate, however, that these listings are not comprehensive, and that other implementations are possible.
- Table 3 the table lists several exemplary implementations of a genome editing system comprising a single gRNA and an optional donor template.
- genome editing systems according to this disclosure can incorporate multiple gRNAs, multiple RNA-guided nucleases, and other components such as proteins, and a variety of implementations will be evident to the skilled artisan based on the principles illustrated in the table.
- [N/A] indicates that the genome editing system does not include the indicated component.
- Table 7 summarizes various delivery methods for the components of genome editing systems, as described herein. Again, the listing is intended to be exemplary rather than limiting.
- Nucleic acids encoding the various elements of a genome editing system according to the present disclosure can be administered to subjects or delivered into cells by art-known methods or as described herein.
- RNA-guided nuclease-encoding and/or gRNA-encoding DNA, as well as donor template nucleic acids can be delivered by, e.g., vectors (e.g., viral or non-viral vectors), non-vector based methods (e.g., using naked DNA or DNA complexes), or a combination thereof
- Nucleic acids encoding elements of a genome editing system can include sequences that encode for one, two, three, four, or more gRNAs.
- the nucleic acids can encode for both a first and a second gRNA molecule, e.g., where the second gRNA has a second targeting domain that is complementary to a second target sequence of the TGFBR2 gene.
- the nucleic acids disclosed herein can further comprise a nucleotide sequence that encodes a third gRNA molecule having a third targeting domain that is complementary to a third target sequence of the TGFBR2 gene.
- the nucleic acid compositions disclosed herein can further comprise a nucleotide sequence that encodes a fourth gRNA molecule described herein having a fourth targeting domain that is complementary to a fourth target sequence of the TGFBR2 gene.
- the second, third and/or fourth gRNA molecule comprises a targeting domain comprising a nucleotide sequence selected SEQ ID NOS: 5036 to 5096.
- Nucleic acids encoding genome editing systems or components thereof can be delivered directly to cells as naked DNA or RNA, for instance by means of transfection or electroporation, or can be conjugated to molecules (e.g., N-acetylgalactosamine) promoting uptake by the target cells (e.g., erythrocytes, HSCs).
- Nucleic acid vectors such as the vectors summarized in Table 4, can also be used.
- Nucleic acid vectors can comprise one or more sequences encoding genome editing system components, such as an RNA-guided nuclease, a gRNA and/or a donor template.
- a vector can also comprise a sequence encoding a signal peptide (e.g., for nuclear localization, nucleolar localization, or mitochondrial localization), associated with (e.g., inserted into or fused to) a sequence coding for a protein.
- a nucleic acid vectors can include a Cas9 coding sequence that includes one or more nuclear localization sequences (e.g., a nuclear localization sequence from SV40).
- the nucleic acid vector can also include any suitable number of regulatory/control elements, e.g., promoters, enhancers, introns, polyadenylation signals, Kozak consensus sequences, or internal ribosome entry sites (IRES). These elements are well known in the art, and are described in Cotta- Ramusino.
- regulatory/control elements e.g., promoters, enhancers, introns, polyadenylation signals, Kozak consensus sequences, or internal ribosome entry sites (IRES). These elements are well known in the art, and are described in Cotta- Ramusino.
- Nucleic acid vectors according to this disclosure include recombinant viral vectors. Exemplary viral vectors are set forth in Table 7, and additional suitable viral vectors and their use and production are described in Cotta-Ramusino.
- the vector is a viral vector, e.g., an adeno-associated virus (AAV) vector or a lentivirus (LV) vector.
- AAV adeno-associated virus
- LV lentivirus
- viral particles can be used to deliver genome editing system components in nucleic acid and/or peptide form. For example, "empty" viral particles can be assembled to contain any suitable cargo.
- Viral vectors and viral particles can also be engineered to incorporate targeting ligands to alter target tissue specificity.
- non-viral vectors can be used to deliver nucleic acids encoding genome editing systems according to the present disclosure.
- One important category of non-viral nucleic acid vectors are nanoparticles, which can be organic or inorganic. Nanoparticles are well known in the art, and are summarized in Cotta-Ramusino. Any suitable nanoparticle design can be used to deliver genome editing system components or nucleic acids encoding such components.
- organic (e.g., lipid and/or polymer) nanoparticles can be suitable for use as delivery vehicles in certain embodiments of this disclosure. Exemplary lipids for use in nanoparticle formulations, and/or gene transfer are shown in Table 8, and Table 9 lists exemplary polymers for use in gene transfer and/or nanoparticle formulations.
- Non-viral vectors optionally include targeting modifications to improve uptake and/or selectively target certain cell types. These targeting modifications can include e.g., cell specific antigens, monoclonal antibodies, single chain antibodies, aptamers, polymers, sugars (e.g., N- acetylgalactosamine (GalNAc)), and cell penetrating peptides.
- Such vectors also optionally use fusogenic and endosome-destabilizing peptides/polymers, undergo acid-triggered conformational changes (e.g., to accelerate endosomal escape of the cargo), and/or incorporate a stimuli-cleavable polymer, e.g., for release in a cellular compartment.
- a stimuli-cleavable polymer e.g., for release in a cellular compartment.
- disulfide-based cationic polymers that are cleaved in the reducing cellular environment can be used.
- nucleic acid molecules other than the components of a genome editing system, e.g., the RNA-guided nuclease component and/or the gRNA component described herein, are delivered.
- the nucleic acid molecule is delivered at the same time as one or more of the components of the Genome editing system.
- the nucleic acid molecule is delivered before or after (e.g., less than about 30 minutes, 1 hour, 2 hours, 3 hours, 6 hours, 9 hours, 12 hours, 1 day, 2 days, 3 days, 1 week, 2 weeks, or 4 weeks) one or more of the components of the Genome editing system are delivered.
- the nucleic acid molecule is delivered by a different means than one or more of the components of the genome editing system, e.g., the RNA-guided nuclease component and/or the gRNA component, are delivered.
- the nucleic acid molecule can be delivered by any of the delivery methods described herein.
- the nucleic acid molecule can be delivered by a viral vector, e.g., an integration-deficient lentivirus, and the RNA-guided nuclease molecule component and/or the gRNA component can be delivered by electroporation, e.g., such that the toxicity caused by nucleic acids (e.g., DNAs) can be reduced.
- the nucleic acid molecule encodes a therapeutic protein, e.g., a protein described herein. In certain embodiments, the nucleic acid molecule encodes an RNA molecule, e.g., an RNA molecule described herein.
- RNPs complexes of gRNAs and RNA-guided nucleases, i.e., ribonucleoprotein complexes
- RNAs encoding RNA-guided nucleases and/or gRNAs can be delivered into cells or administered to subjects by art-known methods, some of which are described in Cotta-Ramusino.
- RNA-guided nuclease-encoding and/or gRNA-encoding RNA can be delivered, e.g., by microinjection, electroporation, transient cell compression or squeezing (see, e.g., Lee 2012).
- Lipid- mediated transfection, peptide-mediated delivery, GalNAc- or other conjugate-mediated delivery, and combinations thereof, can also be used for delivery in vitro and in vivo.
- delivery via electroporation comprises mixing the cells with the RNA encoding RNA-guided nucleases and/or gRNAs, with or without donor template nucleic acid molecules, in a cartridge, chamber or cuvette and applying one or more electrical impulses of defined duration and amplitude.
- Systems and protocols for electroporation are known in the art, and any suitable electroporation tool and/or protocol can be used in connection with the various embodiments of this disclosure
- the RNP complexes of the present disclosure can be used to: (1) improve T cell proliferation; (2) improve T cell survival; and/or (3) improve T cell function.
- two or more RNP complexes comprising distinct gRNAs can be employed concurrently or sequentially to alter TGFBR2 gene expression in a cell, e.g., a T cell.
- Such RNP complexes can comprise distinct gRNAs targeting distinct TGFBR2 gene sequences.
- the RNP complexes can, in certain instances, induce a cleavage event, e.g., a double strand or single strand break.
- the RNP complexes can comprise enzymatically active Cas9 (eaCas9) molecules that form double strand breaks in a target nucleic acid or eaCas9 molecules that form single strand breaks in a target nucleic acid (e.g., nickase molecules).
- eaCas9 enzymatically active Cas9
- eaCas9 molecules that form single strand breaks in a target nucleic acid
- a dual-nickase RNP strategy can be used to form two offset single strand breaks that, in turn, form a single double strand break having an overhang (e.g., a 5' overhang).
- Genome editing systems, or cells altered or manipulated using such systems can be administered to subjects by any suitable mode or route, whether local or systemic.
- Systemic modes of administration include oral and parenteral routes.
- Parenteral routes include, by way of example, intravenous, intramarrow, intrarterial, intramuscular, intradermal, subcutaneous, intranasal, and intraperitoneal routes.
- Components administered systemically can be modified or formulated to target, e.g., HSCs, hematopoietic stem/progenitor cells, or erythroid progenitors or precursor cells.
- Local modes of administration include, by way of example, intramarrow injection into the trabecular bone or intrafemoral injection into the marrow space, and infusion into the portal vein.
- significantly smaller amounts of the components can exert an effect when administered locally (for example, directly into the bone marrow) compared to when administered systemically (for example, intravenously).
- Local modes of administration can reduce or eliminate the incidence of potentially toxic side effects that may occur when therapeutically effective amounts of a component are administered systemically.
- Administration can be provided as a periodic bolus (for example, intravenously) or as continuous infusion from an internal reservoir or from an external reservoir (for example, from an intravenous bag or implantable pump).
- Components can be administered locally, for example, by continuous release from a sustained release drug delivery device.
- a release system can include a matrix of a biodegradable material or a material which releases the incorporated components by diffusion.
- the components can be homogeneously or heterogeneously distributed within the release system.
- a variety of release systems can be useful, however, the choice of the appropriate system will depend upon rate of release required by a particular application. Both non-degradable and degradable release systems can be used. Suitable release systems include polymers and polymeric matrices, non-polymeric matrices, or inorganic and organic excipients and diluents such as, but not limited to, calcium carbonate and sugar (for example, trehalose). Release systems may be natural or synthetic. However, synthetic release systems are preferred because generally they are more reliable, more reproducible and produce more defined release profiles.
- the release system material can be selected so that components having different molecular weights are released by diffusion through or degradation of the material.
- Representative synthetic, biodegradable polymers include, for example: polyamides such as poly(amino acids) and poly(peptides); polyesters such as poly(lactic acid), poly(glycolic acid), poly(lactic-co-glycolic acid), and poly(caprolactone); poly(anhydrides); polyorthoesters; polycarbonates; and chemical derivatives thereof (substitutions, additions of chemical groups, for example, alkyl, alkylene, hydroxylations, oxidations, and other modifications routinely made by those skilled in the art), copolymers and mixtures thereof.
- polyamides such as poly(amino acids) and poly(peptides)
- polyesters such as poly(lactic acid), poly(glycolic acid), poly(lactic-co-glycolic acid), and poly(caprolactone)
- poly(anhydrides) polyorthoesters
- polycarbonates and chemical derivatives thereof (substitutions, additions of chemical groups, for example, alkyl, alkylene, hydroxylation
- Representative synthetic, non-degradable polymers include, for example: polyethers such as poly(ethylene oxide), poly(ethylene glycol), and poly(tetramethylene oxide); vinyl polymers-polyacrylates and polymethacrylates such as methyl, ethyl, other alkyl, hydroxyethyl methacrylate, acrylic and methacrylic acids, and others such as poly( vinyl alcohol), poly(vinyl pyrolidone), and poly( vinyl acetate); poly(urethanes); cellulose and its derivatives such as alkyl, hydroxyalkyl, ethers, esters, nitrocellulose, and various cellulose acetates; polysiloxanes; and any chemical derivatives thereof (substitutions, additions of chemical groups, for example, alkyl, alkylene, hydroxylations, oxidations, and other modifications routinely made by those skilled in the art), copolymers and mixtures thereof.
- polyethers such as poly(ethylene oxide), poly(ethylene glycol), and poly(tetram
- Poly(lactide-co-glycolide) microsphere can also be used.
- the microspheres are composed of a polymer of lactic acid and glycolic acid, which are structured to form hollow spheres.
- the spheres can be approximately 15-30 microns in diameter and can be loaded with components described herein. Multi-modal or differential delivery of components
- Genome editing systems disclosed herein can be delivered together or separately and simultaneously or non- simultaneously. Separate and/or asynchronous delivery of genome editing system components can be particularly desirable to provide temporal or spatial control over the function of genome editing systems and to limit certain effects caused by their activity.
- Different or differential modes as used herein refer to modes of delivery that confer different pharmacodynamic or pharmacokinetic properties on the subject component molecule, e.g., a RNA- guided nuclease molecule, gRNA, template nucleic acid, or payload.
- the modes of delivery can result in different tissue distribution, different half-life, or different temporal distribution, e.g., in a selected compartment, tissue, or organ.
- Some modes of delivery e.g., delivery by a nucleic acid vector that persists in a cell, or in progeny of a cell, e.g., by autonomous replication or insertion into cellular nucleic acid, result in more persistent expression of and presence of a component.
- examples include viral, e.g., AAV or lentivirus, delivery.
- the components of a genome editing system can be delivered by modes that differ in terms of resulting half-life or persistent of the delivered component the body, or in a particular compartment, tissue or organ.
- a gRNA can be delivered by such modes.
- the RNA-guided nuclease molecule component can be delivered by a mode which results in less persistence or less exposure to the body or a particular compartment or tissue or organ.
- a first mode of delivery is used to deliver a first component and a second mode of delivery is used to deliver a second component.
- the first mode of delivery confers a first pharmacodynamic or pharmacokinetic property.
- the first pharmacodynamic property can be, e.g., distribution, persistence, or exposure, of the component, or of a nucleic acid that encodes the component, in the body, a compartment, tissue or organ.
- the second mode of delivery confers a second pharmacodynamic or pharmacokinetic property.
- the second pharmacodynamic property can be, e.g., distribution, persistence, or exposure, of the component, or of a nucleic acid that encodes the component, in the body, a compartment, tissue or organ.
- the first pharmacodynamic or pharmacokinetic property e.g., distribution, persistence or exposure
- the first mode of delivery is selected to optimize, e.g., minimize, a pharmacodynamic or pharmacokinetic property, e.g., distribution, persistence or exposure.
- the second mode of delivery is selected to optimize, e.g., maximize, a pharmacodynamic or pharmacokinetic property, e.g., distribution, persistence or exposure.
- the first mode of delivery comprises the use of a relatively persistent element, e.g., a nucleic acid, e.g., a plasmid or viral vector, e.g., an AAV or lentivirus.
- a relatively persistent element e.g., a nucleic acid, e.g., a plasmid or viral vector, e.g., an AAV or lentivirus.
- the second mode of delivery comprises a relatively transient element, e.g., an RNA or protein.
- the first component comprises gRNA
- the delivery mode is relatively persistent, e.g., the gRNA is transcribed from a plasmid or viral vector, e.g., an AAV or lentivirus. Transcription of these genes would be of little physiological consequence because the genes do not encode for a protein product, and the gRNAs are incapable of acting in isolation.
- the second component a RNA-guided nuclease molecule, is delivered in a transient manner, for example as mRNA or as protein, ensuring that the full RNA-guided nuclease molecule/gRNA complex is only present and active for a short period of time.
- the components can be delivered in different molecular form or with different delivery vectors that complement one another to enhance safety and tissue specificity.
- differential delivery modes can enhance performance, safety, and/or efficacy, e.g., the likelihood of an eventual off-target modification can be reduced.
- Delivery of immunogenic components, e.g., Cas9 molecules, by less persistent modes can reduce immunogenicity, as peptides from the bacterially-derived Cas enzyme are displayed on the surface of the cell by MHC molecules.
- a two-part delivery system can alleviate these drawbacks.
- a first component e.g., a gRNA is delivered by a first delivery mode that results in a first spatial, e.g., tissue, distribution.
- a second component e.g., a RNA-guided nuclease molecule is delivered by a second delivery mode that results in a second spatial, e.g., tissue, distribution.
- the first mode comprises a first element selected from a liposome, nanoparticle, e.g., polymeric nanoparticle, and a nucleic acid, e.g., viral vector.
- the second mode comprises a second element selected from the group.
- the first mode of delivery comprises a first targeting element, e.g., a cell specific receptor or an antibody, and the second mode of delivery does not include that element.
- the second mode of delivery comprises a second targeting element, e.g., a second cell specific receptor or second antibody.
- RNA-guided nuclease molecule When the RNA-guided nuclease molecule is delivered in a virus delivery vector, a liposome, or polymeric nanoparticle, there is the potential for delivery to and therapeutic activity in multiple tissues, when it may be desirable to only target a single tissue.
- a two-part delivery system can resolve this challenge and enhance tissue specificity. If the gRNA and the RNA-guided nuclease molecule are packaged in separated delivery vehicles with distinct but overlapping tissue tropism, the fully functional complex will only be formed in the tissue that is targeted by both vectors.
- the cells include immune cells such as T cells.
- the cells generally are engineered by introducing one or more genetically engineered nucleic acids or products thereof.
- genetically engineered antigen receptors including engineered T cell receptors (TCRs) and functional non-TCR antigen receptors, such as chimeric antigen receptors (CARs), including activating, stimulatory, and costimulatory CARs, and combinations thereof.
- TCRs engineered T cell receptors
- CARs chimeric antigen receptors
- the cells also are introduced, either simultaneously or sequentially, with the nucleic acid encoding the genetically engineered antigen receptor, with an agent (e.g., Cas9/gRNA RNP) that is capable of disrupting a gene encoding TGFBR2.
- an agent e.g., Cas9/gRNA RNP
- the cells can be incubated or cultivated prior to, during and/or subsequent to, introducing the nucleic acid molecule encoding the recombinant receptor and/or the agent (e.g., Cas9/gRNA RNP).
- the cells e.g., T cells
- the cells can be incubated or cultivated prior to, during or subsequent to, the introduction of the nucleic acid molecule encoding the recombinant receptor, such as prior to, during or subsequent to, the transduction of the cells with a viral vector (e.g., a lentiviral vector) encoding the recombinant receptor.
- a viral vector e.g., a lentiviral vector
- the cells e.g., T cells
- the cells can be incubated or cultivated prior to, during or subsequent to, the introduction of the agent (e.g., Cas9/gRNA RNP), such as prior to, during or subsequent to, contacting the cells with the agent or prior to, during or subsequent to, delivering the agent into the cells, e.g., via electroporation.
- the incubation can be both in the context of introducing the nucleic acid molecule encoding the recombinant receptor and introducing the agent, e.g., Cas9/gRNA RNP.
- the incubation can be in the presence of a cytokine, such as IL-2, IL-7 or IL-15, or in the presence of a stimulating or activating agents that induces the proliferation or activation of cells, such as an anti-CD3/anti-CD28 antibodies.
- the method includes activating or stimulating cells with a stimulating or activating agent (e.g., anti-CD3/anti-CD28 antibodies) prior to introducing the nucleic acid molecule encoding the recombinant receptor and the agent, e.g., Cas9/gRNA RNP.
- incubation also can be performed in the presence of a cytokine, such as IL-2 (e.g., 1 U/mL to 500 U/mL, such as 10 U/mL to 200 U/mL, for example at least or about 50 U/mL or 100 U/mL), IL-7 (e.g.
- a cytokine such as IL-2 (e.g., 1 U/mL to 500 U/mL, such as 10 U/mL to 200 U/mL, for example at least or about 50 U/mL or 100 U/mL), IL-7 (e.g.
- 0.5 ng/mL to 50 ng/mL such as 1 ng/mL to 20 ng/mL, for example, at least or about 5 ng/mL or 10 ng/mL) or IL-15 (e.g., 0.1 ng/mL to 50 ng/mL, such as 0.5 ng/mL to 25 ng/mL, for example, at least or about 1 ng/mL or 5 ng/mL).
- the cells are incubated for 6 hours to 96 hours, such as 24-48 hours or 24-36 hours prior to introducing the nucleic acid molecule encoding the recombinant receptor (e.g., via transduction).
- Recombinant receptors that bind to a specific antigen and agents e.g., Cas9/gRNA RNP
- a recombinant receptor is engineered and/or the TGFBR2 target gene is manipulated ex vivo and the resulting genetically engineered cells are administered to a subject.
- Sources of target cells for ex vivo manipulation may include, e.g., the subject's blood, the subject's cord blood, or the subject's bone marrow.
- Sources of target cells for ex vivo manipulation may also include, e.g., heterologous donor blood, cord blood, or bone marrow.
- the cells are eukaryotic cells, such as mammalian cells, e.g., human cells.
- the cells are derived from the blood, bone marrow, lymph, or lymphoid organs, are cells of the immune system, such as cells of the innate or adaptive immunity, e.g., myeloid or lymphoid cells, including lymphocytes, typically T cells and/or NK cells.
- Other exemplary cells include stem cells, such as multipotent and pluripotent stem cells, including induced pluripotent stem cells (iPSCs).
- the cells are human cells. With reference to the subject to be treated, the cells may be allogeneic and/or autologous.
- the cells typically are primary cells, such as those isolated directly from a subject and/or isolated from a subject and frozen.
- the target cell is a T cell, e.g., a CD8+ T cell (e.g., a CD8+ naive T cell, central memory T cell, or effector memory T cell), a CD4+ T cell, a natural killer T cell (NKT cells), a regulatory T cell (Treg), a stem cell memory T cell, a lymphoid progenitor cell a hematopoietic stem cell, a natural killer cell (NK cell) or a dendritic cell.
- a CD8+ T cell e.g., a CD8+ naive T cell, central memory T cell, or effector memory T cell
- a CD4+ T cell e.g., a CD4+ T cell, a natural killer T cell (NKT cells), a regulatory T cell (Treg), a stem cell memory T cell, a lymphoid progenitor cell a hematopoietic stem cell, a natural killer cell (NK cell) or
- the cells are monocytes or granulocytes, e.g., myeloid cells, macrophages, neutrophils, dendritic cells, mast cells, eosinophils, and/or basophils.
- the target cell is an induced pluripotent stem (iPS) cell or a cell derived from an iPS cell, e.g., an iPS cell generated from a subject, manipulated to alter (e.g., induce a mutation in) or manipulate the expression of one or more target genes, and differentiated into, e.g., a T cell, e.g., a CD8+ T cell (e.g., a CD8+ naive T cell, central memory T cell, or effector memory T cell), a CD4+ T cell, a stem cell memory T cell, a lymphoid progenitor cell or a hematopoietic stem cell.
- iPS induced pluripotent stem
- the cells include one or more subsets of T cells or other cell types, such as whole T cell populations, CD4+ cells, CD8+ cells, and subpopulations thereof, such as those defined by function, activation state, maturity, potential for differentiation, expansion, recirculation, localization, and/or persistence capacities, antigen-specificity, type of antigen receptor, presence in a particular organ or compartment, marker or cytokine secretion profile, and/or degree of differentiation.
- T cells or other cell types such as whole T cell populations, CD4+ cells, CD8+ cells, and subpopulations thereof, such as those defined by function, activation state, maturity, potential for differentiation, expansion, recirculation, localization, and/or persistence capacities, antigen-specificity, type of antigen receptor, presence in a particular organ or compartment, marker or cytokine secretion profile, and/or degree of differentiation.
- TN cells naive T cells
- TEFF effector T cells
- memory T cells sub-types thereof, such as stem cell memory T (TSCM), central memory T (TCM), effector memory T (TEM), or terminally differentiated effector memory T cells, tumor-infiltrating lymphocytes (TIL), immature T cells, mature T cells, helper T cells, cytotoxic T cells, mucosa-associated invariant T (MAIT) cells, naturally occurring and adaptive regulatory T (Treg) cells, helper T cells, such as TH1 cells, TH2 cells, TH3 cells, TH17 cells, TH9 cells, TH22 cells, follicular helper T cells, alpha/beta T cells, and delta/gamma T cells.
- TIL tumor-infiltrating lymphocytes
- MAIT mucosa-associated invariant T
- Reg adaptive regulatory T
- helper T cells such as TH1 cells, TH2 cells, TH3 cells, TH17 cells,
- the methods include isolating cells from the subject, preparing, processing, culturing, and/or engineering them.
- preparation of the engineered cells includes one or more culture and/or preparation steps.
- the cells for engineering as described may be isolated from a sample, such as a biological sample, e.g., one obtained from or derived from a subject.
- the subject from which the cell is isolated is one having the disease or condition or in need of a cell therapy or to which cell therapy will be administered.
- the subject in some embodiments is a human in need of a particular therapeutic intervention, such as the adoptive cell therapy for which cells are being isolated, processed, and/or engineered.
- the cells in some embodiments are primary cells, e.g., primary human cells.
- the samples include tissue, fluid, and other samples taken directly from the subject, as well as samples resulting from one or more processing steps, such as separation, centrifugation, genetic engineering (e.g. transduction with viral vector), washing, and/or incubation.
- the biological sample can be a sample obtained directly from a biological source or a sample that is processed.
- Biological samples include, but are not limited to, body fluids, such as blood, plasma, serum, cerebrospinal fluid, synovial fluid, urine and sweat, tissue and organ samples, including processed samples derived therefrom.
- the sample from which the cells are derived or isolated is blood or a blood-derived sample, or is derived from an apheresis or leukapheresis product.
- exemplary samples include whole blood, peripheral blood mononuclear cells (PBMCs), leukocytes, bone marrow, thymus, tissue biopsy, tumor, leukemia, lymphoma, lymph node, gut associated lymphoid tissue, mucosa associated lymphoid tissue, spleen, other lymphoid tissues, liver, lung, stomach, intestine, colon, kidney, pancreas, breast, bone, prostate, cervix, testes, ovaries, tonsil, or other organ, and/or cells derived therefrom.
- Samples include, in the context of cell therapy, e.g., adoptive cell therapy, samples from autologous and allogeneic sources.
- the cells are derived from cell lines, e.g., T cell lines.
- the cells in some embodiments are obtained from a xenogeneic source, for example, from a mouse, a rat, a non- human primate, or a pig.
- isolation of the cells includes one or more preparation and/or non- affinity based cell separation steps.
- cells are washed, centrifuged, and/or incubated in the presence of one or more reagents, for example, to remove unwanted components, enrich for desired components, lyse or remove cells sensitive to particular reagents.
- cells are separated based on one or more properties, such as density, adherent properties, size, sensitivity and/or resistance to particular components.
- cells from the circulating blood of a subject are obtained, e.g., by apheresis or leukapheresis.
- the samples contain lymphocytes, including T cells, monocytes, granulocytes, B cells, other nucleated white blood cells, red blood cells, and/or platelets, and, in some aspects, contains cells other than red blood cells and platelets.
- the blood cells collected from the subject are washed, e.g., to remove the plasma fraction and to place the cells in an appropriate buffer or media for subsequent processing steps.
- the cells are washed with phosphate buffered saline (PBS).
- PBS phosphate buffered saline
- the wash solution lacks calcium and/or magnesium and/or many or all divalent cations.
- a washing step is accomplished a semi-automated "flow-through" centrifuge (for example, the Cobe 2991 cell processor, Baxter) according to the manufacturer's instructions.
- a washing step is accomplished by tangential flow filtration (TFF) according to the manufacturer's instructions.
- the cells are resuspended in a variety of biocompatible buffers after washing, such as, for example, Ca++/Mg++-free PBS.
- components of a blood cell sample are removed and the cells directly resuspended in culture media.
- the methods include density-based cell separation methods, such as the preparation of white blood cells from peripheral blood by lysing the red blood cells and centrifugation through a Percoll or Ficoll gradient.
- the isolation methods include the separation of different cell types based on the expression or presence in the cell of one or more specific molecules, such as surface markers, e.g., surface proteins, intracellular markers, or nucleic acid. In some embodiments, any known method for separation based on such markers may be used. In some embodiments, the separation is affinity- or immunoaffinity-based separation.
- the isolation in some aspects, includes separation of cells and cell populations based on the cells' expression or expression level of one or more markers, typically cell surface markers, for example, by incubation with an antibody or binding partner that specifically binds to such markers, followed generally by washing steps and separation of cells having bound the antibody or binding partner, from those cells having not bound to the antibody or binding partner.
- Such separation steps can be based on positive selection, in which the cells having bound the reagents are retained for further use, and/or negative selection, in which the cells having not bound to the antibody or binding partner are retained. In some examples, both fractions are retained for further use. In some aspects, negative selection can be particularly useful where no antibody is available that specifically identifies a cell type in a heterogeneous population, such that separation is best carried out based on markers expressed by cells other than the desired population.
- the separation need not result in 100% enrichment or removal of a particular cell population or cells expressing a particular marker.
- positive selection of or enrichment for cells of a particular type refers to increasing the number or percentage of such cells, but need not result in a complete absence of cells not expressing the marker.
- negative selection, removal, or depletion of cells of a particular type refers to decreasing the number or percentage of such cells, but need not result in a complete removal of all such cells.
- multiple rounds of separation steps are carried out, where the positively or negatively selected fraction from one step is subjected to another separation step, such as a subsequent positive or negative selection.
- a single separation step can deplete cells expressing multiple markers simultaneously, such as by incubating cells with a plurality of antibodies or binding partners, each specific for a marker targeted for negative selection.
- multiple cell types can simultaneously be positively selected by incubating cells with a plurality of antibodies or binding partners expressed on the various cell types.
- one or more of the T cell populations is enriched for or depleted of cells that are positive for (marker+) or express high levels (marker* 11811 ) of one or more particular markers, such as surface markers, or that are negative for (marker -) or express relatively low levels (marker 10* ) of one or more markers.
- specific subpopulations of T cells such as cells positive or expressing high levels of one or more surface markers, e.g., CD28+, CD62L+, CCR7+, CD27+, CD127+, CD4+, CD8+, CD45RA+, and/or CD45RO+ T cells, are isolated by positive or negative selection techniques.
- such markers are those that are absent or expressed at relatively low levels on certain populations of T cells (such as non-memory cells) but are present or expressed at relatively higher levels on certain other populations of T cells (such as memory cells).
- the cells such as the CD8+ cells or the T cells, e.g., CD3+ cells
- the cells are enriched for (i.e., positively selected for) cells that are positive or expressing high surface levels of CD45RO, CCR7, CD28, CD27, CD44, CD127, and/or CD62L and/or depleted of (e.g., negatively selected for) cells that are positive for or express high surface levels of CD45RA.
- cells are enriched for or depleted of cells positive or expressing high surface levels of CD122, CD95, CD25, CD27, and/or IL7-Ra (CD127).
- CD8+ T cells are enriched for cells positive for CD45RO (or negative for CD45RA) and for CD62L.
- CD3+, CD28+ T cells can be positively selected using CD3/CD28 conjugated magnetic beads (e.g., DYNABEADS® M-450 CD3/CD28 T Cell Expander).
- CD3/CD28 conjugated magnetic beads e.g., DYNABEADS® M-450 CD3/CD28 T Cell Expander
- T cells are separated from a peripheral blood mononuclear cell (PBMC) sample by negative selection of markers expressed on non-T cells, such as B cells, monocytes, or other white blood cells, such as CD 14.
- PBMC peripheral blood mononuclear cell
- a CD4+ or CD8+ selection step is used to separate CD4+ helper and CD8+ cytotoxic T cells.
- Such CD4+ and CD8+ populations can be further sorted into sub-populations by positive or negative selection for markers expressed or expressed to a relatively higher degree on one or more naive, memory, and/or effector T cell subpopulations.
- CD8+ cells are further enriched for or depleted of naive, central memory, effector memory, and/or central memory stem cells, such as by positive or negative selection based on surface antigens associated with the respective subpopulation.
- enrichment for central memory T (TCM) cells is carried out to increase efficacy, such as to improve long-term survival, expansion, and/or engraftment following administration, which in some aspects is particularly robust in such sub-populations. (See Terakura et al. (2012) Blood.1 :72-82; Wang et al. (2012) J Immunother. 35(9):689-701.)
- combining TCM-enriched CD8+ T cells and CD4+ T cells further enhances efficacy.
- memory T cells are present in both CD62L+ and CD62L- subsets of CD8+ peripheral blood lymphocytes.
- PBMC can be enriched for or depleted of CD62L-CD8+ and/or CD62L+CD8+ fractions, such as using anti-CD8 and anti-CD62L antibodies.
- a CD4+ T cell population and a CD8+ T cell sub-population e.g., a sub -population enriched for central memory (TCM) cells.
- the enrichment for central memory T (TCM) cells is based on positive or high surface expression of CD45RO, CD62L, CCR7, CD28, CD3, and/or CD 127; in some aspects, it is based on negative selection for cells expressing or highly expressing CD45RA and/or granzyme B.
- isolation of a CD8+ population enriched for TCM cells is carried out by depletion of cells expressing CD4, CD 14, CD45RA, and positive selection or enrichment for cells expressing CD62L.
- enrichment for central memory T (TCM) cells is carried out starting with a negative fraction of cells selected based on CD4 expression, which is subjected to a negative selection based on expression of CD14 and CD45RA, and a positive selection based on CD62L.
- Such selections are carried out simultaneously and in other aspects are carried out sequentially, in either order.
- the same CD4 expression-based selection step used in preparing the CD8+ cell population or subpopulation also is used to generate the CD4+ cell population or sub -population, such that both the positive and negative fractions from the CD4-based separation are retained and used in subsequent steps of the methods, optionally following one or more further positive or negative selection steps.
- a sample of PBMCs or other white blood cell sample is subjected to selection of CD4+ cells, where both the negative and positive fractions are retained.
- the negative fraction then is subjected to negative selection based on expression of CD14 and CD45RA or CD19, and positive selection based on a marker characteristic of central memory T cells, such as CD62L or CCR7, where the positive and negative selections are carried out in either order.
- CD4+ T helper cells are sorted into naive, central memory, and effector cells by identifying cell populations that have cell surface antigens.
- CD4+ lymphocytes can be obtained by standard methods.
- naive CD4+ T lymphocytes are CD45RO-, CD45RA+, CD62L+, CD4+ T cells.
- central memory CD4+ cells are CD62L+ and CD45RO+.
- effector CD4+ cells are CD62L- and CD45RO.
- a monoclonal antibody cocktail typically includes antibodies to CD14, CD20, CDl lb, CD16, HLA-DR, and CD8.
- the antibody or binding partner is bound to a solid support or matrix, such as a magnetic bead or paramagnetic bead, to allow for separation of cells for positive and/or negative selection.
- a solid support or matrix such as a magnetic bead or paramagnetic bead
- the cells and cell populations are separated or isolated using immunomagnetic (or affinitymagnetic) separation techniques (reviewed in Methods in Molecular Medicine, vol. 58: Metastasis Research Protocols, Vol. 2: Cell Behavior In Vitro and In Vivo, p 17-25 Edited by: S. A. Brooks and U. Schumacher ⁇ Humana Press Inc., Totowa, NJ).
- the cells are incubated and/or cultured prior to or in connection with genetic engineering.
- the incubation steps can include culture, cultivation, stimulation, activation, and/or propagation.
- the compositions or cells are incubated in the presence of stimulating conditions or a stimulatory agent. Such conditions include those designed to induce proliferation, expansion, activation, and/or survival of cells in the population, to mimic antigen exposure, and/or to prime the cells for genetic engineering, such as for the introduction of a recombinant antigen receptor.
- the conditions can include one or more of particular media, temperature, oxygen content, carbon dioxide content, time, agents, e.g., nutrients, amino acids, antibiotics, ions, and/or stimulatory factors, such as cytokines, chemokines, antigens, binding partners, fusion proteins, recombinant soluble receptors, and any other agents designed to activate the cells.
- agents e.g., nutrients, amino acids, antibiotics, ions, and/or stimulatory factors, such as cytokines, chemokines, antigens, binding partners, fusion proteins, recombinant soluble receptors, and any other agents designed to activate the cells.
- the stimulating conditions or agents include one or more agent, e.g., ligand, which is capable of activating an intracellular signaling domain of a TCR complex.
- the agent turns on or initiates TCR/CD3 intracellular signaling cascade in a T cell.
- agents can include antibodies, such as those specific for a TCR component and/or costimulatory receptor, e.g., anti-CD3, anti-CD28, for example, bound to solid support such as a bead, and/or one or more cytokines.
- the expansion method may further comprise the step of adding anti-CD3 and/or anti CD28 antibody to the culture medium (e.g., at a concentration of at least about 0.5 ng/ml).
- the stimulating agents include IL-2 and/or IL-15, for example, an IL-2 concentration of at least about 10 units/mL.
- incubation is carried out in accordance with techniques such as those described in US Patent No. 6,040, 1 77 to Riddell et al., Klebanoff et al. (2012) J Immunother. 35(9): 651-660, Terakura et al. (2012) Blood.1 : 72-82, and/or Wang et al. (2012) J Immunother. 35(9):689- 701.
- the T cells are expanded by adding to the culture -initiating composition feeder cells, such as non-dividing PBMCs (e.g., such that the resulting population of cells contains at least about 5, 10, 20, or 40 or more PBMC feeder cells for each T lymphocyte in the initial population to be expanded), and incubating the culture (e.g., for a time sufficient to expand the numbers of T cells).
- the non-dividing feeder cells can comprise gamma-irradiated PBMC feeder cells.
- the PBMC are irradiated with gamma rays in the range of about 3000 to 3600 rads to prevent cell division.
- the feeder cells are added to culture medium prior to the addition of the populations of T cells.
- the stimulating conditions include temperature suitable for the growth of human T lymphocytes, for example, at least about 25 degrees Celsius, generally at least about 30 degrees Celsius, and generally at or about 37 degrees Celsius.
- the incubation may further comprise adding non-dividing EBV-transformed lymphoblastoid cells (LCLs) as feeder cells.
- LCLs can be irradiated with gamma rays in the range of about 6000 to 10,000 rads.
- the LCL feeder cells in some aspects, are provided in any suitable amount, such as a ratio of LCL feeder cells to initial T lymphocytes of at least about 10: 1.
- the preparation methods include steps for freezing, e.g., cryopreserving, the cells, either before or after isolation, incubation, and/or engineering.
- the freeze and subsequent thaw step removes granulocytes and, to some extent, monocytes in the cell population.
- the cells are suspended in a freezing solution, e.g., following a washing step to remove plasma and platelets. Any of a variety of known freezing solutions and parameters may be used in certain aspects.
- a freezing solution e.g., following a washing step to remove plasma and platelets.
- Any of a variety of known freezing solutions and parameters may be used in certain aspects.
- One example involves using PBS containing 20% DMSO and 8% human serum albumin (HSA), or other suitable cell freezing media. This is then diluted 1 : 1 with media so that the final concentration of DMSO and HSA are 10% and 4%), respectively.
- the cells are generally then frozen to -80° C at a rate of 1° per minute and stored in the vapor phase of
- the methods include re-introducing the engineered cells into the same patient, before or after cryopreservation.
- the cells comprise one or more nucleic acids encoding a recombinant receptor introduced via genetic engineering, and genetically engineered products of such nucleic acids.
- the cells can be produced or generated by introducing into a cell (e.g., via transduction of a viral vector, such as a retroviral or lentiviral vector) a nucleic acid molecule encoding the recombinant receptor.
- the nucleic acids are heterologous, i.e., normally not present in a cell or sample obtained from the cell, such as one obtained from another organism or cell, which for example, is not ordinarily found in the cell being engineered and/or an organism from which such cell is derived.
- the nucleic acids are not naturally occurring, such as a nucleic acid not found in nature, including one comprising chimeric combinations of nucleic acids encoding various domains from multiple different cell types.
- the target cell has been altered to bind to one or more target antigen, such as one or more tumor antigen.
- the target antigen is selected from ROR1, B cell maturation antigen (BCMA), carbonic anhydrase 9 (CAIX), tEGFR, Her2/neu (receptor tyrosine kinase erbB2), LI -CAM, CD19, CD20, CD22, mesothelin, CEA, and hepatitis B surface antigen, anti-folate receptor, CD23, CD24, CD30, CD33, CD38, CD44, EGFR, epithelial glycoprotein 2 (EPG-2), epithelial glycoprotein 40 (EPG-40), EPHa2, erb-B2, erb-B3, erb-B4, erbB dimers, EGFR vIII, folate binding protein (FBP), FCRL5, FCRH5, fetal acetylcholine receptor, GD2,
- the target cell has been altered to bind one or more of the following tumor antigens, e.g., by a TCR or a CAR.
- Tumor antigens may include, but are not limited to, AD034, AKT1, BRAP, CAGE, CDX2, CLP, CT-7, CT8/HOM-TES-85, cTAGE-1, Fibulin-1 , HAGE, HCA587/MAGE-C2, hCAP-G, HCE661, HER2/neu, HLA-Cw, HOM- HD-21/Galectin9, HOM-MEEL-40/S SX2, HOM-RCC-3.1.3/CAXII, HOXA7, HOXB6, Hu, HUB1, KM-HN-3, KM-KN-1, KOC1, KOC2, KOC3, KOC3, LAGE-1, MAGE-1, MAGE-4a, MPP11, MSLN, NNP-1, NY-BR-1, NY-BR-62, NY-BR-85,
- the cells generally express recombinant receptors, such as antigen receptors including functional non-TCR antigen receptors, e.g., chimeric antigen receptors (CARs), and other antigen- binding receptors such as transgenic T cell receptors (TCRs). Also among the receptors are other chimeric receptors.
- antigen receptors including functional non-TCR antigen receptors, e.g., chimeric antigen receptors (CARs), and other antigen- binding receptors such as transgenic T cell receptors (TCRs).
- CARs chimeric antigen receptors
- TCRs transgenic T cell receptors
- Exemplary antigen receptors including CARs, and methods for engineering and introducing such receptors into cells, include those described, for example, in international patent application publication numbers WO200014257, WO2013126726, WO2012/129514, WO2014031687, WO2013/166321, WO2013/071154, WO2013/123061 U.S. patent application publication numbers US2002131960, US2013287748, US20130149337, U.S. Patent Nos. : 6,451,995,
- the antigen receptors include a CAR as described in U.S. Patent No. : 7,446, 190, and those described in International Patent Application Publication No. : WO/2014055668 Al .
- CARs examples include CARs as disclosed in any of the aforementioned publications, such as WO2014031687, US 8,339,645, US 7,446,179, US 2013/0149337, U.S. Patent No. : 7,446,190, US Patent No.: 8,389,282, Kochenderfer et al., 2013, Nature Reviews Clinical Oncology, 10, 267-276 (2013); Wang et al. (2012) J. Immunother. 35(9): 689-701 ; and Brentjens et al., Sci Transl Med. 2013 5(177). See also WO2014031687, US 8,339,645, US 7,446,179, US 2013/0149337, U.S. Patent No.: 7,446, 190, and US Patent No.
- the chimeric receptors such as CARs, generally include an extracellular antigen binding domain, such as a portion of an antibody molecule, generally a variable heavy (VH) chain region and/or variable light (VL) chain region of the antibody, e.g., an scFv antibody fragment.
- VH variable heavy
- VL variable light
- the antigen targeted by the receptor is a polypeptide. In some embodiments, it is a carbohydrate or other molecule. In some embodiments, the antigen is selectively expressed or overexpressed on cells of the disease or condition, e.g., the tumor or pathogenic cells, as compared to normal or non-targeted cells or tissues. In other embodiments, the antigen is expressed on normal cells and/or is expressed on the engineered cells.
- Antigens that may be targeted by the receptors include, but are not limited to, ⁇ 6 integrin (avb6 integrin), B cell maturation antigen (BCMA), B7-H6, carbonic anhydrase 9 (CA9, also known as CAIX or G250), a cancer-testis antigen, cancer/testis antigen IB (CTAG, also known as NY-ESO-1 and LAGE-2), carcinoembryonic antigen (CEA), a cyclin, cyclin A2, C-C Motif Chemokine Ligand 1 (CCL-1), CD19, CD20, CD22, CD23, CD24, CD30, CD33, CD38, CD44, CD44v6, CD44v7/8, CD 123, CD 138, CD171, epidermal growth factor protein (EGFR), truncated epidermal growth factor protein (tEGFR), type III epidermal growth factor receptor mutation (EGFR vIII), epithelial glycoprotein 2 (EPG-2), epithelial glycoprotein
- antigens targeted by the receptors include orphan tyrosine kinase receptor ROR1, tEGFR, Her2, Ll-CAM, CD19, CD20, CD22, mesothelin, CEA, and hepatitis B surface antigen, anti-folate receptor, CD23, CD24, CD30, CD33, CD38, CD44, EGFR, EGP-2, EGP-4, 0EPHa2, ErbB2, 3, or 4, FBP, fetal acetylcholine e receptor, GD2, GD3, HMW-MAA, IL-22R-alpha, IL-13R-alpha2, kdr, kappa light chain, Lewis Y, LI -cell adhesion molecule, MAGE-A1, mesothelin, MUC1, MUC16, PSCA, NKG2D Ligands, NY-ESO-1, MART-1, gplOO, oncofetal antigen, ROR1, tEGFR, Her2, Ll-
- the CAR has binding specificity for a tumor associated antigen, e.g., CD19, CD20, carbonic anhydrase IX (CAIX), CD171, CEA, ERBB2, GD2, alpha-folate receptor, Lewis Y antigen, prostate specific membrane antigen (PSMA) or tumor associated glycoprotein 72 (TAG72).
- a tumor associated antigen e.g., CD19, CD20, carbonic anhydrase IX (CAIX), CD171, CEA, ERBB2, GD2, alpha-folate receptor, Lewis Y antigen, prostate specific membrane antigen (PSMA) or tumor associated glycoprotein 72 (TAG72).
- a tumor associated antigen e.g., CD19, CD20, carbonic anhydrase IX (CAIX), CD171, CEA, ERBB2, GD2, alpha-folate receptor, Lewis Y antigen, prostate specific membrane antigen (PSMA) or tumor associated glycoprotein 72 (TAG72).
- PSMA
- the CAR binds a pathogen-specific antigen.
- the CAR is specific for viral antigens (such as HIV, HCV, HBV, etc.), bacterial antigens, and/or parasitic antigens.
- the chimeric receptors are chimeric antigen receptors (CARs).
- CARs chimeric antigen receptors
- the chimeric receptors such as CARs, generally include an extracellular antigen binding domain, such as a portion of an antibody molecule, generally a variable heavy (VH) chain region and/or variable light (VL) chain region of the antibody, e.g., an scFv antibody fragment.
- VH variable heavy
- VL variable light chain region of the antibody, e.g., an scFv antibody fragment.
- the antibody portion of the recombinant receptor e.g., CAR
- an immunoglobulin constant region such as a hinge region, e.g., an IgG4 hinge region, and/or a CH1/CL and/or Fc region.
- the constant region or portion is of a human IgG, such as IgG4 or IgGl .
- the portion of the constant region serves as a spacer region between the antigen-recognition component, e.g., scFv, and transmembrane domain.
- the spacer can be of a length that provides for increased responsiveness of the cell following antigen binding, as compared to in the absence of the spacer.
- Exemplary spacers e.g., hinge regions, include those described in international patent application publication number WO2014031687.
- the spacer is or is about 12 amino acids in length or is no more than 12 amino acids in length.
- Exemplary spacers include those having at least about 10 to 229 amino acids, about 10 to 200 amino acids, about 10 to 175 amino acids, about 10 to 150 amino acids, about 10 to 125 amino acids, about 10 to 100 amino acids, about 10 to 75 amino acids, about 10 to 50 amino acids, about 10 to 40 amino acids, about 10 to 30 amino acids, about 10 to 20 amino acids, or about 10 to 15 amino acids, and including any integer between the endpoints of any of the listed ranges.
- a spacer region has about 12 amino acids or less, about 119 amino acids or less, or about 229 amino acids or less.
- Exemplary spacers include IgG4 hinge alone, IgG4 hinge linked to CH2 and CH3 domains, or IgG4 hinge linked to the CH3 domain.
- Exemplary spacers include, but are not limited to, those described in Hudecek et al. (2013) Clin. Cancer Res., 19:3153 or international patent application publication number WO2014031687.
- This antigen recognition domain generally is linked to one or more intracellular signaling components, such as signaling components that mimic activation through an antigen receptor complex, such as a TCR complex, in the case of a CAR, and/or signal via another cell surface receptor.
- the antigen-binding component e.g., antibody
- the transmembrane domain is fused to the extracellular domain.
- a transmembrane domain that naturally is associated with one of the domains in the receptor e.g., CAR
- the transmembrane domain is selected or modified by amino acid substitution to avoid binding of such domains to the transmembrane domains of the same or different surface membrane proteins to minimize interactions with other members of the receptor complex.
- the transmembrane domain in some embodiments is derived either from a natural or from a synthetic source. Where the source is natural, the domain, in some aspects, is derived from any membrane-bound or transmembrane protein.
- Transmembrane regions include those derived from (i.e., comprise at least the transmembrane region(s) of) the alpha, beta or zeta chain of the T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD 16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154.
- the transmembrane domain in some embodiments is synthetic.
- the synthetic transmembrane domain comprises predominantly hydrophobic residues such as leucine and valine. In some aspects, a triplet of phenylalanine, tryptophan and valine will be found at each end of a synthetic transmembrane domain. In some embodiments, the linkage is by linkers, spacers, and/or transmembrane domain(s).
- intracellular signaling domains are those that mimic or approximate a signal through a natural antigen receptor, a signal through such a receptor in combination with a costimulatory receptor, and/or a signal through a costimulatory receptor alone.
- a short oligo- or polypeptide linker for example, a linker of between 2 and 10 amino acids in length, such as one containing glycines and serines, e.g., glycine-serine doublet, is present and forms a linkage between the transmembrane domain and the cytoplasmic signaling domain of the CAR.
- the receptor e.g., the CAR
- the receptor generally includes at least one intracellular signaling component or components.
- the receptor includes an intracellular component of a TCR complex, such as a TCR CD3 chain that mediates T-cell activation and cytotoxicity, e.g., CD3 zeta chain.
- the antigen-binding portion is linked to one or more cell signaling modules.
- cell signaling modules include CD3 transmembrane domain, CD3 intracellular signaling domains, and/or other CD transmembrane domains.
- the receptor e.g., CAR
- the receptor further includes a portion of one or more additional molecules such as Fc receptor ⁇ , CD8, CD4, CD25, or CD16.
- the CAR or other chimeric receptor includes a chimeric molecule between CD3-zeta (CD3- or Fc receptor ⁇ and CD8, CD4, CD25 or CD 16.
- the cytoplasmic domain or intracellular signaling domain of the receptor activates at least one of the normal effector functions or responses of the immune cell, e.g., T cell engineered to express the CAR.
- the CAR induces a function of a T cell such as cytolytic activity or T- helper activity, such as secretion of cytokines or other factors.
- a truncated portion of an intracellular signaling domain of an antigen receptor component or costimulatory molecule is used in place of an intact immunostimulatory chain, for example, if it transduces the effector function signal.
- the intracellular signaling domain or domains include the cytoplasmic sequences of the T cell receptor (TCR), and in some aspects, also those of co- receptors that in the natural context act in concert with such receptors to initiate signal transduction following antigen receptor engagement, and/or any derivative or variant of such molecules, and/or any synthetic sequence that has the same functional capability.
- TCR T cell receptor
- full activation generally requires not only signaling through the TCR, but also a costimulatory signal.
- a component for generating secondary or co-stimulatory signal is also included in the CAR.
- the CAR does not include a component for generating a costimulatory signal.
- an additional CAR is expressed in the same cell and provides the component for generating the secondary or costimulatory signal.
- T cell activation is, in some aspects, described as being mediated by two classes of cytoplasmic signaling sequences: those that initiate antigen-dependent primary activation through the TCR (primary cytoplasmic signaling sequences), and those that act in an antigen-independent manner to provide a secondary or co- stimulatory signal (secondary cytoplasmic signaling sequences).
- primary cytoplasmic signaling sequences those that initiate antigen-dependent primary activation through the TCR
- secondary cytoplasmic signaling sequences those that act in an antigen-independent manner to provide a secondary or co- stimulatory signal.
- the CAR includes one or both of such signaling components.
- the CAR includes a primary cytoplasmic signaling sequence that regulates primary activation of the TCR complex.
- Primary cytoplasmic signaling sequences that act in a stimulatory manner may contain signaling motifs which are known as immunoreceptor tyrosine- based activation motifs or ITAMs.
- ITAM containing primary cytoplasmic signaling sequences include those derived from the CD3 zeta chain, FcR gamma, CD3 gamma, CD3 delta and CD3 epsilon.
- cytoplasmic signaling molecule(s) in the CAR contain(s) a cytoplasmic signaling domain, portion thereof, or sequence derived from CD3 zeta.
- the CAR includes a signaling domain and/or transmembrane portion of a costimulatory receptor, such as CD28, 4-1BB, OX40, DAP10, and ICOS.
- a costimulatory receptor such as CD28, 4-1BB, OX40, DAP10, and ICOS.
- the same CAR includes both the activating and costimulatory components.
- the activating domain is included within one CAR, whereas the costimulatory component is provided by another CAR recognizing another antigen.
- the CARs include activating or stimulatory CARs, costimulatory CARs, both expressed on the same cell (see WO2014/055668).
- the cells include one or more stimulatory or activating CAR and/or a costimulatory CAR.
- the cells further include inhibitory CARs (iCARs, see Fedorov et al., Sci. Transl.
- the intracellular signaling domain comprises a CD28 transmembrane and signaling domain linked to a CD3 (e.g., CD3-zeta) intracellular domain.
- the intracellular signaling domain comprises a chimeric CD28 and CD137 (4-1BB, TNFRSF9) co-stimulatory domains, linked to a CD3 zeta intracellular domain.
- the CAR encompasses one or more, e.g., two or more, costimulatory domains and an activation domain, e.g., primary activation domain, in the cytoplasmic portion.
- exemplary CARs include intracellular components of CD3-zeta, CD28, and 4-1BB.
- the CAR or other antigen receptor further includes a marker, such as a cell surface marker, which may be used to confirm transduction or engineering of the cell to express the receptor, such as a truncated version of a cell surface receptor, such as truncated EGFR (tEGFR).
- a marker such as a cell surface marker, which may be used to confirm transduction or engineering of the cell to express the receptor, such as a truncated version of a cell surface receptor, such as truncated EGFR (tEGFR).
- the marker includes all or part (e.g., truncated form) of CD34, a NGFR, or epidermal growth factor receptor (e.g., tEGFR).
- the nucleic acid encoding the marker is operably linked to a polynucleotide encoding for a linker sequence, such as a cleavable linker sequence, e.g., T2A.
- a linker sequence such as a cleavable linker sequence, e.g., T2A.
- introduction of a construct encoding the CAR and EGFRt separated by a T2A ribosome switch can express two proteins from the same construct, such that the EGFRt can be used as a marker to detect cells expressing such construct.
- a marker, and optionally a linker sequence can be any as disclosed in published Application No. WO 2014/031687.
- the marker can be a truncated EGFR (tEGFR) that is optionally linked to a linker sequence, such as a T2A cleavable linker sequence.
- tEGFR truncated EGFR
- the marker is a molecule, e.g., cell surface protein, not naturally found on T cells or not naturally found on the surface of T cells, or a portion thereof.
- the molecule is a non-self molecule, e.g., non-self protein, i.e., one that is not recognized as "self by the immune system of the host into which the cells will be adoptively transferred.
- the marker serves no therapeutic function and/or produces no effect other than to be used as a marker for genetic engineering, e.g., for selecting cells successfully engineered.
- the marker may be a therapeutic molecule or molecule otherwise exerting some desired effect, such as a ligand for a cell to be encountered in vivo, such as a costimulatory or immune checkpoint molecule to enhance and/or dampen responses of the cells upon adoptive transfer and encounter with ligand.
- CARs are referred to as first, second, and/or third generation CARs.
- a first-generation CAR is one that solely provides a CD3 -chain induced signal upon antigen binding;
- a second-generation CARs is one that provides such a signal and costimulatory signal, such as one including an intracellular signaling domain from a costimulatory receptor such as CD28 or CD 137;
- a third generation CAR is one that includes multiple costimulatory domains of different costimulatory receptors.
- the chimeric antigen receptor includes an extracellular portion containing an antibody or antibody fragment. In some aspects, the chimeric antigen receptor includes an extracellular portion containing the antibody or fragment and an intracellular signaling domain. In some embodiments, the antibody or fragment includes an scFv and the intracellular domain contains an ITAM. In some aspects, the intracellular signaling domain includes a signaling domain of a zeta chain of a CD3-zeta ( ⁇ 3 ⁇ ) chain. In some embodiments, the chimeric antigen receptor includes a transmembrane domain linking the extracellular domain and the intracellular signaling domain. In some aspects, the transmembrane domain contains a transmembrane portion of CD28.
- the extracellular domain and transmembrane can be linked directly or indirectly.
- the extracellular domain and transmembrane are linked by a spacer, such as any described herein.
- the chimeric antigen receptor contains an intracellular domain of a T cell costimulatory molecule, such as between the transmembrane domain and intracellular signaling domain.
- the T cell costimulatory molecule is CD28 or 41BB.
- the CAR contains an antibody, e.g., an antibody fragment, a transmembrane domain that is or contains a transmembrane portion of CD28 or a functional variant thereof, and an intracellular signaling domain containing a signaling portion of CD28 or functional variant thereof and a signaling portion of CD3 zeta or functional variant thereof.
- the CAR contains an antibody, e.g., antibody fragment, a transmembrane domain that is or contains a transmembrane portion of CD28 or a functional variant thereof, and an intracellular signaling domain containing a signaling portion of a 4- IBB or functional variant thereof and a signaling portion of CD3 zeta or functional variant thereof.
- the receptor further includes a spacer containing a portion of an Ig molecule, such as a human Ig molecule, such as an Ig hinge, e.g. an IgG4 hinge, such as a hinge-only spacer.
- an Ig molecule such as a human Ig molecule
- an Ig hinge e.g. an IgG4 hinge, such as a hinge-only spacer.
- the transmembrane domain of the receptor e.g., the CAR is a transmembrane domain of human CD28 or variant thereof, e.g., a 27-amino acid transmembrane domain of a human CD28 (Accession No. : P10747.1).
- the chimeric antigen receptor contains an intracellular domain of a T cell costimulatory molecule.
- the T cell costimulatory molecule is CD28 or 41BB.
- the intracellular signaling domain comprises an intracellular costimulatory signaling domain of human CD28 or functional variant or portion thereof, such as a 41 amino acid domain thereof and/or such a domain with an LL to GG substitution at positions 186- 187 of a native CD28 protein.
- the intracellular domain comprises an intracellular costimulatory signaling domain of 4 IBB or functional variant or portion thereof, such as a 42-amino acid cytoplasmic domain of a human 4-1BB (Accession No. Q07011.1) or functional variant or portion thereof.
- the intracellular signaling domain comprises a human CD3 zeta stimulatory signaling domain or functional variant thereof, such as a 112 AA cytoplasmic domain of isoform 3 of human ⁇ )3 ⁇ (Accession No.: P20963.2) or a CD3 zeta signaling domain as described in U.S. Patent No. : 7,446,190 or U.S. Patent No. 8,911,993.
- a human CD3 zeta stimulatory signaling domain or functional variant thereof such as a 112 AA cytoplasmic domain of isoform 3 of human ⁇ )3 ⁇ (Accession No.: P20963.2) or a CD3 zeta signaling domain as described in U.S. Patent No. : 7,446,190 or U.S. Patent No. 8,911,993.
- the spacer contains only a hinge region of an IgG, such as only a hinge of IgG4 or IgGl .
- the spacer is an Ig hinge, e.g., and IgG4 hinge, linked to a CH2 and/or CH3 domains.
- the spacer is an Ig hinge, e.g., an IgG4 hinge, linked to CH2 and CH3 domains.
- the spacer is an Ig hinge, e.g., an IgG4 hinge, linked to a CH3 domain only.
- the spacer is or comprises a glycine- serine rich sequence or other flexible linker such as known flexible linkers.
- the CAR includes an antibody or fragment that specifically binds an antigen, a spacer such as any of the Ig-hinge containing spacers, a CD28 transmembrane domain, a CD28 intracellular signaling domain, and a CD3 zeta signaling domain.
- the CAR includes an antibody or fragment that specifically binds an antigen, a spacer such as any of the Ig-hinge containing spacers, a CD28 transmembrane domain, a CD28 intracellular signaling domain, and a CD3 zeta signaling domain.
- such CAR constructs further include a T2A ribosomal skip element and/or a tEGFR sequence, e.g., downstream of the CAR.
- polypeptide and “protein” are used interchangeably to refer to a polymer of amino acid residues, and are not limited to a minimum length.
- Polypeptides including the provided receptors and other polypeptides, e.g., linkers or peptides, may include amino acid residues including natural and/or non-natural amino acid residues.
- the terms also include post-expression modifications of the polypeptide, for example, glycosylation, sialylation, acetylation, and phosphorylation.
- the polypeptides may contain modifications with respect to a native or natural sequence, as long as the protein maintains the desired activity. These modifications may be deliberate, as through site-directed mutagenesis, or may be accidental, such as through mutations of hosts which produce the proteins or errors due to PCR amplification.
- the genetically engineered antigen receptors include recombinant T cell receptors (TCRs) and/or TCRs cloned from naturally occurring T cells.
- TCRs T cell receptors
- the target cell has been altered to contain specific T cell receptor (TCR) genes (e.g., a TRAC and TRBC gene).
- TCRs or antigen-binding portions thereof include those that recognize a peptide epitope or T cell epitope of a target polypeptide, such as an antigen of a tumor, viral or autoimmune protein.
- the TCR has binding specificity for a tumor associated antigen, e.g., carcinoembryonic antigen (CEA), GP100, melanoma antigen recognized by T cells 1 (MARTI), melanoma antigen A3 (MAGEA3), YESOl or p53.
- a tumor associated antigen e.g., carcinoembryonic antigen (CEA), GP100, melanoma antigen recognized by T cells 1 (MARTI), melanoma antigen A3 (MAGEA3), YESOl or p53.
- a "T cell receptor” or “TCR” is a molecule that contains a variable a and ⁇ chains (also known as TCRa and TCRp, respectively) or a variable ⁇ and ⁇ chains (also known as TCRy and TCR6, respectively), or antigen-binding portions thereof, and which is capable of specifically binding to a peptide bound to an MHC molecule.
- the TCR is in the ⁇ form.
- TCRs that exist in ⁇ and ⁇ forms are generally structurally similar, but T cells expressing them may have distinct anatomical locations or functions.
- a TCR is or can be expressed on the surface of T cells (or T lymphocytes) where it is generally responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules.
- MHC major histocompatibility complex
- the TCR is a full TCR or an antigen-binding portions or antigen- binding fragments thereof.
- the TCR is an intact or full-length TCR, including TCRs in the ⁇ form or ⁇ form.
- the TCR is an antigen-binding portion that is less than a full-length TCR but that binds to a specific peptide bound in an MHC molecule, such as binds to an MHC-peptide complex.
- an antigen-binding portion or fragment of a TCR can contain only a portion of the structural domains of a full-length or intact TCR, but yet is able to bind the peptide epitope, such as MHC-peptide complex, to which the full TCR binds.
- an antigen-binding portion contains the variable domains of a TCR, such as variable a chain and variable ⁇ chain of a TCR, sufficient to form a binding site for binding to a specific MHC-peptide complex.
- the variable chains of a TCR contain complementarity determining regions (CDRs) involved in recognition of the peptide, MHC and/or MHC-peptide complex.
- variable domains of the TCR contain hypervariable loops, or CDRs, which generally are the primary contributors to antigen recognition and binding capabilities and specificity.
- CDRs hypervariable loops
- a CDR of a TCR or combination thereof forms all or substantially all of the antigen-binding site of a given TCR molecule.
- the various CDRs within a variable region of a TCR chain generally are separated by framework regions (FRs), which generally display less variability among TCR molecules as compared to the CDRs (see, e.g., Jores et ah, Proc. Nat'l Acad. Sci. U.S.A. #7:9138, 1990; Chothia et al, EMBO J.
- CDR3 is the main CDR responsible for antigen binding or specificity, or is the most important among the three CDRs on a given TCR variable region for antigen recognition, and/or for interaction with the processed peptide portion of the peptide-MHC complex.
- the CDR1 of the alpha chain can interact with the N- terminal part of certain antigenic peptides.
- CDR1 of the beta chain can interact with the C-terminal part of the peptide.
- CDR2 contributes most strongly to or is the primary CDR responsible for the interaction with or recognition of the MHC portion of the MHC- peptide complex.
- the variable region of the ⁇ -chain can contain a further hypervariable region (CDR4 or HVR4), which generally is involved in super-antigen binding and not antigen recognition (Kotb (1995) Clinical Microbiology Reviews, 8:411-426).
- a TCR contains a variable alpha domain (V a ) and/or a variable beta domain (Vp) or antigen-binding fragments thereof.
- the a-chain and/or ⁇ - chain of a TCR also can contain a constant domain, a transmembrane domain and/or a short cytoplasmic tail (see, e.g., Janeway et al., Immunobiology: The Immune System in Health and Disease, 3 rd Ed., Current Biology Publications, p. 4:33, 1997).
- the a chain constant domain is encoded by the TRAC gene (IMGT nomenclature) or is a variant thereof.
- the ⁇ chain constant region is encoded by TRBC1 or TRBC2 genes (IMGT nomenclature) or is a variant thereof.
- the constant domain is adjacent to the cell membrane.
- the extracellular portion of the TCR formed by the two chains contains two membrane -proximal constant domains, and two membrane-distal variable domains, which variable domains each contain CDRs.
- the CDR1 sequences within a TCR Va chains and/or ⁇ chain correspond to the amino acids present between residue numbers 27-38, inclusive
- the CDR2 sequences within a TCR Va chain and/or ⁇ chain correspond to the amino acids present between residue numbers 56-65, inclusive
- the CDR3 sequences within a TCR Va chain and/or ⁇ chain correspond to the amino acids present between residue numbers 105-117, inclusive.
- the TCR may be a heterodimer of two chains a and ⁇ (or optionally ⁇ and ⁇ ) that are linked, such as by a disulfide bond or disulfide bonds.
- the constant domain of the TCR may contain short connecting sequences in which a cysteine residue forms a disulfide bond, thereby linking the two chains of the TCR.
- a TCR may have an additional cysteine residue in each of the a and ⁇ chains, such that the TCR contains two disulfide bonds in the constant domains.
- each of the constant and variable domains contain disulfide bonds formed by cysteine residues.
- the TCR for engineering cells as described is one generated from a known TCR sequence(s), such as sequences of ⁇ , ⁇ chains, for which a substantially full-length coding sequence is readily available. Methods for obtaining full-length TCR sequences, including V chain sequences, from cell sources are well known.
- nucleic acids encoding the TCR can be obtained from a variety of sources, such as by polymerase chain reaction (PCR) amplification of TCR-encoding nucleic acids within or isolated from a given cell or cells, or synthesis of publicly available TCR DNA sequences.
- the TCR is obtained from a biological source, such as from cells such as from a T cell (e.g.
- the T-cells can be obtained from in vivo isolated cells.
- the T- cells can be a cultured T-cell hybridoma or clone.
- the TCR or antigen-binding portion thereof can be synthetically generated from knowledge of the sequence of the TCR.
- a high-affinity T cell clone for a target antigen e.g., a cancer antigen is identified, isolated from a patient, and introduced into the cells.
- the TCR clone for a target antigen has been generated in transgenic mice engineered with human immune system genes (e.g., the human leukocyte antigen system, or HLA). See, e.g., tumor antigens (see, e.g., Parkhurst et al. (2009) Clin Cancer Res. 15: 169-180 and Cohen et al. (2005) J Immunol. 175 :5799- 5808.
- human immune system genes e.g., the human leukocyte antigen system, or HLA
- tumor antigens see, e.g., Parkhurst et al. (2009) Clin Cancer Res. 15: 169-180 and Cohen et al. (2005) J Immunol. 175 :5799- 5808.
- phage display is used to isolate TCRs against a target antigen (see, e.g., Varela-Rohena et al. (2008) Nat Med. 14: 1390-1395 and Li (2005) Nat Biotechnol. 23
- the TCR or antigen-binding portion thereof is one that has been modified or engineered.
- directed evolution methods are used to generate TCRs with altered properties, such as with higher affinity for a specific MHC-peptide complex.
- directed evolution is achieved by display methods including, but not limited to, yeast display (Holler et al. (2003) Nat Immunol, 4, 55-62; Holler et al. (2000) Proc Natl Acad Sci U S A, 97, 5387-92), phage display (Li et al. (2005) Nat Biotechnol, 23, 349-54), or T cell display (Chervin et al. (2008) J Immunol Methods, 339, 175-84).
- display approaches involve engineering, or modifying, a known, parent or reference TCR.
- a wild-type TCR can be used as a template for producing mutagenized TCRs in which in one or more residues of the CDRs are mutated, and mutants with a desired altered property, such as higher affinity for a desired target antigen, are selected.
- the TCR can contain an introduced disulfide bond or bonds.
- the native disulfide bonds are not present.
- the one or more of the native cysteines (e.g. in the constant domain of the a chain and ⁇ chain) that form a native inter-chain disulfide bond are substituted to another residue, such as to a serine or alanine.
- an introduced disulfide bond can be formed by mutating non-cysteine residues on the alpha and beta chains, such as in the constant domain of the a chain and ⁇ chain, to cysteine. Exemplary non-native disulfide bonds of a TCR are described in published International PCT Nos.
- cysteines can be introduced at residue Thr48 of the a chain and Ser57 of the ⁇ chain, at residue Thr45 of the a chain and Ser77 of the ⁇ chain, at residue TyrlO of the a chain and Serl7 of the ⁇ chain, at residue Thr45 of the a chain and Asp59 of the ⁇ chain and/or at residue Serl5 of the a chain and Glul5 of the ⁇ chain.
- the presence of non-native cysteine residues e.g.
- the TCR chains contain a transmembrane domain.
- the transmembrane domain is positively charged.
- the TCR chain contains a cytoplasmic tail.
- each chain (e.g. alpha or beta) of the TCR can possess one N-terminal immunoglobulin variable domain, one immunoglobulin constant domain, a transmembrane region, and a short cytoplasmic tail at the C-terminal end.
- a TCR for example via the cytoplasmic tail, is associated with invariant proteins of the CD3 complex involved in mediating signal transduction.
- the structure allows the TCR to associate with other molecules like CD3 and subunits thereof.
- a TCR containing constant domains with a transmembrane region may anchor the protein in the cell membrane and associate with invariant subunits of the CD3 signaling apparatus or complex.
- the intracellular tails of CD3 signaling subunits e.g. CD3y, CD36, CD3E and CD3C, chains
- the TCR is a full-length TCR. In some embodiments, the TCR is an antigen-binding portion. In some embodiments, the TCR is a dimeric TCR (dTCR). In some embodiments, the TCR is a single-chain TCR (sc-TCR). A TCR may be cell-bound or in soluble form. In some embodiments, for purposes of the provided methods, the TCR is in cell-bound form expressed on the surface of a cell.
- a dTCR contains a first polypeptide wherein a sequence corresponding to a TCR a chain variable region sequence is fused to the N terminus of a sequence corresponding to a TCR a chain constant region extracellular sequence, and a second polypeptide wherein a sequence corresponding to a TCR ⁇ chain variable region sequence is fused to the N terminus a sequence corresponding to a TCR ⁇ chain constant region extracellular sequence, the first and second polypeptides being linked by a disulfide bond.
- the bond can correspond to the native inter-chain disulfide bond present in native dimeric ⁇ TCRs.
- the inter-chain disulfide bonds are not present in a native TCR.
- one or more cysteines can be incorporated into the constant region extracellular sequences of dTCR polypeptide pair.
- both a native and a non-native disulfide bond may be desirable.
- the TCR contains a transmembrane sequence to anchor to the membrane.
- a dTCR contains a TCR a chain containing a variable a domain, a constant a domain and a first dimerization motif attached to the C-terminus of the constant a domain, and a TCR ⁇ chain comprising a variable ⁇ domain, a constant ⁇ domain and a first dimerization motif attached to the C-terminus of the constant ⁇ domain, wherein the first and second dimerization motifs easily interact to form a covalent bond between an amino acid in the first dimerization motif and an amino acid in the second dimerization motif linking the TCR a chain and TCR ⁇ chain together.
- the TCR is a scTCR, which is a single amino acid strand containing an a chain and a ⁇ chain that is able to bind to MHC-peptide complexes.
- a scTCR can be generated using methods known to those of skill in the art, See e.g., International published PCT Nos. WO 1996/13593, WO 1996/18105, WO 1999/18129, WO 2004/033685, WO 2006/037960, WO 2011/044186; U.S. Patent No. 7,569,664; and Schlueter, C. J. et al. J. Mol. Biol. 256, 859 (1996).
- a scTCR contains a first segment constituted by an amino acid sequence corresponding to a TCR a chain variable region, a second segment constituted by an amino acid sequence corresponding to a TCR ⁇ chain variable region sequence fused to the N terminus of an amino acid sequence corresponding to a TCR ⁇ chain constant domain extracellular sequence, and a linker sequence linking the C terminus of the first segment to the N terminus of the second segment.
- a scTCR contains a first segment constituted by an amino acid sequence corresponding to a TCR ⁇ chain variable region, a second segment constituted by an amino acid sequence corresponding to a TCR a chain variable region sequence fused to the N terminus of an amino acid sequence corresponding to a TCR a chain constant domain extracellular sequence, and a linker sequence linking the C terminus of the first segment to the N terminus of the second segment.
- a scTCR contains a first segment constituted by an a chain variable region sequence fused to the N terminus of an a chain extracellular constant domain sequence, and a second segment constituted by a ⁇ chain variable region sequence fused to the N terminus of a sequence ⁇ chain extracellular constant and transmembrane sequence, and, optionally, a linker sequence linking the C terminus of the first segment to the N terminus of the second segment.
- a scTCR contains a first segment constituted by a TCR ⁇ chain variable region sequence fused to the N terminus of a ⁇ chain extracellular constant domain sequence, and a second segment constituted by an a chain variable region sequence fused to the N terminus of a sequence a chain extracellular constant and transmembrane sequence, and, optionally, a linker sequence linking the C terminus of the first segment to the N terminus of the second segment.
- the a and ⁇ chains must be paired so that the variable region sequences thereof are orientated for such binding.
- Various methods of promoting pairing of an a and ⁇ in a scTCR are well known in the art.
- a linker sequence is included that links the a and ⁇ chains to form the single polypeptide strand.
- the linker should have sufficient length to span the distance between the C terminus of the a chain and the N terminus of the ⁇ chain, or vice versa, while also ensuring that the linker length is not so long so that it blocks or reduces bonding of the scTCR to the target peptide-MHC complex.
- the linker of a scTCRs that links the first and second TCR segments can be any linker capable of forming a single polypeptide strand, while retaining TCR binding specificity.
- the linker sequence may, for example, have the formula - P-AA-P-, wherein P is proline and AA represents an amino acid sequence wherein the amino acids are glycine and serine.
- the first and second segments are paired so that the variable region sequences thereof are orientated for such binding.
- the linker has a sufficient length to span the distance between the C terminus of the first segment and the N terminus of the second segment, or vice versa, but is not too long to block or reduces bonding of the scTCR to the target ligand.
- the linker can contain from or from about 10 to 45 amino acids, such as 10 to 30 amino acids or 26 to 41 amino acids residues, for example 29, 30, 31 or 32 amino acids.
- the linker has the formula -PGGG-(SGGGG)5-P- or -PGGG- (SGGGG)e-P-, wherein P is proline, G is glycine and S is serine.
- the linker has the sequence GS ADDAK D AAK DGKS) .
- a scTCR contains a disulfide bond between residues of the single amino acid strand, which, in some cases, can promote stability of the pairing between the a and ⁇ regions of the single chain molecule (see e.g. U.S. Patent No. 7,569,664).
- the scTCR contains a covalent disulfide bond linking a residue of the immunoglobulin region of the constant domain of the a chain to a residue of the immunoglobulin region of the constant domain of the ⁇ chain of the single chain molecule.
- the disulfide bond corresponds to the native disulfide bond present in a native dTCR.
- the disulfide bond in a native TCR is not present.
- the disulfide bond is an introduced non-native disulfide bond, for example, by incorporating one or more cysteines into the constant region extracellular sequences of the first and second chain regions of the scTCR polypeptide.
- Exemplary cysteine mutations include any as described above. In some cases, both a native and a non-native disulfide bond may be present.
- a scTCR is a non-disulfide linked truncated TCR in which heterologous leucine zippers fused to the C-termini thereof facilitate chain association (see e.g. International published PCT No. WO 1999/60120).
- a scTCR contain a TCRa variable domain covalently linked to a TCRP variable domain via a peptide linker (see e.g., International published PCT No. WO 1999/18129).
- any of the TCRs can be linked to signaling domains that yield an active TCR on the surface of a T cell.
- the TCR is expressed on the surface of cells.
- the TCR does contain a sequence corresponding to a transmembrane sequence.
- the transmembrane domain can be a Ca or ⁇ transmembrane domain.
- the transmembrane domain can be from a non-TCR origin, for example, a transmembrane region from CD3z, CD28 or B7.1.
- the TCR does contain a sequence corresponding to cytoplasmic sequences.
- the TCR contains a CD3z signaling domain.
- the TCR is capable of forming a TCR complex with CD3.
- the TCR or antigen-binding fragment thereof exhibits an affinity with an equilibrium binding constant for a target antigen of between or between about 10 "5 and 10 "12 M and all individual values and ranges therein.
- the target antigen is an MHC- peptide complex or ligand.
- the TCR or antigen binding portion thereof may be a recombinantly produced natural protein or mutated form thereof in which one or more property, such as binding characteristic, has been altered.
- a TCR may be derived from one of various animal species, such as human, mouse, rat, or other mammal.
- the a and ⁇ chains can be PCR amplified from total cDNA isolated from a T cell clone expressing the TCR of interest and cloned into an expression vector.
- the a and ⁇ chains can be synthetically generated.
- the TCR alpha and beta chains are isolated and cloned into a gene expression vector.
- transcription units can be engineered as a bicistronic unit containing an IRES (internal ribosome entry site), which allows co-expression of gene products (e.g. encoding an a and ⁇ chains) by a message from a single promoter.
- IRES internal ribosome entry site
- a single promoter may direct expression of an RNA that contains, in a single open reading frame (ORF), multiple genes (e.g.
- a and ⁇ chains separated from one another by sequences encoding a self-cleavage peptide (e.g., T2A) or a protease recognition site (e.g., furin).
- the ORF thus encodes a single polyprotein, which, either during (in the case of T2A) or after translation, is cleaved into the individual proteins.
- the peptide such as T2A, can cause the ribosome to skip (ribosome skipping) synthesis of a peptide bond at the C-terminus of a 2A element, leading to separation between the end of the 2A sequence and the next peptide downstream.
- Examples of 2A cleavage peptides are T2A, P2A, E2A and F2A.
- the a and ⁇ chains are cloned into different vectors.
- the generated a and ⁇ chains are incorporated into a retroviral, e.g., a lentiviral, vector.
- the TCR alpha and beta genes are linked via a picornavirus 2A ribosomal skip peptide so that both chains are co-expression.
- genetic transfer of the TCR is accomplished via retroviral or lentiviral vectors, or via transposons (see, e.g., Baum et al. (2006) Molecular Therapy: The Journal of the American Society of Gene Therapy. 13 : 1050-1063; Frecha et al. (2010) Molecular Therapy: The Journal of the American Society of Gene Therapy. 18: 1748-1757; anhackett et al. (2010) Molecular Therapy: The Journal of the American Society of Gene Therapy. 18:674-683.
- the provided methods include expressing the recombinant receptors, including CARs or TCRs, for producing the genetically engineered cells expressing such binding molecules.
- the genetic engineering generally involves introduction of a nucleic acid encoding the recombinant or engineered component into the cell, such as by retroviral transduction, transfection, or transformation.
- gene transfer is accomplished by first stimulating the cell, such as by combining it with a stimulus that induces a response such as proliferation, survival, and/or activation, e.g., as measured by expression of a cytokine or activation marker, followed by transduction of the activated cells, and expansion in culture to numbers sufficient for clinical applications.
- a stimulus such as proliferation, survival, and/or activation, e.g., as measured by expression of a cytokine or activation marker
- antigen receptors e.g., CARs
- exemplary methods include those for transfer of nucleic acids encoding the receptors, including via viral, e.g., retroviral or lentiviral, transduction, transposons, and electroporation.
- nucleic acid encoding a recombinant receptor can be cloned into a suitable expression vector or vectors.
- the expression vector can be any suitable recombinant expression vector, and can be used to transform or transfect any suitable host.
- Suitable vectors include those designed for propagation and expansion or for expression or both, such as plasmids and viruses.
- the vector can a vector of the pUC series (Fermentas Life Sciences), the pBluescript series (Stratagene, La Jolla, Calif), the pET series (Novagen, Madison, Wis.), the pGEX series (Pharmacia Biotech, Uppsala, Sweden), or the pEX series (Clontech, Palo Alto, Calif.).
- bacteriophage vectors such as ⁇ , ⁇ ⁇ , ZapII (Stratagene), ⁇ 4, and ⁇ 1149, also can be used.
- plant expression vectors can be used and include pBIO l, pBI101.2, pBI101.3, pBI121 and pBIN19 (Clontech).
- animal expression vectors include pEUK-Cl, pMAM and pMAMneo (Clontech).
- a viral vector is used, such as a retroviral vector.
- the recombinant expression vectors can be prepared using standard recombinant DNA techniques.
- vectors can contain regulatory sequences, such as transcription and translation initiation and termination codons, which are specific to the type of host (e.g., bacterium, fungus, plant, or animal) into which the vector is to be introduced, as appropriate and taking into consideration whether the vector is DNA- or RNA-based.
- the vector can contain a nonnative promoter operably linked to the nucleotide sequence encoding the recombinant receptor.
- the promoter can be a non-viral promoter or a viral promoter, such as a cytomegalovirus (CMV) promoter, an SV40 promoter, an RSV promoter, and a promoter found in the long-terminal repeat of the murine stem cell virus.
- CMV cytomegalovirus
- SV40 SV40 promoter
- RSV RSV promoter
- promoter found in the long-terminal repeat of the murine stem cell virus a promoter found in the long-terminal repeat of the murine stem cell virus.
- Other promoters known to a skilled artisan also are contemplated.
- recombinant nucleic acids are transferred into cells using recombinant infectious virus particles, such as, e.g., vectors derived from simian virus 40 (SV40), adenoviruses, adeno-associated virus (AAV).
- recombinant nucleic acids are transferred into T cells using recombinant lentiviral vectors or retroviral vectors, such as gamma- retroviral vectors (see, e.g., Koste et al. (2014) Gene Therapy 2014 Apr 3. doi: 10.1038/gt.2014.25; Carlens et al.
- the retroviral vector has a long terminal repeat sequence (LTR), e.g., a retroviral vector derived from the Moloney murine leukemia virus (MoMLV), myeloproliferative sarcoma virus (MPSV), murine embryonic stem cell virus (MESV), murine stem cell virus (MSCV), spleen focus forming virus (SFFV), or adeno-associated virus (AAV).
- LTR long terminal repeat sequence
- MoMLV Moloney murine leukemia virus
- MPSV myeloproliferative sarcoma virus
- MMV murine embryonic stem cell virus
- MSCV murine stem cell virus
- SFFV spleen focus forming virus
- AAV adeno-associated virus
- retroviral vectors are derived from murine retroviruses.
- the retroviruses include those derived from any avian or mammalian cell source.
- the retroviruses typically are amphotropic, meaning that they are capable of
- the gene to be expressed replaces the retroviral gag, pol and/or env sequences.
- retroviral systems e.g., U.S. Pat. Nos. 5,219,740; 6,207,453; 5,219,740; Miller and Rosman (1989) BioTechniques 7:980-990; Miller, A. D. (1990) Human Gene Therapy 1 :5-14; Scarpa et al. (1991) Virology 180:849-852; Burns et al. (1993) Proc. Natl. Acad. Sci. USA 90:8033-8037; and Boris-Lawrie and Temin (1993) Cur. Opin. Genet. Develop. 3 : 102-109.
- recombinant nucleic acids are transferred into T cells via electroporation ⁇ see, e.g., Chicaybam et al, (2013) PLoS ONE 8(3): e60298 and Van Tedeloo et al. (2000) Gene Therapy 7(16): 1431-1437).
- recombinant nucleic acids are transferred into T cells via transposition (see, e.g., Manuri et al. (2010) Hum Gene Ther 21(4): 427- 437; Sharma et al. (2013) Molec Ther Nucl Acids 2, e74; and Huang et al. (2009) Methods Mol Biol 506: 115-126).
- the engineered cells include gene segments that cause the cells to be susceptible to negative selection in vivo, such as upon administration in adoptive immunotherapy.
- the cells are engineered so that they can be eliminated as a result of a change in the in vivo condition of the patient to which they are administered.
- the negative selectable phenotype may result from the insertion of a gene that confers sensitivity to an administered agent, for example, a compound.
- Negative selectable genes include the Herpes simplex virus type I thymidine kinase (HSV-I TK) gene (Wigler et al., Cell II :223, 1977) which confers ganciclovir sensitivity; the cellular hypoxanthine phosphribosyltransferase (HPRT) gene, the cellular adenine phosphoribosyltransferase (APRT) gene, bacterial cytosine deaminase, (Mullen et al., Proc. Natl. Acad. Sci. USA. 89:33 (1992)).
- HSV-I TK Herpes simplex virus type I thymidine kinase
- HPRT hypoxanthine phosphribosyltransferase
- APRT cellular adenine phosphoribosyltransferase
- the cells further are engineered to promote expression of cytokines or other factors.
- genes for introduction are those to improve the efficacy of therapy, such as by promoting viability and/or function of transferred cells; genes to provide a genetic marker for selection and/or evaluation of the cells, such as to assess in vivo survival or localization; genes to improve safety, for example, by making the cell susceptible to negative selection in vivo as described by Lupton S. D. et al., Mol.
- compositions containing such cells and/or enriched for such cells such as in which cells expressing the recombinant receptor make up at least 50%, 60%, 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or more of the total cells in the composition or cells of a certain type such as T cells or CD8+ or CD4+ cells.
- pharmaceutical compositions and formulations for administration such as for adoptive cell therapy.
- therapeutic methods for administering the cells and compositions to subjects e.g., patients.
- compositions including the cells for administration including pharmaceutical compositions and formulations, such as unit dose form compositions including the number of cells for administration in a given dose or fraction thereof.
- the pharmaceutical compositions and formulations generally include one or more optional pharmaceutically acceptable carrier or excipient.
- the composition includes at least one additional therapeutic agent.
- composition refers to a preparation which is in such form as to permit the biological activity of an active ingredient contained therein to be effective, and which contains no additional components which are unacceptably toxic to a subject to which the formulation would be administered.
- a "pharmaceutically acceptable carrier” refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject.
- a pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excipient, stabilizer, or preservative.
- the choice of carrier is determined in part by the particular cell and/or by the method of administration. Accordingly, there are a variety of suitable formulations.
- the pharmaceutical composition can contain preservatives. Suitable preservatives may include, for example, methylparaben, propylparaben, sodium benzoate, and benzalkonium chloride. In some aspects, a mixture of two or more preservatives is used.
- the preservative or mixtures thereof are typically present in an amount of about 0.0001% to about 2% by weight of the total composition.
- Carriers are described, e.g., by Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980).
- Pharmaceutically acceptable carriers are generally nontoxic to recipients at the dosages and concentrations employed, and include, but are not limited to: buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride; benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10
- Buffering agents are included in the compositions. Suitable buffering agents include, for example, citric acid, sodium citrate, phosphoric acid, potassium phosphate, and various other acids and salts. In some aspects, a mixture of two or more buffering agents is used. The buffering agent or mixtures thereof are typically present in an amount of about 0.001% to about 4% by weight of the total composition. Methods for preparing administrable pharmaceutical compositions are known. Exemplary methods are described in more detail in, for example, Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins; 21st ed. (May 1, 2005).
- the formulations can include aqueous solutions.
- the formulation or composition may also contain more than one active ingredient useful for the particular indication, disease, or condition being treated with the cells, preferably those with activities complementary to the cells, where the respective activities do not adversely affect one another.
- active ingredients are suitably present in combination in amounts that are effective for the purpose intended.
- the pharmaceutical composition further includes other pharmaceutically active agents or drugs, such as chemotherapeutic agents, e.g., asparaginase, busulfan, carboplatin, cisplatin, daunorubicin, doxorubicin, fluorouracil, gemcitabine, hydroxyurea, methotrexate, paclitaxel, rituximab, vinblastine, and/or vincristine.
- chemotherapeutic agents e.g., asparaginase, busulfan, carboplatin, cisplatin, daunorubicin, doxorubicin, fluorouracil, gemcitabine, hydroxyurea, methotrexate, paclitaxel, rituximab, vinblastine, and/or vincristine.
- the pharmaceutical composition in some embodiments contains the cells in amounts effective to treat or prevent the disease or condition, such as a therapeutically effective or prophylactically effective amount.
- Therapeutic or prophylactic efficacy in some embodiments is monitored by periodic assessment of treated subjects.
- the desired dosage can be delivered by a single bolus administration of the cells, by multiple bolus administrations of the cells, or by continuous infusion administration of the cells.
- the cells and compositions may be administered using standard administration techniques, formulations, and/or devices. Administration of the cells can be autologous or heterologous.
- immunoresponsive cells or progenitors can be obtained from one subject, and administered to the same subject or a different, compatible subject.
- Peripheral blood derived immunoresponsive cells or their progeny e.g., in vivo, ex vivo or in vzYro-derived
- a therapeutic composition e.g., a pharmaceutical composition containing a genetically modified immunoresponsive cell
- it will generally be formulated in a unit dosage injectable form (solution, suspension, emulsion).
- Formulations include those for oral, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual, or suppository administration.
- the cell populations are administered parenterally.
- parenteral includes intravenous, intramuscular, subcutaneous, rectal, vaginal, and intraperitoneal administration.
- the cells are administered to the subject using peripheral systemic delivery by intravenous, intraperitoneal, or subcutaneous injection.
- compositions in some embodiments are provided as sterile liquid preparations, e.g., isotonic aqueous solutions, suspensions, emulsions, dispersions, or viscous compositions, which may, in some aspects, be buffered to a selected pH.
- sterile liquid preparations e.g., isotonic aqueous solutions, suspensions, emulsions, dispersions, or viscous compositions, which may, in some aspects, be buffered to a selected pH.
- Liquid preparations are normally easier to prepare than gels, other viscous compositions, and solid compositions. Additionally, liquid compositions are somewhat more convenient to administer, especially by injection. Viscous compositions, on the other hand, can be formulated within the appropriate viscosity range to provide longer contact periods with specific tissues.
- Liquid or viscous compositions can comprise carriers, which can be a solvent or dispersing medium containing, for example, water, saline, phosphate buffered saline, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol) and suitable mixtures thereof.
- a solvent or dispersing medium containing, for example, water, saline, phosphate buffered saline, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol) and suitable mixtures thereof.
- Sterile injectable solutions can be prepared by incorporating the cells in a solvent, such as in admixture with a suitable carrier, diluent, or excipient such as sterile water, physiological saline, glucose, dextrose, or the like.
- compositions can contain auxiliary substances such as wetting, dispersing, or emulsifying agents (e.g., methylcellulose), pH buffering agents, gelling or viscosity enhancing additives, preservatives, flavoring agents, and/or colors, depending upon the route of administration and the preparation desired. Standard texts may, in some aspects, be consulted to prepare suitable preparations.
- auxiliary substances such as wetting, dispersing, or emulsifying agents (e.g., methylcellulose), pH buffering agents, gelling or viscosity enhancing additives, preservatives, flavoring agents, and/or colors, depending upon the route of administration and the preparation desired. Standard texts may, in some aspects, be consulted to prepare suitable preparations.
- compositions including antimicrobial preservatives, antioxidants, chelating agents, and buffers, can be added.
- antimicrobial preservatives for example, parabens, chlorobutanol, phenol, and sorbic acid.
- Prolonged absorption of the injectable pharmaceutical form can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
- the formulations to be used for in vivo administration are generally sterile. Sterility may be readily accomplished, e.g., by filtration through sterile filtration membranes.
- the cells, populations, and compositions are administered to a subject or patient having the particular disease or condition to be treated, e.g., via adoptive cell therapy, such as adoptive T cell therapy.
- adoptive cell therapy such as adoptive T cell therapy.
- cells and compositions prepared by the provided methods such as engineered compositions and end-of-production compositions following incubation and/or other processing steps, are administered to a subject, such as a subject having or at risk for the disease or condition.
- the methods thereby treat, e.g., ameliorate one or more symptom of, the disease or condition, such as by lessening tumor burden in a cancer expressing an antigen recognized by an engineered T cell.
- a "subject" is a mammal, such as a human or other animal, and typically is human.
- the subject e.g., patient, to whom the cells, cell populations, or compositions are administered is a mammal, typically a primate, such as a human.
- the primate is a monkey or an ape.
- the subject can be male or female and can be any suitable age, including infant, juvenile, adolescent, adult, and geriatric subjects.
- the subject is a non-primate mammal, such as a rodent.
- treatment refers to complete or partial amelioration or reduction of a disease or condition or disorder, or a symptom, adverse effect or outcome, or phenotype associated therewith. Desirable effects of treatment include, but are not limited to, preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis. The terms do not imply complete curing of a disease or complete elimination of any symptom or effect(s) on all symptoms or outcomes.
- delay development of a disease means to defer, hinder, slow, retard, stabilize, suppress and/or postpone development of the disease (such as cancer). This delay can be of varying lengths of time, depending on the history of the disease and/or individual being treated. As is evident to one skilled in the art, a sufficient or significant delay can, in effect, encompass prevention, in that the individual does not develop the disease. For example, a late stage cancer, such as development of metastasis, may be delayed.
- Preventing includes providing prophylaxis with respect to the occurrence or recurrence of a disease in a subject that may be predisposed to the disease but has not yet been diagnosed with the disease.
- the provided cells and compositions are used to delay development of a disease or to slow the progression of a disease.
- an "effective amount" of an agent e.g., a pharmaceutical formulation, cells, or composition, in the context of administration, refers to an amount effective, at dosages/ amounts and for periods of time necessary, to achieve a desired result, such as a therapeutic or prophylactic result.
- a "therapeutically effective amount" of an agent refers to an amount effective, at dosages and for periods of time necessary, to achieve a desired therapeutic result, such as for treatment of a disease, condition, or disorder, and/or pharmacokinetic or pharmacodynamic effect of the treatment.
- the therapeutically effective amount may vary according to factors such as the disease state, age, sex, and weight of the subject, and the populations of cells administered.
- the provided methods involve administering the cells and/or compositions at effective amounts, e.g., therapeutically effective amounts.
- a “prophylactically effective amount” refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired prophylactic result. Typically, but not necessarily, since a prophylactic dose is used in subjects prior to or at an earlier stage of disease, the prophylactically effective amount will be less than the therapeutically effective amount. In the context of lower tumor burden, the prophylactically effective amount, in some aspects, will be higher than the therapeutically effective amount.
- the subject has persistent or relapsed disease, e.g., following treatment with another therapeutic intervention, including chemotherapy, radiation, and/or hematopoietic stem cell transplantation (HSCT), e.g., allogenic HSCT.
- HSCT hematopoietic stem cell transplantation
- the administration effectively treats the subject despite the subject having become resistant to another therapy.
- the cell therapy e.g., adoptive T cell therapy
- the cells are carried out by autologous transfer, in which the cells are isolated and/or otherwise prepared from the subject who is to receive the cell therapy, or from a sample derived from such a subject.
- the cells are derived from a subject, e.g., patient, in need of a treatment and the cells, following isolation and processing are administered to the same subject.
- the cell therapy e.g., adoptive T cell therapy
- the cells are isolated and/or otherwise prepared from a subject other than a subject who is to receive or who ultimately receives the cell therapy, e.g., a first subject.
- the cells then are administered to a different subject, e.g., a second subject, of the same species.
- the first and second subjects are genetically identical.
- the first and second subjects are genetically similar.
- the second subject expresses the same HLA class or super-type as the first subject.
- the subject has been treated with a therapeutic agent targeting the disease or condition, e.g., the tumor, prior to administration of the cells or composition containing the cells.
- a therapeutic agent targeting the disease or condition, e.g., the tumor, prior to administration of the cells or composition containing the cells.
- the subject is refractory or non-responsive to the other therapeutic agent.
- the subject has persistent or relapsed disease, e.g., following treatment with another therapeutic intervention, including chemotherapy, radiation, and/or hematopoietic stem cell transplantation (HSCT), e.g., allogenic HSCT.
- the administration effectively treats the subject despite the subject having become resistant to another therapy.
- the subject is responsive to the other therapeutic agent, and treatment with the therapeutic agent reduces disease burden.
- the subject is initially responsive to the therapeutic agent, but exhibits a relapse of the disease or condition over time.
- the subject has not relapsed.
- the subject is determined to be at risk for relapse, such as at a high risk of relapse, and thus the cells are administered prophylactically, e.g., to reduce the likelihood of or prevent relapse.
- the subject has not received prior treatment with another therapeutic agent.
- the diseases, conditions, and disorders for treatment with the provided compositions, cells, methods and uses are tumors, including solid tumors, hematologic malignancies, and melanomas, and infectious diseases, such as infection with a virus or other pathogen, e.g., HIV, HCV, HBV, CMV, and parasitic disease.
- the disease or condition is a tumor, cancer, malignancy, neoplasm, or other proliferative disease or disorder.
- Such diseases include but are not limited to leukemia, lymphoma, e.g., chronic lymphocytic leukemia (CLL), acute - lymphoblastic leukemia (ALL), non-Hodgkin's lymphoma, acute myeloid leukemia, multiple myeloma, refractory follicular lymphoma, mantle cell lymphoma, indolent B cell lymphoma, B cell malignancies, cancers of the colon, lung, liver, breast, prostate, ovarian, skin, melanoma, bone, and brain cancer, ovarian cancer, epithelial cancers, renal cell carcinoma, pancreatic adenocarcinoma, Hodgkin's lymphoma, cervical carcinoma, colorectal cancer, glioblastoma, neuroblastoma, Ewing's sarcoma, medulloblastoma, osteosarcoma, synovial sarcoma, and/or mesothelio
- the disease or condition is an infectious disease or condition, such as, but not limited to, viral, retroviral, bacterial, and protozoal infections, immunodeficiency, Cytomegalovirus (CMV), Epstein-Barr virus (EBV), adenovirus, BK polyomavirus.
- infectious disease or condition such as, but not limited to, viral, retroviral, bacterial, and protozoal infections, immunodeficiency, Cytomegalovirus (CMV), Epstein-Barr virus (EBV), adenovirus, BK polyomavirus.
- the disease or condition is an autoimmune or inflammatory disease or condition, such as arthritis, e.g., rheumatoid arthritis (RA), Type I diabetes, systemic lupus erythematosus (SLE), inflammatory bowel disease, psoriasis, scleroderma, autoimmune thyroid disease, Grave's disease, Crohn's disease, multiple sclerosis, asthma, and/or a disease or condition associated with transplant.
- arthritis e.g., rheumatoid arthritis (RA), Type I diabetes, systemic lupus erythematosus (SLE), inflammatory bowel disease, psoriasis, scleroderma, autoimmune thyroid disease, Grave's disease, Crohn's disease, multiple sclerosis, asthma, and/or a disease or condition associated with transplant.
- RA rheumatoid arthritis
- SLE systemic lupus erythematosus
- inflammatory bowel disease e.
- antigen associated with the disease, disorder or condition is selected from RORl, B cell maturation antigen (BCMA), carbonic anhydrase 9 (CAIX), tEGFR, Her2/neu (receptor tyrosine kinase erbB2), Ll -CAM, CD19, CD20, CD22, mesothelin, CEA, and hepatitis B surface antigen, anti-folate receptor, CD23, CD24, CD30, CD33, CD38, CD44, EGFR, epithelial glycoprotein 2 (EPG-2), epithelial glycoprotein 40 (EPG-40), EPHa2, erb-B2, erb-B3, erb- B4, erbB dimers, EGFR vIII, folate binding protein (FBP), FCRL5, FCRH5, fetal acetylcholine receptor, GD2, GD3, HMW-MAA, IL-22R-alpha, IL-13R-al
- the antigen associated with the disease or disorder is selected from the group consisting of orphan tyrosine kinase receptor RORl, tEGFR, Her2, Ll-CAM, CD19, CD20, CD22, mesothelin, CEA, and hepatitis B surface antigen, anti-folate receptor, CD23, CD24, CD30, CD33, CD38, CD44, EGFR, EGP-2, EGP-4, 0EPHa2, ErbB2, 3, or 4, FBP, fetal acetylcholine e receptor, GD2, GD3, HMW-MAA, IL-22R-alpha, IL-13R-alpha2, kdr, kappa light chain, Lewis Y, Ll-cell adhesion molecule, MAGE-A1, mesothelin, MUC1, MUC16, PSCA, NKG2D Ligands, NY- ESO-1, MART-1, gpl OO, onco
- the cells are administered at a desired dosage, which, in some aspects, includes a desired dose or number of cells or cell type(s) and/or a desired ratio of cell types.
- the dosage of cells in some embodiments is based on a total number of cells (or number per kg body weight) and a desired ratio of the individual populations or sub-types, such as the CD4+ to CD8+ ratio.
- the dosage of cells is based on a desired total number (or number per kg of body weight) of cells in the individual populations or of individual cell types.
- the dosage is based on a combination of such features, such as a desired number of total cells, desired ratio, and desired total number of cells in the individual populations.
- the populations or sub-types of cells are administered at or within a tolerated difference of a desired dose of total cells, such as a desired dose of T cells.
- the desired dose is a desired number of cells or a desired number of cells per unit of body weight of the subject to whom the cells are administered, e.g., cells/kg.
- the desired dose is at or above a minimum number of cells or minimum number of cells per unit of body weight.
- the individual populations or sub-types are present at or near a desired output ratio (such as CD4 + to CD8 + ratio), e.g., within a certain tolerated difference or error of such a ratio.
- a desired output ratio such as CD4 + to CD8 + ratio
- the cells are administered at or within a tolerated difference of a desired dose of one or more of the individual populations or sub-types of cells, such as a desired dose of CD4+ cells and/or a desired dose of CD8+ cells.
- the desired dose is a desired number of cells of the sub-type or population, or a desired number of such cells per unit of body weight of the subject to whom the cells are administered, e.g., cells/kg.
- the desired dose is at or above a minimum number of cells of the population or sub-type, or minimum number of cells of the population or sub-type per unit of body weight.
- the dosage is based on a desired fixed dose of total cells and a desired ratio, and/or based on a desired fixed dose of one or more, e.g., each, of the individual subtypes or sub-populations.
- the dosage is based on a desired fixed or minimum dose of T cells and a desired ratio of CD4 + to CD8 + cells, and/or is based on a desired fixed or minimum dose of CD4 + and/or CD8 + cells.
- the cells, or individual populations of sub-types of cells are administered to the subject at a range of about one million to about 100 billion cells, such as, e.g., 1 million to about 50 billion cells (e.g., about 5 million cells, about 25 million cells, about 500 million cells, about 1 billion cells, about 5 billion cells, about 20 billion cells, about 30 billion cells, about 40 billion cells, or a range defined by any two of the foregoing values), such as about 10 million to about 100 billion cells (e.g., about 20 million cells, about 30 million cells, about 40 million cells, about 60 million cells, about 70 million cells, about 80 million cells, about 90 million cells, about 10 billion cells, about 25 billion cells, about 50 billion cells, about 75 billion cells, about 90 billion cells, or a range defined by any two of the foregoing values), and in some cases about 100 million cells to about 50 billion cells (e.g., about 120 million cells, about 250 million cells, about 350 million cells, about 450 million cells, about 650 million cells).
- the dose of total cells and/or dose of individual sub-populations of cells is within a range of between at or about 10 4 and at or about 10 9 cells/kilograms (kg) body weight, such as between 10 5 and 10 6 cells / kg body weight, for example, at or about 1 x 10 5 cells/kg, 1.5 x 10 5 cells/kg, 2 x 10 5 cells/kg, or 1 x 10 6 cells/kg body weight.
- the cells are administered at, or within a certain range of error of, between at or about 10 4 and at or about 10 9 T cells/kilograms (kg) body weight, such as between 10 5 and 10 6 T cells / kg body weight, for example, at or about 1 x 10 5 T cells/kg, 1.5 x 10 5 T cells/kg, 2 x 10 5 T cells/kg, or 1 x 10 6 T cells/kg body weight.
- the cells are administered at or within a certain range of error of between at or about 10 4 and at or about 10 9 CD4 + and/or CD8 + cells/kilograms (kg) body weight, such as between 10 5 and 10 6 CD4 + and/or CD8 + cells / kg body weight, for example, at or about 1 x 10 5 CD4 + and/or CD8 + cells/kg, 1.5 x 10 5 CD4 + and/or CD8 + cells/kg, 2 x 10 5 CD4 + and/or CD8 + cells/kg, or 1 x 10 6 CD4 + and/or CD8 + cells/kg body weight.
- the cells are administered at or within a certain range of error of, greater than, and/or at least about 1 x 10 6 , about 2.5 x 10 6 , about 5 x 10 6 , about 7.5 x 10 6 , or about 9 x 10 6 CD4 + cells, and/or at least about 1 x 10 6 , about 2.5 x 10 6 , about 5 x 10 6 , about 7.5 x 10 6 , or about 9 x 10 6 CD8+ cells, and/or at least about 1 x 10 6 , about 2.5 x 10 6 , about 5 x 10 6 , about 7.5 x 10 6 , or about 9 x 10 6 T cells.
- the cells are administered at or within a certain range of error of between about 10 8 and 10 12 or between about 10 10 and 10 11 T cells, between about 10 8 and 10 12 or between about 10 10 and 10 11 CD4 + cells, and/or between about 10 8 and 10 12 or between about 10 10 and 10 11 CD8 + cells.
- the cells are administered at or within a tolerated range of a desired output ratio of multiple cell populations or sub-types, such as CD4+ and CD8+ cells or sub- types.
- the desired ratio can be a specific ratio or can be a range of ratios, for example, in some embodiments, the desired ratio (e.g., ratio of CD4 + to CD8 + cells) is between at or about 5: 1 and at or about 5: 1 (or greater than about 1 :5 and less than about 5: 1), or between at or about 1 :3 and at or about 3 : 1 (or greater than about 1 :3 and less than about 3 : 1), such as between at or about 2: 1 and at or about 1 :5 (or greater than about 1 :5 and less than about 2: 1, such as at or about 5: 1, 4.5 : 1, 4: 1, 3.5: 1, 3 : 1 , 2.5: 1, 2: 1 , 1.9: 1, 1.8: 1, 1.7: 1, 1.6: 1, 1.5:
- the tolerated difference is within about 1%, about 2%, about 3%, about 4% about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, about 50%) of the desired ratio, including any value in between these ranges.
- the appropriate dosage may depend on the type of disease to be treated, the type of cells or recombinant receptors, the severity and course of the disease, whether the cells are administered for preventive or therapeutic purposes, previous therapy, the subject's clinical history and response to the cells, and the discretion of the attending physician.
- the compositions and cells are in some embodiments suitably administered to the subject at one time or over a series of treatments.
- the cells can be administered by any suitable means, for example, by bolus infusion, by injection, e.g., intravenous or subcutaneous injections, intraocular injection, periocular injection, subretinal injection, intravitreal injection, trans-septal injection, subscleral injection, intrachoroidal injection, intracameral injection, subconjectval injection, subconjuntival injection, sub-Tenon's injection, retrobulbar injection, peribulbar injection, or posterior juxtascleral delivery.
- injection e.g., intravenous or subcutaneous injections, intraocular injection, periocular injection, subretinal injection, intravitreal injection, trans-septal injection, subscleral injection, intrachoroidal injection, intracameral injection, subconjectval injection, subconjuntival injection, sub-Tenon's injection, retrobulbar injection, peribulbar injection, or posterior juxtascleral delivery.
- injection e.g., intravenous or subcutaneous injection
- Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration.
- a given dose is administered by a single bolus administration of the cells. In some embodiments, it is administered by multiple bolus administrations of the cells, for example, over a period of no more than 3 days, or by continuous infusion administration of the cells.
- the cells are administered as part of a combination treatment, such as simultaneously with or sequentially with, in any order, another therapeutic intervention, such as an antibody or engineered cell or receptor or agent, such as a cytotoxic or therapeutic agent.
- the cells in some embodiments are co-administered with one or more additional therapeutic agents or in connection with another therapeutic intervention, either simultaneously or sequentially in any order.
- the cells are co-administered with another therapy sufficiently close in time such that the cell populations enhance the effect of one or more additional therapeutic agents, or vice versa.
- the cells are administered prior to the one or more additional therapeutic agents.
- the cells are administered after the one or more additional therapeutic agents.
- the one or more additional agents includes a cytokine, such as IL-2, for example, to enhance persistence.
- the methods comprise administration of a chemotherapeutic agent.
- the biological activity of the engineered cell populations in some embodiments is measured, e.g., by any of a number of known methods.
- Parameters to assess include specific binding of an engineered or natural T cell or other immune cell to antigen, in vivo, e.g., by imaging, or ex vivo, e.g., by ELISA or flow cytometry.
- the ability of the engineered cells to destroy target cells can be measured using any suitable method known in the art, such as cytotoxicity assays described in, for example, Kochenderfer et al., J. Immunotherapy, 32(7): 689-702 (2009), and Herman et al. J. Immunological Methods, 285(1): 25-40 (2004).
- the biological activity of the cells is measured by assaying expression and/or secretion of one or more cytokines, such as CD 107a, IFNy, IL-2, and TNF. In some aspects, the biological activity is measured by assessing clinical outcome, such as reduction in tumor burden or load.
- cytokines such as CD 107a, IFNy, IL-2, and TNF.
- the engineered cells are further modified in any number of ways, such that their therapeutic or prophylactic efficacy is increased.
- the engineered CAR or TCR expressed by the population can be conjugated either directly or indirectly through a linker to a targeting moiety.
- the practice of conjugating compounds, e.g., the CAR or TCR, to targeting moieties is known in the art. See, for instance, Wadwa et al., J. Drug Targeting 3 : 1 1 1 (1995), and U.S. Patent 5,087,616.
- RNAs were screened by complexing commercially synthesized gRNAs with Cas9 in vitro and delivering the gRNA/Cas9 ribonucleoprotein (RNP) to cells via electroporation.
- RNP gRNA/Cas9 ribonucleoprotein
- Fig. 1 depicts %indel frequency of test gRNAs.
- Fig. 2 depicts the genome editing efficiency of certain exemplary gRNA pairs.
- the goal of CRISPR-Cas9 editing of cells is to achieve the highest percent knock out of the target gene using the lowest possible concentration of the gRNA/Cas9 complex and the least number of off-target cutting events.
- seven potential gRNAs were tested. T cells were transfected with gRNA/Cas9 RNPs at various concentrations. The percent indel frequency was then determined using Illumina miSeq analysis. The results show % indel frequency for different gRNAs ranged from 20% to 80%, with all gRNAs achieving their highest % indel frequency at a RNP concentration of 2 ⁇ (Fig. 3). The 2 ⁇ RNP concentration was subsequently used for all experiments.
- gRNAs from the miSeq analysis were chosen for further analysis in BCMA CAR T cells.
- the gRNAs of SEQ ID Nos: 5050, 5052, 5093, and 5043 were used in an RNP re-cutting assay to determine the % editing of the TGFBR2 gene.
- a dual gRNA approach was also tested using the combination of SEQ ID NO5043/5093. While the individual gRNAs of SEQ ID NO: 5043 and 5093 only achieved 20-40%) editing, the combination was found to be more efficacious, producing a % edited value of approximately 80%> (Fig. 4A).
- % indel frequency is a useful measure of a gRNA's effectiveness
- some portion of the indels produced may be in-frame insertions or deletions.
- Such in-frame indels may produce a modified gene whose protein product retains some activity.
- out-of-frame indels are preferred.
- the sequencing results were analyzed for the specific types of indels produced. The results show that while SEQ ID NO: 5052 produced the highest % indel frequency, SEQ ID NO: 5050 generated the higher % out-of-frame indel frequency (Fig. 5).
- Example 3 In vitro efficacy of gene editing in primary and engineered T cells.
- the inhibitory effects of TGFp were analyzed in primary T cells modified to expresses a BCMA-targeting CAR.
- the anti-BCMA CAR T cells were additionally modified to have the TGFBR2 gene edited via the CRISPR-Cas9 system using select gRNAs. AAVSl control for gene editing and anti-BCMA CAR-expressing T cells with unedited TGFBR2 gene were used as controls.
- the cell lines were co-cultured with the RPMI 8226 multiple myeloma cell line with or without 10 ng/ml TGFp.
- primary T cells and anti-BCMA CAR T cells display the expected inhibitory effects of reduced production of Interferon-gamma (IFNy).
- IFNy Interferon-gamma
- the inhibitory effects of TGFp are rescued, restoring IFNy production to levels found without TGFp addition (Fig. 6).
- TGFp signaling TGFp signaling
- anti-BCMA CAR T cell proliferation was monitored in a TGFBR2 gene-edited background using several gRNAs. With the TGFBR2 gene edited, anti-BCMA CAR T cells were able to proliferate in the presence of excess TGFp (Fig. 7).
- T cell activity is influenced by the expression of various stimulatory and inhibitory receptors.
- CD25 expression was assessed in the TGFBR2 gene-edited anti-BCMA CAR T cells.
- TGFBR2 gene- editing lead to higher expression levels of CD25 compared to control cells when exposed to TGFp (Fig. 8A).
- the expression of the inhibitory receptor, PD-1 was also assessed by measuring the % of PD-1+ cells with and without excess TGFp.
- the TGFBR2 gene-edited anti-BCMA CAR T cells showed a lower increase of PD-1 relative to control cells. Surprisingly, the gene-edited cells also led to fewer PD- 1+ cells even when TGFp was not added (Fig. 8B).
- Activation of the TGFp signaling pathway leads to phosphorylation of the Smad 2/3 complex, which in turn regulates many of the downstream processes of the TGFp signaling pathway. For this reason, phosphorylated Smad 2/3 is often used as a measure of TGFp signaling in cells.
- Phospho Smad2/3 was detected in T cells transduced to express different CARs, with or without TGFBR2 gene-editing.
- TGFBR2 CRISPR gene editing approach is effective at silencing the TGFp signaling pathway.
- IFNy production was analyzed in a similar experiment as described in Fig. 10 using a cytokine detection assay. Consistent with the previous results of GzmB expression, IFNy production was maintained and even increased in the TGFBR2 gene-edited background relative to controls in conditions of excess TGFp (Fig. 11).
- Example 4 Comparison of the TGFBR2 Dominant Negative approach to the CRISPR- Cas9 gene editing approach.
- TGFBR2 TGFBR2
- the inhibitory effects of TGFp were analyzed in primary T cells transduced to express anti- BCMA CAR.
- the anti-BCMA CAR-expressing T cells were additionally modified to either express the DN or to have the TGFBR2 gene edited via the CRISPR-Cas9 system.
- the CRISPR-Cas9 edited cells were edited with dual gRNAs of SEQ ID Nos: 5043 and 5093.
- AAVSl control for gene editing, anti-BCMA CAR-expressing T cells with unedited TGFBR2 gene, and anti-BCMA CAR-expressing T cells including AAVSl gene editing control were used as controls.
- the cell lines were co-cultured with the RPMI 8226 multiple myeloma cell line with or without 10 ng/ml TGFp.
- primary T cells and anti-BCMA CAR T cells display the expected inhibitory effects of reduced production of GzmB (Fig. 13 A) and IFNy (Fig. 13B).
- the inhibitory effects of TGFp are rescued, restoring GzmB and IFNy production to levels found without TGFp addition.
- the anti-BCMA CAR T cells in the DN or CRISPR-edited background were analyzed for their killing activity.
- Anti-BCMA CAR-expressing T cells with unedited TGFBR2 gene were used as control.
- Anti-BCMA CAR T cells were co-cultured with RPMI 8226 cells at a ratio of 1 anti-BCMA CAR T cells to 4 RPMI cells, with or without 10 ng/ml TGFp.
- the lytic activity of the anti-BCMA CAR T cells was maintained in both the DN and CRISPR-edited background in the presence of TGFp.
- the CRISPR-edited anti-BCMA CAR T cells displayed superior lytic activity when compared to the DN cells (Fig. 14).
- the lytic activity of the CRISPR-edited anti-BCMA CAR T cells was higher than the control cells even without excess TGFp added.
- IFNy production was analyzed again in the DN and gene-edited background using a single, rather than dual, gRNA approach.
- An additional multiple myeloma cell line, OPM2 was used as a comparison to the previously used RPMI 8226 cell line as described in Fig. 13. Consistent with the previous results, inhibition TGFp signaling effectively maintained IFNy production in the presence of excess TGFp. This held true in both the RPMI (Fig. 16A) and OPM2 (Fig. 16B) cell lines.
- CD25 expression was also analyzed in the DN and gene-edited background using a single gRNA approach. CD25 expression was maintained in the presence of excess TGFp in the RPMI (Fig. 17A) and OPM2 (Fig. 17B) cell lines. Surprisingly, CD25 expression in the DN and gene-edited cells was increased relative to the unmodified T cells.
- PD-1 expression was similarly detected in the DN and gene-edited background. Repression of TGFp signaling with either approach effectively prevented increased PD-1 expression in the presence of excess TGFp (Fig. 18A and 18B).
- TGFp at a set dose of lOng/ml for stimulation.
- a range of 0 to 100 ng/ml TGFp was used. This range is based on previous work demonstrating that TGFp serum levels range from approximately 3 to 88 ng/ml (Aref et al. Hematological Oncology. 35: 51-57. 2017) and approximately 3 to 10 ng/ml in multiple myeloma bone marrow (Bruns et al. Blood. 120: 2620-2630. 2012).
- a cell proliferation assay was used to determine relative fold expansion of cells in the DN and gene-edited background.
- Anti-BCMA CAR T cells were co-cultured with RPMI 8226 cells at a ratio of 1 : 1. Unexpectedly, the positive effects on cell proliferation can be seen at concentrations as low as 0.1 ng/ml TGFp, well below the physiological levels in a normal or cancer setting (Fig. 19A). This responsiveness is further demonstrated by the increased production of IFNy (Fig. 19B).
- the DN TGFBR2 strategy was used to access cell proliferation, Granzyme B expression, and Smad 2/3 phosphorylation in T cells transduced to express three different CARs.
- the DN TGFBR2 was able to rescue the anti-proliferative effects of excess TGFp in the tested CAR T cell backgrounds.
- the DN was able to maintain Granzyme B expression in the presence of excess TGFp in the tested CAR T cell backgrounds.
- the DN was able to effectively suppress TGFp signaling in the presence of excess TGFp as measured by reduced Smad 2/3 phosphorylation in the tested CAR T cell backgrounds (Fig. 22). Data for only one of the three CAR T cell backgrounds is shown, however similar results were observed for the other two CAR T cell backgrounds as well.
- the TGFp immunosuppressive pathway In order for the TGFBR2 gene-editing strategy disclosed herein to form an effective therapy, the TGFp immunosuppressive pathway must be present in an in vivo setting. If said TGFp immunosuppressive pathway is present, then the TGFBR2 gene-editing strategy must effectively release T cells from the TGFp immunosuppressive effects.
- transcriptional profiling of anti-BCMA CAR T cells was performed from anti-BCMA CAR T cells isolated from the tumor microenvironment in mice.
- the tumor xenograft model was generated by implanting mice with the multiple myeloma RPMI 8226 cells and allowed to propagate/form a tumor for 21 days.
- TILs Tumor Infiltrating Leukocytes
- spleen a non-cancerous negative control tissue
- TGFBR2 gene-editing in CAR T cells successfully limits the expression of several TGFp signaling pathway members in the tumor, where TGFp repressive signaling occurs (Fig. 21).
- TGFp signaling pathway members are upregulated in anti-BCMA CAR-expressing T cells isolated from tumor but not spleen.
- TEM tumor micro-environ
- anti-BCMA CAR-expressing T cells with TGFBR2 DN or TGFBR2 gene-edited background reverse the upregulation of TGFp signaling pathway members.
- TGFp signaling pathway was abolished effectively.
- TGFBR2 gene-edited CAR T cells have a selective proliferation advantage
- anti-BCMA CAR T cells were gene edited to produce different % indel frequencies. These separate % indel populations of CAR T cells were co-cultured with RPMI 8226 cells in a 1 : 1 ratio for stimulation, with or without 10 ng/ml TGFp. Cells were evaluated, using high throughput sequencing, approximately every 7 days to determine which portion of the cells are gene-edited and which portion are still wild-type (Fig. 23). Cells are re-stimulated with fresh RPMI cells, and readjusted to 1 : 1 ratio weekly. The results show that TGFBR2 gene-editing confers a selective proliferation advantage over wild-type cells in an immune-stimulatory environment (see Fig. 24). INCORPORATION BY REFERENCE
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Medicinal Chemistry (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Biochemistry (AREA)
- Cell Biology (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Mycology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Hematology (AREA)
- Developmental Biology & Embryology (AREA)
- Virology (AREA)
Abstract
Description




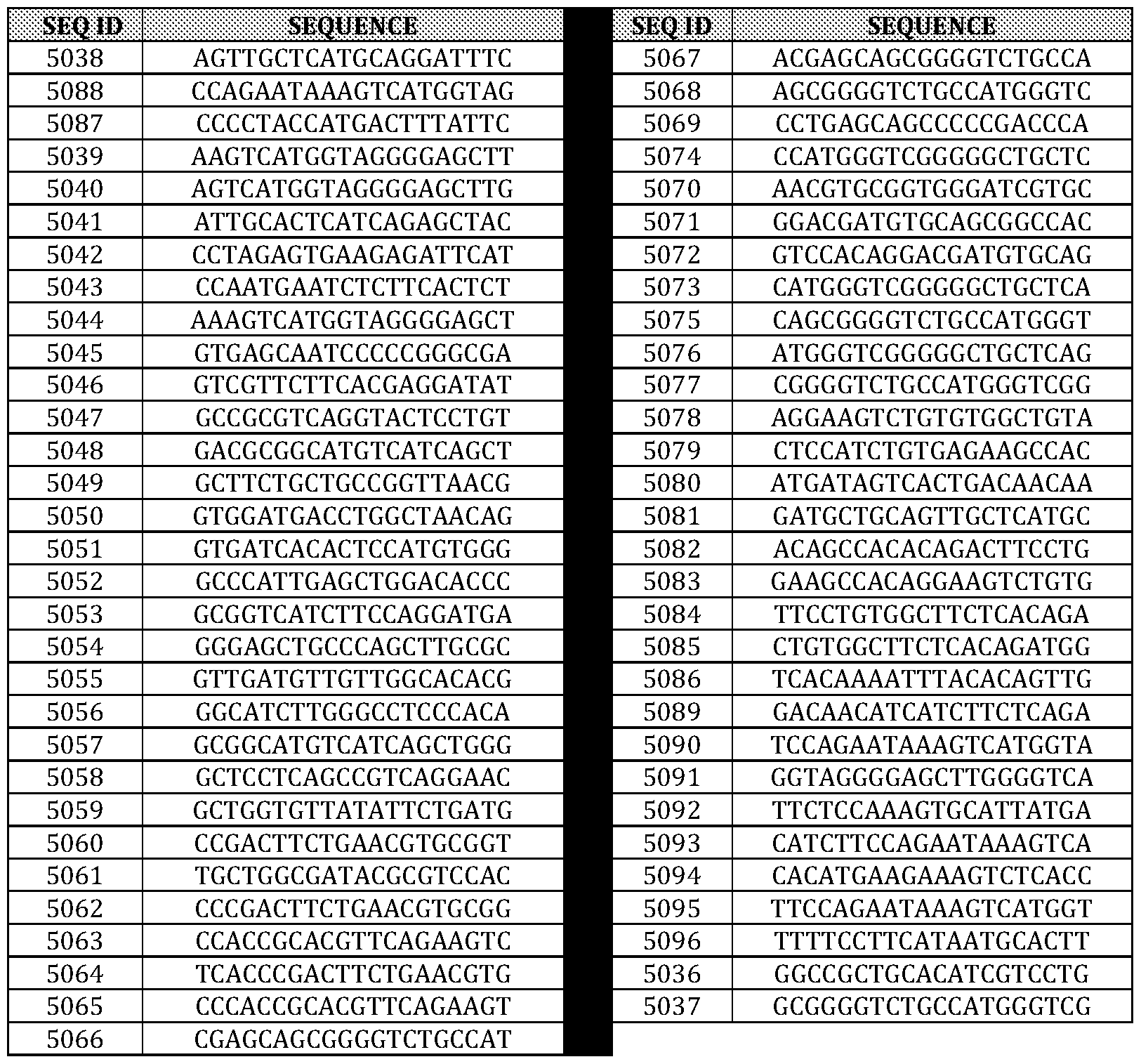







Claims
Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/758,842 US20230061455A1 (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
| BR112020008478-6A BR112020008478A2 (en) | 2017-11-01 | 2018-11-01 | methods, compositions and components for editing crispr-cas9 of tgfbr2 in t cells for immunota-rapy |
| JP2020543733A JP2021502113A (en) | 2017-11-01 | 2018-11-01 | Methods, compositions, and components for CRISPR-CAS9 editing of TGFBR2 in T cells for immunotherapy |
| RU2020117752A RU2798380C2 (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for editing tgfbr2 by crispr-cas9 in t cells for immunotherapy |
| AU2018358250A AU2018358250A1 (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for CRISPR-CAS9 editing of TGFBR2 in T cells for immunotherapy |
| CN201880084188.9A CN111527209A (en) | 2017-11-01 | 2018-11-01 | Methods, compositions, and components of CRISPR-CAS9 editing TGFBR2 in T cells for immunotherapy |
| MX2020004608A MX2020004608A (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy. |
| CA3081456A CA3081456A1 (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
| EP18807785.3A EP3704251A2 (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
| KR1020207015701A KR20200075000A (en) | 2017-11-01 | 2018-11-01 | CRISPR-CAS9 editing method, composition and components of TGFBR2 in T cells for immunotherapy |
| SG11202003862PA SG11202003862PA (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
| IL274299A IL274299A (en) | 2017-11-01 | 2020-04-27 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
| JP2023146660A JP2023179469A (en) | 2017-11-01 | 2023-09-11 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762580320P | 2017-11-01 | 2017-11-01 | |
| US62/580,320 | 2017-11-01 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2019089884A2 true WO2019089884A2 (en) | 2019-05-09 |
| WO2019089884A3 WO2019089884A3 (en) | 2019-06-13 |
Family
ID=64453598
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2018/058635 WO2019089884A2 (en) | 2017-11-01 | 2018-11-01 | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20230061455A1 (en) |
| EP (1) | EP3704251A2 (en) |
| JP (2) | JP2021502113A (en) |
| KR (1) | KR20200075000A (en) |
| CN (1) | CN111527209A (en) |
| AU (1) | AU2018358250A1 (en) |
| BR (1) | BR112020008478A2 (en) |
| CA (1) | CA3081456A1 (en) |
| IL (1) | IL274299A (en) |
| MA (1) | MA50858A (en) |
| MX (1) | MX2020004608A (en) |
| SG (1) | SG11202003862PA (en) |
| WO (1) | WO2019089884A2 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020223535A1 (en) * | 2019-05-01 | 2020-11-05 | Juno Therapeutics, Inc. | Cells expressing a recombinant receptor from a modified tgfbr2 locus, related polynucleotides and methods |
| WO2020243368A1 (en) * | 2019-05-29 | 2020-12-03 | Monsanto Technology Llc | Methods and compositions for generating dominant alleles using genome editing |
| WO2021011864A1 (en) * | 2019-07-18 | 2021-01-21 | Memorial Sloan Kettering Cancer Center | METHODS AND COMPOSITIONS FOR TARGETING TGF-β SIGNALING IN CD4+ HELPER T CELLS FOR CANCER IMMUNOTHERAPY |
| WO2021097521A1 (en) * | 2019-11-20 | 2021-05-27 | Cartherics Pty. Ltd. | Method for providing immune cells with enhanced function |
| WO2021127261A1 (en) * | 2019-12-17 | 2021-06-24 | The General Hospital Corporation | Engineered immune cells with reduced toxicity and uses thereof |
| WO2021127594A1 (en) * | 2019-12-18 | 2021-06-24 | Editas Medicine, Inc. | Engineered cells for therapy |
| WO2021182917A1 (en) | 2020-03-12 | 2021-09-16 | 기초과학연구원 | Composition for inducing apoptosis of cells having genomic sequence variation and method for inducing apoptosis of cells by using composition |
| WO2022064428A1 (en) * | 2020-09-23 | 2022-03-31 | Crispr Therapeutics Ag | Genetically engineered t cells with regnase-1 and/or tgfbrii disruption have improved functionality and persistence |
| EP3854877A4 (en) * | 2018-09-17 | 2022-10-12 | Institute Of Zoology, Chinese Academy Of Sciences | Modified t cell, preparation method therefor and use thereof |
| WO2023081900A1 (en) | 2021-11-08 | 2023-05-11 | Juno Therapeutics, Inc. | Engineered t cells expressing a recombinant t cell receptor (tcr) and related systems and methods |
| WO2023180967A1 (en) * | 2022-03-23 | 2023-09-28 | Crispr Therapeutics Ag | Anti-cd83 car-t cells with regnase-1 and/or tgfbrii disruption |
| WO2023147428A3 (en) * | 2022-01-26 | 2023-12-07 | Orthobio Therapeutics, Inc. | Gene editing to improve joint function |
| EP3980450A4 (en) * | 2019-06-04 | 2024-06-19 | Nkarta, Inc. | Combinations of engineered natural killer cells and engineered t cells for immunotherapy |
| US12024711B2 (en) | 2019-05-29 | 2024-07-02 | Monsanto Technology Llc | Methods and compositions for generating dominant short stature alleles using genome editing |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20240040035A (en) * | 2022-09-16 | 2024-03-27 | 한국과학기술연구원 | Chimeric Antigen Receptor Cell Prepared Using CRISPR/Cas9 Knock―and Uses Thereof |
| CN116515766A (en) * | 2023-06-30 | 2023-08-01 | 上海贝斯昂科生物科技有限公司 | Natural killer cell, preparation method and application thereof |
Citations (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4690915A (en) | 1985-08-08 | 1987-09-01 | The United States Of America As Represented By The Department Of Health And Human Services | Adoptive immunotherapy as a treatment modality in humans |
| US5087616A (en) | 1986-08-07 | 1992-02-11 | Battelle Memorial Institute | Cytotoxic drug conjugates and their delivery to tumor cells |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| WO1996013593A2 (en) | 1994-10-26 | 1996-05-09 | Procept, Inc. | Soluble single chain t cell receptors |
| WO1996018105A1 (en) | 1994-12-06 | 1996-06-13 | The President And Fellows Of Harvard College | Single chain t-cell receptor |
| WO1999018129A1 (en) | 1997-10-02 | 1999-04-15 | Sunol Molecular Corporation | Soluble single-chain t-cell receptor proteins |
| WO1999060120A2 (en) | 1998-05-19 | 1999-11-25 | Avidex Limited | Soluble t cell receptor |
| WO2000014257A1 (en) | 1998-09-04 | 2000-03-16 | Sloan-Kettering Institute For Cancer Research | Fusion receptors specific for prostate-specific membrane antigen and uses thereof |
| US6040177A (en) | 1994-08-31 | 2000-03-21 | Fred Hutchinson Cancer Research Center | High efficiency transduction of T lymphocytes using rapid expansion methods ("REM") |
| US6207453B1 (en) | 1996-03-06 | 2001-03-27 | Medigene Ag | Recombinant AAV vector-based transduction system and use of same |
| US6410319B1 (en) | 1998-10-20 | 2002-06-25 | City Of Hope | CD20-specific redirected T cells and their use in cellular immunotherapy of CD20+ malignancies |
| US6451995B1 (en) | 1996-03-20 | 2002-09-17 | Sloan-Kettering Institute For Cancer Research | Single chain FV polynucleotide or peptide constructs of anti-ganglioside GD2 antibodies, cells expressing same and related methods |
| US20020131960A1 (en) | 2000-06-02 | 2002-09-19 | Michel Sadelain | Artificial antigen presenting cells and methods of use thereof |
| US20030170238A1 (en) | 2002-03-07 | 2003-09-11 | Gruenberg Micheal L. | Re-activated T-cells for adoptive immunotherapy |
| WO2004033685A1 (en) | 2002-10-09 | 2004-04-22 | Avidex Ltd | Single chain recombinant t cell receptors |
| WO2006000830A2 (en) | 2004-06-29 | 2006-01-05 | Avidex Ltd | Cells expressing a modified t cell receptor |
| WO2006037960A2 (en) | 2004-10-01 | 2006-04-13 | Avidex Ltd. | T-cell receptors containing a non-native disulfide interchain bond linked to therapeutic agents |
| US7070995B2 (en) | 2001-04-11 | 2006-07-04 | City Of Hope | CE7-specific redirected immune cells |
| US7446190B2 (en) | 2002-05-28 | 2008-11-04 | Sloan-Kettering Institute For Cancer Research | Nucleic acids encoding chimeric T cell receptors |
| US7446179B2 (en) | 2000-11-07 | 2008-11-04 | City Of Hope | CD19-specific chimeric T cell receptor |
| WO2011044186A1 (en) | 2009-10-06 | 2011-04-14 | The Board Of Trustees Of The University Of Illinois | Human single-chain t cell receptors |
| WO2012129514A1 (en) | 2011-03-23 | 2012-09-27 | Fred Hutchinson Cancer Research Center | Method and compositions for cellular immunotherapy |
| US8324353B2 (en) | 2001-04-30 | 2012-12-04 | City Of Hope | Chimeric immunoreceptor useful in treating human gliomas |
| US8339645B2 (en) | 2008-05-27 | 2012-12-25 | Canon Kabushiki Kaisha | Managing apparatus, image processing apparatus, and processing method for the same, wherein a first user stores a temporary object having attribute information specified but not partial-area data, at a later time an object is received from a second user that includes both partial-area data and attribute information, the storage unit is searched for the temporary object that matches attribute information of the received object, and the first user is notified in response to a match |
| EP2537416A1 (en) | 2007-03-30 | 2012-12-26 | Memorial Sloan-Kettering Cancer Center | Constitutive expression of costimulatory ligands on adoptively transferred T lymphocytes |
| US8398282B2 (en) | 2011-05-12 | 2013-03-19 | Delphi Technologies, Inc. | Vehicle front lighting assembly and systems having a variable tint electrowetting element |
| WO2013071154A1 (en) | 2011-11-11 | 2013-05-16 | Fred Hutchinson Cancer Research Center | Cyclin a1-targeted t-cell immunotherapy for cancer |
| US20130149337A1 (en) | 2003-03-11 | 2013-06-13 | City Of Hope | Method of controlling administration of cancer antigen |
| US8479118B2 (en) | 2007-12-10 | 2013-07-02 | Microsoft Corporation | Switching search providers within a browser search box |
| WO2013123061A1 (en) | 2012-02-13 | 2013-08-22 | Seattle Children's Hospital D/B/A Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| WO2013126726A1 (en) | 2012-02-22 | 2013-08-29 | The Trustees Of The University Of Pennsylvania | Double transgenic t cells comprising a car and a tcr and their methods of use |
| US20130287748A1 (en) | 2010-12-09 | 2013-10-31 | The Trustees Of The University Of Pennsylvania | Use of Chimeric Antigen Receptor-Modified T-Cells to Treat Cancer |
| WO2013166321A1 (en) | 2012-05-03 | 2013-11-07 | Fred Hutchinson Cancer Research Center | Enhanced affinity t cell receptors and methods for making the same |
| WO2014031687A1 (en) | 2012-08-20 | 2014-02-27 | Jensen, Michael | Method and compositions for cellular immunotherapy |
| WO2014055668A1 (en) | 2012-10-02 | 2014-04-10 | Memorial Sloan-Kettering Cancer Center | Compositions and methods for immunotherapy |
| WO2015070083A1 (en) | 2013-11-07 | 2015-05-14 | Editas Medicine,Inc. | CRISPR-RELATED METHODS AND COMPOSITIONS WITH GOVERNING gRNAS |
| US9108442B2 (en) | 2013-08-20 | 2015-08-18 | Ricoh Company, Ltd. | Image forming apparatus |
| WO2015138510A1 (en) | 2014-03-10 | 2015-09-17 | Editas Medicine., Inc. | Crispr/cas-related methods and compositions for treating leber's congenital amaurosis 10 (lca10) |
| WO2016073990A2 (en) | 2014-11-07 | 2016-05-12 | Editas Medicine, Inc. | Methods for improving crispr/cas-mediated genome-editing |
| US9405601B2 (en) | 2012-12-20 | 2016-08-02 | Mitsubishi Electric Corporation | In-vehicle apparatus and program |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014204723A1 (en) * | 2013-06-17 | 2014-12-24 | The Broad Institute Inc. | Oncogenic models based on delivery and use of the crispr-cas systems, vectors and compositions |
| WO2015048577A2 (en) * | 2013-09-27 | 2015-04-02 | Editas Medicine, Inc. | Crispr-related methods and compositions |
| EP3020813A1 (en) * | 2014-11-16 | 2016-05-18 | Neurovision Pharma GmbH | Antisense-oligonucleotides as inhibitors of TGF-R signaling |
| EP3328399B1 (en) * | 2015-07-31 | 2023-12-27 | Regents of the University of Minnesota | Modified cells and methods of therapy |
| CA2997912A1 (en) * | 2015-09-09 | 2017-03-16 | Seattle Children's Hospital (dba Seattle Children's Research Institute) | Genetic engineering of macrophages for immunotherapy |
-
2018
- 2018-11-01 EP EP18807785.3A patent/EP3704251A2/en active Pending
- 2018-11-01 US US16/758,842 patent/US20230061455A1/en active Pending
- 2018-11-01 CN CN201880084188.9A patent/CN111527209A/en active Pending
- 2018-11-01 AU AU2018358250A patent/AU2018358250A1/en active Pending
- 2018-11-01 JP JP2020543733A patent/JP2021502113A/en active Pending
- 2018-11-01 WO PCT/US2018/058635 patent/WO2019089884A2/en unknown
- 2018-11-01 BR BR112020008478-6A patent/BR112020008478A2/en unknown
- 2018-11-01 KR KR1020207015701A patent/KR20200075000A/en not_active Application Discontinuation
- 2018-11-01 CA CA3081456A patent/CA3081456A1/en active Pending
- 2018-11-01 SG SG11202003862PA patent/SG11202003862PA/en unknown
- 2018-11-01 MA MA050858A patent/MA50858A/en unknown
- 2018-11-01 MX MX2020004608A patent/MX2020004608A/en unknown
-
2020
- 2020-04-27 IL IL274299A patent/IL274299A/en unknown
-
2023
- 2023-09-11 JP JP2023146660A patent/JP2023179469A/en active Pending
Patent Citations (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4690915A (en) | 1985-08-08 | 1987-09-01 | The United States Of America As Represented By The Department Of Health And Human Services | Adoptive immunotherapy as a treatment modality in humans |
| US5087616A (en) | 1986-08-07 | 1992-02-11 | Battelle Memorial Institute | Cytotoxic drug conjugates and their delivery to tumor cells |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| US6040177A (en) | 1994-08-31 | 2000-03-21 | Fred Hutchinson Cancer Research Center | High efficiency transduction of T lymphocytes using rapid expansion methods ("REM") |
| WO1996013593A2 (en) | 1994-10-26 | 1996-05-09 | Procept, Inc. | Soluble single chain t cell receptors |
| WO1996018105A1 (en) | 1994-12-06 | 1996-06-13 | The President And Fellows Of Harvard College | Single chain t-cell receptor |
| US6207453B1 (en) | 1996-03-06 | 2001-03-27 | Medigene Ag | Recombinant AAV vector-based transduction system and use of same |
| US6451995B1 (en) | 1996-03-20 | 2002-09-17 | Sloan-Kettering Institute For Cancer Research | Single chain FV polynucleotide or peptide constructs of anti-ganglioside GD2 antibodies, cells expressing same and related methods |
| WO1999018129A1 (en) | 1997-10-02 | 1999-04-15 | Sunol Molecular Corporation | Soluble single-chain t-cell receptor proteins |
| WO1999060120A2 (en) | 1998-05-19 | 1999-11-25 | Avidex Limited | Soluble t cell receptor |
| WO2000014257A1 (en) | 1998-09-04 | 2000-03-16 | Sloan-Kettering Institute For Cancer Research | Fusion receptors specific for prostate-specific membrane antigen and uses thereof |
| US6410319B1 (en) | 1998-10-20 | 2002-06-25 | City Of Hope | CD20-specific redirected T cells and their use in cellular immunotherapy of CD20+ malignancies |
| US20020131960A1 (en) | 2000-06-02 | 2002-09-19 | Michel Sadelain | Artificial antigen presenting cells and methods of use thereof |
| US7446179B2 (en) | 2000-11-07 | 2008-11-04 | City Of Hope | CD19-specific chimeric T cell receptor |
| US7446191B2 (en) | 2001-04-11 | 2008-11-04 | City Of Hope | DNA construct encoding CE7-specific chimeric T cell receptor |
| US7070995B2 (en) | 2001-04-11 | 2006-07-04 | City Of Hope | CE7-specific redirected immune cells |
| US7265209B2 (en) | 2001-04-11 | 2007-09-04 | City Of Hope | CE7-specific chimeric T cell receptor |
| US7354762B2 (en) | 2001-04-11 | 2008-04-08 | City Of Hope | Method for producing CE7-specific redirected immune cells |
| US8324353B2 (en) | 2001-04-30 | 2012-12-04 | City Of Hope | Chimeric immunoreceptor useful in treating human gliomas |
| US20030170238A1 (en) | 2002-03-07 | 2003-09-11 | Gruenberg Micheal L. | Re-activated T-cells for adoptive immunotherapy |
| US7446190B2 (en) | 2002-05-28 | 2008-11-04 | Sloan-Kettering Institute For Cancer Research | Nucleic acids encoding chimeric T cell receptors |
| WO2004033685A1 (en) | 2002-10-09 | 2004-04-22 | Avidex Ltd | Single chain recombinant t cell receptors |
| US7569664B2 (en) | 2002-10-09 | 2009-08-04 | Immunocore Limited | Single chain recombinant T cell receptors |
| US20130149337A1 (en) | 2003-03-11 | 2013-06-13 | City Of Hope | Method of controlling administration of cancer antigen |
| WO2006000830A2 (en) | 2004-06-29 | 2006-01-05 | Avidex Ltd | Cells expressing a modified t cell receptor |
| WO2006037960A2 (en) | 2004-10-01 | 2006-04-13 | Avidex Ltd. | T-cell receptors containing a non-native disulfide interchain bond linked to therapeutic agents |
| US8389282B2 (en) | 2007-03-30 | 2013-03-05 | Memorial Sloan-Kettering Cancer Center | Constitutive expression of costimulatory ligands on adoptively transferred T lymphocytes |
| EP2537416A1 (en) | 2007-03-30 | 2012-12-26 | Memorial Sloan-Kettering Cancer Center | Constitutive expression of costimulatory ligands on adoptively transferred T lymphocytes |
| US8479118B2 (en) | 2007-12-10 | 2013-07-02 | Microsoft Corporation | Switching search providers within a browser search box |
| US8339645B2 (en) | 2008-05-27 | 2012-12-25 | Canon Kabushiki Kaisha | Managing apparatus, image processing apparatus, and processing method for the same, wherein a first user stores a temporary object having attribute information specified but not partial-area data, at a later time an object is received from a second user that includes both partial-area data and attribute information, the storage unit is searched for the temporary object that matches attribute information of the received object, and the first user is notified in response to a match |
| WO2011044186A1 (en) | 2009-10-06 | 2011-04-14 | The Board Of Trustees Of The University Of Illinois | Human single-chain t cell receptors |
| US8911993B2 (en) | 2010-12-09 | 2014-12-16 | The Trustees Of The University Of Pennsylvania | Compositions for treatment of cancer |
| US20130287748A1 (en) | 2010-12-09 | 2013-10-31 | The Trustees Of The University Of Pennsylvania | Use of Chimeric Antigen Receptor-Modified T-Cells to Treat Cancer |
| WO2012129514A1 (en) | 2011-03-23 | 2012-09-27 | Fred Hutchinson Cancer Research Center | Method and compositions for cellular immunotherapy |
| US8398282B2 (en) | 2011-05-12 | 2013-03-19 | Delphi Technologies, Inc. | Vehicle front lighting assembly and systems having a variable tint electrowetting element |
| WO2013071154A1 (en) | 2011-11-11 | 2013-05-16 | Fred Hutchinson Cancer Research Center | Cyclin a1-targeted t-cell immunotherapy for cancer |
| WO2013123061A1 (en) | 2012-02-13 | 2013-08-22 | Seattle Children's Hospital D/B/A Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| WO2013126726A1 (en) | 2012-02-22 | 2013-08-29 | The Trustees Of The University Of Pennsylvania | Double transgenic t cells comprising a car and a tcr and their methods of use |
| WO2013166321A1 (en) | 2012-05-03 | 2013-11-07 | Fred Hutchinson Cancer Research Center | Enhanced affinity t cell receptors and methods for making the same |
| WO2014031687A1 (en) | 2012-08-20 | 2014-02-27 | Jensen, Michael | Method and compositions for cellular immunotherapy |
| WO2014055668A1 (en) | 2012-10-02 | 2014-04-10 | Memorial Sloan-Kettering Cancer Center | Compositions and methods for immunotherapy |
| US9405601B2 (en) | 2012-12-20 | 2016-08-02 | Mitsubishi Electric Corporation | In-vehicle apparatus and program |
| US9108442B2 (en) | 2013-08-20 | 2015-08-18 | Ricoh Company, Ltd. | Image forming apparatus |
| WO2015070083A1 (en) | 2013-11-07 | 2015-05-14 | Editas Medicine,Inc. | CRISPR-RELATED METHODS AND COMPOSITIONS WITH GOVERNING gRNAS |
| WO2015138510A1 (en) | 2014-03-10 | 2015-09-17 | Editas Medicine., Inc. | Crispr/cas-related methods and compositions for treating leber's congenital amaurosis 10 (lca10) |
| WO2016073990A2 (en) | 2014-11-07 | 2016-05-12 | Editas Medicine, Inc. | Methods for improving crispr/cas-mediated genome-editing |
Non-Patent Citations (101)
| Title |
|---|
| "Cell Behavior In Vitro and In Vivo", vol. 2, HUMANA PRESS INC., article "Methods in Molecular Medicine, vol. 58: Metastasis Research Protocols", pages: 17 - 25 |
| "Current Protocols in Molecular Biology", JOHN WILEY & SONS |
| "Remington: The Science and Practice of Pharmacy", 1 May 2005, LIPPINCOTT WILLIAMS & WILKINS |
| "Remington's Pharmaceutical Sciences", 1980 |
| "The T Cell Factsbook", 2001, ACADEMIC PRESS |
| ALONSO-CAMINO ET AL., MOL THER NUCL ACIDS, vol. 2, 2013, pages e93 |
| AREF ET AL., HEMATOLOGICAL ONCOLOGY, vol. 35, 2017, pages 51 - 57 |
| BAE ET AL., BIOINFORMATICS, vol. 30, no. 10, 2014, pages 1473 - 5 |
| BAUM ET AL., MOLECULAR THERAPY: THE JOURNAL OF THE AMERICAN SOCIETY OF GENE THERAPY, vol. 13, 2006, pages 1050 - 1063 |
| BORIS-LAWRIE; TEMIN, CUR. OPIN. GENET. DEVELOP., vol. 3, 1993, pages 102 - 109 |
| BRASH ET AL., MOL. CELL BIOL., vol. 7, 1987, pages 2031 - 2034 |
| BRENTJENS ET AL., SCI TRANSL MED., vol. 5, no. 177, 2013 |
| BRINER ET AL., MOLECULAR CELL, vol. 56, no. 2, 23 October 2014 (2014-10-23), pages 333 - 339 |
| BRUNS ET AL., BLOOD, vol. 120, 2012, pages 2620 - 2630 |
| BURNS ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 8033 - 8037 |
| CARLENS ET AL., EXP HEMATOL, vol. 28, no. 10, 2000, pages 1137 - 46 |
| CAVALIERI ET AL., BLOOD, vol. 102, no. 2, 2003, pages 497 - 505 |
| CHEN ET AL., J. EXPERIMENTAL MED., vol. 198, no. 12, 15 December 2003 (2003-12-15), pages 1875 - 1886 |
| CHERVIN ET AL., J IMMUNOL METHODS, vol. 339, 2008, pages 175 - 84 |
| CHICAYBAM ET AL., PLOS ONE, vol. 8, no. 3, 2013, pages e60298 |
| CHOTHIA ET AL., EMBO J., vol. 7, 1988, pages 3745 |
| COHEN ET AL., J IMMUNOL., vol. 175, 2005, pages 5799 - 5808 |
| COOPER ET AL., BLOOD, vol. 101, 2003, pages 1637 - 1644 |
| CORNISH-BOWDEN A, NUCLEIC ACIDS RES., vol. 13, no. 9, 10 May 1985 (1985-05-10), pages 3021 - 30 |
| DAVILA ET AL., PLOS ONE, vol. 8, no. 4, 2013, pages e61338 |
| DAVIS; MAIZELS, PNAS, vol. 111, no. 10, 11 March 2014 (2014-03-11), pages E924 - 932 |
| FEDOROV ET AL., SCI. TRANSL. MEDICINE, vol. 5, December 2013 (2013-12-01), pages 215 |
| FINE ET AL., SCI REP., vol. 5, 1 July 2015 (2015-07-01), pages 10777 |
| FRECHA ET AL., MOLECULAR THERAPY: THE JOURNAL OF THE AMERICAN SOCIETY OF GENE THERAPY, vol. 18, 2010, pages 1748 - 1757 |
| FRIT ET AL., DNA REPAIR, vol. 17, 2014, pages 81 - 97 |
| FU ET AL., NAT BIOTECHNOL, vol. 32, no. 3, 2014, pages 279 - 84 |
| GARGETT ET AL., MOL. THER., vol. 24, 2016, pages 1135 - 1149 |
| GUILINGER ET AL., NATURE BIOTECHNOLOGY, vol. 32, 2014, pages 577 - 582 |
| HACKETT ET AL., MOLECULAR THERAPY: THE JOURNAL OF THE AMERICAN SOCIETY OF GENE THERAPY, vol. 18, 2010, pages 674 - 683 |
| HEIGWER ET AL., NAT METHODS, vol. 11, no. 2, 2014, pages 122 - 3 |
| HERMAN ET AL., J. IMMUNOLOGICAL METHODS, vol. 285, no. 1, 2004, pages 25 - 40 |
| HOLLER ET AL., NAT IMMUNOL, vol. 4, 2003, pages 55 - 62 |
| HOLLER ET AL., PROC NATL ACAD SCI U S A, vol. 97, 2000, pages 5387 - 92 |
| HSU ET AL., NAT BIOTECHNOL., vol. 31, no. 9, September 2013 (2013-09-01), pages 827 - 832 |
| HUANG ET AL., METHODS MOL BIOL, vol. 506, 2009, pages 115 - 126 |
| HUDECEK ET AL., CLIN. CANCER RES., vol. 19, 2013, pages 3153 |
| IYAMA; WILSON III, DNA REPAIR (AMST., vol. 12, no. 8, August 2013 (2013-08-01), pages 620 - 636 |
| JANEWAY ET AL.: "Immunobiology: The Immune System in Health and Disease", 1997, CURRENT BIOLOGY PUBLICATIONS, pages: 4,33 |
| JIANG ET AL., NAT BIOTECHNOL., vol. 31, no. 3, March 2013 (2013-03-01), pages 233 - 239 |
| JINEK ET AL., SCIENCE, vol. 337, no. 6096, 17 August 2012 (2012-08-17), pages 816 - 821 |
| JOHNSON, L. A. ET AL., BLOOD, vol. 114, 2009, pages 535 - 546 |
| JOHNSTON, NATURE, vol. 346, 1990, pages 776 - 777 |
| JORES ET AL., PROC. NAT'L ACAD. SCI. U.S.A., vol. 87, 1990, pages 9138 |
| KLEBANOFF ET AL., J IMMUNOTHER, vol. 35, no. 9, 2012, pages 651 - 660 |
| KLEINSTIVER ET AL., NAT BIOTECHNOL., vol. 33, no. 12, December 2015 (2015-12-01), pages 1293 - 1298 |
| KLEINSTIVER ET AL., NATURE, vol. 523, no. 7561, 23 July 2015 (2015-07-23), pages 481 - 5 |
| KOCHENDERFER ET AL., J. IMMUNOTHERAPY, vol. 32, no. 7, 2009, pages 689 - 702 |
| KOCHENDERFER ET AL., NATURE REVIEWS CLINICAL ONCOLOGY, vol. 10, 2013, pages 267 - 276 |
| KOCHENDERFER, J. N. ET AL., BLOOD, vol. 119, 2012, pages 2709 - 2720 |
| KOMOR ET AL., NATURE, vol. 533, 19 May 2016 (2016-05-19), pages 420 - 424 |
| KOSTE ET AL., GENE THERAPY, 3 April 2014 (2014-04-03) |
| KOTB, CLINICAL MICROBIOLOGY REVIEWS, vol. 8, 1995, pages 411 - 426 |
| LAMERS, C. H. ET AL., MOL. THER., vol. 21, 2013, pages 904 - 912 |
| LEFRANC ET AL., DEV. COMP. IMMUNOL., vol. 27, 2003, pages 55 |
| LEFRANC ET AL., DEVELOPMENTAL AND COMPARATIVE IMMUNOLOGY, vol. 2, 2003, pages 55 - 77 |
| LI ET AL., NAT BIOTECHNOL, vol. 23, 2005, pages 349 - 54 |
| LI, NAT BIOTECHNOL., vol. 23, 2005, pages 349 - 354 |
| LUPTON S. D. ET AL., MOL. AND CELL BIOL., vol. 11, 1991, pages 6 |
| MAKAROVA ET AL., NAT REV MICROBIOL, vol. 9, no. 6, June 2011 (2011-06-01), pages 467 - 477 |
| MALI ET AL., SCIENCE, vol. 339, no. 6121, 15 February 2013 (2013-02-15), pages 823 - 826 |
| MANURI ET AL., HUM GENE THER, vol. 21, no. 4, 2010, pages 427 - 437 |
| MILLER, A. D., HUMAN GENE THERAPY, vol. 1, 1990, pages 5 - 14 |
| MILLER; ROSMAN, BIOTECHNIQUES, vol. 7, 1989, pages 980 - 990 |
| MULLEN ET AL., PROC. NATL. ACAD. SCI. USA., vol. 89, 1992, pages 33 |
| NATURE, vol. 529, 28 January 2016 (2016-01-28), pages 490 - 495 |
| NISHIMASU ET AL., CELL, vol. 156, 27 February 2014 (2014-02-27), pages 935 - 949 |
| NISHIMASU ET AL., CELL, vol. 162, 27 August 2015 (2015-08-27), pages 1113 - 1126 |
| PARK ET AL., TRENDS BIOTECHNOL, vol. 29, no. 11, November 2011 (2011-11-01), pages 550 - 557 |
| PARKHURST ET AL., CLIN CANCER RES., vol. 15, 2009, pages 169 - 180 |
| RAN; HSU ET AL., CELL, vol. 154, no. 6, 12 September 2013 (2013-09-12), pages 1380 - 1389 |
| RICHARDSON ET AL., NATURE BIOTECHNOLOGY, vol. 34, 2016, pages 339 - 344 |
| RIDDELL ET AL., HUMAN GENE THERAPY, vol. 3, 1992, pages 319 - 338 |
| ROSENBERG, NAT REV CLIN ONCOL, vol. 8, no. 10, 2011, pages 577 - 85 |
| SADELAIN ET AL., CANCER DISCOV., vol. 3, no. 4, April 2013 (2013-04-01), pages 388 - 398 |
| SCARPA ET AL., VIROLOGY, vol. 180, 1991, pages 849 - 852 |
| SCHLUETER, C. J. ET AL., J. MOL. BIOL., vol. 256, 1996, pages 859 |
| SHARMA ET AL., MOLEC THER NUCL ACIDS, vol. 2, 2013, pages e74 |
| SHMAKOV ET AL., MOLECULAR CELL, vol. 60, 5 November 2015 (2015-11-05), pages 385 - 397 |
| TERAKURA ET AL., BLOOD, vol. 1, 2012, pages 72 - 82 |
| THEMELI ET AL., NAT BIOTECHNOL., vol. 31, no. 10, 2013, pages 928 - 933 |
| TIEMESSEN ET AL., INT. IMMUNOL., vol. 15, 2003, pages 1495 - 1504 |
| TSUKAHARA ET AL., BIOCHEM BIOPHYS RES COMMUN, vol. 438, no. 1, 2013, pages 84 - 9 |
| TURTLE ET AL., CURR. OPIN. IMMUNOL., vol. 24, no. 5, October 2012 (2012-10-01), pages 633 - 39 |
| VAN TEDELOO ET AL., GENE THERAPY, vol. 7, no. 16, 2000, pages 1431 - 1437 |
| VARELA-ROHENA ET AL., NAT MED., vol. 14, 2008, pages 1390 - 1395 |
| VERHOEYEN ET AL., METHODS MOL BIOL., vol. 506, 2009, pages 97 - 114 |
| WADWA ET AL., J. DRUG TARGETING, vol. 3, 1995, pages 1 1 1 |
| WANG ET AL., J IMMUNOTHER, vol. 35, no. 9, 2012, pages 689 - 701 |
| WANG ET AL., J. IMMUNOTHER., vol. 35, no. 9, 2012, pages 689 - 701 |
| WARREN, R. S. ET AL., CANCER GENE THER., vol. 5, 1998, pages S1 - S2 |
| WIGLER ET AL., CELL II, 1977, pages 223 |
| WU ET AL., CANCER, vol. 18, no. 2, March 2012 (2012-03-01), pages 160 - 75 |
| XIAO A ET AL., BIOINFORMATICS, vol. 30, no. 8, 2014, pages 1180 - 1182 |
| YAMANO ET AL., CELL, vol. 165, no. 4, 5 May 2016 (2016-05-05), pages 949 - 962 |
| ZETSCHE ET AL., CELL, vol. 163, 22 October 2015 (2015-10-22), pages 759 - 771 |
| ZETSCHE ET AL., NAT BIOTECHNOL., vol. 33, no. 2, February 2015 (2015-02-01), pages 139 - 42 |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3854877A4 (en) * | 2018-09-17 | 2022-10-12 | Institute Of Zoology, Chinese Academy Of Sciences | Modified t cell, preparation method therefor and use thereof |
| WO2020223535A1 (en) * | 2019-05-01 | 2020-11-05 | Juno Therapeutics, Inc. | Cells expressing a recombinant receptor from a modified tgfbr2 locus, related polynucleotides and methods |
| US12024711B2 (en) | 2019-05-29 | 2024-07-02 | Monsanto Technology Llc | Methods and compositions for generating dominant short stature alleles using genome editing |
| WO2020243368A1 (en) * | 2019-05-29 | 2020-12-03 | Monsanto Technology Llc | Methods and compositions for generating dominant alleles using genome editing |
| EP3980450A4 (en) * | 2019-06-04 | 2024-06-19 | Nkarta, Inc. | Combinations of engineered natural killer cells and engineered t cells for immunotherapy |
| WO2021011864A1 (en) * | 2019-07-18 | 2021-01-21 | Memorial Sloan Kettering Cancer Center | METHODS AND COMPOSITIONS FOR TARGETING TGF-β SIGNALING IN CD4+ HELPER T CELLS FOR CANCER IMMUNOTHERAPY |
| EP3999527A4 (en) * | 2019-07-18 | 2024-04-17 | Memorial Sloan Kettering Cancer Center | Methods and compositions for targeting tgf-beta signaling in cd4+ helper t cells for cancer immunotherapy |
| WO2021097521A1 (en) * | 2019-11-20 | 2021-05-27 | Cartherics Pty. Ltd. | Method for providing immune cells with enhanced function |
| CN114729315A (en) * | 2019-11-20 | 2022-07-08 | 卡瑟里克斯私人有限公司 | Method for providing immune cells with enhanced function |
| WO2021127261A1 (en) * | 2019-12-17 | 2021-06-24 | The General Hospital Corporation | Engineered immune cells with reduced toxicity and uses thereof |
| WO2021127594A1 (en) * | 2019-12-18 | 2021-06-24 | Editas Medicine, Inc. | Engineered cells for therapy |
| EP4076479A4 (en) * | 2019-12-18 | 2024-05-22 | Editas Medicine, Inc. | Engineered cells for therapy |
| WO2021182917A1 (en) | 2020-03-12 | 2021-09-16 | 기초과학연구원 | Composition for inducing apoptosis of cells having genomic sequence variation and method for inducing apoptosis of cells by using composition |
| US11497773B2 (en) * | 2020-09-23 | 2022-11-15 | Crispr Therapeutics Ag | Genetically engineered t cells with regnase-1 and/or TGFBRII disruption have improved functionality and persistence |
| US20230263828A1 (en) * | 2020-09-23 | 2023-08-24 | Crispr Therapeutics Ag | Genetically engineered t cells with regnase-1 and/or tgfbrii disruption have improved functionality and persistence |
| US11857574B2 (en) | 2020-09-23 | 2024-01-02 | Crispr Therapeutics Ag | Genetically engineered T cells with Regnase-1 and/or TGFBRII disruption have improved functionality and persistence |
| US11679130B2 (en) | 2020-09-23 | 2023-06-20 | Crispr Therapeutics Ag | Genetically engineered t cells with Regnase-1 and/or TGFBRII disruption have improved functionality and persistence |
| US11679131B2 (en) | 2020-09-23 | 2023-06-20 | Crispr Therapeutics Ag | Genetically engineered T cells with regnase-1 and/or TGFBRII disruption have improved functionality and persistence |
| WO2022064428A1 (en) * | 2020-09-23 | 2022-03-31 | Crispr Therapeutics Ag | Genetically engineered t cells with regnase-1 and/or tgfbrii disruption have improved functionality and persistence |
| EP4397321A3 (en) * | 2020-09-23 | 2024-09-04 | CRISPR Therapeutics AG | Genetically engineered t cells with regnase-1 and/or tgfbrii disruption have improved functionality and persistence |
| WO2023081900A1 (en) | 2021-11-08 | 2023-05-11 | Juno Therapeutics, Inc. | Engineered t cells expressing a recombinant t cell receptor (tcr) and related systems and methods |
| WO2023147428A3 (en) * | 2022-01-26 | 2023-12-07 | Orthobio Therapeutics, Inc. | Gene editing to improve joint function |
| WO2023180967A1 (en) * | 2022-03-23 | 2023-09-28 | Crispr Therapeutics Ag | Anti-cd83 car-t cells with regnase-1 and/or tgfbrii disruption |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112020008478A2 (en) | 2020-10-20 |
| WO2019089884A3 (en) | 2019-06-13 |
| EP3704251A2 (en) | 2020-09-09 |
| KR20200075000A (en) | 2020-06-25 |
| MX2020004608A (en) | 2020-10-05 |
| CN111527209A (en) | 2020-08-11 |
| US20230061455A1 (en) | 2023-03-02 |
| AU2018358250A1 (en) | 2020-05-14 |
| JP2021502113A (en) | 2021-01-28 |
| IL274299A (en) | 2020-06-30 |
| JP2023179469A (en) | 2023-12-19 |
| SG11202003862PA (en) | 2020-05-28 |
| MA50858A (en) | 2020-09-09 |
| RU2020117752A (en) | 2021-12-01 |
| CA3081456A1 (en) | 2019-05-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230061455A1 (en) | Methods, compositions and components for crispr-cas9 editing of tgfbr2 in t cells for immunotherapy | |
| CN112204148B (en) | Modified immune cells with enhanced function and screening method thereof | |
| US20210139583A1 (en) | Production of engineered t-cells by sleeping beauty transposon coupled with methotrexate selection | |
| US11608500B2 (en) | Gene-regulating compositions and methods for improved immunotherapy | |
| CN109843915B (en) | Genetically engineered cell and preparation method thereof | |
| US20230137729A1 (en) | Methods, compositions and components for crispr-cas9 editing of cblb in t cells for immunotherapy | |
| CN112040987A (en) | Gene regulatory compositions and methods for improved immunotherapy | |
| US11786550B2 (en) | gRNA targeting HPK1 and a method for editing HPK1 gene | |
| WO2020163365A2 (en) | Combination gene targets for improved immunotherapy | |
| CN117597358A (en) | Compositions and methods for treating HER2 positive cancers | |
| KR20230090367A (en) | Cells Expressing Chimeric Receptors from Modified Invariant CD3 Immunoglobulin Superfamily Chain Loci and Related Polynucleotides and Methods | |
| RU2798380C2 (en) | Methods, compositions and components for editing tgfbr2 by crispr-cas9 in t cells for immunotherapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 3081456 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2020543733 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018358250 Country of ref document: AU Date of ref document: 20181101 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20207015701 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018807785 Country of ref document: EP Effective date: 20200602 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020008478 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112020008478 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200428 |